JP2008179556A - 4a,9a−架橋−ヘキサヒドロ−1H−インデノピリジン誘導体、その医薬用途、およびその製造方法 - Google Patents
4a,9a−架橋−ヘキサヒドロ−1H−インデノピリジン誘導体、その医薬用途、およびその製造方法 Download PDFInfo
- Publication number
- JP2008179556A JP2008179556A JP2007013599A JP2007013599A JP2008179556A JP 2008179556 A JP2008179556 A JP 2008179556A JP 2007013599 A JP2007013599 A JP 2007013599A JP 2007013599 A JP2007013599 A JP 2007013599A JP 2008179556 A JP2008179556 A JP 2008179556A
- Authority
- JP
- Japan
- Prior art keywords
- carbons
- carbon atoms
- hexahydro
- bridged
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 COc1ccc(C[C@](*)(CCC2)[C@]3(CCNCC4CC4)CC22OCCO2)c3c1 Chemical compound COc1ccc(C[C@](*)(CCC2)[C@]3(CCNCC4CC4)CC22OCCO2)c3c1 0.000 description 2
Images
Landscapes
- Other In-Based Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
【課題】オピオイド受容体に関連する様々な疾患または症状、例えば疼痛の予防または治療に有用な医薬を提供すること。
【解決手段】下記化合物[2-メチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オール]に代表される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体及びその薬理学的に許容される酸付加塩、該化合物を有効成分として含有する医薬、並びにその製造方法を提供した。
【選択図】図1
【解決手段】下記化合物[2-メチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オール]に代表される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体及びその薬理学的に許容される酸付加塩、該化合物を有効成分として含有する医薬、並びにその製造方法を提供した。
【選択図】図1
Description
本発明は、新規な4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体、医薬としてのその使用、およびその製造方法に関する。
古くから鎮痛薬として用いられてきたモルヒネなどが結合する膜タンパクとして、主に三種(μ、δ、κ)のタイプのオピオイド受容体が同定された。オピオイド受容体は鎮痛作用を含む様々な薬理作用に関与していることが報告されており、その受容体のリガンド(アゴニスト、アンタゴニスト)は、疼痛を含む様々な疾患、症状に対する治療薬として利用しうる。
モルヒネが単離構造決定されて以来、より有用なオピオイド医薬の創出を目指して、以下の様に多種多彩な誘導体が合成されてきた(非特許文献1)。
・ 4,5-エポキシモルヒナン類(モルヒネと共通の骨格を有する化合物群)
・ モルヒナン類
・ ベンゾモルファン類
・ アリールモルファン類
・ ペチジン、およびそれに関連する4-フェニルピペリジン類
・ ピペリジン類
・ フェンタニル、およびそれに関連する4-アニリノピペリジン類
・ メサドン類
・ ベンズイミダゾール類
・ テトラヒドロイソキノリン類
・ シクロヘキサン類
・ アミノテトラリン類
・ 4-ピペリジノール類
・ ピペラジン類
・ 4,5-エポキシモルヒナン類(モルヒネと共通の骨格を有する化合物群)
・ モルヒナン類
・ ベンゾモルファン類
・ アリールモルファン類
・ ペチジン、およびそれに関連する4-フェニルピペリジン類
・ ピペリジン類
・ フェンタニル、およびそれに関連する4-アニリノピペリジン類
・ メサドン類
・ ベンズイミダゾール類
・ テトラヒドロイソキノリン類
・ シクロヘキサン類
・ アミノテトラリン類
・ 4-ピペリジノール類
・ ピペラジン類
しかしながら、本願になる4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体はこれらの公知化合物とは構造が全く異なっており、新たに見出された転移反応を介し、新規に得られた該化合物がオピオイド受容体結合性、また鎮痛活性を有し、鎮痛薬をはじめとする有用な医薬となりうることを、これらの公知例はなんら示唆するものではない。
Casy A.F.et.al., Opioid Analgesics, Plenum Press, New York, 1986.
本発明は、オピオイド受容体に関連する様々な疾患、症状、例えば疼痛などの予防または治療に有用である新規な誘導体またはその薬理学的に許容される酸付加塩、該化合物を有効成分として含有する医薬、およびその製造方法を提供することを目的とする。
上記目的を達成するため鋭意検討した結果、新たに見出された転移反応を介して得られた4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体が、予想外にもオピオイド受容体への結合性、および鎮痛活性を有することを見出し、本発明を完成した。
すなわち本発明は、一般式(I)
[式中R1は、水素、炭素数1から5のアルキル、炭素数4から7のシクロアルキルアルキル、炭素数6から8のシクロアルケニルアルキル、炭素数6から12のアリール、炭素数7から13のアラルキル、炭素数3から7のアルケニル、フラニルアルキル(アルキル部の炭素数は1から5)、チエニルアルキル(アルキル部の炭素数は1から5)、またはピリジルアルキル(アルキル部の炭素数は1から5)を表し;
R2、R3は、それぞれ別個に水素、ヒドロキシ、炭素数1から5のアルコキシ、炭素数3から7のアルケニルオキシ、炭素数7から13のアラルキルオキシ、または炭素数1から5のアルカノイルオキシを表す。一般式(I)は(+)体、(−)体、(±)体を含む。]で示される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩を提供する。また、本発明は、上記本発明の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体誘導体又はその薬理学的に許容される酸付加塩を有効成分として含有する医薬を提供する。さらに、本発明は、一般式(II)
