ES2857553T3 - Humanización de anticuerpos basada en ángulo interdominio VH-VL - Google Patents
Humanización de anticuerpos basada en ángulo interdominio VH-VL Download PDFInfo
- Publication number
- ES2857553T3 ES2857553T3 ES15784344T ES15784344T ES2857553T3 ES 2857553 T3 ES2857553 T3 ES 2857553T3 ES 15784344 T ES15784344 T ES 15784344T ES 15784344 T ES15784344 T ES 15784344T ES 2857553 T3 ES2857553 T3 ES 2857553T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- angle
- fragments
- fragment
- variant
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21106—Hepsin (3.4.21.106)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Un procedimiento para seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo no humano que comprende las siguientes etapas: - generar una multitud de fragmentos Fv de anticuerpo variante mediante injerto/transferencia de los residuos determinantes de la especificidad de unión a antígeno del fragmento Fv del anticuerpo no humano en/a una secuencia de aminoácidos de línea germinal de fragmento Fv de anticuerpo humano, con lo que cada fragmento Fv de anticuerpo variante de la multitud de fragmentos Fv de anticuerpo variante difiere de los otros fragmentos Fv de anticuerpo variante en al menos un residuo de aminoácido, - determinar el ángulo VH-VL para el fragmento Fv no humano y para cada uno de los fragmentos Fv de anticuerpo variante de la multitud de fragmentos Fv de anticuerpo variante en base a un conjunto de residuos de interfase VH-VL que comprenden los residuos L44, L46, L87, H45, H62 (numeración de acuerdo con el índice de Chothia) del fragmento Fv de anticuerpo, - seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la diferencia más pequeña en el ángulo VH- VL en comparación con el ángulo VH-VL del anticuerpo no humano y, de este modo, seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo no humano, con lo que el uno o más fragmentos Fv de anticuerpo variante se unen al mismo antígeno que el fragmento Fv de anticuerpo no humano, en el que el ángulo VH-VL se determina calculando los seis parámetros - la longitud de C, dc, - el ángulo de torsión, HL, de H1 a L1 medido alrededor de C, - el ángulo de curvatura, HC1, entre H1 y C, - el ángulo de curvatura, HC2, entre H2 y C, - el ángulo de curvatura, LC1, entre L1 y C, y - el ángulo de curvatura, LC2, entre L2 y C, en el que los planos de la región estructural de referencia se registran i) alineando las coordenadas Cα correspondientes a las ocho posiciones H36, H37, H38, H39, H89, H90, H91 y H92 de VH y ajustando un plano a través de ellas y ii) alineando las coordenadas Cα correspondientes a las ocho posiciones L35, L36, L37, L38, L85, L86, L87 y L88 de VL y ajustando un plano a través de ellas, iii) colocando un colocado en cada plano, con lo que cada estructura tiene puntos de malla equivalentes y pares de punto de malla VH-VL equivalentes, y iv) midiendo la distancia euclidiana para cada par de puntos de malla en cada estructura, con lo que el vector C une el par de puntos con la varianza mínima en su distancia de separación, en el que H1 es el vector que discurre paralelo al primer componente principal del plano de VH, H2 es el vector que discurre paralelo al segundo componente principal del plano de VH, L1 es el vector que discurre paralelo al primer componente principal del plano de VL, L2 es el vector que discurre paralelo al segundo componente principal del plano 0 de VL, el ángulo HL es el ángulo de torsión entre los dos dominios, HC1 y LC1 son los ángulos de curvatura equivalentes a variaciones de tipo inclinación de un dominio con respecto al otro, y los ángulos de curvatura HC2 y LC2 son equivalentes a las variaciones de tipo torsión de un dominio con respecto al otro.
Description
DESCRIPCIÓN
Humanización de anticuerpos basada en ángulo interdominio VH-VL
La presente invención pertenece al campo de la humanización de anticuerpos. En el presente documento se informa de un procedimiento para la humanización de anticuerpos que comprende el injerto de residuos donantes en una región estructural aceptadora en el que la selección de la región estructural aceptadora se realiza dependiendo del ángulo interdominio VH-VL del anticuerpo humanizado y el anticuerpo donante.
Antecedentes
El sitio de unión a antígeno de anticuerpos se forma en la interfase de los dominios variables de cadena pesada y ligera, VH y VL, lo que hace que la orientación de los dominios VH-VL sea un factor que afecta la especificidad y afinidad del anticuerpo. Conservar la orientación del dominio VH-VL en el proceso de genomanipulación y humanización de anticuerpos sería ventajoso para mantener las propiedades del anticuerpo donante. Se ha reconocido que predecir la orientación VH-Vl correcta es un factor importante en la modelización de homología de anticuerpos.
En el documento WO 2011/021009, inmunoglobulinas variantes con capacidad de fabricación mejorada se relacionan con el hallazgo de que la modificación de la secuencia de aminoácidos de las moléculas de inmunoglobulina en determinadas posiciones clave da lugar a mejoras en la capacidad de fabricación y, en particular, a reducciones en la propensión a la agregación y/o incrementos en los niveles de producción.
En el documento WO 2008/003931 se informa de un procedimiento para la selección de regiones estructurales para humanizar anticuerpos, de modo que se puede seleccionar la región estructural de región variable más apropiada teniendo en cuenta la homología de una región estructural aceptadora humana con la secuencia donante, pero lo que es más importante, seleccionar aquellas regiones estructurales de región variable en las que se tienen en cuenta residuos específicos, que son residuos donantes obligatorios, es decir, dándoles una ponderación. Por tanto, cuanto mayor es el número de estos residuos donantes ponderados (importantes) ya presentes en una región estructural humana homóloga, más apropiada es la región estructural humana, independientemente de si la homología global es algo menor que la de otra región estructural con menos residuos ponderados coincidentes.
En el documento WO 2001/027160 (EP 1224224) se informa de un procedimiento de producción de anticuerpos monoclonales y, específicamente, para la optimización simultánea de la afinidad in vitro de múltiples dominios distintos de una región variable de un anticuerpo monoclonal. El injerto se logra generando una colección diversa de fragmentos de región variable injertados en CDR y, a continuación, cribando la colección en busca de actividad de unión similar o mejor que la actividad de unión del donante. Se genera una colección diversa seleccionando posiciones de región estructural aceptadora que difieren en la posición correspondiente en comparación con la región estructural donante y preparando una población de la colección que contiene todos los posibles cambios de residuos de aminoácidos en cada una de esas posiciones conjuntamente con todos los posibles cambios de residuos de aminoácidos en cada posición dentro de las CDR de la región variable.
Dunbar, J. et al. (Prot. Eng. Des. Sel. 26 (2013) 611-620) informan que ABangle caracteriza la orientación VH-VL en anticuerpos. La predicción de la orientación de los dominios VH-VL para la modelización del dominio variable de anticuerpo fue informada por Bujotzek, A. et al. (Proteins: Structure, Function, and Bioinformatics 83 (2015) 681-695). En el capítulo 12 del libro "Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharmaceutical Industry" (2012, Elsevier, ISBN:9781908818096), en las páginas 265-297 se han revisado fragmentos de anticuerpos como agentes terapéuticos. Dunbar, J. et al. informaron sobre SAbDab, la base de datos de anticuerpos estructurales (Nucleic Acids Res. 42 (2013) D1140-D1146).
Sumario de la invención
La invención se define por las reivindicaciones. Todo lo que queda fuera del alcance de las reivindicaciones se presenta solo como referencia.
En el presente documento se informa de un procedimiento basado en secuencia rápida para humanizar un anticuerpo basado en la determinación de la orientación del dominio variable de la cadena ligera y la cadena pesada, orientación (ángulo) interdominio VH-VL. Con los procedimientos como se informan en el presente documento se proporciona una selección mejorada, es decir, más rápida, más económica, que requiere menos recursos y más eficiente de la variante humanizada más adecuada de un anticuerpo no humano.
Con más detalle, el procedimiento como se informa en el presente documento usa un predictor basado en secuencia rápida que predice la orientación interdominio VH-VL. La orientación VH-VL se describe en términos de los seis parámetros absolutos de ABangle para separar con precisión los diferentes grados de libertad de la orientación VH-VL. Se ha descubierto que, con el procedimiento como se informa en el presente documento, se puede lograr una mejora en la selección de anticuerpos humanizados con respecto a la desviación de la orientación VH-VL de los
anticuerpos variantes (humanizados) con respecto al anticuerpo original (no humano). Esto muestra una mejora con respecto a la similitud del ángulo interdominio VH-VL entre el anticuerpo original (no humano) y el variante (humanizado). El procedimiento como se informa en el presente documento (que comprende un procedimiento de injerto) proporciona mejores propiedades de unión de los anticuerpos variantes (humanizados) en comparación con los anticuerpos humanizados obtenidos con diferentes procedimientos. Se pueden combinar otros procedimientos de genomanipulación tales como la reorganización de la región estructural con el procedimiento como se informa en el presente documento, lo que da como resultado una unión mejorada de los anticuerpos variantes obtenidos al intercambiar una región estructural humana por otra para cambiar las propiedades biofísicas del anticuerpo.
Un aspecto de la invención es un procedimiento para seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo original que comprende las siguientes etapas:
- generar una multitud de fragmentos Fv de anticuerpos variantes mediante injerto/transferencia de los residuos determinantes de la especificidad de unión a antígeno del fragmento Fv del anticuerpo original no humano en/a una secuencia de aminoácidos de línea germinal de fragmento Fv de anticuerpo humano, con lo que cada fragmento Fv de anticuerpo variante de la multitud de fragmentos Fv de anticuerpo variante difiere de los otros fragmentos Fv de anticuerpo variante en al menos un residuo de aminoácido,
- determinar la orientación VH-VL para el fragmento Fv no humano y para cada uno de los fragmentos Fv de anticuerpo variante de la multitud de fragmentos Fv de anticuerpo variante en base a un conjunto de residuos de interfase VH-Vl que comprenden los residuos L44, L46, L87, H45, h 62 (numeración de acuerdo con el índice de Chothia) del fragmento Fv de anticuerpo,
- seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la diferencia más pequeña en el ángulo VH-VL en comparación con el ángulo VH-VL del anticuerpo no humano y, de este modo, seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo no humano,
con lo que el uno o más fragmentos Fv de anticuerpo variante se unen al mismo antígeno que el fragmento Fv de anticuerpo no humano,
en el que el ángulo VH-VL se determina calculando los seis parámetros
- la longitud de C, dc,
- el ángulo de torsión, HL, de H1 a L1 medido alrededor de C,
- el ángulo de curvatura, HC1, entre H1 y C,
- el ángulo de curvatura, HC2, entre H2 y C,
- el ángulo de curvatura, LC1, entre L1 y C, y
- el ángulo de curvatura, LC2, entre L2 y C,
en el que los planos de la región estructural de referencia se registran i) alineando las coordenadas Ca correspondientes a las ocho posiciones H36, H37, H38, H39, H89, H90, H91 y H92 de VH y ajustando un plano a través de ellas y ii) alineando las coordenadas Ca correspondientes a las ocho posiciones L35, L36, L37, L38, L85, L86, L87 y L88 de VL y ajustando un plano a través de ellas, iii) colocando un colocado en cada plano, con lo que cada estructura tiene puntos de malla equivalentes y pares de punto de malla VH-VL equivalentes, y iv) midiendo la distancia euclidiana para cada par de puntos de malla en cada estructura, con lo que el vector C une el par de puntos con la varianza mínima en su distancia de separación,
en el que H1 es el vector que discurre paralelo al primer componente principal del plano de VH, H2 es el vector que discurre paralelo al segundo componente principal del plano de VH, L1 es el vector que discurre paralelo al primer componente principal del plano de VL, L2 es el vector que discurre paralelo al segundo componente principal del plano de Vl , el ángulo HL es el ángulo de torsión entre los dos dominios, HC1 y LC1 son los ángulos de curvatura equivalentes a variaciones de tipo inclinación de un dominio con respecto al otro, y los ángulos de curvatura HC2 y LC2 son equivalentes a las variaciones de tipo torsión de un dominio con respecto al otro.
En un modo de realización de la invención, el procedimiento comprende la siguiente etapa:
- seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la mayor similitud (estructural) en el ángulo interdominio v H-VL en comparación con el ángulo interdominio VH-VL del anticuerpo original y, de este modo, seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo original.
En el presente documento se divulga un procedimiento para humanizar un anticuerpo no humano que comprende las
siguientes etapas:
- proporcionar un anticuerpo no humano que se une específicamente a un antígeno,
- generar una multitud de anticuerpos variantes mediante injerto/transferencia de uno o más residuos determinantes de especificidad del anticuerpo no humano en/a una secuencia de anticuerpo aceptador humano o humanizado o secuencia de anticuerpo de línea germinal, con lo que cada anticuerpo variante de la multitud de anticuerpos variantes difiere de los otros anticuerpos variantes en al menos un residuo de aminoácido,
- determinar la orientación VH-VL para el fragmento Fv de anticuerpo no humano y para cada uno de los fragmentos Fv de anticuerpo variante de la multitud de anticuerpos variantes en base a una huella de secuencia del fragmento Fv de anticuerpo,
- seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la diferencia más pequeña en la orientación VH-VL en comparación con la orientación de VH-VL del anticuerpo original y, de este modo, seleccionar uno o más anticuerpos humanizados derivados de uno no humano,
con lo que el uno o más anticuerpos humanizados se unen al mismo antígeno que el anticuerpo no humano.
En el presente documento se divulga un procedimiento para humanizar un anticuerpo no humano que comprende las siguientes etapas:
- proporcionar un anticuerpo no humano que se une específicamente a un antígeno,
- generar una multitud de anticuerpos variantes mediante injerto/transferencia de uno o más residuos determinantes de especificidad del anticuerpo no humano en/a una secuencia de anticuerpo aceptador humano o humanizado o secuencia de anticuerpo de línea germinal, con lo que cada anticuerpo variante de la multitud de anticuerpos variantes difiere de los otros anticuerpos variantes en al menos un residuo de aminoácido,
- determinar la orientación VH-VL para el fragmento Fv de anticuerpo no humano y para cada uno de los fragmentos Fv de anticuerpo variante de la multitud de anticuerpos variantes en base a una huella de secuencia del fragmento Fv de anticuerpo,
- seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la mayor similitud (estructural) en el ángulo interdominio v H-VL en comparación con el ángulo interdominio VH-VL del anticuerpo original y, de este modo, seleccionar uno o más anticuerpos humanizados derivados de un anticuerpo no humano,
con lo que el uno o más anticuerpos humanizados se unen al mismo antígeno que el anticuerpo no humano.
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos H35, H37, H39, H45, H47, H50, H58, H60, H61, H91, H95, H96, H98, H100x-2, H100x-1, H100x, H101, H102, H103, H105, L32, L34, L36, L38, L43, L44, L46, L49, L50, L55, L87, L89, L91, L95x-1, L95x, L96 (numeración de acuerdo con el índice de Chothia).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos H33, H35, H43, H44, H46, H50, H55, H56, H58, H61, H62, H89, H99, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50, L53, L55, L56, L85, L87, L89, L91, L93, L94/L95x-1, L95x, L96, L97, L100 (numeración de acuerdo con el índice de Chothia).
En un modo de realización preferente de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos H33, H35, H37, H39, H43, H44, H45, H46, H47, H50, H55, H56, H58, H60, H61, H62, H89, H91, H95, H96, H98, H99, H100x-2, H100x-1, H100x, H101, H102, H103, H105, L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50, L53, L55, L56, L85, L87, L89, L91, L93, L94/L95x-1, L95x, L96, L97, L100 (numeración de acuerdo con el índice de Chothia).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos H35, H37, H39, H45, H47, H50, H58, H60, H61, H91, H95, H96, H98, H100x-2, H100x-1, H100x, H101, H102, H103, H105, L32, L34, L36, L38, L43, L44, L46, L49, L50, L55, L87, L89, L91, L95x-1, L95x, L96, L98 (numeración de acuerdo con el índice de Chothia).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos H33, H35, H37, H39, H43, H44, H45, H46, H47, H50, H58, H60, H61, H62, H89, H91, H95, H96, H98, H99, H100x-2, H100x-1, H100x, H101, H102, H103, H105, L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50, L53, L55, L56, L85, L87, L89, L91, L93, L94, L95x-1, L95x, L96, L97, L98, L100 (numeración de acuerdo con el índice de Chothia).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos 210,
296, 610, 612, 733 (numeración de acuerdo con el índice de Wolfguy).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos 199, 202, 204, 210, 212, 251, 292, 294, 295, 329, 351, 352, 354, 395, 396, 397, 398, 399, 401, 403, 597, 599, 602, 604, 609, 610, 612, 615, 651, 698, 733, 751, 753, 796, 797, 798 (numeración de acuerdo con el índice de Wolfguy).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos 197, 199, 208, 209, 211, 251, 289, 290, 292, 295, 296, 327, 355, 599, 602, 604, 607, 608, 609, 610, 611, 612, 615, 651, 696, 698, 699, 731, 733, 751, 753, 755, 796, 797, 798, 799, 803 (numeración de acuerdo con el índice de Wolfguy).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos 197, 199, 202, 204, 208, 209, 210, 211, 212, 251, 292, 294, 295, 296, 327, 329, 351, 352, 354, 355, 395, 396, 397, 398, 399, 401, 403, 597, 599, 602, 604, 607, 608, 609, 610, 611, 612, 615, 651, 696, 698, 699, 731, 733, 751, 753, 755, 796, 796, 797, 798, 799, 801, 803 (numeración de acuerdo con el índice de Wolfguy).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos 199, 202, 204, 210, 212, 251, 292, 294, 295, 329, 351, 352, 354, 395, 396, 397, 398, 399, 401, 403, 597, 599, 602, 604, 609, 610, 612, 615, 651, 698, 733, 751, 753, 796, 797, 798, 801 (numeración de acuerdo con el índice de Wolfguy).
En un modo de realización de la invención, el conjunto de residuos de interfase VH-VL comprende los residuos 197, 199, 202, 204, 208, 209, 210, 211, 212, 251, 292, 294, 295, 296, 327, 329, 351, 352, 354, 355, 395, 396, 397, 398, 399, 401, 403, 597, 599, 602, 604, 607, 608, 609, 610, 611, 612, 615, 651, 696, 698, 699, 731, 733, 751, 753, 755, 796, 797, 798, 799, 801, 803 (numeración de acuerdo con el índice de Wolfguy).
En un modo de realización de la invención, la selección se basa en el 80 % superior de los fragmentos Fv de anticuerpo variante con respecto a la orientación VH-VL.
En un modo de realización de la invención, la selección es del 20 % superior de los fragmentos Fv de anticuerpo variante con respecto a la orientación VH-VL.
En un modo de realización de la invención, la selección es una deselección de los 20 % peores fragmentos Fv de anticuerpo variante con respecto a la orientación VH-VL.
En un modo de realización de la invención, la orientación VH-VL se determina calculando los parámetros de orientación VH-VL de ABangle usando un procedimiento de bosque aleatorio.
En un modo de realización de la invención, la orientación VH-VL se determina calculando los parámetros de orientación VH-VL de ABangle usando un procedimiento de bosque aleatorio para cada ABangle.
En un modo de realización de la invención, la orientación VH-VL se determina calculando el ángulo de torsión habitual, los cuatro ángulos de curvatura (dos por cada dominio variable) y la longitud del eje de pivote de VH y VL (HL, HC1, LC1, HC2, LC2, dc) usando un modelo de bosque aleatorio.
En un modo de realización de la invención, el modelo de bosque aleatorio se entrena solo con datos de estructura de anticuerpos complejos.
En un modo de realización de la invención, la diferencia más pequeña es la diferencia más pequeña entre el valor del parámetro de ángulo real y predicho en relación con el valor de Q2 más alto.
En un modo de realización de la invención, la diferencia más pequeña es la diferencia más pequeña entre el valor del parámetro de ángulo de anticuerpo original y el valor del parámetro de ángulo de anticuerpo variante humanizado en relación con el valor de Q2 más alto.
En un modo de realización de la invención, la similitud estructural más alta es la desviación de la media cuadrática (RMSD) promedio más baja. En un modo de realización, la RMSD es la RMSD determinada para todos los átomos de Ca (o átomos de carbonilo) de los residuos de aminoácidos del anticuerpo no humano u original frente a los átomos de Ca correspondientes del anticuerpo variante.
En general, distABangle se mejoró con respecto a la referencia de estructuras usando el predictor de VH-VL. La reducción de d istABangie mediante la reorientación VH-VL se tradujo en general en mejores valores de RMSD, especialmente con respecto a las regiones estructurales. Como promedio, se descubrieron notables mejoras de d istABangle y mejoras de la RMSD de carbonilo para todo el Fv.
En un modo de realización de la invención se usa un modelo ensamblado a partir de estructuras molde alineadas en la región estructural consenso de VH o VL, seguido de la reorientación VH-VL en una región estructural consenso de
Fv para determinar la orientación VH-VL.
En un modo de realización de la invención se usa un modelo alineado en el núcleo de lámina p del Fv completo (VH y VL simultáneamente) para determinar la orientación VH-VL.
En un modo de realización de la invención se usa un modelo en el que el fragmento Fv de anticuerpo se reorienta en una región estructural consenso de Fv para determinar la orientación VH-VL.
En un modo de realización de la invención se usa un modelo que usa estructuras molde alineadas en una región estructural consenso de Fv común y en el que no se ajusta la orientación VH-VL en ninguna forma para determinar la orientación VH-VL.
En un modo de realización de la invención se usa un modelo ensamblado a partir de estructuras molde alineadas en la región estructural consenso de VH o VL, seguido de la reorientación VH-VL en una estructura molde de orientación VH-VL elegida en base a la similitud para determinar la orientación VH-VL.
En el presente documento se divulga un procedimiento para producir un anticuerpo que comprende las siguientes etapas:
- seleccionar uno o más anticuerpos o fragmentos Fv de anticuerpo de acuerdo con un procedimiento como se informa en el presente documento,
- seleccionar entre el uno o más anticuerpos o fragmentos Fv de anticuerpo un solo anticuerpo o fragmento Fv de anticuerpo en base a sus propiedades de unión,
- clonar los ácidos nucleicos que codifican VH y VL en uno o más vectores de expresión,
- transfectar una célula con los vectores de expresión obtenidos en la etapa previa,
- cultivar la célula transfectada y de este modo producir el anticuerpo.
En el presente documento se divulga un procedimiento para producir un anticuerpo que comprende las siguientes etapas:
- seleccionar uno o más anticuerpos o fragmentos Fv de anticuerpo que comprende las siguientes etapas:
■ generar una multitud de anticuerpos variantes mediante injerto/transferencia de uno o más residuos determinantes de especificidad de un anticuerpo no humano en/a una secuencia de anticuerpo aceptador humano o humanizado o secuencia de anticuerpo de línea germinal, con lo que cada anticuerpo variante de la multitud de anticuerpos variantes difiere de los otros anticuerpos variantes en al menos un residuo de aminoácido,
■ determinar la orientación VH-VL para el fragmento Fv de anticuerpo no humano y para cada uno de los fragmentos Fv del anticuerpo variante de la multitud de anticuerpos variantes calculando el ángulo de torsión habitual, los cuatro ángulos de curvatura (dos por cada dominio variable) y la longitud del eje de pivote de VH y VL (HL, HC1, LC1, HC2, LC2, dc) usando un modelo de bosque aleatorio basado en un conjunto de residuos de interfase VH-VL que consiste en los residuos H33, H35, H37, H39, H43, H44, H45, H46, H47, H50, H55, H56, H58, H60, H61, H62, H89, H91, H95, H96, H98, H99, H100x-2, H100x-1, H100x, H101, H102, H103, H105, L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50, L53, L55, L56, L85, L87, L89, L91, L93, L94/L95x-1, L95x, L96, L97, L100 (numeración de acuerdo con el índice de Chothia) del fragmento Fv de anticuerpo,
■ seleccionar aquellos fragmentos Fv de anticuerpo variante que tengan la desviación de la media cuadrática (RMSD) promedio más pequeña determinada para todos los pares de átomos de Ca correspondientes del fragmento Fv de anticuerpo no humano y del fragmento Fv de anticuerpo variante,
- seleccionar entre el uno o más anticuerpos un solo anticuerpo en base a sus propiedades de unión,
- clonar los ácidos nucleicos que codifican VH y VL en uno o más vectores de expresión,
- transfectar una célula con los vectores de expresión obtenidos en la etapa previa,
- cultivar la célula transfectada y de este modo producir el anticuerpo.
En el presente documento se divulga un anticuerpo humanizado que comprende residuos de aminoácidos de un anticuerpo donante no humano en las posiciones de aminoácidos L26-L32, L44, L46, L50-L52, L87, L91-L96, H26-H32, H45, H53-H55, H62 y H96-H101 (numeración de acuerdo con el índice de Chothia) y, en las posiciones restantes del dominio variable de la cadena ligera y pesada, residuos de un anticuerpo aceptador humano o humanizado o una
secuencia de aminoácidos de línea germinal humana aceptadora.
En el presente documento se divulga un anticuerpo humanizado que comprende residuos de aminoácidos de un anticuerpo donante no humano en las posiciones de aminoácidos H26-H32, H35, H37, H39, H45, H47, H50, H53-H55, H58, H60, H61, H91, H95, H96-H101, H102, H103, H105, L26-L32, L34, L36, L38, L43, L44, L46, L49, L50-L52, L55, L87, L89, L91-L96 (numeración de acuerdo con el índice de Chothia) y, en las posiciones restantes del dominio variable de la cadena ligera y pesada, residuos de un anticuerpo aceptador humano o humanizado o una secuencia de aminoácidos de línea germinal humana aceptadora.
En el presente documento se divulga un anticuerpo humanizado que comprende residuos de aminoácidos de un anticuerpo donante no humano en las posiciones de aminoácidos H26-H32, H33, H35, H43, H44, H46, H50, H53-H55, H56, H58, H61, H62, H89, H96-H101, L26-L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50-L52, L53, L55, L56, L85, L87, L89, L91-L96, L97, L100 (numeración de acuerdo con el índice de Chothia) y, en las posiciones restantes del dominio variable de la cadena ligera y pesada, residuos de un anticuerpo aceptador humano o humanizado o una secuencia de aminoácidos de línea germinal humana aceptadora.
En el presente documento se divulga un anticuerpo humanizado que comprende residuos de aminoácidos de un anticuerpo donante no humano en las posiciones de aminoácidos H26-H32, H33, H35, H37, H39, H43, H44, H45, H46, H47, H50, H53-H55, H56, H58, H60, H61, H62, H89, H91, H95, H96-H101, H102, H103, H105, L26-L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50-L52, L53, L55, L56, L85, L87, L89, L91-L96, L97, L100 (numeración de acuerdo con el índice de Chothia) y, en las posiciones restantes del dominio variable de la cadena ligera y pesada, residuos de un anticuerpo aceptador humano o humanizado o una secuencia de aminoácidos de línea germinal humana aceptadora.
En el presente documento se divulga un anticuerpo humanizado que comprende residuos de aminoácidos de un anticuerpo donante no humano en las posiciones de aminoácidos H26-H32, H35, H37, H39, H45, H47, H50, H53-H55, H58, H60, H61, H91, H95, H96-H101, H102, H103, H105, L26-L32, L34, L36, L38, L43, L44, L46, L49, L50-L52, L55, L87, L89, L91-L96, L98 (numeración de acuerdo con el índice de Chothia) y, en las posiciones restantes del dominio variable de la cadena ligera y pesada, residuos de un anticuerpo aceptador humano o humanizado o una secuencia de aminoácidos de línea germinal humana aceptadora.
En el presente documento se divulga un anticuerpo humanizado que comprende residuos de aminoácidos de un anticuerpo donante no humano en las posiciones de aminoácidos H26-H32, H33, H35, H37, H39, H43, H44, H45, H46, H47, H50, H53-H55, H58, H60, H61, H62, H89, H91, H95, H96-H101, H102, H103, H105, L26-L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50-L52, L53, L55, L56, L85, L87, L89, L91-L96, L97, L98, L100 (numeración de acuerdo con el índice de Chothia) y, en las posiciones restantes del dominio variable de la cadena ligera y pesada, residuos de un anticuerpo aceptador humano o humanizado o una secuencia de aminoácidos de línea germinal humana aceptadora.
DESCRIPCIÓN DE LAS FIGURAS
Figura 1 Superposición de tres bucles de CDR-H3 ejemplares con 5, 10 y 15 aminoácidos de longitud, tomados de estructuras cristalinas con PDB ID 1N7M, 1DLF y 3HZM, respectivamente:
a) la numeración de Chothia/Kabat muestra la amplia distribución espacial del residuo 97 en los tres bucles de CDR-H3 representativos;
b) la numeración de Wolfguy muestra una localización espacial compacta del residuo 97, ya que siempre es el antepenúltimo residuo antes del final de CDR-H3, denominado 397 de acuerdo con el índice de Wolfguy;
c) varios aminoácidos de las CDR tienen contactos intercatenarios, especialmente los que se localizan al final de CDR-H3 y CDR-L3 (el residuo 797 de acuerdo con el índice de Wolfguy claramente colocaliza y realiza contactos con el VH).
Figura 2 Parámetros de orientación de ABangle predichos (eje vertical) frente a reales (eje horizontal) para una ejecución ejemplar en el conjunto de datos de prueba solo de estructuras complejas (2/3 de las estructuras complejas se usan como conjunto de entrenamiento, mientras que 1/3 se usa como conjunto de prueba). Las predicciones perfectas estarían en la línea diagonal.
Figura 3 Las 25 posiciones más importantes de la huella 3 para los seis parámetros de ABangle en términos de frecuencia de selección porcentual durante el entrenamiento del predictor. Los valores se promedian para diez ejecuciones con un conjunto de entrenamiento variable elegido aleatoriamente (solo estructuras complejas). Las barras de error corresponden a una desviación estándar. La clasificación de regiones estructurales y CDR sigue la nomenclatura de Wolfguy.
Figura 4 Cambio promedio en la RMSD de carbonilo para la región estructural (FW), las CDR (CDR) y todos los
residuos Fv (Todos) y cambio promedio en distABangie cuando se usa la minimización sin restricciones en lugar de restringida (se muestra para las tres variantes 1, II, III frente a 1, 2, 3).
Figura 5 Cambio promedio en la RMSD de carbonilo para la región estructural (FW), las CDR (CDR) y todos los residuos Fv (Todos) y cambio promedio en distABangle por anticuerpo AMAII entre los modelos originales y los reorientados.
Figura 6 Cambio promedio en la RMSD de carbonilo para la región estructural (FW), las CDR (CDR) y todos los residuos Fv (Todos) y cambio promedio en distABangle por participante AMAII entre los modelos originales y los reorientados.
Figura 7 Los HC (filas de la matriz, izquierda) y los LC (columnas de la matriz, derecha) se ordenan de acuerdo con su distancia angular media. Estas visualizaciones se usan para seleccionar HC/LC "malas".
Figura 8 Matriz con mediciones de ELISA para las diferentes combinaciones HC/LC. Los anticuerpos deseleccionados por los diferentes procedimientos están sombreados; 1: malas combinaciones HC/LC; 2: HC/LC completos rechazados; 3: 20 % peores.
Figura 9 Histogramas apilados de las mediciones de ELISA para los tres procedimientos de selección: "malas combinaciones HC/LC" (izquierda), "HC y LC completos" (centro) y "20 % peores" (derecha). Las regiones de color gris claro de las barras del histograma indican los anticuerpos que se rechazan.
Figura 10 Los HC (filas de la matriz, izquierda) y los LC (columnas de la matriz, derecha) se ordenan de acuerdo con su distancia angular media. Estas visualizaciones se usan para seleccionar HC/LC "malas".
Figura 11 Matriz con mediciones de ELISA para las diferentes combinaciones HC/LC. Los anticuerpos deseleccionados por los diferentes procedimientos están sombreados; 1: malas combinaciones HC/LC; 2: HC/LC completos rechazados; 3: 20 % peores.
Figura 12 Histogramas apilados de las mediciones de ELISA para los tres procedimientos de selección: "malas combinaciones HC/LC" (izquierda), "HC y LC completos" (centro) y "20 % peores" (derecha). Las regiones de color gris claro de las barras del histograma indican los anticuerpos que se rechazan.
Figura 13 Los HC (filas de la matriz, izquierda) y los LC (columnas de la matriz, derecha) se ordenan de acuerdo con su distancia angular media. Estas visualizaciones se usan para seleccionar HC/LC "malas".
Figura 14 Cada una de las tres imágenes muestran la matriz con las mediciones de BL para las diferentes combinaciones HC/LC; los anticuerpos deseleccionados por los diferentes procedimientos están sombreados; 1: malas combinaciones HC/LC; 2: HC/LC completos rechazados; 3: 20 % peores.
Figura 15 Histogramas apilados de las mediciones de ELISA para los tres procedimientos: "malas combinaciones HC/LC" (izquierda), "HC y LC completos" (centro) y "20 % peores" (derecha). Las regiones de color gris claro de las barras del histograma indican los anticuerpos deseleccionados.
Figura 16 Cada una de las tres imágenes muestran la matriz con las mediciones de t1/2 para las diferentes combinaciones HC/LC; los anticuerpos seleccionados por los diferentes procedimientos están sombreados; 1: malas combinaciones HC/LC; 2: HC/LC completos rechazados; 3: 20 % peores.
Figura 17 Histogramas apilados de las mediciones de t1/2 para los tres procedimientos: "malas combinaciones HC/LC" (izquierda), "HC y LC completos" (centro) y "20 % peores" (derecha). Las regiones de color gris claro de las barras del histograma indican los anticuerpos deseleccionados.
DEFINICIONES
Esquema de numeración de Wolfguy
La numeración de Wolfguy define las regiones CDR como la unión de conjuntos de la definición de Kabat y Chothia. Además, el esquema de numeración identifica puntas de bucle de CDR en base a la longitud de CDR (y parcialmente en base a la secuencia), de modo que el índice de una posición de CDR indica si un residuo de CDR es parte del bucle ascendente o descendente. En la tabla 1 se muestra una comparación con los esquemas de numeración establecidos.
Tabla 1: Numeración de CDR-L3 y CDR-H3 usando esquemas de numeración de Chothia/Kabat (Ch-Kb), Honegger y Wolfguy. Este último tiene números crecientes desde la base N terminal hasta el pico de CDR y decrecientes comenzando a partir del extremo C terminal de CDR. Los esquemas de Kabat fijan los dos últimos residuos de CDR e introducen letras para adaptarse a la longitud de CDR. A diferencia de la nomenclatura de Kabat, la numeración de
Honegger no usa letras y es común para VH y VL.

