ES2724112T3 - Adición del 1,4-conjugado asimétrico catalizado por metal de compuestos de vinilboro a 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 que producen prostaglandinas y análogos de prostaglandinas - Google Patents
Adición del 1,4-conjugado asimétrico catalizado por metal de compuestos de vinilboro a 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 que producen prostaglandinas y análogos de prostaglandinas Download PDFInfo
- Publication number
- ES2724112T3 ES2724112T3 ES15819728T ES15819728T ES2724112T3 ES 2724112 T3 ES2724112 T3 ES 2724112T3 ES 15819728 T ES15819728 T ES 15819728T ES 15819728 T ES15819728 T ES 15819728T ES 2724112 T3 ES2724112 T3 ES 2724112T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- compound
- formula
- oxy
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 139
- 150000003180 prostaglandins Chemical class 0.000 title description 37
- 229940094443 oxytocics prostaglandins Drugs 0.000 title description 18
- 238000000034 method Methods 0.000 claims abstract description 113
- 239000000654 additive Substances 0.000 claims abstract description 51
- 230000000996 additive effect Effects 0.000 claims abstract description 49
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 20
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 20
- 230000008569 process Effects 0.000 claims abstract description 20
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 18
- 229910052751 metal Inorganic materials 0.000 claims abstract description 18
- 239000002184 metal Substances 0.000 claims abstract description 18
- 239000002904 solvent Substances 0.000 claims abstract description 18
- 125000003118 aryl group Chemical group 0.000 claims abstract description 16
- 229910052796 boron Inorganic materials 0.000 claims abstract description 14
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims abstract description 11
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 10
- 239000001257 hydrogen Substances 0.000 claims abstract description 10
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims abstract description 10
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 9
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 9
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 9
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 8
- 125000001960 7 membered carbocyclic group Chemical group 0.000 claims abstract description 8
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims abstract description 8
- 125000004104 aryloxy group Chemical group 0.000 claims abstract description 8
- 125000000304 alkynyl group Chemical group 0.000 claims abstract description 7
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 7
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 7
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 claims abstract description 7
- 125000004423 acyloxy group Chemical group 0.000 claims abstract description 6
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 6
- 125000005160 aryl oxy alkyl group Chemical group 0.000 claims abstract description 6
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims abstract description 5
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims abstract description 5
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 5
- 150000002367 halogens Chemical class 0.000 claims abstract description 5
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims abstract description 4
- 125000004043 oxo group Chemical group O=* 0.000 claims abstract description 4
- 125000003341 7 membered heterocyclic group Chemical group 0.000 claims abstract 2
- -1 aryloxy acyloxy Chemical group 0.000 claims description 76
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 42
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 40
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 30
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 claims description 22
- 239000004912 1,5-cyclooctadiene Substances 0.000 claims description 22
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 claims description 21
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 17
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 15
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 15
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 claims description 14
- WSNODXPBBALQOF-VEJSHDCNSA-N tafluprost Chemical compound CC(C)OC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\C(F)(F)COC1=CC=CC=C1 WSNODXPBBALQOF-VEJSHDCNSA-N 0.000 claims description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 12
- 229960004458 tafluprost Drugs 0.000 claims description 12
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 12
- 229960002986 dinoprostone Drugs 0.000 claims description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 11
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 10
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 claims description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 8
- 229960001342 dinoprost Drugs 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 8
- PXGPLTODNUVGFL-YNNPMVKQSA-N prostaglandin F2alpha Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O PXGPLTODNUVGFL-YNNPMVKQSA-N 0.000 claims description 8
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 7
- WGFOBBZOWHGYQH-MXHNKVEKSA-N lubiprostone Chemical compound O1[C@](C(F)(F)CCCC)(O)CC[C@@H]2[C@@H](CCCCCCC(O)=O)C(=O)C[C@H]21 WGFOBBZOWHGYQH-MXHNKVEKSA-N 0.000 claims description 7
- MKPLKVHSHYCHOC-AHTXBMBWSA-N travoprost Chemical compound CC(C)OC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)COC1=CC=CC(C(F)(F)F)=C1 MKPLKVHSHYCHOC-AHTXBMBWSA-N 0.000 claims description 7
- 125000004665 trialkylsilyl group Chemical group 0.000 claims description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 6
- 229960000345 lubiprostone Drugs 0.000 claims description 6
- 229910021645 metal ion Inorganic materials 0.000 claims description 6
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 claims description 6
- 150000003284 rhodium compounds Chemical class 0.000 claims description 6
- 229960002368 travoprost Drugs 0.000 claims description 6
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 claims description 6
- GMVPRGQOIOIIMI-UHFFFAOYSA-N (8R,11R,12R,13E,15S)-11,15-Dihydroxy-9-oxo-13-prostenoic acid Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CCCCCCC(O)=O GMVPRGQOIOIIMI-UHFFFAOYSA-N 0.000 claims description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 5
- 229960000711 alprostadil Drugs 0.000 claims description 5
- AQOKCDNYWBIDND-FTOWTWDKSA-N bimatoprost Chemical compound CCNC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)CCC1=CC=CC=C1 AQOKCDNYWBIDND-FTOWTWDKSA-N 0.000 claims description 5
- 229960002470 bimatoprost Drugs 0.000 claims description 5
- 150000001993 dienes Chemical class 0.000 claims description 5
- GMVPRGQOIOIIMI-DWKJAMRDSA-N prostaglandin E1 Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O GMVPRGQOIOIIMI-DWKJAMRDSA-N 0.000 claims description 5
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 4
