ES2896331T3 - Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol - Google Patents
Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol Download PDFInfo
- Publication number
- ES2896331T3 ES2896331T3 ES18740157T ES18740157T ES2896331T3 ES 2896331 T3 ES2896331 T3 ES 2896331T3 ES 18740157 T ES18740157 T ES 18740157T ES 18740157 T ES18740157 T ES 18740157T ES 2896331 T3 ES2896331 T3 ES 2896331T3
- Authority
- ES
- Spain
- Prior art keywords
- methyl
- imidazo
- thiadiazol
- methoxymethyl
- trifluoromethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- UKFPGWLFAKZWBA-UHFFFAOYSA-N O=C1OCCN1C=1N=C2C(=NNS2)N=1 Chemical class O=C1OCCN1C=1N=C2C(=NNS2)N=1 UKFPGWLFAKZWBA-UHFFFAOYSA-N 0.000 title claims abstract description 7
- 239000000203 mixture Substances 0.000 claims abstract description 58
- 150000003839 salts Chemical class 0.000 claims abstract description 17
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 14
- 125000001424 substituent group Chemical group 0.000 claims abstract description 9
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims abstract description 7
- 125000005843 halogen group Chemical group 0.000 claims abstract description 6
- 150000001875 compounds Chemical class 0.000 claims description 111
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 51
- -1 2,2-difluoropropyl Chemical group 0.000 claims description 43
- UFHFLCQGNIYNRP-VVKOMZTBSA-N Dideuterium Chemical compound [2H][2H] UFHFLCQGNIYNRP-VVKOMZTBSA-N 0.000 claims description 26
- 206010010904 Convulsion Diseases 0.000 claims description 25
- LXQUDOVVQQLLCW-UHFFFAOYSA-N COCC1=NN2C(S1)=NC(=C2CN1C(OC(C1)CC(F)(F)F)=O)C(F)(F)F Chemical compound COCC1=NN2C(S1)=NC(=C2CN1C(OC(C1)CC(F)(F)F)=O)C(F)(F)F LXQUDOVVQQLLCW-UHFFFAOYSA-N 0.000 claims description 16
- ZSJCFJSSOMAZRW-UHFFFAOYSA-N FC(C=1N=C2SC(=NN2C=1CN1C(OC(C1)CCC)=O)COC)F Chemical compound FC(C=1N=C2SC(=NN2C=1CN1C(OC(C1)CCC)=O)COC)F ZSJCFJSSOMAZRW-UHFFFAOYSA-N 0.000 claims description 16
- 238000011282 treatment Methods 0.000 claims description 15
- NHOQHYPBBCUQCW-UHFFFAOYSA-N FC(CC1CN(C(O1)=O)CC1=C(N=C2SC(=NN21)COC)C(F)(F)F)(C)F Chemical compound FC(CC1CN(C(O1)=O)CC1=C(N=C2SC(=NN21)COC)C(F)(F)F)(C)F NHOQHYPBBCUQCW-UHFFFAOYSA-N 0.000 claims description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims description 12
- NZQWBRYRKHLJIQ-UHFFFAOYSA-N COCC1=NN2C(S1)=NC(=C2CN1C(OC(C1)CCC)=O)C(F)(F)F Chemical compound COCC1=NN2C(S1)=NC(=C2CN1C(OC(C1)CCC)=O)C(F)(F)F NZQWBRYRKHLJIQ-UHFFFAOYSA-N 0.000 claims description 11
- 239000003814 drug Substances 0.000 claims description 10
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 10
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims description 9
- 206010015037 epilepsy Diseases 0.000 claims description 9
- QMAFUOFHSCCWCP-UHFFFAOYSA-N FC(C=1N=C2SC(=NN2C=1CN1C(OC(C1)CC(C)(F)F)=O)COC)F Chemical compound FC(C=1N=C2SC(=NN2C=1CN1C(OC(C1)CC(C)(F)F)=O)COC)F QMAFUOFHSCCWCP-UHFFFAOYSA-N 0.000 claims description 8
- FOGARGSYAPLGHB-UHFFFAOYSA-N FC(C=1N=C2SC(=NN2C=1CN1C(OC(C1)CC(F)(F)F)=O)COC)F Chemical compound FC(C=1N=C2SC(=NN2C=1CN1C(OC(C1)CC(F)(F)F)=O)COC)F FOGARGSYAPLGHB-UHFFFAOYSA-N 0.000 claims description 8
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 8
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 8
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 claims description 7
- UVNXNSUKKOLFBM-UHFFFAOYSA-N imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=CSC2=NC=CN21 UVNXNSUKKOLFBM-UHFFFAOYSA-N 0.000 claims description 6
- 229910052801 chlorine Inorganic materials 0.000 claims description 5
- 239000003085 diluting agent Substances 0.000 claims description 5
- 125000004780 2-chloro-2,2-difluoroethyl group Chemical group [H]C([H])(*)C(F)(F)Cl 0.000 claims description 4
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 3
- 230000036461 convulsion Effects 0.000 claims description 3
- 230000008579 epileptogenesis Effects 0.000 claims description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 3
- 125000004778 2,2-difluoroethyl group Chemical group [H]C([H])(*)C([H])(F)F 0.000 claims description 2
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 claims description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 61
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 47
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 38
- 101000584505 Homo sapiens Synaptic vesicle glycoprotein 2A Proteins 0.000 description 38
- 102100030701 Synaptic vesicle glycoprotein 2A Human genes 0.000 description 37
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 26
- 230000015572 biosynthetic process Effects 0.000 description 25
- 238000005160 1H NMR spectroscopy Methods 0.000 description 24
- 239000000243 solution Substances 0.000 description 24
- 238000003786 synthesis reaction Methods 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 21
- 238000000034 method Methods 0.000 description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000007787 solid Substances 0.000 description 17
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- QRDLRQBHRVUZJE-UHFFFAOYSA-N 5-propyl-1,3-oxazolidin-2-one Chemical compound CCCC1CNC(=O)O1 QRDLRQBHRVUZJE-UHFFFAOYSA-N 0.000 description 15
- 238000003556 assay Methods 0.000 description 13
- 235000019439 ethyl acetate Nutrition 0.000 description 13
- 239000012528 membrane Substances 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 12
- 102000004328 Cytochrome P-450 CYP3A Human genes 0.000 description 12
- LCTYULDOLQFVFP-UHFFFAOYSA-N [6-(difluoromethyl)-2-(methoxymethyl)imidazo[2,1-b][1,3,4]thiadiazol-5-yl]methanol Chemical compound FC(C=1N=C2SC(=NN2C=1CO)COC)F LCTYULDOLQFVFP-UHFFFAOYSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 238000004949 mass spectrometry Methods 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- 238000010828 elution Methods 0.000 description 11
- 101000584382 Homo sapiens Synaptic vesicle glycoprotein 2C Proteins 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 238000003818 flash chromatography Methods 0.000 description 10
- 239000012634 fragment Substances 0.000 description 10
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 10
- NEACJRFBJRBDHT-UHFFFAOYSA-N 6-(difluoromethyl)-2-(methoxymethyl)imidazo[2,1-b][1,3,4]thiadiazole Chemical compound C1=C(C(F)F)N=C2SC(COC)=NN21 NEACJRFBJRBDHT-UHFFFAOYSA-N 0.000 description 9
- 102100030637 Synaptic vesicle glycoprotein 2C Human genes 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 239000003795 chemical substances by application Substances 0.000 description 9
- 239000003446 ligand Substances 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- JJLJIZRNYBEBDV-UHFFFAOYSA-N FC(CC1CNC(O1)=O)(C)F Chemical compound FC(CC1CNC(O1)=O)(C)F JJLJIZRNYBEBDV-UHFFFAOYSA-N 0.000 description 8
- DHJRTAQQZSICKT-UHFFFAOYSA-N FC(CC1CNC(O1)=O)(F)F Chemical compound FC(CC1CNC(O1)=O)(F)F DHJRTAQQZSICKT-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 8
- 210000004027 cell Anatomy 0.000 description 8
- 239000012230 colorless oil Substances 0.000 description 8
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 8
- 235000019341 magnesium sulphate Nutrition 0.000 description 8
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 8
- 0 *c([s]1)n[n]2c1nc(*=C)c2CN(CN(*)C1)C1=O Chemical compound *c([s]1)n[n]2c1nc(*=C)c2CN(CN(*)C1)C1=O 0.000 description 7
- ZRUPXAZUXDFLTG-UHFFFAOYSA-N 1-aminopentan-2-ol Chemical compound CCCC(O)CN ZRUPXAZUXDFLTG-UHFFFAOYSA-N 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- FMHQJXGMLMSMLC-WBUYAQKGSA-N azamulin Chemical compound O([C@@H]1C[C@@]([C@H]([C@H](C)[C@]23CCC(=O)[C@H]2[C@@]1(C)[C@@H](CC3)C)O)(C)CC)C(=O)CSC1=NNC(N)=N1 FMHQJXGMLMSMLC-WBUYAQKGSA-N 0.000 description 7
- 229950008762 azamulin Drugs 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 238000004811 liquid chromatography Methods 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- KZNGMHZLGGFBIH-UHFFFAOYSA-N 1-amino-4,4,4-trifluorobutan-2-ol Chemical compound NCC(O)CC(F)(F)F KZNGMHZLGGFBIH-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 6
- 239000001961 anticonvulsive agent Substances 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 210000004556 brain Anatomy 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 150000002367 halogens Chemical group 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 229910000104 sodium hydride Inorganic materials 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 238000004808 supercritical fluid chromatography Methods 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- 229930040373 Paraformaldehyde Natural products 0.000 description 5
- JRUVMZWVOOBALP-UHFFFAOYSA-N [2-(methoxymethyl)-6-(trifluoromethyl)imidazo[2,1-b][1,3,4]thiadiazol-5-yl]methanol Chemical compound OCC1=C(C(F)(F)F)N=C2SC(COC)=NN21 JRUVMZWVOOBALP-UHFFFAOYSA-N 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 210000003494 hepatocyte Anatomy 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 229920002866 paraformaldehyde Polymers 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 5
- MLFNIXAEDKGMPE-UHFFFAOYSA-N 4,4,4-trifluoro-1-nitrobutan-2-ol Chemical compound FC(CC(C[N+](=O)[O-])O)(F)F MLFNIXAEDKGMPE-UHFFFAOYSA-N 0.000 description 4
- IIWPWEAVBKFGSW-UHFFFAOYSA-N 4,4-difluoro-1-nitropentan-2-ol Chemical compound FC(CC(C[N+](=O)[O-])O)(C)F IIWPWEAVBKFGSW-UHFFFAOYSA-N 0.000 description 4
- 101000584515 Homo sapiens Synaptic vesicle glycoprotein 2B Proteins 0.000 description 4
- LHHVJYHCDVZPNM-UHFFFAOYSA-N NCC(CC(C)(F)F)O Chemical compound NCC(CC(C)(F)F)O LHHVJYHCDVZPNM-UHFFFAOYSA-N 0.000 description 4
- CWRVKFFCRWGWCS-UHFFFAOYSA-N Pentrazole Chemical compound C1CCCCC2=NN=NN21 CWRVKFFCRWGWCS-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 102100030700 Synaptic vesicle glycoprotein 2B Human genes 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 238000013537 high throughput screening Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 229960004002 levetiracetam Drugs 0.000 description 4
- HPHUVLMMVZITSG-ZCFIWIBFSA-N levetiracetam Chemical compound CC[C@H](C(N)=O)N1CCCC1=O HPHUVLMMVZITSG-ZCFIWIBFSA-N 0.000 description 4
- 229960005152 pentetrazol Drugs 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- ACNMOTQCLBFDMQ-UHFFFAOYSA-N 1-nitropentan-2-ol Chemical compound CCCC(O)C[N+]([O-])=O ACNMOTQCLBFDMQ-UHFFFAOYSA-N 0.000 description 3
- ZLDOCTTXAVVDHG-UHFFFAOYSA-N 5-(methoxymethyl)-1,3,4-thiadiazol-2-amine Chemical compound COCC1=NN=C(N)S1 ZLDOCTTXAVVDHG-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N Butyraldehyde Chemical compound CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 230000003556 anti-epileptic effect Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 238000002372 labelling Methods 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000005191 phase separation Methods 0.000 description 3
- 239000002798 polar solvent Substances 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 238000002821 scintillation proximity assay Methods 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- MAZRLHAJUPFISK-UHFFFAOYSA-N 3-bromo-1,1-difluoropropan-2-one Chemical compound FC(F)C(=O)CBr MAZRLHAJUPFISK-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 206010053398 Clonic convulsion Diseases 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 229940035676 analgesics Drugs 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000000730 antalgic agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- CODNYICXDISAEA-UHFFFAOYSA-N bromine monochloride Chemical compound BrCl CODNYICXDISAEA-UHFFFAOYSA-N 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- YKPUWZUDDOIDPM-SOFGYWHQSA-N capsaicin Chemical compound COC1=CC(CNC(=O)CCCC\C=C\C(C)C)=CC=C1O YKPUWZUDDOIDPM-SOFGYWHQSA-N 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- 238000004807 desolvation Methods 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 230000001037 epileptic effect Effects 0.000 description 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 2
- 230000021824 exploration behavior Effects 0.000 description 2
- 210000003414 extremity Anatomy 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 2
- 150000004682 monohydrates Chemical class 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 238000006386 neutralization reaction Methods 0.000 description 2
- 239000002547 new drug Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 238000011533 pre-incubation Methods 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 238000007423 screening assay Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- NDVLTYZPCACLMA-UHFFFAOYSA-N silver oxide Chemical compound [O-2].[Ag+].[Ag+] NDVLTYZPCACLMA-UHFFFAOYSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 230000001360 synchronised effect Effects 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- VLPIATFUUWWMKC-SNVBAGLBSA-N (2r)-1-(2,6-dimethylphenoxy)propan-2-amine Chemical compound C[C@@H](N)COC1=C(C)C=CC=C1C VLPIATFUUWWMKC-SNVBAGLBSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 description 1
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 1
- XMGJGSKRRWXOIF-UHFFFAOYSA-N 2-(azepan-1-yl)ethyl 2-cyclohexyl-2-thiophen-3-ylacetate;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C1CCCCC1C(C1=CSC=C1)C(=O)OCCN1CCCCCC1 XMGJGSKRRWXOIF-UHFFFAOYSA-N 0.000 description 1
- KWGSNTNURWIDRM-UHFFFAOYSA-N 2-(methoxymethyl)-6-(trifluoromethyl)imidazo[2,1-b][1,3,4]thiadiazole Chemical compound C1=C(C(F)(F)F)N=C2SC(COC)=NN21 KWGSNTNURWIDRM-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- KWGSNTNURWIDRM-SMZGMGDZSA-N 2-[dideuterio(methoxy)methyl]-6-(trifluoromethyl)imidazo[2,1-b][1,3,4]thiadiazole Chemical compound C1=C(C(F)(F)F)N=C2SC(C([2H])(OC)[2H])=NN21 KWGSNTNURWIDRM-SMZGMGDZSA-N 0.000 description 1
- UTMIEQASUFFADK-UHFFFAOYSA-N 3,3,3-trifluoropropanal Chemical compound FC(F)(F)CC=O UTMIEQASUFFADK-UHFFFAOYSA-N 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- GJSURZIOUXUGAL-UHFFFAOYSA-N Clonidine Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCN1 GJSURZIOUXUGAL-UHFFFAOYSA-N 0.000 description 1
- 206010009346 Clonus Diseases 0.000 description 1
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 description 1
- 208000001654 Drug Resistant Epilepsy Diseases 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- UEQUQVLFIPOEMF-UHFFFAOYSA-N Mianserin Chemical compound C1C2=CC=CC=C2N2CCN(C)CC2C2=CC=CC=C21 UEQUQVLFIPOEMF-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 206010028347 Muscle twitching Diseases 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 238000011785 NMRI mouse Methods 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 229920005439 Perspex® Polymers 0.000 description 1
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- JRUVMZWVOOBALP-SMZGMGDZSA-N [2-[dideuterio(methoxy)methyl]-6-(trifluoromethyl)imidazo[2,1-b][1,3,4]thiadiazol-5-yl]methanol Chemical compound [2H]C(C1=NN2C(S1)=NC(=C2CO)C(F)(F)F)(OC)[2H] JRUVMZWVOOBALP-SMZGMGDZSA-N 0.000 description 1
- JRUVMZWVOOBALP-WNWXXORZSA-N [2-[dideuterio(trideuteriomethoxy)methyl]-6-(trifluoromethyl)imidazo[2,1-b][1,3,4]thiadiazol-5-yl]methanol Chemical compound [2H]C(C1=NN2C(S1)=NC(=C2CO)C(F)(F)F)(OC([2H])([2H])[2H])[2H] JRUVMZWVOOBALP-WNWXXORZSA-N 0.000 description 1
- KWGSNTNURWIDRM-WNWXXORZSA-N [2H]C(C1=NN2C(S1)=NC(=C2)C(F)(F)F)(OC([2H])([2H])[2H])[2H] Chemical compound [2H]C(C1=NN2C(S1)=NC(=C2)C(F)(F)F)(OC([2H])([2H])[2H])[2H] KWGSNTNURWIDRM-WNWXXORZSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000003797 alkaloid derivatives Chemical class 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940025084 amphetamine Drugs 0.000 description 1
- 230000003288 anthiarrhythmic effect Effects 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 230000003561 anti-manic effect Effects 0.000 description 1
- 230000001663 anti-spastic effect Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229960003965 antiepileptics Drugs 0.000 description 1
- 229940082988 antihypertensives serotonin antagonists Drugs 0.000 description 1
- 239000000228 antimanic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000000164 antipsychotic agent Substances 0.000 description 1
- 229940005529 antipsychotics Drugs 0.000 description 1
- 239000003420 antiserotonin agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960000794 baclofen Drugs 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 238000007413 biotinylation Methods 0.000 description 1
- 230000006287 biotinylation Effects 0.000 description 1
- 230000003925 brain function Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229960005069 calcium Drugs 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 229960002504 capsaicin Drugs 0.000 description 1
- 235000017663 capsaicin Nutrition 0.000 description 1
- 229960000623 carbamazepine Drugs 0.000 description 1
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229940112822 chewing gum Drugs 0.000 description 1
- 235000015218 chewing gum Nutrition 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229960001076 chlorpromazine Drugs 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 description 1
- 229960003120 clonazepam Drugs 0.000 description 1
- 229960002896 clonidine Drugs 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000002920 convulsive effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000009109 curative therapy Methods 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229960003914 desipramine Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000007877 drug screening Methods 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 208000028329 epileptic seizure Diseases 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 231100000573 exposure to toxins Toxicity 0.000 description 1
- 238000010304 firing Methods 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 238000002875 fluorescence polarization Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229960002464 fluoxetine Drugs 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N gamma-butyrolactam Natural products O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 229960003878 haloperidol Drugs 0.000 description 1
- 208000021760 high fever Diseases 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 238000012203 high throughput assay Methods 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 102000055511 human SV2A Human genes 0.000 description 1
- 102000056327 human SV2C Human genes 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical class C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 238000007031 hydroxymethylation reaction Methods 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 description 1
- 229960004801 imipramine Drugs 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 231100000566 intoxication Toxicity 0.000 description 1
- 230000035987 intoxication Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000009191 jumping Effects 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 230000003137 locomotive effect Effects 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229960001047 methyl salicylate Drugs 0.000 description 1
- 229960003404 mexiletine Drugs 0.000 description 1
- 229960003955 mianserin Drugs 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 229960003793 midazolam Drugs 0.000 description 1
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 239000004050 mood stabilizer Substances 0.000 description 1
- 230000003533 narcotic effect Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000000014 opioid analgesic Substances 0.000 description 1
- 229940005483 opioid analgesics Drugs 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- PRGUDWLMFLCODA-UHFFFAOYSA-N oxybuprocaine hydrochloride Chemical compound [Cl-].CCCCOC1=CC(C(=O)OCC[NH+](CC)CC)=CC=C1N PRGUDWLMFLCODA-UHFFFAOYSA-N 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 150000002990 phenothiazines Chemical class 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 229960002036 phenytoin Drugs 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- URKOMYMAXPYINW-UHFFFAOYSA-N quetiapine Chemical compound C1CN(CCOCCO)CCN1C1=NC2=CC=CC=C2SC2=CC=CC=C12 URKOMYMAXPYINW-UHFFFAOYSA-N 0.000 description 1
- 229960004431 quetiapine Drugs 0.000 description 1
- 239000002287 radioligand Substances 0.000 description 1
- 230000000384 rearing effect Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 230000001020 rhythmical effect Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960001534 risperidone Drugs 0.000 description 1
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229910001923 silver oxide Inorganic materials 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 238000002603 single-photon emission computed tomography Methods 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000001256 tonic effect Effects 0.000 description 1
- 229960003991 trazodone Drugs 0.000 description 1
- PHLBKPHSAVXXEF-UHFFFAOYSA-N trazodone Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1 PHLBKPHSAVXXEF-UHFFFAOYSA-N 0.000 description 1
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 210000001364 upper extremity Anatomy 0.000 description 1
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 1
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol de acuerdo con la fórmula (I), sus isómeros geométricos, sus enantiómeros, sus diastereómeros, sus isótopos y sus mezclas, o una sal farmacéuticamente aceptable de los mismos, **(Ver fórmula)** en donde R1 es un alquilo(C1-4) opcionalmente sustituido con uno o más sustituyentes de tipo halógeno; R2 es un átomo de halógeno, o un alquilo(C1-4) opcionalmente sustituido con uno o más átomos de halógeno; R3 es un alquilo(C1-4) sustituido con un sustituyente alcoxi.
Description
DESCRIPCIÓN
Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol
Introducción
La presente invención se refiere a derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol, procedimientos para prepararlos, composiciones farmacéuticas que los contienen y su uso como productos farmacéuticos.
Un problema persistente en el control de las convulsiones surge con los pacientes que no responden en absoluto o solo de manera insuficiente a los tratamientos actualmente disponibles. Se considera que esos pacientes son insensibles al tratamiento y representan un desafío considerable para la comunidad médica. Se estima que aproximadamente el 30 % de los pacientes con epilepsia se clasifican entre los pacientes que no responden al tratamiento. Por tanto, existe la necesidad de desarrollar nuevos medicamentos que se dirijan específicamente a esta población de pacientes.
Los compuestos antiepilépticos de la fórmula (A) se describen en la solicitud de patente internacional WO 2008/132139:

Y es O o S;
R1 es hidrógeno o alquilo(C1-6);
R2 es hidrógeno;
R3 es -CONR5R6, -COR7, un resto imidazolilo, un resto imidazopiridinilo, un resto imidazopiridazinilo;
R5, R6 son iguales o diferentes, y se seleccionan independientemente entre hidrógeno y alquilo(C1-6);
R7 es alquilo(C1-6);
Z es un resto heterocíclico monocíclico o bicíclico seleccionado del grupo que consiste en imidazolidín-1-ilo, 1,3-oxazolidín-3-ilo, 2,5-dihidro-1 H-pirrol-1 -ilo, 1,3-tiazol-3(2H)-ilo, 1,3-tiazolidín-3-ilo, piperidín-1-ilo, azepán-1-ilo, 5,6-dihidro-4H-tieno[3,2-b]pirrol-4-ilo, hexahidro-4H-tieno[3,2-b]pirrol-4-ilo, 2,3-dihidro-1 H-tieno[3,4-b]pirrol-1 -ilo, 1,3-benzotiazol-3(2H)-ilo, 1,3-benzoxazol-3(2H)-ilo, pirazolo[1,5-a]piridín-1(2H)-ilo, 3,4-dihidroisoquinolín-2(1H)-ilo, 3,4-dihidroquinolín-1 (2H)-ilo, 1,3,4,5-tetrahidro-2H-2-benzazepín-2-ilo, 1,2,4,5-tetrahidro-3H-3-benzazepín-3-ilo.
En una realización específica de la solicitud de patente internacional WO 2008/132139, el resto Z = Y en la fórmula (A) podría ser:

en donde X es O o S.
Los compuestos antiepilépticos de la fórmula (I) se describen en la solicitud de patente internacional WO 2011/047860: derivados del 2-oxo-1 -pirrolidinilimidazotiadiazol de acuerdo con la fórmula (I)

en donde
R1 es un alquilo(Ci-4) que contiene al menos un sustituyente de tipo halógeno;
R2 es un halógeno (cloro, bromo, yodo) o un alquilo(C1-4) que contiene al menos un sustituyente de tipo halógeno; R3 es un alquilo(C1-4) (por ejemplo, un metilo o etilo) que contiene al menos un sustituyente hidroxi (OH) o alcoxi (por ejemplo, metoxi o etoxi o propoxi).
Los compuestos antiepilépticos de la fórmula (I) se describen en la solicitud de patente internacional WO 2012/143116: derivados del 4-oxo-1-imidazolidinilimidazotiadiazol de acuerdo con la fórmula (I),

R1 es un alquilo(C1-4) opcionalmente sustituido con uno o más (1 a 6, preferiblemente 2, 3 o 5) halógenos, con un fenilo sustituido o sin sustituir o con un cicloalquilo(C1-4); sustituido o sin sustituir;
R2 es un halógeno (cloro, bromo, yodo) o un alquilo(C1-4) que contiene uno o más (es decir, 1,2 o 3) sustituyentes de tipo halógeno;
R3 es un alquilo(C1-4) (por ejemplo, un metilo o etilo) que contiene al menos un sustituyente hidroxi (OH) o alcoxi (por ejemplo, metoxi o etoxi o propoxi).
La invención da a conocer nuevos derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol que tienen la fórmula (I), sus isómeros geométricos, sus enantiómeros, sus diastereoisómeros y sus mezclas, o una sal farmacéuticamente aceptable de los mismos,

Otros aspectos de la invención resultarán evidentes a partir de la descripción detallada.
Descripción detallada de la invención
La presente invención se refiere a derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol de acuerdo con la fórmula (I),

