ES2940263T3 - Compuestos químicos - Google Patents
Compuestos químicos Download PDFInfo
- Publication number
- ES2940263T3 ES2940263T3 ES18795809T ES18795809T ES2940263T3 ES 2940263 T3 ES2940263 T3 ES 2940263T3 ES 18795809 T ES18795809 T ES 18795809T ES 18795809 T ES18795809 T ES 18795809T ES 2940263 T3 ES2940263 T3 ES 2940263T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- oxo
- chloro
- substituted
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 250
- 238000011282 treatment Methods 0.000 claims abstract description 37
- 230000009385 viral infection Effects 0.000 claims abstract description 21
- 208000036142 Viral infection Diseases 0.000 claims abstract description 20
- 150000003839 salts Chemical class 0.000 claims description 69
- 239000008194 pharmaceutical composition Substances 0.000 claims description 29
- 208000002672 hepatitis B Diseases 0.000 claims description 19
- 238000002560 therapeutic procedure Methods 0.000 claims description 17
- 239000003085 diluting agent Substances 0.000 claims description 5
- 241000700721 Hepatitis B virus Species 0.000 abstract description 57
- 239000000427 antigen Substances 0.000 abstract description 20
- 102000036639 antigens Human genes 0.000 abstract description 20
- 108091007433 antigens Proteins 0.000 abstract description 20
- 208000037262 Hepatitis delta Diseases 0.000 abstract description 12
- 241000724709 Hepatitis delta virus Species 0.000 abstract description 12
- 238000000034 method Methods 0.000 abstract description 11
- 239000003112 inhibitor Substances 0.000 abstract description 3
- 239000000203 mixture Substances 0.000 description 215
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 207
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 184
- 239000000243 solution Substances 0.000 description 156
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 154
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 151
- 239000011541 reaction mixture Substances 0.000 description 150
- 239000007787 solid Substances 0.000 description 118
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 117
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 113
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 110
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 109
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 109
- -1 Polycyclic pyridine compounds Chemical class 0.000 description 106
- 125000001072 heteroaryl group Chemical group 0.000 description 100
- 125000000217 alkyl group Chemical group 0.000 description 94
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 93
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 92
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 90
- 125000000753 cycloalkyl group Chemical group 0.000 description 86
- 229910052757 nitrogen Inorganic materials 0.000 description 85
- 239000002253 acid Substances 0.000 description 79
- 235000019439 ethyl acetate Nutrition 0.000 description 77
- 125000003545 alkoxy group Chemical group 0.000 description 75
- 238000005160 1H NMR spectroscopy Methods 0.000 description 71
- 125000005842 heteroatom Chemical group 0.000 description 70
- 235000002639 sodium chloride Nutrition 0.000 description 70
- 125000003118 aryl group Chemical group 0.000 description 68
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 61
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 58
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 58
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 56
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 55
- 235000019253 formic acid Nutrition 0.000 description 55
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 55
- 239000001257 hydrogen Substances 0.000 description 54
- 229910052739 hydrogen Inorganic materials 0.000 description 54
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 53
- 229910052760 oxygen Inorganic materials 0.000 description 52
- 229910052736 halogen Inorganic materials 0.000 description 51
- 150000002367 halogens Chemical class 0.000 description 51
- 238000006243 chemical reaction Methods 0.000 description 45
- 239000012044 organic layer Substances 0.000 description 45
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 45
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 42
- 239000002609 medium Substances 0.000 description 42
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 42
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 41
- 239000012267 brine Substances 0.000 description 41
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 41
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 39
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 39
- 125000003107 substituted aryl group Chemical group 0.000 description 39
- 125000003342 alkenyl group Chemical group 0.000 description 37
- 229910052938 sodium sulfate Inorganic materials 0.000 description 37
- 235000011152 sodium sulphate Nutrition 0.000 description 37
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- 239000002904 solvent Substances 0.000 description 35
- 125000001424 substituent group Chemical group 0.000 description 35
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- 125000000547 substituted alkyl group Chemical group 0.000 description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 30
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 30
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 30
- 238000004587 chromatography analysis Methods 0.000 description 28
- 239000000741 silica gel Substances 0.000 description 28
- 229910002027 silica gel Inorganic materials 0.000 description 28
- 238000003756 stirring Methods 0.000 description 28
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 28
- 125000005415 substituted alkoxy group Chemical group 0.000 description 27
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 26
- 239000000706 filtrate Substances 0.000 description 26
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 26
- 125000005017 substituted alkenyl group Chemical group 0.000 description 26
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 25
- 239000003921 oil Substances 0.000 description 25
- 239000000843 powder Substances 0.000 description 25
- 125000004093 cyano group Chemical group *C#N 0.000 description 24
- 235000019198 oils Nutrition 0.000 description 23
- 239000011734 sodium Substances 0.000 description 23
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 22
- 229910052799 carbon Inorganic materials 0.000 description 22
- 125000000623 heterocyclic group Chemical group 0.000 description 22
- 208000015181 infectious disease Diseases 0.000 description 22
- 230000002829 reductive effect Effects 0.000 description 22
- 229910000027 potassium carbonate Inorganic materials 0.000 description 21
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 20
- 235000011181 potassium carbonates Nutrition 0.000 description 20
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 19
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 19
- 125000004103 aminoalkyl group Chemical group 0.000 description 19
- 239000003795 chemical substances by application Substances 0.000 description 19
- 239000012071 phase Substances 0.000 description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 18
- 125000005843 halogen group Chemical group 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- 238000001914 filtration Methods 0.000 description 17
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 17
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 17
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 16
- 238000009472 formulation Methods 0.000 description 16
- 239000001301 oxygen Substances 0.000 description 16
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 16
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 15
- 239000007832 Na2SO4 Substances 0.000 description 15
- 238000004440 column chromatography Methods 0.000 description 15
- 235000011056 potassium acetate Nutrition 0.000 description 15
- 239000000377 silicon dioxide Substances 0.000 description 15
- 229910052717 sulfur Inorganic materials 0.000 description 15
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 14
- 239000012043 crude product Substances 0.000 description 14
- 125000004404 heteroalkyl group Chemical group 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 12
- 229910021605 Palladium(II) bromide Inorganic materials 0.000 description 12
- 239000008346 aqueous phase Substances 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 12
- INIOZDBICVTGEO-UHFFFAOYSA-L palladium(ii) bromide Chemical compound Br[Pd]Br INIOZDBICVTGEO-UHFFFAOYSA-L 0.000 description 12
- 238000002360 preparation method Methods 0.000 description 12
- 230000002441 reversible effect Effects 0.000 description 12
- 150000002148 esters Chemical class 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 11
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 11
- 239000012453 solvate Substances 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 10
- 239000008280 blood Substances 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- 229920006395 saturated elastomer Polymers 0.000 description 10
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 10
- 230000003612 virological effect Effects 0.000 description 10
- KAATUXNTWXVJKI-NSHGMRRFSA-N (1R)-cis-(alphaS)-cypermethrin Chemical compound CC1(C)[C@@H](C=C(Cl)Cl)[C@H]1C(=O)O[C@H](C#N)C1=CC=CC(OC=2C=CC=CC=2)=C1 KAATUXNTWXVJKI-NSHGMRRFSA-N 0.000 description 9
- DLAPWRLGUCWNIV-UHFFFAOYSA-N 5-(6-chloro-5-methoxypyridin-3-yl)-2,2-dimethylcyclopentan-1-amine Chemical compound ClC1=C(C=C(C=N1)C1CCC(C1N)(C)C)OC DLAPWRLGUCWNIV-UHFFFAOYSA-N 0.000 description 9
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- BOJLXWSIMUEPCI-UHFFFAOYSA-N ethyl 4-oxopyran-3-carboxylate Chemical compound O=C1C(=COC=C1)C(=O)OCC BOJLXWSIMUEPCI-UHFFFAOYSA-N 0.000 description 9
- 229910052740 iodine Inorganic materials 0.000 description 9
- 239000011630 iodine Substances 0.000 description 9
- OHENQANLQNOMAO-UHFFFAOYSA-N oxaborole Chemical group O1B=CC=C1 OHENQANLQNOMAO-UHFFFAOYSA-N 0.000 description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 9
- 230000009467 reduction Effects 0.000 description 9
- 238000006722 reduction reaction Methods 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- 125000004001 thioalkyl group Chemical group 0.000 description 9
- 150000003573 thiols Chemical class 0.000 description 9
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 8
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 8
- GDAHCVXDGPKSFK-UHFFFAOYSA-N 5-(6-chloro-5-methoxypyridin-3-yl)-2,2-dimethylcyclopentan-1-one Chemical compound ClC1=C(C=C(C=N1)C1CCC(C1=O)(C)C)OC GDAHCVXDGPKSFK-UHFFFAOYSA-N 0.000 description 8
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 8
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 8
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 8
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 8
- 239000002244 precipitate Substances 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 125000000999 tert-butyl group Chemical class [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 8
- 125000004149 thio group Chemical group *S* 0.000 description 8
- CEVMYGZHEJSOHZ-UHFFFAOYSA-N 1-bromo-3-methoxypropane Chemical compound COCCCBr CEVMYGZHEJSOHZ-UHFFFAOYSA-N 0.000 description 7
- OAXQOMDBXDGFJJ-UHFFFAOYSA-N 5-(2-amino-3,3-dimethylcyclopentyl)-2-chloro-6-iodopyridin-3-ol Chemical compound NC1C(CCC1(C)C)C=1C=C(C(=NC=1I)Cl)O OAXQOMDBXDGFJJ-UHFFFAOYSA-N 0.000 description 7
- 101710132601 Capsid protein Proteins 0.000 description 7
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 7
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 7
- 239000002245 particle Substances 0.000 description 7
- 230000002265 prevention Effects 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 6
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- 238000002965 ELISA Methods 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 230000004069 differentiation Effects 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 238000001704 evaporation Methods 0.000 description 6
- 230000008020 evaporation Effects 0.000 description 6
- 239000012065 filter cake Substances 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 6
- 239000003208 petroleum Substances 0.000 description 6
- 235000010265 sodium sulphite Nutrition 0.000 description 6
- 239000011593 sulfur Chemical group 0.000 description 6
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 6
- MPEOYZBCMHIGIB-UHFFFAOYSA-N 1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethylbutan-2-one Chemical compound ClC1=C(C=C(C=N1)CC(C(C)(C)C)=O)OC MPEOYZBCMHIGIB-UHFFFAOYSA-N 0.000 description 5
- YNTJKQDWYXUTLZ-UHFFFAOYSA-N 2-(3-chlorophenoxy)propanoic acid Chemical compound OC(=O)C(C)OC1=CC=CC(Cl)=C1 YNTJKQDWYXUTLZ-UHFFFAOYSA-N 0.000 description 5
- FBWMQYIMJMVINO-UHFFFAOYSA-N 5-(2-amino-3,3-dimethylcyclopentyl)-2-chloropyridin-3-ol Chemical compound NC1C(CCC1(C)C)C=1C=C(C(=NC=1)Cl)O FBWMQYIMJMVINO-UHFFFAOYSA-N 0.000 description 5
- GFJMOZYIDADKLJ-UHFFFAOYSA-N 5-bromo-2-chloro-3-methoxypyridine Chemical compound COC1=CC(Br)=CN=C1Cl GFJMOZYIDADKLJ-UHFFFAOYSA-N 0.000 description 5
- OERUXIYQWSPQPV-UHFFFAOYSA-N 6-tert-butyl-2-chloro-3-(cyclopropylmethoxy)-10-oxo-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)C1CC=2C=C(C(=NC=2C=2N1C=C(C(C=2)=O)C(=O)O)Cl)OCC1CC1 OERUXIYQWSPQPV-UHFFFAOYSA-N 0.000 description 5
- 102000014150 Interferons Human genes 0.000 description 5
- 108010050904 Interferons Proteins 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 5
- 239000011230 binding agent Substances 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 238000010828 elution Methods 0.000 description 5
- USUZNEDGQXROIB-UHFFFAOYSA-N ethyl 1,2-dihydropyridine-3-carboxylate Chemical compound CCOC(=O)C1=CC=CNC1 USUZNEDGQXROIB-UHFFFAOYSA-N 0.000 description 5
- CSDMSMQKXPTQLH-UHFFFAOYSA-N ethyl 1-[5-(6-chloro-5-hydroxy-2-iodopyridin-3-yl)-2,2-dimethylcyclopentyl]-4-oxopyridine-3-carboxylate Chemical compound ClC1=C(C=C(C(=N1)I)C1CCC(C1N1C=C(C(C=C1)=O)C(=O)OCC)(C)C)O CSDMSMQKXPTQLH-UHFFFAOYSA-N 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 229940079322 interferon Drugs 0.000 description 5
- 208000019423 liver disease Diseases 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 238000001819 mass spectrum Methods 0.000 description 5
- 125000006574 non-aromatic ring group Chemical group 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 239000011534 wash buffer Substances 0.000 description 5
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 4
- MXBVNILGVJVVMH-UHFFFAOYSA-N 1,7-naphthyridine Chemical compound C1=NC=CC2=CC=CN=C21 MXBVNILGVJVVMH-UHFFFAOYSA-N 0.000 description 4
- OPLCPMGRQXVPJB-UHFFFAOYSA-N 1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethylbutan-2-amine Chemical compound ClC1=C(C=C(C=N1)CC(C(C)(C)C)N)OC OPLCPMGRQXVPJB-UHFFFAOYSA-N 0.000 description 4
- LUCJWTAPFGYJLS-VIFPVBQESA-N 5-[(2S)-2-amino-3,3-dimethylbutyl]-2-chloropyridin-3-ol Chemical compound N[C@@H](CC=1C=C(C(=NC=1)Cl)O)C(C)(C)C LUCJWTAPFGYJLS-VIFPVBQESA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- 229910004373 HOAc Inorganic materials 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- CVQUWLDCFXOXEN-UHFFFAOYSA-N Pyran-4-one Chemical class O=C1C=COC=C1 CVQUWLDCFXOXEN-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 4
- 239000003443 antiviral agent Substances 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- 229940126214 compound 3 Drugs 0.000 description 4
- SACNIGZYDTUHKB-UHFFFAOYSA-N ditert-butyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C(C)(C)C)C(C)(C)C SACNIGZYDTUHKB-UHFFFAOYSA-N 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000012091 fetal bovine serum Substances 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 150000004677 hydrates Chemical class 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 208000014018 liver neoplasm Diseases 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 239000002777 nucleoside Substances 0.000 description 4
- 150000003833 nucleoside derivatives Chemical class 0.000 description 4
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 4
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 4
- 238000004808 supercritical fluid chromatography Methods 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- UGGRMFGKRXDDKI-WHEQGISXSA-N (R)-N-[(2S)-1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethylbutan-2-yl]-2-methylpropane-2-sulfinamide Chemical compound ClC1=C(C=C(C=N1)C[C@@H](C(C)(C)C)N[S@](=O)C(C)(C)C)OC UGGRMFGKRXDDKI-WHEQGISXSA-N 0.000 description 3
- UGGRMFGKRXDDKI-DMZKTXOQSA-N (S)-N-[(2R)-1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethylbutan-2-yl]-2-methylpropane-2-sulfinamide Chemical compound ClC1=C(C=C(C=N1)C[C@H](C(C)(C)C)N[S@@](=O)C(C)(C)C)OC UGGRMFGKRXDDKI-DMZKTXOQSA-N 0.000 description 3
- FLBAYUMRQUHISI-UHFFFAOYSA-N 1,8-naphthyridine Chemical compound N1=CC=CC2=CC=CN=C21 FLBAYUMRQUHISI-UHFFFAOYSA-N 0.000 description 3
- BBENVALKLPPOGP-UHFFFAOYSA-N 1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethyl-4-phenylmethoxybutan-2-one Chemical compound C(C1=CC=CC=C1)OCC(C(CC=1C=NC(=C(C=1)OC)Cl)=O)(C)C BBENVALKLPPOGP-UHFFFAOYSA-N 0.000 description 3
- BUSSOYRVXZNGQX-UHFFFAOYSA-N 1-[2-bromo-6-chloro-5-(cyclopropylmethoxy)pyridin-3-yl]-3,3-dimethylbutan-2-amine Chemical compound BrC1=NC(=C(C=C1CC(C(C)(C)C)N)OCC1CC1)Cl BUSSOYRVXZNGQX-UHFFFAOYSA-N 0.000 description 3
- XLVIOHQIWMLQTO-UHFFFAOYSA-N 1-[[2-bromo-6-chloro-5-(cyclopropylmethoxy)pyridin-3-yl]methyl]cyclobutan-1-amine Chemical compound BrC1=NC(=C(C=C1CC1(CCC1)N)OCC1CC1)Cl XLVIOHQIWMLQTO-UHFFFAOYSA-N 0.000 description 3
- FTGZMZBYOHMEPS-UHFFFAOYSA-N 2,2-dimethylcyclopentan-1-one Chemical compound CC1(C)CCCC1=O FTGZMZBYOHMEPS-UHFFFAOYSA-N 0.000 description 3
- MWXVMYZRFLABRM-UHFFFAOYSA-N 2,3-dimethyl-7-propan-2-yl-7,8-dihydro-1,6-naphthyridine Chemical compound C(C)(C)C1N=CC=2C=C(C(=NC=2C1)C)C MWXVMYZRFLABRM-UHFFFAOYSA-N 0.000 description 3
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 3
- JZKKIEVIOFDWEF-UHFFFAOYSA-N 2-bromo-3-(bromomethyl)-6-chloro-5-(cyclopropylmethoxy)pyridine Chemical compound BrC1=NC(=C(C=C1CBr)OCC1CC1)Cl JZKKIEVIOFDWEF-UHFFFAOYSA-N 0.000 description 3
- FEEKFEGRGFQHQX-UHFFFAOYSA-N 2-bromo-6-chloro-5-(cyclopropylmethoxy)-3-methylpyridine Chemical compound BrC1=NC(=C(C=C1C)OCC1CC1)Cl FEEKFEGRGFQHQX-UHFFFAOYSA-N 0.000 description 3
- BYVPJDZJIHOUHA-UHFFFAOYSA-N 2-chloro-5,6-dimethylpyridine-3-carbonitrile Chemical compound CC1=CC(C#N)=C(Cl)N=C1C BYVPJDZJIHOUHA-UHFFFAOYSA-N 0.000 description 3
- LUCJWTAPFGYJLS-UHFFFAOYSA-N 5-(2-amino-3,3-dimethylbutyl)-2-chloropyridin-3-ol Chemical compound NC(CC=1C=C(C(=NC=1)Cl)O)C(C)(C)C LUCJWTAPFGYJLS-UHFFFAOYSA-N 0.000 description 3
- BIFSEZHXCMPHIP-UHFFFAOYSA-N 5-(2-amino-4-hydroxy-3,3-dimethylbutyl)-2-chloro-6-iodopyridin-3-ol Chemical compound NC(CC=1C=C(C(=NC=1I)Cl)O)C(CO)(C)C BIFSEZHXCMPHIP-UHFFFAOYSA-N 0.000 description 3
- VDXUNISASRVDGG-UHFFFAOYSA-N 5-(2-amino-4-hydroxy-3,3-dimethylbutyl)-2-chloropyridin-3-ol Chemical compound NC(CC=1C=C(C(=NC=1)Cl)O)C(CO)(C)C VDXUNISASRVDGG-UHFFFAOYSA-N 0.000 description 3
- IXLVUUFUDRJUSL-RPBOFIJWSA-N 5-[[4-(3-acetamidophenyl)phenyl]methyl]-n-[(1s,2r)-2-phenylcyclopropyl]-1,3-oxazole-4-carboxamide Chemical compound CC(=O)NC1=CC=CC(C=2C=CC(CC3=C(N=CO3)C(=O)N[C@@H]3[C@H](C3)C=3C=CC=CC=3)=CC=2)=C1 IXLVUUFUDRJUSL-RPBOFIJWSA-N 0.000 description 3
- GNWONFBPVNMWTH-UHFFFAOYSA-N 6-bromo-2-chloro-5-methylpyridin-3-ol Chemical compound Cc1cc(O)c(Cl)nc1Br GNWONFBPVNMWTH-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 239000005695 Ammonium acetate Substances 0.000 description 3
- 208000003322 Coinfection Diseases 0.000 description 3
- 229920000742 Cotton Polymers 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 238000008157 ELISA kit Methods 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 3
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 108010067390 Viral Proteins Proteins 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 235000019257 ammonium acetate Nutrition 0.000 description 3
- 229940043376 ammonium acetate Drugs 0.000 description 3
- 230000000840 anti-viral effect Effects 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- XSCHRSMBECNVNS-UHFFFAOYSA-N benzopyrazine Natural products N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 3
- 239000006227 byproduct Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 208000019425 cirrhosis of liver Diseases 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 239000013058 crude material Substances 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 238000003821 enantio-separation Methods 0.000 description 3
- GSSBCHMKZXJMBT-UHFFFAOYSA-N ethyl 2-(dimethylaminomethylidene)-3-oxo-4-phenylmethoxybutanoate Chemical compound CCOC(=O)C(=CN(C)C)C(=O)COCC1=CC=CC=C1 GSSBCHMKZXJMBT-UHFFFAOYSA-N 0.000 description 3
- ZXJVZLYWKONRFB-UHFFFAOYSA-N ethyl 2-methyl-4-oxopyran-3-carboxylate Chemical compound CCOC(=O)C1=C(C)OC=CC1=O ZXJVZLYWKONRFB-UHFFFAOYSA-N 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 210000003494 hepatocyte Anatomy 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000009169 immunotherapy Methods 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000011221 initial treatment Methods 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 201000007270 liver cancer Diseases 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 235000010446 mineral oil Nutrition 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 235000015424 sodium Nutrition 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000009269 systemic vascular permeability Effects 0.000 description 3
- VCMJCVGFSROFHV-WZGZYPNHSA-N tenofovir disoproxil fumarate Chemical compound OC(=O)\C=C\C(O)=O.N1=CN=C2N(C[C@@H](C)OCP(=O)(OCOC(=O)OC(C)C)OCOC(=O)OC(C)C)C=NC2=C1N VCMJCVGFSROFHV-WZGZYPNHSA-N 0.000 description 3
- FZGJKSXYHIEZRN-NSHDSACASA-N tert-butyl N-[(2S)-1-(6-chloro-5-hydroxy-2-iodopyridin-3-yl)-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C(=N1)I)C[C@@H](C(C)(C)C)NC(OC(C)(C)C)=O)O FZGJKSXYHIEZRN-NSHDSACASA-N 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- UGGRMFGKRXDDKI-XMHCIUCPSA-N (S)-N-[(2S)-1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethylbutan-2-yl]-2-methylpropane-2-sulfinamide Chemical compound ClC1=C(C=C(C=N1)C[C@@H](C(C)(C)C)N[S@@](=O)C(C)(C)C)OC UGGRMFGKRXDDKI-XMHCIUCPSA-N 0.000 description 2
- UWYZHKAOTLEWKK-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline Chemical compound C1=CC=C2CNCCC2=C1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 2
- XEVXYNZWIKBONP-UHFFFAOYSA-N 1-(6-chloro-5-methoxypyridin-3-yl)-3,3-dimethyl-4-phenylmethoxybutan-2-amine Chemical compound C(C1=CC=CC=C1)OCC(C(CC=1C=NC(=C(C=1)OC)Cl)N)(C)C XEVXYNZWIKBONP-UHFFFAOYSA-N 0.000 description 2
- FIMIKKOMVTUONY-UHFFFAOYSA-N 1-[3-(1,3-dioxolan-2-yl)-5,6-dimethylpyridin-2-yl]-3-methylbutan-2-one Chemical compound O1C(OCC1)C=1C(=NC(=C(C=1)C)C)CC(C(C)C)=O FIMIKKOMVTUONY-UHFFFAOYSA-N 0.000 description 2
- NMBGABXLGNXJMJ-UHFFFAOYSA-N 1-[[2-bromo-6-chloro-5-(cyclopropylmethoxy)pyridin-3-yl]methyl]cyclobutane-1-carboxylic acid Chemical compound BrC1=NC(=C(C=C1CC1(CCC1)C(=O)O)OCC1CC1)Cl NMBGABXLGNXJMJ-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 2
- CVSKFRQVNUAQFL-UHFFFAOYSA-N 2,3-dimethoxy-10-oxospiro[5H-pyrido[1,2-h][1,7]naphthyridine-6,1'-cyclobutane]-9-carboxylic acid Chemical compound COC1=NC=2C=3N(C4(CC=2C=C1OC)CCC4)C=C(C(C=3)=O)C(=O)O CVSKFRQVNUAQFL-UHFFFAOYSA-N 0.000 description 2
- UFZBZIWUJMJFJU-UHFFFAOYSA-N 2,3-dimethyl-10-oxo-6-propan-2-yl-5,6-dihydropyrido[2,1-f][1,6]naphthyridine-9-carboxylic acid Chemical compound C1(CC2=NC(=C(C=C2C2=CC(=O)C(C(=O)O)=CN12)C)C)C(C)C UFZBZIWUJMJFJU-UHFFFAOYSA-N 0.000 description 2
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 2
- NMFNDJGDWLVLPN-UHFFFAOYSA-N 2-chloro-3-(1,3-dioxolan-2-yl)-5,6-dimethylpyridine Chemical compound ClC1=NC(=C(C=C1C1OCCO1)C)C NMFNDJGDWLVLPN-UHFFFAOYSA-N 0.000 description 2
- ZEJHNAWMJMJMCZ-UHFFFAOYSA-N 2-chloro-5,6-dimethylpyridine-3-carbaldehyde Chemical compound CC1=CC(C=O)=C(Cl)N=C1C ZEJHNAWMJMJMCZ-UHFFFAOYSA-N 0.000 description 2
- SPGHDBDNNNWOKA-UHFFFAOYSA-N 2-chloro-5-(hydroxymethyl)-6-iodopyridin-3-ol Chemical compound ClC1=NC(=C(C=C1O)CO)I SPGHDBDNNNWOKA-UHFFFAOYSA-N 0.000 description 2
- LITDKXAAVHVYOS-UHFFFAOYSA-N 2-chloro-5-(hydroxymethyl)pyridin-3-ol Chemical compound OCc1cnc(Cl)c(O)c1 LITDKXAAVHVYOS-UHFFFAOYSA-N 0.000 description 2
- DVNAEKRGMBNCFF-UHFFFAOYSA-N 2h-cyclopenta[d][1,3]thiazole Chemical compound C1=CC2=NCSC2=C1 DVNAEKRGMBNCFF-UHFFFAOYSA-N 0.000 description 2
- GOLXRNDWAUTYKT-UHFFFAOYSA-N 3-(1H-indol-3-yl)propanoic acid Chemical compound C1=CC=C2C(CCC(=O)O)=CNC2=C1 GOLXRNDWAUTYKT-UHFFFAOYSA-N 0.000 description 2
- SYBYTAAJFKOIEJ-UHFFFAOYSA-N 3-Methylbutan-2-one Chemical compound CC(C)C(C)=O SYBYTAAJFKOIEJ-UHFFFAOYSA-N 0.000 description 2
- 125000002373 5 membered heterocyclic group Chemical group 0.000 description 2
- JPNFHBHFNKQDLL-UHFFFAOYSA-N 5-[(1-aminocyclobutyl)methyl]-6-bromo-2-chloropyridin-3-ol Chemical compound NC1(CCC1)CC=1C=C(C(=NC=1Br)Cl)O JPNFHBHFNKQDLL-UHFFFAOYSA-N 0.000 description 2
- LUCJWTAPFGYJLS-SECBINFHSA-N 5-[(2R)-2-amino-3,3-dimethylbutyl]-2-chloropyridin-3-ol Chemical compound N[C@H](CC=1C=C(C(=NC=1)Cl)O)C(C)(C)C LUCJWTAPFGYJLS-SECBINFHSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 238000012286 ELISA Assay Methods 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 101710142246 External core antigen Proteins 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- 206010019663 Hepatic failure Diseases 0.000 description 2
- 108700024845 Hepatitis B virus P Proteins 0.000 description 2
- 208000005331 Hepatitis D Diseases 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 241001660687 Xantho Species 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 150000003857 carboxamides Chemical class 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 150000007942 carboxylates Chemical class 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 150000005829 chemical entities Chemical class 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- DZNFQIYYEXFFGV-UHFFFAOYSA-M chloropalladium(1+) 2-phenylaniline tritert-butylphosphane Chemical compound [Pd+]Cl.CC(C)(C)P(C(C)(C)C)C(C)(C)C.NC1=CC=CC=C1C1=CC=CC=[C-]1 DZNFQIYYEXFFGV-UHFFFAOYSA-M 0.000 description 2
- 235000012000 cholesterol Nutrition 0.000 description 2
- 230000007882 cirrhosis Effects 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 230000000875 corresponding effect Effects 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 238000002784 cytotoxicity assay Methods 0.000 description 2
- 231100000263 cytotoxicity test Toxicity 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 230000005860 defense response to virus Effects 0.000 description 2
- 239000012067 demethylated product Substances 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 229960000980 entecavir Drugs 0.000 description 2
- YXPVEXCTPGULBZ-WQYNNSOESA-N entecavir hydrate Chemical compound O.C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)C1=C YXPVEXCTPGULBZ-WQYNNSOESA-N 0.000 description 2
- 238000010931 ester hydrolysis Methods 0.000 description 2
- ADOMFWLVFFNNNF-UHFFFAOYSA-N ethyl 1-[[2-bromo-6-chloro-5-(cyclopropylmethoxy)pyridin-3-yl]methyl]cyclobutane-1-carboxylate Chemical compound BrC1=NC(=C(C=C1CC1(CCC1)C(=O)OCC)OCC1CC1)Cl ADOMFWLVFFNNNF-UHFFFAOYSA-N 0.000 description 2
- DJKUTEOMCLKHDZ-UHFFFAOYSA-N ethyl 4-oxo-5-phenylmethoxypyran-3-carboxylate Chemical compound C(C)OC(=O)C1=COC=C(C1=O)OCC1=CC=CC=C1 DJKUTEOMCLKHDZ-UHFFFAOYSA-N 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 230000003259 immunoinhibitory effect Effects 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 239000012155 injection solvent Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 125000005956 isoquinolyl group Chemical group 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 208000007903 liver failure Diseases 0.000 description 2
- 231100000835 liver failure Toxicity 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000006578 monocyclic heterocycloalkyl group Chemical group 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- WXHIJDCHNDBCNY-UHFFFAOYSA-N palladium dihydride Chemical compound [PdH2] WXHIJDCHNDBCNY-UHFFFAOYSA-N 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical compound C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Chemical group 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 238000006476 reductive cyclization reaction Methods 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 239000012089 stop solution Substances 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- IQFYYKKMVGJFEH-CSMHCCOUSA-N telbivudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 IQFYYKKMVGJFEH-CSMHCCOUSA-N 0.000 description 2
- 229960004556 tenofovir Drugs 0.000 description 2
- PPGSYFBUWKLBGA-UHFFFAOYSA-N tert-butyl N-[1-(2-bromo-6-chloro-5-hydroxypyridin-3-yl)-3,3-dimethylbutan-2-yl]carbamate Chemical compound BrC1=NC(=C(C=C1CC(C(C)(C)C)NC(OC(C)(C)C)=O)O)Cl PPGSYFBUWKLBGA-UHFFFAOYSA-N 0.000 description 2
- CJRHKPTWUDCHGM-UHFFFAOYSA-N tert-butyl N-[1-(6-chloro-5-hydroxypyridin-3-yl)-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C=N1)CC(C(C)(C)C)NC(OC(C)(C)C)=O)O CJRHKPTWUDCHGM-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 230000005570 vertical transmission Effects 0.000 description 2
- XFQNWPYGEGCIMF-HCUGAJCMSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].[Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 XFQNWPYGEGCIMF-HCUGAJCMSA-N 0.000 description 1
- XRLQMTYCACPCCQ-GFCCVEGCSA-N (2R)-1-[6-chloro-5-(cyclopropylmethoxy)-2-iodopyridin-3-yl]-3,3-dimethylbutan-2-amine Chemical compound ClC1=C(C=C(C(=N1)I)C[C@H](C(C)(C)C)N)OCC1CC1 XRLQMTYCACPCCQ-GFCCVEGCSA-N 0.000 description 1
- JFVCUINASNRMTO-HXPMCKFVSA-N (2R,6S)-10-chloro-9-(3-methoxypropoxy)-3,3-dimethyl-15-oxo-1,11-diazatetracyclo[11.4.0.02,6.07,12]heptadeca-7,9,11,13,16-pentaene-16-carboxylic acid Chemical compound ClC1=NC=2C=3N([C@@H]4[C@H](C=2C=C1OCCCOC)CCC4(C)C)C=C(C(C=3)=O)C(=O)O JFVCUINASNRMTO-HXPMCKFVSA-N 0.000 description 1
- GQGLVCKGANQXSC-LBPRGKRZSA-N (2S)-1-[6-chloro-2-iodo-5-(3-methoxypropoxy)pyridin-3-yl]-3,3-dimethylbutan-2-amine Chemical compound ClC1=C(C=C(C(=N1)I)C[C@@H](C(C)(C)C)N)OCCCOC GQGLVCKGANQXSC-LBPRGKRZSA-N 0.000 description 1
- XRLQMTYCACPCCQ-LBPRGKRZSA-N (2S)-1-[6-chloro-5-(cyclopropylmethoxy)-2-iodopyridin-3-yl]-3,3-dimethylbutan-2-amine Chemical compound ClC1=C(C=C(C(=N1)I)C[C@@H](C(C)(C)C)N)OCC1CC1 XRLQMTYCACPCCQ-LBPRGKRZSA-N 0.000 description 1
- XWXLJPKINLDVFP-AWEZNQCLSA-N (2S)-2-(phenylmethoxycarbonylamino)-3-[4-(trifluoromethyl)piperidin-1-yl]propanoic acid Chemical compound C(C1=CC=CC=C1)OC(=O)N[C@H](C(=O)O)CN1CCC(CC1)C(F)(F)F XWXLJPKINLDVFP-AWEZNQCLSA-N 0.000 description 1
- JFVCUINASNRMTO-BLVKFPJESA-N (2S,6R)-10-chloro-9-(3-methoxypropoxy)-3,3-dimethyl-15-oxo-1,11-diazatetracyclo[11.4.0.02,6.07,12]heptadeca-7,9,11,13,16-pentaene-16-carboxylic acid Chemical compound ClC1=NC=2C=3N([C@H]4[C@@H](C=2C=C1OCCCOC)CCC4(C)C)C=C(C(C=3)=O)C(=O)O JFVCUINASNRMTO-BLVKFPJESA-N 0.000 description 1
- BPPKLJZCVAYGCV-CMJOXMDJSA-N (2S,6R)-10-cyclopropyl-9-(3-methoxypropoxy)-3,3-dimethyl-15-oxo-1,11-diazatetracyclo[11.4.0.02,6.07,12]heptadeca-7,9,11,13,16-pentaene-16-carboxylic acid Chemical compound C1(CC1)C1=NC=2C=3N([C@H]4[C@@H](C=2C=C1OCCCOC)CCC4(C)C)C=C(C(C=3)=O)C(=O)O BPPKLJZCVAYGCV-CMJOXMDJSA-N 0.000 description 1
- PZLOINLBOHMXFE-UHFFFAOYSA-N (6-chloro-2-iodo-5-methoxypyridin-3-yl)methanol Chemical compound ClC1=C(C=C(C(=N1)I)CO)OC PZLOINLBOHMXFE-UHFFFAOYSA-N 0.000 description 1
- LJXPRCQPDWSRIT-SLBUAACQSA-N (6S)-2-(difluoromethyl)-11-fluoro-6-methyl-10-oxo-3-(oxolan-3-ylmethoxy)-6-propan-2-yl-5H-pyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound FC(C1=NC=2C=3N([C@](CC=2C=C1OCC1COCC1)(C)C(C)C)C=C(C(C=3F)=O)C(=O)O)F LJXPRCQPDWSRIT-SLBUAACQSA-N 0.000 description 1
- YPFUTXUPLOKMMH-IBGZPJMESA-N (6S)-6-tert-butyl-2-(hydroxymethyl)-3-(3-methoxypropoxy)-10-oxo-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)[C@@H]1CC=2C=C(C(=NC=2C=2N1C=C(C(C=2)=O)C(=O)O)CO)OCCCOC YPFUTXUPLOKMMH-IBGZPJMESA-N 0.000 description 1
- DWDFLWFOAOFQGV-KRWDZBQOSA-N (6S)-6-tert-butyl-2-chloro-3-(3-methoxypropoxy)-8-methyl-10-oxo-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)[C@@H]1CC=2C=C(C(=NC=2C=2N1C(=C(C(C=2)=O)C(=O)O)C)Cl)OCCCOC DWDFLWFOAOFQGV-KRWDZBQOSA-N 0.000 description 1
- UUCAYEMTRZHJRR-KRWDZBQOSA-N (6S)-6-tert-butyl-2-cyclopropyl-11-hydroxy-3-(3-methoxypropoxy)-10-oxo-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)[C@@H]1CC=2C=C(C(=NC=2C=2N1C=C(C(C=2O)=O)C(=O)O)C1CC1)OCCCOC UUCAYEMTRZHJRR-KRWDZBQOSA-N 0.000 description 1
- LQZPBLIRTWNKQM-FQEVSTJZSA-N (6S)-6-tert-butyl-3-(3-methoxypropoxy)-10-oxo-2-prop-1-en-2-yl-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)[C@@H]1CC=2C=C(C(=NC=2C=2N1C=C(C(C=2)=O)C(=O)O)C(=C)C)OCCCOC LQZPBLIRTWNKQM-FQEVSTJZSA-N 0.000 description 1
- SWVCMLTWUWQBIJ-KRWDZBQOSA-N (6S)-6-tert-butyl-3-(3-methoxypropoxy)-2,10-dioxo-5,6-dihydro-1H-pyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)[C@@H]1CC=2C=C(C(=NC=2C=2N1C=C(C(C=2)=O)C(=O)O)O)OCCCOC SWVCMLTWUWQBIJ-KRWDZBQOSA-N 0.000 description 1
- MOOBNOCKGZQCKI-SFHVURJKSA-N (6S)-6-tert-butyl-3-(cyclopropylmethoxy)-2-methoxy-10-oxo-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound C(C)(C)(C)[C@@H]1CC=2C=C(C(=NC=2C=2N1C=C(C(C=2)=O)C(=O)O)OC)OCC1CC1 MOOBNOCKGZQCKI-SFHVURJKSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- JYJVAKTXQGASNS-UHFFFAOYSA-N 1-(6-chloro-2-iodo-5-methoxypyridin-3-yl)-2,3-dimethylbutan-2-amine Chemical compound ClC1=C(C=C(C(=N1)I)CC(C(C)C)(N)C)OC JYJVAKTXQGASNS-UHFFFAOYSA-N 0.000 description 1
- DNZNGVJALFRXGV-UHFFFAOYSA-N 1-[3-(1,3-dioxolan-2-yl)-5,6-dimethylpyridin-2-yl]-3-methylbutan-2-amine Chemical compound O1C(OCC1)C=1C(=NC(=C(C=1)C)C)CC(C(C)C)N DNZNGVJALFRXGV-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- XLUXAKJPHJEFKL-UHFFFAOYSA-N 1-ethylcyclobutane-1-carboxylic acid Chemical compound CCC1(C(O)=O)CCC1 XLUXAKJPHJEFKL-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- JFVCUINASNRMTO-UHFFFAOYSA-N 10-chloro-9-(3-methoxypropoxy)-3,3-dimethyl-15-oxo-1,11-diazatetracyclo[11.4.0.02,6.07,12]heptadeca-7,9,11,13,16-pentaene-16-carboxylic acid Chemical compound ClC1=NC=2C=3N(C4C(C=2C=C1OCCCOC)CCC4(C)C)C=C(C(C=3)=O)C(=O)O JFVCUINASNRMTO-UHFFFAOYSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Substances C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 1
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical group O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 1
- BZMVHLIYOASZTA-UHFFFAOYSA-N 2-[(6-chloro-2-iodo-5-methoxypyridin-3-yl)methyl]-2,3-dimethylbutanoic acid Chemical compound ClC1=C(C=C(C(=N1)I)CC(C(=O)O)(C(C)C)C)OC BZMVHLIYOASZTA-UHFFFAOYSA-N 0.000 description 1
- UHTQHHLSGVOGQR-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-4-ium-1-yl]ethanesulfonate Chemical compound OCCN1CCN(CCS(O)(=O)=O)CC1.OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 UHTQHHLSGVOGQR-UHFFFAOYSA-N 0.000 description 1
- SCVJRXQHFJXZFZ-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound C1=2NC(N)=NC(=S)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)O1 SCVJRXQHFJXZFZ-KVQBGUIXSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- BFTRXCWDCODNCZ-UHFFFAOYSA-N 2-bromo-6-chloro-5-(cyclopropylmethoxy)-3-[(1-isocyanatocyclobutyl)methyl]pyridine Chemical compound BrC1=NC(=C(C=C1CC1(CCC1)N=C=O)OCC1CC1)Cl BFTRXCWDCODNCZ-UHFFFAOYSA-N 0.000 description 1
- HZLWVMLWHXTONK-UHFFFAOYSA-N 2-chloro-3-(cyclopropylmethoxy)-6-methyl-10-oxo-6-propan-2-yl-5H-pyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound ClC1=NC=2C=3N(C(CC=2C=C1OCC1CC1)(C)C(C)C)C=C(C(C=3)=O)C(=O)O HZLWVMLWHXTONK-UHFFFAOYSA-N 0.000 description 1
- JEXYBQCGHFYMDX-UHFFFAOYSA-N 2-chloro-3-hydroxy-6-methyl-10-oxo-6-propan-2-yl-5H-pyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound ClC1=NC=2C=3N(C(CC=2C=C1O)(C)C(C)C)C=C(C(C=3)=O)C(=O)O JEXYBQCGHFYMDX-UHFFFAOYSA-N 0.000 description 1
- XJYCVIAPOBSBLW-UHFFFAOYSA-N 2-chloro-6-(1-hydroxy-2-methylpropan-2-yl)-3-(3-methoxypropoxy)-10-oxo-5,6-dihydropyrido[1,2-h][1,7]naphthyridine-9-carboxylic acid Chemical compound ClC1=NC=2C=3N(C(CC=2C=C1OCCCOC)C(CO)(C)C)C=C(C(C=3)=O)C(=O)O XJYCVIAPOBSBLW-UHFFFAOYSA-N 0.000 description 1
- WOQYIBOZNROQES-UHFFFAOYSA-N 2-cyclopentyl-1,3-thiazole Chemical compound C1CCCC1C1=NC=CS1 WOQYIBOZNROQES-UHFFFAOYSA-N 0.000 description 1
- CESUXLKAADQNTB-SSDOTTSWSA-N 2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@](N)=O CESUXLKAADQNTB-SSDOTTSWSA-N 0.000 description 1
- CESUXLKAADQNTB-ZETCQYMHSA-N 2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](N)=O CESUXLKAADQNTB-ZETCQYMHSA-N 0.000 description 1
- VLRSADZEDXVUPG-UHFFFAOYSA-N 2-naphthalen-1-ylpyridine Chemical compound N1=CC=CC=C1C1=CC=CC2=CC=CC=C12 VLRSADZEDXVUPG-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- GCEWYAUVJWQAGR-UHFFFAOYSA-N 3,3-dimethyl-4-phenylmethoxybutan-2-one Chemical compound CC(=O)C(C)(C)COCC1=CC=CC=C1 GCEWYAUVJWQAGR-UHFFFAOYSA-N 0.000 description 1
- VPMAWSAODAKKSI-UHFFFAOYSA-N 3-chloroprop-2-enoyl chloride Chemical compound ClC=CC(Cl)=O VPMAWSAODAKKSI-UHFFFAOYSA-N 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- CBKDCOKSXCTDAA-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1-benzothiophene Chemical compound C1CCCC2=C1C=CS2 CBKDCOKSXCTDAA-UHFFFAOYSA-N 0.000 description 1
- GVCLNACSYKYUHP-UHFFFAOYSA-N 4-amino-7-(2-hydroxyethoxymethyl)pyrrolo[2,3-d]pyrimidine-5-carbothioamide Chemical compound C1=NC(N)=C2C(C(=S)N)=CN(COCCO)C2=N1 GVCLNACSYKYUHP-UHFFFAOYSA-N 0.000 description 1
- PJOMFEHOLJKQAZ-UHFFFAOYSA-N 4-oxopyran-3-carboxylic acid Chemical compound OC(=O)C1=COC=CC1=O PJOMFEHOLJKQAZ-UHFFFAOYSA-N 0.000 description 1
- GDRVFDDBLLKWRI-UHFFFAOYSA-N 4H-quinolizine Chemical compound C1=CC=CN2CC=CC=C21 GDRVFDDBLLKWRI-UHFFFAOYSA-N 0.000 description 1
- BUGNNHLZTBPABI-UHFFFAOYSA-N 5,6-dimethyl-2-oxo-1h-pyridine-3-carbonitrile Chemical compound CC=1C=C(C#N)C(=O)NC=1C BUGNNHLZTBPABI-UHFFFAOYSA-N 0.000 description 1
- YFNLJPJXBGPZOO-UHFFFAOYSA-N 5-(bromomethyl)-2-chloro-6-iodo-3-methoxypyridine Chemical compound BrCC=1C(=NC(=C(C=1)OC)Cl)I YFNLJPJXBGPZOO-UHFFFAOYSA-N 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- FHKXSCVDIDHPNG-UHFFFAOYSA-N 6,7-dihydrobenzo[a]quinolizin-2-one Chemical class C=1C(C=CN2CCC3=C(C=12)C=CC=C3)=O FHKXSCVDIDHPNG-UHFFFAOYSA-N 0.000 description 1
- KCCYUVIIKCPVDH-UHFFFAOYSA-N 6-bromo-5-methylpyridin-3-ol Chemical compound CC1=CC(O)=CN=C1Br KCCYUVIIKCPVDH-UHFFFAOYSA-N 0.000 description 1
- NJIAKNWTIVDSDA-FQEVSTJZSA-N 7-[4-(1-methylsulfonylpiperidin-4-yl)phenyl]-n-[[(2s)-morpholin-2-yl]methyl]pyrido[3,4-b]pyrazin-5-amine Chemical compound C1CN(S(=O)(=O)C)CCC1C1=CC=C(C=2N=C(NC[C@H]3OCCNC3)C3=NC=CN=C3C=2)C=C1 NJIAKNWTIVDSDA-FQEVSTJZSA-N 0.000 description 1
- 229940126670 AB-836 Drugs 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 235000007652 Arbutus Nutrition 0.000 description 1
- 240000008327 Arbutus unedo Species 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical group [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 206010008909 Chronic Hepatitis Diseases 0.000 description 1
- 208000000419 Chronic Hepatitis B Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical group [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000037319 Hepatitis infectious Diseases 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000001940 Massive Hepatic Necrosis Diseases 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- 229910003827 NRaRb Inorganic materials 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 102000007501 Thymosin Human genes 0.000 description 1
- 108010046075 Thymosin Proteins 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 206010058874 Viraemia Diseases 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- DRBWRJPFNOBNIO-KOLCDFICSA-N [(2r)-1-[(2r)-2-(pyridine-4-carbonylamino)propanoyl]pyrrolidin-2-yl]boronic acid Chemical compound N([C@H](C)C(=O)N1[C@@H](CCC1)B(O)O)C(=O)C1=CC=NC=C1 DRBWRJPFNOBNIO-KOLCDFICSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- GJFNLGKXCWQHIZ-UHFFFAOYSA-N acetic acid;azane;sodium Chemical compound N.[Na].CC(O)=O GJFNLGKXCWQHIZ-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 229960001997 adefovir Drugs 0.000 description 1
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000001062 anti-nausea Effects 0.000 description 1
- 229940125714 antidiarrheal agent Drugs 0.000 description 1
- 239000003793 antidiarrheal agent Substances 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000003096 antiparasitic agent Substances 0.000 description 1
- 229940125687 antiparasitic agent Drugs 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000008122 artificial sweetener Substances 0.000 description 1
- 235000021311 artificial sweeteners Nutrition 0.000 description 1
- 125000005098 aryl alkoxy carbonyl group Chemical group 0.000 description 1
- 239000012911 assay medium Substances 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 150000001602 bicycloalkyls Chemical group 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 239000010836 blood and blood product Substances 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 229910052796 boron Chemical group 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 210000000234 capsid Anatomy 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 231100000357 carcinogen Toxicity 0.000 description 1
- 239000003183 carcinogenic agent Substances 0.000 description 1
- 239000012592 cell culture supplement Substances 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000007541 cellular toxicity Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 150000001840 cholesterol esters Chemical class 0.000 description 1
- WCZVZNOTHYJIEI-UHFFFAOYSA-N cinnoline Chemical compound N1=NC=CC2=CC=CC=C21 WCZVZNOTHYJIEI-UHFFFAOYSA-N 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000013267 controlled drug release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- WVIIMZNLDWSIRH-UHFFFAOYSA-N cyclohexylcyclohexane Chemical group C1CCCCC1C1CCCCC1 WVIIMZNLDWSIRH-UHFFFAOYSA-N 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical group C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- MAWOHFOSAIXURX-UHFFFAOYSA-N cyclopentylcyclopentane Chemical group C1CCCC1C1CCCC1 MAWOHFOSAIXURX-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- OLAMWIPURJGSKE-UHFFFAOYSA-N et2o diethylether Chemical compound CCOCC.CCOCC OLAMWIPURJGSKE-UHFFFAOYSA-N 0.000 description 1
- CKHUMILJZKSHJU-UHFFFAOYSA-N ethane;tin Chemical compound CC[Sn] CKHUMILJZKSHJU-UHFFFAOYSA-N 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- LQSOVGAUOHMPLK-SOFGYWHQSA-N ethyl (2e)-2-(dimethylaminomethylidene)-3-oxobutanoate Chemical compound CCOC(=O)C(\C(C)=O)=C\N(C)C LQSOVGAUOHMPLK-SOFGYWHQSA-N 0.000 description 1
- FNASCUBBFNCFQO-SOFGYWHQSA-N ethyl (2e)-2-(ethoxymethylidene)-3-oxobutanoate Chemical compound CCO\C=C(/C(C)=O)C(=O)OCC FNASCUBBFNCFQO-SOFGYWHQSA-N 0.000 description 1
- SKROEPCBVAJDBZ-UHFFFAOYSA-N ethyl 2,3-dimethyl-10-oxo-6-propan-2-yl-5,6-dihydropyrido[2,1-f][1,6]naphthyridine-9-carboxylate Chemical compound C(C1CC2=NC(C)=C(C)C=C2C=2N1C=C(C(=O)C=2)C(=O)OCC)(C)C SKROEPCBVAJDBZ-UHFFFAOYSA-N 0.000 description 1
- UOLDHHQOKRYISV-UHFFFAOYSA-N ethyl 2,3-dimethylbutanoate Chemical compound CCOC(=O)C(C)C(C)C UOLDHHQOKRYISV-UHFFFAOYSA-N 0.000 description 1
- GAOMYGVMKJWEJG-UHFFFAOYSA-N ethyl 2-[(6-chloro-2-iodo-5-methoxypyridin-3-yl)methyl]-2,3-dimethylbutanoate Chemical compound ClC1=C(C=C(C(=N1)I)CC(C(=O)OCC)(C(C)C)C)OC GAOMYGVMKJWEJG-UHFFFAOYSA-N 0.000 description 1
- OAOWLYJFXFPOPW-UHFFFAOYSA-N ethyl 2h-pyran-3-carboxylate Chemical compound CCOC(=O)C1=CC=COC1 OAOWLYJFXFPOPW-UHFFFAOYSA-N 0.000 description 1
- DXUTWLTWGKEWJA-UHFFFAOYSA-N ethyl 3-oxo-4-phenylmethoxybutanoate Chemical compound CCOC(=O)CC(=O)COCC1=CC=CC=C1 DXUTWLTWGKEWJA-UHFFFAOYSA-N 0.000 description 1
- YRXYFESNWZYWSD-UHFFFAOYSA-N ethyl 4H-pyran-3-carboxylate Chemical compound CCOC(=O)C1=COC=CC1 YRXYFESNWZYWSD-UHFFFAOYSA-N 0.000 description 1
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 208000005252 hepatitis A Diseases 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- LPAGFVYQRIESJQ-UHFFFAOYSA-N indoline Chemical compound C1=CC=C2NCCC2=C1 LPAGFVYQRIESJQ-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000012678 infectious agent Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229960001627 lamivudine Drugs 0.000 description 1
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000013160 medical therapy Methods 0.000 description 1
- 230000008384 membrane barrier Effects 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 1
- KJJSHOHQQHACLE-UHFFFAOYSA-N methyl 5-hydroxypyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC(O)=C1 KJJSHOHQQHACLE-UHFFFAOYSA-N 0.000 description 1
- ZDIHYBCAMKQMID-UHFFFAOYSA-N methyl 6-chloro-5-hydroxypyridine-3-carboxylate Chemical compound COC(=O)C1=CN=C(Cl)C(O)=C1 ZDIHYBCAMKQMID-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- REPVNSJSTLRQEQ-UHFFFAOYSA-N n,n-dimethylacetamide;n,n-dimethylformamide Chemical compound CN(C)C=O.CN(C)C(C)=O REPVNSJSTLRQEQ-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 1
- 125000005542 phthalazyl group Chemical group 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- PJGSXYOJTGTZAV-UHFFFAOYSA-N pinacolone Chemical compound CC(=O)C(C)(C)C PJGSXYOJTGTZAV-UHFFFAOYSA-N 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011253 protective coating Substances 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- CPNGPNLZQNNVQM-UHFFFAOYSA-N pteridine Chemical compound N1=CN=CC2=NC=CN=C21 CPNGPNLZQNNVQM-UHFFFAOYSA-N 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 150000005299 pyridinones Chemical class 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 239000000565 sealant Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 208000037921 secondary disease Diseases 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 230000005582 sexual transmission Effects 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Chemical group 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- YROXIXLRRCOBKF-UHFFFAOYSA-N sulfonylurea Chemical class OC(=N)N=S(=O)=O YROXIXLRRCOBKF-UHFFFAOYSA-N 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229960005311 telbivudine Drugs 0.000 description 1
- 229960004693 tenofovir disoproxil fumarate Drugs 0.000 description 1
- FZGJKSXYHIEZRN-LLVKDONJSA-N tert-butyl N-[(2R)-1-(6-chloro-5-hydroxy-2-iodopyridin-3-yl)-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C(=N1)I)C[C@H](C(C)(C)C)NC(OC(C)(C)C)=O)O FZGJKSXYHIEZRN-LLVKDONJSA-N 0.000 description 1
- CJRHKPTWUDCHGM-GFCCVEGCSA-N tert-butyl N-[(2R)-1-(6-chloro-5-hydroxypyridin-3-yl)-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C=N1)C[C@H](C(C)(C)C)NC(OC(C)(C)C)=O)O CJRHKPTWUDCHGM-GFCCVEGCSA-N 0.000 description 1
- MEODGROJAMYGGN-OAHLLOKOSA-N tert-butyl N-[(2R)-1-[6-chloro-5-(cyclopropylmethoxy)-2-iodopyridin-3-yl]-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C(=N1)I)C[C@H](C(C)(C)C)NC(OC(C)(C)C)=O)OCC1CC1 MEODGROJAMYGGN-OAHLLOKOSA-N 0.000 description 1
- CJRHKPTWUDCHGM-LBPRGKRZSA-N tert-butyl N-[(2S)-1-(6-chloro-5-hydroxypyridin-3-yl)-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C=N1)C[C@@H](C(C)(C)C)NC(OC(C)(C)C)=O)O CJRHKPTWUDCHGM-LBPRGKRZSA-N 0.000 description 1
- SGBGZODJAGQQAD-HNNXBMFYSA-N tert-butyl N-[(2S)-1-[6-chloro-2-iodo-5-(3-methoxypropoxy)pyridin-3-yl]-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C(=N1)I)C[C@@H](C(C)(C)C)NC(OC(C)(C)C)=O)OCCCOC SGBGZODJAGQQAD-HNNXBMFYSA-N 0.000 description 1
- MEODGROJAMYGGN-HNNXBMFYSA-N tert-butyl N-[(2S)-1-[6-chloro-5-(cyclopropylmethoxy)-2-iodopyridin-3-yl]-3,3-dimethylbutan-2-yl]carbamate Chemical compound ClC1=C(C=C(C(=N1)I)C[C@@H](C(C)(C)C)NC(OC(C)(C)C)=O)OCC1CC1 MEODGROJAMYGGN-HNNXBMFYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 description 1
- 125000005887 tetrahydrobenzofuranyl group Chemical group 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- LCJVIYPJPCBWKS-NXPQJCNCSA-N thymosin Chemical compound SC[C@@H](N)C(=O)N[C@H](CO)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CO)C(=O)N[C@H](CO)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@H]([C@H](C)O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@H](CCC(O)=O)C(O)=O LCJVIYPJPCBWKS-NXPQJCNCSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- UORVGPXVDQYIDP-UHFFFAOYSA-N trihydridoboron Substances B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 1
- BPLKQGGAXWRFOE-UHFFFAOYSA-M trimethylsulfoxonium iodide Chemical compound [I-].C[S+](C)(C)=O BPLKQGGAXWRFOE-UHFFFAOYSA-M 0.000 description 1
- MHNHYTDAOYJUEZ-UHFFFAOYSA-N triphenylphosphane Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 MHNHYTDAOYJUEZ-UHFFFAOYSA-N 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 210000000605 viral structure Anatomy 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/438—The ring being spiro-condensed with carbocyclic or heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/20—Spiro-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Compuestos de Fórmula (I), específicamente inhibidores del virus de la hepatitis B y/o del virus de la hepatitis D, más específicamente compuestos que inhiben el antígeno HBe y el antígeno HBs en un sujeto, para el tratamiento de infecciones virales, y métodos para preparar y usar tales compuestos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos químicos
CAMPO DE LA INVENCIÓN
La presente invención se refiere a compuestos útiles para el tratamiento de VHB en animales, y más particularmente para el tratamiento de VHB en seres humanos.
ANTECEDENTES DE LA INVENCIÓN
La hepatitis B es una enfermedad viral transmitida por vía parenteral mediante material contaminado tal como sangre y productos sanguíneos, agujas contaminadas, por transmisión sexual y verticalmente de madres infectadas o portadoras a su descendencia. En aquellas zonas del mundo donde la enfermedad es común, la transmisión vertical a una edad temprana da como resultado que una alta proporción de individuos infectados se conviertan en portadores crónicos de hepatitis B. La Organización Mundial de la Salud estima que más de 2 millones de personas se han infectado en todo el mundo, con aproximadamente 4 millones de casos agudos al año, 1 millón de muertes al año y 350-400 millones de portadores crónicos. Aproximadamente el 25 % de los portadores crónicos mueren por hepatitis crónica, cirrosis o cáncer de hígado y casi el 75 % de los portadores crónicos son asiáticos. El virus de la hepatitis B (VHB) es el segundo carcinógeno más significativo después del tabaco, provocando del 60 % al 80 % de todos los cánceres de hígado primarios. El VHB es 100 veces más contagioso que el VIH.
El VHB se transmite por contacto percutáneo o parenteral con sangre infectada, fluidos corporales y por relaciones sexuales. El VHB es capaz de permanecer sobre cualquier superficie con la que entre en contacto durante aproximadamente una semana, por ejemplo, tableros de mesa, hojas de afeitar, manchas de sangre, sin perder infectividad. Sin embargo, el VHB no puede atravesar la piel o la barrera de la membrana mucosa. Se requiere alguna ruptura en esta barrera, que puede ser mínima e insignificante, para la transmisión.
El VHB es un virus de ADN con envuelta pequeño que pertenece a la familia de hepadnavirus. El virus se replica a través de una forma intermedia de ARN por transcripción inversa, lo que en la práctica los relaciona con los retrovirus, como el VIH. Aunque la replicación tiene lugar en el hígado, el virus se propaga a la sangre donde se encuentran proteínas virales y anticuerpos contra ellas en personas infectadas. El VHB es muchas veces más infeccioso que el VIH debido a las mayores concentraciones del virus VHB que se encuentran en el torrente sanguíneo en cualquier momento dado.
La infección por VHB da como resultado la producción de dos partículas diferentes: 1) el propio virus VHB (o partícula Dane) que incluye una cápside viral ensamblada a partir de la proteína del antígeno central del VHB (HBcAg) y está cubierta por el antígeno de superficie de la hepatitis B (HBsAg) y es capaz de reinfectar células y 2) partículas subvirales (o SVP), que son partículas similares a lipoproteínas de alta densidad compuestas por lípidos, colesterol, ésteres de colesterol y las formas pequeña y mediana del antígeno de superficie de la hepatitis B (HBsAg) que no son infecciosas. Por cada partícula viral producida, se liberan entre 1.000 y 10.000 SVP en la sangre. Como tales, las SVP (y la proteína HBsAg que portan) representan la gran mayoría de las proteínas virales en la sangre. Las células infectadas por VHB también secretan un producto proteolítico soluble de la proteína precentral denominado antígeno e de VHB (HBeAg).
El virus de la hepatitis D (VHD) usa HBsAg para formar su estructura viral (Taylor, 2006, Virology, 344: 71-76) y, como tal, la infección por VHD solo puede ocurrir en sujetos con infección concomitante por VHB. Aunque la incidencia de coinfección por VHD en portadores asintomáticos de VHB y enfermedad hepática crónica relacionada con VHB es baja en países con una baja incidencia de infección por VHB, es una complicación significativa en sujetos infectados por VHB en países con una alta incidencia de infección por VHB, y puede aumentar la tasa de progresión de la enfermedad hepática a hepatitis fulminante. Como tal, la clara necesidad médica insatisfecha en la infección por VHB es aún más apremiante en sujetos coinfectados por VHB/VHD.
Los métodos convencionales actuales de tratamiento para el VHB incluyen inmunoterapias basadas en interferón o timosina 1 y la supresión de la producción viral mediante la inhibición de la polimerasa de VHB (por ejemplo, “nucs”). Los inhibidores de la polimerasa de VHB son eficaces en la reducción de la producción viral, pero tienen poco o ningún efecto en la reducción rápida de los niveles sanguíneos de HBsAg o pueden reducir lentamente el HBsAg con un tratamiento a largo plazo en un número limitado de pacientes (como es el caso del fumarato de disoproxilo de tenofovir). La inmunoterapia basada en interferón puede lograr una reducción tanto de la producción viral como de la eliminación temprana de HBsAg de la sangre, pero solo en un pequeño porcentaje de sujetos tratados. El papel generalmente aceptado de HBsAg en la sangre es secuestrar anticuerpos anti-HBsAg y permitir que las partículas virales infecciosas escapen a la detección inmunitaria, lo que probablemente sea uno de los motivos por los que la infección por VHB sigue siendo una afección crónica. Además, HBsAg, HBeAg y HBcAg tienen todas propiedades inmunoinhibitorias, tal como se comenta a continuación, y es probable que la persistencia de estas proteínas virales
en la sangre de los pacientes después de la administración de cualquiera de los tratamientos actualmente disponibles para el VHB, tal como se describió anteriormente, tenga un impacto significativo al impedir que los pacientes alcancen el control inmunológico de su infección por VHB.
Aunque las tres proteínas primarias del VHB (HBsAg, HBeAg y HBcAg) tienen todas propiedades inmunoinhibitorias (véase a continuación), HBsAg comprende la gran mayoría de la proteína del VHB en la circulación de los sujetos infectados por VHB. Además, mientras que la eliminación (por medio de seroconversión) de HBeAg o las reducciones en la viremia sérica no están correlacionadas con el desarrollo de un control sostenido de la infección por VHB sin tratamiento, la eliminación de HBsAg sérica de la sangre (y la seroconversión) en la infección por VHB es un excelente indicador de pronóstico bien reconocido de la respuesta antiviral en el tratamiento que conducirá al control de la infección por VHB sin tratamiento (aunque esto solo se produce en una pequeña fracción de los pacientes que reciben inmunoterapia). Por tanto, mientras que la reducción de las tres proteínas principales del VHB (HBsAg, HBeAg y HBcAg) puede dar como resultado la eliminación óptima del efecto inhibitorio, la eliminación de HBsAg solo puede ser suficiente por sí sola para eliminar la mayor parte de la inhibición viral de la función inmunitaria en sujetos con infección por VHB.
Por tanto, en ausencia de un régimen de tratamiento actual que pueda restablecer el control inmunológico del VHB en una gran proporción de pacientes, existe la necesidad de que se proporcione un tratamiento eficaz contra la infección por VHB y la coinfección por VHB/VHD que pueda restablecer el control inmunológico en la mayoría de los pacientes.
Las infecciones virales de la hepatitis B, junto con las infecciones virales de la hepatitis D, son un problema médico continuo porque, como cualquier agente infeccioso que se replica rápidamente, existen mutaciones continuas que ayudan a algunas subpoblaciones de VHB a volverse resistentes a los regímenes de tratamiento actuales. En la actualidad, no existen agentes terapéuticos eficaces para el tratamiento de seres humanos infectados con infecciones por VHB y/o virus de la hepatitis D (VHD) que den como resultado la seroconversión al virus en el cuerpo, o que produzcan una reducción del 90 % del antígeno, en comparación con los números basales antes del tratamiento, en personas que padecen una infección viral de hepatitis B. Actualmente, las terapias recomendadas para la infección crónica por VHB y/o VHD por la Asociación Americana para el Estudio de Enfermedades del Hígado (American Association for the Study of Liver Diseases, AASLD) y la Asociación Europea para el Estudio del Hígado (European Association for the Study of the Liver, EASL) incluyen interferón alfa (INF ), interferón pegilado alfa-2a (Peg-IFN2a), entecavir y tenofovir. Sin embargo, la terapia típica con interferón es de 48 semanas y da como resultado efectos secundarios graves y desagradables, y la seroconversión de HBeAg, 24 semanas después de que la terapia haya cesado, oscila entre solo el 27 y el 36 %. La seroconversión de HBsAg es incluso más baja: solo se observa un 3 % inmediatamente después de que cese el tratamiento, con un aumento hasta por encima del 12 % después de 5 años.
Las terapias con nucleósidos y nucleótidos entecavir y tenofovir tienen éxito en la reducción de la carga viral, pero las tasas de seroconversión de HBeAg y pérdida de HBsAg son incluso más bajas que las obtenidas usando terapia con IFNa. También se usan otras terapias similares, incluidos lamivudina (3TC), telbivudina (LdT) y adefovir, pero para las terapias con nucleósidos/nucleótidos en general, la aparición de resistencia limita la eficacia terapéutica.
Investigaciones clínicas recientes han encontrado una correlación entre la seroconversión y las reducciones en HBeAg (Fried et al. (2008) Hepatology 47:428) y las reducciones en HBsAg (Moucari et al. (2009) Hepatology 49:1151). Las reducciones en los niveles de antígeno pueden haber permitido el control inmunológico de la infección por VHB porque se cree que altos niveles de antígenos inducen tolerancia inmunológica. Las terapias actuales con nucleósidos para el VHB son capaces de lograr reducciones drásticas en los niveles séricos de VHB, pero tienen poco impacto sobre los niveles de HBeAg y HBsAg. La terapia antisentido difiere de la terapia con nucleósidos en que puede seleccionar como diana directamente los transcritos de los antígenos y, de ese modo, reducir los niveles séricos de HBeAg y HBsAg. Pero la terapia antisentido es costosa y requiere administración intravenosa.
Por tanto, existe la necesidad en la técnica de descubrir y desarrollar nuevas terapias antivirales. Más particularmente, existe la necesidad de nuevas terapias anti-VHB capaces de aumentar las tasas de seroconversión de HBeAg y HBsAg. Estos marcadores séricos son indicativos del control inmunológico de la infección por VHB y conducen a un mejor pronóstico, es decir, la prevención de la enfermedad hepática y la progresión a la cirrosis, la prevención de la insuficiencia hepática, la prevención del cáncer hepatocelular (CHC), la prevención del trasplante relacionado con enfermedad hepática y la prevención de la muerte.
El documento WO 2015/173164 A1 fue presentado por Hoffman La Roche para “dihidroquinolizinonas para el tratamiento y la profilaxis de la infección por el virus de la hepatitis B”. Hoffman La Roche presentó posteriormente el documento WO 2016/071215 A1 que se dirige a “derivados de 6,7-dihidrobenzo[a]quinolizin-2-ona para el tratamiento y la profilaxis de la infección por el virus de la hepatitis B”. Arbutus Biopharma Inc. presentó el documento WO 2018/085619 A1 dirigido a “compuestos tricíclicos que contienen piridinona sustituida, y métodos de uso de los mismos", y Novartis AG presentó el documento WO 2018/047109 A1 denominado “compuestos de piridina policíclicos como antivirales”.
SUMARIO DE LA INVENCIÓN
Las realizaciones de la presente invención presentan un compuesto seleccionado de:

En algunas realizaciones, la administración de un compuesto tal como se describe en el presente documento inhibe la liberación del antígeno de superficie de la hepatitis B (HBsAg), la proteína de antígeno central de la HB (HBcAg) y/o la proteína precentral de la hepatitis B conocida como antígeno de antígeno e de VHB (HBeAg) de hepatocitos infectados. Solo los dos compuestos anteriores y la materia a la que se hace referencia forman parte de la invención. Las referencias a la materia dada a conocer en los siguientes párrafos debe tomarse como referencia y no como parte de la invención.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
Las realizaciones de la presente invención presentan compuestos que inhiben los niveles de los antígenos HBe y/o HBs en un sujeto infectado con el virus de la hepatitis B y, por tanto, son útiles para tratar infecciones humanas por el virus de la hepatitis B, y la enfermedad y los síntomas asociados con tales infecciones por virus. Las características anteriores de la invención se entenderán más fácilmente por referencia a la siguiente descripción detallada, tomada con referencia a las tablas adjuntas, en las que:
La tabla 1 es una lista de compuestos de fórmula I descritos en el presente documento.
La tabla 2 es una lista de sustituyentes R10 para compuestos de fórmula I tal como se describe en el presente documento.
La tabla 3 es un resumen de células HepAD38 - ELISA de HBsAg y ensayos de citotoxicidad que muestran los valores de CE50 medidos para un compuesto sometido a prueba contra antígenos HBs (HBsAg).
DESCRIPCIÓN DETALLADA DE REALIZACIONES ESPECÍFICAS
A lo largo de esta solicitud, se hace referencia a diversas realizaciones que se refieren a compuestos y composiciones. Las diversas realizaciones descritas pretenden proporcionar una variedad de ejemplos ilustrativos y no deben interpretarse como descripciones de especies alternativas. Más bien, debe indicarse que las descripciones de diversas realizaciones proporcionadas en el presente documento pueden ser de alcance solapante. Las realizaciones comentadas en el presente documento son meramente ilustrativas y no pretenden limitar el alcance de la presente invención que se define mediante las reivindicaciones.
Ha de entenderse que la terminología usada en el presente documento es para el fin de describir realizaciones particulares solo y no pretende limitar el alcance de la presente invención. En esta memoria descriptiva y en las reivindicaciones que siguen, se hará referencia a varios términos que se definirá que tienen los siguientes significados.
Tal como se usa en el presente documento, a menos que se especifique otra cosa, “alquilo" se refiere a un grupo hidrocarbilo alifático saturado monovalente que tiene desde 1 hasta 14 átomos de carbono y, en algunas realizaciones, desde 1 hasta 6 átomos de carbono El término “alquilo" incluye, a modo de ejemplo, grupos hidrocarbilo lineales y ramificados tales como metilo (CH3-), etilo (CH3CH2-), n-propilo (CH3CH2CH2-), isopropilo ((CH3)2CH-), n-butilo (CH3CH2CH2CH2-), isobutilo ((CH3)2CHCH2-), sec-butilo ((CH3)(CH3CH2)CH-), í-butilo ((CH3)3C-), n-pentilo (CH3CH2CH2CH2CH2-) y neopentilo ((CH3)3CCH2-). Los grupos alquilo puede estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Alcoxi" o “alcoxilo" se refiere al grupo -O-alquilo en donde el alquilo se define en el presente documento. Alcoxilo incluye, a modo de ejemplo, metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, í-butoxi, sec-butoxi, n-pentoxi, morpholinilpropoxi, piperidiniletoxi. Los grupos alcoxilo pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Amino" se refiere al grupo -NRaRb donde Ra y Rb se seleccionan independientemente de hidrógeno, hidroxilo, alquilo o alquilo sustituido, alquenilo o alquenilo sustituido, arilo o arilo sustituido, cicloalquilo o cicloalquilo sustituido, heterocicloalquilo o heterocicloalquilo sustituido, heteroarilo o heteroarilo sustituido, y en donde Ra y Rb se unen
opcionalmente junto con el nitrógeno unido a los mismos para formar un grupo heterocíclico. Cuando Ra es hidrógeno y Rb es alquilo, el grupo amino se denomina algunas veces en el presente documento alquilamino o aminoalquilo. Cuando Ra y Rb son alquilo, el grupo amino se denomina algunas veces en el presente documento dialquilamino. Cuando se hace referencia a un amino monosustituido, quiere decirse que o bien Ra o bien Rb es hidrógeno, pero no ambos. Cuando se hace referencia a un amino disustituido, quiere decirse que ni Ra ni R6 son hidrógeno.
“Arilo” se refiere a un grupo aromático de desde 5 hasta 14 átomos de carbono y ningún heteroátomo de anillo y que tiene un solo anillo (por ejemplo, fenilo) o múltiples anillos condensados (fusionados) (por ejemplo, naftilo o antrilo). Para sistemas de múltiples anillos, incluidos sistemas de anillos fusionados, con puente y de espiro que tienen anillos aromáticos y no aromáticos que no tienen heteroátomos de anillo, el término “arilo” o “Ar” se aplica cuando el punto de unión está en un átomo de carbono aromático (por ejemplo, 5,6,7,8-tetrahidronaftalen-2-ilo es un grupo arilo ya que su punto de unión está en la posición 2 del anillo de fenilo aromático). Los grupos arilo pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Cicloalquilo” se refiere a un grupo cíclico saturado o parcialmente saturado de desde 3 hasta 14 átomos de carbono y ningún heteroátomo de anillo y que tiene un solo anillo o múltiples anillos incluidos sistemas de anillos fusionados, con puente y de espiro. Para sistemas de múltiples anillos que tienen anillos aromáticos y no aromáticos que no tienen heteroátomos de anillo, el término “cicloalquilo” se aplica cuando el punto de unión está en un átomo de carbono no aromático (por ejemplo 5,6,7,8-tetrahidronaftalen-5-ilo). El término “cicloalquilo” incluye grupos cicloalquenilo, tales como ciclohexenilo. Los ejemplos de grupos cicloalquilo incluyen, por ejemplo, adamantilo, ciclopropilo, ciclobutilo, ciclohexilo, ciclopentilo, ciclooctilo, ciclopentenilo y ciclohexenilo. Ejemplos de grupos cicloalquilo que incluyen sistemas de múltiples anillos de bicicloalquilo son biciclohexilo, biciclopentilo, biciclooctilo, y similares. Los grupos cicloalquilo pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Halo” o “halógeno” se refiere a flúor, cloro, bromo y yodo.
“Haloalquilo” se refiere a la sustitución de grupos alquilo con de 1 a 9 (por ejemplo, cuando el grupo alquilo tiene 3 átomos de carbono, tal como un grupo t-butilo completamente sustituido con halógeno) o en algunas realizaciones de 1 a 3 grupos halo (por ejemplo trifluorometilo).
“Hidroxi” o “hidroxilo” se refiere al grupo -OH.
“Heteroarilo” se refiere a un grupo aromático de desde 1 hasta 14 átomos de carbono y de 1 a 6 heteroátomos seleccionados de oxígeno, nitrógeno, azufre, fósforo, silicio y boro, e incluye sistemas de un solo anillo (por ejemplo imidazolilo) y de múltiples anillos (por ejemplo bencimidazol-2-ilo y bencimidazol-6-il). Para sistemas de múltiples anillos, incluidos sistemas de anillos fusionados, con puente y de espiro que tienen anillos aromáticos y no aromáticos, el término “heteroarilo” se aplica si hay al menos un heteroátomo de anillo y el punto de unión está en un átomo de un anillo aromático (por ejemplo 1,2,3,4-tetrahidroquinolin-6-ilo y 5,6,7,8-tetrahidroquinolin-3-ilo). En algunas realizaciones, el/los átomo(s) de anillo de nitrógeno y/o azufre del grupo heteroarilo están opcionalmente oxidados para proporcionar los restos N-óxido (N ^O ), sulfinilo o sulfonilo. Más específicamente, el término heteroarilo incluye, pero no se limita a, piridilo, furanilo, tienilo, tiazolilo, isotiazolilo, triazolilo, imidazolilo, imidazolinilo, isoxazolilo, pirrolilo, pirazolilo, piridazinilo, pirimidinilo, purinilo, ftalazilo, naftilo, naftilpiridilo, oxazolilo, quinolilo, benzofuranilo, tetrahidrobenzofuranilo, isobenzofuranilo, benzotiazolilo, benzoisotiazolilo, benzotriazolilo, indolilo, isoindolilo, indolizinilo, dihidroindolilo, indazolilo, indolinilo, benzoxazolilo, quinolilo, isoquinolilo, quinolizilo, quianazolilo, quinoxalilo, tetrahidroquinolinilo, isoquinolilo, quinazolinonilo, bencimidazolilo, bencisoxazolilo, benzotienilo, benzopiridazinilo, pteridinilo, carbazolilo, carbolinilo, fenanthridinilo, acridinilo, fenantrolinilo, fenazinilo, fenoxazinilo, fenotiazinilo y ftalimidilo. Los grupos heteroarilo pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Heterocíclico” o “heterociclo” o “heterocicloalquilo” o “heterociclilo” se refiere a un grupo cíclico saturado o parcialmente saturado que tiene desde 1 hasta 14 átomos de carbono y desde 1 hasta 6 heteroátomos seleccionados de nitrógeno, azufre, fósforo u oxígeno e incluye sistemas de un solo anillo y de múltiples anillos incluidos sistemas de anillos fusionados, con puente y de espiro. Para sistemas de múltiples anillos que tienen anillos aromáticos y/o no aromáticos, los términos “heterocíclico”, “heterociclo”, “heterocicloalquilo” o “heterociclilo” se aplican cuando hay al menos un heteroátomo de anillo y el punto de unión está en un átomo de un anillo no aromático (por ejemplo 1,2,3,4-tetrahidroquinolin-3-ilo, 5,6,7,8-tetrahidroquinolin-6-ilo y decahidroquinolin-6-ilo). En una realización, el/los átomo(s) de nitrógeno, fósforo y/o azufre del grupo heterocíclico están opcionalmente oxidados para proporcionar los restos N-óxido, óxido de fosfinano, sulfinilo, sulfonilo. Más específicamente, el heterociclilo incluye, pero no se limitan a, tetrahidropiranilo, piperidinilo, piperazinilo, 3-pirrolidinilo, 2-pirrolidon-1-ilo, morfolinilo y pirrolidinilo. Un prefijo que indica el número de átomos de carbono (por ejemplo, C3-C10) se refiere al número total de átomos de carbono en la porción del grupo heterociclilo exclusivo del número de heteroátomos. Los grupos heterociclilo o heterocicloalquilo
pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
Los ejemplos de grupos heterociclo y heteroarilo incluyen, pero no se limitan a, azetidina, pirrol, imidazol, pirazol, piridina, pirazina, pirimidina, piridazina, piridona, indolizina, isoindol, indol, dihidroindol, indazol, purina, quinolizina, isoquinolina, quinolina, ftalazina, naftilpiridina, quinoxalina, quinazolina, cinolina, pteridina, carbazol, carbolina, fenantridina, acridina, fenantrolina, isotiazol, fenazina, isoxazol, fenoxazina, fenotiazina, imidazolidina, imidazolina, naftaleno, oxazol, oxopirrolidina, piperidina, piperazina, indolina, ftalimida, quinolina, 1,2,3,4-tetrahidroisoquinolina, 4,5,6,7-tetrahidrobenzo[b]tiofeno, tiazol, ciclopentatiazol, tiazolidina, tiofeno, benzo[b]tiofeno, morfolina, tiomorfolina (también denominada tiamorfolina), piperidina, pirrolidina y tetrahidrofuranilo.
“Heterocíclico fusionado” o “heterociclilo fusionado” se refiere a un sustituyentes cíclico de 3 a 10 miembros formado por el reemplazo de dos átomos de hidrógeno en diferentes átomos de carbono en una estructura de anillo de cicloalquilo, tal como se ejemplifica por la siguiente estructura de ciclopentatiazol:

Los grupos heterociclilo fusionados pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Arilo fusionado y heteroarilo fusionado” se refiere a una estructura de arilo o estructura de heteroarilo de 5 a 6 miembros fusionada con un anillo de arilo, heteroarilo o cicloalquilo de 5 a 6 miembros en diferentes átomos de carbono en la estructura de arilo o la estructura de heteroarilo, que puede estar sustituida en uno de los carbonos en el arilo fusionado o heteroarilo fusionado y conectada a la molécula central en otro de los carbonos, tal como se ejemplifica mediante las siguientes estructuras de ciclopentiltiazol, quinolina o naftaleno:

Los grupos arilo fusionado y heteroarilo fusionado pueden estar también sustituidos, por ejemplo, con uno o más sustituyentes alquilo, cicloalquilo, heterocicloalquilo, alcoxilo, amino, aminoalquilo, tiol, tioalquilo, arilo, heteroarilo, halo o haloalquilo.
“Compuesto”, “compuestos”, “entidad química” y “entidades químicas” tal como se usan en el presente documento se refieren a un compuesto abarcado por las fórmulas genéricas dadas a conocer en el presente documento, cualquier subgénero de esas fórmulas genéricas y cualquier forma de los compuestos dentro de las fórmulas genéricas y subgenéricas, incluidos los racematos, estereoisómeros y tautómeros del compuesto o compuestos.
El término “heteroátomo” significa nitrógeno, oxígeno o azufre e incluye cualquier forma oxidada de nitrógeno, tal como N(O) {N+-O-} y azufre tal como S(O) y S(O)2, y la forma cuaternizada de cualquier nitrógeno básico.
“Oxazol” y “oxazolilo” se refiere a un anillo heterocíclico de 5 miembros que contiene un nitrógeno y un oxígeno como heteroátomos y también contiene tres carbonos y puede estar sustituido en uno de los tres carbonos y puede estar conectado a otra molécula en otro de los tres carbonos, tal como se ejemplifica mediante cualquiera de las siguientes estructuras, en donde los grupos oxazolidinona mostrados en este caso están unidos a una molécula original, lo que se indica mediante una línea ondulada en el enlace a la molécula original:

“Oxopirrolidina” y “oxopirrolidinilo” se refieren a un anillo heterocíclico de 5 miembros que contiene nitrógeno y 4 carbonos que está sustituido en uno de los carbonos en el anillo heterocíclico por un carbonilo y puede estar conectado a otro sustituyente en otro carbono en el anillo heterocíclico, tal como se ejemplifica por la estructura a continuación:

“Piridina” y “piridinilo” se refiere a un anillo de heteroarilo de 6 miembros que contiene un nitrógeno y 5 carbonos que puede estar sustituido también en uno o más de los carbonos en el anillo de heteroarilo, y puede estar conectado a otro sustituyente en otro carbono en el anillo de heteroarilo, tal como se ejemplifica mediante las estructuras a continuación:

“Tiazol” y “tiazolilo” se refiere a un heteroarilo de 5 miembros que contiene un azufre y un nitrógeno en el anillo de heteroarilo y 3 carbonos en el anillo de heteroarilo que puede estar también sustituido en uno o más de los carbonos en el anillo de heteroarilo, y puede estar conectado a otro sustituyente en otro carbono en el anillo de heteroarilo, tal como se ejemplifica mediante las estructuras a continuación:

“Pirimidina” y “pirimidinilo” se refiere a un anillo de heteroarilo de 6 miembros que contiene dos nitrógenos en el anillo de heteroarilo y 4 carbonos en el anillo de heteroarilo que puede estar sustituido en uno o más de los carbonos en el anillo de heteroarilo, y puede estar conectado a otro sustituyente en otro carbono en el anillo de heteroarilo, tal como se ejemplifica mediante las estructuras a continuación:

“Racematos” se refiere a una mezcla de enantiómeros. En una realización de la divulgación, los compuestos de fórmulas I, o sales farmacéuticamente aceptables de los mismos, están enriquecidos enantioméricamente con un enantiómero en donde todos los carbonos quirales a los que se hace referencia están en una configuración. En general, la referencia a un compuesto o sal enriquecido enantioméricamente pretende indicar que el enantiómero especificado comprenderá más del 50 % en peso del peso total de todos los enantiómeros del compuesto o sal.
“Solvato” o “solvatos” de un compuesto se refieren a aquellos compuestos, tal como se definió anteriormente, que están unidos a una cantidad estequiométrica o no estequiométrica de un disolvente. Los solvatos de un compuesto incluyen solvatos de todas las formas del compuesto. En determinadas realizaciones, los disolventes son volátiles, no tóxicos y/o aceptables para su administración a seres humanos en cantidades traza. Los solvatos adecuados incluyen agua.
“Estereoisómero” o “estereoisómeros” se refieren a compuestos que difieren en la quiralidad de uno o más estereocentros. Los estereoisómeros incluyen enantiómeros y diastereomeros.
“Tautómero” se refiere a formas alternativas de un compuesto que difieren en la posición de un protón, tal como tautómeros enol-ceto e imina-enamina, o las formas tautoméricas de grupos heteroarilo que contienen un átomo de anillo unido a tanto un resto -NH- del anillo como un resto =N- del anillo tal como pirazoles, imidazoles, bencimidazoles, triazoles y tetrazoles.
“Tio” o “tiol” se refiere al grupo -SR donde R se selecciona de hidrógeno, alquilo, alquenilo, arilo, cicloalquilo, heterocicloalquilo, heteroarilo, heterocíclico. Cuando R es H, el grupo tio se denomina algunas veces en el presente documento grupo tiol, y cuando R es alquilo, el grupo tio se denomina algunas veces en el presente documento grupo tioalquilo o alquiltio. El azufre puede unirse también a otro carbono o átomo en la misma molécula para formar un
grupo heterocíclico.
“Sal farmacéuticamente aceptable” se refiere a sales farmacéuticamente aceptables derivadas de una variedad de contraiones orgánicos e inorgánicos bien conocidos en la técnica e incluyen, a modo de ejemplo solo, sodio, potasio, calcio, magnesio, amonio y tetraalquilamonio, y cuando la molécula contiene una funcionalidad básica, sales de ácidos orgánicos o inorgánicos, tales como clorhidrato, bromhidrato, tartrato, mesilato, acetato, maleato y oxalato. Las sales adecuadas incluyen las descritas en P. Heinrich Stahl, Camille G. Wermuth (Eds.), Handbook of Pharmaceutical Salts Properties, Selection, and Use; 2002.
“Paciente” o “sujeto” se refiere a mamíferos e incluye seres humanos y mamíferos no humanos.
“Tratar” o “tratamiento” de una enfermedad en un paciente se refiere a 1) prevenir que se produzca la enfermedad en un paciente que está predispuesto o que aún no presenta síntomas de la enfermedad; 2) inhibir la enfermedad o detener su desarrollo; o 3) mejorar o provocar la regresión de la enfermedad.
Dondequiera que aparezcan líneas discontinuas adyacentes a enlaces sencillos indicados por líneas continuas, entonces la línea discontinua representa un doble enlace opcional en esa posición. Asimismo, donde quiera que aparezcan círculos discontinuos dentro de estructuras de anillo indicadas por líneas continuas o círculos continuos, entonces los círculos discontinuos representan de uno a tres dobles enlaces opcionales dispuestos según su valencia apropiada teniendo en cuenta si el anillo tiene cualquier sustitución opcional alrededor del anillo, tal como conocerá un experto en la técnica. Por ejemplo, la línea discontinua en la estructura a continuación podría indicar o bien un doble enlace en esa posición o bien un enlace sencillo en esa posición:

De manera similar, el anillo A a continuación podría ser un anillo de ciclohexilo sin ningún doble enlace o podría ser también un anillo de fenilo que tiene tres dobles enlaces dispuestos en cualquier posición que todavía representa la valencia apropiada para un anillo de fenilo. Asimismo, en el anillo B a continuación, cualquiera de X1-X5 podría seleccionarse de: C, CH o CH2, N, o NH, y el círculo discontinuo significa que el anillo B podría ser un anillo de ciclohexilo o fenilo o un heterociclo que contiene N sin dobles enlaces o un anillo de heteroarilo que contiene N con de uno a tres dobles enlaces dispuestos en cualquier posición que todavía representa la valencia apropiada:

Cuando se dibujan compuestos específicos o fórmulas genéricas que tienen anillos aromáticos, tales como anillos de arilo o heteroarilo, entonces un experto en la técnica entenderá que la ubicación aromática particular de cualquier doble enlace es una combinación de posiciones equivalentes incluso si se dibujan en diferentes ubicaciones de un compuesto a otro o de una fórmula a otra. Por ejemplo, en los dos anillos de piridina (A y B) a continuación, los dobles enlaces se dibujan en diferentes ubicaciones, sin embargo, se sabe que son la misma estructura y compuesto:
A B

A menos que se indique lo contrario, se llega a la nomenclatura de sustituyentes que no se definen explícitamente en el presente documento nombrando la porción terminal de la funcionalidad seguida por la funcionalidad adyacente hacia el punto de unión. Por ejemplo, el sustituyente “arilalquiloxicarbonilo” se refiere al grupo (aril)-(alquil)-O-C(O)-. En un término tal como “-C(Rx)2”, debe entenderse que los dos grupos Rx pueden ser iguales, o pueden ser diferentes si se define que Rx tiene más de una identidad posible. Además, ciertos sustituyentes se dibujan como -RxRy, donde el “-” indica un enlace adyacente a la molécula original y siendo Ry la porción terminal de la funcionalidad. De manera similar, se entiende que las definiciones anteriores no pretenden incluir patrones de sustitución inadmisibles (por ejemplo, metilo sustituido con 5 grupos flúor). Tales patrones de sustitución inadmisibles los conoce bien el experto en la técnica.
Según la presente divulgación, se proporcionan compuestos de fórmula I o una sal o profármaco de los mismos,

en donde
W e Y son independientemente C o N, con la condición de que W e Y no sean ambos C;
en donde
si W es C, entonces R1 es hidrógeno, hidroxilo, halógeno, ciano, amino o amino sustituido, tio o tio sustituido, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; alquenilo o alquenilo sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5, 6; u -OR12; y
si Y es C, entonces R4 es hidrógeno, hidroxilo, halógeno, ciano, amino o amino sustituido, tio o tio sustituido, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; alquenilo o alquenilo sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5, 6; u -OR12;
en donde
si W es N, entonces R1 está ausente; y
si Y es N, entonces R4 está ausente;
R2 y R3 se seleccionan independientemente de hidrógeno, hidroxilo, halógeno, ciano, amino o amino sustituido, tio o tio sustituido, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; alquenilo o alquenilo sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5, 6; u -OR12;
R5 y R6 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R7 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
o R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22', en donde el anillo de heterocicloalquilo de 3 a 8 miembros está opcionalmente sustituido con R13, R13', R14 y/o R14';
o R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22';
o R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R17', R18 y/o R18', en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R9 es un enlace, hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido, heterocicloalquilo o heterocicloalquilo sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5
o 6;
R10 es un sustituyente mostrado en la tabla 2 o un tautómero del mismo;
o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol;
R11 es hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido; alquenilo o alquenilo sustituido; alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; heterocicloalquilo o heterocicloalquilo sustituido, arilo, heteroarilo, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R12 es hidrógeno; alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, heterocicloalquilo o heterocicloalquilo sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido;
R13, R13', R14 y R14' son independientemente hidrógeno, hidroxilo, halógeno, amino, aminoalquilo, ciano, alquilo C1-6, alcoxilo C1-6, carbonilo, carboxamida, amida; o R13 y R13' o R14 y R14' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R15, R15', R16 y R16' son independientemente hidrógeno, hidroxilo, halógeno, amino, ciano, alquilo C1-6 o alcoxilo C1-6; o R15 y R15' o R16 y R16' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R17, R17', R18 y R18' son independientemente hidrógeno, hidroxilo, halógeno, amino, ciano, alquilo C1-6 o alcoxilo C1-6; o R17 y R18 o R17' y R18' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R19, R19' y R19” son independientemente hidrógeno, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6, alcoxilo C1-6, fenilo, alquil C1-6-imidizol, alquil C1-6-triazol, alquil C1-6-tetrazol, alquil C1-6-tiazol, alquil C1-6-oxazol, alquil C1-6-dioxazol; alquil C1-6-oxazolidona; y
R20 y R21 son independientemente hidrógeno, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6, alcoxilo C1-6, fenilo, alquil C1-6-imidizol, alquil C1-6-triazol, alquil C1-6-tetrazol, alquil C1-6-tiazol, alquil C1-6-oxazol, alquil C1-6-dioxazol; alquil C1-6-oxazolidona, o R20 y R21 junto con el nitrógeno al que están unidos forman pirrolidinilo no sustituido, piperidinilo no sustituido o morfolinilo no sustituido; o forman pirrolidinilo sustituido con carboxilo, piperidinilo sustituido con carboxilo o morfolinilo sustituido con carboxilo; y
r22 y r22' se seleccionan independientemente de hidrógeno, oxígeno, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-6 o alquenilo C2-6 sustituido, arilo o arilo sustituido, incluido alquil C1-6-imidizol sustituido o no sustituido, alquil C1-6-triazol sustituido o no sustituido, alquil C1-6-tetrazol, alquil C1-6-tiazol, alquil C1-6-oxazol sustituido o no sustituido, alquil C1-6-dioxazol; alquil C1-6-oxazolidona; -COR19, -COOR19', -CSOR19”, -CONR20R21,
o una sal farmacéuticamente aceptable de los mismos.
Una realización particular proporciona un compuesto según la fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, en donde:
R1 se selecciona de hidrógeno, hidroxilo, halógeno, ciano, amino, pirrolidinilo, alquilo C1-6 no sustituido o alquilo C1-6 sustituido con halo, alcoxilo C1-6 no sustituido o alcoxilo C1-6 sustituido con halo; cicloalquilo C3-7 o cicloalquilo C3-7 sustituido con halo; heterocicloalquilo monocíclico que contiene N, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; u -OR12;
R2 y R3 son independientemente OR12;
R4 se selecciona de hidrógeno, hidroxilo, halógeno, ciano, amino, pirrolidinilo, alquilo C1-6 no sustituido o alquilo C1-6 sustituido con halo, alcoxilo C1-6 no sustituido o alcoxilo C1-6 sustituido con halo; cicloalquilo C3-7 o cicloalquilo C3-7 sustituido con halo; heterocicloalquilo monocíclico que contiene N, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; u -OR12;
R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22'; R11 es hidrógeno; R12 es tal como se describe en el presente documento; y R15 y R16 o R15' y R16' forman juntos un anillo de heterocicloalquilo o anillo de cicloalquilo de 3 a 8 miembros opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heteroalquilo es O, N, NR22, S, SR22 o SR22R22', o una sal farmacéuticamente aceptable del mismo.
Otra realización proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, R9 es tal como se describe en el presente documento y R10 es un sustituyente mostrado en la tabla 2, o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol; o una sal farmacéuticamente aceptable del mismo.
Otra realización particular proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, R9 es tal como se describe en el presente documento y R10 es un sustituyente mostrado en la tabla 2, o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol; y R12 es alquilo C1-6 no sustituido; o una sal farmacéuticamente aceptable del mismo.
Otra realización particular proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, en donde W e Y son tal como se describe en el presente documento, R1, R2, R3 y R4 son independientemente OR12; R9 es tal como se describe en el presente documento y R10 es un sustituyente mostrado en la tabla 2, o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol; y R12 es tal como se describe en el presente documento; o una sal farmacéuticamente aceptable del mismo.
Otra realización de la divulgación proporciona un compuesto de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo, tal como se proporciona en el presente documento, en donde R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol.
Aún otra realización de la divulgación proporciona un compuesto de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo, tal como se describe en el presente documento, en donde
W e Y son tal como se describe en el presente documento;
R1 y R4, son tal como se describe;
R2 y R3 se seleccionan independientemente de hidrógeno, hidroxilo, halógeno, ciano, amino, tio, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido; cicloalquilo C3-8 o cicloalquilo C3-8 sustituido; alquenilo C2-8 o alquenilo C2-8 sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, u -OR12;
R5 y R6 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-8 o alquenilo C2-8 sustituido; arilo o arilo sustituido, heteroarilo o arilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R7 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-8 o alquenilo C2-8 sustituido; arilo o arilo sustituido, heteroarilo o arilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
o R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22', en donde el anillo de heterocicloalquilo de 3 a 8 miembros está opcionalmente sustituido con R13, R13', R14 y/o R14';
o R6 y R7 forman juntos un anillo de heterocicloalquilo o anillo de cicloalquilo de 3 a 8 miembros que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22';
o R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R17', R18 y/o R18', en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R9 es un enlace, hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido, heterocicloalquilo o heterocicloalquilo sustituido,
arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R10 es un sustituyente mostrado en la tabla 2 o un tautómero del mismo;
o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol;
R11 es hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-8 o alquenilo C2-8 sustituido; arilo o arilo sustituido, heteroarilo o arilo sustituido,-CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R12 es hidrógeno; alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, heterocicloalquilo C3-8 o heterocicloalquilo C3-8 sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido; y
r22 y r22' se seleccionan independientemente de hidrógeno, oxígeno, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-6 o alquenilo C2-6 sustituido, arilo o arilo sustituido,-COR19, -COOR19', -CSOR19”, -CONR20R21.
Otra realización de la divulgación proporciona un compuesto de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo, tal como se describe en el presente documento, en donde W e Y son cada uno N.
Una realización particular de la divulgación proporciona un compuesto de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo, tal como se describe en el presente documento, en donde
W e Y son tal como se describe en el presente documento;
R1, R2, R3 y R4 son independientemente H o OR12; y
R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22', en donde el anillo de heterocicloalquilo de 3 a 8 miembros está opcionalmente sustituido con R13, R13', R14 y/o R14'.
Otra realización particular de la divulgación proporciona un compuesto de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo, tal como se describe en el presente documento, en donde
W e Y son tal como se describe en el presente documento;
R2 y R3 son independientemente H o OR12; y
R6 y R7 forman juntos un anillo de heterocicloalquilo o anillo de cicloalquilo de 3 a 8 miembros que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22'. Otra realización particular de la divulgación proporciona un compuesto de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo, tal como se describe en el presente documento, en donde
R1, R2, R3 y R4 son independientemente H o OR12; y
R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17 y R18, en donde el heteroátomo en el anillo de heterocicloalquilo se selecciona de O, N, NR22, S, SR22 o SR22R22'.
Otra realización particular proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, en donde R2 y R3 son independientemente OR12; R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R13 y R14, en donde el heteroátomo en el anillo de heteroalquilo es O, N, NR22, S, SR22 o SR22R22'; R10 es un sustituyente mostrado en la tabla 2, o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol; R11 es H; R12 es alquilo C1-6 no sustituido; y R13 y R14 o R13' y R14' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heteroalquilo es O, N, NR22, S, SR22 o SR22R22'; o una sal farmacéuticamente aceptable del mismo.
Todavía otra realización particular proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, en donde R2 y R3 son independientemente OR12; R7 y R8 forman juntos un anillo de 3 a 8 miembros, opcionalmente sustituido con R17 y R18; R10 es un sustituyente mostrado en la tabla 2 o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol; R11 es H; R12 es alquilo C1-6 no sustituido; y R17 y R18 o R17' y R18' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heteroalquilo es O, N, NR22, S, SR22 o SR22R22'; o una sal farmacéuticamente aceptable del mismo.
Otra realización particular proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, en donde R2 y R3 son independientemente OR12; y R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R13, R14, R13' y/o R14', en donde el heteroátomo en el anillo de heteroalquilo es O, N, NR22, S, SR22 o SR22R22', o una sal farmacéuticamente aceptable del mismo.
Otra realización particular proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento en donde R2 y R3 son independientemente OR12; y R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R18, R17' y/o R18', en donde el heteroátomo en el anillo de heteroalquilo es O, N, NR22, S, SR22 o SR22R22', o una sal farmacéuticamente aceptable del mismo.
Todavía más realizaciones particulares proporcionan compuestos de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable de los mismos tal como se describe en el presente documento, en donde
R1, R2, R3 y R4 son tal como se describe; y
(A) R5 y R6 son independientemente hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido,
-CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; o R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R13, R13', R14 y/o R14', en donde el heteroátomo en el anillo de heterocicloalquilo se selecciona de O, N, NR22, S, SR22 o SR22R22'; y
R7 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; o
(B) R5 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido,
-CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; y
R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22'; o
(C) R5 y R6 son independientemente hidrógeno, hidroxilo, halógeno, ciano, aminoalquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido,
-CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; y
R7 y R8 son independientemente hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; o R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o un anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R18', R17 y/o R18', en donde el heteroátomo en el anillo de heterocicloalquilo se selecciona de O, N, NR22, S, SR22 o SR22R22'.
En otras realizaciones particulares, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe, en donde el compuesto se selecciona de los compuestos de la tabla 1, en donde R10 es tal como se describe en el presente documento o un sustituyente mostrado en la tabla 2, o un tautómero del mismo.
En realizaciones particulares, se proporciona un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo tal como se describe en el presente documento, en donde W es N; Y es C; R1 está ausente; R2 y R3 se seleccionan independientemente de hidrógeno, hidroxilo, halógeno, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-8 o alquenilo C2-8 sustituido, u -OR12, R4 es H; R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R15, R15', R16 y/o R16'; R11 es H; y R15, R15', R16 y/o R16' son independientemente hidrógeno, hidroxilo, halógeno, amino, ciano, alquilo C1-6 o alcoxilo C1-6.
En una realización particular, se proporciona un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo tal como se describe en el presente documento, en donde W es N; Y es C: R1 está ausente; R2 es halógeno; R3 es OR12; y R4 es H.
En otra realización particular, se proporciona un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo tal como se describe en el presente documento, en donde W es N; Y es C; R1 está ausente; R2 es halógeno y R3 es -OR12; R4 es H; R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R15, R15', R16 y/o R16'; R11 es H; y R15, R15', R16 y/o R16' son independientemente alquilo C1-6.
En otra realización particular, se proporciona un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo tal como se describe en el presente documento, en donde: R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R15, R15', R16 y/o R16'.
En otra realización particular, se proporciona un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo tal como se describe en el presente documento, en donde: R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17 y R18, en donde el heteroátomo en el anillo de heterocicloalquilo se selecciona de O, N, NR22, S, SR22 o SR22R22'.
En una realización particular, se proporciona un compuesto seleccionado del grupo que consiste en:
ácido (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (4bS,7aR)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (4bR,7aS)-2-Ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido 2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (7aR)-2-ciclopropil-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a, 11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (7aR)-2-cloro-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (7aR)-2-cloro-4b-metoxi-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (4bR,7aS)-2-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico;
ácido (4bR,7aS)-2-cloro-3-hidroxi-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]nafti ridin-10-carboxílico;
ácido 2-cloro-6-(1 -hidroxi-2-metilpropan-2-il)-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-3-(3-metoxipropoxi)-2-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido (S)-6-(terc-butil)-2-ciclopropil-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido (S)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]nafti ridin-9-carboxílico;
ácido (R)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-hidroxi-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-2-metoxi-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-2-hidroxi-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido (S)-6-(terc-butil)-2-isopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido (S)-6-(terc-butil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido (S)-6-(terc-butil)-2-ciclopropil-11 -hidroxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9- carboxílico;
ácido (2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido (2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido 2-ciclopropil-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido 2-ciclopropil-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico;
ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; ácido 2'-cloro-3'-(ciclopropilmetoxi)-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico;
ácido 2',3'-dimetoxi-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico;
ácido 6-isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxílico;
ácido 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]nafti ridin-10- carboxílico;
ácido 3'-(ciclopropilmetoxi)-2'-(difluorometil)-11'-fluoro-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico;
ácido 2'-(difluorometil)-11'-fluoro-10'-oxo-3'-((tetrahidrofuran-3-il)metoxi)-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico;
ácido (S)-3-(ciclopropilmetoxi)-2-(difluorometil)-11-fluoro-6-isopropil-6-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; y
ácido (6S)-2-(difluorometil)-11-fluoro-6-isopropil-6-metil-10-oxo-3-((tetrahidrofuran-3-il)metoxi)-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico; o una sal farmacéuticamente aceptable o tautómero de los mismos.
En una realización particular, se proporciona un compuesto seleccionado de:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
En una realización particular, se proporciona un compuesto cuya estructura es:

o una sal farmacéuticamente aceptable del mismo.
Según una realización de la presente divulgación, se proporciona un compuesto que tiene la estructura de fórmula I, IA o IB en donde los compuestos se seleccionan de:

en donde W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es tal como se describe en el presente documento o un sustituyente mostrado en la tabla 2 y

heterociclo o heterociclo sustituido, heteroarilo o heteroarilo sustituido, en donde R y R' son independientemente alquilo o alquilo sustituido.
En realizaciones particulares, se proporcionan compuestos de fórmula I, IA o IB en donde los compuestos se seleccionan de:

en donde W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es tal como se describe en el presente documento o un sustituyente mostrado en la tabla 2.
En otras realizaciones particulares, se proporcionan compuestos de fórmula I, IA o IB en donde los compuestos se seleccionan de:

, en donde W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es un sustituyente tal como se describe en el presente documento o un sustituyente mostrado en la tabla 2, y R es H, alquilo o alquilo sustituido.
En otra realización de la presente divulgación, los compuestos de fórmula I, fórmula IA o fórmula IB se seleccionan de:

, en donde W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es un sustituyente tal como se describe en el presente documento o un sustituyente mostrado en la tabla 2, y R es hidrógeno, hidroxilo,

heterocicloalquilo o heterocicloalquilo sustituido, heteroarilo o heteroarilo sustituido, en donde R' y R” son independientemente alquilo o alquilo sustituido.
En otra realización de la presente divulgación, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se muestra:

en donde R1, R2, R3, R4, R9 y R11 son tal como se describe, W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es tal como se describe en el presente documento o seleccionado de los sustituyentes mostrados en la tabla 2, y R' y R se seleccionan independientemente de hidrógeno, hidroxilo, halo, alquilo o alquilo sustituido, alquileno o alquileno sustituido, carbociclo o carbociclo sustituido, heterocicloalquilo o heterocicloalquilo sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, en donde dichos grupos R y R' sustituidos pueden estar sustituidos con hidroxilo, halo, alquilo, alquileno, cicloalquilo, heterociclo, arilo, heteroarilo,

o R y R' forman juntos un anillo carbocíclico o heterocíclico de espiro, fusionado o con puente.
En otra realización de la presente divulgación, se proporciona un compuesto que tiene la estructura de fórmula I, IA o IB, en donde el compuesto se selecciona de:


en donde W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es tal como se describe en el presente documento o se selecciona de los sustituyentes mostrados en la tabla 2, R y R' son independientemente H, hidroxilo, alquilo o alquilo sustituido, acilo, éster, carbamoílo, sulfonilurea o urea, y en donde Z es H, OH, NH2, SH o C, O, N o S sustituido.
En otra realización particular, se proporciona un compuesto de fórmula I, IA o IB tal como se indica:

en donde R4, R5, R6, R9 y R11 son tal como se describe, W e Y son independientemente N o C, con la condición de que W e Y no sean ambos C, y en donde R10 es tal como se describe en el presente documento o se selecciona de los sustituyentes mostrados en la tabla 2, R1 es tal como se describe, R2 y R3 se seleccionan independientemente de:

En una realización se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento o una sal farmacéuticamente aceptable del mismo, en donde:
(A) R5 y R6 son independientemente hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; o R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R13, R13', R14 y/o R14', en donde el heteroátomo en el anillo de heterocicloalquilo se selecciona de O, N, NR22, S, SR22 o SR22R22'; y
R7 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; o
(B) R5 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido,
heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; y
R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22'; o
(C) R5 y R6 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; y
R7 y R8 son independientemente hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6; o R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o un anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R18', R17 y/o R18', en donde el heteroátomo en el anillo de heterocicloalquilo se selecciona de O, N, NR22, S, SR22 o SR22R22'.
En una realización se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento o una sal farmacéuticamente aceptable del mismo, en donde el compuesto se selecciona de los compuestos de la tabla 1, y en donde R10 es tal como se describe en el presente documento o es un sustituyente mostrado en la tabla 2, o un tautómero del mismo.
En una realización, se proporciona una composición farmacéutica que comprende un diluyente farmacéuticamente aceptable y una cantidad terapéuticamente eficaz de un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento.
En una realización, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, para su uso en terapia.
En una realización, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, para su uso en el tratamiento de una infección viral.
En una realización, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, para su uso en el tratamiento de una infección viral, en donde la infección viral es una infección viral de hepatitis B.
En una realización, se proporciona el uso de un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, en la fabricación de un medicamento para su uso en el tratamiento de una infección viral de hepatitis B en un ser humano.
En una realización, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, para su uso en terapia médica.
En una realización, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, para su uso en el tratamiento de prevención de una infección viral de hepatitis B en un ser humano.
En una realización, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB tal como se describe en el presente documento, o una composición farmacéutica del mismo, para su uso en la inhibición del nivel de antígeno HBe o HBs en un HBsAg de mamífero in vitro.
Los compuestos escritos en el presente documento pueden existir en formas geométricas o estereoisoméricas particulares. La invención contempla todos de tales compuestos, incluidos isómeros cis y trans, enantiómeros (-) y (+), enantiómeros (R) y (S), diastereómeros, isómeros (D), isómeros (L), las mezclas racémicas de los mismos, y otras mezclas de los mismos, tales como mezclas enriquecidas enantiomérica o diastereoméricamente, que se encuentran dentro del alcance de la invención. Pueden estar presentes átomos de carbono asimétricos adicionales en un sustituyente tal como un grupo alquilo. Se pretende que todos de tales isómeros, así como mezclas de los mismos, estén incluidos en esta invención.
Pueden prepararse isómeros d y I e isómeros (R) y (S) ópticamente activos usando sintones quirales o reactivos quirales, o resolverse usando técnicas convencionales. Si, por ejemplo, se desea un enantiómero particular de un compuesto de la presente invención, puede prepararse mediante síntesis asimétrica, o mediante derivatización con un auxiliar quiral, donde la mezcla diastereomérica resultante se separa y el grupo auxiliar se escinde para proporcionar los enantiómeros deseados puros. Alternativamente, cuando la molécula contiene un grupo funcional
básico, tal como un grupo amino, o un grupo funcional ácido, tal como un grupo carboxilo, pueden formarse sales diastereoméricas con un ácido o base ópticamente activo apropiado, seguido por resolución de los diastereómeros así formados mediante cristalización fraccionada o medios cromatográficos conocidos en la técnica, y recuperación posterior de los enantiómeros puros. Además, la separación de enantiómeros y diastereómeros se logra frecuentemente usando cromatografía empleando fases estacionarias quirales, opcionalmente en combinación con derivatización química (por ejemplo, formación de carbamatos a partir de aminas).
En otra realización de la divulgación, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB para su uso en terapia.
En otra realización de la divulgación, se proporciona un compuesto de fórmula I, fórmula IA o fórmula IB, para su uso en el tratamiento de una infección viral.
En otra realización de la divulgación, se proporciona un uso de un compuesto de fórmula I, fórmula IA o fórmula IB en la fabricación de un medicamento para su uso en el tratamiento de una infección viral en un ser humano.
En otra realización de la divulgación, se proporciona una composición farmacéutica que comprende un diluyente farmacéuticamente aceptable y una cantidad terapéuticamente eficaz de un compuesto tal como se define en la fórmula I, fórmula IA o fórmula IB.
En una realización, la formulación farmacéutica que contiene un compuesto de fórmula I, fórmula IA o fórmula IB o una sal del mismo es una formulación adaptada para administración parenteral. En otra realización, la formulación es una formulación parenteral de acción prolongada. En una realización adicional, la formulación es una formulación de nanopartículas.
En una realización, la formulación farmacéutica que contiene un compuesto de fórmula I, fórmula IA o fórmula IB o una sal del mismo es una formulación adaptada para formulación oral, rectal, tópica o intravenosa, en donde la formulación farmacéutica comprende opcionalmente uno cualquiera o más de un portador, adyuvante o vehículo farmacéuticamente aceptable.
En una realización, los compuestos de fórmula I, fórmula IA o fórmula IB se formulan para administración oral, y pueden administrarse como una preparación convencional, por ejemplo, como cualquier forma farmacéutica de un agente sólido tal como comprimidos, polvos, gránulos, cápsulas y similares; un agente acuoso; una suspensión oleosa; o un agente líquido tal como jarabe y elixir. En una realización, los compuestos de fórmula I, fórmula IA o fórmula IB se formulan para administración parenteral, y pueden administrarse como una suspensión acuosa u oleosa inyectable, o una gota nasal. Tras la preparación de una formulación parenteral con un compuesto de fórmula I, fórmula IA o fórmula IB, pueden usarse arbitrariamente excipientes, aglutinantes, lubricantes, disolventes acuosos, disolventes oleosos, emulsionantes, agentes de suspensión, conservantes, estabilizadores y similares convencionales. Como fármaco antiviral, particularmente, es preferible un agente oral. Puede prepararse una preparación de un compuesto de fórmula I, fórmula IA o fórmula IB combinando (por ejemplo, mezclando) una cantidad terapéuticamente eficaz de un compuesto de fórmula I, fórmula IA o fórmula IB con un portador o diluyente farmacéuticamente aceptable.
Las formulaciones farmacéuticas adaptadas para administración oral pueden presentarse como unidades diferenciadas tales como cápsulas o comprimidos; polvos o gránulos; disoluciones o suspensiones en líquidos acuosos o no acuosos; espumas o batidos comestibles; o emulsiones líquidas de aceite en agua o emulsiones líquidas de agua en aceite.
Por ejemplo, para administración oral en forma de un comprimido o una cápsula, el compuesto de fórmula I, fórmula IA o fórmula IB puede combinarse con un portador inerte farmacéuticamente aceptable, no tóxico, oral, tal como etanol, glicerol, agua y similares. Se preparan polvos triturando el compuesto de fórmula I, fórmula IA o fórmula IB hasta un tamaño fino adecuado y mezclándolo con un portador farmacéutico triturado de manera similar, tal como un hidrato de carbono comestible, como, por ejemplo, almidón o manitol. También pueden estar presentes agentes aromatizantes, conservantes, dispersantes y colorantes.
Se fabrican cápsulas preparando una mezcla de polvo, tal como se describió anteriormente, y rellenando las vainas de gelatina formadas. Pueden añadirse deslizantes y lubricantes tales como sílice coloidal, talco, estearato de magnesio, estearato de calcio o polietilenglicol sólido a la mezcla de polvo antes de la operación de llenado. También puede añadirse un agente disgregante o solubilizante tal como agar-agar, carbonato de calcio o carbonato de sodio para mejorar la disponibilidad del medicamento cuando se ingiere la cápsula.
Además, cuando se desee o sea necesario, también pueden incorporarse a la mezcla aglutinantes, lubricantes, agentes disgregantes y colorantes adecuados. Los aglutinantes adecuados incluyen almidón, gelatina, azúcares naturales tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares. Los lubricantes usados
en estas formas farmacéuticas incluyen oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y similares. Los disgregantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares. Se formulan comprimidos, por ejemplo, preparando una mezcla de polvo, granulando o golpeando, añadiendo un lubricante y disgregante y aplicando presión para dar comprimidos. Se prepara una mezcla de polvo mezclando el compuesto, adecuadamente triturado, con un diluyente o base tal como se describió anteriormente y, opcionalmente, con un aglutinante tal como carboximetilcelulosa, un aligato, gelatina o polivinilpirrolidona, un retardante de la disolución tal como parafina, un acelerador de la reabsorción tal como una sal cuaternaria y/o un agente de absorción tal como bentonita, caolín o fosfato de dicalcio. La mezcla de polvo puede granularse mojándola con un aglutinante tal como jarabe, pasta de almidón, mucílago de acadia o disoluciones de materiales celulósicos o poliméricos y forzándola a través de un tamiz. Como alternativa a la granulación, la mezcla de polvo puede hacerse pasar a través de la máquina de comprimidos y el resultado son lingotes imperfectamente formados que se rompen en gránulos. Los gránulos pueden lubricarse para evitar que se peguen a los troqueles formadores de comprimidos por medio de la adición de ácido esteárico, una sal de estearato, talco o aceite mineral. La mezcla lubricada se comprime entonces para dar comprimidos. Los compuestos de la presente invención también pueden combinarse con un portador inerte de flujo libre y comprimirse para dar comprimidos directamente sin pasar por las etapas de granulación o golpeo. Puede proporcionarse un recubrimiento protector transparente u opaco que consiste en una capa selladora de goma laca, un recubrimiento de azúcar o material polimérico y un recubrimiento de cera de pulido. Pueden añadirse colorantes a estos recubrimientos para distinguir diferentes dosificaciones unitarias.
Pueden prepararse fluidos orales tales como disoluciones, jarabes y elixires en forma de dosificación unitaria de modo que una cantidad determinada contenga una cantidad predeterminada del compuesto. Pueden prepararse jarabes disolviendo el compuesto en una disolución acuosa adecuadamente aromatizada, mientras que se preparan elixires mediante el uso de un vehículo alcohólico no tóxico. Pueden formularse suspensiones dispersando el compuesto en un vehículo no tóxico. También pueden añadirse solubilizantes y emulsionantes tales como alcoholes isoestearílicos etoxilados y éteres de polioxietilensorbitol, conservantes, aditivos de sabor tales como aceite de menta o edulcorantes naturales o sacarina u otros edulcorantes artificiales, y similares.
Cuando sea apropiado, las formulaciones de dosificación unitaria para administración oral pueden microencapsularse. Las formulaciones de compuestos de fórmula I, fórmula IA o fórmula IB también pueden prepararse para prolongar o sostener la liberación del compuesto, como por ejemplo, recubriendo o incrustando material particulado en polímeros, cera o similares.
Los compuestos de fórmula I, fórmula IA o fórmula IB o sales, solvatos o hidratos de los mismos, también pueden administrarse en forma de sistemas de administración de liposomas, tales como vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Pueden formarse liposomas a partir de una variedad de fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas.
Los compuestos de fórmula I, fórmula IA o fórmula IB o sales, solvatos o hidratos de los mismos, también pueden administrarse mediante el uso de anticuerpos monoclonales como portadores individuales a los que se acoplan las moléculas del compuesto. Los compuestos también pueden acoplarse con polímeros solubles como portadores de fármacos dirigibles. Tales polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietilaspartamida-fenol o poli(óxido de etileno)polilisina sustituida con residuos de palmitoílo. Además, los compuestos pueden acoplarse a una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, poli(ácido láctico), poli(épsilon-caprolactona), poli(ácido hidroxibutírico), poliortoésteres, poliacetales, polidihidropiranos, policianoacrilatos y copolímeros de bloques reticulados o anfipáticos de hidrogeles.
Las formulaciones farmacéuticas adaptadas para administración transdérmica pueden presentarse como parches diferenciados destinados a permanecer en contacto íntimo con la epidermis del receptor durante un período de tiempo prolongado. Por ejemplo, los compuestos de fórmula I, fórmula IA o fórmula IB pueden administrarse desde un parche mediante iontoforesis tal como se describe generalmente en Pharmaceutical Research, 3(6), 318 (1986).
Las formulaciones farmacéuticas adaptadas para administración tópica pueden formularse como ungüentos, cremas, suspensiones, lociones, polvos, disoluciones, pastas, geles, pulverizaciones, aerosoles o aceites. Cuando se formula en una pomada, el principio activo puede emplearse con una base de pomada parafínica o miscible en agua. Alternativamente, el principio activo puede formularse en una crema con una base de crema de aceite en agua o una base de agua en aceite.
Las formulaciones farmacéuticas adaptadas para la administración rectal pueden presentarse como supositorios o como enemas.
Las formulaciones farmacéuticas adaptadas para la administración nasal en las que el portador es un sólido incluyen un polvo grueso que tiene un tamaño de partícula, por ejemplo, en el intervalo de 20 a 500 micrómetros que se administra de la misma manera que se toma el rapé, es decir, por inhalación rápida a través de las fosas nasales
desde un recipiente del polvo sostenido cerca de la nariz. Las formulaciones adecuadas en las que el portador es un líquido, para administración como una pulverización nasal o como gotas nasales, incluyen disoluciones acuosas u oleosas del principio activo.
Las formulaciones farmacéuticas adaptadas para la administración por inhalación incluyen polvos o nieblas de partículas finas, que pueden generarse por medio de diversos tipos de aerosoles, nebulizadores o insufladores presurizados de dosis medida.
Las formulaciones farmacéuticas adaptadas para administración parenteral incluyen disoluciones de inyección estériles acuosas y no acuosas que pueden contener antioxidantes, tampones, bacteriostáticos y solutos que hacen que la formulación sea isotónica con la sangre del receptor previsto; y suspensiones estériles acuosas y no acuosas que pueden incluir agentes de suspensión y agentes espesantes. Las formulaciones pueden presentarse en recipientes de dosis unitaria o multidosis, por ejemplo, ampollas y viales sellados, y pueden almacenarse en un estado secado por congelación (liofilizado) que solo requiere la adición del portador líquido estéril, por ejemplo, agua para inyecciones, inmediatamente antes de su uso. Las disoluciones y suspensiones para inyección extemporánea pueden prepararse a partir de polvos, gránulos y comprimidos estériles.
Debe entenderse que, además de los componentes mencionados anteriormente en particular, las formulaciones descritas en el presente documento pueden incluir otros agentes convencionales en la técnica teniendo en cuenta el tipo de formulación en cuestión, por ejemplo, aquellas adecuadas para administración oral pueden incluir agentes aromatizantes.
Una cantidad terapéuticamente eficaz de un compuesto de fórmula I, fórmula IA o fórmula IB dependerá de varios factores que incluyen, por ejemplo, la edad y el peso del ser humano u otro animal, la afección precisa que requiere tratamiento y su gravedad, la naturaleza de la formulación y la vía de administración, y en última instancia quedará a discreción del médico o veterinario encargado. Una cantidad eficaz de una sal o hidrato de la misma puede determinarse como una proporción de la cantidad eficaz del compuesto de fórmula I, fórmula IA o fórmula IB o sales, solvatos o hidratos del mismo per se.
Las realizaciones de la presente divulgación proporcionan la administración de un compuesto de fórmula I, fórmula IA o fórmula IB a un paciente sano o infectado por virus, o bien como agente único o bien en combinación con (a) otro agente que es eficaz en el tratamiento o la prevención del virus de la hepatitis B o el virus de la hepatitis D, (b) otro agente que mejora la respuesta inmunitaria y la robustez, o (c) otro agente que reduce la inflamación y/o el dolor.
Se cree que los compuestos de fórmula I, fórmula IA o fórmula IB o sales, solvatos o hidratos de los mismos tienen actividad en la prevención, detención o reducción de los efectos del virus de la hepatitis B al inhibir los antígenos HBe y/o HBs, interfiriendo de ese modo con o evitando que el virus permanezca en la célula huésped y haciendo que el virus no pueda replicarse.
En otras realizaciones, los compuestos de la presente invención pueden usarse en combinación con uno o más agentes terapéuticos antivirales o agentes antiinflamatorios útiles en la prevención o el tratamiento de enfermedades virales o la fisiopatología asociada. Por tanto, los compuestos de la presente invención y sus sales, solvatos u otros derivados farmacéuticamente aceptables de los mismos pueden emplearse solos o en combinación con otros agentes terapéuticos antivirales o antiinflamatorios.
Los compuestos de la presente invención y cualquier otro agente farmacéuticamente activo pueden administrarse juntos o por separado y, cuando se administran por separado, la administración puede producirse simultánea o secuencialmente, en cualquier orden. Las cantidades de los compuestos de la presente invención y el/los otro(s) agente(s) farmacéuticamente activo(s) y los tiempos relativos de administración se seleccionarán con el fin de lograr el efecto terapéutico combinado deseado. La administración en combinación de un compuesto de la presente invención y sales, solvatos u otros derivados farmacéuticamente aceptables de los mismos con otros agentes de tratamiento puede ser en combinación mediante la administración concomitante en: (1) una composición farmacéutica unitaria que incluye ambos compuestos; o (2) composiciones farmacéuticas separadas que incluyen uno de los compuestos cada una.
Alternativamente, la combinación puede administrarse por separado de manera secuencial en donde un agente de tratamiento se administra en primer lugar y el otro en segundo lugar o viceversa. Tal administración secuencial puede ser cercana en el tiempo o remota en el tiempo. Las cantidades del/de los compuesto(s) de fórmula I, fórmula IA o fórmula IB o sales de los mismos y el/los otro(s) agente(s) farmacéuticamente activo(s) y los tiempos relativos de administración se seleccionarán con el fin de lograr el efecto terapéutico combinado deseado.
Más particularmente, un agente adicional puede seleccionarse de un agente antiviral, un antibiótico, un analgésico, un agente antiinflamatorio no esteroideo (AINE), un agente antifúngico, un agente antiparasitario, un agente contra las náuseas, un agente antidiarreico agente o un agente inmunosupresor. En ciertas realizaciones, el agente antiviral es
un agente contra la hepatitis B o un agente contra la hepatitis C. Todavía más particularmente, el agente adicional se administra como parte de una forma de dosificación individual de dicha formulación farmacéutica, o como una forma de dosificación separada.
La presente invención se refiere a compuestos, composiciones y composiciones farmacéuticas que tienen utilidad como tratamientos novedosos y/o terapias preventivas para infecciones virales. Sin pretender limitarse a ninguna teoría en particular, se cree que los presentes compuestos pueden inhibir los niveles de antígenos HBe y HBs en un sujeto infectado con el virus de la hepatitis B o que padece una infección viral crónica por hepatitis B. Al reducir los niveles de antígenos HBe y HBs en un sujeto infectado con el virus de la hepatitis B, los compuestos descritos en el presente documento son eficaces en el tratamiento de infecciones por hepatitis B y trastornos secundarios tales como cirrosis hepática, insuficiencia hepática y cáncer de hígado que a menudo están asociados con infecciones por el virus de la hepatitis B.
Se dan a conocer compuestos, métodos y composiciones farmacéuticas para tratar infecciones virales mediante la administración de compuestos de fórmula I, fórmula IA o fórmula IB en cantidades terapéuticamente eficaces. También se dan a conocer métodos para preparar compuestos de fórmula I, fórmula IA o fórmula IB y métodos de uso de los compuestos y composiciones farmacéuticas de los mismos. En particular, se dan a conocer el tratamiento y la profilaxis de infecciones virales tales como las provocadas por hepatitis B y/o hepatitis D.
En otras realizaciones, los compuestos descritos en el presente documento son útiles para tratar infecciones en un sujeto en donde la infección está provocada por una cepa resistente a múltiples fármacos del virus de la hepatitis B y/o un virus de la hepatitis D.
EJEMPLOS
Esquemas de síntesis
Los compuestos de la presente divulgación o invención que tienen la fórmula I, fórmula IA o fórmula IB

