ES2765511T3 - Procesos para la preparación de compuestos de pirimidinilciclopentano - Google Patents
Procesos para la preparación de compuestos de pirimidinilciclopentano Download PDFInfo
- Publication number
- ES2765511T3 ES2765511T3 ES14863015T ES14863015T ES2765511T3 ES 2765511 T3 ES2765511 T3 ES 2765511T3 ES 14863015 T ES14863015 T ES 14863015T ES 14863015 T ES14863015 T ES 14863015T ES 2765511 T3 ES2765511 T3 ES 2765511T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- kred
- process according
- binap
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[C@](C1)c2c(N3CCN(*)CC3)ncnc2[C@]1O Chemical compound C[C@](C1)c2c(N3CCN(*)CC3)ncnc2[C@]1O 0.000 description 7
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/02—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups from isocyanates with formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/06—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups by reactions not involving the formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/16—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing two or more hetero rings
- C12P17/165—Heterorings having nitrogen atoms as the only ring heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Microbiology (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Epidemiology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Pyridine Compounds (AREA)
Abstract
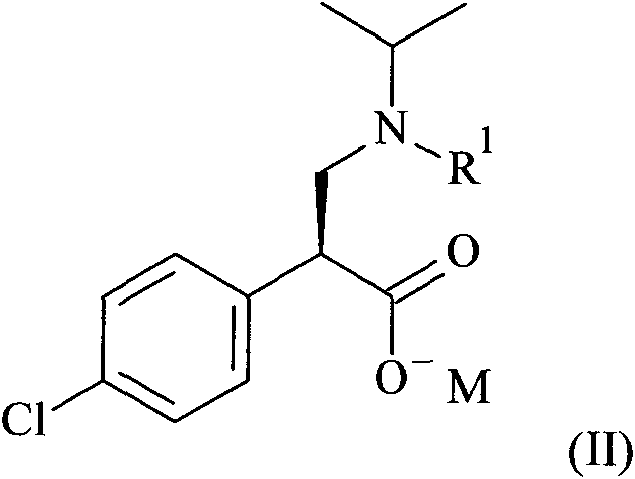
Un proceso para la preparación de un compuesto de fórmula (I) **(Ver fórmula)** o sales del mismo, que comprende la reacción de acoplamiento de un compuesto de fórmula (II) **(Ver fórmula)** con un compuesto de fórmula (III) **(Ver fórmula)** en donde R1 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, tercbutoxicarbonilo (BOC) y trifluoroacetilo. R2 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, terc- butoxicarbonilo (BOC) y trifluoroacetilo. M es un ion metálico seleccionado entre la lista de ion de metal alcalino, ion de metal alcalinotérreo e ion de metal de transición.
Description
DESCRIPCIÓN
Procesos para la preparación de compuestos de pirimidinilcidopentano
Campo de la invención
La presente invención se refiere a procesos para la preparación de compuestos de pirimidinilcidopentano que son útiles como intermedios en la preparación de inhibidores de la proteína cinasa AKT con actividad terapéutica contra enfermedades tales como cáncer.
Antecedentes de la invención
Las enzimas proteína cinasa B/Akt son un grupo de cinasas serina/treonina que están sobreexpresadas en determinados tumores humanos. La solicitud de patente internacional WO 2008/006040 y la patente de Estados Unidos n.° 8.063.050 analizan varios inhibidores de AKT, incluyendo el compuesto (S)-2-(4-clorofenil)-1-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-(isopropilamino)propan-1-ona (ipatasertib, GDC-0068), que se está investigando en ensayos clínicos para el tratamiento de diversos cánceres.
Si bien los procesos descritos en los documentos WO 2008/006040 y US 8.063.050 son útiles para proporcionar compuestos de ciclopenta[d]pirimidina hidroxilados como inhibidores de la proteína cinasa AKT, se necesitan procesos alternativos o mejorados, que incluyen la fabricación a gran escala de estos compuestos. El documento WO 2012/135750 describe una combinación de vemurafenib con un compuesto inhibidor de AKT que tiene un núcleo de ciclopenta[d]pirimidin-4-il piperazin-1-ilo. El documento WO 2012/135781 describe compuestos que tienen un núcleo similar junto con una gama más amplia de agentes quimioterapéuticos. El documento US 2013/0059859 describe un método para predecir la sensibilidad del crecimiento de las células tumorales a inhibición mediante un inhibidor de cinasa PI3K/AKT y describe algunos compuestos adecuados que tienen un núcleo de ciclopenta[d]pirimidin-4-il piperazin-1-ilo.
Sumario de la invención
La presente invención proporciona procesos para la preparación de un compuesto de fórmula (I)

o sales del mismo, que comprenden la reacción de acoplamiento de un compuesto de fórmula (II)

con un compuesto de fórmula (III)

en donde R1, R2 y M son como se describen en el presente documento.
Descripción detallada de la invención
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que el comprendido habitualmente por un experto en la técnica a la que pertenece la presente invención. Aunque en la práctica o ensayo de la invención se pueden usar métodos y materiales similares o equivalentes a los descritos en el presente documento, a continuación se describen métodos y materiales adecuados.
La nomenclatura usada en esta memoria descriptiva se basa en la nomenclatura sistemática de la IUPAC, a menos que se indique lo contrario.
Cualquier valencia abierta que aparezca en un átomo de carbono, oxígeno, azufre o nitrógeno en las estructuras de presente documento indica la presencia de un hidrógeno, a menos que se indique lo contrario.
Cuando se indica el número de sustituyentes, la expresión "uno o más" se refiere al intervalo desde un sustituyente hasta el número de sustitución posible más elevado, es decir sustitución de un hidrógeno hasta sustitución de todos los hidrógenos por los sustituyentes.
El término "opcional" u "opcionalmente" representa que un evento o circunstancia descrito posteriormente puede, pero no necesariamente, ocurrir y que la descripción incluye casos en los que el evento o la circunstancia sucede y casos en los que no.
La expresión "sales farmacéuticamente aceptables" representa sales que no son indeseables biológicamente o de otro modo. Las sales farmacéuticamente aceptables incluyen sales de adición de ácidos y de bases.
La expresión "sal de adición de ácidos farmacéuticamente aceptables" representa las sales farmacéuticamente aceptables formadas con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido carbónico, ácido fosfórico y ácidos orgánicos seleccionados entre las clases alifática, cicloalifática, aromática, aralifática, heterocíclica, carboxílica y sulfónica de ácidos orgánicos, tales como ácido fórmico, ácido acético, ácido propiónico, ácido glicólico, ácido glucónico, ácido láctico, ácido pirúvico, ácido oxálico, ácido málico, ácido maleico, ácido maloneico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido aspártico, ácido ascórbico, ácido glutámico, ácido antranílico, ácido benzoico, ácido cinámico, ácido mandélico, ácido embónico, ácido fenilacético, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico y ácido salicílico.
La expresión "sal de adición de bases farmacéuticamente aceptable" representa las sales farmacéuticamente aceptables formadas con una base orgánica o inorgánica. Los ejemplos de bases inorgánicas adecuadas incluyen sales de sodio, potasio, amonio, calcio, magnesio, hierro, cinc, cobre, manganeso y aluminio. Las sales derivadas de bases no tóxicas farmacéuticamente aceptables incluyen sales de aminas primarias, secundarias y terciarias, aminas sustituidas incluyendo aminas sustituidas de origen natural, aminas cíclicas y resinas de intercambio iónico básicas, tales como isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, etanolamina, 2-dietilaminoetanol, trimetamina, diciclohexilamina, lisina, arginina, histidina, cafeína, procaína, hidrabamina, colina, betaína, etilendiamina, glucosamina, metilglucamina, teobromina, purinas, piperizina, piperidina, N-etilpiperidina y resinas de poliamina.
Las convenciones y definiciones estereoquímicas usadas en el presente documento siguen generalmente a S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, Nueva York; y Eliel, E. y
Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., Nueva York, 1994. En la descripción de un compuesto ópticamente activo, los prefijos D y L o R y S, se usan para indicar la configuración absoluta de la molécula en torno a su centro o centros quirales. Los sustituyentes unidos al centro quiral en consideración se clasifican de acuerdo con la Sequence Rule of Cahn, Ingold and Prelog. (Cahn et al. Angew. Chem. Inter. Edit. 1966, 5, 385; errata 511). Los prefijos D y L o (+) y (-) se emplean para designar el signo de rotación de luz polarizada en el plano por el compuesto, con (-) o L designando que el compuesto es levógiro. Un compuesto con el prefijo (+) o D es dextrógiro.
El término "estereoisómero" representa un compuesto que posee idéntica conectividad molecular y multiplicidad de enlaces, pero que difiere en la disposición de sus átomos en el espacio.
La expresión "centro quiral" representa un átomo de carbono unido a cuatro sustituyentes no idénticos. El término "quiral" representa la capacidad de no superposición con la imagen especular, mientras que el término "aquiral" se refiere a realizaciones que son superponibles con su imagen especular. Las moléculas quirales son ópticamente activas, es decir, tienen la capacidad de rotar el plano de luz polarizada.
Los compuestos de la presente invención pueden tener uno o más centros quirales y pueden existir en la forma de enantiómeros ópticamente puros, mezclas de enantiómeros tales como, por ejemplo, racematos, diaestereoisómeros ópticamente puros, mezclas de diaestereoisómeros, racematos diastereoisoméricos o mezclas de racematos diastereoisoméricos. Cuando en una estructura química está presente un centro quiral, se pretende que la presente invención incluya todos los estereoisómeros asociados a ese centro quiral.
El término "enantiómeros" representa dos estereoisómeros de un compuesto que no son imágenes especulares entre sí.
El término "diastereómero" representa un estereoisómero con dos o más centros de quiralidad y cuyas moléculas no son imágenes especulares entre sí. Los diastereómeros tienen propiedades físicas diferentes, por ejemplo, puntos de fusión, puntos de ebullición, propiedades espectrales y reactividades.
La expresión "exceso diastereomérico" (de) representa la pureza diastereomérica, es decir (diastereómero A -diastereómero B) / (diastereómero A diastereómero B) (en % en área).
La expresión "exceso enantiomérico" (ee) representa la pureza enantiomérica, es decir (enantiómero A -enantiómero B) / (enantiómero A enantiómero B) (en % en área).
Los términos "halo" y "halógeno" se usan de manera intercambiable en el presente documento y se refieren a flúor, cloro, bromo o yodo.
El término "haluro" representa un ion halógeno, en particular fluoruro, cloruro, bromuro o yoduro.
El término "alquilo" representa un grupo hidrocarburo saturado, lineal o ramificado, monovalente, de 1 a 12 átomos de carbono. En realizaciones particulares, el alquilo tiene de 1 a 7 átomos de carbono y en más realizaciones particulares de 1 a 4 átomos de carbono. Los ejemplos de alquilo incluyen metilo, etilo, propilo, isopropilo, n-butilo, iso-butilo, sec-butilo o tere-butilo.
El término "alquenilo" representa un grupo hidrocarburo lineal o ramificado, monovalente, de 2 a 7 átomos de carbono con al menos un doble enlace. En realizaciones particulares, alquenilo tiene de 2 a 4 átomos de carbono con al menos un doble enlace. Los ejemplos de alquenilo incluyen etenilo, propenilo, prop-2-enilo, isopropenilo, nbutenilo e iso-butenilo.
El término "alquinilo" representa un grupo hidrocarburo saturado, lineal o ramificado, monovalente, de 2 a 7 átomos de carbono que comprende uno, dos o tres triples enlaces. En realizaciones particulares el alquinilo tiene de 2 a 4 átomos de carbono que comprende uno o dos triples enlaces. Los ejemplos de alquinilo incluyen etinilo, propinilo y n-butinilo.
El término "alcoxi" representa un grupo de fórmula -O-R', en donde R' es un grupo alquilo. Los ejemplos de restos alcoxi incluyen metoxi, etoxi, isopropoxi y terc-butoxi.
El término "haloalquilo" representa un grupo alquilo en donde al menos uno de los átomos de hidrógeno del grupo alquilo se ha sustituido el mismo o diferentes átomos de halógeno, en particular, átomos de flúor. Los ejemplos de haloalquilo incluyen monofluoro-, difluoro- o trifluoro-metilo, -etilo o -propilo, por ejemplo, 3,3,3-trifluoropropilo, 2-fluoroetilo, 2,2,2-trifluoroetilo, fluorometilo o trifluorometilo. El término "perhaloalquilo" representa un grupo alquilo en donde todos los átomos de hidrógeno del grupo alquilo se han sustituido por el mismo o diferentes átomos de halógeno.
El término "haloalcoxi" representa un grupo alcoxi en donde al menos uno de los átomos de hidrógeno del grupo
alcoxi se ha sustituido por el mismo o diferentes átomos de halógeno, en particular, átomos de flúor. Los ejemplos de haloalcoxilo incluyen monofluoro-, difluoro- o trifluoro-metoxi, - etoxi o -propoxi, por ejemplo 3,3,3-trifluoropropoxi, 2-fluoroetoxi, 2,2,2-trifluoroetoxi, fluorometoxi o trifluorometoxi. El término "perhaloalcoxi" representa un grupo alcoxi en donde todos los átomos de hidrógeno del grupo alcoxi se han sustituido por el mismo o diferentes átomos de halógeno.
El término "cicloalquilo" representa un grupo hidrocarburo monocíclico o bicíclico, saturado, monovalente, de 3 a 10 átomos de carbono en el anillo. En realizaciones particulares cicloalquilo representa un grupo hidrocarburo monocíclico, saturado, monovalente, de 3 a 8 átomos de carbono en el anillo. Bicíclico significa que consiste en dos carbociclos saturados que tienen uno o más átomos de carbono en común. Algunos grupos cicloalquilo particulares son monocíclicos. Son ejemplos de cicloalquilo monocíclico ciclopropilo, ciclobutanilo, ciclopentilo, ciclohexilo o cicloheptilo. Son ejemplos de cicloalquilo bicíclico biciclo[2.2.1]heptanilo o biciclo[2.2.2]octanilo.
El término "heterocicloalquilo" representa un sistema de anillos mono o bicíclico, saturado o parcialmente insaturado, monovalente, de 3 a 9 átomos en el anillo, que comprende 1, 2 o 3 heteroátomos en el anillo seleccionados entre N, O y S, siendo el resto de los átomos del anillo carbono. En realizaciones particulares, heterocicloalquilo es un sistema de anillo monocíclico, saturado, monovalente, de 4 a 7 átomos en el anillo, que comprende 1,2 o 3 heteroátomos en el anillo seleccionados entre N, O y S, siendo el resto de los átomos del anillo carbono. Algunos ejemplos de heterocicloalquilo saturado monocíclico son aziridinilo, oxiranilo, azetidinilo, oxetanilo, pirrolidinilo, tetrahidrofuranilo, tetrahidrotienilo, pirazolidinilo, imidazolidinilo, oxazolidinilo, isoxazolidinilo, tiazolidinilo, piperidinilo, tetrahidropiranilo, tetrahidrotiopiranilo, piperazinilo, morfolinilo, tiomorfolinilo, 1,1-dioxo-tiomorfolin-4-ilo, azepanilo, diazepanilo, homopiperazinilo u oxazepanilo. Algunos ejemplos de heterocicloalquilo saturado bicíclico son 8 -azabiciclo[3.2.1]octilo, quinuclidinilo, 8-oxa-3-aza-biciclo[3.2.1]octilo, 9-aza-biciclo[3.3.1]nonilo, 3-oxa-9-azabiciclo[3.3.1]nonilo o 3-tia-9-aza-biciclo[3.3.1]nonilo. Algunos ejemplos de heterocicloalquilo parcialmente insaturado son dihidrofurilo, imidazolinilo, dihidro-oxazolilo, tetrahidro-piridinilo o dihidropiranilo.
El término "arilo" representa un sistema de anillos mono o bicíclico, carbocíclico, aromático, monovalente, que comprende 6 a 10 átomos de carbono en el anillo. Los ejemplos de restos arilo incluyen fenilo y naftilo. Un arilo particular es fenilo.
El término "heteroarilo" representa un sistema de anillos mono o bicíclico, heterocíclico, aromático, monovalente, de 5 a 12 átomos en el anillo, que comprende 1,2, 3 o 4 heteroátomos seleccionados entre N, O y S, siendo el resto de los átomos del anillo carbono. Los ejemplos de restos heteroarilo incluyen pirrolilo, furanilo, tienilo, imidazolilo, oxazolilo, tiazolilo, triazolilo, oxadiazolilo, tiadiazolilo, tetrazolilo, piridinilo, pirazinilo, pirazolilo, piridazinilo, pirimidinilo, triazinilo, azepinilo, diazepinilo, isoxazolilo, benzofuranilo, isotiazolilo, benzotienilo, indolilo, isoindolilo, isobenzofuranilo, benzoimidazolilo, benzoxazolilo, benzoisoxazolilo, benzotiazolilo, benzoisotiazolilo, benzooxadiazolilo, benzotiadiazolilo, benzotriazolilo, purinilo, quinolinilo, isoquinolinilo, quinazolinilo o quinoxalinilo.
"Grupo saliente" se refiere a una porción de un primer reactivo en una reacción química que se desplaza del primer reactivo en la reacción química. Los ejemplos de grupos salientes incluyen, pero sin limitación, hidrógeno, halógeno, grupos hidroxilo, grupos sulfhidrilo, grupos amino (por ejemplo -NRR, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), grupos sililo (por ejemplo -SiRRR, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), -N(R)OR (en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), grupos alcoxi (por ejemplo -OR, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), grupos tiol (por ejemplo -SR, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), grupos sulfoniloxi (por ejemplo -OS(O)1 -2 R, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), grupos sulfamato (por ejemplo -OS(O)1-2NRR, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente), grupos carbamato (por ejemplo -OC(O)2NRR, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente) y grupos carbonato (por ejemplo -OC(O)2R, en donde R es independientemente alquilo, alquenilo, alquinilo, cicloalquilo, fenilo o heterociclilo y R está opcionalmente sustituido de manera independiente). Los ejemplos de grupos carbonato incluyen carbonato de tere-butilo. Los ejemplos de grupos sulfoniloxi incluyen, pero sin limitación, grupos alquilsulfoniloxi (por ejemplo metilsulfoniloxi (grupo mesilato) y trifluorometilsulfoniloxi (grupo triflato)) y grupos arilsulfoniloxi (por ejemplo p-toluensulfoniloxi (grupos tosilato) y p-nitrosulfoniloxi (grupo nosilato)). Otros ejemplos de grupos salientes incluyen grupos amino sustituidos y sin sustituir, tales como grupos amino, alquilamino, dialquilamino, hidroxilamino, alcoxilamino, N-alquil-N-alcoxiamino, acilamino, sulfonilamino, t-butiloxi y similares.
El término "grupo protector" representa el grupo que bloquea de forma selectiva un sitio reactivo en un compuesto multifuncional tal que puede llevarse a cabo una reacción química de forma selectiva en otro sitio reactivo no protegido, en el significado asociado convencionalmente a la química sintética. Los grupos protectores pueden
eliminarse en el punto apropiado. Los grupos protectores a modo de ejemplo son grupos protectores de amino, grupos protectores de carboxi o grupos protectores de hidroxi.
La expresión "grupo protector de amino" representa grupos destinados a proteger un grupo amino e incluye bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), Fmoc (9-fluorenilmetiloxicarbonilo), p-metoxibenciloxicarbonilo, pnitrobenciloxicarbonilo, ferc-butoxicarbonilo (BOC) y trifluoroacetilo. Otros ejemplos de estos grupos se encuentran en T. W. Greene y P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2a ed., John Wiley & Sons, Inc., Nueva York, NY, 1991, capítulo 7; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. McOmie, Ed., Plenum Press, Nueva York, NY, 1973, capítulo 5 y T. W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, Nueva York, NY, 1981. La expresión "grupo amino protegido" se refiere a un grupo amino sustituido por un grupo protector de amino. Un ejemplo particular de un grupo protector de amino es ferc-butoxicarbonilo (BOC).
El término "desprotección" o "desproteger" representa el proceso por el cual un grupo protector se elimina después de completar la reacción selectiva. Los reactivos de desprotección incluyen ácidos, bases o hidrógeno, en particular carbonato de sodio o potasio, hidróxido de litio en soluciones alcohólicas, cinc en metanol, ácido acético, ácido trifluoroacético, catalizadores de paladio o tribromuro de boro. Un reactivo de desprotección particular es ácido clorhídrico.
El término "tampón" representa un excipiente, que estabiliza el pH de una preparación. Se conocen bien en la técnica tampones adecuados y pueden encontrarse en la bibliografía. Los tampones farmacéuticamente aceptables particulares comprenden tampones de histidina, tampones de arginina, tampones de citrato, tampones de succinato, tampones de acetato y tampones de fosfato. Con independencia del tampón usado, el pH can se puede ajustar con un ácido o una base conocidos en la técnica, por ejemplo ácido clorhídrico, ácido acético, ácido fosfórico, ácido sulfúrico y ácido cítrico, hidróxido sódico e hidróxido potásico.
La expresión "metal alcalino" se refiere a los elementos químicos del grupo 1 de la tabla periódica, es decir litio (Li), sodio (Na), potasio (K), rubidio (Rb), cesio (Cs) y francio (Fr). Son ejemplos particulares de metales alcalinos Li, Na y K, más en particular Na.
El término "metal alcalinotérreo" se refiere a los elementos químicos del grupo 2 de la tabla periódica, es decir berilio (Be), magnesio (Mg), calcio (Ca), estroncio (Sr), bario (Ba) y radio (Ra). Son ejemplos particulares de metales alcalinotérreos Mg y Ca.
La expresión "metal de transición" representa elementos químicos cuyos átomos tienen una subcapa d incompleta.
Abreviaturas
Ac acetilo
AcOH ácido acético
AN acetonitrilo
BINAP 2 ,2 '-bis(difenilfosfin)-1 ,1 '-binaftilo
BINAPHANE 1.2- bis[4,5-dihidro-3H-binafto(1,2-c:2',1'-e)fosfepino]benceno
BIPHEMP (6 ,6 '-dimetilbifenil-2 ,2 '-diil)bis(difenil-fosfina)
BOC ferc-butoxicarbonilo
(Boc)2O dicarbonato de di-ferc-butilo
CBS catalizador de Corey-Bakshi-Shibata
CBZ benciloxicarbonilo, carbobenciloxi
COD 1,5-ciclooctadieno
CPME ciclopentil metil éter
de exceso diastereomérico
DIPEA diisopropiletilamina
DMAP dimetilaminopiridina
DMF N,N-dimetilformamida
DPEN 1.2 - difenil etilendiamina
ee exceso enantiomérico
Et etilo
EtOAc acetato de etilo
Fmoc 9-fluorenilmetiloxicarbonil
(2-furil)-MeOBIPHEP (6 ,6 '-dimetoxibifenil-2 ,2 '-diil)bis[bis(2 -furil)-fosfina]
HAP contaminante peligroso en el aire
HBTU hexafluorofosfato de N,N,N',N'-tetrametil-O-(1H-benzotriazol-1-il)uronio
iBu iso-butilo
ICM International Conference on Harmonisation
IPC en control de proceso
iPr iso-propilo
iPr-DUPHOS 1,2-bs(2,5-di-i-propilfosfolano)benceno
Me metilo
MeOBlPHEP (6 ,6 '-dimetoxibifenil-2 ,2 '-diil)bis(difenil-fosfina)
MES ácido 2-(N-morfolino)etanosulfónico
MTBE metil ferc-butil éter
NAD Nicotinamida adenina dinucleótido
NADP Nicotinamida adenina dinucleótido fosfato
nBu n-butilo
NEM N-etil morfolina
nPr n-propilo
OAc acetato
PBS tampón de dihidrogenofosfato de potasio
pCym p-cimeno
PDE exposición diaria permitida
Ph fenilo
pTol p-tolilo
pTol-Binap 2 ,2 '-bis(di-p-tolilfosfin)-1 ,1 '-binaftilo
S/C relación molar sustrato/catalizador
T3P anhídrido propilfosfónico
tBu ferc-butilo
t-BuOK ferc-butóxido de potasio
TEA trietilamina
TFA trifluoroacetato
THF tetrahidrofurano
TMBTP 2,2',5,5'-tetrametil-4,4'-bis(difenilfosfin)-3,3'-bitiofeno
TPA tri(n-propil)amina
Xyl 3,5-dimetilfenilo
3.5- Xil,4-MeO-MeOBIPHE (P6,6'-dimetoxibifenil-2,2'-diil)bis[bis(3,5-dimetil-4-metoxi-fenil)-fosfina]
2,2'-bis[di(3,5-xilil)fosfina]-1,1'-binaftilo

