ES2895419T3 - Formas sólidas de (1S,4S)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1-metilciclohexan-1-carboxamida y métodos para su uso - Google Patents
Formas sólidas de (1S,4S)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1-metilciclohexan-1-carboxamida y métodos para su uso Download PDFInfo
- Publication number
- ES2895419T3 ES2895419T3 ES17776743T ES17776743T ES2895419T3 ES 2895419 T3 ES2895419 T3 ES 2895419T3 ES 17776743 T ES17776743 T ES 17776743T ES 17776743 T ES17776743 T ES 17776743T ES 2895419 T3 ES2895419 T3 ES 2895419T3
- Authority
- ES
- Spain
- Prior art keywords
- cancer
- compound
- solid
- solid form
- ray powder
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- YQAOJVZAUIUIPT-LBTCCZLRSA-N C[C@](CC(N)=O)(CC1)CC[C@@H]1[n]1c(Nc(c(Cl)cc(Cl)c2)c2Cl)nc2c1nc(N[C@H](CCOC1)[C@@H]1F)nc2 Chemical compound C[C@](CC(N)=O)(CC1)CC[C@@H]1[n]1c(Nc(c(Cl)cc(Cl)c2)c2Cl)nc2c1nc(N[C@H](CCOC1)[C@@H]1F)nc2 YQAOJVZAUIUIPT-LBTCCZLRSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Gastroenterology & Hepatology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Una forma cristalina que comprende el compuesto 1 o un tautómero de este: **(Ver fórmula)** que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,3, 8,5, 18,2 y 21,3º 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 13,8, 19,5, 20,0 y 20,8° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,7, 8,9, 10,3 y 18,3° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,4, 8,7, 10,1 y 18,1° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,8, 14,6, 17,5 y 22,2° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,4, 11,8, 18,0 y 18,3° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,9, 15,3, 18,4 y 22,9° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,7, 8,9, 10,3 y 18,2° 2θ (± 0,2° 2θ) o que tiene un patrón de difracción de rayos X en polvo que comprende picos en 6,3, 15,7, 18,1 y 20,0° 2θ (± 0,2° 2θ).
Description
DESCRIPCIÓN
Formas sólidas de (1 S,4S)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1-metilciclohexan-1-carboxamida y métodos para su uso
Campo
En el presente documento se proporcionan formas sólidas de cis-4-[2-{[(3S,4R)-3-fluorooxan-4-il]amino}-8-(2,4,6-tricloroanilino)-9H-purin-9-il]-1 -metilciclohexan-1 -carboxamida, denominado alternativamente (1 s,4s)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1-metilciclohexan-1-carboxamida y su uso para el tratamiento del cáncer.
Antecedentes
La identificación y selección de una forma sólida de compuesto farmacéutico es compleja, ya que un cambio en la forma sólida puede afectar a una diversidad de propiedades físicas y químicas, lo cual puede brindar beneficios o desventajas en cuanto al procesamiento, la formulación, la estabilidad, la biodisponibilidad, el almacenamiento, la manipulación (p. ej., el envío), entre otras características farmacéuticas importantes. Los sólidos farmacéuticos útiles incluyen sólidos cristalinos y sólidos amorfos, dependiendo del producto y de su forma de administración. Los sólidos amorfos se caracterizan por una falta de orden estructural de gran espectro, mientras que los sólidos cristalinos se caracterizan por periodicidad estructural. La clase deseada de sólido farmacéutico depende de la aplicación específica; algunas veces los sólidos amorfos se seleccionan en función de, p. ej., un mejor perfil de disolución, mientras que los sólidos cristalinos pueden resultar convenientes para propiedades tales como, p. ej., estabilidad química o física (véase, p. ej., S. R. Vippagunta et al., Adv. Drug. Deliv. Rev., (2001) 48: 3-26; L. Yu, Adv. Drug. Deliv. Rev., (2001) 48: 27-42).
Ya sean cristalinas o amorfas, las formas sólidas de un compuesto farmacéutico incluyen sólidos de un único componente y de componentes múltiples. Los sólidos de un único componente consisten esencialmente en el compuesto farmacéutico o principio activo sin otros compuestos. La variedad entre los materiales cristalinos de un único componente puede surgir posiblemente del fenómeno de polimorfismo, en donde existen múltiples disposiciones tridimensionales para un compuesto farmacéutico particular (véase, p. ej., S. R. Byrn et al., Solid State Chemistry of Drugs, (1999) SSCI, West Lafayette). La importancia de descubrir polimorfos se vio resaltada por el caso de Ritonavir™, un inhibidor de la proteasa del VIH que se formuló en forma de cápsulas blandas de gelatina. El producto tuvo que ser retirado del mercado aproximadamente dos años después de su lanzamiento hasta que se pudiera desarrollar una formulación más consistente, debido a la precipitación imprevista de un polimorfo nuevo menos soluble en la formulación (véase S. R. Chemburkar et al., Org. Process Res. Dev., (2000) 4:413-417).
En particular, no es posible predecir a prior.i si existen formas cristalinas de un compuesto, menos aún saber cómo prepararlas de forma exitosa (véase, p. ej., Braga y Grepioni, 2005, "Making crystals from crystals: a green route to crystal engineering and polymorphism," Chem. Commun.: 3635-3645 (con respecto a la ingeniería de cristales, si las instrucciones no son muy precisas y/o si otros factores externos afectan el proceso, el resultado puede ser impredecible); Jones et al., 2006, Pharmaceutical Cocrystals: An Emerging Approach to Physical Property Enhancement", MRS Bulletin 31: 875-879 (actualmente, no es posible predecir con medios informáticos la cantidad de polimorfos observables, incluso en las moléculas más simples); Price, 2004, "The computational prediction of pharmaceutical crystal structures and polymorphism", Advanced Drug Delivery Reviews 56:301-319 ("Price") y Bernstein, 2004, "Crystal Structure Prediction and Polymorphism", ACA Transactions 39: 14-23 (todavía queda mucho por aprender y hacer antes de poder afirmar con algún grado de confianza la capacidad de predecir una estructura cristalina, mucho menos formas polimórficas)).
La variedad de formas sólidas posibles crea un potencial de diversidad en las propiedades químicas y físicas para un compuesto farmacéutico específico. El descubrimiento y la selección de formas sólidas son muy importantes en la creación de un producto farmacéutico eficaz, estable y comercializable.
Desde hace más de 20 años se conoce la vinculación entre la fosforilación de proteínas anómalas y la causa o consecuencia de enfermedades. Por consiguiente, las proteínas cinasas se han vuelto un grupo muy importante de objetivos para los fármacos. (Véase Cohen, Nature, 1: 309-315 (2002), Gaestel et al. Curr. Med. Chem. 14: 2214 223 (2007); Grimminger et al. Nat. Rev. Drug. 9 (12): 956-970 (2010)). A nivel clínico, se han utilizado diversos inhibidores de proteína cinasa en el tratamiento de una gran variedad de enfermedades, tales como el cáncer y enfermedades inflamatorias crónicas, que incluyen artritis reumatoide y psoriasis. (Véase Cohen, Eur. J. Biochem., 268: 5001-5010 (2001); Protein Kinase Inhibitors for the Treatment of Disease: The Promise and the Problems, Handbook of Experimental Pharmacology, Springer Berlin Heidelberg, 167 (2005)).
El cáncer se caracteriza principalmente por aumento en la cantidad de células anormales derivadas de un tejido normal determinado, invasión de tejidos adyacentes por parte de estas células anormales o propagación sanguínea o linfática de las células malignas a los ganglios linfáticos regionales y a puntos distantes (metástasis). Los datos clínicos y los estudios biológicos moleculares indican que el cáncer es un proceso de múltiples etapas que comienza
con cambios preneoplásicos menores que, en determinadas circunstancias, pueden evolucionar hasta una neoplasia. La lesión neoplásica puede evolucionar mediante clonación y desarrollar una mayor capacidad de invasión, crecimiento, metástasis y heterogeneidad, en especial en condiciones en donde las células neoplásicas escapan del control inmunológico del hospedador (Roitt, I., Brostoff, J y Kale, D., Immunology, 17.1-17.12 (3a ed., Mosby, San Luis, Mo., 1993)).
El cáncer se encuentra entre las principales causas de muerte a nivel mundial, con 8,2 millones de muertes por esta enfermedad en 2012. Se prevé que la cantidad de casos de cáncer por año aumente de 14 millones en 2012 a 22 millones en las próximas dos décadas (véase la hoja informativa sobre el cáncer n.° 297 de la Organización Mundial de la Salud, publicada en febrero de 2014 y consultada el 10 de junio de 2014, así como a Globocan 2012, IARC). Los fármacos que se utilizan actualmente para tratar el cáncer son muy tóxicos y a menudo no específicos. Las estrategias de terapias actuales contra el cáncer habitualmente se enfocan en las células de rápida proliferación, lo cual puede reducir los tumores primarios y metastásicos, pero dichos efectos generalmente tienen un carácter transitorio y es habitual que se produzca una recidiva tumoral en la mayoría de los cánceres metastásicos. Una posible razón de este fracaso es la existencia de células madre cancerosas. A diferencia de la mayoría de las células dentro del tumor, las células madre cancerosas son resistentes a la quimioterapia bien definida y, tras el tratamiento, pueden regenerar todos los tipos de células en el tumor mediante un comportamiento típico de las células madre de naturaleza mayormente inactiva y su expresión abundante de transportadores de fármaco.
Existe una enorme variedad de cánceres que se describen con detalle en la bibliografía médica. La incidencia del cáncer continúa aumentando debido al envejecimiento de la población general, debido al surgimiento de nuevos cánceres y debido al crecimiento de poblaciones susceptibles (p. ej., personas infectadas con SIDA o con exposición excesiva a la luz solar). Sin embargo, las opciones para el tratamiento del cáncer son limitadas. Por lo tanto, existe una gran demanda de nuevos métodos y composiciones que puedan utilizarse para tratar a los pacientes con cáncer.
La mención o identificación de cualquier referencia en alguna sección de la presente solicitud no debe interpretarse como una admisión de que dicha referencia constituya técnica previa de la presente solicitud.
Por consiguiente, sigue existiendo una necesidad de terapias contra el cáncer, por ejemplo, moduladores y, en particular formas sólidas.
Sumario
En el presente documento se proporcionan formas sólidas del compuesto 1:

con el nombre cis-4-[2-{[(3S,4R)-3-fluorooxan-4-il]amino}-8-(2,4,6-tricloroanilino)-9H-purin-9-il]-1 -metilciclohexan-1 -carboxamida, denominado alternativamente (1 s,4s)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1 -metilciclohexan-1 -carboxamida, incluidos sus tautómeros.
También se proporcionan métodos para preparar, aislar y caracterizar las formas sólidas.
En determinados aspectos, las formas sólidas del compuesto 1 descritas en el presente documento son útiles para tratar o prevenir una o más enfermedades o afecciones tales como, por ejemplo, cáncer.
En el presente documento se proporcionan formas sólidas del compuesto 1 para su uso en métodos para tratar un cáncer, en particular un tumor sólido o un cáncer hematológico. Las formas sólidas del compuesto 1 que se proporcionan en el presente documento se pueden usar en métodos para tratar o prevenir un cáncer, en particular un tumor sólido o un cáncer hematológico, según se describe en el presente documento. Los métodos comprenden
administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 proporcionada en el presente documento. Además, los métodos pueden ser para tratar y prevenir la metástasis del cáncer, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 proporcionada en el presente documento. Las formas sólidas del compuesto 1 proporcionadas en el presente documento se pueden usar en los métodos para tratar y prevenir la metástasis del cáncer. Asimismo, los métodos pueden ser para erradicar las células madre cancerosas en un sujeto, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 proporcionada en el presente documento. Las formas sólidas del compuesto 1 proporcionadas en el presente documento se pueden usar en los métodos para erradicar las células madre cancerosas en un sujeto. Además, los métodos pueden ser para inducir la diferenciación en células madre cancerosas en un sujeto, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 proporcionada en el presente documento. Las formas sólidas del compuesto 1 proporcionadas en el presente documento se pueden usar en los métodos para inducir la diferenciación en células madre cancerosas en un sujeto. En otro aspecto, los métodos pueden ser para inducir la muerte de las células madre cancerosas en un sujeto, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 proporcionada en el presente documento. Las formas sólidas del compuesto 1 proporcionadas en el presente documento se pueden usar en los métodos para inducir la muerte de las células madre cancerosas en un sujeto.
Los compuestos útiles en los métodos divulgados en el presente documento incluyen las formas sólidas del compuesto 1 descritas en el presente documento o un tautómero, un estereoisómero, un enantiómero, un isotopólogo o una sal farmacéuticamente aceptable de este.
Las realizaciones presentes se pueden comprender de forma más completa en relación con la descripción detallada y los ejemplos, que pretender mostrar a modo de ejemplo realizaciones no limitantes.
Breve descripción de los dibujos
La figura 1 ilustra un gráfico de líneas apiladas de XRPD de las formas cristalinas de base libre A-I.
La figura 2 ilustra un patrón de XRPD de la forma A de base libre.
La figura 3 ilustra una imagen de SEM de la forma A de base libre.
La figura 4 ilustra un termograma de TGA de la forma A de base libre.
La figura 5 ilustra un termograma de DSC de la forma A de base libre.
La figura 6A ilustra una gráfica isotérmica de DVS de la forma A de base libre. La figura 6B ilustra los valores de la gráfica isotérmica de la figura 6A.
La figura 7 ilustra un espectro de RMN 1H de la forma A de base libre.
La figura 8 ilustra un patrón de XRPD de la forma B de base libre.
La figura 9 ilustra un termograma de TGA de la forma B de base libre.
La figura 10 ilustra un termograma de DSC de la forma B de base libre.
La figura 11 ilustra un patrón de XRPD de la forma C de base libre.
La figura 12 ilustra un termograma de TGA de la forma C de base libre.
La figura 13 ilustra un termograma de DSC de la forma C de base libre.
La figura 14 ilustra un espectro de RMN 1H de la forma C de base libre.
La figura 15 ilustra un patrón de XRPD de la forma D de base libre.
La figura 16 ilustra una imagen de SEM de la forma D de base libre.
La figura 17 ilustra un termograma de TGA de la forma D de base libre.
La figura 18 ilustra un termograma de DSC de la forma D de base libre.
La figura 19 ilustra un espectro de RMN 1H de la forma D de base libre.
La figura 20 i ustra un patrón de XRPD de la forma E de base libre.
La figura 21 i ustra un termograma de TGA de la forma E de base libre.
La figura 22 i ustra un termograma de DSC de la forma E de base libre.
La figura 23 i ustra un patrón de XRPD de la forma F de base libre.
La figura 24 i ustra una SEM de la forma F de base libre.
La figura 25 i ustra un termograma de TGA de la forma F de base libre.
La figura 26 i ustra un termograma de DSC de la forma F de base libre.
La figura 27 i ustra un espectro de RMN 1H de la forma F de base libre.
La figura 28 i ustra un patrón de XRPD de la forma G de base libre.
La figura 29 i ustra una imagen de SEM de la forma G de base libre.
La figura 30 i ustra un termograma de TGA de la forma G de base libre.
La figura 31 i ustra una DSC de la forma G de base libre.
La figura 32 i ustra un espectro de RMN 1H de la forma G de base libre.
La figura 33 i ustra un patrón de XRPD de la forma H de base libre.
La figura 34 i ustra un termograma de TGA de la forma H de base libre.
La figura 35 i ustra un termograma de DSC de la forma H de base libre.
La figura 36 i ustra un patrón de XRPD de la forma I de base libre.
La figura 37 i ustra un espectro de RMN 1H de la forma I de base libre.
La figura 38 i ustra un termograma de TGA de la forma I de base libre.
La figura 39 i ustra un termograma de DSC de la forma I de base libre.
La figura 40 i ustra un patrón de XRPD de material amorfo.
La figura 41 i ustra un termograma de DSC de material amorfo.
La figura 42 i ustra un espectro de RMN 1H de material amorfo.
La figura 43A ilustra una gráfica isotérmica de DVS de material amorfo. La figura 43B ilustra los valores de la gráfica isotérmica de DVS de la figura 43A.
La figura 44 i ustra un gráfico de líneas apiladas de XRPD de las formas Y Z de citrato.
La figura 45 i ustra un patrón de XRPD de la forma Y de citrato.
La figura 46 i ustra una imagen de SEM de la forma Y de citrato.
La figura 47 i ustra un termograma de TGA de la forma Y de citrato.
La figura 48 i ustra un termograma de DSC de la forma Y de citrato.
La figura 49A ilustra una gráfica isotérmica de DVS de la forma Y de citrato. La figura 49B ilustra los valores de la gráfica isotérmica de la figura 49A.
La figura 50 ilustra un espectro de RMN 1H de la forma Y de citrato.
La figura 51A ilustra una comparación de patrones de XRPD de la forma Y de citrato antes de la compresión. La figura 51B ilustra una comparación de patrones de XRPD de la forma Y de citrato después de la compresión.
La figura 52 ilustra un patrón de XRPD de la forma Z de citrato.
La figura 53 ilustra una imagen de SEM de la forma Z de citrato.
La figura 54 ilustra un termograma de TGA de  forma Z de citrato.
forma Z de citrato.
La figura 55 ilustra un termograma de DSC de  forma Z de citrato.
forma Z de citrato.
La figura 56A ilustra una gráfica isotérmica de DVS de la forma Z de citrato.
La figura 56B ilustra los valores de la gráfica isotérmica de la figura 56A.
La figura 57 ilustra un espectro de RMN 1H de la forma Z de citrato.
La figura 58 ilustra un termograma de TGA de una forma de hidrato de la forma Z de citrato.
La figura 59 ilustra un termograma de TGA de una forma no estequiométrica de la forma Z de citrato. La figura 60 ilustra un termograma de TGA de una forma de solvato de la forma Z de citrato.
La figura 61 ilustra un espectro de RMN 1H de la forma de solvato de la forma Z de citrato.
Las figuras 62A a 62B ilustran una comparación de patrones de XRPD de la forma Z de citrato. La figura 62A ilustra la XRPD antes de la compresión. La figura 62B ilustra la XRPD tras la compresión.
La figura 63 ilustra un patrón de XRPD de material de partida de sal de HCl.
La figura 64 ilustra un termograma de DSC y TGA de material de partida de sal de HCl.
La figura 65 ilustra una gráfica isotérmica de DVS de material de partida de sal de HCl.
La figura 66 ilustra un patrón de XRPD de la forma 1 de sal de HCl.
La figura 67 ilustra un termograma de DSC y TGA de la forma 1 de sal de HCl.
La figura 68 ilustra un patrón de XRPD de la forma 2 de sal de HCl.
La figura 69 ilustra un termograma de DSC y TGA de la forma 2 de sal de HCl.
La figura 70 ilustra un patrón de XRPD de la forma 3 de sal de HCl.
La figura 71 ilustra un termograma de DSC y TGA de la forma 3 de sal de HCl.
La figura 72 ilustra un patrón de XRPD de la forma 4 de sal de HCl.
La figura 73 ilustra un termograma de DSC y TGA de la forma 4 de sal de HCl.
La figura 74 ilustra un patrón de XRPD de la forma 5 de sal de HCl.
La figura 75 ilustra un termograma de DSC y TGA de la forma 5 de sal de HCl.
La figura 76 ilustra un patrón de XRPD de la forma 6 de sal de HCl.
La figura 77 ilustra un termograma de DSC y TGA de la forma 6 de sal de HCl.
La figura 78 ilustra un termograma de TGA de la forma 6 de sal de HCl.
La figura 79 ilustra un termograma de DSC de la forma 6 de sal de HCl.
La figura 80 ilustra un patrón de XRPD de la forma 7 de sal de HCl.
La figura 81 ilustra un termograma de DSC y TGA de la forma 7 de sal de HCl.
La figura 82 ilustra un termograma de DSC de la forma 7 de sal de HCl.
La figura 83 ilustra un termograma de TGA de la forma 7 de sal de HCl.
La figura 84 ilustra una gráfica isotérmica de DVS de la forma 7 de sal de HCl.
La figura 85 ilustra un patrón de XRPD de la forma 8 de sal de HCl.
La figura 86 ilustra un termograma de DSC y TGA de la forma 8 de sal de HCl.
La figura 87 ilustra un termograma de TGA de la forma 8 de sal de HCl.
La figura 88 ilustra un termograma de DSC de la forma 8 de sal de HCl.
La figura 89 ilustra un patrón de XRPD del compuesto 1.
La figura 90 ilustra un termograma de TGA del compuesto 1.
La figura 91 ilustra un termograma de DSC del compuesto 1.
La figura 92 ilustra una gráfica isotérmica de DVS del compuesto 1.
La figura 93 ilustra una RMN 1H del compuesto 1.
La figura 94 ilustra un patrón de XRPD de la sal de HCl del compuesto 1 aislado del estudio de solubilidad en la SGF.
La figura 95 ilustra un termograma de TGA de la sal de HCl del compuesto 1 aislado del estudio de solubilidad en la SGF.
La figura 96 ilustra un termograma de DSC de la sal de HCl del compuesto 1 aislado del estudio de solubilidad en la SGF.
La figura 97 ilustra una gráfica isotérmica de DVS de la sal de HCl del compuesto 1 aislada del estudio de solubilidad en la SGF.
La figura 98 ilustra una RMN 1H en D6-DMSO de la sal de HCl del compuesto 1 a partir de la solubilidad en la SGF.
La figura 99 ilustra una RMN 1H en D6-DMSO de la sal de sulfato del compuesto 1 a partir de SVSS pocillo n.° H2.
La figura 100 ilustra un termograma de TGA de la sal de sulfato, SVSS pocillo n.° A2.
La figura 101 ilustra un termograma de DSC de la sal de sulfato, SVSS pocillo n.° A2.
La figura 102 ilustra un termograma de TGA de la sal de sulfato, SVSS pocillo n.° D2.
La figura 103 ilustra un termograma de DSC de la sal de sulfato, SVSS pocillo n.° D2.
La figura 104 ilustra un termograma de TGA de la sal de sulfato, SVSS pocillo n.° G2.
La figura 105 ilustra un termograma de DSC de la sal de sulfato, SVSS pocillo n.° A2.
La figura 106 ilustra un patrón de XRPD de sales de mesilato de un estudio de SVSS en EtOAc.
La figura 107 ilustra una RMN 1H en D6-DMSO de la sal de mesilato del compuesto 1 a partir de SVSS pocillo n.° B4.
La figura 108 ilustra un termograma de TGA de la sal de mesilato, SVSS pocillo n.° A4.
La figura 109 ilustra un termograma de DSC de la sal de mesilato, SVSS pocillo n.° A4.
La figura 110 ilustra un termograma de TGA de la sal de mesilato, SVSS pocillo n.° B4.
La figura 111 ilustra una gráfica isotérmica de DVS de la sal de mesilato, SVSS pocillo n.° B4.
La figura 112 ilustra un termograma de TGA de la sal de mesilato, SVSS pocillo n.° E4.
La figura 113 ilustra un termograma de DSC de la sal de mesilato, SVSS pocilio n.° E4.
La figura 114 ilustra un termograma de TGA de  sal de mesilato, SVSS pocillo n.° G4.
sal de mesilato, SVSS pocillo n.° G4.
La figura 115 ilustra un termograma de DSC de la sal de mesilato, SVSS pocillo n.° G4.
La figura 116 ilustra un termograma de TGA de  sal de mesilato, SVSS pocillo n.° H4.
sal de mesilato, SVSS pocillo n.° H4.
La figura 117 ilustra un termograma de DSC de la sal de mesilato, SVSS pocillo n.° H4.
La figura 118 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en etanol.
La figura 119 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en IPA.
La figura 120 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en 3-metil-2-butanol. La figura 121 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en acetonitrilo.
La figura 122 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en MTBE.
La figura 123 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en acetona.
La figura 124 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en agua.
La figura 125 ilustra un patrón de XRPD de la sal de citrato del compuesto 1 de SVSS en EtOAc.
La figura 126 ilustra perfiles de comparación de XRPD de sales de citrato de SVSS en etanol, IPA, MTBA y acetona.
La figura 127 ilustra perfiles de comparación de XRPD de sales de citrato de SVSS en 3-metil-2-butanol y acetonitrilo.
La figura 128 ilustra perfiles de comparación de XRPD de sales de citrato de SVSS en EtOAc e IPA.
La figura 129 ilustra una RMN 1H en D6-DMSO de la sal de citrato del compuesto 1 a partir del SVSS, pocillo n.° D9.
La figura 130 ilustra un termograma de TGA de la sal de citrato a partir del SVSS, pocillo n.° A9.
La figura 131 ilustra un termograma de DSC de la sal de citrato a partir del SVSS, pocillo n.° A9.
La figura 132 ilustra un termograma de TGA de la sal de citrato a partir del SVSS, pocillo n.° B9.
La figura 133 ilustra un termograma de TGA de la sal de citrato a partir del SVSS, pocillo n.° B9.
La figura 134 ilustra un termograma de TGA de la sal de citrato a partir del SVSS, pocillo n.° E9.
La figura 135 ilustra un termograma de DSC de la sal de citrato a partir del SVSS, pocillo n.° D9.
La figura 136 ilustra un termograma de TGA de la sal de citrato a partir del SVSS, pocillo n.° G9.
La figura 137 ilustra un termograma de DSC de la sal de citrato a partir del SVSS, pocillo n.° G9.
La figura 138 ilustra un termograma de TGA de la sal de citrato a partir del SVSS, pocillo n.° H9.
La figura 139 ilustra un termograma de DSC de la sal de citrato a partir del SVSS en EtOAc, pocillo n.° H9. La figura 140 ilustra una RMN 1H en D6-DMSO del compuesto 1 y ácido fosfórico a partir de SVSS, pocillo n.° E7. La figura 141 ilustra una RMN 1H en D6-DMSO del compuesto 1 y ácido málico a partir de SVSS, pocillo n.° G10. La figura 142 ilustra una RMN 1H en D6-DMSO del compuesto 1 y ácido glicólico a partir de SVSS, pocillo n.° G11.
La figura 143 ilustra un patrón de XRPD de sal de HCl (superior) en comparación con el de la solubilidad de la
base libre en SGF (inferior).
La figura 144 ilustra un termograma de TGA de la sal de HCl, monohidrato (1).
La figura 145 ilustra un termograma de DSC de la sal de HCl, monohidrato (1).
La figura 146 ilustra un patrón de XRPD de la sal de HCl.
La figura 147 ilustra un termograma de TGA de la sal de HCl.
La figura 148 ilustra un termograma de DSC de la sal de HCl.
La figura 149 ilustra un patrón de XRPD del compuesto 1 secado a 40 °C al vacío.
La figura 150 ilustra un termograma de TGA del compuesto 1 secado a 40 °C al vacío.
La figura 151 ilustra un termograma de DSC del compuesto 1 secado a 40 °C al vacío.
La figura 152 ilustra un patrón de XRPD del monohidrato de sal de HCl del compuesto 1 tras calentar a 140 °C en la etapa de XRD-DSC.
La figura 153 ilustra un patrón de XRPD del sulfato del compuesto 1.
La figura 154 ilustra un termograma de TGA del sulfato del compuesto 1.
La figura 155 ilustra un termograma de DSC del sulfato del compuesto 1.
La figura 156 ilustra un patrón de XRPD del mesilato del compuesto 1.
La figura 157 ilustra un termograma de TGA del mesilato del compuesto 1.
La figura 158 ilustra un termograma de DSC del mesilato del compuesto 1.
La figura 159 ilustra un patrón de XRPD del mesilato del compuesto 1.
La figura 160 ilustra un termograma de TGA del mesilato del compuesto 1.
La figura 161 ilustra un termograma de DSC del mesilato del compuesto 1.
La figura 162 ilustra un patrón de XRPD del mesilato del compuesto 1 tras la suspensión en agua.
La figura 163 ilustra una gráfica isotérmica de DVS de la sal de mesilato del compuesto 1.
La figura 164 ilustra un patrón de XRPD del mesilato del compuesto 1 tras el estudio de DVS.
La figura 165 ilustra un patrón de XRPD del citrato del compuesto 1 en el sistema de EtOAc-agua.
La figura 166 ilustra un termograma de TGA del citrato del compuesto 1 en el sistema de EtOAc-agua. La figura 167 ilustra un termograma de DSC del citrato del compuesto 1 en el sistema de EtOAc-agua. La figura 168 ilustra un patrón de XRPD del citrato del compuesto 1 en acetona.
La figura 169 ilustra un termograma de TGA del citrato del compuesto 1 en acetona.
La figura 170 ilustra un termograma de DSC del citrato del compuesto 1 en acetona.
La figura 171 ilustra un patrón de XRPD del citrato del compuesto 1.
La figura 172 ilustra un termograma de TGA del citrato del compuesto 1.
La figura 173 ilustra un termograma de DSC del citrato del compuesto 1.
La figura 174 ilustra un patrón de XRPD del citrato del compuesto 1.
La figura 175 ilustra un termograma de TGA del citrato del compuesto 1.
La figura 176 ilustra un termograma de DSC del citrato del compuesto 1.
La figura 177 ilustra un patrón de XRPD del citrato del compuesto 1.
La figura 178 ilustra un termograma de TGA del citrato del compuesto 1.
La figura 179 ilustra un termograma de DSC del citrato del compuesto 1.
La figura 180 ilustra un patrón de XRPD del citrato del compuesto 1.
La figura 181 ilustra un termograma de TGA del citrato del compuesto 1.
La figura 182 ilustra un termograma de DSC del citrato del compuesto 1.
La figura 183 ilustra una gráfica isotérmica de DVS de la sal de citrato del compuesto 1.
La figura 184 ilustra una disolución de base libre (FB, por sus siglas en inglés), citrato y sal de HCl en solución de HCl 0,01 N.
La figura 185 ilustra una disolución de base libre (FB) del compuesto 1, citrato y sal de HCl en solución de HCl 0,001 N.
La figura 186 ilustra una solubilidad cinética de base libre (FB), citrato y sal de HCl en FeSSIF.
La figura 187 ilustra una solubilidad cinética de base libre (FB), citrato y sal de HCl en FaSSIF.
La figura 188 ilustra cómo el tratamiento con el compuesto 1 provoca una inhibición sostenida del sustrato de ERK pRSK1 S380 en células Colo 205 (BRAFV600E mut). Las células Colo 205 se trataron con DMSO o compuesto 10,5 gM durante el tiempo indicado. Se midió el pRSK1 S380 con un ensayo de MSD (superior). DUSP4 y DUSP6 se detectaron mediante transferencia Western (inferior).
Las figuras 189A a 189I ilustran cómo el compuesto 1 inhibe de forma potente la señalización de MAP cinasa y genes objetivo corriente abajo en Colo 205. Se trataron cultivos de la línea celular de cáncer de colon Colo 205 (BRAF V600E) con DMSO o concentraciones en aumento del compuesto 1 durante 2, 8 o 24 h. La figura 189A ilustra proteínas extraídas de células tratadas y analizadas mediante transferencia Western, utilizando anticuerpos contra DUSP4, DUSP6, ciclina D1, c-Myc, YAP o p-actina. Las figuras 189B a 189C ilustran ARN extraídos utilizando el kit de célula a CT y se realizó PCR cuantitativa con sondas específicas para DUSP4, DUSP6, SPRY2, c-Myc y ciclina D1. Para la normalización, se utilizaron sondas específicas para p-actina. Las figuras 189D a 189I ilustran cómo el tratamiento con el compuesto 1 modula los niveles de ARNm impulsados por MAPK en células Colo 205 (BRAFV600E mut) y HT-29 (BRAFV600E mut). Las células Colo 205 o HT-29 se trataron con DMSO o compuesto 10,3 o 1 gM durante 6 h. Se extrajo ARNm utilizando el kit de aislamiento de ARN MagMAX Total y se realizó PCR cuantitativa.
La figura 190A ilustra los efectos del compuesto 1 sobre genes objetivo de la ruta de señalización HIPPO/YAP y WNT/beta-catenina en Colo 205. Se trataron cultivos de la línea celular de cáncer de colon Colo 205 (BRAF V600E) con DMSO o concentraciones en aumento de compuesto 1 durante 2, 8 o 24 h. Se extrajo ARN utilizando el kit de célula a CT y se realizó PCR cuantitativa con sondas específicas para Axina2, CTGF y AREG. Para la normalización, se utilizaron sondas específicas para p-actina. La figura 190B a 190E ilustran cómo el tratamiento con el compuesto 1 regula los niveles de ARNm impulsados por YAP en células Colo 205 (BRAFV600E mut) y hT-29 (BRAFV600E mut). Las células Colo 205 o hT-29 se trataron con DMSO o compuesto 10,3 o 1 gM durante 6 h. Se extrajo ARN utilizando el kit de aislamiento de ARN MagMAX Total y se realizó PCR cuantitativa.
Las figuras 191A a 191B ilustran cómo el compuesto 1 regula en disminución el nivel de PD-L1 en múltiples líneas celulares cancerosas. La figura 191A ilustra transferencia Western de PD-L1 total en Hop66, Karpas-299 y LOX-IMVI. Las células se cultivaron con o sin el compuesto 1 durante el tiempo indicado antes de medir los niveles de expresión de PD-L1, DUSP4 y a-tubulina o a-actina mediante transferencia Western. la figura 191B ilustra la tinción de superficie de PD-L1 con selección de células activadas por fluorescencia (FACS, por sus siglas en inglés). Las células se trataron con DMSO o compuesto 1 en las concentraciones indicadas durante 48 h y se detectó la expresión de PD-L1 en la superficie celular utilizando el análisis FACS con un anticuerpo para PD-L1 marcado con APC (clon 29E.1A3.; BioLegend, San Diego, CA). Se determinó la media geométrica de células positivas para PD-L1 mediante FlowJo 10 (Treestar, Ashland, OR).
Las figuras 192A a 192B ilustran cómo las células KARPAS-299 tratadas con el compuesto 1 aumentan la producción de IL-2 (Figura 192A) e IFNy (Figura 192B) mediante linfocitos T derivados de PBMC y estimulados
con superantígeno (SEB) in vitro. Las células KARPAS-299 se trataron con DMSO (D) o compuesto 1 en las concentraciones indicadas durante 48 h. Se trataron PBMC de donantes saludables con o sin 20 ng/ml de SEB durante 48 h. Tras el lavado con PBS, las PBMC se incubaron con las células cancerosas durante 24 h y se recogieron los sobrenadantes para medir la IL-2 y el IFNy utilizando ensayos de MSD. La figura 192 C ilustra el efecto del tratamiento con el compuesto 1 sobre los niveles de IL-8, determinados en medios de cultivo de PBMC. Se aislaron PBMC de sangre entera y se cultivaron en medio RPMI más FBS al 10 %. Las PBMC se colocaron en placas a 1 x 106 por mililitro en platillos de 10 cm2. Las PBMC se trataron con DMSO al 0,1 % o compuesto 1 0,5 pM. Los tratamientos se retiraron en los puntos temporales indicados. Las PBMC se sedimentaron y utilizaron para análisis de transferencia Western y se tomó 1 ml del medio de cultivo para análisis de IL-8. El análisis de IL-8 se realizó con un kit de IL-8 humana Mesoscale V-Plex, de acuerdo con las instrucciones del fabricante. Se mostró que el compuesto 1 inhibe los niveles de IL-8 en diferentes puntos temporales.
La figura 193 ilustra la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto LOX-IMVI. Se inocularon ratones SCID hembra con 1 x 106 células tumorales LOX-IMVI en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 9/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 13, con tumores con un tamaño aproximado de 240 mm3.
La figura 194 ilustra la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto LOX IMVI. Los ratones hembra con inmunodeficiencia combinada grave (SCID, por sus siglas en inglés) se inocularon con 1 x 106 células tumorales LOX-IMVI en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 13, con tumores con un tamaño aproximado de 300 mm3. Se calculó el porcentaje de inhibición con respecto al control con vehículo el último día del estudio y se incluye entre paréntesis junto al volumen tumoral respectivo para los grupos de tratamiento. La línea punteada es el volumen tumoral al inicio de la dosificación.
La figura 195 ilustra la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto de Colo 205. Se inocularon ratones SCID hembra con 2 x 106 células tumorales Colo 205 en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 10, con tumores con un tamaño aproximado de 160 mm3. Se calculó el porcentaje de inhibición con respecto al control con vehículo el último día del estudio y se incluye entre paréntesis junto al volumen tumoral respectivo para los grupos de tratamiento. La línea punteada es el volumen tumoral al inicio de la dosificación.
La figura 196 ilustra la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto de Colo 205. Se inocularon ratones SCID hembra con 2 x 106 células tumorales Colo 205 en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 10, con tumores con un tamaño aproximado de 130 o 160 mm3. Se calculó el porcentaje de inhibición con respecto al control con vehículo el último día del estudio y se incluye entre paréntesis junto al volumen tumoral respectivo para los grupos de tratamiento. La línea punteada es el volumen tumoral al inicio de la dosificación.
Las figuras 197A a 197 B ilustran la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto PDX146. Se inocularon ratones NSG hembra con 25 gg de tumor PDX146 en una suspensión celular en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 8 a 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 19, con tumores con un tamaño aproximado de 100 a 110 mm3. La figura 197A ilustra el volumen tumoral en función del tiempo. La figura 10B ilustra el volumen tumoral individual el último día del estudio, día 40. Se calculó el porcentaje de inhibición con respecto al control con vehículo el último día del estudio y se incluye entre paréntesis junto al volumen tumoral respectivo para los grupos de tratamiento. La línea punteada es el volumen tumoral al inicio de la dosificación. Camp = camptosar.
La figura 198 ilustra el retraso del crecimiento tumoral con tratamiento continuo con el compuesto 1 en el modelo de xenoinjerto PDX146. Se inocularon ratones NSG hembra con 25 gg de tumor PDX146 en una suspensión celular en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 8 a 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 16, con tumores con un tamaño aproximado de 100 a 110 mm3. La línea punteada de color negro es el volumen tumoral al inicio de la dosificación y la línea punteada de color rojo es el volumen tumoral el día 43, al finalizar el grupo de control con vehículo.
Las figuras 199A a 199D ilustran cómo dosis individuales del compuesto 1 inhiben biomarcadores en las rutas de señalización de Hippo, Wnt y MAPK en el modelo de xenoinjerto PDX146: Modulación de las rutas de Hippo, Wnt y MAPK en tumores PDX146 tratados con el compuesto 1. Se realizaron ensayos qRT-PCR en ARN extraído de tumores PDX146 en los puntos temporales indicados después de la dosis. Los datos se expresan como media ± SEM. Los valores P se derivan de un ANOVA de una sola ruta, con análisis post-hoc de Dunnet.
Las figuras 200A a 200D ilustran cómo el compuesto 1 inhibe los biomarcadores en las rutas de señalización de Hippo, Wnt y MAPK de tumores PDX146 tras una administración de dosis única: Modulación de las rutas de Hippo, Wnt y MAPK en tumores PDX146 tratados con el compuesto 1. Se realizaron ensayos qRT-PCR en ARN extraído de tumores PDX146 en los puntos temporales indicados después de la dosis. Se generaron datos de YAP a partir del análisis de transferencia Western de los tumores del grupo de tratamiento con 5 mg/kg y se expresan en forma de una relación de expresión de proteína YAP a p-actina. Los datos se expresan como media ± SEM. Los valores P se derivan de un ANOVA de una sola ruta, con análisis post-hoc de Dunnet.
Las figuras 201A a 201D ilustran cómo los niveles de proteína fosfo-RSK (pRSK) y fosfo-ERK (pERK), biomarcadores de la ruta de señalización de MAPK, se vieron modulados con una administración de dosis única del compuesto 1. Se realizaron ensayos de transferencia Western (pRSK) o Mesoscale (pERK) en proteínas extraídas de tumores PDX146 en el punto temporal indicado después de la dosis. Los datos de fosfo-RSK se expresan como el % del control con vehículo. Los datos de fosfo-ERK se expresan como media ± SEM.
Las figuras 202A a 202B ilustran la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto colorrectal SW48 con mutación de p-catenina. Se inocularon ratones SCID hembra con 2 x 106 células tumorales SW48 en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó en el día 10, con tumores con un tamaño aproximado de 110 y 105 mm3 (Figura 202A y figura 202B, respectivamente). La línea punteada de color negro es el volumen tumoral al inicio de la dosificación. La gráfica a la izquierda es el estudio de respuesta a la dosis (gráfica A). La gráfica a la derecha es el estudio de tiempo hasta la evolución, en donde los animales recibieron el fármaco durante todo el transcurso del estudio (gráfica B). La línea punteada es el volumen tumoral al día 28, cuando finalizó el grupo de control con vehículo.
La figura 203 ilustra la actividad antitumoral en el xenoinjerto de carcinoma hepatocelular Hep3B2.1-7 ortotópico. Se inocularon ratones SCID hembra de forma ortotópica con 2 x 106 células tumorales Hep3B2.1-7 por animal. Siete días después de la inoculación, los animales se separaron de forma aleatoria en grupos de tratamiento según el peso corporal y comenzó el tratamiento (día 0 del estudio). La evaluación de la tasa de absorción de un grupo satélite confirmó la presencia de tumor en el hígado en el 100 % de los animales. El compuesto 1 se administró por vía oral, QD, durante 21 días. Los tumores se retiraron y pesaron el último día del estudio. Se representan en un gráfico los pesos tumorales individuales y el peso tumoral medio ± SEM de cada grupo. Se calculó el porcentaje de inhibición con respecto al control con vehículo y este supera el peso tumoral respectivo de los grupos de tratamiento. Los valores P se derivan de un ANOVA de una sola vía, con análisis post-hoc de Dunnet. *** = p < 0,001. El compuesto 1 mostró una reducción estadísticamente significativa en el peso tumoral, en comparación con los controles con vehículo.
La figura 204 ilustra la actividad antitumoral del compuesto 1 en el modelo de xenoinjerto derivado de pacientes con carcinoma hepatocelular amplificado C-Met, LI0612. Se inocularon ratones SCID hembra con fragmentos de tumor LI0612 del modelo PDX de carcinoma hepatocelular (2 a 4 mm de diámetro) en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) al inicio del tratamiento. El tratamiento con el artículo de prueba comenzó el día 18, con tumores con un tamaño de aproximadamente 150 mm3. El crecimiento tumoral avanzó en los grupos de tratamiento con el compuesto 1 y control con vehículo durante el período de dosificación. Se observó un cambio en la cinética de crecimiento con la administración del compuesto 1, lo cual produjo una inhibición del crecimiento tumoral (TGI, por sus siglas en inglés) considerable con 30 mg/kg de tratamiento (p = 0,038, en comparación con el control con vehículo).
La figura 205 ilustra la sensibilidad de las líneas celulares con mutaciones de p-catenina con respecto al tratamiento con el compuesto 1 y muestra que las líneas celulares con mutación de p-catenina suelen presentar mayor sensibilidad al tratamiento con el compuesto 1.
Las figuras 206A a 206E ilustran la sensibilidad y resistencia de la línea celular al tratamiento con el compuesto 1. Las figuras 206A a 206C muestran que las líneas celulares con mutaciones de BRAF y CTNNB1 presentan mayor sensibilidad al tratamiento con el compuesto 1 que las líneas celulares con BRAF y CTNNB1 de tipo salvaje. La figura D y la figura E muestran que las líneas celulares con mutaciones en RB y la ruta PI3K/PTEN se encuentran asociadas con la resistencia al tratamiento con el compuesto 1 in vitro.
La figura 207 ilustra cómo el compuesto 1 modula MAPK, p-catenina y YAP en la línea celular SW48 con mutación de BRAF y CTNNB1.
Las figuras 208A a 208B ilustran cómo el compuesto 1 modula la expresión del gen objetivo controlada por MAPK, p-catenina y YAP en la línea celular SW48 con mutación de BRAF y CTNNB1.
La figura 209 ilustra que el compuesto 1 inhibe la expresión de Axina2 en células epiteliales bronquiales humanas. La expresión génica se midió a las 24 horas.
Las figuras 210A a 210D ilustran que el compuesto 1 inhibe la formación de colonias de células con mutación de
b-catenina en niveles mayores que los inhibidores de MEK (trametinib) y ERK (GDC0994). La figura 210A muestra la inhibición de formación de colonias de células SW48 (colo). La figura 210B muestra la inhibición de formación de colonias de células HTC-116 (colo). La figura 210C muestra la inhibición de formación de colonias de células AGS (gástricas). La figura 210D muestra la inhibición de formación de colonias de células Hep3B (HCC).
La figura 211 ilustra que las células AGS resistentes al inhibidor de MEK trametinib presentan sensibilidad al compuesto 1 en un ensayo de formación de colonias.
La figura 212 ilustra la actividad del informante TEAD en células 8xGTIIC-luciferasa WI38 VA13 tratadas con el compuesto 1 y trametinib durante 72 horas. Se analizó la actividad de luciferasa utilizando el ensayo de luciferasa Bright Glo (Promega). El compuesto 1 inhibió la actividad del informante TEAD, con una CI50 promedio de >10 gM en el ensayo de 24 horas y una CI50 promedio de 1,85 gM en el ensayo de 72 horas (datos acumulados de tres experimentos). La viabilidad no se vio afectada de manera reproducible por el compuesto 1 en ninguno de los tres ensayos. Trametinib no inhibió la actividad del informante TEAD a las 24 o 72 horas. Descripción detallada
DEFINICIONES
Tal como se usa en el presente documento y en la memoria descriptiva y las reivindicaciones adjuntas, los artículos indefinidos "un" y "una" y el artículo definido "el/la" incluyen referentes en plural así como en singular, salvo que el contexto indique claramente lo contrario.
Tal como se utilizan en el presente documento y salvo que se especifique lo contrario, las expresiones "aproximadamente el" y "aproximadamente", cuando se utilizan en conexión con dosis, cantidades o porcentajes en peso de ingredientes de una composición o una forma de dosificación, significan una dosis, cantidad o porcentaje en peso que, según reconoce el experto en la técnica, brinda un efecto farmacológico equivalente al obtenido con la dosis, cantidad o porcentaje en peso especificados. En determinadas realizaciones, los términos "aproximadamente el" y "aproximadamente", cuando se utilizan en este contexto, contemplan una dosis, cantidad o porcentaje en peso dentro del 30 %, dentro del 20 %, dentro del 15 %, dentro del 10 % o dentro del 5 % de la dosis, cantidad o porcentaje en peso especificados.
Tal como se usa en el presente documento y a menos que se indique lo contrario, las expresiones "aproximadamente el" y "aproximadamente", en conexión con un valor numérico o un intervalo de valores que se brindan para caracterizar una forma sólida particular, p. ej., una temperatura o un intervalo de temperaturas específicos tal como, por ejemplo, que describe una temperatura de fusión, deshidratación, evaporación de disolvente o transición vítrea; un cambio de masa tal como, por ejemplo, un cambio de masa en función de la temperatura o la humedad; un contenido de disolvente o agua en términos de, por ejemplo, masa o un porcentaje, o una posición pico, tal como, por ejemplo, en análisis mediante, por ejemplo, IR, espectroscopía Raman o XRPD, indican que el valor o intervalo de valores se puede desviar en un grado considerado razonable por un experto en la técnica sin dejar de describir la forma sólida particular. Las técnicas para caracterizar los sólidos de formas cristalinas y formas amorfas incluyen, pero no se limitan a, análisis termogravimétrico (TGA, por sus siglas en inglés), calorimetría de barrido diferencial (DSC, por sus siglas en inglés), difractometría en polvo de rayos X (XRPD, por sus siglas en inglés), difractometría de rayos X con cristal único, espectroscopía vibracional, p. ej., espectroscopía infrarroja (IR) y Raman, espectroscopía de resonancia magnética nuclear (RMN) de estado sólido y solución, microscopía óptica, microscopía óptica en etapa caliente, microscopía electrónica de barrido (SEM, por sus siglas en inglés), análisis cuantitativo y cristalografía de electrones, análisis de tamaño de partícula (PSA, por sus siglas en inglés), análisis de área de superficie, estudios de solubilidad y estudios de disolución. En determinadas realizaciones, las expresiones "aproximadamente el" y "aproximadamente", cuando se usan en este contexto, indican que el valor numérico o intervalo de valores puede variar dentro de un 30 %, 20 %, 15 %, 10 %, 9 %, 8 %, 7 %, 6 %, 5 %, 4 %, 3 %, 2 %, 1,5 %, 1 %, 0,5 % o 0,25 % del valor o intervalo de valores mencionados. Por ejemplo, en algunas realizaciones, el valor de una posición pico de XRPD puede variar en hasta ±0,1° 20 (o ±0,2° 20) e igualmente describir el pico de XRPD particular.
Tal como se utiliza en el presente documento y salvo que se especifique lo contrario, un sólido cristalino "puro", es decir, sustancialmente libre de otros sólidos cristalinos o amorfos, contiene menos de alrededor el 10 % en peso de uno o más de otros sólidos cristalinos o amorfos adicionales, menos aproximadamente el 5 % en peso de uno o más de otros sólidos cristalinos o amorfos, menos de alrededor el 3 % en peso de uno o más de otro sólidos cristalinos o amorfos o menos de alrededor el 1 % en peso de uno o más de otros sólidos cristalinos o amorfos.
Tal como se utiliza en el presente documento y salvo que se especifique lo contrario, una forma sólida que es "sustancialmente pura a nivel físico" se encuentra sustancialmente libre de otras formas sólidas. En determinadas realizaciones, una forma cristalina que es sustancialmente pura a nivel físico contiene menos aproximadamente el 10 %, 9 %, 8 %, 7 %, 6 %, 5 %, 4 %, 3 %, 2 %, 1 %, 0,5 %, 0,4 %, 0,3 %, 0,2 %, 0,1 %, 0,05 %, o 0,01 % de una o más de otras formas sólidas en base al peso. La detección de otras formas sólidas puede lograrse mediante
cualquier método evidente para un experto en la técnica que incluye, pero no se limita a, análisis por difracción, análisis térmico, análisis de combustión elemental y/o análisis espectroscópico.
Tal como se utiliza en el presente documento y salvo que se especifique lo contrario, una forma sólida que es "sustancialmente pura a nivel químico" se encuentra sustancialmente libre de otros compuestos químicos (es decir, impurezas químicas). En determinadas realizaciones, una forma sólida que es sustancialmente pura a nivel químico contiene menos de aproximadamente el 10 %, 9 %, 8 %, 7 %, 6 %, 5 %, 4 %, 3 %, 2 %, 1 %, 0,5 %, 0,4 %, 0,3 %, 0,2 %, 0,1 %, 0,05 %, o 0,01 % de uno o más de otros compuestos químicos en base al peso. La detección de otras formas sólidas puede lograrse mediante cualquier método evidente para un experto en la técnica que incluye, pero no se limita a, métodos de análisis químico tales como, p. ej., análisis de espectrometría de masa, análisis espectroscópico, análisis térmico, análisis de combustión elemental y/o análisis cromatográfico.
Tal como se utiliza en el presente documento y salvo que se indique lo contrario, un compuesto químico, una forma sólida o una composición que está "sustancialmente libre" de otro compuesto químico, forma sólida o composición significa que el compuesto, la forma sólida o la composición contiene, en determinadas realizaciones, menos de aproximadamente el 10 %, 9 %, 8 %, 7 %, 6 %, 5 %, 4 %, 3 %, 2 %, 1 %, 0,5 %, 0,4 %, 0,3 %, 0,2 % 0,1 %, 0,05 % o 0,01 % en peso del otro compuesto, forma sólida o composición.
Salvo que se especifique lo contrario, los términos "solvato" y "solvatado/a" según se utilizan en el presente documento hacen referencia a una forma sólida de una sustancia que contiene disolvente. Los términos "hidrato" e "hidratado" hacen referencia a un solvato en donde el disolvente es agua. "Polimorfos de solvatos" hace referencia a la existencia de más de una forma sólida para una composición de solvatos particular. De forma similar, "polimorfos de hidratos" hace referencia a la existencia de más de una forma sólida para una composición de hidratos particular. La expresión "solvato desolvatado", tal como se usa en el presente documento, hace referencia a una forma sólida de una sustancia que se puede elaborar eliminando el disolvente de un solvato. Los términos "solvato" y "solvatado/a", tal como se utilizan en el presente documento, también pueden hacer referencia a un solvato de una sal, un cocristal o un complejo molecular. Los términos "hidrato" e "hidratado/a", tal como se utilizan en el presente documento, también pueden hacer referencia a un hidrato de una sal, un cocristal o un complejo molecular.
"Tautómeros" hace referencia a formas isoméricas de un compuesto que están en equilibrio entre sí. Las concentraciones de las formas isoméricas dependerán del entorno en donde se encuentre el compuesto y pueden diferir dependiendo, por ejemplo, de si el compuesto es un sólido o se encuentra en una solución orgánica o acuosa. Por ejemplo, en solución acuosa, los pirazoles pueden presentar las siguientes formas isoméricas, que se indican como tautómeros entre sí:

Como comprenderá fácilmente un experto en la técnica, una amplia variedad de grupos funcionales y otras estructuras pueden presentar tautomerismo y todos los tautómeros del compuesto 1 se encuentran dentro del alcance de la presente invención.
Salvo que se especifique lo contrario, el término "composición", tal como se utiliza en el presente documento, pretende incluir un producto que comprende el o los ingredientes especificados (en la o las cantidades especificadas, si se indicaran), así como cualquier producto que resulte, de forma directa o indirecta, de la combinación del o de los ingredientes especificados en la o las cantidades especificadas. Por "farmacéuticamente aceptable" se refiere a un diluyente, excipiente o vehículo en una formulación que debe ser compatible con los demás ingredientes de la formulación y no debe ser perjudicial para sus receptores.
La expresión "forma sólida" hace referencia a una forma física que no está predominantemente en un estado líquido o gaseoso. Las expresiones "tipo sólido" y "tipo" se utilizan de manera intercambiable en el presente documento con "forma sólida". Tal como se usa en el presente documento y salvo que se indique lo contrario, la expresión "forma sólida", cuando se utiliza en el presente documento para hacer referencia al compuesto 1, se refiere a una forma física que comprende el compuesto 1 que no se encuentra predominantemente en un estado líquido o gaseoso. Una forma sólida puede ser una forma cristalina o una mezcla de esta. En determinadas realizaciones, una forma sólida puede ser un cristal líquido. En determinadas realizaciones, la expresión "formas sólidas comprendiendo el compuesto 1" incluye las formas cristalinas comprendiendo el compuesto 1. En determinadas realizaciones, la forma sólida del compuesto 1 es la forma A, la forma B, la forma C, la forma D, la forma E, la forma F, la forma G, la forma H, la forma I, el sólido amorfo o una mezcla de estos. En una realización, la forma sólida del compuesto 1 es una forma Y de sal de citrato o forma Z de sal de citrato. En determinadas realizaciones, la forma sólida del compuesto 1 es la forma 1 de sal de HCl, la forma 2 de sal de HCl, la forma 3 de sal de HCl, la forma 4 de sal de HCl, la forma 5 de sal de HCl, la forma 6 de sal de HCl, la forma 7 de sal de HCl, la forma 8 de sal de HCl, el sólido amorfo o una mezcla de estos.
Tal como se usa en el presente documento y salvo que se indique lo contrario, el término "cristalino", cuando se usa
para describir un compuesto, una sustancia, una modificación, un material, un componente o un producto, salvo que
se indique lo contrario, significa que el compuesto, la sustancia, la modificación, el material, el componente o el
producto es sustancialmente cristalino según se determina mediante difracción de rayos X. (véase, p. ej.,
Remington: The Science and Practice of Pharmacy, 21a edición, Lippincott, Williams &Wilkins, Baltimore, MD (2005);
The United States Pharmacopeia, 23a ed., 1843-1844 (1995).
Las expresiones "forma cristalina" o "forma de cristal" hacen referencia a una forma sólida que es cristalina. En
determinadas realizaciones, una forma cristalina de una sustancia puede encontrarse sustancialmente libre de
sólidos amorfos y/u otras formas cristalinas. En determinadas realizaciones, una forma cristalina de una sustancia
puede contener menos de aproximadamente el 1 %, menos de aproximadamente el 2 %, menos de
aproximadamente el 3 %, menos de aproximadamente el 4 %, menos de aproximadamente el 5 %, men aproximadamente el 6 %, menos de aproximadamente el 7 %, menos de aproximadamente el 8 %, men aproximadamente el 9 %, menos de aproximadamente el 10 %, menos de aproximadamente el 15 %, menos de
aproximadamente el 20 %, menos de aproximadamente el 25 %, menos de aproximadamente el 30 %, men aproximadamente el 35 %, menos de aproximadamente el 40 %, menos de aproximadamente el 45 % o menos de
aproximadamente el 50 % en peso de uno o más sólidos amorfos y/u otras formas cristalinas. En determinadas
realizaciones, una forma cristalina de una sustancia puede ser físicamente y/o químicamente pura. En determinadas
realizaciones, una forma cristalina de una sustancia puede ser aproximadamente el 99 %, aproximadamente el 98
%, aproximadamente el 97 %, aproximadamente el 96 %, aproximadamente el 95 %, aproximadamente el 94 %,
aproximadamente el 93 %, aproximadamente el 92 %, aproximadamente el 91 % o aproximadamente el 90 % física
y/o químicamente pura.
Salvo que se indique lo contrario, la expresión "amorfo" o "sólido amorfo" significa que la sustancia, el componente o
producto en cuestión no es sustancialmente cristalino, según se determina mediante difracción de rayos X. En
particular, la expresión "sólido amorfo" describe una forma sólida desordenada, es decir, una forma sólida que
carece de orden cristalino de amplio alcance. En determinadas realizaciones, un sólido amorfo de una sustancia
puede encontrarse sustancialmente libre de otros sólidos amorfos y/o formas cristalinas. En determinadas
realizaciones, un sólido amorfo de una sustancia puede contener menos aproximadamente el 1 %, menos
aproximadamente el 2 %, menos aproximadamente el 3 %, menos aproximadamente el 4 %, menos
aproximadamente el 5 %, menos aproximadamente el 10 %, menos aproximadamente el 15 %, menos
aproximadamente el 20 %, menos aproximadamente el 25 %, menos aproximadamente el 30 %, menos
aproximadamente el 35 %, menos aproximadamente el 40 %, menos aproximadamente el 45 % o menos
aproximadamente el 50 % en peso de uno o más de otros sólidos amorfos y/o formas cristalinas en base al peso. En
determinadas realizaciones, un sólido amorfo de una sustancia puede ser física y/o químicamente puro. En
determinadas realizaciones, un sólido amorfo de una sustancia puede ser aproximadamente el 99 %,
aproximadamente el 98 %, aproximadamente el 97 %, aproximadamente el 96 %, aproximadamente el 95 %,
aproximadamente el 94 %, aproximadamente el 93 %, aproximadamente el 92 %, aproximadamente el 91 % o
aproximadamente el 90 % física y/o químicamente puro.
"Tratar", tal como se utiliza en el presente documento, significa un alivio, completo o parcial, de un trastorno, una
enfermedad o una afección o de uno o más de los síntomas asociados con un trastorno, una enfermedad o una
afección o la ralentización o detención del avance o empeoramiento de dichos síntomas, así como el alivio o la
erradicación de la o las causas del trastorno, la enfermedad o la afección en sí misma. En una realización, el
trastorno es un cáncer, en particular, un tumor sólido o un cáncer hematológico. En algunas realizaciones, "tratar"
significa un alivio, completo o parcial, de un cáncer o de los síntomas asociados con un cáncer, en particular, un
tumor sólido o un cáncer hematológico, o una ralentización o detención del avance o empeoramiento de dichos
síntomas.
"Prevenir", tal como se utiliza en el presente documento, significa un método para retrasar y/o evitar la aparición,
recurrencia o dispersión, completa o parcial, de un cáncer, en particular, un tumor sólido o un cáncer hematológico,
impedir que un sujeto tenga un cáncer, en particular, un tumor sólido o un cáncer hematológico o reducir el riesgo de
un sujeto de tener un cáncer, en particular, un tumor sólido o un cáncer hematológico.
La expresión "cantidad eficaz", en conexión con una forma sólida del compuesto 1 significa una cantidad que puede
tratar o prevenir un trastorno, una enfermedad o una afección o síntomas de estos, como se divulga en el presente
documento. Una cantidad eficaz hace referencia a una cantidad con capacidad de tratar o prevenir un cáncer, en
particular, un tumor sólido o un cáncer hematológico, o síntomas de este, según se divulga en el presente
documento. La cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento, por
ejemplo, en una composición farmacéutica, puede encontrarse en un nivel que ejerza el efecto deseado, por
ejemplo, aproximadamente el 0,005 mg/kg del peso corporal de un sujeto a aproximadamente el 100 mg/kg del peso
corporal de un paciente en una dosis unitaria para administración parenteral. Como será evidente para los expertos
en la técnica, se espera que la cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente
documento puede variar dependiendo de la gravedad de la afección que se está tratando.
"Paciente" o "sujeto", según su uso en el presente documento, incluye un animal, que incluye, pero no se limita a, un
animal tal como una vaca, un mono, un caballo, una oveja, un cerdo, un pollo, un pavo, una codorniz, un gato, un perro, un ratón, una rata, un conejo o una cobaya, en una realización un mamífero, en otra realización un ser humano. En una realización, un sujeto es un ser humano que padece o se encuentra en riesgo de padecer cáncer, en particular, un tumor sólido o un cáncer hematológico o síntomas de este. En una realización, un paciente es un ser humano que tiene un cáncer hematológico o un tumor sólido histológica o citológicamente confirmado, incluyendo sujetos que hayan experimentado un avance mientras recibían terapia estándar contra el cáncer (o que no la hayan tolerado) o para quienes no exista terapia estándar contra el cáncer.
Tal como se usa en el presente documento y salvo que se especifique lo contrario, el término "cáncer" describe o se refiere a la afección fisiológica en mamíferos que se caracteriza habitualmente por un crecimiento celular no regulado. Los ejemplos de cánceres incluyen tumores sólidos y cáncer hematológico. En algunas realizaciones, el cáncer es un cáncer primario, en otras, el cáncer es una metástasis.
Tal como se usa en el presente documento, "tumores sólidos" incluye, pero no se limita a, cáncer de vejiga (que incluye, pero no se limita a, cáncer de vejiga superficial), cáncer de mama (que incluye, pero no se limita a, cáncer de mama Her2+, PR+, ER+ y de tipo B luminal), cáncer del sistema nervioso central (que incluye, pero no se limita a, glioblastoma multiforme (GBM), glioma, meduloblastoma y astrocitoma), cáncer colorrectal, cáncer gastrointestinal (que incluye, pero no se limita a, cáncer de estómago, cáncer de esófago y cáncer de recto), cáncer endocrino (que incluyen, pero no se limitan a, cáncer de tiroides y cáncer de glándulas adrenales), cáncer oftálmico (que incluye, pero no se limita a, retinoblastoma), cáncer genitourinario femenino (que incluye, pero no se limita a, cáncer de placenta, útero, vulva, ovarios, cuello del útero), cáncer de cabeza y cuello (que incluye, pero no se limita a, cáncer de faringe, esófago y lengua), cáncer de hígado, cáncer de pulmón (que incluye, pero no se limita a, cáncer de pulmón no microcítico (NSCLC, por sus siglas en inglés), cáncer de pulmón microcítico (SCLC, por sus siglas en inglés), mucoepidermoide, broncogénico, carcinoma de células escamosas (SQCC, por sus siglas en inglés) y anaplásico/NSCLC), cáncer de piel (que incluye, pero no se limita a, melanoma y SQCC), cáncer de tejido blando (que incluye, pero no se limita a, sarcoma, sarcoma de Ewing y rabdomiosarcoma), cáncer óseo (que incluye, pero no se limita a, sarcoma, sarcoma de Ewing y osteosarcoma), cáncer de células escamosas (que incluye, pero no se limita a, cáncer de pulmón, esófago, cuello del útero y cabeza y cuello), cáncer de páncreas, cáncer de riñón (que incluye, pero no se limita a, tumor renal de Wilm y carcinoma de células renales) y cáncer de próstata. En una realización, el tumor sólido no es cáncer de mama triple negativo (TNBC, por sus siglas en inglés). En algunas realizaciones, el tumor sólido es cáncer de mama, cáncer de colon, cáncer de pulmón o cáncer de vejiga. En una realización de este tipo, el tumor sólido es cáncer de vejiga superficial. En otra, el tumor sólido es carcinoma de células escamosas de pulmón. En incluso otra realización, el tumor sólido es cáncer de mama de tipo B luminal.
Tal como se usa en el presente documento, "cáncer hematológico" incluye, pero no se limita a, leucemia (que incluye, pero no se limita a, leucemia linfocítica aguda (ALL, por sus siglas en inglés), leucemia mieloide crónica (CML, por sus siglas en inglés), leucemia de linfocitos T aguda, leucemia precursora de linfocitos B, leucemia promielocítica aguda (APML, por sus siglas en inglés), leucemia de células plasmáticas, leucemia mielomonocítica B, mielomonoblástica/T-ALL, eritroleucemia y leucemia mieloide aguda (AML, por sus siglas en inglés)), linfoma (que incluye, pero no se limita a, linfoma de Hodgkin, linfoma no Hodgkin (NHL, por sus siglas en inglés), linfoma de Burkitt (BL, por sus siglas en inglés), linfoma de linfocitos B, linfoma linfoblástico, linfoma folicular (FL, por sus siglas en inglés), linfoma difuso de células B grandes (DLBCL, por sus siglas en inglés), linfoma inmunoblástico de células grandes) y mieloma múltiple.
En el contexto de un cáncer, la inhibición puede evaluarse mediante inhibición del avance de la enfermedad, inhibición del crecimiento tumoral, reducción del tumor primario, alivio de los síntomas relacionados con el tumor, inhibición de los factores secretados por el tumor (incluidas hormonas secretadas por el tumor, como las que contribuyen al síndrome carcinoide), retraso en la aparición de tumores primarios o secundarios, desarrollo ralentizado de tumores primarios o secundarios, menor aparición de tumores primarios o secundarios, ralentización o menor gravedad de los efectos secundarios de la enfermedad, detención del crecimiento tumoral y regresión de tumores, mayor tiempo hasta el avance (TTP, por sus siglas en inglés), mayor supervivencia libre de avance (PFS, por sus siglas en inglés), mayor supervivencia general (OS, por sus siglas en inglés), entre otros. OS, según se utiliza en el presente documento, significa el tiempo desde el inicio del tratamiento hasta la muerte debida a cualquier causa. TTP, según su uso en el presente documento, significa el tiempo desde el inicio del tratamiento hasta el avance del tumor. El TTP no incluye la muerte. Tal como se usa en el presente documento, PFS significa el tiempo desde el inicio del tratamiento hasta el avance del tumor o la muerte. En una realización, las tasas de PFS se calculan utilizando los cálculos estimativos de Kaplan-Meier. En el extremo, la inhibición completa se indica en el presente documento como prevención o quimioprevención. En este contexto, el término "prevención" incluye prevención completa del inicio de cáncer clínicamente evidente o prevención del inicio de una etapa de un cáncer evidente a nivel preclínico. En esta definición también se pretende incluir la prevención de la transformación en células malignas o la detención o regresión del avance de células premalignas a células malignas. Esto incluye el tratamiento profiláctico de quienes se encuentran en riego de desarrollar un cáncer.
En determinadas realizaciones, el tratamiento de un linfoma puede evaluarse mediante los Criterios del Taller Internacional (IWC, por sus siglas en inglés) para linfoma no Hodgkin (NHL) (véase Cheson BD, Pfistner B, Juweid, ME, et. ál. Revised Response Criteria for Malignant Lymphoma. J. Clin. Oncol: 2007: (25) 579-586), utilizando las
definiciones de respuesta y criterio de valoración que se incluyen a continuación:

Abreviaturas: RT, remisión total; FDG, [18F]fluorodeoxiglucosa; PET (por sus siglas en inglés), tomografía por emisión de positrones; TC, tomografía computarizada; RP, remisión parcial; SPD, suma del producto de los diámetros; EE, enfermedad estable; EP, enfermedad progresiva.

En una realización, el criterio de valoración para el linfoma es la evidencia de beneficios clínicos. El beneficio clínico
puede reflejar una mejora en la calidad de vida o una reducción de los síntomas del paciente, los requisitos de transfusión, la frecuencia de las infecciones u otros parámetros. En este criterio de valoración también se puede utilizar el tiempo hasta la reaparición o el avance de síntomas relacionados con el linfoma.
En determinadas realizaciones, el tratamiento de CLL puede evaluarse mediante las directrices del taller internacional para CLL (véase Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood, 2008; (111) 12: 5446-5456) utilizando las definiciones de respuesta y criterio de valoración ilustradas en dicho documento y, en particular:

Los criterios del grupo A definen la carga tumoral; los criterios del grupo B definen la función del sistema hematopoyético (o la médula). RT (remisión total): se deben cumplir todos los criterios y los pacientes deben carecer de síntomas constitucionales relacionados con la enfermedad; RP (remisión parcial): se deben cumplir al menos dos de los criterios del grupo A más uno de los criterios del grupo B; EE es la ausencia de enfermedad progresiva (EP) e incapacidad de alcanzar al menos una RP; EP: se debe cumplir al menos uno de los criterios anteriores del grupo A o el grupo B. Suma de los productos de múltiples ganglios linfáticos (según la evaluación mediante análisis de TC en ensayos clínicos o mediante examen físico general). Estos parámetros son irrelevantes para algunas categorías de respuesta.
En determinadas realizaciones, el tratamiento de mieloma múltiple puede evaluarse mediante los Criterios de Respuesta Uniformes Internacionales (IURC, por sus siglas en inglés) para mieloma múltiple (véase Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia, 2006; (10) 10: 1-7), utilizando las definiciones de respuesta y criterios de valoración ilustrados a continuación:

continuación

Abreviaturas: RT, respuesta total; FLC, cadena ligera libre (por sus siglas en inglés); RP, respuesta parcial; EE, enfermedad estable; RTe, respuesta total estricta; RPMB, respuesta parcial muy buena; aTodas las categorías de respuesta requieren dos evaluaciones consecutivas realizadas en cualquier momento antes de comenzar cualquier nueva terapia; todas las categorías requieren también ausencia de evidencias conocidas de lesiones óseas progresivas o nuevas en caso de haber realizado estudios radiográficos. No se requieren estudios radiográficos para cumplir con estos requisitos de respuesta; bConfirmación con repetición de biopsia de médula ósea no necesaria; cPresencia/ausencia de células clonales basada en la relación de k/A. Una relación anómala de k/A en la inmunohistoquímica y/o inmunofluorescencia requiere un mínimo de 100 células plasmáticas en análisis. Una relación anómala que refleja la presencia de un clon anormal es una k/A de > 4:1 o < 1:2; dEnfermedad mensurable definida mediante al menos una de las siguientes mediciones: Células plasmáticas de médula ósea > 30 %; Proteína M en suero > 1 g/dl (> 10 gm/l) [10 g/l]; Proteína M en orina > 200 mg/24 h; ensayo de FLC en suero: Nivel de FLC implicado > 10 mg/dl (> 100 mg/l); la relación de FLC en suero proporcionada es anormal.
En determinadas realizaciones, el tratamiento de un cáncer puede evaluarse mediante los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST 1.1, por sus siglas en inglés) (véase Thereasse P., et al. New Guidelines to Evaluate the Response to Treatment in Solid Tumors. J. of the National Cancer Institute; 2000; (92) 205-216 y Eisenhauer E.A., Therasse P., Bogaerts J., et al. New response evaluation criteria in solid tumors: Revised RECIST guideline (versión 1.1). European J. Cancer; 2009; (45) 228-247). Las respuestas generales para todas las posibles combinaciones de respuestas tumorales en lesiones objetivo y no objetivo con o sin aparición de nuevas lesiones son las siguientes:

Con respecto a la evaluación de lesiones objetivo, la respuesta total (RT) es la desaparición de todas las lesiones objetivo, la respuesta parcial (RP) es una disminución de al menos el 30 % en la suma del diámetro más prolongado de lesiones objetivo, con el diámetro más prolongado de suma inicial como referencia, la enfermedad progresiva (EP) es un aumento de al menos 20 % en la suma del diámetro más prolongado de lesiones objetivo, con el diámetro más prolongado de suma más pequeño registrado desde el inicio del tratamiento o la aparición de una o más nuevas lesiones como referencia, y la enfermedad estable (EE) es una reducción insuficiente para calificar como respuesta parcial o un aumento insuficiente para calificar como enfermedad progresiva, con el diámetro más prolongado de suma más pequeño desde el inicio del tratamiento como referencia.
Con respecto a la evaluación de lesiones no objetivo, la respuesta total (RT) es la desaparición de todas las lesiones no objetivo y la normalización del nivel de marcador tumoral; la respuesta incompleta/enfermedad estable (EE) es la persistencia de una o más lesiones no objetivo y/o la conservación del nivel de marcador tumoral por encima de los
límites normales, y la enfermedad progresiva (EP) es la aparición de una o más nuevas lesiones y/o el avance inequívoco de lesiones no objetivo existentes.
Los procedimientos, las convenciones y las definiciones que se describen a continuación sirven para orientar la implementación de las recomendaciones del Grupo de Trabajo para evaluación de respuesta neuro-oncológica (RANO, por sus siglas en inglés) sobre criterios de respuesta para gliomas de alto grado (Wen P., Macdonald, DR., Reardon, DA., et al. Updated response assessment criteria for high-grade gliomas: Response assessment in neurooncology working group. J Clin Oncol 2010; 28: 1963-1972). Las modificaciones principales de los criterios RANO para los criterios de respuestas puntuales (TPR, por sus siglas en inglés) pueden incluir la adición de convenciones operativas para definir cambios en la dosis de glucocorticoides y la eliminación del componente de deterioro clínico del sujeto para enfocarse en evaluaciones radiológicas objetivas. El análisis de RM de referencia se define como la evaluación realizada al final del período de reposo tras la cirugía, antes de comenzar o retomar el tratamiento con compuesto. La RM de referencia se utiliza como referencia para evaluar una respuesta total (RT) y una respuesta parcial (RP). Mientras que la menor SPD (suma de los productos de diámetros perpendiculares) obtenida en el punto de referencia o en evaluaciones posteriores se indica como la evaluación del nadir y se utiliza como referencia para determinar el avance. Durante los 5 días anteriores a cualquier análisis de RM definido en el protocolo, los sujetos no reciben glucocorticoides o se encuentran en una dosis estable de glucocorticoides. Una dosis estable se define como la misma dosis diaria durante los 5 días consecutivos previos al análisis de RM. Si la dosis de glucocorticoides indicada cambia en los 5 días previos al análisis de referencia, se requiere un nuevo análisis de referencia con un uso de glucocorticoides que cumpla con los criterios antedichos. Se utilizan las siguientes definiciones.
Lesiones mensurables: Las lesiones mensurables son lesiones realzadas por contraste y que se pueden medir de forma bidimensional. Se realiza una medición del diámetro máximo de tumor realzado (también conocido como el diámetro más prolongado, LD, por sus siglas en inglés). En la misma imagen se mide el mayor diámetro perpendicular. Los puntos de mira de las mediciones bidimensionales deberían cruzarse y se calcula el producto de estos diámetros.
Diámetro mínimo: Imagen con ponderación de T1, en donde las secciones son de 5 mm, con un salto de 1 mm. El LD mínimo de una lesión mensurable se configura en 5 mm por 5 mm. Se pueden requerir diámetros mayores para inclusión y/o designación como lesión objetivo. Tras la referencia, las lesiones objetivo que se hacen más pequeñas que el requisito mínimo de medición o que ya no permiten medición bidimensional se registrarán con el valor predeterminado de 5 mm para cada diámetro por debajo de 5 mm. Las lesiones que desaparecen se registrarán como 0 mm por 0 mm.
Lesiones multicéntricas: Las lesiones que se consideran multicéntricas (a diferencia de las continuas) son lesiones donde existe tejido cerebral normal intermedio entre las dos (o más) lesiones. Para lesiones multicéntricas que representan focos separados de realce, el enfoque es medir por separado cada lesión realzada que cumpla con los criterios de inclusión. Si no existe tejido cerebral normal entre dos (o más) lesiones, estas no se consideran una misma lesión.
Lesiones no mensurables: Todas las lesiones que no cumplen con los criterios de enfermedades mensurables definidos anteriormente se consideran lesiones no mensurables, así como todas las lesiones no realzadas y otras verdaderamente no mensurables. Las lesiones no mensurables incluyen focos de potenciamiento que son inferiores al diámetro más pequeño indicado (es decir, inferiores a 5 mm por 5 mm), lesiones no realzadas (p. ej., según se observa en las imágenes de recuperación de inversión atenuada de fluido (FLAIR, por sus siglas en inglés), con ponderación de T2 o poscontraste con ponderación de T1), lesiones necróticas, hemorrágicas o predominantemente císticas y tumor leptomeníngeo. Las lesiones hemorrágicas suelen presentar hiperintensidad con ponderación de T1 intrínseca que podría interpretarse incorrectamente como un tumor realzado y, por este motivo, se puede examinar la imagen con ponderación de T1 precontraste para excluir la referencia o la hemorragia subaguda del intervalo o el punto de referencia.
En el punto de referencia, se clasifican las lesiones de la siguiente forma: Lesiones objetivo: Se pueden seleccionar hasta 5 lesiones mensurables como lesiones objetivo, cada una con un tamaño de al menos 10 mm por 5 mm, para representar la enfermedad del sujeto. Lesiones no objetivo: Todas las demás lesiones, incluidas las lesiones no mensurables (lo cual incluye efectos de masa y hallazgos de T2/FLAIR), así como cualquier lesión mensurable no seleccionada como una lesión objetivo. Las lesiones objetivo deben medirse en el momento de referencia como se describe en la definición de lesiones mensurables y se debe determinar el SPD de todas las lesiones objetivo. Es necesario documentar la presencia de cualquier otra lesión. En todas las evaluaciones posteriores al tratamiento, se mantendrá la clasificación de referencia de las lesiones como lesiones objetivo y no objetivo y se realizará la documentación y descripción de las lesiones de manera uniforme durante todo el período (p. ej., se registrarán en el mismo orden en documentos originales y eCRF). Todas las lesiones mensurables y no mensurables deben evaluarse utilizando la misma técnica en el momento de la medición de referencia (p. ej., se debe someter a los sujetos a imagenología en el mismo escáner de RM o al menos con la misma fuerza magnética) durante todo el estudio, para reducir las dificultades a la hora de interpretar los cambios. En cada evaluación, se miden las lesiones objetivo y se calcula el SPD. Las lesiones no objetivo se evalúan de forma cualitativa y las nuevas lesiones, si las
hubiera, se documentan por separado. En cada evaluación, se determina una respuesta puntual para ese momento para las lesiones objetivo, lesiones no objetivo y lesiones nuevas. Se puede establecer el avance tumoral incluso evaluando solamente un subconjunto de lesiones. No obstante, salvo que se observe avance, el estado objetivo (enfermedad estable, RP o RT) solamente puede determinarse evaluando todas las lesiones.
Se realizan evaluaciones de confirmación para las respuestas puntuales globales de RT y RP en la siguiente evaluación programada, pero puede no haber confirmación si los análisis se realizan con un intervalo de < 28 días. La mejor respuesta, que incorpore los requisitos de confirmación, se derivará de la serie de puntos temporales. COMPUESTO 1
Las formas sólidas, las formulaciones y los usos que se proporcionan en el presente documento hacen referencia a formas sólidas (p. ej., polimorfos) del compuesto 1:

de nombre (1 s,4s)-4-(8-((2,4,6-triclorofenil)amino)-2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-9H-purin-9-il)-1 -metilciclohexan-1-carboxamida, denominado de forma alternativa como cis-4-[2-{[(3S,4R)-3-fluorooxan-4-il]amino}-8-(2,4,6-tricloroanilino)-9H-purin-9-il]-1 -metilciclohexan-1 -carboxamida, incluidos sus tautómeros.
FORMAS SÓLIDAS DEL COMPUESTO 1
En determinadas realizaciones, en el presente documento se proporcionan formas sólidas del compuesto 1. En determinadas realizaciones, la forma sólida es cristalina. En determinadas realizaciones, la forma sólida es una forma sólida de componente único. En determinadas realizaciones, la forma sólida es un hidrato. En determinadas realizaciones, la forma sólida es un anhidrato. En determinadas realizaciones, la forma sólida es una sal de HCl del compuesto 1. En determinadas realizaciones, la forma sólida es una sal de citrato del compuesto 1. En determinadas realizaciones, la forma sólida es una sal de mesilato. En determinadas realizaciones, la forma sólida es una sal de sulfato. En determinadas realizaciones, la forma sólida es un solvato.
Sin intención de limitarse a ninguna teoría en particular, determinadas formas sólidas se caracterizan por tener propiedades físicas adecuadas para las formas de dosificación terapéuticas y farmacéuticas, p. ej., estabilidad, solubilidad y velocidad de disolución. Asimismo, sin intención de limitarse a ninguna teoría en particular, determinadas formas sólidas se caracterizan por tener propiedades físicas (p. ej., densidad, compresibilidad, dureza, morfología, escisión, adherencia, solubilidad, absorción de agua, propiedades eléctricas, comportamiento térmico, reactividad en estado sólido, estabilidad física y estabilidad química) que afectan procesos particulares (p. ej., rendimiento, filtración, lavado, secado, molienda, mezcla, formación de comprimidos, fluidez, disolución, formulación y liofilización), lo que hace que determinadas formas sólidas sean adecuadas para la fabricación de una forma de dosificación sólida. Dichas propiedades pueden determinarse utilizando técnicas de química analítica particulares, incluidas técnicas analíticas de estado sólido (p. ej., difracción de rayos X, microscopía, espectroscopía y análisis térmico), tal como se describe en el presente documento y se conoce en la técnica.
Las formas sólidas que se proporcionan en el presente documento (p. ej., forma A, forma B, forma C, forma D, forma E, forma F, forma G, forma H, forma I y el sólido amorfo del compuesto 1, así como la forma 1 de sal de HCl, la forma 2 de sal de HCl, la forma 3 de sal de HCl, la forma 4 de sal de HCl, la forma 5 de sal de HCl, la forma 6 de sal de HCl, la forma 7 de sal de HCl, la forma 8 de sal de HCl y el sólido amorfo de sal de HCl del compuesto 1, además de la forma Y de sal de citrato, la forma Z y el sólido amorfo de sal de citrato del compuesto 1) pueden caracterizarse utilizando diversos métodos conocidos por el experto en la técnica y que incluyen, pero no se limitan a, difracción de rayos X de cristal único, difracción de rayos X en polvo (XRPD, por sus siglas en inglés), microscopía (p. ej., microscopía electrónica de barrido (SEM, por sus siglas en inglés)), análisis térmico (p. ej., calorimetría de barrido diferencial (DSC, por sus siglas en inglés), sorción dinámica de vapores (DVS, por sus siglas en inglés), análisis gravimétrico térmico (TGA, por sus siglas en inglés) y microscopía en estado caliente),
espectroscopia (p. ej., infrarroja, Raman y resonancia magnética nuclear de estado sólido), cromatografía líquida de rendimiento ultraelevado (UHPLC, por sus siglas en inglés) y espectro de resonancia magnética nuclear de protones (RMN 1H, por sus siglas en inglés). El tamaño de partícula y la distribución del tamaño de la forma sólida que se proporciona en el presente documento se pueden determinar con métodos convencionales, tales como la técnica de difracción de luz láser.
La pureza de las formas sólidas que se proporcionan en el presente documento se puede determinar con métodos analíticos estándar, tales como cromatografía de capa fina (TLC, por sus siglas en inglés), electroforesis en gel, cromatografía gaseosa, cromatografía líquida de rendimiento ultraelevado (UHPLC) y espectrometría de masa (MS, por sus siglas en inglés).
Se debe comprender que los valores numéricos de los picos de un patrón de difracción de rayos X en polvo pueden variar levemente entre una máquina y otra o entre una muestra y otra y, por lo tanto, los valores que se indican no se deben interpretar como absolutos, sino con cierta variabilidad, como ±0,2 ° 20 o ±0,1° 20 (véase United State Pharmacopoeia, página 2228 (2003)).
En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) obtener una suspensión del compuesto 1 en un disolvente, 2) agitar la suspensión durante un período de tiempo (p. ej., aproximadamente 24 h) a una temperatura determinada (p. ej., aproximadamente 25 °C o aproximadamente 50 °C) y 3) recoger los sólidos de la suspensión mediante filtración y, opcionalmente, secado, en donde los sólidos pueden ser la forma A. En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) obtener una suspensión del compuesto 1 en un disolvente, 2) agitar la suspensión durante aproximadamente 24 h a aproximadamente 25 °C o a aproximadamente 50 °C y 3) recoger los sólidos de la suspensión mediante filtración, por ejemplo, a través de un filtro con jeringa de PTFE de 0,45 pm y, opcionalmente, secado con aire, en donde los sólidos pueden ser la forma A. En determinadas realizaciones, los métodos para realizar una forma sólida del compuesto 1 son experimentos de equilibrio, como experimentos de suspensión.
En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) disolver el compuesto 1 en un disolvente para brindar una solución, 2) filtrar la solución si el compuesto no se disuelve por completo y 3) evaporar la solución con presión de aire determinada (p. ej., aproximadamente 101,325 kPa (1 atm)) a una temperatura determinada (p. ej., aproximadamente 25 °C o aproximadamente 50 °C) para brindar un sólido que sea opcionalmente la forma A. En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida de la forma A, comprendiendo 1) disolver el compuesto 1 en un disolvente para brindar una solución, 2) filtrar la solución (por ejemplo, a través de un filtro con jeringa de PTFE de 0,45 pm) si la forma A no se disuelve por completo y 3) evaporar la solución con una presión de aire de aproximadamente 101,325 kPa (1 atm) a aproximadamente 25 °C o a aproximadamente 50 °C en nitrógeno para brindar un sólido. En determinadas realizaciones, los métodos para realizar una forma sólida del compuesto 1 son experimentos de evaporación.
En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) obtener una solución saturada de la forma A en un disolvente a una primera temperatura (p. ej., aproximadamente 60 °C), 2) agitar la solución a la primera temperatura durante un período de tiempo (p. ej., 10 minutos), 3) filtrar la solución, 4) enfriar la solución lentamente hasta una segunda temperatura (p. ej., de aproximadamente -5 °C a aproximadamente 15 °C) y 5) aislar los sólidos de la solución y, opcionalmente, secar. En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) obtener una solución saturada de la forma A en un disolvente a aproximadamente 60 °C, 2) agitar la solución a aproximadamente 60 °C durante 10 minutos, 3) filtrar la solución (por ejemplo, a través de un filtro con jeringa de PTFE de 0,45 pm), 4) enfriar la solución lentamente hasta aproximadamente 5 °C y 5) aislar los sólidos de la solución y, opcionalmente, secar con aire. En determinadas realizaciones, los métodos para realizar una forma sólida del compuesto 1 son experimentos de recristalización con enfriamiento.
En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) obtener una solución saturada de la forma A en un disolvente a una primera temperatura (p. ej., aproximadamente 60 °C), 2) agregar un antidisolvente a la solución saturada a la primera temperatura, 3) enfriar hasta una segunda temperatura (p. ej., de aproximadamente -5 °C a aproximadamente 15 °C) y 4) recoger un sólido si hay precipitación y evaporar el disolvente para recoger un sólido si no hay precipitación y 5) opcionalmente secar. En determinadas realizaciones, en el presente documento se proporcionan métodos para realizar una forma sólida del compuesto 1, comprendiendo 1) obtener una solución saturada de la forma A en un disolvente a aproximadamente 60 °C, 2) agregar un antidisolvente a la solución saturada a aproximadamente 60 °C, 3) enfriar hasta aproximadamente 5 °C y 4) recoger un sólido si hay precipitación y evaporar el disolvente para recoger un sólido si no hay precipitación y 5) opcionalmente secar con aire. En determinadas realizaciones, la relación en volumen de disolvente y antidisolvente es aproximadamente 1:9. En determinadas realizaciones, los métodos para realizar una forma sólida del compuesto 1 son experimentos de recristalización con antidisolvente.
En determinadas realizaciones, el disolvente es acetona, DCM, EtOAc, EtOH, EtOH/hbO (aproximadamente 1:1), H2O, heptano, IPA, ACN, ACN/H2O (aproximadamente 1:1), MEK, MeOH, MTBE, n-BuOH, THF, THF/H2O (aproximadamente 1:1), tolueno o sulfolano.
En determinadas realizaciones, el antidisolvente es ACN, heptano, MTBE o agua.
Forma A
En determinadas realizaciones, en el presente documento se proporciona la forma A.
En una realización, la forma A es una forma sólida del compuesto 1. En una realización, la forma A es un monohidrato. En una realización, la forma A es una forma sólida de hidrato del canal no estequiométrico del compuesto 1. En una realización, la forma A es una forma de base libre del compuesto 1. En otra realización, la forma A es cristalina.
En determinadas realizaciones, la forma A que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma A se obtiene a partir de determinados sistemas de disolventes, que incluyen heptano/agua, heptanos, agua, tolueno, MeCN, MeCN/agua, EtOH, EtOH/H2O (aproximadamente 1:1), THF/agua (aproximadamente 1:1) e IPA.
En una realización, un método para preparar la forma A comprende las etapas de poner en contacto el compuesto 1 (p. ej., una forma cristalina del compuesto 1, tal como la forma B, la forma C o la forma H) con condiciones ambiente que comprendan más de aproximadamente el 10 %-20 % de humedad relativa (RH, por sus siglas en inglés).
En una realización, un método para preparar la forma A comprende las etapas de enfriar el compuesto 1 en un disolvente a una temperatura inferior a aproximadamente 50 °C y recoger los sólidos.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma A, es la base libre del compuesto 1 y es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, es un patrón de difracción de rayos X en polvo (XRPD) sustancialmente como se muestra en la figura 2 (p. ej., forma A). En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma A, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 3,2, 7,3, 8,5, 10,7, 11,1, 12,7, 13,0, 13,4, 13,8, 14,5, 14,7, 15,9, 16,9, 17,1, 17,3, 17,7, 18,2, 18,7, 20,3, 20,7, 21,0, 21,3, 22,1, 22,7, 22,9, 23,2, 23,6, 24,0, 24,8, 25,5, 26,1, 26,4, 26,8, 27,9, 28.1, 28,8, 29,4, 29,8, 31,4, 31,8, 32,6, 33,1, 33,6, 33,9, 34,2, 34,7, 36,1, 36,5, 37,2, 37,7, 38,9 o 39,5,220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 2. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once o doce picos característicos de difracción de rayos X en polvo aproximadamente a 7,3, 8,5, 10,8, 14,5, 14,7, 15,9, 16,9, 17,1, 18.2, 21,0, 21,3 o 28,8° 20 (± 0,2° 20). En algunas realizaciones, la forma sólida es la forma A. En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,3, 8,5, 18,2 o 21,3° 20 (± 0,2° 20). En otra realización, la forma A tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta, cuarenta y uno, cuarenta y dos, cuarenta y tres, cuarenta y cuatro, cuarenta y cinco, cuarenta y seis, cuarenta y siete, cuarenta y ocho, cuarenta y nueve, cincuenta, cincuenta y uno o cincuenta y dos picos característicos de difracción de rayos X en polvo, como se indica en la tabla 12.
En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma A, tiene una imagen de SEM sustancialmente como se muestra en la figura 3.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma A, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 4. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 2,8 % de la masa total de la muestra de aproximadamente 50 °C a aproximadamente 175 °C cuando se calienta de aproximadamente 50 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 2,8 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma A, que tiene un termograma de DSC sustancialmente como se ilustra en la figura 5, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 94 °C y una temperatura máxima pico de aproximadamente 117 °C. En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma A, que tiene un termograma de DSC sustancialmente como se ilustra en la figura 5, que comprende un evento endotérmico con una
temperatura de inicio de aproximadamente 174 °C y una temperatura máxima pico de aproximadamente 182 °C al calentarse desde aproximadamente 25 °C hasta aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma A, que tiene una gráfica isotérmica de DVS sustancialmente como se ilustra en la figura 6A.
En una realización, en el presente documento se proporciona la forma A que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 7.
En aun otra realización, la forma A es sustancialmente pura. En determinadas realizaciones, la forma A sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la forma A sustancialmente pura se encuentra sustancialmente libre de la forma B, la forma C o la forma H. En determinadas realizaciones, la pureza de la forma A sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma B
En determinadas realizaciones, en el presente documento se proporciona la forma B.
En una realización, la forma B es una forma sólida del compuesto 1. En otra realización, la forma B es cristalina. En una realización, la forma B es una forma de anhidrato del compuesto 1.
En determinadas realizaciones, la forma B que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma B se obtiene a partir de determinados sistemas de disolventes, incluidos heptano/agua, heptanos, agua, tolueno, MeCN, MeCN/agua, EtOH, EtOH/H2O (aproximadamente 1:1), THF/agua (aproximadamente 1:1) e IPA. En determinadas realizaciones, la forma B se obtiene secando o reduciendo la RH de la forma A por debajo de aproximadamente el 10 %.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma B, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma B tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 8. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma B, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 6,9, 8,7, 10,5, 11,6, 12,0, 13.6, 13,8, 14,1, 14,2, 16,3, 16,9, 17,5, 18,0, 18,4, 19,1, 19,5, 20,0, 20,8, 21,1, 22,1, 22,7, 23,3, 25,2, 26,0, 26,7, 27,4, 28,4, 28,8, 29,2, 30,1, 31,0, 31,5 o 31,8a 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 8. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos característicos de difracción de rayos X en polvo aproximadamente a 8,7, 11.6, 12,0, 13,8, 14,1, 17,5, 18,0, 19,5, 20,0 o 20,8° 20 (± 0,2° 20) En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 13,8, 19,5, 20,0 o 20,8° 20 (± 0,2° 20). En una realización, la forma sólida es la forma B. En otra realización, la forma B tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos o treinta y tres picos característicos de difracción de rayos X en polvo, como se indica en la tabla 13.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, p. ej., la forma B, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 9. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 0,1 % de la masa total de la muestra entre aproximadamente 30 °C y aproximadamente 155 °C cuando se calienta de aproximadamente 25 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 0,1 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C. En determinadas realizaciones, la forma cristalina es un anhidrato del compuesto 1 y corresponde a la forma B. En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 10, que comprende un evento endotérmico con temperatura de inicio a aproximadamente 174 °C y temperatura máxima pico a aproximadamente 182 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En aun otra realización, la forma B es sustancialmente pura. En determinadas realizaciones, la forma B sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En otra realización, la forma B se encuentra sustancialmente libre de la forma A. En determinadas realizaciones, la pureza
de la forma B sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma C
En determinadas realizaciones, en el presente documento se proporciona la forma C.
En una realización, la forma C es una forma sólida del compuesto 1. En otra realización, la forma C es cristalina. En una realización, la forma C es una forma solvatada del compuesto 1. En una realización, la forma C es una forma solvatada en acetonitrilo (MeCN) del compuesto 1.
En determinadas realizaciones, la forma C que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación, experimentos de recristalización con enfriamiento y experimentos de recristalización con antidisolvente (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma C se obtiene de determinados sistemas de disolventes que incluyen MeCN o MeCN/H2O (aproximadamente 1:1). En determinadas realizaciones la forma C se obtiene de determinados sistemas de disolventes que incluyen MeCN o MeCN/H2O (aproximadamente 1:1) a una temperatura de aproximadamente 50 °C. En otra realización, la forma C se obtiene de una solución de 2-MeTHF/H2O (aproximadamente 1:1) destilada al vacío a un volumen constante con adición de MeCN.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma C, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma C tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 11. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma C, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 3,1, 7,7, 8,9, 10,3, 13,3, 13.7, 14,2, 14,6, 14,8, 15,0, 15,3, 15,5, 15,7, 16,9, 17,4, 17,8, 18,3, 18,7, 19,5, 19,9, 20,7, 21,1, 21,4, 22,1, 22,4, 22.7, 23,1, 23,9, 24,6, 25,0, 25,5, 25,8, 26,1, 26,7, 26,9, 27,2, 27,7, 28,5, 29,4, 29,8, 30,3, 30,9, 31,3, 32,4, 33,0, 33,6, 34,3, 35,4, 35,9, 36,2, 37,1, 37,9 o 38,9220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 11. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho o nueve picos característicos de difracción de rayos X en polvo aproximadamente a 7,7, 8,9, 10,3, 15,3, 17,4, 18,3, 19,9, 21,4 o 28,5° 20 (± 0,2° 20) En una realización, la forma sólida es la forma C. En otra realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma C, tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,7, 8,9, 10,3 o 18,3° 20 (± 0,2° 20). En otra realización, la forma C tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta, cuarenta y uno, cuarenta y dos, cuarenta y tres, cuarenta y cuatro, cuarenta y cinco, cuarenta y seis, cuarenta y siete, cuarenta y ocho, cuarenta y nueve, cincuenta, cincuenta y uno, cincuenta y dos o cincuenta y tres picos característicos de difracción de rayos X en polvo, como se indica en la tabla 14.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 12. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 6,6 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 175 °C cuando se calienta de aproximadamente 25 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 6,6 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C. El contenido teórico de MeCN del monosolvato de MeCN del compuesto 1 es del 6,7 % en peso, lo cual se corresponde a la pérdida de peso TGA observada.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 13, que comprende un evento endotérmico con una máxima a aproximadamente 165 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C. En otra realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC como se ilustra en la figura 13 que comprende además un evento endotérmico con una máxima a aproximadamente 186 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma C, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 14.
En aun otra realización, la forma C es sustancialmente pura. En determinadas realizaciones, la forma C sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma C sustancialmente pura no es inferior a aproximadamente el 95
%, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma D
En determinadas realizaciones, en el presente documento se proporciona la forma D.
En una realización, la forma D es una forma sólida del compuesto 1. En otra realización, la forma D es cristalina. En una realización, la forma D es una forma solvatada del compuesto 1. En una realización, la forma D es una forma solvatada en IPA del compuesto 1.
En determinadas realizaciones, la forma D que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación, experimentos de recristalización con enfriamiento y experimentos de recristalización con antidisolvente (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma D se obtiene de determinados sistemas de disolventes que incluyen IPA a temperatura ambiente.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma D, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma D tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 15. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma D, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 3,1, 5,9, 7,4, 8,7, 10,1, 11,1, 13,7, 14,8, 15,1, 16,3, 16,6, 17,6, 18,1, 19,2, 19,8, 20,4, 21,5, 22,1, 22,3, 24,0, 24,3, 25,0, 26,2, 26,9, 27,3, 27,6, 28,2, 28,6, 30,9, 31,4, 32,9, 33,6, 34,6 o 37,2220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 15. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,4, 8,7, 10,1 o 18,1° 20 (± 0,2° 20). En una realización, la forma sólida es la forma D. En otra realización, la forma D tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres o treinta y cuatro picos característicos de difracción de rayos X en polvo, como se indica en la tabla 15.
En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma D, tiene una imagen SEM sustancialmente como se muestra en la figura 16.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 17. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 7,4 % de la masa total de la muestra entre aproximadamente 100 °C y aproximadamente 160 °C cuando se calienta de aproximadamente 25 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 7,4 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 18, con un evento endotérmico con una temperatura de inicio de aproximadamente 125 °C y una temperatura máxima pico de aproximadamente 154 °C al calentarse desde aproximadamente 25 °C a aproximadamente 220 °C. En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la 18, que comprende además un evento endotérmico con una temperatura de inicio de aproximadamente 175 °C y una temperatura máxima pico de aproximadamente 185 °C al calentarse desde aproximadamente 25 °C a aproximadamente 220 °C. En una realización, en el presente documento se proporciona una forma sólida, la forma D, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 19.
En aun otra realización, la forma D es sustancialmente pura. En determinadas realizaciones, la forma D sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma D sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma E
En determinadas realizaciones, en el presente documento se proporciona la forma E.
En una realización, la forma E es una forma sólida del compuesto 1. En otra realización, la forma E es cristalina. En una realización, la forma E es una forma solvatada del compuesto 1. La forma E puede ser un solvato en etanol, en donde el solvato contiene opcionalmente agua.
En determinadas realizaciones, la forma E que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio y experimentos de evaporación (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma E se obtiene de determinados sistemas de disolventes que incluyen EtOH o EtOH/agua (aproximadamente 1:1). En determinadas realizaciones, la forma E se obtiene de determinados sistemas de disolventes que incluyen EtOH o EtOH/agua (aproximadamente 1:1) a una temperatura de aproximadamente 50 °C. En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma E, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma E tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 20. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma E, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 3,1, 5,5, 7,8, 11,0, 13,5, 14.6, 15,6, 16,6, 17,5, 18,4, 20,0, 20,7, 22,2, 22,9, 23,5, 24,2, 24,8, 26,1, 26,7, 27,3, 27,8, 28,4, 29,5, 30,0, 31,1, 31.6, 32,1, 32,6, 33,6, 34,0, 34,5, 35,4, 36,3, 37,2, 38,1, 39,4 o 39,8220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 20. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres, cuatro, cinco, seis o siete picos característicos de difracción de rayos X en polvo aproximadamente a 7,8, 14.6, 15,6, 17,5, 22,2, 23,5 o 26,1° 20 (± 0,2° 20) En una realización, la forma sólida es la forma E. En otra realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma E, tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,8, 14,6, 17,5 o 22,2° 20 (± 0,2° 20). En otra realización, la forma E tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis o treinta y siete picos característicos de difracción de rayos X en polvo, como se indica en la tabla 16.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 21. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 13,7 % de la masa total de la muestra entre aproximadamente 35 °C y aproximadamente 175 °C cuando se calienta de aproximadamente 35 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 13,7 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 22, con un evento endotérmico amplio con una temperatura de inicio de aproximadamente 92 °C y un máximo de aproximadamente 104 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En incluso otra realización, la forma E es sustancialmente pura. En determinadas realizaciones, la forma E sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma E sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma F
En determinadas realizaciones, en el presente documento se proporciona la forma F.
En una realización, la forma F es una forma sólida del compuesto 1. En otra realización, la forma F es cristalina. En una realización, la forma F es una forma solvatada del compuesto 1. En una realización, la forma F es una forma solvatada en IPA del compuesto 1.
En determinadas realizaciones, la forma F que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma F se obtiene de determinados sistemas de disolventes que incluyen IPA o IPA/agua a aproximadamente 50 °C.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma F, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma F tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 23. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma F, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 4,9, 7,0, 9,4, 11,1, 11,8, 15,5, 15,8, 17,0, 17,6, 18,0, 18,3, 19,0, 19,7, 20,0, 20,3, 20,9, 22,4, 22,6, 23,2, 23,7, 24,4, 25,1, 25,4, 25,6, 26,4, 26,8,
27,3, 27,7, 28,6, 29,2, 30,0, 30,4, 30,7, 31,2, 32,1, 34,1, 34,4, 35,2, 35,8, 36,5, 38,5, 38,8 o 39,2220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 23. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma F, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho o nueve picos característicos de difracción de rayos X en polvo aproximadamente a 7,0, 9,4, 11,8, 15,5, 18,0, 18,3, 19,7, 20,0 o 20,9° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 9,4, 11,8, 18,0 o 18,3° 20 (± 0,2° 20). En una realización, la forma sólida es la forma F. En otra realización, la forma F tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta, cuarenta y uno, cuarenta y dos o cuarenta y tres picos característicos de difracción de rayos X en polvo, como se indica en la tabla 17.
En una realización, una forma sólida proporcionada en el presente documento, p. ej., la forma F, tiene una imagen de SEM sustancialmente como se muestra en la figura 24.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 25. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 14,3 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 175 °C cuando se calienta de aproximadamente 50 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 14,3 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 26, con un evento endotérmico con una temperatura de inicio de aproximadamente 137 °C y una temperatura máxima pico de aproximadamente 152 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma sólida, la forma F, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 27.
En incluso otra realización, la forma F es sustancialmente pura. En determinadas realizaciones, la forma F sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma F sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma G
En determinadas realizaciones, en el presente documento se proporciona la forma G.
En una realización, la forma G es una forma sólida del compuesto 1. En otra realización, la forma G es cristalina. En una realización, la forma G es una forma solvatada del compuesto 1. En una realización, la forma G es una forma solvatada en MTBE del compuesto 1.
En determinadas realizaciones, la forma G que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma G se obtiene de determinados sistemas de disolventes que incluyen MTBE. En determinadas realizaciones, la forma G se obtiene de determinados sistemas de disolventes que incluyen MTBE a una temperatura de 50 °C.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma G, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma G tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 28. En una realización, la forma G tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 4,5, 8,0, 9,0, 9,9, 10,0, 10,2, 11,6, 11,9, 13,5, 14,4, 14,6, 15,3, 15,9, 16,4, 16,9, 17,5, 17,7, 18,0, 18.4, 18,7, 18,8, 19,4, 19,6, 20,3, 20,8, 21,2, 21,6, 22,0, 22,2, 22,5, 22,9, 23,4, 24,0, 24,5, 24,6, 25,0, 25,2, 25,6, 25.9, 26,0, 26,4, 26,9, 27,3, 27,6, 28,0, 28,2, 28,8, 29,4, 29,9, 30,2, 30,8, 31,4, 31,8, 32,8, 33,2, 34,4, 34,9, 35,7, 36,1,38,2 o 38,9° 20 (± 0,2° 20) o (± 0,1° 20). En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma G, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez u once picos característicos de difracción de rayos X en polvo aproximadamente a 9,0, 9,9, 10,0, 15,3, 17,5, 18,4, 18,7, 19.4, 19,6, 21,2 o 22,9° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 9.9, 15,3, 18,4 o 22,9° 20 (± 0,2° 20). En una realización, la forma sólida es la forma G. En otra realización, la forma
G tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta, cuarenta y uno, cuarenta y dos, cuarenta y tres, cuarenta y cuatro, cuarenta y cinco, cuarenta y seis, cuarenta y siete, cuarenta y ocho, cuarenta y nueve, cincuenta, cincuenta y uno, cincuenta y dos, cincuenta y tres, cincuenta y cuatro, cincuenta y cinco, cincuenta y seis, cincuenta y siete, cincuenta y ocho, cincuenta y nueve, sesenta o sesenta y un picos característicos de difracción de rayos X en polvo, como se indica en la tabla 18.
En una realización, la forma G tiene una imagen de SEM sustancialmente como se ilustra en la figura 29.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 30. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 1,1 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 140 °C. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 8,7 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 180 °C cuando se calienta de aproximadamente 25 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 8,7 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 31, con un evento endotérmico con una temperatura de inicio de aproximadamente 144 °C y una temperatura máxima pico de aproximadamente 148 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 31, con un evento endotérmico con una máxima de aproximadamente 161 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma sólida, la forma G, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 32. En una realización, el espectro de RMN 1H de la forma G muestra que la forma G contiene una cantidad significativa de MBTE.
En incluso otra realización, la forma G es sustancialmente pura. En determinadas realizaciones, la forma G sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma G sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma H
En determinadas realizaciones, en el presente documento se proporciona la forma H.
En una realización, la forma H es una forma sólida del compuesto 1. En otra realización, la forma H es cristalina. En una realización, la forma H es una forma solvatada del compuesto 1. En una realización, la forma H es una forma solvatada en EtOH del compuesto 1. En determinadas realizaciones, la forma H puede convertirse en la forma A mediante contacto con un entorno que comprenda al menos el 20 % de RH.
En determinadas realizaciones, la forma H que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 6, la tabla 7 y la tabla 9). En determinadas realizaciones, la forma H se obtiene de determinados sistemas de disolventes que incluyen EtOH o EtOH/agua (aproximadamente 1:1) o EtOAc. En determinadas realizaciones, la forma H se obtiene de determinados sistemas de disolventes que incluyen EtOH o EtOH/agua (aproximadamente 1:1) o EtOAc a una temperatura de 50 °C.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma H, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma H tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 33. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma H, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 6,1, 7,7, 8,9, 10,3, 10,9, 11,3, 11,6, 13,7, 14,4, 15,0, 15,2, 15,4, 15,6, 15,9, 16,9, 17,2, 17,7, 18,2, 18,7, 19,4, 19,6, 20,6, 20,9, 21,4, 22,5, 23,2, 23,7, 24,6, 24,9, 25,6, 25,9, 26,2, 26,9, 27,4, 28,1, 28,4, 29,0, 29,4, 31,1, 32,2, 33,1, 34,1, 34,7, 35,3, 37,3 o 38,6° 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 33. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma H, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho,
nueve, diez u once picos característicos de difracción de rayos X en polvo aproximadamente a 7,7, 8,9, 10,3, 15,2, 15,4, 15,6, 17,2, 18,2, 19,6, 21,4 o 24,9° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,7, 8,9, 10,3 o 18,2° 20 (± 0,2° 20). En una realización, la forma sólida es la forma H. En otra realización, la forma H tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta, cuarenta y uno, cuarenta y dos, cuarenta y tres, cuarenta y cuatro, cuarenta y cinco o cuarenta y seis picos característicos de difracción de rayos X en polvo, como se indica en la tabla 19.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 34. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 6,5 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 175 °C cuando se calienta de aproximadamente 50 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 6,5 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 35, con un evento endotérmico con una máxima pico de aproximadamente 163 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 35, con un evento endotérmico con una temperatura de inicio de aproximadamente 179 °C y una temperatura máxima pico de aproximadamente 187 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En incluso otra realización, la forma H es sustancialmente pura. En determinadas realizaciones, la forma H sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma H sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma I
En determinadas realizaciones, en el presente documento se proporciona la forma I.
En una realización, la forma I es una forma sólida del compuesto 1. En otra realización, la forma I es cristalina. En una realización, la forma I es una forma solvatada del compuesto 1. En una realización, la forma I es una forma solvatada en MeCN del compuesto 1.
En determinadas realizaciones, la forma I que se proporciona en el presente documento se obtiene mediante experimentos de recristalización con enfriamiento y experimentos de recristalización con antidisolvente. En determinadas realizaciones, la forma I se obtiene de determinados sistemas de disolventes que incluyen MeCN. En determinadas realizaciones, la forma I puede convertir la forma C en una suspensión en MeCN a temperatura ambiente.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma I, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, la forma I tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 36. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma I, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 5,2, 5,5, 6,3, 8,6, 9,3, 10,4, 10,9, 11,5, 11,9, 12,6, 15,7, 16,6, 17,3, 18,1, 18,7, 19,0, 20,0, 20,9, 22,0, 22,5, 23,3, 24,1, 24,6, 25,4, 26,4, 27,6, 28,4, 29,6, 31,0, 31,6, 32,1,33,2, 33,9, 35,3, 35,9 o 38,5° 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 36. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 6,3, 15,7, 18,1 o 20,0° 20 (± 0,2° 20). En una realización, la forma sólida es la forma I. En otra realización, la forma I tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco o treinta y seis picos característicos de difracción de rayos X en polvo, como se indica en la tabla 20.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura
38. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 2,3 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 180 °C cuando se calienta de aproximadamente 50 °C a aproximadamente 220 °C. Por lo tanto, en determinadas realizaciones, la forma cristalina pierde aproximadamente el 2,3 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 39, con un evento endotérmico con una máxima pico de aproximadamente 75 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 39, con un evento endotérmico con una temperatura de inicio de aproximadamente 173 °C y una temperatura máxima pico de aproximadamente 183 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma sólida, la forma I, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 37.
En incluso otra realización, la forma I es sustancialmente pura. En determinadas realizaciones, la forma I sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma I sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Sólido amorfo
En determinadas realizaciones, en el presente documento se proporciona un sólido amorfo del compuesto 1.
En determinadas realizaciones, el sólido amorfo que se proporciona en el presente documento se obtiene mediante tratamiento con calor y/o evaporación de la forma A.
En una realización, el sólido amorfo tiene un espectro de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 40.
En una realización, en el presente documento se proporciona un sólido amorfo del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 41, con una temperatura de transición vitrea de 84 °C al calentarse de aproximadamente 40 °C a aproximadamente 260 °C.
En una realización, en el presente documento se proporciona un sólido amorfo del compuesto 1, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 42.
En una realización, en el presente documento se proporciona un sólido amorfo del compuesto 1 que tiene una gráfica isotérmica de DVS sustancialmente como se ilustra en la figura 43A.
En aun otra realización, el sólido amorfo del compuesto 1 es sustancialmente puro. En determinadas realizaciones, el sólido amorfo sustancialmente puro del compuesto 1 se encuentra sustancialmente libre de otras formas sólidas, p. ej., la forma A, la forma B, la forma C, la forma D, la forma E, la forma F, la forma G, la forma H y la forma I. En determinadas realizaciones, la pureza del sólido amorfo sustancialmente puro no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma Y de sal de citrato
En el presente documento también se proporcionan formas sólidas del compuesto 1 que incluyen sales de citrato. En determinadas realizaciones, en el presente documento se proporciona la forma Y de la sal de citrato.
En una realización, la forma Y de sal de citrato es una forma sólida del compuesto 1. En otra realización, la forma Y de sal de citrato es cristalina. En otra realización, la forma Y de sal de citrato es un anhidrato.
En determinadas realizaciones, la forma Y de sal de citrato que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 23, la tabla 24 y la tabla 25). En determinadas realizaciones, la forma Y de sal de citrato se obtiene de determinados sistemas de disolventes que incluyen acetona, MeCN, n-butanol, EtOH,
EtOH/agua (aproximadamente 1:1), EtOAc, heptanos, IPA, DCM, MeOAc, MTBE, MEK, tolueno, THF, THF/agua (aproximadamente 1:1), 1,4-dioxano, MIBK, IPAc y 2-MeTHF. En determinadas realizaciones, la forma Y de sal de citrato se obtiene de determinados sistemas de disolventes que incluyen acetona, MeCN, n-butanol, EtOH, EtOH/agua (aproximadamente 1:1), EtOAc, heptanos, IPA, DCM, MeOAc, MTBE, MEK, tolueno, THF, THF/agua (aproximadamente 1:1), 1,4-dioxano, MIBK, IPAc y 2-MeTHF a 50 °C. En una realización, la forma Y de sal de citrato es un solvato en EtOH.
En una realización, un método para preparar la forma Y de sal de citrato comprende las etapas de enfriar hasta una temperatura inferior a aproximadamente el 50 °C en THF o THF/agua y recoger los sólidos.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma Y, es una sal de citrato del compuesto 1 y es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma Y, tiene un patrón de difracción de rayos X en polvo (XRPD) sustancialmente como se ilustra en la figura 45. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma Y, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 4,8, 6,6, 9,6, 13.6, 14,4, 15,4, 16,0, 16,9, 18,0, 18,9, 19,2, 19,9, 20,1, 20,9, 21,8, 22,4, 22,7, 23,2, 23,4, 24,0, 24,1, 24,3, 25,1, 26.7, 27,0, 27,9, 28,5, 29,0, 29,6, 30,2, 30,4, 30,8, 31,1, 31,6, 32,3, 33,1, 33,5, 34,0, 34,6 o 35,1° 20 (± 0,2220) o (± 0,1° 20), como se ilustra en la figura 45. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma Y, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez u once picos característicos de difracción de rayos X en polvo aproximadamente a 4,8, 6,6, 9,6, 15,4, 16,0, 16,9, 18,9, 19,2, 19,9, 20,9 o 28,5° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 4,8, 9,6, 18,9 o 19,2° 20 (± 0,2° 20). En una realización, la forma sólida es la forma Y de sal de citrato. En otra realización, la forma Y de sal de citrato tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve o cuarenta picos característicos de difracción de rayos X en polvo, como se indica en la tabla 27.
En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma Y, tiene una imagen de SEM sustancialmente como se muestra en la figura 46.
En una realización, en el presente documento se proporciona una sal de citrato cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 47. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 0,1 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 150 °C al calentarse de aproximadamente 50 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una sal de citrato cristalina del compuesto 1 que tiene un termograma de DSC sustancialmente como se ilustra en la figura 48, con un evento endotérmico con una temperatura de inicio de aproximadamente 213 °C y una temperatura máxima pico de aproximadamente 217 °C al calentarse de aproximadamente 25 °C a aproximadamente 260 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma Y, que tiene una gráfica isotérmica de DVS sustancialmente como se ilustra en la figura 49A.
En una realización, en el presente documento se proporciona una forma sólida, la forma Y, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 50.
En aun otra realización, la forma Y de sal de citrato es sustancialmente pura. En determinadas realizaciones, la forma Y de sal de citrato sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma Y de sal de citrato sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma Z de sal de citrato
En determinadas realizaciones, en el presente documento se proporciona la forma Z de la sal de citrato.
En una realización, la forma Z de sal de citrato es una forma sólida del compuesto 1. En otra realización, la forma Z de sal de citrato es cristalina. En otra realización, la forma Z de sal de citrato es un anhidrato. En otra realización, la forma Z de sal de citrato es un hidrato. En una realización, la forma Z de sal de citrato es un hidrato no estequiométrico. En incluso otra realización, la forma Z de sal de citrato es un hidrato de canal. En incluso otra
realización, la forma Z de sal de citrato es un hidrato de canal no estequiométrico. En incluso otra realización, la forma Z de sal de citrato es un solvato.
En determinadas realizaciones, la forma Z se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 23, la tabla 24 y la tabla 25). En determinadas realizaciones, la forma Z de sal de citrato se obtiene de determinados sistemas de disolventes que incluyen MeCN/agua (aproximadamente 1:1), EtOH, EtOH/agua (aproximadamente 1:1) o MeOH. En determinadas realizaciones, la forma Z de sal de citrato se obtiene de determinados sistemas de disolventes que incluyen MeCN/agua (aproximadamente 1:1), EtOH, EtOH/agua (aproximadamente 1:1) o MeOH a una temperatura de aproximadamente 50 °C.
En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma Z, tiene una imagen de SEM sustancialmente como se muestra en la figura 53.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma Z, es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma Z, tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 52. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma Z, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 4,6, 6,6, 9,4, 13,1, 14,1, 15,3, 15,6, 17,4, 18,8, 19,0, 19,9, 20,4, 21,1, 21,9, 22,2, 22,7, 23,5, 23,9, 25,2, 26,3, 26,8, 27,8, 28,3, 28,7, 29,8, 31,2, 31,9, 32,6, 33,7, 35,1, 35,9, 37,4 o 38,0220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 52. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 9,4, 18,8, 19,0 o 28,7° 20 (± 0,2° 20). En una realización, la forma sólida es la forma Z de sal de citrato. En otra realización, la forma Z de sal de citrato tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos o treinta y tres picos característicos de difracción de rayos X en polvo, como se indica en la tabla 30. En una realización, en el presente documento se proporciona una sal de citrato cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 54. En determinadas realizaciones, la forma cristalina presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 0,1 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 150 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. En determinadas realizaciones, la forma cristalina es un anhidrato del compuesto 1.
En una realización, en el presente documento se proporciona una forma de sal de citrato cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 55, con un evento endotérmico con una temperatura de inicio de aproximadamente 217 °C y una temperatura máxima pico de aproximadamente 221 °C al calentarse de aproximadamente 25 °C a aproximadamente 260 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., la forma Y, que tiene una gráfica isotérmica de DVS sustancialmente como se ilustra en la figura 56A.
En una realización, en el presente documento se proporciona una forma sólida, la forma Z, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 57.
En una realización, en el presente documento se proporciona un hidrato de la sal de citrato del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 58. En determinadas realizaciones, el hidrato presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 2 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 200 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. En determinadas realizaciones, la forma cristalina es un hidrato de la forma de citrato del compuesto 1.
En una realización, en el presente documento se proporciona un hidrato no estequiométrico de la sal de citrato del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 59. En determinadas realizaciones, la forma de hidrato no estequiométrica presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 1,7 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 200 °C al calentarse entre aproximadamente 50 °C y aproximadamente 300 °C. En determinadas realizaciones, la forma cristalina es un hidrato no estequiométrico de la forma de citrato del compuesto 1.
En una realización, en el presente documento se proporciona un solvato de la sal de citrato del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 60. En determinadas realizaciones, el solvato presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 1,3 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente
200 °C al calentarse de aproximadamente 25 °C a aproximadamente 3002C. En determinadas realizaciones, la forma cristalina es un solvato de la forma de citrato del compuesto 1.
En una realización, en el presente documento se proporciona una forma sólida, el solvato de la forma Z, que tiene un espectro de RMN 1H sustancialmente como se ilustra en la figura 61.
En aun otra realización, la forma Z de sal de citrato es sustancialmente pura. En determinadas realizaciones, la forma Z de sal de citrato sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma Z de sal de citrato sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Formas de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona una forma de sal de HCl como material de partida. En una realización, la forma de sal de HCl como material de partida es una forma sólida del compuesto 1. En otra realización, la forma de sal de HCl como material de partida es un anhidrato.
En una realización, la forma de sal de HCl como material de partida se obtiene disolviendo el compuesto 1 en MeOH (p. ej., aproximadamente 10 vol.) a una temperatura de aproximadamente 25 °C a aproximadamente 30 °C. Se agrega HCl en MeOH (~ 1,25 M, 1,10 eq.) para obtener el material de partida en forma de sal de HCl del compuesto 1. La solución puede destilarse al vacío y se puede cambiar el disolvente de MeOH a EtOAc (p. ej., aproximadamente 30-35 vol.), donde la temperatura se mantiene opcionalmente a aproximadamente 25 °C hasta aproximadamente 35 °C. La suspensión puede filtrarse y la torta se lava con EtOAc (p. ej., aproximadamente 5 vol.). La torta puede secarse en un horno de vacío a 50 °C.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma de sal de HCl como material de partida, es sustancialmente cristalina, según se indica, p. ej., a través de las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., una forma de sal de HCl como material de partida, tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la figura 63. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma de sal de HCl como material de partida, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 5,8, 7,1, 8,3, 10,1, 11,3, 11,6, 12,7, 15,5, 16,1, 17,8, 19,2, 19,7, 20,5, 21,1, 23,0, 24,0, 25,5, 26,3, 27,2, 28,4, 31,0 o 35,6° 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 63. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 21,1, 19,2, 20,5 o 17,8° 20 (± 0,2° 20) como se ilustra en la figura 63. En una realización, la forma sólida es una forma de sal de HCl como material de partida. En otra realización, la forma de sal de HCl como material de partida tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno o veintidós picos característicos de difracción de rayos X en polvo, según se indica en la tabla 42. En una realización, en el presente documento se proporciona una forma de sal de HCl como material de partida del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 64. En determinadas realizaciones, la forma de sal de HCl como material de partida presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 1,1 % de la masa total de la muestra entre aproximadamente 24 °C y aproximadamente 100 °C al calentarse de aproximadamente 24 °C a aproximadamente 300 °C. En determinadas realizaciones, la forma cristalina es un anhidrato del compuesto 1.
En una realización, en el presente documento se proporciona una forma de sal de HCl del material de partida del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 64, con un evento endotérmico con una temperatura de inicio de aproximadamente 239 °C y una temperatura máxima pico de aproximadamente 249 °C al calentarse de aproximadamente 24 °C a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., una forma de sal de HCl como material de partida, que tiene una gráfica isotérmica de DVS sustancialmente como se ilustra en la figura 65.
En incluso otra realización, la forma de sal de HCl como material de partida es sustancialmente pura. En determinadas realizaciones, la forma de sal de HCl como material de partida sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma de sal de HCl como material de partida sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 1 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona una forma 1 de sal de HCl del compuesto 1.
En una realización, la forma 1 de sal de HCl es una forma sólida del compuesto 1. En una realización, la forma 1 de sal de HCl es un solvato. En una realización, la forma 1 de sal de HCl es una forma de solvato IPA del compuesto 1. En otra realización, la forma 1 de sal de HCl es cristalina.
En determinadas realizaciones, la forma 1 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente (véase la tabla 23, la tabla 24 y la tabla 25). En determinadas realizaciones, la forma 1 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen IPA.
En una realización, un método para preparar la forma 1 de sal de HCl comprende las etapas de disolver el compuesto 1 en IPA y evaporar lentamente el IPA y recoger los sólidos.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 1 de sal de HCl, es una sal de HCl del compuesto 1 y es sustancialmente cristalina, según se indica, p. ej., mediante las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 1 de sal de HCl, tiene un patrón de difracción de rayos X en polvo (XRPD) sustancialmente como se ilustra en la figura 66. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 1 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 5,5, 5,9, 5,9, 8,8, 9,4, 9,9, 11,5, 12,4, 12,8, 15,6, 15,9, 16,2, 17,4, 18,6, 20,4, 20,7, 21,0, 21,3, 21,7, 22,1, 22,6, 22,8, 22,9, 23,2, 23,5, 23,7, 23,9, 25,5, 25,8, 26,3, 26,8, 27,0, 28,4, 29,3, 30,3, 32,1, 32,2, 33,5, 35,5, 36,6 o 37,6° 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 66. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce o trece picos característicos de difracción de rayos X en polvo aproximadamente a 5,5 5,9, 8,8, 9,4, 15,9, 18,6, 20,7, 22,6, 22,8, 22,9, 29,3, 32,1 o 32,2° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 1 de sal de HCl. En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 8,8, 9,4, 15,9 o 20,7° 20 (± 0,2° 20). En otra realización, la forma 1 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta o cuarenta y un picos característicos de difracción de rayos X en polvo, como se indica en la tabla 34.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 67. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 6,6 % de la masa total de la muestra entre aproximadamente 20 °C y aproximadamente 140 °C al calentarse de aproximadamente 20 °C a aproximadamente 325 °C.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 10 % de la masa total de la muestra entre aproximadamente 150 °C y aproximadamente 200 °C cuando se calienta de aproximadamente 20 °C a aproximadamente 325 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 17 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 325 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC sustancialmente como se ilustra en la figura 67, con un evento endotérmico con una temperatura de inicio de aproximadamente 102 °C y una temperatura máxima pico de aproximadamente 114 °C al calentarse de aproximadamente 25 °C a aproximadamente 350 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC sustancialmente como se ilustra en la figura 67, con un evento endotérmico con una temperatura de inicio de aproximadamente 146 °C y una temperatura máxima pico de aproximadamente 181 °C al calentarse de aproximadamente 25 °C a aproximadamente 350 °C.
En incluso otra realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC sustancialmente como se ilustra en la figura 67, con múltiples eventos endotérmicos, cada uno con una máxima superior a 250 °C al calentarse de aproximadamente 25 °C a
aproximadamente 3502C.
En aun otra realización, la forma 1 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 1 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 1 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 2 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 2 de sal de HCl.
En una realización, la forma 2 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 2 de sal de HCl es cristalina. En una realización, la forma 2 de sal de HCl es una forma de anhidrato del compuesto 1. En una realización, la forma 2 de sal de HCl es una forma solvatada del compuesto 1. En una realización, la forma 2 de sal de HCl es una forma solvatada en IPA del compuesto 1. En una realización, la forma 2 de sal de HCl es una forma solvatada en tolueno del compuesto 1.
En determinadas realizaciones, la forma 2 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente. En determinadas realizaciones, la forma 2 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen IPA/tolueno (aproximadamente 1:1).
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 2 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, la forma 2 de sal de HCl tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la figura 68. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 2 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 5,5, 5,8, 9,0, 9,8, 9,9, 10,7, 11,0, 12,3, 12,6, 14,0, 16,7, 18,1, 19,0, 19,5, 19,9, 20,1, 20,8, 21,5, 21,8, 22,0, 22,8, 23,3, 24,0, 24,3, 24,6, 24,9, 25,3, 26,0, 26,5, 26,8, 27,0, 27,6, 28,4, 29,2, 29,7, 30,7, 33,0 o 35,1° 20 (± 0,2° 20) o (± 0,1° 20) como se ilustra en la figura 68. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma 2 de sal de HCl, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez u once picos característicos de difracción de rayos X en polvo aproximadamente a 5,5, 9,0, 9,8, 9,9, 12,3, 12,6, 16,7, 18,1, 19,0, 21,5 o 22,8° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 9,0, 9,8, 9,9 o 16,7° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 2 de sal de HCl. En otra realización, la forma 2 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete o treinta y ocho picos característicos de difracción de rayos X en polvo, como se indica en la tabla 35.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 69. En determinadas realizaciones, la forma cristalina, forma 2 de sal de HCl, presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 2,6% de la masa total de la muestra entre aproximadamente 20 °C y aproximadamente 140 °C al calentarse de aproximadamente 20 °C a aproximadamente 325 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene una termografía TGA que se corresponde sustancialmente al termograma de TGA representativo ilustrado en la figura 69, donde la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 14 % de la masa total de la muestra entre aproximadamente 140 °C y aproximadamente 200 °C al calentarse de aproximadamente 20 °C a aproximadamente 325 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 17 % de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 325 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC como se ilustra en la figura 69, con un evento endotérmico con una temperatura de inicio de aproximadamente 151 °C y una temperatura máxima pico de aproximadamente 157 °C al calentarse de aproximadamente 25 °C a aproximadamente 350 °C.
En aun otra realización, la forma 2 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 2 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo.
En determinadas realizaciones, la pureza de la forma 2 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 3 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 3 de sal de HCl.
En una realización, la forma 3 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 3 de sal de HCl es cristalina. En una realización, la forma 3 de sal de HCl es una forma solvatada del compuesto 1. En una realización, la forma 3 de sal de HCl es una forma solvatada en n-butanol del compuesto 1. En una realización, la forma 3 de sal de HCl es una forma solvatada en heptano del compuesto 1. En una realización, la forma 3 de sal de HCl es una forma solvatada en n-butanol/heptano del compuesto 1. En una realización, la forma 3 de sal de HCl es una forma hidratada del compuesto 1. En una realización, la forma 3 de sal de HCl es una forma de anhidrato del compuesto 1.
En determinadas realizaciones, la forma 3 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación, experimentos de recristalización con enfriamiento y experimentos de recristalización con antidisolvente. En determinadas realizaciones, la forma 3 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen n-butanol/tolueno (aproximadamente 1:1). En determinadas realizaciones, una forma sólida proporcionada en el presente documento, p. ej., la forma 3 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, la forma 3 de sal de HCl tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la figura 70. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 3 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 5,5, 6,5, 7,7, 10,0, 10,5, 10,9, 12,9, 14,8, 15,9, 16,2, 18,3, 18,9, 19,4, 20,4, 21,0, 21,8, 22,2, 22,5, 24,1, 26,0, 28,8, 29,9, 32,7 o 39,4a 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 70. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma 3 de sal de HCl, tiene uno, dos, tres, cuatro, cinco, seis, siete u ocho picos característicos de difracción de rayos X en polvo aproximadamente a 5,5, 6,5, 10,9, 16,2, 19,4, 20,4, 22,2 o 22,5° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 5,5, 16,2, 19,4 o 20,4° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 3 de sal de HCl. En otra realización, la forma 3 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés o veinticuatro picos característicos de difracción de rayos X en polvo, según se indica en la tabla 36.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 71. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 2,3 % de la masa total de la muestra entre aproximadamente 50 °C y aproximadamente 175 °C al calentarse de aproximadamente 25 °C a aproximadamente 140 °C. En determinadas realizaciones, la forma cristalina del compuesto 1 presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 11 % de la masa total de la muestra entre aproximadamente 140 °C y aproximadamente 210 °C al calentarse de aproximadamente 25 °C a aproximadamente 140 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 13 % de su masa total cuando se calienta de aproximadamente temperatura ambiente a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC como se ilustra en la figura 71, con un evento endotérmico con una temperatura de inicio de aproximadamente 153 °C y una temperatura máxima pico de aproximadamente 168 °C al calentarse de aproximadamente 25 °C a aproximadamente 350 °C.
En aun otra realización, la forma 3 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 3 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 3 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 4 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 4 de sal de HCl.
En una realización, la forma 4 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 4 de sal de HCl es cristalina. En una realización, la forma 4 de sal de HCl es una forma solvatada del compuesto 1. En una realización, la forma 4 de sal de HCl es una forma solvatada en metanol del compuesto 1. En una realización, la forma 4 de sal de HCl es una forma solvatada en IPAc del compuesto 1. En una realización, la forma 4 de sal de HCl es una forma solvatada en MeOH/IPAc del compuesto 1.
En determinadas realizaciones, la forma 4 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación, experimentos de recristalización con enfriamiento y experimentos de recristalización con antidisolvente. En determinadas realizaciones, la forma 4 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen MeOH/IPAc.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 4 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 4 de sal de HCl, tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 72. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 4 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,1, 8,2, 8,4, 10,8, 14,0, 15,3, 15,9, 16,4, 16,8, 17,8, 18,3, 18,9, 19,1, 19,7, 20,3, 21,0, 21,6, 22,1, 23,3, 23,6, 24,9, 25,3, 26,3, 27,6, 28,5, 29,1, 29,9, 30,6, 30,9, 31,9, 32,8, 34,6 o 36,2a 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 72. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma 4 de sal de HCl, tiene uno, dos, tres, cuatro, cinco, seis, siete u ocho picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,1, 8,4, 15,9, 16,8, 18,9, 19,1 o 19,7° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,1, 8,4 o 15,9° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 4 de sal de HCl. En otra realización, la forma 4 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres o treinta y cuatro picos característicos de difracción de rayos X en polvo, como se indica en la tabla 37.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 73. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 4,5 % de la masa total de la muestra entre aproximadamente 20 °C y aproximadamente 140 °C al calentarse de aproximadamente 20 °C a aproximadamente 275 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 4,5 % de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 275 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 73, con un evento endotérmico con una temperatura de inicio de aproximadamente 94 °C y una temperatura máxima pico de aproximadamente 118 °C al calentarse de aproximadamente 25 °C a aproximadamente 275 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 73, que comprende además un evento endotérmico con una temperatura de inicio de aproximadamente 219 °C y una temperatura máxima pico de aproximadamente 236 °C al calentarse de aproximadamente 25 °C a aproximadamente 275 °C.
En aun otra realización, la forma 4 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 4 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 4 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 5 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 5 de sal de HCl.
En una realización, la forma 5 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 5 de sal de HCl es cristalina. En una realización, la forma 5 de sal de HCl es una forma solvatada del compuesto 1. En una realización, la forma 5 de sal de HCl es una forma solvatada en DMF del compuesto 1.
En determinadas realizaciones, la forma 5 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y difusión con vapor. En determinadas realizaciones, la forma 5 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen DMF. En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 5 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 5 de sal de HCl, tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 74. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 5 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,7, 10,0, 11,7, 13,3, 13,6, 15,1, 15,7, 17,2, 17,9, 19,9, 20,6, 21,3, 23,3, 24,2, 25,5, 27,0, 28,5, 29,3, 30,1, 31,7, 32,2, 34,1, 35,4, 37,0 o 38,8° 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 74. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma 5 de sal de HCl, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once o doce picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,7, 10,0, 11,7, 15,1, 15,7, 17,2, 19,9, 20,6, 21,3, 24,2 o 27,0° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 19,9, 20,6 o 27,0° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 5 de sal de HCl. En otra realización, la forma 5 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco o veintiséis picos característicos de difracción de rayos X en polvo, según se indica en la tabla 38.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 75. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 4,2 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 140 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 4,2 % de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 75, con un evento endotérmico con una máxima de aproximadamente 104 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En una realización, en el presente documento se proporciona una forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 75, con un evento endotérmico con una temperatura de inicio de aproximadamente 192 °C y una temperatura máxima pico de aproximadamente 209 °C al calentarse de aproximadamente 25 °C a aproximadamente 220 °C.
En aun otra realización, la forma 5 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 5 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 5 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 6 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 6 de sal de HCl.
En una realización, la forma 6 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 6 de sal de HCl es cristalina. En una realización, la forma 6 de sal de HCl es un hidrato del compuesto 1. En una realización, la forma 6 de sal de HCl es un pentahidrato del compuesto 1.
En determinadas realizaciones, la forma 6 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio. En determinadas realizaciones, la forma 6 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen HCl 0,1 N en agua.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 6 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 6 de sal de HCl, tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 76. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 6 de sal de HCl, tiene
uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 7,0, 8,8, 9,9, 11,5, 12,2, 12,4, 14,7, 16,3, 16,7, 17,5, 17,9, 18,2, 18,6, 19,2, 19,4, 19,7, 19,9, 20,3, 21,0, 21,2, 21,9, 22,7, 22,9, 23,7, 24,6, 25,0, 25,6, 26,3, 26,5, 26,8, 27,2, 27,6, 28,2, 28,7, 29,1, 29,6, 30,3, 30,8, 31,2, 31,7, 32,3, 32,8, 33,3, 34,0, 34,3, 35,3, 36,2 o 37,0° 20 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 76. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma 6 de sal de HCl, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once o doce picos característicos de difracción de rayos X en polvo aproximadamente a 9,9, 12,4, 16,3, 17,5, 19,2, 19,7, 19,9, 21,0, 25,0, 25,6, 27,2 o 32,3° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 9,9, 12,4, 17,9 o 19,7° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 6 de sal de HCl. En otra realización, la forma 6 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco, treinta y seis, treinta y siete, treinta y ocho, treinta y nueve, cuarenta, cuarenta y uno, cuarenta y dos, cuarenta y tres, cuarenta y cuatro, cuarenta y cinco, cuarenta y seis, cuarenta y siete o cuarenta y ocho picos característicos de difracción de rayos X en polvo, como se indica en la tabla 39.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 77. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 11,6 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 100 °C al calentarse de aproximadamente 20 °C a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 78. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 14,3 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 60 °C al calentarse de aproximadamente 20 °C a aproximadamente 300 °C.
En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 1,6 % de la masa total de la muestra entre aproximadamente 60 °C y aproximadamente 125 °C al calentarse de aproximadamente 20 °C a aproximadamente 300 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina en cristal del compuesto 1 pierde aproximadamente el 15,9 % de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 100 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 77, con un evento endotérmico con una temperatura de inicio de aproximadamente 53 °C y una temperatura máxima pico de aproximadamente 88 °C al calentarse de aproximadamente 25 °C a aproximadamente 325 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la 77, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 201 °C y una temperatura máxima pico de aproximadamente 228 °C al calentarse de aproximadamente 25 °C a aproximadamente 325 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 79, con un evento endotérmico con una temperatura de inicio de aproximadamente 59 °C y una temperatura máxima pico de aproximadamente 89 °C al calentarse de aproximadamente 25 °C a aproximadamente 325 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 79, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 211 °C y una temperatura máxima pico de aproximadamente 225 °C al calentarse de aproximadamente 25 °C a aproximadamente 325 °C.
En aun otra realización, la forma 6 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 6 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 6 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 7 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 7 de sal de HCl.
En una realización, la forma 7 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 7 de sal de HCl es cristalina. En una realización, la forma 7 de sal de HCl es un hidrato del compuesto 1. En una realización, la forma 7 de sal de HCl es un monohidrato del compuesto 1.
En determinadas realizaciones, la forma 7 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente. En determinadas realizaciones, la forma 7 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen agua a temperatura ambiente.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 7 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 7 de sal de HCl, tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 80. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 7 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,1, 8,3, 10,8, 13,8, 14,5, 15.3, 15,6, 16,2, 16,6, 17,0, 17,6, 18,3, 18,6, 19,1, 19,6, 19,7, 20,2, 20,7, 21,5, 22,0, 22,9, 24,0, 24,3, 25,2, 26,2, 28.4, 29,0, 29,5, 30,2, 30,8, 31,2, 32,5, 33,1,34,7 o 36,3220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 80. En una realización específica, una forma sólida que se proporciona en el presente documento, p. ej., la forma 7 de sal de HCl, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece o catorce picos característicos de difracción de rayos X en polvo aproximadamente a 7,9, 8,1, 8,3, 10,8, 15,3, 16,2, 18,3, 19,1, 19,6, 19,7, 21,5, 24,0, 24,3 o 25,2° 20 (± 0,2° 20). En otra realización, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 8,1, 8,3, 19,1 o 19,7° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 7 de sal de HCl. En otra realización, la forma 7 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos, treinta y tres, treinta y cuatro, treinta y cinco o treinta y seis picos característicos de difracción de rayos X en polvo, como se indica en la tabla 40.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 81. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 3,8 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 100 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 3,8 % de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 83. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 3,4% de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 100 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 3,4% de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 81, con un evento endotérmico con una temperatura de inicio de aproximadamente 80 °C y una temperatura máxima pico de aproximadamente 111 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 81, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 215 °C y una temperatura máxima pico de aproximadamente 233 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 82, con un evento endotérmico con una temperatura de inicio de aproximadamente 71 °C y una temperatura máxima pico de aproximadamente 98 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 82, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 209 °C y una temperatura máxima pico de aproximadamente 230 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una forma sólida, p. ej., una forma de sal de HCl como material de partida, que tiene una gráfica isotérmica de DVS sustancialmente como se ilustra en la figura 84.
En aun otra realización, la forma 7 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 7 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 7 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
Forma 8 de sal de HCl
En determinadas realizaciones, en el presente documento se proporciona la forma 8 de sal de HCl.
En una realización, la forma 8 de sal de HCl es una forma sólida del compuesto 1. En otra realización, la forma 8 de sal de HCl es cristalina. En una realización, la forma 8 de sal de HCl es un hidrato del compuesto 1. En una realización, la forma 8 de sal de HCl es un monohidrato del compuesto 1.
En determinadas realizaciones, la forma 8 de sal de HCl que se proporciona en el presente documento se obtiene mediante experimentos de equilibrio, experimentos de evaporación y experimentos de recristalización con antidisolvente. En determinadas realizaciones, la forma 8 de sal de HCl se obtiene de determinados sistemas de disolventes que incluyen agua a 50 °C.
En determinadas realizaciones, una forma sólida que se proporciona en el presente documento, p. ej., la forma 8 de sal de HCl, es sustancialmente cristalina, como se indica, p. ej., según las mediciones de difracción de rayos X en polvo. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 8 de sal de HCl, tiene un patrón de difracción de rayos X en polvo sustancialmente como se ilustra en la figura 85. En una realización, una forma sólida que se proporciona en el presente documento, p. ej., la forma 8 de sal de HCl, tiene uno o más picos característicos de difracción de rayos X en polvo aproximadamente a 8,1, 9,8, 10,1, 10,8, 11,1, 11.6, 15,7, 16,2, 16,9, 17,4, 18,0, 18,7, 19,2, 19,6, 21,4, 22,1, 22,9, 23,8, 24,2, 25,1, 25,5, 26,2, 26,7, 28,2, 28,4, 29.6, 30,5, 31,7, 32,0, 33,7, 35,6, 36,4 o 37,4220 (± 0,2° 20) o (± 0,1° 20), como se ilustra en la figura 85. En una realización específica, una forma sólida que se proporciona en el presente documento tiene uno, dos, tres o cuatro picos característicos de difracción de rayos X en polvo aproximadamente a 9,8, 17,4, 18,7 o 26,2° 20 (± 0,2° 20). En una realización, la forma sólida es una forma 8 de sal de HCl. En otra realización, la forma 8 de sal de HCl tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte, veintiuno, veintidós, veintitrés, veinticuatro, veinticinco, veintiséis, veintisiete, veintiocho, veintinueve, treinta, treinta y uno, treinta y dos o treinta y tres picos característicos de difracción de rayos X en polvo, como se indica en la tabla 41.
En una realización, en el presente documento se proporciona una sal de HCl de forma cristalina del compuesto 1, que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo que se ilustra en la figura 86. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 3,1 % de la masa total de la muestra entre aproximadamente 25 °C y aproximadamente 100 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene una termografía TGA sustancialmente correspondiente al termograma de TGA representativo ilustrado en la figura 87. En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA con una pérdida de masa total de aproximadamente el 3,0 % de la masa total de la muestra entre aproximadamente 30 °C y aproximadamente 120 °C al calentarse de aproximadamente 25 °C a aproximadamente 300 °C.
En determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente el 2,6 % de la masa total de la muestra entre aproximadamente 125 °C y aproximadamente 215 °C al calentarse de aproximadamente 30 °C a aproximadamente 300 °C. Por lo tanto, en determinadas realizaciones, la sal de HCl en forma cristalina del compuesto 1 pierde aproximadamente el 5,6 % de su masa total al calentarse de aproximadamente temperatura ambiente a aproximadamente 220 °C. El contenido teórico de agua para la forma 8 de sal de HCl monohidrato es del 2,9 % y se corresponde al porcentaje de masa total perdido por la muestra en el termograma de TGA anterior.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 86, con un evento endotérmico con una temperatura de inicio de aproximadamente 117 °C y una temperatura máxima pico de aproximadamente 148 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 86, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 208 °C y
una temperatura máxima pico de aproximadamente 221 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C.
En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 88, con un evento endotérmico con una máxima de aproximadamente 148 °C al calentarse desde aproximadamente 25 °C a aproximadamente 275 °C. En una realización, en el presente documento se proporciona una sal de HCl en forma cristalina del compuesto 1 que tiene un termograma de DSC según se ilustra en la figura 88, que comprende un evento endotérmico con una temperatura de inicio de aproximadamente 204 °C y una temperatura máxima pico de aproximadamente 224 °C al calentarse desde aproximadamente 25 °C a aproximadamente 300 °C.
En aun otra realización, la forma 8 de sal de HCl es sustancialmente pura. En determinadas realizaciones, la forma 8 de sal de HCl sustancialmente pura se encuentra sustancialmente libre de otras formas sólidas, p. ej., sólido amorfo. En determinadas realizaciones, la pureza de la forma 8 de sal de HCl sustancialmente pura no es inferior a aproximadamente el 95 %, no es inferior a aproximadamente el 96 %, no es inferior a aproximadamente el 97 %, no es inferior a aproximadamente el 98 %, no es inferior a aproximadamente el 98,5 %, no es inferior a aproximadamente el 99 %, no es inferior a aproximadamente el 99,5 % o no es inferior a aproximadamente el 99,8 %.
MÉTODOS DE USO
Las formas sólidas del compuesto 1 descritas en el presente documento son útiles como productos farmacéuticos para tratar, prevenir o mejorar afecciones en animales o seres humanos. Por consiguiente, en el presente documento se proporcionan formas sólidas del compuesto 1 descrito en el presente documento que se pueden utilizar en todos los métodos que se describen en el presente documento. En particular, las formas sólidas del compuesto 1 que se proporciona en el presente documento son para su uso en el tratamiento o la prevención de un cáncer. Los métodos comprenden la administración de una cantidad eficaz de una o más formas sólidas del compuesto 1 descrito en el presente documento a un sujeto que lo necesite. Se debe comprender que los métodos descritos en el presente documento también incluyen tratamiento con una composición farmacéutica, como las que se indican posteriormente, donde la composición farmacéutica incluye una forma sólida del compuesto 1 que se describe en el presente documento y, opcionalmente, al menos un excipiente farmacéuticamente aceptable.
En otro aspecto, los métodos son para tratar o prevenir un cáncer, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 una forma sólida del compuesto 1 descrito en el presente documento. En algunas realizaciones, el cáncer es un tumor sólido o un tumor hematológico. En algunas realizaciones, el cáncer no es melanoma.
En algunas realizaciones, el tumor sólido es melanoma, cáncer colorrectal, cáncer de estómago, cáncer de cabeza y cuello, cáncer de tiroides, cáncer de vejiga, cáncer del SNC, cáncer de pulmón, cáncer pancreático y cáncer de tejido blando. En una realización, el tumor sólido es cáncer endocrino, cáncer de vejiga, cáncer de mama, cáncer de cuello del útero, cáncer de colon, cáncer de duodeno, glioma, cáncer de cabeza y cuello, cáncer de riñón, cáncer de hígado, cáncer de pulmón (p. ej., cáncer de pulmón no microcítico, NSCLC), cáncer de esófago, cáncer de tiroides o cáncer pancreático.
En otra realización, el cáncer es cáncer de vejiga, cáncer de mama (por ejemplo, Her positivo, Her negativo o EGFR positivo), cáncer del SNC (incluidos neuroblastoma y glioma), cáncer de colon, cáncer gastrointestinal (por ejemplo, cáncer de estómago y cáncer de colon), cáncer endocrino (por ejemplo, cáncer de tiroides o cáncer de la glándula adrenal), cáncer genitourinario femenino (por ejemplo, cáncer de cuello del útero, cáncer de ovario de células claras, cáncer de vulva, cáncer de útero o cáncer de ovario), cáncer de cabeza y cuello, cáncer hematopoyético (por ejemplo, leucemia o mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC o SCLC), melanoma, cáncer de páncreas, cáncer de próstata o cáncer de tejido blando (por ejemplo, sarcoma u osteosarcoma).
En otra realización, el cáncer es cáncer de vejiga, cáncer de mama (por ejemplo, Her positivo, Her negativo o EGFR positivo), cáncer del SNC (por ejemplo, glioma o neuroblastoma), cáncer de colon, cáncer gastrointestinal (por ejemplo, cáncer de estómago), cáncer endocrino (por ejemplo, cáncer de tiroides o cáncer de la glándula adrenal), cáncer genitourinario femenino (por ejemplo, cáncer de útero, de cuello del útero, de ovario de células claras o de vulva), cáncer de cabeza y cuello, cáncer hematopoyético (por ejemplo, leucemia o mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC o SCLC), melanoma, cáncer de páncreas, cáncer de próstata o cáncer de tejido blando (por ejemplo, sarcoma u osteosarcoma).
En incluso otra realización, el cáncer es un cáncer indicado en la tabla 44.
Además, los métodos pueden ser para tratar o prevenir el carcinoma hepatocelular (HCC), comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento.
Además, los métodos pueden ser para tratar o prevenir cáncer colorrectal (CRC, por sus siglas en inglés), melanoma, cáncer gástrico, HCC, cáncer de pulmón, cáncer pancreático, leucemia o mieloma múltiple, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 como se describe en el presente documento o una composición farmacéutica de este, como se describe en el presente documento. En una realización, el CRC, cáncer gástrico o HCC es un cáncer caracterizado por una mutación de p-catenina. Además, los métodos pueden ser para tratar o prevenir cáncer colorrectal (CRC), cáncer gástrico, HCC, cáncer de pulmón, cáncer pancreático, leucemia y mieloma múltiple, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1 como se describe en el presente documento.
En otra realización, los métodos son para tratar la leucemia comprendiendo administrar una forma sólida del compuesto 1 como se describe en el presente documento o una composición farmacéutica de este. La leucemia puede ser leucemia mielógena crónica (CML, por sus siglas en inglés). En otra realización, la leucemia es leucemia mielógena aguda (AML, por sus siglas en inglés). En una realización, la leucemia es AML con mutación de FLT-3.
En otra realización, los métodos son para tratar el linfoma, comprendiendo administrar una forma sólida del compuesto 1 como se describe en el presente documento o una composición farmacéutica de este. El linfoma puede ser linfoma de Burkitt. En una realización, la leucemia es linfoma de Hodgkin. En otra realización, la leucemia es un linfoma de linfocitos B. En otra realización, la leucemia es un linfoma de linfocitos T. En incluso otra realización, el linfoma es linfoma de efusión primaria (PEL, por sus siglas en inglés).
Las formas sólidas del compuesto 1 muestran actividad antiproliferativa en diversas líneas celulares cancerosas. (Tabla 44) La actividad antiproliferativa en estas líneas celulares cancerosas indica que las formas sólidas del compuesto 1 son útiles en el tratamiento de cánceres, incluidos tumores sólidos y hematopoyéticos. En una realización, los tumores sólidos y hematopoyéticos se seleccionan de cáncer de vejiga, cáncer de mama, cáncer del SNC (por ejemplo, neuroblastoma, meduloblastoma y glioma), cáncer de colon, cáncer de duodeno, cáncer endocrino (por ejemplo, cáncer de tiroides y cáncer de glándulas adrenales), cáncer genitourinario femenino (por ejemplo, cáncer de útero, cáncer de cuello del útero, cáncer de ovario y cáncer de vulva), cáncer de cabeza y cuello (por ejemplo, cáncer de esófago), cáncer hematopoyético y linfoide (por ejemplo, linfoma, leucemia y mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC y SCLC), cáncer de páncreas, cáncer de próstata, cáncer de piel (por ejemplo, melanoma y carcinoma), cáncer de tejido blando (por ejemplo, sarcoma y osteosarcoma), cáncer de estómago y cáncer de testículos. En una realización, los tumores sólidos y hematopoyéticos se seleccionan de cáncer de vejiga, cáncer de mama, cáncer del SNC (por ejemplo, neuroblastoma, meduloblastoma y glioma), cáncer de colon, cáncer de duodeno, cáncer endocrino (por ejemplo, cáncer de tiroides y cáncer de glándulas adrenales), cáncer genitourinario femenino (por ejemplo, cáncer de útero, cáncer de cuello del útero y cáncer de vulva), cáncer de cabeza y cuello, cáncer hematopoyético y linfoide (por ejemplo, linfoma, leucemia y mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC y SCLC), cáncer de páncreas, cáncer de próstata, cáncer de piel (por ejemplo, melanoma y carcinoma), cáncer de tejido blando (por ejemplo, sarcoma y osteosarcoma), cáncer de estómago y cáncer de testículos.
En otra realización, las formas sólidas del compuesto 1 descrito en el presente documento inducen apoptosis en diversas líneas celulares cancerosas. La inducción de apoptosis indica que las formas sólidas del compuesto 1 descritas en el presente documento son útiles en el tratamiento de cánceres, incluidos tumores sólidos y hematopoyéticos. En una realización, los tumores sólidos y hematopoyéticos se seleccionan de cáncer de vejiga, cáncer de mama, cáncer del SNC (por ejemplo, neuroblastoma y glioma), cáncer de colon, cáncer de duodeno, cáncer endocrino (por ejemplo, cáncer de tiroides y cáncer de glándulas adrenales), cáncer genitourinario femenino (por ejemplo, cáncer de útero, cáncer de cuello del útero, cáncer de ovario y cáncer de vulva), cáncer de cabeza y cuello (por ejemplo, cáncer de esófago), cáncer hematopoyético y linfoide (por ejemplo, linfoma, leucemia y mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC y SCLC), cáncer de páncreas, cáncer de próstata, cáncer de piel (por ejemplo, melanoma y carcinoma), cáncer de tejido blando (por ejemplo, sarcoma y osteosarcoma), cáncer de estómago y cáncer de testículos. En una realización, los tumores sólidos y hematopoyéticos se seleccionan de cáncer de vejiga, cáncer de mama, cáncer del SNC (por ejemplo, neuroblastoma y glioma), cáncer de colon, cáncer de duodeno, cáncer endocrino (por ejemplo, cáncer de tiroides y cáncer de glándulas adrenales), cáncer genitourinario femenino (por ejemplo, cáncer de vulva), cáncer de cabeza y cuello (por ejemplo, cáncer de esófago), cáncer hematopoyético y linfoide (por ejemplo, linfoma y leucemia), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC y SCLC), cáncer de páncreas, cáncer de próstata, cáncer de piel (por ejemplo, melanoma), cáncer de tejido blando (por ejemplo, sarcoma y osteosarcoma), cáncer de estómago y cáncer de testículos. En una realización, los tumores sólidos y hematopoyéticos se seleccionan de cáncer de vejiga, cáncer de mama, cáncer del SNC (por ejemplo, meduloblastoma, neuroblastoma y glioma), cáncer de colon, cáncer de duodeno, cáncer endocrino (por ejemplo, cáncer de tiroides y cáncer de glándulas adrenales), cáncer genitourinario femenino (por ejemplo, cáncer de placenta, cáncer de útero, cáncer de cuello del útero, cáncer de ovario y cáncer de vulva), cáncer de cabeza y cuello (por ejemplo, cáncer de esófago), cáncer hematopoyético y linfoide (por ejemplo, linfoma, leucemia y mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC y SCLC), cáncer de páncreas, cáncer de próstata, cáncer de piel (por ejemplo, melanoma y carcinoma), cáncer de tejido blando (por ejemplo, sarcoma y osteosarcoma), cáncer de
estómago y cáncer de testículos. En incluso otra realización, el cáncer es un cáncer indicado en la tabla 44.
Además los métodos pueden ser para tratar o prevenir un cáncer caracterizado por una mutación de BRAF y/o una mutación de beta-catenina (indicados de forma alternativa como mutación de CTNNB1), comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer se caracteriza por una mutación de BRAF. En otra realización, el cáncer se caracteriza por una mutación de beta-catenina. En incluso otra realización, el cáncer se caracteriza por una ruta activada de beta-catenina. En algunas de dichas realizaciones, el cáncer es CRC o melanoma, caracterizado por una mutación de BRAF. En otras realizaciones, el cáncer es CRC caracterizado por una mutación de beta-catenina, que comprende adicionalmente una mutación del EGFR o mayor actividad del EGFR (por ejemplo, CRC caracterizado por una ruta de beta-catenina activada y una mutación del EGFR o CRC caracterizado por una ruta de beta-catenina activada y mayor actividad del EGFR). En incluso otras realizaciones, el cáncer es cáncer gástrico caracterizado por una mutación de beta-catenina, que comprende adicionalmente una mutación de KRAS (es decir, cáncer gástrico caracterizado por una ruta de betacatenina activada y una mutación de KRAS). En otra realización, el cáncer es HCC, caracterizado por una ruta de beta-catenina activada. En algunas de dichas realizaciones, la mutación de BRAF es BRAF V660E. En otras realizaciones, la mutación de BRAF es una o más de BRAF V600E, BRAF T119S o BRAF G596R. En algunas de dichas realizaciones, la mutación de betacatenina es una o más de beta-catenina S33Y, G34E, S45del o S33C. En algunas de dichas realizaciones, la mutación del EGFR es una o más de EGFR E282K, G719S, P753S o V1011M. En algunas de dichas realizaciones, la mutación de KRAS es A146T, G12C, G12D, G12V, G13D o Q61L.
Además, los métodos pueden ser para tratar o prevenir un cáncer con expresión de PD-L1, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer con expresión de PD-L1 es melanoma, cáncer de pulmón, carcinoma de células renales (RCC) o HCC.
Además, los métodos pueden ser para tratar o prevenir un cáncer caracterizado por una mutación de BRAF, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer caracterizado por una mutación de BRAF es CRC, cáncer de tiroides, melanoma o cáncer de pulmón. En algunas de dichas realizaciones, el cáncer caracterizado por una mutación de BRAF es CRC, cáncer de tiroides o cáncer de pulmón. En algunas de dichas realizaciones, la mutación de BRAF es BRAF V660E. En otras realizaciones, la mutación de BRAF es una o más de BRAF V600E, BRAF T119S o BRAF G596R.
Además, los métodos pueden ser para tratar o prevenir un cáncer caracterizado por una mutación de NRAS, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer caracterizado por una mutación de NRAS es melanoma.
Además, los métodos pueden ser para tratar o prevenir un cáncer caracterizado por una mutación de KRAS, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer caracterizado por una mutación de KRAS es CRC, cáncer de páncreas o cáncer de pulmón.
Además, los métodos pueden ser para tratar o prevenir un cáncer caracterizado por una mutación de beta-catenina, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. Además, los métodos pueden ser para tratar o prevenir un cáncer caracterizado por una ruta de beta-catenina activada, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer caracterizado por una mutación de beta-catenina es CRC, cáncer de estómago, HCC o sarcoma. En algunas de dichas realizaciones, el cáncer caracterizado por una ruta activada de beta-catenina es CRC, cáncer de estómago, HCC o sarcoma.
Además, los métodos pueden ser para tratar o prevenir el carcinoma hepatocelular (HCC), comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el HCC se caracteriza por una mutación de betacatenina y/o mayor expresión de YAP. En algunas de dichas realizaciones, e1HCC se caracteriza por una ruta activada de beta-catenina y/o mayor expresión de amplificación de YAP. En algunas realizaciones, la mayor expresión de YAP se debe a amplificación o mutación.
Además, los métodos pueden ser para tratar o prevenir el cáncer colorrectal (CRC), comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el CRC se caracteriza por una mutación de BRAF y/o mutación de beta-catenina. En algunas de dichas realizaciones, el CRC se caracteriza por una mutación de BRAF y/o una ruta activada de beta-catenina.
Además, los métodos pueden ser para tratar o prevenir el cáncer gástrico, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer gástrico se caracteriza por una mutación de betacatenina. En algunas de dichas realizaciones, el cáncer gástrico se caracteriza por una ruta activada de betacatenina.
Además, los métodos pueden ser para tratar o prevenir el melanoma, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el melanoma se caracteriza por una mutación de BRAF y/o mutación de NRAS.
Además los métodos pueden ser para predecir la respuesta al tratamiento con una forma sólida del compuesto 1 que se describe en el presente documento en un paciente que tiene un cáncer caracterizado por una mutación génica, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener una secuencia génica de uno o más genes seleccionados de BRAF, NRAS, KRAS y/o CTNNB1 en dicha muestra biológica de prueba, c) comparar dicha o dichas secuencias génicas con la o las secuencias génicas de una muestra biológica de tipo salvaje, donde la presencia de una mutación indica una mayor probabilidad de respuesta al tratamiento del cáncer de dicho paciente con una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento.
Además, los métodos pueden ser para predecir la eficacia terapéutica de una forma sólida del compuesto 1 que se describe en el presente documento para tratar a un paciente con un cáncer caracterizado por una mutación génica, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener una o más secuencias génicas de uno o más genes seleccionados de BRAF, NAS, KRAS y/o CTNNB1 en dicha muestra biológica de prueba, c) comparar dicha o dichas secuencias génicas con la o las secuencias génicas de una muestra biológica de tipo salvaje, donde la presencia de una mutación indica una mayor probabilidad de eficacia terapéutica de dicho tratamiento con una forma sólida del compuesto 1 descrito en el presente documento para dicho paciente. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento.
En algunas realizaciones los métodos son para tratar y prevenir la metástasis del cáncer, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, el cáncer es un cáncer metastásico, en particular, un tumor sólido metastásico o cáncer hematológico metastásico, en donde el tumor sólido y el cáncer hematológico son según se describe en el presente documento. En otras realizaciones los métodos son para tratar y prevenir la metástasis del cáncer, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En incluso otro aspecto, en el presente documento se proporcionan métodos para erradicar las células madre cancerosas en un sujeto, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En otras realizaciones, en el presente documento se proporcionan métodos para inducir la diferenciación en células madre cancerosas en un sujeto, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En otras realizaciones, en el presente documento se proporcionan métodos para inducir la muerte de células madre cancerosas en un sujeto, comprendiendo administrar a un sujeto que lo necesite una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas de dichas realizaciones, el cáncer es un tumor sólido o un cáncer hematológico, como se describe en el presente documento.
En una realización, en el presente documento se proporcionan métodos para lograr un criterio de evaluación de respuesta en tumores sólidos (RECIST, por sus siglas en inglés, 1.1) de respuesta total, respuesta parcial o enfermedad estable en un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento. En otra realización, los métodos son para aumentar las tasas de supervivencia libre de avance, según se determinan mediante cálculos estimativos de Kaplan-Meier.
En una realización, los métodos son para prevenir o retrasar un criterio de evaluación de respuesta en tumores sólidos (RECIST 1.1) de enfermedad progresiva en un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que presente un tumor sólido, como se describe en el presente documento. En una realización, la prevención o el retraso de la enfermedad progresiva se caracterizan o logran mediante un cambio en el tamaño global de las lesiones objetivo, por ejemplo, de entre -30 % y 20 %, en comparación con el tamaño antes del tratamiento. En otra realización, el cambio de tamaño de las lesiones objetivo es una reducción del tamaño global de más de 30 %, por ejemplo, más de 50 % de reducción del tamaño de la lesión objetivo, en comparación con el tamaño antes del tratamiento. En otra, la prevención se caracteriza o logra mediante una reducción del tamaño o un retraso en el avance de lesiones no objetivo, en comparación con el estado antes del tratamiento. En una realización, la prevención se logra o caracteriza por una reducción en la cantidad de lesiones objetivo, en comparación con la cantidad antes del tratamiento. En otra, la prevención se logra o caracteriza por una reducción en la cantidad o calidad de lesiones no
objetivo, en comparación con la cantidad o calidad antes del tratamiento. En una realización, la prevención se logra o caracteriza por ausencia o desaparición de lesiones objetivo, en comparación con el estado antes del tratamiento. En otra, la prevención se logra o caracteriza por ausencia o desaparición de lesiones no objetivo, en comparación con el estado antes del tratamiento. En otra realización, la prevención se logra o caracteriza por la prevención de nuevas lesiones, en comparación con el estado antes del tratamiento. En incluso otra realización, la prevención se logra o caracteriza por la prevención de síntomas o signos clínicos de avance de la enfermedad, en comparación con el estado antes del tratamiento, como caquexia relacionada con el cáncer o mayor dolor. En una realización, el cáncer es un cáncer indicado en la tabla 44.
En determinadas realizaciones, los métodos son para disminuir el tamaño de lesiones objetivo en un paciente en comparación con el estado antes del tratamiento, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento.
En determinadas realizaciones, los métodos son para disminuir el tamaño de una lesión no objetivo en un paciente en comparación con el estado antes del tratamiento, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento.
En determinadas realizaciones, los métodos son para para lograr una reducción en la cantidad de lesiones objetivo en un paciente en comparación con el estado antes del tratamiento, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento.
En determinadas realizaciones, los métodos son para para lograr una reducción en la cantidad de lesiones no objetivo en un paciente en comparación con el estado antes del tratamiento, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento.
En determinadas realizaciones, los métodos son para para lograr la desaparición de todas las lesiones objetivo en un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento.
En determinadas realizaciones, los métodos son para para lograr la desaparición de todas las lesiones no objetivo en un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido, como se describe en el presente documento.
En determinadas realizaciones, los métodos son para para tratar un cáncer, en particular un tumor sólido según se describe en el presente documento, en donde los métodos comprenden administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular un tumor sólido, donde el tratamiento produce una respuesta total, una respuesta parcial o una enfermedad estable, según se determine mediante los criterios de evaluación de respuesta en tumores sólidos (RECIST 1.1).
En determinadas realizaciones, los métodos son para para tratar un cáncer, en particular un tumor sólido según se describe en el presente documento, en donde los métodos comprenden administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular un tumor sólido como se describe en el presente documento, donde el tratamiento produce una reducción en el tamaño de la lesión objetivo, una reducción en el tamaño de la lesión no objetivo y/o ausencia de nuevas lesiones objetivo y/o no objetivo, en comparación con el estado antes del tratamiento. En una realización, el cáncer es un cáncer indicado en la tabla 44.
En determinadas realizaciones, los métodos son para para tratar un cáncer, en particular un tumor sólido según se describe en el presente documento, en donde los métodos comprenden administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular un tumor sólido como se describe en el presente documento, donde el tratamiento produce prevención o retraso del avance clínico, como caquexia relacionada con el cáncer o mayor dolor.
En otra realización, los métodos son para para inducir una respuesta terapéutica caracterizada con los Criterios del Taller Internacional (IWC) de NHL (véase Cheson BD, Pfistner B, Juweid, ME, et. ál. Revised Response Criteria for Malignant Lymphoma. J. Clin. Oncol: 2007: (25) 579-586) de un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular cánceres hematológicos, tales como linfoma, como se describe en el presente documento. En otra realización, los métodos son para para lograr una remisión completa, remisión parcial o enfermedad estable, según se determine mediante los criterios del Taller Internacional (IWC) de NHL en un paciente, comprendiendo
administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular cánceres hematológicos tales como linfoma, como se describe en el presente documento. En otra realización, los métodos son para para lograr un aumento en la supervivencia global, la supervivencia libre de avance, la supervivencia sin episodios, el tiempo hasta el avance, la supervivencia libre de enfermedad o la supervivencia libre de linfoma, según se determine mediante los criterios del Taller Internacional (IWC) de NHL en un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular cánceres hematológicos tales como linfoma, como se describe en el presente documento.
En otra realización, los métodos son para para inducir una respuesta terapéutica evaluada a través de los criterios de respuesta uniformes internacionales (IURC, por sus siglas en inglés) para mieloma múltiple (véase Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia, 2006; (10) 10: 1-7) de un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 a un paciente que padezca un cáncer, en particular mieloma múltiple. En otra realización, los métodos son para para lograr una respuesta total estricta, respuesta total, respuesta parcial muy buena o respuesta parcial, según se determine mediante los criterios de respuesta uniformes internacionales para mieloma múltiple (IURC) en un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular mieloma múltiple. En otra realización, los métodos son para para lograr un aumento en la supervivencia global, la supervivencia libre de avance, la supervivencia sin episodios, el tiempo hasta el avance o la supervivencia libre de enfermedad, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular mieloma múltiple.
En otra realización, los métodos son para para inducir una respuesta terapéutica evaluada con la evaluación de respuesta neurooncológica (RANO) del Grupo de Trabajo de GMB (véase Wen P., Macdonald, DR., Reardon, DA., et al. Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J. Clin. Oncol. 2010; 28: 1963-1972) de un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular glioblastoma multiforme (GBM). En una realización, se utiliza RANO para establecer la proporción de sujetos sin avance a los 6 meses desde el día 1 de tratamiento, con respecto a sujetos en los que se puede evaluar la eficacia en el tipo con GBM.
En otra realización, los métodos son para para mejorar el estado de rendimiento del grupo cooperativo oriental de oncología (ECOG, por sus siglas en inglés) de un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido o un cáncer hematológico, como se describe en el presente documento.
En otra realización, los métodos son para para inducir una respuesta terapéutica evaluada por el resultado de tomografía de emisión de positrones (PET) de un paciente, comprendiendo administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular, un tumor sólido o un cáncer hematológico, como se describe en el presente documento. En determinadas realizaciones, los métodos son para para tratar un cáncer, en particular un tumor sólido o cáncer hematológico según se describe en el presente documento, en donde los métodos comprenden administrar una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento a un paciente que padezca un cáncer, en particular un tumor sólido o cáncer hematológico como se describe en el presente documento, donde el tratamiento produce una reducción en la actividad metabólica del tumor, por ejemplo, según la medición a través de imagenología PET.
Adicionalmente, los métodos son para para tratar a pacientes que hayan recibido previamente tratamiento contra un cáncer, en particular un tumor sólido o un cáncer hematológico descrito en el presente documento, así como aquellos que no hayan recibido tratamiento previo. Dichos métodos incluyen la administración de una forma sólida del compuesto 1 descrito en el presente documento. Dado que los pacientes con cáncer presentan manifestaciones clínicas heterogéneas y resultados clínicos variables, el tratamiento que se le administre a un paciente puede variar según su pronóstico. Los expertos en medicina podrán determinar fácilmente, sin experimentación innecesaria, los agentes secundarios específicos, los tipos de cirugía y los tipos de tratamiento estándar no basado en fármacos que se pueden utilizar de forma eficaz para tratar un paciente individual con cáncer.
BIOMARCADORES
En una realización, los métodos son para para modular los niveles de un biomarcador en un sujeto que padezca un cáncer como se describe en el presente documento, comprendiendo administrar a dicho sujeto una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, la modulación del marcador se evalúa en una muestra biológica del sujeto, tal como en sangre en circulación, biopsias epiteliales, biopsias tumorales, células tumorales en circulación, cabello y/u orina. En una realización, la muestra biológica es de células mononucleares de sangre periférica (PBMC, por sus siglas en inglés). En dichas realizaciones, la cantidad de modulación del biomarcador se evalúa comparando la cantidad de biomarcador antes y
después de administrar la forma sólida del compuesto 1 descrito en el presente documento o una composición farmacéutica de este. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia. En algunas realizaciones diferentes, la modulación del biomarcador es un aumento de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia. En algunas realizaciones, el biomarcador es ERK, RSK1, DUSP4, DUSP5, DUSP6, BMF, EFNA1, EGR1, ETV5, FOS, FOSL1, GJA1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1, MAFF, CITED2, ELF3 o PD-L1. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de fosforilación de uno o más de ERK y RSK1. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia. En algunas realizaciones diferentes, la modulación del biomarcador es un aumento de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es uno o más de DUSP4, DUSP6, ciclina D1, c-Myc, SPRY2 y YAP. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de DUSP4, DUSP6, ciclina D1, c-Myc e YAP. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de DUSP4, DUSP6, SPRY2, c-Myc y ciclina D1. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia. En algunas realizaciones, el biomarcador es uno o más de DUSP4, DUSP6, ciclina D1, c-Myc, SPRY2 y YAP. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de DUSP4, DUSP6, ciclina D1, c-Myc e YAP. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de DUSP4, DUSP6, SPRY2, c-Myc y ciclina D1. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia. En algunas realizaciones, el biomarcador es uno o más de DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL8, SPRY2 y SPRY4. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL8, SPRY2 y SPRY4 En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es uno o más de BMF y EFNA. En algunas realizaciones de este tipo, la modulación se mide a través de medición del aumento de los niveles de expresión de ARNm y/o proteína de uno o más de BMF y EFNA1. En algunas realizaciones, la modulación del biomarcador es un aumento de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es GJA1. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la modulación de los niveles de expresión de ARNm y/o proteína de uno o más de GJA1. En algunas realizaciones de este tipo, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia. En algunas realizaciones, la modulación del biomarcador es un aumento de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es uno o más de Axina2, CTGF, Cur61 y AREG. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de Axina2, CTGF y AREG. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es uno o más de CYR61, CXCL1, HAS2, HES1 y MAFF. En algunas realizaciones de este tipo, la modulación se mide a través de medición de la reducción de los niveles de expresión de ARNm y/o proteína de uno o más de CYR61, CXCL1, HAS2, HES1 y MAFF. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es uno o más de CITED2 y ELF3. En algunas realizaciones de este tipo, la modulación se mide a través de medición del aumento de los niveles de expresión de ARNm y/o proteína de uno o más de CITED2 y ELF3. En algunas realizaciones, la modulación del biomarcador es un aumento de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En algunas realizaciones, el biomarcador es PD-L1. En algunas realizaciones, la modulación de los niveles de biomarcador es una reducción en los niveles de expresión en la superficie celular de PD-L1. En algunas realizaciones, la modulación del biomarcador es una reducción de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En otra realización, el biomarcador es IFNy o IL-2. En algunas realizaciones de este tipo, la modulación de los niveles de biomarcador es un aumento en los niveles de expresión de proteína y/o ARNm de IFNy o IL-2. En algunas realizaciones de este tipo, la modulación de los niveles de expresión de proteína y/o ARNm de IFNy o IL-2 es un aumento de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En otra realización, el biomarcador es IL-8. En algunas realizaciones de este tipo, la modulación de los niveles de biomarcador es una disminución en los niveles de expresión de proteína y/o ARNm de IL-8. En algunas realizaciones de este tipo, la modulación de los niveles de expresión de proteína y/o ARNm de IL-8 es una disminución de aproximadamente el 10 %, 20 %, 25 %, 30 %, 40 %, 50 %, 60 %, 70 %, 75 %, 80 %, 90 %, 95 %, 99 % o aproximadamente el 100 %, en comparación con los niveles de referencia.
En una realización, los métodos son para para inhibir la fosforilación de ERK y/o RSK1 en un sujeto que padezca un cáncer como se describe en el presente documento, comprendiendo administrar a dicho sujeto una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones de este tipo, la inhibición de la fosforilación se evalúa en una muestra biológica del sujeto, como en células tumorales y/o sangre en circulación, biopsias epiteliales y/o biopsias tumorales o aspirado. En dichas realizaciones, la cantidad de inhibición de la fosforilación se evalúa comparando la cantidad de fosfo-ERK y/o RSK1 antes y después de administrar la forma sólida del compuesto 1 que se proporciona en el presente documento. En determinadas realizaciones, los métodos son para para medir la inhibición de la fosforilación de ERK y/o RSK1 en un sujeto que padezca cáncer como se describe en el presente documento, comprendiendo administrar a dicho sujeto una cantidad eficaz de una forma sólida del compuesto 1 que se proporciona en el presente documento, medir la cantidad de ERK y/o RSK1 fosforilados en dicho sujeto y comparar dicha cantidad de ERK y/o RSK fosforilados con la de dicho sujeto antes de la administración de la cantidad eficaz de la forma sólida del compuesto 1 que se proporciona en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
En determinadas realizaciones, los métodos son para para inhibir la fosforilación de ERK y/o RSK1 en una muestra biológica de un sujeto que padezca un cáncer como se describe en el presente documento, comprendiendo administrar a dicho sujeto una cantidad eficaz de una forma sólida del compuesto 1 que se proporciona en el presente documento y comparar la cantidad de ERK y/o RSK1 fosforilados en una muestra biológica de un sujeto obtenida antes y después de la administración de dicha forma sólida del compuesto 1 que se proporciona en el presente documento, donde menos ERK y/o RSK1 fosforilados en dicha muestra biológica obtenida tras la administración de dicha forma sólida del compuesto 1 que se proporciona en el presente documento con respecto a la cantidad de ERK y/o RSK1 fosforilados en dicha muestra biológica obtenida antes de la administración de dicha forma sólida del compuesto 1 que se proporciona en el presente documento indica inhibición. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para determinar si un paciente presenta sensibilidad con respecto a una forma sólida del compuesto 1 que se describe en el presente documento, comprendiendo administrar a dicho paciente la forma sólida del compuesto 1 que se describe en el presente documento y determinar si se inhibe o no la fosforilación de ERK y/o RSK1 en dicho paciente midiendo la cantidad de ERK y/o RSK1 fosforilados en una muestra biológica de dicho pacientes antes y después de la administración de una forma sólida del compuesto 1 que se describe en el presente documento a dicho paciente, donde la inhibición de la fosforilación de ERK y/o RSK1 indica que dicho paciente presenta sensibilidad con respecto a dicha forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para determinar la cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento para el tratamiento de un cáncer que se pueda tratar mediante inhibición de la fosforilación
de ERK y/o RSK1 en un paciente, comprendiendo administrar a dicho paciente dosis variables de dicha forma sólida del compuesto 1 que se describe en el presente documento y determinar la cantidad de inhibición de la fosforilación de ERK y/o RSK1 en dicho paciente como resultado de cada dosis de dicha forma sólida del compuesto 1 que se describe en el presente documento midiendo la cantidad de ERK y/o RSK1 fosforilados en una muestra biológica de dicho paciente antes y después de la administración de cada dosis de una forma sólida del compuesto 1 descrito en el presente documento a dicho paciente, donde la inhibición de la fosforilación de ERK y/o RSK1 en al menos aproximadamente el 10 %, aproximadamente el 20 %, aproximadamente el 30 %, aproximadamente el 40 %, aproximadamente el 50 % o más de aproximadamente el 50 % corresponde a una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para predecir la respuesta al tratamiento con una forma sólida del compuesto 1 descrito en el presente documento en un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde una reducción en los niveles de expresión de proteína y/o ARNm en la muestra biológica de prueba de dicho paciente con respecto a dicha muestra biológica de tipo salvaje indica una mayor probabilidad de respuesta al tratamiento del cáncer de dicho paciente con una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para predecir la eficacia terapéutica del tratamiento con una forma sólida del compuesto 1 descrito en el presente documento de un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axin2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde una reducción en los niveles de expresión de proteína y/o ARNm indica una mayor probabilidad de eficacia terapéutica de dicho tratamiento con una forma sólida del compuesto 1 descrito en el presente documento para dicho paciente. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para determinar si un paciente presenta sensibilidad a una forma sólida del compuesto 1 descrito en el presente documento, comprendiendo administrar a dicho paciente la forma sólida del compuesto 1 descrito en el presente documento y determinar si se inhiben o no los niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en dicho paciente midiendo la cantidad de niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de una forma sólida del compuesto 1 que se describe en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para determinar la cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento para el tratamiento de un cáncer que se pueda tratar mediante inhibición de los niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en un paciente, comprendiendo administrar a dicho paciente dosis variables de la forma sólida del compuesto 1 descrito en el presente documento y determinar la cantidad de inhibición de los niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en dicho paciente como resultado de cada dosis de dicha forma sólida del compuesto 1 descrito en el presente documento, midiendo la cantidad de niveles de expresión de proteína y/o ARNm de uno o más de DUSP4, DUSP5, DUSP6, eGr 1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de cada dosis de una forma sólida del
compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la respuesta al tratamiento con una forma sólida del compuesto 1 descrito en el presente documento en un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de uno o más de BMF, EFNA1, CITED2 y ELF3 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde un aumento en los niveles de expresión de proteína y/o ARNm en la muestra biológica de prueba de dicho paciente con respecto a dicha muestra biológica de tipo salvaje indica un mayor probabilidad de respuesta al tratamiento del cáncer de dicho paciente con una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la eficacia terapéutica del tratamiento con una forma sólida del compuesto 1 descrito en el presente documento de un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de uno o más de BMF, EFNA1, CITED2 y ELF3 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde un aumento en los niveles de expresión de proteína y/o ARNm indica una mayor probabilidad de eficacia terapéutica del tratamiento con dicha forma sólida del compuesto 1 descrito en el presente documento para dicho paciente. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar si un paciente presenta sensibilidad a una forma sólida del compuesto 1 descrito en el presente documento, comprendiendo administrar a dicho paciente la forma sólida del compuesto 1 descrito en el presente documento y determinar si aumentan o no los niveles de expresión de proteína y/o ARNm de uno o más de BMF, EFNA1, CITED2 y ELF3 en dicho paciente midiendo la cantidad de niveles de expresión de proteína y/o ARNm de uno o más de BMF, EFNA1, CITED2 y ELF3 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de una forma sólida del compuesto 1 que se describe en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar la cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento para tratamiento de un cáncer que se pueda tratar mediante aumento de los niveles de expresión de proteína y/o ARNm de uno o más de BMF, EFNA1, CITED2 y ELF3 en un paciente, comprendiendo administrar a dicho paciente dosis variables de la forma sólida del compuesto 1 descrito en el presente documento y determinar la cantidad de aumento de los niveles de expresión de proteína y/o ARNm de uno 0 más de BMF, EFNA1, CITED2 y ELF3 en dicho paciente como resultado de cada dosis de dicha forma sólida del compuesto 1 descrito en el presente documento, midiendo la cantidad de niveles de expresión de proteína y/o ARNm de uno o más de BMF, EFNA1, CITED2 y ELF3 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de cada dosis de una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la respuesta al tratamiento con una forma sólida del compuesto 1 descrito en el presente documento en un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de GJA1 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde una reducción en los niveles de expresión de proteína y/o ARNm en la muestra biológica de prueba de dicho paciente con respecto a dicha muestra biológica de tipo salvaje indica un mayor probabilidad de respuesta al tratamiento del cáncer de dicho paciente con una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica
es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la eficacia terapéutica del tratamiento con una forma sólida del compuesto 1 descrito en el presente documento de un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de GJA1 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde una reducción en los niveles de expresión de proteína y/o ARNm indica una mayor probabilidad de eficacia terapéutica del tratamiento con dicha forma sólida del compuesto 1 descrito en el presente documento para dicho paciente. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar si un paciente presenta sensibilidad a una forma sólida del compuesto 1 descrito en el presente documento, comprendiendo administrar a dicho paciente la forma sólida del compuesto 1 descrito en el presente documento y determinar si se inhiben o no los niveles de expresión de proteína y/o ARNm de GJA1 en dicho paciente midiendo la cantidad de niveles de expresión de proteína y/o ARNm de GJA1 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de una forma sólida del compuesto 1 que se describe en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar la cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento para tratamiento de un cáncer que se pueda tratar mediante inhibición de los niveles de expresión de proteína y/o ARNm de GJA1 en un paciente, comprendiendo administrar a dicho paciente dosis variables de la forma sólida del compuesto 1 descrito en el presente documento y determinar la cantidad de inhibición de los niveles de expresión de proteína y/o ARNm de GJA1 en dicho paciente como resultado de cada dosis de dicha forma sólida del compuesto 1 descrito en el presente documento, midiendo la cantidad de niveles de expresión de proteína y/o ARNm de GJA1 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de cada dosis de una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la respuesta al tratamiento con una forma sólida del compuesto 1 descrito en el presente documento en un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de GJA1 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde un aumento en los niveles de expresión de proteína y/o ARNm en la muestra biológica de prueba de dicho paciente con respecto a dicha muestra biológica de tipo salvaje indica un mayor probabilidad de respuesta al tratamiento del cáncer de dicho paciente con una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la eficacia terapéutica del tratamiento con una forma sólida del compuesto 1 descrito en el presente documento de un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión de proteína y/o ARNm de GJA1 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión de proteína y/o ARNm con los niveles de expresión de proteína y/o ARNm de una muestra biológica de tipo salvaje, donde un aumento en los niveles de expresión de proteína y/o ARNm indica una mayor probabilidad de eficacia terapéutica del tratamiento con dicha forma sólida del compuesto 1 descrito en el presente documento para dicho paciente. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar si un paciente presenta sensibilidad a una forma sólida del compuesto 1, comprendiendo administrar a dicho paciente la forma sólida del compuesto 1 descrito en el presente documento y determinar si aumentan o no los niveles de expresión de proteína y/o ARNm de GJA1 en dicho paciente midiendo la cantidad de niveles de expresión de proteína y/o ARNm de GJA1 en una muestra biológica de
dicho paciente antes y después de la administración a dicho paciente de una forma sólida del compuesto 1 que se describe en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar la cantidad eficaz de una forma sólida del compuesto 1 para tratamiento de un cáncer que se pueda tratar mediante un aumento de los niveles de expresión de proteína y/o ARNm de GJA1 en un paciente, comprendiendo administrar a dicho paciente dosis variables de la forma sólida del compuesto 1 descrito en el presente documento y determinar la cantidad de aumento de los niveles de expresión de proteína y/o ARNm de GJA1 en dicho paciente como resultado de cada dosis de dicha forma sólida del compuesto 1 descrito en el presente documento, midiendo la cantidad de niveles de expresión de proteína y/o ARNm de GJA1 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de cada dosis de una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la respuesta al tratamiento con una forma sólida del compuesto 1 descrito en el presente documento en un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión en la superficie celular de PD-L1 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión en superficie de PD-L1 con los niveles de expresión en superficie de PD-L1 de una muestra biológica de tipo salvaje, donde una reducción en los niveles de expresión en superficie de PD-L1 indica un mayor probabilidad de respuesta al tratamiento del cáncer de dicho paciente con una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para predecir la eficacia terapéutica del tratamiento con una forma sólida del compuesto 1 descrito en el presente documento de un paciente que padezca un cáncer, en donde el método comprende: a) obtener una muestra biológica de prueba del cáncer del paciente, b) obtener los niveles de expresión en la superficie celular de PD-L1 en dicha muestra biológica de prueba, c) comparar dichos niveles de expresión en superficie de PD-L1 con los niveles de expresión en superficie de PD-L1 de una muestra biológica de tipo salvaje, donde una reducción en los niveles de expresión en superficie de PD-L1 indica un mayor probabilidad de eficacia terapéutica de dicho tratamiento con una forma sólida del compuesto 1 descrito en el presente documento para dicho paciente. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar si un paciente presenta sensibilidad a una forma sólida del compuesto 1, comprendiendo administrar a dicho paciente la forma sólida del compuesto 1 descrito en el presente documento y determinar si se inhiben los niveles de expresión en superficie celular de PD-L1 en dicho paciente, midiendo la cantidad de niveles de expresión en superficie celular de PD-L1 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de una forma sólida del compuesto 1 que se describe en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
Además los métodos pueden ser para para determinar la cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento para tratamiento de un cáncer que se pueda tratar mediante los niveles de expresión en superficie celular de PD-L1 en un paciente, comprendiendo administrar a dicho paciente dosis variables de la forma sólida del compuesto 1 descrito en el presente documento y determinar la cantidad de inhibición de los niveles de expresión en superficie celular de PD-L1 en dicho paciente como resultado de cada dosis de dicha forma sólida del compuesto 1 descrito en el presente documento, midiendo la cantidad de niveles de expresión en superficie celular de PD-L1 en una muestra biológica de dicho paciente antes y después de la administración a dicho paciente de cada dosis de una forma sólida del compuesto 1 descrito en el presente documento. En algunas de dichas realizaciones, el método comprende además administrar una cantidad eficaz de una forma sólida del compuesto 1, como se describe en el presente documento. En algunas realizaciones, la muestra biológica es una biopsia tumoral. En otra realización, la muestra biológica es PBMC. En incluso otra realización, la muestra biológica es de células tumorales en circulación.
TERAPIA DE COMBINACIÓN
Las formas sólidas del compuesto 1 que se proporciona en el presente documento también se pueden combinar o utilizar en combinación con otros agentes terapéuticos útiles en el tratamiento y/o la prevención de cáncer que se describen en el presente documento.
En una realización, el método es para para tratar, prevenir o controlar el cáncer, comprendiendo administrar a un paciente una forma sólida del compuesto 1 que se proporciona en el presente documento en combinación con uno o más agentes activos secundarios y opcionalmente en combinación con una terapia de radiación, transfusiones de sangre o cirugía. En el presente documento se divulgan ejemplos de agentes activos secundarios.
Tal como se usa en el presente documento, la expresión "en combinación" incluye el uso de más de una terapia (p. ej., uno o más agentes profilácticos y/o terapéuticos). Sin embargo, el uso de la expresión "en combinación" no restringe el orden en el cual se administran las terapias (p. ej., los agentes profilácticos y/o terapéuticos) a un paciente con una enfermedad o un trastorno. Se puede administrar una primera terapia (p. ej., un agente profiláctico o terapéutico tal como una forma sólida del compuesto 1 proporcionado en el presente documento antes (p. ej., 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas o 12 semanas antes), al mismo tiempo o después (p. ej., 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas o 12 semanas después) de la administración de una segunda terapia (p. ej., un agente profiláctico o terapéutico) a un sujeto. También se contempla en el presente documento la terapia triple.
La administración a un paciente de una forma sólida del compuesto 1 que se proporciona en el presente documento y uno o más agentes activos secundarios puede producirse en forma simultánea o en secuencia mediante las mismas vías de administración o por vías diferentes. La adecuación de una vía de administración particular que se utilice para un agente activo particular dependerá del agente activo en sí mismo (p. ej., si este puede administrarse por vía oral sin descomponerse antes de ingresar al torrente sanguíneo) y el cáncer que se trate.
La vía de administración de una forma sólida del compuesto 1 descrito en el presente documento es independiente de la vía de administración de una terapia secundaria. Por los tanto, de acuerdo con estas realizaciones, una forma sólida del compuesto 1 descrito en el presente documento se administra por vía o intravenosa y la terapia secundaria se puede administrar por vía oral, parenteral, intraperitoneal, intravenosa, intraarterial, transdérmica, sublingual, intramuscular, rectal, transbucal, intranasal, liposómica, mediante inhalación, por vía vaginal, intraocular, mediante administración local por medio de catéter o endoprótesis, de forma subcutánea, intraadiposa, intraarticular, intratecal o en una forma de dosificación de liberación lenta. En una realización, una forma sólida del compuesto 1 descrito en el presente documento y una terapia secundaria se administran utilizando el mismo modo de administración, por ejemplo, oral. En otra realización, la forma sólida del compuesto 1 descrito en el presente documento se administra mediante un modo de administración, p. ej., oral, mientras que el agente secundario (un agente contra el cáncer) se administra mediante otro modo de administración, p. ej., IV.
En una realización, el segundo agente activo se administra, por ejemplo, por ruta oral, intravenosa o subcutánea, una o dos veces al día en una cantidad de aproximadamente 1 a aproximadamente 1000 mg, de aproximadamente 5 a aproximadamente 500 mg, de aproximadamente 10 a aproximadamente 350 mg o de aproximadamente 50 a aproximadamente 200 mg, de aproximadamente 1 a aproximadamente 100 mg, de aproximadamente 1 a aproximadamente 200 mg, de aproximadamente 1 a aproximadamente 300 mg, de aproximadamente 1 a aproximadamente 400 mg o de aproximadamente 1 a aproximadamente 500 mg. La cantidad específica del segundo agente activo dependerá del agente específico que se utilice, el tipo de enfermedad que se esté tratando y/o controlando, la gravedad y etapa de la enfermedad y la cantidad la forma sólida del compuesto 1 descrito en el presente documento y de cualquier otro agente activo opcional que se administren al paciente de forma simultánea.
En los métodos y las composiciones que se proporcionan en el presente documento se pueden usar uno o más agentes o principios activos secundarios junto con la forma sólida del compuesto 1 descrito en el presente documento. Los agentes activos secundarios pueden ser moléculas grandes (p. ej., proteínas) o moléculas pequeñas (p. ej., moléculas orgánicas, organometálicas o inorgánicas sintéticas).
Los ejemplos de agentes activos de moléculas grandes incluyen, pero no se limitan a, factores de crecimiento hematopoyéticos, citocinas y anticuerpos monoclonales y policlonales, en particular anticuerpos terapéuticos para antígenos del cáncer. Los agentes activos de moléculas grandes habituales son moléculas biológicas, tales como proteínas recombinantes de origen natural o sintéticas. Las proteínas particularmente útiles en los métodos y las composiciones que se proporcionan en el presente documento incluyen proteínas que estimulan la supervivencia y/o la proliferación in vitro o in vivo de células precursoras hematopoyéticas y células linfopoyéticas. Otras proteínas útiles estimulan la división y la diferenciación de progenitoras hematopoyéticas comprometidas en células in vitro o in vivo. Las proteínas particulares incluyen, pero no se limitan a: interleucinas, tales como IL-2 (incluidas IL-2 recombinante ("rIL2") e IL-2 canaripox), IL-10, IL-12 e IL-18; interferones, tales como interferón alfa-2a, interferón alfa-2b, interferón alfa-n1, interferón alfa-n3, interferón beta-Ia e interferón gamma-I b; GM-CF y GM-CSF y EPO.
En determinadas realizaciones, GM-CSF, G-CSF, SCF o EPO se administran por vía subcutánea durante aproximadamente cinco días en un ciclo de cuatro o seis semanas, en una cantidad que oscila entre aproximadamente 1 y aproximadamente 750 mg/m2/día, entre aproximadamente 25 y aproximadamente 500 mg/m2/día, entre aproximadamente 50 y aproximadamente 250 mg/m2/día o entre aproximadamente 50 y aproximadamente 200 mg/m2/día. En determinadas realizaciones, GM-CSF puede administrarse en una cantidad de aproximadamente 60 a aproximadamente 500 mcg/m2 por vía intravenosa durante 2 horas o de aproximadamente 5 a aproximadamente 12 mcg/m2/día por vía subcutánea. En determinadas realizaciones, G-CSF se puede administrar por vía subcutánea en una cantidad de aproximadamente 1 mcg/kg/día inicialmente y se puede ajustar según el aumento del conteo de granulocitos totales. La dosis de mantenimiento de G-CSF puede administrarse en una cantidad de aproximadamente 300 (en pacientes más pequeños) o 480 mcg por vía subcutánea. En determinadas realizaciones, se puede administrar EPO por vía subcutánea en una cantidad de 10000 unidades 3 veces por semana.
Las proteínas particulares que se pueden utilizar en los métodos y las composiciones incluyen, pero no se limitan a: filgrastim, sargramostim y EPO recombinante.
Las formas recombinantes y mutadas de GM-CSF pueden prepararse como se describe en las patentes estadounidenses n.° 5.391.485, 5.393.870 y 5.229.496, todas las cuales se incorporan en el presente documento mediante esta referencia. Las formas recombinantes y mutadas de G-CSF pueden prepararse como se describe en las patentes estadounidenses n.° 4.810.643, 4.999.291, 5.528.823 y 5.580.755, las cuales se incorporan en el presente documento en su totalidad mediante esta referencia.
Además, para su uso en combinación con una forma sólida del compuesto 1, en el presente documento se describen proteínas recombinantes de origen natural nativas. Además, se encuentran comprendidos los mutantes y derivados (por ejemplo, formas modificadas) de las proteínas de origen natural que presentan, in vivo, al menos parte de la actividad farmacológica de las proteínas en las que se basan. Los ejemplos de mutantes incluyen, pero no se limitan a, proteínas que tienen uno o más residuos de aminoácidos que difieren de los residuos correspondientes en las formas de origen natural de las proteínas. Además, el término "mutaciones" abarca proteínas que carecen de restos de carbohidratos normalmente presentes en sus formas de origen natural (por ejemplo, formas no glucosiladas). Los ejemplos de derivados incluyen, pero no se limitan a, derivados pegilados y proteínas de fusión, tales como proteínas formadas mediante la fusión de IgG1 o IgG3 a la proteína o la parte activa de la proteína de interés. Véase, p. ej., Penichet, M.L. y Morrison, S.L., J. Immunol. Methods 248:91 -101 (2001). Los anticuerpos que pueden usarse en combinación con una forma sólida del compuesto 1 descrito en el presente documento incluyen anticuerpos monoclonales y policlonales. Los ejemplos de anticuerpos incluyen, pero no se limitan a, trastuzumab, rituximab, bevacizumab, pertuzumab, tositumomab, edrecolomab y G250. Las formas sólidas del compuesto 1 descrito en el presente documento también pueden combinarse o usarse en combinación con anticuerpos anti-TNF-a y/o anticuerpos anti-EGFR tales como, por ejemplo, cetuximab o panitumumab.
Los anticuerpos que pueden usarse en combinación con una forma sólida del compuesto 1 descrito en el presente documento incluyen inhibidores del punto de control inmunológico, tales como anticuerpos anti-CTLA4, anti-PD1, anti-PD-L1, anti-Tim-3, anti-Lag-3. En algunas realizaciones de este tipo, los anticuerpos de PD-1 o PD-L1 son, por ejemplo, avelumab, durvalumab, MEDI0680, atezolizumab, BMS-936559, nivolumab, pembrolizumab, pidilizumab o PDR-001. En una realización de este tipo, el anticuerpo anti-Lag-3 es BMS-986016.
Otros anticuerpos que pueden usarse en combinación con una forma sólida del compuesto 1 descrito en el presente documento incluyen los anticuerpos anti-RSPO.
Los agentes activos de moléculas grandes pueden administrarse en forma de vacunas contra el cáncer. Por ejemplo, las vacunas que secretan citocinas, o provocan la secreción de estas, tales como IL-2, G-CSF y GM-CSF, se pueden utilizar en los métodos y composiciones farmacéuticas que se proporcionan en el presente documento. Véase, p. ej., Emens, L.A., et al., Curr. Opinión Mol. Ther. 3(1):77-84 (2001).
También es posible usar agentes activos secundarios que sean moléculas pequeñas para aliviar los efectos adversos asociados a la administración de una forma sólida del compuesto 1 que se describe en el presente documento. Sin embargo, al igual que algunas moléculas grandes, se considera que muchos tienen la capacidad de proporcionar un efecto sinérgico o aditivo cuando se administran con (p. ej., antes, después o en forma simultánea) una forma sólida del compuesto 1 que se describe en el presente documento. Los ejemplos de agentes activos secundarios de molécula pequeña incluyen, pero no se limitan a, agentes contra el cáncer, antibióticos, agentes inmunosupresores y esteroides.
En determinadas realizaciones, el agente secundario es un inhibidor de BRAF, un inhibidor de HSP, un inhibidor de proteasoma, un inhibidor de FLT3, un inhibidor de MEK, un inhibidor de PI3K, un inhibidor de EGFR, un compuesto inmunomodulador o un inhibidor de TOR cinasa. En algunas realizaciones de este tipo, el inhibidor de BRAF es sorafenib, dabrafenib, encorafenib o vemurafenib. En algunas realizaciones de este tipo, el inhibidor de HSP es geldanamicina, gamitrinib, luminespib o radicicol. En algunas realizaciones, el inhibidor de proteasoma es
bortezomib, carfilzomib, ixazomib, disulfiram, oprozomib, delanzomib o ixazomib. En otras realizaciones, el inhibidor de FLT3 es quizartinib, midostaurin, sorafenib, sunitinib o lestaurtinib. En algunas realizaciones de este tipo, el inhibidor de MEK es trametinib, cobimetinib, binimetinib, selumetinib, PD-325901, CI-1040 (PD184352) o Ta K-733. En algunas realizaciones más, el inhibidor de PI3K es AT7867, AZD 8055, BX-912, silmitasertib, pictilisib, MK-2206 o pilaralisib. En otra realización, el inhibidor de EGFR es gefitinib, erlotinib, afatinib, osimertinib (TAGRISSO), rociletinib o lapatinib. En algunas realizaciones más, el inhibidor de TOR cinasa es CC-115, CC-223, OSI-027, AZD8055, sapanisertib, dactolisib, BGT226, voxtalisib (SAR-245409), apitolisib, omipalisib (Gs K-2126458), PF-04691502, gedatolisib o PP242. En algunas realizaciones, el compuesto inmunomodulador es talidomida, lenalidomida, pomalidomida, CC-220 o CC-122.
Los ejemplos de otros agentes contra el cáncer que se pueden utilizar en los métodos o las composiciones que se describen en el presente documento incluyen, pero no se limitan a: acivicina, aclarubicina, clorhidrato de acodazol, acronina, adozelesina, aldesleucina, altretamina, ambomicina, acetato de ametantrona, amsacrina, anastrozol, antramicina, asparaginasa, asperlina, azacitidina, azetepa, azotomicina, batimastat, benzodepa, bicalutamida, clorhidrato de bisantreno, dimesilato de bisnafida, bizelesina, sulfato de bleomicina, brequinar sodio, bropirimina, busulfán, cactinomicina, calusterona, caracemida, carbetimer, carboplatino, carmustina, clorhidrato de carubicina, carzelesina, cedefingol, celecoxib (inhibidor de COX-2), clorambucilo, cirolemicina, cisplatino, cladribina, clofarabina, mesilato de crisnatol, ciclofosfamida, arabinoxilcitosina, dacarbazina, dabrafenib, dactinomicina, clorhidrato de daunorubicina, decitabina, dexormaplatino, dezaguanina, mesilato de dezaguanina, diazicuona, docetaxel, doxorubicina, clorhidrato de doxorubicina, droloxifeno, citrato de droloxifeno, propionato de dromostanolona, duazomicina, edatrexato, clorhidrato de eflornitina, elsamitrucina, enloplatino, enpromato, epipropidina, clorhidrato de epirubicina, erbulozol, clorhidrato de esorubicina, estramustina, fosfato sódico de estramustina, etanidazol, etopósido, fosfato de etopósido, etoprina, clorhidrato de fadrozol, fazarabina, fenretinida, floxuridina, fosfato de fludarabina, fluorouracilo, flurocitabina, fosquidona, fostriecina sodio, gemcitabina, clorhidrato de gemcitabina, hidroxiurea, clorhidrato de idarubicina, ifosfamida, ilmofosina, iproplatino, irinotecano, clorhidrato de irinotecano, acetato de lanreotida, letrozol, acetato de leuprolida, clorhidrato de liarozol, lometrexol sodio, lomustina, clorhidrato de losoxantrona, masoprocol, maitansina, clorhidrato de mecloretamina, acetato de megestrol, acetato de melengestrol, melfalán, menogaril, mercaptopurina, metotrexato, metotrexato sodio, metoprina, meturedepa, mitindomida, mitocarcina, mitocromina, mitogilina, mitomalcina, mitomicina, mitosper, mitotano, clorhidrato de mitoxantrona, ácido micofenólico, nocodazol, nogalamicina, omacetaxina, ormaplatino, oxisurán, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable, (ABRAXANE®) enlazado a albúmina, pegaspargasa, peliomicina, pentamustina, sulfato de peplomicina, perfosfamida, pipobromán, piposulfán, clorhidrato de piroxantrona, plicamicina, plomestano, porfimer sodio, porfiromicina, prednimustina, clorhidrato de procarbazina, puromicina, clorhidrato de puromicina, pirazofurina, riboprina, safingol, clorhidrato de safingol, semustina, simtraceno, sorafenib, esparfosato sodio, esparsomicina, clorhidrato de espirogermanio, espiromustina, espiroplatino, estreptonigrina, estreptozocina, sulofenur, talisomicina, tecogalán sodio, docetaxel, tegafur, clorhidrato de teloxantrona, temoporfina, tenipósido, teroxirona, testolactona, tiamiprina, tioguanina, tiotepa, tiazofurina, tirapazamina, citrato de toremifeno, acetato de trestolona, fosfato de triciribina, trimetrexato, glucuronato de trimetrexato, triptorelina, clorhidrato de tubulozol, mostaza de uracilo, uredepa, vapreotida, vemurafenib, verteporfín, sulfato de vinblastina, sulfato de vincristina, vindesina, sulfato de vindesina, sulfato de vinepidina, sulfato de vinglicinato, sulfato de vinleurosina, tartrato de vinorelbina, sulfato de vinrosidina, sulfato de vinzolidina, vorozol, zeniplatino, zinostatina y clorhidrato de zorubicina.
Otros fármacos contra el cáncer que se pueden incluir en los métodos o las composiciones incluyen, pero no se limitan a: 20-epi-1,25 dihidroxivitamina D3; 5-etiniluracilo; abiraterona; aclarubicina; acilfulveno; adecipenol; adocelesina; aldesleucina; antagonistas de ALL-TK; altretamina; ambamustina; amidox; amifostina; ácido aminolevulínico; amrubicina; amsacrina; anagrelida; anastrozol; andrografolida; inhibidores de angiogénesis; antagonista D; antagonista G; antarelix; proteína 1 morfogenética antidorsal; antiandrógenos, carcinoma prostático; antiestrógeno; antineoplastón; oligonucleótidos antisentido; afidicolín glicinato; moduladores del gen de apoptosis; reguladores de apoptosis; ácido apurínico; ara-CDP-DL-PTBA; arginina desaminasa; asulacrina; atamestano; atrimustina; axinastatina 1; axinastatina 2; axinastatina 3; azasetrona; azatoxina; azatirosina; derivados de bacatina III; balanol; batimastat; antagonistas de BCR/ABL; benzoclorinas; benzoilestaurosporina; derivados de beta lactama; beta-aletina; betaclamicina B; ácido betulínico; inhibidor de bFGF; bicalutamida; bisantreno; bisaziridinilespermina; bisnafida; bistrateno A; bizelesina; breflato; bropirimina; budotitano; butionina sulfoximina; calcipotriol; calfostina C; derivados de camptotecina; capecitabina; carboxamida-amino-triazol; carboxiamidotriazol; CaRest M3; inhibidor derivado de cartílago; carcelesina; inhibidores de caseína cinasa (ICOS); castanoespermina; cecropina B; cetrorelix; clorlnas; cloroquinoxalina sulfonamida; cicaprost; cis-porfirina; cladribina; análogos de clomifeno; clotrimazol; colismicina A; colismicina B; combretastatina A4; análogo de combretastatina; conagenina; crambescidina 816; crisnatol; criptoficina 8; derivados de criptoficina A; curacina A; ciclopentantraquinonas; cicloplatam; cipemicina; ocfosfatode citarabina; factor citolítico; citostatina; dacliximab; decitabina; deshidrodidemnina B; deslorelina; dexametasona; dexifosfamida; dexrazoxano; dexverapamil; diazicuona; didemnina B; didox; dietilnorspermina; dihidro-5-azacitidina; dihidrotaxol, 9-; dioxamicina; difenil espiromustina; docetaxel; docosanol; dolasetrón; doxifluridina; doxorubicina; droloxifeno; dronabinol; duocarmicina SA; ebselén; ecomustina; edelfosina; edrecolomab; eflornitina; elemén; emitefur; epirubicina; epristerida; análogo de estramustina; agonistas de estrógeno; antagosnistas de estrógeno; etanidazol; fosfato de etopósido; exemestano; fadrozol; fazarabiae; fenretinida; filgrastim; finasterida; flavopiridol; flezelastina; fluasterona; fludarabina; clorhidrato de fluorodaunorunicina;
forfenimex; formestano; fostriecina; fotemustina; gadolinio texafirina; nitrato de galio; galocitabina; ganirelix; inhibidores de gelatinasa; gemcitabina; inhibidores de glutationa; hepsulfam; heregulina; hexametileno bisacetamida; hipericina; ácido ibandrónico; idarubicina; idoxifeno; idramantona; ilmofosina; ilomastat; imatinib; imiquimod; péptidos inmunoestimulantes; inhibidor del receptor del factor de crecimiento tipo insulina 1; agonistas de interferón; interferonas; interleucinas; yobenguano; yododoxorubicina; ipomeanol, 4-; iroplact; irsogladina; isobengazol; isohomohalicondrina B; itasetrón; jasplacinolida; cahálalido F; triacetato de lamelarina-N; lanreotida; leinamicina; lenograstim; sulfato de lentinano; leptolstatina; letrozol; factor inhibidor de leucemia; interferón alfa de leucocito; leuprolida+estrógeno+progesterona; leuprorelina; levamisol; liarozol; análogo de poliamina lineal; péptido disacárido lipofílico; compuestos de platino lipofílicos; lisoclinamida 7; lobaplatino; lombricina; lometrexol; lonidamina; losoxantrona; loxoribina; lurtotecán; lutecio texafirina; lisofilina; péptidos líticos; maitansina; manostatina A; marimastat; masoprocol; maspín; inhibidores de matrilisina; inhibidores de metaloproteinasa de matriz; menogaril; merbarona; meterelina; metioninasa; metoclopramida; inhibidor de MIF; mifepristona; miltefosina; mirimostim; mitoguazona; mitolactol; análogos de mitomicina; mitonafida; saporina-factor de crecimiento de fibroblastos mitotoxina; mitoxantrona; mofaroteno; molgramostim; cetuximab, gonadotrofina coriónica humana; lípido monofosforilo A+pared celular de micobateria sk; mopidamol; agente de mostaza contra el cáncer; micaperóxido B; extracto de pared celular micobacteriana; miriaporona; N-acetildinalina; benzamidas N-sustituidas; nafarelina; nagrestip; naloxona+pentazocina; napavina; nafterpina; nartograstim; nedaplatino; nemorubicina; ácido neridrónico; nilutamida; nisamicina; moduladores de óxido nítrico; antioxidante de nitróxido; nitrulina; oblimersén; O6-bencilguanina; octreótido; oquicenona; oligonucleótidos; onapristona; ondansetrón; ondansetrón; oracina; inductor de citocina oral; ormaplatino; osaterona; oxaliplatino; oxaunomicina; paclitaxel; análogos de paclitaxel; derivados de paclitaxel; partículas enlazadas a proteína de paclitaxel para suspensión inyectable, (ABRAXANE®) enlazado a albúmina; palauamina; palmitoilrhizoxina; ácido pamidrónico; panaxitriol; panomifeno; parabactina; pazeliptina; pegaspargasa; peldesina; pentosán polisulfato sódico; pentostatina; pentrozol; perflubrón; perfosfamida; alcohol perilílico; fenazinomicina; fenilacetato; inhibidores de fosfatasa; picibanil; clorhidrato de pilocarpina; pirarubicina; piritrexim; placetina A; placetina B; inhibidor del activador de plasminógenos; complejo de platino; compuestos de platino; complejo de platino-triamina; porfimer sodio; porfiromicina; prednisona; propil bis-acridona; prostaglandina J2; inhibidores de proteasoma; inmunomodulador a base de proteína A; inhibidores de proteína cinasa C, microalgal; inhibidores de proteína tirosina fosfatasa; inhibidores de purina nucleósido fosforilasa; purpurinas; pirazoloacridina; conjugado de polioxietileno de hemoglobina piridoxilada; antagonistas de raf; raltitrexed; ramosetrón; inhibidores de transferasa de proteína farnesilo ras; inhibidores de ras; inhibidor de ras-GAP; reteliptina desmetilada; retidronato de enio Re 186; rizoxina; ribozimas; retinamida RII; rohitucina; romurtida; roquinimex; rubiginona B1; ruboxil; safingol; saintopín; sarmustina; sarcofitol A; sargramostim; miméticos de Sdi 1; semustina; inhibidor 1 derivado de senescence; oligonucleótidos sentido; inhibidores de transducción de señal; sizofirán; sobuzoxano; borocaptato sódico; fenilacetato de sodio; solverol; proteína de unión a somatomedina; sonermin; ácido esparfósico; espicamicina D; espiromustina; esplenopentina; espongistatina 1; escualamina; estipiamida; inhibidores de estromelisina; sulfinosina; antagonista superactivo del péptido intestinal vasoactivo; suradista; suramina; swainsonina; talimustina; tamoxifén metiodida; tauromustina; tazaroteno; tecogalán sodio; tegafur; telurapirilio; inhibidores de telomerasa; temoporfín; tenipósido; tetraclorodecaóxido; tetrazomina; taliblastina; tiocoralina; trombopoyetina; mimético de trombopoyetina; timalfasina; agonista del receptor de timopoyetina; timotrinán; hormona que estimula la tiroides; etil etiopurpurina de estaño; tirapazamina; bicloruro de titanoceno; topsentina; toremifeno; inhibidores de la traducción; tretinoína; triacetiluridina; triciribina; trimetrexato; triptorelina; tropisetrón; turosterida; inhibidores de tirosina cinasa; tirfostinas; inhibidores de UBC; ubenimex; factor inhibidor del crecimiento derivado de senos urogenitales; antagonistas del receptor de urocinasa; vapreotida; variolina B; velaresol; veramina; verdinas; verteporfín; vinorelbina; vinxaltina; vitaxina; vorozol; zanoterona; zeniplatino; zilascorb y zinostatina estimalamer.
Los agentes activos secundarios específicos particularmente útiles en los métodos o las composiciones incluyen, pero no se limitan a, rituximab, oblimersén, infliximab, docetaxel, celecoxib, melfalán, dexametasona, esteroides, gemcitabina, cisplatino, temozolomida, etopósido, ciclofosfamida, temodar, carboplatino, procarbazina, carmustina, tamoxifeno, topotecán, metotrexato, gefitinib, paclitaxel, fluorouracilo, leucovorina, irinotecán, capacitabina, interferón alfa, interferón alfa pegilado, cisplatino, tiotepa, fludarabina, carboplatino, daunorrubicina liposómica, citarabina, vinblastina, IL-2, GM-CSF, dacarbazina, vinorrelbina, ácido zoledrónico, palmitronato, claritromicina, busulfán, prednisona, bisfosfonato, trióxido arsénico, vincristina, doxorrubicina, ganciclovir, fosfato sódico de estramustina, clinoril y etopósido.
Otros agentes activos secundarios específicos que resultan particularmente útiles en los métodos o las composiciones incluyen, pero no se limitan a, sorafenib, dabrafenib, vemurafenib, trametinib, cobimetinib, binimetinib, selumetinib, PD-325901, CI-1040 (PD184352), TAK-733, AT7867, AZD 8055, BX-912, silmitasertib, pictilisib, Mk -2206, pilaralisib, gefitinib, erlotinib, lapatinib, osimertinib, CC-115, CC-223, OSI-027, AZD8055, sapanisertib, dactolisib, BGT226, voxtalisib, apitolisib, omipalisib, PF-04691502, gedatolisib, PP242, lenalidomida, pomalidomida o CC-122.
Otros agentes activos secundarios específicos que resultan particularmente útiles en los métodos o las composiciones incluyen, pero no se limitan a, avelumab, durvalumab, MEDI0680, atezolizumab, BMS-936559, nivolumab, pembrolizumab, pidilizumab, PDR-001, sorafenib, cetuximab, panatumumab, erlotinib, trametinib, trastuzumab, CC-223, CC-122 o lapatinib.
En determinadas realizaciones de los métodos que se proporcionan en el presente documento, el uso de un agente activo secundario en combinación con una forma sólida del compuesto 1 descrito en el presente documento se puede modificar o retrasar durante o inmediatamente después de la administración de una forma sólida del compuesto 1 descrito en el presente documento, según considere adecuado el profesional experto en la técnica. En determinadas realizaciones, los sujetos a los que se les administra una forma sólida del compuesto 1 descrito en el presente documento, solo o en combinación con otras terapias, pueden recibir cuidados complementarios que incluyen antieméticos, factores de crecimiento mieloides y transfusiones de hemoderivados, cuando corresponda. En algunas realizaciones, los sujetos a los que se les administra una forma sólida del compuesto 1 descrito en el presente documento pueden recibir un factor de crecimiento como agente activo secundario según el criterio del profesional experto en la técnica.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con gemcitabina, cisplatino, 5-fluorouracilo, mitomicina, metotrexato, vinblastina, doxorrubicina, carboplatino, tiotepa, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable-con enlace a albúmina ABRAXANE®) o docetaxel a pacientes con carcinoma urotelial metastásico o con avance local.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con un principio activo secundario como se indica a continuación: temozolomida a pacientes pediátricos con neuroblastoma recurrente o tumores cerebrales progresivos o recidivantes; celecoxib, etopósido y ciclofosfamida para cáncer del SNC progresivo o recidivante; temozolomida a pacientes con meningioma progresivo o recurrente, meningioma maligno, hemangiopericitoma, metástasis cerebrales múltiples, tumores cerebrales recidivantes o glioblastoma multiforme recientemente diagnosticado; irinotecán a pacientes con glioblastoma recurrente; carboplatino a pacientes pediátricos con glioma del tronco encefálico; procarbazina a pacientes pediátricos con gliomas malignos progresivos; ciclofosfamida a pacientes con tumores cerebrales malignos con pronóstico negativo, glioblastoma multiforme recurrente o recientemente diagnosticado; carmustina para gliomas malignos recurrentes de alto grado; temozolomida y tamoxifén para astrocitoma anaplásico o topotecán para gliomas, glioblastoma, astrocitoma anaplásico u oligodendroglioma anaplásico.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con metotrexato, ciclofosfamida, 5-fluorouracilo, everolimus, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®), lapatinib, trastuzumab, pamidronato disódico, mesilato de eribulina, everolimus, gemcitabina, palbociclib, ixabepilona, ado-trastuzumab emtansina, pertuzumab, tiotepa, inhibidores de aromatasa, exemestano, moduladores de estrógenos selectivos, antagonistas de los receptores de estrógenos, antraciclinas, emtansina y/o pexidartinib a pacientes con cáncer de mama metastásico. En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con temozolomida, doxorrubicina, everolimus, fluorouracilo, 5-fluorouracilo o estreptozocina a pacientes con tumores neuroendocrinos.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con metotrexato, gemcitabina, cisplatino, cetuximab, 5-fluorouracilo, bleomicina, docetaxel o carboplatino a pacientes con cáncer de cuello o cabeza metastásico o recurrente. En una realización, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento con cetuximab a pacientes con cáncer de cabeza o cuello.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con gemcitabina, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®), 5-fluorouracilo, everolimus, irinotecán, mitomicina C, sunitinib o erlotinib a pacientes con cáncer pancreático.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con cáncer de colon, en combinación con getfitinib, erlotinib, oxaliplatino, 5-fluorouracilo, irinotecán, capecitabina, cetuximab, ramucirumab, panitumumab, bevacizumab, leucovorin calcio, LONSURF, regorafenib, zivaflibercept, trametinib, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®) y/o docetaxel. En determinadas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer de colon, en combinación con bevacizumab, clorhidrato de irinotecán, capecitabina, cetuximab, ramucirumab, oxaliplatino, cetuximab, fluorouracilo, leucovorin calcio, trifluridina y clorhidrato de tipiracilo, panitumumab, regorafenib o zivaflibercept. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer de colon, en combinación con un inhibidor de EGFR (por ejemplo, cetuximab o erlotinib) y/o un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib). En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con capecitabina, cetuximab, erlotinib, trametinib y/o vemurafenib a pacientes con cáncer colorrectal refractario o pacientes que no respondan a la terapia de primera línea o tengan un desempeño deficiente en el adenocarcinoma
rectal o de colon. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer colorrectal refractario o pacientes que no respondan a la terapia de primera línea o tengan un desempeño deficiente en el adenocarcinoma rectal o de colon, en combinación con un inhibidor de EGFR (por ejemplo, cetuximab o erlotinib) y un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib). En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer colorrectal refractario o pacientes que no respondan a la terapia de primera línea o tengan un desempeño deficiente en el adenocarcinoma rectal o de colon, en combinación con un anticuerpo anti-RSPO.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con fluorouracilo, leucovorina, trametinib y/o irinotecán a pacientes con cáncer colorrectal en fase IIIa o IV o a pacientes que hayan sido tratados previamente por cáncer colorrectal metastásico. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer colorrectal en fase IIIa a IV o a pacientes que hayan sido tratados previamente por cáncer colorrectal metastásico, en combinación con un inhibidor de EGFR (por ejemplo, cetuximab o erlotinib) y un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib). En determinadas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer colorrectal refractario, en combinación con capecitabina, xeloda, trametinib, oxaliplatino y/o irinotecán. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer colorrectal refractario, en combinación con un inhibidor de EGFR (por ejemplo, cetuximab o erlotinib) y un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib). En determinadas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento con capecitabina, trametinib y/o irinotecán a pacientes con cáncer colorrectal refractario o a pacientes con carcinoma colorrectal no extirpable o metastásico. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer colorrectal refractario o a pacientes con carcinoma colorrectal no extirpable o metastásico, en combinación con un inhibidor de EGFR (por ejemplo, cetuximab o erlotinib) y un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib).
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento sola o en combinación con interferón alfa, 5-fluorouracilo/leucovorina o capecitabina a pacientes con carcinoma hepatocelular no extirpable o metastásico, o con cisplatino o tiotepa, o con sorafenib a pacientes con cáncer de hígado primario o metastásico. En determinadas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento sola o en combinación con sorafenib, sunitinib, erlotinib y/o sirolimus a pacientes con carcinoma hepatocelular no extirpable o metastásico o con sorafenib, sunitinib, erloinib y/o rapamicina a pacientes con cáncer de hígado primario o metastásico. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento a pacientes con cáncer de hígado primario, no extirpable o metastásico, en combinación con un inhibidor del punto de control inmunológico (por ejemplo, un anticuerpo anti-CTLA4, anti-PD1, anti-PD-L1, anti-Tim-3 o anti-Lag-3) o un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib). En algunas realizaciones de este tipo, el anticuerpo anti-PD-1 o anti-PD-L1 es avelumab, durvalumab, MEDI0680, atezolizumab, BMS-936559, nivolumab, pembrolizumab, pidilizumab o PDR-001. En determinadas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento sola o en combinación con lenalidomida, pomalidomida o CC-122 a pacientes con carcinoma hepatocelular primario, no extirpable o metastásico. En determinadas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento sola o en combinación con CC-223 a pacientes con carcinoma hepatocelular primario, no extirpable o metastásico.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con cisplatino/5-fluorouracilo, ramucirumab, docetaxel, clorhidrato de doxorrubicina, inyección de fluorouracilo, trastuzumab y/o mitomicina C a pacientes con cáncer gástrico (de estómago).
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrita en el presente documento en combinación con un inhibidor del punto de control inmunológico (por ejemplo, un anticuerpo anti-CTLA4, anti-PD1, anti-PD-L1, anti-Tim-3 o anti-Lag-3) y/o un inhibidor de BRAF (por ejemplo, sorafenib, dabrafenib o vemurafenib) a pacientes con diversos tipos o diversas fases de melanoma. En algunas realizaciones, se administra una forma sólida del compuesto 1 según se describe y proporciona en el presente documento en combinación con aldesleucina, cobimetinib, dabrafenib, dacarbazina, IL-2, talimogén laherparepvec, interferón alfa-2b recombinante, ipilimumab, pembrolizumab, lapatinib, trametinib, nivolumab, peginterferón alfa-2b, aldesleucina, dabrafenib y/o vemurafenib a pacientes con diversos tipos o diversas fases de melanoma.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con doxorrubicina, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®), vinblastina o interferón alfa pegilado a pacientes con sarcoma de Kaposi.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con metotrexato, clorhidrato de mecloretamina, dimaleato de afatinib, pemetrexed, bevacizumab, carboplatino, cisplatino, ceritinib, crizotinib, ramucirumab, pembrolizumab, docetaxel, tartrato de vinorelbina, gemcitabina, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®), erlotinib, geftinib y/o irinotecán a pacientes con cáncer de pulmón no microcítico.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con carboplatino e irinotecán a pacientes con cáncer de pulmón no microcítico.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento con docetaxel a pacientes con cáncer de pulmón no microcítico que hayan recibido previamente tratamiento con radioterapia y carboplatino/etopósido.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con carboplatino y/o docetaxel o en combinación con carboplatino, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®) y/o radioterapia toráxica a pacientes con cáncer de pulmón no microcítico.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con docetaxel a pacientes con cáncer de pulmón no microcítico en fase IIIB o IV.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con oblimersén, metotrexato, clorhidrato de mecloretamina, etopósido, topotecán o doxorrubicina a pacientes con cáncer de pulmón microcítico.
En determinadas realizaciones, se administran una forma sólida del compuesto 1 descrito en el presente documento y doxetaxol a pacientes con cáncer de pulmón no microcítico previamente tratados con carbo/VP 16 y radioterapia. En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de cáncer de ovarios, tales como carcinoma peritoneal, carcinoma seroso papilar, cáncer de ovario refractario o cáncer de ovario recurrente, en combinación con carboplatino, doxorrubicina, gemcitabina, cisplatino, capecitabina, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®), dexametasona, avastina, ciclofosfamida, topotecán, olaparib, tiotepa o una combinación de estos.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de cáncer de próstata, en combinación con capecitabina, 5-fluorouracilo más leucovorina, gemcitabina, irinotecán más gemcitabina, ciclofosfamida, vincristina, dexametasona, GM-CSF, celecoxib, ganciclovir, paclitaxel, partículas enlazadas a proteína de paclitaxel para suspensión inyectable con enlace a albúmina (ABRAXANE®), docetaxel, estramustina, denderona, bicalutamida, cabazitaxl, degarelix, enzalutamida, goserelina, acetato de leuprolida, clorhidrato de mitoxantrona, prednisona, sipuleucel-T, dicloruro de radio 223 o una combinación de estos.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de cáncer de células renales, en combinación con capecitabina, IFN, tamoxifén, IL-2, GM-CSF, celecoxib o una combinación de estos.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de cánceres de ginecológico, de útero o sarcoma de tejido blando, en combinación con IFN, dactinomicina, doxorrubicina, mesilato de imatinib, pazopanib, clorhidrato, trabectedina, un inhibidor de COX-2 tal como celecoxib y/o sulindac.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de tumores sólidos, en combinación con celecoxib, etopósido, ciclofosfamida, docetaxel, apecitabina, IFN, tamoxifén, IL-2, GM-CSF o una combinación de estos.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento sola o en combinación con vinorelbina a pacientes con mesotelioma maligno o cáncer de pulmón no microcítico en fase IIIB con implantes pleurales o síndrome de mesotelioma maligno.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con A navitoclax, venetoclax y/u obatoclax a pacientes con linfoma y otros cánceres hematológicos. En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con trióxido arsénico, fludarabina, carboplatino, daunorrubicina, ciclofosfamida, citarabina, doxorrubicina, idarrubicina, clorhidrato de mitoxantrona, tioguanina, vincritina y/o topotecán a pacientes con
leucemia mieloide aguda, incluidas leucemia mieloide aguda de alto riesgo, recidivante o refractaria.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrita en el presente documento en combinación con daunorrubicina liposómica, topotecán y/o citarabina a pacientes con leucemia mieloblástica aguda con cariotipo desfavorable.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento sola o en combinación con un principio activo secundario, tal como vinblastina o fludarabina, clorambucilo, bleomicina, brentuximab vedotina, carmustina, clorambucilo, ciclofosfamida, dacarbazina, doxorrubicina, lomustina, clorhidrato de mecloretamina, prednisona, clorhidrato de procarbazina o vincristina a pacientes con diversos tipos de linfoma, que incluyen, pero no se limitan a, linfoma de Hodgkin, linfoma no Hodgkin, linfoma cutáneo de linfocitos T, linfoma cutáneo de linfocitos B, linfoma difuso de linfocitos B grandes o linfoma folicular de bajo grado recidivante o refractario.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de mieloma múltiple, en combinación con dexametasona, ácido zoledrónico, palmitronato, GM-CSF, claritromicina, vinblastina, melfalán, busulfán, ciclofosfamida, IFN, prednisona, bisfosfonato, celecoxib, trióxido arsénico, peginterferón alfa-2b, vincristina, carmustina, bortezomib, carfilzomib, doxorrubicina, panobinostat, lenalidomida, pomalidomida, talidomida, plerixafor o una combinación de estos.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con diversos tipos o diversas fases de mieloma múltiple, en combinación con linfocitos T del receptor de antígenos quiméricos (CAR, por sus siglas en inglés).
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con mieloma múltiple recidivante o refractario, en combinación con doxorrubicina, vincristina y/o dexametasona.
En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con escleroderma o vasculitis cutánea, en combinación con celecoxib, etopósido, ciclofosfamida, docetaxel, capecitabina, IFN, tamoxifén, IL-2, GM-CSF o una combinación de estos.
En el presente documento también se contempla un método para aumentar la dosificación de un fármaco o agente contra el cáncer que pueda administrarse en forma segura y eficaz a un paciente, el cual comprende administrarle al paciente (p. ej., un ser humano) una forma sólida del compuesto 1 descrito en el presente documento. Los pacientes que pueden beneficiarse con este método son aquellos que presentan probabilidad de padecer un efecto adverso asociado con los fármacos contra el cáncer para el tratamiento de un cáncer específico de la piel, de tejido subcutáneo, de ganglios linfáticos, cerebral, pulmonar, hepático, óseo, intestinal, de colon, de corazón, pancreático, adrenal, renal, prostático, de mama, colorrectal o combinaciones de estos. La administración de una forma sólida del compuesto 1 descrito en el presente documento alivia o reduce los efectos adversos de una gravedad tal que de otro modo limitarían la cantidad de fármaco contra el cáncer.
En una realización, se administra diariamente una forma sólida del compuesto 1 descrito en el presente documento en una cantidad que oscila entre aproximadamente 0,1 y aproximadamente 150 mg, entre aproximadamente 1 y aproximadamente 100 mg, entre aproximadamente 2 y aproximadamente 50 mg o entre aproximadamente 1 y aproximadamente 10 mg antes, durante o después de la aparición del efecto adverso asociado con la administración de un fármaco contra el cáncer a un paciente. En determinadas realizaciones, se administra una forma sólida del compuesto 1 descrito en el presente documento en combinación con agentes específicos tales como heparina, aspirina, cumadina, anti-Factor Xa o G-CSF, para evitar los efectos adversos asociados con los fármacos contra el cáncer, tales como, pero no limitados a, tromboembolia, neutropenia o trombocitopenia.
En una realización, se administra una forma sólida del compuesto 1 descrito en el presente documento a pacientes con enfermedades y trastornos asociados con angiogénesis no deseada o caracterizados por esta, en combinación con principios activos adicionales que incluyen, pero no se limitan a, fármacos contra el cáncer, antiinflamatorios, antihistamínicos, antibióticos y esteroides.
En otra realización, en el presente documento se contempla un método para tratar, prevenir y/o controlar el cáncer, que comprende administrar una forma sólida del compuesto 1 descrito en el presente documento junto con (p. ej., antes, durante o después) terapias convencionales que incluyen, pero no se limitan a, cirugía, inmunoterapia, terapia biológica, terapia de radiación u otras terapias no basadas en fármacos que se utilicen actualmente para tratar, prevenir y/o controlar el cáncer. El uso combinado del compuesto que se proporciona en el presente documento y terapia convencional puede brindar un régimen de tratamiento único con una eficacia inesperada en determinados pacientes. Sin deseo de limitarse a la teoría, se considera que una forma sólida del compuesto 1 descrito en el presente documento puede brindar efectos aditivos o sinérgicos al administrarse en concurrencia con una terapia convencional.
Tal como se menciona en otra parte del presente documento, el método puede ser para reducir, tratar y/o prevenir los efectos adversos o no deseados que se asocian con una terapia convencional que incluye, pero no se limitan a, cirugía, quimioterapia, terapia con radiación, terapia hormonal, terapia biológica e inmunoterapia. Se puede administrar una forma sólida del compuesto 1 que se proporciona en el presente documento y otro principio activo a un paciente antes, durante o después de la aparición del efecto adverso asociado con la terapia convencional.
TERAPIA CÍCLICA
En determinadas realizaciones, los agentes profilácticos o terapéuticos que se proporcionan en el presente documento se administran de forma cíclica a un paciente. La terapia cíclica implica la administración de un agente activo durante un período de tiempo, seguida de un descanso durante un período de tiempo y la repetición de esta administración secuencial. La terapia cíclica puede reducir el desarrollo de resistencia a una o más terapias, evitar o reducir los efectos secundarios de una de las terapias y/o mejorar la eficacia del tratamiento.
Por consiguiente, en determinadas realizaciones, se administra una forma sólida del compuesto 1 que se proporciona en el presente documento de forma diaria en una única dosis o en dosis divididas en un ciclo de cuatro a seis semanas con un período de descanso aproximadamente una semana o dos semanas. En determinadas realizaciones, se administra una forma sólida del compuesto 1 que se proporciona en el presente documento de forma diaria en una dosis única o en dosis divididas durante uno a diez días consecutivos de un ciclo de 28 días, con un período de descanso posterior sin administración durante el resto del ciclo de 28 días. El método cíclico también permite aumentar la frecuencia, cantidad y extensión de los ciclos de dosificación. Por lo tanto, en determinadas realizaciones, el presente documento comprende la administración de una forma sólida del compuesto 1 que se proporciona en el presente documento durante más ciclos de los habituales al administrarse sola. En determinadas realizaciones, se administra una forma sólida del compuesto 1 que se proporciona en el presente documento durante una mayor cantidad de ciclos de los que generalmente provocarían toxicidad que limita la dosis en un paciente que no reciba también un principio activo secundario.
En una realización, se administra una forma sólida del compuesto 1 que se proporciona en el presente documento de forma diaria y continua durante tres o cuatro semanas en una dosis aproximadamente 0,1 a aproximadamente 150 mg/día, seguido por un descanso de una o dos semanas.
En otra realización, se administra una forma sólida del compuesto 1 que se proporciona en el presente documento por vía intravenosa y se administra un principio activo secundario por vía oral, con la administración de la forma sólida del compuesto 1 descrito en el presente documento 30 a 60 minutos antes de un principio activo secundario, durante un ciclo de cuatro a seis semanas. En determinadas realizaciones, la combinación de una forma sólida del compuesto 1 que se proporciona en el presente documento y un principio activo secundario se administra mediante infusión intravenosa durante aproximadamente 90 minutos en cada ciclo. En determinadas realizaciones, un ciclo comprende la administración aproximadamente 0,1 a aproximadamente 150 mg/día de una forma sólida del compuesto 1 que se proporciona en el presente documento y de aproximadamente 50 a aproximadamente 200 mg/m2/día de un principio activo secundario de forma diaria durante tres a cuatro semanas con una o dos semanas de descanso posterior. En determinadas realizaciones, la cantidad de ciclos durante los cuales se administra el tratamiento combinado a un paciente oscila entre aproximadamente uno y aproximadamente 24 ciclos, entre aproximadamente dos y aproximadamente 16 ciclos o entre aproximadamente cuatro y aproximadamente tres ciclos.
COMPOSICIONES FARMACÉUTICAS Y VÍAS DE ADMINISTRACIÓN
Las formas sólidas del compuesto 1 que se describe en el presente documento se pueden administrar a un sujeto por vía oral, tópica o parenteral en la forma convencional de preparaciones, tales como cápsulas, microcápsulas, comprimidos, gránulos, polvo, pastillas, píldoras, supositorios, inyecciones, suspensiones, jarabes, parches, cremas, lociones, ungüentos, geles, aerosoles, soluciones y emulsiones. Las formulaciones adecuadas pueden prepararse con métodos que se suelen utilizar y que emplean aditivos orgánicos o inorgánicos convencionales, tales como un excipiente (p. ej., sacarosa, almidón, manitol, sorbitol, lactosa, glucosa, celulosa, talco, fosfato cálcico o carbonato cálcico), un aglutinante (p. ej., celulosa, metilcelulosa, hidroximetilcelulosa, polipropilpirrolidona, polivinilpirrolidona, gelatina, goma arábiga, polietilenglicol, sacarosa o almidón), un desintegrante (p. ej., almidón, carboximetilcelulosa, hidropropilalmidón, hidroxipropilcelulosa de sustitución baja, bicarbonato de sodio, fosfato cálcico o citrato de calcio), un lubricante (p. ej., estearato de magnesio, ácido silícico anhidro suave, talco o lauril sulfato sódico), un agente saborizante (p. ej., ácido cítrico, mentol, glicina o polvo naranja), un conservante (p. ej., benzoato de sodio, bisulfito de sodio, metilparabeno o propilparabeno), un estabilizante (p. ej., ácido cítrico, citrato de sodio o ácido acético), un agente de suspensión (p. ej., metilcelulosa, polivinil pirrolidona o estearato de aluminio), un agente dispersante (p. ej., hidroxipropilmetilcelulosa), un diluyente (p. ej., agua) y cera base (p. ej., manteca de cacao, vaselina blanca o polietilenglicol). La cantidad eficaz de las formas sólidas del compuesto 1 descrito en el presente documento en la composición farmacéutica puede encontrarse en un nivel que logre el efecto deseado, por ejemplo, de aproximadamente 0,005 mg/kg del peso corporal de un sujeto a aproximadamente 10 mg/kg del peso corporal de un sujeto en una dosificación unitaria para administración oral y parenteral.
La dosis de una forma sólida del compuesto 1 que se administra a un sujeto puede variar ampliamente y puede estar sujeta al criterio de un profesional de salud. En general, las formas sólidas del compuesto 1 se pueden administrar una a cuatro veces al día en una dosis de aproximadamente 0,005 mg/kg del peso corporal de un sujeto a aproximadamente 10 mg/kg del peso corporal de un sujeto en un sujeto, pero la dosificación anterior puede alterarse de forma correcta dependiendo de la edad, el peso corporal, la afección médica del sujeto y el tipo de administración. En una realización, la dosis es de aproximadamente 0,01 mg/kg del peso corporal del sujeto a aproximadamente 10 mg/kg del peso corporal de un sujeto, de aproximadamente 0,1 mg/kg del peso corporal de un sujeto a aproximadamente 10 mg/kg del peso corporal de un sujeto, de aproximadamente 1 mg/kg del peso corporal de un sujeto a aproximadamente 10 mg/kg del peso corporal de un sujeto o de aproximadamente 1 mg/kg del peso corporal de un sujeto a aproximadamente 5 mg/kg del peso corporal de un sujeto. En una realización, se administra una dosis por día. En cualquier caso específico, la cantidad de la forma sólida del compuesto 1 que se administre dependerá de factores como la solubilidad del componente activo, la formulación que se utilice y la vía de administración. En una realización, la aplicación de una concentración tópica brinda concentraciones o exposiciones intracelulares de aproximadamente 0,01 a 10 M.
En otra realización, los métodos son para el tratamiento o la prevención de una enfermedad o un trastorno comprendiendo administrar de aproximadamente 1 mg/día a de aproximadamente 1000 mg/día, de aproximadamente 1 mg/día a aproximadamente 750 mg/día, de aproximadamente 1 mg/día a aproximadamente 500 mg/día, de aproximadamente 1 mg/día a aproximadamente 250 mg/día o de aproximadamente 100 mg/día a aproximadamente 1000 mg/día de una forma sólida del compuesto 1 descrito en el presente documento a un sujeto que lo necesite.
En otra realización, en el presente documento se brindan formulaciones de dosificación unitaria comprendiendo entre aproximadamente 1 mg y 1000 mg, aproximadamente 5 mg y aproximadamente 1000 mg, aproximadamente 10 mg y aproximadamente 1000 mg, aproximadamente 25 mg y aproximadamente 1000 mg, aproximadamente 50 mg y aproximadamente 1000 mg, aproximadamente 100 mg y aproximadamente 1000 mg o aproximadamente 250 mg y aproximadamente 1000 mg de una forma sólida del compuesto 1 descrito en el presente documento. Se pueden administrar formas sólidas del compuesto 1 descrito en el presente documento una, dos, tres, cuatro o más veces al día. En una realización particular, se administran dosis de 600 mg o menos como una dosis diaria y se administran dosis de más de 600 mg dos veces al día en una cantidad igual a la mitad de la dosis diaria total.
En otra realización, en el presente documento se brindan formulaciones de dosificación unitaria que comprenden entre aproximadamente 1 mg y 200 mg, aproximadamente 35 mg y aproximadamente 1400 mg, aproximadamente 125 mg y aproximadamente 1000 mg, aproximadamente 250 mg y aproximadamente 1000 mg o aproximadamente 500 mg y aproximadamente 1000 mg de una forma sólida del compuesto 1 descrito en el presente documento. En una realización particular, en el presente documento se proporcionan formulaciones de dosificación unitaria que comprenden aproximadamente 100 mg o 400 mg de una forma sólida del compuesto 1 descrito en el presente documento.
En otra realización, en el presente documento se proporcionan formulaciones de dosificación unitaria que comprenden 1 mg, 5 mg, 10 mg, 15 mg, 20 mg, 30 mg, 35 mg, 50 mg, 70 mg, 100 mg, 125 mg, 140 mg, 175 mg, 200 mg, 250 mg, 280 mg, 350 mg, 500 mg, 560 mg, 700 mg, 750 mg, 1000 mg o 1400 mg de una forma sólida del compuesto 1 descrito en el presente documento.
Las formas sólidas del compuesto 1 descrito en el presente documento se pueden administrar una, dos, tres, cuatro o más veces al día. En una realización particular, se administran dosis de 600 mg o menos como una dosis diaria y se administran dosis de más de 600 mg dos veces al día en una cantidad igual a la mitad de la dosis diaria total. Las formas sólidas del compuesto 1 descrito en el presente documento pueden administrarse por vía oral por motivos de conveniencia. En una realización, cuando se administra por vía oral, la forma sólida del compuesto 1 se administra con alimento y agua. En otra realización, la forma sólida del compuesto 1 se dispersa en agua o zumo (p. ej., zumo de manzana o zumo de naranja) y se administra por vía oral como una suspensión.
Las formas sólidas del compuesto 1 descrito en el presente documento también se pueden administrar por vía intradérmica, intramuscular, intraperitoneal, percutánea, intravenosa, subcutánea, intranasal, epidural, sublingual, intracerebral, intravaginal, transdérmica, rectal, mucosal, mediante inhalación o por vía tópica en los oídos, la nariz, los ojos o la piel. La forma de administración se deja a criterio del profesional de la salud y puede depender, en parte, de la ubicación de la afección médica.
En una realización, en el presente documento se proporcionan cápsulas que contienen una forma sólida del compuesto 1 descrito en el presente documento sin un portador, excipiente o vehículo adicional.
En otra realización, en el presente documento se proporcionan composiciones que comprenden una cantidad eficaz de una forma sólida del compuesto 1 descrito en el presente documento y un portador o vehículo farmacéuticamente
aceptable, en donde un portador o vehículo farmacéuticamente aceptable puede comprender un excipiente, diluyente o una mezcla de estos. En una realización, la composición es una composición farmacéutica.
Las composiciones pueden encontrarse en forma de comprimidos, comprimidos masticables, cápsulas, soluciones, soluciones parenterales, pastillas, supositorios y suspensiones y similares. Las composiciones pueden formularse para contener una dosis diaria o una fracción conveniente de una dosis diaria, en una unidad de dosificación que puede ser un comprimido o una cápsula individual o un volumen conveniente de un líquido. En una realización, las soluciones se preparan a partir de sales solubles en agua, tales como la sal de clorhidrato. En general, todas las composiciones se elaboran de acuerdo con métodos conocidos en la química farmacéutica. Las cápsulas pueden elaborarse mezclando una forma sólida del compuesto 1 descrito en el presente documento con un portador o diluyente adecuado y rellenando la cantidad adecuada de la mezcla en cápsulas. Los portadores y diluyentes habituales incluyen, pero no se limitan a, sustancias en polvo inertes tales como almidón de muchos tipos diversos, celulosa en polvo, en especial celulosa cristalina y microcristalina, azúcares como fructosa, manitol y sacarosa, harinas de grano y polvos comestibles similares.
Los comprimidos pueden elaborarse mediante compresión directa, granulación húmeda o granulación seca. La compresión de las formas sólidas del compuesto 1 descrito en el presente documento puede no reducir o modular la actividad del fármaco administrado a un paciente. Sus formulaciones suelen incorporar diluyentes, aglutinantes, lubricantes y desintegrantes, así como el compuesto. Los diluyentes habituales incluyen, por ejemplo, diversos tipos de almidón, lactosa, manitol, caolín, sulfato o fosfato cálcico, sales inorgánicas tales como cloruro de sodio y azúcar en polvo. También son útiles los derivados de celulosa en polvo. Los aglutinantes habituales de comprimidos son sustancias tales como almidón, gelatina y azúcares tales como lactosa, fructosa, glucosa y similares. También son convenientes las gomas naturales y sintéticas, que incluyen acacia, alginatos, metilcelulosa, polivinilpirrolidina y similares. También se pueden usar como aglutinantes polietilenglicol, etilcelulosa y ceras.
Puede requerirse un lubricante en la formulación de un comprimido para evitar la adhesión del comprimido y los perforadores en el troquel. El lubricante puede seleccionarse de sólidos deslizables como talco, estearato de calcio y de magnesio, ácido esteárico y aceites vegetales hidrogenados. Los desintegrantes de comprimidos son sustancias que se expanden al humectarse para descomponer el comprimido y liberar el compuesto. Estos incluyen almidones, arcillas, celulosas, alginas y gomas. De manera más particular, se pueden utilizar almidones de maíz y patata, metilcelulosa, agar, bentonita, celulosa de madera, esponja natural en polvo, resinas de intercambio catiónico, ácido algínico, goma guar, pulpa cítrica y carboximetilcelulosa, por ejemplo, así como lauril sulfato de sodio. Los comprimidos pueden recubrirse con azúcar como saborizante y sellante o con agentes protectores formadores de película para modificar las propiedades de disolución del comprimido. Las composiciones también pueden formularse como comprimidos masticables, por ejemplo, utilizando sustancias como manitol en la formulación. Si se desea administrar una forma sólida del compuesto 1 descrito en el presente documento como supositorio, se pueden utilizar las bases habituales. La manteca de cacao es una base de supositorio tradicional, que puede modificarse mediante adición de ceras para aumentar levemente su punto de fusión. Se utilizan ampliamente bases de supositorio miscibles en agua que comprenden, en particular, polietilenglicoles de diversos pesos moleculares. El efecto de la forma sólida del compuesto 1 descrito en el presente documento puede retrasarse o prolongarse mediante la formulación adecuada. Por ejemplo, se puede elaborar un gránulo de solubilidad lenta de la forma sólida del compuesto 1 descrito en el presente documento e incorporarse en un comprimido o una cápsula o como un dispositivo implantable de liberación lenta. La técnica también incluye elaborar gránulos con diversas velocidades de disolución diferentes y rellenar las cápsulas con una mezcla de los gránulos. Los comprimidos o las cápsulas pueden recubrirse con una película que resista la disolución durante un período de tiempo previsible. Incluso las preparaciones parenterales pueden convertirse en preparaciones de acción prolongada disolviendo o suspendiendo la forma sólida del compuesto 1 descrito en el presente documento en vehículos oleosos o emulsionados que le permitan dispersarse lentamente en el suero.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma A, incluida la forma A sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 1 de sal de HCl, incluida la forma de sal de HCl como material de partida sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 1 de sal de HCl, incluida la forma 1 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma B, incluida la forma B sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 2 de sal de HCl, incluida la forma 2 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma C, incluida la forma C sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 3 de sal de HCl, incluida la forma 3 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma D, incluida la forma D sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 4 de sal de HCl, incluida la forma 4 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma E, incluida la forma E sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 5 de sal de HCl, incluida la forma 5 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma F, incluida la forma F sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 6 de sal de HCl, incluida la forma 6 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma G, incluida la forma G sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 7 de sal de HCl, incluida la forma 7 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma H, incluida la forma H sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma 8 de sal de HCl, incluida la forma 8 de sal de HCl sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma I, incluida la forma I sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma Y, incluida la forma Y sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden la forma Z, incluida la forma Z sustancialmente pura.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden un sólido amorfo, p. ej., una base libre, una sal de HCl, una sal de citrato u otra sal descrita en el presente documento, incluido el sólido amorfo sustancialmente puro.
En determinadas realizaciones, las composiciones farmacéuticas que se proporcionan en el presente documento comprenden una mezcla de una o más formas sólidas del compuesto 1, incluidas la forma A, forma B, forma C, forma D, forma E, forma F, forma G, forma H, forma I, forma Y, forma Z, forma 1 de sal de HCl, forma 2 de sal de HCl, forma 3 de sal de HCl, forma 4 de sal de HCl, forma 5 de sal de HCl, forma 6 de sal de HCl, forma 7 de sal de HCl, forma 8 de sal de HCl o un sólido amorfo descrito en el presente documento, donde se permite cualquier combinación posible de las formas sólidas del compuesto 1.
Ejemplos
Los siguientes ejemplos se proporcionan con fines ilustrativos, no taxativos. En las descripciones y los ejemplos se utilizan las siguientes abreviaturas:
ACN: acetonitrilo
Am: amorfo
AmPhos: p-dimetilamino fenilditbutilfosfina
API: principio farmacéutico activo
Boc: terc-butoxicarbonilo
n-BuOH: n-butanol
dba: dibenciliden acetona
DBU: 1,8-diazabiciclo[5.4.0]undec-7-eno
DCM: diclorometano
DIPEA: W,W-diisopropiletilamina
DMAc: W,W-dimetilacetamida
DMF: W,W-dimetilformida
DMSO: dimetilsulfóxido
DSC: calorimetría diferencial de barrido
DVS: sorción dinámica de vapor
EDTA: tetraacetato de etilendiamina
ESI: ionización por electropulverización
EtOAc: acetato de etilo
EtOH: etanol
FTIR: espectroscopia infrarroja con transformada de Fourier
HPLC: cromatografía líquida de alto rendimiento
IPA: 2-propanol
IPAc: acetato de isopropilo
LCMS: cromatografía líquida con espectroscopía de masas
MeCN: acetonitrilo
MEK: metiletilcetona
MeOH: metanol
2-MeTHF: tetrahidrofurano de 2-metilo
mp: punto de fusión
MS: espectrometría de masas
MTBE: terc-butil metil éter
NBS: W-bromosuccinimida
NMP: W-metil-2-pirrolidona
RMN: resonancia magnética nuclear
RH: humedad relativa
RT: temperatura ambiente
Rx: recristalziación
S: disolvente
SDTA: análisis térmico diferencial único
SM: material de partida
S-SegPhos : (S)-(-)-5,5-bis(difenilfosfino)-4,4-bi-1,3-benzodioxol
TA: análisis térmico
Tf: triflato o trifluorometanosulfonilo
TFA: ácido trifluoroacético
TFE: 2,2,2-trifluoroetanol
TGA: análisis termogravimétrico
TGA-MS/TG-MS: análisis termogravimétrico junto con espectroscopia de masas
THF: tetrahidrofurano
TLC: cromatografía en capa fina
XRPD: difracción de rayos X en polvo
EJEMPLOS SINTÉTICOS
Los siguientes ejemplos sintéticos no limitantes muestran métodos para elaborar el compuesto 1. Se utilizaron ACD/NAME (Advanced Chemistry Development, Inc., Ontario, Canadá) y/o Chemdraw (Cambridgesoft, Perkin Elmer, Waltham, MA) para generar nombres para las estructuras químicas y se utilizó Chemdraw para dibujar las estructuras químicas.
En una realización, el compuesto 1 se sintetiza de la forma descrita en el ejemplo 53 de la patente estadounidense n.° 9.512.124, que se incorpora en el presente documento en su totalidad mediante esta referencia.
ANÁLISIS PRELIMINAR Y SELECCIÓN DE LA SAL DEL COMPUESTO 1
Se realizó un análisis preliminar de la forma de sal del compuesto 1 utilizando enfoques con volúmenes pequeños. La pKa del compuesto 1 es de 5,14. Se seleccionaron diversos contraiones para la formación de sales, que incluían los ácidos glicólico, málico, cítrico, tartárico, fosfórico, maleico, bencenosulfónico, metanosulfónico, toluenosulfónico, sulfúrico y clorhídrico con varios disolventes.
El compuesto 1 de base libre es material de hidrato (un monohidrato). La pérdida de peso según el TGA ascendió al 2,9 % antes de la descomposición y la DSC mostró dos picos endotérmicos amplios a temperatura baja, debido a la
deshidratación y luego un pico de fusión a 1822C. La forma cristalina permaneció inalterada tras cualquiera de las suspensiones en agua. La base libre es estable en solución (pH 1,2 a 7,5) a 40 °C. Presenta estabilidad química y física en estado sólido en condiciones de estrés durante hasta siete semanas. En condiciones secas, la forma de hidrato cambió a hemihidratos o hidratos parciales. La forma de sal probablemente mejore las propiedades en estado sólido y la solubilidad dependiente del pH. La forma cristalina de monohidrato permaneció inalterada salvo que se secara (< 5 % RH) o mantuviera a una mayor temperatura (> 60 °C). La base libre de monohidrato es levemente higroscópica.
Las muestras sólidas se examinaron utilizando un difractómetro de rayos X (SmartLab, Rigaku). El detector estaba equipado con un fotomultiplicador con tecnología de detección por rayos X con preamplificador. Las muestras se analizaron de 3 a 40 °26, con una fracción de etapa de 0,02 °26 y un tiempo por etapa de 20 segundos. El voltaje y la corriente del tubo fueron de 40 KV y 44 mA, respectivamente. La muestra se transfirió de un recipiente para muestra a un contenedor de XRD con referencia cero y se molió suavemente.
Se realizaron análisis TGA en un instrumento de TA TGA Q5000. Se colocaron aproximadamente 1,50 mg de las muestras en una bandeja de aluminio o platino tarada, se pesaron de forma automática y se insertaron en el horno de TGA. Las muestras se calentaron a una velocidad de 10 °C/min, hasta una temperatura final de 300 °C. El gas de purga fue nitrógeno, para equilibrio a aproximadamente 10 cc/min y para el horno a aproximadamente 90 cc/min, respectivamente.
Los análisis de DSC se realizaron en un instrumento de TA Q2000. El estándar de calibración fue indio. Se colocó una muestra con un peso de 1,50 mg en una bandeja de TA DSC tarada y se registró correctamente el peso. Se utilizaron bandejas selladas para análisis y las muestras se calentaron con nitrógeno (50 cc/min) a una velocidad de 10 °C/min, hasta una temperatura final de 300 °C. Los datos se procesaron utilizando un analizador térmico (Universal Analyzer 2000, TA Instruments).
Se utilizó RMN protónica para estudiar los desplazamientos químicos del compuesto producido por la formación de sal. La RMN protónica se realizó utilizando Bruker Advance 300 Ultrashield™ equipado con muestra automática (B-ACS 60). Se utilizó sulfóxido de dimetilo-d6 (DMSO-d6) como disolvente para el análisis de RMN. El tiempo de adquisición fue aproximadamente el 16 segundos.
Se midió la adsorción dinámica de vapores (DVS) utilizando DVS advantage (Surface Measurement Systems Ltd). Las muestras se analizaron con isotermia (25 °C) a una RH deseada del 0 a 95 % por ciclo completo en modo de etapas. Para un análisis isotérmico, la temperatura de la cámara se conservó mediante un baño de agua a 25,0 ± 1,0 °C constantes. La humedad relativa en la cámara de muestra se generó combinando diferentes flujos de nitrógeno húmedo y seco con velocidades de flujo variables. El análisis se realizó en aumentos de RH del 10 %. La velocidad de muestreo fue de 1 seg, la velocidad de registro de datos es de 20 seg. El valor de dm/dt (%) se fijó en 0,001, con un espectro de dm/dt de 5 min, un tiempo de duración de estabilidad mínimo de 10 min y un tiempo máximo por etapa de 180 min. Se registró el peso en equilibrio de la muestra correspondiente a cada RH. Se obtuvo la isotermia de sorción mediante una gráfica del contenido de humedad en equilibrio en relación con la RH.
Se disolvió 1,00 gramo de la base libre del compuesto 1 en 10 ml de metanol. Luego se agregaron 100 gl de la solución madre a cada pocillo en una placa de 96 pocillos. Se agregaron soluciones ácidas con una relación molar de 1:1 a cada pocillo de la placa, con un ácido para 8 pocillos en la misma fila. Tras el secado de la placa, se agregaron alícuotas de 400 gl de 8 disolventes diferentes al pocillo en la placa en forma de columnas. Las placas luego se cubrieron y se dejaron evaporar en una campana extractora de laboratorio en funcionamiento en condiciones de temperatura y humedad ambiente. Para el análisis preliminar se utilizaron disolventes que incluyeron etanol, 2-propanol, 3-metil-butanol, acetonitrilo, metil terc-butil éter (MTBE), acetona, agua y acetato de etilo.
La forma no salina inicial de la base libre del compuesto 1 se caracterizó mediante XRPD, TGA y DSC. Es un monohidrato cristalino y en el presente documento se indica como la forma 1.
Se realizó difracción de rayos X en polvo del compuesto 1 y el perfil se muestra en la figura 89.
En la figura 90, el termograma de TGA del compuesto 1 muestra que se observó aproximadamente un 2,9 % de pérdida de peso a una temperatura relativamente baja (< 75 °C), debido a la deshidratación. La pérdida de peso final se debe a la descomposición del compuesto del fármaco.
En la figura 91, el termograma de DSC del compuesto 1 muestra que el sólido cristalino presenta un evento endotérmico amplio a una temperatura relativamente baja correspondiente a la deshidratación/desolvatación y el pico endotérmico con temperatura de inicio y pico de 174,6 y 182,1 °C, respectivamente, con una entalpía de 52,0 J/g, debido a la fusión de la forma deshidratada.
Los perfiles de DVS mostraron que la muestra (el compuesto 1) es levemente higroscópica (< 4,3 %) del 0 al 95 % de RH, con respecto a la base libre de monohidrato ilustrada en la figura 92. La base libre de monohidrato se convirtió en anhidra al colocarse por primera vez en un ciclo de secado a humedad muy baja o igual a cero. Luego
se produjo un incremento del ~ 2,6 % (en peso) de agua al exponer las partículas del sólido a una humedad relativamente mayor de hasta el 25 % de RH y volvió a convertirse a monohidrato en este espectro de humedad. Se produjo un incremento adicional del ~ 1,7 % de sorción de humedad de forma lenta y continua del 25 al 95 % de RH. Durante el ciclo de desorción del 95 al 5 % de RH, la pérdida de contenido acuoso fue muy lenta, ~ 1,5 %, y se mantuvo la estructura de monohidrato. Luego, se liberó repentinamente el ~ 2,8 % en peso de agua restante de la muestra a medida que disminuyó la humedad relativa del 5 % de RH hasta sequedad. El nivel de contenido acuoso del 3,0 % corresponde a un monohidrato.
La adsorción/desorción son casi reversibles por encima del 30 % de RH. Por debajo del 30 % de RH, la liberación de agua durante la desorción es más difícil que la captación durante la sorción.
Se examinó la RMN protónica del compuesto 1 en DMSO y se ilustra en la figura 6. La base libre del compuesto 1 (1,00 gramo) se disolvió en metanol (10,0 ml). Luego se distribuyeron alícuotas de 100 gl de la solución en cada uno de los pocillos de la placa de 96 pocillos (insertos de vidrio transparente y fondo plano de 1,0 ml). Se agregaron a los pocillos once ácidos, incluidos los ácidos glicólico, málico, cítrico, tartárico, fosfórico, maleico, bencensulfónico, metansulfónico, toluensulfónico, sulfúrico y clorhídrico, con una relación molar de 1:1, véase la tabla 1. El disolvente se evaporó en una campana de extracción de laboratorio en funcionamiento en condiciones de temperatura y humedad ambiente. Tras la sequedad, se introdujeron los disolventes para recristalización, véase la tabla 1. Luego, la placa se cubrió con una rejilla de tapa redonda con recubrimiento de silicona/PTFE, para permitir una evaporación y cristalización lenta en un entorno ambiente.
Tabla 1: Contenido de formadores de sal y disolventes en la placa de 96 pocillos.

