ES2917377T3 - Derivados de benzimidazol, procedimientos de preparación y utilizaciones de los mismos - Google Patents
Derivados de benzimidazol, procedimientos de preparación y utilizaciones de los mismos Download PDFInfo
- Publication number
- ES2917377T3 ES2917377T3 ES17882916T ES17882916T ES2917377T3 ES 2917377 T3 ES2917377 T3 ES 2917377T3 ES 17882916 T ES17882916 T ES 17882916T ES 17882916 T ES17882916 T ES 17882916T ES 2917377 T3 ES2917377 T3 ES 2917377T3
- Authority
- ES
- Spain
- Prior art keywords
- fluoro
- methyl
- pyridin
- imidazo
- pyrimidin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
La presente invención se relaciona con los compuestos de bencimidazol útiles en el tratamiento de trastornos asociados a la proteína quinasa. También existe la necesidad de compuestos útiles en el tratamiento o prevención de uno o más síntomas de cáncer, rechazos de trasplante. Además, existe la necesidad de métodos para modular la actividad de las proteínas quinasas, como CDK4 y/o CDK6, utilizando los compuestos proporcionados en este documento. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de benzimidazol, procedimientos de preparación y utilizaciones de los mismos
Sector técnico
La presente invención se refiere a compuestos de benzimidazol que son útiles para inhibir la cinasa dependiente de ciclina. Más especialmente, la presente invención da a conocer compuestos y composiciones farmacéuticas de los mismos que se utilizan como inhibidores de CDK4/6 y procedimientos de tratamiento para enfermedades mediadas por CDK4/6, tales como el cáncer.
Estado de la técnica anterior
Las cinasas dependientes de ciclina (CDK, Cyclin-dependent kinases) median la progresión del ciclo celular, regulando la transición de la fase G1 a la fase S y de la fase G2 a la fase M. La actividad de las CDK está estrictamente controlada a lo largo del ciclo celular por modificaciones postranscrlpcionales, así como por la expresión de ciclinas e Inhibidores de CDK. Existen cuatro CDK proliferativas: la CDK1, que regula predominantemente la transición de la fase G2 a la M, y las CDK2/4/6, que regulan la transición de la fase G1 a la S. La progresión a través del ciclo celular es un proceso altamente regulado. En ausencia de señales de crecimiento adecuadas, una familia de proteínas de bolsillo, que incluye la proteína de retinoblastoma (pRb), impide que las células entren en la fase de replicación del ADN (fase S). El ciclo de replicación comienza cuando los mltógenos desencadenan vías de transducción de señales, lo que conduce a un aumento de los niveles celulares de ciclinas D. Las ciclinas D, a su vez, activan las cinasas dependientes de ciclina 4/6 (CDK4/6), que fosforllan e inactivan la pRb. La proliferación celular descontrolada es una de las características del cáncer, y la inactivación de pRb es el evento clave que permite que las células tumorales progresen a través del ciclo celular sin control. Mientras que algunos tumores eliminan el propio gen pRb, la mayoría mantiene un pRb funcional y en su lugar activa la actividad de la cinasa CDK4/6. La ablación de la actividad de la cinasa CDK4/6 ha conducido a la inhibición completa del crecimiento tumoral en muchos tipos de cáncer, tales como el cáncer de mama HR+, el linfoma de células del manto, el glloblastoma y el cáncer de pulmón escamoso. Además, se ha demostrado que las células de fibroblastos normales superan la ausencia de CDK4/6 debido a la compensación por parte de CDK1, cuya ausencia no se tolera. En conjunto, esta evidencia sugiere que un inhibidor selectivo de CDK4/6 puede tener una ventana terapéutica más amplia que los inhibidores pan-CDK.
Además de los efectos antlneopláslcos directos, se ha descubierto que los inhibidores de CDK4/6 tratan enfermedades Inflamatorias, enfermedades óseas, enfermedades metabóllcas, enfermedades neurológlcas y neurodegenerativas, enfermedades cardiovasculares, alergias y asma, enfermedades de Alzhelmer y enfermedades relacionadas con hormonas. Por consiguiente, ha habido un esfuerzo sustancial en química médica para descubrir inhibidores de CDK4/6 que sean efectivos como agentes terapéuticos.
La CDK1 es un determinante clave de la progresión mitótlca y es la única CDK que puede iniciar el comienzo de la mltosis. Los experimentos de desactivación en ratones han demostrado que se requiere CDK1 para la proliferación de células de mamíferos111. Dado que la CDK1 es fundamental para la proliferación celular, la toxicidad provocada por la inhibición de CDK1 limitará la capacidad de alcanzar el nivel terapéutico, por lo que es necesario mantener la selectividad de CDK1 contra la diana farmacológica CDK4/6.
La CDK2 está estructural y funcionalmente relacionada con la CDK1; tiene un perfil de sustrato considerablemente más amplio que CDK4 y CDK6, y fosforila una gran cantidad de proteínas involucradas en la progresión del ciclo celular (por ejemplo, p27KIP1 y RB), la replicación de ADN (por ejemplo, factores de replicación A y C), la síntesis de histonas (por ejemplo, ejemplo, NPAT), la duplicación del centrosoma (por ejemplo, nucleofosmina (NPM, nudeophosmin)), entre otros procesos. A diferencia de CDK4 y CDK6, la CDK2 no está regulada mediante las proteínas INK4, sino por la clase de inhibidores de CDK que ¡nteractúan con CDK/proteína inhibidora de la cinasa (CIP/KIP, CDK-interacting protein/kinase inhibitory protein), que se enlazan a los complejos CDK2-ciclina y los vuelven inactivos. [1] Cuando los inventores de la presente invención diseñan un inhibidor de CDK4/6 como fármaco para el cáncer, es mejor mantener la selectividad contra CDK2.
Además de las CDK que promueven directamente la progresión del ciclo celular (por ejemplo, CDK4, CDK6, CDK2 y CDK1), se identificó una familia adicional de CDK que regulan la transcripción, que incluyen CDK7, CDK8 y CDK9. La CDK7 tiene una función general en la fosforilación del dominio carboxiterminal de la ARN polimerasa II que contribuye al inicio de la transcripción, y la CDK9 también fosforila la ARN polimerasa II, promoviendo de esta manera el alargamiento de la transcripción. [1] La primera generación de inhibidores de CDK son inhibidores de pan-CDK y no tuvieron éxito debido a toxicidades inmanejables. Por ejemplo, el flavopiridol es el inhibidor de CDK más investigado hasta el momento. Aunque el flavopiridol puede Inducir la detención del ciclo celular en las fases G1 y G2, en ciertos contextos también induce una respuesta cltotóxica, probablemente como resultado de la inhibición
de CDK7 y CDK9 que conduce a la supresión de la transcripción.121 Por lo tanto, es necesario evitar CDK7/9 cuando se diseñan fármacos dirigidos frente a CDK4/6.
Hasta el momento, se han evaluado de manera preclínica y clínica una variedad de inhibidores de CDK. Dada la evidencia descrita anteriormente, muchos grupos de investigación se han embarcado en el descubrimiento de un inhibidor selectivo de CDK4/6, siendo los bien documentados Palbociclib (PD-0332991), Ribociclib (LEE-011) y Abemaciclib (LY2835219). La Patente WO2016/173505A1 da a conocer ciertos compuestos y/o sales farmacéuticamente aceptables de los mismos que pueden inhibir la actividad cinasa de CDK4/6 y pueden ser útiles para el tratamiento de enfermedades hiperproliferativas, como el cáncer y la inflamación. Sin embargo, sigue existiendo la necesidad de dar a conocer inhibidores de CDK4/6 más potentes, selectivos y seguros que puedan utilizarse en el tratamiento de trastornos de proliferación celular, tales como el cáncer.
Referencia:
[1] . Uzma Asghar, Agnieszka K. Witkiewicz, Nicholas C. Turner, et al. Nat Rev Drug Discov. 2015,14(2): 130-146.
[2] , Prithviraj Bose, Gary L Simmons, Steven Grant. Expert Opin Investig Drugs. 2013, 22(6): 723-738.
Características de la invención
La presente invención se refiere a compuestos de benzimidazol que se utilizan como inhibidores de CDK4/6 y para el tratamiento de enfermedades mediadas por CDK4/6. Los compuestos de la presente invención tienen las estructuras generales de la fórmula I. Un compuesto de fórmula I, o un estereoisómero, un tautómero, un polimorfo, un solvato o una sal farmacéuticamente aceptable del mismo,

Fórmula I
en la que,
el anillo A es arilo o heteroarilo;
Z se selecciona entre el grupo que consiste en CH2, NH, O y S;
Ri se selecciona independientemente entre el grupo que consiste en hidrógeno, halógeno, CN, N02, OH, NH2, alquilo Ci-a, alcoxi Ci-8, cicloalquilo C3-8, arilo, heteroarilo, heterociclilo, heterociclilo-(CH2)m-, aril-alquilo C1-6-, heteroaril-alquilo C1-6-, -NRi2Ri3, -NRi2-alquileno Ci -6-NRi2Ri3 y heterociclilo-C(O)-, en los que cada uno de los alquilo Ci-a, alcoxi Ci-8, cicloalquilo C3-8, arilo, heteroarilo, heterociclilo, heterociclilo-(CH2)m-, aril-alquilo C1-6-, heteroaril-alquilo C1-6- o heterociclilo-C(O)- están sin sustituir o sustituidos con, como mínimo, un sustituyeme seleccionado entre halógeno, hidroxilo, alquilo Ci-8, cicloalquilo C3-8, heterociclilo, -NRi2Ri3 o -(CH2)tOH;
R2 y R3 se seleccionan cada uno independientemente entre H, OH, CN, N02, NH2, halógeno, alquilo Ci-8, alcoxi Ci-8, cicloalquilo C3-a, arilo, heteroarilo, heterociclilo; en los que cada uno de los alquilo Ci-8, alcoxi ¿i-a, cicloalquilo C3-8, arilo, heteroarilo, heterociclilo están sin sustituir o sustituidos con, como mínimo, un sustituyeme seleccionado entre halógeno, hidroxilo, alquilo Ci-8, cicloalquilo C3-8 o heterociclilo;
R12 y R13 se seleccionan cada uno independientemente entre H, alquilo Ci-8, arilo, heteroarilo, heterociclilo o cicloalquilo C3-8; en el que cada uno de los alquilo Ci-8, arilo, heteroarilo, heterociclilo o cicloalquilo C3-8 están sin sustituir o sustituidos con, como mínimo, un sustituyeme seleccionado entre halógeno, hidroxilo, alquilo Ci-8, cicloalquilo C3-8 o heterociclilo;
m es 0,1,2, 3 o 4;
n es 0,1, 2, 3 o 4;
t es 0,1,2, 3 o 4;
arilo es un sistema de anillo aromático de 6-10 miembros monocíclico o bicíclico;
heteroarilo es un sistema de anillo aromático monocíclico de cinco o seis miembros estable no sustituido o sustituido o un sistema de anillo heteroaromático benzocondensado de nueve o diez miembros no sustituido o sustituido o un sistema de anillo heteroaromático bicíclico que consiste en átomos de carbono y de uno a cuatro heteroátomos seleccionados entre N, O o S; y
heterociclilo es un sistema de anillo saturado monocíclico de tres a ocho miembros, estable, no sustituido o sustituido, que consiste en átomos de carbono y de uno a tres heteroátomos seleccionados entre N, O o S.
En algunas realizaciones de la fórmula I, Z es CH2.
En algunas realizaciones de la fórmula I, Z es O.
En algunas realizaciones de la fórmula I, el anillo A es un heteroarllo de 6 miembros que comprende uno o dos heteroátomos de N, por ejemplo, plrldllo, pirimidinilo, piridazinilo y similares.
En otras realizaciones de la fórmula I, el anillo A es

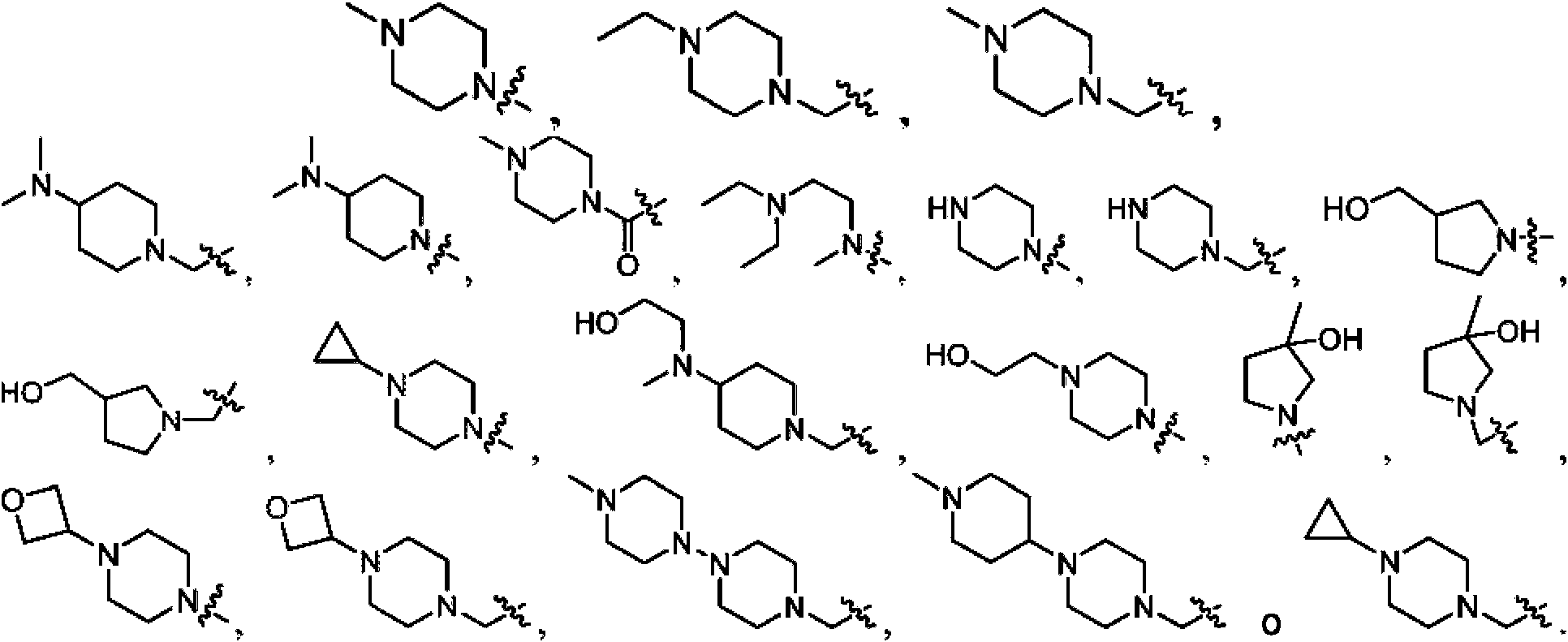
En algunas realizaciones de la fórmula I, Ri es heterociclilo-(CH2)m- o heterociclilo-(CH2)m- sustituido con alquilo Ci-8, NR12R13, heterociclilo de 4 a 6 miembros, cicloalquilo C3-6 o -(CH2)t-OH.
En otras realizaciones de la fórmula I, R1 es heterociclilo-CH2- de 5 a 6 miembros o heterociclilo-CH2- de 5 a 6 miembros sustituido con alquilo C1-3, -N(CH3)2, -N(CH2CH2OI-l)CH3,

-CH2OH, -CH2CH2OH o -OH.
En otras realizaciones de la fórmula I, R1 es heteroclclllo-CH2- de 6 miembros o heterociclilo-CH2- de 6 miembros sustituido con metilo o etilo.
En algunas realizaciones de la fórmula I, R1 es heterociclilo o heterociclilo sustituido con alquilo C1-8, NR12R13, heterociclilo de 4 a 6 miembros, cicloalquilo C3-6 o -(CH2)r OH.
En otras realizaciones de la fórmula I, R1 es heterociclilo de 5 a 6 miembros, o heterociclilo de 5 a 6 miembros sustituido con alquilo C1-3, -N(CH3)2, -N(CH2CH2OH)CH3,