- 150000007942 carboxylates Chemical group 0.000 claims description 4
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 claims description 4
- 239000003446 ligand Substances 0.000 claims description 4
- VBKNTGMWIPUCRF-UHFFFAOYSA-M potassium;fluoride;hydrofluoride Chemical compound F.[F-].[K+] VBKNTGMWIPUCRF-UHFFFAOYSA-M 0.000 claims description 4
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 claims description 4
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 claims description 3
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 claims description 3
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 3
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 3
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 3
- 125000005129 aryl carbonyl group Chemical group 0.000 claims description 3
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 claims description 3
- SJYNFBVQFBRSIB-UHFFFAOYSA-N norbornadiene Chemical compound C1=CC2C=CC1C2 SJYNFBVQFBRSIB-UHFFFAOYSA-N 0.000 claims description 3
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 2
- 239000007836 KH2PO4 Substances 0.000 claims description 2
- 229960003395 carboprost Drugs 0.000 claims description 2
- DLJKPYFALUEJCK-MRVZPHNRSA-N carboprost Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C\CCCC(O)=O DLJKPYFALUEJCK-MRVZPHNRSA-N 0.000 claims description 2
- 150000001869 cobalt compounds Chemical class 0.000 claims description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 239000011737 fluorine Substances 0.000 claims description 2
- 229940035429 isobutyl alcohol Drugs 0.000 claims description 2
- 229960001160 latanoprost Drugs 0.000 claims description 2
- GGXICVAJURFBLW-CEYXHVGTSA-N latanoprost Chemical compound CC(C)OC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1CC[C@@H](O)CCC1=CC=CC=C1 GGXICVAJURFBLW-CEYXHVGTSA-N 0.000 claims description 2
- 229910000402 monopotassium phosphate Inorganic materials 0.000 claims description 2
- 235000019796 monopotassium phosphate Nutrition 0.000 claims description 2
- 150000002816 nickel compounds Chemical class 0.000 claims description 2
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 claims description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 claims description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims 2
- AQTFKGDWFRRIHR-UHFFFAOYSA-L 3-[18-(2-carboxylatoethyl)-8,13-bis(ethenyl)-3,7,12,17-tetramethylporphyrin-21,24-diid-2-yl]propanoate;cobalt(2+);hydron Chemical compound [Co+2].[N-]1C(C=C2C(=C(C)C(C=C3C(=C(C)C(=C4)[N-]3)C=C)=N2)C=C)=C(C)C(CCC(O)=O)=C1C=C1C(CCC(O)=O)=C(C)C4=N1 AQTFKGDWFRRIHR-UHFFFAOYSA-L 0.000 claims 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims 1
- XXUPXHKCPIKWLR-JHUOEJJVSA-N isopropyl unoprostone Chemical group CCCCCCCC(=O)CC[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(=O)OC(C)C XXUPXHKCPIKWLR-JHUOEJJVSA-N 0.000 claims 1
- 229910000027 potassium carbonate Inorganic materials 0.000 claims 1
- 235000015320 potassium carbonate Nutrition 0.000 claims 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 claims 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 claims 1
- 235000019798 tripotassium phosphate Nutrition 0.000 claims 1
- 229950008081 unoprostone isopropyl Drugs 0.000 claims 1
- 238000002360 preparation method Methods 0.000 abstract description 6
- 125000000896 monocarboxylic acid group Chemical group 0.000 abstract description 3
- 238000003786 synthesis reaction Methods 0.000 description 91
- 230000015572 biosynthetic process Effects 0.000 description 90
- 238000005160 1H NMR spectroscopy Methods 0.000 description 63
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 58
- 239000000203 mixture Substances 0.000 description 54
- 239000000243 solution Substances 0.000 description 48
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 44
- 238000004440 column chromatography Methods 0.000 description 42
- 238000006243 chemical reaction Methods 0.000 description 41
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 41
- 239000011541 reaction mixture Substances 0.000 description 41
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 36
- 239000012043 crude product Substances 0.000 description 34
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 31
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 31
- 238000001437 electrospray ionisation time-of-flight quadrupole detection Methods 0.000 description 31
- HHRKFGMMAHZWIM-UHFFFAOYSA-N ethenoxyboronic acid Chemical compound OB(O)OC=C HHRKFGMMAHZWIM-UHFFFAOYSA-N 0.000 description 29
- 235000019439 ethyl acetate Nutrition 0.000 description 29
- KDCIHNCMPUBDKT-UHFFFAOYSA-N hexane;propan-2-one Chemical class CC(C)=O.CCCCCC KDCIHNCMPUBDKT-UHFFFAOYSA-N 0.000 description 27
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- 239000010410 layer Substances 0.000 description 23
- 238000011084 recovery Methods 0.000 description 20
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 19
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 18
- 235000019341 magnesium sulphate Nutrition 0.000 description 18
- 239000012044 organic layer Substances 0.000 description 18
- 239000012267 brine Substances 0.000 description 16
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 16
- 229920002554 vinyl polymer Polymers 0.000 description 16
- 239000003153 chemical reaction reagent Substances 0.000 description 15
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 12
- 238000007259 addition reaction Methods 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 11
- 229910002027 silica gel Inorganic materials 0.000 description 11
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 10
- 238000010438 heat treatment Methods 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- NIRIPZHJYDSGFG-VOTSOKGWSA-N [(e)-2-phenylethenoxy]boronic acid Chemical compound OB(O)O\C=C\C1=CC=CC=C1 NIRIPZHJYDSGFG-VOTSOKGWSA-N 0.000 description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 9
- 239000002243 precursor Substances 0.000 description 9
- PXGPLTODNUVGFL-BRIYLRKRSA-N (E,Z)-(1R,2R,3R,5S)-7-(3,5-Dihydroxy-2-((3S)-(3-hydroxy-1-octenyl))cyclopentyl)-5-heptenoic acid Chemical class CCCCC[C@H](O)C=C[C@H]1[C@H](O)C[C@H](O)[C@@H]1CC=CCCCC(O)=O PXGPLTODNUVGFL-BRIYLRKRSA-N 0.000 description 8
- DEQYTNZJHKPYEZ-UHFFFAOYSA-N ethyl acetate;heptane Chemical compound CCOC(C)=O.CCCCCCC DEQYTNZJHKPYEZ-UHFFFAOYSA-N 0.000 description 8
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 8
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 7
- 101150065749 Churc1 gene Proteins 0.000 description 7
- 235000019270 ammonium chloride Nutrition 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- BZKFMUIJRXWWQK-UHFFFAOYSA-N Cyclopentenone Chemical compound O=C1CCC=C1 BZKFMUIJRXWWQK-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Chemical compound [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 6
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N hexane Substances CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- YDIYEOMDOWUDTJ-UHFFFAOYSA-N 4-(dimethylamino)benzoic acid Chemical compound CN(C)C1=CC=C(C(O)=O)C=C1 YDIYEOMDOWUDTJ-UHFFFAOYSA-N 0.000 description 5
