ES2751948T3 - Nuevo compuesto y proceso - Google Patents
Nuevo compuesto y proceso Download PDFInfo
- Publication number
- ES2751948T3 ES2751948T3 ES17748880T ES17748880T ES2751948T3 ES 2751948 T3 ES2751948 T3 ES 2751948T3 ES 17748880 T ES17748880 T ES 17748880T ES 17748880 T ES17748880 T ES 17748880T ES 2751948 T3 ES2751948 T3 ES 2751948T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- hydrogen
- carbonate
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *C(*c(c(C1=C(*)C(N2CC#N)=N[Al])c3*)c(*)c(O)c3O*)N1*2=O Chemical compound *C(*c(c(C1=C(*)C(N2CC#N)=N[Al])c3*)c(*)c(O)c3O*)N1*2=O 0.000 description 1
- KYGVEPLGHJUKBR-SLEBQGDGSA-N Cc(cc1C)cc(C)c1/N=C(\C=C(c1c2)N3CCc1cc(OC)c2OC)/N(CC#N)C3=O Chemical compound Cc(cc1C)cc(C)c1/N=C(\C=C(c1c2)N3CCc1cc(OC)c2OC)/N(CC#N)C3=O KYGVEPLGHJUKBR-SLEBQGDGSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Un compuesto de nitrilo o una sal del mismo, compuesto de nitrilo que es un compuesto de fórmula (A):**Fórmula** en la que: - R1 y R2 son iguales o diferentes, y cada uno es independientemente un grupo alquilo C1-C6 o un grupo acilo C2- C7, o R1 y R2 forman juntos un grupo alquileno C1-C6; - R3 y R4 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1- C6; - R5 y R6 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1- C6; - X es CHR7, O o NR7, y R7 es hidrógeno o un grupo alquilo C1-C6; y - Ar es un grupo arilo C6-C10 sustituido con de 0 a 4 sustituyentes, siendo cada uno de los sustituyentes independientemente halógeno o un grupo alquilo C1-C6.
Description
DESCRIPCIÓN
Nuevo compuesto y proceso
Campo de la invención
La invención se refiere a un nuevo derivado de nitrilo de un compuesto de pirimido[6,1-a]isoquinolin-4-ona. La presente invención se refiere además a un proceso para producir un derivado de nitrilo de un compuesto de pirimido[6,1-a]isoquinolin-4-ona. La invención también se refiere a procesos adicionales útiles en la producción de compuestos de pirimido[6,1-a]isoquinolin-4-ona.
Antecedentes de la invención
RPL554 (9,10-dimetoxi-2-(2,4,6-trimetilfenilimino)-3-(W-carbamoil-2-aminoetil)-3,4,6,7-tetrahidro-2H-pirimido[6,1-a]isoquinolin-4-ona) es un inhibidor doble de PDE3/PDE4, y se describe en el documento WO 00/58308. Como un inhibidor combinado de PDE3/PDE4, RPL554 tiene actividad antiinflamatoria y broncodilatadora, y es útil en el tratamiento de trastornos respiratorios tales como el asma y la enfermedad pulmonar obstructiva crónica (EPOC). La estructura de RPL554 se muestra más adelante.

El método conocido para producir RPL554 usa química de alquilación de ftalimidoetilo seguida de la desprotección usando hidrazina (véase el documento WO 00/58308). Sin embargo, los rendimientos de RPL554 logrados por esta vía son relativamente bajos y no son adecuados para la producción a gran escala. También hay problemas asociados con el uso del agente desprotector genotóxico hidrazina para eliminar el grupo ftalimida. Además, los agentes de alquilación de ftalimidoetilo son relativamente caros.
Por lo tanto, existe la necesidad de desarrollar una nueva vía para la producción de RPL554. En particular, es deseable desarrollar un proceso con un rendimiento mejorado. También es necesario evitar el uso de compuestos genotóxicos tales como la hidrazina. Además, sería útil desarrollar un método que fuera más eficiente en cuanto a los átomos y evitara la necesidad de usar agentes de alquilación costosos.
Sumario de la invención
Se ha desarrollado un método mejorado para producir compuestos de pirimido[6,1-a]isoquinolin-4-ona que tiene lugar a través de un nuevo producto intermedio de nitrilo. Este nuevo método tiene un rendimiento favorable, es eficiente en cuanto a los átomos y es fácilmente escalable para la producción de grandes cantidades de compuestos de pirimido[6,1-a]isoquinolin-4-ona.
El nuevo proceso tiene lugar a través de un novedoso producto intermedio de nitrilo. Por lo tanto, la invención proporciona además un compuesto de nitrilo o una sal del mismo, compuesto de nitrilo que es un compuesto de fórmula (A):

en la que:
• R1 y R2 son iguales o diferentes, y cada uno es independientemente un grupo alquilo C1-C6 o un grupo acilo C2-C7 , o R1 y R2 forman juntos un grupo alquileno C1-C6 ;
• R3 y R4 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1-Ca;
• R5 y R6 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1-Ca;
• X es CHR7, O o NR7, y R7 es hidrógeno o un grupo alquilo C1-C6 ; y
• Ar es un grupo arilo C6-C10 sustituido con de 0 a 4 sustituyentes, siendo cada uno de los sustituyentes independientemente halógeno o un grupo alquilo C1-C6.
La invención proporciona un proceso para producir un compuesto de nitrilo de fórmula (A):

proceso que comprende hacer reaccionar un compuesto de fórmula (I) con un compuesto de fórmula (II):

en la que:
• R1 y R2 son iguales o diferentes, y cada uno es independientemente un grupo alquilo C1-C6 o un grupo acilo C2-C7 , o R1 y R2 forman juntos un grupo alquileno C1-C6 ;
• R3 y R4 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1-Ca;
• R5 y R6 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1-Ca;
• X es CHR7, O o NR7, y R7 es hidrógeno o un grupo alquilo C1-C6 ;
• Ar es un grupo arilo C6-C10 sustituido con de 0 a 4 sustituyentes, siendo cada uno de los sustituyentes independientemente halógeno o un grupo alquilo C1-C6 ; e
• Y es un grupo saliente.
El compuesto de nitrilo de fórmula (A) se puede convertir fácilmente en un compuesto de amina, que es un importante producto intermedio en los procesos de producción de compuestos de pirimido[6,1-a]isoquinolin-4-ona tales como RPL554. La invención proporciona además un proceso para producir un compuesto de fórmula (B):

cuyo proceso comprende reducir un compuesto de fórmula (A):

en la que cada uno de entre R1, R2, R3, R4, R5, R6, X, Ar e Y es como se define en el presente documento.
Como se ha analizado anteriormente, el proceso de la invención permite la producción eficiente y segura de compuestos de pirimido[6,1-a]isoquinolin-4-ona tales como RPL554. Además, la invención proporciona un proceso para producir un compuesto de fórmula (C):

en la que cada uno de entre R1, R2, R3, R4, R5, R6, X, Ar e Y es como se define en el presente documento, y Q es O, NR8 o CR8, y R8 es hidrógeno o un grupo alquilo C1-C6 , comprendiendo el proceso:
(i) la preparación de un compuesto de fórmula (A) como se define en el presente documento; y/o
(ii) la reducción de un compuesto de fórmula (A) como se define en el presente documento.
El compuesto de fórmula (C) así obtenido puede combinarse con un vehículo, excipiente o diluyente farmacéuticamente aceptable, para obtenerse una composición farmacéutica.
Descripción detallada de la invención
Como se usa en el presente documento, un grupo o una fracción alquilo C1-C6 es un grupo o una fracción alquilo lineal o ramificado que contiene de 1 a 6 átomos de carbono. Por lo general, un grupo o una fracción alquilo C1-C6 es un grupo o una fracción alquilo C1-C4. Un grupo o una fracción alquilo C1-C4 es un grupo o una fracción alquilo lineal o ramificado que contiene de 1 a 4 átomos de carbono. Los ejemplos de grupos y fracciones alquilo C1-C6 incluyen metilo, etilo, n-propilo, /-propilo, n-butilo, /'-butilo, í-butilo y 3-metil-butilo. Los ejemplos de grupos y fracciones alquilo C1-C4 incluyen metilo, etilo, n-propilo, /-propilo, n-butilo, /-butilo y í-butilo. Para disipar cualquier duda, cuando están presentes dos fracciones alquilo en un grupo, las fracciones alquilo pueden ser iguales o diferentes.
Como se usa en el presente documento, un grupo o una fracción alquileno C1-C6 es un grupo o una fracción alquileno lineal o ramificado. Los ejemplos incluyen grupos y fracciones metileno, etileno y n-propileno.
Como se usa en el presente documento, el haluro normalmente es cloro, flúor, bromo o yodo.
Como se usa en el presente documento, un grupo alcoxi C1-C6 normalmente es un dicho grupo alquilo C1-C6 unido a un átomo de oxígeno.
Como se usa en el presente documento, un grupo acilo C2-C7 normalmente es un dicho grupo alquilo C1-C6 unido a un grupo -C(O)-.
Como se usa en el presente documento, un grupo o una fracción arilo C6-C10 normalmente es fenilo o naftilo. Se prefiere fenilo.
Por lo general, en el proceso de la invención:
R1 y R2 son iguales o diferentes, y cada uno es metilo o etilo;
R3 y R4 son iguales o diferentes, y cada uno es independientemente hidrógeno o metilo;
R5 y R6 son iguales o diferentes, y cada uno es independientemente hidrógeno o metilo;
X es CHR7, y R7 es hidrógeno, metilo o etilo;
Ar es un grupo fenilo sustituido con de 0 a 4 sustituyentes, siendo cada uno de los sustituyentes independientemente metilo o etilo; e
• Y es un grupo saliente.
Preferentemente, en el proceso de la invención:
R1 y R2 son metilo;
R3 y R4 son hidrógeno;
R5 y R6 son hidrógeno;
X es CH2 ; y
Ar es 2,4,6-trimetilfenilo.
Por lo tanto, el compuesto de fórmula (I) normalmente es un compuesto de fórmula (la):

