ES2819232T3 - Inhibidores de Bcl-2/Bcl-xL y su uso en el tratamiento de cáncer - Google Patents
Inhibidores de Bcl-2/Bcl-xL y su uso en el tratamiento de cáncer Download PDFInfo
- Publication number
- ES2819232T3 ES2819232T3 ES14741180T ES14741180T ES2819232T3 ES 2819232 T3 ES2819232 T3 ES 2819232T3 ES 14741180 T ES14741180 T ES 14741180T ES 14741180 T ES14741180 T ES 14741180T ES 2819232 T3 ES2819232 T3 ES 2819232T3
- Authority
- ES
- Spain
- Prior art keywords
- cancer
- bcl
- tumors
- lymphoma
- leukemia
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims description 79
- 201000011510 cancer Diseases 0.000 title claims description 44
- 238000011282 treatment Methods 0.000 title claims description 19
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 title description 83
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 title description 83
- 229940122035 Bcl-XL inhibitor Drugs 0.000 title description 27
- 229940123711 Bcl2 inhibitor Drugs 0.000 title description 27
- 150000001875 compounds Chemical class 0.000 claims abstract description 98
- 210000004027 cell Anatomy 0.000 claims description 39
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 201000009030 Carcinoma Diseases 0.000 claims description 8
- 206010025323 Lymphomas Diseases 0.000 claims description 8
- 206010029260 Neuroblastoma Diseases 0.000 claims description 8
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 8
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 6
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 6
- 206010005003 Bladder cancer Diseases 0.000 claims description 6
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 6
- 206010009944 Colon cancer Diseases 0.000 claims description 6
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 6
- 208000000649 small cell carcinoma Diseases 0.000 claims description 6
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 6
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 5
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 4
- 206010006187 Breast cancer Diseases 0.000 claims description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 4
- 208000017604 Hodgkin disease Diseases 0.000 claims description 4
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 4
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 4
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims description 4
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 4
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 4
- 208000025113 myeloid leukemia Diseases 0.000 claims description 4
- 201000008968 osteosarcoma Diseases 0.000 claims description 4
- 201000002528 pancreatic cancer Diseases 0.000 claims description 4
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 4
- 230000004614 tumor growth Effects 0.000 claims description 4
- 239000003981 vehicle Substances 0.000 claims description 4
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 206010027476 Metastases Diseases 0.000 claims description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 3
- 230000001154 acute effect Effects 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 201000005787 hematologic cancer Diseases 0.000 claims description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 3
- 238000012360 testing method Methods 0.000 claims description 3
- 210000004881 tumor cell Anatomy 0.000 claims description 3
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 claims description 2
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 claims description 2
- 206010003445 Ascites Diseases 0.000 claims description 2
- 206010003571 Astrocytoma Diseases 0.000 claims description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 2
- 206010005949 Bone cancer Diseases 0.000 claims description 2
- 208000018084 Bone neoplasm Diseases 0.000 claims description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 2
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 claims description 2
- 208000006402 Ductal Carcinoma Diseases 0.000 claims description 2
- 206010063045 Effusion Diseases 0.000 claims description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 2
- 201000008808 Fibrosarcoma Diseases 0.000 claims description 2
- 206010016935 Follicular thyroid cancer Diseases 0.000 claims description 2
- 208000022072 Gallbladder Neoplasms Diseases 0.000 claims description 2
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 claims description 2
- 208000032612 Glial tumor Diseases 0.000 claims description 2
- 206010018338 Glioma Diseases 0.000 claims description 2
- 208000006050 Hemangiopericytoma Diseases 0.000 claims description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 2
- 206010023347 Keratoacanthoma Diseases 0.000 claims description 2
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 2
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 claims description 2
- 206010061523 Lip and/or oral cavity cancer Diseases 0.000 claims description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 2
- 208000007433 Lymphatic Metastasis Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 206010026673 Malignant Pleural Effusion Diseases 0.000 claims description 2
- 208000032271 Malignant tumor of penis Diseases 0.000 claims description 2
- 206010027406 Mesothelioma Diseases 0.000 claims description 2
- 208000034578 Multiple myelomas Diseases 0.000 claims description 2
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 2
- 206010061309 Neoplasm progression Diseases 0.000 claims description 2
- 208000034179 Neoplasms, Glandular and Epithelial Diseases 0.000 claims description 2
- 206010033128 Ovarian cancer Diseases 0.000 claims description 2
- 208000002471 Penile Neoplasms Diseases 0.000 claims description 2
- 206010034299 Penile cancer Diseases 0.000 claims description 2
- 206010060862 Prostate cancer Diseases 0.000 claims description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 2
- 206010038389 Renal cancer Diseases 0.000 claims description 2
- 201000000582 Retinoblastoma Diseases 0.000 claims description 2
- 206010039491 Sarcoma Diseases 0.000 claims description 2
- 201000010208 Seminoma Diseases 0.000 claims description 2
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 2
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 2
- 208000036765 Squamous cell carcinoma of the esophagus Diseases 0.000 claims description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 2
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 claims description 2
- 206010042971 T-cell lymphoma Diseases 0.000 claims description 2
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 claims description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 2
- 206010057644 Testis cancer Diseases 0.000 claims description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 2
- 206010047741 Vulval cancer Diseases 0.000 claims description 2
- 208000004354 Vulvar Neoplasms Diseases 0.000 claims description 2
- 208000008383 Wilms tumor Diseases 0.000 claims description 2
- 210000003567 ascitic fluid Anatomy 0.000 claims description 2
- 230000003140 astrocytic effect Effects 0.000 claims description 2
- 210000000601 blood cell Anatomy 0.000 claims description 2
- 201000007983 brain glioma Diseases 0.000 claims description 2
- 210000000481 breast Anatomy 0.000 claims description 2
- 210000003169 central nervous system Anatomy 0.000 claims description 2
- 201000007455 central nervous system cancer Diseases 0.000 claims description 2
- 208000025997 central nervous system neoplasm Diseases 0.000 claims description 2
- 208000019065 cervical carcinoma Diseases 0.000 claims description 2
- 210000003679 cervix uteri Anatomy 0.000 claims description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 210000001072 colon Anatomy 0.000 claims description 2
- 208000029742 colonic neoplasm Diseases 0.000 claims description 2
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 claims description 2
- 229960000258 corticotropin Drugs 0.000 claims description 2
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 claims description 2
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 2
- 208000007276 esophageal squamous cell carcinoma Diseases 0.000 claims description 2
- 210000003238 esophagus Anatomy 0.000 claims description 2
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 claims description 2
- 210000005002 female reproductive tract Anatomy 0.000 claims description 2
- 210000000232 gallbladder Anatomy 0.000 claims description 2
- 201000010175 gallbladder cancer Diseases 0.000 claims description 2
- 206010017758 gastric cancer Diseases 0.000 claims description 2
- 208000005017 glioblastoma Diseases 0.000 claims description 2
- 201000009277 hairy cell leukemia Diseases 0.000 claims description 2
- 210000002510 keratinocyte Anatomy 0.000 claims description 2
- 201000010982 kidney cancer Diseases 0.000 claims description 2
- 210000000867 larynx Anatomy 0.000 claims description 2
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 claims description 2
- 210000004185 liver Anatomy 0.000 claims description 2
- 201000007270 liver cancer Diseases 0.000 claims description 2
- 208000014018 liver neoplasm Diseases 0.000 claims description 2
- 210000004072 lung Anatomy 0.000 claims description 2
- 201000005249 lung adenocarcinoma Diseases 0.000 claims description 2
- 201000005202 lung cancer Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- 210000004324 lymphatic system Anatomy 0.000 claims description 2
- 208000003747 lymphoid leukemia Diseases 0.000 claims description 2
- 210000005001 male reproductive tract Anatomy 0.000 claims description 2
- 230000009401 metastasis Effects 0.000 claims description 2
- 230000001394 metastastic effect Effects 0.000 claims description 2
- 208000037819 metastatic cancer Diseases 0.000 claims description 2
- 208000011575 metastatic malignant neoplasm Diseases 0.000 claims description 2
- 210000003205 muscle Anatomy 0.000 claims description 2
- 201000005962 mycosis fungoides Diseases 0.000 claims description 2
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 2
- 230000009826 neoplastic cell growth Effects 0.000 claims description 2
- 201000008026 nephroblastoma Diseases 0.000 claims description 2
- 208000008798 osteoma Diseases 0.000 claims description 2
- 210000001672 ovary Anatomy 0.000 claims description 2
- 210000000496 pancreas Anatomy 0.000 claims description 2
- 201000005528 peripheral nervous system neoplasm Diseases 0.000 claims description 2
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 claims description 2
- 210000002307 prostate Anatomy 0.000 claims description 2
- 210000000664 rectum Anatomy 0.000 claims description 2
- 201000006845 reticulosarcoma Diseases 0.000 claims description 2
- 208000029922 reticulum cell sarcoma Diseases 0.000 claims description 2
- 210000003491 skin Anatomy 0.000 claims description 2
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 2
- 208000017572 squamous cell neoplasm Diseases 0.000 claims description 2
- 210000002784 stomach Anatomy 0.000 claims description 2
- 201000011549 stomach cancer Diseases 0.000 claims description 2
- 208000001608 teratocarcinoma Diseases 0.000 claims description 2
- 201000003120 testicular cancer Diseases 0.000 claims description 2
- 210000001550 testis Anatomy 0.000 claims description 2
- 201000002341 thymus lymphoma Diseases 0.000 claims description 2
- 201000002510 thyroid cancer Diseases 0.000 claims description 2
- 210000001685 thyroid gland Anatomy 0.000 claims description 2
- 208000030901 thyroid gland follicular carcinoma Diseases 0.000 claims description 2
- 206010044412 transitional cell carcinoma Diseases 0.000 claims description 2
- 230000005751 tumor progression Effects 0.000 claims description 2
- 230000002485 urinary effect Effects 0.000 claims description 2
- 206010046766 uterine cancer Diseases 0.000 claims description 2
- 206010046885 vaginal cancer Diseases 0.000 claims description 2
- 208000013139 vaginal neoplasm Diseases 0.000 claims description 2
- 201000005102 vulva cancer Diseases 0.000 claims description 2
- 206010000830 Acute leukaemia Diseases 0.000 claims 1
- 235000015107 ale Nutrition 0.000 claims 1
- 230000009545 invasion Effects 0.000 claims 1
- 230000000527 lymphocytic effect Effects 0.000 claims 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims 1
- 210000002394 ovarian follicle Anatomy 0.000 claims 1
- 230000001573 trophoblastic effect Effects 0.000 claims 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims 1
- 210000002229 urogenital system Anatomy 0.000 claims 1
- -1 Bcl-xL Proteins 0.000 description 57
- 239000000243 solution Substances 0.000 description 55
- 102100026596 Bcl-2-like protein 1 Human genes 0.000 description 52
- 239000000203 mixture Substances 0.000 description 45
- 239000002904 solvent Substances 0.000 description 45
- 238000000034 method Methods 0.000 description 37
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 33
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 32
- 239000002253 acid Substances 0.000 description 30
- 239000003112 inhibitor Substances 0.000 description 26
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 22
- 239000003814 drug Substances 0.000 description 22
- 201000010099 disease Diseases 0.000 description 21
- 238000005160 1H NMR spectroscopy Methods 0.000 description 20
- 230000015572 biosynthetic process Effects 0.000 description 20
- 239000011541 reaction mixture Substances 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- 229940124597 therapeutic agent Drugs 0.000 description 20
- 102000004169 proteins and genes Human genes 0.000 description 16
- 108090000623 proteins and genes Proteins 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- 230000006907 apoptotic process Effects 0.000 description 14
- 238000000746 purification Methods 0.000 description 14
- 239000007858 starting material Substances 0.000 description 14
- 230000027455 binding Effects 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 239000002246 antineoplastic agent Substances 0.000 description 11
- 238000003556 assay Methods 0.000 description 11
- 239000012267 brine Substances 0.000 description 11
- 235000019439 ethyl acetate Nutrition 0.000 description 11
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 11
- 150000003839 salts Chemical class 0.000 description 11
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- HPLNQCPCUACXLM-PGUFJCEWSA-N ABT-737 Chemical compound C([C@@H](CCN(C)C)NC=1C(=CC(=CC=1)S(=O)(=O)NC(=O)C=1C=CC(=CC=1)N1CCN(CC=2C(=CC=CC=2)C=2C=CC(Cl)=CC=2)CC1)[N+]([O-])=O)SC1=CC=CC=C1 HPLNQCPCUACXLM-PGUFJCEWSA-N 0.000 description 10
- 108010090931 Proto-Oncogene Proteins c-bcl-2 Proteins 0.000 description 10
- 102000013535 Proto-Oncogene Proteins c-bcl-2 Human genes 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- 230000005855 radiation Effects 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- 229910052938 sodium sulfate Inorganic materials 0.000 description 10
- 235000011152 sodium sulphate Nutrition 0.000 description 10
- 101001056180 Homo sapiens Induced myeloid leukemia cell differentiation protein Mcl-1 Proteins 0.000 description 9
- 102100026539 Induced myeloid leukemia cell differentiation protein Mcl-1 Human genes 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 9
- 238000004128 high performance liquid chromatography Methods 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- JLYAXFNOILIKPP-KXQOOQHDSA-N navitoclax Chemical compound C([C@@H](NC1=CC=C(C=C1S(=O)(=O)C(F)(F)F)S(=O)(=O)NC(=O)C1=CC=C(C=C1)N1CCN(CC1)CC1=C(CCC(C1)(C)C)C=1C=CC(Cl)=CC=1)CSC=1C=CC=CC=1)CN1CCOCC1 JLYAXFNOILIKPP-KXQOOQHDSA-N 0.000 description 9
- 229950004847 navitoclax Drugs 0.000 description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 9
- 150000003384 small molecules Chemical class 0.000 description 9
- 0 *c1cc(S=O)ccc1N[C@](CCN(CC1)CCC1C(OCCP(O)(O)=O)=O)CSc1ccccc1 Chemical compound *c1cc(S=O)ccc1N[C@](CCN(CC1)CCC1C(OCCP(O)(O)=O)=O)CSc1ccccc1 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 229940127089 cytotoxic agent Drugs 0.000 description 8
- 230000003389 potentiating effect Effects 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 108090000765 processed proteins & peptides Proteins 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- TZPPDWDHNIMTDQ-UHFFFAOYSA-N 2-dimethoxyphosphorylethanol Chemical compound COP(=O)(OC)CCO TZPPDWDHNIMTDQ-UHFFFAOYSA-N 0.000 description 7
- 238000002875 fluorescence polarization Methods 0.000 description 7
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 239000000824 cytostatic agent Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 5
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- SRJOCJYGOFTFLH-UHFFFAOYSA-N isonipecotic acid Chemical compound OC(=O)C1CCNCC1 SRJOCJYGOFTFLH-UHFFFAOYSA-N 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 230000008685 targeting Effects 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 229930012538 Paclitaxel Natural products 0.000 description 4
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 4
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 108700000711 bcl-X Proteins 0.000 description 4
- 102000055104 bcl-X Human genes 0.000 description 4
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 4
- 239000007850 fluorescent dye Substances 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 229940088597 hormone Drugs 0.000 description 4
- 239000005556 hormone Substances 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 4
- 229930014626 natural product Natural products 0.000 description 4
- 229960001592 paclitaxel Drugs 0.000 description 4
- 238000007911 parenteral administration Methods 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 4
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 3
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 3
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 3
- 102000010565 Apoptosis Regulatory Proteins Human genes 0.000 description 3
- 108010063104 Apoptosis Regulatory Proteins Proteins 0.000 description 3
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N N-phenyl amine Natural products NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 3
- 229960004316 cisplatin Drugs 0.000 description 3
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 229960001842 estramustine Drugs 0.000 description 3
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 3
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 3
- 229960002949 fluorouracil Drugs 0.000 description 3
- 229960002074 flutamide Drugs 0.000 description 3
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 3
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 229940043355 kinase inhibitor Drugs 0.000 description 3
- 230000036210 malignancy Effects 0.000 description 3
- 231100000682 maximum tolerated dose Toxicity 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229960002340 pentostatin Drugs 0.000 description 3
- 229940127557 pharmaceutical product Drugs 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000000861 pro-apoptotic effect Effects 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- 229940121649 protein inhibitor Drugs 0.000 description 3
- 239000012268 protein inhibitor Substances 0.000 description 3
- VSIVTUIKYVGDCX-UHFFFAOYSA-M sodium;4-[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)tetrazol-2-ium-5-yl]benzene-1,3-disulfonate Chemical compound [Na+].COC1=CC([N+]([O-])=O)=CC=C1[N+]1=NC(C=2C(=CC(=CC=2)S([O-])(=O)=O)S([O-])(=O)=O)=NN1C1=CC=C([N+]([O-])=O)C=C1 VSIVTUIKYVGDCX-UHFFFAOYSA-M 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- DLMYFMLKORXJPO-FQEVSTJZSA-N (2R)-2-amino-3-[(triphenylmethyl)thio]propanoic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SC[C@H](N)C(O)=O)C1=CC=CC=C1 DLMYFMLKORXJPO-FQEVSTJZSA-N 0.000 description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- WVWOOAYQYLJEFD-UHFFFAOYSA-N 1-(2-nitroimidazol-1-yl)-3-piperidin-1-ylpropan-2-ol Chemical compound C1=CN=C([N+]([O-])=O)N1CC(O)CN1CCCCC1 WVWOOAYQYLJEFD-UHFFFAOYSA-N 0.000 description 2
- OEWYWFJWBZNJJG-UHFFFAOYSA-N 1-(aziridin-1-yl)-3-(2-nitroimidazol-1-yl)propan-2-ol Chemical compound C1=CN=C([N+]([O-])=O)N1CC(O)CN1CC1 OEWYWFJWBZNJJG-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 2
- XPBJPGMCFKYBBV-UHFFFAOYSA-N 2-bromoethyl-[2-hydroxy-3-(2-nitroimidazol-1-yl)propyl]azanium;bromide Chemical compound Br.BrCCNCC(O)CN1C=CN=C1[N+]([O-])=O XPBJPGMCFKYBBV-UHFFFAOYSA-N 0.000 description 2
- MHIITNFQDPFSES-UHFFFAOYSA-N 25,26,27,28-tetrazahexacyclo[16.6.1.13,6.18,11.113,16.019,24]octacosa-1(25),2,4,6,8(27),9,11,13,15,17,19,21,23-tridecaene Chemical class N1C(C=C2C3=CC=CC=C3C(C=C3NC(=C4)C=C3)=N2)=CC=C1C=C1C=CC4=N1 MHIITNFQDPFSES-UHFFFAOYSA-N 0.000 description 2
- NUGLIYXAARVRPQ-UHFFFAOYSA-N 3-(2-nitroimidazol-1-yl)propane-1,2-diol Chemical compound OCC(O)CN1C=CN=C1[N+]([O-])=O NUGLIYXAARVRPQ-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- KISUPFXQEHWGAR-RRKCRQDMSA-N 4-amino-5-bromo-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound C1=C(Br)C(N)=NC(=O)N1[C@@H]1O[C@H](CO)[C@@H](O)C1 KISUPFXQEHWGAR-RRKCRQDMSA-N 0.000 description 2
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 102000051485 Bcl-2 family Human genes 0.000 description 2
- 108700038897 Bcl-2 family Proteins 0.000 description 2
- PINCORMWRWUBAD-UHFFFAOYSA-N CC(C)[n](c(C)c1/S=[O]/C)c(-c(cc2)ccc2Cl)c1-c1cc(F)cc(N(CC2)CCN2c(cc2)ccc2N)c1 Chemical compound CC(C)[n](c(C)c1/S=[O]/C)c(-c(cc2)ccc2Cl)c1-c1cc(F)cc(N(CC2)CCN2c(cc2)ccc2N)c1 PINCORMWRWUBAD-UHFFFAOYSA-N 0.000 description 2
- QODMQNJLDPSQEH-UHFFFAOYSA-N CC(C)[n](c(C)c1/S=[O]/C)c(-c2ccc(C)cc2)c1-c1cc(F)cc(N(CC2)CCN2c(cc2)ccc2N)c1 Chemical compound CC(C)[n](c(C)c1/S=[O]/C)c(-c2ccc(C)cc2)c1-c1cc(F)cc(N(CC2)CCN2c(cc2)ccc2N)c1 QODMQNJLDPSQEH-UHFFFAOYSA-N 0.000 description 2
- AWAYZCQVLIGMOK-QZNUWAOFSA-N CC(C)[n](c(C)c1/S=[O]/C)c(-c2ccc(C)cc2)c1-c1cc(N(CC2)CCN2c(cc2)ccc2NS(c(cc2C)ccc2N[C@H](CCN(CC2)CCC2C(OCP(O)(O)=O)=O)CSc2ccccc2)(=O)=O)cc(F)c1 Chemical compound CC(C)[n](c(C)c1/S=[O]/C)c(-c2ccc(C)cc2)c1-c1cc(N(CC2)CCN2c(cc2)ccc2NS(c(cc2C)ccc2N[C@H](CCN(CC2)CCC2C(OCP(O)(O)=O)=O)CSc2ccccc2)(=O)=O)cc(F)c1 AWAYZCQVLIGMOK-QZNUWAOFSA-N 0.000 description 2
- DFOWAJKZJSZEKA-UHFFFAOYSA-N CC(C)[n](c(C)c1/[S+]=[O]/C)c(-c2ccc(C)cc2)c1-c1cc(F)cc(N(CC2)CCN2c(cc2)ccc2N)c1 Chemical compound CC(C)[n](c(C)c1/[S+]=[O]/C)c(-c2ccc(C)cc2)c1-c1cc(F)cc(N(CC2)CCN2c(cc2)ccc2N)c1 DFOWAJKZJSZEKA-UHFFFAOYSA-N 0.000 description 2
- DOMJMYSMCNTLFU-OAQYLSRUSA-N CSc(cc(cc1)S=O)c1N[C@H](CCN(CC1)CCC1C(OCCCP(O)(O)=O)=O)CSc1ccccc1 Chemical compound CSc(cc(cc1)S=O)c1N[C@H](CCN(CC1)CCC1C(OCCCP(O)(O)=O)=O)CSc1ccccc1 DOMJMYSMCNTLFU-OAQYLSRUSA-N 0.000 description 2
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102000006771 Gonadotropins Human genes 0.000 description 2
- 108010086677 Gonadotropins Proteins 0.000 description 2
- 108010069236 Goserelin Proteins 0.000 description 2
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 2
- RPTUSVTUFVMDQK-UHFFFAOYSA-N Hidralazin Chemical compound C1=CC=C2C(NN)=NN=CC2=C1 RPTUSVTUFVMDQK-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- 108010000817 Leuprolide Proteins 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 108091022875 Microtubule Proteins 0.000 description 2
- 102000029749 Microtubule Human genes 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- 208000025174 PANDAS Diseases 0.000 description 2
- 208000021155 Paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection Diseases 0.000 description 2
- 240000004718 Panda Species 0.000 description 2
- 235000016496 Panda oleosa Nutrition 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229960000473 altretamine Drugs 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000001772 anti-angiogenic effect Effects 0.000 description 2
- 230000002424 anti-apoptotic effect Effects 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 230000000118 anti-neoplastic effect Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 238000010822 cell death assay Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229940044683 chemotherapy drug Drugs 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 235000008504 concentrate Nutrition 0.000 description 2
- NZNMSOFKMUBTKW-UHFFFAOYSA-N cyclohexanecarboxylic acid Chemical compound OC(=O)C1CCCCC1 NZNMSOFKMUBTKW-UHFFFAOYSA-N 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 2
- 229960000452 diethylstilbestrol Drugs 0.000 description 2
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 description 2
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 description 2
- OFDNQWIFNXBECV-VFSYNPLYSA-N dolastatin 10 Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-VFSYNPLYSA-N 0.000 description 2
- 108010045524 dolastatin 10 Proteins 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 230000004064 dysfunction Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 2
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 2
- 229940011871 estrogen Drugs 0.000 description 2
- 239000000262 estrogen Substances 0.000 description 2
- WCDWBPCFGJXFJZ-UHFFFAOYSA-N etanidazole Chemical compound OCCNC(=O)CN1C=CN=C1[N+]([O-])=O WCDWBPCFGJXFJZ-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 229960001751 fluoxymesterone Drugs 0.000 description 2
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 239000002622 gonadotropin Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000005734 heterodimerization reaction Methods 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000013067 intermediate product Substances 0.000 description 2
- 238000007913 intrathecal administration Methods 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 229960002985 medroxyprogesterone acetate Drugs 0.000 description 2
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 2
- 229960004296 megestrol acetate Drugs 0.000 description 2
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229960000282 metronidazole Drugs 0.000 description 2
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 2
- 210000004688 microtubule Anatomy 0.000 description 2
- OBBCSXFCDPPXOL-UHFFFAOYSA-N misonidazole Chemical compound COCC(O)CN1C=CN=C1[N+]([O-])=O OBBCSXFCDPPXOL-UHFFFAOYSA-N 0.000 description 2
- 229950010514 misonidazole Drugs 0.000 description 2
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 2
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical class CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 2
- 229960004918 nimorazole Drugs 0.000 description 2
- MDJFHRLTPRPZLY-UHFFFAOYSA-N nimorazole Chemical compound [O-][N+](=O)C1=CN=CN1CCN1CCOCC1 MDJFHRLTPRPZLY-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 2
- 239000002831 pharmacologic agent Substances 0.000 description 2
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 2
- 235000011007 phosphoric acid Nutrition 0.000 description 2
- 229940109328 photofrin Drugs 0.000 description 2
- 229950010456 pimonidazole Drugs 0.000 description 2
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229960003171 plicamycin Drugs 0.000 description 2
- 230000010287 polarization Effects 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 229960004618 prednisone Drugs 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- OWPCHSCAPHNHAV-QIPOKPRISA-N rhizoxin Chemical compound C/C([C@@H]([C@@H](C)[C@H]1OC(=O)[C@@H]2O[C@H]2C[C@@H]2C[C@@H](OC(=O)C2)[C@H](C)/C=C/[C@H]2O[C@]2(C)[C@@H](O)C1)OC)=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-QIPOKPRISA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229960003440 semustine Drugs 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- QVMPZNRFXAKISM-UHFFFAOYSA-N tirapazamine Chemical compound C1=CC=C2[N+]([O-])=NC(=N)N(O)C2=C1 QVMPZNRFXAKISM-UHFFFAOYSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000011277 treatment modality Methods 0.000 description 2
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 2
- 229950001353 tretamine Drugs 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- AQTQHPDCURKLKT-JKDPCDLQSA-N vincristine sulfate Chemical compound OS(O)(=O)=O.C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 AQTQHPDCURKLKT-JKDPCDLQSA-N 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical class CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- ILQAABNLYHLUON-OAHLLOKOSA-N (2r)-4-[4-(methylsulfanylmethoxy)piperidin-1-yl]-1-phenylsulfanylbutan-2-amine Chemical compound C1CC(OCSC)CCN1CC[C@@H](N)CSC1=CC=CC=C1 ILQAABNLYHLUON-OAHLLOKOSA-N 0.000 description 1
- ITOFPJRDSCGOSA-KZLRUDJFSA-N (2s)-2-[[(4r)-4-[(3r,5r,8r,9s,10s,13r,14s,17r)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H](CC[C@]13C)[C@@H]2[C@@H]3CC[C@@H]1[C@H](C)CCC(=O)N[C@H](C(O)=O)CC1=CNC2=CC=CC=C12 ITOFPJRDSCGOSA-KZLRUDJFSA-N 0.000 description 1
- HONKEGXLWUDTCF-YFKPBYRVSA-N (2s)-2-amino-2-methyl-4-phosphonobutanoic acid Chemical compound OC(=O)[C@](N)(C)CCP(O)(O)=O HONKEGXLWUDTCF-YFKPBYRVSA-N 0.000 description 1
- RTUPYIJYJUYJCV-MRXNPFEDSA-N (3r)-3-(9h-fluoren-9-ylmethoxycarbonylamino)-4-(2-fluorophenyl)sulfanylbutanoic acid Chemical compound C([C@@H](CC(=O)O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21)SC1=CC=CC=C1F RTUPYIJYJUYJCV-MRXNPFEDSA-N 0.000 description 1
- MWWSFMDVAYGXBV-MYPASOLCSA-N (7r,9s)-7-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.O([C@@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-MYPASOLCSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical class CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- GCKMFJBGXUYNAG-UHFFFAOYSA-N 17alpha-methyltestosterone Natural products C1CC2=CC(=O)CCC2(C)C2C1C1CCC(C)(O)C1(C)CC2 GCKMFJBGXUYNAG-UHFFFAOYSA-N 0.000 description 1
- DBPWSSGDRRHUNT-CEGNMAFCSA-N 17α-hydroxyprogesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 DBPWSSGDRRHUNT-CEGNMAFCSA-N 0.000 description 1
- JERGUCIJOXJXHF-DBAXYKBZSA-N 2,2,2-trideuterio-1-[(3s,8r,9s,10r,13s,14s,17r)-3,17-dihydroxy-10,13-dimethyl-1,2,3,4,7,8,9,11,12,14,15,16-dodecahydrocyclopenta[a]phenanthren-17-yl]ethanone Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C([2H])([2H])[2H])(O)[C@@]1(C)CC2 JERGUCIJOXJXHF-DBAXYKBZSA-N 0.000 description 1
- XOHUEYCVLUUEJJ-UHFFFAOYSA-N 2,3-Bisphosphoglyceric acid Chemical compound OP(=O)(O)OC(C(=O)O)COP(O)(O)=O XOHUEYCVLUUEJJ-UHFFFAOYSA-N 0.000 description 1
- LXFQSRIDYRFTJW-UHFFFAOYSA-M 2,4,6-trimethylbenzenesulfonate Chemical compound CC1=CC(C)=C(S([O-])(=O)=O)C(C)=C1 LXFQSRIDYRFTJW-UHFFFAOYSA-M 0.000 description 1
- BIVIXXXEPPIGSS-UHFFFAOYSA-N 2-[amino(propan-2-yloxy)phosphanyl]oxypropane Chemical compound CC(C)OP(N)OC(C)C BIVIXXXEPPIGSS-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- ZFVCONUOLQASEW-UHFFFAOYSA-N 2-hydroxypropylphosphonic acid Chemical compound CC(O)CP(O)(O)=O ZFVCONUOLQASEW-UHFFFAOYSA-N 0.000 description 1
- CTRPRMNBTVRDFH-UHFFFAOYSA-N 2-n-methyl-1,3,5-triazine-2,4,6-triamine Chemical compound CNC1=NC(N)=NC(N)=N1 CTRPRMNBTVRDFH-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- VVPIOJRUCSFBBJ-UHFFFAOYSA-N 3-dimethoxyphosphorylpropan-1-ol Chemical compound COP(=O)(OC)CCCO VVPIOJRUCSFBBJ-UHFFFAOYSA-N 0.000 description 1
- ALRHLSYJTWAHJZ-UHFFFAOYSA-M 3-hydroxypropionate Chemical compound OCCC([O-])=O ALRHLSYJTWAHJZ-UHFFFAOYSA-M 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- CCVCODGZORBOGV-UHFFFAOYSA-N 4-(methylsulfanylmethoxy)piperidine Chemical compound CSCOC1CCNCC1 CCVCODGZORBOGV-UHFFFAOYSA-N 0.000 description 1
- SYZOKHJRIIDLOQ-UHFFFAOYSA-N 4-fluoro-3-(trifluoromethylsulfonyl)benzenesulfonyl chloride Chemical compound FC1=CC=C(S(Cl)(=O)=O)C=C1S(=O)(=O)C(F)(F)F SYZOKHJRIIDLOQ-UHFFFAOYSA-N 0.000 description 1
- NSUDGNLOXMLAEB-UHFFFAOYSA-N 5-(2-formyl-3-hydroxyphenoxy)pentanoic acid Chemical compound OC(=O)CCCCOC1=CC=CC(O)=C1C=O NSUDGNLOXMLAEB-UHFFFAOYSA-N 0.000 description 1
- VCLFZSRULXVGLN-UHFFFAOYSA-N 5-(4-chlorophenyl)-4-[3-[4-[4-[[4-fluoro-3-(trifluoromethylsulfonyl)phenyl]sulfonylamino]phenyl]piperazin-1-yl]phenyl]-2-methyl-n-methylsulfonyl-1-propan-2-ylpyrrole-3-carboxamide Chemical compound CC(C)N1C(C)=C(C(=O)NS(C)(=O)=O)C(C=2C=C(C=CC=2)N2CCN(CC2)C=2C=CC(NS(=O)(=O)C=3C=C(C(F)=CC=3)S(=O)(=O)C(F)(F)F)=CC=2)=C1C1=CC=C(Cl)C=C1 VCLFZSRULXVGLN-UHFFFAOYSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- NMKUAEKKJQYLHK-UHFFFAOYSA-N Allocolchicine Natural products CC(=O)NC1CCC2=CC(OC)=C(OC)C(OC)=C2C2=CC=C(C(=O)OC)C=C21 NMKUAEKKJQYLHK-UHFFFAOYSA-N 0.000 description 1
- MXPOCMVWFLDDLZ-NSCUHMNNSA-N Apaziquone Chemical compound CN1C(\C=C\CO)=C(CO)C(C2=O)=C1C(=O)C=C2N1CC1 MXPOCMVWFLDDLZ-NSCUHMNNSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 1
- 108010070075 Bacteriochlorophyll A Proteins 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- UJKPHYRXOLRVJJ-MLSVHJFASA-N CC(O)C1=C(C)/C2=C/C3=N/C(=C\C4=C(CCC(O)=O)C(C)=C(N4)/C=C4\N=C(\C=C\1/N\2)C(C)=C4C(C)O)/C(CCC(O)=O)=C3C Chemical class CC(O)C1=C(C)/C2=C/C3=N/C(=C\C4=C(CCC(O)=O)C(C)=C(N4)/C=C4\N=C(\C=C\1/N\2)C(C)=C4C(C)O)/C(CCC(O)=O)=C3C UJKPHYRXOLRVJJ-MLSVHJFASA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102100033189 Diablo IAP-binding mitochondrial protein Human genes 0.000 description 1
- 101710101225 Diablo IAP-binding mitochondrial protein Proteins 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 1
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 1
- OFDNQWIFNXBECV-UHFFFAOYSA-N Dolastatin 10 Natural products CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)CC)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-UHFFFAOYSA-N 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 1
- 108010040476 FITC-annexin A5 Proteins 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N Folic acid Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- XYZZKVRWGOWVGO-UHFFFAOYSA-N Glycerol-phosphate Chemical compound OP(O)(O)=O.OCC(O)CO XYZZKVRWGOWVGO-UHFFFAOYSA-N 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 229940125497 HER2 kinase inhibitor Drugs 0.000 description 1
- 101000616438 Homo sapiens Microtubule-associated protein 4 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DOMWKUIIPQCAJU-LJHIYBGHSA-N Hydroxyprogesterone caproate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)CCCCC)[C@@]1(C)CC2 DOMWKUIIPQCAJU-LJHIYBGHSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- KJQFBVYMGADDTQ-CVSPRKDYSA-N L-buthionine-(S,R)-sulfoximine Chemical compound CCCCS(=N)(=O)CC[C@H](N)C(O)=O KJQFBVYMGADDTQ-CVSPRKDYSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229930126263 Maytansine Natural products 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- GCKMFJBGXUYNAG-HLXURNFRSA-N Methyltestosterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 GCKMFJBGXUYNAG-HLXURNFRSA-N 0.000 description 1
- 102100021794 Microtubule-associated protein 4 Human genes 0.000 description 1
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 1
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-UHFFFAOYSA-N N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methylpropanamide Chemical compound C=1C=C(C#N)C(C(F)(F)F)=CC=1NC(=O)C(O)(C)CS(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-UHFFFAOYSA-N 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 1
- XEJUHUKGBUCEQI-UHFFFAOYSA-N OC1CCN(CCCCSC2=CC=CC=C2)CC1 Chemical compound OC1CCN(CCCCSC2=CC=CC=C2)CC1 XEJUHUKGBUCEQI-UHFFFAOYSA-N 0.000 description 1
- AKCUYHXGYNKHAQ-UHFFFAOYSA-N OCCCOP(O)=O Chemical compound OCCCOP(O)=O AKCUYHXGYNKHAQ-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- BYPFEZZEUUWMEJ-UHFFFAOYSA-N Pentoxifylline Chemical compound O=C1N(CCCCC(=O)C)C(=O)N(C)C2=C1N(C)C=N2 BYPFEZZEUUWMEJ-UHFFFAOYSA-N 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 244000088415 Raphanus sativus Species 0.000 description 1
- 235000006140 Raphanus sativus var sativus Nutrition 0.000 description 1
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- PDMMFKSKQVNJMI-BLQWBTBKSA-N Testosterone propionate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 PDMMFKSKQVNJMI-BLQWBTBKSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- RGHILYZRVFRRNK-UHFFFAOYSA-N anthracene-1,2-dione Chemical compound C1=CC=C2C=C(C(C(=O)C=C3)=O)C3=CC2=C1 RGHILYZRVFRRNK-UHFFFAOYSA-N 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000002280 anti-androgenic effect Effects 0.000 description 1
- 229940046836 anti-estrogen Drugs 0.000 description 1
- 230000001833 anti-estrogenic effect Effects 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 239000000051 antiandrogen Substances 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 239000003080 antimitotic agent Substances 0.000 description 1
- 229940045695 antineooplastic colchicine derivative Drugs 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- NOFOAYPPHIUXJR-APNQCZIXSA-N aphidicolin Chemical compound C1[C@@]23[C@@]4(C)CC[C@@H](O)[C@@](C)(CO)[C@@H]4CC[C@H]3C[C@H]1[C@](CO)(O)CC2 NOFOAYPPHIUXJR-APNQCZIXSA-N 0.000 description 1
- SEKZNWAQALMJNH-YZUCACDQSA-N aphidicolin Natural products C[C@]1(CO)CC[C@]23C[C@H]1C[C@@H]2CC[C@H]4[C@](C)(CO)[C@H](O)CC[C@]34C SEKZNWAQALMJNH-YZUCACDQSA-N 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- ZSERVQBSOBTXFV-DHHJBRQQSA-M bacteriochlorophyll a Chemical compound C1([C@H](C(=O)OC)C(=O)C2=C3C)=C2N2C3=CC([C@@H](CC)[C@@H]3C)=[N+]4C3=CC3=C(C(C)=O)C(C)=C5N3[Mg]42[N+]2=C1[C@@H](CCC(=O)OC\C=C(/C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)[C@H](C)C2=C5 ZSERVQBSOBTXFV-DHHJBRQQSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 239000012152 bradford reagent Substances 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 1
- 229950004398 broxuridine Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 238000007816 calorimetric assay Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 230000005880 cancer cell killing Effects 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- UBAZGMLMVVQSCD-UHFFFAOYSA-N carbon dioxide;molecular oxygen Chemical compound O=O.O=C=O UBAZGMLMVVQSCD-UHFFFAOYSA-N 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 229940097647 casodex Drugs 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 238000002701 cell growth assay Methods 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 230000004637 cellular stress Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960002559 chlorotrianisene Drugs 0.000 description 1
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- JNGZXGGOCLZBFB-IVCQMTBJSA-N compound E Chemical compound N([C@@H](C)C(=O)N[C@@H]1C(N(C)C2=CC=CC=C2C(C=2C=CC=CC=2)=N1)=O)C(=O)CC1=CC(F)=CC(F)=C1 JNGZXGGOCLZBFB-IVCQMTBJSA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 108010040008 cytochrome Cy Proteins 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 108010037444 diisopropylglutathione ester Proteins 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- YLFBFPXKTIQSSY-UHFFFAOYSA-N dimethoxy(oxo)phosphanium Chemical compound CO[P+](=O)OC YLFBFPXKTIQSSY-UHFFFAOYSA-N 0.000 description 1
- OIERWUPLBOKSRB-UHFFFAOYSA-N dimethoxyphosphorylmethanol Chemical compound COP(=O)(CO)OC OIERWUPLBOKSRB-UHFFFAOYSA-N 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- YEWZQCDRZRYAEB-UHFFFAOYSA-M ditert-butyl phosphate Chemical compound CC(C)(C)OP([O-])(=O)OC(C)(C)C YEWZQCDRZRYAEB-UHFFFAOYSA-M 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 230000005670 electromagnetic radiation Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 229960004750 estramustine phosphate Drugs 0.000 description 1
- ADFOJJHRTBFFOF-RBRWEJTLSA-N estramustine phosphate Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OP(O)(O)=O)[C@@H]4[C@@H]3CCC2=C1 ADFOJJHRTBFFOF-RBRWEJTLSA-N 0.000 description 1
- 239000000328 estrogen antagonist Substances 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethane-1,2-disulfonate Chemical compound [O-]S(=O)(=O)CCS([O-])(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical class C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 229960002474 hydralazine Drugs 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 229960002899 hydroxyprogesterone Drugs 0.000 description 1
- 229950000801 hydroxyprogesterone caproate Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 239000000367 immunologic factor Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 239000013038 irreversible inhibitor Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 238000012792 lyophilization process Methods 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 description 1
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- NMKUAEKKJQYLHK-KRWDZBQOSA-N methyl (7s)-7-acetamido-1,2,3-trimethoxy-6,7-dihydro-5h-dibenzo[5,3-b:1',2'-e][7]annulene-9-carboxylate Chemical compound CC(=O)N[C@H]1CCC2=CC(OC)=C(OC)C(OC)=C2C2=CC=C(C(=O)OC)C=C21 NMKUAEKKJQYLHK-KRWDZBQOSA-N 0.000 description 1
- FHTOGDZUTZFFEO-CYBMUJFWSA-N methyl 1-[(3r)-3-amino-4-phenylsulfanylbutyl]-3-methylazetidine-3-carboxylate Chemical compound C1C(C(=O)OC)(C)CN1CC[C@@H](N)CSC1=CC=CC=C1 FHTOGDZUTZFFEO-CYBMUJFWSA-N 0.000 description 1
- WCWOZXZUAGSNAO-OAHLLOKOSA-N methyl 1-[(3r)-3-amino-4-phenylsulfanylbutyl]-4-methylpiperidine-4-carboxylate Chemical compound C1CC(C(=O)OC)(C)CCN1CC[C@@H](N)CSC1=CC=CC=C1 WCWOZXZUAGSNAO-OAHLLOKOSA-N 0.000 description 1
- FNWKNOBPYWRMPL-UHFFFAOYSA-N methyl 3-methylazetidine-3-carboxylate Chemical compound COC(=O)C1(C)CNC1 FNWKNOBPYWRMPL-UHFFFAOYSA-N 0.000 description 1
- FFKGMXGWLOPOAO-UHFFFAOYSA-N methyl 4-aminocyclohexane-1-carboxylate Chemical compound COC(=O)C1CCC(N)CC1 FFKGMXGWLOPOAO-UHFFFAOYSA-N 0.000 description 1
- KMROKLMQVGAOMJ-UHFFFAOYSA-N methyl 4-methylpiperidine-4-carboxylate Chemical compound COC(=O)C1(C)CCNCC1 KMROKLMQVGAOMJ-UHFFFAOYSA-N 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 229960001566 methyltestosterone Drugs 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 201000010225 mixed cell type cancer Diseases 0.000 description 1
- 208000029638 mixed neoplasm Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- CMEGANPVAXDBPL-INIZCTEOSA-N n-[(7s)-1,2,3-trimethoxy-10-methylsulfanyl-9-oxo-6,7-dihydro-5h-benzo[a]heptalen-7-yl]acetamide Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(SC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC CMEGANPVAXDBPL-INIZCTEOSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229950006344 nocodazole Drugs 0.000 description 1
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 description 1
- 239000003956 nonsteroidal anti androgen Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical group C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960001476 pentoxifylline Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 238000002428 photodynamic therapy Methods 0.000 description 1
- 239000003504 photosensitizing agent Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 208000022131 polyp of large intestine Diseases 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 238000010837 poor prognosis Methods 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000036515 potency Effects 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 102000009076 src-Family Kinases Human genes 0.000 description 1
- 108010087686 src-Family Kinases Proteins 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical class O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- UDACWKHNECHEBB-UHFFFAOYSA-N tert-butyl 2-chloro-2-oxoacetate Chemical compound CC(C)(C)OC(=O)C(Cl)=O UDACWKHNECHEBB-UHFFFAOYSA-N 0.000 description 1
- MVSANBPTBLEQMT-UHFFFAOYSA-N tert-butyl 4-hydroxycyclohexane-1-carboxylate Chemical compound CC(C)(C)OC(=O)C1CCC(O)CC1 MVSANBPTBLEQMT-UHFFFAOYSA-N 0.000 description 1
- FUYBPBOHNIHCHM-UHFFFAOYSA-N tert-butyl piperidine-4-carboxylate Chemical compound CC(C)(C)OC(=O)C1CCNCC1 FUYBPBOHNIHCHM-UHFFFAOYSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 229960001712 testosterone propionate Drugs 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 150000003918 triazines Chemical class 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- 229940066528 trichloroacetate Drugs 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- 208000029387 trophoblastic neoplasm Diseases 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 239000002525 vasculotropin inhibitor Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- 229960004982 vinblastine sulfate Drugs 0.000 description 1
- KDQAABAKXDWYSZ-PNYVAJAMSA-N vinblastine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KDQAABAKXDWYSZ-PNYVAJAMSA-N 0.000 description 1
- KDQAABAKXDWYSZ-JKDPCDLQSA-N vincaleukoblastine sulfate Chemical compound OS(O)(=O)=O.C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 KDQAABAKXDWYSZ-JKDPCDLQSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960002110 vincristine sulfate Drugs 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 229940033942 zoladex Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/30—Zinc; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61N—ELECTROTHERAPY; MAGNETOTHERAPY; RADIATION THERAPY; ULTRASOUND THERAPY
- A61N5/00—Radiation therapy
- A61N5/10—X-ray therapy; Gamma-ray therapy; Particle-irradiation therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Radiology & Medical Imaging (AREA)
- Pathology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Compuesto que tiene una estructura **(Ver fórmula)**
Description
DESCRIPCIÓN
Inhibidores de Bcl-2/Bcl-xL y su uso en el tratamiento de cáncer
Campo de la invención
La presente invención se refiere a inhibidores de Bcl-2/Bcl-xL y al uso de los mismos en el tratamiento de cáncer.
Antecedentes de la invención
La resistencia a la apoptosis es un sello distintivo de cáncer humano (1-3). Las células cancerosas deben superar un bombardeo continuo de tensiones celulares, tales como el daño del ADN, la activación de oncogenes, la progresión aberrante del ciclo celular y los microentornos rigurosos, lo que provocaría que las células normales experimentaran la apoptosis. Uno de los principales medios mediante el cual las células cancerosas evitan la apoptosis es mediante la regulación por incremento de proteínas antiapoptóticas de la familia de Bcl-2. La selección como diana de reguladores clave de la apoptosis para superar la resistencia a la apoptosis y fomentar la apoptosis de células tumorales es una nueva estrategia terapéutica contra el cáncer (4,5).
Las proteínas Bcl-2 funcionan como reguladores críticos de la apoptosis en células tanto cancerosas como normales (6-10). Las proteínas Bcl-2 sirven como control sobre la apoptosis, lo que permite la supervivencia de células sanas y útiles. Esta familia de proteínas incluye proteínas antiapoptóticas, tales como Bcl-2, Bcl-xL y Mcl-1, y moléculas proapoptóticas, incluyendo Bid, Bim, Bad, Bak y Bax (6-10). Mientras que las células normales presentan bajos niveles de expresión de las proteínas Bcl-2 y Bcl-xL antiapoptóticas, estas proteínas se encuentran altamente sobreexpresadas en muchos tipos diferentes de tumores humanos (6-10). Esta sobreexpresión se ha relacionado con el mal pronóstico en diversos tipos de cáncer y con la resistencia clínica a los agentes quimioterápicos y a la radiación (6-10). De acuerdo con las observaciones clínicas, los estudios de laboratorio han establecido que la sobreexpresión de Bcl-2 o Bcl-xL provoca que las células cancerosas se vuelvan más resistentes a los agentes quimioterápicos in vitro e in vivo (6-10). La inhibición de la apoptosis por Bcl-2 contribuye al cáncer mediante la inhibición de la muerte celular. Por tanto, la selección como diana de Bcl-2 y/o Bcl-xL se ha perseguido como estrategia terapéutica contra el cáncer (11-34). La inhibición de la actividad de Bcl-2 en células cancerosas puede reducir la resistencia a los quimioterápicos y aumentar la destrucción de células cancerosas.
Las proteínas Bcl-2 y Bcl-xL inhiben la apoptosis mediante la heterodimerización con las proteínas proapoptóticas de la familia Bcl-2, tales como Bak, Bax, Bim, Bid, Puma y Bad (6-10). Las estructuras tridimensionales determinadas experimentalmente de Bcl-xL y Bcl-2 han mostrado que estas proteínas poseen un surco bien definido, que interacciona con el dominio BH3 (homología 3 de Bcl-2) de las proteínas Bcl-2 proapoptóticas (38-42). Se ha propuesto que moléculas pequeñas que no son péptidos diseñadas para bloquear la heterodimerización de proteínas Bcl-2/Bcl-xL con sus parejas de unión que provocan la muerte pueden ser eficaces como antagonistas de Bcl-2/Bcl-xL y que tales inhibidores de molécula pequeña pueden tener un gran potencial terapéutico para el tratamiento de cánceres humanos en los que Bcl-2 y/o Bcl-xL están altamente expresadas (18-37).
Se han notificado inhibidores de molécula pequeña que no son péptidos de Bcl-2/Bcl-xL. El documento “Structure-Based Discovery of BM-957 as a Potent Small-Molecule Inhibitor of Bcl-2 and Bcl-xL Capable of Achieving Complete Tumor Regression” (J. Med. Chem 20128502-8514) da a conocer los compuestos 27-29 como inhibidores de Bcl-2 y Bcl-xL, así como el documento WO2012103059 da a conocer los compuestos 28-29 i como inhibidores de Bcl-2 y Bcl-xL.
La mayoría de los inhibidores tienen afinidades de débiles a moderadas por estas proteínas y carecen de un modo de acción bien definido para su actividad celular (18-37). Las excepciones son ABT-737, ABT-263 y sus análogos (26-34). ABT-737 y ABT-263 se unen a Bcl-2, Bcl-xL y Bcl-w con afinidades muy altas (Ki <1 nM) y tienen una alta especificidad con respecto a Mcl-1 y A1, otras dos proteínas Bcl-2 antiapoptóticas (26, 32, 34). ABT-263 ha avanzado en ensayos clínicos en fase I/II y muestra una actividad antitumoral prometedora en la práctica clínica (45).
A pesar del descubrimiento de ABT-737 y ABT-263, el diseño de potentes inhibidores que no sean péptidos de Bcl-2/Bcl-xL sigue siendo un reto significativo en el descubrimiento de fármacos modernos. Por consiguiente, sigue existiendo la necesidad en la técnica de inhibidores de Bcl-2/Bcl-xL que tengan propiedades físicas y farmacológicas que permitan el uso de los inhibidores en aplicaciones terapéuticas. La presente invención proporciona compuestos diseñados para unirse a Bcl-2/Bcl-xL e inhibir la actividad de Bcl-2/Bcl-xL.
Sumario de la invención
La presente invención se refiere a inhibidores de Bcl-2/Bcl-xL, tal como se define en reivindicación 1, a composiciones que comprenden los inhibidores y al uso de los inhibidores en un tratamiento terapéutico de cáncer. Los presentes compuestos son potentes inhibidores de la activación de Bcl-2/Bcl-xL e inducen la apoptosis de células cancerosas que expresan Bcl-2 y/o Bcl-xL.
La presente invención se refiere a compuestos específicos que tienen una fórmula estructural:


Los compuestos dados a conocer anteriormente pueden usarse para tratar cáncer.
Otra realización de la presente invención es proporcionar una composición que comprende los compuestos dados a conocer anteriormente.
Esta composición puede usarse para tratar cáncer.
Descripción detallada de las realizaciones preferidas
La presente invención se describe en relación con realizaciones preferidas.
El término “Bcl-2/Bcl-xL”, tal como se usa en el presente documento, significa Bcl-2, Bcl-xL o Bcl-2 y Bcl-xL, es decir,
Bcl-2 y/o Bcl-xL.
El término “una enfermedad o un estado en el que la inhibición de Bcl-2 y/o Bcl-xL proporciona un beneficio” se refiere a un estado en el que Bcl-2 y/o Bcl-xL, y/o una acción de Bcl-2 y/o Bcl-xL, es importante o necesaria, por ejemplo, para la aparición, el progreso, la expresión de esa enfermedad o ese estado, o una enfermedad o un estado que se sabe que va a tratarse por un inhibidor de Bcl-2/Bcl-xL, tal como ABT-737 o ABT-263. Un ejemplo de un estado de este tipo es un cáncer. Un experto habitual en la técnica puede determinar fácilmente si un compuesto trata una enfermedad o un estado mediado por Bcl-2/Bcl-xL para cualquier tipo de célula particular, por ejemplo, mediante ensayos que pueden usarse de manera conveniente para evaluar la actividad de compuestos particulares.
El término “segundo agente terapéutico” se refiere a un agente terapéutico diferente de un inhibidor de Bcl-2 y/o BclxL dado a conocer anteriormente y que se sabe que trata la enfermedad o el estado de interés. Por ejemplo, cuando un cáncer es la enfermedad o el estado de interés, el segundo agente terapéutico puede ser un fármaco quimioterápico conocido, como taxol o radiación, por ejemplo.
El término “enfermedad” o “estado” indica alteraciones y/o anomalías que, como regla, se considera que son estados o funciones patológicas y que pueden manifestarse por sí solas en forma de signos, síntomas y/o disfunciones particulares. Tal como se demuestra a continuación, los compuestos dados a conocer anteriormente son potentes inhibidores de Bcl-2/Bcl-xL y pueden usarse en el tratamiento de enfermedades y estados en los que la inhibición de Bcl-2/Bcl-xL proporciona un beneficio.
Tal como se usa en el presente documento, los términos “tratar”, “que trata”, “tratamiento” y similares se refieren a la eliminación, reducción o mejora de una enfermedad o un estado, y/o los síntomas asociados con los mismos. Aunque no se excluye, el tratamiento de una enfermedad o un estado no requiere que la enfermedad, el estado o los síntomas asociados con los mismos se eliminen por completo. Tal como se usa en el presente documento, los términos “tratar”, “que trata”, “tratamiento” y similares pueden incluir “tratamiento profiláctico”, que se refiere a la reducción de la probabilidad de volver a desarrollar una enfermedad o un estado, o de una recaída de una enfermedad o un estado previamente controlado, en un sujeto que no tiene, pero está en riesgo de o es susceptible a, volver a desarrollar una enfermedad o un estado o una recaída de la enfermedad o el estado. El término “tratamiento” también incluye profilaxis de recidiva o profilaxis de fase, así como el tratamiento de signos, síntomas y/o disfunciones agudos o crónicos. El tratamiento puede orientarse de manera sintomática, por ejemplo, para suprimir los síntomas. Puede realizarse a lo largo de un corto periodo, orientarse a medio plazo o puede ser un tratamiento a largo plazo, por ejemplo, dentro del contexto de una terapia de mantenimiento.
El término “cantidad terapéuticamente eficaz” o “dosis eficaz”, tal como se usa en el presente documento, se refiere a una cantidad del/de los principio(s) activo(s) que es suficiente, cuando se administra, para administrar eficazmente el/los principio(s) activo(s) para el tratamiento del estado o la enfermedad de interés a un individuo que lo necesita. En el caso de un cáncer u otro trastorno de proliferación, la cantidad terapéuticamente eficaz del agente puede reducir (es decir, retardar hasta cierto punto y preferiblemente detener) la proliferación celular no deseada; reducir el número de células cancerosas; reducir el tamaño del tumor; inhibir (es decir, retardar hasta cierto punto y preferiblemente detener) la infiltración de células cancerosas en los órganos periféricos; inhibir (es decir, retardar hasta cierto punto y preferiblemente detener) la metástasis del tumor; inhibir, hasta cierto punto, el crecimiento del tumor; reducir la señalización de Bcl-2/Bcl-xL en las células diana; y/o aliviar, hasta cierto punto, uno o más de los síntomas asociados con el cáncer. Hasta el punto en el que el compuesto o la composición administrados evita el crecimiento y/o destruye las células cancerosas existentes, y puede ser citostática y/o citotóxica.
El término “recipiente” significa cualquier receptáculo y cierre para el mismo adecuado para almacenar, enviar, dispensar y/o manipular un producto farmacéutico.
El término “prospecto” significa la información que acompaña a un producto farmacéutico que proporciona una descripción de cómo administrar el producto, junto con los datos de seguridad y eficacia requeridos para permitir que el médico, el farmacéutico y el paciente tomen una decisión informada sobre el uso del producto. El prospecto del envase se considera generalmente como la “etiqueta” de un producto farmacéutico.
“Administración concurrente”, “administrado en combinación”, “administración simultánea” y frases similares significan que dos o más agentes se administran de manera concurrente al sujeto que va a tratarse. Por “de manera concurrente” quiere decirse que cada agente se administra o bien simultáneamente o bien secuencialmente en cualquier orden en momentos de tiempo diferentes. Sin embargo, si no se administran simultáneamente, quiere decirse que se administran a un individuo en una secuencia y lo suficientemente próximo en el tiempo para proporcionar el efecto terapéutico deseado y pueden actuar de manera concertada. Por ejemplo, un inhibidor de Bcl-2/Bcl-xL según la presente invención puede administrarse al mismo tiempo o secuencialmente en cualquier orden en momentos de tiempo diferentes como segundo agente terapéutico. Un inhibidor de Bcl-2/Bcl-xL presente y el segundo agente terapéutico pueden administrarse por separado, en cualquier forma apropiada y por cualquier vía adecuada. Cuando un inhibidor de Bcl-2/Bcl-xL presente y el segundo agente terapéutico no se administran de manera concurrente, se entiende que pueden administrarse en cualquier orden a un sujeto que lo necesita. Por ejemplo, un inhibidor de Bcl-2/Bcl-xL presente puede administrarse antes de (por ejemplo, 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas,
1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas o 12 semanas antes), de manera concomitante con, o posterior a (por ejemplo, 5 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 12 horas, 24 horas, 48 horas, 72 horas, 96 horas, 1 semana, 2 semanas, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 8 semanas o 12 semanas después) la administración de una modalidad de tratamiento con un segundo agente terapéutico (por ejemplo, radioterapia) a un individuo que lo necesita. Un inhibidor de Bcl-2/Bcl-xL y el segundo agente terapéutico se administran con 1 minuto de diferencia, 10 minutos de diferencia, 30 minutos de diferencia, menos de 1 hora de diferencia, 1 hora de diferencia, de 1 hora a 2 horas de diferencia, de 2 horas a 3 horas de diferencia, de 3 horas a 4 horas de diferencia, de 4 horas a 5 horas de diferencia, de 5 horas a 6 horas de diferencia, de 6 horas a 7 horas de diferencia, de 7 horas a 8 horas de diferencia, de 8 horas a 9 horas de diferencia, de 9 horas a 10 horas de diferencia, de 10 horas a 11 horas de diferencia, de 11 horas a 12 horas de diferencia, no más de 24 horas de diferencia o no más de 48 horas de diferencia. Los componentes de las terapias de combinación pueden administrarse con de 1 minuto a 24 horas de diferencia.
El uso de los términos “un”, “una”, “el/la” y referentes similares en el contexto de describir la invención (especialmente en el contexto de las reivindicaciones) debe interpretarse como que cubre tanto el singular como el plural, a menos que se indique de otro modo. La recitación de intervalos de valores en el presente documento están destinados a servir simplemente como método de abreviatura para hacer referencia individualmente a cada valor por separado que se encuentra dentro del intervalo, a menos que se indique de otro modo en el presente documento, y cada valor por separado se incorpora en la memoria descriptiva como si se recitaran individualmente en el presente documento. Durante la última década, la investigación sobre la apoptosis ha establecido que la selección como diana de Bcl-2 y/o Bcl-xL usando inhibidores de molécula pequeña es una estrategia terapéutica viable contra el cáncer (35-37). El descubrimiento de ABT-737 y ABT-263 y los primeros datos clínicos sobre ABT-263 han demostrado que los inhibidores de molécula pequeña que no son péptidos de Bcl-2 y/o Bcl-xL tienen un gran potencial terapéutico para el tratamiento de muchos tipos de cáncer humano en los que Bcl-2 y/o Bcl-xL están sobreexpresados y para los cuales los agentes anticancerígenos actuales son bastante ineficaces (26-36).
A pesar del descubrimiento de ABT-737 y ABT-263, se han notificado pocas clases nuevas de inhibidores de molécula pequeña de Bcl-2/Bcl-xL altamente potentes con afinidades por Bcl-2/Bcl-xL y potencias celulares próximas a las logradas por ABT-737/ABT-263. Esto se debe a que el diseño de inhibidores de molécula pequeña de Bcl-2/Bcl-xL implica la selección como diana y el bloqueo de las interacciones de las proteínas Bcl-2/Bcl-xL con sus parejas de unión proapoptóticas, una tarea que se ha demostrado que es muy desafiante por al menos tres motivos principales. En primer lugar, en comparación con los sitios de unión típicos en enzimas y receptores, las superficies de contacto entre Bcl-2 o Bcl-xL y sus parejas de unión son muy grandes (38-42). La interacción de Bcl-2/Bcl-xL con sus parejas de unión, tales como las proteínas BAD y Bim, está mediada por un dominio BH3 de 20-25 residuos en BAD y Bim y un gran surco de unión en Bcl-2/Bcl-xL. En segundo lugar, los surcos de unión en Bcl-2/Bcl-xL son muy hidrófobos por naturaleza, lo que dificulta el diseño de moléculas pequeñas de tipo fármaco (26, 38-42). En tercer lugar, Bcl-2 y Bcl-xL son extremadamente flexibles de manera conformacional y pueden adoptar conformaciones bastante distintas en la estructura libre de ligando y cuando se unen a ligandos diferentes (26, 38-42). Algunos de los bolsillos de unión observados para Bcl-xL en las estructuras cristalinas de sus complejos con BAD (41), Bim (43) y ABT-737(44) se inducen por la unión de ligandos y no están presentes en una estructura cristalino libre de ligando (38). Estos tres factores hacen que el diseño de inhibidores de molécula pequeña potentes y similares a fármaco de Bcl-2/Bcl-xL sea un reto fundamental en el descubrimiento de fármacos modernos.
La presente invención se refiere a compuestos que son inhibidores potentes y específicos de Bcl-2/Bcl-xL. Los presentes compuestos pueden unirse a Bcl-2 y/o Bcl-xL con valores de Ki <10 nM y funcionar como potentes antagonistas de Bcl-2 y Bcl-xL en ensayos funcionales libres de células. Los compuestos inducen potencialmente la apoptosis en células cancerosas y tienen un mecanismo de acción que es muy coherente con la selección como diana de Bcl-2 y Bcl-xL. Un compuesto sometido a prueba demuestra una robusta inducción de la apoptosis in vivo en tejidos tumorales y muestra una fuerte actividad antitumoral contra tumores de xenoinjerto H146.
Por tanto, los inhibidores de Bcl-2/Bcl-xL de la presente invención son útiles en el tratamiento de células proliferantes no deseadas, incluyendo cánceres y precánceres, en sujetos que necesitan tal tratamiento. Los compuestos de la presente invención pueden reducir la proliferación de células no deseadas induciendo la apoptosis en esas células.
La presente invención se refiere a inhibidores de Bcl-2/Bcl-xL que tienen una fórmula estructural