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 5
- 239000005749 Copper compound Substances 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- KEXBLKHKABOPSR-UHFFFAOYSA-N [K]C=C.FB(F)F Chemical compound [K]C=C.FB(F)F KEXBLKHKABOPSR-UHFFFAOYSA-N 0.000 description 5
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 5
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- COCAUCFPFHUGAA-MGNBDDOMSA-N n-[3-[(1s,7s)-5-amino-4-thia-6-azabicyclo[5.1.0]oct-5-en-7-yl]-4-fluorophenyl]-5-chloropyridine-2-carboxamide Chemical compound C=1C=C(F)C([C@@]23N=C(SCC[C@@H]2C3)N)=CC=1NC(=O)C1=CC=C(Cl)C=N1 COCAUCFPFHUGAA-MGNBDDOMSA-N 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- LMPPOYVKJVJRTH-UHFFFAOYSA-N propan-2-yl hept-5-enoate Chemical compound C(C)(C)OC(CCCC=CC)=O LMPPOYVKJVJRTH-UHFFFAOYSA-N 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 150000003254 radicals Chemical class 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- DZUXGQBLFALXCR-UHFFFAOYSA-N (+)-(9alpha,11alpha,13E,15S)-9,11,15-trihydroxyprost-13-en-1-oic acid Chemical class CCCCCC(O)C=CC1C(O)CC(O)C1CCCCCCC(O)=O DZUXGQBLFALXCR-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 229940006138 antiglaucoma drug and miotics prostaglandin analogues Drugs 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 229910052681 coesite Inorganic materials 0.000 description 4
- 229910052906 cristobalite Inorganic materials 0.000 description 4
- KPSZWAJWFMFMFF-UHFFFAOYSA-N hept-5-enoic acid Chemical compound CC=CCCCC(O)=O KPSZWAJWFMFMFF-UHFFFAOYSA-N 0.000 description 4
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 4
- 239000012041 precatalyst Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 150000003283 rhodium Chemical class 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 235000012239 silicon dioxide Nutrition 0.000 description 4
- 229910052682 stishovite Inorganic materials 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 229910052905 tridymite Inorganic materials 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 229910000085 borane Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 239000007857 degradation product Substances 0.000 description 3
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 150000002739 metals Chemical class 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 239000003586 protic polar solvent Substances 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 239000010948 rhodium Substances 0.000 description 3
- BLSRGJPGRJBHQK-BUSXIPJBSA-N (2s)-2-amino-1-(2-diphenoxyphosphorylpyrrolidin-1-yl)propan-1-one Chemical compound C[C@H](N)C(=O)N1CCCC1P(=O)(OC=1C=CC=CC=1)OC1=CC=CC=C1 BLSRGJPGRJBHQK-BUSXIPJBSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 2
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo[3.3.1]nonane Substances C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- LMPPOYVKJVJRTH-PLNGDYQASA-N C\C=C/CCCC(=O)OC(C)C Chemical compound C\C=C/CCCC(=O)OC(C)C LMPPOYVKJVJRTH-PLNGDYQASA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical group [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 102100034741 Cyclin-dependent kinase 20 Human genes 0.000 description 2
- 238000006546 Horner-Wadsworth-Emmons reaction Methods 0.000 description 2
- 101500014379 Lymnaea stagnalis Ovulation hormone Proteins 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- QBBFLVILAPIVET-MDZDMXLPSA-N [(E)-3-(oxan-2-yloxy)oct-1-enyl]boronic acid Chemical compound O1C(CCCC1)OC(/C=C/B(O)O)CCCCC QBBFLVILAPIVET-MDZDMXLPSA-N 0.000 description 2
- OSSUPWTWJFJXDD-VAWYXSNFSA-N [(E)-3-phenylmethoxyoct-1-enyl]boronic acid Chemical compound C(C1=CC=CC=C1)OC(/C=C/B(O)O)CCCCC OSSUPWTWJFJXDD-VAWYXSNFSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 125000005336 allyloxy group Chemical group 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Inorganic materials [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- 150000001879 copper Chemical class 0.000 description 2
- HOXDXGRSZJEEKN-UHFFFAOYSA-N cycloocta-1,5-diene;rhodium Chemical compound [Rh].C1CC=CCCC=C1 HOXDXGRSZJEEKN-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- RWMJRMPOKXSHHI-UHFFFAOYSA-N ethenylboron Chemical compound [B]C=C RWMJRMPOKXSHHI-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- JYGYEBCBALMPDC-UHFFFAOYSA-N heptane;propan-2-one Chemical compound CC(C)=O.CCCCCCC JYGYEBCBALMPDC-UHFFFAOYSA-N 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- 231100001231 less toxic Toxicity 0.000 description 2
- 238000006138 lithiation reaction Methods 0.000 description 2
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 2
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 2
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- WGJJROVFWIXTPA-OALUTQOASA-N prostanoic acid Chemical compound CCCCCCCC[C@H]1CCC[C@@H]1CCCCCCC(O)=O WGJJROVFWIXTPA-OALUTQOASA-N 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical class [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 229910052723 transition metal Inorganic materials 0.000 description 2
- 150000003624 transition metals Chemical class 0.000 description 2
- 125000005106 triarylsilyl group Chemical group 0.000 description 2
- 125000004306 triazinyl group Chemical group 0.000 description 2
- ZGBHPZUKKNSAOS-UHFFFAOYSA-N tris(ethenyl)borane Chemical compound C=CB(C=C)C=C ZGBHPZUKKNSAOS-UHFFFAOYSA-N 0.000 description 2
- PLIQWXNVJOPJOT-UXMRNZNESA-N (2s)-2-amino-1-(2-diphenoxyphosphorylpyrrolidin-1-yl)-3-phenylpropan-1-one Chemical compound C([C@H](N)C(=O)N1C(CCC1)P(=O)(OC=1C=CC=CC=1)OC=1C=CC=CC=1)C1=CC=CC=C1 PLIQWXNVJOPJOT-UXMRNZNESA-N 0.000 description 1
- BNUHTPCULLFDEA-UHFFFAOYSA-N 1,2,2-trifluoroethenoxyboronic acid Chemical class OB(O)OC(F)=C(F)F BNUHTPCULLFDEA-UHFFFAOYSA-N 0.000 description 1
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 description 1
- YSDWCSDIGREWSD-UHFFFAOYSA-N 1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)oct-1-en-3-ol Chemical compound CCCCCC(O)C=CB1OC(C)(C)C(C)(C)O1 YSDWCSDIGREWSD-UHFFFAOYSA-N 0.000 description 1
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- LAGGTOBQMQHXON-UHFFFAOYSA-N 2,6-octadiene Chemical compound CC=CCCC=CC LAGGTOBQMQHXON-UHFFFAOYSA-N 0.000 description 1
- NXFFJDQHYLNEJK-UHFFFAOYSA-N 2-[4-[(4-chlorophenyl)methyl]-7-fluoro-5-methylsulfonyl-2,3-dihydro-1h-cyclopenta[b]indol-3-yl]acetic acid Chemical compound C1=2C(S(=O)(=O)C)=CC(F)=CC=2C=2CCC(CC(O)=O)C=2N1CC1=CC=C(Cl)C=C1 NXFFJDQHYLNEJK-UHFFFAOYSA-N 0.000 description 1
- DPGSPRJLAZGUBQ-UHFFFAOYSA-N 2-ethenyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical class CC1(C)OB(C=C)OC1(C)C DPGSPRJLAZGUBQ-UHFFFAOYSA-N 0.000 description 1
- FWJRSOGVYOIVOC-UHFFFAOYSA-N 2-methylprop-1-enylboronic acid Chemical compound CC(C)=CB(O)O FWJRSOGVYOIVOC-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 description 1
- JWYDHPFGQBATPA-BUHFOSPRSA-N 4,4,5,5-tetramethyl-2-[(E)-3-(oxan-2-yloxy)oct-1-enyl]-1,3,2-dioxaborolane Chemical compound O1C(CCCC1)OC(/C=C/B1OC(C(O1)(C)C)(C)C)CCCCC JWYDHPFGQBATPA-BUHFOSPRSA-N 0.000 description 1
