ES2776353T3 - Inhibidores de la fosfodiesterasa novedosos y usos de los mismos - Google Patents
Inhibidores de la fosfodiesterasa novedosos y usos de los mismos Download PDFInfo
- Publication number
- ES2776353T3 ES2776353T3 ES14826002T ES14826002T ES2776353T3 ES 2776353 T3 ES2776353 T3 ES 2776353T3 ES 14826002 T ES14826002 T ES 14826002T ES 14826002 T ES14826002 T ES 14826002T ES 2776353 T3 ES2776353 T3 ES 2776353T3
- Authority
- ES
- Spain
- Prior art keywords
- hydrogen
- alkyl
- mmol
- compound
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000002571 phosphodiesterase inhibitor Substances 0.000 title description 4
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 159
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 146
- 239000001257 hydrogen Substances 0.000 claims abstract description 145
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims abstract description 109
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 82
- 150000002367 halogens Chemical group 0.000 claims abstract description 82
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 38
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 32
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 claims abstract description 27
- 150000003839 salts Chemical class 0.000 claims abstract description 16
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims abstract description 12
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 claims abstract description 12
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 10
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 3
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims abstract description 3
- 101000869592 Daucus carota Major allergen Dau c 1 Proteins 0.000 claims abstract description 3
- 101000650136 Homo sapiens WAS/WASL-interacting protein family member 3 Proteins 0.000 claims abstract description 3
- 102100027539 WAS/WASL-interacting protein family member 3 Human genes 0.000 claims abstract description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract 38
- 101150020251 NR13 gene Proteins 0.000 claims abstract 4
- 239000000203 mixture Substances 0.000 claims description 81
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 32
- 230000004770 neurodegeneration Effects 0.000 claims description 31
- 208000024827 Alzheimer disease Diseases 0.000 claims description 30
- 230000015654 memory Effects 0.000 claims description 18
- 230000027928 long-term synaptic potentiation Effects 0.000 claims description 15
- 125000001188 haloalkyl group Chemical group 0.000 claims description 10
- 239000000460 chlorine Substances 0.000 claims description 9
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- 230000001965 increasing effect Effects 0.000 claims description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 5
- 201000010099 disease Diseases 0.000 claims description 4
- 125000006526 (C1-C2) alkyl group Chemical group 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 102
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 84
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 78
- 238000005160 1H NMR spectroscopy Methods 0.000 description 69
- 239000000243 solution Substances 0.000 description 62
- 239000012074 organic phase Substances 0.000 description 55
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 55
- 239000011541 reaction mixture Substances 0.000 description 53
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 51
- 229910052938 sodium sulfate Inorganic materials 0.000 description 51
- 235000011152 sodium sulphate Nutrition 0.000 description 51
- 239000007832 Na2SO4 Substances 0.000 description 50
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 48
- 241000699670 Mus sp. Species 0.000 description 45
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 41
- 238000000034 method Methods 0.000 description 41
- 230000002829 reductive effect Effects 0.000 description 40
- 238000003818 flash chromatography Methods 0.000 description 39
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 34
- -1 heteroalkylene quinolinamine Chemical compound 0.000 description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 32
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 30
- 102000011016 Type 5 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 description 30
- 108010037581 Type 5 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 description 30
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 29
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 28
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 28
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 28
- 239000002904 solvent Substances 0.000 description 28
- 101710137189 Amyloid-beta A4 protein Proteins 0.000 description 27
- 101710151993 Amyloid-beta precursor protein Proteins 0.000 description 27
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 27
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 27
- 230000000694 effects Effects 0.000 description 26
- 239000000047 product Substances 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 238000001914 filtration Methods 0.000 description 24
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 23
- 238000001704 evaporation Methods 0.000 description 23
- 230000008020 evaporation Effects 0.000 description 23
- 239000002244 precipitate Substances 0.000 description 23
- 238000006243 chemical reaction Methods 0.000 description 21
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 239000008346 aqueous phase Substances 0.000 description 19
- 235000002639 sodium chloride Nutrition 0.000 description 19
- 239000003981 vehicle Substances 0.000 description 19
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 18
- 125000006239 protecting group Chemical group 0.000 description 18
- 238000011830 transgenic mouse model Methods 0.000 description 18
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 17
- 230000005764 inhibitory process Effects 0.000 description 17
- 238000012360 testing method Methods 0.000 description 17
- IKWWOZCEHOYKAO-UHFFFAOYSA-N (3-chloro-4-methoxyphenyl)methanamine;hydrochloride Chemical compound Cl.COC1=CC=C(CN)C=C1Cl IKWWOZCEHOYKAO-UHFFFAOYSA-N 0.000 description 16
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 16
- 238000010992 reflux Methods 0.000 description 16
- 235000017557 sodium bicarbonate Nutrition 0.000 description 16
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 16
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 14
- BJVCSICIEDHBNI-UHFFFAOYSA-N benzo[b][1,8]naphthyridine Chemical class N1=CC=CC2=CC3=CC=CC=C3N=C21 BJVCSICIEDHBNI-UHFFFAOYSA-N 0.000 description 13
- 241000699660 Mus musculus Species 0.000 description 12
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- 239000003795 chemical substances by application Substances 0.000 description 11
- 235000019439 ethyl acetate Nutrition 0.000 description 11
- 229940123333 Phosphodiesterase 5 inhibitor Drugs 0.000 description 10
- 210000004556 brain Anatomy 0.000 description 10
- 230000007547 defect Effects 0.000 description 10
- 230000006390 fear memory Effects 0.000 description 10
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 125000005843 halogen group Chemical group 0.000 description 9
- 230000003956 synaptic plasticity Effects 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 8
- 229910019213 POCl3 Inorganic materials 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 230000003750 conditioning effect Effects 0.000 description 7
- 239000003480 eluent Substances 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 229960003742 phenol Drugs 0.000 description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 7
- 230000000638 stimulation Effects 0.000 description 7
- 0 *c1bc2nc(*)c(*)c(**c3cc(*)*(*)c(*)c3)c2cc1* Chemical compound *c1bc2nc(*)c(*)c(**c3cc(*)*(*)c(*)c3)c2cc1* 0.000 description 6
- HQNZNIJPAYTWJK-UHFFFAOYSA-N 2-amino-5-cyanobenzoic acid Chemical compound NC1=CC=C(C#N)C=C1C(O)=O HQNZNIJPAYTWJK-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 235000015243 ice cream Nutrition 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000003977 synaptic function Effects 0.000 description 6
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 5
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 5
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 5
- 206010043376 Tetanus Diseases 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 229910052794 bromium Inorganic materials 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- 150000002431 hydrogen Chemical class 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 238000012549 training Methods 0.000 description 5
- HUUPVABNAQUEJW-UHFFFAOYSA-N 1-methylpiperidin-4-one Chemical compound CN1CCC(=O)CC1 HUUPVABNAQUEJW-UHFFFAOYSA-N 0.000 description 4
- RWZYAGGXGHYGMB-WGGUOBTBSA-N 2-aminobenzoic acid Chemical class NC1=CC=CC=C1[14C](O)=O RWZYAGGXGHYGMB-WGGUOBTBSA-N 0.000 description 4
- LGOZNQPHTIGMQJ-UHFFFAOYSA-N 4-bromo-2-ethylaniline Chemical compound CCC1=CC(Br)=CC=C1N LGOZNQPHTIGMQJ-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- 208000026139 Memory disease Diseases 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical compound NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- SWJXWSAKHXBQSY-UHFFFAOYSA-N benzo(c)cinnoline Chemical compound C1=CC=C2C3=CC=CC=C3N=NC2=C1 SWJXWSAKHXBQSY-UHFFFAOYSA-N 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 239000000835 fiber Substances 0.000 description 4
- 238000002875 fluorescence polarization Methods 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 238000002194 freeze distillation Methods 0.000 description 4
- 238000007710 freezing Methods 0.000 description 4
- 230000008014 freezing Effects 0.000 description 4
- 238000007490 hematoxylin and eosin (H&E) staining Methods 0.000 description 4
- 230000000971 hippocampal effect Effects 0.000 description 4
- 239000005457 ice water Substances 0.000 description 4
- 238000012744 immunostaining Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 231100000161 signs of toxicity Toxicity 0.000 description 4
- 229960003310 sildenafil Drugs 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 230000000946 synaptic effect Effects 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- SJZKULRDWHPHGG-UHFFFAOYSA-N 1-benzylpiperidin-4-one Chemical compound C1CC(=O)CCN1CC1=CC=CC=C1 SJZKULRDWHPHGG-UHFFFAOYSA-N 0.000 description 3
- UUFQTNFCRMXOAE-UHFFFAOYSA-N 1-methylmethylene Chemical compound C[CH] UUFQTNFCRMXOAE-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 101100167062 Caenorhabditis elegans chch-3 gene Proteins 0.000 description 3
- 102000005636 Cyclic AMP Response Element-Binding Protein Human genes 0.000 description 3
- 108010045171 Cyclic AMP Response Element-Binding Protein Proteins 0.000 description 3
- 101100351286 Dictyostelium discoideum pdeE gene Proteins 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 102100039289 Glial fibrillary acidic protein Human genes 0.000 description 3
- 101710193519 Glial fibrillary acidic protein Proteins 0.000 description 3
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 3
- 229940099471 Phosphodiesterase inhibitor Drugs 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 230000006399 behavior Effects 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 230000008602 contraction Effects 0.000 description 3
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 229910052805 deuterium Inorganic materials 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000002612 dispersion medium Substances 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 210000005046 glial fibrillary acidic protein Anatomy 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 230000006993 memory improvement Effects 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 101150037969 pde-6 gene Proteins 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 230000035939 shock Effects 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 238000003828 vacuum filtration Methods 0.000 description 3
- VSOSXKMEQPYESP-UHFFFAOYSA-N 1,6-naphthyridine Chemical compound C1=CN=CC2=CC=CN=C21 VSOSXKMEQPYESP-UHFFFAOYSA-N 0.000 description 2
- CQPLNIMGZGYFNB-UHFFFAOYSA-N 1,6-naphthyridine-8-carbonitrile Chemical compound C1=CN=C2C(C#N)=CN=CC2=C1 CQPLNIMGZGYFNB-UHFFFAOYSA-N 0.000 description 2
- UMKSAURFQFUULT-UHFFFAOYSA-N 2-Amino-5-methoxybenzoic acid Chemical compound COC1=CC=C(N)C(C(O)=O)=C1 UMKSAURFQFUULT-UHFFFAOYSA-N 0.000 description 2
- CUKXRHLWPSBCTI-UHFFFAOYSA-N 2-amino-5-bromobenzoic acid Chemical compound NC1=CC=C(Br)C=C1C(O)=O CUKXRHLWPSBCTI-UHFFFAOYSA-N 0.000 description 2
- HLCPWBZNUKCSBN-UHFFFAOYSA-N 2-aminobenzonitrile Chemical class NC1=CC=CC=C1C#N HLCPWBZNUKCSBN-UHFFFAOYSA-N 0.000 description 2
- XQVCBOLNTSUFGD-UHFFFAOYSA-N 3-chloro-4-methoxyaniline Chemical compound COC1=CC=C(N)C=C1Cl XQVCBOLNTSUFGD-UHFFFAOYSA-N 0.000 description 2
- XPONRKKKLFCCTR-UHFFFAOYSA-N 8-bromo-10-chloro-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridine Chemical compound Clc1c2CNCCc2nc2ccc(Br)cc12 XPONRKKKLFCCTR-UHFFFAOYSA-N 0.000 description 2
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 description 2
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 238000011740 C57BL/6 mouse Methods 0.000 description 2
- OHVGSHPEDMCTQL-UHFFFAOYSA-N COC(=O)c1c(C)nc2ccc(OC)cc2c1Cl Chemical compound COC(=O)c1c(C)nc2ccc(OC)cc2c1Cl OHVGSHPEDMCTQL-UHFFFAOYSA-N 0.000 description 2
- SSJXDXYVNVDJSJ-UHFFFAOYSA-N COC(=O)c1c(C)nc2ccc(cc2c1Cl)C(F)(F)F Chemical compound COC(=O)c1c(C)nc2ccc(cc2c1Cl)C(F)(F)F SSJXDXYVNVDJSJ-UHFFFAOYSA-N 0.000 description 2
- BZRRQRJPHAYBPS-UHFFFAOYSA-N COC(=O)c1c(Cl)c2cc(ccc2nc1CBr)C(F)(F)F Chemical compound COC(=O)c1c(Cl)c2cc(ccc2nc1CBr)C(F)(F)F BZRRQRJPHAYBPS-UHFFFAOYSA-N 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- 101100407341 Drosophila melanogaster Pde9 gene Proteins 0.000 description 2
- 208000010228 Erectile Dysfunction Diseases 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- 206010018341 Gliosis Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 102000011990 Sirtuin Human genes 0.000 description 2
- 108050002485 Sirtuin Proteins 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 2
- DDSZWBCJXDRQDU-UHFFFAOYSA-N [N].C1CCNCC1 Chemical group [N].C1CCNCC1 DDSZWBCJXDRQDU-UHFFFAOYSA-N 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- IUKQLMGVFMDQDP-UHFFFAOYSA-N azane;piperidine Chemical compound N.C1CCNCC1 IUKQLMGVFMDQDP-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 125000005997 bromomethyl group Chemical group 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- RNFNDJAIBTYOQL-UHFFFAOYSA-N chloral hydrate Chemical compound OC(O)C(Cl)(Cl)Cl RNFNDJAIBTYOQL-UHFFFAOYSA-N 0.000 description 2
- 229960002327 chloral hydrate Drugs 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 239000000544 cholinesterase inhibitor Substances 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000007405 data analysis Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 230000004761 fibrosis Effects 0.000 description 2
- 230000009969 flowable effect Effects 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 230000007387 gliosis Effects 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 239000008241 heterogeneous mixture Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000003018 immunoassay Methods 0.000 description 2
- 201000001881 impotence Diseases 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- JXDYKVIHCLTXOP-UHFFFAOYSA-N isatin Chemical compound C1=CC=C2C(=O)C(=O)NC2=C1 JXDYKVIHCLTXOP-UHFFFAOYSA-N 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 description 2
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 230000008599 nitrosative stress Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 230000036542 oxidative stress Effects 0.000 description 2
- 230000000149 penetrating effect Effects 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 230000001954 sterilising effect Effects 0.000 description 2
- 238000004659 sterilization and disinfection Methods 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- 229960001685 tacrine Drugs 0.000 description 2
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical compound C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 description 2
- 229960000835 tadalafil Drugs 0.000 description 2
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 2
- OVQBGGSESVPMIZ-UHFFFAOYSA-N tert-butyl 8-bromo-10-[(3-chloro-4-methoxyphenyl)methylamino]-3,4-dihydro-1H-benzo[b][1,6]naphthyridine-2-carboxylate Chemical compound COc1ccc(CNc2c3CN(CCc3nc3ccc(Br)cc23)C(=O)OC(C)(C)C)cc1Cl OVQBGGSESVPMIZ-UHFFFAOYSA-N 0.000 description 2
- FYEUKOROYHHAHI-UHFFFAOYSA-N tert-butyl 8-bromo-10-chloro-3,4-dihydro-1H-benzo[b][1,6]naphthyridine-2-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCc2nc3ccc(Br)cc3c(Cl)c2C1 FYEUKOROYHHAHI-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 229960002381 vardenafil Drugs 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- NPIFZAZFJJSDAP-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 2-chloro-4-[(3-chloro-4-methoxyphenyl)methylamino]-6-cyanoquinoline-3-carboxylate Chemical compound COc1ccc(CNc2c(C(=O)Oc3c(F)c(F)c(F)c(F)c3F)c(Cl)nc3ccc(cc23)C#N)cc1Cl NPIFZAZFJJSDAP-UHFFFAOYSA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- LQAUXDMGRBWDIU-UHFFFAOYSA-N (3-chloro-4-fluorophenyl)methanamine Chemical compound NCC1=CC=C(F)C(Cl)=C1 LQAUXDMGRBWDIU-UHFFFAOYSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- BDVKAMAALQXGLM-UHFFFAOYSA-N 1-ethylpiperidin-4-one Chemical compound CCN1CCC(=O)CC1 BDVKAMAALQXGLM-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- PMBQHJFQTGZRDS-UHFFFAOYSA-N 10-[(3-chloro-4-methoxyphenyl)methylamino]-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridine-8-carbonitrile Chemical compound C1=C(Cl)C(OC)=CC=C1CNC1=C(CNCC2)C2=NC2=CC=C(C#N)C=C12 PMBQHJFQTGZRDS-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 1
- OUVJCSFLLVRFRW-UHFFFAOYSA-N 2,4-dioxo-1h-3,1-benzoxazine-6-carbonitrile Chemical compound C1=C(C#N)C=C2C(=O)OC(=O)NC2=C1 OUVJCSFLLVRFRW-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- GLCQUPLYYXSPQB-UHFFFAOYSA-N 2-amino-5-(trifluoromethyl)benzoic acid Chemical compound NC1=CC=C(C(F)(F)F)C=C1C(O)=O GLCQUPLYYXSPQB-UHFFFAOYSA-N 0.000 description 1
- JHHRXLLPQQOJJW-UHFFFAOYSA-N 2-amino-5-bromo-3-ethylbenzoic acid Chemical compound CCC1=CC(Br)=CC(C(O)=O)=C1N JHHRXLLPQQOJJW-UHFFFAOYSA-N 0.000 description 1
- SGUJJNTURMLHNJ-UHFFFAOYSA-N 2-amino-5-cyano-3-ethylbenzoic acid Chemical compound CCC1=CC(C#N)=CC(C(O)=O)=C1N SGUJJNTURMLHNJ-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- ZFQUJOLQDNNAJH-UHFFFAOYSA-N 2-chloro-4-[(3-chloro-4-methoxyphenyl)methylamino]-6-cyanoquinoline-3-carboxylic acid Chemical compound C1=C(Cl)C(OC)=CC=C1CNC1=C(C(O)=O)C(Cl)=NC2=CC=C(C#N)C=C12 ZFQUJOLQDNNAJH-UHFFFAOYSA-N 0.000 description 1
- PNGPMNXTYUTTTK-UHFFFAOYSA-N 4-[[4-[3-benzoyl-8-(trifluoromethyl)quinolin-4-yl]phenoxy]methyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1COC1=CC=C(C=2C3=CC=CC(=C3N=CC=2C(=O)C=2C=CC=CC=2)C(F)(F)F)C=C1 PNGPMNXTYUTTTK-UHFFFAOYSA-N 0.000 description 1
- DQAZPZIYEOGZAF-UHFFFAOYSA-N 4-ethyl-n-[4-(3-ethynylanilino)-7-methoxyquinazolin-6-yl]piperazine-1-carboxamide Chemical compound C1CN(CC)CCN1C(=O)NC(C(=CC1=NC=N2)OC)=CC1=C2NC1=CC=CC(C#C)=C1 DQAZPZIYEOGZAF-UHFFFAOYSA-N 0.000 description 1
- NBFPSAKAPKBKNF-UHFFFAOYSA-N 5-(6,7-dimethoxyquinolin-4-yl)oxypyridin-2-amine Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC1=CC=C(N)N=C1 NBFPSAKAPKBKNF-UHFFFAOYSA-N 0.000 description 1
- CTGQHGOEVARVKX-UHFFFAOYSA-N 5-bromo-7-ethyl-1h-indole-2,3-dione Chemical compound CCC1=CC(Br)=CC2=C1NC(=O)C2=O CTGQHGOEVARVKX-UHFFFAOYSA-N 0.000 description 1
- HAMDXJUAHDROHP-UHFFFAOYSA-N 6-(trifluoromethyl)-1h-3,1-benzoxazine-2,4-dione Chemical compound N1C(=O)OC(=O)C2=CC(C(F)(F)F)=CC=C21 HAMDXJUAHDROHP-UHFFFAOYSA-N 0.000 description 1
- FECZHVISRNXUCS-UHFFFAOYSA-N 6-[(3-chloro-4-methoxyphenyl)methylamino]-5-oxo-1,2,3,4-tetrahydro-[1,4]diazepino[5,6-b]quinoline-8-carbonitrile Chemical compound C1=C(Cl)C(OC)=CC=C1CNC1=C2C(=O)NCCNC2=NC2=CC=C(C#N)C=C12 FECZHVISRNXUCS-UHFFFAOYSA-N 0.000 description 1
- JFAFNQOODJCVGT-UHFFFAOYSA-N 6-methoxy-1h-3,1-benzoxazine-2,4-dione Chemical compound N1C(=O)OC(=O)C2=CC(OC)=CC=C21 JFAFNQOODJCVGT-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- XNXGPHBXUMZYND-UHFFFAOYSA-N 8-bromo-n-[(3-chloro-4-methoxyphenyl)methyl]-1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridin-10-amine Chemical compound C1=C(Cl)C(OC)=CC=C1CNC1=C(CNCC2)C2=NC2=CC=C(Br)C=C12 XNXGPHBXUMZYND-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 238000000035 BCA protein assay Methods 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 1
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 description 1
- LBZWFBCIDIEGIX-UHFFFAOYSA-N CCN1Cc2nc3ccc(OC)cc3c(Cl)c2C1=O Chemical compound CCN1Cc2nc3ccc(OC)cc3c(Cl)c2C1=O LBZWFBCIDIEGIX-UHFFFAOYSA-N 0.000 description 1
- JGFAJHTUXZKCEQ-UHFFFAOYSA-N CCN1Cc2nc3ccc(OC)cc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O Chemical compound CCN1Cc2nc3ccc(OC)cc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O JGFAJHTUXZKCEQ-UHFFFAOYSA-N 0.000 description 1
- NRTLPQVBGOXGRW-UHFFFAOYSA-N CCN1Cc2nc3ccc(cc3c(Cl)c2C1=O)C#N Chemical compound CCN1Cc2nc3ccc(cc3c(Cl)c2C1=O)C#N NRTLPQVBGOXGRW-UHFFFAOYSA-N 0.000 description 1
- CXMJWJJNVWXSJV-UHFFFAOYSA-N CCN1Cc2nc3ccc(cc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O)C#N Chemical compound CCN1Cc2nc3ccc(cc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O)C#N CXMJWJJNVWXSJV-UHFFFAOYSA-N 0.000 description 1
- VDANUPKSDIDCJU-UHFFFAOYSA-N CCN1Cc2nc3ccc(cc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O)C(F)(F)F Chemical compound CCN1Cc2nc3ccc(cc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O)C(F)(F)F VDANUPKSDIDCJU-UHFFFAOYSA-N 0.000 description 1
- CPPUEDNXMVGJTE-UHFFFAOYSA-N CCN1Cc2nc3ccccc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O Chemical compound CCN1Cc2nc3ccccc3c(NCc3ccc(OC)c(Cl)c3)c2C1=O CPPUEDNXMVGJTE-UHFFFAOYSA-N 0.000 description 1
- LDADSMCZGLMSBT-UHFFFAOYSA-N COC(=O)C1=C(C)Nc2ccc(cc2C1O)C#N Chemical compound COC(=O)C1=C(C)Nc2ccc(cc2C1O)C#N LDADSMCZGLMSBT-UHFFFAOYSA-N 0.000 description 1
- GYCPMVXZHJTPHU-UHFFFAOYSA-N COC(=O)c1c(C)[nH]c2ccc(Cl)cc2c1=O Chemical compound COC(=O)c1c(C)[nH]c2ccc(Cl)cc2c1=O GYCPMVXZHJTPHU-UHFFFAOYSA-N 0.000 description 1
- MZXSVMOQKRQACI-UHFFFAOYSA-N COC(=O)c1c(C)[nH]c2ccc(OC)cc2c1=O Chemical compound COC(=O)c1c(C)[nH]c2ccc(OC)cc2c1=O MZXSVMOQKRQACI-UHFFFAOYSA-N 0.000 description 1
- GIWXAMCDXKEIPR-UHFFFAOYSA-N COC(=O)c1c(C)[nH]c2ccc(cc2c1=O)C(F)(F)F Chemical compound COC(=O)c1c(C)[nH]c2ccc(cc2c1=O)C(F)(F)F GIWXAMCDXKEIPR-UHFFFAOYSA-N 0.000 description 1
- JHOLWAFCXNMHFQ-UHFFFAOYSA-N COC(=O)c1c(C)nc2ccc(Cl)cc2c1Cl Chemical compound COC(=O)c1c(C)nc2ccc(Cl)cc2c1Cl JHOLWAFCXNMHFQ-UHFFFAOYSA-N 0.000 description 1
- KCGMBTQQCALFIX-UHFFFAOYSA-N COC(=O)c1c(C)nc2ccc(cc2c1Cl)C#N Chemical compound COC(=O)c1c(C)nc2ccc(cc2c1Cl)C#N KCGMBTQQCALFIX-UHFFFAOYSA-N 0.000 description 1
- ZTJDVVKXJAQEFC-UHFFFAOYSA-N COC(=O)c1c(CBr)nc2ccccc2c1Cl Chemical compound COC(=O)c1c(CBr)nc2ccccc2c1Cl ZTJDVVKXJAQEFC-UHFFFAOYSA-N 0.000 description 1
- ZUHCVUDQBPYDIY-UHFFFAOYSA-N COC(=O)c1c(Cl)c2cc(ccc2nc1CBr)C#N Chemical compound COC(=O)c1c(Cl)c2cc(ccc2nc1CBr)C#N ZUHCVUDQBPYDIY-UHFFFAOYSA-N 0.000 description 1
- DQIPBGOJKRTACL-UHFFFAOYSA-N COC(c(c(CBr)nc(cc1)c2cc1OC)c2Cl)=O Chemical compound COC(c(c(CBr)nc(cc1)c2cc1OC)c2Cl)=O DQIPBGOJKRTACL-UHFFFAOYSA-N 0.000 description 1
- PNDCCASNKRUOMF-AXUFZUPDSA-N CO[C@H]1[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1CO[C@@H](O[C@@H]2[C@@H](O)[C@H](O)CO[C@H]2N2[C@@H](CC(N)=O)C(=O)\C(=C(/O)\C=C\C=C\C=C\C=C\C=C(/Cl)\C=C\C=C(/Cl)[C@H]3O[C@H](C)C[C@@H]3Cl)C2=O)[C@@H](O)[C@@H]1O Chemical compound CO[C@H]1[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1CO[C@@H](O[C@@H]2[C@@H](O)[C@H](O)CO[C@H]2N2[C@@H](CC(N)=O)C(=O)\C(=C(/O)\C=C\C=C\C=C\C=C\C=C(/Cl)\C=C\C=C(/Cl)[C@H]3O[C@H](C)C[C@@H]3Cl)C2=O)[C@@H](O)[C@@H]1O PNDCCASNKRUOMF-AXUFZUPDSA-N 0.000 description 1
- OCNMSDZALRAYEX-UHFFFAOYSA-N COc1ccc(CN)cc1Cl Chemical compound COc1ccc(CN)cc1Cl OCNMSDZALRAYEX-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- NBUUUJWWOARGNW-UHFFFAOYSA-N Cc(cc1C(O)=O)ccc1N Chemical compound Cc(cc1C(O)=O)ccc1N NBUUUJWWOARGNW-UHFFFAOYSA-N 0.000 description 1
- UIEBVPYLBIGKNL-UHFFFAOYSA-N Cc1c(cc(cc2)Br)c2nc2c1CN(Cc1ccccc1)CC2 Chemical compound Cc1c(cc(cc2)Br)c2nc2c1CN(Cc1ccccc1)CC2 UIEBVPYLBIGKNL-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- FSBZWPQOJWCYEC-UHFFFAOYSA-N ClC1=C2C(=NC=3C=CC(=CC1=3)C(F)(F)F)CN(C2=O)CC Chemical compound ClC1=C2C(=NC=3C=CC(=CC1=3)C(F)(F)F)CN(C2=O)CC FSBZWPQOJWCYEC-UHFFFAOYSA-N 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- 229940126062 Compound A Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 208000016192 Demyelinating disease Diseases 0.000 description 1
- 206010012305 Demyelination Diseases 0.000 description 1
- 101100296720 Dictyostelium discoideum Pde4 gene Proteins 0.000 description 1
- 101100135868 Dictyostelium discoideum pde3 gene Proteins 0.000 description 1
- 101100407335 Dictyostelium discoideum pde7 gene Proteins 0.000 description 1
- 101100135859 Dictyostelium discoideum regA gene Proteins 0.000 description 1
- 101001117089 Drosophila melanogaster Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1 Proteins 0.000 description 1
- 101001072031 Drosophila melanogaster Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11 Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108010076876 Keratins Proteins 0.000 description 1
- 102000011782 Keratins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 244000062730 Melissa officinalis Species 0.000 description 1
- 235000010654 Melissa officinalis Nutrition 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 238000012347 Morris Water Maze Methods 0.000 description 1
- 206010048654 Muscle fibrosis Diseases 0.000 description 1
- 101000909851 Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) cAMP/cGMP dual specificity phosphodiesterase Rv0805 Proteins 0.000 description 1
- 229940099433 NMDA receptor antagonist Drugs 0.000 description 1
- 102000008299 Nitric Oxide Synthase Human genes 0.000 description 1
- 108010021487 Nitric Oxide Synthase Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 101710176384 Peptide 1 Proteins 0.000 description 1
- 101100082606 Plasmodium falciparum (isolate 3D7) PDEbeta gene Proteins 0.000 description 1
- 101100082610 Plasmodium falciparum (isolate 3D7) PDEdelta gene Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- 108091027981 Response element Proteins 0.000 description 1
- 101100135860 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) PDE2 gene Proteins 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- 102000005918 Ubiquitin Thiolesterase Human genes 0.000 description 1
- 108010005656 Ubiquitin Thiolesterase Proteins 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- OVFDZNSGNUXERC-UHFFFAOYSA-N [3-chloro-4-(trifluoromethoxy)phenyl]methanamine Chemical compound NCC1=CC=C(OC(F)(F)F)C(Cl)=C1 OVFDZNSGNUXERC-UHFFFAOYSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 238000002679 ablation Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000035508 accumulation Effects 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 231100000215 acute (single dose) toxicity testing Toxicity 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229940039856 aricept Drugs 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 229960000307 avanafil Drugs 0.000 description 1
- WEAJZXNPAWBCOA-INIZCTEOSA-N avanafil Chemical compound C1=C(Cl)C(OC)=CC=C1CNC1=NC(N2[C@@H](CCC2)CO)=NC=C1C(=O)NCC1=NC=CC=N1 WEAJZXNPAWBCOA-INIZCTEOSA-N 0.000 description 1
- YEJAJYAHJQIWNU-UHFFFAOYSA-N azelastine hydrochloride Chemical compound Cl.C1CN(C)CCCC1N1C(=O)C2=CC=CC=C2C(CC=2C=CC(Cl)=CC=2)=N1 YEJAJYAHJQIWNU-UHFFFAOYSA-N 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- LCGGZWDDXGLOAO-UHFFFAOYSA-N benzo[b][1,6]naphthyridine Chemical class C1=CN=CC2=CC3=CC=CC=C3N=C21 LCGGZWDDXGLOAO-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 239000003833 bile salt Substances 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 230000007370 cognitive improvement Effects 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 150000001975 deuterium Chemical group 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229960003530 donepezil Drugs 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 229940108366 exelon Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- IECPWNUMDGFDKC-MZJAQBGESA-N fusidic acid Chemical class O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C(O)=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C IECPWNUMDGFDKC-MZJAQBGESA-N 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000000762 glandular Effects 0.000 description 1
- 210000000585 glomerular basement membrane Anatomy 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 102000027411 intracellular receptors Human genes 0.000 description 1
- 108091008582 intracellular receptors Proteins 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000000865 liniment Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- MVYUCRDXZXLFSB-UHFFFAOYSA-N lodenafil Chemical compound CCCC1=NN(C)C(C(N=2)=O)=C1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N(CC1)CCN1CCOC(=O)OCCN(CC1)CCN1S(=O)(=O)C(C=1)=CC=C(OCC)C=1C(N1)=NC(=O)C2=C1C(CCC)=NN2C MVYUCRDXZXLFSB-UHFFFAOYSA-N 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- XVICOGIQWUAIAV-UHFFFAOYSA-N methyl 2-amino-5-cyanobenzoate Chemical compound COC(=O)C1=CC(C#N)=CC=C1N XVICOGIQWUAIAV-UHFFFAOYSA-N 0.000 description 1
- ZSPADABVCNESSU-UHFFFAOYSA-N methyl 2-chloro-4-[(3-chloro-4-methoxyphenyl)methylamino]-6-cyanoquinoline-3-carboxylate Chemical compound COC(=O)C1=C(Cl)N=C2C=CC(C#N)=CC2=C1NCC1=CC=C(OC)C(Cl)=C1 ZSPADABVCNESSU-UHFFFAOYSA-N 0.000 description 1
- XSWODDQKGOXFFU-UHFFFAOYSA-N methyl 2-methyl-4-oxo-1h-quinoline-3-carboxylate Chemical compound C1=CC=C2C(=O)C(C(=O)OC)=C(C)NC2=C1 XSWODDQKGOXFFU-UHFFFAOYSA-N 0.000 description 1
- UTBCRHAMJFMIIR-UHFFFAOYSA-N methyl 3-chloro-3-oxopropanoate Chemical compound COC(=O)CC(Cl)=O UTBCRHAMJFMIIR-UHFFFAOYSA-N 0.000 description 1
- ISRJCWLLGCDNPO-UHFFFAOYSA-N methyl 4-chloro-2-methylquinoline-3-carboxylate Chemical compound C1=CC=CC2=C(Cl)C(C(=O)OC)=C(C)N=C21 ISRJCWLLGCDNPO-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960001047 methyl salicylate Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 229950002245 mirodenafil Drugs 0.000 description 1
- MIJFNYMSCFYZNY-UHFFFAOYSA-N mirodenafil Chemical compound C1=C(C=2NC=3C(CCC)=CN(CC)C=3C(=O)N=2)C(OCCC)=CC=C1S(=O)(=O)N1CCN(CCO)CC1 MIJFNYMSCFYZNY-UHFFFAOYSA-N 0.000 description 1
- 150000004712 monophosphates Chemical class 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 description 1
- 229940033872 namenda Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 239000006218 nasal suppository Substances 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 230000007171 neuropathology Effects 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- CMFNMSMUKZHDEY-UHFFFAOYSA-N peroxynitrous acid Chemical compound OON=O CMFNMSMUKZHDEY-UHFFFAOYSA-N 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 1
- 208000002815 pulmonary hypertension Diseases 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 229960004136 rivastigmine Drugs 0.000 description 1
- HJORMJIFDVBMOB-UHFFFAOYSA-N rolipram Chemical compound COC1=CC=C(C2CC(=O)NC2)C=C1OC1CCCC1 HJORMJIFDVBMOB-UHFFFAOYSA-N 0.000 description 1
- 229950005741 rolipram Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000021317 sensory perception Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 231100000873 signs of neurotoxicity Toxicity 0.000 description 1
- MNWBNISUBARLIT-UHFFFAOYSA-N sodium cyanide Chemical compound [Na+].N#[C-] MNWBNISUBARLIT-UHFFFAOYSA-N 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000003976 synaptic dysfunction Effects 0.000 description 1
- 230000005062 synaptic transmission Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 102000013498 tau Proteins Human genes 0.000 description 1
- 108010026424 tau Proteins Proteins 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 229960000438 udenafil Drugs 0.000 description 1
- IYFNEFQTYQPVOC-UHFFFAOYSA-N udenafil Chemical compound C1=C(C=2NC=3C(CCC)=NN(C)C=3C(=O)N=2)C(OCCC)=CC=C1S(=O)(=O)NCCC1CCCN1C IYFNEFQTYQPVOC-UHFFFAOYSA-N 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 238000009777 vacuum freeze-drying Methods 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 229950005371 zaprinast Drugs 0.000 description 1
- REZGGXNDEMKIQB-UHFFFAOYSA-N zaprinast Chemical compound CCCOC1=CC=CC=C1C1=NC(=O)C2=NNNC2=N1 REZGGXNDEMKIQB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compuesto de fórmula (I), **(Ver fórmula)** en la que A es NR4; B es CR16 o N; V es un enlace o C(O); W es un enlace o NR13; X es alquilo (C1-C3), alquilo (C1-C3) sustituido con al menos un D; Y es NR5; Z es C; R1 es halógeno o haloalquilo (C1-C6); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R4 es hidrógeno o alquilo (C1-C3); R5 es hidrógeno, alquilo (C1-C3), cicloalquilo (C3-C5) o -C(O)R7; R6 es alquilo (C1-C3) o haloalquilo (C1-C3); R7 es independientemente alquilo (C1-C3); R8 es hidrógeno o alquilo (C1-C3); R13 es hidrógeno o alquilo (C1-C3); R14 es hidrógeno, halógeno o haloalquilo (C1-C6); R15 es hidrógeno, -OR17, -OH o halógeno; R16 es hidrógeno, -OR17, -OH o halógeno; R17 es hidrógeno, alquilo (C1-C6), haloalquilo (C1-C6) o cicloalquilo (C3-C8); m y n son cada uno 0, 1 ó 2, siempre que la suma de m + n sea un número entero de desde 2-3; o una sal o tautómero farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Inhibidores de la fosfodiesterasa novedosos y usos de los mismos
Referencia cruzada a solicitud relacionada
Esta solicitud reivindica el beneficio y prioridad de la solicitud de patente provisional estadounidense n.° 61/847.280, presentada el 17 de julio de 2013.
Esta divulgación de patente contiene material sujeto a protección de propiedad intelectual. El propietario de la propiedad intelectual no tiene ninguna objeción a la reproducción facsímil del documento de patente o de la divulgación de la patente tal como aparece en el archivo o registros de patentes de la Oficina de Patentes y Marcas de EE.UU., pero se reserva de otro modo todos los derechos de propiedad intelectual.
Apoyo gubernamental
Esta invención se realizó con apoyo gubernamental bajo la subvención NIH/NIA n.° 1U01AG032973 concedida por el Instituto Nacional de Salud. El gobierno tiene determinados derechos en la invención.
Antecedentes de la invención
La enfermedad de Alzheimer (EA) es un trastorno neurodegenerativo que es la causa más frecuente de demencia entre personas de edad avanzada. Se caracteriza por una pérdida progresiva de la memoria, disfunción sináptica y acumulación de péptidos p-amiloides (Ap). Está provocada en parte por niveles aumentados de péptido p-amiloide 1 42 (Ap42). Los fármacos aprobados para tratar la EA incluyen inhibidores de la colinesterasa tales como Cognex® (tacrina), Aricept® (donepezil), Exelon® (rivastigmina) y Razdyne® (galantamina); y el antagonista del receptor de N-metil-d-aspartato Namenda® (memantina). Sin embargo, varios de estos medicamentos padecen de eficacia limitada y producen efectos secundarios adversos.
Los inhibidores de la fosfodiesterasa 5 (PDE5) se usan ampliamente como agentes terapéuticos para la disfunción eréctil y la hipertensión pulmonar. Se cree que estos inhibidores aumentan los niveles de GMPc, lo que potencia la fosforilación del factor de la transcripción y la unión del elemento de respuesta a AMPc (CREB) de la molécula que afecta a la memoria a través de la activación de las proteína cinasas dependientes de GMPc. El documento US 4 843 079 A da a conocer una heteroalquilenquinolinamina fusionada que es útil para potenciar la memoria. El documento WO 2006/094237 A2 da a conocer compuestos que modulan la sirtuina y métodos de uso de los mismos. El documento WO2006/094237A2 enseña que los compuestos que modulan la sirtuina pueden usarse para tratar y/o prevenir una amplia variedad de enfermedades y trastornos que incluyen, por ejemplo, enfermedades neurodegenerativas. Musial & al., 2007 dan a conocer desarrollos en inhibidores de la colinesterasa para rratamiento de la enfermedad de Alzheimer, incluyendo análogos de los fármacos existentes, tales como inhibidores duales del sitio de unión. El documento WO 2013/109738 A1 da a conocer derivados de benzonaftiridina, tales como inhibidores de la fosfodiesterasa, y composiciones que comprenden derivados novedosos de benzonaftiridina. El documento WO 2013/109738 A1 también da a conocer métodos para el tratamiento de enfermedades neurodegenerativas, para aumentar la memoria o la potenciación a largo plazo con derivados de benzonaftiridina o composiciones que comprenden derivados novedosos de benzonaftiridina.
El monofosfato de adenosina cíclico (AMPc) y el monofosfato de guanosina cíclico (GMPc), segundos mensajeros biológicos de nucleótidos, regulan diversos procesos biológicos, tales como regulación de la circulación sanguínea, contracción del miocardio, diferenciación celular, transmisión neuronal, secreción glandular y expresión génica. Los receptores intracelulares para estas moléculas incluyen nucleótido cíclico fosfodiesterasas (PDE), proteína cinasas dependientes de nucleótido cíclico (PGK) y canales activados por nucleótido cíclico. Las PDE son una gran familia de proteínas que catalizan la hidrólisis de nucleótidos cíclicos 3',5' a los correspondientes monofosfatos 5'. Existen once grupos de genes de PDE humanos relacionados, pero bioquímicamente distintos. Algunas PDE son específicas para la hidrólisis de AMPc (tales como PDE4, PDE7, y p DE8) y algunas son específicas para GMPc (tales como PDE5, PDE6 y PDE9), mientras que algunas PDE tienen especificidad mixta (tales como p DE1, PDE2, PDE3, PDE10 y PDE11).
Los inhibidores de PDE 5 representativos son inhibidores de la PDE de GMPc tipo cinco de 3',5'-monofosfato de guanosina cíclico, también conocidos como inhibidores de la PDE-5, que incluyen, por ejemplo, sildenafilo, tadalafilo, zaprinast y vardenafilo. Los inhibidores de la PDE5 aumentan los niveles de GMPc inhibiendo la acción degradante de PDE5 sobre GMPc. Los inhibidores de la PDE5 actuales padecen inconvenientes tales como selectividad limitada con respecto a otros subtipos de PDE. Sigue existiendo la necesidad de inhibidores de la PDE5 estructuralmente novedosos.
Sumario de la invención
En un aspecto, la invención se refiere a una clase de derivados de benzo[b][1,6]naftirid¡na derivados de fórmula (I)

