ES2824774T3 - Nuevos derivados de quinazolinona que inhiben PI3K y composición farmacéutica que los contiene - Google Patents
Nuevos derivados de quinazolinona que inhiben PI3K y composición farmacéutica que los contiene Download PDFInfo
- Publication number
- ES2824774T3 ES2824774T3 ES17827977T ES17827977T ES2824774T3 ES 2824774 T3 ES2824774 T3 ES 2824774T3 ES 17827977 T ES17827977 T ES 17827977T ES 17827977 T ES17827977 T ES 17827977T ES 2824774 T3 ES2824774 T3 ES 2824774T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- methyl
- amino
- cyclopropyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*([C@@](C1CC1)C(*(COCc1c(C)cccc1C)C1=CCCC=C1)=C)C1C(*(C)C=*2)=C2*=C*1 Chemical compound C*([C@@](C1CC1)C(*(COCc1c(C)cccc1C)C1=CCCC=C1)=C)C1C(*(C)C=*2)=C2*=C*1 0.000 description 3
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/91—Oxygen atoms with aryl or aralkyl radicals attached in position 2 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un compuesto de Fórmula 1 o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** en donde X es hidrógeno, halógeno o metilo; e Y es cicloalquilo C3-4.
Description
DESCRIPCIÓN
Nuevos derivados de quinazolinona que inhiben PI3K y composición farmacéutica que los contiene
[Campo técnico]
La presente invención se refiere a nuevos derivados de quinazolinona que inhiben PI3K y a un método para preparar estos derivados.
Además, la presente invención proporciona una composición farmacéutica para tratar una neoplasia sanguínea, una hepatopatía o una enfermedad autoinmunitaria, que incluye los derivados de quinazolinona.
[Antecedentes de la técnica]
El cáncer es la segunda causa principal de muerte después de las cardiopatías en los Estados Unidos (Cancer Facts and Figures 2005, American Cancer Society, Inc.). En la etapa inicial del desarrollo del cáncer, se puede seleccionarse un método para extirpar tumores o destruir células cancerosas mediante quimioterapia, radioterapia y similares, pero en el caso de pacientes con cáncer terminal, los efectos secundarios debidos a las terapias agresivas son relativamente grandes y la tasa de respuesta después del tratamiento es baja y, por tanto, se pueden seleccionar terapias para reducir los efectos secundarios al retardar la progresión del cáncer y mejorar la calidad de vida. En estos aspectos, los fármacos antineoplásicos están destinados no solo a prevenir la recaída del cáncer al destruir células cancerosas, pero también prolongan el periodo de supervivencia al inhibir el crecimiento y la proliferación de células cancerosas, cuando es difícil esperar una recuperación completa.
La quimioterapia existente para cánceres metastásicos no proporciona tratamiento a largo plazo debido a su eficacia reducida. Además, aunque se han introducido nuevas quimioterapias en el campo médico, todavía existe la necesidad de nuevos medicamentos eficaces como terapias primarias, secundarias y terciarias en tratamiento individual o tratamiento conjunto con agentes existentes, para el tratamiento de tumores resistentes.
Además, incluso los fármacos antineoplásicos con gran potencia no son aplicables a todos los cánceres, por lo que existe una necesidad urgente de desarrollar medicamentos para mejorar la eficacia del tratamiento.
Las terapias dirigidas son ventajosas porque son específicas del tumor, eficaces y tienen un efecto mucho menor sobre las células normales en comparación con los tratamientos antineoplásicos sistémicos existentes. La desregulación de proteínas cinasas se encuentra habitualmente en células cancerosas y, por tanto, es una diana atractiva para el desarrollo de fármacos antineoplásicos.
Entre las lípido cinasas, la estructura y función de las PI3K (isómeros de fosfatidilinositol-3-cinasa) se han verificado gradual y claramente en los últimos años. Se sabe que las PI3K pertenecen a una familia de enzimas que desempeñan una función vital en rutas de señalización intracelular y están implicadas en funciones celulares importantes tales como crecimiento celular, proliferación, diferenciación, motilidad, supervivencia y tráfico intracelular.
Durante los últimos 20 años, se ha revelado constantemente que cuando las PI3K pierden su función reguladora, se producen problemas tales como sobreactivación y similares en rutas de señalización intracelular para inducir muchos tipos de enfermedades.
Las PI3K se clasifican en clase I, clase II y clase III. La clase I se divide de nuevo en subclases: clase IA y clase IB. La PI3K de clase I está en forma de dímero y el dímero se divide en subunidades catalíticas y reguladoras. La PI3K de clase 1A es un dímero que consta de una subunidad catalítica p110 y una subunidad reguladora p85 y, en este sentido, la subunidad catalítica p110 incluye tres isoformas, es decir, pllOa, p110p y p1105. Por tanto, las isoformas de PI3K se denominan PI3Ka, PI3KP y PI3K5.
Por otra parte, la PI3K de clase IB es un dímero que consta de una subunidad catalítica p110Y y una subunidad reguladora p101, y la PI3K se denomina en general PI3Ky.
PI3K5 es inducida principalmente por tirosina cinasas receptoras (RTK) para fosforilar PIP2 a PIP3 y PI3Ky es inducida principalmente por receptores acoplados a proteína G (GPCR) para fosforilar PIP2 a PIP3. PIP3 activa la proteína cinasa B (Akt/PKB) y conduce continuamente a señalización corriente abajo, participando de este modo en la regulación de las funciones celulares principales, tal como crecimiento celular, proliferación, diferenciación, motilidad, supervivencia y tráfico intracelular. Una de las mayores preocupaciones de los últimos años ha sido que diversas enfermedades que van desde la inflamación y la autoinmunidad hasta la neoplasia maligna hematológica y el cáncer sólido se producen cuando PI3K5 y PI3Ky tienen una disfunción en la regulación de la transducción de señales intracelulares y, en consecuencia, se han realizado intensos esfuerzos para desarrollar fármacos para tratar la inflamación, la autoinmunidad, la neoplasia maligna hematológica y el cáncer sólido al inhibir PI3K5 y PI3Ky que pierden sus funciones reguladoras.
Un ejemplo de fármacos representativos que se están desarrollando en este campo es idelalisib, una sustancia que fue desarrollada por Gilead Calistoga e inhibe selectivamente PI3K5. Este fármaco tiene eficacia excelente contra diversos tipos de neoplasias hematológicas y, por tanto, ha llamado la atención como un fármaco revolucionario que aborda problemas (especialmente la citotoxicidad contra las células normales) de los fármacos antineoplásicos citotóxicos existentes y también compensa problemas de eficacia de los fármacos antineoplásicos existentes (Q. YANG et al., Clinical Cancer Research, 2015, 21 (7), págs. 1537-1542). Sin embargo, en Europa, se han producido algunos casos de toxicidad grave durante los ensayos clínicos, en los que los pacientes murieron de neumonía, por lo que ahora se ha suspendido el desarrollo de este fármaco. Según un informe, la razón es que la actividad inhibidora de este fármaco fue suficientemente selectiva y potente para PI3K6 en lugar de para PI3Ka y PI3Kp, pero no suficientemente selectiva en comparación con PI3Ky. Duvelisib presentó actividad inhibidora doble sobre PI3K6 y PI3Ky y, por tanto, tenía la posibilidad de desarrollarse como un fármaco muy prometedor para el tratamiento de neoplasias hematológicas, inflamación y enfermedad autoinmunitaria. Sin embargo, durante los ensayos clínicos, el desarrollo de Duvelisib se interrumpió debido a problemas similares a los de idelalisib. Se sabe que la eficacia inhibidora de esta sustancia no es suficientemente selectiva para PI3K6 y PI3Ky en comparación con PI3Kp. Por lo tanto, existe la necesidad de desarrollar un fármaco capaz de inhibir de forma más selectiva PI3K6 en lugar de al menos idelalisib.
Por otra parte, los derivados de quinazolinona son estructuras especiales presentes en muchos compuestos biológicamente activos tales como metacualona, que es un fármaco hipnótico-sedante, clorocualona, que es un antitusivo, y piricualona, que es un anticonvulsivo. La quinazolinona y derivados de la misma tienen una amplia gama de propiedades biológicas tales como hipnosis, analgesia, inhibición de convulsiones, inhibición de la tos y actividad antiinflamatoria.
En particular, se utilizan derivados de quinazolinona en el tratamiento de enfermedades proliferativas celulares, incluyendo el cáncer, y son uno de los agentes terapéuticos que se han utilizado de forma amplia recientemente. Por ejemplo, los registros de patente de los Estados Unidos n.° 5.747.498 y 5.773.476 desvelan derivados de quinazolinona utilizados para el tratamiento de cáncer que es inducido por la sobreactivación o activación aberrante de tirosina cinasas receptoras. Además, el documento WO 2014/023083 desvela derivados de quinazolinona como inhibidores de PI3K6 útiles para el tratamiento de tumores. Por lo tanto, es necesario estudiar y desarrollar derivados de quinazolinona mediante diversos enfoques para el tratamiento de enfermedades proliferativas celulares.
[Divulgación]
[Problema técnico]
Un objeto de la presente invención es proporcionar nuevos derivados de quinazolinona que inhiban las PI3K y métodos para preparar estos derivados.
Otro objeto de la presente invención es proporcionar una composición farmacéutica que comprenda los derivados de quinazolinona para prevenir o tratar un tumor sanguíneo, una hepatopatía o una enfermedad autoinmunitaria.
[Solución técnica]
De acuerdo con un aspecto de la presente solicitud, se proporciona un compuesto representado por la Fórmula 1 a continuación o una sal farmacéuticamente aceptable del mismo:
[Fórmula 1]

en donde
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
De acuerdo con otro aspecto de la presente invención, se proporciona una composición farmacéutica que comprende
un compuesto representado por la Fórmula 1 a continuación o una sal farmacéuticamente aceptable del mismo como ingrediente activo para prevenir o tratar un cáncer de sangre, una enfermedad hepática o una enfermedad autoinmune.
[Fórmula 1]

en donde,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
La neoplasia sanguínea puede ser leucemia o linfoma.
La hepatopatía se puede seleccionar del grupo que consiste en esteatosis hepática no alcohólica (EHNA), esteatohepatitis no alcohólica (ENA), esteatosis hepática, hepatocirrosis, hepatitis, adenoma hepático, hipersensibilidad a la insulina y cáncer de hígado.
La enfermedad autoinmunitaria se puede seleccionar del grupo que consiste en rinitis alérgica, asma, enfermedad pulmonar obstructiva crónica (EPOC) y artritis reumatoide.
De acuerdo con aún otro aspecto de la presente invención, se proporciona un método para preparar un compuesto representado por la Fórmula 1 a continuación, incluyendo el método:
hacer reaccionar un compuesto representado por la Fórmula 2 a continuación con un compuesto representado por la Fórmula 3 a continuación para preparar un compuesto representado por la Fórmula 4 a continuación; desproteger el compuesto representado por la Fórmula 4 a continuación para preparar un compuesto representado por la Fórmula 5 a continuación; y
hacer reaccionar un compuesto representado por la Fórmula 5 a continuación con un compuesto representado por la Fórmula 6 para preparar el compuesto de la Fórmula 1.
[Fórmula 1]