La presente invención proporciona procesos para la preparación de un compuesto de fórmula (I) o sales del mismo, que comprenden la reacción de acoplamiento de un compuesto de fórmula (II) con un compuesto de fórmula (III), en donde R1, R2 y M son como se describen en el presente documento (esquema 1 a continuación).
Un aspecto más de la presente invención se refiere al proceso para fabricar compuestos de fórmula (II) que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV) usando un catalizador complejo de metal (C) (esquema 1 a continuación).
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (III) que comprende la reducción asimétrica del compuesto de fórmula (V) catalizado por una oxidorreductasa (esquema 1 a continuación).
Un aspecto más de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (VI) o sales farmacéuticamente aceptables de los mismos, en donde se desprotege un compuesto de fórmula (I) (Esquema 1 a continuación).
Esquema 1

En una realización de la invención, R1 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, pnitrobenciloxicarbonilo, ferc-butoxicarbonilo (BOC) y trifluoroacetilo.
En una realización particular de la invención, R1 es ferc-butoxicarbonilo (BOC).
En una realización de la invención, R2 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, pnitrobenciloxicarbonilo, ferc-butoxicarbonilo (BOC) y trifluoroacetilo.
En una realización particular de la invención, R2 es ferc-butoxicarbonilo (BOC).
En una realización de la invención, M es un ion metálico seleccionado entre la lista de ion de metal alcalino, ion de metal alcalinotérreo e ion de metal de transición.
En una realización particular de la invención, M es un ion metálico, en particular un ion de metal alcalino, ion de metal alcalinotérreo o ion de metal de transición con la condición de que no sea K+.
En una realización particular de la invención, M es un ion de metal alcalino.
En una realización particular de la invención, M es Li+, K+ o Na+.
En una realización particular de la invención, M no es K+.
En la realización más particular de la invención, M es Na+.
El documento WO 2008/006040 divulga aminoácidos de fórmula (II-pa) y métodos para la fabricación de los mismos, en donde R6 y R9 pueden tener diversas alternativas y t es de 0 a 4.

Los procesos divulgados en el presente documento para la fabricación de los compuestos de fórmula (II-pa) implican o bien a) la reacción no enantioselectiva de una alquilamina con 2-arilacrilato para producir una mezcla racémica o bien b) la adición asimétrica de una alcoximetanamina a 2-fenilacetato que contiene un auxiliar quiral apropiado. Ambos procesos no implican una hidrogenación asimétrica sino una reacción de adición. Por consiguiente, ambos procesos requieren etapas adicionales para la adición, escisión y separación del auxiliar.
La síntesis de acuerdo con el método a) anterior, se lleva a cabo mediante la formación de un intermedio de éster racémico que se hidroliza adicionalmente al ácido racémico, se acopla con un auxiliar quiral (como solamente, por ejemplo, el enantiómero S) para genera una mezcla 50:50 de diastereómeros aminoácido R/auxiliar S y aminoácido S/auxiliar S. Los diastereómeros tienen que separarse por cromatografía. El rendimiento del intermedio S-S deseado es solamente del 38 %. Además, el intermedio S-S tiene que hidrolizarse para proporcionar el ácido S-II (con pérdida del otro componente quiral, el auxiliar quiral). Este procedimiento es largo y poco eficaz, ya que en una etapa se pierde en 72 % del material. En resumen, la adición no enantioselectiva de amina y acrilato muestra el problema intrínseco de la falta de estereoselectividad y, por lo tanto, la separación obligatoria de la mezcla racémica mediante, por ejemplo, cromatografía. Por consiguiente, el rendimiento es al menos el 100 % inferior en comparación con una secuencia estereoselectiva.
Además, la adición asimétrica a un intermedio que contiene un auxiliar quiral (método b) anterior) requiere etapas adicionales para la adición, escisión y separación del auxiliar. Un precursor en la síntesis del ácido diana se combina con un auxiliar quiral y el intermedio resultante se acopla con una alcoximetanamina. El producto consiste, en el mejor de los casos, en una mezcla ligeramente enriquecida de los diastereómeros R/S y S/S, si no una mezcla 1:1, que tienen que tratarse adicionalmente tal como se ha mencionado anteriormente para aislar el isómero (S) del compuesto de fórmula (II-pa) con un, en el mejor de los casos, modesto rendimiento.
Existe, por lo tanto, una necesidad no reconocida de procesos mejorados para la preparación de compuestos de fórmula (II) que proporcionen una mejor estereoselectividad evitando la posterior cromatografía quiral, que requieran menos etapas de reacción, que proporcionen un rendimiento más elevado y que sean, por lo tanto, más eficientes, más ecológicos y menos costosos.
Los inventores de la presente invención han encontrado un nuevo proceso para la fabricación de compuestos de fórmula (II) que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV) usando un catalizador de complejo de metal (C).
Este nuevo proceso para la fabricación de compuestos de fórmula (II) presenta diversos beneficios relevantes en comparación con los procesos conocidos en la técnica:
Se introduce una reacción altamente estereoselectiva en la síntesis;
Se evita la posterior purificación usando cromatografía quiral;
Se disminuye el número de etapas de reacción;
Se aumenta el rendimiento general;
La reacción general es más eficaz, más ecológica y menos costosa.
Se ha descubierto que los catalizadores de complejo de metal particulares de la presente invención son mucho más eficaces y mucho más activos y selectivos que otros catalizadores conocidos, en el sentido de que, en condiciones de reacción similares (es decir, sin aditivos), se pueden emplear en una relación molar sustrato/catalizador (S/C) de hasta 10.000 mientras otros catalizadores necesitan usarse en una S/C de 200-250. Por lo tanto, el uso de 40-50 veces menos catalizador tiene un impacto sustancial en eficacia, coste y ecologismo.
Ciertos catalizadores conocidos requieren una gran cantidad de LiBF4 como aditivo (hasta 5,8 % en moles para el sustrato de hidrogenación, hasta 100 equivalentes molares para el catalizador) para aumentar la actividad de los catalizadores. Elevadas cantidades de LiBF4 son desventajosas para un proceso industrial, debido a que la presencia de esta gran cantidad de iones fluoruro (hasta un 23,2 % del sustrato de hidrogenación) plantea un problema en cuanto a la corrosión de los reactores a presión de acero a gran escala. Por otro lado, incluso con el aditivo LiBF4 el catalizador no alcanza la actividad de nuestros nuevos catalizadores (por ejemplo, hasta una S/C de
10.000).
Las reacciones catalizadas de forma homogénea tales como, por ejemplo, hidrogenaciones asimétricas, tal como se conocen en la técnica, requieren procedimientos de tratamiento muy ventajosos, que comprenden muchos ciclos de extracción y concentración de soluciones. Además, las hidrogenaciones asimétricas, tal como se conocen en la técnica, requieren la eliminación de catalizadores metálicos con un eliminador (por ejemplo, resinas de tiol) en grandes cantidades (hasta el 6 % en peso para el sustrato de hidrogenación; hasta 193 veces el peso del catalizador). Dicha eliminación de los contaminantes de rutenio usando resinas eliminadoras es, de lejos, difícil y bastante costosa. Además, el contenido de rutenio se reduce solamente en parte (por ejemplo, a aproximadamente 50 ppm) y se lleva a cabo durante la etapa siguiente, aumentando así el potencial para la formación de subproductos. Esto añade costes de material y mano de obra y abre el debate sobre posibles impurezas.
En conclusión, la eficacia de los procesos de purificación y aislamiento del producto de hidrogenación a partir de los catalizadores y aditivos es baja.
En contraste, el proceso de acuerdo con la invención proporciona sales del compuesto de fórmula (II) que se precipitan directamente de la mezcla de hidrogenación y que se pueden eliminar fácilmente por filtración. Dichos aislamiento y purificación del producto de hidrogenación proporcionan altos rendimientos (> 94 %) con el 100 % de ee y con un contenido de rutenio por debajo del límite de detección de 5 ppm. El tratamiento del producto de reacción de la hidrogenación asimétrica, tal como han descubierto los presentes inventores, es por tanto sustancialmente más simple, más barata y más útil que los procesos convencionales.
Un aspecto de la presente invención se refiere a un compuesto de fórmula (II)

en donde R1 y M son como se definen en el presente documento.
Un aspecto de la presente invención se refiere a un compuesto de fórmula (II) que es (S)-3-(tercbutoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio.
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (II)
que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV)

usando un catalizador de complejo de metal (C) en donde R1 y M son como se definen en el presente documento. En una realización de la invención, el catalizador de complejo de metal (C) es un catalizador de complejo de rutenio. En una realización de la invención, el catalizador de complejo de rutenio comprende rutenio caracterizado por el número de oxidación II.
En una realización de la invención, el catalizador de complejo de rutenio comprende un ligando de fosfina quiral (D). En una realización de la invención, el catalizador de complejo de rutenio comprende ligandos, en particular ligandos neutros (L) y/o ligandos aniónicos (Z).
Son ejemplos de ligandos neutros (L) olefinas tales como etileno o propileno, cicloocteno, 1,3-hexadieno, norbornadieno, 1,5-ciclooctadieno, benceno, hexametilbenceno, 1,3,5-trimetilbenceno y p-cimeno o también disolventes tales como tetrahidrofurano, dimetilformamida, acetonitrilo, benzonitrilo, acetona, tolueno y metanol. Son ejemplos de ligandos aniónicos (Z) acetato (CH3COO-), trifluoroacetato (CF3COO-), n5-2,4-pentadienilo, n5-2,4-dimetil-pentadienilo e iones de halógeno tales como fluoruro, cloruro, bromuro o yoduro.
Si el catalizador de complejo de rutenio está cargado, este comprende además aniones no coordinantes (Y). Son ejemplos de aniones no coordinantes (Y) iones de halógeno tales como fluoruro, cloruro, bromuro o yoduro, BF4-, ClO4-, SbF6-, PF6-, B(fenilo)4-, B(3,5-di-trifluorometil-fenilo)4-, CF3SO3- y C6H5SO3-.
Opcionalmente, el catalizador de complejo de rutenio se puede coordinar adicionalmente con un ácido de Lewis, tal como AlCl3.
En una realización de la invención, el catalizador de complejo de rutenio se selecciona entre un compuesto de fórmula (C1), (C2) o (C3):
Ru(Z)3D (C1)
[Ru(Z)2-p(D)(L)m](Y)p (C2)
Ru(E)(E')(D)(F) (C3)
en donde:
D es un ligando de fosfina quiral;
L es un ligando neutro seleccionado entre alqueno C2-7, cicloocteno, 1,3-hexadieno, norbornadieno, 1,5-ciclooctadieno,
benceno, hexametilbenceno, 1,3,5-trimetilbenceno, p-cimeno, tetrahidrofurano, dimetilformamida, acetonitrilo, benzonitrilo, acetona, tolueno y metanol;
Z es un ligando aniónico seleccionado entre hidruro, fluoruro, cloruro, bromuro, n5-2,4-pentadienilo, n5-2,4-dimetil-pentadienilo o el grupo A-COO-,
con la condición de que cuando dos Z se unen al átomo de Ru estos pueden ser tanto iguales como diferentes;
A es alquilo C1-7, haloalquilo C1-7, arilo o haloarilo;
Y es un anión no coordinante seleccionado entre fluoruro, cloruro, bromuro, BF4-, ClO4-, SbF6-, PF6-, B(fenilo4-, B(3,5-di-trifluorometil-fenilo)4-, CF3SO3-, y C6H5SO3-;
F es una diamina opcionalmente quiral;
E y E' son ambos iones halógeno o E es hidruro y E' es BH4-;
m es 1,2, 3 o 4;
p es 1 o 2.
En una realización particular de la invención, el catalizador de complejo de rutenio se selecciona entre un compuesto de fórmula (C1) o (C2) en donde Z, D, L, Y, m y p son como se describen en el presente documento.
En una realización particular de la invención, el catalizador de complejo de rutenio se selecciona entre un compuesto de fórmula (C1), en donde Z y D son como se describen en el presente documento.
En una realización particular de la invención, el catalizador de complejo de rutenio es Ru(Z)2D, en donde Z y D son como se describen en el presente documento.
En una realización particular de la invención, el catalizador de complejo de rutenio se selecciona entre un compuesto de fórmula (C2), en donde Z, D, L, Y, m y p son como se describen en el presente documento.
En una realización particular de la invención, el catalizador de complejo de rutenio se selecciona entre un compuesto de fórmula (C3), en donde E, E', D y F son como se describen en el presente documento.
En una realización particular de la invención, el ligando aniónico (Z) se selecciona independientemente entre cloruro, bromuro, yoduro, OAc y TFA.
En una realización particular de la invención, el ligando aniónico (Z) es A-COO-.
En una realización particular de la invención, A es -CF3.
En una realización particular de la invención, el ligando aniónico (Z) es trifluoroacetato (TFA).
En una realización particular de la invención, el ligando neutro (L) se selecciona independientemente entre benceno (C6H6), p-cimeno (pCym) y acetonitrilo (AN).
En una realización particular de la invención, el ligando neutro (L) es benceno (C6H6).
En una realización particular de la invención, el anión no coordinante (Y) se selecciona entre cloruro, bromuro, yoduro y BF4-.
En una realización particular de la invención, el anión no coordinante (Y) es BF4". En una realización particular de la invención, m es 1 o 4.
En una realización particular de la invención, m es 1.
En una realización particular de la invención, m es 4.
En una realización particular de la invención, p es 1.
En una realización particular de la invención, p es 2.
En una realización particular de la invención, E y E' son ambos cloruro;
En una realización particular de la invención, la diamina quiral F es (1S,2S)-1,2-difeniletilendiamina (S,S-DPEN). En una realización particular de la invención, el catalizador de complejo de rutenio está coordinado con un ácido de Lewis, en particular AlCh.
En una realización de la invención, el ligando de fosfina quiral D se selecciona entre un compuesto de fórmula (D1) a (D12):


R11 es alquilo C1-7, alcoxi C1-7, benciloxi, hidroxi o alquilo Ci-7-C(O)O-;
cada uno de R12 y R13 es independientemente hidrógeno, alquilo C1-7, alcoxi C1-7 o dialquilamino (C1-7); o
R11 y R12, que están unidos al mismo grupo fenilo, o R12 y R13 que están unidos al mismo grupo fenilo, tomados juntos, son -X-(CH)rY-, en donde X es -O- o -C(O)O-, Y es -O-, -N(alquilo inferior)- o -CF2- y r es un número entero de 1 a 6; o
dos R11 tomados juntos son -O-(CH2)s-O- u O-CH(CH3)-(CH2)s-CH(CH3)-O-, en donde s es un número entero de 1 a 6; o R11 y R12 o R12 y R13, junto con los átomos de carbono a los que están unidos, forman un anillo naftilo, tetrahidronaftilo o dibenzofurano;
R14 y R15 son cada uno independientemente alquilo C1-7, cicloalquilo C3-8, fenilo, naftilo o heteroarilo, opcionalmente sustituidos con de 1 a 7 sustituyentes seleccionados independientemente entre el grupo que consiste en alquilo C1-7, alcoxi C1-7, dialquilamino (C1-7), morfolinilo, fenilo, trisilil(alquilo C1-7), alcoxicarbonilo C1-7, hidroxicarbonilo, hidroxisulfonilo, (CH2)t-OH y (CH2)t-NH2, en donde t es un número entero de 1 a 6;
R16 es alquilo C1-7;
R17 es alquilo C1-7; y
R18 independientemente es arilo, heteroarilo, cicloalquilo C3-8 o alquilo C1-7.
En una realización particular de la invención, el ligando de fosfina quiral (D) se selecciona entre el compuesto de fórmula (D1), en donde R11 a R15 son como se describen en el presente documento.
En una realización particular de la invención, el ligando de fosfina quiral (D) se selecciona entre (R)-3,5-Xyl-BINAP, (R) -BINAP, (S)-2-furil-MeOBIPHEP, (S)-BINAP, (S)-BINAPHANE, (S)-BIPHEMP, (S)-MeOBIPHEP, (S)-pTol-BINAP), (S) -TMBTP y (S,S)-iPr-DUPHOS.
En una realización particular de la invención, el ligando de fosfina quiral (D) se selecciona entre (S)-BIPHEMP, (S)-BINAP y (S)-MeOBIPHEP.
En una realización particular de la invención, el ligando de fosfina quiral (D) es (S)-BINAP.
En una realización particular de la invención, el ligando de fosfina quiral (D) es (S)-2,2'-bis(difenilfosfino)-1,1'-binaftilo.
En una realización particular de la invención, el ligando de fosfina quiral (D) es