El compuesto de fórmula (I) se puede obtener mediante métodos sintéticos conocidos, por ejemplo, los descritos en el documento WO 00/58308.
Los grupos salientes, por ejemplo, la fracción Y en la fórmula (II), son bien conocidos por el experto. Por lo general, un grupo saliente corresponde a la base conjugada de un ácido que tiene una pKa inferior a 10,0, o inferior a 5,0, en agua a 25 °C. Los ejemplos de grupos salientes incluyen haluros, perfluoroalquilsulfonatos, arilsulfonatos, alquilsulfonatos, carboxilatos, alquiltiolatos, nitratos, fosfatos ésteres de fosfato.
Y, por ejemplo, se puede seleccionar entre Cl, Br, I, -OC(O)RY, -OS(O)2RY y - S(O)2RY, en las que RY es hidrógeno, un grupo alquilo C1-C6 , un grupo alcoxi C1-C6 o un grupo arilo Ca-Ci0 , estando el grupo alquilo, alcoxi o arilo opcionalmente sustituido con de 1 a 6 grupos seleccionados entre nitrato y haluro.
Por lo general, Y es un grupo saliente que es Br, I, mesilato (CH3SO3", OMs), tosilato (CH3CaH4SO3", OTs), nosilato (O2NCaH4SO3-, ONs), acetato (CH3C(O)O-, OAc) o triflato (CF3SO3", OTf). Preferentemente, Y es un Br o I. Por ejemplo, el compuesto de fórmula (II) puede ser bromoacetonitrilo o yodoacetonitrilo. Más preferentemente, Y es Br.
En la reacción entre los compuestos de fórmulas (I) y (II), la cantidad del compuesto de fórmula (II) normalmente es de 0,5 a 10,0 equivalentes del compuesto de fórmula (I). Preferentemente, la cantidad del compuesto de fórmula (II) es de 1,0 a 2,5 equivalentes del compuesto de fórmula (I). Por ejemplo, la cantidad de compuesto (II) puede ser de 2,0 a 2,5 equivalentes del compuesto de fórmula (I).
Por lo general, la reacción entre los compuestos de fórmulas (I) y (II) comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) en presencia de una base. Las bases adecuadas para catalizar dichas reacciones de sustitución nucleófila son bien conocidas por el experto.
Por lo general, la base comprende carbonato, hidrogenocarbonato, hidróxido, un alcóxido, un carboxilato o una amina. Los ejemplos de una base que comprende carbonato incluyen carbonatos metálicos tales como carbonato de litio, carbonato de sodio, carbonato de potasio, carbonato de rubidio, carbonato de cesio, carbonato de magnesio, carbonato de calcio, carbonato de estroncio y carbonato de bario. Los ejemplos de una base que comprende hidrogenocarbonato incluyen hidrogenocarbonato de litio, hidrogenocarbonato de sodio, hidrogenocarbonato de potasio, hidrogenocarbonato de rubidio, hidrogenocarbonato de cesio, hidrogenocarbonato de magnesio, hidrogenocarbonato de calcio, hidrogenocarbonato de estroncio e hidrogenocarbonato de bario. Los ejemplos de una base que comprende hidróxido incluyen hidróxidos metálicos de fórmula MOH y M(OH)2 , en la que M es un metal seleccionado entre litio, sodio, potasio, rubidio, cesio, magnesio, calcio, estroncio y bario. Los ejemplos de una base que comprende un alcóxido incluyen alcóxidos metálicos de fórmula MOR y M(OR)2 , en la que M es un metal seleccionado entre litio, sodio, potasio, rubidio, cesio, magnesio, calcio, estroncio y bario, y R es un grupo alquilo C1-Ca, por ejemplo, un MOEt, MOiPr y MOtBu. Los ejemplos de una base que comprende un carboxilato incluyen carboxilatos metálicos de fórmula m Or o M(OR)2 , en la que M es un metal como se define en el presente documento y R es un grupo acilo C2-C7 , por ejemplo, MOAc o M(OAc)2. Los ejemplos de una base que comprende una amina incluyen alquilaminas de fórmula NR3 , en la que cada R es independientemente H o un grupo alquilo C1-Ca.
Preferentemente, la base comprende carbonato, hidrogenocarbonato o un carboxilato (por ejemplo, acetato). Más preferentemente, la base es carbonato de litio, carbonato de sodio, carbonato de potasio, carbonato de cesio, acetato de litio, acetato de sodio o acetato de potasio. Más preferentemente, la base es carbonato de litio o acetato de sodio.
Por lo general, la base se usa en una cantidad de 1,0 a 10,0 equivalentes del compuesto de fórmula (I). Preferentemente, la cantidad de la base es de 1,5 a 4,0 equivalentes del compuesto de fórmula (I).
El proceso normalmente comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) en
a
presencia de un disolvente o mezclas de disolventes. El disolvente puede ser cualquier disolvente adecuado, como los conocidos por el experto. Los ejemplos de disolventes incluyen agua y disolventes orgánicos polares. El disolvente puede ser un disolvente polar prótico o un disolvente polar aprótico. El disolvente orgánico puede ser un alcohol (por ejemplo, metanol, etanol, propanol o butanol), una cetona (por ejemplo, acetona o metiletilcetona), un disolvente halogenado (por ejemplo, cloroformo, diclorometano, clorobenceno), un éter (por ejemplo, éter dietílico o tetrahidrofurano (THF)), una amida (por ejemplo, dimetilformamida (DMF) o dimetilacetamida (DMA)), y éster (por ejemplo, etilacetato), un compuesto de nitrilo (por ejemplo, acetonitrilo (ACN) o propionitrilo) o un sulfóxido (por ejemplo, dimetilsulfóxido).
Preferentemente, el disolvente comprende un disolvente aprótico polar. Más preferentemente, el disolvente comprende acetonitrilo, tetrahidrofurano o dioxano. Más preferentemente, el disolvente comprende acetonitrilo.
Por lo general, el disolvente comprende menos del 10 % en volumen de agua, por ejemplo, menos del 5,0 % en volumen, o menos o igual al 1,0 % en volumen de agua.
El proceso normalmente comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) durante al menos 5 horas, preferentemente, de 5 a 48 horas.
Por lo general, el proceso comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) a una temperatura de 50 a 100 °C. Por ejemplo, los compuestos pueden hacerse reaccionar a una temperatura de 70 a 90 °C.
Preferentemente, el proceso comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) en presencia de un disolvente a reflujo. La temperatura requerida para alcanzar el reflujo dependerá del disolvente y normalmente es a o más/menos 5 °C del punto de ebullición de ese disolvente a presión atmosférica. El disolvente puede ser como se define en el presente documento, por ejemplo, acetonitrilo o THF.
En el proceso para producir el compuesto de fórmula (A), el compuesto de fórmula (II) puede ser bromoacetonitrilo o yodoacetonitrilo, y la base puede comprender carbonato (por ejemplo, carbonato de litio o carbonato de sodio) o acetato (por ejemplo, acetato de sodio o acetato de potasio) y los compuestos de fórmula (I) y (II) puede hacerse reaccionar en un disolvente aprótico polar (por ejemplo, THF, dioxano o acetonitrilo). Se puede usar un catalizador de transferencia de fase. En algunos casos, el disolvente puede comprender acetonitrilo y la base puede ser carbonato de litio.
En un proceso ilustrativo de acuerdo con la invención, el compuesto de fórmula (I), bromoacetonitrilo, carbonato de litio y acetonitrilo se combinan en un recipiente para formar una mezcla de reacción. La mezcla de reacción se puede calentar luego a reflujo (por ejemplo, de 60 a l00 °C) durante 4 a 48 horas. La cantidad en peso de carbonato de litio normalmente es del 20 al 100 % en peso con respecto al peso del compuesto de fórmula (I). El peso del acetonitrilo puede ser de 10 a 100 veces el peso del compuesto de fórmula (I).
El compuesto de fórmula (A) puede recuperarse siguiendo el proceso para producir un compuesto de fórmula (A) mediante el enfriamiento de la mezcla de reacción y su filtración, opcionalmente, a temperatura elevada, seguido de la evaporación de la solución resultante para recuperar el compuesto de fórmula (A). En algunos casos, el proceso para producir el compuesto de fórmula (A) comprende además cristalizar el compuesto de fórmula (A) en un disolvente tal como tetrahidrofurano.
Además, la invención proporciona un proceso para producir un compuesto de fórmula (B), cuyo proceso comprende reducir un compuesto de fórmula (A) como se define en el presente documento. La reducción del compuesto de fórmula (A) normalmente comprende reducir el grupo nitrilo (-CN) del compuesto de fórmula (A), normalmente, para formar un grupo amina primaria (-CH2NH2) en lugar del grupo nitrilo.
La reducción de un compuesto de fórmula (A) normalmente comprende: tratar el compuesto de fórmula (A) con un agente reductor; o hidrogenar el compuesto de fórmula (A). Los ejemplos de agentes reductores incluyen hidruros (tales como hidruro de litio y aluminio, y borohidruro de litio), boranos (tales como diborano) y metales alcalinos (tales como sodio o potasio, normalmente, en un disolvente alcohólico). La hidrogenación del compuesto de fórmula (A) normalmente comprende la hidrogenación catalítica del compuesto de fórmula (A), por ejemplo, mediante el tratamiento del compuesto de fórmula (A) con gas hidrógeno en presencia de un catalizador metálico. Los ejemplos de catalizadores metálicos incluyen catalizadores que comprenden níquel, níquel Raney, paladio, negro de paladio, hidróxido de paladio, platino y dióxido de platino.
El catalizador metálico normalmente se carga con del 1 al 100 % en peso del metal, por ejemplo, del 10 al 100 % en peso, del 20 al 100 % en peso o del 50 al 100 % en peso. El peso activo del catalizador normalmente es del 5 % en peso al 100 % en peso, por ejemplo, del 20 al 40 % en peso o del 80 al 100 % en peso.
Por lo general, la reducción del compuesto de fórmula (A) comprende hidrogenar el compuesto de fórmula (A) en presencia de níquel Raney. El níquel Raney es bien conocido por el experto, y normalmente comprende una aleación
en polvo de níquel y aluminio. El níquel Raney está disponible en el mercado. Por lo general, el níquel Raney se usa como una suspensión en agua.
La hidrogenación de un compuesto de fórmula (A) normalmente se lleva a cabo en presencia de gas hidrógeno a presión y un catalizador metálico. La presión del hidrógeno puede ser de 0,1 MPa a 5 MPa (1 bar a 50 bar), por ejemplo, de 0,2 MPa a 2 MPa (2 bar a 20 bar). Por lo general, la presión de hidrógeno es de 0,5 MPa a 1,5 MPa (5 a 15 bar), por ejemplo, de 0,8 MPa a 1 MPa (8 a 10 bar). La hidrogenación del compuesto de fórmula (A) normalmente se lleva a cabo durante al menos 10 minutos o al menos 1 hora, por ejemplo, de 1 a 24 horas.
A menudo, hay presente amoníaco disuelto o amoníaco gaseoso durante la hidrogenación. Por lo general, la hidrogenación se lleva a cabo en un compuesto de fórmula (A) en un disolvente que comprende amoníaco disuelto. El amoníaco normalmente se disuelve en un disolvente prótico polar, por ejemplo, agua, metanol, etanol o propanol. La concentración de amoníaco en el disolvente normalmente es de 1 a 20 M, por ejemplo, de 2 a 10 M. Preferentemente, la concentración de amoníaco en el disolvente (por ejemplo, metanol o metanol y agua) es de 5 a 9 M.
La reducción del compuesto de fórmula (A) puede comprender, por ejemplo, hidrogenar el compuesto de fórmula (A) en presencia de níquel Raney y amoníaco metanólico (opcionalmente, con una concentración de amoníaco de 2 a 10 M) en hidrógeno a una presión de 0,5 a 2 MPa (5 a 20 bar) durante al menos 1 hora.
En algunos casos, el proceso de producción un compuesto de fórmula (B) como se describe en el presente documento comprende además, antes de la reducción del compuesto de fórmula (A), la producción del compuesto de fórmula (A) mediante un proceso como se define en el presente documento.
La invención también proporciona un proceso de producción de un compuesto de fórmula (C) como se define en el presente documento, proceso que comprende un proceso para producir un compuesto de fórmula (A) como se define en el presente documento o un proceso para producir un compuesto de fórmula (B) como se define en el presente documento. La etapa (b) es una reacción de ureación
R1, R2, R3, R4, R5, R6, X, Ar, Y y Q en el compuesto de fórmula (C) pueden ser como se definen en el presente documento. Preferentemente: R1 y R2 son metilo; R3 y R4 son hidrógeno; R5 y R6 son hidrógeno; X es CH2 ; Ar es 2,4,6-trimetilfenilo; y Q es O. Por consiguiente, el compuesto de fórmula (C) es preferentemente RPL554, cuya estructura se muestra anteriormente.
Por lo general, la reacción adicional del compuesto de fórmula (B) para dar un compuesto de fórmula (C) comprende realizar una reacción de ureación. Una reacción de ureación es una reacción que conduce a la formación de un grupo urea, por ejemplo, mediante la conversión de un grupo amina. Un grupo urea es un grupo de fórmula -NHC(O)NH2.
Por lo general, el proceso para producir un compuesto de fórmula (C) comprende hacer reaccionar el compuesto de fórmula (B) con un compuesto de fórmula (III):