en la que
A es NR4 ;
B es CR16 o N;
V es un enlace o C(O);
W es un enlace o NR13;
X es alquilo (C1-C3 ), alquilo (C1-C3) sustituido con al menos un D;
Y es NR5 ;
Z es C o N;
R1 es halógeno o haloalquilo (C1-C6);
R2 es -OR6;
R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;
R4 es hidrógeno o alquilo (C1 -C3);
R5 es hidrógeno, alquilo (C1 -C3) o cicloalquilo (C3-C5), -C(O)R7 ;
R6 es alquilo (C1-C3 ) o haloalquilo (C1 -C3);
R7 es independientemente alquilo (C1-C3 );
R8 es hidrógeno o alquilo (C1 -C3);
R13 es hidrógeno o alquilo (C1 -C3);
R14 es hidrógeno, halógeno o haloalquilo (C1 -C6);
R15 es hidrógeno, -OR17, -OH o halógeno;
R16 es hidrógeno, -OR17, -OH o halógeno;
R17 es hidrógeno, alquilo (C1 -C6), haloalquilo (C1 -C6) o cicloalquilo (C3-C8);
m y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3;
o una sal o tautómero farmacéuticamente aceptable de los mismos.
En algunas realizaciones, los compuestos de la invención disminuyen la actividad de la PDE en al menos aproximadamente el 10%, al menos aproximadamente el 20%, al menos aproximadamente el 30%, al menos aproximadamente el 40%, al menos aproximadamente el 50%, al menos aproximadamente el 60%, al menos aproximadamente el 70%, al menos aproximadamente el 75%, al menos aproximadamente el 80%, al menos
aproximadamente el 85%, al menos aproximadamente el 90%, al menos aproximadamente el 95%, al menos aproximadamente el 97%, al menos aproximadamente el 99% o el 100%.
En algunas realizaciones, los compuestos de la invención inhiben la fosfodiesterasa. En algunas realizaciones, la fosfodiesterasa es fosfodiesterasa tipo V (PDE5).
En algunas realizaciones, los compuestos de la invención tienen una CI50 para fosfodiesterasa de al menos aproximadamente 0,1 nM, al menos aproximadamente 1 nM, al menos aproximadamente 5 nM, al menos aproximadamente 10 nM, al menos aproximadamente 25 nM, al menos aproximadamente 50 nM, al menos aproximadamente 100 nM, al menos aproximadamente 200 nM, al menos aproximadamente 300 nM, al menos aproximadamente 400 nM, al menos aproximadamente 500 nM, al menos aproximadamente 600 nM, al menos aproximadamente 700 nM, al menos aproximadamente 800 nM, al menos aproximadamente 900 nM, o al menos aproximadamente 1000 nM.
En otro aspecto, la invención se refiere a composiciones que comprenden un compuesto de fórmula (I) y un portador farmacéuticamente aceptable.
La divulgación se refiere a un método de inhibición de una fosfodiesterasa que comprende poner en contacto una fosfodiesterasa con un compuesto de fórmula (I) o una composición que comprende un compuesto de fórmula (I). La fosfodiesterasa puede ser PDE5.
La divulgación se refiere a un método de tratamiento de una enfermedad neurodegenerativa en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I). La enfermedad puede ser enfermedad de Alzheimer. En un aspecto de la invención, se proporciona un compuesto o composición de fórmula (I), fórmula (Ia), fórmula (Ib), fórmula o (Ic), para su uso en el tratamiento de una enfermedad neurodegenerativa en un sujeto.
La divulgación se refiere a un método de aumento de la potenciación a largo plazo en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I). En un aspecto de la invención, se proporciona un compuesto o composición de fórmula (I), fórmula (Ia), fórmula (Ib), fórmula o (Ic), para su uso en el tratamiento de una enfermedad neurodegenerativa en un sujeto, en el que se aumenta la potenciación a largo plazo en el sujeto.
La divulgación se refiere a un método de mejora de la memoria en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer. Por tanto, en un aspecto de la invención, se proporciona un compuesto o una composición de fórmula (I), fórmula (Ia), fórmula (Ib), fórmula o (Ic), para su uso en el tratamiento de una enfermedad neurodegenerativa en un sujeto, en el que se mejora la memoria en el sujeto.
La divulgación se refiere a un método de mejora de la función sináptica en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
Todavía otros objetos y ventajas de la invención serán evidentes para los expertos en la técnica a partir de la divulgación en el presente documento, que es simplemente ilustrativa y no restrictiva. Por tanto, otras realizaciones se reconocerán por el experto en la técnica sin alejarse del espíritu y alcance de la invención.
Breve descripción de las figuras
La figura 1 muestra los efectos del compuesto H sobre los niveles hipocámpicos de GMPc en ratones.
La figura 2 muestra los efectos del compuesto H en ratones silvestres y APP/PS1, un modelo de ratón transgénico de EA, sobre la potenciación residual registrada durante los últimos 5 minutos de un registro de 2 h tras la estimulación tetánica de las fibras colaterales de Schaffer en la conexión CA3-CA1.
La figura 3 muestra los efectos del compuesto H en ratones silvestres y APP/PS1, un modelo de ratón transgénico de EA, sobre la potenciación residual registrada durante los últimos 5 minutos de un registro de 2 h tras la estimulación tetánica de las fibras colaterales de Schaffer en la conexión CA3-CA1. El tratamiento diario con el compuesto H (3 mg/kg, i.p.) durante 3 semanas a la edad de 3-4 meses restableció la potenciación normal cuando se registraron cortes a los 6-7 meses de edad.
La figura 4 muestra los efectos del compuesto H sobre los defectos en la memoria de referencia en ratones silvestres y APP/PS1, un modelo de ratón transgénico de EA, en el laberinto de agua de brazo radial de 2 días. El tratamiento diario con el compuesto durante 3 semanas a la edad de 3-4 meses redujo el número de errores con el laberinto de agua de brazo radial de 2 días en ratones APP/PS1.
La figura 5 muestra los efectos del compuesto H sobre los defectos en la memoria del miedo en ratones silvestres y APP/PS1, un modelo de ratón transgénico de EA, en la memoria del miedo contextual. El tratamiento diario con el
compuesto durante 3 semanas a la edad de 3-4 meses restableció la congelación normal en una prueba para la memoria del miedo contextual en ratones APP/PS1.
La figura 6 muestra los efectos del compuesto H sobre los defectos en la memoria de referencia en ratones silvestres y APP/PS1, un modelo de ratón transgénico de EA, en el laberinto de agua de brazo radial de 2 días. El tratamiento diario con el compuesto (3 mg/kg, i.p.) durante 3 semanas a la edad de 3-4 meses redujo el número de errores con el laberinto de agua de brazo radial de 2 días cuando se examinaron los ratones a los 6-7 meses de edad.
La figura 7 muestra los efectos del compuesto H sobre los defectos en la memoria del miedo en ratones silvestres y APP/PS1, un modelo de ratón transgénico de EA, en la memoria del miedo contextual. El tratamiento diario con el compuesto (3 mg/kg, i.p.) durante 3 semanas a la edad de 3-4 meses restableció la congelación normal en una prueba para la memoria del miedo contextual cuando se examinaron los ratones a los 6-7 meses de edad.
La figura 8 muestra el transcurso de tiempo frente a las concentraciones del compuesto H en el cerebro y plasma de los ratones tras la administración i.p. de una dosis de 25 mg/kg.
La figura 9 muestra la tinción con H&E para tres grupos (A: vehículo; B: 120 mg/kg de compuesto H; C: 240 mg/kg de compuesto H); y una inmunotinción con GFAP para tres grupos (D: vehículo; E: 120 mg/kg de compuesto H, F: 240 mg/kg de compuesto H).
Descripción detallada de la invención
El tratamiento de la EA sigue siendo un objetivo principal en la comunidad médica. Para tal fin, están explorándose varias dianas biológicas para el tratamiento de la EA tales como Tau, beta-secretasa y gamma-secretasa. La EA comienza supuestamente como un trastorno sináptico producido, al menos en parte, por Ap (Selkoe, D.J. Science 2002, 298, 789-791). La reducción inducida por Ap en la potenciación a largo plazo (LTP), un indicador fisiológico de plasticidad sináptica que se piensa que forma la base del aprendizaje y la memoria, y la fosforilación del factor de transcripción de la memoria CREB, se mejoran mediante donadores de óxido nítrico (NO) y análogos de GMP (Puzzo, et al, J. Neurosci 2005, 25, 6887-6897). Al revés, la ablación genética de NO-sintasa 2 (NOS2) da como resultado un empeoramiento del fenotipo de EA en ratones que expresan proteína precursora de amiloide mutada (APP) (Colton et al. Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 12867-12872). En conjunto, estos hallazgos muestran que la regulación por incremento de la ruta de NO puede ser protectora en la EA.
A pesar de la función neuroprotectora de NO, también se ha considerado al gas como un agente principal de neuropatología y muerte celular cuando se produce en grandes cantidades. Grandes cantidades de NO conducen a la generación de una cantidad significativa de peroxinitritos que son responsables del estrés oxidativo y nitrosativo en la muerte celular inducida por Ap. La liberación de pequeñas cantidades de NO por las formas constitutivas de NOS que incluyen las isoformas tanto neuronales como endoteliales, n-NOS y e-NOS, promueve la plasticidad sináptica y el aprendizaje, mientras que la producción descontrolada de grandes cantidades del gas por la forma inducible de NOS (i-NOS) puede promover estrés oxidativo y nitrosativo por medio de la producción de peroxinitrito. Por tanto, ambas regulación por disminución inducida por Ap de la cascada de NO que bloquea la plasticidad y la memoria y la generación de peroxinitritos que conducen a muerte celular, puede desempeñar papeles en la EA. Las estrategias que pueden sortear la producción de NO se centran en las etapas aguas abajo de la generación de NO. Los agentes que potencian la señalización de NO/GMPc/CREB pueden rescatar la reducción inducia por Ab de la plasticidad sináptica y memoria (véase el documento WO 2010/074783 y las referencias citadas en el mismo; Prickaerts, et al., Eur. J. Pharmacol. 1999, 10, 731-737; y Neuroscience 2002, 113, 351-361).
En un aspecto, la invención se refiere a derivados de fórmula (I):

más particularmente a unos derivados de fórmula (la):

todavía más particularmente a derivados de fórmula (Ib):

en la que
A es NR4;
B es CR16 o N;
V es un enlace o C(O);
W es un enlace o NR13;
X es alquilo (C1-C3) sustituido con al menos un D;
Y es NR5 , O S;
Z es C o N;
R1 es halógeno o haloalquilo (C1-C6);
R2 es -OR6;
R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;
R4 es hidrógeno o alquilo (C1 -C3);
R5 es hidrógeno, alquilo (C1 -C3), cicloalquilo (C3-C5) o -C(O)R7 ;
R6 es hidrógeno, alquilo (C1 -C6), haloalquilo (C1 -C6) o cicloalquilo (C3-C8);
R7 es independientemente hidrógeno, alquilo (C1-C6), haloalquilo (C1 -C6) o arilo;
R8 es hidrógeno, alquilo (C1 -C6), haloalquilo (C1 -C6), cicloalquilo (C3-C8), -NR9R10, -S(O)2R11 o heterociclilo; R13 es hidrógeno o alquilo (C1 -C3);
R14 es hidrógeno, halógeno o haloalquilo (C1 -C6);
R15 es hidrógeno, -OR17, -OH o halógeno;
R16 es hidrógeno, -OR17, -OH o halógeno;
R17 es hidrógeno, alquilo (C1 -C6), haloalquilo (C1 -C6) o cicloalquilo (C3-C8);
m y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3;
o una sal o tautómero farmacéuticamente aceptable de los mismos.
En otro aspecto, la invención se refiere a derivados de fórmula (Ic):