R1 es un alquilo(C1-4) opcionalmente sustituido con uno o más sustituyentes de tipo halógeno;
R2 es un átomo de halógeno o un alquilo(C1-4) opcionalmente sustituido con uno o más átomos de halógeno;
R3 es un alquilo(Ci-4) sustituido con un sustituyente de tipo alcoxi.
También se incluyen tautómeros, isómeros geométricos, enantiómeros, diastereómeros, isótopos y mezclas, o una sal farmacéuticamente aceptable de los compuestos de la fórmula (I), así como cualquier variante deuterada.
En una realización específica, R1 es un resto de i-butilo, n-propilo, 2,2-difluoropropilo, 2-cloro-2,2-difluoroetilo, 2,2-difluoroetilo, 2,2,2-trifluoroetilo o 2-fluoroetilo.
En una realización más específica, R1 es un resto de n-propilo, 2-cloro-2,2-difluoroetilo, 2,2-difluoropropilo o 2,2,2-trifluoroetilo.
Preferiblemente, R1 es un resto de 2,2-difluoropropilo, 2,2,2-trifluoroetilo o n-propilo.
En otra realización específica, R2 es un átomo de cloro, un resto difluorometilo o un resto trifluorometilo.
En una realización preferida, R2 es un resto difluorometilo o un resto trifluorometilo.
En otra realización específica, R3 es un resto de metoximetilo, [(2H3)metiloxi]metilo, metoxi(2H2)metilo, o [(2H3)metiloxi](2H2)metilo.
En una realización preferida, R3 es un resto metoximetilo.
En una realización específica adicional, los compuestos de la fórmula (I) son aquellos en los que:
R1 es un resto n-propilo, un resto de 2-cloro-2,2-difluoroetilo, 2,2-difluoropropilo o 2,2,2-trifluoroetilo;
R2 es un átomo de cloro, un resto de difluorometilo o trifluorometilo;
R3 es un resto de metoximetilo, [(2H3)metiloxi]metilo, metoxi(2H2)metilo, o [(2H3)metiloxi](2H2)metilo.
En una realización específica adicional, los compuestos de la fórmula (I) son aquellos en los que:
R1 es un resto n-propilo; un resto de 2,2-difluoropropilo o 2,2,2-trifluoroetilo
R2 es un resto de difluorometilo o trifluorometilo;
R3 es un resto de metoximetilo, [(2H3)metiloxi]metilo, metoxi(2H2)metilo o [(2H3)metiloxi](2H2)metilo.
En una realización preferida, los compuestos de la fórmula (I) son aquellos en los que:
R1 es un resto n-propilo; un resto de 2,2-difluoropropilo o 2,2,2-trifluoroetilo;
R2 es un resto de difluorometilo o trifluorometilo;
R3 es un resto de metoximetilo.
Los compuestos específicos de la presente invención son los seleccionados del grupo que consiste en:
(+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
(-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
(+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
(-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
(+)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona; (-)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona; (+)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
(-)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
(+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona; (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona;
• (+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona;
• (-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona;
• (-)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona;
• (+)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona;
• 3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2-difluoropropil)oxazolidín-2-ona, enantiómero 1;
• 3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2-difluoropropil)oxazolidín-2-ona, enantiómero 2.
Los compuestos de la presente invención son para uso como un medicamento, en el tratamiento de la epilepsia, epileptogénesis, trastornos convulsivos, convulsiones, en concreto, para las crisis comiciales que no responden al tratamiento.
Los siguientes párrafos proporcionan definiciones de los diversos restos químicos que forman los compuestos de acuerdo con la invención y se pretende que se apliquen uniformemente a lo largo de la memoria descriptiva y de las reivindicaciones, a menos que una definición establecida expresamente de otro modo proporcione una definición más amplia.
"Alquilo(C1-4)" se refiere a los grupos alquilo que tienen de 1 a 4 átomos de carbono. Este término está ejemplificado por grupos tales como metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, ferf-butilo. Los grupos “alquilo(C1-4)” pueden estar sustituidos con uno o más sustituyentes seleccionados entre halógeno o alcoxi.
Cualquier resto "H" en la fórmula (I) puede ser el isótopo de hidrógeno, deuterio o tritio.
"Alcoxi" se refiere al grupo -O-R en donde R incluye "alquilo(C1-4)".
"Halógeno" se refiere a átomos de flúor, cloro, bromo y yodo, preferiblemente flúor y cloro.
Las “sales farmacéuticamente aceptables” de acuerdo con la invención incluyen formas de sales de ácido o de base no tóxicas y terapéuticamente activas que los compuestos de la fórmula (I) son capaces de formar.
La forma de sal de adición de ácido de un compuesto de fórmula (I) que se presenta en su forma libre como una base puede obtenerse al tratar la base libre con un ácido apropiado, tal como un ácido inorgánico, por ejemplo, un hidrohaluro, tal como clorhídrico o bromhídrico, sulfúrico, nítrico, fosfórico y similares; o un ácido orgánico, tal como, por ejemplo, acético, trifluoroacético, hidroxiacético, propanoico, láctico, pirúvico, malónico, succínico, maleico, fumárico, málico, tartárico, cítrico, metanosulfónico, etanosulfónico, bencenosulfónico, p-toluenosulfónico, ciclámico, salicílico, p-aminosalicílico, pamoico y similares.
Los compuestos de la fórmula (I) que contienen protones ácidos se pueden convertir en sus formas de sales de adición de base no tóxicas y terapéuticamente activas, p. ej., sales de metales o de amina, mediante el tratamiento con las bases orgánicas e inorgánicas apropiadas. Las formas de sal de base apropiadas incluyen, por ejemplo, sales de amonio, sales de metales alcalinos y alcalinotérreos, p. ej., sales de litio, sodio, potasio, magnesio, calcio y similares, sales con bases orgánicas, p. ej., N-metil-D-glucamina, sales de hidrabamina, y sales con aminoácidos tales como, por ejemplo, arginina, lisina y similares.
Y a la inversa, dichas formas de sales se pueden convertir en las formas libres mediante el tratamiento con una base o ácido apropiados.
Los compuestos de la fórmula (I) y sus sales pueden estar en forma de solvato, que está incluido dentro del alcance de la presente invención. Dichos solvatos incluyen, por ejemplo, hidratos, alcoholatos y similares.
Los compuestos de la fórmula (I) y/o sus intermedios pueden tener al menos un centro estereogénico en su estructura. Este centro estereogénico puede estar presente en una configuración R o S, en donde dicha notación R y S se utiliza en correspondencia con las reglas descritas en Pure Appl. Chem, 45 (1976) 11-30. Por lo tanto, la invención también se refiere a todas las formas estereoisoméricas, tales como las formas enantioméricas y diastereoisoméricas de los compuestos de la fórmula (I) o las mezclas de los mismos (incluidas todas las posibles mezclas de estereoisómeros). Con respecto a la presente invención, la referencia a un compuesto o compuestos pretende abarcar ese compuesto en cada una de sus posibles formas isoméricas y mezclas de las mismas, a menos que se mencione específicamente la forma isomérica concreta. La expresión "enantioméricamente puro", tal y como se usa en la presente memoria, se refiere a los compuestos que tienen un exceso enantiomérico (ee) superior al 95 %.
Los compuestos de acuerdo con la presente invención pueden existir en diferentes formas polimórficas. Aunque no se
indica explícitamente en la fórmula anterior, se pretende que tales formas estén incluidas dentro del alcance de la presente invención.
Los compuestos de la fórmula (I) de acuerdo con la invención se pueden preparar de forma análoga a los métodos convencionales como los entiende el experto en la técnica de la química orgánica sintética.
Según una realización, los compuestos que tienen la fórmula general (I) pueden prepararse mediante la reacción de un compuesto de fórmula (II) con una urea de la fórmula (III) según la ecuación:
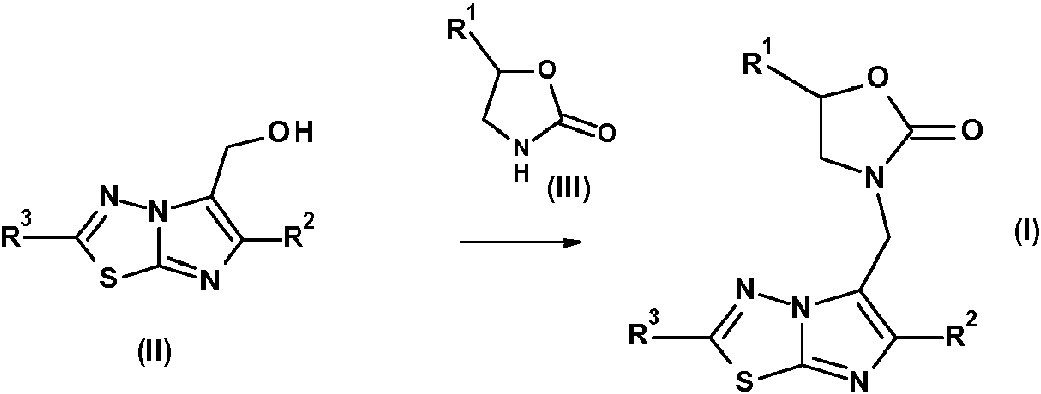
en donde R1, R2 y R3 tienen las mismas definiciones que las definidas anteriormente para los compuestos de la fórmula (I). Esta reacción se puede llevar a cabo con un ácido, tal como el ácido p-toluensulfónico en un solvente aprótico como el sulfolano a alta temperatura.
Los compuestos de la fórmula (II) se pueden preparar mediante la hidroximetilación de un compuesto de la fórmula (IV) de acuerdo con la ecuación:

en donde R2 y R3 tienen la misma definición que la definida anteriormente para los compuestos de la fórmula (I). Esta reacción se puede realizar con un agente formilante, tal como el paraformaldehído, en condiciones ácidas en un solvente polar, tal como el dioxano a 100°C, o de acuerdo con cualquier otro método conocido por el experto en la técnica. Los compuestos de la fórmula (IV) se pueden sintetizar mediante la reacción de un compuesto de la fórmula (V) con un derivado del bromo de la fórmula (VI) de acuerdo con la ecuación:

en donde R2 y R3 tienen la misma definición que la descrita anteriormente para los compuestos de la fórmula (I). Esta reacción se puede realizar con los procedimientos descritos en la bibliografía o conocidos por el experto en la técnica. Los compuestos de la fórmula (V) y los compuestos de la fórmula (VI) están disponibles comercialmente o pueden sintetizarse de acuerdo con algún método conocido por el experto en la técnica.
Según una realización, los compuestos de la fórmula (III) pueden prepararse mediante la ciclación de un compuesto de la fórmula (VII) según la ecuación:

Esta reacción se puede realizar con el 1,1'-carbonildiimidazol en un solvente polar, tal como el tetrahidrofurano, en presencia de una base, tal como el hidruro de sodio, a temperatura ambiente o mediante el uso de los procedimientos conocidos por el experto en la técnica.
Los compuestos de la fórmula (VII) se pueden preparar mediante la reducción de un compuesto de la fórmula (VIII) de acuerdo con la ecuación

