FR2751973A1 - Nouveaux sels n proteges d'acide alfa-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent - Google Patents
Nouveaux sels n proteges d'acide alfa-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent Download PDFInfo
- Publication number
- FR2751973A1 FR2751973A1 FR9609845A FR9609845A FR2751973A1 FR 2751973 A1 FR2751973 A1 FR 2751973A1 FR 9609845 A FR9609845 A FR 9609845A FR 9609845 A FR9609845 A FR 9609845A FR 2751973 A1 FR2751973 A1 FR 2751973A1
- Authority
- FR
- France
- Prior art keywords
- group
- salt
- chosen
- alkyl
- intermediate compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 33
- BUUPQKDIAURBJP-UHFFFAOYSA-N sulfinic acid Chemical compound OS=O BUUPQKDIAURBJP-UHFFFAOYSA-N 0.000 title 1
- 150000003839 salts Chemical class 0.000 claims abstract description 55
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 29
- 150000001875 compounds Chemical class 0.000 claims abstract description 29
- 125000002252 acyl group Chemical group 0.000 claims abstract description 23
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 17
- 239000001257 hydrogen Substances 0.000 claims abstract description 17
- 125000003118 aryl group Chemical group 0.000 claims abstract description 16
- 150000002431 hydrogen Chemical group 0.000 claims abstract description 13
- 150000001768 cations Chemical class 0.000 claims abstract description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 4
- 229910052799 carbon Inorganic materials 0.000 claims description 17
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 12
- 150000001408 amides Chemical class 0.000 claims description 10
- 238000006243 chemical reaction Methods 0.000 claims description 10
- XMYQHJDBLRZMLW-UHFFFAOYSA-N methanolamine Chemical compound NCO XMYQHJDBLRZMLW-UHFFFAOYSA-N 0.000 claims description 10
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical class OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 claims description 9
- 150000001721 carbon Chemical group 0.000 claims description 8
- 150000002148 esters Chemical group 0.000 claims description 8
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 claims description 8
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 claims description 7
- 150000002576 ketones Chemical class 0.000 claims description 7
- 125000006239 protecting group Chemical group 0.000 claims description 7
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 6
- 150000002367 halogens Chemical class 0.000 claims description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 6
- 150000007854 aminals Chemical class 0.000 claims description 5
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 claims description 5
- 230000006378 damage Effects 0.000 claims description 5
- XWGJFPHUCFXLBL-UHFFFAOYSA-M rongalite Chemical compound [Na+].OCS([O-])=O XWGJFPHUCFXLBL-UHFFFAOYSA-M 0.000 claims description 5
- 125000000565 sulfonamide group Chemical group 0.000 claims description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 4
- 125000000962 organic group Chemical group 0.000 claims description 4
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 claims description 4
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 4
- 125000004122 cyclic group Chemical group 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 150000004715 keto acids Chemical class 0.000 claims description 3
- 229910006069 SO3H Inorganic materials 0.000 claims description 2
- 150000001412 amines Chemical class 0.000 claims description 2
- 150000001450 anions Chemical class 0.000 claims description 2
- 239000007864 aqueous solution Substances 0.000 claims description 2
- XBPCUCUWBYBCDP-UHFFFAOYSA-O dicyclohexylazanium Chemical compound C1CCCCC1[NH2+]C1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-O 0.000 claims description 2
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical group OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 claims description 2
- 125000006575 electron-withdrawing group Chemical group 0.000 claims description 2
- 125000001033 ether group Chemical group 0.000 claims description 2
- 238000001704 evaporation Methods 0.000 claims description 2
- 230000008020 evaporation Effects 0.000 claims description 2
- 150000007530 organic bases Chemical group 0.000 claims description 2
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 claims description 2
- 238000005576 amination reaction Methods 0.000 abstract description 6
- 239000002253 acid Substances 0.000 abstract description 4
- -1 hydroxyacid salt Chemical class 0.000 abstract description 3
- 230000015572 biosynthetic process Effects 0.000 description 14
- 238000003786 synthesis reaction Methods 0.000 description 13
- 230000008018 melting Effects 0.000 description 11
- 238000002844 melting Methods 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 239000000243 solution Substances 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 5
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 238000000605 extraction Methods 0.000 description 4
- DOUHZFSGSXMPIE-UHFFFAOYSA-N hydroxidooxidosulfur(.) Chemical group [O]SO DOUHZFSGSXMPIE-UHFFFAOYSA-N 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 150000002466 imines Chemical class 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 150000003456 sulfonamides Chemical class 0.000 description 3
- 230000007704 transition Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 150000001371 alpha-amino acids Chemical group 0.000 description 2
- JXUFISIHEZOBPI-UHFFFAOYSA-N amidosulfurous acid Chemical group NS(O)=O JXUFISIHEZOBPI-UHFFFAOYSA-N 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 238000000806 fluorine-19 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- VGJWVEYTYIBXIA-UHFFFAOYSA-N 2,2,2-trifluoroethane-1,1-diol Chemical compound OC(O)C(F)(F)F VGJWVEYTYIBXIA-UHFFFAOYSA-N 0.000 description 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000003295 alanine group Chemical class N[C@@H](C)C(=O)* 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 125000003275 alpha amino acid group Chemical group 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- DQPBABKTKYNPMH-UHFFFAOYSA-N amino hydrogen sulfate Chemical compound NOS(O)(=O)=O DQPBABKTKYNPMH-UHFFFAOYSA-N 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 150000001733 carboxylic acid esters Chemical group 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000002026 chloroform extract Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000004030 hiv protease inhibitor Substances 0.000 description 1
- 150000001261 hydroxy acids Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- QLOAVXSYZAJECW-UHFFFAOYSA-N methane;molecular fluorine Chemical compound C.FF QLOAVXSYZAJECW-UHFFFAOYSA-N 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 150000003017 phosphorus Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 125000001010 sulfinic acid amide group Chemical group 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/31—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atoms of the sulfonamide groups bound to acyclic carbon atoms
- C07C311/32—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups having the sulfur atoms of the sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/45—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups at least one of the singly-bound nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom, e.g. N-acylaminosulfonamides
- C07C311/47—Y being a hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C313/00—Sulfinic acids; Sulfenic acids; Halides, esters or anhydrides thereof; Amides of sulfinic or sulfenic acids, i.e. compounds having singly-bound oxygen atoms of sulfinic or sulfenic groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C313/02—Sulfinic acids; Derivatives thereof
- C07C313/04—Sulfinic acids; Esters thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C313/00—Sulfinic acids; Sulfenic acids; Halides, esters or anhydrides thereof; Amides of sulfinic or sulfenic acids, i.e. compounds having singly-bound oxygen atoms of sulfinic or sulfenic groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C313/02—Sulfinic acids; Derivatives thereof
- C07C313/06—Sulfinamides
Abstract
L'invention concerne un procédé d'obtention de sels N protégés d'acide alpha -aminosulfinique de formule Y - NH - CR'R - SO2 **-, M**+. On fait réagir un composé intermédiaire Y - NH - CR'R - R1 en milieu fortement basique avec un sel d'hydroxyacide sulfinique. L'invention concerne aussi les sels obtenus où Y est un groupement acyle distinct de R'' SO2 , R'' étant le méthyle ou PCH3 C6 H4 , R est un groupe carboné halogène, un alkyle ou un aryle, R' est l'hydrogène, un alkyle ou un aryle, M**+ est un cation. L'invention concerne aussi des sulfonopeptides stables dérivant de ces sels par amination électrophile.