en la que
A es NR4;
B es CR16 o N;
V es un enlace o C(O);
W es un enlace o NR13;
X es alquilo (C1-C3 ), alquilo (C1-C3) sustituido con al menos un D;
Y es NR5 ;
R1 es halógeno o haloalquilo (C1-C6);
R2 es -OR6;
R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;
R4 es hidrógeno o alquilo (C1 -C3);
R5 es hidrógeno, alquilo (C1 -C3), cicloalquilo (C3-C5) o -C(O)R7 ;
R6 es alquilo (C1-C3 ) o haloalquilo (C1 -C3);
R7 es independientemente alquilo (C1-C3 );
R8 es hidrógeno o alquilo (C1 -C3);
R13 es hidrógeno o alquilo (C1 -C3);
R15 es hidrógeno, -OR17, -OH o halógeno;
R16 es hidrógeno, -OR17, -OH o halógeno;
R17 es hidrógeno, alquilo (C1 -C6), haloalquilo (C1 -C6) o cicloalquilo (C3-C8);
m y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3;
o una sal o tautómero farmacéuticamente aceptable de los mismos.
Los compuestos y/o composiciones de la invención pueden ser eficaces en el tratamiento, la reducción y/o supresión de complicaciones relacionadas con PDE, incluyendo PDE5, tal como, por ejemplo, disfunción eréctil.
Abreviaturas y definiciones
Los términos “inhibidor de la fosfodiesterasa” o “inhibidor de la PDE” se refieren a compuestos y sales o solvatos de los mismos que actúan inhibiendo la actividad de la enzima fosfodiesterasa. Una fosfodiesterasa a modo de ejemplo es la fosfodiesterasa tipo 5 (PDE5). Un inhibidor de la PDE puede ser un compuesto que disminuye la actividad de PDE in vivo y/o in vitro. Pueden encontrarse inhibidores de la PDE5 a modo de ejemplo en las patentes estadounidenses n.os 5.250.534; 5.859.006; 6.362.178; y 7.378.430.
El término “compuesto de la invención” tal como se usa en el presente documento significa un compuesto de fórmula (I) o cualquier subgénero o especie del mismo. El término también se pretende que abarque sales, hidratos y solvatos del mismo.
El término “composición/composiciones de la invención” tal como se usa en el presente documento significa composiciones que comprenden un compuesto de la invención, y sales, hidratos, o solvatos del mismo. Las composiciones de la invención pueden comprender además otros agentes tales como, por ejemplo, portadores, excipientes, estabilantes, lubricantes, disolventes, y similares.
El término “alquilo”, tal como se usa en el presente documento, a menos que se indique lo contrario, se refiere a un radical de hidrocarburo alifático monovalente que tiene una cadena lineal o cadena ramificada. Los ejemplos de grupos “alquilo” incluyen metilo, etilo, propilo, isopropilo, butilo, /so-butilo, sec-butilo, tere-butilo, pentilo, hexilo, heptilo, y similares.
El término “D” se refiere a un átomo de deuterio, y se conoce en la técnica que hace referencia a una especie enriquecida en deuterio, es decir, en la que D está presente por encima de su abundancia isotópica natural.
El término “haloalquilo”, tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con uno o más átomos de halógeno. El término también abarca grupos haloalquilo que contienen más de una especie de átomo de halógeno, por ejemplo -CF2C y similares.
El término “halógeno”, tal como se usa en el presente documento, significa cloro (Cl), flúor (F), yodo (I) o bromo (Br). El término “solvato” tal como se usa en el presente documento significa un compuesto, o una sal farmacéuticamente
aceptable del mismo, en el que las moléculas de un disolvente adecuado se incorporan en la red cristalina. Un disolvente adecuado es fisiológicamente tolerable a la dosificación administrada. Los ejemplos de disolventes adecuados son etanol, agua y similares. Cuando el agua es el disolvente, la molécula se denomina “hidrato.” Una “composición farmacéutica” se refiere a una mezcla de uno o más de los compuestos descritos en el presente documento, o sales, solvatos o hidratos farmacéuticamente aceptables de los mismos, con otros componentes químicos, tales como portadores y excipientes fisiológicamente aceptables. El fin de una composición farmacéutica es facilitar la administración de un compuesto a un organismo o sujeto.
El término “sal farmacéuticamente aceptable” se pretende que incluya sales derivadas de ácidos inorgánicos u orgánicos incluyendo, por ejemplo, ácido clorhídrico, bromhídrico, sulfúrico, nítrico perclórico, fosfórico, fórmico, acético, láctico, maleico, fumárico, succínico, tartárico, glicólico, salicílico, cítrico, metanosulfónico, bencenosulfónico, benzoico, malónico, trifluroacético, tricloroacético, naftalen-2-sulfónico y otros ácidos; y sales derivadas de bases inorgánicas u orgánicas incluyendo, por ejemplo, sodio, potasio, calcio, amonio o tetrafluoroborato. Sales farmacéuticamente aceptable a modo de ejemplo se encuentran, por ejemplo, en Berge, et al. (J. Pharm. Sci. 1977, 66(1), 1).
Tal como se usa en el presente documento el término “aproximadamente” se usa en el presente documento para significar de manera aproximada, más o menos, alrededor de o en la región de. Cuando el término “aproximadamente” se usa junto con un intervalo numérico, modifica ese intervalo ampliando los límites por encima y por debajo de los valores numéricos expuestos. En general, el término “aproximadamente” se usa en el presente documento para modificar un valor numérico por encima y por debajo del valor declarado mediante una variación del 20% más o menos (mayor o menor).
El término “portador” se refiere a un diluyente, adyuvante, excipiente o vehículo con el que se administra un compuesto. Los ejemplos no limitativos de tales portadores farmacéuticos incluyen líquidos, tales como agua y aceites, incluyendo los de origen de petróleo, animal, vegetal o sintético, tales como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Los portadores farmacéuticos también pueden ser solución salina, goma arábiga, gelatina, pasta de almidón, talco, queratina, sílice coloidal, urea, y similares. Además, pueden usarse agentes auxiliares, estabilizantes, espesantes, lubricantes y colorantes. Otros ejemplos de portadores farmacéuticos adecuados se describen en Remington's Pharmaceutical Sciences (Alfonso Gennaro ed., Krieger Publishing Company (1997); Remington's: The Science and Practice de Pharmacy, 21a ed. (Lippincot, Williams & Wilkins (2005); Modern Pharmaceutics, vol. 121 (Gilbert Banker y Christopher Rhodes, CRC Press (2002)).
Se describen inhibidores de la PDE5, por ejemplo, en las patentes estadounidenses n.os 5.250.534; 5.859.006; 6.362.178; y 7.378.430; publicación de patente internacional n.os WO/2008/095835, WO/2009/050554, WO/2009/124119, WO/2010/015589, WO/2010/074783, y WO/2011/015523; y Uthayathas et al en Pharmacol. Rep.
2007, 59(2), 150-63; y referencias citadas en el mismo. Los inhibidores de la PDE5 incluyen, por ejemplo, sildenafilo, tadalafilo, vardenafilo, avanafilo, lodenafilo, udenafilo, mirodenafilo, P20066 (Etipharm), SLx-2101 (Kadmon Pharmaceuticals), PF00489791 (Pfizer), INT007 (IntelGenx Technologies) y dasantafilo. Se ha descubierto una familia novedosa de inhibidores de la PDE5 y se describe en el presente documento.
Se describen derivados de benzonaftiridina, por ejemplo, en las patentes estadounidenses n.os 3.674.790; 4.742.061; 6.294.547; 6.384.047; 6.436.952; y la publicación de patente internacional n.° WO/1998/055481. En el presente documento, los inventores describen compuestos que son derivados novedosos de benzonaftiridina. En algunas realizaciones, los compuestos inhiben la PDE5.
En algunas realizaciones, A es NR4. En algunas realizaciones, A es N-alquilo (C1 -C3). En algunas realizaciones, A es N-metilo. En algunas realizaciones, A es NH.
En algunas realizaciones, X es alquilo (C1-C3 ). En algunas realizaciones, X es alquilo (C1-C2 ). En algunas realizaciones, X es CH2.
En algunas realizaciones, V es un enlace. En algunas realizaciones, V es C(O).
En algunas realizaciones, W es un enlace. En algunas realizaciones, W es NR13.
En algunas realizaciones, R1 es un halógeno. En algunas realizaciones, R1 es hidrógeno, halógeno o haloalquilo (C1 -C3 ). En algunas realizaciones, R1 es halógeno o haloalquilo (C1-C6). En algunas realizaciones, R1 es halógeno o haloalquilo (C1 -C3). En algunas realizaciones, R1 es halógeno. En algunas realizaciones, R1 es flúor, cloro o bromo. En algunas realizaciones, R1 es cloro o bromo. En algunas realizaciones, R1 es cloro. En algunas realizaciones, R1 es haloalquilo (C1 -C3). En algunas realizaciones, R1 es trifluorometilo.
En algunas realizaciones, R2 es -OR6. En algunas realizaciones, R2 es -O-alquilo (C1 -C3). En algunas realizaciones, R2 es -O-metilo o -O-etilo. En algunas realizaciones, R2 es -O-metilo.
En algunas realizaciones, R3 es hidrógeno, -OMe, -CF3, -CN o halógeno. En algunas realizaciones, R3 es -CN. En algunas realizaciones, R3 es halógeno. En algunas realizaciones, R3 es flúor, cloro o bromo. En algunas
realizaciones, R3 es flúor. En algunas realizaciones, R3 es cloro. En algunas realizaciones, R3 es bromo. En algunas realizaciones, R3 es hidrógeno. En otras realizaciones, R3 es -OMe. En algunas realizaciones, R3 es -CF3.
En algunas realizaciones, R5 es hidrógeno, alquilo (C1-C3) o cicloalquilo (C3-C5). En algunas realizaciones, R5 es hidrógeno o alquilo (C1-C3 ). En algunas realizaciones, R5 es alquilo (C1-C3 ) o cicloalquilo (C3-C5). En algunas realizaciones, R5 es hidrógeno. En algunas realizaciones, R5 es alquilo (C1 -C3). En algunas realizaciones, R5 es metilo o etilo. En algunas realizaciones, R5 es metilo. En algunas realizaciones, R5 es etilo. En algunas realizaciones,
R5 es cicloalquilo (C3-C5). En algunas realizaciones, R5 es ciclopropilo o ciclobutilo. En algunas realizaciones, R5 es ciclopropilo.
En algunas realizaciones, R6 es cicloalquilo (C3-C8). En algunas realizaciones, R6 es alquilo (C1-C3 ) o haloalquilo (C1 -C3 ). En algunas realizaciones, R6 es alquilo (C1-C3).
En algunas realizaciones, R7 es independientemente hidrógeno o alquilo (C1-C3 ).
En algunas realizaciones, R8 es hidrógeno o alquilo (C1 -C6). En algunas realizaciones, R8 es hidrógeno o alquilo (C1 -C3 ). En algunas realizaciones, R8 es hidrógeno o alquilo (C1-C2 ). En algunas realizaciones, R8 es hidrógeno o etilo.
En algunas realizaciones, R8 es hidrógeno.
En algunas realizaciones, R13 es hidrógeno o alquilo (C1 -C3 ). En algunas realizaciones, R13 es hidrógeno. En algunas realizaciones, R13 es alquilo (C1 -C3 ). En algunas realizaciones, R13 es etilo.
En algunas realizaciones, m es 0, 1 ó 2. En algunas realizaciones, m es 0 ó 1. En algunas realizaciones, m es 1 ó 2.
En algunas realizaciones, m es 0. En algunas realizaciones, m es 1. En algunas realizaciones, m es 2.
En algunas realizaciones, n es 0, 1 ó 2. En algunas realizaciones, n es 0 ó 1. En algunas realizaciones, n es 1 ó 2.
En algunas realizaciones, n es 0. En algunas realizaciones, n es 1. En algunas realizaciones, n es 2.
En algunas realizaciones, m es 1 y n es 1. En algunas realizaciones, m es 2 y n es 1.  lizaciones, m es 1 y n es 2. En algunas realizaciones, m es 0 y n es 1. En algunas realizaciones, m es 0 y n es 2.
lizaciones, m es 1 y n es 2. En algunas realizaciones, m es 0 y n es 1. En algunas realizaciones, m es 0 y n es 2.
En algunas realizaciones, Y es NR5. En algunas realizaciones, Y es NH.
En algunas realizaciones, V es un enlace, W es un enlace, X es alquilo (C1-C3 ); Y es  son cada uno 1 ó 2.
son cada uno 1 ó 2.
En algunas realizaciones, A es NR4 , V es un enlace o C(O), W es un enlace o NR13, X es alquilo (C1 -C3); Y es NR5 ;
R1 es halógeno o haloalquilo (C1-C3 ); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno, alquilo (C1 -C3), cicloalquilo (C1 -C3) o -C(O)R7 ; R6 es alquilo (C1 -C3) o haloalquilo (C1 -C3); R7 es alquilo (C1-C3 ); R8 es hidrógeno; R13 es hidrógeno o alquilo (C1 -C3); m es 0 ó 1 ; y n es 2 , siempre que la suma de m n sea un número entero de desde 2-3.
En algunas realizaciones, A es NR4 , V es un enlace o C(O), W es un enlace o NR13, X es alquilo (C1 -C3); Y es NR5 ;
R1 es halógeno; R2 es -OR6 ; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3 ) o -C(O)R7; R6 es alquilo (C1-C3 ); R7 es alquilo (C1 -C3); R8 es hidrógeno; R13 es hidrógeno; m es 0 ó 1; y n es 2, siempre que la suma de m n sea un número entero de desde 2-3.
La divulgación se refiere a en la que A es O, V es un enlace o C(O), W es un enlace o NH, X es -CH2-; Y es NR5 ; R1 es halógeno; R2 es -OCH3 ; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno, alquilo (C1 -C3) o -C(O)CH3; R8 es hidrógeno; m es 0 ó 1; y n es 2, siempre que la suma de m n sea un número entero de desde 2-3.
En algunas realizaciones, X es CH2 ; Y es NR5 ; R1 es un halógeno o haloalquilo (C1-C3 ); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3) o cicloalquilo (C3-C5); R6 es hidrógeno, alquilo (C1 -C3) o haloalquilo (C1 -C3); y R8 es hidrógeno, alquilo (C1-C3) o haloalquilo (C1-C3).
En algunas realizaciones, V es un enlace, W es un enlace, X es CH2 ; Y es NR5; R1 es un halógeno o haloalquilo (C1-C3 ); R2 es -OR6 ; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3) o cicloalquilo (C3-C5 ); R6 es hidrógeno, alquilo (C1-C3) o haloalquilo (C1 -C3); y R8 es hidrógeno, alquilo (C1 -C3) o haloalquilo (C1 -C3).
En algunas realizaciones, X es CH2 ; Y es NR5 ; R1 es un halógeno o haloalquilo (C1-C3 ); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3) o cicloalquilo (C3-C5); R6 es hidrógeno, alquilo (C1-C3 ) o haloalquilo (C1-C3 ); R6 es hidrógeno, alquilo (C1 -C3) o haloalquilo (C1 -C3); R8 es hidrógeno, alquilo
(C1 -C3) o haloalquilo (C1 -C3); y m y n son cada uno 1 ó 2.
En algunas realizaciones, V es un enlace, W es un enlace, X es CH2 ; Y es NR5; R1 es un halógeno o haloalquilo (C1-C3 ); R2 es -OR6 ; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3) o cicloalquilo (C3-C5 ); R6 es hidrógeno, alquilo (C1 -C3) o haloalquilo (C1 -C3); R6 es hidrógeno, alquilo (C1-C3 ) o haloalquilo (C1-C3 ); R8 es hidrógeno, alquilo (C1-C3 ) o haloalquilo (C1-C3); y m y n son cada uno 1 ó 2.
En algunas realizaciones, V es un enlace, W es un enlace, A es NH; X es CH2 ; Y es NR5 ; R1 es halógeno o haloalquilo (C1 -C3); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno o alquilo (C1 -C3); y R6 es hidrógeno, -(C1-C2 )-alquilo o haloalquilo (C1-C2 ).
En algunas realizaciones, A es NH; X es CH2 ; Y es NR5; R1 es halógeno o haloalquilo (C1 -C3); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno o alquilo (C1-C3 ); y R6 es alquilo (C1-C2 ) o haloalquilo (C1 -C2).
En algunas realizaciones, A es NH; X es CH2 ; Y es NR5; R1 es halógeno o haloalquilo (C1 -C3); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno o alquilo (C1 -C3); R6 es alquilo (C1 -C2) o haloalquilo (C1-C2 ); m es 1 ; y n es 2.
En algunas realizaciones, A es NH; X es CH2 ; Y es NR5 ; R1 es halógeno; R2 es -OCH3; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno o alquilo (C1-C3); m es 1; y n es 2.
La divulgación se refiere a en la que, R1 y R2 no son ninguno hidrógeno cuando V y W son cada uno un enlace, Y es NR5 , A es NR4 , X es CO, n = 2 y m = 1.
En algunas realizaciones, A es NR4 ; V es un enlace o C(O); W es un enlace o NR13; X es alquilo (C1 -C3); Y es NR5 ; R1 es halógeno o haloalquilo (C1-C3 ); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno, alquilo (C1 -C3), cicloalquilo (C1 -C3) o -C(O)R7 ; R6 es alquilo (C1 -C3) o haloalquilo (C1 -C3); R7 es alquilo (C1-C3 ); R8 es hidrógeno o alquilo (C1-C3); R13 es hidrógeno o alquilo (C1-C3 ); ym y n son independientemente 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3.
En algunas realizaciones, A es NH; V es un enlace o C(O); W es un enlace o NR13; X es alquilo (C1 -C3); Y es NR5 ; R1 es halógeno; R2 es -OR6 ; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3 ) o -C(O)R7 ; R6 es alquilo (C1-C3 ); R7 es alquilo (C1 -C3); R8 es hidrógeno o alquilo (C1 -C3); R13 es hidrógeno; m es 0 ó 1; y n es 2.
En algunas realizaciones, A es NH; V es un enlace o C(O); W es un enlace o NH; X es -CH2-; Y es NR5 ; R1 es halógeno; R2 es -OCH3; R3 es hidrógeno, -OMe, -CF3 , -CN o halógeno; R5 es hidrógeno, alquilo (C1 -C3) o -C(O)CH3 ; R8 es hidrógeno o alquilo (C1 -C3); m es 0 ó 1; y n es 2.
En algunas realizaciones, V y W son cada uno un enlace, o V es C(O) y W es NR13; X es alquilo (C1-C3); Y es NR5 ; y m y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3.
En algunas realizaciones, A es NH; V es C(O); W es NH; X es CH2 ; Y es NR5; R1 es halógeno o haloalquilo (C1 -C3); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3 , -c N o halógeno; R5 es hidrógeno, alquilo (C1-C3), o C(O)R7; R6 es alquilo (C1 -C3) o haloalquilo (C1 -C3); R7 es hidrógeno o alquilo (C1-C3 ); R8 es hidrógeno, alquilo (C1 -C3), haloalquilo (C1-C3 ), cicloalquilo (C3-C5), -Nr9R10; m es 0 ó 1 ; y n es 2.
En algunas realizaciones, A es NH; V es C(O); W es NH; X es CH2 ; Y es NR5 ; R1 es halógeno; R2 es -OCH3 ; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1 -C3), o C(O)R7; R7 es alquilo (C1-C3); R8 es hidrógeno o alquilo (C1-C3); m es 0; y n es 2.
En algunas realizaciones, A es NH; V y W son cada uno un enlace, o V es C(O) y W es NH; X es CH2 ; Y es NR5; R1 es halógeno o haloalquilo (C1 -C3); R2 es -OR6; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R5 es hidrógeno, alquilo (C1-C3 ), o C(O)R7; R6 es hidrógeno, alquilo (C1 -C2 ) o haloalquilo (C1 -C2); R7 es alquilo (C1-C3); R8 es hidrógeno, alquilo (C1-C3 ); m es 0 ó 1 ; y n es 2.
En algunas realizaciones, A es NH; V y W son cada uno un enlace; X es CH2 ; Y es NR5 ; R1 es cloro; R2 es -OCH3 ; R3 es -CN; R5 es hidrógeno o alquilo (C1 -C3); R8 es hidrógeno o alquilo (C1-C3 ); m es 1; y n es 2.
En algunas realizaciones, A es NH; V y W son cada uno un enlace; X es CH2 ; Y es NC(O)CH3 ; R1 es halógeno; R2 es -OCH3; R3 es hidrógeno, -OMe, -CF3, -CN o halógeno; R8 es hidrógeno; m es 1; y n es 2.
En otro aspecto, la invención se refiere a composiciones que comprenden un compuesto de fórmula (I), (la), (Ib) o (Ic) y un portador farmacéuticamente aceptable.
La divulgación se refiere a un método de inhibición de fosfodiesterasa que comprende poner en contacto una fosfodiesterasa con un compuesto de fórmula (I), (la), (Ib) o (Ic) o una composición que comprende un compuesto de fórmula (I), (Ia), (Ib), o (Ic). La fosfodiesterasa puede ser PDE5.
La divulgación se refiere a un método de tratamiento de una enfermedad neurodegenerativa en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I), (Ia), (Ib), o (Ic). La enfermedad puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de tratamiento de una enfermedad neurodegenerativa en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de una composición que comprende un
compuesto de fórmula (I), (la), (Ib) o (Ic). La enfermedad puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de aumento de la potenciación a largo plazo en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I), (Ia), (Ib) o (Ic). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de aumento de la potenciación a largo plazo en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de una composición que comprende un compuesto de fórmula (I), (Ia), (Ib) o (Ic). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de mejora de la memoria en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I), (Ia), (Ib) o (Ic). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de mejora de la memoria en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de una composición que comprende un compuesto de fórmula (I), (Ia), (Ib) o (Ic). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de mejora de la función sináptica en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I), (Ia), (Ib) o (Ic). Función sináptica comprende plasticidad sináptica. Plasticidad sináptica puede comprender aprendizaje, memoria o una combinación de los mismos. Plasticidad sináptica puede comprender potenciación a largo plazo (LTP). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método de mejora de la función sináptica en un sujeto que comprende la administración de una cantidad terapéuticamente eficaz de una composición que comprende un compuesto de fórmula (I), (Ia), (Ib) o (Ic) Función sináptica comprende plasticidad sináptica. Plasticidad sináptica comprende aprendizaje, memoria o una combinación de los mismos. Plasticidad sináptica comprende potenciación a largo plazo (LTP). El sujeto puede tener una enfermedad neurodegenerativa. La enfermedad neurodegenerativa puede ser enfermedad de Alzheimer.
La divulgación se refiere a un método para aumentar la retención de la memoria en un sujeto que padece una enfermedad neurodegenerativa, comprendiendo el método administrar a un sujeto una cantidad terapéutica de un compuesto de fórmula (I), (Ia), (Ib) o (Ic) o una composición que comprende un compuesto de fórmula (I), (Ia), (Ib) o (Ic).
Puede administrarse un compuesto de fórmula (I), (Ia), (Ib) o (Ic). Puede administrarse una composición que comprende un compuesto de fórmula (I), (Ia), (Ib) o (Ic).
Enfermedades neurodegenerativas y métodos de tratamiento de las mismas a modo de ejemplo también se describen en los documentos en WO 2010/074783, WO2011/072243 y WO2012/088420
Pueden incorporarse compuestos de fórmula (I), (Ia), (Ib) o (Ic) en composiciones farmacéuticas adecuadas para administración. Tales composiciones pueden comprender un compuesto de fórmula (I), (Ia), (Ib) o (Ic) y un portador farmacéuticamente aceptable. Por tanto, en algunas realizaciones, los compuestos de la invención están presentes en una composición farmacéutica.
Según la invención, un portador farmacéuticamente aceptable puede comprender cualquier disolvente, medio de dispersión, recubrimiento, agente antibacteriano y antifúngico, agente isotónico y de retardo de la absorción, y similares, compatibles con la administración farmacéutica. El uso de tales medios y agentes para sustancias farmacéuticamente activas se conoce bien en la técnica. Puede usarse cualquier medio o agente convencional que es compatible con el compuesto activo. También pueden incorporarse compuestos activos complementarios en las composiciones.
Cualquiera de las aplicaciones terapéuticas descritas en el presente documento puede aplicarse a cualquier sujeto que necesita tal terapia, incluyendo, por ejemplo, un mamífero tal como un ratón, una rata, un conejo, un gato, una vaca, un caballo, un conejo, un mono, un cerdo, una oveja, una cabra o un ser humano. En algunas realizaciones, el sujeto es un ratón, rata o ser humano. En algunas realizaciones, el sujeto es un ratón. En algunas realizaciones, el sujeto es una rata. En algunas realizaciones, el sujeto es un ser humano.
Una composición farmacéutica de la invención se formula para ser compatible con su vía prevista de administración. Los ejemplos de vías de administración incluyen administración parenteral, por ejemplo, intravenosa, intradérmica, subcutánea, oral (por ejemplo, inhalación), transdérmica (tópica), transmucosa y rectal. Las disoluciones o suspensiones usadas para aplicación parenteral, intradérmica o subcutánea pueden incluir los siguientes
componentes: un diluyente estéril tal como agua para inyección, solución salina, aceites fijos, polietilenglicoles, glicerina, propilenglicol u otros disolventes sintéticos; agentes antibacterianos tales como alcohol bencílico o metilparabenos; antioxidantes tales como ácido ascórbico o bisulfito de sodio; agentes quelantes tales como ácido etilendiaminotetraacético; tampones tales como acetatos, citratos o fosfatos y agentes para el ajuste de la tonicidad tales como cloruro de sodio o dextrosa. El pH puede ajustarse con ácidos o bases, tales como ácido clorhídrico o hidróxido de sodio. La preparación parenteral puede contenerse en ampollas, jeringas desechables o viales de múltiples dosis elaborados de vidrio o plástico.
Las composiciones farmacéuticas adecuadas para uso inyectable incluyen disoluciones acuosas estériles (cuando son solubles en agua) o dispersiones y polvos estériles para la preparación extemporánea de dispersiones o disoluciones inyectables estériles. Para administración intravenosa, los portadores adecuados incluyen solución salina fisiológica, agua bacteriostática, Cremophor EM™ (BASF, Parsippany, N.J.) o solución salina tamponada con fosfato (PBS). En todos los casos, la composición debe ser estéril y debe ser fluida en la medida en que exista una fácil inyectabilidad. Debe ser estable en las condiciones de fabricación y almacenamiento y debe conservarse contra la acción contaminante de microorganismos tales como bacterias y hongos. El portador puede ser un disolvente o medio de dispersión que contiene, por ejemplo, agua, etanol, un poliol farmacéuticamente aceptable como glicerol, propilenglicol, polieteilenglicol líquido, y mezclas adecuadas de los mismos. Puede mantenerse la fluidez adecuada, por ejemplo, mediante el uso de un recubrimiento tal como lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersión y mediante el uso de tensioactivos. Puede lograrse el impedimento de la acción de microorganismos mediante diversos agentes antibacterianos y antifúngicos, por ejemplo, parabenos, clorobutanol, fenol, ácido ascórbico y timerosal. En muchos casos, puede ser útil incluir agentes isotónicos, por ejemplo, azúcares, polialcoholes tales como manitol, sorbitol, cloruro de sodio en la composición. Puede lograrse la absorción prolongada de las composiciones inyectables incluyendo en la composición un agente que retrasa la absorción, por ejemplo, monoestearato de aluminio y gelatina.
Pueden prepararse disoluciones inyectables estériles incorporando el compuesto inhibidor de la PDE5 en la cantidad requerida en un disolvente apropiado con uno o una combinación de componentes enumerados en el presente documento, según se requiera, seguido por filtración por esterilización. Generalmente, las dispersiones se preparan incorporando el compuesto activo en un vehículo estéril que contiene un medio básico de dispersión y los otros componentes requeridos de aquellos enumerados en el presente documento. En el caso de polvos estériles para la preparación de disoluciones inyectables estériles, ejemplos de métodos de preparación útiles son secado a vacío y secado por congelación que proporciona un polvo del principio activo más cualquier componente deseado adicional de una disolución filtrada previamente por esterilización del mismo.
Las composiciones orales incluyen generalmente un diluyente inerte o un portador comestible. Pueden contenerse en cápsulas de gelatina o comprimirse para dar comprimidos. Para el fin de administración terapéutica oral, el compuesto activo puede incorporarse con excipientes y usarse en forma de comprimidos, pastillas para chupar o cápsulas. También pueden prepararse composiciones orales usando un portador fluido para su uso como enjuague bucal, en las que el compuesto en el portador fluido se aplica por vía oral y puede enjugarse y expectorarse o tragarse.
Pueden incluirse materiales adyuvantes y/o agentes de unión farmacéuticamente compatibles como parte de la composición. Los comprimidos, pastillas, cápsulas, pastillas para chupar y similares pueden contener cualquiera de los siguientes componentes o compuestos de naturaleza similar: un aglutinante tal como celulosa microcristalina, goma tragacanto o gelatina; un excipiente tal como almidón o lactosa, un agente disgregante tal como ácido algínico, Primogel, o almidón de maíz; un lubricante tal como estearato de magnesio o sterotes; un deslizante tal como dióxido de silicio coloidal; un edulcorante tal como sacarosa o sacarina; o un agente saborizante tal como menta piperita, salicilato de metilo o sabor a naranja.
La administración sistémica también puede ser mediante medios transmucosos o transdérmicos. Para la administración transmucosa o transdérmica, se usan agentes penetrantes apropiados para la barrera que va a permearse en la formulación. Tales agentes penetrantes se conocen generalmente en la técnica, e incluyen, por ejemplo, para administración transmucosa, detergentes, sales biliares y derivados de ácido fusídico. La administración transmucosa puede lograrse a través del uso de aerosoles nasales o supositorios. Para la administración transdérmica, los compuestos activos se formulan en pomadas, bálsamos, geles o cremas tal como se conoce generalmente en la técnica.
Se sintetizan derivados de benzonaftiridina mediante métodos dentro del campo del experto común en la técnica. Métodos a modo de ejemplo mediante los cuales pueden sintetizarse derivados de benzonaftiridina son los siguientes.
Método A: pueden sintetizarse derivados de benzonaftiridina, por ejemplo, a partir de derivado de ácido 2-aminobenzoico (esquema 1 ).