Esta reacción se puede llevar a cabo con un reductor, tal como, pero sin limitarse a ellos, hidrógeno en presencia de un catalizador tal como el paladio sobre carbón en un solvente polar tal como el etanol, o por otros procedimientos conocidos por el experto en la técnica.
Los compuestos de la fórmula (VIII) se pueden preparar a partir de aldehídos o alcoholes disponibles en el mercado, en los que R1 tiene la misma definición que la definida anteriormente según cualquier secuencia de reacción conocida por el experto en la técnica.
Los compuestos de la presente invención son para uso como medicamento, para el tratamiento de la epilepsia, epileptogénesis, trastornos convulsivos, convulsiones, en concreto, para las crisis comiciales que no responden al tratamiento.
Las convulsiones se pueden clasificar como insensibles cuando un paciente no se mantiene sin convulsiones durante 12 meses o más con el tratamiento más avanzado con dos o más fármacos antiepilépticos a las dosis máximas toleradas. La Liga Internacional Contra la Epilepsia (ILAE, por su nombre en inglés) ha definido la epilepsia farmacorresistente como "el fracaso de los ensayos adecuados de dos pautas de FAE tolerados, elegidos y utilizados apropiadamente (ya sea como monoterapias o en politerapias) para lograr una ausencia sostenida de convulsiones". Los métodos de la invención comprenden la administración de un compuesto según la invención a un mamífero (preferiblemente a un ser humano) que padece las afecciones o trastornos mencionados anteriormente, en una cantidad suficiente para aliviar o prevenir el trastorno o la afección.
El compuesto se administra por comodidad en cualquier forma de dosificación unitaria adecuada, que incluye, pero no se limita a ellas, una que contiene de 1 a 2000 mg, preferiblemente de 1 a 1000 mg, más preferiblemente de 1 a 500 mg, del ingrediente activo por forma de dosificación unitaria.
El término "tratamiento", tal y como se usa en la presente memoria, incluye un tratamiento curativo y un tratamiento profiláctico.
Por "curativo" se entiende la eficacia para el tratamiento de un episodio sintomático actual de un trastorno o afección. Por "profiláctico" se entiende la prevención de la aparición o recurrencia de un trastorno o afección.
El término "epilepsia", tal y como se usa en la presente memoria, se refiere a una afección neurológica crónica caracterizada por crisis epilépticas recurrentes no provocadas. Una crisis epiléptica es la manifestación de una descarga sincronizada anómala y excesiva de un conjunto de neuronas cerebrales; sus manifestaciones clínicas son repentinas y transitorias. El término "epilepsia" tal y como se usa en la presente memoria también puede referirse a un trastorno del funcionamiento cerebral caracterizado por la aparición periódica de convulsiones. Las convulsiones pueden ser "no epilépticas" cuando se desencadenan en un cerebro normal por afecciones tales como fiebre alta o la exposición a toxinas, o "epilépticas" cuando aparecen sin una provocación evidente.
El término "convulsión", tal y como se usa en la presente memoria, se refiere a una alteración transitoria del comportamiento debido a la descarga desordenada, sincronizada y rítmica de poblaciones de neuronas cerebrales. Otro aspecto de la presente invención se refiere a una composición farmacéutica que comprende una cantidad eficaz de un compuesto de la fórmula (I) en combinación con un diluyente o vehículo farmacéuticamente aceptable.
Por supuesto, la actividad en cualquiera de las indicaciones mencionadas anteriormente puede determinarse cuando se llevan a cabo ensayos clínicos adecuados de una manera conocida por un experto en la técnica relevante para la indicación en concreto y/o en el diseño de los ensayos clínicos en general.
Para el tratamiento de las enfermedades, los compuestos de la fórmula (I) o sus sales farmacéuticamente aceptables pueden emplearse a una dosis diaria eficaz y se pueden administrar en forma de una composición farmacéutica. Por lo tanto, otra realización de la presente invención se refiere a una composición farmacéutica que comprende una cantidad eficaz de un compuesto de la fórmula (I) o una sal farmacéuticamente aceptable del mismo en combinación con un diluyente o vehículo farmacéuticamente aceptable.
Para preparar una composición farmacéutica de acuerdo con la invención, uno o más de los compuestos de la fórmula (I) o una sal farmacéuticamente aceptable de los mismos se mezclan íntimamente con un diluyente o vehículo farmacéutico de acuerdo con las técnicas convencionales de composición farmacéutica conocidas por el facultativo experto.
Los diluyentes y los vehículos adecuados pueden tomar una amplia variedad de formas dependiendo de la vía de administración deseada, por ejemplo, oral, rectal, parenteral o intranasal.
Las composiciones farmacéuticas que comprenden los compuestos de acuerdo con la invención pueden, por ejemplo, administrarse por vía oral, parenteral, es decir, intravenosa, intramuscular o subcutánea, intratecal, transdérmica (parche), por inhalación o por vía intranasal.
Las composiciones farmacéuticas adecuadas para la administración oral pueden ser sólidas o líquidas y pueden, por ejemplo, estar en forma de comprimidos, píldoras, grageas, cápsulas de gelatina, soluciones, jarabes, chicles y similares.
Con este fin, el ingrediente activo se puede mezclar con un diluyente inerte o un vehículo no tóxico farmacéuticamente aceptable, tal como el almidón o la lactosa. Opcionalmente, estas composiciones farmacéuticas también pueden contener un aglutinante, tal como la celulosa microcristalina, la goma de tragacanto o la gelatina, un disgregante, tal como el ácido algínico, un lubricante como el estearato de magnesio, un deslizante tal como el dióxido de silicio coloidal, un edulcorante como la sacarosa o la sacarina, o colorantes o saborizantes, tales como la menta piperita o el salicilato de metilo.
La invención también contempla las composiciones que pueden liberar la sustancia activa de una forma controlada.
Las composiciones farmacéuticas que se pueden usar para la administración parenteral están en forma convencional, tales como soluciones o suspensiones acuosas u oleosas, por lo general contenidas en ampollas, jeringuillas desechables, viales de vidrio o plástico, o recipientes para infusión.
Además del ingrediente activo, estas soluciones o suspensiones pueden contener opcionalmente también un diluyente estéril, tal como agua para inyección, una solución salina fisiológica, aceites, polietilenglicoles, glicerina, propilenglicol u otros solventes sintéticos, antibacterianos tales como el alcohol bencílico, antioxidantes tales como el ácido ascórbico o el bisulfito de sodio, quelantes como el ácido etilendiaminotetraacético, tamponantes tales como acetatos, citratos o fosfatos, y agentes para ajustar la osmolaridad, tales como el cloruro de sodio o la dextrosa.
Estas formas farmacéuticas se preparan con los métodos que los farmacéuticos utilizan habitualmente.
La cantidad de ingrediente activo en las composiciones farmacéuticas puede caer dentro de un amplio margen de concentraciones y depende de numerosos factores, tales como el sexo, la edad, el peso y el estado médico del paciente, así como del método de administración. Por tanto, la cantidad del compuesto de la fórmula (I) en las composiciones para la administración oral es de al menos el 0,5 % en peso y puede llegar al 80 % en peso con respecto al peso total de la composición.
De acuerdo con la invención, también se ha encontrado que los compuestos de la fórmula (I) o las sales farmacéuticamente aceptables de los mismos pueden administrarse solos o en combinación con otros ingredientes farmacéuticamente activos. Los ejemplos no limitantes de tales compuestos adicionales que pueden citarse para su uso en combinación con los compuestos de acuerdo con la invención son antivíricos, antiespásticos (por ejemplo, baclofeno), antieméticos, agentes estabilizadores del estado de ánimo, antimaníacos, analgésicos (por ejemplo, aspirina, ibuprofeno, paracetamol), analgésicos narcóticos, anestésicos tópicos, analgésicos opioides, sales de litio, antidepresivos (p. ej., mianserina, fluoxetina, trazodona), antidepresivos tricíclicos (p. ej., imipramina, desipramina), anticonvulsivos (p. ej., ácido valproico, carbamazepina, fenitoína), antipsicóticos (p. ej., risperidona, haloperidol), neurolépticos, benzodiazepinas (por ejemplo, diazepam, clonazepam), fenotiazinas (por ejemplo, clorpromazina), bloqueadores de los canales de calcio, anfetamina, clonidina, lidocaína, mexiletina, capsaicina, cafeína, quetiapina, antagonistas de la serotonina, betabloqueantes, antiarrítmicos, triptanos, derivados del alcaloide del cornezuelo y amantadina.
Para las composiciones orales, la dosis diaria está en el intervalo de 1 mg a 2000 mg de los compuestos de la fórmula (I). Preferiblemente en el intervalo de 1 mg a 1000 mg de los compuestos de la fórmula (I), lo más preferiblemente de 1 mg a 500 mg.
En las composiciones para la administración parenteral, la cantidad del compuesto de la fórmula (I) presente es de al menos el 0,5 % en peso y puede llegar al 33 % en peso con respecto al peso total de la composición. Para las composiciones parenterales preferidas, la unidad de dosificación está en el intervalo de 1 mg a 2000 mg de los compuestos de la fórmula (I).
La dosis diaria puede caer dentro de un amplio margen de unidades de dosificación del compuesto de la fórmula (I) y generalmente está en el margen de 1 a 2000 mg, preferiblemente de 1 a 1000 mg. Sin embargo, debe entenderse que las dosis específicas se pueden adaptar a casos particulares en función de los requisitos individuales, a criterio del médico.
Los compuestos de fijación a las proteínas SV2 que dan a conocer en esta invención y los derivados marcados de los mismos pueden ser útiles como patrones y reactivos a la hora de determinar la capacidad que tienen los compuestos analizados (por ejemplo, un producto farmacéutico potencial) para unirse a las proteínas SV2.
Los derivados marcados de los ligandos de las proteínas SV2 que dan a conocer en esta invención también pueden ser útiles como radiotrazadores para la formación de imágenes por tomografía por emisión de positrones (PET) o para la tomografía computarizada de emisión monofotónica (SPECT).
Por lo tanto, la presente invención da a conocer además ligandos marcados como herramientas para cribar quimiotecas para el descubrimiento de posibles sustancias farmacéuticas, en concreto, para el tratamiento y la prevención de las afecciones presentadas en la presente memoria, sobre la base de una fijación más potente a las proteínas SV2, para localizar las proteínas SV2 en los tejidos y para caracterizar las proteínas SV2 purificadas. Las proteínas SV2 incluyen SV2A, SV2B y SV2C, por lo que SV2A es el sitio de unión para el fármaco anticonvulsivo levetiracetam y sus análogos. Las isoformas de SV2, SV2A, SV2B o SV2C, pueden obtenerse de tejidos, especialmente del cerebro, de cualquier especie de mamífero, incluidos los humanos, las ratas o los ratones. Como alternativa, las isoformas pueden ser versiones clonadas de cualquier especie de mamífero, incluidos los humanos, las ratas y los ratones, expresadas heterólogamente y utilizadas para los ensayos. El método de cribado comprende exponer las membranas cerebrales, tales como las membranas cerebrales de mamíferos o humanos, o las líneas celulares que expresan las proteínas SV2 o fragmentos de las mismas, sobre todo SV2A y SV2C, pero que incluyen SV2B, a un posible agente e incubar las membranas o las proteínas o los fragmentos y el agente con un compuesto marcado de la fórmula (I). El método comprende además determinar si la unión del compuesto de la fórmula (I) a la proteína está inhibida por el posible agente, con lo que así se identifican los compañeros de unión para la proteína. Por lo tanto, los ensayos de cribado permiten la identificación de nuevos fármacos o compuestos que interactúan con las proteínas SV2. La presente invención también da a conocer los ligandos fotoactivables de las proteínas SV2.
Los ligandos marcados también se pueden usar como herramientas para valorar el estado conformacional de las proteínas SV2 después de la solubilización, purificación y cromatografía. Los ligandos marcados pueden estar marcados directa o indirectamente. Los ejemplos de las etiquetas adecuadas incluyen una radiomarcación, tal como 3H, un marcador fluorescente, una enzima, europio, biotina y otros marcadores convencionales para los ensayos de este tipo.
Los compuestos marcados de la fórmula (I) son útiles en los métodos como sondas en los ensayos para identificar nuevos compuestos o agentes que se unen a las proteínas SV2 (SV2A, SV2B y SV2C). En tales realizaciones del ensayo, los ligandos pueden usarse sin modificación o pueden modificarse de diversas formas; por ejemplo, mediante marcación, tal como la unión covalente o no covalente a un resto que proporciona directa o indirectamente una señal detectable. En cualquiera de estos ensayos, los materiales pueden estar marcados directa o indirectamente. Las posibilidades de etiquetado directo incluyen grupos de etiquetas tales como: radiomarcadores que incluyen, entre otros, [3H], [14C], [32P], [35S] o [125I], enzimas tales como peroxidasa y fosfatasa alcalina, y marcadores fluorescentes capaces de monitorizar el cambio en la intensidad de la fluorescencia, el desplazamiento de la longitud de onda o la polarización de la fluorescencia, que incluyen, entre otros, fluoresceína o rodamina. Las posibilidades de marcación indirecta incluyen la biotinilación de un constituyente seguida de la unión a la avidina acoplada a uno de los grupos marcadores anteriores, o el uso de anticuerpos contra el ligando. Los compuestos también pueden incluir espaciadores o conectores en los casos en que los compuestos deban unirse a un soporte sólido. Para identificar los agentes o compuestos que compiten o interaccionan con los ligandos marcados de acuerdo con la invención por unirse a las proteínas SV2 (sobre todo SV2A y SV2C), se pueden usar células intactas, fragmentos celulares o de membrana que contienen SV2A o SV2C, o la proteína SV2 completa o un fragmento de la misma. El agente o compuesto se puede incubar con las células, membranas, proteína SV2, o fragmentos, antes, al mismo tiempo o después de la incubación con levetiracetam marcado o un análogo o derivado del mismo. Los ensayos pueden modificarse o prepararse en cualquier formato disponible, incluidos los ensayos de cribado de alto rendimiento (HTS, por su nombre en inglés) que controlan la unión del levetiracetam o la unión de derivados o análogos del mismo a las proteínas SV2 o a los fragmentos de las mismas. En muchos programas de selección de fármacos que analizan colecciones de compuestos, son deseables los ensayos de alto rendimiento para elevar al máximo el número de compuestos examinados en un período de tiempo dado. Dichos ensayos de cribado pueden usar células intactas, fragmentos celulares o de membrana que contienen SV2, así como sistemas acelulares o sin membrana, como los que se pueden derivarse con proteínas purificadas o semipurificadas. La ventaja del ensayo con fragmentos de membrana que contienen SV2 o proteínas y péptidos de SV2 purificados es que los efectos de la intoxicación celular y/o la biodisponibilidad del compuesto problema suelen poder ignorarse, para que el ensayo se pueda centrar principalmente en el efecto del fármaco en la diana molecular como puede ponerse de manifiesto en una inhibición de, por ejemplo, la unión entre dos moléculas. El ensayo puede formularse para detectar la capacidad que tiene un agente o compuesto problema para inhibir la unión del ligando marcado según la invención a SV2 o a un fragmento de SV2 o del levetiracetam marcado, o derivados o análogos del mismo, a SV2 o a un fragmento de la proteína SV2. La inhibición de la formación de los complejos puede detectarse mediante una serie de técnicas diferentes, tales como los ensayos de filtración, FlashPlate® (Perkin Elmer), ensayos de centelleo por proximidad (SPA, GE). Para los cribados de alto rendimiento (HTS), el ensayo de centelleo por proximidad que utiliza microesferas recubiertas con membranas biológicas o FlashPlate® recubiertas con membranas biológicas son métodos poderosos que no requieren etapas de separación ni de lavado.
Un problema que se puede afrontar al desarrollar compuestos para su uso en algún tratamiento es la capacidad que tienen ciertos compuestos (fármacos desencadenantes), que podrían coadministrarse junto con los compuestos de la presente invención (fármacos víctimas), para inducir las enzimas de tipo CYP450, en concreto el CYP3A4/5. La inducción de tales enzimas por los fármacos desencadenantes podría afectar la exposición del fármaco víctima, cuando es metabolizado principalmente por las enzimas de tipo CYP450 y el CYP3A4/5 en concreto, con lo que se podría alterar su perfil de eficacia. Por lo tanto, es deseable desarrollar compuestos que tangan pocas probabilidades de ser metabolizados por las enzimas de tipo CYP3A4/5.
La contribución del CYP3A4/5 al metabolismo total de los compuestos de acuerdo con la presente invención se ha evaluado al calcular la relación entre la eliminación de hepatocitos humanos en ausencia y presencia de un inhibidor selectivo del CYP3A4/5, tal como la azamulina.
Cuando se comprueban en este ensayo de acuerdo con el protocolo descrito en la presente solicitud de patente, una fracción de los compuestos de acuerdo con la presente invención acaban metabolizados por el CYP3A4/5 (Fm.CYP3A4/5), normalmente inferior al 40 %, lo que disminuye al mínimo el riesgo de interacciones farmacológicas cuando se coadministra con los inductores de los CYP450.
Apartado experimental
Abreviaturas/reactivos recurrentes
Ac: acetilo
ACN: acetonitrilo
Salmuera: solución acuosa saturada de cloruro de sodio
nBu: n-butilo
tBu: tert-butilo
Bz: benzoílo
VC: volúmenes de columna
DCM: diclorometano
DMF: W,W-Dimetilformamida
DMSO: sulfóxido de dimetilo
Et: etilo
EtOH: etanol
Et2O: éter dietílico
EtOAc: acetato de etilo
h: Hora
HPLC: cromatografía líquida de alta presión
LC: cromatografía líquida
LCMS: espectrometría de masas (MS) acoplada a cromatografía líquida (LC)
MeOH: metanol
min: minutos
RMN: resonancia magnética nuclear
iPrOH: isopropanol
PTSA: ácido p-toluensulfónico
TA: temperatura ambiente
SFC: Cromatografía de fluidos supercríticos
THF: Tetrahidrofurano
TLC: Cromatografía en capa fina
En la presente solicitud, se emplea el nombre de la IUPAC (o el nombre generado a partir de Accelerys Draw 4.0) de los compuestos.
Métodos analíticos
Todas las reacciones en las que intervienen reactivos sensibles a la humedad o al aire se realizaron en una atmósfera de nitrógeno o argón con el uso de solventes y material de vidrio secos. Los experimentos que requieren la irradiación de microondas se realizan en un horno microondas Biotage Initiator Sixty actualizado con la versión 2.0 del programa operativo. Los experimentos se llevan a cabo de manera que se alcance la temperatura necesaria lo más rápido posible (potencia máxima de irradiación: 400 W, sin refrigeración externa). Los solventes y reactivos comerciales se utilizaron generalmente sin purificación adicional, incluidos los solventes anhidros cuando sea apropiado (generalmente productos Sure-Seal™ de Aldrich Chemical Company, o AcroSeal™ de ACROS Organics). En general, tras las reacciones se realizaron análisis por cromatografía en capa fina, HPLC o espectrometría de masas.
Los análisis por HPLC se realizan con un sistema de HPLC de la serie 1100 de Agilent montado con una columna XBridge MS C18, 5 pm, 150 x 4,6 mm de Waters. El gradiente va desde el 100 % del solvente A (agua/ACN/solución de formiato de amonio 85/5/10 (v/v/v)) hasta el 100 % del solvente B (agua/ACN/solución de formiato de amonio 5/85/10 (v/v/v) durante 6 min, con retención de 5 minutos cuando B está al 100 % . El flujo del caudal se establece en 8 ml/min durante 6 min, luego se incrementa a 3 ml/min durante 2 min con una retención a los 3 ml/min durante 3 minutos. Se utiliza una fracción de 1/25 justo antes de la fuente de API. La cromatografía se lleva a cabo a 45°C. La solución de formiato de amonio (pH de aproximadamente 8,5) se prepara mediante la disolución del formiato de amonio (630 mg) en agua (1 l) y la adición del hidróxido de amonio al 30 % (500 gl).
Resultará evidente para el experto en la técnica que pueden obtenerse diferentes tiempos de retención para los datos de LC si se utilizan diferentes condiciones analíticas.
Las mediciones de espectrometría de masas en modo LCMS se realizan de la siguiente manera:
- Para la elución básica, los análisis se realizan con:
Para el análisis por LCMS se utiliza un espectrómetro de masas con cuadrupolo simple QDA de Waters. Este espectrómetro está equipado con una fuente de ESI y una UPLC Acquity Hclass con un detector de matriz de diodos (200 a 400 nm). Los datos se adquieren en un barrido completo de la MS desde m/z 70 a 800 en modo positivo con una elución básica. La separación de la fase inversa se lleva a cabo a 45°C en una columna Acquity UPLC BEHC18 de 1,7 gm (2,1 x 50 mm) de Waters para la elución básica. El gradiente de elución se realiza con agua/ACN/formiato de amonio (95/5/63 mg/l) (solvente A) y ACN/agua/formiato de amonio (95/5/63 mg/l) (solvente B). Volumen de inyección: 1 gl. Flujo total en MS.
Programa básico "4 m in"