Wolfguy está diseñado de modo que los residuos estructuralmente equivalentes (es decir, los residuos que son muy similares en términos de localización espacial conservada en la estructura de Fv) se numeran con índices equivalentes en la medida de lo posible. Esto se ilustra en la figura 1.
En la tabla 2 se puede encontrar un ejemplo de una secuencia de VH y VL de longitud completa numerada según Wolfguy.
Tabla 2: Secuencia de VH (izquierda) y VL (derecha) de la estructura cristalina con PDB ID 3PP4 (21), numerada según Wolfguy, Kabat y Chothia. En la numeración de Wolfguy, CDR-H1-H3, CDR-L2 y CDR-L3 se numeran dependiendo solo de la longitud, mientras que CDR-L1 se numera dependiendo de la longitud de bucle y de la pertenencia a la agrupación canónica. Este último se determina calculando similitudes de secuencia con diferentes secuencias consenso. Aquí solo se da un solo ejemplo de numeración de CDR-L1, ya que no tiene importancia para generar la huella de secuencia de orientación VH-VL.







El concepto ABangle (7)
Al hacer una comparación entre dos estructuras basadas en aminoácidos, en general se usan métricas basadas en la distancia, tal como la desviación de la media cuadrática (RMSD) de átomos equivalentes.
Para caracterizar la orientación entre dos objetos tridimensionales cualesquiera, es necesario definir:
- una región estructural de referencia en cada objeto.
- ejes sobre los que medir los parámetros de orientación.
- terminología para describir y cuantificar estos parámetros.
El concepto ABangle es un procedimiento que caracteriza completamente la orientación VH-VL en un sentido absoluto y consecuente usando cinco ángulos (HL, HC1, LC1, HC2 y LC2) y una distancia (dc). El par de dominios variables de un anticuerpo, VH y VL, se indica conjuntamente como un fragmento Fv de anticuerpo.
En una primera etapa, las estructuras de anticuerpo se extrajeron de un banco de datos (por ejemplo, el banco de datos de proteínas, PDB). Se aplicó la numeración de anticuerpos de Chothia (Chothia y Lesk, 1987) a cada una de las cadenas de anticuerpo. Las cadenas que se numeraron correctamente se emparejaron para formar regiones Fv. Esto se hizo aplicando la restricción de que la coordenada Ca de la posición H37 de la cadena pesada (átomo de carbono alfa del residuo de aminoácido en la posición 37 del dominio variable de la cadena pesada) debe estar dentro de los 20 A de la coordenada Ca de la posición L87 de la cadena ligera. Se creó un conjunto no redundante de anticuerpos usando CDHIT (Li, W. y Godzik, A. Bioinformatics, 22 (2006) 1658-1659), aplicando un corte de identidad de secuencia sobre la región estructural de la región Fv del 99 %.
Se usaron las posiciones de residuos más estructuralmente conservadas en los dominios pesado y ligero para definir la localización del dominio. Estas posiciones se indican como los núcleos de VH y VL. Estas posiciones están localizadas predominantemente en las hebras p de la región estructural y forman el núcleo de cada dominio. Las posiciones de los núcleos se dan en la tabla 3 siguiente:


Las posiciones de los núcleos se usaron para registrar regiones estructurales de referencia en los dominios de la región Fv del anticuerpo.
Los dominios VH en el conjunto de datos no redundantes se agruparon usando CDHIT, aplicando un corte de identidad de secuencia del 80 % sobre las posiciones de la región estructural en el dominio. Se eligió de forma aleatoria una estructura de cada una de las 30 agrupaciones más grandes. Este conjunto de dominios se alineó sobre las posiciones de los núcleos de VH usando Mammoth-mult (Lupyan, D. et al., Bioinf. 21 (2005) 3255-3263). A partir de esta alineación, se extrajeron las coordenadas Ca correspondientes a las ocho posiciones estructuralmente conservadas H36, H37, H38, H39, H89, H90, H91 y H92 en la interfase de lámina p. A través de las 240 coordenadas resultantes se ajustó un plano. Para el dominio VL se usaron las posiciones L35, L36, L37, L38, L85, L86, L87 y L88 para ajustar el plano.
El procedimiento descrito anteriormente permite mapear los dos planos de la región estructural de referencia en cualquier estructura de Fv. Por lo tanto, la medición de la orientación VH-VL se puede hacer equivalente a la medición de la orientación entre los dos planos. Para hacer esto completamente y en un sentido absoluto, se requieren al menos seis parámetros: una distancia, un ángulo de torsión y cuatro ángulos de curvatura. Estos parámetros se deben medir en torno a un vector definido de forma consecuente que conecta los planos. Este vector se indica como C a continuación. Para identificar C, los planos de la región estructural de referencia se registraron en cada una de las estructuras en el conjunto no redundante como se describe anteriormente y se colocó una malla en cada plano. Por lo tanto, cada estructura tenía puntos de malla equivalentes y, por tanto, pares de puntos de malla VH-VL equivalentes. La distancia euclidiana se midió para cada par de puntos de malla en cada estructura. Se identificó el par de puntos con la mínima varianza en su distancia de separación. El vector que une estos puntos se define como C.
El sistema de coordenadas está completamente definido usando vectores, que se encuentran en cada plano y están centrados en los puntos correspondientes a C. H1 es el vector que discurre paralelo al primer componente principal del plano de VH, mientras que H2 discurre paralelo al segundo componente principal. L1 y L2 se definen de forma similar en el dominio VL. El ángulo HL es un ángulo de torsión entre los dos dominios. Los ángulos de curvatura HC1 y LC1 son equivalentes a variaciones de tipo inclinación de un dominio con respecto al otro. Los ángulos de curvatura HC2 y LC2 describen variaciones de tipo torsión de un dominio con respecto al otro.
Para describir la orientación VH-VL se usan seis medidas, una distancia y cinco ángulos. Estos se definen en el sistema de coordenadas como sigue:
- la longitud de C, dc,
- el ángulo de torsión, HL, de H1 a L1 medido alrededor de C,
- el ángulo de curvatura, HC1, entre H1 y C,
- el ángulo de curvatura, HC2, entre H2 y C,
- el ángulo de curvatura, LC1, entre L1 y C, y
- el ángulo de curvatura, LC2, entre L2 y C.
El término "orientación VH-VL" se usa de acuerdo con su significado común en la técnica, como lo entendería un experto en la técnica (véase, por ejemplo, Dunbar et al., Prot. Eng. Des. Sel. 26 (2013) 611-620; y Bujotzek, A. et al., Proteins, Struct. Funct. Bioinf. 83 (2015) 681-695). Indica cómo se orientan los dominios VH y VL uno con respecto al otro.
Por tanto, la orientación VH-VL se define por
- la longitud de C, dc,
- el ángulo de torsión, HL, de H1 a L1 medido alrededor de C,
- el ángulo de curvatura, HC1, entre H1 y C,
- el ángulo de curvatura, HC2, entre H2 y C,
- el ángulo de curvatura, LC1, entre L1 y C, y
- el ángulo de curvatura, LC2, entre L2 y C,
en el que los planos de la región estructural de referencia se registran i) alineando las coordenadas Ca correspondientes a las ocho posiciones H36, H37, H38, H39, H89, H90, H91 y H92 de VH y ajustando un plano a través de ellas y ii) alineando las coordenadas Ca correspondientes a las ocho posiciones L35, L36, L37, L38, L85, L86, L87 y L88 de VL y ajustando un plano a través de ellas, iii) colocando un colocado en cada plano, con lo que cada estructura tiene puntos de malla equivalentes y pares de punto de malla VH-VL equivalentes, y iv) midiendo la distancia euclidiana para cada par de puntos de malla en cada estructura, con lo que el vector C une el par de puntos con la varianza mínima en su distancia de separación,
en el que H1 es el vector que discurre paralelo al primer componente principal del plano de VH, H2 es el vector que discurre paralelo al segundo componente principal del plano de VH, L1 es el vector que discurre paralelo al primer componente principal del plano de VL, L2 es el vector que discurre paralelo al segundo componente principal del plano de Vl, el ángulo HL es el ángulo de torsión entre los dos dominios, HC1 y LC1 son los ángulos de curvatura equivalentes a variaciones de tipo inclinación de un dominio con respecto al otro, y los ángulos de curvatura HC2 y LC2 son equivalentes a las variaciones de tipo torsión de un dominio con respecto al otro.
Las posiciones se determinan de acuerdo con el índice de Chothia.
Se eligió el vector C para tener la longitud más conservada del conjunto de estructuras no redundante. La distancia, dc, es esta longitud. Tiene un valor medio de 16,2 A y una desviación estándar de solo 0,3 A.
La tabla 4 enumera las 10 posiciones y residuos superiores identificados por el algoritmo de bosque aleatorio como importantes para determinar cada una de las medidas angulares de la orientación VH-VL.
Tabla 4: X representa la variable L36Va/L38Eb/L42Ha/L43La/L44Fa,b/L45T/L46Gb/L49G/L95H


(para obtener información más detallada, véase la referencia 7 y Bujotzek, A. et al., Prot. Struct. Funct. Bioinf. 83 (2015) 681-695).
Definiciones adicionales:
Una "región estructural humana aceptadora" para los propósitos en el presente documento es una región estructural que comprende la secuencia de aminoácidos de una región estructural del dominio variable de la cadena ligera (VL) o una región estructural del dominio variable de la cadena pesada (VH) derivada de una región estructural de inmunoglobulina humana o una región estructural consenso humana, como se define a continuación. Una región estructural humana aceptadora "derivada de" una región estructural de inmunoglobulina humana o una región estructural consenso humana puede comprender la misma secuencia de aminoácidos de la misma, o puede contener cambios en la secuencia de aminoácidos. En algunos modos de realización, el número de cambios aminoacídicos es de 10 o menos, 9 o menos, 8 o menos, 7 o menos, 6 o menos, 5 o menos, 4 o menos, 3 o menos o 2 o menos. En algunos modos de realización, la región estructural humana aceptadora de VL tiene una secuencia idéntica a la secuencia de la región estructural de inmunoglobulina humana de VL o la secuencia de la región estructural consenso humana.
"Afinidad" se refiere a la fuerza de la suma total de interacciones no covalentes entre un único sitio de unión de una molécula (por ejemplo, un anticuerpo) y su ligando de unión (por ejemplo, un antígeno). A menos que se indique de otro modo, como se usa en el presente documento, "afinidad de unión" se refiere a la afinidad de unión intrínseca que refleja una interacción 1:1 entre los miembros de un par de unión (por ejemplo, anticuerpo y antígeno). La afinidad de una molécula X por su compañero Y se puede representar en general por la constante de disociación (Kd). Se puede medir la afinidad por procedimientos comunes conocidos en la técnica, incluyendo los descritos en el presente documento.
El término "anticuerpo" en el presente documento se usa en el sentido más amplio y engloba diversas estructuras de anticuerpo, incluyendo pero sin limitarse a anticuerpos monoclonales, anticuerpos policlonales, anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos) y fragmentos de anticuerpo siempre que presenten la actividad de unión a antígeno deseada.
Un "fragmento de anticuerpo" se refiere a una molécula distinta de un anticuerpo intacto que comprende una parte de un anticuerpo intacto que se une al antígeno al que se une el anticuerpo intacto. Los ejemplos de fragmentos de anticuerpo incluyen, pero no se limitan a, Fv, Fab, Fab', Fab'-SH, F(ab')2; diacuerpos; anticuerpos lineales; moléculas de anticuerpo monocatenario (por ejemplo, scFv) y anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpo.
El término anticuerpo "quimérico" se refiere a un anticuerpo en el que una parte de la cadena pesada y/o ligera se deriva de una fuente o especie particular, mientras que el resto de la cadena pesada y/o ligera se deriva de una fuente o especie diferente.
La "clase" de un anticuerpo se refiere al tipo de dominio constante o región constante que posee su cadena pesada. Existen cinco clases principales de anticuerpos: IgA, IgD, IgE, IgG e IgM, y varias de estas se pueden dividir además en subclases (isotipos), por ejemplo, IgG1, IgG2, IgG3, IgG4, IgA1 e IgA2. Los dominios constantes de la cadena pesada que se corresponden con las diferentes clases de inmunoglobulinas se llaman a, 6, £, y, y H, respectivamente.
El término "región Fc" en el presente documento se usa para definir una región C terminal de una cadena pesada de inmunoglobulina que contiene al menos una parte de la región constante. El término incluye regiones Fc de secuencia natural y regiones Fc variantes. En un modo de realización, una región Fc de la cadena pesada de IgG humana se extiende de Cys226, o de Pro230, al extremo carboxilo de la cadena pesada. Sin embargo, la lisina C terminal (Lys447) de la región Fc puede estar o no estar presente. A menos que se especifique de otro modo en el presente documento, la numeración de los residuos aminoacídicos de la región Fc o región constante se realiza de acuerdo con el sistema de numeración EU, también denominado índice EU, como se describe en Kabat, E.A. et al., Sequences of Proteins of Immunological Interest, 5a ed., Public Health Service, National Institutes of Health, Bethesda, MD (1991), NIH Publication 91-3242.
"Región estructural" o "FR" se refiere a residuos del dominio variable distintos de los residuos de la región hipervariable (HVR). La FR de un dominio variable consiste en general en cuatro dominios de FR: FR1, FR2, FR3 y FR4. En consecuencia, las secuencias de HVR y FR aparecen, en general, en la siguiente secuencia en VH (o VL): FR1 -H1(L1 )-FR2-H2(L2)-FR3-H3(L3)-FR4.
Los términos "anticuerpo de longitud completa", "anticuerpo intacto" y "anticuerpo completo" se usan en el presente documento de manera intercambiable para referirse a un anticuerpo que tiene una estructura sustancialmente similar
a una estructura de anticuerpo natural o que tiene cadenas pesadas que contienen una región Fc como se define en el presente documento.
Un "anticuerpo humano" es uno que posee una secuencia de aminoácidos que se corresponde con la de un anticuerpo producido por un humano o una célula humana o derivado de una fuente no humana que utiliza repertorios de anticuerpos humanos u otras secuencias que codifican anticuerpos humanos. Esta definición de un anticuerpo humano excluye específicamente un anticuerpo humanizado que comprende residuos de unión a antígeno no humanos.
Una "región estructural consenso humana" es una región estructural que representa los residuos de aminoácido que se producen lo más comúnmente en una selección de secuencias de la región estructural de VL o VH de inmunoglobulina humana. En general, la selección de secuencias de VL o VH de inmunoglobulina humana es de un subgrupo de secuencias de dominio variable. En general, el subgrupo de secuencias es un subgrupo como en Kabat, E.A. et al., Sequences of Proteins of Immunological Interest, 5.a ed., Bethesda MD (1991), NIH Publication 91-3242, vol. 1-3. En un modo de realización, para el VL, el subgrupo es el subgrupo kappa I como en Kabat et al., supra. En un modo de realización, para el VH, el subgrupo es el subgrupo III como en Kabat et al., supra.
Un anticuerpo "humanizado" se refiere a un anticuerpo quimérico que comprende residuos de aminoácido de HVR no humanas y residuos de aminoácido de FR humanas. En determinados modos de realización, un anticuerpo humanizado comprenderá sustancialmente todos de al menos uno, y típicamente dos, dominios variables, en los que todas o sustancialmente todas las HVR (por ejemplo, CDR) corresponden a las de un anticuerpo no humano, y todas o sustancialmente todas las FR corresponden a las de un anticuerpo humano. Un anticuerpo humanizado opcionalmente puede comprender al menos una parte de una región constante de anticuerpo derivada de un anticuerpo humano. Una "forma humanizada" de un anticuerpo, por ejemplo, un anticuerpo no humano, se refiere a un anticuerpo que se ha sometido a humanización.
El término "región hipervariable" o "HVR", como se usa en el presente documento, se refiere a cada una de las regiones de un dominio variable de anticuerpo que son hipervariables en secuencia ("regiones determinantes de la complementariedad" o "CDR") y/o forman bucles estructuralmente definidos ("bucles hipervariables") y/o contienen los residuos de contacto con antígeno ("contactos con antígeno"). En general, los anticuerpos comprenden seis HVR; tres en el VH (H1, H2, H3) y tres en el VL (L1, L2, L3).
Las HVR en el presente documento incluyen
(a) los bucles hipervariables que se producen en los residuos de aminoácido 26-32 (L1), 50-52 (L2), 91-96 (L3), 26-32 (H1), 53-55 (H2) y 96-101 (H3) (Chothia, C. y Lesk, A.M., J. Mol. Biol. 196 (1987) 901-917);
(b) las CDR que se producen en los residuos de aminoácido 24-34 (L1), 50-56 (L2), 89-97 (L3), 31-35b (H1), 50-65 (H2) y 95-102 (H3) (Kabat E.A. et al., Sequences of Proteins of Immunological Interest, 5.a ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991), NIH Publication 91-3242);
(c) los contactos con antígeno que se producen en los residuos de aminoácido 27c-36 (L1), 46-55 (L2), 89-96 (L3), 30-35b (H1), 47-58 (H2) y 93-101 (H3) (MacCallum et al., J. Mol. Biol. 262:732-745 (1996)); y
(d) combinaciones de (a), (b) y/o (c), incluyendo los residuos de aminoácido de HVR46-56 (L2), 47-56 (L2), 48-56 (L2), 49-56 (L2), 26-35 (H1), 26-35b (H1), 49-65 (H2), 93-102 (H3) y 94-102 (H3).
A menos que se indique de otro modo, los residuos de HVR y otros residuos en el dominio variable (por ejemplo, residuos de FR) se numeran en el presente documento de acuerdo con Kabat et al., supra.
El término "residuo determinante de especificidad" se usa de acuerdo con su significado en la técnica. Define los residuos de un anticuerpo que están directamente implicados en la interacción con el antígeno (véase, por ejemplo, Padlan, E.A. et al., FASEB J. 9 (1995) 133-139).
Un anticuerpo "aislado" es uno que se ha separado de un componente de su entorno natural. En algunos modos de realización, se purifica un anticuerpo a más de un 95 % o 99 % de pureza como se determina, por ejemplo, por electroforesis (por ejemplo, SDS-PAGE, isoelectroenfoque (IEF), electroforesis capilar) o cromatografía (por ejemplo, HPLC de intercambio iónico o de fase inversa). Para una revisión de los procedimientos para la evaluación de la pureza de los anticuerpos, véase, por ejemplo, Flatman, S. et al., J. Chromatogr. B 848 (2007) 79-87.
El término "anticuerpo monoclonal", como se usa en el presente documento, se refiere a un anticuerpo obtenido de una población de anticuerpos sustancialmente homogéneos, es decir, los anticuerpos individuales que comprende la población son idénticos y/o se unen al mismo epítopo, excepto por posibles anticuerpos variantes, por ejemplo, que contienen mutaciones naturales o que surgen durante la producción de una preparación de anticuerpos monoclonales, estando presentes dichas variantes en general en cantidades insignificantes. En contraste con las preparaciones de anticuerpos policlonales, que típicamente incluyen anticuerpos diferentes dirigidos frente a diferentes determinantes (epítopos), cada anticuerpo monoclonal de una preparación de anticuerpos monoclonales se dirige frente a un único
determinante en un antígeno. Por tanto, el modificador "monoclonal" indica el carácter del anticuerpo como que se ha obtenido de una población sustancialmente homogénea de anticuerpos, y no se ha de interpretar como que requiere la producción del anticuerpo por ningún procedimiento particular. Por ejemplo, los anticuerpos monoclonales que se van a usar de acuerdo con la presente invención se pueden preparar por una variedad de técnicas, incluyendo pero sin limitarse al procedimiento de hibridoma, procedimientos de ADN recombinante, procedimientos de presentación en fagos y procedimientos que utilizan animales transgénicos que contienen todos o parte de los locus de inmunoglobulina humana, describiéndose en el presente documento dichos procedimientos y otros procedimientos ejemplares para preparar anticuerpos monoclonales.
El término "región variable" o "dominio variable" se refiere al dominio de una cadena pesada o ligera de anticuerpo que está implicado en la unión del anticuerpo al antígeno. Los dominios variables de la cadena pesada y de la cadena ligera (VH y VL, respectivamente) de un anticuerpo natural tienen, en general, estructuras similares, comprendiendo cada dominio cuatro regiones estructurales (FR) conservadas y tres regiones hipervariables (HVR). (Véase, por ejemplo, Kindt, T.J. etal., Kuby Immunology, 6.a ed., W.H. Freeman and Co., N.Y. (2007), página 91). Un único dominio VH o VL puede ser suficiente para conferir especificidad de unión a antígeno. Además, los anticuerpos que se unen a un antígeno particular se pueden aislar usando un dominio VH o VL de un anticuerpo que se une al antígeno para cribar una colección de dominios VL o VH complementarios, respectivamente. Véanse, por ejemplo, Portolano, S. et al., J. Immunol. 150 (1993) 880-887; Clackson, T. et al., Nature 352 (1991) 624-628).
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
En el presente documento se informa de un predictor basado en secuencia rápida que predice la orientación interdominio VH-VL. Se logran valores de Q2 que oscilan entre 0,67 y 0,80. La orientación VH-VL se describe en términos de los seis parámetros absolutos de ABangle para separar con precisión los diferentes grados de libertad de la orientación VH-VL. Se evaluó el impacto de la orientación VH-VL en diferentes estructuras de anticuerpo. Se ha descubierto que, con el procedimiento como se informa en el presente documento, se puede lograr una mejora con respecto a la desviación de la orientación VH-VL de los anticuerpos variantes (humanizados) con respecto al anticuerpo original (no humano). Esto se muestra por la desviación de la media cuadrática (RMSD) promedio de los átomos de carbonilo de la cadena principal de aminoácidos. Esto muestra una mejora con respecto a la similitud del ángulo interdominio VH-VL entre el anticuerpo original (no humano) y el variante (humanizado). El procedimiento como se informa en el presente documento (que comprende un procedimiento de injerto) proporciona mejores propiedades de unión de los anticuerpos variantes (humanizados). Se pueden combinar otros procedimientos de genomanipulación tales como la reorganización de la región estructural con el procedimiento como se informa en el presente documento, lo que da como resultado una unión mejorada de los anticuerpos variantes obtenidos al intercambiar una región estructural humana por otra para cambiar las propiedades biofísicas del anticuerpo. Esto da como resultado la provisión de un procedimiento para seleccionar mejores anticuerpos humanizados de una multitud de anticuerpos variantes derivados de un anticuerpo original.
El uso de anticuerpos en los tratamientos y en el diagnóstico clínico creó una demanda de modelos precisos de homología de estructuras de anticuerpos que permitan la genomanipulación racional de anticuerpos siempre que no se disponga de una estructura cristalina. Por lo tanto, se ha desarrollado una multitud de procedimientos informáticos para el diseño de anticuerpos asistido por ordenador (1), entre ellos una serie de enfoques de modelización de homología que se evalúan regularmente mediante estudios de modelización enmascarada (8, 2).
Debido al número de estructuras de anticuerpo derivadas experimentalmente (la base de datos de anticuerpos estructurales SAbDab3 cuenta con 1841 entradas a mayo de 2014), la calidad de los modelos de homología de anticuerpos es excelente en comparación con los modelos de homología de otras biomoléculas. Los seis bucles de unión a antígeno de los dos fragmentos variables de anticuerpo (Fv) son de secuencia hipervariable (regiones hipervariables, HVR). Cinco de ellos son propensos a adoptar conformaciones canónicas que se pueden predecir a partir de secuencias basadas en estructuras molde existentes. Esto no es válido para el tercer bucle de la región variable de la cadena pesada, HVR-H3. El HVR-H3 es el bucle más variable con respecto a la secuencia y la longitud, y típicamente es el principal sitio determinante de la especificidad de interacción del antígeno.
El sitio de unión a antígeno de un anticuerpo se forma en la interfase de los dos Fv (dominio variable de cadena pesada (VH) y dominio variable de cadena ligera (VL)). Cada dominio variable comprende tres HVR. La orientación relativa de los dominios VH y VL se suma a la topología del sitio de unión a antígeno.
En su reciente estudio de evaluación de la modelización de anticuerpos 2 (AMAII), Teplyakov et al. (2) usaron una única medida angular para describir la orientación VH-VL. La diferencia en el ángulo de inclinación VH-VL con respecto a una estructura de referencia se calcula como el ángulo k en el sistema angular esférico (w, $, k) de la transformación de coordenadas lograda por la superposición secuencial de los dominios VL y VH usando un conjunto de posiciones de núcleo de lámina p estructuralmente conservadas. Narayanan et al. (6) usaron una métrica basada en RMSD (desviación de la media cuadrática) para entrenar y evaluar un predictor basado en energía de la orientación VH-VL. Chailyan et al. (5) identificaron grupos de estructuras de Fv de orientación VH-VL similar y determinaron posiciones de secuencia influyentes midiendo la RMSD de superposición de Ca de determinados residuos conservados. Otros estudios aumentan los valores de RMSD al proporcionar la cantidad de rotación necesaria para reorientar el VH o VL
de una estructura cristalina hacia otra (10-12).
Abhinandan y Martin (4) definieron el ángulo de empaquetamiento VH-VL, una métrica absoluta para comparar la orientación VH-VL. El ángulo de empaquetamiento VH-VL es el ángulo de torsión abarcado por un vector ajustado a través de los ejes principales de un conjunto altamente conservado de posiciones Ca en cada uno de los dos dominios. A diferencia de los valores relativos de RMSD, el ángulo de empaquetamiento VH-VL permite describir cada estructura de Fv individual en términos de su orientación VH-VL en el espacio estructural. Junto con la definición del ángulo de empaquetamiento VH-VL, los autores identificaron un conjunto de posiciones influyentes y proporcionaron un predictor basado en secuencia del empaquetamiento VH-VL aprendido con una red neuronal.
En base a las observaciones anteriores, que son al menos en partes inconsistentes con respecto a las posiciones de la secuencia Fv que se considera que tienen un impacto en la orientación VH-VL (4, 5), Dunbar et al. (7) sugirieron que la orientación VH-VL está sujeta a múltiples grados de libertad, y que cada grado de libertad está determinado por un conjunto diferente de posiciones de secuencia influyentes. En consecuencia, los autores, además del ángulo de torsión habitual, definieron cuatro ángulos de curvatura (dos por cada dominio de variable), así como la longitud del eje de pivote de VH y VL, y, usando un modelo de bosque aleatorio, identificaron las posiciones de secuencia más influyentes para cada uno de los cinco parámetros de ángulo (ABangle), así como para la longitud del eje de pivote entre VH y VL
En el presente documento se informa de un procedimiento basado en ABangle para la caracterización y explotación de la orientación VH-VL durante la humanización de un anticuerpo. En el presente documento se informa de un predictor basado en secuencia de la orientación VH-VL para cada una de las seis medidas de ABangle. También se informa de un procedimiento para ajustar la orientación VH-VL en modelos reales de homología de anticuerpos.
En el presente documento se informa de un procedimiento basado en ABangle para la caracterización y explotación de la orientación VH-VL durante la transferencia de residuos determinantes de unión desde un anticuerpo donante a una región estructural de anticuerpo aceptador.
En el presente documento se informa de un procedimiento basado en ABangle para la caracterización y explotación de la orientación VH-VL durante el intercambio de partes o regiones estructurales completas de un anticuerpo (reorganización de región estructural).
Predictor de orientación VH-VL
Tabla 5: Los valores de Q2 y RMSE (error cuadrático medio) para la predicción de los seis parámetros de ABangle promediaron más de 50 ejecuciones. El número de árboles por cada modelo de bosque aleatorio se ajustó manualmente para maximizar Q2. Los valores entre paréntesis especifican la desviación estándar.