en la que R1, R2, R3, R4, R5, R6, X, Ar, Y y Q son como se definen en el presente documento, y Z+ es un catión metálico, por ejemplo, sodio o potasio.
Por lo general, Q es O y el compuesto de fórmula (III) es un compuesto de cianato. Por ejemplo, el compuesto de fórmula (III) puede ser cianato de sodio. Los procesos para producir un compuesto de fórmula (C) a partir del compuesto de fórmula (B) se describen en el documento WO 00/58308.
Como alternativa, se puede producir un compuesto de fórmula (C) haciendo reaccionar un compuesto de fórmula (B) con un compuesto que comprende un grupo cloroformiato, por ejemplo, cloroformiato de 4-nitrofenilo, y la posterior
reacción con amoníaco. Por ejemplo, el proceso para producir un compuesto de fórmula (C) puede comprender hacer reaccionar un compuesto de fórmula (B) con un compuesto de fórmula (IV)

en la que RIV es un grupo arilo opcionalmente sustituido con uno o más sustituyentes seleccionados entre grupos nitro y grupos flúor, y luego hacer reaccionar el producto con amoníaco.
La invención también proporciona un proceso para producir una composición farmacéutica que comprende (a) un compuesto de fórmula (C) y (b) un vehículo, diluyente o excipiente farmacéuticamente aceptable. Los vehículos, diluyentes y excipientes farmacéuticamente aceptables son conocidos por el experto.
Preferentemente, la composición farmacéutica es adecuada para la inhalación. Más preferentemente, es un polvo seco, una solución o una suspensión adecuada para la inhalación. Más preferentemente, es una suspensión como se describe en el documento WO 2016/0423143.
La invención también proporciona un compuesto de nitrilo o una sal del mismo, cuyo compuesto de nitrilo es un compuesto de fórmula (A).