- PWTHRCFGLFZGHD-FOCLMDBBSA-N 4,4,5,5-tetramethyl-2-[(E)-3-phenylmethoxyoct-1-enyl]-1,3,2-dioxaborolane Chemical compound C(C1=CC=CC=C1)OC(/C=C/B1OC(C(O1)(C)C)(C)C)CCCCC PWTHRCFGLFZGHD-FOCLMDBBSA-N 0.000 description 1
- YFBXUHFVLMHTAI-OUKQBFOZSA-N 4,4,5,5-tetramethyl-2-[(E)-3-prop-2-enoxyoct-1-enyl]-1,3,2-dioxaborolane Chemical compound CCCCCC(OCC=C)\C=C\B1OC(C)(C)C(C)(C)O1 YFBXUHFVLMHTAI-OUKQBFOZSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- HSYSCPBUWMIKMI-UHFFFAOYSA-N 4-silyloxycyclopent-2-en-1-one Chemical group [SiH3]OC1CC(=O)C=C1 HSYSCPBUWMIKMI-UHFFFAOYSA-N 0.000 description 1
- 125000004199 4-trifluoromethylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C(F)(F)F 0.000 description 1
- AQOKCDNYWBIDND-ABRBVVEGSA-N 5-trans-17-phenyl trinor Prostaglandin F2alpha ethyl amide Chemical compound CCNC(=O)CCC\C=C\C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)CCC1=CC=CC=C1 AQOKCDNYWBIDND-ABRBVVEGSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- AMKGKYQBASDDJB-UHFFFAOYSA-N 9$l^{2}-borabicyclo[3.3.1]nonane Chemical compound C1CCC2CCCC1[B]2 AMKGKYQBASDDJB-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- LGPBIFFEKVLFHT-VOTSOKGWSA-N B(O)(O)O/C=C/C1=CC=CC=C1C Chemical compound B(O)(O)O/C=C/C1=CC=CC=C1C LGPBIFFEKVLFHT-VOTSOKGWSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 0 CC(*)[C@]1C=CC=CC1 Chemical compound CC(*)[C@]1C=CC=CC1 0.000 description 1
- VWEZAKLFZNZUKP-BQYQJAHWSA-N CCCCCC/C=C/B1OCCO1 Chemical compound CCCCCC/C=C/B1OCCO1 VWEZAKLFZNZUKP-BQYQJAHWSA-N 0.000 description 1
- QCEMGXGRSCHQTM-UHFFFAOYSA-N COC(CCCCCCC(C(C1)=O)=CC1OC1OCCCC1)O Chemical compound COC(CCCCCCC(C(C1)=O)=CC1OC1OCCCC1)O QCEMGXGRSCHQTM-UHFFFAOYSA-N 0.000 description 1
- CLEQYDZLNUXUFR-VOTSOKGWSA-N COC1=CC=C(C=C1)/C=C/OB(O)O Chemical compound COC1=CC=C(C=C1)/C=C/OB(O)O CLEQYDZLNUXUFR-VOTSOKGWSA-N 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 206010010774 Constipation Diseases 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- 206010018307 Glaucoma and ocular hypertension Diseases 0.000 description 1
- 239000002879 Lewis base Substances 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- QSWXHURYWXJJIV-AATRIKPKSA-N [(E)-2-[4-(trifluoromethyl)phenyl]ethenoxy]boronic acid Chemical compound FC(C1=CC=C(C=C1)/C=C/OB(O)O)(F)F QSWXHURYWXJJIV-AATRIKPKSA-N 0.000 description 1
- VTMFXCDGVGRMAX-VAWYXSNFSA-N [(E)-3-[tert-butyl(dimethyl)silyl]oxyoct-1-enyl]boronic acid Chemical compound [Si](C)(C)(C(C)(C)C)OC(/C=C/B(O)O)CCCCC VTMFXCDGVGRMAX-VAWYXSNFSA-N 0.000 description 1
- ZKQSEPNVMYZKEI-CMDGGOBGSA-N [(E)-3-prop-2-enoxyoct-1-enyl]boronic acid Chemical compound CCCCCC(OCC=C)\C=C\B(O)O ZKQSEPNVMYZKEI-CMDGGOBGSA-N 0.000 description 1
- VKBIRAUSKUGVLY-VUSFMPOISA-N [(E,3S)-3-[tert-butyl(dimethyl)silyl]oxy-5-phenylpent-1-enyl]boronic acid Chemical compound [Si](C)(C)(C(C)(C)C)O[C@H](/C=C/B(O)O)CCC1=CC=CC=C1 VKBIRAUSKUGVLY-VUSFMPOISA-N 0.000 description 1
- VTMFXCDGVGRMAX-YLSINNKHSA-N [(E,3S)-3-[tert-butyl(dimethyl)silyl]oxyoct-1-enyl]boronic acid Chemical compound [Si](C)(C)(C(C)(C)C)O[C@H](/C=C/B(O)O)CCCCC VTMFXCDGVGRMAX-YLSINNKHSA-N 0.000 description 1
- RBTAJLKAPFBZDQ-BQYQJAHWSA-N [(e)-oct-1-enyl]boronic acid Chemical compound CCCCCC\C=C\B(O)O RBTAJLKAPFBZDQ-BQYQJAHWSA-N 0.000 description 1
- CCXHEPSOPWJNJB-ONEGZZNKSA-N [(z)-but-2-en-2-yl]boronic acid Chemical compound C\C=C(/C)B(O)O CCXHEPSOPWJNJB-ONEGZZNKSA-N 0.000 description 1
- QBBRWBVKFOXJCQ-UHFFFAOYSA-N [Cu]C=C Chemical class [Cu]C=C QBBRWBVKFOXJCQ-UHFFFAOYSA-N 0.000 description 1
- WLLIXJBWWFGEHT-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl] trifluoromethanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OS(=O)(=O)C(F)(F)F WLLIXJBWWFGEHT-UHFFFAOYSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 229940040386 amitiza Drugs 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000001231 benzoyloxy group Chemical group C(C1=CC=CC=C1)(=O)O* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Chemical group 0.000 description 1
- XIXSBTKGTZXZFJ-UHFFFAOYSA-N bis(2,5-dimethylhex-4-en-3-yl)borane Chemical compound CC(C)=CC(C(C)C)BC(C=C(C)C)C(C)C XIXSBTKGTZXZFJ-UHFFFAOYSA-N 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 125000005621 boronate group Chemical class 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- ZDQWVKDDJDIVAL-UHFFFAOYSA-N catecholborane Chemical compound C1=CC=C2O[B]OC2=C1 ZDQWVKDDJDIVAL-UHFFFAOYSA-N 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 150000001804 chlorine Chemical class 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- XZWQKJXJNKYMAP-UHFFFAOYSA-N cyclohexen-1-ylboronic acid Chemical compound OB(O)C1=CCCCC1 XZWQKJXJNKYMAP-UHFFFAOYSA-N 0.000 description 1
- 150000001940 cyclopentanes Chemical group 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- WVJGKRMLCSNRKG-UHFFFAOYSA-N dibromoborane Chemical compound BrBBr WVJGKRMLCSNRKG-UHFFFAOYSA-N 0.000 description 1
- GFKJMQOPKRDGGL-UHFFFAOYSA-N dibromoboron Chemical compound Br[B]Br GFKJMQOPKRDGGL-UHFFFAOYSA-N 0.000 description 1
- LHCGBIFHSCCRRG-UHFFFAOYSA-N dichloroborane Chemical compound ClBCl LHCGBIFHSCCRRG-UHFFFAOYSA-N 0.000 description 1
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 1
- XNYOSXARXANYPB-UHFFFAOYSA-N dicyclohexylborane Chemical compound C1CCCCC1BC1CCCCC1 XNYOSXARXANYPB-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- HXJFQNUWPUICNY-UHFFFAOYSA-N disiamylborane Chemical compound CC(C)C(C)BC(C)C(C)C HXJFQNUWPUICNY-UHFFFAOYSA-N 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- YFXCNIVBAVFOBX-UHFFFAOYSA-N ethenylboronic acid Chemical class OB(O)C=C YFXCNIVBAVFOBX-UHFFFAOYSA-N 0.000 description 1
- BADWIIDKTXQYLW-UHFFFAOYSA-N ethenylstannane Chemical class [SnH3]C=C BADWIIDKTXQYLW-UHFFFAOYSA-N 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 235000019256 formaldehyde Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- JAGYXYUAYDLKNO-UHFFFAOYSA-N hepta-2,5-diene Chemical compound CC=CCC=CC JAGYXYUAYDLKNO-UHFFFAOYSA-N 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 238000009434 installation Methods 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000007527 lewis bases Chemical class 0.000 description 1
- LZWQNOHZMQIFBX-UHFFFAOYSA-N lithium;2-methylpropan-2-olate Chemical compound [Li+].CC(C)(C)[O-] LZWQNOHZMQIFBX-UHFFFAOYSA-N 0.000 description 1
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- NIOJULSEJPAEJV-UHFFFAOYSA-N methyl 7-[3-[tert-butyl(dimethyl)silyl]oxy-5-oxocyclopenten-1-yl]heptanoate Chemical compound COC(=O)CCCCCCC1=CC(O[Si](C)(C)C(C)(C)C)CC1=O NIOJULSEJPAEJV-UHFFFAOYSA-N 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 1
- GDDAJHJRAKOILH-UHFFFAOYSA-N octa-2,5-diene Chemical compound CCC=CCC=CC GDDAJHJRAKOILH-UHFFFAOYSA-N 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 150000002900 organolithium compounds Chemical class 0.000 description 1