-CHzOH, -CH2CH2OH u OH.
En otras realizaciones de la fórmula I, R1 es heterociclilo de 6 miembros o heterociclilo de 6 miembros sustituido con metilo o etilo.
En algunas realizaciones de la fórmula I, R1 es heterociclilo-C(O) de 6 miembros o heterociclilo-C(O) de 6 miembros sustituido con alquilo C1-3.
En otras realizaciones de la fórmula I, R1 es heterociclilo-C(O)- de 6 miembros sustituido con metilo.
En algunas realizaciones de la fórmula I, el heterociclilo comprende uno o dos heteroátomos de N u O como átomos del anillo.
En algunas realizaciones de la Fórmula I, el heterociclilo comprende uno o dos heteroátomos de N como átomos del anillo.
En algunas realizaciones de la fórmula I, R1 es -N R i2-alquileno C1-3-NR12R13.
En algunas realizaciones de la fórmula I, R12 y R13 son cada uno independientemente H, -(CH2)r OH o alquilo C1-3.
Preferentemente, R12 y R13 son cada uno independientemente OH, CH2CH2OH, metilo o etilo.
En algunas realizaciones de la fórmula I, R1 es
En algunas realizaciones de la fórmula I, m es 1.
En algunas realizaciones de la fórmula I, n es 1.
En algunas realizaciones de la fórmula I, t es 0,1 o 2.
En algunas realizaciones de la fórmula I, R2 y R3 son cada uno independientemente H, OH, halógeno, alquilo C1-6, alquilo C1-6 sustituido con halógeno, alcoxl Ci-6, alcoxi Ci-6 sustituido con halógeno.
En otras realizaciones de la fórmula I, R2 y R3 son cada uno independientemente H, OH, F, Cl, CH3, CH2CH3, CF3, -OCH3 o -OCF3.
En otras realizaciones de la fórmula I, R2 y R3 son ambos F.
La presente Invención da a conocer además algunas soluciones técnicas preferentes con respecto al compuesto de la fórmula I, y el compuesto es:
1) 4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(5-(4-metilpiperazin-1 -il)piridin-2-il)pirimidin-2-amina;
2) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
3) 5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(5-(4-metilpiperazin-1-i I) pi ridi n-2-i I) pi rim idi n-2-am i na;
4) 5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(6-((4-metilpiperazin-1-il)metil)piridin-3-il)pirimidin-2-amina;
5) 5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(5-((4-metilpiperazin-1-il)metil)pirimidin-2-il)pirimidin-2-amina;
6) N-(5-((4-(dimetilamino)piperidin-1-il)metil)pirimidin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
7) N-(5-((4-(dimetilamino)piperidin-1-il)metil)piridin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
8) N-(5-(4-(dimetilamino)piperidin-1-il)piridin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
9) (2-((5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-il)amino)pirimidin-5-il)(4-metilpiperazin-1-il)metanona;
10) (6-((5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-11) amino)piridin-3-il)(4-metilpiperazin-1-il)metanona;
11) N5-(2-(dietilamino)etil)-N2-(5-fluoro-4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-il)-N5-metilpiridin-2,5-diamina;
12) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-4-(6-fluoro-1-metil-1,2,3,4,4a,5-hexahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-5-(trifluorometil)pirimidin-2-amina;
13) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-4-(6-fluoro-1-metil-1,2,3,4,4a,5-hexahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-5-metilpirimidin-2-amina;
14) 5-cloro-N-(5-((4-etilpiperazin-1 -il)metil)piridin-2-il)-4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
15) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-(4-metilpiperazin-1-i I) pi ridi n-2-i I) pi rim idi n-2-am i na;
16) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-1-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
17) N-(5-(4-(dimetilamino)piperidin-1 -il)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
18) N-(5-((4-(dimetilamino)piperidin-1 -il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-1 -3,4-dihidro-1 H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
19) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina;
20) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-(piperazin-1-ilmetil)piridin-2-il)pirimidin-2-amina;
21) N-(5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)-6-(4-m etil pi perazi n-1 -i I) pi ridazi n-3-am ina;
22) é-((4-etilpiperazin-1-il)metil)-N-(5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)pi ridazi n-3-amina;
23) (1-(6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)pirrolidin-3-il)metanol;
24) (1 -((6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)metil)pirrolidin-3-il)metanol;
25) N-(5-(4-ciclopropilpiperazin-1-il)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
26) N-(5-((4-ciclopropilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
27) 2-((1 -((6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)metil)piperidin-4-il)(metil)amino)etan-1-ol;
28) 1-(6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)-3-metilpirrolidin-3-ol;
29) 1 -((6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)metil)-3-metilpirrolidin-3-ol;
30) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)-N-(5-((4-(oxetan-3-il)piperazin-1 -il)metil)piridin-2-il)pirimidin-2-amina;
31) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)-N-(5-(4-(oxetan-3-i I) pi perazi n-1 -i I) pi ridi n-2-i I) pi rim idi n-2-am ina;
32) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]pirazin-7-il)pirimidin-2-amina;
33) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-((4'-metil-[1,1'-bipiperazin]-4-il)metil)piridin-2-il)pirimidin-2-amina;
34) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-((4-(1-metilpiperidin-4-il)piperazin-1-il)metil)piridin-2-il)pirimidin-2-amina.
Sorprendentemente, el enantiómero (-) altamente purificado del compuesto de la fórmula I es ventajoso sobre el enantiómero (+) en actividad biológica. Por ejemplo, el enantiómero (-) ópticamente puro del compuesto 2-(N-(5-((4-etilpiperazin-1 -il)metil)piridin-2-il)-5-fluoro-4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-i I) pi ri m idi n-2-am i na) es más potente que su enantiómero (+).
A menos que se indique lo contrario, "(-)" en la presente invención significa que la rotación óptica es un valor negativo; y "(+)" significa que la rotación óptica es un valor positivo. El presente compuesto descrito en la presente memoria descriptiva puede ser un isómero (-) del compuesto y/o un isómero (+) del compuesto.
La presente invención da a conocer también composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto de la fórmula I o una sal terapéuticamente aceptable del mismo y un excipiente farmacéuticamente aceptable.
En algunas realizaciones, dicho compuesto en una proporción en peso con respecto a dicho excipiente dentro del intervalo de, aproximadamente, 0,001, a, aproximadamente,10.
La presente invención da a conocer adicionalmente un compuesto de la presente invención, una sal farmacéuticamente aceptable del mismo o una composición farmacéutica mencionada anteriormente para la preparación de un medicamento.
En algunas realizaciones, el medicamento se utiliza para el tratamiento del cáncer, tal como cáncer de colon, cáncer de recto, llnfoma de células del manto, mleloma múltiple, cáncer de mama, cáncer de próstata, glloblastoma, cáncer de esófago de células escamosas, llposarcoma, llnfoma de células T, melanoma, cáncer de páncreas, cáncer de cerebro o cáncer de pulmón.
En algunas realizaciones, el medicamento se utiliza como inhibidor de CDK, preferentemente, de CDK4 y/o CDK6. La presente Invención da a conocer compuestos para su utilización en un procedimiento de tratamiento del cáncer en un Individuo, que comprende administrar al Individuo que la necesita una cantidad terapéuticamente eficaz de un compuesto de la presente Invención, una sal farmacéuticamente aceptable del mismo o la composición farmacéutica mencionada anteriormente. En particular, el cáncer se selecciona entre el grupo que consiste en cáncer de colon, cáncer de recto, llnfoma de células del manto, mleloma múltiple, cáncer de mama, cáncer de próstata, glloblastoma, cáncer de esófago de células escamosas, llposarcoma, llnfoma de células T, melanoma, cáncer de páncreas, cáncer de cerebro o cáncer de pulmón.
La presente Invención da a conocer además compuestos para su utilización en un procedimiento de tratamiento de una enfermedad mediada por CDK, por ejemplo, CDK4 y/o CDK6 en un individuo, que comprende administrar al Individuo que la necesita una cantidad terapéuticamente eficaz de un compuesto de la presente Invención, una sal farmacéuticamente aceptable del mismo o la composición farmacéutica mencionada anteriormente.
Los términos químicos generales utilizados en la fórmula anterior tienen sus significados habituales. Por ejemplo, el término "halógeno", tal como se utiliza en la presente memoria descriptiva, a menos que se Indique lo contrario, significa flúor, cloro, bromo o yodo. Entre los grupos halógeno preferentes se incluyen F, Cl y Br.
Tal como se utiliza en la presente memoria descriptiva, a menos que se Indique lo contrarío, alquilo Incluye radicales hldrocarbonados monovalentes saturados que tienen restos lineales o ramificados. Por ejemplo, entre los radicales alquilo se Incluyen metilo, etilo, propilo, ¡sopropllo, n-butllo, ¡sobutllo, sec-butllo, t-butllo, n-pentllo, 3-(2-metll)butllo, 2-pentilo, 2-metilbutilo, neopentilo, n-hexilo, 2-hexilo, 2-metilpentilo y similares. De manera similar, Ci-8, tal como en alquilo C1-8, se define para Identificar el grupo que tiene 1, 2, 3, 4, 5, 6, 7 u 8 átomos de carbono en una disposición lineal o ramificada.
Entre los grupos alquenllo y alquinilo se Incluyen alquenos y alquinos de cadena lineal o ramificada. De Igual manera, "alquenilo C2-8" y "alquinilo C2-8" significan un radical alquenilo o alquinilo que tiene 2, 3, 4, 5, 6, 7 u 8 átomos de carbono en una disposición lineal o ramificada.
Los alcoxl son éteres de oxígeno formados a partir de los grupos alquilo de cadena lineal o ramificada descritos anteriormente, es decir -O-alquilo.
Tal como se utilizan en la presente memoria descriptiva, "un", “uno” "el", "como mínimo, uno" y "uno o más" se utilizan indistintamente. De este modo, por ejemplo, una composición que comprende "un" excipiente farmacéuticamente aceptable se puede Interpretar en el sentido de que la composición Incluye "uno o más" excipientes farmacéuticamente aceptables.
El término "arllo", tal como se utiliza en la presente memoria descriptiva, a menos que se Indique lo contrarío, se refiere a un sistema de anillo monocícllco o policíclico no sustituido o sustituido que contiene átomos de carbono en el anillo. Los arllos preferentes son sistemas de anillos aromáticos monocícllcos o bicíclicos de 6-10 miembros. Los arllos preferentes son fenllo y naftllo. El arllo más preferente es fenllo.
El término "heteroclclllo", tal como se utiliza en la presente memoria descriptiva, a menos que se Indique lo contrarío, representa un sistema de anillo saturado monocícllco de tres a ocho miembros estable no sustituido o sustituido que consiste en átomos de carbono y de uno a tres heteroátomos seleccionados entre N, O o S, y en el que los heteroátomos de nitrógeno o azufre pueden estar opclonalmente oxidados, y el heteroátomo de nitrógeno puede estar opclonalmente cuaternlzado. El grupo heteroclclllo se puede enlazar a cualquier heteroátomo o átomo de carbono que dé como resultado la creación de una estructura estable. Entre los ejemplos de dichos grupos heteroclclllo se Incluyen, sin limitación, azetidinilo, pirrolidinilo, piperidinilo, piperazinilo, oxopiperazinilo, oxopiperidinilo, tetrahldrofuranllo, dioxolanilo, tetrahidroimidazolilo, tetrahldrotlazolllo, tetrahldrooxazolllo, tetrahidropiranilo, morfolinilo, tiomorfolinilo, tiamorfolinilsulfóxido, tiazolinilsulfona y tetrahidrooxadiazolilo.
El término "heteroarilo", tal como se utiliza la presente memoria descriptiva, a menos que se indique lo contrario, representa un sistema de anillo aromático monocícllco estable de cinco o seis miembros no sustituido o sustituido o un sistema de anillo heteroaromátlco benzocondensado de nueve o diez miembros no sustituido o sustituido o un sistema de anillo heteroaromátlco bicíclico que consiste en átomos de carbono y de uno a cuatro heteroátomos seleccionados entre N, O o S, y en el que los heteroátomos de nitrógeno o azufre pueden estar opclonalmente oxidados, y el heteroátomo de nitrógeno puede estar opclonalmente cuaternlzado. El grupo heteroarilo se puede enlazar a cualquier heteroátomo o átomo de carbono que dé como resultado la creación de una estructura estable.
Entre los ejemplos de grupos heteroarllo se Incluyen, sin limitación, tlenllo, furanllo, imidazolilo, ¡soxazolllo, oxazolllo, pirazolilo, plrrolllo, tlazolllo, tiadiazolilo, trlazolllo, piridilo, piridazinilo, ¡ndolllo, azalndolllo, ¡ndazolllo, benzimidazolilo, benzofuranllo, benzotlenllo, benzlsoxazolllo, benzoxazolllo, benzoplrazolllo, benzotlazolllo, benzotiadiazolilo, benzotriazolilo adenlnllo, quinolinilo o isoquinolinilo.
El término "clcloalqullo" se refiere a una cadena de alquilo saturada cíclica que tiene de 3 a 12 átomos de carbono, por ejemplo, clclopropllo, clclobutllo, ciclopentilo o ciclohexilo.
El término "sustituido" se refiere a un grupo en el que uno o más átomos de hidrógeno se sustituyen cada uno Independientemente con el mismo o diferentes sustltuyentes. Entre los sustltuyentes normales se Incluyen, sin limitación, halógeno (F, Cl, Br o I), alquilo Ci-a, cicloalquilo C3-12, -OR1, SR1, =0, =S, -C(O)R1, -C(S)R1, =NR1, -C(O)0R1, -C(S)OR1, -NR1RZ, -C(O)NR1R2, ciano, nitro, -S(0)2R1, -0S(02)0R1, -0S(0)2R1, -OP(Ó)(OR1)(ORz); en los que R1 y Rz se seleccionan independientemente entre -H, alquilo C1-6, haloalqullo Ci-6. En algunas realizaciones, los sustltuyentes se seleccionan independientemente entre el grupo que consiste en -F, -Cl, -Br, -I, -OH, trifluorometoxi, etoxi, propiloxi, isopropiloxi, n-butiloxi, isobutiloxi, t-butiloxi, -SCH3, -SC2H5, grupo formaldehído, -C(OCH3), ciano, nitro, CF3, -OCF3, amino, dlmetllamlno, metiltio, sulfonilo y acetilo.
Entre los ejemplos de grupos alquilo sustituidos se Incluyen, sin limitación, 2-amlnoetllo, 2-hldroxletllo, pentacloroetllo, trlfluorometllo, metoxlmetllo, pentafluoroetllo y piperazinilmetilo.
Entre los ejemplos de grupos alcoxl sustituidos se Incluyen, sin limitación, amlnometoxl, trifluorometoxi, 2-dletllamlnoetoxl, 2-etoxlcarbonlletoxl, 3-hidroxipropoxi.
La expresión "sales farmacéuticamente aceptables" se refiere a sales preparadas a partir de bases o ácidos no tóxicos farmacéuticamente aceptables. Cuando el compuesto de la presente Invención es ácido, su sal correspondiente se puede preparar convenientemente a partir de bases no tóxicas farmacéuticamente aceptables, Incluyendo bases Inorgánicas y bases orgánicas. Entre las sales derivadas de dichas bases Inorgánicas se Incluyen sales de aluminio, amonio, caldo, cúpricas y cuprosas, férricas, ferrosas, de litio, magnesio, mangánlcas y manganosas, de potasio, sodio, zinc y similares. Son particularmente preferentes las sales de amonio, caldo, magnesio, potasio y sodio. Entre las sales derivadas de bases orgánicas no tóxicas farmacéuticamente aceptables se Incluyen sales de aminas primarlas, secundarías y terciarlas, así como aminas cíclicas y aminas sustituidas, tales como aminas sustituidas naturales y sintéticas. Entre otras bases orgánicas no tóxicas farmacéuticamente aceptables a partir de las cuales se pueden formar sales se Incluyen resinas de Intercambio Iónico tales como, por ejemplo, arginina, betaína, cafeína, colina, A/'.A/'-dibenziletilendiamina, dietilamina, 2-dietilaminoetanol, 2-dimetilaminoetanol, etanolamlna, etllendlamlna, N-etilmorfolina, N-etilpiperidina, glucamlna, glucosamlna, histidina, hldrabamlna, isopropilamina, Usina, metllglucamlna, morfolina, plperazlna, pi peridi na, resinas de pollamlna, procaína, purinas, teobromina, trietilamina, trimetilamina, tripropilamina, trometamina y similares.
Cuando el compuesto de la presente Invención es básico, su sal correspondiente se puede preparar convenientemente a partir de ácidos no tóxicos farmacéuticamente aceptables, Incluyendo ácidos Inorgánicos y orgánicos. Entre dichos ácidos se Incluyen, por ejemplo, ácido acético, bencenosulfónlco, benzoico, canforsulfónlco, cítrico, etanosulfónico, fórmico, fumárico, glucónico, glutámico, bromhídrico, clorhídrico, isetiónico, láctico, maleico, málico, mandélico, metanosulfónico, múcico, nítrico, pamoico, pantoténico, fosfórico, succínico, sulfúrico, tartárico, p-toluenosulfónico y similares. Son preferentes los ácidos cítrico, bromhídrico, fórmico, clorhídrico, maleico, fosfórico, sulfúrico y tartárico, siendo particularmente preferentes los ácidos fórmico y clorhídrico. Dado que los compuestos de la fórmula I están destinados para su utilización farmacéutica, se proporcionan preferentemente en forma sustancialmente pura, por ejemplo, como mínimo, con el 60 % de pureza, más adecuadamente, como mínimo, el 75 % de pureza, especialmente, como mínimo, el 98 % de pureza (% son sobre una base en peso).
Los compuestos de la presente Invención también pueden estar presentes en forma de sales farmacéuticamente aceptables. Para su utilización en medicina, las sales de los compuestos de la presente Invención se refieren a "sales farmacéuticamente aceptables" no tóxicas. Entre las formas de sal farmacéuticamente aceptables se Incluyen sales ácldas/anlónlcas o básicas/catiónicas farmacéuticamente aceptables. La sal áclda/anlónlca farmacéuticamente aceptable adopta generalmente una forma en la que el nitrógeno básico se protona con un ácido Inorgánico u orgánico. Entre los ácidos orgánicos o inorgánicos representativos se incluyen ácido clorhídrico, bromhídrico, yodhídrico, perclórico, sulfúrico, nítrico, fosfórico, acético, propiónico, glicólico, láctico, succínico, maleico, fumárico, málico, tartárico, cítrico, benzoico, mandélico, metanosulfónico, hidroxietanosulfónico, bencenosulfónico, oxálico, pamoico, 2-naftalenosulfónlco, p-toluenosulfónico, clclohexanosulfámlco, salicílico, sacarínico o trifluoroacético. Entre las sales básicas/catiónicas farmacéuticamente aceptables se Incluyen, sin limitación, sales de aluminio, caldo, cloroprocaína, colina, dletanolamlna, etllendlamlna, litio, magnesio, potasio, sodio y zinc.
