ES2927283T3 - Derivados del ácido borónico - Google Patents
Derivados del ácido borónico Download PDFInfo
- Publication number
- ES2927283T3 ES2927283T3 ES18755481T ES18755481T ES2927283T3 ES 2927283 T3 ES2927283 T3 ES 2927283T3 ES 18755481 T ES18755481 T ES 18755481T ES 18755481 T ES18755481 T ES 18755481T ES 2927283 T3 ES2927283 T3 ES 2927283T3
- Authority
- ES
- Spain
- Prior art keywords
- ethyl
- benzofuran
- formula
- acid
- formamido
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001642 boronic acid derivatives Chemical class 0.000 title description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 230
- 238000011282 treatment Methods 0.000 claims abstract description 22
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 21
- 101001136986 Homo sapiens Proteasome subunit beta type-8 Proteins 0.000 claims abstract description 15
- 102100035760 Proteasome subunit beta type-8 Human genes 0.000 claims abstract description 15
- 201000011510 cancer Diseases 0.000 claims abstract description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 14
- 201000010099 disease Diseases 0.000 claims abstract description 11
- 230000002265 prevention Effects 0.000 claims abstract description 6
- -1 bicyclic hydrocarbon Chemical class 0.000 claims description 127
- 239000002253 acid Substances 0.000 claims description 100
- 239000000203 mixture Substances 0.000 claims description 94
- 150000003839 salts Chemical class 0.000 claims description 76
- 238000000034 method Methods 0.000 claims description 56
- 229910052760 oxygen Inorganic materials 0.000 claims description 33
- 239000012453 solvate Substances 0.000 claims description 33
- 125000001424 substituent group Chemical group 0.000 claims description 33
- 229910052801 chlorine Inorganic materials 0.000 claims description 31
- 229910052731 fluorine Inorganic materials 0.000 claims description 30
- QUPDWYMUPZLYJZ-UHFFFAOYSA-N ethyl Chemical compound C[CH2] QUPDWYMUPZLYJZ-UHFFFAOYSA-N 0.000 claims description 29
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 28
- 239000004480 active ingredient Substances 0.000 claims description 25
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 24
- 239000003814 drug Substances 0.000 claims description 23
- 239000008194 pharmaceutical composition Substances 0.000 claims description 22
- 229910052717 sulfur Inorganic materials 0.000 claims description 22
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 21
- 229910052757 nitrogen Inorganic materials 0.000 claims description 20
- 125000000623 heterocyclic group Chemical group 0.000 claims description 18
- 125000003118 aryl group Chemical group 0.000 claims description 16
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 16
- 229910052799 carbon Inorganic materials 0.000 claims description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims description 15
- VMWJCFLUSKZZDX-UHFFFAOYSA-N n,n-dimethylmethanamine Chemical compound [CH2]N(C)C VMWJCFLUSKZZDX-UHFFFAOYSA-N 0.000 claims description 14
- 230000005764 inhibitory process Effects 0.000 claims description 13
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 12
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 claims description 12
- 229960001124 trientine Drugs 0.000 claims description 12
- 125000004429 atom Chemical group 0.000 claims description 11
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 10
- 229920006395 saturated elastomer Polymers 0.000 claims description 10
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 9
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 9
- 238000002360 preparation method Methods 0.000 claims description 9
- 125000000217 alkyl group Chemical group 0.000 claims description 8
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 8
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 claims description 8
- 125000004122 cyclic group Chemical group 0.000 claims description 8
- 125000004434 sulfur atom Chemical group 0.000 claims description 8
- 230000005856 abnormality Effects 0.000 claims description 7
- 125000004432 carbon atom Chemical group C* 0.000 claims description 7
- 230000004957 immunoregulator effect Effects 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 7
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 6
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 claims description 6
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 claims description 6
- 125000005620 boronic acid group Chemical group 0.000 claims description 6
- 229930195733 hydrocarbon Natural products 0.000 claims description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 6
- 239000004215 Carbon black (E152) Substances 0.000 claims description 5
- 206010066476 Haematological malignancy Diseases 0.000 claims description 5
- 208000002250 Hematologic Neoplasms Diseases 0.000 claims description 5
- 208000034578 Multiple myelomas Diseases 0.000 claims description 5
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 5
- 125000002947 alkylene group Chemical group 0.000 claims description 5
- 208000010159 IgA glomerulonephritis Diseases 0.000 claims description 4
- 229910052794 bromium Inorganic materials 0.000 claims description 4
- 201000006417 multiple sclerosis Diseases 0.000 claims description 4
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 3
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 206010018364 Glomerulonephritis Diseases 0.000 claims description 3
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 3
- 208000005777 Lupus Nephritis Diseases 0.000 claims description 3
- 201000004681 Psoriasis Diseases 0.000 claims description 3
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 3
- 206010052779 Transplant rejections Diseases 0.000 claims description 3
- 206010047115 Vasculitis Diseases 0.000 claims description 3
- 208000006673 asthma Diseases 0.000 claims description 3
- 208000037976 chronic inflammation Diseases 0.000 claims description 3
- 208000037893 chronic inflammatory disorder Diseases 0.000 claims description 3
- 201000005787 hematologic cancer Diseases 0.000 claims description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 3
- 208000014018 liver neoplasm Diseases 0.000 claims description 3
- 206010028417 myasthenia gravis Diseases 0.000 claims description 3
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 2
- 206010003827 Autoimmune hepatitis Diseases 0.000 claims description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 2
- 206010009944 Colon cancer Diseases 0.000 claims description 2
- 201000004331 Henoch-Schoenlein purpura Diseases 0.000 claims description 2
- 206010019617 Henoch-Schonlein purpura Diseases 0.000 claims description 2
- 208000031814 IgA Vasculitis Diseases 0.000 claims description 2
- 208000005726 Inflammatory Breast Neoplasms Diseases 0.000 claims description 2
- 206010021980 Inflammatory carcinoma of the breast Diseases 0.000 claims description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 2
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 claims description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 201000003791 MALT lymphoma Diseases 0.000 claims description 2
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 claims description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 2
- 201000002481 Myositis Diseases 0.000 claims description 2
- RFQDLTYXNINJON-OYNZBZHQSA-N O1C=C(C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O Chemical compound O1C=C(C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O RFQDLTYXNINJON-OYNZBZHQSA-N 0.000 claims description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 2
- 208000007452 Plasmacytoma Diseases 0.000 claims description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 2
- 206010060862 Prostate cancer Diseases 0.000 claims description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 2
- 206010038389 Renal cancer Diseases 0.000 claims description 2
- 206010039710 Scleroderma Diseases 0.000 claims description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 2
- MWNDPMUFCDVOCD-WYUUTHIRSA-N [(1R)-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]-2-thiophen-3-ylethyl]boronic acid Chemical compound OB(O)[C@H](CC1=CSC=C1)NC(=O)[C@@H]1C[C@H]2CC[C@@H]1O2 MWNDPMUFCDVOCD-WYUUTHIRSA-N 0.000 claims description 2
- XSFAXJUIENANOX-KBXIAJHMSA-N [(1R)-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]-3-phenylpropyl]boronic acid Chemical compound [C@@H]12[C@@H](C[C@@H](CC1)O2)C(=O)N[C@@H](CCC2=CC=CC=C2)B(O)O XSFAXJUIENANOX-KBXIAJHMSA-N 0.000 claims description 2
- RFQDLTYXNINJON-YUDUHTQSSA-N [(1R)-2-(1-benzofuran-3-yl)-1-[[(1R,2S,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@H](NC(=O)[C@@H]1[C@H]2CC[C@@H](C1)O2)B(O)O RFQDLTYXNINJON-YUDUHTQSSA-N 0.000 claims description 2
- RFQDLTYXNINJON-XTBFLFJDSA-N [(1R)-2-(1-benzofuran-3-yl)-1-[[(1S,2S,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@H](NC(=O)[C@@H]1[C@@H]2CC[C@H](C1)O2)B(O)O RFQDLTYXNINJON-XTBFLFJDSA-N 0.000 claims description 2
- YDOSWWDHHDEBJM-WCVJEAGWSA-N [(1R)-2-(2,4-dimethylphenyl)-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound CC1=C(C=CC(=C1)C)C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O YDOSWWDHHDEBJM-WCVJEAGWSA-N 0.000 claims description 2
- CXYWNICDMTXIDH-MQYQWHSLSA-N [(1R)-2-cyclohexyl-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound C1(CCCCC1)C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O CXYWNICDMTXIDH-MQYQWHSLSA-N 0.000 claims description 2
- ZAZQGGQECKMHPA-ZNSHCXBVSA-N [(1R)-3-methyl-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]butyl]boronic acid Chemical compound CC(C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O)C ZAZQGGQECKMHPA-ZNSHCXBVSA-N 0.000 claims description 2
- RFQDLTYXNINJON-DDUYRFODSA-N [(1S)-2-(1-benzofuran-3-yl)-1-[[(1R,2S,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@@H](NC(=O)[C@@H]1[C@H]2CC[C@@H](C1)O2)B(O)O RFQDLTYXNINJON-DDUYRFODSA-N 0.000 claims description 2
- RFQDLTYXNINJON-XABYNWHXSA-N [(1S)-2-(1-benzofuran-3-yl)-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O RFQDLTYXNINJON-XABYNWHXSA-N 0.000 claims description 2
- RFQDLTYXNINJON-SHPPOPEUSA-N [(1S)-2-(1-benzofuran-3-yl)-1-[[(1S,2S,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@@H](NC(=O)[C@@H]1[C@@H]2CC[C@H](C1)O2)B(O)O RFQDLTYXNINJON-SHPPOPEUSA-N 0.000 claims description 2
- 230000001363 autoimmune Effects 0.000 claims description 2
- 208000037979 autoimmune inflammatory disease Diseases 0.000 claims description 2
- 125000002619 bicyclic group Chemical group 0.000 claims description 2
- 230000001684 chronic effect Effects 0.000 claims description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000029742 colonic neoplasm Diseases 0.000 claims description 2
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 claims description 2
- 201000003444 follicular lymphoma Diseases 0.000 claims description 2
- 125000002541 furyl group Chemical group 0.000 claims description 2
- 206010017758 gastric cancer Diseases 0.000 claims description 2
- 201000010536 head and neck cancer Diseases 0.000 claims description 2
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 2
- 230000002871 immunocytoma Effects 0.000 claims description 2
- 208000015446 immunoglobulin a vasculitis Diseases 0.000 claims description 2
- 201000004653 inflammatory breast carcinoma Diseases 0.000 claims description 2
- 201000010982 kidney cancer Diseases 0.000 claims description 2
- 201000005202 lung cancer Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 2
- 201000002528 pancreatic cancer Diseases 0.000 claims description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 2
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 2
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 2
- 201000011549 stomach cancer Diseases 0.000 claims description 2
- 125000001544 thienyl group Chemical group 0.000 claims description 2
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 2
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims 2
- 230000000694 effects Effects 0.000 abstract description 26
- 230000002401 inhibitory effect Effects 0.000 abstract description 5
- 208000023275 Autoimmune disease Diseases 0.000 abstract description 4
- 230000004770 neurodegeneration Effects 0.000 abstract description 4
- 208000015122 neurodegenerative disease Diseases 0.000 abstract description 4
- 208000027866 inflammatory disease Diseases 0.000 abstract description 2
- 230000002757 inflammatory effect Effects 0.000 abstract description 2
- 230000002062 proliferating effect Effects 0.000 abstract description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 125
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 105
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 99
- 239000000243 solution Substances 0.000 description 92
- 239000011541 reaction mixture Substances 0.000 description 75
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 70
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 64
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 62
- 235000002639 sodium chloride Nutrition 0.000 description 52
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 49
- 238000006243 chemical reaction Methods 0.000 description 45
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 42
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 40
- 239000007787 solid Substances 0.000 description 40
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 38
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 36
- 238000005160 1H NMR spectroscopy Methods 0.000 description 32
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 32
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 30
- 239000000460 chlorine Substances 0.000 description 28
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 27
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 25
- 239000000706 filtrate Substances 0.000 description 25
- 235000019439 ethyl acetate Nutrition 0.000 description 24
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 22
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 21
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 21
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 21
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 20
- 239000012044 organic layer Substances 0.000 description 20
- 229910052938 sodium sulfate Inorganic materials 0.000 description 19
- 235000011152 sodium sulphate Nutrition 0.000 description 19
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 239000007788 liquid Substances 0.000 description 18
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 18
- 239000002904 solvent Substances 0.000 description 17
- 239000007832 Na2SO4 Substances 0.000 description 16
- 239000002585 base Substances 0.000 description 16
- 239000000872 buffer Substances 0.000 description 16
- 239000012299 nitrogen atmosphere Substances 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 239000000543 intermediate Substances 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 14
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 14
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 14
- 235000019198 oils Nutrition 0.000 description 14
- 239000003208 petroleum Substances 0.000 description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 13
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 13
- 238000003818 flash chromatography Methods 0.000 description 13
- 239000012071 phase Substances 0.000 description 13
- 239000003826 tablet Substances 0.000 description 13
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 13
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 239000012267 brine Substances 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 239000000843 powder Substances 0.000 description 12
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 12
- 101100054666 Streptomyces halstedii sch3 gene Proteins 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 11
- 125000006239 protecting group Chemical group 0.000 description 11
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 10
- 230000000875 corresponding effect Effects 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 10
- UYLYISCHTFVYHN-PBXRRBTRSA-N (1r,3r,4s)-7-oxabicyclo[2.2.1]heptane-3-carboxylic acid Chemical compound C1C[C@H]2[C@H](C(=O)O)C[C@@H]1O2 UYLYISCHTFVYHN-PBXRRBTRSA-N 0.000 description 9
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 9
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 9
- 238000004128 high performance liquid chromatography Methods 0.000 description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 8
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 8
- 150000001721 carbon Chemical group 0.000 description 8
- 239000012230 colorless oil Substances 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- 150000007513 acids Chemical class 0.000 description 7
- 125000002252 acyl group Chemical group 0.000 description 7
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 7
- 229960001467 bortezomib Drugs 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 229940002612 prodrug Drugs 0.000 description 7
- 239000000651 prodrug Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- IHCCAYCGZOLTEU-UHFFFAOYSA-N 3-furoic acid Chemical compound OC(=O)C=1C=COC=1 IHCCAYCGZOLTEU-UHFFFAOYSA-N 0.000 description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- 102000014150 Interferons Human genes 0.000 description 6
- 108010050904 Interferons Proteins 0.000 description 6
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 229940124639 Selective inhibitor Drugs 0.000 description 6
- 239000013543 active substance Substances 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 238000004296 chiral HPLC Methods 0.000 description 6
- 239000013058 crude material Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 229910052805 deuterium Inorganic materials 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 239000012458 free base Substances 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 239000012442 inert solvent Substances 0.000 description 6
- 229910052744 lithium Inorganic materials 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 230000004783 oxidative metabolism Effects 0.000 description 6
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 6
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 5
- NKBWMBRPILTCRD-UHFFFAOYSA-N 2-Methylheptanoic acid Chemical compound CCCCCC(C)C(O)=O NKBWMBRPILTCRD-UHFFFAOYSA-N 0.000 description 5
- 239000007821 HATU Substances 0.000 description 5
- 102000035195 Peptidases Human genes 0.000 description 5
- 108091005804 Peptidases Proteins 0.000 description 5
- 239000004365 Protease Substances 0.000 description 5
- 239000012317 TBTU Substances 0.000 description 5
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical class NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 description 5
- 239000002246 antineoplastic agent Substances 0.000 description 5
- 239000012300 argon atmosphere Substances 0.000 description 5
- 230000003197 catalytic effect Effects 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 239000006071 cream Substances 0.000 description 5
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 229940079322 interferon Drugs 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 5
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 239000011592 zinc chloride Substances 0.000 description 5
- 235000005074 zinc chloride Nutrition 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- 108010073038 Penicillin Amidase Proteins 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- 229940079156 Proteasome inhibitor Drugs 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 4
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 4
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 230000000269 nucleophilic effect Effects 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 4
- 239000003207 proteasome inhibitor Substances 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 230000002797 proteolythic effect Effects 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 229960002930 sirolimus Drugs 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000004808 supercritical fluid chromatography Methods 0.000 description 4
- 239000000454 talc Substances 0.000 description 4
- 229910052623 talc Inorganic materials 0.000 description 4
- 235000012222 talc Nutrition 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 239000001993 wax Substances 0.000 description 4
- UYLYISCHTFVYHN-HCWXCVPCSA-N (1s,3s,4r)-7-oxabicyclo[2.2.1]heptane-3-carboxylic acid Chemical compound C1C[C@@H]2[C@@H](C(=O)O)C[C@H]1O2 UYLYISCHTFVYHN-HCWXCVPCSA-N 0.000 description 3
- QDPKEELEKYHEFO-UHFFFAOYSA-N (7-methyl-1-benzofuran-3-yl)methanol Chemical compound CC1=CC=CC2=C1OC=C2CO QDPKEELEKYHEFO-UHFFFAOYSA-N 0.000 description 3
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 3
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- RIHILKGXXNZBMI-UHFFFAOYSA-N 2-[(2,4-dimethylphenyl)methyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound CC1=CC(C)=CC=C1CB1OC(C)(C)C(C)(C)O1 RIHILKGXXNZBMI-UHFFFAOYSA-N 0.000 description 3
- ZMEABXOZCQGPON-UHFFFAOYSA-N 2-[(7-chloro-1-benzofuran-3-yl)methyl]-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound ClC1=CC=CC=2C(=COC=21)CB1OC(C(O1)(C)C)(C)C ZMEABXOZCQGPON-UHFFFAOYSA-N 0.000 description 3
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 3
- QIYTYRZCWHULDO-UHFFFAOYSA-N 3-(bromomethyl)-7-chloro-1-benzofuran Chemical compound Clc1cccc2c(CBr)coc12 QIYTYRZCWHULDO-UHFFFAOYSA-N 0.000 description 3
- XQVHUDUCKCDWPH-UHFFFAOYSA-N 3-(bromomethyl)-7-methyl-1-benzofuran Chemical compound BrCC1=COC2=C1C=CC=C2C XQVHUDUCKCDWPH-UHFFFAOYSA-N 0.000 description 3
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 3
- 108010022579 ATP dependent 26S protease Proteins 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- QUGUMFRYSPCESK-UHFFFAOYSA-N CC1=CC=CC=2C(=COC=21)CB1OC(C(O1)(C)C)(C)C Chemical compound CC1=CC=CC=2C(=COC=21)CB1OC(C(O1)(C)C)(C)C QUGUMFRYSPCESK-UHFFFAOYSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 3
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 3
- 229910002651 NO3 Inorganic materials 0.000 description 3
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 3
- DAWHFKLQNUXZBM-UHFFFAOYSA-N OCC1=COC2=C(Cl)C=CC=C12 Chemical compound OCC1=COC2=C(Cl)C=CC=C12 DAWHFKLQNUXZBM-UHFFFAOYSA-N 0.000 description 3
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 3
- 239000012159 carrier gas Substances 0.000 description 3
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 3
- 229960001231 choline Drugs 0.000 description 3
- 229960002436 cladribine Drugs 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 229960004397 cyclophosphamide Drugs 0.000 description 3
- 229960003957 dexamethasone Drugs 0.000 description 3
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 3
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 3
- 229940113088 dimethylacetamide Drugs 0.000 description 3
- CEVCBRGAPBYKPG-UHFFFAOYSA-N ethyl 7-chloro-1-benzofuran-3-carboxylate Chemical compound C1=CC=C2C(C(=O)OCC)=COC2=C1Cl CEVCBRGAPBYKPG-UHFFFAOYSA-N 0.000 description 3
- BESBVJZMGXFOFR-UHFFFAOYSA-N ethyl 7-methyl-1-benzofuran-3-carboxylate Chemical compound C1=CC=C2C(C(=O)OCC)=COC2=C1C BESBVJZMGXFOFR-UHFFFAOYSA-N 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000001307 helium Substances 0.000 description 3
- 229910052734 helium Inorganic materials 0.000 description 3
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 3
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 3
- 238000007327 hydrogenolysis reaction Methods 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 230000001976 improved effect Effects 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 230000001965 increasing effect Effects 0.000 description 3
- 239000003456 ion exchange resin Substances 0.000 description 3
- 229920003303 ion-exchange polymer Polymers 0.000 description 3
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 229960003194 meglumine Drugs 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 3
- 208000033808 peripheral neuropathy Diseases 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 238000003825 pressing Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 235000015424 sodium Nutrition 0.000 description 3
- 239000011343 solid material Substances 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Substances C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 3
- 229960000281 trometamol Drugs 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- MOILFCKRQFQVFS-OORONAJNSA-N (1s,3r,4s,5s)-4,6,6-trimethylbicyclo[3.1.1]heptane-3,4-diol Chemical compound C1[C@H]2C(C)(C)[C@@H]1C[C@@H](O)[C@]2(O)C MOILFCKRQFQVFS-OORONAJNSA-N 0.000 description 2
- WAPNOHKVXSQRPX-SSDOTTSWSA-N (R)-1-phenylethanol Chemical compound C[C@@H](O)C1=CC=CC=C1 WAPNOHKVXSQRPX-SSDOTTSWSA-N 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- NTWWUWHQMMQLSI-UHFFFAOYSA-N 1-benzofuran-3-ylmethanol Chemical compound C1=CC=C2C(CO)=COC2=C1 NTWWUWHQMMQLSI-UHFFFAOYSA-N 0.000 description 2
- ZVNCBRKXMRSTRI-UHFFFAOYSA-N 2-(1-benzofuran-3-ylmethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1CC1=COC2=CC=CC=C12 ZVNCBRKXMRSTRI-UHFFFAOYSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- ABFPKTQEQNICFT-UHFFFAOYSA-M 2-chloro-1-methylpyridin-1-ium;iodide Chemical compound [I-].C[N+]1=CC=CC=C1Cl ABFPKTQEQNICFT-UHFFFAOYSA-M 0.000 description 2
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 2
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 2
- NVHDYYQPGLLKJC-UHFFFAOYSA-N 2-methylpropyl n-(2-methylpropoxycarbonylimino)carbamate Chemical compound CC(C)COC(=O)N=NC(=O)OCC(C)C NVHDYYQPGLLKJC-UHFFFAOYSA-N 0.000 description 2
- ZAZPDOYUCVFPOI-UHFFFAOYSA-N 2-methylpropylboronic acid Chemical compound CC(C)CB(O)O ZAZPDOYUCVFPOI-UHFFFAOYSA-N 0.000 description 2
- AEWASAYNGKXLBK-UHFFFAOYSA-N 3-(bromomethyl)-1-benzofuran Chemical compound C1=CC=C2C(CBr)=COC2=C1 AEWASAYNGKXLBK-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- 125000006275 3-bromophenyl group Chemical group [H]C1=C([H])C(Br)=C([H])C(*)=C1[H] 0.000 description 2
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 2
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 2
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- LLKFNPUXQZHIAE-UHFFFAOYSA-N 5-(3-aminopropyl)-8-bromo-3-methyl-2h-pyrazolo[4,3-c]quinolin-4-one Chemical compound O=C1N(CCCN)C2=CC=C(Br)C=C2C2=C1C(C)=NN2 LLKFNPUXQZHIAE-UHFFFAOYSA-N 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 102100025566 Chymotrypsin-like protease CTRL-1 Human genes 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000206602 Eukaryota Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 101000856199 Homo sapiens Chymotrypsin-like protease CTRL-1 Proteins 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 102000003996 Interferon-beta Human genes 0.000 description 2
- 108090000467 Interferon-beta Proteins 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 101100073357 Streptomyces halstedii sch2 gene Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- 108090000848 Ubiquitin Proteins 0.000 description 2
- 102000044159 Ubiquitin Human genes 0.000 description 2
- DVUWGERKEWTPTD-ZRJCITRHSA-N [(1R)-1-phenylethyl] (1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carboxylate Chemical compound C1(=CC=CC=C1)[C@@H](C)OC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2 DVUWGERKEWTPTD-ZRJCITRHSA-N 0.000 description 2
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 229960001220 amsacrine Drugs 0.000 description 2
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000030741 antigen processing and presentation Effects 0.000 description 2
- 229940034982 antineoplastic agent Drugs 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- 229960002170 azathioprine Drugs 0.000 description 2
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 2
- 239000003637 basic solution Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940050390 benzoate Drugs 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 229960003237 betaine Drugs 0.000 description 2
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 239000003560 cancer drug Substances 0.000 description 2
- 150000001718 carbodiimides Chemical class 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 2
- 229960002438 carfilzomib Drugs 0.000 description 2
- 108010021331 carfilzomib Proteins 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 2
- 229960002023 chloroprocaine Drugs 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 229940001468 citrate Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 239000007822 coupling agent Substances 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- 125000004431 deuterium atom Chemical group 0.000 description 2
- 150000004683 dihydrates Chemical class 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- YVPJCJLMRRTDMQ-UHFFFAOYSA-N ethyl diazoacetate Chemical compound CCOC(=O)C=[N+]=[N-] YVPJCJLMRRTDMQ-UHFFFAOYSA-N 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 229940012356 eye drops Drugs 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 229960000556 fingolimod Drugs 0.000 description 2
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 2
- GAKMQHDJQHZUTJ-ULHLPKEOSA-N fluocortolone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)CO)[C@@]2(C)C[C@@H]1O GAKMQHDJQHZUTJ-ULHLPKEOSA-N 0.000 description 2
- 229960003973 fluocortolone Drugs 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 125000004005 formimidoyl group Chemical group [H]\N=C(/[H])* 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 229940050410 gluconate Drugs 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 230000005283 ground state Effects 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 239000002050 international nonproprietary name Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 2
- 230000005445 isotope effect Effects 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 229910003002 lithium salt Inorganic materials 0.000 description 2
- 159000000002 lithium salts Chemical class 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 150000004682 monohydrates Chemical class 0.000 description 2
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 2
- 229960004866 mycophenolate mofetil Drugs 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 2
- 229940049964 oleate Drugs 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 2
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 235000019371 penicillin G benzathine Nutrition 0.000 description 2
- 238000011458 pharmacological treatment Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 229960005205 prednisolone Drugs 0.000 description 2
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 230000000630 rising effect Effects 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- UVFAEQZFLBGVRM-MSMWPWNWSA-N succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)CCC(O)=O)CC(C)C)C(=O)NC=1C=C2OC(=O)C=C(C)C2=CC=1)C1=CC=C(O)C=C1 UVFAEQZFLBGVRM-MSMWPWNWSA-N 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 229960001967 tacrolimus Drugs 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229960003433 thalidomide Drugs 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 239000013638 trimer Substances 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 230000001810 trypsinlike Effects 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 238000003828 vacuum filtration Methods 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- GFYBIVJQTILVMY-AMWBNRJOSA-N (1R)-2-(2,4-dimethylphenyl)-1-[(1S,2S,6R,8S)-2,9,9-trimethyl-3,5-dioxa-4-boratricyclo[6.1.1.02,6]decan-4-yl]-N,N-bis(trimethylsilyl)ethanamine Chemical compound CC1=C(C=CC(=C1)C)C[C@H](N([Si](C)(C)C)[Si](C)(C)C)B1O[C@]2([C@@H]3C([C@H](C[C@H]2O1)C3)(C)C)C GFYBIVJQTILVMY-AMWBNRJOSA-N 0.000 description 1
- LWDBMUAJGMXQAY-GSEQGPDBSA-L (1r,2r)-cyclohexane-1,2-diamine;platinum(2+);tetradecanoate;hydrate Chemical compound O.[Pt+2].N[C@@H]1CCCC[C@H]1N.CCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCC([O-])=O LWDBMUAJGMXQAY-GSEQGPDBSA-L 0.000 description 1
- WCWUXEGQKLTGDX-LLVKDONJSA-N (2R)-1-[[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-5-methyl-6-pyrrolo[2,1-f][1,2,4]triazinyl]oxy]-2-propanol Chemical compound C1=C2NC(C)=CC2=C(F)C(OC2=NC=NN3C=C(C(=C32)C)OC[C@H](O)C)=C1 WCWUXEGQKLTGDX-LLVKDONJSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- FWFGIHPGRQZWIW-SQNIBIBYSA-N (2S)-2-[[(2R)-2-[(1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-2-phenylacetic acid cyclopentyl ester Chemical compound O=C([C@@H](NC(=O)[C@@H]([C@H](O)C(=O)NO)CC(C)C)C=1C=CC=CC=1)OC1CCCC1 FWFGIHPGRQZWIW-SQNIBIBYSA-N 0.000 description 1
- UUBHZHZSIKRVIV-KCXSXWJSSA-N (2e,6e,10e)-3,7,11,15-tetramethylhexadeca-2,4,6,10,14-pentaenoic acid Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\C=C\C(\C)=C\C(O)=O UUBHZHZSIKRVIV-KCXSXWJSSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- JUSWZYFYLXTMLJ-JTQLQIEISA-N (2s)-1-(benzenesulfonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1S(=O)(=O)C1=CC=CC=C1 JUSWZYFYLXTMLJ-JTQLQIEISA-N 0.000 description 1
- RQYKQWFHJOBBAO-JTQLQIEISA-N (2s)-1-benzoylpyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)C1=CC=CC=C1 RQYKQWFHJOBBAO-JTQLQIEISA-N 0.000 description 1
- KCOYQXZDFIIGCY-CZIZESTLSA-N (3e)-4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinolin-2-one Chemical compound C1CN(C)CCN1C1=CC=C(N\C(N2)=C/3C(=C4C(F)=CC=CC4=NC\3=O)N)C2=C1 KCOYQXZDFIIGCY-CZIZESTLSA-N 0.000 description 1
- ZMKGDQSIRSGUDJ-VSROPUKISA-N (3s,6s,9s,12r,15s,18s,21s,24s,30s,33s)-33-[(e,1r,2r)-1-hydroxy-2-methylhex-4-enyl]-1,4,7,10,12,15,19,25,28-nonamethyl-6,9,18,24-tetrakis(2-methylpropyl)-3,21-di(propan-2-yl)-30-propyl-1,4,7,10,13,16,19,22,25,28,31-undecazacyclotritriacontane-2,5,8,11,14,1 Chemical compound CCC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O ZMKGDQSIRSGUDJ-VSROPUKISA-N 0.000 description 1
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 description 1
- MWTUOSWPJOUADP-XDJHFCHBSA-N (5z)-5-(4-hydroxy-6-oxo-3-propan-2-ylcyclohexa-2,4-dien-1-ylidene)-4-(1-methylindol-5-yl)-1,2,4-triazolidin-3-one Chemical compound O=C1C=C(O)C(C(C)C)=C\C1=C\1N(C=2C=C3C=CN(C)C3=CC=2)C(=O)NN/1 MWTUOSWPJOUADP-XDJHFCHBSA-N 0.000 description 1
- MWWSFMDVAYGXBV-FGBSZODSSA-N (7s,9s)-7-[(2r,4s,5r,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydron;chloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-FGBSZODSSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- GETTZEONDQJALK-UHFFFAOYSA-N (trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC=C1 GETTZEONDQJALK-UHFFFAOYSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- WGLUZJWOTTXZIC-UHFFFAOYSA-N 1-(bromomethyl)-2,4-dimethylbenzene Chemical compound CC1=CC=C(CBr)C(C)=C1 WGLUZJWOTTXZIC-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- SPMVMDHWKHCIDT-UHFFFAOYSA-N 1-[2-chloro-4-[(6,7-dimethoxy-4-quinolinyl)oxy]phenyl]-3-(5-methyl-3-isoxazolyl)urea Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC=1C=C(C)ON=1 SPMVMDHWKHCIDT-UHFFFAOYSA-N 0.000 description 1
- XUMWJQJGCFTJOE-UHFFFAOYSA-N 1-benzofuran-3-carbaldehyde Chemical compound C1=CC=C2C(C=O)=COC2=C1 XUMWJQJGCFTJOE-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- IDINUJSAMVOPCM-UHFFFAOYSA-N 15-Deoxyspergualin Natural products NCCCNCCCCNC(=O)C(O)NC(=O)CCCCCCN=C(N)N IDINUJSAMVOPCM-UHFFFAOYSA-N 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- 125000005808 2,4,6-trimethoxyphenyl group Chemical group [H][#6]-1=[#6](-[#8]C([H])([H])[H])-[#6](-*)=[#6](-[#8]C([H])([H])[H])-[#6]([H])=[#6]-1-[#8]C([H])([H])[H] 0.000 description 1
- UZYQSNQJLWTICD-UHFFFAOYSA-N 2-(n-benzoylanilino)-2,2-dinitroacetic acid Chemical compound C=1C=CC=CC=1N(C(C(=O)O)([N+]([O-])=O)[N+]([O-])=O)C(=O)C1=CC=CC=C1 UZYQSNQJLWTICD-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical group COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- PIMQWRZWLQKKBJ-SFHVURJKSA-N 2-[(2S)-1-[3-ethyl-7-[(1-oxido-3-pyridin-1-iumyl)methylamino]-5-pyrazolo[1,5-a]pyrimidinyl]-2-piperidinyl]ethanol Chemical compound C=1C(N2[C@@H](CCCC2)CCO)=NC2=C(CC)C=NN2C=1NCC1=CC=C[N+]([O-])=C1 PIMQWRZWLQKKBJ-SFHVURJKSA-N 0.000 description 1
- PDWUPXJEEYOOTR-UHFFFAOYSA-N 2-[(3-iodophenyl)methyl]guanidine Chemical compound NC(=N)NCC1=CC=CC(I)=C1 PDWUPXJEEYOOTR-UHFFFAOYSA-N 0.000 description 1
- QXLQZLBNPTZMRK-UHFFFAOYSA-N 2-[(dimethylamino)methyl]-1-(2,4-dimethylphenyl)prop-2-en-1-one Chemical compound CN(C)CC(=C)C(=O)C1=CC=C(C)C=C1C QXLQZLBNPTZMRK-UHFFFAOYSA-N 0.000 description 1
- RZHKDBRREKOZEW-AAXZNHDCSA-N 2-[4-[2-[[(2r)-1-[[(4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-4-[[(2r,3r)-1,3-dihydroxybutan-2-yl]carbamoyl]-7-[(1r)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl] Chemical compound C([C@H](C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)NC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1)C1=CC=CC=C1 RZHKDBRREKOZEW-AAXZNHDCSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- VOXBZHOHGGBLCQ-UHFFFAOYSA-N 2-amino-3,7-dihydropurine-6-thione;hydrate Chemical compound O.N1C(N)=NC(=S)C2=C1N=CN2.N1C(N)=NC(=S)C2=C1N=CN2 VOXBZHOHGGBLCQ-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- 125000006276 2-bromophenyl group Chemical group [H]C1=C([H])C(Br)=C(*)C([H])=C1[H] 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- SMNDYUVBFMFKNZ-UHFFFAOYSA-N 2-furoic acid Chemical class OC(=O)C1=CC=CO1 SMNDYUVBFMFKNZ-UHFFFAOYSA-N 0.000 description 1
- NJRXVEJTAYWCQJ-UHFFFAOYSA-L 2-mercaptosuccinate Chemical compound OC(=O)CC([S-])C([O-])=O NJRXVEJTAYWCQJ-UHFFFAOYSA-L 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004362 3,4,5-trichlorophenyl group Chemical group [H]C1=C(Cl)C(Cl)=C(Cl)C([H])=C1* 0.000 description 1
- 125000004361 3,4,5-trifluorophenyl group Chemical group [H]C1=C(F)C(F)=C(F)C([H])=C1* 0.000 description 1
- 125000004189 3,4-dichlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(Cl)C([H])=C1* 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- 125000003762 3,4-dimethoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 1
- AXRCEOKUDYDWLF-UHFFFAOYSA-N 3-(1-methyl-3-indolyl)-4-[1-[1-(2-pyridinylmethyl)-4-piperidinyl]-3-indolyl]pyrrole-2,5-dione Chemical compound C12=CC=CC=C2N(C)C=C1C(C(NC1=O)=O)=C1C(C1=CC=CC=C11)=CN1C(CC1)CCN1CC1=CC=CC=N1 AXRCEOKUDYDWLF-UHFFFAOYSA-N 0.000 description 1
- UZFPOOOQHWICKY-UHFFFAOYSA-N 3-[13-[1-[1-[8,12-bis(2-carboxyethyl)-17-(1-hydroxyethyl)-3,7,13,18-tetramethyl-21,24-dihydroporphyrin-2-yl]ethoxy]ethyl]-18-(2-carboxyethyl)-8-(1-hydroxyethyl)-3,7,12,17-tetramethyl-22,23-dihydroporphyrin-2-yl]propanoic acid Chemical compound N1C(C=C2C(=C(CCC(O)=O)C(C=C3C(=C(C)C(C=C4N5)=N3)CCC(O)=O)=N2)C)=C(C)C(C(C)O)=C1C=C5C(C)=C4C(C)OC(C)C1=C(N2)C=C(N3)C(C)=C(C(O)C)C3=CC(C(C)=C3CCC(O)=O)=NC3=CC(C(CCC(O)=O)=C3C)=NC3=CC2=C1C UZFPOOOQHWICKY-UHFFFAOYSA-N 0.000 description 1
- NHFDRBXTEDBWCZ-ZROIWOOFSA-N 3-[2,4-dimethyl-5-[(z)-(2-oxo-1h-indol-3-ylidene)methyl]-1h-pyrrol-3-yl]propanoic acid Chemical compound OC(=O)CCC1=C(C)NC(\C=C/2C3=CC=CC=C3NC\2=O)=C1C NHFDRBXTEDBWCZ-ZROIWOOFSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- DOHOPUBZLWVZMZ-UHFFFAOYSA-N 3-chloro-2-hydroxybenzaldehyde Chemical compound OC1=C(Cl)C=CC=C1C=O DOHOPUBZLWVZMZ-UHFFFAOYSA-N 0.000 description 1
- IPPQNXSAJZOTJZ-UHFFFAOYSA-N 3-methylsalicylaldehyde Chemical compound CC1=CC=CC(C=O)=C1O IPPQNXSAJZOTJZ-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 1
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 1
- AFLWPAGYTPJSEY-CODXZCKSSA-N 4-[2-[(8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-10,13-dimethyl-3-oxo-2,6,7,8,9,11,12,14,15,16-decahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-oxoethoxy]-4-oxobutanoic acid;hydrate Chemical compound O.O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)COC(=O)CCC(O)=O)[C@@H]4[C@@H]3CCC2=C1 AFLWPAGYTPJSEY-CODXZCKSSA-N 0.000 description 1
- ZLHFILGSQDJULK-UHFFFAOYSA-N 4-[[9-chloro-7-(2-fluoro-6-methoxyphenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino]-2-methoxybenzoic acid Chemical compound C1=C(C(O)=O)C(OC)=CC(NC=2N=C3C4=CC=C(Cl)C=C4C(=NCC3=CN=2)C=2C(=CC=CC=2F)OC)=C1 ZLHFILGSQDJULK-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 1
- 125000006306 4-iodophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1I 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical compound [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- PQGCEDQWHSBAJP-TXICZTDVSA-N 5-O-phosphono-alpha-D-ribofuranosyl diphosphate Chemical compound O[C@H]1[C@@H](O)[C@@H](O[P@](O)(=O)OP(O)(O)=O)O[C@@H]1COP(O)(O)=O PQGCEDQWHSBAJP-TXICZTDVSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- UPALIKSFLSVKIS-UHFFFAOYSA-N 5-amino-2-[2-(dimethylamino)ethyl]benzo[de]isoquinoline-1,3-dione Chemical compound NC1=CC(C(N(CCN(C)C)C2=O)=O)=C3C2=CC=CC3=C1 UPALIKSFLSVKIS-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- OZPFIJIOIVJZMN-SFHVURJKSA-N 6-[(7s)-7-hydroxy-5,6-dihydropyrrolo[1,2-c]imidazol-7-yl]-n-methylnaphthalene-2-carboxamide Chemical compound C1=CC2=CC(C(=O)NC)=CC=C2C=C1[C@]1(O)C2=CN=CN2CC1 OZPFIJIOIVJZMN-SFHVURJKSA-N 0.000 description 1
- GWKQSBXDFHODNU-UQPSFNCESA-N 6z83r2hm9l Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)CCC(O)=O)[C@@]1(C)C[C@@H]2O GWKQSBXDFHODNU-UQPSFNCESA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- UYLYISCHTFVYHN-UHFFFAOYSA-N 7-oxabicyclo[2.2.1]heptane-3-carboxylic acid Chemical class C1CC2C(C(=O)O)CC1O2 UYLYISCHTFVYHN-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- MXPOCMVWFLDDLZ-NSCUHMNNSA-N Apaziquone Chemical compound CN1C(\C=C\CO)=C(CO)C(C2=O)=C1C(=O)C=C2N1CC1 MXPOCMVWFLDDLZ-NSCUHMNNSA-N 0.000 description 1
- 241000203069 Archaea Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- 108010037003 Buserelin Proteins 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- RVTSJTSTBCGFIX-UHFFFAOYSA-N C(C(C)C)B(O)O.CO Chemical compound C(C(C)C)B(O)O.CO RVTSJTSTBCGFIX-UHFFFAOYSA-N 0.000 description 1
- 125000005947 C1-C6 alkylsulfonyloxy group Chemical group 0.000 description 1
- QWOJMRHUQHTCJG-UHFFFAOYSA-N CC([CH2-])=O Chemical compound CC([CH2-])=O QWOJMRHUQHTCJG-UHFFFAOYSA-N 0.000 description 1
- SJDJFKQVTOWGBZ-UHFFFAOYSA-N CC12OB(OC1(C1(CC(C2)C1)C)C)CC1=COC2=C1C=CC=C2C Chemical compound CC12OB(OC1(C1(CC(C2)C1)C)C)CC1=COC2=C1C=CC=C2C SJDJFKQVTOWGBZ-UHFFFAOYSA-N 0.000 description 1
- WYLMQYPGUDTTOS-JNIAIEOXSA-N CC1=C(C=CC(=C1)C)C[C@@H](B1O[C@]2([C@@H]3C([C@H](C[C@H]2O1)C3)(C)C)C)N Chemical compound CC1=C(C=CC(=C1)C)C[C@@H](B1O[C@]2([C@@H]3C([C@H](C[C@H]2O1)C3)(C)C)C)N WYLMQYPGUDTTOS-JNIAIEOXSA-N 0.000 description 1
- VSEIDZLLWQQJGK-CHOZPQDDSA-N CCC1=C(C)C2=N\C\1=C/C1=C(C)C(C(O)=O)=C(N1)\C(CC(=O)N[C@@H](CC(O)=O)C(O)=O)=C1/N=C(/C=C3\N/C(=C\2)C(C=C)=C3C)[C@@H](C)[C@@H]1CCC(O)=O Chemical compound CCC1=C(C)C2=N\C\1=C/C1=C(C)C(C(O)=O)=C(N1)\C(CC(=O)N[C@@H](CC(O)=O)C(O)=O)=C1/N=C(/C=C3\N/C(=C\2)C(C=C)=C3C)[C@@H](C)[C@@H]1CCC(O)=O VSEIDZLLWQQJGK-CHOZPQDDSA-N 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 229920002284 Cellulose triacetate Polymers 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- ITRJWOMZKQRYTA-RFZYENFJSA-N Cortisone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)CC2=O ITRJWOMZKQRYTA-RFZYENFJSA-N 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 108010036941 Cyclosporins Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- GJKXGJCSJWBJEZ-XRSSZCMZSA-N Deslorelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CNC2=CC=CC=C12 GJKXGJCSJWBJEZ-XRSSZCMZSA-N 0.000 description 1
- VEXZGXHMUGYJMC-DYCDLGHISA-N Deuterium chloride Chemical compound [2H]Cl VEXZGXHMUGYJMC-DYCDLGHISA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- HHJIUUAMYGBVSD-YTFFSALGSA-N Diflucortolone valerate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)COC(=O)CCCC)[C@@]2(C)C[C@@H]1O HHJIUUAMYGBVSD-YTFFSALGSA-N 0.000 description 1
- WYQPLTPSGFELIB-JTQPXKBDSA-N Difluprednate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2CC[C@@](C(=O)COC(C)=O)(OC(=O)CCC)[C@@]2(C)C[C@@H]1O WYQPLTPSGFELIB-JTQPXKBDSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 description 1
- 108700038672 Edotreotide Proteins 0.000 description 1
- 102400001047 Endostatin Human genes 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- OBMLHUPNRURLOK-XGRAFVIBSA-N Epitiostanol Chemical compound C1[C@@H]2S[C@@H]2C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 OBMLHUPNRURLOK-XGRAFVIBSA-N 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical compound [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 description 1
- WJOHZNCJWYWUJD-IUGZLZTKSA-N Fluocinonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)C)[C@@]2(C)C[C@@H]1O WJOHZNCJWYWUJD-IUGZLZTKSA-N 0.000 description 1
- WHZRCUIISKRTJL-YTZKRAOUSA-N Fluocortolone caproate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)COC(=O)CCCCC)[C@@]2(C)C[C@@H]1O WHZRCUIISKRTJL-YTZKRAOUSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- POPFMWWJOGLOIF-XWCQMRHXSA-N Flurandrenolide Chemical compound C1([C@@H](F)C2)=CC(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O POPFMWWJOGLOIF-XWCQMRHXSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 108010081348 HRT1 protein Hairy Proteins 0.000 description 1
- 102100021881 Hairy/enhancer-of-split related with YRPW motif protein 1 Human genes 0.000 description 1
- MUQNGPZZQDCDFT-JNQJZLCISA-N Halcinonide Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CCl)[C@@]1(C)C[C@@H]2O MUQNGPZZQDCDFT-JNQJZLCISA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001063392 Homo sapiens Lymphocyte function-associated antigen 3 Proteins 0.000 description 1
- 101001124792 Homo sapiens Proteasome subunit beta type-10 Proteins 0.000 description 1
- 101001136981 Homo sapiens Proteasome subunit beta type-9 Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 206010021263 IgA nephropathy Diseases 0.000 description 1
- 102000012214 Immunoproteins Human genes 0.000 description 1
- 108010036650 Immunoproteins Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108010008212 Integrin alpha4beta1 Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- NHTGHBARYWONDQ-JTQLQIEISA-N L-α-methyl-Tyrosine Chemical compound OC(=O)[C@](N)(C)CC1=CC=C(O)C=C1 NHTGHBARYWONDQ-JTQLQIEISA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 1
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 1
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 1
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 1
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 1
- 239000002139 L01XE22 - Masitinib Substances 0.000 description 1
- 239000002137 L01XE24 - Ponatinib Substances 0.000 description 1
- 239000002176 L01XE26 - Cabozantinib Substances 0.000 description 1
- UCEQXRCJXIVODC-PMACEKPBSA-N LSM-1131 Chemical compound C1CCC2=CC=CC3=C2N1C=C3[C@@H]1C(=O)NC(=O)[C@H]1C1=CNC2=CC=CC=C12 UCEQXRCJXIVODC-PMACEKPBSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 1
- 229920001491 Lentinan Polymers 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 102100030984 Lymphocyte function-associated antigen 3 Human genes 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- HZQDCMWJEBCWBR-UUOKFMHZSA-N Mizoribine Chemical compound OC1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 HZQDCMWJEBCWBR-UUOKFMHZSA-N 0.000 description 1
- 229920000715 Mucilage Polymers 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- ADBUEVSHEJICCM-LZNHGOJSSA-N N-[(2R)-1-[[(2S)-3-(1H-indol-3-yl)-1-[[(2S)-1-[(2R)-2-methyloxiran-2-yl]-1-oxo-3-phenylpropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]-3-methyl-1H-indene-2-carboxamide Chemical compound C[C@@H](NC(=O)C1=C(C)c2ccccc2C1)C(=O)N[C@@H](Cc3c[nH]c4ccccc34)C(=O)N[C@@H](Cc5ccccc5)C(=O)[C@@]6(C)CO6 ADBUEVSHEJICCM-LZNHGOJSSA-N 0.000 description 1
- VIUAUNHCRHHYNE-JTQLQIEISA-N N-[(2S)-2,3-dihydroxypropyl]-3-(2-fluoro-4-iodoanilino)-4-pyridinecarboxamide Chemical compound OC[C@@H](O)CNC(=O)C1=CC=NC=C1NC1=CC=C(I)C=C1F VIUAUNHCRHHYNE-JTQLQIEISA-N 0.000 description 1
- 150000007945 N-acyl ureas Chemical class 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- HIEKJRVYXXINKH-ADVKXBNGSA-N N1([C@H]2CC[C@H](C[C@H]2OC)/C=C(\C)[C@H]2OC(=O)[C@@H]3CCCCN3C(=O)C(=O)[C@]3(O)O[C@@H]([C@H](C[C@H]3C)OC)[C@@H](OC)C[C@@H](C)C/C(C)=C/[C@H](C(C[C@H](O)[C@H]2C)=O)CC)C=NN=N1 Chemical compound N1([C@H]2CC[C@H](C[C@H]2OC)/C=C(\C)[C@H]2OC(=O)[C@@H]3CCCCN3C(=O)C(=O)[C@]3(O)O[C@@H]([C@H](C[C@H]3C)OC)[C@@H](OC)C[C@@H](C)C/C(C)=C/[C@H](C(C[C@H](O)[C@H]2C)=O)CC)C=NN=N1 HIEKJRVYXXINKH-ADVKXBNGSA-N 0.000 description 1
- LYPFDBRUNKHDGX-SOGSVHMOSA-N N1C2=CC=C1\C(=C1\C=CC(=N1)\C(=C1\C=C/C(/N1)=C(/C1=N/C(/CC1)=C2/C1=CC(O)=CC=C1)C1=CC(O)=CC=C1)\C1=CC(O)=CC=C1)C1=CC(O)=CC=C1 Chemical compound N1C2=CC=C1\C(=C1\C=CC(=N1)\C(=C1\C=C/C(/N1)=C(/C1=N/C(/CC1)=C2/C1=CC(O)=CC=C1)C1=CC(O)=CC=C1)\C1=CC(O)=CC=C1)C1=CC(O)=CC=C1 LYPFDBRUNKHDGX-SOGSVHMOSA-N 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- ZMKGDQSIRSGUDJ-UHFFFAOYSA-N NVa2 cyclosporine Natural products CCCC1NC(=O)C(C(O)C(C)CC=CC)N(C)C(=O)C(C(C)C)N(C)C(=O)C(CC(C)C)N(C)C(=O)C(CC(C)C)N(C)C(=O)C(C)NC(=O)C(C)NC(=O)C(CC(C)C)N(C)C(=O)C(C(C)C)NC(=O)C(CC(C)C)N(C)C(=O)CN(C)C1=O ZMKGDQSIRSGUDJ-UHFFFAOYSA-N 0.000 description 1
- 108010021717 Nafarelin Proteins 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 108010016076 Octreotide Proteins 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 101800000628 PDH precursor-related peptide Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- HYRKAAMZBDSJFJ-LFDBJOOHSA-N Paramethasone acetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)COC(C)=O)(O)[C@@]2(C)C[C@@H]1O HYRKAAMZBDSJFJ-LFDBJOOHSA-N 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- LRJOMUJRLNCICJ-JZYPGELDSA-N Prednisolone acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O LRJOMUJRLNCICJ-JZYPGELDSA-N 0.000 description 1
- 102100029081 Proteasome subunit beta type-10 Human genes 0.000 description 1
- 102100035764 Proteasome subunit beta type-9 Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000005464 Radotinib Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- YJDYDFNKCBANTM-QCWCSKBGSA-N SDZ PSC 833 Chemical compound C\C=C\C[C@@H](C)C(=O)[C@@H]1N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C(=O)[C@H](C(C)C)NC1=O YJDYDFNKCBANTM-QCWCSKBGSA-N 0.000 description 1
- 229910006124 SOCl2 Inorganic materials 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 229920000519 Sizofiran Polymers 0.000 description 1
- 206010050207 Skin fibrosis Diseases 0.000 description 1
- OCOKWVBYZHBHLU-UHFFFAOYSA-N Sobuzoxane Chemical compound C1C(=O)N(COC(=O)OCC(C)C)C(=O)CN1CCN1CC(=O)N(COC(=O)OCC(C)C)C(=O)C1 OCOKWVBYZHBHLU-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GKLVYJBZJHMRIY-OUBTZVSYSA-N Technetium-99 Chemical compound [99Tc] GKLVYJBZJHMRIY-OUBTZVSYSA-N 0.000 description 1
- NCLGDOBQAWBXRA-PGRDOPGGSA-N Telotristat Chemical compound N1=C(C)C=CN1C1=CC(Cl)=CC=C1[C@H](C(F)(F)F)OC1=CC(C=2C=CC(C[C@H](N)C(O)=O)=CC=2)=NC(N)=N1 NCLGDOBQAWBXRA-PGRDOPGGSA-N 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 108010078233 Thymalfasin Proteins 0.000 description 1
- 108010066702 Thyrotropin Alfa Proteins 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- YCPOZVAOBBQLRI-WDSKDSINSA-N Treosulfan Chemical compound CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O YCPOZVAOBBQLRI-WDSKDSINSA-N 0.000 description 1
- XGMPVBXKDAHORN-RBWIMXSLSA-N Triamcinolone diacetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](OC(C)=O)[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O XGMPVBXKDAHORN-RBWIMXSLSA-N 0.000 description 1
- TZIZWYVVGLXXFV-FLRHRWPCSA-N Triamcinolone hexacetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)CC(C)(C)C)[C@@]1(C)C[C@@H]2O TZIZWYVVGLXXFV-FLRHRWPCSA-N 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- 108010050144 Triptorelin Pamoate Proteins 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 108010005705 Ubiquitinated Proteins Proteins 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- XOOGCKRYCICQOH-SNVBAGLBSA-N [(1R)-1-phenylethyl] furan-3-carboxylate Chemical compound C1(=CC=CC=C1)[C@@H](C)OC(=O)C1=COC=C1 XOOGCKRYCICQOH-SNVBAGLBSA-N 0.000 description 1
- RFQDLTYXNINJON-ZCDTZLGTSA-N [(1R)-2-(1-benzofuran-3-yl)-1-[[(1R,2R,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@H]2CC[C@@H](C1)O2)B(O)O RFQDLTYXNINJON-ZCDTZLGTSA-N 0.000 description 1
- BNRPDTGWHIDEDH-HIOOUATESA-N [(1R)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1R,2R,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@H]2CC[C@@H](C1)O2)B(O)O BNRPDTGWHIDEDH-HIOOUATESA-N 0.000 description 1
- BNRPDTGWHIDEDH-LTDJIEQWSA-N [(1R)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1R,2S,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@@H]1[C@H]2CC[C@@H](C1)O2)B(O)O BNRPDTGWHIDEDH-LTDJIEQWSA-N 0.000 description 1
- BNRPDTGWHIDEDH-LTWQWCQGSA-N [(1R)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-LTWQWCQGSA-N 0.000 description 1
- BNRPDTGWHIDEDH-AYCVOAMHSA-N [(1R)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2S,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-AYCVOAMHSA-N 0.000 description 1
- BNRPDTGWHIDEDH-PDGPNTQRSA-N [(1R)-2-[(3S)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1R,2R,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@H]2CC[C@@H](C1)O2)B(O)O BNRPDTGWHIDEDH-PDGPNTQRSA-N 0.000 description 1
- BNRPDTGWHIDEDH-GKRFPXGYSA-N [(1R)-2-[(3S)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-GKRFPXGYSA-N 0.000 description 1
- BNRPDTGWHIDEDH-VWHJJCLCSA-N [(1R)-2-[(3S)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2S,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@H](C2=C1C=CC=C2)C[C@H](NC(=O)[C@@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-VWHJJCLCSA-N 0.000 description 1