Programa básico "10 m in"

- Para la elución ácida, se realizan los análisis con:
Para el análisis por LCMS se utiliza un espectrómetro de masas cuadrupolo simple QDA de Waters. Este espectrómetro está equipado con una fuente de ESI y una UPLC Acquity Hclass con un detector de matriz de diodos (200 a 400 nm). Los datos se adquieren en un barrido completo de la MS desde m/z 70 a 800 en modo positivo con una elución ácida. La separación de fase inversa se lleva a cabo a 45°C en una columna Acquity UPLC HSS T3 de 1,8 pm (2,1 x 50 mm) de Waters para la elución ácida. El gradiente de elución se realiza con agua/ACN/TFA (95/5/0,5 ml/l) (solvente A) y ACN (solvente B). Volumen de inyección: 1 pl. Flujo total en MS.
Programa ácido "4 m in"

Programa ácido "10 min "

Los materiales brutos se pudieron purificar mediante cromatografía en fase normal, cromatografía en fase inversa (ácida o básica), separación quiral o retrocristalización.
La cromatografía de fase inversa normal se realiza con columnas de gel de sílice (gel de sílice de malla 100:200 o columnas Puriflash®-50SIHC-JP de Interchim).
La cromatografía preparativa en fase inversa se realiza de la siguiente manera:
• La purificación por LCMS (modo básico, LCMS preparativa) con un espectrómetro de masas de triple cuadrupolo SQD o QM de Waters se usa para la purificación por LCMS. Este espectrómetro está equipado con una fuente de ESI y una bomba cuaternaria de Waters con controlador de LC preparativa con un detector de matriz de diodos (210 a 400 nm).
Parámetros de la MS: Voltaje capilar de la ESI 3 kV. Voltaje del cono y del extractor 10. Temperatura del bloque de la fuente 120°C. Temperatura de desolvatación 300°C. Flujo de gas del cono 30 l/h (nitrógeno), flujo del gas de desolvatación 650 l/h. Los datos se adquieren en un barrido completo de la MS desde m/z 100 a 700 en modo positivo con una elución ácida o básica.
Parámetros de la LC: La separación en fase inversa se lleva a cabo a TA en una columna XBridge prep OBD C18 (5 pm, 30 x 50 mm) (elución básica). La elución en gradiente se realiza con agua (solvente A), ACN (solvente B), bicarbonato de amonio en agua 8 g/l 500 pl/l de NH4OH al 30 % (solvente C) (pH 8,5). Velocidad de flujo de la HPLC: 35 ml/min a 60 ml/min, volumen de la inyección: 1 ml. La relación de la división se ajusta a ±1/6000 para la MS.


Las separaciones cromatográficas quirales preparativas se realizan con el uso de instrumentos de cromatografía en fase líquida o cromatografía de fluidos supercríticos (SFC) con diversas mezclas de alcoholes inferiores y alcanos lineales, ramificados o cíclicos de C5 a C8 a 360 ml/min. Las mezclas de los solventes y las columnas se describen en cada uno de los procedimientos. Los productos se suelen secar al vacío antes de los análisis finales y se someten a pruebas biológicas.
Los espectros de la RMN se registran en un espectrómetro de RMN AVANCEIII 400 MHz-Ultrashield de BRUKER equipado con una estación de trabajo de Windows 7 Professional en la que se ejecuta el programa informático Topspin 3.2 y una sonda de banda ancha de doble resonancia de 5 mm (PABBI 1H/19F-BB Z-g Rd Z82021/0075) o una sonda de resonancia triple de 1 mm (PATXI 1H/D-13C/15N Z-GRD Z868301/004). Los compuestos se estudiaron en una solución de DMSO-cfe, o CDCh a una temperatura de la sonda de 300 K y a una concentración de 10 mg/ml. El instrumento está bloqueado en la señal de deuterio de DMSO-afe o CDCl3. Los desplazamientos químicos se dan en ppm campo abajo del TMS (tetrametilsilano) tomado como patrón interno.
Las rotaciones ópticas ([a]D) se midieron en un polarímetro 341 de PERKIN-ELMER en una cubeta (/ = 1 dm) a una concentración de 10 mg/ml, a una temperatura mencionada en los ejemplos específicos, a 589 nm (lámpara de sodio).
Los siguientes ejemplos ilustran cómo se pueden sintetizar los compuestos cubiertos por la fórmula (I). Se dan a conocer únicamente con fines ilustrativos y no pretenden, ni deben interpretarse, como limitantes de la invención de ninguna manera.
Ejemplo 1. Síntesis de la (+)-3-[[2-(metox¡met¡l)-6-(tr¡fluoromet¡l)¡m¡dazo[2,1-b][1,3,4]t¡ad¡azol-5-¡l]met¡l]-5-propiloxazolidín-2-ona 1 y de la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-prop¡loxazol¡dín-2-ona 2

1.1 Síntes¡s del 1-n¡tropentán-2-ol I
Una mezcla de butiraldehído (CAS: 123-72-8, 1 eq., 5,0 g, 69,3 mmol) y de carbonato de potasio (1 eq., 9,68 g, 69,3 mmol) en nitrometano (95 ml) se agitó a 50°C durante 5 h. La mezcla se enfrió a temperatura ambiente y se le añadió acetato de etilo (100 ml). La capa orgánica se lavó con agua (dos veces), se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad para dar un aceite marrón. La mezcla bruta se purificó por cromatografía ultrarrápida Biotage Isolera Four (columna de gel de sílice KP-SNAP de 100 g con diclorometano al 100 % durante 12 VC) y las fracciones puras se evaporaron hasta la sequedad para dar el 1-nitropentán-2-ol I (4,7 g, 35,0 mmol) como un aceite incoloro.
Rendimiento: 51 %
RMN 1H (400 MHz, DMSO-cfe): 55,26 (d, J = 6,2 Hz, 1 H), 4,64 (dd, J = 12,2, 3,2 Hz, 1 H), 4,31 (dd, J = 12,1,9,3 Hz, 1 H), 4,09 (ddt, J = 8,9, 5,9, 3,2 Hz, 1 H), 1,47-1,23 (m, 4 H), 0,95-0,77 (m, 3 H).
1.2 Síntes¡s del 1-am¡nopentán-2-ol II
Una solución de 1-nitropentán-2-ol I (1 eq., 200 mg, 1,5 mmol) en etanol (8 ml) se hidrogenó en un cartucho de Pd/C
al 10 %, a 50°C, con presión de H2 (50 bar) en un reactor H-Cube durante 1 h (flujo: 1 ml/min, 7,5 ciclos). La mezcla bruta se evaporó hasta la sequedad y se secó a alto vacío para dar el 1-aminopentán-2-ol II (140 mg, 1,22 mmol) como un aceite incoloro. El compuesto bruto obtenido se usó directamente en el siguiente paso sin ninguna purificación adicional.
Rendimiento: 81 %
RMN 1H (400 MHz, DMSO-afe) 53,37-3,25 (m, 1 H), 2,52 (d, J = 4,1 Hz, 1 H), 2,37 (dd, J = 12,6, 7,3 Hz, 1 H), 1,45 1,20 (m, 4 H), 0,93-0,79 (m, 3 H).
1.3 Síntesis de la 5-propiloxazolidín-2-ona III
A una solución de 1-aminopentán-2-ol II (1 eq., 1,0 g, 9,2 mmol) y 1,1'-carbonildiimidazol (1 eq., 1,6 g, 9,7 mmol) en THF (20 ml) se le añadió hidruro de sodio (0,1 eq., 23 mg, 0,92 mmol) y la mezcla se agitó a 25°C durante 24 h. Se añadieron acetato de etilo y agua a la mezcla de reacción y la capa acuosa se extrajo con acetato de etilo (dos veces), se lavó con agua y salmuera, se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad para dar un aceite amarillo. El compuesto bruto se purificó mediante cromatografía ultrarrápida Biotage Isolera Four (50 g de columna de gel de sílice KP-SNAP en un gradiente del 0 % al 10 % de metanol en diclorometano a lo largo de 10 VC) y las fracciones puras se evaporaron hasta la sequedad para dar la 5-propiloxazolidín-2-ona III (990 mg, 7,59 mmol) como un sólido blanco.
Rendimiento: 82 %
LC/MS:[M H]+ = 130,1
RMN 1H (400 MHz, CDCla): 55,98 (s, 1 H), 4,73-4,55 (m, 1 H), 3,65 (td, J = 8,4, 0,8 Hz, 1 H), 3,22 (ddd, J = 8,2, 7,2, 0,9 Hz, 1 H), 1,87-1,70 (m, 1 H), 1,69-1,32 (m, 3 H), 0,96 (t, J = 7,3 Hz, 3 H).
1.4 Síntesis del [2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol V
En un tubo sellado, se mezclaron 2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol IV (CAS: 1294001 -16 3, solicitud de patente internacional WO 2012/143117, 1 eq., 2,0 g, 8,4 mmol), paraformaldehído (6,0 eq., 1,52 g, 50,6 mmol) y una solución acuosa de ácido clorhídrico (2 N) (1,0 equiv., 4,2 ml, 8,4 mmol) en 1,4-dioxano (4 ml). La mezcla se agitó a 100°C durante 18 h y la reacción se comprobó por LC/MS. La mezcla bruta se enfrió a TA y se le añadió una solución acuosa saturada de NaHCO3 hasta que el pH = 6-7. La capa acuosa se extrajo con acetato de etilo (tres veces) y la combinación de las capas orgánicas se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad. El compuesto bruto se purificó por cromatografía ultrarrápida Biotage Isolera Four (columna de gel de sílice KP-SNAP de 100 g en un gradiente de metanol del 0 % al 10 % en diclorometano a lo largo de 15 VC) para dar el [2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol (1,06 g, 3,5 mmol) V como un sólido beis. El compuesto se utilizó directamente en el siguiente paso sin más purificación.
Rendimiento: 41 %
LC/MS: [M H]+ = 268,2
RMN 1H (400 MHz, CDCla): 55,04 (d, J = 5,2 Hz, 2 H), 4,77 (s, 2 H), 3,53 (s, 3 H), 2,32 (t, J = 6,3 Hz, 1 H).
1.5 Síntesis de la (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 1 y la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2 ona 2
A una mezcla de [2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol V (1 eq., 200 mg, 0,74 mmol) y 5-propiloxazolidín-2-ona III (2 eq., 190 mg, 1,48 mmol) en sulfolano (3 ml), se le añadió monohidrato de ácido p-toluensulfónico (1 eq., 152 mg, 0,74 mmol) y la mezcla se agitó a 110°C durante 16 h. La mezcla bruta se purificó directamente mediante HPLC preparativa en fase inversa (condiciones básicas) para dar un sólido beis (155 mg del racemato) que se purificó mediante SFC quiral (fase WhelkO1 (R, R), 50 x 227 mm / Cosolvente de CO2/EtOH al 20% / 30°C / 360 ml/min). Las fracciones más puras se evaporaron hasta la sequedad para dar la (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 1 (primer eluido, 56 mg, 0,15 mmol) y la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 2 (segundo eluido, 56 mg, 0,15 mmol) como aceites incoloros.
Rendimientos: 20 %
LC/MS: [M H]+ = 379,4
RMN 1H (400 MHz, DMSO-afe): 54,84 (s, 2 H), 4,74 (qd, J = 15,8, 1,2 Hz, 2 H), 4,44 (dtd, J = 8,3, 7,0, 5,4 Hz, 1 H), 3,54 (t, J = 8,3 Hz, 1 H), 3,43 (s, 3 H), 3,08 (dd, J = 8,4, 6,9 Hz, 1 H), 1,62-1,45 (m, 2 H), 1,28 (dddd, J = 16,1, 13,6, 8,4, 4,0 Hz, 1 H), 0,85 (t, J = 7,4 Hz, 3 H).
Alfa-D (1, MeOH, 10 mg/ml, 26,5°C) = 27,0
Alfa-D (2, MeOH, 10 mg/ml, 26,5°C) = -26,2
Ejemplo 2. (+)-3-[[6-(Difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 3 y (-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 4