El modelo de bosque aleatorio se entrenó una vez en el conjunto de datos completo de estructuras apo y complejas (tabla 5, columna central) y una vez solo en las estructuras complejas (tabla 5, columna derecha). Aunque el conjunto de entrenamiento se redujo en casi 550 estructuras, los valores de Q2 y RMSE mejoraron cuando solo se usaron las estructuras complejas. Para HL, LC2 y dc, el valor de Q2 es de aproximadamente 0,68, mientras que HC1, LC1 y LC2 tienen valores de Q2 de 0,75 y superiores (cuando se consideran las estructuras complejas).
De forma alternativa, para asegurarse de incluir la máxima diversidad de huellas de orientación diferentes en el conjunto de entrenamiento, se puede usar CD-HIT para agrupar las huellas de orientación al 100 % de identidad y, para cada grupo, se puede agregar al menos un representante al conjunto de entrenamiento, hasta que % de las estructuras disponibles se asignen al conjunto de entrenamiento. El 14 restante se puede usar para los ensayos. Debido al hecho de que el conjunto de prueba consistiría, a continuación, en huellas de orientación que también se incluyen en el conjunto de entrenamiento, los valores de Q2 resultantes, por ejemplo que oscilan entre 0,71 y 0,88 para el conjunto de datos actual dependiendo del parámetro de ABangle respectivo, exagerarían las capacidades reales del predictor cuando se enfrenta a una huella de orientación desconocida. En ese caso, se podrían encontrar valores de Q2 en el intervalo de 0,54 a 0,73, aproximadamente, para el conjunto de datos actual.
La figura 2 muestra gráficos de regresión ejemplares para los parámetros de ABangle predichos frente a los reales en el conjunto de datos formado solo por estructuras complejas.
La correlación mejora en comparación, por ejemplo, con la informada por Abhinandan y Martin (4). Sin quedar vinculado a esta teoría, la mejora se puede atribuir a una descripción más ajustada de los grados de libertad de la orientación VH-VL en términos de los seis parámetros de ABangle y al uso del esquema de numeración de Wolfguy que reduce o incluso evita ambigüedades en la numeración de residuos de HVR.
La clasificación de importancia de las posiciones de las huellas como descriptores de los diferentes parámetros de ABangle se representa en la figura 3.
Basado en el hallazgo de la clasificación de importancia de la posición de la huella, se ha descubierto que cada parámetro de ABangle está influenciado por un conjunto muy diferente de posiciones de interfase tanto en VH como en VL. Para todos los parámetros excepto HC2, una posición de región estructural fue el descriptor más importante. No obstante, en cada caso, al menos dos residuos de HVR-H3 estaban entre los descriptores más importantes. Las posiciones que se han clasificado entre las diez variables de entrada más importantes en la publicación original de ABangle (7) también se monitorizaron en la clasificación que se presenta en el presente documento. Pero, mientras que Dunbar et al. (7) descubren que HC1 está determinado exclusivamente por residuos de la cadena pesada, y LC1 está determinado exclusivamente por residuos de la cadena ligera, los diez descriptores superiores determinados con un procedimiento como se informa en el presente documento para HC1 y LC1 implican posiciones de huellas en ambas cadenas. En el presente documento, las posiciones de las huellas se clasifican independientemente de la especificidad de los aminoácidos.
Las 25 posiciones de huellas superiores también contienen varios miembros de los conjuntos de posiciones determinantes de la orientación VH-VL identificadas por Chailyan et al. (5) (L41, L42, L43, L44) y por Abhinandan y Martin (4) (L41, L44, L46, L87, H33, H45, H60, H62, H91, H105). Se ha descubierto que L87 es el descriptor superior para HL, L46 para HC1, H45 para LC1, H62 para HC2 y L44 para LC2.
Modelización de homología de anticuerpos con reorientación VH-VL
Modelos MoFvAb
Bujotzek, A. et al. (mAbs 7 (2015) 838-852) han publicado una descripción detallada del procedimiento de MoFvAb (modelización del Fv para anticuerpo). Los resultados obtenidos para la construcción de modelos Variante 1 (modelos ensamblados a partir de estructuras molde alineadas en la región estructural consenso de VH o VL, seguido de la reorientación VH-VL en una región estructural consenso de Fv) se muestran en la tabla 6.
Tabla 6: Modelos AMAII construidos con Variante 1 de MoFvAb. Los valores indican la RMSD de carbonilo para los fragmentos como se define por Teplyakov et al. (7) después de la alineación en cadena en el núcleo de lámina p.


Para tener en cuenta el desplazamiento de carbonilo causado por desviaciones en la orientación VH-VL, los mismos modelos se alinearon en el núcleo de lámina p del Fv completo (VH y VL simultáneamente) y se volvieron a calcular los valores. Los resultados se muestran en la tabla 7.
Tabla 7: Modelos AMAII construidos con Variante 1 de MoFvAb. Los valores indican la RMSD de carbonilo para los fragmentos como se define por Teplyakov et al. (7) después de la alineación en el núcleo de lámina p del Fv completo. Las tres columnas más a la derecha especifican la RMSd de carbonilo para la región estructural (FW), las HVR (CDR) y todos los residuos de Fv (Todos) en base a la definición del fragmento de Wolfguy y la definición de CDR de Kabat.

La comparación entre la tabla 6 y la tabla 7 revela cómo los valores de RMSD se deterioran en el momento en que se considera la estructura de Fv completa. La RMSD de carbonilo media para el núcleo de lámina p aumentó de 0,37 A a 0,57 A para VL, y de 0,47 A a 0,69 A para VH. Esta tendencia no se limitó a la región estructural, sino que se extendió a las HVR. La RMSD de carbonilo media para HVR-L3, por ejemplo, aumentó de 1,02 A a 1,45 A, y de 3,20 A a 3,32 A para HVR-H3. La desviación en la orientación VH-VL al observar directamente los seis parámetros de ABangle y las diferencias con respecto a las estructuras de referencia se muestra en la tabla 8.
Tabla 8: Desviación en la orientación VH-VL con respecto a la estructura de referencia en términos de los seis parámetros de ABangle para los modelos AMAII construidos con Variante 1 de MoFvAb.


Los modelos enumerados en la tabla 8 se han reorientado en la misma región estructural consenso de Fv y, por tanto, comparten esencialmente la misma orientación de ABangle de aproximadamente Qcons := (-59,45, 71,65, 120,49, 117,46, 82,77, 16,11). Se muestra una gran diversidad en la orientación VH-VL que era inherente a las estructuras AMAII. Las mayores desviaciones se produjeron para los parámetros HL y HC2, no solo entre diferentes estructuras, sino también para estructuras de secuencia idéntica de la misma unidad asimétrica: el parámetro HL para 4MA3_B_A y 4MA3_H_L se desvía 5,87 grados. Esto confirma que la orientación VH-VL, aunque está guiada por determinados rasgos característicos de la secuencia (véase la figura 3), también está sujeta a una variabilidad intrínseca, no dirigida. Esto es especialmente pronunciado para los anticuerpos de unión a proteína en la forma libre (7).
Todos los modelos se reconstruyeron con la Variante 2 de construcción de modelos (modelos ensamblados a partir de estructuras molde alineadas en la región estructural consenso de VH o VL, seguido de reorientación VH-VL en una estructura molde de orientación VH-VL elegida en base a la similitud con los parámetros de ABangle predichos) usando la misma elección de estructuras molde. Los resultados se muestran en la tabla 9 (los valores se refieren a pares modelo-referencia alineados en el núcleo de lámina p del Fv completo).
Tabla 9: Modelos AMAII construidos con Variante 2 de MoFvAb.

Los valores medios de RMSD de carbonilo por fragmento calculados para los modelos de la Variante 2 con orientación VH-VL optimizada mostraron una mejora de aproximadamente 0,05 A en comparación con los modelos que usan una orientación VH-VL genérica (véase la tabla 7). Las desviaciones de ABangle revelaron que los modelos de la Variante 2 se han acercado más a la orientación VH-VL real de las estructuras de referencia (véase la tabla 10).
Tabla 10: Desviación en la orientación VH-VL con respecto a la estructura de referencia en términos de los seis parámetros de ABangle para los modelos AMAII construidos con Variante 2 de MoFvAb. La estructura molde de orientación VH-VL seleccionada en base a los parámetros de ABangle predichos se da en la columna más a la derecha.

La distABangle media mejoró de 5,53 para los modelos de orientación genérica a 4,78 para las versiones con orientación optimizada. En la columna más a la derecha de la tabla se muestran los moldes de orientación VH-VL elegidos en base a la distancia ponderada distABangle a los parámetros de ABangle predichos. Los moldes de orientación VH-VL no se seleccionaron en base a la similitud de las huellas, sino por la similitud en el espacio de orientación de ABangle.
Todos los modelos se reconstruyeron con la Variante 3 de construcción de modelos, usando estructuras molde alineadas en una región estructural consenso de Fv común en lugar de una estructura consenso por cadena, y la orientación VH-VL no se ajustó de ninguna forma. Debido al hecho de que todas las estructuras molde se alinearon por Fv, la RMSD de carbonilo en cadena (véase la tabla 6) aumentó de 0,37 A a 0,43 A para VL y de 0,47 A a 0,55 A para VH (datos no mostrados). Los valores de RMSD de carbonilo para los pares modelo-referencia alineados en el Fv completo se enumeran en la tabla 11.
Tabla 11: Modelos AMAII construidos con Variante 3 de MoFvAb.


Los resultados de la Variante 3 no fueron tan buenos como los de las otras dos variantes. A pesar del hecho de que en la Variante 3 se mezclaron fragmentos molde de estructuras de Fv con orientación VH-VL completamente no relacionada, no parecía existir un efecto particularmente dañino sobre la calidad del modelo. Las desviaciones de ABangle correspondientes se muestran en la tabla 12.
Tabla 12: Desviación en la orientación VH-VL con respecto a la estructura de referencia en términos de los seis parámetros de ABangle para los modelos AMAII construidos con Variante 3 de MoFvAb.

La distABangle media de los modelos de la Variante 3 no fue tan buena como para los modelos optimizados de orientación VH-VL, pero fue mejor que para los modelos con la orientación de Fv consenso producida por la Variante 1. Sin quedar vinculado a esta teoría, los fragmentos molde extraídos de estructuras alineadas en una región estructural consenso de Fv común codifican alguna información de orientación VH-VL que de otro modo se perdería.
Todos los datos mostrados anteriormente se refieren a modelos minimizados en presencia de restricciones de posición en todos los residuos, excepto aquellos que fueron remodelizados o situados en los extremos de los fragmentos con residuos contiguos que se originan en diferentes estructuras molde. Por tanto, se conservó un máximo de información de orientación VH-VL conferida por las estructuras molde y/o la reorientación VH-VL.
A modo de comparación, se repitió el proceso de construcción del modelo y todos los modelos se minimizaron usando la misma combinación de campo de fuerza y modelo de agua implícita (CHARMm y GBSW) mientras se omitían las restricciones de posición. Para simplificar, solo se resume el cambio promedio en RMSD de carbonilo y distABangle cuando se cambia de minimización restringida a minimización completamente flexible (véase la figura 4). Para los modelos construidos con Variante 1 y 2 de MoFvAb, la RMSD de carbonilo media con respecto a las estructuras de referencia se vuelve ligeramente mayor. Los modelos de Variante 3, debido a la configuración de su estructura molde, probablemente los más afectados por las imprecisiones estéricas, se benefician de la minimización de energía sin restricciones por un pequeño margen. Las tres variantes de modelo mejoran en términos de distABangle con respecto a las estructuras de referencia. Sin quedar vinculado a esta teoría, parece que la minimización de energía sin
restricciones induce una igualación de la calidad del modelo con respecto a las diferentes variantes de construcción del modelo (combinación de campo de fuerza/modelo de agua implícita).
Modelos AMAII originales
El enfoque de mejorar un modelo de homología de Fv dado reorientándolo en un molde de orientación VH-VL como se informa en el presente documento se integró en el programa informático de modelización de última generación actual. Los modelos AMAII originales se obtuvieron de http://www.3dabmod.com, las estructuras se anotaron con la numeración de Wolfguy para facilitar la integración, y los modelos se reorientaron en los moldes de orientación VH-VL como se enumeran en la tabla 8. En la figura 5 se muestra el cambio medio en la RMSD de carbonilo y la distABangle por anticuerpo después de la reorientación, promediado sobre los modelos respectivos de todos los participantes de AMAII.
Después de la reorientación en cadena en una estructura de Fv diferente (en particular sin ningún procesamiento posterior) para ocho de los once conjuntos de modelos de anticuerpos (constituidos por todas las estructuras enviadas por acc, ccg, jef, joa, mmt, pig y sch para el Ab02 correspondiente), la RMSD de carbonilo para la cadena principal de Fv completa se mejora mediante la reorientación VH-Vl . Además, los conjuntos de modelos Ab01, Ab10 y Ab11 mejoran en la orientación VH-VL y la RMSD estructural.
Finalmente, el conjunto de modelos se dividió para evaluar en qué medida el procedimiento de reorientación VH-VL, como se informa en el presente documento, concuerda con los modelos de anticuerpos construidos con diferentes enfoques. En la figura 6 se muestra el cambio medio en la RMSD de carbonilo y la distABangle después de la reorientación, promediado sobre todos los modelos del respectivo participante de AMAII.
Se mejoró la distABangle media con respecto a la referencia de estructuras para los modelos de todos los participantes. La reducción de distABangle por reorientación VH-VL se tradujo en mejores valores de RMSD en cinco de los siete casos, especialmente con respecto a las regiones estructurales.
Como promedio, se descubrieron mejoras notables de distABangle y pequeñas mejoras de la RMSD de carbonilo para todo el Fv.
Por tanto, como se informa en el presente documento, el concepto de predicción de orientación VH-VL basada en rasgos característicos de secuencia se puede ampliar pasando de un solo ángulo de empaquetamiento de VH-VL a una descripción más ajustada de la orientación v H-VL en términos de los seis parámetros de ABangle definidos por Dunbar et al. (7).
Para cada parámetro de ABangle, se entrenó un modelo de bosque aleatorio en un conjunto actualizado de estructuras de Fv conocidas. Los valores de Q2 para los seis predictores oscilan entre 0,67 y 0,80 cuando se entrenan en un conjunto que consiste en solo estructuras complejas.
Un análisis de los descriptores superiores de los modelos de bosque aleatorio reveló una serie de residuos de HVR-H3 que no se conocían como tales anteriormente. El esquema de numeración de anticuerpos "Wolfguy" informado en el presente documento contribuyó a la identificación de estos residuos, ya que está diseñado de modo que los residuos estructuralmente equivalentes se numeran con índices equivalentes en la medida de lo posible, también (y en particular) en las regiones hipervariables.
Se compararon dos variantes de construcción de modelos sin ajuste y predicción de orientación VH-VL (Variantes 1 y 3) con una variante de construcción de modelos que predice la orientación VH-VL más probable en términos de los seis parámetros de ABangle, busca automáticamente la estructura de Fv orientada con mayor similitud en una base de datos de moldes de anticuerpos, y reorienta el modelo sin procesar en este molde de orientación VH-VL antes de su procesamiento adicional (Variante 2).
Se esperan efectos de sinergia con respecto a la modelización de bucles HVR-H3 debido a una mejorada orientación previa de VH y VL. Además, el coste computacional de optimizar la orientación VH-VL basada en un predictor basado en secuencia y la reorientación posterior en una estructura molde es insignificante (por ejemplo, cuando se compara con el trabajo sintético).
La invención actual
Se ha descubierto que la diferencia total en la orientación VH-VL entre dos variantes de anticuerpos (humanizados) que se unen al mismo epítopo de un antígeno en relación con su anticuerpo no humano original se correlaciona con la diferencia respectiva en la capacidad de unión a antígeno de los anticuerpos.
La orientación VH-VL se predice en el presente documento a partir de un subconjunto (significativo) de posiciones de secuencia de Fv (una "huella de secuencia") en lugar de a partir de secuencias de Fv completas. En base al supuesto de que la orientación VH-VL se rige por residuos en o cerca de la interfase VH-VL, se ha identificado un conjunto de
residuos de interfase en el que un residuo se define como parte de la interfase VH-VL si sus átomos de cadena lateral son átomos vecinos de la cadena opuesta con una distancia menor o igual a 4 A en al menos el 90 % de todas las estructuras de Fv superpuestas en la base de datos, por ejemplo, en RAB3D. Los resultados se resumen en la tabla 29, que también establece si una posición de secuencia se ha relacionado previamente con ser un determinante de la orientación VH-VL en base a análisis estadísticos (4, 5, 7).
Tabla 29: Residuos de interfase VH-VL donde un residuo es parte de la interfase si sus átomos de cadena lateral son átomos vecinos de la cadena opuesta con una distancia menor o igual a 4 A en al menos el 90 % de todas las estructuras de Fv superpuestas en RAB3D.
*Numeración dependiendo de la longitud del bucle
+Parte de la interfase VH-VL como se define por Chothia et al. (13)


Al conjunto anterior de residuos de interfase le faltan algunas de las posiciones de secuencia que se habían enumerado entre las "10 variables de entrada importantes superiores" para la orientación VH-VL por Dunbar et al. (7). Esas posiciones de secuencia se enumeran en la siguiente tabla 30.
Tabla 30: Posiciones de secuencia adicionales enumeradas entre las "10 variables de entrada importantes superiores" para la orientación VH-VL por Dunbar et al. (7).
*Numeración dependiendo de la longitud del bucle
+Parte de la interfase VH-VL como se define por Chothia et al. (13)

Para seleccionar una variante de anticuerpo (humanizada) apropiada de un anticuerpo original, la orientación VH-VL se describe en términos de los seis parámetros de orientación de "ABangle", que consisten en un ángulo de torsión, cuatro ángulos de curvatura (dos por cada dominio variable), así como la longitud del eje de pivote de VH y VL.
Se ha descubierto que la orientación relativa entre los dominios VH y VL (orientación VH-VL) se puede usar para (pre)seleccionar el anticuerpo o anticuerpos variantes con la mejor afinidad de unión. Esto es aplicable dentro de un grupo de anticuerpos humanizados, así como entre un grupo de anticuerpos humanizados y el anticuerpo no humano original.
Además, se ha descubierto que cada vez que un residuo de región estructural o una región estructural completa tiene que (inter)cambiarse, la unión de la nueva variante a su antígeno se puede evaluar en base al procedimiento que se informa en el presente documento.
La invención se ejemplifica a continuación con anticuerpos específicos que se pretende que sirvan como ejemplo y que no se deben interpretar como limitantes del alcance de la invención. El procedimiento como se informa en el presente documento es un procedimiento de aplicación en general.
La huella de secuencia consiste en 54 aminoácidos, 29 en la región de VH y 25 en la región de VL. Véase la siguiente tabla 13.
Tabla 13: Huella de secuencia.


