JP2007169657A - ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 - Google Patents
ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 Download PDFInfo
- Publication number
- JP2007169657A JP2007169657A JP2007025660A JP2007025660A JP2007169657A JP 2007169657 A JP2007169657 A JP 2007169657A JP 2007025660 A JP2007025660 A JP 2007025660A JP 2007025660 A JP2007025660 A JP 2007025660A JP 2007169657 A JP2007169657 A JP 2007169657A
- Authority
- JP
- Japan
- Prior art keywords
- polymer
- copolymer
- group
- mol
- sulfonated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 C*C*(C1)C*(*I)C*(C2)C(*)(C3)CCCC(*C)(CC4)*1C(*C)C*2C34C(C)C Chemical compound C*C*(C1)C*(*I)C*(C2)C(*)(C3)CCCC(*C)(CC4)*1C(*C)C*2C34C(C)C 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N C1CCCCC1 Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- CLLHDRWCFDBGPG-UHFFFAOYSA-N CC(C)(C(C=C1)=CCC1Sc1ccc(C)cc1)Oc1cc(C)c(C(C(F)(F)F)(C(F)(F)F)c(cc2)ccc2Oc(cc2)ccc2S(c2ccc(C(C)(C)Cl)cc2)=O)cc1 Chemical compound CC(C)(C(C=C1)=CCC1Sc1ccc(C)cc1)Oc1cc(C)c(C(C(F)(F)F)(C(F)(F)F)c(cc2)ccc2Oc(cc2)ccc2S(c2ccc(C(C)(C)Cl)cc2)=O)cc1 CLLHDRWCFDBGPG-UHFFFAOYSA-N 0.000 description 1
- SYISJYJRFCXAHO-UHFFFAOYSA-N CC(C)(c(cc1)ccc1Sc(cc1)ccc1Cl)Oc(cc1)ccc1S(c(cc1)ccc1Oc(cc1)ccc1S(c1ccc(C(C)(C)Cl)cc1)=O)(=O)=O Chemical compound CC(C)(c(cc1)ccc1Sc(cc1)ccc1Cl)Oc(cc1)ccc1S(c(cc1)ccc1Oc(cc1)ccc1S(c1ccc(C(C)(C)Cl)cc1)=O)(=O)=O SYISJYJRFCXAHO-UHFFFAOYSA-N 0.000 description 1
Images




















Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polyethers (AREA)
Abstract
【課題】主鎖中に屈曲性構造を有するために靭性が高く、スルホン化しても上記の機械的性質及び熱的性質が低下しにくい重合体、該重合体をスルホン化して得られるスルホン酸基含有重合体、及び該スルホン酸基含有重合体からなる機械的強度に優れ、耐久性に優れるプロトン伝導膜を提供する。
【解決手段】一般式(1m):
で表されるハロゲン化芳香族化合物、該化合物の屈曲性構造を主鎖中に有する重合体又は共重合体、これをスルホン化したスルホン化共重合体重合体、及び該スルホン化共重合体からなるプロトン伝導膜。
【選択図】なし
【解決手段】一般式(1m):
で表されるハロゲン化芳香族化合物、該化合物の屈曲性構造を主鎖中に有する重合体又は共重合体、これをスルホン化したスルホン化共重合体重合体、及び該スルホン化共重合体からなるプロトン伝導膜。
【選択図】なし
Description
本発明は、新規なハロゲン化芳香族化合物、それをモノマー成分として重合したポリフェニレン系重合体、及び該重合体のスルホン化物からなるプロトン伝導膜に関する。プロトン伝導膜は、一次電池用電解質、二次電池用電解質、燃料電池用高分子固体電解質、表示素子、各種センサー、信号伝達媒体、固体コンデンサー、イオン交換膜などに有用であることが知られている。
電解質は、通常、(水)溶液で用いられることが多い。しかし、近年、これを固体系に置き替えていく傾向が高まってきている。その第1の理由としては、例えば、上記の電気・電子材料に応用する場合のプロセッシングの容易さであり、第2の理由としては、軽薄短小・省電力化への移行である。
従来、プロトン伝導性材料としては、無機物からなるもの、有機物からなるものの両方が知られている。無機物の例としては、例えば水和化合物であるリン酸ウラニルが挙げられるが、これら無機化合物は界面での接触が十分でなく、伝導層を基板あるいは電極上に形成するには問題が多い。
従来、プロトン伝導性材料としては、無機物からなるもの、有機物からなるものの両方が知られている。無機物の例としては、例えば水和化合物であるリン酸ウラニルが挙げられるが、これら無機化合物は界面での接触が十分でなく、伝導層を基板あるいは電極上に形成するには問題が多い。
一方、有機化合物の例としては、いわゆる陽イオン交換樹脂に属するポリマー、例えばポリスチレンスルホン酸などのビニル系ポリマーのスルホン化物、ナフィオン(デュポン社製)を代表とするパーフルオロアルキルスルホン酸ポリマー、パーフルオロアルキルカルボン酸ポリマーや、ポリベンズイミダゾールやポリエーテルエーテルケトンなどの耐熱性高分子にスルホン酸基やリン酸基を導入したポリマー〔Polymer Preprints,Japan,Vol.42,No.7,p.2490〜2492(1993)、Polymer Preprints,Japan,Vol.43,No.3,p.735〜736(1994)、Polymer Preprints,Japan,Vol.42,No.3,p.730(1993)〕などの有機系ポリマーが挙げられる。
これら有機系ポリマーは、通常、フィルム状で用いられるが、溶媒に可溶性であること、または熱可塑性であることを利用し、電極上に伝導膜を接合加工できる。しかしながら、これら有機系ポリマーの多くは、プロトン伝導性がまだ十分でないことに加え、耐久性や高温(100℃以上)でプロトン伝導性が低下してしまうこと、スルホン化により脆化し、機械的強度が低下すること、湿度条件下の依存性が大きいこと、あるいは電極との密着性が十分満足のいくものとはいえなかったり、含水ポリマー構造に起因する稼働中の過度の膨潤による強度の低下や形状の崩壊に至るという問題がある。したがって、これらの有機ポリマーは、上記の電気・電子材料などに応用するには種々問題がある。
米国特許第5,403,675号明細書では、スルホン化された剛直ポリフェニレンからなる固体高分子電解質が提案されている。このポリマーは、フェニレン連鎖からなる芳香族化合物を重合して得られるポリマー(同明細書カラム9記載の構造)を主成分とし、これをスルホン化剤と反応させてスルホン酸基を導入している。しかしながら、スルホン酸基の導入量の増加によって、プロトン伝導度が向上するものの、同時に得られるスルホン化ポリマーの機械的性質、例えば破断伸び、耐折り曲げ性等の靭性や耐熱水性は著しく損なわれる。
そこで、本発明の課題は、主鎖中に屈曲性構造を有するために靭性が高く、スルホン化しても上記の靭性および耐熱水性が低下しにくい重合体、該重合体をスルホン化して得られるスルホン酸基含有重合体、及び該スルホン酸基含有重合体からなる靭性に優れ、耐久性に優れるプロトン伝導膜を提供することにある。
即ち、本発明は、第一に、重合体に屈曲性構造を導入するのに有効なモノマーとして有用な化合物を提供するもので、該化合物は、
一般式(1m):
一般式(1m):
[式中、Aは独立に電子吸引性の基であり、Bは独立に電子供与性の原子又は基であり、Xは塩素原子、ヨウ素原子または臭素原子であり、R1〜R8は同一でも異なってもよく、水素原子、フッ素原子またはアルキル基であり、nは2以上、好ましくは2〜100、特に好ましくは2〜80の整数である。]
で表されるハロゲン化芳香族化合物を提供する。
このハロゲン化芳香族化合物は、重合体中に屈曲性構造をもたらし、重合体の靭性を向上させる。
そこで、本発明は、第二に、一般式(1):
[式中、A、B、R1〜R8及びnは一般式(1m)に関して定義の通りである。]
で表される繰返し単位を有するポリフェニレン系重合体を提供する。
該ポリフェニレン系重合体は単独重合体でもよいし、他の繰返し単位を含む共重合体であってもよい。
即ち、本発明は、第三に、一般式(1)で表される繰返し単位と、別の2価の芳香族基からなる繰返し単位とを有するポリフェニレン系共重合体を提供する。
本発明は、第四に、かかる共重合体の一種として、上記2価の芳香族基からなる繰返し単位が、一般式(2)〜(5):
本発明は、第四に、かかる共重合体の一種として、上記2価の芳香族基からなる繰返し単位が、一般式(2)〜(5):
[式中、A及びBは前記一般式(1m)に関して定義の通りであり、R9〜R15は同一でも異なってもよく、水素原子、フッ素原子またはアルキル基を示し、Zはアリール基である。mは0〜2の整数である。]
〔一般式(3)〜(5)中、R17〜R24は、同一または異なり、水素原子、アルキル基又はアリール基を示す。〕
からなる群から選ばれる少なくとも1種の単位であることを特徴とするポリフェニレン系共重合体を提供する。
該共重合体は容易にスルホン化してプロトン伝導性を付与することができる。
そこで、本発明は、第五に、さらにスルホン酸基を有する上記の共重合体を提供する。
このスルホン酸基含有共重合体は、プロトン伝導膜の材料として有用である。
そこで、本発明は、第六に、上記スルホン酸基含有共重合体からなるプロトン伝導膜を提供する。
このスルホン酸基含有共重合体は、プロトン伝導膜の材料として有用である。
そこで、本発明は、第六に、上記スルホン酸基含有共重合体からなるプロトン伝導膜を提供する。
本発明のハロゲン化芳香族化合物は重合体の分子中に屈曲性構造を導入するのに有用であり、得られるより得られる芳香族系の重合体は主鎖中に屈曲性構造を有するために靭性が高く、スルホン化しても上記の機械的性質及び熱的性質が低下しにくい。該重合体をスルホン化して得られるスルホン酸基含有重合体はプロトン伝導膜材料として有用であり、得られるプロトン伝導膜は機械的強度に優れ、耐久性に優れる。
また、さらに一般式(2)で表される繰返し単位をも有する本発明の好ましい実施形態による共重合体はスルホン化の際にスルホン酸基の導入量を容易に制御することができる。得られるスルホン基含有共重合体は、伝導膜として、広い温度範囲にわたって高いプロントン伝導性を有し、かつ基板、電極に対する密着性が優れ、脆くなく強度において優れており、さらに温水耐性に優れている。
従って、本発明のプロトン伝導膜は、一次電池用電解質、二次電池用電解質、燃料電池用高分子固体電解質、表示素子、各種センサー、信号伝達媒体、固体コンデンサー、イオン交換膜などの伝導膜として利用可能であり、この工業的意義は極めて大である。
また、さらに一般式(2)で表される繰返し単位をも有する本発明の好ましい実施形態による共重合体はスルホン化の際にスルホン酸基の導入量を容易に制御することができる。得られるスルホン基含有共重合体は、伝導膜として、広い温度範囲にわたって高いプロントン伝導性を有し、かつ基板、電極に対する密着性が優れ、脆くなく強度において優れており、さらに温水耐性に優れている。
従って、本発明のプロトン伝導膜は、一次電池用電解質、二次電池用電解質、燃料電池用高分子固体電解質、表示素子、各種センサー、信号伝達媒体、固体コンデンサー、イオン交換膜などの伝導膜として利用可能であり、この工業的意義は極めて大である。
以下、本発明を詳細に説明する。
[ハロゲン化芳香族化合物]
本発明の、一般式(1m)で表されるハロゲン化芳香族化合物(以下、「モノマー(1m)」という)は、これをモノマー単位として含む重合体に屈曲性構造を付与し、重合体の靭性、その他の機械的強度などを向上させる作用を有する。
以下、一般式(1m)について説明する。
Xとしては、塩素原子、臭素原子、及びヨウ素原子が挙げられる。
Aは、電子吸引性の基であり、例えば、−CO−、−CONH−、−(CF2)p−(ここで、pは1〜10の整数である)、−C(CF3)2−、−COO−、−SO−、−SO2−などが挙げられる。なお、電子吸引性の基とは、ハメット(Hammett)置換基常数がフェニル基のm位の場合、0.06以上、p位の場合、0.01以上の値となる基をいう。