R2、R3は、それぞれ別個に水素、ヒドロキシ、炭素数1から5のアルコキシ、炭素数3から7のアルケニルオキシ、炭素数7から13のアラルキルオキシ、または炭素数1から5のアルカノイルオキシを表す。一般式(I)は(+)体、(−)体、(±)体を含む。]で示される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩を提供する。また、本発明は、上記本発明の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体誘導体又はその薬理学的に許容される酸付加塩を有効成分として含有する医薬を提供する。さらに、本発明は、一般式(II)
[式中R1、R2、R3は前記一般式(I)中のR1、R2、R3の定義にそれぞれ同じ、Rは炭素数1から5のアルキル、または両者が結合してエチレンまたはプロピレンを表す]で示される化合物を、塩基の共存下、脱ヒドロキシ反応剤と反応させることで、一般式(III)
[式中R1、R2、R3、Rは前記定義に同じ]
で示される転移生成物を得た後、還元、脱保護することを特徴とする一般式(I)で示される上記本発明の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体の製造方法を提供する。
で示される転移生成物を得た後、還元、脱保護することを特徴とする一般式(I)で示される上記本発明の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体の製造方法を提供する。
本発明により新規化合物及びその製造方法が提供された。本発明の新規化合物及びその薬理学的に許容される酸付加塩は、オピオイド受容体に関連する様々な疾患、症状、例えば疼痛などの治療または予防効果を有する。
上記の通り、本発明は、前記一般式(I)で表される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩、該化合物を有効成分として含有する医薬、およびその製造方法を提供する。一般式(I)としては、(+)体、(−)体及び(±)体が含まれる。
一般式(I)で表される化合物の中で、R1としては、水素、炭素数1から5のアルキル、炭素数4から7のシクロアルキルアルキル、炭素数6から8のシクロアルケニルアルキル、または炭素数3から7のアルケニルが好ましく、中でも水素、メチル、シクロプロピルメチル、シクロブチルメチル、またはアリルが好ましい。
R2、R3としては、それぞれ別個に水素、ヒドロキシ、炭素数1から5のアルコキシ、または炭素数1から5のアルカノイルオキシが好ましく、中でも水素、ヒドロキシ、メトキシ、またはアセトキシが好ましい。
薬理学的に好ましい酸付加塩としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩、酢酸塩、乳酸塩、クエン酸塩、シュウ酸塩、グルタル酸塩、リンゴ酸塩、酒石酸塩、フマル酸塩、マンデル酸塩、マレイン酸塩、安息香酸塩、フタル酸塩等の有機カルボン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩、カンファ−スルホン酸塩等の有機スルホン酸塩等があげられ、中でも、塩酸塩、臭化水素酸塩、リン酸塩、酒石酸塩、メタンスルホン酸塩等が好ましく用いられるが、これらに限られるものではない。
本発明になる一般式(I)の化合物の具体例を表1に示す。表中CPMはシクロプロピルメチル基を表す。
また一般式(I)の化合物のうち、R1がメチル、R2が水素、R3がヒドロキシである下記の化合物を、
2-メチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オールと命名する。
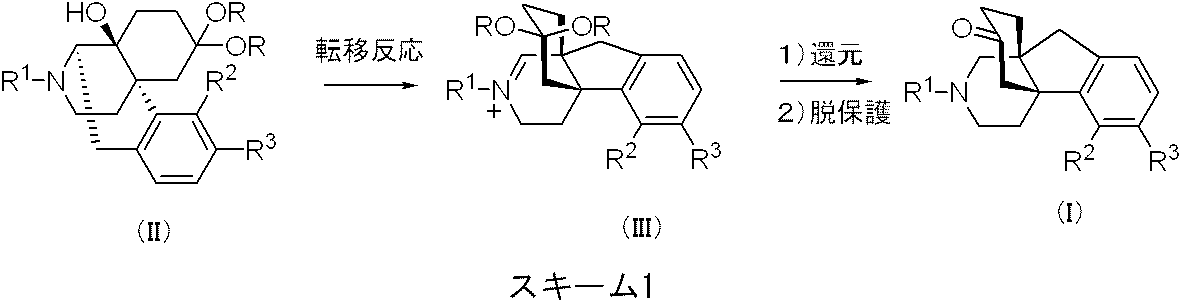
一般式(I)で示される、4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩は、具体的にはスキーム1の方法によって製造することができる。
すなわち、公知の方法(例えばCasy A.F.et.al., Opioid Analgesics, Plenum Press, New York, 1986.に記載の方法)で得られる一般式(II)[式中R1、R2、R3は上記した一般式(I)における定義に同じ、Rは炭素数1から5のアルキル、または両者が結合してエチレンまたはプロピレンを表す]で示される化合物を、脱ヒドロキシ条件、具体的には塩化チオニルなどと反応させることで、一般式(III)[式中R1、R2、R3、Rは前記定義に同じ]で示される転移生成物を得た後、還元、脱保護を行う方法である。
転移反応工程では、通常ヒドロキシ基の脱離に用いうる反応剤はすべて適用可能であるが、中でも、メタンスルホニルクロリド、塩化チオニルが好ましく用いられる。ヒドロキシ基脱離剤は1〜50当量を用いることが可能であるが、2〜20当量を用いることが好ましく、中でも5〜15当量で良好な結果が得られる。反応には塩基を共存させると好ましい結果が得られ、用いる塩基としては、水素化ナトリウム、水素化カリウムなどの水素化金属、ピリジン、ジメチルアミノピリジンなどの窒素系塩基、炭酸カリウム、炭酸ナトリウムなどの無機塩基などが好ましいが、水素化金属、特に水素化ナトリウムを用いると著しく好ましい結果が得られる。塩基は1〜100当量を用いることが可能であるが、2〜50当量を用いることが好ましく、中でも5〜30当量で良好な結果が得られる。溶媒としては、DMF、ジメチルアセトアミド、DMSOなどの非プロトン性極性溶媒、ジエチルエーテル、THF、DME、ジオキサンなどのエーテル系溶媒、ベンゼン、トルエン、キシレンなどの炭化水素系溶媒、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン系溶媒を用いることができるが、中でもTHF、DMEなどが好ましく用いられる。反応温度としては通常-20〜200 ℃の範囲で実施可能であり、好ましくは-10〜100 ℃の範囲で満足すべき結果が得られる。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、10分から30時間程度で満足すべき結果が得られる。また反応系中の基質の濃度は、特に限定されるものではないが、通常、1 mmol/L〜1 mol/L程度が好ましい。