o sales farmacéuticamente aceptables correspondientes de los mismos, se preparan usando síntesis orgánicas convencionales, en donde:
C* es un estereocentro de átomo de carbono que tiene una configuración que es (R) o (S);
W e Y son independientemente C o N, con la condición de que W e Y no sean ambos C;
en donde
si W es C, entonces R1 es hidrógeno, hidroxilo, halógeno, ciano, amino o amino sustituido, tio o tio sustituido, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; alquenilo o alquenilo sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5, 6; u -OR12; y
si Y es C, entonces R4 es hidrógeno, hidroxilo, halógeno, ciano, amino o amino sustituido, tio o tio sustituido, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; alquenilo o alquenilo sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5, 6; u -OR12;
en donde
si W es N, entonces R1 está ausente; y
si Y es N, entonces R4 está ausente;
R2 y R3 se seleccionan independientemente de hidrógeno, hidroxilo, halógeno, ciano, amino o amino sustituido, tio o tio sustituido, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; alquenilo o alquenilo sustituido; heterocicloalquilo de 3 a 8 miembros o heterocicloalquilo de 3 a 8 miembros sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, pirrolidinilo, -CxH2x-fenilo, -O-CxH2x-fenilo o -(alquil C1-6)N-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5, 6; u -OR12;
R5 y R6 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo o arilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R7 y R8 son independientemente hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido; arilo o arilo sustituido, heteroarilo, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
o R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22', en donde el anillo de heterocicloalquilo de 3 a 8 miembros está opcionalmente sustituido con R13, R13', R14 y/o R14';
o R6 y R7 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22';
o R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R17', R18 y/o R18', en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R9 es un enlace, hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, alquenilo o alquenilo sustituido, heterocicloalquilo o heterocicloalquilo sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R10 es un sustituyente mostrado en la tabla 2 o un tautómero del mismo;
o R9 es un enlace y R9 y R10 forman juntos un anillo de oxaborol;
R11 es hidrógeno, hidroxilo, halógeno, ciano, amino, alquilo o alquilo sustituido; alquenilo o alquenilo sustituido; alcoxilo o alcoxilo sustituido; cicloalquilo o cicloalquilo sustituido; heterocicloalquilo o heterocicloalquilo sustituido, arilo, heteroarilo, -CxH2x-fenilo u -O-CxH2x-fenilo en donde x es 0, 1,2, 3, 4, 5 o 6;
R12 es hidrógeno; alquilo o alquilo sustituido, alcoxilo o alcoxilo sustituido, cicloalquilo o cicloalquilo sustituido, heterocicloalquilo o heterocicloalquilo sustituido, arilo o arilo sustituido, heteroarilo o heteroarilo sustituido;
R13, R13', R14 y R14' son independientemente hidrógeno, hidroxilo, halógeno, amino, aminoalquilo, ciano, alquilo C1-6, alcoxilo C1-6, carbonilo, carboxamida, amida; o R13 y R13' o R14 y R14' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R15, R15', R16 y R16' son independientemente hidrógeno, hidroxilo, halógeno, amino, ciano, alquilo C1-6 o alcoxilo C1-6; o R15 y R15' o R16 y R16' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R17, R17', R18 y R18' son independientemente hidrógeno, hidroxilo, halógeno, amino, ciano, alquilo C1-6 o alcoxilo C1-6; o R17 y R18 o R17' y R18' forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con oxígeno, halógeno, hidroxilo, amino, ciano, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6 o alcoxilo C1-6, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22';
R19, R19' y R19” son independientemente hidrógeno, alquilo C1-6, cicloalquilo C3-8, alquenilo C2-6, alcoxilo C1-6, fenilo, alquil C1-6-imidizol, alquil C1-6-triazol, alquil C1-6-tetrazol, alquil C1-6-tiazol, alquil C1-6-oxazol, alquil C1-6-dioxazol; alquil C1-6-oxazolidona; y
R20 y R21 son independientemente hidrógeno, alquilo Ci-6, cicloalquilo C3-8, alquenilo C2-6, alcoxilo C1-6, fenilo, alquil Ci-6-imidizol, alquil C1-6-triazol, alquil C1-6-tetrazol, alquil C1-6-tiazol, alquil C1-6-oxazol, alquil C1-6-dioxazol; alquil C1-6-oxazolidona, o R20 y R21 junto con el nitrógeno al que están unidos forman pirrolidinilo no sustituido, piperidinilo no sustituido o morfolinilo no sustituido; o forman pirrolidinilo sustituido con carboxilo, piperidinilo sustituido con carboxilo o morfolinilo sustituido con carboxilo; y
r22 y r22' se seleccionan independientemente de hidrógeno, oxígeno, alquilo C1-6 o alquilo C1-6 sustituido, alcoxilo C1-6 o alcoxilo C1-6 sustituido, cicloalquilo C3-8 o cicloalquilo C3-8 sustituido, alquenilo C2-6 o alquenilo C2-6 sustituido, arilo o arilo sustituido, incluidos alquil C1-6-imidizol sustituido o no sustituido, alquil C1-6-triazol sustituido o no sustituido, alquil C1-6-tetrazol, alquil C1-6-tiazol, alquil C1-6-oxazol sustituido o no sustituido, alquil C1-6-dioxazol; alquil C1-6-oxazolidona; -COR19, -COOR19', -CSOR19”,-CONR20R21,
o una sal farmacéuticamente aceptable de los mismos.
Otras realizaciones proporcionan compuestos de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable del mismo tal como se describe en el presente documento, en donde:
R5 y R6 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22', en donde el anillo de heterocicloalquilo de 3 a 8 miembros está opcionalmente sustituido con R13, R13', R14 y/o R14'.
Otras realizaciones particulares proporcionan compuestos de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable de los mismos tal como se describe en el presente documento, en donde:
R6 y R7 forman juntos un anillo de heterocicloalquilo o anillo de cicloalquilo de 3 a 8 miembros que comprende un heteroátomo o dos o más heteroátomos, opcionalmente sustituido con R15, R15', R16 y/o R16', en donde el heteroátomo en el anillo de heteroalquilo es NR20 y los dos o más heteroátomos se seleccionan de N, NR22, O, S, SR22 y SR22R22'.
Otras realizaciones particulares proporcionan compuestos de fórmula I, fórmula IA o fórmula IB o una sal farmacéuticamente aceptable de los mismos tal como se describe en el presente documento, en donde:
R7 y R8 forman juntos un anillo de cicloalquilo de 3 a 8 miembros o anillo de heterocicloalquilo de 3 a 8 miembros, opcionalmente sustituido con R17, R17', R18 y/o R18', en donde el heteroátomo en el anillo de heterocicloalquilo es O, N, NR22, S, SR22 o SR22R22'.
A continuación se representan rutas de síntesis adecuadas en los siguientes esquemas de reacción generales. El experto en la técnica apreciará que si un sustituyente descrito en el presente documento no es compatible con los métodos de síntesis descritos en el presente documento, el sustituyente puede protegerse con un grupo protector adecuado que es estable en las condiciones de reacción. El grupo protector puede eliminarse en un punto adecuado de la secuencia de reacción para proporcionar un compuesto diana o producto intermedio deseado. Los expertos en la técnica conocen bien grupos protectores adecuados y los métodos para proteger y desproteger diferentes sustituyentes usando tales grupos protectores adecuados; ejemplos de los cuales pueden encontrarse en T. Greene y P. Wuts, Protecting Groups in Chemical Synthesis (35 ed.), John Wiley & Sons, NY (1999). En algunos casos, puede seleccionarse específicamente un sustituyente para que sea reactivo en las condiciones de reacción usadas. En estas circunstancias, las condiciones de reacción convierten el sustituyente seleccionado en otro sustituyente que o bien es útil como compuesto intermedio o bien es un sustituyente deseado en un compuesto diana.
Abreviaturas
En la descripción de los ejemplos, los elementos químicos se identifican según la tabla periódica de los elementos. Las abreviaturas y símbolos utilizados en el presente documento son según el uso común de tales abreviaturas y símbolos por parte de los expertos en las técnicas químicas. Las siguientes abreviaturas se usan en el presente documento:
AcOH ácido acético
Ac2O anhídrido acético
ac. acuoso
B4 ácido (S)-2-(((benciloxi)carbonil)amino)-3-(4-(trifluorometil)piperidin-1-il)propanoico
BOC (Boc) N-terc-butoxicarbonilo o terc-butiloxicarbonilo
CBz carboxibencilo
dba dibencilidenacetona o dibenzalacetona
DCE dicloroetano
DCM diclorometano
DCM/EA diclorometano/etanol
DDQ 2,3-dicloro-5,6-dicianobenzoquinona
DIPEA (o DIEA) N,N-diisopropiletilamina, o base de Hünig
DME dimetoxietano
DMEM medio de Eagle modificado por Dulbecco
DMF dimetilformamida
DMP peryodinano de Dess-Martin
DMSO-d6 dimetilsulfóxido deuterado
DMSO dimetilsulfóxido
DPPA difenilfosforilazida
CE50 concentración eficaz al 50 %
EDTA ácido etilendiaminatetraacético
Et etilo
Et2O dietil éter
EtOH etanol
EtOAc, EA, AcOEt acetato de etilo
GlutaMAX™ suplemento de cultivo celular de Life Technologies h hora(s)
HEPES ácido 4-(2-hidroxietil)-1 -piperazinetanosulfónico HPLC cromatografía de líquidos de alta resolución
CI50 concentración de inhibición al 50 %
iPrOH alcohol isopropílico o isopropanol
CLEM cromatografía de líquidos-espectrometría de masas LDA di-isopropilamida de litio
Me metilo
MeOH metanol
NBS N-bromosuccinimida
NCS N-clorosuccinimida
NIS N-yodosuccinimida
NXS N-halosuccinimida
NaBH(OAc)3 triacetoxiborohidruro de sodio
RMN espectroscopía de resonancia magnética nuclear
Pd2(dba)3 Tris(dibencilidenacetona)dipaladio (0)
PE éter de petróleo
PPh3 trifenilfosfina
Pr propilo
FR fondo redondo
ta o t.a. temperatura ambiente
TR tiempo de retención
SFC cromatografía de fluidos supercríticos
SO3pyr complejo de trióxido de azufre y piridina - fórmula C5H5NSO3
SPhos 2-diciclohexilfosfina-2',6'-dimetoxibifenilo o diciclohexil(2',6'-dimetoxi-[1,1'-bifenil]-2-ilo) t-BuOMe metil t-butil éter
T3P disolución de anhídrido 1-propanofosfónico, 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosforinano-2,4,6-trióxido
TFA ácido trifluoroacético
THF tetrahidrofurano
uv ultravioleta
A menos que se indique lo contrario, todos los materiales de partida se obtuvieron de proveedores comerciales y se usaron sin purificación adicional. A menos que se indique lo contrario, todas las temperaturas se expresan en °C (grados centígrados). A menos que se indique lo contrario, todas las reacciones se llevan a cabo bajo una atmósfera inerte a temperatura ambiental.
Todas las temperaturas se dan en grados Celsius, todos los disolventes tienen la mayor pureza disponible y todas las reacciones se realizan en condiciones anhidras en una atmósfera de argón (Ar) o nitrógeno (N2) cuando sea necesario. Los siguientes ejemplos ilustran la invención. Estos ejemplos no pretenden limitar el alcance de la presente invención, sino más bien proporcionar orientación al experto en la técnica para preparar y usar los compuestos y las composiciones de la presente invención.
Tal como se usan en el presente documento, los símbolos y convenciones usados en estos procesos, esquemas y ejemplos son consecuentes con los usados en la bibliografía científica contemporánea, por ejemplo, la Journal of the American Chemical Society o la Journal of Biological Chemistry. A menos que se indique lo contrario, todos los materiales de partida se obtuvieron de proveedores comerciales y se usaron sin purificación adicional.
Todas las referencias a éter son a dietil éter; salmuera se refiere a una solución acuosa saturada de NaCl. A menos que se indique lo contrario, todas las temperaturas se expresan en °C (grados centígrados). Todas las reacciones se llevan a cabo bajo una atmósfera inerte a temperatura ambiente a menos que se indique lo contrario, y todos los disolventes tienen la mayor pureza disponible a menos que se indique lo contrario.
Los espectros de 1H-RMN (a continuación en el presente documento también “RMN”) se registraron en un espectrómetro Varian Unity-400. Los desplazamientos químicos se expresan en partes por millón (ppm, unidades d).
Las constantes de acoplamiento están en unidades de hercios (Hz). Los patrones de división describen multiplicidades aparentes y se designan como s (singlete), d (doblete), t (triplete), q (cuarteto), quin (quinteto), m (multiplete), a (ancho).
Se realizó cromatografía ultrarrápida sobre gel de sílice Merck 60 (230 - 400 de malla), o usando un instrumento Teledyne Isco Combiflash® Companion con columnas ultrarrápidas Redi-Sep® de fase normal, desechables. Los espectros de masas se realizaron en un sistema de CL-EM de acceso abierto usando ionización por electropulverización. Los espectros de masas (EM) analíticos de baja resolución se registraron en un instrumento Waters SQD con análisis de UPLC realizado en una columna Phenomenex® Kinetex® XB-C18 de 1,7 um, 2,1 x 50 mm a 40 CSQ usando un método de elución en gradiente. Disolvente A: ácido fórmico (FA) al 0,2 % en agua; disolvente B: FA al 0,15 % en acetonitrilo; gradiente del 1 % - 99 % de disolvente B durante 1,1 minutos y manteniéndose constante en el 99 % de disolvente B durante otros 0,4 minutos, a una velocidad de flujo de 1 ml/min.
Preparación sintética de compuestos
A continuación se muestran protocolos generales para producir compuestos tal como se describe en el presente documento en los Esquemas 1- 15. Solo los compuestos 220 y 253 son parte de la invención. El resto debe entenderse como compuestos de referencia.

Los compuestos particulares dados a conocer en el presente documento pueden producirse según el Esquema 1. Por ejemplo, el compuesto 4 puede prepararse haciendo reaccionar el compuesto 1 con una 4-piranona apropiadamente sustituida, tal como se muestra, en ácido acético o etanol, con calor. La ciclación reductora de 2 mediante el tratamiento del compuesto 2 con catalizador de Pd(II) con una base apropiada tal como acetato de potasio en un disolvente apropiado tal como N,N-dimetilacetamida (DMA) o N,N-dimetilformamida (DMF) a 90-120 °C produce el compuesto de carboxilato tricíclico fusionado 3. El compuesto 3 se somete entonces a hidrólisis del éster en condiciones de ácido o base para producir el ácido carboxílico 4. La manipulación química adicional del ácido carboxílico puede conducir a grupos R10 adicionales.
Alternativamente, los compuestos descritos en el presente documento pueden prepararse según el Esquema 2.

Tal como se muestra en el Esquema 2, el compuesto 2 puede prepararse haciendo reaccionar el compuesto 1 con una 4-piranona apropiadamente sustituida, tal como se muestra, en ácido acético o etanol, con calor. La ciclación reductora de 2 mediante el tratamiento del compuesto 2 con catalizador de Pd(II) con una base apropiada tal como acetato de potasio en un disolvente apropiado tal como N,N-dimetilacetamida (DMA) o N,N-dimetilformamida (DMF) a 90-120 °C produce el compuesto de carboxilato tricíclico fusionado 3. El compuesto 3 se somete entonces a hidrólisis del éster en condiciones de ácido o base para producir el ácido carboxílico 4. Tal como con el Esquema 1, la manipulación química adicional del ácido carboxílico puede conducir a grupos R10 adicionales.
Esquema 3 para la preparación del compuesto del ejemplo 1
(Compuesto 220)
Pdiídbaj:

.
Etapa 4 Etapa 6
Ejemplo 1 Preparación A (Compuesto 220)
Ácido_____________________________ (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarf|piridor1,2-hir1,71naftiridin-10-carboxílico

Etapa 1: 5-(6-Cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentanona

Se cargó un matraz con una barra agitadora, 5-bromo-2-cloro-3-metoxipiridina (16,5 g, 74,2 mmol), tris(dibencilidenacetona)dipaladio (0) (1,02 g, 1,11 mmol), xantphos (1,16 g, 2,00 mmol) y terc-butóxido de sodio (12,6 g, 131 mmol). Se purgó el matraz con una corriente de nitrógeno durante 30 minutos. Se desgasificó tetrahidrofurano (THF) (300 ml) con nitrógeno durante 30 minutos. Se añadió 2,2-dimetilciclopentan-1-ona (11,2 ml, 89 mmol) al tetrahidrofurano desgasificado (THF) (300 ml) y se añadió esta disolución al matraz purgado con nitrógeno que contenía el sustrato inicial. Se calentó la mezcla hasta reflujo bajo nitrógeno durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se filtró a través de un tapón de sílice sobre nitrógeno. Se lavó el tapón con tetrahidrofurano y se concentró el filtrado. Se disolvió el residuo en diclorometano mínimo y se inyectó sobre una columna de sílice. Se eluyó la columna 1 minuto con hexanos y luego un gradiente rápido (3 minutos) hasta el 20 % de acetato de etilo / hexanos, y luego isocrático al 20 % de acetato de etilo / hexanos hasta que eluyó el producto. Se concentraron las fracciones para dar 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-ona (10,7 g, 42,1 mmol, rendimiento del 56,8 %) como un aceite. CLEM (ESI) m/z 254,2 (M+1).
Etapa 2: 5-(6-Cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentanamina (mezcla trans / cis)

Una mezcla en agitación de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-ona (mezcla trans / cis) (10,7 g, 42,2 mmol) y acetato de amonio (32,5 g, 422 mmol) en metanol (200 ml) se desgasificó con una corriente de nitrógeno durante 25 minutos. Se añadió cianoborohidruro de sodio (5,30 g, 84 mmol) y se calentó la mezcla a 65 °C a lo largo del fin de semana. Se retiró el condensador de reflujo y se calentó el baño de aceite hasta 80 °C, permitiendo que la mezcla de reacción se concentrara hasta ~ 70 % del volumen original a lo largo de 4 horas. Se unió de nuevo el condensador de reflujo y se calentó la mezcla a reflujo fuerte durante la noche (baño de aceite a 80 °C). Se concentró la mezcla y se suspendió el residuo en -300 ml de diclorometano. Se agitó la mezcla vigorosamente durante -30 minutos. Se separaron los sólidos por filtración y se lavó la torta de filtro con diclorometano. Se concentró el filtrado para dar 22 g de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1 -amina en bruto (mezcla trans / cis). Se purificó el material en bruto mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 10 % de amoniaco 2 M / metanol en diclorometano. Se concentraron las fracciones para dar 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-amina (mezcla trans / cis) (9,04 g, 35,5 mmol, rendimiento del 84 %). CLEM (ESI) m/z 255,2 (M+1).
Etapa 3: 5-(2-Amino-3,3-dimetilciclopentil)-2-cloro-6-vodopiridin-3-ol (mezcla trans / cis)

Se añadió lentamente tribromuro de boro (6,71 ml, 71,0 mmol) gota a gota a una disolución en agitación vigorosa de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-amina (mezcla trans / cis) (9,04 g, 35,5 mmol) en 1,2-dicloroetano (DCE) (175 ml). Se agitó la mezcla a temperatura ambiente durante 30 minutos y luego se calentó a 70 °C durante la noche. Se enfrió la mezcla en un baño de hielo y luego se extinguió cuidadosamente con adición lenta de metanol. Se añadió metanol adicional (100 ml). Se calentó la mezcla hasta temperatura ambiente, se agitó durante 30 minutos y se concentró para dejar -17 g de 5-(2-amino-3,3-dimetilciclopentil)-2-cloropiridin-3-ol en bruto (mezcla trans / cis). Se añadió agua (200 ml) al material en bruto y se agitó con una barra agitadora. Se añadió lenta y cuidadosamente carbonato de potasio (24,5 g, 177 mmol) en porciones con el fin de controlar la efervescencia. Se agitó la mezcla durante 10 minutos después de la adición completa del carbonato de potasio. Se sometió a prueba la mezcla acuosa con papel de pH para garantizar que la mezcla era básica. Se extrajo la mezcla acuosa 1 vez con 100 ml de diclorometano. La CL-EM indicó solo impurezas en la capa orgánica y solo 5-(2-amino-3,3-dimetilciclopentil)-2-cloropiridin-3-ol (mezcla trans / cis) en la capa acuosa. Se desechó la fase orgánica y se transfirió la capa acuosa desde el embudo de decantación hasta un matraz de fondo redondo antes de que se añadiera yodo (18,0 g, 71,0 mmol). Se agitó la mezcla durante la noche a temperatura ambiente. Se añadió sulfito de sodio en exceso (~4 eq.) en porciones. Se añadió 2-metiltetrahidrofurano (100 ml) y se agitó la mezcla. Se añadió ácido acético en exceso cuidadosamente para neutralizar la capa acuosa al tiempo que se controlaba la efervescencia. Se añadió cloruro de sodio sólido a la fase acuosa. Se extrajo la mezcla 3 veces con 2-metiltetrahidrofurano. Se lavaron las capas orgánicas combinadas 2 veces con salmuera. Se secó la fase orgánica sobre sulfato de sodio y se concentró para dar 12,6 g de material en bruto. Se purificó el material mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 20 % de amoniaco 2 M / metanol en diclorometano. Se concentraron las fracciones para dar 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (mezcla trans / cis) (6,6 g, 18 mmol, rendimiento del 50,7 %). CLEM (ESI) m/z 367,1 (M+1).
Etapa 4: 1 -(5-(6-Cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (mezcla trans / cis)

Se agitaron 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (mezcla trans / cis) (6,6 g, 18 mmol) y 4-oxo-4H-piran-3-carboxilato de etilo (3,94 g, 23,4 mmol) en ácido acético (150 ml) a 100 °C durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se concentró hasta sequedad a vacío. Se añadió tolueno y se
sometió a evaporación rotatoria varias veces para ayudar a eliminar el ácido acético restante. La evaporación final dio 1-(5-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo en bruto. CLEM (ESI) m/z 517,1 (M+1). Se disolvió 1-(5-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo en bruto en N,N-dimetilformamida (DMF) (75 ml) antes de que se añadieran carbonato de potasio (12,4 g, 90 mmol) y 1-bromo-3-metoxipropano (4,05 ml, 36,0 mmol). Se permitió que la mezcla se agitara a temperatura ambiente durante 15 minutos y luego se calentó hasta 60 °C durante 1 hora. Se enfrió la mezcla de reacción en un baño de hielo y se extinguió con agua. Precipitó un sólido. Se extrajo la mezcla 3 veces con acetato de etilo. Estaba flotando un sólido en la superficie de contacto de las fases y así se extrajo la mezcla 1 vez con diclorometano. Se disolvieron los sólidos y se observó el producto en la fase orgánica. Se lavaron las capas orgánicas combinadas 2 veces con cloruro de litio al 5 %, se lavaron 1 vez con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se suspendió el residuo en etil éter y se recogió el precipitado mediante filtración. Se secó el sólido al aire para dar 1-(5-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (trans racémico) (5,21 g, 8,85 mmol, rendimiento del 49,1 %) como un sólido de color tostado pálido. CLEM (ESI) m/z 589,2 (M+1). Se concentró el filtrado y se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 10 % de metanol en diclorometano. Se concentraron las fracciones para dar 1 -(5-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (mezcla trans / cis) (2,59 g, 3,08 mmol, rendimiento del 17,1 %, el 70 % puro). CLEM (ESI) m/z 589,2 (M+1).
Etapa 5: 2-Cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hiri,71naftiridin-10-carboxilato de etilo (mezcla trans / cis)

Un matraz de fondo redondo que contenía una barra agitadora, 1-(5-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (mezcla trans / cis) (5,57 g, 9,46 mmol), acetato de potasio (4,64 g, 47,3 mmol) y bromuro de paladio (II) (0,504 g, 1,89 mmol) se purgó con nitrógeno durante 20 minutos usando un tabique con aguja de entrada / salida. Se purgó N,N-dimetilformamida (DMF) (75 ml) con nitrógeno durante 20 minutos antes de añadirse al recipiente de reacción. Se colocó el recipiente de reacción en un baño de aceite que se precalentó hasta 100 °C y se agitó la mezcla durante la noche. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se filtró a través de un lecho de celite. Se lavó el lecho de celite con diclorometano y se concentró el filtrado. Se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 10 % de metanol en diclorometano. Se concentraron las fracciones para dar 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo (mezcla trans / cis) (3,15 g, 6,83 mmol, rendimiento del 72,2 %) como un sólido de color tostado pálido. CLEM (ESI) m/z 461,3 (M+1).
Etapa_______ 6:_______ Ácido_______ (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopentarflpiridoH ,2-hlH ,71naftiridin-10-carboxílico

A una disolución de 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo (mezcla trans / cis) (3,15 g, 6,83 mmol) en 1,4-dioxano (40 ml) se le añadió una disolución de hidróxido de litio monohidratado (1,434 g, 34,2 mmol) en agua (30 ml). Se calentó la mezcla a 70 °C durante 3 horas. La CL-EM indicó la conversión completa en el producto cis racémico deseado. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se diluyó con ácido clorhídrico 0,5 M (100 ml). Se recogió el precipitado mediante filtración y se lavó la torta de filtro minuciosamente con agua. Se secó la torta de filtro al aire durante varias horas con vacío en un embudo Buchner hasta que los sólidos ya no parecían húmedos mediante inspección visual. Se disolvió la torta de filtro en 150 ml de diclorometano. La disolución completa requirió agitar durante varios minutos. Se secó la disolución sobre sulfato de sodio y se concentró para dar ácido 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (cis racémico) (2,6 g, 6,01 mmol, rendimiento del 88 %).
Se purificó el racemato en varios lotes usando las siguientes condiciones: Columna = Chiralpak IC, 10 mm x 250 mm (5 u); fase móvil = MeOH/EtOH 3:1 TFA al 0,1 %; velocidad de flujo = 10 ml/min; volumen de inyección = 500 ul (conc. de 30 mg/ml, DCM usado como disolvente de inyección); longitud de onda de recogida = 254 nm. Se concentraron las fracciones correspondiente al pico 1. Se suspendieron los residuos en etil éter, se sonicaron, se agitaron durante 10 minutos y luego se enfrió la mezcla en agitación en un baño de hielo. Se recogieron los sólidos mediante filtración a vacío, se secaron al aire y se secaron a alto vacío para dar ácido (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (1,24 g, 2,87 mmol, rendimiento del 42 %) como sólidos blancos. CLEM (ESI) m/z 433,2 (M+1). 1H-RMN (400 MHz, DMSO-ds) " ppm 8,62 (s, 1 H), 7,63 (s, 1 H), 7,43 (s, 1 H), 4,72 (d, J=8,98 Hz, 1 H), 4,19 - 4,39 (m, 2 H), 3,94 (td, J=8,68, 3,32 Hz, 1 H), 3,51 (t, J=6,05 Hz, 2 H), 3,26 (s, 3 H), 2,35 - 2,46 (m, 1 H), 2,19 - 2,31 (m, 1 H), 2,03 (quin, J=6,25 Hz, 2 H), 1,55 - 1,67 (m, 1 H), 1,40 - 1,50 (m, 1 H), 1,15 (s, 3 H), 0,40 (s, 3 H).
Esquema 4 - Síntesis alternativa del compuesto 220, ejemplo 1

Ejemplo 1 Preparación B (Compuesto 220)
Ácido (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico
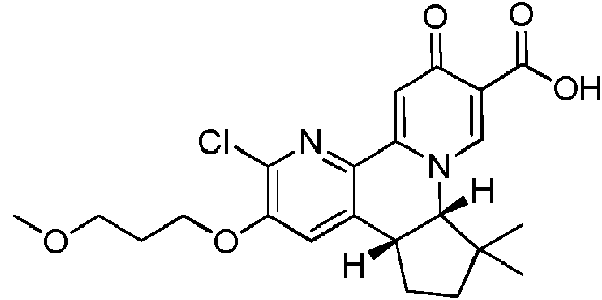
Etapa 1: 5-(6-Cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-ona

A una disolución de 5-bromo-2-cloro-3-metoxipiridina (100 g, 449 mmol) en THF (2 l) se le añadió NaOtBu (76,02 g, 791 mmol), Xantphos (7,01 g, 12,1 mmol) y 2,2-dimetilciclopentan-1-ona (65,56 g, 584,6 mmol). Se desgasificó la mezcla de reacción con nitrógeno durante 30 minutos. Se añadió Pd2(dba)3 a la reacción y se agitó la mezcla de reacción a 70 °C durante 3 h. Después de la finalización de la reacción, se filtró la mezcla de reacción a través de un lecho de gel de sílice (60-120) y se lavó con THF. Se eliminó el disolvente a presión reducida y se purificó el producto en bruto mediante cromatografía en columna (gel de sílice 230-400) usando el 0-30 % de acetato de etilo en éter de petróleo como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título
(61 g, rendimiento del 54 %), CLEM (ESI) m/z 253,9 (M+1).
Etapa 2:5-(6-Cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-amina (mezcla trans / cis)

A una disolución de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-ona (60 g, 236,5 mmol) en metanol (1,2 l) se le añadió NH4ÜAc (182 g, 2365 mmol). Se desgasificó la mezcla de reacción con nitrógeno durante 30 minutos. Se añadió NaBH3CN (29,7 g, 473 mmol) a la mezcla de reacción y se agitó la mezcla de reacción a 65 °C durante 3 días. Después de la finalización de la reacción, se eliminó el disolvente a presión reducida para proporcionar el compuesto del título en bruto (55 g). CLEM (ESI) m/z: 255,7(M 1). Se llevó esto a la siguiente etapa sin purificación adicional.
Etapa 3: Preparación de 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-vodopiridin-3-ol (mezcla trans / cis)

A una disolución de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-amina (60 g, 236 mmol) en 1,2-dicloroetano (DCE) (1300 ml), se le añadió BBr3 (22,27 ml, 236 mmol) y se agitó la mezcla de reacción a 70 °C durante 16 h. Después de la finalización de la reacción, se añadió metanol (1,5 l) a la mezcla de reacción lentamente gota a gota a 0 °C y se agitó a temperatura ambiente durante 30 minutos. Se concentró la mezcla de reacción y se llevó el residuo a agua (1 l) y se ajustó el pH a básico usando carbonato de potasio (325 g, 2355 mmol). Se lavó la capa acuosa con DCM (200 ml). A la capa acuosa se le añadió yodo (120 g, 471 mmol) y se agitó a temperatura ambiente durante 16 h. Después de la finalización de la reacción, se añadió sulfito de sodio (200 g) para extinguir el yodo en exceso. Se añadió ácido acético (250 ml) y se extrajo con 2-metil.THF (2*1 l). Se separó la capa orgánica y se secó sobre sulfato de sodio. Se eliminó el disolvente a presión reducida y se trituró el producto en bruto con diclorometano al 20 % en hexano (2*600 ml). Se filtró el sólido obtenido y se secó para proporcionar el compuesto del título (35 g, rendimiento del 21,89 %). CLEM (54 %) (ESI) m/z: 367 (M 1).
Etapa 4: 1 -(5-(6-Cloro-5-hidroxi-2-vodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (mezcla trans / cis)

Una suspensión de 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (87 g, 237 mmol), 4-oxo-4H-piran-3-carboxilato de etilo (45,9 g, 273 mmol) y ácido acético (1,8 l) se agitó a 100 °C durante 3 h. Después de la finalización de la reacción, se concentró la mezcla de reacción y se coevaporó con tolueno (4*200 ml) y se purificó el residuo mediante cromatografía en columna (gel de sílice 230-400) usando el 10-15 % de MeOH en DCM como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título (51 g, rendimiento del 22 %) como un sólido marrón oscuro. CLEM (53 %) (ESI) m/z: 517 (M 1).
Etapa 5: 1 -(5-(6-Cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (mezcla trans / cis)

A una disolución de 1-(5-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (51 g, 99 mmol) en N,N-dimetilformamida (580 ml) se le añadió K2CO3 (68,2 g, 493 mmol) y 1-bromo-3-metoxipropano (30,2 g, 197 mmol). Se agitó la mezcla de reacción a 60 °C durante 1 h. Después de la finalización de la reacción, se extinguió la mezcla de reacción con agua helada (1500 ml) y se extrajo con DCM (2 l). Se lavaron las capas orgánicas combinadas con salmuera (500 ml), se secaron sobre Na2SO4 y se concentraron. Se purificó el producto en bruto mediante cromatografía en columna (gel de sílice 230-400) usando el 0-10 % de metanol en DCM como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título (52 g, rendimiento del 84 %). CLEM (ESI) m/z: 588,8 (M+1).
Etapa______ 6:______ 2-Cloro-3-(3-metoxipropox¡)-7,7-d¡met¡l-11 -oxo-4b,5,6,7,7a,11 -hexahidroc¡clopenta[f1p¡rido[1,2-h1[1,71naftirid¡n-10-carboxilato de etilo (mezcla trans / cis)

A una disolución de 1 -(5-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (52 g, 88 mmol) en N,N-dimetilformamida (550 ml) se le añadió acetato de potasio (43,3 g, 442 mmol). Se desgasificó la mezcla de reacción con nitrógeno durante 20 minutos. Se añadió bromuro de paladio (II) (4,70 g, 17,66 mmol) a la mezcla de reacción y se agitó la mezcla de reacción a 100 °C durante 16 h. Después de la finalización de la reacción, se filtró la mezcla de reacción a través de celite y se lavó con DCM. Se lavó la fase orgánica con agua (250 ml), se secó sobre Na2SO4 y se concentró. Se purificó el producto en bruto mediante cromatografía en columna (gel de sílice 230-400) usando el 2-5 % de MeOH en DCM como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título (20 g, rendimiento del 42 %). CLEM (ESI) m/z: 461,1 (M 1).
Etapa_______ 7:_______ Ácido_______ (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarrflpiridoH,2-hiri Jlnaftiridin-10-carboxílico

A una disolución de 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo (17,5 g, 38 mmol) en 1,4-dioxano (150 ml) y agua (150 ml) se le añadió hidróxido de litio.H2O (7,9 g, 190,14 mmol). Se calentó la mezcla de reacción hasta 70 °C durante 2 h. Después de la finalización de la reacción, se concentró la mezcla de reacción y se llevó el residuo a DCM (250 ml). Se añadió HCI 1,5 N a esta mezcla hasta pH = 6. Se separó la capa orgánica y se secó sobre Na2SO4 y se concentró para conseguir 15 g (rendimiento: 91 %) de mezcla racémica. Se purificó esta mezcla racémica mediante HPLC prep. usando para proporcionar el compuesto del título (5,5 g, rendimiento del 73 %) como un sólido amarillo pálido. Método de HPLC quiral: Chiral pak IC (21*250), 5 mic, fase móvil A: TFA al 0,1 % en metanol, B: TFA al 0,1 % en etanol; A:B: 75:25, velocidad de flujo: 25 ml/min. Primer pico, tr: 5,4. 1H-RMN 400 MHz, DMSO-afe: 8,62 (s, 1H), 7,63 (s, 1H), 7,43 (s, 1H), 4,73 (d, J = 8,96 Hz, 1H), 4,33 - 4,28 (m, 2H), 3,95 - 3,94 (m, 1H), 3,53 - 3,50 (m, 2H), 3,27 (s, 3H), 2,39 - 2,38 (m, 1H), 2,28 - 2,27 (m, 1H), 2,04-2,01 (m, 2H), 1,61-1,60 (m, 1H), 1,47-1,44 (m, 1H), 1,15 (s, 3H), 0,40 (s, 3H). CLEM (ESI) m/z: 432,9 (M 1).
Ejemplo 2 (Compuesto 221)
Ácido_____________________________ (4bS,7aR)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hlH ,71naftiridin-10-carboxílico

Etapa 1: Acido_______ (4bS,7aR)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hl[1,7lnaftiridin-10-carboxílico
Se purificó ácido 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (cis racémico) (500 mg) mediante cromatografía quiral. Columna = Chiralpak IC, 10 mm x 250 mm (5 u), fase móvil = MeOH/EtOH 3:1 TFA al 0,1 %, velocidad de flujo = 10 ml/min, volumen de inyección = 500 ul (conc. de 30 mg/ml); se usó DCM como disolvente de inyección, longitud de onda de recogida = 254 nm. Se concentró el pico 2 para dar acido (4bS,7aR)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (190 mg, 0,438 mmol, rendimiento del 38 %) como un sólido blanco. CLEM (ESI) m/z 433,3 (M+1). 1H-RMN (400 MHz, DMSO-ds) " ppm 8,59 (s, 1 H), 7,60 (s, 1 H), 7,40 (s, 1 H), 4,69 (d, J=8,98 Hz, 1 H), 4,18 - 4,36 (m, 2 H), 3,84 - 3,96 (m, 1 H), 3,48 (t, J=6,25 Hz, 2 H), 3,23 (s, 3 H), 2,30 -2,43 (m, 1 H), 2,18 - 2,28 (m, 1 H), 2,00 (quin, J=6,15 Hz, 2 H), 1,53 - 1,64 (m, 1 H), 1,37 - 1,48 (m, 1 H), 1,12 (s, 3 H), 0,37 (s, 3 H).
Ejemplo 3: (Compuesto 222)
Ácido________________________ (4bR,7aS)-2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hl[1,7lnaftiridin-10-carboxílico

Etapa 1: Acido_____ (4bR,7aS)-2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hl[1,7lnaftiridin-10-carboxílico
Un vial de reacción que contenía una barra agitadora, ácido (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (81 mg, 0,19 mmol), carbonato de potasio (103 mg, 0,748 mmol), ácido ciclopropilborónico (32,1 mg, 0,374 mmol) y tetrakis(trifenilfosfina)paladio (0) (43,2 mg, 0,037 mmol) se purgó concienzudamente con una corriente de nitrógeno (aguja de entrada / salida). Se añadió 1,4-dioxano (2 ml) y se colocó el vial de reacción en un bloque de calentamiento que se precalentó hasta 100 °C. Se calentó la mezcla a 100 °C durante la noche. Se permitió que la mezcla de reacción se enfriara hasta temperatura ambiente y luego se diluyó con 2-metiltetrahidrofurano y agua. Se añadió lenta y cuidadosamente ácido acético (efervescencia) hasta que se neutralizó la fase acuosa. Se extrajo la mezcla 2 veces con 2-metiltetrahidrofurano. Se lavaron las capas orgánicas combinadas con salmuera y se concentraron. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se combinaron las fracciones y se concentraron hasta que se observó un precipitado blanco. Se añadió una pequeña cantidad de acetonitrilo y la disolución se volvió transparente. Se liofilizó la disolución para dar ácido (4bR,7aS)-2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (67 mg, 0,151 mmol, rendimiento del 81 %) como un polvo blanco. CLEM (ESI) m/z 439,1 (M+1). 1H-RMN (400 MHz, DMSO-afe) " ppm 8,54 (s, 1 H), 7,46 (s, 1 H), 7,31 (s, 1 H), 4,64 (d, J=8,98 Hz, 1 H), 4,08 - 4,33 (m, 2 H), 3,79 - 3,91 (m, 1 H), 3,50 (t, J=6,25 Hz, 2 H), 3,24 (s, 3 H), 2,39 -2,45 (m, 1 H), 2,16 - 2,38 (m, 2 H), 2,01 (quin, J=6,15 Hz, 2 H), 1,51 - 1,63 (m, 1 H), 1,32 - 1,45 (m, 1 H), 1,12 (s, 3 H), 0,92 - 1,05 (m, 4 H), 0,34 (s, 3 H).
Ejemplo 4: (Compuesto 223)
Ácido 2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hlH,7lnaftiridin-10-carboxílico (cis racémico)

Etapa 1: Ácido________ 2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hl[1 Jlnaftiridin-10-carboxílico (cis racémico)
Un vial de reacción que contenía una barra agitadora, 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo (cis racémico) (200 mg, 0,434 mmol), carbonato de potasio (240 mg, 1,74 mmol), ácido ciclopropilborónico (74,5 mg, 0,868 mmol) y tetrakis(trifenilfosfina)paladio (0) (100 mg, 0,087 mmol) se purgó concienzudamente con una corriente de nitrógeno (aguja de entrada / salida). Se añadió 1,4-dioxano (4 ml) y se colocó el vial de reacción en un bloque de calentamiento que se precalentó hasta 100 °C. Se calentó la mezcla a 100 °C durante 3 horas. Se añadieron ácido ciclopropilborónico (74,5 mg, 0,868 mmol) y tetrakis(trifenilfosfina)paladio (0) (100 mg, 0,087 mmol) adicionales y la mezcla continuó calentándose a 100 °C durante 2 horas. Se permitió que la mezcla se enfriara y se filtró a través de un tapón de celite. Se lavó el tapón de celite con acetato de etilo. Se concentró el filtrado para dar 2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo en bruto. Se disolvió el producto intermedio en bruto en metanol (2 ml) antes de que se añadiera una disolución de hidróxido de litio monohidratado (182 mg, 4,34 mmol) en agua (2 ml). Se calentó la mezcla a 60 °C durante 3 horas y se enfrió hasta temperatura ambiente. Se purificó la mezcla mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido 2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (cis racémico) (50 mg, 0,114 mmol, rendimiento del 26,3 %) como un polvo de color tostado pálido. CLEM (ESI) m/z 439,3 (M+1). 1H-RMN (400 MHz, DMSO-dfe) " ppm 8,54 (s, 1 H), 7,46 (s, 1 H), 7,31 (s, 1 H), 4,64 (d, J=8,98 Hz, 1 H), 4,11 -4,28 (m, 2 H), 3,79 - 3,91 (m, 1 H), 3,50 (t, J=6,25 Hz, 2 H), 3,24 (s, 3 H), 2,39 - 2,45 (m, 1 H), 2,16 - 2,38 (m, 2 H), 1,96 - 2,04 (m, 2 H), 1,51 - 1,64 (m, 1 H), 1,34 - 1,44 (m, 1 H), 1,12 (s, 3 H), 0,93 - 1,06 (m, 4 H), 0,34 (s, 3 H).
Esquema 5 - Preparación de compuestos tales como el ejemplo 5

donde R5 = OR y R6 = distinto de hidrógeno y donde R6 puede ciclarse
con R7 o R8 para formar un anillo
Ejemplo 5: (Compuesto 224)
Ácido__________________ (7aR)-2-ciclopropil-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hl[1,7lnaftiridin-10-carboxílico

Etapa 1: Ácido (7aR)-2-ciclopropil-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hlH Jlnaftiridin-10-carboxílico
Se añadió terc-butóxido de potasio (13,3 mg, 0,119 mmol) a una disolución de ácido (4bR,7aS)-2-ciclopropil-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (13 mg, 0,030 mmol) en dimetilsulfóxido (DMSO) (0,5 ml) a temperatura ambiente y se agitó durante la noche. Se
inyectó la mezcla sobre una columna de fase inversa a presión media y se eluyó (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido (7aR)-2-ciclopropil-4bhidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (isómero individual) (6 mg, 0,013 mmol, rendimiento del 43,6 %) como un polvo blanquecino. La CL-EM y RMN fueron consecuentes con el producto deseado. CLEM (ESI) m/z 455,3 (M+1). 1H-RMN (400 MHz, DMSO-afe) " ppm 8,68 (s, 1 H), 7,50 (s, 2 H), 6,11 (s, 1 H), 4,62 (s, 1 H), 4,12 - 4,35 (m, 2 H), 3,51 (t, J=6,05 Hz, 2 H), 3,24 (s, 3 H), 2,57 - 2,68 (m, 1 H), 2,11 - 2,24 (m, 1 H), 2,02 (quin, J=6,15 Hz, 2 H), 1,56 - 1,70 (m, 1 H), 1,13 - 1,26 (m, 4 H), 0,97 -1,10 (m, 4 H), 0,24 (s, 3 H).
Ejemplo 6: (Compuesto 225)
Ácido_______________________(7aR)-2-cloro-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hlH Jlnaftiridin-10-carboxílico

Etapa 1: Ácido_____ (7aR)-2-cloro-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hlH ,7lnaftiridin-10-carboxílico
Se añadió terc-butóxido de potasio (15,5 mg, 0,139 mmol) a una disolución de ácido (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (15 mg, 0,035 mmol) en dimetilsulfóxido (DMSO) (1 ml) a temperatura ambiente y se agitó durante 2 horas. Se inyectó la mezcla sobre una columna de fase inversa a presión media y se eluyó (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido (7aR)-2-cloro-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (7 mg, 0,015 mmol, rendimiento del 44,6 %) como un polvo blanco. CLEM (e S i) m/z 449,1 (M+1). 1H-RMN (400 MHz, DMSO-ds) " ppm 8,74 (s, 1 H), 7,78 (s, 1 H), 7,44 (s, 1 H), 6,28 (s, 1 H), 4,70 (s, 1 H), 4,18 - 4,43 (m, 2 H), 3,49 (t, J=6,25 Hz, 2 H), 3,24 (s, 3 H), 2,59 - 2,73 (m, 1 H), 2,21 (dt, J=13,47, 8,88 Hz, 1 H), 2,01 (quin, J=6,15 Hz, 2 H), 1,58 - 1,71 (m, 1 H), 1,21 - 1,29 (m, 1 H), 1,18 (s, 3 H), 0,27 (s, 3 H).
Ejemplo 7: (Compuesto 226)
Ácido_______________________ (7aR)-2-cloro-4b-metoxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hlH ,7lnaftiridin-10-carboxílico

Etapa 1: Ácido______(7aR)-2-cloro-4b-metoxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,1-hexahidrociclopentarflpiridoH ,2-hlH ,7lnaftiridin-10-carboxílico
Se añadió hidruro de sodio (al 60 % en aceite mineral) (1,87 mg, 0,047 mmol) a una disolución en agitación de ácido (7aR)-2-cloro-4b-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (7 mg, 0,016 mmol) y yoduro de metilo (2,9 pl, 0,047 mmol) en N,N-dimetilformamida (DMF) (0,5 ml). Se agitó la mezcla durante 2 horas. Se calentó la mezcla hasta 80 °C durante 2 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se extinguió con agua. Se agitó la mezcla durante 20 minutos. Se inyectó la mezcla sobre una cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido (4bS,7aR)-2-cloro-4b-metoxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico como un polvo blanquecino. CLEM (ESI) m/z 463,2 (M+1). 1H-RMN (400 MHz, DMSO-afe) " ppm 8,79 (s, 1 H), 7,78 (s, 1 H), 7,46 (s, 1 H), 4,92 (s, 1 H), 4,26 - 4,42 (m, 2 H), 3,49 (t, J=6,25 Hz, 2 H), 3,24 (s, 3 H), 2,94 (s, 3 H), 2,66 (dt, J=13,56, 6,68 Hz, 1 H), 2,19 - 2,33 (m, 1 H), 2,01 (quin, J=6,15 Hz, 2 H), 1,63 - 1,74 (m, 1 H), 1,28 - 1,40 (m, 1 H), 1,19 (s, 3 H),
0,29 (s, 3 H).
Ejemplo 8 (compuesto 227) y ejemplo 9 (compuesto 228)
Ácido___________________________(4bR,7aS)-2-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidroc¡clopentarf1piridori,2-hiri Jlnaftiridin-10-carboxílico
Ácido_________ (4bR,7aS)-2-cloro-3-hidroxi-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-h1T1,71naftiridin-10-carboxílico