- RFQDLTYXNINJON-KSZJFAHPSA-N [(1S)-2-(1-benzofuran-3-yl)-1-[[(1R,2R,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C=C(C2=C1C=CC=C2)C[C@@H](NC(=O)[C@H]1[C@H]2CC[C@@H](C1)O2)B(O)O RFQDLTYXNINJON-KSZJFAHPSA-N 0.000 description 1
- BNRPDTGWHIDEDH-GBGXWIRYSA-N [(1S)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1R,2R,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@@H](NC(=O)[C@H]1[C@H]2CC[C@@H](C1)O2)B(O)O BNRPDTGWHIDEDH-GBGXWIRYSA-N 0.000 description 1
- BNRPDTGWHIDEDH-WKYPKFPVSA-N [(1S)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-WKYPKFPVSA-N 0.000 description 1
- BNRPDTGWHIDEDH-AREYPJKDSA-N [(1S)-2-[(3R)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2S,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@@H](C2=C1C=CC=C2)C[C@@H](NC(=O)[C@@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-AREYPJKDSA-N 0.000 description 1
- BNRPDTGWHIDEDH-VUBORPHZSA-N [(1S)-2-[(3S)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1R,2R,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@H](C2=C1C=CC=C2)C[C@@H](NC(=O)[C@H]1[C@H]2CC[C@@H](C1)O2)B(O)O BNRPDTGWHIDEDH-VUBORPHZSA-N 0.000 description 1
- BNRPDTGWHIDEDH-JOCHCUSFSA-N [(1S)-2-[(3S)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1R,2S,4S)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@H](C2=C1C=CC=C2)C[C@@H](NC(=O)[C@@H]1[C@H]2CC[C@@H](C1)O2)B(O)O BNRPDTGWHIDEDH-JOCHCUSFSA-N 0.000 description 1
- BNRPDTGWHIDEDH-CQSRIBRNSA-N [(1S)-2-[(3S)-2,3-dihydro-1-benzofuran-3-yl]-1-[[(1S,2R,4R)-7-oxabicyclo[2.2.1]heptane-2-carbonyl]amino]ethyl]boronic acid Chemical compound O1C[C@H](C2=C1C=CC=C2)C[C@@H](NC(=O)[C@H]1[C@@H]2CC[C@H](C1)O2)B(O)O BNRPDTGWHIDEDH-CQSRIBRNSA-N 0.000 description 1
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 1
- XJXKGUZINMNEDK-GPJOBVNKSA-L [(4r,5r)-5-(aminomethyl)-2-propan-2-yl-1,3-dioxolan-4-yl]methanamine;platinum(2+);propanedioate Chemical compound [Pt+2].[O-]C(=O)CC([O-])=O.CC(C)C1O[C@H](CN)[C@@H](CN)O1 XJXKGUZINMNEDK-GPJOBVNKSA-L 0.000 description 1
- FPVRUILUEYSIMD-RPRRAYFGSA-N [(8s,9r,10s,11s,13s,14s,16r,17r)-9-fluoro-11-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(OC(C)=O)[C@@]1(C)C[C@@H]2O FPVRUILUEYSIMD-RPRRAYFGSA-N 0.000 description 1
- XSMVECZRZBFTIZ-UHFFFAOYSA-M [2-(aminomethyl)cyclobutyl]methanamine;2-oxidopropanoate;platinum(4+) Chemical compound [Pt+4].CC([O-])C([O-])=O.NCC1CCC1CN XSMVECZRZBFTIZ-UHFFFAOYSA-M 0.000 description 1
- WBNNIURTBZHJTI-WDCKKOMHSA-N [2-[(8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-17-yl]-2-oxoethyl] dihydrogen phosphate;2-[(2-propan-2-ylphenoxy)methyl]-4,5-dihydro-1h-imidazole Chemical compound CC(C)C1=CC=CC=C1OCC1=NCCN1.O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)COP(O)(O)=O)[C@@H]4[C@@H]3CCC2=C1 WBNNIURTBZHJTI-WDCKKOMHSA-N 0.000 description 1
- DNSAKUGJOSFARZ-FOMYWIRZSA-N [2-[(8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-17-yl]-2-oxoethyl] hexanoate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)CCCCC)(O)[C@@]1(C)C[C@@H]2O DNSAKUGJOSFARZ-FOMYWIRZSA-N 0.000 description 1
- 229960002184 abarelix Drugs 0.000 description 1
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 1
- 108010023617 abarelix Proteins 0.000 description 1
- 229960000853 abiraterone Drugs 0.000 description 1
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- MGVGMXLGOKTYKP-ZFOBEOMCSA-N acetic acid;(6s,8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-17-(2-hydroxyacetyl)-6,10,13-trimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-3-one Chemical compound CC(O)=O.C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)CO)CC[C@H]21 MGVGMXLGOKTYKP-ZFOBEOMCSA-N 0.000 description 1
- ZDGWGNDTQZGISB-UHFFFAOYSA-N acetic acid;perchloric acid Chemical compound CC(O)=O.OCl(=O)(=O)=O ZDGWGNDTQZGISB-UHFFFAOYSA-N 0.000 description 1
- 108010052004 acetyl-2-naphthylalanyl-3-chlorophenylalanyl-1-oxohexadecyl-seryl-4-aminophenylalanyl(hydroorotyl)-4-aminophenylalanyl(carbamoyl)-leucyl-ILys-prolyl-alaninamide Proteins 0.000 description 1
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 1
- 229960004176 aclarubicin Drugs 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 229960005310 aldesleukin Drugs 0.000 description 1
- 108700025316 aldesleukin Proteins 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 229950009447 alisertib Drugs 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 229940061720 alpha hydroxy acid Drugs 0.000 description 1
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 1
- AEMOLEFTQBMNLQ-BKBMJHBISA-N alpha-D-galacturonic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-BKBMJHBISA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229960003099 amcinonide Drugs 0.000 description 1
- ILKJAFIWWBXGDU-MOGDOJJUSA-N amcinonide Chemical compound O([C@@]1([C@H](O2)C[C@@H]3[C@@]1(C[C@H](O)[C@]1(F)[C@@]4(C)C=CC(=O)C=C4CC[C@H]13)C)C(=O)COC(=O)C)C12CCCC1 ILKJAFIWWBXGDU-MOGDOJJUSA-N 0.000 description 1
- LDPIQRWHBLWKPR-UHFFFAOYSA-N aminoboronic acid Chemical class NB(O)O LDPIQRWHBLWKPR-UHFFFAOYSA-N 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 229960004701 amonafide Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229960002550 amrubicin Drugs 0.000 description 1
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229950002465 apaziquone Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 229960004669 basiliximab Drugs 0.000 description 1
- 229950000210 beclometasone dipropionate Drugs 0.000 description 1
- 229960003094 belinostat Drugs 0.000 description 1
- NCNRHFGMJRPRSK-MDZDMXLPSA-N belinostat Chemical compound ONC(=O)\C=C\C1=CC=CC(S(=O)(=O)NC=2C=CC=CC=2)=C1 NCNRHFGMJRPRSK-MDZDMXLPSA-N 0.000 description 1
- LNHWXBUNXOXMRL-VWLOTQADSA-N belotecan Chemical compound C1=CC=C2C(CCNC(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 LNHWXBUNXOXMRL-VWLOTQADSA-N 0.000 description 1
- 229950011276 belotecan Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 229950004221 besilate Drugs 0.000 description 1
- 229950010559 besilesomab Drugs 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 229960004648 betamethasone acetate Drugs 0.000 description 1
- AKUJBENLRBOFTD-QZIXMDIESA-N betamethasone acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(C)=O)(O)[C@@]1(C)C[C@@H]2O AKUJBENLRBOFTD-QZIXMDIESA-N 0.000 description 1
- 229960001102 betamethasone dipropionate Drugs 0.000 description 1
- CIWBQSYVNNPZIQ-XYWKZLDCSA-N betamethasone dipropionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CIWBQSYVNNPZIQ-XYWKZLDCSA-N 0.000 description 1
- 229960005354 betamethasone sodium phosphate Drugs 0.000 description 1
- PLCQGRYPOISRTQ-LWCNAHDDSA-L betamethasone sodium phosphate Chemical compound [Na+].[Na+].C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COP([O-])([O-])=O)(O)[C@@]1(C)C[C@@H]2O PLCQGRYPOISRTQ-LWCNAHDDSA-L 0.000 description 1
- 229960004311 betamethasone valerate Drugs 0.000 description 1
- SNHRLVCMMWUAJD-SUYDQAKGSA-N betamethasone valerate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(OC(=O)CCCC)[C@@]1(C)C[C@@H]2O SNHRLVCMMWUAJD-SUYDQAKGSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- JESWDXIHOJGWBP-UHFFFAOYSA-N bicyclo[2.2.1]heptane-3-carboxylic acid Chemical compound C1CC2C(C(=O)O)CC1C2 JESWDXIHOJGWBP-UHFFFAOYSA-N 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- LHHCSNFAOIFYRV-DOVBMPENSA-N boceprevir Chemical compound O=C([C@@H]1[C@@H]2[C@@H](C2(C)C)CN1C(=O)[C@@H](NC(=O)NC(C)(C)C)C(C)(C)C)NC(C(=O)C(N)=O)CC1CCC1 LHHCSNFAOIFYRV-DOVBMPENSA-N 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 1
- 229960003736 bosutinib Drugs 0.000 description 1
- 150000005693 branched-chain amino acids Chemical class 0.000 description 1
- 229960000455 brentuximab vedotin Drugs 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- CUWODFFVMXJOKD-UVLQAERKSA-N buserelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](COC(C)(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 CUWODFFVMXJOKD-UVLQAERKSA-N 0.000 description 1
- 229960002719 buserelin Drugs 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960001573 cabazitaxel Drugs 0.000 description 1
- BMQGVNUXMIRLCK-OAGWZNDDSA-N cabazitaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OC)C(=O)C1=CC=CC=C1 BMQGVNUXMIRLCK-OAGWZNDDSA-N 0.000 description 1
- 229960001292 cabozantinib Drugs 0.000 description 1
- ONIQOQHATWINJY-UHFFFAOYSA-N cabozantinib Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 ONIQOQHATWINJY-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 229960001921 calcium levofolinate Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- KVUAALJSMIVURS-QNTKWALQSA-L calcium;(2s)-2-[[4-[[(6s)-2-amino-5-formyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl]methylamino]benzoyl]amino]pentanedioate Chemical compound [Ca+2].C([C@@H]1N(C=O)C=2C(=O)N=C(NC=2NC1)N)NC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 KVUAALJSMIVURS-QNTKWALQSA-L 0.000 description 1
- IVFYLRMMHVYGJH-PVPPCFLZSA-N calusterone Chemical compound C1C[C@]2(C)[C@](O)(C)CC[C@H]2[C@@H]2[C@@H](C)CC3=CC(=O)CC[C@]3(C)[C@H]21 IVFYLRMMHVYGJH-PVPPCFLZSA-N 0.000 description 1
- 229950009823 calusterone Drugs 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 description 1
- 150000001719 carbohydrate derivatives Chemical class 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229950005499 carbon tetrachloride Drugs 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000009787 cardiac fibrosis Effects 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 229960000419 catumaxomab Drugs 0.000 description 1
- 229960002412 cediranib Drugs 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229950001357 celmoleukin Drugs 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- JQXXHWHPUNPDRT-YOPQJBRCSA-N chembl1332716 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CCN(C)CC1 JQXXHWHPUNPDRT-YOPQJBRCSA-N 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 229960002559 chlorotrianisene Drugs 0.000 description 1
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229940114081 cinnamate Drugs 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004703 clobetasol propionate Drugs 0.000 description 1
- CBGUOGMQLZIXBE-XGQKBEPLSA-N clobetasol propionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CBGUOGMQLZIXBE-XGQKBEPLSA-N 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229960005527 combretastatin A-4 phosphate Drugs 0.000 description 1
- WDOGQTQEKVLZIJ-WAYWQWQTSA-N combretastatin a-4 phosphate Chemical compound C1=C(OP(O)(O)=O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 WDOGQTQEKVLZIJ-WAYWQWQTSA-N 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- BMCQMVFGOVHVNG-TUFAYURCSA-N cortisol 17-butyrate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)CO)(OC(=O)CCC)[C@@]1(C)C[C@@H]2O BMCQMVFGOVHVNG-TUFAYURCSA-N 0.000 description 1
- ALEXXDVDDISNDU-JZYPGELDSA-N cortisol 21-acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O ALEXXDVDDISNDU-JZYPGELDSA-N 0.000 description 1
- 229960003290 cortisone acetate Drugs 0.000 description 1
- 229960005061 crizotinib Drugs 0.000 description 1
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229940109275 cyclamate Drugs 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 108010019249 cyclosporin G Proteins 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- LVXJQMNHJWSHET-AATRIKPKSA-N dacomitinib Chemical compound C=12C=C(NC(=O)\C=C\CN3CCCCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 LVXJQMNHJWSHET-AATRIKPKSA-N 0.000 description 1
- 229950002205 dacomitinib Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- POZRVZJJTULAOH-LHZXLZLDSA-N danazol Chemical compound C1[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=CC2=C1C=NO2 POZRVZJJTULAOH-LHZXLZLDSA-N 0.000 description 1
- 229960000766 danazol Drugs 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960002272 degarelix Drugs 0.000 description 1
- MEUCPCLKGZSHTA-XYAYPHGZSA-N degarelix Chemical compound C([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)NC(=O)[C@H](CC=1C=CC(NC(=O)[C@H]2NC(=O)NC(=O)C2)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(NC(N)=O)C=C1 MEUCPCLKGZSHTA-XYAYPHGZSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 229960002923 denileukin diftitox Drugs 0.000 description 1
- 108010017271 denileukin diftitox Proteins 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 108700025485 deslorelin Proteins 0.000 description 1
- 229960005408 deslorelin Drugs 0.000 description 1
- 229960003657 dexamethasone acetate Drugs 0.000 description 1
- 229960000524 dexamethasone isonicotinate Drugs 0.000 description 1
- 229960004833 dexamethasone phosphate Drugs 0.000 description 1
- VQODGRNSFPNSQE-CXSFZGCWSA-N dexamethasone phosphate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COP(O)(O)=O)(O)[C@@]1(C)C[C@@H]2O VQODGRNSFPNSQE-CXSFZGCWSA-N 0.000 description 1
- RPBJOYICBFNIMN-RDWMNNCQSA-M dexamethasone sodium m-sulfobenzoate Chemical compound [Na+].O=C([C@]1(O)[C@@]2(C)C[C@H](O)[C@]3(F)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)COC(=O)C1=CC=CC(S([O-])(=O)=O)=C1 RPBJOYICBFNIMN-RDWMNNCQSA-M 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 229960003970 diflucortolone valerate Drugs 0.000 description 1
- 229960004875 difluprednate Drugs 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- IPZJQDSFZGZEOY-UHFFFAOYSA-N dimethylmethylene Chemical group C[C]C IPZJQDSFZGZEOY-UHFFFAOYSA-N 0.000 description 1
- 229950009859 dinaciclib Drugs 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical compound CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229950005778 dovitinib Drugs 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 229950006595 edotreotide Drugs 0.000 description 1
- 238000001493 electron microscopy Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229960004137 elotuzumab Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 description 1
- 229950005837 entinostat Drugs 0.000 description 1
- 229950002189 enzastaurin Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229960003265 epirubicin hydrochloride Drugs 0.000 description 1
- 229950002973 epitiostanol Drugs 0.000 description 1
- 229950009760 epratuzumab Drugs 0.000 description 1
- 229950006835 eptaplatin Drugs 0.000 description 1
- 229960003649 eribulin Drugs 0.000 description 1
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- WCDWBPCFGJXFJZ-UHFFFAOYSA-N etanidazole Chemical compound OCCNC(=O)CN1C=CN=C1[N+]([O-])=O WCDWBPCFGJXFJZ-UHFFFAOYSA-N 0.000 description 1
- 229950006566 etanidazole Drugs 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229950011548 fadrozole Drugs 0.000 description 1
- 229950009929 farletuzumab Drugs 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000005454 flavour additive Substances 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- SYWHXTATXSMDSB-GSLJADNHSA-N fludrocortisone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O SYWHXTATXSMDSB-GSLJADNHSA-N 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- JWRMHDSINXPDHB-OJAGFMMFSA-N flumethasone pivalate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)COC(=O)C(C)(C)C)(O)[C@@]2(C)C[C@@H]1O JWRMHDSINXPDHB-OJAGFMMFSA-N 0.000 description 1
- 229940042902 flumethasone pivalate Drugs 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 229960001347 fluocinolone acetonide Drugs 0.000 description 1
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 1
- 229960000785 fluocinonide Drugs 0.000 description 1
- 229960005283 fluocortolone pivalate Drugs 0.000 description 1
- XZBJVIQXJHGUBE-HZMVJJPJSA-N fluocortolone pivalate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)COC(=O)C(C)(C)C)[C@@]2(C)C[C@@H]1O XZBJVIQXJHGUBE-HZMVJJPJSA-N 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229960003336 fluorocortisol acetate Drugs 0.000 description 1
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 description 1
- 229960001048 fluorometholone Drugs 0.000 description 1
- FAOZLTXFLGPHNG-KNAQIMQKSA-N fluorometholone Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@]2(F)[C@@H](O)C[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FAOZLTXFLGPHNG-KNAQIMQKSA-N 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 1
- 229960001751 fluoxymesterone Drugs 0.000 description 1
- DEFOZIFYUBUHHU-IYQKUMFPSA-N fluprednidene acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC(=C)[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O DEFOZIFYUBUHHU-IYQKUMFPSA-N 0.000 description 1
- 229960002650 fluprednidene acetate Drugs 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960000289 fluticasone propionate Drugs 0.000 description 1
- WMWTYOKRWGGJOA-CENSZEJFSA-N fluticasone propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O WMWTYOKRWGGJOA-CENSZEJFSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960004421 formestane Drugs 0.000 description 1
- OSVMTWJCGUFAOD-KZQROQTASA-N formestane Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1O OSVMTWJCGUFAOD-KZQROQTASA-N 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 229960002258 fulvestrant Drugs 0.000 description 1
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 1
- 229950004161 ganetespib Drugs 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 229960005144 gemcitabine hydrochloride Drugs 0.000 description 1
- 229960000578 gemtuzumab Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 125000003147 glycosyl group Chemical group 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- PPGHYTPFGILTSZ-PPJQWWMSSA-M gpe7477yek Chemical compound [Na+].O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C([O-])C=2\C=N\N1CCN(C)CC1 PPGHYTPFGILTSZ-PPJQWWMSSA-M 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229960002383 halcinonide Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 235000015220 hamburgers Nutrition 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- VUVZASHBYYMLRC-UHFFFAOYSA-N heptane-2,3-diol Chemical compound CCCCC(O)C(C)O VUVZASHBYYMLRC-UHFFFAOYSA-N 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- HHXHVIJIIXKSOE-QILQGKCVSA-N histrelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC(N=C1)=CN1CC1=CC=CC=C1 HHXHVIJIIXKSOE-QILQGKCVSA-N 0.000 description 1
- 108700020746 histrelin Proteins 0.000 description 1
- 229960002193 histrelin Drugs 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 229960001067 hydrocortisone acetate Drugs 0.000 description 1
- 229960001524 hydrocortisone butyrate Drugs 0.000 description 1
- 229950006240 hydrocortisone succinate Drugs 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 229960005236 ibandronic acid Drugs 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- JBFYUZGYRGXSFL-UHFFFAOYSA-N imidazolide Chemical compound C1=C[N-]C=N1 JBFYUZGYRGXSFL-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 229940124622 immune-modulator drug Drugs 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 229950002133 iniparib Drugs 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229910052500 inorganic mineral Chemical class 0.000 description 1
- 229950004101 inotuzumab ozogamicin Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229960001388 interferon-beta Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229960003795 iobenguane (123i) Drugs 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 1
- 229960002014 ixabepilone Drugs 0.000 description 1
- 229960003648 ixazomib Drugs 0.000 description 1
- MXAYKZJJDUDWDS-LBPRGKRZSA-N ixazomib Chemical compound CC(C)C[C@@H](B(O)O)NC(=O)CNC(=O)C1=CC(Cl)=CC=C1Cl MXAYKZJJDUDWDS-LBPRGKRZSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- 229960000681 leflunomide Drugs 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 1
- 229940115286 lentinan Drugs 0.000 description 1
- 229960003784 lenvatinib Drugs 0.000 description 1
- WOSKHXYHFSIKNG-UHFFFAOYSA-N lenvatinib Chemical compound C=12C=C(C(N)=O)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC1CC1 WOSKHXYHFSIKNG-UHFFFAOYSA-N 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 229960001614 levamisole Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 1
- 229950002216 linifanib Drugs 0.000 description 1
- 229950001762 linsitinib Drugs 0.000 description 1
- PKCDDUHJAFVJJB-VLZXCDOPSA-N linsitinib Chemical compound C1[C@](C)(O)C[C@@H]1C1=NC(C=2C=C3N=C(C=CC3=CC=2)C=2C=CC=CC=2)=C2N1C=CN=C2N PKCDDUHJAFVJJB-VLZXCDOPSA-N 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 210000001853 liver microsome Anatomy 0.000 description 1
- 229950008991 lobaplatin Drugs 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 229960003538 lonidamine Drugs 0.000 description 1
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- MMIPFLVOWGHZQD-UHFFFAOYSA-N manganese(3+) Chemical compound [Mn+3] MMIPFLVOWGHZQD-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229960004655 masitinib Drugs 0.000 description 1
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 1
- 229950008001 matuzumab Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960001786 megestrol Drugs 0.000 description 1
- JBVNBBXAMBZTMQ-CEGNMAFCSA-N megestrol Chemical compound C1=CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 JBVNBBXAMBZTMQ-CEGNMAFCSA-N 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 229960001810 meprednisone Drugs 0.000 description 1
- PIDANAQULIKBQS-RNUIGHNZSA-N meprednisone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)CC2=O PIDANAQULIKBQS-RNUIGHNZSA-N 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- XBYZJUMTKHUJIY-UHFFFAOYSA-N methyl 5-methylfuran-2-carboxylate Chemical compound COC(=O)C1=CC=C(C)O1 XBYZJUMTKHUJIY-UHFFFAOYSA-N 0.000 description 1
- OJLOPKGSLYJEMD-URPKTTJQSA-N methyl 7-[(1r,2r,3r)-3-hydroxy-2-[(1e)-4-hydroxy-4-methyloct-1-en-1-yl]-5-oxocyclopentyl]heptanoate Chemical compound CCCCC(C)(O)C\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(=O)OC OJLOPKGSLYJEMD-URPKTTJQSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 229960001293 methylprednisolone acetate Drugs 0.000 description 1
- IMBXEJJVJRTNOW-XYMSELFBSA-N methylprednisolone succinate Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC(O)=O)CC[C@H]21 IMBXEJJVJRTNOW-XYMSELFBSA-N 0.000 description 1
- 229950009831 methylprednisolone succinate Drugs 0.000 description 1
- 229960001980 metirosine Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 229950010895 midostaurin Drugs 0.000 description 1
- BMGQWWVMWDBQGC-IIFHNQTCSA-N midostaurin Chemical compound CN([C@H]1[C@H]([C@]2(C)O[C@@H](N3C4=CC=CC=C4C4=C5C(=O)NCC5=C5C6=CC=CC=C6N2C5=C43)C1)OC)C(=O)C1=CC=CC=C1 BMGQWWVMWDBQGC-IIFHNQTCSA-N 0.000 description 1
- 229960005225 mifamurtide Drugs 0.000 description 1
- 108700007621 mifamurtide Proteins 0.000 description 1
- 229960003775 miltefosine Drugs 0.000 description 1
- PQLXHQMOHUQAKB-UHFFFAOYSA-N miltefosine Chemical compound CCCCCCCCCCCCCCCCOP([O-])(=O)OCC[N+](C)(C)C PQLXHQMOHUQAKB-UHFFFAOYSA-N 0.000 description 1
- 239000011707 mineral Chemical class 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 229960005249 misoprostol Drugs 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229950000844 mizoribine Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 229950007699 mogamulizumab Drugs 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- RAHBGWKEPAQNFF-UHFFFAOYSA-N motesanib Chemical compound C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 RAHBGWKEPAQNFF-UHFFFAOYSA-N 0.000 description 1
- 229950003968 motesanib Drugs 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229960003816 muromonab-cd3 Drugs 0.000 description 1
- 201000006938 muscular dystrophy Diseases 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- BLUYEPLOXLPVCJ-INIZCTEOSA-N n-[(1s)-2-[4-(3-aminopropylamino)butylamino]-1-hydroxyethyl]-7-(diaminomethylideneamino)heptanamide Chemical compound NCCCNCCCCNC[C@H](O)NC(=O)CCCCCCNC(N)=N BLUYEPLOXLPVCJ-INIZCTEOSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- XNSAINXGIQZQOO-UHFFFAOYSA-N n-[1-(2-carbamoylpyrrolidin-1-yl)-3-(1h-imidazol-5-yl)-1-oxopropan-2-yl]-5-oxopyrrolidine-2-carboxamide Chemical compound NC(=O)C1CCCN1C(=O)C(NC(=O)C1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- RWHUEXWOYVBUCI-ITQXDASVSA-N nafarelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 RWHUEXWOYVBUCI-ITQXDASVSA-N 0.000 description 1
- 229960002333 nafarelin Drugs 0.000 description 1
- 229960004719 nandrolone Drugs 0.000 description 1
- NPAGDVCDWIYMMC-IZPLOLCNSA-N nandrolone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 NPAGDVCDWIYMMC-IZPLOLCNSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 229960000513 necitumumab Drugs 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 229950008835 neratinib Drugs 0.000 description 1
- JWNPDZNEKVCWMY-VQHVLOKHSA-N neratinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960001346 nilotinib Drugs 0.000 description 1
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229960004918 nimorazole Drugs 0.000 description 1
- MDJFHRLTPRPZLY-UHFFFAOYSA-N nimorazole Chemical compound [O-][N+](=O)C1=CN=CN1CCN1CCOCC1 MDJFHRLTPRPZLY-UHFFFAOYSA-N 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- 229960004378 nintedanib Drugs 0.000 description 1
- XZXHXSATPCNXJR-ZIADKAODSA-N nintedanib Chemical compound O=C1NC2=CC(C(=O)OC)=CC=C2\C1=C(C=1C=CC=CC=1)\NC(C=C1)=CC=C1N(C)C(=O)CN1CCN(C)CC1 XZXHXSATPCNXJR-ZIADKAODSA-N 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 229960003347 obinutuzumab Drugs 0.000 description 1
- 229950009090 ocaratuzumab Drugs 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 229960002700 octreotide Drugs 0.000 description 1
- 229960002450 ofatumumab Drugs 0.000 description 1
- 229950009057 oportuzumab monatox Drugs 0.000 description 1
- 229950006354 orantinib Drugs 0.000 description 1
- 229950007283 oregovomab Drugs 0.000 description 1
- 229950004023 orteronel Drugs 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 150000004866 oxadiazoles Chemical class 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 1
- 238000007248 oxidative elimination reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229950007318 ozogamicin Drugs 0.000 description 1
- 125000003232 p-nitrobenzoyl group Chemical group [N+](=O)([O-])C1=CC=C(C(=O)*)C=C1 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 1
- 229960003978 pamidronic acid Drugs 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 229960005184 panobinostat Drugs 0.000 description 1
- FPOHNWQLNRZRFC-ZHACJKMWSA-N panobinostat Chemical compound CC=1NC2=CC=CC=C2C=1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FPOHNWQLNRZRFC-ZHACJKMWSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229960000865 paramethasone acetate Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 1
- 229960001744 pegaspargase Drugs 0.000 description 1
- 108010001564 pegaspargase Proteins 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 229950010307 peretinoin Drugs 0.000 description 1
- SZFPYBIJACMNJV-UHFFFAOYSA-N perifosine Chemical compound CCCCCCCCCCCCCCCCCCOP([O-])(=O)OC1CC[N+](C)(C)CC1 SZFPYBIJACMNJV-UHFFFAOYSA-N 0.000 description 1
- 229950010632 perifosine Drugs 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 239000003504 photosensitizing agent Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 229950005566 picoplatin Drugs 0.000 description 1
- IIMIOEBMYPRQGU-UHFFFAOYSA-L picoplatin Chemical compound N.[Cl-].[Cl-].[Pt+2].CC1=CC=CC=N1 IIMIOEBMYPRQGU-UHFFFAOYSA-L 0.000 description 1
- 229950002592 pimasertib Drugs 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229960004403 pixantrone Drugs 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- 239000011505 plaster Substances 0.000 description 1
- 150000003058 platinum compounds Chemical class 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229950008499 plitidepsin Drugs 0.000 description 1
- UUSZLLQJYRSZIS-LXNNNBEUSA-N plitidepsin Chemical compound CN([C@H](CC(C)C)C(=O)N[C@@H]1C(=O)N[C@@H]([C@H](CC(=O)O[C@H](C(=O)[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(OC)=CC=2)C(=O)O[C@@H]1C)C(C)C)O)[C@@H](C)CC)C(=O)[C@@H]1CCCN1C(=O)C(C)=O UUSZLLQJYRSZIS-LXNNNBEUSA-N 0.000 description 1
- 108010049948 plitidepsin Proteins 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 1
- 229960000688 pomalidomide Drugs 0.000 description 1
- PHXJVRSECIGDHY-UHFFFAOYSA-N ponatinib Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2N3N=CC=CC3=NC=2)=C1 PHXJVRSECIGDHY-UHFFFAOYSA-N 0.000 description 1
- 229960001131 ponatinib Drugs 0.000 description 1
- 229960004293 porfimer sodium Drugs 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- OGSBUKJUDHAQEA-WMCAAGNKSA-N pralatrexate Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CC(CC#C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OGSBUKJUDHAQEA-WMCAAGNKSA-N 0.000 description 1
- 229960000214 pralatrexate Drugs 0.000 description 1
- 229960001297 prednazoline Drugs 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 229960002800 prednisolone acetate Drugs 0.000 description 1
- JDOZJEUDSLGTLU-VWUMJDOOSA-N prednisolone phosphate Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)COP(O)(O)=O)[C@@H]4[C@@H]3CCC2=C1 JDOZJEUDSLGTLU-VWUMJDOOSA-N 0.000 description 1
- 229960002943 prednisolone sodium phosphate Drugs 0.000 description 1
- 229950004954 prednisolone sulfobenzoate Drugs 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960001917 prednylidene Drugs 0.000 description 1
- WSVOMANDJDYYEY-CWNVBEKCSA-N prednylidene Chemical group O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](C(=C)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 WSVOMANDJDYYEY-CWNVBEKCSA-N 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 238000000039 preparative column chromatography Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- XYWJNTOURDMTPI-UHFFFAOYSA-N procodazole Chemical compound C1=CC=C2NC(CCC(=O)O)=NC2=C1 XYWJNTOURDMTPI-UHFFFAOYSA-N 0.000 description 1
- 229950000989 procodazole Drugs 0.000 description 1
- 201000008752 progressive muscular atrophy Diseases 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011241 protective layer Substances 0.000 description 1
- 230000004844 protein turnover Effects 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 1
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 description 1
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- DUPWHXBITIZIKZ-UHFFFAOYSA-N radotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3N=CC=NC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 DUPWHXBITIZIKZ-UHFFFAOYSA-N 0.000 description 1
- 229950004043 radotinib Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 238000009790 rate-determining step (RDS) Methods 0.000 description 1
- BMKDZUISNHGIBY-UHFFFAOYSA-N razoxane Chemical compound C1C(=O)NC(=O)CN1C(C)CN1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-UHFFFAOYSA-N 0.000 description 1
- 229960000460 razoxane Drugs 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 229960004836 regorafenib Drugs 0.000 description 1
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- OIRUWDYJGMHDHJ-AFXVCOSJSA-N retaspimycin hydrochloride Chemical compound Cl.N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(O)C1=CC(O)=C2NCC=C OIRUWDYJGMHDHJ-AFXVCOSJSA-N 0.000 description 1
- 229960001302 ridaforolimus Drugs 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 229950006764 rigosertib Drugs 0.000 description 1
- OWBFCJROIKNMGD-BQYQJAHWSA-N rigosertib Chemical compound COC1=CC(OC)=CC(OC)=C1\C=C\S(=O)(=O)CC1=CC=C(OC)C(NCC(O)=O)=C1 OWBFCJROIKNMGD-BQYQJAHWSA-N 0.000 description 1
- 229950003238 rilotumumab Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 1
- 229960003452 romidepsin Drugs 0.000 description 1
- 108010091666 romidepsin Proteins 0.000 description 1
- OHRURASPPZQGQM-UHFFFAOYSA-N romidepsin Natural products O1C(=O)C(C(C)C)NC(=O)C(=CC)NC(=O)C2CSSCCC=CC1CC(=O)NC(C(C)C)C(=O)N2 OHRURASPPZQGQM-UHFFFAOYSA-N 0.000 description 1
- 229960000215 ruxolitinib Drugs 0.000 description 1
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 1
- HNMATTJJEPZZMM-BPKVFSPJSA-N s-[(2r,3s,4s,6s)-6-[[(2r,3s,4s,5r,6r)-5-[(2s,4s,5s)-5-[acetyl(ethyl)amino]-4-methoxyoxan-2-yl]oxy-6-[[(2s,5z,9r,13e)-13-[2-[[4-[(2e)-2-[1-[4-(4-amino-4-oxobutoxy)phenyl]ethylidene]hydrazinyl]-2-methyl-4-oxobutan-2-yl]disulfanyl]ethylidene]-9-hydroxy-12-(m Chemical compound C1[C@H](OC)[C@@H](N(CC)C(C)=O)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@@](C/3=C/CSSC(C)(C)CC(=O)N\N=C(/C)C=3C=CC(OCCCC(N)=O)=CC=3)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HNMATTJJEPZZMM-BPKVFSPJSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- MOODSJOROWROTO-UHFFFAOYSA-N salicylsulfuric acid Chemical compound OC(=O)C1=CC=CC=C1OS(O)(=O)=O MOODSJOROWROTO-UHFFFAOYSA-N 0.000 description 1
- 229950006896 sapacitabine Drugs 0.000 description 1
- LBGFKUUHOPIEMA-PEARBKPGSA-N sapacitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](C#N)[C@H](O)[C@@H](CO)O1 LBGFKUUHOPIEMA-PEARBKPGSA-N 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000002412 selectin antagonist Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229960003323 siltuximab Drugs 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229950001403 sizofiran Drugs 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229950010372 sobuzoxane Drugs 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- SARBMGXGWXCXFW-GJHVZSAVSA-M sodium;2-[[(2s)-2-[[(4r)-4-[[(2s)-2-[[(2r)-2-[(3r,4r,5s,6r)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxypropanoyl]amino]propanoyl]amino]-5-amino-5-oxopentanoyl]amino]propanoyl]amino]ethyl [(2r)-2,3-di(hexadecanoyloxy)propyl] phosphate;hydrate Chemical compound O.[Na+].CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP([O-])(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(C)=O SARBMGXGWXCXFW-GJHVZSAVSA-M 0.000 description 1
- RWFZSORKWFPGNE-VDYYWZOJSA-M sodium;3-[2-[(8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-17-yl]-2-oxoethoxy]carbonylbenzenesulfonate Chemical compound [Na+].O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)C1=CC=CC(S([O-])(=O)=O)=C1 RWFZSORKWFPGNE-VDYYWZOJSA-M 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 208000002320 spinal muscular atrophy Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 229940071103 sulfosalicylate Drugs 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 239000005061 synthetic rubber Substances 0.000 description 1
- 229950010924 talaporfin Drugs 0.000 description 1
- MUTNCGKQJGXKEM-UHFFFAOYSA-N tamibarotene Chemical compound C=1C=C2C(C)(C)CCC(C)(C)C2=CC=1NC(=O)C1=CC=C(C(O)=O)C=C1 MUTNCGKQJGXKEM-UHFFFAOYSA-N 0.000 description 1
- 229950010130 tamibarotene Drugs 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229960003102 tasonermin Drugs 0.000 description 1
- 229950001899 tasquinimod Drugs 0.000 description 1
- ONDYALNGTUAJDX-UHFFFAOYSA-N tasquinimod Chemical compound OC=1C=2C(OC)=CC=CC=2N(C)C(=O)C=1C(=O)N(C)C1=CC=C(C(F)(F)F)C=C1 ONDYALNGTUAJDX-UHFFFAOYSA-N 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229950001699 teceleukin Drugs 0.000 description 1
- 229950002246 telotristat Drugs 0.000 description 1
- 229960002197 temoporfin Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 229960000331 teriflunomide Drugs 0.000 description 1
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 description 1
- NBRKLOOSMBRFMH-UHFFFAOYSA-N tert-butyl chloride Chemical compound CC(C)(C)Cl NBRKLOOSMBRFMH-UHFFFAOYSA-N 0.000 description 1
- MODVSQKJJIBWPZ-VLLPJHQWSA-N tesetaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3CC[C@@]2(C)[C@H]2[C@@H](C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C(=CC=CN=4)F)C[C@]1(O)C3(C)C)O[C@H](O2)CN(C)C)C(=O)C1=CC=CC=C1 MODVSQKJJIBWPZ-VLLPJHQWSA-N 0.000 description 1
- 229950009016 tesetaxel Drugs 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- FYOWZTWVYZOZSI-UHFFFAOYSA-N thiourea dioxide Chemical compound NC(=N)S(O)=O FYOWZTWVYZOZSI-UHFFFAOYSA-N 0.000 description 1
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 1
- 229960004231 thymalfasin Drugs 0.000 description 1
- 229960000902 thyrotropin alfa Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- 229950002376 tirapazamine Drugs 0.000 description 1
- ORYDPOVDJJZGHQ-UHFFFAOYSA-N tirapazamine Chemical compound C1=CC=CC2=[N+]([O-])C(N)=N[N+]([O-])=C21 ORYDPOVDJJZGHQ-UHFFFAOYSA-N 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 229950005976 tivantinib Drugs 0.000 description 1
- 229960000940 tivozanib Drugs 0.000 description 1
- BISFDZNIUZIKJD-XDANTLIUSA-N tixocortol pivalate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)CSC(=O)C(C)(C)C)(O)[C@@]1(C)C[C@@H]2O BISFDZNIUZIKJD-XDANTLIUSA-N 0.000 description 1
- 229960003114 tixocortol pivalate Drugs 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229940100611 topical cream Drugs 0.000 description 1
- 229940100615 topical ointment Drugs 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- 229950005801 tosedostat Drugs 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 1
- 229960000977 trabectedin Drugs 0.000 description 1
- FNCMIJWGZNHSBF-UHFFFAOYSA-N trabedersen Chemical compound CC1=CN(C2CC(O)C(COP(=O)(S)OC3CC(OC3COP(=O)(S)OC4CC(OC4COP(=O)(S)OC5CC(OC5COP(=O)(S)OC6CC(OC6COP(=O)(S)OC7CC(OC7COP(=O)(S)OC8CC(OC8COP(=O)(S)OC9CC(OC9COP(=O)(S)OC%10CC(OC%10COP(=O)(S)OC%11CC(OC%11COP(=O)(S)OC%12CC(OC%12COP(=O)(S)OC%13CC(OC%13COP(=O)(S)OC%14CC(OC%14COP(=O)(S)OC%15CC(OC%15CO)N%16C=CC(=NC%16=O)N)n%17cnc%18C(=O)NC(=Nc%17%18)N)n%19cnc%20C(=O)NC(=Nc%19%20)N)N%21C=CC(=NC%21=O)N)n%22cnc%23c(N)ncnc%22%23)N%24C=C(C)C(=O)NC%24=O)n%25cnc%26C(=O)NC(=Nc%25%26)N)N%27C=C(C)C(=O)NC%27=O)N%28C=CC(=NC%28=O)N)N%29C=C(C)C(=O)NC%29=O)n%30cnc%31c(N)ncnc%30%31)N%32C=C(C)C(=O)NC%32=O)N%33C=C(C)C(=O)NC%33=O)O2)C(=O)NC1=O.CC%34=CN(C%35CC(OP(=O)(S)OCC%36OC(CC%36OP(=O)(S)OCC%37OC(CC%37OP(=O)(S)OCC%38OC(CC%38O)n%39cnc%40c(N)ncnc%39%40)N%41C=C(C)C(=O)NC%41=O)n%42cnc%43C(=O)NC(=Nc%42%43)N)C(COP(=O)S)O%35)C(=O)NC%34=O FNCMIJWGZNHSBF-UHFFFAOYSA-N 0.000 description 1
- 229950002824 trabedersen Drugs 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229960004066 trametinib Drugs 0.000 description 1
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M trans-cinnamate Chemical compound [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229960001612 trastuzumab emtansine Drugs 0.000 description 1
- 108010075758 trebananib Proteins 0.000 description 1
- 229950001210 trebananib Drugs 0.000 description 1
- 229960003181 treosulfan Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- GUYPYYARYIIWJZ-CYEPYHPTSA-N triamcinolone benetonide Chemical compound O=C([C@]12[C@H](OC(C)(C)O1)C[C@@H]1[C@@]2(C[C@H](O)[C@]2(F)[C@@]3(C)C=CC(=O)C=C3CC[C@H]21)C)COC(=O)C(C)CNC(=O)C1=CC=CC=C1 GUYPYYARYIIWJZ-CYEPYHPTSA-N 0.000 description 1
- 229950006782 triamcinolone benetonide Drugs 0.000 description 1
- 229960004320 triamcinolone diacetate Drugs 0.000 description 1
- 229960004221 triamcinolone hexacetonide Drugs 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- UBOXGVDOUJQMTN-UHFFFAOYSA-N trichloroethylene Natural products ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 1
- WTVXIBRMWGUIMI-UHFFFAOYSA-N trifluoro($l^{1}-oxidanylsulfonyl)methane Chemical group [O]S(=O)(=O)C(F)(F)F WTVXIBRMWGUIMI-UHFFFAOYSA-N 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- VXKHXGOKWPXYNA-PGBVPBMZSA-N triptorelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 VXKHXGOKWPXYNA-PGBVPBMZSA-N 0.000 description 1
- 229960004824 triptorelin Drugs 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 238000007738 vacuum evaporation Methods 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 229950010938 valspodar Drugs 0.000 description 1
- 108010082372 valspodar Proteins 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 1
- 230000004218 vascular function Effects 0.000 description 1
- 229940099259 vaseline Drugs 0.000 description 1
- 229960003862 vemurafenib Drugs 0.000 description 1
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229960004449 vismodegib Drugs 0.000 description 1
- BPQMGSKTAYIVFO-UHFFFAOYSA-N vismodegib Chemical compound ClC1=CC(S(=O)(=O)C)=CC=C1C(=O)NC1=CC=C(Cl)C(C=2N=CC=CC=2)=C1 BPQMGSKTAYIVFO-UHFFFAOYSA-N 0.000 description 1
- XZAFZXJXZHRNAQ-STQMWFEESA-N vosaroxin Chemical compound C1[C@H](OC)[C@@H](NC)CN1C1=CC=C2C(=O)C(C(O)=O)=CN(C=3SC=CN=3)C2=N1 XZAFZXJXZHRNAQ-STQMWFEESA-N 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229950008250 zalutumumab Drugs 0.000 description 1
- 229950009002 zanolimumab Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
- 229960004276 zoledronic acid Drugs 0.000 description 1
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0031—Rectum, anus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0048—Eye, e.g. artificial tears
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
- A61K9/2826—Sugars or sugar alcohols, e.g. sucrose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
- A61K47/183—Amino acids, e.g. glycine, EDTA or aspartame
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/24—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing atoms other than carbon, hydrogen, oxygen, halogen, nitrogen or sulfur, e.g. cyclomethicone or phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Dermatology (AREA)
- Inorganic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
La presente invención se refiere a derivados del ácido a-amino borónico. Estos compuestos son útiles para inhibir la actividad del inmunoproteasoma (LMP7) y para el tratamiento y/o prevención de condiciones médicas afectadas por la actividad del inmunoproteasoma tales como enfermedades inflamatorias y autoinmunes, enfermedades neurodegenerativas, enfermedades proliferativas y cáncer. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados del ácido borónico
Campo de la invención
La presente invención se refiere a derivados del ácido a-amino borónico. Estos compuestos son útiles para inhibir la actividad del inmunoproteosoma (LMP7) y para el tratamiento y/o prevención de enfermedades que se ven afectadas por la actividad del inmunoproteosoma como enfermedades inflamatorias y autoinmunitarias, enferme dades neurodegenerativas, enfermedades proliferativas y el cáncer. En especial, los compuestos de la presente invención son inhibidores selectivos del inmunoproteosoma.
Antecedentes de la invención
El proteosoma (también conocido como macropaína, proteasa multicatalítica y proteasa 20S) es una proteasa multisubunidad de alto peso molecular que se ha identificado en todas las especies examinadas desde arqueobacterias a seres humanos. La enzima tiene un peso molecular nativo de aproximadamente 650 000 y, como se mostró mediante microscopía electrónica, una morfología característica en forma de cilindro (Rivett, [1989] Arch. Biochem. Biophys. 268:1-8; y Orlowski, (1990) Biochemistry 29:10289-10297). Las subunidades del proteosoma tienen un peso molecular que oscila de 20000 a 35 000 y son homólogas entre sí pero no con ninguna otra proteasa conocida.
El proteosoma 20S es un complejo proteasa multicatalítico con forma cilíndrica de 700 kDa compuesto de 28 subunidades, clasificadas como de tipo a y p, que se disponen en 4 anillos heptaméricos apilados. En levadu ras y otros eucariotas, los anillos externos están formados por 7 subunidades a diferentes y los anillos internos por 7 subunidades p diferentes. Las subunidades a sirven como sitios de unión para los complejos reguladores 19S (PA700) y 1 IS (PR68), así como una barrera física de la cámara proteolítica interna formada por los dos anillos de subunidades p. Por tanto, in vivo, se considera que el proteosoma aparece como una partícula 26S («el proteosoma 26S»). Experimentos in vivo han mostrado que la inhibición de la forma 20S del proteosoma puede correlacionarse fácilmente con la inhibición del proteosoma 26S.
La escisión de prosecuencias aminoterminales de subunidades p durante la formación de partículas deja expues tos restos de treonina aminoterminales, que sirven como los nucleófilos catalíticos. Las subunidades responsables de la actividad catalítica en el proteosoma poseen, por tanto, un resto nucleófilo amino terminal, y estas subuni dades pertenecen a la familia de hidrolasas nucleófilas N-terminales (Ntn) (en las que el resto nucleófilo N-terminal es, por ejemplo, Cys, Ser, Thr y otros restos nucleófilos). Esta familia incluye, por ejemplo, penicilina G acilasa (PGA), penicilina V acilasa (PVA), glutamina PRPP amidotransferasa (GAT) y glicosilasparaginasa bacteriana. Además de las subunidades p expresadas de forma ubicua, los vertebrados superiores también poseen tres subunidades p inducibles por interferón-Y (LMP7, LMP2 y MECLl), que sustituyen a sus homólogos normales, p5, p1 y p2, respectivamente. Cuando están presentes las tres subunidades inducibles por IFN-y, el proteosoma se denomina «inmunoproteosoma». Por tanto, las células eucariotas pueden poseer dos formas de proteosomas en diversas proporciones.
Mediante el uso de diferentes sustratos peptídicos, se han definido tres actividades proteolíticas principales del proteosoma 20S eucariota: la actividad de tipo quimiotripsina (CT-L), que escinde detrás de restos hidrófobos grandes; la actividad de tipo tripsina (T-L), que escinde detrás de restos básicos; y la actividad que hidroliza el péptido peptidilglutamilo (PGPH), que escinde detrás de restos ácidos. También se han atribuido al proteosoma dos actividades adicionales menos caracterizadas: la actividad BrAAP, que escinde detrás de aminoácidos de cadena ramificada; y la actividad SNAAP, que escinde detrás de aminoácidos neutros pequeños. Aunque ambas formas del proteosoma poseen las cinco actividades enzimáticas, se han descrito diferencias en el grado de acti vidad entre las formas en función de sustratos específicos. En ambas formas del proteosoma, las principales actividades proteolíticas del proteosoma parecen suponer la aportación de diferentes sitios catalíticos dentro del núcleo 20S.
En eucariotas, la degradación de proteínas está mediada predominantemente a través de la vía de la ubiquitina en la que las proteínas destinadas a la destrucción se ligan al polipéptido de 76 aminoácidos ubiquitina. Una vez dirigidas, las proteínas ubiquitinadas sirven a continuación como sustratos para el proteosoma 26S, que escinde las proteínas en péptidos cortos mediante la acción de sus tres principales actividades proteolíticas. Aunque tiene una función general en el recambio de proteínas intracelulares, la degradación mediada por proteosomas también tiene un papel importante en muchos procesos como la presentación en el complejo principal de histocompati bilidad (MHC) de clase I, la apoptosis y la viabilidad celular, el procesamiento de antígenos, la activación de NF-kB y la transducción de señales proinflamatorias.
La actividad del proteosoma es alta en las enfermedades de atrofia muscular progresiva que implican degradación de proteínas, como distrofia muscular, cáncer y SIDA. Las evidencias también sugieren una posible función del proteosoma en el procesamiento de antígenos para las moléculas MHC de clase I (Goldberg y col. (1992) Nature 357:375-379).
Los proteosomas están implicados en las enfermedades y trastornos neurodegenerativos como la esclerosis lateral amiotrófica (ELA) (J Biol Chem 2003, Allen S y cols., Exp Neurol 2005, Puttaparthi K y cols.), síndrome de Sjogren (Arthritis & Rheumatism, 2006, Egerer T y cols.), lupus eritematoso sistémico y nefritis lúpica (LES/NL), (Arthritis & rheuma 2011, Ichikawa y cols., J Immunol, 20 l0, Lang VR y cols., Nat Med, 2008, Neubert K y cols.), glomerulonefritis (J Am Soc Nephrol 2011, Bontscho y cols.), artritis reumatoide (Clin Exp Rheumatol, 2009, Van der Heiden JW y cols.), enfermedad inflamatoria intestinal (ElI), colitis ulcerosa, enfermedad de Crohn (Gut 2010, Schmidt N y cols., J Immunol 2010, Basler M y cols., Clin Exp Immunol, 2009, Inoue S y cols.), esclerosis múltiple (Eur J Immunol 2008, Fissolo N y cols., J Mol Med 2003, Elliott PJ y cols., J Neuroimmunol 2001, Hosseini y cols., J Autoimmun 2000, Vanderlugt Cl y cols.), esclerosis lateral amiotrófica (ELA), (Exp Neurol 2005, Puttaparthi k y cols., J Biol Chem 2003, Allen S y cols.), artrosis (Pain 2011, Ahmed S y cols., Biomed Mater Eng 2008, Etienne S y cols.), ateroesclerosis (J Cardiovasc Pharmacol 2010, Feng B y cols.), psoriasis (Genes & Immunity, 2007, Kramer U y cols.), miastenia grave (J Immunol, 2011, Gomez AM y cols.), fibrosis cutánea (Thorax 2011, Mutlu GM y cols., Inflammation 2011, Koca SS y cols., Faseb J 2006, Fineschi S y cols.), fibrosis renal (Nephrology 2011 Sakairi T y cols.), fibrosis cardíaca (Biochem Pharmacol 2011, Ma Y y cols.) fibrosis hepática (Am J Physiol Gastrointest Liver Physiol 2006, Anan A y cols.), fibrosis pulmonar (Faseb J 2006, Fineschi S y cols.), nefropatía por inmunoglobulina A (nefropatía por IgA), (Kidney Int, 2009, Coppo R y cols.), vasculitis (J Am Soc nephrol 2011, Bontscho y cols.), rechazo de trasplante (Nephrol Dial Transplant 2011, Waiser J y cols.), neoplasias malignas hematológicas (Br J Haematol 2011, Singh AV y cols., Curr Cancer Drug Target 2011, Chen D y cols.) y asma.
Aún más, debe apreciarse que los inhibidores de proteosomas disponibles en el mercado inhiben tanto la forma constitutiva como la inmunitaria del proteosoma. Incluso bortezomib, el inhibidor del proteosoma autorizado por la FDA para el tratamiento de pacientes con mieloma múltiple recidivante, no distingue entre las dos formas (Altun y cols., Cancer Res 65:7896, 2005). Adicionalmente, el uso de bortezomib se asocia con neuropatía periférica (NP) dolorosa surgida durante el tratamiento; esta neurodegeneración inducida por bortezomib in vitro se produce a través de un mecanismo independiente del proteosoma y bortezomib inhibe varias dianas no proteosómicas in vitro e in vivo (Clin. Cancer Res, 17[9], 1 de mayo de 2011).
Además de los inhibidores de proteosomas convencionales, una estrategia novedosa puede ser dirigirse especí ficamente al inmunoproteosoma específico hematológico, aumentando de este modo la eficacia general y redu ciendo los efectos negativos fuera de la diana. Se ha demostrado que el inhibidor específico del inmunoproteo soma podría mostrar una mejora de la eficacia en células de origen hematológico (Curr Cancer Drug Targets, 11 [3], marzo, 2011).
Por tanto, existe la necesidad de proporcionar nuevos inhibidores de proteosomas que sean selectivos para una forma específica del proteosoma. En especial, existe la necesidad de proporcionar inhibidores selectivos del inmunoproteosoma que puedan utilizarse como fármacos para el tratamiento de, por ejemplo, el LES u otros trastornos inmunitarios o autoinmunitarios en el contexto de la artritis reumatoide. Los inhibidores selectivos del inmunoproteosoma son útiles para minimizar los efectos secundarios no deseados mediados por la inhibición del proteosoma constitutivo u otras dianas no proteosómicas.
En el documento WO 2013/092979 A1 se describen derivados del ácido borónico que muestran selectividad por la inhibición de la actividad LMP7. No obstante, el grado de selectividad que se puede conseguir con los tipos de compuestos descritos es limitado, especialmente con respecto a la división de la actividad inhibidora del proteosoma constitutivo.
Se ha demostrado el valor clínico en la indicación de mieloma múltiple de los inhibidores inespecíficos del proteosoma y el inmunoproteosoma como bortezomib y carfilzomib. Aunque este perfil inespecífico, que afecta a los componentes principales del inmunoproteosoma, así como al proteosoma constitutivo, se considera beneficioso en términos de inhibición de la diana y efectividad clínica, este perfil inespecífico limita la aplicabilidad clínica de estos fármacos al inducir efectos secundarios pronunciados como trombocitopenia, neutropenia y neuropatía periférica. A un determinado grado, este perfil de efectos secundarios podría atribuirse a la amplia inhibición de la actividad catalítica, especialmente la inhibición combinada de las subunidades p5 del proteosoma constitutivo y el inmunoproteosoma. La estrategia para obtener inhibidores más selectivos del inmunoproteosoma (y específica mente de la subunidad p5i del inmunoproteosoma), para reducir los principales efectos secundarios fue descrita, por ejemplo, en 2011 por Singh y cols. (Br. J. Hematology 152(2): 155-163) para PR-924, un inhibidor 100 veces más selectivo de la subunidad l MP7 del inmunoproteosoma. Los autores demostraron la presencia de altos nive les de expresión del inmunoproteosoma en el mieloma múltiple. Los autores también describieron el efecto de un inhibidor selectivo de la subunidad LMP7 sobre la inducción de la muerte celular en líneas celulares de MM, así
como en células de pacientes de MM primario CD138+ sin disminución de la viabilidad de PBMC control de vo luntarios sanos que puede considerarse como prueba de concepto. Además del concepto de un perfil de reducción de efectos secundarios para los inhibidores selectivos de p5i, otro grupo demostró la eficacia de los inhibidores selectivos de p5i sobre la viabilidad de líneas celulares resistentes a bortezomib, lo que evidencia el valor y la posible perspectiva de la aplicación de inhibidores selectivos de LMP7 para las neoplasias malignas hematológicas (D. Niewerth y cols. Biochemical Pharmacology 89 [2014] 43-51).
En los documentos WO 2016/050356, WO 2016/050355, WO 2016/050359 y WO 2016/050358 se describen com puestos que inhiben la actividad del inmunoproteosoma (LMP7) y proporcionan una división significativa en la actividad inhibidora del proteosoma constitutivo.
Sorprendentemente se encontró que los derivados del ácido amino borónico de la presente invención proporcio nan una división especialmente alta en la actividad inhibidora del proteosoma constitutivo. Además, muestran buenos resultados en vista de la unión a proteínas plasmáticas, inhibición de CYP, perfil FC y biodisponibilidad oral.
Descripción de la invención
La presente invención proporciona compuestos de fórmula (I)