得られたイミニウムを還元する工程では、水素化金属還元剤で還元するか、金属触媒(場合によっては酸を添加)の存在下、水素添加することで目的物を得ることができる。水素化金属還元剤を用いる場合の反応溶媒としては、メタノール、エタノール等のアルコール系溶媒、特にメタノールを用いると好ましい結果が得られる。水素化金属還元剤としては、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム、水素化ホウ素亜鉛、水素化トリアセトキシホウ素ナトリウム、水素化トリアセトキシホウ素テトラメチルアンモニウム、ボランーピリジン錯体、水素化アルミニウムリチウムなどを用いることができるが、特に水素化ホウ素ナトリウムが好ましく用いられる。水素化金属還元剤は0.5〜50当量を用いることが可能であるが、通常1〜20当量、好ましくは1〜10当量が用いられる。反応温度は、通常-40〜150 ℃、好ましくは-30〜80 ℃で満足すべき結果が得られる。反応時間は反応温度等の条件に応じて適宜選択されるが、通常5分から30時間程度で満足すべき結果が得られる。また反応系中の基質の濃度は、特に限定されるものではないが、通常、1 mmol/L〜1 mol/Lが好ましい。金属触媒(場合によっては酸を添加)の存在下水素添加する場合には、反応溶媒としてはメタノール、エタノール等のアルコール系溶媒、またはTHF、エーテル等のエーテル系溶媒、特にメタノール、エタノール等のアルコール系溶媒で好ましい結果が得られる。酸を共存させる場合には、塩酸、臭化水素酸、硫酸、リン酸等の無機酸、メタンスルホン酸、p-トルエンスルホン酸などのスルホン酸、安息香酸、酢酸、シュウ酸などのカルボン酸等、通常アミン類と塩を形成する酸は何でも用いることができるが、塩酸、硫酸、メタンスルホン酸、p-トルエンスルホン酸、安息香酸が好ましく用いられる。共存させる酸の量は特に限定されないが、0.5〜50当量の範囲で実施可能であり、通常は1〜30当量、好ましくは1〜10当量で満足すべき結果が得られる。金属触媒としては、酸化白金、水酸化パラジウム、パラジウム-炭素など、通常の水素添加反応に用いられる触媒はすべて使用可能であるが、酸化白金、パラジウム-炭素が好ましく用いられる。反応温度は-30〜80 ℃、好ましくは10〜50 ℃で、水素圧は1気圧〜100気圧、好ましくは1気圧〜30気圧で実施可能であるが、通常は室温、常圧で好ましい結果が得られる。反応時間は反応条件によって適宜選択されるが、通常10分〜30時間で満足すべき結果が得られる。また反応系中の基質の濃度は、特に限定されるものではないが、通常、1 mmol/L〜1 mol/Lが好ましい。
カルボニル部分の脱保護工程は通常の酸性加水分解条件を用いることで行うことができる。酸としては塩酸、硫酸、リン酸などの無機酸、酢酸、プロピオン酸、安息香酸などのカルボン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸などのスルホン酸を用いることができるが、塩酸が好ましく用いられる。また用いる酸の量としては基質に対して、1〜30当量、好ましくは1〜10当量が用いられる。反応溶媒としては、DMF、ジメチルアセトアミド、DMSOなどの非プロトン性極性溶媒、ジエチルエーテル、THF、DME、ジオキサンなどのエーテル系溶媒、ベンゼン、トルエン、キシレンなどの炭化水素系溶媒、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン系溶媒、水などを用いることができるが、酸の水溶液中で好ましい結果が得られる。反応温度としては0〜200 ℃の範囲で実施可能であり、好ましくは50〜120 ℃の範囲で満足すべき結果が得られる。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、5分から30時間程度で満足すべき結果が得られる。また反応系中の基質の濃度は、特に限定されるものではないが、通常、1 mmol/L〜1 mol/L程度が好ましい。
なお、得られたフリー塩基の塩化は、水、メタノールまたは種々の有機溶媒中で薬理学的に許容される酸と混合し、濃縮乾固、再沈殿、再結晶などをすることで行うことができる。
一般式(I)で示される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩がオピオイド受容体に結合することは、実施例に記載したモルモット脳ホモジネートを用いた結合アッセイ法によって、また、当該化合物が鎮痛活性を有することは、実施例に示した酢酸ライジング試験によって確認することができる。
本発明になる医薬を臨床で使用する際には、薬剤はフリーの塩基またはその塩自体でもよく、また賦形剤、安定化剤、保存剤、緩衝剤、溶解補助剤、乳化剤、希釈剤、等張化剤などの添加剤が適宜混合されていてもよい。また、当該薬剤は、これらの薬剤用担体を適宜用いて通常の方法によって製造することができる。投与形態としては、錠剤・カプセル剤・顆粒剤・散剤・シロップ剤などによる経口剤、注射剤・座剤・液剤などによる非経口剤、あるいは軟膏剤・クリーム剤・貼付剤などによる局所投与等を挙げることができる。
本発明になる医薬は上記有効成分を0.00001〜90重量%、より好ましくは0.0001〜70重量%含有することが望ましい。その使用量は症状、年齢、体重、投与方法等に応じて適宜選択されるが、成人に対する1日の有効成分量としては、注射剤の場合0.1μg〜1g、経口剤の場合1μg〜10gであり、それぞれ1回または数回に分けて投与することができる。
本発明になる医薬は、オピオイド受容体に関連する種々の疾患又は症状の予防又は治療に用いることができる。ここで、「オピオイド受容体に関連する疾患又は症状」とは、オピオイド受容体へのリガンドの結合が症状の発現に関与している疾患又は症状のことを言う。より具体的には、オピオイド受容体へのリガンドの結合により症状の発現が促進される疾患等、又は該結合により症状の発現が抑制される疾患等を言う。下記実施例に記載するとおり、本発明の化合物は、オピオイド受容体に対し選択的な結合性を有する。従って、例えば、オピオイド受容体へのリガンドの結合によって症状の発現が促進される疾患の場合、本発明の医薬を患者に投与すれば、患者体内で該リガンドと本発明の化合物とが競合することにより、本発明の化合物が受容体−リガンドの結合に拮抗的に作用することになるため、結果として疾患の症状を改善し得る。この場合には、本発明の化合物はアンタゴニストとして作用する。また、例えば、オピオイド受容体へのリガンドの結合により症状の発現が抑制される疾患の場合、本発明の医薬を患者に投与すれば、患者体内で本発明の化合物がオピオイド受容体に結合するので、それにより症状を緩和し得る。この場合、本発明の化合物はアゴニストとして作用する。そのようなオピオイド受容体に関連する疾患又は症状としては、特に限定されないが、例えば疼痛、咳嗽、掻痒、虚血性脳疾患、薬物依存、虚血性心疾患、頻尿、尿失禁、機能性腸障害などの他、WO 05/015242にオピオイドκ受容体に関連するとして、WO 05/015203にオピオイドμ受容体に関連するとして、WO 05/005478にオピオイドδ受容体に関連するとしてあげられた疾患などを挙げることができる。
以下、実施例をあげて本発明を具体的に説明する。なお、これらはあくまでも例示したものに過ぎず、いかなる意味においても限定的に解釈されるべきものではない。
実施例1
転移生成物 2(塩酸塩)の合成
転移生成物 2(塩酸塩)の合成