Inicialmente, se encontró HCl durante el estudio de solubilidad de la base libre del compuesto 1 en solución de fluido gástrico simulado (SGF, por sus siglas en inglés). Se pesaron aproximadamente 30 mg de la base libre del compuesto 1 en un matraz de vidrio y se introdujo 1 ml de SGF. La mezcla rápidamente se convirtió en una solución transparente. La precipitación se produjo durante la noche. Las partículas sólidas se recogieron mediante filtración y se caracterizaron. El perfil de XRPD es diferente al de la base libre, como se muestra en la figura 94. El TGA mostró aproximadamente el 3,3 % de pérdida de peso a una temperatura relativamente baja (< 70 °C) antes de la descomposición (figura 95). El perfil de DSC presentó dos eventos endotérmicos 1) a temperatura relativamente baja, debido a deshidratación y la forma deshidratada en fusión, con temperaturas de inicio y pico de 209,3 y 230,5 °C, respectivamente (figura 96). Se realizó sorción dinámica de vapores de esta muestra en la isotermia y se muestra en la figura 97. La RMN 1H mostró que se observaron desplazamientos químicos en el hidrógeno en el anillo purina y benceno, lo cual sugiere la formación de sal (figura 98).
Tabla 2: Análisis preliminar de un pequeño volumen de sal del compuesto 1

