ES2946785T3 - Linfocitos T de memoria central anti-terceros modificados genéticamente y uso de los mismos en inmunoterapia - Google Patents
Linfocitos T de memoria central anti-terceros modificados genéticamente y uso de los mismos en inmunoterapia Download PDFInfo
- Publication number
- ES2946785T3 ES2946785T3 ES16750269T ES16750269T ES2946785T3 ES 2946785 T3 ES2946785 T3 ES 2946785T3 ES 16750269 T ES16750269 T ES 16750269T ES 16750269 T ES16750269 T ES 16750269T ES 2946785 T3 ES2946785 T3 ES 2946785T3
- Authority
- ES
- Spain
- Prior art keywords
- cell
- cells
- antigen
- tmc
- party
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 210000003071 memory t lymphocyte Anatomy 0.000 title claims abstract description 28
- 238000009169 immunotherapy Methods 0.000 title description 12
- 210000004027 cell Anatomy 0.000 claims abstract description 423
- 238000000034 method Methods 0.000 claims abstract description 114
- 108091008874 T cell receptors Proteins 0.000 claims abstract description 79
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims abstract description 79
- 230000011664 signaling Effects 0.000 claims abstract description 37
- 238000002054 transplantation Methods 0.000 claims abstract description 36
- 230000001939 inductive effect Effects 0.000 claims abstract description 30
- 210000001165 lymph node Anatomy 0.000 claims abstract description 23
- 102000000844 Cell Surface Receptors Human genes 0.000 claims abstract description 20
- 108010001857 Cell Surface Receptors Proteins 0.000 claims abstract description 20
- 239000000427 antigen Substances 0.000 claims description 251
- 108091007433 antigens Proteins 0.000 claims description 250
- 102000036639 antigens Human genes 0.000 claims description 250
- 206010028980 Neoplasm Diseases 0.000 claims description 74
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims description 64
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 claims description 43
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 36
- 230000000139 costimulatory effect Effects 0.000 claims description 34
- 201000010099 disease Diseases 0.000 claims description 33
- 230000014509 gene expression Effects 0.000 claims description 29
- 239000013598 vector Substances 0.000 claims description 29
- 230000003612 virological effect Effects 0.000 claims description 28
- 230000027455 binding Effects 0.000 claims description 27
- 108091033319 polynucleotide Proteins 0.000 claims description 27
- 102000040430 polynucleotide Human genes 0.000 claims description 27
- 239000002157 polynucleotide Substances 0.000 claims description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims description 23
- 239000012634 fragment Substances 0.000 claims description 21
- 102000003812 Interleukin-15 Human genes 0.000 claims description 20
- 102100021592 Interleukin-7 Human genes 0.000 claims description 20
- 108010002586 Interleukin-7 Proteins 0.000 claims description 20
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 claims description 20
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 claims description 19
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 claims description 18
- 238000000338 in vitro Methods 0.000 claims description 18
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 claims description 16
- 208000024908 graft versus host disease Diseases 0.000 claims description 15
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 claims description 14
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 claims description 14
- 230000001580 bacterial effect Effects 0.000 claims description 14
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 claims description 12
- 230000035755 proliferation Effects 0.000 claims description 12
- 230000009261 transgenic effect Effects 0.000 claims description 11
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 10
- 238000012258 culturing Methods 0.000 claims description 10
- 108091008036 Immune checkpoint proteins Proteins 0.000 claims description 8
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 claims description 7
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 claims description 7
- 230000003211 malignant effect Effects 0.000 claims description 7
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 claims description 6
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 claims description 6
- 101001018097 Homo sapiens L-selectin Proteins 0.000 claims description 6
- 101000679851 Homo sapiens Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 claims description 6
- 102100033467 L-selectin Human genes 0.000 claims description 6
- 230000002538 fungal effect Effects 0.000 claims description 6
- 208000035143 Bacterial infection Diseases 0.000 claims description 5
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 claims description 5
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 claims description 5
- 239000006285 cell suspension Substances 0.000 claims description 5
- 230000000779 depleting effect Effects 0.000 claims description 5
- 230000001575 pathological effect Effects 0.000 claims description 5
- 208000022362 bacterial infectious disease Diseases 0.000 claims description 4
- 101000738335 Homo sapiens T-cell surface glycoprotein CD3 zeta chain Proteins 0.000 claims description 3
- 102100037906 T-cell surface glycoprotein CD3 zeta chain Human genes 0.000 claims description 3
- 244000045947 parasite Species 0.000 claims description 3
- 230000002463 transducing effect Effects 0.000 claims description 3
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 claims 1
- 238000013519 translation Methods 0.000 abstract description 4
- 210000001744 T-lymphocyte Anatomy 0.000 description 92
- 241000699670 Mus sp. Species 0.000 description 59
- 210000004698 lymphocyte Anatomy 0.000 description 58
- 108090000765 processed proteins & peptides Proteins 0.000 description 50
- 108090000623 proteins and genes Proteins 0.000 description 50
- 102000004196 processed proteins & peptides Human genes 0.000 description 34
- 239000000203 mixture Substances 0.000 description 32
- 210000004443 dendritic cell Anatomy 0.000 description 30
- 150000002632 lipids Chemical class 0.000 description 29
- 230000000735 allogeneic effect Effects 0.000 description 27
- 102000004169 proteins and genes Human genes 0.000 description 27
- 235000018102 proteins Nutrition 0.000 description 26
- 230000003750 conditioning effect Effects 0.000 description 25
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 24
- -1 CD137 Proteins 0.000 description 23
- 230000001086 cytosolic effect Effects 0.000 description 23
- 239000003446 ligand Substances 0.000 description 23
- 230000000694 effects Effects 0.000 description 22
- 150000007523 nucleic acids Chemical class 0.000 description 22
- 241000700605 Viruses Species 0.000 description 21
- 239000003814 drug Substances 0.000 description 21
- 229920001184 polypeptide Polymers 0.000 description 21
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 20
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 20
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 20
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 20
- 230000001363 autoimmune Effects 0.000 description 19
- 230000037396 body weight Effects 0.000 description 19
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 18
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 18
- 208000023275 Autoimmune disease Diseases 0.000 description 17
- 238000003556 assay Methods 0.000 description 17
- 201000011510 cancer Diseases 0.000 description 17
- 239000003795 chemical substances by application Substances 0.000 description 17
- 102000039446 nucleic acids Human genes 0.000 description 17
- 108020004707 nucleic acids Proteins 0.000 description 17
- 230000007170 pathology Effects 0.000 description 16
- 241000712461 unidentified influenza virus Species 0.000 description 16
- 241000699666 Mus <mouse, genus> Species 0.000 description 15
- 239000004480 active ingredient Substances 0.000 description 15
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 15
- 238000011282 treatment Methods 0.000 description 15
- 238000011740 C57BL/6 mouse Methods 0.000 description 14
- 230000004913 activation Effects 0.000 description 14
- 230000003834 intracellular effect Effects 0.000 description 14
- 239000002502 liposome Substances 0.000 description 14
- 210000005259 peripheral blood Anatomy 0.000 description 14
- 239000011886 peripheral blood Substances 0.000 description 14
- 238000013459 approach Methods 0.000 description 13
- 210000004369 blood Anatomy 0.000 description 13
- 239000008280 blood Substances 0.000 description 13
- 229940079593 drug Drugs 0.000 description 13
- 230000006870 function Effects 0.000 description 13
- 238000002347 injection Methods 0.000 description 13
- 239000007924 injection Substances 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 102000005962 receptors Human genes 0.000 description 13
- 108020003175 receptors Proteins 0.000 description 13
- 241000701022 Cytomegalovirus Species 0.000 description 12
- 208000026935 allergic disease Diseases 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 208000009329 Graft vs Host Disease Diseases 0.000 description 11
- 206010020751 Hypersensitivity Diseases 0.000 description 11
- 241001465754 Metazoa Species 0.000 description 11
- 230000007815 allergy Effects 0.000 description 11
- 150000001413 amino acids Chemical group 0.000 description 11
- 210000000612 antigen-presenting cell Anatomy 0.000 description 11
- 239000012636 effector Substances 0.000 description 11
- 108020004414 DNA Proteins 0.000 description 10
- 238000004113 cell culture Methods 0.000 description 10
- 208000015181 infectious disease Diseases 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 230000004083 survival effect Effects 0.000 description 10
- 238000012546 transfer Methods 0.000 description 10
- 239000003981 vehicle Substances 0.000 description 10
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 9
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 9
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 9
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 9
- 241000725303 Human immunodeficiency virus Species 0.000 description 9
- 102100030703 Interleukin-22 Human genes 0.000 description 9
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 9
- 230000001464 adherent effect Effects 0.000 description 9
- 210000003719 b-lymphocyte Anatomy 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- 230000004936 stimulating effect Effects 0.000 description 9
- 210000001519 tissue Anatomy 0.000 description 9
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 8
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 8
- 206010009944 Colon cancer Diseases 0.000 description 8
- 208000035473 Communicable disease Diseases 0.000 description 8
- 241000282412 Homo Species 0.000 description 8
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 8
- 108010002350 Interleukin-2 Proteins 0.000 description 8
- 102000000588 Interleukin-2 Human genes 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 238000010322 bone marrow transplantation Methods 0.000 description 8
- 230000001413 cellular effect Effects 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 238000002955 isolation Methods 0.000 description 8
- 231100000518 lethal Toxicity 0.000 description 8
- 230000001665 lethal effect Effects 0.000 description 8
- 239000002609 medium Substances 0.000 description 8
- 230000001400 myeloablative effect Effects 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 102000004127 Cytokines Human genes 0.000 description 7
- 108090000695 Cytokines Proteins 0.000 description 7
- 241000711549 Hepacivirus C Species 0.000 description 7
- 206010025323 Lymphomas Diseases 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 108091028043 Nucleic acid sequence Proteins 0.000 description 7
- 108700008625 Reporter Genes Proteins 0.000 description 7
- 230000001154 acute effect Effects 0.000 description 7
- 238000011467 adoptive cell therapy Methods 0.000 description 7
- 235000001014 amino acid Nutrition 0.000 description 7
- 238000001802 infusion Methods 0.000 description 7
- 230000004068 intracellular signaling Effects 0.000 description 7
- 208000032839 leukemia Diseases 0.000 description 7
- 238000010369 molecular cloning Methods 0.000 description 7
- 201000006417 multiple sclerosis Diseases 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 230000009257 reactivity Effects 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 241000894007 species Species 0.000 description 7
- 238000013518 transcription Methods 0.000 description 7
- 239000013603 viral vector Substances 0.000 description 7
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 6
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- 238000002965 ELISA Methods 0.000 description 6
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 6
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 6
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 6
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 6
- 208000036142 Viral infection Diseases 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 210000001185 bone marrow Anatomy 0.000 description 6
- 229940127089 cytotoxic agent Drugs 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 230000028993 immune response Effects 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 238000002826 magnetic-activated cell sorting Methods 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 230000035897 transcription Effects 0.000 description 6
- 241001430294 unidentified retrovirus Species 0.000 description 6
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 5
- 102100027207 CD27 antigen Human genes 0.000 description 5
- 208000015943 Coeliac disease Diseases 0.000 description 5
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 5
- 241000588724 Escherichia coli Species 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 5
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 5
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 5
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 5
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 5
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 5
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 5
- 108090000978 Interleukin-4 Proteins 0.000 description 5
- 102000057297 Pepsin A Human genes 0.000 description 5
- 108090000284 Pepsin A Proteins 0.000 description 5
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 5
- 102100038358 Prostate-specific antigen Human genes 0.000 description 5
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 5
- 229940024606 amino acid Drugs 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 229960004397 cyclophosphamide Drugs 0.000 description 5
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 5
- 239000008298 dragée Substances 0.000 description 5
- 239000013604 expression vector Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 239000003018 immunosuppressive agent Substances 0.000 description 5
- 230000001965 increasing effect Effects 0.000 description 5
- 201000001441 melanoma Diseases 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 206010039073 rheumatoid arthritis Diseases 0.000 description 5
- 230000019491 signal transduction Effects 0.000 description 5
- 210000000952 spleen Anatomy 0.000 description 5
- 210000004988 splenocyte Anatomy 0.000 description 5
- 230000002269 spontaneous effect Effects 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- 241000701161 unidentified adenovirus Species 0.000 description 5
- 102100034540 Adenomatous polyposis coli protein Human genes 0.000 description 4
- 102000006306 Antigen Receptors Human genes 0.000 description 4
- 108010083359 Antigen Receptors Proteins 0.000 description 4
- 206010006187 Breast cancer Diseases 0.000 description 4
- 101150013553 CD40 gene Proteins 0.000 description 4
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 4
- 229940045513 CTLA4 antagonist Drugs 0.000 description 4
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 4
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 4
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 4
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 4
- 241000700721 Hepatitis B virus Species 0.000 description 4
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 4
- 101000924577 Homo sapiens Adenomatous polyposis coli protein Proteins 0.000 description 4
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 4
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 4
- 241000701806 Human papillomavirus Species 0.000 description 4
- 241000713666 Lentivirus Species 0.000 description 4
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 4
- 108010037274 Member 9 Tumor Necrosis Factor Receptor Superfamily Proteins 0.000 description 4
- 108010052285 Membrane Proteins Proteins 0.000 description 4
- 206010029260 Neuroblastoma Diseases 0.000 description 4
- 208000030852 Parasitic disease Diseases 0.000 description 4
- 108020004511 Recombinant DNA Proteins 0.000 description 4
- 206010039491 Sarcoma Diseases 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 208000006011 Stroke Diseases 0.000 description 4
- 206010052779 Transplant rejections Diseases 0.000 description 4
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 4
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 4
- 102000013529 alpha-Fetoproteins Human genes 0.000 description 4
- 230000000890 antigenic effect Effects 0.000 description 4
- 230000005784 autoimmunity Effects 0.000 description 4
- 210000000988 bone and bone Anatomy 0.000 description 4
- 229960002092 busulfan Drugs 0.000 description 4
- 239000011651 chromium Substances 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 4
- 238000012217 deletion Methods 0.000 description 4
- 230000037430 deletion Effects 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 238000004520 electroporation Methods 0.000 description 4
- 230000002255 enzymatic effect Effects 0.000 description 4
- 230000001506 immunosuppresive effect Effects 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 206010025135 lupus erythematosus Diseases 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 239000000693 micelle Substances 0.000 description 4
- 210000001616 monocyte Anatomy 0.000 description 4
- 201000000050 myeloid neoplasm Diseases 0.000 description 4
- 229940111202 pepsin Drugs 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 229920003023 plastic Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 208000011580 syndromic disease Diseases 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 4
- 230000009385 viral infection Effects 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 3
- 208000031212 Autoimmune polyendocrinopathy Diseases 0.000 description 3
- 241000193738 Bacillus anthracis Species 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 208000023328 Basedow disease Diseases 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- 102100038078 CD276 antigen Human genes 0.000 description 3
- 102100035793 CD83 antigen Human genes 0.000 description 3
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 3
- 206010008027 Cerebellar atrophy Diseases 0.000 description 3
- 108020004705 Codon Proteins 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 208000011231 Crohn disease Diseases 0.000 description 3
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 3
- 108010036949 Cyclosporine Proteins 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 241000702421 Dependoparvovirus Species 0.000 description 3
- 101150029707 ERBB2 gene Proteins 0.000 description 3
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 3
- 241000282324 Felis Species 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108060003393 Granulin Proteins 0.000 description 3
- 208000015023 Graves' disease Diseases 0.000 description 3
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 3
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 3
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 3
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 3
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 3
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 3
- 239000000232 Lipid Bilayer Substances 0.000 description 3
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 3
- 102100022430 Melanocyte protein PMEL Human genes 0.000 description 3
- 102000018697 Membrane Proteins Human genes 0.000 description 3
- 102000003735 Mesothelin Human genes 0.000 description 3
- 108090000015 Mesothelin Proteins 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 3
- 206010028665 Myxoedema Diseases 0.000 description 3
- 208000012902 Nervous system disease Diseases 0.000 description 3
- 208000025966 Neurological disease Diseases 0.000 description 3
- 206010033128 Ovarian cancer Diseases 0.000 description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 201000011152 Pemphigus Diseases 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 description 3
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 3
- 206010038389 Renal cancer Diseases 0.000 description 3
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 3
- 101800001271 Surface protein Proteins 0.000 description 3
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 description 3
- 241000223109 Trypanosoma cruzi Species 0.000 description 3
- 206010047115 Vasculitis Diseases 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 208000010668 atopic eczema Diseases 0.000 description 3
- 230000000599 auto-anti-genic effect Effects 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 239000001506 calcium phosphate Substances 0.000 description 3
- 229910000389 calcium phosphate Inorganic materials 0.000 description 3
- 235000011010 calcium phosphates Nutrition 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 238000002659 cell therapy Methods 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- 108700010039 chimeric receptor Proteins 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 208000029742 colonic neoplasm Diseases 0.000 description 3
- 230000001143 conditioned effect Effects 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 230000003828 downregulation Effects 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 229960000390 fludarabine Drugs 0.000 description 3
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 3
- 108020001507 fusion proteins Proteins 0.000 description 3
- 102000037865 fusion proteins Human genes 0.000 description 3
- 238000001476 gene delivery Methods 0.000 description 3
- 102000054766 genetic haplotypes Human genes 0.000 description 3
- 239000003102 growth factor Substances 0.000 description 3
- 201000005787 hematologic cancer Diseases 0.000 description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 3
- 229940125721 immunosuppressive agent Drugs 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 201000010982 kidney cancer Diseases 0.000 description 3
- 238000002372 labelling Methods 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 238000001638 lipofection Methods 0.000 description 3
- 229920006008 lipopolysaccharide Polymers 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 208000014018 liver neoplasm Diseases 0.000 description 3
- 230000000527 lymphocytic effect Effects 0.000 description 3
- 230000036210 malignancy Effects 0.000 description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 3
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 3
- 229960001924 melphalan Drugs 0.000 description 3
- 230000009401 metastasis Effects 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 231100000052 myelotoxic Toxicity 0.000 description 3
- 230000002556 myelotoxic effect Effects 0.000 description 3
- 208000003786 myxedema Diseases 0.000 description 3
- 210000000822 natural killer cell Anatomy 0.000 description 3
- 201000001119 neuropathy Diseases 0.000 description 3
- 230000007823 neuropathy Effects 0.000 description 3
- 230000002611 ovarian Effects 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 239000005017 polysaccharide Substances 0.000 description 3
- 150000004804 polysaccharides Chemical class 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000001177 retroviral effect Effects 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 210000000130 stem cell Anatomy 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 3
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 238000011830 transgenic mouse model Methods 0.000 description 3
- 230000032258 transport Effects 0.000 description 3
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 3
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 3
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- PXFBZOLANLWPMH-UHFFFAOYSA-N 16-Epiaffinine Natural products C1C(C2=CC=CC=C2N2)=C2C(=O)CC2C(=CC)CN(C)C1C2CO PXFBZOLANLWPMH-UHFFFAOYSA-N 0.000 description 2
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 2
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 2
- 102100038222 60 kDa heat shock protein, mitochondrial Human genes 0.000 description 2
- 102100023990 60S ribosomal protein L17 Human genes 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- 102100036601 Aggrecan core protein Human genes 0.000 description 2
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 2
- 102100035526 B melanoma antigen 1 Human genes 0.000 description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 2
- 208000003950 B-cell lymphoma Diseases 0.000 description 2
- 108010077805 Bacterial Proteins Proteins 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- 102100032912 CD44 antigen Human genes 0.000 description 2
- 102100025221 CD70 antigen Human genes 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 241000282465 Canis Species 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 108010058432 Chaperonin 60 Proteins 0.000 description 2
- 101710098119 Chaperonin GroEL 2 Proteins 0.000 description 2
- 206010009900 Colitis ulcerative Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 201000004624 Dermatitis Diseases 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 102100039554 Galectin-8 Human genes 0.000 description 2
- 108010061711 Gliadin Proteins 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 2
- 102000008214 Glutamate decarboxylase Human genes 0.000 description 2
- 108091022930 Glutamate decarboxylase Proteins 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 2
- 241000606768 Haemophilus influenzae Species 0.000 description 2
- 101710154606 Hemagglutinin Proteins 0.000 description 2
- 208000005176 Hepatitis C Diseases 0.000 description 2
- 208000009889 Herpes Simplex Diseases 0.000 description 2
- 241000228402 Histoplasma Species 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 101000874316 Homo sapiens B melanoma antigen 1 Proteins 0.000 description 2
- 101100165850 Homo sapiens CA9 gene Proteins 0.000 description 2
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 2
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 2
- 101000608769 Homo sapiens Galectin-8 Proteins 0.000 description 2
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 2
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 2
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 2
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 2
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 2
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 2
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 102100037850 Interferon gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical group CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- 241000222722 Leishmania <genus> Species 0.000 description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 2
- 108091054437 MHC class I family Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 102000000380 Matrix Metalloproteinase 1 Human genes 0.000 description 2
- 108010016113 Matrix Metalloproteinase 1 Proteins 0.000 description 2
- 102100030200 Matrix metalloproteinase-16 Human genes 0.000 description 2
- 201000005505 Measles Diseases 0.000 description 2
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 description 2
- 102000003792 Metallothionein Human genes 0.000 description 2
- 108090000157 Metallothionein Proteins 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 208000029578 Muscle disease Diseases 0.000 description 2
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 2
- 208000031888 Mycoses Diseases 0.000 description 2
- 108010000123 Myelin-Oligodendrocyte Glycoprotein Proteins 0.000 description 2
- 102100023302 Myelin-oligodendrocyte glycoprotein Human genes 0.000 description 2
- 201000002481 Myositis Diseases 0.000 description 2
- 101710163270 Nuclease Proteins 0.000 description 2
- 102000011931 Nucleoproteins Human genes 0.000 description 2
- 108010061100 Nucleoproteins Proteins 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- 108010038807 Oligopeptides Proteins 0.000 description 2
- 102000015636 Oligopeptides Human genes 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 2
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 2
- 108010058846 Ovalbumin Proteins 0.000 description 2
- 108090000526 Papain Proteins 0.000 description 2
- 108010081690 Pertussis Toxin Proteins 0.000 description 2
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- 101710176177 Protein A56 Proteins 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 206010037742 Rabies Diseases 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 208000025747 Rheumatic disease Diseases 0.000 description 2
- 241000714474 Rous sarcoma virus Species 0.000 description 2
- 108020004459 Small interfering RNA Proteins 0.000 description 2
- 206010072148 Stiff-Person syndrome Diseases 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- 230000005867 T cell response Effects 0.000 description 2
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 2
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 2
- 206010043376 Tetanus Diseases 0.000 description 2
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 2
- 208000002474 Tinea Diseases 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 241000223996 Toxoplasma Species 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 241000893966 Trichophyton verrucosum Species 0.000 description 2
- 102100040247 Tumor necrosis factor Human genes 0.000 description 2
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 description 2
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 2
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 2
- 102000003425 Tyrosinase Human genes 0.000 description 2
- 108060008724 Tyrosinase Proteins 0.000 description 2
- 201000006704 Ulcerative Colitis Diseases 0.000 description 2
- 208000024780 Urticaria Diseases 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Chemical group CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 108010067390 Viral Proteins Proteins 0.000 description 2
- 238000010317 ablation therapy Methods 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 230000000172 allergic effect Effects 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000000469 anti-sperm effect Effects 0.000 description 2
- 229940034982 antineoplastic agent Drugs 0.000 description 2
- 239000003443 antiviral agent Substances 0.000 description 2
- 238000002617 apheresis Methods 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 230000006472 autoimmune response Effects 0.000 description 2
- 229960002170 azathioprine Drugs 0.000 description 2
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000002458 cell surface marker Substances 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 208000018631 connective tissue disease Diseases 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 229930182912 cyclosporin Natural products 0.000 description 2
- 210000005220 cytoplasmic tail Anatomy 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- RNPXCFINMKSQPQ-UHFFFAOYSA-N dicetyl hydrogen phosphate Chemical compound CCCCCCCCCCCCCCCCOP(O)(=O)OCCCCCCCCCCCCCCCC RNPXCFINMKSQPQ-UHFFFAOYSA-N 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 206010013023 diphtheria Diseases 0.000 description 2
- 239000000428 dust Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 206010014599 encephalitis Diseases 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 210000003238 esophagus Anatomy 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 201000003444 follicular lymphoma Diseases 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000012239 gene modification Methods 0.000 description 2
- 238000001415 gene therapy Methods 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 230000005017 genetic modification Effects 0.000 description 2
- 235000013617 genetically modified food Nutrition 0.000 description 2
- 230000000762 glandular Effects 0.000 description 2
- 208000005017 glioblastoma Diseases 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- 239000000185 hemagglutinin Substances 0.000 description 2
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 2
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 2
- 208000005252 hepatitis A Diseases 0.000 description 2
- 208000002672 hepatitis B Diseases 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 229940124589 immunosuppressive drug Drugs 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 208000000509 infertility Diseases 0.000 description 2
- 230000036512 infertility Effects 0.000 description 2
- 231100000535 infertility Toxicity 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 206010022000 influenza Diseases 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 229960000681 leflunomide Drugs 0.000 description 2
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 231100000355 lymphocytotoxic Toxicity 0.000 description 2
- 230000001391 lymphocytotoxic effect Effects 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 238000012737 microarray-based gene expression Methods 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 210000005087 mononuclear cell Anatomy 0.000 description 2
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 206010028417 myasthenia gravis Diseases 0.000 description 2
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 2
- 230000002956 necrotizing effect Effects 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 238000005457 optimization Methods 0.000 description 2
- 229940092253 ovalbumin Drugs 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 201000001976 pemphigus vulgaris Diseases 0.000 description 2
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 2
- 239000002831 pharmacologic agent Substances 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 2
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 2
- 208000017805 post-transplant lymphoproliferative disease Diseases 0.000 description 2
- 239000000955 prescription drug Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 201000007094 prostatitis Diseases 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 230000001850 reproductive effect Effects 0.000 description 2
- 230000004043 responsiveness Effects 0.000 description 2
- WVYADZUPLLSGPU-UHFFFAOYSA-N salsalate Chemical compound OC(=O)C1=CC=CC=C1OC(=O)C1=CC=CC=C1O WVYADZUPLLSGPU-UHFFFAOYSA-N 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 208000017520 skin disease Diseases 0.000 description 2
- 239000004055 small Interfering RNA Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 230000002483 superagonistic effect Effects 0.000 description 2
- 210000002437 synoviocyte Anatomy 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 229960001967 tacrolimus Drugs 0.000 description 2
- 101150047061 tag-72 gene Proteins 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229960004964 temozolomide Drugs 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 206010043778 thyroiditis Diseases 0.000 description 2
- 241001529453 unidentified herpesvirus Species 0.000 description 2
- 229960001055 uracil mustard Drugs 0.000 description 2
- 239000004474 valine Chemical group 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- RDJGLLICXDHJDY-NSHDSACASA-N (2s)-2-(3-phenoxyphenyl)propanoic acid Chemical compound OC(=O)[C@@H](C)C1=CC=CC(OC=2C=CC=CC=2)=C1 RDJGLLICXDHJDY-NSHDSACASA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- HMLGSIZOMSVISS-ONJSNURVSA-N (7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2C(=C(C=C)CSC21)C(O)=O)=O)C(=O)\C(=N/OCOC(=O)C(C)(C)C)C1=CSC(N)=N1 HMLGSIZOMSVISS-ONJSNURVSA-N 0.000 description 1
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 description 1
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- 101150084750 1 gene Proteins 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- XOQABDOICLHPIS-UHFFFAOYSA-N 1-hydroxy-2,1-benzoxaborole Chemical compound C1=CC=C2B(O)OCC2=C1 XOQABDOICLHPIS-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- LDGWQMRUWMSZIU-LQDDAWAPSA-M 2,3-bis[(z)-octadec-9-enoxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)C)OCCCCCCCC\C=C/CCCCCCCC LDGWQMRUWMSZIU-LQDDAWAPSA-M 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- LKKMLIBUAXYLOY-UHFFFAOYSA-N 3-Amino-1-methyl-5H-pyrido[4,3-b]indole Chemical compound N1C2=CC=CC=C2C2=C1C=C(N)N=C2C LKKMLIBUAXYLOY-UHFFFAOYSA-N 0.000 description 1
- HVCOBJNICQPDBP-UHFFFAOYSA-N 3-[3-[3,5-dihydroxy-6-methyl-4-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyoxan-2-yl]oxydecanoyloxy]decanoic acid;hydrate Chemical compound O.OC1C(OC(CC(=O)OC(CCCCCCC)CC(O)=O)CCCCCCC)OC(C)C(O)C1OC1C(O)C(O)C(O)C(C)O1 HVCOBJNICQPDBP-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- 102000002627 4-1BB Ligand Human genes 0.000 description 1
- 108010082808 4-1BB Ligand Proteins 0.000 description 1
- 108020005029 5' Flanking Region Proteins 0.000 description 1
- 102100030310 5,6-dihydroxyindole-2-carboxylic acid oxidase Human genes 0.000 description 1
- 101710163881 5,6-dihydroxyindole-2-carboxylic acid oxidase Proteins 0.000 description 1
- 101710163573 5-hydroxyisourate hydrolase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-FOQJRBATSA-N 59096-14-9 Chemical compound CC(=O)OC1=CC=CC=C1[14C](O)=O BSYNRYMUTXBXSQ-FOQJRBATSA-N 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 208000002008 AIDS-Related Lymphoma Diseases 0.000 description 1
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 1
- 102100030840 AT-rich interactive domain-containing protein 4B Human genes 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 241000588626 Acinetobacter baumannii Species 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 208000026872 Addison Disease Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 101710192389 Aggrecan core protein Proteins 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 102100038910 Alpha-enolase Human genes 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 206010001935 American trypanosomiasis Diseases 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 101100118004 Arabidopsis thaliana EBP1 gene Proteins 0.000 description 1
- 101100446590 Arabidopsis thaliana FIM5 gene Proteins 0.000 description 1
- 241000712891 Arenavirus Species 0.000 description 1
- 208000008037 Arthrogryposis Diseases 0.000 description 1
- 239000000592 Artificial Cell Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 241000714230 Avian leukemia virus Species 0.000 description 1
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 1
- 208000025324 B-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 241000304886 Bacilli Species 0.000 description 1
- 101000840545 Bacillus thuringiensis L-isoleucine-4-hydroxylase Proteins 0.000 description 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 1
- 102000015735 Beta-catenin Human genes 0.000 description 1
- 108060000903 Beta-catenin Proteins 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 241000701822 Bovine papillomavirus Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 1
- 108700012439 CA9 Proteins 0.000 description 1
- 101150052583 CALM1 gene Proteins 0.000 description 1
- 229940123189 CD40 agonist Drugs 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 102100037904 CD9 antigen Human genes 0.000 description 1
- 101001039256 Caenorhabditis elegans Low-density lipoprotein receptor-related protein Proteins 0.000 description 1
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 1
- 101100297347 Caenorhabditis elegans pgl-3 gene Proteins 0.000 description 1
- 101100510617 Caenorhabditis elegans sel-8 gene Proteins 0.000 description 1
- 102100025580 Calmodulin-1 Human genes 0.000 description 1
- 102100039510 Cancer/testis antigen 2 Human genes 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 101100495352 Candida albicans CDR4 gene Proteins 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 101710132601 Capsid protein Proteins 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- 102000012406 Carcinoembryonic Antigen Human genes 0.000 description 1
- 102100025466 Carcinoembryonic antigen-related cell adhesion molecule 3 Human genes 0.000 description 1
- 208000010667 Carcinoma of liver and intrahepatic biliary tract Diseases 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100035882 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 1
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 1
- 101000926196 Cercopithecine herpesvirus 9 (strain DHV) Envelope glycoprotein E Proteins 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 208000024699 Chagas disease Diseases 0.000 description 1
- 108091007741 Chimeric antigen receptor T cells Proteins 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 1
- 108091007403 Cholesterol transporters Proteins 0.000 description 1
- 206010008748 Chorea Diseases 0.000 description 1
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 1
- 241001533384 Circovirus Species 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000193163 Clostridioides difficile Species 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000016918 Complement C3 Human genes 0.000 description 1
- 108010028780 Complement C3 Proteins 0.000 description 1
- 102100032768 Complement receptor type 2 Human genes 0.000 description 1
- 108010062580 Concanavalin A Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 241000711573 Coronaviridae Species 0.000 description 1
- 102000004420 Creatine Kinase Human genes 0.000 description 1
- 108010042126 Creatine kinase Proteins 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241001337994 Cryptococcus <scale insect> Species 0.000 description 1
- 240000005109 Cryptomeria japonica Species 0.000 description 1
- 108010060385 Cyclin B1 Proteins 0.000 description 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 1
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 1
- 108010072210 Cyclophilin C Proteins 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 101100459256 Cyprinus carpio myca gene Proteins 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- VVNCNSJFMMFHPL-VKHMYHEASA-N D-penicillamine Chemical compound CC(C)(S)[C@@H](N)C(O)=O VVNCNSJFMMFHPL-VKHMYHEASA-N 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 206010013700 Drug hypersensitivity Diseases 0.000 description 1
- 208000010975 Dystrophic epidermolysis bullosa Diseases 0.000 description 1
- 201000011001 Ebola Hemorrhagic Fever Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010014596 Encephalitis Japanese B Diseases 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 241000588914 Enterobacter Species 0.000 description 1
- 101001095863 Enterobacteria phage T4 RNA ligase 1 Proteins 0.000 description 1
- 241000588921 Enterobacteriaceae Species 0.000 description 1
- 241000194031 Enterococcus faecium Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010014950 Eosinophilia Diseases 0.000 description 1
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 1
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 1
- 102100038595 Estrogen receptor Human genes 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 101150106011 FIM2 gene Proteins 0.000 description 1
- 101150048576 FIM3 gene Proteins 0.000 description 1
- 102000008857 Ferritin Human genes 0.000 description 1
- 108050000784 Ferritin Proteins 0.000 description 1
- 238000008416 Ferritin Methods 0.000 description 1
- 102100028073 Fibroblast growth factor 5 Human genes 0.000 description 1
- 108090000380 Fibroblast growth factor 5 Proteins 0.000 description 1
- 229920001917 Ficoll Polymers 0.000 description 1
- 241000711950 Filoviridae Species 0.000 description 1
- 108010040721 Flagellin Proteins 0.000 description 1
- 241000710831 Flavivirus Species 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 208000004262 Food Hypersensitivity Diseases 0.000 description 1
- 206010016946 Food allergy Diseases 0.000 description 1
- 108010058643 Fungal Proteins Proteins 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- 102100039717 G antigen 1 Human genes 0.000 description 1
- 102100032340 G2/mitotic-specific cyclin-B1 Human genes 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- 101710087459 Gamma-gliadin Proteins 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 208000034951 Genetic Translocation Diseases 0.000 description 1
- 241000699694 Gerbillinae Species 0.000 description 1
- 201000004311 Gilles de la Tourette syndrome Diseases 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 101710088083 Glomulin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- BCCRXDTUTZHDEU-VKHMYHEASA-N Gly-Ser Chemical compound NCC(=O)N[C@@H](CO)C(O)=O BCCRXDTUTZHDEU-VKHMYHEASA-N 0.000 description 1
- 102000019058 Glycogen Synthase Kinase 3 beta Human genes 0.000 description 1
