JP2006141292A - 微生物の破砕・核酸抽出方法、この方法を用いたキット、及びその製造方法 - Google Patents
微生物の破砕・核酸抽出方法、この方法を用いたキット、及びその製造方法 Download PDFInfo
- Publication number
- JP2006141292A JP2006141292A JP2004336245A JP2004336245A JP2006141292A JP 2006141292 A JP2006141292 A JP 2006141292A JP 2004336245 A JP2004336245 A JP 2004336245A JP 2004336245 A JP2004336245 A JP 2004336245A JP 2006141292 A JP2006141292 A JP 2006141292A
- Authority
- JP
- Japan
- Prior art keywords
- beads
- container
- nucleic acid
- treatment
- cell disruption
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 103
- 108020004707 nucleic acids Proteins 0.000 title claims abstract description 74
- 102000039446 nucleic acids Human genes 0.000 title claims abstract description 74
- 150000007523 nucleic acids Chemical class 0.000 title claims abstract description 74
- 238000000605 extraction Methods 0.000 title claims description 55
- 238000004519 manufacturing process Methods 0.000 title claims description 7
- 230000000813 microbial effect Effects 0.000 title description 2
- 239000011324 bead Substances 0.000 claims abstract description 200
- 238000011282 treatment Methods 0.000 claims abstract description 135
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 claims abstract description 108
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims abstract description 96
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims abstract description 66
- 239000004094 surface-active agent Substances 0.000 claims abstract description 56
- 244000005700 microbiome Species 0.000 claims abstract description 52
- 239000007788 liquid Substances 0.000 claims abstract description 40
- 238000004090 dissolution Methods 0.000 claims abstract description 17
- 210000004027 cell Anatomy 0.000 claims description 131
- 239000000243 solution Substances 0.000 claims description 67
- 239000000203 mixture Substances 0.000 claims description 21
- 210000002421 cell wall Anatomy 0.000 claims description 11
- 239000011259 mixed solution Substances 0.000 claims description 11
- 239000000463 material Substances 0.000 claims description 10
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 9
- 230000008569 process Effects 0.000 claims description 9
- 229910052726 zirconium Inorganic materials 0.000 claims description 9
- 230000005484 gravity Effects 0.000 claims description 7
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 claims description 4
- 229910001928 zirconium oxide Inorganic materials 0.000 claims description 4
- 229910001220 stainless steel Inorganic materials 0.000 claims description 3
- 239000010935 stainless steel Substances 0.000 claims description 3
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 3
- 229910052721 tungsten Inorganic materials 0.000 claims description 3
- 239000010937 tungsten Substances 0.000 claims description 3
- 239000000523 sample Substances 0.000 claims 4
- 239000012488 sample solution Substances 0.000 claims 1
- 108090000623 proteins and genes Proteins 0.000 abstract description 21
- 238000010438 heat treatment Methods 0.000 abstract description 20
- 230000035945 sensitivity Effects 0.000 abstract description 16
- 238000012545 processing Methods 0.000 abstract description 14
- 238000012360 testing method Methods 0.000 abstract description 13
- 238000001514 detection method Methods 0.000 abstract description 12
- 238000007796 conventional method Methods 0.000 abstract description 11
- 238000012869 ethanol precipitation Methods 0.000 abstract description 11
- 239000006228 supernatant Substances 0.000 abstract description 11
- 241000192125 Firmicutes Species 0.000 abstract description 10
- 241000233866 Fungi Species 0.000 abstract description 7
- 230000002068 genetic effect Effects 0.000 abstract description 6
- 108020004414 DNA Proteins 0.000 description 51
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- 230000004544 DNA amplification Effects 0.000 description 20
- 239000012139 lysis buffer Substances 0.000 description 20
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 19
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 19
- 241000894006 Bacteria Species 0.000 description 18
- 230000001580 bacterial effect Effects 0.000 description 18
- 230000000694 effects Effects 0.000 description 17
- 239000011521 glass Substances 0.000 description 16
- 102000004169 proteins and genes Human genes 0.000 description 16
- 238000007400 DNA extraction Methods 0.000 description 12
- FFYPMLJYZAEMQB-UHFFFAOYSA-N diethyl pyrocarbonate Chemical compound CCOC(=O)OC(=O)OCC FFYPMLJYZAEMQB-UHFFFAOYSA-N 0.000 description 12
- 238000002156 mixing Methods 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 239000002244 precipitate Substances 0.000 description 10
- 239000012153 distilled water Substances 0.000 description 9
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 244000000010 microbial pathogen Species 0.000 description 8
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000006872 improvement Effects 0.000 description 6
- 238000011084 recovery Methods 0.000 description 6
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- -1 Labiase Proteins 0.000 description 5
- 241000191967 Staphylococcus aureus Species 0.000 description 5
- 229920004890 Triton X-100 Polymers 0.000 description 5
- 239000013504 Triton X-100 Substances 0.000 description 5
- 230000003321 amplification Effects 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 238000012790 confirmation Methods 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 238000003199 nucleic acid amplification method Methods 0.000 description 5
- 238000001556 precipitation Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 102000016943 Muramidase Human genes 0.000 description 4
- 108010014251 Muramidase Proteins 0.000 description 4
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 4
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 239000002223 garnet Substances 0.000 description 4
- ZJYYHGLJYGJLLN-UHFFFAOYSA-N guanidinium thiocyanate Chemical compound SC#N.NC(N)=N ZJYYHGLJYGJLLN-UHFFFAOYSA-N 0.000 description 4
- 235000010335 lysozyme Nutrition 0.000 description 4
- 108700004121 sarkosyl Proteins 0.000 description 4
- KSAVQLQVUXSOCR-UHFFFAOYSA-M sodium lauroyl sarcosinate Chemical compound [Na+].CCCCCCCCCCCC(=O)N(C)CC([O-])=O KSAVQLQVUXSOCR-UHFFFAOYSA-M 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000004925 denaturation Methods 0.000 description 3
- 230000036425 denaturation Effects 0.000 description 3
- 239000004325 lysozyme Substances 0.000 description 3
- 229960000274 lysozyme Drugs 0.000 description 3