en donde, en la Fórmula 1,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
[Fórmula 2]

en donde, en la Fórmula 2,
X es -H, halógeno o -CH3.
[Fórmula 3]

en donde, en la Fórmula 3,
Y es cicloalquilo C3-4.
[Fórmula 4]

en donde, en la Fórmula 4,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
[Fórmula 5]

en donde, en la Fórmula 5,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
[Fórmula 6]

en donde, en la Fórmula 6,
Z es halógeno.
[Efectos ventajosos]
Los nuevos derivados de quinazolinona según la presente invención son eficaces en el tratamiento de la neoplasia sanguínea o hepatopatías.
En particular, en comparación con los inhibidores de PI3K5 existentes, los derivados de quinazolinona de la presente invención pueden inhibir PI3K5 con alta selectividad para reducir la inmunotoxicidad de forma significativa o inhibir PI3K5 y PI3Ky simultáneamente, permitiendo de este modo no solo el tratamiento antineoplásico de neoplasias hematológicas y similares, sino también el tratamiento de enfermedades autoinmunitarias. Estos agentes terapéuticos dirigidos pueden abordar problemas tales como los efectos secundarios de los tratamientos antineoplásicos existentes con citotoxicidad grave.
[Descripción de los dibujos]
La fig. 1 ilustra el principio de un experimento de análisis de cinasa Adapta del ejemplo experimental 1.
La fig. 2 ilustra los resultados del ejemplo experimental 2 para confirmar un efecto sobre la reducción de la fosforilación de AKT (Ser473) en líneas celulares de leucemia y linfoma.
La fig. 3 ilustra los resultados del ejemplo experimental 3 para confirmar un efecto inhibidor sobre el crecimiento de células de leucemia y linfoma.
La fig. 4 ilustra los resultados del análisis SDS-PAGE del ejemplo experimental 4 para confirmar un efecto sobre la apoptosis de células de linfoma difuso de linfocitos B grandes (DLBCL) y células de leucemia linfoblástica aguda (LLA).
La fig. 5 ilustra los resultados de la citometría de flujo del ejemplo experimental 5 para confirmar un efecto sobre la apoptosis de células de linfoma difuso de linfocitos B grandes (DLBCL) y células de leucemia linfoblástica aguda (LLA).
La fig. 6 ilustra imágenes microscópicas de fluorescencia de Cytation™ 5 (BioTek) que muestran los resultados de la formación de tubos del ejemplo experimental 6 para confirmar un efecto inhibidor sobre la angiogénesis. La fig. 7 es un gráfico que muestra un efecto inhibidor sobre la angiogénesis en el ejemplo experimental 6 para confirmar un efecto inhibidor sobre la angiogénesis.
La fig. 8 es un gráfico que muestra el número de HUVEC viables tratadas con un compuesto en el ejemplo experimental 6 para confirmar un efecto inhibidor sobre la angiogénesis.
La fig. 9 es una tabla para confirmar la toxicidad de acuerdo con las concentraciones de un compuesto de fórmula 7 y un compuesto de Fórmula 8 en el ejemplo experimental 7, que es una prueba de toxicidad de dosis única en ratas.
[Modo de la invención]
A continuación en el presente documento, se describirá en detalle la presente invención. Los términos o palabras utilizados en la presente memoria descriptiva y las reivindicaciones no deben interpretarse como limitados a significados ordinarios o de diccionario y deben interpretarse como significados y conceptos coherentes con el espíritu de la presente invención basándose en el principio de que un inventor puede definir de manera adecuada conceptos de términos para explicar la invención del inventor de la mejor manera. Por tanto, las configuraciones descritas en las realizaciones expuestas en el presente documento son únicamente realizaciones ilustrativas de la presente invención y no representan todas las ideas técnicas de la presente invención y, por tanto, debe entenderse que se pueden realizar diversos equivalentes y modificaciones que pueden reemplazar estas realizaciones en el momento de presentación de la presente solicitud.
La presente invención se refiere a nuevos compuestos de derivados de quinazolinona como inhibidores de PI3K. Los inhibidores de PI3K bloquean la ruta de señalización de PI3K-AKT al acoplarse con un sitio de unión a ATP de p1105, y la activación de las rutas de PI3K está mediada por isotipos catalíticos de PI3K, es decir, p110a, p110p, p1105 y
p110Y-p1105 desempeña una función vital en la neoplasia sanguínea y el desarrollo de linfocitos B y se expresa predominantemente en células madre hematopoyéticas y se expresa en muchos cánceres, incluyendo leucemia, linfoma, cáncer colorrectal, cáncer de vejiga, glioma maligno y similares. Regula la proliferación celular mediante la estimulación de citocinas y quimiocinas relacionadas a través de la ruta de señalización PI3K-AKT.
Además, los nuevos derivados de quinazolinona según la presente invención superan los problemas de toxicidad existentes, incluyendo la hepatotoxicidad. Ya que PI3K p1105 podría expresarse en gran medida incluso en el carcinoma hepatocelular avanzado, los nuevos derivados de quinazolinona según la presente invención también son eficaces como agentes terapéuticos para el carcinoma hepatocelular, que es un cáncer sólido.
En vista de los problemas de los fármacos antineoplásicos a base de quinazolinona existentes, los nuevos derivados de quinazolinona deberían inhibir de forma suficiente y selectiva PI3K5. Preferentemente, la selectividad entre isómeros de PI3K debe satisfacer los criterios de que, usando valores de CI50, cada una de las proporciones de PI3Ka/PI3K8 y PI3Kp/PI3K8 supera 150, y una proporción de PI3Ky/PI3K8 es mayor que al menos la de idelalisib. Además, cuando PI3K8 y PI3Ky se inhiben simultáneamente, es preferible que las proporciones de PI3Kp/PI3K8 y PI3Kp/PI3KY sean mayores que las de duvelisib.
La presente invención proporciona un compuesto representado por la Fórmula 1 o una sal farmacéuticamente aceptable del mismo.
[Fórmula 1]

en donde,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
El término "halógeno", como se usa en el presente documento, se refiere a flúor (F), bromo (Br), cloro (Cl) o yodo (I). En la Fórmula 1, Y puede estar unido en forma de isómero (S) o isómero (R), pero preferentemente en forma de isómero (S).
Los ejemplos específicos del compuesto de Fórmula 1 incluyen compuestos representados por las siguientes Fórmulas 7 a 14, pero la presente invención no se limita a los mismos.
También se muestran los compuestos de referencia 9 - 11.
El compuesto de Fórmula 7 de acuerdo con la presente invención es el compuesto de Fórmula 1 en donde X es -F e Y es ciclopropilo.
[Fórmula 7]

Además, el compuesto de Fórmula 8 de acuerdo con la presente invención es el compuesto de Fórmula 1 en donde
X es metilo e Y es ciclopropilo.
[Fórmula 8]

Además, el compuesto de Fórmula 9 es el compuesto de Fórmula 1 en donde X es -NH2 e Y es ciclopropilo.
[Fórmula 9 - Referencia]

Además, el compuesto de Fórmula 10 es el compuesto de Fórmula 1 en donde X es -NH2 e Y es metilo.
[Fórmula 10 - Referencia]

Además, el compuesto de Fórmula 11 es el compuesto de Fórmula 1 en donde X es -NH2 e Y es etilo.
[Fórmula 11 - Referencia]

Además, el compuesto de Fórmula 12 de acuerdo con la presente invención es el compuesto de Fórmula 1 en donde X es -Cl e Y es ciclopropilo.
[Fórmula 12]

Además, el compuesto de Fórmula 13 de acuerdo con la presente invención es el compuesto de Fórmula 1 en donde X es -F e Y es ciclobutilo.
[Fórmula 13]

Además, el compuesto de Fórmula 14 de acuerdo con la presente invención es el compuesto de Fórmula 1 en donde X es -Cl e Y es ciclobutilo.
[Fórmula 14]

El compuesto representado por la Fórmula 1 de la presente invención puede usarse en forma de una sal farmacéuticamente aceptable y la sal puede ser una sal de adición de ácido formada por un ácido libre farmacéuticamente aceptable. La sal de adición de ácido se obtiene a partir de: ácidos inorgánicos, tales como ácido clorhídrico, ácido nítrico, ácido fosfórico, ácido sulfúrico, ácido bromhídrico, ácido yodhídrico, ácido nitroso, ácido fosforoso y similares; ácidos orgánicos no tóxicos, tales como mono y dicarboxilatos alifáticos, alcanoatos, hidroxi alcanoatos y alcandioato sustituidos con fenilo, ácidos aromáticos, ácidos sulfónicos alifáticos y aromáticos y similares; o ácidos orgánicos, tales como ácido acético, ácido benzoico, ácido cítrico, ácido láctico, ácido maleico, ácido glucónico, ácido metansulfónico, ácido 4-toluenosulfónico, ácido tartárico, ácido fumárico y similares. Ejemplos de estas sales farmacéuticamente no tóxicas incluyen sulfatos, pirosulfatos, bisulfatos, sulfitos, bisulfitos, nitratos, fosfatos, monohidrogenofosfatos, dihidrogenofosfatos, metafosfatos, pirofosfatocloruros, bromuros, yoduros, fluoruros, acetatos, propionatos, decanoatos, caprilatos, acrilatos, formiatos, isobutiratos, capratos, heptanoatos, propiolatos, oxalatos, malonatos, succinatos, suberatos, sebacatos, fumaratos, maleatos, butina-1,4-dioatos, hexano-1,6-dioatos, benzoatos, clorobenzoatos, metilbenzoatos, dinitrobenzoatos, hidroxibenzoatos, metoxibenzoatos, ftalatos, tereftalatos, bencenosulfonatos, toluenosulfonatos, clorobencenosulfonatos, xilenosulfonatos, fenilacetatos, fenilpropionatos, fenilbutiratos, citratos, lactatos, p-hidroxibutiratos, glicolatos, malatos, tartratos, metanosulfonatos, propanosulfonatos, naftaleno-1-sulfonatos, naftaleno-2-sulfonatos, mandelatos y similares.
Las sales de adición de ácido de acuerdo con la presente invención pueden prepararse usando un método convencional. Por ejemplo, estas sales de adición de ácido pueden prepararse disolviendo el derivado de Fórmula 1
en un disolvente orgánico, tales como metanol, etanol, acetona, diclorometano, acetonitrilo o similares, añadiendo un ácido orgánico o un ácido inorgánico a las misma para producir un precipitado y filtrando y secando el precipitado, o puede prepararse destilando un disolvente y un exceso de un ácido a presión reducida y después secando la solución resultante, seguido de cristalización en presencia de un disolvente orgánico.
Además, las sales metálicas farmacéuticamente aceptables pueden prepararse usando bases. Las sales de metales alcalinos o de metales alcalinotérreos se obtienen, por ejemplo, disolviendo un compuesto en un exceso de una solución de hidróxido de metal alcalino o hidróxido de metal alcalinotérreo, filtrando una sal de compuesto insoluble y evaporando y secando el filtrado. En este momento, es farmacéuticamente preferible que se prepare una sal de sodio, una sal de potasio o una sal de calcio como una sal metálica. Además, las sales correspondientes se obtienen haciendo reaccionar un metal alcalino o una sal de metal alcalinotérreo con una sal de plata adecuada (por ejemplo, nitrato de plata).
Además, la presente invención incluye no solo el compuesto representado por la Fórmula 1 y sus sales farmacéuticamente aceptables, sino también solvatos, estereoisómeros, hidratos y similares que se pueden preparar a partir de ellos.
La presente invención también proporciona una composición farmacéutica para prevenir o tratar un cáncer de sangre, una enfermedad hepática y una enfermedad autoinmune, que incluye un compuesto representado por la Fórmula 1 a continuación o una sal farmacéuticamente aceptable del mismo como un principio activo.
[Fórmula 1]