El compuesto de fórmula (A) puede ser como se define en el presente documento. Por ejemplo, en el compuesto de fórmula (A), los sustituyentes pueden definirse de la siguiente manera:
R1 y R2 son iguales o diferentes, y cada uno es metilo o etilo;
R3 y R4 son iguales o diferentes, y cada uno es independientemente hidrógeno o metilo;
R5 y R6 son iguales o diferentes, y cada uno es independientemente hidrógeno o metilo;
X es CHR7, y R7 es hidrógeno, metilo o etilo; y
Ar es un grupo fenilo sustituido con de 0 a 4 sustituyentes, siendo cada uno de los sustituyentes independientemente metilo o etilo.
Preferentemente, el compuesto de nitrilo es un compuesto de fórmula (D):
La invención puede describirse con más detalle haciendo referencia a los siguientes ejemplos.
Ejemplos
Se comparó la vía de alquilación de ftalimidoetilo conocida con el proceso de la invención que tiene lugar a través de un producto intermedio de nitrilo. Los dos procesos se resumen en el esquema de reacción (Esquema 1) que se presenta a continuación.
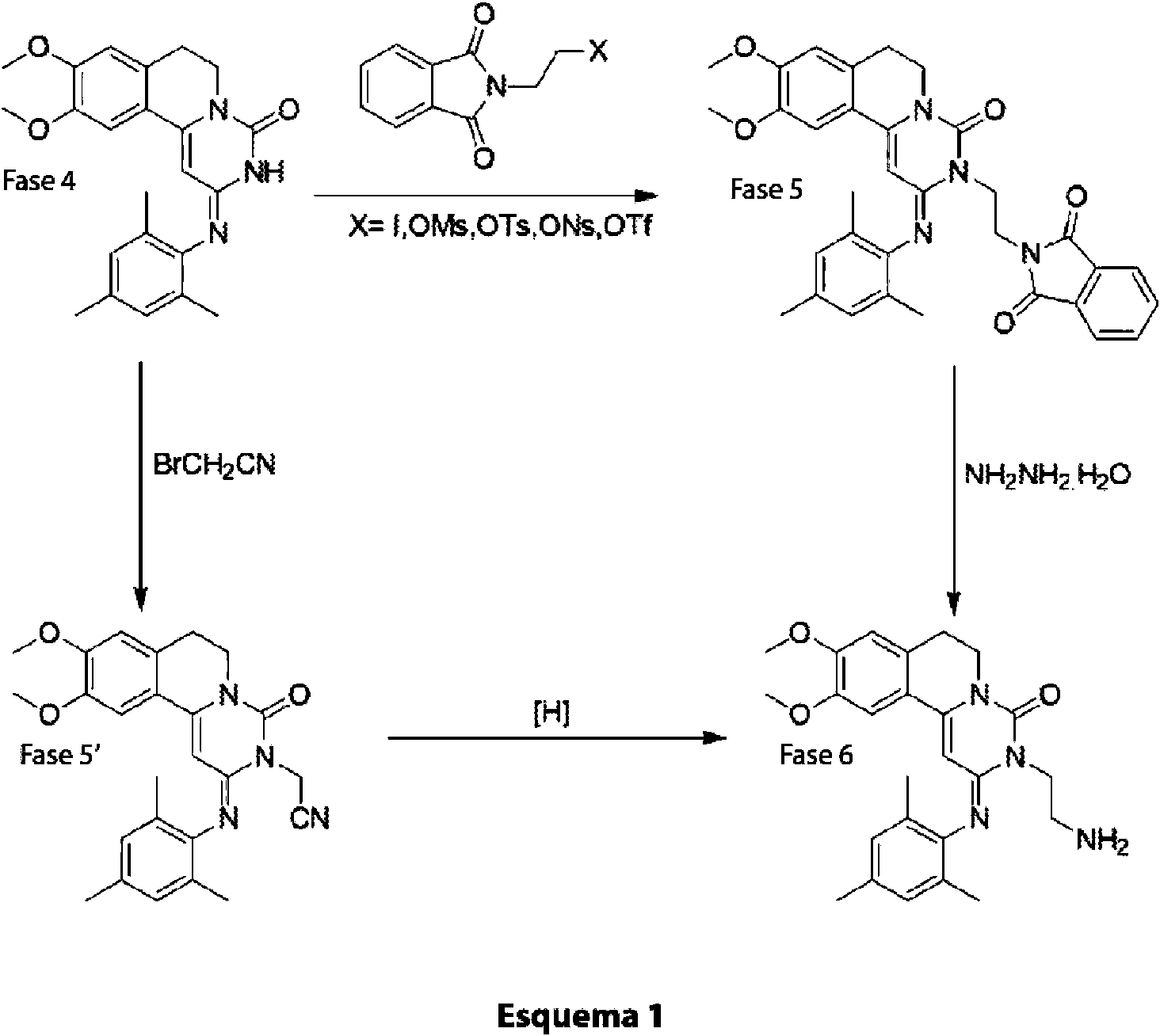
Ejemplo comparativo 1: Alquilación de ftalimidoetilo
Preparación de 9,10-dimetoxi-2-(2,4,6-trimetilfenilimino)-3-(2-N-ftalimidoetil)-3,4,6,7-tetrahidro-2H-pirimido[6,1-a]isoquinolin-4-ona:
Se agitó una mezcla de 9,10-dimetoxi-2-(2,4,6-trimetilfenilimino)-3,4,6,7-tetrahidro-2H-pirimido[6,1-a]isoquinolin-4-ona (Fase 4 del Esquema 1,60,0 g, 0,153 mol), carbonato potásico (191 g, 1,38 mol), yoduro sódico (137 g, 0,92 mol) y N-(2-bromoetil) ftalimida (234 g, 0,92 mol) en 2-butanona (1.500 ml) y se calentó a reflujo en una atmósfera de nitrógeno durante 4 días. Tras enfriar hasta la temperatura ambiente, la mezcla se filtró y el filtrado se evaporó al vacío. Se trató el residuo con metanol (1.000 ml) y se retiró el sólido mediante filtración, se lavó con metanol y se recristalizó en acetato de etilo, obteniéndose el compuesto de la Fase 5 del Esquema 1 en forma de un sólido de color amarillo pálido en un rendimiento de 40,0 g, 46 %. La evaporación de las aguas madres y la cromatografía en columna del residuo sobre gel de sílice (CH2Ch / MeOH 95:5) proporcionaron 11,7 g más de producto, 13,5 %.
Un trabajo adicional en una etapa posterior identificó que los rendimientos para la etapa de alquilación de ftalimidoetilo cayeron hasta aproximadamente una media del 34 % cuando se realizaron en una escala de varios kilogramos.
Ejemplo 1: Alquilación de bromoacetonitrilo
Una reacción de alquilación inicial, usando un gran exceso (8 equivalentes) de bromoacetonitrilo en presencia de carbonato de sodio en ciclopentanona, dio el consumo completo del material de partida (compuesto de fórmula (la), Fase 4 del Esquema 1), produciendo dos regioisómeros de producto según lo indicado por LC, 72 % y 12 %, respectivamente. Las condiciones de reacción se modificaron para dar un mejor perfil de reacción. Los equivalentes de bromoacetonitrilo se redujeron de 8 a 2,2, y la reacción se calentó hasta 110 °C durante 2 h. La LC indicó dos
regioisómeros del 87 % y 8 %, que luego se identificaron como el producto deseado de la Fase 5' (N-alquilado) (87 % de isómero principal) en el Esquema 1 y el isómero O-alquilado (8 % de isómero menor, que se muestra a continuación).

Se usó DMF para eluir el producto. Se aisló un total de 6,5 g con una pureza del 96 % según la LC (contenía 3,5 % del isómero O-alquilado). La confirmación de que este material era realmente el regiosiómero deseado se obtuvo mediante reducción y comparación del producto resultante con una muestra auténtica del producto de amino (Fase 6 del Esquema 1).
Ejemplo 2
Se realizaron varias reacciones usando bromoacetonitrilo para mejorar el perfil de reacción. El primer conjunto de reacciones de prueba implicó la reacción de RPL554, Fase 4, con bromoacetonitrilo en THF con varias bases. Se escogió THF frente a la ciclopentanona (usada anteriormente), ya que es considerablemente más barato, se podía usar con bases fuertes y podía cambiarse fácilmente el disolvente por metanol, que se usa en la siguiente fase. Los resultados se muestran en la Tabla 1. "LC" significa cromatografía líquida.
Tabla 1.

Las bases que dieron los resultados más favorables fueron el carbonato de litio y sodio, y el acetato de sodio y potasio. Las bases fuertes, en general, favorecieron la formación del isómero O-alquilado no deseado. Se dejaron agitar las cuatro reacciones más favorables durante la noche a 65 °. Los resultados de estas reacciones continuas se muestran en la Tabla 2.
Tabla 2.

Como muestran los resultados de la Tabla 2, los dos mejores conjuntos de condiciones fueron aquellas que usaron acetato de sodio y carbonato de litio.
Ejemplo 3
El siguiente conjunto de reacciones usó esas bases, acetato de sodio y carbonato de litio, variando la cantidad de disolvente y variando el número de equivalentes de bromoacetonitrilo para examinar la posibilidad de una reacción de mayor rendimiento. Los resultados se muestran en la Tabla 3.
Tabla 3.

A partir de la Tabla 3, se puede ver que la disminución de los volúmenes de disolvente puede tener un efecto negativo en la velocidad de reacción con acetato de sodio como base. Este efecto no es tan alto cuando se usa carbonato de litio como base. Un mayor número de equivalentes de bromoacetonitrilo (2,2 eq frente a 1,1 eq) también tiene un efecto positivo en la reacción. Las reacciones que usaron carbonato de litio se dejaron agitar durante otras 24 h. Los resultados se muestran en la Tabla 4. La reacción en THF es una conversión de suspensión a suspensión; Las reacciones más lentas a concentraciones aparentemente superiores pueden atribuirse al hecho de que, en realidad, hay menos material de partida (MP) de la Fase 4 en solución y disponible para reaccionar.
Tabla 4.

Todas las reacciones que se continuaron durante otras 24 h siguieron dando terminaciones similares a las observadas con 30 vol de disolvente, 2,2 eq de bromoacetonitrilo con carbonato de litio tras 16 h. Este conjunto de reacciones demuestra que se pueden lograr muy buenos resultados usando menos bromoacetonitrilo (1,1 eq frente a 2,2 eq) y menos disolvente (10 vol frente a 30 vol), siempre que se pueda adaptar un período de agitación más largo.
Ejemplo 4
El siguiente conjunto de reacciones consideró el uso de yodoacetonitrilo como agente de alquilación. Los resultados se muestran en la Tabla 5.
Tabla 5.

Ejemplo 5
Un conjunto adicional de reacciones examinó el carbonato de litio (la base más eficaz observada en experimentos anteriores) en 2-MeTHF (un disolvente de mayor ebullición para facilitar el logro de una temperatura de reacción más alta y, en principio, reacciones más rápidas) con cantidades variables de bromoacetonitrilo y disolvente. Los resultados se muestran en la Tabla 6.
Tabla 6.

Estos resultados sugieren que la solubilidad del material de partida en 2-MeTHF no es tan buena como en THF y, por lo tanto, la conversión en producto es más lenta a pesar de la temperatura de reacción más alta.
Ejemplo 6
Volviendo al bromoacetonitrilo como agente de alquilación, se realizaron dos reacciones a pequeña escala usando carbonato de litio y acetato de litio en agua al 5 %:THF como disolvente. La reacción usando acetato de litio fue menos favorable en términos de conversión en producto y la reacción usando carbonato de litio dio niveles más altos de lo esperado del isómero O-alquilado no deseado. Estas reacciones también sugieren que el agua puede tener un efecto negativo en la reacción. Los resultados se muestran en la Tabla 7.
Tabla 7.

Ejemplo 7
Se realizaron dos reacciones de alquilación de 10 g en THF usando acetato de litio y carbonato de litio respectivamente como base. Las reacciones fueron más lentas de lo esperado basándose en los experimentos previos (por ejemplo, Tabla 9). Las conversiones tras 16 h se muestran en la Tabla 8. Se postuló que un efecto de molienda de la barra agitadora en las reacciones anteriores a menor escala puede haber acelerado la reacción al reducir las partículas de la base inorgánica y, por lo tanto, aumentar el área superficial disponible para la reacción.
Tabla 8.

Se añadieron más THF (10 vol) y bromoacetonitrilo (1,1 eq.) a las reacciones en un intento por aumentar la velocidad de reacción. Los resultados después de 48 h se muestran en la Tabla 9.
Tabla 9.