- 239000013642 negative control Substances 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 229940016590 sarkosyl Drugs 0.000 description 3
- 238000007790 scraping Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 229910021642 ultra pure water Inorganic materials 0.000 description 3
- 239000012498 ultrapure water Substances 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 108010053229 Lysyl endopeptidase Proteins 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 206010036790 Productive cough Diseases 0.000 description 2
- 238000002123 RNA extraction Methods 0.000 description 2
- 102000006382 Ribonucleases Human genes 0.000 description 2
- 108010083644 Ribonucleases Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 239000000919 ceramic Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000012568 clinical material Substances 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- ZPIRTVJRHUMMOI-UHFFFAOYSA-N octoxybenzene Chemical compound CCCCCCCCOC1=CC=CC=C1 ZPIRTVJRHUMMOI-UHFFFAOYSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 210000003802 sputum Anatomy 0.000 description 2
- 208000024794 sputum Diseases 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- 108020004465 16S ribosomal RNA Proteins 0.000 description 1
- VLEIUWBSEKKKFX-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;2-[2-[bis(carboxymethyl)amino]ethyl-(carboxymethyl)amino]acetic acid Chemical compound OCC(N)(CO)CO.OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O VLEIUWBSEKKKFX-UHFFFAOYSA-N 0.000 description 1
- 239000005725 8-Hydroxyquinoline Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 241000304886 Bacilli Species 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- 108020000946 Bacterial DNA Proteins 0.000 description 1
- 241000222122 Candida albicans Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- GZDFHIJNHHMENY-UHFFFAOYSA-N Dimethyl dicarbonate Chemical compound COC(=O)OC(=O)OC GZDFHIJNHHMENY-UHFFFAOYSA-N 0.000 description 1
- 239000003109 Disodium ethylene diamine tetraacetate Substances 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- 241000186359 Mycobacterium Species 0.000 description 1
- 241000186367 Mycobacterium avium Species 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 206010033101 Otorrhoea Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 108010059712 Pronase Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 238000010802 RNA extraction kit Methods 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- PPWHTZKZQNXVAE-UHFFFAOYSA-N Tetracaine hydrochloride Chemical compound Cl.CCCCNC1=CC=C(C(=O)OCCN(C)C)C=C1 PPWHTZKZQNXVAE-UHFFFAOYSA-N 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N anhydrous guanidine Natural products NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- 229940095731 candida albicans Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000003693 cell processing method Methods 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- YMKDRGPMQRFJGP-UHFFFAOYSA-M cetylpyridinium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 YMKDRGPMQRFJGP-UHFFFAOYSA-M 0.000 description 1
- 230000003196 chaotropic effect Effects 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- DTPCFIHYWYONMD-UHFFFAOYSA-N decaethylene glycol Polymers OCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO DTPCFIHYWYONMD-UHFFFAOYSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229960003964 deoxycholic acid Drugs 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 235000010300 dimethyl dicarbonate Nutrition 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 235000019301 disodium ethylene diamine tetraacetate Nutrition 0.000 description 1
- UQGFMSUEHSUPRD-UHFFFAOYSA-N disodium;3,7-dioxido-2,4,6,8,9-pentaoxa-1,3,5,7-tetraborabicyclo[3.3.1]nonane Chemical compound [Na+].[Na+].O1B([O-])OB2OB([O-])OB1O2 UQGFMSUEHSUPRD-UHFFFAOYSA-N 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 238000003505 heat denaturation Methods 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229940051921 muramidase Drugs 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229960003540 oxyquinoline Drugs 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 1
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- FHHPUSMSKHSNKW-SMOYURAASA-M sodium deoxycholate Chemical compound [Na+].C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 FHHPUSMSKHSNKW-SMOYURAASA-M 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- 239000004328 sodium tetraborate Substances 0.000 description 1
- VGTPCRGMBIAPIM-UHFFFAOYSA-M sodium thiocyanate Chemical compound [Na+].[S-]C#N VGTPCRGMBIAPIM-UHFFFAOYSA-M 0.000 description 1
- 239000002689 soil Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- 238000009210 therapy by ultrasound Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- UONOETXJSWQNOL-UHFFFAOYSA-N tungsten carbide Chemical compound [W+]#[C-] UONOETXJSWQNOL-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images

Landscapes
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
【課題】 遺伝子検査のための検体処理を行うに際し、従来法では十分に破砕・核酸抽出を行い難かったカビ・グラム陽性菌等の菌類を含む微生物を、簡便、迅速、かつ高効率に破砕し、その核酸を抽出する方法等を提供することにより、少数の微生物に対しても遺伝子検出感度を上げる方法等を提供すること。
【解決手段】 微生物の有無を確認するための検体に、検体溶解処理用液を混合して処理液とし、この処理液と大小2種類の大きさのジルコニアビーズとを空き空間容量を充分に確保したチューブに混合し、このチューブに物理的衝撃を加えて細胞破砕処理を行う。この破砕処理後の処理液に、界面活性剤を含む溶液を混合、加熱処理し、フェノール/クロロホルム処理を行った後に、上清を回収し、エタノール沈殿処理により核酸を回収する。
【選択図】 図1
【解決手段】 微生物の有無を確認するための検体に、検体溶解処理用液を混合して処理液とし、この処理液と大小2種類の大きさのジルコニアビーズとを空き空間容量を充分に確保したチューブに混合し、このチューブに物理的衝撃を加えて細胞破砕処理を行う。この破砕処理後の処理液に、界面活性剤を含む溶液を混合、加熱処理し、フェノール/クロロホルム処理を行った後に、上清を回収し、エタノール沈殿処理により核酸を回収する。
【選択図】 図1
Description
本発明は、微生物を効率よく破砕し、その核酸を抽出する方法等に関するものである。
細菌、カビ等の微生物の細胞を破砕する技術の現状は、次の(1)〜(3)の通りである。すなわち、(1)細胞の破砕にガラスビーズ、又はジルコニウム(Zirconium)ビーズを使用した破砕方法は既に研究者の間では一般的に実施されており、ガラスビーズキットは国外では既に販売されている。(2)米国においては、2種類の粒子サイズの異なった小さなガラスビーズとテトラフルオロエチレンで表面をコートした大きな磁性ビーズを混在させて破砕する製品が販売されている。(3)ガラスビーズより密度が高いジルコニウムビーズはガラスより破砕効率が良いというのは周知の事実で、破砕キットが日本国内で販売(例:安井機器、大阪)されている。
これらの技術は大量の菌を使用して破砕する技術として認識されており、臨床材料のような微量に病原体が存在する材料については、物理的破砕法を導入したキットは販売されていない。
これらの技術は大量の菌を使用して破砕する技術として認識されており、臨床材料のような微量に病原体が存在する材料については、物理的破砕法を導入したキットは販売されていない。
上記従来法とその問題点については、次の通りである。カビ、グラム陽性菌のように細胞壁が強固な細胞については、グラム陰性菌に適用される界面活性剤を用いた方法では、十分な RNA/DNA の回収ができない。そこで特定の Achromopeptidaseや Labiase, Lysozymeといったグラム陽性菌を溶解する酵素が使用されるが、これらの酵素は特定の構造をもった菌種には有効であるが、すべての微生物にたいしては有効ではない。
そこで微生物の細胞から RNA/DNA を同時に抽出する方法として、高濃度のGuanidium塩、及び SDS、 Triton x100などの界面活性剤の存在下で細胞を変性させる方法が使われている。これらの方法は、グラム陰性菌には極めて有効であるが、グラム陽性菌やカビには十分な溶菌効果を発揮できない。
そこでどの菌にも有効な方法として物理的な破砕方法が開発され、超音波破砕や、ガラスビーズによる破砕が推奨されてきた。特にガラスビーズは DNAを吸着することから破砕後、ビーズに破砕した菌の DNAを吸着させて精製する方法が一般に利用されている。