en donde
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
En la Fórmula 1, Y puede estar unido en forma de isómero (S) o isómero (R), pero es preferible que Y esté unido en forma de isómero (S). En la composición farmacéutica de acuerdo con la presente invención, el compuesto representado por la Fórmula 1 o una sal farmacéuticamente aceptable del mismo puede administrarse por vía oral o parenteral en diversas formas de dosificación durante la administración clínica, y puede formularse usando diluyentes o excipientes de uso común, tales como rellenos, expansores, aglutinantes, agentes humectantes, agentes disgregantes, tensioactivos y similares.
Las formulaciones para administración oral pueden incluir, por ejemplo, comprimidos, píldoras, cápsulas duras/blandas, líquidos, suspensiones, emulsiones, jarabes, gránulos, elixires, suspensiones, trociscos y similares. Estas formulaciones incluyen, además del principio activo, un diluyente (p. ej., lactosa, dextrosa, sacarosa, manitol, sorbitol, celulosa y/o glicina) o un lubricante (p. ej., sílice, talco, ácido esteárico y sales de magnesio o calcio del mismo y/o polietilenglicol). Los comprimidos pueden incluir un aglutinante tal como silicato de magnesio y aluminio, pasta de almidón, gelatina, metilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona y, en algunos casos, pueden incluir un agente disgregante tal como almidón, agar, ácido algínico o sales sódicas del mismo, o una mezcla en ebullición y/o un absorbente, un agente colorante, un agente aromatizante y un agente edulcorante.
La composición farmacéutica que incluye el compuesto representado por fórmula 1 como principio activo puede administrarse por vía parenteral, y la administración parenteral se realiza mediante inyección subcutánea, inyección intravenosa, inyección intramuscular o inyección intratorácica.
A este respecto, para preparar formulaciones para administración parenteral, el compuesto representado por la fórmula 1 o una sal farmacéuticamente aceptable del mismo se mezcla con un estabilizador o un tampón en agua para preparar una solución o una suspensión, seguido de la preparación en una forma de dosificación unitaria de ampolla o vial. La composición puede esterilizarse y/o puede incluir un adyuvante tal como un conservante, un estabilizador, polvo humectable, una sal para osmorregulación y/o un tampón, y otros materiales terapéuticamente eficaces, y pueden formularse utilizando un método convencional, tal como mezclado, granulación o recubrimiento.
La composición de la presente invención puede incluir adicionalmente, además del compuesto de quinazolinona, uno o más ingredientes eficaces que presentan funciones idénticas o similares.
Puede seleccionarse adecuadamente una dosis adecuada de la composición farmacéutica de la presente invención dependiendo de la condición y el peso corporal de los pacientes, la gravedad de los síntomas, la forma de dosificación, la vía de administración y el periodo de administración. En la composición de la presente invención, es preferible que el ingrediente o los ingredientes eficaces se administren en una cantidad de 0,2 mg/kg a 200 mg/kg al día para una eficacia óptima. La composición se puede administrar una vez al día o en múltiples dosis al día, pero la presente invención no se limita a lo mismo.
Según la presente invención, la neoplasia sanguínea puede ser leucemia o linfoma.
La leucemia puede seleccionarse de leucemia linfocítica aguda (LLA), leucemia mielógena aguda (LMA), leucemia linfocítica crónica (LLC) y linfoma linfocítico pequeño (LLP) y la leucemia linfocítica aguda también se conoce como leucemia linfoblástica aguda.
El linfoma puede ser una neoplasia de linfocitos B maduros (periféricos) y, más en particular, puede seleccionarse de leucemia linfocítica crónica de linfocitos B/linfoma linfocítico pequeño; leucemia prolinfocítica de linfocitos B; linfoma linfoplasmacítico; linfoma de la zona marginal, por ejemplo, linfoma de linfocitos B de la zona marginal esplénica (+/-linfocitos vellosos), linfoma ganglionar de la zona marginal (+/- linfocitos B monocitoides) y linfoma extraganglionar de linfocitos B de la zona marginal de tipo tejido linfoide asociado a la mucosa (MALT); tricoleucemia; mieloma de células plasmáticas/plasmacitoma; linfoma folicular; linfoma del centro del folículo; linfoma de células del manto; linfoma difuso de linfocitos B grandes (incluyendo linfoma mediastínico de linfocitos B grandes, linfoma intravascular de linfocitos B grandes y linfoma de efusión primario); y linfoma de Burkitt/linfoma de células de Burkitt.
Además, el linfoma puede seleccionarse de mieloma múltiple (MM), linfoma no hodgkiniano (LNH), linfoma de células del manto (LCM), linfoma folicular, macroglobulinemia de Waldenstrom (MW), linfoma de linfocitos B y linfoma difuso de linfocitos B grandes (DLBCL).
La hepatopatía de la presente invención se puede seleccionar del grupo que consiste en esteatosis hepática no alcohólica (EHNA), esteatohepatitis no alcohólica (ENA), esteatosis hepática, hepatocirrosis, hepatitis, adenoma hepático, hipersensibilidad a la insulina y cáncer de hígado.
El cáncer de hígado puede ser, por ejemplo, un tumor de hígado, adenoma hepatocelular o carcinoma hepatocelular. La enfermedad autoinmunitaria se puede seleccionar del grupo que consiste en rinitis alérgica, asma, enfermedad pulmonar obstructiva crónica (EPOC) y artritis reumatoide.
La presente invención también proporciona un método para preparar un compuesto representado por la Fórmula 1 a continuación, en donde el método comprende:
hacer reaccionar un compuesto representado por la Fórmula 2 a continuación con un compuesto representado por la Fórmula 3 a continuación para preparar un compuesto representado por la Fórmula 4 a continuación; desproteger el compuesto de Fórmula 4 para preparar un compuesto representado por la Fórmula 5 a continuación; y
hacer reaccionar un compuesto representado por la Fórmula 5 a continuación con un compuesto representado por la Fórmula 6 para preparar el compuesto representado por la Fórmula 1.
[Fórmula 1]

en donde, en la Fórmula 1
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
[Fórmula 2]

en donde, en la Fórmula 2,
X es -H, halógeno o -CH3
[Fórmula 3]

en donde, en la Fórmula 3,
Y es cicloalquilo C3-4
[Fórmula 4]

en donde, en la Fórmula 4,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
[Fórmula 5]
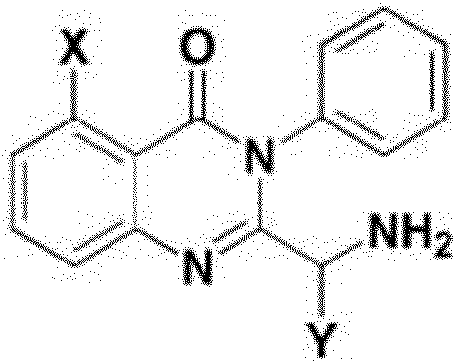
en donde, en la Fórmula 5,
X es -H, halógeno o -CH3 ; e
Y es cicloalquilo C3-4.
[Fórmula 6]

en donde, en la Fórmula 6,
Z es halógeno.
La etapa 1 es un proceso de preparación del compuesto representado por la Fórmula 4 haciendo reaccionar el compuesto representado por la Fórmula 2 con el compuesto representado por la Fórmula 3.
Por ejemplo, se puede añadir trifenilfosfito a una solución en la cual el compuesto representado por la Fórmula 2 y el compuesto representado por la Fórmula 3 se mezclan juntos en presencia de un disolvente de piridina mientras se agita a temperatura ambiente.
En este momento, la temperatura no está particularmente limitada, pero la mezcla se puede agitar a una temperatura de 30 °C a 100 °C, preferentemente de 45 °C a 80 °C, y más preferentemente de 55 °C a 60 °C.
El tiempo de agitación no está particularmente limitado, pero el proceso de agitación puede realizarse durante 5 horas a 20 horas, preferentemente 8 horas a 16 horas y más preferentemente 10 horas a 14 horas.
Posteriormente, puede añadirse anilina para permitir que ocurra una reacción. En este momento, la temperatura no está particularmente limitada, pero la reacción puede ocurrir a una temperatura de 50 °C a 200 °C, preferentemente de 90 °C a 150 °C, y más preferentemente de 100 °C a 120 °C.
En este momento, el tiempo de reacción no está particularmente limitado, pero la reacción puede tener lugar durante 1 hora a 20 horas, preferentemente de 3 horas a 15 horas y más preferentemente de 5 horas a 10 horas.
La etapa 2 es un proceso de preparación del compuesto representado por la Fórmula 5 desprotegiendo el compuesto representado por la Fórmula 4.
Por ejemplo, el compuesto representado por la Fórmula 5 puede prepararse como sigue a continuación: El compuesto representado por la Fórmula 4 se agrega a una solución de diclorometano en la cual se disuelve ácido trifluoroacético (CF3COOH) y después se hace reaccionar a temperatura ambiente durante 0,1 a 2 horas, preferentemente de 0,2 horas a 1,5 horas y más preferentemente 0,5 horas a 1 hora.
La etapa 3 es un proceso de preparación del compuesto representado por la Fórmula 1 haciendo reaccionar el compuesto representado por la Fórmula 5 con el compuesto representado por la Fórmula 6.
Por ejemplo, se añade el compuesto representado por la Fórmula 5 a ferc-butanol, se añade N,N-diisopropiletilamina a la misma, y después, se añade el compuesto representado por la Fórmula 6 a la solución resultante y la solución de reacción puede agitarse mientras se somete a reflujo de 10 horas a 48 horas, preferentemente de 15 horas a 30 horas y más preferentemente de 20 horas a 26 horas.
La presente invención proporciona un alimento funcional para la salud para prevenir o aliviar un tumor sanguíneo o una enfermedad hepática, que incluye un nuevo compuesto de quinazolinona o una sal farmacéuticamente aceptable del mismo como principio activo.
El alimento funcional saludable se puede preparar en forma de, pero no se limita a, diversos tipos de bebidas, gomas, té, productos de confitería, complejos vitamínicos y suplementos para la salud.
En lo sucesivo en el presente documento, la presente invención se describirá en detalle con referencia a ejemplos y ejemplos experimentales.
Sin embargo, estos ejemplos y ejemplos experimentales se proporcionan únicamente con fines ilustrativos y no pretenden limitar el alcance de la presente invención.
<Ejemplo 1> Preparación de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-fluoro-3-fenilquinazolin-4(3H)-ona
(Fórmula 7)
[Esquema de reacción 1]