Bは、電子供与性の基又は原子であり、例えば、−O−、−S−、−CH=CH−、−C≡C−、
[ハロゲン化芳香族化合物]
本発明の、一般式(1m)で表されるハロゲン化芳香族化合物(以下、「モノマー(1m)」という)は、これをモノマー単位として含む重合体に屈曲性構造を付与し、重合体の靭性、その他の機械的強度などを向上させる作用を有する。
以下、一般式(1m)について説明する。
Xとしては、塩素原子、臭素原子、及びヨウ素原子が挙げられる。
Aは、電子吸引性の基であり、例えば、−CO−、−CONH−、−(CF2)p−(ここで、pは1〜10の整数である)、−C(CF3)2−、−COO−、−SO−、−SO2−などが挙げられる。なお、電子吸引性の基とは、ハメット(Hammett)置換基常数がフェニル基のm位の場合、0.06以上、p位の場合、0.01以上の値となる基をいう。
Bは、電子供与性の基又は原子であり、例えば、−O−、−S−、−CH=CH−、−C≡C−、
などが挙げられる。
本発明のモノマー(1m)としては、例えば、2,2-ビス[4-{4-(4-クロロベンゾイル)フェノキシ}フェニル]-1,1,1,3,3,3-ヘキサフルオロプロパン、ビス[4-{4-(4-クロロベンゾイル)フェノキシ}フェニル]スルホン、及び下記の化学式で示される化合物が挙げられる。
〔上記式中、Xは一般式(1m)に関して定義した通りである。〕
モノマー(1m)は、例えば、次のような反応により、合成することができる。
まず電子吸引性基で連結されたビスフェノールを対応するビスフェノールのアルカリ金属塩とするために、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、スルホラン、ジフェニルスルホン、ジメチルスルホキサイドのような誘電率の高い極性溶媒中でリチウム、ナトリウム、カリウムなどのアルカリ金属、水素化アルカリ金属、水酸化アルカリ金属、アルカリ金属炭酸塩などを加える。
まず電子吸引性基で連結されたビスフェノールを対応するビスフェノールのアルカリ金属塩とするために、N-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、スルホラン、ジフェニルスルホン、ジメチルスルホキサイドのような誘電率の高い極性溶媒中でリチウム、ナトリウム、カリウムなどのアルカリ金属、水素化アルカリ金属、水酸化アルカリ金属、アルカリ金属炭酸塩などを加える。
通常、アルカリ金属はフェノールの水酸基に対し、過剰気味で反応させ、通常、1.1ないしは2倍当量を使用する。好ましくは、1.2〜1.5倍当量の使用である。この際、ベンゼン、トルエン、キシレン、ヘキサン、シクロヘキサン、オクタン、クロロベンゼン、ジオキサン、テトラヒドロフラン、アニソール、フェネトールなどの水と共沸する溶媒を共存させて、電子吸引性基で活性化されたフッ素、塩素等のハロゲン原子で置換された芳香族ジハライド化合物、例えば、4,4'-ジフルオロベンゾフェノン、4,4'-ジクロロベンゾフェノン、4,4'-クロロフルオロベンゾフェノン、ビス(4-クロロフェニル)スルホン、ビス(4-フルオロフェニル)スルホン、4-フルオロフェニル-4'-クロロフェニルスルホン、ビス(3-ニトロ-4-クロロフェニル)スルホン、2,6-ジクロロベンゾニトリル、2,6-ジフルオロベンゾニトリル、ヘキサフルオロベンゼン、デカフルオロビフェニル、2,5-ジフルオロベンゾフェノン、1,3-ビス(4-クロロベンゾイル)ベンゼンなどを反応させる。反応性から言えば、フッ素化合物が好ましいが、次の芳香族カップリング反応を考慮した場合、末端が塩素原子となるように芳香族求核置換反応を組み立てる必要がある。活性芳香族ジハライドはビスフェノールに対し、2から4倍モル、好ましくは2.2から2.8倍モルの使用である。芳香族求核置換反応の前に予め、ビスフェノールのアルカリ金属塩としていてももよい。反応温度は60℃から300℃で、好ましくは80℃〜250℃の範囲である。反応時間は15分から100時間、好ましくは1時間から24時間の範囲である。最も好ましい方法としては、式(6):
[式中、Aは一般式(1m)に関して定義した通りである。]
で示される活性芳香族ジハライドとして反応性の異なるハロゲン原子を一個づつ有するクロロフルオロ体を用いることであり、フッ素原子が優先してフェノキシドと求核置換反応が起きるので、目的の活性化された末端クロロ体を得るのに好都合である。
または特開平2−159号公報に記載のように求核置換反応と親電子置換反応を組み合わせ、目的の電子吸引性基、電子供与性基からなる屈曲性化合物の合成方法がある。
具体的には電子吸引性基で活性化された芳香族ビスハライド、例えば、ビス(4-クロロフェニル)スルホンをフェノールとで求核置換反応させてビスフェノキシ置換体とする。次いで、この置換体を例えば、4-クロロ安息香酸クロリドとのフリーデルクラフト反応から目的の化合物を得る。ここで用いる電子吸引性基で活性化された芳香族ビスハライドは上記で例示した化合物が適用できる。フェノール化合物は置換されていても良いが、耐熱性や屈曲性の観点から、無置換化合物が好ましい。なお、フェノールの置換反応にはアルカリ金属塩とするのが、好ましく、使用可能なアルカリ金属化合物は上記に例示した化合物を使用できる。使用量はフェノール1モルに対し、1.2〜2倍モルである。反応に際し、上述した極性溶媒や水との共沸溶媒を用いることができる。ビスフェノキシ化合物を塩化アルミニウム、3フッ化ホウ素、塩化亜鉛などのルイス酸のフリーデルクラフト反応の活性化剤存在下に、アシル化剤として、クロロ安息香酸クロライドを反応させる。クロロ安息香酸クロライドはビスフェノキシ化合物に対し、2〜4倍モル、好ましくは2.2〜3倍モルの使用である。フリーデルクラフト活性化剤は、アシル化剤のクロロ安息香酸などの活性ハライド化合物1モルに対し、1.1から2倍当量使用する。反応時間は15分から10時間の範囲で、反応温度は−20℃から80℃の範囲である。使用溶媒は、フリ−デルクラフト反応に不活性な、クロロベンゼンやニトロベンゼンなどを用いることが出来る。
このようにして得られる本発明のモノマー(1m)は、IR、NMR、元素分析などにより、その構造を確認することができる。
本発明で使用できる一般式(1m)で示されるハロゲン化合物はn=2で示される単量体の他、nが2よりも大きなオリゴマーないしポリマーも使用できる。
これらのオリゴマーないしポリマーは、例えば、一般式(1m)において電子供与性基Bであるエーテル性酸素の供給源となるビスフェノールと、電子吸引性基Aである、>C=O、−SO2−、および/または>C(CF3)2とを組み合わした、具体的には2,2-ビス(4-ヒドロキシフェニル)-1,1,1,3,3,3-ヘキサフルオロプロパン、2,2-ビス(4-ヒドロキシフェニル)ケトン、2,2-ビス(4-ヒドロキシフェニル)スルホンなどのビスフェノールのアルカリ金属塩と過剰の4,4-ジクロロベンゾフェノン、ビス(4-クロロフェニル)スルホンなどの活性芳香族ハロゲン化合物との置換反応をN-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、スルホランなどの極性溶媒存在下で前記単量体の合成手法に順次重合して得られる。得られたオリゴマーないしポリマーはポリマーの一般的な精製方法、例えば、溶解−沈殿の操作によって行うことができる。分子量の調整は、過剰の芳香族ジクロライドとビスフェノールとの反応モル比によって行う。芳香族ジクロライドが過剰にあるため、得られるオリゴマー、ポリマーの分子末端は芳香族クロライドになっている。得られたオリゴマー、ポリマーの分子量はGPC、また、オリゴマーであれば、NMRからは数平均分子量を求めることができる。
これらのオリゴマーないしポリマーは、例えば、一般式(1m)において電子供与性基Bであるエーテル性酸素の供給源となるビスフェノールと、電子吸引性基Aである、>C=O、−SO2−、および/または>C(CF3)2とを組み合わした、具体的には2,2-ビス(4-ヒドロキシフェニル)-1,1,1,3,3,3-ヘキサフルオロプロパン、2,2-ビス(4-ヒドロキシフェニル)ケトン、2,2-ビス(4-ヒドロキシフェニル)スルホンなどのビスフェノールのアルカリ金属塩と過剰の4,4-ジクロロベンゾフェノン、ビス(4-クロロフェニル)スルホンなどの活性芳香族ハロゲン化合物との置換反応をN-メチル-2-ピロリドン、N,N-ジメチルアセトアミド、スルホランなどの極性溶媒存在下で前記単量体の合成手法に順次重合して得られる。得られたオリゴマーないしポリマーはポリマーの一般的な精製方法、例えば、溶解−沈殿の操作によって行うことができる。分子量の調整は、過剰の芳香族ジクロライドとビスフェノールとの反応モル比によって行う。芳香族ジクロライドが過剰にあるため、得られるオリゴマー、ポリマーの分子末端は芳香族クロライドになっている。得られたオリゴマー、ポリマーの分子量はGPC、また、オリゴマーであれば、NMRからは数平均分子量を求めることができる。
具体的な分子末端に芳香族クロライドを有したオリゴマー、またはポリマーの構造として以下のものを挙げることができる。
[重合体]
本発明の重合体は、上記一般式(1)で表される繰り返し単位(以下、繰返し単位(1)という)のみから構成される単独重合体でもよいし、繰返し単位(1)と他の繰返し単位とから構成される共重合体でもよい。いずれの場合でも、重合体のゲルパーミエーションクロマトグラフィーで測定したポリスチレン換算の重量平均分子量(以下、単に「重量平均分子量」という)は1万〜100万、好ましくは2万〜80万である。
該重合体が他の繰返し単位を有する場合には、繰返し単位(1)の含有量は10〜80モル%であることが好ましい。繰返し単位(1)が10モル%未満では、得られる重合体の靭性の向上が期待できない。
ここで、繰返し単位(1)は、本発明の上記モノマー(1m)から構成される。
本発明の重合体は、上記一般式(1)で表される繰り返し単位(以下、繰返し単位(1)という)のみから構成される単独重合体でもよいし、繰返し単位(1)と他の繰返し単位とから構成される共重合体でもよい。いずれの場合でも、重合体のゲルパーミエーションクロマトグラフィーで測定したポリスチレン換算の重量平均分子量(以下、単に「重量平均分子量」という)は1万〜100万、好ましくは2万〜80万である。
該重合体が他の繰返し単位を有する場合には、繰返し単位(1)の含有量は10〜80モル%であることが好ましい。繰返し単位(1)が10モル%未満では、得られる重合体の靭性の向上が期待できない。
ここで、繰返し単位(1)は、本発明の上記モノマー(1m)から構成される。
本発明の重合体が繰返し単位(1)以外の他の繰返し単位(以下「他の繰返し単位」ともいう)を有する場合、他の繰返し単位としては、ポリマーに求められる特性、機能等に応じて種々の単位を選択することができるが、プロトン伝導性を有する重合体を得ようとする場合は、例えば、前記一般式(2)〜(5)で表される単位(以下、それぞれ、単位(2)、(3)、(4)及び(5)といい、単位(2)〜(5)を総じて「単位(A)」ともいう)が挙げられる。この繰返し単位(1)と単位(A)とを含む共重合体は、スルホン化してプロトン伝導膜材料として用い得ることができる。
単位(A)としては、特に、単位(2)が該重合体をスルホン化してスルホン酸基を導入する際にその量を制御し易い点で有利であり好ましい。
単位(2)を表す一般式(2)において、R9〜R15は水素原子、フッ素原子またはアルキル基を示し、アルキル基としてはメチル基、エチル基、プロピル基、ブチル基、ヘキシル基などが例示される。アルキル基はフッ素化されていてもよく、トリフルオロメチル基、ペンタフルオルエチル基などのパーフルオロアルキル基であってもよい。
また、Zにより表されるアリール基としては、例えば、フェニル基、ナフチル基、式:
単位(A)としては、特に、単位(2)が該重合体をスルホン化してスルホン酸基を導入する際にその量を制御し易い点で有利であり好ましい。
単位(2)を表す一般式(2)において、R9〜R15は水素原子、フッ素原子またはアルキル基を示し、アルキル基としてはメチル基、エチル基、プロピル基、ブチル基、ヘキシル基などが例示される。アルキル基はフッ素化されていてもよく、トリフルオロメチル基、ペンタフルオルエチル基などのパーフルオロアルキル基であってもよい。
また、Zにより表されるアリール基としては、例えば、フェニル基、ナフチル基、式:
[式中、R25〜R33は同一又は異なり水素原子、フッ素原子又はアルキル基であり、アルキル基としては一般式(1)中のR9〜R15について例示したものが例示される。]
で表されるビフェニリル基が挙げられる。
単位(3)〜(5)を表す一般式(3)〜(5)において、R17〜R24は、各々、水素原子、アルキル基、フッ素原子、フロロアルキル基及びアリール基を示すが、アルキル基としてはメチル基、エチル基、プロピル基、ブチル基、ヘキシル基などが例示され、フロロアルキル基としては、パーフルオロメチル基、パーフルオロエチル基等が例示され、アリール基としてはフェニル基、トリル基、キシリル基等が例示される。