Se pretende que la definición de cualquier sustltuyente o variable en una ubicación particular en una molécula sea Independiente de sus definiciones en cualquier otra parte de esa molécula. Se entiende que los sustltuyentes y los patrones de sustitución de los compuestos de la presente Invención pueden ser seleccionados por un experto en la materia para proporcionar compuestos que sean químicamente estables y que se puedan sintetizar fácilmente
medíante técnicas conocidas en la materia, así como con los procedimientos establecidos a continuación en la presente memoria descriptiva.
La presente invención incluye compuestos descritos en la presente memoria descriptiva que pueden contener uno o más centros asimétricos y, de este modo, pueden dar lugar a diastereómeros y enantiómeros ópticos. La presente invención incluye todos los díastereoísómeros posibles así como sus mezclas racémicas, sus enantiómeros resueltos sustancíalmente puros, todos los enantiómeros geométricos posibles y las sales aceptables farmacéuticamente de los mismos.
Se ha descubierto ahora que el enantíómero (-) ópticamente puro del compuesto de la presente invención es un inhibidor de CDK4/6 más potente. La presente invención incluye procedimientos para tratar una enfermedad mediada por CDK4/6 en un individuo, que comprende administrar a dicho individuo una cantidad de enantíómero (-) o una sal farmacéuticamente aceptable del mismo, sustancíalmente líbre de su enantíómero (+), siendo dicha cantidad suficiente para aliviar la enfermedad, pero insuficiente para provocar dichos efectos adversos.
La expresión "sustancíalmente líbre de su enantíómero (+)", tal como se utiliza en la presente memoria descriptiva, significa que la composición contiene una mayor proporción o porcentaje del enantíómero (-) en relación con el enantíómero (+), basándose dicho porcentaje en la cantidad total de la mezcla. En una realización, la expresión "sustancialmente libre de su enantíómero (+)" significa que la composición contiene, como mínimo, el 60 % en peso de enantíómero (-) y el 40 % en peso o menos de enantíómero (+). En una realización preferente, la expresión "sustancialmente libre de su enantíómero (+)" significa que la composición contiene, como mínimo, el 70 % en peso de enantíómero (-) y el 30 % en peso o menos de enantíómero (+). En otra realización, la expresión "sustancialmente libre de su enantíómero (+)" significa que la composición contiene, como mínimo, el 80 % en peso de enantíómero (-) y el 20 % en peso o menos de enantíómero (+). Además, la expresión "sustancialmente libre de su enantíómero (+)" significa que la composición contiene, como mínimo, el 90 % en peso de enantíómero (-) y el 10 % en peso o menos de enantíómero (+). Aún además, la expresión "sustancialmente libre de su enantíómero (+)" significa que la composición contiene, como mínimo, el 95% en peso de enantíómero (-) y el 5% en peso o menos de enantíómero (+). Además, la expresión "sustancialmente libre de su enantíómero (+)" significa que la composición contiene, como mínimo, el 99 % en peso de enantíómero (-) y el 1 % en peso o menos de enantíómero (+).
La fórmula I anterior se muestra sin una estereoquímica definitiva en ciertas posiciones. La presente invención incluye todos los estereoísómeros de la fórmula I y sus sales farmacéuticamente aceptables. Además, también se incluyen mezclas de estereoísómeros así como estereoísómeros específicos aislados. Durante el curso de los procedimientos sintéticos utilizados para preparar dichos compuestos, o al utilizar procedimientos de racemizacíón o epimerización conocidos por los expertos en la materia, los productos de estos procedimientos pueden ser una mezcla de estereoísómeros.
Cuando existe un tautómero del compuesto de la fórmula I, la presente invención incluye cualquier posible tautómero y sus sales farmacéuticamente aceptables, y mezclas de los mismos, excepto cuando se indique específicamente lo contrario.
Cuando el compuesto de la fórmula I y sus sales farmacéuticamente aceptables existen en forma de solvatos o formas polímórficas, la presente invención incluye cualquier posible solvato y forma polímórfica. El tipo de disolvente que forma el solvato no está particularmente limitado siempre que el disolvente sea farmacológicamente aceptable. Por ejemplo, se puede utilizar agua, etanol, propanol, acetona o similares.
En la presente memoria descriptiva, se pretende que el término "composición" incluya productos que incluyen las cantidades especificadas de los ingredientes especificados, así como cualquier producto que se produzca, directa o indirectamente, a partir de la combinación especificada de ingredientes especificados. Por lo tanto, las composiciones farmacéuticas que contienen los compuestos de la presente invención como principios activos y los procedimientos para preparar los compuestos de la presente invención también forman parte de la presente invención. Además, algunas de las formas cristalinas de los compuestos pueden existir como polimorfos, y estos polimorfos se incluyen en la presente invención. Además, algunos de los compuestos pueden formar solvatos con agua (es decir, hidratos) o disolventes orgánicos comunes, y estos solvatos también quedan dentro del alcance de la presente invención.
Las composiciones farmacéuticas de la presente invención comprenden un compuesto representado por la fórmula I (o una sal farmacéuticamente aceptable del mismo) como principio activo, un vehículo farmacéuticamente aceptable y, opcionalmente, otros ingredientes terapéuticos o adyuvantes. Las composiciones incluyen composiciones adecuadas para la administración oral, rectal, tópica y parenteral (incluyendo la subcutánea, intramuscular e intravenosa), aunque en cualquier caso la vía más adecuada dependerá del huésped particular, y de la naturaleza y gravedad de las afecciones para las que se administra el principio activo. Las composiciones farmacéuticas se pueden presentar convenientemente en forma de dosificación unitaria y prepararse medíante cualquiera de los procedimientos bien conocidos en la técnica farmacéutica.
En la práctica, los compuestos representados por la fórmula I, o sus sales farmacéuticamente aceptables, de la presente Invención, se pueden combinar como principio activo en una mezcla íntima con un vehículo farmacéutico, según las técnicas convencionales de preparación de compuestos farmacéuticos. El vehículo puede adoptar una amplia variedad de formas, dependiendo de la forma de preparación deseada para la administración, por ejemplo, oral o parenteral (Incluida la Intravenosa). De este modo, las composiciones farmacéuticas de la presente Invención se pueden presentar como unidades discretas adecuadas para la administración oral, tales como cápsulas, sellos o comprimidos, cada uno de los cuales contiene una cantidad predeterminada del principio activo. Además, las composiciones se pueden presentar en forma de polvo, gránulos, solución, suspensión en un líquido acuoso, líquido no acuoso, emulsión de aceite en agua o emulsión líquida de agua en aceite. Además de las formas de dosificación comunes expuestas anteriormente, el compuesto representado por la fórmula I, o una sal farmacéuticamente aceptable del mismo, también se puede administrar mediante medios de liberación y/o dispositivos de administración controlada. Las composiciones se pueden preparar mediante cualquiera de los procedimientos de farmacia. En general, dichos procedimientos Incluyen una etapa de asociación del principio activo con el vehículo que constituye uno o más Ingredientes necesarios. En general, las composiciones se preparan mezclando de manera uniforme e íntima el principio activo con vehículos líquidos o vehículos sólidos finamente divididos o ambos. A continuación, el producto se puede moldear convenientemente en la presentación deseada.
De este modo, las composiciones farmacéuticas de la presente Invención pueden Incluir un vehículo farmacéuticamente aceptable y un compuesto, un estereoisómero, un tautómero, un polimorfo, un solvato o una sal farmacéuticamente aceptable de la fórmula I. Los compuestos de la fórmula I o las sales farmacéuticamente aceptables de los mismos, también se pueden Incluir en composiciones farmacéuticas en combinación con uno o más compuestos terapéuticamente activos.
El vehículo farmacéutico utilizado puede ser, por ejemplo, un sólido, un líquido o un gas. Entre los ejemplos de vehículos sólidos se Incluyen lactosa, térra alba, sacarosa, talco, gelatina, agar, pectlna, goma arábiga, estearato de magnesio y ácido esteárico. Entre los ejemplos de vehículos líquidos se Incluyen jarabe de azúcar, aceite de cacahuete, aceite de oliva y agua. Entre los ejemplos de vehículos gaseosos se Incluyen dióxido de carbono y nitrógeno. Al preparar las composiciones para formas de dosificación oral, se puede utilizar cualquier medio farmacéutico conveniente. Por ejemplo, se pueden utilizar agua, gllcoles, aceites, alcoholes, agentes aromatizantes, conservantes, agentes colorantes y similares para formar preparaciones líquidas orales, tales como suspensiones, elixires y soluciones; mientras que vehículos tales como almidones, azúcares, celulosa mlcrocrlstallna, dlluyentes, agentes de granulación, lubricantes, aglutinantes, agentes disgregantes y similares se pueden utilizar para formar preparaciones sólidas orales tales como polvos, cápsulas y comprimidos. Debido a su facilidad de administración, los comprimidos y las cápsulas son las unidades de dosificación oral preferentes en las que se utilizan vehículos farmacéuticos sólidos. Opclonalmente, los comprimidos se pueden revestir mediante técnicas estándar acuosas o no acuosas.
Un comprimido que contiene la composición de la presente Invención se puede preparar por compresión o moldeo, opclonalmente con uno o más Ingredientes auxiliares o adyuvantes. Los comprimidos sometidos a compresión se pueden preparar comprimiendo, en una máquina adecuada, el principio activo en una forma de flujo libre, tal como polvo o gránulos, opclonalmente mezclado con un aglutinante, lubricante, dlluyente Inerte, surfactante o agente dispersante. Los comprimidos moldeados se pueden fabricar moldeando en una máquina adecuada una mezcla del compuesto en polvo humedecido con un diluyente líquido inerte. Cada comprimido contiene preferentemente de, aproximadamente, 0,05 mg a, aproximadamente, 5 g del principio activo y cada sello o cápsula contiene preferentemente de, aproximadamente, 0,05 mg a, aproximadamente, 5 g del principio activo. Por ejemplo, una formulación destinada a la administración oral a seres humanos puede contener de, aproximadamente, 0,5 mg a, aproximadamente, 5 g de agente activo, combinado con una cantidad adecuada y conveniente de material de vehículo que puede variar de, aproximadamente, el 5 a, aproximadamente, el 95 por ciento de la composición total. Las formas de dosificación unitarias contendrán generalmente entre, aproximadamente, 1 mg y, aproximadamente, 2 g del principio activo, normalmente, 25 mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800 mg o 1.000 mg.
Las composiciones farmacéuticas de la presente Invención adecuadas para administración parenteral se pueden preparar como soluciones o suspensiones de los compuestos activos en agua. Se puede Incluir un surfactante adecuado tal como, por ejemplo, hldroxlpropllcelulosa. Las dispersiones también se pueden preparar en gllcerol, polietilenglicoles líquidos y mezclas de los mismos en aceites. Además, se puede Incluir un conservante para evitar el crecimiento perjudicial de microorganismos.
Las composiciones farmacéuticas de la presente Invención adecuadas para utilización Inyectable Incluyen soluciones o dispersiones acuosas estériles. Además, las composiciones se pueden presentar en forma de polvos estériles para la preparación extemporánea de dichas soluciones o dispersiones Inyectables estériles. En todos los casos, la forma Inyectable final debe ser estéril y debe ser efectivamente fluida para facilitar el paso a través de la jeringa. Las composiciones farmacéuticas deben ser estables en las condiciones de fabricación y almacenamiento; por tanto, preferentemente, se deben ser conservar contra la acción contaminante de microorganismos, tales como bacterias y hongos. El vehículo puede ser un disolvente o medio de dispersión que contenga, por ejemplo, agua,
etanol, poliol (por ejemplo, gllcerol, propilenglicol y polietilenglicol líquido), aceites vegetales y mezclas adecuadas de los mismos.
Las composiciones farmacéuticas de la presente Invención pueden estar en una forma adecuada para utilización tópica tal como, por ejemplo, un aerosol, crema, pomada, loción, polvo para espolvorear o similares. Además, las composiciones pueden estar en una forma adecuada para su utilización en dispositivos transdérmlcos. Estas formulaciones se pueden preparar utilizando un compuesto representado por la fórmula I de la presente Invención, o una sal farmacéuticamente aceptable del mismo, mediante procedimientos de procesamiento convencionales. Como ejemplo, se prepara una crema o pomada mezclando material hidrofílico y agua, junto con, aproximadamente, de un 5% en peso a, aproximadamente, un 10% en peso del compuesto, para producir una crema o pomada que tenga la consistencia deseada.
Las composiciones farmacéuticas de la presente Invención pueden estar en una forma adecuada para la administración rectal en la que el vehículo es un sólido. Es preferente que la mezcla se conforme en supositorios de dosis unitaria. Los vehículos adecuados Incluyen manteca de cacao y otros materiales comúnmente utilizados en la técnica. Los supositorios se pueden conformar convenientemente mezclando en primer lugar la composición con el o los vehículos ablandados o fundidos seguido de enfriamiento y moldeado en moldes.
Además de los Ingredientes de vehículo mencionados anteriormente, las formulaciones farmacéuticas descritas anteriormente pueden Incluir, según corresponda, uno o más Ingredientes de vehículo adicionales, tales como dlluyentes, tampones, agentes aromatizantes, aglutinantes, agentes surfactantes, espesantes, lubricantes, conservantes (Incluidos los antloxldantes) y similares. Además, se pueden Incluir otros adyuvantes para hacer que la formulación sea ¡sotónica con la sangre del receptor previsto. Las composiciones que contienen un compuesto descrito por la fórmula I, o sus sales farmacéuticamente aceptables, también se pueden preparar en forma de concentrado líquido o en polvo.
Generalmente, los niveles de dosificación del orden de, aproximadamente, 0,01 mg/kg a, aproximadamente, 150 mg/kg de peso corporal por día son útiles en el tratamiento de las afecciones Indicadas anteriormente, o alternativamente de, aproximadamente, 0,5 mg a, aproximadamente, 7 g por paciente por día. Por ejemplo, se pueden tratar eficazmente el cáncer de colon, el cáncer de recto, el llnfoma de células del manto, el mleloma múltiple, el cáncer de mama, el cáncer de próstata, el glloblastoma, el cáncer de esófago de células escamosas, el llposarcoma, el llnfoma de células T, el melanoma, el cáncer de páncreas, el glloblastoma o el cáncer de pulmón mediante la administración de, aproximadamente, 0,01 a 50 mg del compuesto por kilogramo de peso corporal por día o, alternativamente de, aproximadamente, 0,5 mg a, aproximadamente, 3,5 g por paciente por día. Sin embargo, se entiende que pueden ser necesarias dosis más bajas o más elevadas que las Indicadas anteriormente. El nivel de dosis específico y los regímenes de tratamiento para cualquier Individuo en particular dependerán de una variedad de factores, Incluida la actividad del compuesto específico utilizado, la edad, el peso corporal, la salud general, el sexo, la dieta, el momento de la administración, la vía de administración, la tasa de la excreción, la combinación de fármacos, la gravedad y el curso de la enfermedad particular que se somete a la terapia, la disposición del Individuo a la enfermedad y el juicio del médico tratante.
Estos y otros aspectos resultarán evidentes a partir de la siguiente descripción escrita de la presente Invención. Ejemplos
Debe entenderse que la descripción general anterior y la descripción detallada siguiente son solo Ilustrativas y explicativas y no son restrictivas de ningún objeto reivindicado. Todas las partes y porcentajes son en peso y todas las temperaturas en grados Celsius, a menos que se Indique explícitamente lo contrario. Los compuestos descritos en la presente memoria descriptiva se pueden obtener a partir de fuentes comerciales o sintetizar mediante procedimientos convencionales, tales como los que se muestran a continuación, utilizando materiales de partida y reactivos disponibles en el mercado. En los ejemplos se han utilizado las siguientes abreviaturas:
ATP: trifosfato de adenosina;
B0C2O: dicarbonato de di-terc-butilo;
con-H2S04 : ácido sulfúrico concentrado;
Crk: CT10 (retrovirus tumoral de pollo 10);
DCM: diclorometano;
DEA: dletllamlna;
DEAD: azodicarboxilato de dietilo;
DIEA: N,N-diisopropiletilamina;
DMEM: medio Eagle modificado por Dulbecco;
DMF: N,N-dimetilformamida;
DMA: N,N-dlmetllacetam¡da;
DMAP: 4-N,N-dimetilaminopiridina;
DMSO: dimetilsulfóxido;
DTT: DL-ditiotreitol;
EA: acetato de etilo;
EDC: 1-etil-3-(3-dimetilaminopropil)carbodiimida;
EDTA: ácido etilendiaminotetraacético;
EtOH: alcohol etílico;
FBS: suero bovino fetal;
GSR: glutatión-S-transferasa;
HATU: hexafluorofosfato de 0-(7-Azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio;
HEPES: ácido 4-(2-hidroxietil)piperazin-1-etanosulfónico;
Hex: n-hexano;
h o hr: hora;
IPA: isopropanol
KOAc: acetato de potasio;
KTB: terc-butóxido de potasio;
MeOH: metanol;
min: minuto;
MsCI: cloruro de metllsulfonilo;
MTS: 3-(4,5-dimetiltiazol-2-il)-5-(3-carboximetoxifenil)-2-(4-sulfofenil)-2H-tetrazolio;
NaBH4: borohidruro de sodio;
NaBH(OAc)3: triacetoxiborohidruro de sodio;
P(Cy)3: triciclohexilfosfina;
Pd2(dba)3: tris(dibenzilidenacetona)dipaladio
Pd(dppf)CÍ2: [1,1 '-bis(difenilfosfino)ferroceno]dicloropaladio;
Pd(OAc)2: acetato de paladio;
PE: éter de petróleo;
PMS: metosulfato de fenazina;
POCI3: oxicloruro de fósforo;
P/S: solución de penicilina/estreptomicina;
T.A. o t.a.: temperatura ambiente;
SDS: dodecilsulfato de sodio;
SDS-PAGE: gel de electroforesls de pollacrilamida con dodecilsulfato de sodio;
TBAB: bromuro de tetrabutilamonio;
TE: trietilamina;
THF: tetrahidrofurano;
TLC: cromatografía en capa fina;
Tol: tolueno.
Preparación 1: 5-(4-metilpiperazin-1-il)piridin-2-amina (Intermedio M1)