*Numeración dependiendo de la longitud del bucle
+Parte de la interfase VH-VL como se define por Chothia et al. (13)
En el primer ejemplo se evalúan dos anticuerpos murinos, CD81K04 y CD81K13 que se unen al bucle extracelular grande (LEL) del dominio extracelular del receptor CD81 (ECD) y variantes humanizadas de los mismos de acuerdo con los procedimientos como se informa en el presente documento.
En el segundo ejemplo se evalúan un anticuerpo de conejo que reconoce un segmento peptídico de la proteína pTau (que incluye la fosforilación de S422) y sus variantes humanizadas de acuerdo con los procedimientos como se informa en el presente documento.
En el tercer ejemplo se evalúan un anticuerpo antihepsina y sus variantes humanizadas de acuerdo con los procedimientos como se informa en el presente documento.
En las alineaciones de secuencia, las HVR están marcadas con un fondo gris. La definición de HVR usada corresponde a la unión de conjuntos de la definición de CDR de Kabat y Chothia. Las posiciones de secuencia que forman parte de la huella de secuencia usada para predecir la orientación VH-VL están marcadas con un fondo negro. Las posiciones de huellas que no están pobladas en un anticuerpo dado están marcadas con una 'X'.
Secuencias de VH originales de CD81K04, CD81K13 (murino) y Rb86:

FR2
w|k|rpg^ H ig
w0 k@rPAMrtwarci:g
wjRBAPGf^ffllG


Secuencias de VL originales de CD81K04, CD81K13 (murino) y Rb86:


FR3
GVPARFSGSGSGTDFTLN1HPVEEEDAA0 Y0C GVPSRFSGSGSGTQYYLKINSLQSEDFGHYfflC
GVPSRFSASGSGTQFTLTISDVQCDDAAjJygc

Variantes de humanización de VH CD81K04 (la secuencia murina original se muestra en la parte superior):
FR3
KAT LTADKSS STAYMQLSS LTSEDSA CAR KATITADE STSTAYMELSSLRSEDTA CAR KATITADE STSTAYMELSSLRS EDT CAR RATITADE STSTAYMELSSLRS EDT cAR
RVTITADE STSTAYMELSSLRS EDT CAR RVTITADE STSTAYMELSSLRS EDT cAR
RVTITADE STSTAYMELSSLRSEDT CAR RVTITADE STSTAYMELSSLRS EDTA CAR
qv tisad ksis taylq lsslk asdta car
QVTISADKSISTAYLQLS SLKASDTa AR
RVTMTTDTSTSTAYMELRSLRSDDTA AR RATITADTSASTAYMELS SLRS EDT CAR RVTITADTSASTAYMELSSLRSEDTAf CAR
RVTITADTSASTAYMELS SLRSEDT CAR
rv titrd tsas tayme lsslr sedta ca r
RVTITADTSASTAYMELS S LRS EDT AR
AR

Variantes de humanización de VL CD81K04 (la secuencia murina original se muestra en la parte superior):


FR3
GVPARFSGSGSGTDFTLN1HPVEEED.
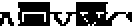
GVPDRFSGSGSGTDFTLTISSLQAEDVA0Y0C GVPDRFSGSGSGTDFTLTISSLQAEDVA0YHC gvpdrfsgsgsgtdftltisslqaedvaByHc GVPDRFSGSGSGTDFTLTISSLQAEDVAHYgC GVPDRFSGSGSGTDFTLTISSLQAEDVaEyBc GVPDRFSGSGSGTDFTLTISSLQAEDV.
giparfsgsgsgtdftltisslepedfaKy Y gvpsrfsgsgsgtdftltisslqpedfaHy Y gvparfsgsgsgtdftltinpveandaaSy Y

Variantes de humanización de VH CD81K13 (la secuencia murina original se muestra en la parte superior):


FR3 _
KatittDIPSMTAYLHLSNLTSEDTaHyJcVP
RVTITADTSTDTAYMELSS LRS EDTAgY@CAP
RVTITTDTSASTAYMELS S LRS EDTaByHCAP
RVTITADESTSTAYMELS S LRS EDTAHYWCAP RVTITADESTSTAYMELS SLR SEDTAHYHCVP RVTITTDESTSTAYMELS S LRS EDTAgYgCVP
RVTITTDESTSTAYMELS S LRSEDTA0Y0CVP
RVTITTDESTSTAYMELS S LRS EDTA0Y0CVP
RVTITTDTSASTAYMELS S LRS EDTA0Y@CAP
RVTITTDTSASTAYMELS S LRSEDTA0Y0CAP

Variantes de humanización de VL CD81K13 (la secuencia murina original se muestra en la parte superior):


FR3
GVPSRFSGSGSGTQYYLKINSLQSEDFG0Y¡jjC GIPARFSGSGSGTEFTLTISSLQSEDFGEy0C GIPARFSGSGSGTEFTLTISSLQSEDFA0Y0C GIPARFSGSGSGTEFTLTISSLQSEDFG^ySc GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFG0Y0C GVPSRFSGSGSGTDFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTDFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTDFTLTISSLQPEDFA|¡Y0C gvpsrfsgsgsgtdftltisslqpedfgByHc GVPSRFSGSGSGTDFTLTISSLQPEDVG¡YHC
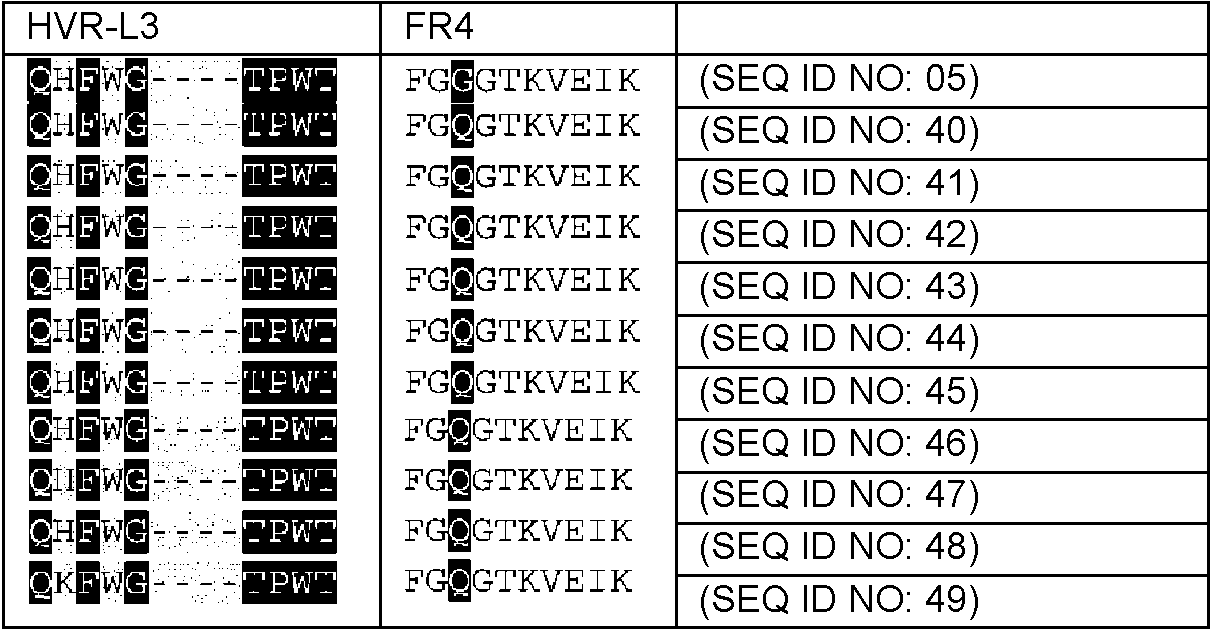
Variantes de humanización de VH Rb86 (la secuencia de conejo original se muestra en la parte superior):


FR3
RFTISKAS--TTVDLKMTSPTAEDTG
RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA
RFTISRDS--TTLYLQMNSLRAEDTA
RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RVTMTTDTSTSTAYMELRSLRSDDTA RVTMTKAS--STAYMELRSLRSDDTA
RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA RFTISRDNSKNTLYLQMNSLRAEDTA

Variantes de humanización de VL Rb86 (la secuencia de conejo original se muestra en la parte superior):

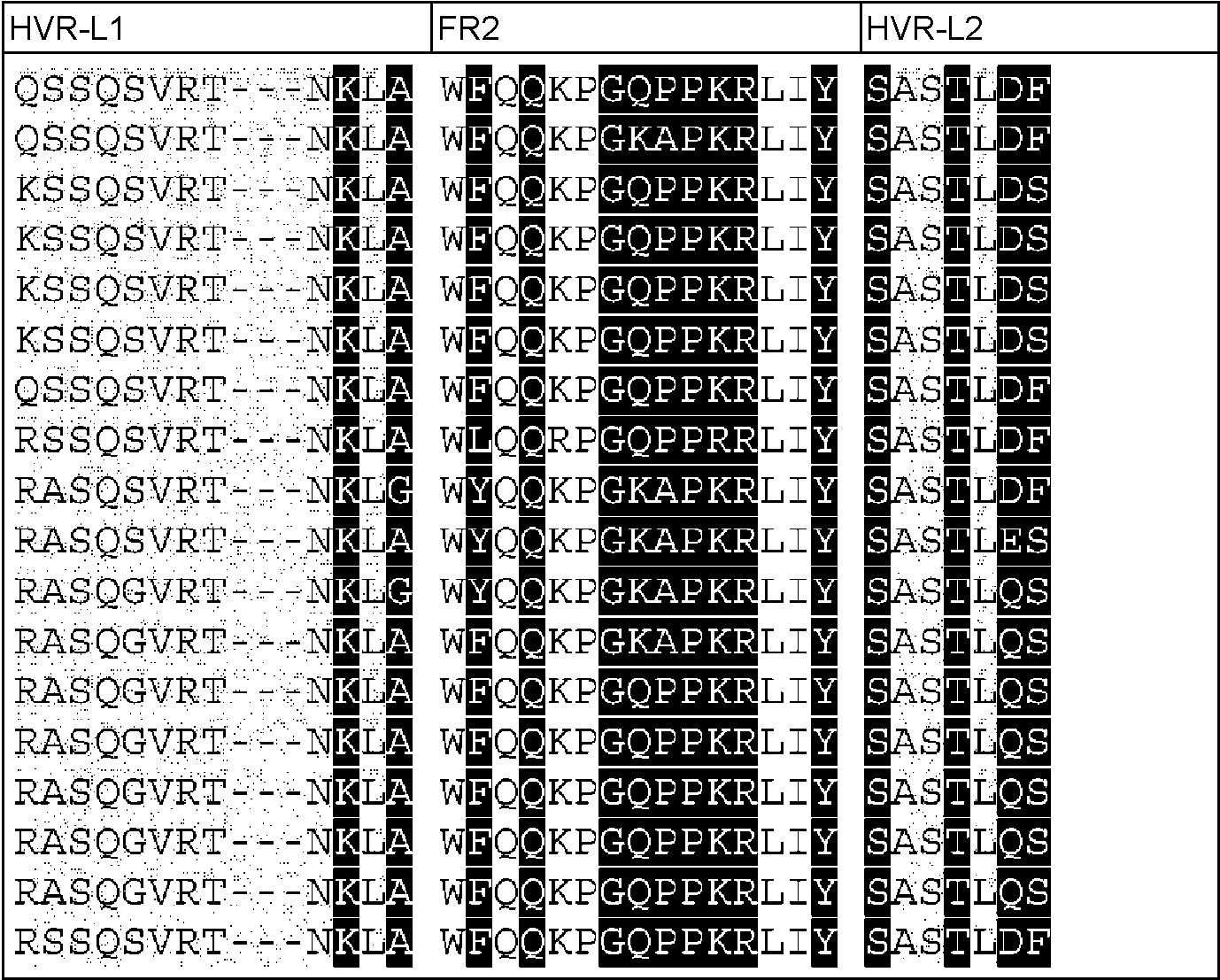
FR3
GVPSRFSASGSGTQFTLTISDVQCDDAAgYgC GVPSRFSGSGSGTEFTLTISSLQPDDFA0Y0C GVPDRFSGSGSGTDFTLTISSLQAEDVA0Y0C GVPDRFSGSGSGTDFTLTISSLQAEDVA0Y0C GVPDRFSGSGSGTDFTLTISSLQAEDVA0Y0C GVPDRFSGSGSGTDFTLTISSLQAEDVA0Y0C GVPARFSGSGSGTDFTLTINPVEANDTA¡J¡Y0C gvpdrfsgsgagtdftlkisrveaedvgEy^c GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPDDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQSEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C GVPSRFSGSGSGTEFTLTISSLQPEDFA0Y0C

Las variantes de secuencia se han diseñado usando el principio general de injerto. El injerto, en general, se desarrolló para producir anticuerpos humanizados. Además, el injerto también se puede usar para obtener anticuerpos compatibles con otras especies, o simplemente para intercambiar la región estructural de un anticuerpo para obtener otras propiedades biofísicas para este anticuerpo o fragmento de anticuerpo.
Después de la clonación de las regiones variables humanizadas en el homólogo de la región constante humana, los anticuerpos se expresan en una "matriz" combinando todos los plásmidos de cadena pesada con todos los plásmidos de cadena ligera. La primera fila y la primera columna son, a continuación, anticuerpos semihumanizados, mientras que la primera célula es el anticuerpo original murino o de conejo en su forma quimérica, y el resto de la matriz son anticuerpos completamente humanizados.
Para los anticuerpos anti-CD81 CD81K04 y CD81K13, los datos de unión son datos de ELISA bioquímicos (unión celular) como se resumen en la tabla 14 y la tabla 15 a continuación, respectivamente.
Tabla 14: Datos de ELISA de matriz de humanización CD81K04. El anticuerpo de referencia CD81K04 se enumera en la columna más a la izquierda de la fila superior.


La forma quimérica de CD81K04 está cerca del valor de 1,15 y las variantes humanizadas se unen de manera ligeramente menos eficaz. Para algunas de las variantes, la afinidad cae más drásticamente.
Tabla 15: Datos de ELISA de matriz de humanización CD81K13. El anticuerpo de referencia CD81K13 se enumera en la columna más a la izquierda de la fila superior.


Para el anticuerpo de conejo Rb86, se analizó el material micropurificado de los sobrenadantes en el primer cribado para determinar su capacidad para tener parámetros de asociación y disociación que no se desvíen demasiado de los del anticuerpo original. Las RU (unidad de respuesta en experimentos SPR/BIAcore al final de la fase de asociación) de unión tardía (BL) se compilan para cada variante y para el anticuerpo de conejo de referencia, así como la constante de disociación kd [1/s] que se puede traducir en semivida del anticuerpo en su diana (t1/2 = ln2/kd), véanse la tabla 16 y la tabla 17, respectivamente. Para algunas variantes que se asociaron muy deficientemente (RU en la fase de asociación cercanas a cero o negativas), tampoco existe una manera de determinar un valor de kd; el valor de la semivida se establece en 0.
Tabla 16: Valores de BL de SPR/BIAcore de matriz de humanización de Rb86. El anticuerpo de referencia Rb86 se enumera en la columna más a la izquierda de la fila superior.


Tabla 17: Valores de semivida (t1/2) de SPR/BIAcore de matriz de humanización de Rb86. El anticuerpo de referencia Rb86 se enumera en la columna más a la izquierda de la fila superior.

Las distancias de ABangle predichas de las variantes de humanización con respecto al anticuerpo de referencia se enumeran en la tabla 19 (c D81 K04), tabla 20 (CD81K13) y tabla 21 (Rb86), de manera consecuente con el orden de los datos de ELISA y SPR/BIAcore indicados anteriormente (tabla 14 a 17).
Tabla 19: Distancias de ABangle de matriz de humanización de CD81K04 con respecto al anticuerpo de referencia CD81K04, enumerado en la columna más a la izquierda de la fila superior.

Tabla 20: Distancias de ABangle de matriz de humanización de CD81K13 con respecto al anticuerpo de referencia CD81K13, enumerado en la columna más a la izquierda de la fila superior.

Tabla 21: Distancias de ABangle de matriz de humanización de Rb86 con respecto al anticuerpo de referencia Rb86, enumerado en la columna más a la izquierda de la fila superior.


Tabla 22: Distancias de ABangle no ponderadas de matriz de humanización de anticuerpo antihepsina con respecto al anticuerpo de referencia Hepsina 35, enumerado en la columna más a la izquierda de la fila superior.


Correlación entre distancia de ABangle y unión
Las matrices se pueden correlacionar usando el coeficiente RV u otros coeficientes tales como el coeficiente de correlación del procedimiento PROTEST. Estos procedimientos evalúan esencialmente la correlación entre dos conjuntos de datos, donde se dispone de no una sino varias mediciones para cada muestra y, por lo tanto, son, hasta cierto punto, extensiones del coeficiente de correlación univariante estándar. El coeficiente RV se usa a continuación. En la tabla 23 se muestra el coeficiente RV y sus valores de p para los tres conjuntos de datos diferentes desde la perspectiva de los HC y los LC.
Tabla 23: Coeficientes RV y valores de p correspondientes para los cuatro conjuntos de datos. El coeficiente RV se calcula desde la perspectiva de los HC como muestras y, por tanto, los LC como mediciones multivariante y viceversa. Los valores de p se calculan por medio de una prueba de permutación e indican la probabilidad de alcanzar un coeficiente RV tan alto como o superior al calculado.

Una vista menos restringida del conjunto de datos sería ver cada mAh como un individuo. En ese caso, tiene sentido vectorizar ambas matrices y calcular la correlación de Pearson. La tabla 24 muestra el coeficiente de correlación y el valor de p correspondiente para los diferentes conjuntos de datos.
Tabla 24: Coeficiente de correlación de Pearson para los tres conjuntos de datos y valor de p correspondiente. La correlación se calcula en versiones vectorizadas de las matrices de unión y distancia angular. El valor de p indica la probabilidad de alcanzar la correlación calculada bajo la hipótesis nula de no tener correlación.

Todos los conjuntos de datos muestran una correlación. Por tanto, se ha descubierto que se pueden usar procedimientos que rechazan anticuerpos individuales en base únicamente a la distancia angular para seleccionar variantes de anticuerpos humanizados.
Rechazo de subconjuntos de anticuerpos
A continuación se analizan tres procedimientos para la selección de subconjuntos de anticuerpos en cuanto a su rendimiento. Estos procedimientos son la selección de los 20 % peores, la selección de malas combinaciones HC/LC y la selección de HC o LC completos como se describe en el presente documento.
Para evaluar el rendimiento de los diferentes procedimientos de selección se calcularon diferentes estadísticas. La primera es la proporción de la mediana de la longitud de unión entre los subconjuntos conservados y rechazados. Para esta proporción también se calculó un valor de p usando una prueba de permutación. Además, se inspeccionó visualmente la distribución de longitudes de unión o para valores de IC50 de un conjunto de datos en los dos subconjuntos usando histogramas apilados.
CD81K04
Para ambos procedimientos que usan información de HC/LC, se deben elegir los HC/LC que se espera que tengan un rendimiento peor que el resto. La figura 7 representa la distancia angular promedio para los HC (filas de la matriz, izquierda) y los LC (columnas de la matriz, derecha). Se deseleccionaron los anticuerpos que comprenden los HC 6, 7, 8, 9, 12, 13 y 14 y los LC 7 y 9.
En la figura 8 se muestra qué anticuerpos se eliminan mediante el procedimiento de selección respectivo (sombreado) y cuáles se conservan.
La tabla 25 muestra los resultados de la comparación de los subconjuntos de anticuerpos en cuanto a su longitud de unión. Se calculó la mediana de la longitud de unión en ambos conjuntos y se determinó la proporción de ambos. En la tabla se muestran los resultados de los tres procedimientos conjuntamente con el valor de p, lo que indica la probabilidad de obtener una proporción al menos tan baja.
,
de la medición de ELISA entre los anticuerpos rechazados y conservados. Adicionalmente, se calculó un valor de p por medio de un procedimiento de permutación, que muestra la probabilidad de alcanzar un valor tan bajo como o más bajo que la mediana de la proporción encontrada.