重合体中における繰返し単位(A)の割合は共重合体中に10〜90モル%の範囲であることが好ましく、より好ましくは20〜80モル%である。単位(A)が少なすぎると、スルホン化により共重合体に導入されるスルホン酸基量が不十分となりがちで得られるプロトン伝導性が十分でない。
重合体中における繰返し単位(A)の割合は共重合体中に10〜90モル%の範囲であることが好ましく、より好ましくは20〜80モル%である。単位(A)が少なすぎると、スルホン化により共重合体に導入されるスルホン酸基量が不十分となりがちで得られるプロトン伝導性が十分でない。
本発明の重合体は、例えば、本発明のモノマー(1m)を、必要に応じて他の繰返し単位に対応するモノマー、例えば上記の繰返し単位(2)、(3)、(4)及び(5)のそれぞれに対応する一般式(2m)、(3m)、(4m)及び(5m):
[式中、X,A及びBは一般式(1m)に関して定義の通りであり、R9〜R15は同一でも異なってもよく、水素原子、フッ素原子またはアルキル基を示し、mおよびZは一般式(2)に関して定義の通りである。]
[一般式(3m)〜(5m)中、R17〜R24は一般式(3)〜(5)に関して定義した通りであり、R及びR′は独立にフッ素以外のハロゲン原子又は式:−OSO2 Y(ここで、Yはアルキル基、フロロアルキル基もしくはアリール基を示す。)で表される基である。]
で表されるモノマー(それぞれ、モノマー(2m)、(3m)、(4m)及び(5m)といい、これらモノマーを総じてモノマー(A)ともいう。)からなる群から選ばれる少なくとも1種のモノマーとを、遷移金属化合物を含む触媒の存在下で重合又は共重合することにより得られる。
一般式(3m)〜(5m)において、R及びR’で表されるハロゲン原子、並びにY中のアルキル基、フロロアルキル基及びアリール基は、いずれも一般式(3)〜(5)についてR17〜R24に関して例示したものと同様のものを例示できる
さらに、本発明のスルホン酸基含有重合体は、こうして得られた重合体を前駆体としてスルホン化剤を用いてスルホン化することにより得られる。
モノマー(2m)としては、例えば、以下の式で表される化合物が挙げられる。
さらに、本発明のスルホン酸基含有重合体は、こうして得られた重合体を前駆体としてスルホン化剤を用いてスルホン化することにより得られる。
モノマー(2m)としては、例えば、以下の式で表される化合物が挙げられる。
[式中、X及びZは一般式(2m)に関して前記のとおりである。]
さらに具体的には、モノマー(2m)の例としては下記式で表される化合物があげられる。
モノマー(2m)としては、溶解性、高分子量化の面から、ジクロロ安息香酸誘導体、例えば2.5-ジクロロ-4'-フェノキシベンゾフェノン、2,4-ジクロロ-4'-フェノキシベンゾフェノン、4'-フェノキシフェニル2,5-ジクロロベンゾエート、4'-フェノキシフェニル2,4-ジクロロベンゾエートを使用することが好ましい。
モノマー(2m)は、例えば、2,5-ジクロロ-4'-[4-(4-フェノキシ)フェノキシ]ベンゾフェノンを例にとると、次のような反応により、合成することができる。
すなわち、化合物(2m)’(2,5-ジクロロ-4'-フルオロベンゾフェノン)と化合物(2m)”(4-フェノキシフェノール)とを、炭酸カリウムなどの存在下で、反応溶媒として、ジメチルアセトアミド、N-メチル-2-ピロリドン、ジメチルスルホキシドなどの非プロトン系双極子極性溶媒などを用い、生成する水を共沸することにより反応液から除去するための共沸溶媒、例えばベンゼン、トルエン、キシレンなどを併用して反応温度80〜200℃で0.5から30時間反応させて、合成することができる。化合物(2m)’1モル当り化合物(2m)”を、通常ほぼ等モルで反応させる。
このようにして得られる本発明のモノマー(1)は、IR、NMR、元素分析などにより、その構造を確認することができる。
モノマー(3m)の具体例としては、p-ジクロロベンゼン、p-ジブロモベンゼン、p-ジヨードベンゼン、p-ジメチルスルフォニロキシベンゼン、2,5-ジクロロトルエン、2,5-ジブロモトルエン、2,5-ジヨードトルエン、2,5-ジメチルスルフォニロキシベンゼン、2,5-ジクロロ-p-キシレン、2,5-ジブロモ-p-キシレン、2,5-ジヨード-p-キシレン、2,5-ジクロロベンゾトリフルオライド、2,5-ジブロモベンゾトリフルオライド、2,5-ジヨードベンゾトリフルオライド、1,4-ジクロロ-2,3,5,6-テトラフルオロベンゼン、1,4-ジブロモ-2,3,5,6-テトラフルオロベンゼン、1,4-ジヨード-2,3,5,6-テトラフルオロベンゼンなどが挙げられ、好ましくはp-ジクロロベンゼン、p-ジメチルスルフォニロキシベンゼン、2,5-ジクロロトルエン、2,5-ジクロロベンゾトリフルオライドである。
モノマー(3m)の具体例としては、p-ジクロロベンゼン、p-ジブロモベンゼン、p-ジヨードベンゼン、p-ジメチルスルフォニロキシベンゼン、2,5-ジクロロトルエン、2,5-ジブロモトルエン、2,5-ジヨードトルエン、2,5-ジメチルスルフォニロキシベンゼン、2,5-ジクロロ-p-キシレン、2,5-ジブロモ-p-キシレン、2,5-ジヨード-p-キシレン、2,5-ジクロロベンゾトリフルオライド、2,5-ジブロモベンゾトリフルオライド、2,5-ジヨードベンゾトリフルオライド、1,4-ジクロロ-2,3,5,6-テトラフルオロベンゼン、1,4-ジブロモ-2,3,5,6-テトラフルオロベンゼン、1,4-ジヨード-2,3,5,6-テトラフルオロベンゼンなどが挙げられ、好ましくはp-ジクロロベンゼン、p-ジメチルスルフォニロキシベンゼン、2,5-ジクロロトルエン、2,5-ジクロロベンゾトリフルオライドである。
モノマー(4m)の具体例としては、4,4'-ジメチルスルフォニロキシビフェニル、4,4'-ジメチルスルフォニロキシ-3,3'-ジプロペニルビフェニル、4,4'-ジブロモビフェニル、4,4'-ジヨードビフェニル、4,4'-ジメチルスルフォニロキシ-3,3'-ジメチルビフェニル、4,4'-ジメチルスルフォニロキシ-3,3'-ジフルオロビフェニル、4,4'-ジメチルスルフォニロキシ-3,3'5,5'-テトラフルオロビフェニル、4,4'-ジブロモオクタフルオロビフェニル、4,4'-ジメチルスルフォニロキシオクタフルオロビフェニルなどが挙げられ、好ましくは4,4'-ジメチルスルフォニロキシビフェニル、4,4'-ジブロモビフェニル、4,4'-ジヨードビフェニル、4,4'-ジメチルスルフォニロキシ-3,3'-ジプロペニルビフェニルである。
モノマー(5m)の具体例としては、m-ジクロロベンゼン、m-ジブロモベンゼン、m-ジヨードベンゼン、m-ジメチルスルフォニロキシベンゼン、2,4-ジクロロトルエン、2,4-ジブロモトルエン、2,4-ジヨードトルエン、3,5-ジクロロトルエン、3,5-ジブロモトルエン、3,5-ジヨードトルエン、2,6-ジクロロトルエン、2,6-ジブロモトルエン、2,6-ジヨードトルエン、3,5-ジメチルスルフォニロキシトルエン、2,6-ジメチルスルフォニロキシトルエン、2,4-ジクロロベンゾトリフルオライド、2,4-ジブロモベンゾトリフルオライド、2,4-ジヨードベンゾトリフルオライド、3,5-ジクロロベンゾトリフルオライド、3,5-ジブロモトリフルオライド、3,5-ジヨードベンゾトリフルオライド、1,3-ジブロモ-2,4,5,6-テトラフルオロベンゼンなどが挙げられ、好ましくはm-ジクロロベンゼン、2,4-ジクロロトルエン、3,5-ジメチルスルフォニロキシトルエン、2,4-ジクロロベンゾトリフルオライドである。
繰返し単位(1)と繰返し単位(A)とを含有する共重合体を合成する場合には、上記一般式(1m)で表されるモノマー(1)と、上記一般式(2m)〜(5m)で表される化合物からなる群から選ばれる少なくとも1種のモノマー(A)との割合は、重合体中における単位(1)と単位(A)との割合と同様である。すなわち、モノマー(1)の使用量は、全モノマーの40〜3モル%が好ましく、より好ましくは35〜5モル%の範囲である。モノマー(A)の使用量は全モノマーの60〜97モル%が好ましく、より好ましくは65〜95モル%である。
特に、モノマー(A)としてモノマー(2m)を用いる場合には、その割合は、全モノマー中に、好ましくは10モル%以上、さらに好ましくは20モル%以上である。この範囲内であると、良好な溶解性、高分子量体が得られる。
特に、モノマー(3m)を用いる場合には、その割合は、全モノマー中に、好ましくは10モル%以上、さらに好ましくは20モル%以上である。この範囲内であると、良好な溶解性、高分子量体が得られる。
また、モノマー(4m)を用いる場合には、その割合は、全モノマー中に、好ましくは50モル%以下、さらに好ましくは30モル%以下である。この範囲内であると、良好な溶解性、高分子量体が得られる。
さらに、モノマー(5m)を用いる場合には、その割合は、全モノマー中に、好ましくは50モル%以下、さらに好ましくは30モル%以下である。
特に、モノマー(3m)を用いる場合には、その割合は、全モノマー中に、好ましくは10モル%以上、さらに好ましくは20モル%以上である。この範囲内であると、良好な溶解性、高分子量体が得られる。
また、モノマー(4m)を用いる場合には、その割合は、全モノマー中に、好ましくは50モル%以下、さらに好ましくは30モル%以下である。この範囲内であると、良好な溶解性、高分子量体が得られる。
さらに、モノマー(5m)を用いる場合には、その割合は、全モノマー中に、好ましくは50モル%以下、さらに好ましくは30モル%以下である。
本発明の共重合体を製造する際に使用される触媒は、遷移金属化合物を含む触媒系であり、この触媒系としては、(1)遷移金属塩および配位子となる化合物(以下、配位子成分という)、または配位子が配位された遷移金属錯体(銅塩を含む)、ならびに(2)還元剤を必須成分とし、さらに、重合速度を上げるために、「塩」を添加してもよい。
ここで、遷移金属塩としては、塩化ニッケル、臭化ニッケル、ヨウ化ニッケル、ニッケルアセチルアセトナートなどのニッケル化合物、塩化パラジウム、臭化パラジウム、ヨウ化パラジウムなどのパラジウム化合物、塩化鉄、臭化鉄、ヨウ化鉄などの鉄化合物、塩化コバルト、臭化コバルト、ヨウ化コバルトなどのコバルト化合物などが挙げられる。これらのうち特に、塩化ニッケル、臭化ニッケルなどが好ましい。
また、配位子成分としては、トリフェニルホスフィン、2,2'-ビピリジン、1,5-シクロオクタジエン、1,3-ビス(ジフェニルホスフィノ)プロパンなどが挙げられるが、トリフェニルホスフィン、2,2'-ビピリジンが好ましい。上記配位子成分である化合物は、1種単独で、あるいは2種以上を併用することができる。
ここで、遷移金属塩としては、塩化ニッケル、臭化ニッケル、ヨウ化ニッケル、ニッケルアセチルアセトナートなどのニッケル化合物、塩化パラジウム、臭化パラジウム、ヨウ化パラジウムなどのパラジウム化合物、塩化鉄、臭化鉄、ヨウ化鉄などの鉄化合物、塩化コバルト、臭化コバルト、ヨウ化コバルトなどのコバルト化合物などが挙げられる。これらのうち特に、塩化ニッケル、臭化ニッケルなどが好ましい。
また、配位子成分としては、トリフェニルホスフィン、2,2'-ビピリジン、1,5-シクロオクタジエン、1,3-ビス(ジフェニルホスフィノ)プロパンなどが挙げられるが、トリフェニルホスフィン、2,2'-ビピリジンが好ましい。上記配位子成分である化合物は、1種単独で、あるいは2種以上を併用することができる。
さらに、あらかじめ配位子が配位された遷移金属錯体としては、例えば、塩化ニッケルビス(トリフェニルホスフィン)、臭化ニッケルビス(トリフェニルホスフィン)、ヨウ化ニッケルビス(トリフェニルホスフィン)、硝酸ニッケルビス(トリフェニルホスフィン)、塩化ニッケル(2,2'-ビピリジン)、臭化ニッケル(2,2'-ビピリジン)、ヨウ化ニッケル(2,2'-ビピリジン)、硝酸ニッケル(2,2'-ビピリジン)、ビス(1,5-シクロオクタジエン)ニッケル、テトラキス(トリフェニルホスフィン)ニッケル、テトラキス(トリフェニルホスファイト)ニッケル、テトラキス(トリフェニルホスフィン)パラジウムなどが挙げられるが、塩化ニッケルビス(トリフェニルホスフィン)、塩化ニッケル(2,2'-ビピリジン)が好ましい。
上で用いられる触媒系に使用することができる上記還元剤としては、例えば、鉄、亜鉛、マンガン、アルミニウム、マグネシウム、ナトリウム、カルシウムなどを挙げることできるが、亜鉛、マグネシウム、マンガンが好ましい。これらの還元剤は、有機酸などの酸に接触させることにより、より活性化して用いることができる。
また、触媒系において使用することのできる「塩」としては、フッ化ナトリウム、塩化ナトリウム、臭化ナトリウム、ヨウ化ナトリウム、硫酸ナトリウムなどのナトリウム化合物、フッ化カリウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、硫酸カリウムなどのカリウム化合物、フッ化テトラエチルアンモニウム、塩化テトラエチルアンモニウム、臭化テトラエチルアンモニウム、ヨウ化テトラエチルアンモニウム、硫酸テトラエチルアンモニウムなどのアンモニウム化合物などが挙げられるが、臭化ナトリウム、ヨウ化ナトリウム、臭化カリウム、臭化テトラエチルアンモニウム、ヨウ化テトラエチルアンモニウムが好ましい。