En una realización particular de la invención, el catalizador de complejo de rutenio se selecciona entre el grupo de:
Ru(TFA)2((R)-3,5-Xyl-BINAP),
Ru(OAc)2((S)-2-furil-MeOBIPHEP),
Ru(OAc)2((S)-BINAP),
[Ru(OAc)2((S)-BINAP)]AlCla,
Ru(TFA)2((S)-BINAP),
Ru(TFA)2((S)-BINAPHANE),
Ru(TFA)2((S)-BIPHEMP),
Ru(OAc)2((S)-MeOBIPHEP),
Ru(TFA)2((S)-TMBTP),
Ru(TFA)2((S,S)-iPr-DUPHOS),
[Ru((R)-BINAP)(pCym)(AN)](BF4)2,
[RuBr((S)-BINAP)(CaH6)]Br,
[RuCl((S)-BINAP)(C6H6)]BF4,
[RuCl((S)-BINAP)(C6H6)]Cl,
[RuI((S)-BINAP)(C6H6)]I,
[Ru((S)-BINAP)(AN))4](BF4)2 y
RuCl2((S)-pTol-BINAP)(S,S-DPEN).
En una realización particular de la invención, el catalizador de complejo de rutenio se selecciona entre el grupo de:
Ru(TFA)2((R)-3,5-Xyl-BINAP),
Ru(OAc)2((S)-2-furil-MeOBIPHEP),
Ru(OAc)2((S)-BINAP),
[Ru(OAc)2((S)-BINAP)]AlCla,
Ru(TFA)2((S)-BINAP),
Ru(TFA)2((S)-BINAPHANE),
Ru(TFA)2((S)-BIPHEMP),
Ru(OAc)2((S)-MeOBIPHEP),
Ru(TFA)2((S)-TMBTP),
Ru(TFA)2((S,S)-iPr-DUPHOS),
[Ru((R)-BINAP)(pCym)(AN)](BF4)2,
[RuBr((S)-BINAP)(C6H6)]Br,
[RuCl((S)-BINAP)(C6H6)]BF4,
[RuI((S)-BINAP)(C6H6)]I,
[Ru((S)-BINAP)(AN))4](BF4)2 y
RuCl2((S)-pTol-BINAP)(S,S-DPEN).
En una realización particular de la invención, el catalizador de complejo de rutenio es un compuesto de fórmula (C1) seleccionado entre el grupo de:
Ru(TFA)2((R)-3,5-Xyl-BINAP),
Ru(OAc)2((S)-2-furil-MeOBIPHEP),
Ru(OAc)2((S)-BINAP),
[Ru(OAc)2((S)-BINAP)]AlCl3,
Ru(TFA)2((S)-BINAP),
Ru(TFA)2((S)-BINAPHANE),
Ru(TFA)2((S)-BIPHEMP),
Ru(OAc)2((S)-MeOBIPHEP),
Ru(TFA)2((S)-TMBTP) y
Ru(TFA)2((S,S)-iPr-DUPHOS).
En una realización particular de la invención, el catalizador de complejo de rutenio es un compuesto de fórmula (C2) seleccionado entre el grupo de:
[Ru((R)-BINAP)(pCym)(AN)](BF4)2,
[RuBr((S)-BINAP)(C6H6)]Br,
[RuCl((S)-BINAP)(C6H6)]BF4,
[RuCl((S)-BINAP)(CaHa)]Cl,
[RuI((S)-BINAP)(C6H6)]I y
[Ru((S)-BINAP)(AN)4](BF4)2.
En una realización particular de la invención, el catalizador de complejo de rutenio es un compuesto de fórmula (C3), en particular RuCl2((S)-pTol-BINAP)(S,S-DPEN)).
En una realización particular de la invención,  el catalizador de complejo de rutenio es Ru(TFA)2((S)-BINAP). En una realización particular de
el catalizador de complejo de rutenio es Ru(TFA)2((S)-BINAP). En una realización particular de  invención,
invención,  el catalizador de complejo de rutenio es [RuCl(S-BINAP)(CaHa)]Cl. En una realización particular de la invención,
el catalizador de complejo de rutenio es [RuCl(S-BINAP)(CaHa)]Cl. En una realización particular de la invención,  el catalizador de complejo de rutenio no es [RuCl(S-BINAP)(CaHa)]Cl. En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se re en un disolvente seleccionado entre alcoholes, hidrocarburos, hidrocarburos clorados, hidrocarburos alifáticos o aromáticos, fluorados o polifluorados, dióxido de carbono supercrítico o líquido, THF, agua o mezclas de los mismos.
el catalizador de complejo de rutenio no es [RuCl(S-BINAP)(CaHa)]Cl. En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se re en un disolvente seleccionado entre alcoholes, hidrocarburos, hidrocarburos clorados, hidrocarburos alifáticos o aromáticos, fluorados o polifluorados, dióxido de carbono supercrítico o líquido, THF, agua o mezclas de los mismos.
Son disolventes particulares para la hidrogenación asimétrica alcoholes, hidrocarburos clorados y THF.
Los disolventes particulares para la hidrogenación asimétrica se seleccionan entre la lista de MeOH, EtOH, i-PrOH, EtOH/ciclopentil metil éter, EtOH/CH2Cl2, EtOH/EtOAc, EtOH/THF, EtOH/H2O, CH2Cl2 y THF.
El disolvente más particular para la hidrogenación asimétrica es etanol (EtOH).
Los disolventes pueden usarse solos o como la mezcla de disolventes mencionada anteriormente.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se realiza
a una concentración del compuesto de fórmula (IV) del 1 al 50 % en peso, en particular del 5 % en peso, el 10 % en
peso, el 20 % en peso o el 30 %en peso.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se realiza
a una concentración del 10 al 25 % en peso del compuesto de fórmula (IV).
Sorprendentemente, se ha encontrado que, en casos especiales, la adición de ciertos aditivos mejora la hidrogenación asimétrica de un compuesto de fórmula (IV). Se teoriza que la actividad, así como la estabilidad, del catalizador de rutenio se mejora sustancialmente y, por lo tanto, la cantidad de catalizador requerida se reduce.
Cantidades menores de catalizador empleadas dan como resultado tratamientos simplificados y costes reducidos.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) comprende además uno o más aditivos seleccionados entre la lista de LiBF4, LiPFa, LO 3SCF3, NaCl, NaBr, NaI, KCl,
KBr, KI, LiCl, LiBr, LiI, HBF4, HCl, HBr, H2SO4 y CH3SO3H.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) no comprende LiBF4, LiPFa o LO3SCF3 como aditivo. En vista de su carácter altamente corrosivo, dicho fluoruro que contiene aditivos es difícil de manejar y, por lo tanto, no se prefiere.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) comprende además uno o más aditivos seleccionados entre la lista de NaCl, NaBr, KCl, KBr, HCl y HBr.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) comprende además uno o más aditivos seleccionados entre la lista de LiBF4, HBF4, HCl, H2SO4 y CH3SO3H.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) comprende además uno o más aditivos seleccionados entre la lista de LiBF4, NaCl, NaBr, LiCl, LiBr, LiI, HBF4, HCl,
HBr, H2SO4 y CH3SO3H.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se lleva a
cabo con hidrógeno como fuente de hidrógeno.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se lleva a cabo a presión de hidrógeno de 100 a 15000 kPa (1 a 150 bar), en particular de 1000 a 3000 kPa (10 a 30 bar), más en particular de 1700 a 2100 kPa (17 a 21 bar).
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se realiza a temperatura de 10 a 120 °C, en particular de 20 a 90 °C.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se realiza durante un periodo de tiempo de 5 a 30 h, en particular de 6 a 25 h, más en particular de 6 a 23 h.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se lleva a cabo a una relación sustrato/catalizador (S/C) de 5 a 100.000, en particular de 100 a 15.000, más en particular de 100 a 10.000.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se realiza por lotes.
En una realización particular de la invención, la hidrogenación asimétrica de un compuesto de fórmula (IV) se realiza de un modo continuo.
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (II)

que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV)

usando un catalizador de complejo de metal (C), seguido de la formación de una sal añadiendo a la mezcla de reacción de hidrogenación una solución alcohólica de un alcóxido de metal de fórmula alquil C1-7-OM, en donde R1 y M son como se definen en el presente documento.
Un aspecto de la presente invención se refiere al proceso para la fabricación de los compuestos de fórmula (II) que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV) que usa un catalizador de complejo de metal (C), seguido de la formación de una sal añadiendo a la mezcla de reacción de hidrogenación una solución alcohólica de un alcóxido de metal de fórmula alquil C1-7-OM, sin purificación o purificación previos del ácido intermedio, en donde R1 y M son como se definen en el presente documento.
En una realización particular de la invención, el alcóxido de metal empleado en la etapa de formación de la sal es MeOM, EtOM, iPrOM, nPrOM, nBuOM, iBuOM o tBuOM, más en particular EtOM.
En una realización particular de la invención, el alcohol usado como disolvente en la etapa de formación de la sal es alquil C1-7-OH, más en particular MeOH, EtOH, iPrOH, nPrOH, nBuOH, iBuOH o tBuOH, la más particular EtOH. Un aspecto de la presente invención se refiere al proceso para la fabricación de los compuestos de fórmula (II) que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV) que usa un catalizador de complejo de
metal (C), seguido de formación de una sal añadiendo a la mezcla de reacción de hidrogenación una solución etanólica de etóxido sódico.
Los compuestos de fórmula (IV) pueden prepararse de acuerdo con los métodos conocidos por los expertos en la técnica. En el esquema 2 se representa un método particular de preparación general de los compuestos de fórmula (IV). Para una descripción más detallada de etapas de reacción individuales, véase la sección de Ejemplos a continuación.
Esquema 2

Un compuesto de fórmula (IVa), en donde R3 es alquilo Ci-7 opcionalmente sustituido, en particular etilo, se condensa en condiciones básicas con un compuesto HCO2R4, en donde R4 es alquilo C1-7 opcionalmente sustituido, en particular etilo, para formar un compuesto de fórmula (IVb). La condensación adicional de los compuestos de fórmula (IVb) con una amina HN(isopropil)R5, en donde R5 es hidrógeno, alquilo C1-7 o un grupo protector de amino, forma compuestos de fórmula (IVc). Cuando R5 es hidrógeno en los compuestos de fórmula (IVc), puede realizarse protección adicional de la amina para proteger los compuestos de fórmula (IVc) (por ejemplo, donde R5 es un grupo protector de amino, tal como Boc). La hidrólisis del éster del compuesto (IVc) proporciona compuestos de fórmula (IV).
Los catalizadores de complejo de rutenio de la invención se pueden preparar, en principio, de una forma conocida per se. Se pueden aislar o usar directamente (preparación in situ), por ejemplo, de acuerdo con B. Heiser et al., Tetrahedron: Asymmetry 1991, 2, 51; o N. Feiken et al., Organometallics 1997, 16, 537; o J.-P. Genet, Acc. Chem. Res. 2003, 36, 908; o K. Mashima et al., J. Org. Chem. 1994, 53, 3064; Angew. Chem. Int. Ed. 1998, 37, 1703-1707; o MP. Fleming et al., documentos US 6.545.165 B1 y referencias citadas en ese documento; así como O. Briel et al. en Catalysis of Organic Reactions, CRC Press, Boca Ratón, 2009 específicamente para complejos de Ru basados en ferroceno.
La síntesis de [Ru(TFA)2((S)-BINAP)] se divulga en B. Heiser et al., Tetrahedron: Asymmetry 1991,2, 51.
Los catalizadores de complejo de rutenio pueden prepararse in situ, es decir justo antes de su uso y sin aislamiento. La solución en la que dicho catalizador se prepara puede contener ya el sustrato para la hidrogenación enantioselectiva o la solución puede se puede mezclar con el sustrato justo antes de que la reacción de hidrogenación se inicie.
El documento WO 2008/006040 divulga 5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-7-oles de fórmula (71) y métodos para fabricarlos, en donde R5 puede tener diversas alternativas.

En particular, el documento WO 2008/006040 divulga la reducción asimétrica de 5-metil-5,6-dihidrociclopenta[d]pirimidin-7-onas a (R) o (S)-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-7-oles usando un
catalizador quiral en presencia de hidrógeno, un catalizador Corey-Bakshi-Shibata (CBS), un agente reductor de borohidruro en presencia de un ligando quiral o un agente reductor no quiral (por ejemplo, H2, Pd/C).
Los métodos conocidos en la técnica para producir los compuestos de fórmula (III) muestran los inconvenientes intrínsecos de aquellos que requieren condiciones de reacción drásticas (por ejemplo, presiones elevadas), el uso de metales pesados y auxiliares quirales y la diastereoselectividad obtenida es solo limitada (es decir 88 % de) lo que requiere etapas adicionales de purificación.
Los inventores de la presente invención han encontrado procesos enzimáticos nuevos para la fabricación de compuestos de fórmula (III), en donde R2 es como se ha descrito en el presente documento.

Estos procesos nuevos para la fabricación de compuestos de fórmula (III) de acuerdo con la presente invención presentan diversos beneficios relevantes en comparación con los procesos conocidos en la técnica. Las ventajas de la reducción enzimática son su naturaleza catalítica, la diastereoselectividad muy alta que evita la necesidad potencial de una resolución posterior de los diastereómeros formados y las condiciones de reacción suaves. Además, no se requieren metales pesados ni auxiliares quirales.
La reducción enzimática de la presente invención simplifica los requisitos técnicos, reduce el número y la cantidad de ingredientes y posibilita un rendimiento espacio/tiempo más elevado. Las ventajas de la presente invención se ejemplifican como los criterios técnicos relevantes mejorados, tales como concentración del sustrato aumentada (hasta un 25 %), concentración del producto aumentada (hasta un 25 %), carga de cofactor disminuida (hasta 1/3000 del compuesto de fórmula (V)) y un sistema de regeneración de cofactor más simple con un 2-propanol como reductor final. El sistema de regeneración de cofactor con 2-propanol como reductor final evita una segunda enzima, reduce la viscosidad, evita la neutralización continua del ácido glucónico como el cosustrato oxidado y permite la eliminación continua de la acetona formada.
Un aspecto de la presente invención se refiere al proceso para la fabricación de los compuestos de fórmula (III)

que comprende la reducción asimétrica del compuesto de fórmula (V)

catalizado por una oxidorreductasa, en donde R2 es como se define en el presente documento.
En un aspecto de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una cetorreductasa.
En un aspecto de la invención, la oxidorreductasa cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) con una diastereoselectividad de al menos un 95 % de exceso diastereomérico (de), en particular con una diastereoselectividad de al menos el 98 % de, más en particular con una diastereoselectividad de al menos el 99 % de.
En un aspecto de la invención, la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se cataliza mediante una oxidorreductasa en presencia de un cofactor.
En un aspecto de la invención, el cofactor que se oxida en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es NADH o NADPH.
En un aspecto de la invención, el cofactor que se oxida en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se regenera in situ aplicando o bien la regeneración del cofactor acoplado a enzima (por ejemplo, basándose en glucosa como reductor final y glucosa deshidrogenasa) o bien la regeneración acoplada a sustrato (por ejemplo, usando un alcohol secundario como cosustrato).
En un aspecto de la invención, el cofactor que se oxida en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se regenera in situ mediante regeneración de cofactor acoplado a enzima usando glucosa y glucosa deshidrogenasa como cosustrato.
En un aspecto de la invención, el cofactor que se oxida en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se regenera in situ mediante regeneración acoplada a sustrato usando un alcohol secundario como cosustrato.
En un aspecto de la invención, el alcohol secundario como cosustrato para la regeneración acoplada a sustrato se selecciona entre 2-propanol, 2-butanol, butan-1,4-diol, 2-pentanol, pentan-1,5-diol, 4-metil-2-pentanol, 2-hexanol, hexan-1,5-diol, 2-heptanol o 2-octanol, en particular 2-propanol.
El 2-propanol es particularmente útil para la regeneración del cofactor en la misma enzima que también cataliza la reacción diana y la eliminación continua de la acetona formada.
En un aspecto de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva.
En un aspecto de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de:
KRED-NADPH-111 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-112 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-113 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-114 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-115 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-121 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-123 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-145 (de Codexis Inc., Redwood City, CA, EEUU),
KRED-NADPH-155 (de Codexis Inc., Redwood City, CA, EEUU),
A231 (de Almac Group Ltd. Craigavon, Reino Unido) y
KRED-NADPH-136 (de Enzysource, Hangzhou, China).
Otra oxidorreductasa adecuada que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de:
KRED-X1, una cetorreductasa genéticamente modificada de Lactobacillus kefir tal como se divulga en la publicación int. PCT n.° WO2010/025238A2 e identificada como SEQ. ID. NO. 34 y
KRED-X2, una cetorreductasa genéticamente modificada de Sporobolomyces salmonicolor como se divulga en la publicación int. PCT n.° WO2009/029554A2 e identificada como SEQ. ID. NO. 138.
Otra oxidorreductasa adecuada que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) son variantes de KRED-X1 que están disponibles en el mercado (de Codexis Inc., Redwood City, CA, Estados Unidos).
Es particularmente útil la cetorreductasa genéticamente modificada "KRED-X1-P1B06", una variante KRED "P1B06" del producto de placa especial de Codexis KRED "KRED-X1-SPECIALTY-PLT".
Otra oxidorreductasa adecuada que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) son variantes de KRED-X1 que están disponibles en el mercado (de Codexis Inc., Redwood City, CA, Estados Unidos). Son particularmente útiles las siguientes cetorreductasas genéticamente modificadas del producto de placa especial de Codexis KRED "KRED-X1.1-B06-SPECIALTY-PLT":
"KRED-X1.1-P1F01" (variante KRED P1F01),
"KRED-X1.1-P1H10" (variante KRED P1H10),
"KRED-X1.1-P1G11" (variante KRED P1G11),
"KRED-X1.1 -P1C04" (variante KRED P1C04),
"KRED-X1.1-P1C11" (variante KRED P1C11) y
"KRED-X1.1 -P1C08" (variante KRED P1C08).
Son particularmente útiles las cetorreductasas genéticamente modificadas "KRED-X1.1-P1C04" y "KRED-X1.1-P1F01". La cetorreductasa más particular es la cetorreductasa modificada genéticamente "KRED-X1.1-P1F01". Las publicaciones int. PCT n.° WO2010/025085A2 y WO2009/029554A2 se señalan como relevantes, en particular los aspectos de las mismas relacionados con la preparación y uso de oxidorreductasas.
Todas las enzimas anteriormente mencionadas podrían usar también el cofactor NADH.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-NADPH-111, KRED-NADPH-112, KRED-NADPH-113, KRED-NADPH-114, KRED-NADPH-115, KRED-NADPH-121, KRED-NADPH-123, KRED-NADPH-145, KRED-NADPH-155, A231, KRED-NADPH-136, KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11 y KRED-X1.1-P1C08.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED- X1.1-P1C04, KRED-X1.1-P1C11 y KRED-X1.1-P1C08.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1C04 y KRED-X1.1-P1F01. En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1, KRED-X1-P1B06, KRED-X1.1-P1C04 y KRED-X1.1-P1F01.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1 y KRED-X2.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1 y KRED-X1-P1B06.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción de un compuesto de fórmula (V) a un compuesto de fórmula (III) es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1.1-P1C04 y KRED-X1.1-P1F01.
En un aspecto particular de la invención, la oxidorreductasa que cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es la oxidorreductasa dependiente de NADPH diastereoselectiva KRED-X1.1-P1F01.
En un aspecto de la invención, la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un medio acuoso en presencia de uno o más codisolventes orgánicos.
En un aspecto de la invención, la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un medio acuoso en presencia de uno o más codisolventes orgánicos, en donde los codisolventes orgánicos están presentes en una concentración total del 1 al 50 % V, en particular del 4 al 40 % V.
En un aspecto de la invención, los codisolventes presentes en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se seleccionan entre la lista de glicerol, 2-propanol, dietiléter, ferc-butilmetiléter, éter diisopropílico, éter dibutílico, metil tetrahidrofurano, acetato de etilo, acetato de butilo, tolueno, heptano, hexano, ciclohexeno y mezclas de los mismos; en particular 2 -propanol.
El 2-propanol es particularmente útil como codisolvente, ya que puede servir como reductor final para la regeneración de cofactor acoplado a sustrato.
En un aspecto de la invención, la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza a una temperatura de la reacción entre 1 °C y 50 °C, en particular entre 20 °C y 45 °C.
Las temperaturas en el intervalo superior aumentan la velocidad de reacción y facilitan la eliminación de la acetona. En un aspecto de la invención, la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza a un pH de entre 5,5 y 8,5.
En un aspecto de la invención, la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un tampón acuoso. Los especialistas en la técnica conocen los tampones adecuados. Son tampones particulares el ácido 2-(N-morfolino)etanosulfónico (MES) o dihidrogenofosfato de potasio (PBS).
El intervalo de pH óptimo y por lo tanto cualquier tampón adecuado dependen de la oxidorreductasa particular empleada.
Un aspecto de la invención se refiere a la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III), en donde el compuesto de fórmula (V) está presente inicialmente a una concentración del 1 al 25 % en peso, en particular del 10 al 20 % en peso.
Un aspecto de la invención se refiere a la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III), en donde la concentración de la reacción (concentración total de la cetona de fórmula (V) y el alcohol quiral de fórmula (III) en la mezcla de reacción) es entre el 1 y el 25 % en peso, en particular entre el 10 y el 20 % en peso.
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (III) que comprende la reducción asimétrica del compuesto de fórmula (V) catalizado por una oxidorreductasa seguido de tratamiento por extracción o por filtración.
Un aspecto de la invención se refiere a la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) catalizado por una oxidorreductasa, en donde el producto se trata de manera convencional por extracción o por filtración.
La pureza del producto en bruto podría incrementarse más por cristalización o usarse tal cual en la siguiente secuencia de reacción para la fabricación de compuestos de fórmula (I).
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (III) que comprenden la reducción asimétrica del compuesto de fórmula (V) catalizado por una oxidorreductasa, seguido de tratamiento por extracción o por filtración y adicionalmente por cristalización.
Un aspecto de la invención se refiere a la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III), en donde el producto se trata de manera convencional por extracción o por filtración y adicionalmente por cristalización.
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (IVc):

o una de sus sales, en donde R1 y R3 se definen en el presente documento, que comprende poner en contacto un compuesto de fórmula (IVd):

o una de sus sales, con R1-X, en donde X es un grupo saliente, en condiciones suficientes para dar un compuesto de fórmula IVc o una sal del mismo.
En una realización, el proceso comprende fabricar (E)-3-(ferc-butoxicarbonil(isopropil)-amino)-2-(4-clorofenil)acrilato de etilo o una sal del mismo, en donde R1 es el grupo protector BOC, R3 es etilo y en donde R1-X es (BOC)20.
En una realización particular, el proceso comprende poner en contacto un compuesto de fórmula IVd o una sal del mismo con menos de aproximadamente 8 equivalentes de (BOC)2O, en particular menos de aproximadamente 4 equivalentes, más en particular aproximadamente 3 equivalentes en condiciones que dan un compuesto de fórmula IVc o una sal del mismo en rendimientos de más de aproximadamente el 50 %, en particular de aproximadamente el 75 % o más de rendimiento, en una mezcla de disolvente polar que comprende DMF.
En una realización más particular, las condiciones comprenden poner en contacto un compuesto de fórmula IVd o una sal del mismo con aproximadamente 3 equivalentes de (BOC)2O y una mezcla básica que comprende aproximadamente 2 equivalentes de tributilamina y dimetilaminopiridina cada uno (DMAP), en una mezcla de disolvente polar que comprende DMF. En una realización, el proceso comprende además la eliminación de una porción del líquido de la mezcla de reacción al vacío durante la adición del (BOC)2O.
Los compuestos de fórmula (V) puede prepararse de acuerdo con métodos conocidos por los expertos en la técnica. Un método particular de preparación general de compuestos de fórmula (V) se representa en el esquema 3. Para una descripción más detallada de etapas de reacción individuales, véase la sección de Ejemplos a continuación.
Esquema 3