Description
NOUVEAUX SELS N PROTEGES D'ACIDE a-AMINO SULFINIQUE, LEUR
PROCEDE D'OBTENTION, ET LES SULFONOPEPTIDES QUI EN DERIVENT
Les analogues de l'étant de transition de l'hydrolyse de la liaison peptidique (rentrant dans la classe des pseudopeptides) peuvent agir comme faux substrats non dégradables ou comme inhibiteurs de protéases, et ont donc un grand intérêt thérapeutique, notamment pour l'obtention d'antibiotiques, d'antihypertenseurs, d'analgésiques, d'inhibiteurs de la protéase du VIH...
PROCEDE D'OBTENTION, ET LES SULFONOPEPTIDES QUI EN DERIVENT
Les analogues de l'étant de transition de l'hydrolyse de la liaison peptidique (rentrant dans la classe des pseudopeptides) peuvent agir comme faux substrats non dégradables ou comme inhibiteurs de protéases, et ont donc un grand intérêt thérapeutique, notamment pour l'obtention d'antibiotiques, d'antihypertenseurs, d'analgésiques, d'inhibiteurs de la protéase du VIH...
On a déjà réussi la synthèse d'analogues phosphorés de peptides. Néanmoins, ces analogues sont imparfaits compte tenu de la fragilité en milieu acide de la liaison P-N.
En outre, on considérait jusqu'à maintenant que la synthèse des a sulfonopeptides (enchaînement d'a-aminoacides comprenant au moins un résidu d'acide a-aminosulfonique) était impossible du fait de l'instabilité fondamentale des a aminosulfonamides (par exemple "Azasulfonamidopeptides as Peptide Bond Hydrolysis
Transition State Analogues. Part 1. Synthetic Approaches,
Thimothy J. Cheeseright et al, J. Chem. Soc. Perkin
Trans. 1, 1994, p. 1595 ; "Some Studies on peptide
Analogues Involving the sulphinamide Group", David Merricks et al, J. Chem. Soc. Perkin Trans. 1., 1991, p. 2169 "Synthesis of Sulfonamido-Pseudopeptides : New Chiral
Unnatural Oligomers", Cesare Gennari et al, Angew. Chem.
Transition State Analogues. Part 1. Synthetic Approaches,
Thimothy J. Cheeseright et al, J. Chem. Soc. Perkin
Trans. 1, 1994, p. 1595 ; "Some Studies on peptide
Analogues Involving the sulphinamide Group", David Merricks et al, J. Chem. Soc. Perkin Trans. 1., 1991, p. 2169 "Synthesis of Sulfonamido-Pseudopeptides : New Chiral
Unnatural Oligomers", Cesare Gennari et al, Angew. Chem.
Int. Ed. Engl. 1994, 33, nO 20, p. 2067...)
Certains ss ou y sulfonopeptides ont été decrits (par exemple "Synthesis of Peptides containing a
Sulfinamide or a Sulfonamide Transition-State Isostere",
W.J. MOREE et al, Tetrahedron, 1993, vol. 49, nO 5, p 1133). Néanmoins, ces composes n'étant pas exactement des analogues de l'étant de transition de l'hydrolyse de la liaison peptidique, c'est-à-dire du groupement

ils n'ont pas ou peu d'activité thérapeutique.
Certains ss ou y sulfonopeptides ont été decrits (par exemple "Synthesis of Peptides containing a
Sulfinamide or a Sulfonamide Transition-State Isostere",
W.J. MOREE et al, Tetrahedron, 1993, vol. 49, nO 5, p 1133). Néanmoins, ces composes n'étant pas exactement des analogues de l'étant de transition de l'hydrolyse de la liaison peptidique, c'est-à-dire du groupement
ils n'ont pas ou peu d'activité thérapeutique.
La publication "Synthese de sels d'aaminoacides sulfiniques substitués à l'azote par un groupe
Méthane ou p-Toluène-Sulfonyle", M. MULLIEZ et C. NAUDY,
Tetrahedron, 1994, vol. 50, NO 18, p 5401, enseigne la synthèse des composés R' 'SO2 - NH - CHR - SO2Na, R'' étant
Me ou pMeC6H4. il était déjà envisagé dans ce document que ces composés devaient permettre la synthèse d'analogues peptidiques a sulfonamides par amination électrophile.
Méthane ou p-Toluène-Sulfonyle", M. MULLIEZ et C. NAUDY,
Tetrahedron, 1994, vol. 50, NO 18, p 5401, enseigne la synthèse des composés R' 'SO2 - NH - CHR - SO2Na, R'' étant
Me ou pMeC6H4. il était déjà envisagé dans ce document que ces composés devaient permettre la synthèse d'analogues peptidiques a sulfonamides par amination électrophile.
Néanmoins, le groupement sulfonyle R''SO2 est délicat à éliminer, de sorte que l'obtention des a sulfonopeptides à partir de ces composés, se heurte au problème de la destruction simultanée de la fonction a sulfonamide pouvant être formée.
Par ailleurs, les rares dérivés d'aaminoacides sulfiniques décrits ne sont pas protégés sur l'azote par un groupement protecteur acyle, ne sont pas substitués en a, et sont instables.
L'invention vise donc à pallier ces inconvénients en proposant des sels N protégés d'acide a-amino sulfinique synthétisés et stables et leur procédé d'obtention. Par stables, on entend que l'on peut les garder au moins 8 heures -notamment plusieurs jours- sans altération.
Plus particulièrement, l'invention vise à proposer un procédé permettant l'obtention desdits sels dont le substituant de l'azote peut être librement choisi, notamment parmi les groupements protecteurs traditionnels acyles (Cbz, Boc, acétyle, ...), et en particulier distinct d'un groupe sulfonyle, de sorte que lesdits sels puissent servir de précurseurs d'a sulfonopeptides.
L'invention vise encore plus particulièrement à proposer des sels tels que mentionnés ci-dessus dont le carbone a est substitue, notamment par un groupe carboné halogéné, et leur procédé d'obtention.
L'invention vise ainsi à proposer ces sels qui sont des précurseurs de sulfonopeptides, voire même de sulfinopeptides, mais qui peuvent aussi être isolés et utilisés en tant que tels, notamment comme analogues
N acyles des acides a aminés carboxyliques naturels.