また、触媒系において使用することのできる「塩」としては、フッ化ナトリウム、塩化ナトリウム、臭化ナトリウム、ヨウ化ナトリウム、硫酸ナトリウムなどのナトリウム化合物、フッ化カリウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、硫酸カリウムなどのカリウム化合物、フッ化テトラエチルアンモニウム、塩化テトラエチルアンモニウム、臭化テトラエチルアンモニウム、ヨウ化テトラエチルアンモニウム、硫酸テトラエチルアンモニウムなどのアンモニウム化合物などが挙げられるが、臭化ナトリウム、ヨウ化ナトリウム、臭化カリウム、臭化テトラエチルアンモニウム、ヨウ化テトラエチルアンモニウムが好ましい。
触媒系における各成分の使用割合は、遷移金属塩または遷移金属錯体が、上記モノマーの総計1モルに対し、通常、0.0001〜10モル、好ましくは0.01〜0.5モルである。0.0001モル未満では、重合反応が十分に進行せず、一方、10モルを超えると、分子量が低下するという問題がある。
触媒系において、遷移金属塩および配位子成分を用いる場合、この配位子成分の使用割合は、遷移金属塩1モルに対し、通常、0.1〜100モル、好ましくは1〜10モルである。0.1モル未満では、触媒活性が不十分となり、一方、100モルを超えると、分子量が低下するという問題がある。
また、触媒系における還元剤の使用割合は、上記モノマーの総計1モルに対し、通常、0.1〜100モル、好ましくは1〜10モルである。0.1モル未満では、重合が十分進行せず、一方、100モルを超えると、得られる重合体の精製が困難になるという問題がある。
さらに、触媒系に「塩」を使用する場合、その使用割合は、上記モノマーの総計1モルに対し、通常、0.001〜100モル、好ましくは0.01〜1モルである。0.001モル未満では、重合速度を上げる効果が不十分であり、一方、100モルを超えると、得られる重合体の精製が困難となるという問題がある。
触媒系において、遷移金属塩および配位子成分を用いる場合、この配位子成分の使用割合は、遷移金属塩1モルに対し、通常、0.1〜100モル、好ましくは1〜10モルである。0.1モル未満では、触媒活性が不十分となり、一方、100モルを超えると、分子量が低下するという問題がある。
また、触媒系における還元剤の使用割合は、上記モノマーの総計1モルに対し、通常、0.1〜100モル、好ましくは1〜10モルである。0.1モル未満では、重合が十分進行せず、一方、100モルを超えると、得られる重合体の精製が困難になるという問題がある。
さらに、触媒系に「塩」を使用する場合、その使用割合は、上記モノマーの総計1モルに対し、通常、0.001〜100モル、好ましくは0.01〜1モルである。0.001モル未満では、重合速度を上げる効果が不十分であり、一方、100モルを超えると、得られる重合体の精製が困難となるという問題がある。
本発明で使用することのできる重合溶媒としては、例えば、テトラヒドロフラン、シクロヘキサノン、ジメチルスルホキシド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、γ-ブチロラクトン、γ-ブチロラクタムなどが挙げられ、テトラヒドロフラン、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドンが好ましい。これらの重合溶媒は、十分に乾燥してから用いることが好ましい。
重合溶媒中における上記モノマーの総計の濃度は、通常、1〜90重量%、好ましくは5〜40重量%である。
また、本発明の共重合体を重合する際の重合温度は、通常、0〜200℃、好ましくは50〜120℃である。また、重合時間は、通常、0.5〜100時間、好ましくは1〜40時間である。
重合溶媒中における上記モノマーの総計の濃度は、通常、1〜90重量%、好ましくは5〜40重量%である。
また、本発明の共重合体を重合する際の重合温度は、通常、0〜200℃、好ましくは50〜120℃である。また、重合時間は、通常、0.5〜100時間、好ましくは1〜40時間である。
ここで、例えば、モノマー(1m)とモノマー(2m)を用いて上記の条件で重合させることにより、一般式:
(ここで、A,B,Z,R1〜R15,m及びnは前記の通りであり、p及びqは独立にそれぞれの繰返し単位の数を示し、p/qの比(即ち、上記二つの繰返し単位のモル比)は99/1〜20/80である。)
で表される共重合体が得られる。
本発明の共重合体の構造は、例えば、赤外線吸収スペクトルによって、1,230〜1,250cm-1のC−O−C吸収、1,640〜1,660cm-1のC=O吸収などにより確認でき、また、核磁気共鳴スペクトル(1H-NMR)により、6.8〜8.0ppmの芳香族プロトンのピークから、その構造を確認することができる。
で表される共重合体が得られる。
本発明の共重合体の構造は、例えば、赤外線吸収スペクトルによって、1,230〜1,250cm-1のC−O−C吸収、1,640〜1,660cm-1のC=O吸収などにより確認でき、また、核磁気共鳴スペクトル(1H-NMR)により、6.8〜8.0ppmの芳香族プロトンのピークから、その構造を確認することができる。
次に、本発明の伝導膜に用いられる、スルホン酸基を有する共重合体は、スルホン酸基を有しない上記共重合体に、スルホン化剤を用い、常法によりスルホン酸基導入することにより得ることができる。
スルホン酸基を導入する方法としては、例えば、上記スルホン酸基を有しない共重合体を、無水硫酸、発煙硫酸、クロルスルホン酸、硫酸、亜硫酸水素ナトリウムなどの公知のスルホン化剤を用いて、公知の条件でスルホン化することができる〔Polymer Preprints,Japan,Vol.42,No.3,p.730(1993);Polymer Preprints,Japan,Vol.43,No.3,p.736(1994);Polymer Preprints,Japan,Vol.42,No.7,p.2490〜2492(1993)〕。
スルホン酸基を導入する方法としては、例えば、上記スルホン酸基を有しない共重合体を、無水硫酸、発煙硫酸、クロルスルホン酸、硫酸、亜硫酸水素ナトリウムなどの公知のスルホン化剤を用いて、公知の条件でスルホン化することができる〔Polymer Preprints,Japan,Vol.42,No.3,p.730(1993);Polymer Preprints,Japan,Vol.43,No.3,p.736(1994);Polymer Preprints,Japan,Vol.42,No.7,p.2490〜2492(1993)〕。
すなわち、このスルホン化の反応条件としては、上記スルホン酸基を有しない共重合体を、無溶剤下、あるいは溶剤存在下で、上記スルホン化剤と反応させる。溶剤としては、例えばn-ヘキサンなどの炭化水素溶剤、テトラヒドロフラン、ジオキサンなどのエーテル系溶剤、ジメチルアセトアミド、ジメチルホルムアミド、ジメチルスルホキシドのような非プロトン系極性溶剤のほか、テトラクロロエタン、ジクロロエタン、クロロホルム、塩化メチレンなどのハロゲン化炭化水素などが挙げられる。反応温度は特に制限はないが、通常、−50〜200℃、好ましくは−10〜100℃である。また、反応時間は、通常、0.5〜1,000時間、好ましくは1〜200時間である。
このようにして得られる、本発明のスルホン酸基含有共重合体中の、スルホン酸基量は、0.5〜3ミリグラム当量/g、好ましくは0.8〜2.8ミリグラム当量/gである。0.5ミリグラム当量/g未満では、プロトン伝導性が上がらず、一方3ミリグラム当量/gを超えると、親水性が向上し、水溶性ポリマーとなってしまうか、また水溶性に至らずとも耐久性が低下する。
上記のスルホン酸基量は、モノマー(1)とモノマー(A)の使用割合、さらにモノマー(A)の種類、組合せを変えることにより、容易に調整することができる。
上記のスルホン酸基量は、モノマー(1)とモノマー(A)の使用割合、さらにモノマー(A)の種類、組合せを変えることにより、容易に調整することができる。
また、このようにして得られる本発明のスルホン酸基含有共重合体のスルホン化前の前駆体のポリマーの分子量は、ポリスチレン換算重量平均分子量で、1万〜100万、好ましくは2万〜80万である。1万未満では、成形フィルムにクラックが発生するなど、塗膜性が不十分であり、また強度的性質にも問題がある。一方、100万を超えると、溶解性が不十分となり、また溶液粘度が高く、加工性が不良になるなどの問題がある。
なお、本発明のスルホン基含有共重合体の構造は、赤外線吸収スペクトルによって、1,030〜1,045cm-1、1,160〜1,190cm-1のS=O吸収、1,130〜1,250cm-1のC−O−C吸収、1,640〜1,660cm-1のC=O吸収などにより確認でき、これらの組成比は、スルホン酸の中和滴定や、元素分析により知ることができる。また、核磁気共鳴スペクトル(1H-NMR)により、6.8〜8.0ppmの芳香族プロトンのピークから、その構造を確認することができる。
次に、本発明の伝導膜は、上記スルホン酸基含有共重合体からなるが、上記スルホン酸基含有共重合体以外に、硫酸、リン酸などの無機酸、カルボン酸を含む有機酸、適量の水などを併用しても良い。
本発明の伝導膜を製造するには、例えば本発明のスルホン酸基含有共重合体を溶剤に溶解したのち、キャスティングによりフィルム状に成形するキャスティング法や、溶融成形法などが挙げられる。
ここで、キャスティング法における溶剤としては、ジメチルアセトアミド、ジメチルホルムアミド、N-メチル-2-ピロリドン、ジメチルスルホキシドなどの非プロトン系極性溶剤などが挙げられる。これらの溶剤にはさらにメタノールなどのアルコール系溶剤が混合されていてもよい。
ここで、キャスティング法における溶剤としては、ジメチルアセトアミド、ジメチルホルムアミド、N-メチル-2-ピロリドン、ジメチルスルホキシドなどの非プロトン系極性溶剤などが挙げられる。これらの溶剤にはさらにメタノールなどのアルコール系溶剤が混合されていてもよい。
本発明の伝導膜は、例えば一次電池用電解質、二次電池用電解質、燃料電池用高分子固体電解質、表示素子、各種センサー、信号伝達媒体、固体コンデンサー、イオン交換膜などに利用可能なプロトン伝導性の伝導膜に利用可能である。
以下、実施例を挙げ本発明をさらに具体的に説明するが、本発明は以下の実施例に限定されるものではない。
なお、実施例中の各種の測定項目は、下記のようにして求めた。
なお、実施例中の各種の測定項目は、下記のようにして求めた。
[重量平均分子量]
スルホン化前の前駆体ポリマーの重量平均分子量は、溶媒にテトラヒドロフラン(THF)を用い、ゲルパーミエーションクロマトグラフィー(GPC)によって、ポリスチレン換算の分子量を求めた。
スルホン化前の前駆体ポリマーの重量平均分子量は、溶媒にテトラヒドロフラン(THF)を用い、ゲルパーミエーションクロマトグラフィー(GPC)によって、ポリスチレン換算の分子量を求めた。
[スルホン酸基の量]
得られたスルホン化ポリマーの水洗水がpH4〜6になるまで洗浄して、フリーの残存している酸を除去後、十分に水洗し、乾燥後、所定量を秤量し、THF/水の混合溶剤に溶解し、フェノールフタレインを指示薬とし、NaOHの標準液にて滴定し、中和点から、スルホン酸基の量(ミリグラム当量/g)を求めた。
得られたスルホン化ポリマーの水洗水がpH4〜6になるまで洗浄して、フリーの残存している酸を除去後、十分に水洗し、乾燥後、所定量を秤量し、THF/水の混合溶剤に溶解し、フェノールフタレインを指示薬とし、NaOHの標準液にて滴定し、中和点から、スルホン酸基の量(ミリグラム当量/g)を求めた。
[引張強度特性]
厚さ50μmに製膜したスルホン化ポリマーの幅3mm×長さ65mm(チャック間距離25mm)の試験片を作成し、引張試験機を用い室温の弾性率、破断強度、降伏強度及び伸びを測定した。
厚さ50μmに製膜したスルホン化ポリマーの幅3mm×長さ65mm(チャック間距離25mm)の試験片を作成し、引張試験機を用い室温の弾性率、破断強度、降伏強度及び伸びを測定した。
[耐折り曲げ性]
厚さ50μmに製膜したスルホン化ポリマーフィルムを耐折曲げ性試験機を用い屈曲回数166回/分、荷重200g、屈曲変形角度135℃の条件で破損までの折り曲げ回数500回以上のものを良とし、500回未満のものを不良とした。
厚さ50μmに製膜したスルホン化ポリマーフィルムを耐折曲げ性試験機を用い屈曲回数166回/分、荷重200g、屈曲変形角度135℃の条件で破損までの折り曲げ回数500回以上のものを良とし、500回未満のものを不良とした。
[プロトン伝導度の測定]