5-bromo-2-nitropiridina 1-metilpiperazina
1-metil-4-(6-nitropiridin-3-il)piperazina

THF 5-(4-metilpiperazin-1 -il)piridin-2-amina
Se añadió 1-metilpiperazina (1,180 g) y K2CO3 (2,720 g) sucesivamente a una solución de 5-bromo-2-nitropiridina (2,010 g) en DMSO (20 mi). Se dejó la reacción agitando a 82 °C durante 15 horas en un baño de aceite. Se añadió agua (50 mi), se extrajo con DCM (20 mi x 8), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró a presión reducida, se purificó mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 1,940 g de 1 -metil-4-(6-nitropiridin-3-il)piperazina.
Se añadió Pd/C (0,194 g) a una solución de 1-metil-4-(6-nitropiridin-3-il)piperazina (1,940 g) en THF (25 mi) bajo hidrógeno durante 2 horas a T.A. El filtrado se recogió por filtración y, a continuación, se concentró para dar 1,480 g de 5-(4-metilpiperazin-1-il)piridin-2-amina.
MS(ES+): m/z= 193,1 (M+H)+.
Se prepararon los siguientes Intermedios (que se muestran en la tabla 1) esencialmente tal como se ha descrito para la 5-(4-met¡lp¡peraz¡n-1-¡l)plr¡d¡n-2-am¡na (denominada en la presente memoria descriptiva Intermedio M1) utilizando el derivado de plperazlna correspondiente.

Preparación 4: 6-((4-metilpiperazin-1-il)metil)piridin-3-amina (Intermedio M4)

Se añadió NaBH4 (1,220 g) a una solución de 5-bromopicolinaldehído (2,010 g) en MeOH (30 mi) a 0 °C en un baño de hielo, después de completar la adición de NaBH4, se retiró el baño de hielo y se calentó a temperatura ambiente de forma natural. Después de agitar durante 2 horas a T.A., la mezcla de reacción se inactivó con agua (50 mi) a 0 °C. Se extrajo con EA (50 mi x 2), la fase orgánica combinada se lavó con solución saturada de NaCI y se secó sobre Na2S04 anhidro, se concentró para dar 1,940 g de (5-bromopiridin-2-il)metanol.
Una solución de (5-bromopiridin-2-il)metanol (1,940 g) en THF (20 mi) se enfrió a 0 °C en un baño de hielo, a continuación, se añadió gota a gota a la solución cloruro de metllsulfonllo (1,780 g). Una vez completada la adición de cloruro de metllsulfonllo, se retiró el baño de hielo y se calentó hasta temperatura ambiente de forma natural. Después de agitar durante 2 horas a T.A., la mezcla de reacción se inactivó con agua (50 mi). Se extrajo con EA (50 mi x 2), la fase orgánica combinada se lavó con solución saturada de NaCI (50 mi) y se secó sobre Na2S04 anhidro, se concentró para dar 2,750 g de producto metanosulfonato de (5-bromopiridin-2-il)metilo en bruto.
Se añadió K2CO3 (2,870 g) y 1-metilpiperazina (1,560 g) sucesivamente a una solución de metanosulfonato de (5-bromopiridin-2-il)metilo (2,750 g) en acetonitrilo (30 mi), se calentó a 50 °C en un baño de aceite y se dejó reaccionar durante 2 horas. A continuación, se enfrió hasta temperatura ambiente, se añadió agua, se extrajo con EA (50 mi x 3), la fase orgánica combinada se lavó con solución saturada de NaCI (50 mi) y se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 2,010 g de 1-((5-bromopiridin-2-il)metil)-4-metilpiperazina.
Se añadió MeOH (20 mi) a un tubo sellado de 100 mi bajo amoníaco a -78 °C, a continuación, se añadió 1-((5-bromopiridin-2-il)metil)-4-metilpiperazina (1,000 g) y óxido cuproso (0,532 g) sucesivamente hasta que el volumen de solución subió a 30 mi. Se retiró el baño exterior, se calentó hasta temperatura ambiente de forma
natural, a continuación se calentó a 70 °C y se dejó reaccionar durante 12 horas. El filtrado se recogió por filtración, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 15/1) para dar 0,730 g de 6-((4-metilpiperazin-1 -il)metil)piridin-3-amina.
MS(ES+): m/z= 207,2 (M+H)+.
Se prepararon los siguientes intermedios (que se muestran en la tabla 2) esencialmente tal como se ha descrito para la 6-((4-metilpiperazin-1-il)metil)piridin-3-amina (denominada en la presente memoria descriptiva Intermedio M4) utilizando el derivado de piperazina correspondiente.

Preparación 7: 5-((4-(dim etilam ino)piperidin-1-il)metil)piridin-2-amina (Intermedio M7)

Se añadió NaBH4 (1,640 g) a una solución de 2-bromo-5-formilpiridina (2,010 g) en THF (20 mi) a 0 °C en un baño de hielo, después de completar la adición de NaBH4, se retiró el baño de hielo, se calentó hasta temperatura ambiente de forma natural. Después de agitar durante 2 horas a temperatura ambiente, la mezcla de reacción se inactivó con agua (50 mi), se extrajo con EA (50 mi x 2), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (PE/EA= 5/1) para dar 1,900 g de (6-bromopi ridin-3-i l)m etanol.
Se enfrió a 0 °C una solución de (6-bromopiridin-3-il)metanol (1,000 g) en DCM (10 mi) en un baño de hielo y se añadió gota a gota a cloruro de tionilo (1,260 g), una vez completada la adición, se retiró el baño de hielo, la solución se calentó hasta temperatura ambiente de forma natural con agitación durante 2 h, a continuación, se concentró directamente para dar 1,050 g de 2-bromo-5-(clorometil)piridina.
Se añadió A/,A/-dimetilpiperidin-4-amina (0,586 g) y K2CO3 (1,160 g) a una solución de 2-bromo-5-(clorometil)piridina (0,853 g) en acetonitrilo (10 mi). Se añadió agua (30 mi), se extrajo con EA (50 mi x 3), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 0,730 g de 1-((6-bromopiridin-3-il)metil)-A/,A/-dimetilpiperidin-4-amina.
Se añadió MeOH (20 mi) a un tubo sellado de 100 mi en amoníaco a -78 °C, a continuación se añadió 1-((6-bromopiridin-3-il)metil)-A/,A/-dimetilpiperidin-4-amina (0,35 mg) y óxido cuproso (0,168 g) sucesivamente hasta que el volumen de solución subió a 30 mi. Se retiró el baño exterior, se calentó hasta temperatura ambiente de forma natural, a continuación se calentó a 70 °C y se hizo reaccionar durante 12 horas. El filtrado se recogió por filtración, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 0,260 g de 5-((4-(dimetilamino)piperidin-1 -il)metil)piridin-2-amina.
MS(ES+): m/z= 235,2 (M+H)+.
Se prepararon los siguientes Intermedios (que se muestran en la tabla 3) esencialmente tal como se ha descrito para5-((4-(dimetilamino)piperidin-1-il)metil)piridin-2-amina (en la presente memoria descriptiva, denominada Intermedio M7) utilizando el derivado de plperldlna correspondiente.
Tabla 3

Preparación 11: clorhidrato de 5-((4-metilpiperazin-1-il)metil)pirim idin-2-amonio (Intermedio M11)

Se añadió amoníaco acuoso (25 %) (1,200 g) a una solución de 2-cloropirimidina-5-carbaldehído (0,500 g) en THF (50 mi), agitando durante 12 horas. Se añadió agua (80 mi), se extrajo con DCM (80 mi x 8), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró para dar 0,540 g del producto 2-aminopirimidina-5-carbaldehído en bruto.
Se añadió B0C2O (2,817 g), trietilamina (1,310 g) y DMAP (0,054 g) sucesivamente a una solución de 2-aminopirimidina-5-carbaldehído (0,540 g) en THF (30 mi) con agitación durante 2 horas. Se añadió agua, se extrajo con EA (50 mi x 2), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (PE/EA= 5/1) para dar 0,514 g del compuesto MH2-12.
Se añadió 1-metilpiperazina (0,109 g) y sulfato de magnesio anhidro (0,216 g) sucesivamente a una solución del compuesto MH2-12 (0,290 g) en DCM (10 mi) con agitación durante 2 h, a continuación, se hizo reaccionar durante 3 h a T.A. después de añadir triacetoxiborohidruro de sodio, la mezcla de reacción se inactivo con agua
(20 mi), se extrajo con DCM (20 mi x 3), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 0,350 g de compuesto MH2-13.
Se hizo reaccionar una solución del compuesto MH2-13 (0,350 g) en DCM durante 2 h bajo ácido clorhídrico gaseoso a temperatura ambiente, la mezcla de reacción se concentró para dar 0,210 g de clorhidrato 5-((4-metilpiperazin-1-il)metil)pirimidin-2-amina.
MS(ES+): m/z= 244,1 (M+H)+.
Se preparó el siguiente producto intermedio (que se muestra en la tabla 4) esencialmente tal como se ha descrito para el clorhidrato de 5-((4-metilpiperazin-1-il)metil)pirimidin-2-amina (en la presente memoria descriptiva, denominado intermedio M11) utilizando el derivado de piperidina correspondiente en lugar del derivado de piperazina.
Tabla 4

Preparación 13: (2-aminopirim idin-5-il)(4-metilpiperazin-1-il)metanona (Intermedio M13)

metilpiperazin-1 -il)metanona
Se añadió oxona (1,810 g) a una mezcla del compuesto MH2-12 (0,315 g) en acetona (10 mi) y agua (3 mi) con agitación durante 2 horas a temperatura ambiente. Se añadió agua (20 mi), se extrajo con DCM (25 mi x 3), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró para dar 0,290 g del compuesto MH8-01.
Se añadió HATU (0,488 g) y DIEA (0,221 g) sucesivamente a una solución del compuesto MH8-01 (0,290 g) en DCM (10 mi) con agitación durante 1 h a T.A., la solución se hizo reaccionar durante 2 horas, después de añadir 1-metilpiperazina (0,105 g) a T.A. Se añadió agua (20 mi), se extrajo con DCM (20 mi x 3), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 50/1) para dar 0,250 g del compuesto MH8-02.
Se hizo reaccionar una solución del compuesto MH8-02 (0,150 g) en DCM (10 mi) durante 2 h bajo gas de ácido clorhídrico a T.A., se añadió agua (10 mi), a continuación se ajustó el pH a 8~9 con Na2C03, la solución acuosa resultante se extrajo con mezcla de disolventes (DCM/MeOH= 10/1) (20 mi x 5). La fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró para dar 0,060 g de (2-aminopirimidin-5-il)(4-metilpiperazin-1-il)metanona. MS(ES+): m/z= 223,1 (M+H)+.
Se preparó el siguiente Intermedio (que se muestra en la tabla 5) esencialmente tal como se ha descrito para (2-aminopirimidin-5-il)-(4-metilpiperazin-1-il)metanona (en la presente memoria descriptiva, denominada Intermedio M13) utilizando el derivado de piridina correspondiente en lugar del derivado de plrlmldlna.
Tabla 5

Preparación 15: A/5-(2-(dietilamino)etil)-A/5-metilpiridina-2,5-diamina (intermedio M15)

5-bromo-2-nitropiridina A/'.A/'-dietil-A^-metil-A/2-(6 nitropiridin 3 ¡Ijetano

A/5-(2-(d¡et¡lamino)etil-A/5-met¡lpir¡d¡na-2,5-diamina
Se añadió /V,/V-dietil-/V-metiletilendiamina (0,305 g) y K2CO3 (0,679 g) sucesivamente a una solución de 2-nitro-5-bromopiridina (0,500 g) en acetonitrilo (10 mi). Se dejó reaccionar con agitación a 82 °C durante 15 horas en un baño de aceite. Se añadió agua (50 mi), se extrajo con DCM (80 mi x 3), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 0,400 g de 1-metil-4-(6-nitropiridin-3-il)-piperazina.
Se añadió Pd/C (0,040 g) a una solución de 1 -metiI-4-(6-nitropiridin-3-iI)piperazina en THF (15 mi) con agitación durante 2 horas a temperatura ambiente bajo gas hidrógeno. El filtrado se recogió por filtración y a continuación se concentró para dar 0,350 g de A/5-(2-(dietilamino)etil)-A/5-metilpiridina-2,5-diamina.
MS(ES+): m/z= 223,2 (M+H)+.
Se preparó el siguiente intermedio (que se muestra en la tabla 6) esencialmente tal como se ha descrito para /V*-(2-(dietilamino)etil)-A/5-metilpiridina-2,5-diamina (en la presente memoria descriptiva, denominada intermedio M15) utilizando A/'.A/-dietil-A^.A^-dimetiletano-l^-diamina en lugar de A/,A/-dietil-A/-metiletilendiamina.
Tabla 6

Preparación 17: 6-((4-(dimetilamino)piperidin-1-il)metil)piridazin-3-amina (Intermedio M17)

Se añadió ácido tricloroisocianúrico (0,189 g) a una solución de 3-cloro-6-metilpiridazina (0,208 g) en CHCI3 (10 mi), se calentó a 60 °C durante 12 horas en un baño de aceite. Se enfrió a temperatura ambiente, el filtrado se recogió por filtración, se concentró y se purificó mediante cromatografía en columna (PE/EA= 10/1) para dar 0,201 g de 3-cloro-6-(clorometil)piridazina.
Se añadió K2CO3 (0,578 g), Kl (0,070 g) y A/,A/-dimetilpiperidin-4-amina (0,322 g) sucesivamente a una solución de 3-cloro-6-(clorometil)piridazina (0,340 g) en DMF (15 mi), se calentó a 50 °C durante 1 h en un baño de aceite. Se enfrió a temperatura ambiente, se añadió DCM (50 mi), se lavaron las fases orgánicas combinadas con solución saturada de NaCI y se secaron sobre Na2S04 anhidro, se concentraron y se purificaron mediante cromatografía en columna (DCM/MeOH= 10/1) para dar 0,370 g de 1-((6-cloropiridazin-3-il)metil)-A/,A/-dimetilpiperidin-4-amina.
Se añadió MeOH (20 mi) a un tubo sellado de 100 mi en amoníaco a -78 °C, a continuación se añadió 1-((6-cloropiridazin-3-il)metil)-A/,A/-dimetilpiperidin-4-amina (0,370 g) y óxido cuproso (0,532 g) sucesivamente hasta que el volumen de solución subió a 30 mi. Se retiró el baño exterior, se calentó hasta temperatura ambiente de forma natural, a continuación se calentó a 70 °C y se hizo reaccionar durante 12 horas. El filtrado se recogió por filtración, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH= 15/1) para dar 0,230 g de 6-((4-(dimetilamino)pipeñdin-1-il)metil)piñdazin-3-amina.
MS(ES+): m/z= 236,2(M+H)+.
Se preparó el siguiente producto intermedio (que se muestra en la tabla 7) esencialmente tal como se ha descrito para la 6-((4-(dimetilamino)piperidin-1-il)metil)piridazin-3-amina (en la presente memoria descriptiva, denominada Intermedio M17) utilizando el derivado de plperazina en lugar del derivado de plpeñdlna.
Tabla 7