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 102100028967 HLA class I histocompatibility antigen, alpha chain G Human genes 0.000 description 1
- 108010026122 HLA-A*33 antigen Proteins 0.000 description 1
- 108010035452 HLA-A1 Antigen Proteins 0.000 description 1
- 108010074032 HLA-A2 Antigen Proteins 0.000 description 1
- 102000025850 HLA-A2 Antigen Human genes 0.000 description 1
- 108010013476 HLA-A24 Antigen Proteins 0.000 description 1
- 108010029526 HLA-A28 antigen Proteins 0.000 description 1
- 108010018475 HLA-A31 antigen Proteins 0.000 description 1
- 108010028938 HLA-B45 antigen Proteins 0.000 description 1
- 108010091938 HLA-B7 Antigen Proteins 0.000 description 1
- 108010037618 HLA-C*08 antigen Proteins 0.000 description 1
- 108010024164 HLA-G Antigens Proteins 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 208000001204 Hashimoto Disease Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 241000590002 Helicobacter pylori Species 0.000 description 1
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 208000027761 Hepatic autoimmune disease Diseases 0.000 description 1
- 206010073069 Hepatic cancer Diseases 0.000 description 1
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 1
- 101710083479 Hepatitis A virus cellular receptor 2 homolog Proteins 0.000 description 1
- 208000007514 Herpes zoster Diseases 0.000 description 1
- 102100035616 Heterogeneous nuclear ribonucleoproteins A2/B1 Human genes 0.000 description 1
- 102000006479 Heterogeneous-Nuclear Ribonucleoproteins Human genes 0.000 description 1
- 108010019372 Heterogeneous-Nuclear Ribonucleoproteins Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 1
- 108010027412 Histocompatibility Antigens Class II Proteins 0.000 description 1
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000694288 Homo sapiens 40S ribosomal protein SA Proteins 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101000792935 Homo sapiens AT-rich interactive domain-containing protein 4B Proteins 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 1
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 1
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 description 1
- 101000889345 Homo sapiens Cancer/testis antigen 2 Proteins 0.000 description 1
- 101000914337 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 3 Proteins 0.000 description 1
- 101000941929 Homo sapiens Complement receptor type 2 Proteins 0.000 description 1
- 101000954709 Homo sapiens Doublecortin domain-containing protein 2 Proteins 0.000 description 1
- 101000886137 Homo sapiens G antigen 1 Proteins 0.000 description 1
- 101000985516 Homo sapiens Hermansky-Pudlak syndrome 5 protein Proteins 0.000 description 1
- 101000854026 Homo sapiens Heterogeneous nuclear ribonucleoproteins A2/B1 Proteins 0.000 description 1
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 1
- 101001103039 Homo sapiens Inactive tyrosine-protein kinase transmembrane receptor ROR1 Proteins 0.000 description 1
- 101001037256 Homo sapiens Indoleamine 2,3-dioxygenase 1 Proteins 0.000 description 1
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 description 1
- 101000614481 Homo sapiens Kidney-associated antigen 1 Proteins 0.000 description 1
- 101000984189 Homo sapiens Leukocyte immunoglobulin-like receptor subfamily B member 2 Proteins 0.000 description 1
- 101000984186 Homo sapiens Leukocyte immunoglobulin-like receptor subfamily B member 4 Proteins 0.000 description 1
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 1
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 1
- 101001014223 Homo sapiens MAPK/MAK/MRK overlapping kinase Proteins 0.000 description 1
- 101000991061 Homo sapiens MHC class I polypeptide-related sequence B Proteins 0.000 description 1
- 101000934372 Homo sapiens Macrosialin Proteins 0.000 description 1
- 101001030069 Homo sapiens Major vault protein Proteins 0.000 description 1
- 101001011886 Homo sapiens Matrix metalloproteinase-16 Proteins 0.000 description 1
- 101001109503 Homo sapiens NKG2-C type II integral membrane protein Proteins 0.000 description 1
- 101001103036 Homo sapiens Nuclear receptor ROR-alpha Proteins 0.000 description 1
- 101000904196 Homo sapiens Pancreatic secretory granule membrane major glycoprotein GP2 Proteins 0.000 description 1
- 101001043564 Homo sapiens Prolow-density lipoprotein receptor-related protein 1 Proteins 0.000 description 1
- 101001065830 Homo sapiens Protein LRATD1 Proteins 0.000 description 1
- 101001062222 Homo sapiens Receptor-binding cancer antigen expressed on SiSo cells Proteins 0.000 description 1
- 101000738769 Homo sapiens Receptor-type tyrosine-protein phosphatase alpha Proteins 0.000 description 1
- 101000973629 Homo sapiens Ribosome quality control complex subunit NEMF Proteins 0.000 description 1
- 101000665137 Homo sapiens Scm-like with four MBT domains protein 1 Proteins 0.000 description 1
- 101100207070 Homo sapiens TNFSF8 gene Proteins 0.000 description 1
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 description 1
- 101000626163 Homo sapiens Tenascin-X Proteins 0.000 description 1
- 101000847107 Homo sapiens Tetraspanin-8 Proteins 0.000 description 1
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 1
- 101000597785 Homo sapiens Tumor necrosis factor receptor superfamily member 6B Proteins 0.000 description 1
- 101001047681 Homo sapiens Tyrosine-protein kinase Lck Proteins 0.000 description 1
- 101000671653 Homo sapiens U3 small nucleolar RNA-associated protein 14 homolog A Proteins 0.000 description 1
- 101000955999 Homo sapiens V-set domain-containing T-cell activation inhibitor 1 Proteins 0.000 description 1
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 1
- 241000829111 Human polyomavirus 1 Species 0.000 description 1
- 108010052919 Hydroxyethylthiazole kinase Proteins 0.000 description 1
- 108010027436 Hydroxymethylpyrimidine kinase Proteins 0.000 description 1
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 1
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 1
- 102100039615 Inactive tyrosine-protein kinase transmembrane receptor ROR1 Human genes 0.000 description 1
- 102100040061 Indoleamine 2,3-dioxygenase 1 Human genes 0.000 description 1
- 102100021317 Inducible T-cell costimulator Human genes 0.000 description 1
- 101710205775 Inducible T-cell costimulator Proteins 0.000 description 1
- 208000006877 Insect Bites and Stings Diseases 0.000 description 1
- 102100023915 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 1
- 108010030506 Integrin alpha6beta4 Proteins 0.000 description 1
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 1
- 108700021006 Interleukin-1 receptor antagonist Proteins 0.000 description 1
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 241000701372 Iridovirus Species 0.000 description 1
- 208000000209 Isaacs syndrome Diseases 0.000 description 1
- 108020003285 Isocitrate lyase Proteins 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 201000005807 Japanese encephalitis Diseases 0.000 description 1
- 241000710842 Japanese encephalitis virus Species 0.000 description 1
- 208000003456 Juvenile Arthritis Diseases 0.000 description 1
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 1
- 102000002698 KIR Receptors Human genes 0.000 description 1
- 108010043610 KIR Receptors Proteins 0.000 description 1
- 102100034872 Kallikrein-4 Human genes 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 241000588747 Klebsiella pneumoniae Species 0.000 description 1
- 102100031413 L-dopachrome tautomerase Human genes 0.000 description 1
- 101710093778 L-dopachrome tautomerase Proteins 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 201000010743 Lambert-Eaton myasthenic syndrome Diseases 0.000 description 1
- 208000007811 Latex Hypersensitivity Diseases 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 108010028275 Leukocyte Elastase Proteins 0.000 description 1
- 102100025583 Leukocyte immunoglobulin-like receptor subfamily B member 2 Human genes 0.000 description 1
- 102100025578 Leukocyte immunoglobulin-like receptor subfamily B member 4 Human genes 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 206010025312 Lymphoma AIDS related Diseases 0.000 description 1
- 102100031520 MAPK/MAK/MRK overlapping kinase Human genes 0.000 description 1
- 102000016200 MART-1 Antigen Human genes 0.000 description 1
- 108010010995 MART-1 Antigen Proteins 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 102100030301 MHC class I polypeptide-related sequence A Human genes 0.000 description 1
- 102100030300 MHC class I polypeptide-related sequence B Human genes 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 description 1
- 102100025136 Macrosialin Human genes 0.000 description 1
- MQHWFIOJQSCFNM-UHFFFAOYSA-L Magnesium salicylate Chemical compound [Mg+2].OC1=CC=CC=C1C([O-])=O.OC1=CC=CC=C1C([O-])=O MQHWFIOJQSCFNM-UHFFFAOYSA-L 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 108090000561 Matrix metalloproteinase-16 Proteins 0.000 description 1
- 101710085938 Matrix protein Proteins 0.000 description 1
- SBDNJUWAMKYJOX-UHFFFAOYSA-N Meclofenamic Acid Chemical compound CC1=CC=C(Cl)C(NC=2C(=CC=CC=2)C(O)=O)=C1Cl SBDNJUWAMKYJOX-UHFFFAOYSA-N 0.000 description 1
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 1
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 1
- 101710127721 Membrane protein Proteins 0.000 description 1
- 206010027260 Meningitis viral Diseases 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 208000005647 Mumps Diseases 0.000 description 1
- 241000714177 Murine leukemia virus Species 0.000 description 1
- 101100207071 Mus musculus Tnfsf8 gene Proteins 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 206010028372 Muscular weakness Diseases 0.000 description 1
- 206010028424 Myasthenic syndrome Diseases 0.000 description 1
- 102000047918 Myelin Basic Human genes 0.000 description 1
- 102000006386 Myelin Proteins Human genes 0.000 description 1
- 108010083674 Myelin Proteins Proteins 0.000 description 1
- 101710107068 Myelin basic protein Proteins 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 102000003505 Myosin Human genes 0.000 description 1
- 108060008487 Myosin Proteins 0.000 description 1
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 description 1
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 description 1
- 230000004988 N-glycosylation Effects 0.000 description 1
- 102100022683 NKG2-C type II integral membrane protein Human genes 0.000 description 1
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 102100024964 Neural cell adhesion molecule L1 Human genes 0.000 description 1
- 102000005348 Neuraminidase Human genes 0.000 description 1
- 108010006232 Neuraminidase Proteins 0.000 description 1
- 206010029240 Neuritis Diseases 0.000 description 1
- 206010072359 Neuromyotonia Diseases 0.000 description 1
- 102100033174 Neutrophil elastase Human genes 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 102000007999 Nuclear Proteins Human genes 0.000 description 1
- 108010089610 Nuclear Proteins Proteins 0.000 description 1
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 1
- 102000004473 OX40 Ligand Human genes 0.000 description 1
- 108010042215 OX40 Ligand Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 208000003435 Optic Neuritis Diseases 0.000 description 1
- 241000713112 Orthobunyavirus Species 0.000 description 1
- 240000007019 Oxalis corniculata Species 0.000 description 1
- 108060006580 PRAME Proteins 0.000 description 1
- 102000036673 PRAME Human genes 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 208000016222 Pancreatic disease Diseases 0.000 description 1
- 102100024019 Pancreatic secretory granule membrane major glycoprotein GP2 Human genes 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 206010034277 Pemphigoid Diseases 0.000 description 1
- 208000027086 Pemphigus foliaceus Diseases 0.000 description 1
- 102100024968 Peptidyl-prolyl cis-trans isomerase C Human genes 0.000 description 1
- 208000031845 Pernicious anaemia Diseases 0.000 description 1
- 102000007982 Phosphoproteins Human genes 0.000 description 1
- 108010089430 Phosphoproteins Proteins 0.000 description 1
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 241000224016 Plasmodium Species 0.000 description 1
- 241000223960 Plasmodium falciparum Species 0.000 description 1
- 101710183389 Pneumolysin Proteins 0.000 description 1
- 208000000474 Poliomyelitis Diseases 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 101710182846 Polyhedrin Proteins 0.000 description 1
- 241001505332 Polyomavirus sp. Species 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000034943 Primary Sjögren syndrome Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 208000024777 Prion disease Diseases 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 102100021923 Prolow-density lipoprotein receptor-related protein 1 Human genes 0.000 description 1
- 102100023832 Prolyl endopeptidase FAP Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 101710194807 Protective antigen Proteins 0.000 description 1
- 102100032095 Protein LRATD1 Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 108010010974 Proteolipids Proteins 0.000 description 1
- 102000016202 Proteolipids Human genes 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 1
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 1
- 206010037549 Purpura Diseases 0.000 description 1
- 241001672981 Purpura Species 0.000 description 1
- 206010071141 Rasmussen encephalitis Diseases 0.000 description 1
- 208000004160 Rasmussen subacute encephalitis Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101001039269 Rattus norvegicus Glycine N-methyltransferase Proteins 0.000 description 1
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 102100029165 Receptor-binding cancer antigen expressed on SiSo cells Human genes 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 241000702263 Reovirus sp. Species 0.000 description 1
- 206010061494 Rhinovirus infection Diseases 0.000 description 1
- 102100022213 Ribosome quality control complex subunit NEMF Human genes 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 241000702670 Rotavirus Species 0.000 description 1
- 206010039251 Rubber sensitivity Diseases 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 101001037255 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) Indoleamine 2,3-dioxygenase Proteins 0.000 description 1
- 206010061934 Salivary gland cancer Diseases 0.000 description 1
- 241000242678 Schistosoma Species 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010048908 Seasonal allergy Diseases 0.000 description 1
- 101710173693 Short transient receptor potential channel 1 Proteins 0.000 description 1
- 101710173694 Short transient receptor potential channel 2 Proteins 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- ABBQHOQBGMUPJH-UHFFFAOYSA-M Sodium salicylate Chemical compound [Na+].OC1=CC=CC=C1C([O-])=O ABBQHOQBGMUPJH-UHFFFAOYSA-M 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 208000006045 Spondylarthropathies Diseases 0.000 description 1
- 241000295644 Staphylococcaceae Species 0.000 description 1
- 206010042033 Stevens-Johnson syndrome Diseases 0.000 description 1
- 231100000168 Stevens-Johnson syndrome Toxicity 0.000 description 1
- 241000272534 Struthio camelus Species 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 108010002687 Survivin Proteins 0.000 description 1
- 208000027522 Sydenham chorea Diseases 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 208000018359 Systemic autoimmune disease Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 230000006052 T cell proliferation Effects 0.000 description 1
- 229940126547 T-cell immunoglobulin mucin-3 Drugs 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 101150031162 TM4SF1 gene Proteins 0.000 description 1
- 208000001106 Takayasu Arteritis Diseases 0.000 description 1
- 108010017842 Telomerase Proteins 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 102100038126 Tenascin Human genes 0.000 description 1
- 108010008125 Tenascin Proteins 0.000 description 1
- 102100024549 Tenascin-X Human genes 0.000 description 1
- 108010055044 Tetanus Toxin Proteins 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 102100032802 Tetraspanin-8 Human genes 0.000 description 1
- 208000002903 Thalassemia Diseases 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 208000024799 Thyroid disease Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 208000000323 Tourette Syndrome Diseases 0.000 description 1
- 208000016620 Tourette disease Diseases 0.000 description 1
- 241000159243 Toxicodendron radicans Species 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 102100034902 Transmembrane 4 L6 family member 1 Human genes 0.000 description 1
- 102000005937 Tropomyosin Human genes 0.000 description 1
- 108010030743 Tropomyosin Proteins 0.000 description 1
- LVTKHGUGBGNBPL-UHFFFAOYSA-N Trp-P-1 Chemical compound N1C2=CC=CC=C2C2=C1C(C)=C(N)N=C2C LVTKHGUGBGNBPL-UHFFFAOYSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102100032100 Tumor necrosis factor ligand superfamily member 8 Human genes 0.000 description 1
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 1
- 102100035284 Tumor necrosis factor receptor superfamily member 6B Human genes 0.000 description 1
- 102100024036 Tyrosine-protein kinase Lck Human genes 0.000 description 1
- 102100040099 U3 small nucleolar RNA-associated protein 14 homolog A Human genes 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 1
- HDOVUKNUBWVHOX-QMMMGPOBSA-N Valacyclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCOC(=O)[C@@H](N)C(C)C)C=N2 HDOVUKNUBWVHOX-QMMMGPOBSA-N 0.000 description 1
- WPVFJKSGQUFQAP-GKAPJAKFSA-N Valcyte Chemical compound N1C(N)=NC(=O)C2=C1N(COC(CO)COC(=O)[C@@H](N)C(C)C)C=N2 WPVFJKSGQUFQAP-GKAPJAKFSA-N 0.000 description 1
- 241000700647 Variola virus Species 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 206010047642 Vitiligo Diseases 0.000 description 1
- 206010047741 Vulval cancer Diseases 0.000 description 1
- 208000004354 Vulvar Neoplasms Diseases 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 208000000260 Warts Diseases 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- 208000003152 Yellow Fever Diseases 0.000 description 1
- 241000907316 Zika virus Species 0.000 description 1
- 208000035472 Zoonoses Diseases 0.000 description 1
- HIHOWBSBBDRPDW-PTHRTHQKSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] n-[2-(dimethylamino)ethyl]carbamate Chemical compound C1C=C2C[C@@H](OC(=O)NCCN(C)C)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HIHOWBSBBDRPDW-PTHRTHQKSA-N 0.000 description 1
- ATBOMIWRCZXYSZ-XZBBILGWSA-N [1-[2,3-dihydroxypropoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] (9e,12e)-octadeca-9,12-dienoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCC\C=C\C\C=C\CCCCC ATBOMIWRCZXYSZ-XZBBILGWSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 108060000200 adenylate cyclase Proteins 0.000 description 1
- 102000030621 adenylate cyclase Human genes 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- 208000004631 alopecia areata Diseases 0.000 description 1
- 239000002160 alpha blocker Substances 0.000 description 1
- 229940124308 alpha-adrenoreceptor antagonist Drugs 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 229960004238 anakinra Drugs 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 230000000919 anti-host Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 102000025171 antigen binding proteins Human genes 0.000 description 1
- 108091000831 antigen binding proteins Proteins 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 208000025150 arthrogryposis multiplex congenita Diseases 0.000 description 1
- 239000000823 artificial membrane Substances 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 208000037896 autoimmune cutaneous disease Diseases 0.000 description 1
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 description 1
- 201000004339 autoimmune neuropathy Diseases 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 239000003012 bilayer membrane Substances 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 201000000053 blastoma Diseases 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 230000006931 brain damage Effects 0.000 description 1
- 231100000874 brain damage Toxicity 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- 229940112133 busulfex Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 101150091339 cam-1 gene Proteins 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 210000001715 carotid artery Anatomy 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000009134 cell regulation Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 208000019069 chronic childhood arthritis Diseases 0.000 description 1
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 1
- 229960000724 cidofovir Drugs 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 230000007699 co-inhibitory pathway Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000001246 colloidal dispersion Methods 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 208000030499 combat disease Diseases 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000003436 cytoskeletal effect Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229940093541 dicetylphosphate Drugs 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 229960000616 diflunisal Drugs 0.000 description 1
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 208000010643 digestive system disease Diseases 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 201000007850 distal arthrogryposis Diseases 0.000 description 1
- 201000005311 drug allergy Diseases 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 229960000284 efalizumab Drugs 0.000 description 1
- 201000008184 embryoma Diseases 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 230000002327 eosinophilic effect Effects 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 208000004298 epidermolysis bullosa dystrophica Diseases 0.000 description 1
- 201000010063 epididymitis Diseases 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 229960005293 etodolac Drugs 0.000 description 1
- XFBVBWWRPKNWHW-UHFFFAOYSA-N etodolac Chemical compound C1COC(CC)(CC(O)=O)C2=N[C]3C(CC)=CC=CC3=C21 XFBVBWWRPKNWHW-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 238000002710 external beam radiation therapy Methods 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 229960001419 fenoprofen Drugs 0.000 description 1
- 231100000562 fetal loss Toxicity 0.000 description 1
- 229960002390 flurbiprofen Drugs 0.000 description 1
- SYTBZMRGLBWNTM-UHFFFAOYSA-N flurbiprofen Chemical compound FC1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-UHFFFAOYSA-N 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 235000020932 food allergy Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 229960005102 foscarnet Drugs 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 210000000973 gametocyte Anatomy 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 208000018685 gastrointestinal system disease Diseases 0.000 description 1
- 229940014259 gelatin Drugs 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002343 gold Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 229940037467 helicobacter pylori Drugs 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000007475 hemolytic anemia Diseases 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 208000010710 hepatitis C virus infection Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229940084986 human chorionic gonadotropin Drugs 0.000 description 1
- 235000003642 hunger Nutrition 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 1
- 229960004171 hydroxychloroquine Drugs 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 208000009326 ileitis Diseases 0.000 description 1
- 210000001822 immobilized cell Anatomy 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000003308 immunostimulating effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 108700032552 influenza virus INS1 Proteins 0.000 description 1
- 108700015453 influenza virus PB2 Proteins 0.000 description 1
- 108010064513 influenza virus polymerase basic protein 1 Proteins 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 201000006334 interstitial nephritis Diseases 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 238000012977 invasive surgical procedure Methods 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 1
- 108010024383 kallikrein 4 Proteins 0.000 description 1
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 229960004752 ketorolac Drugs 0.000 description 1
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 201000005391 latex allergy Diseases 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 238000005339 levitation Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 201000002250 liver carcinoma Diseases 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 238000001325 log-rank test Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000005249 lung adenocarcinoma Diseases 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 230000005427 lymphocyte apoptotic process Effects 0.000 description 1
- 208000019420 lymphoid neoplasm Diseases 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229940072082 magnesium salicylate Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 229960003762 maribavir Drugs 0.000 description 1
- KJFBVJALEQWJBS-XUXIUFHCSA-N maribavir Chemical compound CC(C)NC1=NC2=CC(Cl)=C(Cl)C=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O KJFBVJALEQWJBS-XUXIUFHCSA-N 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- 229940013798 meclofenamate Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000010445 mica Substances 0.000 description 1
- 229910052618 mica group Inorganic materials 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- QXYYYPFGTSJXNS-UHFFFAOYSA-N mitozolomide Chemical compound N1=NN(CCCl)C(=O)N2C1=C(C(=O)N)N=C2 QXYYYPFGTSJXNS-UHFFFAOYSA-N 0.000 description 1
- 229950005967 mitozolomide Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 208000010805 mumps infectious disease Diseases 0.000 description 1
- 230000000869 mutational effect Effects 0.000 description 1
- 230000036473 myasthenia Effects 0.000 description 1
- 229940014456 mycophenolate Drugs 0.000 description 1
- 229960004866 mycophenolate mofetil Drugs 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- 229940090009 myleran Drugs 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- AEMBWNDIEFEPTH-UHFFFAOYSA-N n-tert-butyl-n-ethylnitrous amide Chemical compound CCN(N=O)C(C)(C)C AEMBWNDIEFEPTH-UHFFFAOYSA-N 0.000 description 1
- 229960004270 nabumetone Drugs 0.000 description 1
- 239000002088 nanocapsule Substances 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229960005027 natalizumab Drugs 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 210000005044 neurofilament Anatomy 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 231100001160 nonlethal Toxicity 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- GSSMIHQEWAQUPM-AOLPDKKJSA-N ovalbumin peptide Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)[C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CN=CN1 GSSMIHQEWAQUPM-AOLPDKKJSA-N 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- 201000002628 peritoneum cancer Diseases 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 108010021711 pertactin Proteins 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 229960002895 phenylbutazone Drugs 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 201000004338 pollen allergy Diseases 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 239000000186 progesterone Substances 0.000 description 1
- 229960003387 progesterone Drugs 0.000 description 1
- 229940072288 prograf Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 108010002730 protein S precursor Proteins 0.000 description 1
- 238000012514 protein characterization Methods 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 229940112971 protopic Drugs 0.000 description 1
- 208000028172 protozoa infectious disease Diseases 0.000 description 1
- 230000001698 pyrogenic effect Effects 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000009342 ragweed pollen Substances 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 229940116176 remicade Drugs 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 201000003068 rheumatic fever Diseases 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229960001302 ridaforolimus Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 201000005404 rubella Diseases 0.000 description 1
- 201000003804 salivary gland carcinoma Diseases 0.000 description 1
- 229960000953 salsalate Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000010187 selection method Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000000405 serological effect Effects 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 201000010153 skin papilloma Diseases 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 229960004025 sodium salicylate Drugs 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 201000005671 spondyloarthropathy Diseases 0.000 description 1
- 210000003046 sporozoite Anatomy 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 208000017572 squamous cell neoplasm Diseases 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000003153 stable transfection Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 230000037351 starvation Effects 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000007801 sublethal irradiation Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 229960000894 sulindac Drugs 0.000 description 1
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940118376 tetanus toxin Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 230000002992 thymic effect Effects 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 208000021510 thyroid gland disease Diseases 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 230000024664 tolerance induction Effects 0.000 description 1
- 230000003614 tolerogenic effect Effects 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 1
- 229960004380 tramadol Drugs 0.000 description 1
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 108010020589 trehalose-6-phosphate synthase Proteins 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 229950002860 triplatin tetranitrate Drugs 0.000 description 1
- 190014017283 triplatin tetranitrate Chemical compound 0.000 description 1
- FQCQGOZEWWPOKI-UHFFFAOYSA-K trisalicylate-choline Chemical compound [Mg+2].C[N+](C)(C)CCO.OC1=CC=CC=C1C([O-])=O.OC1=CC=CC=C1C([O-])=O.OC1=CC=CC=C1C([O-])=O FQCQGOZEWWPOKI-UHFFFAOYSA-K 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 208000012991 uterine carcinoma Diseases 0.000 description 1
- 229940093257 valacyclovir Drugs 0.000 description 1
- 229960002149 valganciclovir Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 201000010044 viral meningitis Diseases 0.000 description 1
- 244000052613 viral pathogen Species 0.000 description 1
- 201000005102 vulva cancer Diseases 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 206010048282 zoonosis Diseases 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/001—Preparations to induce tolerance to non-self, e.g. prior to transplantation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39566—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against immunoglobulins, e.g. anti-idiotypic antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4632—T-cell receptors [TCR]; antibody T-cell receptor constructs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464411—Immunoglobulin superfamily
- A61K39/464412—CD19 or B4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/464838—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
- C12N5/0637—Immunosuppressive T lymphocytes, e.g. regulatory T cells or Treg
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0648—Splenocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K2035/122—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells for inducing tolerance or supression of immune responses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5158—Antigen-pulsed cells, e.g. T-cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/26—Universal/off- the- shelf cellular immunotherapy; Allogenic cells or means to avoid rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/38—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/57—Skin; melanoma
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2307—Interleukin-7 (IL-7)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2315—Interleukin-15 (IL-15)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2321—Interleukin-21 (IL-21)
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Abstract
Una célula aislada que tiene un fenotipo de linfocito T de memoria central (Tcm), siendo la célula una célula inductora de tolerancia y capaz de dirigirse a los ganglios linfáticos después del trasplante, siendo la célula transducida para expresar un receptor de superficie celular que comprende un módulo de señalización del receptor de células T se revela. También se describen métodos para generarlos y usarlos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Linfocitos T de memoria central anti-terceros modificados genéticamente y uso de los mismos en inmunoterapia
La presente invención, en algunos aspectos de la misma, se refiere a la tolerancia modificada genéticamente que inducen los linfocitos T de memoria central transducidos para expresar un receptor de la superficie celular y, más particularmente, pero no exclusivamente, al uso de los mismos en inmunoterapia.
La terapia celular adoptiva (ACT) es un procedimiento terapéutico en el que se administran linfocitos (por ejemplo, linfocitos T) a pacientes para tratar el cáncer o infecciones virales.
Este enfoque requiere la generación ex-vivo generación de linfocitos T específicos de tumor o virus y la infusión de los mismos a los pacientes. Para apoyar la aceptación de los linfocitos T, el paciente también suele ser tratado con protocolos de acondicionamiento, por ejemplo, protocolos de preacondicionamiento (p. ej., irradiación o quimioterapia) y/o administración de factores de crecimiento de linfocitos (como IL-2). Se han descrito muchos métodos para generar linfocitos específicos de tumores, siendo los dos enfoques principales la expansión de linfocitos T específicos de antígeno o la redirección de linfocitos T usando ingeniería genética.
De acuerdo con un enfoque, los linfocitos infiltrantes de tumores (TIL) se aíslan de la masa tumoral del propio paciente (por ejemplo, melanoma o cáncer renal), se expanden ex-vivo y se vuelven a infundir en el paciente. Los TIL son una fuente prometedora de células, ya que son un conjunto mixto de células propias del paciente que tienen receptores de linfocitos T (TCR) específicos de los antígenos asociados al tumor (TAA) presentes en el tumor. Sin embargo, solo son aplicables en los casos en que los linfocitos T pueden aislarse de una masa tumoral.
Este enfoque ha sido prometedor en el tratamiento del melanoma metastásico.
De acuerdo con otro enfoque, la modificación genética se utiliza para redirigir los linfocitos contra los tumores mediante el uso de cadenas de TCR transgénicos o receptores quiméricos. En la actualidad, la transferencia retroviral, lentiviral o electroporativa de receptores de antígenos quiméricos (CAR) cuyo reconocimiento de diana depende de un dominio de región variable de cadena única de un anticuerpo monoclonal (scFv) o de un receptor de linfocitos T (TCR) se usa normalmente para la producción estable de linfocitos T terapéuticos (linfocitos T-CAR o linfocitos T-TCR, respectivamente) [Fujiwara, Pharmaceuticals (2014) 7: 1049-1068].
Los linfocitos con TCR transgénico (T-TCR) requieren una molécula HLA específica para el reconocimiento del antígeno diana (es decir, restricción HLA) y tienen la capacidad de reconocer proteínas intracelulares, proporcionando una amplia gama de antígenos diana asociados a tumores o antígenos virales. La calidad terapéutica de los linfocitos T-TCR depende de su avidez. Para crear una mayor avidez se han implementado varias estrategias, que incluyen, el uso de TCR seleccionados de ratones transgénicos para HLA humano inmunizados con epítopos relevantes y/o la inserción de mutaciones dirigidas en las regiones CDR 2 o 3 de las regiones variables de las cadenas a/p del TCR que interactúan con el complejo HLNepítopo [Fujiwara, Pharmaceuticals (2014) supra].
De forma alternativa, los linfocitos CAR-T no están restringidos por el HLA. La construcción del receptor quimérico (receptor de antígeno quimérico - CAR) generalmente se compone de un dominio de unión a antígeno extracelular, un dominio transmembrana y un dominio de señalización citoplasmático. El receptor quimérico original (es decir, de "primera generación") estaba compuesto por un fragmento scFv fusionado con un dominio intracelular de la cadena ^ de CD3.
También se generó un receptor quimérico de "segunda generación" que agrega un dominio de señalización intracelular, de varios receptores de proteínas coestimuladoras (por ejemplo, CD28, CD137, 4-1BB, ICOS), a la cola citoplasmática del CAR para proporcionar señales adicionales al linfocito T. Los estudios preclínicos han indicado que los CAR de "segunda generación" mejoran la actividad antitumoral de los linfocitos T. Recientemente se generó un CAR de "tercera generación" que combina múltiples dominios de señalización, tales como CD3zeta-CD28-4-1BB o CD3zeta-CD28-OX40, para aumentar aún más la potencia.
Los CAR específicos de tumores que se dirigen a una variedad de antígenos tumorales se están probando en la clínica para el tratamiento de una variedad de cánceres diferentes. Los ejemplos de estos cánceres y sus antígenos a los que se dirigen incluyen linfoma folicular (CD20 o GD2), neuroblastoma (CD171), linfoma no Hodgkin (CD20), linfoma (CD19), glioblastoma (IL13Ra2), leucemia linfocítica crónica o LLC y leucemia linfocítica aguda o LLA (ambas CD19). Los CAR que demuestran actividad contra tumores sólidos, incluidos los de ovario, próstata, mama, renal, colon, neuroblastoma y otros están en investigación. También se han desarrollado CAR específicos de virus para atacar células que albergan virus como el VIH. Por ejemplo, se inició un ensayo clínico utilizando un CAR específico de Gp100 para el tratamiento del VIH (Chicaybam, Ibid).
Un objetivo importante es aplicar ACT, incluidos los linfocitos T modificados genéticamente, utilizando células alogénicas total o parcialmente incompatibles sin recurrir al trasplante de médula ósea.
Se han contemplado varios enfoques para modificar los linfocitos T para la terapia celular adoptiva, algunos se describen en Gilham et al., Human Gene Therapy (2015) 26:276-285; en Sharpe and Mount, Disease Models and Mechanisms (2015) 8:337-350 y en Gouble et al. Blood (2014) 124(21) 4689.
Se han contemplado varios enfoques para la generación de células inductoras de tolerancia desprovistas de actividad de injerto contra hospedador (ICH) y el uso de las mismas para el trasplante de injertos, algunos de los cuales se resumen a continuación.
Reisner y colaboradores describieron un enfoque desarrollado para generar LTC de veto desprovistos de actividad ICH, en el que se estimularon los LTC contra estimuladores de terceros en ausencia de IL-2 exógena. Este enfoque se basó en la observación de que solo los precursores de linfocitos T citotóxicos (LTCp) activados eran capaces de sobrevivir a la privación de IL-2 en el cultivo primario (la ausencia de IL-2 da como resultado la apoptosis de los linfocitos T no inducidos). Este método demostró in vitro e in vivo que agotaba la reactividad de ICH de los LCT de veto anti-terceros [Publicación PCT N.° WO 2001/049243, Bachar-Lustig et al., Blood. 2003;102:1943-1950; Aviner et al., Hum Immunol. (2005) 66:644-652]. La introducción de estos LCT de veto anti-terceros en un receptor (junto con un trasplante) impidieron el rechazo del injerto sin inducir la enfermedad de injerto contra hospedador (EICH) (Publicación PCT N.° WO 2001/049243).
La publicación PCT N.° WO 2010/049935 divulga una población aislada de células que comprende células anti terceros no inductoras de EICH que tienen un fenotipo de linfocito T de memoria central (Tmc), siendo las células, células inductoras de tolerancia y capaces de anidar en los ganglios linfáticos después del trasplante.
La publicación PCT N.° WO 2013/035099 divulga nuevos métodos para generar una población aislada de células que comprenden células anti-terceros que tienen un fenotipo de linfocito T de memoria central (Tmc), siendo las células, células inductoras de tolerancia y/o dotadas de actividad anti-enfermedad, y capaces de anidar en los ganglios linfáticos después del trasplante.
El documento WO 2014059173 se refiere a métodos y composiciones para modificar linfocitos T en los que PD1 y/o CTLA-4 se reprimen y/o inactivan usando proteínas de fusión tales como factores de transcripción artificiales y nucleasas.
El documento WO 2012129514 proporciona métodos y composiciones para conferir y/o aumentar las respuestas inmunitarias mediadas por inmunoterapia celular, como la transferencia adoptiva de linfocitos T CD8+ específicos de tumor modificados genéticamente en presencia de linfocitos T específicos de tumor, subconjunto específico de linfocitos T CD4+ modificados genéticamente, en donde los linfocitos T CD4+ confieren y/o aumentan la capacidad de los linfocitos T CD8+ para mantener la reactividad antitumoral y aumentar y/o maximizar la proliferación específica de tumor de los linfocitos T CD8+ específicos de tumor de interés. También se describen formulaciones farmacéuticas producidas por el método y métodos de uso de las mismas.
El documento WO 2014039044 proporciona métodos para producir una población aislada de células madre T de memoria, comprendiendo el método a) aislar linfocitos T vírgenes de un mamífero, en donde el mamífero no es un ratón; b) activar los linfocitos T vírgenes y expandir el número de linfocitos T vírgenes en presencia de uno o más estímulos de linfocitos T no específicos, una o más citocinas y un inhibidor de GSK-3beta. También se proporcionan métodos para producir una población aislada de células madre T de memoria, comprendiendo el método a) aislar linfocitos de un mamífero; b) clasificar los linfocitos usando citometría de flujo en una población que comprende un fenotipo que comprende i) CD95+, CD45RO- y CCR7+; y ii) CD62L+ o uno o más de CD27+, c D28+, CD45RA+ y CD127+ para producir una población aislada de células madre T de memoria. Otras realizaciones proporcionan células relacionadas, poblaciones de células, composiciones farmacéuticas y métodos para tratar o prevenir el cáncer.
El documento WO 2013035099 se refiere a un método para generar una población aislada de células que comprende células anti-terceros que tienen un fenotipo de linfocito T de memoria central (Tmc), siendo las células, células inductoras de tolerancia y/o dotadas de actividad anti-enfermedad, y capaces de anidar en los ganglios linfáticos después del trasplante. El método comprende: (a) poner en contacto células mononucleares de sangre periférica (PBMC) con un antígeno o antígenos de terceros en presencia de IL-21 para permitir el enriquecimiento de células reactivas al antígeno; y (b) cultivar las células resultantes de la etapa (a) en presencia de IL-21, IL-15 e IL-7 en un entorno libre de antígenos para permitir la proliferación de células que comprenden el fenotipo de linfocitos T de memoria central (Tmc).
El documento WO 2014152177 se refiere a células, por ejemplo, linfocitos T que expresan polipéptidos de muerte celular artificial que causan la muerte de una célula, por ejemplo, células (por ejemplo, linfocitos T) que expresan el polipéptido de muerte celular, cuando el polipéptido de muerte celular está multimerizado o dimerizado. También se proporciona en este documento el uso de tales células, por ejemplo, linfocitos T, para tratar enfermedades tales como el cáncer.
Sumario de la invención
La invención se expone en el conjunto de reivindicaciones adjuntas.
Breve descripción de las diversas vistas de los dibujos
Algunos aspectos de la divulgación se describen en el presente documento, a modo de ejemplo solamente, con referencia a los dibujos adjuntos. Con referencia específica ahora a los dibujos en detalle, se enfatiza que los detalles mostrados son a modo de ejemplo y con fines de análisis ilustrativo de los aspectos de la invención. En este sentido, la descripción tomada junto con los dibujos hace evidente a los expertos en la materia cómo pueden ponerse en práctica aspectos de la divulgación.
En los dibujos:
Las figuras 1A-C son ilustraciones esquemáticas de modelos para estudiar la capacidad de los linfocitos Tmc para inducir tolerancia en ausencia de las propiedades inductoras de la MO alogénica. La supervivencia y proliferación de células Tmc se analizó a través de FACS, mientras que la regulación a la baja de la actividad de los LTC del hospedador por células Tmc se probó utilizando el ensayo de 51Cr.
Las figuras 2A-B son gráficos que ilustran la persistencia de células F1-Tmc transferidas adoptivamente en el contexto de trasplante de médula ósea (TMO) singénico. Se trasplantaron ratones C57BL/6 (H-2b) como se describe en la figura 1A. Diagrama de dispersión representativo de un ratón en cada grupo, que muestra el porcentaje de células Tmc en sangre completa periférica de ratones, analizada 60 días después del trasplante por FACS usando aH2Dd (Donante) y aH2Kb para identificar las células F1(H2DdXH2Kb)-Tmc.