La reacción de carbonato de litio continuó durante otras 24 h, dando un 85 % de producto, 7 % de material de partida, 3,1 % del isómero O-alquilado no deseado y una impureza nunca antes vista del 2,7 % (la LCMS mostró una masa de MH+ de 470, lo que sugiere bis-alquilación - véase más adelante). El sobrenadante de reacción se enriqueció con impurezas y contenía muy poco producto como se ve según la LC, lo que sugiere que el producto deseado puede haberse cristalizado. El sólido se aisló por filtración; el análisis indicó un 90,5 % de producto deseado, 6 % de material de partida, <1 % del regioisómero O-alquilado no deseado y <1 % de la otra impureza principal (Tr = 14,5).
Ejemplo 8
Se investigó el uso de más disolventes polares. Se realizaron dos reacciones de 10 g usando carbonato de litio en acetonitrilo y dioxano. Estas reacciones fueron más rápidas, aunque debe tenerse en cuenta que el material de partida de la fase 4 y el carbonato de litio se molieron antes del uso en estas reacciones. Los resultados se muestran en la Tabla 10.
Tabla 10.

Las reacciones se concentraron hasta casi secarse, y se diluyeron con THF (20 vol) y se suspendieron durante 30 minutos antes de filtrarlas. El análisis del sólido aislado se muestra en la Tabla 11.
Tabla 11.

Ejemplo 9
Las tres reacciones de alquilación de bromoacetonitrilo de 10 g usando THF, acetonitrilo y dioxano con carbonato de litio como base se combinaron tras las suspensiones de THF. Se disolvió el residuo orgánico en diclorometano:metanol (500 ml:50 ml) y se filtró la mezcla para retirar los materiales insolubles (principalmente, carbonato de litio). Se concentraron kas fases orgánicas hasta casi secarlas antes de suspenderlas en THF (400 ml).
Se filtró el sólido y se secó, dando 30 g de producto activo (91 % de rendimiento basado en el material de partida de la Fase 4). La LC indicó una pureza del 97,5 % con una sola impureza del 2,5 %, que se creía que era una especie dialquilada.
Ejemplo 10
Se realizaron otras tres reacciones de alquilación de 10 g de bromoacetonitrilo en acetonitrilo con carbonato de litio, carbonato de sodio y carbonato de potasio como bases. Cabe señalar que, en este conjunto de reacciones, ni el material de partida de la Fase 4 ni la base se molieron antes de su uso. Los resultados se muestran en la Tabla 12.
Tabla 12.

Se puede ver claramente en la Tabla 12 que el carbonato de litio dio los mejores resultados.
Ejemplo 11
Se realizó una reacción a gran escala usando acetonitrilo como disolvente y carbonato de litio como base.
Se realizó una reacción de alquilación de 10 g en acetonitrilo con carbonato de litio mediante la que la cantidad de bromoacetonitrilo se redujo de 2,2 eq. a 1,1 eq, la LC después de 18 h indicó un 75 % de producto, un 19 % de material de partida y un 2 % de dialquilado. La reacción continuó durante otras 24 h. El análisis de LC indicó un 90 % de producto y un 7 % de material de partida y un 2,7 % de dialquilado. Se enfrió la reacción hasta la temperatura ambiente
y se filtró para retirar principalmente carbonato de litio (el sólido contenía solo una pequeña cantidad de producto arrastrado). Se concentró el filtrado al vacío y se añadió THF (200 ml) al residuo. Se calentó la suspensión resultante a reflujo durante 20 minutos, y se filtró y se secó en el horno el sólido durante la noche. El análisis de LC del material seco indicó el 98,2 % de pureza. Se obtuvo un total de 11,8 g (11,02 g = rendimiento del 100 %). El análisis de RMN de 1H (usando un patrón interno) indicó una actividad del 77 %. La muestra contenía 9 % de THF y, por lo tanto, el análisis de RMN sugiere que el sólido contiene aproximadamente el 14 % p/p de compuestos inorgánicos (sales de litio). El rendimiento activo para la reacción se calculó en el 83 %.
Ejemplo 12
Se realizó una reacción de alquilación a escala de 100 g, usando 2,2 eq. del bromoacetonitrilo (nivel más alto para reducir el tiempo de reacción general). Se agitó la reacción durante la noche en acetonitrilo a reflujo. La finalización de la LC después de este tiempo indicó un 91 % de producto y un 4 % de material de partida. Se enfrió la reacción hasta la temperatura ambiente y se filtró para retirar la base. Se concentró el filtrado y se suspendió el residuo en THF caliente y se filtró; el producto se secó, dando 123 g de producto que tenía un ensayo del 77 % según la RMN, lo que sugiere el arrastre de material inorgánico. El ensayo de RMN del material resultante indicó una actividad de > 95 %, que fue confirmada mediante análisis de HPLC usando el método de LC de RPL554 de la fase 5, que indicó una pureza del 96 % con ninguna impureza individual >0,7 %.
Ejemplo 13
Se trató una alícuota del sólido del 77 % de pureza del Ejemplo 11 con agua, y se midió el pH a pH 3-4, que tal vez era indicativo de trazas de ácido (HBr). Por lo tanto, se realizó un procesamiento adicional para eliminar las sustancias inorgánicas. Se disolvió el lote a mayor escala del Ejemplo 12 (Li2CO3/MeCN) en diclorometano (20 vol) y se lavó con una solución de carbonato de potasio al 10 % (20 vol). Se secaron las sustancias orgánicas, se filtraron y se concentraron al vacío. El material se analizó mediante RMN en este punto y esto indicó una pureza > 95 %. Se analizó una muestra de este material, indicando el método de LC de RPL554 de la fase 5 una pureza del 94 % con la única impureza superior al 1 %, que era el material de partida de la fase 4 (3,4 %), es decir, el compuesto de fórmula (Ia). Se obtuvo un total de 95 g (sólido verde) para esta reacción (rendimiento del 86 %).
Ejemplo 14: Proceso de alto rendimiento
A un recipiente con camisa de 2 l, se añadió RPL554 de la fase 4 (100 g), acetonitrilo (2.000 ml), carbonato de litio (43,4 g) y bromoacetonitrilo (39,2 ml). Se calentó la reacción hasta 81 °C (reflujo) durante la noche. El análisis de LC indicó un 91 % de producto y un 4 % de material de partida. La reacción se enfrió hasta la temperatura ambiente y se filtró para retirar el carbonato de litio. Se concentró el filtrado al vacío. Se suspendió el residuo en THF (2 l) durante 3 h a temperatura ambiente. Se filtró el sólido, dando 123 g, el análisis de RMN de 1H indicó una actividad del 77 %. Se disolvió el sólido en diclorometano (2,4 l) y se lavó con carbonato de potasio al 10 % (2,4 l). Se separaron las fases orgánicas, se secaron, se filtraron y se concentraron. Se obtuvo un total de 95 g en forma de un sólido verde (86 %). El análisis de LC indicó un 93,9 % de producto y un 3,4 % de material de partida.
Ejemplo 15: Reducción del compuesto intermedio de nitrilo
Se sometió una parte del material de una reacción de alquilación de bromoacetonitrilo satisfactoria a una hidrogenación atmosférica usando Ni Raney y amoníaco metanólico 7 N (es decir, para producir la Fase 6 del Esquema 1). Posteriormente, se añadió THF para mejorar la solubilidad. La reacción se analizó mediante LC tras 2 h, y esta indicó la formación de un compuesto con un tiempo de retención similar al compuesto conocido de la Fase 63-(2-aminoetil)-2-(mesitilimino)-9,10-dimetoxi-2,3,6,7-tetrahidro-4H-pirimido[6,1-a]isoquinolin-4-ona (9,589 minutos frente a 9,657 minutos). Se agitó la reacción bajo una atmósfera de hidrógeno durante la noche. Se observó que gran parte del disolvente se había evaporado durante la noche, entonces se añadieron más THF y amoníaco metanólico 7 N. El análisis de LC indicó un 8 % del producto de la Fase 6. Se presurizó la mezcla a 0,5 MPa (5 bar) y se agitó durante 3 h, la LC indicó un 42 % del compuesto de la Fase 6 con dos impurezas del 8 %. La reacción continuó durante la noche, sin embargo, la LC no mostró más cambios.
Ejemplo 16
Se repitió la reacción del Ejemplo 15 usando 50 vol de amoníaco metanólico 7 N (20 vol usados anteriormente) y una mayor cantidad de Ni Raney (100 % en peso frente al 14 % en peso). Se dejó agitar la reacción durante la noche bajo 0,9 MPa (9 bar) de hidrógeno, la LC indicó el 92,6 % del producto. Se tomó una muestra y se analizó mediante RMN de 1H. Esto mostró un perfil similar a la amina de la Fase 6 deseada. Se trató la reacción con un tratamiento ácido. Se hizo precipitar el producto usando NaHCO3 y se filtró. Se secó una parte del sólido y analizó mediante RMN de 1H, coincidiendo con el producto de la Fase 6.
Ejemplo 17
Se hizo reaccionar adicionalmente una parte del producto obtenido en el Ejemplo 16, formando RPL554, es decir,
reacción con cloroformiato de 4-nitrofenMo seguido de amoníaco, dando RPL554. La LC indicó una pureza del 97 % con una impureza individual más grande del 1,5 %. Los tiempos de retención de RMN de 1H, RMN de 13C y LC fueron idénticos a los del RPL554 producido previamente, demostrando así que esta vía puede usarse para sintetizar con éxito RPL554.
Ejemplo 18
También se investigó el uso de una mezcla de amoníaco acuoso y metanol como alternativa al amoníaco metanólico. Los resultados se muestran en la Tabla 13.