Ejemplo 1: Síntesis del compuesto 1

1. Compuesto 1-01
Se agitó una mezcla de 2-metilciclopentanona (5,200 g), clorhidrato de hidroxilamina (9,200 g) y trietilamina (16,080 g) en etanol anhidro (70 mi) a 85 °C durante toda la noche en un baño de aceite. A continuación, la solución de reacción se concentró; el residuo se lavó con EA. El filtrado se recogió por filtración y a continuación se concentró para dar 5,820 g del compuesto 1-01 en bruto.
2. Compuesto 1-02
Se disolvió el compuesto 1-01 en bruto (5,820 g) en solución de ácido sulfúrico (con-H2S04 :H20 = 20 ml:5 mi), la mezcla resultante se agitó a 90 °C en un baño de aceite durante 90 min, se añadió agua (10 mi), a continuación se ajustó el pH a 8~9 con Na2C03 , la solución acuosa resultante se extrajo con DCM (20 mi x 5), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró a presión reducida para dar 4,110 g del compuesto 1-02 en bruto.
3. Compuesto 1-03
A una mezcla del compuesto 1-02 en bruto (4,110 g) y 4-bromo-2,6-difluoroanilina (3,780 g) en metilbenceno (40 mi) se le añadió POCI3 (4,180 g) y se calentó en un baño de aceite, se añadió TEA (2,770 g ) cuando la temperatura se elevó a 110 °C, se hizo reaccionar a la mezcla resultante a 110 °C durante 20 min. Se eliminó una parte del metilbenceno, a continuación se ajustó el pH a 8~9 con Na^Os, se extrajo con EA, la fase orgánica combinada se lavó con una solución saturada de NaCI y se secó sobre Na2SÜ4 anhidro, se concentró a presión reducida para dar 6,550 g del compuesto 1-03 en bruto.
4. Compuesto 1-04
Se agitó una mezcla del compuesto 1-03 en bruto (6,100 g) y terc-butóxido de potasio (4,520 g) en DMF (60 mi) a 100°C durante 20 min en un baño de aceite y a continuación se extrajo con 300 mi de EA, la fase orgánica combinada se lavó con solución saturada de NaCI (120 mi x 3) y se secó sobre Na2S04 anhidro, se concentró y a continuación se purificó mediante cromatografía en columna (PE/EA= 1/5) para dar 0,705 g del compuesto 1-04-A y 1,500 g del compuesto 1-04-C en bruto.
5. Compuesto 1-05
Se agitó una mezcla de compuesto 1-04-C en bruto (0,957 g), bis(pinacolato)diboro (1,290 g), triciclohexilfosfina (0,047 g), acetato de paladio (0,038 g) en DMSO (20 mi) bajo nitrógeno durante 1 h a 90 °C en baño de aceite. La mezcla resultante se extrajo con EA (60 mi), la fase orgánica combinada se lavó con solución saturada de NaCI (30 mi x 3) y se secó sobre Na2S04 anhidro, se concentró a presión reducida para dar 2,290 g del compuesto 1-05 en bruto.
6. Compuesto 1-06
Se agitó una mezcla del compuesto 1-05 en bruto (2,290 g), 2,4-dicloropirimidina (0,755 g), K2C03 (1,400 g) y Pd(dppf)CI2-DCM (0,138 g) en 1,4-dioxano (30 mi) y agua (3 mi) bajo nitrógeno durante 2 h a 60 °C en un baño de aceite. La mezcla resultante se extrajo con EA (30 mi x 2), la fase orgánica combinada se lavó con solución saturada
de NaCI (30 mi x 1) y se secó sobre Na2S04 anhidro, se concentró y a continuación se purificó mediante cromatografía en columna (PE/EA= 1/1) para dar 0,927 g del compuesto 1-06.
7. Compuesto 1
Se agitó una mezcla del compuesto 1-06 (0,400 g), intermedio M1 (0,291 g), CS2CO3 (0,822 g), Xanphos (0,017 g) y Pd2(dba)3 (0,027 g) en 1,4-dioxano (12 mi) bajo nitrógeno gaseoso durante 1 hora a 110 °C en un baño de aceite, se continuó la reacción bajo microondas durante 0,5 horas a 110 °C. A la mezcla resultante se le añadió agua (10 mi), a continuación, se extrajo con DCM (20 mi x 3), la fase orgánica combinada se lavó con solución saturada de NaCI (30 mi x 1) y se secó sobre Na2S04 anhidro, se concentró y a continuación se purificó mediante cromatografía en columna (DCM/MeOH= 20/1), el sólido se lavó con metil terc-butiléter (10 mi) y n-hexano (10 mi) para dar 290 mg del compuesto 1.
MS(ES+): m/z= 473,2 (M+H)+.
RMN de 1H (CDCI3): 88,498-8,511 (d, 1H, CH), 8,373-8,396 (d, 1H, CH), 8,167 (s, 1H, CH), 8,040-8,047 (d, 1H, CH), 7,963 (s, 1H, CH), 7,631-7,660 (d, 1H, CH), 7,346-7,660 (dd, 1H, CH), 7,177-7,190 (d, 1H, CH), 4,687-4,719 (m, 1H, CH), 3,190-3,10 (m, 4H, CH2), 2,651-2,675 (m, 4H, CH2), 2,999-3,024 (m, 1H, CH2), 2,403 (s, 3H, CH3), 2,337-2,356 (m, 1H, CH2), 2,267-2,357 (m, 1H, CH2), 2,219-2,248 (m, 1H, CH2), 2,010-2,056 (m, 2H, CH2), 1,602-1,618 (d, 3H, CHs).
Ejemplo 1-1 Separación quiral del compuesto 1-06
Las técnicas útiles para la separación de isómeros, por ejemplo, enantiómeros, están dentro de la experiencia en la materia y se describen en Eliel, E. L.; Wilen, S. H.; Mander, L. N. stereochemistry of Organic Compounds, Wiley Interscience, Nueva York, 1994. Por ejemplo, el compuesto 1, 2 o 15 se puede resolver con un elevado exceso enantiomérico (por ejemplo, del 60 %, 70 %, 80 %, 90 %, 95 %, 99 % o más) mediante cromatografía líquida de alta resolución utilizando una columna quiral. En algunas realizaciones, el compuesto 1-06 en bruto del ejemplo 1 se purifica directamente en una columna quiral para proporcionar un compuesto enriquecido enantioméricamente. Condiciones de HPLC quiral:
Columna  CHIRALPAK IE
CHIRALPAK IE
Tamaño de columna 2 cm * 25 cm
Inyección 0,7 mi
Fase móvil Hex: EtOH = 65:35 (v/v)
Caudal 19 ml/min
Longitud de onda JV 220nm
Temperatura 35 °C
Solución de muestra 80 mg/ml en fase móvil
Equipo de HPLC-Prep Prep-Gilson-HPLC
Nombre de la muestra Compuesto 1-06
Ejemplo 1-2 Síntesis del compuesto 1a y el compuesto 1b
Se purifica el compuesto 1-06 en bruto mediante columna quiral en las condiciones anteriores para dar el compuesto 1-06-A y el compuesto 1-06-B.
Se preparó el compuesto 1a y el compuesto 1b esencialmente tal como se ha descrito en la etapa 7 del ejemplo 1 utilizando, respectivamente, el compuesto 1-06-Ay el compuesto 1-06-B.
Además, se midieron 3 veces las rotaciones ópticas para cada compuesto que se muestra a continuación en un polarímetro Rudolf.
Condiciones:
Longitud del tubo del polarímetro 100 mm
Temperatura 20 °C
Solución de muestra 3,0 mg/ml en EtOH
Nombre de la muestra Compuesto 1 a y Compuesto 1 b
Resultados:
1 e(°) 2° (o) 3e (°) Promedio (°)
Compuesto 1a 30,357 30,711 29,906 30,325
Compuesto 1b -35,430 -35,453 -35,298 -35,394
Ejemplo 2: Síntesis del compuesto 2

1. Compuesto 1-01
Se agitó una mezcla de 2-metilciclopentanona (5,200 g), clorhidrato de hidroxilamina (9,200 g) y trietilamina (16,080 g) en etanol anhidro (70 mi) a 85 °C durante la noche en un baño de aceite. A continuación, la solución de reacción se concentró; el residuo se lavó con EA. El filtrado se recogió por filtración y, a continuación, se concentró para dar 5,820 g del compuesto 1-01 en bruto.
2. Compuesto 1-02
Se disolvió el compuesto 1-01 en bruto (5,820 g) en solución de ácido sulfúrico (con-H2S04 :H20 = 20 ml:5 mi), la mezcla resultante se agitó a 90 °C en un baño de aceite durante 90 min, se añadió agua (10 mi), a continuación, se ajustó el pH a 8~9 con Na2C03 , la solución acuosa resultante se extrajo con DCM (20 mi x 5), la fase orgánica combinada se secó sobre Na2S04 anhidro, se concentró a presión reducida para dar 4,110 g del compuesto 1-02 en bruto.
3. Compuesto 1-03
A una mezcla del compuesto 1-02 bruto (4,110 g) y 4-bromo-2,6-difluoroanilina (3,780 g) en metilbenceno (40 mi) se le añadió POCI3 (4,180 g) y se calentó en un baño de aceite, se añadió TEA (2,770 g ) cuando la temperatura se elevó a 110 °C, y se hizo reaccionar la mezcla resultante a 110 °C durante 20 min. Se eliminó una parte del metilbenceno, a continuación, se ajustó el pH a 8~9 con Na2C0 3 , se extrajo con EA, la fase orgánica combinada se lavó con una solución saturada de NaCI y se secó sobre Na2SÜ4 anhidro, se concentró a presión reducida para dar 6,550 g del compuesto 1-03 en bruto.
4. Compuesto 1-04
Se agitó una mezcla del compuesto 1-03 en bruto (6,100 g) y terc-butóxido de potasio (4,520 g) en DMF (60 mi) a 100 °C durante 20 min en un baño de aceite y, a continuación, se extrajo con 300 mi de EA, la fase orgánica combinada se lavó con solución saturada de NaCI (120 mi x 3) y se secó sobre Na2S04 anhidro, se concentró y, a continuación, se purificó mediante cromatografía en columna (PE/EA= 1/5) para dar 0,705 g del compuesto 1-04-A y 1,500 g del compuesto 1-04-C en bruto.
5. Compuesto 1-05
Se agitó una mezcla de compuesto 1-04-C en bruto (0,200 g), bis(pinacolato)diboro (0,270 g), triciclohexilfosfina (0,039 g), acetato de paladio (0,031 g) en DMSO (5 mi) bajo nitrógeno durante 1 h a 90 °C en un baño de aceite. La
mezcla resultante se extrajo con EA (50 mi), la fase orgánica combinada se lavó con solución saturada de NaCI (20 mi x 3) y se secó sobre Na2S04 anhidro, se concentró a presión reducida para dar 0,398 g del compuesto 1-05 en bruto.
6. Compuesto 2-01
Se agitó una mezcla del compuesto 1-05 en bruto (0,398 g), 2,4-dicloropirimidina (0,304 g), K2CO3 (0,502 g) y Pd(dppf)Cl2-DCM (0,050 g) en 1,4-dioxano (10 mi) y agua (1 mi) bajo nitrógeno durante 80 min a 60 °C en un baño de aceite. A la mezcla resultante se le añadió agua (10 mi) y a continuación se extrajo con EA (20 mi x 2), la fase orgánica combinada se lavó con solución saturada de NaCI (20 mi x 1) y se secó sobre Na2S04 anhidro, se concentró y a continuación se purificó mediante cromatografía en columna (PE /EA= 1/1) para dar 0,165 g del compuesto 2-01.
7. Compuesto 2
Se agitó una mezcla del compuesto 2-01 (0,030 g), intermedio M5 (0,025 g), CS2CO3 (0,067 g), Xanphos (0,005 g) y Pd2(dba)3 (0,005 g) en 1,4-dioxano (2 mi) bajo nitrógeno gaseoso durante 3,5 horas a 110 °C en un baño de aceite. A la mezcla resultante se le añadieron 20 mi de mezcla de disolventes (DCM/MeOH = 10/1), a continuación se filtró, se concentró y se purificó mediante cromatografía en columna (DCM/MeOH = 8/1), el sólido se lavó con 1,4-dioxano (5 mi) para dar 0,024 g del compuesto 2.
MS(ES+): m/z= 519,3 (M+H)+.
RMN de 1H (CDCI3): 88,587 (s, 1H, NH), 8,448-8,457 (d, 1H, CH), 8,383-8,405 (d, 1H, CH), 8,280-8,286 (d, 1H, CH), 8,009-8,013 (d, 1H, CH), 7,804-7,837 (dd, 1H, CH), 7,686-7,714 (dd, 1H, CH), 4,681-4,728 (m, 1H, CH), 3,532 (S, 2H, CH2), 3,189-3,256 (m, 1H, CH2), 3,008-3,091 (m, 1H, CH2), 2,654 (s, 8H, CH2), 2,571-2,608 (m, 2H, CH2), 2,226-2,299 (m, 1H,CH2), 2,111-2,15 (m, 1H, CH2), 2,012-2,053 (m, 2H, CH2), 1,596-1,612 (d, 3H, CH3), 1,174-1,210 (t, 3H, CHs).
Ejemplo 2-1 Separación quiral del compuesto 2-01
En esta realización, el compuesto 2-01 en bruto del ejemplo 2 se purifica directamente en una columna quiral para proporcionar el compuesto 2-01-A y el compuesto 2-01-B en las siguientes condiciones.
Condiciones de HPLC quiral:
Columna CHIRALPAK AD-H
Tamaño de columna 2 cm * 25 cm, 5 pm
Inyección 1,0 mi
Fase móvil Hex: IPA = 90:10 (v/v)
Caudal 20 ml/min
Longitud de onda JV 220 nm
Temperatura 25 °C
Solución de muestra 25,9 mg/ml en EtOH:CAN= 3:1
Equipo de HPLC-Prep Prep-YMC-HPLC
Nombre de la muestra Compuesto 2-01
Ejemplo 2-2 Síntesis del compuesto 2a y el compuesto 2b
El compuesto 2-01 en bruto se purifica mediante columna quiral en las condiciones anteriores para dar el compuesto 2-01-A y el compuesto 2-01-B.
Se preparó el compuesto 2a y el compuesto 2b esencialmente tal como se ha descrito en la etapa 7 del ejemplo 2 utilizando, respectivamente, el compuesto 2-01-A y el compuesto 2-01-B.
Además, se midieron 3 veces las rotaciones ópticas para cada compuesto que se muestra a continuación en un polarímetro Rudolf.
Condiciones:
Longitud del tubo del polarímetro 100 mm
Temperatura 20 °C
Solución de muestra 4,0 mg/ml en DCM
Nombre de la muestra compuesto 2a y compuesto 2b
Resultados:
1“ (°) 2e (°) 3e (°) Promedio (°)
Compuesto 2a 38,074 38,111 38,078 38,088
Compuesto 2b -32,070 -32,134 -31,903 -32,036
Se prepararon los siguientes ejemplos (que se muestran en la tabla 8) esencialmente tal como se ha descrito para el ejemplo 2 utilizando los Intermedios correspondientes. En los casos en los que los dos enantlómeros de cada compuesto se separan en una columna qulral, entonces se prueba, respectivamente, su rotación óptica esencialmente tal como se ha descrito para el ejemplo 2-2.
Tabla 8
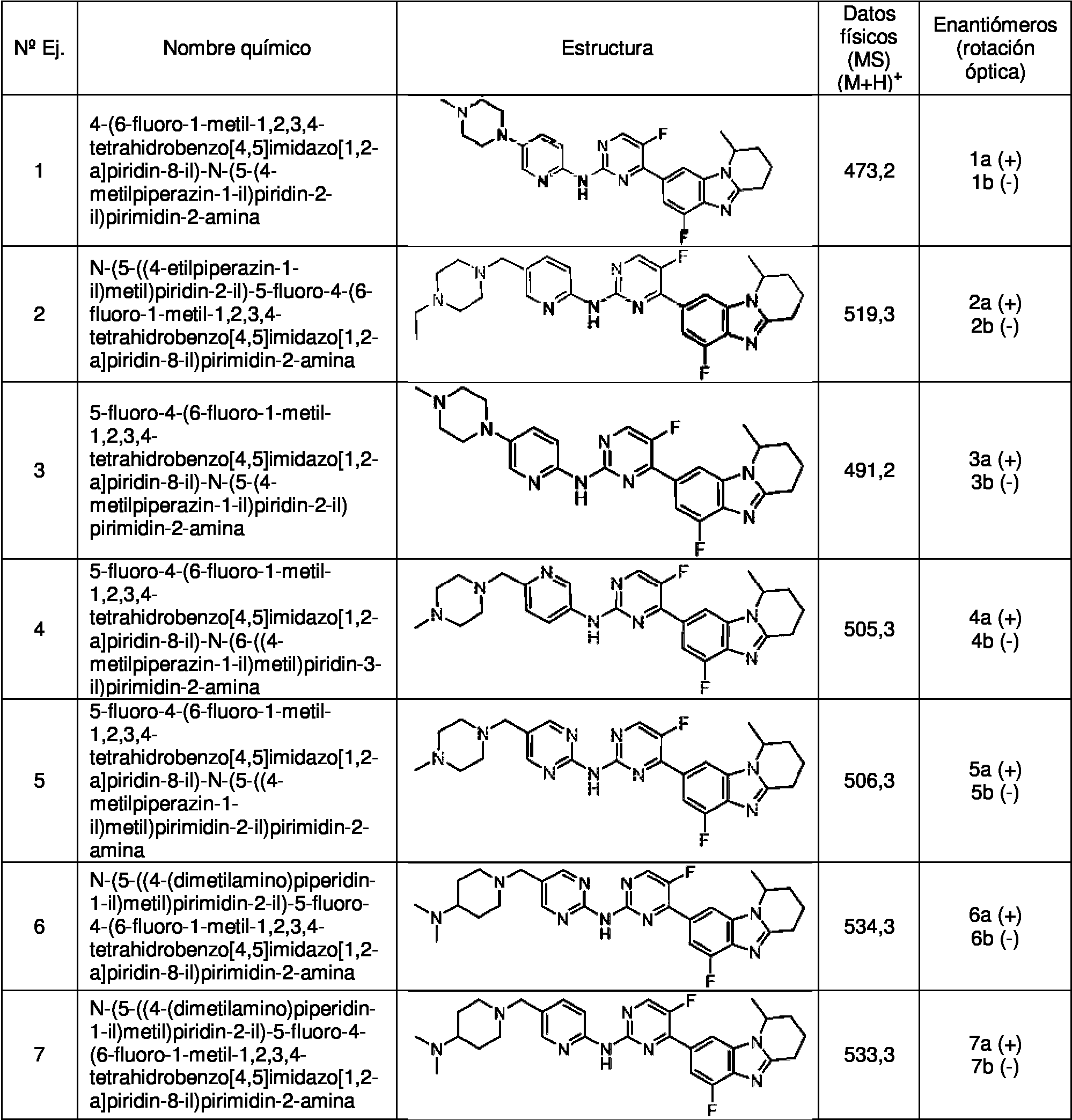