Pueden calentarse una mezcla de derivado de ácido 2-aminobenzoico y derivado de aminocetona cíclica (que contiene opcionalmente un grupo alquilo u otro grupo protector en el átomo de nitrógeno) en, por ejemplo, POCh para obtener una benzonaftiridina. La reinstalación del grupo protector (si se retira durante la formación de benzonaftiridina) puede lograrse mediante métodos conocidos tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006). El tratamiento posterior con una fenilalquilamina y una sal tal como, por ejemplo, un haluro de sodio, en un disolvente tal como un fenol puede calentarse para obtener el derivado de benzonftiridina elaborado. Puede lograrse protección opcional del átomo de nitrógeno de la fenilalquilamina mediante métodos conocidos, tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006). El experto en la técnica reconocerá la selección de un grupo protector apropiado basándose en ortogonalidad y propiedades deseadas con otros grupos protectores en, por ejemplo, el átomo de nitrógeno de piperidina. Puede lograrse desprotección opcional del nitrógeno de piperidina mediante métodos conocidos, tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006), seguido por la incorporación del grupo R5 deseado por medio de aminación reductora de un R5-CHO en presencia de un agente reductor tal como un borohidruro o por medio de alquilación de R5-LG (en el que LG es un grupo saliente tal como, por ejemplo, halógeno, sulfonato, etc.). Puede lograrse retirada de los grupos protectores mediante métodos conocidos, tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006).
Método B: pueden sintetizarse derivados de benzonaftiridina, por ejemplo, a partir de derivado de ácido 2-aminobenzoico (esquema 2).
Esquema 2:

Puede tratarse un derivado de anilina en un disolvente tal como agua con hidrato de cloral, ácido (tal como HCl), un sulfato de grupo I (tal como sulfato de sodio), hidroxilamina y se calienta para generar una hidroxilimina. La hidroxilimina puede tratarse con un ácido (tal como ácido sulfúrico) y calentarse para generar una indolin-2,3-diona. El posterior tratamiento con un oxidante tal como peróxido de hidrógeno en una base tal como hidróxido de sodio puede generar un ácido 2-aminobenzoico. Puede lograrse conversión opcional de R3 a -CN por medio de tratamiento con cianuro de cobre en un disolvente tal como NMP antes del procesamiento adicional.
Pueden calentarse el derivado de ácido 2-aminobenzoico y derivado de aminocetona cíclica (que contiene opcionalmente un grupo alquilo u otro grupo protector en el átomo de nitrógeno) en POCl3 para obtener una benzonaftiridina. La reinstalación del grupo protector (si se retira durante la formación de benzonaftiridina) puede lograrse mediante métodos conocidos tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006). El posterior tratamiento con una fenilalquilamina y una sal tal como, por ejemplo, un haluro de sodio, en un disolvente tal como un fenol puede calentarse para obtener el derivado de benzonftiridina elaborado. Puede lograrse protección opcional del átomo de nitrógeno de la fenilalquilamina mediante métodos conocidos, tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006). El experto en la técnica reconocerá la selección de un grupo protector apropiado basándose en ortogonalidad y propiedades deseadas con otros grupos protectores en, por ejemplo, el átomo de nitrógeno de piperidina. Puede lograrse desprotección opcional del nitrógeno de piperidina mediante métodos conocidos, tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006), seguido por la incorporación del grupo R5 deseado por medio de aminación reductora de un R5-CHO en presencia de un agente reductor tal como un borohidruro o por medio de alquilación de R5-LG (en el que LG es un grupo saliente tal como, por ejemplo, halógeno, sulfonato, etc.). Puede lograrse retirada de los grupos protectores mediante métodos conocidos, tales como los descritos en, por ejemplo, Protecting Groups in Organic Synthesis, 4a edición, por Peter Wuts & Theodora Greene (Wiley 2006).
Método C: también pueden sintetizarse derivados de benzonaftiridina, por ejemplo, a partir de derivado de 2-aminobenzonitrilo (esquema 3).

Puede calentarse una mezcla de derivado de 2-aminobenzonitrilo y derivado de aminocetona cíclica en presencia de un ácido fuerte. Ácidos fuertes empleados en esta transformación pueden ser cualquier ácido orgánico o inorgánico, tal como, por ejemplo, ácido polifosfórico, trifluoroacético, acético y sulfónico. El tratamiento posterior con un resto fenilaquilo que contiene un grupo saliente (LG) y una base tal como, por ejemplo, una trialquilamina, puede formar el derivado de benzonftiridina elaborado.
Además de los métodos generales mencionados anteriormente, también pueden usarse otros métodos tales como los descritos en, por ejemplo, las patentes estadounidenses n.os 3.674.790 y 6.294.547 para obtener los derivados de benzonaftiridina. El experto común en la técnica reconocerá variaciones de los métodos descritos en el presente documento y de los métodos descritos en las referencias en el presente documento citadas para sintetizar otros derivados de benzonaftiridina dentro del alcance de la invención.
Un inhibidor de la PDE5 puede disminuir la actividad de una molécula de PDE5 in vivo y/o in vitro. En una realización, un inhibidor de la PDE5 puede disminuir la actividad de PDE5 en al menos aproximadamente el 10%, al menos aproximadamente el 20%, al menos aproximadamente el 30%, al menos aproximadamente el 40%, al menos aproximadamente el 50%, al menos aproximadamente el 60%, al menos aproximadamente el 70%, al menos aproximadamente el 75%, al menos aproximadamente el 80%, al menos aproximadamente el 85%, al menos aproximadamente el 90%, al menos aproximadamente el 95%, al menos aproximadamente el 97%, al menos aproximadamente el 99% o el 100%.
En algunas realizaciones, los compuestos de la invención presentan inhibición de la PDE5 con una CI50 de menos de aproximadamente 1 |iM. En algunas realizaciones, los compuestos de la invención presentan inhibición de la PDE5 con una CI50 de menos de aproximadamente 500 nM. En algunas realizaciones, la CI50 es de menos de aproximadamente 250 nM, menos de aproximadamente 100 nM, menos de aproximadamente 50 nM, menos de aproximadamente 25 nM, menos de aproximadamente 10 nM, menos de aproximadamente 5 nM, o menos de aproximadamente 1 nM.
En algunas realizaciones, los compuestos de fórmula (I), (Ia), (Ib) o (Ic) son inhibidores selectivos de la PDE5. En algunas realizaciones, los compuestos presentan inhibición de la PDE5 a concentraciones menores que a las que inhiben otros subtipos de PDE. En algunas realizaciones, otros subtipos de PDE pueden incluir cualquiera de PDE1-PDE4 y PDE6-PDE11 o cualquier combinación de los mismos. En algunas realizaciones, el otro subtipo de PDE es PDE1. En algunas realizaciones, el otro subtipo de PDE es PDE6. En algunas realizaciones, el otro subtipo de PDE es PDE9.
Se reconocerá que una o más características de cualquier realización dada a conocer en el presente documento puede combinarse y/o reorganizarse dentro del alcance de la invención para producir realizaciones adicionales que están también dentro del alcance de la invención. Los expertos en la técnica reconocerán, o podrán determinar usando tan sólo experimentación de rutina, muchos equivalentes a las específicas
realizaciones de la invención descritas en el presente documento. Se pretende que tales equivalentes estén dentro del alcance de la presente invención.
A menos que se defina lo contrario, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que entiende comúnmente un experto habitual en la técnica a la que pertenece esta invención. A continuación se describen métodos y materiales a modo de ejemplo, aunque también pueden usarse métodos y materiales similares o equivalentes a los descritos en el presente documento en la práctica o las pruebas de la presente invención.
La invención se describe adicionalmente mediante los siguientes ejemplos no limitativos.
Ejemplos
A continuación se proporcionan ejemplos para facilitar una comprensión más completa de la invención. Los siguientes ejemplos ilustran los modos a modo de ejemplo de elaborar y practicar la invención. Sin embargo, el alcance de la invención no se limita a realizaciones específicas dadas a conocer en estos ejemplos, que son con fines de ilustración solamente, dado que pueden utilizarse métodos para obtener resultados similares.
Ejemplo 1: 8-bromo-A/-(3-cloro-4-metoxibencil)-2-metil-1,2,3,4-tetrahidrobenzo[M1,6lnaftiridin-10-amina

8-Bromo-10-cloro-2-metil-1,2,3,4-tetrahidrobenzo[6l[1,6lnaftiridina, 2
Se calentó una mezcla de 1 (9,25 mmol) y 1-metilpiperidin-4-ona (9,25 mmol) en POCh (10 ml) a 60°C durante 6 h. Se eliminó por evaporación el POCl3 en exceso; se trató el residuo con H2O helada y NaHCO3 y se extrajo con AcOEt (3x50 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. Se purificó el compuesto deseado triturando con Et2O (42% de rendimiento). 1H RMN (300 MHz, CDCh) 88,33 (d, 1H, J= 2,1 Hz), 7,86 (d, 1H, J= 9,0 Hz), 7,77 (dd, 1H, Ji= 2,1, J2= 9,0 Hz), 3,99 (s, 2 h), 3,34 (t, 2 h, J= 5,7 Hz), 3,03 (t, 2 h, J= 5,7 Hz), 2,69 (s, 3H).
Se calentó una mezcla de 2 (1,92 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (2,11 mmol), Nal (0,1 mmol) y
fenol (3,84 mmol) a 130°C durante 1,5 h. Después de enfriar la reacción, se añadió Et2O (20 ml) y se lavó con NaOH 1 N (3x20 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. Se purificó el residuo mediante cromatografía ultrarrápida (AcOEt: MeOH 8:2) para dar el compuesto deseado A (38% de rendimiento). 1H RMN (300 MHz, CDCls) 88,04 (d, 1H, J = 2,1 Hz), 7,80 (d, 1H, J = 8,7 Hz), 7,64 (dd, 1H, Ji = 2,1, J2 = 9,0 Hz), 7,38 (d, 1H, J = 2,1 Hz), 7,16 (dd, 1H, Ji = 2,1, J2 = 8,4 Hz), 6,91 (d, 1H, J = 8,4 Hz), 4,47 (d, 2 h, J= 5,7 Hz), 3,91 (s, 3H), 3,54 (s, 2 h), 3,18 (t, 2 h, J= 6,3 Hz), 2,81 (t, 2 h, J= 6,3 Hz), 2,50 (s, 3H).
Ejemplo 2: 10-rí3-cloro-4-metox¡benc¡l)am¡nol-2-met¡l-1.2.3.4-tetrah¡drobenzorM1.6lnaft¡r¡d¡na-8-carbon¡tr¡lo

Ácido 2-amino-5-cianobenzoico, 3
Se calentaron ácido 2-amino-5-bromobenzoico (2,31 mmol) y CuCN (2,78 mmol) hasta reflujo en NMP (5 ml) durante 3 h. Se vertió la mezcla de reacción en una disolución de FeCh 3H2O (3,0 g) en H2O (3 ml) y HCl (0,5 ml) y se agitó a 60°C durante 1 h. Después de enfriar la reacción, se añadió Et2O (100 ml) y se separaron las dos fases. Se lavó la fase orgánica con HCl 1 N (50 ml) y H2O (2x50 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducida. Se obtuvo ácido 2-amino-5-cianobenzoico (3) (57% de rendimiento) triturando a partir de CH2Cl2.1H RMN (300 MHz, CDCla) 88,32 (d, 1H, J= 1,8 Hz), 7,71 (dd, 1H, Ji= 1,8, J2= 8,7 Hz), 7,32 (s a, 1H), 6,75 (d, 1H, J= 8,7 Hz).
10-Cloro-2-metil-1,2,3,4-tetrah¡drobenzo[6][1,6]naft¡r¡d¡na-8-carbon¡tr¡lo, 4
Se calentó una mezcla de 1 (1,23 mmol) y 1-metilpiperidin-4-ona (1,23 mmol) en POCh (2 ml) a 60°C durante 6 h. Se eliminó por evaporación el POCh en exceso; se trató el residuo con H2O helada y NaHCO3 y se extrajo con CH2Cl2 (3x25 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. La purificación mediante cromatografía ultrarrápida (5% de MeOH en AcOEt) dio el compuesto deseado (55% de rendimiento). 1H RMN (300 MHz, CDCh) 88,58 (s, 1H), 8,07 (d, 1H, J= 8,7 Hz), 7,83 (d, 1H, J= 8,7 Hz), 3,85 (s, 2 h), 3,30 (t, 2 h, J= 6,0 Hz), 2,89 (t, 2 h, J= 6,0 Hz), 2,60 (s, 3H).
10-Cloro-2-etil-1,2,3,4-tetrah¡drobenzo[6][1,6]naft¡r¡d¡na-8-carbon¡tr¡lo, 5
Se calentó una mezcla de 1 (1,23 mmol) y 1-etilpiperidin-4-ona (1,23 mmol) en POCh (2 ml) a 60°C durante 6 h. Se eliminó por evaporación el POCh en exceso; se trató el residuo con H2O helada y NaHCO3 y se extrajo con CH2Cl2 (3x25 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. La purificación mediante cromatografía ultrarrápida (5% de MeOH en CH2Ch) dio el compuesto deseado (26% de rendimiento). 1H RMN (300 MHz, CDCh) 88,58 (d, 1H, J= 1,8 Hz), 8,06 (d, 1H, J= 8,7 Hz), 7,82 (dd, 1H, Ji= 1,8, J2= 8,4 Hz), 3,91 (s, 2 h), 3,30 (t, 2 h, J= 5,7 Hz), 2,93 (t, 2 h, J= 5,7 Hz), 2,75 (q, 2 h, J= 7,2 Hz), 1,26 (t, 3H, J= 7,2 Hz).
10-[(3-Cloro-4-metox¡benc¡l)am¡no]-2-met¡l-1,2,3,4-tetrah¡drobenzo[6][1,6]naft¡r¡d¡na-8-carbon¡tr¡lo.
Se calentó una mezcla de 4 (0,12 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,12 mmol), Nal (0,006 mmol) y fenol (0,12 mmol) a 130°C durante 2,5 h. Se diluyó la mezcla de reacción con Et2O (10 ml) y se lavó con NaOH 1 N
(3x5 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. Se purificó el residuo mediante cromatografía ultrarrápida (AcOEt: MeOH 8:2) para dar el compuesto deseado B (33% de rendimiento). EM ESI (m/z) 394 (M+H)+, 1H RMN (300 MHz, CDCla) 88,32 (d, 1H, J= 1,2 Hz), 7,97 (d, 1H, J= 8,7 Hz), 7,71 (dd, 1H, Ji= 1,8, J2= 8,7 Hz), 7,37 (d, 1H, J= 2,1 Hz), 7,18 (dd, 1H, Ji= 2,4, J2= 8,7 Hz), 6,94 (d, 1H, J= 8,4 Hz), 4,57 (d, 2 h, J= 6,0 Hz), 4,11 (s a, 1H), 3,92 (s, 3H), 3,54 (s, 2 h), 3,22 (t, 2 h, J= 6,3 Hz), 2,83 (t, 2 h, J= 6,3 Hz), 2,52 (s, 3H). Ejemplo 3: 10-r(3-cloro-4-metoxibencil)aminol-2-etil-1,2,3,4-tetrahidrobenzorM1,6lnaftiridina-8-carbonitrilo
Se calentó una mezcla de 5 (0,18 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,18 mmol), Nal (0,009 mmol) y fenol (0,18 mmol) a 130°C durante 2,5 h (esquema 4). Se diluyó la mezcla de reacción con Et2O (30 ml) y se lavó con NaOH 1 N (3x1 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. Se purificó el residuo mediante cromatografía ultrarrápida (5% de MeOH en AcOEt) para dar el compuesto deseado C (38% de rendimiento). EM ESI (m/z) 407 (M+H)+, 1H RMN (300 MHz, CDCla) 88,32 (d, 1H, J= 1,2 Hz), 7,97 (d, 1H, J= 8,7 Hz), 7,71 (dd, 1H, Ji= 1,8, J2= 8,7 Hz), 7,37 (d, 1H, J= 2,1 Hz), 7,17 (dd, 1H, Ji= 2,1, J2= 8,4 Hz), 6,94 (d, 1H, J= 8,4 Hz), 4,56 (d, 2 h, J= 5,7 Hz), 4,12 (s a, 1H), 3,92 (s, 3H), 3,59 (s, 2 h), 3,22 (t, 2 h, J= 6,0 Hz), 2,87 (t, 2 h, J= 6,0 Hz), 2,66 (q, 2 h, J= 7,2 Hz), 1,18 (t, 3H, J= 7,2 Hz).
Ejemplo 4: 10-r(3-cloro-4-metoxibencil)aminol-6-etil-2-metil-1,2,3,4-tetrahidrobenzorM1,6lnaftiridina-8-carbonitrilo

W-(4-Bromo-2-etilfenil)-2-(hidroxiimino)acetamida, 7
A una suspensión de 4-bromo-2-etilanilina (6) (5,0 mmol) en 50 ml de H2O se le añadió HCl conc. (0,5 ml), 4,4 g de Na2SO4 y clorhidrato de NH2OH (14,9 mmol), seguido por adición de hidrato de cloral (5,5 mmol). Se calentó la mezcla de reacción hasta 90°C durante 1 h. Tras enfriar hasta temperatura ambiente, se extrajo la fase acuosa con AcOEt (3x50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron para dar el compuesto 7. 1H RMN (300 MHz, CDCh) 88,22 (s a, 1H), 7,93-7,90 (m, 2 h), 7,60 (s, 1H), 7,37-7,34 (m, 2 h), 2,60 (q, 2 h, J= 7,5 Hz), 1,25 (t, 3H, J= 7,2 Hz).
5-Bromo-7-etilindolin-2,3-diona, 8
A una disolución de ácido sulfúrico (10 ml) y H2O (1 ml) a 80°C se le añadió 7 (3,68 mmol) en porciones pequeñas a lo largo de 20 minutos. Se agitó la mezcla de reacción durante 15 min a 80°C. Después de enfriar la reacción, se
añadió 20 ml de agua helada y se extrajo la mezcla con AcOEt (3x50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron para dar el producto deseado. 1H RMN (300 MHz, CDCh) 88,85 (s, 1H), 7,58 (d, 1H, J= 1,5 Hz), 7,58-7,54 (m, 1H), 2,60 (q, 2 h, J= 7,5 Hz), 1,29 (t, 3H, J= 7,5 Hz).
Ácido 2-amino-5-bromo-3-etilbenzoico, 9
A una suspensión de 8 (1,38 mmol) en 10 ml de NaOH al 10% se le añadió gota a gota una disolución de H2O2 (0,5 ml) en 4,5 ml de H2O. Se agitó la mezcla durante la noche a temperatura ambiente. Se filtró la mezcla de reacción y se acidificó el filtrado añadiendo HCl conc., se filtró el precipitado resultante (9) y se secó. 1H RMN (300 MHz, CDCla) 87,95 (d, 1H, J= 2,1 Hz), 7,32 (d, 1H, J= 2,4 Hz), 2,49 (q, 2 h, J= 7,2 Hz), 1,28 (t, 3H J= 7,2 Hz). Ácido 2-amino-5-ciano-3-etilbenzoico, 10
Se sometió a reflujo una mezcla de 9 (3,3 mmol) y CuCN (3,9 mmol) en 3 ml de NMP durante 4 h. Se vertió la mezcla de reacción en una disolución templada de NaCN (33% p/v) y se agitó vigorosamente. Después de enfriar, se extrajo la reacción con AcOEt (50 ml) y se descartó la fase orgánica. Se acidificó la fase acuosa añadiendo HCl conc. y se recogió el precipitado resultante 10 por filtración. 1H RMN (300 MHz, CDCla) 88,18 (d, 1H, J= 1,8 Hz), 7,26 (s, 1H), 6,49 (s a, 1H), 2,51 (q, 2 h, J= 7,2 Hz), 1,30 (t, 3H, J= 7,2 Hz).
10-Cloro-6-etil-2-metil-1,2,3,4-tetrahidrobenzo[6][1,6]naftiridina-8-carbonitrilo, 11
Se calentó una mezcla de 10 (0,64 mmol) y 1-metilpiperidin-4-ona (0,64 mmol) en POCh (2 ml) a 60°C durante 6 h. Se eliminó por evaporación el POCh en exceso; se trató el residuo con H2O helada y NaHCO3 y se extrajo con CH2Cl2 (3x10 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. Se purificó el compuesto deseado triturando con Et2O. 1H RMN (300 MHz, CDCh) 88,42 (d, 1H, J= 1,8 Hz), 7,65 (m, 1H), 3,82 (s, 2 h), 3,31-3,23 (m, 4 h), 2,87 (t, 2 h J= 5,7 Hz), 2,58 (s, 3H), 1,34 (t, 3H J= 7,2 Hz).
10-[(3-Cloro-4-metoxibencil)amino]-6-etil-2-metil-1,2,3,4-tetrahidrobenzo[6][1,6]naftiridina-8-carbonitrilo
Se calentó una mezcla de 11 (0,18 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,18 mmol), Nal (0,009 mmol) y fenol (0,18 mmol) a 130°C durante 4 h. Se diluyó la mezcla de reacción con Et2O (30 ml) y se lavó con NaOH 1 N (3x10 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. Se obtuvo el producto final D mediante cromatografía ultrarrápida (AcOEt: MeOH 9:1). EM ESI (m/z) 421 (M+H)+, 1H RMN (300 MHz, CDCh) 88,16 (d, 1H, J= 1,5 Hz), 7,56 (s, 1H), 7,36 (d, 1H, J= 2,1 Hz), 7,18-7,16 (m, 1H), 6,93 (d, 1H, J= 8,4 Hz), 4,48 (s, 2 h), 3,92 (s, 3H), 3,54 (s, 2 h), 3,25-3,21 (m, 4 h), 2,89 (t, 2 h, J= 6,0 Hz), 2,51 (s, 3H), 1,35 (t, 3H, J= 7,5 Hz).
Ejemplo 5: 6-[(3-cloro-4-metox¡benc¡l)am¡no1-5-oxo-2.3.4.5-tetrah¡dro-1H-[1.41d¡azep¡no[5.6-b1quinol¡na-8-carbon¡tr¡lo

A una disolución de 2-amino-5-cianobenzoato de metilo (5,68 mmol) en DCM (10 ml) se le añadió trietiamina (8,52 mmol) seguido por 3-cloro-3-oxopropionato de metilo (7,38 mmol). Se agitó la mezcla de reacción resultante a temperatura ambiental durante la noche. Se repartió la mezcla de reacción entre EtOAc y HCl 1 M y luego se separaron las fases. Se lavó la fase orgánica secuencialmente con NaHCO3 acuoso saturado seguido por salmuera. Se secó la fase orgánica sobre Na2SO4, se filtró y se concentró a vacío. Se continuó el residuo resultante sin purificación adicional
Se trató el residuo de lo anterior con NaOMe 0,5 M en MeOH (6,83 mmol). Se agitó la mezcla heterogénea a temperatura ambiental durante 30 min. Se añadió entonces dietil éter a la mezcla de reacción y se recogió el sólido por medio de filtración a vacío, lavando con éter. Se continuó el sólido obtenido sin purificación adicional.
A un matraz que contenía el sólido de lo anterior enfriado hasta 0°C se le añadió POCh (15 ml). La mezcla se templó y se hizo burbujear. Se calentó la mezcla de reacción resultante hasta 90°C durante 3 h. Se enfrió la mezcla de reacción oscura hasta temperatura ambiental y se vertió muy lentamente en NaHCO3 acuoso saturado con agitación hasta 0°C. Se añadió entonces NaHCO3 sólido hasta que la disolución fue básica. Se extrajo la disolución acuosa con EtOAc (3x). Se secaron las fases orgánicas combinadas sobre Na2SO4, se filtraron y se concentraron a vacío. Se purificó el residuo obtenido por medio de cromatografía ultrarrápida en columna, eluyendo con 9:1/hexanos:EtOAc, para dar el producto intermedio 13 (29% a lo largo de 3 etapas). EM ESI (m/z) 281 (MH)+; 1H RMN (300 MHz, CDCls) 88,63 (d, 1H, J=1,8 Hz), 8,16 (d, 1H, J=8,4 Hz), 8,00 (dd, 1H, Ji=1,8, J2=8,4 Hz), 4,08 (s, 3H); 13C RMN (CDCla, 75MHz) 8163,4, 148,9, 148,1, 141,8, 133,3, 130,8, 130,6, 128,8, 124,3, 117,7, 112,8, 54,1.
2-Cloro-4-[(3-cloro-4-metoxibencil)amino]-6-cianoquinolina-3-carboxilato de metilo, 14
A una mezcla heterogénea de quinolina 13 (3,56 mmol) y clorhidrato de 3-cloro-4-metoxibencilamina (3,92 mmol) en
NMP (15 ml) se le añadió 'P^NEt (8,90 mmol). Se calentó la mezcla de reacción hasta 80°C durante 3 h. Posteriormente, se enfrió la mezcla de reacción hasta temperatura ambiental y se añadió HCl 1 M acuoso y EtOAc. Se separaron las fases resultantes y se lavó la fase orgánica con agua (3x) seguido por salmuera (1x). Se secaron las fases orgánicas combinadas sobre Na2SO4, se filtraron y se concentraron a vacío. Se disolvió el residuo en una cantidad mínima de EtOAc caliente y se permitió enfriar hasta temperatura ambiental. Entonces se añadió hexanos a la disolución y se precipitó el producto intermedio 14 (80% de rendimiento) y se recogió mediante filtración a vacío.
1H RMN (300 MHz, CDCls) 88,20 (s, 1H), 7,95 (d, 1H, J=8,4 Hz), 7,82 (d, 1H, J=7,5 Hz), 7,38 (s, 1H), 7,27-7,22 (m, 1H), 6,98 (d, 1H, J=8,4 Hz), 6,26 (s, 1H), 4,54 (d, 2 h J=4,8 Hz), 3,95 (s, 3H), 3,93 (s, 3H).
Ácido 2-cloro-4-[(3-cloro-4-metoxibencil)amino]-6-cianoquinolina-3-carboxílico, 15
A una disolución de éster 14 (2,20 mmol) en 1,4-dioxano (70 ml) se le añadió LiOH (4,40 mmol) y agua (20 ml). Se agitó la disolución resultante a temperatura ambiental durante la noche. Se repartió la mezcla de reacción entre éter y NaOH 1 M acuoso, se separaron las fases y se acidificó la fase acuosa hasta un pH = 2 usando HCl concentrado. Se recogió el sólido precipitado por medio de filtración a vacío para dar ácido 15 (79% de rendimiento) que se usó sin purificación adicional. 1H RMN (300 MHz, DMSO) 89,02 (s, 1H), 8,04-8,01 (m, 2 h), 7,86 (d, 1H, J=8,7 Hz), 7,44 (s, 1H), 7,28 (d, 1H, J=8,4 Hz), 7,12 (d, 1H, J= 8,4 H), 4,57 (d, 2 h, J= 5,7 Hz), 3,84 (s, 3H).
2-Cloro-4-[(3-cloro-4-metoxibencil)amino]-6-cianoquinolina-3-carboxilato de perfluorofenilo, 16
A una disolución de ácido 15 (0,833 mmol) y pentafluorofenol (1,67 mmol) en DMF (5,6 ml) y 1,4-dioxano (2,2 ml), se le añadió DCC (1,25 mmol). Se agitó la mezcla de reacción a temperatura ambiental durante la noche. Se extinguió la mezcla de reacción mediante la adición de HCl 1 M acuoso y se extrajo la fase acuosa con EtOAc (2x). Se lavaron las fases orgánicas combinadas con NaOH 1 M y salmuera. Se secaron las fases orgánicas combinadas sobre Na2SO4, se filtraron y se concentraron a vacío. Se purificó el residuo obtenido por medio de cromatografía ultrarrápida, eluyendo con 4:1/hexanos:EtOAc, para dar el éster 16 (56% de rendimiento). 1H RMN (300 MHz, CDCla) 88,32 (d, 1H, J= 1,2 Hz), 7,99 (d, 1H, J= 8,7 Hz), 7,89 (dd, 1H, Ji=1,2, Ja=8,7 Hz), 7,40 (d, 1H, J=2,4 Hz), 7,30-7,24 (m, 1H), 7,01-6,98 (m, 2 h), 4,69 (d, 2 h, J=4,8 Hz), 3,94 (s, 3H).
W-(2-Aminoetil)-2-cloro-4-[(3-cloro-4-metoxibencil)amino]-6-cianoquinolina-3-carboxamida, 17
A una disolución de éster 16 (77,6 |imol) en THF (1,5 ml) se le añadió trietiamina (0,116 mmol) seguido por etilendiamina (0,10 mmol). Se agitó la mezcla de reacción resultante a temperatura ambiental durante la noche. Se repartió la mezcla de reacción entre EtOAc y NaHCO3 acuoso saturado, se separaron las fases, y se lavó la fase orgánica con HCl 1 M (1x). Se basificó la fase acuosa ácida hasta un pH=10 mediante la adición de NaHCO3 sólido y entonces se extrajo con EtOAc (2x). Se secaron las fases orgánicas combinadas sobre Na2SO4, se filtraron y se concentraron a vacío para dar 0,012 g (35% de rendimiento) de amina 17 que se usó sin purificación adicional. EM ESI (m/z) 444 (M+H)+; 1H RMN (300 MHz, DMSO) 88,92 (s, 1H), 8,52 (t, 1H, J=5,1 Hz), 7,93 (d, 1H, J=8,7 Hz), 7,81 (s, 1H), 7,76 (d, 1H, J=8,7 Hz), 7,33 (s, 1H), 7,19 (d, 1H, J=8,4 Hz), 7,03 (d, 1H, J=8,4 Hz), 4,52 (s, 2 h), 3,75 (s, 3H), 3,10-3,01 (m, 2 h), 2,54 (t, 2 h, J=6,3 Hz).
6-[(3-Cloro-4-metoxibencil)amino]-5-oxo-2,3,4,5-tetrahidro-1H-[1,4]diazepino[5,6-b]quinolina-8-carbonitrilo, E
A una disolución de amina 17 (27,0 |imol) en NMP (1,0 ml) se le añadió TEA (40,5 |imol). Se calentó la mezcla de reacción hasta 100°C durante la noche. Se enfrió hasta temperatura ambiental y se repartió entre EtOAc y HCl 1 M. Se separaron las fases y se basificó la fase acuosa hasta un pH=10 mediante la adición de NaHCO3 sólido y luego se extrajo con EtOAc (2x). Se secaron las fases orgánicas combinadas sobre Na2SO4, se filtraron y se concentraron a vacío. Se purificó el residuo obtenido por medio de cromatografía ultrarrápida, eluyendo con 19:1/CH2Ch:MeOH, para dar el producto deseado (18% de rendimiento). EM ESI (m/z) 408 (M+H)+; 1H RMN (300 MHz, DMSO) 88,72 (s, 1H), 8,28 (s, 1H), 7,78 (s, 1H), 7,68 (dd, 1H, Ji=1,5, Ja=8,4 Hz), 7,34 (d, 1H, J=9), 7,31 (d, 1H, J=2,1 Hz), 7,19 (dd, 1H, Ji=2,1, J2=8,4 Hz), 7,07 (d, 1H, J = 8,4 Hz), 6,55 (s, 1H), 4,32 (d, 2 h, J=5,7 Hz), 3,81 (s, 3H), 3,1-3,05 (m, 4 h).
Ejemplo 6: 8-bromo-N-(3-cloro-4-metoxibencil)-1,2,3,4-tetrahidrobenzo[M1,6]naftiridin-10-amina