Casy A.F.et.al., Opioid Analgesics, Plenum Press, New York, 1986.に記載の方法により得た14-ヒドロキシモルヒナン誘導体 1(1 g, 2.59 mmol)をピリジン(20mL)に溶解し、アルゴン雰囲気下-10 ℃にて攪拌しつつ、塩化チオニル(1.9 mL, 25.9 mmol)を滴下した。そのまま氷冷下で1時間保ち、その後室温に戻して3.5時間攪拌した。
反応液に炭酸水素ナトリウム及び飽和炭酸水素ナトリウム水溶液を加えてpH 9とし、ピリジンを除去後にクロロホルムで抽出した。有機層を飽和食塩水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、濃縮して1.01 gの粗生成物を得た。得られた粗生成物をシリカゲルカラムクロマトグラフィーにて精製し、淡茶色アモルファスとして表題化合物 2(298 mg,28.4%)を得た。
IR (film) νmax 2939, 1610, 1491, cm-1
1H NMR (CDCl3, 300 MHz) δ: 0.27-0.37 (1H, m), 0.41-0.66 (2H, m), 0.96-1.08 (1H, m), 1.26-1.33 (1H, m), 1.69-1.86 (4H, m), 1.98 (1H, ddd, J = 5.0, 11.0, 14.0 Hz), 2.16 (1H, dd, J = 3.0, 15.0 Hz), 2.44 (1H, dt, J = 3.0, 14.0 Hz), 3.34-3.56 (4H, m), 3.76 (3H, s), 3.83-4.01 (6H, m), 6.58 (1H, d, J = 2.5 Hz), 6.74 (1H, dd, J = 2.5, 8.5 Hz), 7.18 (1H, d, J = 8.5 Hz), 10.30 (1H, s)
MS (FAB) m/z 368 [M-Cl-]+
1H NMR (CDCl3, 300 MHz) δ: 0.27-0.37 (1H, m), 0.41-0.66 (2H, m), 0.96-1.08 (1H, m), 1.26-1.33 (1H, m), 1.69-1.86 (4H, m), 1.98 (1H, ddd, J = 5.0, 11.0, 14.0 Hz), 2.16 (1H, dd, J = 3.0, 15.0 Hz), 2.44 (1H, dt, J = 3.0, 14.0 Hz), 3.34-3.56 (4H, m), 3.76 (3H, s), 3.83-4.01 (6H, m), 6.58 (1H, d, J = 2.5 Hz), 6.74 (1H, dd, J = 2.5, 8.5 Hz), 7.18 (1H, d, J = 8.5 Hz), 10.30 (1H, s)
MS (FAB) m/z 368 [M-Cl-]+
実施例2
還元体 3の合成
還元体 3の合成
実施例1で得られた転移生成物 2(100 mg, 0.248 mmol)をメタノール(3 mL)に溶解し、氷冷下で水素化ホウ素ナトリウム(28 mg, 0.774 mmol)を加えた。
反応液に50%酢酸を加えた後、飽和炭酸水素ナトリウム水溶液でpH 9とし、クロロホルムで抽出した。有機層を飽和食塩水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、濃縮して表題化合物 3(83.9 mg, 91.7%)を茶色油状物質として得た。
IR (film) νmax 2914, 1609, 1489 cm-1
1H NMR (CDCl3, 300 MHz) δ: 0.00-0.05 (2H, m), 0.41-0.46 (2H, m), 0.73-0.86 (1H, m), 1.54-1.79 (6H, m), 2.02-2.20 (6H, m), 2.46-2.94 (3H, m), 3.77 (3H, s), 3.79-4.02 (4H, m), 6.60 (1H, d, J = 2.5 Hz), 6.66 (1H, dd, J = 2.5, 8.0 Hz), 7.10 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 370 [M+H]+
1H NMR (CDCl3, 300 MHz) δ: 0.00-0.05 (2H, m), 0.41-0.46 (2H, m), 0.73-0.86 (1H, m), 1.54-1.79 (6H, m), 2.02-2.20 (6H, m), 2.46-2.94 (3H, m), 3.77 (3H, s), 3.79-4.02 (4H, m), 6.60 (1H, d, J = 2.5 Hz), 6.66 (1H, dd, J = 2.5, 8.0 Hz), 7.10 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 370 [M+H]+
実施例3
還元体 3からオキソ体 4への脱保護
還元体 3からオキソ体 4への脱保護
実施例2で得られた還元体 3(226 mg, 0.612 mmol)を1N 塩酸(10 mL)に溶解し、アルゴン雰囲気下80 ℃で10分間反応させた。
冷後、炭酸水素ナトリウムでpH 9とし、クロロホルムで抽出した。有機層を飽和食塩水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、濃縮して表題化合物 4 (208 mg, quant.)を茶色油状物質として得た。
IR (film) νmax 1715 cm-1
1H NMR (CDCl3, 300 MHz) δ: 0.01-0.06 (2H, m), 0.43-0.49 (2H, m), 0.72-0.86 (1H, m), 1.71-1.99 (3H, m), 2.06-2.20 (4H, m), 2.26-2.68 (8H, m), 3.08 (1H, d, J = 15.0 Hz), 3.78 (3H, s), 6.59 (1H, d, J = 2.0 Hz), 6.71 (1H, dd, J = 2.0, 8.0 Hz), 7.16 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 326 [M+H]+
1H NMR (CDCl3, 300 MHz) δ: 0.01-0.06 (2H, m), 0.43-0.49 (2H, m), 0.72-0.86 (1H, m), 1.71-1.99 (3H, m), 2.06-2.20 (4H, m), 2.26-2.68 (8H, m), 3.08 (1H, d, J = 15.0 Hz), 3.78 (3H, s), 6.59 (1H, d, J = 2.0 Hz), 6.71 (1H, dd, J = 2.0, 8.0 Hz), 7.16 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 326 [M+H]+
実施例4
2-シクロプロピルメチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オール 5 の合成