Ejemplo 15: Síntesis del compuesto 15b

1. Compuesto 15-01
A una mezcla de enantiómero (-) de 5-metilmorfolin-3-ona (0,200 g) y 4-bromo-2,6-difluoroanilina (0,210 g) en metilbenceno (20 mi) se le añadió POCI3 (0,510 g) y se calentó en un baño de aceite, se añadió TEA (0,210 g) cuando la temperatura se elevó a 110 °C, se hizo reaccionar la mezcla resultante a 110 °C durante 40 min. Se eliminó una parte del metilbenceno, se ajustó el pH a 8~9 con Na2C03 después de añadir agua (10 mi). A continuación, se extrajo con EA (20 mi x 2), la fase orgánica combinada se lavó con solución saturada de NaCI (20 mi x 1) y se secó sobre Na2S04 anhidro, se concentró para dar 0,280 g del compuesto 15-01.
2. Compuesto 15-02
Se añadió terc-butóxido de potasio (0,482 g) a una mezcla del compuesto 15-01 (0,275 g) en DMF (5 mi), a continuación se agitó durante 30 min a 70 °C en un baño de aceite. La mezcla resultante se extrajo con EA (20 mi x 3), la fase orgánica combinada se lavó con solución saturada de NaCI (30 mi x 4) y se secó sobre Na2S04 anhidro, se concentró y se purificó mediante cromatografía en columna (PE/EA= 5/1) para dar 0,260 g del compuesto 15-02.
3. Compuesto 15-03
Se agitó una mezcla del compuesto 15-02 (0,260 g), bis(pinacolato)diboro (0,320 g), triciclohexilfosfina (0,050 g), acetato de paladio (0,040 g) en DMSO (6 mi) bajo nitrógeno durante 50 min a 90 °C en un baño de aceite. La mezcla resultante se extrajo con EA (20 mi x 2), la fase orgánica combinada se lavó con solución saturada de NaCI (10 mi x 3) y se secó sobre Na2S04 anhidro, se concentró para dar 0,258 g del compuesto 15-03.
4. Compuesto 15-04
Se agitó una mezcla del compuesto 15-03 (0,120 g), 2,4-dicloro-5-fluoropirimidina (0,060 g), K2CO3 (0,099 g) y Pd(dppf)CÍ2-DCM (0,012 g) en 1,4-dioxano (10 mi) y agua (1 mi) bajo nitrógeno durante 1 hora a 60 °C en un baño de aceite. La mezcla resultante se extrajo con EA (20 mi x 2), la fase orgánica combinada se lavó con solución saturada de NaCI (20 mi x 1) y se secó sobre Na2S04 anhidro, se concentró y, a continuación, se purificó mediante HPLC preparativa (DCM/MeOH= 30/1) para dar 0,052 g del enantiómero (-) del compuesto 15-04.
5. Compuesto 15b
Se hizo reaccionar enantiómero (-) del compuesto 15-04 (20 mg), intermedio M1 (0,017 g), CS2CO3 (0,056 g), Xanphos (0,005 g) y Pd2(dba)3 (0,005 g) en 1,4-dioxano (2 mi) bajo microondas a 110 °C durante 1,5 h a través de una corriente de nitrógeno. A la mezcla resultante se le añadieron 20 mi de mezcla de disolventes (DCM/MeOH=10/1), el filtrado se recogió por filtración y, a continuación, se concentró, se purificó mediante HPLC preparativa (DCM/MeOH= 20/1), el sólido resultante se lavó con n-hexano (10 mi) para dar 0,014 g del compuesto 15b.
MS(ES+): m/z= 493,2 (M+H)+.
RMN de 1H (CDCI3): 88,409-8,418 (d, 1H, CH), 8,211-8,288 (m, 2H, CH), 8,070-8,077 (d, 1H, CH), 8,019-8,023 (d,
4,111-4,168 (m, 1H, CH2), 4,039-4,075 (m, 1H, CH2), 3,249-3,275 (m, 4H, CH2), 2,724-2,748 (m, 4H, CH2), 2,462 (s, 3H, CH3), 1,662-1,678 (d, 3H, CH3).
Ejemplo 15-1 Síntesis del compuesto 15a y el compuesto 15
El compuesto 15a y el compuesto 15b son enantlómeros. Se preparó el compuesto 15a esencialmente tal como se ha descrito en el ejemplo 15 utilizando el enantiómero (+) de 5-metilmorfolin-3-ona como material de partida.
En otra realización, se utiliza 5-metilmorfolin-3-ona en bruto como material de partida; se obtendrá al final el compuesto 15 en bruto que Incluye el compuesto 15a y el compuesto 15b.
Además, se midieron 3 veces las rotaciones ópticas para cada compuesto que se muestra a continuación en un polarímetro Rudolf.
Condiciones:
Longitud del tubo del polarímetro 100 mm
Temperatura 20 °C
Solución de muestra 4,6 mg/ml en DCM/MeOH (1:1)
Nombre de la muestra compuesto 15a y compuesto 15b Resultados:
1 e(°) 2° (o) 3e (°) Promedio (°)
Compuesto 1a 32,859 32,818 32,809 32,829
Compuesto 1b -28,979 -28,840 -28,967 -28,929
Se prepararon los siguientes ejemplos (que se muestran en la tabla 9) esencialmente tal como se ha descrito en el ejemplo 15 utilizando los intermedios correspondientes y utilizando el enantiómero (+) y/o (-) de 5-metilmorfolin-3-ona como material de partida. Sus rotaciones ópticas se probaron esencialmente tal como se ha descrito en el ejemplo 15-1.
Tabla 9



EJEMPLOS PARA COMPARACIÓN
Se prepararon los siguientes ejemplos de comparación, esencialmente tal como se ha descrito para el ejemplo 1, 2 o 15 utilizando los correspondientes productos intermedios o materiales de partida. Por ejemplo, se prepararon los siguientes ejemplos de comparación 8, 9 y 10 (que se muestran en la tabla 10) esencialmente tal como se ha descrito en el ejemplo 2 utilizando

en lugar de

Y se preparó el siguiente ejemplo de comparación 7 esencialmente tal como se ha descrito en el ejemplo 1 utilizando

en lugar de

Tabla 10


En la tabla anterior, los ejemplos de comparación números 3b y 4b son síntesis esencialmente tal como se ha descrito en el ejemplo 15 y, a continuación, se probó respectivamente su rotación óptica esencialmente tal como se ha descrito en el ejemplo 15-1. Los ejemplos de comparación números 3b y 4b muestran ambos rotación óptica negativa.
PRUEBAS FARMACOLÓGICAS
Los resultados de las siguientes pruebas demuestran evidencia de que los compuestos Indicados como ejemplo en la presente memoria descriptiva son útiles como inhibidores específicos de CDK4/6 y como agentes anticancerígenos. Tal como se utiliza la presente memoria descriptiva, "IC50" se refiere a la concentración de un agente que produce el 50 % de la respuesta Inhibidora máxima posible para el agente.
A modo Ilustrativo, se muestra a continuación la siguiente estructura general. Sorprendentemente, se descubrió que "R" tiene una Influencia crítica en la actividad biológica, la selectividad y la seguridad.

Prueba 1 Comparación de diferentes sustituyentes por ensayo de proliferación celular
Se midieron los efectos de los compuestos de prueba sobre la proliferación in vitro mediante el ensayo de viabilidad celular MTS.
Cultivo de células
Se expanden células de cáncer colorrectal humano (colo-205) en cultivo (colo-205 se cultiva en medio DMEM con 12 % de FBS, 1 % de P/S y 1 % de L-glutamina).
Ensayo de viabilidad celular MTS:
1. Sembrar células a una densidad de 4x103 células por pocilio de placas de 96 pocilios, dejar crecer durante 24 h; 2. Añadir concentraciones variables de compuestos de prueba a las células;
3. Incubar durante 7 días de exposición;
4. Preparar los reactivos siguiendo las Instrucciones del kit de ensayo de proliferación celular (Promega);
5. Cambiar a medio sin suero con un volumen final de 100 pJ/pocillo. Preparar un conjunto de pocilios únicamente con medio para la sustracción del fondo;
6. Añadir 20 pJ de solución de MTS que contiene PMS a cada pocilio (la concentración final de MTS será de 0,33 mg/ml);
7. Incubar de 1 a 4 h a 37 °C en una atmósfera humidificada, con 5 % de CO2;
8. Registrar la absorbancia a 490 nm con el lector de placas VICTOR® X5 (PerkinElmer).
Todos los puntos experimentales se establecieron en tres pocilios y todos los experimentos se repitieron, como mínimo, tres veces. El valor de ICso se calcula a partir de la curva dosis-respuesta mediante el software (Graphpad prism 6), y los resultados se muestran en la tabla 11.
Tabla 11

Los compuestos Indicados como ejemplo anteriormente muestran actividad antltumoral en este modelo, tal como se muestra en la tabla 11, demostrando de este modo que los compuestos Indicados como ejemplo de la presente invención tienen una actividad in vivo más potente contra tumores Rb+. En comparación con el compuesto conocido LY2835219 (Abemaciclib), el compuesto de la presente invención, por ejemplo, el compuesto del ejemplo 1 o 2, tiene una inhibición más potente contra los tumores Rb+. En comparación con el ejemplo de comparación 1 (denominado a continuación Ej. Com. 1; R es espiro) y el ejemplo de comparación 2 (denominado a continuación Ej. Com. 2; R es H), el compuesto de la presente invención, por ejemplo, el compuesto del ejemplo 1 o 2 (R es metilo) tiene una Inhibición mucho más potente contra los tumores Rb+.
Los compuestos indicados como ejemplo anteriormente también muestran que, cuando R es metilo pero no H o espiro, tienen una actividad biológica mucho más potente en este modelo. A partir de los resultados anteriores, se puede observar que el tipo de sustituyeles tiene una Influencia significativa en la Inhibición contra los tumores Rb+. Prueba 2 Comparación de diferentes sustituyeles por prueba de seguridad
Se probó la seguridad de compuestos seleccionados preparados tal como se ha descrito anteriormente, según el cambio en el peso corporal y si se producía mortalidad, y los procedimientos se han descrito en la presente memoria descriptiva. El compuesto de prueba se prepara en un vehículo apropiado y se administra a ratones BALB/c (22-23 g) por sonda oral. El peso corporal y la mortalidad se toman como medidas generales de toxicidad. La pérdida de peso (cambio de peso corporal en %) se calcula dos veces por semana comparando los grupos tratados con el grupo de control del vehículo durante el curso del tratamiento. Los compuestos 2 y 16 demuestran una pérdida de peso muy pequeña, en estos modelos, cuando se dosifican a 200 mg/kg (qd). Pero a la misma dosis, los ejemplos de comparación, por ejemplo, Ej. Com. 3b y Ej. Com. 4b son capaces de provocar mucha más pérdida de peso, Incluso provocar una elevada mortalidad con 4 y 5 ratones muertos en cada grupo (6 ratones) después del tratamiento durante 2 semanas.
Los resultados se dan en la tabla 12. significa "pérdida de peso inferior al 5%"; "**" significa "pérdida de peso superior al 5 % e inferior al 10 %"; "***" significa "pérdida de peso superior al 10 % e inferior al 30 %"; "****" significa "pérdida de peso superior al 30 %"."+" significa "se produjo m ortalidad";significa "no se produjo mortalidad".
Tabla 12

Sorprendentemente, se descubrió que el Ej. Com. 3b (R es isopropilo) o el Ej. Com. 4b (R es etilo) tiene más efectos secundarios y toxicidad. Sin embargo, los compuestos indicados como ejemplo de la presente invención, por ejemplo, el compuesto 2b o 16b (R es metilo), son mucho más seguros, lo que demuestra que el tipo de sustituyeles tiene una influencia significativa en la seguridad.
Prueba 3 Efecto del número de sustituyentes por ensayos de cinasa CDK
Para demostrar que los compuestos presentan afinidad por las cinasas CDK (CDK2/CycA2, CDK4/CycD3, CDK6/cycD3), se realizaron ensayos de cinasa CDK.
Se prepararon los tampones de reacción tal como se describe a continuación: tampón base de cinasa para CDK2,6 (HEPES 50 mM, pH 7,5; Brij-35 al 0,0015%; MgC^ 10 mM; DTT 2 mM); Tampón de base de cinasa para CDK4
(HEPES 20 mM, pH 7,5; Tritón X-100 al 0,01 %; MgCÍ2 10 mM; DTT 2 mM); Tampón de parada (HEPES 100 mM, pH 7,5; Brij-35 al 0,015 %; Reactivo de recubrimiento ne 3 al 0,2 %; EDTA 50 mM).
Protocolo de reacción enzimática:
1) Diluir el compuesto a 50X de la concentración máxima final deseada en la reacción con DMSO al 100%. Transferir 100 pl de esta dilución de compuesto a un pocilio en una placa de 96 pocilios. A continuación, diluir en serle el compuesto transfiriendo 30 pl a 60 pl de DMSO al 100 % en el pocilio siguiente y así sucesivamente hasta un total de 10 concentraciones. Añadir 100 pl de DMSO al 100% a dos pocilios vacíos para un control sin compuesto y control sin enzimas en la misma placa de 96 pocilios. Marcar la placa como placa de origen.
2) Preparar la placa intermedia transfiriendo 10 pl de compuesto de la placa de origen a una nueva placa de 96 pocilios que contiene 90 pl de tampón de cinasa como placa intermedia.
3) Transferir 5 pl de compuesto de la placa intermedia de 96 pocilios a una placa de 384 pocilios por duplicado. 4) Añadir 10 pl de solución de enzima 2,5x a cada pocilio de la placa de ensayo de 384 pocilios.
5) Incubar a temperatura ambiente durante 10 minutos.
6) Añadir 10 pl de solución de sustrato 2,5x preparada añadiendo péptido marcado con FAM y ATP en el tampón de base de cinasa.
Las concentraciones de reacción para los enzimas y sustratos son como en la siguiente tabla (tabla 13):
Tabla 13

7) Incubar a 28 °C durante un período de tiempo específico.
8) Añadir 25 pl de tampón de parada para detener la reacción.
9) Recopilar los datos en el calibrador. A continuación, convertir los valores de conversión a valores de inhibición.
Porcentaje de inhibición = (max - conversión) / (max - min) *100.
"max" significa control de DMSO y "min" significa aquí control inferior.
10) Ajustar la curva utilizando el porcentaje de inhibición en el complemento XLFit excel versión 4.3.1 para obtener los valores de ICso- La ecuación utilizada es: Y= Pie+(Cima-Pie) /(1+(ICso/X) A Pendiente). En la que Y es el porcentaje de Inhibición (%); X es la concentración del compuesto de prueba.
Los resultados se expresan como valor ICso que se muestra en la tabla 14.
Tabla 14

Tal como se muestra en la tabla anterior, se puede observar que el número de metilos tiene una Influencia muy importante en la selectividad. Sorprendentemente, los compuestos indicados como ejemplo de la presente Invención muestran una ICso de >0,3 pM en el ensayo de inhibición de la cinasa CDK2 anterior y una ICso de SO,04 pM en el ensayo de Inhibición de la cinasa CDK4/6 anterior, tal como se muestra en la tabla 14. Se demuestra que los compuestos indicados como ejemplo de la presente Invención son Inhibidores más selectivos de la actividad de la cinasa CDK4/6. De este modo, se demuestra que los compuestos indicados como ejemplo (R representa solo un metilo) son inhibidores más específicos de CDK4/6, en comparación con el compuesto conocido LY2835219 y el ejemplo de comparación, por ejemplo, el Ej. Com. 5 o el Ej. Com. 6 (R representa dos metilos).
Prueba 4 Efecto del sitio del sustituyente
Los compuestos seleccionados preparados tal como se ha descrito anteriormente se probaron, según los procedimientos biológicos descritos en esta memoria descriptiva. Los resultados se mostraron como en la tabla siguiente.