N acyles des acides a aminés carboxyliques naturels.
L'invention vise en outre à proposer pour la première fois des sulfonopeptiques stables, à savoir, les a sulfonopeptides stables synthétisés et isolés ou pouvant être isolés, dérivant de ces sels.
Dans toute la présente demande, on utilise les terminologies et les abréviations suivantes pour désigner les groupes substituants
- sulfonopeptide : tout enchaînement d'a-amino acides comprenant au moins un résidu d'acide a-amino sulfonique
- acyle : tout groupement ou fonction de type acyle dérivé d'un oxoacide en général, c'est-à-dire non seulement un acyle carboxylique ou thiocarboxylique stricto sensu, mais également les groupements ou fonctions de type acyle issus d'acides minéraux tels que les acides sulfoniques, phosphoriques, thiophosphoriques, phosphoniques, ou phosphiniques
- ester : les esters issus d'acyles tels que mentionnés ci-dessus
- amide : les amides issus d'acyles tels que mentionnés ci-dessus
- CBz ou Z : Benzyloxycarbonyle
- Boc : tertiobutyloxycarbonyle
- Tos : tosyle
- Me : méthyle
- Et : éthyle
- Ac : acétyle
L'invention concerne donc un procédé d'obtention de sels N protégés d'acide a-amino sulfinique de formule
Y - NH - CR'R - Si2 , M+ ou
Y est un acyle -notamment un groupement protecteur adapté pour pouvoir être substitue par un groupement peptidique, et/ou un groupement acyle peptidique ou pseudopeptidique (par exemple sulfonopeptidique) lui-même
N protégé-,
R peut être choisi parmi l'hydrogène, un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, un alkyle ou un aryle,
R' est l'hydrogène, un alkyle ou un aryle,
M+ est un cation, caractérisé en ce que
on choisit ou on prépare un composé intermédiaire répondant à la formule linéaire
Y - NH - CR'R - R1 où R1 est un groupe électro-attracteur,
ou aux analogues cycliques de cette formule, tels que

- sulfonopeptide : tout enchaînement d'a-amino acides comprenant au moins un résidu d'acide a-amino sulfonique
- acyle : tout groupement ou fonction de type acyle dérivé d'un oxoacide en général, c'est-à-dire non seulement un acyle carboxylique ou thiocarboxylique stricto sensu, mais également les groupements ou fonctions de type acyle issus d'acides minéraux tels que les acides sulfoniques, phosphoriques, thiophosphoriques, phosphoniques, ou phosphiniques
- ester : les esters issus d'acyles tels que mentionnés ci-dessus
- amide : les amides issus d'acyles tels que mentionnés ci-dessus
- CBz ou Z : Benzyloxycarbonyle
- Boc : tertiobutyloxycarbonyle
- Tos : tosyle
- Me : méthyle
- Et : éthyle
- Ac : acétyle
L'invention concerne donc un procédé d'obtention de sels N protégés d'acide a-amino sulfinique de formule
Y - NH - CR'R - Si2 , M+ ou
Y est un acyle -notamment un groupement protecteur adapté pour pouvoir être substitue par un groupement peptidique, et/ou un groupement acyle peptidique ou pseudopeptidique (par exemple sulfonopeptidique) lui-même
N protégé-,
R peut être choisi parmi l'hydrogène, un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, un alkyle ou un aryle,
R' est l'hydrogène, un alkyle ou un aryle,
M+ est un cation, caractérisé en ce que
on choisit ou on prépare un composé intermédiaire répondant à la formule linéaire
Y - NH - CR'R - R1 où R1 est un groupe électro-attracteur,
ou aux analogues cycliques de cette formule, tels que

(R1 étant confondu avec Y)
on fait réagir ce composé intermédiaire en milieu fortement basique avec un sel d'hydroxyacide sulfinique de formule
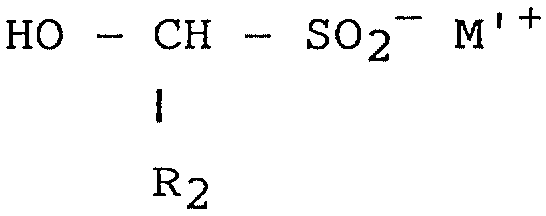
où R2 est l'hydrogène ou un groupe organique lié par un atome de carbone dans le sel d'hydroxyacide sulfinique, et M'+ est un cation, identique ou non à M+.
Avantageusement et selon l'invention, R1 est choisi pour favoriser la formation de l'imine correspondante Y - N = CR'R en milieu basique lors de la réaction dudit composé intermédiaire avec la solution basique.
Il suffit alors de choisir Y, R, et R' pour que cette imine réagisse uniquement sur la fonction sulfinate. En particulier, on a constaté qu'il est possible de choisir Y, R et R' pour que l'imine réagisse sélectivement sur la fonction sulfinate, même en milieu aqueux.
Avantageusement et selon l'invention, R1 est choisi parmi
- une fonction ester -O-acyle issue d'un acyle tel que défini ci-dessus, notamment une fonction ester carboxylique telle que

ou -O-SO3 , notamment lorsque R est -CF3,
- une fonction éther -O-alkyle, notamment lorsque R est l'hydrogène,
- une fonction amide -N-acyle issue d'un acyle tel que défini ci-dessus, c'est-à-dire amide carboxylique ou issue d'un oxoacide,
- un halogene.
- une fonction ester -O-acyle issue d'un acyle tel que défini ci-dessus, notamment une fonction ester carboxylique telle que
ou -O-SO3 , notamment lorsque R est -CF3,
- une fonction éther -O-alkyle, notamment lorsque R est l'hydrogène,
- une fonction amide -N-acyle issue d'un acyle tel que défini ci-dessus, c'est-à-dire amide carboxylique ou issue d'un oxoacide,
- un halogene.
Avantageusement et selon l'invention, R est un groupe carboné fluoré ou chloré, notamment -CF3.
Pour faire réagir le composé intermédiaire, plusieurs variantes sont possibles.
Dans une première variante selon l'invention, on ajoute ledit composé intermédiaire goutte à goutte dans une solution aqueuse de rongalite HO - CH2 S 2 Na.
Dans une autre variante selon l'invention,
R2 est un groupe organique choisi parmi un alkyle ; un aryle ; ou un groupe carboné halogéné, notamment un groupe carboné fluoré ou chloré, plus particulièrement un groupe carboné halogéné lié par un carbone au carbone du sel d'hydroxyacide sulfinique, avantageusement -CF3.
R2 est un groupe organique choisi parmi un alkyle ; un aryle ; ou un groupe carboné halogéné, notamment un groupe carboné fluoré ou chloré, plus particulièrement un groupe carboné halogéné lié par un carbone au carbone du sel d'hydroxyacide sulfinique, avantageusement -CF3.