そこでどの菌にも有効な方法として物理的な破砕方法が開発され、超音波破砕や、ガラスビーズによる破砕が推奨されてきた。特にガラスビーズは DNAを吸着することから破砕後、ビーズに破砕した菌の DNAを吸着させて精製する方法が一般に利用されている。
しかしながら、ガラスビーズを用いても、黄色ブドウ球菌、芽胞を形成する菌、或いはカビの細胞を効率良く破砕することは困難であり、しかもこの方法は菌が大量にある場合の破砕方法であって、臨床材料に微量に存在する黄色ブドウ球菌や芽胞を形成する菌、あるいはカビの細胞を効率良く破砕することはできず、遺伝子検査の感度を上げる障害になっていた。
更に、ある病気が発生した場合に、その原因となる微生物を特定するのに際しては、病原微生物が不明な状態から同定を開始しなければならない。従って、標的候補として遺伝子増幅可能なプライマーセットを網羅的に準備しておき、病原微生物の核酸を抽出した後に、これを増幅反応液中に添加してPCR等遺伝子増幅反応を行うことになる。こうした場合、病原微生物毎の核酸抽出効率に差があるために、混入数が同程度であっても、ある病原微生物では検出できるが、別の病原微生物では、検出できないといった事態が起こり得る。すると、不明な病原微生物を特定しようとする場合、その微生物の破砕及び核酸抽出操作を一律に行えないことになってしまう。これでは、広く網羅的に病原微生物を特定できる遺伝子操作の可能性に対して、足枷をはめることになってしまう。この様な事態を少なくし、菌種が異なったとしても、高い効率で微生物を破砕し、核酸を抽出可能な方法を確立することが望ましい。
更に、ある病気が発生した場合に、その原因となる微生物を特定するのに際しては、病原微生物が不明な状態から同定を開始しなければならない。従って、標的候補として遺伝子増幅可能なプライマーセットを網羅的に準備しておき、病原微生物の核酸を抽出した後に、これを増幅反応液中に添加してPCR等遺伝子増幅反応を行うことになる。こうした場合、病原微生物毎の核酸抽出効率に差があるために、混入数が同程度であっても、ある病原微生物では検出できるが、別の病原微生物では、検出できないといった事態が起こり得る。すると、不明な病原微生物を特定しようとする場合、その微生物の破砕及び核酸抽出操作を一律に行えないことになってしまう。これでは、広く網羅的に病原微生物を特定できる遺伝子操作の可能性に対して、足枷をはめることになってしまう。この様な事態を少なくし、菌種が異なったとしても、高い効率で微生物を破砕し、核酸を抽出可能な方法を確立することが望ましい。
基礎研究では、上記問題への対策として、検体容量を大きくすることが可能であるものの、単に検体容量を大きくすると取り扱いの簡便さが失われてしまうという欠点が生じる。すなわち、検体容量が大きくなると、遠心濃縮するにしても機械が大掛かりなものとなり、簡便な微量高速遠心機では扱いきれなくなる。その結果、多数の検体を扱うにはたいへん不向きになってしまう。また、PCRの反応液量は今や 25μL以下なので、あまりに大量の検体容量からその液量までに濃縮するには操作が煩雑になることは避けられない。臨床検査では、扱う検体容量の多さや操作の煩雑さは運用コスト増にもつながるため、これを避けることが望まれる。そこで、検体容量の増加をできるだけ回避しつつ、核酸の抽出効率を上げ、最終的な遺伝子検出感度を上げる事が望まれている。
この問題に対処する為、これまで多くの試みが為されている。その一つとして、ガラスビーズよりも密度の高いジルコニウムを用いる方法が開発されてきている。 Hurley, S. S.らは核酸抽出のフェノールとジルコニウムビーズを加え、激しく震盪して細胞壁の物理的破砕を行うことを組み合わせた方法を記載している(非特許文献1)。彼女らが使用したのはバイオスペック・プロダクツ社のMini−Beadbeaterという機械で震盪させるもので、1.5mLのスクリューキャップ付チューブにミコバクテリア属の菌の50%懸濁液を500〜600マイクロリットル、及び等量のトリス−EDTA飽和フェノール溶液を加え(即ち1.0〜1.2mLになる)、これに直径0.1mmのジルコニウムビーズをチューブに満たし、3分間振盪後、遠心して水層のDNAを回収するものである。
これは、バイオスペック・プロダクツ社のMini−Beadbeaterの使用法を基に容器を遠心チューブにし、フェノール溶液を加える改良を施したもので、基のバイオスペック・プロダクツ社(http://www.biospec.com/)の操作方法では、2mLスクリューキャップ付バイアルに1/2〜3/4量のビーズを詰め、残りの空間をバッファーと検体で埋め、できるだけ空気を除くとされている。ビーズの粒径は、細菌には0.1mm、酵母には0.5mm、植物や動物組織片には1.0mmか2.5mmのものが適切なサイズと書かれている。)
また、Shah, J. S. らは、グアニジンチオシアン酸塩で熱処理してジルコニアビーズで物理的破砕を行っている(非特許文献2)。この論文では、0.5gの酸化ジルコニウム(ジルコニア)ビーズが入ったプロセシング・チューブ内に、250μLの検体と400μLの5Mグアニジンチオシアン酸塩、100mMトリスpH7.8、40mM EDTA、1.0%Sarkosylが入ったプロセシング・バッファーを容れて、又はその2倍量(合計約1.6mL)で、GENE−TRAK Sample Processorという機械を使って、6分間振盪させている。彼らはこの方法で、コピー数の多いRNAを抽出することに成功している。
しかし、RNAを抽出する方法は、DNAを抽出する方法に比べると、簡便性と安定性に欠けたものと成らざるを得ない。一方、RNAより取り扱い易いものの、個体に1コピーしかないDNAに関しては該論文の目的外で記載されていない。同じGENE−TRAK社の1991年の論文(非特許文献3)では、DNAを抽出しているが、0.2mLのトリス−マグネシウム−塩化カリウムのバッファーに懸濁した検体、0.1mLの0.1mmガラスビーズ、10μLのSDS、および約200μLの上述バッファー飽和フェノールを、500μLのマイクロ遠心チューブ一杯に容れ、それを2mLチューブに入れてMini−Beadbeaterで振盪している。
また、特開平5−68553(特許文献1)では、ガラスビーズの存在下で超音波処理する方法を開示されている。しかし、その実施例は、菌数7×108/mLの溶液を1mL使用して行われたものであり、少ない菌数に対する抽出効率を示したものではない。更に、ガラスビーズを用いた微生物破砕方法が開示されているものの、十分に満足のできる効果を上げられるものは知られていない(特許文献2、3)。
しかし、一方で破砕効率を上げるために、大小2種類の破砕用物体を添加する例もある( QBIOGENE: http://qbiogene.com/technical/protocols/fastprep/, FastDNAR Kit、FastRNAR Pro Blue Kit、FastRNARPro Red Kit、及び http://qbiogene.com/fastprep/lysing_tubes.shtml)。
しかしながら、上記各方法によっても、黄色ブドウ球菌、芽胞を形成する菌、或いはカビの細胞を効率良く破砕することは困難であり、遺伝子検査の感度を上昇させるための障害であることに変わりがなかった。
特にジルコニウムは、DNAの吸着能力がガラスビーズに比べると非常に低いことから、従来のガラスビーズとは異なるDNAの精製方法が必要となる。また、非常に高い感度が必要となる臨床検査では、従来のジルコニウムを用いた方法を更に改良する必要があった。
特にジルコニウムは、DNAの吸着能力がガラスビーズに比べると非常に低いことから、従来のガラスビーズとは異なるDNAの精製方法が必要となる。また、非常に高い感度が必要となる臨床検査では、従来のジルコニウムを用いた方法を更に改良する必要があった。
本発明は、上記した事情に鑑みてなされたものであり、その目的は、遺伝子検査のための検体処理を行うに際し、従来法では十分に破砕・核酸抽出を行い難かったカビ・グラム陽性菌等の菌類を含む微生物を、簡便、迅速、かつ高効率に破砕し、その核酸を抽出する方法等を提供することにより、少数の微生物に対しても遺伝子検出感度を上げる方法等を提供することである。
更に、本発明によれば、核酸抽出効率を上げることで、病原微生物が解らない段階であっても、一定量の検体を処理し、遺伝子増幅法で検出可能なレベルまで効率良く核酸抽出できるので、遺伝子診断の有用性が高められる。
更に、本発明によれば、核酸抽出効率を上げることで、病原微生物が解らない段階であっても、一定量の検体を処理し、遺伝子増幅法で検出可能なレベルまで効率良く核酸抽出できるので、遺伝子診断の有用性が高められる。
本発明者らは、鋭意検討の結果、微生物からの核酸抽出操作中において、微生物の種類により効率が著しく変化する細胞破砕の段階を飛躍的に上昇させることに成功し、少ない病原体数でも遺伝子検出感度を上げることに成功した。
特に、下記5つの要件が、微生物を効率良く破砕し、その核酸抽出効率を増加させることを見出した。
すなわち、(1)処理液(検体に、必要に応じて検体溶解処理用液を混合した液)とビーズとを混合し、物理的衝撃を加えて破砕処理を行う際に、細胞破砕用ビーズとして、通常サイズの大きさのものと、それよりも大きい約4mm〜約7mmのサイズのものとの少なくとも二種類を用いる(以下、「要件(1)」という)。
特に、下記5つの要件が、微生物を効率良く破砕し、その核酸抽出効率を増加させることを見出した。
すなわち、(1)処理液(検体に、必要に応じて検体溶解処理用液を混合した液)とビーズとを混合し、物理的衝撃を加えて破砕処理を行う際に、細胞破砕用ビーズとして、通常サイズの大きさのものと、それよりも大きい約4mm〜約7mmのサイズのものとの少なくとも二種類を用いる(以下、「要件(1)」という)。
(2)物理的衝撃を加える際には、処理液/ビーズの容量比が1よりも小さくなる状態とする(すなわち、ビーズ容量が処理液容量よりも大きくなる状態とする)(以下、「要件(2)」という)。
(3)処理液及びビーズを加えた合計容量が、容器の全容量(以下、「容器容量」という)の少なくとも3/8以下とし、細胞破砕処理時の容器の空間容量(容器容量−処理液及びビーズの合計容量)を容器容量の5/8以上にする(以下、「要件(3)」という)。
(3)処理液及びビーズを加えた合計容量が、容器の全容量(以下、「容器容量」という)の少なくとも3/8以下とし、細胞破砕処理時の容器の空間容量(容器容量−処理液及びビーズの合計容量)を容器容量の5/8以上にする(以下、「要件(3)」という)。
(4)処理液とビーズとを混合して、物理的衝撃を加える際には、実質的に界面活性剤を含ませない状態とする(以下、「要件(4)」という)。「実質的に」とは、検体中に内在性の界面活性剤成分があったとしても無視できるほど、使用する試薬類から界面活性剤を持ち込まないことにより、実質的に界面活性剤を含ませない条件で、細胞破砕処理を行うことが可能である。数値的な限定条件を示すことは困難であるが、いわゆる遺伝子工学的に用意された試薬を用いることにより、実質的に界面活性剤を含ませない上程で、細胞破砕処理を行うことが可能である。
(5)上記要件(4)を実施する際には、細胞破砕後に、界面活性剤を添加し、核酸抽出操作を行う(以下、「要件(5)」という)。
以上5つの要件は、一つを用いた場合であっても従来の方法に比べて有効であるが、二つ以上を任意に組み合わせて実施することもできる。
(5)上記要件(4)を実施する際には、細胞破砕後に、界面活性剤を添加し、核酸抽出操作を行う(以下、「要件(5)」という)。
以上5つの要件は、一つを用いた場合であっても従来の方法に比べて有効であるが、二つ以上を任意に組み合わせて実施することもできる。
これらの改良を加えた結果、微生物からの核酸の抽出効率が、従来の方法に比較すると、約1000倍〜10000倍にも上昇した。本発明に係る核酸抽出法、細胞破砕用ビーズ充填容器キット、及び細胞破砕用ビーズ充填容器を含めた核酸抽出キットの詳細を以下に説明する。
本発明において、微生物とは、グラム陽性菌またはグラム陰性菌を含む細菌、ウィルス、カビ、細菌・ウィルス等の宿主細胞である動物細胞、及びヒト細胞を含む。
本発明において、微生物とは、グラム陽性菌またはグラム陰性菌を含む細菌、ウィルス、カビ、細菌・ウィルス等の宿主細胞である動物細胞、及びヒト細胞を含む。
細胞破砕とは、細胞内容物が外部に取り出せる程度に細胞壁を破壊することを意味する。但し、その指標は、便宜上DNA又はRNAの核酸を抽出した回収率で見積もる。一般的に、本発明の属する技術分野においては、ビーズによる細胞破砕は、容器容量が2mL程度の容器で行うのが効率が良いとされていることから、本明細書中に記載された数値は、特に断らない限り、このスケールで打ち出されたものである。本発明の実施において、スケールを変更する場合には、当業者であれば、適当な修正、変更を加えることは、極めて容易に行えるであろう。
物理的衝撃とは、検体とビーズとを物理的に接触させる結果として、細胞を破砕させることを意味しており、具体的には、検体とビーズとを混合した容器全体を震盪する方法、又は検体とビーズとを混合した容器を超音波処理する方法が含まれる。
検体とは、特定の微生物の有無を確認するために用いられるものであり、例えば、便、喀痰、血液、土壌、水、食品、咽頭擦過物、結膜擦過物、尿道分泌物、耳漏、分泌液、髄液、関節液、腹水などが含まれる。
検体とは、特定の微生物の有無を確認するために用いられるものであり、例えば、便、喀痰、血液、土壌、水、食品、咽頭擦過物、結膜擦過物、尿道分泌物、耳漏、分泌液、髄液、関節液、腹水などが含まれる。
検体溶解処理用液とは、検体の種類に応じて、適当に添加される溶液のことを意味している。この検体溶解処理用液には、必要に応じて、消化酵素(タンパク分解酵素、多糖類分解酵素を含む)や変性剤(グアニジン塩を含む)などを含有させることが好ましい。但し、要件(4)を共に満足させるためには、実質的に界面活性剤を含まないことが好ましい。
次に、要件(1)について、詳細に説明する。処理液にビーズを加え、物理的衝撃を加えて細胞を破砕する際のビーズの大きさは、通常サイズのものは、約0.1mm〜約0.2mm程度である。本発明では、通常サイズのものに、約4mm〜約7mmのより大きなビーズを少なくとも1個加える。こうして、二種類の大きさのものを用いることで、細胞の破砕効果が顕著に高くなる。
また、ビーズの種類としては、ガラス、ガーネット、ジルコニアなどを用いることができる。大小二種類のビーズの種類は、同種のものでも良く、異種のものでも良い。また、上記種類のうち、大小二種類共に、ジルコニアビーズを用いることが特に好ましい。後述する実施例では、直径5mmのジルコニアビーズを加えることにより、核酸の抽出効率(抽出後のDNAのPCRによる検出感度)が2桁以上も上昇した。