Se calentó una mezcla de 1 (13,9 mmol) y 1-bencilpiperidin-4-ona (13,9 mmol) en POCI3 (50 ml) a 60°C durante 6 h. Se eliminó por evaporación el exceso de POCh; se trató el residuo con H2O helada y NaOH al 10% (50 ml) y se extrajo con CH2O2 (3x50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida. Se purificó el compuesto deseado (60% de rendimiento) mediante cromatografía ultrarrápida (5% de MeOH en DCM). EM ESI (m/z) 387 (M+H)+. 1H RMN (300 MHz, CDCh) 88,32 (d, 1H, J= 1,8 Hz), 7,86 (d, 1H, J= 9,0 Hz), 7,75 (dd, 1H, Ji=2,1, J2=8,7 Hz), 7,43-7,31 (m, 5 h), 3,92 (s, 2 h), 3,82 (s, 2 h), 3,21 (t, 2 h, J= 6,0 Hz), 2,90 (t, 2 h, J= 6,0 Hz).
8-Bromo-10-cloro-1,2,3,4-tetrahidrobenzo[b][1,6]naftiridina, 19
Se añadió gota a gota cloroformiato de 1 -cloroetilo (0,39 mmol) a una disolución de 18 (0,26 mmol) en DCE (2 ml) a 0°C. Se calentó la mezcla hasta reflujo durante 2 h. Se eliminó por evaporación DCE; se disolvió el residuo en MeOH (15 ml) y se sometió a reflujo durante 1 h. Se observó la formación de un precipitado. Después de que la mezcla de reacción se enfriase hasta t.a., se recogió el precipitado mediante filtración, se repartió entre NaOH 1 N (10 ml) y AcOEt (10 ml) y se extrajo la fase acuosa dos veces con AcOEt (10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida para proporcionar el compuesto deseado 19 (77% de rendimiento). 1H RMN (300 MHz, CD3OD) 88,38 (s, 1H), 7,89 (s, 2 h), 4,36 (s, 2 h), 3,41 (t, 2 h, J=6,0 Hz), 3,21 (t, 2 h, J=6,0 Hz).
8-Bromo-10-cloro-3,4-dihidrobenzo[b][1,6]naftiridina-2(1H)-carboxilato de terc-butilo, 20
Se añadió dicarbonato de di-terc-butilo (0,7 mmol) a una disolución de 19 (0,84 mmol) en THF (5 ml) a 0°C. Se agitó la reacción a t.a. durante 1 h. Se eliminó por evaporación THF, se repartió el residuo entre DCM (25 ml) y disolución saturada de NaHCO3 (25 ml). Se extrajo la porción acuosa con DCM (2x25 ml) y se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida para obtener el producto intermedio 20 (80% de rendimiento). EM ESI (m/z) 397 (M+H)+. 1H RMN (300 MHz, CDCh) 88,34 (d, 1H, J= 2,1 Hz), 7,86 (d, 1H, J= 9,0 Hz), 7,77 (dd, 1H, Ji=2,1, J2=8,7 Hz), 4,82 (s, 2 h), 3,83 (t, 2 h, J= 6,0 Hz), 3,16 (t, 2 h, J= 6,0 Hz), 1,52 (s, 9H). 8-Bromo-10-((3-cloro-4-metoxibencil)amino)-3,4-dihidrobenzo[b][1,6]naftiridina-2(1H)-carboxilato de terc-butilo, 21 Se calentó una mezcla de 20 (0,075 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,38 mmol), TEA (0,38 mmol) y Nal (0,0037 mmol) en NMP (1 ml) hasta 130°C y se agitó durante la noche. Se diluyó la reacción con Et2O (10 ml) y se lavó con H2O (2x20 ml) y salmuera (20 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida para dar el producto intermedio 21 (40% de rendimiento). 1H RMN (300 MHz, CDCl3) 88,08 (s, 1H), 7,84 (s, 1H), 7,68 (d, 1H, J=9,0 Hz), 7,35 (s, 1H), 7,19 (d, 1H, J=8,1 Hz), 6,93 (d, 1H, J=8,1 Hz), 4,57 (s, 4 h), 3,91 (s, 3H), 3,76 (t, 2 h, J= 6,0 Hz), 3,19 (t, 2 h, J= 5,7 Hz), 1,49 (s, 9H).
8-Bromo-N-(3-cloro-4-metoxibencil)-1,2,3,4-tetrahidrobenzo[b][1,6]naftiridin-10-amina, F
Se añadió gota a gota una disolución de Et2O/HCl 2 M a una disolución de 21 en DCM:Dioxano (1:2) (1,5 ml). Se
agitó la mezcla a t.a. durante la noche. Se diluyó la mezcla de reacción con HCl 1 N y se descartó la fase orgánica. Entonces se basificó la fase acuosa usando NaHCO3 y se extrajo con DCM (3x10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida. La cromatografía ultrarrápida (DCM:MeOH 1:1) dio el producto deseado (15% de rendimiento). EM ESI (m/z) 432 (M+H)+. 1H RMN (300 MHz, CDCh) 88,04 (s, 1H), 7,84-7,73 (m, 2 h), 7,65-7,62 (m, 2 h), 7,33 (s, 1H), 7,14 (d, 1H, J=8,4 Hz), 6,89 (d, 1H, J=8,4 Hz), 4,45 (d, 2 h, J=4,5 Hz), 3,96 (s, 2 h), 3,90 (s, 3H), 3,22 (t, 2 h, J=6,0 Hz), 3,04 (t, 2 h, J=6,0 Hz).
Ejemplo 7: 10-íí3-cloro-4-metox¡benc¡l)am¡no)-1.2.3.4-tetrah¡drobenzorb1H.61naft¡r¡d¡na-8-carbon¡tr¡lo

Se calentó una mezcla de 3 (12,3 mmol) y 1-bencilpiperidin-4-ona (12,3 mmol) en POCl3 (50 ml) a 60°C durante 6 h. Se eliminó por evaporación el exceso de POCh; se trató el residuo con H2O helada y NaOH al 10% (50 ml) y se extrajo con CH2O2 (3x50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida. Se purificó el residuo mediante cromatografía ultrarrápida (Hex: AcOEt, 1:2) para dar el producto deseado (54% de rendimiento). EM ESI (m/z) 334 (M+H)+; 1H RMN (300 MHz, CDCh) 88,56 (d, 1H, J=1,8 Hz), 8,04 (d, 1H, J= 8,7 Hz), 7,82 (dd, 1H, Ji=1,8, J¿=8,7 Hz), 7,43-7,31 (m, 5 h), 3,93 (s, 2 h), 3,83 (s, 2 h), 3,26 (t, 2 h, J= 6,0 Hz), 2,92 (t, 2 h, J=6,0 Hz).
10-Cloro-1,2,3,4-tetrah¡drobenzo[b][1,6]naft¡r¡dina-8-carbon¡tr¡lo, 23
Se añadió gota a gota cloroformiato de 1 -cloroetilo (9,0 mmol) a una disolución de 22 (6,0 mmol) en DCE (15 ml) a 0°C. Se calentó la mezcla hasta reflujo durante 2 h. Se eliminó por evaporación DCE y se disolvió el residuo en MeOH (15 ml) y se sometió a reflujo durante 1 h. Se observó la formación de un precipitado. Después de que la mezcla de reacción se enfriase hasta t.a., se recogió el precipitado mediante filtración y se repartió entre NaOH 1 N (50 ml) y AcOEt (50 ml) y se extrajo la fase acuosa dos veces con AcOEt (50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida para dar el producto intermedio 23 (62% de rendimiento). EM ESI (m/z) 244 (M+H)+; 1H RMN (300 MHz, CDCla) 88,57 (d, 1H, J= 1,2 Hz), 8,04 (d, 1H, J=8,7 Hz), 7,82 (dd, 1H, Ji=1,8, J2=8,4 Hz), 4,28 (s, 2 h), 3,30 (t, 2 h, J= 6,0 Hz), 3,17 (t, 2 h, J= 6,0 Hz), 1,73 (s, 1H).
10-Cloro-8-c¡ano-3,4-d¡h¡drobenzo[b][1,6]naftir¡d¡na-2(1H)-carbox¡lato de terc-butilo, 24
Se añadió dicarbonato de di-terc-butilo (3,5 mmol) a una disolución de 23 (3,5 mmol) en DCM (10 ml) a 0°C. Se agitó la reacción a t.a. durante 1 h. Se lavó la reacción con disolución saturada de NaHCO3 (2x30 ml) y se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida para obtener el producto intermedio 24 (94% de rendimiento). EM ESI (m/z) 344 (M+H)+.
10-((3-Cloro-4-metox¡benc¡l)am¡no)-8-c¡ano-3,4-d¡hidrobenzo[b][1,6]naft¡r¡d¡na-2(1H)-carbox¡lato de terc-butilo, 25 Se calentó una mezcla de 24 (0,58 mmol), clorhidrato de 3-cloro-4metoxibencilamina (2,90 mmol), TEA (2,90 mmol) y NaI (0,03 mmol) en NMP (3 ml) hasta 130°C y se agitó durante la noche. Se diluyó la reacción con Et2O (20 ml) y se lavó con H2O (2x20 ml) y salmuera (20 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se
evaporaron a presión reducida. La cromatografía ultrarrápida (Hex:AcOEt 1:2) dio el producto intermedio deseado 25 (49% de rendimiento). EM ESI (m/z) 479 (M+H)+; 1H RMN (300 MHz, CDCl3) 88,33 (d, 1H, J=1,5 Hz), 7,94 (d, 1H, J=8,7 Hz), 7,70 (dd, 1H, Ji=1,8, J2=8,7 Hz), 7,31 (s, 1H), 7,17 (dd, 1H, Ji=2,1, J2=8,4 Hz), 6,90 (d, 1H, J=8,1 Hz), 4,60-4,55 (m, 4 h), 4,36 (s, 1H), 3,89 (2, 3H), 3,75 (t, 2 h, J=6,0Hz), 3,12 (t, 2 h, J=6,0Hz), 1,47 (s, 9H).
10-((3-Cloro-4-metoxibencil)amino)-1,2,3,4-tetrahidrobenzo[b][1,6]naftiridina-8-carbonitrilo, G
Se añadió gota a gota una disolución de Et2O/HCl 2 M a una disolución de 25 en DCM:Dioxano (1:2) (1,0 ml). Se agitó la mezcla a t.a. durante la noche. Se diluyó la mezcla de reacción con HCl 1 N y se descartó la fase orgánica. Entonces se basificó la fase acuosa usando NaHCO3 y se extrajo con DCM (3x10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida. La cromatografía ultrarrápida (DCM:MeOH 1:1) dio el producto deseado (20% de rendimiento). EM ESI (m/z) 379 (M+H)+; 1H RMN (300 MHz, CDCl3) 88,32 (s, 1H), 7,97 (d, 1H, J=9,7 Hz), 7,71 (d, 1H, J=9,0 Hz), 7,33 (d, 1H, J=1,8 Hz), 7,17 (d, 1H, J=8,4 Hz), 6,93 (d, 1H, J=8,7 Hz), 4,56 (d, 2 h, J=5,4 Hz), 4,09 (s, 1H), 3,97 (s, 2 h), 3,92 (s, 3H), 3,25 (t, 2 h, J=6,0 Hz), 3,11 (t, 2 h, J=6,0 Hz).
Ejemplo 8: 2-acet¡l-10-[(3-cloro-4-metox¡benc¡l)am¡no1-1.2.3.4-tetrah¡drobenzo[b1[1,61naft¡r¡d¡na-8-carbon¡tr¡lo

Se añadió lentamente anhídrido acético (0,41 mmol) a una disolución con agitación de 23 (0,205 mmol; preparada tal como anteriormente) en DCM (1 ml) a 0°C. Se agitó la reacción a t.a. durante 1 h. Luego se añadió agua helada a la reacción y se lavó la fase orgánica con H2O (2x5 ml) y NaHCO3 (5 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducida para dar el compuesto 26 (77% de rendimiento). EM ESI (m/z) 286 (M+H)+; 1H Rm N (300 MHz, CDCl3) 88,59 (s, 2 h), 8,12-8,06 (m, 2 h), 7,88-7,84 (m, 2 h), 5,01 (s, 2 h, 62%), 4,87 (s, 2 h, 38%), 4,01 (t, 2 h, J=6,0 Hz, 32%), 3,88 (t, 2 h, J=6,0 Hz, 68%), 3,30-3,21 (m, 4 h), 2,27-2,25 (m, 6H).
2-Acet¡l-10-[(3-cloro-4-metox¡benc¡l)am¡no]-1,2,3,4-tetrah¡drobenzo[b][1,6]naft¡rid¡na-8-carbon¡tr¡lo, H
Se calentó una mezcla de 26 (0,14 mmol), clorhidrato de 3-cloro-4metoxibencilamina (0,70 mmol), TEA (0,70 mmol) y Nal (0,007 mmol) en NMP (2 ml) hasta 130°C y se agitó durante la noche. Se diluyó la mezcla con C ^C h (10 ml) y se lavó con H2O (2x10 ml) y salmuera (10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida. La cromatografía ultrarrápida (AcOEt: MeOH 9:1) dio el producto deseado (20% de rendimiento). EM ESI (m/z) 421 (M+H)+; 1H RMN (300 MHz, CDCl3) 88,34 (s, 1H), 7,96 (d, 1H, J=8,7 Hz), 7,73 (dd, 1H, Ji=1,5, J2=9,0 Hz), 7,31 (d, 1H, J=2,1 Hz), 7,20 (dd, 1H, Ji=2,1, J2=8,4 Hz), 6,93 (d, 1H, J=8,1 Hz), 4,70 (s, 2 h), 4,64 (d, 2 h, J=6,0 Hz), 4,42 (s, 1H), 3,91 (s, 3H), 3,82 (t, 2 h, J=6,0 Hz), 3,20 (t, 2 h, J=6,0 Hz), 2,21 (s, 3H).
Ejemplo 9: ensayo de inhibición de la PDE5
Materiales y métodos: se sometió a ensayo la inhibición de la PDE5 en BPS Bioscience (San Diego, CA) usando kits de ensayo de PDE de BPS (número de catálogo de BPS 60300, lote de enzima 090810). Se proporcionaron compuestos de prueba como disoluciones líquidas de concentración 10 mM en DMSO. Se disolvió sildenafilo, usado como patrón en DMSO. Las diluciones intermedias fueron DMSO al 10% en tampón de ensayo de PDE y pruebas a intervalos de desde 0,1 hasta 100 nM. Se usó 0,1 ng de enzima por reacción, y el sustrato fue FAM-GMPc 100 nM.
Condiciones del ensayo: se preparó una serie de diluciones de los compuestos de prueba con DMSO al 10% en tampón de ensayo y 5 |il de la dilución se añadió a una reacción de 50 |il de modo que la concentración final de DMSO es del 1% en todas las reacciones. Las reacciones enzimáticas se llevaron a cabo a temperatura ambiente durante 60 minutos en una mezcla de 50 |il que contiene tampón de ensayo de PDE, FAM-AMPc 100 nM o FAM-GMPc 100 nM, una enzima PDE y el compuesto de prueba. Después de la reacción enzimática, 100 |il de una disolución de unión (dilución 1:100 del agente de unión con el diluyente de agente de unión) se añadió a cada reacción y se realizó la reacción a temperatura ambiente durante 60 minutos. Se midió intensidad de fluorescencia a una excitación de 485 nm y una emisión de 528 nm usando un lector de microplacas M1000 de Tecan Infinite.
Análisis de los datos: se realizaron ensayos de actividad de la PDE por duplicado en cada concentración. La intensidad de fluorescencia se convierte en polarización de fluorescencia usando el software Magellan6 de Tecan. Los datos de polarización de fluorescencia se analizaron usando el software informático, Graphpad Prism. Se definió la polarización de fluorescencia (FPt) en ausencia del compuesto en cada conjunto de datos como el 100% actividad. En ausencia de PDE y el compuesto, el valor de polarización fluorescente (FPb) en cada conjunto de datos se definió como el 0% de actividad. El porcentaje de actividad en presencia del compuesto se calculó según la siguiente ecuación: % de actividad = (FP-FPb)/(FPt-FPb)x100%, donde FP= la polarización de fluorescencia en presencia del compuesto.
Los valores del % de actividad frente a una serie de concentraciones del compuesto se representaron entonces gráficamente usando análisis de regresión no lineal de curva de respuesta a la dosis sigmoidea generada con la ecuación Y=B+(T-B)/1 10((LogEC50'X)xpendiente de Hill), donde Y=porcentaje de actividad, B=porcentaje mínimo de actividad, T=porcentaje máximo de actividad, X= logaritmo del compuesto y pendiente de Hill = factor de pendiente o coeficiente de Hill. El valor de CI50 se determinó mediante la concentración que provocaba un porcentaje de actividad medio máximo.
Los compuestos presentaron inhibición de la PDE5 en el intervalo nanomolar o inferior. Se muestra inhibición a modo de ejemplo de compuestos representativos en la tabla 1.
Tabla 1. Inhibición de la PDE5 y PDE6 de compuestos representativos.