Etapa 1: Preparación de c¡cloprop¡l(5-fluoro-4-oxo-3-fen¡l-3.4-d¡hidroqu¡nazol¡n-2-¡l)met¡lcarbamato de (S)-ferc-butilo Se añadió fosfito de trifenilo (1.4 equiv.) a una solución. en la cual se mezclaron ácido 2-amino-6-fluorobenzoico (1.0 equiv.) y ácido (S)-2-(ferc-butoxicarbon¡lam¡no)-2-c¡cloprop¡lacét¡co (1.0 equiv.) en un disolvente de piridina, mientras que la solución se agitó a temperatura ambiente. La mezcla resultante se agitó de 55 °C a 60 °C durante 12 horas. Se añadió anilina (1.4 equiv.) a la misma. y después se hizo reaccionar a aproximadamente 110 °C durante 7 horas. Después de eso. la solución de reacción mixta se enfrió a temperatura ambiente y se extrajo con acetato de etilo y agua. La capa orgánica obtenida se deshidrató con sulfato de magnesio anhidro (MgSO4) y se concentró a presión reducida. Se añadió n-heptano al residuo. seguido de agitación durante 30 minutos para precipitar un sólido. el sólido se filtró y se lavó con n-heptano. y después el sólido resultante se secó para dar ciclopropil(5-fluoro-4-oxo-3-fen¡l-3.4-dih¡droqu¡nazolin-2-¡l)met¡lcarbamato de (S)-ferc-butilo con un rendimiento del 65% al 80%.
RMN 1H (300 MHz. CDCla): 87.66-7.73 (m. 1H). 7.50-7.61 (m. 4H). 7.32-7.40 (m. 2H). 7.09-7.15 (t. J=18 Hz. 1H). 5.53 5.56 (d. J = 9 Hz. 1H). 4.18-4.23 (t. J=15 Hz. 1H). 1.42 (s. 9H). 1.08-1.16 (m. 1H). 0.38-0.42 (m. 2H). 0.24-0.30 (m.
1H). 0.01-0.11 (m. 1H).
Etapa 2: Preparación de (S)-2-(am¡no(c¡cloprop¡l)metil)-5-fluoro-3-fen¡lqu¡nazol¡n-4(3H)-ona
Se añadió ácido trifluoroacético (aproximadamente 8 veces el peso de ciclopropil(5-fluoro-4-oxo-3-fenil-3.4-dih¡droqu¡nazolin-2-¡l)met¡lcarbamato de (S)-ferc-butilo) a una solución de diclorometano (aproximadamente 15 veces el peso de cicloprop¡l(5-fluoro-4-oxo-3-fen¡l-3.4-d¡h¡droqu¡nazol¡n-2-¡l)met¡lcarbamato de (S)-ferc-butilo). en la cual se disolvió cicloprop¡l(5-fluoro-4-oxo-3-fen¡l-3.4-d¡h¡droqu¡nazol¡n-2-¡l)met¡lcarbamato de (S)-ferc-butilo. La solución de reacción se agitó a temperatura ambiente durante aproximadamente 0.5 horas a aproximadamente 1 hora. y después el pH de la solución de reacción se ajustó a aproximadamente 7 usando una solución acuosa de carbonato sódico. La solución de diclorometano se separó. se deshidrato usando sulfato de magnesio (MgSO4) y se filtró. y se retiró sulfato de magnesio (MgSO4) y después. el filtrado se concentró a presión reducida para dar (S)-2-(amino(cicloprop¡l)met¡l)-5-fluoro-3-fenilquinazolin-4(3H)-ona con un rendimiento del 80 % al 95 %.
RMN 1H (400 MHz. CDCla): 87.66-7.71 (m. 1H). 7.47-7.57 (m. 4H). 7.27-7.31 (m. 2H). 7.08-7.12 (t. J=16 Hz. 1H). 2.97 2.99 (d. J = 8 Hz. 1H). 1.87 (s. 2H). 1.22-1.31 (m. 1H). 0.39-0.53 (m. 2H). 0.01-0.15 (m. 2H).
Etapa 3: Preparación de (S)-2-(((7H-pur¡n-6-¡l)am¡no)(c¡cloprop¡l)metil)-5-fluoro-3-fen¡lqu¡nazol¡n-4(3H)-ona
La (S)-2-(amino(c¡cloprop¡l)met¡l)-5-fluoro-3-fen¡lquinazol¡n-4(3H)-ona obtenida en la etapa 2 se añadió a ferc-butanol (aproximadamente 15 veces el peso de (S)-2-(amino(c¡cloprop¡l)met¡l)-5-fluoro-3-fen¡lquinazol¡n-4(3H)-ona). se añadieron N.N-diisopropilamina (aproximadamente 2 equivalentes de (S)-2-(amino(cicloprop¡l)met¡l)-5-fluoro-3-fenilquinazolin-4(3H)-ona) y 6-bromo-9H-purina a la misma. y después. la solución de reacción se agitó mientras se sometió a reflujo durante 24 horas.
La mezcla de reacción se enfrió y se concentró a presión reducida para retirar ferc-butanol. Se añadió acetato de etilo al concentrado y se lavó secuencialmente con una solución de ácido clorhídrico diluido y una solución de carbonato potásico diluido. La capa de acetato de etilo se deshidrató con sulfato de magnesio anhidro (MgSO4) y se filtró, y el filtrado se concentró a presión reducida, para dar (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-fluoro-3-fenilquinazolina-4(3H)-ona (Fórmula 7) en forma de un sólido con un rendimiento del 60 % al 80 %.
RMN 1H (300 MHz, CDCh): 813,02 (s, 1H), 8,03 (s, 1H), 7,98 (s, 1H), 7,50-7,71 (m, 6H), 7,39-7,42 (dd, J=9 Hz, 1H), 7,08-7,14 (t, J=18 Hz, 1H), 6,76-6,79 (d, J = 9 Hz, 1H), 4,93 (s a, 1H), 1,72 (s a, 1H), 1,33-1,44 (m, 1H), 0,49-0,53 (m, 2H), 0,37-0,46 (m, 1H), 0,21-0,27 (m, 1H).
IEN-EM m/z 428,45 [M+H]+
<Ejemplo 2> Preparación de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-metil-3-fenilquinazolin-4(3H)-ona (Fórmula 8)
[Esquema de reacción 2]

Etapa 1: Preparación de c¡cloprop¡l(5-met¡l-4-oxo-3-fen¡l-3.4-d¡hidroqu¡nazol¡n-2-¡l)met¡lcarbamato de (S)-ferc-butilo Se preparó ciclopropil(5-metil-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)metilcarbamato de (S)-ferc-butilo de la misma manera como en el Ejemplo 1, excepto que se usó ácido 2-amino-6-metilbenzoico en lugar de ácido 2-amino-6-fluorobenzoico.
RMN 1H (400 MHz, CDCla): 88,33 (s a, 3H), 7,50 (d, 4H, J = 7,9 Hz), 7,28 (t, 11H, J = 7,7 Hz), 7,07 (t, 2H, J = 7,3 Hz), 5,35 (s a, 1H), 3,61 (s a, 3H), 1,32-1,51 (m, 12H), 1,17-1,30 (m, 1H), 0,51-0,73 (m, 4H), 0,47 (td, 3H, J=4,7, 9,6 Hz). Etapa 2: Preparación de (S)-2-(am¡no(c¡cloprop¡l)metil)-5-met¡l-3-fen¡lqu¡nazol¡n-4(3H)-ona
Se preparó (S)-2-(amino(ciclopropil)metil)-5-metil-3-fenilquinazolin-4(3H)-ona de la misma manera como en el Ejemplo 1.
RMN 1H (400 MHz, DMSO-d6): 88,41 (s a, 2H), 7,79 (t, 1H, J = 7,7 Hz), 7,54-7,73 (m, 2H), 7,31-7,46 (m, 1H), 2,74 (s, 3H), 1,23 (s a, 1H), 1,18 (tt, 1H, J = 4,4, 8,7 Hz), 0,51 (s, 1H), 0,32-0,41 (m, 1H, J=4,8, 10, 10 Hz).
Etapa 3: Preparación de (S)-2-(((7H-pur¡n-6-¡l)am¡no)(c¡cloprop¡l)metil)-5-met¡l-3-fen¡lqu¡nazol¡n-4(3H)-ona
Se preparó (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-metil-3-fenilquinazolin-4(3H)-ona (Fórmula 8) de la misma manera como en el Ejemplo 1, excepto que se usó (S)-2-(amino(ciclopropil)metil)-5-metil-3-fenilquinazolin-4(3H)-ona en lugar de (S)-2-(amino(ciclopropil)metil)-5-fluoro-3-fenilquinazolin-4(3H)ona.
RMN 1H (400 MHz, CDCh): 88,29 (s, 1H), 7,96 (s a, 1H), 7,36-7,71 (m, 7H), 7,19-7,25 (m, 1H), 6,83 (d, 1H, J=6,6 Hz),
4,96 (t, 1H, J = 8,1 Hz), 2,82 (s, 3H), 1,24-1,43 (m, 2H), 0,29-0,67 (m, 3H), 0,24 (s, 1H), 0,07 (s, 1H).
IEN-EM m/z 424,48 [M+H]+
<Ejemplo de referencia 3> Preparación de (S)-2-(((7H-purin-6-il)aminoXciclopropil)metil)-5-amino-3-fenil-quinazolin-4(3H)-ona (Fórmula 9)
[Esquema de reacción 3]