La figura 3 es un gráfico que ilustra que los linfocitos Tmc eliminan específicamente los linfocitos T anti-donante de una población policlonal de linfocitos T del hospedador (HTC), mientras que evita que otros HTC muestren actividad citotóxica. Los ratones se trasplantaron como se describe en la figura 1A. Sesenta días después del trasplante, se sacrificaron los ratones, se recolectaron bazos y ganglios linfáticos (LN) y se seleccionaron las células CD8+ (y se seleccionaron negativamente H-2Dd para excluir las T mc). Estos HTC vírgenes fueron probados para determinar su capacidad de eliminar las dianas C3H (H-2k) o BALB/c (H-2d) en un ensayo de liberación de cromo. Las barras muestran el efecto de la eliminación de la siguiente manera: La eliminación de la diana C3H por HTC de ratones que reciben solo MO (barras negras, "MO solo C3H") o por células de ratones que reciben también Tmc (barras grises oscuras, "MO+Tmc solo C3H") o la eliminación de la diana BALB/c por HTC de ratones que recibieron solo MO (barras blancas, "MO solo BALB") o por linfocitos T de ratones que recibieron también Tmc (barras grises brillantes, "MO+Tmc BALB"). Los resultados se presentan como media ± DE del porcentaje de eliminación de 12 pocillos para cada grupo. Se muestra el experimento representativo de 2 experimentos independientes realizados. (**) Representa un valor de p inferior a 0,01, (***) Representa un valor de p inferior a 0,001.
La figura 4 es un gráfico que ilustra que los linfocitos Tmc derivados de CB6 F1 persisten en ratones que recibieron 5.5 Gy TBI subletal con médula ósea singénica empobrecida en linfocitos T (TDBMT). Los ratones Balb/c (H2Dd) se irradiaron de forma subletal (5,5 Gy) y se trasplantaron como se describe en la figura 1B. La sangre periférica se analizó 40, 80 y 132 días después del trasplante por FACS usando aH2Dd (Hospedador) y aH2Kb para identificar las células H2db F1-Tmc. Gráfico de dispersión que muestra el porcentaje de células Tmc de CB6 en cada ratón, cada punto representa la población de células Tmc en un ratón que pertenece al grupo apropiado, mostrando la media y la DE de cada grupo.
La figura 5 es un gráfico que ilustra la calibración de la dosis de irradiación que permite la supervivencia de células Tmc sin TDBMT. Los ratones Balb/c (H2Dd) se irradiaron subletalmente con 2/4/5,5/6,5 Gy el día -1. El día 0, los ratones recibieron 5 x 106 o 9 x 106 células F1 CB6 (H-2db) Tmc transferidas adoptivamente en la vena de la cola de los ratones. Gráfico de dispersión que representa el porcentaje de células Tmc de CB6 en sangre completa periférica de ratones hospedadores Balb/c, analizada 42 días después del trasplante por FACS usando aH2Dd (Hospedador) y aH2Kb para identificar células H2db F1-Tmc. Se muestran la media y la De de cada grupo.
Las figuras 6A-B son gráficos que ilustran que los linfocitos Tmc completamente alogénicos persisten en ratones Balb/c 5,5 Gy durante un período prolongado. Los ratones Balb/c (H-2d) se trasplantaron como se describe en la figura 1C. Las células Tmc eran de origen CB6-F1 (H-2db) o C57BL/6(H-2b). Los ratones se sangraron en los días indicados y la población de células Tmc se analizó mediante FACS usando aH2Dd (Hospedador) y aH2Kb (Donante) para identificar células H2db F1-Tmc y H2b alo-Tmc. La Figura 6A es un diagrama de dispersión que representa el porcentaje de células Tmc en hospedadores Balb/c. Cada punto representa la población de células Tmc en un ratón que pertenece al grupo apropiado, mostrando la media y la DE de cada grupo. La figura 6B es un gráfico de curva de tiempo que ilustra la disminución de la población de células Tmc en sangre periférica durante un período de tiempo prolongado.
La figura 7 es un gráfico que ilustra que los linfocitos Tmc completamente alogénicos persisten en ratones Balb/c 5.5 Gy y facilitan el injerto de linfocitos T de donantes adicionales. Los ratones Balb/c (H-2d) recibieron 5,5 Gy TBI el día -1 y 5 x 106 células Tmc derivadas de CB6 (H-2db) o C57BL/6(H-2b) el día 0. 89 días después de la inyección de células Tmc, los ratones fueron irradiados con 2 Gy TBI, al día siguiente recibieron 2 x 106 células CD45.1+, OT1+, RAG+ CD8+. Gráfico de dispersión que muestra el sangrado el día 120 después del trasplante de células Tmc y 30 días después del trasplante de células OT-1.
La figura 8 es un gráfico que ilustra un análisis de células OT-1 en la sangre periférica de ratones Balb/c irradiados subletalmente. Los ratones Balb/c (H-2d) recibieron 5,25 Gy TBI el día -1 y seguidamente recibieron células vírgenes CD8+ OT-1+CD45.1+RAG' el día 0, con o sin células Tmc derivadas de CB6 (H-2db) o C57BL/6 (H-2b) en
los números indicados. Sesenta días después de la inyección de células Tmc, se analizó la sangre periférica de los ratones para detectar la presencia de células OT-1 usando análisis FACS. Gráfico de dispersión que muestra el porcentaje de células OT-1 de diferentes grupos de las células CD8+H-2b+ totales. La figura 9 es un gráfico que ilustra el injerto y la supervivencia de células Tmc preparadas a partir de ratones OT-1 trasplantados en un modelo de ratón Balb/c de acondicionamiento de intensidad reducida.
Descripción de aspectos específicos de la invención
La presente invención, en algunos aspectos de la misma, se refiere a la tolerancia modificada genéticamente que inducen los linfocitos T de memoria central transducidos para expresar un receptor de la superficie celular y, más particularmente, pero no exclusivamente, al uso de los mismos en inmunoterapia.
Los principios y el funcionamiento de la presente divulgación pueden entenderse mejor en referencia a los dibujos y las descripciones adjuntas.
Antes de explicar al menos un aspecto de la divulgación en detalle, ha de comprenderse que la divulgación no está limitada necesariamente en su aplicación a los detalles expuestos en la siguiente descripción o ejemplificados mediante los Ejemplos. La divulgación es susceptible de otros aspectos o de ponerse en práctica o de llevarse a cabo de diversas maneras. También, debe entenderse que la fraseología y la terminología empleadas en el presente documento son con fines descriptivos y no deberían interpretarse como limitantes.
Las terapias basadas en células con linfocitos y células presentadoras de antígenos son enfoques prometedores para la inmunoterapia. La transferencia de células adoptivas (ACT), incluida la transferencia de células de origen inmunitario, de una fuente autóloga o no autóloga ofrece el objetivo de transferir la funcionalidad y las características inmunológicas al hospedador. Un método empleado anteriormente para la ACT comprende linfocitos T modificados genéticamente (por ejemplo, que expresan un receptor de linfocitos T o un receptor de antígeno quimérico), en donde la especificidad de las células se redirige hacia el antígeno diana. Sin embargo, el problema del rechazo del injerto (por parte del receptor del trasplante) y/o la enfermedad del injerto contra el hospedador (por parte de las células trasplantadas) es un problema continuo que debe superarse para aprovechar el potencial terapéutico de estas células.
Al trasladar la presente divulgación a la práctica, los presentes inventores han descubierto que los linfocitos T de memoria central (Tmc) anti-terceros, que carecen de reactividad de injerto contra hospedador, están dotados de una actividad intrínseca inductora de tolerancia al veto y pueden inducir tolerancia por sí mismos, en ausencia de progenitores hematopoyéticos. Los presentes inventores descubrieron además que los linfocitos Tmc anti-terceros pueden modificarse genéticamente para expresar un receptor de linfocitos T (por ejemplo, un receptor de linfocitos T transgénico o un receptor de antígeno quimérico) y pueden usarse para combatir enfermedades a la vez que inducen actividad de veto y carecen de potencial injerto contra hospedador.
Como se muestra a continuación en el presente documento y en la sección de Ejemplos que sigue, los presentes inventores han demostrado que los linfocitos Tmc anti-terceros de tipo donante alogénico pueden sobrevivir en un hospedador durante un tiempo prolongado con o sin un trasplante de médula ósea concomitante (por ejemplo, más de 120 días, figuras 2A-B y figuras 6A-B, respectivamente). Además, los linfocitos Tmc anti-terceros ejercieron actividad de veto (figura 3). Por tanto, la aplicación de linfocitos Tmc anti-terceros solos (es decir, en ausencia de precursores de MO) ofrece una herramienta útil para la inmunoterapia, particularmente para dirigirse a antígenos tumorales, patógenos (por ejemplo, antígenos virales) y autoantígenos. Por tanto, estos resultados confirman aún más la modificación genética de los linfocitos Tmc anti-terceros tolerogénicos, de cualquier donante de células, para expresar funciones efectoras de linfocitos T heterólogas, dando como resultado un producto universal para la inmunoterapia dirigida a un antígeno patológico y evitando el rechazo del injerto y la enfermedad de injerto contra hospedador (EICH).
Consideradas en conjunto, estas células ofrecen la solución al estar desprovistas de potencial injerto contra hospedador, rechazo de injertos y direccionamiento a antígenos específicos, todo en una sola célula. Estas células eliminan la necesidad de usar células autólogas para el tratamiento o la necesidad de trasplantar células hematopoyéticas con las mismas. Además, estas células superan la necesidad de fabricar las terapias basadas en células "por paciente" y permiten la fabricación de un producto "listo para usar" para la terapia.
Por tanto, de acuerdo con un aspecto de la presente divulgación, se proporciona una célula aislada que tiene un fenotipo de linfocito T de memoria central (Tmc), siendo la célula, una célula inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, siendo transducida la célula para expresar un receptor de la superficie celular que comprende un módulo de señalización del receptor de linfocitos T.
Como se usa en el presente documento, la expresión "célula aislada" se refiere a una célula que se ha separado de su entorno natural (por ejemplo, de un tejido, por ejemplo, de un cuerpo humano).
La expresión "fenotipo de linfocito T de memoria central (Tmc)", como se usa en el presente documento, se refiere a un subconjunto de linfocitos T citotóxicos que anidan en los ganglios linfáticos. Las células que tienen el fenotipo T mc,
en los seres humanos, normalmente comprenden una firma CD3+/CD8+/CD62L+/CD45RO+/CD45RA. Se apreciará que los linfocitos Tmc pueden expresar todos los marcadores de la firma en una sola célula o pueden expresar solo una parte de los marcadores de la firma en una sola célula.
Un linfocito Tmc normalmente se aloja en los ganglios linfáticos después del trasplante.
De acuerdo con algunos aspectos, el linfocito Tmc de la presente divulgación puede anidar en cualquiera de los ganglios linfáticos después del trasplante, como, por ejemplo, los ganglios linfáticos periféricos y los ganglios linfáticos mesentéricos. La naturaleza de anidamiento de estas células les permite ejercer su efecto de tolerancia de manera rápida y eficiente.
La expresión "células inductoras de tolerancia", como se usa en el presente documento, se refiere a células que provocan una disminución de la capacidad de respuesta de las células del receptor (por ejemplo, los linfocitos T del receptor) cuando entran en contacto con las células del donante en comparación con la capacidad de respuesta de las células del receptor en ausencia de células inductoras de tolerancia. Las células inductoras de tolerancia incluyen células veto (es decir, linfocitos T que conducen a la apoptosis de los linfocitos T del hospedador al entrar en contacto con los mismos) como se describió previamente en las Publicaciones PCT números WO 2001/049243 y WO 2002/102971.
De acuerdo con un aspecto, los linfocitos Tmc de la divulgación también son células inductoras de no EICH.
La expresión "no EICH", como se usa en el presente documento, se refiere a tener una reactividad inductora de injerto contra hospedador sustancialmente reducida o nula. Por tanto, las células de la presente divulgación se generan de manera que no causen de manera significativa la enfermedad de injerto contra hospedador (EICH) como lo demuestra la supervivencia, peso y aspecto general del sujeto trasplantado 30-100 días después del trasplante.
De acuerdo con un aspecto, las células de la presente divulgación tienen al menos un 20 %, al menos un 30 %, al menos un 40 %, al menos un 50 %, al menos un 55 %, al menos un 60 %, al menos un 65 %, al menos un 70 %, al menos un 75 %, al menos un 80 %, al menos un 85 %, al menos un 90 %, al menos un 95 % o incluso un 100 % de menor reactividad contra un hospedador respecto al trasplante de linfocitos T que no son linfocitos Tmc anti-terceros.
De acuerdo con un aspecto, la célula de la presente divulgación que comprende un fenotipo Tmc está modificada genéticamente.
De acuerdo con un aspecto, la célula de la divulgación se transduce para expresar un receptor de la superficie celular que comprende un módulo de señalización del receptor de linfocitos T.
Como se usa en el presente documento, el término "transducido" se puede usar de forma intercambiable con los términos "transfectado" o "transformado" y se refiere a un proceso mediante el cual se transfiere o introduce un ácido nucleico exógeno (heterólogo) en una célula. Una célula "transfectada" o "transformada" o "transducida" es una que se ha transfectado, transformado o transducido con ácido nucleico exógeno. La célula incluye la célula primaria y su progenie, o líneas celulares de la misma.
La expresión "receptor de la superficie celular", como se usa en el presente documento, se refiere a una molécula sintética o recombinante presentada en una membrana celular que se une a un ligando (p. ej., un antígeno) y media en la activación de la célula.
El término "antígeno" o "Ag" como se usa en el presente documento, se define como una molécula que provoca una respuesta inmunitaria. El experto en la materia entenderá que cualquier macromolécula, incluyendo virtualmente todas las proteínas o péptidos, así como los hidratos de carbono, los lípidos y el ADN pueden servir como antígeno.
De acuerdo con algunos aspectos de la invención, el antígeno está asociado con una enfermedad maligna, es decir, antígeno tumoral (p. ej., antígeno específico de tumor o un antígeno asociado a tumor), un antígeno de proteína viral, un antígeno de proteína bacteriana, un antígeno de proteína fúngica, antígenos asociados con una reacción alérgica (es decir, antígenos alérgicos) o un antígeno asociado autoinmunitario (por ejemplo, un antígeno "propio"), como se describe con más detalle a continuación.
El receptor de la superficie celular de la divulgación comprende un módulo de señalización del receptor de linfocitos T.
La expresión "módulo de señalización del receptor de linfocitos T" se refiere a una porción intracelular del receptor responsable de la activación de al menos una de las funciones efectoras normales del linfocito T en el que se ha colocado el receptor. Las funciones efectoras normales de un linfocito T pueden incluir, por ejemplo, secreción de citocinas inmunoestimuladoras (por ejemplo, IFN-gamma, IL-2, TNF-alfa), citotoxicidad específica de antígeno y proliferación celular. Por tanto, el módulo de señalización del receptor de linfocitos T de la divulgación se refiere a la parte de una proteína que transduce la señal de función efectora y dirige a la célula para que realice una función
especializada.
De acuerdo con un aspecto, el receptor de la superficie celular comprende un receptor de linfocitos T transgénico (tg-TCR) o un receptor de antígeno quimérico (CAR).
Como se usa en el presente documento, la expresión "receptor de linfocitos T transgénico" o "tg-TCR" se refiere a una molécula recombinante o sintética que comprende la especificidad de un receptor de linfocitos T (TCR), es decir, reconocimiento de péptidos antigénicos (es decir, antígeno) presentados por proteínas del complejo principal de histocompatibilidad (MHC).
El tg-TCR de la divulgación normalmente comprende dos cadenas (es decir, cadenas polipeptídicas), tal como, una cadena alfa de un receptor de linfocitos T (TCR), una cadena beta de un TCR, una cadena gamma de un TCR, una cadena delta de un TCR, o una combinación de las mismas (por ejemplo, cadenas ap o cadenas y6). Los polipéptidos del tg-TCR pueden comprender cualquier secuencia de aminoácidos, con la condición de que el tg-TCR tenga especificidad antigénica y funciones efectoras de linfocitos T como se ha descrito anteriormente en el presente documento. Se apreciará que la especificidad del antígeno está determinada por el heterodímero de TCR (es decir, por las cadenas ap o y6).
Se apreciará que cada una de las dos cadenas se compone normalmente de dos dominios extracelulares, es decir, la región variable (V) y la región constante (C).
De acuerdo con un aspecto, el tg-TCR comprende las regiones variables de un TCR. De acuerdo con un aspecto específico, el tg-TCR comprende las regiones variables de las cadenas a y p de un TCR. De acuerdo con otro aspecto específico, el tg-TCR comprende las regiones variables de las cadenas y y 6 de un TCR.
De acuerdo con algunos aspectos de la invención, la región variable del tg-TCR comprende regiones determinantes de la complementariedad (CDR) que son capaces de unirse específicamente al antígeno. Las CDR se pueden seleccionar de cualquiera de CDR1, CDR2, c DR3 y/o CDR4. De acuerdo con un aspecto específico, las CDR están presentes en una sola cadena, preferentemente, las CDR están presentes en ambas cadenas del tg-TCR.
De acuerdo con un aspecto, el tg-TCR comprende las regiones constantes de un TCR. De acuerdo con un aspecto específico, el tg-TCR comprende las regiones constantes de las cadenas a y p de un TCR. De acuerdo con otro aspecto específico, el tg-TCR comprende las regiones constantes de las cadenas y y 6 de un TCR.
Para evitar la formación de dímeros mixtos entre los TCR endógenos (es decir, los TCR que se originan dentro de la célula transducida) y las cadenas de tg-TCR, el tg-TCR de la divulgación puede comprender la región constante de un TCR murino (por ejemplo, de ratón). Otro enfoque que se puede utilizar para aumentar el emparejamiento específico de cadenas del tg-TCR es introducir residuos de cisteína adicionales dentro de la región constante de las cadenas del tg-TCR (por ejemplo, cadenas a y p), esto da como resultado la formación de un enlace disulfuro adicional. De forma alternativa, pueden introducirse inversiones mutacionales de los aminoácidos críticos que interactúan en las regiones constantes de la cadena del tg-TCR (p. ej., cadena a y p) que favorecen el emparejamiento de las cadenas del tg-TCR y también aumentan la reactividad del tg-TCR. Como alternativa o adicionalmente, la regulación a la baja del TCR endógeno puede implementarse utilizando, por ejemplo, ARN de interferencia pequeño (ARNip) que se utiliza para regular a la baja específicamente el TCR endógeno. Para más detalles, véase por ej. Zhang y Morgan, Adv Drug Deliv Rev. (2012) 64(8): 756-762.
Tal como se ha mencionado, el tg-TCR reconoce un antígeno de manera dependiente del MHC.
Como se usa en el presente documento, la expresión "complejo principal de histocompatibilidad" o "MHC" se refiere a un complejo de antígenos codificados por un grupo de loci vinculados, que se denominan colectivamente H-2 en el ratón y antígeno leucocitario humano (HLA) en humanos. Las dos clases principales de antígenos MHC, clase I y clase II, comprenden cada uno un conjunto de glicoproteínas de la superficie celular que desempeñan un papel en la determinación del tipo de tejido y la compatibilidad del trasplante.
Las principales moléculas del MHC de clase I se contemplan en el presente documento.
Las moléculas del complejo mayor de histocompatibilidad (MHC) de clase I se expresan en la superficie de casi todas las células. Estas moléculas funcionan presentando péptidos que se derivan principalmente de proteínas sintetizadas endógenamente a los linfocitos T CD8+ a través de una interacción con la cadena ap del receptor de linfocitos T. En los seres humanos, hay varios haplotipos del MHC, tal como, por ejemplo, HLA-A2, HLA-A1, h La -A3, HLA-A24, HLA-A28, HLA-A31, HLA-A33, HLA-A34, HLA-B7, HLA-B45 y HLA-Cw8, cuyas secuencias se pueden encontrar en la base de datos kabbat, en www.immuno(dot)bme(dot)nwu(dot)edu. Se puede encontrar más información sobre los haplotipos del MHC en Paul, B. Fundamental Immunology Lippincott-Raven Press.
La elección del tg-TCR depende del tipo y número de antígenos que definen la superficie de una célula diana. Por ejemplo, el tg-TCR puede elegirse para reconocer un antígeno que actúa como un marcador de superficie celular en
una célula diana asociada con una patología particular. Por tanto, por ejemplo, los marcadores de superficie celular que pueden actuar como antígenos para el reconocimiento por parte del tg-TCR pueden incluir aquellos asociados con infecciones víricas, bacterianas y parasitarias, enfermedad autoinmunitaria y células cancerosas. A continuación, se proporcionan ejemplos.
Para generar un tg-TCR con éxito, primero se debe identificar una secuencia diana apropiada. Por consiguiente, un TCR puede aislarse de un linfocito T reactivo al antígeno (p. ej., linfocito T reactivo al tumor) o, cuando esto no es posible, pueden emplearse tecnologías alternativas. De acuerdo con un aspecto a modo de ejemplo, un animal transgénico (p. ej., conejo o ratón, preferentemente un ratón transgénico para HLA humano) se inmuniza con péptidos de antígenos humanos (p. ej., antígenos tumorales o virales) para generar linfocitos T que expresan TCR contra los antígenos humanos [como se describe, p. ej., en Stanislawski et al., Nat Immunol. (2001) 2(10):962-70]. De acuerdo con otro aspecto a modo de ejemplo, los linfocitos T específicos de antígeno (p. ej., linfocitos T específicos de tumor) se aíslan de un paciente que experimenta remisión de la enfermedad (p. ej., tumor) y las secuencias del TCR reactivo se aíslan del mismo [como se describe, p. ej., en de Witte et al., Blood (2006) 108(3):870].
De acuerdo con otro aspecto a modo de ejemplo, se emplean tecnologías in vitro para alterar la secuencia de un TCR existente para potenciar la avidez de un t Cr específico de antígeno débilmente reactivo con un antígeno diana (dichos métodos se describen a continuación).
De acuerdo con un aspecto, el tg-TCR de la divulgación se selecciona para reconocer el complejo péptido antigénico-HLA con gran avidez (es decir, la fuerza física de la interacción monomérica entre el TCR y un complejo péptido-MHC).
La producción de células con una alta avidez funcional (es decir, que respondan eficazmente a los antígenos) se puede lograr utilizando cualquier método conocido por un experto en la materia. Por tanto, de acuerdo con un ejemplo, el aumento de la avidez del tg-TCR se logra aumentando la afinidad (es decir, la fuerza de unión de un TCR a su ligando) del tg-TCR o aumentando la expresión del tg-TCR en la superficie celular. De acuerdo con un aspecto ilustrativo, el aumento de la afinidad por el TCR se lleva a cabo mediante la modificación de los genes del tg-TCR. Por ejemplo, una posible modificación de los genes del tg-TCR incluye modificaciones en una región determinante de la complementariedad (CDR), p.ej. tercera CDR (CDR3), del tg-TCR. Por consiguiente, se pueden utilizar sustituciones de aminoácidos simples o dobles en las cadenas de la CDR (por ejemplo, cadenas a o p) para aumentar la afinidad del tg-TCR y potenciar la reactividad específica de antígeno en las células transducidas. De acuerdo con otro aspecto a modo de ejemplo, el aumento de la avidez funcional del tg-TCR se lleva a cabo mediante la eliminación de motivos de N-glicosilación definidos en los dominios constantes de las cadenas del tg-TCR. De acuerdo con otro aspecto a modo de ejemplo, el aumento de la afinidad se lleva a cabo mediante optimización de codones.
Por consiguiente, los codones raros del tg-TCR son reemplazados por codones distribuidos con mayor frecuencia en genes humanos altamente expresados. Durante el proceso de optimización, los estiramientos de secuencia rica en AT o GC que actúan en cis, también se pueden eliminar motivos de inestabilidad de ARN y corte y empalme críptico. Para información adicional, véase por ej. Zhang y Morgan, Adv Drug Deliv Rev. (2012), supra.
De acuerdo con un aspecto, el módulo de señalización del tg-TCR puede comprender una sola subunidad o una pluralidad de unidades de señalización. Por consiguiente, el tg-TCR de la divulgación puede usar correceptores que actúan en concierto con un TCR para iniciar la transducción de señales después de la interacción con el receptor del antígeno, así como cualquier derivado o variante del mismo que tenga la misma capacidad funcional.
De acuerdo con un aspecto, el módulo de señalización de TCR comprende el complejo CD3 (p. ej., cadenas de CD3, p. ej., cadenas CD38/e, CD3y/£ y/o zeta, p. ej., Z/Z o Z/n).
Adicionalmente, o como alternativa, el módulo de señalización de TCR puede comprender receptores de proteínas coestimuladoras para proporcionar señales adicionales al linfocito T. Estos se analizan en detalle a continuación.
De acuerdo con un aspecto, el tg-TCR puede comprender un dominio transmembrana como se describe en detalle a continuación.
Los métodos para transducir una célula con un TCR se describen en detalle a continuación.
Como se usa en el presente documento, la expresión "receptor de antígeno quimérico (CAR)" se refiere a una molécula recombinante o sintética que combina la especificidad por un antígeno deseado con un dominio intracelular activador del receptor de linfocitos T (es decir, módulo de señalización del receptor de linfocitos T) para generar una proteína quimérica que presenta actividad inmunitaria celular al antígeno específico.
Por tanto, el CAR de la divulgación generalmente comprende un dominio extracelular que comprende un resto de unión a antígeno, un dominio transmembrana y un dominio intracelular (es decir, el dominio citoplasmático) que se requiere para una respuesta eficaz del linfocito T al antígeno.
Resto de unión a antígeno
En un aspecto, el CAR de la divulgación comprende un elemento de unión específico a la diana, también denominado resto de unión a antígeno. La elección del resto depende del tipo y número de ligandos (es decir, antígenos) que definen la superficie de una célula diana. Por ejemplo, el dominio de unión a antígeno puede elegirse para reconocer un ligando (es decir, un antígeno) que actúa como un marcador de la superficie celular en células diana con una patología particular. Por lo tanto, los ejemplos de marcadores de la superficie celular que pueden actuar como ligandos para el dominio del resto de unión a antígeno en el CAR de la divulgación incluyen aquellos asociados con infecciones víricas, bacterianas y parasitarias, enfermedad autoinmunitaria y células cancerosas.
De acuerdo con algunos aspectos de la invención, el resto de unión al anticuerpo comprende regiones determinantes de la complementariedad (CDR) que son capaces de unirse específicamente al antígeno. Tales CDR se pueden obtener a partir de un anticuerpo.
El término "anticuerpo" como se usa en la presente divulgación incluye moléculas intactas, así como fragmentos funcionales de las mismas, tales como Fab, Fab', F(ab')2, Fv, anticuerpos lineales, anticuerpos scFv y anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpos que son capaces de unirse al antígeno. Estos fragmentos de anticuerpos funcionales se definen como se indica a continuación: (1) Fab, el fragmento que contiene un fragmento de unión a antígeno monovalente de una molécula de anticuerpo, puede producirse mediante la digestión del anticuerpo completo con la enzima papaína para producir una cadena ligera intacta y una porción de una cadena pesada; (2) Fab', el fragmento de una molécula de anticuerpo que puede obtenerse tratando el anticuerpo completo con pepsina, seguido de reducción, para producir una cadena ligera inalterada y una porción de la cadena pesada; se obtienen dos fragmentos Fab' por molécula de anticuerpo; (3) (Fab')2, el fragmento del anticuerpo que se puede obtener tratando el anticuerpo completo con la enzima pepsina sin reducción posterior; F(ab')2 es un dímero de dos fragmentos Fab' unidos entre sí por dos enlaces disulfuro; (4) Fv, definido como un fragmento modificado genéticamente que contiene la región variable de la cadena ligera y la región variable de la cadena pesada expresadas como dos cadenas; (5) Anticuerpo monocatenario ("SCA"), una molécula modificada genéticamente que contiene la región variable de la cadena ligera y la región variable de la cadena pesada, unidas por un enlazador polipeptídico adecuado tal como una molécula monocatenaria fusionada genéticamente; (6) el péptido CDR es un péptido que codifica una sola región determinante de la complementariedad (CDR); y (7) anticuerpos de un solo dominio (también llamados nanocuerpos), un dominio de anticuerpo variable monomérico único modificado genéticamente que se une selectivamente a un antígeno específico. Los nanocuerpos tienen un peso molecular de solo 12-15 kDa, que es mucho más pequeño que un anticuerpo común (150-160 kDa).
Una "cadena pesada de anticuerpo", como se usa en el presente documento, se refiere al mayor de los dos tipos de cadenas polipeptídicas presentes en todas las moléculas de anticuerpo en sus conformaciones naturales.
Una "cadena ligera de anticuerpo", como se usa en el presente documento, se refiere al más pequeño de los dos tipos de cadenas polipeptídicas presentes en todas las moléculas de anticuerpo en sus conformaciones naturales. Las cadenas ligeras kappa y lambda se refieren a los dos isotipos principales de cadena ligera de los anticuerpos.
Por la expresión "anticuerpo sintético", como se usa en el presente documento, se entiende un anticuerpo que se genera utilizando tecnología de ADN recombinante, tal como, por ejemplo, un anticuerpo expresado por un bacteriófago como se describe en el presente documento. La expresión también debe interpretarse en el sentido de un anticuerpo que se ha generado mediante la síntesis de una molécula de ADN que codifica el anticuerpo, molécula de ADN que expresa una proteína de anticuerpo, o una secuencia de aminoácidos que especifica el anticuerpo, en donde la secuencia de aminoácidos o ADN se ha obtenido utilizando tecnología de secuencia de aminoácidos o ADN sintética que está disponible y se conoce bien en la técnica.
Los métodos para producir anticuerpos policlonales y monoclonales, así como fragmentos de los mismos, son bien conocidos en la técnica (véase, por ejemplo, Harlow y Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, Nueva York, 1988).
Los fragmentos de anticuerpo de acuerdo con la presente divulgación se pueden preparar mediante hidrólisis proteolítica del anticuerpo o mediante expresión en células de E. coli o de mamífero (p. ej., cultivo de células de ovario de hámster chino u otros sistemas de expresión de proteínas) del ADN que codifica el fragmento. Los fragmentos de anticuerpo pueden obtenerse mediante digestión con pepsina o papaína de anticuerpos completos mediante métodos convencionales. Por ejemplo, se pueden producir fragmentos de anticuerpos por escisión enzimática de anticuerpos con pepsina para proporcionar un fragmento 5S denominado F(ab')2. Este fragmento puede escindirse adicionalmente usando un agente reductor de tiol y opcionalmente un grupo de bloqueo para los grupos sulfhidrilo resultantes de la escisión de enlaces disulfuro, para producir fragmentos monovalentes Fab' 3,5S. De forma alternativa, una escisión enzimática con pepsina produce dos fragmentos Fab' monovalentes y un fragmento Fc directamente. Estos métodos se describen, por ejemplo, por Goldenberg, en las patentes de Estados Unidos N.° 4.036.945 y 4.331.647 y las referencias contenidas en las mismas. Véase también Porter, R. R. [Biochem. J. 73: 119-126 (1959)]. También pueden usarse otros métodos para escindir anticuerpos, tales como la separación de cadenas pesadas para formar fragmentos monovalentes de cadenas ligeras y pesadas, la escisión adicional de fragmentos u otras técnicas enzimáticas, químicas o genéticas, siempre que los fragmentos se unan al antígeno que es reconocido por el anticuerpo intacto.
Los fragmentos Fv comprenden una asociación de cadenas VH y VL. Esta asociación puede ser no covalente, como se describe en Inbar et al. [Proc. Nat'l Acad. Sci. USA 69:2659-62 (19720]. De forma alternativa, las cadenas variables pueden estar unidas por un enlace disulfuro intermolecular o reticuladas por productos químicos tal como el glutaraldehído. Preferentemente, los fragmentos Fv comprenden cadenas VH y Vl conectadas por un enlazador peptídico. Se prepararon estas proteínas de unión a antígeno monocatenarias (sFv) construyendo un gen estructural que comprende secuencias de ADN que codifican los dominios VH y VL conectados mediante un oligonucleótido. El gen estructural se inserta en un vector de expresión, que posteriormente se introduce en una célula hospedadora tal como E. coli. Las células hospedadoras recombinantes sintetizan una única cadena polipeptídica con un péptido enlazador que une los dos dominios V. Se describen métodos para producir sFv, por ejemplo, por [Whitlow y Filpula, Methods 2: 97-105 (1991); Bird et al., Science 242:423-426 (1988); Pack et al., Bio/Technology 11:1271-77 (1993); y la Patente de Estados Unidos N.° 4.946.778.
Los péptidos de CDR ("unidades de reconocimiento mínimas") se pueden obtener mediante la construcción de genes que codifican la CDR de un anticuerpo de interés. Dichos genes se preparan, por ejemplo, mediante el uso de la reacción en cadena de la polimerasa para sintetizar la región variable a partir del ARN de células productoras de anticuerpos. Véase, por ejemplo, Larrick y Fry [Métodos, 2: 106-10 (1991)].
Una vez que se identifican las CDR de un anticuerpo, usando técnicas convencionales de ingeniería genética, los polinucleótidos expresables que codifican cualquiera de las formas o fragmentos de anticuerpos descritos en el presente documento se pueden sintetizar y modificar de una de muchas maneras para producir un espectro de productos relacionados.
De acuerdo con algunos aspectos de la invención, las CDR se derivan de un receptor de linfocitos T (TCR) ap que se une específicamente al antígeno.
De acuerdo con algunos aspectos de la invención, las CDR se derivan de un receptor de linfocitos T (TCR) y§ que se une específicamente al antígeno.
De acuerdo con algunos aspectos de la invención, las CDR se derivan de un receptor de linfocitos T (TCR) ap o receptor de linfocitos T y§ modificado con afinidad mejorada que se une específicamente al antígeno (como se ha descrito en detalle anteriormente en el presente documento).
De acuerdo con algunos aspectos de la invención, las CDR se derivan de un receptor de linfocitos T (TCR) ap o un receptor de linfocitos T y§ modificado con estabilidad mejorada o cualquier otra propiedad biofísica.
De acuerdo con algunos aspectos de la invención, las CDR se derivan de un anticuerpo similar al receptor de linfocitos T (TCRL) que se une específicamente al antígeno. Los ejemplos de TCRL y métodos para generar los mismos se describen en los documentos WO03/068201, WO2008/120203, WO2012/007950, WO2009125395, WO2009/125394.
De acuerdo con algunos aspectos de la invención, el dominio de unión a antígeno comprende una molécula Fv monocatenaria (scFv).
Dominio citoplasmático
El dominio citoplásmico (también denominado "dominio de señalización intracelular" o "módulo de señalización del receptor de linfocitos T") de la molécula CAR de la divulgación es responsable de la activación de al menos una de las funciones efectoras normales de la célula en la que se ha colocado el CAR.
Si bien habitualmente se puede emplear todo el dominio de señalización intracelular, en muchos casos no es necesario usar toda la cadena. En la medida en que se use una porción truncada del dominio de señalización intracelular, dicha porción truncada puede usarse en lugar de la cadena intacta siempre que transduzca la señal de función efectora. La expresión dominio de señalización intracelular pretende incluir cualquier porción truncada del dominio de señalización intracelular suficiente para transducir la señal de la función efectora.
Los ejemplos preferidos de dominios de señalización intracelular para su uso en la molécula CAR de la divulgación incluyen las secuencias citoplásmicas del receptor de linfocitos T (TCR) y los correceptores que actúan en concierto para iniciar la transducción de señales después del acoplamiento del receptor del antígeno, así como cualquier derivado o variante de estas secuencias y cualquier secuencia sintética que tenga la misma capacidad funcional.
Se sabe que las señales generadas a través del TCR solo son insuficientes para la activación completa del linfocito T y que también se requiere una señal secundaria o coestimuladora. Por tanto, la activación de los linfocitos T puede estar mediada por dos clases distintas de secuencias de señalización citoplasmática: las que inician la activación primaria dependiente de antígeno a través del TCR (secuencias de señalización citoplasmáticas primarias) y las que actúan de forma independiente del antígeno para proporcionar una señal secundaria o coestimuladora (secuencias de señalización citoplásmicas secundarias).
Las secuencias de señalización citoplásmica primarias regulan la activación primaria del complejo TCR ya sea de forma estimuladora o de forma inhibidora. Las secuencias de señalización citoplásmicas primarias que actúan de manera estimulante pueden contener motivos de señalización que se conocen como motivos de activación del inmunorreceptor basados en tirosina (ITAM).
Los ejemplos de ITAM que contienen secuencias de señalización citoplasmáticas primarias que son de uso particular en la divulgación incluyen los derivados de TCR zeta, FcR gamma, FcR beta, CD3 gamma, CD3 delta, CD3 épsilon, CD5, CD22, CD79a, CD79b y CD66d. Se prefiere particularmente que la molécula de señalización citoplasmática en el CAR de la divulgación comprenda una secuencia de señalización citoplasmática derivada de CD3 zeta.