1.5 Síntesis del 6-(difluorometil)-2-(metoximetil)imidazo[2,1b][1,3,4]tiadiazol VII
A una solución de 5-(metoximetil)-1,3,4-tiadiazol-2-amina VI (CAS: 15884-86-3, 1,0 eq., 6,5 g, 45 mmol) en DMF (100 ml), a 100°C, se le agregó gota a gota una solución de 3-bromo-1,1-difluoro-propán-2-ona (CAS: 883233-85-0, 1,05 eq., 8,1 g, 47 mmol) en DMF (5 ml). La mezcla de reacción se calentó a 100°C durante 3 h y se comprobó que se había completado por LC/MS. Se le añadió una solución acuosa saturada de NaHCO3 y la capa orgánica se extrajo con acetato de etilo (tres veces). La combinación de las capas orgánicas se lavó con agua (cinco veces), se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad para dar un sólido marrón (7,6 g). El compuesto bruto se purificó por cromatografía ultrarrápida Biotage Isolera Four (columna de gel de sílice KP-SNAP de 100 g en un gradiente del 0 % al 5 % de metanol en diclorometano a lo largo de 12 VC) y las fracciones puras se evaporaron a alto vacío para dar el 6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol VII (3,95 g, 17,8 mmol) como un sólido naranja.
Rendimiento: 40 %
LC/MS: [M H]+ = 220,2
RMN 1H (400 MHz, DMSO-afe): 58,53 (t, J = 2,2 Hz, 1 H), 7,01 (t, J = 54,6 Hz, 1 H), 4,83 (s, 2 H), 3,43 (s, 3 H).
1.6 Síntesis del [6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol VIII
En un tubo sellado, se mezclaron 6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol VII (1,0 eq., 3,95 g, 18,0 mmol), paraformaldehído (6,0 eq., 3,24 g, 108 mmol) y una solución acuosa de ácido clorhídrico (2 N) (0,9 equiv., 8,1 ml, 16,2 mmol) en 1,4-dioxano (8 ml). La mezcla se agitó a 100°C durante 3,5 h y la reacción se comprobó mediante LC/MS. La mezcla bruta se enfrió a TA y se le añadió una solución acuosa saturada de NaHCO3 hasta que el pH = 6 7. La capa acuosa se extrajo con acetato de etilo (tres veces) y la combinación de las capas orgánicas se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad. El compuesto bruto se purificó mediante cromatografía ultrarrápida Biotage Isolera Four (columna de gel de sílice KP-SNAP de 100 g en un gradiente de metanol del 0 % al 10 % en diclorometano a lo largo de 15 VC) para dar un aceite amarillo (3 g) que se purificó por segunda vez por HPLC en fase inversa (KROMASIL-Eternity XT C1810 gm / gradiente de ACN/H2O/NH4OH de 20/80/0,1 a 50/50/0,1). Las fracciones más puras se evaporaron hasta la sequedad para dar el [6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol VIII (2 g, 8,02 mmol) como un sólido blanco.
Rendimiento: 45 %
LC/MS: [M H]+ = 250,2
RMN 1H (400 MHz, DMSO-afe): 57,11 (t, J = 53,6 Hz, 1 H), 5,47 (t, J = 5,4 Hz, 1 H), 4,84 (s, 2 H), 4,79 (d, J = 5,5, Hz, 2 H), 3,44 (d, J = 0,9 Hz, 3 H).
1.7. Síntesis de la (+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 3 y la (-)--3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2 -ona 4
A una mezcla de [6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol VIII (1 eq., 200 mg, 0,8 mmol) y 5-propiloxazolidín-2-ona III (2 eq., 206 mg, 1,6 mmol) en sulfolano (3 ml), se le añadió monohidrato de ácido p-toluensulfónico (1 eq., 152 mg, 0,8 mmol) y la mezcla se agitó a 110°C durante 3,5 h. La mezcla bruta se purificó directamente mediante HPLC preparativa en fase inversa (condiciones básicas) para dar un sólido beis (216 mg del racemato) que se purificó mediante SFC quiral (fase WhelkO1 (R, R), 50 x 227 mm / Cosolvente de CO2/EtOH al 20 %
/ 30°C / 360 ml/min). Las fracciones más puras se evaporaron hasta la sequedad para dar la (+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 3 (primer eluido, 54 mg, 0,15 mmol, 19 % de rendimiento) y la (-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 4 (segundo eluido, 52 mg, 0,14 mmol, rendimiento del 18 %) como aceites incoloros.
Rendimientos: 19 % (3) y 18 % (4)
LC/MS: [M H]+ = 361,4
RMN 1H (400 MHz, DMSO-afe): 57,12 (t, J = 53,5 Hz, 1 H), 4,83 (d, J = 0,9 Hz, 2 H), 4,81-4,66 (m, 2 H), 4,46 (dtd, J = 8,4, 7,0, 5,5 Hz, 1 H), 3,56 (t, J = 8,4 Hz, 1 H), 3,44 (s, 3 H), 3,09 (dd, J = 8,4, 7,0 Hz, 1 H), 1,65-1,42 (m, 2 H), 1,39 1,17 (m, 2H), 0,86 (t, J = 7,3 Hz, 3H).
Alfa-D (3, MeOH, 10 mg/ml, 27,7 ° C) = 17.2
Ejemplo 3. Síntesis de la (+)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 5 y la (-)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 6

3.1 Síntesis del [2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol X
Una solución de 2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol IX (solicitud de patente internacional WO2011047860, 1,0 eq., 1,25 g, 5,24 mmol), paraformaldehído (6,0 equiv., 943 mg, 31,45 mmol) y ácido clorhídrico (1,0 eq., 2,6 ml, 5,24 mmol) en 1,4-dioxano (2,6 ml) se agitó a 100°C durante 18 h. Se le añadió una solución acuosa saturada de NaHCO3 y la capa acuosa se extrajo con acetato de etilo (3 veces). La combinación de las capas orgánicas se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad para dar un aceite amarillo. El compuesto bruto obtenido se purificó mediante cromatografía ultrarrápida Biotage Isolera Four (50 g de columna de gel de sílice KP-SNAP desde diclorometano al 100 % a metanol al 10 % en diclorometano a lo largo de 15 VC) para dar un sólido marrón que se purificó mediante cromatografía de fase inversa en modo básico. La fracción más pura se evaporó hasta la sequedad para dar el [2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol X (475 mg, 1,73 mmol) como un sólido blanco.
Rendimiento: 33 %
LC/MS: [M H]+ = 270,01
RMN 1H (400 MHz, DMSO-afe): 55,56 (s, 1 H), 4,77 (s, 2 H), 3,44 (s, 3 H).
3.2 Síntesis de la (+)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 5 y la (-)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 6
A una mezcla de [2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol X (1,0 eq., 200 mg, 0,74 mmol) y 5-propiloxazolidín-2-ona III (2,0 eq., 190 mg, 1,47 mmol) en sulfolano (0,2 M, 3,5 ml), se le añadió monohidrato de ácido p-toluensulfónico (1,0 eq., 140 mg, 0,73 mmol) y la mezcla se agitó a 110°C durante 16 h. El compuesto bruto se purificó directamente mediante cromatografía en fase inversa en condiciones básicas para dar un aceite amarillo (172 mg). La mezcla de enantiómeros se purificó mediante SFC quiral (W (3 pm) * EtOH al 50 %-heptano al 50 %-DEA al 0,1 %) para dar la (+)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 5 (75 mg, 0,20 mmol) y la (-)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 6 (72 mg, 0,19 mmol) como aceites incoloros.
Rendimiento global: 52 % (26,5 % 25,5 %)
LC/MS: [M H]+ = 381,11
RMN 1H (400 MHz, CDCla): 55,00-4,75 (m, 2 H), 4,51-4,40 (m, 1 H), 3,51 (s, 4 H), 3,04 (dd, J = 8,4, 7,4 Hz, 1 H), 1,76 1,66 (m, 1 H), 1,60-1,50 (m, 1 H), 1,48-1,29 (m, 2 H), 0,92 (t, J = 7,3 Hz, 3 H).
Alfa-D (5, MeOH, 10 mg/ml, 25°C) = 17,6
Ejemplo 4. Síntesis de la (+)-3-[[2-[dideuterio(trideuteriometoxi)metil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 7 y la (-)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 8

4.1 Síntesis del -[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol XII
A una solución de dideuterio-[6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-2-il]metanol XI (solicitud de patente internacional WO2011047860, 1,0 eq., 3,25 g, 13,0 mmol) en metanol (100 ml), a temperatura ambiente, se le añadió óxido de plata (1,3 eq., 3,9 g, 16,9 mmol) y yodometano-c/3 (4,0 eq., 3,23 ml, 52,0 mmol). La mezcla de reacción se agitó a 40°C durante 20 h. Después, la mezcla se filtró a través de un lecho filtrante de Celite y se evaporó hasta la sequedad. Se le añadió una solución acuosa saturada de NaHCO3 y la capa acuosa se extrajo con acetato de etilo (3 veces). La combinación de las capas orgánicas se secó sobre MgSO4 , se filtró y se evaporó hasta la sequedad para dar un aceite amarillo que se purificó por cromatografía ultrarrápida Biotage Isolera Four (100 g de columna de gel de sílice KP-SNAP desde diclorometano al 100 % a metanol al 5 % en diclorometano a lo largo de 15 VC) para dar el 2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol XII (2,11 g, 7,4 mmol) como un sólido blanco.
Rendimiento: 57 %
LC/MS: [M H]+ = 243,08
RMN 1H (400 MHz, DMSO-afe): 58,90 (s, 1 H).
4.2 Síntesis del [2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol XIII
Una solución de 2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol XII (1,0 eq., 1,0 g, 4,1 mmol), paraformaldehído (6,0 eq., 743 mg, 24,7 mmol) y ácido clorhídrico (1,0 eq., 4,1 mmol, 2,0 ml) en 1,4-dioxano (2 ml) se agitó a 100°C durante 18 h. Se le añadió una solución acuosa saturada de NaHCO3 y la capa acuosa se extrajo con acetato de etilo (3 veces). La combinación de las capas orgánicas se lavó con salmuera, se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad para dar un aceite amarillo que se purificó mediante cromatografía en fase inversa en modo básico. La fracción más pura se evaporó hasta la sequedad para dar el [2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol XIII (560 mg, 1,97 mmol) como un sólido blanco.
Rendimiento: 48 %
LC/MS: [M H]+ = 270,01
RMN 1H (400 MHz, DMSO-afe): 55,30-6,30 (s ancho, 1 H) 4,77 (d, J = 1,3 Hz, 2 H).
4.3 Síntesis de la (+)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 7 y la (-)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 8
A una mezcla de [2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol XIII (1,0 eq., 200 mg, 0,73 mmol) y 5-propiloxazolidín-2-ona III (2,0 eq., 190 mg, 1,47 mmol) en sulfolano
(0,2 M, 3,5 ml), se le añadió monohidrato de ácido p-toluensulfónico (1,0 eq., 101 mg, 0,53 mmol) y la mezcla se agitó a 110°C durante 16 h. El bruto se purificó mediante cromatografía en fase inversa en condiciones básicas para dar un aceite amarillo (175 mg). La mezcla de enantiómeros se purificó mediante SFC quiral (W (3 gm) * EtOH al 50 %-heptano al 50 %-DEA al 0,1 %) para dar la (+)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 7 (47 mg, 0,12 mmol) y la (-)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona 8 (51 mg, 0,13 mmol) como aceites transparentes.
Rendimiento global: 35 % (17 % 18 %)
LC/MS: [M H]+ = 384,14
RMN 1H (400 MHz, CDCla): 54,95 (dd, J = 15,7, 1,2 Hz, 1 H), 4,79 (dd, J = 15,6, 1,2 Hz, 1 H), 4,47 (qd, J = 7,7, 5,2 Hz, 1 H), 3,49 (t, J = 8,4 Hz, 1 H), 3,13-2,95 (m, 1 H), 1,70 (dddd, J = 12,8, 9,6, 7,5, 5,2 Hz, 1 H), 1,57-1,49 (m, 1 H), 1,48-1,28 (m, 2 H), 0,93 (t, J = 7,3 Hz, 3 H).
Alfa-D (7, MeOH, 10 mg/ml, 25°C) = 26,9
Ejemplo 5. Síntesis de la (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 9 y la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 10