Etapa 1: Preparación de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-((4-metoxibencil)amino)-3-fenilquinazolin-4(3H)-ona
A un tubo cerrado herméticamente en el cual se preparó una solución añadiendo secuencialmente la (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-fluoro-3-fenilquinazolin-4(3H)-ona (1,0 equiv.) preparada en el Ejemplo 1 y se acomodó trietilamina (5,0 equiv.) a etanol (15 veces el volumen de trietilamina), se añadió adicionalmente 4-metoxibencilamina. Posteriormente, el tubo se reemplazó con nitrógeno y se cerró herméticamente, y después la mezcla de reacción se calentó a 180 °C y se hizo reaccionar durante un día. Después de enfriar hasta la temperatura ambiente, el disolvente de etanol se retiró a presión reducida. Después de eso, una mezcla cruda se sometió a cromatografía en columna sobre gel de sílice (diclorometano/metanol 20:1) para dar (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-((4-metoxibencil)amino)-3-fenilquinazolin-4(3H)-ona en forma de un sólido de color amarillo (rendimiento: 38 %).
RMN 1H (300 MHz, CDCla): 813,67 (s, 1H), 8,80-8,84 (t, J=12 Hz, 1H), 8,31 (s, 1H), 7,96 (s, 1H), 7,52-7,62 (m, 4H), 7,39-7,48 (m, 2H), 7,23-7,25 (d, J = 6 Hz, 2H), 6,84-6,92 (t, J=24 Hz, 2H), 6,80-6,84 (d, J = 12 Hz, 2H), 6,46-6,49 (d, J = 9 Hz, 1H), 4,92 (s, 1H), 4,31-4,33 (d, J = 6 Hz, 2H), 3,76 (s, 3H), 1,37-1,39 (m, 1H), 0,43-0,50 (m, 2H), 0,38-0,40 (m, 1H), 0,20-0,25 (m, 1H).
Etapa 2: Preparación de (S)-2-(((7H-pur¡n-6-¡l)am¡no)(c¡cloprop¡l)metil)-5-am¡no-3-fen¡lqu¡nazol¡n-4(3H)-ona
A una solución en la cual se disolvió (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-(4-metoxibencilamino)-3-fenilquinazolin-4(3H)-ona (1,0 equiv.) en diclorometano (6 veces el volumen de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-(4-metoxibencilamino)-3-fenilquinazolin-4(3H)-ona), se le añadió ácido trifluoroacético (2 veces el volumen de diclorometano) y la solución resultante se agitó a temperatura ambiente durante 0,5 horas a 2 horas. Después de eso, el pH de una mezcla en bruto se ajustó a 7 con una solución 1 M de NaOH a 0 °C. La solución resultante se extrajo tres veces con diclorometano, y las fases orgánicas combinadas se deshidrataron con sulfato de magnesio anhidro (MgSO4) y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice para obtener (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-amino-3-fenilquinazolin-4(3H)-ona (Fórmula 9) en forma de un sólido de marfil (rendimiento: 21 %).
RMN 1H (300 MHz, CDCI3): 513,16 (s, 1H), 8,30 (s, 1H), 7,97 (s, 1H), 7,53-7,64 (m, 4H), 7,40-7,45 (t, J=15 Hz, 2H), 6,92-6,95 (d, J = 9 Hz, 1H), 6,84-6,86 (d, J = 6 Hz, 1H), 6,54-6,56 (d, J = 6 Hz, 1H), 6,15 (s, 2H), 4,93 (s, 1H), 1,32 1,41 (m, 1H), 0,47-0,48 (m, 2H), 0,38-0,43 (m, 1H), 0,22-0,25 (m, 1H).
<Ejemplo de referencia 4> Preparación de (S)-2-(1-((7H-purin-6-il)amino)etil)-5-amino-3-fenilquinazolin-4(3H)-ona (Fórmula 10)
[Esquema de reacción 4]
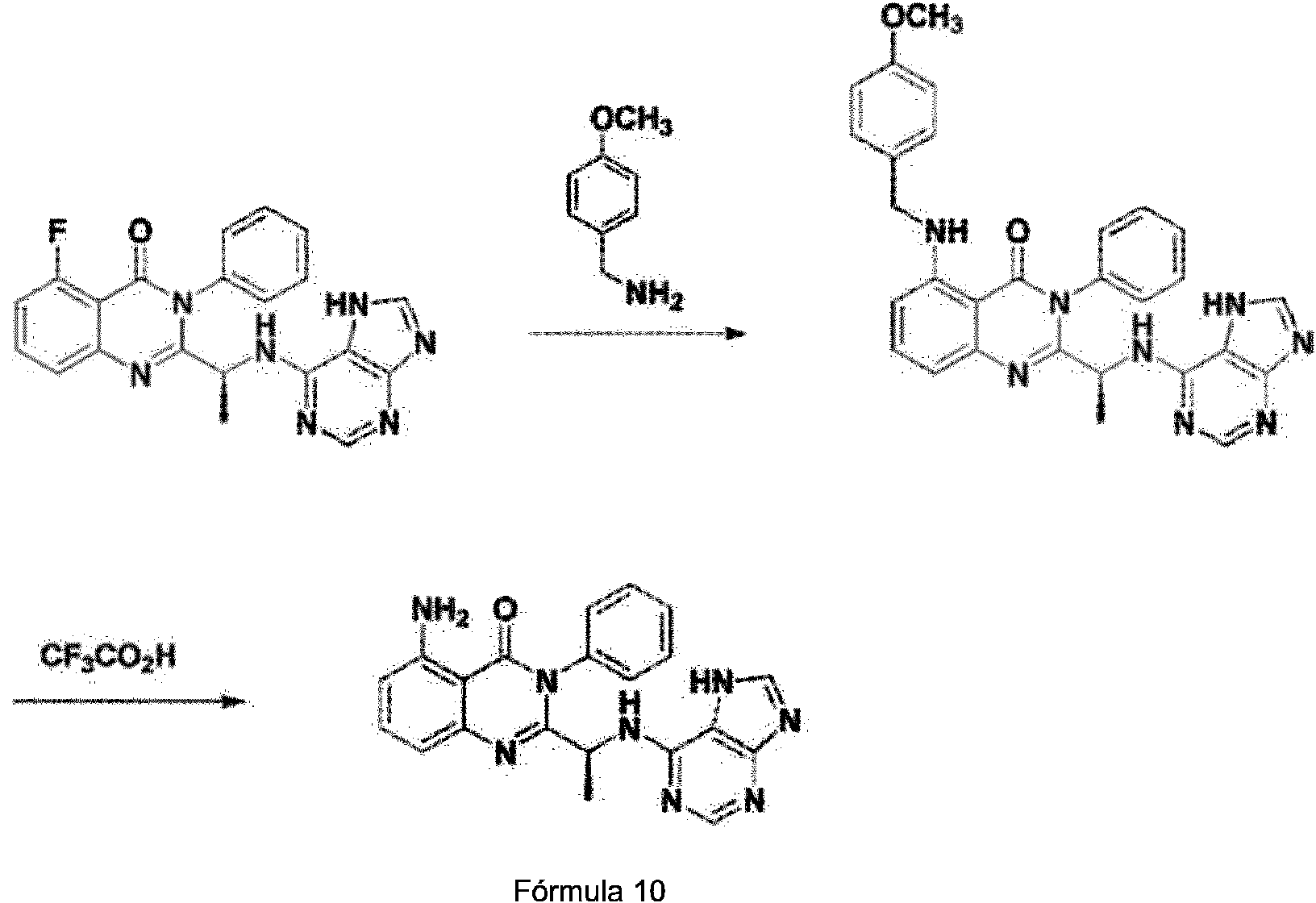
Se preparó (S)-2-(1-((7H-purin-6-il)amino)etil)-5-amino-3-fenilquinazolin-4(3H)-ona (Fórmula 10) de la misma manera como en el Ejemplo 3, excepto que se usó (S)-2-(1-((7H-purin-6-il)amino)etil)-5-fluoro-3-fenilquinazolin-4(3H)-ona en lugar de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-fluoro-3-fenilquinazolin-4(3H)-ona.
RMN 1H (500 MHz, DMSO-d6): 58,00-8,22 (m, 1H), 7,32-7,78 (m, 4H), 7,06 (s a, 1H), 6,55-6,70 (m, 1H), 1,99 (s, 1H), 1,06-1,55 (m, 4H), 0,70-0,93 (m, 2H).
<Ejemplo de referencia 5> Preparación de (S)-2-(1-(7H-purin-6-ilamino)propil)-5-amino-3-fenilquinazolin-4(3H)-ona (Fórmula 11)
[Esquema de reacción 5]