La reacción de un compuesto de fórmula (Va) con un agente de yodación (por ejemplo, sal de yoduro, tal como Nal y opcionalmente con un ácido) proporciona una diyodopirimidina de fórmula (Vb), que además puede reaccionar con una piperazina monoprotegida para proporcionar un compuesto de fórmula (Vc). El compuesto de fórmula (Vc) se somete a metalación con un agente metalante, tal como un reactivo de Grignard (por ejemplo, un haluro de alquilmagnesio C1-7, tal como iPrMgCl) para formar un compuesto de fórmula (Vd) que se vuelve a ciclar para formar una ciclopentil cetona de fórmula (V), en donde
R2 es como se ha descrito en el presente documento,
G es Li o Mg,
R6 es Cl u OH,
R7 es -CN, -COORa o -CONRaRb, en donde Ra y Rb se seleccionan independientemente entre la lista de hidrógeno, -OH, alcoxi C1-7, alquilo C1-7, alquenilo C2-7, alquinilo C2-7, cicloalquilo C3-8, fenilo o heterocicloalquilo de 3 a 12 miembros; o Ra y Rb se toman junto con el átomo de nitrógeno al que están unidos para formar un heterocicloalquilo de 3-7 miembros.
El documento WO 2008/006040 divulga métodos para fabricar compuestos de fórmula (73, en donde los compuestos de fórmula (71) después de la desprotección usando un ácido se acilan con el aminoácido apropiado, en donde R y R5 pueden tener diversas alternativas.

Las reacciones de acilación tal como se describen en la técnica anterior muestran los inconvenientes siguientes:
• Un proceso que emplea HBTU como reactivo de acoplamiento no es adecuado para un proceso de fabricación comercial a gran escala. El HBTU plantea graves problemas de higiene industrial ya que se describen en la bibliografía casos de anafilaxia y dermatitis por contacto alérgica ocupacional (Hannu T. et al, Occup Med, 2006, 56 (6), 430-433 y M. A. Aleer et al, Contact Dermatitis, 201062, 2123).
• Un proceso que emplea diclorometano como disolvente no es adecuado para un proceso de fabricación comercial a gran escala dado que está clasificado como contaminante peligroso en el aire (HAP) en los EEUU. Además, el diclorometano está clasificado por la International Conference on Harmonisation (ICH) como solvente de clase 2 con una escasa exposición diaria permitida (PDE) debido a su toxicidad inherente.
• La purificación de un producto usando cromatografía no es un método de purificación aceptable para la fabricación de moléculas pequeñas a gran escala, debido a su consumo de disolventes muy alto y su bajo rendimiento.
• En el caso de que un proceso de reacción a gran escala comercial implique más de un disolvente en una mezcla, los disolventes requieren distintos puntos de ebullición suficientemente separados entre sí como para permitir la separación entre ellos y el reciclado usando destilación. Los procesos, que implican en la misma etapa cuatro disolventes (por ejemplo, con ciclopentil metil éter (CPME)) que no son reciclables debido a que la mezcla es inseparable, no son adecuados para la fabricación a gran escala.
• El tratamiento de los productos requiere numerosas (por ejemplo, seis) extracciones acuosas, todas ellas con sales inorgánicas concentradas, dando como resultado cantidades significativas de agua residual contaminada. Dichas condiciones de proceso dan como resultado procesos de producción ambientalmente desventajosos.
Los inventores de la presente invención han descubierto un proceso nuevo mejorado para la fabricación de un compuesto de fórmula (I), que comprende el acoplamiento de un compuesto de fórmula (II), que es una sal, en particular sal sódica, a un compuesto de fórmula (III). Se ha encontrado que el uso del compuesto de fórmula (II) en forma de sal, en particular una sal sódica, facilita y simplifica dicho proceso de manera sustancial, en comparación con el uso de un aminoácido libre.
El proceso para la fabricación de compuestos de fórmula (I) de acuerdo con la invención presenta una serie de ventajas relevantes en comparación con los procesos descritos en la técnica, entre otras, por ejemplo:
• El tratamiento del compuesto de fórmula (I) se mejora de manera considerable. Solamente se emplean tres disolventes (isopropanol, tolueno y heptano), que se separan con facilidad.
• El anhídrido propilfosfónico (T3P) es un agente de acoplamiento no tóxico sin propiedades alergénicas ni sensibilizantes.
• Los subproductos de la reacción son solubles en agua y, por lo tanto, pueden eliminarse con facilidad mediante, por ejemplo, extracción acuosa triple.
Un aspecto de la presente invención proporciona un proceso para la preparación de un compuesto de fórmula (I)

o sales del mismo, que comprende la reacción de acoplamiento de un compuesto de fórmula (II)

con un compuesto de fórmula (III)
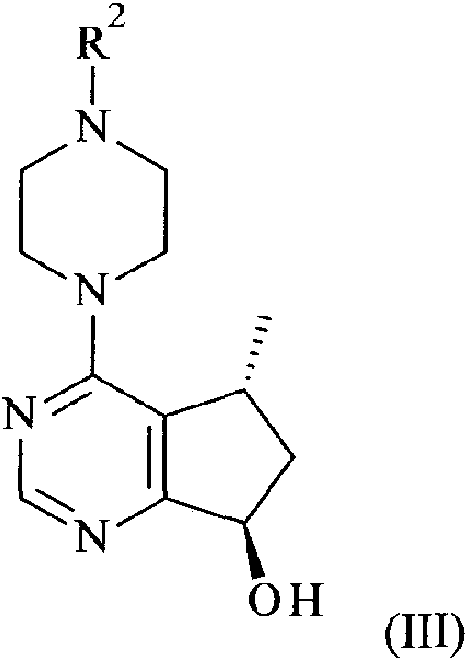
en donde R1, R2 y M son como se definen en el presente documento.
Un aspecto de la presente invención proporciona un proceso para la preparación de un compuesto de fórmula (I) o sales del mismo, que comprende la reacción de acoplamiento de un compuesto de fórmula (II) con un compuesto de fórmula (III) en donde R1, R2 y M son como se definen en el presente documento, que comprende las etapas de reacción siguientes:
a) Desprotección del compuesto de fórmula (III) en un disolvente en condiciones ácidas;
b) Ajuste a pH alcalino usando una base;
c) Adición de una solución que comprende el compuesto de fórmula (II) en un disolvente;
d) Adición de una solución que comprende un agente de acoplamiento en un disolvente.
En un aspecto de la invención, la desprotección en la etapa a) se realiza usando ácido clorhídrico, ácido sulfúrico, ácido trifluoroacético o ácido bromhídrico.
En un aspecto particular de la invención, la desprotección en la etapa a) se realiza usando ácido clorhídrico.
En un aspecto de la invención, el disolvente usado para la desprotección en la etapa a) se selecciona entre agua, metanol, etanol, n-propanol, isopropanol, n-butanol y ferc-butanol.
En un aspecto particular de la invención, el disolvente usado para la desprotección en la etapa a) se selecciona entre n-propanol o isopropanol.
En un aspecto de la invención, la desprotección en la etapa a) se realiza a una temperatura desde 50 a 100 °C, en particular a 80 °C.
En un aspecto de la invención, la desprotección en la etapa a) se realiza durante un tiempo de reacción de 0,1 a 24 horas, en particular durante un tiempo de reacción de 1 a 2 horas.
En un aspecto de la invención, la base en la etapa b) es una base líquida seleccionada entre N-etil morfolina (NEM), trietilamina (TEA), tri(n-propil)amina (TPA), diisopropiletilamina (DIPEA), piridina y lutidina.
En un aspecto de la invención, la base en la etapa b) es N-etil morfolina (NEM).
En un aspecto de la invención, en la etapa b) se añaden de 4 a 8 equivalentes de base en relación con el compuesto de fórmula (III), en particular de 6 a 7 equivalentes de base, más en particular 6,5 equivalentes de base.
En un aspecto de la invención, el disolvente usado en la etapa c) es idéntico al disolvente usado en la etapa a). En un aspecto de la invención, el disolvente usado en la etapa c) se selecciona entre agua, metanol, etanol, npropanol, isopropanol, n-butanol y ferc-butanol.
En un aspecto particular de la invención, el disolvente en la etapa c) se selecciona entre n-propanol o isopropanol. En un aspecto de la invención, el agente de acoplamiento usado en la etapa d) es anhídrido propilfosfónico (T3P). En un aspecto de la invención, el disolvente usado en la etapa d) se selecciona entre metanol, etanol, n-propanol, isopropanol, n-butanol, ferc-butanol, tolueno, acetonitrilo, tetrahidrofurano, W,W-dimetilformamida, cloroformo, cloruro de metileno, diclorometano, dicloroetano, éter dietílico, acetona, metil etil cetona, dimetilsulfóxido, N,N-dimetil acetamida, N-metil pirrolidinona, dioxano, tetrahidropirano, piridina, 2-propanona, 2-butanona, etilenglicol dimetil éter, acetato de etilo, acetato de butilo, acetato de isopropilo y mezclas de los anteriores.
En un aspecto particular de la invención, el disolvente usado en la etapa d) se selecciona entre una mezcla de npropanol y tolueno o isopropanol y tolueno, más en particular una mezcla de n-propanol y tolueno.
En un aspecto de la invención, la reacción de acoplamiento en la etapa d) se realiza a una temperatura desde -10 a 50 °C, en particular desde 0 a 25 °C.
En un aspecto de la invención, la reacción de acoplamiento en la etapa d) se realiza durante un tiempo de reacción de 0,1 a 24 horas, en particular durante un tiempo de reacción de 1 a 4 horas.
Un aspecto de la presente invención se refiere a la reacción de acoplamiento de un compuesto de fórmula (II) con un compuesto de fórmula (III), en donde, después de la etapa d) el producto se trata por extracción acuosa.
En un aspecto particular de la invención, el tratamiento del producto después de la etapa d) comprende de una a seis extracciones con agua, en particular, tres extracciones con agua.
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (VI)

o sales farmacéuticamente aceptables de los mismos, en donde un compuesto de fórmula (I) se desprotege

en donde R1 es como se define en el presente documento.
Un aspecto de la presente invención se refiere al proceso para la fabricación de compuestos de fórmula (VI) o sales farmacéuticamente aceptables los mismos, en donde un compuesto de fórmula (I) se desprotege, en donde R1 es como se define en el presente documento, que comprende las etapas de reacción siguientes:
i) La desprotección del compuesto de fórmula (I) en un disolvente en condiciones ácidas;
ii) Ajuste del pH usando una base en un disolvente;
iii) Opcionalmente cristalizar el compuesto de fórmula (VI).
En un aspecto de la invención, la desprotección en la etapa i) se realiza usando ácido clorhídrico, ácido sulfúrico, ácido trifluoroacético o ácido bromhídrico.
En un aspecto particular de la invención, la desprotección en la etapa i) se realiza usando ácido clorhídrico.
En un aspecto de la invención, el disolvente usado para la desprotección en la etapa i) se selecciona entre agua, metanol, etanol, n-propanol, isopropanol y ferc-butanol o mezclas de los mismos.
En un aspecto particular de la invención, el disolvente usado para desprotección en la etapa i) se selecciona entre npropanol, isopropanol y una mezcla 1 :1 de n-propanol/agua.
En un aspecto de la invención, la desprotección en la etapa i) se realiza a una temperatura desde 30 a 100 °C, en particular a 80 °C.
En un aspecto de la invención, la desprotección en la etapa i) se realiza durante un tiempo de reacción de 1 a 24 horas, en particular durante un tiempo de reacción de 1 a 4 horas.
En un aspecto de la invención, la base en la etapa ii) es NaOH en una mezcla 1:1 de n-propanol/agua.
En un aspecto de la invención, la base en la etapa ii) es amoniaco.
En un aspecto de la invención, el disolvente usado en la etapa ii) es idéntico al disolvente usado en la etapa i).
En un aspecto de la invención, el disolvente usado en la etapa ii) se selecciona entre agua, metanol, etanol, npropanol, isopropanol, n-butanol y ferc-butanol o mezclas de los mismos.
En un aspecto particular de la invención, el disolvente en la etapa ii) se selecciona entre n-propanol, isopropanol y una mezcla 1:1 de n-propanol/agua.
En un aspecto particular de la invención, el ajuste del pH se realiza mediante la adición gota a gota de una solución de amoniaco (2-4 % en peso, en particular 3,8 % en peso) en isopropanol o, de una solución de NaOH (5-10 M, en particular 7 M) en una mezcla 1:1 de n-propanol/agua.
En un aspecto particular de la invención, el pH final después del ajuste de la etapa ii) es aproximadamente pH 6 , en particular entre pH 6 y 7.
En un aspecto de la invención, la cristalización en la etapa iii) se realiza mediante un cambio de disolvente a un disolvente de cristalización adecuado para la cristalización del compuesto de fórmula (VI).
En un aspecto particular de la invención, el disolvente de cristalización en la etapa iii) se selecciona entre tolueno, heptano, tetrahidrofurano, 2 -propanona, 2 -butanona, etilenglicol dimetil éter, acetato de etilo, acetato de butilo, acetato de isopropilo y mezclas de los mismos.
En un aspecto particular de la invención, el disolvente de cristalización en la etapa iii) es acetato de etilo.
Un aspecto de la divulgación se refiere a compuestos que se obtienen mediante cualquier proceso tal como se describe en el presente documento.
Un aspecto de la divulgación se refiere a composiciones farmacéuticas que comprenden compuestos que se obtienen mediante cualquier proceso tal como se describe en el presente documento.
Un aspecto de la divulgación se refiere a un compuesto de fórmula (VI) tal como se describe en el presente documento que comprende entre 1 ppb and 100 ppm del compuesto de fórmula (I), en donde R1 es como se define en el presente documento.
Un aspecto de la divulgación se refiere a un compuesto de fórmula (VI) tal como se describe en el presente documento que comprende entre 1 ppb and 1 ppm del compuesto de fórmula (I), en donde R1 es como se define en el presente documento.
Un aspecto de la divulgación se refiere a una composición farmacéutica que comprende compuestos de fórmula (VI) tal como se describen en el presente documento.
Un aspecto de la invención se refiere a un compuesto de fórmula (I) tal como se describe en el presente documento que comprende entre 1 ppb y 100 ppm del compuesto de fórmula (II), en donde R1 y M son como se definen en el presente documento.
Un aspecto de la invención se refiere a un compuesto de fórmula (I) tal como se describe en el presente documento que comprende entre 1 ppb y 1 ppm del compuesto de fórmula (II), en donde R1 y M son como se definen en el presente documento.
Un aspecto de la invención se refiere a un compuesto de fórmula (I) tal como se describe en el presente documento que comprende entre 1 ppb y 100 ppm del compuesto de fórmula (III), en donde R1 y R2 son como se definen en el presente documento.
Un aspecto de la invención se refiere a un compuesto de fórmula (I) tal como se describe en el presente documento que comprende entre 1 ppb y 1 ppm del compuesto de fórmula (III), en donde R1 y R2 son como se definen en el presente documento.
Un aspecto de la invención se refiere a un compuesto de fórmula (I) tal como se describe en el presente documento que comprende entre 1 ppb y 100 ppm del compuesto de fórmula (II) y entre 1 ppb y 100 ppm del compuesto de fórmula (III), en donde R1, R2 y M son como se definen en el presente documento.
Un aspecto de la invención se refiere a un compuesto de fórmula (I) tal como se describe en el presente documento que comprende entre 1 ppb y 1 ppm del compuesto de fórmula (II) y entre 1 ppb y 1 ppm del compuesto de fórmula (III), en donde R1, R2 y M son como se definen en el presente documento.
Ejemplos
Los siguientes ejemplos 1-15 se proporcionan para ilustración de la invención. No deben considerarse como limitantes del alcance de la invención, sino meramente como representativos de la misma.
Ejemplo 1
Ácido (E)-3-(íerc-butoxicarbonil(isopropil)amino)-2-(4-dorofenil)acrílico

En una solución de formiato de etilo (123,9 l, 1538,9 mol) en MTBE (189 l) se añadió 4-clorofenilacetato de etilo (120 kg, 604,1 mol). La mezcla se agitó a 15-30 °C durante 30 min y después se añadió una mezcla de t-BuOK (136,8 kg, 1219,1 mol) en MTBE (1215,8 l) mientras se mantenía la temperatura interna por debajo de 5 °C. La mezcla se agitó entre 0-10 °C durante 1,5 h. La mezcla de reacción se añadió a una solución acuosa de ácido clorhídrico (35%, 99,8 l en 560 l de H2O) manteniendo la temperatura interna por debajo de 10 °C. La mezcla se agitó durante 30 min entre 0-10 °C hasta que se observó un pH final = 2. Las capas se separaron y la capa orgánica se lavó con solución al 25 % de NaCl (496 l).
La mezcla se enfrió a -5 °C y después se añadieron lentamente isopropilamina (107,2 l, 1251,9 mol) y AcOH (70,5 l, 1233,3 mol) manteniendo la temperatura <10 °C. La mezcla se agitó durante 3 h a 0-10 °C y después la capa orgánica se lavó con H2O (760 l), Na2CO3 acuoso al 15 % (424 l) y después NaCl acuoso al 25 % (650 l). La capa acuosa se separó y se añadieron DMF (443 l) y DMAP (14,4 kg, 117,9 mol) a la solución orgánica. Después, la mezcla se calentó a 60-65 °C seguido de adición lenta de (Boc)2O (951,6 l, 4142 mol), DMF (228,6 l) y trietilamina (263,0 l, 1821,8 mol) durante 24 h. Después de agitar ~6 h, la mezcla se enfrió a temperatura ambiente y se añadieron MTBE (1434 l), agua (1010 l) y ácido cítrico acuoso al 10 % (938 l). La capa acuosa se separó y la mezcla se lavó mediante NaCl acuoso al 25 % (984 l). Después se concentró la capa orgánica mediante destilación hasta un volumen de trabajo mínimo (~240 l) mientras se mantenía la temperatura por debajo de 50 °C. La capa orgánica se agitó después durante 5 h a 0-5 °C y se filtró. La torta de filtro se lavó con heptano (20.6 l) y se secó para proporcionar 3-((ferc-butoxicarbonil)(iso-propil)amino)-2-(4-clorofenil)acrilato de (E)-etilo (148,55 kg, 63% de rendimiento en tres etapas) en forma de un sólido de color blanco.
Se añadió el 3-((terc-butoxicarbonil)(isopropil)amino)-2-(4-clorofenil)acrilato de (E)-etilo (133,5 kg, 362,9 mol) en una mezcla de H2O (252 l), NaOH (58,25 kg, 1456 mol) y EtOH (383,5 l) con agitación a temperatura ambiente. La mezcla se calentó a 40-45 °C durante 2,5 h hasta que se formó una solución transparente. La mezcla se concentró a un volumen de trabajo mínimo manteniendo la temperatura por debajo de 50 °C. Después, la mezcla se enfrió a 10 25 °C y se añadió una solución de HCl (842 l de HCl 2 N y 11 l de HCl al 35 %) hasta que se obtuvo un pH final = 2~4. La capa acuosa se separó y la capa orgánica se lavó con NaCl acuoso al 25 % (810 l). Se añadió n-heptano mientras se destilaba para formar una suspensión. El producto se recogió y se lavó con n-heptano y se secó a 40 45 °C durante -10 h para proporcionar 110,7 kg (90,5 % de rendimiento) de ácido (E)-3-(fercbutoxicarbonil(isopropil)amino)-2-(4-clorofenil)acrílico que tiene una pureza del 99,9 % mediante HPLC. Se confirmó la configuración E usando análisis de rayos X de cristal único.
Ejemplo 1a
3-((íerc-butoxicarbonM)-(isopropM)ammo)-2-(4-dorofeml)acrMato de (E)-etilo