Además, en la siguiente tabla se muestra una comparación directa del ejemplo 2 y su ejemplo de comparación 8, 9 o 10. Los resultados se dan en la tabla 16. "++++" significa "valor de IC50 inferior a 0,2 pM ";"+++" significa "valor de IC50 superior a 0,2 pM e inferior a 1,0 pM"; "++" significa "valor de IC50 superior a 1,0 pM e inferior a 2,0 pM"; "+" significa "valor IC50 superior a 2,0 pM".

Tal como se muestra en las tablas 15 y 16 anteriores, se puede observar que el sitio del sustituyente también desempeña una fundón importante en la actividad biológica. El efecto sobre la actividad biológica resultante del cambio de sitio de los sustituyeles es significativamente inesperado.
Tal como se muestra en los resultados presentados anteriormente, se puede saber que "R" del anillo heterocíclico de 6 miembros tiene una influencia crítica sobre la actividad biológica, la selectividad y la seguridad. Sorprendentemente, cuando hay uno y solo un metilo en dicho anillo heterocíclico de 6 miembros, los efectos se obtendrán, como mínimo, de la siguiente manera:
© actividad biológica mejorada;
© buena selectiva; y
© efectos secundarios bajos.
Prueba 5 Efecto de la actividad óptica
Se ensayaron los compuestos seleccionados preparados tal como se ha descrito anteriormente, según los procedimientos biológicos descritos como Prueba 3 (ensayos de cinasa CDK). Los resultados se mostraron como en la siguiente tabla.
Tabla 17

Tal como se muestra en la tabla anterior, se puede observar que los compuestos 1b, 2b, 15b, 16b son más potentes que los compuesto 1a, 2a, 15a, 16a para inhibir CDK4/6 respectivamente, lo que demuestra que los enantiómeros (-) de los presentes compuestos son ventajosos sobre los enantiómeros (+).
En algunos casos, el compuesto dado a conocer en la presente memoria descriptiva se administra cuando un enantiómero [por ejemplo, el enantiómero (-) o el enantiómero (+)] está presente en un alto exceso enantiomérico. En un caso, el enantiómero del compuesto 1b que tiene una rotación óptica negativa, por ejemplo, -35,394° (c=3,0 mg/ml, EtOH) tiene mayor actividad contra la enzima CDK4/6 que el enantiómero (compuesto 1a) que tiene una rotación óptica positiva de 30,325° (c= 3,0mg/ml, EtOH). En otro caso, el enantiómero del compuesto 2b que tiene una rotación óptica negativa, por ejemplo, -32,036° (c= 4,0 mg/ml, DCM) tiene mayor actividad contra la enzima CDK4/6 que el enantiómero que tiene una rotación óptica positiva de 38,088° (c= 4,0 mg/ml, DCM). En otros casos, el enantiómero del compuesto 15b que tiene una rotación óptica negativa, por ejemplo, -28,929° (c= 4,6 mg/ml, DCM/MeOH= 1:1) tiene mayor actividad contra la enzima CDK4/6 que el enantiómero que tiene una rotación óptica positiva de 32,829° (c= 4,6 mg/ml, DCM/MeOH= 1:1).
Prueba 6 Prueba de actividad inhibidora y de selectividad sobre otros subtipos de cinasa CDK a nivel molecular
El compuesto representativo 2b de la presente invención se utilizó como compuesto de prueba y se comparó con el fármaco de control positivo LY2835219 (Abemaciclib) para comparar la actividad inhibidora de la cinasa CDK y la especificidad selectiva entre ellos.
El mecanismo de este procedimiento se muestra en la fórmula (II). La cinasa cataliza la fosforilación del sustrato proteico para marcar el 33P en el ATP marcado con 33P (y-33P-ATP) al sustrato proteico en el sistema de reacción; el sistema de reacción se colocó en una membrana de intercambio iónico P81 y la membrana se lavó extensamente con tampón de fosfato al 0,75 %; el sustrato fosforilado radiactivamente quedó en la membrana y la actividad de la cinasa se reflejó registrando la intensidad del radiomarcador de la proteína del sustrato.
Sustrato [y-^Pj-ATP _________ Enzima______ 33P-Sustrato ADP
Fórmula (II)
Los datos se procesaron con el software Prism4 (GraphPad), y la fórmula de ajuste de la curva fue:
Y = Pie (Cima - Pie) / (1 10 A Log (IC50 -X ) * Pendiente));
en la que, Y es el porcentaje de inhibición (%); X es el logaritmo de la concentración del inhibidor.
Resultados: a través de la selección de varías cinasas CDK, se descubrió que el compuesto representativo 2, 2a y 2b tienen una IC50 superior a 0,4 pM para inhibir CDK1/2/7/9, que es de decenas a miles de veces mayor que la de CDK4/6. (Véase la tabla 18)
Tabla 18: Actividad inhibidora de cinasa CDK
Cinasa IC50 (nM)
LY2835219 Compuesto 2 Compuesto 2a Compuesto 2b CDK1/ciclina B 308 2.350 3.573 1.683 CDK2/ciclina E 90 474 596 441
CDK7/ciclina H 2.071 1.050 2.370 664
CDK9/ciclina T1 111 572 779 649
Conclusión: A nivel molecular, el compuesto 2 y el compuesto 2b representativos de la presente invención mostraron un efecto inhibidor fuerte sobre CDK4/6 y un efecto inhibidor débil sobre CDK1/2/7/9, lo que indica que el compuesto 2 y el compuesto 2b son inhibidores de la cinasa CDK4/6 con excelente selectividad. El compuesto 2a mostró un efecto inhibidor fuerte sobre CDK4, un efecto inhibidor leve sobre CDK6 y un efecto inhibidor muy débil sobre CDK1/2/7/9, lo que indica que el compuesto 2a es un inhibidor de la cinasa CDK4 con extrema selectividad y
un inhibidor de la cinasa CDK6 con buena selectividad. Además, la selectividad de los compuestos representativos de la presente invención entre CDK1/2/9 y CDK4/6 fue significativamente mayor que la de LY2835219 (Abemaciclib). Prueba 7 Efecto de regresión tumoral en modelo animal de xenoinjerto JeKo-1
Las células JeKo-1 se cultivaron en medio RPMI 1640 que contenía suero bovino fetal al 20%. Se recogieron células JeKo-1 en crecimiento exponencial y se resuspendieron en PBS a una concentración adecuada para la inoculación de tumores subcutáneos en ratones NOD/SCID. Setenta ratones hembra fueron inoculados por vía subcutánea a la derecha con 5x106 células JeKo-1, resuspendidas en PBS y matrigel (1:1). Cuando el volumen promedio del tumor alcanzó los 134 mm3, los ratones se agruparon aleatoriamente según el tamaño del tumor y se les administró. Cuarenta y ocho ratones se dividieron en el grupo experimental y los veintidós ratones restantes no se utilizaron para el experimento. El volumen del tumor se calcula como: diámetro largo x diámetro cortoz/2. La prueba se dividió en un grupo de control de disolvente, compuesto 2b representativo del fármaco de prueba (10mg/kg), compuesto 2b representativo del fármaco de prueba (25 mg/kg), compuesto 2b representativo del fármaco de prueba (50 mg/kg), compuesto 2b representativo del fármaco de prueba (100 mg/kg), un total de 6 grupos con 8 ratones cada uno, y a los ratones se les administró por vía oral por sonda una vez al día y a continuación la administración continuó durante 19 días. La eficacia se evalúa según la tasa relativa de Inhibición del crecimiento tumoral TGI (tumor growth inhibition).
La fórmula de cálculo es la siguiente: TGI (%) = (C-T)/C x 100 % (C y T son el peso promedio del tumor del grupo de control con disolvente y el peso promedio del tumor del grupo de tratamiento, respectivamente). Cuanto mayor sea el valor de TGI (%) mejor será la potencia; y viceversa.
Resultados: el compuesto 2b demuestra una excelente actividad antltumoral.
Tabla 19 Evaluación de la eficacia antitumoral del compuesto 2b representativo en el modelo de xenoinjerto JeKo-1
Grupo Dosis (mg/kg) Tasa relativa de Inhibición del crecimiento tumoral Valor de pa (TGI (%))
Control de disolventes — — Compuesto 2b 10 42,7 0,087 Compuesto 2b 25 73,8 0,003 Compuesto 2b 50 98,3 0,001 Compuesto 2b 100 104,5 0,001 Nota: a: el valor de p es el análisis comparativo del volumen del tumor para el grupo de tratamiento y el grupo de control con disolvente.______________________________________________________________ Por consiguiente, en algunos casos, es beneficioso administrar a un individuo un compuesto 1, 2 o 15 que tiene un elevado exceso enantlomérico del enantlómero que tiene una rotación óptica negativa para tratar una enfermedad. Inesperadamente, el enantiómero (-) ópticamente puro del presente compuesto, incluyendo, sin limitación, los compuestos 1b, 2b o 15b, es un fármaco más potente para tratar una enfermedad mediada por CDK4/6 en un individuo.
Claims (34)
1. Compuesto de fórmula I, o un estereoisómero, un tautómero, un polimorfo, un solvato o una sal farmacéuticamente aceptable del mismo,

en la que,
el anillo A es arllo o heteroarllo;
Z se selecciona entre el grupo que consiste en CH2, NH, O y S;
Ri se selecciona independientemente entre el grupo que consiste en hidrógeno, halógeno, CN, N02, OH, NH2, alquilo C1-8, alcoxi Ci-a, cicloalquilo C3-8, arilo, heteroarilo, heterociclilo, heterociclilo-(CH2)m-, aril-alquilo C1-6-, heteroaril-alquilo C1-6-, -NRi2Ri3, -NRi2-alquileno Ci -6-NRi2Ri3 y heterociclilo-C(O)-, en los que cada uno de los alquilo C1-8, alcoxi C1-8, cicloalquilo C3-a, arilo, heteroarilo, heterociclilo, heterociclilo-(CH2)m-, aril-alquilo C1-6-, heteroaril-alquilo C1-6- o heterociclilo-C(O)- están sin sustituir o sustituidos con, como mínimo, un sustituyeme seleccionado entre halógeno, hidroxilo, alquilo C1-8, cicloalquilo C3-a, heterociclilo, -NRi2Ri3 o -(CH2)tOH;
R2 y R3 se seleccionan cada uno independientemente entre H, OH, CN, N02, NH2, halógeno, alquilo C1-8, alcoxi C1-8, cicloalquilo C3-8, arilo, heteroarilo o heterociclilo; en los que cada uno de los alquilo C1-8, alcoxi C1-8, cicloalquilo C3-8, arilo, heteroarilo, heterociclilo están sin sustituir o sustituidos con, como mínimo, un sustituyeme seleccionado entre halógeno, hidroxilo, alquilo C1-8, cicloalquilo C3-a o heterociclilo;
R12 y R13 se seleccionan cada uno Independientemente entre H, alquilo C1-8, arilo, heteroarilo, heterociclilo o cicloalquilo C3-a; en el que cada uno de los alquilo C1-8, arilo, heteroarilo, heterociclilo o cicloalquilo C3-a están sin sustituir o sustituidos con, como mínimo, un sustituyeme seleccionado entre halógeno, hidroxilo, alquilo C1-8, cicloalquilo C3-a o heterociclilo;
m es 0,1,2, 3 o 4;
n es 0,1, 2, 3 o 4;
t es 0,1,2, 3 o 4;
arilo es un sistema de anillo aromático de 6-10 miembros monocícllco o bicíclico;
heteroarilo es un sistema de anillo aromático monocícllco de cinco o seis miembros estable no sustituido o sustituido o un sistema de anillo heteroaromátlco benzocondensado de nueve o diez miembros no sustituido o sustituido o un sistema de anillo heteroaromátlco bicíclico que consiste en átomos de carbono y de uno a cuatro heteroátomos seleccionados entre N, O o S; y
heterociclilo es un sistema de anillo saturado monocícllco de tres a ocho miembros, estable, no sustituido o sustituido, que consiste en átomos de carbono y de uno a tres heteroátomos seleccionados entre N, O o S.
2. Compuesto, según la reivindicación 1, en el que Z es CH2.
3. Compuesto, según la reivindicación 1, en el que Z es O.
4. Compuesto, según cualquiera de las reivindicaciones 1-3, en el que el anillo A es un heteroarilo de 6 miembros que comprende uno o dos heteroátomos de N.
5. Compuesto, según cualquiera de las reivindicaciones 1-4, en el que el anillo A es piridilo, pirimidinilo o piridazinilo.
6. Compuesto, según cualquiera de las reivindicaciones 1-5, en el que el anillo A es

7. Compuesto, según cualquiera de las reivindicaciones 1 -6 , en el que Ri es heterociclilo-(CH2)m- o heterociclilo-(CH2)m- sustituido con alquilo Ci-a, NRi2Ri3, heterociclilo de 4 a 6 miembros, cicloalquilo C3-6 o -(CH2)r OH.
8. Compuesto, según cualquiera de las reivindicaciones 1-7, en el que R1 es heterociclilo-CH2- de 5 a 6 miembros o heterociclilo-CH2- de 5 a 6 miembros sustituido con alquilo C1-3, -N(CH3)2, -N(CH2CH2OH)CH3,

-CH2OH, -CHsCHsOH u -OH.
9. Compuesto, según cualquiera de las reivindicaciones 1-8, en el que R1 es heterociclilo-CH2- de 6 miembros o heterociclilo-CH2- de 6 miembros sustituido con metilo o etilo.
10. Compuesto, según cualquiera de las reivindicaciones 1-6, en el que R1 es heterociclilo o heterociclilo sustituido con alquilo C1-8, NRi2Ri3, heterociclilo de 4 a 6 miembros, cicloalquilo C3-6 o -(CH2)t-OH.
11. Compuesto, según cualquiera de las reivindicaciones 1 -6 o 10 , en el que R1 es heterociclilo de 5 a 6 miembros o heterociclilo de 5 a 6 miembros sustituido con alquilo Ci-3, -N(CH3)2, -N(CH2CH2OH)CH3,

-CH2OH, -CH2CH2OH u OH.
12. Compuesto, según cualquiera de las reivindicaciones 1 -6 ,10, u 11, en el que R1 es heterociclilo de 6 miembros o heterociclilo de 6 miembros sustituido con metilo o etilo.
13. Compuesto, según cualquiera de las reivindicaciones 1-6, en el que R1 es heterociclilo-C(O) de 6 miembros o heterociclilo-C(O) de 6 miembros sustituido con alquilo C1-3.
14. Compuesto, según cualquiera de las reivindicaciones 1 -6 o 13, en el que R1 es heterociclilo-C(O)- de 6 miembros sustituido con metilo.
15. Compuesto, según cualquiera de las reivindicaciones 1-14, en el que el heterociclilo comprende uno o dos heteroátomos de N u O como átomos del anillo.
16. Compuesto, según cualquiera de las reivindicaciones 1-14, en el que el heterociclilo comprende uno o dos heteroátomos de N como átomos del anillo.
17. Compuesto, según cualquiera de las reivindicaciones 1-6, en el que R1 es -NRi2-alquileno Ci ^-NRi2Ri3.
18. Compuesto, según cualquiera de las reivindicaciones 1-17, en el que Ri2 y Ri3 son cada uno independientemente H, -(CH2)r OH o alquilo C1-3.
19. Compuesto, según cualquiera de las reivindicaciones 1-18, en el que Ri2 y Ri3 son cada uno independientemente OH, CH2CH2OH, metilo o etilo.
20. Compuesto, según la reivindicación 1, en el que R1 es