La sorción dinámica de vapores (DVS) mostró que la sal de HCl tiene una higroscopicidad baja (< 1,0 %) entre el 0 y el 95 % de RH, con respecto al HCl monohidrato, como se muestra en la figura 97. El monohidrato de HCl se convirtió en anhidro al colocarse por primera vez en un ciclo de secado a humedad muy baja o igual a cero. Luego se produjo un incremento del ~ 2,9 % (en peso) de agua al exponer las partículas del sólido a una humedad relativamente mayor de hasta el 25 % de RH y volvió a convertirse a monohidrato en este espectro de humedad. Se produjo un incremento adicional del ~ 1,7 % de adsorción de humedad de forma lenta y continua entre el 25 y el 95 % de RH. Durante el ciclo de desorción del 95 hasta el 5 % de RH, la pérdida de contenido acuoso fue muy lenta ~ 1,5 % y se mantuvo la estructura de monohidrato. Luego, se liberó rápidamente el ~2,8 % en peso de agua restante de la muestra a medida que disminuyó la humedad relativa del 5 % de RH hasta sequedad. El nivel de contenido acuoso de 3,0 % corresponde a un monohidrato.
La adsorción/desorción fueron casi reversibles por encima del 30 % de RH. Por debajo del 30 % de RH, la liberación de agua durante la desorción fue más difícil que la captación durante la adsorción. Se observó histéresis entre las curvas de adsorción y desorción por debajo del 25 % de RH.
Se analizó la muestra del pocillo n.° H2 que contenía base libre del compuesto 1 y ácido sulfúrico mediante RMN 1H en DMSO-d6, figura 99. La RMN 1H mostró que se observaron cambios químicos en el hidrógeno en el anillo purina y benceno, lo cual sugiere la formación de sal. Las muestras seleccionadas se analizaron mediante TGA y DSC, figura 100 a figura 105. Todos los perfiles de TGA mostraron pérdidas de peso iniciales (2,5 a 3,2 %) a temperatura relativamente baja. No obstante, los comportamientos no fueron los mismos. Los perfiles de DSC de estos pocillos también mostraron eventos endotérmicos amplios a temperaturas relativamente bajas, pero no fueron el mismo perfil. Las sales de sulfato son solvatos, dependiendo del uso del disolvente para la cristalización.
Se hallaron sales de mesilato cristalinas de SVSS en diversos disolventes y los perfiles de XRPD de las sales de citrato cristalinas de diversos disolventes son muy similares. En la figura 106 se muestra un perfil de XRPD representativo de las sales de mesilato cristalinas de SVSS en EtOAc. Se analizó la muestra con base libre del compuesto 1 y ácido metanosulfúrico mediante RMN 1H en DMSO-d6, figura 107. La RMN 1H mostró que se observaron cambios químicos en el hidrógeno en el anillo purina y benceno, lo cual sugiere la formación de sal. Las muestras seleccionadas se analizaron mediante TGA y DSC, figura 108 a figura 117. Todos los perfiles de TGA f mostraron pérdidas de peso iniciales (1,4 a 2,2 %) a temperatura relativamente baja, así como comportamientos levemente diferentes. Los perfiles de DSC de estos pocillos también mostraron eventos endotérmicos amplios a temperaturas relativamente bajas, pero perfiles diferentes tras la evaporación del disolvente.
Se hallaron sales de citrato cristalinas de SVSS en diversos disolventes y se muestran perfiles de XRPD de las sales de citrato cristalinas de SVSS en la figura 118 a la figura 125. Los perfiles de XRPD de etanol (A9), IPA (B9), 3-metil-2-butanol (C9), acetonitrilo (D9), MTBE (E9) y acetona (F9) parecen similares (forma 1 de citrato), como se muestra en la figura 126 y la figura 127. El perfil de XRPD de SVSS en EtOAc (H9) fue diferente (figura 128, forma 2 de citrato). El perfil de XRPD de SVSS en agua (figura 124) fue muy similar a los de la figura 126. Se analizó la muestra con base libre del compuesto 1 y ácido cítrico mediante RMN 1H en DMSO-d6, figura 129. La RMN 1H mostró que no se observaron cambios químicos en el hidrógeno en el anillo de purina y benceno, lo cual sugirió que la formación de sal tiene una interacción débil en la fase de solución y puede interpretarse como un cocristal. Las muestras seleccionadas se analizaron mediante TGA y DSC, figura 130 a figura 139. Todos los perfiles de TGA mostraron pérdidas de peso iniciales mínimas (< 0,5 %) a temperatura relativamente baja antes de la descomposición. Los perfiles de DSC de estos pocillos también mostraron una única endotermia debida a la fusión, con temperaturas de inicio y pico de ~ 203 y ~ 211 °C, respectivamente.
Se analizó la muestra con base libre del compuesto 1 y ácido fosfórico mediante RMN 1H en DMSO-d6, figura 140. La RMN 1H mostró que no se observaron cambios químicos en el hidrógeno en el anillo de purina y benceno, lo cual sugirió que la formación de sal puede no ser probable en la fase de solución. Se hallaron diferentes estructuras cristalinas en SVSS, lo cual sugirió formación de cocristales de fosfato con el compuesto 1.
Los patrones de difracción de rayos X en polvo de los pocillos B4 y B6 fueron similares, indicados como la forma 1A de fosfato, figura 141.
La difracción de rayos X en polvo de los pocillos B7 y B10 fue diferente, así como diferente a la forma 1A, y se indica como las formas 1B y 1C, respectivamente, como se muestra en la figura 142.
Monohidrato de sal de HCl (1): Se pesaron 240 mg del compuesto 1 en un matraz de vidrio de 4 ml y luego se introdujeron 4,60 ml de HCl 0,1 N. La mezcla rápidamente se volvió transparente. La solución se filtró con un filtro de 0,22 pm y el sobrenadante se colocó en una campana para cristalización. La precipitación se produjo rápidamente. El sólido se recogió mediante filtración.
El análisis de la muestra de sólido determinó que era un monohidrato (1), con un perfil de XRPD similar al del estudio de solubilidad de la base libre en SGF, pero con mejor cristalinidad en el matraz de vidrio en ml y con posterior adición de 3,10 ml de HCl 0,5 N en agua. La mezcla rápidamente se volvió transparente. Se agregaron 5,0 ml más de agua. La solución se filtró con un filtro de 0,22 pm. El sobrenadante se colocó en una campana para cristalización. La precipitación se produjo rápidamente. El sólido se recogió mediante filtración.
Se pesaron 303,7 mg del compuesto 1 en un matraz de vidrio de 20 ml y luego se agregaron 10 ml de SGF. La mezcla rápidamente se volvió transparente. Se agregaron partículas sólidas de sal de HCl del compuesto 1 al matraz en forma de semillas. La suspensión se mantuvo con agitación en rotación en LabQuake durante 24 horas. Las partículas sólidas se recogieron mediante filtración.
No se observó la forma anhidra en el proceso de precipitación de solución. La deshidratación se realizó en la etapa de XRD-DSC.
Se pesaron 170 mg del compuesto 1 en un matraz de vidrio de 4 ml y luego se introdujeron 3,3 ml de H2SO40,1 M en EtOAc. La mezcla se volvió inmediatamente un material gomoso/gelificado. Tras el secado, se recogió y analizó el sólido mediante XRPD, TGA y DSC, como se muestra en la figura 153 a la figura 155.
Se pesaron 105 mg del compuesto 1 en un matraz de vidrio de 4 ml y se introdujeron 2,0 ml de H2SO4 0,1 M en agua. La mezcla se convirtió en un material tipo gel y se agregó una adición de 1 ml de agua. El material seguía siendo pegajoso y aceitoso.
Se pesaron 138 mg del compuesto 1 en un matraz de vidrio de 4 ml y luego se agregó 1,0 ml de EtOAc para disolver el material en primer lugar. Luego, se introdujeron 2,60 ml de ácido metanosulfónico 0,1 M en EtOAc e inmediatamente apareció un precipitado. El sólido se recogió mediante filtración y se secó a 40 °C en vacío durante la noche, tras lo cual se analizó mediante XRPD, TGA y DSC, como se muestra en la figura 156 a la figura 158. Se pesaron 34 mg del compuesto 1 en un matraz de vidrio de 4 ml y luego se agregaron 0,65 ml de ácido metanosulfónico 0,1 M en acetonitrilo. No se logró una solución transparente. No obstante, se observó claramente una nueva fase sólida. El sólido se recogió mediante filtración y se secó a 40 °C en vacío durante la noche, tras lo cual se analizó mediante XRPD, TGA y DSC, como se muestra en la figura 159 a la figura 161.
Forma de sal de mesilato: Tras la suspensión en agua, el perfil de XRPD es levemente diferente, como se ilustra en la figura 162.
También se estudió la sal de mesilato con humedad, utilizando sorción dinámica de vapores (DVS). Tras el ciclo de
sorción y desorción (figura 163), el patrón de XRPD es levemente diferente al material de partida, como se muestra en la figura 164.
Se pesaron 114 mg del compuesto 1 en un matraz de vidrio y luego se agregaron 0,6 ml de disolvente de EtOAc. Tras la agitación, la mezcla se volvió una suspensión transparente. Se agregaron a la solución 2,20 ml de cítrico 0,1 N en agua e inmediatamente aparecieron precipitados tipo algodón. El sólido se recogió mediante filtración y se caracterizó, como se muestra en la figura 165 a la figura 167. El TGA mostró una pérdida de peso escasa (< 0,2 %) a temperatura relativamente baja antes de la descomposición. La DSC mostró un único pico endotérmico debido a la fusión, con temperaturas de inicio y pico de 205,2 y 209,5 °C, respectivamente, con una entalpia de 240,7 J/g. Se pesaron 95 mg del compuesto 1 en un recipiente de vidrio y luego se agregaron 0,5 ml de disolvente de acetona para disolver el material. Luego se agregaron 1,8 ml de cítrico 0,1 N en acetona. La solución transparente se colocó en una campana para cristalización. Los precipitados aparecieron rápidamente.
El sólido se recogió mediante filtración y se caracterizó, como se muestra en la figura 168 a la figura 170. El perfil de XRPD fue similar al de la forma 1. El TGA mostró una pérdida de peso escasa (< 0,4%) a temperatura relativamente baja antes de la descomposición. La DSC mostró un único pico endotérmico debido a la fusión, con temperaturas de inicio y pico de 205,3 y 209,5 °C, respectivamente, con una entalpia de 250,2 J/g.
Se pesaron 208 mg del compuesto 1 en un recipiente de vidrio y luego se agregó 1,0 ml de disolvente de acetona para disolver el material. Luego se agregaron 4,0 ml de cítrico 0,1 N en agua. La solución transparente se colocó en una campana para cristalización. Los precipitados aparecieron rápidamente. El sólido se recogió mediante filtración y se caracterizó. El TGA mostró una pérdida de peso escasa (< 0,4%) a temperatura relativamente baja antes de la descomposición. La DSC mostró un único pico endotérmico, debido a la fusión con temperaturas de inicio y pico de 206,0 y 211,4 °C, respectivamente, con una entalpia de 257,7 J/g.
El TGA mostró que prácticamente no hubo pérdida de peso antes de la descomposición. No obstante, tanto el análisis de RMN como el de GC mostraron presencia de 4000 a 5000 ppm de acetona.
Se pesaron 43,02 mg del compuesto 1 en un vidrio y luego se agregó 1,0 ml de disolvente de etanol para disolver el material (no por completo). Luego se agregaron 0,82 ml de cítrico 0,1 N en agua y la mezcla se volvió transparente. La solución transparente se colocó en una campana para cristalización. Los precipitados aparecieron rápidamente. Se agregó 1,0 ml más de agua. El sólido se recogió mediante filtración y se caracterizó, como se muestra en la figura 174 a la figura 176.
El perfil de XRPD fue diferente al de las formas 1, 2 y 3. El TGA mostró una pérdida de peso escasa (< 0,3%) a temperatura relativamente baja antes de la descomposición. La DSC mostró un único pico endotérmico, debido a la fusión con temperaturas de inicio y pico de 211,2 y 214,8 °C, respectivamente, con una entalpia de 277,1 J/g.
Se pesaron 45,74 mg del compuesto 1 en un recipiente de vidrio y luego se agregó 1,0 ml de disolvente de IPA para disolver el material (turbio). Luego se agregaron 0,87 ml de cítrico 0,1 N en agua y la mezcla se volvió transparente. La solución transparente se colocó en una campana para cristalización. Los precipitados aparecieron rápidamente. El sólido se recogió mediante filtración y se caracterizó. El TGA mostró una pérdida de peso escasa (< 0,3 %) a una temperatura relativamente baja antes de la descomposición. No obstante, la DSC mostró un pico endotérmico amplio a una temperatura relativamente baja, debido posiblemente a la evaporación de disolventes y el pico de fusión con temperaturas de inicio y pico de 207,9 y 212,7 °C, respectivamente, con una entalpía de 190,7 J/g.
Se pesaron 51,4 mg del compuesto 1 en un recipiente de vidrio y luego se agregó 1,0 ml de ácido cítrico 0,1 N en agua. La suspensión se mantuvo con agitación en el ambiente para conversión y cristalización. (Adición de 1 ml de agua). El sólido se recogió mediante filtración y se caracterizó, como se muestra en la figura 180 a la figura 182. El TGA mostró una pérdida de peso escasa (< 0,1%) a temperatura relativamente baja antes de la descomposición. La DSC mostró un único pico de fusión con temperaturas de inicio y pico de 204,2 y 207,6 °C, respectivamente, con una entalpía de 249,9 J/g.
La sal de citrato presentó una higroscopicidad baja, según demostró el estudio de sorción dinámica de vapores (DVS) (figura 183). En función del análisis preliminar de SVSS, las formas sólidas con ácido cítrico en diversos disolventes produjeron un perfil de XRPD similar y pueden ser solvatos isoestructurales. El TGA mostró una pérdida de peso leve (< 0,5 %) a una temperatura relativamente baja. La DSC mostró un único pico endotérmico con temperaturas de inicio y pico de 205,3 y 209,5 °C. La sal de citrato produjo diversos disolventes, incluidos etanol, IPA, acetona y EtOAc.
Se determinó la solubilidad de la forma de hidrato de HCl (2), la forma Z de sal de citrato (2) y la base libre en agua, fluido gástrico simulado (SGF), fluido intestinal simulado (SIF, por sus siglas en inglés) y HPMC al 0,5 % en Tween 80 al 0,25 %. La solubilidad en agua varió dependiendo del pH. En la tabla 3 puede observarse que el monohidrato de sal de HCl tiene la mayor solubilidad en agua (moderadamente soluble en agua) a un pH de 3,65. La solubilidad
de la sal de citrato y la base libre en agua es de 0,252 y 0,003 mg/ml, respectivamente, dependiendo del pH. El pH en medio acuoso se determinó mediante contraiones y solubilidad. Las sales de HCl produjeron el pH más bajo de 3,65 en agua, mientras que el citrato produjo un pH relativamente elevado de 4,61. La solubilidad de la forma de sal de HCl en SGF tiene efecto de iones comunes. No obstante, la base libre tiene una solubilidad cinética elevada significativa en SGF, seguida por la sal de citrato. La solubilidad de estas sales, así como la base libre en SIF, es considerablemente baja, prácticamente insoluble en SIF.
Tabla 3: Solubilidad de diversas formas de sal en a ua SGF SIF HPMC al 05 % en Tween80 2