Una mezcla de ácido (4bR,7aS)-2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (61 mg, 0,141 mmol) en hidróxido de sodio (2 M) (4 ml, 8 mmol) se calentó a 150 °C en un reactor de microondas durante 1 hora. Se inyectó la mezcla sobre una columna de fase inversa a presión media y se eluyó (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se liofilizaron 2 conjuntos de fracciones por separado para dar:
Ejemplo 8: Ácido (4bR,7aS)-2-hidroxi-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (20 mg, 0,048 mmol, rendimiento del 34 %). CLEM (ESI) m/z 415,2 (M+1). 1H-RMN (400 MHz, DMSO-ak) 5 ppm 11,15 - 11,71 (m, 1 H), 8,53 (s, 1 H), 7,44 (s, 1 H), 6,97 - 7,27 (m, 1 H), 4,61 (d, J=8,98 Hz, 1 H), 4,00 - 4,19 (m, 2 H), 3,73 (s. a., 1 H), 3,45 (t, J=6,05 Hz, 2 H), 3,23 (s, 3 H), 2,11 -2,33 (m, 2 H), 1,96 (quin, J=6,25 Hz, 2 H), 1,50 - 1,61 (m, 1 H), 1,31 - 1,45 (m, 1 H), 1,11 (s, 3 H), 0,42 (s, 3 H).
Ejemplo 9: Ácido (4bR,7aS)-2-cloro-3-hidroxi-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (8 mg, 0,022 mmol, rendimiento del 15,6 %). CLEM (ESI) m/z 361,1 (M+1). 1H-RMN (400 MHz, DMSO-afe) 5 ppm 11,85 (s. a., 1 H), 8,57 (s, 1 H), 7,37 (s, 1 H), 7,24 (s, 1 H), 4,66 (d, J=8,98 Hz, 1 H), 3,80 - 3,94 (m, 1 H), 2,25 - 2,43 (m, 2 H), 1,94 - 2,08 (m, 1 H), 1,52 - 1,64 (m, 1 H), 1,38 - 1,49 (m, 1 H), 1,11 (s, 3 H), 0,37 (s, 3 H).
Ejemplo 10: (Compuesto 229)
Ácido_________ 2-cloro-6-(1-hidroxi-2-metilpropan-2-il)-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-h1H ,71naftiridin-9-carboxílico

Etapa 1: 4-(Benciloxi)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona

Se cargó un matraz con una barra agitadora, 5-bromo-2-cloro-3-metoxipiridina (5,8 g, 26 mmol), tris(dibencilidenacetona)dipaladio (0) (0,358 g, 0,391 mmol) y terc-butóxido de sodio (4,41 g, 45,9 mmol). Se purgó el matraz con una corriente de nitrógeno antes de que se añadiera 4-(benciloxi)-3,3-dimetilbutan-2-ona (5,45 g, 26,4 mmol) en tetrahidrofurano (THF) (100 ml). Se calentó la mezcla hasta reflujo durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se diluyó con agua. Se extrajo la mezcla 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 50 % de acetato de etilo en hexanos. Se concentraron las fracciones para dar 4-(benciloxi)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona (5,62 g, 16,2 mmol, rendimiento del 62 %) como un aceite amarillo pálido. CLEM (ESI) m/z 348,2 (M+1). 1H-RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,70 (d, J=1,95 Hz, 1 H), 7,21 - 7,39 (m, 5 H), 7,01 (d, J=1,56 Hz, 1 H), 4,49 (s, 2 H), 3,79 (s, 5 H), 3,52 (s, 2 H), 1,22 (s, 6 H).
Etapa 2: 4-(Benciloxi)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina

Una mezcla de 4-(benciloxi)-1 -(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona (3 g, 8,6 mmol) y acetato de amonio (9,97 g, 129 mmol) en metanol (50 ml) se agitó a temperatura ambiente durante la noche. Se añadió cianoborohidruro de sodio (1,08 g, 17,3 mmol) y se calentó la mezcla a 60 °C durante la noche. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se concentró hasta -15 ml, se extinguió con hidróxido de sodio 1 M y se extrajo 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar 4-(benciloxi)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina en bruto (2,84 g, 5,62 mmol, rendimiento del 65 %). CLEM (ESI) m/z 349,3 (M+1).
Etapa 3: 5-(2-Amino-4-hidroxi-3,3-dimetilbutil)-2-cloropiridin-3-ol

Se añadió tribromuro de boro (2,66 ml, 28,1 mmol) gota a gota a una disolución de 4-(benciloxi)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina (2,84 g, 5,62 mmol) en 1,2-dicloroetano (DCE) (100 ml) a 0 °C. Se permitió que la mezcla se calentara hasta temperatura ambiente y se agitó durante 4 horas. Precipitó un sólido oleoso en el fondo del matraz. Se retiraron los sólidos por raspado de las paredes del matraz y se agitó la mezcla vigorosamente a temperatura ambiente durante la noche. Un precipitado sólido oleoso recubrió el fondo del matraz. Se decantó la fase líquida y se vertió en hielo. Se suspendió el precipitado en el matraz de reacción original en 1,2-dicloroetano (DCE) nuevo (100 ml) y se agitó mientras que se añadía 1 ml (~2 eq.) de tribromuro de boro nuevo gota a gota a temperatura ambiente. Se calentó la mezcla a 70 °C durante 4 horas y entonces se permitió que se enfriara hasta temperatura ambiente. Se enfrió la mezcla con un baño de hielo, se extinguió con adición cuidadosa lenta de 100 ml de metanol, se calentó hasta temperatura ambiente y se agitó durante 1 hora. Se concentró la mezcla y se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 /acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 20 %). Se liofilizaron las fracciones para dar 5-(2-amino-4-hidroxi-3,3-dimetilbutil)-2-cloropiridin-3-ol (910 mg, 3,72 mmol, rendimiento del 66,2 %) como un polvo blanco. CLEM (ESI) m/z 245,2 (M+1). 1H-RMN (400 MHz, DMSO-ds) 5 ppm 7,80 (d, J=1,95 Hz, 1 H), 7,59 (s. a., 3 H), 7,25 (d, J=1,95 Hz, 1 H), 3,31 - 3,38 (m, 2 H), 3,19 -3,29 (m, 1 H), 2,92 - 3,02 (m, 1 H), 2,53 - 2,63 (m, 1 H), 0,95 (s, 3 H), 0,90 (s, 3 H).
Etapa 4: 5-(2-Amino-4-hidroxi-3,3-dimetilbutil)-2-cloro-6-yodopiridin-3-ol

Se añadió yodo (233 mg, 0,919 mmol) a una mezcla en agitación de 5-(2-amino-4-hidroxi-3,3-dimetilbutil)-2-cloropiridin-3-ol (225 mg, 0,919 mmol) y carbonato de potasio (381 mg, 2,76 mmol) en agua (10 ml). Se agitó la mezcla a temperatura ambiente durante 2 horas. Se añadió sulfito de sodio sólido en porciones a la mezcla hasta que se disipó el color. Se inyectó la mezcla acuosa sobre una columna C18 de fase inversa a presión media y luego se eluyó con acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 50 %. Se liofilizaron las fracciones combinadas para dar 5-(2-amino-4-hidroxi-3,3-dimetilbutil)-2-cloro-6-yodopiridin-3-ol (164 mg, 0,443 mmol, rendimiento del 48,1 %) como un sólido blanco. CLEM (ESI) m/z 371,0 (M+1). 1H-RMN (400 MHz, DMSO-ak) " ppm 8,23 (s, 1 H), 7,20 (s, 1 H), 3,13 - 3,41 (m, 2 H), 2,70 - 2,88 (m, 2 H), 2,29 - 2,39 (m, 1 H), 0,92 (s, 3 H), 0,84 (s, 3 H).
Etapa 5: 1-(4-Acetoxi-1-(6-cloro-5-hidroxi-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Se agitaron 5-(2-amino-4-hidroxi-3,3-dimetilbutil)-2-cloro-6-yodopiridin-3-ol (164 mg, 0,443 mmol) y 4-oxo-4H-piran-3-carboxilato de etilo (83 mg, 0,494 mmol) en ácido acético (4 ml) a 100 °C durante 4 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se concentró. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se concentraron 2 conjuntos de fracciones por separado para dar 1-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-4-hidroxi-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (83 mg, 0,16 mmol, rendimiento del 36 %) y 1 -(4-acetoxi-1 -(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (27 mg, 0,048 mmol, rendimiento del 11 %). CLEM (ESI) m/z 563,2 (M+1).
Etapa 6: 1-(4-Acetoxi-1-(6-cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Se agitaron 1 -(4-acetoxi-1 -(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (27 mg, 0,048 mmol), carbonato de potasio (26,5 mg, 0,192 mmol) y 1-bromo-3-metoxipropano (19 mg, 0,124 mmol) a temperatura ambiente durante la noche. Se extinguió la mezcla con agua y se extrajo 2 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se lavaron con cloruro de litio al 5 % (ac.) y se concentraron. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar 1-(4-acetoxi-1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (15 mg, 0,024 mmol, rendimiento del 49,2 %) como un polvo blanco. CLEM (ESI) m/z 635,9 (M+1).
Etapa 7: Ácido 2-cloro-6-(1 -hidroxi-2-metilpropan-2-il)-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-hiri,71naftiridin-9-carboxílico

Un matraz de fondo redondo que contenía una barra agitadora, 1-(4-acetoxi-1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (14 mg, 0,022 mmol), acetato de potasio (4,33 mg, 0,044 mmol) y bromuro de paladio (II) (1,2 mg, 4,4 pmol) se purgó con nitrógeno durante 15 minutos. Se purgó N,N-dimetilacetamida (DMA) (1 ml) con nitrógeno durante 5 minutos antes de añadirse al recipiente de reacción. Se colocó el recipiente de reacción en un baño de aceite que se precalentó hasta 90 °C y se agitó la mezcla durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se filtró a través de celite y se purificó el filtrado mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se combinaron las fracciones y se basificaron con hidróxido de sodio 1 M. Se calentó la mezcla a 60 °C durante la noche, se enfrió hasta temperatura ambiente y se concentró. Se disolvió el residuo en agua y se acidificó a pH = 3-4 con ácido clorhídrico 1 M. Se purificó la mezcla mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se liofilizaron las fracciones para dar ácido 2-cloro-6-(1-hidroxi-2-metilpropan-2-il)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (3 mg, 6,9 pmol, rendimiento del 31 %). CLEM (ESI) m/z 437,2 (M+1).
Esquema general 6 para la preparación de compuestos de los ejemplos
11-25

Ejemplo 11 (Compuesto 230)
Ácido______ (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-h1H,71naftiridin-9-carboxílico

Etapa 1: (R)-N-((S)-1-(6-Cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida

Una disolución de 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona (2,54 g, 10,50 mmol), (R)-2-metilpropano-2-sulfinamida (2,55 g, 2 1 ,00 mmol) y Ti(OEt)4 (5,99 g, 26,3 mmol) en tolueno (8,5 ml) se agitó durante 10 min a 60 °C y entonces se evacuó el recipiente. Se agitó la mezcla de reacción a 60 °C a vacío durante 24 h. Volvió a presurizarse el recipiente con nitrógeno y se añadieron tolueno (5 ml) y THF (35 ml). Se añadió lentamente una disolución de LiBH4 (15,8 ml, 2 M en THF, 31,5 mmol). Se agitó la mezcla de reacción a TA durante la noche. Se diluyó la mezcla de reacción con THF (130 ml) y salmuera (3 ml), se agitó durante 30 min y luego se filtró a través de celite. Se evaporó el filtrado y se purificó mediante cromatografía en gel de sílice (EtOAc al 0-100 %/hexanos) para proporcionar (R)-N-((S)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida (2,58 g, 71 %) como diastereómero principal. CLEM (m/z, ES+) = 346,9, 348,1 (M+1).
Etapa 2: (S)-5-(2-Amino-3,3-dimetilbutil)-2-cloropiridin-3-ol

Una disolución de (R)-N-((S)-1 -(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida (2,56 g, 7,38 mmol) en 1 ,2-dicloroetano (36,9 ml) se agitó a 0 °C. Se añadió tribromuro de boro (4,88 ml, 51,7 mmol) lentamente. Se retiró la mezcla de reacción del baño de enfriamiento y se agitó durante la noche a TA. Se enfrió la disolución hasta 0 °C y se extinguió mediante la adición cuidadosa de MeOH. Se evaporó la suspensión resultante. Se añadió EtOAc y se recogió el sólido mediante filtración, se lavó con EtOAc y se secó para proporcionar (S)-5-(2-amino-3,3-dimetilbutil)-2-cloropiridin-3-ol, como sal de bis HBr. (2,89 g, cuant.) CLEM (m/z, ES+) = 229,1,231,1 (M+1).
Etapa 3: (S)-(1-(6-Cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Se añadió trietilamina (0,95 ml, 6,85 mmol) a una suspensión agitada de (S)-5-(2-amino-3,3-dimetilbutil)-2-cloropiridin-3-ol. 2HBr (1,36 g, 3,42 mmol) y anhídrido Boc (0,87 ml, 3,73 mmol) en THF (34,2 ml) y se agitó la mezcla de reacción a 60 °C durante 1 h y luego se evaporó hasta sequedad. Se suspendió el sólido en dietil éter, se aisló mediante filtración y luego se repartió entre EtOAc y agua. Se extrajo la capa acuosa con CH2Cl2 (4x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-(1-(6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (cuant. supuesto). CLEM (m/z, ES+) = 329,6, 331,2 (M+1).
Etapa 4: (S)-(1-(6-Cloro-5-hidroxi-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Se añadió yodo (0,87 g, 3,42 mmol) a una disolución agitada de (S)-(1-(6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2 -il)carbamato de terc-butilo (1,13 g, 3,42 mmol) y K2CO3 (1,42 g, 10,26 mmol) en agua (8 ,6 ml) y 1 ,4-dioxano (8 ,6 ml). Se agitó la mezcla de reacción durante la noche a ta. Se añadieron más yodo (0 , 20 g, 0,79 mmol) y K2CO3 (0,40 g, 7,23 mmol) y se calentó la mezcla de reacción a 40 °C durante 5 h. Se añadió Na2SO3 sólido mientras se agitaba hasta que la disolución ya no era de color marrón oscuro. Se diluyó la disolución con salmuera y EtOAc. Se extrajo la fase acuosa con EtOAc. Se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo como una espuma amarilla (1,56 g, cuant.). CLEM (m/z, ES+) = 455,1,457,1 (M+1).
Etapa 5: (SM1-(6-Cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Una disolución de (S)-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,83 g, 1,84 mmol), K2CO3 (0,76 g, 5,51 mmol) y 1-bromo-3-metoxipropano (0,56 g, 3,67 mmol) en DMF (12,2 ml) se calentó a 80 °C durante 3 h. Se evaporó la mezcla de reacción hasta sequedad y se llevó el residuo a CH2Cl2 y H2O. Se extrajo la fase acuosa CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-(1 -(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,97 g, cuant.) como un sólido blanquecino. CLEM (m/z, ES+) = 527,2, 529,2 (M+1).
Etapa______ 6 _______(S)-1 -(1 -(6-Cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Se añadió cloruro de hidrógeno 4 M en dioxano (6,89 ml, 27,5 mmol) a una disolución de (S)-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (967 mg, 1,84 mmol) en CH2Cl2 (7 ml). Se agitó la mezcla de reacción a ta durante 3 h y se evaporó hasta sequedad. Se disolvió el sólido en CH2Cl2 (1 ml) y Et3N (1 ml) y luego se evaporó hasta sequedad. Se llevó el residuo a NaHCO3 saturado y CH2Cl2. Se extrajo la fase acuosa con CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-amina. Una disolución de la amina anterior y 4-oxo-4H-piran-3-carboxilato de etilo (340 mg, 2,02 mmol) en ácido acético (18,4 ml) se agitó a 100 °C durante 7 h. Se evaporó la mezcla de reacción hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 10-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar (S)-1-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (593 mg, 56 %) como un sólido de color tostado. CLEM (m/z, ES+) = 577,7, 579,2 (M+1).
Etapa 7: (S)-6-(terc-Butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-hir1,71naftiridin-9-carboxilato de etilo

Un matraz que contenía (S)-1-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (0,59 g, 1,03 mmol), acetato de potasio (0,20 g, 2,06 mmol) y bromuro de paladio (II) (0,055 g, 0,21 mmol) se purgó con nitrógeno. Se añadió N,N-dimetilacetamida (DMA) desgasificada (10,3 ml) y se calentó la mezcla de reacción a 90 °C durante 24 h. Se eliminó el disolvente mediante evaporación y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (326 mg, 71 %) como un sólido blanquecino. 1H-RMN (400 MHz, CDCh) 5 ppm 8,18 (s, 1 H), 7,49 (s, 1 H), 7,02 (s, 1 H), 4,40 (q, J=7,0 Hz, 2 H), 4,13 - 4,26 (m, 2 H), 3,93 (d, J=6,6 Hz, 1 H), 3,62 (m, 2 H), 3,42 - 3,51 (m, 1 H), 3,38 (s, 3 H), 3,17 - 3,25 (m, 1 H), 2,15 (quin, J=6,0 Hz, 2 H), 1,41 (t, J=7,2 Hz, 3 H), 0,86 (s, 9 H); CLEM (m/z, ES+) = 449,3, 451,3 (M+1).
Etapa 8: Ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-hir1,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (42 mg, 0,094 mmol) en LiOH 1 M (1,7 ml) y MeOH (1,7 ml) se agitó a 50 °C durante 1,5 h. Se añadió ácido cítrico 1 M (2 ml) y se agitó la mezcla de reacción durante varios min. Se recogió el sólido blanco mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (35,5 mg, 90 %) como un sólido blanco. 1H-RMN (400 MHz, DMSO-a6) " ppm 16,31 (s, 1 H), 8,81 (s, 1 H), 7,75 (s, 1 H), 7,30 (s, 1 H), 4,68 (d, J=6,3 Hz, 1 H), 4,19 - 4,32 (m, 2 H), 3,40 - 3,60 (m, 4 H), 3,26 (s, 3 H), 2,04 (quin, J=6,2 Hz, 2 H), 0,75 (s, 9 H); CLEM (m/z, ES+) = 421,3, 423,2 (M+1).
Ejemplo 12 (compuesto 231)
Ácido_____ (S)-6-(terc-butil)-3-(ciclopropMmetoxi)-2-metil-10-oxo-6,10-dihidro-5H-piridoH,2-hiri ,71naftiridin-9-carboxílico

Etapa 1: (S)-(1-(6-Cloro-5-(ciclopropilmetoxi)-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Una disolución de (S)-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,73 g, 1,60 mmol), K2CO3 (0 , 66 g, 4,78 mmol) y (bromometil)ciclopropano (0,43 g, 3,19 mmol) en DMF (10,6 ml) se calentó a 80 °C durante 3 h. Se evaporó la mezcla de reacción hasta sequedad y se llevó el residuo a CH2Cl2 y H2O. Se extrajo la fase acuosa con CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,81 g, cuant.) como un sólido blanquecino. CLEM (m/z, ES+) = 508,8, 511,1 (M+1).
Etapa______ 2 ______ (S)-1 -(1 -(6-Cloro-5-(ciclopropilmetoxi)-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Se añadió cloruro de hidrógeno 4 M en dioxano (5,98 ml, 23,9 mmol) a una disolución de (S)-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (812 mg, 1,60 mmol) en CH2Cl2 (6 ml). Se agitó la mezcla de reacción a ta durante 3 h y se evaporó hasta sequedad. Se disolvió el sólido en CH2Cl2 (1 ml) y Et3N (1 ml) y luego se evaporó hasta sequedad. Se llevó el residuo a NaHCO3 saturado y CH2Cl2. Se extrajo la fase acuosa con CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-1-(6-cloro-2-yodo-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-amina. CLEM (m/z, ES+) = 409,1,411,1 (M+1).
Una disolución de la amina anterior y 4-oxo-4H-piran-3-carboxilato de etilo (295 mg, 1,76 mmol) en ácido acético (16,0 ml) se agitó a 100 °C durante 7 h. Se evaporó la mezcla de reacción hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2Ü (ácido fórmico al 0,1 %)) para proporcionar (S)-1-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (453 mg, 51 %) como un sólido de color tostado. CLEM (m/z, ES+) = 559,4, 561,1 (M+1).
Etapa 3: (S)-6-(terc-Butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-hiri,71naftiridin-9-carboxilato de etilo

Un matraz que contenía (S)-1-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (0,45 g, 0,81 mmol), acetato de potasio (0,16 g, 1,62 mmol) y bromuro de paladio (II) (0,043 g, 0,16 mmol) se purgó con nitrógeno. Se añadió N,N-dimetilacetamida (DMA) desgasificada (8,1 ml) y se calentó la mezcla de reacción a 90 °C durante 24 h. Se eliminó el disolvente mediante evaporación y se llevó el residuo a CH2Cl2 y agua y se filtró a través de celite. Se evaporó la fase orgánica hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 10-100 % de CHaCN/H2Ü (ácido fórmico al 0,1 %)) para proporcionar (S)-6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (216 mg, 62 %) como un sólido blanquecino. CLEM (m/z, ES+) = 431,2, 433,2 (M+1); >97 % de e.e. mediante HPLC quiral.
Etapa 4: Ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metil-10-oxo-6,10-dihidro-5H-piridoH,2-hiri,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (52,7 mg, 0,12 mmol), Pd(PPh3)4 (28 mg, 0,024 mmol), carbonato de potasio (34 mg, 0,25 mmol) y trimetilboroxina (46 mg, 0,37 mmol) en 1,4-dioxano (0,61 ml) se calentó a 100 °C durante la noche. Se diluyó la mezcla de reacción con CH2Cl2 y se filtró a través de celite. Se evaporó el filtrado y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2Ü (ácido fórmico al 0,1 %)) para proporcionar (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (36,5 mg, 73 %).
1H-RMN (400 MHz, CDCh) 5 ppm 8,18 (s, 1 H), 7,56 (s, 1 H), 6,80 (s, 1 H), 4,40 (q, J=7,0 Hz, 2 H), 3,87 (s, 3 H), 3,39 - 3,48 (m, 1 H), 3,10 - 3,18 (m, 1 H), 2,50 (s, 3 H), 1,41 (t, J=7,0 Hz, 3 H), 0,69 (m, 2 H), 0,40 (m, 2 H); CLEM (m/z, ES+) = 411,4 (M+1).
Una disolución del éster anterior en LiOH 1 M (0,9 ml) y MeOH (0,9 ml) se calentó a 50 °C durante 1,5 h. Se añadió ácido cítrico 1 M (1,2 ml) y se agitó la mezcla de reacción durante 15 min. Se recogió el sólido mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (27,7 mg, 59 % durante 2 etapas) como un sólido blanco. 1H-RMN (400 MHz, DMSO-ds) 5 ppm 16,53 (s. a., 1 H), 8,77 (s, 1 H), 7,44 (s., 1 H), 7,42 (s, 1 H), 4,58 - 4,69 (m, 1 H), 3,98 (m, 2 H), 3,46 - 3,55 (m, 2 H), 2,43 (s, 3H), 1,22 - 1,36 (m, 1 H), 0,73 (s, 9 H), 0,65-0,55 (m, 2 H), 0,33 - 0,44 (m, 2 H); CLEM (m/z, ES+) = 383,2 (M+1).
Ejemplo 13 (compuesto 232)
Ácido______ (S)-6-(terc-butM)-3-(3-metoxipropoxi)-2-metil-10-oxo-6,10-dihidro-5H-piridoH,2-h1H,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (47,7 mg, 0,11 mmol), Pd(PPh3)4 (25 mg, 0,021 mmol), carbonato de potasio (29 mg, 0,21 mmol) y trimetilboroxina (40 mg, 0,32 mmol) en 1,4-dioxano (0,53 ml) se calentó a 100 °C durante la noche. Se diluyó la mezcla de reacción con CH2Cl2 y se filtró a través de celite. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-3-(3-metoxipropoxi)-2-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (41,3 mg, 91 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 8,17 (s, 1 H), 7,27 (s, 1 H), 6,86 (s, 1 H), 4,39 (q, J=7,0 Hz, 2 H), 4,05 - 4,17 (m, 2 H), 3,91 (d, J=6,6 Hz, 1 H), 3,63-3,55 (m, 2 H), 3,49-3,41 (m, 1 H), 3,38 (s, 3 H), 3,15 (d, J=16,8 Hz, 1 H), 2,46 (s, 3 H), 2,12 (quin, J=6,1 Hz, 2 H), 1,40 (t, J=7,0 Hz, 3 H), 0,84 (s, 9 H); CLEM (m/z, ES+) = 429,4 (M+1).
Una disolución del éster anterior en LiOH 1 M (1 ml) y MeOH (1 ml) se calentó a 50 °C durante 1,5 h. Se añadió ácido cítrico 1 M (1,2 ml) y se agitó la mezcla de reacción durante 15 min. Se recogió el sólido mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-3-(3-metoxipropoxi)-2-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (27,6 mg, 65 % durante 2 etapas) como un sólido blanco. 1H-RMN (400 MHz, DMSO-ds) 5 ppm 16,53 (s, 1 H), 8,77 (s, 1 H), 7,46 (s, 1 H), 7,44 (s, 1 H), 4,64 (d, J=6,3 Hz, 1 H), 4,09 - 4,23 (m, 2 H), 3,47 - 3,56 (m, 3 H), 3,38-3,32 (m, 1H), 3,26 (s, 3 H), 2,42 (s, 3 H), 2,02 (quin, J=6,2 Hz, 2 H), 0,74 (s, 9 H); CLEM (m/z, ES+) = 401,2 (M+1).
Ejemplo 14 (compuesto 233)
Ácido (S)-6-(terc-butil)-2-ciclopropil-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-h1T1,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (53 mg, 0,12 mmol), Pd(PPh3)4 (28 mg, 0,025 mmol), carbonato de potasio (51 mg, 0,37 mmol) y ácido ciclopropilborónico (21 mg, 0,25 mmol) en 1,4-dioxano (1,2 ml) se calentó a 100 °C durante la noche. Se diluyó la mezcla de reacción con CH2Cl2 y se filtró a través de celite. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-2-ciclopropil-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (53,3 mg, 99 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 8,14 (s, 1 H), 7,42 (s, 1 H), 6,77 (s, 1 H), 4,38 (q, J=7,3 Hz, 2 H), 3,88 (m, J=6,8, 2,1 Hz, 2 H), 3,43 (dd, J=16,8, 7,0 Hz, 1 H), 3,11 (d, J=16,8 Hz, 1 H), 2,56 - 2,47 (m, 1 H), 1,39 (t, J=7,0 Hz, 3 H), 1,35 - 1,29 (m, 1 H), 1,22 - 1,29 (m, 1 H), 1,04 - 1,19 (m, 2 H), 0,93 - 1,01 (m, 2 H), 0,82 (s, 9 H), 0,63 - 0,73 (m, 2 H), 0,44 - 0,38 (m, 2 H); CLEM (m/z, ES+) = 437,4 (M+1).
Una disolución del éster anterior en LiOH 1 M (1,2 ml) y MeOH (1,2 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (1,5 ml) y se agitó la mezcla de reacción durante 15 min. Se recogió el sólido mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-2-ciclopropil-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (38,8 mg, 77 % durante 2 etapas) como un sólido blanco. 1H-RMN (400 MHz, DMSO-afe) 5 ppm 16,53 (s, 1 H), 8,76 (s, 1 H), 7,38 (s, 1 H), 7,35 (s, 1 H), 4,62 (d, J=6,3 Hz, 1 H), 4,05 -3,94 (m, 2 H), 3,42 - 3,55 (m, 1 H), 3,30 - 3,28 (m, 1H), 1,26 - 1,44 (m, 1 H), 0,93 - 1,06 (m, 4 H), 0,72 (s, 9 H), 0,65 -0,59 (m, 2 H), 0,43 - 0,5 (m, 2 H); CLEM (m/z, ES+) = 409,2 (M+1).
Ejemplo 15 (compuesto 234)
Ácido (S)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-h1H ,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (46 mg, 0,10 mmol), Pd(PPh3)4, (24 mg, 0,020 mmol), carbonato de potasio (42 mg, 0,31 mmol) y ácido ciclopropilborónico (18 mg, 0 ,21 mmol) en 1,4-dioxano (1,0 ml) se calentó a 100 °C durante la noche. Se diluyó la mezcla de reacción con CH2Cl2 y se filtró a través de celite. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (43,6 mg, 94 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 8,16 (s, 1 H), 7,45 (s, 1 H), 6,83 (s, 1 H), 4,39 (q, J=7,0 Hz, 2 H), 4,13 (m, 2 H), 3,89 (d, J=6 ,6 Hz, 1 H), 3,62 (m, 2 H), 3,39 - 3,46 (m, 1 H), 3,38 (s, 3 H), 3,13 (d, J=16,8 Hz, 1 H), 2,40 - 2,50 (m, 1 H), 2,15 (quin, J=6,1 Hz, 2 H), 1,40 (t, J=7,0 Hz, 3 H), 1,14 - 1,22 (m, 1 H), 1,04 - 1,12 (m, 1 H), 0,93 - 1,02 (m, 2 H), 0,83 (s, 9 H); CLEM (m/z, ES+) = 455,5 (M+1).
Una disolución del éster anterior en LiOH 1 M (1,2 ml) y MeOH (1,2 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (1,5 ml) y se agitó la mezcla de reacción durante 15 min. Se recogió el sólido mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (27,2 mg, 62 %) como un sólido blanco. 1H-RMN (400 MHz, DMSO-ds) 5 ppm 16,53 (s, 1 H), 8,76 (s, 1 H), 7,43 (s, 1 H), 7,35 (s, 1 H), 4,62 (d, J=6,3 Hz, 1 H), 4,11 - 4,24 (m, 2 H), 3,42 -3,57 (m, 4 H), 3,26 (s, 3 H), 2,41 - 2,46 (m, 1 H), 2,05 (quin, J=6,2 Hz, 2 H), 0,94 - 1,06 (m, 4 H), 0,73 (s, 9 H); CLEM (m/z, ES+) = 427,2 (M+1).
Ejemplo 16 (compuesto 235)
Ácido (R)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-6,10-dihidro-5H-piridoH ,2-h1T1,71naftiridin-9-carboxílico

Etapa 1: (S)-N-((R)-1-(6-Cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida y (S)-N-((S)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida

Una disolución de 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona (2,56 g, 10,57 mmol), (S)-2-metilpropano-2-sulfinamida (2,56 g, 21,14 mmol), Ti(OEt)4 (6,03 g, 26,4 mmol) en tolueno (8,5 ml) se agitó durante 10 min a 60 °C y entonces se evacuó el recipiente. Se agitó la mezcla de reacción a 60 °C a vacío durante 24 h. Volvió a presurizarse el recipiente con nitrógeno, se añadieron tolueno (5 ml) y Ti(OEt)4 (2,4 g, 10,5 mmol) y se agitó la mezcla de reacción a vacío durante 4 h. Volvió a presurizarse el recipiente con nitrógeno, se añadió tolueno (5 ml) y se agitó la mezcla de reacción a vacío durante otras 3 h. Entonces se llevó la mezcla de reacción espesa a THF (70,5 ml), se añadió Ti(OEt)4 adicional (2,4 g, 10,5 mmol) y se enfrió la disolución hasta -30 °C. Se añadió NaBH4 (1,20 g, 31,7 mmol) en porciones. Entonces se permitió que la mezcla de reacción se calentara lentamente hasta ta durante la noche. Se diluyó la mezcla de reacción con THF (130 ml) y salmuera (3 ml), se agitó durante 30 min y luego se filtró a través de celite. Se evaporó el filtrado y se purificó mediante cromatografía en gel de sílice para proporcionar los dos diastereómeros.
El diastereómero principal es un sólido blanco: (S)-N-((R)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida (1,8 g, 50 %). 1H-RMN (400 MHz, CDCh) 5 ppm 7,81 (d, J=2,0 Hz, 1 H), 7,40 (d, J=2,0 Hz, 1 H), 3,97 (s, 3 H), 3,26 (m, 1 H), 3,06 - 3,15 (m, 2 H), 2,72 (dd, J=14,8, 8,2 Hz, 1 H), 1,17 (s, 9 H), 0,99 (s, 9 H); CLEM
(m/z, ES+) = 346,8, 348,5 (M+1).
El diastereómero minoritario es un aceite transparente: (S)-N-((S)-1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida (1,2 g, 33 %). 1H-RMN (400 MHz, CDCh) " ppm 7,77 (d, J=1,6 Hz, 1 H), 7,07 (d, J=1,6 Hz, 1 H), 3,93 (s, 3 H), 3,19 - 3,33 (m, 2 H), 3,02 (dd, J=14,1,2,7 Hz, 1 H), 2,55 (dd, J=14,1, 10,5 Hz, 1 H), 1,08 (s, 9 H), 0,98 (s, 9 H); CLEM (m/z, ES+) = 346,8, 348,0 (M+1).
Etapa 2: (R)-5-(2-Amino-3,3-dimetilbutil)-2-cloropiridin-3-ol

Una disolución de (S)-N-((R)-1 -(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-il)-2-metilpropano-2-sulfinamida (1,82 g, 5,26 mmol) en 1,2-dicloroetano (26,3 ml) se agitó a 0 °C. Se añadió tribromuro de boro (3,48 ml, 36,8 mmol) lentamente. Se retiró la mezcla de reacción del baño de enfriamiento y se agitó durante la noche a TA. Se enfrió la disolución hasta 0 °C y se extinguió mediante la adición cuidadosa de MeOH. Se evaporó la suspensión resultante. Se añadió EtOAc y se recogió el sólido mediante filtración, se lavó con EtOAc y se secó para proporcionar (S)-5-(2-amino-3,3-dimetilbutil)-2-cloropiridin-3-ol, como sal de bis HBr. (2,1 g, cuant.) CLEM (m/z, ES+) = 229,2, 231,2 (M+1).
Etapa 3: (R)-(1-(6-Cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Una suspensión agitada de (R)-5-(2-amino-3,3-dimetilbutil)-2-cloropiridin-3-ol.2HBr (1,0 g, 2,51 mmol) y anhídrido Boc (0,80 g, 3,67 mmol) en THF (25,1 ml) se agitó a 60 °C durante 1,5 h. Se añadieron THF (10 ml) y trietilamina (0,35 ml, 2,51 mmol) y se agitó la mezcla de reacción durante otros 30 min. Se añadió trietilamina adicional (0,35 ml, 2,51 mmol) y se agitó la mezcla de reacción a 60 °C durante 1 h. Entonces se evaporó la disolución hasta sequedad. Se suspendió el sólido en dietil éter, se aisló mediante filtración y se lavó con dietil éter adicional. Se llevó el sólido a EtOAc y se lavó con NaHCO3 acuoso diluido y salmuera. Se secó la fase orgánica (Na2SO4), se filtró y se evaporó para proporcionar (R)-(1-(6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (1,01 g, 99 %) como un sólido amarillo claro. CLEM (m/z, ES+) = 329,2, 331,2 (M+1).
Etapa 4: (R)-(1-(6-Cloro-5-h¡drox¡-2-vodop¡r¡d¡n-3-il)-3,3-d¡met¡lbutan-2-¡l)carbamato de terc-butilo

Se añadió yodo (0,64 g, 2,51 mmol) a una disolución agitada de (R)-(1-(6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,83 g, 2,51 mmol) y K2CO3 (1,04 g, 7,53 mmol) en agua (6,3 ml) y 1,4-dioxano (6,3 ml). Se agitó la mezcla de reacción a TA durante 1,5 h.
Se añadió Na2SO3 sólido mientras se agitaba hasta que la disolución ya no era de color marrón oscuro. Se diluyó la disolución con salmuera y EtOAc. Se extrajo la fase acuosa con EtOAc. Se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (R)-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo como una espuma amarilla (0,38 g, 34 %). CLEM (m/z, ES+) = 455,4, 457,1 (M+1).
Etapa 5: (R)-(1-(6-Cloro-5-(ciclopropilmetoxi)-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Una disolución de (R)-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,3819 g, 0,840 mmol), K2CO3 (0,35 g, 2,52 mmol) y (bromometil)ciclopropano (0,23 g, 1,68 mmol) en DMF (5,6 ml) se calentó a 80 °C durante 3 h. Se evaporó la mezcla de reacción hasta sequedad y se llevó el residuo a CH2Cl2 y H2O. Se extrajo la fase acuosa CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (R)-(1 -(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (415,4 mg, 97 %) como un sólido blanquecino. CLEM (m/z, ES+) = 509,2, 511,2 (M+1).
Etapa______ 6:______ (R)-1-(1-(6-Cloro-5-(ciclopropilmetoxi)-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Se añadió cloruro de hidrógeno 4 M en dioxano (3,06 ml, 12,25 mmol) a una disolución de (R)-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (0,42 g, 0,82 mmol) en CH2Cl2 (3 ml). Se agitó la mezcla de reacción a ta durante 3 h y se evaporó hasta sequedad. Se disolvió el sólido en CH2Cl2 (1 ml) y Et3N (1 ml), se agitó y luego se evaporó hasta sequedad. Se llevó el residuo a NaHCO3 saturado y CH2Cl2. Se extrajo la fase acuosa con CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (R)-1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-amina. Una disolución de la amina anterior y 4-oxo-4H-piran-3-carboxilato de etilo (151 mg, 0,90 mmol) en ácido acético (8,2 ml) se agitó a 100 °C durante 7 h. Se evaporó la mezcla de reacción hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar (R)-1-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (389 mg, 85 %) como un sólido de color tostado claro. CLEM (m/z, ES-) = 575,2, 577,2 (M-1).
Etapa 7: (R)-6-(terc-Butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-piridor1,2-h1H,71naftiridin-9-carboxilato de etilo

Una matraz que contenía (R)-1-(1-(6-cloro-5-(ciclopropilmetoxi)-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (0,39 g, 0,70 mmol), acetato de potasio (0,14 g, 1,39 mmol) y bromuro de paladio (II) (0,037 g, 0,14 mmol) se purgó con nitrógeno. Se añadió N,N-dimetilacetamida (DMA) desgasificada (7,0 ml) y la mezcla de reacción se calentó a 90 °C durante 24 h. Se purificó la disolución mediante cromatografía de fase inversa (el 10-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar (R)-6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (167 mg, 56 %). 1H-RMN (400 MHz, CDCh) " ppm 8,18 (s, 1 H), 7,49 (s, 1 H), 6,96 (s, 1 H), 4,40 (q, J=7,3 Hz, 2 H), 3,96 (d, J=7,0 Hz, 2 H), 3,93 (d, J=6,6 Hz, 1 H), 3,46 (dd, J=17,0, 6,8 Hz, 1 H), 3,19 (d, J=16,8 Hz, 1 H), 1,41 (t, J=7,0 Hz, 3 H), 1,30 - 1,37 (m, 1 H), 0,85 (s, 9 H), 0,76 - 0,70 (m, 2 H), 0,41 - 0,48 (m, 2 H); CLEM (m/z, ES-) = 429,4, 431,4 (M-1).
Etapa 8: Ácido (R)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-6,10-dihidro-5H-piridoH,2-hin,71naft¡r¡d¡n-9-carboxíl¡co

Una disolución de (R)-6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9
carboxilato de etilo (33 mg, 0,077 mmol), NaB(OMe)4 (36 mg, 0,23 mmol), tBuXPhos (6,5 mg, 0,015 mmol) y Pd2(dba)3 (7,0 mg, 7,66 pmol) en DMF (0,77 ml) se calentó a 80 °C durante 2 h. Se filtró la mezcla de reacción a través de celite y se enjuagó la celite con EtOAc y CH2Cl2. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (R)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (27 mg, 83 %). 1H-RMN (400 MHz, CDCh) " ppm 8,17 (s, 1 H), 7,42 (s, 1 H), 6,80 (s, 1 H), 4,39 (q, J=7,0 Hz, 2 H), 4,05 (s, 3 H), 3,94 - 3,85 (m, 3 H), 3,42 (dd, J=16,6, 6,8 Hz, 1 H), 3,09 (d, J=16,8 Hz, 1 H), 1,37 - 1,40 (t, J=7,2 Hz, 4 H), 1,27 - 1,37 (m, 1 H), 0,85 (s, 9 H), 0,66 - 0,75 (m, 2 H), 0,42 - 0,37 (m, 2 H); CLEM (m/z, ES+) = 427,4 (M+1).
Una disolución del éster anterior en LiOH 1 M (1 ml) y MeOH (1 ml) se calentó a 50 °C durante 2 h. Se filtró la mezcla de reacción a través de un filtro Acrodisc para eliminar materiales particulados finos y se añadió ácido cítrico 1 M (1 ml) al filtrado transparente. Se recogió el precipitado mediante filtración, se lavó con agua y se secó para proporcionar ácido (R)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (19 mg, 62 % durante 2 etapas) como un sólido blanquecino. 1H-RMN (400 MHz, DMSO-ds) " ppm 16,53 (s, 1 H), 8,77 (s, 2 H), 7,40 (s, 2 H), 7,36 (s, 2 H), 4,62 (d, J=6,8 Hz, 1 H), 3,97 (s, 3 H), 3,96 - 3,89 (m, 2 H), 3,49 - 3,40 (m, 1 H), 1,22 - 1,32 (m, 1 H), 0,75 (s, 9 H), 0,57 - 0,65 (m, 2 H), 0,31 - 0,40 (m, 2 H); CLEM (m/z, ES+) = 399,2 (M+1).
Ejemplo 17 (compuesto 236)
Ácido (S)-6-(terc-butil)-3-(ciclopropMmetoxi)-2-metoxi-10-oxo-6,10-dihidro-5H-piridoH,2-hiri,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (52 mg, 0,12 mmol), NaB(OMe)4 (57 mg, 0,36 mmol), tBuXPhos (10,3 mg, 0,024 mmol) y Pd2(dba)3 (11,0 mg, 0,012 mmol) en DMF (1,2 ml) se calentó a 80 °C durante 2 h. Se filtró la mezcla de reacción a través de celite y se enjuagó la celite con EtOAc y CH2Cl2. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (32 mg, 62 %). 1H-RMN (400 MHz, CDCh) " ppm 8,17 (s, 1 H), 7,44 (s, 1 H), 6,80 (s, 1 H), 4,40 (q, J=7,2 Hz, 2 H), 4,06 (s, 3 H), 3,89 (m, 3 H), 3,42 (dd, J=16,8, 7,0 Hz, 1 H), 3,09 (d, J=16,4 Hz, 1 H), 1,40 (t, J=7,0 Hz, 3 H), 1,31 - 1,37 (m, 1 H), 0,84 (s, 9 H), 0,68 - 0,75 (m, 2 H), 0,36 - 0,43 (m, 2 H); CLEM (m/z, ES+) = 427,4 (M+1).
Una disolución del éster anterior en LiOH 1 M (0,75 ml) y MeOH (0,75 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (0,75 ml) a la mezcla de reacción y se recogió el precipitado mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (26,5 mg, 55 % durante 2 etapas) como un sólido blanco. 1H-RMN (400 MHz, CDCh) " ppm 16,11 (s, 1 H), 8,45 (s, 1 H), 7,65 (s, 1 H), 6,83 (s, 1 H), 4,08 (s, 3 H), 4,04 (d, J=6,8 Hz, 1 H), 3,92 (m, 2 H), 3,48 (dd, J=17,1,7,3 Hz, 1 H), 3,16 (d, J=17,1 Hz, 1 H), 1,31 - 1,43 (m, 1 H), 0,86 (s, 9 H), 0,69 - 0,78 (m, 2 H), 0,41 (m, 2 H); CLEM (m/z, ES+) = 399,3 (M+1).
Ejemplo 18 (compuesto 237)
Ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-hidroxi-10-oxo-6,10-dihidro-5H-piridoH,2-hir1,71naftiridin-9-carboxílico