Avantageusement et selon l'invention, on fait réagir ledit composé intermédiaire en milieu organique anhydre avec un sel d'hydroxyacide sulfinique de formule

où B est une base organique telle qu'une amine.
où B est une base organique telle qu'une amine.
Avantageusement et selon l'invention, B est une amine aliphatique, telle que NH(C6H11)2.
Selon l'invention, pour préparer ledit composé intermédiaire, on fait tout d'abord réagir l'aldéhyde ou la cétone

avec l'amide Y - NH2.
avec l'amide Y - NH2.
R' est de préférence l'hydrogène, l'aldéhyde étant en général plus réactif que la cétone.
A la suite de cette réaction, deux situations (1) et 2) ci-apres), peuvent se présenter, selon les produits stables obtenus, dont la nature dépend des groupements R, R' et Y considérés.
1) Lorsque la réaction de l'aldéhyde ou la cétone avec l'amide fournit une carbinolamine stable
Y - NH - CR'R - OH, on active dans une deuxième étape cette carbinolamine en la faisant réagir sur un composé choisi parmi un halogène, un acyle ou un alkyle, de façon à obtenir ledit composé intermédiaire où R1 est un halogène, une fonction ester ou une fonction éther. En particulier et selon l'invention, on active la carbinolamine en la faisant réagir sur l'anhydride trifluoroacétique
CF3 - CO - O - CO - CF3, de façon à obtenir ledit composé intermédiaire où R1 est l'ester

Y - NH - CR'R - OH, on active dans une deuxième étape cette carbinolamine en la faisant réagir sur un composé choisi parmi un halogène, un acyle ou un alkyle, de façon à obtenir ledit composé intermédiaire où R1 est un halogène, une fonction ester ou une fonction éther. En particulier et selon l'invention, on active la carbinolamine en la faisant réagir sur l'anhydride trifluoroacétique
CF3 - CO - O - CO - CF3, de façon à obtenir ledit composé intermédiaire où R1 est l'ester
En variante et selon l'invention, on active la carbinolamine en la faisant réagir sur un acyle sulfonique de façon à obtenir ledit composé intermédiaire où R1 est un ester sulfonique - O - SO3H ou l'anion correspondant - O - Su3 . En particulier, on utilise le complexe SO3 - Py où Py représente la Pyridine. Avec cette dernière variante, après réaction, on sépare le sel N protégé d'acide a-amino sulfinique par addition d'un sel soluble dans l'eau de dicyclohexylammonium (notamment un citrate), extraction sélective dans un solvant organique et évaporation. En effet, le sel de dicyclohexylammonium formé avec l'aminosulfinatè est extrait sélectivement dans un solvant organique d'extraction (tel que le chloroforme), alors que les autres sels restent dans la phase aqueuse.
Cette première situation et les variantes correspondantes du procédé selon l'invention sont en particulier applicables lorsque Y est un groupement protecteur, distinct d'un groupe sulfonyle, et choisi pour pouvoir être éliminé ultérieurement (et substitué par un groupement peptidique) sans destruction de la fonction sulfonamide pouvant être formée à partir du sel, notamment dans des conditions plus douces qu'un radical sulfonyle, et en particulier lorsque Y est choisi parmi le benzyloxycarbonyle ou le tertiobutyloxycarbonyle.
2) Lorsque la réaction de l'aldéhyde ou de la cétone avec l'amide fournit un diacyle aminal linéaire, tel que Y - NH - CR'R - NH - Y, ou cyclique, on utilise ce diacyle aminal directement comme composé intermédiaire (avec R1 étant une fonction -NH-acyle). Cette situation se rencontre notamment lorsque Y est le groupement tosyle.
Le procédé selon l'invention permet d'obtenir des sels stables synthétisés N protégés d'acide a-amino sulfinique, et en particulier les nouveaux sels stables synthétisés selon l'invention de formule
Y - NH - CR'R - Si2 , M+ ou
Y est un groupe acyle, distinct de R'' SO2, R'' étant le méthyle ou pCH3C6H4,
R est un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, ou un alkyle ou un aryle,
R' est l'hydrogène, un alkyle ou un aryle,
M+ est un cation.
Y - NH - CR'R - Si2 , M+ ou
Y est un groupe acyle, distinct de R'' SO2, R'' étant le méthyle ou pCH3C6H4,
R est un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, ou un alkyle ou un aryle,
R' est l'hydrogène, un alkyle ou un aryle,
M+ est un cation.
Avantageusement et selon l'invention, R est distinct de l'hydrogène, et est un groupe carboné fluoré ou chloré, notamment -CF3. En outre, avantageusement et selon l'invention, Y est un groupement protecteur distinct d'un groupement sulfonyle, choisi pour pouvoir être éliminé (et substitué ultérieurement par un groupement peptidique) sans destruction de la fonction sulfonamide pouvant être formée à partir du sel, en particulier dans des conditions plus douces qu ' un groupe sulfonyle. Y est notamment un groupement choisi parmi le benzyloxycarbonyle ou le tertiobutyloxycarbonyle.
Il est aussi à noter que Y est un groupement acyle qui peut lui-même incorporer un résidu d'a-aminoacide N protégé par une fonction acyle ou même une chaîne peptidique ou pseudo-peptidique (par exemple sulfonopeptidique) N protégée par une fonction acyle.
Ainsi, les sels selon l'invention sont alors déjà eux-mêmes de nature sulfonopeptique.
Les nouveaux sels selon l'invention sont stables et peuvent être isolés. Ils constituent des analogues N acylés des acides a aminés carboxyliques naturels et possèdent en eux-mêmes les propriétés, notamment thérapeutiques, correspondant à cette structure.
Les nouveaux sels selon l'invention permettent aussi d'obtenir pour la première fois des sulfonopeptides stables synthétisés. Pour ce faire, les sels stables selon l'invention sont soumis à une amination électrophile du type Y-NH-CR'R-SO2 ,M+ + H2N-O-S02-OH
Y-NH-CR'R-S02-NH2 + HSO4 ,M+
Y-NH-CR'R-S02-NH2 est utilisé ensuite dans les réactions de synthèse peptidique conventionnelles.
Y-NH-CR'R-S02-NH2 + HSO4 ,M+
Y-NH-CR'R-S02-NH2 est utilisé ensuite dans les réactions de synthèse peptidique conventionnelles.
L'invention permet ainsi d'obtenir les sulfonopeptides stables synthétisés incorporant au moins un résidu d'a-aminoacide sulfonique
- NH - CR'R - S02 où R est choisi parmi l'hydrogène, un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, un alkyle ou un aryle ; et R' est l'hydrogène, ou un alkyle ou un aryle. En particulier, R est un groupe carboné fluoré ou chloré, notamment -CF3. De même, R' est en particulier l'hydrogène de sorte que le sulfonopeptide est un analogue de peptide naturel.