Independientemente del procedimiento, el subconjunto de anticuerpos conservados tiene una longitud de unión de 3 a 5 veces mayor que la de los anticuerpos deseleccionados. Además, estos resultados son significativos (p <0,05). En la figura 9 se muestran los histogramas apilados de las mediciones de ELISA para los tres procedimientos de selección. Los histogramas confirman los resultados de la mediana de la proporción. Los tres procedimientos rechazan los anticuerpos monoclonales de baja unión.
CD81K13
Se realizó el mismo enfoque explicado anteriormente para las variantes humanizadas del anticuerpo CD81K13. Los resultados se muestran en las figuras 10 a 12 y en la tabla 26 siguiente.
Se deseleccionaron los anticuerpos que comprenden los HC 3, 4 y 9 y los LC 2, 5, 6, 7, 10 y 11.
Los tres procedimientos dan lugar a un subconjunto con una longitud de unión 1,6 a 2 veces mayor que los anticuerpos del subconjunto deseleccionado (véase la tabla 26).
Tabla 26: Para los tres procedimientos diferentes de selección de anticuerpos, se calculó la proporción de la mediana de la medición de ELISA entre los anticuerpos rechazados y conservados. Adicionalmente, se calculó un valor de p por medio de un procedimiento de permutación, que muestra la probabilidad de alcanzar un valor tan bajo como o más bajo que la mediana de la proporción encontrada.

La figura 12 muestra que se rechazan predominantemente los anticuerpos con menor longitud de unión.
Rb86
Se realizó el mismo enfoque explicado anteriormente para las variantes humanizadas del anticuerpo Rb86. Los resultados se muestran en las figuras 13 a 15 y en la tabla 27 siguiente.
Se deseleccionaron los anticuerpos que comprenden los HC 9, 10 y 11 y los LC 2 y 8.
Para las variantes del anticuerpo Rb86, los datos de SPR se usan en la etapa de selección/deselección. Están disponibles dos mediciones diferentes con respecto al comportamiento de unión de los diferentes anticuerpos. En base a los datos de BL, se deseleccionaron diferentes anticuerpos (véase la figura 14).
Los tres procedimientos de selección seleccionan anticuerpos que se unen de 3 a 5,5 veces mejor como promedio (véase la tabla 27) con respecto a “BL”. El valor de p indica que este resultado no es casual, sino que se debe a la forma beneficiosa de seleccionar anticuerpos en los diferentes procedimientos.
Tabla 27: Para los tres procedimientos diferentes de rechazo de anticuerpos se calcula la proporción de la mediana de I a medición de BL entre los anticuerpos rechazados y conservados. Adicionalmente, se calcula un valor de p por medio de un procedimiento de permutación, que muestra la probabilidad de alcanzar un valor tan bajo como o más bajo que la mediana de la proporción encontrada.

Esto se subraya en la figura 15, en la que los histogramas apilados muestran que se deseleccionaron predominantemente anticuerpos con una longitud de unión baja.
Como enfoque alternativo basado en los datos de t1/2, se deseleccionaron anticuerpos (véase la figura 16). Como se puede observar, se seleccionan los mismos anticuerpos en base a los datos de BL.
La mediana de la proporción para los diferentes procedimientos de selección muestra que el subconjunto de anticuerpos deseleccionado es siempre peor que el subconjunto conservado (véase la tabla 28). Con el procedimiento de “mala combinación HC/LC”, solo se deseleccionaron unos pocos anticuerpos, pero existen varios anticuerpos sin semivida en el conjunto. Esta es la razón por la que, para este procedimiento, el valor de p no es significativo. Para los otros procedimientos, los valores de p indican que el subconjunto deseleccionado se eligió bien.
Tabla 28: Para los tres procedimientos diferentes de selección de anticuerpos, se calculó la proporción de la mediana de la medición de t1/2 entre los anticuerpos deseleccionados y seleccionados. Adicionalmente, se calculó un valor de p por medio de un procedimiento de permutación, que muestra la probabilidad de alcanzar un valor tan bajo como o más bajo que la mediana de la proporción encontrada.

Esto también se muestra en los histogramas apilados de la figura 17. Todos los procedimientos deseleccionan una buena cantidad de los muchos anticuerpos con una semivida muy baja.
Sumario
Se ha descubierto que, cuando se empleó la predicción de orientación VH-VL (ángulo VH-VL) en variantes de humanización que se derivan todas de un anticuerpo original común, las buenas variantes humanizadas respetan más los parámetros de ángulo del anticuerpo original. Los tres procedimientos usados para rechazar anticuerpos con una orientación VH-VL subóptima, es decir, rechazar los "20 % peores" o rechazar un conjunto de HC/LC completos o rechazar las malas combinaciones HC/LC, dieron como resultado subconjuntos de anticuerpos similares. Los histogramas apilados y el análisis de correlación confirmaron que la distancia angular es un buen indicador del comportamiento de unión. Se ha descubierto que, mediante el uso de un procedimiento de selección como se informa en el presente documento, la calidad de dicha matriz de humanización filtrada se puede incrementar drásticamente.
En un modo de realización de la invención, la matriz de confianza se incorpora como una etapa adicional, por ejemplo, primero se elige el subconjunto deseleccionado y. a continuación, se elige el subconjunto de alta confianza.
En un modo de realización de la invención se usa la información de distancia entre todos los anticuerpos para calcular grupos e identificar grupos de anticuerpos que están lo más alejados del grupo que incorpora la referencia.
Los siguientes son ejemplos de procedimientos y composiciones de la invención. Se entiende que se pueden poner en práctica otros modos de realización diversos, dada la descripción general proporcionada anteriormente. No se debe entender que los ejemplos limitan la invención. El verdadero alcance se establece en las reivindicaciones.
Ejemplos
Ejemplo 1
Materiales y procedimientos
Base de datos 3D de anticuerpos de Roche (RAB3D)
La base de datos de estructuras de anticuerpos RAB3D contiene principalmente estructuras de Fv disponibles públicamente. Las estructuras de Fv se procesan y anotan con el esquema de numeración interno "Wolfguy" (véase la siguiente sección). Todas las estructuras de Fv anotadas se superponen en una región estructural consenso de Fv, en una región estructural consenso de VH y en una región estructural consenso de VL. Las estructuras consenso se calculan usando un subconjunto de estructuras de alta resolución del PDB. Las estructuras de Fv anotadas y reorientadas sirven como depósito de moldes para la modelización de homología.
Esquema de numeración de Wolfguy
La numeración de Wolfguy define las regiones CDR como la unión de conjuntos de la definición de Kabat y Chothia. Además, el esquema de numeración identifica puntas de bucle de CDR en base a la longitud de CDR (y parcialmente en base a la secuencia), de modo que el índice de una posición de CDR indica si un residuo de CDR es parte del bucle ascendente o descendente. En la siguiente tabla 1 se muestra una comparación con los esquemas de numeración establecidos.
Tabla 1: Numeración de CDR-L3 y CDR-H3 usando esquemas de numeración de Chothia/Kabat (Ch-Kb), Honegger y Wolfguy. Este último tiene números crecientes desde la base N terminal hasta el pico de CDR y decrecientes comenzando a partir del extremo C terminal de CDR. Los esquemas de Kabat fijan los dos últimos residuos de CDR e introducen letras para adaptarse a la longitud de CDR. A diferencia de la nomenclatura de Kabat, la numeración de Honegger no usa letras y es común para VH y VL.

Wolfguy está diseñado de modo que los residuos estructuralmente equivalentes (es decir, los residuos que son muy similares en términos de localización espacial conservada en la estructura de Fv) se numeran con índices equivalentes en la medida de lo posible. Esto se ilustra en la figura 1.
En la siguiente tabla 2 se puede encontrar un ejemplo de una secuencia de VH y VL de longitud completa numerada según Wolfguy.
Tabla 2: Secuencia de VH (izquierda) y VL (derecha) de la estructura cristalina con PDB ID 3PP4 (21), numerada según Wolfguy, Kabat y Chothia. En la numeración de Wolfguy, CDR-H1-H3, CDR-L2 y CDR-L3 se numeran dependiendo solo de la longitud, mientras que CDR-L1 se numera dependiendo de la longitud de bucle y de la pertenencia a la agrupación canónica. Este último se determina calculando similitudes de secuencia con diferentes secuencias consenso. Aquí solo se da un solo ejemplo de numeración de CDR-L1, ya que no tiene importancia para generar la huella de secuencia de orientación VH-VL.


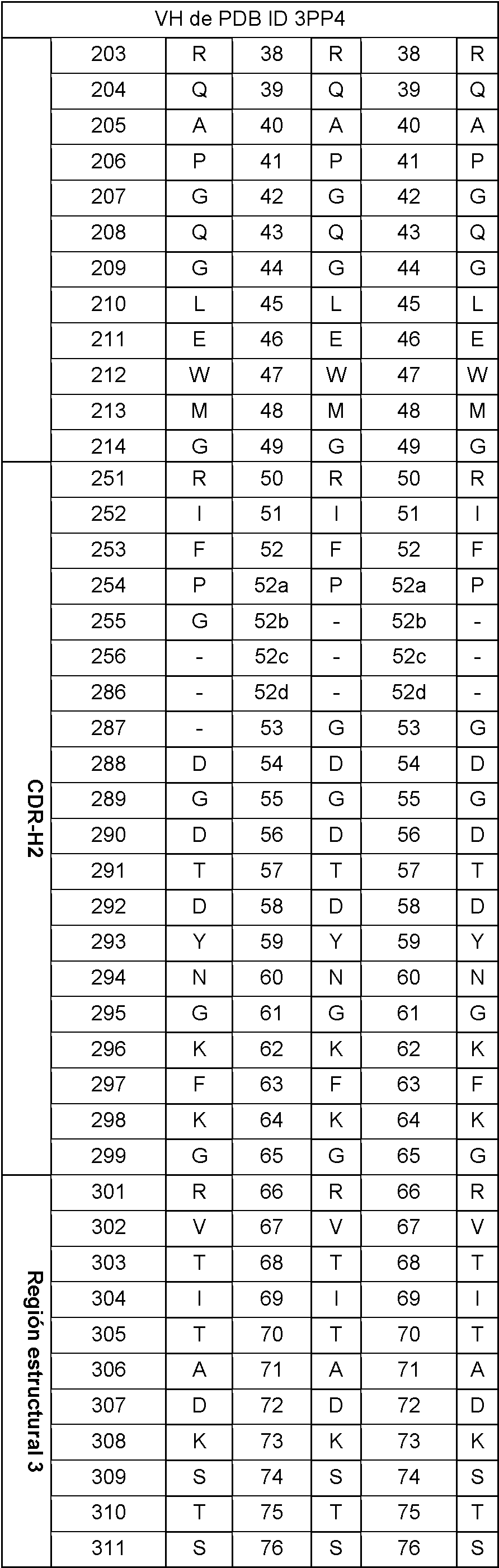
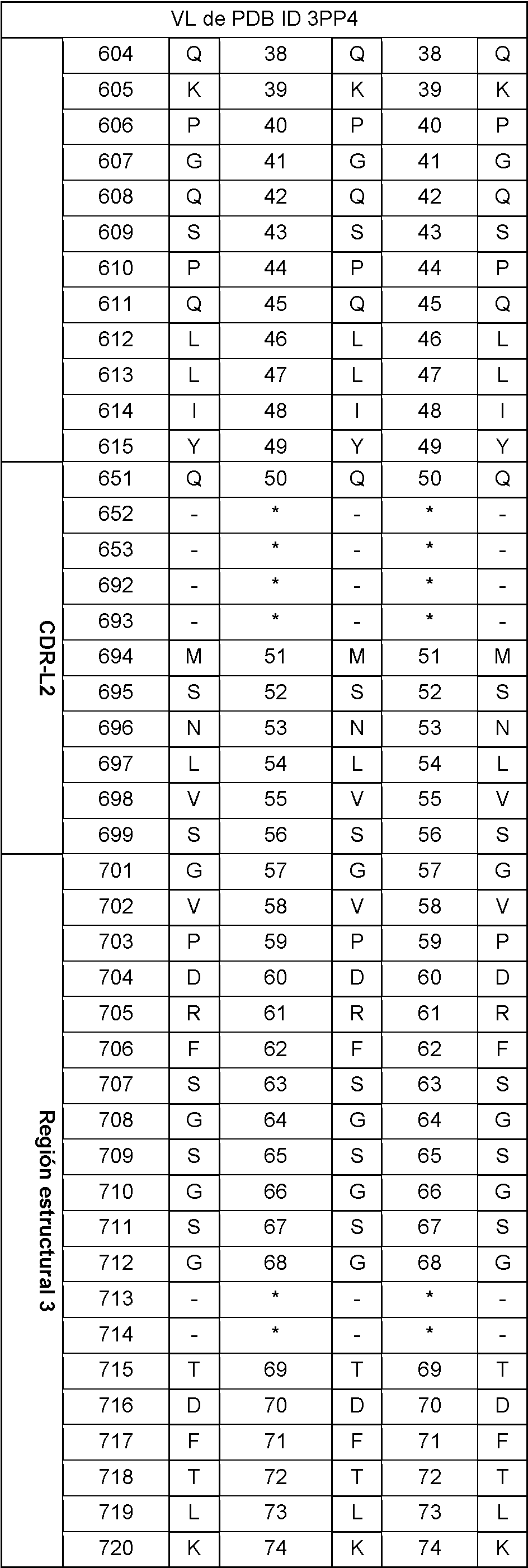



Ejemplo 1
Selección de huellas de orientación VH-VL
La orientación VH-VL se predice en el presente documento a partir de un subconjunto (significativo) de posiciones de secuencia de Fv (una "huella de secuencia") en lugar de a partir de secuencias de Fv completas. En base al supuesto de que la orientación VH-VL se rige por residuos en o cerca de la interfase VH-VL, se ha identificado un conjunto de residuos de interfase en el que un residuo se define como parte de la interfase VH-VL si sus átomos de cadena lateral son átomos vecinos de la cadena opuesta con una distancia menor o igual a 4 A en al menos el 90 % de todas las estructuras de Fv superpuestas en la base de datos, por ejemplo, en RAB3D. Los resultados se resumen en la tabla 29, que también establece si una posición de secuencia se ha relacionado previamente con ser un determinante de la orientación VH-VL en base a análisis estadísticos (4, 5, 7).
Tabla 29: Residuos de interfase VH-VL donde un residuo es parte de la interfase si sus átomos de cadena lateral son átomos vecinos de la cadena opuesta con una distancia menor o igual a 4 A en al menos el 90 % de todas las estructuras de Fv superpuestas en RAB3D.
*Numeración dependiendo de la longitud del bucle
+Parte de la interfase VH-VL como se define por Chothia et al. (13)


Al conjunto anterior de residuos de interfase le faltan algunas de las posiciones de secuencia que se habían enumerado entre las "10 variables de entrada importantes superiores" para la orientación VH-VL por Dunbar et al. (7). Esas posiciones de secuencia se enumeran en la siguiente tabla 30.
Tabla 30: Posiciones de secuencia adicionales enumeradas entre las "10 variables de entrada importantes superiores" para la orientación VH-VL por Dunbar et al. (7).
*Numeración dependiendo de la longitud del bucle
+Parte de la interfase VH-VL como se define por Chothia et al. (13)

A partir de esta colección de posiciones de secuencia potencialmente determinantes de orientación VH-VL, se reunieron tres huellas de secuencia para su evaluación estadística:
La huella 1 contiene todas las posiciones de secuencia que se ha descubierto que forman parte de la interfase VH-VL como se indica en la tabla 29, descartándose la posición 801 (L98) dado su alto grado de conservación de secuencia. La huella 2 contiene todas las posiciones de secuencia enumeradas entre las "10 variables de entrada importantes superiores" de ABangle (7), es decir, 197, 199, 208, 209, 211, 251, 292, 295, 296, 327, 355, 599, 602, 604, 607, 608, 609, 610, 611, 612, 615, 651, 696, 698, 699, 731, 733, 751, 753, 755, 796, 797, 798, 799, 803 (H33, H35, H43, H44, H46, H50, H58, H61, H62, H89, H99, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50, L53, L55, L56, L85, L87, L89, L91, L93, L94/L95x-1, L95x, L96, L97, L100) y las posiciones 289 y 290 (H55 y H56).
La huella 3 es la unión de conjuntos de la huella 1 y la huella 2.
Para evaluar hasta qué punto es posible predecir la orientación VH-VL en base únicamente a la secuencia de la región estructural, se generaron dos variantes reducidas para cada una de las tres huellas, a saber
a: con solo los residuos de CDR más externos en el borde de la región estructural, y
b: 5 sin ningún residuo de CDR.
Ejemplo 2
Entrenamiento del predictor de orientación VH-VL
Las estructuras cristalinas de Fv de anticuerpo disponibles públicamente hasta octubre de 2013 se acumularon del PDB de RCSB (www.rcsb.org) (24) y, para cada estructura, se calcularon los parámetros de orientación VH-VL de ABangle como se describe en el presente documento (véase el ejemplo 4). Además, para cada estructura, se generó la huella de secuencia de orientación VH-VL como se ilustra anteriormente (resaltado en negro en las secuencias). La huella de secuencia consiste en 54 aminoácidos, 29 en la región de VH y 25 en la región de VL. La huella de secuencia también contiene residuos que pertenecen a las regiones hipervariables que, dependiendo de la longitud del bucle, pueden no estar presentes en una secuencia de anticuerpo dada. En este caso, la posición de la huella de secuencia desocupada se indica con una 'X', en lugar de la descripción regular de aminoácidos en un código de una letra. Después de calcular los parámetros de ABangle, así como la huella de secuencia, el conjunto de datos (n = 2249) se clasificó en estructuras complejas (n_complejas = 1468) y apo (n_apo = 781).
El procedimiento de "bosque aleatorio" resultó ser el mejor predictor estadísticamente significativo para cada uno de
los parámetros de orientación de ABangle, seguido de "red neuronal" y "árbol de decisión". El procedimiento de "árbol reforzado" fue el de menor rendimiento en el conjunto de datos (datos no mostrados).
Para cada parámetro de ABangle se realizaron 50 ejecuciones (cada una de las cuales consistió en una fase de entrenamiento y una de prueba) mientras se variaba el número de árboles de decisión en el bosque aleatorio de 10 a 100 para determinar el número óptimo de árboles con respecto al valor de Q2 del conjunto de prueba. Para cada ejecución individual, el conjunto de datos de entrada se dividió aleatoriamente en un 70 % para el conjunto de entrenamiento y en un 30 % para el conjunto de prueba. El modelo de bosque aleatorio se implementó usando Accelrys Pipeline Pilot 8.5 (19) con el componente "Modelo de bosque de RP (partición aleatoria) de aprendizaje" en modo de "Regresión". En la tabla 31 se incluye una lista de la configuración de los parámetros del modelo de bosque.
Tabla 31: Configuración de parámetros para la regresión usando el componente "Modelo de bosque de RP (partición aleatoria) de aprendizaje" en Accelrys Pipeline Pilot 8.5.

La tabla 32 muestra los valores de Q2 y del error de la media cuadrática (RMSE) para la predicción de los seis parámetros de ABangle promediados en 50 ejecuciones con conjunto de entrenamiento y de prueba elegidos aleatoriamente.
Tabla 32: Los valores de Q2 y RMSE para la predicción de los seis parámetros de ABangle promediaron más de 50 ejecuciones. El número de árboles por cada modelo de bosque aleatorio se ajustó manualmente para maximizar Q2. Los valores entre paréntesis especifican la desviación estándar.