donde
LY indica (CH2)m, donde de 1 a 4 átomos de H pueden estar sustituidos por Hal, R3a y/u OR4a, y/o donde un grupo CH2 puede estar sustituido por O, S, SO y/o SO2;
X indica un heterobiciclo o heterotriciclo de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) o (xi) cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, NO2, CN, R5a, OR5a, CONR5aR5b, NR5aCOR5b, SO2R5a, SOR5a, SO2NR5aR5b, NR5aSO2R5b, NR5aR5b, (CH2)q-R6, COR5a y/o SO2R5a, y donde 1,2 o 3 de los grupos CH2 cíclicos pueden estar sustituidos por CR4aR4b, C=O, O, S, NR5a, SO y/o SO2;

Y indica P1, P2 o P3;
P1 indica un alquilo C1-C6 o cicloalquilo C3-C8 lineal o ramificado, independientemente entre sí, no sus tituido o mono-, di-, tri- o tetrasustituido por Hal, CN, R3a, OR3a, y/o (CH2)q-R6;
P2 indica fenilo o un heterociclo aromático monocíclico de 5, 6 o 7 átomos, cada uno no sustituido o mono, di, tri, tetra o pentasustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6, donde el sistema heterocíclico contiene 1, 2 o 3 átomos de N, O y/o S;
P3 indica un heterociclo o hidrocarburo bicíclico de 8, 9 o 10 átomos, cada uno independientemente entre sí no sustituido o mono, di, tri, tetra o pentasustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6, donde al menos un anillo del heterociclo o hidrocarburo bicíclico es aromático y donde el sistema heterocíclico contiene 1,2 o 3 átomos de N, O y/o S;
Cy1, Cy2, Cy3, Cy4 y Cy5 indican cada uno, independientemente entre sí, Ar1 o Het1;
R1, R2 indican cada uno, independientemente entre sí, H o alquilo C1-C6, o R1 y R2 forman juntos un resto según la fórmula (CE)
R3a, R3b indican cada uno, independientemente entre sí, un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8, donde de 1 a 5 átomos de H pueden estar sustituidos por Hal, CN, OH y/u OAlq; R4a, R4b indican cada uno, independientemente entre sí, H o R3a;
o
R4a y R4b forman juntos un grupo alquileno C3-C8;
R5a, R5b indican cada uno, independientemente entre sí, H, R3a, Ar2 o Het2;
R6 indica OH u OR3a;
T1, T2, T3, T4, T5, T6, T7, T8 y T9 indican cada uno, independientemente entre sí, O, SO, C=O;
Alq indica alquilo C1-C6 lineal o ramificado;
Ar1 representa un carbociclo aromático de 6 átomos;
Het1 representa un heterociclo saturado, insaturado o aromático de 5 o 6 átomos con 1 a 4 átomos de N, O y/o S;
Ar2 indica fenilo, que está no sustituido o mono o disustituido por Hal, NO2, CN, R3a, OR3a, CONHR3a,
NR3aCOR3b, SO2R3a, SOR3a, NH2, NHR3a, N(R3a)2 y/o (CH2)q-R6;
Het2 indica un heterociclo de 5 o 6 átomos saturado, insaturado o aromático con 1 a 4 átomos de N, O y/o S, que no está sustituido o está mono o disustituido por Hal, NO2, CN, R3a, OH, OR3a, CONHR3a, NR3aCOR3b, SO2R3a, SOR3a, NH2, NHR3a, N(R3a)2, (CH2)q-R6 y/u oxo (=O);
q indica 1, 2, 3, 4, 5 o 6;
m indica 0, 1 o 2;
Hal indica F, Cl, Br o I;
y solvatos, tautómeros, oligómeros y estereoisómeros de los mismos, así como las sales farmacéuticamente acep tables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
Los compuestos de la presente invención son inhibidores de la subunidad LMP7 del inmunoproteosoma. Estos muestran una selectividad especialmente alta sobre LMP7 con respecto a Beta5 (cP) y buenas propiedades en términos de solubilidad, unión a proteínas plasmáticas, inhibición del CYP, perfil farmacocinético y biodisponibilidad oral.
Es sabido que los derivados del ácido borónico como los compuestos de fórmula (I), donde R1 y R2 indican H, forman oligómeros (Boronic Acids. Editado por Dennis G. Hall, Copyright © 2005 WÍLEY-VCH Verlag, GmbH & Co. KGaA, Weinheim, ISBN 3-527-30991-8). Estos oligómeros (en especial, pero sin limitaciones, dímeros o trí meros) de compuestos de fórmula (I) se incluyen dentro de esta invención. Los trímeros cíclicos de ácidos borónicos conocidos tienen, por ejemplo, la siguiente estructura:

También se sabe que los derivados del ácido borónico, tales como los compuestos de fórmula (I), donde R1 y R2 indican H, forman aductos mediante reacción con alcoholes alifáticos o aromáticos, dioles, azúcares, alcoholes de azúcar, ácidos a-hidroxi o nucleófilos que contienen uno, dos o tres grupos funcionales que contienen N-/O-(p. ej., -NH2, -CONH2 o C=NH, -OH, -COOH) donde, en caso de que tres grupos funcionales estén presentes, uno de los tres heteroátomos podría formas un enlace de coordinación («Boronic Acids», editado por Dennis G. Hall, 2.a edición, Copyright © 2011 WILEY-VCH Verlag, GmbH & Co. KGaA, Weinheim, ISBN 978-3-527-32598-6; do cumentos WO2013128419 y WO2009154737). La formación de aductos es especialmente rápida con dioles pre organizados. La presente invención incluye dichos aductos (en particular, ésteres o derivados heterocíclicos) de compuestos de ácido borónico de fórmula (I).
Es de destacar que los compuestos de la presente invención contienen un centro estereogénico en el átomo de carbono adyacente al resto de ácido borónico; este se ha indicado con un asterisco (*) en la fórmula (I)* a conti nuación:
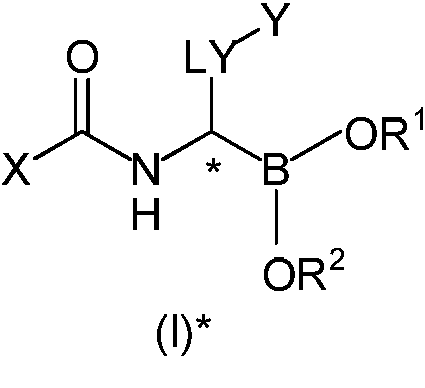
Los compuestos según la fórmula (I) muestran, por tanto, dos configuraciones diferentes en este centro estereo génico, es decir, la configuración (R) y la configuración (S). Por tanto, los compuestos de la presente invención pueden estar presentes enantiopuros o como una mezcla racémica (1:1) de los dos enantiómeros de fórmulas (R)-(I) y (S)-(I).

R-(I) S-(I)
Los compuestos de fórmula (I) también pueden estar presentes en una mezcla en la que uno de los enantiómeros (R)-(I) o (S)-(I) esté presente en exceso con respecto al otro, por ejemplo, 60:40, 70:30, 80:20, 90:10, 95:5 o similares. En una realización en particular de la presente invención, el estereoisómero de fórmula (R)-(I) del com puesto de fórmula (I) y el estereoisómero de fórmula (S)-(I) del compuesto de fórmula (Ia) están presentes en una relación de (R)-(I) con respecto a (S)-(I) de al menos 90 partes de (R)-(I) por no más de 10 partes de (S)-(I), preferiblemente de al menos 95 de (R)-(I) por no más de 5 de (S)-(I), más preferiblemente de al menos 99 de (R)-(I) por no más de 1 de (S)-(I), incluso más preferiblemente de al menos 99,5 de (R)-(I) por no más de 0,5 de (S)-(I). En otra realización en particular de la presente invención, el estereoisómero de fórmula (S)-(I) del compuesto de fórmula (Ia) y el estereoisómero de fórmula (R)-(I) del compuesto de fórmula (Ia) están presentes en una rela ción de (S)-(I) con respecto a (R)-(I) de al menos 90 partes de (S)-(I) por no más de 10 de (R)-(I), preferiblemente
de al menos 95 de (S)-(I) por no más de 5 (R)-(I), más preferiblemente de al menos 99 de (S)-(I) por no más de 1 de (R)-(I), incluso más preferiblemente de al menos 99,5 de (S)-(I) por no más de 0,5 de (R)-(l).
Pueden obtenerse estereoisómeros enriquecidos o puros de fórmulas (R)-(I) y (S)-(I) mediante métodos habituales conocidos en la técnica y métodos específicos descritos a continuación en este documento. Un método en parti cular para su obtención es la cromatografía preparativa en columna, como HPLC o SFC, usando material quiral en columna.
Realizaciones preferidas en particular de la presente invención comprenden compuestos de fórmula (R)-(I), donde el centro estereogénico en el átomo de carbono adyacente al resto de ácido borónico muestra una configuración (R):

Los compuestos según la fórmula (I) también podrían contener además de centros estereogénicos localizados en átomos de carbono distintos al átomo de carbono adyacente al resto de ácido borónico. Todos estos centros estereogénicos pueden presentarse en configuración (R) o (S).
En particular, los compuestos de la presente invención contienen, además, centros estereogénicos en el átomo de carbono del sustituyente X (que está unido directamente al átomo de carbono del grupo amida CONH que se muestra en la fórmula (I)) y en los átomos de carbono adyacentes al átomo puente; estos centros estereogénicos se muestran, por ejemplo, a continuación en la fórmula (xa*), indicados con un asterisco (*):

(xa*):
(sustituyentes opcionales de (xa*) no mostrados)
A continuación se muestran los posibles estereoisómeros de (xa*):

Por tanto, los compuestos según la fórmula (I) muestran también dos configuraciones diferentes en estos centros estereogénicos, es decir, la configuración (R) y la configuración (S). Los compuestos de la presente invención pueden tener una configuración (R) o una configuración (S) en cada uno de estos centros estereogénicos o estar presentes los compuestos en una mezcla racémica (1:1) de dos estereoisómeros. Los compuestos de fórmula (I) también pueden estar presentes en una mezcla en la que uno de los estereoisómeros esté presente en exceso con respecto al otro, por ejemplo, 60:40, 70:30, 80:20, 90:10, 95:5 o similares.
Anteriormente y a continuación, en esos casos, donde se muestra una estructura química con un centro estereo génico y no se indica una estereoquímica específica, la estructura incluye todos los posibles estereoisómeros, así como sus mezclas. Por ejemplo, la presente invención incluye los estereoisómeros ácido [(1 R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]-heptan-2-il]-formamido}etil]borónico, ácido [(1r )-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido
[(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1s)-2-[(3S)-2,3-dihidro-1-benzofuran-3-N]-1-{[(1S,2s,4R)-7-oxabiddo[2.2.1]heptan-2-N]formamido}etN]-borónico, ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]-borónico, ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo-[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]-borónico, ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]-heptan-2-il]formamido}etil]borónico, ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(lR,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]-borónico, ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1 R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico, ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]-borónico, así como ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]-borónico. Los diferentes estereoisómeros de un compuesto determinado son útiles para la caracteriza ción analítica de una muestra específica (p. ej., para fines de control de calidad) mediante RMN, HPLC, SFC o cualquier otro método analítico adecuado. De este modo, otro aspecto de la presente invención se refiere al uso de estereoisómeros de compuestos según la fórmula (I) en métodos analíticos de caracterización.
En general, todos los restos de los compuestos descritos en este documento que aparecen más de una vez pueden ser idénticos o diferentes, es decir, son independientes entre sí. Anteriormente y a continuación, los restos y parámetros tienen el significado indicado para la fórmula (I), siempre que no se indique expresamente otra cosa. Por consiguiente, la invención se refiere, en particular, a los compuestos de fórmula (I) en los que al menos uno de dichos restos tiene uno de los significados preferidos indicados a continuación. Adicionalmente, todas las rea lizaciones específicas descritas a continuación deberían incluir derivados, solvatos, tautómeros o estereoisómeros de las mismas, así como las sales fisiológicamente aceptables de cada uno de los anteriores, incluidas sus mez clas en todas las proporciones.
Los heterobiciclos o heterotriciclos de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) y (xi) pueden no estar sustituido o estar mono, di o trisustituidos por Hal, NO2, C n , R5a, OR5a, CONR5aR5b, NR5aCOR5b, SO2R5a, SOR5a, SO2NR5aR5b, NR5aSO2R5b, NR5aR5b, (CH2)q-R6, COR5a y/o SO2R5a. En caso de que los heterobiciclos o heterotriciclos de fórmula (xe), (xf), (xg), (xh) y (xi) no estén sustituidos, el uno o más sustituyentes pueden estar unidos a uno de los anillos en puente o a uno de los anillos fusionados Cy1, Cy2, Cy3, Cy4 y Cy5. Esto incluye, por ejemplo, compuestos en los que un sustituyente está unido al anillo en puente o un sustituyente está unido al anillo fusio nado Cy1, Cy2, Cy3, Cy4 o Cy5. En caso de que uno de los anillos fusionados Cy1, Cy2, Cy3, Cy4 y Cy5 contengan uno o más grupos CH2, se entiende que estos grupos forman parte de los «grupos CH2 cíclicos» de heterobiciclos o heterotriciclos de fórmula (xe), (xf), (xg), (xh) y (xi), que pueden sustituirse por CR4aR4b, C=O, O, S, NR5a, SO o SO2. De este modo, si 1, 2 o 3 de los grupos CH2 cíclicos de heterobiciclos o heterotriciclos de fórmula (xe), (xf), (xg), (xh) y (xi) están sustituidos por CR4aR4b, C=O, O, S, NR5a, SO o SO2, estos CH2 cíclicos pueden formar parte del anillo con puente y/o los anillos fusionados Cy1, Cy2, Cy3, Cy4 y Cy5. Esto incluye, por ejemplo, compuestos en los que está sustituido un grupo CH2 del anillo en puente y un CH2 del anillo fusionado Cy1, Cy2, Cy3, Cy4 o Cy5.
En caso de que Y sea P3, donde al menos uno de los dos anillos del heterociclo o hidrocarburo bicíclico es un anillo aromático, el otro anillo puede ser un anillo saturado, insaturado o aromático. En ejemplos específicos de estas realizaciones, P3 y el grupo LY adyacente están unidos entre sí por medio del anillo aromático de P3 En otras realizaciones, P3 y el grupo LY adyacente están unidos entre sí por medio del anillo saturado o insaturado de P3 En caso de que P3 sea un heterociclo bicíclico, contiene preferiblemente 1 o 2 heteroátomos seleccionado entre N, O y/o S. En caso de que P2 sea un heterociclo monocíclico aromático, contiene preferiblemente 1 o 2 heteroátomos seleccionados entre N, O y/o S.
En el caso de que Y sea P2 y P2 sea fenilo, preferiblemente no está sustituido o está mono, di o trisustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6. Son especialmente preferidas las realizaciones en las que P2 indica un fenilo di o trisustituido. En aquellas realizaciones en las que P2 indica un fenilo monosustituido, el sustituyente está preferiblemente en la posición 3 o 4. En aquellas realizaciones en las que P2 indica un fenilo disustituido, los dos sustituyentes están preferiblemente en posición 2,3-, 2,4-, 2,5- o 3,4- (más preferiblemente en posición 2,4- o 3,4-). Y en aquellas realizaciones en las que P2 indica un fenilo trisustituido, los tres sustituyentes están preferiblemente en posición 2,3,4- del anillo aromático.
En caso de que P2 indique un heterociclo monocíclico, este heterociclo puede estar saturado, insaturado o ser aromático.
En realizaciones en las que m indica 0, LY está ausente.
En el contexto de la presente invención «alquilo C1-C6» significa un resto alquilo con 1, 2, 3, 4, 5 o 6 átomos de carbono y de cadena recta o ramificada. El término «cicloalquilo C3-C6» se refiere a grupos de hidrocarburo cíclico saturado con 3, 4, 5 o 6 átomos de carbono.
El término «no sustituido» significa que el radical, grupo o resto correspondiente no tiene sustituyentes distintos de H; el término «sustituido», que se aplica a uno o más hidrógenos que son implícitos o explícitos de la estructura, significa que el radical, grupo o resto correspondiente tiene uno o más sustituyentes distintos de H. Cuando un radical tiene varios sustituyentes, es decir, al menos dos, y se especifica una selección de diversos sustituyentes, los sustituyentes se seleccionan independientemente entre sí y no es necesario que sean idénticos.
El término «carbociclo» significa un sistema de anillo, en el que todos los átomos del anillo son átomos de carbono. El término «heterociclo» significa un sistema de anillos, en el que alguno de los átomos del anillo son heteroátomos como N, O o S.
El grupo «NRR'» es un grupo amino, donde R y R' son, por ejemplo, cada uno independientemente entre sí H o alquilo C1-C6 lineal o ramificado (especialmente metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o tercbutilo, pentilo, hexilo).
El grupo «SO», como por ejemplo cuando se incluye en el grupo SOR5a, es un grupo donde S y O están unidos a través de un enlace doble (S=O).
El grupo «CO», como por ejemplo cuando se incluye en el grupo COR4a, es un grupo donde C y O están unidos a través de un enlace doble (C=O).
El término «alquileno» se refiere a un grupo alquilo divalente. Un «grupo alquileno» es un grupo (poli)metileno (-(CH2)x-).
El grupo oxo (=O) es un sustituyente que puede darse, por ejemplo, en restos cíclicos saturados o, en la medida de lo posible, en un anillo (parcialmente) insaturado, como en particular Het1 y Het2 En realizaciones preferidas, los heterociclos Het1 y Het2 opcionalmente portan uno o dos grupos oxo.
Según se usa en este documento, el término «insaturado» significa que un resto tiene una o más unidades de insaturación. Según se usa en este documento en referencia a cualquier anillo, los sistemas cíclicos, restos cícli cos y similares, el término «parcialmente insaturado» se refiere a un resto cíclico que incluye al menos un enlace doble o triple. El término «parcialmente insaturado» pretende abarcar restos cíclicos que tienen más de un enlace doble o triple.
En el contexto de la presente invención, notaciones como «O-CH3» y «OCH3» o «CH2CH2» y «-CH2-CH2-» tienen el mismo significado y se usan indistintamente.
Según se usa en este documento, en las fórmulas estructurales, las flechas o enlaces con líneas discontinuas verticales se usan para indicar el punto de unión a un grupo adyacente. Por ejemplo, la flecha en (xa) muestra el punto de unión al grupo C=O adyacente.
Realizaciones importantes especiales de la presente invención incluyen compuestos de fórmula (I) donde R1, R2 indican cada uno, independientemente entre sí, H o alquilo C1-C4, o R1 y R2 forman juntos un resto según la fórmula (CE); y
LY indica CH2 o CH2CH2, donde 1 o 2 átomos de H pueden estar sustituidos por Hal, R3a, OR4a (pre feriblemente, 1 o 2 átomos de H pueden estar sustituidos por F, Cl, CH3, CH2CF3, CH2CHF2, CH2F, CHF2, CF3, OCH3 y/u OCF3, y más preferiblemente, LY indica CH2 o CH2CH2).
Realizaciones específicas incluyen compuestos de fórmula (I), donde T1, T2, T3, T4, T5, T6, T7, T8 y T9 indican O. Otras realizaciones específicas comprenden compuestos según la fórmula (I), donde
P1 indica un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8, independientemente entre sí, no sustituido o mono-, di- o trisustituido por Hal, CN, R3a, OR3a y/o (C^)q-R6;
P2 indica fenilo, piridilo, pirrolilo, furanilo, tiofenilo, pirimidilo, piranzinilo o piridazinilo, cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, CN, R3a, OH, OR3a,
CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; y
P3 indica un resto bicíclico de fórmula (ya), (yb), (yc), (yd), (ye), (yf), (yg), (yh), (yi), (yj), (yk), (yl), (ym),
(yn), (yp) o (yp), cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6:

(sustituyentes opcionales de (ya)-(yp) no mostrados)
donde
Ea indica O, S, N(Alq) o CH=CH;
Eb indica O, S, N(Alq), CH2, CH2CH2, OCH2, SCH2 o N(Alq)CH2.
En realizaciones adicionales de la invención, los restos de un compuesto de fórmula (I) se definen como sigue: R3a, R3b indican cada uno, independientemente entre sí, un alquilo C1-C4 lineal o ramificado o cicloalquilo C3-C6, donde de 1 a 3 átomos de H pueden estar sustituidos por F, Cl y/o donde 1 o 2 átomos de H pueden estar sustituidos por CN, OH, OCH3 y/o OC2H5.
Realizaciones adicionales de la invención comprenden compuestos según la fórmula (I), donde Y indica P2 o P3. Otras realizaciones específicas comprenden compuestos según la fórmula (I), donde
X es un heterobiciclo o heterotriciclo de fórmula (xa1), (xb1), (xc1), (xd1), (xe1), (xf1), (xg1), (xh1) o (xi1) cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, NO2, CN, R5a, OR5a, CONR5aR5b, NR5aCOR5b, SO2R5a, SOR5a, SO2NR5aR5b, NR5aSO2R5b, NR5aR5b, (CH2)q-R6, COR5a y/o SO2R5a, y donde 1 de los grupos CH2 cíclicos puede estar sustituido por CR4aR4b, C=O, O, S, NR5a, SO y/o SO2:

Otras realizaciones específicas comprenden compuestos según la fórmula (I), donde
X es un heterobiciclo o heterotriciclo de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) o (xi) cada uno, independientemente entre sí, no sustituido o mono o disustituido por F, Cl, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 y/o N(C2H5)2, y donde 1 o 2 de los grupos CH2 cíclicos pueden estar sustituidos por (CH3)2, C(C2H5)2, C=O, O, S, NH, NR3a, SO y/o SO2 (donde R3a es preferiblemente metilo, etilo, propilo, isopropilo o ciclopropilo). Las realizaciones importantes comprenden compuestos de fórmula (I), donde X es un heterobiciclo o heterotriciclo de fórmula (xa1), (xb1), (xc1), (xd1), (xe1), (xf1), (xg1), (xh1) o (xi 1), cada uno, independientemente entre sí, no sustituido o mono o disustituido por F, Cl, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 y/o N(C2H5).
Las realizaciones muy específicas comprenden compuestos de fórmula (I), donde X es un heterobiciclo o hetero triciclo seleccionado entre el siguiente grupo:

Otras realizaciones comprenden compuestos de fórmula (I), donde
P3 indica 1 o 2-naftilo no sustituido o mono o disustituido, donde los sustituyentes opcionales se selec cionan entre un grupo compuesto por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6,
o
(P3 es) un resto según la fórmula (Ra) o (Rb):

donde
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a, CONH2, NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;
Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a,
CONH2, NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;
Ea indica O, S, N(Alq) o CH=CH;
Eb indica O, S, N(Alq), CH2, CH2-CH2, O-CH2, S-CH2 o N(Alq)CH2.
El resto según la fórmula (Rb) contiene un centro estereogénico en el átomo de carbono próximo a LY; este se ha indicado con un asterisco (*) en la fórmula (Rb)* a continuación:

(Rb)*
Los restos según la fórmula (Rb) muestran, por tanto, dos configuraciones diferentes en este centro estereogénico, es decir, la configuración (R) y la configuración (S). Por tanto, los compuestos de la presente invención pueden estar presentes enantiopuros o como una mezcla racémica (1:1) de los dos enantiómeros de fórmula (R)-(Rb) y (S)-(Rb).
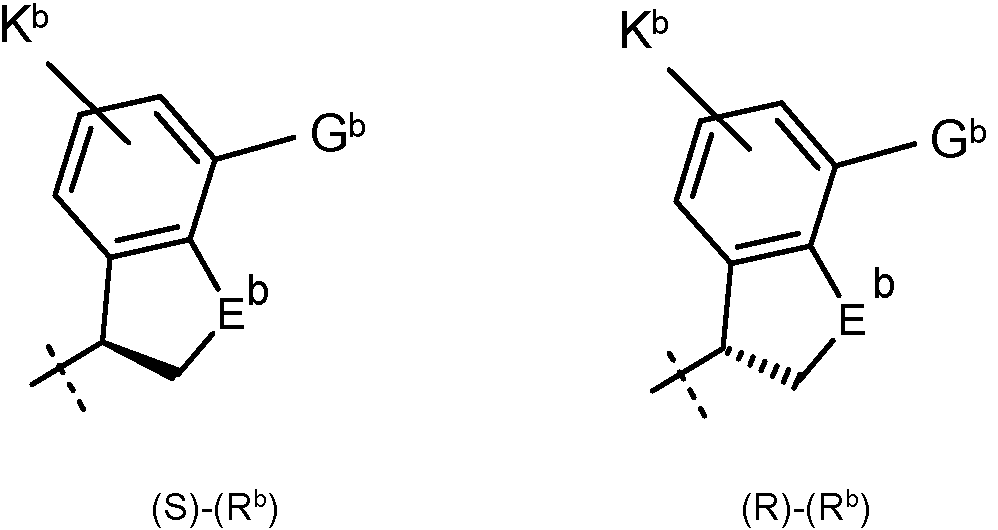
Los compuestos de fórmula (I) que incluyen restos según la fórmula (Rb) también pueden estar presentes en una mezcla en la que uno de los enantiómeros (R)-(Rb) o (S)-(Rb) está presente en exceso con respecto al otro, por ejemplo, 60:40, 70:30, 80:20, 90:10, 95:5 o similares. En una realización en particular de la presente invención, el estereoisómero de fórmula (R)-(Rb) del compuesto de fórmula (I) y el estereoisómero de fórmula (S)-(Rb) del com puesto de fórmula (I) están presentes en una relación de (R)-(Rb) con respecto a (S)-(Rb) de al menos 90 partes de (R)-(Rb) por no más de 10 partes de (S)-(Rb), preferiblemente de al menos 95 de (R)-(Rb) por no más de 5 de (S)-(Rb), más preferiblemente de al menos 99 de (R)-(Rb) por no más de 1 de (S)-(Rb), incluso más preferiblemente de al menos 99,5 de (R)-(Rb) por no más de 0,5 de (S)-(Rb). En otra realización en particular de la presente invención, el estereoisómero de fórmula (S)-(Rb) del compuesto de fórmula (Rb) y el estereoisómero de fórmula (R)-(Rb) del compuesto de fórmula (I) están presentes en una relación de (S)-(Rb) con respecto a (R)-(Rb) de al menos 90 partes de (S)-(Rb) por no más de 10 de (R)-(Rb), preferiblemente de al menos 95 de (S)-(Rb) por no más de 5 (R)-(Rb), más preferiblemente de al menos 99 de (S)-(Rb) por no más de 1 de (R)-(Rb), incluso más preferi blemente de al menos 99,5 de (S)-(Rb) por no más de 0,5 de (R)-(Rb).
Realizaciones preferidas en particular de la presente invención comprenden compuestos de fórmula (I), donde P3 es un resto de fórmula (S)-(Rb) (que tiene una configuración (S) en el carbono unido a LY).
Realizaciones específicas comprenden compuestos según la fórmula (I), donde
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-, 2.3- , 2,4-, 2,5-, 3,4- o 2,3,4, donde, en cada caso, los sustituyentes opcionales se seleccionan inde pendientemente entre un grupo compuesto por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;
P3 indica 1 o 2-naftilo no sustituido o mono o disustituido, donde los sustituyentes opcionales se selec cionan entre un grupo compuesto por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6,
o
(P3 es) un resto según la fórmula (Ra) o (Rb) (preferiblemente P3 es un resto según la fórmula (Ra) o (S)-(Rb));
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a,
CONH2, NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a,
CONH2, NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; Ea indica O, S, N(Alq) o CH=CH;
Eb indica O, S, N(Alq), CH2, CH2-CH2, OCH2, SCH2 o N(Alq)CH2.
Realizaciones adicionales específicas comprenden compuestos de fórmula (I), donde:
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-,
2.3- , 2,4-, 2,5-, 3,4- o 2,3,4, donde, en cada caso, los sustituyentes opcionales se seleccionan inde pendientemente entre un grupo compuesto por Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a, CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8;
P3 es un resto según la fórmula (Ra) o (Rb) (preferiblemente P3 es un resto según la fórmula (Ra) o (S)-(Rb));
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; R7a, R7b indican cada uno, independientemente entre sí, un alquilo C1-C3 lineal o ramificado, donde de 1 a 3 átomos de H pueden estar sustituidos por Hal; y
R8 indica OH u OR7a; y
p indica 1 o 2.
Otras realizaciones incluso más específicas comprenden compuestos según la fórmula (I), donde:
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-,
2.3- , 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre un grupo compuesto por F, Cl, CN, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2;
P3 es un resto según la fórmula (Ra) o (Rb) (preferiblemente P3 es un resto según la fórmula (Ra) o (S)-(Rb));
Ga, Gb indican cada uno, independientemente entre sí, H, F, Cl, CN, CH3, C2H5, CF3, OCH3, OC2H5, COCF3,
SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2;
Ka, Kb indican cada uno, independientemente entre sí, H, F, Cl, CH3, C2H5, CF3, OCH3, OC2H5, COCF3,
SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2.
Realizaciones en particular comprenden compuestos de fórmula (I), donde P3 es un resto según la fórmula (Ra) o (Rb) y donde Ea, Eb indican cada uno, independientemente entre sí, O o S. Realizaciones preferidas en particular comprenden compuestos de fórmula (I), donde P3 es un resto según la fórmula (Fa) o (Fb):

(Fa) (Fb)
En dichas realizaciones, el centro estereogénico en el átomo de carbono en posición 3 del resto dihidrofuranilo (Fb) muestra preferiblemente una configuración (S), es decir, el resto es un resto (3S)-2,3-dihidrobenzofuran-3-il (opcionalmente sustituido) (S)-(Fb)*:

(S)-(Fb)*
(sustituyentes opcionales no mostrados)
Por tanto, realizaciones adicionales muy específicas de la presente invención comprenden compuestos según la fórmula (I), donde
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-,
2.3- , 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre un grupo compuesto por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;
P3 indica un resto según la fórmula (Fa) o (S)-(Fb);
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OH, OR3a, CONR4aR4b,
NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; y Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OH, OR3a, CONR4aR4b,
NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6.
Otras realizaciones muy específicas de la presente invención comprenden compuestos según la fórmula (I), donde:
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-,
2.3- , 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre el grupo com puesto por H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a, CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8;
P3 indica un resto según la fórmula (Fa) o (S)-(Fb);

(S)-(Fb);
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; R7a, R7b indican cada uno, independientemente entre sí, un alquilo C1-C3 lineal o ramificado, donde de 1 a 3 átomos de H pueden estar sustituidos por Hal; y
R8 indica OH u OR7a; y
p indica 1 o 2.
Otras realizaciones muy específicas de la presente invención comprenden compuestos según la fórmula (I), donde:
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-,
2.3- , 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre el grupo com puesto por F, Cl, CN, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2;
P3 indica un resto según la fórmula (Fa) o (S)-(Fb),
Ga, Gb indican cada uno, independientemente entre sí, H, F, Cl, CN, CH3, C2H5, CF3, OCH3, OC2H5, COCF3,
SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2; y
Ka, Kb indican cada uno, independientemente entre sí, H, F, Cl, CH3, C2H5, CF3, OCH3, OC2H5, COCF3,
SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2.
Realizaciones en particular de la presente invención comprenden compuestos según la fórmula (I), donde Y indica P2 o P3, preferiblemente Y indica P3.
Realizaciones específicas de la presente invención comprenden compuestos según la fórmula (I), donde
LY indica CH2 o CH2CH2, donde 1 o 2 átomos de H pueden estar sustituidos por Hal, R7a, OH y/u OR7a, y/o donde un grupo CH2 puede estar sustituido por O o S;
X es un heterobiciclo o heterotriciclo de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) o (xi) cada uno, independientemente entre sí, no sustituido o mono o disustituido por F, Cl, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 y/o N(C2H5)2, y donde 1 de los grupos CH2 cíclicos puede estar sustituido por C(CH3)2, C(C2H5)2, C=O, O, S, NCH3, SO o SO2; Y indica P2 o P3 (preferiblemente P3);
P2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-,
2.3- , 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre el grupo com puesto por H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a, CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8;
P3 indica un resto según la fórmula (Fa) o (S)-(Fb);

(S)-(Fb);
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; R7a, R7b indican cada uno, independientemente entre sí, un alquilo C1-C3 lineal o ramificado, donde de 1 a 3 átomos de H pueden estar sustituidos por Hal; y
R8 indica OH u OR7a; y
p indica 1 o 2.
Cy1, Cy2, Cy3, Cy4, Cy5 indican cada uno, independientemente entre sí, Ar1 o Het1;
R1, R2 indican cada uno, independientemente entre sí, H o alquilo C1-C6, o R1 y R2 forman juntos un resto según la fórmula (CE);
T1, T2, T3, T4, T5, T6, T7, T8 y T9 indican cada uno O;
Hal indica F, Cl o Br.
Otras realizaciones muy específicas de la presente invención comprenden compuestos según la fórmula (R)-(I)-(S)-(Fb) o (R)-(I)-(Fa):
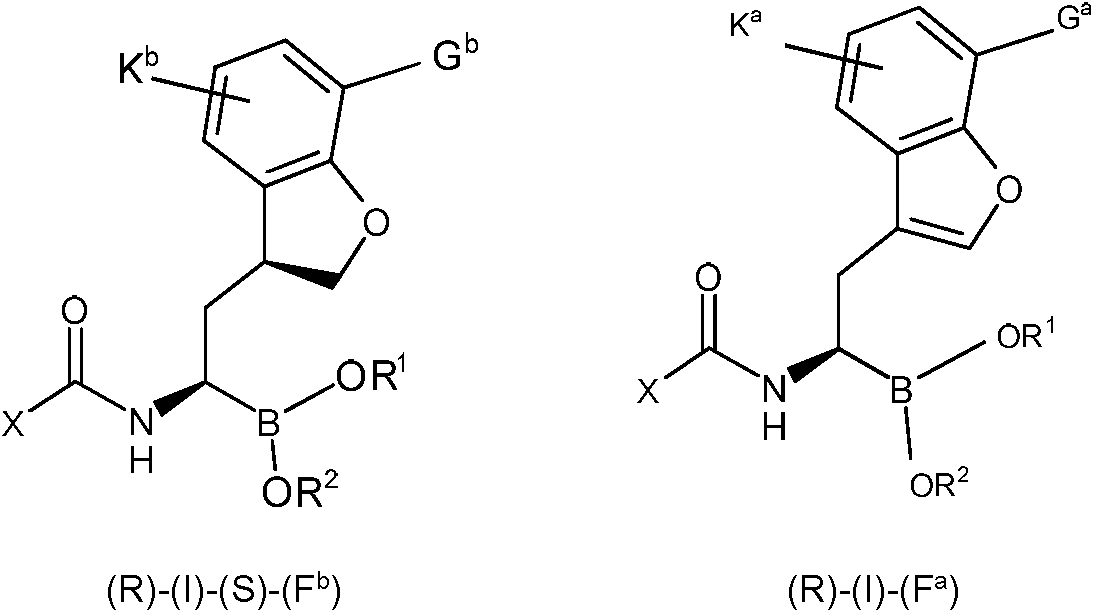
donde
Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R7a, OR7a, CONHR7a, CONR7bR7a,
CONH2, NR7aCOR7b, SO2R7a, SOR7a, NHR7a, N(R7a)2, (CH2)p-SR7a, (CH2)p-N(R7a)2 y/o (CH2)p-R8; X es un heterobiciclo o heterotriciclo de fórmula (xa1), (xb1), (xc1), (xd1), (xe1), (xf1), (xg1), (xh1) o (xi1) cada uno, independientemente entre sí, no sustituido o mono o disustituido por F, Cl, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 y/o N(C2H5)2,
donde 1 de los grupos CH2 cíclicos puede estar sustituido por C(CH3)2, C(C2H5)2, C=O, O, S, NCH3, SO o SO2;
R1, R2 indican H o alquilo C1-C4 o R1 y R2 forman juntos un resto según la fórmula (CE);
R7a, R7b indican cada uno, independientemente entre sí, un alquilo C1-C3 lineal o ramificado, donde de 1 a 3 átomos de H pueden estar sustituidos por Hal;
R8 indica OH u OR7a; y
p indica 1 o 2.
En general, los restos incluidos en los compuestos según la fórmula (I) según se describe anteriormente puede tener el siguiente significado:
LY indica preferiblemente -CH2- o -CH2-CH2-, donde de 1 a 4 átomos de H pueden estar sustituidos por Hal y/o 1 átomo de H puede estar sustituido por Hal, R3a y/o OR4a, y/o donde 1 o 2 grupos CH2 no adyacentes pueden estar sustituidos por O, SO y/o SO2. Más preferiblemente, LY indica -CH2- o -CH2-CH2-, donde de 1 a 4 átomos de H pueden estar sustituidos por F o Cl y/o 1 o 2 átomos de H pueden estar sustituidos por OH, metilo, etilo, isopropilo, CF3, CF2CF3, OCH3, OCH2CH3, OCH2CH2OH y/o CH2OCH3 y/o donde 1 grupo CH2 de LY puede estar sustituido por O.
R1, R2 indican preferiblemente cada uno, independientemente entre sí, H o metilo, etilo, n-propilo o isopropilo, o R1 y R2 forman juntos un resto según la fórmula (CE) como se describe anteriormente. Más preferiblemente R1, R2 indican H, metilo o etilo y preferiblemente en particular R1, R2 indican H.
En realizaciones en las que R3a o R3b representan un alquilo C1-C6 lineal o ramificado, indican preferiblemente cada una, independientemente entre sí, metilo, etilo, n-propilo o isopropilo lineal o ramificado, donde de 1 a 5 átomos de H pueden estar sustituidos por F, Cl, CN, OH y OAlq, donde Alq es preferiblemente metilo o etilo. Más preferiblemente, R3a y R3b indican cada uno, independientemente entre sí, metilo, etilo, n-propilo o isopropilo, donde 1, 2 o 3 átomos de H están sustituidos por F, Cl, OH, OCH3, OC2H5 u OCH(CH3)2.
En realizaciones en las que R3a o R3b representan independientemente entre sí un grupo alquilo cíclico (cicloalquilo), preferiblemente indican, independientemente entre sí, ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo, cada uno no sustituido o mono, di o trisustituido por Hal (preferiblemente F o Cl), metilo, etilo, n-propilo, OH, CN, OCH3 u OC2H5.
R4a y R4b indican cada uno, independientemente entre sí, preferiblemente H, metilo, adicionalmente etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, ferc-butilo o pentilo, donde 1,2 o 3 átomos de H están sustituidos por F, Cl, OH, OCH3, OC2H5 u OCH(CH3)2 o R4a y R4b forman juntos un grupo alquileno C3-C6.
Y puede indicar fenilo, 1-o 2-naftilo, 4- o 5- indanilo, 1-, 2-, 3-, 4-, 5-, 6- o 7-indolilo, 1-, 2-, 4-, 5- o 6- azulenilo, 1-o 2-tetrahidronaftalin-5- o -6-il, 2- o 3-furilo, 2-, 3-, 4-, 5-, 6- o 7-benzofurilo, 2,3-dihidrobenzofuran-2- o -3-ilo, 2- o 3-tienilo, 2- o 3-benzotienilo, 2-, 3-, 4-, 5-, 6- o 7-benzotiofenilo, metilendioxifenilo, benzodioxan-6- o -7-ilo o 3,4-dihidro-1,5-benzodioxepin-6- o -7-ilo, cada uno, independientemente entre sí, no sustituido, mono, di o trisustituido por Hal (preferiblemente F o Cl), CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6 En particular, Y puede indicar fenilo, 1- o 2-naftilo, 2-, 3-, 4-, 5-, 6- o 7-benzofurilo, 2,3-dihidrobenzofuran-2- o -3-ilo, 2- o 3-tienilo, 2- o 3-benzotienilo o benzodioxan-6- o -7-ilo, cada uno, independientemente entre sí, no sustituido, mono, di o trisustituido por F, Cl, CN, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2. En caso de que Y indique un fenilo disustituido, los sustituyentes están preferiblemente en posición 2,4-, 2,5- o 3,4-, más preferiblemente en posición 2.4- o 3,4-. En el caso de que Y indique un fenilo trisustituido, los sustituyentes están preferiblemente en posición 2.3.4- .
En particular, Y puede indicar o-, m- o p-tolilo, o-, m- o p-etilfenilo, o-, m- o p-propilfenilo, o-, m- o p-isopropilfenilo, o-, m- o p-ferc-butilfenilo, o-, m- o p-acetamidofenilo, o-, m- o p-metoxifenilo, o-, m- o p-etoxifenilo, o-, m- o p-fluorofenilo o-, m- o p-bromofenilo, o-, m- o p-clorofenilo, o-, m- o p-trifluorometil-fenilo, o-, m- o p-triclorometil-fenilo, o-, m- o p-(metilsulfonil)fenilo, o-, m- o p-fenoxifenilo, o-, m- o p-metoximetil-fenilo, además preferiblemente 2,4-, 2,5-, 2,6- o 3,4-dimetilfenilo, 2,4-, 2,5- o 3,4-difluorofenilo, 2,4-, 2,5- o 3,4-diclorofenilo, 2,4-, 2,5- o 3,4-dibromofenilo, 2,5- o 3,4-dimetoxifenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-triclorofenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-trifluorofenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-trimetilfenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-tristrifluorometilfenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-tristriclormetil-fenilo, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- o 3,4,5-trimetoximetilfenilo, 2,4,6-trimetoxifenilo, p-yodofenilo, 2-fluoro-3-clorofenilo, 2-fluoro-3-bromofenilo, 2,3-difluoro-4-bromofenilo,
3- bromo-3-metoxifenilo, 2-cloro-3-metoxifenilo, 2-fluoro-3-metoxifenilo, 2-cloro-3-acetamidofenilo, 2-fluoro-3-metoxifenilo, 2-cloro-3-acetamidofenilo, 2,3-dimetil-4-dorofenilo, 2,3-dimetil-4-fluorofenilo.
Y también puede indicar 1- o 2-naftilo, 4- o 5- indanilo, 1-, 2-, 4-, 5- o 6- azulenilo, 1- o 2-tetrahidronaftalin-5- o 6-ilo, 2- o 3-furilo, 2-, 3-, 4-, 5-, 6- o 7-benzofurilo, 2-, 3-, 4-, 5-, 6- o 7-benzotiofenilo, metilendioxifenilo, benzodioxan-6- o 7-ilo o 3,4-dihidro-1,5-benzodioxepin-6- o -7-ilo. En particular, los sustituyentes preferidos de Y se seleccionan entre el grupo compuesto por Cl, CN, CH3, C2H5, CF3, OCH3, OC2H5, COCF3, SCH3, SC2H5, CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2.
Ar2 indica preferiblemente fenilo, que no está sustituido o está mono o disustituido por Hal, CN, R3a, OR3a, CONHR3a, NH2, NHR3a y/o N(R3a)2. Por tanto, Ar2 preferiblemente indica, por ejemplo, fenilo, o-, m- o p-tolilo, o-, m- o p-etilfenilo, o-, m- o p-propilfenilo, o-, m- o p-isopropilfenilo, o-, m- o p-ferc-butilfenilo, o-, m- o p-hidroxifenilo, o-, m- o p-nitrofenilo, o-, m- o p-aminofenilo, o-, m- o p-(N-metilamino)fenilo, o-, m- o p-(N-metilaminocarbonil)fenilo, o-, m- o p-acetamidofenilo, o-, m- o p-metoxifenilo, o-, m- o p-etoxifenilo, o-, m- o p-(N,N-dimetilamino)fenilo, o-, m- o p-(N-etilamino)fenilo, o-, m- o p-(N,N-dietilamino)fenilo, o-, m- o p-fluorofenilo, o-, m- o p-bromofenilo, o-, m- o p-clorofenilo, o-, m- o p-cianofenilo.
Het2 indica preferiblemente un heterociclo de 5 o 6 átomos saturado, insaturado o aromático que tiene de 1 a 4 átomos de N, O y/o S, que no está sustituido o está mono o disustituido por Hal, CN, R3a, OR3a, CONHR3a, NH2, NHR3a y/o N(R3a)2. Por tanto, Het2 por ejemplo puede indicar 2- o 3-furilo, 2- o 3-tienilo, 1-, 2- o 3-pirrolilo, 1-, 2-, 4- o 5-imidazolilo, 1-, 3-, 4- o 5-pirazolilo, 2-, 4- o 5-oxazolilo, 3-, 4- o 5-isoxazolilo, 2-, 4- o 5-tiazolilo, 3-, 4- o 5- isotiazolilo, 2-, 3- o 4-piridilo, 2-, 4-, 5- o 6-pirimidinilo, imidazolilo, morfolinilo o piperazinilo.
Alq indica preferiblemente metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o ferc-butilo, pentilo o hexilo, más preferiblemente, metilo, etilo, propilo o isopropilo, más preferiblemente metilo, etilo, n-propilo o isopropilo. Hal indica preferiblemente F, Cl o Br, más preferiblemente F o Cl.
m indica preferiblemente 0, 1 o 2, más preferiblemente 1 o 2 y lo más preferiblemente 1.
q indica preferiblemente 0, 1,2, 3 o 4 e incluso más preferiblemente 0, 1 o 2.
Realizaciones en particular de la presente invención comprenden compuestos seleccionados entre el grupo compuesto por:
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}-2-(tiofen-3-il)etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;
ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;
ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(7-cloro-1-benzofuran-3-il)-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-(7-doro-1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-[(3R)-7-metil-2,3-dihidro-1-benzofuran-3-N]-1-([(1S,2R,4R)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-7-metN-2,3-dihidro-1-benzofuran-3-il]-1-([(1S,2R,4R)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,8S)-11-oxatriddo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,6S,7R)-3-ciclopropil-4-oxo-10-oxa-3-azatriciclo[5.2.1.01,5]dec-8-en-6-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(7-metil-1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2,4,6-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(7-metil-1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2,4,6-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,8R)-8-metil-11-oxatriciclo[6.2.1.02,7]undeca-2,4,6-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-9-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,8S)-8-metil-11-oxatriciclo[6.2.1.02,7]undeca-2,4,6-trien-1-il]formamido}etil]borónico;
ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-9-il]formamido}etil]borónico;
ácido [(1R)-2-(2,4-dimetilfenil)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-ciclohexil-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}-3-fenilpropil]borónico;
ácido [(1R)-3-metil-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}butil]borónico;
ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-H[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-H[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;
y solvatos, tautómeros, oligómeros o estereoisómeros de los mismos, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
El término solvatos de los compuestos se refiere a aducciones de moléculas solventes inertes en los compuestos que se forman gracias a su fuerza de atracción mutua. Los solvatos son, por ejemplo, mono o dihidratos o alcóxidos. Se entiende que la invención también se refiere a los solvatos de las sales.
El término derivados farmacéuticamente aceptables se considera que significa, por ejemplo, las sales de los compues tos según la invención y también los denominados compuestos profármacos.
Según se usa en este documento y a menos que se indique lo contrario, el término «profármaco» significa un derivado de un compuesto de fórmula (I) que se puede hidrolizar, oxidar o hacer reaccionar de alguna otra forma en condiciones biológicas (in vitro o in vivo) para proporcionar un compuesto activo, especialmente un compuesto de fórmula (I). Entre los ejemplos de profármacos se incluyen, pero sin limitaciones, derivados y metabolitos de un compuesto de fórmula (I) que incluyen restos biohidrolizables tales como amidas biohidrolizables, ésteres biohidrolizables, carbamatos biohidrolizables, carbonatos biohidrolizables, ureidos biohidrolizables y análogos de fosfato biohidrolizables. En ciertas rea lizaciones, los profármacos de compuestos con grupos funcionales carboxilo son los ésteres alquilo menores del ácido carboxílico. Los ésteres de carboxilato se forman convenientemente mediante esterificación de cualquiera de los restos de ácido carboxílico presentes en la molécula. Los profármacos se pueden preparar normalmente usando métodos bien conocidos, como los descritos en Burger's Medicinal Chemistry and Drug Discovery 6.a ed. (Donald J. Abraham ed., 2001, Wiley) y en Design and Application of Prodrugs (H. Bundgaard ed., 1985, Harwood Academic Publishers Gmfh).
La expresión «cantidad eficaz» indica la cantidad de un medicamento o de un principio activo farmacéutico que causa en un tejido, sistema, animal o ser humano la respuesta biológica o médica que busca o desea, por ejemplo, un inves tigador o un médico.
Además, la expresión «cantidad terapéuticamente eficaz» indica una cantidad que, comparada con un sujeto concreto que no ha recibido esta cantidad, tiene las siguientes consecuencias: mejora del tratamiento, curación, prevención o eliminación de una enfermedad, síndrome, afección, dolencia, trastorno o efectos secundarios, o también la reducción de la progresión de una enfermedad, dolencia o trastorno.
La expresión «cantidad terapéuticamente eficaz» también abarca las cantidades que son eficaces para aumentar la función fisiológica normal.
La invención también se refiere al uso de mezclas de los compuestos de fórmula (I), por ejemplo, mezclas de dos diastereoisómeros, por ejemplo, en la proporción 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 o 1:1000.
El término «tautómeros» se refiere a formas isoméricas de un compuesto que están en equilibrio entre sí. Las concen traciones de las formas isoméricas dependerán del entorno en el que se encuentre el compuesto y puede ser diferente dependiendo de, por ejemplo, si el compuesto es un sólido o está en una solución orgánica o acuosa.
La invención además comprende un proceso para la preparación de un compuesto de fórmula (I) según se des cribe anteriormente y las sales, tautómeros y estereoisómeros farmacéuticamente aceptables del mismo, carac terizados porque un compuesto de fórmula (III)
O
XA OH
se acopla con un compuesto de fórmula (VI)

donde todos los restos de fórmula (III) y fórmula (IV) son como se define anteriormente y donde el compuesto obtenido de fórmula (Ib) pueden convertirse posteriormente en un compuesto de fórmula (Ia) mediante el trata miento con HCl, HBr, HI y/o TFA, en presencia o ausencia de un exceso de un ácido borónico de bajo peso molecular

Las siguientes abreviaturas se refieren a las abreviaturas que se utilizan a continuación:
AcOH (ácido acético), ACN (acetonitrilo), BINAP (2,2'-bis(disfenilfosfino)-1,1'-binaftaleno), dba (dibencilidenacetona), tBu (ferc-butilo), tBuOK (ferc-butóxido de potasio), CDI (1,1'-carbonildiimidazol), d Bu (1,8-dizabiciclo[5.4.0]undec-7-eno), DCC (diciclohexilcarbodiimida), DCM (diclorometano), DIAD (diisobutilazodicarboxilato), DIC (diisopropilcarbodiimida), DIEA (di-isopropil etilamina), DMA (dimetil acetamida), DMAP (4-dimetilaminopiridina), DMSO (dimetilsulfóxido), DMF (N,N-dimetilformamida), EDC.HCl (clohidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida), EM (espectrometría de masas), ÉterPet (éter de petróleo), EtOAc o EE (acetato de etilo), EtOH (etanol), g (gramo), cHex (ciclohexano), HATU (hexafluorofosfato de dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio), HOBt (N-hidroxibenzotriazol), HPLC (cromatografía líquida de alta resolución), h (hora), MHz (megahercio), MeOH (metanol), min (minuto), ml (mililitro), mmol (milimol), mM (milimolar), MO (mi croondas), NMM (N-metil morfolina), NBS (N-bromo succinimida), PBS (solución salina tamponada con fosfato), pf (punto de fusión), PMB (para-metoxibencilo), PyBOP (hexafluorofosfato de benzotri azol-1 -il-oxitri pirrolidinofosfonio), RMN (resonancia magnética nuclear), TA (temperatura ambiente), TBAF (fluoruro de tetra-butilamonio), TBTU (tetrafluoroborato de N,N,N',N'-tetrametil-O-(benzotriazol-1-il)uronio), T3P (anhídrido del ácido propanofosfónico), TEA (trietilamina), TFA (ácido trifluoroacético), THF (tetrahidrofurano), TBME (éter de metil ferc-butilo), TLC (cromatografía en capa fina), TMS (trimetilsililo), TMSI (yoduro de trimetilsililo), UV (ultravioleta).
En general, los compuestos de fórmula (I), donde todos los restos se definen como anteriormente, pueden obte nerse a partir de un compuesto de fórmula (III) como se describe en el esquema 1.
Esquema 1:

como sal d e TFA o HCl
(III) (IV ) (I)
El primer paso consiste en la reacción de un compuesto de fórmula (III), donde X es como se define anteriormente, con un compuesto de fórmula (IV), donde R1, R2, LY e Y son como se define anteriormente. La reacción se realiza utilizando condiciones y métodos bien conocidos por los expertos en la materia para la preparación de amidas a partir de un ácido carboxílico con agentes de acoplamiento convencionales, como por ejemplo, pero sin limitacio nes, HATU, TBTU, sal de 1 -alquil-2-cloropiridinio con soporte de polímero (reactivo de Mukaiyama con soporte de polímero), yoduro de 1 -metil-2-cloropiridinio (reactivo de Mukaiyama), una carbodiimida (como DCC, DIC, EDC) y HOBt, PyBOP® y otros reactivos bien conocidos por los expertos en la materia, preferiblemente TBTU, en pre sencia o ausencia de bases como TEA, DIEA, n Mm , morfolina con soporte de polímero, preferiblemente DIEA, en un solvente adecuado como DCM, THF o DMF, a una temperatura de entre -10 °C y 50 °C, preferiblemente a 0 °C, durante algunas horas, por ejemplo, de una hora a 24 h. Alternativamente, los compuestos de fórmula (III) pueden convertirse en derivados de ácido carboxílico como haluros o anhídridos de acilo, mediante métodos bien conocidos por los expertos en la materia, como por ejemplo, pero sin limitaciones, el tratamiento con SOCl2, POCb, PCl5, (COCl)2, en presencia o ausencia de cantidades catalíticas de DMF, en presencia o ausencia de un solvente adecuado como tolueno, DCM, THF, a una temperatura que se eleva de 20 °C a 100 °C, preferiblemente a 50 °C, durante algunas horas, por ejemplo de una hora a 24 h. La conversión de los derivados del ácido carboxílico en compuestos de fórmula (I) puede conseguirse utilizando condiciones y métodos bien conocidos por los expertos en la materia para la preparación de amidas a partir de un derivado de ácido carboxílico (p. ej., cloruro de acilo) con alquilaminas, en presencia de bases como TEA, DIEA, NMM en un solvente adecuado como DCM, THF o DMF, a una temperatura que se eleva de 20 °C a 100 °C, preferiblemente a 50 °C, durante algunas horas, por ejemplo de una hora a 24 h.
En el proceso descrito anteriormente, la reacción entre el compuesto de fórmula (III) y el compuesto de fórmula (IV) se realiza en presencia de un agente de acoplamiento seleccionado entre HATU, TBTU, sal de 1 -alquil-2-cloropiridinio con soporte de polímero (reactivo de Mukaiyama con soporte de polímero), yoduro de 1-metil-2-cloropiridinio (reactivo de Mukaiyama) y una carbodiimida.
Los compuestos de fórmula (Ia), donde X, LY e Y se definen como anteriormente o donde R1 y R2 son H, pueden prepararse a partir de compuestos de fórmula (Ib) usando métodos bien conocidos por los expertos en la materia para la hidrólisis de ésteres borónicos, como por ejemplo, pero sin limitaciones, el tratamiento con HCl, HBr, HI, TFA, en presencia o ausencia de un exceso de un ácido borónico de bajo peso molecular, como por ejemplo, pero sin limitaciones, iBuB(OH)2 (esquema 2).
Esquema 2:

Los compuestos de fórmula (III) o (IV) están disponibles en el mercado o pueden prepararse mediante métodos bien conocidos por los expertos en la materia.
En general, los compuestos de fórmula (IV) son accesibles, por ejemplo, mediante el siguiente esquema 3a:
Éster boronato

La síntesis de compuestos de fórmula (IV) se describe adicionalmente en los documentos WO 2016/050356, WO 2016/050355, WO 2016/050359 y WO 2016/050358.
Los compuestos de fórmula (III) son accesibles, por ejemplo, mediante las rutas descritas en los esquemas 4, 5 y 6:
Esquema 4:

rac endo/exo rac endo/exo

exclusivamente rac exo
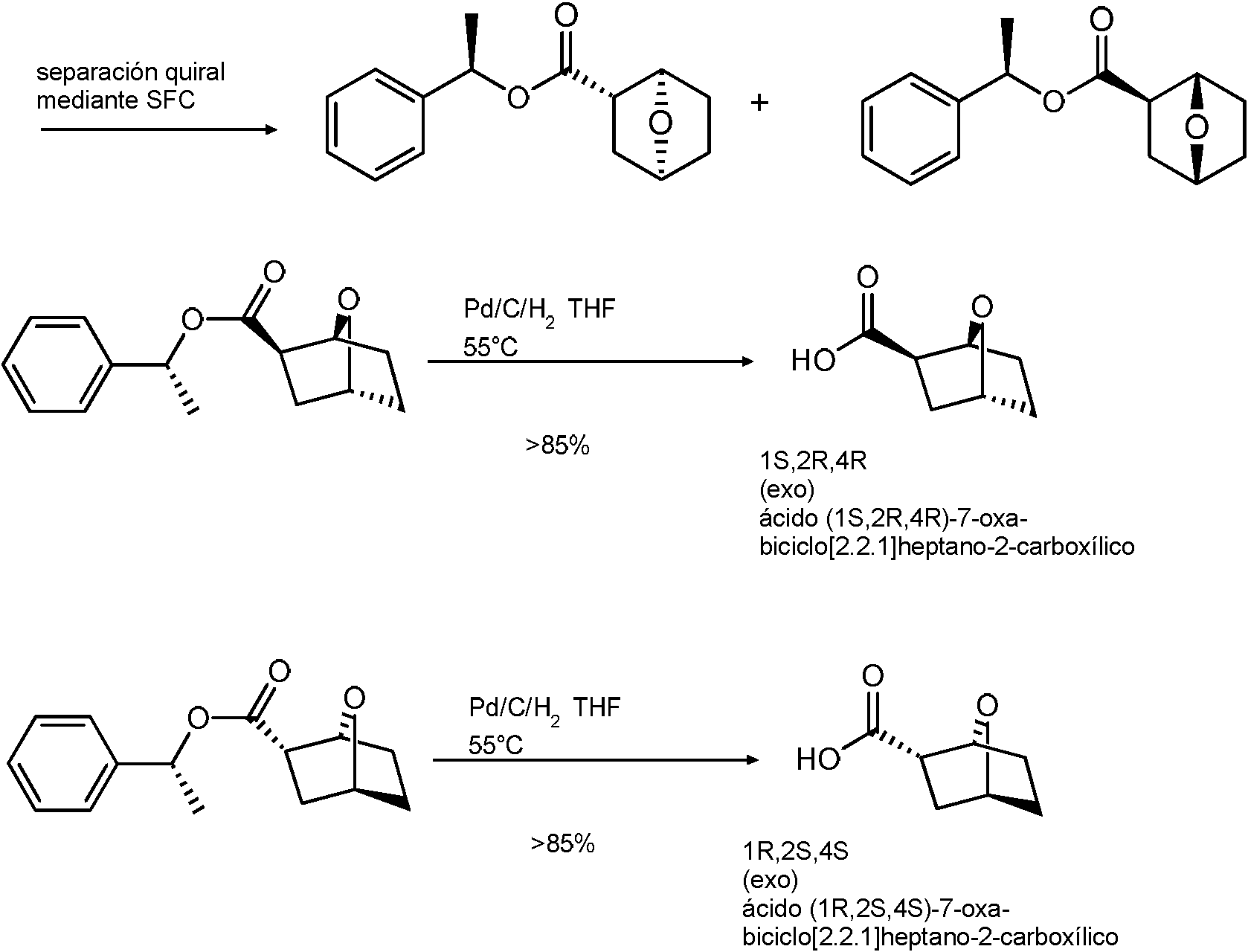
Mediante métodos similares también se pueden sintetizar ácidos 7-oxa-biciclo[2.2.1]heptano-2-carboxílicos susti tuidos.
Esquema 5:



Mediante métodos similares también se pueden sintetizar ácidos 11-oxatricido[6.2.1.02,7]undeca-2,4,6,9-tetraeno-9-carboxílicos sustituidos.
Esquema 6:


Mediante métodos similares, partiendo de ácidos furano-2-carboxílicos sustituidos, se pueden sintetizar ácidos 11-oxatricido[6.2.1.02,7]undeca-2,4,6-tetraeno-1-carboxílicos sustituidos. También se pueden utilizar ácidos antranílicos sustituidos para sintetizar los correspondientes compuestos que portan sustituyentes adicionales en el resto aromático.
Si los métodos de síntesis generales establecidos anteriormente no fueran aplicables para la obtención de com puestos según la fórmula (I) y/o compuestos intermedios necesarios para la síntesis de compuestos de fórmula (I), podrán usarse métodos de preparación adecuados conocidos por un experto en la materia.
En general, la ruta de síntesis para cualquier compuesto individual de fórmula (I) dependerá de los sustituyentes específicos de cada molécula y de la fácil disponibilidad de los productos intermedios necesarios, siendo una vez más apreciados estos factores por los expertos en la materia. Para obtener información sobre todos los métodos de protección y desprotección, consulte Philip J. Kocienski, en «Protecting Groups», Georg Thieme Verlag Stuttgart, Nueva York, 1994 y Theodora W. Greene and Peter G. M. Wuts en «Protective Groups in Organic Synthesis», Wiley Interscience, 3a Edición 1999.
Los compuestos de esta invención pueden aislarse asociados con moléculas de solvente mediante cristalización a partir de la evaporación de un solvente apropiado. Las sales de adición de ácido farmacéuticamente aceptables de los compuestos de fórmula (I), que contienen un centro básico, pueden prepararse de forma convencional. Por ejemplo, una solución de la base libre puede tratarse con un ácido adecuado, bien puro o en una solución adecuada, y la sal resultante puede aislarse mediante filtración o evaporación al vacío del solvente de la reacción. Las sales de adición de base farmacéuticamente aceptables pueden obtenerse de forma análoga tratando una solución de compuestos de fórmula (I), que contiene un centro ácido, con una base adecuada. Ambos tipos de sales pueden obtenerse o interconvertirse usando técnicas de resina de intercambio iónico.
Dependiendo de las condiciones usadas, los tiempos de reacción generalmente están entre algunos minutos y 14 días, y la temperatura de reacción está entre aproximadamente -30 °C y 140 °C, normalmente entre -10 °C y 90 °C, en particular entre aproximadamente 0 °C y aproximadamente 70 °C.
Los compuestos de fórmula (I) se pueden obtener además mediante la liberación de compuestos de fórmula (I) a partir de uno de sus derivados funcionales mediante el tratamiento con un agente solvolizante o hidrogenolizante.
Los materiales de partida preferidos para la solvólisis o hidrogenólisis son aquellos que conforman la fórmula (I), pero que contienen los correspondientes grupos amino y/o hidroxilo protegidos, en lugar de uno o más grupos amino y/o hidroxilo libres, preferiblemente aquellos que contienen un grupo protector de amino en lugar de un átomo de H unido a un átomo de N, en particular aquellos que llevan un grupo R'-N, donde R' indica un grupo protector de amino, en lugar de un grupo HN, y/o aquellos que contienen un grupo protector de hidroxilo en lugar del átomo de H de un grupo hidroxilo, por ejemplo aquellos que conforman la fórmula (I), pero que contienen un grupo -COOR'', donde R'' indica un grupo protector de hidroxilo, en lugar de un grupo -COOH.
También es posible que en la molécula del material de partida se encuentren diversos grupos (idénticos o diferen tes) amino y/o hidroxilo protegidos. Si los grupos protectores presentes son diferentes entre sí, en muchos casos, estos pueden escindirse de forma selectiva.
El término «grupo protector de amino» se conoce en términos generales y se refiere a grupos que son adecuados para proteger (bloquear) un grupo amino frente a reacciones químicas, pero que se eliminan fácilmente después de que se haya llevado a cabo la reacción química deseada en cualquier otra parte de la molécula. Son típicos entre estos grupos, en particular, los grupos acilo, arilo, aralcoximetilo o aralquilo sustituidos o no sustituidos. Puesto que los grupos protectores de amino se eliminan tras la reacción deseada (o secuencia de reacciones), su tipo y tamaño no son por otra parte cruciales; sin embargo, se da preferencia a aquellos que tienen de 1 a 20, en particular de 1 a 8 átomos de carbono. El término «grupo acilo» debe entenderse en el sentido más amplio en conexión con el presente proceso. Incluye grupos acilo derivados de ácidos sulfónicos o ácidos carboxílicos alifáticos, aralifáticos, aromáticos o heterocíclicos, y en particular, grupos alcoxicarbonilo, ariloxicarbonilo y especial mente aralcoxicarbonilo. Ejemplos de estos grupos acilo son alcanoílo, como acetilo, propionilo y butirilo; aralcanoílo, como fenilacetilo; aroílo, como benzoílo y tolilo; ariloxialcanoílo, como POA; alcoxicarbonilo, como metoxicarbonilo, etoxicarbonilo, 2,2,2-tricloroetoxicarbonilo, BOC (ferc-butoxicarbonilo) y 2-yodoetoxicarbonilo; aralcoxi carbonilo, como CBZ («carbobenzoxi»), 4-metoxibenciloxicarbonilo y FMOC; y arilsulfonilo, como Mtr. Los grupos protectores de amino preferidos son BOC y Mtr, además de CBZ, FMOC, bencilo y acetilo.
El término «grupo protector de hidroxilo» es igualmente conocido en términos generales y se refiere a grupos que son adecuados para proteger un grupo hidroxilo frente a reacciones químicas, pero que se elimina fácilmente después de que se ha llevado a cabo la reacción química deseada en cualquier otra parte de la molécula. Son típicos de estos grupos los grupos arilo, aralquilo o acilo sustituidos o no sustituidos mencionados anteriormente, además de los grupos alquilo. La naturaleza y tamaño de los grupos protectores de hidroxilo no son cruciales, ya que se eliminan de nuevo después de la reacción química (o secuencia de reacciones) deseada; se da preferencia a grupos que tienen de 1 a 20, en particular de 1 a 10 átomos de carbono. Ejemplos de grupos protectores de hidroxilo son, entre otros, bencilo, 4-metoxibencilo, p-nitrobenzoílo, p-toluenosulfonilo, ferc-butilo y acetilo, donde bencilo y ferc-butilo son especialmente preferidos.
El término «solvatos de los compuestos» se refiere a aducciones de moléculas solventes inertes en los compues tos que se forman gracias a su fuerza de atracción mutua. Los solvatos son, por ejemplo, mono o dihidratos o alcoholatos.
Los compuestos de fórmula (I) se liberan a partir de sus derivados funcionales (dependiendo del grupo protector utilizado), por ejemplo, usando ácidos fuertes, usando de forma ventajosa TFA o ácido perclórico, pero también usando otros ácidos inorgánicos fuertes, como ácido clorhídrico o ácido sulfúrico, ácidos carboxílicos orgánicos fuertes, como ácido tricloroacético, o ácidos sulfónicos, como ácido benceno- o p-toluenosulfónico. Es posible la presencia de un solvente inerte adicional, pero no siempre es necesario. Los solventes inertes adecuados son, preferiblemente, orgánicos, por ejemplo ácidos carboxílicos, como ácido acético, éteres, como THF o dioxano, amidas, como DMF, hidrocarburos halogenados, como DCM, además también alcoholes como metanol, etanol o isopropanol, y agua. Adicionalmente, son adecuadas mezclas de los solventes mencionados anteriormente. Se usa preferiblemente TFA en exceso sin adición de otro solvente, y el ácido perclórico se usa preferiblemente en forma de mezcla de ácido acético y ácido perclórico al 70 % en la proporción 9:1. Las temperaturas de reacción para la escisión están, de forma ventajosa, entre aproximadamente 0 y aproximadamente 50 °C, preferiblemente entre 15 y 30 °C (TA).
Los grupos BOC, OBut y Mtr pueden, por ejemplo, escindirse preferiblemente usando TFA en DCM o usando HCl a aproximadamente 3 a 5 N en dioxano a 15-30 °C, y el grupo FMOC puede escindirse usando una solución de aproximadamente el 5 al 50% de dimetilamina, dietilamina o piperidina en DMF a 15-30 °C.
Los grupos protectores que pueden eliminarse hidrogenolíticamente (por ejemplo, CBZ, bencilo o la liberación del grupo amidino del derivado oxadiazol de los mismos) pueden escindirse, por ejemplo, mediante tratamiento con hidrógeno en presencia de un catalizador (por ejemplo, un catalizador de un metal noble, como paladio, de forma ventajosa sobre un soporte como carbono). Los solventes adecuados aquí son aquellos indicados anteriormente, en particular, por ejemplo, alcoholes, como metanol o etanol, o amidas, como DMF. Generalmente, la hidrogenólisis se realiza a temperaturas de entre aproximadamente 0 y 100 °C y presiones de entre aproximadamente 1 y 200 bares, preferiblemente a 20-30 °C y 1-10 bares. La hidrogenólisis del grupo CBZ se produce bien, por ejem plo, entre el 5 y el 10 % de Pd/C en metanol o usando formato de amonio (en lugar de hidrógeno) en Pd/C en metanol/DMF a 20-30 °C.
Son ejemplos de solventes inertes adecuados hidrocarburos, como hexano, éter de petróleo, benceno, tolueno o xileno; hidrocarburos clorados, como tricloroetileno, 1,2-dicloroetano, tetraclorometano, trifluorometilbenceno, clo roformo o DCM; alcoholes, como metanol, etanol, isopropanol, n-propanol, n-butanol o terc-butanol; éteres, como éter dietílico, éter diisopropílico, tetrahidrofurano (THF) o dioxano; éteres glicólicos, como éter monometílico o monoetílico de etilenglicol o éter dimetílico de etilenglicol (diglima); cetonas, como acetona o butanona; amidas, como acetamida, dimetilacetamida, N-metilpirrolidona (NMP) o dimetilformamida (DMF); nitrilos, como acetonitrilo; sulfóxidos, como dimetilsulfóxido (DMSO); disulfuro de carbono; ácidos carboxílicos, como ácido fórmico o ácido acético; compuestos nitrogenados, como nitrometano o nitrobenceno; ésteres, como EtOAc, o mezclas de dichos solventes.
Los ésteres pueden saponificarse, por ejemplo, usando LiOH, NaOH o KOH en agua, agua/THF, agua/THF/etanol o agua/dioxano, a temperaturas de entre 0 y 100 °C. Además, el éster puede hidrolizarse, por ejemplo, usando ácido acético, TFA o HCl.
Los grupos amino libres pueden además acilarse de forma convencional usando un cloruro o un anhídrido de acilo o alquilarse usando un haluro de alquilo sin sustituir o sustituido o hacerse reaccionar con CH3-C(=NH)-OEt, de forma ventajosa en un solvente inerte, como DCM o THF y/o en presencia de una base, como trietilamina o piridina, a temperaturas de entre -60 °C y 30 °C.
A lo largo de la memoria descriptiva, el término grupo saliente preferiblemente indica Cl, Br, I o un grupo OH modificado reactivamente, como por ejemplo, un éster activado, una imidazolida o un grupo alquilsulfoniloxi con 1 a 6 átomos de carbono (preferiblemente metilsulfoniloxi o trifluorometilsulfoniloxi) o arilsulfoniloxi con 6 a 10 áto mos de carbono (preferiblemente fenil- o p-tolilsulfoniloxi).
En la literatura (por ejemplo en trabajos convencionales, como Houben-Weyl, Methoden der organischen Chemie [Métodos de química orgánica], Georg-Thieme-Verlag, Stuttgart) se describen radicales de este tipo para la acti vación del grupo carboxilo en reacciones típicas de acilación.
Los ésteres activados se forman ventajosamente in situ, por ejemplo, mediante la adición de HOBt o N-hidroxisuccinimida.
Sales farmacéuticas y otras formas
Dichos compuestos de fórmula (I) pueden usarse en su forma final no sal. Por otro lado, la presente invención también se refiere al uso de estos compuestos en forma de sus sales farmacéuticamente aceptables, que pueden derivar de diversos ácidos y bases orgánicos e inorgánicos mediante procedimientos conocidos en la técnica. Las formas salinas farmacéuticamente aceptables de los compuestos de fórmula (I) se preparan, en su mayor parte, por métodos convencionales. Si el compuesto de fórmula (I) contiene un centro ácido, como un grupo carboxilo, puede formarse una de sus sales adecuadas haciendo reaccionar el compuesto con una base adecuada para obtener la correspondiente sal de adición de base. Dichas bases son, por ejemplo, hidróxidos de metales alcalinos, como hidróxido de potasio e hidróxido de sodio; hidróxidos de metales alcalinotérreos, como hidróxido de magne sio e hidróxido de calcio; y diversas bases orgánicas como piperidina, dietanolamina y N-metil-glucamina (meglumina), benzatina, colina, dietanolamina, etilendiamina, benetamina, dietilamina, piperazina, lisina, L-arginina, amo níaco, trietanolamina, betaína, etanolamina, morfolina y trometamina. En el caso de determinados compuestos de fórmula I, que contienen un centro básico, se pueden formar sales de adición de ácido tratando estos compuestos con ácidos orgánicos e inorgánicos farmacéuticamente aceptables, por ejemplo, haluros de hidrógeno, como clo ruro de hidrógeno o bromuro de hidrógeno, otros ácidos minerales y sus sales correspondientes, como sulfato, nitrato o fosfato y similares, y sulfonatos de alquilo y monoarilo, como metanosulfonato, etanosulfonato, toluenosulfonato y bencenosulfonato, y otros ácidos orgánicos y sus sales correspondientes, como carbonato, acetato, trifluoroacetato, tartrato, maleato, succinato, citrato, benzoato, salicilato, ascorbato y similares. Por consiguiente, entre las sales de adición de ácido farmacéuticamente aceptables de los compuestos de fórmula (I) se incluyen
las siguientes: acetato, adipato, alginato, aspartato, benzoato, bencenosulfonato (besilato), bisulfato, bisulfito, bro muro, alcanforato, alcanforsulfonato, caprato, caprilato, cloruro, clorobenzoato, citrato, ciclamato, cinamato, digluconato, dihidrogenofosfato, dinitrobenzoato, dodecilsulfato, etanosulfonato, formato, glicolato, fumarato, galacterato (a partir de ácido múcico), galacturonato, glucoheptanoato, gluconato, glutamato, glicerofosfato, hemisuccinato, hemisulfato, heptanoato, hexanoato, hipurato, clorhidrato, bromhidrato, yodhidrato, 2-hidroxi-etano-sulfonato, yoduro, isetionato, isobutirato, lactato, lactobionato, malato, maleato, malonato, mandelato, metafosfato, metanosulfonato, metilbenzoato, monohidrógeno-fosfato, 2-naftalenosulfonato, nicotinato, nitrato, oxalato, oleato, palmoato, pectinato, persulfato, fenilacetato, 3-fenilpropionato, fosfato, fosfonato, ftalato, aunque esto no repre senta una limitación. Ambos tipos de sales pueden obtenerse o interconvertirse preferiblemente usando técnicas de resina de intercambio iónico.
Además, entre las sales de bases de los compuestos de fórmula I se incluyen las sales de aluminio, amonio, calcio, cobre, hierro(III), hierro(II), litio, magnesio, manganeso(III), manganeso(II), potasio, sodio y cinc, aunque esto no pretende representar una limitación. De las sales mencionadas anteriormente, se da preferencia a las de amonio, a las sales de metales alcalinos de sodio y potasio y a las sales de metales alcalinotérreos de calcio y magnesio. Las sales de los compuestos de fórmula (I) que derivan de bases orgánicas no tóxicas farmacéutica mente aceptables incluyen sales de aminas primarias, secundarias y terciarias, aminas sustituidas, también inclu yen aminas naturales sustituidas, aminas cíclicas y resinas básicas de intercambio iónico, por ejemplo, arginina, betaína, cafeína, cloroprocaína, colina, N,N'-dibenciletilenediamina (benzatina), diciclohexilamina, dietanolamina, dietilamina, 2-dietil-amino-etanol, 2-dimetil-amino-etanol, etanolamina, etilendiamina, N-etilmorfolina, N-etil-piperidina, glucamina, glucosamina, histidina, hidrabamina, isopropilamina, lidocaína, lisina, meglumina (N-metil-D-glucamina), morfolina, piperazina, piperidina, resinas de poliamina, procaína, purinas, teobromina, trietanolamina, trietilamina, trimetilamina, tripropilamina y tris(hidroximetil)-metilamina (trometamina), aunque esto no pretende representar una limitación.
Los compuestos de fórmula (I) de la presente invención que contienen grupos básicos con nitrógeno pueden cuaternizarse usando agentes como haluros de alquilo (C1-C4), por ejemplo cloruro, bromuro y yoduro de metilo, etilo, isopropilo y tere-butilo; sulfatos de dialquilo (C1-C4), por ejemplo, sulfato de dimetilo, dietilo y diamilo; haluros de alquilo (C10-C18), por ejemplo, cloruro, bromuro y yoduro de decilo, dodecilo, laurilo, miristilo y estearilo, y ha luros de aril-alquilo (C1-C4), por ejemplo cloruro de bencilo y bromuro de fenetilo. Tanto los compuestos hidrosolubles como los liposolubles de fórmula (I) pueden prepararse usando estas sales.
Entre las sales farmacéuticas mencionadas anteriormente preferidas se incluyen acetato, trifluoroacetato, besilato, citrato, fumarato, gluconato, hemisuccinato, hipurato, clorhidrato, bromhidrato, isetionato, mandelato, meglumina, nitrato, oleato, fosfonato, pivalato, fosfato sódico, estearato, sulfato, sulfosalicilato, tartrato, tiomalato, tosilato y trometamina, aunque esto no pretende representar una limitación.
Las sales de adición de ácido de compuestos básicos de fórmula (I) se preparan poniendo la forma de base libre en contacto con una cantidad suficiente del ácido deseado, provocando la formación de la sal de manera conven cional. La base libre puede regenerarse poniendo la forma salina en contacto con una base y aislando la base libre de manera convencional. Las formas de base libre difieren en cierto modo de sus formas salinas correspon dientes con respecto a determinadas propiedades físicas, como la solubilidad en solventes polares; sin embargo, para los fines de la invención, las sales se corresponden de otro modo con sus respectivas formas de base libre.
Como se ha mencionado, las sales de adición de base farmacéuticamente aceptables de los compuestos de fórmula (I) se forman con metales o aminas, como metales alcalinos y metales alcalinotérreos o aminas orgánicas. Los metales preferidos son sodio, potasio, magnesio y calcio. Las aminas orgánicas preferidas son N,N'-dibenciletilendiamina, cloroprocaína, colina, dietanolamina, etilendiamina, N-metil-D-glucamina y procaína.
Las sales de adición de base de compuestos ácidos de fórmula (I) se preparan poniendo la forma de ácido libre en contacto con una cantidad suficiente de la base deseada, lo que provoca la formación de la sal de manera convencional. El ácido libre puede regenerarse poniendo la forma salina en contacto con un ácido y aislando el ácido libre de manera convencional. Las formas de ácido libre difieren en cierto modo de sus formas salinas correspondientes con respecto a determinadas propiedades físicas, como la solubilidad en solventes polares; sin embargo, para los fines de la invención, las sales se corresponden por lo demás con sus respectivas formas de ácido libre.
Si un compuesto de fórmula (I) contiene más de un grupo que es capaz de formar sales farmacéuticamente acep tables de este tipo, la fórmula (I) abarca también sales múltiples. Entre las formas de sales múltiples típicas se incluyen, por ejemplo, bitartrato, diacetato, difumarato, dimeglumina, difosfato, disodio y triclorhidrato, aunque esto no pretende representar una limitación.
Con respecto a lo indicado anteriormente, puede observarse que el término «sal farmacéuticamente aceptable» en el presente contexto se entiende como un principio activo que comprende un compuesto de fórmula (I) en
forma de una de sus sales, en particular si esta forma de sal aporta propiedades farmacocinéticas mejoradas al principio activo en comparación con la forma libre de dicho principio activo o cualquier otra forma de sal del prin cipio activo utilizado anteriormente. La forma de sal farmacéuticamente aceptable del principio activo también puede proporcionar este principio activo por primera vez con una propiedad farmacocinética deseada que no tenía antes y puede incluso tener una influencia positiva sobre la farmacodinámica de este principio activo con respecto a su eficacia terapéutica en el organismo.
Debido a su estructura molecular, los compuestos de fórmula (I) son quirales y pueden, en consecuencia, aparecer en diversas formas enantioméricas. Por tanto, pueden existir en forma racémica u ópticamente activa.
Puesto que la actividad farmacéutica de los racematos o estereoisómeros de los compuestos según la invención puede diferir, sería deseable usar los enantiómeros. En estos casos, el producto final, o incluso los productos intermedios, pueden separarse en compuestos enantioméricos por medios químicos o físicos conocidos por el experto en la materia o incluso emplearse tal cual en la síntesis.
En el caso de aminas racémicas, los diastereómeros se forman a partir de la mezcla mediante reacción con un agente de resolución ópticamente activo. Son ejemplos de agentes de resolución adecuados los ácidos óptica mente activos, como las formas (R) y (S) del ácido tartárico, ácido diacetiltartárico, ácido dibenzoiltartárico, ácido mandélico, ácido málico, ácido láctico, aminoácidos N-protegidos adecuados (p. ej., N-benzoilprolina o N-bencenosulfonilprolina) o los diversos ácidos alcanforsulfónicos ópticamente activos. También resulta ventajosa la reso lución cromatográfica de enantiómeros con la ayuda de un agente de resolución ópticamente activo (por ejemplo, dinitrobenzoilfenilglicina, triacetato de celulosa u otros derivados de hidratos de carbono o polímeros de metacrilato quiralmente derivatizados inmovilizados en gel de sílice). Los eluyentes adecuados para este objetivo son mezclas de solventes acuosos o alcohólicos como, por ejemplo hexano/isopropanol/acetonitrilo, por ejemplo en una proporción 82:15:3.
Isótopos
Se pretende además que un compuesto de fórmula (I) incluya sus formas marcadas con isótopo. Una forma mar cada con isótopo de un compuesto de fórmula (I) es idéntica a este compuesto excepto por el hecho de que uno o más átomos del compuesto se han sustituido por un átomo o átomos con una masa atómica o un número másico que difiere de la masa atómica o el número másico del átomo natural. Entre los ejemplos de isótopos que se encuentran fácilmente en el mercado y que pueden incorporarse a un compuesto de fórmula (I) mediante métodos bien conocidos se incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, por ejemplo, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36Cl, respectivamente. Se pretende que un compuesto de la fórmula (I), un profármaco del mismo o una sal farmacéuticamente aceptable de cualquiera de los dos que con tenga uno o más de los isótopos mencionados anteriormente y/u otros isótopos de otros átomos sea parte de la presente invención. Se puede usar un compuesto de fórmula (I) marcado con isótopo de diversas formas benefi ciosas. Por ejemplo, un compuesto de fórmula (I) marcado con isótopo dentro del cual se ha incorporado, por ejemplo, un radioisótopo, como 3H o 14C, es adecuado para ensayos de distribución en tejidos de medicamentos y/o sustratos. Estos radioisótopos, es decir, tritio (3H) y carbono 14 (14C), son especialmente preferidos debido a su sencilla preparación y a la excelente capacidad de detección. La incorporación de isótopos más pesados, por ejemplo deuterio (2H), a un compuesto de fórmula (I) tiene ventajas terapéuticas debido a la mayor estabilidad metabólica de este compuesto marcado con el isótopo. Una mayor estabilidad metabólica se traduce directamente en dosis más bajas o un aumento de la semivida in vivo, lo que en la mayoría de las circunstancias representaría una realización preferida de la presente invención. Un compuesto de fórmula (I) marcado con isótopo puede pre pararse habitualmente realizando los procedimientos descritos en los esquemas de síntesis y la descripción rela cionada, en la parte de ejemplos y en la parte de preparación en el presente texto, sustituyendo un reactivo no marcado con isótopo por un reactivo marcado con isótopo fácilmente disponible.
También se puede incorporar deuterio (2H) en un compuesto de fórmula (I) con el fin de manipular el metabolismo oxidativo del compuesto por medio del efecto isotópico cinético primario. El efecto isotópico cinético primario es un cambio de la velocidad de una reacción química que es consecuencia del intercambio de núcleos isotópicos, lo que a su vez está causado por el cambio en las energías del estado fundamental para la formación de enlaces covalentes después de este intercambio isotópico. El intercambio de un isótopo más pesado normalmente tiene como consecuencia una reducción de la energía del estado fundamental para un enlace químico y causa, por tanto, una reducción de la velocidad de rotura de enlaces limitantes de la velocidad. Si la rotura de enlaces se produce en o en las proximidades de una región de punto de silla a lo largo de la coordenada de una reacción multiproducto, los cocientes de distribución de productos se pueden alterar de forma sustancial. Como explicación: si el deuterio se une a un átomo de carbono en una posición no intercambiable, son típicas diferencias de velocidad de kM/kü = 2-7. Si esta diferencia de velocidad se aplica con éxito a un compuesto de fórmula (I) que es susceptible a la oxidación, el perfil de este compuesto in vivo se puede modificar considerablemente y tiene como resultado una mejora de las propiedades farmacocinéticas.
Cuando se descubren y desarrollan agentes terapéuticos, la persona experta en la materia intenta optimizar los parámetros farmacocinéticos, conservando a la vez las propiedades in vitro deseables. Es razonable asumir que muchos compuestos con malos perfiles farmacocinéticos son susceptibles del metabolismo oxidativo. Los ensa yos de microsomas hepáticos in vitro actualmente disponibles proporcionan información valiosa sobre el curso del metabolismo oxidativo de este tipo, lo que a su vez permite el diseño racional de compuestos deuterados de fórmula (I) con mejor estabilidad a través de la resistencia a dicho metabolismo oxidativo. Se obtienen de este modo mejoras significativas en los perfiles farmacocinéticos de compuestos de fórmula (I), y se pueden expresar cuantitativamente en términos de aumentos en la semivida (ti/2) in vivo, concentración en el efecto terapéutico máximo (Cmáx), área bajo la curva de dosis-respuesta (AUC) y F; y en términos de reducción del aclaramiento, dosis y costes de materiales.
La siguiente explicación está destinada a ilustrar lo anterior: un compuesto de fórmula (I) que tiene múltiples sitios potenciales de ataque para el metabolismo oxidativo, por ejemplo, átomos de hidrógeno bencílico y átomos de hidrógeno unidos a un átomo de nitrógeno, se prepara como una serie de análogos en los que diversas combina ciones de átomos de hidrógeno se sustituyen por átomos de deuterio, de modo que algunos, la mayoría o todos estos átomos de hidrógeno se han sustituido por átomos de deuterio. Las determinaciones de la semivida permiten la determinación favorable y precisa del grado al cual ha mejorado la resistencia al metabolismo oxidativo. De este modo, se determina que la semivida del compuesto original se puede extender hasta el 100 % como consecuencia de un intercambio de hidrógeno-deuterio de este tipo.
El intercambio de hidrógeno-deuterio en un compuesto de fórmula (I) también se puede usar para conseguir una modificación favorable del espectro del metabolito del compuesto de partida con el fin de disminuir o eliminar los metabolitos tóxicos no deseados. Por ejemplo, si aparece un metabolito tóxico por escisión oxidativa del enlace carbono-hidrógeno (C-H), puede ser razonable asumir que el análogo deuterado disminuirá o eliminará en gran medida la producción del metabolito no deseado, incluso si la oxidación en particular no es un paso determinante de la velocidad. Se puede encontrar más información sobre el estado de la técnica con respecto al intercambio de hidrógeno-deuterio por ejemplo en Hanzlik y cols., J. Org. Chem. 55, 3992-3997, 1990, Reider y cols., J. Org. Chem. 52, 3326-3334, 1987, Foster, Adv. Drug Res. 14, 1-40, 1985, Gillette y cols., Biochemistry 33(10) 2927 2937, 1994, y Jarman y cols. Carcinogenesis 16(4), 683-688, 1993.
La presente invención se refiere a una formulación farmacéutica (preferiblemente para su uso en el tratamiento de una anomalía inmunorreguladora o un cáncer) que comprende al menos un compuesto de fórmula (I) (espe cialmente una cantidad terapéuticamente eficaz de un compuesto de fórmula (I)), y/o un solvato, tautómero, oligómero o estereoisómero del mismo, así como una sal farmacéuticamente aceptable de cada uno de los anterio res, incluidas sus mezclas en todas las proporciones, como principio activo, junto con un vehículo farmacéutica mente aceptable.
A los fines de la presente invención, el término «formulación farmacéutica» se refiere a una composición o pro ducto que comprende uno o más principios activos, y uno o más excipientes que constituyen el vehículo, así como cualquier producto que sea el resultado, directo o indirecto, de la combinación, formación de complejos o agrega ción de cualesquiera dos o más de los componentes, o de la disociación de uno o más de los componentes, o de otros tipos de reacciones o interacciones de uno o más de los componentes. Por consiguiente, las formulaciones farmacéuticas de la presente invención abarcan cualquier composición obtenida mezclando al menos un com puesto de la presente invención y un transportador, excipiente o vehículo farmacéuticamente aceptable.
Las formulaciones farmacéuticas de la presente invención también abarcan cualquier composición que además comprende un segundo principio activo y/o un solvato del mismo, así como una sal farmacéuticamente aceptable de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones, en la que el segundo principio activo es distinto de un compuesto de fórmula (I) donde todos los restos se definen como anteriormente.
Las formulaciones farmacéuticas según la presente invención pueden usarse como medicamentos en medicina humana y veterinaria.
Para los fines de la presente invención, una anomalía inmunorreguladora es preferiblemente una enfermedad inflamatoria crónica o autoinmune seleccionada entre el grupo compuesto por: lupus eritematoso sistémico, artritis reumatoide crónica, enfermedad inflamatoria intestinal, esclerosis múltiple, esclerosis lateral amiotrófica (ELA), ateroesclerosis, escleroderma, hepatitis autoinmune, síndrome de Sjogren, nefritis lúpica, glomerulonefritis, artritis reumatoide, psoriasis, miastenia grave, nefropatía por inmunoglobulina A, vasculitis, rechazo de trasplante, miositis, púrpura de Henoch-Schonlein y asma; el cáncer es preferiblemente una neoplasia maligna hematológica o un tumor sólido, donde la neoplasia maligna hematológica es preferiblemente una enfermedad seleccionada entre el grupo compuesto por linfoma no Hodgkin de células B y T/NK malignas, como: mieloma múltiple, linfoma de células del manto, linfoma difuso de células B grandes, plasmocitoma, linfoma folicular, inmunocitoma, leucemia linfoblástica aguda, leucemia linfocítica crónica y leucemia mieloide; y donde el tumor sólido es preferiblemente una enfermedad seleccionada entre el grupo compuesto por: cáncer inflamatorio de mama, hígado y colon, cáncer
de pulmón, cáncer de cabeza y cuello, cáncer de próstata, cáncer de páncreas, cáncer de vejiga, cáncer renal, cáncer hepatocelular y cáncer gástrico.
Las formulaciones farmacéuticas pueden administrarse en forma de unidades de dosis, que comprenden una cantidad predeterminada de principio activo por unidad de dosis. Dicha unidad puede comprender, por ejemplo, de 0,5 mg a 1 g, preferiblemente de 1 mg a 700 mg, en especial preferiblemente de 5 mg a 100 mg, de un com puesto según la invención, dependiendo de la enfermedad tratada, el método de administración y la edad, peso y estado del paciente, o las formulaciones farmacéuticas se pueden administrar en forma de unidades de dosis que comprendan una cantidad predeterminada de principio activo por unidad de dosis. Las formulaciones de unidad de dosis preferidas son aquellas que comprenden una dosis diaria, o parte de la dosis, como se indica anterior mente, o una fracción correspondiente de la misma de un principio activo. Además, las formulaciones farmacéu ticas de este tipo pueden prepararse usando un proceso, que generalmente es conocido en la técnica farmacéu tica.
Las formulaciones farmacéuticas pueden adaptarse para su administración mediante cualquier método adecuado deseado, por ejemplo, mediante métodos orales (incluyendo bucal o sublingual), rectales, nasales, tópicos (inclu yendo bucal, sublingual o transdérmico), vaginales o parenterales (incluyendo subcutáneo, intramuscular, intrave noso o intradérmico). Estas formulaciones pueden prepararse usando todos los procesos conocidos en la técnica farmacéutica mediante, por ejemplo, combinación del principio activo con excipientes o adyuvantes.
Las formulaciones farmacéuticas adaptadas para la administración oral pueden administrarse como unidades in dependientes como, por ejemplo, cápsulas o comprimidos; polvos o gránulos; soluciones o suspensiones en lí quidos acuosos o no acuosos; espumas comestibles o alimentos en forma de espuma; o emulsiones líquidas de aceite en agua o emulsiones líquidas de agua en aceite.
Por tanto, por ejemplo, en el caso de administración oral en forma de comprimido o cápsula, el principio activo puede combinarse con un excipiente inerte, oral, no tóxico y farmacéuticamente aceptable, como por ejemplo, etanol, glicerol, agua y similar. Los polvos se preparan triturando el compuesto hasta un tamaño fino adecuado y mezclándolo con un excipiente farmacéutico triturado de forma similar, como por ejemplo, un hidrato de carbono comestible, como por ejemplo, almidón o manitol. También pueden estar presente un agente aromatizante, un conservante, un dispersante y un colorante.
Las cápsulas se producen preparando una mezcla en polvo como se describe anteriormente y rellenando el en voltorio de gelatina conformado con la mezcla. Antes de la operación de llenado pueden añadirse a la mezcla en polvo agentes deslizantes y lubricantes, como por ejemplo, ácido silícico altamente disperso, talco, estearato de magnesio, estearato de calcio o polietilenglicol en forma sólida. Del mismo modo, puede añadirse un agente de sintegrante o solubilizante, como por ejemplo, agar-agar, carbonato de calcio o carbonato sódico, para mejorar la disponibilidad del medicamento después de que se haya tomado la cápsula.
Además, si se desea o es necesario, pueden incorporarse también a la mezcla agentes aglutinantes, lubricantes y desintegrantes adecuados, así como colorantes. Entre los aglutinantes idóneos se incluyen almidón, gelatina, azúcares naturales, como por ejemplo, glucosa o beta-lactosa, edulcorantes a base de maíz, caucho natural y sintético, como por ejemplo, de acacia, tragacanto o alginato sódico, carboximetilcelulosa, polietilenglicol, ceras y similares. Entre los agentes lubricantes utilizados en estas formas farmacéuticas se incluyen oleato sódico, estea rato sódico, estearato de magnesio, benzoato sódico, acetato sódico, cloruro sódico y similares. Entre los agentes desintegrantes se incluyen, sin restricciones, almidón, metilcelulosa, agar, bentonita, goma de xantano y similares. Los comprimidos se formulan, por ejemplo, preparando una mezcla en polvo, granulando o prensando en seco la mezcla, añadiendo un lubricante y un desintegrante y prensando la mezcla completa para obtener los comprimi dos. Se prepara una mezcla en polvo mezclando el compuesto triturado de forma adecuada con un diluyente o una base, como se describe anteriormente, y opcionalmente con un aglutinante, como por ejemplo, carboximetil celulosa, un alginato, gelatina o polivinilpirrolidona, un retardante de disolución, como por ejemplo, parafina, un acelerador de la absorción, como por ejemplo, una sal cuaternaria, y/o un absorbente, como por ejemplo, bento nita, caolina o fosfato dicálcico. La mezcla en polvo puede granularse humedeciéndola con un aglutinante, como por ejemplo, sirope, pasta de almidón, mucílago de acadia o soluciones de celulosa o materiales poliméricos y haciéndola pasar a través de un tamiz. Como alternativa a la granulación, la mezcla en polvo puede hacerse pasar a través de una máquina de comprimidos, dando lugar a trozos de forma no uniforme que se rompen para formar los gránulos. Los gránulos pueden lubricarse mediante la adición de ácido esteárico, una sal de estearato, talco o aceite mineral para evitar que se pegue a los moldes de vaciado de comprimidos. La mezcla con lubricante se prensa a continuación para obtener los comprimidos. Los principios activos pueden también combinarse con un excipiente inerte de flujo libre y, a continuación, prensarse directamente para obtener los comprimidos sin realizar las etapas de granulación o prensado en seco. Puede haber una capa protectora transparente u opaca compuesta por una capa de sellado de laca shellac, una capa de azúcar o material polimérico y una capa brillante de cera. Pueden añadirse colorantes a estos recubrimientos para diferenciar entre distintas unidades de dosis.
Pueden prepararse líquidos orales, como por ejemplo, soluciones, jarabes y elixires, en forma de unidades de dosis, de modo que una cantidad determinada comprenda una cantidad preespecificada de los compuestos. Los jarabes pueden prepararse disolviendo los compuestos en una solución acuosa con un aromatizante adecuado, mientras que los elixires se preparan usando un vehículo alcohólico no tóxico. Las suspensiones pueden formu larse mediante dispersión de los compuestos en un vehículo no tóxico. Pueden, asimismo, añadirse solubilizantes y emulsionantes, como por ejemplo, alcoholes de isostearilo etoxilados y éteres de polioxietilensorbitol, conser vantes, aditivos aromatizantes, como por ejemplo, aceite de menta o edulcorantes naturales o sacarina, u otros aromatizantes artificiales y similares.
Las formulaciones de unidad de dosis para administración oral pueden, si se desea, encapsularse en microcápsulas. La formulación también puede prepararse de manera que se prolongue o retrase la liberación, por ejemplo, recubriendo o incluyendo el material particulado en polímeros, cera y similares.
Los compuestos de fórmula (I) y sales, solvatos y derivados fisiológicamente funcionales de los mismos y los demás principios activos pueden también administrarse en forma de sistemas de administración de liposomas, como por ejemplo, vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas pueden formarse a partir de varios fosfolípidos como, por ejemplo, colesterol, estearilamina y fosfatidilcolinas.
Los compuestos de fórmula (I) y sus sales, solvatos y los demás principios activos también pueden administrarse usando anticuerpos monoclonales como vehículos individuales a los que están acopladas las moléculas del com puesto. Los compuestos también pueden acoplarse con polímeros solubles como vehículos que dirigen el medi camento. Estos polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropil-metacrilamidofenol, polihidroxietilaspartamido-fenol u óxido de polietileno-polilisina, sustituidos por radicales palmitoílo. Los compuestos pueden además estar conjugados con una clase de polímeros biodegradables que son adecuados para conseguir la liberación controlada de un medicamento, por ejemplo, ácido poliláctico, poliépsilon-caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidroxipiranos, policianoacrilatos y copolímeros de hidrogeles entrecruzados o de bloque anfipáticos.
Las formulaciones farmacéuticas adaptadas para administración transdérmica pueden administrarse como yesos independientes para un contacto próximo y prolongado con la epidermis del receptor. Por tanto, por ejemplo, el principio activo puede administrarse a partir del yeso mediante iontoforesis, como se describe en términos gene rales en Pharmaceutical Research, 3(6), 318 (1986).
Los compuestos farmacéuticos adaptados para la administración tópica pueden formularse como pomadas, cre mas, suspensiones, lociones, polvos, soluciones, pastas, geles, pulverizadores, aerosoles o aceites.
Para el tratamiento de los ojos u otros tejidos externos, por ejemplo, la boca y la piel, las formulaciones se aplican preferiblemente como una pomada o crema tópica. En el caso de la formulación para obtener una pomada, el principio activo puede emplearse con una base de crema parafínica o miscible con agua. Alternativamente, el principio activo puede formularse para obtener una crema con una base de crema de aceite en agua o una base de agua en aceite.
Las formulaciones farmacéuticas adaptadas para aplicación tópica en los ojos incluyen colirios, en los que el principio activo se disuelve o suspende en un vehículo adecuado, en particular, un solvente acuoso.
Las formulaciones farmacéuticas adaptadas para aplicación tópica en la boca incluyen pastillas para chupar, pas tillas y colutorios.
Las formulaciones farmacéuticas adaptadas para la administración rectal pueden administrarse en forma de su positorios o enemas.
Las formulaciones farmacéuticas adaptadas para administración nasal en las que la sustancia vehículo es un sólido comprenden un polvo grueso con un tamaño de partícula, por ejemplo, en el intervalo de 20 a 500 micrómetros, que se administra de manera que se aspira, es decir, mediante inhalación rápida a través de las fosas nasales a partir de un recipiente que contiene el polvo mantenido cerca de la nariz. Las formulaciones adecuadas para su administración como aerosol nasal o gotas nasales con un líquido como sustancia vehículo incluyen so luciones de principios activos en agua o aceite.
Las formulaciones farmacéuticas adaptadas para administración mediante inhalación incluyen vapores o polvos finamente particulados, que pueden generarse mediante diversos tipos de dispensadores presurizados con aero soles, nebulizadores o insufladores.
Las formulaciones farmacéuticas adaptadas para la administración vaginal pueden administrarse como óvulos vaginales, tampones, cremas, geles, pastas, espumas o aerosol.
Entre las formulaciones farmacéuticas adaptadas para administración parenteral se incluyen soluciones acuosas y no acuosas estériles para inyección que comprenden antioxidantes, tampones, bacteriostáticos y solutos, me diante las cuales la formulación se hace isotónica con la sangre del receptor que se va a tratar, y suspensiones acuosas y no acuosas estériles, que pueden comprender medios de suspensión y espesantes. Las formulaciones pueden administrarse en recipientes de dosis única o multidosis, por ejemplo, en ampollas y viales sellados, y conservarse liofilizadas, de modo que solo sea necesaria la adición del líquido vehículo estéril, por ejemplo agua para inyección, inmediatamente antes de que sea necesario su uso.
Las soluciones y suspensiones para inyección preparadas según la fórmula pueden prepararse a partir de polvos, gránulos y comprimidos estériles.
Resulta evidente que, además de los constituyentes especialmente mencionados anteriormente, las formulacio nes también pueden comprender otros agentes normales en la técnica con respecto al tipo de formulación en particular; por tanto, por ejemplo, las formulaciones que son adecuadas para la administración oral pueden com prender aromatizantes.
Las composiciones/formulaciones farmacéuticas según la invención pueden usarse como medicamentos en me dicina humana y veterinaria.
Una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) y del otro principio activo depende de varios factores, como por ejemplo, la edad y el peso del animal, la enfermedad precisa que requiere tratamiento y su gravedad, la naturaleza de la formulación y el método de administración, y la determina finalmente el médico o veterinario responsable del tratamiento. Sin embargo, una cantidad eficaz de un compuesto está generalmente en el intervalo de 0,1 a 100 mg/kg de peso corporal del receptor (mamífero) al día y, en particular, normalmente en el intervalo de 1 a 10 mg/kg de peso corporal al día. Por tanto, la cantidad real al día para un mamífero adulto que pesa 70 kg normalmente está entre 70 y 700 mg, donde esta cantidad puede administrarse como una dosis individual al día o normalmente en una serie de dosis divididas (como, por ejemplo, dos, tres, cuatro, cinco o seis) al día, de modo que la dosis total diaria sea la misma. Una cantidad eficaz de una sal o solvato o de un derivado fisiológicamente funcional de los mismos se puede determinar como la fracción de la cantidad efectiva del com puesto per se.
La invención además se refiere a un compuesto según la fórmula (I) o cualquier realización específica descrita anteriormente y/o los solvatos, tautómeros, oligómeros o estereoisómeros del mismo, así como las sales farma céuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones, para su uso en la prevención y/o tratamiento de enfermedades que se ven afectadas por la inhibición de LMP7.
La invención se refiere a un compuesto según la fórmula (I) o cualquier realización específica descrita anterior mente y/o sus solvatos, tautómeros, oligómeros o estereoisómeros, así como las sales farmacéuticamente acep tables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones, para su uso en el trata miento y/o profilaxis (prevención) de una anomalía inmunorreguladora o cáncer (incluidas en particular neoplasias malignas hematológicas y tumores sólidos).
En la presente invención además se describe un método para tratar a un sujeto que padece una anomalía inmu norreguladora o un cáncer, que comprende la administración a dicho sujeto de un compuesto de fórmula (I) en una cantidad que es eficaz para tratar dicha anomalía inmunorreguladora o cáncer. En la presente invención en particular se describe un método para tratar a un sujeto que sufre una enfermedad inflamatoria crónica o autoinmune, una neoplasia maligna hematológica o un tumor sólido.
Los compuestos divulgados de fórmula (I) se pueden administrar y/o usar en combinación con otros agentes terapéuticos conocidos (principios activos), incluidos agentes antineoplásicos. Como se usa aquí, el término «agente antineoplásico» se refiere a cualquier agente que se administre a un paciente con cáncer para los fines de tratamiento del cáncer. El
El tratamiento antineoplásico definido anteriormente se puede aplicar como monoterapia o puede implicar, ade más de los compuestos de fórmula (I) descritos en este documento, cirugía convencional o radioterapia o trata miento farmacológico. Dicho tratamiento farmacológico, por ejemplo, una quimioterapia o una terapia dirigida, puede incluir uno o más, pero preferiblemente uno, de los siguientes agentes antitumorales:
Agentes alquilantes
como altretamina, bendamustina, busulfán, carmustina, clorambucilo, clormetina, ciclofosfamida, dacarbazina, ifosfamida, improsulfán, tosilato, lomustina, melfalán, mitobronitol, mitolactol, nimustina, ranimustina, temozolomida, tiotepa, treosulfán, mecloretamina, carbocuona,
apazicuona, fotemustina, glufosfamida, palifosfamida, pipobromán, trofosfamida, uramustina, TH-3024, VAL-0834. Compuestos de platino
como carboplatino, cisplatino, eptaplatino, hidrato de miriplatino, oxaliplatino, lobaplatino, nedaplatino, picoplatino, satraplatino.
Agentes que alteran el ADN
como amrubicina, bisantreno, decitabina, mitoxantrona, procarbazina, trabectedina, clofarabina, amsacrina, brostalicina, pixantrona, laromustina13.
Inhibidores de la topoisomerasa
como etopósido, irinotecán, razoxano, sobuzoxano, tenipósido, topotecán,
amonafida, belotecán, acetato de eliptinio, voreloxina.
Modificadores de microtúbulos
como cabazitaxel, docetaxel, eribulina, ixabepilona, paclitaxel, vinblastina, vincristina, vinorelbina, vindesina, vinflunina,
fosbretabulina, tesetaxel.
Antimetabolitos
como asparaginasa3, azacitidina, levofolinato de calcio, capecitabina, cladribina, citarabina, enocitabina, floxuridina, fludarabina, fluorouracilo, gemcitabina, mercaptopurina, metotrexato, nelarabina, pemetrexed, pralatrexato, azatioprina, tioguanina, carmofur; doxifluridina, elacitarabina, raltitrexed, sapacitabina, tegafur23, trimetrexato. Antibióticos antineoplásicos
como bleomicina, dactinomicina, doxorubicina, epirubicina, idarubicina, levamisol, miltefosina, mitomicina C, romidepsina, estreptozocina, valrubicina, zinostatina, zorubicina, daunorubicina, plicamicina; aclarubicina, peplomicina, pirarubicina.
Hormonas/antagonistas
como abarelix, abiraterona, bicalutamida, buserelina, calusterona, clorotrianiseno, degarelix, dexametasona, estradiol, fluocortolona; fluoximesterona, flutamida, fulvestrant, goserelina, histrelina, leuprorelina, megestrol, mitotano, nafarelina, nandrolona, nilutamida, octreotida, prednisolona, raloxifeno, tamoxifeno, tirotropina alfa, toremifeno, trilostano, triptorelina, dietilestilbestrol;
acolbifeno, danazol, deslorelina, epitiostanol, orteronel, enzalutamida13.
Inhibidores de la aromatasa
como aminoglutetimida, anastrozol, exemestano, fadrozol, letrozol, testolactona; formestano.
Moléculas pequeñas inhibidoras de quinasas
como crizotinib, dasatinib, erlotinib, imatinib, lapatinib, nilotinib, pazopanib, regorafenib, ruxolitinib, sorafenib, sunitinib, vandetanib, vemurafenib, bosutinib, gefitinib, axitinib,
afatinib, alisertib, dabrafenib, dacomitinib, dinaciclib, dovitinib, enzastaurin, nintedanib, lenvatinib, linifanib, linsitinib, masitinib, midostaurin, motesanib, neratinib, orantinib, perifosine, ponatinib, radotinib, rigosertib, tipifarnib, tivantinib, tivozanib, trametinib, pimasertib, brivanib alaninato, cediranib, apatinib4, cabozantinib S-malato13, ibrutinib13, icotinib4, buparlisib2, cipatinib4, cobimetinib13 idelalisib13 fedratinib1, XL-6474.
Fotosensibilizadores
como metoxsaleno3, porfímero de sodio, talaporfina, temoporfina.
Anticuerpos
como alemtuzumab, besilesomab, brentuximab vedotina, cetuximab, denosumab, ipilimumab, ofatumumab, panitumumab, rituximab, tositumomab, trastuzumab, bevacizumab, pertuzumab23, catumaxomab, elotuzumab, epratuzumab, farletuzumab, mogamulizumab, necitumumab, nimotuzumab, obinutuzumab, ocaratuzumab, oregovomab, ramucirumab, rilotumumab, siltuximab, tocilizumab, zalutumumab, zanolimumab, matuzumab, dalotuzumab123, onartuzumab13, racotumomab1, tabalumab13, EMD-5257974, nivolumab13.
Citoquinas
como aldesleuquina, interferón alfa2, interferón alfa2a3, interferón alfa2b23, celmoleuquina, tasonermina, teceleuquina, oprelvequina13, interferón beta-1a recombinante4.
Fármacos conjugados
como denileuquina diftitox, ibritumomab tiuxetán, iobenguano 123I, prednimustina, trastuzumab emtansina, estramustina, gemtuzumab, ozogamicina, aflibercept; cintredequina besudotox, edotreotida, inotuzumab ozogamicina, naptumomab estafenatox, oportuzumab monatox, tecnecio (99mTc) arcitumomab13 vintafolida13 Vacunas
como sipuleucel3; vitespen3, emepepimut-S3, oncoVAX4, rindopepimut3, troVax4, MGN-16014, MGN-17034. Varios
alitretinoína, bexaroteno, bortezomib, everolimús, ácido ibandrónico, imiquimod, lenalidomida, lentinano, metirosina, mifamurtida, ácido pamidrónico, pegaspargasa, pentostatina, sipuleucel3, sizofirano, tamibaroteno, temsirolimús, talidomida, tretinoína, vismodegib, ácido zoledrónico, vorinostat, celecoxib, cilengitida, entinostat, etanidazol, ganetespib, idronoxilo, iniparib, ixazomib, lonidamina, nimorazol, panobinostat, peretinoína, plitidepsina, pomalidomida, procodazol, ridaforolimús, tasquinimod, telotristat, timalfasina, tirapazamina, tosedostat, trabedersen, ubenimex, valspodar, gendicina4, picibanil4, reolisina4, clorhidrato de retaspimicina13, trebananib23, virulizina4, carfilzomib13, endostatina4, immucothel4, belinostat3, MGN-17034.
1 DCI Prop. (Denominación Común Internacional Propuesta)
2 DCI Rec. (Denominación Común Internacional Recomendada)
3 USAN (United States A dopted Name [Nombre adoptado en Estados Unidos])
4 no DCI.
Se divulga el uso de compuestos de fórmula (I) y formulas relacionadas en combinación con al menos un principio activo de medicamento adicional, preferiblemente los medicamentos utilizados en el tratamiento de la esclerosis múltiple, como cladribina u otros coagentes, como interferón, por ejemplo, interferones pegilados o no pegilados, preferiblemente interferón beta y/o con compuestos que mejoran la función vascular o en combinación con fárma cos inmunomoduladores, por ejemplo, fingolimod; ciclosporinas, rapamicinas o ascomicinas, o sus análogos inmunodepresores, por ejemplo, ciclosporina A, ciclosporina G, FK-506, ABT-281, ASM981, rapamicina, 40-O-(2-hidroxi)etil-rapamicina, etc.; corticoesteroides; ciclofosfamida; azatiopreno; metotrexato; leflunomida; mizoribina; ácido micofenólico, micofenolato de mofetilo; 15-deoxispergualina; valerato de diflucortolona; difluprednato; dipropionato de alclometasona; amcinonida; amsacrina; asparaginasa; azatioprina; basiliximab; dipropionato de beclometasona; betametasona; acetato de betametasona; dipropionato de betametasona; fosfato sódico de betametasona; valerato de betametasona; budesonida; captoprilo; clorhidrato de clormetina; cladribina; propionato de clobetasol; acetato de cortisona; cortivazol; ciclofosfamida; citarabina; daclizumab; dactinomicina; desonida; desoximetasona; dexametasona; acetato de dexametasona; isonicotinato de dexametasona; metasulfobenzoato sódico de dexametasona; fosfato de dexametasona; tebutato de dexametasona; acetato de diclorisona; clorhidrato de
doxorubicina; clorhidrato de epirubicina; acetonida de fluclorolona; acetato de fludrocortisona; fludroxicortida; pivalato de flumetasona; flunisolida; acetonida de fluocinolona; fluocinonida; fluocortolona; hexanoato de fluocortolona; pivalato de fluocortolona; fluorometolona; acetato de fluprednideno; propionato de fluticasona; clorhidrato de gemcitabina; halcinonida; hidrocortisona, acetato de hidrocortisona, butirato de hidrocortisona, hemisuccinato de hidrocortisona; melfalán; meprednisona; mercaptopurina; metilprednisolona; acetato de metilprednisolona; hemi succinato de metilprednisolona; misoprostol; muromonab-cd3; micofenolato de mofetilo; acetato de parametasona; prednazolina, prednisolona; acetato de prednisolona; caproato de prednisolona; metasulfobenzoato sódico de prednisolona; fosfato sódico de prednisolona; prednisona; prednilideno; rifampicina; rifampicina sódica; tacrolimús; teriflunomida; talidomida; tiotepa; pivalato de tixocortol; triamcinolona; hemisuccinato acetónido de triamcinolona; benetónido de triamcinolona; diacetato de triamcinolona; hexacetónido de triamcinolona; anticuerpos monoclonales inmunodepresores, por ejemplo, anticuerpos monoclonales frente a receptores de leucocitos, como MHC, CD2, CD3, Cd 4, CD7, c D25, Cd 28, B7, CD40, CD45 o CD58, o sus ligandos; u otros compuestos inmunomoduladores, por ejemplo CTLA41g, u otros inhibidores de moléculas de adhesión, por ejemplo AcM o inhibi dores de bajo peso molecular como antagonistas de selectinas y antagonistas de VLA-4. Una composición preferida es con ciclosporina A, FK506, rapamicina o 40-(2-hidroxi)etil-rapamicina y fingolimod. Estos medicamentos adicionales, como el interferón beta, pueden administrarse concomitante o secuencialmente, por ejemplo, a través de las vías subcutánea, intramuscular u oral.
Se describe el uso de compuestos de fórmula (I) y fórmulas relacionadas en combinación con al menos un com puesto activo de un medicamento adicional, preferiblemente medicamentos utilizados en el tratamiento del cáncer (como en particular, los agentes anticancerosos y/o antitumorales descritos anteriormente).
La presente invención también se refiere a un set (kit) compuesto de envases independientes de
(a) una cantidad eficaz de un compuesto de la fórmula (I) y/o un solvato, tautómero, oligómero o estereoisómero del mismo, así como una sal farmacéuticamente aceptable de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
y
(b) una cantidad eficaz de un principio activo adicional de un medicamento.
Los compuestos de la presente invención pueden prepararse según los procedimientos de los siguientes esque mas y ejemplos, usando materiales apropiados y se ejemplifican adicionalmente mediante los siguientes ejemplos específicos.
Además, mediante la utilización de los procedimientos descritos en la presente memoria, junto con los expertos en la técnica, se pueden preparar fácilmente compuestos adicionales de la presente invención reivindicados en este documento. Sin embargo, los compuestos ilustrados en los ejemplos no deben interpretarse como que forman el único género que se considera como la invención. Los ejemplos ilustran adicionalmente detalles para la preparación de los compuestos de la presente invención. Los expertos en la técnica entenderán fácilmente que pueden utilizarse variaciones conocidas de las condiciones y procesos de los siguientes procedimientos preparativos para preparar estos compuestos.
Los materiales de partida para la preparación de compuestos de la presente invención pueden prepararse me diante métodos como los que se describen en los ejemplos o mediante métodos conocidos per se, según se describe en la literatura sobre química orgánica sintética y conocidos por el experto, o pueden obtenerse en el mercado.
La síntesis de compuestos de fórmula (IV) se describe en los documentos WO 2016/050356, WO 2016/050355, WO 2016/050359 y WO 2016/050358.
Ejemplos
CL/EM:
Método A: Agilización 70108359 - Chromolith Speed Rod RP18e 50-4,6 mm; polar.m; 2,4 ml/min; 220 nm; tampón A: HCOOH al 0,05 %/H2O, tampón B: HCOOH al 0,04 %/ACN; 0,0-2,8 min 4-100 % de tampón B; 2,8-3,3 min 100 % de tampón B; 3,3-3,4 min 100-4 % de tampón B.
Método B: Waters XBrigde C83,5 |jm; 4,6 * 50 mm; EliteLa Chrom 70173815; 8,1 min; 2 ml/min; 215 nm; tampón A: TFA al 0,05 %/H2O, tampón B: TFA al 0,04 %/ACN; 0,0-0,2 min 5 % de tampón B; 0,2-8,5 min 5-100 % de tampón B; 8,5-10,0 min 99-5 % de tampón.
TR: Tiempo de retención.
La invención se ilustrará, pero sin limitaciones, en referencia a las realizaciones específicas descritas en los si guientes ejemplos. Siempre que no se indique lo contrario en los esquemas, las variables tienen el mismo signifi cado que el descrito anteriormente.
Siempre que no se especifique lo contrario, todos los materiales de partida se obtienen a partir de proveedores comerciales y se utilizan sin purificación adicional. Siempre que no se especifique lo contrario, todas las temperaturas se expresan en °C y todas las reacciones se realizan a TA. Los compuestos se pueden purificar mediante métodos comunes como, en particular, cromatografía en columna de sílice o HPLC preparativa.
A menos que se establezca otra cosa, todas las estructuras indicadas a continuación, en las que se indica la estereoquímica específica, se refieren a mezclas de los estereoisómeros.
Compuesto intermedio 1:

Etapa 1: 2-(2,4-dimetil-bencil)-4,4,5,5-tetrametil-[1,3,2]dioxaborolano
A una solución de 1-bromometil-2,4-dimetil-benceno (25,00 g; 114,40 mmol; 1,00 eq.) en dioxano degaseado (250,00 ml) se añaden bis(pinacolato)diboro (35,21 g; 137,28 mmol; 1,20 eq.), K2CO3 seco (47,91 g; 343,19 mmol; 3,00 eq.) y tetrakis(trifenilfosfina)paladio(0) (6623 mg; 5,72 mmol; 0,05 eq.). A continuación, la mezcla de reacción se calienta a 100 °C bajo atmósfera de nitrógeno durante 16 h. La mezcla de reacción se diluye con diclorometano y se pasa a través de celite. El filtrado se concentra. El residuo se disuelve en acetato de etilo y se lava con salmuera. La capa orgánica se seca sobre Na2SO4 anhidro, se filtra y se concentra. El material sin procesar se purifica mediante cromatografía en columna utilizando acetato de etilo al 1 % en éter de petróleo para obtener 2 (2,4-dimetil-bencil)-4,4,5,5-tetrametil-[1,3,2]dioxaborolano (11,50 g; 37,84 mmol; 33,1 %) como un líquido inco loro.
RMN 1H (400 MHz, CDCla) 57,04-7,02 (m, 1H), 6,95-6,93 (m, 1H), 6,92-6,90 (m, 1H), 2,28 (s, 3H), 2,25 (s, 3H), 2,23 (s, 2H), 1,24 (s, 12H).
Etapa 2: (1S,2S,6R,8S)-4-(2,4-dimetN-bencN)-2,9,9-trimetM-3,5-dioxa-4-bora-tricid o[6.1.1.02'6]decano
A una solución enfriada en hielo de 2-(2,4-dimetil-bencil)-4,4,5,5-tetrametil-[1,3,2]dioxaborolano (24,00 g; 79,3 mmol; 1,0 eq.) en éter dietílico (240,00 ml) bajo atmósfera de nitrógeno se añade (1S,2S,3R,5S)-2,6,6-trimetil-bicido[3.1.1]heptano-2,3-diol (20,68 g; 119,07 mmol; 1,50 eq.) y la mezcla de reacción se agita a TA durante 14 h. El análisis mediante TLC mostró la finalización de la reacción. La mezcla de reacción se lava con salmuera. La capa orgánica se seca sobre Na2SO4 anhidro y se concentra. El material sin procesar se purifica mediante cromatografía ultrarrápida en columna utilizando acetato de etilo al 2 % en éter de petróleo para obtener (1S,2S,6R,8S)-4-(2,4-dimetil-bencil)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (28,00 g; 82,96 mmol; 90,0 %) como un aceite incoloro.
RMN 1H (400 MHz, CDCb): 57,05-7,03 (m, 1H), 6,95-6,94 (m, 1H), 6,92-6,90 (m, 1H), 4,27-4,25 (m, 1H), 2,33 2,30 (m, 9H), 2,27-2,17 (m, 1H), 2,05 (t, J = 5,76 Hz, 1H), 1,90-1,89 (m, 1H), 1,84-1,80 (m, 1H), 1,38 (s, 3H), 1,28 (s, 3H), 1,11-1,09 (m, 1H), 0,91 (s, 3H)
CG/EM: m/z: 298,3.
Etapa 3: (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(2,4-dimetil-fenil)-etil]-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1.026]decano
Se toma diclorometano (37,33 ml; 583,45 mmol; 3,00 eq.) en tetrahidrofurano (140,00 ml) en un matraz RB (ma traz de fondo redondo) a presión positiva de nitrógeno y se enfría a -99 °C usando una mezcla de nitrógeno líquido y etanol. A esta se añade n-butilo de litio (1,6 M en THF) (133,71 ml; 213,93 mmol) gota a gota por las paredes laterales del matraz RB (a una velocidad media, la adición llevó aproximadamente 35 min) de modo que la tem peratura interna se mantenga entre -92 °C y -102 °C. Tras la adición, la mezcla de reacción se agita durante 25 minutos. Durante el transcurso de la reacción se forma un precipitado de color blanco (la temperatura interna se mantiene entre -90 °C y -96 °C). A continuación se añade una solución de (1S,2S,6R,8S)-4-(2,4-dimetil-bencil)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (58,00 g; 194,5 mmol) en THF (300,00 ml) gota a gota por las paredes laterales del matraz RB (aproximadamente 40 min) de modo que la temperatura interna se mantiene entre -94 °C y -100 °C. Tras la adición, la mezcla de reacción se agita durante 10 min. A continuación, se añade cloruro de cinc (0,5 M en THF) (388,97 ml; 194,48 mmol) gota a gota por las paredes laterales del matraz RB (a una velocidad media, la adición llevó aproximadamente 35 min) de modo que la temperatura interna se mantiene entre -94 °C y -99 °C. A continuación, se deja que la mezcla de reacción alcance lentamente los 20 °C y se agita a 20 °C durante 2,5 h. Se procesa una alícuota de la mezcla de reacción y se analiza mediante RMN 1H que mostró la finalización de la reacción. La mezcla de reacción se concentra (temperatura del baño 30 °C). El residuo se reparte entre éter dietílico y solución saturada de NH4CL La capa orgánica se seca sobre Na2SO4 anhidro y se concentra (temperatura del baño 30 °C) para obtener (1S,2S,6R,8s)-4-[(S)-1-cloro-2-(2,4-dimetil-fenil)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (75,70 g; 154,83 mmol; 79,6 %) como un sólido de color blanco.
RMN 1H (400 MHz, CDCla): □ 7,12 (d, J = 7,64 Hz, 1H), 6,98 (s, 1H), 6,96 (d, J = 7,68 Hz, 1H), 4,38-4,36 (m, 1H), 3,67-3,62 (m, 1H), 3,18-3,11 (m, 2H), 2,40-2,36 (m, 2H), 2,32 (s, 3H), 2,30 (s, 3H), 2,23-2,20 (m, 1H), 2,08 (t, J = 5,96 Hz, 1H), 1,93-1,87 (m, 2H), 1,36 (s, 3H), 1,30 (s, 3H), 1,14-1,11 (m, 1H), 0,84 (s, 3H). 7.18-7.08 (m, 5H), 4.37 (dd, J = 1.32, 8.74 Hz, 1H), 3.77-3.75 (m, 1H), 3.67-3.63 (m, 1H), 3.19-3.17 (m, 1H), 3.10-3.08 (m, 1H), 2.36-2.31 (m, 5H), 2.09 (t, J = 5.84 Hz, 1H), 1.93-1.86 (m, 4H), 1.39 (s, 3H), 1.30 (s, 3H), 1.13-1.10 (m, 1H), 0.84 (s, 3H).
CG/EM: m/z: 346,3.
Etapa 4: (1S,2S,6R,8S)-4-[(R)-2-(2,4-dimetil-fenil)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-d ioxa-4-bora-triciclo[6.1.1.02,6]decano
Una solución de (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(2,4-dimetil-fenil)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (75,70 g; 218,35 mmol; 1,00 eq.) en THF (400,00 ml) a presión positiva de atmósfera de ni trógeno se enfría hasta -78 °C. A esta solución se añade (bistrimetilsilil)amida de litio (1,0 M en THF) (262 ml; 262 mmol; 1,20 eq.) gota a gota durante un periodo de 30 minutos. Se deja que la mezcla de reacción alcance la TA y se agita a TA durante 18 h. La mezcla de reacción se evapora a una temperatura de 30 °C. El residuo se tritura con hexano y el sólido formado se filtra. Se deja reposar el filtrado durante algún tiempo al vacío y se filtra de nuevo si se forma material sólido. El filtrado se concentra a una temperatura de 30 °C para obtener (1S,2S,6R,8S)-4-[(R)-2-(2,4-dimetil-fenil)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (80,10 g; 169,84 mmol; 77,8 %; aceite de color marrón).
RMN 1H (400 MHz, CDCb): 5: 7,06 (d, J = 7,64 Hz, 1H), 6,94 (s, 1H), 6,90 (d, J = 7,80 Hz, 1H), 4,29-4,27 (m, 1H), 3,15-3,10 (m, 1H), 2,87-2,83 (m, 1H), 2,58-2,53 (m, 1H), 2,34-2,32 (m, 2H), 2,30 (s, 3H), 2,28 (s, 3H), 2,15-2,13
(m, 1H), 2,03 (t, J = 5,88 Hz, 1H), 1,90-1,88 (m, 1H), 1,81-1,77 (m, 1H), 1,39 (s, 3H), 1,32 (s, 3H), 1,01-0,98 (m, 1H), 0,93 (s, 3H), 0,85 (s, 3H), 0,09 (s, 18H).
Etapa 5: clorhidrato de (R)-2-(2,4-dimetil-fenil)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1.02,6]dec-4-il)-etilamina
Una solución en agitación de (1S,2S,6R,8S)-4-[(R)-2-(2,4-dimetil-fenil)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (80,10 g; 169,84 mmol; 1,00 eq.) en éter dietílico (400,00 ml) bajo atmósfera de nitrógeno se enfría hasta -10 °C. A esta solución se añade gota a gota ácido clor hídrico 2 M en éter dietílico (212,30 ml; 424,59 mmol; 2,50 eq.). La mezcla de reacción se agita a TA durante 2 h. La mezcla de reacción se evapora a presión reducida para obtener clorhidrato de (R)-2-(2,4-dimetil-fenil)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026 ]dec-4-i)-etilamina (63,00 g; 72,61 mmol; 42,8 %; sólido de color marrón).
RMN 1H (400 MHz, DMSO-d6 ): 5 8,19 (s, 3H), 7,05 (d, J = 7,68 Hz, 1H), 6,95 (s, 1H), 6,90 (d, J = 8,16 Hz, 1H), 4,31 (dd, J = 1,80, 8,76 Hz, 1H), 3,02-3,00 (m, 1H), 2,99-2,92 (m, 1H), 2,87-2,84 (m, 1H), 2,26-2,24 (m, 3H), 2,26 (s, 3H), 2,24 (s, 3H), 2,03-2,00 (m, 1H), 1,91 (t, J = 5,68 Hz, 1H), 1,82-1,80 (m, 1H), 1,71-1,66 (m, 1H), 1,31 (s, 3H), 1,21 (s, 3H), 0,98-0,96 (m, 1H), 0,77 (s, 3H).
Mediante secuencias similares a las descritas para el compuesto intermedio 1 pueden prepararse los siguientes compuestos

donde el grupo Y indica uno de los siguientes grupos:

Compuesto intermedio 2:



Etapa 1: benzofuran-3-ilmetanol
Una solución de 1-benzofurano-3-carbaldehído (5 g; 34,2 mmol) en metanol (50 ml) se enfría con hielo y se añade borohidruro de sodio (1,9 g; 51,3 mmol) en porciones. La mezcla de reacción se agita a temperatura ambiente durante 1 h. La mezcla de reacción se concentra y el residuo se reparte entre cloruro de amonio saturado y etilacetato. La capa orgánica se separa, se seca sobre sulfato de sodio y se concentra (5,0 g; líquido incoloro, 98 %). RMN 1H (400 MHz, CDCla): 57,70-7,68 (m, 1H), 7,62 (s, 1H), 7,52-7,50 (m, 1H), 7,36-7,26 (m, 2H), 4,86 (s, 2H).
Etapa 2: 3-(bromometil)benzofurano
Una solución fría (0 °C) de benzofuran-3-ilmetanol (5,0 g; 33,7 mmol) en éter dietílico (50 ml) se trata con tri bro muro de fósforo (1,1 ml; 11,2 mmol) y la mezcla de reacción se agita a 0 °C durante 30 min. A continuación, la mezcla de reacción se vierte en hielo y se extrae con éter. La capa orgánica se seca sobre sulfato de sodio y se concentra (7,1 g; líquido de color amarillo, 100 %).
RMN 1H (400 MHz, CDCla): 57,74-7,71 (m, 2H), 7,53 (s, 1H), 7,39-7,31 (m, 2H), 4,65 (s, 2H).
Etapa 3: 2-(benzofuran-3-ilmetil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano
Una solución de 3-(bromometil)benzofurano (7,1 g; 33,8 mmol) en 1,4-dioxano degaseado (70 ml) se trata con bis(pinacolato)diboro (10,3 g; 40,5 mmol), carbonato de potasio (13,9 g; 101,0 mmol) y tetrakis(trifenilfosfina) paladio(0) (1,9 g; 1,7 mmol) y la mezcla se calienta a 100 °C durante 12 horas. El contenido del matraz se enfría hasta temperatura ambiente y se filtra a través de un lecho de celite. El filtrado se concentra y el producto sin procesar se purifica mediante cromatografía en columna ultrarrápida de gel de sílice, que se eluye con el 2-5 % de acetato de etilo en éter de petróleo para obtener el compuesto del título (6,1 g; 69 %) como un aceite de color amarillo.
RMN 1H (400 MHz, CDCla) 57,57-7,52 (m, 2H), 7,46-7,44 (m, 1H), 7,30-7,21 (m, 2H), 2,23 (s, 2H), 1,29 (s, 12H).
Etapa 4: éster de (+)-pinanediol del ácido 2-(benzofuran-3-ilmetil)borónico
Una solución de 2-(benzofuran-3-ilmetil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (6,1 g; 23,6 mmol) en éter dietílico (60 ml) se trata con (1S, 2S, 3R, 5S)-(+)-pinanediol (6,0 g; 35,4 mmol). La mezcla de reacción se agita a tempe ratura ambiente durante 12 h, a continuación la mezcla se lava dos veces con agua, después con salmuera, se seca sobre sulfato sódico anhidro y se concentra. El producto sin procesar se purifica mediante cromatografía ultrarrápida en columna de gel de sílice, que se eluye con un 5 % de acetato de etilo en éter de petróleo, para obtener el compuesto del título (6,3 g; 82 %).
RMN 1H (400 MHz, CDCla): 5 7,58-7,56 (m, 1H), 7,55-7,53 (m, 1H), 7,46-7,44 (m, 1H), 7,28-7,23 (m, 2H), 4,33 (dd, J = 1,88, 8,76 Hz, 1H), 2,34-2,32 (m, 1H), 2,28 (s, 2H), 2,22-2,21 (m, 1H), 2,08 (t, J = 5,88 Hz, 1H), 1,42 (s, 3H), 1,29 (s, 3H), 1,13 (d, J = 10,92 Hz, 1H), 0,85 (s, 3H). CG/EM: m/z: 310.
Etapa 5: éster de (+)-pinanediol del ácido [(1S)-1-cloro-2-(benzofuran-3-ilmetil)borónico
A una mezcla enfriada (-100 °C) de diclorometano (6,3 ml; 60,9 mmol) y tetrahidrofurano anhidro (36 ml) se añade n-butilo de litio (1,6 M en hexanos; 14,0 ml; 22,3 mmol) durante 20 min. Tras agitar durante 20 min a -100 °C, se añade una solución de éster de (+)-pinanediol del ácido 2-(benzofuran-3-ilmetil)borónico (6,3 g; 20,3 mmol) en THF anhidro (22 ml) durante 20 min. A continuación se añade una solución de cloruro de cinc (0,5 M en THF; 36,5 ml; 18,2 mmol) a -100 °C durante 30 min. Se permite que la mezcla alcance la temperatura ambiente, se agita durante 18 h y se concentra. Al aceite resultante se añade éter dietílico y cloruro de amonio saturado. La capa orgánica se seca sobre sulfato de sodio anhidro y se concentra al vacío (residuo: 7,3 g; 99 %).
RMN 1H (400 MHz, DMSO-d6): 57,60-7,57 (m, 2H), 7,49-7,47 (m, 1H), 7,31-7,25 (m, 2H), 4,36-4,34 (m, 1H), 3,31 3,29 (m, 1H), 3,24-3,22 (m, 1H), 2,35-2,31 (m, 1H), 2,14-2,12 (m, 1H), 2,06 (t, J = 5,84 Hz, 1H), 1,90-1,86 (m, 2H), 1,42 (s, 3H), 1,04 (d, J = 11,04 Hz, 1H), 0,85 (s, 3H). CG/EM: m/z: 358,2.
Etapa 6: éster de (+)-pinanediol del ácido [(1R)-1-[bis(trimetilsiNl)ammo]-2-(benzofuran-3-MmetM) borónico
A una solución enfriada (-78 °C) de éster de (+)-pinanediol del ácido [(1S)-1-cloro-2-(benzofuran-3-ilmetil)borónico (7,3 g; 20,3 mmol) en 40 ml de tetrahidrofurano anhidro se añade bis(trimetilsilil)amida de litio (1 M en THF; 25,5 ml; 25,5 mmol). Se permite que la mezcla alcance la temperatura ambiente, se agita durante 18 h y se con centra hasta sequedad. Al residuo resultante se añade hexano y a continuación el sólido precipitado se recoge por filtración. El filtrado se concentra para obtener el producto sin procesar requerido (6,7 g, 68 %).
RMN 1H (400 MHz, CDCla): 57,60-7,59 (m, 1H), 7,50-7,45 (m, 2H), 7,28-7,24 (m, 2H), 4,31 (dd, J = 1,56, 8,70 Hz, 1H), 3,18-3,14 (m, 1H), 2,92-2,90 (m, 1H), 2,75-2,72 (m, 1H), 2,34-2,30 (m, 1H), 2,15-2,14 (m, 1H), 2,03 (t, J = 5,68 Hz, 1H), 1,88-1,80 (m, 2H), 1,39 (s, 3H), 1,30 (s, 3H), 1,01 (d, J = 10,88 Hz, 1H), 0,84 (s, 3H), 0,09 (s, 18H).
Etapa 7: trifluroacetato del éster de (+)-pinanediol del ácido [(1R)-1-amino-2-(benzofuran-3-ilmetil) borónico
Una solución enfriada (0 °C) de éster de (+)-pinanediol del ácido [(1R)-1-[bis(trimetilsilil)amino]-2-(benzofuran-3-ilmetil)borónico (6,7 g; 13,9 mmol) en éter dietílico (30 ml) se trata con ácido trifluoroacético (3,2 ml; 41,7 mmol)
añadido gota a gota. La mezcla de reacción se agita a continuación a TA durante 3 horas. Se observa precipita ción. La mezcla de reacción se enfría hasta 0 °C y se filtra. El sólido filtrado se lava con éter frío y se seca al vacío para obtener el compuesto del título (2,3 g; sólido de color blanco; 36 %).
RMN 1H (400 MHz, DMSO-da): 57,66 (s, 1H), 7,61-7,60 (m, 1H), 7,47-7,45 (m, 1H), 7,29-7,20 (m, 2H), 4,30-4,28 (m, 1H), 3,27-3,16 (m, 3H), 2,25-2,13 (m, 3H), 1,94 (t, J = 5,56 Hz, 1H), 1,86-1,81 (m, 2H), 1,25 (s, 6H), 1,01 (d, J = 8,00 Hz, 1H), 0,75 (s, 3H).
Compuesto intermedio 3: clorhidrato de 2-(7-metil-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-d ioxa-4-bora-triciclo[6.1.1.02,6]dec-4-il)-eti lamina

CH 3 C H 3

Etapa 8 
Etapa 1: éster etílico del ácido 7-metil-benzofurano-3-carboxílico
A una solución de 2-hidroxi-3-metil-benzaldehído (20,00 g; 139,55 mmol; 1,00 eq.) en diclorometano (120 ml) se añade complejo de ácido tetrafluorobórico y éter dietílico (1,88 ml; 13,96 mmol; 0,10 eq.). A la mezcla de color rojo oscuro resultante se añade etildiazoacetato (31,70 ml; 300,04 mmol; 2,15 eq.) en diclorometano (80 ml) gota a gota lentamente a 25-30 °C (temperatura interna) durante aproximadamente 50 min. Después de 16 h, se añade H2SO4 concentrado. La mezcla de reacción se agita durante 30 min. A continuación, la mezcla de reacción se neutraliza con NaHCO3 sólido, se filtra a través de celite y el filtrado se concentra para obtener un residuo sin procesar. El residuo se purifica mediante cromatografía en columna usando acetato de etilo al 2 % en éter de petróleo para obtener éster etílico del ácido 7-metil-benzofurano-3-carboxílico (19,00 g; 86,83 mmol; 62,2 %; aceite de color amarillo).
HPLC (método A): TR 4,98 min (pureza en HPLC 93 %).
RMN 1H, 400 MHz, CDCb: 8,27 (s, 1H), 7,88-7,90 (m, 1H), 7,25-7,29 (m, 1H), 7,17 (d, J = 7,32 Hz, 1H), 4,39-4,45 (m, 2H), 2,55 (s, 3H), 1,44 (t, J = 7,16 Hz, 3H).
Etapa 2: (7-metil-benzofuran-3-il)-metanol
A una solución de éster etílico del ácido 7-metil-benzofurano-3-carboxílico (19,00 g; 86,83 mmol; 1,00 eq.) en diclorometano (190,00 ml) bajo atmósfera de nitrógeno se añade hidruro de diisobutilaluminio (1,0 M en tolueno) (191,03 ml; 191,03 mmol; 2,20 eq.) gota a gota a -78 °C. Se permite que la mezcla de reacción llegara a TA y se agitó durante 1 h. La mezcla de reacción se enfría en un baño de hielo y se detiene con una solución acuosa de HCl 1,5 N. La mezcla resultante (que era una masa sólida pegajosa resuspendida en solvente) se diluye con acetato de etilo y se filtra a través de celite. El lecho de celite se lava abundantemente con acetato de etilo y diclorometano. El filtrado se evapora para obtener un residuo sin procesar. Se recoge el sólido que queda en el lecho de celite, se tritura con acetato de etilo y se filtra. El filtrado se mezcla junto con el residuo sin procesar y se evapora. De este modo, el residuo obtenido se recoge en acetato de etilo y se lava con una solución acuosa de HCl 1,5 N y salmuera. La capa orgánica se seca sobre Na2SO4 anhidro y se concentra. El residuo obtenido se purifica mediante cromatografía ultrarrápida en columna usando acetato de etilo al 40-50 % en éter de petróleo como eluyente para obtener (7-metil-benzofuran-3-il)-metanol (8,20 g; 48,40 mmol; 55,7 %; aceite de color amarillo claro).
HPLC (método A): TR 3,33 min, (pureza en HPLC 95,7 %).
RMN 1H, 400 MHz, CDCb: 7,64 (s, 1H), 7,50-7,52 (m, 1H), 7,17-7,21 (m, 1H), 7,14 (d, J = 7,20 Hz, 1H), 4,86-4,86 (m, 2H), 2,54 (s, 3H).
Etapa 3: 3-(bromometil)-7-metil-benzofurano
A una solución enfriada en hielo de (7-metil-benzofuran-3-il)-metanol (8,20 g; 48,40 mmol; 1,00 eq.) en éter dietí lico (82,00 ml) bajo atmósfera de nitrógeno se añade gota a gota tribromuro de fósforo (1,53 ml; 16,12 mmol; 0,33 eq.) y la mezcla de reacción se agita en condiciones de enfriamiento en hielo durante 30 minutos. La mezcla de reacción se vierte sobre hielo y se extrae con éter dietílico. La capa orgánica se seca sobre Na2SO4 anhidro y se concentra para obtener 3-bromometil-7-metil-benzofurano (10,00 g; 44,43 mmol; 91,8 %; aceite incoloro).
RMN 1H, 400 MHz, CDCla: 7,71 (s, 1H), 7,53-7,55 (m, 1H), 7,21-7,25 (m, 1H), 7,16 (d, J = 7,32 Hz, 1H), 4,65 (s, 2H), 2,48 (s, 3H).
Etapa 4: 7-metil-3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-ilmetil)-benzofurano
A una solución de 3-bromometil-7-metil-benzofurano (10,00 g; 44,43 mmol; 1,00 eq.) en dioxano-1,4 degaseado (100,00 ml) se añade bis(pinacolato)diboro (13,68 g; 53,31 mmol; 1,20 eq.), K2CO3 seco (18,61 g; 133,28 mmol; 3,00 eq.) y tetrakis(trifenilfosfina)paladio(0) (2,57 g; 2,22 mmol; 0,05 eq.). A continuación, la mezcla de reacción se calienta a 100 °C bajo atmósfera de nitrógeno durante 16 h. La mezcla de reacción se diluye con diclorometano y se filtra a través de celite. El filtrado se concentra. El residuo se disuelve en acetato de etilo y se lava con salmuera. La capa orgánica se seca sobre Na2SO4 anhidro y se concentra. El material sin procesar se purifica mediante cromatografía en columna utilizando acetato de etilo al 2 % en éter de petróleo para obtener 7-metil-3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-ilmetil)-benzofurano (5,00 g; 18,37 mmol; 41,4 %; líquido incoloro).
RMN 1H, 400 MHz, DMSO-d6: 7,65 (s, 1H), 7,33-7,35 (m, 1H), 7,07-7,13 (m, 2H), 2,43 (s, 3H), 2,13 (s, 2H), 1,16 (s, 12H).
Etapa 5: trimetil-4-(7-metil-benzofuran-3-ilmetil)-3,5-dioxa-4-bora-triciclo[6.1.1.02,6]decano
A una solución enfriada en hielo de 7-metil-3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-ilmetil)-benzofurano (5,00 g; 18,37 mmol; 1,00 eq.) en Et2O (50,00 ml) bajo atmósfera de nitrógeno se añade 1S,2S,3R, 5S-(+)-2,3-pinanodiol (4,69 g; 27,56 mmol; 1,50 eq.) y la mezcla de reacción se agitó a TA durante 14 h. El análisis mediante TLC mostró la finalización de la reacción. La mezcla de reacción se lava con salmuera. La capa orgánica se seca sobre Na2SO4 anhidro y se concentra. El material sin procesar se purifica mediante cromatografía ultrarrápida en columna utili zando acetato de etilo al 2 % en éter de petróleo para obtener (1S,2S,6R,8S)-2,9,9-trimetil-4-(7-metil-benzofuran-3-ilmetil)-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (5,00 g; 13,00 mmol; 70,7 %; líquido incoloro).
CG/EM: m/z: 324,2
RMN 1H, 400 MHz, CDCb: 7,53-7,55 (m, 1H), 7,39-7,40 (m, 1H), 7,12-7,27 (m, 1H), 7,06-7,08 (m, 1H), 4,31-4,34 (m, 1H), 2,53 (s, 3H), 2,30-2,37 (m, 1H), 2,26 (s, 2H), 2,18-2,23 (m, 1H), 2,07 (t, J = 5,76 Hz, 1H), 1,84-1,93 (m, 2H), 1,42 (s, 3H), 1,29 (s, 3H), 1,12-1,15 (m, 1H), 0,85 (s, 3H).
Etapa 6: (1S,2S,6R,8S)-4-[1-cloro-2-(7-met¡l-benzofuran-3-¡l)-et¡l]-2,9,9-tr¡metM-3,5-d¡oxa-4-boratric iclo[6.1.1.026]decano
Se toma diclorometano (2,96 ml; 46,26 mmol; 3,00 eq.) en THF (40 ml) en un matraz RB a presión positiva de nitrógeno y se enfría a -95 °C usando una mezcla de nitrógeno líquido y etanol. A esta se añade n-butilo de litio (1,6 M en hexanos) (10,60 ml; 16,96 mmol; 1,10 eq.) gota a gota por las paredes laterales del matraz RB (a una velocidad media, la adición llevó aproximadamente 30 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. Tras la adición, la mezcla de reacción se agita durante 20 minutos. Durante el transcurso de la reacción se forma un precipitado de color blanco (la temperatura interna se mantiene entre -95 °C y -100 °C). A continuación se añade una solución de (1S,2S,6R,8S)-2,9,9-trimetil-4-(7-metil-benzofuran-3-ilmetil)-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (5,00 g; 15,42 mmol; 1,00 eq.) en THF (20 ml) gota a gota por las paredes laterales del matraz Rb (aproximadamente 25 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. Tras la adición, se añade inmediatamente cloruro de cinc (0,5 M en THF) (27,76 ml; 13,88 mmol; 0,90 eq.) gota a gota por las paredes laterales del matraz RB (a una velocidad media, la adición llevó aproximadamente 45 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. A continuación, se deja que la mezcla de reacción alcance lentamente la TA y se agita a TA durante 16 h. La mezcla de reacción se concentra (temperatura del baño 30 °C). El residuo se reparte entre éter dietílico y solución saturada de NH4CL La capa orgánica se separa, se seca sobre Na2SO4 anhidro y se concentra (temperatura del baño 30 °C) para obtener (1S,2S,6R,8S)-4-[1-cloro-2-(7-metil-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.0,2,6]decano (5,90 g; 15,83 mmol; 102,7 %; líquido de color marrón).
RMN 1H, 400 MHz, CDCb: 7,57 (s, 1H), 7,42-7,44 (m, 1H), 7,27 (s, 1H), 7,09-7,18 (m, 1H), 4,34-4,36 (m, 1H), 3,74-3,76 (m, 1H), 3,28-3,30 (m, 1H), 3,20-3,22 (m, 1H), 2,52 (s, 3H), 2,32-2,34 (m, 1H), 2,07 (t, J = 5,88 Hz, 1H), 1,85-1,91 (m, 2H), 1,42 (s, 3H), 1,29 (s, 3H), 1,06-1,09 (m, 1H), 0,85 (s, 3H).
Etapa 7: ((1S,2S,6R,8S)-4-[1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-2-(7-metil-benzofuran-3-il)-etil]-2,9,9-tr¡metil-3,5-d¡oxa-4-bora-tr¡c¡clo[6.1.1.02'6]decano
Una solución de (1S,2S,6R,8S)-4-[1-cloro-2-(7-metil-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (5,90 g; 15,83 mmol; 1,00 eq.) en THF (40,00 ml) a presión positiva de atmósfera de nitró geno se enfría hasta -78 °C. A esta solución se añade (bistrimetilsilil)amida de litio (1,0 M en THF) (17,41 ml; 17,41 mmol; 1,10 eq.) gota a gota durante un periodo de 30 minutos. Se deja que la mezcla de reacción alcance la TA y se agita a TA durante 18 h. La mezcla de reacción se evapora a 30 °C. El residuo se tritura con n-hexano y el sólido formado se filtra. El filtrado se concentra a 30 °C para obtener (1S,2S,6R,8S)-4-[1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-2-(7-metil-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (6,00 g; 12,06 mmol; 76,2 %; aceite de color marrón oscuro).
RMN 1H, 400 MHz, CDCb: 7,50 (s, 1H), 7,41-7,43 (m, 1H), 7,12-7,16 (m, 1H), 7,06-7,08 (m, 1H), 4,29-4,32 (m, 1H), 3,17-3,09 (m, 1H), 2,70-2,89 (m, 1H), 2,52-2,70 (m, 1H), 2,52 (s, 3H), 2,28-2,31 (m, 1H), 2,14-2,14 (m, 1H), 2,03 (t, J = 5,68 Hz, 1H), 1,78-1,89 (m, 2H), 1,39 (s, 3H), 1,31 (s, 3H), 1,01-1,04 (m, 1H), 0,90-0,92 (m, 2H), 0,88 (s, 3H), 0,12 (s, 18H).
Etapa 8: clorhidrato de 2-(7-met¡l-benzofuran-3-¡l)-1-((1S,2S,6R,8S)-2,9,9-tr¡met¡l-3,5-d¡oxa-4-boratr¡c¡clo[6.1.1.02,6]dec-4-¡l)-et¡lam¡na
Una solución en agitación de (1S,2S,6R,8S)-4-[1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-2-(7-metil-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (6,00 g; 12,06 mmol; 1,00 eq.) en éter dietílico (60,00 ml) bajo atmósfera de nitrógeno se enfría hasta -10 °C. A esta solución se añade ácido clorhídrico 2 M en éter dietílico (15,07 ml; 30,14 mmol; 2,50 eq.) gota a gota. La mezcla de reacción se agita a TA durante 2 h. La mezcla de reacción se evapora a 30 °C. Se añade éter dietílico (20 ml) al residuo y el sólido formado se recoge
mediante filtración, se lava con éter dietílico frío y se seca al vacío para obtener clorhidrato de 2-(7-metil-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026 ]dec-4-il)-etilamina (3,50 g; 8,98 mmol; 74,5 %; sólido de color marrón anaranjado).
RMN 1H, 400 MHz, DMSO-d6 : 8,09 (s, 3H), 7,83 (s, 1H), 7,52-7,53 (m, 1H), 7,12-7,19 (m, 2H), 4,39 (dd, J = 1,84, 8,62 Hz, 1H), 3,07-3,13 (m, 1H), 3,03-3,07 (m, 2H), 2,43 (s, 4H), 2,28-2,30 (m, 1H), 2,07-2,08 (m, 1H), 1,92 (t, J = 5,68 Hz, 1H), 1,82-1,84 (m, 1H), 1,71-1,75 (m, 1H), 1,19-1,25 (m, 8 H), 1,00-1,08 (m, 1H), 0,78 (s, 3H).
Compuesto intermedio 4: clorhidrato de 2-(7-cloro-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-d ioxa-4-bora-triciclo[6.1.1.02,6]dec-4-il)-eti lamina