- NH - CR'R - S02 où R est choisi parmi l'hydrogène, un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, un alkyle ou un aryle ; et R' est l'hydrogène, ou un alkyle ou un aryle. En particulier, R est un groupe carboné fluoré ou chloré, notamment -CF3. De même, R' est en particulier l'hydrogène de sorte que le sulfonopeptide est un analogue de peptide naturel.
Il est à noter que l'utilisation, selon l'invention, d'un groupe R carboné fluoré tel que -CF3 procure de nombreux avantages : il ne perturbe pas l'encombrement stérique du sel ou du sulfonopeptide ; il peut- être facilement suivi par résonance magnétique nucléaire et constitue donc un élément traceur ; il confère à la molécule une aptitude à traverser les membranes cellulaires et améliore son efficacité thérapeutique...
L'invention concerne aussi un procédé d'obtention des nouveaux sels et des sulfonopeptides comprenant en combinaison tout ou partie des caractéristiques mentionnées ci-dessus ou ci-après.
La description qui suit fournit des exemples de mise en oeuvre du procédé selon l'invention, et de nouveaux sels et de sulfonopeptides conformes à l'invention.
Dans les exemples, toutes les synthèses ont été réalisées sous argon et avec des solvants dégazés. Les produits ont été entièrement caractérisés par RMN (appareil
Bruker AC 80) de 1H, 13C, éventuellement 19F et par IR (appareil Perkin-elmer 257). Les analyses élémentaires ont été effectuées sur un appareil Carlo Erba 11-06. Les points de fusion ont été mesurés en tubes capillaires (appareil Büchi du Dr. Tottoli) et ne sont pas corrigés.
Bruker AC 80) de 1H, 13C, éventuellement 19F et par IR (appareil Perkin-elmer 257). Les analyses élémentaires ont été effectuées sur un appareil Carlo Erba 11-06. Les points de fusion ont été mesurés en tubes capillaires (appareil Büchi du Dr. Tottoli) et ne sont pas corrigés.
EXEMPLE 1 : Synthèse du sel Z-NH-CHCF3SO2-H2N+(C6H11)2
Dans ce sel, Y est Z, R est CF3, R' est H,
M+ est H2N+(C6H11)2
A une solution de 0,32 g (1,28 mmol) de carbinolamine Z-NH-CHCF30H (Chem. Ber. 1966, 99, 1933) dans 3 g de pyridine anhydre, on ajoute goutte à goutte, en 3 min, à 40 C, sous bonne agitation magnétique, 0,2 cm3 (1,41 mmol, 10 % d'excès) d'anhydride trifluoroacétique.
Dans ce sel, Y est Z, R est CF3, R' est H,
M+ est H2N+(C6H11)2
A une solution de 0,32 g (1,28 mmol) de carbinolamine Z-NH-CHCF30H (Chem. Ber. 1966, 99, 1933) dans 3 g de pyridine anhydre, on ajoute goutte à goutte, en 3 min, à 40 C, sous bonne agitation magnétique, 0,2 cm3 (1,41 mmol, 10 % d'excès) d'anhydride trifluoroacétique.
La solution obtenue du composé intermédiaire Z-NH-CHCF3-OCOCF3 obtenu, gardée à 40 C, est ajoutée sous argon, goutte à goutte, en 7 min, à une solution alcaline de 0,56 g (3,63 mmol) de rongalite dans 9 g d'eau, fortement agitée à 40 C. On ajoute alors 0,26 g (1,43 mmol) de dicyclohexylamine et on réalise quatre extractions avec environ 20 cmJ de chloroforme. La RMN de 19F montre que le produit est contaminé par environ 50 % de trifluoroacétate. Les extraits CHCl3 rassemblés sont ainsi lavés quatre fois avec environ 20 cm3 d'eau. Après séchage (Na2SO4) et concentration à sec, on obtient 0,49 g (rendement 79 %) de produit qui cristallise, dont le point de fusion est de 1200 C à 1230 C après recristallisation dans MeCN.
Analyse : C22H33F3N204St M = 478,564 % théoriques calculés : C : 55,21 ; H : 6,95 ; N : 5,86 % expérimentaux trouvés : C : 54,8 ; H : 6,9 ; N : 5,8.
EXEMPLE 2 : Synthèse du sel Boc-NH-CHCF3SO2 H2N+(C6H11)2
Dans ce sel, Y est Boc ; R est -CF3 ; R' est H ; M+ est H2N+(C6H11)2.
Dans ce sel, Y est Boc ; R est -CF3 ; R' est H ; M+ est H2N+(C6H11)2.
A une solution de 0,86 g (3,98 mmol) de carbinolamine BocNH-CHCF30H (Chem. Ber. 1967, 3838) dans 3 g de pyridine anhydre, on ajoute 0,95 g (6 mmol) de complexe S03 pyridine. Après trois jours d'agitation, la solution finalement obtenue du composé intermédiaire Boc NH-CHCF3-O-SO3 PyH+ est ajoutée goutte à goutte, en 2 min, à une solution bien agitée, à 40 C de 0,74 g (4,80 mmol 1,2 équivalent) de rongalite dans 12 g d'eau.
Après traitement comme dans l'exemple 1, mais avec seulement deux lavages à l'eau, dans laquelle le produit est partiellement soluble, on obtient par cristallisation du résidu dans 10 g d'alcool à 60 %, 1,12 g (rendement 63 %) de produit cristallisé, dont le point de fusion est de 1570 C à 1590 C après recristallisation dans 1' eau.
Analyse : C19H5F3N2O4S, M = 444,554 % théoriques calculés : C : 51,33 ; H : 7,94 ; N : 6,30 % expérimentaux trouvés : C : 51,1 ; H : 8 ; N : 6,2.
Lorsqu'on opère, non pas avec le complexe
S03 pyridine, mais avec de l'anhydride trifluoroacétique comme dans l'exemple 1, on n'obtient que 37 % de produit en raison des pertes lors des lavages aqueux nécessaires pour éliminer le trifluoroacétate.
S03 pyridine, mais avec de l'anhydride trifluoroacétique comme dans l'exemple 1, on n'obtient que 37 % de produit en raison des pertes lors des lavages aqueux nécessaires pour éliminer le trifluoroacétate.
EXEMPLE 3 : Synthèse du sel Tos-NH-CHCF3SO2 H2N+(C6H11)2
Dans ce sel, Y est Tos ; R est CF3 ; R' est
H ; M+ est H2N+(C6H11)2.
Dans ce sel, Y est Tos ; R est CF3 ; R' est
H ; M+ est H2N+(C6H11)2.
A 0,41 g (1,19 mmol, 1,5 éq.) de l'hydroxyacide sulfinique HOCHCF3SO2H2N+(C6H11)2 (R2 est -CF3 et M' est identique à M ; Tetrahedron 1993, 2469) et 0,29 g (0,75 mmol) du diacyle aminal (TosNH)2CHCF3 (Chem.