Dichos compuestos anteriores inhiben Bcl-2/Bcl-xL y son útiles en el tratamiento de cáncer.
Dichos compuestos anteriores pueden administrarse con un segundo agente terapéutico. El segundo agente terapéutico se selecciona de fármacos que se sabe que son útiles en el tratamiento de la enfermedad o el estado que aflige al individuo que lo necesita, por ejemplo, un agente quimioterápico y/o radiación que se sabe que son útiles en el tratamiento de un cáncer particular.
De manera adicional, también pueden usarse sales, hidratos y solvatos de los presentes compuestos. La presente invención incluye además todos los posibles estereoisómeros e isómeros geométricos de los compuestos. La
presente invención incluye tanto compuestos racémicos como isómeros ópticamente activos. Cuando se desea un compuesto de la invención como único enantiómero, puede obtenerse o bien mediante resolución del producto final o bien mediante síntesis estereoespecífica a partir de o bien el material de partida isoméricamente puro o bien el uso de un reactivo auxiliar quiral, por ejemplo, véase Z. Ma et al., Tetrahedron: Asymmetry, 8(6), páginas 883-888 (1997). La resolución del producto final, un producto intermedio o un material de partida puede lograrse mediante cualquier método adecuado conocido en la técnica. De manera adicional, en situaciones en las que son posibles tautómeros de dichos compuestos anteriores, la presente invención pretende incluir todas las formas tautoméricas de los compuestos.
Los compuestos de la invención pueden existir como sales. Pueden usarse sales farmacéuticamente aceptables de los compuestos de la invención. Tal como se usa en el presente documento, el término “sales farmacéuticamente aceptables” se refiere a sales o formas zwitteriónicas de dichos compuestos anteriores. Las sales de dichos compuestos anteriores pueden prepararse durante el aislamiento y la purificación finales de los compuestos o haciendo reaccionar por separado el compuesto con un ácido que tiene un catión adecuado. Las sales farmacéuticamente aceptables de los compuestos pueden ser sales de adición de ácido formadas con ácidos farmacéuticamente aceptables. Los ejemplos de ácidos que pueden emplearse para formar sales farmacéuticamente aceptables incluyen ácidos inorgánicos tales como nítrico, bórico, clorhídrico, bromhídrico, sulfúrico y fosfórico, y ácidos orgánicos tales como oxálico, maleico, succínico y cítrico. Los ejemplos no limitativos de sales incluyen, pero no se limitan a, las sales de clorhidrato, bromhidrato, yodhidrato, sulfato, bisulfato, 2-hidroxietanosulfonato, fosfato, hidrogenofosfato, acetato, adipato, alginato, aspartato, benzoato, bisulfato, butirato, canforato, canforsulfonato, digluconato, glicerolfosfato, hemisulfato, heptanoato, hexanoato, formiato, succinato, fumarato, maleato, ascorbato, isetionato, salicilato, metanosulfonato, mesitilenosulfonato, naftilenosulfonato, nicotinato, 2-naftalenosulfonato, oxalato, pamoato, pectinato, persulfato, 3-fenilproprionato, picrato, pivalato, propionato, tricloroacetato, trifluoroacetato, fosfato, glutamato, bicarbonato, para-toluenosulfonato, undecanoato, lactato, citrato, tartrato, gluconato, metanosulfonato, etanodisulfonato, bencenosulfonato y p-toluenosulfonato. Además, los grupos amino disponibles presentes en los compuestos de la invención pueden cuaternizarse con cloruros, bromuros y yoduros de metilo, etilo, propilo y butilo; sulfatos de dimetilo, dietilo, dibutilo y diamilo; cloruros, bromuros y yoduros de decilo, laurilo, miristilo y estearilo; y bromuros de bencilo y fenetilo.
Los compuestos específicos de la presente invención incluyen los compuestos que tienen la estructura expuesta a continuación.