2-シクロプロピルメチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オール 5 の合成
実施例3で得られたオキソ体 4(208 mg, 0.639 mmol)をジクロロメタン(5 mL)に溶解し、アルゴン雰囲気下で氷冷しつつ1M 三臭化ホウ素/ジクロロメタン溶液(4.5 mL, 4.5 mmol)を滴下した。その後室温に戻し、1時間反応させた。
反応液に6%アンモニア水溶液(15 mL)を加え、5分間攪拌した。有機層を分離し、水層をジクロロメタンで洗った後これを合わせ、飽和食塩水溶液で洗浄した。有機層を無水硫酸ナトリウムで乾燥後濃縮し、粗生成物203 mgを得た。得られた粗生成物をシリカゲルカラムクロマトグラフィーにて精製し、淡茶色油状物質として表題化合物 5(122 mg, 61.4%)を得た。薬理評価には定法にしたがって塩酸塩としたものを用いた。
IR (film) νmax 1708 cm-1
1H NMR (CDCl3, 300 MHz) δ: 0.02-0.07 (2H, m), 0.43-0.49 (2H, m), 0.73-1.95 (1H, m), 1.66-1.95 (4H, m), 2.01-2.22 (4H, m), 2.29-2.65 (6H, m), 2.69 (1H, d, J = 15.0 Hz), 3.01 (1H, d, J = 15.0 Hz), 6.53 (1H, d, J = 2.5 Hz), 6.62 (1H, dd, J = 2.5, 8.0 Hz), 7.08 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 312 [M+H]+
1H NMR (CDCl3, 300 MHz) δ: 0.02-0.07 (2H, m), 0.43-0.49 (2H, m), 0.73-1.95 (1H, m), 1.66-1.95 (4H, m), 2.01-2.22 (4H, m), 2.29-2.65 (6H, m), 2.69 (1H, d, J = 15.0 Hz), 3.01 (1H, d, J = 15.0 Hz), 6.53 (1H, d, J = 2.5 Hz), 6.62 (1H, dd, J = 2.5, 8.0 Hz), 7.08 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 312 [M+H]+
実施例5
転移生成物 2(メタンスルホン酸塩)の別法合成
水素化ナトリウム(2.4 g)を窒素気流下、無水ヘキサンで洗浄し、アルゴン雰囲気下THF(10 mL)に懸濁した。氷冷下で塩化メタンスルホニル(2 mL, 25.9 mmol)を滴下した。2時間後、14-ヒドロキシモルヒナン誘導体 1 (1 g, 2.59 mmol)をTHF(15 mL)に溶解して懸濁液に加え、室温に戻して21時間反応させた。
転移生成物 2(メタンスルホン酸塩)の別法合成
水素化ナトリウム(2.4 g)を窒素気流下、無水ヘキサンで洗浄し、アルゴン雰囲気下THF(10 mL)に懸濁した。氷冷下で塩化メタンスルホニル(2 mL, 25.9 mmol)を滴下した。2時間後、14-ヒドロキシモルヒナン誘導体 1 (1 g, 2.59 mmol)をTHF(15 mL)に溶解して懸濁液に加え、室温に戻して21時間反応させた。
炭酸カリウム及び飽和炭酸水素ナトリウム水溶液を加えてpH 9とし、酢酸エチルで抽出した。有機層を飽和食塩水溶液で洗浄し、無水硫酸ナトリウムで乾燥後濃縮して粗生成物1.46 gを得た。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製し、淡茶色アモルファスとして表題化合物 2(1.12 g, 93.1%)を得た。
IR (film) νmax 2943, 1685, 1611 cm-1
1H NMR (CDCl3, 300 MHz) δ: 0.23-0.32 (1H, m), 0.37-0.53 (2H, m), 0.55-0.64 (1H, m), 0.92-1.06 (1H, m), 1.70-1.85 (4H, m), 2.01 (1H, ddd, J = 5.5, 11.0, 15.0 Hz), 2.15 (1H, ddd, J = 2.5, 4.5, 15.0 Hz), 2.38 (1H, dt, J = 4.0, 14.0 Hz), 2.69-2.81 (4H, m), 3.35 (2H, dd, J = 16.5, 21.5 Hz), 3.46-3.58 (1H, m), 3.78 (3H, s), 3.80-4.00 (6H, m), 6.58 (1H, d, J = 2.5 Hz), 6.73 (1H, dd, J = 2.5, 8.5 Hz), 7.18 (1H, d, J = 8.5 Hz), 9.42 (1H, s)
MS (FAB) m/z 368 [M-MeSO3-]+
1H NMR (CDCl3, 300 MHz) δ: 0.23-0.32 (1H, m), 0.37-0.53 (2H, m), 0.55-0.64 (1H, m), 0.92-1.06 (1H, m), 1.70-1.85 (4H, m), 2.01 (1H, ddd, J = 5.5, 11.0, 15.0 Hz), 2.15 (1H, ddd, J = 2.5, 4.5, 15.0 Hz), 2.38 (1H, dt, J = 4.0, 14.0 Hz), 2.69-2.81 (4H, m), 3.35 (2H, dd, J = 16.5, 21.5 Hz), 3.46-3.58 (1H, m), 3.78 (3H, s), 3.80-4.00 (6H, m), 6.58 (1H, d, J = 2.5 Hz), 6.73 (1H, dd, J = 2.5, 8.5 Hz), 7.18 (1H, d, J = 8.5 Hz), 9.42 (1H, s)
MS (FAB) m/z 368 [M-MeSO3-]+
実施例6
N-メチル体 7の合成
N-メチル体 7の合成
実施例5で得られた 2(メタンスルホン酸塩)から、実施例2と同様にして得られる還元体 3(520 mg, 1.41 mmol)を1,2-ジクロロエタン(10 mL)に溶解し、1,8-ビス(N,N-ジメチルアミノ)ナフタレン (915 mg, 4.27 mmol)およびクロロギ酸2,2,2-トリクロロエチル(600 μL, 4.36 mmol)を加えて、アルゴン雰囲気下0 ℃で12.5時間反応させた。
反応液にクロロホルムを加えて1N塩酸で素早く洗い、次いで飽和炭酸水素ナトリウム水溶液で洗浄した。有機層を飽和食塩水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、濃縮して、トリクロロエトキシカルボニル体 6 (972 mg ,quant.)を淡茶色油状物質として得た。
IR (film) νmax 1715 cm-1