Ber. 1964, 97, 483), on ajoute 7 g du solvant CH2Cl2 et, à la solution obtenue, 0,45 g (4,5 mmol) de NEt3. Le spectre de RMN de 19F montre que la réaction est pratiquement achevée en quelques heures. On concentre à sec et on dissout le résidu avec léger chauffage dans 3 g d'EtOH absolu. Le produit cristallise après quelques jours à 150 C : 0,43 g (rendement 73 %) dont le point de fusion est de 1620 C à 1650 C (à comparer avec le rendement de 16 %, et avec le point de fusion de 1680 C à 1690 C décrits antérieurement dans la publication précitée).
Lorsqu'o utilise comme sulfinate la rongalite, en milieu aqueux, les spectres de RMN de 19F montrent la formation du produit puis sa conversion en quelques heures en hydrate de fluoral. Après extraction à l'AcOEt, on récupère un seul équivalent de TosNH2. Après addition d'un équivalent de citrate de DCHA, TosNHCH2SO2 H2N(C6H11)2 cristallise, dont le point de fusion est de 1410 C à 1450 C (à comparer avec le point de fusion décrit antérieurement de 1420 C à 1430 C) et est isolé par filtration avec un rendement de 62 %.
On note ainsi l'intérêt qu'il y a à utiliser soit l'hydroxysulfinate fluoré, soit l'hydroxysulfinate non substitué selon le sel final que l'on veut obtenir.
EXEMPLE 4 : Synthèse du monosulfonopeptide protégé Tos
NHCH2SO2NH2 par amination électrophile
A une solution refroidie à 40 C de 4,53 g (15,6 mmol) de TosNHCH2SO2Na monohydraté (obtenu à partir du sel de l'exemple 3) dans 10 g d'eau, on ajoute 1,54 g (17,2 mmol) de NH2OSO3H sous agitation, en 2 min. Après 5 min, le produit commence à précipiter. Après quelques heures, on filtre la suspension épaisse, on rince l'insoluble abondamment à l'eau et on le sèche en présence de P205. On isole 3,8 g (rendement 92 %) de produit dont le point de fusion est de 870 C.
NHCH2SO2NH2 par amination électrophile
A une solution refroidie à 40 C de 4,53 g (15,6 mmol) de TosNHCH2SO2Na monohydraté (obtenu à partir du sel de l'exemple 3) dans 10 g d'eau, on ajoute 1,54 g (17,2 mmol) de NH2OSO3H sous agitation, en 2 min. Après 5 min, le produit commence à précipiter. Après quelques heures, on filtre la suspension épaisse, on rince l'insoluble abondamment à l'eau et on le sèche en présence de P205. On isole 3,8 g (rendement 92 %) de produit dont le point de fusion est de 870 C.
Analyse : C8H12N2O4S2, M = 264,32 % théoriques calculés : C : 36,75 ; H : 4,58 ; N : 10,60
S : 24,26 % expérimentaux trouvés : C : 36,3, ; H : 4,5 ; N : 10,7
S : 24,4.
S : 24,26 % expérimentaux trouvés : C : 36,3, ; H : 4,5 ; N : 10,7
S : 24,4.
Le produit peut être recristallisé, sans chauffage, dans un mélange MeCN-CHCL3. Par contre, dans le mélange MeCN-H2O le produit est hydrolysé en moins d'une heure et après addition de dicyclohexylamine, on isole avec un rendement quasi quantitatif, WosNHCH2SO3 H2N(C6H11)2 dont le point de fusion est de 1800 C à 1810 C (le point de fusion décrit antérieurement étant de 1800 C à 1830 C). On constate ainsi que le monosulfonopeptide protégé est dans ce cas peu stable.
EXEMPLE 5 : Synthèse du monosulfonopeptide protégé Z-NH
CHCF3SO2NH2 par amination électrophile
En procédant comme pour le dérivé de l'exemple 4 mais avec le sel Z-NH-CHCF3SO2Na obtenu à partir du sel de l'exemple 1, on obtient le sulfonamide avec un rendement de 82 %, dont le point de fusion est de 1480 C.
CHCF3SO2NH2 par amination électrophile
En procédant comme pour le dérivé de l'exemple 4 mais avec le sel Z-NH-CHCF3SO2Na obtenu à partir du sel de l'exemple 1, on obtient le sulfonamide avec un rendement de 82 %, dont le point de fusion est de 1480 C.
Analyse : C10H11F3N204St M = 312,274 % théoriques calculés : C : 38,46 ; H : 3,55 ; N : 8,97 % expérimentaux trouvés : C : 38,0, ; H : 3,5 ; N : 8,8.
Le produit est stable en solution MeCN-H2O et est reconnu quantitativement après 24 h par addition d'un excès d'eau.
Le groupement protecteur Z est facile à éliminer, par exemple par hydrogénation catalytique, et l'on obtient alors le nouveau sulfonopeptide selon l'invention sous forme de sel
N+H3 - CH CF3 - SO2 - NH2, A
Cet analogue de l'alanine présente un intérêt thérapeutique majeur, notamment à titre d'antibiotique.
N+H3 - CH CF3 - SO2 - NH2, A
Cet analogue de l'alanine présente un intérêt thérapeutique majeur, notamment à titre d'antibiotique.
Ces exemples 4 et 5 démontrent que l'on peut réaliser une amination électrophile pour obtenir les monosulfonopeptides protégés issus des sels sulfinates obtenus selon l'invention.
En outre, on constate que les nouveaux monosulfonopeptides de l'invention sont stables, en particulier quand on choisit -CF3 pour le groupement R.
Claims (30)
1/ - Procédé d'obtention de sels N protégés d'acide a-amino sulfinique de formule
Y - NH - CR'R - Si2 , M+ où:
Y est un groupement acyle,
R peut être choisi parmi l'hydrogène, un groupe carboné halogéné lié directement par un atome de carbone au carbone a du sel, un alkyle ou un aryle,
R' est l'hydrogène, un alkyle ou un aryle,
M+ est un cation, caractérisé en ce que
on choisit ou on prépare un composé intermédiaire répondant à la formule linéaire
Y - NH - CR'R - R1 où Rî est un groupe électro-attracteur,
ou aux analogues cycliques de cette formule,
on fait réagir ce composé intermédiaire en milieu fortement basique avec un sel d'hydroxyacide sulfinique de formule

où R2 est l'hydrogène ou un groupe organique lié par un atome de carbone dans le sel d'hydroxyacide sulfinique, et M '+ est un cation.
2/ - Procédé selon la revendication 1, caractérisé en ce que R est un groupe carboné fluoré ou chloré.
3/ - Procédé selon l'une des revendications 1 et 2, caractérisé en ce que R est -CF3.
4/ - Procédé selon l'une des revendications 1 à 3, caractérisé en ce que R1 et choisi parmi une fonction ester -O-acyle, une fonction éther -O-alkyle, une fonction amide -N-acyle carboxylique ou issue d'oxoacide, ou un halogène.