交流抵抗は5mm幅の短冊状膜試料の表面に白金線(直径0.5mm)を押し当て、恒温恒湿装置中に試料を保持し、白金線間の交流インピーダンス測定から求めた。85℃、相対湿度90%の環境下で交流10kHzにおけるインピーダンスを測定した。
抵抗測定装置として(株)NF回路設計ブロック社製ケミカルインピーダンス測定システムを、恒温恒湿器には(株)ヤマト化学社製のJW241を使用した。白金線は5mm間隔に5本押し当てて極間距離を5〜20mmに変化させ交流抵抗を測定した。
極間距離と抵抗の勾配から下記式に従って膜の比抵抗を算出し、比抵抗の逆数から交流インピーダンスを算出した。
比抵抗R[Ω・cm]=0.5[cm]×膜厚[cm]×抵抗極間勾配[Ω/cm]
交流抵抗は5mm幅の短冊状膜試料の表面に白金線(直径0.5mm)を押し当て、恒温恒湿装置中に試料を保持し、白金線間の交流インピーダンス測定から求めた。85℃、相対湿度90%の環境下で交流10kHzにおけるインピーダンスを測定した。
抵抗測定装置として(株)NF回路設計ブロック社製ケミカルインピーダンス測定システムを、恒温恒湿器には(株)ヤマト化学社製のJW241を使用した。白金線は5mm間隔に5本押し当てて極間距離を5〜20mmに変化させ交流抵抗を測定した。
極間距離と抵抗の勾配から下記式に従って膜の比抵抗を算出し、比抵抗の逆数から交流インピーダンスを算出した。
比抵抗R[Ω・cm]=0.5[cm]×膜厚[cm]×抵抗極間勾配[Ω/cm]
[熱的性質]
熱分解温度:
TGA(窒素下、20℃/分の昇温速度)により測定されたスルホン化ポリマーの分解温度を熱分解温度とした。
ガラス転移温度:
DSC(窒素下、20℃/分の昇温速度)により熱容量変化を示す温度をガラス転移温度とした。
耐熱水性:
厚さ50μmのスルホン化ポリマーフィルムを95℃の熱水に5時間浸漬し、浸漬後の寸法変化50%未満の場合を良、寸法変化が50%以上およびフィルムが溶解した場合を不良とした。
熱分解温度:
TGA(窒素下、20℃/分の昇温速度)により測定されたスルホン化ポリマーの分解温度を熱分解温度とした。
ガラス転移温度:
DSC(窒素下、20℃/分の昇温速度)により熱容量変化を示す温度をガラス転移温度とした。
耐熱水性:
厚さ50μmのスルホン化ポリマーフィルムを95℃の熱水に5時間浸漬し、浸漬後の寸法変化50%未満の場合を良、寸法変化が50%以上およびフィルムが溶解した場合を不良とした。
−実施例1−
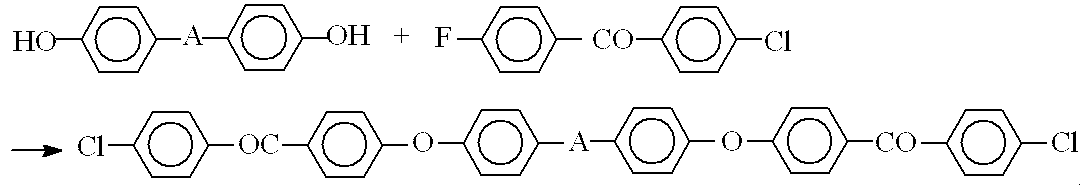
−−2,2-ビス[4-{4-(4-クロロベンゾイル)フェノキシ}フェニル]-1,1,1,3,3,3-ヘキサフルオロプロパン(BCPAF)の合成−−
2,2-ビス(4-ヒドロキシフェニル)-1,1,1,3,3,3-ヘキサフルオロプロパン(ビスフェノールAF)33.6g(100mmol)を撹拌機、Dean-stark管、冷却管、三方コック、温度計をつけた1L三口フラスコに計りとり、乾燥窒素置換した。ここにN,N-ジメチルアセトアミド150mL、トルエン75mLを加えて撹拌し溶解した。炭酸カリウム30.4g(220mmol)を加えたのち、オイルバスで反応液を130℃に加熱還流した。反応によって生成する水をトルエンと共沸させ、Dean-Starkトラップから系外に除きながら、反応温度を徐々に150℃まで上げた。約1時間後に大部分のトルエンが除去されたところで、反応液が80〜90℃になるまで冷却した。次に、4-クロロ-4'-フルオロベンゾフェノン58.7g(250mmol)を加え、反応温度115〜120℃で、7時間反応させた。
放冷後、ろ過により無機物を除いた。ろ液をメタノール500mLに注ぎ、生成した沈殿をろ過、メタノールで洗浄後、乾燥した。得られた粗生成物75gをトルエン165mLで再結晶し、目的物65g(85%)を得た。融点168〜170℃。
赤外吸収スペクトルを図1、NMRスペクトルを図2に示す。
−−2,2-ビス[4-{4-(4-クロロベンゾイル)フェノキシ}フェニル]-1,1,1,3,3,3-ヘキサフルオロプロパン(BCPAF)の合成−−
2,2-ビス(4-ヒドロキシフェニル)-1,1,1,3,3,3-ヘキサフルオロプロパン(ビスフェノールAF)33.6g(100mmol)を撹拌機、Dean-stark管、冷却管、三方コック、温度計をつけた1L三口フラスコに計りとり、乾燥窒素置換した。ここにN,N-ジメチルアセトアミド150mL、トルエン75mLを加えて撹拌し溶解した。炭酸カリウム30.4g(220mmol)を加えたのち、オイルバスで反応液を130℃に加熱還流した。反応によって生成する水をトルエンと共沸させ、Dean-Starkトラップから系外に除きながら、反応温度を徐々に150℃まで上げた。約1時間後に大部分のトルエンが除去されたところで、反応液が80〜90℃になるまで冷却した。次に、4-クロロ-4'-フルオロベンゾフェノン58.7g(250mmol)を加え、反応温度115〜120℃で、7時間反応させた。
放冷後、ろ過により無機物を除いた。ろ液をメタノール500mLに注ぎ、生成した沈殿をろ過、メタノールで洗浄後、乾燥した。得られた粗生成物75gをトルエン165mLで再結晶し、目的物65g(85%)を得た。融点168〜170℃。
赤外吸収スペクトルを図1、NMRスペクトルを図2に示す。
−実施例2−
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンとの50:50共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン22.8g(35mmol)、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパン26.7g(35mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.57g(2.4mmol)、よう化ナトリウム1.56g(10.4mmol)、トリフェニルホスフィン8.39g(32mmol)、亜鉛12.6g(192mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン(NMP)100mlを加え、70℃に加熱し、3時間撹拌し、重合反応を行った。反応液をメタノール:濃塩酸(容積比9:1)の混合液3,000mlに注ぎ、生成物を凝固沈殿させた。沈殿物を濾過、メタノールで洗浄後、真空乾燥し目的の共重合体35g(95%)を得た。得られた共重合体のIRスペクトルを図3に示す。GPCで求めた重合体の数平均分子量は29,400、重量平均分子量は60,500であった。ガラス転移温度168℃、窒素下での熱分解開始温度は336℃であった。また、フィルムは、弾性率2.6GPa、降伏応力94MPa、降伏伸び6%、引張強度87MPa、破断伸び10%を示す延性的な引張り特性を示した。
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンとの50:50共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン22.8g(35mmol)、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパン26.7g(35mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.57g(2.4mmol)、よう化ナトリウム1.56g(10.4mmol)、トリフェニルホスフィン8.39g(32mmol)、亜鉛12.6g(192mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン(NMP)100mlを加え、70℃に加熱し、3時間撹拌し、重合反応を行った。反応液をメタノール:濃塩酸(容積比9:1)の混合液3,000mlに注ぎ、生成物を凝固沈殿させた。沈殿物を濾過、メタノールで洗浄後、真空乾燥し目的の共重合体35g(95%)を得た。得られた共重合体のIRスペクトルを図3に示す。GPCで求めた重合体の数平均分子量は29,400、重量平均分子量は60,500であった。ガラス転移温度168℃、窒素下での熱分解開始温度は336℃であった。また、フィルムは、弾性率2.6GPa、降伏応力94MPa、降伏伸び6%、引張強度87MPa、破断伸び10%を示す延性的な引張り特性を示した。
(2)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンとの50:50共重合体のスルホン化物の調製−−
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図4に示す。
得られたスルホン化ポリマーの特性を表1に示す。
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図4に示す。
得られたスルホン化ポリマーの特性を表1に示す。
−実施例3−
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンとの70:30共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン24.4g(56mmol)、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパン18.3g(24mmol)に代えた他は、実施例1と同様に重合を実施し、共重合体35g(95%)を得た。GPCで求めた重合体の数平均分子量は27,800、重量平均分子量は60,200であった。得られた共重合体のIRスペクトルを図5に示す。ガラス転移温度155℃、窒素下での熱分解開始温度は384℃であった。
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンとの70:30共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン24.4g(56mmol)、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパン18.3g(24mmol)に代えた他は、実施例1と同様に重合を実施し、共重合体35g(95%)を得た。GPCで求めた重合体の数平均分子量は27,800、重量平均分子量は60,200であった。得られた共重合体のIRスペクトルを図5に示す。ガラス転移温度155℃、窒素下での熱分解開始温度は384℃であった。
(2)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンとの70:30共重合体のスルホン化物の調製−−
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して23g(93%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図6に示す。
得られたスルホン化ポリマーの特性を表1に示す。
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して23g(93%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図6に示す。
得られたスルホン化ポリマーの特性を表1に示す。
−比較例1−
(1)−−2,5-ジクロロ-4'-フェノキシベンゾフェノンの単独重合体の調製−−
実施例1で用いた2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン22.8g(35mmol)、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパン26.7g(35mmol)の代わりに2,5-ジクロロ-4'-フェノキシベンゾフェノン24.0g(70mmol)のみを用い、その他は実施例1と同様の重合、後処理操作をおこなった。
得られたポリマーのGPCで測定した分子量はMn34,800、Mw95,100であった。ガラス転移温度152℃、窒素下での5%熱分解温度は404℃であった。また、フィルムの引張り特性は、弾性率2.2GPa、引張強度2.1MPa、破断伸び3%で折り曲げると破損した。
(1)−−2,5-ジクロロ-4'-フェノキシベンゾフェノンの単独重合体の調製−−
実施例1で用いた2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン22.8g(35mmol)、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパン26.7g(35mmol)の代わりに2,5-ジクロロ-4'-フェノキシベンゾフェノン24.0g(70mmol)のみを用い、その他は実施例1と同様の重合、後処理操作をおこなった。
得られたポリマーのGPCで測定した分子量はMn34,800、Mw95,100であった。ガラス転移温度152℃、窒素下での5%熱分解温度は404℃であった。また、フィルムの引張り特性は、弾性率2.2GPa、引張強度2.1MPa、破断伸び3%で折り曲げると破損した。