Tabla 13.

Estos resultados indican que el amoníaco metanólico y la mezcla acuosa de amoníaco:metanol dieron conversiones similares al producto. Sin embargo, los perfiles de LC de las reacciones de amoníaco metanólico fueron más limpios, con menos productos secundarios observados. La segunda reacción en la tabla se escaló a 1 g, usando agitación por encima durante 18 h. La finalización de la LC tras 18 h se muestra en la Tabla 14.
Tabla 14.

La finalización de la LC fue similar a la de la reacción a menor escala, a pesar de la agitación más eficaz. Por lo tanto, se decidió calentar la reacción hasta 60 °C durante la noche bajo 0,9 MPa (9 bar) de hidrógeno. Esto efectuó una reacción completa; el resultado también se resume en la Tabla 14. Se filtró el catalizador y se concentraron las fases orgánicas. Se disolvió el residuo en HCl 1 M y se lavó con diclorometano (x3). Luego se basificó la fase acuosa con hidrogenocarbonato de sodio para hacer precipitar el producto. Se filtró el sólido y se secó al vacío a 50 °C, dando 655 mg de producto de amina (65 % de rendimiento). La LC indicó una pureza del 97,5 % sin impurezas individuales > 1 %. La modesta recuperación puede atribuirse a los lavados de diclorometano que la LC mostró que contenían algo de producto.
Ejemplo 19
Se realizaron varias reacciones de reducción a pequeña escala mediante las que se varió la cantidad de disolvente junto con la variación de la molaridad del amoníaco metanólico. Las reacciones se calentaron hasta 60 °C bajo 0,9 MPa (9 bar) de hidrógeno durante 18 h. Los resultados se muestran en la Tabla 15.
Tabla 15.
Se realizaron otras reacciones agitando primero a temperatura ambiente bajo 0,9 MPa (9 bar) de hidrógeno durante 18 h, luego agitando a 60 °C bajo 0,9 MPa (9 bar) de hidrógeno durante 18 h. La Tabla 16 muestra los resultados tras 18 h a temperatura ambiente ( T a ). La Tabla 17 muestra los resultados de la reacción tras calentar posteriormente a presión durante otras 24 h.
Tabla 16.

Tabla 17.

Se puede ver que las dos mejores reacciones están usando 50 vol de metanol (1 N y 7 N, resultados 1 y 6 de la Tabla 17) y, dentro del error experimental, este resultado es esencialmente similar al de la reacción de 1 g realizada previamente usando un agitador superior (Tabla 14).
Ejemplo 20
Se realizaron experimentos para investigar si otros disolventes podrían facilitar la reacción al conferir una mayor solubilidad al material de partida, y tal vez permitir el uso de volúmenes de disolvente más bajos. Por lo tanto, se realizaron varias reducciones de nitrilo a pequeña escala (60 mg) en una mezcla de amoníaco metanólico 2 N con THF, DMF y DMA. Los resultados de estas reacciones se muestran en la Tabla 18.
Tabla 18.

Ejemplo 21
Se llevaron a cabo reacciones similares a las detalladas en el Ejemplo 20 usando el compuesto de la fase 5' purificado (2-(2 (mesitilimino)-9,10-dimetoxi-4-oxo-6,7-dihidro-2H-pirimido [6,1-a]isoquinolin-3(4H)-il)acetonitrilo) para investigaciones posteriores. Se redujo el número de volúmenes de disolvente. Los resultados que usan este material de partida se muestran en la Tabla 19.
Tabla 19.
Las primeras cuatro reacciones de la tabla dieron alguna conversión a producto y producto intermedio de imina, pero fueron incompletas.
Ejemplo 22
Se repitieron las reacciones del Ejemplo 21, excepto que se llevaron a cabo a 60 °C bajo 0,9 MPa ( 9 bar) de hidrógeno. Los resultados se muestran en la Tabla 20. Se puede ver claramente que, a medida que la temperatura se eleva en estas reacciones, la cantidad de dimerización aumenta significativamente.
Tabla 20.

Ejemplo 23
Para mitigar la formación de dímeros, se repitieron los procesos del Ejemplo 22 a una mayor dilución. Se realizó una reacción de níquel Raney de 1 g en 2-(2-(mesitilimino)-9,10-dimetoxi-4-oxo-6,7-dihidro-2H-pirimido[6,1-a]isoquinolin-3(4H)-il)acetonitrilo purificado (Fase 5') usando 50 % en peso de catalizador en 50 volúmenes de MeOH:NH37 N. Se agitó la reacción durante 2 días bajo 0,9 MPa (9 bar) de hidrógeno, La LC indicó el 71 % de producto, 3,9 % de imina, 11,6 % de material de partida.
Ejemplo 24
Se investigó el efecto de tres parámetros sobre la hidrogenación en una escala intermedia: cantidad de disolvente (30 volúmenes o 50 volúmenes); molaridad de amoníaco metanólico (1 N frente a 7 N); y la cantidad de catalizador de níquel Raney (50 % en peso, 75 % en peso del 100 % en peso). Después de las dos primeras reacciones usando 30 y 50 volúmenes respectivamente con el 50 % en peso de catalizador activo, quedó claro que 50 volúmenes de disolvente eran mejores que 30 volúmenes.
Tabla 21.

El segundo conjunto de reacciones usó el 100 % en peso de catalizador activo con 50 volúmenes de amoníaco metanólico 1 N y 7 N. Posteriormente, se realizó una reacción con 50 volúmenes de amoníaco metanólico 7 N con el mantenimiento activo de la temperatura a 20 °C durante la noche usando un baño de aceite. La reacción se completó (91,7 % de producto) durante la noche. Los detalles se muestran en la Tabla 22.
Tabla 22.

Ejemplo 25
Se realizó una reacción de reducción con el 75 % en peso de catalizador con 50 volúmenes de amoníaco metanólico 7 N. La reacción se mantuvo activamente a 20 °C durante la noche. La reacción dio lugar a un 84,5 % de producto (se esperaría una conversión adicional al finalizar con un tiempo de agitación suficientemente largo). En la Tabla 23, se muestra el resultado. Esta reacción mostró que una reducción en la carga del catalizador puede compensarse con un aumento de la temperatura.
Tabla 23.

Ejemplo 26
Se realizaron dos reacciones adicionales con una carga de catalizador más baja (catalizador activo al 50 % en peso) con agitación a 30 °C usando 50 volúmenes de amoníaco metanólico 7 N y 1 N. La reacción 7 N se calentó a 30 °C durante un fin de semana.
Tabla 24.

La segunda reacción usando amoníaco metanólico 1 N dio un 77 % de producto (4 % de dímero) después de 18 h, sin embargo, la agitación adicional provocó la erosión del producto a través de la reacción con la imina intermedia, produciendo mayores niveles de dímero. Esto puede ser atribuible a la pérdida de amoníaco a una temperatura ligeramente elevada. Los resultados de la reacción de amoníaco metanólico 1 N se muestran en la Tabla 25.
Tabla 25.