- 150000002902 organometallic compounds Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- WCONKKYQBKPMNZ-UHFFFAOYSA-N prop-1-en-2-ylboronic acid Chemical compound CC(=C)B(O)O WCONKKYQBKPMNZ-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003166 prostaglandin E2 derivatives Chemical class 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 230000001012 protector Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000012713 reactive precursor Substances 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- KTEDZFORYFITAF-UHFFFAOYSA-K rhodium(3+);trihydroxide Chemical class [OH-].[OH-].[OH-].[Rh+3] KTEDZFORYFITAF-UHFFFAOYSA-K 0.000 description 1
- SONJTKJMTWTJCT-UHFFFAOYSA-K rhodium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Rh+3] SONJTKJMTWTJCT-UHFFFAOYSA-K 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- SUBJHSREKVAVAR-UHFFFAOYSA-N sodium;methanol;methanolate Chemical compound [Na+].OC.[O-]C SUBJHSREKVAVAR-UHFFFAOYSA-N 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 125000005504 styryl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- UGSDKGSWOAVCBG-IWLLYFRBSA-N tert-butyl-dimethyl-[(E,3S)-1-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)oct-1-en-3-yl]oxysilane Chemical compound C(C)(C)(C)[Si](O[C@H](/C=C/B1OC(C(O1)(C)C)(C)C)CCCCC)(C)C UGSDKGSWOAVCBG-IWLLYFRBSA-N 0.000 description 1
- MMKOSVMWXHDGEI-UHFFFAOYSA-N tert-butyl-dimethyl-oct-1-yn-3-yloxysilane Chemical compound CCCCCC(C#C)O[Si](C)(C)C(C)(C)C MMKOSVMWXHDGEI-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 229940113006 travatan Drugs 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- TVHAZVBUYQMHBC-SNHXEXRGSA-N unoprostone Chemical compound CCCCCCCC(=O)CC[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O TVHAZVBUYQMHBC-SNHXEXRGSA-N 0.000 description 1
- 229960004317 unoprostone Drugs 0.000 description 1
- 238000006886 vinylation reaction Methods 0.000 description 1
- 229940018148 zioptan Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/1892—Preparation; Treatments not provided for in C07F7/20 by reactions not provided for in C07F7/1876 - C07F7/1888
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/48—Separation; Purification; Stabilisation; Use of additives
- C07C67/62—Use of additives, e.g. for stabilisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/738—Esters of keto-carboxylic acids or aldehydo-carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Un procedimiento de preparación de un compuesto de 4-oxi-ciclopentan-1-ona 2,3-disustituido de fórmula I**Fórmula** el procedimiento comprende poner en contacto un compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II**Fórmula** un compuesto de fórmula III**Fórmula** en un disolvente con un aditivo metálico, opcionalmente en presencia de un aditivo básico, para dar el compuesto de fórmula I; en la que R1, R3, R4 y R5 representan independientemente hidrógeno, arilo, heteroarilo, alquilo, arilalquilo, ariloxialquilo, alquenilo o alquinilo; o R3 y R5 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, que tiene opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S; o R3 y R4 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, teniendo opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S, y en la que cada uno de R1, R3, R4 y R5 están opcionalmente sustituidos con uno a tres miembros seleccionados del grupo que consiste en halógeno, hidroxi, alquilo C1-4, haloalquilo C1-4, alcoxi C1-4, sililoxi, ariloxi, aciloxi, un anillo heterocíclico de 5 a 7 miembros, oxo, COOH, CONH2, CONH alquilo C1-4, C(O)OCH2 arilo C6-10, C(O)O arilo C6-10 y C(O)O alquilo C1-4; R2 representa hidrógeno o un grupo protector de hidroxilo; X representa un grupo que contiene boro.
Description
DESCRIPCIÓN
Adición del 1,4-conjugado asimétrico catalizado por metal de compuestos de vinilboro a 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 que producen prostaglandinas y análogos de prostaglandinas
Referencias cruzadas a aplicaciones relacionadas
La presente solicitud reivindica prioridad a la solicitud de patente provisional U.S. No. 62/022.797 presentada el 10 de julio de 2014.
Antecedentes de la invención
Las prostaglandinas naturales tienen una estructura química única con base en el ácido prostanoico (ácido 7-[(1S,2S)-2-octilciclopentil]heptanoico) y exhiben un amplio intervalo de actividades fisiológicas incluso cuando están presentes en cantidades extremadamente pequeñas. Se ha desarrollado una gran cantidad de prostaglandinas y fármacos basados en análogos de prostaglandinas para una variedad de indicaciones médicas. Por ejemplo, tafluprost (ZIOPTAN), travoprost (TRAVATAN) y bimatoprost (LUMIGAN) se usan por vía tópica (como gotas para los ojos) para tratar el glaucoma y la hipertensión ocular. Lubiprostone (AMITIZA) se usa en el manejo del estreñimiento crónico. La dinoprostona es una prostaglandina de origen natural (PGE2) que se usa en la clínica para la inducción del trabajo de parto en humanos. Por lo tanto, dada la importancia farmacéutica de estos compuestos y sus análogos, se han desarrollado y divulgado numerosos procedimientos tanto en la academia como en la industria para la fabricación de prostaglandinas y análogos estructurales de las prostaglandinas.
Una metodología temprana útil para la síntesis del análogo de prostaglandina F2a y la prostaglandina E2 fue divulgado por E. J. Corey en 1969 (J. Am. Chem. Soc., 1969, 91, 5675-5676). Esta metodología se conoce como el procedimiento Corey, y la bien conocida Corey lactona -que a su vez requiere alrededor de 10 etapas sintéticas y contiene los tres centros estereoquímicos de prostaglandina E (PGE) ya existentes- es fundamental para el procedimiento Corey. Las cadenas laterales basadas en hidrocarburos w y a se agregan secuencialmente por las reacciones de Horner-Wadsworth-Emmons y Wittig (véase Figura 1). El procedimiento Corey y sus modificaciones y permutaciones más recientes son probablemente las metodologías sintéticas más utilizadas e informadas para la fabricación industrial de prostaglandinas y análogos de prostaglandinas. Las desventajas de esta metodología incluyen, sin embargo, el alto coste de la lactona Corey y la pesada purificación cromatográfica en columna que a menudo se requieren para eliminar los isómeros y/o impurezas no deseados.
Otra metodología que se puede usar para preparar prostaglandinas y sus análogos a veces se conoce como la metodología de dos componentes (J. Am. Chem. Soc., 1972, 94, 3643-3644 y J. Am. Chem. Soc., 1972, 94, 7827 7832). La característica clave de esta metodología es la instalación de la cadena lateral w utilizando una reacción de adición de 1,4-conjugado de un organocobre de vinilo o reactivo de organocuprato a un sistema de ciclopentenona en el que ya está presente la cadena lateral a (véase Figura 2). Este procedimiento, que utiliza reactivos de organocobre, se refiere aquí como la metodología convencional de dos componentes. Hay muchos procedimientos conocidos para hacer ciclopentenonas sustituidas en la cadena lateral a que son útiles en la metodología convencional de dos componentes. La metodología convencional de dos componentes se ha utilizado para la síntesis de una variedad de prostaglandinas y sus análogos.