En un aspecto preferido, el dominio citoplasmático del CAR puede diseñarse para comprender el dominio de señalización de CD3-zeta por sí mismo o combinado con cualquier otro dominio citoplasmático deseado útil en el contexto del CAR de la invención. Por ejemplo, el dominio citoplasmático del CAR puede comprender una porción de la cadena CD3 zeta y una región de señalización coestimuladora. La región de señalización coestimuladora se refiere a una porción del CAR que comprende el dominio intracelular de una molécula coestimuladora. Una molécula coestimuladora es una molécula de superficie celular distinta de un receptor de antígeno o sus ligandos que es necesaria para una respuesta eficiente de los linfocitos a un antígeno. Los ejemplos de dichas moléculas incluyen CD27, Cd 28, 4-1BB (CD137), OX40, CD30, CD40, PD-1, ICOS, antígeno-1 asociado a la función de linfocitos (LFA-1), CD2, CD7, LIGh T, NKg 2c , B7-H3 y un ligando que se une específicamente con CD83 y similares. Por tanto, mientras que la divulgación se ilustra principalmente con 4-1BB como elemento de señalización coestimulador, otros elementos coestimuladores están dentro del alcance de la invención.
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende una región de señalización coestimuladora y una porción de cadena zeta. La región de señalización coestimuladora se refiere a una porción de la molécula de CAR que comprende el dominio intracelular de una molécula coestimuladora. Las moléculas coestimuladoras son moléculas de la superficie celular distintas de los receptores antigénicos o sus ligandos que se requieren para una respuesta eficiente de los linfocitos al antígeno.
"Ligando coestimulador", como se usa la expresión en el presente documento, incluye una molécula en una célula presentadora de antígeno [por ejemplo, una aAPC (célula presentadora de antígeno artificial), célula dendrítica, linfocito B, y similares] que se une específicamente a una molécula coestimuladora afín en un linfocito T, proporcionando de este modo una señal que, además de la señal primaria proporcionada mediante, por ejemplo, la unión de un complejo de TCR/CD3 con una molécula del MHC cargada con péptido, media una respuesta de linfocitos T, que incluyen, aunque no de forma limitativa, proliferación, activación, diferenciación y similares. Un ligando coestimulador puede incluir, pero sin limitación, c D7, B7-1 (CD80), B7-2 (CD86), PD-L1, Pd -L2, 4-1BBL, OX40L, ligando coestimulador inducible (ICOS-L), molécula de adhesión intercelular (ICAM, CD30L, CD40, CD70, CD83, HLA-G, MICA, MICB, HVEM, receptor de linfotoxina beta, 3/TR6, ILT3, ILT4, HVEM, un agonista o anticuerpo que se une con el receptor de ligando Toll y un ligando que se une específicamente con B7-H3. Un ligando coestimulador también abarca, entre otros, un anticuerpo que se une específicamente con una molécula coestimuladora presente en un linfocito T, tal como, aunque no de forma limitativa, CD27, CD28, 4-1BB, OX40, CD30, CD40, PD-1, ICOS, antígeno-1 asociado a la función de linfocitos (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3 y un ligando que se une específicamente con CD83.
Una "molécula coestimuladora" se refiere al compañero de unión afín en un linfocito T que se une específicamente con un ligando coestimulador, mediando de ese modo una respuesta coestimuladora por el linfocito T, tal como, aunque no de forma limitativa, proliferación. Las moléculas coestimuladoras incluyen, pero sin limitación, una molécula del MHC de clase I, BTLA y un receptor de ligando Toll.
Una "señal coestimuladora", como se usa en el presente documento, se refiere a una señal, que, en combinación con la señal primaria, tal como el ligamiento de TCR/CD3, conduce a la proliferación de linfocitos T y/o la regulación al alza o a la baja de las moléculas clave.
Por el término "estimulación", se entiende una respuesta primaria inducida por la unión de una molécula estimuladora (p. ej., un complejo TCR/CD3) con su ligando afín, mediando de este modo un acontecimiento de transducción de señal, tal como, aunque no de forma limitativa, transducción de señales a través del complejo TCR/CD3. La estimulación puede mediar en la expresión alterada de ciertas moléculas, tales como la regulación a la baja de TGF-p y/o la reorganización de las estructuras del citoesqueleto y similares.
Una "molécula estimuladora", como se usa la expresión en el presente documento, significa una molécula en un linfocito T que se une específicamente con un ligando estimulador afín presente en una célula presentadora de antígeno.
Un "ligando estimulador", como se usa en el presente documento, significa un ligando que cuando está presente en una célula presentadora de antígeno (por ejemplo, una aAPC, una célula dendrítica, un linfocito B y similares) puede unirse específicamente con un compañero de unión afín (denominado en el presente documento como una "molécula
estimuladora") en un linfocito T, mediando de ese modo una respuesta primaria por el linfocito T, que incluyen, aunque no de forma limitativa, activación, inicio de una respuesta inmunitaria, proliferación y similares. Los ligandos estimulantes son bien conocidos en la técnica y abarcan, entre otros, una molécula MHC de clase I cargada con un péptido, un anticuerpo anti-CD3, un anticuerpo superagonista anti-CD28 y/o un anticuerpo superagonista anti-CD2.
Con respecto al dominio citoplasmático, la molécula CAR de algunos aspectos de la divulgación puede diseñarse para comprender el dominio de señalización CD28 y/o 4-1BB por sí mismo o combinarse con cualquier otro dominio o dominios citoplasmáticos deseados útiles en el contexto de la molécula CAR de algunos aspectos de la invención. En un aspecto, el dominio citoplasmático del CAR puede diseñarse para comprender además el dominio de señalización de CD3-zeta. Por ejemplo, el dominio citoplasmático del CAR puede incluir, entre otros, los módulos de señalización CD3-zeta, 4-1BB y CD28 y combinaciones de los mismos.
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende al menos uno, por ejemplo, al menos dos, al menos tres, al menos cuatro, al menos cinco, por ejemplo, al menos seis de los polipéptidos seleccionados del grupo que consiste en: CD3Z (CD247, CD3z), CD27, CD28, 4-1BB/CD137, ICOS, OX40/CD134, DAP10, receptor del factor de necrosis tumoral (TNFr) y Lsk.
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende la cadena CD3Z [molécula CD247, también conocidos como "CD3-ZETA" y "CD3z", Números de acceso de GenBank: NP_000725.1 y NP_932170.1], que es el principal transmisor de señales de los TCR endógenos.
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende varios receptores proteicos coestimuladores de la cola citoplásmica del CAR para proporcionar señales adicionales al linfocito T (CAR de "segunda generación"). Los ejemplos incluyen, pero sin limitación, CD28 [por ejemplo, los números de acceso de GenBank NP_001230006.1, NP_001230007.1, Np_006130.1], 4-1BB [superfamilia del receptor del factor de necrosis tumoral, miembro 9 (TNFRSF9), también conocido como "CD137", por ejemplo, n.° de acceso de GenBank NP_001552.2, ICOS [coestimulador inducible de linfocitos T, por ejemplo, n.° de acceso de GenBank NP_036224.1, DAP10 [transductor de señal de células hematopoyéticas, por ejemplo, los números de acceso de GenBank NP_001007470, NP_055081.1] y Lsk [protooncogén LCK, tirosina cinasa de la familia Src, por ejemplo, los números de acceso de GenBank NP_001036236.1, NP_005347.3]. Los estudios preclínicos han indicado que la segunda generación de diseños de los CAR mejora la actividad antitumoral de los linfocitos T.
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende múltiples dominios de señalización, tales como CD3z-CD28-4-1BB o CD3z-CD28-OX40, para aumentar aún más la potencia. El término "OX40" se refiere a la superfamilia de receptores del factor de necrosis tumoral, miembro 4 (TNFRSF4), por ejemplo, N.° de acceso de GenBank NP_003318.1 (CAR de "tercera generación").
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende CD28-CD3z, CD3z, CD28-CD137-CD3z. El término "CD137" se refiere a la superfamilia de receptores del factor de necrosis tumoral, miembro 9 (TNFRSF9), por ejemplo, N.° de referencia de GenBank NP_001552.2.
De acuerdo con algunos aspectos de la invención, el dominio intracelular comprende CD3z, CD28 y un receptor del factor de necrosis tumoral (TNFr).
De acuerdo con algunos aspectos de la invención, el CAR comprende una cadena zeta CD3.
De acuerdo con algunos aspectos de la invención, el CAR comprende al menos un dominio coestimulador seleccionado del grupo que consiste en CD28, CD134/OX40, CD137/4-1BB, Lck, ICOS y DAP10.
De acuerdo con algunos aspectos de la invención, el CAR comprende al menos dos dominios coestimuladores seleccionados del grupo que consiste en CD28, CD134/OX40, CD137/4-1 BB, Lck, ICOS y DAP10.
Dominio transmembrana
El dominio transmembrana del CAR puede proceder de una fuente natural o sintética. Cuando la fuente es natural, el dominio puede derivar de cualquier proteína unida a la membrana o transmembrana. Las regiones transmembrana de uso particular en esta divulgación pueden derivar de (es decir, comprender al menos la región o regiones transmembrana de) la cadena alfa, beta o zeta del receptor de linfocitos T, CD28, CD3 épsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154. Como alternativa, el dominio transmembrana puede ser sintético, en cuyo caso comprenderá predominantemente restos hidrófobos tales como leucina y valina. Preferentemente se encontrará un triplete de fenilalanina, triptófano y valina en cada extremo de un dominio transmembrana sintético.
Opcionalmente, un enlazador de oligopéptido o polipeptídico corto, preferentemente entre 2 y 10 aminoácidos de longitud puede formar el enlace entre el dominio transmembrana y el dominio de señalización citoplasmático del CAR. Un doblete glicina-serina proporciona un enlazador en particular adecuado.
De acuerdo con algunos aspectos de la invención, el dominio transmembrana comprendido en la molécula CAR de algunos aspectos de la divulgación es un dominio transmembrana que está asociado naturalmente con uno de los dominios del CAR. De acuerdo con algunos aspectos de la invención, el dominio transmembrana se puede seleccionar o modificar mediante sustitución de aminoácidos para evitar la unión de dichos dominios a los dominios transmembrana de las mismas o diferentes proteínas de membrana de la superficie para minimizar las interacciones con otros miembros del complejo de receptor.
De acuerdo con algunos aspectos de la invención, el dominio transmembrana es el dominio bisagra CD8a.
De acuerdo con algunos aspectos, entre el dominio extracelular y el dominio transmembrana de la molécula CAR, o entre el dominio citoplasmático y el dominio transmembrana de la molécula CAR, puede haber incorporado un dominio espaciador. Como se usa en el presente documento, la expresión "dominio espaciador" generalmente significa cualquier oligo o polipéptido que funcione para unir el dominio transmembrana a, ya sea el dominio extracelular o, el dominio citoplasmático en la cadena polipeptídica. Un dominio espaciador puede comprender hasta 300 aminoácidos, preferentemente de 10 a 100 aminoácidos y lo más preferentemente de 25 a 50 aminoácidos.
Tal como se ha mencionado, el receptor de la superficie celular de la célula de la divulgación (p. ej., tg-TCR y/o CAR) se une a un antígeno (p. ej., en una célula diana).
De acuerdo con un aspecto, el antígeno puede comprender un antígeno asociado a un tumor, un antígeno viral, un antígeno bacteriano, un antígeno fúngico, un antígeno protozoario, un antígeno del parásito, un antígeno asociado a alergia y/o un antígeno autoinmunitario.
Como se usa en el presente documento, la expresión "antígeno tumoral" se refiere a un antígeno que es común a trastornos hiperproliferativos específicos tales como el cáncer. Los antígenos tumorales son proteínas producidas por las células tumorales que provocan una respuesta inmunitaria, particularmente las respuestas inmunitarias mediadas por linfocitos T. La selección del resto de unión a antígeno de la divulgación dependerá del tipo particular de cáncer a tratar.
De acuerdo con un aspecto, el antígeno tumoral está asociado con un tumor sólido.
De acuerdo con un aspecto, el antígeno tumoral está asociado con una neoplasia hematológica.
El tipo de antígeno tumoral al que se hace referencia en la divulgación incluye un antígeno específico de tumor (TSA) o un antígeno asociado a tumor (TAA). Un "TSA" se refiere a una proteína o antígeno polipeptídico exclusivo de las células tumorales y que no se encuentra en otras células del cuerpo. Un "TAA" se refiere a una proteína o antígeno polipeptídico que es expresado por una célula tumoral. Por ejemplo, un TAA puede ser una o más proteínas o polipéptidos de superficie, proteínas nucleares o glicoproteínas, o fragmentos de las mismas, de una célula tumoral.
Los antígenos descritos en este documento se incluyen simplemente a modo de ejemplo. No se pretende que la lista sea exclusiva y los ejemplos adicionales serán fácilmente evidentes para los expertos en la materia.
Los antígenos tumorales son bien conocidos en la técnica e incluyen, por ejemplo, un antígeno asociado a glioma, antígeno carcinoembrionario (CEA), gonadotropina coriónica humana p, alfafetoproteína (AFP), AFP sensible a lectina, tiroglobulina, RAGE-1, MN-CAIX, transcriptasa inversa telomerasa humana, RU1, RU2 (AS), carboxilo esterasa intestinal, mut hsp70-2, M-CSF, prostasa, antígeno específico de la próstata (PSA), PAP, n Y-e So -1, LAGE-1a, p53, prosteína, PSMA, Her2/neu, survivina y telomerasa, antígeno 1 de tumor de carcinoma prostático (PCTA-1), MAGE, ELF2M, elastasa de neutrófilos, ephrinB2, CD22, factor de crecimiento de insulina (IGF)-I, IGF-II, receptor de IGF-I y mesotelina.
Estas moléculas incluyen, pero sin limitación, antígenos específicos de tejido como MART-1, tirosinasa y GP 100 en melanoma y fosfatasa ácida prostática (PAP) y antígeno específico de la próstata (PSA) en cáncer de próstata. Otras moléculas diana pertenecen al grupo de moléculas relacionadas con la transformación, como el oncogén HER-2/Neu/ErbB-2. Otro grupo más de antígenos diana son los antígenos oncofetales tales como el antígeno carcinoembrionario (CEA). En el linfoma de linfocitos B, la inmunoglobulina idiotípica específica de tumor constituye un antígeno de inmunoglobulina verdaderamente específico de tumor que es único para el tumor individual. Los antígenos de diferenciación de linfocitos B como CD19, CD20 y CD37 son otros candidatos para antígenos diana en el linfoma de linfocitos B. Algunos de estos antígenos (CEA, HER-2, CD19, CD20, idiotipo) se han utilizado como dianas para la inmunoterapia pasiva con anticuerpos monoclonales con un éxito limitado.
Los ejemplos no limitantes de antígenos TSA o TAA incluyen los siguientes: Antígenos de diferenciación como MART-1/MelanA (MART-1), gp100 (Pmel 17), tirosinasa, TRP-1, TRP-2 y antígenos multilinaje específicos de tumores como MAGE-1, MAGE-3, BAGE, GAGE-1, GAGE-2, p15; antígenos embrionarios sobreexpresados tales como CEA; oncogenes sobreexpresados y genes supresores de tumores mutados como p53, Ras, HER-2/neu; antígenos tumorales únicos resultantes de translocaciones cromosómicas; tales como BCR-ABL, E2A-PRL, H4-RET, IGH-IGK,
MYL-RAR; y antígenos virales, tales como los antígenos del virus de Epstein Barr EBVA y los antígenos del virus del papiloma humano (HPV) E6 y E7. Otros antígenos basados en proteínas grandes incluyen TSP-180, MAGE-4, MAGE-5, MAGE-6, RAGE, NY-ESO, p185erbB2, p180erbB-3, c-met, nm-23H1, PSA, TAG-72, CA 19-9, CA 72-4, CAM 17,1, NuMa, K-ras, beta-catenina, CDK4, Mum-1, p 15, p 16, 43-9F, 5T4, 791Tgp72, alfa-fetoproteína, beta-HCG, BCA225, BTAA, CA 125, CA 15-3\CA 27.291\BCAA, CA 195, CA 242, CA-50, CAM43, CD68\P1, CO-029, FGF-5, G250, Ga733\EpCAM, HTgp-175, M344, MA-50, MG7-Ag, MOV18, NB/70K, NY-CO-1, RCAS1, SDCCAG16, TA-90\proteína de unión a Mac-2\proteína asociada a ciclofilina C), TAAL6, TAG72, TLP y TPS.
Otros ejemplos de antígenos tumorales incluyen, pero sin limitación, A33, BAGE, Bcl-2, p-catenina, CA125, CA19-9, CD5, CD19, CD20, CD21, CD22, CD33, CD37, CD45, CD123, CEA, c-Met, CS-1, ciclina B1, DAGE, EBNA, EGFR, ephrinB2, receptor de estrógenos, FAP, ferritina, proteína de unión a folato, GAGE, G250, GD-2, GM2, gp75, gp100 (Pmel 17), HER-2/neu, HPV E6, HPV E7, Ki-67, LRP, mesotelina, p53 y PRAME. Se proporcionan antígenos tumorales adicionales en van der Bruggen P, Stroobant V., Vigneron N, Van den Eynde B. Peptide database: T cell-defined tumor antigens. Cáncer Immun (2013), www(dot)cancerimmunity(dot)org/peptide/.
De acuerdo con un aspecto específico, el antígeno tumoral incluye, pero sin limitación, CD19, CD20, CD22, ROR1, mesotelina, CD33/IL3Ra, c-Met, PSMA, Glicolípido F77, Eg Frv III, Her2, GD-2, gp100, p53, antígeno carcinoembrionario (CEA), MY-ESO-1, MART-1, MAGE A3, y similares.
De acuerdo con un aspecto, el antígeno diana es CD19.
De acuerdo con algunos aspectos de la invención, el antígeno es un antígeno viral. El antígeno viral puede derivar de cualquier virus, tal como, pero sin limitación, virus de la inmunodeficiencia humana (VIH), virus de la gripe, citomegalovirus (CMV), virus de la leucemia de linfocitos T tipo 1 (TAX), virus de la hepatitis C (VHC), (VHB), virus de Epstein Barr (VEB), Adenovirus (Adv), virus del resfriado, virus de la gripe, virus de la hepatitis A, B y C, herpes simple, encefalitis japonesa, sarampión, polio, rabia, sincitial respiratorio, rubéola, viruela, varicela zóster, rotavirus, virus del Nilo Occidental, poliomavirus (por ejemplo, virus BK) y/o virus zika.
De acuerdo con algunos aspectos de la invención, los antígenos virales incluyen, pero sin limitación, epítopos virales de un polipéptido seleccionado del grupo que consiste en: factor de transcripción (TAX) del virus linfotrópico de linfocitos T humanos tipo I (HTLV-1), epítopo de proteína de matriz del virus de la gripe, epítopo derivado del virus de Epstein-Bar (EBV), TI del VIH-1, Gag del VIH, Pol del VIH, proteína de membrana M1 del virus de la gripe, hemaglutinina del virus de la gripe, neuraminidasa del virus de la gripe, nucleoproteína del virus de la gripe, nucleoproteína del virus de la gripe, proteína de matriz (M1) del virus de la gripe, canal de iones del virus de la gripe (M2), proteína no estructural del virus de la gripe NS-1, proteína no estructural del virus de la gripe NS-2, PA del virus de la gripe, PB1 del virus de la gripe, PB2 del virus de la gripe, proteína BM2 del virus de la gripe, proteína NB del virus de la gripe, proteína de la nucleocápside del virus de la gripe, proteína de matriz fosforilada del citomegalovirus (CMV) (pp65), TAX, virus de la hepatitis C (VHC), proteína pre-S del VHB 85-66, tax del HTLV-1 11-19, antígeno de superficie del VHB 185-194.
De acuerdo con algunos aspectos de la invención, el antígeno es un antígeno bacteriano. El antígeno bacteriano puede derivar de cualquier bacteria, tal como, pero sin limitación, carbunco; bacilos gramnegativos, clamidia, difteria, Haemophilus influenza, Helicobacterpylori, paludismo, Tuberculosis por Mycobacterium, toxina pertúsica, neumococo, rickettsias, estafilococos, estreptococos y tétanos.
De acuerdo con algunos aspectos de la invención, los antígenos bacterianos incluyen, pero sin limitación, los antígenos del carbunco incluyen, pero sin limitación, el antígeno protector del carbunco; los antígenos de bacilos gramnegativos incluyen, pero sin limitación, lipopolisacáridos; los antígenos del virus Haemophilus influenza incluyen, pero sin limitación, polisacáridos capsulares; los antígenos de la difteria incluyen, pero sin limitación, la toxina diftérica; Los antígenos de Mycobacterium tuberculosis incluyen, pero sin limitación, ácido micólico, proteína de choque térmico 65 (HSP65), la principal proteína secretada de 30 kDa y el antígeno 85A; los antígenos de la toxina pertussis incluyen, pero sin limitación, hemaglutinina, pertactina, FIM2, FIM3 y adenilato ciclasa; los antígenos neumocócicos incluyen, pero sin limitación, neumolisina y polisacáridos capsulares neumocócicos; los antígenos de Rickettsiae incluyen, pero sin limitación, rompA; los antígenos estreptocócicos incluyen, pero sin limitación, proteínas M; y los antígenos del tétanos incluyen, pero sin limitación, toxina tetánica.
De acuerdo con algunos aspectos de la invención, el antígeno es un antígeno de superbacteria (por ejemplo, bacterias resistentes a múltiples fármacos). Los ejemplos de superbacterias incluyen, pero sin limitación, Enterococcus faecium, Clostridium difficile, Acinetobacter baumannii, Pseudomonas aeruginosa y Enterobacteriaceae (incluyendo Escherichia coli, Klebsiella pneumoniae, Enterobacter spp.).
De acuerdo con algunos aspectos de la invención, el antígeno es un antígeno fúngico. Ejemplos de hongos incluyen, pero sin limitación, Candida, Coccidiodes, Cryptococcus, Histoplasma, Leishmania, Plasmodium, protozoos, parásitos, Schistosomae, los hongos causantes de la tiña, Toxoplasma y Tripanosoma cruzi.
De acuerdo con algunos aspectos de la invención, los antígenos fúngicos incluyen, pero sin limitación, los antígenos
de coccidiodes incluyen, pero sin limitación, antígenos de esférulas; los antígenos criptocócicos incluyen, pero sin limitación, polisacáridos capsulares; los antígenos de histoplasma incluyen, pero sin limitación, proteína de choque térmico 60 (HSP60); los antígenos de Leishmania incluyen, pero sin limitación, gp63 y lipofosfoglicano; los antígenos de Plasmodium falciparum incluyen, pero sin limitación, antígenos de superficie de merozαtos, antígenos de superficie de esporozoítos, antígenos de circumsporozoíto, gametocitos/antígenos de superficie de gametos, antígenos de protozoos y otros parasitarios, incluido el antígeno del estadio sanguíneo pf 155/RESA; los antígenos de Schistosoma incluyen, pero sin limitación, glutatión-S-transferasa y paramiosina; los antígenos fúngicos de la tiña incluyen, pero sin limitación, tricofitina; los antígenos de Toxoplasma incluyen, pero sin limitación, SAG-1 y p30; y los antígenos de Trypanosoma cruzi incluyen, pero sin limitación, el antígeno de 75-77 kDa y el antígeno de 56 kDa.
De acuerdo con algunos aspectos de la invención, el antígeno es un antígeno expresado por células asociadas con una afección alérgica. Los ejemplos de antígenos alérgicos incluyen, pero sin limitación, antígenos de polen tales como antígenos de polen de cedro japonés, antígenos del polen de ambrosía, antígenos del polen de lolio, antígenos derivados de animales (como antígenos de ácaros del polvo y antígenos felinos), antígenos de histocompatibilidad, penicilina y otros fármacos terapéuticos.
De acuerdo con algunos aspectos de la invención, el antígeno es un autoantígeno asociado con un trastorno autoinmunitario.
La expresión "enfermedad autoinmunitaria", como se usa en el presente documento, se define como un trastorno que resulta de una respuesta autoinmunitaria. Una enfermedad autoinmunitaria es el resultado de una respuesta inadecuadamente excesiva a un autoantígeno.
Ejemplos de enfermedades autoinmunitarias incluyen, pero sin limitación, enfermedad de Addison, alopecia areata, espondilitis anquilosante, hepatitis autoinmunitaria, parotiditis autoinmunitaria, enfermedad de Crohn, enfermedad inflamatoria intestinal (IBD), enfermedad celíaca, dermatitis (incluyendo dermatitis atópica y dermatitis eccematosa), diabetes de tipo I, epidermólisis ampollosa distrófica, epididimitis, glomerulonefritis, enfermedad de Graves, síndrome de Guillain-Barr, enfermedad de Hashimoto, anemia hemolítica, lupus eritematoso sistémico (LES), esclerosis múltiple (EM), miastenia grave, pénfigo vulgar, psoriasis, fiebre reumática, artritis (incluyendo artritis reumatoide, artritis reumatoide juvenil, artrosis, artritis psoriásica), sarcoidosis, esclerodermia, síndrome de Sjogren, síndrome de Stevens-Johnson, granulomatosis de Wegener, espondiloartropatías, tiroiditis, vasculitis, vitíligo, mixedema, anemia, asma, anemia perniciosa, colitis ulcerosa y accidente cerebrovascular, entre otros.
Como se usa en el presente documento, la expresión "péptido autoantigénico" se refiere a un antígeno derivado de una proteína endógena (es decir, propia) o consumida (por ejemplo, con los alimentos) contra la que se provoca una respuesta inflamatoria como parte de una respuesta inflamatoria autoinmunitaria.
Cabe señalar que los términos "endógena", "auto" son expresiones relativas que se refieren al individuo en el que se provoca la respuesta autoinmunitaria.
Los autoantígenos comprenden, pero sin limitación, proteínas celulares, fosfoproteínas, proteínas de la superficie celular, lípidos celulares, ácidos nucleicos, glucoproteínas, incluidos los receptores de la superficie celular.
De acuerdo con algunos aspectos de la divulgación, el péptido autoantigénico está asociado con una enfermedad seleccionada del grupo que consiste en diabetes, esclerosis múltiple, artritis reumatoide, enfermedad celíaca y accidente cerebrovascular.
Los autoantígenos de la esclerosis múltiple incluyen, pero sin limitación, proteínas de mielina como la proteína básica de mielina (MBP), proteína proteolipídica (PLP) y glicoproteína de oligodendrocitos de mielina (MOG).
Los autoantígenos asociados con la artritis reumatoide incluyen, pero sin limitación, péptidos autoantigénicos derivados del colágeno II (COL2A1), metaloproteinasa de matriz-1 (MMP1), precursor de la proteína del núcleo de agrecano (ACAN), metaloproteinasa de matriz-16 (MMP16), tenascina (TNXB) y ribonucleoproteína nuclear heterogénea A2 (HNRNPA2B1).
Los autoantígenos de la diabetes tipo 1 (T1D) incluyen, pero sin limitación, antígenos expresados en los islotes pancreáticos, incluyendo ácido glutámico descarboxilasa (GAD65) y péptido autoantigénico de células beta.
Los autoantígenos celíacos (celiaquía) incluyen, pero sin limitación, gliadina (p. ej., alfa gliadina, gamma gliadina) y proteína de choque térmico 20.
enfermedad de Crohn, colitis ulcerosa o enfermedad inflamatoria intestinal (IBD), los autoantígenos incluyen, pero sin limitación, FAM84A, glicoproteína de membrana granular 2 (GP2), dominios CUB y tipo zona pelúcida 1 (CUZD1), complemento C3, catalasa y alfa-enolasa.
De acuerdo con algunos aspectos de la invención, los autoantígenos asociados con el accidente cerebrovascular
incluyen, pero sin limitación, péptidos autoantigénicos derivados de un antígeno cerebral como la proteína básica de mielina, neurofilamentos y el subtipo NR2N2B del receptor de N-metil-D-aspartato (MOG-35-55).
De acuerdo con un aspecto de algunos aspectos de la divulgación, se proporciona un método para generar la célula aislada de algunos aspectos de la invención, comprendiendo el método transducir una célula que tiene un fenotipo de linfocito T de memoria central (Tmc), siendo la célula, una célula inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, con un polinucleótido que codifica un receptor de la superficie celular que comprende un módulo de señalización del receptor de linfocitos T.
De acuerdo con un aspecto, la célula que tiene el fenotipo de linfocito T de memoria central (Tmc), al ser una célula inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, es una célula anti-terceros.
La expresión "células anti-terceros" como se usa en el presente documento se refiere a linfocitos (es decir, linfocitos T) que se dirigen (es decir, mediante el reconocimiento de linfocitos T) contra un antígeno o antígenos de terceros.
Como se usa en el presente documento, la expresión "antígeno o antígenos de terceros" se refiere a un antígeno o antígenos solubles o no solubles (como los asociados a la membrana) que no están presentes ni en el donante ni en el receptor, como se muestra en detalle a continuación.
Por ejemplo, los antígenos de terceros pueden ser células de terceros, antígenos celulares (por ejemplo, antígenos de la superficie celular), antígenos de virus (es decir, antígeno viral), tal como, por ejemplo, virus de Epstein-Barr (EBV) o citomegalovirus (CMV), o antígenos de bacterias (es decir, antígeno bacteriano), como la flagelina. Los antígenos virales o bacterianos pueden ser presentados por células (p. ej., líneas celulares) infectadas con estos o preparadas de otro modo para expresar proteínas virales/bacterianas.
Las células presentadoras de antígeno autólogas o no autólogas, o un vehículo artificial o células presentadoras de antígeno artificial, se pueden usar para presentar péptidos sintéticos cortos fusionados o cargados en las mismas o para presentar extractos de proteínas o proteínas purificadas. Tales péptidos cortos, extractos de proteínas o proteínas purificadas pueden ser péptidos derivados de virus o bacterias o péptidos que representan cualquier otro antígeno.
Se puede usar un software especializado para analizar secuencias virales u otras para identificar péptidos cortos inmunogénicos, es decir, péptidos presentables en el contexto del MHC de clase I o MHC de clase II.
Las células de terceros pueden ser alogénicas o xenogénicas con respecto al receptor (explicado con más detalle a continuación). En el caso de células alogénicas de terceros, tales células tienen antígenos HLA diferentes a los del donante pero que no tienen reactividad cruzada con los antígenos HLA del receptor, de manera que las células anti terceros generadas contra tales células no sean reactivas contra un trasplante o antígenos del receptor.
De acuerdo con un aspecto de la presente divulgación, las células de terceros alogénicas o xenogénicas son células estimulantes seleccionadas del grupo que consiste en células purificadas de linfocitos de sangre periférica (PBL), bazo o ganglios linfáticos, PBL movilizados por citocinas, células presentadoras de antígeno (APC) expandidasin vitro, células dendríticas (DC) expandidas in vitro y células presentadoras de antígenos artificiales.
La APC artificial de la presente divulgación puede diseñarse para exhibir MHC autólogo con un péptido de terceros o un MHC de terceros sin ser pulsado con un péptido exógeno. Por tanto, de acuerdo con un aspecto, la APC artificial comprende células tumorales K562 transfectadas con un determinante MHC de terceros y una molécula coestimuladora [como ha descrito anteriormente, p. ej., Suhoski MM et al., Mol Ther. (2007) 15(5): 981-8], o fibroblastos transfectados con el mismo.
Los antígenos de terceros se pueden presentar en las superficies celulares, virales o bacterianas o derivadas y/o purificadas de las mismas. Adicionalmente, un antígeno viral o bacteriano se puede mostrar en una célula infectada y un antígeno celular se puede mostrar en un vehículo artificial como un liposoma o una célula presentadora de antígeno artificial (por ejemplo, una línea celular leucémica o de fibroblastos transfectada con el antígeno o antígenos de terceros).
El antígeno de terceros puede comprender además un péptido sintético presentado por células presentadoras autólogas, células presentadoras no autólogas o en un vehículo artificial o en células presentadoras de antígenos artificiales.
Asimismo, los antígenos de terceros pueden, por ejemplo, ser proteínas extraídas o purificadas de una variedad de fuentes. Un ejemplo de una proteína purificada que puede servir como antígeno de terceros de acuerdo con la presente divulgación es la ovoalbúmina. Se prevén otros ejemplos.
La utilización de células, células infectadas por virus, células infectadas por bacterias, células presentadoras de péptidos virales o células presentadoras de péptidos bacterianos como antígenos de terceros es particularmente ventajosa, ya que dichos antígenos de terceros incluyen una serie diversa de determinantes antigénicos y, como tales,
dirigen la formación de células anti-terceros de una población diversa, lo que puede servir además para la reconstitución más rápida de los linfocitos T en los casos en que se requiera tal reconstitución, por ejemplo, después de un procedimiento de quimioterapia o irradiación letal o subletal.
Es más, cuando las células anti-terceros se dirigen contra antígenos de terceros, las células están dotadas de actividad anti-enfermedad. La expresión "actividad anti-enfermedad" se refiere a la actividad (p. ej., capacidad de eliminación) de los linfocitos Tmc contra una célula enferma (p. ej., célula cancerosa, como la actividad injerto contra leucemia, GVL). Esta actividad se debe normalmente a la eliminación independiente del TCR mediada por la unión de LFA1-I/CAM1 [Arditti et al., Blood (2005) 105(8):3365-71. publicación electrónica 6 jul 2004).
De acuerdo con un aspecto, las células de terceros comprenden células dendríticas.
De acuerdo con un aspecto, las células de terceros comprenden células dendríticas maduras.
Los métodos de generación de células dendríticas de terceros, que pueden usarse como células estimuladoras para inducir linfocitos Tmc, son bien conocidos en la técnica. Por tanto, como un ejemplo no limitativo, las células mononucleares de sangre periférica (PBMC) pueden obtenerse de terceros como un donante de células no singénicas [p. ej., en caso de que los linfocitos Tmc sean singénicos, p.ej., las células dendríticas (DC) autólogas pueden ser no singénicas, p. ej., alogénicas, con respecto al sujeto; mientras que si los linfocitos Tmc son no singénicos, p. ej., alogénicos, las Dc se seleccionan de un donante que es no singénico, p.ej., alogénico y no existe compatibilidad h La ni con el sujeto ni con los linfocitos Tmc]. A continuación, los monocitos pueden aislarse mediante adherencia plástica y cultivarse (p. ej., en placas de cultivo celular) utilizando medio celular DC (p. ej., medio Cellgro DC) suplementado con suero humano (p. ej., suero humano al 1 %), penicilina/estreptomicina y GM-CSF (p. ej., 800 UI/ml) e IL-4 (p. ej., 20 ng/ml) (comercializado por, p. ej., Peprotech, Hamburgo, Alemania). Después de aproximadamente 24-72 h (por ejemplo, 48 h) de cultivo, se puede añadir el medio DC que comprende GM-CSF (p. ej., 1600 UI/ml) e IL-4 (p. ej., 20 ng/ml). Aproximadamente 12-36 h (por ejemplo, 24 h) más tarde, las células no adherentes pueden recolectarse y las células grandes (principalmente DC inmaduras) pueden resuspenderse en medio fresco que contiene GM-CSF (por ejemplo, 800 UI/ml), IL-4 (por ejemplo, 20 ng/ml), LPS (por ejemplo, de E. coli O55:B5 a, por ejemplo, 10 ng/ml) e IFNy (por ejemplo, 100 UI/ml) (comercializado por, por ejemplo, Peprotech, Hamburgo, Alemania), sembrarse e incubarse durante la noche. Al día siguiente, las células no adherentes se pueden desechar y las DC adherentes se pueden eliminar suavemente utilizando, p. ej., PBS frío/HS al 1 % después de la incubación en hielo durante aproximadamente 15-30 minutos (por ejemplo, 20 minutos), obteniendo de este modo células grandes que consisten en DC maduras.
De acuerdo con un aspecto, las células de terceros comprenden células dendríticas irradiadas.
Por tanto, de acuerdo con un aspecto, las DC se irradian con aproximadamente 5-10 Gy, aproximadamente 10-20 Gy, aproximadamente 20-30 Gy, aproximadamente 20-40 Gy, aproximadamente 20-50 Gy, aproximadamente 10-50 Gy. De acuerdo con un aspecto específico, las DC se irradian con aproximadamente 10-50 Gy (por ejemplo, 30 Gy).
Se puede usar cualquier método para producir linfocitos Tmc anti-terceros de acuerdo con la presente divulgación, como se describió previamente en las Publicaciones PCT números WO 2010/049935, WO 2012/032526 y WO 2013/035099.
De acuerdo con un aspecto, la generación de una célula anti-terceros que tenga un fenotipo Tmc puede llevarse a cabo mediante un método que comprende: (a) poner en contacto células mononucleares de sangre periférica (PBMC) con un antígeno o antígenos de terceros en presencia o ausencia de IL-21 para permitir el enriquecimiento de células reactivas al antígeno; y (b) cultivar las células resultantes de la etapa (a) en presencia de IL-21, IL-15 e IL-7 en un entorno libre de antígenos para permitir la proliferación de células que comprenden el fenotipo de linfocitos T de memoria central (Tmc).
De acuerdo con un aspecto, las PBMC en la etapa (a) se ponen en contacto con un antígeno o antígenos de terceros en ausencia de IL-21.
De acuerdo con un aspecto, las PBMC en la etapa (a) se ponen en contacto con un antígeno o antígenos de terceros en presencia de IL-21.