また、ビーズの種類としては、ガラス、ガーネット、ジルコニアなどを用いることができる。大小二種類のビーズの種類は、同種のものでも良く、異種のものでも良い。また、上記種類のうち、大小二種類共に、ジルコニアビーズを用いることが特に好ましい。後述する実施例では、直径5mmのジルコニアビーズを加えることにより、核酸の抽出効率(抽出後のDNAのPCRによる検出感度)が2桁以上も上昇した。
次に、要件(2)について説明する。従来の方法では、ビーズを用いて細胞破砕を行う場合には、処理液容量がビーズ容量よりも多くなっている。しかし、本発明者らの検討によれば、処理液容量の絶対値を小さくした方が、細胞の破砕効率が良いことを見出した。具体的な目安としては、処理液容量がビーズ容量よりも小さくすることが好ましい。しかしこれは、必ずしも処理液容量がビーズ容量より大きくなってはいけないということではない。後述する実施例では処理液容量が200μL以下の領域では、ビーズ容量が減った場合であっても、従来法(すなわち、処理液容量が1mL〜600μL程度で細胞破砕を行う方法)に比べると、高い核酸抽出効率を確保できた。処理液容量が少ない場合には、処理液中の微生物がビーズに衝突する頻度が大きくなるためではないかと考えられる。
次に、要件(3)について説明する。この要件は、処理液及びビーズを加えた合計容量を小さくすることにより、細胞破砕時の容器の空間容量を大きくすることを示している。具体的には、空間容量として、容器容量の5/8以上、好ましくは2/3以上、更に好ましくは3/4以上、更に好ましくは4/5以上を確保する。空間容量を大きく確保することにより、物理的衝撃を加える際に、細胞破砕用のビーズが、容器の中で極めて動きやすい状態となるので、細胞破砕効果が向上するものと考えられる。空間容量を容器容量の3/4以上確保するには、処理液にビーズを加えた合計容量を容器容量の1/4以下、より好ましくは1/5以下とする必要がある。
ここで、処理液にビーズを加えた合計容量が容器容量の1/4以下、又は1/5以下という条件を設定する際に、要件(2)と要件(3)とを共に満足させるためには、処理液の容量が容器容量の1/8以下、又は1/10以下とする必要がある。何故ならば、要件(2)の処理液容量がビーズ容量を越えないという条件を満足させようとすると、処理液容量にビーズ容量を加えた合計容量が容器容量の1/4以下、又は1/5以下の更に半分以下の量とする必要があるからである。この条件を一般的に使用されている2mLの容量容器に当てはめると、1/8は250μL、1/10は200μLとなる。これが処理液容量の目安である。この容量が細胞破砕用ビーズの容量を下回る(要件(2))には、ビーズ容量の最低値として、200μL程度が必要となる。
更に、要件(1)を満足させるためには、大小二種類のビーズとして以下の容量を用いる。具体的には、直径が約0.2mmのジルコニアビーズ0.5gの容量は約140μL程度であり、直径が約5mmのジルコニアビーズの容量は約65μLである。大小のビーズ容量を合わせると、約200μL程度となる。この程度のビーズ容量を用い、処理液容量が約200μL程度のものを混合して、破砕処理することが望ましい。
次に、要件(4)について説明する。物理的衝撃を加えて破砕処理を行う際には、実質的に、SDS、Triton X−100、ザルコシル等の界面活性剤を含ませないことが好ましい。界面活性剤は、分子内に親水性原子団と疎水性原子団とを備えた両親媒性物質であり、少量で界面又は表面の諸性質を変化させる性質を有し、基本的には、石鹸の類である。物理的衝撃を加える際に、界面活性剤が存在すると、微生物が、ビーズ同士及び/又はビーズと容器壁とに挟まれたときに、表面を滑りやすくしてしまうため、破砕効率が下がっていると考えられる。後述する実施例では、物理的衝撃を加える際に、実質的に界面活性剤を含ませない状態としておくことで、従来法に比べると、細胞破砕効率が10倍〜100倍に上昇したことから、要件(4)の効果が明らかである。
次に、要件(5)について説明する。要件(4)を満足させて細胞破砕処理を行った後に、界面活性剤を加えてから、通常の核酸抽出操作を続けることを示している。具体的には、フェノール/クロロホルム抽出法を用いることができるが、この方法に限られず、他の方法を用いても良い。界面活性剤は、次に行うフェノール/クロロホルム抽出を行う際、核酸とそれに結合するタンパクとを解離させるのにも役立ち、これを除くことは核酸の抽出効率を下げることにつながる。従って、細胞破砕処理の後、フェノール/クロロホルム抽出の前に界面活性剤を加える。細胞破砕処理後の処理液(以下、「細胞破砕液」という)とフェノール/クロロホルムを激しく混合すると、フェノールは殆ど水とは混ざらず分離し、且つタンパクを変性させ、水層とフェノール層との中間層に変性タンパクを生じさせる。フェノール層が酸性でなければ、即ち中性〜弱アルカリ性の範囲で、核酸は水層に抽出される。クロロホルムはフェノールと水の分離を高める。イソアミルアルコールをフェノール/クロロホルムに少量加えることが好ましい。これは、イソアミルアルコールの消泡作用により、フェノールと水の分離を助けるためである。
界面活性剤を含む溶液(以下、「界面活性剤溶液」という)の容量と処理液及びビーズを合わせた容量との合計が、容器容量の4/20〜7/20程度となることが望ましい。上記の範囲を満足させるには、要件(2)と要件(3)とを満足させるための条件において、処理液にビーズを加えた合計容量が容器容量の1/6に設定した場合に、界面活性剤溶液の容量は、容器容量の4/20にするためには1/30、7/20にするためには11/60となる。界面活性剤溶液を加えて容器容量の4/20〜7/20にするということは、フェノール/クロロホルム混合液を等量加えて核酸を抽出するので、フェノール/クロロホルム混合液を加えたときには、全体の容量が容器容量の2/5〜7/10になることを意味する。フェノール/クロロホルム混合液を加えて激しく混合し、タンパク変性、核酸抽出を行うに適した量は容器容量の半分から半分強程度である。この量が多すぎると容器の空間容量が少なくなり、激しく混合する際に、混合が不十分となり、タンパク変性効果が弱くなる可能性がある。
従って、加える界面活性剤溶液の容量は、2mL容器を用いる場合には、1/30〜11/60、即ち約70μL〜約350μL(好ましくは、100μL〜200μL)となる。更に、処理液とビーズとを加えた容量を少なく抑えることにより、加える界面活性剤溶液の容量を増加することが可能になる。界面活性剤溶液の容量を増加させると、フェノール/クロロホルムと共に激しく混合した後に上層の水層を回収する際、水層が多くなり、中間層の変性タンパクが混入してしまうのを防ぐという効果がある。従って、処理液容量を少なくし、界面活性剤の容量を多くする方が、操作上有利になる。また、フェノール/クロロホルム混液を添加する場合には、細胞破砕用ビーズが完全に浸漬する程度の量を加えることが好ましい。
界面活性剤溶液の容量は、フェノール/クロロホルム抽出後の水層の回収に影響する。微量な検体を小さな容量で処理した場合、余りに水層の全量が少ないために、核酸が含まれる水層を分離することが非常に困難となる事態が生じ得る。容器の全容量が、約2mLのものを用いる場合、水層を分離する際には、中間層を吸い込むことなく0.2mL以上を回収しやすい設定とすることが好ましい。検体の処理量を極力少量に抑えると水層量が少なくなるが、新たに界面活性剤を含む溶液を加えなかった場合には、実質的に水層を回収することが不可能となることもあり得る。そこで、界面活性剤を含む溶液を加えて水層量をマイクロピペットで回収し易い量とする為には、約2mL容器で液の高さ(深さ)が、最低でも3.5mm程度となることが好ましい。代表的な2mL容器の内径は、8.3mm〜9.2mmの範囲であることから、0.2mLの界面活性剤溶液を加えた場合の高さ上昇は、3.0mm〜3.7mmになる。界面活性剤を含む溶液を加える容量を、2mL容器の深さ(39mm〜43mm)の4/20(最大8.6mm)以下に抑えることにより、処理液とビーズを加えた容量(約1/10〜約1/4、即ち2mL容器中の0.2mL〜0.5mL)に加えて、界面活性剤を含む溶液を0.2mL程度(約4mmの深さ)から0.4mL程度(処理液とビーズを加えた容量が少ない、0.2mLの場合;約8.5mmの深さ)に制限することで、処理液、ビーズ及び界面活性剤溶液を加えた容量合計が0.4mL〜0.7mL程度となり、後のフェノール/クロロホルム処理、及び水層分離処理を円滑に行うことができる。
こうして、上記課題を解決するための第1の発明に係る微生物の細胞処理方法は、微生物の有無を確認するための検体と検体溶解処理用液を混合した処理液と、ビーズとを容器に混合し、この容器に物理的衝撃を加えて、前記微生物の細胞を破砕処理する方法であって、前記処理液とビーズとを加えた合計容量が、前記容器の全容量の少なくとも3/8以下とし、細胞破砕処理時の容器の空間容量を容器の全容量の5/8以上としたことを特徴とする。
上記発明においては、前記細胞破砕処理時には、前記処理液の容量が、前記ビーズの容量よりも小さいことが好ましい。また、前記細胞破砕処理時には、前記容器内には、実質的に界面活性剤を含ませない状態とすることが好ましい。
上記発明においては、前記細胞破砕処理時には、前記処理液の容量が、前記ビーズの容量よりも小さいことが好ましい。また、前記細胞破砕処理時には、前記容器内には、実質的に界面活性剤を含ませない状態とすることが好ましい。
また、前記ビーズは、大きさ及び重さが異なる少なくとも二種類以上のビーズを同時に用いることが好ましい。このときには、大きさ及び重さが異なる少なくとも二種類以上のビーズは、同じ材質からなるものであることが好ましい。または、大きさ及び重さが異なる少なくとも二種類以上のビーズは、異なる材質からなるものでも良い。更に、大きさ及び重さが異なる少なくとも二種類以上のビーズのうち、より小さいビーズについては多数使用する一方、より大きいビーズについては、1個だけ使用することがより好ましい。
また、第1の発明において、前記ビーズの材質の比重は、5以上であることが好ましい。ガラスビーズは比重が約2.5、アルミナのそれは3〜4、ジルコニアでは5.5〜6強、ステンレススチールでは7.9前後、タングステン・カーバイドは約15、金およびタングステン単独では19.3であるが、基本的にビーズは、柔らかくない限り、重いほうが破砕効果は高くなる。第1の発明の効果は、少なくとも比重5以上のジルコニアビーズで確認されている。
また、第1の発明において、前記ビーズの材質の比重は、5以上であることが好ましい。ガラスビーズは比重が約2.5、アルミナのそれは3〜4、ジルコニアでは5.5〜6強、ステンレススチールでは7.9前後、タングステン・カーバイドは約15、金およびタングステン単独では19.3であるが、基本的にビーズは、柔らかくない限り、重いほうが破砕効果は高くなる。第1の発明の効果は、少なくとも比重5以上のジルコニアビーズで確認されている。
第2の発明に係る核酸抽出方法は、第1の発明に係る方法で前記微生物の細胞壁を破砕し、その微生物の核酸を抽出することを特徴とする。
また、第1の発明に係る方法で前記微生物の細胞壁を破砕した後、界面活性剤を添加し、前記微生物の核酸を抽出することが好ましい。また、第1の発明に係る方法で前記微生物の細胞壁を破砕した後、前記ビーズが存在する状態で界面活性剤を添加し、フェノール/クロロホルム混液を混合することが好ましい。また、第1の発明に係る方法で前記微生物の細胞壁を破砕した後、前記容器中に界面活性剤を加え、その容器中にフェノール/クロロホルム混液を前記ビーズが浸漬するまで添加することが好ましい。
また、第1の発明に係る方法で前記微生物の細胞壁を破砕した後、界面活性剤を添加し、前記微生物の核酸を抽出することが好ましい。また、第1の発明に係る方法で前記微生物の細胞壁を破砕した後、前記ビーズが存在する状態で界面活性剤を添加し、フェノール/クロロホルム混液を混合することが好ましい。また、第1の発明に係る方法で前記微生物の細胞壁を破砕した後、前記容器中に界面活性剤を加え、その容器中にフェノール/クロロホルム混液を前記ビーズが浸漬するまで添加することが好ましい。
第2の発明においては、添加する界面活性剤を含む溶液の容量は、前記処理液及びビーズを加えたときの容量が前記容器の全容量の4/20〜7/20であることが好ましい。
第3の発明に係る細胞破砕用キットは、第1の発明に係る方法を用いることを特徴とする。このキットにおいて、大小2種類のビーズを用いる方法を使用する場合には、前記2種類以上のビーズを封入した容器を備えることが好ましい。また、少なくとも1種類のビーズの材質の比重が5以上であることが好ましく、前記ビーズが、ジルコニウム、ジルコニア(酸化ジルコニウム)、ステンレススチール又はタングステンであることがより好ましい。
第3の発明に係る細胞破砕用キットは、第1の発明に係る方法を用いることを特徴とする。このキットにおいて、大小2種類のビーズを用いる方法を使用する場合には、前記2種類以上のビーズを封入した容器を備えることが好ましい。また、少なくとも1種類のビーズの材質の比重が5以上であることが好ましく、前記ビーズが、ジルコニウム、ジルコニア(酸化ジルコニウム)、ステンレススチール又はタングステンであることがより好ましい。
第4の発明に係るキットの製造方法は、第3の発明に係るキットであることを特徴とする。この場合に、前記ビーズを容器の全容量の7%〜15%として封入することが好ましい。
第5の発明に係る核酸抽出キットは、第2の発明に係る核酸抽出方法を用いることを特徴とする。この場合に、前記検体溶解処理用液には、実質的に界面活性剤を含まないことが好ましい。また、少なくとも、(1)ビーズを封入した容器、(2)検体溶解処理用液、(3)界面活性剤を含む溶液、及び(4)フェノール/クロロホルムを含む混合液を、それぞれ独立して備えることが好ましい。
第5の発明に係る核酸抽出キットは、第2の発明に係る核酸抽出方法を用いることを特徴とする。この場合に、前記検体溶解処理用液には、実質的に界面活性剤を含まないことが好ましい。また、少なくとも、(1)ビーズを封入した容器、(2)検体溶解処理用液、(3)界面活性剤を含む溶液、及び(4)フェノール/クロロホルムを含む混合液を、それぞれ独立して備えることが好ましい。
第6の発明に係るキットの製造方法は、第5の発明に係るキットであることを特徴とする。
こうして、本発明によれば、従来法では十分に破砕・核酸抽出を行い難かったカビ・グラム陽性菌等の菌類を含む微生物を、簡便、迅速、かつ高効率に破砕する方法、キット等を提供できる。また、その方法を用いて、抽出効率の良好な核酸抽出方法、キット等を提供できる。こうして、本発明の方法によれば、遺伝子検査を行うに際し、多種類の微生物を含み得る検体に対して、少数の微生物であっても、非常に高感度な遺伝子検出を行うことが可能となる。