También se determinó la solubilidad de la sal de citrato y HCl en medios biorrelevantes, en comparación con la base libre. En presencia de tensioactivos (lecitina y taurocolato de sodio), los valores de solubilidad de la sal de HCl, el citrato y la base libre son similares tanto en FeSSIF como en FaSSIF (tabla 4).
Tabla 4: Solubilidad de las sales de citrato HCl en medios biorrelevantes en com aración con la base libre

Entre la base libre, la sal de citrato y HCl no hubo una diferencia significativa en las propiedades en estado sólido. Todas son química y físicamente estables en estado sólido. Los datos en el presente documento sugieren que la solubilidad de la base libre, la sal de citrato y HCl depende del pH, el efecto de los iones comunes y la presencia de tensioactivos. No se obtuvieron resultados de PK concluyentes de un único estudio de comparación PK en perros. Todas las formas sólidas mostraron perfiles de disolución similares tanto en FeSSIF como en FaSSIF (figura 186 y figura 187) y demostraron perfiles de disolución similares de BIC en HCl 0,01 N y HCl 0,001 N.
Tabla 5: Estabilidad en estado sólido de formas de sal base libre

FORMAS SÓLIDAS
MÉTODOS ANALÍTICOS - BASE LIBRE
Se realizó un análisis polimórfico del compuesto 1 para investigar si las distintas formas sólidas se podían generar en diversas condiciones, como diferentes disolventes, cambios de humedad y temperatura.
Los disolventes utilizados en el análisis polimórfico fueron de grado de reactivo o HPLC, incluyendo acetonitrilo (MeCN), MeCN/agua (1:1), n-butanol (n-BuOH), etanol absoluto (EtOH), etanol/agua (1:1), metanol (MeOH), 2-propanol (IPA), acetato de etilo (EtOAc), acetato de metilo (MeOAc), diclorometano (DCM), metil etil cetona (MEK), metil t-butil éter (MTBE), heptano, tolueno, acetato de metilo (MeOAc), acetato de isopropilo (IPAc), metil isobutil cetona (MIBK), 2-metiltetrahidrofurano (2-MeTHF), 1,4-dioxano, tetrahidrofurano (THF), THF/agua (1:1) y agua. Se trató una muestra pesada del compuesto 1 con un volumen conocido de un disolvente de prueba. La mezcla resultante se agitó durante aproximadamente 1 día a temperatura ambiente. Si todos los sólidos parecían disueltos en la inspección visual, se calculaba la solubilidad estimada según el volumen total de disolvente utilizado para brindar una solución completa. En caso de haber sólidos presentes, se evaporaba un volumen conocido de filtrado hasta sequedad y se medía el peso del residuo para calcular la solubilidad.
Todas las muestras de sólido generadas en el análisis polimórfico se analizaron mediante XRPD. El análisis de XRPD se realizó en un difractómetro de rayos X en polvo PANalytical Empyrean utilizando radiación de Cu Ka a 1,54 Á.
El instrumento PANalytical Empyrean estaba equipado con un tubo de rayos X de foco fino. El voltaje y el amperaje del generador de rayos X se fijaron en 45 kV y 40 mA, respectivamente. Las ranuras de divergencia se fijaron a 1/16° y 1/8° y las ranuras de recepción se fijaron a 1/16°. La radiación difractada se midió con un detector 2D Pixel. Se configuró un barrido continuo theta-dos theta con una fracción de etapa de 0,013 entre 3° y 40° 26, con una velocidad de rotación de muestra de 4. Se utilizó un estándar de alúmina sinterizada para verificar las posiciones de los picos.
Los análisis de DSC se llevaron a cabo en un calorímetro de barrido diferencial de TA Discovery. Se utilizó indio como estándar de calibración. Se colocaron aproximadamente 1-5 mg de muestra en una bandeja de DSC. La muestra se calentó en nitrógeno a una velocidad de 10 °C/min, hasta una temperatura final de 220 °C. Los puntos de fusión se indicaron como temperaturas de inicio extrapoladas.
Los análisis TGA se llevaron a cabo en un analizador termogravimétrico de TA Discovery. Se colocaron aproximadamente 2-10 mg de muestra ponderada con precisión sobre una bandeja y se cargaron en el horno de TGA. La muestra se calentó en nitrógeno a una velocidad de 10 °C/min, hasta una temperatura final de 220 °C. Se llevó a cabo un análisis morfológico de las muestras en un Evex Mini-SEM. Se dispersaron cantidades pequeñas de muestra en un contenedor de muestra, se recubrieron con oro utilizando un dispositivo de recubrimiento por pulverización Evex Mini Au y se realizó imagenología con un aumento de 300x a 1000x.
Se determinó la higroscopicidad en un DVS con sistema de medición de superficie. Se cargó un tamaño de muestra de 5 a 20 mg en la bandeja de muestras del instrumento de DVS y la muestra se analizó en un analizador de sorción automático de DVS a temperatura ambiente. La humedad relativa aumentó del 0 % al 90 % de RH en etapas del 10 % de RH, luego disminuyó de manera similar para lograr un ciclo completo de adsorción/desorción.
Se obtuvieron espectros de RMN 1H en un espectrómetro de RMN Bruker de 300 MHz. Las muestras se disolvieron en DMSO-D6 y se analizaron con 8-64 barridos.
El contenido de agua Karl Fischer (KF) se midió con un titulador para horno Metrohm KF colométrico, equipado con un procesador de muestras en horno. La temperatura del horno se fijó en 100 °C.
EXPERIMENTOS DE EQUILIBRIO/SUSPENSIÓN Y EVAPORACIÓN
Experimentos de equilibrio y evaporación realizados a temperatura ambiente. En caso de haber sólidos presentes tras 1 día, estos se filtraron utilizando un filtro de PTFE de 0,45 ^m y se secaron con aire antes del análisis. El sobrenadante restante se evaporó hasta sequedad y los sólidos se aislaron para análisis.
Se realizaron experimentos de equilibrio y evaporación a 50 °C mediante adición de un exceso de compuesto 1 sólido hasta 1 ml de un disolvente de prueba. La mezcla resultante se agitó durante 1 día a temperatura ambiente y 1 día a 50 °C, por separado. Después de alcanzar el equilibrio, se retiró la solución de sobrenadante saturada, se filtró utilizando filtros de PTFE de 0,45 ^m y se dejó evaporar en un vial abierto con nitrógeno a temperatura ambiente y 50 °C, respectivamente. El sólido resultante del equilibrio se aisló y se secó con aire antes del análisis.
Tabla 6: Resumen de resultados de e uilibrio E eva oración EV
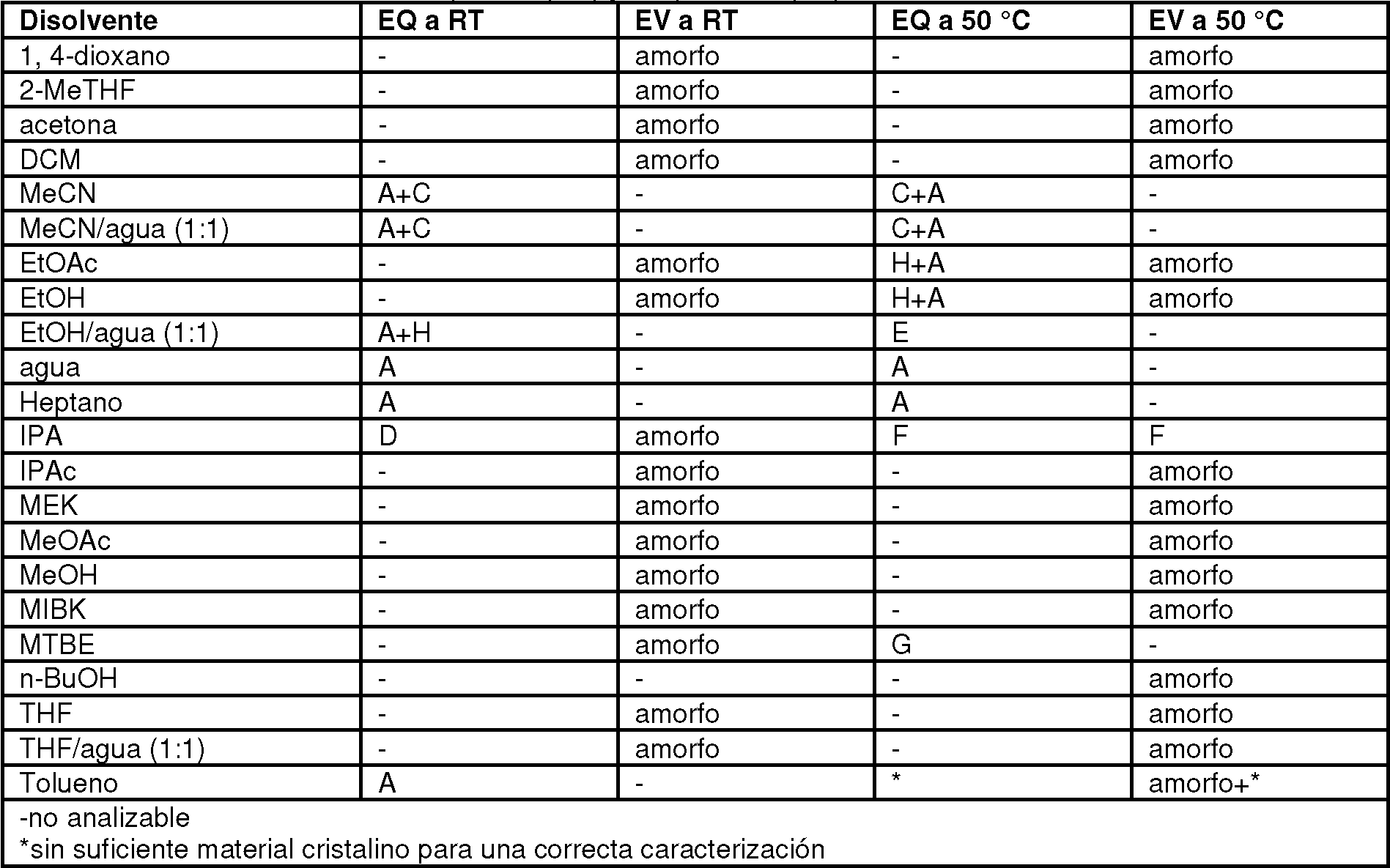
EXPERIMENTOS DE RECRISTALIZACIÓN CON ANTIDISOLVENTE Y RECRISTALIZACIÓN CON ENFRIAMIENTO
Para la recristalización con enfriamiento, cada uno de los disolventes seleccionados se saturó con el compuesto 1 sólido a 65 °C. Los disolventes incluyeron MeCN, MeCN/agua (1:1), EtOH, EtOH/agua (1:1), IPA y THF/agua (1:1). La solución se agitó durante aproximadamente 10 minutos, se filtró utilizando un filtro de jeringa de PTFE de 0,45 gm y luego se enfrió hasta aproximadamente -15 °C colocando los matraces en un congelador. El sólido resultante de la recristalización se aisló y se secó con aire antes del análisis. Para la recristalización con enfriamiento, cada uno de los disolventes seleccionados (MeOH, EtOH y EtOH/agua) se saturó con el compuesto 1 a 60 °C. La solución se agitó a 60 °C durante 10 minutos, se filtró utilizando un filtro de jeringa de PTFE de 0,45 gm y luego se enfrió hasta temperatura ambiente de forma natural y se colocó en un refrigerador. El sólido resultante de la recristalización se aisló y se secó con aire antes del análisis.
Tabla 7: Resultados de recristalización con enfriamiento

Para la recristalización con antidisolvente, se saturaron los disolventes seleccionados, MeCN y MeOH, con el compuesto 1 sólido a temperatura ambiente. Tras la disolución total del sólido, se filtró una parte de la solución en un matraz con un antidisolvente seleccionado (agua). La mezcla se enfrió hasta 4 °C colocando los matraces en un refrigerador. El sólido resultante de la recristalización se aisló y se secó con aire antes del análisis. Para la recristalización con antidisolvente, los disolventes seleccionados (MeOH, EtOH, IPA y EtOAc) se saturaron con el compuesto 1 a 60 °C. Tras la disolución total del sólido, se filtró una parte de la solución en un matraz previamente calentado y se agregó un antidisolvente seleccionado (agua, MTBE o heptano) a 60 °C. La mezcla se enfrió hasta temperatura ambiente de forma natural y luego se colocó en un refrigerador. El sólido resultante de la recristalización se aisló y se secó con aire antes del análisis.
Ta l Ex rim n r n r r m ri l r r riz i n


Se utilizaron MeOH, EtOH, EtOH/agua, IPA y EtOAc como disolventes únicos o primarios. Se utilizaron agua, MTBE y heptanos como antidisolvente. Los resultados se resumen en la tabla 6. Solamente las cristalizaciones que usaron agua como antidisolvente generaron la forma A. Todos los demás disolventes o las combinaciones de disolventes brindaron formas de solvato similares, tal como se observó durante el experimento de equilibrio.
Tabla 9: Resultados de recristalización con antidisolvente

RESUMEN DE FORMAS POLIMÓRFICAS
Durante este estudio de análisis polimórfico se hallaron un total de nueve formas cristalinas y una forma amorfa del compuesto 1 como base libre. El gráfico de líneas apiladas de patrones de XRPD de las nueve formas cristalinas se muestra en la figura 1 y las características físicas se resumen en la tabla 10. El patrón de XRPD de la forma amorfa se muestra en la figura 40.
Tabla 10: Resumen de formas sólidas forma amorfa de la base libre del com uesto 1

FORMA A
Se calculó la solubilidad aproximada de la forma A de base libre en diversos disolventes a temperatura ambiente, como se describió anteriormente. Los resultados se resumen en la tabla 11. La forma A de base libre se mostró como la más soluble (> 100 mg/ml) en acetona, EtOAc, MeOAc y THF. La forma A fue muy soluble (> 50 mg/ml) en 1,4-dioxano, 2-MeTHF, DCM, MeCN/agua (1:1), IPAc, MEK, MeOH, MIBK y THF/agua (1:1). La forma A demostró cierta solubilidad (> 20 mg/ml) en EtOH, Mt b E, n-BuOH (> 10 mg/ml) en IPA, tolueno, (> 3 mg/ml) en MeCN y EtOH/agua (1:1). La forma A demostró solubilidad baja (< 1 mg/ml) en agua y heptano.
T abla 11 l ili r xim l f rm A li r l m 1 m r r m i n

Los experimentos de equilibrio a 50 °C generaron la forma A en agua y heptano. Se obtuvo una forma única, indicada como la forma E, a partir de la forma A en EtOH/agua (1:1). Se obtuvo una forma única, indicada como la forma F, a partir de la forma A en IPA. Se obtuvo una forma única, indicada como la forma G, a partir de la forma A en MTBE. Se obtuvo una mezcla de la forma A y la forma C en MeCN y MeCN/agua (1:1). Se obtuvo una mezcla de la forma A y la forma H en EtOAc y EtOH. También se obtuvo la forma F a partir de la forma A con evaporación a 50 °C a partir de IPA. La evaporación en tolueno generó una mezcla del material amorfo y poco cristalino (forma desconocida). Todos los demás experimentos de evaporación a 50 °C produjeron la forma amorfa del compuesto 1. Los experimentos de recristalización con enfriamiento se realizaron como se describió anteriormente. Los disolventes incluyeron MeCN, MeCN/agua (1:1), EtOH, EtOH/agua (1:1), THF/agua (1:1) e IPA. Los resultados se resumen en la tabla 7. Los sólidos obtenidos a partir de MeCN/agua (1:1) se confirmaron como la forma C. Los sólidos obtenidos a partir de MeCN se confirmaron como una forma única, indicada como la forma I. El resto de los disolventes no precipitó tras 14 días a -15 °C.
Las recristalizaciones con antidisolventes se realizaron como se describió anteriormente. Se utilizaron MeCN y MeOH como disolventes primarios. Se utilizó agua como antidisolvente. Los resultados se resumen en la tabla 10. Utilizando XRPD, se confirmó que los sólidos obtenidos a partir de MeCN/agua eran una mezcla de la forma C y la forma A. Los sólidos obtenidos a partir de MeOH/agua se confirmaron como amorfos.
La forma A es un monohidrato. Esta forma se obtuvo principalmente a partir de experimentos de recristalización o suspensión en sistemas de disolventes acuosos o "ricos en agua".
La forma A también puede obtenerse mediante conversión de la forma B, la forma C y la forma H mediante exposición a condiciones ambiente con más de aproximadamente el 20 % de humedad relativa (RH).
La forma A se convierte en la forma B anhidra tras secado con menos del 10 % de RH o a temperatura elevada. La forma A tiene un patrón de XRPD cristalina como se ilustra en la figura 2. En la figura 4 y la figura 5, respectivamente, se ilustran los termogramas TGA y DSC de la forma A. El termograma de DSC mostró dos eventos, en los que el primero presenta una temperatura de inicio de aproximadamente 94 °C y una máxima de aproximadamente 117 °C, lo cual se atribuye a la deshidratación, y el segundo presenta una temperatura de inicio de aproximadamente 174 °C y una máxima de aproximadamente 182 °C, lo cual corresponde a la
fusión/descomposición. Se observó una pérdida de peso en el TGA del 2,8 % hasta la fusión. El espectro de RMN 1H de la forma A fue coherente con la estructura del compuesto 1, sin degradación significativa o disolvente residual (véase la figura 7).
El comportamiento de sorción/desorción de humedad de la forma A se determinó mediante DVS. Los resultados se resumen en la figura 6A-figura 6B. Se observó un cambio marcado en el peso de más del 3 % entre el 10 y el 30 % de RH. Se observó un cambio de peso similar entre el 10 % y el 0 % de RH tras la desorción, lo cual se condice con un hidrato. Se observó una absorción de agua adicional de aproximadamente el 1,4 % en peso entre el 30 y el 90 % de RH, lo cual sugiere que el hidrato es levemente higroscópico.
La figura 1 proporciona un patrón de XRPD de la forma A. A continuación, en la tabla 12 se proporciona una lista de picos de difracción de rayos X para la forma A.
Tabla 12: Pico

(continuación)

La figura 3 es una imagen de SEM de la forma A.
FORMA B
La forma B se obtuvo secando la forma A a aproximadamente 0 °C con vacío. La forma B también se puede obtener secando la forma C a 50-60 °C con vacío. La forma B se convierte en la forma A en condiciones ambiente que incluyen más de aproximadamente el 20 % de RH. La forma B tuvo un patrón de XRPD cristalina como se ilustra en la figura 8. En la figura 9 y la figura 10, respectivamente, se ilustran los termogramas TGA y DSC de la forma B obtenidos a partir de acetona. La pérdida de peso del TGA del 0,1 % en peso se corresponde con un pico de DSC con un inicio aproximado de aproximadamente 174 °C y una máxima de aproximadamente 182 °C y se corresponde con la fusión/descomposición. Estas observaciones sugirieron que la forma B es un anhidrato del compuesto 1. En la tabla 13 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma B.
Tabla 13: Picos de dif

FORMA C
La forma C se obtuvo a partir del equilibrio de la forma A en MeCN o MeCN/agua a temperatura ambiente o a 50 °C. La forma C también se puede obtener a partir de un proceso de solución del compuesto 1 en MeTHF. Se destiló
MeTHF (10 vol) al vacío en un volumen constante, con adición de MeCN (~ 20 vol), para eliminar el MeTHF (30,6641 kPa (230 torr)/46 °C). Al final no había más del 5 % en vol de MeTHF en el lote. Los sólidos se cristalizaron durante la destilación. Se enfrió el lote, se dejó madurar, se filtró y se secó al vacío a no más de 30 °C. La forma C tuvo un patrón de XRPD cristalina tal como se ilustra en la figura 11. En la figura 12 y la figura 13, respectivamente, se ilustran los termogramas TGA y DSC de la forma C obtenidos a partir de MeCN/agua. La pérdida de peso del TGA de 6,6 % en peso corresponde a un pico de DSC amplio a aproximadamente 165 °C y puede atribuirse a la evaporación de los disolventes en la forma C. El pico de DSC con temperatura de inicio de 180 °C y máxima de aproximadamente 186 °C se corresponde a la fusión/descomposición. Se obtuvo el espectro de RMN 1H para la muestra de la forma C y este fue coherente con la estructura, si bien presentó una mayor cantidad de MeCN (figura 14). El contenido teórico de MeCN de un monosolvato del compuesto 1 es del 6,7 % en peso, lo cual se corresponde con la pérdida de peso observada en el TGA. Estas observaciones sugirieron que la forma C es un monosolvato de acetonitrilo del compuesto 1. La forma C a temperaturas ambiente con más de aproximadamente el 20 % de RH produjo conversión en la forma A.
En la tabla 14 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma C.
Tabla 14: Picos de di

(continuación)

FORMA D
La forma D se obtuvo a partir del equilibrio con recristalización de la forma A en IPA a temperatura ambiente. La forma D tuvo un patrón de XRPD cristalina como se ilustra en la figura 15. En la figura 17 y la figura 18, respectivamente, se ilustran los termogramas TGA y DSC de la forma D. La pérdida de peso del TGA de aproximadamente el 7,4 % en peso corresponde a un pico de DSC amplio de aproximadamente 154 °C y puede atribuirse a la pérdida de disolventes en la forma D. El menor pico de DSC con temperatura de inicio de 175 °C y máxima de aproximadamente 185 °C se corresponde a fusión/descomposición. Se obtuvo el espectro de RMN 1H para la muestra de la forma D y este fue coherente con la estructura y presentó IPA (figura 19). Estas observaciones sugirieron que la forma D probablemente sea un solvato en IPA del compuesto 1.
En la tabla 15 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma D.
Tabla 15: Picos de dif

(continuación)

FORMA E
La forma E se obtuvo a partir del equilibrio de la forma A en EtOH/agua (1:1) a 502C. La forma E tuvo un patrón de XRPD cristalina tal como se muestra en la figura 20. En la figura 21 y la figura 22, respectivamente, se ilustran los termogramas TGA y DSC de la forma E. La pérdida de peso del TGA de 13,7 % en peso se corresponde con un pequeño pico de DSC amplio a aproximadamente 104 °C y puede atribuirse a la pérdida de disolvente en la forma E. Estas observaciones sugirieron que la forma E es un solvato o un hidrato que contiene etanol.
En la tabla 16 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma E.
Tabla 16: Picos de difr i n r X r l f rm E

FORMAF
La forma F se obtuvo a partir del equilibrio de la forma A en IPA a 50 °C. La forma F tuvo un patrón de XRPD
cristalina tal como se muestra en la figura 23. En la figura 24 se proporciona una imagen de SEM de la forma F. En la figura 25 y la figura 26, respectivamente, se ilustran los termogramas TGA y DSC de la forma F. La pérdida de peso del TGA del 14,3 % en peso corresponde a un pico de DSC amplio con inicio a aproximadamente 137 °C y puede atribuirse a la pérdida de disolventes en la forma F. El pico de DSC con temperatura máxima de 153 °C se corresponde a la fusión/descomposición. El espectro de RMN 1H obtenido para la muestra de la forma F fue coherente con la estructura, pero incluyó IPA. Véase la figura 27. Estas observaciones sugirieron que la forma F es un solvato en IPA del compuesto 1.
En la tabla 17 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma F.
Tabla 17: Picos de difr i n r X r l f rm F