El modelo de bosque aleatorio se ha entrenado una vez en el conjunto de datos completo de estructuras apo y complejas (tabla 32, columna central) y una vez solo en las estructuras complejas (tabla 32, arriba, columna derecha). A pesar del hecho de que el conjunto de entrenamiento se reduce en casi 550 estructuras, los valores de Q2 y RMSE mejoran cuando solo se consideran estructuras complejas. Para HL, LC2 y dc, los valores de Q2 son de aproximadamente 0,68, mientras que HC1, LC1 y LC2 tienen valores de Q2 de 0,75 y superiores (cuando se consideran estructuras complejas). La figura 2 muestra gráficos de regresión ejemplares para los parámetros de ABangle predichos frente a los reales en el conjunto de datos formado solo por estructuras complejas.
Además, se ha evaluado si el tamaño del conjunto de validación (ya sea 1/3 o 1/2 del conjunto de datos) tiene un impacto en las predicciones de bosque aleatorio. Para todos los parámetros de ABangle, se ha descubierto una diferencia en R2, como era de esperar, que favorece la validación más pequeña y el conjunto de entrenamiento más grande (datos no mostrados).
Finalmente, se evaluó el rendimiento de predicción de las tres huellas y sus variantes reducidas para los seis parámetros de ABangle diferentes en tres repeticiones (véase la tabla 33).
Tabla 33: Los valores medios de R2 para la predicción de los seis parámetros de ABangle HL, HC1, LC1, HC2, LC2 y dc en tres repeticiones usando los tres huellas de secuencia y sus variantes con solo los residuos de CDR más exteriores en el borde de la región estructural (a) y sin ningún residuo de CDR (b).
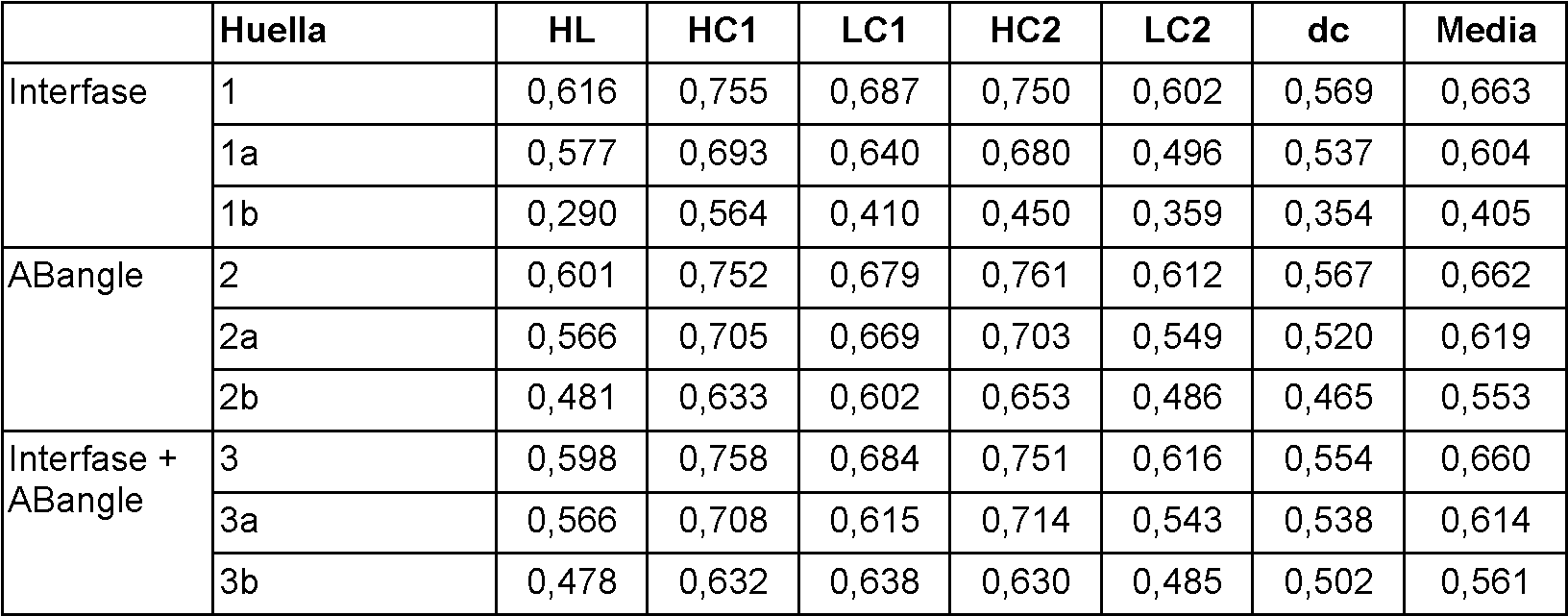
La huella 1, basada en el conjunto de residuos de interfase, y la huella 2, basada en las posiciones variables de entrada superiores de ABangle, fueron igualmente buenas, mientras que la combinación de las dos (huella 3) no parece conferir poder predictivo adicional ni afectar a los resultados. En los tres casos, las dos variantes de huellas reducidas empeoran, lo que confirma que la información de la secuencia de región estructural sola es insuficiente para determinar la orientación de los dominios VH-VL.
Se ha elegido la huella 3 para una evaluación adicional y un modelo de bosque aleatorio para el aprendizaje. Para incorporar el componente de predicción en la solución de modelización de homología, se implementó y entrenó el modelo de bosque aleatorio usando Accelrys Pipeline Pilot 8.5(19) con el componente “Modelo de bosque de RP (partición aleatoria) de aprendizaje” en modo de “Regresión”. Para todas las estructuras de Fv disponibles en RAB3D (n = 2249), se calcularon los parámetros de orientación VH-VL de ABangle, así como la huella 3, y los miembros del conjunto de datos se clasificaron en estructuras complejas (n = 1468) y apo (n = 781). Para cada parámetro de ABangle se realizaron 50 ejecuciones mientras se variaba el número de árboles de decisión en el bosque aleatorio de 10 a 100 para determinar el número óptimo de árboles con respecto al valor de Q2 del conjunto de prueba. Para cada ejecución individual, el conjunto de datos de entrada se dividió aleatoriamente en un 70 % para el conjunto de prueba y en un 30 % para el conjunto de entrenamiento.
Ejemplo 3
Algoritmo de modelización de homología de anticuerpos con ajuste de orientación VH-VL
El programa informático de modelización para modelizar la región Fv de anticuerpos (MoFvAb) usa las estructuras anotadas y reorientadas, por ejemplo, de la base de datos RAB3D, como depósito de moldes. Un par dado de secuencia de entrada de cadena pesada y ligera se anotó con Wolfguy y se redujo a VH y VL, respectivamente. Tanto VH como VL se dividieron a continuación en siete segmentos funcionales, es decir, Región estructural 1, CDR1, Región estructural 2, CDR2, Región estructural 3, CDR3 y Región estructural 4 (véase la tabla 10). A diferencia de otros enfoques de modelización de homología, no se seleccionó un molde de región estructural común por cada Fv o por cada cadena, pero cada fragmento se buscó/alineó/determinó de forma independiente en base a la homología de secuencia. Por ejemplo, un solo modelo de MoFvAb se podría ensamblar a partir de catorce estructuras molde diferentes, y posiblemente incluso más, ya que también es factible reconstruir la sección ascendente y descendente de los bucles de CDR a partir de diferentes estructuras molde. Los resultados positivos del molde de fragmento se clasificaron en el siguiente orden por
1) similitud de secuencia (puntuación de matriz BLOSUM62),
2) número de cadenas laterales incompletas,
3) resolución de la estructura molde, y
4) RMSD de alineación de la estructura molde frente a la región estructural consenso de RAB3D.
Opcionalmente, es posible aumentar (o incluso reemplazar en su totalidad) la selección de molde disponible para un fragmento dado por un segmento de novo.
Todas las estructuras molde se han alineado en una región estructural consenso común y, por lo tanto, comparten el mismo sistema de coordenadas. Por tanto, las coordenadas molde se transfirieron a un archivo de modelo sin procesar sin ajustes adicionales. A continuación, se procesó el modelo sin procesar: Se intercambiaron cadenas laterales no homólogas, se remodelizaron las cadenas laterales molde incompletas y se eliminaron los choques estéricos mediante optimización con rotámero. Debido al hecho de que cada fragmento se seleccionó de forma independiente, el número de intercambios de cadena lateral necesarios por cada modelo es manejable. El modelo procesado se parametrizó para el campo de fuerza CHARMm y se minimizó usando el modelo de agua implícita GBSW (Generalized Born with a simple Switching), primero por el procedimiento de descenso más pronunciado y a continuación por el procedimiento de gradiente conjugado. Para preservar un máximo de información conformacional de las estructuras molde, todos los residuos que no se habían remodelizado y que no estaban situados en los bordes de los fragmentos (con residuos contiguos que se originan en diferentes estructuras molde) se restringieron durante la minimización. MoFvAb está disponible como un servicio web basado en un protocolo implementado en Accelrys Pipeline Pilot 8.5 (19) usando la interfase Accelrys Discovery Studio 3.5 (20).
Se compararon tres variantes de la construcción del modelo MoFvAb para evaluar el impacto del ajuste de los dominios VH-VL:
Variante 1: los modelos se construyeron a partir de estructuras molde alineadas por cadena, es decir, en una región estructural consenso de VH y en una región estructural consenso de VL, respectivamente, y, antes del procesamiento del modelo y la minimización de energía, la orientación VH-VL del modelo se ajustó por alineación en cadena en una estructura consenso de Fv. Esta variante produjo modelos que tienen una orientación VH-VL promedio, genérica, no relacionada con la secuencia.
Variante 2: la orientación VH-VL del modelo se predijo en base a una huella de secuencia como se describe anteriormente, y se buscó el molde de Fv más similar en la base de datos en términos de sus parámetros de ABangle. La orientación VH-VL del modelo se ajustó a continuación por alineación en cadena en el denominado molde de orientación.
Tanto en la variante 1 como en la variante 2, la alineación en cadena en el Fv consenso o el molde de orientación se realizó mediante la superposición de Ca de los 35 residuos del conjunto esencial de ABangle definidos por Dunbar et al. (7).
Variante 3: los modelos se construyeron a partir de estructuras molde alineadas en una región estructural consenso de Fv común en lugar de una estructura consenso por cadena y la orientación VH-VL no se ajustó.
Para crear un conjunto de prueba representativo, se usó el MoFvAb para construir las 11 estructuras de Fv de anticuerpo de AMAII. Las estructuras de AMAII eran diversas con respecto a las especies (conejo, ratón, humano) y consisten principalmente en anticuerpos de unión a proteína, siendo la excepción el Fab A52 anti-ADN (PDB ID 4M61). Todas las estructuras de referencia de AMAII se cristalizaron en forma no unida. En el momento de la construcción del modelo, fue posible el acceso a más estructuras molde que los “candidatos” originales de AMAII, incluyendo varias estructuras de anticuerpos de conejo. Por lo tanto, los resultados de la modelización en términos de RMSD presentados en el presente documento no se pueden comparar directamente con los resultados presentados por los estudios originales de modelización enmascarada. Para simular al menos un escenario de modelización enmascarada, no se usaron fragmentos molde de estructuras con una identidad de secuencia de CDR mayor o igual al 95 % por cadena, que obviamente incluía las estructuras cristalinas originales de AMAII, así como variantes de secuencia de las mismas. La identificación de estructuras molde de secuencia idéntica para excluir de la construcción del modelo se realizó usando el programa informático CD-HIT (15, 16).
Ejemplo 4
Cálculo de la distancia de ABangle
Para comparar la similitud en el espacio de ABangle, se definió un conjunto de parámetros de ABangle como la tupla
0 :=(HL, HC1, LC1, HC2, LC2, dc) :=(01, f l2,03,fl4,05/06)
. La distancia euclidiana entre dos conjuntos de parámetros de ABangle es entonces
distAB„ Ble(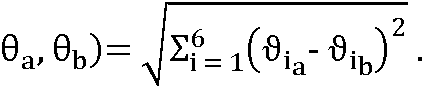
Un conjunto predicho de parámetros de ABangle 9 viene con un conjunto de desviaciones estándar asociadas
0,dd=v :=(a(HL),CT(HC1),CT(LC1),CT(HC2),CT(LC2),CT(dc)) := (° i, 02 ,03 ,04 ,05 ,06 ).
Al calcular la distancia entre un conjunto predicho de parámetros de ABangle 9 con desviaciones estándar 9stddev y un conjunto medido de parámetros de ABangle 9, la incertidumbre de la predicción se factorizó usando una función de distancia ponderada
dütABaJe, 0 ) := Jzf= ! ( ($ - S O M )2.
La función de distancia ponderada se usó para encontrar moldes de orientación en la base de datos que mejor se ajustan a un conjunto predicho de parámetros de ABangle. Como y disí^sangie mezclan medidas de distancias angulares (HL, HC1, LC1, HC2, LC2) con medidas de distancias lineales (dc), no se pueden interpretar como una distancia real en el espacio angular, sino que sirven solo como una medida de distancia abstracta.
Para calcular los parámetros de orientación de ABangle, se usó el código de programa publicado por Dunbar et al. (7) disponible en http://www.stats.ox.ac.uk/~dunbar/abangle/ en una versión ligeramente modificada que funciona en estructuras numeradas según Wolfguy.
Ejemplo 5
Correlaciones
Coeficiente de correlación de Pearson
El coeficiente de correlación de Pearson mide la correlación lineal entre dos variables X e Y. Se calcula como cov(X, Y)
r = ------------ ,CxCy
Siendo cov(X, Y) la covarianza entre X e Y y c la desviación estándar. Se usó el procedimiento estándar cor.test en R(25) para calcular el coeficiente de correlación y el valor de p para evaluar si difiere significativamente de cero. Coeficiente RV
El coeficiente RV fue introducido por Escoufier para medir la similitud entre matrices simétricas cuadradas (26). La definición se puede ampliar fácilmente a matrices rectangulares (27). Para dos matrices X e Y, el coeficiente RV se puede calcular como
RV traza{STT}
j (traza{STT}xtraza{TTT})
con S = XXT y T = YYT
Para calcular un valor de p asociado para el coeficiente RV, es decir, si es tan alto como lo sería por azar, se usó el procedimiento coeffRV del paquete FactoMine® (28) en R, que implementa una prueba de permutación como se describe en Josse et al. (29).
Procedimientos para rechazar anticuerpos basados en distancias angulares a un anticuerpo de referencia Suponiendo que las distancias angulares mayores apuntan a un peor comportamiento de unión de los anticuerpos en comparación con una referencia, son concebibles muchos procedimientos diferentes para rechazar anticuerpos. i) Rechazo de los 20 % peores
En el presente documento, un determinado porcentaje de anticuerpos se rechaza directamente en base a su distancia angular a la referencia. Como ejemplo, aquí se decide rechazar el 20 %. Por tanto, las etapas en este algoritmo son 1. ordenar la matriz de distancia angular y recordar los índices
2. usar los índices del 20 % de las distancias angulares más altas para rechazar los peores anticuerpos ii) Rechazo de HC y LC completos
En el presente documento, los HC o LC completos se rechazan y solo producen las otras combinaciones HC/LC. Para ello, se propone el siguiente algoritmo
1. calcular la distancia angular promedio para cada HC/LC
2. visualizar estas distancias y seleccionar un subconjunto de HC/LC para su rechazo
iii) Rechazo de malas combinaciones HC/LC
Una variante del procedimiento posterior es simplemente rechazar los anticuerpos que tienen "malas" combinaciones HC/LC. Este procedimiento podría funcionar bien si la correlación de la distancia angular con el anticuerpo no es tan fuerte para los anticuerpos individuales, pero se conserva mejor que en los HC y CL completos. El algoritmo es: 1. calcular la distancia angular promedio para cada HC/LC
2. visualizar estas distancias y seleccione un subconjunto de HC/LC para rechazar solo todas las combinaciones posibles entre estos.
Ejemplo 6
RMSD de carbonilo
En aras de la coherencia con AMAII, se usaron la RMSD de carbonilo y la definición de núcleo de lámina p y bucles de CDR de acuerdo con Teplyakov et al. (2). Para determinar la RMSD de carbonilo para un fragmento dado, primero se superpuso el modelo a la estructura cristalina usando los átomos de Ca del núcleo de lámina p con el procedimiento de superposición proporcionado en Accelrys Discovery Studio 3.5 (20). A continuación, se calculó la RMSD de carbonilo como la desviación de los átomos del grupo carbonilo de la cadena principal del segmento dado con respecto a la estructura cristalina. En comparación con la RMSD de Ca o RMSD de la cadena principal completa usadas habitualmente, la RMSD de carbonilo es más sensible con respecto a las desviaciones en la conformación de la cadena principal. Mientras que en AMAII todos los valores de RMSD de carbonilo se calcularon en base a una superposición del núcleo de lámina p de VH o de VL solamente, en el presente documento se calculó adicionalmente la RMSD de carbonilo basada en una superposición del núcleo de lámina p de VH y VL simultáneamente. La superposición en todo el Fv da lugar a peores valores de RMSD, ya que tiene en cuenta los defectos en la orientación VH-VL.
Al volver a calcular los valores de la RMSD de carbonilo de los modelos AMAII originales descargados de http://www.3dabmod.com, no todos los valores de la referencia original se pudieron reproducir exactamente, lo que se atribuyó a diferencias menores en el algoritmo de superposición o a inexactitudes numéricas.
Ejemplo 7
ELISA celular de unión con variantes de anticuerpos anti-CD81 humanizados
Para el ensayo de ELISA celular de unión, se propagaron HuH7-Rluc-H3 (línea celular positiva que expresa CD81) y HuH7-Rluc-L1 (línea celular de control negativo) en medio F-12 DMEM con FCS al 10 % a 37 °C y CO2 al 5 %. El día 1, las células se trataron con tripsina en aproximadamente el 90 % de confluencia y se volvieron a suspender a 4 x 105 células/ml. Se sembraron 2 x 104 células/pocillo de HuH7-Rluc-H3 y HuH7-Rluc-L1 (línea celular de control negativo) en 50 pl de medio DMEM y se permitió que se adhirieran a la placa de 96 pocillos de poli-D-lisina (Greiner, n.° de cat. 655940) durante 24 horas a 37 °C y CO2 al 5 %. El día 2, las muestras de anticuerpos que se iban a someter a prueba se prepararon en una placa de fondo redondo de polipropileno separada con dos veces la concentración deseada con un volumen final de 120 pl. Todas las muestras de ensayo se diluyeron en medio de cultivo celular. Se añadieron 50 pl de cada muestra de anticuerpo (pocillos duplicados) a las células para dar un volumen final de 100 pl/pocillo y se incubaron durante 2 horas a 4 °C. Después de la incubación primaria, las muestras se retiraron por aspiración y las células se fijaron con glutaraldehído al 0,05 % en solución de PBS (Roche Diagnostics GmbH, Mannheim, Alemania, n.° de cat. 1666789) durante 10 minutos a temperatura ambiente. Después de la fijación, cada pocillo se lavó 3 veces con 200 pl de PBS/Tween al 0,05 %. La etapa de incubación secundaria para la detección de anticuerpos anti-CD81 unidos se realizó durante 2 horas a temperatura ambiente en un agitador recíproco. Para anticuerpos CD81K humanizados, la detección se realizó usando un anticuerpo específico contra cadena gamma de IgG humana de oveja conjugado con peroxidasa (The Binding Site, n.° de cat. AP004) y se usó un conjugado de cabra anti-IgG de ratón, (H+L)-HRP (BIORAD, n.° de cat. 170-6516) para el anticuerpo de control positivo de ratón JS81 (BD Biosciences, n.° de cat. 555675), ambos diluidos a 1:1000 en tampón de bloqueo de PBS al 10 %. Cada pocillo se lavó tres veces con 200 pl de PBS/Tween al 0,05 % para eliminar los anticuerpos no unidos. La actividad de HRP se detectó usando 50 pl de solución TMB lista para usar (Roche Diagnostics GmbH, Mannheim, Alemania, n-° de cat.
1432559) y la reacción se detuvo después de aproximadamente 7-10 minutos con 50 pl por pocillo de H2SO41 M. La absorbancia se leyó usando el lector ELISA Tecan a 450 nm con una longitud de onda de referencia de 620 nm.
Ejemplo 8
Cribado cinético