こうして、本発明によれば、従来法では十分に破砕・核酸抽出を行い難かったカビ・グラム陽性菌等の菌類を含む微生物を、簡便、迅速、かつ高効率に破砕する方法、キット等を提供できる。また、その方法を用いて、抽出効率の良好な核酸抽出方法、キット等を提供できる。こうして、本発明の方法によれば、遺伝子検査を行うに際し、多種類の微生物を含み得る検体に対して、少数の微生物であっても、非常に高感度な遺伝子検出を行うことが可能となる。
次に、本発明の実施形態について、図面を参照しつつ説明するが、本発明の技術的範囲は、これらの実施形態によって限定されるものではなく、発明の要旨を変更することなく様々な形態で実施することができる。また、本発明の技術的範囲は、均等の範囲にまで及ぶものである。
図1には、本発明を適用して、細胞を破砕し、核酸を抽出するための基本操作手順を示した。図中最上段の「B)」は、液体状の検体にライシスバッファーを混合して細胞破砕工程に進むスキームを示し、次段の「A)」は、沈殿状の検体にライシスバッファーを混合して細胞破砕工程に進むスキームを示している。つまり、「A)」又は「B)」のいずれかの前処理は、検体の種類に応じて選択的である。なお、図1は、通常の生物工学分野で使用されるチューブ(容器の全容量が、約500μL〜約2mL)を使用して、本発明を実施する方法を示すものであり、当業者であれば、この方法を適当に拡大・又は縮小して実施できる。
微生物の有無を確認するための検体については、必要に応じて前処理しておくことが好ましい。この前処理は、従来の微生物検体からの核酸抽出方法にも用いられていたもので実施することができる。前処理は、多くの場合に、微生物を濃縮する微生物濃縮工程が含まれる。具体的な前処理方法としては、例えば検体を遠心チューブに入れて遠心沈降させる(なお、沈殿の生成を促進させるために、ポリエチレングリコール等の沈殿促進剤を添加する場合がある)、酵素処理して分散させる(特に、喀痰検体の場合に有効な方法となる)、フィルターを用いて遠心濾過して濃縮する方法等がある。
検体が懸濁液等のように沈殿処理を要する場合には、容量2mLのチューブに検体を入れた後、約15,000rpmにて、約3分間〜約10分間の遠心処理を行い、上清を除いてから、次の処理「A)」に移る。なお、沈殿促進剤を添加した場合には、約15分間〜約20分間程度まで、遠心時間を延長することもできる。また、検体が微生物の浮遊液或いは混入液の場合には、約200μLの検体を次の処理「B)」に用いる。
こうして、適当な前処理を施した沈殿(図中「A」))、又は液体(図中「B」))に、150μLのライシスバッファーを加え、よく混ぜて懸濁させる。ライシスバッファーは、本発明の検体溶解処理用液に該当しており、検体中の微生物を溶解、或いは変性させるために添加される。ライシスバッファーには、例えばグアニジンチオシアン酸塩等の変性剤、ラビアーゼ(Labiase)・リゾチーム(Lysozyme)等の酵素、メルタプトエタノール等の還元剤、pH調整用の塩(例えば、クエン酸緩衝液、トリス緩衝液など)が含まれている。なお、本発明を実施するに際しては、ライシスバッファー中には、実質的に界面活性剤を含ませないことが好ましい。
次に、検体とライシスバッファーを混合した処理液を細胞破砕用のチューブに移す。ここで処理液は、約150μL〜約350μLの容量となっている。チューブには、大小二種類のビーズが添加されている。チューブは、本発明における容器に該当しており、その全容量は約2mLである。また、二種類のビーズの材質は、いずれも比重が5以上のジルコニア(酸化ジルコニウム)である。小ビーズの大きさは、約0.1mm〜約0.2mm程度のものであり、チューブには約500mg(容量として約140μL程度)が含まれている。また、大ビーズの大きさは、約4mm〜約7mmのものであり、チューブには1個のみ(重量として約200mg〜約1100mg、容量として約30μL〜約200μL)が含まれている。つまり、チューブ中には、約180μL〜約350μLのビーズが含まれている。なお、本発明の実施においては、処理液の容量が、ビーズの容量よりも小さくなるように設定することが好ましい。
こうして、チューブに処理液とビーズとを混合した後、フタをし、約70℃(場合により、約90℃まで上昇させてもよい)にて、約10分間程度の加熱処理を行うことにより、微生物の溶菌を促進させる。なお、対象とする核酸が、DNA又はRNAのいずれであっても、次に行う破砕処理操作を行う限り、約70℃の加熱処理で、十分な抽出処理が行えることが確認されている。また、微生物として、グラム陽性菌のみを対象とするDNA抽出の場合には、約90℃で加熱処理を行うことが好ましい。
加熱処理の後、念のためにフタをしっかり締め直し、チューブ全体に物理的衝撃(例えば、機械的な震盪処理)を加えることにより、微生物の破砕処理を行う。細胞破砕処理時には、処理液とビーズとの合計容量は、約330μL〜約700μLとされており、容器の全容量(2mL)の約3/20〜約7/20を占めている。約60秒間の物理的衝撃(具体的には、 multibeads shocker(安井器械)を用いる場合には、2500rpm、60秒間、Fast Prep(フナコシ)を用いる場合には、強度4.5、60秒間、Smart Smash(トミー)を用いる場合には、5000rpm、60秒間)を加える。なお、ボルテックスミキサーにて60秒間の処理を行った場合でも、RNA抽出処理、又はグラム陰性菌のDNA抽出処理を行うことは可能であるが、グラム陽性菌の核酸抽出効率は減少することがある。
物理的衝撃を加えた後、フタを外して、約200μLの界面活性剤(例えば、SDS)を含む溶液を添加し、再度フタを締めた後、約70℃〜約90℃にて約10分間の加熱処理を行う。
その後、フタを外して、細胞破砕液に約400μLのフェノール混合液(本発明におけるフェノール/クロロホルム混液に該当する)を混合し、再度フタをしっかり閉めて約1分間振盪又は混合した後、15,000rpm、3分間の遠心処理を行い、タンパク質を変性させ中間層に集める。フェノール混合液は、フェノール:クロロホルム=約0.8〜約1.2(好ましくは、約1)の比率(v/v)で混合させたものであり、好ましくは約1/40〜約1/60(好ましくは、約1/49)のイソアミルアルコールを添加したものを用いる。すなわち、フェノール/クロロホルム/イソアミルアルコール混液とすることが、より好ましい。
その後、フタを外して、細胞破砕液に約400μLのフェノール混合液(本発明におけるフェノール/クロロホルム混液に該当する)を混合し、再度フタをしっかり閉めて約1分間振盪又は混合した後、15,000rpm、3分間の遠心処理を行い、タンパク質を変性させ中間層に集める。フェノール混合液は、フェノール:クロロホルム=約0.8〜約1.2(好ましくは、約1)の比率(v/v)で混合させたものであり、好ましくは約1/40〜約1/60(好ましくは、約1/49)のイソアミルアルコールを添加したものを用いる。すなわち、フェノール/クロロホルム/イソアミルアルコール混液とすることが、より好ましい。
次に、上層(水層)を滅菌した別のチューブに回収する。こうして、核酸(粗精製物)の抽出処理を完了するが、場合に応じて、エタノール沈殿処理を2回行うことにより、核酸の精製度を向上させる。具体的には、粗精製核酸抽出物に、約1mLの99%エタノールを添加し、適当に振盪後、15,000rpm、3分間の遠心処理を行い、上清部分を除く(1回目のエタノール沈殿処理)。更に、沈殿に70%エタノールを添加し、適当に振盪後、再度15,000rpm、3分間の遠心処理を行う(2回目のエタノール沈殿処理)。上清を除いた後、適当な溶液(例えば、DEPC(Diethyl Pyrocarbonate)処理水、又はTris-EDTA溶液(10mM Tris, 1mM EDTA, pH8.0)))に懸濁する。
本発明による細胞破砕用キットおよび核酸抽出用キットを作製するには、次のようにすることが好ましい。これらの方法は、本発明を実施するための事例として提示するものでは、本発明の技術的範囲を限定するものではない。
直径0.2mm程度の小ジルコニアビーズ、及び直径5mm程度の大ジルコニアビーズ(1個)をpH1.5〜2の酸で90℃以上で熱処理して、核酸を分解する。熱処理は、121℃、20分間のオートクレーブ(以下、「オートクレーブ処理」という)で行うこともできる。以降の操作は、クリーンルーム又はクリーンベンチ等、無菌的条件で行うことが望ましい。
次いで、酸で熱処理したジルコニアビーズをジエチルピロカーボネート(DEPC)処理水で洗浄する。DEPC処理水は、密閉可能な蓋付容器に、超純水及びDEPCを0.1%になるように加え、混合して37℃に2時間から1晩静置後、蓋を弛めてオートクレーブすることでDEPCの残存物を分解して使用する。DEPC処理はRNaseを不活化させる操作であり、DEPCの代わりにジメチルピロカーボネートを使用しても良く、また、洗浄には他の方法でRNaseを不活化させた水を用いても良い。
洗浄したジルコニアビーズを乾熱滅菌機で160℃以上で乾燥させ、必要に応じて、更にクリーンベンチ内で乾燥させる。乾燥させた0.2mm程度のジルコニアビーズを約0.5g又は約140μL、2mL容量の遠心可能なスクリューキャップ付容器(以下、「遠心チューブ」という)に容れる。この操作は、0.2mm程度のジルコニアビーズが0.5g入る柄付容器・用具に擦り切りで掬い取る、又は注ぐ(落とす)、乃至吸引して一定量取ったものを遠心チューブに入れることにより行う。これに直径5mm程度のジルコニアビーズを1個加え、蓋を閉めて、ジルコニアビーズ充填チューブを作製することができる。
検体溶解処理用液は、尿素、グアニジンチオシアン酸塩やチオシアン酸ナトリウム等カオトロピック塩、エチレンジアミン四酢酸二ナトリウム塩、トリス(ヒドロキシメチル)アミノメタン(Tris)、クエン酸、グリシン、コハク酸、酢酸、塩化アンモニウム、四ホウ酸ナトリウム、2 -メルカプトエタノール、ジチオスレイトール、N−アセチル−L−システイン、アクロモペプチダーゼ、ラビアーゼ、β−N−アセチル−D−グルコサミニダーゼ、ムラミダーゼ、リゾチーム、プロナーゼ、サチリシン、セミアルカリプロテアーゼ、塩化ナトリウム、酢酸カリウムのうち、少なくとも2つ以上を含むもので構成する。これらは再蒸留水、または超純水若しくはDEPC処理水に溶解し、滅菌容器又は準無菌容器に無菌的に容れて製造する。準無菌容器とは滅菌されてはいないが、清潔な環境で無菌的に準ずる操作で作られた容器を意味する。分注に使用する器具類も滅菌されたものを使用する(以降の操作についても、同様である)。検体溶解処理用液は基本的に遮光して保存する。
界面活性剤を含む溶液は、SDS(ドデシル硫酸ナトリウム;ラウリル硫酸ナトリウム)、N−ラウロイルサルコシン酸ナトリウム、デオキシコール酸ナトリウム、ポリオキシエチレン(10)オクチルフェニルエーテル、ポリオキシエチレン(9)オクチルフェニルエーテル、ポリオキシエチレン(20)ソルビタンモノラウレート、ザルコシル、サポニン、3−〔(3−コラミドプロピル)ジメチルアンモニオ〕プロパンスルホン酸、ヘキサデシルピリジニウムクロリド、塩化ベンザルコニウムまたはこれらの類似誘導体からなるもの、エチレンジアミン四酢酸二ナトリウム塩、トリス(ヒドロキシメチル)アミノメタン、クエン酸、グリシン、コハク酸、酢酸のなかから、少なくとも1つまたは2つ以上含み、これ又はこれらは再蒸留水、または超純水若しくはDEPC処理水に溶解して、滅菌容器又は準無菌容器に無菌的に分注して製造する。
フェノール/クロロホルム/イソアミルアルコール(25:24:1)を含む混合液は、従来から行われている作製方法により作成することができる。一例を挙げれば、フェノールを60℃の水浴で加温融解し、0.5Mトリス塩酸pH8.0を加えて良く撹拌し、静置後、フェノール層と分離して上層に来る水層だけを出来るだけ除く。次に0.1Mトリス塩酸pH8.0を加えて良く撹拌し、クロロホルム、イソアミルアルコールを加えて更に撹拌する。静置後、フェノール層と水層が分離したら、0.1Mトリス塩酸pH8.0を少量容れておいた滅菌容器又は準無菌容器に、フェノール層のみを無菌的に分注して製造する。先に容れておく0.1Mトリス塩酸pH8.0の量は、容器中で高さ4mm〜5mm程度になる量から容れるフェノール層の量の1/5程度、またはそれ以上入れば良い。これはフェノール層が空気に触れて酸化しないようにするためであり、保存中に水が蒸発してフェノール層を空気と遮断するのに足りなくならない量が入っていれば良い。
フェノールを加温融解した時に、8−ヒドロキシキノリンを最終濃度0.05%になるように加えることが、酸化防止する上でも、フェノール層が黄色に着色して水層と見分けやすくなる点からも好ましい。又、フェノール層の酸化が進むと黄色から褐色更には赤身を帯びる様になるので使用すべきでない指標になる。尚、クロロホルム、イソアミルアルコールを加えるタイミングを、分離する0.5Mトリス塩酸pH8.0を除く前に早めても良いし、0.1Mトリス塩酸pH8.0をフェノール層に加えて良く撹拌する工程を省略しても良い。フェノール/クロロホルム/イソアミルアルコール(25:24:1)混液を、RNAの抽出専用にする場合には、RNAの分解を防ぐために、加えるトリス塩酸のpHを7.4〜7.6程度に下げても良い。また、DNA専用にする場合はpHを8.3〜8.8まで上げても良い。フェノールとクロロホルム/イソアミルアルコールを別に分けて、破砕処理液及びSDS溶液と混合、遠心、上清回収する操作を2度に分けるようにしても良い。通常はこれらを一緒にした1度の操作で充分である。フェノール/クロロホルム/イソアミルアルコール(25:24:1)混液は遮光して冷蔵保存する。
DEPC処理水は、0.1%DEPC水を容器に分注し、蓋をしっかり締めて1回以上転倒させた後、37℃に2時間以上静置させる。その後、蓋を弛めてオートクレーブ処理し、無菌的に取り出して蓋を閉めて製造する。細胞破砕用キットとは、前述のように製造した、2種類のビーズを充填したチューブ(ビーズ充填チューブ)、又は該ビーズ充填チューブおよび検体溶解処理用液を含むキットであり、検体溶解処理用液が遮光されていれば室温保存が可能である。核酸抽出用キット(DNA/RNA抽出キット)とは、該ビーズ充填チューブ、検体溶解処理用液、界面活性剤を含む溶液、およびフェノール/クロロホルム/イソアミルアルコール(25:24:1)混合液を含むキットであり、キット全体では冷蔵保存が必要である。これに上清回収する1.5〜2.0mL遠心チューブやエタノールを含んでも良いが、エタノール沈殿を行う為のエタノールはユーザーが自前で用意できるだろう。キット全体では冷蔵保存が必要である。しかし、フェノール/クロロホルム/イソアミルアルコール(25:24:1)混合液を単独で遮光して冷蔵保存すれば、残りは、検体溶解処理用液を遮光する限り、室温保存で良い。
ライシスバッファー(溶解液:本発明における検体溶解処理用液に該当する)の液量がDNA抽出効率に与える影響の確認試験