5/ - Procédé selon l'une des revendications 1 à 3, caractérisé en ce que R1 est

6/ - Procédé selon l'une des revendications 1 à 3, caractérisé en ce que R1 est -O- Su3 .
7/ - Procédé selon l'une des revendications 1 à 6, caractérisé en ce qu'on ajoute ledit composé intermédiaire goutte à goutte dans une solution aqueuse de rongalite HO - CH2 - S02 Na.
8/ - Procédé selon l'une des revendications 1 à 6, caractérisé en ce que R2 est un groupe organique choisi parmi un alkyle, un aryle, ou un groupe carboné halogéné.
9/ - Procédé selon l'une des revendications 1 à 6, caractérisé en ce que R2 est un groupe carboné fluoré ou chloré.
10/ - Procédé selon l'une des revendications 1 à 6, caractérisé en ce que R2 est -CF3.
11/ - Procédé selon l'une des revendications 1 à 6, caractérisé en ce qu'on fait réagir ledit composé intermédiaire en milieu organique anhydre avec un sel d'hydroxyacide sulfinique de formule

où B est une base organique telle qu'une amine.
12/ - Procédé selon la revendication 11, caractérisé en ce que B est une amine aliphatique, telle que NH(C6H11)2.
13/ - Procédé selon l'une des revendications 1 à 12, caractérisé en ce qu'on prépare ledit composé intermédiaire en faisant réagir l'aldéhyde ou la cétone

avec l'amide Y - NH2.
14/ - Procédé selon la revendication 13, caractérisé en ce que lorsque la réaction de l'aldéhyde ou de la cétone avec l'amide fournit une carbinolamine stable
Y - NH - CR'R - OH, on active dans une deuxième étape cette carbinolamine en la faisant réagir sur un composé choisi parmi un halogène, un acyle ou un alkyle, de façon à obtenir ledit composé intermédiaire où R1 est un halogène, une fonction ester ou une fonction éther.
15/ - Procédé selon la revendication 14, caractérisé en ce que 1 on active la carbinolamine en la faisant réagir sur l'anhydride trifluoroacétique
CF3 - CO - O - CO - CF3, de façon à obtenir ledit composé intermédiaire où R1 est la fonction ester

16/ - Procédé selon la revendication 14, caractérisé en ce que 1 on active la carbinolamine en la faisant réagir sur un acyle sulfinique de façon à obtenir ledit composé intermédiaire où R1 est un ester sulfinique - O - SO3H ou l'anion correspondant - O - SO3
17/ - Procédé selon la revendication 16, caractérisé en ce qu'on utilise l'acyle sulfinique SO3 - Py où Py représente la Pyridine.
18/ - Procédé selon l'une des revendications 16 et 17, caractérisé en ce qu'après réaction, on sépare le sel N protégé d'acide a-amino sulfinique par addition d'un sel soluble dans l'eau de dicyclohexylammonium et évaporation.
19/ - Procédé selon l'une des revendications 1 à 18, caractérisé en ce que Y est un groupement protecteur distinct d'un groupe sulfonyle, choisi pour pouvoir être éliminé ultérieurement sans destruction de la fonction sulfonamide pouvant être formée à partir du sel.
20/ - Procédé selon l'une des revendications 1 à 19, caractérisé en ce que Y est choisi parmi le benzyloxycarbonyle ou le tertiobutyloxycarbonyle.
21/ - Procédé selon la revendication 13, caractérisé en ce que lorsque la réaction de l'aldéhyde ou de la cétone avec l'amide fournit un diacyle aminal, on utilise ce diacyle aminal directement comme composé intermédiaire.
22/ - Procédé selon la revendication 21, caractérisé en ce que Y est le groupement tosyle.
23/ - Nouveaux sels stables synthétisés N protégés d'acide a-amino sulfinique de formule
Y - NH - CR'R - Si2 , M+ ou
Y est un groupement acyle, distinct de R' ' SO2, R'' étant le méthyle ou pCH3C6H4,
R est un groupe carboné halogéné lié directement par un atome de carbone ou carbone a du sel, un alkyle ou un aryle,
R' est l'hydrogène, un alkyle, ou un aryle,
M+ est un cation.
24/ - Nouveaux sels selon la revendication 23, caractérisés en ce que R est un groupe carboné fluoré ou chloré.
25/ - Nouveaux sels selon la revendication 23, caractérisés en ce que R est -CF3.
26/ - Nouveaux sels selon l'une des revendications 23 à 25, caractérisés en ce que Y est un groupement, distinct d'un groupe sulfonyle, choisi pour pouvoir être éliminé ultérieurement sans destruction de la fonction sulfonamide pouvant être formée à partir du sel.
27/ - Nouveaux sels selon l'une des revendications 23 à 26, caractérisés en ce que Y est un radical choisi parmi le benzyloxycarbonyle ou le tertiobutyloxycarbonyle.
28/ - Sulfonopeptides stables synthétisés incorporant au moins un résidu d'a-aminoacide sulfonique
- NH - CR'R - SO2 où R est choisi parmi l'hydrogène, un groupe carboné halogéné lié directement par un atome de carbone au carbone
X du sel, un alkyle ou un aryle et R' est l'hydrogène, un alkyle ou un aryle.
29/ - Sulfonopeptides selon la revendication 28, caractérisés en ce que R est un groupe carboné fluoré ou chloré.