Ejemplo 27
Todas las reacciones de hidrogenación de nitrilo descritas hasta ahora habían usado níquel Raney (suspensión al 50 % en agua, Acros). Se investigó el uso de catalizadores alternativos, comenzando con el catalizador de níquel Raney MC512 de Evonik, un catalizador desarrollado específicamente para reducir al mínimo la formación de impurezas diméricas en las reducciones de nitrilo. El catalizador de Evonik dio un 56 % de producto con un 31 % de material de partida restante (50 % en peso de catalizador en MeOH/NH3 1 N, 24 h, 20 °C).
Se realizó otra reacción usando níquel Raney 3202 suministrado por Aldrich; este dio un 60 % de producto, un 8 % de material de partida y un 18 % de dímero (50 % en peso de catalizador en MeOH/NH3 1 N, 48 h, 30 °C).
El mejor procedimiento actual para la reducción del producto intermedio de nitrilo usa 50 volúmenes de amoníaco metanólico 7 N con 50-100 % en peso de níquel Raney mantenido a 20 °C durante la noche bajo 0,9 MPa (9 bar) de hidrógeno para dar una conversión completa al producto (91,7 %, Tabla 22).
Ejemplo 28: Proceso de alto rendimiento
Se añadió a un hidrogenador Parr el 50 % de catalizador de níquel Raney húmedo (2 g), amoníaco metanólico 7 N (50 ml) y 2-(2-(mesitilimino)-9,10-dimetoxi-4-oxo-6,7-dihidro-2H-pirimido[6,1-a]isoquinolin-3(4H)-il)acetonitrilo (producto intermedio de la fase 5', 1 g, > 95 % de pureza según el ensayo de RMN). Se calentó la reacción hasta 20 °C y se presurizó a 0,9 MPa (9 bar) de hidrógeno durante la noche. La Le indicó el consumo completo de material de partida (91,7 % del producto).
Se filtró la mezcla de reacción y se lavó con metanol (25 ml). Se concentró el filtrado al vacío. Se disolvió el residuo en HCl 1 M (30 ml) y se lavó con diclorometano (DCM, 3 x 15 ml). Se basificó la fase acuosa con NaHCO3 a pH 8 y se filtró el sólido. Se secó el sólido sobre sulfato de magnesio, dando 700 mg. El análisis de RMN de 1H (que se muestra a continuación) indicó el producto de amina deseado con una pureza > 95 %. El rendimiento molar fue del 69 %. Se usó este material para preparar RPL554 con una pureza > 97 %.
La nueva vía a través del producto intermedio de nitrilo (Esquema 1) tiene los siguientes beneficios.
- La bromoetilftalimida (8 equivalentes) puede reemplazarse por el bromoacetonitrilo, mucho más barato y más fácilmente obtenible (2,2 equivalentes).
- El disolvente para la reacción de alquilación puede cambiarse de la carísima ciclopentanona (que contribuye significativamente al coste de los bienes) al acetonitrilo.
El largo y difícil trabajo, de bajo rendimiento (~40 %) del procedimiento antiguo puede reemplazarse por un procedimiento más directo, y el rendimiento de la nueva reacción de alquilación es superior al 85 %.
- El nuevo proceso da una reacción más rápida, más barata y de mayor rendimiento con un rendimiento mucho mejor.
- El desenmascaramiento de la funcionalidad de la amina latente en el proceso original requería el uso de la hidrazina altamente tóxica (y genotóxica) para eliminar el grupo protector de ftalimida (rendimiento típico del 85 %). El nuevo proceso de desenmascaramiento de aminas implica la reducción de un grupo nitrilo con níquel Raney para dar la amina en un rendimiento del ~70 % con una pureza similar al producto derivado de la vía de desprotección de ftalimida.
- Se pueden lograr fácilmente conversiones de sustrato superiores al 90 %. La alta carga de catalizador no es necesariamente una preocupación importante, pues el níquel Raney es barato y, si es necesario, puede reciclarse.
El rendimiento combinado de las nuevas reacciones de alquilación y reducción usando la química del bromoacetonitrilo es actualmente ~60 %, lo que se compara muy favorablemente con el rendimiento de aproximadamente el 40 % obtenido usando el enfoque anterior de alquilación de ftalimidoetilo que se usaba antes para la fabricación de RPL554.
Método analítico
Condiciones de LC:
Sistema: Cromatografía de líquidos de serie 1100 de Agilent o equivalente
Columna: Acquity BEH Fenil 4,6 x 30 mm; Tamaño de partícula de 1,7 pm (Antes Waters n.°
186004644)
Fase móvil A: Agua:TFA (100:0,03)
Fase móvil B: Acetonitrilo:TFA (100:0,03)
Caudal: 2,0 ml.min-1
Volumen de 5 pl
inyección:
Detección: Detección UV (defecto 254 nm, dependiente del proyecto de longitud de onda)
Temp. de la columna: 40 °C
Tras ejecución: 2,3 min
Gradiente:

Condiciones de la espectrometría de masas:
Sistema EM de trampa iónica Esquire 3000 Plus de Bruker Polaridad iónica Positiva
Tipo de fuente iónica ESI
Nebulizador 344,738 kPa (50 psi)
Gas seco 10 l/min
Temperatura de secado 350 °C
Masa diana : 400 m/z
Intervalo de exploración 50 m/z -1.000 m/z
Preparación de las muestras:
La preparación predeterminada es de aproximadamente 0,5 mg.ml-1 en MeCN:agua (1:1), usando ultrasonidos para disolver la muestra por completo.
Claims (15)
1. Un compuesto de nitrilo o una sal del mismo, compuesto de nitrilo que es un compuesto de fórmula (A):

en la que:
• R1 y R2 son iguales o diferentes, y cada uno es independientemente un grupo alquilo C1-C6 o un grupo acilo C2-C7 , o R1 y R2 forman juntos un grupo alquileno C1-C6 ;
• R3 y R4 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1-Ca;
• R5 y R6 son iguales o diferentes, y cada uno es independientemente hidrógeno, halógeno o un grupo alquilo C1-Ca;
• X es CHR7, O o NR7, y R7 es hidrógeno o un grupo alquilo C1-C6 ; y
• Ar es un grupo arilo C6-C10 sustituido con de 0 a 4 sustituyentes, siendo cada uno de los sustituyentes independientemente halógeno o un grupo alquilo C1-C6.
2. Un compuesto de nitrilo de acuerdo con la reivindicación 1 o una sal del mismo, compuesto de nitrilo que es un compuesto de fórmula (D):

3. Un proceso de producción de un compuesto de nitrilo de fórmula (A) como se ha definido en la reivindicación 1 o 2, proceso que comprende hacer reaccionar un compuesto de fórmula (I) con un compuesto de fórmula (II):

en la que R1, R2, R3, R4, R5, R6, X y Ar son como se han definido en la reivindicación 1, e Y es un grupo saliente.
4. Un proceso de acuerdo con la reivindicación 3, en el que:
• R1 y R2 son metilo;
• R3 y R4 son hidrógeno;
• R5 y R6 son hidrógeno;
• X es CH2 ; y
• Ar es 2,4,6-tri metilfenilo.
5. Un proceso de acuerdo con la reivindicación 3 o 4, en el que Y es un grupo saliente que es Cl, Br, I, -OC(O)RY, -OS(O)2Ry y -S(O)2Ry, en las que RY es hidrógeno, un grupo alquilo C1-C6 , un grupo alcoxi C1-C6 o un grupo arilo C6-C10, estando el grupo alquilo, alcoxi o arilo opcionalmente sustituido con de 1 a 6 grupos seleccionados entre nitrato y haluro, preferentemente, en el que Y es un grupo saliente que es Br, I, OMs, OTs, ONs, OAc u OTf, más preferentemente, en el que Y es un grupo saliente que es Br.
6. Un proceso de acuerdo con una cualquiera de las reivindicaciones 3 a 5, en el que la cantidad del compuesto de fórmula (II) es de 0,5 a 10,0 equivalentes del compuesto de fórmula (I), preferentemente, en el que la cantidad del compuesto de fórmula (II) es de 1,0 a 2,5 equivalentes del compuesto de fórmula (I).
7. Un proceso de acuerdo con una cualquiera de las reivindicaciones 3 a 6, en el que el proceso comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) en presencia de una base, preferentemente, en el que la base comprende carbonato, hidrogenocarbonato, un alcóxido, un carboxilato o una amina, más preferentemente, en el que la base es carbonato de litio, carbonato de sodio, carbonato de potasio, carbonato de cesio, acetato de litio, acetato de sodio o acetato de potasio, lo más preferentemente, en el que la base es carbonato de litio o acetato de sodio.
8. Un proceso de acuerdo con la reivindicación 7, en el que la cantidad de la base es de 1,0 a 10,0 equivalentes del compuesto de fórmula (I), preferentemente, en el que la cantidad de la base es de 1,5 a 3,0 equivalentes del compuesto de fórmula (I).
9. Un proceso de acuerdo con una cualquiera de las reivindicaciones 3 a 8, proceso que comprende:
hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) en presencia de un disolvente, preferentemente, en el que el disolvente comprende un disolvente aprótico polar, más preferentemente, en el que el disolvente comprende acetonitrilo, tetrahidrofurano o dioxano; y/o
hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) durante al menos 5 horas; y/o hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) a una temperatura de 50 a 100 °C, preferentemente, proceso que comprende hacer reaccionar el compuesto de fórmula (I) y el compuesto de fórmula (II) en presencia de un disolvente a reflujo.
10. Un proceso de producción de un compuesto de fórmula (B):

proceso que comprende reducir un compuesto de fórmula (A) como se ha definido en la reivindicación 1 o 2, en la que R1, R2, R3, R4, R5, R6, X y Ar son como se han definido en una cualquiera de las reivindicaciones 1 a 4.
11. Un proceso de acuerdo con la reivindicación 10, en el que la reducción del compuesto de fórmula (A) comprende hidrogenar el compuesto de fórmula (A) en presencia de níquel, níquel Raney, paladio, negro de paladio, hidróxido de paladio, platino y dióxido de platino, preferentemente, níquel Raney.
12. Un proceso de acuerdo con la reivindicación 10 u 11, proceso que comprende además producir el compuesto de fórmula (A) mediante un proceso como se ha definido en una cualquiera de las reivindicaciones 3 a 9.
13. Un proceso de producción de un compuesto de fórmula (C):