Ejemplo 10: ensayo de GMPc hipocámpico
Se inyectaron ratones macho y hembra de 2-3 meses de edad (20-25 g; ratones C57Bl6) con el compuesto H (3 mg/kg y 10 mg/kg, DMSO al 2% y Tween al 2%, i.p.) o vehículo (DMSO al 2% y Tween al 2%, i.p.). 30 min después de la administración del vehículo o compuesto H, los ratones se sometieron a descarga eléctrica aplicada a las patas y se sacrificaron 10 s, 1 min y 3 min después de la descarga eléctrica mediante luxación cervical y decapitación. Se extrajeron las muestras del hipocampo y se ultracongelaron en nitrógeno líquido. Los niveles GMPc se cuantificaron mediante procedimiento de inmunoensayo enzimático (Cayman Chemical Company, n.° de artículo 581021) siguiendo las pautas del fabricante por duplicado. Se normalizaron los niveles de GMPc con la concentración de proteína calculada usando reactivo de ensayo de proteína BCA (Thermo Scientific).
Se midieron los niveles de GMPc en ratones adultos después del tratamiento con el compuesto H. En una serie de experimentos preliminares, la descarga eléctrica aplicada a las patas induce un aumento inmediato en niveles de GMPc en el hipocampo (figura 1). Se midió la concentración de GMPc mediante inmunoensayo enzimático. Basal representa niveles de GMPc sin descarga eléctrica aplicada a las patas. Los valores son la media de determinaciones duplicadas. Las barras de error muestran EEM (n=3 por grupo); *p<0,01; *p= 0,033. El compuesto H (3 mg/kg y 10 mg/kg, i.p., 30 minutos antes de la descarga eléctrica) potenció adicionalmente los niveles de GMPc a los 10 s, 60 s y 180 s (0,48 ± 0,014, 0,58 ± 0,033 y 0,58 ± 0,044 pmol/mg, respectivamente, a la concentración de 3 mg/kg; 0,56 ± 0,033, 0,70 ± 0,028 y 0,63 ± 0,013 pmol/mg, respectivamente, a la concentración de 10 mg/kg) en comparación con el vehículo (0,32 ± 0,015, 0,41 ± 0,028 y 0,46 ± 0,038 pmol/mg después de 10 s, 60 s y 3 min, respectivamente).
Ejemplo 11: estudios electrofisiológicos de la potenciación a largo plazo
Sección experimental
Se cortaron cortes transversos del hipocampo (400 |im) con un cortador de tejidos (EMS, PA) y se mantuvieron en una cámara de superficie de contacto a 29°C durante 90 min antes del registro, tal como se describe en Vitolo et al, Proc. Natl. Acad. Sci. USA 2002, 13217-13221. La disolución de baño extracelular consistió en NaCl 124,0 mM, KCl 4,4 mM, Na2HPO41,0 mM, NaHCO325,0 mM, CaCl22,0 mM, MgSO42,0 mM y glucosa 10,0 mM, se aireó continuamente con el 95% de 02/el 5% de CO2 hasta un pH final de 7,4. Se registraron las respuestas postsinápticas extracelulares de campo (fEPSP) colocando los electrodos de estimulación y registro en el estrato radiado CA1. se
usó un electrodo bipolar de tungsteno (FHC, Bowdoin, ME) como electrodo de estimulación, y se usó una pipeta de vidrio llena con disolución de baño como electrodo de registro. La transmisión sináptica basal se evaluó en primer lugar representando gráficamente los voltajes de estímulo (V) contra pendientes de fEPSP para generar relaciones de entrada-salida. Se registró en primer lugar un nivel inicial de 20-30 min cada minuto a una intensidad que suscitó una respuesta a aproximadamente el 35% de la respuesta suscitada máxima. Se indujo LTP usando una estimulación de estallido theta (4 pulsos a 100 Hz, con los estallidos repetidos a 5 Hz, y consistiendo cada contracción tetánica en 3 trenes de diez estadillos separados por 15 s). Se midieron las respuestas como pendientes de fEPSP expresadas como porcentaje de nivel inicial.
Se determinó si el compuesto H (50 nM, a través de la perfusión de baño, durante 5 min antes de la contracción tetánica) rescata el defecto en LTP en animales APP/PS1 de 3-4 meses de edad. Se encontró el 201,76% ± 14,09 de potenciación en cortes de ratones transgénicos tratados con compuesto en comparación con el 137,30% ± 10,83 de potenciación en cortes de ratones APP/PS1 tratados con vehículo (figura 2). El compuesto H rescata el defecto en LTP de cortes de APP/PS1. Potenciación residual registrada durante los últimos 5 minutos de un registro de 2 h tras la estimulación tetánica de las fibras colaterales de Schaffer en la conexión CA3-CA1. El compuesto no tuvo ningún efecto sobre cortes de WT. P<0,05 que compara cortes tratados con el compuesto frente a cortes tratados con el vehículo en ratones APP/PS1.
Estudios preliminares con sildenafilo han demostrado que la inhibición de la PDE5 tiene efectos beneficiosos prolongados sobre las anomalías sinápticas y cognitivas en ratones APP/PS1 que persiste más allá de la administración del inhibidor. Este hallazgo sugiere la posibilidad de usar estos fármacos para interferir con la progresión de los déficits de memoria. Se decidió investigar si esta importante posibilidad terapéutica se produce con el compuesto H. En estos experimentos, tanto los ratones APP/PS1 como WT de 3 meses de edad se inyectaron i.p. con 3 mg/kg/día con el compuesto H durante 3 semanas, entonces se detuvo el tratamiento durante 9-12 semanas antes de las pruebas. Cortes de ratones transgénicos tratados con el compuesto tuvieron el 226,23% ± 6,72 de potenciación en comparación con el 164,12% ± 10,37 en cortes de ratones transgénicos tratados con vehículo (figura3). Potenciación residual registrada durante los últimos 5 minutos de un registro de 2 h tras la estimulación tetánica de las fibras colaterales de Schaffer en la conexión CA3-CA1. El tratamiento diario con el compuesto H (3 mg/kg, i.p.) durante 3 semanas a la edad de 3-4 meses restableció la potenciación normal cuando se registraron cortes a los 6-7 meses de edad.
Ejemplo 12: estudios de comportamiento
Sección experimental
Se realizó la tarea de laberinto de agua de brazo radial, un hibrido del clásico laberinto de agua de Morris y el laberinto de tierra de brazo radial, tal como se describe en J. Alamed, D.M. Wilcock, D.M. Diamond, M.N. Gordon, D. Morgan, Two-day radial-arm water maze learning and memoria task; robust resolution of amyloid-related memory deficits in transgenic mice, Nat. Prot. 1 (2006) 1671-1679. El ratón tenía que nadar en 6 calles (brazos) que radiaban desde un área central hasta que encontrara una plataforma oculta (sumergida) al final de uno de los brazos, basándose en pistas visuales colocadas en la sala. El primer día del protocolo fue un día de entrenamiento en el que los ratones se entrenaron para identificar la ubicación de la plataforma alternando entre una plataforma visible y una oculta en un brazo objetivo. Los 3 ensayos finales en ese día y todos los 15 ensayos en el día 2 usaron una plataforma de escape oculta para forzar a los ratones a usar pistas espaciales para identificar la ubicación del brazo objetivo. Para evitar limitaciones de aprendizaje impuestas por la práctica agotadora y para evitar la fatiga que puede resultar de ensayos consecutivos, se estableció un entrenamiento de práctica espaciado separando los ratones en cohortes de 4 y alternando diferentes cohortes a través de los 15 ensayos de entrenamiento a lo largo de periodos de prueba de 3 horas cada día. Se contó el número de entradas de brazo incorrecto (entradas a brazos sin plataforma). Si el animal entró en el brazo incorrecto, se le retiró con cuidado al brazo de inicio. La imposibilidad de seleccionar un brazo después de 15 s se contó como error y se devolvió el ratón al brazo de inicio. Cada ensayo duró hasta 1 min. Después de 1 min, si la plataforma no se había ubicado, el ratón fue guiado con cuidado a través del agua colocando una mano detrás suya para dirigirlo hacia la plataforma. El ratón descansó sobre la plataforma durante 15 s. La ubicación de la plataforma objetivo fue diferente para cada ratón. El día 2, se repitió el mismo procedimiento que en el día 1 para los 15 ensayos usando únicamente la plataforma oculta. Para el análisis de los datos, se calcularon los promedios para cada ratón usando bloques de 3 ensayos.
Se evaluó el condicionamiento del medio tal como se describe en B. Gong, O.V. Vitolo, F. Trinchese, S. Liu, M. Shelanski, O. Arancio, Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment, J. Clin. Invest. 114 (2004) 1624-1634; F. Trinchese, S. Liu, F. Battaglia, S. Walter, P.M. Mathews, O. Arancio, Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice, Ann. Neurol. 55 (2004) 801-814; y B. Gong, Z. Cao, P. Zheng, O.V. Vitolo, S. Liu, A. Staniszewski, D. Moolman, H. Zhang, M. Shelanski, O. Arancio, Ubiquitin hidrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory, Cell 126 (2006) 775-788. En primer lugar, se examinó la percepción sensorial de descarga eléctrica aplicada a las patas en diferentes grupos de ratones a través de la prueba de evaluación umbral. Los animales se colocaron en la cámara de condicionamiento y la corriente eléctrica (0,1 mA durante 1 s) se aumentó a intervalos de 30 s desde 0,1 mA hasta 0,7 mA. El umbral de retroceso (primera respuesta visible a la descarga eléctrica), salto (primera respuesta motora extrema y respuesta vocalizada se cuantificaron para cada animal
obteniendo el promedio de la intensidad de descarga eléctrica a la que cada animal mostró la respuesta de comportamiento para ese tipo de descarga eléctrica. El entrenamiento del condicionamiento del miedo se realizó colocando el ratón en una cámara de condicionamiento durante 2 min antes de la aparición de un tono (estímulo condicionado (CS), 30 s, 85 dB sonido a 2800 Hz). En los últimos 2 s del CS, los ratones recibieron una leve descarga eléctrica aplicada a las patas de 2 s, 0,7 mA (estímulo no condicionado, (US)) a través de las barras del suelo. Después del Us , los ratones se dejaron en la cámara durante otros 30 s. El comportamiento de congelación, definido como la ausencia de movimientos a excepción de los movimientos respiratorios, se puntuó usando el software Freezeview (Med Associates, St. Albans, v T). Se evaluó el aprendizaje de miedo contextual 24 h después del entrenamiento midiendo las respuestas de congelación durante 5 min en la misma cámara en la que se entrenaron los ratones. Se evaluó el aprendizaje de miedo acondicionado 24 h después de las pruebas contextuales. Los ratones se colocaron en un contexto novedoso durante 2 min (prueba pre-CS), después del cual recibieron un CS durante 3 min (prueba de CS), y se midió el comportamiento de congelación durante los primeros 30 s que imitan el condicionamiento de CS-US y los 2,5 min restantes.
Se determinó si el compuesto H (3 mg/kg, i.p, durante 3 semanas) rescata el defecto en la memoria de referencia en animales APP/PS1 de 3-4 meses de edad. Los ratones transgénicos tratados con el compuesto cometieron entre 1 y 2 errores en el último ensayo de la prueba RAWM de 2 días en contraposición a crías transgénicas de la camada tratadas con vehículo que cometieron aproximadamente 3 errores (figura 4). El compuesto H rescata el defecto en la memoria de referencia de ratones APP/PS1. El tratamiento diario con el compuesto durante 3 semanas a la edad de 3-4 meses redujo el número de errores con el laberinto de agua de brazo radial de 2 días en ratones APP/PS1. El compuesto no tuvo ningún efecto sobre ratones WT. p<0,05 que compara ratones transgénicos tratados con el compuesto frente a ratones APP/PS1 tratados con vehículo.
También se determinó si el compuesto H (3 mg/kg, i.p, durante 3 semanas) rescata el defecto en la memoria del miedo en animales APP/PS1 de 3-4 meses de edad. Los ratones transgénicos tratados con el compuesto se congelaron ~30% del tiempo cuando se expusieron después de 24 h al mismo contexto en el que recibieron una descarga eléctrica en comparación con aproximadamente el 15% para ratones APP/PS1 tratados con vehículo (figura 5). El compuesto H rescata el defecto en la memoria del miedo de ratones APP/PS1. El tratamiento diario con el compuesto durante 3 semanas a la edad de 3-4 meses restableció la congelación normal en una prueba para la memoria del miedo contextual en ratones APP/PS1. El compuesto no tuvo ningún efecto sobre ratones WT. p<0,05 que compara ratones transgénicos tratados con el compuesto frente a ratones APP/PS1 tratados con vehículo.
Se investigó la posibilidad del compuesto H de interferir con la progresión de déficits de memoria. Tanto ratones APP/PS1 como WT de 3-4 meses de edad se inyectaron i.p. con el compuesto H (3 mg/kg/día) durante 3 semanas, entonces se detuvo el tratamiento durante 9-12 semanas antes de las pruebas. Los ratones se sometieron a continuación a RAWM de 2 días y condicionamiento del miedo. Los ratones transgénicos tratados con el compuesto cometieron ~1 error en la prueba de RAWM de 2 días en comparación con ~2 errores en ratones transgénicos tratados con vehículo (figura 6). El tratamiento diario con el compuesto H (3 mg/kg, i.p.) durante 3 semanas a la edad de 3-4 meses redujo el número de errores con el laberinto de agua de brazo radial de 2 días cuando se examinaron los ratones a los 6-7 meses de edad. Los experimentos de memoria contextual revelaron ~40% de congelación en ratones transgénicos tratados con el compuesto en comparación con ~15% en ratones transgénicos tratados con vehículo (figura 7). El tratamiento diario con el compuesto H (3 mg/kg, i.p.) durante 3 semanas a la edad de 3-4 meses restableció la congelación normal en una prueba para la memoria del miedo contextual cuando se examinaron los ratones a los 6-7 meses de edad. Por tanto, las mejoras sinápticas y cognitivas persisten más allá de la administración del inhibidor.
Ejemplo 13: estudios farmacocinéticos
El estudio PK del compuesto H (25mg/kg, i.p.) se llevó a cabo en ratones macho C57Bl6. Se muestran concentraciones en plasma y en cerebro del compuesto H en diferentes puntos temporales en la figura 8.
Tras la administración i.p. de 25 mg/kg a ratones C57Bl6, el compuesto H se absorbió rápidamente tal como se indica mediante la concentración en plasma máxima que se produce a las 0,25 h después de la dosificación (tabla 2). La distribución del compuesto H al cerebro fue rápida con un valor de Tmáx similar en el cerebro. La exposición del compuesto H en el cerebro fue menor que en plasma con una razón de Cmáx de 0,31. Las semividas de eliminación del compuesto H en el cerebro y el plasma fueron de 0,65 y 1,07 h, respectivamente.
Tabla 2. Parámetros farmacocinéticos para el compuesto H en ratones.

Cmáx: la concentración máxima observada; Tmáx: tiempo correspondiente a Cmáxi
MRT: tiempo medio de residencia; t-i^: la semivida de eliminación.Ejemplo 14: estudios de toxicidad aguda
Los ratones recibieron el compuesto H a dos concentraciones diferentes (120 ó 240 mg/kg, i.p.) o vehículo, y se sacrificaron después de eso (el compuesto H a una concentración de 480 mg/kg, i.p. fue letal, sin embargo, la dosis eficaz del compuesto H fue de 3 mg/kg). Se recogieron el cerebro, músculo, riñón e hígado de cada ratón, y un patólogo acreditado examinó histológicamente tejido de cada órgano para determinar signos de toxicidad. Las secciones de cerebro no mostraron ningún signo de toxicidad mediante H&E, y una inmunotinción con GFAP no mostró signos de gliosis (figura 9). Tinciones adicionales en secciones de cerebro con LFB-PAS no revelaron signos de desmielinización, y una inmunotinción para CD45 no mostró signos de inflamación. Las secciones de músculo no mostraron signos de toxicidad mediante H&E, y una tinción tricrómica no mostró fibrosis muscular. Las secciones de riñón no mostraron toxicidad mediante tinción con H&E. La tinción adicional de secciones de riñón con PAS no mostró ninguna anomalía en la membrana basal glomerular, y la tinción tricrómica no mostró fibrosis. Las secciones de hígado no mostraron ningún signo de toxicidad mediante tinción con H&E, y una tinción tricrómica no mostró fibrosis o enfermedad de hígado cirrótico. El compuesto no mostró signos de neurotoxicidad en el cerebro. La tinción con H&E no muestra diferencia entre los tres grupos (figura 9A: vehículo; figura 9B: 120 mg/kg; figura 9C: 240 mg/kg). Además, una inmunotinción con GFAP no muestra signos de gliosis en ninguno de los tres grupos (figura 9D: vehículo; figura 9E: 120 mg/kg, figura 9F: 240 mg/kg).
Ejemplo 15: 1-(10-((3-cloro-4-metoxibencil)amino)-3,4-dihidrobenzo[;b1[1,61naftiridin-2(1H)-il)etan-1-ona

Se calentó una mezcla de ácido 2-aminobenzoico 27 (7,3 mmol) y 1-metilpiperidin-4-ona (7,3 mmol) en POCh (5 ml) a 60°C durante 6 h. Se eliminó por evaporación el POCh en exceso; se trató el residuo con H2O helada y NaHCO3 y se extrajo con CH2Ch (3x25 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. La purificación mediante cromatografía ultrarrápida (5% de MeOH en AcOEt) dio el compuesto deseado 28. EM ESI (m/z) 309 (M+H)+; 1H RMN (400 MHz, CDCla) 88,16 (dd, 1H, Ji=1,2, J2=8,4 Hz), 8,0 (d, 1H, J=8,4 Hz), 7,71-7,67 (m, 1H), 7,58-7,54 (m, 1H), 7,44-7,42 (m, 2 h), 7,39-7,35 (m, 2 h), 7,33-7,30 (m, 1H), 3,95 (s, 2 h), 3,84 (s, 2 h), 3,25 (t, 2 h, J=5,6 Hz), 2,93 (t, 2 h, J=5,6 Hz).

10-Cloro-1,2,3,4-tetrah¡drobenzo[6][1,6]naftirid¡na 29
Se añadió gota a gota cloroformiato de 1 -cloroetilo (6,3 mmol) a una disolución de 28 (4,2 mmol) en DCE (5 ml) a 0°C. Se calentó la mezcla hasta reflujo durante 2 h. Se eliminó por evaporación DCE y se disolvió el residuo en MeOH (5 ml) y se sometió a reflujo durante 1 h. Se observó la formación de un precipitado. Después de que la mezcla de reacción se enfriase hasta t.a., se recogió el precipitado mediante filtración y se repartió entre NaOH 1 N (50 ml) y AcOEt (50 ml) y se extrajo la fase acuosa dos veces con AcOEt (50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida para dar 29. EM ESI (m/z) 219 (M+H)+.

1-(10-Cloro-3,4-dih¡drobenzo[6][1,6]naft¡rid¡n-2(1H)-¡l)etan-1-ona 30
Se añadió lentamente anhídrido acético (3,65 mmol) a una disolución con agitación de 29 (1,83 mmol) en DCM (4 ml) a 0°C. Se agitó la reacción a t.a. durante 1 h. Luego se añadió agua helada a la reacción y se lavó la fase orgánica con H2O (2x10 ml) y NaHCO3 (10 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducida para dar el compuesto 30.

1-(10-((3-Cloro-4-metox¡benc¡l)am¡no)-3,4-d¡h¡drobenzo[;b][1,6]naft¡r¡d¡n-2(1H)-¡l)etan-1-ona, I
Se calentó una disolución de 30 (1,15 mmol), clorhidrato de 3-cloro-4-metox¡benc¡lam¡na (3,45 mmol), Nal (0,06 mmol) y trietilamina (3,45 mmol) en NMP (5,0 ml) a 130°C durante 6 h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante cromatografía ultrarrápida (AcOEt: MeOH 8:2) para dar I. EM ESI (m/z) 396 (M+H)+; 1H RMN (400 MHz, CDCla) 88,05-8,02 (m, 1H), 7,92 (d, 1H, J=9,2 Hz), 7,67-7,63 (m, 1H), 7,45-7,40 (m, 1H), 7,35 (d, 1H, J=2,4 Hz), 7,22 (dd, 1H, Ji=2,0, J2=8,4 Hz), 6,92 (d, 1H, J=8,4 Hz), 4,75 (s, 2 h), 4,65 (d, 2 h, J=5,2 Hz), 3,90 (s, 4 h), 3,81 (t, 2 h, J=6,0 Hz), 3,27 (t, 2 h, J=6,4 Hz), 2,20 (s, 3H).
Ejemplo 16: 1-í10-íí3-cloro-4-metox¡benc¡l)am¡no)-8-metox¡-3.4-d¡h¡drobenzo^M1.61naft¡r¡d¡n-2í1H)-¡l)etan-1-ona

2-Benc¡l-10-cloro-8-metox¡-1,2,3,4-tetrah¡drobenzo[6][1,6]naft¡r¡d¡na 62
Se calentó una mezcla de ácido 2-amino-5-metoxibenzoico 61 (1,2 mmol) y 1-met¡lp¡per¡d¡n-4-ona (1,2 mmol) en POCla (2,0 ml) a 60°C durante 3 h. Se eliminó por evaporación el POCl3 en exceso; se trató el residuo con NH3 ac. helado y se extrajo con AcOEt (3x 20 ml). Se secó la fase orgánica sobre Na2SO4, se filtró y se evaporó a presión reducida. La purificación mediante cromatografía ultrarrápida (AcOEt) dio el compuesto deseado 62. EM ESl (m/z) 339 (M+H)+; 1H RMN (400 MHz, CDCh) 87,86 (d, 1H, J=9,2 Hz), 7,41-7,23 (m, 7H), 3,93 (s, 3H), 3,90 (s, 2 h), 3,80 (s, 2 h), 3,18 (t, 2 h, J=6,0 Hz), 2,87 (t, 2 h, J=5,6 Hz).

10-Cloro-8-metoxM,2,3,4-tetrah¡drobenzo[6][1,6]naftmd¡na 63
Se añadió gota a gota cloroformiato de 1 -cloroetilo (0,62 mmol) a una disolución de 62 (0,41 mmol) en DCE (3,0 ml) a 0°C. Se calentó la mezcla hasta reflujo durante 2 h. Se eliminó por evaporación DCE y se disolvió el residuo en MeOH (5,0 ml) y se sometió a reflujo durante 1 h. Se observó la formación de un precipitado. Después de que la mezcla de reacción se enfriase hasta t.a., se recogió el precipitado mediante filtración y se repartió entre NaOH 1 N (20 ml) y AcOEt (20 ml) y se extrajo la fase acuosa dos veces con AcOEt (20 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducida para dar 63. Em ESI (m/z) 249 (M+H)+; 1H RMN (400 MHz, CDCla) 87,89 (d, 1H, J=8,8 Hz), 7,39 (d, 1H, J=2,8 Hz), 7,34 (dd, 1H, Ji=2,4, J2=8,8 Hz), 4,25 (s, 2 h), 3,96 (s, 3H), 3,28 (t, 2 h, J=6,0 Hz), 3,11 (t, 2 h, J=6,4 Hz).

1-(10-Cloro-8-metox¡-3,4-d¡h¡drobenzo[;b][1,6]naft¡r¡d¡n-2(1H)-¡l)etan-1-ona 64
Se añadió lentamente anhídrido acético (0,56 mmol) a una disolución con agitación de 63 (0,28 mmol) en DCM (1,0 ml) a 0°C. Se agitó la reacción a t.a. durante 1 h. Luego se añadió agua helada a la reacción y se lavó la fase orgánica con H2O (2x 10 ml) y NaHCO3 (10 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducida para dar el compuesto 64. EM ESI (m/z) 291 (M+H)+; 1H RMN (400 MHz, CDCla) 87,91 (d, 1H, J=9,2 Hz), 7,42-7,40 (m, 1H), 7,39-7,36 (m, 1H), 4,98 (s, 2 h), 3,97 (s, 3H), 3,86 (t, 2 h, J=6,0 Hz), 3,22 (t, 2 h, J=6,0 Hz), 2,25 (s, 3H).

1-(10-((3-Cloro-4-metox¡benc¡l)am¡no)-8-metox¡-3,4-d¡h¡drobenzo[b][1,6]naft¡r¡d¡n-2(1H)-¡l)etan-1-ona J
Se calentó una disolución de 64 (0,15 mmol), clorhidrato de 3-cloro-4-metox¡benc¡lam¡na (0,69 mmol), Nal (0,007 mmol) y trietilamina (0,69 mmol) en NMP (3,0 ml) a 130°C durante 6 h. Se eliminó el disolvente por evaporación y Se purificó el residuo mediante cromatografía ultrarrápida (AcOEt: MeOH 8:2) para dar el producto J. EM ESI (m/z) 426 (M+H)+. 1H RMN (400 MHz, CDCls) 87,86 (d, 1H, J=8,8 Hz), 7,37 (d, 1H, J=2,0 Hz), 7,29 (dd, 1H, Ji=2,8, J2=9,6 Hz), 7,23 (dd, 1H, Ji=2,4, J2=8,0 Hz), 7,15 (d, 1H, J=2,8 Hz), 6,91 (d, 1H, J=8,4 Hz), 4,73 (s, 2 h), 4,53 (d, 2 h, J=5,6 Hz), 3,90 (s, 4 h), 3,80 (t, 2 h, J=6,4 Hz), 3,75 (s, 3H), 3,18 (t, 2 h, J=6,0 Hz), 2,21 (s, 3H).
Puede usarse el siguiente esquema general para preparar compuestos adicionales según determinados aspectos de la invención:

2,4-Dioxo-1,4-dihidro-2 h-benzo[d][1,3]oxazina-6-carbonitrilo 33
Se añadió (C^CO^CO (3,08 mmol) a una suspensión de ácido 2-amino-5-cianobenzoico 3 (9,25 mmol) en 1,4-dioxano a 0°C. Se calentó la mezcla de reacción homogénea hasta 90°C durante 5 h y luego se enfrió. Se aisló el precipitado resultante 33 mediante filtración. 1H RMN (400 MHz, DMSO-d6) 812,15 (s, 1H), 8,38 (d, 1H, J=1,6 Hz), 8,11 (dd, 1H, J í=1,6, J2=8,8 Hz), 7,25 (d, 1H, J=8,4 Hz)

6-Ciano-4-hidroxi-2-metil-1,4-dihidroquinolina-3-carboxilato de metilo 34
Se añadió en porciones NaH (6,38 mmol) a una disolución de acetoacetato de metilo (6,38 mmol) en DMA (2 ml). Se añadió el compuesto 33 (5,32 mmol) y se agitó la mezcla de reacción hasta 120°C durante 30 min. Se redujo el
disolvente y se añadió agua. Se recogió el precipitado resultante 34 mediante filtración. 1H RMN (400 MHz, DMSO-d6) 812,25 (s, 1H), 8,39 (d, 1H, J=2,0 Hz), 8,01 (dd, 1H, Ji=2,0, J2=8,8 Hz), 7,66 (d, 1H, J=8,8 Hz), 3,70 (s, 3H), 2,41 (s, 3H)

4-Cloro-6-ciano-2-metilquinolina-3-carboxilato de metilo 35
Se suspendió el compuesto 34 (2,0 mmol) en POCh (4 ml) y se calentó hasta 110°C durante 20 min. Se vertió lentamente la mezcla de reacción homogénea en NH3 ac. helado. Se extrajo la fase acuosa con AcOEt (3x 50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir para dar el compuesto desear 35. EM ESI (m/z) 261 (M+H)+; 1H RMN (400 MHz, CDCls) 88,61 (d, 1H, J=2,0 Hz), 8,12 (d, 1H, J=8,8 Hz), 7,92 (dd, 1H, Ji=1,6, J2=8,4 Hz), 4,05 (s, 3H), 2,75 (s, 3H).

2-(Bromometil)-4-cloro-6-cianoquinolina-3-carboxilato de metilo 36
Se calentó una disolución de 35 (0,14 mmol), N-bromosuccinimida (0,22 mmol) y 2,2'-azobis(2-metilpropionitrilo) (0,03 mmol) en CCU (1 ml) hasta reflujo durante 4 h. Se enfrió la mezcla de reacción y se eliminó el sólido por filtración. Se concentró el filtrado y se purificó el residuo mediante cromatografía ultrarrápida (hexanos: AcOEt 9:1) para dar el producto deseado 36.

9-Cloro-2-etil-1-oxo-2,3-dihidro-1H-pirrolo[3,4-b]quinolina-7-carbonitrilo 37
A una disolución de 36 (0,15 mmol) en etanol se le añadió EtNH2 (2 M en THF, 0,46 mmol). Se calentó la mezcla de reacción hasta reflujo durante 2 h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante cromatografía ultrarrápida (Hexanos:AcOEt 3:7) para dar el producto deseado 37. e M ESI (m/z) 272 (M+H)+; 1H RMN (400 MHz, CDCla) 88,84 (d, 1H, J=2,0 Hz), 8,23 (d, 1H, J=1,4 Hz), 7,99 (dd, 1H, Ji=1,6, J2=8,8 Hz), 4,57 (s, 2 h), 3,78 (q, 2 h, J=7,2 Hz), 1,35 (t, 3H, J=6,8 Hz)
9-[(3-Cloro-4-metoxibencil)amino]-2-etil-1-oxo-2,3-dihidro-1H-pirrolo[3,4-b]quinolina-7-carbonitrilo, K Se calentó una disolución de 37 (0,037 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,055 mmol) y ('pr)2NEt (0,22 mmol) en n-propanol (1 ml) hasta 90°C durante 1 h. Después de enfriar, el precipitado resultante K se recogió mediante filtración. EM ESI (m/z) 407 (M+H)+; 1H RMN (400 MHz, CDCls) 88,58 (t, 1H, J=6,0 Hz), 8,53 (d, 1H, J=1,6 Hz), 7,94 (d, 1H, J=9,2 Hz), 7,75 (dd, 1H, Ji=1,6, J2=8,4 Hz), 7,42 (d, 1H, J=1,6 Hz), 7,30 (dd, 1H, Ji=2,4, J2=8,4 Hz), 6,96 (d, 1H, J=8,4 Hz), 4,93 (d, 2 h, J=5,6 Hz), 4,38 (s, 2 h), 3,89 (s, 3H), 3,63 (q, 2 h, J=7,2 Hz), 1,27 (t, 3H, J=7,2 Hz)
Ejemplo 18: 9-((3-cloro-4-metox¡benc¡l)am¡no)-2-et¡l-7-metox¡-2.3-d¡h¡dro-1H-p¡rrolo[3,4-b1qu¡nol¡n-1-ona

6-Metoxi-2 h-benzo[d][1,3]oxazina-2,4(1H)-diona 39
Se añadió (Cl3CO)2CO (1,0 mmol) a una suspensión de ácido 2-amino-5-metoxibenzoico 38 (3,0 mmol) en 1,4-dioxano (3 ml) a 0°C. Se calentó la mezcla de reacción homogénea hasta 90°C durante 2 h y luego se enfrió. Se aisló el precipitado resultante 39 mediante filtración. EM ESI (m/z) 192 (M-1); 1H RMN (400 MHz, DMSO-d6) 811,61 (s, 1H), 7,38 (dd, 1H, Ji=2,8, J2=8,4 Hz), 7,34 (d, 1H, J=2,4 Hz), 7,11 (d, 1H, J=8,4 Hz), 3,80 (s, 3H)

6-Metoxi-2-metil-4-oxo-1,4-dihidroquinolina-3-carboxilato de metilo 40
Se añadió en porciones NaH (3,1 mmol) a una disolución de acetoacetato de metilo (3,1 mmol) en DMA (3 ml). Se añadió el compuesto 39 (2,6 mmol) y se agitó la mezcla de reacción hasta 120°C durante 30 min. Se redujo el disolvente y se añadió agua. Se recogió el precipitado resultante 40 mediante filtración. 1H RMN (400 MHz, DMSO-d6) 811,87 (s, 1H), 7,49 (d, 1H, J=8,8 Hz), 7,45 (d, 1H, J=2,8 Hz), 7,31 (dd, 1H, Ji=3,2, J2=8,8 Hz), 3,82 (s, 3H), 3,75 (s, 3H), 2,37 (s, 3H)

4-Cloro-6-metoxi-2-metilquinolina-3-carboxilato de metilo 41
Se suspendió el compuesto 40 (1,62 mmol) en POCl3 (3 ml) y se calentó hasta 110°C durante 30 min. Se vertió lentamente la mezcla de reacción homogénea en NH3 ac. helado. Se extrajo la fase acuosa con AcOEt (3x 50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir para dar el compuesto desear 41. 1H RMN (400 MHz, CDCla) 87,93 (d, 1H, J=9,6 Hz), 7,43-7,41 (m, 2 h), 4,03 (s, 3H), 3,97 (s, 3H), 2,67 (s, 3H).

2-(Bromometil)-4-doro-6-metoxiquinolina-3-carboxilato de metilo 42
Se calentó una disolución de 41 (1,47 mmol), W-bromosuccinimida (2,20 mmol) y 2,2'-azobis(2-metilpropionitrilo) (0,3 mmol) en CCU (3 ml) hasta reflujo durante 5 h. Se enfrió la mezcla de reacción y se eliminó el sólido por filtración. Se concentró el filtrado y Se purificó el residuo mediante cromatografía ultrarrápida (hexanos: AcOEt 9:1) para dar el producto deseado 42. 1H RMN (400 MHz, CDCls) 87,98 (d, 1H, J=10,4 Hz), 7,47-7,44 (m, 2 h), 4,77 (s, 2 h), 4,07 (s, 3H), 3,99 (s, 3H).