30/ - Sulfonopeptides selon la revendication 26, caractérisés en ce que R est -CF3.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9609845A FR2751973B1 (fr) | 1996-07-31 | 1996-07-31 | Nouveaux sels n proteges d'acide alfa-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent |
| PCT/FR1997/001185 WO1998004522A1 (fr) | 1996-07-31 | 1997-07-03 | NOUVEAUX SELS N PROTEGES D'ACIDE α-AMINO SULFINIQUE, LEUR PROCEDE D'OBTENTION, ET LES SULFONOPEPTIDES QUI EN DERIVENT |
| EP97931852A EP0925280A1 (fr) | 1996-07-31 | 1997-07-03 | Nouveaux sels n proteges d'acide alpha-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9609845A FR2751973B1 (fr) | 1996-07-31 | 1996-07-31 | Nouveaux sels n proteges d'acide alfa-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2751973A1 true FR2751973A1 (fr) | 1998-02-06 |
| FR2751973B1 FR2751973B1 (fr) | 1998-09-11 |
Family
ID=9494811
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9609845A Expired - Fee Related FR2751973B1 (fr) | 1996-07-31 | 1996-07-31 | Nouveaux sels n proteges d'acide alfa-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP0925280A1 (fr) |
| FR (1) | FR2751973B1 (fr) |
| WO (1) | WO1998004522A1 (fr) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387671A (en) * | 1990-12-27 | 1995-02-07 | Abbott Laboratories | Hexa- and heptapeptide anaphylatoxin-receptor ligands |
-
1996
- 1996-07-31 FR FR9609845A patent/FR2751973B1/fr not_active Expired - Fee Related
-
1997
- 1997-07-03 EP EP97931852A patent/EP0925280A1/fr not_active Withdrawn
- 1997-07-03 WO PCT/FR1997/001185 patent/WO1998004522A1/fr not_active Application Discontinuation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387671A (en) * | 1990-12-27 | 1995-02-07 | Abbott Laboratories | Hexa- and heptapeptide anaphylatoxin-receptor ligands |
Non-Patent Citations (10)
| Title |
|---|
| E. FROMM: "Über die niedersten Oxyde des Schwefelwasserstoffs", BERICHTE DER DEUTSCHEN CHEMISCHEN GESELLSCHAFT, vol. 41, 1908, WEINHEIM, DE, pages 3397 - 3425, XP002029796 * |
| F. LEURQUIN, ET AL.: "Etude du couplage de la rongalite avec divers composés aminés", PHOSPHORUS, SULFUR, AND SILICON, vol. 92, no. 1-4, 1994, NEW YORK, US, pages 201 - 211, XP000671063 * |
| G.W. HUFFMANN, ET AL.: "Substrate specificity of isopennicillin N synthase", JOURNAL OF MEDICINAL CHEMISTRY, vol. 35, no. 10, 15 May 1992 (1992-05-15), WASHINGTON, DC, US, pages 1897 - 1914, XP002029802 * |
| J.J. STEZOWSKI, ET AL.: "A crystal structure determination for Tyr-D-Nle-Gly-Phe-NleS [NleS = MeCH2CH2CH2CH(NH2)SO3H]: an active synthetic encephalin analogue", JOURNAL OF THE CHEMICAL SOCIETY, CHEMICAL COMMUNICATIONS, no. 11, 1985, LETCHWORTH, GB, pages 681 - 682, XP002029803 * |
| M. FRANKEL, ET AL.: "Synthesis of amino alkyl sulphonic acids and their peptide analogues", TETRAHEDRON, vol. 9, 1960, OXFORD, GB, pages 289 - 294, XP002029800 * |
| M. MULLIEZ, ET AL.: "Synthèse d'alpha-hydroxysulfintes", TETRAHEDRON, vol. 49, no. 12, 19 March 1993 (1993-03-19), OXFORD, GB, pages 2469 - 2476, XP002029797 * |
| M. MULLIEZ, ET AL.: "Synthèse de sels d'alpha-aminoacides sufiniques substitués à l'azote par un groupe méthane- ou p-toluène-sulfonyle", TETRAHEDRON, vol. 50, no. 18, 2 May 1994 (1994-05-02), OXFORD, GB, pages 5401 - 5412, XP002029798 * |
| S. PAIK, ET AL.: "alpha-Aminosulphonopeptides as possible functional analogues of pennicillin: evidence for their extreme instability", TETRAHEDRON, vol. 52, no. 15, 8 April 1996 (1996-04-08), OXFORD, GB, pages 5303 - 5318, XP000671264 * |
| T. SHIBA, ET AL.: "Synthesis of alanyl-1-aminoethanesulphonic acid", BULLETIN OF THE CHEMICAL SOCIETY OF JAPAN, vol. 50, no. 1, January 1977 (1977-01-01), TOKYO, JP, pages 254 - 257, XP002029801 * |
| W.F. GILMORE, ET AL.: "Synthesis of alpha-amino sulphonamides", JOURNAL OF ORGANIC CHEMISTRY, vol. 43, no. 23, 10 November 1978 (1978-11-10), WASHINGTON, DC, US, pages 4535 - 4537, XP002029799 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO1998004522A1 (fr) | 1998-02-05 |
| EP0925280A1 (fr) | 1999-06-30 |
| FR2751973B1 (fr) | 1998-09-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2486020B1 (fr) | Procede de synthese de l'ergothioneine et analogues | |
| CA1213896A (fr) | Phosphines chirales sulfonees, leur preparation et leur emploi en catalyse asymetrique | |
| JPS581105B2 (ja) | 光学活性アミノ酸−マンデル酸複合体及びその製造法 | |
| CH636344A5 (fr) | Composes analogues de dipeptides. | |
| EP0297641B1 (fr) | Composés guanidiniques comprenant un ion tétraphénylborate substitué, procédé d'obtention de ces composés et utilisation des composés lors de la synthése peptidique | |
| BE1012819A6 (fr) | Procede de preparation du maleate de fluvoxamine. | |
| FR2751973A1 (fr) | Nouveaux sels n proteges d'acide alfa-amino sulfinique, leur procede d'obtention, et les sulfonopeptides qui en derivent | |
| EP2084125B1 (fr) | Derives d'aminobenzocycloheptene, leurs procedes de preparation et leur utilisation en therapeutique | |
| BE1005720A3 (fr) | Procede de synthese peptidique et nouveaux intermediaires de synthese. | |
| JPS6013775A (ja) | 光学活性3−(p−アルコキシフエニル)グリシツド酸アルカリ金属塩の製造法 | |
| JP2002515004A (ja) | アミノ酸誘導ジアミノプロパノール | |
| CA2524489A1 (fr) | Procede de synthese de la 4-hydroxyisoleucine et de ses derives | |
| FR2950890A1 (fr) | Procede de synthese de la 2-thiohistidine et analogues | |
| EP0659175B1 (fr) | Derives d'(amino-3 phenyl)-1 ethanesulfonate d'alkylammonium optiquement actifs, leur preparation et leur utilisation | |
| FR2760452A1 (fr) | Procede d'obtention d'enantiomeres d'acides alpha-amines et sels diastereoisomeriques intermediaires | |
| FR2676059A1 (fr) | Nouveaux derives de peptides utilisables comme inhibiteurs de collagenases bacteriennes. | |
| RU2741389C1 (ru) | Способ получения промежуточного соединения для синтеза лекарственного средства | |
| JP3888402B2 (ja) | 光学活性N−カルボベンゾキシ−tert−ロイシンの製造法 | |
| JPH021446A (ja) | N‐ヒドロキシ‐α‐アミノ酸およびその誘導体の製造法、ならびにこうして得られた化合物 | |
| CA3119871A1 (fr) | Nouvelle voie de synthese de diazirines, enrichies ou non en azote-15 | |
| FR2504525A1 (fr) | Procede de preparation d'oligopeptides lysine-tryptophane, de leurs derives et de leurs sels physiologiquement utilisables | |
| EP1440977B1 (fr) | Procédé de synthèse amélioré de dérivés diamides du tripeptide KPV | |
| FR2654430A1 (fr) | Nouveaux derives de peptides, utilisables comme inhibiteurs des collagenases bacteriennes. | |
| FR2571055A1 (fr) | Procede de preparation de la 5-(2-chlorobenzyl)-4,5,6,7-tetrahydrothieno(3,2-c)pyridine | |
| WO1990010009A1 (fr) | Sels de phosphonium comme reactifs de couplage peptidique |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |
Effective date: 20130329 |