Frito, et. al. (J. Am. Chem. Soc. 1972, 94, 7827-7832), Lipshutz et al. (J. Am. Chem. Soc. 1988, 110, 2641-2643), Lipshutz, et. al.. . (J. Am. Chem. Soc. 1990, 112, 7440-7441) y Van Hijfte et al. (Tetrahedron 1992, 48, 6393-6402) han divulgado procedimientos para preparar prostaglandina E1 (PGE1) utilizando la metodología de dos componentes, en el que diversos organocupratos, como agentes de vinilación, se acoplaron con ciclopentenonas por adición de 1,4-conjugado bajo temperaturas criogénicas. Los reactivos de organoestaño, los reactivos de orangolitio o los reactivos de organozirconio se requieren como precursores sintéticos de los compuestos organocobre mencionados anteriormente utilizados en el procedimiento.
La Patente U.S. No. 7.897.795 (la patente '795) y la Patente de U.S. No. 8.846.958 (la patente '958) que fueron divulgadas por el solicitante, describen la utilización de la metodología convencional de dos componentes (véase Figura 2) para las síntesis de ciertos análogos de la prostaglandina (por ejemplo, travoprost, bimatoprost y lubiprostona). En ciertos procedimientos descritos en la patente '795 y en la patente '958, un producto intermedio II 4-oxi-ciclopent-2-en-1-ona sustituido en 2 reacciona con un cuprato de orden superior mediante adición de 1,4-conjugado para dar un compuesto I de 4-oxi-ciclopentan-1-ona 2,3-disustituido. Este compuesto, I, puede modificarse y desprotegerse opcionalmente para proporcionar diversos análogos de prostaglandina E y prostaglandina F.
Sin embargo, el uso de una metodología de dos componentes está asociado con un número de limitaciones y desventajas que incluyen: la necesidad de temperaturas criogénicas (aproximadamente -50 a -78 °C) en la etapa de adición de 1,4-conjugado; el uso de compuestos organometálicos como precursores de los compuestos de organocobre , tales como los compuestos de organoestaño, que se consideran tóxicos y difíciles de purificar, o los compuestos de organozirconio, que son sensibles a la humedad y que pueden requerir temperaturas criogénicas para su preparación; el uso de compuestos de organolitio reactivos y difíciles de manejar para la preparación de los compuestos de organocobre; y la necesidad de etapas múltiples para la conversión de los materiales de partida de
alquino a través de los compuestos de organocobre requeridos. Además, el cianuro en algunos de los reactivos de cuprato es tóxico. Los cupratos no están disponibles comercialmente, debido en parte a su reactividad e inestabilidad bajo condiciones ambientales, incluida su sensibilidad al aire, lo que requiere su uso inmediato en la síntesis. La reactividad del cuprato está modulada, e incluso puede estar limitada, por la naturaleza electrónica de los sustituyentes en la estructura de carbono adyacente al átomo de cobre. De hecho, en algunos casos (por ejemplo, tafluprost; véase Figura 4), la reacción de adición de 1,4-conjugado deseada a ciclopentenonas sustituidas en la cadena lateral a no funciona. Adicionalmente, la sal de cobre utilizada para hacer los compuestos de organocobre se requiere en cantidades estequiométricas.
Dados los aspectos ineficientes y las dificultades operativas asociadas con el uso de los procedimientos convencionales en el campo al que se refiere la presente invención, existe la necesidad de desarrollar un procedimiento más suave, menos tóxico, económico y fácil de usar para enantioselectivamente y diastereoselectivamente preparar análogos de prostaglandinas con buenos rendimientos. Sorprendentemente, la presente invención proporciona soluciones a este y otros problemas en el campo relevante al que pertenece la presente invención.
El documento WO 2010/104344 A2 divulga un procedimiento para preparar derivados de prostaglandina (4-oxiciclopentan-1-ona 2,3-disustituido), en el que el 4-oxi-ciclopentan-1-ona 2,3-disustituido se prepara por adición de 1,4-conjugados de cupratos obtenidos después de la litiación y adición de cobre a compuestos de vinilestannano. David A. Evans et al.: "Studies directed toward the synthesis of prostaglandins. Useful boron-mediated olefin syntheses", J. Org. Chem., vol. 41, 1976, páginas 3947-3953, XP055251140 divulga el uso de vinilborantos para el acoplamiento C-C en 1,4-ciclopent-2-en-1,4-dioles.
El documento US 2009/259058 A1 divulga la adición de 1,4-conjugados de vinilcupratos a andamios de ciclopent-2-enona, y en los que los vinilcupratos se obtienen de vinilestannanos después de la litiación y el tratamiento con sales de cobre.
El documento WO 2012/048447 A1 divulga la adición de 1,4-conjugados de cupratos a los andamios de ciclopent-2-enona.
David A. Evans et al.: "Stereospecific olefin synthesis via boronic esters. Studies related to prostaglandin synthesis", Tetrahedron Letters, no. 8, 1976, páginas 1427-1430, XP000957068 divulga la formación de enlaces C-C en la posición 2 de ciclopentanos sustituidos con 1 -alquilo.
Breve sumario de la invención
En un aspecto, la presente invención proporciona un procedimiento para preparar un compuesto de 4-oxiciclopentan-1-ona 2,3-disustituido de fórmula I

El procedimiento incluye poner en contacto un compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II

con un compuesto de fórmula III

en un disolvente en presencia de un aditivo metálico, opcionalmente en presencia de un aditivo básico, para dar el compuesto de fórmula I;
en la que
R1, R3, R4 y R5 representan independientemente hidrógeno, arilo, heteroarilo, alquilo, arilalquilo, ariloxialquilo, alquenilo o alquinilo;
o R3 y R5 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, que tiene opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S;
o R3 y R4 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, que tiene opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S,
y en la que cada uno de R1, R3, R4 y R5 están opcionalmente sustituidos con uno a tres miembros seleccionados del grupo que consiste en halógeno, hidroxi, alquilo C1-4, haloalquilo C1-4, alcoxi C1-4, sililoxi, ariloxi, aciloxi, anillo heterocíclico, oxo, COOH, CONH2, CONH alquilo C1-4, C(O)OCH2 arilo C6-10, C(O)O arilo C6-10 y C(O)O alquilo C1-4;
R2 representa hidrógeno o un grupo protector de hidroxilo;
X representa un grupo que contiene boro.
Diversos rasgos que caracterizan la invención se señalan con particularidad en las reivindicaciones adjuntas y que forman parte de la divulgación. Para una mejor comprensión de la invención, sus ventajas operativas y los objetos específicos alcanzados por su uso, debe hacerse referencia al dibujo y al material descriptivo en el que se ilustran y divulgan las realizaciones preferidas de la invención.
Breve descripción de los dibujos
La Figura 1 muestra el procedimiento Corey para la síntesis de prostaglandinas.
La Figura 2 muestra la metodología convencional de dos componentes para la síntesis de prostaglandinas.
La Figura 3 muestra la metodología mejorada de dos componentes para la síntesis de prostaglandinas y análogos de prostaglandinas como se describe por el presente solicitante.
La Figura 4 muestra algunas prostaglandinas y análogos de prostaglandinas de importancia industrial y farmacéutica.
La Figura 5 muestra la síntesis de dinoprost utilizando la presente invención.
La Figura 6 muestra la síntesis de tafluprost utilizando la presente invención.