Se agitó una mezcla de 3,3,3-trifluoropropanal (1,0 eq., 5,0 g, 44,62 mmol), carbonato de potasio (1,0 eq., 6,2 g, 44 mmol) y nitrometano (30 eq., 60,2 ml, 1340 mmol) a 50°C durante 5 h. La mezcla se enfrió a temperatura ambiente y se diluyó con agua y acetato de etilo. La capa acuosa se extrajo con acetato de etilo (3 veces) y la combinación de las capas orgánicas se lavó con agua (2 veces), se secó sobre MgSO4, se filtró y se evaporó hasta la sequedad. El compuesto bruto se purificó mediante cromatografía ultrarrápida Biotage Isolera Four (columna en gel de sílice KP-SNAP de 100 g con gradiente de diclorometano al 100 % a lo largo de 10 VC). Las fracciones más puras se evaporaron hasta la sequedad para dar el 4,4,4-trifluoro-1 -nitrobután-2-ol XIV (3,8 g, 22 mmol) como un aceite incoloro.
Rendimiento: 49 %
RMN 1H (400 MHz, CDCla): 54,73 (dddd, J = 12,7, 7,6, 5,0, 3,8 Hz, 1 H), 4,58-4,43 (m, 2 H), 3,01 (d, J = 5,1 Hz, 1 H), 2,60-2,28 (m, 2 H).
5.2 Síntesis del 1-amino-4,4,4-trifluorobután-2-ol XV
Una mezcla de 4,4,4-trifluoro-1-nitrobután-2-ol XIV (1,0 eq., 4,8 g, 28 mmol) en etanol (140 ml) se hidrogenó con un reactor H-cube equipado con un cartucho de Ni Raney (flujo de 1 ml/min, 50°C, 50 bar). La mezcla se evaporó hasta la sequedad para dar el 1-amino-4,4,4-trifluorobután-2-ol XV (2,5 g, 17 mmol) como un aceite incoloro.
Rendimiento: 63 %
RMN 1H (400 MHz, CDCh): 53,89 (tt, J = 8,0, 3,9 Hz, 1 H), 2,86 (dd, J = 12,8, 3,5 Hz, 1 H), 2,62 (dd, J = 12,8, 8,3 Hz, 1 H), 2,35-2,07 (m, 2 H).
5.3 Síntesis de la 5-(2,2,2-trifluoroetil)oxazolidín-2-ona XVI
A una mezcla de 1-amino-4,4,4-trifluorobután-2-ol XV (1,0 eq., 2,5 g, 17 mmol) y 1, 1 '-carbonildiimidazol (1,0 eq., 2,9 g, 18 mmol) en THF (35 ml) se le añadió hidruro de sodio (0,1 eq., 42 mg, 1,75 mmol) y la mezcla se agitó a 25°C durante 16 h. Se le agregaron unas gotas de agua (neutralización del NaH) y la mezcla se evaporó directamente hasta la sequedad y se purificó por cromatografía en fase inversa en modo básico para dar la 5-(2,2,2-trifluoroetil)oxazolidín-2-ona XVI (2,25 g, 13,2 mmol) como un sólido blanco.
Rendimiento: 75 %
LC/MS: [M H]+ = 170,11
RMN 1H (400 MHz, DMSO-afe): 57,61 (s, 1 H), 4,81 (qd, J = 7,6, 4,9 Hz, 1 H), 3,62 (t, J = 8,7 Hz, 1 H), 3,20 (dd, J = 8,9, 7,2 Hz, 1 H), 2,89-2,68 (m, 2 H).
5.4 Síntesis de la (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 9 y la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 10
A una mezcla de [2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol V (1,0 eq., 200 mg, 0,75 mmol) y 5-(2,2,2-trifluoroetil)oxazolidín-2-ona XVI (1,8 eq., 227 mg, 1,34 mmol) en sulfolano (0,5 M, 3,7 ml), se le añadió monohidrato de ácido p-toluensulfónico (1,0 eq., 142 mg, 0,75 mmol) y la mezcla se agitó a 110°C durante 3 h y 30 min. La mezcla se purificó directamente mediante cromatografía en fase inversa en condiciones básicas para dar un aceite amarillo (250 mg). La mezcla de enantiómeros obtenida se purificó mediante HPLC preparativa quiral (AS * EtOH al 50 %-heptano al 50 %-DEA al 0,1 %) para dar la (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 9 (29 mg, 0,07 mmol) y la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo [2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 10 (30 mg, 0,07 mmol). Rendimiento global: 19 % (9,3 % 9,6 %)
LC/MS: [M H]+ = 419,05
RMN 1H (400 MHz, CDCla): 55,02-4,66 (m, 5H), 3,66 (t, J = 8,7 Hz, 1 H), 3,52 (s, 3 H), 3,33-3,17 (m, 1 H), 2,76-2,58 (m, 1 H), 2,51-2,35 (m, 1 H).
Alfa-D (9, MeOH, 10 mg/ml, 25°C) = 13,0
La (+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 11 y la (-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 12 se sintetizan según el mismo procedimiento a partir de la 5-(2,2,2-trifluoroetil)oxazolidín-2-ona XVI y el [6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol V iIi. Rendimiento global: 14 % (7,8 % 6,2 %)
LC/MS: [M H]+ = 401,06
RMN 1H (400 MHz, DMSO-afe): 57,14 (t, J = 53,5 Hz, 1 H), 4,90-4,60 (m, 5 H), 3,67 (t, J = 8,6 Hz, 1 H), 3,43 (s, 3 H), 3,25 (dd, J = 8,6, 7,0 Hz, 1 H), 2,91-2,63 (m, 2 H).
Alfa-D (11, MeOH, 10 mg/ml, 25°C) = 3,6
Ejemplo 6. Síntesis de la (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 13 y la (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona 14

A una solución de 3,3-difluorobután-1-ol (1,0 eq., 1,5 g, 9,8 mmol) en diclorometano (50 ml) a 0°C se le añadió peryodinano de Dess-Martin (1,2 eq., 5,1 g, 12 mmol) y la mezcla de reacción se agitó a 0°C durante 2 h. Después, la mezcla bruta se enfrió a -78°C, se filtró y el sólido obtenido se lavó con diclorometano frío (-78°C). Al filtrado obtenido calentado a temperatura ambiente se le añadió carbonato de potasio (15,0 eq., 21 g, 150 mmol) y nitrometano (30,0 eq., 18 g, 290 mmol), y la mezcla se agitó a 45°C durante 2 h. La mezcla bruta se filtró sobre Celite y el filtrado obtenido se concentró al vacío a 30°C para dar una goma amarilla. La goma obtenida se purificó mediante cromatografía ultrarrápida Biotage Isolera Four (columna de gel de sílice KP-SNAP de 100 g en un gradiente de diclorometano al 100 % a lo largo de 6 VC y luego del 0 % al 10 % de metanol en diclorometano a lo largo de 10 VC) para dar el 4,4-difluoro-1-nitropentán-2-ol XVII (678 mg, 4,0 mmol) como un aceite amarillo.
Rendimiento: 41 %
RMN 1H (400 MHz, CDCla): 54,71 (tt, J = 7,9, 4,0 Hz, 1 H), 4,61-4,36 (m, 2 H), 2,79 (dd, J = 4,6, 1,7 Hz, 1 H), 2,32 2,03 (m, 2 H), 1,71 (t, J = 18,9 Hz, 3 H).
6.2 Síntesis del 1-amino-4,4-difluoropentán-2-ol XVIII
Una solución de 4,4-difluoro-1-nitropentán-2-ol XVII (1,0 eq., 1 g, 5,91 mmol) en etanol (40 ml) se hidrogenó en un H-Cube equipado con un cartucho de Pd 10 % (flujo de 1 ml/min, 50°C, 50 bar). La mezcla bruta se evaporó hasta la sequedad para dar el 1-amino-4,4-difluoropentán-2-ol XVIII (760 mg, 4,9 mmol) como un aceite incoloro.
Rendimiento: 83 %
RMN 1H (400 MHz, CDCla): 53,95-3,76 (m, 1 H), 2,88 (dd, J = 12,7, 3,6 Hz, 1 H), 2,58 (dd, J = 12,7, 8,2 Hz, 1 H), 2,16 1,88 (m, 2 H), 1,69 (t, J = 18,9 Hz, 3 H).
6.3 Síntesis de la 5-(2,2-difluoropropil)oxazolidín-2-ona XIX
A una solución de 1 -amino-4,4-difluoropentán-2-ol XVIII (1,0 eq., 760 mg, 5,46 mmol) en THF (18 ml) a temperatura ambiente se le añadieron 1,1 '-carbonildiimidazol (1,0 eq., 876 mg, 5,30 mmol) e hidruro de sodio (0,1 eq., 21 mg, 0,53 mmol). La reacción se agitó a temperatura ambiente durante 16 h. Se agregaron unas gotas de agua a la solución (neutralización del NaH) y la mezcla se evaporó hasta la sequedad. El compuesto bruto se purificó directamente mediante cromatografía ultrarrápida Biotage Isolera Four (columna de gel de sílice KP-SNAP de 100 g en un gradiente del 0 % al 10 % de metanol en diclorometano a lo largo de 10 VC, y luego 10 % de metanol a lo largo de 4 VC) para dar la 5-(2,2-difluoropropil)oxazolidín-2-ona XIX (675 mg, 4,08 mmol) como un sólido beis.
Rendimiento: 75 %
RMN 1H (400 MHz, CDCta): 55,34 (d, J = 37,0 Hz, 1 H), 4,96-4,85 (m, 1 H), 3,78 (td, J = 8,5, 8,0, 0,9 Hz, 1 H), 3,37 (t, J = 8,2 Hz, 1 H), 2,56-2,18 (m, 2 H), 1,71 (t, J = 18,9 Hz, 3 H).
6.4 Síntesis de la (-)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona 13 y la (+)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona 14
A una mezcla de [2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol V (1,0 eq., 150 mg, 0,56 mmol) y 5-(2,2-difluoropropil)oxazolidín-2-ona XIX (1,5 eq., 139 mg, 0,84 mmol) en sulfolano (3 ml), se le añadió monohidrato de ácido p-toluensulfónico (1,0 eq., 106 mg, 0,56 mmol). La mezcla se agitó durante 16 h a 100°C. La mezcla se purificó directamente mediante cromatografía en fase inversa en modo básico para dar un aceite amarillo. La mezcla de enantiómeros se purificó mediante SFC quiral (AS * EtOH al 50 %-heptano al 50 %-DEA al 0,1 %) para dar la (-)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona 13 (19,1 mg, 0,05 mmol) y la (+)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona 14 (19,9 mg, 0,05 mmol) como sólidos blancos.
Rendimiento global: 16,7 % (8,2 % 8,5 %)
LC/MS: [M H]+ = 415,02
RMN 1H (400 MHz, DMSO-afe): 54,90-4,55 (m, 5 H), 3,63 (t, J = 8,4 Hz, 1 H), 3,45 (s, 3 H), 3,22 (t, J = 8,0 Hz, 1 H), 2,42-2,26 (m, 2 H), 1,63 (t, J = 19,3 Hz, 3 H).
Alfa-D (13, MeOH, 10 mg/ml, 28,6°C) = -13,3
La 3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1 -b][1,3,4]tiadiazol-5-il]metil]-5-(2,2-difluoropropil)oxazolidín-2-ona 15 y la 3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2-difluoropropil)oxazolidín-2-ona 16 se sintetizan según el mismo procedimiento a partir de la 5-(2,2-difluoropropil)oxazolidín-2-ona XIX y el [6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metanol VIII.
Rendimiento global: 8 % (4 % 4 %)
LC/MS: [M H]+ = 397,04
RMN 1H (400 MHz, DMSO-afe): 57,13 (t, J = 53,5 Hz, 1 H), 4,89-4,60 (m, 5 H), 3,64 (t, J = 8,5 Hz, 1 H), 3,43 (s, 3 H), 3,21 (t, J = 8,0 Hz, 1 H), 2,41-2,17 (m, 2 H), 1,62 (t, J = 19,3 Hz, 3 H).
Cromatografía quiral (SFC, fase W, CO2-MeOH al 15 %, 3 ml/min): pico 1 (15): 6,64 min y pico 2 (16): 7,72 min. La tabla (I) indica el nombre de la IUPAC (o el nombre generado a partir de Accelerys Draw 4.0) del compuesto, el pico de iones observado en la espectroscopía de masas y la descripción de la RMN 1H.
Tabla I: Caracterización física de los compuestos de los ejemplos.