グラム陽性球菌(Staphylococcus aureus)、グラム陽桿菌(Bacillus subtilis, Mycobacterium avium)、グラム陰性桿菌(Salmonella typimurium)、及び酵母(カビ類の一種であるCandida albicans)をそれぞれ所定の濃度に培養し、懸濁液とした(括弧内はここで使用した菌種名である)。これを遠心して、沈殿としたものにライシスバッファーの液量を100、200、400、600、800、及び1000μLに変化させて混合し、ビーズ入りチューブ(全容量2mL)に移した。ビーズとして、約0.2mm径のジルコニアビーズを約0.3gと、約5mm径のジルコニアビーズ1個を用いた。
グラム陽性球菌(Staphylococcus aureus)、グラム陽桿菌(Bacillus subtilis, Mycobacterium avium)、グラム陰性桿菌(Salmonella typimurium)、及び酵母(カビ類の一種であるCandida albicans)をそれぞれ所定の濃度に培養し、懸濁液とした(括弧内はここで使用した菌種名である)。これを遠心して、沈殿としたものにライシスバッファーの液量を100、200、400、600、800、及び1000μLに変化させて混合し、ビーズ入りチューブ(全容量2mL)に移した。ビーズとして、約0.2mm径のジルコニアビーズを約0.3gと、約5mm径のジルコニアビーズ1個を用いた。
チューブに蓋をし、約70℃にて10分間加熱処理後、蓋をしっかりと閉め直し、 multibeads shocker(安井器械)を用いて、2500rpm、60秒間の破砕処理を行った。卓上ミニ遠心機で蓋に付いた処理液を落とし、これに、200μLのSDS含有溶液を混合し、更に約10分間、約70℃にて加熱処理を行った。その後、400μLのフェノール/クロロホルム混液(フェノール:クロロホルム:イソアミルアルコール=25:24:1)を混合し、ボルテックスミキサーにて約60秒間混合した後、15,000rpm、3分間遠心した。上澄み液を200μL回収して、別のチューブに移し、1mLの99%エタノールを用いて1回目の沈殿処理(15,000rpm、3分間)を行った後、沈殿に70%のエタノールを加えて2回目の沈殿処理を行った(15,000rpm、3分間)。抽出したDNAを100μLの蒸留水に懸濁し、濃度を計測した。
結果を表1に示した。
結果を表1に示した。
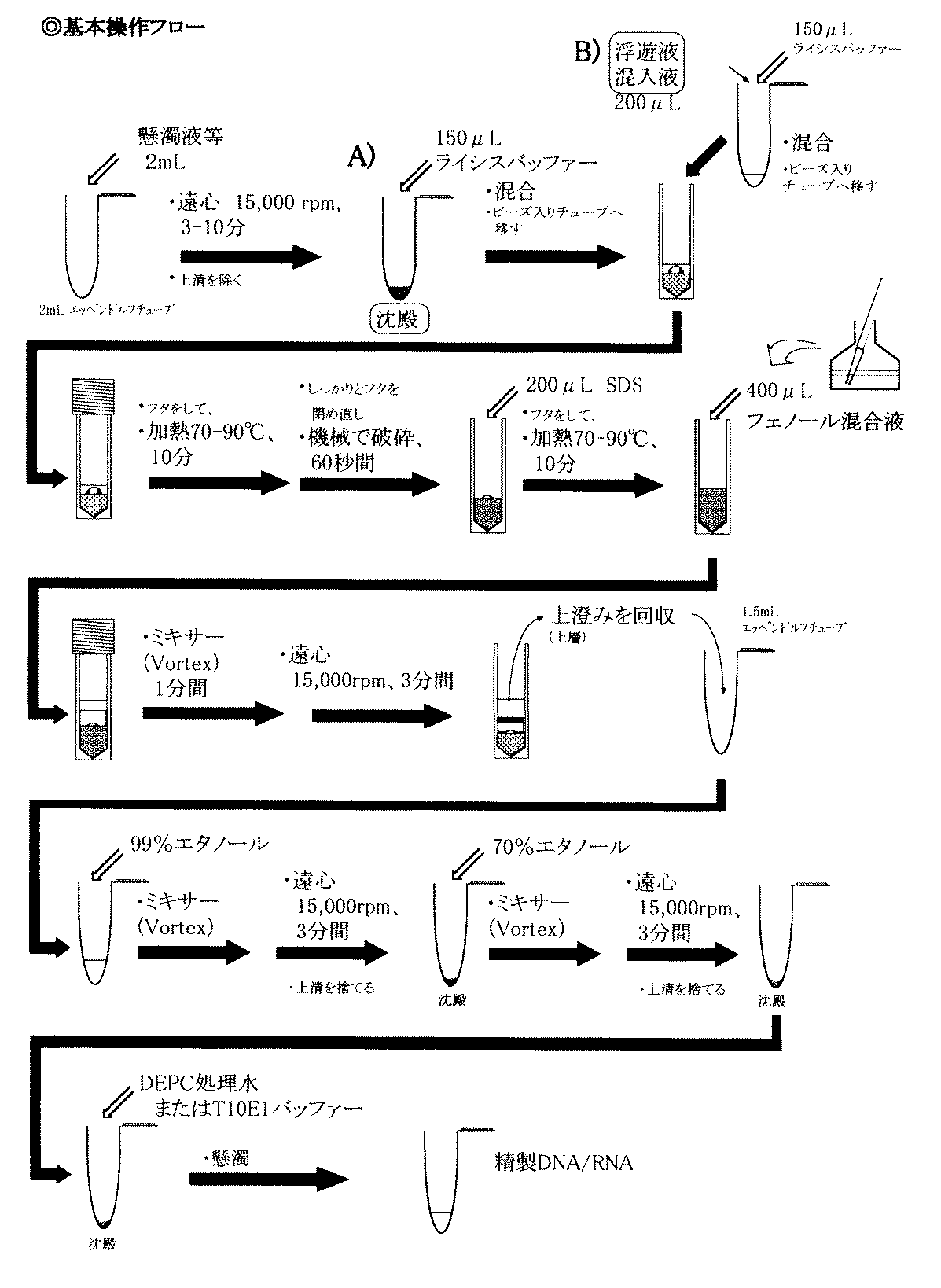
表1中の「A値」(菌種名の横に示す数値)は、DNA抽出操作に用いた菌体量(Ax108cfu/tube)を示している。また、「N.D.」は、計測限界以下を示す。
表より、本実施形態の方法は、各種の細胞(グラム陽性菌、グラム陰性菌、及び酵母)から良好に核酸(DNA)を抽出できることが確認された。また、最初に用いるライシスバッファーの液量が少ない方が、菌体からの核酸の抽出効率が良好であることが確認された。この時、DNAの回収を確認できたライシスバッファーの容量は最大で600μLであり、0.2mm径のジルコニアビーズ0.3gと5mm径のジルコニアビーズの容量は合わせて約150μLなので、合計約750μLとなり、2mL容器の空き空間は容器容量の8分の5になる。すべての菌でDNAの回収を確認できたライシスバッファーの容量は、200μLであり、0.2mm径のジルコニアビーズと5mm径のジルコニアビーズと合計すると約350μLとなり、2mL容器の空き空間は容器容量の100分の83になる。
表より、本実施形態の方法は、各種の細胞(グラム陽性菌、グラム陰性菌、及び酵母)から良好に核酸(DNA)を抽出できることが確認された。また、最初に用いるライシスバッファーの液量が少ない方が、菌体からの核酸の抽出効率が良好であることが確認された。この時、DNAの回収を確認できたライシスバッファーの容量は最大で600μLであり、0.2mm径のジルコニアビーズ0.3gと5mm径のジルコニアビーズの容量は合わせて約150μLなので、合計約750μLとなり、2mL容器の空き空間は容器容量の8分の5になる。すべての菌でDNAの回収を確認できたライシスバッファーの容量は、200μLであり、0.2mm径のジルコニアビーズと5mm径のジルコニアビーズと合計すると約350μLとなり、2mL容器の空き空間は容器容量の100分の83になる。
ビーズ量がDNA抽出効率に与える影響の確認試験
次に、小さいビーズとして、約0.2mm径のジルコニアビーズを約0.6g使用したことを除き、上記実施例1と同じ操作を行い、小さいビーズ量がDNA抽出効率に与える影響を確認した。
結果を表2に示した。
次に、小さいビーズとして、約0.2mm径のジルコニアビーズを約0.6g使用したことを除き、上記実施例1と同じ操作を行い、小さいビーズ量がDNA抽出効率に与える影響を確認した。
結果を表2に示した。
表2中の「A値」、及び「N.D.」は、表1と同じ意味であるため、説明を省略する。
表1及び表2を比較すると、いずれの方法においても、良好に菌体の核酸を抽出できたことが確認された。このことから、小さいビーズの使用量は、全容量2mLのチューブ当り、約0.3g〜約0.6gにおいて、特に核酸の抽出効率に影響を与えることなく、良好に使用できることが分かった。また、この時DNAの回収を確認できたライシスバッファーの容量も実施例1同様、最大で600μLであり、0.2mm径のジルコニアビーズ0.6gと5mm径のジルコニアビーズの容量は合わせて約230μLなので、合計約830μLとなり、2mL容器の空き空間は容器容量の100分の59になる。すべての菌でDNAの回収を確認できたライシスバッファーの容量は、200μLであり、0.2mm径のジルコニアビーズ0.6gと5mm径のジルコニアビーズの容量は合わせて約230μLなので、合計約430μLとなり、2mL容器の空き空間は容器容量の100分の79になる。
表1及び表2を比較すると、いずれの方法においても、良好に菌体の核酸を抽出できたことが確認された。このことから、小さいビーズの使用量は、全容量2mLのチューブ当り、約0.3g〜約0.6gにおいて、特に核酸の抽出効率に影響を与えることなく、良好に使用できることが分かった。また、この時DNAの回収を確認できたライシスバッファーの容量も実施例1同様、最大で600μLであり、0.2mm径のジルコニアビーズ0.6gと5mm径のジルコニアビーズの容量は合わせて約230μLなので、合計約830μLとなり、2mL容器の空き空間は容器容量の100分の59になる。すべての菌でDNAの回収を確認できたライシスバッファーの容量は、200μLであり、0.2mm径のジルコニアビーズ0.6gと5mm径のジルコニアビーズの容量は合わせて約230μLなので、合計約430μLとなり、2mL容器の空き空間は容器容量の100分の79になる。
細胞破砕処理時における界面活性剤の有無が破砕効率に与える影響の確認試験
次に、グラム陽性球菌の Staphylococcus aureus(S.aureus)を用いて、細胞破砕処理時における界面活性剤の有無が細胞の破砕効率、及び核酸抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の4種類の異なる条件で細胞破砕処理、及び核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度の計測、及びPCR法による遺伝子増幅実験を行った。
次に、グラム陽性球菌の Staphylococcus aureus(S.aureus)を用いて、細胞破砕処理時における界面活性剤の有無が細胞の破砕効率、及び核酸抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の4種類の異なる条件で細胞破砕処理、及び核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度の計測、及びPCR法による遺伝子増幅実験を行った。
(1)各菌体溶液1mLを遠心分離した後、上清を除いた沈殿に150μLのライシスバッファー、及び200μLのSDSを混合し、加熱処理(70℃、10分間)の後、大小2種類のジルコニアビーズ(約0.2mm径ビーズを2mLチューブ当り約0.5g、及び約5mm径ビーズを1個)を用いて、実施例1と同様に細胞破砕処理を行い、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(2)各菌体溶液1mLを遠心分離した後、上清を除いた沈殿に150μLのライシスバッファーを混合し、熱処理(70℃、10分間)の後、大小2種類のジルコニアビーズ(上記(1)と同じ組合せ)を用いて、実施例1と同様に細胞破砕処理を行った後に、200μLのSDSを添加し、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(2)各菌体溶液1mLを遠心分離した後、上清を除いた沈殿に150μLのライシスバッファーを混合し、熱処理(70℃、10分間)の後、大小2種類のジルコニアビーズ(上記(1)と同じ組合せ)を用いて、実施例1と同様に細胞破砕処理を行った後に、200μLのSDSを添加し、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(3)上記(1)において、SDSに代えて、TritonX−100を用いた以外は、同じ処理を行い、DNAを抽出した。
(4)上記(2)において、SDSに代えて、TritonX−100を用いた以外は、同じ処理を行い、DNAを抽出した。
(1)〜(4)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)によりDNA濃度を測定した後、PCR法による遺伝子増幅処理を行った。
なお、PCRに用いたプライマーは、 S. aureus の16S rDNA部分(約414bp)を増幅するためのユニバーサルプライマーを使用した。PCRの条件は、92℃、20秒間の熱変性の後、55℃、20秒間のアニーリング、72℃、30秒間の合成反応を1サイクルとし、これを35サイクル行った。また、PCRには、タカラ EX Taq;コードRR001Aを用い、全量が20μLの試験系を用いた。
結果を表3に示した。
(4)上記(2)において、SDSに代えて、TritonX−100を用いた以外は、同じ処理を行い、DNAを抽出した。
(1)〜(4)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)によりDNA濃度を測定した後、PCR法による遺伝子増幅処理を行った。
なお、PCRに用いたプライマーは、 S. aureus の16S rDNA部分(約414bp)を増幅するためのユニバーサルプライマーを使用した。PCRの条件は、92℃、20秒間の熱変性の後、55℃、20秒間のアニーリング、72℃、30秒間の合成反応を1サイクルとし、これを35サイクル行った。また、PCRには、タカラ EX Taq;コードRR001Aを用い、全量が20μLの試験系を用いた。
結果を表3に示した。
表3中、DNAが計測できたサンプルについては、PCR処理を行わなかった。また、「N.D.」は、計測限界以下を示し、PCR法による「+」は増幅バンドが認められたことを、「−」は増幅バンドが認められなかったことをそれぞれ示す。
表より、ビーズを用いた細胞破砕処理を行う際に、界面活性剤が存在する場合には、界面活性剤が存在しない場合に比べて、核酸の抽出効率が10倍〜100倍程度も低下することが分かった。また、SDSとTritonX−100では、いずれの界面活性剤を用いた場合にも、ほぼ同等の結果を示したことから、界面活性剤の種類による大きな影響は認め難かった。但し、PCR法を用いた遺伝子増幅試験においては、SDSを用いた場合には、23個/mLの濃度で増幅バンドが検出できた一方、TritonX−100を用いた場合には、23個/mLの濃度では増幅バンドが検出されなかったことから、菌体の種類によっては、界面活性剤の種類に応じて、検出感度が変わるのかも知れない。
表より、ビーズを用いた細胞破砕処理を行う際に、界面活性剤が存在する場合には、界面活性剤が存在しない場合に比べて、核酸の抽出効率が10倍〜100倍程度も低下することが分かった。また、SDSとTritonX−100では、いずれの界面活性剤を用いた場合にも、ほぼ同等の結果を示したことから、界面活性剤の種類による大きな影響は認め難かった。但し、PCR法を用いた遺伝子増幅試験においては、SDSを用いた場合には、23個/mLの濃度で増幅バンドが検出できた一方、TritonX−100を用いた場合には、23個/mLの濃度では増幅バンドが検出されなかったことから、菌体の種類によっては、界面活性剤の種類に応じて、検出感度が変わるのかも知れない。
大小2種類のビーズがDNA抽出効率に与える影響の確認試験
次に、 S.aureus を用いて、大小2種類のビーズが細胞の破砕効率、及び核酸抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の2種類の異なる条件で細胞破砕処理、及び核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度測定、及びPCR法による遺伝子増幅実験を行った。また、陰性コントロールとして、菌体を含まない溶液を用いて、下記(1)及び(2)の処理を行い、DNA濃度、及びPCR法を用いた遺伝子増幅処理を行った。
次に、 S.aureus を用いて、大小2種類のビーズが細胞の破砕効率、及び核酸抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の2種類の異なる条件で細胞破砕処理、及び核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度測定、及びPCR法による遺伝子増幅実験を行った。また、陰性コントロールとして、菌体を含まない溶液を用いて、下記(1)及び(2)の処理を行い、DNA濃度、及びPCR法を用いた遺伝子増幅処理を行った。
(1)各菌体溶液1mLを遠心分離した後、上清を除いた沈殿に150μLのライシスバッファーを混合し、熱処理(70℃、10分間)の後、1種類のジルコニアビーズ(約0.2mm径ビーズを2mLチューブ当り約0.5g)を用いて、実施例1と同様に細胞破砕処理を行った後に、200μLのSDSを添加し、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(2)上記(1)において、ジルコニアビーズとして、2種類のもの(約0.2mm径ビーズを2mLチューブ当り約0.5g、及び約5mm径ビーズを1個)を用いた以外は、同じ処理を行い、DNAを抽出した。
(2)上記(1)において、ジルコニアビーズとして、2種類のもの(約0.2mm径ビーズを2mLチューブ当り約0.5g、及び約5mm径ビーズを1個)を用いた以外は、同じ処理を行い、DNAを抽出した。