La presente invención proporciona los inhibidores de Bcl-2/Bcl-xL de los ejemplos 12, 14, 15, 17, 19, 21, 27.
Los compuestos de la invención pueden usarse como compuesto puro o como composición farmacéutica. El uso de una composición farmacéutica, o un compuesto puro de la invención, puede realizarse durante o después de la aparición de la enfermedad o el estado de interés. Normalmente, las composiciones farmacéuticas son estériles y contienen compuestos no tóxicos, carcinógenos o mutagénicos que provocarían una reacción adversa cuando se administraran.
Dicho compuesto anterior puede usarse junto con un segundo agente terapéutico útil en el tratamiento de cáncer. El segundo agente terapéutico es diferente de dichos compuestos anteriores y puede administrarse de manera simultánea o secuencial para lograr el efecto deseado. Además, dicho compuesto anterior y el segundo agente terapéutico pueden administrarse a partir de una sola composición o dos composiciones por separado.
Un compuesto de la invención y el segundo agente terapéutico pueden usarse juntos como dosis unitaria individual o por separado como múltiples dosis unitarias, en las que el compuesto se usa antes del segundo agente terapéutico o viceversa. Pueden usarse una o más dosis del compuesto y/o una o más dosis del segundo agente terapéutico. Por tanto, los compuestos de la invención pueden usarse junto con uno o más segundos agentes terapéuticos, por ejemplo, pero sin limitarse a, agentes anticancerígenos.
Las enfermedades y los estados que pueden tratarse según la invención incluyen cánceres. Puede tratarse una variedad de cánceres, incluyendo, pero sin limitarse a: carcinomas, incluyendo vejiga (incluyendo cáncer de vejiga acelerado y metastásico), mama, colon (incluyendo cáncer colorrectal), riñón, hígado, pulmón (incluyendo cáncer de pulmón de células pequeñas y no pequeñas y adenocarcinoma de pulmón), ovario, próstata, testículos, aparato genitourinario, sistema linfático, recto, laringe, páncreas (incluyendo carcinoma de páncreas exocrino), esófago, estómago, vesícula biliar, cuello uterino, tiroides, renal y piel (incluyendo carcinoma de células escamosas); tumores hematopoyéticos de linaje linfoide, incluyendo leucemia, leucemia linfocítica aguda, leucemia linfoblástica aguda, linfoma de células B, linfoma de células T, linfoma de Hodgkin, linfoma no Hodgkin, linfoma de células pilosas, linfoma histiocítico y linfoma de Burkett, tumores hematopoyéticos de linaje mieloide, incluyendo leucemias mielógenas agudas y crónicas, síndrome mielodisplásico, leucemia mieloide y leucemia promielocítica; tumores del sistema nervioso central y periférico, incluyendo astrocitoma, neuroblastoma, glioma y nuerilemomas; tumores de origen mesenquimatoso, incluyendo fibrosarcoma, rabdomioscarcoma y osteosarcoma; y otros tumores, incluyendo melanoma, xenoderma pigmentoso, queratoacantoma, seminoma, cáncer folicular de tiroides, teratocarcinoma, carcinoma de células renales (CCR), cáncer de páncreas, mieloma, leucemia mieloide y linfoblástica, neuroblastoma y glioblastoma.
Las formas adicionales de cáncer que pueden tratarse mediante los inhibidores de Bcl-2/Bcl-xL de la presente invención incluyen, por ejemplo, oncología de adultos y pediátrica, crecimiento de tumores sólidos/neoplasias malignas, carcinoma mixoide y de células redondas, tumores localmente avanzados, cáncer metastásico, sarcomas de tejidos blandos humanos, incluyendo sarcoma de Ewing, metástasis de cánceres, incluyendo metástasis linfáticas, carcinoma de células escamosas, particularmente de la cabeza y el cuello, carcinoma de células escamosas del esófago, carcinoma oral, neoplasias malignas de glóbulos sanguíneos, incluyendo mieloma múltiple, leucemias, incluyendo leucemia linfocítica aguda, leucemia no linfocítica aguda, leucemia linfocítica crónica, leucemia mielocítica crónica y leucemia de células pilosas, linfomas de derrame (linfomas basados en las cavidades
corporales), linfoma tímico cáncer de pulmón (incluyendo carcinoma de células pequeñas, linfoma cutáneo de células T, linfoma de Hodgkin, linfoma no Hodgkin, cáncer de la corteza suprarrenal, tumores que producen ACTH, cánceres no microcíticos, cáncer de mama, incluyendo carcinoma de células pequeñas y carcinoma ductal), cánceres gastrointestinales (incluyendo cáncer de estómago, cáncer de colon, cáncer colorrectal y pólipos asociados con neoplasia colorrectal), cáncer de páncreas, cáncer de hígado, cánceres urinarios (incluyendo cáncer de vejiga, tal como tumores primarios de vejiga superficial, carcinoma invasivo de células de transición de la vejiga y cáncer de vejiga invasivo del músculo), cáncer de próstata, neoplasias malignas del aparato genital femenino (incluyendo carcinoma de ovario, neoplasias epiteliales peritoneales primarias, carcinoma de cuello uterino, cánceres endometriales uterinos, cáncer de vagina, cáncer de la vulva, cáncer de útero y tumores sólidos en el folículo ovárico), neoplasias malignas del aparato genital masculino (incluyendo cáncer de testículo y cáncer de pene), cáncer de riñón (incluyendo carcinoma de células renales, cáncer de cerebro (incluyendo tumores de cerebro intrínsecos, neuroblastoma, tumores de cerebro astrocíticos, gliomas e invasión de células de tumor metastásico en el sistema nervioso central), cánceres de hueso (incluyendo osteomas y osteosarcomas), cánceres de piel (incluyendo melanoma maligno, progresión tumoral de queratinocitos cutáneos humanos y cáncer de células escamosas), cáncer de tiroides, retinoblastoma, neuroblastoma, derrame peritoneal, derrame pleural maligno, mesotelioma, tumores de Wilms, cáncer de vesícula biliar, neoplasias trofoblásticas, hemangiopericitoma y sarcoma de Kaposi.
Un compuesto de la invención puede usarse por cualquier vía adecuada, por ejemplo por administración oral, bucal, inhalación, sublingual, rectal, vaginal, suboccipital o intratecal a través de punción lumbar, transuretral, nasal, percutánea, es decir, transdérmica, o parenteral (incluyendo inyección intravenosa, intramuscular, subcutánea, intracoronaria, intradérmica, intramamaria, intraperitoneal, intraarticular, intratecal, retrobulbar, intrapulmonar y/o implantación quirúrgica en un sitio particular). La administración parenteral puede lograrse usando una aguja y una jeringa o usando una técnica de alta presión.
Las composiciones farmacéuticas incluyen aquellas en las que se usa un compuesto de la invención en una cantidad eficaz para lograr su propósito previsto. La formulación, la vía de administración y la dosificación exactas las determina un médico individual en vista del estado condición o la enfermedad diagnosticados.
La toxicidad y la eficacia terapéutica de los compuestos de la presente invención pueden determinarse mediante procedimientos farmacéuticos convencionales en cultivos de células o animales de experimentación, por ejemplo, para determinar la dosis máxima tolerada (DMT) de un compuesto, que se define como la mayor dosis que no provoca toxicidad en animales. La razón de dosis entre la dosis máxima tolerada y los efectos terapéuticos (por ejemplo, inhibición del crecimiento del tumor) es el índice terapéutico. La dosificación puede variar dentro de este intervalo dependiendo de la forma de dosificación empleada y de la vía de administración utilizada. La determinación de una cantidad terapéuticamente eficaz se encuentra dentro de la capacidad de los expertos en la técnica, especialmente a la luz de la divulgación detallada proporcionada en el presente documento.
Una cantidad terapéuticamente eficaz de un compuesto de la presente invención requerida para su uso en terapia varía con la naturaleza del estado que va a tratarse, la duración de tiempo que se desea actividad y la edad y el estado del paciente y, en última instancia, se determina por el médico tratante. Las cantidades y los intervalos de dosificación pueden ajustarse individualmente para proporcionar niveles en plasma del inhibidor de Bcl-2/Bcl-xL que sean suficientes para mantener los efectos terapéuticos deseados. La dosis deseada puede administrarse de manera conveniente en una sola dosis, o como múltiples dosis administradas en intervalos apropiados, por ejemplo como una, dos, tres, cuatro o más subdosis al día. A menudo se desean o se requieren múltiples dosis. Por ejemplo, un inhibidor de Bcl-2/Bcl-xL presente puede administrarse a una frecuencia de: una dosis al día durante 2 días con un descanso de 5 días durante 2 semanas; una dosis al día durante 3 días con un descanso de 4 días durante 3 semanas; dosificación semanalmente durante 2 semanas; dosificación semanalmente durante 4 semanas; o cualquier pauta posológica que se considere apropiada para la circunstancia.
Un compuesto de la presente invención puede usarse en una cantidad de aproximadamente 0,005 a aproximadamente 500 miligramos por dosis, de aproximadamente 0,05 a aproximadamente 250 miligramos por dosis o de aproximadamente 0,5 a aproximadamente 100 miligramos por dosis. Por ejemplo, un compuesto de fórmula estructural (I), (II) o (III) puede administrarse, por dosis, en una cantidad de aproximadamente 0,005, 0,05, 0,5, 5, 10, 20, 30, 40, 50, 100, 150, 200, 250, 300, 350, 400, 450 ó 500 miligramos, incluyendo todas las dosis entre 0,005 y 500 miligramos.
La dosificación de una composición que contiene un inhibidor de Bcl-2/Bcl-xL de la presente invención o una composición que contiene el mismo puede ser de desde aproximadamente 1 ng/kg hasta aproximadamente 200 mg/kg, desde aproximadamente 1 pg/kg hasta aproximadamente 100 mg/kg o desde aproximadamente 1 mg/kg hasta aproximadamente 50 mg/kg. La dosificación de una composición puede ser cualquier dosificación incluyendo, pero sin limitarse a, aproximadamente 1 pg/kg. La dosificación de una composición puede ser cualquier dosificación incluyendo, pero sin limitarse a, aproximadamente 1 pg/kg, 10 pg/kg, 25 pg/kg, 50 pg/kg, 75 pg/kg, 100 pg/kg, 125 pg/kg, 150 pg/kg, 175 pg/kg, 200 pg/kg, 225 pg/kg, 250 pg/kg, 275 pg/kg, 300 pg/kg, 325 pg/kg, 350 pg/kg, 375 pg/kg, 400 pg/kg, 425 pg/kg, 450 pg/kg, 475 pg/kg, 500 pg/kg, 525 pg/kg, 550 pg/kg, 575 pg/kg, 600 pg/kg, 625 pg/kg, 650 pg/kg, 675 pg/kg, 700 pg/kg, 725 pg/kg, 750 pg/kg, 775 pg/kg, 800 pg/kg, 825 pg/kg, 850 pg/kg,
875 pg/kg, 900 pg/kg, 925 pg/kg, 950 pg/kg, 975 pg/kg, 1 mg/kg, 5 mg/kg, 10 mg/kg, 15 mg/kg, 20 mg/kg, 25 mg/kg, 30 mg/kg, 35 mg/kg, 40 mg/kg, 45 mg/kg, 50 mg/kg, 60 mg/kg, 70 mg/kg, 80 mg/kg, 90 mg/kg, 100 mg/kg, 125 mg/kg, 150 mg/kg, 175 mg/kg o 200 mg/kg.
Muchos protocolos de tratamiento de cáncer emplean actualmente radiosensibilizadores activados por radiación electromagnética, por ejemplo, rayos X. Los ejemplos de radiosensibilizadores activados por rayos X incluyen, pero no se limitan a, metronidazol, misonidazol, desmetilmisonidazol, pimonidazol, etanidazol, nimorazol, mitomicina C, RSU 1069, SR 4233, EO9, RB 6145, nicotinamida, 5-bromodesoxiuridina (BUdR), 5-yododesoxiuridina (IUdR), bromodesoxicitidina, fluorodesoxiuridina (FUdR), hidroxiurea, cisplatino y análogos y derivados terapéuticamente eficaces de los mismos.
La terapia fotodinámica (PDT) de cánceres emplea luz visible como activador de radiación del agente sensibilizador. Los ejemplos de radiosensibilizadores fotodinámicos incluyen los siguientes, pero no se limitan a: derivados de hematoporfirina, PHOTOFRIN®, derivados de benzoporfirina, NPe6, etioporfirina de estaño (SnET2), feoborbida-a, bacterioclorofila-a, naftalocianinas, ftalocianinas, ftalocianina de zinc y análogos y derivados terapéuticamente eficaces de los mismos.
Los radiosensibilizadores pueden administrarse junto con una cantidad terapéuticamente eficaz de uno o más compuestos además de un inhibidor de Bcl-2/Bcl-xL presente, incluyendo tales compuestos, pero sin limitarse a, compuestos que fomentan la incorporación de radiosensibilizadores en las células diana, compuestos que controlan el flujo de productos terapéuticos, nutrientes y/u oxígeno hacia las células diana, agentes quimioterápicos que actúan sobre el tumor con o sin radiación adicional u otros compuestos terapéuticamente eficaces para tratar el cáncer u otra enfermedad. Los ejemplos de agentes terapéuticos adicionales que pueden usarse junto con radiosensibilizadores incluyen, pero no se limitan a, 5-fluorouracilo (5-FU), leucovorina, oxígeno, carbógeno, transfusiones de glóbulos rojos, perfluorocarbonos (por ejemplo, FLUOSOLW®-DA), 2,3-DPG, BW12C, bloqueadores de los canales de calcio, pentoxifilina, compuestos antiangiogénicos, hidralazina y L-BSO.
El agente quimioterápico puede ser cualquier agente farmacológico o compuesto que induzca la apoptosis. El agente farmacológico o compuesto puede ser, por ejemplo, una molécula orgánica pequeña, un péptido, un polipéptido, un ácido nucleico o un anticuerpo. Los agentes quimioterápicos que pueden usarse incluyen, pero no se limitan a, agentes alquilantes, antimetabolitos, hormonas y antagonistas de las mismas, productos naturales y sus derivados, radioisótopos, anticuerpos, así como productos naturales y combinaciones de los mismos. Por ejemplo, un inhibidor de Bcl-2/Bcl-xL de la presente invención puede administrarse con antibióticos, tales como doxorrubicina y otros análogos de antraciclinas, mostazas de nitrógeno, tales como ciclofosfamida, análogos de pirimidina tales como 5-fluorouracilo, cisplatino, hidroxiurea, taxol y sus derivados naturales y sintéticos, y similares. Como otro ejemplo, en el caso de tumores mixtos, tales como adenocarcinoma de mama, en los que los tumores incluyen células dependientes de la gonadotropina e independientes de la gonadotropina, el compuesto puede administrarse junto con leuprolida o goserelina (análogos peptídicos sintéticos de LH-RH). Otros protocolos antineoplásicos incluyen el uso de un compuesto inhibidor con otra modalidad de tratamiento, por ejemplo, cirugía o radiación, también denominadas en el presente documento “modalidades antineoplásicos adyuvantes”. Los agentes quimioterápicos adicionales útiles en la invención incluyen hormonas y antagonistas de las mismas, radioisótopos, anticuerpos, productos naturales y combinaciones de los mismos.
Los ejemplos de agentes quimioterápicos útiles se enumeran en la siguiente tabla.
Tabla 1
Agentes alquilantes Productos naturales Mostazas nitrogenadas Fármacos antimitóticos mecloretamina
ciclofosfamida Taxanos ifosfamida paclitaxel melfalán alcaloides de la vinca clorambucilo vinblastina (VLB) mostaza de uracilo vincristina temozolomida vinorelbina vindesina
Nitro soureas Taxotere® (docetaxel) carmustina (BCNU) estramustina lomustina (CCNU) fosfato de estramustina semustina (metil-CCNU)
clormetina Epipodofilotoxinas estreptozocina etopósido
tenipósido
Etilenimina/Metil-melamina
trietilenmelamina (TEM) Antibióticos trietilentiofosforamida actimomicina D
(tiotepa) daunomicina (rubidomicina) hexametilmelamina doxorrubicina (adriamicina) (HMM, altretamina) mitoxantronaidarrubicina bleomicina
Sultanatos de alquilo splicamicina (mitramicina) busulfán mitromicina-C
pipobromán dactinomicina
afidicolina
Triazinas epirrubicina
dacarbazina (DTIC) idarrubicina
daunorrubicina Antimetabolitos mitramicina
Análogos de ácido fólico desoxi-coformicina metotrexato
trimetrexato Enzimas
pemetrexed L-asparaginasa
(antifolato multidirigido) L-arginasa
Análogos de pirimidina Radiosensibilizadores
5-fluorouracllo metronidazol fluorodesoxiuridina misonidazol
gemcitabina desmetilmisonidazol arabinósido de cítosina pimonidazol
(AraC, citarabina) etanidazol
5- azac¡t¡d¡na nimorazol
2,2’-difluorodesoxicit¡dina RSU 1069
floxurídina EOS
pentostatina RB 6145
Análogos de purina Antiandrógenos no esteroideos 6- mercaptopurina SR4233
6-t¡oguan¡na flutamida
azatioprína nicotinamida
2’-desoxicoformicina 5-bromodesoziuridina (pentostatina) 5-yododesoxiuridina eritrohidroxinoniladenina (EHNA) bromodesoxicitidina
fosfato de fludarabina
2-clorodesox¡adenosina Agentes misceláneos (cladribina, 2-CdA) Complejos de coordinación de platino cisplatino
Inhibidores de la topoisomerasa de tipo I carbop latino
camptotecina oxaiip latino
topotecán antracenodiona
¡rinotecán mitoxantrona
Modificadores de la respuesta biológica Urea sustituida
G-CSF bidroxiurea
GM-CSF
Derivados de metilhidrazina
Agentes de diferenciación N-metilhidrazina (MIH)
derivados de ácido retinoico procarbazlna
Hormonas y antagonistas Depresores adrenocorticales
Adrenocorticoesteroides/antagonistas mitotano (o,p'-DDD)
prednisona y equivalentes ainoglutetimida
dexametasona
ainoglutetimida Citocinas
interferón (a, p, y)
Proeestinas interleucina-2
caproato de hidroxiprogesterona
acetato de medroxiprogesterona Fotosensibilizadores
acetato de megestrol derivados de hematoporfinna
PHOTOFRIN®
Estróqenos derivados de benzoporfirina
dietilestilbestrol Npe6
etinilestradiol/equivalentes etioporfirina de estaño(SnET2)
feoborida-a
Antiestróaeno bactenoclorofila-a
tamoxifeno naftalocianinas
ftalocianinas
Andróqenos ftalocianinas de zinc
propionato de testosterona
fluoximesterona/equivalentes Radiación
rayos X
Antiandrógenos luz ultravioleta
flutamida radiación gamma
análogos de hormonas liberadores de gonadotropina luz visible
leuproiida radiación infrarroja
radiación microondas
Los agentes que afectan a los microtúbulos interfieren con la mitosis celular y se conocen bien en la técnica por su actividad citotóxica. Los agentes que afectan a los microtúbulos incluyen, pero no se limitan a, alocolquicina (NSC 406042), halicondrina B (NSC 609395), colquicinas (NSC 757), derivados de colquicinas (por ejemplo, Ns C 33410), dolastatina 10 (NSC 376128), maitansina (NSC 153858), rizoxina (NSC 332598), paclitaxel (Ns C 125973), derivados de TAXOL® (por ejemplo, nSc 608832), tiocolquicina NSC 361792), tritilcisteína (NSC 83265), sulfato de vinblastina (NSC 49842), sulfato de vincristina (NSC 67574), epotilonas naturales y sintéticas incluyendo, pero sin limitarse a,
epotilona A, epotilona B y discodermolida (véase Service, (1996) Science, 274:2009) estramustina, nocodazol, MAP4 y similares. Los ejemplos de tales agentes también se describen en Bulinski (1997) J. Cell Sci. 110:3055 3064; Panda (1997) Proc. Natl. Acad. Sci. USA 94:10560-10564; Muhlradt (1997) Cancer Res. 57:3344-3346; Nicolaou (1997) Nature 397:268-272; Vasquez (1997) Mol. Biol. Cell. 8:973-985; y Panda (1996) J. Biol. Chem.
271:29807-29812.
Los agentes citostáticos que pueden usarse incluyen, pero no se limitan a, hormonas y esteroides (incluyendo análogos sintéticos): 17-a-etinilestradiol, dietilestilbestrol, testosterona, prednisona, fluoximesterona, propionato de dromostanolona, testolactona, acetato de megestrol, metilprednisolona, metil-testosterona, prednisolona, triamcinolona, clorotrianiseno, hidroxiprogesterona, aminoglutimida, estramustina, acetato de medroxiprogesterona, leuprolida, flutamida, toremifeno y zoladex.
Otros agentes citostáticos son antiangiogénicos, tales como inhibidores de metaloproteinasa de la matriz y otros inhibidores de VEGF, tales como anticuerpos anti-VEGF y moléculas pequeñas tales como ZD6474 y SU668. También pueden utilizarse anticuerpos anti-Her2. Un inhibidor de EGFR es EKB-569 (un inhibidor irreversible). También se incluyen anticuerpo C225 inmunoespecífico para los inhibidores de EGFR y Src.
También es adecuado para su uso como agente citostático CASODEX® (bicalutamida, Astra Zeneca) que hace que los carcinomas dependientes de andrógenos no proliferen. Aún otro ejemplo de un agente citostático es el antiestrógeno TAMOXIFEN® que inhibe la proliferación o el crecimiento de cáncer de mama dependiente de estrógenos. Los inhibidores de la transducción de señales proliferativas celulares son agentes citostáticos. Los ejemplos representativos incluyen inhibidores del factor de crecimiento epidérmico, inhibidores de Her-2, inhibidores de cinasa MEK-1, inhibidores de cinasa MAPK, inhibidores de PI3, inhibidores de cinasa Src e inhibidores de PDGF.
Los segundos agentes terapéuticos adicionales que pueden administrarse con un inhibidor de Bcl-2/Bcl-xL de la presente invención se dan a conocer en la publicación de patente estadounidense 2007/0027135; la patente estadounidense n.° 7.432.304; la publicación de patente estadounidense n.° 2010/0278921; el documento WO 2012/017251.
Los compuestos de la presente invención se usan normalmente en una mezcla con un portador farmacéutico seleccionado con respecto a la vía de administración prevista y la práctica farmacéutica convencional. Las composiciones farmacéuticas para su uso según la presente invención se formulan de manera convencional usando uno o más portadores fisiológicamente aceptables que comprenden excipientes y sustancias auxiliares que facilitan el procesamiento de los compuestos de la invención.
Estas composiciones farmacéuticas pueden fabricarse, por ejemplo, mediante procedimientos de mezclado, disolución, granulación, elaboración de grageas, emulsificación, encapsulación, atrapamiento o liofilización convencionales. La formulación apropiada depende de la vía de administración elegida. Cuando se usa una cantidad terapéuticamente eficaz del compuesto de las invenciones por vía oral, la composición normalmente está en forma de un comprimido, una cápsula, un polvo, una disolución o un elixir. Cuando se usa en forma de comprimido, la composición puede contener adicionalmente un portador sólido, tal como una gelatina o un adyuvante. El comprimido, la cápsula y el polvo contienen de aproximadamente el 0,01% a aproximadamente el 95%, y preferiblemente desde aproximadamente el 1% hasta aproximadamente el 50%, de un compuesto de la invención. Cuando se administra en forma líquida, puede añadirse un portador líquido, tal como agua, vaselina o aceites de origen animal o vegetal. La forma líquida de la composición puede contener además solución salina fisiológica, dextrosa u otras disoluciones de sacáridos, o glicoles. Cuando se usa en forma líquida, la composición contiene de aproximadamente el 0,1% a aproximadamente el 90%, y preferiblemente de aproximadamente el 1% a aproximadamente el 50%, en peso, de un compuesto de la invención. Cuando se usa una cantidad terapéuticamente eficaz de un compuesto de la invención mediante inyección intravenosa, cutánea o subcutánea, la composición está en forma de una disolución acuosa parenteralmente aceptable libre de pirógenos. La preparación de tales disoluciones parenteralmente aceptables, teniendo debidamente en cuenta el pH, la isotonicidad, la estabilidad y similares, se encuentra dentro de la habilidad en la técnica. Una composición preferida para inyección intravenosa, cutánea o subcutánea contiene normalmente un vehículo isotónico. Los compuestos de la invención pueden combinarse fácilmente con portadores farmacéuticamente aceptables bien conocidos en la técnica. Tales portadores permiten que los agentes activos se formulen como comprimidos, pastillas, grageas, cápsulas, líquidos, geles, jarabes, pastas, suspensiones y similares, para ingestión oral por un paciente que va a tratarse. Las preparaciones farmacéuticas para uso oral pueden obtenerse añadiendo el compuesto de la invención a un excipiente sólido, opcionalmente triturando la mezcla resultante y procesando la mezcla de gránulos, después de añadir sustancias auxiliares adecuadas, si se desea, para obtener núcleos de grageas o comprimidos. Los excipientes adecuados incluyen, por ejemplo, cargas y preparaciones de celulosa. Si se desea, pueden añadirse agentes disgregantes.
Un compuesto de la invención puede formularse para administración parenteral mediante inyección, por ejemplo, mediante inyección en bolo o infusión continua. Las formulaciones para inyección pueden presentarse en forma de dosificación unitaria, por ejemplo, en ampollas o en recipientes multidosis, con un conservante añadido. Las composiciones pueden tomar formas tales como suspensiones, disoluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilización y/o dispersión.
Las composiciones farmacéuticas para administración parenteral incluyen disoluciones acuosas del agente activo en forma soluble en agua. De manera adicional, pueden prepararse suspensiones de un compuesto de la presente invención como suspensiones oleosas para inyección apropiadas. Los disolventes o vehículos lipófilos adecuados incluyen aceites grasos o ésteres de ácidos grasos sintéticos. Las suspensiones acuosas para inyección pueden contener sustancias que aumentan la viscosidad de la suspensión. Opcionalmente, la suspensión también puede contener estabilizadores adecuados o agentes que aumentan la solubilidad de los compuestos y permiten la preparación de disoluciones altamente concentradas. Alternativamente, una composición presente puede estar en forma de polvo para su constitución con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, antes de su uso.
Un compuesto de la presente invención también puede formularse en composiciones rectales, tales como supositorios o enemas de retención, por ejemplo, que contienen bases de supositorio convencionales. Además de las formulaciones descritas anteriormente, el compuesto de la presente invención también puede formularse como una preparación de liberación lenta. Tales de formulaciones de larga actuación pueden administrarse mediante implantación (por ejemplo, por vía subcutánea o intramuscular) o mediante inyección intramuscular. Por tanto, por ejemplo, los compuestos de la presente invención pueden formularse con materiales poliméricos o hidrófobos adecuados (por ejemplo, como una emulsión en un aceite aceptable) o resinas de intercambio iónico.
En particular, los compuestos de la invención pueden administrarse por vía oral, bucal o sublingual en forma de comprimidos que contienen excipientes, tales como almidón o lactosa, o en cápsulas u óvulos, o bien solos o bien en una mezcla con excipientes, o en forma de elixires o suspensiones que contienen agentes aromatizantes o colorantes. Tales preparaciones líquidas pueden prepararse con aditivos farmacéuticamente aceptables, tales como agentes de suspensión. Los compuestos de la invención también pueden inyectarse por vía parenteral, por ejemplo, por vía intravenosa, intramuscular, subcutánea o intracoronaria. Para la administración parenteral, los inhibidores de Bcl-2/Bcl-xL se usan mejor en forma de una disolución acuosa estéril que puede contener otras sustancias, por ejemplo, sales o monosacáridos, tales como manitol o glucosa, para hacer que la disolución sea isotónica con la sangre.
Los inhibidores de Bcl-2/Bcl-xL anteriores presentaban propiedades que dificultaban su desarrollo como agentes terapéuticos. Los compuestos de la invención se sintetizaron y evaluaron como inhibidores para Bcl-2/Bcl-xL. Por ejemplo, los compuestos de la presente invención tienen normalmente una afinidad de unión (CI50) a Bcl-2/Bcl-xL de menos de 100 nM.
Síntesis de compuestos
Los compuestos de la presente invención se prepararon de la siguiente manera. Los siguientes esquemas de síntesis son representativos de las reacciones usadas para sintetizar compuestos tales como los compuestos dados a conocer a continuación con los siguientes números 12 (equivalente al compuesto 14), 15-17, 19 y 27 que forman parte de la invención.
Modificaciones y esquemas alternativos para prepare inhibidores de Bcl-2/Bcl-xL se encuentran fácilmente dentro de las capacidades de los expertos en la técnica.
Los disolventes y reactivos se obtuvieron comercialmente y se usaron sin purificación adicional. Los desplazamientos químicos (8) de los espectros de RMN se notificaron como valores de 8 (ppm) de campo bajo con respecto a un patrón interno, notificándose las multiplicidades de la manera habitual.
A menos que se indique de otro modo, todas las temperaturas están en grados Celsius (°C).
Pueden sintetizarse determinados productos intermedios clave para la síntesis de los compuestos mediante los métodos tal como se establece en el documento WO 2012/103059, seguido por su conversión en su derivado de fosfato de la siguiente manera:
Esquema 1. Síntesis del compuesto 1