Se formó el producto desmetilado como un subproducto de la reacción que produjo (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-metoxi-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo y se recogió como fracciones mixtas que se combinaron. La repurificación mediante cromatografía de fase inversa (el 10-100 % de
CHaCN/H2O (ácido fórmico al 0,1 %)) proporcionó (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-hidroxi-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (8,3 mg, 17 %). 1H-RMN (400 MHz, CDCl3) 5 ppm 8,18 (s, 1 H), 6,85 (s, 1 H), 6,55 (s, 1 H), 4,37 (q, J=7,0 Hz, 2 H), 3,83 - 3,97 (m, 3 H), 3,33 (dd, J=16,8, 6,6 Hz, 1 H), 2,95 (d, J=17,2 Hz, 1 H), 1,33 - 1,42 (m, 4 H), 0,65 - 0,74 (m, 2 H), 0,40 (m, 2 H); CLEM (m/z, ES+) = 413,3 (M+1).
Una disolución del éster anterior en LiOH 1 M (0,5 ml) y MeOH (0,5 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (0,5 ml) a la mezcla de reacción y se purificó la disolución mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar ácido (S)-6-(terc-butil)-3-(ciclopropilmetoxi)-2-hidroxi-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (6,4 mg, 14 % durante 2 etapas) como un sólido amarillo. 1H-RMN (400 MHz, CDCh) 5 ppm 15,72 (s. a., 1 H), 8,44 (s, 1 H), 7,38 (s, 1 H), 6,66 (s, 1 H), 4,04 (d, J=4,9 Hz, 1 H), 3,96 (d, J=6,8 Hz, 2 H), 3,40 - 3,53 (m, 1 H), 3,04 (d, J=17,1 Hz, 1 H), 1,34 - 1,45 (m, 1 H), 0,89 (s, 9 H), 0,77 - 0,72 (m, 2 H), 0,47 - 0,42 (m, 2 H); CLEM (m/z, ES+) = 385,2 (M+1).
Ejemplo 19 (compuesto 238)
Ácido (S)-6-(terc-butil)-2-metoxi-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-hiri,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (46,8 mg, 0,10 mmol), NaB(OMe)4 (49 mg, 0,31 mmol), tBuXPhos (8,9 mg, 0,021 mmol) y Pd2(dba)3 (9,6 mg, 0,010 mmol) en DMF (1,0 ml) se calentó a 80 °C durante 2 h. Se filtró la mezcla de reacción a través de celite y se enjuagó la celite con CH2Cl2. Se evaporó el filtrado y volvió a someterse el residuo a las condiciones de reacción con más reactivos: NaB(OMe)4 (49 mg, 0,31 mmol), tBuXPhos (8,9 mg, 0,021 mmol) y Pd2(dba)3 (9,6 mg, 0,010 mmol) en DMF (1,0 ml) a 80 °C durante otras 3 h. Se filtró la mezcla de reacción a través de celite y se enjuagó la celite con CH2Cl2. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-2-metoxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo como un sólido de color tostado. 1H-RMN (400 MHz, CDCh) 5 ppm 8,18 (s, 1 H), 7,44 (s, 1 H), 6,86 (s, 1 H), 4,40 (q, J=7,3 Hz, 2 H), 4,19 - 4,12 (m, 2 H), 4,05 (s, 3 H), 3,90 (d, J=6,6 Hz, 1 H), 3,61 - 3,55 (m, 2 H), 3,42 (dd, J=16,8, 7,0 Hz, 1 H), 3,37 (s, 3 H), 3,11 (d, J=16,8 Hz, 1 H), 2,15 (quin, J=6,2 Hz, 2 H), 1,40 (t, J=7,0 Hz, 3 H), 0,86 (s, 9 H); CLEM (m/z, ES+) = 445,0 (M+1).
Una disolución del éster anterior en LiOH 1 M (0,75 ml) y MeOH (0,75 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (0,75 ml) a la mezcla de reacción y se purificó la disolución mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar ácido (S)-6-(terc-butil)-2-metoxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (19,6 mg, 45 % durante 2 etapas) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 16,12 (s, 1 H), 8,46 (s, 1 H), 7,65 (s, 1 H), 6,90 (s, 1 H), 4,24 - 4,13 (m, 2 H), 4,06 (s, 1 H), 3,63 - 3,55 (m, 2 H), 3,49 (dd, J=16,6, 6,3 Hz, 1 H), 3,38 (s, 3 H), 3,18 (d, J=17,1 Hz, 1 H), 2,16 (quin, J=6,1 Hz, 2 H), 0,87 (s, 9 H); CLEM (m/z, ES+) = 417,2 (M+1).
Ejemplo 20 (compuesto 239)
Ácido (S)-6-(terc-butil)-2-hidroxi-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-h1HJ1naftiridin-9-carboxílico

Se formó el producto desmetilado como un subproducto de la reacción que produjo (S)-6-(terc-butil)-2-metoxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo y se recogió como fracciones mixtas que se combinaron y se sometieron a condiciones de hidrólisis sin purificación adicional. Una disolución del éster en LiOH 1 M (0,5 ml) y MeOH (0,5 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (0,5 ml) a la
mezcla de reacción y se purificó la disolución mediante cromatografía de fase inversa (el 5-100 % de CH3CN/H2O (ácido fórmico al 0,1 %)) para proporcionar ácido (S)-6-(terc-butil)-2-hidroxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (3,4 mg, 8 % durante 2 etapas) como un sólido amarillo. 1H-RMN (400 MHz, CDCl3) 5 ppm 15,76 (s. a., 1 H), 8,42 (s, 1 H), 7,47 (s, 1 H), 6,70 (s, 1 H), 4,20 (t, J=6,1 Hz, 2 H), 4,04 (d, J=6,3 Hz, 1 H), 3,61 - 3,76 (m, 2 H), 3,48 (dd, J=17,1,6,8 Hz, 1 H), 3,41 (s, 3 H), 3,04 (d, J=17,1 Hz, 1 H), 2,21 (quin, J=6,0 Hz, 2 H), 0,89 (s, 9 H); CLEM (m/z, ES+) = 403,2 (M+1).
Ejemplo 21 (compuesto 240)
Ácido______________ (S)-6-(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-6,10-dihidro-5H-piridoH,2-hiri,71naftiridin-9-carboxílico

Etapa______ 1 ______ (S)-6-(terc-Butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-6,10-dihidro-5H-piridoH,2-h lHJInaftirid in-9-carboxila to de etilo

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (88,4 mg, 0,20 mmol), isopropeniltrifluoroborato de potasio (58 mg, 0,39 mmol), carbonato de sodio (63 mg, 0,59 mmol) y Pd(PPh3)4 (23 mg, 0,020 mmol) en etanol (2,0 ml) se agitó a 80 °C durante la noche. Se diluyó la mezcla de reacción con CH2Cl2 y se filtró a través de celite. Se evaporó el filtrado y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (87,5 mg, 98 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 8 , 18 (s, 1 H), 7,54 (s, 1 H), 7,00 (s, 1 H), 5,91 (s, 1 H), 5,54 (s, 1 H), 4,39 (q, J=7,0 Hz, 2 H), 4,07 - 4,22 (m, 2 H), 3,93 (d, J=6 ,6 Hz, 1 H), 3,61 - 3,56 (m, 2 H), 3,48 (dd, J=17,0, 6 , 8 Hz, 1 H), 3,37 (s, 3 H), 3,19 (d, J=16,8 Hz, 1 H), 2,25 (s, 3 H), 2,13 (quin, J=6,1 Hz, 2 H), 1,40 (t, J=7,2 Hz, 3 H), 0,85 (s, 9 H); CLEM (m/z, ES+) = 455,9 (M+1).
,
Etapa 2: Ácido (S)-6-(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-6,10-dihidro-5H-piridoH,2-hin,71naft¡r¡d¡n-9-carboxíl¡co

Se aisló el compuesto del título como un producto secundario mediante hidrólisis promovida por base durante una reacción de oxidación de (S)-6-(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (30,3 mg, 0,067 mmol) con yoduro de trimetilsulfoxonio (31 mg, 0,14 mmol) y KOtBu (15 mg, 0,13 mmol) en DMSO (0,64 ml) y THF (0,27 ml). La purificación de la mezcla de reacción mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) proporcionó ácido (S)-6 -(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1-en-2-il)-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (4,8 mg, 17 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 16,16 (s, 1 H), 8,47 (s, 1 H), 7,78 (s, 1 H), 7,04 (s, 1 H), 5,95 (s, 1 H), 5,59 (s, 1 H), 4,26 - 4,12 (m, 2 H), 4,07 (d, J=6 ,8 Hz, 1 H), 3,62 - 3,57 (m, 2 H), 3,53 (dd, J=17,3, 7,1 Hz, 1 H), 3,38 (s, 3 H), 3,26 (d, J=17,1 Hz, 1 H), 2,26 (s, 3 H), 2,15 (quin, J=6,0 Hz, 2 H), 0,87 (s, 9 H); CLEM (m/z, ES+) = 427,3 (M+1).
Ejemplo 22: (compuesto 241)
Ácido (S)-6-(terc-butil)-2-isopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-h1T1,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-3-(3-metoxipropoxi)-10-oxo-2-(prop-1 -en-2-il)-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (11,9 mg, 0,026 mmol) y Pd al 10 %/C (catalítico) en MeOH (1 ml) se agitó bajo 60 psi de H2 durante 1,5 h. Se filtró la mezcla de reacción a través de celite y se evaporó hasta sequedad para proporcionar el (S)-6-(terc-butil)-2-isopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo en bruto, que se usó sin purificación adicional.
Una disolución del éster anterior en LiOH 1 M (0,7 ml) y MeOH (0,7 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (0,8 ml) y se agitó la mezcla de reacción durante 15 min. Se recogió el precipitado mediante filtración, se lavó con agua y se secó para proporcionar ácido (S)-6-(terc-butil)-2-isopropil-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (9,3 mg, 83 % durante dos etapas) como un sólido blanquecino. 1H-RMN (400 MHz, CDCh) 5 ppm 16,26 (s, 1 H), 8,46 (s, 1 H), 7,83 (s, 1 H), 6,91 (s, 1 H), 4,08 - 4,21 (m, 2 H), 4,05 (d, J=6,8 Hz, 1 H), 3,66 - 3,58 (m, 2 H), 3,41 - 3,55 (m, 2 H), 3,38 (s, 3 H), 3,22 (d, J=17,1 Hz, 1 H), 2,14 (quin, J=6,0 Hz, 2 H), 1,27 (d, J=6,8 Hz, 6 H), 0,85 (s, 9 H); CLEM (m/z, ES+) = 429,3 (M+1).
Ejemplo 23: (Compuesto 243)
Ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-6,10-dihidro-5H-piridoH ,2-h1H ,71naftiridin-9-carboxílico

Etapa 1: 2-Metil-4-oxo-4H-piran-3-carboxilato de etilo

Una disolución de 3-oxobutanoato de etilo (3,64 g, 28 mmol) en 20 ml de THF se añadió mediante una bomba de jeringa (gota a gota) a un recipiente que contenía NaH (dispersión al 60 % en aceite mineral) (1,176 g, 29,4 mmol) que se enfrió a 0 °C durante la adición. Se agitó la mezcla de reacción a TA durante 1 h después de completarse la adición, y entones se enfrió de nuevo hasta 0 °C. Una disolución de cloruro de 3-cloroacriloílo (3,50 g, 28 mmol) en 20 ml de THF se añadió lentamente a la mezcla de reacción enfriada mediante una bomba de jeringa a lo largo de 1 h. Después de completarse la adición, se agitó la mezcla de reacción a TA durante la noche y luego se calentó bajo reflujo durante 2,5 h. Se diluyó la mezcla de reacción con agua y se extrajo con Et2O (4x) y CH2Cl2 (2x). Se secaron los extractos orgánicos combinados (Na2SO4), se filtraron, se evaporaron y se purificaron mediante cromatografía en gel de sílice (el 0-100 % de EtOAc/hexanos) para proporcionar 2-metil-4-oxo-4H-piran-3-carboxilato de etilo (2,65 g, 52 %) como un líquido marrón. 1H-RMN (400 MHz, CDCh) 5 ppm 7,66 (d, J=5,9 Hz, 1 H), 6,36 (d, J=5,9 Hz, 1 H), 4,39 (q, J=7,3 Hz, 2 H), 2,38 (s, 3 H), 1,38 (t, J=7,2 Hz, 3 H); CLEM (m/z, ES+) = 183,1 (M+1).
Etapa 2: (S)-1-(1-(6-Cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-2-metil-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Una disolución de (S)-1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-amina, clorhidrato (196 mg, 0,42 mmol) y 2-metil-4-oxo-4H-piran-3-carboxilato de etilo (0,23 g, 1,27 mmol) en HOAc (4,2 ml) se agitó a 100 °C durante 10 h. Se evaporó la mezcla de reacción hasta sequedad, se llevó a CH2Cl2 y NaHCO3 saturado y se agitó durante 1 h. Se aisló la capa orgánica, se secó (Na2SO4), se filtró y se evaporó. Se agitó el residuo con 2-metil-4-oxo-4H-piran-3-carboxilato de etilo adicional (200 mg, 1,10 mmol) en HOAc (4,2 ml) a 100 °C durante la noche. Se evaporó la mezcla de reacción hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar (S)-1-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-2-metil-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (67,4 mg, 27 %) como un sólido marrón. CLEM (m/z, ES+) = 591,2, 593,2 (M+1).
Etapa_______ 3:_______ (S)-6-(terc-Butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-6,10-dihidro-5H-piridoH,2-hlHJlnaftiridin-9-carboxilato de etilo

Un matraz que contenía (S)-1-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-2-metil-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (67,4 mg, 0,11 mmol), acetato de potasio (22 mg, 0,23 mmol) y bromuro de paladio (II) (6,1 mg, 0,023 mmol) se purgo con nitrógeno. Se añadió DMF desgasificadoa (1,1 ml) y se calentó la mezcla de reacción a 90 °C durante 24 h. Se enfrió la disolución hasta TA, se añadió bromuro de paladio (II) adicional (6,1 mg, 0,023 mmol) y se calentó la mezcla de reacción a 90 °C durante otras 6 h. Se eliminó el disolvente mediante evaporación y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc/EtOH 3:1) en hexanos) para proporcionar (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (12,1 mg, 23 %) como un aceite marrón. 1H-RMN (400 MHz, CDCh) 5 ppm 7,48 (s, 1 H), 7,00 (s, 1 H), 4,34 - 4,45 (m, 3 H), 4,13 - 4,24 (m, 2 H), 3,65 - 3,58 (m, 2 H), 3,31 - 3,41 (m, 4 H), 3,15 (d, J=16,8 Hz, 1 H), 2,45 (s, 3 H), 2,12 - 2,19 (m, 2 H), 1,38 (t, J=7,2 Hz, 3 H), 0,80 (s, 9 H); CLEM (m/z, ES+) = 463,3, 465,3 (M+1).
Etapa 4: Ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-6,10-dihidro-5H-piridoH,2-hiri,71naftiridin-9-carboxílico

Una disolución de (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (12,1 mg, 0,026 mmol) en LiOH 1 M (0,5 ml) y MeOH (0,5 ml) se calentó a 50 °C durante 2 h y luego 60 °C durante 2 h. Se añadió ácido cítrico 1 M (0,7 ml) a la mezcla de reacción. Se evaporó la disolución hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 5-90 % de CHaCN/H2O (ácido fórmico al 0,1 %)) para proporcionar ácido (S)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-8-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (7,0 mg, 62 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 17,61 (s. a., 1H), 7,82 (s, 1 H), 7,05 (s, 1 H), 4,81 (d, J=5,4 Hz, 1 H), 4,15 - 4,28 (m, 2 H), 3,67 - 3,57 (m, 2 H), 3,49 - 3,41 (m, 1 H), 3,38 (s, 3 H), 3,25 (d, J=17,6 Hz, 1 H), 3,18 (s, 3 H), 2,17 (quin, J=6,0 Hz, 2 H), 0,81 (s, 9 H); CLEM (m/z, ES+) = 435,2, 437,2 (M+1).
Ejemplo 24: (Compuesto 243)
Ácido (S)-6-(terc-butil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-piridoH,2-h1HJ1naftiridin-9-carboxílico

Etapa 1: (S)-(1-(5-(Benciloxi)-6-cloro-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Una disolución de (S)-(1-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (2,18 g, 4,79 mmol), K2CO3 (1,99 g, 14,38 mmol), (bromometil)benceno (0,86 ml, 7,19 mmol) en DMF (32,0 ml) se calentó a 80 °C durante 3 h. Se evaporó la mezcla de reacción hasta sequedad y se llevó el residuo a CH2Cl2 y agua. Se extrajo la capa acuosa con CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para proporcionar (S)-(1-(5-(benciloxi)-6-cloro-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (cuant. supuesto), que se llevó a la siguiente etapa sin purificación adicional. CLEM (m/z, eS+) = 545,4, 547,0 (M+1).
Etapa 2: (S)-1-(1-(5-(Benciloxi)-6-cloro-2-vodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Se añadió HCI (4 M en dioxano) (11,98 ml, 47,9 mmol) a una disolución de (S)-(1-(5-(benciloxi)-6-cloro-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (2,61 g, 4,79 mmol) en CH2Cl2 (6 ml) y se agitó la mezcla de reacción a TA durante 3 h. Se evaporó la mezcla hasta sequedad y se llevó el residuo a NaHCO3 saturado y CH2Cl2. Se extrajo la capa acuosa con CH2Cl2 (2x) y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron para dar un aceite marrón.
Una disolución de la amina anterior y 4-oxo-4H-piran-3-carboxilato de etilo (0,89 g, 5,27 mmol) en HOAc (24 ml) se agitó a 100 °C durante 7 h. Se evaporó la mezcla de reacción, se llevó el residuo a CH2Cl2 y NaHCO3 saturado y se agitó la disolución vigorosamente durante 30 min. Se extrajo la fase acuosa con CH2Cl2 (5x10 ml), y se secaron las fases orgánicas combinadas (Na2SO4), se filtraron, se evaporaron y se purificaron mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc:EtOH 3:1) en hexanos) para proporcionar (S)-1-(1-(5-(benciloxi)-6-cloro-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (1,39 g, 49 %) como un sólido marrón. CLEM (m/z, ES+) = 596,1,597,2 (M+1).
Etapa 3: (S)-3-(Benciloxi)-6-(terc-butil)-2-cloro-10-oxo-5,10-dihidro-6H-piridor1,2-hir171naftiridin-9-carboxilato de etilo

Un matraz que contenía (S)-1-(1-(5-(benciloxi)-6-cloro-2-yodopiridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (1,39 g, 2,34 mmol), acetato de potasio (460 mg, 4,68 mmol) y bromuro de paladio (II) (125 mg, 0,47 mmol) se purgó con nitrógeno. Se añadió DMF desgasificada (23,4 ml) y se calentó la mezcla de reacción a 90 °C durante 24 h. Se eliminó el disolvente mediante evaporación y se disolvió el residuo en CH2Cl2, se filtró a través de celite y se purificó mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc/EtOH 3:1) en hexanos) para proporcionar (S)-3-(benciloxi)-6-(terc-butil)-2-cloro-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (706 mg, 65 %) como un sólido marrón. CLEM (m/z, ES+) = 467,3, 469,3 (M+1).
Etapa 4: (S)-3-(Benciloxi)-6-(terc-butil)-10-oxo-2-vinil-5,10-dihidro-6H-piridori,2-hirU1naftiridin-9-carboxilato de etilo

Una disolución de (S)-3-(benciloxi)-6-(terc-butil)-2-cloro-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (0,34 g, 0,74 mmol), sal de potasio de trifluoro(vinil)-I4-borano (0,20 g, 1,48 mmol), carbonato de sodio (0,24 g, 2,22 mmol) y Pd(PPh3)4 (85 mg, 0,074 mmol) en EtOH (7,4 ml) se agitó a 80 °C durante la noche. Se diluyó la mezcla de reacción con CH2Cl2, se filtró a través de celite y se evaporó. Se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc/EtOH 3:1) en hexanos) para proporcionar (S)-3-(benciloxi)-6-(terc-butil)-10-oxo-2-vinil-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (251 mg, 74 %) como una espuma blanquecina. 1H-RMN (400 MHz, CDCh) 5 ppm 8,21 (s, 1 H), 7,39 - 7,73 (m, 7 H), 7,00 (s, 1 H), 6,59 (d, J=6,6 Hz, 1 H), 5,54 (d, J=10,5 Hz, 1 H), 5,09 - 5,24 (m, 2 H), 4,40 (q, J=7,0 Hz, 2 H), 3,90 - 4,01 (m, 1 H), 3,39 - 3,55 (m, 1 H), 3,13 - 3,24 (m, 1 H), 1,41 (t, J=7,0 Hz, 3 H), 0,84 (s., 9 H); CLEM (m/z, ES+) = 459,1,460,4 (M+1).
Etapa 5: (S)-6-(terc-Butil)-3-hidroxi-2-(hidroximetil)-10-oxo-6,10-dihidro-5H-piridoH,2-hir1,71naftiridin-9-carboxilato de etilo

A una disolución de (S)-3-(benciloxi)-6-(terc-butil)-10-oxo-2-vinil-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (251 mg, 0,55 mmol) en THF (5,5 ml) y agua (1,4 ml) a 0 °C se le añadió osmiato de potasio dihidratado (20,2 mg, 0,055 mmol) seguido por peryodato de sodio (468 mg, 2,19 mmol). Después de completarse la adición, se agitó la mezcla de reacción a TA durante 2 h. Se diluyó la mezcla de reacción con CH2Cl2 y agua, se agitó y se filtró a través de celite. Se secaron las fases orgánicas combinadas (Na2SO4), se filtraron, se evaporaron y se purificaron mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc/EtOH 3:1) en CH2Cl2) para proporcionar (S)-3-(benciloxi)-6-(terc-butil)-2-formil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (cuant. supuesto). CLEM (m/z, ES+) = 461,3 (M+1).
Una disolución del aldehído anterior y Pd al 10 %/C (catalítico) en EtOH (20 ml) se agitó bajo 60 psi de H2 durante la noche. Se filtró la mezcla de reacción a través de celite y se evaporó para proporcionar (S)-6-(terc-butil)-3-hidroxi-2-(hidroximetil)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (122,1 mg, 60 %), que se llevó a la siguiente etapa sin purificación. CLEM (m/z, e S+) = 373,3 (M+1).
Etapa_______6 ______ (S)-6-(terc-Butil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-piridoH,2-h1H,71naftiridin-9-carboxilato de etilo

Se añadió 1-bromo-3-metoxipropano (44,3 pl, 0,39 mmol) a una disolución de (S)-6-(terc-butil)-3-hidroxi-2-(hidroximetil)-l 0-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (122,1 mg, 0,33 mmol) y carbonato de potasio (136 mg, 0,98 mmol) en DMF (3,3 ml) y se agitó la mezcla de reacción a TA durante la noche. Se añadió carbonato de potasio (100 mg, 0,72 mmol) y 1-bromo-3-metoxipropano (44,3 pl, 0,39 mmol) adicionales y se calentó la mezcla de reacción a 40 °C durante 2,5 h, y luego se agitó a TA durante la noche. Se evaporó el disolvente y se diluyó el residuo con CH2Cl2 y salmuera. Se secó la capa orgánica (Na2SÜ4), se filtró, se evaporó y se purificó mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc/EtOH 3:1) en CH2Cl2) para proporcionar (S)-6-(tercbutil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (62,8 mg, 43 %) como un aceite amarillo. 1H-RMN (400 MHz, CDCh) " ppm 8,18 (s, 1 H), 7,44 (s, 1 H), 6,98 (s, 1 H), 4,72 (s, 2 H), 4,39 (q, J=7,2 Hz, 2 H), 4,22 - 4,12 (m, 2 H), 3,90 - 4,06 (m, 2 H), 3,61 - 3,53 (m, 3 H), 3,37 (s, 3 H), 3,21 (d, J=16,8 Hz, 1 H), 2,11 (quin, J=6,0 Hz, 2 H), 1,40 (t, J=7,2 Hz, 3 H), 0,83 (s, 9 H); CLEM (m/z, ES+) = 445,3 (M+1).
Etapa 7: Ácido (S)-6-(terc-butil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-piridoH,2-h1'ri,71naft¡r¡d¡n-9-carboxíl¡co

Una disolución de (S)-6-(terc-butil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (29 mg, 0,065 mmol) en LiOH 1 M (0,65 ml) y MeOH (0,65 ml) se calentó a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (1 ml) y se evaporó la mezcla de reacción hasta sequedad. Se purificó el residuo mediante cromatografía de fase inversa (el 5-90 % de CH3CN/H2O (ácido fórmico al 0,1 %)) para proporcionar ácido (S)-6-(terc-butil)-2-(hidroximetil)-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (18,5 mg, 68 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) " ppm 15,96 (s, 1 H), 8,49 (s, 1 H), 7,74 (s, 1 H), 7,02 (s, 1 H), 4,79 (d, J=4,4 Hz, 2 H), 4,14 - 4,27 (m, 2 H), 4,10 (d, J=6,3 Hz, 1 H), 3,90 (t, J=4,9 Hz, 1 H), 3,62 - 3,53 (m, 3 H), 3,37 (s, 3 H), 3,29 (d, J=17,1 Hz, 1 H), 2,13 (quin, J=5,9 Hz, 2 H), 0,86 (s, 9 H); CLEM (m/z, ES-) = 415,3 (M-1).
Ejemplo 25: (Compuesto 244)
Ácido________ (S)-6-(terc-butil)-2-ciclopropil-11 -hidroxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-piridoH ,2-hiri,71naftiridin-9-carboxílico

Etapa 1: 4-(Benciloxi)-2-((dimetilamino)metilen)-3-oxobutanoato de etilo

Se añadió DMF-DMA (5,70 ml, 42,5 mmol) a una disolución de 4-(benciloxi)-3-oxobutanoato de etilo (6,70 g,
28,4 mmol) en tolueno (30 ml) y se agitó la mezcla de reacción durante la noche y se evaporó hasta sequedad. Se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de EtOAc en hexanos) para proporcionar 4-(benciloxi)-2-((dimetilamino)metilen)-3-oxobutanoato de etilo (6,78 g, 82 %) como un aceite amarillo. CLEM (m/z, ES+) = 292,6 (M+1).
Etapa 2: 5-(Benciloxi)-4-oxo-4H-piran-3-carboxilato de etilo

Se añadió formiato de etilo (10,2 ml, 126 mmol) lentamente a una suspensión de terc-butóxido de potasio (3,54 g, 31,6 mmol) en 24 ml de THF a 0 °C. Después de completarse la adición, se agitó la mezcla de reacción durante otros 15 min a 0 °C y se añadió una disolución de 4-(benciloxi)-2-((dimetilamino)metilen)-3-oxobutanoato de etilo (4,6 g, 15,8 mmol) en 24 ml de THF gota a gota usando un embudo de adición. Después de completarse la adición, se retiró la mezcla de reacción del baño de hielo y se agito a TA durante la noche. Se añadió HCI 1 M (50 ml) y se extrajo la disolución con EtOAc (2x). Se secaron las fases orgánicas combinadas (Na2SO4), se filtraron, se evaporaron y se purificaron mediante cromatografía en gel de sílice (el 0-100 % de EtOAc en hexanos) para proporcionar 5-(benciloxi)-4-oxo-4H-piran-3-carboxilato de etilo (1,48 g, 34 %) como un sólido amarillo claro. 1H-RMN (400 MHz, CDCh) " ppm 8,41 (s, 1 H), 7,55 (s, 1 H), 7,32 - 7,43 (m, 5 H), 5,12 (s, 2 H), 4,37 (q, J=7,0 Hz, 2 H), 1,38 (t, J=7,2 Hz, 3 H); CLEM (m/z, ES+) = 275,5 (M+1).
Etapa 3: (S)-5-(Benciloxi)-1-(1-(6-cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

Una disolución de la (S)-1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-amina, clorhidrato (0,402 g, 0,87 mmol) y 5-(benciloxi)-4-oxo-4H-piran-3-carboxilato de etilo (0,38 g, 1,37 mmol) en HOAc (8,7 ml) se agitó a 100 °C durante 10 h. Se evaporó la mezcla de reacción hasta sequedad, se llevó a CH2Cl2 y NaHCO3 saturado y se agitó durante 1 h. Se extrajo la capa acuosa con CH2Cl2 y EtOAc (2x). Se secaron las fases orgánicas combinadas (Na2SO4), se filtraron y se evaporaron. Se disolvió el residuo en EtOH (8 ml) y se calentó a 80 °C durante la noche. Se evaporó la mezcla de reacción hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CH3CN/H2O (ácido fórmico al 0,1 %)) para proporcionar (S)-5-(benciloxi)-1-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (89,8 mg, 15 %) como un sólido marrón. CLEM (m/z, ES+) = 683,7, 685,7 (M+1).
Etapa 4: (S)-11-(Benciloxi)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-h1HJ1naftiridin-9-carboxilato de etilo

Un matraz que contenía (S)-5-(benciloxi)-1-(1-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (89,8 mg, 0,13 mmol), acetato de potasio (26 mg, 0,26 mmol) y bromuro de paladio (II) (7,0 mg, 0,03 mmol) se purgó con nitrógeno. Se añadió DMF desgasificada (1,3 ml) y se calentó la mezcla de reacción a 90 °C durante 24 h. Se filtró la mezcla de reacción a través de celite, se eliminó el disolvente mediante evaporación y se purificó el residuo mediante cromatografía en gel de sílice (el 0-100 % de (EtOAc/EtOH 3:1) en hexanos) para proporcionar (S)-11-(benciloxi)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (47,9 mg, 66 %) como un aceite marrón. 1H-RMN (400 MHz, CDCh)
5 ppm 8,15 (s, 1 H), 7,73 (d, J= 6,2 Hz, 2 H), 7,28 - 7,36 (m, 3 H), 6,94 (s, 1 H), 5,26 (s. a., 2 H), 4,35 (q, J=6,4 Hz, 2 H), 4,21 - 4,10 (m, 2 H), 3,82 - 3,92 (m, 1 H), 3,56 - 3,67 (m, 2 H), 3,46 - 3,37 (m, 1 H), 3,39 (s, 3 H), 2,98 (d, J=16,0 Hz, 1 H), 2,13 (quin, J=5,9 Hz, 2 H), 1,40 - 1,31 (m, 3 H), 0,71 (s, 9 H); CLEM (m/z, ES+) = 555,4, 557,3 (M+1).
Etapa 5: (S)-11-(Benciloxi)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-piridoH,2-h1HJ1naftiridin-9-carboxilato de etilo

Una disolución de (S)-11 -(benciloxi)-6-(terc-butil)-2-cloro-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (47,9 mg, 0,086 mmol), Pd(PPh3)4 (20 mg, 0,017 mmol), carbonato de potasio (36 mg, 0,26 mmol) y ácido ciclopropilborónico (15 mg, 0,17 mmol) en 1,4-dioxano (0,9 ml) se calentó a 100 °C durante 2 días. Se diluyó la mezcla de reacción con CH2Cl2 y se filtró a través de celite. Se evaporó el filtrado y se purificó el residuo mediante cromatografía de fase inversa (el 5-100 % de CH3CN/H2O (ácido fórmico al 0,1 %)) para proporcionar (S)-11 -(benciloxi)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (14,7 mg, 30 %) como un sólido blanco. 1H-RMN (400 MHz, CDCh) 5 ppm 8,18 (s, 1 H), 7,66 (d, J=7,0 Hz, 2 H), 7,27 - 7,20 (m, 3 H), 6,82 (s, 1 H), 5,45 (d, J=10,9 Hz, 1 H), 5,21 (d, J=10,9 Hz, 1 H), 4,46 - 4,38 (m, 2 H), 4,04 - 4,20 (m, 2 H), 3,89 (d, J=5,5 Hz, 1 H), 3,65 - 3,56 (m, 2 H), 3,38 (s, 3 H), 3,30 (dd, J=16,0, 6,2 Hz, 1 H), 3,03 (d, J=16,0 Hz, 1 H), 2,39 - 2,49 (m, 1 H), 2,13 (quin, J=6,1 Hz, 2 H), 1,42 (t, J=7,2 Hz, 3 H), 1,07 - 1,15 (m, 1 H), 0,96 - 1,04 (m, 1 H), 0,88 - 0,96 (m, 1 H), 0,80 - 0,86 (m, 1 H), 0,77 (s, 9 H); CLEM (m/z, ES+) = 561,8 (M+1).
Etapa 6: Ácido (S)-6-(terc-butil)-2-ciclopropil-11 -hidroxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-piridoH ,2-hir1,71naftiridin-9-carboxílico

Una disolución de (S)-11 -(benciloxi)-6-(terc-butil)-2-ciclopropil-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (14,7 mg, 0,026 mmol) y Pd al 10 %/C (catalítico) en MeOH (5 ml) se agitó bajo 1 atm de H2 durante 1,5 h. Se filtró la mezcla de reacción a través de celite y se evaporó hasta sequedad. Se llevó el residuo a MeOH (0,5 ml) y LiOH 1 M (0,5 ml) y se calentó la disolución a 50 °C durante 2 h. Se añadió ácido cítrico 1 M (1,5 ml), se evaporó la mezcla hasta sequedad y se purificó el residuo mediante cromatografía de fase inversa (el 5-90 % de CH3CN/H2O (ácido fórmico al 0,1 %)) para proporcionar ácido (S)-6-(terc-butil)-2-ciclopropil-11-hidroxi-3-(3-metoxipropoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (6,3 mg, 54 %) como un sólido blanco.
1H-RMN (400 MHz, CDCh) 5 ppm 15,79 (s, 1 H), 13,96 (s, 1 H), 8,28 (s, 1 H), 7,08 (s, 1 H), 4,25 - 4,16 (m, 2 H), 4,06 (d, J=6,3 Hz, 1 H), 3,58 - 3,69 (m, 2 H), 3,51 (dd, J=17,1,6,8 Hz, 1 H), 3,39 (s, 3 H), 3,25 (d, J=17,1 Hz, 1 H), 2,49 -2,59 (m, 1 H), 2,18 (quin, J=6,1 Hz, 2 H), 1,06 - 1,19 (m, 4 H), 0,84 (s, 9 H); CLEM (m/z, ES+) = 443,7 (M+1).
Ċ

63
Esquema de síntesis específico 8 para compuestos del ejemplo 26,2

Ejemplo 26: (Compuesto 245)
Ácido (2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-piridoí1 ,2-h1H ,71naftiridin-9-carboxílico - isómero-1
Ejemplo 27: (Compuesto 246)
Ácido (2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-piridoí1 ,2-h1H ,71naftiridin-9-carboxílico - isómero-2

Etapa 1: 6-C loro-5-hidroxinicotinato de metilo

A una disolución de 5-hidroxinicotinato de metilo (100 g, 0,65 mol) en DMF (1000 ml), se le añadió NCS (130,5 g, 0,97 mol) y se calentó la mezcla de reacción hasta 80 °C durante 16 h. Se enfrió la mezcla de reacción hasta temperatura ambiente y se eliminó el disolvente a presión reducida. Se llevó el residuo a EtOAc (2 l) y se lavó con disolución saturada de cloruro de sodio (500 ml). Se secó la capa orgánica sobre Na2SO4 y se concentró. Se purificó el producto en bruto mediante cromatografía en columna sobre gel de sílice 230-400 usando el 0-30 % de acetato de
etilo en éter de petróleo como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título como un aceite amarillo (60 g, rendimiento del 49 %), CLEM (ESI) m/z 187,9 (M+1).
Etapa 2: 2-Cloro-5-(hidroximetil)piridin-3-ol

Una disolución de 6-cloro-5-hidroxinicotinato de metilo (60 g, 0,32 mol) en THF (600 ml) se añadió gota a gota a hidruro de aluminio y litio (160 ml, 0,32 mol, 2,0 M en THF) a -50 °C bajo atmósfera de nitrógeno. Se agitó la mezcla de reacción a -25 °C durante 2 h. Se añadieron EtOAc (1000 ml), agua (50 ml) y disolución ac. saturada de tartrato de potasio y sodio (500 ml) a la mezcla de reacción gota a gota a -25 °C. Se filtró la mezcla a través de un lecho de celite, se lavó con el 20 % de MeOH en DCM (500 ml). Se concentró el filtrado a presión reducida para proporcionar el compuesto del título (40 g, rendimiento del 80 %) como un sólido blanquecino. CLEM (ESI) m/z 157,9 (M-H).
Etapa 3: 2-Cloro-5-(hidroximetil)-6-yodopiridin-3-ol

A una disolución de 2-cloro-5-(hidroximetil)piridin-3-ol (3,5 g, 22,01 mmol) en THF (35 ml), se le añadieron agua (35 ml), K2CO3 (6,1 g, 44,24 mmol) y yodo (5,84 g , 23,11 mmol) y se agitó la mezcla de reacción a temperatura ambiente durante 2 h. Se enfrió la mezcla de reacción hasta 0 °C y se extinguió con disolución ac. de sulfito de sodio (50 ml). Se añadió acetato de etilo (150 ml) a la misma, se separó la capa orgánica y se desechó. Se acidificó la capa acuosa con disolución de HCl ac. 1,5 N hasta pH = 6 y se extrajo con acetato de etilo (150 ml). Se separó la capa orgánica, se lavó con agua (25 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a presión reducida para proporcionar el compuesto del título como un sólido blanquecino (4 g, rendimiento del 64,5 %). CLEM (ESI) m/z: 283,7 (M-H).
Etapa 4: (6-Cloro-2-vodo-5-metoxipiridin-3-il)metanol

A una disolución de 2-cloro-5-(hidroximetil)-6-yodopiridin-3-ol (4 g, 14,03 mmol) en acetonitrilo (50 ml), se le añadieron K2CO3 (3,87 g, 28,06 mmol) y yoduro de metilo (Mel) (5,97 g, 42,10 mmol) y se calentó la mezcla de reacción hasta 80 °C durante 2 h en un tubo sellado. Se extinguió la mezcla de reacción con agua (100 ml), se acidificó con HCI 1,5 N (pH = 6) y se extrajo con acetato de etilo (150 ml). Se lavó la capa orgánica con salmuera, se secó sobre Na2SO4 y se concentró. Se purificó el producto en bruto mediante cromatografía en columna sobre gel de sílice 230-400 usando el 0-30 % de acetato de etilo en éter de petróleo como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título como un sólido blanquecino (3,3 g, rendimiento del 80 %). CLEM (ESI) m/z: 299,8 (M 1).
Etapa 5: 3-(Bromometil)-6-cloro-2-vodo-5-metoxipiridina

A una disolución de (6-cloro-2-yodo-5-metoxipiridin-3-il)metanol (2,2 g, 7,35 mmol) en DCM (20 ml) se le añadió CBr4 (3,6 g, 11,35 mmol) y PPh3 (2,3 g, 8,82 mmol) en THF (20 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 6 h. Se eliminó el disolvente a presión reducida para conseguir el producto en bruto. Se purificó este mediante cromatografía en columna sobre gel de sílice 60-120 de malla usando el 0-20 % de EtOAc en éter de petróleo como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título como un sólido blanquecino (2,4 g, rendimiento del 92 %). CLEM (ESI) m/z: 363,6 (M+H).
Etapa 6: 2-((6-Cloro-2-vodo-5-metoxipiridin-3-il)metil)-2.3-dimetilbutanoato de etilo

A una disolución de diisopropilamina (2,43 ml, 17,38 mmol) en THF seco (20 ml) se le añadió n-BuLi (10,86 ml, 17,38 mmol, 1,6 M en hexano) gota a gota a -78 °C. Se agitó la mezcla de reacción a la misma temperatura durante 30 min. Se añadió 2,3-dimetilbutanoato de etilo (2,3 g, 16,57 mmol) en THF (20 ml) a la mezcla de reacción y se agitó la mezcla de reacción a -78 °C durante 1 h. Se añadió 3-(bromometil)-6-cloro-2-yodo-5-metoxipiridina (3 g, 8,28 mmol) en THF (20 ml) gota a gota a la misma temperatura y se calentó la mezcla de reacción hasta temperatura ambiente y se agitó a temperatura ambiente durante 1 h. Se enfrió la mezcla de reacción hasta 0 °C, se extinguió con disolución saturada de NH4Cl (50 ml), se diluyó con agua (20 ml) y se extrajo con EtOAc (3X50 ml). Se lavaron las capas orgánicas combinadas con salmuera (20 ml), se secaron sobre Na2SO4 anhidro y se filtraron. Se eliminaron los disolventes a presión reducida para conseguir el producto en bruto. Se purificó el producto en bruto mediante cromatografía en columna sobre gel de sílice 230-400 de malla usando el 0-10 % de EtOAc en éter de petróleo como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título como un aceite amarillo (3 g, rendimiento del 85 %). CLEM (ESI) m/z: 426 (M+H).
Etapa 7: Ácido 2-((6-cloro-2-vodo-5-metoxipiridin-3-il)metil)-2,3-dimetilbutanoico

A una disolución de 2-((6-cloro-2-yodo-5-metoxipiridin-3-il)metil)-2,3-dimetilbutanoato de etilo (3 g, 7,05 mmol) en DMSO (9 ml), se le añadió disolución de NaOH 5 M (21,6 ml, 98,11 mmol) y se agitó la mezcla de reacción a 120 °C durante 6 h. Se enfrió la mezcla de reacción hasta 0 °C, se acidificó con h C i concentrado hasta pH = 1. Tras diluir con agua (50 ml), se extrajo con EtOAc (2x100 L). Se lavaron las capas orgánicas combinadas con salmuera (50 ml), se secaron sobre Na2SO4 anhidro y se filtraron. Se eliminó el disolvente a presión reducida para conseguir el producto en bruto. Se purificó el producto en bruto mediante cromatografía en columna sobre gel de sílice 230-400 usando el 0-10 % de MeOH en DCM para proporcionar ácido 2-((6-cloro-2-yodo-5-metoxipiridin-3-il)metil)-2,3-dimetilbutanoico (500 mg, rendimiento del 18 %) (Cl EM (ESI) m/z: 397,8 (M+1)).
Etapa 8: 1 -(6-Cloro-2-vodo-5-metox¡p¡r¡d¡n-3-¡l)-2,3-d¡met¡lbutan-2-am¡na

A una disolución de ácido 2-((6-cloro-2-yodo-5-metoxipiridin-3-il)metil)-2,3-dimetilbutanoico (1 g, 2,5 mmol) en tolueno (30 ml), se le añadieron trietilamina (0,57 g, 5,5 mmol) y difenilfosforilazida (1,52 g, 5,5 mmol) y se agitó la mezcla de reacción a 80 °C durante 2 h. Se enfrió la mezcla de reacción hasta temperatura ambiente y se añadió HCI concentrado (8,4 ml) a la mezcla de reacción gota a gota a lo largo de 30 min a 0 °C. Se agitó la mezcla de reacción a 60 °C durante 2 h. Se enfrió la mezcla de reacción hasta 0 °C y se basificó con disolución acuosa de NaOH 1 N hasta pH = 10. Se diluyó la mezcla de reacción con agua (20 ml) y se extrajo con EtOAc (2X50 ml). Se lavaron las capas orgánicas combinadas con salmuera (20 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron para conseguir un sólido gomoso marrón. Se purificó este mediante cromatografía en columna sobre sílice 230-400 usando el 0-3 % de MeOH en DCM para proporcionar el compuesto del título como un aceite amarillo (800 mg, rendimiento del 87 %). CLEM (ESI) m/z: 368,8 (M+1).
Etapa 9: 1-(1 -(6-Cloro-2-vodo-5-metoxipiridin-3-il)-2,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo

A una disolución de 1-(6-cloro-2-yodo-5-metoxipiridin-3-il)-2,3-dimetilbutan-2-amina (800 mg, 2,17 mmol) en ácido acético (25 ml), se le añadió 4-oxo-4H-piran-3-carboxilato de etilo (0,547 g, 3,2 mmol) y se agitó la mezcla de reacción a 100 °C durante 8 h. Se enfrió la mezcla de reacción hasta temperatura ambiente, se eliminó el disolvente a presión reducida y se purificó el residuo mediante cromatografía en columna sobre gel de sílice 230-400 usando el 0-10 % de MeOH en DCM como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título (400 mg, rendimiento del 35 %) como un sólido marrón. CLEM (ESI) m/z: 518,7 (M 1).
Etapa 10: 2-Cloro-6-isopropil-3-metoxi-6-metil-10-oxo-5,10-dihidro-6H-piridori,2-hiri,71naftiridin-9-carboxilato de etilo