De acuerdo con un aspecto, las células resultantes de la etapa (a) se cultivan en un entorno libre de antígenos (p. ej., sin la adición de un antígeno al cultivo celular) en presencia únicamente de IL-15. Se pueden añadir opcionalmente IL-21 y/o IL-7.
Los linfocitos Tmc anti-terceros de la presente divulgación se generan normalmente poniendo en contacto primero células mononucleares de sangre periférica (PBMC) singénicas (p. ej., autólogas) o no singénicas (p. ej., no autólogas como alogénicas o xenogénicas, como se describe con más detalle a continuación) con un antígeno o antígenos de terceros (como se ha descrito anteriormente) en un cultivo suplementado con IL-21 (p. ej., en un cultivo libre de citocinas, es decir, sin la adición de citocinas adicionales). Esta etapa se lleva a cabo normalmente durante aproximadamente 12-24 horas, aproximadamente 12-36 horas, aproximadamente 12-72 horas, 24-48 horas, 24-36
horas, aproximadamente 24-72 horas, aproximadamente 48-72 horas, 1-2 días, 2-3 días, 1-3 días, 2-4 días, 1-5 días, 2-5 días, 2-6 días, 1-7 días, 5-7 días, 2-8 días, 8-10 días o 1-10 días y permite el enriquecimiento de células reactivas al antígeno.
De acuerdo con un aspecto específico, el contacto de PBMC singénicas o no singénicas con un antígeno o antígenos de terceros (tal como se ha descrito anteriormente) en un cultivo suplementado con IL-21 (o de otra forma, cultivo sin citocinas) se efectúa durante 1-5 días (por ejemplo, 3 días).
El contacto de las PBMC singénicas o no singénicas con un antígeno o antígenos de terceros (como los descritos anteriormente) en un cultivo suplementado con IL-21 se lleva a cabo normalmente en presencia de aproximadamente 0,001-3000 UI/ml, 0,01-3000 UI/ml, 0,1-3000 UI/ml, 1-3000 UI/ml, 10-3000 UI/ml, 100-3000 UI/ml, 1000-3000 UI/ml, 0,001-1000 UI/ml, 0,01-1000 UI/ml, 0,1-1000 UI/ml, 1-1000 UI/ml, 10-1000 UI/ml, 100-1000 UI/ml, 250-1000 UI/ml, 500-1000 UI/ml, 750-1000 UI/ml, 10-500 UI/ml, 50-500 UI/ml, 100-500 UI/ml, 250-500 UI/ml, 100-250 UI/ml, 0,1-100 UI/ml, 1-100 UI/ml, 10-100 UI/ml, 30-100 UI/ml, 50-100 UI/ml, 1-50 UI/ml, 10-50 UI/ml, 20-50 UI/ml, 30-50 UI/ml, 1-30 UI/ml, 10-30 UI/ml, 20-30 UI/ml, 10-20 UI/ml, 0,1-10 UI/ml o 1-10 UI/ml de IL-21.
De acuerdo con un aspecto específico, la concentración de IL-21 es de 50-150 UI/ml (por ejemplo, 100 UI/ml).
De acuerdo con un aspecto específico, el contacto de las PBMC singénicas o no singénicas con un antígeno o antígenos de terceros se realiza en un cultivo libre de citocinas (por ejemplo, complementado solo con IL-21), tal condición de cultivo permite la supervivencia y el enriquecimiento solo de aquellas células que se someten a estimulación y activación por el antígeno o antígenos de terceros (es decir, de células reactivas al antígeno) ya que estas células secretan citocinas (por ejemplo, IL-2) que permiten su supervivencia (el resto de las células mueren en estas condiciones de cultivo).
La proporción entre antígeno o antígenos de terceros (p. ej., células dendríticas) y PBMC es normalmente de aproximadamente 1:2 a aproximadamente 1:10, como aproximadamente 1:4, aproximadamente 1:6, aproximadamente 1:8 o aproximadamente 1:10.
De acuerdo con un aspecto específico, la proporción entre antígeno o antígenos de terceros (por ejemplo, células dendríticas) y PBMC es de aproximadamente 1:2 a aproximadamente 1:8 (por ejemplo, 1:5).
A continuación, las células anti-terceros se cultivan en presencia de IL-21, IL-15 y/o IL-7 en un ambiente libre de antígeno para permitir la proliferación de células que comprenden el fenotipo Tmc. Esta etapa se lleva a cabo normalmente durante aproximadamente 12-24 horas, aproximadamente 12-36 horas, aproximadamente 12-72 horas, 24-48 horas, 24-36 horas, aproximadamente 24-72 horas, aproximadamente 48-72 horas, 1-20 días, 1-15 días, 1-10 días, 1-5 días, 5-20 días, 5-15 días, 5-10 días, 1-2 días, 2-3 días, 1-3 días, 2-4 días, 2-5 días, 2-8 días, 2-10 días, 4 10 días, 4-8 días, 6-8 días, 8-10 días, 7-9 días, 7-11 días, 7-13 días, 7-15 días, 10-12 días, 10-14 días, 12-14 días, 14 16 días, 14-18 días, 16-18 días o 18-20 días. De acuerdo con un aspecto específico, las células anti-terceros se cultivan en presencia de IL-21, IL-15 e IL-7 en un entorno libre de antígenos durante aproximadamente 7-11 días (p. ej., 8 días).
Esta etapa normalmente se lleva a cabo en presencia de IL-21 a una concentración de aproximadamente 0,001-3000 UI/ml, 0,01-3000 UI/ml, 0,1-3000 UI/ml, 1-3000 UI/ml, 10-3000 UI/ml, 100-3000 UI/ml, 1000-3000 UI/ml, 0,001-1000 UI/ml, 0,01-1000 UI/ml, 0,1-1000 UI/ml, 1-1000 UI/ml, 10-1000 UI/ml, 100-1000 UI/ml, 250-1000 UI/ml, 500-1000 UI/ml, 750-1000 UI/ml, 10-500 UI/ml, 50-500 UI/ml, 100-500 UI/ml, 250-500 UI/ml, 100-250 UI/ml, 0,1-100 UI/ml, 1-100 UI/ml, 10-100 UI/ml, 30-100 UI/ml, 50-100 UI/ml, 1-50 UI/ml, 10-50 UI/ml, 20-50 UI/ml, 30-50 UI/ml, 1-30 UI/ml, 10-30 UI/ml, 20-30 UI/ml, 10-20 UI/ml, 0,1-10 UI/ml o 1-10 UI/ml de IL-21.
De acuerdo con un aspecto específico, la concentración de IL-21 es de 50-150 UI/ml (por ejemplo, 100 UI/ml).
Esta etapa se lleva a cabo además en presencia de IL-15 a una concentración de aproximadamente 0,001-3000 UI/ml, 0,01-3000 UI/ml, 0,1-3000 UI/ml, 1-3000 UI/ml, 10-3000 UI/ml, 100-3000 UI/ml, 125-3000 UI/ml, 1000-3000 UI/ml, 0,001-1000 UI/ml, 0,01-1000 UI/ml, 0,1-1000 UI/ml, 1-1000 UI/ml, 10-1000 UI/ml, 100-1000 UI/ml, 125-1000 UI/ml, 250-1000 UI/ml, 500-1000 UI/ml, 750-1000 UI/ml, 10-500 UI/ml, 50-500 UI/ml, 100-500 UI/ml, 125-500 UI/ml, 250-500 UI/ml, 250-500 UI/ml, 125-250 UI/ml, 100-250 UI/ml, 0,1-100 UI/ml, 1-100 UI/ml, 10-100 UI/ml, 30-100 UI/ml, 50-100 UI/ml, 1-50 UI/ml, 10-50 UI/ml, 20-50 UI/ml, 30-50 UI/ml, 1-30 UI/ml, 10-30 UI/ml, 20-30 UI/ml, 10-20 UI/ml, 0,1-10 UI/ml o 1-10 UI/ml de IL-15. De acuerdo con un aspecto específico, la concentración de IL-15 es de 100-150 UI/ml (por ejemplo, 125 UI/ml).
Esta etapa se lleva a cabo además en presencia de IL-7 a una concentración de aproximadamente 0,001-3000 UI/ml, 0,01-3000 UI/ml, 0,1-3000 UI/ml, 1-3000 UI/ml, 10-3000 UI/ml, 30-3000 UI/ml, 100-3000 UI/ml, 1000-3000 UI/ml, 0,001 1000 UI/ml, 0,01-1000 UI/ml, 0,1-1000 UI/ml, 1-1000, 10-1000, 30-1000, 100-1000 UI/ml, 250-1000 UI/ml, 500-1000 UI/ml, 750-1000 UI/ml, 10-500 UI/ml, 30-500 UI/ml, 50-500 UI/ml, 100-500 UI/ml, 250-500 UI/ml, 100-250 UI/ml, 0,1 100 UI/ml, 1-100 UI/ml, 10-100 UI/ml, 30-100 UI/ml, 50-100 UI/ml, 1-50 UI/ml, 10-50 UI/ml, 20-50 UI/ml, 30-50 UI/ml, 1-30 UI/ml, 10-30 UI/ml, 20-30 UI/ml, 10-20 UI/ml, 0,1-10 UI/ml o 1-10 UI/ml de IL-7. De acuerdo con un aspecto
específico la concentración de IL-7 es de 10-50 Ul/ml (30 Ul/ml).
Los presentes inventores han recopilado a través de una laboriosa experimentación y selección una serie de criterios que pueden aprovecharse para mejorar la proliferación de células anti-terceros que comprenden un fenotipo de linfocitos T de memoria central (Tmc), sin células reactivas injerto contra hospedador (GVH) y/o potenciando las células reactivas antienfermedad (p. ej., GVL).
De acuerdo con un aspecto, las PBMC se agotan de células adherentes antes de ponerse en contacto con un antígeno o antígenos de terceros en presencia de IL-21.
De acuerdo con un aspecto, las PBMC se agotan de células CD4 y/o CD56+ antes de ponerse en contacto con un antígeno o antígenos de terceros en presencia de IL-21.
De acuerdo con un aspecto, las PBMC se seleccionan para las células CD45RA+ antes de ponerlas en contacto con un antígeno o antígenos de terceros en presencia de IL-21.
El agotamiento de las células CD4 y/o CD56 se puede llevar a cabo utilizando cualquier método conocido en la técnica, como por purificación basada en afinidad (por ejemplo, como mediante el uso de perlas MACS, clasificador FACS y/o marcado con ELISA de captura). Tal etapa puede ser beneficiosa para aumentar la pureza de las células CD8+ dentro del cultivo (es decir, eliminar otros linfocitos dentro del cultivo celular, por ejemplo, T CD4+ o linfocitos NK) o para aumentar el número de linfocitos T CD8+.
De acuerdo con un aspecto, las PBMC comprenden células no adherentes.
De acuerdo con un aspecto, las PBMC comprenden linfocitos T CD8+.
De acuerdo con un aspecto, las PBMC comprenden linfocitos T CD8+ vírgenes.
La selección de linfocitos T CD8+ vírgenes puede efectuarse mediante la selección de células que expresan CD45RA+ y/o células que expresan CD45RO- y puede llevarse a cabo utilizando cualquier método conocido en la técnica, como por purificación basada en afinidad (por ejemplo, como mediante el uso de perlas MACS, clasificador FACS y/o marcado con ELISA de captura).
De acuerdo con un aspecto, las PBMC comprenden células CD45RA+.
Una etapa adicional que puede llevarse a cabo de acuerdo con las presentes enseñanzas incluye cultivar las células PBMC con un antígeno o antígenos de terceros en presencia de iL-21, IL-15 e IL-7 antes de eliminar el antígeno o antígenos de terceros del cultivo celular (es decir, antes de generar un entorno libre de antígenos).
Esta etapa se lleva a cabo normalmente durante aproximadamente 12-24 horas, aproximadamente 12-36 horas, aproximadamente 12-72 horas, 24-48 horas, 24-36 horas, aproximadamente 24-72 horas, aproximadamente 48 72 horas, 1-2 días, 2-3 días, 1-3 días, 2-4 días, 1-5 días o 2-5 días, y se efectúa a las mismas dosis de IL-21, IL-15 e IL-7 indicadas anteriormente. De acuerdo con un aspecto específico, el cultivo de las células PBMC con un antígeno o antígenos de terceros en presencia de IL-21, IL-15 e IL-7 se lleva a cabo durante 12 horas a 4 días (por ejemplo, 1 2 días).
Adicionalmente, o como alternativa, puede llevarse a cabo un proceso adicional de dos etapas que permite la selección y el aislamiento de las células activadas. Tal etapa de selección facilita la eliminación de posibles linfocitos T reactivos del hospedador (por ejemplo, células alorreactivas) en situaciones en las que las PBMC no son singénicas con respecto al sujeto (como se describe con más detalle a continuación).
Por tanto, el aislamiento de células activadas puede llevarse a cabo en un enfoque de dos etapas. En la primera etapa, las células activadas se seleccionan antes de cultivar las células en presencia de IL-15 e IL-7. Esta primera etapa normalmente se lleva a cabo después del contacto inicial de las PBMC con un antígeno o antígenos de terceros en presencia de IL-21. Este proceso de selección selecciona solo aquellas células que fueron activadas por el antígeno de terceros (por ejemplo, expresa marcadores de activación como se describe a continuación) y generalmente se efectúa entre 12 y 24 horas, aproximadamente 24-36 horas, aproximadamente 12-36 horas, aproximadamente 36 48 horas, aproximadamente 12-48 horas, aproximadamente 48-60 horas, aproximadamente 12-60 horas, aproximadamente 60-72 horas, aproximadamente 12-72 horas, aproximadamente 72-84 horas, aproximadamente 12 84 horas, aproximadamente 84-96 horas, aproximadamente 12-96 horas, después del contacto inicial de las PBMC con un antígeno o antígenos de terceros.
De acuerdo con un aspecto específico, el proceso de selección se efectúa aproximadamente 12-24 horas (por ejemplo, 14 horas) después del contacto inicial de las PBMC con un antígeno o antígenos de terceros.
El aislamiento de células activadas puede efectuarse mediante purificación basada en afinidad (por ejemplo, mediante
el uso de perlas MACS, clasificador FACS y/o marcado con ELISA de captura) y puede efectuarse con cualquier marcador de activación, incluidos los marcadores de la superficie celular, tales como, aunque no de forma limitativa, CD69, CD44, CD25, CFSE, CD137 o marcadores no de superficie celular tales como, aunque no de forma limitativa, IFN-y e IL-2. El aislamiento de células activadas también puede efectuarse mediante purificación basada en la morfología (p. ej., aislamiento de células grandes) utilizando cualquier método conocido en la técnica (p. ej., mediante FACS). Normalmente, las células activadas también se seleccionan para la expresión de células CD8+. Es más, se puede utilizar cualquier combinación de los métodos anteriores para aislar eficazmente las células activadas.
De acuerdo con un aspecto de la presente invención, la selección de células activadas se efectúa mediante la selección de células CD137+ y/o CD25+.
La segunda etapa de aislamiento de las células activadas normalmente se lleva a cabo al final del cultivo (es decir, después del cultivo en un entorno libre de antígenos con IL-21, IL-15 y IL-7). Esta etapa agota las células alorreactivas mediante el agotamiento de aquellas células que se activaron después del contacto del linfocito T de memoria central (T mc) con células presentadoras de antígeno del hospedador irradiadas (APC, por ejemplo, células dendríticas). Como se ha mencionado anteriormente, el aislamiento de células activadas puede efectuarse mediante purificación basada en afinidad (por ejemplo, mediante el uso de perlas MACS, clasificador FACS y/o marcado con ELISA de captura) y puede efectuarse con cualquier marcador de activación, incluidos los marcadores de la superficie celular, tales como, aunque no de forma limitativa, CD69, CD44, CD25, CFSE, CD137 o marcadores no de superficie celular tales como, aunque no de forma limitativa, IFN-y e IL-2.
De acuerdo con un aspecto de la presente invención, el agotamiento de las células alorreactivas se efectúa mediante el agotamiento de las células CD137+ y/o CD25+ y/o la captura de IFNy.
Los siguientes son protocolos de ejemplo que se pueden usar de acuerdo con algunos aspectos de la invención.
Aquí se proporciona un método para generar una célula aislada que tiene un fenotipo de memoria central, siendo la célula, una célula inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, comprendiendo el método: (a) poner en contacto células mononucleares de sangre periférica (PBMC) con un antígeno o antígenos de terceros en presencia de IL-21 (por ejemplo, durante 12 horas a 5 días) para permitir el enriquecimiento de células reactivas al antígeno; y (b) cultivar las células resultantes de la etapa (a) en presencia de IL-21, IL-15 e IL-7 en un entorno libre de antígenos (por ejemplo, durante 5-20 días) para permitir la proliferación de células anti-terceros que comprenden el fenotipo de linfocitos T de memoria central (Tmc).
De acuerdo con un aspecto, el método comprende además (c) separar las células resultantes de la etapa (b) en suspensiones de células individuales.
De acuerdo con un aspecto, el método comprende además agotar las células adherentes de las PBMC antes de la etapa (a).
De acuerdo con un aspecto, el método comprende además agotar las células CD4+ y/o CD56+ de las PBMC antes de la etapa (a).
De acuerdo con un aspecto, el método comprende además seleccionar las células activadas después de la etapa (a) y antes de la etapa (b).
De acuerdo con un aspecto, el método comprende además seleccionar las células activadas que se efectúa mediante la selección de células CD137+ y/o CD25+.
Aquí se proporciona un método para generar una célula aislada que tiene un fenotipo de memoria central, siendo la célula, una célula inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, comprendiendo el método: (a) tratar células mononucleares de sangre periférica (PBMC) no adherentes con un agente capaz de reducir los linfocitos CD4+ y/o CD56+ para obtener linfocitos T CD8+; (b) poner en contacto los linfocitos T c D8+ con células dendríticas de terceros en presencia de IL-21 (por ejemplo, durante 12 horas a 5 días) para permitir el enriquecimiento de células reactivas al antígeno; (c) cultivar las células resultantes de la etapa (b) con las células dendríticas de terceros en presencia de IL-21, IL-15 e IL-7 (por ejemplo, durante 12 horas a 3 días); y (d) cultivar las células resultantes de la etapa (c) en presencia de IL-21, IL-15 e IL-7 en un entorno libre de antígenos (por ejemplo, durante 5-20 días) para permitir la proliferación de células que comprenden el fenotipo de linfocitos T de memoria central (Tmc).
De acuerdo con un aspecto, el método comprende además separar las células resultantes de la etapa (d) en suspensiones de células individuales.
De acuerdo con un aspecto, las células anti-terceros que comprenden el fenotipo Tmc comprenden una firma CD3+, CD8+, CD62L+, CD45RA', CD45RO+.
Se valorará que al menos un 30 %, al menos un 40 %, al menos un 50 %, al menos un 55 %, al menos un 60 %, al menos un 65 %, al menos un 70 %, al menos un 75 %, al menos un 80 %, al menos un 85 %, al menos un 90 %, al menos un 95 % o incluso un 100 % de las células anti-terceros son células CD3+CD8+. De acuerdo con un aspecto específico, las células anti-terceros comprenden aproximadamente el 70-90 % de células CD3+CD8+.
Se valorará que al menos un 30 %, al menos un 40 %, al menos un 50 %, al menos un 55 %, al menos un 60 %, al menos un 65 %, al menos un 70 %, al menos un 75 %, al menos un 80 %, al menos un 85 %, al menos un 90 %, al menos un 95 % o incluso un 100 % de las células CD3+CD8+ tienen la firma de linfocito Tmc. De acuerdo con un aspecto específico, aproximadamente un 30-80 % de las células CD3+CD8+ tienen la firma de linfocito Tmc (por ejemplo, 40-50 %).
De acuerdo con un aspecto, al menos un 50 % de las células son linfocitos CD3+CD8+ de los cuales al menos un 50 % tienen la firma.
Por tanto, las células de la divulgación que tienen un fenotipo de linfocito T de memoria central (T mc) no se producen de forma natural y no son un producto de la naturaleza. Estas células son normalmente producidas por manipulación ex-vivo (es decir, exposición a un antígeno o antígenos de terceros en presencia de citocinas específicas).
Tal como se ha mencionado, el linfocito Tmc de la divulgación se transduce con un polinucleótido que codifica un receptor de la superficie celular que comprende un módulo de señalización del receptor de linfocitos T.
Como se usa en el presente documento, el término "polinucleótido" se refiere a una secuencia de ácido nucleico monocatenaria o bicatenaria que se aísla y se proporciona en forma de una secuencia de ARN, una secuencia polinucleotídica complementaria (ADNc), una secuencia polinucleotídica genómica y/o una secuencia polinucleotídica compuesta (p. ej., una combinación de las anteriores).
El término "aislado" se refiere al menos parcialmente separado del entorno natural, por ejemplo, de una célula, o de un tejido, por ejemplo, de un cuerpo humano.
El polinucleótido aislado se puede obtener utilizando métodos recombinantes conocidos en la técnica, tal como, por ejemplo, cribado de bibliotecas de células que expresan el gen, derivando el gen de un vector que se sabe que lo incluye, o aislándolo directamente de células y tejidos que lo contienen, usando técnicas convencionales. De forma alternativa, el gen de interés se puede producir sintéticamente, en lugar de clonado.
El polinucleótido de acuerdo con algunos aspectos de la divulgación puede comprender un solo polinucleótido que comprende una secuencia de ácido nucleico que codifica el dominio extracelular, el dominio transmembrana y/o el módulo de señalización del receptor de la superficie celular (por ejemplo, tg-TCR y/o CAR). De forma alternativa, se pueden usar dos o más polinucleótidos en donde un polinucleótido puede comprender una secuencia de ácido nucleico que codifica, por ejemplo, el dominio extracelular y el dominio transmembrana y otro polinucleótido puede comprender una secuencia de ácido nucleico que codifica el módulo de señalización.
De acuerdo con un aspecto de algunos aspectos de la divulgación, se proporciona una construcción de ácido nucleico que comprende un polinucleótido aislado que comprende una secuencia de ácido nucleico que codifica la molécula de algunos aspectos de la divulgación y un elemento regulador que actúa en cis para dirigir la transcripción del polinucleótido aislado en una célula hospedadora.
Por tanto, la expresión de ácidos nucleicos naturales o sintéticos que codifican el receptor de la superficie celular (p. ej., tg-TCR o molécula CAR) de la divulgación se logra normalmente mediante la unión operativa de un ácido nucleico que codifica el polipéptido o porciones del receptor de la superficie celular (p. ej., tg-TCR o CAR) del mismo a un elemento regulador que actúa en cis (p. ej., una secuencia promotora), y la incorporación de la construcción en un vector de expresión.
La construcción de ácido nucleico de la divulgación también puede incluir un potenciador, una secuencia de iniciación de la transcripción y la traducción, terminador de la transcripción y la traducción y, opcionalmente, una señal de poliadenilación, una LTR en 5', un sitio de unión de ARNt, una señal de empaquetamiento, un origen de la síntesis de la segunda cadena de ADN y una LTR en 3' o una porción de la misma; secuencias de polinucleótidos adicionales que permiten, por ejemplo, la traducción de varias proteínas a partir de un solo ARNm, como un sitio interno de entrada al ribosoma (IRES) y secuencias para la integración genómica del polipéptido promotor quimérico; secuencias diseñadas para aumentar la estabilidad, producción, purificación, rendimiento o toxicidad del péptido expresado.
Los potenciadores regulan la frecuencia de iniciación transcripcional. Normalmente, los elementos promotores están ubicados en la región de 30-110 pb en dirección 5' del sitio de inicio, aunque recientemente se ha visto que varios promotores también contienen elementos funcionales en dirección 3' del sitio de inicio. El espaciado entre los elementos promotores frecuentemente es flexible, de modo que la función del promotor se conserva cuando los elementos se invierten o mueven unos con respecto a otros. En el promotor de la timidina cinasa (tk), el espaciado entre los elementos promotores puede aumentarse hasta 50 pb entre sí antes de que la actividad empiece a disminuir.
Dependiendo del promotor, parece que elementos individuales pueden funcionar cooperativa o independientemente para activar la transcripción.
Un ejemplo de un promotor adecuado es la secuencia promotora temprana inmediata del citomegalovirus (CMV). Esta secuencia promotora es una secuencia promotora constitutiva fuerte capaz de impulsar altos niveles de expresión de cualquier secuencia polinucleotídica operativamente unida a la misma. Otro ejemplo de un promotor adecuado es el factor de crecimiento por elongación 1 alfa. (EF-1 alfa). Sin embargo, también se pueden usar otras secuencias promotoras constitutivas, que incluyen, pero sin limitarse al promotor temprano del virus del simio 40 (SV40), virus de tumor mamario de ratón (MMTV), promotor de la repetición terminal larga (LTR) del virus de la inmunodeficiencia humana (VIH), promotor MoMuLV, un promotor del virus de la leucemia aviar, un promotor temprano inmediato del virus de Epstein-Barr, un promotor del virus del sarcoma de Rous, así como promotores de genes humanos tales como, aunque no de forma limitativa, el promotor de actina, el promotor de miosina, el promotor de hemoglobina y el promotor de creatina cinasa. Además, la divulgación no debe limitarse al uso de promotores constitutivos. Los promotores inducibles también se contemplan como parte de la invención. El uso de un promotor inducible proporciona un interruptor molecular capaz de activar la expresión de la secuencia polinucleotídica a la que está unida operativamente cuando se desea dicha expresión, o desactivar la expresión cuando no se desea la expresión. Los ejemplos de promotores inducibles incluyen, pero no se limitan a un promotor de metalotioneína, un promotor de glucocorticoides, un promotor de progesterona y un promotor de tetraciclina.
El polinucleótido aislado de la divulgación se puede clonar en varios tipos de vectores. Por ejemplo, el polinucleótido aislado se puede clonar en un vector que incluye, pero sin limitación, un plásmido, un fagémido, un derivado de fago, un virus animal y un cósmido. Los vectores de particular interés incluyen vectores de expresión, vectores de replicación, vectores de generación de sondas y vectores de secuenciación.
Los ejemplos de vectores de expresión de mamífero incluyen, pero sin limitación, pcDNA3, pcDNA3.1(+/-), pGL3, pZeoSV2(+/-), pSecTag2, pDisplay, pEHmyc/cyto, pCMV/myc/cyto, pCR3.1, pSinRep5, DH26S, DHBB, pNMT1, pNMT41, pNMT81, que están comercializados por Invitrogen, pCI que está comercializado por Promega, μMbac, pPbac, pBK-RSV y pBK-CMV que están comercializados por Strategene, pTRES que está comercializado por Clontech y sus derivados.
También se pueden utilizar vectores de expresión que contienen elementos reguladores de virus eucariotas tales como retrovirus. Los vectores SV40 incluyen pSVT7 y μMT2. Los vectores procedentes del virus del papiloma bovino incluyen pBV-1MTHA, y los vectores procedentes del virus Epstein Bar incluyen pHEBO y p2O5. Otros vectores ilustrativos incluyen μMSG, pAV009/A+, μMTO10/A+, μMAMneo-5, baculovirus pDSVE, y cualquier otro vector que permita la expresión de proteínas bajo la dirección del promotor temprano SV-40, promotor posterior SV-40, promotor de metalotioneína, promotor del virus del tumor mamario murino, promotor del virus del sarcoma de Rous, promotor de polihedrina u otros promotores que se hayan mostrado eficaces para la expresión en células eucariotas.
Las técnicas de transferencia de ácidos nucleicos in vivo o in vitro actualmente preferidas incluyen la transfección con construcciones virales o no virales, tales como adenovirus, lentivirus, Virus del herpes simple 1, o virus adenoasociado (AAV). Los vectores virales recombinantes ofrecen ventajas como infección lateral y especificidad de direccionamiento. La introducción de ácidos nucleicos por infección viral ofrece varias ventajas sobre otros métodos como la lipofección y la electroporación, ya que se puede obtener una mayor eficiencia de transfección debido a la naturaleza infecciosa de los virus.
La tecnología de vectores virales es bien conocida en la técnica y se describe, por ejemplo, en Sambrook et al. (2001, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, New York), y en otros manuales de virología y biología molecular. Los virus, que son útiles como vectores incluyen, pero sin limitación, retrovirus, adenovirus, virus adenoasociados, virus del herpes y lentivirus. En general, un vector adecuado contiene un origen de replicación funcional en al menos un organismo, una secuencia promotor, sitios de endonucleasas de restricción convenientes, y uno o más marcadores seleccionables, (por ejemplo, los documentos WO 01/96584; WO 01/29058; y la patente de Estados Unidos N.° 6.326.193).
De acuerdo con algunos aspectos de la invención, la construcción de ácido nucleico de la divulgación es un vector viral.
Los vectores derivados de retrovirus tales como el lentivirus son herramientas adecuadas para lograr la transferencia de genes a largo plazo, ya que permiten la transferencia de genes a largo plazo, la integración estable de un transgén y su propagación en células hijas. Los vectores lentivirales tienen la ventaja añadida sobre los vectores procedentes de onco-retrovirus, tales como los virus de la leucemia murina, de que pueden transducir células que no proliferan, como los hepatocitos.
Es más, los vectores lentivirales ofrecen una mayor capacidad de inserción de genes y también tienen la ventaja añadida de una baja inmunogenicidad. De forma alternativa, pueden usarse vectores gamma-retrovirales. Los vectores gamma-retrovirales tienen una buena eficiencia de transducción y no tienen toxicidad asociada al vector [véase, p. ej., Zhang y Morgan, Adv Drug Deliv Rev. (2012) supra].
Por ejemplo, los retrovirus proporcionan una plataforma conveniente para los sistemas de administración de genes. Un gen seleccionado puede insertarse en un vector y empaquetarse en partículas retrovirales utilizando técnicas conocidas en la técnica. A continuación, el virus recombinante se puede aislar y administrar a las células del sujeto, ya sea in vivo o ex-vivo.
Para evaluar la expresión de un polipéptido del receptor de la superficie celular (por ejemplo, tg-TCR o CAR) o porciones del mismo, la construcción de ácido nucleico que se va a introducir en una célula también puede contener un gen marcador seleccionable o un gen indicador o ambos para facilitar la identificación y selección de células de expresión de la población de células que se busca transfectar o infectar a través de vectores virales. En otros aspectos, el marcador seleccionable puede llevarse en una porción separada de ADN y usarse en un procedimiento de cotransfección. Tanto los marcadores seleccionables como los genes indicadores pueden estar flanqueados por secuencias reguladoras apropiadas para permitir la expresión en las células del hospedador. Los marcadores seleccionables útiles incluyen, por ejemplo, genes de resistencia a antibióticos, tales como neo y similares.
Los genes indicadores se utilizan para identificar células potencialmente transfectadas y para evaluar la funcionalidad de las secuencias reguladoras. En general, un gen indicador es un gen que no está presente ni es expresado por el organismo o tejido receptor y que codifica un polipéptido cuya expresión se manifiesta por alguna propiedad fácilmente detectable, por ejemplo, actividad enzimática. La expresión del gen indicador se analiza en un momento adecuado después de que el ADN se haya introducido en las células receptoras. Los genes indicadores adecuados pueden incluir genes que codifican luciferasa, beta-galactosidasa, cloranfenicol acetil transferasa, fosfatasa alcalina secretada, o el gen de la proteína fluorescente verde (p. ej., Ui-Tel et al., 2000 FEBS Letters 479: 79-82). Los sistemas de expresión adecuados son bien conocidos y pueden prepararse utilizando técnicas conocidas u obtenerse comercialmente. En general, la construcción con la región flanqueante en 5' mínima que muestra el mayor nivel de expresión del gen indicador se identifica como el promotor. Dichas regiones promotoras pueden estar unidas a un gen indicador y usarse para evaluar la capacidad de los agentes para modular la transcripción impulsada por el promotor.
Se pueden usar varios métodos para introducir la construcción de ácido nucleico de la divulgación en una célula hospedadora, por ejemplo, una célula de mamífero, bacteriana, de levadura o de insecto. Tales métodos se describen en general en Sambrook et al., Molecular Cloning: A Laboratory Manual, Cold Springs Harbor Laboratory, Nueva York (1989, 1992), en Ausubel et al., Current Protocols in Molecular Biology, John Wiley and Sons, Baltimore, Md. (1989), Chang et al., Somatic Gene Therapy, CRC Press, Ann Arbor, Mich. (1995), Vega et al., Gene Targeting, CRC Press, Ann Arbor Mich. (1995), Vectors: A Survey of Molecular Cloning Vectors and Their Uses, Butterworths, Boston Mass. (1988) y Gilboa et al. [Biotechniques 4 (6): 504-512, 1986] e incluyen, medios físicos, químicos o biológicos (por ejemplo, transfección estable o transitoria, lipofección, electroporación e infección con vectores virales recombinantes). Asimismo, véanse las patentes de EE.UU. números 5.464.764 y 5.487.992 para métodos de selección positivanegativa.
Los métodos físicos para introducir un polinucleótido en una célula hospedadora incluyen la precipitación con fosfato de calcio, lipofección, bombardeo de partículas, microinyección, electroporación, y similares. Los métodos para producir células que comprenden vectores y/o ácidos nucleicos exógenos son bien conocidos en la técnica. Véase, por ejemplo, Sambrook et al. (2001, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Nueva York). Un método preferido para la introducción de un polinucleótido en una célula hospedadora es la transfección con fosfato de calcio.
Los medios químicos para introducir un polinucleótido en una célula hospedadora incluyen sistemas de dispersión coloidal, como complejos de macromoléculas, nanocápsulas, microesferas, microesferas y sistemas basados en lípidos, incluidas emulsiones de aceite en agua, micelas, micelas mixtas y liposomas. Un sistema coloidal ilustrativo para su uso como vehículo de administración in vitro e in vivo es un liposoma (p. ej., una vesícula de membrana artificial).
Los métodos biológicos para introducir un polinucleótido de interés en una célula hospedadora incluyen el uso de vectores de ADN y ARN (como se ha descrito anteriormente). Los vectores virales, y especialmente los vectores retrovirales, se han convertido en el método más utilizado para insertar genes en mamíferos, por ejemplo, células humanas. Otros vectores virales pueden derivarse de lentivirus, poxvirus, virus del herpes simple 1, adenovirus y virus adenoasociados, y similares. Véase, por ejemplo, patentes de EE. UU. números 5.350.674 y 5.585.362.
En el caso de que se utilice un sistema de administración no viral, un vehículo de administración ilustrativo es un liposoma.
"Liposoma" es un término genérico que abarca una variedad de vehículos lipídicos simples y multilaminares formados por la generación de agregados o bicapas lipídicas encerradas. Los liposomas se pueden caracterizar por tener estructuras vesiculares con una membrana bicapa de fosfolípidos y un medio acuoso interno. Los liposomas multilamelares tienen múltiples capas lipídicas separadas por medio acuoso. Se forman espontáneamente cuando se suspenden fosfolípidos en un exceso de solución acuosa.
Los componentes lipídicos se reorganizan antes de la formación de estructuras cerradas, y atrapan agua y solutos disueltos entre las bicapas lipídicas (Ghosh et al., 1991 Glycobiology 5: 505-10). Sin embargo, también se incluyen composiciones que tienen estructuras diferentes en solución que la estructura vesicular normal. Por ejemplo, los lípidos pueden adoptar una estructura micelar o simplemente existir como agregados no uniformes de moléculas lipídicas. También se contemplan los complejos de lipofectamina-ácido nucleico.
Se contempla el uso de formulaciones lipídicas para la introducción de los ácidos nucleicos en una célula hospedadora (in vitro, ex vivo o in vivo). En otro aspecto, el ácido nucleico puede estar asociado con un lípido. El ácido nucleico asociado a un lípido puede encapsularse en el interior acuoso de un liposoma, intercalarse dentro de la bicapa lipídica de un liposoma, unirse a un liposoma a través de una molécula de enlace que está asociada tanto con el liposoma como con el oligonucleótido, atraparse en un liposoma, formar un complejo con un liposoma, dispersarse en una solución que contiene un lípido, mezclarse con un lípido, combinarse con un lípido, estar contenido como una suspensión en un lípido, estar contenido o formando un complejo con una micela, o asociarse de otro modo con un lípido. Las composiciones asociadas a lípido, lípido/ADN o lípido/vector de expresión no se limitan a ninguna estructura particular en solución. Por ejemplo, pueden estar presentes en una estructura bicapa, como micelas, o con una estructura "colapsada". También pueden estar simplemente entremezcladas en una solución, posiblemente formando agregados que no son uniformes en tamaño o forma. Los lípidos son sustancias grasas que pueden ser lípidos naturales o sintéticos. Por ejemplo, los lípidos incluyen las gotitas de grasa que se encuentran de forma natural en el citoplasma, así como la clase de compuestos que contienen hidrocarburos alifáticos de cadena larga y sus derivados, tal como ácidos grasos, alcoholes, aminas, amino alcoholes y aldehídos.