Se preparó (S)-2-(1-((7H-purin-6-il)amino)propil)-5-amino-3-fenilquinazolin-4(3H)-ona (Fórmula 11) de la misma manera como en el Ejemplo 3, excepto que se usó (S)-2-(1-((7H-purin-6-il)amino)propil)-5-fluoro-3-fenilquinazolin-4(3H)-ona en lugar de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-fluoro-3-fenilquinazolin-4(3H)-ona.
RMN 1H (300 MHz, CDCla): 88,31 (s, 1H), 7,97 (s, 1H), 7,35-7,68 (m, 6H), 6,91-6,94 (d, J = 9 Hz, 1H), 6,82-6,84 (d, J = 6 Hz, 1H), 6,53-6,56 (d, J = 9 Hz, 1H), 6,15 (s, 2H), 5,16 (s, 1H), 1,91-2,05 (m, 1H), 1,74-1,84 (m, 1H), 0,84-0,89 (t, J=15 Hz, 3H).
<Ejemplo 6> Preparación de (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-cloro-3-fenilquinazolin-4(3H)-ona (Fórmula 12)
[Esquema de reacción 6]
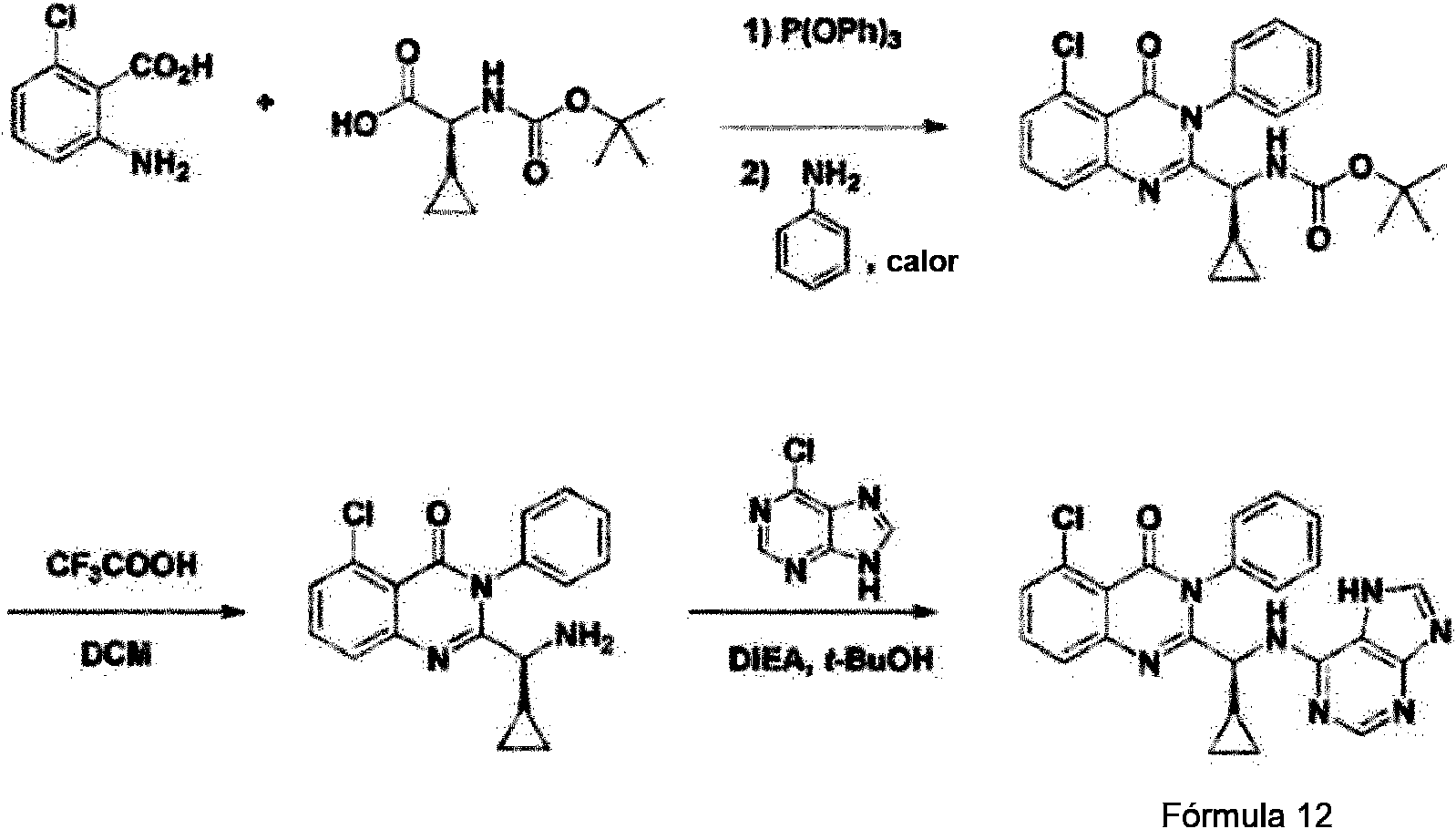
Etapa 1: Preparación de (5-cloro-4-oxo-3-fen¡l-3.4-d¡h¡droqu¡nazolin-2-¡l)(c¡cloprop¡l)met¡l-carbamato de (SWerc-butilo
Se preparó (5-doro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)(cidopropil)metilcarbamato de (S)-ferc-butilo de la misma manera como en el Ejemplo 1, excepto que se usó ácido 2-amino-6-clorobenzoico en lugar de ácido 2-amino-6-fluorobenzoico.
Etapa 2: Preparación de (S)-2-(am¡no(c¡cloprop¡l)met¡l)-5-cloro-3-fen¡lqu¡nazol¡n-4(3H)-ona
Se preparó (S)-2-(amino(ciclopropil)metil)-5-cloro-3-fenilquinazolin-4(3H)-ona de la misma manera como en el Ejemplo 1.
Etapa 3: Preparación de (S)-2-(((7H-pur¡n-6-¡l)am¡no)(c¡cloprop¡l)met¡l)-5-cloro-3-fen¡lqu¡nazol¡n-4(3H)-ona
Se preparó (S)-2-(((7H-purin-6-il)amino)(ciclopropil)metil)-5-cloro-3-fenilquinazolin-4(3H)-ona (Fórmula 12) de la misma manera como en el Ejemplo 1, excepto que se usó (S)-2-(amino(ciclopropil)metil)-5-cloro-3-fenilquinazolin-4(3H)-ona en lugar de (S)-2-(amino(ciclopropil)metil)-5-fluoro-3-fenilquinazolin-4(3H)-ona.
RMN 1H (300 MHz, CDCla): 813,02 (s, 1H), 8,02 (s, 1H), 7,98 (s, 1H), 7,15-7,68 (m, 8H), 6,76-6,79 (d, J = 9 Hz, 1H), 4,93 (s a, 1H), 1,72 (s a, 1H), 1,33-1,44 (m, 1H), 0,49-0,53 (m, 2H), 0,37-0,46 (m, 1H), 0,21-0,27 (m, 1H).
IEN-EM m/z 444,40 [M+H]+
<Ejemplo 7> Preparación de (S)-2-(((7H-purin-6-il)amino)(ciclobutil)metil)-5-fluoro-3-fenilquinazolin-4(3H)-ona (Fórmula 13)

Se preparó (S)-2-(((7H-purin-6-il)amino)(ciclobutil)metil)-5-flouro-3-fenilquinazolin-4(3H)-ona (Fórmula 13) de la misma manera como en el Ejemplo 1, excepto que se usó ácido (S)-2-(ferc-butoxicarbonilamino)-2-ciclobutilacético en lugar de ácido (S)-2-(ferc-butoxicarbonilamino)-2-ciclopropilacético.
RMN 1H (300 MHz, DMSO-d6): 812,95 (s, 1H), 8,13 (s a, 1H), 7,85 (s a, 1H), 7,24-7,60 (m, 8H), 5,18 (s a, 1H), 3,05 (s a, 1H), 1,64-2,01 (m, 7H).
<Ejemplo 8> Preparación de (S)-2-(((7H-purin-6-il)amino)(ciclobutil)metil)-5-cloro-3-fenilquinazolin-4(3H)-ona (Fórmula 14)

Se preparó (S)-2-(((7H-purin-6-il)amino)(cidobutil)metil)-5-doro-3-fenilquinazolin-4(3H)-ona (Fórmula 14) de la misma manera como en el Ejemplo 1, excepto que se usó ácido 2-amino-6-clorobenzoico en lugar de ácido 2-amino-6-fluorobenzoico y ácido (S)-2-(terc-butoxicarbonilamino)-2-ciclobutilacético se usó en lugar de ácido (S)-2-(terc-butoxicarbonilamino)-2-ciclopropilacético.
RMN 1H (300 MHz, DMSO-da): 812,88 (s, 1H), 8,17 (s a, 1H), 8,00 (s, 1H), 7,12-7,87 (m, 8H), 5,18 (s a, 1H), 3,06 (s a, 1H), 1,62-1,99 (m, 7H)
Las estructuras de los compuestos de Fórmulas 7 a 14 se muestran en la Tabla 1 a continuación. (N.b. Las fórmulas 9-11 de la Tabla 1 no forman parte de la presente invención)
T l 11

continuación

<Ejemplo experimental 1> Prueba de actividad de cinasa PI3K
(1) Método experimental
Se realizó un experimento utilizando un inmunoensayo basado en fluorescencia, homogéneo, que es el ensayo de cinasa Adapta.
Se llevaron a cabo servicios SelectScreen™ disponibles de Thermo Fisher Scientific Inc. Un principio del experimento se muestra en la fig. 1.
(2) Resultados experimentales
Los resultados del experimento se muestran en la tabla 2 a continuación.
Como se muestra en la tabla 2, el compuesto de fórmula 7 y el compuesto de fórmula 8 presentaron mayor actividad que el fármaco de control, idelalisib. En particular, presentaron actividad inhibidora específica sobre p1108 y también presentaron excelente actividad con respecto a p11üY.
En particular, se mostró que las proporciones de P13Ka/PI3K8 y PI3Kp/PI3K8, como valores de CI50, eran 412 y 210 respectivamente en el caso del compuesto de fórmula 7 y 1488 y 1800 respectivamente en el caso del compuesto de fórmula 8.
Además, se confirmó que el compuesto de fórmula 7 y el compuesto de fórmula 8 tienen alta selectividad delta (8). Cada uno de ellos presentó una proporción de PI3Ky/PI3K8 de 51 y 95 respectivamente, mientras que idelalisib presentó la proporción de aproximadamente 25. A partir de estos resultados, se confirmó que los compuestos de fórmulas 7 y 8 podrían tener actividad eficaz y suficiente con respecto a cánceres dependientes de delta (8).
T l 21