A una solución concentrada de 2-(4-dorofenil)-3-(isopropilamino)acrilato de (E)-etilo (preparada como anteriormente en el ejemplo 1 a partir de 120 kg de 2-(4-clorofenil)acetato de etilo, 0,604 kmol) se le añadió DMF (354 kg) y el lote se concentró hasta 3 volúmenes. Se añadieron DMAP (14,0 kg, 114,6 mol) y n-Bu3N (224,21 kg, 1,21 kmol) y la mezcla se calentó a 70-75 °C y se añadió una solución de (BOC)2O (330 kg, 1,51 kmol) en solución de DMF (169 kg) durante 2 h a 70-75 °C. Después de completar la adición, se eliminaron 200 L de DMF al vacío durante 3 h por debajo de 75 °C. La adición de solución de (BOC)2O (6 8 , 6 kg, 0,314 kmol) en DMF (32,4 kg) continuó durante 0,5 h a 70-75 °C. Después de completar la adición, el lote se concentró a una temperatura inferior a 75 °C y después se enfrió a aproximadamente 23,5 °C. Se cargó MTBE (899,6 kg) y después se enfrió la mezcla a aproximadamente 12,6 °C. Se añadió solución de ácido cítrico monohidrato (197,4 kg) en agua (702 kg) a 10-20 °C. Las capas se separaron y la capa orgánica se lavó con NaCl acuoso al 5 % (582 kg). Las capas se cortaron y la capa orgánica se concentró a 240-360 l a menos de 50 °C. Después de que se cargara n-heptano (77 kg), la mezcla se concentró a 240-360 l a menos de 50 °C. Después de que se cargara n-heptano (70 kg), la suspensión se agitó durante 4 h a 0 10 °C y el producto se recogió por centrifugación. La torta se lavó con n-heptano (28,2 kg) y se obtuvo 3-((terc-butoxicarbonil)(isopropil)amino)-2-(4-clorofenil)acrilato de (E)-etilo (170,6 kg, 77% de rendimiento, 99,8A% h PlC), que se puede usar como anteriormente en este ejemplo 1 para preparar ácido (E)-3-(terc-butoxicarbonil(isopropil)amino)-2-(4-clorofenil)acrílico.
Ejemplo 2
(S)-3-(íerc-butoxicarboml(isopropM)ammo)-2-(4-dorofeml)propanoato de sodio

En una cámara de atmósfera inerte (contenido de O2 s 2 ppm), se cargó un autoclave de 50 ml con 6 ,8 g (20,0 mmol) de ácido (E)-2-(4-clorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico, 34 ml de etanol y 4 ,81 mg (0,0051 mmol, S/C 4.000) de [Ru(TFA)2((S)-BINAP]. Se llevó a cabo la hidrogenación asimétrica durante 7 h a 60 °C a 1800 kPa (18 bar) de hidrógeno. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave y se analizó una muestra de la solución de reacción de color amarillo para mostrar conversión > 99 % a ácido (S)-3-(terc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico con una relación enantiomérica S/R de 99,3 a 0,7. La mezcla de hidrogenación se transfirió con ayuda de 100 ml de terc-butil metil éter en un reactor de vidrio 1 1 en atmósfera de argón que contenía la mezcla de reacción en bruto de 6 experimentos de hidrogenación análogos (en total 47,9 g de ácido (E)-2-(4-clorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico), después se añadió gota a gota una solución en etanol de etóxido de sodio (52,3 ml, 140 mmol) a 50 °C con agitación. Se formó un precipitado de color amarillento que se agitó a la misma temperatura y después a temperatura ambiente durante una noche. El producto precipitado se eliminó por filtración, se lavó con una mezcla 4:1 de t-butil metil éter/etanol (300 ml) y con t-butil metil éter (200 ml), se secó al vacío hasta peso constante para producir (S)-3-(terc-butoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio con un 96,3 % de rendimiento (49,03 g) y con una relación enantiomérica S/R de >99,9 a < 0,1 % en forma de cristales de color blanco. RMN 1H (D2O): 8 7,32 (d, 2H), 7,22 (d, 2H), 3,65-3,85 dos s a, aril-CH y N-CHÍCH^)?). 3,55 (m, 2H), 1,29 (s, 9H), 1,00 (d, 3H), 0,80 (s a, 3H).
La relación enantiomérica se determinó mediante HPLC usando una columna Chiralpak-AD-3, 150mm*4,6mm. Eluyentes: A) n-heptano con ácido trifluoroacético al 0,10%, B) etanol, velocidad: 1,25 ml/min, 25 °C, 5 pl de
volumen de inyección, 220 nm. Tiempos de retención: Ácido (S)-3-(ferc-butoxicarbonil-isopropil-amino)-2-(4-clorofenil)-propiónico 2,48 min, ácido (R)-3-(ferc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico 2,77 min, ácido (£)-2-(4-dorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico 3,16 min.
Ejemplo 3
(S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)propanoato de sodio
En una cámara de atmósfera inerte (contenido de O2 á 2 ppm), se cargó un autoclave de 185 ml con 17,0 g (50,0 mmol) de ácido (£)-2-(4-clorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico, 70 ml de etanol y 4,74 mg (0,005 mmol, S/C 10.000) de [Ru(TFA)2((S)-BINAP]. Se llevó a cabo la hidrogenación asimétrica durante 22 h a 70 °C a 1800 kPa (18 bar) de hidrógeno. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave y se analizó una muestra de la solución de reacción de color amarillo para mostrar conversión > 99 % a ácido (S)-3-(ferc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico con una relación enantiomérica S/R de 98,2 a 1,8. La mezcla de hidrogenación se transfirió con ayuda de 200 ml de ferc-butil metil éter en un reactor de vidrio de 400 ml en atmósfera de argón, después se añadió gota a gota una solución en etanol de etóxido de sodio (18,7 ml, 50 mmol) a 50 °C con agitación. Se formó un precipitado de color amarillento que se agitó a la misma temperatura y después a temperatura ambiente durante un total de 3,5 h. El producto precipitado se eliminó por filtración, se lavó con una mezcla 4:1 de t-butil metil éter/etanol (80 ml) y con t-butil metil éter (20 ml), se secó al vacío hasta peso constante para producir (S)-3-(ferc-butoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio con un 94,6 % de rendimiento (17,35 g) y con una relación enantiomérica S/R de 100 a 0 % en forma de cristales de color blanco.
Ejemplo 3a
(S)-3-(ferc-butoxicarboml(isopropM)ammo)-2-(4-clorofeml)propanoato de sodio
En una cámara de atmósfera inerte (contenido de O2 á 2 ppm), se cargó un autoclave de 185 ml con 10,0 g (29,4 mmol) de ácido (£)-2-(4-clorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico, 125 ml de etanol, 0,118 ml de solución de NaBr en agua (1 M) and 5,77 mg (0,006 mmol, S/C 5.000) de [Ru(TFA)2((S)-BINAP]. Se llevó a cabo la hidrogenación asimétrica durante 22 h a 60 °C a 1800 kPa (18 bar) de hidrógeno. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave y se analizó una muestra de la solución de reacción de color amarillo para mostrar conversión > 99 % a ácido (S)-3-(ferc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico con una relación enantiomérica S/R de 98 a 2. La mezcla de hidrogenación se transfirió con ayuda de 10 ml de etanol a un reactor de 0,5 l. La mezcla de reacción se evaporó a 45 °C al vacío hasta un volumen residual de 65 ml.
Se añadieron 70 ml de ferc-butil metil éter a 45 °C. Después se añadió gota a gota una solución en etanol de etóxido sódico (11,4 g, 35 mmol) a 45 °C con agitación. El embudo se aclaró con 1,3 g etanol. Se formó un precipitado de color amarillento que se agitó a la misma temperatura durante 1 h y después a temperatura ambiente durante 1 h. El producto precipitado se eliminó por filtración, se lavó con una mezcla 1:1 de t-butil metil éter/etanol (13,6 g) y con tbutil metil éter (16 g), se secó al vacío hasta peso constante para producir (S)-3-(ferc-butoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio con un 89 % de rendimiento (9,52 g) y con una relación enantiomérica S/R de 100 a 0 % en forma de cristales de color blanco.
Ejemplo 3b
(S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)propanoato de sodio
En una cámara de atmósfera inerte (contenido de O2 á 2 ppm), se cargó un autoclave de 185 ml con 10,0 g (29,4 mmol) de ácido (E)-2-(4-clorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico, 120 ml de etanol (destilado en atmósfera de Ar), 0,118 ml de solución de NaCl en agua (1 M) y 5,8 mg (0,006 mmol, S/C 5.000) de [Ru(TFA)2((S)-BINAP]. Se llevó a cabo la hidrogenación asimétrica durante 12 h a 60 °C a 1800 kPa (18 bar) de hidrógeno. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave y se analizó una muestra de la solución de reacción de color amarillo para mostrar conversión > 99 % a ácido (S)-3-(ferc-butoxicarbonilisopropil-amino)-2-(4-cloro-fenil)-propiónico con una relación enantiomérica S/R de 99 a 1. La mezcla de hidrogenación se transfirió con ayuda de 10 ml de etanol a un reactor de 0,5 l. La mezcla de reacción se evaporó a 45 °C al vacío hasta un volumen residual de 65 ml.
Se añadieron 70 ml de ferc-butil metil éter a 20 °C. Después se añadió gota a gota una solución de etóxido sódico (21 % (m/m), 9,5 g, 29,4 mmol) en etanol a 45 °C con agitación. El embudo se aclaró con 1,3 g etanol. Se formó un precipitado que se agitó a la misma temperatura durante 1 h y después a temperatura ambiente durante 1 h. El producto precipitado se eliminó por filtración, se lavó con una mezcla 1:1 de t-butil metil éter/etanol (13,6 g) y con tbutil metil éter (16 g), se secó al vacío hasta peso constante para producir (S)-3-(ferc-butoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio con un 89 % de rendimiento (9,5 g) y con una relación enantiomérica S/R de 100 a 0% en forma de cristales de color blanco.
Ejemplo 3c
Ácido (S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)-propiómco
En una cámara de atmósfera inerte (contenido de O2 á 2 ppm), se cargó un autoclave de 185 ml con 17,1 g (50,0 mmol) de ácido (E)-2-(4-dorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico y 75 ml de etanol. En un matraz separado se trató una mezcla de 4,79 mg (0,005 mmol, S/C 10.000) de [Ru(TFA)2((S)-BINAP] y 8,3 ml de etanol con 1,67 ml (0,10 mmol) de una solución 60 milimolar de HCl en agua, la suspensión resultante se agitó durante 30 min y después se añadió al autoclave. Después de cerrar herméticamente el autoclave la hidrogenación asimétrica se llevó a cabo durante 12 h a 60 °C a 1800 kPa (18 bar) de hidrógeno. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave y se analizó una muestra de la solución de reacción de color amarillo para mostrar conversión del 99,5 % a ácido (S)-3-(terc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico con una relación enantiomérica S/R de 98,7 a 1,3.
Ejemplo 3d
Ácido (S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)-propiómco
Se repitió el procedimiento del ejemplo 3c usando HBr como aditivo. La hidrogenación se llevó a cabo con un 99,8 % de conversión, el (S)-ácido deseado se aisló en rendimiento cuantitativo con una relación enantiomérica S/R de 98,7:1,3.
Ejemplo 3e
Ácido (S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)-propiómco
En una cámara de atmósfera inerte (contenido de O2 á 2 ppm), un autoclave de 185 ml se cargó con 17,0 g (50,0 mmol) de ácido (E)-2-(4-clorofenil)-3-[(2-metilpropan-2-il)oxicarbonil-propan-2-ilamino]prop-2-enoico y 75 ml de etanol. En un matraz separado se trató una mezcla de 9,59 mg (0,010 mmol, S/C 5.000) de [Ru(TFA)2((S)-BINAP] y 9,8 ml de etanol con 0,20 ml (0,20 mmol) de una solución 1 molar de HCl en agua, la suspensión resultante se agitó durante 30 min y después se añadió al autoclave. Después de cerrar herméticamente el autoclave la hidrogenación asimétrica se llevó a cabo durante 12 h a 60 °C a 1800 kPa (18 bar) de hidrógeno. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave y se analizó una muestra de la solución de reacción de color amarillo para mostrar una conversión del 99,7 % a ácido (S)-3-(terc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico con una relación enantiomérica S/R de 99,0 a 1,0.
Ejemplo 3f
Ácido (S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)-propiómco
Se repitió el procedimiento del ejemplo 3f usando LiBr como aditivo. La hidrogenación se llevó a cabo con un 98,9 % de conversión, el (S)-ácido deseado se aisló en rendimiento cuantitativo con una relación enantiomérica S/R de 98,5:1,5.
Ejemplo 4
Ácido (S)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-clorofeml)-propiómco
En una cámara de atmósfera inerte (contenido de O2 á 2 ppm), se cargó un autoclave de 35 ml, equipado con un inserto de vidrio y una barra de agitación magnética, con 400 mg (1,18 mmol) de ácido (E)-2-(4-clorofenil)-3-[(2-metilpropan-2 -il)oxicarbonil-propan-2 -ilamino]prop-2 -enoico, 5,92 mg (0,00589 mmol) de [Ru(t Fa )2((S)-BINAP)] (S/C 2 0 0 ) y 4 ml de etanol. El autoclave se cerró herméticamente y se presurizó con 2 00 0 kPa ( 20 bar) de hidrógeno, la hidrogenación se llevó a cabo con agitación durante 14 horas a 60 °C. Después de enfriar a temperatura ambiente, se liberó la presión del autoclave, se evaporó al vacío la solución de etanol para dar ácido (S)-3-(terc-butoxicarbonilisopropil-amino)-2-(4-cloro-fenil)-propiónico con rendimiento cuantitativo y con una relación enantiomérica S/R de 99:1. La conversión fue >= 99,9 %.
Ejemplos 5.1 a 5.17 ácido (S) o (R)-3-(íerc-butoxicarbonil(isopropil)amino)-2-(4-clorofenil)propiónico
El procedimiento del ejemplo 4 se repitió usando diferentes catalizadores de rutenio quirales para producir los correspondientes isómeros (R) y (S) de ácido 3-(terc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico. Los resultados se muestran en la tabla 1, junto con el catalizador, el % de conversión y la relación enantiomérica S/R. La escala de la reacción en todos los experimentos fue 400 mg (a menos que se indique específicamente lo contrario en una nota al pie), la temperatura fue de 60 °C, la presión de hidrógeno fue de 2000 kPa (20 bar) con una relación S/C de 200, el tiempo de reacción fue de 14 h. El reactor fue un autoclave de 35 ml. La cantidad de aditivo indicada
pretende estar en relación con la cantidad de catalizador metálico.
T l 1

Autoclave de 35 ml, 1,7 g de escala; S/C 250, 22 h. b) 1,7 g de escala, S/C 250,14 h; c) 6 ,8 g de sustrato en autoclave de 50 ml, S/C 1500, 5 h. d) Catalizador preparado in situ por agitación de 2,56 mg [Ru(COD)(TFA)2]2 y 2,2 equivalentes molares de difosfina quiral en una cámara de atmósfera inerte en 3 ml de etanol durante 3 h a 50 °C.
Ejemplos 6.1 a 6.8
Ácido (S) o (R)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-dorofenM)propiómco
De un modo análogo a los ejemplos 5.1-5.16 se realizaron las hidrogenaciones siguientes usando diversas sustancias como aditivos para proporcionar los isómeros (R) y (S) de ácido 3-(terc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico con la pureza y la pureza enantiomérica indicadas en la tabla 2. La escala de la reacción en todos los experimentos fue 400 mg (a menos que se indique específicamente lo contrario en una nota al pie), la temperatura fue de 60 °C, la presión de hidrógeno fue de 1800-2000 kPa (18-20 bar) durante 4 h a una relación S/C de 200. El reactor fue un autoclave de 35 ml. La cantidad de aditivo indicada pretende estar en relación con la cantidad de catalizador metálico.
T l 2

Para a) Tiempo de reacción 14 h; b) S/C 4000, 18 h, escala de 6 ,8 g en un autoclave de 185 ml; c) S/C 200, 14 h, escala de 1,18 g, Autoclave de 30 ml, 2000 kPa (20 bar) de H2.
Ejemplos 7.1 a 7.11
Ácido (S) o (R)-3-(ferc-butoxicarboml(isopropil)ammo)-2-(4-dorofeml)propiómco
Se repitió el procedimiento de los ejemplos 5.1-5.16 pero las condiciones de reacción se variaron en los términos de presión de hidrógeno, concentración y disolvente para producir los correspondientes isómeros (R) y (S) de ácido 3-(terc-butoxicarbonil-isopropil-amino)-2-(4-cloro-fenil)-propiónico. Los resultados se muestran en la tabla 3, La escala
de reacción en todos los experimentos fue (a menos que se indique específicamente en una nota de pie) de 400 mg en 4 ml de disolvente, la temperatura fue de 60 °C a una relación S/C de 200, el tiempo de reacción fue de 14 h. El reactor fue un autoclave de 35 ml, el catalizador fue Ru(TFA)2((S)-BINAP).
T l