El cribado cinético se realizó de acuerdo con Schraeml et al. (Schraeml, M. y M. Biehl, Methods Mol. Biol. 901 (2012) 171-181) en un instrumento BIAcore 4000, montado con un sensor BIAcore CM5. El instrumento BIAcore 4000 estaba bajo el control de la versión del programa informático VI. 1. Se montó un chip BIAcore CM5 serie S en el instrumento y se dirigió hidrodinámicamente y se preacondicionó de acuerdo con las instrucciones del fabricante. El tampón del instrumento era un tampón de HBS-EP (HEPES 10 mM (pH 7,4), NaCl 150 mM, EDTA 1 mM, P20 al 0,05 % (p/v)). Se preparó un sistema de captura de anticuerpos en la superficie del sensor. Se inmovilizó un anticuerpo policlonal anti humano caprino con especificidad de Fc de IgG humana (Jackson Lab.) a 30 pg/ml en tampón de acetato de sodio 10 mM (pH 5) en los puntos 1, 2, 4 y 5 en las cubetas de lectura del instrumento 1, 2, 3 y 4 a 10.000 RU usando química de NHS/EDC. En cada cubeta de lectura se capturaron los anticuerpos en el punto 1 y el punto 5. El punto 2 y el punto 4 se usaron como puntos de referencia. Se desactivó el sensor con una solución de etanolamina 1 M. Se aplicaron derivados de anticuerpos humanizados a concentraciones entre 44 nM y 70 nM en tampón del instrumento complementado con 1 mg/ml de CMD (carboximetildextrano). Se inyectaron los anticuerpos durante 2 min a un caudal de 30 pl/min. Se midió el nivel de captura (CL) de los anticuerpos presentados en la superficie en unidades de respuesta rel. (RU). Se inyectaron los analitos en solución, la proteína tau humana fosforilada, la proteína tau humana no fosforilada y la proteína tau humana fosforilada con la mutación T422S, a 300 nM durante 3 min, a un caudal de 30 pl/min. Se supervisó la disociación durante 5 min. Se regeneró el sistema de captura mediante una inyección de 1 min de tampón de glicina 10 mM pH 1,7 a 30 pl/min sobre todas las cubetas de lectura. Se usaron dos puntos de
informe, la señal registrada poco antes del final de la inyección de analito, indicada como unión tardía (BL), y la señal registrada poco antes del final del tiempo de disociación, estabilidad tardía (SL), para caracterizar el rendimiento del cribado cinético. Además, se calculó la constante de disociación kd (1/s) de acuerdo con un modelo de Langmuir y se calculó la semivida del complejo anticuerpo/antígeno en minutos de acuerdo con la fórmula ln(2)/(60*kd). Se calculó la proporción molar (MR) de acuerdo con la fórmula MR = (Unión tardía (RU)) / (Nivel de captura (RU)) * (PM(anticuerpo) / (PM(antígeno)). En caso de que el sensor se hubiera configurado con una cantidad adecuada de nivel de captura de ligando del anticuerpo, cada anticuerpo se debería poder unir funcionalmente al menos a un analito en solución, que está representado por una proporción molar de MR = 1,0. Entonces, la proporción molar también es un indicador del modo de valencia de la unión del analito. La valencia máxima puede ser MR = 2 para un anticuerpo que se une dos analitos, uno con cada valencia de Fab.
En otro modo de realización, las velocidades cinéticas se determinaron a 25 °C y 37 °C usando la misma configuración experimental, pero usando series de concentraciones múltiples de cada analito en solución a 0 nM (tampón), 1,2 nM, 3,7 nM, 11,1 nM, 33,3 nM, 100 nM y 300 nM. A partir del comportamiento de unión dependiente de la concentración, se calcularon los datos cinéticos usando el programa informático de evaluación BIAcore de acuerdo con las instrucciones del fabricante y un modelo de Langmuir 1.1 con RMAX global.
BIBLIOGRAFÍA
1. Kuroda, D. et al., Protein Eng. Des. Sel., 25 (2012) 507-521.
2. Teplyakov, A. et al., Proteins, 2014; DOI: 10.1002/prot.24554
3. Dunbar, J. et al., Nuc. Acids Res., 42 (2014) D1140-D1146.
4. Abhinandan, K.R. y Martin, A.C.R., Protein Eng. Des. Sel., 23 (2010) 689-697.
5. Chailyan, A. et al., FEBS J., 278 (2011) 2858-2866
6. Narayanan, A. et al., J. Mol. Biol. 388 (2009) 941-953.
7. Dunbar, J. et al., Protein Eng. Des. Sel., 26 (2013) 611-620.
8. Almagro, J.C. et al., Proteins 79 (2011) 3050-3066.
9. Jayaram, N. et al., Protein Eng. Des. Sel. 25 (2012) 523-529.
10. Banfield, M.J. et al., Proteins, 29 (1997) 161-171.
11. Li, Y. et al., Biochem., 39 (2000) 6296-6309.
12. Teplyakov, A. et al., Acta Cryst., F67 (2011) 1165-1167.
13. Chothia, C. et al., J. Mol. Biol., 278 (1998) 457-479.
14. Chothia, C. y Lesk, A.M., J. Mol. Biol. 196 (1987) 901-917.
15. Li, W. y Godzik, A., Bioinformatics, 22 (2006) 1658-1659.
16. Fu, L. et al., Bioinformatics, 28 (2012) 3150-3152.
17. Pan, R. et al., J. Virol., 87 (2013) 10221-10231.
18. Kaas, Q. et al., Brief Funct. Genom. Prot., 6 (2007) 253-264.
19. Accelrys Software Inc., Pipeline Pilot, versión 8.5.0.200, San Diego: Accelrys Software Inc., 2011
20. Accelrys Software Inc., Discovery Studio Modeling Environment, versión 3.5.0.12158, San Diego: Accelrys Software Inc., 2012.
21. Niederfellner, G. et al., Blood, 118 (2011) 358-367.
22. Kaas, Q. et al., Brief Funct. Genom. Prot., 6 (2007) 253-264.
23. Accelrys Software Inc., Pipeline Pilot, versión 8.5.0.200, San Diego: Accelrys Software Inc., 2011.
24. Berman, H.M. et al., Nucl. Acids Res., 28 (2000) 235-242.
25. R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Viena, Austria. URL http://www.R-project.org/.
26. Robert, P. y Escoufier, Y., Appl. Stat. 25 (1967) 257-265
27. Abdi, H., (2007). RV coefficient and congruence coefficient, en N.J. Salkind (Ed.): Encyclopedia of measurement and statistics. Thousand Oaks: Sage.
28. Husson, F. et al., (2014). FactoMineR: Multivariate Exploratory Data Analysis and Data Mining with R. R package, versión 1.26. http://CRAN.R-project.org/package=FactoMineR
29. Josse, J. et al., Comp. Stat. Data Anal., 53 (2008) 82-91.
Claims (9)
1. Un procedimiento para seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo no humano que comprende las siguientes etapas:
- generar una multitud de fragmentos Fv de anticuerpo variante mediante injerto/transferencia de los residuos determinantes de la especificidad de unión a antígeno del fragmento Fv del anticuerpo no humano en/a una secuencia de aminoácidos de línea germinal de fragmento Fv de anticuerpo humano, con lo que cada fragmento Fv de anticuerpo variante de la multitud de fragmentos Fv de anticuerpo variante difiere de los otros fragmentos Fv de anticuerpo variante en al menos un residuo de aminoácido,
- determinar el ángulo VH-VL para el fragmento Fv no humano y para cada uno de los fragmentos Fv de anticuerpo variante de la multitud de fragmentos Fv de anticuerpo variante en base a un conjunto de residuos de interfase VH-Vl que comprenden los residuos L44, L46, L87, H45, H62 (numeración de acuerdo con el índice de Chothia) del fragmento Fv de anticuerpo,
- seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la diferencia más pequeña en el ángulo VH-VL en comparación con el ángulo VH-VL del anticuerpo no humano y, de este modo, seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo no humano,
con lo que el uno o más fragmentos Fv de anticuerpo variante se unen al mismo
antígeno que el fragmento Fv de anticuerpo no humano,
en el que el ángulo VH-VL se determina calculando los seis parámetros
- la longitud de C, dc,
- el ángulo de torsión, HL, de H1 a L1 medido alrededor de C,
- el ángulo de curvatura, HC1, entre H1 y C,
- el ángulo de curvatura, HC2, entre H2 y C,
- el ángulo de curvatura, LC1, entre L1 y C, y
- el ángulo de curvatura, LC2, entre L2 y C,
en el que los planos de la región estructural de referencia se registran i) alineando las coordenadas Ca correspondientes a las ocho posiciones H36, H37, H38, H39, H89, H90, H91 y H92 de VH y ajustando un plano a través de ellas y ii) alineando las coordenadas Ca correspondientes a las ocho posiciones L35, L36, L37, L38, L85, L86, L87 y L88 de VL y ajustando un plano a través de ellas, iii) colocando un colocado en cada plano, con lo que cada estructura tiene puntos de malla equivalentes y pares de punto de malla VH-VL equivalentes, y iv) midiendo la distancia euclidiana para cada par de puntos de malla en cada estructura, con lo que el vector C une el par de puntos con la varianza mínima en su distancia de separación,
en el que H1 es el vector que discurre paralelo al primer componente principal del plano de VH, H2 es el vector que discurre paralelo al segundo componente principal del plano de VH, L1 es el vector que discurre paralelo al primer componente principal del plano de VL, L2 es el vector que discurre paralelo al segundo componente principal del plano de Vl , el ángulo HL es el ángulo de torsión entre los dos dominios, HC1 y LC1 son los ángulos de curvatura equivalentes a variaciones de tipo inclinación de un dominio con respecto al otro, y los ángulos de curvatura HC2 y LC2 son equivalentes a las variaciones de tipo torsión de un dominio con respecto al otro.
2. El procedimiento de acuerdo con la reivindicación 1, que comprende la siguiente etapa:
- seleccionar aquellos fragmentos Fv de anticuerpo variante que tienen la mayor similitud en el ángulo interdominio VH-VL en comparación con el ángulo interdominio VH-VL del anticuerpo original y, de este modo, seleccionar uno o más fragmentos Fv de anticuerpo variante derivados de un fragmento Fv de anticuerpo original.
3. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 a 2, en el que el conjunto de residuos de interfase VH-VL comprende los residuos H33, H35, H37, H39, H43, H44, H45, H46, H47, H50, H55, H56, H58, H60, H61, H62, H89, H91, H95, H96, H98, H99, H100x-2, H100x-1, H100x, H101, H102, H103, H105, L32, L34, L36, L38, L41, L42, L43, L44, L45, L46, L49, L50, L53, L55, L56, L85, L87, L89, L91, L93, L94/L95x-1, L95x, L96, L97, L100 (numeración de acuerdo con el índice de Chothia).
4. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en el que el ángulo VH-VL se
determina calculando los parámetros del ángulo VH-VL usando un procedimiento de bosque aleatorio para cada parámetro.
5. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en el que el ángulo VH-VL se determina calculando el ángulo de torsión, los cuatro ángulos de curvatura (dos por cada dominio variable) y la longitud del eje de pivote de VH y VL (HL, HC1, LC1, HC2, LC2, dc) usando un modelo de bosque aleatorio.
6. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 4 a 5, en el que el modelo de bosque aleatorio se entrena solo con datos de estructuras de anticuerpo complejas.
7. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 a 6, en el que la diferencia más pequeña es el valor de Q2 más alto.
8. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 a 7, en el que la similitud más alta es la desviación de la media cuadrática (RMSD) promedio más baja.
9. El procedimiento de acuerdo con una cualquiera de las reivindicaciones 1 a 8, en el que se usa un modelo ensamblado a partir de estructuras molde alineadas en la región estructural consenso de VH o VL, seguido de la reorientación VH-VL en una estructura molde de orientación VH-VL elegida en base a la similitud para determinar el ángulo VH-VL.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14190307 | 2014-10-24 | ||
| PCT/EP2015/074294 WO2016062734A1 (en) | 2014-10-24 | 2015-10-21 | Vh-vl-interdomain angle based antibody humanization |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2857553T3 true ES2857553T3 (es) | 2021-09-29 |
Family
ID=51799006
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15784344T Active ES2857553T3 (es) | 2014-10-24 | 2015-10-21 | Humanización de anticuerpos basada en ángulo interdominio VH-VL |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US20170204182A1 (es) |
| EP (1) | EP3209684B1 (es) |
| JP (1) | JP6829194B2 (es) |
| KR (1) | KR20170070070A (es) |
| CN (1) | CN107074933B (es) |
| BR (1) | BR112017005451A2 (es) |
| CA (1) | CA2960445A1 (es) |
| DK (1) | DK3209684T3 (es) |
| ES (1) | ES2857553T3 (es) |
| MX (1) | MX2017004001A (es) |
| RU (1) | RU2017115212A (es) |
| WO (1) | WO2016062734A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201829463A (zh) | 2016-11-18 | 2018-08-16 | 瑞士商赫孚孟拉羅股份公司 | 抗hla-g抗體及其用途 |
| AU2018247796A1 (en) | 2017-04-04 | 2019-08-29 | F. Hoffmann-La Roche Ag | Novel bispecific antigen binding molecules capable of specific binding to CD40 and to FAP |
| CN117050176A (zh) | 2017-07-31 | 2023-11-14 | 豪夫迈·罗氏有限公司 | 基于三维结构的人源化方法 |
| TWI829667B (zh) | 2018-02-09 | 2024-01-21 | 瑞士商赫孚孟拉羅股份公司 | 結合gprc5d之抗體 |
| AR114789A1 (es) | 2018-04-18 | 2020-10-14 | Hoffmann La Roche | Anticuerpos anti-hla-g y uso de los mismos |
| AR115052A1 (es) | 2018-04-18 | 2020-11-25 | Hoffmann La Roche | Anticuerpos multiespecíficos y utilización de los mismos |
| EP3861025A1 (en) | 2018-10-01 | 2021-08-11 | F. Hoffmann-La Roche AG | Bispecific antigen binding molecules with trivalent binding to cd40 |
| NZ773887A (en) | 2018-10-01 | 2024-11-29 | Hoffmann La Roche | Bispecific antigen binding molecules comprising anti-fap clone 212 |
| JP7354306B2 (ja) | 2019-06-27 | 2023-10-02 | エフ・ホフマン-ラ・ロシュ・アクチェンゲゼルシャフト | 新規icos抗体及びそれらを含む腫瘍標的化抗原結合分子 |
| AR119382A1 (es) | 2019-07-12 | 2021-12-15 | Hoffmann La Roche | Anticuerpos de pre-direccionamiento y métodos de uso |
| SG11202112491WA (en) | 2019-07-31 | 2021-12-30 | Hoffmann La Roche | Antibodies binding to gprc5d |
| JP2022543551A (ja) | 2019-07-31 | 2022-10-13 | エフ・ホフマン-ラ・ロシュ・アクチェンゲゼルシャフト | Gprc5dに結合する抗体 |
| AR121706A1 (es) | 2020-04-01 | 2022-06-29 | Hoffmann La Roche | Moléculas de unión a antígeno biespecíficas dirigidas a ox40 y fap |
| JP7326584B2 (ja) | 2020-12-17 | 2023-08-15 | エフ・ホフマン-ラ・ロシュ・アクチェンゲゼルシャフト | 抗hla-g抗体及びその使用 |
| AR126009A1 (es) | 2021-06-02 | 2023-08-30 | Hoffmann La Roche | Moléculas agonistas de unión al antígeno cd28 que se dirigen a epcam |
| WO2023078420A1 (en) * | 2021-11-04 | 2023-05-11 | Vibrant Pharma Limited | Methods for antibody optimization |
| EP4720120A1 (en) | 2023-06-01 | 2026-04-08 | F. Hoffmann-La Roche AG | Bispecific antibodies targeting bcma and cd28 |
| EP4720121A1 (en) | 2023-06-01 | 2026-04-08 | F. Hoffmann-La Roche AG | Immunostimulatory antigen binding molecules that specifically bind to bcma |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5530101A (en) * | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US6849425B1 (en) * | 1999-10-14 | 2005-02-01 | Ixsys, Inc. | Methods of optimizing antibody variable region binding affinity |
| MXPA05000511A (es) * | 2001-07-12 | 2005-09-30 | Jefferson Foote | Anticuepros super humanizados. |
| SI2000481T1 (sl) * | 2003-02-01 | 2016-08-31 | Tanox, Inc. | Anti-humana protitelesa IgE z visoko afiniteto |
| GB0613209D0 (en) * | 2006-07-03 | 2006-08-09 | Ucb Sa | Methods |
| EP2250198B1 (en) * | 2008-01-23 | 2016-03-09 | Department of Biotechnology | A humanized high affinity recombinant antibody against hepatitis b surface antigen |
| GB0914691D0 (en) * | 2009-08-21 | 2009-09-30 | Lonza Biologics Plc | Immunoglobulin variants |
| AU2010290844B2 (en) * | 2009-09-06 | 2015-04-09 | Protab Ltd. | Humanized antibodies specific for HSP65-derived peptide-6 methods and uses thereof |
| US20130108641A1 (en) * | 2011-09-14 | 2013-05-02 | Sanofi | Anti-gitr antibodies |
-
2015
- 2015-10-21 ES ES15784344T patent/ES2857553T3/es active Active
- 2015-10-21 RU RU2017115212A patent/RU2017115212A/ru not_active Application Discontinuation
- 2015-10-21 EP EP15784344.2A patent/EP3209684B1/en active Active
- 2015-10-21 JP JP2017522101A patent/JP6829194B2/ja active Active
- 2015-10-21 KR KR1020177010887A patent/KR20170070070A/ko not_active Withdrawn
- 2015-10-21 DK DK15784344.2T patent/DK3209684T3/da active
- 2015-10-21 BR BR112017005451A patent/BR112017005451A2/pt not_active Application Discontinuation
- 2015-10-21 MX MX2017004001A patent/MX2017004001A/es unknown
- 2015-10-21 CN CN201580056696.2A patent/CN107074933B/zh active Active
- 2015-10-21 WO PCT/EP2015/074294 patent/WO2016062734A1/en not_active Ceased
- 2015-10-21 CA CA2960445A patent/CA2960445A1/en not_active Abandoned
-
2017
- 2017-03-31 US US15/475,440 patent/US20170204182A1/en not_active Abandoned
-
2020
- 2020-03-16 US US16/820,094 patent/US20200377590A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| WO2016062734A1 (en) | 2016-04-28 |
| US20200377590A1 (en) | 2020-12-03 |
| CA2960445A1 (en) | 2016-04-28 |
| MX2017004001A (es) | 2017-06-30 |
| JP6829194B2 (ja) | 2021-02-10 |
| BR112017005451A2 (pt) | 2018-01-02 |
| JP2017531443A (ja) | 2017-10-26 |
| US20170204182A1 (en) | 2017-07-20 |
| KR20170070070A (ko) | 2017-06-21 |
| RU2017115212A3 (es) | 2019-04-24 |
| EP3209684B1 (en) | 2020-12-23 |
| RU2017115212A (ru) | 2018-11-26 |
| CN107074933A (zh) | 2017-08-18 |
| DK3209684T3 (da) | 2021-02-22 |
| CN107074933B (zh) | 2021-03-19 |
| EP3209684A1 (en) | 2017-08-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2857553T3 (es) | Humanización de anticuerpos basada en ángulo interdominio VH-VL | |
| ES2387585T3 (es) | Procedimiento de uso en anticuerpos monoclonales que se adaptan al ser humano | |
| CN104640879B (zh) | Il-18结合分子 | |
| ES2779977T3 (es) | Anticuerpos monoclonales inmunoestimuladores contra interleucina 2 humana | |
| BR112021004723A2 (pt) | anticorpos de receptor anti-il4 para uso veterinário | |
| Bujotzek et al. | VH-VL orientation prediction for antibody humanization candidate selection: A case study | |
| JP7217772B2 (ja) | 三次元構造に基づくヒト化方法 | |
| TWI882142B (zh) | 抗FXI/FXIa抗體、其抗原結合片段及醫藥用途 | |
| CN114341184A (zh) | 抗FcRn抗体、其抗原结合片段及其医药用途 | |
| CN113677701A (zh) | 产生亲合结合多特异性抗体的方法 | |
| Teplyakov et al. | Structural insights into humanization of anti-tissue factor antibody 10H10 | |
| JP7511561B2 (ja) | エピトープおよびパラトープを同定する方法 | |
| HK1242352B (zh) | 基於vh-vl域间角的抗体人源化 | |
| HK1242352A1 (en) | Vh-vl-interdomain angle based antibody humanization | |
| Scire | Structure and Function of MUC1 Specific Monoclonal Antibodies | |
| HK40023878A (en) | Three-dimensional structure-based humanization method | |
| EA048641B1 (ru) | Способы определения эпитопов и паратопов | |
| HK40063170A (en) | Methods for identifying epitopes and paratopes | |
| HK40062039A (en) | Method for generating avid-binding multispecific antibodies | |
| JP2022514362A (ja) | 抗vegf抗体のvegf-r1への結合阻害を改善する方法 | |
| Almagro et al. | Humanization of antibodies |