Br
Etapa 3  Etapa 4
Etapa 4 
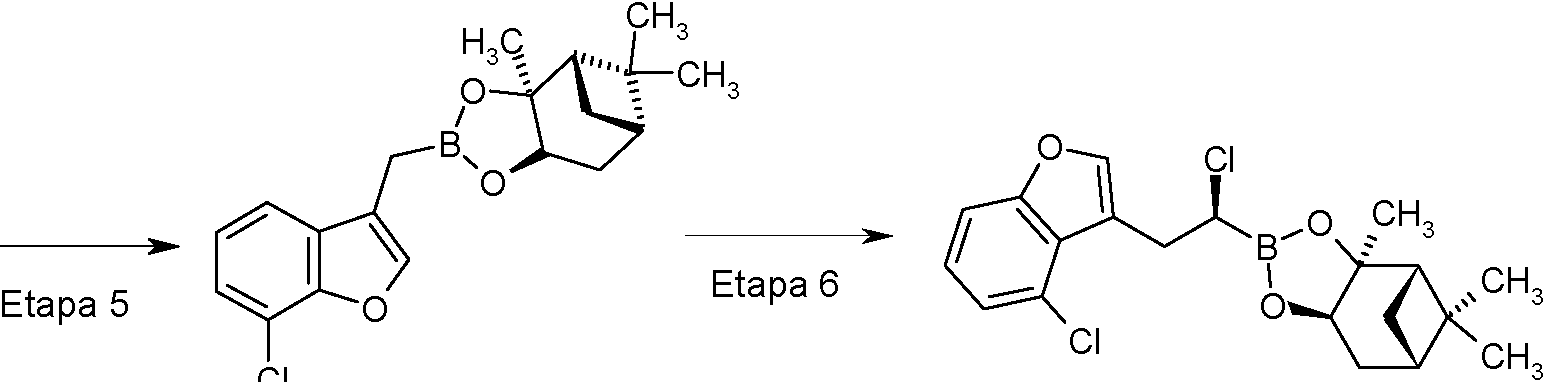

Etapa 1: éster etílico del ácido 7-cloro-benzofurano-3-carboxílico
A una solución de 3-cloro-2-hidroxi-benzaldehído (25,00 g; 156,48 mmol; 1,00 eq.) en diclorometano (250 ml) se añade complejo de ácido tetrafluorobórico y éter dietílico (2,11 ml; 15,65 mmol; 0,10 eq.). A la mezcla de color rojo oscuro resultante se añade etildiazoacetato (35,55 ml; 336,44 mmol; 2,15 eq.) en diclorometano (50 ml) gota a gota lentamente a 25-30 °C (temperatura interna) durante aproximadamente 50 min. Después de 16 h, se añade H2SO4 concentrado. La mezcla de reacción se agita durante 15 minutos. A continuación, la mezcla de reacción se neutralizó con NaHCO3 sólido, se filtra a través de celite y el filtrado se concentra para obtener un residuo sin procesar. El residuo se purifica mediante cromatografía en columna usando acetato de etilo al 2 % en éter de petróleo para obtener éster etílico del ácido 7-cloro-benzofurano-3-carboxílico 2 (18,20 g; 81,02 mmol; 51,8 %; líquido incoloro).
RMN 1H, 400 MHz, DMSO-d6: 8,88 (s, 1H), 7,95-7,93 (m, 1H), 7,57-7,55 (m, 1H), 7,42 (t, J = 7,8 Hz, 1H), 4,38 4,33 (m, 2H), 1,35 (t, J = 7,1 Hz, 3H).
Etapa 2: (7-cloro-benzofuran-3-il)-metanol
A una solución en agitación de éster etílico del ácido 7-cloro-benzofurano-3-carboxílico (450 g; 2,0089 mol; 1,00 eq.) en DCM (4500 ml) a -78 °C se añade hidruro de diisobutilaluminio 1,0 M en tolueno (4017 ml; 4,0178 mol; 2,20 eq.). A continuación, se deja que la mezcla de reacción alcance lentamente la TA y se agita a TA durante 2 h. Una vez confirmada la finalización de la reacción mediante TLC, la mezcla de reacción se detiene con HCl 1,5 N (500 ml), se pasa a través de celite y se lava con DCM (2000 ml). El filtrado se lava con una solución de salmuera (1 * 2000 ml). La capa orgánica se separa, se seca sobre Na2SO4, se filtra y se concentra al vacío. El producto sin procesar se somete a cromatografía en columna y se eluye con acetato de etilo al 15 % en éter de petróleo para obtener (7-cloro-benzofuran-3-il)-metanol 3 (365 g; 2,0054 mol; 99,8 %; espuma sólida de color blanco).
RMN 1H, 400 MHz, DMSO-d6: 7,99 (s, 1H), 7,66 (dd, J = 1,0, 7,8 Hz, 1H), 7,41 (dd, J = 0,8, 7,8 Hz, 1H), 7,26 (t, J = 7,8 Hz, 1H), 5,24 (t, J = 5,6 Hz, 1H), 4,63-4,62 (m, 2H).
Etapa 3: 3-(bromometil)-7-cloro-benzofurano
A una solución enfriada en hielo de (7-cloro-benzofuran-3-il)-metanol (365 g; 2,0054 mol; 1,00 eq.) en éter dietílico (3650 ml) se añade tribromuro de fósforo (62,2 ml; 0,6618 mol; 0,33 eq.) gota a gota bajo atmósfera de nitrógeno. La mezcla de reacción se agita enfriando en un baño de hielo durante 30 minutos. Posteriormente, la mezcla de reacción se vierte sobre hielo y se extrae con éter dietílico. La capa orgánica se seca sobre Na2SO4 anhidro, se filtra y se concentra para obtener 3-bromometil-7-cloro-benzofurano (480 g; 1,9591 mol; 97,71 %; sólido de color blanco).
RMN 1H, 400 MHz, DMSO-d6: 8,29 (s, 1H), 7,72 (dd, J = 1,0, 7,8 Hz, 1H), 7,49 (dd, J = 0,8, 7,8 Hz, 1H), 7,36 (t, J = 7,8 Hz, 1H), 4,90 (s, 2H).
Etapa 4: 7-cloro-3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-ilmetil)-benzofurano
A una solución de 3-bromometil-7-cloro-benzofurano (480 g; 1,9591 mol; 1,00 eq.) en dioxano-1,4 degaseado (4800 ml) se añade bis(pinacolato)diboro (596,9 g; 2,3510 mol; 1,20 eq.), acetato de potasio seco (576,8 g; 5,877 mol; 3,00 eq.) y [1,1-bis(difenilfosfino)ferroceno] dicloropaladio(II), en complejo con diclorometano (70,33 g; 0,0979 mol; 0,05 eq.). A continuación, la mezcla de reacción se calienta hasta 100 °C bajo atmósfera de nitrógeno durante toda la noche. La mezcla de reacción se diluye con diclorometano y se pasa a través de celite. El filtrado se concentra. El residuo se disuelve en acetato de etilo y se lava con salmuera (1000 ml * 1). La capa orgánica se seca sobre Na2SO4 anhidro, se filtra y se concentra al vacío. El material sin procesar se purifica mediante cromatografía en columna utilizando acetato de etilo al 2 % en éter de petróleo para obtener 7-cloro-3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-ilmetil)-benzofurano (480 g; 1,6438 mol; 83,9 %; semisólido de color amarillo).
CG/EM: m/z: 292 (Columna: DB-5 ms (15 m * 0,25 mm * 0,25 |jm); gas portador: helio, caudal: 2,0 ml/min).
RMN 1H, 400 MHz, DMSO-d6: 7,79 (s, 1H), 7,52 (dd, J = 1,0, 7,8 Hz, 1H), 7,38 (dd, J = 0,8, 7,8 Hz, 1H), 7,27-7,23 (m, 1H), 2,17 (s, 2H), 1,18 (s, 12H).
Etapa 5: (1S,2S,6R,8S)-4-(7-cloro-benzofuran-3-il-metil)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo [6.1.1.02'6]decano
A una solución enfriada en hielo de 7-cloro-3-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-ilmetil)-benzofurano (480 g; 1,6438 mol; 1,00 eq.) en éter dietílico (5000 ml) bajo atmósfera de nitrógeno se añade 1S, 2S, 3R, 5S-(+)-2,3-pinanodiol (335,7 g; 1,9726 mol; 1,20 eq.). La mezcla de reacción se agita a TA durante 16 h. La mezcla de reac ción se lava con agua (2000 ml * 1) y salmuera (1500 ml * 1). La capa orgánica se seca sobre Na2SO4 anhidro, se filtra y se concentra. El material sin procesar se purifica mediante cromatografía ultrarrápida utilizando acetato
de etilo al 1 % en éter de petróleo para obtener (1S,2S,6R,8S)-4-(7-cloro-benzofuran-3-il-metil)-2,9,9-trimetil-3,5-dioxa-4-bora-tricido [6.1.1.026]decano (520 g; 1,510 mol; 91,9 %; semisólido de color amarillo pálido).
CG/EM: m/z: 344 (Columna: HP-5MS (12 m * 0,20 D mm * 0,33 |jm); gas portador: helio, caudal: 2,0 ml/min)
RMN 1H, 400 MHz, DMSO-d6: 7,80 (s, 1H), 7,54 (dd, J = 0,9, 7,8 Hz, 1H), 7,38 (dd, J = 0,7, 7,8 Hz, 1H), 7,24 (t, J = 7,8 Hz, 1H), 4,33 (t, J = 6,9 Hz, 1H), 2,29-2,24 (m, 3H), 2,14-2,10 (m, 1H), 1,93 (t, J = 5,4 Hz, 1H), 1,84-1,81 (m, 1H), 1,70-1,65 (m, 1H), 1,31 (s, 3H), 1,21 (s, 3H), 0,98 (d, J = 10,72 Hz, 1H), 0,78 (s, 3H).
Etapa 6: (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(7-cloro-benzofuran-3-M)-etM]-2,9,9-trimetM-3,5-dioxa-4-boratriciclo[6.1.1.026]decano
Se toma diclorometano (95,7 ml; 1,499 mol; 3,00 eq.) en THF (1200 ml) en un matraz RB a presión positiva de nitrógeno y se enfría a -95 °C usando una mezcla de nitrógeno líquido y etanol. A esta se añade n-butilo de litio (1,6 M en THF) (343,6 ml; 0,549 mol; 1,10 eq.) gota a gota por el cuello lateral del matraz RB (a una velocidad media, la adición llevó aproximadamente 45 min) de modo que la temperatura interna se mantenga entre -95 °C y -100 °C. Tras la adición, la mezcla de reacción se agita durante 30 minutos. Durante el transcurso de la reacción se forma un precipitado de color blanco (la temperatura interna se mantiene entre -95 °C y -100 °C). A continuación se añade una solución de (1S,2S,6R,8S)-4-(7-cloro-benzofuran-3-ilmetil)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (172 g; 0,4999 mol; 1,00 eq.) en THF (500 ml) gota a gota por el cuello lateral del matraz RB (aproximadamente 25 min) de modo que la temperatura interna se mantiene de nuevo entre -95 °C y -100 °C. Tras finalizar la adición, se añade inmediatamente cloruro de cinc (0,5 M en THF) (1599,6 ml; 0,7998 mol; 1,6 eq.) gota a gota por el cuello lateral del matraz RB (a una velocidad media, la adición llevó aproximadamente 40 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. A continuación, se deja que la mezcla de reacción llegue a -5 °C y se agita a -5 °C durante 1,5 h. La mezcla de reacción se detiene mediante la adición de solución de NH4Cl saturada (500 ml). La mezcla de reacción se concentra al vacío (temperatura del baño 30 °C). El residuo se reparte entre éter dietílico y solución saturada de NH4Cl. La capa orgánica se separa, se seca sobre Na2SO4 anhidro y se concentra (temperatura del baño 30 °C) para obtener (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(7-cloro-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (205 g; 0,5214 mol; 104,5 %; aceite de color naranja).
CG/EM: m/z: 392 (Columna: ZB-1MS (10 m * 0,101D mm * 0,1 jm ); gas portador: helio, caudal: 2,0 ml/min)
RMN 1H, 400 MHz, CDCb: 7,64 (s, 1H), 7,50 (d, J = 8,00 Hz, 1H), 7,33-7,31 (m, 1H), 7,23-7,21 (m, 1H), 4,36-4,34 (m, 1H), 3,29-3,27 (m, 1H), 3,22-3,20 (m, 1H), 2,34-2,32 (m, 1H), 2,15-2,14 (m, 1H), 2,06 (t, J = 5,60 Hz, 1H), 1,91 1,83 (m, 7H), 1,36 (s, 3H), 1,29 (s, 3H), 1,05-1,02 (m, 1H), 0,85 (s, 3H).
Etapa 7: (1S,2S,6R,8S)-4-[(R)-2-(7-cloro-benzofuran-3-il)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.02'6]decano
Una solución de (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(7-cloro-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (205 g; 0,5214 mol; 1,00 eq.) en THF (2050 ml) a presión positiva de atmósfera de nitrógeno se enfría hasta -78 °C. A esta solución se añade (bistrimetilsilil)amida de litio (1,0 M en THF) (625 ml; 0,6257 mol; 1,20 eq.) gota a gota durante un periodo de 30 minutos. Se deja que la mezcla de reacción alcance la TA y se agita a TA durante 18 h. El solvente de la mezcla de reacción se evapora a 30 °C. El residuo se tritura con hexano y el sólido formado se filtra. Se deja reposar el filtrado durante algún tiempo al vacío y se filtra de nuevo si se forma material sólido. El filtrado se concentra a 30 °C para obtener (1S,2S,6R,8S)-4-[(R)-2-(7-cloro-benzofuran-3-il)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (180 g; 0,3481 mol; 66,7 %; aceite de color naranja).
RMN 1H, 400 MHz, CDCb: 7,63 (s, 1H), 7,51-7,49 (m, 1H), 7,29-7,27 (m, 1H), 7,19-7,15 (m, 1H), 4,32-4,29 (m, 1H), 3,63-3,61 (m, 1H), 3,14-3,12 (m, 1H), 2,87-2,85 (m, 1H), 2,26-2,24 (m, 1H), 2,14-2,12 (m, 1H), 1,88-1,86 (m, 1H), 1,88-1,76 (m, 2H), 1,33 (s, 3H), 1,30 (s, 3H), 1,02-0,99 (m, 1H), 0,85 (s, 3H), 0,07 (s, 18H).
Etapa 8: clorhidrato de (R)-2-(7-cloro-benzofuran-3-N)-1-((1S,2S,6R,8S)-2,9,9-trimetN-3,5-dioxa-4-boratriciclo[6.1.1.02,6]dec-4-il)-etilamina
Una solución en agitación de (1S,2S,6R,8S)-4-[(R)-2-(7-cloro-benzofuran-3-il)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-tridclo[6.1.1.026]decano (180 g; 0,348 mol) en éter dietílico (1800 ml) bajo atmósfera de nitrógeno se enfría hasta -10 °C. A esta solución se añade gota a gota ácido clorhídrico en éter dietílico (concentración 2,0 M; 435,2 ml; 0,870 mol; 2,50 eq.). La mezcla de reacción se agita a TA durante 2 h (se observa precipitación de un sólido durante el transcurso de la reacción). La mezcla de reacción se evapora hasta sequedad y el sólido obtenido se tritura con éter dietílico (500 ml) y, posteriormente, se filtra. La torta de filtración se lava con éter dietílico (3 * 300 ml) y se seca bajo atmósfera al vacío para obtener clorhidrato de (R)-2-(7-clorobenzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.02,6]dec-4-il)-etilamina (81,5 g; 0,1992 mol; 57,2 %; sólido blanquecino).
RMN 1H, 400 MHz, CDCla: 8,09 (s, 3H), 7,97 (s, 1H), 7,73 (dd, J = 1,52 Hz, 7,76 Hz, 1H), 7,44 (d, J = 7,76 Hz, 1H), 7,31 (d, J = 7,80 Hz, 1H), 4,42-4,40 (m, 1H), 3,16-3,07 (m, 3H), 2,32-2,27 (m, 1H), 2,10-2,04 (m, 1H), 1,93 (t, J = 5,56 Hz, 1H), 1,83-1,71 (m, 2H), 1,27 (s, 3H), 1,25 (s, 3H), 1,08-1,02 (m, 1H), 0,79 (s, 3H).
Compuesto intermedio 5: clorhidrato de (R)-2-(2,3-dihidro-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-tricido[6.1.1.02'6]dec-4-il)-etilamina
Etapa 1: (1S,2S,6R,8S)-4-(2,3-dihidro-benzofuran-3-ilmetil)-2,9,9-trimetil-3,5-dioxa-4-boratric iclo[6.1.1.026]decano

A una solución de (1S,2S,6R,8S)-4-benzofuran-3-¡lmet¡l-2,9,9-tr¡met¡l-3,5-d¡oxa-4-bora-tr¡ciclo[6.1.1.02’6]decano (5,00 g; 10,72 mmol; 1,00 eq.) en metanol (100,00 ml) en un minirreactor a presión se añade paladio sobre car bono (10 % en peso) (2,28 g; 2,14 mmol; 0,20 eq.). El contenido se hidrogena a presión de H2 de 490 kPa (5 kg/cm2) durante 3 h. El análisis mediante TLC mostró una conversión completa. La mezcla de reacción se filtra a través de celite y el filtrado se evapora. El producto sin procesar se purifica mediante cromatografía en columna Biotage-Isolera (columna C18; fase móvil: ACN/H2O; 50:50 ¡socrático) para obtener (1S,2S,6R,8S)-4-(2,3-dihidrobenzofuran-3-ilmetil)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (4,10 g; 13,13 mmol; 122,5 %; lí quido de color amarillo pálido).
CG/EM: m/z: 312,3.
Etapa 2: (1S,2S,6R,8S)-4-[1-cloro-2-(7-metil-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1.02'6]decano

Se toma diclorometano (2,46 ml; 38,44 mmol; 3,00 eq.) en THF (40,00 ml) en un matraz RB a presión positiva de nitrógeno y se enfría a -95 °C usando una mezcla de nitrógeno líquido y etanol. A esta se añade n-butilo de litio (1,6 M en THF) (8,81 ml; 14,09 mmol; 1,10 eq.) gota a gota por las paredes laterales del matraz RB (a una velo cidad media, la adición llevó aproximadamente 20 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. Tras la adición, la mezcla de reacción se agita durante 25 minutos. Durante el transcurso de la reacción se forma un precipitado de color blanco (la temperatura interna se mantiene entre -95 °C y -100 °C). A continuación se añade una solución de (1S,2S,6R,8S)-4-(2,3-dihidro-benzofuran-3-ilmetil)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (4,00 g; 12,81 mmol; 1,00 eq.) en THF (15,00 ml) gota a gota por las paredes la terales del matraz RB (aproximadamente 25 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. Tras la adición, se añade inmediatamente cloruro de cinc (0,5 M en THF) (25,62 ml; 12,81 mmol; 1,00 eq.) gota a gota por las paredes laterales del matraz RB (a una velocidad media, la adición llevó aproximadamente 25 min) de modo que la temperatura interna se mantiene entre -95 °C y -100 °C. A continuación, se deja que la mezcla de reacción alcance lentamente la TA y se agita a TA durante 18 h. La mezcla de reacción se concentra (temperatura del baño 30 °C). El residuo se reparte entre éter dietílico y solución saturada de NH4Cl. La capa orgánica se seca sobre Na2SO4 anhidro y se concentra (temperatura del baño 30 °C) para obtener (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(2,3-dihidro-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (4,60 g; 12,75 mmol; 99,5 %; aceite de color amarillo).
RMN 1H, 400 MHz, CDCla: 7,29 (d, J = 6,72 Hz, 1H), 7,21-7,10 (m, 1H), 6,90-6,77 (m, 2H), 4,68-4,65 (m, 1H), 4,32-4,29 (m, 2H), 3,65-3,60 (m, 1H), 2,40-2,08 (m, 4H), 1,94-1,85 (m, 2H), 1,42 (s, 3H), 1,33 (s, 3H), 1,22 (s, 3H), 1,17-1,15 (m, 1H), 0,86 (s, 3H).
Etapa 3: (1S,2S,6R,8S)-4-[(R)-2-(2,3-dihidro-benzofuran-3-il)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-tr¡metil-3,5-d¡oxa-4-bora-tr¡c¡clo[6.1.1.02'6]decano

Una solución de (1S,2S,6R,8S)-4-[(S)-1-cloro-2-(2,3-dihidro-benzofuran-3-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1.026]decano (4,60 g; 12,75 mmol; 1,00 eq.) en THF (45,00 ml) a presión positiva de atmósfera de ni trógeno se enfría hasta -78 °C. A esta solución se añade (bistrimetilsilil)amida de litio (1,0 M en THF) (16,58 ml; 16,58 mmol; 1,30 eq.) gota a gota durante un periodo de 30 minutos. Se deja que la mezcla de reacción alcance la TA y se agita a t A durante 18 h. La mezcla de reacción se evapora a 30 °C. El residuo se tritura con hexano y el sólido formado se filtra. Se deja reposar el filtrado durante algún tiempo al vacío y se filtra de nuevo si se forma material sólido. El filtrado se concentra a 30 °C para obtener (1S,2S,6R,8S)-4-[(R)-2-(2,3-dihidro-benzofuran-3-il)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (3,77 g; 7,76 mmol; 60,9 %; aceite de color amarillo).
RMN 1H, 400 MHz, CDCla: 7,22-7,10 (m, 2H), 6,90-6,79 (m, 2H), 4,62-4,59 (m, 1H), 4,33-4,27 (m, 1H), 2,34-2,20 (m, 2H), 2,07-2,05 (m, 1H), 1,94-1,84 (m, 2H), 1,40 (s, 3H), 1,30 (s, 3H), 1,15-1,13 (m, 1H), 0,86 (s, 3H), 0,10 (s, 18H).
Etapa 4: clorhidrato de (R)-2-(2,3-d¡h¡dro-benzofuran-3-¡l)-1-((1S,2S,6R,8S)-2,9,9-tr¡met¡l-3,5-d¡oxa-4-boratr¡c¡clo[6.1.1.02,6]dec-4-¡l)-et¡lam¡na

Una solución en agitación de (1S,2S,6R,8S)-4-[(R)-2-(2,3-dihidro-benzofuran-3-il)-1-(1,1,1,3,3,3-hexametil-disilazan-2-il)-etil]-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]decano (3,77 g; 7,76 mmol; 1,00 eq.) en Et2O (35,00 ml) bajo atmósfera de nitrógeno se enfría hasta -10 °C. A esta solución se añade gota a gota ácido clorhídrico 2 N en éter dietílico (9,70 ml; 19,41 mmol; 2,50 eq.). La mezcla de reacción se agita a TA durante 2 h. La mezcla de reacción se evapore hasta sequedad a presión reducida para obtener un sólido. El sólido formado se tritura con éter dietílico, se filtra, se lava con éter dietílico y se seca al vacío para obtener clorhidrato de (R)-2-(2,3-dihidro-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]dec-4-il)-etilamina (2,30 g; 5,25 mmol; 67,7 %; sólido de color marrón pálido). El análisis mostró la presencia de isómeros (~65,50 % 20,75 %) en la posición indicada (*).
CL/EM: 4,73 min; 86,25 % (máx.); 80,47 % (220 nm), 342,20 (M+1).
RMN 1H, 400 MHz, DMSO-d6: 8,11 (s, 3H), 7,23-7,19 (m, 1H), 7,13-7,10 (m, 1H), 6,85 (t, J = 7,40 Hz, 1H), 6,77 (d, J = 8,04 Hz, 1H), 4,61-4,57 (m, 1H), 4,48-4,45 (m, 1H), 4,25-4,22 (m, 1H), 3,68-3,62 (m, 1H), 2,90-2,85 (m, 1H), 2,34-2,32 (m, 1H), 2,19-2,17 (m, 1H), 2,02-1,99 (m, 2H), 1,89-1,77 (m, 3H), 1,39 (s, 3H), 1,25 (s, 3H), 1,17 1,14 (m, 1H), 0,82 (s, 3H).
Mediante secuencias similares a las descritas para los compuestos intermedios 2-4, se pueden preparar otros ejemplos de los siguientes restos

como por ejemplo, en particular, compuestos en los que el grupo Y indica uno de los siguientes grupos:

Compuesto intermedio ácido 1: ácido (1S,2R,4R)-7-oxabiciclo[2.2.1]heptano-2-carboxílico
Etapa 1: éster (R)-l-fenil-etílico del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico

A una solución de ácido 7-oxa-bicido[2.2.1]heptano-2-carboxílico (4,680 g; 31,276 mmol, racémico) en diclorometano seco (máx. 0,005 % de H2O) SeccoSolv® (100 ml) se añade bajo atmósfera de argón (R)-1-fenil-etanol (4,623 ml; 37,531 mmol), 4-(dimetilamino)piridina para síntesis (DMAP) (3,821 g; 31,276 mmol) y clorhidrato (3-dimetilamino-propil)-etil-carbodiimida (EDCl) (6,730 g; 34,404 mmol) en agitación a 0 °C. Posteriormente, la solu ción de reacción transparente se agita durante toda la noche a temperatura ambiente. Tras completarse la forma ción del éster, la reacción se detiene mediante la adición de solución sat. de NH4Cl (ac.). A continuación, la mezcla se extrae dos veces con CH2Cl2. La capa orgánica se lava dos veces con una solución sat. de NaHCO3 (ac.) y salmuera, se seca sobre Na2SO4, se filtra y se evapora hasta sequedad. El producto sin procesar se purifica mediante cromatografía ultrarrápida (gel de sílice; n-heptano/acetato de etilo al 0-30 %) para obtener 7,496 g (30,43 mmol; 97,3 %) de un aceite incoloro (HPLC: mezcla pura al 100 % de diastereómeros).
La mezcla de diastereómeros se separa mediante HPLC quiral preparativa (Chiralcel OD-H; n-heptano/2-propanol 95:5; 220 nm) para obtener éster (R)-1 -fenil-etílico del ácido (1R,2S,4S)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (3,22 g; aceite incoloro, rendimiento 41,8 %, HPLC quiral 100 %) y éster (R)-1 -fenil-etílico del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (3,14 g, aceite, rendimiento 40,7 % HPLC quiral 100 %).
Etapa 2: ácido (1S,2R,4R)-7-oxabiciclo[2.2.1]heptano-2-carboxílico
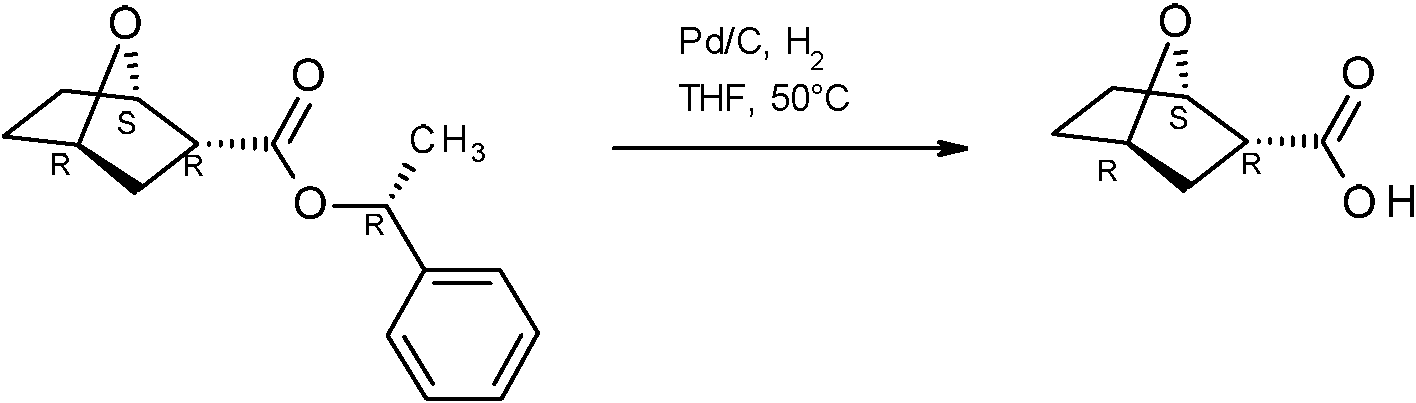
A una solución de éster (R)-1 -fenil-etílico del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (46,74 g; 182,75 mmol; 1,00 eq.) en THF (233,70 ml), se añade paladio sobre carbono (10 % p/p) (1,94 g; 1 , 8 3 mmol; 0,01 eq.). Los contenidos se hidrogenan bajo atmósfera de H2 a 50 °C y 5 bares de presión durante 16 h. Tras finalizar la hidrogenación, la mezcla de reacción se filtra a través de celite, el filtrado se evapora hasta sequedad y se recoge en pentano. La capa orgánica se extrae tres veces con agua. Posteriormente, la capa acuosa se liofiliza para obtener ácido (1S,2R,4R)-7-oxabiciclo[2.2.1]heptano-2-carboxílico (22,62 g; 159,09 mmol; rendi miento 87,1 %) como un sólido incoloro.
TLC: cloroformo/metanol (9,5/0,5) Rf 0,5. RMN 1H, 400 MHz, DMSO-d6: 12,16 (s, 1H), 4,66 (d, J = 4,4 Hz, 1H), 4,54 (t, J = 4,4 Hz, 1H), 2,57 (d, J = 35,2 Hz, 1H), 1,91-1,86 (m, 1H), 1,65-1,37 (m, 4H), 1,34-1,33 (m, 1H). Rotación opcional [D]20 D = 31,9°; DD20 = 0,0644° (etanol, 20,16 mg/10 ml).
Compuesto intermedio ácido 2: ácido (1R,2S,4S)-7-oxabiciclo[2.2.1]heptano-2-carboxílico

A una solución de éster (R)-l-fenil-etílico del ácido (1R,2S,4S)-7-oxa-bicido[2.2.1]heptano-2-carboxílico (4,52 g; 17,98 mmol; 1,00 eq.) en THF (22,60 ml), se añade paladio sobre carbono (10 % p/p) (0,19 g; 0 , 1 8 mmol; 0,01 eq.). El contenido se hidrogena bajo atmósfera de H2 a 50 °C durante 12 h. El análisis mediante TLC mostró que el inicio se completó. La mezcla de reacción se filtra a través de celite y el filtrado se evapora para obtener ácido (1R,2S,4S)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (2,10 g; 14,77 mmol; 82,1 %; sólido blanquecino).
RMN 1H, 400 MHz, DMSO-d6 : 12,16 (s, 1H), 4,66 (d, J = 4,4 Hz, 1H), 4,54 (t, J = 4,4 Hz, 1H), 2,57 (d, J = 35,2 Hz, 1H), 1,91-1,86 (m, 1H), 1,65-1,37 (m, 4H), 1,34-1,33 (m, 1H).
Compuesto intermedio ácido 3: (sal de) litio del ácido (1S,8R)-8-metiM1-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-1 -carboxílico

Etapa 1: éster metílico del ácido 8-metiM1-oxa-triciclo[6.2.1.02'7]undeca-2,4,6,9-tetraeno-1-carboxílico

Se añade nitrito de isopentilo (3,64 ml; 27,12 mmol) a una solución de 3,72 g de ácido antranílico (27,12 mmol) y ácido trifluoroacético (0,26 ml; 3,39 mmol) en 45 ml de THF seco a 0 °C. La solución resultante se agita enérgica mente durante unos minutos a 0 °C y luego se calienta hasta TA. Tras agitar durante 1 h a TA, el color de la suspensión se vuelve amarilla. El sólido de color marrón se recoge mediante filtración, se lava con THF seco y se transfiere a un matraz que contiene una solución de éster metílico del ácido 5-metil-furan-2-carboxílico (2,00 g; 13,56 mmol) en éter dimetílico de etilenglicol (45,00 ml). A continuación, la mezcla resultante se calienta gradual mente hasta 100 °C hasta completarse la descomposición y se agita durante otra hora más a 100 °C. Tras la evaporación del solvente, la mezcla de reacción se purifica mediante cromatografía ultrarrápida (gel de sílice; gradiente de EE/heptano; 0-25 % de EE) para obtener éter metílico del ácido 8-metil-11-oxa-triciclo[6.2.1.027 ]undeca-2,4,6,9-tetraeno-1-carboxílico (1,82 g; 53.7 %; goma de color amarillo) como una mezcla 1:1 de estereoisómeros.
Método A de CLEM: (M+H) 217,0; tR 2,03 min.
Etapa 2: éster metílico del ácido (1S,8R)-8-metiM1-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-1-carboxflico y éster metílico del ácido (1R,8S)-8-metiM1-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-1-carboxflico

Una solución de éster metílico del ácido 8-metiM1-oxa-triddo[6.2.1.027]undeca-2,4,6,9-tetraeno-1-carboxílico (1,82 g; 7,28 mmol) en 18 ml de EE se hidrogena a TA y presión normal usando 500 mg de Pd/C (54 % de agua) hasta completarse la reacción. La mezcla de reacción se filtra y el filtrado se concentra. El aceite pegajoso residual (mezcla de estereoisómeros) se separa usando SFC quiral preparativa (Chiral Cel OD-H; CO2/2-propanol 58.5: 1,5; 220 nm) para obtener éster metílico del ácido (1S,8R)-8-metiM1-oxa-tricido[6.2.1.027]undeca-2,4,6-trieno-1-carboxílico (439 mg, rendimiento 25,3 %) y éster metílico del ácido (1R,8S)-8-metiM1-oxa-tridclo[6.2.1.027]undeca-2,4,6-trieno-1-carboxílico (449 mg, rendimiento 25,9 %), ambos como aceites incoloros.
Método A de CLEM: (M+H) no detectado; tR 2,09 min (el mismo para ambos compuestos).
Etapa 3: (sal de) litio del ácido (1S,8R)-8-metiM1-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-1-carboxflico

Se recoge éster metílico del ácido (1S,8R)-8-metiM1-oxa-tridclo[6.2.1.027]undeca-2,4,6-trieno-1-carboxílico (0,439 g; 1,84 mmol) en 5 ml de agua desionizada y 2,5 ml de THF. Se añade LiOH (44 mg; 1,84 mmol), la mezcla se agita bajo atmósfera de argón a TA durante 1 h y se evapora para obtener el compuesto del título, que se utiliza sin purificación adicional.
Método A de CLEM: (M-Li+H-18) 187; tR 1,71 min.
Compuesto intermedio ácido 4: litio del ácido (1R,8S)-8-metiM1-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-1 -carboxíl ico

Se recoge éster metílico del ácido (1R,8S)-8-metil-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-1-carboxílico (pu reza 91 %; 0,449 g; 1,88 mmol) en 5 ml de agua desionizada y 2,5 ml de THF. Se añade LiOH (45 mg; 1,88 mmol), la mezcla se agita bajo atmósfera de argón a TA durante 1 h y se evapora para obtener el compuesto del título. Método A de CLEM: (M-Li+H-18) 187; tR 1,71 min.
Compuesto intermedio ácido 5: ácido (1S,8R)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-9-carboxflico

Etapa 1: éster (R )-l-fenil-etílico del ácido furano-3-carboxílico
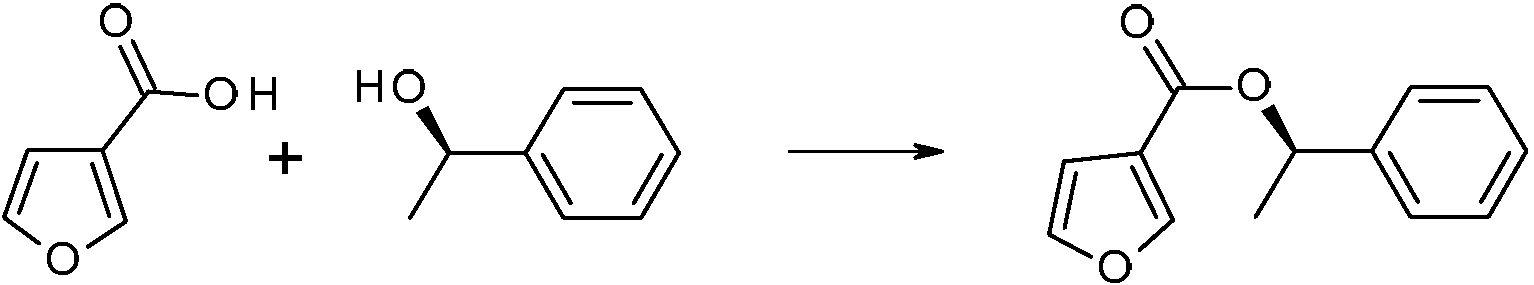
A una solución de ácido 3-furanocarboxílico (2,00 g; 17,84 mmol) en 40 ml de diclorometano seco se añaden (R)-1-fenil-etanol (2,64 ml; 21,41 mmol), 4-dimetilamino-piridina (2,18 g; 17,85 mmol) y clorhidrato de (3-dimetilaminopropil)-etil-carbodiimida (3,84 g; 19,63 mmol) bajo atmósfera de argón a 0 °C. La solución de reacción transpa rente se agita durante 3 h sin enfriamiento adicional. La solución de reacción se detiene con solución sat. de NH4Cl y se extrae con diclorometano. La capa orgánica se lava tres veces con una solución sat. de NaHCO3 y salmuera, se seca sobre Na2SO4 y se filtra. Tras la evaporación del solvente, la mezcla de reacción se purifica mediante cromatografía ultrarrápida (gel de sílice; gradiente de EE/heptano; 0-30 % de EE) para obtener éster (R)-1-feniletílico del ácido furano-3-carboxílico (3,55 g; rendimiento 90 %; aceite de color amarillo).
Método A de CLEM: (M+H) no detectado; tR 2,46 min.
Etapa 2: éster (R)-l-fenil-etílico del ácido (1R,8R)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6,9-tetraeno-9-carboxílico y éster (R)-1-fenil-etílico del ácido (1S,8S)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6,9-tetraeno-9-carboxílico

Se añade nitrito de isopentilo (1,91 ml; 14,18 mmol) a una solución de ácido antranílico (1,94 g; 14,18 mmol) y ácido trifluoroacético (0,137 ml; 1,77 mmol) en 22 ml de THF seco a 0 °C. La solución resultante se agita enérgi camente durante unos minutos a 0 °C y luego se calienta hasta TA. Tras agitar durante 1 h a TA, el color de la suspensión se vuelve amarillo. El líquido se elimina mediante decantación y el sólido de color marrón restante se lava con THF seco antes de transferirlo a un matraz que contiene una solución de éster (R)-1 -fenil-etílico del ácido furano-3-carboxílico (1,60 g; 7,09 mmol) en éter dimetílico de etilenglicol (22 ml). A continuación, a mezcla resul tante se calienta gradualmente hasta 100 °C hasta completarse la descomposición y se agita durante otra hora más a 100 °C. Tras la evaporación del solvente, la mezcla de reacción se purifica mediante cromatografía ultra rrápida (gel de sílice; gradiente de EE/heptano; 0-35 % de EE) para obtener 677 mg de éster (R)-1 -fenil etílico del ácido 11-oxa-triciclo[6.2.1.027]undeca-2,4,6,9-tetraeno-9-carboxílico (677 mg, aceite incoloro) como una mezcla de diastereómeros. Esta mezcla se separa mediante HPLC quiral preparativa (ChiralPak AD; n-heptano/etanol 1:1; 220 nm) para obtener éster (R)-1 -fenil-etílico del ácido (1R,8R)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6,9-tetraeno-9-carboxílico (190 mg, HPLC quiral >97 %; tR 7,73 min) y éster (R)-1 -fenil-etílico del ácido (1S,8S)-11-oxatriciclo[6.2.1.027]undeca-2,4,6,9-tetraeno-9-carboxílico (180 mg; HPLC quiral >96 %; tR 12,53 min).
Etapa 3: éster (R )-l-fenil-etílico del ácido (1S,8R)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-tNeno-9-carboxílico

Una solución de éster (R)-l-fenil-etílico del ácido (1S,8S)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6,9-tetraeno-9-carboxílico (0,470 g; 1,190 mmol) en 10 ml de THF se hidrogena a TA y presión normal usando 500 mg de Pd/C (54 % de agua) hasta completarse la reacción. La mezcla de reacción se filtra, el filtrado se concentra y se purifica mediante cromatografía ultrarrápida (gel de sílice; gradiente de EE/heptano; 0-60 % de EE) para obtener el éster (R)-1-fenil-etílico del ácido (1S,8R)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-9-carboxílico (184 mg) como un aceite incoloro.
Método A de CLEM: (M+H) no detectado; tR 2,51 min.
Etapa 4: ácido (1S,8R)-11-oxa-tnciclo[6.2.1.02'7]undeca-2,4,6-trieno-9-carboxflico

Una solución de éster (R)-1 -fenil-etílico del ácido (1S,8R)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-9-carboxílico (0,184 g; 0,626 mmol) en 10 ml de THF se hidrogena a TA y presión normal usando 200 mg de Pd/C (54 % de agua) durante toda la noche. La mezcla de reacción se filtra y el filtrado se evapora para obtener 130 mg de ácido (1S,8R)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-9-carboxícilo como un sólido de color blanco.
Método A de CLEM: (M-18) 174; tR 1,42 min.
Compuesto intermedio ácido 6: ácido (1R,8S)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-9-carboxílico

Etapa 1: éster (R)-1-fenil-etílico del ácido (1R,8S)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-9-carboxílico

Una solución de éster (R)-l-fenil-etílico del ácido (1R,8R)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6,9-tetraeno-9-carboxílico (0,470 g; pureza 76 %; 1,22 mmol) en 10 ml de THF se hidrogena a TA y presión normal usando 100 mg de Pd/C (54 % de agua) hasta completarse la reacción (5 min). La mezcla de reacción se filtra, el filtrado se concentra y se purifica mediante cromatografía ultrarrápida (gel de sílice; gradiente de EE/heptano; 0-50 % de EE) para obtener el éster (R)-1 -fenil-etílico del ácido (1R,8S)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-9-carboxílico (107 mg; rendimiento 30 %) como una cera incolora.
Método A de CLEM: (M+H) no detectado; tR 2,52 min.
Etapa 2: ácido (1R,8S)-11-oxa-triciclo[6.2.1.02'7]undeca-2,4,6-trieno-9-carboxílico
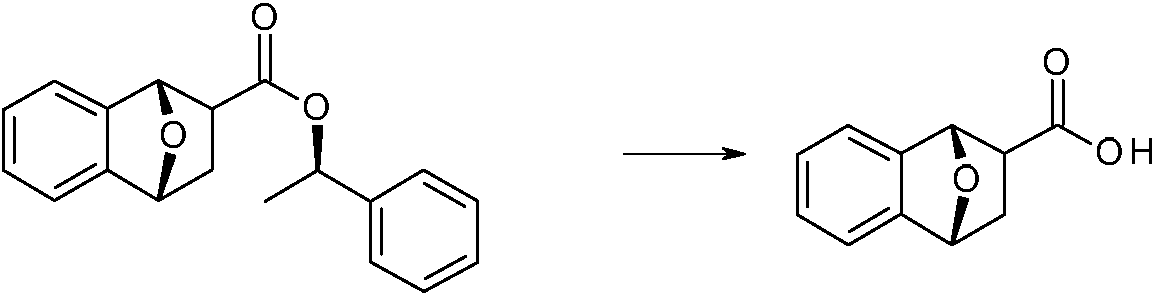
Una solución de éster (R)-1 -fenil-etílico del ácido (1S,8R)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-9-carboxílico (0,107 g; 0,364 mmol) en 10 ml de THF se hidrogena a TA y presión normal usando 100 mg de Pd/C (54 % de agua) durante toda la noche. La mezcla de reacción se filtra y el filtrado se evapora para obtener ácido (1 R,8S)-11-oxa-triciclo[6.2.1.027]undeca-2,4,6-trieno-9-carboxícilo (69 mg; rendimiento 92 %) como un sólido de color blanco.
Método A de CLEM: (M+H) no detectado; tR 1,36 min.
Ejemplo 1: ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-M]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]form am ido}etil]borónico (compuesto n.° 1)

Etapa 1: [(R)-2-(S)-2,3-dihidro-benzofuran-3-iM-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1.02,6]dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico

A una solución de clorhidrato de (R)-2-(S)-2,3-dihidro-benzofuran-3-il-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.02,6]dec-4-il)-etilamina (0,250 g; 0,66 mmol) en 5 ml de DMF seco se añade, a -15 °C y bajo atmósfera de argón, ácido (1S,2R,4R)-7-oxa-biddo[2.2.1]heptano-2-carboxílico (0,113 g; 0,79 mmol), etil-diisopropil-amina (0,34 ml; 1,99 mmol) y TBTU (0,70 g; 2,18 mmol). La solución de reacción de color amarillo se agita durante 1 h a -10 °C y, a continuación, durante otra hora a TA. La solución de reacción se enfría con hielo y se diluye con acetato de etilo. Tras la separación de la fase orgánica, se lava con salmuera y solución sat. de NaHCO3, se seca sobre sulfato sódico, se filtra y se concentra al vacío (temperatura del baño 30 °C). El aceite de color naranja obtenido se purifica primero mediante cromatografía ultrarrápida (gel de sílice; gradiente de heptano/EE, 0-100 % de EE) para obtener una mezcla de diastereómeros, que se separan mediante SFC quiral (ChiralPak AD, CO2:metanol [88:12]). Se obtienen 122 mg del compuesto del título (rendimiento 39,6 %) como un aceite incoloro.
Método A de CLEM: (M-H) 466,2; tR 2,49 min.
Etapa 2: ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-N]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]form am ido}etil]borónico (compuesto n.° 1)