(2)−−2,5-ジクロロ-4'-フェノキシベンゾフェノンの単独重合体のスルホン化物の調製−−
得られた単独重合体20gに濃硫酸200mlを加え、室温で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して23g(93%)のスルホン化ポリマーを得た。
得られたスルホン化ポリマーの特性を表1に示す。
得られた単独重合体20gに濃硫酸200mlを加え、室温で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して23g(93%)のスルホン化ポリマーを得た。
得られたスルホン化ポリマーの特性を表1に示す。
−実施例4−
−4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホン(BCPES)の製造法−
4,4'-ジクロロジフェニルスルホン(Bis-S)25.0g(100mmol)を撹拌機、Dean-stark管、冷却管、三方コック、温度計をつけた1L三口フラスコにとり、乾燥窒素置換した。ここにN,N-ジメチルアセトアミド150mL、トルエン75mLを加えて撹拌し溶解した。炭酸カリウム30.4g(220mmol)を加えたのち、オイルバスで反応液を130℃に加熱還流した。反応によって生成する水をトルエンと共沸させ、Dean-starkトラップから系外に除きながら、反応温度を徐々に150℃まで上げた。約1時間後に大部分のトルエンが除去されたところで、反応液が80〜90℃になるまで冷却した。次に、4-クロロ-4'-フルオロベンゾフェノン58.7g(250mmol)を加え、反応温度140〜150℃で、7時間反応させた。
放冷後、ろ過により無機物を除いた。ろ液をメタノール500mLに注ぎ、生成した沈殿をろ過、メタノールで洗浄後、乾燥した。得られた粗生成物66gをトルエン160mLで再結晶し、目的物58g(85%)を得た。融点191〜195℃。
赤外吸収スペクトルを図7に、NMRスペクトルを図8に示す。
−4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホン(BCPES)の製造法−
4,4'-ジクロロジフェニルスルホン(Bis-S)25.0g(100mmol)を撹拌機、Dean-stark管、冷却管、三方コック、温度計をつけた1L三口フラスコにとり、乾燥窒素置換した。ここにN,N-ジメチルアセトアミド150mL、トルエン75mLを加えて撹拌し溶解した。炭酸カリウム30.4g(220mmol)を加えたのち、オイルバスで反応液を130℃に加熱還流した。反応によって生成する水をトルエンと共沸させ、Dean-starkトラップから系外に除きながら、反応温度を徐々に150℃まで上げた。約1時間後に大部分のトルエンが除去されたところで、反応液が80〜90℃になるまで冷却した。次に、4-クロロ-4'-フルオロベンゾフェノン58.7g(250mmol)を加え、反応温度140〜150℃で、7時間反応させた。
放冷後、ろ過により無機物を除いた。ろ液をメタノール500mLに注ぎ、生成した沈殿をろ過、メタノールで洗浄後、乾燥した。得られた粗生成物66gをトルエン160mLで再結晶し、目的物58g(85%)を得た。融点191〜195℃。
赤外吸収スペクトルを図7に、NMRスペクトルを図8に示す。
−実施例5−
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホンとの60:40共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン24.4g(48mmol)、4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホン16.3g(32mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.57g(2.4mmol)、よう化ナトリウム1.56g(10.4mmol)、トリフェニルホスフィン8.39g(32mmol)、亜鉛12.6g(192mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン(NMP)100mlを加え、70℃に加熱し、3時間撹拌し、重合反応を行った。反応液をメタノール:濃塩酸(容積比9:1)の混合液3,000mlに注ぎ、生成物を凝固沈殿させた。沈殿物を濾過、メタノールで洗浄後、真空乾燥し目的の共重合体35g(95%)を得た。得られた共重合体のIRスペクトルを図9に示す。GPCで求めた重合体の数平均分子量は29,400、重量平均分子量は60,500であった。ガラス転移温度168℃、窒素下での熱分解開始温度は336℃であった。また、フィルムは、弾性率2.6GPa、降伏応力94MPa、降伏伸び6%、引張強度87MPa、破断伸び10%を示す延性的な引張り特性を示した。
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホンとの60:40共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン24.4g(48mmol)、4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホン16.3g(32mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.57g(2.4mmol)、よう化ナトリウム1.56g(10.4mmol)、トリフェニルホスフィン8.39g(32mmol)、亜鉛12.6g(192mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン(NMP)100mlを加え、70℃に加熱し、3時間撹拌し、重合反応を行った。反応液をメタノール:濃塩酸(容積比9:1)の混合液3,000mlに注ぎ、生成物を凝固沈殿させた。沈殿物を濾過、メタノールで洗浄後、真空乾燥し目的の共重合体35g(95%)を得た。得られた共重合体のIRスペクトルを図9に示す。GPCで求めた重合体の数平均分子量は29,400、重量平均分子量は60,500であった。ガラス転移温度168℃、窒素下での熱分解開始温度は336℃であった。また、フィルムは、弾性率2.6GPa、降伏応力94MPa、降伏伸び6%、引張強度87MPa、破断伸び10%を示す延性的な引張り特性を示した。
(2)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホンとの60:40共重合体のスルホン化物の調製−−
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図10に示す。
得られたスルホン化ポリマーの特性を表2に示す。
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図10に示す。
得られたスルホン化ポリマーの特性を表2に示す。
−実施例6−
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホンとの50:50共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン15.2g(35mmol)、4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホン22.8g(35mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.37g(2.1mmol)、よう化ナトリウム1.36g(9.1mmol)、トリフェニルホスフィン7.34g(28mmol)、亜鉛110g(168mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン(NMP)88mlを加え、70℃に加熱し、3時間撹拌し、重合反応を行った。反応液をメタノール:濃塩酸(容積比9:1)の混合液3,000mlに注ぎ、生成物を凝固沈殿させた。沈殿物を濾過、メタノールで洗浄後、真空乾燥し目的の共重合体32g(95%)を得た。得られた共重合体のIRスペクトルを図11に示す。GPCで求めた重合体の数平均分子量は29,400、重量平均分子量は60,500であった。ガラス転移温度168℃、窒素下での熱分解開始温度は336℃であった。また、フィルムは、弾性率2.6GPa、降伏応力94MPa、降伏伸び6%、引張強度87MPa、破断伸び10%を示す延性的な引張り特性を示した。
(1)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホンとの50:50共重合体の調製−−
2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン15.2g(35mmol)、4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホン22.8g(35mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.37g(2.1mmol)、よう化ナトリウム1.36g(9.1mmol)、トリフェニルホスフィン7.34g(28mmol)、亜鉛110g(168mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン(NMP)88mlを加え、70℃に加熱し、3時間撹拌し、重合反応を行った。反応液をメタノール:濃塩酸(容積比9:1)の混合液3,000mlに注ぎ、生成物を凝固沈殿させた。沈殿物を濾過、メタノールで洗浄後、真空乾燥し目的の共重合体32g(95%)を得た。得られた共重合体のIRスペクトルを図11に示す。GPCで求めた重合体の数平均分子量は29,400、重量平均分子量は60,500であった。ガラス転移温度168℃、窒素下での熱分解開始温度は336℃であった。また、フィルムは、弾性率2.6GPa、降伏応力94MPa、降伏伸び6%、引張強度87MPa、破断伸び10%を示す延性的な引張り特性を示した。
(2)−−2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンと4,4'-ビス[(4-クロロベンゾイル)フェノキシ]ジフェニルスルホンとの50:50共重合体のスルホン化物の調製−−
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図12に示す。
得られたスルホン化ポリマーの特性を表2に示す。
得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図12に示す。
得られたスルホン化ポリマーの特性を表2に示す。
−実施例7−
(1)実施例2において、2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンの使用量を27.36g(42mmol)に、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンの使用量を10.68g(14mmol)に変え、さらに4,4'-ジクロロベンゾフェノン3.51g(14mmol)を加えた以外は実施例2と同様にして、重合反応を行い、共重合体34.2g(94%)を得た。
GPCで求めた重合体の重量平均分子量は109,800であった。得られた赤外線吸収スペクトルを図13に示す。
(2)得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図14に示す。
得られたスルホン化ポリマーの特性を表2に示す。
(1)実施例2において、2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノンの使用量を27.36g(42mmol)に、2,2'-ビス[4-(4-クロロベンゾイル)フェノキシ]ジフェニル-1,1,1,3,3,3-ヘキサフルオロプロパンの使用量を10.68g(14mmol)に変え、さらに4,4'-ジクロロベンゾフェノン3.51g(14mmol)を加えた以外は実施例2と同様にして、重合反応を行い、共重合体34.2g(94%)を得た。
GPCで求めた重合体の重量平均分子量は109,800であった。得られた赤外線吸収スペクトルを図13に示す。
(2)得られた共重合体20gに濃硫酸200mlを加え、60℃で5時間撹拌した。反応液を水に注ぎ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して25g(96%)のスルホン化ポリマーを得た。赤外線吸収スペクトルを図14に示す。
得られたスルホン化ポリマーの特性を表2に示す。
−実施例8−
(オリゴマーの合成)