9-Cloro-2-etil-7-metoxi-2,3-dihidro-1H-pirrolo[3,4-b]quinolin-1-ona 43
A una disolución de 42 (0,17 mmol) en etanol (1 ml) se le añadió EtNH2 (2 M en THF, 0,52 mmol). Se calentó la mezcla de reacción hasta reflujo durante 3h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante cromatografía ultrarrápida (Hexanos: AcOEt 3:7) para dar el producto deseado 43. 1H RMN (400 MHz, CDCla) 88,01 (d, 1H, J=8,8 Hz), 7,64 (d, 1H, J=2,8 Hz), 7,50 (dd, 1H, Ji=2,8, J2=9,6 Hz), 4,49 (s, 2 h), 4,01 (s, 3H), 3,76 (q, 2 h, J=7,2 Hz), 1,33 (t, 3H, J=7,2 Hz)

9-((3-Cloro-4-metoxibencil)amino)-2-etil-7-metoxi-2,3-dihidro-1H-pirrolo[3,4-b]quinolin-1-ona L
Se calentó una disolución de 43 (0,11 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,16 mmol) y ('pr)2NEt (0,66 mmol) en n-propanol (1,5 ml) hasta 90°C durante 1 h. Se eliminó el disolvente por evaporación y Se purificó el residuo mediante cromatografía ultrarrápida (AcOEt: MeOH 19:1) para dar el producto final L. EM ESI (m/z) 412 (M+H)+; 1H RMN (400 MHz, CDCh) 88,14 (t, 1H, J=6,4 Hz), 7,85 (d, 1H, J=9,6 Hz), 7,48 (d, 1H, J=2,4 Hz), 7,37-7,29 (m, 3H), 6,95 (d, 1H, J=8,8 Hz), 4,95 (d, 2 h, J=6,8 Hz), 4,38 (s, 2 h), 3,90 (s, 3H), 3,66 (q, 2 h, J=7,2 Hz), 3,59 (s, 3H), 1,28 (t, 3H, J=7,2 Hz)
Ejemplo 19: 9-[(3-cloro-4-metox¡benc¡l)am¡no1-2-et¡l-7-(tr¡fluoromet¡l)-2.3-d¡h¡dro-1H-p¡rrolo[3,4-b1qu¡nol¡n-1-ona
6-(Trifluorometil)-2H-benzo[d][1,3]oxazina-2,4(1H)-diona 45
Se añadió (C^CO^CO (1,3 mmol) a una suspensión de ácido 2-amino-5-(trifluorometil)benzoico 44 (3,9 mmol) en 1,4-dioxano (3 ml) a 0°C. Se calentó la mezcla de reacción homogénea hasta 90°C durante 2 h y luego se enfrió. Se aisló el precipitado resultante 45 mediante filtración. 1H RMN (400 MHz, DMSO-d6) 812,09 (s, 1H), 8,14 (d, 1H, J=1,2 Hz), 8,07 (dd, 1H, Ji=1,6, J2=8,8 Hz), 7,32 (d, 1H, J=8,8 Hz)

2-Metil-4-oxo-6-(trifluorometil)-1,4-dihidroquinolina-3-carboxilato de metilo 46
Se añadió en porciones NaH (3,1 mmol) a una disolución de acetoacetato de metilo (3,1 mmol) en DMA (2 ml). Se añadió el compuesto 45 (2,6 mmol) y se agitó la mezcla de reacción hasta 120°C durante 30 min. Se redujo el disolvente y se añadió agua. Se recogió el precipitado resultante 46 mediante filtración. EM ESI (m/z) 286 (M+H)+.

4-Cloro-2-metil-6-(trifluorometil)quinolina-3-carboxilato de metilo 47
Se suspendió el compuesto 46 (1,6 mmol) en POCl3 (1 ml) y se calentó hasta 110°C durante 20 min. Se vertió lentamente la mezcla de reacción homogénea en NH3 ac. helado. Se extrajo la fase acuosa con AcOEt (3x 50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir para dar el compuesto desear 47. EM ESI (m/z) 304 (M+H)+.
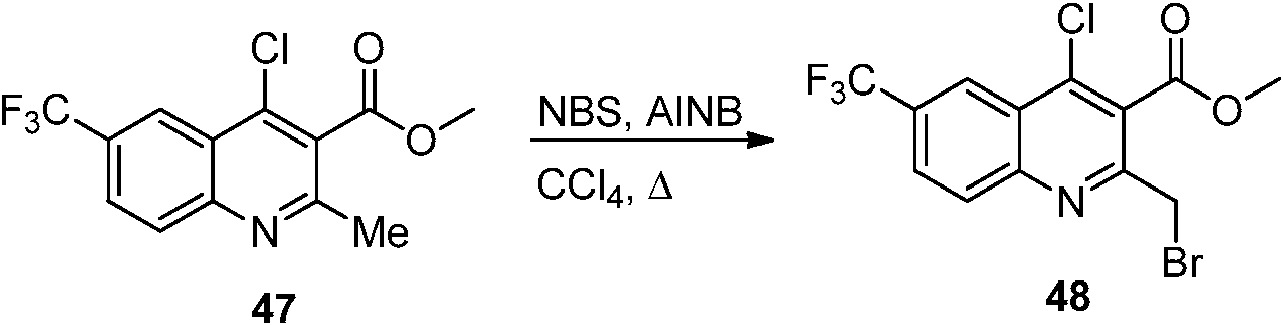
2-(Bromometil)-4-cloro-6-(trifluorometil)quinolina-3-carboxilato de metilo 48
Se calentó una disolución de 47 (0,43 mmol), N-bromosuccinimida (0,64 mmol) y 2,2'-azobis(2-metilpropionitrilo) (0,03 mmol) en CCU (2,0 ml) hasta reflujo durante 4 h. Se enfrió la mezcla de reacción y se eliminó el sólido por filtración. Se concentró el filtrado y se purificó el residuo mediante cromatografía ultrarrápida (hexanos: AcOEt 9:1) para dar el producto deseado 48. EM ESI (m/z) 384 (M+H)+; 1H RMN (400 MHz, CDCh) 88,58 (d, 1H, J=1,2 Hz), 8,22 (d, 1H, J=8,8 Hz), 8,01 (dd, 1H, Ji=2,0, J2=8,8 Hz), 4,77 (s, 2 h), 4,06 (s, 3H).

9-Cloro-2-etil-7-(trifluorometil)-2,3-dihidro-1H-pirrolo[3,4-b]quinolin-1-ona 49
A una disolución de 48 (0,21 mmol) en etanol se le añadió EtNH2 (2 M en THF, 0,63 mmol). Se calentó la mezcla de reacción hasta reflujo durante 2,5 h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante
cromatografía ultrarrápida (Hexanos:AcOEt 1:1) para dar el producto deseado 49. EM ESI (m/z) 315 (M+H)+; 1H RMN (400 MHz, CDCl3) 88,76 (s, 1H), 8,26 (d, 1H, J=8,8 Hz), 8,03 (dd, 1H, Ji=2,0, J2=8,8 Hz), 4,57 (s, 2 h), 3,79 (q, 2 h, J=6,8 Hz), 1,35 (t, 3H, J=6,8 Hz)

9-[(3-Cloro-4-metoxibencil)amino]-2-etil-7-(trifluorometil)-2,3-dihidro-1H-pirrolo[3,4-b]quinolin-1-ona M
Se calentó una disolución de 49 (0,095 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,14 mmol) y ('pr)2NEt (0,57 mmol) en n-propanol (1,0 ml) hasta 90°C durante 1 h. Se eliminó el disolvente por evaporación y Se purificó el residuo mediante cromatografía ultrarrápida (AcOEt) para dar el producto final M. EM ESI (m/z) 450 (M+H)+; 1H RMN (400 MHz, CDCla) 88,58 (t, 1H, J=6,0 Hz), 8,46 (s, 1H), 8,0 (d, 1H, J=8,8 Hz), 7,80 (dd, 1H, Ji=2,0, J2=9,2 Hz), 7,45 (d, 1H, J=2,0 Hz), 7,32 (dd, 1H, Ji=2,4, J2=8,4 Hz), 6,95 (d, 1H, J=8,0 Hz), 4,95 (d, 2 h, J=6,0 Hz), 4,41 (s, 2 h), 3,90 (s, 3H), 3,67 (q, 2 h, J=7,2 Hz), 1,3 (t, 3H, J=7,2 Hz)
Ejemplo 20: 7-cloro-9-[(3-cloro-4-metoxibencil)amino1-2-etil-2,3-dihidro-1H-pirrolo[3,4-¿)1quinolin-1-ona

6-Cloro-2-metil-4-oxo-1,4-dihidroquinolina-3-carboxilato de metilo 51
Se añadió en porciones NaH (9,1 mmol) a una disolución de acetoacetato de metilo (9,1 mmol) en DMA (2 ml). Se añadió el compuesto 50 (7,6 mmol) y se agitó la mezcla de reacción hasta 120°C durante 30 min. Se redujo el disolvente y se añadió agua. Se recogió el precipitado resultante 51 mediante filtración. 1H RMN (400 MHz, DMSO-d6) 812,07 (s, 1H), 7,98 (d, 1H, J=2,4 Hz), 7,71 (dd, 1H, Ji=2,4, J2=8,4 Hz), 7,57 (d, 1H, J=8,8 Hz), 3,76 (s, 3H), 2,39 (s, 3H).

4,6-Dicloro-2-metilquinolina-3-carboxilato de metilo 52
Se suspendió el compuesto 51 (3,0 mmol) en POCl3 (4 ml) y se calentó hasta 110°C durante 20 min. Se vertió lentamente la mezcla de reacción homogénea en NH3 ac. helado. Se extrajo la fase acuosa con AcOEt (3x 50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir para dar el compuesto desear 52. 1H RMN (400 MHz, DMSO-d6) 88,21 (d, 1H, J=2,4 Hz), 8,08 (d, 1H, J=9,2 Hz), 7,95 (dd, 1H, Ji=2,4, J2=8,8 Hz), 4,01 (s, 3H), 2,63 (s, 3H).

2-(Bromometil)-4,6-didoroquinolina-3-carboxilato de metilo 53
Se calentó una disolución de 52 (1,5 mmol), W-bromosuccinimida (3,0 mmol) y 2,2'-azobis(2-metilpropionitrilo) (0,3 mmol) en CCU (1 ml) hasta reflujo durante 4 h. Se enfrió la mezcla de reacción y se eliminó el sólido por filtración. Se concentró el filtrado y se purificó el residuo mediante cromatografía ultrarrápida (hexanos: AcOEt 2:1) para dar el producto deseado 53. 1H RMN (400 MHz, CDCls) 88,26 (d, 1H, J=2,0 Hz), 8,04 (d, 1H, J=8,4 Hz), 7,78 (dd, 1H, Ji=2,4, J2=8,8 Hz), 4,78 (s, 2 h), 4,08 (s, 3H).

7,9-Dicloro-2-etil-2,3-dihidro-1H-pirrolo[3,4-£)1quinolin-1-ona 54
A una disolución de 53 (0,41 mmol) en etanol se le añadió EtNH2 (2 M en THF, 1,24 mmol). Se calentó la mezcla de reacción hasta reflujo durante 2 h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante cromatografía ultrarrápida (Hexanos: AcOEt 3:7) para dar el producto deseado 54. EM ESI (m/z) 281 (M+H)+; 1H RMN (400 MHz, CDCla) 88,43 (d, 1H, J=2,0 Hz), 8,07 (d, 1H, J=8,8 Hz), 7,79 (dd, 1H, Ji=2,4, J2=8,8 Hz), 4,52 (s, 2 h), 3,78 (q, 2 h, J=7,6 Hz), 1,33 (t, 3H, J=7,6 Hz).

7-Cloro-9-[(3-cloro-4-metoxibencil)amino]-2-etil-2,3-dihidro-1H-pirrolo[3,4-6]quinolin-1-ona N
Se calentó una disolución de 54 (0,18 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,27 mmol) y ('pr)2NEt (1,08 mmol) en n-propanol (1 ml) hasta 90°C durante 2 h. Después de enfriar, el precipitado resultante N se recogió mediante filtración. EM ESI (m/z) 416 (M+H)+; 1H RMN (400 MHz, CDCh) 88,29 (t, 1H, J=5,6 Hz), 8,14 (d, 1H, J=2,0 Hz), 7,86 (d, 1H, J=9,2 Hz), 7,58 (dd, 1H, Ji=2,0, J2=9,2 Hz), 7,45 (d, 1H, J=1,2 Hz), 7,32 (d, 1H, J=8,8 Hz), 6,95 (d, 1H, J=8,4 Hz), 4,93 (d, 2 h, J=6,0 Hz), 4,37 (s, 2 h), 3,91 (s, 3H), 3,64 (q, 2 h, J=7,6 Hz), 1,28 (t, 3H, J=7,2 Hz). Ejemplo 21: 9-[(3-cloro-4-metox¡benc¡l)am¡no1-2-et¡l-2.3-d¡h¡dro-1H-p¡rrolo[3,4-b1qu¡nol¡n-1-ona
2-Metil-4-oxo-1,4-dihidroquinolina-3-carboxilato de metilo 56
Se añadió en porciones NaH (7,35 mmol) a una disolución de acetoacetato de metilo (7,35 mmol) en DMA (2,0 ml). Se añadió anhídrido W-metilisatoico 55 (6,13 mmol) y se agitó la mezcla de reacción hasta 120°C durante 30 min. Se redujo el disolvente y se añadió agua. Se recogió el precipitado resultante 56 mediante filtración. EM ESI (m/z) 218 (M+H)+; 1H RMN (400 MHz, DMSO-d6) 811,88 (s, 1H), 8,05 (d, 1H, J=8,0 Hz), 7,69-7,65 (m, 1H), 7,53 (d, 1H, J=8,4 Hz), 7,36 (m, 1H), 3,76 (s, 3H), 2,39 (s, 3H).

4-Cloro-2-metilquinolina-3-carboxilato de metilo 57
Se suspendió el compuesto 56 (3,9 mmol) en POCl3 (3,0 ml) y se calentó hasta 110°C durante 30 min. Se vertió lentamente la mezcla de reacción homogénea en NH3 ac. helado. Se extrajo la fase acuosa con AcOEt (3x 50 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir para dar el compuesto desear 57. EM ESI (m/z) 236 (M+H)+; 1H RMN (400 MHz, CDCla) 88,4 (d, 1H, J=8,4 Hz), 8,03 (d, 1H, J=8,4 Hz), 7,79 (td, 1H, Ji=1,2, J2=7,2), 7,63 (t, 1H, J=8,4), 4,04 (s, 3H), 2,72 (s, 3H).

2-(Bromometil)-4-cloroquinolina-3-carboxilato de metilo 58
Se calentó una disolución de 57 (3,44 mmol), W-bromosuccinimida (5,15 mmol) y 2,2'-azobis(2-metilpropionitrilo) (0,7 mmol) en CCU (4,0 ml) hasta reflujo durante 4 h. Se enfrió la mezcla de reacción y se eliminó el sólido por filtración. Se concentró el filtrado y se purificó el residuo mediante cromatografía ultrarrápida (hexanos: AcOEt 2:1) para dar el producto deseado 58. EM ESI (m/z) 315 (M+H)+; 1H RMN (400 MHz, CDCla) 88,28 (dd, 1H, Ji=1,2, J2=8,0 Hz), 8,09 (d, 1H, J=8,0 Hz), 7,86-7,82 (m, 1H), 7,73-7,68 (m, 1H), 4,80 (s, 2 h), 4,08 (s, 3H).

9-Cloro-2-etil-2,3-dihidro-1H-pirrolo[3,4-6]quinolin-1-ona 59
A una disolución de 58 (0,26 mmol) en etanol (1 ml) se le añadió EtNH2 (2 M en THF, 0,76 mmol). Se calentó la mezcla de reacción hasta reflujo durante 2 h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante cromatografía ultrarrápida (Hexanos: AcOEt 3:7) para dar el producto deseado 59. Em ESI (m/z) 247 (M+H)+; 1H RMN (400 MHz, CDCla) 88,46 (d, 1H, J=8,4 Hz), 8,13 (d, 1H, J=8,4 Hz), 7,87 (m, 1H), 7,72 (m, 1H), 4,53 (s, 2 h), 3,77 (q, 2 h, J=7,2 Hz), 1,34 (t, 3H, J=7,2 Hz).

9-[(3-Cloro-4-metoxibencil)amino]-2-etil-2,3-dihidro-1H-pirrolo[3,4-b]quinolin-1-ona O
Se calentó una disolución de 59 (0,22 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,33 mmol) y ('pr)2NEt (1,31 mmol) en n-propanol (2,0 ml) hasta 90°C durante 2 h. Después de enfriar, el precipitado resultante O se recogió mediante filtración. EM ESI (m/z) 382 (M+H)+; 1H RMN (400 MHz, CDCls) 88,26 (t, 1H, J=6,0 Hz), 8,14 (dd, 1H, Ji=1,2, J2=8,0 Hz), 7,94 (dd, 1H, Ji=1,2, J2=8,8 Hz), 7,68-7,64 (m, 1H), 7,45 (d, 1H, J=2,4 Hz), 7,33-7,29 (m, 1H), 6,95 (d, 1H, J=8,4 Hz), 4,98 (d, 2 h, J=5,6 Hz), 4,39 (s, 2 h), 3,90 (s, 3H), 3,65 (q, 2 h, J=7,6 Hz), 1,29 (t, 3H, J=7,6 Hz).
Ejemplo 22: 2-(terc-but¡l)-9-[(3-cloro-4-metox¡benc¡l)am¡no1-2.3-d¡h¡dro-1H-p¡rrolo[3,4-b1qu¡nol¡n-1-ona

2-(Terc-butil)-9-cloro-2,3-dihidro-1H-pirrolo[3,4-b1quinolin-1-ona 60
A una disolución de 58 (0,32 mmol) en etanol (2,0 ml) se le añadió ferc-butilamina (2 M en THF, 0,95 mmol). Se calentó la mezcla de reacción hasta reflujo durante 2 h. Se eliminó el disolvente por evaporación y se purificó el residuo mediante cromatografía ultrarrápida (Hexanos: AcOEt 3:7) para dar el producto deseado 60. 1H RMN (400 MHz, CDCla) 88,46 (dd, 1H, Ji=1,2, J2=8,4 Hz), 8,12 (d, 1H, J=8,8 Hz), 7,88-7,84 (m, 1H), 7,73-7,69 (m, 1H), 4,60 (s, 2 h), 1,63 (s, 9H).

2-(Terc-but¡l)-9-[(3-cloro-4-metoxibenc¡l)am¡no]-2,3-d¡h¡dro-1H-p¡rrolo[3,4-b]qu¡nol¡n-1-ona P
Se calentó una disolución de 60 (0,07 mmol), clorhidrato de 3-cloro-4-metoxibencilamina (0,11 mmol) y ('pr)2NEt (0,44 mmol) en n-propanol (1,0 ml) hasta 90°C durante 2 h. Después de enfriar, se recogió el precipitado resultante P mediante filtración. EM ESI (m/z) 410 (M+H)+; 1H RMN (400 MHz, CDCh) 88,53 (t, 1H, J=6,4 Hz), 8,10 (d, 1H, J=8,4 Hz), 7,92 (d, 1H, J=8,0 Hz), 7,63 (t, 1H, J=7,6 Hz), 7,45 (d, 1H, J=2,0 Hz), 7,33-7,25 (m, 2 h), 6,94 (d, 1H, J=8,8 Hz), 4,95 (d, 2 h, J=6,8 Hz), 4,46 (s, 2 h), 1,57 (s, 9H).
Los compuestos presentaron inhibición de la PDE5 en el intervalo nanomolar o inferior. Se muestra inhibición a modo de ejemplo de los compuestos representativos en la tabla 3.
Tabla 3. Inhibición de la PDE5 de compuestos representativos.

Ejemplo 23:

Se calentó una mezcla de 26 (0,35 mmol), 3-cloro-4-(trifluorometoxi)bencilamina (1,05 mmol), TEA (1,05 mmol) y Nal (0,035 mmol) en NMP (2 ml) hasta 130°C y se agitó durante 24 h. Se diluyó la mezcla con AcOEt (10 ml) y se lavó con H2O (2x 10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir. La cromatografía ultrarrápida (eluyente: 5% de MeOH en AcOEt) dio el producto deseado Q (37% de rendimiento). EM ESI (m/z) 475 (M+H)+; 1H RMN (400 MHz, CDCla) 88,38 (s, 1H), 8,07 (s, 1H), 7,77 (dd, 1H, Ji=1,6, J2=8,4 Hz), 7,49 (d, 1H, J=2,0 Hz), 7,35 (dd, 1H, Ji=0,8, J2=8,4 Hz), 7,30 (dd, 1H, Ji=2,4, J2=8,4 Hz), 4,75 (s, 5 h), 3,83 (t, 2 h, J=6,4 Hz), 3,27 (s, 2 h), 2,2 (s, 3H).
Ejemplo 24:

Se calentó una mezcla de 26 (0,28 mmol), 3-cloro-4-fluorobencilamina (0,84 mmol), TEA (0,84 mmol) y NaI
(0,028 mmol) en NMP (2 ml) hasta 130°C y se agitó durante 24 h. Se diluyó la mezcla con AcOEt (10 ml) y se lavó con H2O (2x 10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir. La cromatografía ultrarrápida (eluyente: 5% de MeOH en AcOEt) dio el producto deseado R (18% de rendimiento). EM ESI (m/z) 409 (M+H)+; 1H RMN (400 MHz, CDCla) 88,34 (s, 1H), 8,03 (s, 1H), 7,74-7,71 (m, 1H), 7,36 (dd, 1H, J í=2,4, J2=7,2 Hz), 7,20-7,12 (m, 2 h), 4,70 (s, 4 h), 4,63 (s, 1H), 3,79 (t, 2 h, J=6 Hz), 3,24 (s, 2 h), 2,18 (s, 3H). Ejemplo 25:

Se calentó una mezcla de 26 (0,27 mmol), 3-cloro-4-metoxianilina (0,82 mmol), TEA (0,82 mmol) y NaI (0,027 mmol) en NMP (2 ml) hasta 130°C y se agitó durante la noche. Se diluyó la mezcla con AcOEt (10 ml) y se lavó con H2O (2x 10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir. La cromatografía ultrarrápida (eluyente: 5% de MeOH en AcOEt) dio el producto deseado S (14% de rendimiento). EM ESI (m/z) 407 (M+H)+.
Ejemplo 26:

Se calentó una mezcla de 26 (0,25 mmol), 3-cloro-4-metoxianilina (0,75 mmol), TEA (0,75 mmol) y Nal (0,025 mmol) en NMP (2 ml) hasta 130°C y se agitó durante la noche. Se diluyó la mezcla con AcOEt (10 ml) y se lavó con H2O (2x 10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir. La cromatografía ultrarrápida (eluyente: 5% de MeOH en AcOEt) dio el producto deseado T (10% de rendimiento). EM ESI (m/z) 407 (M+H)+.
Ejemplo 27:

Se añadió lentamente LiAlD4 a una disolución de 65 en THF (2 ml) a 0°C. Se calentó la disolución hasta 70°C durante 2 h. Se extinguió la mezcla de reacción añadiendo NaOH 1 M a 0°C. Se extrajo la fase acuosa con AcOEt (3x 10 ml), se secó Na2SO4, se filtró y se evaporó a presión reducir. Se trató el residuo con HCl 2 M en dietil éter y se recogió el precipitado resultante (66) mediante filtración (80% de rendimiento). 1H RMN (400 MHz, DMSO-d6) 8 8,37 (s, 3H), 7,61 (d, 1H, J=2,4 Hz), 7,45-7,42 (m, 1H), 7,18 (d, 1H, J=8,8 Hz), 3,95 (s, 2 h), 3,86 (s, 3H).

Se calentó una mezcla de 26 (0,07 mmol), 66 (0,21 mmol), TEA (0,21 mmol) y Nal (0,007 mmol) en NMP (1 ml) hasta 130°C y se agitó durante la noche. Se diluyó la mezcla con AcOEt (10 ml) y se lavó con H2O (2x 10 ml). Se secaron las fases orgánicas sobre Na2SO4, se filtraron y se evaporaron a presión reducir. La cromatografía ultrarrápida (eluyente: 2% de MeOH en DCM) dio el producto deseado U (12% de rendimiento). EM ESI (m/z) 423 (M+H)+; 1H RMN (400 MHz, CDCls) 88,35 (s, 1H), 7,97 (d, 1H, J=8,8 Hz), 7,74 (dd, 1H, Ji=1,8, J2=9,0 Hz), 7,31 (d, 1H, J=2,0 Hz), 7,21 (dd, 1H, Ji=2,0, J2=8,4 Hz), 6,94 (d, 1H, J=8,0 Hz), 4,71 (s, 2 h, CH2), 4,56 (s, 1H, NH), 3,92 (s, 3H, OCH3), 3,82 (t, 2 h, J=6,4 Hz, CH2), 3,21 (t, 2 h, J=6,4 Hz, CH2), 2,22 (s, 3H, COCH3).
La sustitución de H con D tal como se muestra en el compuesto U condujo a la formación de sólo 1 metabolito en lugar de tres.
Ejemplo 28:

Se añadió NaBH3CN (10,52 mmol) a una disolución de 67 (5,26 mmol) y NH4OAc (52,6 mmol) en MeOH (15 ml). Se agitó la mezcla de reacción durante la noche a 60°C y luego se enfrió hasta temperatura ambiente y se repartió entre AcOEt (50 ml) y H2O (50 ml). Se descartó la fase acuosa y se lavó la fase orgánica con agua (2x 50 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducir para dar el compuesto 68 (61%). 1H RMN (400 MHz, CDCh) 8 7,37 (d, 1H, J=2,0 Hz), 7,20 (dd, 1H, Ji=2,0, J2=8,4 Hz), 6,88 (d, 1H, J=8,0 Hz), 4,07 (q, 1H, J=6,4 Hz, CHCH3), 3,88 (s, 3H, OCH3), 1,84 (s a, 2 h, NH2), 1,36 (d, 3H, J=6,4 Hz, CHCH3).

Se calentó una mezcla de 26 (0,175 mmol), 68 (0,526 mmol), TEA (0,7 mmol) y Nal (0,017 mmol) en NMP (1 ml) hasta 130°C y se agitó durante la noche. Se enfrió la mezcla y se añadió AcOEt (10 ml). Se lavó la fase orgánica con H2O (2x 10 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducir. La cromatografía ultrarrápida (eluyente: 2% de MeOH en DCM) dio el producto deseado V (17% de rendimiento). EM ESI (m/z) 435 (M+H)+; 1H RMN (400 MHz, CDCla) 88,29 (S, 1H), 7,94 (d, 1H, J=8,4 Hz), 7,72 (d, 1H, J=8,8 Hz), 7,25 (s, 1H), 7,16 (d, 1H, J=8,8 Hz), 6,88 (d, 1H, J=8,4 Hz), 4,86 (t, 1H, J=6,8 Hz, NH), 4,71 (s, 2 h, CH2), 3,88 (s, 3H, OCH3), 3,84-3,72 (m, 3H, CH2y CH), 3,18 (t, 2 h, J=5,6 Hz, CH2), 2,22 (s, 3H, COCH3), 1,68 (d, 3H, J=5,2 Hz, CHCH3).
Ejemplo 29:

Se calentó una mezcla de 26 (0,175 mmol), 69 (0,526 mmol), TEA (0,7 mmol) y Nal (0,017 mmol) en NMP (1 ml) hasta 130°C y se agitó durante la noche. Se enfrió la mezcla y se añadió AcOEt (10 ml). Se lavó la fase orgánica con H2O (2x 10 ml), se secó sobre Na2SO4, se filtró y se evaporó a presión reducir. La cromatografía ultrarrápida (eluyente: 2% de MeOH en DCM) dio el producto deseado W (21% de rendimiento). EM ESI (m/z) 435 (M+H)+; 1H RMN (400 MHz, CDCla) 88,19 (s, 1H), 8,01 (s, 1H), 7,73 (d, 1H, J=8,8 Hz), 7,17 (s, 1H), 7,11 (dd, 1H, Ji=2,0, J2=8,4 Hz), 6,92 (d, 1H, J=9,0 Hz), 4,62 (s, 2 h), 3,91-3,85 (m, 6H), 3,81 (t, 2 h, J=6,0 Hz), 3,23 (s, 2 h), 2,95 (t, 2 h, J=6,0 Hz), 2,22 (s, 3H).
Los compuestos presentaron inhibición de la PDE5 en el intervalo nanomolar o inferior. Se muestra inhibición a modo de ejemplo de los compuestos representativos en la tabla 4.
Tabla 4. Inhibición de la PDE5 de compuestos representativos.