1H NMR (CDCl3, 300 MHz) δ: 1.54-1.84 (5H, m), 1.91-2.08 (1H, m), 2.32 (1H, dd, J = 8.0, 16.0 Hz), 2.40-2.53 (1H, m), 2.79-3.40 (2H, m), 3.65 (1H, dd, J = 4.0, 13.5 Hz), 3.79 (3H, s), 3.81-4.03 (4H, m), 4.65-4.86 (3H, m),4.88 (2H, s), 6.62 (1H, d, J = 2.5 Hz), 6.70 (1H, dd, J = 2.5, 8.0 Hz), 7.14 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 490 [M+H]+
1H NMR (CDCl3, 300 MHz) δ: 1.54-1.84 (5H, m), 1.91-2.08 (1H, m), 2.32 (1H, dd, J = 8.0, 16.0 Hz), 2.40-2.53 (1H, m), 2.79-3.40 (2H, m), 3.65 (1H, dd, J = 4.0, 13.5 Hz), 3.79 (3H, s), 3.81-4.03 (4H, m), 4.65-4.86 (3H, m),4.88 (2H, s), 6.62 (1H, d, J = 2.5 Hz), 6.70 (1H, dd, J = 2.5, 8.0 Hz), 7.14 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 490 [M+H]+
水素化アルミニウムリチウム(122 mg, 3.21 mmol)をアルゴン雰囲気下、THF(1 mL)に懸濁した。上記で得られたトリクロロエトキシカルボニル体 6 (260 mg, 0.530 mmol)をTHF(1 mL)に溶解して氷冷下で懸濁液に加え、その後室温に戻して2.5時間反応させた。
反応後酢酸エチルを加え、次いで飽和酒石酸ナトリウムカリウム水溶液を加えて攪拌した。セライト濾過し、飽和炭酸水素ナトリウム水溶液及び飽和食塩水溶液で洗浄した。有機層を無水硫酸ナトリウムで乾燥後濃縮し、粗生成物82 mgを得た。得られた粗生成物をPLCにて精製し、淡黄色油状物質として表題化合物 7(26.2 mg, 15.0%)を得た。
IR (film) νmax 2789, 1610, 1586 cm-1
1H NMR (CDCl3, 300 MHz) δ: 1.55-1.79 (7H, m), 1.92-2.06 (2H, m), 2.15 (3H, s), 2.29-2.44 (4H, m), 2.84-2.88 (1H, m), 378 (3H, s), 3.82-3.99 (4H, m), 6.60 (1H, d, J = 2.5 Hz), 6.68 (1H, dd, J = 2.5, 8.0 Hz), 7.10 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 330 [M+H]+
1H NMR (CDCl3, 300 MHz) δ: 1.55-1.79 (7H, m), 1.92-2.06 (2H, m), 2.15 (3H, s), 2.29-2.44 (4H, m), 2.84-2.88 (1H, m), 378 (3H, s), 3.82-3.99 (4H, m), 6.60 (1H, d, J = 2.5 Hz), 6.68 (1H, dd, J = 2.5, 8.0 Hz), 7.10 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 330 [M+H]+
実施例7
オキソ化合物 8の合成
オキソ化合物 8の合成
実施例6で得られたN-メチル体 7 (24 mg, 0.0729 mmol)を1N 塩酸に溶解し、アルゴン雰囲気下80 ℃で10分間反応させた。
冷後、炭酸カリウムを加えてpH 9-10とし、クロロホルムで抽出した。有機層を飽和食塩水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、濃縮して、表題化合物 8 (18.7 mg, 89.9%)を無色油状物質として得た。
IR (film) νmax 1714 cm-1
1H NMR (CDCl3, 300 MHz) δ: 1.78-2.08 (6H, m), 2.16 (3H, s), 2.26-2.57 (6H, m), 2.64 (1H, d, J = 15.5 Hz), 3.06 (1H, d, J = 15.5 Hz), 3.78 (3H, s), 6.59 (1H, d, J = 2.5 Hz), 6.71 (1H, dd, J = 2.5, 8.0 Hz), 7.16 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 286 [M+H]+
1H NMR (CDCl3, 300 MHz) δ: 1.78-2.08 (6H, m), 2.16 (3H, s), 2.26-2.57 (6H, m), 2.64 (1H, d, J = 15.5 Hz), 3.06 (1H, d, J = 15.5 Hz), 3.78 (3H, s), 6.59 (1H, d, J = 2.5 Hz), 6.71 (1H, dd, J = 2.5, 8.0 Hz), 7.16 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 286 [M+H]+
実施例8
2-メチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オール 9の合成
2-メチル-2,3,4,4a,9,9a-ヘキサヒドロ-4a,9a-(テトラメチレン-2-オキソ)-1H-インデノ[2,1-c]ピリジン-6-オール 9の合成
実施例7で得られたオキソ化合物 8 (404 mg, 1.42 mmol)をジクロロメタン(5 mL)に溶解し、アルゴン雰囲気下で氷冷しつつ1M 三臭化ホウ素/ジクロロメタン溶液(9.94 mL, 9.94 mmol)を滴下した。その後室温に戻し、1時間反応させた。
反応後、反応液に6%アンモニア水溶液(30 mL)を加え攪拌した。有機層を分離し、飽和食塩水溶液で洗浄した。無水硫酸ナトリウムで乾燥後濃縮し、粗生成物378 mgを得た。得られた粗生成物をシリカゲルカラムクロマトグラフィーにて精製し、茶色油状物質(133.2 mg, 34.6%)を得た。これを酢酸エチルから再結晶して表題化合物 9(76.9 mg)を得た。薬理評価には定法に従って塩酸塩としたものを用いた。
mp : 211-213 ℃
IR (film) νmax 1707 cm-1
1H NMR (CDCl3, 300 MHz) δ: 1.69-1.96 (4H, m), 2.06-2.16 (2H, m), 2.18 (3H, s), 2.29-2.55 (6H, m), 2.68 (1H, d, J = 15.5 Hz), 3.01 (1H, d, J = 15.5 Hz), 6.53 (1H, d, J = 2.0 Hz), 6.63 (1H, dd, J = 2.0, 8.0 Hz), 7.09 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 272 [M+H]+