Para a) Tiempo de reacción 4 h; b) 200 mg de sustrato en 4 ml de etanol; c) 600 mg de sustrato en 2 ml de etanol; d) Catalizador preparado in situ por agitación de 2,56 mg de [Ru(COD)(TFA)2]2 y 4,0 mg de (S)-BINAP en una cámara de atmósfera inerte en 3 ml de etanol durante 3 h a 50 °C.
Ejemplo 8
4-(5-metM-7-oxo-5,6-dihidro-5H-cidopenta[d]pirimidm-4-M)piperazm-1-carboxMato de (R)-íerc-butilo
3-(4.6-d¡clorop¡rim¡d¡n-5-¡l)butanoato de (R)-metilo
En una mezcla de 3-(4,6-dihidroxipirimidin-5-il)butanoato de (R)-metilo (1,00 kg, 4,70 mol), tolueno (4,00 l) y 2,6-lutidina (0,550 l, 4,70 mol) se añadió oxicloruro de fósforo (0,960 l, 10,6 mol) a 50 °C lentamente. La mezcla se agitó a 70 °C durante 24 h. La solución se enfrió a 0 °C. A la mezcla se le añadió lentamente hidróxido sódico acuoso al 20 % (aproximadamente 40,0 mol, 1,60 kg en 8,00 l de H2O) mientras se mantenía la temperatura interna por debajo de 30 °C, para obtener un valor final de pH entre 5 y 6. Se añadió acetato de etilo (2,50 l), se agitó durante 0,5 h y después se separaron las capas. La fase acuosa se extrajo con acetato de etilo (3 x 1,00 l). Los extractos orgánicos se combinaron y se lavaron con ácido clorhídrico 1 N (2 x 2,50 l) y salmuera (2,50 l). Las capas orgánicas se combinaron y se secaron sobre sulfato sódico y se filtraron a través de un filtro de fibra de vidrio. La solución se concentró a aproximadamente 3,00 ml/g y se diluyó con acetonitrilo a aproximadamente 7,00 ml/g. La secuencia se repitió dos veces para eliminar el acetato de etilo y el tolueno residuales (confirmado por análisis de RMN 1H). La solución en bruto restante se usó directamente en la etapa siguientes sin más purificación o aislamiento.
3-(4.6-d¡vodop¡r¡m¡d¡n-5-¡l)butanoato de (R)-metilo
En una solución de 3-(4,6-dicloropirimidin-5-il)butanoato de (R)-metilo (36,0 g, 145 mmol) en acetonitrilo (540 ml) se añadió yoduro de sodio (152 g, 1,02 mol). La mezcla se agitó a 25 °C durante 30 min y después se enfrió a aproximadamente 5 °C. Se añadió ácido metanosulfónico (9,41 ml, 1,00 equiv.) durante 5 min. La mezcla se agitó a aproximadamente 5 °C durante 3 h. El reactor se enfrió a aproximadamente 5 °C y se añadió N,N-diisopropiletilamina (20,3 ml, 116 mmol). La mezcla se agitó durante 1 h mientras se calentaba la mezcla a 20 °C. Se añadió solución saturada de sulfito de sodio hasta que no se observó más cambio de color para eliminar el yodo. Se
añadió agua (540 ml) y se ajustó el pH a entre aproximadamente 5 y 7. La mezcla bifásica se concentró a presión reducida a una temperatura de menos de 40 °C para eliminar el acetonitrilo. La suspensión acuosa se filtró para dar 48,8 g (78 % de rendimiento) de un producto sólido de color blanquecino.
4-(6-vodo-5-(4-metox¡-4-oxobutan-2-¡l)p¡rim¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de (R)-ferc-butilo
En una solución de 3-(4,6-diyodopirimidin-5-il)butanoato de (R)-metilo (212 g, 491 mmol) y Boc-piperazina (101 g, 540 mmol) en metanol (424 ml) se añadió N,N-diisopropiletilamina (94,3 ml, 540 mmol). La mezcla se calentó a 60 °C durante 24 h. El metanol se eliminó por destilación a presión reducida por debajo de 40 °C. A la mezcla se le añadió 318 ml de tetrahidrofurano. El proceso anterior de intercambio de disolvente se repitió dos veces. A la mezcla se le añadió 424 ml de tetrahidrofurano, 212 ml de cloruro de amonio acuoso saturado y 21,2 ml de agua. La capa orgánica se lavó con 212 ml (1,00 vol.) de cloruro de amonio acuoso saturado. Esta solución de tetrahidrofurano se usó en la etapa siguiente sin más purificación (91 % de rendimiento del ensayo en peso).
Ácido (R)-3-(4-(4-(ferc-butoxicarbonil)piperazin-1-il)-6-yodo-pirimidin-5-il)butanoico
En una solución de 4-(6-yodo-5-(4-metoxi-4-oxobutan-2-il)pirimidin-4-il)piperazin-1-carboxilato de (R)-ferc-butilo (219 g, 0,447 mol) en tetrahidrofurano (657 ml) se añadió una solución de hidróxido de litio monohidrato (56,2 g, 1,34 mol) en 329 ml de agua a 25 °C. La mezcla se agitó a 25 °C durante 5 h. La capa acuosa inferior se descartó. La mezcla se acidificó con ácido clorhídrico 1 N a 5 °C para dar un valor final de pH de entre aproximadamente 1 a 2. Las capas se separaron. Después se extrajo la capa superior con acetato de isopropilo (440 ml x 3), se combinó con la capa inferior y se lavó con agua (220 ml x 2). El disolvente se eliminó por destilación a presión reducida por debajo de 50 °C. El acetato de isopropilo residual se eliminó por destilación azeotrópica con heptano a presión reducida por debajo de 50 °C. El producto precipitó de forma gradual y se filtró para dar un polvo de color blanquecino a amarillo claro (196 g, 84 % de rendimiento).
4-(6-vodo-5-(4-(metox¡(met¡l)am¡no)-4-oxobutan-2-¡l)p¡r¡m¡d¡n-4-¡l)p¡peraz¡n-1-carbox¡lato de (R)-terc-butilo
En una solución de ácido (R)-3-(4-(4-(ferc-butoxicarbonil)piperazin-1-il)-6-yodo-pirimidin-5-il)butanoico (100 g, 210 mmol) en tetrahidrofurano (700 ml) se añadió 1,1'-carbonildiimidazol (40,9 g, 252 mmol) en porciones. La mezcla de reacción se agitó a 20 °C durante 1 h y se enfrió a 5 °C. Se añadió clorhidrato de N,O-dimetilhidroxiamina (41,0 g, 420 mmol) en porciones seguido de N-metilmorfolina (6,94 ml, 63,0 mmol). La mezcla se agitó a 5 °C durante aproximadamente 1 h, lentamente se calentó a temperatura ambiente y se agitó durante 24 h. Se añadieron cloruro de amonio (500 ml) y agua (150 ml) para conseguir una separación de fases clara. La capa orgánica se lavó con cloruro de amonio acuoso saturado (500 ml) y salmuera (200 ml). El agua residual se eliminó por destilación azeotrópica hasta menos de 500 ppm por coevaporación con tetrahidrofurano. El producto, en forma de una solución en tetrahidrofurano se usó en la etapa siguiente sin más purificación o aislamiento (rendimiento del ensayo en peso: > 99 %.
4-(5-metil-7-oxo-6,7-dihidro-5H-ciclopenta[d1pirimidin-4-il)piperazin-1-carboxilato de (R)-terc-butilo
Una solución de 4-(6-yodo-5-(4-(metoxi(metil)amino)-4-oxobutan-2-il)pirimidin-4-il)piperazin-1-carboxilato de (R)-fercbutilo (109 g, 210 mmol) en tetrahidrofurano (600 ml) se purgó con nitrógeno durante 30 min. Se añadió gota a gota solución de cloruro de isopropil magnesio (159 ml, 210 mmol, 1,32 M en tetrahidrofurano) a -15 °C. La mezcla se agitó a -10 °C durante 1 h y se transfirió lentamente en cloruro de amonio acuoso al 20 % en peso frío (600 ml) con agitación mientras se mantenía la temperatura interna por debajo de 10 °C. Después se lavó la capa orgánica con cloruro de amonio acuoso saturado (500 ml). El tetrahidrofurano se eliminó por destilación a presión reducida por debajo de 40 °C. Se añadió metil ferc-butil éter (350 ml) lentamente mientras se mantenía la temperatura interna entre 35 °C y 40 °C, seguido de heptano (350 ml). La mezcla se enfrió lentamente a 20 °C y se precipitó producto de forma gradual durante el proceso. La suspensión se filtró y la torta se secó a 40 °C al vacío para dar un sólido de color gris (52,3 g, 75 % de rendimiento en dos etapas). RMN 1H (300 MHz, CDCla) 8 8,73 (s, 1H), 3,92-3,83 (m, 2H), 3,73-3,49 (m, 7H), 2,96 (dd, J = 16,5, 7,2 Hz, 1H), 2,33 (dd, J = 16,5, 1,8 Hz, 1H), 1,50 (s, 9H), 1,32 (d, J = 6,9 Hz, 3H). HRMS calc. Para C17H25N403 [M+H]+: 333,1921, observada 333,1924.
Ejemplo 9
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de íerc-butilo

Se formó una suspensión de color amarillento de 3 g de 4-[(5R)-5-metil-7-oxo-5,6-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo en 21 ml de tampón acuoso (ácido 2-(W-morfolino)etanosulfónico 100 mM pH 5,8), 6 ml de 2-propanol y 3 mg de cofactor NADP oxidado [Roche] con agitación vigorosa. La solución de reacción se calentó a 40 °C y se agitó durante 5 min. y posteriormente, la reducción comenzó mediante la adición de 30 mg de KRED-X1-P1B06. El pH se ajustó de 5,6 a 5,8. Durante el curso de la reacción a 40 °C durante 21,5 h logrando una conversión casi completa (iPC: 0,6 % de área de educto) el pH aumentó a 6,4. En la reacción se añadieron 30 ml de acetato de iso-propilo y se agitó vigorosamente durante 15 min. La división de las fases sucedió de forma espontánea. La fase acuosa separada se extrajo dos veces con 50 ml de acetato de iso-propilo, total 100 ml de acetato de iso-propilo. Las fases orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío a 50 °C produciendo 3,07 g (102 %) de una espuma de color rojo claro como producto en bruto del compuesto del título que contenía alrededor del 4 % de acetato de isopropilo. GC-EI-MS: 334,2 (M+H)+; HPLC quiral: 99,88 % (R,R), 0,12 % (R,S) [254 nm; Chiralpak IC-3; 150*4,6 mm, 3 pm, caudal 0,8 ml, 30 °C, A: n-heptano al 60 %, B: EtOH al 40 % 0,1 DEA, 0-15 min 100 % de B, 15-17 min 100 % de B, 17,1 min 40 % de B]; pureza química HPLC: 99,2 % en área (contiene 0,6 % de área de educto). RMN 1H (600 MHz, CDCla) 8 ppm 1,17 -1,22 (m, 3H) 1,45 -1,51 (m, 9H) 2,02 (s, 1H) 2,12 - 2,24 (m, 2H) 3,43 - 3,83 (m, 9H) 3,85 - 4,08 (m, 1H) 5,12 (t, J = 7,2 Hz, 1H) 8,53(s, 1H) (contiene ~4 % de acetato de iso-propilo).
Ejemplos 10.1-10.6
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de íerc-butilo Para los ejemplos 9.1-9.6, se repitió el procedimiento del ejemplo 9 pero la proporción del cofactor (NADP [Roche]) se varió como se indica en la tabla siguiente y se aplicó una variante diferente de cetorreductasa, denominada KRED-X1.
T l 4
Ejemplo 11
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de íerc-butilo Se formó una suspensión de color amarillento de 6 g de 4-[(5R)-5-metil-7-oxo-5,6-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo en 18 ml de tampón acuoso (ácido 2-(W-morfolino)etanosulfónico 100 mM pH 5,8), 6 ml de 2-propanol y 6 mg de cofactor NADP oxidado [Roche] con agitación vigorosa. La solución de reacción se calentó a 40 °C y se agitó durante 5 min. y posteriormente, la reducción comenzó mediante la adición de 60 mg de KRED-X1-P1B06. El pH se ajustó de 5,5 a 5,8. Durante el curso de la reacción a 40 °C con 2 d logrando casi conversión completa (IPC: 1 d 1,3 % de área de educto, 2 d 1 ,2 % de área de educto) el pH aumentó a 6 ,0. En la reacción se añadieron 30 ml de acetato de iso-propilo y se agitó vigorosamente durante 15 min. La división de las fases sucedió de forma espontánea. La fase acuosa separada se extrajo dos veces con 50 ml de acetato de iso
propilo, total 100 ml de acetato de iso-propilo. Las fases orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío a 50 °C produciendo 6,02 g (99,7 %) de una espuma de color rojo claro como producto en bruto del compuesto del título que contenía alrededor del 4 % de acetato de isopropilo. GC-EI-MS: 334,2 (M+H)+; HPLC quiral: 99,88 % (R,R), 0,12 % (R,S) [254 nm; Chirapakl IC-3; 150*4,6 mm, 3 pm, caudal 0,8 ml, 30 °C, A: nheptano al 60 %, B: EtOH al 40 % 0,1 DEA, 0-15 min 100 % de B, 15-17 min 100 % de B, 17,1 min 40 % de B]; pureza química HPLC: 98,4 % de área (contiene 1,3 % de área de educto). RMN 1H (600 MHz, CDC3) 5 ppm 1,2 (d, J = 7,1 Hz 3H) 1,49 (s, 9H) 2,14 - 2,23 (m, 2H) 3,46 - 3,59 (m, 5H) 3,64 (ddd, J = 13,1, 6,9, 3,3 Hz, 2H) 3,78 (ddd J=13,1, 7,2, 3,3 Hz, 2H) 5,12 (t, J = 7,2 Hz, 1H) 8,53 (s, 1H) (contiene ~4 % de acetato de iso-propilo).
Ejemplo 12
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo Se formó una suspensión de color amarillento de 3 g de 4-[(5R)-5-metil-7-oxo-5,6-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo en 21 ml de tampón acuoso (dihidrogenofosfato de potasio 100 mM pH 7,2; cloruro de magnesio 2 mM), 6 ml de 2-propanol y 3 mg de cofactor NADP oxidado [Roche] con agitación vigorosa. La solución de reacción se calentó a 40 °C y se agitó durante 5 min. y posteriormente, la reducción comenzó mediante la adición de 30 mg de KRED-X1-P1B06. El pH se ajustó de 7,5 a 7,2. Durante el curso de la reacción a 40 °C durante 18,5 h logrando una conversión casi completa (iPC: 0,8 % de área de educto) el pH disminuyó a 7,15. En la reacción se añadieron 30 ml de acetato de iso-propilo y se agitó vigorosamente durante 15 min. La división de las fases sucedió de forma espontánea. La fase acuosa separada se extrajo dos veces con 50 ml de acetato de isopropilo, total 100 ml de acetato de iso-propilo. Las fases orgánicas combinadas se secaron sobre MgSO4, se filtraron y se evaporaron al vacío a 50 °C produciendo 3,06 g (102 %) de una espuma de color rojo claro como producto en bruto del compuesto del título que contenía alrededor del 4 % de acetato de isopropilo. GC-EI-MS: 334,2 (M+H)+; HPLC quiral: 99,76 % (R,R), 0,24 % (R,S) [254 nm; Chirapakl IC-3; 150*4,6 mm, 3 pm, caudal 0,8 ml, 30 °C, A: nheptano al 60 %, B: EtOH al 40 % 0,1 DEA, 0-15 min 100 % de B, 15-17 min 100 % de B, 17,1 min 40 % de B]; pureza química HPLC: 98,9 % de área (contiene un 0,8 % de área de educto). RMN 1H (600 MHz, CDC3) 5 ppm 1,16-1,22 (m, 3H) 1,45-1,53 (m, 9H) 2,12 - 2,25 (m, 2H) 3,42 - 3,86 (m, 9H) 4,13 (s a, 1H) 5,12 (t, J = 7,2 Hz, 1H) 8,44 - 8,59 (m, 1 H) (contiene ~4 % de acetato de iso-propilo).
Ejemplos 13.1-13.7
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrocictopenta[d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo Se añadieron 10 mg de 4-[(5R)-5-metil-7-oxo-5,6-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de terebutilo disueltos en una mezcla de 50 pl de DMSO y 50 pl de 2-propanol en cada pocillo de una placa de pocillos profundos que contenía 300 pl de tampón (MES 100 mM, MgCh 2 mM; pH 5,8) 1 mg de NADP y variantes de KRED-X1. Después de agitar durante 1,5 h a temperatura ambiente se añadieron en cada pocillo 0,5 ml de MeOH y se analizó por HPLC. Los resultados de las mejores variantes se enumeran en la tabla siguiente.
T l

Ejemplo 13a
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de tere-butilo Se formó una suspensión de 50 g (150mmol) de 4-[(5R)-5-metil-7-oxo-5,6-dihidrociclopenta[d]-pirimidin-4-il]piperazin-1-carboxilato de tere-butilo en 100 ml de tampón acuoso (dihidrogenofosfato de potasio 100 mM pH 7,2), 78 g de 2-propanol y 50 mg NAD (75 pmol) con agitación vigorosa. La reducción comenzó mediante la adición de 500 mg de KRED-X1.1-P1F01. La mezcla de reacción se rocía con nitrógeno y se calienta a 40 °C durante 22 horas. Después de que la reacción se complete, se añaden 174 g de acetato de isopropilo, se agitó, las fases se separaron y la fase acuosa se eliminó. La fase acuosa se extrajo otra vez con 174 g de acetato de isopropilo. La fase acuosa se eliminó y las fases orgánicas se combinaron y se concentraron a 35 °C al vacío hasta un volumen final de 115 ml. A la misma temperatura se añaden 212 g de heptano durante 1 hora, la suspensión se envejece durante 1 hora y se enfría a 10 °C durante 6 horas. La suspensión se filtra y se lava con 68 g de heptano. Después se seca de la torta de filtro durante 4 horas a 50 °C y se obtienen 41,1 g (82 % de rendimiento, pureza del 100 % de área) de cristales de
color blanco.
Ejemplo 13b
4-[(5R,7R)-7-hidroxi-5-metil-6,7-dihidrociclopenta[d]pirimidin-4-il]piperazin-1-carboxilato de íerc-butilo
Se formó una suspensión de 40 g (150mmol) de 4-[(5R)-5-metil-7-oxo-5,6-dihidrociclopenta[d]-pirimidin-4-il]piperazin-1-carboxilato de ferc-butilo en 240 ml de tampón acuoso (que contiene 3,3 g de KH2PO4 y 8,4 g de K2HPO4), 26 g de glucosa y 40 mg de NAD con agitación vigorosa. La reducción se calentó a 35 °C y comenzó mediante la adición de 400 mg de KRED-X1.1-P1F01 y 400 mg de GDH-101. Durante el curso de la reacción (26 horas) el pH se mantiene a 7,0 usando 58,8 ml de KOH ac. (10% (m/m)). Después de que se completara la reacción, se añadieron 290 g de acetato de isopropilo y 117 g de NaSCN, se agitó, las fases se separaron y la fase acuosa se eliminó. La fase orgánica se lava con 200 g agua y se filtra usando una placa de filtro filtrox, la fase acuosa se lavó con 175 g de acetato de isopropilo. Las fases orgánicas combinadas se concentran at 25 °C al vacío hasta un volumen final de 100 ml. A 25 °C se añaden 383 g de heptano y durante 1 hora. La suspensión se enfría a 0 °C durante 30 minutos y se envejece durante 30 minutos, La suspensión se filtra y se lava con 91 g de heptano. Después se seca de la torta de filtro durante 16 horas a 50 °C y se obtienen 30,9 g (76 % de rendimiento, pureza del 100 % de área) de cristales de color blanco.
Ejemplo 14
((S)-2-(4-clorofenN)-3-(4-((5R,7R)-7-hidroxi-5-metN-6,7-dihidro-5H-ciclopenta[d]pirimidm-4-N)piperazm-1-N)-3-oxopropil)(isopropil)carbamato de íerc-butilo

A un reactor de tres bocas de 500 ml, equipado con un agitador mecánico, una entrada de nitrógeno y un termómetro se le cargó 4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-carboxilato de ferc-butilo (16,7 g, 52,5 mmol) y 2-propanol (65 ml). La solución se calentó a 55 °C. Después se añadió HCl al 20,8 % (m/m) en 2-propanol (24,6 g, 140 mmol) durante 10 minutos a 55 °C. La suspensión se agitó hasta que se completó la reacción. La mezcla de reacción se enfrió a 10 °C y se añadió 4-metilmorfolina (32,9 g, 325 mmol). La mezcla se agitó a 15 °C durante 30 min. Se añadió sal sódica de ácido (S)-3-((ferc-butoxicarbonil)(isopropil)amino)-2-(4-clorofenil)propanoico (19,1 g, 52,5 mmol) y 2-propanol (73 g) y la mezcla de reacción se enfrió a 5 °C. Se añadió anhídrido propanofosfónico (T3P) (50 % en peso (m/m) en tolueno) (35 g, 57,3 mmol) a una velocidad manteniendo la temperatura a 5 °C. Después de que se completara la reacción, se añadieron 20 g de agua. La solución se concentró por destilación a 45 °C y 15 kPa (150 mbar) hasta un volumen final de 100 ml. Se añadió tolueno (260 g). La solución se concentró otra vez por destilación a 45 °C y 15 kPa (150 mbar) hasta un volumen final de 300 ml. Se añadió agua (150 g) y la suspensión se agitó durante 15 minutos. Las fases se separaron durante 15 minutos y la fase acuosa se eliminó. Se añadió agua (100 g) y la suspensión se agitó durante 15 minutos. Las fases se separaron durante 15 minutos y la fase acuosa se eliminó. Se añadió agua (100 g) otra vez y la suspensión se agitó durante 15 minutos. Las fases se separaron durante 15 minutos y la fase acuosa se eliminó. La solución se concentró por destilación a 45 °C y 15 kPa (150 mbar) hasta un volumen final de 100 ml. Se añadió n-heptano (34 g), la solución se enfrió a 0 °C durante 1 hora para permitir al ((S)-2-(4-clorofenil)-3-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-oxopropil)(isopropil)carbamato de ferc-butilo cristalizar. Se añadió más n-heptano (170 g). La suspensión se envejeció durante 2 horas, se filtró y se lavó con una mezcla de tolueno (6,4 g) y n-heptano (29,2 g) seguido dos veces de heptano (cada una 68,4 g). La torta de filtro se secó a < 55 °C para dar ((S)-2-(4-clorofenil)-3-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-oxopropil)(isopropil)carbamato de ferc-butilo en forma de un sólido de color blanquecino, se aislaron 23,9 g, 86 % de rendimiento. (RMN 1H (600 MHz, CDCls) 5 ppm 0,68 (s a, 3H) 0,94 - 1,08 (m, 3H) 1,14 (d, J = 7,0 Hz, 3H) 1,47
(s, 10H) 2,06 -2,27 (m, 2H) 3,30 (s a, 1H) 3,38 - 3,53 (m, 5H) 3,56 -3,73 (m, 4H) 3,78 (s a, 3H) 4,62 (s a, 1H) 5,10 (t, J = 7,1 Hz, 1H) 7,24 (s, 1H) 8,49 (s, 1H).
Ejemplo 15
Monoclorhidrato de (S)-2-(4-dorofenN)-1-(4-((5R,7R)-7-hidroxi-5-metN-6,7-dihidro-5H-ddopenta[d]pirimidm-4-il)piperazin-1 -il)-3-(isopropilamino)propan-1-ona