Sección experimental: 2-aminoacetato de (R)-1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-¡lo (1). Se agitó una disolución de BM-1197 (113 mg, 0,10 mmol) y Fmoc-Gly-OSu (43 mg, 0,11 mmol) en CH2Cl2 (2 ml) a temperatura ambiente durante 1 hora hasta que no se observó BM-1197 mediante CCF. Se concentró la disolución a vacío para proporcionar el precursor en bruto de 1 que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en acetonitrilo (5 ml) y se siguió por la adición de dietilamina (0,2 ml, 2 mmol). Se agitó la mezcla a temperatura ambiente durante la noche hasta que no se observó material de partida mediante CCF y se concentró a vacío. Se purificó el residuo mediante HPLC para dar el producto 1 (sal con TFA, 83 mg, rendimiento del 70% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. EM (ESI) m/z 1189,08 (M H)+.
Esquema 2. Síntesis de 2

Sección experimental: ácido (R)-2-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-iloxi)-2-oxoacético (2). A una disolución de BM-1197 (113 mg, 0,10 mmol), DMAP (2 mg, 0,02 mmol), Et3N (42 ul, 0,3 mmol) en CH2Cl2 (2 ml) se le añadió 2-cloro-2-oxoacetato de terc-butilo (33 mg, 0,2 mmol). Se agitó la disolución a temperatura ambiente durante 1 hora hasta que no se observó BM-1197 mediante CCF y se concentró a vacío. Se sometió a cromatografía ultrarrápida el residuo en bruto sobre gel de sílice con el 5% de MeOH/CH2Cl2 para proporcionar el precursor de 2. Se disolvió el precursor en CH2Cl2 (3 ml) y se siguió por la adición de TFA (3 ml). Se agitó la mezcla a temperatura ambiente durante 1 hora hasta que no se observó material de partida mediante CCF y se concentró a vacío. Se purificó el residuo mediante HPLC para dar el producto 2 (sal con TFA, 66 mg, rendimiento del 55% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. EM (ESI) m/z 1189,08 (M H)+.
Esquema 3. Preparación de producto intermedio clave B y D

Sección experimental: N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)-4-fluoro-3-(trifluorometilsulfonil)bencenosulfonamida (B). A una disolución de A (3,0 g, 4,9 mmol) en 150 ml de metanol se le añadió Pd al 10% en peso/C (300 mg, 0,1 eq. m/m). Se agitó la disolución bajo una atmósfera de hidrógeno a temperatura ambiente durante aproximadamente 20 min hasta que no se observó A mediante CCF. Se filtró la mezcla de reacción y se concentró el filtrado a vacío. Se usó el residuo en la siguiente etapa directamente sin purificación. A la disolución de esta anilina en piridina, se le añadió cloruro de 4-fluoro-3-(trifluorometilsulfonil)benceno-1-sulfonilo (1,8 g, 5,4 mmol) a 0C. Se agitó la mezcla at 0°C hasta temperatura ambiente durante 1 hora hasta que no se observó anilina mediante CCF. Se añadió agua (10 ml) y se extrajo con acetato de etilo (200 ml * 2). Se lavó la disolución combinada de acetato de etilo con salmuera (150 ml), se secó sobre sulfato de sodio y se concentró a vacío. Se sometió a cromatografía ultrarrápida el concentrado sobre gel de sílice con el 40% de EtOA/hexano para proporcionar el producto intermedio B (3,2 g, rendimiento del 75% en dos etapas). EM (ESI) m/z 931,75 (M K)+.
Procedimiento general I. 4-Oxo-1-(feniltio)butan-2-ilcarbamato de (R)-(9H-fluoren-9-il)metilo (D). A una disolución de C (5,0 g, 11,5 mmol) en THF (100 ml) se le añadió trietilamina (4,8 ml, 34,5 mmol) y cloroformiato de etilo (3,3 ml, 34,5 mmol) a -10°C bajo una atmósfera de argón. Se agitó la mezcla a -10°C durante 1 h y se añadió NaBH4 (1,7 g, 46,1 mmol) en agua (60 ml) gota a gota a -10°C. Se agitó la mezcla a -10°C durante 1 h, luego a temperatura ambiente durante 2 h. Se extinguió la reacción con KHSO4 acuoso 1 M (200 ml) y se extrajo la mezcla con EtOAc (3x200 ml). Se lavaron los extractos con salmuera (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a vacío. Se sometió a cromatografía ultrarrápida el concentrado sobre gel de sílice con el 50% de EtOA/hexano para proporcionar el alcohol correspondiente (4,3 g, rendimiento del 90%). A una disolución de cloruro de oxalilo (2,6 ml, 31,1 mmol) en DCM (100 ml) a -78°C, se le añadió dimetilsulfóxido (3,7 ml, 51,8 mmol). Se calentó la disolución hasta -40°C durante 5 min y volvió a enfriarse hasta -78°C, y luego se añadió gota a gota una disolución del alcohol resultante de la etapa previa (4,3 g, 10,4 mmol) en DCM (50 ml). Se agitó la disolución durante 40 min adicionales y se siguió por un exceso de trietilamina (25 ml) y se agitó durante otros 30 min. Se calentó la mezcla de reacción hasta temperatura ambiente seguido por la adición de disolución acuosa saturada de cloruro de amonio (100 ml) y se extrajo con DCM (2x200 ml). Se lavó la disolución combinada en DCM con salmuera (150 ml), se secó sobre sulfato de sodio y se concentró a vacío. Se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 20% EtOA/hexano para proporcionar el producto intermedio D (3,7 g, rendimiento del 85%). EM (ESI) m/z 418,25 (M H)+.
Esquema 4. Síntesis de 3

Sección experimental: fosfato de di-terc-butilo y piperidin-4-ilo (F). Se agitó la disolución de fosforamidito de di-tbutilo y di-isopropilo (832 mg, 3,0 mmol) y tetrazol (6,6 ml, 0,45 M en acetanitrilo) en THF (15 ml) bajo N2 a temperatura ambiente durante aproximadamente 10 min. Luego se añadió el compuesto E (626 mg, 2,0 mmol) en THF seco (2 ml) a la reacción a lo largo de 15 minutos y se agitó a temperatura ambiente bajo N2 durante 2 horas hasta que no se observó E mediante CCF. Luego se enfrió la mezcla de reacción hasta 0°C y se añadió una disolución acuosa al 14% de peróxido de t-butilo (3,0 ml, 4,6 mmol). Luego se dejó que la temperatura aumentara hasta temperatura ambiente y se agitó la mezcla durante la noche. Se extinguió la reacción con disolución acuosa saturada de NaHCÜ3 (2 ml). Se añadió agua (50 ml) en la mezcla de reacción, que luego se extrajo con acetato de etilo (2x50 ml). Se lavó la disolución combinada de acetato de etilo con salmuera (50 ml), se secó sobre sulfato de sodio y se concentró a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en acetonitrilo (20 ml) y se siguió por la adición de dietilamina (4,1 ml, 40 mmol). Se agitó la mezcla a temperatura ambiente durante la noche hasta que no se observó material de partida mediante CCF y se concentró a vacío. Se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 5% de MeOH/DCM para proporcionar el producto intermedio F (452 mg, rendimiento del 77% en dos etapas). EM (ESI) m/z 295,17 (M H)+.
Procedimiento general II. Fosfato de (R)-1-(3-amino-4-(feniltio)butil)piperidin-4-ilo y di-terc-butilo (G). A una disolución de F (293 mg, 1,0 mmol) y producto intermedio D (500 mg, 1,2 mmol) en DCE (10 ml) se le añadió NaBH(OAc)3 (636 mg, 3,0 mmol) y se agitó la mezcla a temperatura ambiente durante la noche hasta que no se observó F mediante CCF. Se diluyó la mezcla con DCM (50 ml), se lavó con salmuera (50 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en acetonitrilo (10 ml) y se siguió por la adición de dietilamina (2,1 ml, 20 mmol). Se agitó la mezcla a temperatura ambiente durante la noche hasta que no se observó material de partida mediante CCF y se concentró a vacío. Se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 10% de MeOH/DCM para proporcionar el producto intermedio G (307 mg, rendimiento del 65% en dos etapas). EM (ESI) m/z 474,00 (M H)+.
Procedimiento general III. Dihidrogenofosfato de (R)-1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1Hpirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-ilo (3). A una disolución de B (100 mg, 0,11 mmol) y G (65 mg, 0,14 mmol) en DMF (2 ml) se le añadió D I P E a (1 ml). Se agitó la disolución durante 4 horas a temperatura ambiente hasta que no se observó B mediante CCF. Se concentró la mezcla de reacción a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en DCM (5 ml) y se siguió por la adición de TFA (2,5 ml). Se agitó la disolución a temperatura ambiente durante 1 h hasta que no se observó material mediante CCF. Se concentró la mezcla de reacción a vacío y se purificó el residuo mediante HPLC para dar el producto 3 puro (sal con TFA, 88 mg, rendimiento del 66% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. 1H-RMN (300 MHz, CD3OD): 8 7,96 (s, 1H), 7,73 (d, J = 8,9 Hz, 1H), 7,32-7,07 (m, 13H), 6,93-6,41 (m, 4H), 4,61-4,41 (m, 2H), 3,99 (s, 1H), 3,55 3,11 (m, 16H), 2,84 (s, 3H), 2,74 (s, 3H), 2,26-1,80 (m, 6H), 1,43 (d, J = 7,0 Hz, 6H). EM (ESI): m/z 1212,67 (M H)+.
Esquema 5. Síntesis de 4