Los lípidos adecuados para su uso se pueden obtener de fuentes comerciales. Por ejemplo, la dimiristil fosfatidilcolina ("DMPc ") se puede obtener en Sigma, St. Louis, Mo.; el fosfato de dicetilo ("DCP") se puede obtener en K & K Laboratories (Plainview, N.Y.); el colesterol ("Choi") se puede obtener en Calbiochem-Behring; el dimiristil fosfatidilglicerol ("DM-PG"); y otros lípidos se pueden obtener en Avanti Polar Lipids, Inc, (Birmingham, Ala.). Adicionalmente, o como alternativa, se pueden usar los lípidos DOTMA, DOPE y DC-Chol [Tonkinson et al., Cancer Investigation, 14(1): 54-65 (1996)]. Las soluciones madre de lípidos en cloroformo o cloroformo/metanol se pueden almacenar a aproximadamente -20 °C. El cloroformo se usa como único disolvente ya que se evapora más fácilmente que el metanol.
Otro sistema de administración no viral ilustrativo que se puede usar de acuerdo con la presente divulgación es un sistema de administración de genes no virales basado en transposones, como p. ej., Sleeping Beauty o PiggyBac.
Independientemente del método utilizado para introducir ácidos nucleicos exógenos en una célula hospedadora, para confirmar la presencia de la secuencia de ADN recombinante en la célula hospedadora, se puede realizar una variedad de ensayos. Tales ensayos incluyen, por ejemplo, ensayos de "biología molecular" bien conocidos por los expertos en la materia, como transferencia Southern y Northern, RT-PCR y PCR; ensayos "bioquímicos", tales como la detección de la presencia o ausencia de un péptido particular, por ejemplo, por medios inmunológicos (ELISA y transferencias Western) o mediante ensayos descritos en el presente documento para identificar agentes que entran dentro del alcance de la invención.
Se apreciará que la célula transducida con el receptor de la superficie celular (p. ej., tg-TCR y/o CAR) puede modificarse genéticamente adicionalmente para reprimir la expresión de al menos un gen de punto de control inmunitario endógeno en la célula.
El gen del punto de control inmunitario puede comprender un gen PD o CTLA.
Como se usa en el presente documento, la expresión "gen de punto de control inmunitario" se refiere a cualquier gen que esté involucrado en un proceso inhibidor (p. ej., bucle de retroalimentación) que actúa para regular la amplitud de una respuesta inmunitaria, por ejemplo, un bucle de retroalimentación inhibidor inmunitario que mitiga la propagación descontrolada de respuestas inmunitarias dañinas.
Los ejemplos no limitativos de genes de puntos de control inmunitario incluyen miembros de la familia ampliada de receptores CD28 y sus ligandos, así como genes implicados en vías co-inhibitorias (p. ej., CTLA-4 y PD-1).
Por tanto, de acuerdo con un aspecto, se pueden utilizar nucleasas o represores de transcripción dirigidos a PD1 y/o CTLA-4 como se describe en la solicitud de patente de EE. UU. N.° 20140120622.
Adicionalmente, o como alternativa, las proteínas de punto de control inmunitario, que regulan la activación o función de un linfocito T, incluyendo, por ejemplo, PD1, PDL-1, B7H2, B7H4, CTLA-4, CD80, CD86, LAG-3, TIM-3, KIR, IDO, CD19, OX40, 4-1BB (CD137), CD27, CD70, CD40, GITR, CD28 y/o ICOS (CD278), pueden modularse (por ejemplo, regularse al alza o a la baja según sea necesario) en la célula transducida mediante el uso de un regulador del punto de control inmunitario.
De acuerdo con aspectos específicos, el regulador del punto de control inmunitario se selecciona del grupo que consiste en anti-CTLA4, anti-PD-1, anti-PDL-1, agonista de CD40, agonista de 4-1BB, agonista de GITR y agonista
de OX40.
De acuerdo con un aspecto de algunos aspectos de la divulgación, se proporciona una población aislada de células que comprende la célula aislada de algunos aspectos de la invención.
La célula aislada o la población de células de algunos aspectos de la divulgación se pueden administrar a un organismo per se, o en una composición farmacéutica donde se mezcla con vehículos o excipientes adecuados.
Como se usa en el presente documento, una "composición farmacéutica" se refiere a una preparación de uno o más de los principios activos descritos en el presente documento con otros componentes químicos tales como vehículos y excipientes fisiológicamente adecuados. El fin de una composición farmacéutica es facilitar la administración de un compuesto a un organismo.
En el presente documento, la expresión "principio activo" se refiere a las células de algunos aspectos de la descripción responsables del efecto biológico.
En lo sucesivo en el presente documento, las expresiones "vehículo fisiológicamente aceptable" y "vehículo farmacéuticamente aceptable", que pueden usarse indistintamente, se refieren a un vehículo o un diluyente que no provoca irritación significativa a un organismo y no anula la actividad biológica y las propiedades del compuesto administrado. En estas expresiones se incluye un adyuvante.
En el presente documento el término "excipiente" se refiere a una sustancia inerte añadida a una composición farmacéutica para facilitar adicionalmente la administración de un principio activo. Los ejemplos, sin limitación, de excipientes incluyen carbonato de calcio, fosfato de calcio, diversos azúcares y tipos de almidón, derivados de celulosa, gelatina, aceites vegetales y polietilenglicoles.
Pueden encontrarse técnicas para la formulación y administración de fármacos en "Remington's Pharmaceutical Sciences", Mack Publishing Co., Easton, Pensilvania, última edición.
Las vías de administración adecuadas pueden, por ejemplo, incluir la administración oral, rectal, transmucosa, especialmente transnasal, intestinal o parenteral, incluyendo las inyecciones intramusculares, subcutáneas e intramedulares, así como intratecales, intraventricular directa, intracardíaca, por ejemplo, en la cavidad ventricular derecha o izquierda, en la arteria coronaria común, intravenosa, intraperitoneal, intranasal o intraocular.
Los enfoques convencionales para la administración de fármacos al sistema nervioso central (SNC) incluyen: estrategias neuroquirúrgicas (por ejemplo, inyección intracerebral o infusión intracerebroventricular); manipulación molecular del agente (por ejemplo, producción de una proteína de fusión quimérica que comprende un péptido de transporte que tiene una afinidad por una molécula de superficie celular endotelial en combinación con un agente que en sí mismo es incapaz de cruzar la BHE) en un intento de aprovechar una de las rutas de transporte endógenas de la BHE; estrategias farmacológicas diseñadas para aumentar la solubilidad en lípidos de un agente (por ejemplo, conjugación de agentes hidrosolubles con transportadores lipídicos o de colesterol); y la alteración transitoria de la integridad de la BHE mediante alteración hiperosmótica (que es el resultado de la infusión de una solución de manitol en la arteria carótida o del uso de un agente biológicamente activo tal como un péptido de angiotensina). Sin embargo, cada una de estas estrategias tiene limitaciones, tales como los riesgos intrínsecos asociados con un procedimiento quirúrgico invasivo, una limitación de tamaño impuesta por una limitación intrínseca a los sistemas de transporte endógeno, efectos secundarios biológicos potencialmente indeseables asociados con la administración sistémica de una molécula quimérica compuesta por un motivo transportador que podría estar activo fuera del SNC y el posible riesgo de daño cerebral dentro de las regiones del cerebro donde se altera la BHE, lo que lo convierte en un método de administración deficiente.
Como alternativa, puede administrarse la composición farmacéutica de una manera local en lugar de sistémica, por ejemplo, mediante inyección de la composición farmacéutica directamente en una región tisular de un paciente.
De acuerdo con un aspecto, la vía de administración incluye, por ejemplo, una inyección, ingestión, transfusión, implante o trasplante. Las composiciones descritas en el presente documento pueden administrarse a un paciente por vía subcutánea, intradérmica, intratumoral, intraganglionar, intramedular, intramuscular, por inyección intravenosa (i.v.), o por vía intraperitoneal. En un aspecto, la composición farmacéutica de la presente divulgación se administra a un paciente mediante inyección intradérmica o subcutánea. En otro aspecto, la composición farmacéutica de la presente divulgación se administra preferentemente mediante inyección i.v. La composición farmacéutica se puede inyectar directamente en un tumor, ganglio linfático o sitio de infección.
Las composiciones farmacéuticas de algunos aspectos de la divulgación pueden fabricarse mediante procesos bien conocidos en la técnica, por ejemplo, por medio de procesos convencionales de mezcla, disolución, granulación, preparación de grageas, levitación, emulsionado, encapsulación, atrapamiento o liofilización.
Las composiciones farmacéuticas para su uso de acuerdo con algunos aspectos de la divulgación pueden formularse
de esta forma de manera convencional usando uno o más vehículos fisiológicamente aceptables que comprenden excipientes y adyuvantes, que facilitan el procesamiento de los principios activos en preparaciones que, pueden usarse farmacéuticamente. La formulación adecuada depende de la vía de administración elegida.
Para inyección, los principios activos de la composición farmacéutica pueden formularse en soluciones acuosas, preferentemente, en tampones fisiológicamente compatibles, tales como solución de Hank, solución de Ringer o tampón salino fisiológico. Para la administración transmucosa, se usan en la formulación penetrantes apropiados para la barrera a permear. Dichos penetrantes son generalmente conocidos en la técnica.
Para administración oral, la composición farmacéutica puede formularse fácilmente combinando el compuesto activo con vehículos farmacéuticamente aceptables bien conocidos en la técnica. Tales vehículos permiten que la composición farmacéutica se formule como comprimidos, píldoras, grageas, cápsulas, líquidos, geles, jarabes, pastas, suspensiones y similares, para la ingesta oral por un paciente. Pueden prepararse preparaciones farmacológicas para su uso oral usando un excipiente sólido, opcionalmente moliendo la mezcla resultante y procesando la mezcla de gránulos, después de añadir auxiliares adecuados si se desea, para obtener comprimidos o núcleos de grageas. Son excipientes adecuados, en particular, cargas, tales como azúcares, entre las que se incluyen lactosa, sacarosa, manitol o sorbitol; preparaciones de celulosa tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma de tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carbometilcelulosa de sodio; y/o polímeros fisiológicamente aceptables tales como polivinilpirrolidona (PVP). Si se desea, pueden añadirse agentes disgregantes, tales como polivinilpirrolidona reticulada, agar o ácido algínico o una sal del mismo, tal como alginato de sodio.
Los núcleos de grageas se proporcionan con recubrimientos adecuados. Para este fin, pueden usarse soluciones concentradas de azúcar que opcionalmente pueden contener goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol, dióxido de titanio, soluciones de laca y disolventes orgánicos o mezclas de disolventes adecuados. Pueden añadirse colorantes o pigmentos a los comprimidos o recubrimientos de grageas para la identificación o para caracterizar distintas combinaciones de dosis de compuestos activos.
Las composiciones farmacéuticas que se pueden usar por vía oral, incluyen cápsulas de encaje a presión hechas de gelatina, así como cápsulas blandas selladas hechas de gelatina y un plastificante, tal como glicerol o sorbitol. Las cápsulas de ajuste por presión pueden contener los principios activos en mezcla con una carga tal como lactosa, aglutinantes tales como almidones, lubricantes tales como talco o estearato de magnesio y, opcionalmente, estabilizantes. En las cápsulas blandas, los principios activos pueden disolverse o suspenderse en líquidos adecuados, tales como aceites grasos, parafina líquida o polietilenglicoles líquidos. Asimismo, pueden añadirse estabilizantes. Todas las formulaciones para administración oral deben estar en dosificaciones adecuadas para la vía de administración elegida.
Para administración bucal, las composiciones pueden adoptar la forma de comprimidos o grageas formulados de forma convencional.
Para la administración mediante inhalación nasal, los principios activos para su uso de acuerdo con algunos aspectos de la divulgación se suministran convenientemente en forma de una presentación de pulverización en aerosol de un envase presurizado o un nebulizador con el uso de un propulsor adecuado, por ejemplo, diclorodifluorometano, triclorofluorometano, dicloro-tetrafluoroetano o dióxido de carbono. En el caso de un aerosol presurizado, la unidad de dosificación puede determinarse dotándolo de una válvula para suministrar una cantidad medida. Las cápsulas y cartuchos de, por ejemplo, gelatina, para su uso en un dosificador pueden formularse conteniendo una mezcla de polvo del compuesto y una base de polvo adecuada, tal como lactosa o almidón.
La composición farmacéutica descrita en el presente documento puede formularse para administración parenteral, por ejemplo, mediante inyección intravenosa rápida o infusión continua.
Las formulaciones para inyección pueden presentarse en una forma farmacéutica unitaria, por ejemplo, en ampollas o en recipientes multidosis con, opcionalmente, un conservante añadido. Las composiciones pueden ser suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos y pueden comprender agentes de formulación, tales como agentes de suspensión, estabilizantes y/o dispersantes.
Las composiciones farmacéuticas para administración parenteral incluyen soluciones acuosas de la preparación de principio activo en forma hidrosoluble. Adicionalmente, pueden prepararse suspensiones de los principios activos como suspensiones para inyección de base oleaginosa o acuosa apropiadas. Los disolventes o vehículos lipófilos adecuados incluyen aceites grasos tales como aceite de sésamo o ésteres de ácidos grasos sintéticos tales como oleato de etilo, triglicéridos o liposomas.
Las suspensiones de inyección acuosas pueden contener sustancias, que aumentan la viscosidad de la suspensión, tales como carboximetilcelulosa sódica, sorbitol o dextrano. Opcionalmente, la suspensión también puede contener estabilizantes o agentes adecuados que aumenten la solubilidad de los principios activos para permitir la preparación de soluciones altamente concentradas.
De forma alternativa, el principio activo puede estar en forma de polvo para constitución con un vehículo adecuado, por ejemplo, solución basada en agua estéril, apirógena, antes de su uso.
La composición farmacéutica de algunos aspectos de la divulgación también puede formularse en composiciones rectales tales como supositorios o enemas de retención, usando, por ejemplo, bases de supositorio convencionales tales como manteca de cacao u otros glicéridos.
Las composiciones farmacéuticas adecuadas para su uso en el contexto de la divulgación incluyen composiciones en donde los principios activos están contenidos en una cantidad eficaz para lograr el fin pretendido. Más específicamente, una cantidad terapéuticamente eficaz significa una cantidad de principios activos eficaces para prevenir, aliviar o mejorar síntomas de una patología o prolongar la supervivencia del sujeto que se está tratando.
La determinación de una cantidad terapéuticamente eficaz se encuentra dentro de la capacidad de los expertos en la materia, especialmente a la luz de la divulgación detallada proporcionada en el presente documento.
Cuando se indica "cantidad terapéutica", la cantidad precisa de las composiciones de la presente divulgación que se administrarán se puede determinar por un médico teniendo en cuenta las diferencias individuales de edad, peso, patología, p.ej. tamaño del tumor, extensión de la infección o metástasis, y la condición del paciente (sujeto). En general, se puede afirmar que una composición farmacéutica que comprende las células descritas en el presente documento se puede administrar a una dosis de 104 a 109 células/kg de peso corporal, incluyendo todos los valores enteros dentro de esos intervalos.
Por ejemplo, el número de células infundidas a un receptor debe ser superior a 1 x 104/kg de peso corporal. El número de células infundidas a un receptor normalmente debe estar en el intervalo de 1 x 103/kg de peso corporal a 1 x 104/kg de peso corporal, intervalo de 1 x 104/kg de peso corporal a 1 x 105/kg de peso corporal, intervalo de 1 x 104/kg de peso corporal a 1 x 106/kg de peso corporal, intervalo de 1 x 104 /kg de peso corporal a 10 x 107/kg de peso corporal, intervalo de 1 x 104/kg de peso corporal a 1 x 108 /kg de peso corporal, intervalo de 1 x 103/kg de peso corporal a 1 x 105/kg de peso corporal, intervalo de 1 x 104/kg de peso corporal a 1 x 106/kg de peso corporal, intervalo de 1 x 106 /kg de peso corporal a 10 x 107/kg de peso corporal, intervalo de 1 x 105 /kg de peso corporal a 10 x 107/kg de peso corporal, intervalo de 1 x 106/kg de peso corporal a 1 x 108/kg de peso corporal, o intervalo de 1 x 106/kg de peso corporal a 1 x 109/kg de peso corporal. De acuerdo con un aspecto específico, el número de células infundidas a un receptor debe estar en el intervalo de 1 x 106/kg de peso corporal a 10 x 108/kg de peso corporal.
Las composiciones celulares de algunos aspectos de la divulgación también pueden administrarse múltiples veces en estas dosis. Las células se pueden administrar usando técnicas de infusión que se conocen comúnmente en inmunoterapia (véase, por ejemplo, Rosenberg et al., New Eng. J. of Med. 319:1676, 1988). Un experto en la materia de la medicina puede determinar fácilmente la dosificación y el régimen de tratamiento óptimos para un paciente particular mediante el seguimiento del paciente en busca de signos de enfermedad y ajustando el tratamiento en consecuencia.
Por ejemplo, el efecto de los principios activos (p. ej., las células de algunos aspectos de la invención) sobre la patología puede evaluarse controlando el nivel de marcadores, por ejemplo, hormonas, glucosa, péptidos, hidratos de carbono, etc. en una muestra biológica del sujeto tratado utilizando métodos bien conocidos (por ejemplo, ELISA, FACS, etc) o controlando el tamaño del tumor utilizando métodos bien conocidos (por ejemplo, ultrasonido, TC, RM, etc.).
Para cualquier preparación utilizada en los métodos de la invención, la cantidad o dosis terapéuticamente eficaz puede estimarse inicialmente a partir de ensayos de cultivo celular e in vitro. Por ejemplo, puede formularse una dosis en modelos animales para conseguir una concentración o un título deseado. Dicha información puede usarse para determinar de forma más precisa las dosis útiles en seres humanos.
La toxicidad y la eficacia terapéutica de los principios activos descritos en el presente documento pueden determinarse mediante procedimientos farmacéuticos convencionales in vitro, en cultivos celulares o animales experimentales. Los datos obtenidos de estos ensayos in vitro y de cultivo celular y estudios animales pueden utilizarse en la formulación de un intervalo de dosificación para su uso en el ser humano.
La dosificación puede variar dependiendo de la forma de dosificación empleada y la vía de administración utilizada. La formulación exacta, la vía de administración y la dosificación pueden ser elegidas por el médico individual a la vista del estado del paciente. (Véase, p. ej., Fingl, et al., 1975, en "The Pharmacological Basis of Therapeutics", cap. 1 pág.
1).
La cantidad y el intervalo de dosificación pueden ajustarse individualmente para proporcionar niveles del principio activo que sean suficientes para inducir o suprimir el efecto biológico (concentración eficaz mínima, CEM). La CEM variará para cada preparación, pero puede estimarse a partir de datos in vitro. Las dosificaciones necesarias para lograr la CEM dependerán de las características individuales y de la vía de administración. Pueden usarse ensayos
de detección para determinar las concentraciones plasmáticas.
Dependiendo de la gravedad y el grado de respuesta de la afección a tratar, la dosificación puede ser de una única administración o una pluralidad de administraciones, durando el ciclo de tratamiento de varios días a varias semanas o hasta que se efectúe la curación o se logre la disminución de la patología.
La cantidad de una composición a administrar será, por supuesto, dependiente del sujeto que se esté tratando, la gravedad de la afección, la forma de administración, el criterio del médico que la receta, etc.
De acuerdo con algunos aspectos de la invención, el agente terapéutico de la divulgación se puede proporcionar al sujeto junto con otro(s) fármaco(s) diseñado(s) para tratar la patología [terapia de combinación, (por ejemplo, antes, simultáneamente o después)].
En ciertos aspectos de la presente invención, las células de algunos aspectos de la divulgación se administran a un paciente junto con cualquier número de modalidades de tratamiento relevantes, incluyendo pero sin limitarse al tratamiento con agentes tales como agentes antivirales (por ejemplo, ganciclovir, valaciclovir, aciclovir, valganciclovir, foscarnet, cidofovir, maribavir, leflunomida); agentes quimioterapéuticos (por ejemplo, agentes antineoplásicos, tales como, entre otros, agentes alquilantes que incluyen, p. ej., ciclofosfamida, busulfán, mecloretamina o mostina (HN2), uramustina o mostaza de uracilo, melfalán, clorambucilo, ifosfamida, bendamustina, nitrosoureas carmustina, lomustina, estreptozocina, tiotepa, cisplatino, carboplatino, nedaplatino, oxaliplatino, satraplatino, triplatino tetranitrato, procarbazina, altretamina, triacenos (dacarbazina, mitozolomida, temozolomida), dacarbazina, temozolomida, Myleran, Busulfex, fludarabina, dimetilmilerano o citarabina); agentes para el tratamiento de la EM (por ejemplo, natalizumab); o agentes para el tratamiento de la psoriasis (por ejemplo, efalizumab).
En aspectos adicionales, las células de algunos aspectos de la divulgación pueden usarse en combinación con quimioterapia, radiación, agentes inmunosupresores (por ejemplo, ciclosporina, azatioprina, metotrexato, micofenolato y FK506), anticuerpos u otros agentes inmunosupresores (descritos con más detalle a continuación).
En un aspecto adicional, las composiciones de células de algunos aspectos de la divulgación se administran a un paciente junto con (por ejemplo, antes, simultáneamente o después de) un trasplante de médula ósea.
En un aspecto adicional, las composiciones de células de algunos aspectos de la divulgación se administran a un paciente después de una terapia de ablación de linfocitos T usando, por ejemplo, agentes de quimioterapia tales como, fludarabina, radioterapia de haz externo (XRT), ciclofosfamida o anticuerpos, tales como OKT3 o CAMPATH.
En otro aspecto, las composiciones de células de la presente divulgación se administran después de la terapia ablativa de linfocitos B, tales como agentes que reaccionan con CD20, por ejemplo, Rituxan.
La terapia de combinación puede aumentar el efecto terapéutico del agente de la divulgación en el sujeto tratado.
Las composiciones de algunos aspectos de la divulgación pueden, si se desea, presentarse en un envase o dispositivo dosificador, tal como un kit aprobado por la FDA, que puede contener una o más formas de dosificación unitaria que contienen el principio activo. El envase puede, por ejemplo, comprender papel metálico o de plástico, tal como un envase de tipo blíster. El envase o dispositivo dosificador puede estar acompañado de instrucciones para la administración. El envase o dosificador también puede estar acompañado de una nota asociada con el recipiente en una forma prescrita por un organismo público que regule la fabricación, el uso o la venta de productos farmacéuticos, reflejando dicha nota la aprobación por parte del organismo de la forma de las composiciones o la administración humana o veterinaria. Dicha nota, por ejemplo, puede ser un etiquetado aprobado por la Administración de Fármacos y Alimentos de EE. UU. para fármacos con receta o un prospecto de producto aprobado. Además, pueden prepararse composiciones que comprenden una preparación de la divulgación formulada en un vehículo farmacéutico compatible, colocarse en un recipiente adecuado y marcarse para el tratamiento de una afección indicada, como se ha detallado anteriormente de manera adicional.
El kit puede, por ejemplo, comprender papel metálico o de plástico, tal como un envase de tipo blíster. El envase o dispositivo dosificador puede estar acompañado de instrucciones para la administración. El envase o dosificador también puede estar acompañado de una nota asociada con el recipiente en una forma prescrita por un organismo público que regule la fabricación, el uso o la venta de productos farmacéuticos, reflejando dicha nota la aprobación por parte del organismo de la forma de las composiciones o la administración humana o veterinaria. Dicha nota, por ejemplo, puede ser un etiquetado aprobado por la Administración de Fármacos y Alimentos de EE. UU. para fármacos con receta o un prospecto de producto aprobado. Además, pueden prepararse composiciones que comprenden una preparación de la divulgación formulada en un vehículo farmacéutico compatible, colocarse en un recipiente adecuado y marcarse para el tratamiento de una afección indicada, como se ha detallado anteriormente de manera adicional.
De acuerdo con un aspecto, el kit comprende además un agente quimioterapéutico (p. ej., un agente antineoplásico, como se ha descrito en detalle anteriormente).
De acuerdo con un aspecto, el kit comprende además un agente antiviral (como se ha descrito en detalle anteriormente).
De acuerdo con un aspecto que no forma parte de la invención, se proporciona un método para tratar una enfermedad en un sujeto que lo necesita, comprendiendo el método administrar al sujeto una cantidad terapéuticamente eficaz de la población de células de algunos aspectos de la invención, tratando de esta manera al sujeto.
De acuerdo con un aspecto de algunos aspectos de la invención, se proporciona una cantidad terapéuticamente eficaz de la población de células de algunos aspectos de la divulgación para su uso en el tratamiento de una enfermedad en un sujeto que lo necesite.
El término "tratar" se refiere a inhibir, prevenir o detener el desarrollo de una patología (enfermedad, trastorno o afección) y/o provocar la reducción, remisión o regresión de una patología. Los expertos en la materia entenderán que pueden usarse diversas metodologías y ensayos para evaluar el desarrollo de una patología y, de manera similar, pueden utilizarse diversas metodologías y ensayos para evaluar la reducción, remisión o regresión de una patología.
Como se usa en el presente documento, el término "sujeto" incluye mamíferos, preferentemente seres humanos de cualquier edad o sexo que padezcan la patología.
La patología puede ser, pero sin limitación, una enfermedad maligna (cáncer), una enfermedad infecciosa (por ejemplo, infección viral, infección bacteriana, infección fúngica, infección por protozoos o infecciones parasitarias), una alergia y/o una enfermedad autoinmunitaria.
Enfermedades cancerosas
Las enfermedades malignas (también denominadas cánceres) que pueden tratarse mediante el método de algunos aspectos de la divulgación pueden ser cualquier tumor sólido o no sólido y/o metástasis tumoral.
Los ejemplos de cáncer incluyen, pero sin limitación, carcinoma, linfoma, blastoma, sarcoma y leucemia. Los ejemplos más particulares de dichos cánceres incluyen cáncer de células escamosas, sarcoma de partes blandas, sarcoma de Kaposi, melanoma, cáncer de pulmón (incluyendo cáncer de pulmón microcítico, cáncer de pulmón no microcítico, adenocarcinoma de pulmón y carcinoma epidermoide de pulmón), cáncer de peritoneo, cáncer hepatocelular, cáncer gástrico o de estómago (incluyendo cáncer gastrointestinal), cáncer de páncreas, glioblastoma, cáncer de cuello uterino, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama, cáncer de colon, cáncer colorrectal, cáncer rectal, carcinoma de endometrio o uterino, carcinoma carcinoide, carcinoma de las glándulas salivales, cáncer de riñón o renal, cáncer de hígado, cáncer de próstata, cáncer de vulva, cáncer de tiroides, carcinoma hepático, mesotelioma, mieloma múltiple, trastorno linfoproliferativo postrasplante (PTLD), y varios tipos de cáncer de cabeza y cuello (por ejemplo, tumor cerebral). Las afecciones cancerosas susceptibles de tratamiento de la divulgación incluyen cánceres metastásicos.
De acuerdo con un aspecto, la enfermedad maligna es una neoplasia hematológica. Las neoplasias hematológicas ilustrativas incluyen, pero sin limitación, leucemia [por ejemplo, linfática aguda, linfoblástica aguda, leucemia linfoblástica aguda de linfocitos pre-B, leucemia linfoblástica aguda de linfocitos T, aguda - megacarioblástica, monocítica, mielógena aguda, mieloide aguda, mieloide aguda con eosinofilia, de linfocitos B, basófila, mieloide crónica, crónica, de linfocitos B, eosinofílica, de Friend, granulocítica o mielocítica, de células pilosas, linfocítica, megacarioblástica, monocítica, monocítica/macrófagos, mieloblástica, mieloide, mielomonocítica, de plasmocitos, de linfocitos pre-B, promielocítica, subaguda, de linfocitos T, neoplasia linfoide, predisposición a la malignidad mieloide, leucemia no linfocítica aguda, leucemia linfocítica aguda de linfocitos T (LLA-T) y leucemia linfocítica crónica de linfocitos B (LLC-B)] y linfoma [p. ej., enfermedad de Hodgkin, linfoma no hodgkiniano, de Burkitt, cutáneo de linfocitos T, histiocítico, linfoblástico, de linfocitos T, tímico, de linfocitos B, incluyendo bajo grado/folicular; NHL linfocítico de células pequeñas (SL); NHL de grado intermedio/folicular; NHL difuso de grado intermedio; NHL inmunoblástico de alto grado; NHL linfoblástico de alto grado; NHL de células no escindidas pequeñas de alto grado; NHL con neoplasia maligna; linfoma de células del manto; linfoma relacionado con SIDA; y macroglobulinemia de Waldenstrom].
De acuerdo con un aspecto específico, la enfermedad maligna es una leucemia, un linfoma, un mieloma, un melanoma, un sarcoma, un neuroblastoma, un cáncer de colon, un cáncer colorrectal, un cáncer de mama, un cáncer de ovario, un cáncer de esófago, un cáncer de células sinoviales o un cáncer de páncreas.
De acuerdo con algunos aspectos de la invención, la patología es un tumor sólido. De acuerdo con algunos aspectos de la invención, la patología es una metástasis tumoral. De acuerdo con algunos aspectos de la invención, la patología es una neoplasia hematológica.
De acuerdo con algunos aspectos de la invención, la patología es una leucemia o un linfoma.
Se enumeran ejemplos de enfermedades malignas que son tratables mediante los métodos de algunos aspectos de la divulgación en las Tablas 1 y 2, a continuación.
Tabla 1: Aplicaciones clínicas que utilizan células transducidas con tg-TCR con regímenes de preacondicionamiento o cionales
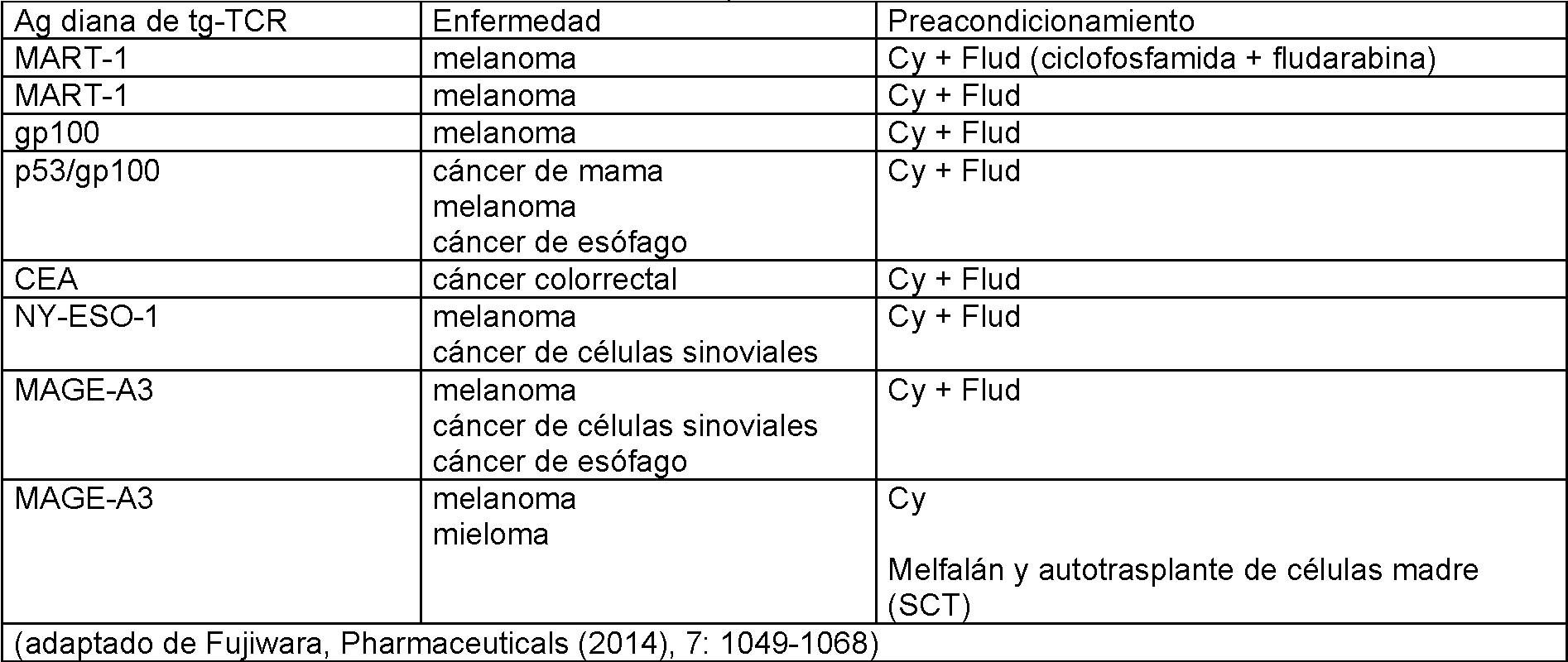
Tabla 2: Aplicaciones clínicas que utilizan células transducidas con CAR con regímenes de preacondicionamiento o cionales

De acuerdo con un aspecto específico, la enfermedad maligna es una leucemia, un linfoma, un mieloma, un melanoma, un sarcoma, un neuroblastoma, un cáncer de colon, un cáncer colorrectal, un cáncer de mama, un cáncer de ovario, un cáncer de esófago, un cáncer de células sinoviales y un cáncer de páncreas.
Enfermedades infecciosas
Los ejemplos de enfermedades infecciosas incluyen, pero sin limitación, enfermedades infecciosas crónicas, enfermedades infecciosas subagudas, enfermedades infecciosas agudas, enfermedades víricas, enfermedades bacterianas, enfermedades por protozoos, enfermedades parasitarias, enfermedades fúngicas, enfermedades por micoplasma y enfermedades por priones.
Los tipos específicos de patógenos virales que causan enfermedades infecciosas tratables de acuerdo con las enseñanzas de la presente divulgación incluyen, pero sin limitación, retrovirus, circovirus, parvovirus, papovavirus, adenovirus, herpesvirus, iridovirus, poxvirus, hepadnavirus, picornavirus, calicivirus, togavirus, flavivirus, reovirus, ortomixovirus, paramixovirus, rabdovirus, bunyavirus, coronavirus, arenavirus y filovirus.
Los ejemplos específicos de infecciones virales que pueden tratarse de acuerdo con las enseñanzas de la presente divulgación incluyen, pero sin limitación, virus de la inmunodeficiencia humana (VIH) o síndrome de inmunodeficiencia adquirida (SIDA), virus de la gripe, infección por rinovirus, meningitis vírica, infección por virus de Epstein Barr (EBV), infección por virus de la hepatitis A, B o C, sarampión, infección por el virus del papiloma/verrugas, infección por citomegalovirus (CMV), infecciones por el virus Herpes simplex, fiebre amarilla, infección por el virus del Ébola y rabia.
De acuerdo con un aspecto específico, la enfermedad viral se selecciona del grupo que consiste en una infección por el virus de la inmunodeficiencia (VIH), virus de la gripe, citomegalovirus (CMV), virus de la leucemia de linfocitos T tipo 1 (TAX), virus de la hepatitis C (VHC) y virus de la hepatitis B (VHB).
Enfermedades alérgicas
Ejemplos de enfermedades alérgicas incluyen, pero sin limitación, asma, urticaria, urticaria, alergia al polen, alergia a los ácaros del polvo, alergia al veneno, alergia a los cosméticos, alergia al látex, alergia química, alergia a fármacos, alergia a las picaduras de insectos, alergia a la caspa animal, alergia a las plantas urticantes, alergia a la hiedra venenosa y alergia alimentaria.
Enfermedades autoinmunitarias
Incluyen, pero sin limitación, enfermedades cardiovasculares, enfermedades reumáticas, enfermedades glandulares, gastroenteropatías, enfermedades cutáneas, enfermedades hepáticas, enfermedades neurológicas, enfermedades musculares, enfermedades nefríticas, enfermedades relacionadas con la reproducción, enfermedades del tejido conjuntivo y enfermedades sistémicas.
Los ejemplos de enfermedades cardiovasculares autoinmunitarias incluyen, pero sin limitación, ateroesclerosis (Matsuura E. et al., Lupus. 1998;7 Suμl. 2:S135), infarto de miocardio (Vaarala O. Lupus. 1998;7 Suμl. 2:S132), trombosis (Tincani A. et al., Lupus 1998;7 Suμl. 2:S 107-9), granulomatosis de Wegener, arteritis de Takayasu, síndrome de Kawasaki (Praprotnik S. et al., Wien Klin Wochenschr 25 de agosto de 2000;112 (15-16):660), enfermedad autoinmunitaria anti-factor VIII (Lacroix-Desmazes S. et al., Semin Thromb Hemost.2000;26 (2):157), vasculitis necrosante de vasos pequeños, poliangitis microscópica, síndrome de Churg y Strauss, glomerulonefritis necrosante focal pauci-inmunitaria y con semilunas (Noel LH. Ann Med Interne (París). mayo de 2000;151 (3): 178), síndrome antifosfolípido (Flamholz R. et al., J Clin Apheresis 1999;14 (4):171), insuficiencia cardiaca inducida por anticuerpos (Wallukat G. et al., Am J Cardiol. 17 de junio de 1999;83 (12A):75H), púrpura tromobocitopénica (Moccia F. Ann Ital Med Int. abril-junio de 1999;14 (2):114); Semple JW. et al., Blood 15 de mayo de 1996;87(10):4245), anemia hemolítica autoinmunitaria (Efremov DG. et al., Leuk Lymphoma 1998 Jan;28(3-4):285; Sallah S. et al., Ann Hematol marzo de 1997;74 (3):139), autoinmunidad cardíaca en la enfermedad de Chagas (Cunha-Neto E. et al., J Clin Invest 15 de octubre de 1996; 98 (8): 1709) y autoinmunidad anti-linfocitos T cooperadores (Caporossi AP. et al., Viral Immunol 1998; 11 (1):9).