A un reactor de 500 ml, equipado con un agitador mecánico, una entrada de nitrógeno, un termómetro y un medidor de pH se le añadió ((S)-2-(4-clorofenil)-3-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-oxopropil)(isopropil)carbamato de ferc-butilo (50 g) y 2-propanol (128 g). La solución se calentó a 50 °C. Se añadió una solución de HCl en 2-propanol (21 % en peso (m/m), 46,7 g) a 50 °C. La solución se mantuvo a 50 °C hasta que se completó la reacción y la mezcla se enfrió a 25 °C. Se añadió solución de amoniaco en 2 -propanol (2 M, 66 , 6 g, 1,66 equiv.) durante aprox. 1 hora hasta que se alcanzó pH 6,7. La suspensión se enfrió a 0 °C y se filtró. La torta se lavó con 2-propanol (39 g). El filtrado se concentró por destilación a 50 °C y 15 kPa (150 mbar) hasta un volumen final de 100 ml. Se añadió acetato de etilo (130 g) a la solución. La lechada se cambió de disolvente a 40 °C a volumen constante (300 ml) usando acetato de etilo (670 g). La suspensión se enfrió a 5 °C y la lechada se filtró. La torta de filtro se lavó con EtOAc (105 ml) y se secó al vacío a 100 °C durante 16 horas para proporcionar monoclorhidrato de (S)-2-(4-clorofenil)-1-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-(isopropilamino)propan-1-ona en forma de un sólido de color blanquecino: 36,4 g (82 % de rendimiento). (RMN 1H (600 MHz, D2O) 5 ppm 0,92 (d, J = 7,1 Hz, 3H) 1,23 (t, J = 6,4 Hz, 6 H) 1,89 -2,15 (m, 2H) 2,85 -3,06 (m, 1H) 3,17 -3,59 (m, 10H) 3,83 (d, J = 10,5 Hz, 2H) 4,33(dd, J = 8,5, 4,9 Hz, 1H) 4,98 (t, J = 7,0 Hz, 1H) 7,23 (d, J = 8,5 Hz, 2H) 7,36 (d, J = 8,7 Hz, 2H) 8,10 - 8,35 (m, 1H). LCMS [M+H]+ 458,2).
Ejemplo 16
Monoclorhidrato de (S)-2-(4-dorofenN)-1-(4-((5R,7R)-7-hidroxi-5-metN-6,7-dihidro-5H-ddopenta[d]pirimidm-4-il)piperazin-1 -il)-3-(isopropilamino)propan-1-ona
A un reactor de 500 ml, equipado con un agitador mecánico, una entrada de nitrógeno, un termómetro y un medidor de pH se le añadió ((S)-2-(4-clorofenil)-3-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-oxopropil)(isopropil)carbamato de ferc-butilo (50 g) y 1-propanol (131 g). La solución se calentó a 60 °C. Se añadió una solución de HCl en 1-propanol (22 % en peso (m/m), 38,0 g) a 60 °C. La solución se mantuvo a 50 °C hasta que se completó la reacción y la mezcla se enfrió a 25 °C. Se añadió NaOH ac. (28 %) (16 g) hasta que se alcanzó pH 6. La suspensión se concentró a 60 °C al vacío hasta un volumen final de 100 ml. La suspensión se enfría a 20 °C, se añaden 90 g de acetato de etilo y se filtra con una placa filtrox. El reactor y la unidad de filtración se lavan con 41 g de 1-propanol/acetato de etilo. La solución se filtra a 20 °C a través de almohadillas de filtro de carbón, El reactor y el filtro se aclaran con 82 g de 1-propanol/acetato de etilo. A 60 °C la solución se concentra al vacío hasta un volumen final de 300 ml. La destilación se continúa a 60 °C y se añaden de forma simultánea 1260 g de acetato de etilo manteniendo el volumen constante.
La suspensión se enfrió a 5 °C y la lechada se filtró. La torta de filtro se lavó con EtOAc (105 ml) y se secó al vacío a 60 °C durante 16 horas para proporcionar monoclorhidrato de (S)-2-(4-clorofenil)-1-(4-((5R,7R)-7-hidroxi-5-metil-6,7-dihidro-5H-ciclopenta[d]pirimidin-4-il)piperazin-1-il)-3-(isopropilamino)propan-1-ona en forma de un sólido de color blanquecino: 36,5 g (81 % de rendimiento, 91,4 % de pureza (m/m), 99,9 % (área) ensayo).
Claims (1)
- REIVINDICACIONES1. Un proceso para la preparación de un compuesto de fórmula (I)
 o sales del mismo, que comprende la reacción de acoplamiento de un compuesto de fórmula (II)
o sales del mismo, que comprende la reacción de acoplamiento de un compuesto de fórmula (II) con un compuesto de fórmula (III)
con un compuesto de fórmula (III) en dondeR1 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, terc-butoxicarbonilo (BOC) y trifluoroacetilo.R2 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, terc butoxicarbonilo (BOC) y trifluoroacetilo.M es un ion metálico seleccionado entre la lista de ion de metal alcalino, ion de metal alcalinotérreo e ion de metal de transición.2. El proceso de acuerdo con la reivindicación 1, en donde R1 es ferc-butoxicarbonilo (BOC).3. El proceso de acuerdo con cualquiera de las reivindicaciones 1 o 2, en donde R2 es ferc-butoxicarbonilo (BOC).4. El proceso de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde M es Na+.5. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 4, que comprende las etapas de reacción siguientes:a) Desprotección del compuesto de fórmula (III) en un disolvente en condiciones ácidas;b) Ajuste a pH alcalino usando una base;c) Adición de una solución que comprende el compuesto de fórmula (II) en un disolvente;d) Adición de una solución que comprende un agente de acoplamiento en un disolvente.6. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 5, en donde la base en la etapa b) se selecciona entre N-etil morfolina (NEM), trietilamina (TEA), tri(n-propil)amina (TPA), diisopropiletilamina (DIPEA), piridina y lutidina.7. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 6 , en donde el agente de acoplamiento usado en la etapa d) es anhídrido propilfosfónico (T3P).8. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 7, que comprende además un proceso para la fabricación de compuestos de fórmula (II)
en dondeR1 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, terc-butoxicarbonilo (BOC) y trifluoroacetilo.R2 es un grupo protector de amino seleccionado entre la lista de bencilo, benciloxicarbonilo (carbobenciloxi, CBZ), 9-fluorenilmetiloxicarbonilo (Fmoc), p-metoxibenciloxicarbonilo, p-nitrobenciloxicarbonilo, terc butoxicarbonilo (BOC) y trifluoroacetilo.M es un ion metálico seleccionado entre la lista de ion de metal alcalino, ion de metal alcalinotérreo e ion de metal de transición.2. El proceso de acuerdo con la reivindicación 1, en donde R1 es ferc-butoxicarbonilo (BOC).3. El proceso de acuerdo con cualquiera de las reivindicaciones 1 o 2, en donde R2 es ferc-butoxicarbonilo (BOC).4. El proceso de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde M es Na+.5. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 4, que comprende las etapas de reacción siguientes:a) Desprotección del compuesto de fórmula (III) en un disolvente en condiciones ácidas;b) Ajuste a pH alcalino usando una base;c) Adición de una solución que comprende el compuesto de fórmula (II) en un disolvente;d) Adición de una solución que comprende un agente de acoplamiento en un disolvente.6. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 5, en donde la base en la etapa b) se selecciona entre N-etil morfolina (NEM), trietilamina (TEA), tri(n-propil)amina (TPA), diisopropiletilamina (DIPEA), piridina y lutidina.7. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 6 , en donde el agente de acoplamiento usado en la etapa d) es anhídrido propilfosfónico (T3P).8. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 7, que comprende además un proceso para la fabricación de compuestos de fórmula (II) que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV)
que comprende la hidrogenación asimétrica de un compuesto de fórmula (IV) usando un catalizador de complejo de metal (C) en donde R1 y M son como se define en cualquiera de las reivindicaciones 1 a 4.9. El proceso de acuerdo con las reivindicaciones 8 , en donde el catalizador de complejo de metal (C) es un catalizador de complejo de rutenio seleccionado entre un compuesto de fórmula (C1), (C2) o (C3):Ru(Z)2D (C1)[Ru(Z)2-p(D)(L)m](Y)p (C2)Ru(E)(E')(D)(F) (C3)en donde:D es un ligando de fosfina quiral;L es un ligando neutro seleccionado entre alqueno C2-7, cicloocteno, 1,3-hexadieno, norbornadieno, 1,5-ciclooctadieno, benceno, hexametilbenceno, 1,3,5-trimetilbenceno, p-cimeno, tetrahidrofurano, dimetilformamida, acetonitrilo, benzonitrilo, acetona, tolueno y metanol;Z es un ligando aniónico seleccionado entre hidruro, fluoruro, cloruro, bromuro, n5-2,4-pentadienilo, n5-2,4-dimetil-pentadienilo o el grupo A-COO-, con la condición de que cuando dos Z se unen al átomo de Ru estos pueden ser tanto iguales como diferentes;A es alquilo C1-7, haloalquilo C1-7, arilo o haloarilo;Y es un anión no coordinante seleccionado entre fluoruro, cloruro, bromuro, BF4-, ClO4-, SbF6-, PF6-, B(fenilo)4-, B(3 ,5 -di-trifluorometil-fenilo)4-, CF3SO3-, y C6H5SO3-;F es una diamina opcionalmente quiral;E y E' son ambos iones de halógeno o E es hidruro y E' es BH4-;m es 1,2, 3 o 4; yp es 1 o 2.10. El proceso de acuerdo con la reivindicación 9, en donde el ligando aniónico (Z) se selecciona independientemente entre cloruro, bromuro, yoduro, OAc y TFA.11. El proceso de acuerdo con la reivindicación 9, en donde el anión no coordinante (Y) es BF4-.12. El proceso de acuerdo con la reivindicación 9, en donde la diamina quiral F es (1S,2S)-1,2-difeniletilendiamina (S,S-DPEN).13. El proceso de acuerdo con la reivindicación 9, en donde el ligando de fosfina quiral D se selecciona entre un compuesto de fórmulas (D1) a (D12):
usando un catalizador de complejo de metal (C) en donde R1 y M son como se define en cualquiera de las reivindicaciones 1 a 4.9. El proceso de acuerdo con las reivindicaciones 8 , en donde el catalizador de complejo de metal (C) es un catalizador de complejo de rutenio seleccionado entre un compuesto de fórmula (C1), (C2) o (C3):Ru(Z)2D (C1)[Ru(Z)2-p(D)(L)m](Y)p (C2)Ru(E)(E')(D)(F) (C3)en donde:D es un ligando de fosfina quiral;L es un ligando neutro seleccionado entre alqueno C2-7, cicloocteno, 1,3-hexadieno, norbornadieno, 1,5-ciclooctadieno, benceno, hexametilbenceno, 1,3,5-trimetilbenceno, p-cimeno, tetrahidrofurano, dimetilformamida, acetonitrilo, benzonitrilo, acetona, tolueno y metanol;Z es un ligando aniónico seleccionado entre hidruro, fluoruro, cloruro, bromuro, n5-2,4-pentadienilo, n5-2,4-dimetil-pentadienilo o el grupo A-COO-, con la condición de que cuando dos Z se unen al átomo de Ru estos pueden ser tanto iguales como diferentes;A es alquilo C1-7, haloalquilo C1-7, arilo o haloarilo;Y es un anión no coordinante seleccionado entre fluoruro, cloruro, bromuro, BF4-, ClO4-, SbF6-, PF6-, B(fenilo)4-, B(3 ,5 -di-trifluorometil-fenilo)4-, CF3SO3-, y C6H5SO3-;F es una diamina opcionalmente quiral;E y E' son ambos iones de halógeno o E es hidruro y E' es BH4-;m es 1,2, 3 o 4; yp es 1 o 2.10. El proceso de acuerdo con la reivindicación 9, en donde el ligando aniónico (Z) se selecciona independientemente entre cloruro, bromuro, yoduro, OAc y TFA.11. El proceso de acuerdo con la reivindicación 9, en donde el anión no coordinante (Y) es BF4-.12. El proceso de acuerdo con la reivindicación 9, en donde la diamina quiral F es (1S,2S)-1,2-difeniletilendiamina (S,S-DPEN).13. El proceso de acuerdo con la reivindicación 9, en donde el ligando de fosfina quiral D se selecciona entre un compuesto de fórmulas (D1) a (D12): R11 es alquilo C1-7, alcoxi C1-7, hidroxi o alquilo C1-7-C(O)O-;cada uno de R12 y R13 es independientemente hidrógeno, alquilo C1-7, alcoxi C1-7 o di(C1-7 alquil)amino; o R11 y R12, que están unidos al mismo grupo fenilo, o R12 y R13 que están unidos al mismo grupo fenilo, tomados juntos, son -X-(CH2)r-Y-, en donde X es -O- o -C(O)O-, Y es -O-, -N(alquilo inferior)- o -CF2- y r es un número entero de 1 a 6 ; odos R11 tomados juntos son -O-(CH2)s-O- u O-CH(CH3)-(CH2)s-CH(CH3)-O-, en donde s es un número entero de 1 a 6 ; o R11 y R12 o R12 y R13, junto con los átomos de carbono a los que están unidos, forman un anillo naftilo, tetrahidronaftilo o dibenzofurano;R14 y R15 son cada uno independientemente alquilo C1-7, cicloalquilo C3-8, fenilo, naftilo o heteroarilo, opcionalmente sustituidos con de 1 a 7 sustituyentes seleccionados independientemente entre el grupo que consiste en alquilo C1-7, alcoxi C1-7, di(alquil C1-7)amino, morfolinilo, fenilo, trisilil(alquilo C1-7), alcoxicarbonilo C1-7, hidroxicarbonilo, hidroxisulfonilo, (CH2)t-OH y (CH2)t-NH2, en donde t es un número entero de 1 a 6 ;R16 es alquilo C1-7;R17 es alquilo C1-7; yR18 independientemente es arilo, heteroarilo, cicloalquilo C3-8 o alquilo C1-7.14. El proceso de acuerdo con las reivindicaciones 9 o 13, en donde el ligando de fosfina quiral (D) es (S)-BINAP.15. El proceso de acuerdo con la reivindicación 9, en donde el catalizador de complejo de rutenio se selecciona entre el grupo de:Ru(TFA)2((R)-3 ,5 -Xyl-BINAP),Ru(OAc)2((S)-2 -furil-MeOBIPHEP),Ru(OAc)2((S)-BINAP),[Ru(OAc)2((S)-BINAP)]AlCl3,Ru(TFA)2((S)-BINAP),Ru(TFA)2((S)-BINAPHANE),Ru(TFA)2((S)-BIPHEMP),Ru(OAc)2((S)-MeOBIPHEP),Ru(TFA)2((S)-TMBTP),Ru(TFA)2((S,S)-iPr-DUPHOS),[Ru((R)-BINAP)(pCym)(AN)](BF4)2,[RuBr((S)-BINAP)(C6H6)]Br,[RuCl((S)-BINAP)(C6H6)]BF4,[RuI((S)-BINAP)(C6H6)]I,[Ru((S)-BINAP)(AN))4](BF4)2 yRuCl2((S)-pTol-BINAP)(S,S-DPEN).16. El proceso de acuerdo con la reivindicación 9, en donde el catalizador de complejo de rutenio es Ru(TFA)2((S)-BINAP).17. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 7, que comprende además un proceso para la fabricación de compuestos de fórmula (III)
R11 es alquilo C1-7, alcoxi C1-7, hidroxi o alquilo C1-7-C(O)O-;cada uno de R12 y R13 es independientemente hidrógeno, alquilo C1-7, alcoxi C1-7 o di(C1-7 alquil)amino; o R11 y R12, que están unidos al mismo grupo fenilo, o R12 y R13 que están unidos al mismo grupo fenilo, tomados juntos, son -X-(CH2)r-Y-, en donde X es -O- o -C(O)O-, Y es -O-, -N(alquilo inferior)- o -CF2- y r es un número entero de 1 a 6 ; odos R11 tomados juntos son -O-(CH2)s-O- u O-CH(CH3)-(CH2)s-CH(CH3)-O-, en donde s es un número entero de 1 a 6 ; o R11 y R12 o R12 y R13, junto con los átomos de carbono a los que están unidos, forman un anillo naftilo, tetrahidronaftilo o dibenzofurano;R14 y R15 son cada uno independientemente alquilo C1-7, cicloalquilo C3-8, fenilo, naftilo o heteroarilo, opcionalmente sustituidos con de 1 a 7 sustituyentes seleccionados independientemente entre el grupo que consiste en alquilo C1-7, alcoxi C1-7, di(alquil C1-7)amino, morfolinilo, fenilo, trisilil(alquilo C1-7), alcoxicarbonilo C1-7, hidroxicarbonilo, hidroxisulfonilo, (CH2)t-OH y (CH2)t-NH2, en donde t es un número entero de 1 a 6 ;R16 es alquilo C1-7;R17 es alquilo C1-7; yR18 independientemente es arilo, heteroarilo, cicloalquilo C3-8 o alquilo C1-7.14. El proceso de acuerdo con las reivindicaciones 9 o 13, en donde el ligando de fosfina quiral (D) es (S)-BINAP.15. El proceso de acuerdo con la reivindicación 9, en donde el catalizador de complejo de rutenio se selecciona entre el grupo de:Ru(TFA)2((R)-3 ,5 -Xyl-BINAP),Ru(OAc)2((S)-2 -furil-MeOBIPHEP),Ru(OAc)2((S)-BINAP),[Ru(OAc)2((S)-BINAP)]AlCl3,Ru(TFA)2((S)-BINAP),Ru(TFA)2((S)-BINAPHANE),Ru(TFA)2((S)-BIPHEMP),Ru(OAc)2((S)-MeOBIPHEP),Ru(TFA)2((S)-TMBTP),Ru(TFA)2((S,S)-iPr-DUPHOS),[Ru((R)-BINAP)(pCym)(AN)](BF4)2,[RuBr((S)-BINAP)(C6H6)]Br,[RuCl((S)-BINAP)(C6H6)]BF4,[RuI((S)-BINAP)(C6H6)]I,[Ru((S)-BINAP)(AN))4](BF4)2 yRuCl2((S)-pTol-BINAP)(S,S-DPEN).16. El proceso de acuerdo con la reivindicación 9, en donde el catalizador de complejo de rutenio es Ru(TFA)2((S)-BINAP).17. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 7, que comprende además un proceso para la fabricación de compuestos de fórmula (III) que comprende la reducción asimétrica del compuesto de fórmula (V)
que comprende la reducción asimétrica del compuesto de fórmula (V) catalizado por una oxidorreductasa, en donde R2 es como se define en cualquiera de las reivindicaciones 1 y 3. 18. El proceso de acuerdo con la reivindicación 17, en donde la oxidorreductasa cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) con una diastereoselectividad de al menos un 95 % de exceso diastereomérico (de).19. El proceso de acuerdo con la reivindicación 17, en donde la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es catalizada por una oxidorreductasa en presencia de un cofactor.20. El proceso de acuerdo con la reivindicación 19, el cofactor que se oxida en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es NADH o NADPH.21. El proceso de acuerdo con la reivindicación 19, en donde el cofactor se regenera in situ mediante: (a) regeneración de cofactor acoplado a enzima usando glucosa y glucosa deshidrogenasa como cosustrato o (b) regeneración de cofactor acoplado a sustrato usando un alcohol secundario como cosustrato.22. El proceso de acuerdo con la reivindicación 21, en donde el alcohol secundario como cosustrato para la regeneración acoplada a sustrato se selecciona entre 2-propanol, 2-butanol, butan-1,4-diol, 2-pentanol, pentan-1,5-diol, 4-metil-2-pentanol, 2-hexanol, hexan-1,5-diol, 2-heptanol o 2-octanol.23. El proceso de acuerdo con la reivindicación 17, en donde la oxidorreductasa es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-NADPH-111, KRED-NADPH-112, KRED-NADPH-113, KRED-NADPH-114, KRED-NADPH-115, KRED-NADPH-121, KRED-NADPH-123, KRED-NADPH-145, KRED-NADPH-155, A231, KRED-NADPH-136, KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11 y KRED-X1.1-P1C08.24. El proceso de acuerdo con la reivindicación 17, en donde la oxidorreductasa es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11 y KRED-X1.1-P1C08. 25. El proceso de acuerdo con la reivindicación 17, en donde la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un medio acuoso en presencia de uno o más codisolventes orgánicos.26. El proceso de acuerdo con la reivindicación 25, en donde los codisolventes orgánicos están presentes en una concentración total del 1 al 50 % V.27. El proceso de acuerdo con la reivindicación 25, en donde los codisolventes orgánicos se seleccionan entre la lista de glicerol, 2 -propanol, dietiléter, terc-butilmetiléter, éter diisopropílico, éter dibutílico, metil tetrahidrofurano, acetato de etilo, acetato de butilo, tolueno, heptano, hexano, ciclohexeno y mezclas de los mismos.28. El proceso de acuerdo con la reivindicación 17, en donde la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un tampón acuoso.29. El proceso de acuerdo con la reivindicación 28, en donde el tampón es ácido 2-(N-morfolino)etanosulfónico (MES) o dihidrogenofosfato de potasio (PBS).30. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 7, que comprende además el proceso para la fabricación de compuestos de fórmula (VI)