(1)及び(2)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)により濃度を測定した後、PCR法による遺伝子増幅処理を行った。なお、PCR法は、実施例3と同じであるため、記載を省略する。
結果を表4に示した。
結果を表4に示した。
表4中の「N.D.」、並びにPCR法による「+」、及び「−」の意味は、表3と同じである。
表より、1種類のみ(約0.2mm径)のジルコニアビーズを用いた場合には、2.3x103個/mLの菌体濃度までにしかPCR法による遺伝子増幅バンドが認められなかったが、大小2種類のジルコニアビーズを用いた場合には、2.3x10個/mLの菌体濃度までバンドが認められた。このことより、大小2種類のジルコニアビーズを用いることにより、1種類のみ(約0.2mm径)のビーズを用いる場合に比べると、約100倍以上の検出感度の向上が認められることが分かった。
表より、1種類のみ(約0.2mm径)のジルコニアビーズを用いた場合には、2.3x103個/mLの菌体濃度までにしかPCR法による遺伝子増幅バンドが認められなかったが、大小2種類のジルコニアビーズを用いた場合には、2.3x10個/mLの菌体濃度までバンドが認められた。このことより、大小2種類のジルコニアビーズを用いることにより、1種類のみ(約0.2mm径)のビーズを用いる場合に比べると、約100倍以上の検出感度の向上が認められることが分かった。
ビーズを用いた細胞破砕処理がDNA抽出効率に与える影響の確認試験
次に、 S.aureus を用いて、ビーズを用いた細胞破砕処理が、DNA抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の2種類の異なる条件を用いて、核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度測定、及びPCR法による遺伝子増幅実験を行った。また、陰性コントロールとして、菌体を含まない溶液を用いて、下記(1)及び(2)の処理を行い、DNA濃度測定、及びPCR法を用いた遺伝子増幅処理を行った。
次に、 S.aureus を用いて、ビーズを用いた細胞破砕処理が、DNA抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の2種類の異なる条件を用いて、核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度測定、及びPCR法による遺伝子増幅実験を行った。また、陰性コントロールとして、菌体を含まない溶液を用いて、下記(1)及び(2)の処理を行い、DNA濃度測定、及びPCR法を用いた遺伝子増幅処理を行った。
(1)各菌体溶液1mLを遠心分離した後、上清を除いた沈殿に150μLのライシスバッファーを混合し、加熱処理(70℃、10分間)の後、2種類のジルコニアビーズ(約0.2mm径ビーズを2mLチューブ当り約0.5g、約5mm径ビーズを1個)を用いて、実施例1と同様に細胞破砕処理を行った後に、200μLのSDSを添加し、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(2)上記(1)において、ジルコニアビーズを用いた細胞破砕処理を行わず、ライシスバッファーの混合・加熱処理(70℃、10分間)の後、200μLのSDSを添加し、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(1)及び(2)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)により濃度を測定した後、PCR法による遺伝子増幅処理を行った。なお、PCR法は、実施例3と同じであるため、記載を省略する。結果を表5に示した。
(1)及び(2)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)により濃度を測定した後、PCR法による遺伝子増幅処理を行った。なお、PCR法は、実施例3と同じであるため、記載を省略する。結果を表5に示した。
表5中の「N.D.」、並びにPCR法による「+」、及び「−」の意味は、表3と同じである。
表より、ジルコニアビーズを用いない場合(Lysisのみ)には、2.3x109個/mLの菌体濃度のみにおいて、DNA濃度が計測され(26.7ng/μL)、2.3x105個/mLの菌体濃度までにおいて、PCR法による遺伝子増幅バンドが認められた。一方、大小2種類のジルコニアビーズを用いた場合には、2.3x108個/mLの菌体濃度までDNA濃度が計測され(3.7ng/μL)、2.3x10個/mLの菌体濃度までPCR法による遺伝子増幅バンドが認められた。このことより、大小2種類のジルコニアビーズを用いることにより、ビーズを用いない場合に比べて、約10000倍以上の検出感度の向上が認められることが分かった。
表より、ジルコニアビーズを用いない場合(Lysisのみ)には、2.3x109個/mLの菌体濃度のみにおいて、DNA濃度が計測され(26.7ng/μL)、2.3x105個/mLの菌体濃度までにおいて、PCR法による遺伝子増幅バンドが認められた。一方、大小2種類のジルコニアビーズを用いた場合には、2.3x108個/mLの菌体濃度までDNA濃度が計測され(3.7ng/μL)、2.3x10個/mLの菌体濃度までPCR法による遺伝子増幅バンドが認められた。このことより、大小2種類のジルコニアビーズを用いることにより、ビーズを用いない場合に比べて、約10000倍以上の検出感度の向上が認められることが分かった。
ビーズを用いた細胞破砕処理の後、フェノール/クロロホルム抽出処理の際のビーズの有無がDNA抽出効率に与える影響の確認試験
次に S.aureus を用いて、ビーズを用いた細胞破砕処理の後、フェノール/クロロホルム混液を用いたタンパク除去処理を行う際に、ビーズの除去又は非除去が、DNA抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の2種類の異なる条件を用いて、核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度測定、及びPCR法による遺伝子増幅実験を行った。また、陰性コントロールとして、菌体を含まない溶液を用いて、下記(1)及び(2)の処理を行い、DNA濃度測定、及びPCR法を用いた遺伝子増幅処理を行った。
次に S.aureus を用いて、ビーズを用いた細胞破砕処理の後、フェノール/クロロホルム混液を用いたタンパク除去処理を行う際に、ビーズの除去又は非除去が、DNA抽出効率に与える影響を確認した。2.3個/mL〜2.3x109個/mLの濃度に調製した S.aureus を用い、以下の2種類の異なる条件を用いて、核酸抽出処理を行った後に、DNAを100μLの蒸留水に懸濁し、DNA濃度測定、及びPCR法による遺伝子増幅実験を行った。また、陰性コントロールとして、菌体を含まない溶液を用いて、下記(1)及び(2)の処理を行い、DNA濃度測定、及びPCR法を用いた遺伝子増幅処理を行った。
(1)各菌体溶液1mLを遠心分離した後、上清を除いた沈殿に150μLのライシスバッファーを混合し、加熱処理(70℃、10分間)の後、2種類のジルコニアビーズ(約0.2mm径ビーズを2mLチューブ当り約0.5g、約5mm径ビーズを1個)を用いて、実施例1と同様に細胞破砕処理を行った後に、200μLのSDSを添加し、加熱処理(70℃、10分間)の後、フェノール/クロロホルム混液によるタンパク質除去処理、及びエタノール沈殿処理を行い、DNAを抽出した。
(2)上記(1)において、フェノール/クロロホルム混液によるタンパク質除去処理を行う前に、ジルコニアビーズを除去し、それ以降は、同様の処理を行った。
(1)及び(2)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)により濃度を測定した後、PCR法による遺伝子増幅処理を行った。なお、PCR法は、実施例3と同じであるため、記載を省略する。結果を表6に示した。
(1)及び(2)の処理によって抽出されたDNAは、100μLの蒸留水に懸濁し、紫外吸光度(260nm)により濃度を測定した後、PCR法による遺伝子増幅処理を行った。なお、PCR法は、実施例3と同じであるため、記載を省略する。結果を表6に示した。
表6中の「N.D.」、並びにPCR法による「+」、及び「−」の意味は、表3と同じである。
表より、フェノール/クロロホルム処理の前にジルコニアビーズを除去した場合には、2.3x109個/mLの菌体濃度のみにおいて、DNA濃度が計測され(60.7ng/μL)、2.3x103個/mLの菌体濃度までにおいて、PCR法による遺伝子増幅バンドが認められた。一方、ジルコニアビーズを除去することなく、フェノール/クロロホルム処理を行った場合には、2.3x108個/mLの菌体濃度までDNA濃度が計測され(3.7ng/μL)、2.3x10個/mLの菌体濃度までPCR法による遺伝子増幅バンドが認められた。このことより、フェノール/クロロホルム処理を行う際には、細胞破砕処理に用いたジルコニアビーズを除去することなく、チューブ内に存在させておく方が、ビーズを除去した場合に比べて、約100倍以上の検出感度の向上が認められることが分かった。
表より、フェノール/クロロホルム処理の前にジルコニアビーズを除去した場合には、2.3x109個/mLの菌体濃度のみにおいて、DNA濃度が計測され(60.7ng/μL)、2.3x103個/mLの菌体濃度までにおいて、PCR法による遺伝子増幅バンドが認められた。一方、ジルコニアビーズを除去することなく、フェノール/クロロホルム処理を行った場合には、2.3x108個/mLの菌体濃度までDNA濃度が計測され(3.7ng/μL)、2.3x10個/mLの菌体濃度までPCR法による遺伝子増幅バンドが認められた。このことより、フェノール/クロロホルム処理を行う際には、細胞破砕処理に用いたジルコニアビーズを除去することなく、チューブ内に存在させておく方が、ビーズを除去した場合に比べて、約100倍以上の検出感度の向上が認められることが分かった。
<実施例のまとめ、及び考察>
上記各実施例より、以下(1)〜(6)のことが判明した。
(1)本実施例の方法によれば、各種の細胞(グラム陽性菌、グラム陰性菌、及び酵母)から良好に核酸(DNA)を抽出できることが確認された。また、最初に用いるライシスバッファーの液量が少なく、ビーズ容量を考慮した容器の空き空間容量が容器容量の2分の1以上且つ大きい方が、菌体からの核酸の抽出効率が良好である。
(2)大小2種類のビーズを用いて、細胞破砕処理を行う場合において、小さいビーズの使用量は、全容量2mLのチューブ当り、約0.3g〜約0.6gにおいて、特に核酸の抽出効率に影響を与えることなく、良好に使用できる。
上記各実施例より、以下(1)〜(6)のことが判明した。
(1)本実施例の方法によれば、各種の細胞(グラム陽性菌、グラム陰性菌、及び酵母)から良好に核酸(DNA)を抽出できることが確認された。また、最初に用いるライシスバッファーの液量が少なく、ビーズ容量を考慮した容器の空き空間容量が容器容量の2分の1以上且つ大きい方が、菌体からの核酸の抽出効率が良好である。
(2)大小2種類のビーズを用いて、細胞破砕処理を行う場合において、小さいビーズの使用量は、全容量2mLのチューブ当り、約0.3g〜約0.6gにおいて、特に核酸の抽出効率に影響を与えることなく、良好に使用できる。
(3)ビーズを用いた細胞破砕処理を行う際に、界面活性剤が存在する場合には、界面活性剤が存在しない場合に比べて、核酸の抽出効率が10倍〜100倍程度も低下する。
(4)大小2種類のジルコニアビーズを用いることにより、一般的に用いられる約0.2mm径の小ビーズのみを用いる場合に比べると、約100倍以上の検出感度の向上が認められる。
(4)大小2種類のジルコニアビーズを用いることにより、一般的に用いられる約0.2mm径の小ビーズのみを用いる場合に比べると、約100倍以上の検出感度の向上が認められる。
(5)大小2種類のジルコニアビーズを用いることにより、ビーズを用いない場合に比べて、約10000倍以上の検出感度の向上が認められる。
(6)フェノール/クロロホルム処理を行う際には、細胞破砕処理に用いたジルコニアビーズを除去することなく、チューブ内に存在させておく方が、ビーズを除去した場合に比べて、約100倍以上の検出感度の向上が認められる。
(6)フェノール/クロロホルム処理を行う際には、細胞破砕処理に用いたジルコニアビーズを除去することなく、チューブ内に存在させておく方が、ビーズを除去した場合に比べて、約100倍以上の検出感度の向上が認められる。
従来にも、大小2種類の破砕用物体を添加する例が認められる( QBIOGENE(前出))。しかし、この例では、小さい物体は、ビーズではなくガーネットの破片様の尖った物質であることに加え、大小2種類の破砕用物体が同一材質から形成されたものではなかった。本実施例によれば、大小2種類のビーズを用い、特にそれらが同じジルコニアから形成された場合には、各種菌体から良好に核酸を抽出できることが確認された。
また、従来例には、ビーズを容器の半分まで入れる方法がある(バイオスペック・プロダクツ社、 Mini-BeadBeator)。この例では、検体およびフェノールを入れて震盪し、該検体およびフェノールの合計量を容器の半分として、容器に残りの空間容量を作らない但し書き付きの方法による。しかしながら、実際に検証したところ、この方法では余り破砕効果が良くない。一方、本実施例のフェノール/クロロホルム処理の前に破砕処理を行い、容器の空き空間を充分にとる方法によれば、破砕効果が非常に高い。更にフェノール/クロロホルム処理の際には、全容量が容器容量(2mL)の約7/10程度以下に収まるので、激しく混合することが可能となり、タンパク変性効果も良好にできる。
また、従来例においては、ビーズではないガーネット・マトリクスが入った2.0mLチューブに、使用時に1/4インチのセラミック粒子を使用時に加えるものがある(FastDNAR Kit)。しかしながら、裏を返せば、これは予めガーネット・マトリクスとセラミック粒子とを共にチューブに入れておくことは、輸送に堪えないことを示唆しており、使用感の良好なキットを調製することは困難である。一方、本実施例によれば、細胞破砕処理用のチューブに、大小2種類のジルコニアビーズを封入した状態で、キットとすることができるので、微生物の核酸抽出を行おうとする者に対して、使用感の良好なキットを提供することができる。
Claims (23)
- 微生物の有無を確認するための検体と検体溶解処理用液を混合した処理液と、ビーズとを容器に混合し、この容器に物理的衝撃を加えて、前記微生物の細胞を破砕処理する方法であって、
前記処理液とビーズとを加えた合計容量が、前記容器の全容量の少なくとも3/8以下とし、細胞破砕処理時の容器の空間容量を容器の全容量の5/8以上としたことを特徴とする微生物の細胞破砕処理方法。 - 前記細胞破砕処理時には、前記処理液の容量が、前記ビーズの容量よりも小さいことを特徴とする請求項1に記載の細胞破砕処理方法。