en la que R1, R2, R3, R4, R5, R6, X y Ar son como se han definido en una cualquiera de las reivindicaciones 1 a 4, y Q es O, NR8 o CR8, y R8 es hidrógeno o un grupo alquilo C1-C6 ,
proceso que comprende:
(i) un proceso de producción de un compuesto de fórmula (A), como se ha definido en una cualquiera de las reivindicaciones 3 a 9; y/o
(ii) un proceso de preparación de un compuesto de fórmula (B) mediante la reducción de un compuesto de fórmula (A), como se ha definido en una cualquiera de las reivindicaciones 10 a 12.
14. Un proceso de acuerdo con la reivindicación 13, proceso que comprende:
(a) preparar un compuesto de fórmula (B) mediante la reducción de un compuesto de fórmula (A), como se ha definido en una cualquiera de las reivindicaciones 13 a 15; y
(b) hacer reaccionar además el compuesto de fórmula (B), dando un compuesto de fórmula (C),
opcionalmente, en el que la etapa (b) es una reacción de ureación, preferentemente, en el que la reacción de ureación se realiza haciendo reaccionar un compuesto de fórmula (B) con un compuesto O=C=N- Z+, en el que Z+ es un catión metálico, más preferentemente, en el que Z+ es sodio o potasio.
15. Un proceso de acuerdo con la reivindicación 13 u 14, en el que
R1 y R2 son metilo;
R3 y R4 son hidrógeno; R5 y R6 son hidrógeno; X es CH2 ;
Ar es 2,4,6-trimetilfenilo; y Q es O.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1613054.4A GB201613054D0 (en) | 2016-07-28 | 2016-07-28 | New compound and process |
| PCT/GB2017/052190 WO2018020249A1 (en) | 2016-07-28 | 2017-07-27 | New compound and process |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2751948T3 true ES2751948T3 (es) | 2020-04-02 |
Family
ID=56936838
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17748880T Active ES2751948T3 (es) | 2016-07-28 | 2017-07-27 | Nuevo compuesto y proceso |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US10710998B2 (es) |
| EP (1) | EP3490988B1 (es) |
| JP (1) | JP6985367B2 (es) |
| KR (1) | KR102396059B1 (es) |
| CN (1) | CN109641891B (es) |
| CY (1) | CY1122138T1 (es) |
| DK (1) | DK3490988T3 (es) |
| ES (1) | ES2751948T3 (es) |
| GB (1) | GB201613054D0 (es) |
| HR (1) | HRP20192109T1 (es) |
| HU (1) | HUE046509T2 (es) |
| LT (1) | LT3490988T (es) |
| PL (1) | PL3490988T3 (es) |
| PT (1) | PT3490988T (es) |
| RS (1) | RS59566B1 (es) |
| SI (1) | SI3490988T1 (es) |
| TW (1) | TWI739871B (es) |
| WO (1) | WO2018020249A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2015261239B2 (en) | 2014-05-12 | 2020-02-27 | Verona Pharma Plc | New treatment |
| GB201502260D0 (en) | 2015-02-11 | 2015-04-01 | Verona Pharma Plc | Salt of Pyrimido[6,1-A]Isoquinolin-4-one compound |
| JP2024517583A (ja) | 2021-04-29 | 2024-04-23 | ▲蘇▼州盛迪▲亞▼生物医▲薬▼有限公司 | イソキノリノン系化合物及びその用途 |
| CA3241037A1 (en) * | 2021-12-14 | 2023-06-22 | Xizang Haisco Pharmaceutical Co., Ltd. | Tricyclic fused heterocyclic pde3/4 dual inhibitor and use thereof |
| EP4378942A1 (en) | 2022-12-02 | 2024-06-05 | Sandoz AG | Crystal form of a pde3/4 inhibitor |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ185900A (en) | 1976-12-10 | 1981-04-24 | Hoechst Ag | Pyrimido (6,1-a) isoquinolin-4-ones medicaments intermediate isoquinolines |
| FI64370C (fi) | 1977-05-05 | 1983-11-10 | Hoechst Ag | Foerfarande foer framstaellning av terapeutiskt anvaendbara pyimido-(6,1-a)isokinolin-2-imino-4-onderivat |
| DE2847693A1 (de) | 1978-11-03 | 1980-05-22 | Hoechst Ag | Verfahren zur herstellung von pyrimido-(6,1-a)-isochinolin-2-onen |
| DE3135831A1 (de) | 1981-09-10 | 1983-04-28 | Hoechst Ag, 6230 Frankfurt | 9,10-substituierte 2-mesitylimino-3-alkyl-3,4,6,7-tetrahydro-2h-pyrimido(6,1-a)isochinolin-4-one, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| CA1237429A (en) | 1983-05-05 | 1988-05-31 | Frank Kienzle | Pyrimidone derivatives |
| ES2208310T3 (es) | 1999-03-31 | 2004-06-16 | Vernalis Limited | Derivados de pirimido(6,1-a) isoquinolin-4-ona. |
| PT2603509E (pt) | 2010-08-09 | 2014-08-29 | Verona Pharma Plc | Forma cristalina de um composto pirimidio[6,1-a]isoquinolin-4-ona |
| LT2968313T (lt) | 2013-03-15 | 2018-05-10 | Verona Pharma Plc | Vaistų derinys |
| AU2015261239B2 (en) | 2014-05-12 | 2020-02-27 | Verona Pharma Plc | New treatment |
| SI3193835T1 (en) | 2014-09-15 | 2018-06-29 | Verona Pharma PLC Bradley Court | Liquid inhalation preparation containing rpl554 |
| GB201502260D0 (en) | 2015-02-11 | 2015-04-01 | Verona Pharma Plc | Salt of Pyrimido[6,1-A]Isoquinolin-4-one compound |
-
2016
- 2016-07-28 GB GBGB1613054.4A patent/GB201613054D0/en not_active Ceased
-
2017
- 2017-07-27 SI SI201730098T patent/SI3490988T1/sl unknown
- 2017-07-27 RS RS20191474A patent/RS59566B1/sr unknown
- 2017-07-27 KR KR1020197004436A patent/KR102396059B1/ko active IP Right Grant
- 2017-07-27 LT LTEP17748880.6T patent/LT3490988T/lt unknown
- 2017-07-27 PT PT177488806T patent/PT3490988T/pt unknown
- 2017-07-27 US US16/309,435 patent/US10710998B2/en active Active
- 2017-07-27 TW TW106125312A patent/TWI739871B/zh active
- 2017-07-27 CN CN201780046788.1A patent/CN109641891B/zh active Active
- 2017-07-27 WO PCT/GB2017/052190 patent/WO2018020249A1/en unknown
- 2017-07-27 EP EP17748880.6A patent/EP3490988B1/en active Active
- 2017-07-27 HU HUE17748880A patent/HUE046509T2/hu unknown
- 2017-07-27 ES ES17748880T patent/ES2751948T3/es active Active
- 2017-07-27 DK DK17748880T patent/DK3490988T3/da active
- 2017-07-27 JP JP2019503748A patent/JP6985367B2/ja active Active
- 2017-07-27 PL PL17748880T patent/PL3490988T3/pl unknown
-
2019
- 2019-10-04 CY CY20191101046T patent/CY1122138T1/el unknown
- 2019-11-22 HR HRP20192109TT patent/HRP20192109T1/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20192109T1 (hr) | 2020-02-21 |
| EP3490988A1 (en) | 2019-06-05 |
| JP6985367B2 (ja) | 2021-12-22 |
| CY1122138T1 (el) | 2020-11-25 |
| JP2019527228A (ja) | 2019-09-26 |
| US20190330206A1 (en) | 2019-10-31 |
| KR102396059B1 (ko) | 2022-05-10 |
| GB201613054D0 (en) | 2016-09-14 |
| TWI739871B (zh) | 2021-09-21 |
| CN109641891B (zh) | 2021-05-04 |
| RS59566B1 (sr) | 2019-12-31 |
| SI3490988T1 (sl) | 2019-11-29 |
| US10710998B2 (en) | 2020-07-14 |
| KR20190034231A (ko) | 2019-04-01 |
| PT3490988T (pt) | 2019-10-28 |
| DK3490988T3 (da) | 2019-10-28 |
| TW201806951A (zh) | 2018-03-01 |
| LT3490988T (lt) | 2019-10-10 |
| WO2018020249A1 (en) | 2018-02-01 |
| HUE046509T2 (hu) | 2020-03-30 |
| CN109641891A (zh) | 2019-04-16 |
| EP3490988B1 (en) | 2019-08-28 |
| PL3490988T3 (pl) | 2020-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2751948T3 (es) | Nuevo compuesto y proceso | |
| CN107771178B (zh) | 杂环化合物的合成 | |
| ES2769255T3 (es) | Métodos para fabricar inhibidores de la proteína desacetilasa | |
| KR20180136953A (ko) | 단일쇄 핵산 분자용 모노머의 제조 방법 | |
| US20080275265A1 (en) | Process for the Preparation of (Aminoalkylamino)Alkyl Halides and Conversion to Amifostine | |
| KR20180038461A (ko) | 세포독성 벤조다이아제핀 유도체의 제조 방법 | |
| KR102593509B1 (ko) | 질소 머스타드 유도체의 제조 방법 | |
| CA2813571A1 (en) | Substituted 2-(9h-purin-9-yl) acetic acid analogues as inhibitors of stat3 | |
| US20230416243A1 (en) | Process for Preparing Heterocyclic Methanone Compounds and AZA-Bicyclo Intermediates Thereof | |
| AU2014389984A1 (en) | Process of preparing a quinazoline derivative | |
| ES2386758T3 (es) | Procedimiento para preparar un derivado de BENZOILBENCENOACETAMIDA | |
| ES2950143T3 (es) | Síntesis de 3-metil-1,2,4-tiadiazol-5-carbohidrazida o de la forma deuterada metil-d3 de esta | |
| AU2022424485A1 (en) | Method for producing oligonucleic acid compound | |
| WO2017043563A1 (ja) | ピリジンカルボキサミドの製造方法 | |
| CN116406365A (zh) | 用于制备1-甲基-6-[6-R2-5-甲基-8-(甲基氨基)-4-[(3aS,6aS)-5-甲基-2,3,3a,4,6,6a-六氢吡咯并[2,3-c]吡咯-1-基]-9H-吡啶并[2,3-b]吲哚-3-基]-4-氧代-1,8-萘啶-3-甲酸盐酸盐的方法 | |
| CN111943894A (zh) | 一种4,7-二氮杂螺[2.5]辛烷类化合物的合成方法 | |
| NZ736564B2 (en) | Process for preparation of nitrogen mustard derivatives | |
| CN101627041A (zh) | 合成(3-烷基-5-哌啶-1-基-3,3a-二氢-吡唑并[1,5-a]嘧啶-7-基)-氨基衍生物和中间体的方法和中间体 |