FORMA G
La forma G se obtuvo a partir del equilibrio de la forma A en MTBE a 50 °C. La forma G tuvo un patrón de XRPD cristalina tal como se muestra en la figura 28. En la figura 29 se proporciona una imagen de SEM de la forma G. En la figura 30 y la figura 31, respectivamente, se ilustran los termogramas TGA y DSC de la forma G. La pérdida de peso del TGA de 8,7 % en peso corresponde a un pico de DSC a aproximadamente 147 °C y puede atribuirse a la pérdida de disolventes en la forma G. El pico de DSC con una máxima de aproximadamente 161 °C se corresponde
a la fusión/descomposición. El espectro de RMN 1H obtenido para la muestra de la forma G fue coherente con la estructura, pero incluyó MTBE (véase la figura 32). Estas observaciones sugirieron que la forma G es un solvato en MTBE del compuesto 1.
En la tabla 18 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma G.
Tabla 18: Picos de difr

(continuación)

FORMA H
La forma H se obtuvo a partir de la forma A en EtOH/agua (1:1), EtOH o EtOAc a 502C. La forma H tuvo un patrón de XRPD cristalina tal como se muestra en la figura 33. En la figura 34 y la figura 35, respectivamente, se ilustran los termogramas TGA y DSC de la forma H. La pérdida de peso del termograma de TGA del 6,5 % en peso corresponde a un pico de DSC amplio de aproximadamente 163 °C y puede atribuirse a la pérdida de disolventes en la forma H. El pico de DSC con temperatura de inicio de aproximadamente 179 °C y máxima de aproximadamente 187 °C se corresponde a la fusión/descomposición. El contenido teórico de EtOH de un monosolvato del compuesto 1 es del 7,5 %, lo cual se corresponde con la pérdida de peso observada en el TGA. Estas observaciones sugirieron que la forma H es un solvato o hidrato del compuesto 1. El experimento de transferencia de forma mostró que la exposición de la forma H a más del 20 % de RH produce la forma A.
En la tabla 19 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma H.
Tabla 19: Picos de difr

(continuación)

FORMA I
La forma I se obtuvo a partir de recristalización con enfriamiento de la forma A en MeCN. La forma I tuvo un patrón de XRPD cristalina como se ilustra en la figura 36. En la figura 38 y la figura 39, respectivamente, se ilustran los termogramas TGA y DSC de la forma I. La pérdida de peso del TGA de 2,3 % en peso corresponde a un pico de DSC amplio de aproximadamente 75 °C y puede atribuirse a la pérdida de disolventes en la forma I. El pico de DSC con temperatura de inicio de aproximadamente 173 °C y máxima de aproximadamente 183 °C se corresponde a la fusión/descomposición. El experimento de transferencia de forma demostró que una suspensión con MeCN a RT produce la forma C. Estas observaciones sugirieron que la forma I es un solvato o un hidrato del compuesto 1. En la tabla 20 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma I.
Tabla 20: Picos de difr

(continuación)

BASE LIBRE SÓLIDA AMORFA
El sólido amorfo del compuesto 1 se obtuvo a partir de la mayoría de los experimentos de evaporación a temperatura ambiente o a 50 °C, como se muestra en la tabla 6.
El sólido amorfo tuvo un espectro de XRPD según se ilustra en la figura 40. El termograma de DSC de la muestra de sólido amorfo se ilustra en la figura 41. El sólido amorfo tiene una temperatura de transición vitrea de aproximadamente 84 °C. En la figura 43A se muestra la gráfica isotérmica de DVS del sólido amorfo. Se observó un cambio de peso reversible de aproximadamente el 3,5 % entre el 10 y el 50 % de RH.
T abla 21: Resumen de ex erimentos de conversión de forma

FORMAS SÓLIDAS MÉTODOS ANALÍTICOS - FORMAS DE SAL DE CITRATO
Se realizó un análisis polimórfico del compuesto 1 de sal de citrato para investigar si las distintas formas sólidas se podían generar en diversas condiciones, como diferentes disolventes, cambios de humedad y temperatura.
Los disolventes utilizados en el análisis polimórfico fueron de grado de reactivo o HPLC, incluyendo acetonitrilo (MeCN), MeCN/agua (1:1), n-butanol (n-BuOH), etanol absoluto (EtOH), etanol/agua (1:1), metanol (MeOH), 2-propanol (IPA), acetato de etilo (EtOAc), acetato de metilo (MeOAc), diclorometano (DCM), metil etil cetona (MEK), metil t-butil éter (MTBE), heptano, tolueno, acetato de metilo (MeOAc), acetato de isopropilo (IPAc), metil isobutil cetona (MIBK), 2-metiltetrahidrofurano (2-MeTHF), 1,4-dioxano, tetrahidrofurano (THF), THF/agua (1:1), agua, sulfóxido de dimetilo (DMSO), dimetilacetamida (dMa , Dm Ac) y N-metilpirrolidona (n Mp).
Se trató una muestra pesada del citrato del compuesto 1 con un volumen conocido de un disolvente de prueba. La mezcla resultante se agitó durante 1 día a temperatura ambiente. Si todos los sólidos parecían disueltos en la inspección visual, se calculaba la solubilidad estimada según el volumen total de disolvente utilizado para brindar una solución completa. En caso de haber sólidos presentes, se evaporaba un volumen conocido de filtrado hasta sequedad y se medía el peso del residuo para calcular la solubilidad.
Todas las muestras de sólido generadas en el análisis polimórfico se analizaron mediante XRPD. El análisis de XRPD se realizó en un difractómetro de rayos X en polvo PANalytical Empyrean utilizando radiación de Cu Ka a 1,54 Á.
El instrumento PANalytical Empyrean estaba equipado con un tubo de rayos X de foco fino. El voltaje y el amperaje del generador de rayos X se fijaron en 45 kV y 40 mA, respectivamente. Las ranuras de divergencia se fijaron a 1/16° y 1/8 ° y las ranuras de recepción se fijaron a 1/16°. La radiación difractada se midió con un detector 2D Pixel. Se configuró un barrido continuo theta-dos theta con un tamaño de etapa de 0,013 o 0,026 entre 3° y 40° 20, con una velocidad de rotación de muestra de 4. Se utilizó un estándar de alúmina sinterizada para verificar las posiciones de los picos.
Los análisis de DSC se llevaron a cabo en un calorímetro de barrido diferencial de TA Discovery. Se utilizó indio como estándar de calibración. Se colocaron aproximadamente de 1 a 5 mg de muestra en una bandeja de DSC. La muestra se calentó en nitrógeno a una velocidad de 10 °C/min, hasta una temperatura final de 260 °C. Los puntos de fusión se indicaron como temperaturas de inicio extrapoladas.
Los análisis TGA se llevaron a cabo en un analizador termogravimétrico de TA Discovery. Se colocaron aproximadamente de 2 a 10 mg de muestra ponderada con precisión sobre una bandeja y se cargaron en el horno de TGA. La muestra se calentó en nitrógeno a una velocidad de 10 °C/min, hasta una temperatura final de 300 °C. Se llevó a cabo un análisis morfológico de las muestras en un Evex Mini-SEM. Se dispersaron cantidades pequeñas de muestra en un contenedor de muestra, se recubrieron con oro utilizando un dispositivo de recubrimiento por pulverización Evex Mini Au y se realizó imagenología con un aumento de 500x a 1000x.
Se determinó la higroscopicidad en un DVS con sistema de medición de superficie. Se cargó un tamaño de muestra de 5 a 20 mg en la bandeja de muestras del instrumento de DVS y la muestra se analizó en un analizador de adsorción automático de DVS a temperatura ambiente. La humedad relativa aumentó del 0 % al 90 % de RH en etapas del 10 % de RH, luego disminuyó de manera similar para lograr un ciclo completo de adsorción/desorción. Se obtuvieron espectros de RMN 1H en un espectrómetro de RMN Bruker de 300 MHz. Las muestras se disolvieron en DMSO-D6 y se analizaron con 128 barridos.
Se determinó la solubilidad de la forma A y la forma B en disolventes orgánicos seleccionados mezclando las formas sólidas individuales con disolventes seleccionados a temperatura ambiente. Se obtuvieron alícuotas en múltiples momentos (18 h, 4 días, 8 días o 12 días), se filtraron y cuantificaron con un método de HPLC. Los sólidos recuperados se analizaron mediante XRPD para confirmar las formas sólidas.
EXPERIMENTOS DE EQUILIBRIO/SUSPENSIÓN Y EVAPORACIÓN
Se realizaron experimentos de equilibrio y evaporación a temperatura ambiente y 50 °C mediante adición de un exceso de sólido de citrato del compuesto 1 hasta 1 ml de un disolvente de prueba. La mezcla resultante se agitó durante 1 día a temperatura ambiente y 1 día a 50 °C, por separado. Luego de alcanzar el equilibrio, se retiró la solución de sobrenadante saturada, se filtró utilizando filtros de PTFE de 0,45 gm y se dejó evaporar en un matraz abierto con nitrógeno a temperatura ambiente y 50 °C, respectivamente. El sólido resultante del equilibrio se aisló y se secó con aire antes del análisis.
EXPERIMENTOS DE RECRISTALIZACIÓN CON ANTIDISOLVENTE Y RECRISTALIZACIÓN CON ENFRIAMIENTO
Para la recristalización con enfriamiento, cada uno de los disolventes seleccionados se saturó con citrato del compuesto 1 a 65 °C. Los disolventes incluyeron MeCN/agua (1:1), EtOH, EtOH/agua (1:1), MeOH, THF/agua (1:1) y THF. La solución se agitó durante 10 minutos, se filtró utilizando un filtro de jeringa de PTFE de 0,45 gm y luego se enfrió hasta -15 °C colocando los viales en un congelador. El sólido resultante de la recristalización se aisló y se secó con aire antes del análisis.
Para la recristalización con antidisolvente, el disolvente DMA seleccionado se saturó con el material de citrato del compuesto 1 a temperatura ambiente. Tras la disolución total del sólido, se filtró una parte de la solución en un matraz con un antidisolvente seleccionado (MeCN, MeOH, heptano, EtOAc, tolueno y agua). La mezcla se enfrió hasta -15 °C y 4 °C colocando los viales en un congelador o un refrigerador. El sólido resultante de la recristalización se aisló y se secó con aire antes del análisis.
RESUMEN DE FORMAS POLIMÓRFICAS
Durante este estudio de análisis polimórfico se hallaron dos formas cristalinas de la sal de citrato del compuesto 1. El gráfico de líneas apiladas de patrones de XRPD de estas formas se muestra en la figura 44 y las características físicas se resumen en la tabla 29.
FORMA Y
La forma Y se obtuvo disolviendo el material de partida de compuesto 1 en 5 vol de acetona a 25 C. Se cargaron aproximadamente 1,15 eq. de ácido cítrico en agua (~ 0,2 M) en el lote para formar la sal de citrato del compuesto 1. La sal de citrato del compuesto 1 se dejó madurar a 25 °C hasta que la concentración de agua madre se encontrara por debajo de 1 mg/ml. La suspensión se retiró mediante filtración y se lavó utilizando ~ 4 vol (1:1) de acetona/H2O para lavar la torta. La torta se secó en un horno de vacío a 50 °C hasta que la RMN ya no detectó más acetona. Se calculó la solubilidad aproximada de la forma Y de citrato del compuesto 1 en diversos disolventes a temperatura ambiente, como se describió anteriormente. Los resultados se resumen en la tabla 22. Se descubrió que la forma Y de citrato del compuesto 1 era más soluble (> 50 mg/ml) en DMSO, DMA y NMP. La forma Y de citrato del compuesto 1 mostró cierta solubilidad (> 20 mg/ml) en THF/agua, (> 5 mg/ml) en THF, (> 3 mg/ml) en MeCN/agua (1:1) y MeOH, (> 2 mg/ml) en 1,4-dioxano. La forma Y de citrato del compuesto 1 demostró una solubilidad baja (< 1~2 mg/ml) en todos los demás disolventes analizados, incluidos acetona, n-BuOH, MeCN, EtOH, EtOH/agua (1:1),
IPA, EtOAc, MeOAc, DCM, MTBE, MEK, heptano, tolueno, 2-MeTHF y agua.
Tabla 22 l ili r xim l f rm Y ir l m 1 m r r m i n
Se realizaron experimentos de equilibrio y evaporación a temperatura ambiente y 50 °C utilizando la forma Y de citrato del compuesto 1 como material de partida, como se describió anteriormente. Los resultados se resumen en la tabla 23. El equilibrio en MeOH y MeCN/agua a 50 °C brindó una forma única, indicada como forma Z de sal de citrato. Todos los demás experimentos de equilibrio brindaron la forma Y de citrato del compuesto 1 o la forma Y de citrato del compuesto 1 mezclada con la forma Z de citrato de compuesto 1. Debido a la solubilidad relativamente baja, la mayoría de los experimentos de evaporación no brindaron un sólido analizable. La evaporación a partir de EtOH y EtOH/agua brindó la mezcla de las formas Y y Z de citrato del compuesto 1. Los sólidos de la evaporación de MeOH brindaron la forma Z de citrato del compuesto 1.
Tabla 23: Resumen de los resultados de e uilibrio eva oración

(continuación

Los experimentos de recristalización con enfriamiento se realizaron como se describió anteriormente. Los disolventes incluyeron MeCN/agua (1:1), EtOH/agua (1:1), THF/agua (1:1), EtOH, MeOH y THF. Los resultados se resumen en la tabla 24. Los sólidos obtenidos de THF y THF/agua se confirmaron como la forma Y de citrato del compuesto 1. Los sólidos obtenidos de MeOH y MeCN/agua se confirmaron como la forma Z de citrato del compuesto 1. Los sólidos obtenidos de EtOH y EtOH/agua se confirmaron como una mezcla de las formas Y y Z de citrato del compuesto 1.
Tabla 24: Resultados de la recristalización con enfriamiento

Las recristalizaciones con antidisolventes se realizaron como se describió anteriormente. Se usó DMA como disolvente principal. Se usaron MeCN, MeOH, heptano, EtOAc, tolueno y agua como antidisolventes. Los resultados se resumen en la tabla 25. Utilizando XRPD, se confirmó que los sólidos obtenidos de DMA/MeCN, DMA/MeOH y DMA/agua eran la forma Z de citrato del compuesto 1. Los sólidos obtenidos de DMA/EtOAc se confirmaron como la forma Y de citrato del compuesto 1 y los sólidos obtenidos de DMA/tolueno se confirmaron como una mezcla de las formas Y y Z de citrato del compuesto 1. No se observó precipitación del experimento de recristalización con
DMA/heptano.
Tabla 25: Resultados de la recristalización con antidisolvente

La forma Y se indicó como la forma cristalina de la muestra de DSD utilizada como material de partida para este análisis. La forma Y tiene un patrón de XRPD cristalina como se ilustra en la figura 45. En la figura 46 se ilustra la imagen de SEM. En la figura 47 y la figura 48, respectivamente, se ilustran los termogramas de TGA y DSC de la forma Y. No se observó pérdida de peso en el TGA hasta 150 °C para la forma Y. Se observó una leve pérdida de peso adicional hasta la temperatura de fusión de la forma Y. El termograma de DSC mostró un episodio de fusión con una temperatura de inicio de 213 °C y una máxima de 217 °C. La forma Y es levemente higroscópica, con aproximadamente el 2,1 % p/p de absorción de agua entre el 0 y el 90 % de RH. El espectro de RMN 1H es coherente con la estructura de una sal de citrato, con aproximadamente el 0,2 % p/p de acetona residual (figura 50). El contenido de ácido cítrico fue del 25,1 % en peso, según se determinó mediante la HPLC, lo cual es coherente con una sal 1:1 (teóricamente con el 25,2 % en peso de ácido cítrico). Estas observaciones sugieren que la forma Y probablemente sea un anhidrato del citrato del compuesto 1.
La estabilidad de la forma Y se caracterizó adicionalmente mediante un análisis de compresión y experimentos de transferencia de forma. Luego de aplicar una presión de 13,790 MPa (2000 psi) durante aproximadamente 1 minuto, el material seguía siendo la forma Y (figura 51A y figura 51B).
Tabla 26: Solubilidad de la forma Z y la forma Y de citrato del compuesto 1 en disolventes seleccionados a tem eratura ambiente mediante HPLC

Nota: todos los sólidos recuperados de los análisis de solubilidad permanecieron como la forma inicial en la XRPD. En la tabla 27 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma Y.
Tabla 27: Picos de ifr i n r X r l f rm Y

(continuación)

FORMA Z
La forma Z de citrato del compuesto 1 se generó mediante un experimento de equilibrio en MeOH y MeCN/agua (1:1) a 50 °C, así como diversos experimentos de recristalización, que incluyen la recristalización con enfriamiento a partir de MeCN/agua y recristalización con antidisolvente a partir de DMA/MeCN, DMA/MeOH y DMA/agua. La forma Z tiene un patrón de XRPD cristalina como se ilustra en la figura 52. En la figura 53 se ilustra la imagen de SEM. En la figura 54 y la figura 55, respectivamente, se ilustran los termogramas TGA y DSC de la forma Z. No se observó pérdida de peso significativa para la forma Z en el TGA hasta 150 °C. Se observó una pérdida de peso adicional (de un porcentaje bajo) hasta la temperatura de fusión de la forma Z. El termograma de DSC mostró un episodio de fusión con una temperatura de inicio de 217 °C y una máxima de 221 °C. La forma Z es levemente higroscópica, con aproximadamente el 1,6 % p/p de absorción de agua entre 0 y 90 % de RH. El espectro de RMN 1H de la muestra para equilibrio de la forma Z en MeCN/agua a 50 °C es coherente con la estructura de la sal de citrato del compuesto 1, con aproximadamente el 1,0 % p/p de MeCN residual (figura 57). El contenido de ácido cítrico fue del 24,6 % en peso, según se determinó mediante la HPLC, lo cual es coherente con una sal 1:1 (teóricamente con el 25,2 % en peso de ácido cítrico). Estas observaciones sugieren que la forma Z probablemente sea un anhidrato del citrato del compuesto 1.
Ta

Se realizó un estudio adicional de secado para comprender la pérdida de peso observada por encima de 150 °C. Se secaron alícuotas de la muestra de forma Z en un horno KF (con barrido de N2) a 150 y 180 °C, respectivamente. El contenido de ácido cítrico de los sólidos recuperados fue del 24,2 y el 17,8 % en peso, respectivamente, lo cual sugiere pérdida de ácido cítrico. El disolvente residual en las muestras secas también fue considerablemente menor que en la muestra de la forma Z "en estado actual".
La estabilidad de la forma Z se caracterizó además mediante un análisis de compresión y experimentos de transferencia de forma. Después de aplicar una presión de 13,790 MPa (2000 psi) durante aproximadamente 1 minuto, el material seguía estando en forma Z (figuras 62A-B).
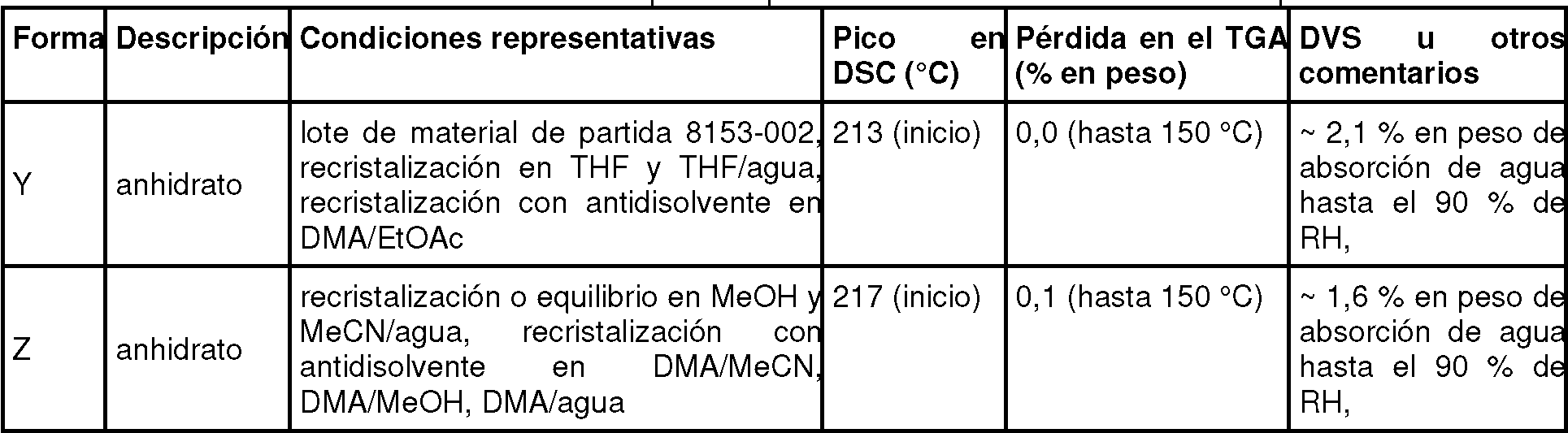
En la tabla 30 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma Z.
Tabla 30: Picos d

Se exploró la relación termodinámica entre las dos formas a través de experimentos de conversión de forma (tabla 31). Las suspensiones de competencia a partir de mezclas de las formas Y y Z produjeron resultados específicos del disolvente. La forma Y surgió de suspensiones en THF a temperatura ambiente y de suspensiones en THF y EtOAc a 50 °C. La forma Z surgió de suspensiones en EtOH, agua, MEK y MeCN a temperatura ambiente y 50 °C.
Se exploró además la relación termodinámica entre las dos formas a través de experimentos de solubilidad (tabla 26). Estos experimentos se diseñaron de modo de determinar si los resultados de las suspensiones de competencia se debían a diferentes cinéticas de crecimiento/disolución de cada forma en un disolvente específico o a la termodinámica global. Tal como se muestra en la tabla 26, la solubilidad de la forma Z fue inferior a la de la forma Y en EtOH, mientras que la solubilidad de la forma Y fue inferior en acetona, MeOAc y 2-MeTHF. Estos resultados parecen ser coherentes con las observaciones de suspensiones de competencia, lo cual sugiere que la cinética de
crecimiento/disolución no era la causa de la conversión de forma específica del disolvente.
Tabla 31: Resumen de los ex erimentos de transferencia de formas

FORMAS SÓLIDAS MÉTODOS ANALÍTICOS - FORMAS DE SAL DE HCL
Se realizó un análisis polimórfico del compuesto 1 para investigar si las distintas formas sólidas se podían generar en diversas condiciones, como diferentes disolventes, cambios de humedad y temperatura.
Se generó material de partida disolviendo el compuesto 1 en 10 vol de MeOH a 25-30 °C. Luego se cargaron 1,10 equiv. de HCl en MeOH (~ 1,25 M) en el lote para formar la sal de HCl del compuesto 1. Se realizó destilación constante con vacío para cambiar el disolvente de MeOH a EtOAc (~ 30-35 vol) y la temperatura del lote de mantuvo a 25-35 °C. La suspensión se retiró mediante filtración y se utilizaron ~ 5 vol (1:1) de EtOAc para lavar la torta, que se secó en un horno de vacío a 50 °C.
El material de partida tiene una cristalinidad relativamente baja, con una pérdida de peso del 1,1 % en peso hasta 100 °C en el TGA y un pico de fusión a 238,5 °C (temperatura de inicio) en la DSC.
Se observó un cambio de masa del 3,6 % en peso para el material de partida del 0 % de RH al 95 % de RH a 25 °C. La muestra es moderadamente higroscópica.
El contenido teórico de Cl para una sal de HCl 1:1 es del 5,84 % en peso. El contenido de Cl de la sal de HCl del compuesto 1 fue del 5,70 % en peso.
Se determinó la solubilidad del material de partida en disolventes orgánicos seleccionados a través de mezcla con disolventes seleccionados a temperatura ambiente.
Tabl

(continuación

Durante el análisis polimórfico se hallaron ocho formas cristalinas y se indican en el presente documento como forma 1 de sal de HCl a forma 8 de sal de HCl. En la tabla 33 se brindan las características generales de las formas cristalinas.

Forma 1 de sal de HCl
La forma 1 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 66. En la figura 67 se muestran los termogramas de TGA y DSC de la forma 1 de sal de HCl. El termograma de DSC mostró múltiples eventos térmicos, en los que el primer evento presenta una temperatura de inicio de aproximadamente 102 °C y una máxima de aproximadamente 114 °C, lo cual se atribuye a la deshidratación, y un segundo evento presenta una temperatura de inicio de aproximadamente 146 °C y una máxima de aproximadamente 181 °C, lo cual corresponde a la fusión/descomposición. La pérdida de peso en el TGA fue del 6,6 % antes de los 140 °C y otro del 10 % de pérdida de peso antes de los 205 °C. Se obtuvo la forma 1 de sal de HCl a partir de evaporación lenta con IPA. La
forma 1 de sal de HCl es un solvato de IPA de la sal de HCl del compuesto 1.
En la tabla 34 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 1 de sal de HCl.
Tabla 34: Picos de^ifr in r X r l f rm 1 l H l

(continuación)

FORMA 2 DE SAL DE HCL
La forma 2 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 68. En la figura 69 se muestran los termogramas de TGA y DSC de la forma 2 de sal de HCl. El termograma de DSC mostró un evento térmico amplio con un inicio de aproximadamente 151 °C, lo cual se atribuye a la fusión, descomposición y desproporción. La pérdida de peso en el TGA fue del 2,6 % antes de los 140 °C y otro del 14 % de pérdida de peso antes de los 200 °C. Se obtuvo la forma 2 de sal de HCl a partir de difusión líquida de vapores con IPA/tolueno. En la tabla 35 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 2 de sal de HCl.
Tabla 35: Picos d

(continuación)

FORMA 3 DE SAL DE HCL
La forma 3 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 70. En la figura 71 se muestran los termogramas de TGA y DSC de la forma 3 de sal de HCl. El termograma de DSC mostró un evento térmico amplio a 153 °C (inicio) lo cual se atribuye probablemente a la fusión, descomposición y desproporción. La pérdida de peso en el TGA fue del 2,3 % antes de los 140 °C y otro del 11 % de pérdida de peso antes de los 210 °C. Se obtuvo la forma 3 de sal de HCl a partir de múltiples condiciones relacionadas con n-BuOH.
En la tabla 36 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 3 de sal de HCl.
Tabla 36: Picos de ifr i n r X r ^ l f rm l H l

(continuación)

FORMA 4 DE SAL DE HCL
La forma 4 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 72. En la figura 73 se muestran los termogramas de TGA y DSC de la forma 4 de sal de HCl. El termograma de DSC mostró una endotermia con desolvatación a 94 °C, exotermia posterior y otra endotermia a 219 °C. La pérdida de peso del TGA fue del 4,5 % antes de 140 °C. Se obtuvo la forma 4 de sal de HCl a partir de difusión líquida de vapores con MeOH/IPAc. La forma 4 de sal de HCl puede ser un solvato de la sal de HCl del compuesto 1.
En la tabla 37 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 4 de sal de HCl.
Tabla 37: Picos de^ ifr i n r X r l f rm 4 l H l

(continuación)

FORMA 5 DE SAL DE HCL
La forma 5 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 74. En la figura 75 se muestran los termogramas de TGA y DSC de la forma 5 de sal de HCl. El termograma de DSC mostró una endotermia con desolvatación a aproximadamente 104 °C y aproximadamente 162 °C, seguida por una posible endotermia de fusión con inicio a 192 °C y una máxima a 209 °C. La pérdida de peso del TGA fue del 4,2 % antes de 140 °C. Se obtuvo la forma 5 de sal de HCl a partir de difusión sólida de vapores con DMF. La forma 5 de sal de HCl puede ser un solvato de la sal de HCl del compuesto 1. La forma 5 de sal de HCl puede ser un hidrato de la sal de HCl del compuesto 1.
En la tabla 38 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 5 de sal de HCl.
Tabla 38: Picos d

FORMA 6 DE SAL DE HCL
La forma 6 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 76. En la figura 77 se muestran los termogramas de TGA y DSC de la forma 6 de sal de HCl. El termograma de DSC mostró una endotermia con deshidratación a 88 °C y otra endotermia con inicio a 201 °C y una máxima a 228 °C. La pérdida de peso del TGA fue del 11,6 % antes de 100 °C. Un termograma del segundo procesamiento de DSC mostró endotermia a 89 °C y otra endotermia con inicio a 211 °C y una máxima a 225 °C (figura 79). La pérdida de peso del TGA corresponde al 15,9 % de pérdida de peso antes de 120 °C (figura 78). La muestra parecía contener agua cristalina y superficial. El contenido de agua teórico para un pentahidrato y hexahidrato es del 12,9 % y el 15,1 %, respectivamente. La forma 6 de sal de HCl se obtuvo a partir de una suspensión en HCl 0,1 N en agua. La forma 6 de sal de HCl es un pentahidrato o un hexahidrato de la sal de HCl del compuesto 1.
En la tabla 39 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 6 de sal de HCl.
Tabla 39: Picos ^

(continuación)

FORMA 7 DE SAL DE HCL
La forma 7 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 80. En la figura 81 se muestran los termogramas de TGA y DSC de la forma 7 de sal de HCl. El termograma de DSC mostró una endotermia con deshidratación con inicio a 80 °C y una máxima a 110 °C y otra endotermia con inicio a 215 °C y una máxima a 233 °C. La pérdida de peso del TGA fue del 3,8 % antes de 100 °C. Un termograma del segundo procesamiento de DSC mostró endotermia con inicio a 71 °C y una máxima a 98 °C y otra endotermia con inicio a 209 °C y una máxima a 230 °C (figura 83). La pérdida de peso del TGA corresponde al 3,4 % de pérdida de peso antes de aproximadamente 90 °C (figura 82). El contenido de agua teórico para un monohidrato es del 2,9 %. Se obtuvo la forma 7 de sal de HCl a partir de una suspensión en agua a RT y se secó con aire durante 2 semanas. La forma 7 de sal de HCl es un monohidrato de la sal de HCl del compuesto 1.
En la tabla 40 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 7 de sal de HCl.
Tabla 40: Pico

(continuación)

FORMA 8 DE SAL DE HCL
La forma 8 de sal de HCl tiene un patrón de XRPD cristalina según se ilustra en la figura 85. En la figura 86 se muestran los termogramas de TGA y DSC de la forma 8 de sal de HCl. El termograma de DSC mostró una endotermia con deshidratación con inicio a 117 °C y una máxima a 146 °C y otra endotermia con inicio a 208 °C y una máxima a 221 °C. La pérdida de peso del TGA fue del 3,2 % antes de 100 °C. Un termograma del segundo procesamiento de DSC mostró endotermia a 148 °C (máxima) y otra endotermia con inicio a 204 °C y una máxima a 224 °C (figura 88). La pérdida de peso del TGA corresponde al 3,0% de pérdida de peso antes de 120 °C (figura 87). El contenido de agua teórico para un monohidrato es del 2,9 %. Se obtuvo la forma 8 de sal de HCl de una suspensión en agua a 50 °C y se secó con aire durante 2 semanas. La forma 8 de sal de HCl es un monohidrato de la sal de HCl del compuesto 1.
En la tabla 41 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma 8 de sal de HCl.
Tabla 41: Picos ^

(continuación)

FORMA DE SAL DE HCL COMO MATERIAL DE PARTIDA
La forma de sal de HCl como material de partida tiene un patrón de XRPD cristalina según se ilustra en la figura 63. En la figura 64 se muestran los termogramas de TGA y DSC de la forma de sal de HCl como material de partida. El termograma de DSC mostró una endotermia con inicio a aproximadamente 238 °C y una máxima a aproximadamente 248 °C. La pérdida de peso del TGA se correspondió con aproximadamente el 1,0 % de pérdida de peso antes de 100 °C. Se observó un cambio de masa del 3,6 % en peso para el material de partida del 0 % de RH al 95 % de RH a 25 °C. El contenido de Cl fue del 5,70 % en peso, lo cual se condice con el contenido teórico de Cl para una sal de HCl 1:1 del 5,84 % en peso. La muestra es moderadamente higroscópica. La forma de sal de HCl como material de partida puede ser una forma de anhidrato de la sal de HCl del compuesto 1.
En la tabla 42 a continuación se proporciona una lista de los picos de difracción de rayos X para la forma de sal de HCl como material de partida.
Tabla 42: Pico

(continuación)

ENSAYOS CELULARES
Ensayo de citotoxicidad en formato múltiple. Las células se cultivan en RPMI1640, FBS al 10 %, L-alanina-L-glutamina 2 mM, piruvato de Na 1 mM o un medio especial en una atmósfera humidificada de CO2 al 5 % a 37 °C. Se colocan las células en placas de 384 pocillos y se incuban en una atmósfera humidificada de CO2 al 5 % a 37 °C. Los compuestos se agregan 24 h tras el cultivo celular. Al mismo tiempo, se genera una placa con células sin tratamiento para el momento cero. Tras un período de incubación de 72 horas, las células se fijan y tiñen con anticuerpos con etiqueta fluorescente y tinte nuclear, para permitir la visualización de los núcleos, las células apoptóticas y las células mitóticas. Las células apoptóticas se detectan utilizando un anticuerpo anti-caspasa-3 activa. Las células mitóticas se detectan utilizando un anticuerpo anti-fosfo-histona-3. Los compuestos se diluyen en serie 3,16 veces y se analizan en 10 concentraciones con una concentración de análisis final de DMSO al 0,1 %, a partir de la concentración de análisis más elevada de 10 pM. Se realizó microscopía de fluorescencia automática utilizando un captador de imágenes de gran contenido ImageXpress Micro XL de Molecular Devices y las imágenes se registran con un objetivo de 4X.
Análisis de datos. Se obtienen imágenes TIFF de dieciséis bits y se analizan con software MetaXpress 5.1.0.41. La proliferación celular se mide según la intensidad de señal del tinte nuclear incorporado. El producto del ensayo de proliferación celular se indica como conteo celular relativo. Para determinar el criterio de valoración de la proliferación celular, se transforma el producto de datos de proliferación celular en el porcentaje de control (POC, por sus siglas en inglés) utilizando la siguiente fórmula:
POC = conteo celular relativo (pocillos con compuesto) / conteo celular relativo (pocillos con vehículo) x 100 La CI50 del conteo celular relativo es la concentración de compuesto de prueba a un 50 % de respuesta máxima posible con respecto al control con DMSO. La GI50 es la concentración necesaria para reducir a la mitad el crecimiento observado. Esta es la concentración que inhibe el crecimiento hasta un nivel intermedio entre el crecimiento en células sin tratamiento y la cantidad de células cultivadas en el pocillo (valor en el momento cero). Los valores de CI50 se calculan utilizando regresión no lineal para ajustar los datos a un modelo sigmoideo de respuesta a la dosis en un solo lugar, con 4 parámetros y 4 puntos, en donde:
y (ajuste) = A [(B - A) / (1 ((C/x) A D))].
El marcador de caspasa-3 activada etiqueta las células desde las primeras etapas hasta las etapas avanzadas de apoptosis. Las concentraciones de compuesto de prueba que provocan una inducción quíntuple en la señal de caspasa-3 (Cal_X5) indican una inducción significativa de apoptosis. La inducción máxima de caspasa 3 por compuesto en comparación con el control con DMSO se indica como cambio_múltiplo_máx.
Tabla 43: Líneas celulares utilizadas en ensa os de citotoxicidad en formato múlti le

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Las formas sólidas del compuesto 1 que se describen en el presente documento muestran o se demostrará que presentan actividad antiproliferativa en diversas líneas celulares cancerosas. La actividad antiproliferativa en estas líneas celulares cancerosas indica que los compuestos de aminopurina pueden ser útiles en el tratamiento de cánceres, incluidos tumores sólidos, por ejemplo, melanoma, cáncer colorrectal, cáncer de estómago, cáncer de cabeza y cuello, cáncer de tiroides, cáncer de vejiga, cáncer del SNC, cáncer de pulmón, cáncer de páncreas y cáncer de tejido blando.
En otra realización, las formas sólidas del compuesto 1 descrito en el presente documento muestran o se demostrará que inducen apoptosis en diversas líneas celulares cancerosas. La inducción de la apoptosis indica que las formas sólidas del compuesto 1 descrito en el presente documento pueden ser útiles en el tratamiento de cánceres, incluidos tumores sólidos, por ejemplo, cáncer de vejiga, cáncer de mama, cáncer del SNC (incluido neuroblastoma y glioma), cáncer de colon, cáncer gastrointestinal (por ejemplo, cáncer de estómago o cáncer de colon), cáncer endocrino (por ejemplo, cáncer de tiroides o cáncer de la glándula adrenal), cáncer genitourinario femenino (por ejemplo, cáncer de cuello del útero o cáncer de ovario de células claras, cáncer de vulva, cáncer de útero o cáncer de ovario), cáncer de cabeza y cuello, cáncer hematopoyético (por ejemplo, leucemia o mieloma), cáncer de riñón, cáncer de hígado, de pulmón (por ejemplo, NSCLC o SCLC), melanoma, cáncer de páncreas, cáncer de próstata o cáncer de tejido blando (por ejemplo, sarcoma u osteosarcoma).
En otra realización, las formas sólidas del compuesto 1 descrito en el presente documento muestran o se demostrará que producen detención de G1/S en diversas líneas celulares cancerosas. La provocación de una detención de G1/S en estas líneas celulares cancerosas indica que los compuestos pueden ser útiles en el tratamiento de cánceres, incluidos tumores sólidos, por ejemplo, cáncer de vejiga, cáncer de mama, cáncer del SNC (por ejemplo, glioma o neuroblastoma), cáncer de colon, cáncer gastrointestinal (por ejemplo, cáncer de estómago), cáncer endocrino (por ejemplo, cáncer de tiroides o cáncer de glándula adrenal), cáncer genitourinario femenino (por ejemplo, cáncer de útero, cáncer de cuello del útero, cáncer de ovario de células claras o cáncer de vulva), cáncer de cabeza y cuello, cáncer hematopoyético (por ejemplo, leucemia o mieloma), cáncer de riñón, cáncer de hígado, cáncer de pulmón (por ejemplo, NSCLC o SCLC), melanoma, cáncer de páncreas, cáncer de próstata o cáncer de tejido blando (sarcoma u osteosarcoma).
Ensayo de citotoxicidad en formato múltiple. En otro experimento, las células se cultivaron en RPMI1640, FBS al 10 %, L-alanina-L-glutamina 2 mM, piruvato de Na 1 mM o un medio especial en una atmósfera humidificada de CO2 al 5 % a 37 °C. Se colocaron las células en placas de 384 pocillos y se incubaron en una atmósfera humidificada de CO2 al 5 % a 37 °C. Los compuestos se agregaron 24 h tras el cultivo celular. Al mismo tiempo, se generó una placa con células sin tratamiento para el momento cero. Tras un período de incubación de 72 horas, las células se fijaron y tiñeron con anticuerpos con etiqueta fluorescente y tinte nuclear, para permitir la visualización de los núcleos, las células apoptóticas y las células mitóticas. Las células apoptóticas se detectaron utilizando un anticuerpo anticaspasa-3 activa. Las células mitóticas se detectaron utilizando un anticuerpo anti-fosfo-histona-3. Los compuestos se diluyeron en serie 3,16 veces y se analizaron en 10 concentraciones con una concentración de análisis final de DMSO al 0,1 %, a partir de la concentración de análisis más elevada de 10 pM. Se realizó microscopía de fluorescencia automática utilizando un captador de imágenes de gran contenido ImageXpress Micro XL de Molecular Devices y las imágenes se registraron con un objetivo de 4X.
Análisis de datos. Se obtuvieron imágenes TIFF de dieciséis bits y se analizaron con software MetaXpress 5.1.0.41. La proliferación celular se midió según la intensidad de señal del tinte nuclear incorporado. El producto del ensayo de proliferación celular se indicó como conteo celular relativo. Para determinar el criterio de valoración de la
proliferación celular, se transformó el producto de datos de proliferación celular en el porcentaje de control (POC) utilizando la siguiente fórmula:
POC = conteo celular relativo (pocillos con compuesto) / conteo celular relativo (pocillos con vehículo) x 100
La CI50 del conteo celular relativo fue la concentración de compuesto de prueba a un 50 % de respuesta máxima posible con respecto al control con DMSO. La GI50 se refiere a la concentración necesaria para reducir a la mitad el crecimiento observado. Esta corresponde a la concentración que inhibe el crecimiento hasta un nivel intermedio entre el crecimiento en células sin tratamiento y la cantidad de células cultivadas en el pocillo (valor en el momento cero). Los valores de CI50 se calcularon utilizando regresión no lineal para ajustar los datos a un modelo sigmoideo de respuesta a la dosis en un solo lugar, con 4 parámetros y 4 puntos, en donde:
y (ajuste) = A [(B - A) / (1 ((C/x) A D))].
El marcador de caspasa-3 activada etiqueta las células desde las primeras etapas hasta las etapas avanzadas de apoptosis. Las concentraciones de compuesto de prueba que provocan una inducción doble (Cal-X2) o inducción quíntuple en la señal de caspasa-3 (Cal_X5) indicaron una inducción significativa de apoptosis. La inducción máxima de caspasa 3 por compuesto en comparación con el control con DMSO se indicó como cambio_múltiplo_máx.














Efecto sobre la proliferación de HCC. Las líneas celulares de HCC se trataron con DMSO o concentraciones en aumento del compuesto 1 durante 72 h. De forma específica, se colocó el compuesto 1 en diversas concentraciones en sulfóxido de dimetilo (DMSO) mediante un dispensador acústico (EDC ATS-100) en una placa vacía de 384 pocillos. La colocación del compuesto 1 se realizó en forma de dilución en serie de 10 puntos (dilución triple) en duplicado en la placa. Se realizaron réplicas de las placas que contenían compuesto 1 para uso con diferentes líneas celulares. Tras la replicación de las placas con compuesto, se sellaron todas las placas (Agilent ThermoLoc) y se almacenaron a -20 °C durante hasta 1 mes. Una vez que estaban listas para el análisis, estas se retiraron del congelador, se descongelaron y se abrieron inmediatamente antes de la adición de las células de prueba.
Antes del análisis, las células se cultivaron y expandieron en matraces de cultivo, para proporcionar cantidades suficientes de material de partida. Las células luego se diluyeron hasta las densidades adecuadas y se agregaron directamente a las placas de 384 pocillos que contenían compuesto. Luego se dejaron crecer durante 72 h a 37 °C/CO2 al 5 %. Al momento de agregar el compuesto (tü), se evaluó la cantidad inicial de células mediante un ensayo de viabilidad (Cell Titer-Glo) cuantificando el nivel de luminiscencia generado por la ATP presente en las células viables. Tras 72 h, se evaluó la viabilidad celular de las células tratadas con compuesto mediante Cell Titer-Glo y medición de luminiscencia. Se evaluó la respuesta apoptótica del compuesto 1 cuantificando las actividades de caspasa 3 y caspasa 7 (caspasa 3/7-Glo) en células tratadas y células de control con DMSO.
Determinación de valores de GI50 y CI50. Se utilizó un modelo logístico de cuatro parámetros (modelo sigmoideo de respuesta a la dosis) para determinar el valor de GI50 del compuesto.
y = (A+ ((B-A)/ (1+ ((C/x)AD))))
A = Y Mín
B = Y Máx
C = CE50
D = pendiente de Hill
GI50 es la concentración del compuesto cuando Y = (YMáx+Yt0)/2
CI50 es la concentración del compuesto cuando Y = 50 % del control con DMSO
Y = viabilidad celular medida como unidad de luminiscencia
t0 = momento de adición del compuesto
La proliferación y la apoptosis se midieron utilizando CellTiter-Glo y Caspase 3/7-Glo. Los valores de CalX2 son la menor concentración con la cual el compuesto 1 induce un aumento doble de caspasa 3/7 escindida, en comparación con el control con DMSO. Los datos de proliferación y apoptosis son el promedio de 3 experimentos. Tabla 45: Efecto de

(continuación)

Conclusión: El compuesto 1 inhibe la proliferación e induce la apoptosis en múltiples líneas de HCC.
Actividad antiproliferativa en un panel de 64 líneas celulares cancerosas. Las células se trataron con DMSO o concentraciones en aumento del compuesto 1 durante 72 h. La proliferación se midió utilizando CellTiter-Glo según lo descrito. Los resultados se muestran en la tabla 46.
Tabla 46: Actividad anti roliferativa del com uesto 1 en un anel de 64 líneas celulares cancerosas

(continuación

El compuesto 1 demostró inhibición de la proliferación de múltiples líneas celulares cancerosas derivadas de CRC, melanoma, cáncer gástrico, HCC, cáncer de pulmón, cáncer de páncreas, leucemia y mieloma múltiple.
Actividad antiproliferativa y apoptótica en líneas celulares cancerosas con BRAF y beta-catenina mutantes o activas. Se evaluó el estado de mutación de BRAF, CTNNB1, KRAS y EGFR en cinco líneas celulares en función de datos públicos (COSMIC y CCLE) y confirmados a nivel interno. Se evaluó el estado de p-catenina utilizando el sistema informante TOP Flash con transfección transitoria. Se definió que una línea celular era activa en p-catenina si la relación del informante Top Flash con respecto al informante Fop Flash es superior a 2. N/D: No disponible. La eficacia de transfección en Colo 205 (BRAF V600E) fue demasiado baja para acceder a su actividad de p-catenina utilizando este enfoque. La actividad antiproliferativa y apoptótica del compuesto 1 en las cinco líneas celulares se midió como se describió anteriormente.
Tabla 47: Actividad antiproliferativa y apoptótica del compuesto 1 en líneas celulares con BRAF y beta-catenina mutantes o activas
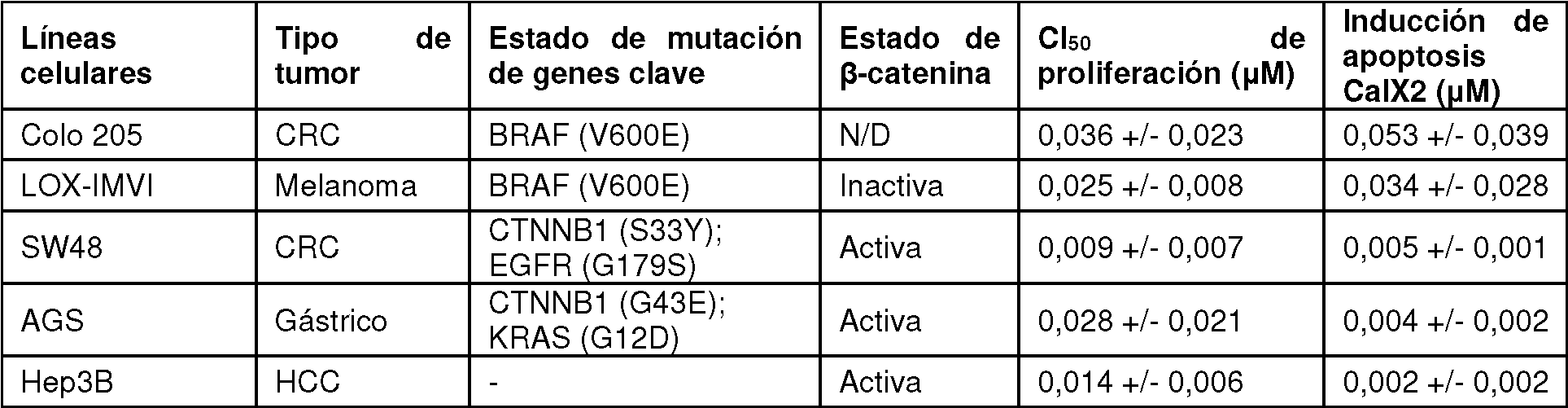
El compuesto 1 inhibe de forma potente la proliferación e induce la apoptosis en líneas celulares cancerosas con BRAF y beta-catenina mutantes o activas, incluidas CRC con mutación de BRAF, melanoma con mutación de BRAF, CRC con mutación de EGFR/con mutación de beta-catenina (es decir, CRC con mutación de EGFR/activo para beta-catenina), cáncer gástrico con mutación de KRAS/con mutación de beta-catenina (es decir, cáncer gástrico con mutación de KRAS/activo para beta-catenina) y HCC.
Inhibición de la ruta oncogénica. Efecto sobre la señalización de MAPK. Las células cancerosas se cultivaron a una densidad de 25000 células por pocillo en una placa de cultivo de 96 pocillos y se incubaron a 37 °C en una incubadora de CO2 durante la noche. Tras el tratamiento con el compuesto 1 a 37 °C durante 2 h, las células se lisaron con amortiguador de lisis de Mesoscale y se midieron los niveles de pRSK S380 en cada lisado mediante tecnología de ELISA de Mesoscale.
Conclusión. El compuesto 1 inhibió pRSK1 de forma potente en múltiples líneas celulares cancerosas (tabla 48).
Tabla 48: Valores de CI50 de pRSK1 S380 para el compuesto 1 en líneas celulares cancerosas Colo 205 y LOX-IMVI con mutación de BRAF