撹拌機、温度計、冷却管、Dean-Stark管、窒素導入の三方コックをとりつけた1Lの三つ口のフラスコに、2,2-ビス(4-ヒドロキシフェニル)-1,1,1,3,3,3-ヘキサフルオロプロパン(ビスフェノールAF)67.3g(0.20モル)、4,4'-ジクロロベンゾフェノン(4,4'-DCBP)60.3g(0.24モル)、炭酸カリウム71.9g(0.52モル)、N,N-ジメチルアセトアミド(DMAc)300mL、トルエン150mLをとり、オイルバス中、窒素雰囲気下で加熱し撹拌下130℃で反応させた。反応により生成する水をトルエンと共沸させ、Dean-Stark管で系外に除去しながら反応させると、約3時間で水の生成がほとんど認められなくなった。反応温度を130℃から徐々に150℃まで上げた。その後、反応温度を徐々に150℃まで上げながら大部分のトルエンを除去し、150℃で10時間反応を続けた後、4,4'-DCBP10.0g(0.40モル)を加え、さらに5時間反応した。得られた反応液を放冷後、副生した無機化合物の沈殿物を濾過除去し、濾液を4Lのメタノール中に投入した。沈殿した生成物を濾別、回収し乾燥後、テトラヒドロフラン300mLに溶解した。これをメタノール4Lに再沈殿し、目的の化合物95g(収率85%)を得た。
(オリゴマーの合成)
撹拌機、温度計、冷却管、Dean-Stark管、窒素導入の三方コックをとりつけた1Lの三つ口のフラスコに、2,2-ビス(4-ヒドロキシフェニル)-1,1,1,3,3,3-ヘキサフルオロプロパン(ビスフェノールAF)67.3g(0.20モル)、4,4'-ジクロロベンゾフェノン(4,4'-DCBP)60.3g(0.24モル)、炭酸カリウム71.9g(0.52モル)、N,N-ジメチルアセトアミド(DMAc)300mL、トルエン150mLをとり、オイルバス中、窒素雰囲気下で加熱し撹拌下130℃で反応させた。反応により生成する水をトルエンと共沸させ、Dean-Stark管で系外に除去しながら反応させると、約3時間で水の生成がほとんど認められなくなった。反応温度を130℃から徐々に150℃まで上げた。その後、反応温度を徐々に150℃まで上げながら大部分のトルエンを除去し、150℃で10時間反応を続けた後、4,4'-DCBP10.0g(0.40モル)を加え、さらに5時間反応した。得られた反応液を放冷後、副生した無機化合物の沈殿物を濾過除去し、濾液を4Lのメタノール中に投入した。沈殿した生成物を濾別、回収し乾燥後、テトラヒドロフラン300mLに溶解した。これをメタノール4Lに再沈殿し、目的の化合物95g(収率85%)を得た。
得られた重合体のGPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は4,200、重量平均分子量は8,300であった。赤外線吸収スペクトルを図15に示す。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは110℃、熱分解温度は498℃であった。
得られた重合体は式(7):
得られた重合体は式(7):
で表される構造を有することが推定され、該構造と上記の数平均分子量とから、nの平均値は7.8と求められた。
−実施例9−
(オリゴマーの合成)
実施例8で用いたモノマーの最初の仕込量をビスフェノールAF67.3g(0.20モル)、4,4'-DCBP58.3g(0.232モル)、後添加する4,4'-DCBPの仕込み量を2g(0.029モル)に変えた以外は、実施例8と同様に重合を行った。重合体は88%の収率で71g得られた。GPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は7,300、重量平均分子量は16,400であった。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは129℃、熱分解温度は516℃であった。得られた重合体は式(7)で表され、n=13.9(平均値)のものである。
(オリゴマーの合成)
実施例8で用いたモノマーの最初の仕込量をビスフェノールAF67.3g(0.20モル)、4,4'-DCBP58.3g(0.232モル)、後添加する4,4'-DCBPの仕込み量を2g(0.029モル)に変えた以外は、実施例8と同様に重合を行った。重合体は88%の収率で71g得られた。GPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は7,300、重量平均分子量は16,400であった。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは129℃、熱分解温度は516℃であった。得られた重合体は式(7)で表され、n=13.9(平均値)のものである。
−実施例10−
(オリゴマーの合成)
実施例8で用いたモノマーの最初の仕込量をビスフェノールAF67.3g(0.20モル)、4,4'-DCBP53.5g(0.214モル)、後添加する4,4'-DCBPの仕込み量を3.3g(0.0133モル)、炭酸カリウムの使用量を34.6g(0.251モル)に変えた以外は、実施例8と同様に重合を行った。重合体は93%の収率で98g得られた。
GPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は9,900、重量平均分子量は22,000あった。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは151℃、熱分解温度は524℃であった。得られた重合体は式(7)で表され、n=18.9(平均値)のものである。
(オリゴマーの合成)
実施例8で用いたモノマーの最初の仕込量をビスフェノールAF67.3g(0.20モル)、4,4'-DCBP53.5g(0.214モル)、後添加する4,4'-DCBPの仕込み量を3.3g(0.0133モル)、炭酸カリウムの使用量を34.6g(0.251モル)に変えた以外は、実施例8と同様に重合を行った。重合体は93%の収率で98g得られた。
GPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は9,900、重量平均分子量は22,000あった。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは151℃、熱分解温度は524℃であった。得られた重合体は式(7)で表され、n=18.9(平均値)のものである。
−実施例11−
(オリゴマーの合成)
攪拌機、温度計、冷却管、デイーンスターク管、窒素導入の三方コックをとりつけた1Lの三ツ口フラスコに、ビスフェノールAF67.3g(0.20モル)、4,4'-DCPB50.2g(0.20モル)、炭酸カリウム71.9g(0.52モル)、スルホラン300ml、トルエン150mlをとり、オイルバス中、窒素雰囲気下で加熱し攪拌下130℃で反応させた。反応のより生成する水をトルエンと共沸させ、Dean-Stark管で系外に除去しながら反応させると、約3時間で水の生成が認められなくなった。反応温度を130℃から徐々に160℃まで上げた。その後、反応温度を180℃まで上げながら大部分のトルエンを除去し、180℃で16時間反応を続けたあと、4,4'-DCPB10.0g(0.040モル)を加え、さらに4時間反応した。得られた反応液を放冷後、副生した無機化合物の沈殿物を濾過除去し、濾液を4Lのメタノール中に投入した。沈殿した生成物を濾別、回収し乾燥後、THF300mlに溶解した。これをメタノール4Lに再沈殿し、目的の化合物82.5g(収率80.2%)を得た。
(オリゴマーの合成)
攪拌機、温度計、冷却管、デイーンスターク管、窒素導入の三方コックをとりつけた1Lの三ツ口フラスコに、ビスフェノールAF67.3g(0.20モル)、4,4'-DCPB50.2g(0.20モル)、炭酸カリウム71.9g(0.52モル)、スルホラン300ml、トルエン150mlをとり、オイルバス中、窒素雰囲気下で加熱し攪拌下130℃で反応させた。反応のより生成する水をトルエンと共沸させ、Dean-Stark管で系外に除去しながら反応させると、約3時間で水の生成が認められなくなった。反応温度を130℃から徐々に160℃まで上げた。その後、反応温度を180℃まで上げながら大部分のトルエンを除去し、180℃で16時間反応を続けたあと、4,4'-DCPB10.0g(0.040モル)を加え、さらに4時間反応した。得られた反応液を放冷後、副生した無機化合物の沈殿物を濾過除去し、濾液を4Lのメタノール中に投入した。沈殿した生成物を濾別、回収し乾燥後、THF300mlに溶解した。これをメタノール4Lに再沈殿し、目的の化合物82.5g(収率80.2%)を得た。
得られた重合体のGPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は16,400、重量平均分子量は37,400であった。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは162℃、熱分解温度は535℃であった。得られた重合体は式(7)で表され、n=31.6(平均値)のものである。
−実施例12−
実施例8において4,4'-DCBPに代えて、ビス(4-クロロフェニル)スルホン(BCPS)を使用し、その最初の仕込量を53.5g(0.214モル)とし、後添加する仕込み量を3.3g(0.0133モル)としたこと、また炭酸カリウムの使用量を58.0g(0.42モル)に変えた以外は、実施例8と同様にして重合を行った。重合体は96%の収率で120g得られた。
GPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は4,600、重量平均分子量は7,600であった。赤外吸収スペクトルを図16に示す。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは158℃、熱分解温度は513℃であった。
実施例8において4,4'-DCBPに代えて、ビス(4-クロロフェニル)スルホン(BCPS)を使用し、その最初の仕込量を53.5g(0.214モル)とし、後添加する仕込み量を3.3g(0.0133モル)としたこと、また炭酸カリウムの使用量を58.0g(0.42モル)に変えた以外は、実施例8と同様にして重合を行った。重合体は96%の収率で120g得られた。
GPC(THF溶媒)で求めたポリスチレン換算の数平均分子量は4,600、重量平均分子量は7,600であった。赤外吸収スペクトルを図16に示す。また、得られた重合体はTHF、NMP、DMAc、スルホランなどに可溶で、Tgは158℃、熱分解温度は513℃であった。
得られた重合体は式(8):
で表される構造を有するものと推定され、実施例8の場合の同様の手法でnは平均値で8.0と求められた。
−実施例13−
(1)重合体の合成
実施例10で得られたオリゴマー28.4g(2.87mmol)、2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン(DCPPB)29.2g(67.1mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.37g(2.1mmol)、よう化ナトリウム1.36g(9.07mmol)、トリフェニルホスフィン7.34g(28.0mmol)、亜鉛末11.0g(168mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン130mlを加え、80℃に加熱し、4時間攪拌し、重合をおこなった。重合溶液をTHFで希釈し、塩酸/メタノールで凝固回収し、メタノール洗滌を繰り返し、THFで溶解、メタノールへ再沈殿による精製し、濾集したポリマーを真空乾燥し目的の共重合体50.7g(96%)を得た。 GPC(THF)で求めたポリスチレン換算の数平均分子量は40,000、重量平均分子量は145,000であった。赤外吸収スペクトルを図17に示す。
(1)重合体の合成
実施例10で得られたオリゴマー28.4g(2.87mmol)、2,5-ジクロロ-4'-(4-フェノキシ)フェノキシベンゾフェノン(DCPPB)29.2g(67.1mmol)、ビス(トリフェニルホスフィン)ニッケルジクロリド1.37g(2.1mmol)、よう化ナトリウム1.36g(9.07mmol)、トリフェニルホスフィン7.34g(28.0mmol)、亜鉛末11.0g(168mmol)をフラスコにとり、乾燥窒素置換した。N-メチル-2-ピロリドン130mlを加え、80℃に加熱し、4時間攪拌し、重合をおこなった。重合溶液をTHFで希釈し、塩酸/メタノールで凝固回収し、メタノール洗滌を繰り返し、THFで溶解、メタノールへ再沈殿による精製し、濾集したポリマーを真空乾燥し目的の共重合体50.7g(96%)を得た。 GPC(THF)で求めたポリスチレン換算の数平均分子量は40,000、重量平均分子量は145,000であった。赤外吸収スペクトルを図17に示す。
(2)スルホン化ポリマーの調製
上記の(1)で得た共重合体25gを500mlのセパラブルフラスコに入れ、96%硫酸250mlを加え、窒素気流下で24時間攪拌した。得られた溶液を大量のイオン交換水の中に注ぎ入れ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して、29g(96%)のスルホン化ポリマーを得た。赤外吸収スペクトルを図18に示す。