A un sistema de dos fases de [(R)-2-(S)-2,3-dihidro-benzofuran-3-il-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (0,206 g; 0,44 mmol) en 27 ml de n-pentano y 20,6 ml de metanol se añade a 0 °C ácido isobutilborónico (0,180 g; 1,77 mmol) y HCl 2 N (1,99 ml; 3,98 mmol). La reacción se agita a TA durante toda la noche. Se separa la fase de pentano y la fase metanólica acuosa se lava cinco veces con pentano. La fase metanólica se concentra al vacío, se diluye con agua en hielo y se alcaliniza con NaOH 1 N. Posteriormente, se extrae con DCM (2x). La fase acuosa se acidifica con HCl 1 N y se extrae de nuevo con DCM (5x). Esta fase orgánica se seca sobre Na2SO4, se filtra y se evapora. El residuo se disuelve en acetonitrilo/agua y se liofiliza para obtener 104 mg (rendimiento: 71 %) del compuesto del título como un sólido de color blanco.
RMN 1H (500 MHz, DMSO-d6/D2O) d 7,23 - 7,20 (m, 1H), 7,13 - 7,09 (m, 1H), 6,86 (td, J = 7,4, 1,0 Hz, 1H), 6,76 (d, J = 7,8 Hz, 1H), 4,60 (d, J = 4,7 Hz, 1H), 4,59 - 4,53 (m, 2H), 4,21 (dd, J = 9,0, 6,7 Hz, 1H), 3,47 - 3,38 (m, 1H), 2,94 - 2,89 (m, 1H), 2,59 (dd, J = 9,0, 4,9 Hz, 1H), 1,91 - 1,84 (m, 2H), 1,71 (dd, J = 12,0, 9,1 Hz, 1H), 1,64 - 1,42 (m, 5H).
Método A de CLEM: (M-H) 314,2; tR 1,57 min.
Ejemplo 2: ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formam ido}etil]borónico (compuesto n.° 9)

etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico

A una solución de ácido (1S,2R,4R)-7-oxa-bicido[2.2.1]heptano-2-carboxíNco (1,87 g; 13,18 mmol), hexafluorofosfato de [dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio (HATU) (4,62 g; 14,37 mmol) y 4-metil-morfolina (3,29 ml; 29,94 mmol) en 70 ml de DMF seco se añade, bajo atmósfera de argón y enfriamiento en hielo, clorhidrato de (R)-2-(benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1.026] dec-4-il)-etilamina (4,50 g; 11,98 mmol). La solución de color amarillo se agitó durante 2,5 h a TA. La mezcla de reacción se vierte en 500 ml de solución saturada de NaHCO3 enfriada en hielo y con agitación durante 15 min. El precipitado se recoge mediante filtración al vacío y se lava con agua. El sólido obtenido se tritura con acetonitrilo, se diluye con MTB-éter y se succiona para obtener [(R)-2-(benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (3,26 g; rendimiento: 58,8 %) como un sólido de color blanco (pureza 100 %).
Método A de CLEM: (M-H) 464,2; tR 2,57 min.
Etapa 2: ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}-etil] borónico

A un sistema de dos fases de [(R)-2-benzofuran-3-il-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (ee=97 %; 3,45 mmol; 1,60 g) en 150 ml de n-pentano y 50 ml de metanol se añade a 0 °C ácido isobutilborónico (13,81 mmol; 1,41 g) y ácido clorhídrico 1 N (15,54 ml; 15,54 mmol). La reacción se agita a TA durante toda la noche. La fase de pentano se separa y la fase metanólica se lava con pentano (3x 80 ml). La fase metanólica se
concentra (temperatura del baño 30 °C) al vacío, se diluye con agua con hielo y se alcaliniza con NaOH 1 N (pH 11-12). Esta solución básica se extrae con DCM (3x 80 ml). La fase acuosa se acidifica con HCl 1 N (pH 2) y se extrae de nuevo con DCM (5x 80 ml). Las fases orgánicas combinadas se secan sobre Na2SO4, se filtran y se evaporan. El residuo se disuelve en acetonitrilo/agua y se liofiliza para obtener 0,697 g (rendimiento 61,3 %) del compuesto del título como un sólido de color blanco.
Datos analíticos: véase la tabla 2
Ejemplo 3: ácido [(1R)-2-(7-cloro-1-benzofuran-3-M)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formam ido}etil]borónico (compuesto n.° 13)

Etapa 1: [(R)-2-(7-cloro-benzofuran-3-M)-1-((1S,2S,6R,8S)-2,9,9-trimetM-3,5-dioxa-4-bora-triciclo[6.1.1.02'6] dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-bicido[2.2.1]heptano-2-carboxílico

A una solución de ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (3,77 g; 26,55 mmol), hexafluorofosfato de [dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio (HATU) (9,30 g; 28,97 mmol) y 4-metil-morfolina (6,63 ml; 60,34 mmol) en 148 ml de DMF seco se añade, bajo atmósfera de argón y enfriamiento en hielo, clorhidrato de (R)-2-(7-cloro-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026]dec-4-il)-etilamina (9,90 g; 24,14 mmol). La solución de color amarillo se agita durante 3 h a TA. La mezcla de reacción se vierte en 1 litro de solución saturada de NaHCO3 enfriada en hielo y se agita durante 15 min. El precipitado se recoge mediante filtración al vacío y se lava con agua. El sólido obtenido se tritura con acetonitrilo, se diluye con MTB-éter y se aspira para obtener [(R)-2-(7-cloro-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-bora-triciclo[6.1.1.026] dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (6,9 g; rendimiento: 56,6 %) como un sólido de color blanco (pureza 95 %).
Método A de CLEM: (M-H) 498,2; tR 2,70 min.
Etapa 2: ácido [(1R)-2-(7-cloro-1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]form am ido}etil]borónico (compuesto n.° 13)
A un sistema de dos fases de [(R)-2-(7-cloro-benzofuran-3-il)-1-((1S,2S,6R,8S)-2,9,9-trimetil-3,5-dioxa-4-boratriciclo[6.1.1 026]dec-4-il)-etil]-amida del ácido (1S,2R,4R)-7-oxa-biciclo[2.2.1]heptano-2-carboxílico (ee=99 %; 12,39 mmol; 6,26 g) en 220 ml de n-pentano y 125 ml de metanol se añade a 0 °C ácido isobutilborónico (37,17 mmol; 3,79 g) y ácido clorhídrico 1 N (55,760 mmol; 55,760 ml). La reacción se agita a TA durante toda la noche. La fase de pentano se separa y la fase metanólica se lava con pentano (3x 200 ml). La fase metabólica se concentra (temp, del baño por debajo de 30 °C) al vacío, se diluye con 200 ml de agua en hielo y se alcaliniza con NaOH 1 N (pH 12-13). Esta solución básica se extrae con DCM (3x 200 ml). La fase acuosa se acidifica con HCl
1 N (pH 2-3) y se extrae de nuevo con DCM (5x 220 ml). Las fases orgánicas combinadas se secan sobre Na2SO4, se filtran y se evaporan. El residuo se disuelve en acetonitrilo/agua y se liofiliza para obtener 3,277 g del com puesto del título como un sólido de color blanco.
RMN 1H (500 MHz, DMSO-da/D2O) d 7,78 (s, 1H), 7,64 - 7,60 (m, 1H), 7,42 - 7,39 (m, 1H), 7,29 (t, J = 7,8 Hz, 1H), 4,54 - 4,50 (m, 1H), 4,48 - 4,45 (m, 1H), 3,15 - 3,09 (m, 1H), 2,89 (dd, J = 14,9, 5,8 Hz, 1H), 2,77 (dd, J = 14,9, 8,4 Hz, 1H), 2,50 (dd, J = 9,0, 4,9 Hz, 1H), 1,82 - 1,75 (m, 1H), 1,65 (dd, J = 11,9, 9,0 Hz, 1H), 1,58 - 1,49 (m, 2H), 1,49 - 1,39 (m, 2H).
Waters XBridge C83,5 |jm 4,6 x 50 mm (A19/533 - La Chrom Elite; 70173815); 8,1 min; 2 ml/min; 215 nm; tampón A: TFA al 0,05 %/H2O; tampón B: TFA al 0,04 %/ACN; 0,0-0,2 min tampón B al 5 %; 0.2-8,1 min 5 %-100 % de tampón B; 8,1-10,0 min 100 %-5 % de tampón B: %; tR 4,24 min.
UPLC MS Waters Acquity UPLC; CORTECS C18 1,6 jm 50-2,1 mm; A: H2O HCOOH al 0,05 %; B: MeCN HCOOH al 0,04 %; T: 30 °C; flujo: 0,9 ml/min; 2 % -> B al 100 %: 0 -> 1,0 min; B al 100 %: 1,0 -> 1,3 min: 346,1 [M+H-H2O]; TR 0,61 min.
Partiendo de ácidos disponibles en el mercado (o ácidos sintetizados mediante saponificación de ésteres disponibles en el mercado) o ácidos descritos en la literatura, se han sintetizado los siguientes compuestos según los pasos 1 y 2 del ejemplo 1 o 2:
Tabla 1: Lista de compuestos de ejemplo





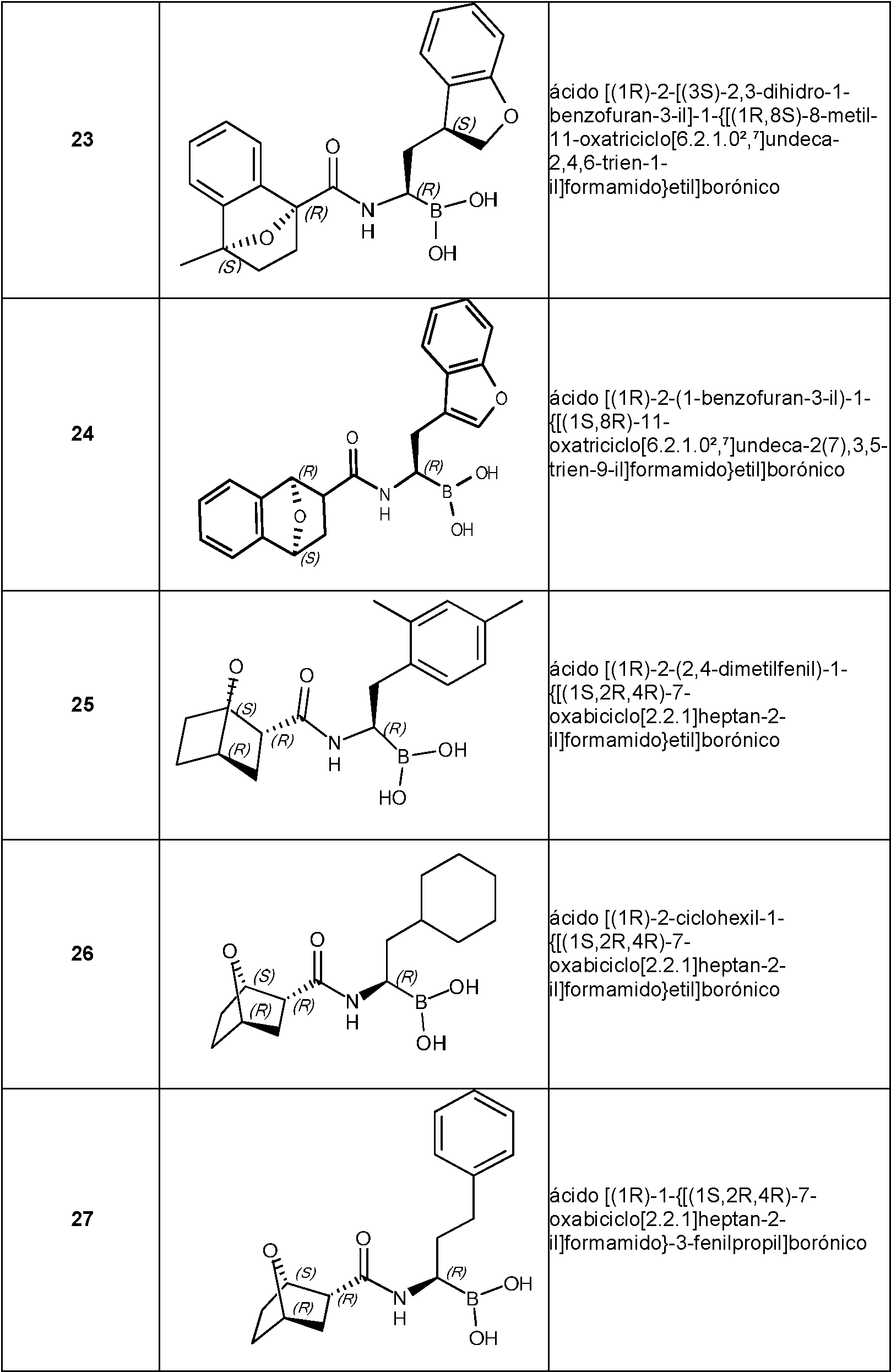

Tabla 2: Datos analíticos





Actividad biológica
Determinación de la actividad LMP7:
La medida de la inhibición de LMP7 se realiza en un formato de 384 pocillos basado en un ensayo de intensidad de fluorescencia.
El inmunoproteosoma humano purificado (0,25 nM) y los compuestos diluidos en serie en DMSO (intervalo de concentraciones de 30 |jM a 15 pM) o los controles se incubaron durante 20 minutos o 120 minutos (incubación larga) a 25 °C en tampón de ensayo que contenía Tris 50 mM pH 7,4, SDS al 0,03 %, EDTA 1 mM y DMSO al 1 %. La reacción se inicia con la adición del sustrato peptídico fluorogénico Suc-LLVY-AMC (Bachem I-1395) a una concentración de 40 j M. Después de 60 minutos de incubación a 37 °C, se mide la intensidad de fluorescen cia a Xex = 350 nm y Xem = 450 nm con un lector de fluorescencia (lector Perkin Elmer Envision o equivalente). La actividad LMP7 de los compuestos se resume en la tabla 3. A menos que se indique otra cosa, los resultados se obtienen tras la incubación durante 20 minutos.
Determinación de la actividad Beta5:
La medida de la inhibición de Beta5 se realiza en un formato de 384 pocillos basado en un ensayo de intensidad de fluorescencia.
El proteosoma constitutivo humano purificado (1,25 nM) y los compuestos diluidos en serie en DMSO (intervalo de concentraciones de 30 j M a 15 pM) o los controles se incubaron durante 20 minutos o 120 minutos (incubación larga) a 25 °C en tampón de ensayo que contenía Tris 50 mM pH 7,4, SDS al 0,03 %, EDTA 1 mM y DMSO al 1 %. La reacción se inicia con la adición del sustrato peptídico fluorogénico Suc-LLVY-AMC (Bachem I-1395) a una concentración de 40 j M. Después de 60 minutos de incubación a 37 °C, se mide la intensidad de fluorescen cia a Xex = 350 nm y Xem = 450 nm con un lector de fluorescencia (lector Perkin Elmer Envision o equivalente). En la tabla 3 se muestra la actividad Beta5 de los compuestos según la invención y su selectividad por LMP7 frente a Beta5. A menos que se indique otra cosa, los resultados se obtienen tras la incubación durante 20 minu tos.
Tabla 3:



*: 5 |iM < IC50 < 3,0*10-5M, **: 0,5 |iM < IC50 < 5 |iM, ***: 0,05 |iM < IC50 < 0,5 |iM, ****: IC50 < 0,05 |iM, : selecti vidad < 100, +: 100 < selectividad < 300, ++: 300 < selectividad < 500, +++: 500 < selectividad < 700, ++++: selectividad > 700; a. n. c.: actividad no cuantificable en el intervalo de concentración dado; según el método descrito anteriormente, «incubación larga» significa que la muestra se incuba durante 120 min.
Los ejemplos siguientes hacen referencia a medicamentos:
Ejemplo A: viales para inyección
Una solución de 100 g de un principio activo de fórmula (I) y 5 g de hidrogenofosfato disódico en 3 litros de agua bidestilada se ajusta a pH 6,5 usando ácido clorhídrico 2 N, se esteriliza por filtración, se transfiere a viales para inyección, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada vial para inyección contiene 5 mg de principio activo.
Ejemplo B: supositorios
Se funde una mezcla de 20 g de un principio activo de fórmula (I) con 100 g de lecitina de soja y 1400 g de manteca de cacao, se vierte en los moldes y se deja enfriar. Cada supositorio contiene 20 mg de principio activo.
Ejemplo C: solución
Se prepara una solución de 1 g de un principio activo de fórmula I, 9,38 g de NaH2PO4-2 H2O, 28,48 g de Na2HPO4-12 H2O y 0,1 g de cloruro de benzalconio en 940 ml de agua bidestilada. El pH se ajusta a 6,8, la solución se lleva a 1 l y se esteriliza mediante radiación. Esta solución puede usarse en forma de colirio.
Ejemplo D: pomada
Se mezclan 500 mg de un principio activo de fórmula (I) con 99,5 g de vaselina en condiciones asépticas.
Ejemplo E: comprimidos
Una mezcla de 1 kg de principio activo de fórmula (I), 4 kg de lactosa, 1,2 kg de almidón de patata, 0,2 kg de talco y 0,1 kg de estearato de magnesio se prensa de forma habitual para obtener comprimidos, de manera que cada comprimido contiene 10 mg de principio activo.
Ejemplo F: grageas
Los comprimidos se prensan de forma análoga al ejemplo E y, posteriormente, se recubren de forma habitual con un recubrimiento de sacarosa, almidón de patata, talco, goma de tragacanto y colorante.
Ejemplo G: cápsulas
Se introducen 2 kg de principio activo de fórmula (I) dentro de cápsulas duras de gelatina de forma habitual, de modo que cada cápsula contiene 20 mg de principio activo.
Ejemplo H: ampollas
Una solución de 1 kg de principio activo de fórmula (I) en 60 litros de agua bidestilada se esteriliza por filtración, se transfiere a ampollas, se liofiliza en condiciones estériles y se sella en condiciones estériles. Cada ampolla contiene 10 mg del principio activo.
Claims (23)
- REIVINDICACIONESUn compuesto de fórmula (I)
 dondeLY indica (CH2)m, donde de 1 a 4 átomos de H pueden estar sustituidos por Hal, R3a y/u OR4a, y/o donde un grupo CH2 puede estar sustituido por O, S, SO y/o SO2 ;X indica un heterobiciclo o heterotriciclo de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) o (xi) cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, NO2 , CN, R5a, OR5a, CONR5aR5b, NR5aCOR5b, SO2R5a, SOR5a, SO2NR5aR5b, NR5aSO2R5b, NR5aR5b, (CH2)q-R6, COR5a y/o SO2R5a, y donde 1,2 o 3 de los grupos CH2 cíclicos pueden estar sustituidos por CR4aR4b, C=O, O, S, NR5a, SO y/o SO2:Y
dondeLY indica (CH2)m, donde de 1 a 4 átomos de H pueden estar sustituidos por Hal, R3a y/u OR4a, y/o donde un grupo CH2 puede estar sustituido por O, S, SO y/o SO2 ;X indica un heterobiciclo o heterotriciclo de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) o (xi) cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, NO2 , CN, R5a, OR5a, CONR5aR5b, NR5aCOR5b, SO2R5a, SOR5a, SO2NR5aR5b, NR5aSO2R5b, NR5aR5b, (CH2)q-R6, COR5a y/o SO2R5a, y donde 1,2 o 3 de los grupos CH2 cíclicos pueden estar sustituidos por CR4aR4b, C=O, O, S, NR5a, SO y/o SO2:Y P1 indica un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8 , independientemente entre sí, no sustituido o mono-, di-, tri- o tetrasustituido por Hal, CN, R3a, OR3a y/o (CH2)q-R6;P2 indica fenilo o un heterociclo aromático monocíclico de 5, 6 o 7 átomos, cada uno no sustituido o mono, di, tri, tetra o pentasustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6, donde el sistema heterocíclico contiene 1, 2 o 3 átomos de N, O y/o S;P3 indica un heterociclo o hidrocarburo bicíclico de 8, 9 o 10 átomos, cada uno independientemente entre sí no sustituido o mono, di, tri, tetra o pentasustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6, donde al menos un anillo del heterociclo o hidrocarburo bicíclico es aromático y donde el sistema heterocíclico contiene 1,2 o 3 átomos de N, O y/o S;Cy1, Cy2, Cy3, Cy4 y Cy5 indican cada uno, independientemente entre sí, Ar1 o Het1;R1, R2 indican cada uno, independientemente entre sí, H o alquilo C1-C6 , o R1 y R2 forman juntos un resto según la fórmula (CE)
P1 indica un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8 , independientemente entre sí, no sustituido o mono-, di-, tri- o tetrasustituido por Hal, CN, R3a, OR3a y/o (CH2)q-R6;P2 indica fenilo o un heterociclo aromático monocíclico de 5, 6 o 7 átomos, cada uno no sustituido o mono, di, tri, tetra o pentasustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6, donde el sistema heterocíclico contiene 1, 2 o 3 átomos de N, O y/o S;P3 indica un heterociclo o hidrocarburo bicíclico de 8, 9 o 10 átomos, cada uno independientemente entre sí no sustituido o mono, di, tri, tetra o pentasustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6, donde al menos un anillo del heterociclo o hidrocarburo bicíclico es aromático y donde el sistema heterocíclico contiene 1,2 o 3 átomos de N, O y/o S;Cy1, Cy2, Cy3, Cy4 y Cy5 indican cada uno, independientemente entre sí, Ar1 o Het1;R1, R2 indican cada uno, independientemente entre sí, H o alquilo C1-C6 , o R1 y R2 forman juntos un resto según la fórmula (CE) R3a, R3b indican cada uno, independientemente entre sí, un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8 , donde de 1 a 5 átomos de H pueden estar sustituidos por Hal, CN, OH y/u OAlq; R4a, R4b indican cada uno, independientemente entre sí, H o R3a;oR4a y R4b forman juntos un grupo alquileno C3-C8 ;R5a, R5b indican cada uno, independientemente entre sí, H, R3a, Ar2 o Het2;R6 indica OH u OR3a;T1, T2, T3, T4, T5, T6, T7, T8 y T9 indican cada uno, independientemente entre sí, O, SO, C=O;Alq indica alquilo C1-C6 lineal o ramificado;Ar1 representa un carbociclo aromático de 6 átomos;Het1 representa un heterociclo saturado, insaturado o aromático de 5 o 6 átomos con 1 a 4 átomos de N, O y/o S;Ar2 indica fenilo, que está no sustituido o mono o disustituido por Hal, NO2 , CN, R3a, OR3a, CONHR3a, NR3aCOR3b, SO2R3a, SOR3a, NH2 , NHR3a, N(R3a)2 y/o (CH2)q-R6;Het2 indica un heterociclo de 5 o 6 átomos saturado, insaturado o aromático con 1 a 4 átomos de N, O y/o S, que no está sustituido o está mono o disustituido por Hal, NO2 , CN, R3a, OH, OR3a, CONHR3a, NR3aCOR3b, SO2R3a, SOR3a, NH2 , NHR3a, N(R3a)2 , (CH2)q-R6 y/u oxo (=O);q indica 1,
R3a, R3b indican cada uno, independientemente entre sí, un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8 , donde de 1 a 5 átomos de H pueden estar sustituidos por Hal, CN, OH y/u OAlq; R4a, R4b indican cada uno, independientemente entre sí, H o R3a;oR4a y R4b forman juntos un grupo alquileno C3-C8 ;R5a, R5b indican cada uno, independientemente entre sí, H, R3a, Ar2 o Het2;R6 indica OH u OR3a;T1, T2, T3, T4, T5, T6, T7, T8 y T9 indican cada uno, independientemente entre sí, O, SO, C=O;Alq indica alquilo C1-C6 lineal o ramificado;Ar1 representa un carbociclo aromático de 6 átomos;Het1 representa un heterociclo saturado, insaturado o aromático de 5 o 6 átomos con 1 a 4 átomos de N, O y/o S;Ar2 indica fenilo, que está no sustituido o mono o disustituido por Hal, NO2 , CN, R3a, OR3a, CONHR3a, NR3aCOR3b, SO2R3a, SOR3a, NH2 , NHR3a, N(R3a)2 y/o (CH2)q-R6;Het2 indica un heterociclo de 5 o 6 átomos saturado, insaturado o aromático con 1 a 4 átomos de N, O y/o S, que no está sustituido o está mono o disustituido por Hal, NO2 , CN, R3a, OH, OR3a, CONHR3a, NR3aCOR3b, SO2R3a, SOR3a, NH2 , NHR3a, N(R3a)2 , (CH2)q-R6 y/u oxo (=O);q indica 1, - 2,
- 3,
- 4, 5 o 6;m indica 0, 1 o 2;Hal indica F, Cl, Br o I;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.Un compuesto de fórmula (I) según la reivindicación 1, dondeR1, R2 indican cada uno, independientemente entre sí, H o alquilo C1-C4 , o R1 y R2 forman juntos un resto según la fórmula (CE); yLY indica CH2 o CH2CH2 , donde de 1 a 2 átomos de H pueden estar sustituidos por Hal, R3a, OR4a; y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.Un compuesto de fórmula (I) según la reivindicación 1 o 2, dondeT1, T2, T3, T4, T5, T6, T7, T8 y T9 indican O;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1, 2 o 3, dondeP1 indica un alquilo C1-C6 lineal o ramificado o cicloalquilo C3-C8 , independientemente entre sí, no sustituido o mono-, di- o trisustituido por Hal, CN, R3a, OR3a y/o (CH2)q-R6;P2 indica fenilo, piridilo, pirrolilo, furanilo, tiofenilo, pirimidilo, piranzinilo o piridazinilo, cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; yP3 indica un resto bicíclico de fórmula (ya), (yb), (yc), (yd), (ye), (yf), (yg), (yh), (yi), (yj), (yk), (yl), (ym), (yn), (yo) o (yp), cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6:
 dondeEa indica O, S, N(Alq) o CH=CH;Eb indica O, S, N(Alq), CH2 , CH2-CH2, O-CH2 , S-CH2 o N(Alq)CH2 ;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
dondeEa indica O, S, N(Alq) o CH=CH;Eb indica O, S, N(Alq), CH2 , CH2-CH2, O-CH2 , S-CH2 o N(Alq)CH2 ;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones. - 5. Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 4, dondeR3a, R3b indican cada uno, independientemente entre sí, un alquilo C1-C4 lineal o ramificado o cicloalquilo C3-C6 , donde de 1 a 3 átomos de H pueden estar sustituidos por F, Cl y/o donde 1 o 2 átomos de H pueden estar sustituidos por CN, OH, OCH3 y/o OC2H5 ;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 6. Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 5, donde Y indica P2 o P3 y solvatos, tautómeros, oligómeros y estereoisómeros de los mismos, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 7. Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 6, en el que el centro estereogénico en el átomo de carbono adyacente al resto de ácido borónico muestra una configuración (R) según la fórmula (R)-(I)/ YO LY■ OR1X ^ NH ^ B ' 'OR 2 (R)-(I).y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 8. Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 7, dondeX es un heterobiciclo o heterotriciclo de fórmula (xa1), (xb1), (xc1), (xd1), (xe1), (xf1), (xg1), (xh1) o (xi1) cada uno, independientemente entre sí, no sustituido o mono, di o trisustituido por Hal, NO2 , CN, R5a, OR5a, CONR5aR5b, NR5aCOR5b, SO2R5a, SOR5a, SO2NR5aR5b, NR5aSO2R5b, NR5aR5b, (CH2)q-R6, COR5a y/o SO2R5a, y donde 1 de los grupos CH2 cíclicos puede estar sustituido por CR4aR4b, C=O, O, S, NR5a, SO y/o SO2 :
 aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones. - 9. Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 7, donde X es un heterobiciclo o heterotriciclo de fórmula (xa), (xb), (xc), (xd), (xe), (xf), (xg), (xh) o (xi), cada uno, independientemente entre sí, no sustituido o mono o disustituido por F, Cl, CH3 , C2H5 , CF3 , OCH3 , OC2H5 , COCF3 , SCH3 , SC2H5 , CH2OCH3, N(CH3)2, CH2N(CH3)2 y/o N(C2H5); y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 10. Un compuesto de fórmula (I) según la reivindicación 8, donde X es un heterobiciclo o heterotriciclo de fórmula (xa1), (xb1), (xc1), (xd1), (xe1), (xf1), (xg1), (xh1) o (xi 1), cada uno, independientemente entre sí, no sustituido o mono o disustituido por F, Cl, CH3 , C2H5 , CF3 , OCH3 , OC2H5 , COCF3 , SCH3 , SC2H5 , CH2OCH3, N(CH3)2, CH2N(CH3)2 y/o N(C2H5); y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 11. Un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 10, dondeP3 indica 1 o 2-naftilo no sustituido o mono o disustituido, donde los sustituyentes opcionales se selec cionan entre un grupo compuesto por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6,oP3 es un resto según la fórmula (Ra) o (Rb):
 dondeGa, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a, CONH2 , NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2 , (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a, CONH2 , NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2 , (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; Ea indica O, S, N(Alq) o CH=CH; yEb indica O, S, N(Alq), CH2 , CH2-CH2, O-CH2 , S-CH2 o N(Alq)CH2.
dondeGa, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a, CONH2 , NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2 , (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OR3a, CONHR3a, CONR3bR3a, CONH2 , NR3aCOR3b, SO2R3a, SOR3a, NHR3a, N(R3a)2 , (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; Ea indica O, S, N(Alq) o CH=CH; yEb indica O, S, N(Alq), CH2 , CH2-CH2, O-CH2 , S-CH2 o N(Alq)CH2. - 12. Un compuesto de fórmula (I) según la reivindicación 11, donde Ea, Eb indican cada uno O o S, y solvatos, tautómeros, oligómeros y estereoisómeros de los mismos, así como las sales farmacéuticamente acepta bles de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 13. Un compuesto de fórmula (I) según la reivindicación 11 o 12, donde P3 es un resto según la fórmula (Fa) o (Fb):
 (Fa) (Fb)y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
(Fa) (Fb)y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones. - 14. Un compuesto de fórmula (I) según la reivindicación 13, dondeP2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-, 2,3-, 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre un grupo compuesto por Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;P3 indica un resto según la fórmula (Fa) o (S)-(Fb)
 (S)-(Fb);Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
(S)-(Fb);Ga, Gb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6;Ka, Kb indican cada uno, independientemente entre sí, H, Hal, CN, R3a, OH, OR3a, CONR4aR4b, NR3aCOR3b, SO2R3a, SOR3a, NR4aR4b, Ar2, Het2, (CH2)q-SR3a, (CH2)q-N(R4a)2 y/o (CH2)q-R6; y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones. - 15. Un compuesto de fórmula (I) según la reivindicación 14, dondeP2 indica 2- o 3-tienilo no sustituido o mono o disustituido o fenilo no sustituido o sustituido en 3-, 4-, 2,3-, 2,4-, 2,5-, 3,4- o 2,3,4-, donde los sustituyentes opcionales se seleccionan entre un grupo compuesto por F, Cl, CN, CH3, C2H5 , CF3 , OCH3 , OC2H5 , COCF3 , SCH3 , SC2H5 , CH2OCH3, N(CH3)2, CH2N(CH3)2 o N(C2H5)2;P3 indica un resto según la fórmula (Fa) o (S)-(Fb),Ga, Gb indican cada uno, independientemente entre sí, H, F, Cl, CN, CH3 , C2H5 , CF3 , OCH3 , OC2H5 , COCF3 , SCH3 , SC2H5 , CH2OCH3 , N(CH3)2, CH2N(CH3)2 o N(C2H5)2;Ka, Kb indican cada uno, independientemente entre sí, H, F, Cl, CH3 , C2H5 , CF3 , OCH3 , OC2H5 , COCF3 , SCH3 , SC2H5 , CH2OCH3 , N(CH3)2, CH2N(CH3)2 o N(C2H5)2;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 16. Un compuesto según la reivindicación 1, seleccionado entre el grupo compuesto por:ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}-2-(tiofen-3-il)etil]borónico; ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,2R,4S)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1R,2R,4S)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(7-don>1-benzofuran-3-il)-1-{[(1R,2S,4S)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-(7-doro-1-benzofuran-3-N)-1-{[(1S,2R,4R)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3R)-7-metil-2,3-dihidro-1-benzofuran-3-N]-1-([(1S,2R,4R)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-7-metN-2,3-dihidro-1-benzofuran-3-il]-1-([(1S,2R,4R)-7-oxabicido[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(1-benzofuran-3-N)-1-{[(1S,6S,7R)-3-cidopropN-4-oxo-10-oxa-3-azatnddo[5.2.1.01,5]dec-8-en-6-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(7-metil-1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2,4,6-trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(7-metil-1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2,4,6-trien-1-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,8R)-8-metil-11-oxatriciclo[6.2.1.02,7]undeca-2.4.6- trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1R,8S)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-9-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,8S)-8-metil-11-oxatriciclo[6.2.1.02,7]undeca-2.4.6- trien-1-il]formamido}etil]borónico;ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,8R)-11-oxatriciclo[6.2.1.02,7]undeca-2(7),3,5-trien-9-il]formamido}etil]borónico;ácido [(1R)-2-(2,4-dimetilfenil)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-ciclohexil-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}-3-fenilpropil]borónico;ácido [(1R)-3-metil-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}butil]borónico;ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-(1-benzofuran-3-il)-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1S)-2-(1-benzofuran-3-il)-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico; ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-H[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-([(1S,2S,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-([(1R,2R,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3S)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1R)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-{[(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-([(1S,2R,4R)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;ácido [(1S)-2-[(3R)-2,3-dihidro-1-benzofuran-3-il]-1-([(1R,2S,4S)-7-oxabiciclo[2.2.1]heptan-2-il]formamido}etil]borónico;y solvatos, tautómeros, oligómeros y estereoisómeros del mismo, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones.
- 17. Proceso para la preparación de un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 16 y sales farmacéuticamente aceptables, tautómeros y estereoisómeros del mismo, caracterizado porque un compuesto de fórmula (III)
 se acopla con un compuesto de fórmula (VI)
se acopla con un compuesto de fórmula (VI) donde todos los restos de fórmula (III) y fórmula (IV) son como se define en cualquiera de las reivindica ciones 1 a 15 y donde el compuesto obtenido de fórmula (Ib) puede convertirse posteriormente en un compuesto de fórmula (la) mediante el tratamiento con HCl, HBr, HI y/o TFA, en presencia o ausencia de un exceso de ácido borónico de bajo peso molecular
donde todos los restos de fórmula (III) y fórmula (IV) son como se define en cualquiera de las reivindica ciones 1 a 15 y donde el compuesto obtenido de fórmula (Ib) puede convertirse posteriormente en un compuesto de fórmula (la) mediante el tratamiento con HCl, HBr, HI y/o TFA, en presencia o ausencia de un exceso de ácido borónico de bajo peso molecular
- 18. Una formulación farmacéutica que comprende al menos un compuesto de fórmula (I) como se define en cualquiera de las reivindicaciones 1 a 16 y/o un solvato, tautómero, oligómero o estereoisómero del mismo, así como una sal farmacéuticamente aceptable de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones, como principio activo, junto con un vehículo farmacéuticamente aceptable.
- 19. Una formulación farmacéutica según la reivindicación 18 que además comprende un segundo principio activo, en el que el segundo principio activo es diferente de un compuesto de fórmula (I), como se define en una cualquiera de las reivindicaciones 1 a 16.
- 20. Un compuesto de fórmula (I) como se define en una cualquiera de las reivindicaciones precedentes o sus solvatos, tautómeros, oligómeros o estereoisómeros, así como las sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones, para su uso en la prevención y/o tratamiento de enfermedades que se ven afectadas por la inhibición de LMP7.
- 21. Un compuesto de fórmula (I) como se define en una cualquiera de las reivindicaciones precedentes para su uso en el tratamiento y/o prevención de una anomalía inmunorreguladora o un cáncer como, en parti cular, una neoplasia maligna hematológica o un tumor sólido.
- 22. Un compuesto para su uso según la reivindicación 21, donde la anomalía inmunorreguladora es una en fermedad inflamatoria crónica o autoinmune seleccionada entre el grupo compuesto por: lupus eritematoso sistémico, artritis reumatoide crónica, enfermedad inflamatoria intestinal, esclerosis múltiple, esclerosis la teral amiotrófica (ELA), ateroesclerosis, escleroderma, hepatitis autoinmune, síndrome de Sjogren, nefritis lúpica, glomerulonefritis, artritis reumatoide, psoriasis, miastenia grave, nefropatía por inmunoglobulina A, vasculitis, rechazo de trasplante, miositis, púrpura de Henoch-Schonlein y asma; y donde la neoplasia maligna hematológica es una enfermedad seleccionada entre el grupo compuesto por: mieloma múltiple, linfoma de células del manto, linfoma difuso de células B grandes, plasmocitoma, linfoma folicular, inmunocitoma, leucemia linfoblástica aguda, leucemia linfocítica crónica y leucemia mieloide; y donde el tumor sólido se selecciona entre el grupo compuesto por: cáncer inflamatorio de mama y colon, cáncer de pulmón, cáncer de cabeza y cuello, cáncer de próstata, cáncer de páncreas, cáncer de vejiga, cáncer renal, cáncer hepatocelular y cáncer gástrico.
- 23. Set (kit) compuesto por envases independientes de(a) una cantidad eficaz de un compuesto de la fórmula (I) y/o un solvato, tautómero, oligómero o este reoisómero del mismo, así como una sal farmacéuticamente aceptable de cada uno de los anteriores, incluidas sus mezclas en todas las proporciones como se reivindica en la reivindicación 1,y(b) una cantidad eficaz de un principio activo adicional de un medicamento.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17187636 | 2017-08-24 | ||
| PCT/EP2018/072485 WO2019038250A1 (en) | 2017-08-24 | 2018-08-21 | BORONIC ACID DERIVATIVES |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2927283T3 true ES2927283T3 (es) | 2022-11-03 |
Family
ID=59699569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18755481T Active ES2927283T3 (es) | 2017-08-24 | 2018-08-21 | Derivados del ácido borónico |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US11274109B2 (es) |
| EP (1) | EP3672977B1 (es) |
| JP (1) | JP7189203B2 (es) |
| KR (1) | KR20200040295A (es) |
| CN (1) | CN111183142B (es) |
| AU (1) | AU2018321118B2 (es) |
| CA (1) | CA3073611A1 (es) |
| DK (1) | DK3672977T3 (es) |
| ES (1) | ES2927283T3 (es) |
| IL (1) | IL272806B2 (es) |
| MX (1) | MX2020001891A (es) |
| PL (1) | PL3672977T3 (es) |
| PT (1) | PT3672977T (es) |
| TW (1) | TWI791595B (es) |
| WO (1) | WO2019038250A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3198048A1 (en) | 2020-10-05 | 2022-04-14 | Merck Patent Gmbh | Lmp7-selective inhibitors for the treatment of blood disorders and solid tumors |
| IL310390A (en) * | 2021-07-29 | 2024-03-01 | Merck Patent Gmbh | Crystalline forms of [(R1)-2-benzofuran-3-yl)-1-{[(R4, R2, S1)-7-oxabicyclo[2.2.1]heptan-2-yl]formamido}ethyl]boronic acid, Their adducts and their use |
| WO2023061445A1 (zh) * | 2021-10-14 | 2023-04-20 | 首药控股(北京)股份有限公司 | 硼酸衍生物 |
| WO2023232830A1 (en) | 2022-06-02 | 2023-12-07 | Merck Patent Gmbh | Boronic acid adducts |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006040646A1 (en) * | 2004-10-14 | 2006-04-20 | Pfizer, Inc. | Benzimidazole or indole amides as inhibitors of pin1 |
| US20090325903A1 (en) | 2008-06-17 | 2009-12-31 | Millennium Pharmaceuticals, Inc. | Boronate ester compounds and pharmaceutical compositions thereof |
| MX352652B (es) | 2011-12-22 | 2017-12-04 | Ares Trading Sa | Inhibidores de inmunoproteasoma selectivos de derivados del acido alfa-amino boronico. |
| EP2819673B1 (en) | 2012-03-02 | 2016-09-07 | Dr. Reddy's Laboratories Ltd. | Pharmaceutical compositions comprising boronic acid compounds |
| CN104822689B (zh) | 2012-12-03 | 2016-11-02 | 弗·哈夫曼-拉罗切有限公司 | 取代的三唑硼酸化合物 |
| CA2903288C (en) * | 2013-03-15 | 2021-09-21 | Deciphera Pharmaceuticals, Llc | N-acyl-n'-(pyridin-2-yl) ureas and analogs exhibiting anti-cancer and anti-proliferative activities |
| MX370336B (es) * | 2014-10-01 | 2019-12-10 | Merck Patent Gmbh | Derivados de acido boronico. |
| CA2963198A1 (en) | 2014-10-01 | 2016-04-07 | Merck Patent Gmbh | Boronic acid derivatives |
| WO2016050355A1 (en) | 2014-10-01 | 2016-04-07 | Merck Patent Gmbh | Boronic acid derivatives |
| SG11201702628XA (en) * | 2014-10-01 | 2017-04-27 | Merck Patent Gmbh | Boronic acid derivatives |
| CN106008572B (zh) * | 2016-05-23 | 2018-08-17 | 成都千禧莱医药科技有限公司 | 一类二肽硼酸化合物及制备方法和用途 |
-
2018
- 2018-08-21 CN CN201880054929.9A patent/CN111183142B/zh active Active
- 2018-08-21 PT PT187554811T patent/PT3672977T/pt unknown
- 2018-08-21 AU AU2018321118A patent/AU2018321118B2/en active Active
- 2018-08-21 US US16/640,669 patent/US11274109B2/en active Active
- 2018-08-21 KR KR1020207008149A patent/KR20200040295A/ko not_active Application Discontinuation
- 2018-08-21 ES ES18755481T patent/ES2927283T3/es active Active
- 2018-08-21 CA CA3073611A patent/CA3073611A1/en active Pending
- 2018-08-21 DK DK18755481.1T patent/DK3672977T3/da active
- 2018-08-21 IL IL272806A patent/IL272806B2/en unknown
- 2018-08-21 MX MX2020001891A patent/MX2020001891A/es unknown
- 2018-08-21 EP EP18755481.1A patent/EP3672977B1/en active Active
- 2018-08-21 JP JP2020511174A patent/JP7189203B2/ja active Active
- 2018-08-21 PL PL18755481.1T patent/PL3672977T3/pl unknown
- 2018-08-21 WO PCT/EP2018/072485 patent/WO2019038250A1/en unknown
- 2018-08-23 TW TW107129390A patent/TWI791595B/zh active
-
2022
- 2022-01-31 US US17/649,411 patent/US11858951B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| TWI791595B (zh) | 2023-02-11 |
| RU2020111001A (ru) | 2021-09-24 |
| AU2018321118A1 (en) | 2020-04-09 |
| US11858951B2 (en) | 2024-01-02 |
| JP2020531510A (ja) | 2020-11-05 |
| CA3073611A1 (en) | 2019-02-28 |
| IL272806A (en) | 2020-04-30 |
| BR112020003368A2 (pt) | 2020-08-18 |
| US11274109B2 (en) | 2022-03-15 |
| AU2018321118B2 (en) | 2023-06-15 |
| RU2020111001A3 (es) | 2022-02-28 |
| IL272806B2 (en) | 2023-09-01 |
| IL272806B1 (en) | 2023-05-01 |
| MX2020001891A (es) | 2020-03-24 |
| US20200354382A1 (en) | 2020-11-12 |
| CN111183142A (zh) | 2020-05-19 |
| JP7189203B2 (ja) | 2022-12-13 |
| EP3672977B1 (en) | 2022-06-22 |
| EP3672977A1 (en) | 2020-07-01 |
| WO2019038250A1 (en) | 2019-02-28 |
| CN111183142B (zh) | 2023-06-16 |
| US20220153761A1 (en) | 2022-05-19 |
| PL3672977T3 (pl) | 2022-09-26 |
| DK3672977T3 (da) | 2022-09-26 |
| KR20200040295A (ko) | 2020-04-17 |
| TW201920208A (zh) | 2019-06-01 |
| PT3672977T (pt) | 2022-09-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2944573T3 (es) | Antagonistas de TLR7/8 y usos de los mismos | |
| ES2927283T3 (es) | Derivados del ácido borónico | |
| ES2811273T3 (es) | Derivados del ácido borónico | |
| ES2968443T3 (es) | Derivados de ácido borónico | |
| ES2873000T3 (es) | Derivados del ácido borónico. | |
| ES2876287T3 (es) | Derivados del ácido borónico. | |
| US20140206673A1 (en) | Therapeutically active compositions and their methods of use | |
| EP3842437B1 (en) | High activity sting protein agonist | |
| US20210300940A1 (en) | Tlr7/8 antagonists and uses thereof | |
| EP3848366A1 (en) | Highly active sting protein agonist compound | |
| RU2793315C2 (ru) | Производные бороновой кислоты | |
| BR112020003368B1 (pt) | Derivados de ácido borônico, seu processo de preparação e seus usos, e formulação farmacêutica | |
| WO2023232830A1 (en) | Boronic acid adducts |