IR (film) νmax 1707 cm-1
1H NMR (CDCl3, 300 MHz) δ: 1.69-1.96 (4H, m), 2.06-2.16 (2H, m), 2.18 (3H, s), 2.29-2.55 (6H, m), 2.68 (1H, d, J = 15.5 Hz), 3.01 (1H, d, J = 15.5 Hz), 6.53 (1H, d, J = 2.0 Hz), 6.63 (1H, dd, J = 2.0, 8.0 Hz), 7.09 (1H, d, J = 8.0 Hz)
MS (FAB) m/z 272 [M+H]+
実施例9
モルモット脳ホモジネートを用いる受容体結合試験
モルモット前脳および小脳を氷冷バッファー(50 mM Tris-HCl、pH 7.4)にてホモジナイズした後、遠心分離(12,000×g、20分、4℃)し、上清を捨てた。この操作を3回繰り返し、得られた沈渣をバッファーにて再懸濁させ、膜標本とした。
モルモット脳ホモジネートを用いる受容体結合試験
モルモット前脳および小脳を氷冷バッファー(50 mM Tris-HCl、pH 7.4)にてホモジナイズした後、遠心分離(12,000×g、20分、4℃)し、上清を捨てた。この操作を3回繰り返し、得られた沈渣をバッファーにて再懸濁させ、膜標本とした。
ポリプロピレンチューブに膜標本(0.2-0.6 mg/チューブ)、放射性リガンド(μ受容体:0.5 nM [3H]-DAMGO、δ受容体:0.1 nM [3H]-NTI、κ受容体:0.5 nM [3H]-U69593)、被験化合物を添加し、25℃にて120分間インキュベートした。反応終了後、0.1%ポリエチレンイミンに少なくとも2時間浸したGF/Bフィルター(Whatman社製)を通して、迅速に吸引濾過し、氷冷バッファーにて洗浄した(3 mLを5回)。フィルターをバイアルに移し、シンチレーションカクテルを5 mLずつ添加し、バイアル中の放射活性(dpm)を液体シンチレーションカウンター(Packard社製)にて計測した。
特異的結合(SB)は、総結合(TB)から各受容体に対する過剰量の非放射性リガンド添加によって得られる非特異的結合(NSB)を差し引くこと(TB−NSB)により算出し、種々の被験化合物の存在下におけるSBを、対照SBの百分率で表した。放射性リガンドによる特異的結合を50%阻害する被験化合物の濃度(IC50)を表計算ソフトMicrosoft Excel(Microsoft社製)により算出した(50%を挟む2点の直線回帰)。
その結果、化合物 5あるいは化合物 9は、オピオイド受容体に対して結合性を有することが明らかとなった。オピオイド受容体に関連する疾患の治療または予防剤として有用である。
実施例10
マウス酢酸ライジング法による鎮痛活性試験
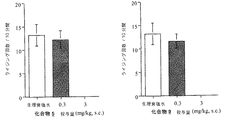
ddY系雄性マウスを用い、投与溶媒または被験化合物を0.1(mL/10g体重)の投与容量で皮下投与した。その15分後に0.6%(v/v)酢酸溶液を0.1(mL/10g体重)の投与容量で腹腔内に投与し、その10分後から10分間に生じたライジング反応(体を反らしたり、ひねったりする行動)の発現回数を測定し、この回数を痛みの指標にした。投与溶媒群におけるライジング反応の回数が減少するかどうかを観測することで鎮痛活性を評価した。結果を図1に示す。
マウス酢酸ライジング法による鎮痛活性試験
ddY系雄性マウスを用い、投与溶媒または被験化合物を0.1(mL/10g体重)の投与容量で皮下投与した。その15分後に0.6%(v/v)酢酸溶液を0.1(mL/10g体重)の投与容量で腹腔内に投与し、その10分後から10分間に生じたライジング反応(体を反らしたり、ひねったりする行動)の発現回数を測定し、この回数を痛みの指標にした。投与溶媒群におけるライジング反応の回数が減少するかどうかを観測することで鎮痛活性を評価した。結果を図1に示す。
その結果、化合物5, 9は、3 mg/kgの皮下投与で、全個体においてほぼ完全にライジング行動を抑制した。このように、化合物 5, 9は、顕著な鎮痛活性を示し、鎮痛剤の有効成分として有用である。
Claims (8)
- 一般式(I)
R2、R3は、それぞれ別個に水素、ヒドロキシ、炭素数1から5のアルコキシ、炭素数3から7のアルケニルオキシ、炭素数7から13のアラルキルオキシ、または炭素数1から5のアルカノイルオキシを表す。]で示される4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩。 - 一般式(I)において、R1が水素、炭素数1から5のアルキル、炭素数4から7のシクロアルキルアルキル、炭素数6から8のシクロアルケニルアルキル、または炭素数3から7のアルケニルであり、R2、R3が、それぞれ別個に水素、ヒドロキシ、炭素数1から5のアルコキシ、または炭素数1から5のアルカノイルオキシである請求項1に記載の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩。
- 一般式(I)において、R1が、水素、メチル、シクロプロピルメチル、シクロブチルメチル、またはアリルであり、R2 、R3が、それぞれ別個に水素、ヒドロキシ、メトキシ、またはアセトキシである請求項2に記載の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩。
- 請求項1から3のいずれか1項に記載の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体またはその薬理学的に許容される酸付加塩を有効成分として含有する医薬。
- オピオイド受容体に関連する疾患又は症状の治療または予防剤である請求項4に記載の医薬。
- 前記オピオイド受容体に関連する疾患又は症状が疼痛である請求項5に記載の医薬。
- 一般式(II)
で示される転移生成物を得た後、還元、脱保護することを特徴とする一般式(I)で示される請求項1から3のいずれか1項に記載の4a,9a-架橋-ヘキサヒドロ-1H-インデノピリジン誘導体の製造方法。 - 前記脱ヒドロキシ反応剤がメタンスルホニルクロリド又は塩化チオニルであり、前記塩基が水素化ナトリウム又は水素化カリウムである請求項7に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007013599A JP2008179556A (ja) | 2007-01-24 | 2007-01-24 | 4a,9a−架橋−ヘキサヒドロ−1H−インデノピリジン誘導体、その医薬用途、およびその製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007013599A JP2008179556A (ja) | 2007-01-24 | 2007-01-24 | 4a,9a−架橋−ヘキサヒドロ−1H−インデノピリジン誘導体、その医薬用途、およびその製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2008179556A true JP2008179556A (ja) | 2008-08-07 |