Derivados de benzo[6][1,6]naftiridina - Fórmula Ia
Ejemplo 30:

Ejemplo 31:

Tabla 5. Inhibición de la PDE5 de compuestos representativos - Fórmula la.


Derivados 2,3-d¡h¡dro-1H-p¡rrolo[3,4-6]qumol¡n-1-ona, con deuterio (D)
Ejemplo 33:

AD

Los siguientes ejemplos proféticos se proporcionan para ilustrar otros aspectos de la presente invención:

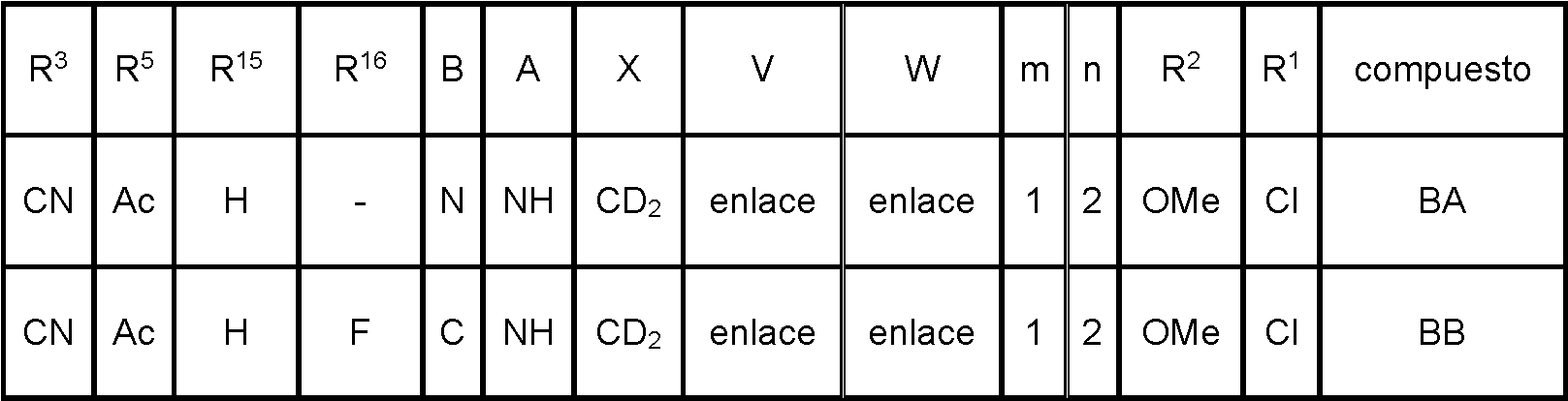

Ejemplo 34:

Ejemplo 36:

Ejemplo 37:
Ċ

Ejemplo 38:

Aunque la invención se ha descrito e ilustrado en las realizaciones ilustrativas anteriores, se entiende que la presente divulgación se ha realizado únicamente a modo de ejemplo, y que pueden realizarse numerosos cambios en los detalles de la implementación de la invención sin alejarse del espíritu y alcance de la invención, que está limitada únicamente por las reivindicaciones que siguen a continuación. Las características de las realizaciones dadas a conocer pueden combinarse y/o reorganizarse de diversos modos dentro del alcance y espíritu de la invención para producir realizaciones adicionales que están también dentro del alcance de la invención. Los expertos en la técnica podrán determinar usando tan sólo experimentación de rutina, numerosos equivalentes de las realizaciones específicas descritas específicamente en esta divulgación. Se pretende que tales equivalentes se abarquen en el alcance de las siguientes reivindicaciones.
Claims (1)
- REIVINDICACIONESCompuesto de fórmula (I),
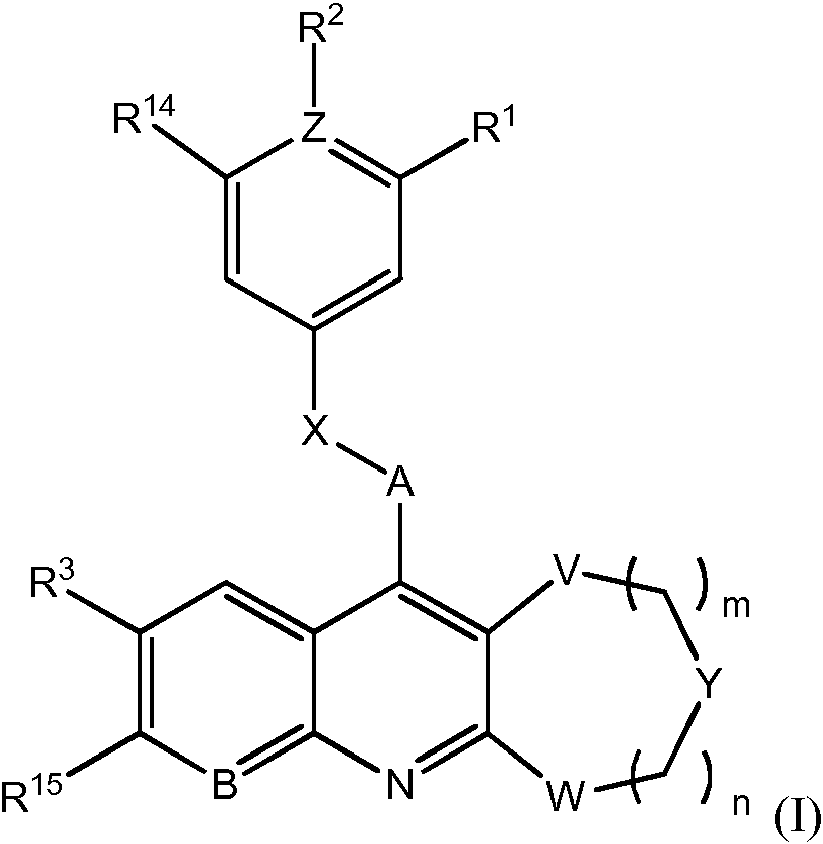 en la queA es NR4;B es CR16 o N;V es un enlace o C(O);W es un enlace o NR13;X es alquilo (C1-C3), alquilo (C1-C3) sustituido con al menos un D;Y es NR5;Z es C;R1 es halógeno o haloalquilo (C1-C6);R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R4 es hidrógeno o alquilo (C1-C3);R5 es hidrógeno, alquilo (C1-C3), cicloalquilo (C3-C5) o -C(O)R7;R6 es alquilo (C1-C3) o haloalquilo (C1-C3);R7 es independientemente alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);R13 es hidrógeno o alquilo (C1-C3);R14 es hidrógeno, halógeno o haloalquilo (C1-C6);R15 es hidrógeno, -OR17, -OH o halógeno;R16 es hidrógeno, -OR17, -OH o halógeno;R17 es hidrógeno, alquilo (C1-C6), haloalquilo (C1-C6) o cicloalquilo (C3-C8);m y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3; o una sal o tautómero farmacéuticamente aceptable del mismo.2. Compuesto según la reivindicación 1, que tiene la fórmula (la):
en la queA es NR4;B es CR16 o N;V es un enlace o C(O);W es un enlace o NR13;X es alquilo (C1-C3), alquilo (C1-C3) sustituido con al menos un D;Y es NR5;Z es C;R1 es halógeno o haloalquilo (C1-C6);R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R4 es hidrógeno o alquilo (C1-C3);R5 es hidrógeno, alquilo (C1-C3), cicloalquilo (C3-C5) o -C(O)R7;R6 es alquilo (C1-C3) o haloalquilo (C1-C3);R7 es independientemente alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);R13 es hidrógeno o alquilo (C1-C3);R14 es hidrógeno, halógeno o haloalquilo (C1-C6);R15 es hidrógeno, -OR17, -OH o halógeno;R16 es hidrógeno, -OR17, -OH o halógeno;R17 es hidrógeno, alquilo (C1-C6), haloalquilo (C1-C6) o cicloalquilo (C3-C8);m y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3; o una sal o tautómero farmacéuticamente aceptable del mismo.2. Compuesto según la reivindicación 1, que tiene la fórmula (la): 3. Compuesto según la reivindicación 1, que tiene la fórmula (Ib):
3. Compuesto según la reivindicación 1, que tiene la fórmula (Ib): 4. Compuesto según la reivindicación 1, que tiene la fórmula (Ic):
4. Compuesto según la reivindicación 1, que tiene la fórmula (Ic): 5. Compuesto según cualquiera de las reivindicaciones 1 - 4, en el que X es alquilo (C1-C3) sustituido con al menos un D.Compuesto según cualquiera de las reivindicaciones 1-4, en el queA es NH;V es un enlace o C(O);W es un enlace o NR13;X es alquilo (C1-C3) o alquilo (C1-C3) sustituido con al menos un D;Y es NR5;R1 es halógeno;R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R6 es alquilo (C1-C3);R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);R13 es hidrógeno;m es 0 ó 1 ; yn es 2 ; oen el queA es NH;V es un enlace o C(O);W es un enlace o NH;X es CH2 o CD2;Y es NR5;R1 es halógeno;R2 es -OCH3;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)CH3;R8 es hidrógeno o alquilo (C1-C3);m es 0 ó 1 ; yn es 2 ; oen el queV y W son cada uno un enlace, o V es C(O) y W es NR13;X es alquilo (C1-C3) o alquilo (C1-C3) sustituido con al menos un D;Y es NR5; ym y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3; o en el queA es NH;V es C(O);W es NH;X es CH2 o CD2;Y es NR5;R1 es halógeno o haloalquilo (C1-C3);R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R6 es alquilo (C1-C3) o haloalquilo (C1-C3);R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 0 ó 1 ; yn es 2 ; oen el queA es NH;V es C(O);W es NH;X es CH2 o CD2;Y es NR5;R1 es halógeno;R2 es -OCH3;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 0; yn es 2 ; oen el queA es NH;V y W son cada uno un enlace, o V es C(O) y W es NH; X es CH2 o CD2;Y es NR5;R1 es halógeno o haloalquilo (C1-C3);R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R6 es alquilo (C1-C2) o haloalquilo (C1-C2);R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 0 ó 1 ; yn es 2 ; oen el queA es NH;V y W son cada uno un enlace;X es CH2 o CD2;Y es NR5;R1 es cloro;R2 es -OCH3;R3 es -CN;R5 es hidrógeno o alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 1 ; yn es 2 ; oen el queA es NH;V y W son cada uno un enlace;X es CH2 o CD2;Y es NC(O)CH3;R1 es halógeno;R2 es -OCH3;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R8 es hidrógeno;m es 1 ; yn es 2.7. Composición que comprende un compuesto según cualquiera de las reivindicaciones 1-6, y un portador farmacéuticamente aceptable.8. Compuesto según cualquiera de las reivindicaciones 1-6 o composición según la reivindicación 7, para su uso en el tratamiento de una enfermedad neurodegenerativa en un sujeto.9. Compuesto o composición para su uso según la reivindicación 8, en el que la enfermedad es enfermedad de Alzheimer.10. Compuesto o composición para su uso según la reivindicación 8 o la reivindicación 9, en el que se aumenta la potenciación a largo plazo en el sujeto.11. Compuesto o composición para su uso según la reivindicación 10, en el que el sujeto tiene una enfermedad neurodegenerativa; y opcionalmente en el que la enfermedad neurodegenerativa es enfermedad de Alzheimer.12. Compuesto o composición para su uso según la reivindicación 8 o la reivindicación 9, en el que se mejora la memoria en el sujeto.13. Compuesto o composición para su uso según la reivindicación 12, en el que el sujeto tiene una enfermedad neurodegenerativa; y opcionalmente en el que la enfermedad neurodegenerativa es enfermedad de Alzheimer.
5. Compuesto según cualquiera de las reivindicaciones 1 - 4, en el que X es alquilo (C1-C3) sustituido con al menos un D.Compuesto según cualquiera de las reivindicaciones 1-4, en el queA es NH;V es un enlace o C(O);W es un enlace o NR13;X es alquilo (C1-C3) o alquilo (C1-C3) sustituido con al menos un D;Y es NR5;R1 es halógeno;R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R6 es alquilo (C1-C3);R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);R13 es hidrógeno;m es 0 ó 1 ; yn es 2 ; oen el queA es NH;V es un enlace o C(O);W es un enlace o NH;X es CH2 o CD2;Y es NR5;R1 es halógeno;R2 es -OCH3;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)CH3;R8 es hidrógeno o alquilo (C1-C3);m es 0 ó 1 ; yn es 2 ; oen el queV y W son cada uno un enlace, o V es C(O) y W es NR13;X es alquilo (C1-C3) o alquilo (C1-C3) sustituido con al menos un D;Y es NR5; ym y n son cada uno 0, 1 ó 2, siempre que la suma de m n sea un número entero de desde 2-3; o en el queA es NH;V es C(O);W es NH;X es CH2 o CD2;Y es NR5;R1 es halógeno o haloalquilo (C1-C3);R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R6 es alquilo (C1-C3) o haloalquilo (C1-C3);R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 0 ó 1 ; yn es 2 ; oen el queA es NH;V es C(O);W es NH;X es CH2 o CD2;Y es NR5;R1 es halógeno;R2 es -OCH3;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 0; yn es 2 ; oen el queA es NH;V y W son cada uno un enlace, o V es C(O) y W es NH; X es CH2 o CD2;Y es NR5;R1 es halógeno o haloalquilo (C1-C3);R2 es -OR6;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R5 es hidrógeno, alquilo (C1-C3) o -C(O)R7;R6 es alquilo (C1-C2) o haloalquilo (C1-C2);R7 es alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 0 ó 1 ; yn es 2 ; oen el queA es NH;V y W son cada uno un enlace;X es CH2 o CD2;Y es NR5;R1 es cloro;R2 es -OCH3;R3 es -CN;R5 es hidrógeno o alquilo (C1-C3);R8 es hidrógeno o alquilo (C1-C3);m es 1 ; yn es 2 ; oen el queA es NH;V y W son cada uno un enlace;X es CH2 o CD2;Y es NC(O)CH3;R1 es halógeno;R2 es -OCH3;R3 es hidrógeno, -OMe, -CF3, -CN o halógeno;R8 es hidrógeno;m es 1 ; yn es 2.7. Composición que comprende un compuesto según cualquiera de las reivindicaciones 1-6, y un portador farmacéuticamente aceptable.8. Compuesto según cualquiera de las reivindicaciones 1-6 o composición según la reivindicación 7, para su uso en el tratamiento de una enfermedad neurodegenerativa en un sujeto.9. Compuesto o composición para su uso según la reivindicación 8, en el que la enfermedad es enfermedad de Alzheimer.10. Compuesto o composición para su uso según la reivindicación 8 o la reivindicación 9, en el que se aumenta la potenciación a largo plazo en el sujeto.11. Compuesto o composición para su uso según la reivindicación 10, en el que el sujeto tiene una enfermedad neurodegenerativa; y opcionalmente en el que la enfermedad neurodegenerativa es enfermedad de Alzheimer.12. Compuesto o composición para su uso según la reivindicación 8 o la reivindicación 9, en el que se mejora la memoria en el sujeto.13. Compuesto o composición para su uso según la reivindicación 12, en el que el sujeto tiene una enfermedad neurodegenerativa; y opcionalmente en el que la enfermedad neurodegenerativa es enfermedad de Alzheimer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361847280P | 2013-07-17 | 2013-07-17 | |
| PCT/US2014/047031 WO2015009930A2 (en) | 2013-07-17 | 2014-07-17 | Novel phosphodiesterase inhibitors and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2776353T3 true ES2776353T3 (es) | 2020-07-30 |
Family
ID=52346846
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14826002T Active ES2776353T3 (es) | 2013-07-17 | 2014-07-17 | Inhibidores de la fosfodiesterasa novedosos y usos de los mismos |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US10899756B2 (es) |
| EP (1) | EP3022205B1 (es) |
| ES (1) | ES2776353T3 (es) |
| WO (1) | WO2015009930A2 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3022205B1 (en) * | 2013-07-17 | 2020-02-05 | The Trustees of Columbia University in the City of New York | Novel phosphodiesterase inhibitors and uses thereof |
| CN106278918B (zh) * | 2016-08-12 | 2018-05-11 | 合肥久诺医药科技有限公司 | 一种溴芬酸钠杂质标准品2-氨基-3-(4-溴苯甲酰基)苯甲酸的合成方法 |
| WO2024173675A1 (en) * | 2023-02-16 | 2024-08-22 | University Of Rochester | Improving glymphatic-lymphatic efflux |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3674790A (en) | 1970-05-01 | 1972-07-04 | American Home Prod | 2,10-disubstituted-1,2,3,4-tetrahydrobenzo(bhq (1,6)naphthyridines useful as cns depressants |
| US4742061A (en) | 1985-03-13 | 1988-05-03 | Hoechst-Roussel Pharmaceuticals Inc. | Benzoc[c]-1,5-naphthyridines and related compounds as memory enhancing agents |
| US4843079A (en) * | 1987-04-23 | 1989-06-27 | Hoechst-Roussel Pharmaceuticals, Inc. | Fused heteroalkylene quinolinamines |
| US4816464A (en) * | 1987-06-22 | 1989-03-28 | American Home Products Corporation | 10-Substituted benzo[b][1,6]naphthyridines as inhibitors of interleukin 1 |
| US5250534A (en) | 1990-06-20 | 1993-10-05 | Pfizer Inc. | Pyrazolopyrimidinone antianginal agents |
| PT100905A (pt) | 1991-09-30 | 1994-02-28 | Eisai Co Ltd | Compostos heterociclicos azotados biciclicos contendo aneis de benzeno, ciclo-hexano ou piridina e de pirimidina, piridina ou imidazol substituidos e composicoes farmaceuticas que os contem |
| US5294612A (en) | 1992-03-30 | 1994-03-15 | Sterling Winthrop Inc. | 6-heterocyclyl pyrazolo [3,4-d]pyrimidin-4-ones and compositions and method of use thereof |
| US5565325A (en) | 1992-10-30 | 1996-10-15 | Bristol-Myers Squibb Company | Iterative methods for screening peptide libraries |
| CA2180526A1 (en) | 1994-01-05 | 1995-07-13 | Joseph C. Hogan, Jr. | Method of identifying chemical compounds having selected properties for a particular application |
| GB9401090D0 (en) | 1994-01-21 | 1994-03-16 | Glaxo Lab Sa | Chemical compounds |
| US5712171A (en) | 1995-01-20 | 1998-01-27 | Arqule, Inc. | Method of generating a plurality of chemical compounds in a spatially arranged array |
| US6008226A (en) | 1995-03-10 | 1999-12-28 | Sanofi-Synthelabo | Substituted N-arylmethyl and heterocylylmethyl-1H-pyrazolo[3,4-b]quinolin-4-amines and compositions and methods of use thereof |
| SK285991B6 (sk) | 1997-04-25 | 2008-01-07 | Pfizer Inc. | Pyrazolopyrimidinóny, spôsob a medziprodukty na ich výrobu, ich použitie a farmaceutické a veterinárne kompozície na ich báze |
| DK0988302T3 (da) | 1997-06-03 | 2003-06-02 | Altana Pharma Ag | Benzonaphthyridiner |
| KR20010014262A (ko) * | 1997-06-30 | 2001-02-26 | 닛뽄카야쿠가부시키가이샤 | 신규 나프티리딘 유도체 또는 그 염 |
| TW589189B (en) | 1997-08-04 | 2004-06-01 | Scras | Kit containing at least one double-stranded RNA combined with at least one anti-viral agent for therapeutic use in the treatment of a viral disease, notably of viral hepatitis |
| UA46166C2 (uk) | 1997-11-12 | 2002-05-15 | Баєр Акцієнгезельшафт | 2-фенілзаміщені імідазотриазинони як інгібітори фосфодіестерази, спосіб їх одержання та лікарський засіб на їх основі |
| US6506559B1 (en) | 1997-12-23 | 2003-01-14 | Carnegie Institute Of Washington | Genetic inhibition by double-stranded RNA |
| CN1134442C (zh) | 1998-04-20 | 2004-01-14 | 美国辉瑞有限公司 | 吡唑并嘧啶酮cGMP PDE5抑制剂、其制备方法及用途及中间体 |
| DE59909760D1 (de) | 1998-08-31 | 2004-07-22 | Altana Pharma Ag | Benzonaphthyridin-n-oxide mit pde3 und pde4 inhibierender aktivität |
| TWI265925B (en) | 1999-10-11 | 2006-11-11 | Pfizer | Pyrazolo[4,3-d]pyrimidin-7-ones useful in inhibiting type 5 cyclic guanosine 3',5'-monophosphate phosphodiesterases(cGMP PDE5), process and intermediates for their preparation, their uses and composition comprising them |
| AU7547900A (en) | 1999-10-11 | 2001-04-23 | Pfizer Inc. | 5-(2-substituted-5-heterocyclylsulphonylpyrid-3-yl)- dihydropyrazolo(4,3-d)pyrimidin-7-ones as phosphodiesterase inhibitors |
| US20030199693A1 (en) | 2000-07-28 | 2003-10-23 | Ing-Jun Chen | Theophylline and 3-isobutyl-1-methylxanthine based N-7 substituted derivatives displaying inhibitory activies on PDE-5 phosphodiesterase |
| US6576644B2 (en) | 2000-09-06 | 2003-06-10 | Bristol-Myers Squibb Co. | Quinoline inhibitors of cGMP phosphodiesterase |
| US6642244B2 (en) | 2001-03-16 | 2003-11-04 | Bristol-Myers Squibb Co. | Pyrazolopyridopyrimidine inhibitors of cGMP phosphodiesterase |
| US7884189B2 (en) | 2003-01-10 | 2011-02-08 | The Trustees Of Columbia University In The City Of New York | Carboxyltransferase domain of acetyl-CoA carboxylase |
| WO2005083069A1 (en) | 2004-01-30 | 2005-09-09 | Pfizer Products Inc. | Pde2 crystal structures for structure based drug design |
| WO2006094237A2 (en) | 2005-03-03 | 2006-09-08 | Sirtris Pharmaceuticals, Inc. | Acridine and quinoline dervatives as sirtuin modulators |
| EP1953159A1 (en) | 2007-02-05 | 2008-08-06 | Nycomed GmbH | 6-Benzyl-2,3,4,7-tetrahydro-indolo[2,3-c]quinoline compounds useful as PDE5 inhibitors |
| WO2009050554A2 (en) | 2007-10-19 | 2009-04-23 | Pfizer Inc. | Treatment of central nervous system disorders |
| WO2009124119A2 (en) | 2008-04-01 | 2009-10-08 | The Trustes of Columbia University in the City of New York | Phosphodiesterase inhibitors and uses thereof |
| WO2010015589A1 (en) | 2008-08-05 | 2010-02-11 | Nycomed Gmbh | Benzyl-substituted tetracyclic heterocyclic compounds as pde5 inhibitors |
| WO2010074783A1 (en) | 2008-12-23 | 2010-07-01 | The Trustees Of Columbia University In The City Of New York | Phosphodiesterase inhibitors and uses thereof |
| WO2011015523A1 (en) | 2009-08-03 | 2011-02-10 | Nycomed Gmbh | Benzyl-substituted tetracyclic heterocyclic compounds |
| WO2011072243A1 (en) | 2009-12-10 | 2011-06-16 | The Trustees Of Columbia University In The City Of New York | Histone acetyltransferase activators and uses thereof |
| WO2012088420A1 (en) | 2010-12-22 | 2012-06-28 | The Trustees Of Columbia University In The City Of New York | Histone acetyltransferase modulators and usese thereof |
| WO2013109738A1 (en) * | 2012-01-17 | 2013-07-25 | The Trustees Of Columbia University In The City Of New York | Novel phosphodiesterase inhibitors and uses thereof |
| EP3022205B1 (en) * | 2013-07-17 | 2020-02-05 | The Trustees of Columbia University in the City of New York | Novel phosphodiesterase inhibitors and uses thereof |
-
2014
- 2014-07-17 EP EP14826002.9A patent/EP3022205B1/en active Active
- 2014-07-17 US US14/905,246 patent/US10899756B2/en active Active
- 2014-07-17 WO PCT/US2014/047031 patent/WO2015009930A2/en active Application Filing
- 2014-07-17 ES ES14826002T patent/ES2776353T3/es active Active
-
2020
- 2020-12-30 US US17/137,665 patent/US11851427B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US11851427B2 (en) | 2023-12-26 |
| WO2015009930A2 (en) | 2015-01-22 |
| EP3022205A2 (en) | 2016-05-25 |
| EP3022205A4 (en) | 2016-11-23 |
| EP3022205B1 (en) | 2020-02-05 |
| US20210130349A1 (en) | 2021-05-06 |
| US10899756B2 (en) | 2021-01-26 |
| WO2015009930A3 (en) | 2015-03-19 |
| US20160152612A1 (en) | 2016-06-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2809974T3 (es) | Compuestos de indol carboxamida útiles como inhibidores de cinasas | |
| ES2793239T3 (es) | Inhibidores del bromodominio | |
| NL1029596C2 (nl) | Nieuwe piperidylderivaten van chinazoline en isochinoline. | |
| ES2797252T3 (es) | Dihidronaftiridinas y compuestos relacionados útiles como inhibidores de quinasas para el tratamiento de enfermedades proliferativas | |
| ES2451348T3 (es) | Dihidro benzocicloalquiloximetil-oxazolopirimidinonas sustituidas, preparación y uso de las mismas | |
| TW201906834A (zh) | 人類免疫不全病毒複製之抑制劑 | |
| US11851427B2 (en) | Phosphodiesterase inhibitors and uses thereof | |
| JP6755922B2 (ja) | 新規な化合物、それらの調製及びそれらの使用 | |
| JP7493586B2 (ja) | Rip1阻害化合物ならびにそれを作製および使用するための方法 | |
| ES2314933T3 (es) | Compuestos de naftiridina. | |
| ES2733502T3 (es) | Compuestos de imidazopiridazina | |
| EA012110B1 (ru) | Пирролодигидроизохинолины как ингибиторы pde10 | |
| JP2014522837A (ja) | 治療用化合物としてのキナゾリン及び関連の使用方法 | |
| TW201623285A (zh) | 新穎雜芳基化合物及其作為藥物之用途 | |
| ES2734268T3 (es) | Derivados de urea y amida de aminoalquilpiperazinas y uso de los mismos | |
| ES2554788T3 (es) | Compuestos Heterocíclicos de Fenoximetilo | |
| PL180679B1 (en) | Derivatives of pyridazinoquinoline | |
| ES2924371T3 (es) | Compuestos tricíclicos y su uso como inhibidores de la fosfodiesterasa | |
| KR20160104729A (ko) | 암 또는 염증성 질환의 치료를 위한 치환 피롤로피리딘 및 피롤로피라진 | |
| ES2260300T3 (es) | Compuestos tetraciclicos sustituidos con amino utiles como agentes antiinflamatorios y composiciones farmaceuticas que comprenden los mismos. | |
| WO2017025058A1 (zh) | 取代的吲哚化合物及其使用方法和用途 | |
| JP6837072B2 (ja) | 6,7−ジヒドロ−5H−ピラゾロ[5,1−b][1,3]オキサジン−2−カルボキサミド化合物 | |
| CN110272425B (zh) | 吡啶酰基八氢吡咯并[3,4-c]吡咯衍生物及其用途 | |
| TW201300390A (zh) | 喹□啉化合物 | |
| US10626113B2 (en) | Phosphodiesterase inhibitors and uses thereof |