Descripción detallada de la invención
I. General
La presente invención proporciona un novedoso procedimiento que utiliza una reacción de adición de 1,4-conjugado asimétrico catalizado por metal de compuestos de vinilboro y 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 para producir compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituido que son precursores sintéticos útiles de prostaglandinas y análogos de prostaglandinas de importancia industrial y farmacológica. Este procedimiento utiliza reactivos menos tóxicos y más fáciles de manipular, y puede realizarse bajo condiciones de reacción más leves que el procedimiento convencional de dos componentes y es capaz de producir un intervalo diverso de precursores de prostaglandinas y de análogos de prostaglandinas de forma enantio- y diastereoselectiva con alto rendimiento. II. Definiciones
Como se usa en el presente documento, el término "contactar" se refiere al procedimiento de poner en contacto al menos dos especies distintas, de tal manera que puedan reaccionar. Sin embargo, debe apreciarse que el producto de reacción resultante se puede producir directamente a partir de una reacción entre los reactivos agregados o a partir de un producto intermedio de uno o más de los reactivos agregados que se pueden producir en la mezcla de reacción.
Como se usa en el presente documento, el término "aditivo metálico" se refiere a un catalizador o precatalizador que se usa en cantidades subestequiométricas en una reacción química. Un catalizador es una sustancia química que aumenta la tasa de una reacción química de uno o más reactivos. Debe apreciarse que, a diferencia de otros reactivos en la reacción química, la reacción no consume un catalizador. Un pre-catalizador es una sustancia química que en sí misma podría no ser químicamente activa, o es menos activa químicamente, en dicha reacción y
se convierte en la reacción en un catalizador por la acción de un aditivo. Adicionalmente, el término "subestequiométrico" se refiere a una cantidad que es menor que una cantidad estequiométrica. Por ejemplo, cuando 1 mol del compuesto II se combina con 1 mol o más del compuesto III, una cantidad subestequiométrica de un aditivo metálico es menor que aproximadamente 1 mol; en general, menos de aproximadamente 0,5 moles; en algunas realizaciones, menos de aproximadamente 0,1 moles; en realizaciones preferidas, de aproximadamente 0,05 a aproximadamente 0,0001 moles.
Como se usa en este documento, el término "básico" es un adjetivo que se refiere a una sustancia química que es una base. Un aditivo básico se refiere a un aditivo que es una base.
Como se usa en este documento, el término "alquilo" por sí mismo o como parte de otro sustituyente, significa, a menos que se indique otra cosa, un radical hidrocarburo de cadena lineal o ramificada. Los sustituyentes alquilo, así como otros sustituyentes hidrocarbonados, pueden contener designadores numéricos que indican el número de átomos de carbono en el sustituyente (es decir, C1-8 significa de uno a ocho carbonos), aunque tales designadores pueden omitirse. A menos que se especifique otra cosa, los grupos alquilo de la presente invención contienen de 1 a 12 átomos de carbono. Por ejemplo, un grupo alquilo puede contener 1-2, 1-3, 1-4, 1-5, 1-6, 1-7, 1-8, 1-9, 1-10, 1-11, 1-12, 2-3, 2-4, 2-5, 2-6, 3-4, 3-5, 3-6, 4-5, 4-6 o 5-6 átomos de carbono. Ejemplos de grupos alquilo incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, t-butilo, isobutilo, sec-butilo, n-pentilo, n-hexilo, n-heptilo, n-octilo, y similares. Los términos "alquenilo" y "alquinilo" se refieren a un grupo alquilo como se definió anteriormente, que tienen uno o más enlaces dobles carbono-carbono (alquenilo) o enlaces triples carbono-carbono (alquinilo), respectivamente. Para aquellos grupos que tienen un doble enlace carbono-carbono y un triple enlace carbono-carbono, se utiliza el término "alquinilo".
Los términos "halo" o "halógeno", por sí mismos o como parte de otro sustituyente, significan, a menos que se indique otra cosa, un átomo de flúor, cloro, bromo o yodo. Adicionalmente, los términos tales como "haloalquilo" están destinados a incluir monohaloalquilo y polihaloalquilo. Por ejemplo, el término "haloalquilo C1-4" significa que incluye trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo y similares.
El término "arilo" significa, a menos que se indique otra cosa, un grupo hidrocarburo poliinsaturado, típicamente aromático, que puede ser un anillo simple o anillos múltiples (hasta tres anillos) que se fusionan entre sí o se unen covalentemente. El término "heteroarilo" se refiere a grupos arilo (o anillos) que contienen de uno a cinco heteroátomos seleccionados de N, O y S, en los que los átomos de nitrógeno y azufre están opcionalmente oxidados, y los átomos de nitrógeno está opcionalmente cuaternizados. Un grupo heteroarilo puede estar unido al resto de la molécula a través de un heteroátomo. Ejemplos no limitantes de grupos arilo incluyen fenilo, naftilo y bifenilo, mientras que los ejemplos no limitantes de grupos heteroarilo incluyen piridilo, piridazinilo, pirazinilo, triazinilo, triazinilo, quinolinilo, quinoxalinilo, quinazolinilo, cinnolinilo, ftalazinilo, benzotriazinilo, purinilo, benzimidazolilo, benzopirazolilo, benzotriazolilo, benzisoxazolilo, isobenzofurilo, isoindolilo, indolizinilo, benzotriazinilo, tienopiridinilo, tienopirimidinilo, pirazolopirimidinilo, imidazopiridines, benzotiaxolilo, benzofuranilo, benzotienilo, indolilo, quinolilo, isoquinolilo, isotiazolilo, pirazolilo, indazolilo, pteridinilo, imidazolilo, triazolilo, tetrazolilo, oxazolilo, isoxazolilo, tiadiazolilo, pirrolilo, tiazolilo, furilo, tienilo y similares. Los sustituyentes para cada uno de los sistemas de anillo arilo y heteroarilo indicados anteriormente se seleccionan del grupo de sustituyentes aceptables descritos a continuación.
El término "arilalquilo" pretende incluir aquellos radicales en los que un grupo arilo está unido a un grupo alquilo (por ejemplo, bencilo, fenetilo y similares). De manera similar, el término "ariloxialquilo" pretende incluir aquellos radicales en los que un grupo arilo está unido a un grupo alquilo a través de un enlace éter (-O-), por ejemplo, fenoxipropilo, fenoxipentilo y fenoxiheptilo.
Como se usa en el presente documento, el término "sililoxi" se refiere a una fracción que tiene la fórmula -OSiR3, en la quje cada R es independientemente un grupo alquilo o grupo arilo como se define en el presente documento. Ejemplos de grupos sililoxi incluyen, pero no se limitan a, trimetilsililoxi, trietilsililoxi y (terc-butil)dimetilsililoxi.
Como se usa en el presente documento, el término "aciloxi" se refiere a una fracción que tiene la fórmula -OC(O)R, en la que R es un grupo alquilo, un grupo arilo o un grupo arilalquilo como se define en el presente documento. Ejemplos de grupos aciloxi incluyen, pero no se limitan a, acetiloxi y benzoiloxi.
Como se usa en el presente documento, el término "grupo protector" se refiere a una fracción que se forma para hacer que una fracción funcional no sea reactiva, o menos reactiva. El grupo protector se puede eliminar para restaurar la fracción funcional a su estado original. Diversos grupos protectores y reactivos protectores, incluidos los grupos protectores de hidroxilo, son bien conocidos por los expertos en la técnica e incluyen compuestos que se describen en Protective Groups in Organic Synthesis, 4a edición, TW Greene y PGM Wuts, John Wiley & Sons. Nueva York, 2006.