Sección experimental: 4-(metiltiometoxi)piperidina (H). A una disolución de alcohol E (1,0 g, 3,1 mmol) y sulfuro de metilo (1,8 ml, 24,8 mmol) en acetonitrilo (31 ml) a 0°C se le añadió peróxido de benzoílo (3,0 g, 12,4 mmol) en cuatro porciones iguales a lo largo de 10 min y se agitó la mezcla a 0°C durante 1 h y luego a temperatura ambiente durante 1 h hasta que no se observó E mediante CCF. Se diluyó la mezcla con acetato de etilo (100 ml), se lavó con Na2CÜ3 al 10% (100 ml) y luego salmuera (100 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en acetonitrilo (10 ml) y se siguió por la adición de dietilamina (6,2 ml, 60 mmol). Se agitó la mezcla a temperatura ambiente durante la noche hasta que no se observó material de partida mediante CCF y se concentró a vacío. Se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 5% de MeOH/DCM para proporcionar el producto intermedio H (270 mg, rendimiento del 54% en dos etapas). EM (ESI) m/z 162,83 (M H)+.
(R)-4-(4-(metiltiometoxi)piperidin-1-il)-1-(feniltio)butan-2-amina (I). Se preparó I a partir de H y D según el procedimiento general II. EM (ESI) m/z 341,58 (m H)+.
Dihidrogenofosfato de (R)-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-iloxi)metilo (4). A una disolución de B (200 mg, 0,23 mmol) e I (86 mg, 0,25 mmol) en DMF (4 ml) se le añadió DIPEA (2 ml). Se agitó la disolución durante 4 horas a temperatura ambiente hasta que no se observó B mediante CCF. Se concentró la mezcla de reacción a vacío. Se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 5% de MeOH/DCM para dar el tioéter correspondiente (241 mg, rendimiento del 88%). A una disolución del tioéter de la primera etapa (200 mg, 0,17 mmol), ácido fosfórico (117 mg, 1,2 mmol) y tamices moleculares (4 A, 500 mg) en THF (6 ml) a 0°C se le añadió W-yodosuccinimida (57 mg, 0,26 mmol) y se agitó la mezcla a temperatura ambiente durante 1 h hasta que no se observó material de partida mediante CCF. Se filtró la mezcla de reacción a través de Celite y se lavaron los sólidos con metanol. Se concentró el filtrado a vacío y se purificó el residuo mediante HPLC para dar el producto 4 puro (sal con TFA, 93 mg, rendimiento del 44%). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. EM (ESI): m/z 1242,08 (M H)+.
Esquema 6. Síntesis de los compuestos 5, 6, 7

Sección experimental: procedimiento general IV. Ácido (R)-3-((5-(4-clorofenil)-1-etil-4-(3-(4-(4-(4-((4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-il)amino)-3-((trifluorometil)sulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-2-metil-1H-pirrol-3-carbonil)oxi)propanoico (5). A una disolución de 957 (100 mg, 0,09 mmol), d Ic (18 mg, 0,14 mmol) y DMAP (20 mg, 0,14 mmol) en DCM (2 ml) se le añadió 3-hidroxipropanoato de terc-butilo (41 mg, 0,28 mmol). Se agitó la disolución durante 6 horas a temperatura ambiente hasta que no se observó BM-957 mediante CCF. Se diluyó la mezcla de reacción con acetato de etilo (50 ml), se lavó con disolución saturada de NaHCO3 (50 ml),
salmuera (50 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en DCM (5 ml) y se siguió por la adición de TFA (2,5 ml). Se agitó la disolución a temperatura ambiente durante 3 h hasta que no se observó material de partida mediante CCF. Se concentró la mezcla de reacción a vacío y se purificó el residuo mediante HPLC para dar el producto 5 puro (sal con TFA, 75 mg, rendimiento del 70% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. EM (ESI): m/z 1238,17 (M H)+.
Ácido (R)-4-((5-(4-clorofenil)-1-etil-4-(3-(4-(4-(4-((4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-il)amino)-3-((trifluorometil)sulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-2-metil-1H-pirrol-3-carbonil)oxi)benzoico (6). Se preparó 6 a partir de BM-957 y 4-hidroxibenzoato de terc-butilo según el procedimiento general IV. EM (ESl): m/z 1186,00 (M H)+.
Ácido (R)-4-((5-(4-clorofenil)-1-etil-4-(3-(4-(4-(4-((4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-il)amino)-3-((trifluorometil)sulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-2-metil-1H-pirrol-3-carbonil)oxi)ciclohexanocarboxílico (7). Se preparó 7 a partir de BM-957 y 4-hidroxiciclohexanocarboxilato de tercbutilo según el procedimiento general IV. e M (ESI): m/z 1192,25 (M H)+.
Esquema 7. Síntesis de 8, 9

Procedimiento general V. Ácido (R)-(((5-(4-clorofenil)-1-etil-4-(3-(4-(4-(4-((4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-il)amino)-3-((trifluorometil)sulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-2-metil-1H-pirrol-3-carbonil)oxi)metil)fosfónico (8). A una disolución de BM-957 (100 mg, 0,09 mmol), DIC (18 mg, 0,14 mmol) y DMAP (20 mg, 0,14 mmol) en DCM (2 ml) se le añadió (hidroximetil)fosfonato de dimetilo (40 mg, 0,28 mmol). Se agitó la disolución durante 6 horas a temperatura ambiente hasta que no se observó BM-957 mediante CCF. Se diluyó la mezcla de reacción con acetato de etilo (50 ml), se lavó con disolución saturada de NaHCÜ3 (50 ml), salmuera (50 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en DCM (5 ml) y se siguió por la adición de TMSBr (248 ul, 1,9 mmol). Se agitó la disolución a temperatura ambiente durante 20 h hasta que no se observó material de partida mediante EM. Se concentró la mezcla de reacción a vacío y se purificó el residuo mediante HPLC para dar el producto 8 puro (sal con TFA, 74 mg, rendimiento del 68% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. 1H-RMN (300 MHz, CD3OD): 87,92 (s, 1H), 7,73-7,70 (m, 2H), 7,34-6,82 (m, 17H), 4,28 (d, J = 8,6 Hz, 2H), 4,06-3,35 (m, 14H), 3,20-2,92 (m, 5H), 2,65 (s, 3H), 2,24-1,67 (m, 6H), 1,10 (t, J = 7,0 Hz, 3H). EM (ESI): m/z 1259,50 (M H)+.
Ácido (R)-(2-((5-(4-clorofenil)-1-etil-4-(3-(4-(4-(4-((4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-il)amino)-3-((trifluorometil)sulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-2-metil-1H-pirrol-3-carbonil)oxi)etil)fosfónico (9). Se preparó 9 a partir de BM-957 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. EM (ESI): m/z 1173,42 (M H)+.
Esquema 8. Síntesis de 10

Ácido ((R)-4-(5-(4-clorofenil)-1-etil-4-(3-(4-(4-(4-(4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-ilamino)-3-(trifluorometilsulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-2-metil-1H-pirrol-3-carboxamido)ciclohexanocarboxílico (10). A una disolución de BM-957 (100 mg, 0,09 mmol), EDCI (27 mg, 0,14 mmol) y HOBT (19 mg, 0,14 mmol) en DCM (2 ml) se le añadió 4-aminociclohexanocarboxilato de metilo (44 mg, 0,28 mmol). Se agitó la disolución durante 2 horas a temperatura ambiente hasta que no se observó BM-957 mediante CCF. Se diluyó la mezcla de reacción con acetato de etilo (50 ml), se lavó con disolución saturada de NaHCO3 (50 ml), salmuera (50 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en H2O y MeOH (5 ml y 5 ml, respectivamente) y se siguió por la adición de NaOH (76 mg, 1,9 mmol). Se agitó la disolución a temperatura ambiente durante 20 h hasta que no se observó material de partida mediante CCF. Se concentró la mezcla de reacción a vacío y se purificó el residuo mediante HPLC para dar el producto 10 puro (sal con TFA, 61 mg, rendimiento del 55% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. EM (ESI): m/z 1191,17 (M H)+.
Esquema 9. Síntesis de 11
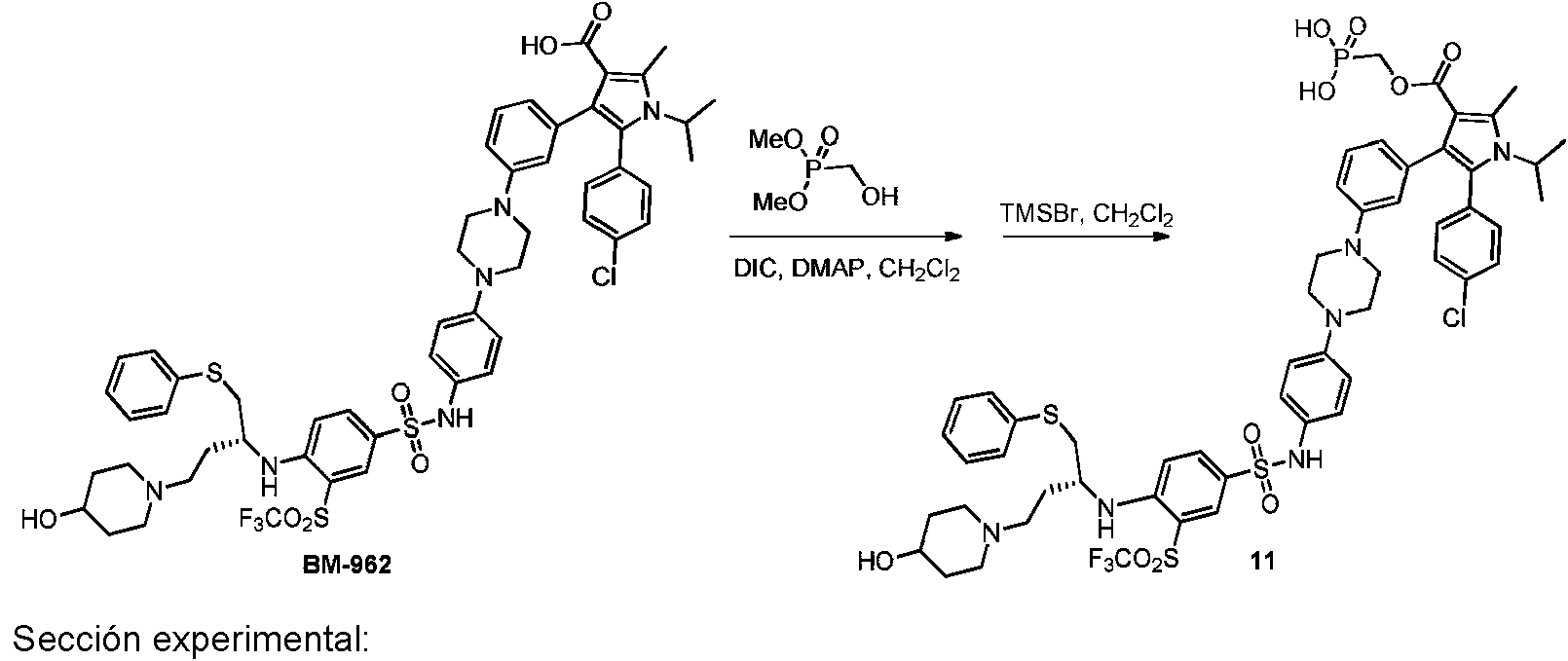
Ácido (R)-(((5-(4-clorofenil)-4-(3-(4-(4-(4-((4-(4-hidroxipiperidin-1-il)-1-(feniltio)butan-2-il)amino)-3-((trifluorometil)sulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-1-isopropil-2-metil-1H-pirrol-3-carbonil)oxi)metil)fosfónico (11). Se preparó 11 a partir de BM-962 e (hidroximetil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 88,00 (s, 1H), 7,80-7,71 (m, 2H), 7,38-6,83 (m, 17H), 4,50 4,41 (m, 1H), 4,29 (d, J = 8,7 Hz, 2H), 4,11-3,59 (m, 12H), 3,25-3,01 (m, 6H), 2,77 (s, 3H), 2,28-1,70 (m, 6H), 1,47 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1174,25 (M H)+.
Esquema 9. Síntesis de 12

(R)-1-(3-amino-4-(feniltio)butil)piperidin-4-carboxilato de terc-butilo (K). Se preparó K a partir de piperidin-4-carboxilato de terc-butilo y D según el procedimiento general II. EM (ESI): m/z 365,50 (M H)+.
Ácido (R)-1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboxílico (13). Se preparó 13 a partir de K y B según el procedimiento general III. e M (ESI): m/z 365,50 (M H)+.
Esquema 11. Síntesis de 14, 15, 16, 17

Ácido (R)-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)metilfosfónico (14). Se preparó 14 a partir de 13 y (2-hidroximetil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,94 (s, 1H), 7,72 (d, J = 9,1 Hz, 1H), 7,30-7,09 (m, 13H), 6,91-6,42 (m, 4H), 4,49-4,40 (m, 1H), 3,99 (s, 1H), 3,55-2,90 (m, 16H), 2,84 (s, 3H), 2,72 (s, 3H), 2,63-2,55 (m, 1H), 2,23-1,81 (m, 6H), 1,41 (d, J = 4,3 Hz, 6H). EM (ESI): m/z 1160,34 (M H)+.
Ácido (R)-2-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)etilfosfónico (15). Se preparó 15 a partir de 13 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,93 (d, J = 1,9 Hz, 1H), 7,72 (dd, J = 9,2, 1,8 Hz, 1H), 7,30 7,12 (m, 12H), 6,83-6,42 (m, 5H), 4,46-4,33 (m, 3H), 3,96 (s, 1H), 3,54-2,93 (m, 16H), 2,82 (s, 3H), 2,72 (s, 3H), 2,71-2,55 (m, 1H), 2,24-1,65 (m, 8H), 1,41 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1268,58 (M H)+.
Ácido (R)-3-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)propilfosfónico (16). Se preparó 16 a partir de 13 y 3-hidroxipropilfosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,95 (d, J = 2,0 Hz, 1H), 7,73 (dd, J = 9,2, 2,1 Hz, 1H), 7,33 7,12 (m, 12H), 6,92-6,43 (m, 5H), 4,51-4,41 (m, 1H), 4,18-3,98 (m, 3H), 3,56-2,92 (m, 16H), 2,85 (s, 3H), 2,73 (s, 3H), 2,67-2,50 (m, 1H), 2,25-1,70 (m, 10H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1282,34 (M H)+.
Ácido 2-(1-((R)-3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)propilfosfónico (17). Se preparó 17 a partir de 13 y 2-hidroxipropilfosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,97 (d, J = 2,1 Hz, 1H), 7,73 (d, J = 9,2 Hz, 1H), 7,36-7,08 (m, 13H), 6,85-6,43 (m, 4H), 5,26 (s, 1H), 4,54-4,44 (m, 1H), 4,01 (s, 1H), 3,58-2,92 (m, 16H), 2,87 (s, 3H), 2,76 (s, 3H), 2,70-2,55 (m, 1H), 2,26-1,85 (m, 8H), 1,46 (d, J = 7,1 Hz, 6H), 1,38 (d, J = 5,9 Hz, 3H). EM (ESI): m/z 1281,34 (M H)+.
Esquema 12. Síntesis de 18

(R)-1-(3-amino-4-(feniltio)butil)-4-metilpiperidin-4-carboxilato de metilo (M). Se preparó M a partir de 4-metilpiperidin-4-carboxilato de metilo y D según el procedimiento general II. EM (ESI): m/z 337,55 (M H)+.
Ácido (R)-1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)-4-metilpiperidin-4-carboxílico (18). A una disolución de B (100 mg, 0,11 mmol) y M (47 mg, 0,14 mmol) en DMF (2 ml) se le añadió DIPEA (1 ml). Se agitó la disolución durante 4 horas a temperatura ambiente hasta que no se observó B mediante CCF. Se concentró la mezcla de reacción a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en H2O y MeOH (5 ml y 5 ml, respectivamente) y se siguió por la adición de NaOH (88 mg, 2,2 mmol). Se agitó la disolución a temperatura ambiente durante 20 h hasta que no se observó material de partida mediante CCF. Se concentró la mezcla de reacción a vacío y se purificó el residuo mediante HPLC para dar el producto 18 puro (sal con TFA, 75 mg, rendimiento del 58% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min. 1H-RMN (300 MHz, CD3OD): 87,99 (d, J = 1,6 Hz, 1H), 7,76 (dd, J = 9,1, 1,9 Hz, 1H), 7,37-6,84 (m, 14H), 6,68-6,45 (m, 3H), 4,55-4,45 (m, 1H), 4,02 (s, 1H), 3,58-2,92 (m, 17H), 2,88 (s, 3H), 2,77 (s, 3H), 2,41-1,86 (m, 5H), 1,47 (d, J = 7,1 Hz, 6H), 1,31 (s, 3H). EM (ESI): m/z 1173,73 (M H)+.
Esquema 13. Síntesis de 19

Ácido (R)-2-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)-4-metilpiperidin-4-carboniloxi)etilfosfónico (19). Se preparó 19 a partir de 18 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,98 (d, J = 1,6 Hz, 1H), 7,73 (dd, J = 9,2, 2,0 Hz, 1H), 7,35 6,83 (m, 14H), 6,65-6,44 (m, 3H), 4,52-4,38 (m, 3H), 4,01 (s, 1H), 3,44-2,92 (m, 17H), 2,87 (s, 3H), 2,77 (s, 3H), 2,45-2,11 (m, 5H), 1,71 (t, J = 14,4 Hz, 2H), 1,46 (d, J = 7,1 Hz, 6H), 1,30 (s, 3H). EM (ESI): m/z 1281,92 (M H)+.
Esquema 14. Síntesis del compuesto 20

Ácido (R)-3-(((9H-fluoren-9-il)metoxi)carbonilamino)-4-(2-fluorofeniltio)butanoico (O). Se trató una disolución de Bu3P (0,8 ml, 3,3 mmol) y ADDP (833 mg, 3,3 mmol) en THF (30 ml) con N (1,2 g, 3,0 mmol) y tiofenol (320 ul, 3,0 mmol), se agitó durante 4 h hasta que no se observó N mediante CCF. Se diluyó la mezcla con acetato de etilo (100 ml), se lavó con HCl acuoso 1 M (100 ml), salmuera (100 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en DCM (10 ml) y se siguió por la adición de TFA (5 ml). Se agitó la disolución a temperatura ambiente durante 1 h hasta que no se observó material de partida mediante CCF. Se concentró la mezcla de reacción a vacío y se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 5% de MeOH/DCM para proporcionar el producto intermedio O (840 mg, rendimiento del 62% en dos etapas). EM (ESI) m/z 452,86 (M H)+.
(R)-1-(2-fluorofeniltio)-4-oxobutan-2-ilcarbamato de (9H-fluoren-9-il)metilo (P). Se preparó P a partir de O según el procedimiento general I. EM (ESI) m/z 437,00 (M H)+.
(R)-1-(3-amino-4-(2-fluorofeniltio)butil)piperidin-4-carboxilato de terc-butilo (Q). Se preparó Q a partir de P y J según el procedimiento general II. EM (ESI) m/z 383,38 (M H)+.
Ácido (R)-1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(2-fluorofeniltio)butil)piperidin-4-carboxílico (20). Se preparó 20 a partir de Q y B según el procedimiento general III. 1H-RMN (300 MHz, CD3OD): 87,97 (d, J = 1,9 Hz, 1H), 7,76 (dd, J = 9,2, 2,0 Hz, 1H), 7,39-6,87 (m, 13H), 6,65-6,43 (m, 3H), 4,54-4,45 (m, 1H), 4,01 (s, 1H), 3,67-2,93 (m, 17H), 2,87 (s, 3H), 2,77 (s, 3H), 2,29-1,86 (m, 6H), 1,46 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1177,92 (M H)+.
Esquema 15. Síntesis de 21
Ácido (R)-2-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(2-fluorofeniltio)butil)piperidin-4-carboniloxi)etilfosfónico (21). Se preparó 21 a partir de 20 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,95 (d, J = 1,7 Hz, 1H), 7,77 (dd, J = 9,0, 2,0 Hz, 1H), 7,36 6,86 (m, 13H), 6,66-6,44 (m, 3H), 4,51-4,33 (m, 3H), 4,01 (s, 1H), 3,58-2,93 (m, 16H), 2,85 (s, 3H), 2,74 (s, 3H), 2,70-2,58 (m, 1H), 2,27-1,84 (m, 8H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1286,58 (M H)+.
Esquema 16. Síntesis de 22