En un experimento temporal, las células cancerosas Colo-205 se trataron con el compuesto 1 0,5 |uM durante diversos períodos de tiempo. Se midió el efecto del compuesto 1 sobre pRSK S380 según lo descrito. Se midió el efecto del compuesto 1 sobre otros marcadores de la ruta de MAPK (DUSP4 y DUSP6) mediante transferencia Western con anticuerpos específicos. Los datos temporales en la figura 188 indican que el compuesto 1 provoca una inhibición sostenida (durante hasta 72 h) de los siguientes objetivos de ERK: pRSK1, DUSP4 y DUSP6. Los inhibidores de BRAF (BRAFi) no provocan una inhibición sostenida de ERK en las líneas de CRC con mutación de BRAF (Corcoran et al., Cáncer Discov. 2012, 2:227-35). Una inhibición suficiente y sostenida de ERK parece ser fundamental para la eficacia clínica de los inhibidores de BRAFi y MEK (MEKi) en pacientes con melanoma (Bollag et al., Nat Rev Drug Disc. 2012; 11, 873-886) y CRC con mutaciones de b Ra F (Corcoran et al., Cáncer Discov.
2012, 2:227-35). La falta de inhibición sostenida de ERK por parte de BRAFi puede contribuir a la carencia de actividad clínica de BRAFi en pacientes con CRC con mutación de BRAF. La inhibición sostenida de ERK por parte del compuesto 1 puede brindar una ventaja con respecto a BRAFi en pacientes con CRC con mutación de BRAF.
Se evaluó la capacidad del compuesto 1 de inhibir la señalización de MAPK determinando la expresión de proteína DUSP4 y DUSP6. Se trataron cultivos de línea de células de cáncer de colon 205 (BRAF V600E) tratadas con DMSO o concentraciones en aumento del compuesto 1 durante 2, 8 o 24 h. Las proteínas se extrajeron de células tratadas y se analizaron mediante transferencia Western utilizando anticuerpos contra DUSP4, DUSP6, ciclina D1, cMyc, YAP o p-actina. Se extrajeron ARN utilizando el kit de célula a CT y se realizó PCR cuantitativa con sondas específicas para DUSP4, DUSP6, SPRY2, c-Myc y ciclina D1. Para la normalización, se utilizaron sondas específicas para p-actina.
En Colo 205 (BRAF V600E), el compuesto 1 redujo considerablemente DUSP4 y DUSP6 ya a las 2 h y la reducción se sostuvo hasta las 24 h (figura 189A). El tratamiento con el compuesto 1 condujo a reducción de la transcripción de SPRY2 de forma dependiente de la concentración en Colo 205 (figura 189B), lo cual se condice con una potente inhibición de ERK. Se evaluaron los niveles de ciclina D1 y c-Myc, que se encuentran corriente abajo de la señalización canónica de MAPK y Wnt. El compuesto 1 redujo considerablemente los niveles de proteína y ARN de c-Myc y ciclina D1 en células Colo 205 (figuras 189A-C). El tratamiento con el compuesto 1 produjo menos proteína YAP a las 24 h en Colo 205 (figura 189A). En su conjunto, estos datos celulares son coherentes con una inhibición fuerte y sostenida de la ruta de MAPK.
Para evaluar adicionalmente la capacidad del compuesto 1 de inhibir la señalización de MAPK, se evaluó la expresión de ARN de otros objetivos de MAPK (BMF, DUSP5, DUSP6, EFNA1, EGR1, ETV5, FOS, FOSL1, GJA1, IL-8, SPRY2 y SPRY4). Se trataron cultivos de las líneas celulares de cáncer de colon Colo 205 (caracterizadas por una mutación de BRAF V600E) y HT-29 (caracterizadas por una mutación de BRAF V600E) con DMSO o compuesto 1 a 0,3 o 1 pM durante 6 h. Se extrajeron ARN utilizando el kit de aislamiento de ARN MagMAX Total y se realizó PCR cuantitativa con sondas específicas de BMF, DUSP5, DUSP6, EFNA1, EGR1, ETV5, FOS, FOSL1, GJA1, IL-8, SPRY2, SPRY4. Para la normalización, se utilizaron sondas específicas de ARNr 18S.
En ambas líneas celulares, el compuesto 1 redujo los niveles de ARNm de DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, SPRY2, SPRY4 (figuras 189D-I), lo cual es coherente con inhibición de ERK. El hallazgo de reducción de los niveles de ARNm de GJA1 en células Colo205 y aumento en HT29 puede estar vinculado con el hallazgo de que el compuesto 1 es citotóxico en Colo205 y citostático en HT29. El tratamiento con el compuesto 1 produjo mayores niveles de ARNm de BMF y EFNA1 a las 6 h en Colo 205 y HT-29. En su conjunto, estos datos celulares son coherentes con una inhibición de la ruta de MAPK.
Efecto sobre la señalización de YAP y beta-catenina. Se evaluó la actividad celular del compuesto 1 contra genes objetivo de YAP y betacatenina. Se trataron cultivos de la línea celular de cáncer de colon Colo 205 (BRAF V600E) con DMSO o concentraciones en aumento de compuesto 1 durante 2, 8 o 24 h. Se extrajeron ARN utilizando el kit de célula a CT y se realizó PCR cuantitativa con sondas específicas para Axin2, CTGF y AREG. Para la normalización, se utilizaron sondas específicas para p-actina.
El tratamiento con el compuesto 1 condujo a mayor ARN de Axina2 (figura 190A). El compuesto 1 redujo considerablemente la expresión de genes objetivo de Hippo/YAP (CTGF, a ReG) en Colo 205 (BRAF V600E) a las 2, 8 y 24 h (figura 190A). En su conjunto, estos datos sugieren que el compuesto 1 tiene impacto sobre la señalización de Wnt y bloquea la señalización de Hippo en células cancerosas de Colo 205.
Se evaluó la actividad celular del compuesto 1 contra otros genes objetivo YAP (figuras 190B-E). Se trataron cultivos de las líneas celulares de cáncer de colon Colo 205 y HT-29 con DMSO o compuesto 1 a 0,3 o 1 pM durante 6 h. Se extrajeron ARN utilizando el kit de aislamiento de ARN MagMAX Total y se realizó PCR cuantitativa con sondas específicas de CYR61, CITED2, CXCL1, ELF3, HAS2, HES1 y MAFF. Para la normalización, se utilizaron sondas específicas de ARNr 18S.
En ambas líneas celulares, el compuesto 1 redujo los niveles de ARNm de CYR61, CXCL1, HAS2, HES1 y MAFF. El hallazgo de reducción de los niveles de ARNm de CYR61 en células Colo205, pero no en HT29, así como aumento de los niveles de ARNm de CITED2 en HT29, pero no en Colo 205, puede estar vinculados con el hallazgo de que el compuesto 1 es citotóxico en Colo205 y citostático en HT29. El tratamiento con el compuesto 1 produjo mayores niveles de ARNm de CITED2 y ARNm de ELF3 a las 6 h en Colo 205 y HT-29. (figura 190B). En su conjunto, estos datos celulares son coherentes con una inhibición de la ruta de YAP.
Evaluación de la sensibilidad en líneas celulares con mutaciones de beta-catenina. Se evaluó el efecto del compuesto 1 sobre líneas celulares con mutaciones de p-catenina. (figura 205 y figura 206A-B). El compuesto 1 mostró eficacia contra líneas celulares con mutación de p-catenina. Estas líneas celulares demuestran que los cánceres caracterizados por mutación de p-catenina tienen mayor sensibilidad al tratamiento con el compuesto 1. El compuesto 1 demostró adicionalmente modulación de p-catenina, así como YAP en las líneas celulares con mutación de BRAF y CTNNB1, como se ilustra en la figura 207. El compuesto 1 también modula la expresión de genes objetivo controlada por MAPK, p-catenina y YAP en líneas celulares con mutación de BRAF y CTNNB1, como se ilustra en las figuras 208A-B.
Transferencia Western. Se evaluó la modulación por parte del compuesto 1 de los marcadores de las rutas de MAPK, WNT/p-catenina e Hippo/YAP, mediante transferencia Western estándar. Se colocaron células LOX-IMVI, SW48 y Colo-205 en placas de 6 pocillos a una densidad de 250 000 células por pocillo y se permitió la unión durante la noche. El compuesto 1 se agregó a las células a concentraciones de 0,03, 0,1,0,3, 1 y 3 pM durante 2, 8
y 24 horas. Las células se extrajeron y lisaron en amortiguador RIPA (Tris-HCl 50 mM, pH 7,4, cloruro de sodio [NaCl] 150 mM, ácido desoxicólico 0,25 %, Nonidet P-40 al 1 %, ácido etilendiaminatetraacético [EDTA] 1 mM, inhibidores de proteasa y fosfatasa). Los lisados celulares se calentaron en amortiguador de muestra- dodecil sulfato sódico (SDS), se cargaron 40 gg de lisado celular por condición en geles y se separaron utilizando electroforesis en gel de poliacrilamida SDS (PAGE). Se transfirió proteína a una membrana de nitrocelulosa y se realizó inmunotransferencia con anticuerpos anti-DUSP4, DUSP6, cMyc, Ciclina D1, YAP, AXIN2, HDAC5 (fosfo S498) y pactina. Las membranas se escanearon en el sistema Licor Odyssey.
Reacción en cadena de la polimerasa cuantitativa. Se evaluó la modulación por parte del compuesto 1 de los genes de las rutas de MAPK, WNT/p-catenina e Hippo/YAP, mediante qPCR en tiempo real (RT). Se colocaron células de lisil oxidasa IMVI, SW48 y Colo-205 en placas de 96 pocillos a una densidad de 20000 células por pocillo y se permitió la unión durante la noche. El compuesto 1 se agregó a las células a concentraciones semilogarítimas de 1 nM a 10 gM durante 2, 8 y 24 horas. Las células se extrajeron utilizando el kit de células a CT para expresión génica TaqMan de acuerdo con el manual del producto. Luego, se realizó RT-PCR y se utilizó el ADNc resultante en reacciones de qPCR en el sistema de PCR en tiempo real ViiA7 (Thermo Fisher Scientific). Se utilizaron sondas TaqMan para supervisar los cambios en genes DUSP4, DUSP6, SPRY2, MYC, CCND1, AXINA2, CTGF, Cyr61, AREG y ACTB. Todos los genes se normalizaron en función de la expresión de ACTB y se indicó como porcentaje de control solamente con DMSO.
Análisis de expresión génica: Se cultivaron células epiteliales bronquiales humanas en matraces T-150 en medio de cultivo BEpiCM y se permitió que alcanzaran un 80 % de confluencia. Las células se colocaron en placas de cultivo plásticas de 12 pocillos a 150000 células por pocillo en medio BEpiCM durante 24 horas. Tras 24 h de incubación, las células se trataron con sulfóxido de dimetilo (DMSO) como control, compuesto 1 a 0,1, 1, 10 gM durante 30 minutos. Las células luego se estimularon con 100 ng/ml de Wnt3a recombinante (formulado en solución salina amortiguada con fosfato [PBS, por sus siglas en inglés]), RSPO3 350 pM (formulado en PBS) o una combinación de Wnt3 y RSPO3 durante 24 horas. Se aisló ácido ribonucleico (ARN) utilizando el kit Rneasy Mini de Qiagen, de acuerdo con las instrucciones del fabricante. Se determinó la expresión génica y de axina2 utilizando ensayos Taq-Man de reacción en la cadena de polimerasa con transcripción inversa (RT-PCR, por sus siglas en inglés). Se realizó PCR cuantitativa (qPCR, por sus siglas en inglés) utilizando el sistema de RT-PCR SuperScript® III One-Step y se ejecutó en un sistema de PCR en tiempo real Viia 7. Los datos se normalizaron en función de la gliceraldehído 3-fosfato deshidrogenasa. El compuesto 1 inhibe la expresión de axina2 en células epiteliales bronquiales humanas. La expresión génica se midió a las 24 horas. En función de estos resultados, se demostró que el compuesto 1 inhibe la expresión de axina2 en células epiteliales bronquiales humanas. (Figura 209).
Ensayo de colonias a largo plazo. Se evaluó el compuesto 1 para determinar su capacidad de inhibir la formación de colonias de células cancerosas mediante un ensayo de formación de colonias a largo plazo. Se agregaron células y compuestos a placas de 96 pocillos y se supervisaron durante hasta 8 semanas para determinar la formación de colonias. Se volvió a rellenar el compuesto y el medio cada 1 semana durante todo el transcurso del ensayo. La formación de colonias se detectó mediante imagenología con 4x en el sistema IncuCyte ZOOM. El compuesto 1 demostró inhibición de la formación de colonias de células con mutación de p-catenina a un mayor nivel que los inhibidores de MEK (trametinib) y los inhibidores de ERK (GDC0994). Se trataron células SW48 (colo), células HCT-116 (colo), células AGS (gástricas) y Hep3B (HCC) con el compuesto 1 y estas demostraron mayores niveles de inhibición que los observados con el tratamiento con inhibidores de MEK o inhibidores de ERK. (Figuras 210A a 210D). El compuesto 1 demostró adicionalmente inhibición sorpresiva de la formación de colonias de células de AGS resistentes al tratamiento de inhibición de MEK con trametinib. Estos resultados sugieren que el compuesto 1 puede ser útil para el tratamiento de cánceres resistentes a otros tratamientos.
Evaluación de efectos inmunomoduladores. Se evaluó el efecto del compuesto 1 sobre los niveles de expresión de PD-L1. Las células se cultivaron con o sin el compuesto 1 durante el tiempo indicado antes de medir los niveles de expresión de PD-L1, DUSP4 y a-tubulina o a-actina mediante transferencia Western. Para detectar los niveles superficiales de PD-L1, las células se trataron con DMSO o compuesto 1 en las concentraciones indicadas durante 48 h y se detectó la expresión de PD-L1 en la superficie celular utilizando el análisis de citometría de flujo (FACS) con un anticuerpo para PD-L1 marcado con APC (clon 29E.1A3.; BioLegend, San Diego, CA). Se determinó la media geométrica de células positivas para PD-L1 mediante FlowJo 10 (Treestar, Ashland, OR).
Conclusión. El compuesto 1 inhibe directamente la expresión de PD-L1 en múltiples células cancerosas, incluidas HOP62, KARPAS-299, y LOX-IMVI (BRAF V600E) (figura 191A). El análisis FACS indica también inhibición de los niveles de PD-L1 en superficie por parte del compuesto 1 en múltiples líneas celulares cancerosas (figura 191B).
Para determinar si la regulación en disminución de PD-L1 por parte del compuesto 1 potencia la activación de linfocitos T, se cultivaron células cancerosas KARPAS-299 tratadas con compuesto en conjunto con linfocitos T derivados de PBMC estimulados con concentraciones bajas de superantígeno (SEB). Las células KARPAS-299 se trataron con DMSO (D) o compuesto 1 en las concentraciones indicadas durante 48 h. Se trataron PBMC de donantes saludables con o sin 20 ng/ml de SEB durante 48 h. Tras el lavado con PBS, las PBMC se incubaron con las células cancerosas durante 24 h y se recogieron los sobrenadantes para medir la IL-2 y el IFNy utilizando ensayos de Mesoscale.
Los niveles de sobrenadante de IL-2 e IFNy se utilizaron como marcadores funcionales de activación de linfocitos T. En ausencia de SEB, las PBMC cultivadas en conjunto con células KARPAS-299 tratadas con el compuesto 1 produjeron poca cantidad de IL-2 o IFNy. En presencia de concentraciones bajas de SEB (20 ng/ml), las células cancerosas tratadas con el compuesto 1 cultivadas en conjunto con PBMC demostraron mayores niveles de producción de IL-2 e IFNy (figuras 192A-B). Los mayores niveles de IL-2 e IFNy en las células cancerosas tratadas con el compuesto 1 fueron similares a los niveles observados con el tratamiento con anti-PD-L1 (Ultra-LEAF™ de Biolegend).
Se determinó el efecto del tratamiento con el compuesto 1 sobre los niveles de IL-8 en medios de cultivo de PBMC. Se aislaron PBMC de sangre entera y se cultivaron en medio RPMI más FBS al 10 %. Las PBMC se colocaron en placas a 1x106 por mililitro en platillos de 10 cm2 Las PBMC se trataron con DMSO al 0,1 % o compuesto 10,5 qM. Los tratamientos se retiraron en los puntos temporales indicados. Se utilizó el medio de cultivo (1 ml) para el análisis de IL-8. El análisis de IL-8 se realizó con un kit de IL-8 humana Mesoscale V-Plex, de acuerdo con las instrucciones del fabricante. Se mostró que el compuesto 1 inhibe los niveles de IL-8 en diferentes momentos (figura 192C).
Ensayo con informante TEAD. Se analizó la actividad de informante TEAD utilizando estabilidad de células WI38 VA13 con expresión de un promotor sintético con respuesta a YAP/TAZ que impulsa la expresión de luciferasa (8xGTIIC-luciferasa). Se cultivaron 10000 células por pocillo en placas de paredes blancas de 96 pocillos y se dejaron durante toda la noche. Tras 16 a 20 horas, las células se trataron con compuesto y se midió la actividad de informante TEAD 24 o 72 horas después utilizando el ensayo de luciferasa Bright Glo (Promega), de acuerdo con las instrucciones del fabricante. Este ensayo se realizó 3 veces para el compuesto 1 y dos veces para Trametinib. Véase la figura 212.
Ensayo de viabilidad. Se cultivaron en paralelo 10000 células WI38 VA13 con expresión de 8xGTIIC-luciferasa en cada pocillo de una placa con paredes negras de 96 pocillos. Tras 16 a 20 horas, las células se trataron con compuesto durante 24 o 72 horas. En ese momento, se extrajo el medio con suero y compuesto y se sustituyó con 100 |ul de medio libre de suero y 100 |ul de flúor de titulación celular (Promega). La placa se incubó durante 2 horas a 37 °C antes de realizar la lectura del producto de fluorescencia. Este ensayo se basa en la medición de actividad de proteasa en células vivas. Se realizó el ensayo de viabilidad para confirmar que los efectos de los compuestos sobre el informante TEAD no se debían a los efectos del compuesto sobre la viabilidad. Este ensayo se realizó 3 veces para el compuesto 1 y dos veces para Trametinib.
Conclusión. Estos datos brindan una hipótesis terapéutica adicional que sugiere que el tratamiento con el compuesto 1 potencia la activación de linfocitos T. Los datos in vitro sugieren que el compuesto 1 puede potenciar la inmunidad de los linfocitos T contra células cancerosas inhibiendo rutas oncogénicas clave como la ruta de MAPK y regulando en disminución la expresión de la molécula PD-L1 del punto de control inmunológico en el microentorno tumoral. Por lo tanto, los tipos de cánceres con expresión de niveles elevados de PD-L1 (por ejemplo, melanoma, de pulmón, RCC o HCC) pueden presentar sensibilidad con respecto al compuesto 1.
MODELOS ANIMALES
Modelos de xenoinjerto. Para los estudios de modelos de xenoinjerto, se inyectaron líneas celulares cancerosas humanas en ratones SCID (inmunodeficiencia combinada grave, por sus siglas en inglés). Las líneas celulares cancerosas se propagaron en cultivo in vitro. Se generaron animales con tumor mediante inyección de cantidades determinadas con exactitud de células en ratones. Tras la inoculación de los animales, se permitió el crecimiento de los tumores hasta un tamaño determinado antes de la separación aleatoria. Los ratones con tumores de xenoinjerto entre tamaños predeterminados se agruparon y asignaron de forma aleatoria a diversos grupos de tratamiento. Un diseño de estudio de eficacia típico implicó administración de uno o más compuestos en diversos niveles de dosis a ratones con tumor. Asimismo, también se administraron y conservaron de forma similar agentes quimioterapéuticos de referencia (control positivo) y controles negativos. Durante el estudio se tomaron mediciones tumorales y de peso corporal.
Los ratones recibieron anestesia con isoflurano inhalado y luego se inocularon con células tumorales LOX-IMVI por ruta subcutánea sobre la pata trasera derecha con 0,1 ml de una suspensión unicelular en PBS, utilizando una jeringa de 1 ml estéril con aguja de calibre 26. Tras la inoculación de los animales, se permitió que los tumores crecieran hasta aproximadamente 75 a 125 mm3 o, en algunos casos, 250 a 400 mm3 antes de la separación aleatoria de los ratones. Se midió el tumor de cada animal y los animales con tumores dentro del espectro adecuado se incluyeron en el estudio. Los animales del grupo de estudio luego se distribuyeron de forma aleatoria en diversas jaulas, las cuales se asignaron de forma aleatoria a grupos con vehículo, control positivo o artículo de prueba. Todos los ratones se marcaron con etiquetas metálicas en la oreja derecha. Un grupo típico consiste en 8 a 10 animales. Para un estudio de xenoinjerto típico, los ratones SCID con tumores se separaron de forma aleatoria y recibieron dosis de compuestos, por ejemplo, entre 100 mg/kg y 0,1 mg/kg, con un cronograma de dosis diferente que incluye, pero no se limita a, qd, q2d, q3d, q5d, q7d y bid. Los ratones recibieron la administración durante 1 a 4 semanas. Los tumores se midieron dos veces por semana utilizando pinzas y se calcularon los volúmenes tumorales utilizando la fórmula W2 x L / 2.
El objetivo de estos estudios era analizar la eficacia del compuesto 1 en modelos de xenoinjerto derivado de líneas celulares, LOX-IMVI (melanoma) y Colo205 (colorrectal) y el modelo de xenoinjerto PDX1994060146 (xenoinjerto derivado de paciente [PDX146]) colorrectal. Estos modelos se seleccionaron por contener la mutación de BRAF V600E. Se realizó un análisis de PK/PD adicional para examinar la inhibición mediada por el compuesto 1 de los biomarcadores de rutas en el modelo de xenoinjerto de PDX146.
Modelo de xenoinjerto de melanoma subcutáneo LOX-IMVI. El objetivo de este estudio era confirmar la eficacia del compuesto 1 en el modelo de xenoinjerto de melanoma LOX-IMVI. Un estudio (figura 193) en el modelo de xenoinjerto LOX-IMVI con análisis de dos niveles de dosis del compuesto 1 (15 y 30 mg/kg) demostró una reducción significativa del volumen tumoral, en comparación con el control con vehículo (p<0,001 para ambos niveles de dosis). Se observó regresión tumoral en 9 de 9 animales para ambos niveles de dosis y 1 de 9 animales de cada grupo se encontraba libre de tumor al final del estudio.
En un experimento separado, se administró compuesto 1 por vía oral, QD durante 8 días a 0,2, 1, 5, 10 y 15 mg/kg. Se observó actividad antitumoral dependiente de la dosis con el tratamiento con el compuesto 1 en el modelo de xenoinjerto LOX-IMVI (figura 194). Se observó regresión tumoral en los niveles de dosis de 10 y 15 mg/kg.
Modelo de xenoinjerto colorrectal subcutáneo de Colo 205. Modelo de xenoinjerto colorrectal subcutáneo de Colo 205. El objetivo de estos estudios era analizar la eficacia del compuesto 1 en el modelo de xenoinjerto de cáncer colorrectal Colo 205 y determinar si una dosificación de dos veces al día (BID) tenía impacto en la actividad antitumoral. En el primer experimento, se administró compuesto 1 por vía oral, QD durante 15 días a 0,2, 1, 5, 10 y 15 mg/kg. Se observó actividad antitumoral dependiente de la dosis con el tratamiento con el compuesto 1 en el modelo de xenoinjerto Colo 205 (figura 195). Se realizó un estudio con cronograma para determinar si una dosificación BID aumentaba la actividad antitumoral del compuesto 1. Se observó actividad antitumoral dependiente de la dosis con el tratamiento con el compuesto 1 en el modelo de xenoinjerto Colo 205 (figura 196).
Modelo de xenoinjerto derivado de paciente colorrectal subcutáneo PDX1994060146. El objetivo de estos estudios era analizar la eficacia del compuesto 1 en el modelo de xenoinjerto de cáncer colorrectal PDX1994060146 (PDX146) y determinar si una dosificación BID tenía impacto en la actividad antitumoral. Se realizó un estudio de tiempo hasta el avance (TTP) para determinar el efecto de una duración de tratamiento más prolongada sobre el crecimiento tumoral.
En el primer experimento, se administró el compuesto 1 por vía oral, QD, a 1, 5 y 15 mg/kg o 5 y 15 mg/kg BID durante 22 días. Se observó actividad antitumoral dependiente de la dosis con el tratamiento con el compuesto 1 en el modelo de xenoinjerto PDX146 (figuras 197A-B). La dosis de 15 mg/kg BID parece aumentar la actividad antitumoral del compuesto 1, en comparación con la administración de 15 mg/kg QD.
En el estudio de TTP, el compuesto 1 se administró por vía oral, 1, 5 y 15 mg/kg BID durante 49 a 77 días. Los grupos de tratamiento con el compuesto 1 recibieron la dosificación durante todo el estudio hasta que la media del grupo alcanzó un criterio de valoración predeterminado de aproximadamente 1200 mm3 o hasta la finalización de estudio. Se calculó el retraso en el crecimiento tumoral (TGD, por sus siglas en inglés) como el tiempo entre la finalización del grupo con control con vehículo (día 43) y los grupos de tratamiento con el compuesto 1. El TGD fue de 8, 12 y >37 días para los grupos de tratamiento con 1,5 y 15 mg/kg, respectivamente. (Figura 198).
Hubo inhibición de los biomarcadores que representan la actividad de tres rutas diferentes, MAPK, Wnt e Hippo, en el modelo de xenoinjerto PDX146. Se observó inhibición sostenida de estos biomarcadores de rutas durante 24 h. Actividad antitumoral del compuesto 1 en el modelo de xenoinjerto colorrectal SW48 con mutación de pcatenina. Se inocularon ratones SCID hembra con 2 x 106 células tumorales SW48 en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) inicio el tratamiento. El tratamiento con el artículo de prueba comenzó en el día 10, con tumores con un tamaño aproximado de 110 y 105 mm3. (Véanse las figuras 202A-B). La línea punteada de color negro es el volumen tumoral al inicio de la dosificación. La gráfica a la izquierda es el estudio de respuesta a la dosis. La gráfica a la derecha es el estudio de tiempo hasta la evolución, en donde los animales recibieron el fármaco durante todo el transcurso del estudio. La línea punteada es el volumen tumoral al día 28, cuando finalizó el grupo de control con vehículo.
Actividad antitumoral en el xenoinjerto de carcinoma hepatocelular Hep3B2.1-7 ortotópico. Se inocularon ratones SCID hembra de forma ortotópica con 2 x 106 células tumorales Hep3B2.1--7 por animal. Siete días tras la inoculación, los animales se separaron de forma aleatoria en grupos de tratamiento según el peso corporal y comenzó el tratamiento (día 0 del estudio). La evaluación de la tasa de absorción de un grupo satélite confirmó la presencia de tumor en el hígado en el 100 % de los animales. Se comenzó el tratamiento con el compuesto 1 y dicho compuesto 1 se administró por vía oral, QD, durante 21 días. Se observó una pérdida de peso corporal media significativa prevista para este modelo en el grupo de control con vehículo. Los animales tratados con 15 mg/kg de compuesto 1 mostraron una pérdida de peso corporal mínima y se observó un aumento significativo de peso corporal medio en el grupo con tratamiento con 30 mg/kg de compuesto 1. Los tumores se retiraron y pesaron el
último día del estudio. Se graficaron los pesos tumorales individuales y el peso tumoral medio ± SEM de cada grupo (figura 203). Se calculó el porcentaje de inhibición con respecto al control con vehículo. Los valores P se derivaron de un ANOVA de una sola vía, con análisis post-hoc de Dunnet. *** = p < 0,001.
Actividad antitumoral del compuesto 1 en el modelo de xenoinjerto derivado de paciente con carcinoma hepatocelular amplificado con C-Met, LI0612. Se inocularon ratones SCID hembra con fragmentos de tumor LI0612 del modelo PDX de carcinoma hepatocelular (2 a 4 mm de diámetro) en el costado derecho. Los ratones se separaron de forma aleatoria en grupos de tratamiento (n = 10/grupo) al inicio del tratamiento. El tratamiento con artículo de prueba comenzó el día 18, cuando los tumores eran de un tamaño de aproximadamente 150 mm3. El crecimiento tumoral avanzó en los grupos de tratamiento con el compuesto 1 y control con vehículo durante el período de dosificación. Se observó un cambio en la cinética de crecimiento con la administración del compuesto 1, lo cual produjo una inhibición del crecimiento tumoral (TGI) considerable con 30 mg/kg de tratamiento (p = 0,038, en comparación con el control con vehículo). Véase la figura 204.
Datos farmacocinéticos/farmacodinámicos en un modelo de xenoinjerto derivado de paciente con mutación de BRAF. En función de las cinasas conocidas (ERK 1/2, NLK y SIK) que se ven inhibidas por el compuesto 1, se evaluó el impacto del tratamiento con compuesto sobre biomarcadores de las rutas de MAPK, p-catenina e Hippo en tumores PDX146 de ratones con xenoinjertos. Los ratones con tumor (tumores de ~ 400 mm3) recibieron tratamiento con una única dosis de 1 o 5 mg/kg del compuesto 1. Se recogió tejido tumoral 1,2, 4, 8 y 24 h después de la dosis. Se evaluó la modulación de la ruta de MAPK mediante examen de los niveles de ARNm de DUSP4, DUSP6 y Sprouty (SPRY2) y los niveles de proteína pRSK y pERK en el tumor. Los niveles de ARNm de DUSP6 fueron considerablemente inferiores con el tratamiento con compuesto a partir de 2 h después de la dosis y permanecieron suprimidos hasta las 24 h con ambos niveles de dosis (Figura 199A). Se observó un patrón similar en los niveles de ARNm de DUSP4 y SPRY2 (Figuras 200A-B). El tratamiento con el compuesto 1 moduló los niveles de proteína fosfo-RSK (pRSK) y fosfo-ERK (pERK) de forma dependiente de la dosis y el tiempo (Figuras 201A-D). Los niveles de cMyc (Figura 199B) y ciclina D1 (Figura 200C), que están corriente abajo de las rutas de señalización de MAPK y Wnt, se vieron inhibidos por el tratamiento con el compuesto 1. El tratamiento con el compuesto 1 reguló en aumento el gen objetivo de Wnt, Axina2. El tratamiento con el compuesto 1 en ambos niveles de dosis demostró un aumento considerable en los niveles de ARNm de Axina2 24 h después de la dosis. Se observó una inhibición sostenida de los niveles de ARNm de AREG (un gen objetivo corriente abajo en la ruta de Hippo) durante 24 h. Asimismo, el compuesto 1 inhibió los niveles de proteína YAP de forma dependiente del tiempo (no estadísticamente significativa (véase la figura 200D), lo cual podría deberse a la inhibición de SIK y la regulación de la ruta de Hippo o podría ser un efecto indirecto como resultado de la inhibición de MAPK.
Estos datos sugieren que el compuesto 1 tiene impacto en tres rutas diferentes, MAPK, Wnt e Hippo, en este modelo de PDX colorrectal con mutación de BRAF, tras una administración de dosis única.
Otros datos del modelo de eficacia: se realizó un perfil del compuesto 1 en modelos de xenoinjerto adicionales, incluidos modelos de mutación de p-catenina (SW48, colorrectal) y p-catenina activada (Hep3B ortotópico, hepatocelular) y un modelo de PDX hepatocelular con amplificación de c-met (LI0612). Se observó actividad antitumoral significativa en todos los modelos.
Conclusión: se observó actividad antitumoral dependiente de la dosis significativa en los tres modelos de xenoinjerto con mutación de BRAF (véanse las figuras 202A-B, la figura 203 y la figura 204). Se observó regresión tumoral con el tratamiento con el compuesto 1 en todos los modelos y hubo un retraso significativo en el crecimiento con el tratamiento a largo plazo en el modelo de PDX146.
Enriquecimiento de pacientes e indicaciones tumorales. En función de los datos in vitro e in vivo del compuesto 1, en la tabla 49 y la tabla 50 se presentan las hipótesis de enriquecimiento de pacientes y las indicaciones tumorales.
Tabla 4

Tabla 50:


Claims (15)
1. Una forma cristalina que comprende el compuesto 1 o un tautómero de este:

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,3, 8,5, 18,2 y 21,3° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 13,8, 19,5, 20,0 y 20,8° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,7, 8,9, 10,3 y 18,3° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,4, 8,7, 10,1 y 18,1° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,8, 14,6, 17,5 y 22,2° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,4, 11,8, 18,0 y 18,3° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,9, 15,3, 18,4 y 22,9° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,7, 8,9, 10,3 y 18,2° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 6,3, 15,7, 18,1 y 20,0° 20 (± 0,2° 20).
2. Una forma cristalina que comprende una sal citrato del compuesto 1 o un tautómero del mismo:

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 4,8, 9,6, 18,9 y 19,2° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,4, 18,8, 19,0 y 28,7° 20 (± 0,2° 20).
3. Una forma cristalina que comprende una sal clorhidrato del compuesto 1 o un tautómero de este:

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 8,7, 9,4, 15,9 y 20,7° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,0, 9,8, 9,9 y 16,7° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 5,5, 16,2, 19,4 y 20,4° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,9, 8,1,8,4 y 15,9° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 7,9, 19,9, 20,6 y 27,0° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,9, 12,4, 17,9 y 19,7° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 8,1, 8,3, 19,1 y 19,7° 20 (± 0,2° 20) o
que tiene un patrón de difracción de rayos X en polvo que comprende picos en 9,8, 17,4, 18,7 y 26,2° 20 (± 0,2° 20).
4. Un sólido amorfo que comprende el compuesto 1 o un tautómero de este:

5. Una composición farmacéutica que comprende una forma sólida de una cualquiera de las reivindicaciones 1 a 4 y un portador farmacéutico; opcionalmente en donde la composición farmacéutica comprende al menos un excipiente farmacéuticamente aceptable.
6. Una forma sólida de una cualquiera de las reivindicaciones 1 a 4 o una composición farmacéutica de la reivindicación 5 para su uso en un método para tratar o prevenir un cáncer, en donde el cáncer es un tumor sólido o un cáncer hematológico.
7. La forma sólida o la composición farmacéutica para su uso de la reivindicación 6, en donde el tumor sólido es melanoma, cáncer colorrectal, cáncer de estómago, cáncer de cabeza y cuello, cáncer de tiroides, cáncer de vejiga, cáncer del SNC, cáncer de pulmón, cáncer de páncreas y cáncer de tejido blando o
en donde el cáncer es cáncer de vejiga, cáncer de mama, cáncer del SNC, cáncer de colon, cáncer gastrointestinal,
cáncer endocrino, cáncer genitourinario femenino, cáncer de cabeza y cuello, cáncer hematopoyético, cáncer de riñón, cáncer de hígado, cáncer de pulmón, melanoma, cáncer de páncreas, cáncer de próstata o cáncer de tejido blando o
en donde el cáncer es glioma, neuroblastoma, cáncer de estómago, cáncer de tiroides, cáncer de glándula adrenal, cáncer de útero, de cuello del útero, de ovario de células claras o de vulva, leucemia, mieloma, NSCLC, SCLC, sarcoma u osteosarcoma.
8. Una forma sólida de cualquiera de las reivindicaciones 1 a 4 o una composición farmacéutica de la reivindicación 5 para su uso en un método para tratar o prevenir el carcinoma hepatoceular (HCC), opcionalmente en donde el HCC está caracterizado por una mutación de beta-catenina y/o expresión de YAP aumentada o
cáncer colorrectal (CRC), melanoma, cáncer gástrico, cáncer de pulmón, cáncer de páncreas, leucemia o mieloma múltiple, opcionalmente en donde el melanoma está caracterizado por una mutación BRAF y/o mutación NRAS o un cáncer con expresión de PD-L1, opcionalmente en donde el cáncer con expresión de PD-L1 es melanoma, cáncer de pulmón, carcinoma de células renales (RCC) o HCC o
un cáncer caracterizado por una mutación de BRAF, opcionalmente en donde el cáncer caracterizado por una mutación de BRAF es CRC, cáncer de tiroides, melanoma o cáncer de pulmón y/u opcionalmente en donde la mutación de BRAF es BRAF V660E o
un cáncer caracterizado por una mutación de NRAS o
un cáncer caracterizado por una mutación de KRAS, en donde el cáncer caracterizado por una mutación de KRAS es CRC, cáncer de páncreas o cáncer de pulmón.
9. Una forma sólida de una cualquiera de las reivindicaciones 1 a 4 o una composición farmacéutica de la reivindicación 5 para su uso en un método para tratar o prevenir un cáncer caracterizado por una mutación de beta-catenina, opcionalmente en donde la mutación de beta-catenina es una o más de beta-catenina S33Y, G34E, S45del o S33C y
opcionalmente en donde el cáncer además está caracterizado por una mutación del EGFR o un aumento en la actividad del EGFR preferentemente en donde la mutación del EGFR es una o más de EGFR E282K, G719S, P753S o V1011M y/o
opcionalmente en donde el cáncer está además caracterizado por una mutación de BRAF, preferentemente en donde la mutación de BRAF comprende una mutación BRAF V660E, BRAF T119S o BRAF G596R.
10. La forma sólida o la composición farmacéutica para su uso de la reivindicación 9, en donde el cáncer caracterizado por una mutación de beta-catenina es CRC, cáncer de estómago, HCC o sarcoma; opcionalmente en donde el HCC además está caracterizado por expresión de YAP u
opcionalmente en donde el melanoma está caracterizado por una mutación en BRAF y/o una mutación de NRAS.
11. Una forma sólida de una cualquiera de las reivindicaciones 1 a 4 o una composición farmacéutica de la reivindicación 5 para su uso en un método para tratar o prevenir un cáncer caracterizado por una ruta de betacatenina activada,
opcionalmente en donde el cáncer caracterizado por la ruta de beta-catenina activada es CRC, cáncer de estómago, HCC o sarcoma.
12. Una forma sólida de una cualquiera de las reivindicaciones 1 a 4 o una composición farmacéutica de la reivindicación 5 para su uso en un método para tratar o prevenir el cáncer gástrico;
opcionalmente en donde el cáncer gástrico está caracterizado por una mutación de beta-catenina o una ruta de beta-catenina activada.
13. Una forma sólida de una cualquiera de las reivindicaciones 1 a 4 o la composición farmacéutica de la reivindicación 5 para su uso en un método para modular los niveles de un biomarcador en un sujeto que padece cáncer, opcionalmente en donde el cáncer es un cáncer como se ha definido en una cualquiera de las reivindicaciones 6 a 12.
14. La forma sólida o la composición farmacéutica para su uso de la reivindicación 13, en donde la modulación del biomarcador se evalúa en una muestra biológica del sujeto, seleccionada entre sangre en circulación, biopsias de piel, biopsias tumorales, células tumorales en circulación, pelo u orina y/o
en donde el biomarcador es ERK, RSK1, DUSP4, DUSP5, DUSP6, BMF, EFNA1, EGR1, ETV5, FOS, FOSL1, GJA1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1, MAFF, CITED2, ELF3 o PD-L1.
15. La forma sólida de la composición farmacéutica para su uso en la reivindicación 13 o 14, en donde la modulación se mide a través de medición de la reducción en los niveles de fosforilación de uno o más de ERK y RSK1 o en donde la modulación se mide a través de medición de la reducción en los niveles de expresión de ARNm o proteína de uno o más de DUSP4, DUSP5, DUSP6, EGR1, ETV5, FOS, FOSL1, IL-8, cMyc, Ciclina D1, YAP, SPRY2, SPRY4, Axina2, CTGF, AREG, CYR61, CXCL1, HAS2, HES1 y MAFF o
o en donde la modulación se mide a través de medición del aumento en los niveles de expresión de ARNm o proteína de uno o más de BMF, EFNA1, CITED2 y ELF3.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662317468P | 2016-04-01 | 2016-04-01 | |
| PCT/US2017/025289 WO2017173218A1 (en) | 2016-04-01 | 2017-03-31 | Solid forms of (1s,4s)-4-(2-(((3s4r)-3-fluorotetrahydro-2h-pyran-4-yl) amino)-8-((2,4,6-trichlorophenyl) amino)-9h-purin-9-yl)-1-methylcyclohexane-1-carboxamide and methods of their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2895419T3 true ES2895419T3 (es) | 2022-02-21 |
Family
ID=59960229
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17776743T Active ES2895419T3 (es) | 2016-04-01 | 2017-03-31 | Formas sólidas de (1S,4S)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1-metilciclohexan-1-carboxamida y métodos para su uso |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US10047090B2 (es) |
| EP (2) | EP3436019B1 (es) |
| JP (1) | JP6995058B2 (es) |
| CN (1) | CN109819649B (es) |
| AU (1) | AU2017240050B2 (es) |
| CA (1) | CA3019105A1 (es) |
| CY (1) | CY1124677T1 (es) |
| DK (1) | DK3436019T3 (es) |
| ES (1) | ES2895419T3 (es) |
| HR (1) | HRP20211546T1 (es) |
| HU (1) | HUE056631T2 (es) |
| LT (1) | LT3436019T (es) |
| MX (1) | MX379439B (es) |
| PL (1) | PL3436019T3 (es) |
| PT (1) | PT3436019T (es) |
| RS (1) | RS62496B1 (es) |
| SI (1) | SI3436019T1 (es) |
| SM (1) | SMT202100604T1 (es) |
| WO (1) | WO2017173218A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK3204386T3 (da) | 2014-10-06 | 2021-05-25 | Signal Pharm Llc | Substituerede aminopurinforbindelser, sammensætninger deraf og fremgangsmåder til behandling dermed |
| NZ746554A (en) * | 2016-04-01 | 2023-03-31 | Signal Pharm Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| ES2836050T3 (es) | 2016-06-21 | 2021-06-23 | Teva Pharmaceuticals Int Gmbh | Formas en estado sólido de citrato de ixazomib |
| CN111417634A (zh) * | 2017-10-04 | 2020-07-14 | 细胞基因公司 | 用于制备顺式-4-[2-{[(3s,4r)-3-氟噁烷-4-基]氨基}-8-(2,4,6-三氯苯胺基)-9h-嘌呤-9-基]-1-甲基环己烷-1-甲酰胺的方法 |
| JP7287955B2 (ja) | 2017-10-04 | 2023-06-06 | セルジーン コーポレイション | シス-4-[2-{[(3s,4r)-3-フルオロオキサン-4-イル]アミノ}-8-(2,4,6-トリクロロアニリノ)-9h-プリン-9-イル]-1-メチルシクロヘキサン-1-カルボキサミドの組成物及び使用方法 |
| US12122787B2 (en) | 2019-09-20 | 2024-10-22 | Shanghai Jemincare Pharmaceuticals Co., Ltd | Fused pyridone compound, and preparation method therefor and use thereof |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5391485A (en) | 1985-08-06 | 1995-02-21 | Immunex Corporation | DNAs encoding analog GM-CSF molecules displaying resistance to proteases which cleave at adjacent dibasic residues |
| US4810643A (en) | 1985-08-23 | 1989-03-07 | Kirin- Amgen Inc. | Production of pluripotent granulocyte colony-stimulating factor |
| JPS63500636A (ja) | 1985-08-23 | 1988-03-10 | 麒麟麦酒株式会社 | 多分化能性顆粒球コロニー刺激因子をコードするdna |
| IL99699A (en) | 1990-10-10 | 2002-04-21 | Autoimmune Inc | Drug with the option of oral, intra-intestinal, or inhaled dosing for suppression of autoimmune response associated with type I diabetes |
| FI109088B (fi) | 1997-09-19 | 2002-05-31 | Leiras Oy | Tabletti ja menetelmä sen valmistamiseksi |
| US7521446B2 (en) | 2005-01-13 | 2009-04-21 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
| US7723340B2 (en) | 2005-01-13 | 2010-05-25 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
| US7759342B2 (en) | 2005-01-13 | 2010-07-20 | Signal Pharmaceuticals, Llc | Methods of treatment and prevention using haloaryl substituted aminopurines |
| WO2007062338A2 (en) | 2005-11-18 | 2007-05-31 | Astrazeneca Ab | Solid formulations |
| JO3235B1 (ar) | 2006-05-26 | 2018-03-08 | Astex Therapeutics Ltd | مركبات بيررولوبيريميدين و استعمالاتها |
| AU2007318132B2 (en) | 2006-10-27 | 2012-11-08 | Signal Pharmaceuticals, Llc | Solid forms comprising 4-[9-(tetrahydro-furan-3-yl)-8-(2,4,6-trifluoro-phenylamino)-9H-purin-2-ylamino]-cyclohexan-1-ol, compositions thereof, and use therewith |
| WO2009130342A1 (es) | 2008-04-23 | 2009-10-29 | Farmasierra Manufacturing, S.L. | Formulación farmacéutica mejorada a base de ibuprofeno y codeína |
| US20130034495A1 (en) | 2009-12-09 | 2013-02-07 | Marie Georges Beauchamps | Isotopologues of 4-[9-(tetrahydro-furan-3-yl)-8-(2,4,6-trifluoro-phenylamino)-9h-purin-2-ylamino]-cyclohexan-1-ol |
| US8680076B2 (en) * | 2010-10-25 | 2014-03-25 | Signal Pharmaceuticals, Llc | Methods of treatment, improvement and prevention using haloaryl substituted aminopurines |
| US8603527B2 (en) | 2010-10-25 | 2013-12-10 | Signal Pharmaceuticals, Llc | Pharmaceutical formulations of a substituted diaminopurine |
| US9466042B2 (en) | 2012-01-24 | 2016-10-11 | International Business Machines Corporation | Facilitating the design of information technology solutions |
| WO2014172616A2 (en) | 2013-04-18 | 2014-10-23 | President And Fellows Of Harvard College | Methods, compositions and kits for promoting motor neuron survival and treating and diagnosing neurodegenerative disorders |
| GB201321737D0 (en) | 2013-12-09 | 2014-01-22 | Ucb Pharma Sa | Therapeutic Agents |
| DK3204386T3 (da) | 2014-10-06 | 2021-05-25 | Signal Pharm Llc | Substituerede aminopurinforbindelser, sammensætninger deraf og fremgangsmåder til behandling dermed |
| NZ746554A (en) | 2016-04-01 | 2023-03-31 | Signal Pharm Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
-
2017
- 2017-03-31 HU HUE17776743A patent/HUE056631T2/hu unknown
- 2017-03-31 JP JP2018551324A patent/JP6995058B2/ja active Active
- 2017-03-31 PT PT177767431T patent/PT3436019T/pt unknown
- 2017-03-31 SI SI201730930T patent/SI3436019T1/sl unknown
- 2017-03-31 US US15/475,836 patent/US10047090B2/en active Active
- 2017-03-31 EP EP17776743.1A patent/EP3436019B1/en active Active
- 2017-03-31 PL PL17776743T patent/PL3436019T3/pl unknown
- 2017-03-31 RS RS20211323A patent/RS62496B1/sr unknown
- 2017-03-31 EP EP20212433.5A patent/EP3845536A1/en not_active Withdrawn
- 2017-03-31 CN CN201780032755.1A patent/CN109819649B/zh active Active
- 2017-03-31 HR HRP20211546TT patent/HRP20211546T1/hr unknown
- 2017-03-31 DK DK17776743.1T patent/DK3436019T3/da active
- 2017-03-31 SM SM20210604T patent/SMT202100604T1/it unknown
- 2017-03-31 AU AU2017240050A patent/AU2017240050B2/en active Active
- 2017-03-31 CA CA3019105A patent/CA3019105A1/en active Pending
- 2017-03-31 MX MX2018011970A patent/MX379439B/es unknown
- 2017-03-31 LT LTEPPCT/US2017/025289T patent/LT3436019T/lt unknown
- 2017-03-31 WO PCT/US2017/025289 patent/WO2017173218A1/en active Application Filing
- 2017-03-31 ES ES17776743T patent/ES2895419T3/es active Active
-
2018
- 2018-07-05 US US16/027,503 patent/US10513519B2/en active Active
-
2021
- 2021-10-27 CY CY20211100932T patent/CY1124677T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20211546T1 (hr) | 2022-01-07 |
| AU2017240050A1 (en) | 2018-10-11 |
| HUE056631T2 (hu) | 2022-02-28 |
| EP3436019A4 (en) | 2019-08-28 |
| JP6995058B2 (ja) | 2022-02-21 |
| PL3436019T3 (pl) | 2022-01-10 |
| RS62496B1 (sr) | 2021-11-30 |
| CY1124677T1 (el) | 2022-07-22 |
| DK3436019T3 (da) | 2021-10-11 |
| EP3845536A1 (en) | 2021-07-07 |
| EP3436019B1 (en) | 2021-07-28 |
| SMT202100604T1 (it) | 2021-11-12 |
| EP3436019A1 (en) | 2019-02-06 |
| AU2017240050B2 (en) | 2021-12-16 |
| MX2018011970A (es) | 2019-05-20 |
| JP2019515889A (ja) | 2019-06-13 |
| CA3019105A1 (en) | 2017-10-05 |
| LT3436019T (lt) | 2021-10-25 |
| US10513519B2 (en) | 2019-12-24 |
| CN109819649A (zh) | 2019-05-28 |
| US10047090B2 (en) | 2018-08-14 |
| MX379439B (es) | 2025-03-10 |
| SI3436019T1 (sl) | 2021-12-31 |
| US20180362529A1 (en) | 2018-12-20 |
| CN109819649B (zh) | 2023-02-28 |
| WO2017173218A1 (en) | 2017-10-05 |
| PT3436019T (pt) | 2021-11-04 |
| US20170283418A1 (en) | 2017-10-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7514005B2 (ja) | がん、炎症性疾患、ラソパシー、及び線維性疾患の治療のためのrasスーパーファミリーと相互作用する化合物 | |
| KR102356433B1 (ko) | 치환된 아미노퓨린 화합물, 이의 조성물, 및 그것에 의한 치료 방법 | |
| ES2895419T3 (es) | Formas sólidas de (1S,4S)-4-(2-(((3S,4R)-3-fluorotetrahidro-2H-piran-4-il)amino)-8-((2,4,6-triclorofenil)amino)-9H-purin-9-il)-1-metilciclohexan-1-carboxamida y métodos para su uso | |
| CN109311868B (zh) | 用于治疗癌症和炎性疾病的化合物 | |
| ES2938061T3 (es) | Profármacos moduladores de la vía integrada del estrés | |
| ES2881220T3 (es) | Métodos de tratamiento del cáncer usando 3-(4-((4-(morfolinometil)bencil)oxi)-1-oxoisoindolin-2-il)piperidin-2,6-diona | |
| ES2562332T3 (es) | Métodos para tratar el cáncer usando 3-(5-amino-2-metil-4-oxo-4H-quinazolin-3-il)-piperidina-2,6-diona | |
| AU2012341028B2 (en) | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof | |
| CA3081750A1 (en) | Anticancer agents | |
| EP3283482B1 (en) | Plk4 inhibitors | |
| US11084829B2 (en) | Ubiquitin-specific-processing protease 7 (USP7) modulators and uses thereof | |
| KR20180088401A (ko) | 3-(5-아미노-2-메틸-4-옥소-4h-퀴나졸린-3-일)-피페리딘-2,6-다이온을 사용한 주기 요법 | |
| CN119013266A (zh) | 新颖gspt1化合物以及新颖化合物的使用方法 | |
| US20180258064A1 (en) | Solid forms of 3-(5-amino-2-methyl-4-oxo-4h-quinazolin-3-yl)-piperidine-2,6-dione, and their pharmaceutical compositions and uses |