Family
ID=39723749
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007013599A Pending JP2008179556A (ja) | 2007-01-24 | 2007-01-24 | 4a,9a−架橋−ヘキサヒドロ−1H−インデノピリジン誘導体、その医薬用途、およびその製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2008179556A (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014115853A1 (ja) * | 2013-01-25 | 2014-07-31 | 学校法人北里研究所 | プロペラン誘導体 |
| WO2015097546A1 (en) * | 2013-12-26 | 2015-07-02 | Purdue Pharma L.P. | Propellane-based compounds and their use as opioid receptor modulators |
-
2007
- 2007-01-24 JP JP2007013599A patent/JP2008179556A/ja active Pending
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014115853A1 (ja) * | 2013-01-25 | 2014-07-31 | 学校法人北里研究所 | プロペラン誘導体 |
| WO2015097546A1 (en) * | 2013-12-26 | 2015-07-02 | Purdue Pharma L.P. | Propellane-based compounds and their use as opioid receptor modulators |
| US9340542B2 (en) | 2013-12-26 | 2016-05-17 | Purdue Pharma L.P. | Propellane-based compounds and the use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2015342887B2 (en) | Substituted pyrazolo(1,5-a)pyrimidines and their use in the treatment of medical disorders | |
| CA2730111C (en) | Synthesis of metabolically stable agents for alcohol and drug abuse | |
| EP0712402B1 (en) | Hydroisoquinoline derivatives | |
| JP2004520383A (ja) | カッパオピオイド受容体リガンド | |
| JP7397095B2 (ja) | オピオイド受容体アンタゴニストプロドラッグ | |
| EP0915094A1 (en) | Morphinane derivatives and medicinal use thereof | |
| JP2016175910A (ja) | オピオイド受容体リガンドとしてのカルボキサミド基を含有するモルヒネ誘導体 | |
| JP5252391B2 (ja) | オキサビシクロ[2.2.2]オクタンを有するモルヒナン誘導体およびその医薬用途 | |
| JP2015044819A (ja) | 大型置換基を有する非フェノール性アミンオピオイド | |
| JP2008179554A (ja) | ピロール縮合モルヒナン誘導体およびその医薬用途 | |
| JP6013345B2 (ja) | 痛み治療用の4,5a−エポキシモルフィナンの6−アミド誘導体 | |
| EP3077398A1 (en) | Novel opioid compounds and their uses | |
| AU2018359336A1 (en) | Opioid receptor antagonist prodrugs | |
| JPH1087667A (ja) | モルフィナンヒドロキサム酸化合物 | |
| EP1342723B1 (en) | Indole derivatives and use thereof in medicines | |
| JP2008179556A (ja) | 4a,9a−架橋−ヘキサヒドロ−1H−インデノピリジン誘導体、その医薬用途、およびその製造方法 | |
| EP0596897A1 (en) | Hydroisoquinoline derivatives | |
| US10807995B2 (en) | Thienothiophene compounds for long-acting injectable compositions and related methods | |
| JP5213157B2 (ja) | 6,14−エポキシモルヒナン誘導体およびその医薬用途 | |
| US20250051332A1 (en) | Opioid receptor agonist and methods of use thereof | |
| CN101492451B (zh) | 8α取代芳基-4,5-环氧吗啡喃衍生物或其盐、制备方法和用途 | |
| CZ324997A3 (cs) | 1-[omega-(3,4-Dihydro-2-naftalenyl)alkyl]cyklický amin, způsob jeho výroby, farmaceutický prostředek ho obsahující a činidlo pro léčení častého močení a močové inkontinence | |
| US20220204449A1 (en) | Biased agonists of opioid receptors | |
| WO2025174540A1 (en) | Novel morphine framework derivatives | |
| WO2020094634A1 (en) | Thiophene prodrugs of naltroxene for long-acting injectable compositions and related methods |