Sección experimental:
(R)-1-(4-(feniltio)-3-(4-sulfamoil-2-(trifluorometilsulfonil)fenilamino)butil)piperidin-4-carboxilato de terc-butilo (S). A una disolución de K (1,1 g, 3,0 mmol) y R (922 mg, 3,0 mmol) en DMF (15 ml) se le añadió DIPEA (3 ml). Se agitó la disolución durante 4 horas a temperatura ambiente hasta que no se observó K mediante CCF. Se concentró la mezcla de reacción a vacío y se sometió a cromatografía ultrarrápida el residuo sobre gel de sílice con el 5% de MeOH/DCM para proporcionar el producto intermedio S (1,7 g, rendimiento del 88% en dos etapas). EM (ESI) m/z 653,21 (M H)+.
Ácido (R)-1-(3-(4-(N-(4-(4-((2-(4-clorofenil)-5,5-dimetilciclohex-1-enil)metil)piperazin-1-il)benzoil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboxílico (22). A una disolución de T (438 mg, 1.0 mmol), EDCI (386 mg, 2,0 mmol) y DmAp (121 mg, 1,0 mmol) en DCM (10 ml) se le añadió S (718 mg, 1.1 mmol). Se agitó la disolución durante 2 horas a temperatura ambiente hasta que no se observó T mediante CCF. Se diluyó la mezcla de reacción con acetato de etilo (50 ml), se lavó con disolución saturada de NaHCO3 (50 ml), salmuera (50 ml) y se secó sobre sulfato de sodio. Se eliminó el disolvente a vacío para dar un producto en bruto que se usó en la siguiente etapa sin purificación. Se disolvió el residuo resultante en DCM (10 ml) y se siguió por la adición de TFA (5 ml). Se agitó la disolución a temperatura ambiente durante 1 h hasta que no se observó material de partida mediante CCF. Se concentró la mezcla de reacción a vacío y se purificó el residuo mediante HPLC para dar el producto 22 puro (sal con TFA, 742 mg, rendimiento del 73% en dos etapas). El gradiente se desarrolló desde el 60% de disolvente A y el 40% de disolvente B hasta el 20% de disolvente A y el 80% de disolvente B en 40 min.
1H-RMN (300 MHz, CD3OD): 88,30 (d, J = 2,1 Hz, 1H), 8,02 (dd, J = 9,2, 2,5 Hz, 1H), 7,70 (d, J = 8,9 Hz, 2H), 7,40 6,88 (m, 12H), 4,04 (s, 1H), 3,67-2,82 (m, 19H), 2,58 (t, J = 14,4 Hz, 1H), 2,37-1,81 (m, 10H), 1,53 (t, J = 6,2 Hz, 2H), 1,03 (s, 6H). EM (ESI): m/z 1017,50 (M H)+.
Esquema 17. Síntesis de 23, 24, 25
Ácido (R)-(1-(3-(4-(N-(4-(4-((2-(4-clorofenil)-5,5-dimetilciclohex-1-enil)metil)piperazin-1-il)benzoil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)metilfosfónico (23). Se preparó 23 a partir de 22 y (2-hidroximetil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 88,35 (s, 1H), 8,09 (d, J = 6,7 Hz, 1H), 7,79 (d, J = 7,7 Hz, 2H), 7,44-6,82 (m, 12H), 4,30-4,10 (m, 3H), 3,74-2,73 (m, 19H), 2,43-1,44 (m, 12H), 1,10 (s, 6H). EM (ESI): m/z 1110,58 (M H)+.
Ácido (R)-2-(1-(3-(4-(N-(4-(4-((2-(4-clorofenil)-5,5-dimetilciclohex-1-enil)metil)piperazin-1-il)benzoil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)etilfosfónico (24). Se preparó 24 a partir de 22 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 88,29 (d, J = 2,0 Hz, 1H), 8,02 (dd, J = 9,2, 2,0 Hz, 1H), 7,71 (d, J = 8,8 Hz, 2H), 7,37-6,84 (m, 12H), 4,34-4,30 (m, 2H), 4,03 (s, 1H), 3,66-2,88 (m, 18H), 2,62 (t, J = 14,4 Hz, 1H), 2,36-1,82 (m, 12H), 1,53 (t, J = 6,1 Hz, 2H), 1,03 (s, 6H). EM (ESI): m/z 1025,64 (M H)+.
Ácido (R)-3-(1-(3-(4-(N-(4-(4-((2-(4-clorofenil)-5,5-dimetilciclohex-1-enil)metil)piperazin-1-il)benzoil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboniloxi)propilfosfónico (25). Se preparó 25 a partir de 22 y 3-hidroxipropilfosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,95 (d, J = 2,0 Hz, 1H), 7,73 (dd, J = 9,2, 2,1 Hz, 1H), 7,33-7,12 (m, 12H), 6,92-6,43 (m, 5H), 4,51-4,41 (m, 1H), 4,18-3,98 (m, 3H), 3,56-2,92 (m, 16H), 2,85 (s, 3H), 2,73 (s, 3H), 2,67-2,50 (m, 1H), 2,25-1,70 (m, 10H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1282,34 (M H)+.

5-(4-Clorofenil)-4-(3-(4-(4-(4-fluoro-3-(trifluorometilsulfonil)fenilsulfonamido)fenil)piperazin-1-il)fenil)-1-isopropil-2-metil-N-(metilsulfonil)-1H-pirrol-3-carboxamida (V). Se preparó V a partir de U según el procedimiento descrito para la preparación del compuesto B.
Ácido (R)-1-(3-(4-(W-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonilcarbamoil)-1H-pirrol-3-il)fenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4-carboxílico (26) (BM-1077): se preparó 26 a partir de K y V según el procedimiento general III. 1H-RMN (300 MHz, CD3OD): 87,94 (d, J = 1,7 Hz, 1H), 7,71 (dd, J = 2,0, 9,2 Hz, 1H), 7,39-7,28 (m, 4H), 7,26-7,14 (m, 6H), 7,09-6,96 (m, 5H), 6,93-6,85 (m, 2H), 6,81 (d, J = 9,3 Hz, 1H), 6,75 (d, J = 7,6 Hz, 1H), 4,41 (quint., J = 7,0 Hz, 1H), 4,06-3,88 (m, 1H), 3,66-3,33 (m, 8H), 3,25-2,79 (m, 10H), 2,63 (s, 3H), 2,36-1,71 (m, 8H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1184,42 (M H)+.
Ácido (R)-2-(1-(3-(4-(N-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonilcarbamoil)-1H-pirrol-3-il)fenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)piperidin-4
carboniloxi)etilfosfónico (27) (BM-1080): se preparó 27 a partir de 26 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,95 (d, J = 1,9 Hz, 1H), 7,69 (dd, J = 1,8, 9,3 Hz, 1H), 7,39 7,28 (m, 4H), 7,27-7,12 (m, 6H), 7,08-6,76 (m, 8H), 6,70 (d, J = 7,5 Hz, 1H), 4,49-4,27 (m, 3H), 4,04-3,89 (m, 1H), 3,65-3,48 (m, 2H), 3,29-2,84 (m, 15H), 2,63 (s, 3H), 2,37-1,74 (m, 11H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1292,00 (M H)+.

(R)-1-(3-amino-4-(feniltio)butil)-3-metilazetidine-3-carboxilato de metilo (X). Se preparó X a partir de 3-metilazetidin-3-carboxilato de metilo (W) y D según el procedimiento general II.
Ácido (R)-1-(3-(4-(W-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)-3-metilazetidin-3-carboxílico (28) (BM-1082): se preparó 28 a partir de X y B según el procedimiento descrito para la preparación del compuesto 18. 1H-RMN (300 MHz, CD3OD): 87,94 (d, J = 1,9 Hz, 1H), 7,70 (dd, J = 2,1, 9,1 Hz, 1H), 7,35-7,24 (m, 4H), 7,23-7,12 (m, 5H), 7,07-6,91 (m, 4H), 6,87 (d, J = 9,0 Hz, 1H), 6,81 (d, J = 9,3 Hz, 1H), 6,63-6,47 (m, 2H), 6,41 (d, J = 9,0 Hz, 1H), 4,55 4,38 (m, 2H), 3,97 (s a, 3H), 3,29-3,08 (m, 13H), 2,84 (s, 3H), 2,74 (s, 3H), 2,12-1,81 (m, 2H), 1,56 (s a, 3H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1144,75 (M H)+.
Ácido (R)-2-(1-(3-(4-(W-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)-3-metilazetidin-3-carboniloxi)etilfosfónico (29) (BM-1083): se preparó 29 a partir de 28 y (2-hidroxietil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,94 (d, J =1,8 Hz, 1H), 7,72 (dd, J = 2,0, 9,1 Hz, 1H), 7,36 7,26 (m, 4H), 7,25-7,15 (m, 5H), 7,10-7,00 (m, 4H), 6,92-6,83 (m, 1H), 6,63 (s, 1H), 6,57 (d, J = 12,0 Hz, 1H), 6,42 (d, J = 9,2 Hz, 1H) 4,58-4,35 (m, 5H), 4,12-3,82 (m, 3H), 3,29-3,05 (m, 11H), 2,84 (s, 3H), 2,74 (s, 3H), 2,25-1,83 (m, 5H), 1,50 (s a, 3H), 1,43 (d, J = 7,1 Hz, 6H). EM (ESI): m/z 1252,83 (M H)+.
Ácido (R)-3-(1-(3-(4-(W-(4-(4-(3-(2-(4-clorofenil)-1-isopropil-5-metil-4-(metilsulfonil)-1H-pirrol-3-il)-5-fluorofenil)piperazin-1-il)fenil)sulfamoil)-2-(trifluorometilsulfonil)fenilamino)-4-(feniltio)butil)-3-metilazetidin-3-carboniloxi)propilfosfónico (30) (BM-1084): se preparó 30 a partir de 28 y (3-hidroxipropil)fosfonato de dimetilo según el procedimiento general V. 1H-RMN (300 MHz, CD3OD): 87,94 (s, 1H), 7,71 (dd, 1,5, 9,0 Hz, 1H), 7,36-7,26 (m, 4H), 7,24-7,15 (m, 5H), 7,08-6,97 (m, 4H), 6,90-6,79 (m, 2H), 6,62 (s, 1H), 6,56 (d, J = 11,8 Hz, 1H), 6,41 (d, J = 8,8 Hz, 1H), 4,54-4,37 (m, 3H), 4,33-4,21 (m, 2H), 3,99 (s a, 3H), 3,28-3,05 (m, 11H), 2,84 (s, 3H), 2,74 (s, 3H), 2,15 1,71 (m, 7H), 1,57 (s, 3H), 1,43 (d, J = 7,0 Hz, 6H). EM (ESI): m/z 1266,92 (M H)+.
Ensayos de unión basados en polarización de fluorescencia para las proteínas Bcl-2/Bcl-xL/Mcl-1
Se desarrollaron y optimizaron ensayos basados en polarización de fluorescencia (PF) sensibles y cuantitativos para determinar las afinidades de unión de los inhibidores de proteínas de la familia de Bcl-2 a las proteínas Bcl-2, Bcl-xL y Mcl-1 recombinantes.
Determinación de los valores de Kd de sondas fluorescentes para proteínas
Se usaron los péptidos BIM (81-106), Bak (72-87) y BID (79-99) marcados con fluoresceína de propia elaboración, denominados Flu-BIM, Flu-BAK y Flu-BID, como sondas fluorescentes en ensayos de PF para Bcl-2, Bcl-xL y Mcl-1, respectivamente. Mediante la monitorización de la polarización de fluorescencia total de la mezclas que se componen de sondas fluorescentes en concentraciones fijas y proteínas con concentraciones crecientes de hasta saturación completa, se determinó que los valores de Kd de Flu-BIM para Bcl-2, de Flu-BAK para Bcl-xL y de Flu-BID para Mcl-1 eran de 0,55±0,15 nM, 4,4±0,8 y 6,8±1,5 nM, respectivamente. Los valores de polarización de fluorescencia se midieron usando el lector de placas multimodal Infinite M-1000 (Tecan U.S., Research Triangle Park, NC) en placas Microfluor 2 de fondo redondo negras de 96 pocillos (Thermo Scientific). A cada pocillo se le añadieron 1 nM de Flu-BIM o 2 nM de Flu-BAK o 2 nM de Flu-BID y concentraciones crecientes de Bcl-2 o Bcl-xL o Mcl-1 hasta un volumen final de 125 pl en el tampón de ensayo (fosfato de potasio 100 mM, pH 7,5, y-globulina bovina 100 pg/ml, azida de sodio al 0,02%, Invitrogen, con Triton X-100 al 0,01% y DMSO al 4%). Se incubaron las
placas a temperatura ambiente durante 2 horas con agitación suave para garantizar el equilibrio. Se midieron los valores de polarización en miliunidades de polarización (mP) a una longitud de onda de excitación de 485 nm y una longitud de onda de emisión de 530 nm. Luego se calcularon las constantes de disociación en equilibrio (Kd) ajustando los aumentos sigmoideos de PF dependientes de la dosis en función de las concentraciones de proteína usando el software Graphpad Prism 5.0 (Graphpad Software, San Diego, CA).
Determinación de los valores de Ki de inhibidores de proteínas de la familia de Bcl-2
Se determinaron los valores de Ki de los inhibidores de proteína de la familia de Bcl-2 para las proteínas Bcl-2/BclxL/Mcl-1 a través de un experimento de unión competitiva dependiente de la dosis en el que diluciones en serie de los inhibidores compitieron contra la sonda fluorescente con concentración fija por la unión a una concentración fija de la proteína. Se añadieron mezclas de 5 pl del inhibidor sometido a prueba en DMSO y 120 pl de complejo de proteína/sonda incubado previamente en el tampón de ensayo a placas de ensayo y se incubaron a temperatura ambiente durante 2 horas con agitación suave. Las concentraciones finales de la proteína y la sonda son de 1,5 nM y 1 nM para el ensayo de Bcl-2, 10 nM y 2 nM para el ensayo de Bcl-xL y 20 nM y 2 nM para el ensayo de Mcl-1, respectivamente. Se incluyeron controles negativos que contenían complejo de proteína/sonda solo (equivalente al 0% de inhibición) y controles positivos que contenían sonda libre sola (equivalentes al 100% de inhibición) en cada placa de ensayo. Se midieron los valores de PF tal como se describió anteriormente. Se determinaron los valores de CI50 mediante ajuste por regresión no lineal de las curvas de competición. Se calcularon los valores de Ki de los inhibidores usando la ecuación derivada de origen descrita anteriormente (Z. Nikolovska-Coleska et al., Analytical Biochemistry, 2004, 332, 261-273.), basándose en los valores de CI50 obtenidos, los valores de Kd de las sondas a las proteínas y las concentraciones de las proteínas y las sondas en los ensayos competitivos. También se calcularon los valores de Ki usando otra ecuación muy usada comúnmente presente en la bibliografía (X. Y. Huang, Journal of Biomolecular Screening, 2003, 8, 34-38.), cuyos resultados coincidieron extremadamente bien con los presentes resultados.
Ensayo de crecimiento celular
Se sembraron células RS4;11 y H146 en placas de cultivo de células de 96 pocillos a una densidad de 10.000 células/pocillo con los compuestos diluidos en serie y se incubaron a 37°C en una atmósfera del 95% de aire y el 5% de CO2 durante 4 días. Se determinó la viabilidad celular usando el kit Cell Counting-8 basado en WST-8 (2-(2-metoxi-4-nitrofenil)-3-(4-nitrofenil)-5-(2,4-disulfofenil)-2H-tetrazolio, sal de monosodio) (Dojindo Molecular Technologies, Inc., Rockville, MD) según la instrucción del fabricante. En resumen, se añadió WST-8 a cada pocillo a una concentración final del 10% (v/v) y luego se incubaron las placas a 37°C durante 1-2 horas para el desarrollo del color. Se midió la absorbancia a 450 nm usando un lector de placas SPECTRAmax PLUS (Molecular Devices, Sunnyvale, CA). Se calculó la concentración inhibidora máxima media (CI50) usando el software GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
Ensayo de muerte celular
Se realizó un ensayo de muerte celular usando una prueba de exclusión de azul de tripano de la viabilidad celular. Se sembraron un millón de células en placas de 6 pocillos y se incubaron a 37°C en una atmósfera del 95% de aire y el 5% de CO2 con o sin compuestos durante los puntos de tiempo indicados. Al final del tratamiento, se recogieron las células y se centrifugaron a 1000 rpm durante 5 minutos. Volvieron a suspenderse los sedimentos celulares en PBS y se mezclaron con azul de tripano al 0,4% (Invitrogen) a una dilución 1:1 para determinar la viabilidad celular usando un microscopio Olympus CKX41 (Olympus, Center Valley, PA).
Ensayo de apoptosis
Se realizó un ensayo de apoptosis usando el kit de tinción Annexin-V-FLUOS (Roche Diagnostics, Indianápolis, IN) según la instrucción del fabricante. En resumen, se trataron las células con los compuestos durante los puntos de tiempo indicados, se recogieron y se lavaron con PBS. Se tiñeron las células con Annexin V-FITC y yoduro de propidio durante 15 minutos a temperatura ambiente en la oscuridad antes de analizarse con un dispositivo FACSCaliburs de BD Biosciences (Becton Dickinson).
Análisis de inmunotransferencia de tipo Western
Se lisaron las células con tampón de lisis (PBS que contenía NP40 al 1%, desoxicolato de Na al 0,5% y SDS al 0,1%) complementado con inhibidores de proteasa (a-completos, Roche). Se cuantificaron los extractos de proteínas usando un ensayo calorimétrico (Bradford Reagent) (BioRad, Hercules, CA). Se sometieron a electroforesis las proteínas sobre geles de SDS-PAGE al 4-20% (Invitrogen) y se transfirieron sobre membranas de poli(difluoruro de vinilideno) (Bio-Rad). Después del bloqueo en leche al 5%, se incubaron las membranas con un anticuerpo primario específico, se lavaron y se incubaron con anticuerpo secundario unido a peroxidasa del rábano (Pierce). Se visualizaron las señales con el reactivo de detección de anticuerpo de peroxidasa del rábano quimioluminiscente (Denville Scientific).
Ensayo de liberación de citocromo c y Smac
Se trataron cuatro millones de células H146 o RS4;11 con los compuestos a 37°C en una atmósfera del 95% de aire y el 5% de CO2 durante los puntos de tiempo indicados, se lavaron con PBS y volvieron a suspenderse en 100 ml de tampón de digitonina (NaCl 75 mM, Na2HPO48 mM, NaH2PO41 mM, EDTA 1 mM, digitonina 350 pg/ml y sacarosa 250 mM). Se separaron las fracciones citosólicas de la fracción de membrana con orgánulos mediante centrifugación a 13.000 rpm durante 1 min. Se resolvieron las fracciones citosólicas en SDS-PAGE al 12% y se sometieron a ensayo usando anticuerpo anti-citocromo c (BD Biosciences) y anticuerpo anti-Smac (Cell Signaling Technology, Danvers, MA).
En particular, se sometió a ensayo un compuesto de la invención para determinar su afinidad por Bcl-2, Bcl-xL y Mcl-1. Los resultados del ensayo se compararon con los resultados de un ensayo para ABT-737, un conocido inhibidor de Bcl-2/Bcl-xL patentado, y para estos péptidos. Los resultados se resumen en la tabla 1.