Los ejemplos de enfermedades reumáticas autoinmunitarias incluyen, pero no se limitan a artritis reumatoide (Krenn V. et al., Histol Histopathol (2000) 15 (3):791; Tisch R y McDevitt HO. Proc Natl Acad Sci USA (1994) 18; 91(2): 437 438] y espondilitis anquilosante [Jan Voswinkel et al., Arthritis Res (2001) 3 (3): 189].
Los ejemplos de enfermedades glandulares autoinmunitarias incluyen, pero sin limitación, enfermedad pancreática, diabetes de tipo I, enfermedad tiroidea, enfermedad de Graves, tiroiditis, tiroiditis autoinmunitaria espontánea, tiroiditis de Hashimoto, mixedema idiopático, autoinmunidad ovárica, infertilidad autoinmunitaria anti-espermatozoides, prostatitis autoinmunitaria y síndrome poliglandular autoinmunitario de tipo I. Las enfermedades incluyen, pero sin limitación, enfermedades autoinmunitarias del páncreas, diabetes de tipo 1 (Castano L. y Eisenbarth GS. Ann. Rev. Immunol. 8:647; Zimmet P. Diabetes Res Clin Pract octubre de 1996;34 Suppl:S125), enfermedades autoinmunitarias tiroideas, enfermedad de Graves (Orgiazzi J. Endocrinol Metab Clin North Am junio de 2000;29 (2): 339; Sakata S. et al., Mol Cell Endocrinol marzo de 1993;92 (1):77), tiroiditis autoinmunitaria espontánea (Braley-Mullen H. y Yu S, J Immunol 15 de diciembre de 2000;165 (12):7262), tiroiditis de Hashimoto (Toyoda N. et al., Nippon Rinsho agosto de 1999; 57 (8):1810), mixedema idiopático (Mitsuma T. Nippon Rinsho. agosto de 1999; 57 (8):1759), autoinmunidad ovárica (Garza KM. et al., J Reprod Immunol febrero de 1998;37 (2):87), infertilidad autoinmunitaria anti espermatozoides (Diekman AB. et al., Am J Reprod Immunol. marzo de 2000;43-3): 134), prostatitis autoinmunitaria (Alexander RB. et al., Urology diciembre de 1997;50 (6): 893) y síndrome poliglandular autoinmunitario de Tipo I (Hara T. et al., Blood. 1 de marzo de 1991;77 (5):1127).
Los ejemplos de enfermedades gastrointestinales autoinmunitarias incluyen, pero sin limitación, enfermedades inflamatorias intestinales crónicas (Garcia Herola A. et al., Gastroenterol Hepatol. enero de 2000;23 (1):16) enfermedad celíaca (Landau YE. y Shoenfeld Y. Harefuah 16 de enero de 2000;138 (2):122), colitis, ileítis y enfermedad de Crohn.
Los ejemplos de enfermedades cutáneas autoinmunitarias incluyen, pero sin limitación, enfermedades cutáneas ampollosas autoinmunitarias, tal como, pero sin limitación, pénfigo vulgar, penfigoide ampolloso y pénfigo foliáceo.
Los ejemplos de enfermedades hepáticas autoinmunitarias incluyen, pero sin limitación, hepatitis, hepatitis activa crónica autoinmunitaria (Franco A. et al., Clin Immunol Immunopathol marzo de 1990;54(3):382), cirrosis biliar primaria (Jones DE). Clin Sci (Colch) noviembre de 1996;91 (5):551; Strassburg CP. et al., Eur J Gastroenterol Hepatol. junio de 1999;11 (6):595) y hepatitis autoinmunitaria (Manns MP. J Hepatol agosto de 2000;33 (2):326).
Los ejemplos de enfermedades neurológicas autoinmunitarias incluyen, pero sin limitación, esclerosis múltiple (Cross
AH. et al., J Neuroimmunol 1 enero de 2001 ;112 (1-2):1), enfermedad de Alzheimer (Oron L. et al., J Neural Transm Suμl. 1997;49:77), miastenia grave (Infante AJ. y Kraig E, Int Rev Immunol 1999;18 (1-2):83; Oshima M. et al., Eur J Immunol diciembre de 1990;20 (12):2563), neuropatías, neuropatías motoras (Kornberg AJ. J Clin Neurosci. mayo de 2000; 7 (3): 191); síndrome de Guillain-Barre y neuropatías autoinmunitarias (Kusunoki S. Am J Med Sci. abril de 2000;319 (4):234), miastenia, síndrome miasténico de Lambert-Eaton (Takamori M. Am J Med Sci. abril de 2000;319 (4):204); enfermedades neurológicas paraneoplásicas, atrofia cerebelosa, atrofia cerebelosa paraneoplásica y síndrome de la persona rígida (Hiemstra HS. et al., Proc Natl Acad Sci USA 27 de marzo de 2001;98(7):3988); síndrome de la persona rígida no paraneoplásico, atrofias cerebelosas progresivas, encefalitis, encefalitis de Rasmussen, esclerosis lateral amiotrófica, corea de Sydenham, síndrome de Gilles de la Tourette y poliendocrinopatías autoinmunitarias (Antoine JC. y Honnorat J. Rev Neurol (París) enero de 2000;156 (1):23); neuropatías disinmunitarias (Nobile-Orazio E. et al., Electroencephalogr Clin Neurophysiol Suμl. 1999;50:419); neuromiotonía adquirida, artrogriposis múltiple congénita (Vincent A. et al., Ann NY Acad Sci. 13 de mayo de 1998;841:482), neuritis, neuritis óptica (Soderstrom M. et al., J Neurol Neurosurg Psychiatry mayo de 1994;57 (5):544) y enfermedades neurodegenerativas.
Los ejemplos de enfermedades musculares autoinmunitarias incluyen, pero sin limitación, miositis, miositis autoinmunitaria y síndrome de Sjogren primario (Feist E. et al., Int Arch Allergy Immunol septiembre de 2000;123 (1):92) y enfermedad autoinmunitaria del músculo liso (Zauli D. et al., Biomed Pharmacother junio de 1999;53 (5- 6): 234).
Los ejemplos de enfermedades nefríticas autoinmunitarias incluyen, pero sin limitación, nefritis y nefritis intersticial autoinmunitaria (Kelly CJ. J Am Soc Nephrol, agosto de 1990;1 (2):140).
Los ejemplos de enfermedades autoinmunitarias relacionadas con la reproducción incluyen, pero sin limitación, pérdidas fetales repetidas (Tincani A. et al., Lupus 1998;7 Suppl 2:S107-9).
Los ejemplos de enfermedades del tejido conjuntivo autoinmunitarias incluyen, pero sin limitación, enfermedades del αdo, enfermedades autoinmunitarias del αdo (Yoo TJ. et al., Cell Immunol Agosto de 1994;157 (1):249) y enfermedades autoinmunitarias del αdo interno (Gloddek B. et al., Ann N Y Acad Sci 29 de diciembre de 1997;830:266).
Los ejemplos de enfermedades sistémicas autoinmunitarias incluyen, pero sin limitación, lupus eritematoso sistémico (Erikson J. et al., Immunol Res 1998;17 (1-2):49) y esclerosis sistémica (Renaudineau Y. et al., Clin Diagn Lab Immunol. marzo de 1999;6 (2):156); Chan Ot . et al., Immunol Rev junio de 1999;169:107).
De acuerdo con un aspecto específico, la enfermedad autoinmunitaria se selecciona del grupo que consiste en una diabetes tipo 1, esclerosis múltiple, artritis reumatoide, celíaquía y accidente cerebrovascular.
Tal como se ha mencionado, las células de la divulgación se pueden obtener de cualquier donante de células. Por tanto, el sujeto a tratar puede ser un sujeto humano mientras que las células pueden obtenerse de un donante singénico (p. ej., autólogo) o no singénico (p. ej., alogénico o xenogénico con respecto al receptor).
Como se usa en el presente documento, el término células "singénicas" se refiere a células que son esencialmente genéticamente idénticas al sujeto o esencialmente a todos los linfocitos del sujeto. Los ejemplos de células singénicas incluyen células derivadas del sujeto (también denominadas en la técnica como "autólogas"), de un clon del sujeto, o de un gemelo idéntico del sujeto.
Como se usa en el presente documento, el término células "no singénicas" se refiere a células que no son esencialmente genéticamente idénticas al sujeto o esencialmente a todos los linfocitos del sujeto, tales como células alogénicas o células xenogénicas.
Como se usa en el presente documento, el término "alogénico" se refiere a células que derivan de un donante que es de la misma especie que el sujeto, pero que es sustancialmente no clonal con el sujeto. Normalmente, los mamíferos gemelos no cigóticos exogámicos de la misma especie son alogénicos entre sí. Se apreciará que una célula alogénica puede ser HLA idéntica, parcialmente HLA idéntica o HLA no idéntica (es decir, que muestra uno o más determinantes HLA dispares) con respecto al sujeto.
Como se usa en el presente documento, el término "xenogénico" se refiere a una célula que expresa sustancialmente antígenos de una especie diferente con respecto la especie de una proporción sustancial de los linfocitos del sujeto. Normalmente, los mamíferos exogámicos de diferentes especies son xenogénicos entre sí.
La presente divulgación prevé que las células xenogénicas procedan de una diversidad de especies. Por tanto, de acuerdo con un aspecto, las células pueden proceder de cualquier mamífero. Los orígenes de especies adecuados para las células comprenden los principales animales domésticos o ganado y primates. Tales animales incluyen, pero sin limitación, porcinos (por ejemplo, cerdo), bovinos (por ejemplo, vaca), equinos (por ejemplo, caballo), ovinos (por ejemplo, cabra, oveja), felinos (por ejemplo, Felis domestica), caninos (por ejemplo, Canis domestica), roedores (por
ejemplo, ratón, rata, conejo, cobaya, jerbo, hámster) o primates (por ejemplo, chimpancé, macaco de la India, mono macaco, tití).
Las células de origen xenogénico (por ejemplo, origen porcino) se obtienen preferentemente de una fuente que se sabe que está libre de zoonosis, tales como los retrovirus endógenos porcinos. De manera similar, las células o tejidos derivados de humanos se obtienen preferentemente de fuentes sustancialmente libres de patógenos.
De acuerdo con un aspecto, las células no son singénicas con el sujeto.
De acuerdo con un aspecto, las células son alogénicas con el sujeto.
De acuerdo con un aspecto, las células son singénicas con el sujeto (por ejemplo, autólogas).
De acuerdo con un aspecto de la presente invención, el sujeto es un ser humano y las células son de origen humano (por ejemplo, no autólogas).
De acuerdo con un aspecto, el sujeto es un ser humano y las células son de origen xenogénico (por ejemplo, origen porcino).
Puede emplearse cualquier método conocido en la técnica para obtener células para trasplante. Por tanto, por ejemplo, las células inmunitarias (por ejemplo, linfocitos T, linfocitos B, linfocitos NK, DC) se pueden obtener recogiendo sangre periférica de un donante. Los métodos para recoger sangre periférica son bien conocidos en la técnica e incluyen, pero sin limitación, extracción de hasta 500-1000 ml de sangre total de un donante y recolegida en un recipiente que contenga un anticoagulante (por ejemplo, heparina o citrato) o por aféresis, un procedimiento en el que la sangre periférica de un individuo se pasa a través de un aparato, produciendo un constituyente predominante (por ejemplo, células mononucleares como linfocitos, monocitos o células dendríticas), y devolver los otros constituyentes a la circulación del sujeto. De forma alternativa, las células se pueden obtener por cultivo de células in vitro o ex-vivo. Se apreciará que las células de la divulgación pueden ser preparaciones frescas o congeladas (p. ej., crioconservadas).
Dependiendo del contexto del trasplante, para facilitar el injerto de las células, el método puede comprender además ventajosamente acondicionar al sujeto en condiciones subletales, condiciones letales o supraletales previas al trasplante.
Como se usa en el presente documento, los términos "subletal", "letal" y "supraletal", cuando están relacionados con el acondicionamiento de sujetos de la presente invención, se refieren a tratamientos mielotóxicos y/o linfocitotóxicos que, cuando se aplican a una población representativa de los sujetos, respectivamente, son normalmente: no letales para prácticamente todos los miembros de la población; letales para algunos pero no para todos los miembros de la población; o letales para prácticamente todos los miembros de la población en condiciones normales de esterilidad.
De acuerdo con algunos aspectos de la invención, el acondicionamiento subletal, letal o supraletal comprende la irradiación corporal total (TBI), irradiación linfoide total (TLI, es decir, exposición de todos los ganglios linfáticos, el timo y el bazo), irradiación corporal parcial (por ejemplo, exposición específica del colon, mama, etc.), acondicionamiento mieloablativo y/o acondicionamiento no mieloablativo, p.ej., con diferentes combinaciones incluyendo, aunque no de forma limitativa, bloqueo coestimulador, agente quimioterapéutico y/o inmunoterapia con anticuerpos. De acuerdo con algunos aspectos de la invención, el acondicionamiento comprende una combinación de cualquiera de los protocolos de acondicionamiento descritos anteriormente (por ejemplo, agente quimioterapéutico y TBI, bloqueo coestimulador y agente quimioterapéutico, inmunoterapia con anticuerpos y agente quimioterapéutico, etc.).
De acuerdo con un aspecto, la TBI comprende una dosis de irradiación única o fraccionada dentro del intervalo de 0,5 1 Gy, 0,5-1,5 Gy, 0,5-2,5 Gy, 0,5-5 Gy, 0,5-7,5 Gy, 0,5-10 Gy, 0,5-15 Gy, 1-1,5 Gy, 1-2 Gy, 1-2,5 Gy, 1-3 Gy, 1-3,5 Gy, 1-4 Gy, 1-4,5 Gy, 1-1,5 Gy, 1-7,5 Gy, 1-10 Gy, 2-3 Gy, 2-4 Gy, 2-5 Gy, 2-6 Gy, 2-7 Gy, 2-8 Gy, 2-9 Gy, 2-10 Gy, 3-4 Gy, 3-5 Gy, 3-6 Gy, 3-7 Gy, 3-8 Gy, 3-9 Gy, 3-10 Gy, 4-5 Gy, 4-6 Gy, 4-7 Gy, 4-8 Gy, 4-9 Gy, 4-10 Gy, 5-6 Gy, 5 7 Gy, 5-8 Gy, 5-9 Gy, 5-10 Gy, 6-7 Gy, 6-8 Gy, 6-9 Gy, 6-10 Gy, 7-8 Gy, 7-9 Gy, 7-10 Gy, 8-9 Gy, 8-10 Gy, 10-12 Gy o 10-15 Gy.
De acuerdo con un aspecto específico, la TBI comprende una dosis de irradiación única o fraccionada dentro del intervalo de 1-7,5 Gy.
De acuerdo con un aspecto, la etapa de acondicionamiento se efectúa acondicionando al sujeto en condiciones supraletales, como en condiciones mieloablativas.
De forma alternativa, la etapa de acondicionamiento puede efectuarse acondicionando al sujeto en condiciones letales o subletales, tal como acondicionando al sujeto en condiciones mielorreductoras o condiciones no mieloablativas.
De acuerdo con un aspecto, la etapa de acondicionamiento se efectúa acondicionando al sujeto con un fármaco mieloablativo (p. ej., busulfán o melfalán) o un fármaco no mieloablativo (p. ej., ciclofosfamida o fludarabina).
Los ejemplos de agentes acondicionadores que se pueden usar para acondicionar al sujeto incluyen, sin limitación, irradiación, agentes farmacológicos y células inductoras de tolerancia (como se describe en el presente documento).
Ejemplos de agentes farmacológicos incluyen fármacos mielotóxicos, fármacos linfocitotóxicos y fármacos inmunosupresores (descritos en detalle a continuación).
Los ejemplos de fármacos mielotóxicos incluyen, sin limitación, busulfán, dimetil mileran, melfalán y tiotepa.
Adicionalmente, o como alternativa, el método puede comprender además acondicionar al sujeto con un régimen inmunosupresor antes de, concomitantemente con, o después del trasplante de las células.
Los ejemplos de tipos adecuados de regímenes inmunosupresores incluyen la administración de fármacos inmunosupresores y/o irradiación inmunosupresora.
En la bibliografía de la técnica se proporciona una amplia orientación para seleccionar y administrar regímenes inmunosupresores adecuados para el trasplante (por ejemplo, consúltese: Kirkpatrick CH. y Rowlands DT Jr., 1992. JAMA. 268, 2952; Higgins RM. et al., 1996. Lancet 348, 1208; Suthanthiran M. y Strom TB., 1996. New Engl. J. Med.
331, 365; Midthun DE. et al., 1997. Mayo Clin Proc. 72, 175; Morrison VA. et al., 1994. Am J Med. 97, 14; Hanto DW., 1995. Annu Rev Med. 46, 381; Senderowicz AM. et al., 1997. Ann Intern Med. 126, 882; Vincenti F. et al., 1998. New Engl. J. Med. 338, 161; Dantal J. et al. 1998. Lancet 351, 623).
Los ejemplos de agentes inmunosupresores incluyen, pero sin limitación, tacrolimus (también conocido como FK-506 o fujimicina, nombres comerciales: Prograf, Advagraf, Protopic), micofenolato de mofetilo, micofenolato de sodio, prednisona, metotrexato, ciclofosfamida, ciclosporina, ciclosporina A, cloroquina, hidroxicloroquina, sulfasalazina (sulfasalazopirina), sales de oro, D-penicilamina, leflunomida, azatioprina, anakinra, infliximab (REMICADE), etanercept, bloqueadores de TNF alfa, un agente biológico que se dirige a una citocina inflamatoria y un fármaco antiinflamatorio no esteroideo (AINE). Los ejemplos de AINE incluyen, pero sin limitación, ácido acetilsalicílico, salicilato de magnesio y colina, diflunisal, salicilato de magnesio, salsalato, salicilato de sodio, diclofenaco, etodolaco, fenoprofeno, flurbiprofeno, indometacina, cetoprofeno, cetorolaco, meclofenamato, naproxeno, nabumetona, fenilbutazona, piroxicam, sulindaco, tolmetina, paracetamol, ibuprofeno, inhibidores de Cox-2, tramadol, rapamicina (sirolimus) y análogos de rapamicina (como CCI-779, RAD001, AP23573). Estos agentes pueden administrarse individualmente o en combinación.
Como se usa en el presente documento, el término "aproximadamente" se refiere a ±10 %.
Las expresiones "comprende", "que comprende", "incluye", "que incluye", "que tiene" y sus conjugaciones significan "que incluye pero sin limitación".
La expresión "que consiste en" significa "que incluye y se limita a".
La expresión "que consiste esencialmente en" significa que la composición, el método o la estructura puede incluir ingredientes, etapas y/o partes adicionales, pero solo si los ingredientes, las etapas y/o las partes adicionales no alteran materialmente las características básicas y novedosas de la composición, el método o la estructura reivindicados.
Como se usa en el presente documento, la forma en singular "un", "uno/a" y "el/la" incluyen referencias en plural, salvo que el contexto indique claramente otra cosa. Por ejemplo, la expresión "un compuesto" o "al menos un compuesto" puede incluir una pluralidad de compuestos, incluyendo mezclas de los mismos.
A lo largo de la presente solicitud, diversos aspectos de esta divulgación pueden presentarse en un formato de intervalo. Debe entenderse que la descripción en formato de intervalo es únicamente por conveniencia y brevedad y no debería interpretarse como una limitación inflexible sobre el alcance de la invención. Por consiguiente, debería considerarse que la descripción de un intervalo ha divulgado específicamente todos los posibles subintervalos así como valores numéricos individuales en ese intervalo. Por ejemplo, debe considerarse que la descripción de un intervalo tal como de 1 a 6 ha divulgado específicamente subintervalos tales como de 1 a 3, de 1 a 4, de 1 a 5, de 2 a 4, de 2 a 6, de 3 a 6, etc., así como números individuales dentro de ese intervalo, por ejemplo, 1, 2, 3, 4, 5 y 6. Esto es aplicable independientemente de la amplitud del intervalo.
Siempre que se indique un intervalo numérico en el presente documento, se pretende incluir cualquier número citado (fraccionario o entero) dentro del intervalo indicado. Las expresiones "que varía/varía entre" un primer número indicador y un segundo número indicador y "que varía/varía de" un primer número indicador "a" un segundo número indicador se usan indistintamente en el presente documento y se entiende que incluyen los números indicadores primero y segundo, y todos los números fraccionarios y enteros entre ellos.
Como se utiliza en el presente documento, el término "método" se refiere a formas, medios, técnicas y procedimientos
para llevar a cabo una tarea dada incluyendo, aunque no de forma limitativa, aquellas formas, medios, técnicas y procedimientos tanto conocidos como desarrollados a partir de formas, medios, técnicas y procedimientos conocidos por los especialistas de las técnicas química, farmacológica, biológica, bioquímica y médica.
Se aprecia que determinadas características de la invención, que están, por claridad, descritas en el contexto de realizaciones separadas, también pueden proporcionarse en combinación en un único aspecto. Por el contrario, diversas características de la invención, que están, por brevedad, descritas en el contexto de un único aspecto, también pueden proporcionarse por separado o en cualquier subcombinación adecuada o según sea adecuado en cualquier otro aspecto descrito de la invención. Determinadas características descritas en el contexto de diversos aspectos no han de considerarse características esenciales de esos aspectos, a menos que el aspecto no funcione sin esos elementos.
Diversos aspectos y aspectos de la presente divulgación como se han delineado anteriormente en el presente documento y como se reivindica en la sección de reivindicaciones a continuación encuentran soporte experimental en los siguientes ejemplos.
Ejemplos
A continuación, se hace referencia a los siguientes ejemplos, que, junto con las descripciones anteriores, ilustran la divulgación de manera no limitante.
Generalmente, la nomenclatura utilizada en el presente documento y los procedimientos de laboratorio utilizados en la presente divulgación incluyen técnicas moleculares, bioquímicas, microbiológicas y de ADN recombinante. Dichas técnicas se explican exhaustivamente en la bibliografía. Véase, por ejemplo, "Molecular Cloning: A laboratory Manual" Sambrook et al., (1989); "Current Protocols in Molecular Biology" Volúmenes I-III Ausubel, R. M., ed. (1994); Ausubel et al., "Current Protocols in Molecular Biology", John Wiley and Sons, Baltimore, Maryland (1989); Perbal, "A Practical Guide to Molecular Cloning", John Wiley & Sons, Nueva York (1988); Watson et al., "Recombinant DNA", Scientific American Books, Nueva York; Birren et al. (eds) "Genome Analysis: A Laboratory Manual Series", Vol. 1-4, Cold Spring Harbor Laboratory Press, Nueva York (1998); metodologías expuestas en las patentes de los Estados Unidos números 4.666.828; 4.683.202; 4.801.531; 5.192.659 y 5.272.057; "Cell Biology: A Laboratory Handbook", volúmenes I-III Cellis, J. E., ed. (1994); "Current Protocols in Immunology" volúmenes I-III Coligan J. E., ed. (1994); Stites et al. (eds), "Basic and Clinical Immunology" (8a edición), Appleton y Lange, Norwalk, CT (1994); Mishell y Shiigi (eds), "Selected Methods in Cellular Immunology", W. H. Freeman and Co., Nueva York (1980); se describen ampliamente inmunoensayos disponibles en la bibliografía científica y de patentes, véase, por ejemplo, las patentes de los EE.UU. números 3.791.932; 3.839.153; 3.850.752; 3.850.578; 3.853.987; 3.867.517; 3.879.262; 3.901.654; 3.935.074; 3.984.533; 3.996.345; 4.034.074; 4.098.876; 4.879.219; 5.011.771 y 5.281.521; "Oligonucleotide Synthesis" Gait, M. J., ed. (1984); "Nucleic Acid Hybridization" Hames, B. D. y Higgins S. J., eds. (1985); "Transcription and Translation" Hames, B. D. y Higgins S. J., Eds. (1984); "Animal Cell Culture" Freshney, R. I., ed. (1986); "Immobilized Cells and Enzymes" IRL Press, (1986); "A Practical Guide to Molecular Cloning" Perbal, B., (1984) y "Methods in Enzymology" vol. 1-317, Academic Press; "PCR Protocols: A Guide To Methods And Applications", Academic Press, San Diego, CA (1990); Marshak et al., "Strategies for Protein Purification and Characterization - A Laboratory Course Manual" CSHL Press (1996). Se proporcionan otras referencias generales a lo largo del presente documento. Se cree que los procedimientos de los mismos son bien conocidos en la técnica y se proporcionan para la comodidad del lector.
MATERIALES GENERALES Y PROCEDIMIENTOS EXPERIMENTALES
Animales
Los ratones BALB/c, CB6 (F1) y C57BL/6 hembra de 6 a 12 semanas se obtuvieron en Harlan Laboratories o crecieron en las instalaciones para animales del Weizmann Institute of Science. Todos los ratones se mantuvieron en jaulas pequeñas (5 animales en cada jaula) y se les alimentó con alimentos estériles y agua ácida. Todos los estudios fueron aprobados por el Comité Institucional de Cuidado y Uso de Animales del Instituto Weizmann de Ciencias.
Preparación de Tmc anti-terceros de ratón no reactivos en el hospedador
Se prepararon Tmc anti-terceros como se ha descrito previamente [Ophir E et al., Blood (2010) 115: 2095-2104] brevemente, los esplenocitos de los ratones donantes se cultivaron frente a esplenocitos de terceros irradiados durante 60 horas con privación de citocinas. Posteriormente, las células CD8+ se seleccionaron positivamente utilizando partículas magnéticas (BD Pharmingen) y se cultivaron en un entorno libre de Ag. rhIL-15 (20 ng/ml; R&D Systems) se agregó cada dos días. Para lograr una población purificada al final del cultivo (día 16), los linfocitos Tmc se seleccionaron positivamente para la expresión de CD62L mediante clasificación de células activadas magnéticamente [MACS, Miltenyi, Bergisch Gladbach, Alemania].
Trasplante de médula ósea
1. Se recogieron los huesos largos de ratones Balb/c o C57BL/6 [desnudos o de tipo silvestre (WT)]. La médula
ósea se extrajo lavando o triturando los huesos para obtener una suspensión de células individuales. Algunas preparaciones recogidas de ratones WT se sometieron a agotamiento de linfocitos T mediante clasificación de células activadas magnéticamente. Se contó la médula ósea y se llevó a la concentración correcta y luego se inyectó a ratones i.v. a la vena de la cola o intraorbitariamente.
2. Antes del trasplante, los ratones se sometieron a un régimen de acondicionamiento. El acondicionamiento de intensidad reducida (RIC) comprendía someter a los ratones a dosis de irradiación subletales (es decir, dosis de irradiación de la que los ratones receptores podían recuperarse espontáneamente) o someter a los ratones a dosis bajas de fármacos mieloablativo (p. ej., busulfán) o no mieloablativo (p. ej., ciclofosfamida). La irradiación corporal total (TBI) se administró usando una máquina de rayos gamma o rayos X (por ejemplo, XRAD-320). Los fármacos se administraron por vía i.v., s.c., i.p. o por vía oral.
Trasplante de células OT1
Se recogieron ganglios linfáticos y/o bazos de ratones transgénicos OT-1. Los ratones eran ratones OT-1 portadores del gen CD45.1 y/o con la constitución de una mutación RAG-/-. De forma alternativa, los ratones OT-1 eran ratones F1-OT1, progenie de ratones hostXOT-1, útil para la eliminación de fenómenos alogénicos. Se crearon suspensiones de células individuales y luego se sometieron a purificación de linfocitos T mediante clasificación de células activadas magnéticamente [MACS, Miltenyi, Bergisch Gladbach, Alemania]. La pureza de la población de linfocitos T OT-1 resultante se analizó mediante FACS. A continuación, las células se inyectaron como células "frescas" o se cultivaron de otro modo ex-vivo para producir linfocitos Tmc como se ha descrito anteriormente, es decir, por activación de terceros hacia esplenocitos irradiados de un ratón que expresa ovoalbúmina. A continuación se inyectaron linfocitos Tmc OT-1 como se describe en el presente documento.
Análisis de citometría de flujo
El análisis de clasificación de células activadas por fluorescencia (FACS) se realizó usando un Becton Dickinson FAC-Scan modificado. Las células se tiñeron con anticuerpos marcados específicos para Va2, Vp5, H2Dd, H2Kb, CD45.1, CD45.2, CD8a, CD4, CD25, CD69, CD19 (Biolegend; BD; Miltenyi).
Ensayo de actividad CTL (ensayo de Cr51)
Se sacrificó a los ratones, se recogieron bazos y GL y se seleccionaron células CD8+ (y seleccionado negativamente para H-2Dd para excluir "Tmc"). Estos HTC vírgenes fueron probados para determinar su capacidad de eliminar las dianas C3H (H-2k) o BALB/c (H-2d) en un ensayo de liberación de cromo. Los esplenocitos BALB/c y C3H, utilizados como células diana, fueron pretratados con 2 μg/ml de concanavalina A (Sigma, St. Louis, MO) durante 48 horas y se expusieron a 70 μCi de 51Cr (Perkin Elmer, Wellesley, MA) durante 1 hora. Las células efectoras se prepararon a partir de células CD8+ seleccionadas de ratones C57BL/6 y se incubaron durante 6 días en diferentes diluciones frente a esplenocitos BALB/c o C3H en 12 réplicas para cada dilución en una placa de 96 pocillos con IL-2 (20 U/ml). El día 6, números titulados de células efectoras y 5 x 103 dianas marcadas con 51Cr se mezclaron en placas con fondo en forma de V en varias proporciones de efector/diana (E:D). La actividad citotóxica se midió en un ensayo de liberación de Cr51 de 4 h. El porcentaje de lisis específica se calculó como (liberación experimental - liberación espontánea)/(liberación máxima - liberación espontánea) x 100. La liberación de 51Cr por las células diana cultivadas en medio solo, o lisadas con SDS al 1 %, se definió como liberación espontánea o liberación total, respectivamente.
Células mononucleares de sangre periférica (PBMC)
Se aislaron PBMC de sangre total de pacientes y de voluntarios sanos mediante centrifugación en gradiente de densidad de Ficoll. Cuando se indicó, las células se tipificaron para HLA de clase I mediante métodos serológicos como se ha descrito previamente [Manual of Tissue Typing Techniques. Washington DC, Instituto Nacional de Alergias y Enfermedades Infecciosas, NIH DHEW Publicación 76-545, 1976, pág. 22].
Generación de células dendríticas
Los monocitos se aislaron mediante adherencia plástica y se cultivaron en placas de 6 pocillos utilizando 3 ml de medio Cellgro DC suplementado con suero humano al 1 % y penicilina/estreptomicina más GM-CSF (800 UI/ml) e IL-4 (20 ng/ml) (Peprotech, Hamburgo, Alemania). Después de 48 horas de cultivo, se añadieron 1,5 ml de medio (GM-CSF a 1600 UI/ml e IL4 a 20 ng/ml). 24 h más tarde, se recogieron las células no adherentes y se contaron las células grandes (principalmente DC inmaduras), resuspendidas en medio fresco que contiene GM-CSf 800 UI/ml, IL-420 ng/ml, LPS de E. coli O55:B5 a 10 ng/ml (Sigma, Deisenhofen, Alemania) e IFNy (Peprotech, 100 UI/ml), y se sembraron a aproximadamente 106 DC por pocillo en 2 ml y se incubaron durante la noche. Al día siguiente, las células no adherentes se descartaron y las DC adherentes se eliminaron suavemente con PBS frío/HS al 1 % después de incubar en hielo durante 20 minutos. Se contaron las células grandes que consistían en DC maduras. Las células se irradiaron con 30 Gy para evitar el crecimiento de pocos linfocitos NK o T de memoria potencialmente contaminantes y luego se usaron para la estimulación de linfocitos T.
Aislamiento de linfocitos T CD8 vírgenes de PBMC
Los linfocitos T CD8 vírgenes se aislaron mediante selección negativa inicial utilizando un kit de selección negativa CD8 (Miltenyi, Bergisch Gladbach, Alemania) de acuerdo con las instrucciones del fabricante. A continuación, se agotaron los linfocitos T CD8+ experimentadas con antígeno utilizando perlas de CD45RO y en una columna LD.
Generación de linfocitos T CD8 humanos de memoria central anti-terceros
Se aislaron linfocitos T CD8 vírgenes y se resuspendieron en medio de linfocitos T complementado con IL-21 (Peprotech, 30 ng/ml). Se añadieron DC irradiadas en una proporción de 1:4 DC:linfocitos T con 4 x 105 linfocitos T por pocillo de una placa de 48 pocillos. El volumen total de cada pocillo fue de 500 μl.
72 h después de iniciado el cultivo, se añadieron 500 μl de medio de linfocitos T con IL-7 e IL-15 (Peprotech, concentraciones finales de 5 ng/ml) y las células se alimentaron posteriormente cada 2-3 días como se describe en la sección de resultados.
Análisis estadístico
El análisis de los datos de supervivencia se realizó mediante curvas de Kaplan-Meier (prueba de rangos logarítmicos). La comparación de medias se realizó mediante la prueba t de Student.
EJEMPLO 1
Los linfocitos Tmc MHC no compatibles sobreviven en ratones hospedadores en un contexto de médula ósea singénica y ejercen actividad de veto específica
Teniendo en cuenta que el trasplante singénico de médula ósea (BMT), incluso cuando se administra en el contexto de la irradiación corporal total (TBI) letal es mucho más seguro en humanos en comparación con el BMT alogénico, los presentes inventores buscaron en primer lugar determinar si las células F1-Tmc transferidas adoptivamente sobreviven al ataque de HTC anti-donante del hospedador cuando se infunden junto con TDBMT singénico (figuras 1A-B). Tal como puede observarse en las figuras 2A-B, F1-Tmc persistió en la sangre periférica el día 60 después del trasplante. Los linfocitos Tmc comprendían aproximadamente 13 % ± 10 del compartimento total de CD8+ (datos no mostrados). A continuación, para evaluar la capacidad de los linfocitos Tmc para inducir la eliminación de clones específicos de antígeno dentro de la población de HTC policlonal de tipo silvestre y para verificar que los HTC restantes conservan su funcionalidad, se empleó un ensayo de eliminación de liberación de cromo. Los resultados muestran que los HTC H2bCD8 de ratones tratados con Tmc mostraban una eliminación significativamente menor de dianas H-2d y retenían la capacidad de eliminar las dianas H-2k, mientras que los ratones no tratados con T mc (es decir, el grupo de MO solo) mostraron niveles similares de eliminación para ambos tipos de células (figura 3). Estos resultados indican que Tmc ejerce una actividad de veto específica sobre una población de HTC policlonal y confirma que los clones no eliminados por Tmc, conservan su funcionalidad. Posteriormente, estos experimentos se repitieron en ratones acondicionados con condicionamiento de intensidad reducida (RIC), más adecuado para la aplicación clínica. Por lo tanto, los estudios en ratones Balb/c irradiados subletalmente con 5,5 Gy TBI, inyectados con médula ósea empobrecida en linfocitos T singénicos (TDBMT) y Tmc alogénicos (Balb x Black) F1 (ilustrado en la figura 1C), arrojaron resultados similares (figura 4). Los linfocitos Tmc estuvieron presentes en la sangre periférica de estos ratones durante más de 15 meses (cuando finalizó el experimento, no se muestran los datos). Por tanto, la supervivencia de Tmc MHC incompatibles se induce mediante un procedimiento muy seguro que implica el acondicionamiento con TBI subletal de 5,5 Gy y BMT autólogo.