- 前記細胞破砕処理時には、前記容器内には、実質的に界面活性剤を含ませない状態とすることを特徴とする請求項1または2に記載の細胞破砕処理方法。
- 前記ビーズは、大きさ及び重さが異なる少なくとも二種類以上のビーズを同時に用いることを特徴とする請求項1〜3のいずれかに記載の細胞破砕処理方法。
- 大きさ及び重さが異なる少なくとも二種類以上のビーズは、同じ材質からなるものであることを特徴とする請求項4に記載の細胞破砕処理方法。
- 大きさ及び重さが異なる少なくとも二種類以上のビーズは、異なる材質からなるものであることを特徴とする請求項4に記載の細胞破砕処理方法。
- 大きさ及び重さが異なる少なくとも二種類以上のビーズのうち、より小さいビーズについては多数使用する一方、より大きいビーズについては、1個だけ使用することを特徴とする請求項4〜6のいずれかに記載の細胞破砕処理方法。
- 前記ビーズの材質の比重は、5以上であることを特徴とする請求項1〜7のいずれかに記載の細胞破砕処理方法。
- 請求項1〜請求項8のいずれかに記載の方法で前記微生物の細胞壁を破砕し、その微生物の核酸を抽出することを特徴とする核酸抽出方法。
- 請求項1〜請求項8のいずれかに記載の方法で前記微生物の細胞壁を破砕した後、界面活性剤を添加し、前記微生物の核酸を抽出することを特徴とする核酸抽出方法。
- 請求項1〜請求項8のいずれかに記載の方法で前記微生物の細胞壁を破砕した後、前記ビーズが存在する状態で界面活性剤を添加し、フェノール/クロロホルム混液を混合することを特徴とする核酸抽出方法。
- 請求項1〜請求項8のいずれかに記載の方法で前記微生物の細胞壁を破砕した後、前記容器中に界面活性剤を加え、その容器中にフェノール/クロロホルム混液を前記ビーズが浸漬するまで添加することを特徴とする核酸抽出方法。
- 請求項10〜請求項12のいずれかに記載の方法において、添加する界面活性剤を含む溶液の容量は、前記処理液及びビーズを加えたときの容量が前記容器の全容量の4/20〜7/20であることを特徴とする核酸抽出方法。
- 請求項1〜8のいずれかに記載の方法を用いることを特徴とする細胞破砕用キット。
- 請求項4〜7のいずれかに記載の方法を用いると共に、前記二種類以上のビーズを封入した容器を備えることを特徴とする細胞破砕用キット。
- 少なくとも1種類のビーズの材質の比重が5以上であることを特徴とする請求項14または15に記載の細胞破砕用キット。
- 前記ビーズが、ジルコニウム、ジルコニア(酸化ジルコニウム)、ステンレススチール、又はタングステンであることを特徴とする請求項16に記載の細胞破砕用キット。
- 請求項14〜請求項17のいずれかに記載のキットの製造方法。
- 前記ビーズを容器の全容量の7%〜15%として封入したことを特徴とする請求項14〜請求項18のいずれかに記載のキットの製造方法。
- 請求項9〜13のいずれかに記載の方法を用いることを特徴とする核酸抽出キット。
- 前記検体溶解処理用液には、実質的に界面活性剤を含まないことを特徴とする請求項20に記載の核酸抽出キット。
- 少なくとも、(1)ビーズを封入した容器、(2)検体溶解処理用液、(3)界面活性剤を含む溶液、及び(4)フェノール/クロロホルムを含む混合液を、それぞれ独立して備えたことを特徴とする請求項20または21に記載の核酸抽出キット。
- 請求項20〜22のいずれかに記載のキットを製造する方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004336245A JP2006141292A (ja) | 2004-11-19 | 2004-11-19 | 微生物の破砕・核酸抽出方法、この方法を用いたキット、及びその製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004336245A JP2006141292A (ja) | 2004-11-19 | 2004-11-19 | 微生物の破砕・核酸抽出方法、この方法を用いたキット、及びその製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2006141292A true JP2006141292A (ja) | 2006-06-08 |
Family
ID=36621656
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004336245A Pending JP2006141292A (ja) | 2004-11-19 | 2004-11-19 | 微生物の破砕・核酸抽出方法、この方法を用いたキット、及びその製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2006141292A (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008109864A (ja) * | 2006-10-30 | 2008-05-15 | Hitachi Ltd | 遺伝子配列解析システム |
| JP2009011210A (ja) * | 2007-07-03 | 2009-01-22 | Keio Gijuku | Dna抽出方法及びそのためのキット |
| JP2010110234A (ja) * | 2008-11-04 | 2010-05-20 | Tokai Univ | 乾式破砕を用いた微生物細胞からの核酸抽出法 |
| JP2010528643A (ja) * | 2007-06-07 | 2010-08-26 | ビオメリュー | 環境または臨床試料中に存在する微生物を溶解し、前記微生物から核酸を分析用に抽出する装置 |
| JP2013021959A (ja) * | 2011-07-20 | 2013-02-04 | Sony Corp | 核酸抽出方法及び核酸抽出用カートリッジ |
| US8986986B2 (en) | 2010-10-29 | 2015-03-24 | Samsung Electronics Co., Ltd. | Cell lysis device and methods of lysing cells or viruses |
| WO2016079981A1 (ja) * | 2014-11-18 | 2016-05-26 | 和光純薬工業株式会社 | 検体の破砕装置およびその方法 |
| CN113817721A (zh) * | 2021-10-21 | 2021-12-21 | 湖北轻工职业技术学院(湖北啤酒学校) | 从多孔吸附材料中提取微生物dna的试剂盒及提取方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002512001A (ja) * | 1997-09-23 | 2002-04-23 | ベーイーオー・メリュー | 微生物の溶解方法 |
| JP2003164282A (ja) * | 2001-11-29 | 2003-06-10 | Rakan:Kk | 微生物の検出方法、及び微生物の検出用プライマーセット |
-
2004
- 2004-11-19 JP JP2004336245A patent/JP2006141292A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002512001A (ja) * | 1997-09-23 | 2002-04-23 | ベーイーオー・メリュー | 微生物の溶解方法 |
| JP2003164282A (ja) * | 2001-11-29 | 2003-06-10 | Rakan:Kk | 微生物の検出方法、及び微生物の検出用プライマーセット |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008109864A (ja) * | 2006-10-30 | 2008-05-15 | Hitachi Ltd | 遺伝子配列解析システム |
| JP2010528643A (ja) * | 2007-06-07 | 2010-08-26 | ビオメリュー | 環境または臨床試料中に存在する微生物を溶解し、前記微生物から核酸を分析用に抽出する装置 |
| JP2009011210A (ja) * | 2007-07-03 | 2009-01-22 | Keio Gijuku | Dna抽出方法及びそのためのキット |
| JP2010110234A (ja) * | 2008-11-04 | 2010-05-20 | Tokai Univ | 乾式破砕を用いた微生物細胞からの核酸抽出法 |
| US8986986B2 (en) | 2010-10-29 | 2015-03-24 | Samsung Electronics Co., Ltd. | Cell lysis device and methods of lysing cells or viruses |
| JP2013021959A (ja) * | 2011-07-20 | 2013-02-04 | Sony Corp | 核酸抽出方法及び核酸抽出用カートリッジ |
| WO2016079981A1 (ja) * | 2014-11-18 | 2016-05-26 | 和光純薬工業株式会社 | 検体の破砕装置およびその方法 |
| EP3222988A4 (en) * | 2014-11-18 | 2017-11-22 | Wako Pure Chemical Industries, Ltd. | Specimen fragmentation device and method for same |
| US10760115B2 (en) | 2014-11-18 | 2020-09-01 | Fujifilm Wako Pure Chemical Corporation | Specimen disrupting method and specimen disrupting apparatus |
| CN113817721A (zh) * | 2021-10-21 | 2021-12-21 | 湖北轻工职业技术学院(湖北啤酒学校) | 从多孔吸附材料中提取微生物dna的试剂盒及提取方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3928973B2 (ja) | 細胞成分を単離する方法、装置及び試薬 | |
| US6992182B1 (en) | Method for isolating DNA from biological materials | |
| KR0148693B1 (ko) | 핵산 분리방법 | |
| JP4358618B2 (ja) | 核酸の放出及びそれらを検出するための細胞を迅速に溶解せしめるための万能な方法及び組成物 | |
| JP3787354B2 (ja) | 核酸の単離方法 | |
| JP5965843B2 (ja) | 微生物核酸および必要に応じて追加のウイルス核酸を選択的に富化し単離するための方法 | |
| US5643767A (en) | Process for isolating cellular components | |
| US7893251B2 (en) | Methods for selective isolation of nucleic acids from microbial cells present in samples containing higher eukaryotic cells and/or tissues | |
| JP2022033906A (ja) | ビーズ破砕用チューブならびに微生物からデオキシリボ核酸および/またはリボ核酸を抽出する方法 | |
| JP2008142083A (ja) | ポリドカノールおよび誘導体を用いた核酸単離 | |
| JP2006517225A (ja) | 核酸抽出のための生物学的試料の化学処理および該処理用のキット | |
| JP5714291B2 (ja) | 抗酸菌dnaの抽出精製法 | |
| JP2006141292A (ja) | 微生物の破砕・核酸抽出方法、この方法を用いたキット、及びその製造方法 | |
| JP6713007B2 (ja) | 核酸の単離 | |
| JP2006311803A (ja) | 核酸精製方法、及び核酸精製器具 | |
| EP3971290A1 (en) | Method for producing a lysate from cells contained in a liquid sample | |
| JP5599013B2 (ja) | 血液検体からの微生物核酸の抽出方法 | |
| EP3408388B1 (en) | Method for producing a lysate from cells contained in a liquid sample | |
| RU2807254C1 (ru) | Универсальный способ выделения ДНК и лизирующая смесь для его осуществления | |
| CN118406740B (zh) | 通用型样本核酸释放液、核酸提取试剂盒及提取方法 | |
| TWI875469B (zh) | 磁珠式核酸萃取系統 | |
| JP7216652B2 (ja) | ビーズ破砕用チューブ並びに微生物からデオキシリボ核酸及び/又はリボ核酸を抽出する方法 | |
| CN118995383A (zh) | 一种磁珠法微生物基因组提取试剂盒及微生物基因组提取方法 | |
| HK40070989A (en) | Method for producing a lysate from cells contained in a liquid sample | |
| JP2010110234A (ja) | 乾式破砕を用いた微生物細胞からの核酸抽出法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Effective date: 20071019 Free format text: JAPANESE INTERMEDIATE CODE: A621 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071212 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20101019 |
|
| A02 | Decision of refusal |
Effective date: 20110412 Free format text: JAPANESE INTERMEDIATE CODE: A02 |