21. Compuesto, según cualquiera de las reivindicaciones 1-20, en el que m es 1.
22. Compuesto, según cualquiera de las reivindicaciones 1-21, en el que n es 1.
23. Compuesto, según cualquiera de las reivindicaciones 1-22, en el que t es 0,1 o 2.
24. Compuesto, según cualquiera de las reivindicaciones 1-23, en el que R2 y R3 son cada uno independientemente H, OH, halógeno, alquilo C1-6, alquilo C1-6 sustituido con halógeno, alcoxi Ci-6, alcoxi Ci-6 sustituido con halógeno.
25. Compuesto, según cualquiera de las reivindicaciones 1-24, en el que R2 y R3 son cada uno independientemente H, OH, F, Cl, CH3, CH2CH3, CF3, OCH3 u OCF3.
26. Compuesto, según cualquiera de las reivindicaciones 1-25, en el que R2 y R3 son ambos F.
27. Compuesto, según la reivindicación 1, en el que el compuesto es:
1) 4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(5-(4-metilpiperazin-1 -il)piridin-2-il)pirimidin-2-amina;
2) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
3) 5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(5-(4-metilpiperazin-1-i I) pi ridi n-2-i I) pi rim idi n-2-am i na;
4) 5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(6-((4-metilpiperazin-1-il)metil)piridin-3-il)pirimidin-2-amina;
5) 5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-N-(5-((4-metilpiperazin-1-il)metil)pirimidin-2-il)pirimidin-2-amina;
6) N-(5-((4-(dimetilamino)piperidin-1-il)metil)pirimidin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
7) N-(5-((4-(dimetilamino)piperidin-1-il)metil)piridin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
8) N-(5-(4-(dimetilamino)piperidin-1-il)piridin-2-il)-5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
9) (2-((5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-il)amino)pirimidin-5-il)(4-metilpiperazin-1-il)metanona;
10) (6-((5-fluoro-4-(6-fluoro-1-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-11) amino)piridin-3-il)(4-metilpiperazin-1-il)metanona;
11) N5-(2-(dietilamino)etil)-N2-(5-fluoro-4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-11) pirimidin-2-il)-N5-metilpiridin-2,5-diamina;
12) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-4-(6-fluoro-1-metil-1,2,3,4,4a,5-hexahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-5-(trifluorometil)pirimidin-2-amina;
13) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-4-(6-fluoro-1-metil-1,2,3,4,4a,5-hexahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)-5-metilpirimidin-2-amina;
14) 5-cloro-N-(5-((4-etilpiperazin-1 -il)metil)piridin-2-il)-4-(6-fluoro-1 -metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]piridin-8-il)pirimidin-2-amina;
15) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-(4-metilpiperazin-1-i I) pi ridi n-2-i I) pi rim idi n-2-am i na;
16) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
17) N-(5-(4-(dimetilamino)piperidin-1 -il)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
18) N-(5-((4-(dimetilamino)piperidin-1 -il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-1 -3,4-dihidro-1 H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
19) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina;
20) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-(piperazin-1-ilmetil)piridin-2-il)pirimidin-2-amina;
21) N-(5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)-6-(4-m etil pi perazi n-1 -i I) pi ridazi n-3-am ina;
22) 6-((4-etilpiperazin-1-il)metil)-N-(5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)pi ridazi n-3-amina;
23) (1-(6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)pirrolidin-3-il)metanol;
24) (1 -((6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)metil)pirrolidin-3-il)metanol;
25) N-(5-(4-ciclopropilpiperazin-1-il)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
26) N-(5-((4-ciclopropilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-amina;
27) 2-((1 -((6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)metil)piperidin-4-il)(metil)amino)etan-1 -ol;
28) 1-(6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)-3-metilpirrolidin-3-ol;
29) 1 -((6-((5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)pirimidin-2-il)amino)piridin-3-il)metil)-3-metilpirrolidin-3-ol;
30) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)-N-(5-((4-(oxetan-3-il)piperazin-1-il)metil)piridin-2-il)pirimidin-2-amina;
31) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1 H-benzo[4,5]imidazo[2,1 -c][1,4]oxazin-7-il)-N-(5-(4-(oxetan-3-i I) pi perazi n-1 -i I) pi ridi n-2-i I) pi rim idi n-2-am ina;
32) N-(5-((4-etilpiperazin-1-il)metil)piridin-2-il)-5-fluoro-4-(9-fluoro-4-metil-1,2,3,4-tetrahidrobenzo[4,5]imidazo[1,2-a]pirazin-7-il)pirimidin-2-amina;
33) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-((4'-metil-[1,1'-bipiperazin]-4-il)metil)piridin-2-il)pirimidin-2-amina;
34) 5-fluoro-4-(9-fluoro-4-metil-3,4-dihidro-1H-benzo[4,5]imidazo[2,1-c][1,4]oxazin-7-il)-N-(5-((4-(1-metilpiperidin-4-il)piperazin-1 -il)metil)piridin-2-il)pirimidin-2-amina.
28. Compuesto, según cualquiera de las reivindicaciones 1-27, en el que el compuesto es el enantlómero (-) del compuesto.
29. Compuesto, según cualquiera de las reivindicaciones 1-27, en el que el compuesto es el enantiómero (+) del compuesto.
30. Composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto, según cualquiera de las reivindicaciones 1-29, y un excipiente farmacéuticamente aceptable.
31. Composición farmacéutica, según la reivindicación 30, en la que la proporción en peso de dicho compuesto respecto a dicho excipiente está en el Intervalo de, aproximadamente, 0,001 a, aproximadamente, 10.
32. Composición farmacéutica, según la reivindicación 30 o 31, o compuesto, según cualquiera de las reivindicaciones 1-29, para su utilización como medicamento.
33. Composición farmacéutica, según la reivindicación 30 o 31, o compuesto, según cualquiera de las reivindicaciones 1-29, para su utilización en el tratamiento o prevención del cáncer.
34. Composición farmacéutica o compuesto para su utilización, según la reivindicación 33, en los que el cáncer es cáncer de colon, cáncer de recto, linfoma de células del manto, mieloma múltiple, cáncer de mama, cáncer de próstata, glloblastoma, cáncer de esófago de células escamosas, llposarcoma, linfoma de células T, melanoma, cáncer de páncreas, cáncer de cerebro o cáncer de pulmón.
REFERENCIAS CITADAS EN LA DESCRIPCIÓN
Esta lista de referencias citada por el solicitante es únicamente para mayor comodidad del lector. No forman parte del documento de la Patente Europea. Incluso teniendo en cuenta que la compilación de las referencias se ha efectuado con gran cuidado, los errores u omisiones no pueden descartarse; la EPO se exime de toda responsabilidad al respecto.
Documentos de patentes citados en la descripción
• WO 2016173505 A1
Literatura no patente citada en la descripción
• BLIN et al. NucletoAcids Res., 1976, vol. 3, 2303 • Korenarawakaru Kagakunotameno Toukeisyuhou -• FAVALORO et al. Methods Enzymol., 1980, vol. 65, Tadasii Data no Atukalkata. MAKOTO NIWA. Statistical 718 method for chemistry that ¡s easy to understand
• Wetting Technology Handbook. Techno System Co., • How to handle data correctly. Kagaku-Dojin Publishing Ltd, 2001 Company, INC, 2008,101
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2016111457 | 2016-12-22 | ||
| CN2017080661 | 2017-04-14 | ||
| PCT/CN2017/117950 WO2018113771A1 (en) | 2016-12-22 | 2017-12-22 | Benzimidazole derivatives, preparation methods and uses theirof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2917377T3 true ES2917377T3 (es) | 2022-07-08 |
Family
ID=62624617
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17882916T Active ES2917377T3 (es) | 2016-12-22 | 2017-12-22 | Derivados de benzimidazol, procedimientos de preparación y utilizaciones de los mismos |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US11053238B2 (es) |
| EP (1) | EP3559000B1 (es) |
| JP (1) | JP7105781B2 (es) |
| KR (1) | KR102543603B1 (es) |
| CN (1) | CN110234652B (es) |
| AU (1) | AU2017379041B2 (es) |
| CA (1) | CA3047876C (es) |
| DK (1) | DK3559000T3 (es) |
| ES (1) | ES2917377T3 (es) |
| TW (1) | TWI732083B (es) |
| WO (1) | WO2018113771A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106608879A (zh) * | 2015-10-27 | 2017-05-03 | 甘李药业股份有限公司 | 一种蛋白激酶抑制剂及其制备方法和医药用途 |
| CN111315379A (zh) | 2017-08-15 | 2020-06-19 | 北京轩义医药科技有限公司 | Cdk4/6抑制剂及其用途 |
| JP2021514359A (ja) | 2018-02-15 | 2021-06-10 | ニューベイション・バイオ・インコーポレイテッドNuvation Bio Inc. | キナーゼ阻害剤としての複素環式化合物 |
| EA202190036A1 (ru) | 2018-06-21 | 2021-03-24 | Бетта Фармасьютикалз Ко., Лтд | Кристаллическая форма соединения для ингибирования активности cdk4/6 и ее применение |
| SG11202112229UA (en) * | 2019-05-05 | 2021-12-30 | Qilu Regor Therapeutics Inc | Cdk inhibitors |
| CN111138455B (zh) * | 2020-01-16 | 2022-03-04 | 郑州大学 | 一种手性四氢呋喃并恶嗪稠合苯并咪唑衍生物及其制备方法和用途 |
| WO2021218939A1 (zh) * | 2020-04-28 | 2021-11-04 | 贝达药业股份有限公司 | 稠环化合物及其在医药上的应用 |
| KR102269420B1 (ko) | 2020-09-18 | 2021-06-24 | 전용석 | 긴급대피용 마스크장치 |
| WO2022113003A1 (en) | 2020-11-27 | 2022-06-02 | Rhizen Pharmaceuticals Ag | Cdk inhibitors |
| WO2022149057A1 (en) | 2021-01-05 | 2022-07-14 | Rhizen Pharmaceuticals Ag | Cdk inhibitors |
| US20240140963A1 (en) * | 2021-02-03 | 2024-05-02 | Tuojie Biotech (Shanghai) Co., Ltd. | Fused tricyclic cyclin-dependent kinase inhibitor, and preparation method therefor and pharmaceutical use thereof |
| CN112979567B (zh) * | 2021-03-05 | 2023-07-18 | 中国医科大学 | Cdk12小分子抑制剂的化合物及其应用 |
| WO2022199656A1 (zh) * | 2021-03-24 | 2022-09-29 | 贝达药业股份有限公司 | 药物组合、包含其的试剂盒及其用途 |
| WO2023131179A1 (zh) * | 2022-01-05 | 2023-07-13 | 贝达药业股份有限公司 | 苯并咪唑衍生物或其盐在治疗白血病中的用途 |
| WO2024027631A1 (zh) * | 2022-08-01 | 2024-02-08 | 江苏恒瑞医药股份有限公司 | 一种稠三环类衍生物的晶型及制备方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PA8852901A1 (es) | 2008-12-22 | 2010-07-27 | Lilly Co Eli | Inhibidores de proteina cinasa |
| EP2594555B1 (en) * | 2010-07-02 | 2018-03-07 | ASKA Pharmaceutical Co., Ltd. | HETEROCYCLIC COMPOUND, AND p27 KIP1 DEGRADATION INHIBITOR |
| UA111756C2 (uk) | 2011-11-03 | 2016-06-10 | Ф. Хоффманн-Ля Рош Аг | Сполуки гетероарилпіридону та азапіридону як інгібітори тирозинкінази брутона |
| SG10202004716RA (en) | 2014-07-24 | 2020-06-29 | Beta Pharma Inc | 2-h-indazole derivatives as cyclin-dependent kinase (cdk) inhibitors and therapeutic uses thereof |
| EP3288939A1 (en) * | 2015-04-17 | 2018-03-07 | AbbVie Inc. | Tricyclic modulators of tnf signaling |
| TWI696617B (zh) * | 2015-04-28 | 2020-06-21 | 大陸商上海復尚慧創醫藥研究有限公司 | 特定蛋白質激酶抑制劑 |
| CN105153119B (zh) | 2015-09-11 | 2019-01-01 | 广州必贝特医药技术有限公司 | 吡啶嘧啶胺类化合物或吡啶吡啶胺类化合物及其应用 |
| CN106608879A (zh) | 2015-10-27 | 2017-05-03 | 甘李药业股份有限公司 | 一种蛋白激酶抑制剂及其制备方法和医药用途 |
-
2017
- 2017-12-22 TW TW106145306A patent/TWI732083B/zh active
- 2017-12-22 ES ES17882916T patent/ES2917377T3/es active Active
- 2017-12-22 CA CA3047876A patent/CA3047876C/en active Active
- 2017-12-22 WO PCT/CN2017/117950 patent/WO2018113771A1/en unknown
- 2017-12-22 US US16/471,415 patent/US11053238B2/en active Active
- 2017-12-22 AU AU2017379041A patent/AU2017379041B2/en active Active
- 2017-12-22 EP EP17882916.4A patent/EP3559000B1/en active Active
- 2017-12-22 KR KR1020197020604A patent/KR102543603B1/ko active IP Right Grant
- 2017-12-22 CN CN201780078241.XA patent/CN110234652B/zh active Active
- 2017-12-22 JP JP2019534709A patent/JP7105781B2/ja active Active
- 2017-12-22 DK DK17882916.4T patent/DK3559000T3/da active
Also Published As
| Publication number | Publication date |
|---|---|
| US11053238B2 (en) | 2021-07-06 |
| AU2017379041A1 (en) | 2019-07-25 |
| KR20190114971A (ko) | 2019-10-10 |
| WO2018113771A1 (en) | 2018-06-28 |
| CN110234652B (zh) | 2020-11-10 |
| DK3559000T3 (da) | 2022-06-07 |
| JP7105781B2 (ja) | 2022-07-25 |
| EP3559000A4 (en) | 2020-05-27 |
| EP3559000A1 (en) | 2019-10-30 |
| CN110234652A (zh) | 2019-09-13 |
| JP2020504747A (ja) | 2020-02-13 |
| CA3047876C (en) | 2023-08-29 |
| KR102543603B1 (ko) | 2023-06-14 |
| RU2019121937A3 (es) | 2021-02-09 |
| EP3559000B1 (en) | 2022-03-23 |
| AU2017379041B2 (en) | 2021-08-26 |
| TW201829406A (zh) | 2018-08-16 |
| TWI732083B (zh) | 2021-07-01 |
| CA3047876A1 (en) | 2018-06-28 |
| RU2019121937A (ru) | 2021-01-22 |
| US20210130345A1 (en) | 2021-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2917377T3 (es) | Derivados de benzimidazol, procedimientos de preparación y utilizaciones de los mismos | |
| ES2904544T3 (es) | Compuestos de indazol como inhibidores de la cinasa FGFR, preparación y uso de los mismos | |
| ES2936827T3 (es) | Moduladores alostéricos positivos del receptor muscarínico de acetilcolina M4 | |
| RU2636589C2 (ru) | Аминопиримидиновые ингибиторы киназ | |
| EP2170827B1 (en) | Indolin-2-ones and aza-indolin-2-ones | |
| US8076338B2 (en) | Kinase modulators and methods of use | |
| US8685967B2 (en) | Substituted triazolopyridines and analogs thereof | |
| EP2283024B1 (en) | 4-aryl-2-anilino-pyrimidines as plk kinase inhibitors | |
| KR101828187B1 (ko) | 신규 축합 피리미딘 화합물 또는 그 염 | |
| CN113166156B (zh) | 酪氨酸激酶抑制剂、组成及其方法 | |
| BR112019011074A2 (pt) | compostos heterocíclicos tricíclicos úteis como inibidores de hiv-integrase | |
| JP2022517085A (ja) | ハロゲン化アリルアミン系化合物及びその適用 | |
| CA3083624A1 (en) | Exo-aza spiro inhibitors of menin-mll interaction | |
| US10647727B2 (en) | Substituted pyrazolo/imidazolo bicyclic compounds as PDE2 inhibitors | |
| JP6785876B2 (ja) | ピリド[3,4−d]ピリミジン誘導体及びその薬学的に許容される塩 | |
| ES2746007T3 (es) | Derivados de pirimidina como inhibidores de cinasa y sus aplicaciones terapéuticas | |
| EP3854793A1 (en) | Aromatic heterocyclic compound with kinase inhibitory activity | |
| RU2779534C2 (ru) | Производные бензимидазола, способы их получения и применения | |
| WO2023033740A1 (en) | Compounds useful in modulation of ahr signalling | |
| WO2023230477A1 (en) | Pyridine checkpoint kinase 1 (chk1) inhibitors and uses thereof | |
| EP3720860A1 (en) | Positive allosteric modulators of the muscarinic acetylcholine receptor m4 |