catalizado por una oxidorreductasa, en donde R2 es como se define en cualquiera de las reivindicaciones 1 y 3. 18. El proceso de acuerdo con la reivindicación 17, en donde la oxidorreductasa cataliza la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) con una diastereoselectividad de al menos un 95 % de exceso diastereomérico (de).19. El proceso de acuerdo con la reivindicación 17, en donde la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es catalizada por una oxidorreductasa en presencia de un cofactor.20. El proceso de acuerdo con la reivindicación 19, el cofactor que se oxida en la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) es NADH o NADPH.21. El proceso de acuerdo con la reivindicación 19, en donde el cofactor se regenera in situ mediante: (a) regeneración de cofactor acoplado a enzima usando glucosa y glucosa deshidrogenasa como cosustrato o (b) regeneración de cofactor acoplado a sustrato usando un alcohol secundario como cosustrato.22. El proceso de acuerdo con la reivindicación 21, en donde el alcohol secundario como cosustrato para la regeneración acoplada a sustrato se selecciona entre 2-propanol, 2-butanol, butan-1,4-diol, 2-pentanol, pentan-1,5-diol, 4-metil-2-pentanol, 2-hexanol, hexan-1,5-diol, 2-heptanol o 2-octanol.23. El proceso de acuerdo con la reivindicación 17, en donde la oxidorreductasa es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-NADPH-111, KRED-NADPH-112, KRED-NADPH-113, KRED-NADPH-114, KRED-NADPH-115, KRED-NADPH-121, KRED-NADPH-123, KRED-NADPH-145, KRED-NADPH-155, A231, KRED-NADPH-136, KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11 y KRED-X1.1-P1C08.24. El proceso de acuerdo con la reivindicación 17, en donde la oxidorreductasa es una oxidorreductasa dependiente de NADPH diastereoselectiva seleccionada entre la lista de KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11 y KRED-X1.1-P1C08. 25. El proceso de acuerdo con la reivindicación 17, en donde la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un medio acuoso en presencia de uno o más codisolventes orgánicos.26. El proceso de acuerdo con la reivindicación 25, en donde los codisolventes orgánicos están presentes en una concentración total del 1 al 50 % V.27. El proceso de acuerdo con la reivindicación 25, en donde los codisolventes orgánicos se seleccionan entre la lista de glicerol, 2 -propanol, dietiléter, terc-butilmetiléter, éter diisopropílico, éter dibutílico, metil tetrahidrofurano, acetato de etilo, acetato de butilo, tolueno, heptano, hexano, ciclohexeno y mezclas de los mismos.28. El proceso de acuerdo con la reivindicación 17, en donde la reducción asimétrica de un compuesto de fórmula (V) a un compuesto de fórmula (III) se realiza en un tampón acuoso.29. El proceso de acuerdo con la reivindicación 28, en donde el tampón es ácido 2-(N-morfolino)etanosulfónico (MES) o dihidrogenofosfato de potasio (PBS).30. El proceso de acuerdo con cualquiera de las reivindicaciones 1 a 7, que comprende además el proceso para la fabricación de compuestos de fórmula (VI)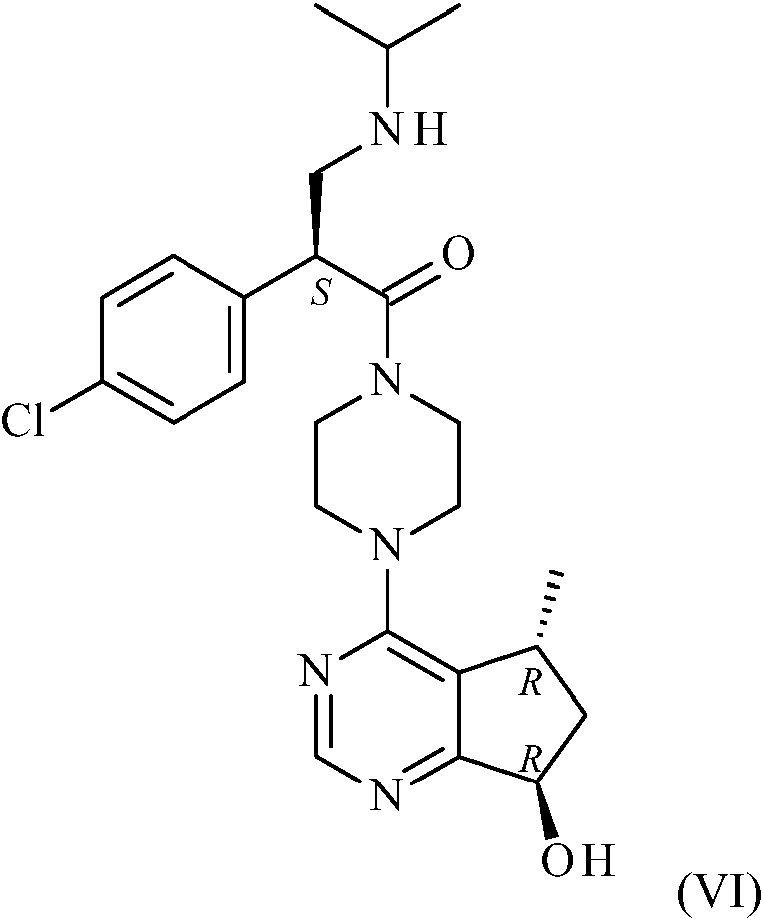 o sales farmacéuticamente aceptables de los mismos, en donde se desprotege un compuesto de fórmula (I)
o sales farmacéuticamente aceptables de los mismos, en donde se desprotege un compuesto de fórmula (I) en donde R1 es como se define en cualquiera de las reivindicaciones 1 a 2.31. El proceso de acuerdo con la reivindicación 30, que comprende las etapas de reacción siguientes:i) La desprotección del compuesto de fórmula (I) en un disolvente en condiciones ácidas;ii) Ajuste del pH usando una base en un disolvente;iii) Opcionalmente cristalizar el compuesto de fórmula (VI).32. El proceso de acuerdo con la reivindicación 31, en donde la desprotección en la etapa i) se realiza usando ácido clorhídrico, ácido sulfúrico, ácido trifluoroacético o ácido bromhídrico.33. El proceso de acuerdo con la reivindicación 31, en donde el disolvente usado para la desprotección en la etapa i) se selecciona entre n-propanol, isopropanol y una mezcla 1:1 de n-propanol/agua.34. El proceso de acuerdo con la reivindicación 31, en donde la cristalización en la etapa iii) se realiza mediante un cambio de disolvente a disolvente de cristalización adecuado para la cristalización del compuesto de fórmula (VI). 35. El proceso de acuerdo con la reivindicación 34, en donde el disolvente de cristalización en la etapa iii) se selecciona entre tolueno, heptano, tetrahidrofurano, 2 -propanona, 2 -butanona, etilenglicol dimetil éter, acetato de etilo, acetato de butilo, acetato de isopropilo y mezclas de los mismos.36. Un proceso de acuerdo con una cualquiera de las reivindicaciones 1-35, que comprende además formular el compuesto en una composición farmacéutica.37. Un compuesto de fórmula (II)
en donde R1 es como se define en cualquiera de las reivindicaciones 1 a 2.31. El proceso de acuerdo con la reivindicación 30, que comprende las etapas de reacción siguientes:i) La desprotección del compuesto de fórmula (I) en un disolvente en condiciones ácidas;ii) Ajuste del pH usando una base en un disolvente;iii) Opcionalmente cristalizar el compuesto de fórmula (VI).32. El proceso de acuerdo con la reivindicación 31, en donde la desprotección en la etapa i) se realiza usando ácido clorhídrico, ácido sulfúrico, ácido trifluoroacético o ácido bromhídrico.33. El proceso de acuerdo con la reivindicación 31, en donde el disolvente usado para la desprotección en la etapa i) se selecciona entre n-propanol, isopropanol y una mezcla 1:1 de n-propanol/agua.34. El proceso de acuerdo con la reivindicación 31, en donde la cristalización en la etapa iii) se realiza mediante un cambio de disolvente a disolvente de cristalización adecuado para la cristalización del compuesto de fórmula (VI). 35. El proceso de acuerdo con la reivindicación 34, en donde el disolvente de cristalización en la etapa iii) se selecciona entre tolueno, heptano, tetrahidrofurano, 2 -propanona, 2 -butanona, etilenglicol dimetil éter, acetato de etilo, acetato de butilo, acetato de isopropilo y mezclas de los mismos.36. Un proceso de acuerdo con una cualquiera de las reivindicaciones 1-35, que comprende además formular el compuesto en una composición farmacéutica.37. Un compuesto de fórmula (II) (II)en donde R1 y M son como se definen en cualquiera de las reivindicaciones 1 a 4.38. El compuesto de fórmula (II) de acuerdo con la reivindicación 37 que es (S)-3-(tercbutoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio.
(II)en donde R1 y M son como se definen en cualquiera de las reivindicaciones 1 a 4.38. El compuesto de fórmula (II) de acuerdo con la reivindicación 37 que es (S)-3-(tercbutoxicarbonil(isopropil)amino)-2-(4-clorofenil)propanoato de sodio.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13193030 | 2013-11-15 | ||
| PCT/US2014/065567 WO2015073739A1 (en) | 2013-11-15 | 2014-11-13 | Processes for the preparation of pyrimidinylcyclopentane compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2765511T3 true ES2765511T3 (es) | 2020-06-09 |
Family
ID=49582648
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19204562T Active ES2957314T3 (es) | 2013-11-15 | 2014-11-13 | Procesos para la preparación de compuestos de pirimidinilciclopentano |
| ES14863015T Active ES2765511T3 (es) | 2013-11-15 | 2014-11-13 | Procesos para la preparación de compuestos de pirimidinilciclopentano |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19204562T Active ES2957314T3 (es) | 2013-11-15 | 2014-11-13 | Procesos para la preparación de compuestos de pirimidinilciclopentano |
Country Status (22)
| Country | Link |
|---|---|
| US (3) | US9862689B2 (es) |
| EP (3) | EP4282973A3 (es) |
| JP (4) | JP6374503B2 (es) |
| KR (2) | KR20220139440A (es) |
| CN (3) | CN105899492B (es) |
| AR (1) | AR098427A1 (es) |
| AU (4) | AU2014348570B2 (es) |
| CA (2) | CA2930870C (es) |
| ES (2) | ES2957314T3 (es) |
| HK (1) | HK1223102A1 (es) |
| HR (2) | HRP20231100T1 (es) |
| HU (1) | HUE063095T2 (es) |
| IL (3) | IL245636B (es) |
| MX (3) | MX367620B (es) |
| MY (2) | MY191759A (es) |
| NZ (1) | NZ720805A (es) |
| PL (2) | PL3656764T3 (es) |
| RU (2) | RU2702355C1 (es) |
| SG (1) | SG10201901578UA (es) |
| SI (1) | SI3068770T1 (es) |
| WO (1) | WO2015073739A1 (es) |
| ZA (2) | ZA201603434B (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL3656764T3 (pl) | 2013-11-15 | 2024-01-22 | F. Hoffmann-La Roche Ag | Sposoby przygotowania związków pirymidynylocyklopentanu |
| WO2016049414A1 (en) | 2014-09-26 | 2016-03-31 | F. Hoffmann-La Roche Ag | PROCESSES FOR PREPARING (CYCLOPENTYL[d]PYRIMIDIN-4-YL)PIPERAZINE COMPOUNDS |
| CN115350192A (zh) | 2016-08-10 | 2022-11-18 | 豪夫迈·罗氏有限公司 | 包含Akt蛋白激酶抑制剂的药物组合物 |
| GB201802383D0 (en) * | 2018-02-14 | 2018-03-28 | Givaudan Sa | Process |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6545165B1 (en) | 2000-02-04 | 2003-04-08 | Roche Colorado Corporation | Synthesis of 3,6-dialkyl-5,6-dihydro-4-hydroxy-pyran-2-one |
| ES2313672T3 (es) * | 2005-05-24 | 2009-03-01 | F. Hoffmann-La Roche Ag | Praparacion de (s)-4-fluorometil-dihidro-furan-2-ona. |
| CN101198594A (zh) * | 2005-06-22 | 2008-06-11 | 尼科梅德有限责任公司 | 用于制备生产三环苯并咪唑类的中间体的方法 |
| US8063050B2 (en) | 2006-07-06 | 2011-11-22 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| CN103288753A (zh) * | 2006-07-06 | 2013-09-11 | 阵列生物制药公司 | 作为akt蛋白激酶抑制剂的环戊二烯并[d]嘧啶 |
| UA95641C2 (en) * | 2006-07-06 | 2011-08-25 | Эррей Биофарма Инк. | Hydroxylated cyclopenta [d] pyrimidines as akt protein kinase inhibitors |
| US7977078B2 (en) | 2007-08-24 | 2011-07-12 | Codexis, Inc. | Ketoreductase polypeptides for the production of (R)-3-hydroxythiolane |
| AU2009204025B2 (en) * | 2008-01-09 | 2014-02-20 | Array Biopharma Inc. | Hydroxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| NZ586720A (en) | 2008-01-09 | 2012-11-30 | Array Biopharma Inc | Hydroxylated pyrimidyl cyclopentane as akt protein kinase inhibitor |
| PE20091364A1 (es) * | 2008-01-17 | 2009-10-13 | Novartis Ag | Proceso para la preparacion de inhibidores de nep |
| EP2329013B1 (en) | 2008-08-27 | 2015-10-28 | Codexis, Inc. | Ketoreductase polypeptides for the production of a 3-aryl-3-hydroxypropanamine from a 3-aryl-3-ketopropanamine |
| CN102186972B (zh) | 2008-08-29 | 2014-08-20 | 科德克希思公司 | 用于立体选择性生产(4s)-3-[(5s)-5-(4-氟苯基)-5-羟基戊酰基]-4-苯基-1,3-噁唑烷-2-酮的酮还原酶多肽 |
| US20110054174A1 (en) * | 2009-08-28 | 2011-03-03 | Stephan Bachmann | Process for the preparation of a glucokinase activator compound |
| RU2012148699A (ru) * | 2010-04-16 | 2014-05-27 | Дженентек, Инк. | Foxo3a как прогностический биомаркер для эффективности ингибитора пути киназы pi3k/акт |
| PT2694072T (pt) | 2011-04-01 | 2018-02-26 | Genentech Inc | Combinação de composto inibidor de akt e abiraterona para utilização em tratamentos terapêuticos |
| BR112014028593A2 (pt) * | 2012-05-17 | 2017-12-19 | Genentech Inc | forma amorfa de um composto de pirimidinil-ciclopentano inibidor de akt, composições e métodos dos mesmos" |
| CN104487430B (zh) | 2012-05-17 | 2016-08-24 | 阵列生物制药公司 | 用于制造羟基化的环戊基嘧啶化合物的方法 |
| NZ702950A (en) * | 2012-05-17 | 2016-09-30 | Genentech Inc | Process of making hydroxylated cyclopentapyrimidine compounds and salts thereof |
| US9278917B2 (en) * | 2012-05-17 | 2016-03-08 | Genentech, Inc. | Process for making amino acid compounds |
| RU2643811C2 (ru) * | 2012-05-17 | 2018-02-06 | Аррэй Байофарма Инк. | Способ получения гидроксилированных циклопентилпиримидиновых соединений |
| PL3656764T3 (pl) | 2013-11-15 | 2024-01-22 | F. Hoffmann-La Roche Ag | Sposoby przygotowania związków pirymidynylocyklopentanu |
| WO2016049414A1 (en) | 2014-09-26 | 2016-03-31 | F. Hoffmann-La Roche Ag | PROCESSES FOR PREPARING (CYCLOPENTYL[d]PYRIMIDIN-4-YL)PIPERAZINE COMPOUNDS |
-
2014
- 2014-11-13 PL PL19204562.3T patent/PL3656764T3/pl unknown
- 2014-11-13 HR HRP20231100TT patent/HRP20231100T1/hr unknown
- 2014-11-13 HU HUE19204562A patent/HUE063095T2/hu unknown
- 2014-11-13 US US15/036,741 patent/US9862689B2/en active Active
- 2014-11-13 EP EP23182382.4A patent/EP4282973A3/en active Pending
- 2014-11-13 ES ES19204562T patent/ES2957314T3/es active Active
- 2014-11-13 CN CN201480072954.1A patent/CN105899492B/zh active Active
- 2014-11-13 RU RU2016123365A patent/RU2702355C1/ru active
- 2014-11-13 CN CN201910735465.4A patent/CN110590606B/zh active Active
- 2014-11-13 AU AU2014348570A patent/AU2014348570B2/en active Active
- 2014-11-13 EP EP14863015.5A patent/EP3068770B1/en active Active
- 2014-11-13 NZ NZ720805A patent/NZ720805A/en unknown
- 2014-11-13 MY MYPI2019002660A patent/MY191759A/en unknown
- 2014-11-13 SG SG10201901578UA patent/SG10201901578UA/en unknown
- 2014-11-13 MY MYPI2016000911A patent/MY174153A/en unknown
- 2014-11-13 KR KR1020227034215A patent/KR20220139440A/ko active IP Right Grant
- 2014-11-13 CA CA2930870A patent/CA2930870C/en active Active
- 2014-11-13 CA CA3207199A patent/CA3207199A1/en active Pending
- 2014-11-13 PL PL14863015T patent/PL3068770T3/pl unknown
- 2014-11-13 RU RU2019130505A patent/RU2019130505A/ru unknown
- 2014-11-13 CN CN202110168758.6A patent/CN112898210A/zh active Pending
- 2014-11-13 KR KR1020167015742A patent/KR102493603B1/ko active IP Right Grant
- 2014-11-13 EP EP19204562.3A patent/EP3656764B1/en active Active
- 2014-11-13 WO PCT/US2014/065567 patent/WO2015073739A1/en active Application Filing
- 2014-11-13 MX MX2016006299A patent/MX367620B/es active IP Right Grant
- 2014-11-13 MX MX2020009462A patent/MX2020009462A/es unknown
- 2014-11-13 JP JP2016530862A patent/JP6374503B2/ja active Active
- 2014-11-13 ES ES14863015T patent/ES2765511T3/es active Active
- 2014-11-13 SI SI201431455T patent/SI3068770T1/sl unknown
- 2014-11-14 AR ARP140104291A patent/AR098427A1/es unknown
-
2016
- 2016-05-13 MX MX2019006959A patent/MX2019006959A/es active IP Right Grant
- 2016-05-15 IL IL245636A patent/IL245636B/en active IP Right Grant
- 2016-05-19 ZA ZA2016/03434A patent/ZA201603434B/en unknown
- 2016-09-30 HK HK16111431.6A patent/HK1223102A1/zh unknown
-
2017
- 2017-11-20 JP JP2017222867A patent/JP6634428B2/ja active Active
- 2017-11-30 US US15/827,985 patent/US10435378B2/en active Active
-
2019
- 2019-04-09 AU AU2019202461A patent/AU2019202461B2/en active Active
- 2019-08-28 JP JP2019155667A patent/JP6857219B2/ja active Active
- 2019-08-29 US US16/555,842 patent/US10858324B2/en active Active
- 2019-12-11 IL IL271331A patent/IL271331B/en active IP Right Grant
- 2019-12-30 HR HRP20192340TT patent/HRP20192340T1/hr unknown
-
2020
- 2020-05-11 ZA ZA2020/02611A patent/ZA202002611B/en unknown
-
2021
- 2021-02-24 IL IL281072A patent/IL281072B/en unknown
- 2021-03-19 JP JP2021045573A patent/JP7377828B2/ja active Active
- 2021-04-12 AU AU2021202196A patent/AU2021202196A1/en not_active Abandoned
-
2022
- 2022-11-24 AU AU2022275477A patent/AU2022275477A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10858324B2 (en) | Processes for the preparation of pyrimidinylcyclopentane compounds | |
| ES2749100T3 (es) | Procesos para preparar compuestos de (ciclopentil[d]pirimidin-4-il)piperazina | |
| CN110452158A (zh) | 具有光学活性的常山酮及其中间体的合成方法 | |
| BR112016011048B1 (pt) | Processo para preparar um composto e composto | |
| BR122023011034B1 (pt) | Processo para fabricar compostos pirimidinilciclopentano | |
| BR122020009898B1 (pt) | Processos para fabricar compostos | |
| AU2017376599B2 (en) | Azepane inhibitors of menin-MLL interaction |