<Ejemplo experimental 2> Experimento para confirmar el efecto sobre la reducción de la fosforilación de AKT(Ser473) en líneas celulares de leucemia y linfoma
(1) Método experimental
Se sometieron líneas celulares (SUDHL 5, SUDHL 10, CCRF-SB y MOLT4) a agotamiento del suero durante 2 horas y después se trataron con 1 pM de cada uno del compuesto de fórmula 7, el compuesto de fórmula 8, idelalisib (fármaco comparativo 1), TGR1202 (fármaco comparativo 2) y dimetilsulfóxido (DMSO) durante 1 hora. Posteriormente, las células se lisaron y fraccionaron según el tamaño, seguido de inmunotransferencia con anticuerpos dirigidos contra fosfo-Akt (Ser473).
(2) Resultados experimentales
Los resultados del experimento se muestran en la fig. 2.
Como se ilustra en la fig. 2, se confirmó que el compuesto de fórmula 7 y el compuesto de fórmula 8 inducían la reducción de la fosforilación de AKT en diversas células de linfoma difuso de linfocitos B grandes (DLBCL) y leucemia linfocítica aguda (LLA).
<Ejemplo experimental 3> Experimento para confirmar el efecto sobre la inhibición del crecimiento de células de leucemia y linfoma
(1) Método experimental
PI3K p1108 se expresa en gran medida en líneas celulares de leucemia y linfoma, y el crecimiento celular se inhibe suprimiendo PI3K p1108.
Por tanto, se llevó a cabo un experimento para confirmar los efectos de los compuestos sobre la inhibición del crecimiento celular.
Se cultivaron células derivadas de linfoma difuso de linfocitos B grandes (DLBCL) y células derivadas de leucemia linfocítica aguda (LLA) con el compuesto de fórmula 7, el compuesto de fórmula 8, o idelalisib, junto con un medio de control durante 48 horas.
Los efectos inhibidores del crecimiento celular sobre las células derivadas de DLBCL y las células derivadas de LLA se evaluaron midiendo la absorbancia del colorante del equipo de recuento celular-8 (CCK-8). Durante las últimas 3 horas de las 48 horas, se añadieron 10 pM de un colorante CCK-8 a cada placa y después se cultivaron.
Todos los datos se expresan como la media (± DT) de tres experimentos independientes.
(2) Resultados experimentales
Los resultados del experimento se muestran en la fig. 3.
Como se ilustra en la fig. 3, el crecimiento de las células derivadas de DLBCL y la línea celular derivada de LLA se redujo a una concentración que variaba de 0,625 pM a 20 pM.
En este momento, la concentración letal 50 (CL50), que es la concentración de compuestos que son letales para el 50 % de las células, se muestra en la tabla 3, y se confirmó que el compuesto de fórmula 7 y el compuesto de fórmula 8 presentaron un valor de CL50 menor que el de idelalisib.
T l

<Ejemplo experimental 4> Experimento para confirmar el efecto sobre la apoptosis de linfoma difuso de linfocitos B grandes (DLBCL) y células de leucemia linfocítica aguda (LLA) mediante s DS-PAGE
(1) Método experimental
Se cultivaron 2,6 x 106 células con idelalisib (fármaco comparativo), el compuesto de fórmula 7 o el compuesto de fórmula 8 a una concentración de 50 pM durante 36 horas, y después se realizó análisis de proteínas mediante electroforesis en gel de poliacrilamida-dodecilsulfato de sodio (SDS-PAGE).
Las proteínas PARP caspasa 3 y 9, que son proteínas implicadas en la apoptosis, existen normalmente como precursores inactivos (FL) y se activan al escindirse (CL) cuando reciben una señal estimulante de la apoptosis. Se realizó análisis de inmunotransferencia usando anticuerpos de estos, y se usó gliceraldehído-3-fosfato deshidrogenasa (GAPDH) como control de carga.
(2) Resultados experimentales
Los resultados del experimento se muestran en la fig. 4.
Como se ilustra en la fig. 4, el compuesto de fórmula 7 y el compuesto de fórmula 8 indujeron apoptosis en linfomas
difusos de linfocitos B grandes (DLBCL) y células de leucemia linfocítica aguda (LLC).
Además, se produjo apoptosis más activamente con el compuesto de fórmula 7 y el compuesto de fórmula 8 que con el fármaco comparativo (idelalisib).
A partir de estos resultados, se confirmó que el compuesto de fórmula 7 y el compuesto de fórmula 8 inhibían el crecimiento celular mediante apoptosis.
<Ejemplo experimental 5> Experimento para confirmar el efecto sobre la apoptosis de células de linfoma difuso de linfocitos B grandes (DLBCL) y leucemia linfocítica aguda (LLA) mediante citometría de flujo
(1) Método experimental
Se cultivaron 1 x 106 células de linfoma difuso de linfocitos B grandes (DLBCL) y 1 x 106 células de leucemia linfocítica aguda (LLA) y se trataron con cada uno de los fármacos comparativos (Idelalisib), el compuesto de fórmula 7 y el compuesto de fórmula 8 a una concentración de 50 pM durante 24 horas. Las células se lavaron con PBS y después se suspendieron en un tampón de unión. Se añadieron a las mismas 5 pl de una solución de reserva de Anexina V-FITC (Becton Dickinson Science, Inc) y 5 pl de PI (20 pg/ml), seguido de incubación a temperatura ambiente durante 15 minutos con protección contra la luz, y después se cuantificó el material diana en FACScan TM (Becton Dickinson) mediante citometría de flujo.
(2) Resultados experimentales
Los resultados del experimento se muestran en la fig. 5.
Como se ilustra en la fig. 5, se confirmó que el compuesto de fórmula 7 y el compuesto de fórmula 8 inducían apoptosis más eficazmente que el fármaco comparativo (Idelalisib).
<Ejemplo experimental 6> Experimento para confirmar el efecto inhibidor sobre la angiogénesis
(1) Método experimental
Para comparar los niveles de inhibición de la angiogénesis por el compuesto de fórmula 7, el compuesto de fórmula 8 y un fármaco comparativo (Idelalisib), se obtuvieron células endoteliales de la vena umbilical humana (HUVEC), que son células endoteliales vasculares, y medios de cultivo de células endoteliales de Life Technologies.
Las células endoteliales de la vena umbilical humana (HUVEC) se cultivaron junto con el compuesto de fórmula 7, el compuesto de fórmula 8 o el fármaco comparativo (Idelalisib) en la matriz de la membrana basal a 37 °C. Después de 18 horas, la formación del tubo se fotografió utilizando un microscopio de fluorescencia Cytation™ 5 (BioTek) y se contó el número de áreas formadas por puntos de ramificación utilizando programas informáticos (véase fig. 6). (2) Resultados experimentales
Los resultados del experimento se muestran en las fig. 7 y 8.
Como se ilustra en la fig. 7, el compuesto de fórmula 7 y el compuesto de fórmula 8 inhibieron la angiogénesis más que el fármaco comparativo (Idelalisib).
Además, como se ilustra en la fig. 8, el compuesto de fórmula 7 y el compuesto de fórmula 8 no afectaron significativamente a la citotoxicidad de las células endoteliales de la vena umbilical humana (HUVEC).
<Ejemplo experimental 7> Prueba de toxicidad de dosis única en ratas
(1) Método experimental
El compuesto de fórmula 7 y el compuesto de fórmula 8 se administraron por vía oral a ocho ratas hembra de seis semanas de edad, para observar la toxicidad oral de dosis única de los mismos y obtener una dosis letal aproximada. La dosis se fijó en 10 ml/kg y la dosis para cada rata se calculó en función del peso corporal. Cada compuesto se administró a dosis de 100 mg/kg, 300 mg/kg, 900 mg/kg y 1500 mg/kg, y se observaron los síntomas generales una vez al día desde el día 1 al día 2 después de la administración.
(2) Resultados experimentales
Los resultados del experimento se muestran en la fig. 9.
La dosis letal del compuesto de fórmula 7 es superior a 1.500 mg/kg y la del compuesto de fórmula 8 es de 1500 mg/kg.
Por otra parte, el compuesto representado por fórmula 1 según la presente invención se puede formular en diversas formas. A continuación se proporcionan varios métodos de formulación que utilizan el compuesto representado por la fórmula 1 según la presente invención como principio activo solo con fines ilustrativos, pero no se pretende que limiten la presente invención.
<Ejemplo de preparación 1> Formulación de preparaciones farmacéuticas
1-1. Preparación de polvo
Compuesto de fórmula 1 500 mg
Lactosa 100 mg
Talco 10 mg
Los ingredientes anteriores se mezclaron y se llenaron paquetes herméticos con ellos para preparar el polvo.
1-2. Preparación de comprimidos
Compuesto de fórmula 1 500 mg
Almidón de maíz 100 mg
Lactosa 100 mg
Estearato de magnesio 2 mg
Se mezclaron los ingredientes anteriores y después se prepararon comprimidos según un método general de preparación de comprimidos.
1-3. Preparación de cápsulas
Compuesto de fórmula 1 500 mg
Almidón de maíz 100 mg
Lactosa 100 mg
Estearato de magnesio 2 mg
Los ingredientes anteriores se mezclaron y después se llenaron cápsulas de gelatina con ellos según un método general de preparación de cápsulas para preparar cápsulas.
1-4. Preparación de inyecciones
Compuesto de fórmula 1 500 mg
Agua destilada estéril para inyección cantidad adecuada
Ajustador del pH cantidad adecuada
Según un método general de preparación de una inyección, se prepararon ampollas con los ingredientes anteriores incluidos en una sola ampolla (2 ml).
1-5. Preparación de líquidos
Compuesto de fórmula 1 100 mg
Azúcar isomerizado 10 g
Manitol 5 g
Agua purificada cantidad adecuada
Cada ingrediente se añadió y se disolvió en agua purificada según un método general de preparación de un líquido. Se añadió un aroma de limón en una cantidad adecuada y se mezclaron los ingredientes anteriores. Se le añadió agua purificada de manera que la cantidad total de la solución resultante se ajuste a 100 ml. Se llena una botella marrón con ella y se esteriliza para preparar líquidos.
Claims (7)
1. Un compuesto de Fórmula 1 o una sal farmacéuticamente aceptable del mismo:
[Fórmula 1]

en donde
X es hidrógeno, halógeno o metilo;
e Y es cicloalquilo C3-4.
2. El compuesto de la reivindicación 1,
en donde X es flúor e Y es ciclopropilo.
3. El compuesto de la reivindicación 1,
en donde X es metilo e Y es ciclopropilo.
4. El compuesto de la reivindicación 1,
en donde X es flúor e Y es ciclobutilo.
5. El compuesto de la reivindicación 1,
en donde X es cloro e Y es ciclobutilo.
6. Un compuesto de Fórmula 1 o una sal farmacéuticamente aceptable del mismo para su uso en la prevención o el tratamiento de una enfermedad o afección,
[Fórmula 1]