A una disolución de 1-(1-(6-cloro-2-yodo-5-metoxipiridin-3-il)-2,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (300 mg, 0,57 mmol) en N,N-dimetilformamida (7,5 ml) se le añadió acetato de potasio (283 mg, 2,89 mmol). Se desgasificó la mezcla de reacción con nitrógeno durante 20 minutos. Se añadió bromuro de paladio (II) (30 mg, 0,11 mmol) y se agitó la mezcla de reacción a 90 °C durante 16 h. Se enfrió la mezcla de reacción hasta temperatura ambiente, se filtró a través de celite y se lavó con DCM. Se eliminaron los disolventes a presión reducida y se purificó el producto en bruto mediante cromatografía en columna sobre gel de sílice 230-400 usando el 0-10 % de MeOH en d Cm como eluyente. Se recogieron las fracciones y se concentraron para proporcionar el compuesto del título (150 mg, rendimiento del 68 %) como un sólido marrón. CLEM (ESI) m/z: 390,9 (M 1).
Etapa 11: Ácido 2-cloro-3-hidroxi-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-piridoH,2-h1r1,71naftiridin-9-carboxílico

A una disolución de 2-cloro-6-isopropil-3-metoxi-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (150 mg, 0,38 mmol) en 1,2-dicloroetano (DCE) (6,45 ml), se le añadió BBr3 (288 mg, 1,15 mmol) y se agitó la mezcla de reacción a 70 °C durante 16 h. Se enfrió la mezcla de reacción hasta -10 °C, se añadió metanol (15 ml) a la mezcla de reacción gota a gota y luego se agitó a temperatura ambiente durante 30 minutos. Se concentró la mezcla de reacción para conseguir el compuesto en bruto (150 mg) como un sólido marrón. CLEM (ESI) m/z: 348,9 (M+1). Se llevó este producto en bruto a la siguiente etapa.
Etapa 12: 2-Cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-piridor1,2-h1HJ1naftiridin-9-carboxilato de metilo-isómero-1 y 2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-piridolH ,2-hir1,71naftiridin-9-carboxilato de metilo-isómero-2

A una disolución de ácido 2-cloro-3-hidroxi-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (150 mg, 0,43 mmol) en DMF (2 ml), se le añadieron K2CO3 (594 mg, 4,3 mmol) y (bromometil)ciclopropano (464 mg, 3,44 mmol) y se agitó la mezcla de reacción a 80 °C durante 5 h. Se enfrió la mezcla de reacción hasta temperatura ambiente, se eliminó el disolvente a presión reducida y se agitó el residuo con MeOH (10 ml) a temperatura ambiente durante 30 min. Se eliminó el disolvente a presión reducida y se diluyó con DCM (20 ml). Se
lavó la capa orgánica con agua (15 ml) y se secó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida para conseguir el compuesto como una mezcla de isómeros que se separó mediante cromatografía de fluidos supercríticos quiral (SFC). Las fracciones recogidas de isómero-1 (primer pico de elución) e isómero-2 (segundo pico de elución) se concentraron a presión reducida para conseguir 2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-1 (50 mg, rendimiento del 28 %) como un sólido blanquecino CLEM (ESI) m/z: 417,9 (M+1) y 2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-2 (50 mg, rendimiento del 28 %) como un sólido blanquecino. CLEM (ESI) m/z: 417,9 (M+1).
Etapa 13: Ácido (2-cloro-3-(c¡cloprop¡lmetox¡)-6-¡soprop¡l-6-met¡l-10-oxo-5.10-d¡h¡dro-6H-p¡r¡doH.2-h1f1.7lnaftir¡d¡n-9-carboxíl¡co-¡sómero-1

A una disolución de 2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-1 (50 mg, 0,11 mmol) en metanol (2 ml) y H2O (0,5 ml), se le añadió hidróxido de litio (25 mg, 0,59 mmol) y se agitó la mezcla de reacción a temperatura ambiente durante 2 h. Se eliminaron los disolventes a presión reducida y se llevó el residuo a agua (3 ml) y se acidificó con HCI 1,5 N hasta pH ~ 3. Se filtró el sólido obtenido, se lavó con agua (5 ml), se secó a vacío y se purificó mediante HPLC prep. para conseguir el compuesto del título (15 mg, rendimiento del 31,2 %) como un sólido blanquecino. CLEM (ESl) m/z: 403,1 (M+1). 1H-RMN; 400 MHz, DMSO-d6: 5 ppm 8,57 (s, 1 H), 7,72 (s, 1 H), 7,39 (s, 1H), 4,10 - 4,07 (m, 2H), 3,36 - 3,35 (m, 2H), 1,91 - 1,84 (m, 1H), 1,70 (s, 3H), 1,35 - 1,25 (m, 1H), 0,85 - 0,82 (m, 3H), 0,65 - 0,63 (m, 5H), 0,42 - 0,39 (m, 2H).
Etapa 13 A: Ác¡do 2-cloro-3-(c¡cloprop¡lmetox¡)-6-¡soprop¡l-6-met¡l-10-oxo-5.10-d¡h¡dro-6H-p¡r¡dof1.2-hlH .71naft¡r¡d¡n-9-carboxíl¡co-¡sómero-2

A una disolución de 2-cloro-3-(ciclopropilmetoxi)-6-isopropil-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-2 (50 mg, 0,11 mmol) en metanol (2 ml) y H2O (0,5 ml), se le añadió hidróxido de litio (25 mg, 0,59 mol) y se agitó la mezcla de reacción a temperatura ambiente durante 2 h. Se eliminaron los disolventes a presión reducida y se llevó el residuo a agua (3 ml) y se acidificó con HCI 1,5 N hasta pH ~ 3. Se filtró el sólido obtenido, se lavó con agua (5 ml), se secó a vacío durante 16 h para conseguir el compuesto del título como un sólido blanquecino (30 g, rendimiento del 62 %). CLEM (ESI) m/z: 402,9 (M+1). 1H-RMN; 400 MHz, DMSO-d6: 5 ppm 8,57 (s, 1H), 7,71 (s, 1H), 7,39 (s, 1 H), 4,09 - 4,08 (m, 2H), 3,39 - 3,38 (m, 2H), 1,90 - 1,87 (m, 1H), 1,70 (s, 3H), 1,32 - 1,28 (m, 1H), 1,11 - 1,07 (m, 3H), 0,83 - 0,81 (m, 5H), 0,63 - 0,62 (m, 2H).
Ejemplo 28: (Compuesto 247)
Ác¡do 2-c¡cloprop¡l-6-¡soprop¡l-3-(3-metox¡propox¡)-6-met¡l-10-oxo-5.10-d¡h¡dro-6H-p¡r¡do n .2-h1PI .71naft¡r¡d¡n-9-carboxíl¡co-¡sómero-1

Ejemplo 29: (Compuesto 248)
Ácido 2-c¡cloprop¡l-6-¡sopropil-3-(3-metox¡propox¡)-6-met¡l-10-oxo-5.10-d¡h¡dro-6H-p¡r¡doH ,2-hlH Jlnaftiridin-9-carboxílico-¡sómero-2

Esquema de síntesis general 9 para compuestos tales como el ejemplo 28. 29:

Esquema de síntesis específico 10 para compuestos del ejemplo 28. 29:

Etapa_______1 _______2-C¡cloprop¡l-6-¡soprop¡l-3-(3-metox¡propox¡)-6-met¡l-10-oxo-5.10-dih¡dro-6H-p¡r¡dori .2-hiri.71naft¡r¡din-9-carbox¡lato de metilo-isómero-1 y 2-c¡cloprop¡l-6-¡soprop¡l-3-(3-metox¡propoxi)-6-met¡l-10-oxo-5.10-dih¡dro-6H-p¡r¡dof1.2-h1f1 ■71naft¡r¡din-9-carbox¡lato de metilo-isómero-2

A una disolución agitada de 2-cloro-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo (570 mg, 1,311 mmol) en 1,4-dioxano (11,4 ml), se le añadió carbonato de potasio (362 mg, 2,62 mmol) y se purgó con nitrógeno durante 5 min. Se añadió ácido ciclopropilborónico (225 mg, 2,62 mmol) seguido por Pd(PPh3)4 (303 mg, 0,262 mmol) y se purgó con nitrógeno durante 5 min. Se calentó la mezcla de reacción hasta 100 °C en un tubo sellado durante 16 h. Se concentró la mezcla de reacción completamente a presión reducida y se diluyó el residuo con DCM (100 ml), se lavó con agua (40 ml), salmuera (40 ml). Se separó la capa orgánica, se secó sobre sulfato de sodio y se concentró y se purificó el producto en bruto mediante cromatografía en sílice Isolera™ y se eluyó con el 3 % de MeOH en DCM. Se concentraron las fracciones recogidas completamente para conseguir el compuesto como una mezcla de isómeros. Entonces, se purificó el compuesto mediante HPLC
preparativa seguido por SFC quiral. Se concentraron las fracciones recogidas de isómero-1 (primer pico de elución) e isómero-2 (segundo pico de elución) por separado a presión reducida para conseguir 2-ciclopropil-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-1 (60 mg, rendimiento el 10,3 %) como un aceite incoloro y 2-ciclopropil-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-2(90 mg, rendimiento del 13,97 %) como un aceite incoloro. Cl EM (ESI) m/z: 441,0 (M+1).
Etapa 2: Ácido 2-c¡cloprop¡l-6-¡soprop¡l-3-(3-metox¡propox¡)-6-met¡l-10-oxo-5,10-dih¡dro-6H-p¡r¡dori ,2-h1f1.7lnaft¡r¡d¡n-9-carboxílico-¡sómero-1

A una disolución de 2-ciclopropil-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo, isómero-1 (60 mg, 0,136 mmol) en metanol (2,4 ml) se le añadió LiOH (16,31 mg, 0,681 mmol) y agua (0,60 ml) a temperatura ambiente y se agitó la mezcla de reacción a ta durante 2 h. Se eliminaron los disolventes de la mezcla de reacción a presión reducida y se disolvió el residuo en agua (2 ml). Se acidificó esta disolución con HCI 1,5 N hasta pH 6 a 0 °C. Se filtró el sólido obtenido, se lavó con agua, n-pentano y se secó para conseguir el compuesto del título como un sólido marrón pálido (15 mg, 25,3 %). CLEM (ESI) m/z: 427,2 (M+1). 1H-RMN (400 MHz, DMSO-d6): 5 ppm 8,54 (s, 1H), 7,44 (s, 2H), 4,19 - 4,18 (m, 2H), 3,53 (t, J =6 Hz, 2H), 3,29 - 3,27 (m, 5H), 2,05 (t, J = 6 Hz, 2H), 1,84 -1,82 ( m, 1H) , 1,68 (s, 3H), 1,03 - 1,01 (m, 4H), 0,82 (d, J =6,5 Hz 3H), 0,61 (d, J = 6,6Hz, 3H)
Etapa 2A: Ác¡do 2-c¡cloprop¡l-6-¡soprop¡l-3-(3-metox¡propox¡)-6-met¡l-10-oxo-5.10-d¡h¡dro-6H-p¡r¡dorr1.2-hlM .71naft¡r¡d¡n-9-carboxíl¡co-¡sómero-2

A una disolución de 2-ciclopropil-6-isopropil-3-(3-metoxipropoxi)-6-metil-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de metilo-isómero-2 (90 mg, 0,204 mmol) en metanol (3,6 ml) se le añadió LiOH (24,46 mg, 1,021 mmol) y agua (0,90 ml) a temperatura ambiente y se agitó la mezcla de reacción a ta durante 2 h. Se eliminaron los disolventes de la mezcla de reacción a presión reducida y se disolvió el residuo en agua (2 ml). Se acidificó esta disolución con HCI 1,5 N hasta pH 6 a 0 °C. Se filtró el sólido obtenido, se lavó con agua, n-pentano y se secó para conseguir el compuesto del título como un sólido marrón pálido (26 mg, 29,3 %). CLEM (ESI) m/z: 427,2 (M+1). 1H-RMN (400MHz , DMSO-d6): 5 ppm 8,53 (s, 1H), 7,44 (s, 2H), 4,25-4,12 (m, 2H), 3,56-3,52 (m, 2H), 3,23 3,18 (m, 5H), 2,10-2,01 (m, 3H), 1,81-1,62 (m, 4H), 1,05-0,99 (m, 4H), 0,85-0,81 (m, 3H), 0,65-0,61 (m, 3H).
Esquema general 11 - Preparación de compuestos de tipo tal como el compuesto 249

El compuesto 249 puede prepararse según el procedimiento de síntesis general explicado de manera resumida en el Esquema 11 anterior, y tal como se describe en detalle a continuación.
Ejemplo 30 (compuesto 249 (racémico))
Ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico

Etapa 1: 1-(6-Cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona

Se cargó un matraz con una barra agitadora, xantphos (0,225 g, 0,388 mmol), tris(dibencilidenacetona)dipaladio (0) (0,198 g, 0,216 mmol) y terc-butóxido de sodio (2,43 g, 25,3 mmol). Se purgó el matraz con una corriente de nitrógeno antes de que se añadiera una disolución de 5-bromo-2-cloro-3-metoxipiridina (3,2 g, 14,4 mmol) y 3,3-dimetilbutan-2-ona (2,16 ml, 17,3 mmol) en tetrahidrofurano (THF) (50 ml). Se calentó la mezcla a reflujo durante la noche. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se diluyó con agua. Se extrajo la mezcla 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona (3,07 g, 12,7 mmol, rendimiento del 88 %). 1H-RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,74 (d, J=1,56 Hz, 1 H), 7,10 (d, J=1,56 Hz, 1 H), 3,89 (s, 3 H), 3,77 (s, 2 H), 1,20 (s, 9 H).
Etapa 2: 1-(6-Cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina

Una mezcla de 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-ona (3,07 g, 12,7 mmol) y acetato de amonio (14,7 g, 191 mmol) en metanol (50 ml) se agitó durante 1 hora a temperatura ambiente antes de que se añadiera cianoborohidruro de sodio (2,00 g, 31,8 mmol) en porciones. Se agitó la mezcla a lo largo del fin de semana a temperatura ambiente antes de que se basificara con hidróxido de sodio 1 M. Se extrajo la mezcla 3 veces con
diclorometano. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina (2,82 g) como un aceite transparente. Se purificó el material mediante cromatografía a presión media de fase inversa (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se combinaron las fracciones, se basificaron con hidróxido de sodio 1 M y se extrajeron 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina (1,45 g, 5,97 mmol, rendimiento del 47,0 %) como un aceite transparente. 1H-RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,83 (d, J=1,56 Hz, 1 H), 7,08 (d, J=1,56 Hz, 1 H), 3,90 (s, 3 H), 2,91 (dd, J=13,48, 2,15 Hz, 1 H), 2,62 (dd, J=10,94, 1,95 Hz, 1 H), 2,22 (dd, J=13,48, 11,13 Hz, 1 H), 1,12 - 1,36 (m, 2 H), 0,98 (s, 9 H). CLEM (ES+) (m/z): 243, 245 (M+1).
Etapa 3: 5-(2-Amino-3,3-dimetilbutil)-2-cloropiridin-3-ol

Se añadió tribromuro de boro (2,82 ml, 29,9 mmol) gota a gota a una disolución de 1-(6-cloro-5-metoxipiridin-3-il)-3,3-dimetilbutan-2-amina (1,45 g, 5,97 mmol) en 1,2-dicloroetano (DCE) (30 ml) a 0 °C. Se permitió que la mezcla se calentara hasta temperatura ambiente y se agitó durante la noche. La CL-EM mostró una conversión significativa en el producto deseado, pero todavía estaba presente material de partida. Se rompieron los sólidos que se habían formado durante la noche y se agitó la mezcla 24 horas adicionales a temperatura ambiente. Se enfrió la mezcla hasta 0 °C y se extinguió con adición gota a gota cuidadosa de metanol. Tras haberse extinguido completamente el BBr3 en exceso, se añadió metanol adicional (100 ml). Se agitó la mezcla durante 1 hora y se concentró. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se liofilizaron las fracciones para dar 5-(2-amino-3,3-dimetilbutil)-2-cloropiridin-3-ol (1,33 g, 5,81 mmol, rendimiento del 97 %) como un sólido blanco. 1H-RMN (400 MHz, DMSO-ds) 5 ppm 8,21 (s, 1 h ), 7,83 -8,16 (m, 2 H), 7,74 (d, J=1,95 Hz, 1 H), 7,21 (d, J=1,56 Hz, 1 H), 2,82 - 3,02 (m, 2 H), 2,35 - 2,43 (m, 1 H), 0,94 (s, 9 H). CLEM (ES+) (m/z): 229, 231 (M+1).
Etapa 4: (1-(6-Cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Se añadió dicarbonato de di-terc-butilo (1,52 g, 6,98 mmol) a una mezcla en agitación de 5-(2-amino-3,3-dimetilbutil)-2-cloropiridin-3-ol (1,33 g, 5,81 mmol) y trietilamina (2,43 ml, 17,4 mmol) en N,N-dimetilformamida (DMF) (30 ml). Se agitó la mezcla a temperatura ambiente durante 1 hora. La CL-EM mostró una conversión mínima. Los sólidos nunca se disolvieron completamente. Se añadió tetrahidrofurano (THF) (50 ml) y los sólidos pasaron a disolución. Se agitó la mezcla durante 2 horas, se concentró y se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron las fracciones y luego se liofilizó el residuo (acetonitrilo / agua) para dar (1-(6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (1,04 g, 3,16 mmol, rendimiento del 54,4 %). 1H-RMN (400 MHz, temp. = 80 °C, DMSO-ds) 5 ppm 9,81 - 10,14 (m, 1 H), 7,68 (d, J=1,56 Hz, 1 H), 7,15 (d, J=1,56 Hz, 1 H), 6,13 - 6,46 (m, 1 H), 3,24 - 3,46 (m, 1 H), 2,76 (dd, J=14,05, 2,34 Hz, 1 H), 2,31 - 2,42 (m, 1 H), 1,21 (s. a., 9 H), 0,91 (s, 9 H). CLEM (ES+) (m/z): 329, 331 (M+1).
Etapa 5: (1-(2-Bromo-6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo

Bajo nitrógeno, una disolución de N-bromosuccinimida (471 mg, 2,65 mmol) en 2 ml de DMF se añadió lentamente gota a gota a una disolución a 0 °C de (1-(6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (791 mg, 2,405 mmol) en N,N-dimetilformamida (DMF) (25 ml) y se agitó la mezcla en un vial de reacción que se
protegió e la luz mediante papel de aluminio. Se mantuvo el baño frío entre -25 °C y -20 °C durante 30 minutos. Se permitió que la mezcla se calentara hasta temperatura ambiente y se agitó durante la noche. Se extinguió la mezcla con agua y se extrajo 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera y se concentraron. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 60 %, luego isocrático al 60 %, luego gradiente del 60 % al 100 %). Se concentraron las fracciones para dar (1-(2-bromo-6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de tercbutilo (485 mg, 1,19 mmol, rendimiento del 49,5 %). 1H-RMN (400 MHz, DMSO-afe) " ppm 10,81 (s. a., 1 H), 7,23 (s, 1 H), 6,60 (d, J= 10,15 Hz, 1 H), 3,35 - 3,50 (m, 1 H), 2,73 - 2,89 (m, 1 H), 2,18 - 2,37 (m, 1 H), 1,15 (s, 9 H), 0,83 - 0,91 (m, 9 H). CLEM (ES+) (m/z): 407, 409, 411 (M+1).
Etapa 6: 1-(2-Bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-amina

Se añadió (bromometil)ciclopropano (0,231 ml, 2,38 mmol) a una mezcla en agitación de (1-(2-bromo-6-cloro-5-hidroxipiridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo (485 mg, 1,19 mmol) y carbonato de potasio (658 mg, 4,76 mmol) en N,N-dimetilformamida (DMF) (5 ml). Se agitó la mezcla a temperatura ambiente durante la noche. Se extinguió la mezcla con agua y se extrajo 2 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con cloruro de litio al 5 %, se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar (1-(2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-il)carbamato de terc-butilo en bruto. Se disolvió el producto intermedio en diclorometano (DCM) (5,00 ml) antes de que se añadiera cloruro de hidrógeno (4 M en dioxano) (5 ml, 20 mmol). Se agitó la mezcla durante 4 horas y se concentró para dar un sólido blanco. Se añadió bicarbonato de sodio saturado y se extrajo la mezcla 2 veces con acetato de etilo. Se secaron las fases orgánicas combinadas sobre sulfato de sodio y se concentraron para dar 1 -(2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2- amina (423 mg, 1,17 mmol, rendimiento del 98 %) como un aceite transparente. 1H-RMN (400 MHz, DMSO-afe) " ppm 7,62 (s, 1 H), 3,96 (d, J=7,03 Hz, 2 H), 2,89 (dd, J=12,88, 1,95 Hz, 1 H), 2,43 - 2,46 (m, 1 H), 2,20 - 2,33 (m, 1 H), 1,19 - 1,36 (m, 3 H), 0,92 (s, 9 H), 0,52 - 0,66 (m, 2 H), 0,26 - 0,42 (m, 2 H). CLEM (ES+) (m/z): 361,363, 365 (M+1).
Etapa 7: 1-(1-(2-Bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3- carboxilato de etilo

Se agitaron 1 -(2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-amina (375 mg, 1,04 mmol) y 4-oxo-4H-piran-3-carboxilato de etilo (174 mg, 1,04 mmol) en ácido acético (10 ml) a 100 °C durante 4 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se concentró. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se concentraron las fracciones y se liofilizó el residuo (agua / acetonitrilo) para dar 1-(1-(2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (420 mg, 0,821 mmol, rendimiento del 79 %) como un polvo blanco. CLEM (ES+) (m/z): 511,513, 515 (M+1).
Etapa 8: 6-(terc-Butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-piridoH ,2-hlH ,71 naftiridin-9-carboxilato de etilo

Un vial de reacción que contenía una barra agitadora, 1-(1-(2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)-3,3-dimetilbutan-2-il)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (395 mg, 0,772 mmol), acetato de potasio (151 mg, 1,543 mmol) y cloro[(tri-terc-butilfosfina)-2-(2-aminobifenil)]paladio (II) (79 mg, 0,15 mmol) se purgó con nitrógeno durante 15 minutos. Se purgó N,N-dimetilacetamida (DMA) (8 ml) con nitrógeno durante 5 minutos antes de añadirse al vial de reacción. Se colocó el vial de reacción en un bloque de calentamiento que se precalentó hasta 90 °C y se agitó la mezcla durante la noche. Se permitió que la mezcla de reacción se enfriara hasta temperatura ambiente, se filtró a través de un tapón de algodón y se purificó mediante cromatografía a presión media de fase inversa (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron las fracciones y se sometió el residuo a destilación azeotrópica con acetonitrilo para ayudar a eliminar el agua restante. Se disolvió el residuo en diclorometano y hexanos antes de concentrarse para dar 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (140 mg, 0,325 mmol, rendimiento del 42,1 %) como un sólido amarillo pálido. 1H-RMN (400 MHz, DMSO-d6) 5 ppm 8,38 (s, 1 H), 7,66 (s, 1 H), 6,90 (s, 1 H), 4,40 (d, J=6,25 Hz, 1 H), 4,21 (q, J=7,03 Hz, 2 H), 3,99 - 4,10 (m, 2 H), 3,37 - 3,50 (m, 1 H), 3,31 - 3,37 (m, 1 H), 1,22 - 1,37 (m, 4 H), 0,74 (s, 9 H), 0,59 - 0,67 (m, 2 H), 0,34 - 0,45 (m, 2 H). CLEM (ES+) (m/z): 431,433 (M+1).
Etapa 9: Ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico

Una disolución de hidróxido de litio monohidratado (1,6 mg, 0,039 mmol) en agua (1 ml) se añadió a una disolución de 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (2,0 mg, 3,9 pmol) en metanol (1 ml) y se calentó la mezcla a 60 °C durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se purificó mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron las fracciones y se secó el residuo para dar ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (1,2 mg, 2,9 pmol, rendimiento del 77 %) como un sólido blanco. 1H-RMN (400 MHz, METANOL-a4) 5 ppm 8,70 (s. a., 1 H), 7,54 (s. a., 1 H), 7,47 (s. a., 1 H), 4,44 - 4,55 (m, 1 H), 3,97 - 4,14 (m, 2 H), 3,39 - 3,61 (m, 2 H), 1,33 (s. a., 1 H), 1,12 (s. a., 1 H), 0,83 (s, 9 H), 0,63 - 0,71 (m, 2 H), 0,34 - 0,50 (m, 2 H). CLEM (ES+) (m/z): 403, 405 (M+1).
Ejemplo 30-a (compuesto 249 isómero 1)
Ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (isómero 1)

Etapa 1: 6-(terc-Butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,71naftiridin-9-carboxilato de etilo (isómero 1)

Se purificó 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo racémico (153 mg) mediante cromatografía quiral usando las siguientes condiciones: Columna = Chiralpak IC, 10 mm x 250 mm (5 u); fase móvil = MeCN/H2O 95:5 ácido fórmico al 0,1 %; velocidad de flujo = 10 ml/min: volumen de inyección = 300 ul (conc. de 18 mg/ml); longitud de onda de recogida: 254 nm. Se concentraron las fracciones para dar 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (isómero 1) (48 mg). CLEM (ES+) (m/z): 431,433 (M+1).
Etapa 2: Ácido 6-(terc-but¡l)-2-cloro-3-(c¡cloprop¡lmetox¡)-10-oxo-6.10-d¡h¡dro-5H-p¡r¡do[1.2-hl[1.7lnaft¡r¡d¡n-9-carboxílico (isómero 1)

Una disolución de hidróxido de litio monohidratado (38,8 mg, 0,925 mmol) en agua (2 ml) se añadió a una disolución de 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (isómero 1) (48 mg, 0,092 mmol) en metanol (2 ml) y se calentó la mezcla a 60 °C durante 3 horas. Se enfrió la mezcla hasta temperatura ambiente y se acidificó con 3 ml de ácido clorhídrico 1 M. Se recogieron los sólidos mediante filtración. Se inyectó el filtrado sobre una columna de fase inversa a presión media. Se disolvió el sólido recogido en DMF y se inyectó sobre la columna de fase inversa. Se eluyó la columna (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (isómero 1) (33 mg, 0,082 mmol, rendimiento del 89 %) como un polvo blanco. 1H-RMN (400 MHz, DMSO-d6) 5 ppm 8,75 (s. a., 1 H), 7,66 (s, 1 H), 7,24 (s. a., 1 H), 4,55 - 4,68 (m, 1 H), 3,93 - 4,10 (m, 2 H), 3,39 - 3,54 (m, 2 H), 1,27 (d, J=5,08 Hz, 1 H), 0,70 (s, 9 H), 0,55 - 0,62 (m, 2 H), 0,30 - 0,39 (m, 2 H). CLEM (ES+) (m/z): 403, 405 (M+1).
Ejemplo 30-b (compuesto 249 ¡sómero 2)
Ác¡do 6-(terc-but¡l)-2-cloro-3-(c¡cloprop¡lmetox¡)-10-oxo-6,10-d¡h¡dro-5H-p¡r¡do[1,2-hl[1,7lnaft¡r¡d¡n-9-carboxíl¡co (¡sómero 2)

Etapa 1: 6-(terc-But¡l)-2-cloro-3-(c¡cloprop¡lmetox¡)-10-oxo-5.10-d¡h¡dro-6H-p¡r¡do[1.2-hl[1.7lnaft¡r¡d¡n-9-carbox¡lato de et¡lo (¡sómero 2)

Se purificó 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo racémico (153 mg) mediante cromatografía quiral usando las siguientes condiciones: Columna = Chiralpak IC, 10 mm x 250 mm (5 u); fase móvil = MeCN/H2O 95:5 ácido fórmico al 0,1 %; velocidad de flujo = 10 ml/min: volumen de inyección = 300 ul (conc. de 18 mg/ml); longitud de onda de recogida: 254 nm. Se concentraron las fracciones para dar 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (isómero 2) (46 mg). CLEM (ES+) (m/z): 431,433 (M+1).
Etapa 2: Ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-6,10-dihidro-5H-pirido[1,2-h][1,71naftiridin-9-carboxílico (isómero 2)

Una disolución de hidróxido de litio monohidratado (37,2 mg, 0,886 mmol) en agua (2 ml) se añadió a una disolución de 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxilato de etilo (isómero 2) (46 mg, 0,089 mmol) en metanol (2 ml) y se calentó la mezcla a 60 °C durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se acidificó con 3 ml de ácido clorhídrico 1 M. Se recogieron los sólidos mediante filtración. Se inyectó el filtrado sobre una columna de fase inversa a presión media. Se disolvió el sólido recogido en DMF y también se inyectó sobre la columna de fase inversa. Se eluyó la columna (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido 6-(terc-butil)-2-cloro-3-(ciclopropilmetoxi)-10-oxo-5,10-dihidro-6H-pirido[1,2-h][1,7]naftiridin-9-carboxílico (isómero 2) (34 mg, 0,084 mmol, rendimiento del 95 %) como un polvo blanco. 1H-RMN (400 MHz, DMSO-d6) " ppm 8,76 (s, 1 H), 7,67 (s, 1 H), 7,25 (s, 1 H), 4,63 (d, J=6,64 Hz, 1 H), 3,96 - 4,11 (m, 2 H), 3,33 - 3,54 (m, 2 H), 1,19 - 1,33 (m, 1 H), 0,70 (s, 9 H), 0,56 - 0,62 (m, 2 H), 0,30 - 0,42 (m, 2 H). CLEM (ES+) (m/z): 403, 405 (M+1).

Ejemplo 31 (compuesto 250)
Ácido 2'-cloro-3'-(ciclopropilmetoxi)-10'-oxo-5\10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico

Etapa 1: 6-Bromo-2-cloro-5-metilpiridin-3-ol

Se calentaron 6-bromo-5-metilpiridin-3-ol (10,4 g, 55,3 mmol) y NCS (8,12 g, 60,8 mmol) en N,N-dimetilformamida (DMF) (150 ml) a 80 °C durante 2 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se extinguió con salmuera y se extrajo 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con cloruro de litio al 5 %, se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 50 % de acetato de etilo en hexanos. Se concentraron las fracciones para dar 6-bromo-2-cloro-5-metilpiridin-3-ol (7,85 g, 35,3 mmol, rendimiento del 63,8 %) como un polvo blanco. CLEM (ES+) (m/z): 222, 224, 226 (M+1).
Etapa 2: 2-Bromo-6-cloro-5-(ciclopropilmetoxi)-3-metilpiridina

Se agitaron 6-bromo-2-cloro-5-metilpiridin-3-ol (1,1 g, 4,9 mmol), carbonato de potasio (2,73 g, 19,8 mmol) y (bromometil)ciclopropano (1,01 ml, 10,4 mmol) en N,N-dimetilformamida (DMF) (10 ml) a temperatura ambiente durante la noche. Se diluyó la mezcla con agua y se extrajo 2 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con cloruro de litio al 5 %, se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar 2-bromo-6-cloro-5-(ciclopropilmetoxi)-3-metilpiridina (1,18 g, 4,27 mmol, rendimiento del 86 %) como un sólido blanco. CLEM (ES+) (m/z): 276, 278, 280 (M+1).
Etapa 3: 2-Bromo-3-(bromometil)-6-cloro-5-(ciclopropilmetoxi)piridina

Se añadieron N-bromosuccinimida (0,801 g, 4,50 mmol) y AIBN (0,049 g, 0,30 mmol) a una disolución en agitación de 2-bromo-6-cloro-5-(ciclopropilmetoxi)-3-metilpiridina (1,0 g, 3,0 mmol) en tetracloruro de carbono (15 ml). Se calentó la mezcla a 85 °C durante 5 horas. Se añadió AIBN adicional (0,049 g, 0,300 mmol) y se permitió que la mezcla se calentara durante la noche. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se recogieron los sólidos mediante filtración. Se extinguió el filtrado con agua y se extrajo 2 veces con diclorometano. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía en sílice eluyendo con el 10 % de acetato de etilo en hexanos. Se concentraron las fracciones para dar 2-bromo-3-(bromometil)-6-cloro-5-(ciclopropilmetoxi)piridina (730 mg, 1,23 mmol, 60 % puro, rendimiento del 41 %). El material era una mezcla ~60:40 de producto deseado / material de partida. CLEM (ES+) (m/z): 354, 356, 358 (M+1).
Etapa 4: 1-((2-Bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutanocarboxilato de etilo

Se añadió n-butil-litio (2,5 M en hexanos) (1,04 ml, 2,59 mmol) gota a gota a una disolución a -78 °C de diisopropilamina (0,369 ml, 2,59 mmol) en tetrahidrofurano (THF) (10 ml). Se permitió que la mezcla se calentara hasta temperatura ambiente y se agitó durante 15 minutos. Se enfrió la mezcla hasta -78 °C antes de que se añadiera ciclobutanocarboxilato de etilo (0,317 ml, 2,46 mmol) gota a gota. Se calentó la mezcla hasta 0 °C y se agitó durante 15 minutos. Se añadió una disolución de 2-bromo-3-(bromometil)-6-cloro-5-(ciclopropilmetoxi)piridina (730 mg, 1,23 mmol) en tetrahidrofurano (THF) (3,33 ml) gota a gota. Se agitó la mezcla 10 minutos adicionales a 0 °C y luego se extinguió con cloruro de amonio saturado. Se extrajo la mezcla 2 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron las fracciones para dar 1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3
il)metil)ciclobutano-1-carboxilato de etilo (350 mg, 0,869 mmol, rendimiento del 70,5 %) como un aceite. CLEM (ES+) (m/z): 402, 404, 406 (M+1).
Etapa 5: Ácido 1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutanocarboxílico

Una disolución de hidróxido de litio monohidratado (365 mg, 8,69 mmol) en agua (3 ml) se añadió a una disolución de 1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutano-1-carboxilato de etilo (350 mg, 0,869 mmol) en 1,4-dioxano (5 ml) y se calentó la mezcla a 60 °C durante 4 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se extinguió con cloruro de amonio saturado y se extrajo 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron para dar ácido 1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutano-1-carboxílico (325 mg, 0,867 mmol, cuantitativo) como un sólido blanco. CLEM (ES+) (m/z): 374, 376, 378 (M+1).
Etapa 6: 1 -((2-Bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutanamina
y
-5-ííl: aminociclobutil)metil)-6-bromo-2-cloropiridin-3-ol

Se añadió fosforazidato de difenilo (0,394 ml, 1,83 mmol) gota a gota a una mezcla en agitación a 0 °C de ácido 1 -((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutano-1-carboxílico (311 mg, 0,830 mmol) y trietilamina (0,255 ml, 1,83 mmol) en tolueno (10 ml). Se permitió que la mezcla se calentara hasta temperatura ambiente y se agitó durante 15 minutos antes de calentarse a 80 °C durante 1 hora. La CL-EM mostró una conversión limpia en 2-bromo-6-cloro-5-(ciclopropilmetoxi)-3-((1 -isocianatociclobutil)metil)piridina. Se permitió que la mezcla se enfriara hasta temperatura ambiente antes de que se añadieran cloruro de hidrógeno 5 N (5 ml, 25 mmol) y 1,4-dioxano (10 ml). Se calentó la mezcla a 80 °C con agitación vigorosa durante 3 horas. Se concentró la mezcla y se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron dos conjuntos de fracciones por separado para dar los siguientes productos:
5-((1-Aminociclobutil)metil)-6-bromo-2-cloropiridin-3-ol (170 mg, 0,583 mmol, rendimiento del 70,2 %). CLEM (ES+) (m/z): 291,293, 295 (M+1).
1-((2-Bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutan-1-amina (50 mg, 0,145 mmol, rendimiento del 17,43 %). CLEM (ES+) (m/z): 345, 347, 349 (M+1).
Etapa 7: 4-Oxo-4H-piran-3-carboxilato de etilo

Se añadió lentamente formiato de etilo (23,6 ml, 292 mmol) a una suspensión de KOtBu (6,06 g, 54,0 mmol) en 50 ml de THF a 0 °C. Se agitó la mezcla de reacción durante 15 min a 0 °C y se añadió una disolución enfriada previamente (0 °C) de 2-((dimetilamino)metilen)-3-oxobutanoato de etilo (5 g, 27,0 mmol) en 45 ml de THF mediante transferencia con cánula. Después de completarse la adición, se retiró la disolución del baño de enfriamiento, se agitó a ta durante la noche y se extinguió mediante la adición de HCI acuoso 1 M (80 ml). Se extrajo la mezcla con EtOAc (3 x 25 ml) y CH2Cl2 (5 x 25 ml). Se agruparon las capas orgánicas, se secaron (Na2SO4), se filtraron, se evaporaron y se purificaron mediante cromatografía en gel de sílice (el 0-100 % de EtOAc/hexanos) para proporcionar 4-oxo-4H-piran-3-carboxilato de etilo (1,8994 g, rendimiento del 41,8 %) como un aceite marrón. 1H-RMN (400 MHz, CDCh) " ppm 8,47 (d, J=0,78 Hz, 1 H), 7,72 (dd, J=5,86, 0,78 Hz, 1 H), 6,47 (d, J=5,86 Hz, 1 H), 4,37 (q, J=7,29 Hz, 2 H), 1,38 (t, J=7,03 Hz, 3 H). CLEM (ESI) m/z: 169,05 (M+1)+.
Etapa 8: 1-(1 -((2-Bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutil)-4-oxo-1,4-dihidropiridin-3-
carboxilato de etilo

Se agitaron 5-((1-aminociclobutil)metil)-6-bromo-2-cloropiridin-3-ol (170 mg, 0,583 mmol) y 4-oxo-4H-piran-3-carboxilato de etilo (98 mg, 0,58 mmol) en ácido acético (5 ml) a 100 °C durante 4 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se concentró. Se diluyó el residuo en tolueno y se concentró la mezcla para eliminar el ácido acético restante. Se disolvió el residuo en N,N-dimetilformamida (DMF) (2 ml) antes de que se añadieran carbonato de potasio (322 mg, 2,33 mmol) y (bromometil)ciclopropano (0,113 ml, 1,17 mmol). Se agitó la mezcla a temperatura ambiente durante 6 horas para dar una mezcla en bruto de 1-(1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo en DMF. En un matraz de reacción separado, se agitaron 1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutan-1-amina (50 mg, 0,145 mmol) y 4-oxo-4H-piran-3-carboxilato de etilo (24,3 mg, 0,145 mmol) en ácido acético (1 ml) a 100 °C durante la noche. Se permitió que la mezcla se enfriara hasta temperatura ambiente y luego se combinó con la mezcla en bruto anterior de 1 -(1 -((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo en DMF dada a conocer anteriormente. Se diluyó la mezcla con agua y se extrajo 2 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con cloruro de litio al 5 %, se lavaron con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron las fracciones hasta que eran turbias. Se añadió acetonitrilo para disolver completamente y se liofilizó la disolución para dar 1 -(1 -((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (100 mg, 0,202 mmol, rendimiento del 26 %) como un sólido amarillo pálido. CLEM (ES+) (m/z): 495, 497, 499 (M+1).
Etapa_______ 9:_______ 2,-Cloro-3,-(ciclopropilmetoxi)-10,-oxo-5,,10,-dihidroespirorciclobutano-1,6,-piridon ,2-hirUlnaft¡r¡d¡nl-9'-carbox¡lato de etilo

Un vial de reacción que contenía una barra agitadora, 1-(1-((2-bromo-6-cloro-5-(ciclopropilmetoxi)piridin-3-il)metil)ciclobutil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (100 mg, 0,202 mmol), acetato de potasio (39,6 mg, 0,403 mmol) y cloro[(tri-terc-butilfosfina)-2-(2-aminobifenil)]paladio (II) (20,7 mg, 0,040 mmol) se purgó con nitrógeno durante 15 minutos. Se purgó N,N-dimetilacetamida (DMA) (2 ml) con nitrógeno durante 5 minutos antes de añadirse al vial de reacción. Se colocó el vial de reacción en un bloque de calentamiento que se precalentó hasta 90 °C y se agitó la mezcla durante la noche. Se permitió que la mezcla de reacción se enfriara hasta temperatura ambiente, se filtró a través de un tapón de algodón y se purificó mediante cromatografía a presión media de fase inversa (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar 2'-cloro-3'-(ciclopropilmetoxi)-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxilato de etilo (30 mg, 0,072 mmol, rendimiento del 35,9 %) como un sólido blanquecino. CLEM (ES+) (m/z): 415, 417 (M+1).
Etapa 10: Ácido 2,-cloro-3,-(ciclopropilmetoxi)-10'-oxo-5',10'-dihidroespirorciclobutano-1,6'-piridor1,2-hir1,71naftiridin1-9'-carboxílico

Una disolución de hidróxido de litio monohidratado (25,2 mg, 0,600 mmol) en agua (1 ml) se añadió a una disolución de 2'-cloro-3'-(ciclopropilmetoxi)-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxilato de etilo (30 mg, 0,060 mmol) en metanol (1 ml) y se calentó la mezcla a 60 °C durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente y se acidificó con ácido clorhídrico 1 M. Se diluyó la mezcla con DMF para disolver la muestra y luego se purificó mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones combinadas para dar ácido 2'-cloro-3'-(ciclopropilmetoxi)-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico (22 mg, 0,056 mmol, rendimiento del 94 %) como un polvo blanquecino. 1H-RMN (400 MHz, DMSO-ak) 5 ppm 8,79 (s, 1 H), 7,68 (s, 1 H), 7,27 (s, 1 H), 4,07 (d, J=7,03 Hz, 2 H), 3,44 (s, 2 H), 2,61 - 2,75 (m, 2 H), 2,00 - 2,12 (m, 2 H), 1,78 - 1,95 (m, 2 H), 1,25 - 1,37 (m, 1 H), 0,57 - 0,68 (m, 2 H), 0,33 - 0,40 (m, 2 H). CLEM (ES+) (m/z): 387, 389 (M+1).
Ejemplo 32 (compuesto 251)
,
Ácido 2',3'-dimetoxi-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico

Etapa 1: Ácido 2',3'-dimetoxi-10'-oxo-5M0'-dihidroespirorciclobutano-1,6'-piridoH,2-hir1,71naftiridin1-9'-carboxílico

Se calentaron 2'-cloro-3'-(ciclopropilmetoxi)-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico (10 mg, 0,026 mmol) y metóxido de sodio (0,5 M en metanol) (0,517 ml, 0,259 mmol) a 65 °C durante 2 horas. Se añadió dioxano (0,5 ml) y se calentó la mezcla hasta 100 °C. Se dejó el vial sin tapar inicialmente de modo que la mayor parte del metanol se eliminó por destilación. Se tapó el vial de reacción y se agitó a 100 °C durante 1,5 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se acidificó con HCl 1 N y se purificó mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se liofilizaron las fracciones para dar ácido 2',3'-dimetoxi-10'-oxo-5',10'-dihidroespiro[ciclobutano-1,6'-pirido[1,2-h][1,7]naftiridin]-9'-carboxílico (1,5 mg, 4,03 pmol, rendimiento del 15,6 %) como un sólido blanco. 1H-RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,90 (s, 1 H), 7,65 (s, 1 H), 6,93 (s, 1 H), 4,08 (s, 3 H), 3,97 (s, 3 H), 3,26 (s, 2 H), 2,57 - 2,75 (m, 2 H), 2,15 - 2,29 (m, 2 H), 2,00 - 2,12 (m, 2 H). CLEM (ES-) (m/z): 341 (M-1)-.
Ejemp

el ejemplo 33 (compuesto 252)