Bibliografía
1. D. Hanahan, et al., Cell 2000;100:57-70.
2. S.W. Lowe, et al., Carcinogenesis 2000, 21, 485-495.
3. C.B. Thompson, Science 1995, 267, 1456-1462.
4. J.C. Reed, Nat Rev Drug Discov 2002;1:111-121.
5. D.W. Nicholson, Nature 2000, 407, 810-816.
6. D.T. Chao, et al., Annu Rev Immunol 1998;16:395-419.
7. J.C. Reed, Advances in Pharmacology 1997;41:501-553.
8. J.C. Reed, et al. J Cell Biochem 1996;60:23-32.
9. A.J. Minn, et al., Advances in Immunology 1998;70:245-279.
10. J.M. Adams, et al, Science 1998;281:1322-1326.
11. A. Ziegler, et al., J Natl Cancer Inst 1997;89:1027-1036.
12. U. Zangemeister-Wittke, et al., Br. J. Cancer 1998;78:1035-1042.
13. B. Jansen, et al., Nature Medicine 1998;4:232-234.
14. U. Zangemeister-Wittke, et al., Br J Cancer 1998;78:1035-1042.
15. O. Gautschi, et al, J Natl Cancer Inst 2001;93:463-471.
16. M. Strasberg Rieber M, et al., Clin Cancer Res 2001;7;1446-1451.
17. S. Hopkins-Donaldson, et al., Int J Cancer 2003;106:160-166.
18. G. Wang, et al, Proc Natl Acad Sci USA 2000;97:7124-7129.
19. A. Degterev, et al, Nat Cell Biol 2001;3:173-182.
20. S.P. Tzung, et al., Nat Cell Biol 2001;3:183-191.
21. I.J. Enyedy, et al., J Med Chem 2001;44:4313-4324.
22. O. Kutzki, et al, J Am Chem Soc 2002;124:11838-11839.
23. G. Wang, et al, J Med Chem. 2006;49:6139-6142.
Claims (1)
1. Compuesto que tiene una estructura



Compuesto según la reivindicación 1, que tiene una estructura

Composición farmacéutica que comprende un compuesto según la reivindicación 1 ó 2, y un portador o vehículo farmacéuticamente aceptable.
Compuesto según cualquiera de las reivindicaciones 1 a 2, o composición farmacéutica según la reivindicación 3, para su uso en el tratamiento de cáncer.
Compuesto o composición farmacéutica para su uso según la reivindicación 4, en los que el cáncer se selecciona de un cáncer seleccionado en el grupo que consiste en carcinomas, incluyendo vejiga (incluyendo cáncer de vejiga acelerado y metastásico), mama, colon (incluyendo cáncer colorrectal), riñón, hígado, pulmón (incluyendo cáncer de pulmón de células pequeñas y no pequeñas y adenocarcinoma de pulmón), ovario, próstata, testículos, aparato genitourinario, sistema linfático, recto, laringe, páncreas (incluyendo carcinoma de páncreas exocrino), esófago, estómago, vesícula biliar, cuello uterino, tiroides, renal y piel (incluyendo carcinoma de células escamosas); tumores hematopoyéticos de linaje linfoide, incluyendo leucemia, leucemia linfocítica aguda, leucemia linfoblástica aguda, linfoma de células B, linfoma de células T, linfoma de Hodgkin, linfoma no Hodgkin, linfoma de células pilosas, linfoma histiocítico y linfoma de Burkett, tumores hematopoyéticos de linaje mieloide, incluyendo leucemias mielógenas agudas y crónicas, síndrome mielodisplásico, leucemia mieloide y leucemia promielocítica; tumores del sistema nervioso central y periférico, incluyendo astrocitoma, neuroblastoma, glioma y nuerilemomas; tumores de origen mesenquimatoso, incluyendo fibrosarcoma, rabdomioscarcoma y osteosarcoma; y otros tumores, incluyendo melanoma, xenoderma pigmentoso, queratoacantoma, seminoma, cáncer folicular de tiroides, teratocarcinoma, carcinoma de células renales (CCR), cáncer de páncreas, mieloma, leucemia mieloide y linfoblástica, neuroblastoma y glioblastoma, oncología de adultos y pediátrica, crecimiento de tumores sólidos/neoplasias malignas, carcinoma mixoide y de células redondas, tumores localmente avanzados, cáncer metastásico, sarcomas de tejidos blandos humanos, incluyendo sarcoma de Ewing, metástasis de cánceres, incluyendo metástasis linfáticas, carcinoma de células escamosas, particularmente de la cabeza y el cuello, carcinoma de células escamosas del esófago, carcinoma oral, neoplasias malignas de glóbulos sanguíneos, incluyendo mieloma múltiple, leucemias, incluyendo leucemia linfocítica aguda, leucemia no
linfocítica aguda, leucemia linfocítica crónica, leucemia mielocítica crónica y leucemia de células pilosas, linfomas de derrame (linfomas basados en las cavidades corporales), linfoma tímico cáncer de pulmón (incluyendo carcinoma de células pequeñas, linfoma cutáneo de células T, linfoma de Hodgkin, linfoma no Hodgkin, cáncer de la corteza suprarrenal, tumores que producen ACTH, cánceres no microcíticos, cáncer de mama, incluyendo carcinoma de células pequeñas y carcinoma ductal), cánceres gastrointestinales (incluyendo cáncer de estómago, cáncer de colon, cáncer colorrectal y pólipos asociados con neoplasia colorrectal), cáncer de páncreas, cáncer de hígado, cánceres urinarios (incluyendo cáncer de vejiga, tal como tumores primarios de vejiga superficial, carcinoma invasivo de células de transición de la vejiga y cáncer de vejiga invasivo del músculo), cáncer de próstata, neoplasias malignas del aparato genital femenino (incluyendo carcinoma de ovario, neoplasias epiteliales peritoneales primarias, carcinoma de cuello uterino, cánceres endometriales uterinos, cáncer de vagina, cáncer de la vulva, cáncer de útero y tumores sólidos en el folículo ovárico), neoplasias malignas del aparato genital masculino (incluyendo cáncer de testículo y cáncer de pene), cáncer de riñón (incluyendo carcinoma de células renales, cáncer de cerebro (incluyendo tumores de cerebro intrínsecos, neuroblastoma, tumores de cerebro astrocíticos, gliomas e invasión de células de tumor metastásico en el sistema nervioso central), cánceres de hueso (incluyendo osteomas y osteosarcomas), cánceres de piel (incluyendo melanoma maligno, progresión tumoral de queratinocitos cutáneos humanos y cáncer de células escamosas), cáncer de tiroides, retinoblastoma, neuroblastoma, derrame peritoneal, derrame pleural maligno, mesotelioma, tumores de Wilms, cáncer de vesícula biliar, neoplasias trofoblásticas, hemangiopericitoma y sarcoma de Kaposi.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361753066P | 2013-01-16 | 2013-01-16 | |
| PCT/US2014/011571 WO2014113413A1 (en) | 2013-01-16 | 2014-01-15 | Bcl-2bcl-xl inhibitors and therapeutic methods using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2819232T3 true ES2819232T3 (es) | 2021-04-15 |
Family
ID=51165292
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14741180T Active ES2819232T3 (es) | 2013-01-16 | 2014-01-15 | Inhibidores de Bcl-2/Bcl-xL y su uso en el tratamiento de cáncer |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US9096625B2 (es) |
| EP (2) | EP3689886A1 (es) |
| JP (1) | JP6347793B2 (es) |
| KR (2) | KR102210316B1 (es) |
| CN (3) | CN110305162A (es) |
| AU (1) | AU2014207716B2 (es) |
| CA (1) | CA2897055C (es) |
| ES (1) | ES2819232T3 (es) |
| HK (1) | HK1219734A1 (es) |
| NZ (1) | NZ709635A (es) |
| SG (1) | SG11201505525UA (es) |
| WO (1) | WO2014113413A1 (es) |
| ZA (1) | ZA201504902B (es) |
Families Citing this family (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3689886A1 (en) * | 2013-01-16 | 2020-08-05 | The Regents of The University of Michigan | Bcl-2/bcl-xl inhibitors and their use in the treatment of cancer |
| SG10201805670QA (en) | 2014-01-28 | 2018-08-30 | Buck Inst Res Aging | Methods and compositions for killing senescent cells and for treating senescence-associated diseases and disorders |
| EP3139942B1 (en) | 2014-05-05 | 2019-12-18 | Bioventures, Llc | COMPOSITIONS AND METHODS FOR INHIBITING ANTIAPOPTOTIC Bcl-2 PROTEINS AS ANTI-AGING AGENTS |
| WO2016014625A1 (en) | 2014-07-22 | 2016-01-28 | Board Of Trustees Of The University Of Arkansas | Compositions and methods for selectively depleting senescent cells |
| US20180000816A1 (en) * | 2015-02-06 | 2018-01-04 | Unity Biotechnology, Inc. | Use of a Heterocyclic Bcl-xL Inhibitor and Related Analogs for Removing Senescent Cells in the Treatment of Eye Diseases and Other Age-Related Conditions |
| CA2981753A1 (en) * | 2015-02-06 | 2016-08-11 | Unity Biotechnology, Inc. | Compounds and uses in treatment of senescence-associated conditions |
| WO2017101851A1 (en) * | 2015-12-18 | 2017-06-22 | Unity Biotechnology, Inc. | Acylsulfonamide derivatives for treating senescence-associated diseases and disorders |
| KR102545392B1 (ko) | 2016-03-28 | 2023-06-20 | 프레시지 바이오싸이언시스, 인크. | 암 치료를 위한 제약학적 조합물 |
| CA3018991A1 (en) * | 2016-04-21 | 2017-10-26 | Bioventures, Llc | Compounds that induce degradation of anti-apoptotic bcl-2 family proteins and the uses thereof |
| CN106177955B (zh) * | 2016-08-18 | 2018-03-16 | 广州威溶特医药科技有限公司 | Bcl‑xL抑制剂和溶瘤病毒在制备抗肿瘤药物中的应用 |
| WO2018098534A1 (en) * | 2016-12-02 | 2018-06-07 | Garvan Institute Of Medical Research | Methods of treating cancer and reagents thereof |
| WO2019033122A1 (en) * | 2017-08-11 | 2019-02-14 | Unity Biotechnology, Inc. | TREATMENT OF PULMONARY DISEASES USING PHARMACEUTICAL AGENTS THAT ELIMINATE SENESCENT CELLS |
| US20200354336A9 (en) * | 2017-08-11 | 2020-11-12 | Unity Biotechnology, Inc. | Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent Cells |
| EP3441069B1 (en) | 2017-08-11 | 2023-04-05 | Unity Biotechnology, Inc. | Treatment of diabetic retinopathy using pharmaceutical agents that eliminate senescent cells |
| US10588916B2 (en) | 2017-10-31 | 2020-03-17 | Unity Biotechnology, Inc. | Technology to inhibit vascular changes that lead to vision loss in the eye |
| EP3548504B1 (en) | 2017-12-30 | 2021-07-21 | Unity Biotechnology, Inc. | Peptide-based proteasome inhibitors for treating conditions mediated by senescent cells and for treating cancer |
| EP3740487A4 (en) | 2018-01-10 | 2021-11-10 | Recurium IP Holdings, LLC | BENZAMIDE COMPOUNDS |
| WO2019144117A1 (en) * | 2018-01-22 | 2019-07-25 | Bioventures, Llc | Bcl-2 proteins degraders for cancer treatment |
| WO2019210828A1 (en) | 2018-04-29 | 2019-11-07 | Beigene, Ltd. | Bcl-2 INHIBITORS |
| CA3056878C (en) | 2018-04-30 | 2021-03-30 | Unity Biotechnology | Phospholidines that are bcl family antagonists for use in clinical management of conditions caused or mediated by senescent cells and for treating cancer |
| CN112469411B (zh) * | 2018-04-30 | 2023-10-13 | 联合生物科技公司 | 供在由衰老细胞引起或介导的状况的临床管理中使用和用于治疗癌症的作为Bcl家族拮抗剂的氨基膦酸酯 |
| US10738042B2 (en) | 2018-04-30 | 2020-08-11 | Unity Biotechnology, Inc. | Phosphonamidates that are Bcl family antagonists for use in clinical management of conditions caused or mediated by senescent cells and for treating cancer |
| US10717722B2 (en) | 2018-06-13 | 2020-07-21 | Unity Biotechnology, Inc. | Acyl sulfonamides that are Bcl family antagonists for use in clinical management of conditions caused or mediated by senescent cells and for treating cancer |
| EP3672595B1 (en) | 2018-07-31 | 2021-07-21 | Ascentage Pharma (Suzhou) Co., Ltd. | Combination product of bcl-2 inhibitor and chemotherapeutic agent and use thereof in the prevention and/or treatment of diseases |
| BR112020018978A2 (pt) | 2018-07-31 | 2021-03-02 | Ascentage Pharma (Suzhou) Co., Ltd. | produto de combinação que compreende um inibidor de bcl-2 e um inibidor de mdm2 e uso dos ditos inibidores na prevenção e/ou tratamento de câncer |
| CN110960537B (zh) * | 2018-07-31 | 2021-04-06 | 苏州亚盛药业有限公司 | Bcl-2/Bcl-xL抑制剂与化疗药物的组合及其应用 |
| US11491168B2 (en) | 2018-07-31 | 2022-11-08 | Ascentage Pharma (Suzhou) Co., Ltd. | Combination of Bcl-2/Bcl-xL inhibitors and chemotherapeutic agent and use thereof |
| GEP20237460B (en) | 2018-07-31 | 2023-01-25 | Ascentage Pharma Suzhou Co Ltd | Synergistic antitumor effect of bcl-2 inhibitor combined with rituximab and/or bendamustine or bcl-2 inhibitor combined with chop |
| CN110772521A (zh) | 2018-07-31 | 2020-02-11 | 苏州亚盛药业有限公司 | Bcl-2抑制剂或Bcl-2/Bcl-xL抑制剂与BTK抑制剂的组合产品及其用途 |
| WO2020041406A1 (en) | 2018-08-22 | 2020-02-27 | Newave Pharmaceutical Inc. | Bcl-2 inhibitors |
| US11872237B2 (en) * | 2018-12-28 | 2024-01-16 | Ascentage Pharma (Suzhou) Co., Ltd. | Pharmaceutical composition and preparation method thereof |
| WO2020140957A1 (en) * | 2019-01-04 | 2020-07-09 | Ascentage Pharma (Suzhou) Co., Ltd. | Method for preparing sulfonamides drugs |
| WO2020140956A1 (en) * | 2019-01-04 | 2020-07-09 | Ascentage Pharma (Suzhou) Co., Ltd. | Process for preparing sulfonamide compounds |
| TWI770503B (zh) * | 2019-05-13 | 2022-07-11 | 大陸商蘇州亞盛藥業有限公司 | 用於預測bcl-2/bcl-xl抑制劑對癌症的功效的方法和組合物 |
| CN112168830B (zh) * | 2019-07-02 | 2022-01-25 | 苏州亚盛药业有限公司 | 一种含有mTOR抑制剂的药物组合及其应用 |
| EP3843751B1 (en) * | 2019-07-31 | 2024-03-27 | Ascentage Pharma (Suzhou) Co., Ltd. | Combination product of a bcl-2/bcl-xl inhibitor and a chemotherapeutic agent and use thereof in the prevention and/or treatment of diseases |
| US20220372042A1 (en) | 2019-10-03 | 2022-11-24 | Newave Pharmaceutical Inc. | Condensed heterocycles as bcl-2 inhibitors |
| CN112707900B (zh) * | 2019-10-24 | 2022-06-10 | 上海科技大学 | 蛋白降解剂及其在疾病治疗中的应用 |
| US20220317127A1 (en) * | 2019-11-27 | 2022-10-06 | Ascentage Pharma (Suzhou) Co., Ltd. | Method and compositions for predicting anti-cancer efficacy of compounds targeting apoptosis pathway |
| EP4069233A4 (en) * | 2019-12-04 | 2024-03-13 | Ascentage Pharma Suzhou Co Ltd | PHARMACEUTICAL COMBINATION AND USE THEREOF |
| US20230025865A1 (en) * | 2019-12-11 | 2023-01-26 | The Regents Of The University Of Michigan | COMPOSITIONS AND METHODS FOR SYSTEMIC DELIVERY OF Bcl-2 AND Bcl-xL ANTAGONISTS |
| CN113350281B (zh) * | 2020-03-02 | 2023-08-15 | 苏州亚盛药业有限公司 | 载药聚合物胶束及其制剂和制备方法 |
| TWI776451B (zh) * | 2020-04-10 | 2022-09-01 | 大陸商蘇州亞盛藥業有限公司 | Bcl-2/bcl-xl抑制劑之組合及相關用途 |
| WO2021233948A1 (en) | 2020-05-19 | 2021-11-25 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method to treat a pathogen lung infection |
| EP4175644A1 (en) | 2020-07-06 | 2023-05-10 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Combination of antineoplastic antibiotics and bcl-2 inhibitors for the treatment of npm-1-driven acute myeloid leukemia (aml) |
| CN113929715A (zh) * | 2020-07-13 | 2022-01-14 | 苏州亚盛药业有限公司 | Bcl-2/Bcl-xL抑制剂化合物或其盐的结晶形式或无定形形式 |
| WO2022022706A1 (en) * | 2020-07-31 | 2022-02-03 | Ascentage Pharma (Suzhou) Co., Ltd. | Compositions and methods for treating lung diseases |
| EP4244231A1 (en) * | 2020-11-10 | 2023-09-20 | Unity Biotechnology, Inc. | Crystalline solid meglumine salt inhibitor of bcl and methods of making and using same |
| WO2022111647A1 (en) * | 2020-11-27 | 2022-06-02 | Ascentage Pharma (Suzhou) Co., Ltd. | Formulation combination containing freeze-dried formulation and reconstituted solvent, preparation method and application thereof |
| WO2023011488A1 (zh) * | 2021-08-02 | 2023-02-09 | 苏州亚盛药业有限公司 | 一种药物组合及其用途 |
| WO2024027706A1 (en) * | 2022-08-02 | 2024-02-08 | Beijing Neox Biotech Limited | Bcl-xl degrading compounds |
| WO2024051741A1 (zh) * | 2022-09-06 | 2024-03-14 | 西藏海思科制药有限公司 | 一种抑制Bcl-2或Bcl-xL的化合物及其在医药上的应用 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7432304B2 (en) | 2001-05-30 | 2008-10-07 | The Regents Of The University Of Michigan | Small molecule antagonists of Bcl-2 family proteins |
| US7973161B2 (en) * | 2003-11-13 | 2011-07-05 | Abbott Laboratories | Apoptosis promoters |
| WO2006023778A2 (en) | 2004-08-20 | 2006-03-02 | The Regents Of The University Of Michigan | Small molecule inhibitors of anti-apoptotic bcl-2 family members and the uses thereof |
| MX2007014049A (es) | 2005-05-12 | 2008-02-11 | Abbott Lab | Activadores de apoptosis. |
| WO2007061923A2 (en) | 2005-11-18 | 2007-05-31 | Takeda San Diego, Inc. | Glucokinase activators |
| CA2662320C (en) * | 2006-09-05 | 2014-07-22 | Abbott Laboratories | Bcl inhibitors treating platelet excess |
| BRPI0916356B1 (pt) | 2008-07-24 | 2022-08-23 | Nerviano Medical Sciences S.R.L | 3,4-diaril pirazois como inibidores da proteína quinase |
| US20100278921A1 (en) | 2009-04-30 | 2010-11-04 | Fischer Cristina M | Solid oral formulation of abt-263 |
| TWI535712B (zh) | 2010-08-06 | 2016-06-01 | 阿斯特捷利康公司 | 化合物 |
| AU2012209295B2 (en) * | 2011-01-25 | 2016-06-30 | The Regents Of The University Of Michigan | Bcl-2/Bcl-xL inhibitors and therapeutic methods using the same |
| EP3689886A1 (en) * | 2013-01-16 | 2020-08-05 | The Regents of The University of Michigan | Bcl-2/bcl-xl inhibitors and their use in the treatment of cancer |
-
2014
- 2014-01-15 EP EP20163914.3A patent/EP3689886A1/en not_active Withdrawn
- 2014-01-15 WO PCT/US2014/011571 patent/WO2014113413A1/en active Application Filing
- 2014-01-15 US US14/155,809 patent/US9096625B2/en active Active
- 2014-01-15 SG SG11201505525UA patent/SG11201505525UA/en unknown
- 2014-01-15 JP JP2015553788A patent/JP6347793B2/ja active Active
- 2014-01-15 KR KR1020157021823A patent/KR102210316B1/ko active IP Right Grant
- 2014-01-15 KR KR1020207037597A patent/KR102318204B1/ko active IP Right Grant
- 2014-01-15 NZ NZ70963514A patent/NZ709635A/en unknown
- 2014-01-15 ES ES14741180T patent/ES2819232T3/es active Active
- 2014-01-15 CN CN201910634723.XA patent/CN110305162A/zh active Pending
- 2014-01-15 CN CN201480015733.0A patent/CN105246882A/zh active Pending
- 2014-01-15 CN CN201910634742.2A patent/CN110302205B/zh active Active
- 2014-01-15 AU AU2014207716A patent/AU2014207716B2/en active Active
- 2014-01-15 CA CA2897055A patent/CA2897055C/en active Active
- 2014-01-15 EP EP14741180.5A patent/EP2945940B1/en active Active
-
2015
- 2015-07-08 ZA ZA2015/04902A patent/ZA201504902B/en unknown
- 2015-08-03 US US14/816,191 patent/US9403856B2/en active Active
-
2016
- 2016-07-05 HK HK16107756.1A patent/HK1219734A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016506916A (ja) | 2016-03-07 |
| KR102318204B1 (ko) | 2021-10-26 |
| US20150336994A1 (en) | 2015-11-26 |
| KR20210002126A (ko) | 2021-01-06 |
| CN110302205A (zh) | 2019-10-08 |
| US20140199234A1 (en) | 2014-07-17 |
| NZ709635A (en) | 2019-10-25 |
| EP2945940A4 (en) | 2016-07-06 |
| KR102210316B1 (ko) | 2021-01-29 |
| CA2897055A1 (en) | 2014-07-24 |
| CN110305162A (zh) | 2019-10-08 |
| SG11201505525UA (en) | 2015-08-28 |
| ZA201504902B (en) | 2016-07-27 |
| US9403856B2 (en) | 2016-08-02 |
| AU2014207716A1 (en) | 2015-07-23 |
| WO2014113413A1 (en) | 2014-07-24 |
| EP3689886A1 (en) | 2020-08-05 |
| EP2945940A1 (en) | 2015-11-25 |
| CN110302205B (zh) | 2020-06-09 |
| US9096625B2 (en) | 2015-08-04 |
| HK1219734A1 (zh) | 2017-04-13 |
| KR20150104631A (ko) | 2015-09-15 |
| AU2014207716B2 (en) | 2017-07-27 |
| EP2945940B1 (en) | 2020-07-15 |
| JP6347793B2 (ja) | 2018-06-27 |
| CA2897055C (en) | 2021-04-20 |
| CN105246882A (zh) | 2016-01-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2819232T3 (es) | Inhibidores de Bcl-2/Bcl-xL y su uso en el tratamiento de cáncer | |
| EP2668180B1 (en) | Bcl-2/bcl-xl inhibitors for use in the treatment of cancer | |
| AU2013306087B2 (en) | Bivalent inhibitors of IAP proteins and therapeutic methods using the same | |
| ES2916449T3 (es) | Inhibidores de DCN1 de molécula pequeña covalentes y métodos terapéuticos de uso de los mismos | |
| AU2012209295A1 (en) | Bcl-2/Bcl-xL inhibitors and therapeutic methods using the same | |
| US20110319362A1 (en) | Stat3 ligands and therapeutic uses thereof | |
| NZ613504B2 (en) | Bcl-2/bcl-xl inhibitors and therapeutic methods using the same |