en donde X es hidrógeno, halógeno o metilo,
en donde Y es cicloalquilo C3-4, y
en donde la enfermedad o afección se selecciona del grupo que consiste en malignidad hematológica, enfermedad hepática y enfermedad autoinmune.
7. El compuesto para su uso según la reivindicación 6 ,
en donde la neoplasia maligna hematológica se selecciona de leucemia o linfoma.
8. El compuesto para su uso según la reivindicación 6 ,
en donde la hepatopatía se selecciona del grupo que consiste en EHNA, ENA, esteatosis hepática, hepatocirrosis, hepatitis, adenoma hepático, hipersensibilidad a la insulina y cáncer de hígado.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20160089417 | 2016-07-14 | ||
| KR1020170086691A KR101932146B1 (ko) | 2016-07-14 | 2017-07-07 | Pi3k를 억제하는 신규한 퀴나졸리논 유도체 및 이를 포함하는 약학적 조성물 |
| PCT/KR2017/007535 WO2018012907A1 (ko) | 2016-07-14 | 2017-07-13 | Pi3k를 억제하는 신규한 퀴나졸리논 유도체 및 이를 포함하는 약학적 조성물 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2824774T3 true ES2824774T3 (es) | 2021-05-13 |
Family
ID=60953246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17827977T Active ES2824774T3 (es) | 2016-07-14 | 2017-07-13 | Nuevos derivados de quinazolinona que inhiben PI3K y composición farmacéutica que los contiene |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US10577369B2 (es) |
| EP (1) | EP3453393B1 (es) |
| JP (2) | JP6614741B2 (es) |
| KR (1) | KR101932146B1 (es) |
| CN (1) | CN109475559B (es) |
| AU (2) | AU2017297096B2 (es) |
| BR (1) | BR112019000614A2 (es) |
| CA (1) | CA3030692C (es) |
| ES (1) | ES2824774T3 (es) |
| GB (2) | GB2576120B (es) |
| RU (2) | RU2019131147A (es) |
| WO (1) | WO2018012907A1 (es) |
| ZA (1) | ZA201900421B (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101932146B1 (ko) * | 2016-07-14 | 2018-12-24 | 주식회사 바이오웨이 | Pi3k를 억제하는 신규한 퀴나졸리논 유도체 및 이를 포함하는 약학적 조성물 |
| AU2019209960B2 (en) | 2018-01-20 | 2023-11-23 | Sunshine Lake Pharma Co., Ltd. | Substituted aminopyrimidine compounds and methods of use |
| KR20210095495A (ko) | 2020-01-23 | 2021-08-02 | 주식회사 바이오웨이 | 신규한 퀴나졸리논 화합물 및 이를 포함하는 약학적 조성물 |
| CN111440173B (zh) * | 2020-03-27 | 2021-05-14 | 山东大学 | 一种pi3k抑制剂的制备方法 |
| EP4171569A4 (en) * | 2020-06-29 | 2024-07-03 | Council Scient Ind Res | QUINAZOLINONE DERIVATIVES FOR THE TREATMENT OF NON-ALCOHOLIC FATTY LIVER DISEASE, MANUFACTURE AND USE THEREOF |
| KR20230065591A (ko) * | 2021-11-05 | 2023-05-12 | 연세대학교 산학협력단 | 피부-특이적 T 세포(skin-specific T cell)의 억제제를 유효성분으로 포함하는 아토피피부염의 예방 또는 치료용 조성물 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6667300B2 (en) | 2000-04-25 | 2003-12-23 | Icos Corporation | Inhibitors of human phosphatidylinositol 3-kinase delta |
| US20050054614A1 (en) * | 2003-08-14 | 2005-03-10 | Diacovo Thomas G. | Methods of inhibiting leukocyte accumulation |
| US20050043239A1 (en) * | 2003-08-14 | 2005-02-24 | Jason Douangpanya | Methods of inhibiting immune responses stimulated by an endogenous factor |
| PT3153514T (pt) * | 2004-05-13 | 2021-06-25 | Icos Corp | Quinazolinonas como inibidoras da fosfatidilinositol 3-quinase delta humana |
| CN101123968A (zh) * | 2004-06-04 | 2008-02-13 | 艾科斯有限公司 | 肥大细胞病的治疗方法 |
| EP1885356A2 (en) * | 2005-02-17 | 2008-02-13 | Icos Corporation | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| CN101575333B (zh) * | 2008-05-09 | 2011-06-22 | 和记黄埔医药(上海)有限公司 | 一种喹唑啉衍生物及其医药用途 |
| KR20170060179A (ko) * | 2008-11-13 | 2017-05-31 | 길리아드 칼리스토가 엘엘씨 | 혈액 종양에 대한 요법 |
| US20130143902A1 (en) * | 2011-12-02 | 2013-06-06 | Gilead Calistoga Llc | Compositions and methods of treating a proliferative disease with a quinazolinone derivative |
| WO2014023083A1 (zh) * | 2012-08-08 | 2014-02-13 | 山东亨利医药科技有限责任公司 | PI3Kδ抑制剂 |
| US9657007B2 (en) | 2013-09-22 | 2017-05-23 | Calitor Sciences, Llc | Substituted aminopyrimidine compounds and methods of use |
| WO2015061204A1 (en) * | 2013-10-21 | 2015-04-30 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| ES2752552T3 (es) * | 2013-12-20 | 2020-04-06 | Gilead Calistoga Llc | Métodos de proceso para inhibidores de fosfatidilinositol 3-quinasa |
| MX2016016538A (es) | 2014-06-13 | 2017-05-01 | Gilead Sciences Inc | Derivados de quinazolinona como inhibidores de fosfatidilinositol 3-cinasa. |
| KR101932146B1 (ko) * | 2016-07-14 | 2018-12-24 | 주식회사 바이오웨이 | Pi3k를 억제하는 신규한 퀴나졸리논 유도체 및 이를 포함하는 약학적 조성물 |
-
2017
- 2017-07-07 KR KR1020170086691A patent/KR101932146B1/ko active IP Right Grant
- 2017-07-13 EP EP17827977.4A patent/EP3453393B1/en active Active
- 2017-07-13 GB GB1915530.8A patent/GB2576120B/en not_active Expired - Fee Related
- 2017-07-13 CN CN201780043603.1A patent/CN109475559B/zh not_active Expired - Fee Related
- 2017-07-13 AU AU2017297096A patent/AU2017297096B2/en not_active Ceased
- 2017-07-13 CA CA3030692A patent/CA3030692C/en active Active
- 2017-07-13 BR BR112019000614-1A patent/BR112019000614A2/pt not_active IP Right Cessation
- 2017-07-13 RU RU2019131147A patent/RU2019131147A/ru unknown
- 2017-07-13 JP JP2019513840A patent/JP6614741B2/ja not_active Expired - Fee Related
- 2017-07-13 ES ES17827977T patent/ES2824774T3/es active Active
- 2017-07-13 GB GB1819957.0A patent/GB2570397B/en not_active Expired - Fee Related
- 2017-07-13 RU RU2019103684A patent/RU2702904C1/ru active
- 2017-07-13 WO PCT/KR2017/007535 patent/WO2018012907A1/ko unknown
-
2019
- 2019-01-10 US US16/244,368 patent/US10577369B2/en not_active Expired - Fee Related
- 2019-01-21 ZA ZA2019/00421A patent/ZA201900421B/en unknown
- 2019-06-14 AU AU2019204181A patent/AU2019204181B2/en not_active Ceased
- 2019-11-01 JP JP2019199810A patent/JP2020040957A/ja active Pending
-
2020
- 2020-01-20 US US16/747,140 patent/US20200148684A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| GB2570397B (en) | 2019-12-11 |
| AU2019204181B2 (en) | 2020-07-16 |
| CA3030692C (en) | 2020-04-14 |
| GB2576120A (en) | 2020-02-05 |
| GB201915530D0 (en) | 2019-12-11 |
| JP2019522678A (ja) | 2019-08-15 |
| AU2017297096B2 (en) | 2019-03-14 |
| AU2019204181A1 (en) | 2019-07-04 |
| CA3030692A1 (en) | 2018-01-18 |
| KR20180008299A (ko) | 2018-01-24 |
| BR112019000614A2 (pt) | 2019-04-24 |
| EP3453393A4 (en) | 2019-06-26 |
| EP3453393A1 (en) | 2019-03-13 |
| US20200148684A1 (en) | 2020-05-14 |
| GB2570397A (en) | 2019-07-24 |
| JP6614741B2 (ja) | 2019-12-04 |
| EP3453393B1 (en) | 2020-09-09 |
| GB201819957D0 (en) | 2019-01-23 |
| CN109475559B (zh) | 2020-06-23 |
| JP2020040957A (ja) | 2020-03-19 |
| RU2702904C1 (ru) | 2019-10-14 |
| ZA201900421B (en) | 2020-05-27 |
| GB2576120B (en) | 2020-07-29 |
| RU2019131147A (ru) | 2020-02-19 |
| US10577369B2 (en) | 2020-03-03 |
| KR101932146B1 (ko) | 2018-12-24 |
| CN109475559A (zh) | 2019-03-15 |
| US20190144453A1 (en) | 2019-05-16 |
| WO2018012907A1 (ko) | 2018-01-18 |
| AU2017297096A1 (en) | 2019-01-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2824774T3 (es) | Nuevos derivados de quinazolinona que inhiben PI3K y composición farmacéutica que los contiene | |
| ES2635386T3 (es) | Inhibidores de indazol de las vías de señalización WNT y usos terapéuticos de los mismos | |
| KR20110110297A (ko) | 불소 함유 화합물 및 이의 사용방법 | |
| US20170015672A1 (en) | Substituted pyrrolo[2,3-d]pyrimidines for selectively targeting tumor cells with fr-alpha and fr-beta type receptors | |
| ES2385850T3 (es) | Potenciador de radioterapia | |
| JP2022501344A (ja) | 新規キナゾリンegfr阻害剤 | |
| US20210040115A1 (en) | Fused ring heteroaryl compounds as ripk1 inhibitors | |
| WO2020051572A1 (en) | Brd4-jak2 inhibitors | |
| US10745433B2 (en) | Compounds for inhibiting cancer and virus | |
| ES2312978T3 (es) | Potenciador del efecto antitumoral y agente antitumoral. | |
| WO2023040810A1 (en) | Methods of cancer treatment using a combination of btk inhibitors and pi3 kinase inhibitors | |
| MXPA04001905A (es) | Compuestos anti-cancer de ciclopenta[g]quinazolina. | |
| AU2017280412A1 (en) | Substituted pyrrolo (2, 3-D) pyridazin-4-ones and pyrazolo (3, 4-D) pyridazin-4-ones as protein kinase inhibitors | |
| KR20220035143A (ko) | 사이클릭 디옥시리보뉴클레오티드 화합물 | |
| WO2024209717A1 (ja) | 腫瘍治療用医薬組成物 | |
| KR20180136930A (ko) | Pi3k를 억제하는 신규한 퀴나졸리논 유도체 및 이를 포함하는 약학적 조성물 | |
| WO2023059893A1 (en) | Yap1 inhibitors | |
| US20220395575A1 (en) | Combination therapy with protein kinase b activation inhibitor to treat cancer | |
| WO2022266418A1 (en) | Triazine inhibitors of cyclin-dependent kinases | |
| WO2024030529A1 (en) | Prodrugs of myc inhibitors | |
| WO2018106118A1 (en) | Treatment of diffuse intrinsic pontine glioma | |
| ES2702698T3 (es) | Polimorfo del derivado de dihidropirazolopirimidinona como inhibidor de la cinasa Wee1 |