Esquema 14 - Preparación del ejemplo 33 (compuesto 252)

Ejemplo 33 (compuesto 252)
Ácido 6-isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxílico

Etapa 1: 2-Cloro-5,6-dimetilnicotinonitrilo

Una mezcla de 2-hidroxi-5,6-dimetilnicotinonitrilo (10 g, 67,5 mmol) en POCI3 (100 ml) se sometió a reflujo durante 10 h y se concentró. Se basificó el residuo con disolución acuosa de NaOH al 10 % y se filtró. Se lavó la torta de filtro con H2O. Se extrajo el filtrado con DCM (200 ml x 2). Se combinaron las capas orgánicas, se lavaron con salmuera (200 ml), se secaron sobre Na2SO4 anhidro y se concentraron para dar 9,8 g de 2-cloro-5,6-dimetilnicotinonitrilo en
bruto como un sólido amarillo. CLEM (ESI) m/z: 167,1, 169,1 (M/M+2)+.
Etapa 2: 2-Cloro-5.6-d¡metiln¡cot¡naldehído

A una disolución a -78 °C de 2-cloro-5,6-dimetilnicotinonitrilo (1,6 g, 9,6 mmol) en DCM (20 ml), se le añadió DIBAL-H (5,76 ml, 2 mol/l en hexano). Se agitó la mezcla resultante a la misma temperatura durante 2 h. Se extinguió la reacción añadiendo agua helada. Entonces se acidificó la mezcla con HCI 1 N hasta pH = ~6 y se extrajo con DCM (100 ml x 2). Se combinaron las capas orgánicas, se lavaron con salmuera (50 ml), se secaron sobre Na2SÜ4 anhidro y se concentraron para dar 1,5 g de 2-cloro-5,6-dimetilnicotinaldehído en bruto como un sólido blanco. CLEM (ESI) m/z: 170,1, 172,1 (M/M+2)+.
Etapa 3: 2-Cloro-3-(1.3-d¡oxolan-2-¡l)-5.6-d¡met¡lp¡r¡d¡na

Una mezcla de 2-cloro-5,6-dimetilnicotinaldehído (1,5 g, 8,84 mmol), etano-1,2-diol (1,1 g, 17,68 mmol), PPTS (444,3 mg, 1,768 mmol) en tolueno (20 ml) se sometió a reflujo durante 12 h. Se enfrió la mezcla de reacción hasta t.a. y se extrajo con EtOAc (100 ml x 2). Se combinaron las capas orgánicas, se lavaron con disolución acuosa de NaHCÜ3 (50 ml), se secaron sobre Na2SO4 anhidro y se concentraron. Se purificó el residuo mediante cromatografía en columna ultrarrápida (gel de sílice, el 0-50 % de EtOAc en PE) para proporcionar 2-cloro-3-(1,3-dioxolan-2-il)-5,6-dimetilpiridina (1,35 g, rendimiento del 71,5 %) como un aceite incoloro. CLEM (ESI) m/z: 214,1,216,1 (M/M+2)+.
Etapa 4: 1-(3-(1.3-D¡oxolan-2-¡l)-5.6-d¡met¡lp¡r¡d¡n-2-¡l)-3-met¡lbutan-2-ona

Una mezcla de 2-cloro-3-(1,3-dioxolan-2-il)-5,6-dimetilpiridina (1,35 g, 6,32 mmol), 3-metilbutan-2-ona (653 mg, 7,58 mmol), Pd(dtbpf)Cl2 (411,9 mg, 0,632 mmol) y t-BuONa (1,52 g, 15,8 mmol) en THF (20 ml) se calentó a 60 °C durante 3 h. Se enfrió la mezcla de reacción hasta t.a. y se filtró. Se concentró el filtrado y se purificó el residuo mediante la cromatografía en columna ultrarrápida (gel de sílice, el 0-70 % de EtOAc en PE) para proporcionar 1 -(3-(1,3-dioxolan-2-il)-5,6-dimetilpiridin-2-il)-3-metilbutan-2-ona (231 mg, rendimiento del 14 %) como un sólido amarillo. CLEM (ESI) m/z: 264,3 (M+1)+.
Etapa 5: 1-(3-(1.3-D¡oxolan-2-¡l)-5.6-d¡met¡lp¡r¡d¡n-2-¡l)-3-met¡lbutan-2-am¡na

A una disolución de 1-(3-(1,3-dioxolan-2-il)-5,6-dimetilpiridin-2-il)-3-metilbutan-2-ona (231 mg, 0,878 mmol) en MeOH (10 ml) se le añadió NH4OAc (683,1 mg, 8,78 mmol) a temperatura ambiente. Tras agitarse la mezcla durante 30 min, se añadió NaBHaCN (82,8 mg, 1,317 mmol) a la mezcla a 0 °C. Entonces se agitó la mezcla resultante a 50 °C durante 10 h. Se enfrió la mezcla de reacción hasta t.a. y se concentró. Se diluyó el residuo con H2O (20 ml), se basificó hasta pH~10 con disolución de NaOH al 10 %. Se extrajo la mezcla acuosa con DCM (2X). Se combinaron las capas
orgánicas, se lavaron con salmuera (30 ml), se secaron sobre Na 2 SÜ 4 anhidro y se concentraron a presión reducida para proporcionar 236 mg de 1 -(3-(1,3-dioxolan-2-il)-5,6-dimetilpiridin-2-il)-3-metilbutan-2-amina en bruto. CLEM (ESI) m/z: 265,3 (M+1)+.
Etapa 6: 7-Isopropil-2,3-dimetil-7,8-dihidro-1,6-naftiridina

A una disolución de 1-(3-(1,3-dioxolan-2-il)-5,6-dimetilpiridin-2-il)-3-metilbutan-2-amina (70 mg, 0,264 mmol) en THF (10 ml), se le añadió disolución acuosa de HCI al 10 % (2 ml). Se calentó la mezcla resultante a 50 °C durante 10 h. Tras enfriarse hasta ta, se concentró la mezcla. Se purificó el residuo mediante cromatografía en columna (gel de sílice, el 0-10 % de MeOH en DCM) para dar 7-isopropil-2,3-dimetil-7,8-dihidro-1,6-naftiridina (20 mg, rendimiento del 37,3 %). CLEM (ESI) m/z: 203,27 (M+1)+.
Etapa 7: 6-Isopropil-2,3-dimetil-10-oxo-5,10,11,11 a-tetrahidro-6H-piridor2,1 -/iH ,61naftiridin-9-carboxilato de etilo

Una disolución de 7-isopropil-2,3-dimetil-7,8-dihidro-1,6-naftiridina (20 mg, 0,099 mmol) y 2-(etoximetilen)-3-oxobutanoato de etilo (55,86 mg, 0,3 mmol) en EtOH (5 ml) se calentó a 85 °C durante 10 h. Tras enfriarse hasta temperatura ambiente, se concentró la mezcla. Se purificó el residuo mediante cromatografía en columna (gel de sílice, el 0-10 % de MeOH en DCM) para dar 6-isopropil-2,3-dimetil-10-oxo-5,10,11,11a-tetrahidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxilato de etilo (20 mg, rendimiento del 58,9 %). CLEM (ESI) m/z: 343,2 (M+1)+.
Etapa 8: 6-Isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-piridor2,1-/ir1,61naftiridin-9-carboxilato de etilo

A una disolución de 6-isopropil-2,3-dimetil-10-oxo-5,10,11,11 a-tetrahidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxilato de etilo (20 mg, 0,058 mmol) en DME (5 ml) se le añadió p-cloranilo (12,3 mg, 0,05 mmol). Se calentó la mezcla a 80 °C durante 3 h. Tras enfriarse hasta t.a., se concentró la mezcla. Se purificó el residuo mediante cromatografía en columna (gel de sílice, el 0-10 % de MeOH en DCM) para proporcionar 6-isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxilato de etilo (15 mg, rendimiento del 76 %). CLEM (ESI) m/z: 341,2 (M+1)+.
,
Etapa 9: Ácido 6-isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-piridor2,1-f1H,61naftiridin-9-carboxílico

A una disolución de 6-isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxilato de etilo (15 mg, 0,044 mmol) en MeOH (5 ml), se le añadió NaOH (7 mg, 0,176 mmol) disuelto en H
2
O (1 ml). Se agitó la mezcla resultante a temperatura ambiente durante 2 h. Se acidificó la mezcla de reacción hasta pH~5 con HCl 1 N.
Entonces se diluyó la mezcla con EtOAc (20 ml) y H2O (20 ml), y luego se extrajo con EtOAc (20 ml x 2). Se secaron las fases orgánicas combinadas sobre Na2SO4 anhidro y se concentraron. Se purificó el residuo mediante HPLC de fase inversa (columna C18, el 5 %-100 % de MeCN en H2O, con ácido fórmico al 0,1 % en H2O) para dar ácido 6-isopropil-2,3-dimetil-10-oxo-5,10-dihidro-6H-pirido[2,1-f][1,6]naftiridin-9-carboxílico (5 mg, rendimiento del 36,3 %) como un sólido blanco. 1H-RMN (400 MHz, DMSO-d6) 58,87 (s, 1H), 8,25 (s, 1H), 7,43 (s, 1H), 4,63 - 4,58 (m, 1H), 3,56 - 3,50 (m, J = 16,8, 5,3 Hz, 1H), 3,18 (d, J = 16,6 Hz, 1H), 2,48 (s, 3H), 2,31 (s, 3H), 1,64 - 1,57 (m, 1H), 0,84 (d, J = 6,6 Hz, 3H), 0,69 (d, J = 6,7 Hz, 3H). CLEM (ESI) m/z: 313,2 (M 1)+.
Ejemplo 34 (Compuesto 253)
Ácido 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (cis racémico)

Etapa 1: 5-(6-Cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentanona

Se cargó un matraz con una barra agitadora, 5-bromo-2-cloro-3-metoxipiridina (9,9 g, 44 mmol), tris(dibencilidenacetona)dipaladio (0) (0,611 g, 0,668 mmol), xantphos (0,695 g, 1,202 mmol) y terc-butóxido de sodio (7,5 g, 78 mmol). Se purgó el matraz con una corriente de nitrógeno antes de que se añadiera 2,2-dimetilciclopentan-1-ona (8,38 ml, 66,8 mmol) en tetrahidrofurano (THF) (200 ml). Se calentó la mezcla hasta reflujo durante 3 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se filtró y se lavó la torta de filtro con acetato de etilo. Se diluyó el filtrado con acetato de etilo adicional, se lavó con salmuera, se concentró y se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 30 % de acetato de etilo en hexanos. Se concentraron las fracciones para dar 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-ona (4,6 g, 18 mmol, rendimiento del 41 %) como un aceite. 1H-RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,82 (d, J=1,56 Hz, 1 H), 7,13 (d, J=1,95 Hz, 1 H), 3,91 (s, 3 H), 3,36 - 3,47 (m, 1 H), 2,45 (m, 1 H), 1,96 - 2,17 (m, 2 H), 1,79 - 1,92 (m, 1 H), 1,18 (s, 3 H), 1,07 (s, 3 H). CLEM (ES+) (m/z): 254,2 (M+1).
Etapa 2: 5-(6-Cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentanamina (isómeros cis / trans)

Una mezcla de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1 -ona (4,6 g, 18 mmol) y acetato de amonio (21 g, 270 mmol) en metanol (75 ml) se agitó a temperatura ambiente durante 1 hora. Se añadió cianoborohidruro de sodio (2,28 g, 36,3 mmol) y se calentó la mezcla a 60 °C durante la noche y luego a 65 °C durante 24 horas adicionales. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se concentró hasta -20 ml, se extinguió con hidróxido de sodio 1 M y se extrajo 3 veces con 2-metiltetrahidrofurano. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se concentraron las fracciones para dar 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-amina (2,63 g, 10,3 mmol, rendimiento del 57 %) como una mezcla de isómeros cis / trans. CLEM (ES+) (m/z): 255,2 (M+1).
s)
Etapa 3: 5-(2-Amino-3,3-dimetilciclopentil)-2-cloropiridin-3-ol (isómeros cis / tran

Se añadió tribromuro de boro (4,82 ml, 51,0 mmol) lentamente gota a gota a una disolución en agitación vigorosa de 5-(6-cloro-5-metoxipiridin-3-il)-2,2-dimetilciclopentan-1-amina (2,6 g, 10,2 mmol) (isómeros cis/trans). Se calentó la mezcla a 70 °C durante la noche. Se permitió que la mezcla se enfriara hasta temperatura ambiente y luego se enfrió hasta 0 °C. Se extinguió cuidadosamente la mezcla con adición lenta de metanol. Se añadieron 50 ml adicionales de metanol y se agitó la mezcla durante 30 minutos antes de concentrarse. Se purificó el residuo mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %). Se liofilizaron las fracciones para dar 5-(2-amino-3,3-dimetilciclopentil)-2-cloropiridin-3-ol (isómeros cis/trans) (2,17 g, 9,01 mmol, rendimiento del 88 %) como un sólido blanco. CLEM (ES+) (m/z): 241,2 (M+1).
Etapa 4: 5-(2-Amino-3,3-dimetilciclopentil)-2-cloro-6-vodopiridin-3-ol (cis racémico) y 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (trans racémico)

Se añadió yodo (2,29 g, 9,01 mmol) a una mezcla en agitación de 5-(2-amino-3,3-dimetilciclopentil)-2-cloropiridin-3-ol (2,17 g, 9,01 mmol) y carbonato de potasio (3,74 g, 27,0 mmol) en agua (40 ml). Se agitó la mezcla a temperatura ambiente durante 2 horas. Se añadió sulfito de sodio sólido (2,39 g, 18,9 mmol) en porciones y se agitó la mezcla durante 30 minutos. Se ajustó la mezcla acuosa a pH = ~3 con ácido clorhídrico 3 N. Se filtró la mezcla. La CL-EM mostró que el sólido contenía una pequeña cantidad de producto, pero se desechó. Se concentró el lavado hasta 50 ml y se inyectó sobre una columna C18 ISCO de fase inversa a presión media que se eluyó con acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 0 % al 100 %. Se liofilizaron 2 conjuntos de fracciones por separado.
Pico 1 = 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (trans racémico) (1,17 g, 3,19 mmol, rendimiento del 35 %). 1H-RMN (400 MHz, DMSO-d6) 5 ppm 8,23 (s, 1 H), 7,26 (s, 1 H), 2,99 - 3,15 (m, 2 H), 2,05 - 2,19 (m, 1 H), 1,51 - 1,64 (m, 2 H), 1,22 - 1,35 (m, 1 H), 1,09 (s, 3 H), 0,95 (s, 3 H). CLEM (ES+) (m/z): 367,1 (M+1).
Pico 2 = 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (cis racémico contaminado con ~20 % de trans racémico) (924 mg, 2,01 mmol, rendimiento del 22 %). 1H-RMN (400 MHz, DMSO-d6) 5 ppm 8,27 (s, 1 H), 7,20 (s, 1 H), 3,41 - 3,53 (m, 1 H), 3,28 (d, J=5,86 Hz, 1 H), 2,07 - 2,19 (m, 1 H), 1,81 - 1,95 (m, 1 H), 1,68 - 1,77 (m, 1 H), 1,47 - 1,57 (m, 1 H), 1,14 (s, 3 H), 1,08 (s, 3 H). CLEM (ES+) (m/z): 367,1 (M+1).
Etapa 5: 1 -(5-(6-Cloro-5-hidroxi-2-yodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (cis racémico)

Se agitaron 5-(2-amino-3,3-dimetilciclopentil)-2-cloro-6-yodopiridin-3-ol (cis racémico) (924 mg, 2,02 mmol) (contaminado con ~20 % de trans) y 4-oxo-4H-piran-3-carboxilato de etilo (466 mg, 2,77 mmol) en ácido acético (13 ml) a 100 °C durante 4 horas. Se permitió que la mezcla se enfriara hasta temperatura ambiente, se diluyó con agua y se extrajo 2 veces con diclorometano. Se añadió salmuera a la fase acuosa y se extrajo la mezcla 2 veces más con diclorometano. Se lavaron las capas orgánicas combinadas con salmuera, se secaron sobre sulfato de sodio y se concentraron. Se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 10 % de metanol en diclorometano. Se concentraron las fracciones para dar 1-(5-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (cis racémico) (346 mg, 0,670 mmol, rendimiento del 33 %). 1H-RMN (400 MHz, DMSO-d6) 5 ppm 10,92 (s. a., 1 H), 7,78 (d, J=1,95 Hz, 1 H), 7,43 (dd, J=7,81,2,34 Hz, 1 H), 7,07 (s, 1 H), 6,04 (d, J=7,42 Hz, 1 H), 4,54 (d, J=7,42 Hz, 1 H), 4,03 - 4,17 (m, 2 H), 3,82 - 3,95 (m, 1 H), 2,15 -2,27 (m, 2 H), 1,99 (dt, J=13,66, 7,22 Hz, 1 H), 1,74 - 1,87 (m, 1 H), 1,24 - 1,31 (m, 3 H), 1,14 - 1,23 (m, 3 H), 0,92 (s, 3 H). CLEM (ES+) (m/z): 517,1 (M+1).
Etapa 6: 1 -(5-(6-Cloro-2-vodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (cis racémico)

Se agitaron 1-(5-(6-cloro-5-hidroxi-2-yodopiridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (cis racémico) (346 mg, 0,670 mmol), carbonato de potasio (463 mg, 3,35 mmol) y 1-bromo-3-metoxipropano (205 mg, 1,34 mmol) en N,N-dimetilformamida (DMF) (5 ml) a temperatura ambiente durante la noche. Se extinguió la mezcla con agua y se extrajo 3 veces con acetato de etilo. Se lavaron las capas orgánicas combinadas con salmuera y se concentraron. Se purificó el residuo mediante cromatografía para dar 1-(5-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (341 mg, 0,579 mmol, rendimiento del 86 %). CLEM (ES+) (m/z): 589,2 (M+1).
Etapa 7: 2-Cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hirU1naftiridin-10-carboxilato de etilo (cis racémico)

Un matraz de fondo redondo tapado con un tabique que contenía una barra agitadora, 1-(5-(6-cloro-2-yodo-5-(3-metoxipropoxi)piridin-3-il)-2,2-dimetilciclopentil)-4-oxo-1,4-dihidropiridin-3-carboxilato de etilo (341 mg, 0,579 mmol), acetato de potasio (114 mg, 1,16 mmol) y bromuro de paladio (II) (30,8 mg, 0,116 mmol) se purgó con nitrógeno durante 15 minutos. Se purgó N,N-dimetilacetamida (DMA) (5 ml) con nitrógeno durante 5 minutos antes de añadirse al recipiente de reacción. Se colocó el recipiente de reacción en un baño de aceite que se precalentó hasta 90 °C y se agitó la mezcla durante 4 horas. Se permitió que la mezcla de reacción se enfriara hasta temperatura ambiente, se diluyó con acetato de etilo y se filtró a través de un tapón de algodón. Se lavó la mezcla con salmuera, se secó sobre sulfato de sodio y se concentró. Se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 10 % de metanol en diclorometano. Se concentraron las fracciones. Se aisló el producto deseado, pero quedaba una cantidad significativa de DMA. Se purificó adicionalmente el producto mediante cromatografía de fase inversa a presión media (C18 / acetonitrilo / agua / ácido fórmico al 0,1 % / gradiente del 10 % al 100 %). Se concentraron las fracciones para dar 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo (cis racémico) (150 mg, 0,325 mmol, rendimiento del 56 %). 1H-RMN (400 MHz, DMSO-ds) " ppm 8,21 (s, 1 H), 7,55 (s, 1 H), 7,00 (s, 1 H), 4,43 (d, J=8,98 Hz, 1 H), 4,15 - 4,31 (m, 4 H), 3,79 - 3,88 (m, 1 H), 3,47 (t, J=6,05 Hz, 2 H), 3,23 (s, 3 H), 2,27 - 2,38 (m, 1 H), 2,12 - 2,25 (m, 1 H), 1,99 (quin, J=6,15 Hz, 2 H), 1,50 - 1,60 (m, 1 H), 1,35 - 1,45 (m, 1 H), 1,24 (t, J=7,22 Hz, 3 H), 1,11 (s, 3 H), 0,39 (s, 3 H). CLEM (ES+) (m/z): 461,3 (M+1).
Etapa 8: Ácido 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopentarflpiridoH ,2-hirU1naftiridin-10-carboxílico (cis racémico)

Una disolución de hidróxido de litio monohidratado (113 mg, 2,70 mmol) en agua (2 ml) se añadió a una disolución de 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11 -oxo-4b,5,6,7,7a,11 -hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxilato de etilo (cis racémico) (150 mg, 0,270 mmol) en metanol (3 ml) y se calentó la mezcla a 65 °C durante 1
hora. Se permitió que la mezcla se enfriara, se diluyó con agua, se acidificó con HCl 1 M y se extrajo 3 veces con 2-metiltetrahidrofurano. Se lavaron las capas orgánicas combinadas con salmuera y se concentraron. Se purificó el residuo mediante cromatografía en sílice eluyendo con un gradiente del 0 % al 10 % de metanol en diclorometano. Se concentraron las fracciones para dar ácido 2-cloro-3-(3-metoxipropoxi)-7,7-dimetil-11-oxo-4b,5,6,7,7a,11-hexahidrociclopenta[f]pirido[1,2-h][1,7]naftiridin-10-carboxílico (cis racémico) (95 mg, 0,219 mmol, rendimiento del 81 %) como un sólido blanco. 1H-RMN (400 MHz, DMSO-afe) " ppm 8,62 (s, 1 H), 7,63 (s, 1 H), 7,43 (s, 1 H), 4,72 (d, J=9,37 Hz, 1 H), 4,21 - 4,39 (m, 2 H), 3,89 - 3,99 (m, 1 H), 3,51 (t, J=6,05 Hz, 2 H), 3,26 (s, 3 H), 2,33 - 2,44 (m, 1 H), 2,19 - 2,31 (m, 1 H), 1,96 - 2,08 (m, 2 H), 1,55 - 1,66 (m, 1 H), 1,40 - 1,51 (m, 1 H), 1,15 (s, 3 H), 0,40 (s, 3 H). CLEM (ES+) (m/z): 433,3 (M+1).
A continuación se muestran en el Esquema 15 protocolos de síntesis general adicionales para producir compuestos tal como se describe en el presente documento.
Esquema 15 - Compuestos de compuestos tipo en donde
R10 es de la tabla 2)

Ejemplos biológicos
Ejemplo 35 - Tratamiento con los compuestos en hepatocitos humanos primarios infectado con VHB
Inhibidores de Ag de VHB
Se colocaron viales de hepatocitos humanos primarios crioconservados en un baño de agua a 37 °C justo hasta que se descongelaron. Se agruparon las células, se resuspendieron suavemente en medio de diferenciación (medio Williams que contiene suplemento de diferenciación, GlutaMax-1™ y penicilina/estreptomicina) y se contaron usando un hemocitómetro. Se sedimentaron las células mediante centrifugación a 1000 X g durante 10 min y se resuspendieron hasta una densidad de 5,5 x 105 células/ml en medio de diferenciación. Se sembraron 100 pl de suspensión de células en cada pocillo de placas de 96 pocillos recubiertos con colágeno. Se incubaron las placas a 37 °C y el 5 % de CO2 durante 2 días antes de la infección.
Se prepararon disoluciones madre de VHB mediante ultrafiltración de medios de cultivos celulares de HepG2.2.15. Para preparar una disolución madre de virus de trabajo con una multiplicidad de infección (MOI) de 100, se añadió la disolución madre de VHB a medio de diferenciación que contenía polietilenglicol al 4 % para lograr una concentración de 5,5 x 107 copias de ADN de VHB/ml. Se reemplazó el medio celular por 100 pl de la disolución madre de virus de trabajo en las columnas 1-11 y con medio de diferenciación en la columna 12. Se incubaron las placas a 37 °C y el 5 % de CO2 durante aproximadamente 24 h.
Se resuspendieron los compuestos en DMSO y se diluyeron en serie 3 veces en DMSO para preparar una serie de dilución de 10 puntos a 200X las concentraciones deseadas finales. Las columnas 11 y 12 contenían DMSO. Usando un instrumento Biomek FX, se estamparon 2,5 pl de cada dilución de compuesto en placas con fondo en U de 96 pocillos, preparando 3 copias. Se sellaron las placas de compuesto y se almacenaron a -20 °C. Después de equilibrar hasta temperatura ambiente, se diluyeron las placas de compuesto 200 veces con medio de ensayo (medio de diferenciación más ABT 1 mM). Se reemplazó el medio de las placas de células por 150 pl de los compuestos diluidos. La concentración de compuesto más alta final fue de 20 pM. Se incubaron las placas a 37 °C y el 5 % de CO2. Los
tratamientos con compuestos se repitieron los días 4 y 9 tras el tratamiento inicial. Se realizaron las lecturas de los ELISA de HBsAg y HBeAg los días 9 y 14.
A los 9 días después del tratamiento inicial, se transfirió el medio de cada placa de células a una placa con fondo en U y se almacenaron a -80 °C. El día 14 después del tratamiento inicial, se lavaron las placas de células una vez con PBS y se almacenaron a -80 °C.
ELISA de HBs Ag
Se equilibraron placas congeladas que contenían los medios recogidos hasta temperatura ambiente en una cabina de bioseguridad durante aproximadamente 30 min. Se usó el kit de ELISA de HBsAg según las directrices del fabricante. En resumen, se equilibraron las placas de ELISA y las disoluciones hasta temperatura ambiente durante aproximadamente 1 h y se lavaron las placas una vez con 300 pl de tampón de lavado 1X. En cada pocillo se colocaron 100 pl de disolución de conjugado enzimático 1X, 120 pl de PBS que contenía FBS al 10 % y 30 pl de medios recogidos. Se sellaron las placas de ensayo y se incubaron a 37 °C durante aproximadamente 2 h. Se lavaron las placas 4 veces con 195 ml de tampón de lavado 1X y se secaron completamente invertidas sobre toallas de papel. Se añadieron 195 pl de disolución de cromógeno/sustrato a cada pocillo y se incubó a temperatura ambiente durante aproximadamente 5 min. Se añadieron 100 pl de disolución de detención a cada pocillo y se leyeron las placas en un lector de microplacas SpectraMax® 384 Plus de Molecular Devices a 450 nm. Se determinaron las CI50 usando GraphPad Prism: Curva logística de cuatro parámetros con la ecuación Y=Inferior (Superior-Inferior)/(1 10A((LogCI50-X)*pendiente de Hill)).
ELISA de HBe Ag
Se equilibraron placas congeladas que contenían los medios recogidos hasta temperatura ambiente en una cabina de bioseguridad durante aproximadamente 30 min. Se usó el kit de ELISA de HBeAg según las directrices del fabricante. En resumen, se equilibraron las placas de ELISA y las disoluciones hasta temperatura ambiente durante aproximadamente 1 h y se lavaron las placas una vez con 300 pl de tampón de lavado 1X. En cada pocillo se colocaron 80 pl de PBS que contenía FBS al 10 %, y 20 pl de medios recogidos. Se sellaron las placas de ensayo y se incubaron a 37 °C durante aproximadamente 1 h. Se lavaron las placas dos veces con 300 ml de tampón de lavado 1X y se secaron completamente invertidas sobre toallas de papel. En cada pocillo se colocaron 100 pl de disolución de conjugado enzimático 1X. Se sellaron las placas de ensayo y se incubaron a 37 °C durante al menos 1 h. Se lavaron las placas 3 veces con 300 ml de tampón de lavado 1X y se secaron completamente invertidas sobre toallas de papel. Se añadieron 100 pl de disolución de cromógeno/sustrato a cada pocillo y se incubaron a temperatura ambiente durante aproximadamente 5 min. Se añadieron 100 pl de disolución de detención a cada pocillo y se leyeron las placas en un lector de microplacas SpectraMax 384 Plus de Molecular Devices a 450 nm.
Análisis de datos
Se determinaron las CI50 usando GraphPad Prism: Curva logística de cuatro parámetros con la ecuación Y=Inferior (Superior-Inferior)/(1+10A((LogCI50-X)*pendiente de Hill)). En la tabla 1 se muestran los valores de CI50 para un 50 % de reducción de los antígenos HBs y HBe para los compuestos sometidos a prueba para los días 9 y 14. Tal como puede observarse a partir de los valores de CI50, los compuestos sometidos a prueba presentaban un 50 % de inhibición de los antígeno HBe y HBs a valores de entre menos de ~0,22 pM y 0,0069 pM.
Ejemplo 36 Células HepAD38 - ELISA de HBsAg y ensayos de citotoxicidad
Se mantienen células HepAD38 en matraces recubiertos con colágeno en medio de cultivo celular (DMEM/F12 que contiene suero bovino fetal (FBS) al 10 %, GlutaMax-1, penicilina/estreptomicina, aminoácidos no esenciales, piruvato de Na, geneticina 250 pg/ml y doxiciclina 1 pg/ml). Se preparan disoluciones de compuesto en DMSO y se diluye en serie el compuesto para concentraciones finales de 4000, 1000, 250, 62,5, 15,6, 3,91,0,977, 0,244, 0,061 y 0,015 $M. Entonces se tripsinizan las células y se siembran las células a 10.000 células por pocillo. Las placas de incubación se incuban a 37 °C, el 5 % de CO2 durante 4 días. Se reemplaza el medio por medio nuevo con tratamiento con compuesto. Entonces se incuban las placas a 37 °C, el 5 % de CO2 durante 3 días adicionales para un tiempo de tratamiento total de 7 días. Para la respuesta antiviral, se midió HBsAg usando el kit de ELISA de HBsAg (International Immuno-diagnostics) con instrucciones proporcionadas. Se usaron 100 ul de muestras de medios celulares para el ELISA. Se leyó la absorbancia en el lector de placas Spectramax® 384 (Molecular Devices) a 450 nm. Para la toxicidad celular, se usaron las células para el reactivo de ensayo de viabilidad celular luminiscente CellTiter-Glo® (Promega). Se leyó la luminiscencia en un lector Envision Multilabel (Perkin Elmer). Se analizaron los datos en comparación con el control de DMSO.
% de inhibición = {1 -(desconocido/control alto ))*l00
Entonces, se representan gráficamente los valores de % de inhibición promedio de placas de ensayo por duplicado
en GraphPad Prism para determinar un valor de CI: Curva logística de cuatro parámetros con la ecuación Y=Inferior (Superior-Inferior)/(1+10A((LogCI50-X)*pendiente de Hill))
pCl5o= log(-CUo en M)
TABLA 1





TABLA 2



Ċ

97

TABLA 3






en donde:

Claims (6)
- REIVINDICACIONESi. Compuesto seleccionado de:
 o una sal farmacéuticamente aceptable del mismo.
o una sal farmacéuticamente aceptable del mismo. - 2. Compuesto según la reivindicación 1 cuya estructura es:
 , o una sal farmacéuticamente aceptable del mismo.
, o una sal farmacéuticamente aceptable del mismo. - 3. Composición farmacéutica que comprende un diluyente farmacéuticamente aceptable y una cantidad terapéuticamente eficaz del compuesto según la reivindicación 1 o 2.
- 4. Compuesto según la reivindicación 1 o 2, o composición farmacéutica según la reivindicación 3, para su uso en terapia.
- 5. Compuesto según la reivindicación 1 o 2, o composición farmacéutica según la reivindicación 3, para su uso en el tratamiento de una infección viral.
- 6. Compuesto o composición farmacéutica según la reivindicación 5 para su uso en el tratamiento de una infección viral, en donde la infección viral es una infección viral de hepatitis B.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762568633P | 2017-10-05 | 2017-10-05 | |
| US201762570509P | 2017-10-10 | 2017-10-10 | |
| US201862681146P | 2018-06-06 | 2018-06-06 | |
| US201862683859P | 2018-06-12 | 2018-06-12 | |
| PCT/IB2018/057767 WO2019069293A1 (en) | 2017-10-05 | 2018-10-05 | CHEMICAL COMPOUNDS |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2940263T3 true ES2940263T3 (es) | 2023-05-04 |
Family
ID=64049476
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18795809T Active ES2940263T3 (es) | 2017-10-05 | 2018-10-05 | Compuestos químicos |
Country Status (26)
| Country | Link |
|---|---|
| US (3) | US20200255428A1 (es) |
| EP (2) | EP4180431A1 (es) |
| JP (1) | JP7248665B2 (es) |
| KR (1) | KR102705376B1 (es) |
| CN (2) | CN116942663A (es) |
| AU (2) | AU2018344926A1 (es) |
| CA (1) | CA3078312A1 (es) |
| CL (1) | CL2020000912A1 (es) |
| CO (1) | CO2020004202A2 (es) |
| DK (1) | DK3692040T3 (es) |
| ES (1) | ES2940263T3 (es) |
| FI (1) | FI3692040T3 (es) |
| HR (1) | HRP20230274T1 (es) |
| HU (1) | HUE061380T2 (es) |
| IL (1) | IL273769B2 (es) |
| LT (1) | LT3692040T (es) |
| MX (1) | MX2020003382A (es) |
| MY (1) | MY201605A (es) |
| PH (1) | PH12020550145A1 (es) |
| PL (1) | PL3692040T3 (es) |
| PT (1) | PT3692040T (es) |
| RS (1) | RS64043B1 (es) |
| SG (2) | SG10202111648YA (es) |
| SI (1) | SI3692040T1 (es) |
| SM (1) | SMT202300089T1 (es) |
| WO (1) | WO2019069293A1 (es) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10738035B2 (en) | 2015-05-13 | 2020-08-11 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| BR112018067964B1 (pt) | 2016-03-07 | 2024-01-16 | Enanta Pharmaceuticals, Inc | Composto, composição farmacêutica que o compreende e uso do referido composto |
| US10821103B2 (en) * | 2016-11-07 | 2020-11-03 | Arbutus Biopharma Corporation | Substituted pyridinone-containing trycyclic compounds, and methods using same |
| IL272941B2 (en) | 2017-08-28 | 2023-03-01 | Enanta Pharm Inc | Antiviral agents for viral hepatitis b |
| US11058678B2 (en) | 2018-01-22 | 2021-07-13 | Enanta Pharmaceuticals, Inc. | Substituted heterocycles as antiviral agents |
| WO2019191166A1 (en) | 2018-03-29 | 2019-10-03 | Enanta Pharmaceuticals, Inc. | Hepatitis b antiviral agents |
| SG11202011137SA (en) | 2018-06-05 | 2020-12-30 | Hoffmann La Roche | Tetrahydro-1 h-pyrazino[2,1 -ajisoindolylquinoline compounds for the treatment of autoimmune disease |
| WO2019246427A1 (en) * | 2018-06-22 | 2019-12-26 | Arbutus Biopharma Corporation | Crystalline forms of pyridinone-containing tricyclic compounds and methods of preparing and using same |
| WO2020043080A1 (en) | 2018-08-28 | 2020-03-05 | Sunshine Lake Pharma Co., Ltd. | Fused tricyclic compounds and uses thereof in medicine |
| WO2020061435A1 (en) | 2018-09-21 | 2020-03-26 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| JP2022511378A (ja) | 2018-09-30 | 2022-01-31 | サンシャイン・レイク・ファーマ・カンパニー・リミテッド | 縮合四環系化合物および医薬品におけるそれらの使用 |
| KR102819704B1 (ko) | 2018-11-21 | 2025-06-11 | 이난타 파마슈티칼스, 인코포레이티드 | 항바이러스제로서의 작용화된 헤테로사이클 |
| CN111217811B (zh) * | 2018-11-26 | 2024-01-16 | 广东东阳光药业股份有限公司 | 稠合三环类化合物及其在药物中的应用 |
| TW202104214A (zh) | 2019-04-05 | 2021-02-01 | 英商葛蘭素史密斯克藍智慧財產發展有限公司 | 化合物 |
| US11236111B2 (en) | 2019-06-03 | 2022-02-01 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11472808B2 (en) | 2019-06-04 | 2022-10-18 | Enanta Pharmaceuticals, Inc. | Substituted pyrrolo[1,2-c]pyrimidines as hepatitis B antiviral agents |
| US11760755B2 (en) | 2019-06-04 | 2023-09-19 | Enanta Pharmaceuticals, Inc. | Hepatitis B antiviral agents |
| US11738019B2 (en) | 2019-07-11 | 2023-08-29 | Enanta Pharmaceuticals, Inc. | Substituted heterocycles as antiviral agents |
| WO2021055425A2 (en) | 2019-09-17 | 2021-03-25 | Enanta Pharmaceuticals, Inc. | Functionalized heterocycles as antiviral agents |
| WO2021188414A1 (en) | 2020-03-16 | 2021-09-23 | Enanta Pharmaceuticals, Inc. | Functionalized heterocyclic compounds as antiviral agents |
| CN113173911A (zh) * | 2021-04-20 | 2021-07-27 | 梯尔希(南京)药物研发有限公司 | 吡虫啉代谢物5-羟基吡虫啉的合成方法 |
| JP2025504017A (ja) | 2022-01-28 | 2025-02-06 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッド | B型肝炎ウイルス感染症を処置するための組合せ療法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015113990A1 (en) * | 2014-01-30 | 2015-08-06 | F. Hoffmann-La Roche Ag | Novel dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection |
| EP3143020B1 (en) * | 2014-05-13 | 2019-08-21 | F. Hoffmann-La Roche AG | Dihydroquinolizinones for the treatment and prophylaxis of hepatitis b virus infection |
| US9637485B2 (en) * | 2014-11-03 | 2017-05-02 | Hoffmann-La Roche Inc. | 6,7-dihydrobenzo[a]quinolizin-2-one derivatives for the treatment and prophylaxis of hepatitis B virus infection |
| WO2016128335A1 (en) * | 2015-02-11 | 2016-08-18 | F. Hoffmann-La Roche Ag | Novel 2-oxo-6,7-dihydrobenzo[a]quinolizine-3-carboxylic acid derivatives for the treatment and prophylaxis of hepatitis b virus infection |
| WO2017108630A1 (en) * | 2015-12-21 | 2017-06-29 | F. Hoffmann-La Roche Ag | Combination therapy of an hbsag inhibitor and an hbv capsid assembly inhibitor |
| TW201811788A (zh) * | 2016-09-09 | 2018-04-01 | 瑞士商諾華公司 | 作為抗病毒劑之多環吡啶酮化合物 |
| US10821103B2 (en) | 2016-11-07 | 2020-11-03 | Arbutus Biopharma Corporation | Substituted pyridinone-containing trycyclic compounds, and methods using same |
| CN109369640B (zh) * | 2017-01-13 | 2020-03-27 | 苏州爱科百发生物医药技术有限公司 | 一种二氢异喹啉类化合物的制备方法 |
| CN108727378A (zh) | 2017-04-19 | 2018-11-02 | 银杏树药业(苏州)有限公司 | 新型异喹啉类化合物及其医药用途 |
-
2018
- 2018-10-05 CA CA3078312A patent/CA3078312A1/en active Pending
- 2018-10-05 PL PL18795809.5T patent/PL3692040T3/pl unknown
- 2018-10-05 LT LTEPPCT/IB2018/057767T patent/LT3692040T/lt unknown
- 2018-10-05 CN CN202310780889.9A patent/CN116942663A/zh active Pending
- 2018-10-05 ES ES18795809T patent/ES2940263T3/es active Active
- 2018-10-05 IL IL273769A patent/IL273769B2/en unknown
- 2018-10-05 MY MYPI2020001721A patent/MY201605A/en unknown
- 2018-10-05 EP EP22214773.8A patent/EP4180431A1/en active Pending
- 2018-10-05 PT PT187958095T patent/PT3692040T/pt unknown
- 2018-10-05 SI SI201830862T patent/SI3692040T1/sl unknown
- 2018-10-05 HR HRP20230274TT patent/HRP20230274T1/hr unknown
- 2018-10-05 WO PCT/IB2018/057767 patent/WO2019069293A1/en not_active Ceased
- 2018-10-05 MX MX2020003382A patent/MX2020003382A/es unknown
- 2018-10-05 KR KR1020207012236A patent/KR102705376B1/ko active Active
- 2018-10-05 AU AU2018344926A patent/AU2018344926A1/en not_active Abandoned
- 2018-10-05 DK DK18795809.5T patent/DK3692040T3/da active
- 2018-10-05 FI FIEP18795809.5T patent/FI3692040T3/fi active
- 2018-10-05 SG SG10202111648YA patent/SG10202111648YA/en unknown
- 2018-10-05 CN CN201880078910.8A patent/CN111433206B/zh active Active
- 2018-10-05 EP EP18795809.5A patent/EP3692040B1/en active Active
- 2018-10-05 SG SG11202002771XA patent/SG11202002771XA/en unknown
- 2018-10-05 US US16/753,645 patent/US20200255428A1/en not_active Abandoned
- 2018-10-05 JP JP2020519410A patent/JP7248665B2/ja active Active
- 2018-10-05 SM SM20230089T patent/SMT202300089T1/it unknown
- 2018-10-05 RS RS20230207A patent/RS64043B1/sr unknown
- 2018-10-05 HU HUE18795809A patent/HUE061380T2/hu unknown
-
2020
- 2020-03-25 PH PH12020550145A patent/PH12020550145A1/en unknown
- 2020-04-03 CL CL2020000912A patent/CL2020000912A1/es unknown
- 2020-04-03 CO CONC2020/0004202A patent/CO2020004202A2/es unknown
-
2021
- 2021-06-04 AU AU2021203650A patent/AU2021203650B9/en active Active
- 2021-10-25 US US17/509,769 patent/US12221444B2/en active Active
-
2024
- 2024-11-11 US US18/943,115 patent/US20250066361A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2940263T3 (es) | Compuestos químicos | |
| JP7716110B2 (ja) | ピリミジン-4(3h)-オンヘテロ環式化合物、その調製方法、およびその医薬的使用 | |
| CN109790169A (zh) | 具有作为usp30抑制剂活性的氰基吡咯烷衍生物 | |
| CN114269748A (zh) | 作为用于治疗癌症的PRC2抑制剂的咪唑并[1,2-c]嘧啶衍生物 | |
| TWI815887B (zh) | 經取代的2,2'-雙嘧啶基化合物、其類似物及其使用方法 | |
| EP4358954A1 (en) | Cdk2 inhibitors and methods of using the same | |
| CN106414443B (zh) | 吡啶并嘧啶二酮衍生物 | |
| CN110981876B (zh) | 8-取代的苯乙烯基黄嘌呤衍生物及其用途 | |
| US10807983B2 (en) | Imidazo-fused heterocycles and uses thereof | |
| CN107759620A (zh) | 八氢吡咯并[3,4‑c]吡咯衍生物及其使用方法和用途 | |
| WO2021218997A1 (zh) | 取代的氮杂五元环类化合物及其在药物中的应用 | |
| RU2800292C2 (ru) | Химические соединения | |
| BR112020006456B1 (pt) | Compostos químicos, composição farmacêutica e seus usos para tratar uma infecção por vírus da hepatite b e para inibir o nível do antígeno hbe ou hbs em um hbsag de mamífero in vitro | |
| HK40028203A (zh) | 化合物 | |
| WO2024104244A1 (zh) | 草酸酰胺化合物、包含其的药物组合物及其制备方法和用途 | |
| HK40028203B (zh) | 化合物 | |
| WO2025067417A1 (zh) | 并内酰胺环类化合物及其用途 | |
| KR20260025059A (ko) | 신규한 치환된 피리딘 유도체 화합물, 이의 제조방법, 및 이를 유효성분으로 포함하는 호흡기 질환의 예방 또는 치료용 약학적 조성물 | |
| WO2025113705A1 (zh) | N-取代酰胺类衍生物抑制剂、其制备方法和应用 | |
| WO2025108406A1 (zh) | 一种靶向降解雄激素受体的双功能嵌合体的杂环化合物及其用途 | |
| WO2025247379A1 (zh) | 一种苯并咪唑类化合物、其制备方法及应用 |