EJEMPLO 2
Los linfocitos Tmc MHC no compatibles sobreviven en ratones hospedadores en ausencia de un trasplante de médula ósea
A la luz de los datos anteriores, se evaluó la capacidad de los linfocitos Tmc solos para inducir tolerancia en ausencia de MO. El alcance de tal protocolo sería mucho mayor. Específicamente, la inducción de inmunotolerancia mediante la administración de linfocitos Tmc anti-terceros solo, en condiciones seguras, sería una ventaja no solo para las personas inmunocomprometidas, sino que posiblemente podría permitir el tratamiento de enfermedades hematológicas no malignas (por ejemplo, anemia y talasemia), enfermedades autoinmunitarias y podría proporcionar una plataforma para la administración de terapia celular. Inicialmente, los presentes inventores intentaron definir la dosis mínima de irradiación con la cual los linfocitos Tmc de origen F1 injertan, con el fin de establecer el modelo en el que se puede probar la inducción de tolerancia en los hospedadores. Para este fin, se expuso a ratones Balb/c a una variedad de dosis de acondicionamiento subletales con y sin transferencia adoptiva de linfocitos F1-Tmc de CB6. El análisis de sangre periférica total para linfocitos Tmc H2db positivo mostró que la dosis de irradiación mínima con la que se podía detectar Tmc (es decir, cuando no se rechazaban los linfocitos Tmc) era de 5,5 Gy TBI (Figura 5). En consecuencia, se probó la sostenibilidad de los Tmc derivados de C57BL/6 completamente alogénicos para sobrevivir con una dosis subletal de TBI de 5,5 Gy (como se muestra en la figura 1C). Este experimento pretendía verificar que las células de origen alogénico no inducen EICH y que la eliminación de linfocitos T anti-donante no está mediada por
alorreactividad sino por la actividad de veto. Además, una vez traducido a humanos y con el fin de producir tolerancia "disponible" que induzca linfocitos Tmc, lo más probable es que las células se deriven de fuentes alogénicas, no compatibles. Los resultados mostraron que los Tmc alogénicos derivados de C57BL/6 eran capaces de sobrevivir dentro de huéspedes Balb/c irradiados con 5,5 Gy (como se muestra en la figura 1C), mostrando porcentajes de Tmc ligeramente más bajos en la sangre periférica que los de los ratones que recibieron F1 Tmc de CB6 (figuras 6A- B). Este resultado puede atribuirse a un proceso de rechazo muy lento de los linfocitos Tmc. Aunque los linfocitos Tmc persisten en la sangre durante más de un año, la reducción en su número durante los primeros meses posteriores a la inyección, tomado junto con la eliminación de clones anti-hospedador detectados en el ensayo de liberación de cromo, sugiere fuertemente que los Tmc inducen tolerancia periférica.
Por lo tanto, la aplicación de Tmc solos se utiliza para crear una ventana de oportunidad, al menos durante unos meses, para la administración de tratamientos, como la terapia celular.
EJEMPLO 3
Los linfocitos Tmc MHC no compatibles admiten la transferencia adoptiva de células del mismo donante
Para probar la hipótesis de que los linfocitos Tmc se pueden usar para la terapia celular adoptiva, los presentes inventores utilizaron ratones OT1 transgénicos que llevan un TCR contra el péptido de ovoalbúmina. La motivación para usar células transgénicas OT1 en este contexto provino de la idea de que estas células pueden usarse como modelo para terapias celulares conocidas como infusión de linfocitos de donante (DLI) con toda la población de linfocitos T de donante o con linfocitos T específicos de antígeno dirigidos contra antígenos virales o tumorales.
Inicialmente, se infundieron linfocitos T CD8+OT1+CD45.1 vírgenes en ratones con Tmc quiméricos, 90 días después de la transferencia adoptiva inicial de los Tmc. El objetivo principal era definir si la población de Tmc superviviente puede facilitar el injerto de células alogénicas recién infundidas. Antes de la inyección de linfocitos Tg vírgenes, la población de T mc en ratones quiméricos se analizó mediante FACS. Por tanto, 2/5 y 9/11 ratones que habían recibido Tmc de C57BL/6 o Tmc de CB6, respectivamente, mantuvieron su población Tmc (figuras 6A-B).
Estos ratones fueron acondicionados adicionalmente en el día 90 después del trasplante con 2 Gy TBI (con el fin de agotar algunos linfocitos T para permitir la introducción de nuevos linfocitos T) y luego se les infundió a los ratones 2 x 106 células OT1 (H-2b). Curiosamente, cuando se evaluaron el día 120 (30 días después del trasplante de células OT1), las células OT1 se pudieron detectar solo en aquellos ratones que habían mostrado una población de T mc antes del trasplante (Figura 7). Estos resultados preliminares, que muestran que en ratones que presentan una población de T mc se puede aceptar la adición de células del mismo origen del donante, se corroboraron adicionalmente utilizando células OT-1 transgénicas, de la siguiente manera: Las células CD8+OT-1 se trasplantaron junto con los Tmc el día 0, para evitar la necesidad de un acondicionamiento secundario (2 Gy TBI empleados previamente en el día 90), y se controló la presencia de células OT 1 en la sangre periférica en diferentes momentos después de la infusión de células.
Como se muestra en la figura 8, los resultados de este experimento ilustran que:
1. Tmc de C57BL/6 así como Tmc (BalbxC57BL)F1 pueden persistir en receptores alogénicos.
2. Tmc de C57BL/6 puede conferir protección a las células CD8 OT-1 vírgenes cultivadas con la constitución C57BL/6 (OT-1+CD45.1+RAG-) cuando se co-inyecta, mientras que las células OT-1 por sí solas desaparecen de la circulación.
3. CB6(F1) Tmc que expresan el haplotipo MHC de los ratones C57BL/6 (H-2b) también pueden conferir protección a las células OT-1 vírgenes.
EJEMPLO 4
Las células de veto Tmc anti-terceros preparadas a partir de células de ratones OT-1 se injertan y sobreviven in vivo
Los experimentos se llevaron a cabo en ratones acondicionados con acondicionamiento de intensidad reducida (RIC), adecuado para la aplicación clínica. Por lo tanto, los ratones Balb/c irradiados subletalmente con 5,5 Gy TBI, fueron inyectados con diferentes concentraciones de linfocitos Tmc no singénicos con origen de ratón OT-1 (ilustrado en la Figura 9). Los linfocitos Tmc estuvieron presentes en la sangre periférica de estos ratones al menos durante 30 días. Por tanto, la supervivencia de linfocitos Tmc MHC no compatible se induce mediante un procedimiento RIC seguro.
Claims (17)
1. Una célula aislada que tiene un fenotipo de linfocito T de memoria central (Tmc), en donde dicho fenotipo Tmc comprende una firma CD3+/CD8+/CD62L7CD45RAVCD45RO+, siendo dicha célula, una célula anti-terceros no inductora de reacción de injerto contra hospedador (EICH), que es inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, generándose dicha célula mediante un método que comprende:
(a) poner en contacto células mononucleares de sangre periférica (PBMC) con un antígeno o antígenos de terceros en presencia de IL-21 para permitir el enriquecimiento de células reactivas al antígeno; y
(b) cultivar dichas células resultantes de la etapa (a) en presencia de IL-21, IL-15 e IL-7 para permitir la proliferación de células anti-terceros que comprenden dicho fenotipo de linfocito T de memoria central (Tmc),
transduciéndose dicha célula para expresar un receptor de la superficie celular que comprende un módulo de señalización del receptor de linfocitos T y un dominio extracelular dirigido contra un antígeno patológico.
2. Una célula aislada que tiene un fenotipo de linfocito T de memoria central (Tmc), en donde dicho fenotipo Tmc comprende una firma CD3+/CD8+/CD62L+/CD45RAVCD45RO+, siendo dicha célula, una célula anti-terceros no inductora de reacción de injerto contra hospedador (EICH), que es inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, generándose dicha célula mediante un método que comprende:
(a) poner en contacto células mononucleares de sangre periférica (PBMC) con un antígeno o antígenos de terceros en presencia de IL-21 para permitir el enriquecimiento de células reactivas al antígeno; y
(b) cultivar dichas células resultantes de la etapa (a) en presencia de IL-21, IL-15 e IL-7 para permitir la proliferación de células anti-terceros que comprenden dicho fenotipo de linfocito T de memoria central (Tmc),
transduciéndose dicha célula para expresar un receptor de antígeno quimérico (CAR) y un dominio extracelular dirigido contra un antígeno patológico.
3. Una célula aislada que tiene un fenotipo de linfocito T de memoria central (Tmc), en donde dicho fenotipo Tmc comprende una firma CD3+/CD8+/CD62L+/CD45RAVCD45RO+, siendo dicha célula, una célula anti-terceros no inductora de reacción de injerto contra hospedador (EICH), que es inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, generándose dicha célula mediante un método que comprende:
(a) poner en contacto células mononucleares de sangre periférica (PBMC) con un antígeno o antígenos de terceros en presencia de IL-21 para permitir el enriquecimiento de células reactivas al antígeno; y
(b) cultivar dichas células resultantes de la etapa (a) en presencia de IL-21, IL-15 e IL-7 para permitir la proliferación de células anti-terceros que comprenden dicho fenotipo de linfocito T de memoria central (Tmc),
transduciéndose dicha célula para expresar un receptor de antígeno quimérico (CAR), en donde que dicho CAR comprende un dominio coestimulador o al menos dos dominios coestimuladores y un dominio extracelular dirigido contra un antígeno patológico.
4. Un método in vitro para generar la célula aislada de una cualquiera de las reivindicaciones 1-3, comprendiendo el método transducir una célula que tiene un fenotipo de linfocito T de memoria central (Tmc), siendo dicha célula una célula inductora de tolerancia y capaz de anidar en los ganglios linfáticos después del trasplante, con un polinucleótido que codifica dicho receptor de la superficie celular que comprende un módulo de señalización del receptor de linfocitos T o un receptor de antígeno quimérico (CAR).
5. El método in vitro de la reivindicación 4, en donde la célula se transduce con un vector que comprende dicho polinucleótido.
6. El método in vitro de la reivindicación 4 o 5, en donde dicho polinucleótido codifica un receptor de linfocitos T transgénico (tg-TCR) o un receptor de antígeno quimérico (CAR).
7. La célula aislada de la reivindicación 1 o el método in vitro de la reivindicación 4, en donde dicho receptor de la superficie celular comprende un receptor de linfocitos T transgénico (tg-TCR) o un receptor de antígeno quimérico (CAR).
8. La célula aislada de una cualquiera de las reivindicaciones 2-3 o 7, o el método in vitro de la reivindicación 4 o 7, en donde dicho CAR comprende:
un dominio de unión a antígeno que es un anticuerpo o un fragmento de unión a antígeno; o
un CD3 Z; o
al menos un dominio coestimulador seleccionado del grupo que consiste en CD28, CD134/OX40, CD137/4-1BB, Lck, ICOS y DAP10; o
al menos dos dominios coestimuladores seleccionados del grupo que consiste en CD28, CD134/OX40, CD137/41BB, Lck, ICOS y DAP10.
9. La célula aislada de una cualquiera de las reivindicaciones 1-3 o 7-8, o el método in vitro de una cualquiera de las reivindicaciones 4-8, en donde dicha célula se modifica genéticamente adicionalmente para reprimir la expresión de al menos un gen de punto de control inmunitario endógeno en dicha célula.
10. La célula aislada de una cualquiera de las reivindicaciones 1-3 o 7-9, en donde dicho antígeno patológico se selecciona del grupo que consiste en un antígeno tumoral, un antígeno viral, un antígeno bacteriano, un antígeno fúngico, un antígeno protozoario y un antígeno de parásito.
11. La célula aislada de una cualquiera de las reivindicaciones 1-3, en donde dicho método comprende además el agotamiento de células CD4+ y/o CD56+ antes de la etapa (a).
12. La célula aislada de una cualquiera de las reivindicaciones 1-3 u 11, en donde dicho método comprende además: (c) separar dichas células resultantes de la etapa (b) en suspensiones de células individuales.
13. La célula aislada de una cualquiera de las reivindicaciones 1-3 u 11-12, en donde dicho método comprende además seleccionar las células activadas después de la etapa (a) y antes de la etapa (b), y opcionalmente en donde dicha selección de células activadas se efectúa mediante la selección de células CD137+ y/o CD25+.
14. La célula aislada de una cualquiera de las reivindicaciones 1-3 u 11-13, en donde al menos el 50 % de las células aisladas son células CD3+CD8+ de las cuales al menos el 50 % tienen dicha firma.
15. Una población aislada de células que comprende la célula aislada de una cualquiera de las reivindicaciones 1-3 o 7-14.
16. Una composición farmacéutica que comprende la población de células de la reivindicación 15 y un vehículo farmacéuticamente activo.
17. Una cantidad terapéuticamente eficaz de la población de células de la reivindicación 15 para su uso en el tratamiento de una enfermedad maligna, una enfermedad viral o una enfermedad bacteriana en un sujeto que lo necesite.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562193207P | 2015-07-16 | 2015-07-16 | |
| US201562193229P | 2015-07-16 | 2015-07-16 | |
| PCT/IL2016/050775 WO2017009853A1 (en) | 2015-07-16 | 2016-07-14 | Genetically modified anti-third party central memory t cells and use of same in immunotherapy |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2946785T3 true ES2946785T3 (es) | 2023-07-26 |
Family
ID=56555512
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16745186T Active ES2963038T3 (es) | 2015-07-16 | 2016-07-14 | Uso de células T de memoria central antitercero |
| ES16750269T Active ES2946785T3 (es) | 2015-07-16 | 2016-07-14 | Linfocitos T de memoria central anti-terceros modificados genéticamente y uso de los mismos en inmunoterapia |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16745186T Active ES2963038T3 (es) | 2015-07-16 | 2016-07-14 | Uso de células T de memoria central antitercero |
Country Status (12)
| Country | Link |
|---|---|
| US (3) | US11179448B2 (es) |
| EP (2) | EP3322424B1 (es) |
| JP (1) | JP7057748B2 (es) |
| CN (3) | CN114457039A (es) |
| AU (1) | AU2016291825B2 (es) |
| CA (1) | CA2991690A1 (es) |
| DK (1) | DK3322425T3 (es) |
| ES (2) | ES2963038T3 (es) |
| HK (1) | HK1255063A1 (es) |
| IL (1) | IL256916B2 (es) |
| PL (2) | PL3322424T3 (es) |
| WO (2) | WO2017009853A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2506311C2 (ru) | 2008-10-30 | 2014-02-10 | Йеда Рисёрч Энд Девелопмент Ко. Лтд. | Т-клетки центральной памяти против третьей стороны, способы их получения и их применение в трансплантации и лечении заболеваний |
| SG11201400513PA (en) | 2011-09-08 | 2014-06-27 | Yeda Res & Dev | Anti third party central memory t cells, methods of producing same and use of same in transplantation and disease treatment |
| KR20230097209A (ko) * | 2014-03-12 | 2023-06-30 | 예다 리서치 앤드 디벨럽먼트 캄파니 리미티드 | Cns의 질환 및 손상을 치료하기 위한 전신적 조절 t 세포 수준 또는 활성의 감소 |
| EP3322424B1 (en) | 2015-07-16 | 2023-10-11 | Yeda Research and Development Co., Ltd. | Use of anti third party central memory t cells |
| EP3475414A1 (en) | 2016-06-27 | 2019-05-01 | Yeda Research and Development Co. Ltd | Veto cells generated from memory t cells |
| CN110392736A (zh) | 2017-01-18 | 2019-10-29 | 耶达研究及发展有限公司 | 遗传修饰的反抑细胞及其在免疫治疗中的用途 |
| US10751368B2 (en) | 2017-01-18 | 2020-08-25 | Yeda Research And Development Co. Ltd. | Methods of transplantation and disease treatment |
| CN107573419A (zh) * | 2017-01-24 | 2018-01-12 | 深圳市体内生物医药科技有限公司 | 一种增强t细胞抗肿瘤活性的核酸分子 |
| CN107254489B (zh) * | 2017-06-19 | 2021-06-18 | 河南省华隆生物技术有限公司 | 一种检测嵌合抗原受体或基因修饰的t细胞受体表达的方法和应用 |
| AU2019215110A1 (en) * | 2018-02-02 | 2020-08-13 | The Trustees Of The University Of Pennsylvania | Modified monocytes/macrophages/dendritic cells expressing chimeric antigen receptors and uses in diseases and disorders associated with protein aggregates |
| BR112021004465A2 (pt) * | 2018-09-10 | 2021-05-25 | Atara Biotherapeutics, Inc. | métodos para expansão de células car-t específicas de antígeno, composições e usos relacionados às mesmas |
| CN109536444B (zh) * | 2018-12-11 | 2022-06-28 | 吉林省拓华生物科技有限公司 | 一种适用于恶性实体瘤肿瘤浸润t淋巴细胞的分离诱导方法 |
| EP3959305A1 (en) * | 2019-04-26 | 2022-03-02 | Allogene Therapeutics, Inc. | Methods of manufacturing allogeneic car t cells |
| CN110358734B (zh) | 2019-06-13 | 2020-08-25 | 首都医科大学宣武医院 | 以Tcm为主要效应成分的CAR-T制备方法及其应用 |
| US20230398214A1 (en) * | 2019-08-06 | 2023-12-14 | Yeda Research And Development Co. Ltd. | Anti-viral central memory cd8+ veto cells in haploidentical stem cell transplantation |
| CN110628717B (zh) * | 2019-10-28 | 2020-10-30 | 上海科医联创生物科技有限公司 | 一种浸润性t细胞的培养方法 |
| IL298693A (en) | 2020-06-04 | 2023-02-01 | Carisma Therapeutics Inc | New structures for chimeric antigen receptors |
| US20240001142A1 (en) * | 2020-10-07 | 2024-01-04 | Board Of Regents, The University Of Texas System | Induction by low dose radiation of cancer cell targets for cell-based or small molecule therapy |
Family Cites Families (96)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL154600B (nl) | 1971-02-10 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van specifiek bindende eiwitten en hun corresponderende bindbare stoffen. |
| NL154598B (nl) | 1970-11-10 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van laagmoleculire verbindingen en van eiwitten die deze verbindingen specifiek kunnen binden, alsmede testverpakking. |
| NL154599B (nl) | 1970-12-28 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van specifiek bindende eiwitten en hun corresponderende bindbare stoffen, alsmede testverpakking. |
| US3901654A (en) | 1971-06-21 | 1975-08-26 | Biological Developments | Receptor assays of biologically active compounds employing biologically specific receptors |
| US3853987A (en) | 1971-09-01 | 1974-12-10 | W Dreyer | Immunological reagent and radioimmuno assay |
| US3867517A (en) | 1971-12-21 | 1975-02-18 | Abbott Lab | Direct radioimmunoassay for antigens and their antibodies |
| NL171930C (nl) | 1972-05-11 | 1983-06-01 | Akzo Nv | Werkwijze voor het aantonen en bepalen van haptenen, alsmede testverpakkingen. |
| US3850578A (en) | 1973-03-12 | 1974-11-26 | H Mcconnell | Process for assaying for biologically active molecules |
| US3935074A (en) | 1973-12-17 | 1976-01-27 | Syva Company | Antibody steric hindrance immunoassay with two antibodies |
| US3996345A (en) | 1974-08-12 | 1976-12-07 | Syva Company | Fluorescence quenching with immunological pairs in immunoassays |
| US4034074A (en) | 1974-09-19 | 1977-07-05 | The Board Of Trustees Of Leland Stanford Junior University | Universal reagent 2-site immunoradiometric assay using labelled anti (IgG) |
| US3984533A (en) | 1975-11-13 | 1976-10-05 | General Electric Company | Electrophoretic method of detecting antigen-antibody reaction |
| US4036945A (en) | 1976-05-03 | 1977-07-19 | The Massachusetts General Hospital | Composition and method for determining the size and location of myocardial infarcts |
| US4098876A (en) | 1976-10-26 | 1978-07-04 | Corning Glass Works | Reverse sandwich immunoassay |
| US4331647A (en) | 1980-03-03 | 1982-05-25 | Goldenberg Milton David | Tumor localization and therapy with labeled antibody fragments specific to tumor-associated markers |
| US4879219A (en) | 1980-09-19 | 1989-11-07 | General Hospital Corporation | Immunoassay utilizing monoclonal high affinity IgM antibodies |
| US5011771A (en) | 1984-04-12 | 1991-04-30 | The General Hospital Corporation | Multiepitopic immunometric assay |
| US4666828A (en) | 1984-08-15 | 1987-05-19 | The General Hospital Corporation | Test for Huntington's disease |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4801531A (en) | 1985-04-17 | 1989-01-31 | Biotechnology Research Partners, Ltd. | Apo AI/CIII genomic polymorphisms predictive of atherosclerosis |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US5192553A (en) | 1987-11-12 | 1993-03-09 | Biocyte Corporation | Isolation and preservation of fetal and neonatal hematopoietic stem and progenitor cells of the blood and methods of therapeutic use |
| US5004681B1 (en) | 1987-11-12 | 2000-04-11 | Biocyte Corp | Preservation of fetal and neonatal hematopoietic stem and progenitor cells of the blood |
| US5272057A (en) | 1988-10-14 | 1993-12-21 | Georgetown University | Method of detecting a predisposition to cancer by the use of restriction fragment length polymorphism of the gene for human poly (ADP-ribose) polymerase |
| US5464764A (en) | 1989-08-22 | 1995-11-07 | University Of Utah Research Foundation | Positive-negative selection methods and vectors |
| US5585362A (en) | 1989-08-22 | 1996-12-17 | The Regents Of The University Of Michigan | Adenovirus vectors for gene therapy |
| US5192659A (en) | 1989-08-25 | 1993-03-09 | Genetype Ag | Intron sequence analysis method for detection of adjacent and remote locus alleles as haplotypes |
| US5851832A (en) | 1991-07-08 | 1998-12-22 | Neurospheres, Ltd. | In vitro growth and proliferation of multipotent neural stem cells and their progeny |
| US5281521A (en) | 1992-07-20 | 1994-01-25 | The Trustees Of The University Of Pennsylvania | Modified avidin-biotin technique |
| US5350674A (en) | 1992-09-04 | 1994-09-27 | Becton, Dickinson And Company | Intrinsic factor - horse peroxidase conjugates and a method for increasing the stability thereof |
| DE69534151T2 (de) | 1994-02-22 | 2006-01-12 | Nippon Telegraph And Telephone Corp. | Gefriergetrocknete Blutzellen, Stammzellen und Plättchen und Verfahren zu deren Herstellung |
| US5955257A (en) | 1997-10-21 | 1999-09-21 | Regents Of The University Of Minnesota | Infusible grade short-term cell storage medium for mononuclear cells |
| US6803036B1 (en) | 1998-03-03 | 2004-10-12 | University Of Southern California | Use of cytokines, cells and mitogens to inhibit graft versus host disease |
| EP1061949B1 (en) | 1998-03-03 | 2009-07-15 | University of Southern California | Cytokines and mitogens to inhibit graft-versus-host disease |
| WO2000039294A1 (en) | 1998-12-24 | 2000-07-06 | Novartis Ag | Porcine cells incapable of expressing cd40 antigen, for xenotransplantation |
| AU1086501A (en) | 1999-10-15 | 2001-04-30 | Carnegie Institution Of Washington | Rna interference pathway genes as tools for targeted genetic interference |
| US6326193B1 (en) | 1999-11-05 | 2001-12-04 | Cambria Biosciences, Llc | Insect control agent |
| US6544506B2 (en) | 2000-01-05 | 2003-04-08 | Yeda Research & Development Co. Ltd. | Veto cells effective in preventing graft rejection and devoid of graft versus host potential |
| CA2405806A1 (en) | 2000-04-11 | 2001-10-18 | University Of Southern California | A method to prevent graft rejection using tgf-.beta. to induce t suppressor cells |
| US7094874B2 (en) | 2000-05-26 | 2006-08-22 | Bristol-Myers Squibb Co. | Soluble CTLA4 mutant molecules |
| WO2001096584A2 (en) | 2000-06-12 | 2001-12-20 | Akkadix Corporation | Materials and methods for the control of nematodes |
| AR035037A1 (es) | 2000-07-03 | 2004-04-14 | Bristol Myers Squibb Co | Metodos para tratar enfermedades reumaticas al usar una molecula ctla4 soluble |
| US20040022787A1 (en) | 2000-07-03 | 2004-02-05 | Robert Cohen | Methods for treating an autoimmune disease using a soluble CTLA4 molecule and a DMARD or NSAID |
| IL155941A0 (en) | 2000-11-30 | 2003-12-23 | Yeda Res & Dev | Methods of utilizing cultured non-gvhd inducing t lymphocytes to treat disease |
| WO2002094202A2 (en) | 2001-05-23 | 2002-11-28 | Bristol-Myers Squibb Company | Methods for protecting allogeneic islet transplant using soluble ctla4 mutant molecules |
| US20030147859A1 (en) | 2001-06-18 | 2003-08-07 | Yair Reisner | Methods of utilizing cultured hematopoietic progenitor cells for inducing immunological tolerance |
| US20040136972A1 (en) * | 2001-09-07 | 2004-07-15 | Yeda Research And Development Co. Ltd. | Methods of treating disease by transplantation of developing allogeneic or xenogeneic organs or tissues |
| US7504099B2 (en) | 2001-12-27 | 2009-03-17 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | Methods of inducing or enhancing connective tissue repair |
| US7745140B2 (en) | 2002-01-03 | 2010-06-29 | The Trustees Of The University Of Pennsylvania | Activation and expansion of T-cells using an engineered multivalent signaling platform as a research tool |
| US6992176B2 (en) | 2002-02-13 | 2006-01-31 | Technion Research & Development Foundation Ltd. | Antibody having a T-cell receptor-like specificity, yet higher affinity, and the use of same in the detection and treatment of cancer, viral infection and autoimmune disease |
| US20030215942A1 (en) | 2002-02-14 | 2003-11-20 | Stemcyte, Inc. | Undesignated allogeneic stem cell bank |
| EP1549277A4 (en) | 2002-05-22 | 2007-09-05 | Cleveland Clinic Foundation | INDUCING AND MAINTAINING COMPLEX TISSUE ALLOGRAFT TOLERANCE |
| IL165425A0 (en) | 2004-11-28 | 2006-01-15 | Yeda Res & Dev | Methods of treating disease by transplantation of developing allogeneic or xenogeneic organs or tissues |
| WO2005016266A2 (en) | 2003-08-04 | 2005-02-24 | Bristol-Myers Squibb Company | Methods for treating cardiovascular disease using a soluble ctla4 molecule |
| JP2007522109A (ja) | 2004-01-15 | 2007-08-09 | ノボ ノルディスク アクティーゼルスカブ | 自己免疫疾患の治療およびil−21での同種異系移植片拒絶反応 |
| EP1732600A2 (en) | 2004-03-26 | 2006-12-20 | Pfizer Products Incorporated | Uses of anti-ctla-4 antibodies |
| WO2006041763A1 (en) | 2004-10-04 | 2006-04-20 | Novartis Ag | Renin inhibitors for treating transplantation induced diseases |
| WO2006040763A2 (en) | 2004-10-12 | 2006-04-20 | Technion Research & Development Foundation Ltd. | Isolated primate embryonic cells and methods of generating and using same |
| LT1814580T (lt) | 2004-11-24 | 2016-12-12 | Fred Hutchinson Cancer Research Center | Il 21 naudojimo pritaikomajai imunoterapijai ir naviko antigenų identifikavimo metodai |
| WO2006108035A1 (en) | 2005-04-06 | 2006-10-12 | Bristol-Myers Squibb Company | Methods for treating immune disorders associated with graft transplantation with soluble ctla4 mutant molecules |
| EP1928479B1 (en) | 2005-08-24 | 2016-06-08 | Yeda Research And Development Co., Ltd. | Universal donor-derived tolerogenic cells for inducing non-syngeneic transplantation tolerance |
| GB0605217D0 (en) | 2006-03-15 | 2006-04-26 | Novartis Ag | Method and compositions for assessing acute rejection |
| GB0606776D0 (en) | 2006-04-03 | 2006-05-10 | Novartis Pharma Ag | Predictive biomarkers for chronic allograft nephropathy |
| US20080131415A1 (en) | 2006-11-30 | 2008-06-05 | Riddell Stanley R | Adoptive transfer of cd8 + t cell clones derived from central memory cells |
| US8361473B2 (en) | 2007-03-29 | 2013-01-29 | Technion Research & Development Foundation Ltd. | Antibodies and their uses for diagnosis and treatment of cytomegalovirus infection and associated diseases |
| US20100254958A1 (en) | 2007-10-24 | 2010-10-07 | Anne Letsch | Antigen-Specific T-Cell Preparations from Bone Marrow |
| WO2009125394A1 (en) | 2008-04-09 | 2009-10-15 | Technion Research & Development Foundation Ltd. | Anti human immunodeficiency antibodies and uses thereof |
| EP2297200A1 (en) | 2008-04-09 | 2011-03-23 | Technion Research & Development Foundation Ltd. | Anti influenza antibodies and uses thereof |
| US8200915B2 (en) | 2008-08-22 | 2012-06-12 | Cadence Design Systems, Inc. | Management of very large streaming data sets for efficient writes and reads to and from persistent storage |
| RU2506311C2 (ru) | 2008-10-30 | 2014-02-10 | Йеда Рисёрч Энд Девелопмент Ко. Лтд. | Т-клетки центральной памяти против третьей стороны, способы их получения и их применение в трансплантации и лечении заболеваний |
| CN102597222B (zh) | 2009-10-27 | 2015-07-15 | 因缪尼卡姆股份公司 | 用于增殖抗原特异性t细胞的方法 |
| ES2686424T5 (es) * | 2010-05-04 | 2023-03-27 | Yeda Res & Dev | Inmunoterapia con células alogénicas redireccionadas |
| US20130189284A1 (en) | 2010-07-15 | 2013-07-25 | Technion Research & Development Foundation Ltd. | Antibodies with t-cell receptor like specificity towards native complexes of mhc class ii and diabetes-associated autoantigenic peptides |
| BR112013005756A2 (pt) | 2010-09-08 | 2018-12-26 | Yeda Research And Development Co. Ltd | use of anti third party central memory t cells for anti-leukemia/lymphoma treatment |
| JP2013540731A (ja) | 2010-09-08 | 2013-11-07 | イェダ リサーチ アンド デベロップメント カンパニー リミテッド | 安定かつ長期間の生着のための免疫抑制薬物組合せ |
| BR112013024395B1 (pt) * | 2011-03-23 | 2021-10-26 | Fred Hutchinson Cancer Research Center | Composições adotivas de imunoterapia celular e método para fabricação da dita composição |
| CN103517707A (zh) * | 2011-04-29 | 2014-01-15 | 西莱克塔生物科技公司 | 从合成纳米载体中控制释放免疫抑制剂 |
| SG11201400513PA (en) * | 2011-09-08 | 2014-06-27 | Yeda Res & Dev | Anti third party central memory t cells, methods of producing same and use of same in transplantation and disease treatment |
| JP2015500279A (ja) | 2011-12-08 | 2015-01-05 | イェダ リサーチ アンド デベロップメント カンパニー リミテッド | 哺乳動物胎児肺細胞および該細胞の治療的使用 |
| SG11201403456UA (en) * | 2011-12-22 | 2014-07-30 | Yeda Res & Dev | A combination therapy for a stable and long term engraftment using specific protocols for t/b cell depletion |
| BR112015002816A8 (pt) | 2012-08-20 | 2023-04-18 | Hutchinson Fred Cancer Res | Método e composições para imunoterapia celular |
| US10316289B2 (en) | 2012-09-06 | 2019-06-11 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methods of producing T memory stem cell populations |
| EP2906684B8 (en) * | 2012-10-10 | 2020-09-02 | Sangamo Therapeutics, Inc. | T cell modifying compounds and uses thereof |
| US9499855B2 (en) * | 2013-03-14 | 2016-11-22 | Elwha Llc | Compositions, methods, and computer systems related to making and administering modified T cells |
| CN105518018B (zh) * | 2013-03-15 | 2020-04-03 | 细胞基因公司 | 修饰的t淋巴细胞 |
| DK2981607T3 (da) | 2013-04-03 | 2020-11-16 | Memorial Sloan Kettering Cancer Center | Effektiv generering af tumormålrettede t-celler afledt af pluripotente stamceller |
| MA47849A (fr) | 2014-05-28 | 2020-01-29 | Agenus Inc | Anticorps anti-gitr et leurs procédés d'utilisation |
| JP7005346B2 (ja) | 2014-10-27 | 2022-02-04 | フレッド ハッチンソン キャンサー リサーチ センター | 養子細胞免疫療法の有効性を増強させるための組成物および方法 |
| EP4219725A3 (en) | 2014-10-31 | 2023-08-30 | The Trustees of the University of Pennsylvania | Altering gene expression in modified t cells and uses thereof |
| IL295224A (en) | 2015-05-28 | 2022-10-01 | Kite Pharma Inc | Methods for training patients for t-cell therapy |
| EP3322424B1 (en) | 2015-07-16 | 2023-10-11 | Yeda Research and Development Co., Ltd. | Use of anti third party central memory t cells |
| EP3402512A4 (en) | 2016-01-11 | 2019-09-25 | Armo Biosciences, Inc. | INTERLEUKIN-10 FOR USE IN THE PRODUCTION OF ANTIGEN-SPECIFIC CD8 + T LYMPHOCYTES AND METHODS OF USE THEREOF |
| CN109475581A (zh) | 2016-05-22 | 2019-03-15 | 耶达研究及发展有限公司 | 为了移植及诱导耐受性而使用肺细胞的方法 |
| EP3475414A1 (en) | 2016-06-27 | 2019-05-01 | Yeda Research and Development Co. Ltd | Veto cells generated from memory t cells |
| US10751368B2 (en) * | 2017-01-18 | 2020-08-25 | Yeda Research And Development Co. Ltd. | Methods of transplantation and disease treatment |
| CN110392736A (zh) | 2017-01-18 | 2019-10-29 | 耶达研究及发展有限公司 | 遗传修饰的反抑细胞及其在免疫治疗中的用途 |
-
2016
- 2016-07-14 EP EP16745186.3A patent/EP3322424B1/en active Active
- 2016-07-14 US US15/744,905 patent/US11179448B2/en active Active
- 2016-07-14 CN CN202210206490.5A patent/CN114457039A/zh active Pending
- 2016-07-14 PL PL16745186.3T patent/PL3322424T3/pl unknown
- 2016-07-14 CA CA2991690A patent/CA2991690A1/en active Pending
- 2016-07-14 WO PCT/IL2016/050775 patent/WO2017009853A1/en active Application Filing
- 2016-07-14 CN CN201680053580.8A patent/CN108135938A/zh active Pending
- 2016-07-14 EP EP16750269.9A patent/EP3322425B1/en active Active
- 2016-07-14 IL IL256916A patent/IL256916B2/en unknown
- 2016-07-14 PL PL16750269.9T patent/PL3322425T3/pl unknown
- 2016-07-14 CN CN201680053579.5A patent/CN108025026A/zh active Pending
- 2016-07-14 ES ES16745186T patent/ES2963038T3/es active Active
- 2016-07-14 AU AU2016291825A patent/AU2016291825B2/en active Active
- 2016-07-14 WO PCT/IL2016/050774 patent/WO2017009852A1/en active Application Filing
- 2016-07-14 JP JP2018501339A patent/JP7057748B2/ja active Active
- 2016-07-14 ES ES16750269T patent/ES2946785T3/es active Active
- 2016-07-14 DK DK16750269.9T patent/DK3322425T3/da active
- 2016-07-14 US US15/744,881 patent/US10933124B2/en active Active
-
2018
- 2018-11-07 HK HK18114191.8A patent/HK1255063A1/zh unknown
-
2021
- 2021-11-22 US US17/531,897 patent/US11844827B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN108025026A (zh) | 2018-05-11 |
| US11179448B2 (en) | 2021-11-23 |
| EP3322425B1 (en) | 2023-03-15 |
| US11844827B2 (en) | 2023-12-19 |
| IL256916A (en) | 2018-03-29 |
| PL3322424T3 (pl) | 2024-03-04 |
| EP3322425A1 (en) | 2018-05-23 |
| EP3322424C0 (en) | 2023-10-11 |
| IL256916B2 (en) | 2024-04-01 |
| HK1255063A1 (zh) | 2019-08-02 |
| CN108135938A (zh) | 2018-06-08 |
| AU2016291825B2 (en) | 2022-08-11 |
| AU2016291825A1 (en) | 2018-03-08 |
| ES2963038T3 (es) | 2024-03-25 |
| US20180207272A1 (en) | 2018-07-26 |
| EP3322424B1 (en) | 2023-10-11 |
| JP2018524987A (ja) | 2018-09-06 |
| WO2017009853A1 (en) | 2017-01-19 |
| US10933124B2 (en) | 2021-03-02 |
| DK3322425T3 (da) | 2023-06-12 |
| WO2017009852A1 (en) | 2017-01-19 |
| PL3322425T3 (pl) | 2023-10-09 |
| CA2991690A1 (en) | 2017-01-19 |
| US20220080034A1 (en) | 2022-03-17 |
| CN114457039A (zh) | 2022-05-10 |
| US20180207247A1 (en) | 2018-07-26 |
| EP3322424A1 (en) | 2018-05-23 |
| IL256916B1 (en) | 2023-12-01 |
| JP7057748B2 (ja) | 2022-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2946785T3 (es) | Linfocitos T de memoria central anti-terceros modificados genéticamente y uso de los mismos en inmunoterapia | |
| US20230181643A1 (en) | Use of trans-signaling approach in chimeric antigen receptors | |
| US11180550B2 (en) | Compositions and methods for treating cancer | |
| ES2700966T3 (es) | Utilización de linfocitos T modificados con receptores de antígeno quiméricos para tratar el cáncer | |
| ES2959443T3 (es) | Uso del dominio de señalización de CD2 en receptores de antígenos quiméricos de segunda generación | |
| ES2733525T3 (es) | Métodos para evaluar la adecuación de los linfocitos T transducidos para su administración | |
| ES2786263T3 (es) | Mejora de la actividad de los CAR de linfocitos T mediante la introducción conjunta de un anticuerpo biespecífico | |
| US11555178B2 (en) | Genetically modified veto cells and use of same in immunotherapy | |
| US20230321235A1 (en) | Veto car-t cells |