得られたスルホン化ポリマーをNMPに溶解させ、キャスト法にてフィルムを作成した。スルホン化ポリマーのスルホン化当量は1.72ミリグラム当量/gであった。スルホン化ポリマーの特性を表3に示す。
上記の(1)で得た共重合体25gを500mlのセパラブルフラスコに入れ、96%硫酸250mlを加え、窒素気流下で24時間攪拌した。得られた溶液を大量のイオン交換水の中に注ぎ入れ、ポリマーを沈殿させた。洗浄水のpHが5になるまでポリマーの洗浄を繰り返した。乾燥して、29g(96%)のスルホン化ポリマーを得た。赤外吸収スペクトルを図18に示す。
得られたスルホン化ポリマーをNMPに溶解させ、キャスト法にてフィルムを作成した。スルホン化ポリマーのスルホン化当量は1.72ミリグラム当量/gであった。スルホン化ポリマーの特性を表3に示す。
−実施例14−
(1)重合体の合成
実施例12で得られたオリゴマー13.8gとDCPPB11.75g(27mmol)をモノマー、ビス(トリフェニルホスフィン)ニッケルクロライド0.589g(0.9mmol)、ヨウ化ナトリウム0.585g(3.9mmol)、トリフェニルホスフィン3.148g(12mmol)、亜鉛末4.701g(72mmol)を環流管、三方コックを取り付けた三口フラスコに入れ、70℃のオイルバスにつけ、3回窒素置換を行った後、減圧下で1時間おいた。その後、窒素雰囲気下に戻し、N-メチル-2-ピロリドン60ml加え、80℃で重合を行った。10時間反応後、N-メチル-2-ピロリドン50mlで希釈し、1:10塩酸/メタノールへ再沈しポリマーを析出させ白色の粉末を得た。ポリマーを回収し60℃で真空乾燥した。収量は22.5g(収率96%)であった。GPC(THF)で求めたポリスチレン換算の数平均分子量は33,000、重量平均分子量は138,000であった。赤外吸収スペクトルを図19に示す。
(1)重合体の合成
実施例12で得られたオリゴマー13.8gとDCPPB11.75g(27mmol)をモノマー、ビス(トリフェニルホスフィン)ニッケルクロライド0.589g(0.9mmol)、ヨウ化ナトリウム0.585g(3.9mmol)、トリフェニルホスフィン3.148g(12mmol)、亜鉛末4.701g(72mmol)を環流管、三方コックを取り付けた三口フラスコに入れ、70℃のオイルバスにつけ、3回窒素置換を行った後、減圧下で1時間おいた。その後、窒素雰囲気下に戻し、N-メチル-2-ピロリドン60ml加え、80℃で重合を行った。10時間反応後、N-メチル-2-ピロリドン50mlで希釈し、1:10塩酸/メタノールへ再沈しポリマーを析出させ白色の粉末を得た。ポリマーを回収し60℃で真空乾燥した。収量は22.5g(収率96%)であった。GPC(THF)で求めたポリスチレン換算の数平均分子量は33,000、重量平均分子量は138,000であった。赤外吸収スペクトルを図19に示す。
(2)スルホン化ポリマーの調製
上記の(1)で得られたポリマー25gに濃硫酸250mlを加え窒素雰囲気下、室温で24h攪拌しスルホン化を行った。これを純水に再沈しスルホン化ポリマーを析出させた。数回水を換え、pH5になるまでポリマーの洗浄を行った。スルホン化ポリマーを回収し、80℃で熱風乾燥した。スルホン化ポリマーの収量は29g(95%)であった。赤外吸収スペクトルを図20に示す。得られたスルホン化ポリマーをNMPに溶解させ、キャスト法にてフィルムを作成した。スルホン化ポリマーのスルホン化当量は1.95ミリグラム当量/gであった。スルホン化ポリマーの特性を表3に示す。
上記の(1)で得られたポリマー25gに濃硫酸250mlを加え窒素雰囲気下、室温で24h攪拌しスルホン化を行った。これを純水に再沈しスルホン化ポリマーを析出させた。数回水を換え、pH5になるまでポリマーの洗浄を行った。スルホン化ポリマーを回収し、80℃で熱風乾燥した。スルホン化ポリマーの収量は29g(95%)であった。赤外吸収スペクトルを図20に示す。得られたスルホン化ポリマーをNMPに溶解させ、キャスト法にてフィルムを作成した。スルホン化ポリマーのスルホン化当量は1.95ミリグラム当量/gであった。スルホン化ポリマーの特性を表3に示す。
Claims (5)
- 一般式:
[式中、nは1〜50の整数である。]
で表されるハロゲン化芳香族化合物。 - 請求項1に記載の化合物である、2,2-ビス[4-{4-(4-クロロベンゾイル)フェノキシ}フェニル]-1,1,1,3,3,3-ヘキサフルオロプロパン。
-
[式中、nは1〜50の整数である。]
で表されるハロゲン化芳香族化合物。 -
[式中、nは1〜50の整数である。]
で表されるハロゲン化芳香族化合物。 -
[式中、nは1〜50の整数である。]
で表されるハロゲン化芳香族化合物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007025660A JP2007169657A (ja) | 2001-03-30 | 2007-02-05 | ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001101586 | 2001-03-30 | ||
| JP2001230650 | 2001-07-30 | ||
| JP2007025660A JP2007169657A (ja) | 2001-03-30 | 2007-02-05 | ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001303964A Division JP3956661B2 (ja) | 2001-03-30 | 2001-09-28 | ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2007169657A true JP2007169657A (ja) | 2007-07-05 |
Family
ID=38296609
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007025660A Pending JP2007169657A (ja) | 2001-03-30 | 2007-02-05 | ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2007169657A (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010070657A (ja) * | 2008-09-19 | 2010-04-02 | Kaneka Corp | ポリエーテル類の製造方法 |
| CN101643548B (zh) * | 2008-11-13 | 2012-05-16 | 南京理工大学 | 直接交联型质子交换膜的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6465129A (en) * | 1987-09-04 | 1989-03-10 | Mitsubishi Gas Chemical Co | Production of aromatic polyether |
| JPH02159A (ja) * | 1987-11-17 | 1990-01-05 | Imperial Chem Ind Plc <Ici> | 芳香族化合物 |
| JPH10338745A (ja) * | 1997-06-10 | 1998-12-22 | Sumitomo Chem Co Ltd | 芳香族ポリエーテル系重合体の製造方法 |
-
2007
- 2007-02-05 JP JP2007025660A patent/JP2007169657A/ja active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6465129A (en) * | 1987-09-04 | 1989-03-10 | Mitsubishi Gas Chemical Co | Production of aromatic polyether |
| JPH02159A (ja) * | 1987-11-17 | 1990-01-05 | Imperial Chem Ind Plc <Ici> | 芳香族化合物 |
| JPH10338745A (ja) * | 1997-06-10 | 1998-12-22 | Sumitomo Chem Co Ltd | 芳香族ポリエーテル系重合体の製造方法 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010070657A (ja) * | 2008-09-19 | 2010-04-02 | Kaneka Corp | ポリエーテル類の製造方法 |
| CN101643548B (zh) * | 2008-11-13 | 2012-05-16 | 南京理工大学 | 直接交联型质子交换膜的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3956661B2 (ja) | ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 | |
| KR100734458B1 (ko) | 전자 흡인성기 및 전자 공여성기를 포함하는 단량체, 이를이용한 공중합체, 및 양성자 전도막 | |
| JP4131216B2 (ja) | ポリアリーレンおよびその製造方法、ならびに高分子固体電解質およびプロトン伝導膜 | |
| US6555626B2 (en) | Polyarylene copolymers and proton-conductive membrane | |
| JP4428181B2 (ja) | ニトリル型疎水性ブロックを有するスルホン化ポリマーおよび固体高分子電解質 | |
| KR100911970B1 (ko) | 할로겐화 방향족 화합물, 이 화합물의 (공)중합체, 및 이 (공)중합체를 포함하는 프로톤 전도막 | |
| JP2006299080A (ja) | 多スルホン化芳香族化合物、スルホン化ポリマー、固体高分子電解質およびプロトン伝導膜 | |
| JP3852108B2 (ja) | ポリアリーレン系重合体およびプロトン伝導膜 | |
| JP3840942B2 (ja) | 分岐状ポリアリーレン系共重合体の製法、スルホン化分岐状ポリアリーレン系共重合体の製法およびスルホン化分岐状ポリアリーレン系共重合体からなるプロトン伝導膜 | |
| JP3841168B2 (ja) | 新規な含リン芳香族ジハロゲン化合物、ポリアリーレン重合体、スルホン化ポリアリーレン重合体およびこれら重合体の製造方法、ならびにプロトン伝導膜 | |
| JP2004244517A (ja) | ポリアリーレン系共重合体およびその製造方法、ならびにプロトン伝導膜 | |
| JP2007169657A (ja) | ハロゲン化芳香族化合物、該化合物の重合体、及び該重合体からなるプロトン伝導膜 | |
| KR100793560B1 (ko) | 할로겐화 방향족 화합물, 이 화합물의 중합체 및 이중합체를 포함하는 양성자 전도막 | |
| JP4107090B2 (ja) | ハロゲン化芳香族化合物、該化合物の(共)重合体、および該(共)重合体からなるプロトン伝導膜 | |
| JP2006335816A (ja) | プロトン伝導体組成物およびプロトン伝導性複合膜 | |
| JP2006299081A (ja) | 多核芳香族スルホン酸誘導体、スルホン化ポリマー、固体高分子電解質およびプロトン伝導膜 | |
| JP3879541B2 (ja) | スルホン酸基含有重合体の製造方法 | |
| JP2005149810A (ja) | 高分子電解質、その製造方法およびプロトン伝導膜 | |
| JP2006335814A (ja) | プロトン伝導体組成物およびプロトン伝導性複合膜 | |
| JP2006328430A (ja) | スルホン酸基含有重合体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A977 | Report on retrieval |
Effective date: 20101206 Free format text: JAPANESE INTERMEDIATE CODE: A971007 |
|
| A131 | Notification of reasons for refusal |
Effective date: 20110111 Free format text: JAPANESE INTERMEDIATE CODE: A131 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20110913 |