Ejemplo 7. Ensayos de fijación a SV2A y SV2C.
Las proteínas SV2A y SV2C humanas se expresaron en las células de riñón embrionario humano (HEK). Las preparaciones de las membranas de HEK con SV2A y de HEK con SV2C se prepararon como se describe en Gillard y col. (Eur. J. Pharmacol. 2006, 536, 102-108). Para medir la afinidad de los compuestos no marcados, los experimentos de competición se realizaron de la siguiente manera: las membranas que expresan las proteínas SV2 (de 5 a 15 pg de proteínas por ensayo) se incubaron durante 60 min a 37°C con [3H]-2-[4-(3-azidofenil)-2-oxo-1-pirrolidiniljbutanamida (5 nM) y/o [3H]-4R-(2-cloro-2,2-difluoroetil)-1-{[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil}pirrolidín-2-ona (25 nM) en 0,2 ml de un tampón de Tris-HCl a 50 mM (pH 7,4) que contiene MgCl2 a 2 mM, dimetilsulfóxido al 0,1 % y diez concentraciones crecientes del compuesto problema no marcado (0,1 nM a 10 pM). Al final del período de incubación, el radioligando fijado a la membrana se recuperó mediante filtración rápida a través de filtros de fibra de vidrio GF/C empapados previamente en polietilenimina al 0,1 %. Las membranas se lavaron con al menos 4 veces el volumen del ensayo del tampón de Tris HCl a 50 mM enfriado en hielo (pH 7,4). Los filtros se secaron y se determinó la radiactividad mediante centelleo en líquido. La etapa de filtración completa no superó los 10 segundos. Los valores de los pIC50 de afinidad medidos se corrigieron con el pKi según Cheng y Prusoff (Biochem. Pharmacol. 1973, 22 (23), 3099-3108).
Los compuestos de la fórmula (I) según la invención muestran típicamente valores de pKi para SV2A de al menos 6,5 y valores de pKi para SV2C de al menos 6,0.
Ejemplo 8. Modelos de convulsiones.
En todos los experimentos se utilizan ratones macho NMRI (Charles River, Alemania) que pesan entre 22 y 32 g. Los animales se mantienen en un ciclo de luz/oscuridad de 12/12 h con el encendido de las luces a las 6:00 am y se alojan con una temperatura mantenida a 20-21°C y una humedad de aproximadamente el 40 %. Los ratones se alojan en grupos de 10 por jaula (Tipo III). Todos los animales tienen acceso libre al agua y a pienso en gránulos estándares antes de la asignación aleatoria a los grupos experimentales que constan de 10 ratones cada uno. Todos los experimentos con animales se realizan de acuerdo con las Normas Nacionales sobre Experimentos con Animales y se llevan a cabo de acuerdo con las directrices de la directiva 2010/63/EU del Consejo de la Unión Europea. Un comité de ética local aprobó los protocolos experimentales.
8.1 Modelo de convulsión a 6 Hz
El modelo de 6 Hz se realiza según un protocolo descrito anteriormente (Kaminski y col., Epilepsia (2004), 45, 864 867). En pocas palabras, la estimulación de la córnea (44 mA, pulsos rectangulares monopolares de duración de 0,2 ms a 6 Hz durante 3 s) se administra mediante un dispositivo de corriente constante (unidad ECT 57800; Ugo Basile, Comerio, Italia). Se coloca una gota de hidrocloruro de oxibuprocaína al 0,4 % (Unicaine, Thea, Francia) en los ojos antes de la estimulación eléctrica. Durante la estimulación, los ratones se inmovilizan manualmente y se liberan en la jaula de observación (38 x 26 x 14 cm) inmediatamente después de la aplicación de la corriente. Las convulsiones suelen ir precedidas de un breve período (~2-3 s) de intensa agitación locomotora (corren y saltan en forma salvaje). Los animales luego exhiben una postura "aturdida" asociada con alzarse a dos patas, movimientos automáticos de las patas delanteras y clono, contracciones de las vibrisas y cola de Strub. Al final de la crisis convulsiva, los animales reanudan su comportamiento exploratorio normal. El criterio de valoración experimental es la protección contra la crisis convulsiva. Se considera que el animal está protegido si reanuda su comportamiento exploratorio normal en menos
de 7 s tras la estimulación.
Las actividades in vivo determinadas para los compuestos de prueba están comprendidas típicamente entre 0,05 mg/kg y 20 mg/kg después de una dosis única i.p.
8.2 Modelo de convulsiones con pentilentetrazol (PTZ)
El pentilentetrazol se utiliza en la dosis de CD97 previamente establecida de 89 mg/kg; una dosis convulsiva que induce convulsiones clónicas de las cuatro extremidades en el 97 % de los ratones (Klitgaard et al., Eur. J. Pharmacol. (1998), 353, 191-206). Inmediatamente después de la inyección del pentilentetrazol, los ratones se colocan individualmente en jaulas de Perspex y se observan para detectar la presencia de convulsiones clónicas en las cuatro extremidades y la extensión tónica de las patas traseras durante un período de 60 minutos.
Las actividades in vivo determinadas para los compuestos de prueba están comprendidas típicamente entre 0,5 mg/kg y 30 mg/kg después de una dosis única i.p.
Ejemplo 9. Ensayo con azamulina
Los hepatocitos humanos criopreservados (grupo de 20 donantes, lote de BSU de Celsis/IVT/Bioreclamation) se descongelaron según la información del proveedor. La viabilidad (exclusión del azul tripano) fue superior al 75 %. Se llevaron a cabo preincubaciones (250 pl de suspensión de hepatocitos a 2 x 106 hepatocitos/ml) con medio de William, que contiene 2 mM de glutamina y 15 mM de Hepes, en placas de 48 pocillos a 37°C, en una incubadora (5 % de CO2), con agitación suave (agitador vibratorio, Titramax 100, en torno a 300 rpm) durante 30 min. Después de la preincubación, la incubación se inició al agregar a los hepatocitos 250 pl del medio de cultivo (véase la composición anterior) que contenía el compuesto UCB (1 pM) o midazolam (control positivo) con o sin azamulina (6 pM, inhibidor específico del CYP3A4/5). La concentración final del compuesto UCB y de la azamulina en los incubados es de 0,5 pM y 3 pM, respectivamente. Las suspensiones celulares se volvieron a homogeneizar rápidamente mediante 2 pipeteos. Después de 0, 30, 60, 120, 180 y 240 minutos de incubación, las reacciones se detuvieron transfiriendo 50 pl de incubados al pocillo idóneo de una placa de 96 pocillos que contenía 50 pl de acetonitrilo helado con ketoconazol a 1 pM como estándar interno. Antes de cada muestreo, las células incubadas se vuelven a homogeneizar mediante dos pipeteos como antes.
Una vez finalizada la incubación, las placas de 96 pocillos se centrifugan a aproximadamente 3700 rpm, 4°C, durante 15 minutos. Se transfieren 50 pl de sobrenadantes a los pocillos de otras placas de pocillos profundos en los que se había añadido 150 pl de H2O de calidad Millipore. Estas muestras se analizaron mediante micro UPLC/HR-MS para determinar la desaparición de los compuestos originales y el seguimiento de la formación de metabolitos.
La contribución de CYP3A4/5 conocida como fracción metabolizada por CYP3A4/5 (fm,cYP3A4/5) se calculó para cada compuesto a partir de la relación entre CLint (basado en la desaparición del fármaco original) en ausencia y en presencia de azamulina, mediante la siguiente ecuación:
r , .
CL¿nt con azam ulina
F m CYP3A4/5 = 1 “ 77 ----------------------------------------------------------- L,Lint sin azam ulina
Fracción metabolizada por el CYP3A4/5 (fm,cYP3A4/5) de los compuestos problema está comprendida típicamente entre 0 y el 40 %.
Claims (11)
1. Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol de acuerdo con la fórmula (I), sus isómeros geométricos, sus enantiómeros, sus diastereómeros, sus isótopos y sus mezclas, o una sal farmacéuticamente aceptable de los mismos,

R1 es un alquilo(C1-4) opcionalmente sustituido con uno o más sustituyentes de tipo halógeno;
R2 es un átomo de halógeno, o un alquilo(C1-4) opcionalmente sustituido con uno o más átomos de halógeno;
R3 es un alquilo(C1-4) sustituido con un sustituyente alcoxi.
2. Un compuesto según cualquiera de la reivindicación 1, en el que R1 es un resto i-butilo, n-propilo, 2,2-difluoropropilo, 2-cloro-2,2-difluoroetilo, 2,2-difluoroetilo, 2,2,2-trifluoroetilo o 2-fluoroetilo.
3. Un compuesto según cualquiera de la reivindicación 1, en el que R1 es un resto n-propilo, 2,2-difluoropropilo o 2,2,2-trifluoroetilo.
4. Un compuesto según cualquiera de la reivindicación 1, en el que R2 es un átomo de cloro, un resto difluorometilo o un resto trifluorometilo.
5. Un compuesto según cualquiera de la reivindicación 1, en el que R2 es un resto difluorometilo o un resto trifluorometilo.
6. Un compuesto según cualquiera de las reivindicaciones anteriores, en el que R3 es un resto metoximetilo, [(2H3)metiloxi]metilo, metoxi(2H2)metilo, o [(2H3)metiloxi](2H2)metilo.
7. Un compuesto según cualquiera de las reivindicaciones anteriores, en el que
R1 es un resto n-propilo, 2,2-difluoropropilo o 2,2,2-trifluoroetilo;
R2 es un resto difluorometilo o trifluorometilo;
R3 es un resto metoximetilo, [(2H3)metoxi]metilo, [(2H3)metoxi](2H2)metilo o metoxi(2H2)metilo.
8. Un compuesto de acuerdo con cualquiera de las reivindicaciones anteriores seleccionado del grupo que comprende:
• (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
• (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
• (+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona;
• (-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona.
• (+)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona • (-)-3-[[2-[dideuterio(metoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona • (+)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona
• (-)-3-[[2-[dideuterio(trideuteriometoxi)metil]-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-propiloxazolidín-2-ona
• (+)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona • (-)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona • (+)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona
• (-)-3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2,2-trifluoroetil)oxazolidín-2-ona • (-)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona • (+)-5-(2,2-difluoropropil)-3-[[2-(metoximetil)-6-(trifluorometil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]oxazolidín-2-ona • 3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2-difluoropropil)oxazolidín-2-ona, enantiómero 1
• 3-[[6-(difluorometil)-2-(metoximetil)imidazo[2,1-b][1,3,4]tiadiazol-5-il]metil]-5-(2,2-difluoropropil)oxazolidín-2-ona, enantiómero 2
9. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 para su uso como medicamento.
10. Una composición farmacéutica que comprende una cantidad eficaz de un compuesto según las reivindicaciones 1 a 8 en combinación con un diluyente o vehículo farmacéuticamente aceptable.
11. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 para su uso en el tratamiento de la epilepsia, epileptogénesis, trastornos convulsivos, convulsiones, en concreto para crisis comiciales que no responden al tratamiento.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17180432 | 2017-07-10 | ||
| PCT/EP2018/068207 WO2019011770A1 (en) | 2017-07-10 | 2018-07-05 | DERIVATIVES OF 2-OXO-1,3-OXAZOLIDINYL IMIDAZOTHIADIAZOLE |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2896331T3 true ES2896331T3 (es) | 2022-02-24 |
Family
ID=59313086
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18740157T Active ES2896331T3 (es) | 2017-07-10 | 2018-07-05 | Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US11155565B2 (es) |
| EP (1) | EP3652183B1 (es) |
| JP (1) | JP7126545B2 (es) |
| CN (1) | CN110869374B (es) |
| BR (1) | BR112019028078A2 (es) |
| CA (1) | CA3069040A1 (es) |
| ES (1) | ES2896331T3 (es) |
| MA (1) | MA49559A (es) |
| RU (1) | RU2020104753A (es) |
| WO (1) | WO2019011770A1 (es) |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101037445A (zh) * | 2007-04-14 | 2007-09-19 | 西北师范大学 | 一种具有潜在生物活性的咪唑并[2,1-b]-1,3,4-噻二唑衍生物及其合成方法 |
| US20100222326A1 (en) * | 2007-04-27 | 2010-09-02 | Ucb Pharma, S.A. | New Heterocyclic Derivatives Useful For The Treatment of CNS Disorders |
| EP2162132A4 (en) * | 2007-05-21 | 2013-01-16 | Sgx Pharmaceuticals Inc | Heterocyclic kinase modulators |
| HUE028668T2 (en) | 2009-10-23 | 2016-12-28 | Ucb Biopharma Sprl | 2-oxo-1-pyrrolidinyl imidazothiadiazole derivatives |
| WO2012143116A1 (en) * | 2011-04-18 | 2012-10-26 | Ucb Pharma, S.A. | 4-oxo-1-imidazolidinyl imidazothiadiazole derivatives |
| PL2699581T3 (pl) * | 2011-04-18 | 2016-05-31 | Ucb Biopharma Sprl | Pochodne 2-okso-1-imidazolidynyloimidazotiadiazolu |
-
2018
- 2018-07-05 CN CN201880046128.8A patent/CN110869374B/zh not_active Expired - Fee Related
- 2018-07-05 EP EP18740157.5A patent/EP3652183B1/en active Active
- 2018-07-05 CA CA3069040A patent/CA3069040A1/en active Pending
- 2018-07-05 JP JP2020500838A patent/JP7126545B2/ja active Active
- 2018-07-05 BR BR112019028078-2A patent/BR112019028078A2/pt not_active Application Discontinuation
- 2018-07-05 MA MA049559A patent/MA49559A/fr unknown
- 2018-07-05 WO PCT/EP2018/068207 patent/WO2019011770A1/en not_active Ceased
- 2018-07-05 RU RU2020104753A patent/RU2020104753A/ru not_active Application Discontinuation
- 2018-07-05 ES ES18740157T patent/ES2896331T3/es active Active
- 2018-07-05 US US16/629,776 patent/US11155565B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN110869374A (zh) | 2020-03-06 |
| US11155565B2 (en) | 2021-10-26 |
| US20200231599A1 (en) | 2020-07-23 |
| MA49559A (fr) | 2021-05-05 |
| EP3652183A1 (en) | 2020-05-20 |
| EP3652183B1 (en) | 2021-09-08 |
| RU2020104753A (ru) | 2021-08-10 |
| JP2020526532A (ja) | 2020-08-31 |
| BR112019028078A2 (pt) | 2020-07-28 |
| JP7126545B2 (ja) | 2022-08-26 |
| CN110869374B (zh) | 2022-08-30 |
| CA3069040A1 (en) | 2019-01-17 |
| WO2019011770A1 (en) | 2019-01-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2565515T3 (es) | Derivados de 2-oxo-1-pirrolidinil imidazotiadiazol | |
| WO2012143117A1 (en) | 2-oxo-1-imidazolidinyl imidazothiadiazole derivatives | |
| WO2012143116A1 (en) | 4-oxo-1-imidazolidinyl imidazothiadiazole derivatives | |
| ES2947618T3 (es) | Derivados de 1-imidazotiadiazolo-2H-pirrol-5-ona | |
| ES2912612T3 (es) | Derivados de 2-oxo-1-pirrolidinil imidazotiadiazol | |
| ES2896331T3 (es) | Derivados del 2-oxo-1,3-oxazolidinilimidazotiadiazol | |
| ES2895373T3 (es) | Derivados de 2-oxo-1-imidazolidinil imidazotiadiazol | |
| AU2012244614B2 (en) | 2-oxo-1-imidazolidinyl imidazothiadiazole derivatives |