El término "ligando de dieno" se refiere a un compuesto orgánico que contiene dos dobles enlaces carbono-carbono, que juntos forman un sistema de dieno, que puede coordinarse con las sales de rodio (I). El ligando dieno puede ser, pero no se limita a, norbornadieno, 1,5-ciclooctadieno, sistema biciclo[2.2.2]octa-2,5-dieno, sistema biciclo [2.2.1]hepta-2,5-dieno y sistema biciclo[2.2.0]octa-2,6-dieno.
La presente invención se describirá ahora más específicamente con referencia a las siguientes realizaciones. Debe observarse que las siguientes descripciones de las realizaciones preferidas de esta invención se presentan en el presente documento únicamente con fines de ilustración y descripción; no pretende ser exhaustivo ni limitarse a la forma precisa divulgada. Los siguientes esquemas se proporcionan como realizaciones para ilustrar, pero no para limitar la presente invención.
MI. Realizaciones de la invención
La presente invención proporciona un procedimiento para preparar un compuesto de 4-oxi-ciclopentan-1-ona 2,3-disustituido de fórmula I

El procedimiento incluye poner en contacto un compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II

con un compuesto de fórmula III
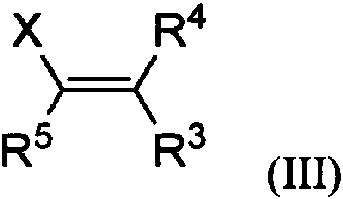
en un disolvente en presencia de un aditivo metálico, opcionalmente en presencia de un aditivo básico, para dar el compuesto de fórmula I;
en la que
R1, R3, R4 y R5 representan independientemente hidrógeno, arilo, heteroarilo, alquilo, arilalquilo, ariloxialquilo, alquenilo o alquinilo;
o R3 y R5 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, que tienen opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S;
o R3 y R4 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, que tiene opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S,
y en la que cada uno de R1, R3, R4 y R5 están opcionalmente sustituidos con uno a tres miembros seleccionados del grupo que consiste en halógeno, hidroxi, alquilo C1-4, haloalquilo C1-4, alcoxi C1-4, sililoxi, ariloxi, aciloxi, anillo heterocíclico, oxo, COOH, CONH2, CONH alquilo C1-4, C(O)OCH2 arilo C6-10, C(O)O arilo C6-10 y C(O)O alquilo C1-4;
R2 representa hidrógeno o un grupo protector de hidroxilo;
X representa un grupo que contiene boro.
Sin desear estar limitado por ninguna teoría particular, se cree que los productos de fórmula I se forman a través de una reacción de adición de 1,4-conjugado. El compuesto III que contiene boro es un compuesto de vinilboro, en el que el átomo de boro está presente en el radical X y se une covalentemente a uno de los átomos de carbono del doble enlace carbono-carbono del compuesto III. El procedimiento de la invención proporciona compuestos de fórmula I que pueden usarse para la preparación de prostaglandinas y análogos de prostaglandinas. Las prostaglandinas y los análogos de prostaglandinas incluyen, pero no se limitan a, compuestos de las series de prostaglandinas E (PGE) y prostaglandinas F (PGF) y sus análogos. Ejemplos de los compuestos de las series PGE y PGF (véase Figura 4) o análogos de estos incluyen, pero no están limitados a, travoprost (un análogo PGF2a),
bimatoprost (un análogo PGF2a), dinoprost (PGF2a; véase también la Figura 5), dinoprostona ( PGE2), lubiprostona (un análogo de PGE), tafluprost (un análogo de PGF2a; véase también la Figura 6), carboprost (un análogo de pGF2a), alprostadil (PGEi), latanoprost (un análogo de PGF2a) y unoprostona (un análogo de PGFa).
En algunas realizaciones, el compuesto de fórmula I tiene la fórmula I'

Las reacciones en el procedimiento de la invención se pueden llevar a cabo a cualquier temperatura adecuada. En algunas realizaciones, la etapa de reacción de adición de 1,4-conjugado se puede realizar a temperatura ambiente (20-30 °C), bajo condiciones de enfriamiento (aproximadamente 0 °C) o bajo condiciones de calentamiento moderado (aproximadamente 50 °C). Ejemplos de instrumentos utilizados para proporcionar calor a la reacción incluyen, pero no se limitan a, equipos de calentamiento convencionales (tales como un baño de aceite, una camisa de calentamiento o una manta de calefacción) o reactores de microondas. Típicamente, no se requieren temperaturas bajo cero o criogénicas cuando se opera este procedimiento. En contraste, los procedimientos conocidos que implementan la metodología convencional de dos componentes usualmente requieren temperaturas criogénicas en la etapa correspondiente de la reacción de adición del 1,4-conjugado.
Las temperaturas de reacción, tales como aproximadamente 30 °C, pueden proporcionar rendimientos más altos de compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituidos que las temperaturas más altas, tales como 50 °C, 60 °C u 80 °C, porque a temperaturas más altas se pueden formar cantidades significativas de compuestos VIII 2,3,4-trisustituido-ciclopentan-1-ona. Por ejemplo, desde aproximadamente 3 % hasta aproximadamente 15 % (basado en4-oxi-ciclopent-2-en-1-onas sustituido en 2 II) de los compuestos VIII de productos secundarios pueden formarse en ciertos casos.

Sin desear estar limitado por ninguna teoría particular, se cree que los compuestos VIII se forman a partir de los precursores reactivos V, que en sí mismos son productos de degradación de los compuestos I, y se forman por eliminación del sustituyente 4-oxi.

En apoyo de esto, cuando el producto de adición de 1,4-conjugado, prostaglandina E2 análogo 10.1, se sometió a las condiciones de adición de 1,4-conjugado estándar de esta invención ([RhCl(COD)]2, KOH, 60 °C en presencia de ácido trans-2-fenilvinilboroico (3.1) durante un tiempo de reacción prolongado de 18 h, se formó el compuesto 6.1 como el producto de reacción principal.

Además de la temperatura de reacción, las cantidades de los compuestos VIII formados pueden depender de otros parámetros de reacción, tales como el tipo de aditivo básico, el disolvente de reacción y el tiempo de reacción (tal como cuando se aplican tiempos de reacción prolongados).
En general, para cada conjunto de sustratos II y III y el solvente de reacción y el aditivo básico, se selecciona una temperatura de reacción que proporciona el mayor rendimiento de compuestos I 4-oxi-ciclopentan-1-ona 2,3-disustituidos y que no proporciona ninguno, o un nivel bajo, de los compuestos VIII 2,3,4-trisustituidos-ciclopentan-1-ona. Como ejemplos no limitantes, la Tabla 1, entradas 1-3 muestran que las temperaturas más bajas pueden proporcionar mayores rendimientos del compuesto I 4-oxi-ciclopentan-1-ona 2,3-disustituido para reacciones conducidas en metanol usando hidróxido de potasio como aditivo básico. Cuando se usó la irradiación de microondas (pw) y la temperatura de reacción se mantuvo a 50 °C, 40 °C o 30 °C, el nivel de 2,3,4-trisustituidociclopentan-1-ona 6.1 se redujo del 9 % al 3 % a 0 % de rendimiento, respectivamente. Mientras tanto, el compuesto de 2,3- disustituido-4,1-oxi-ciclopentan-1-ona deseado 10.1, aumentó de 65 % a 73 % a 83 %, respectivamente.


Se puede usar cualquier disolvente adecuado en la etapa de reacción de adición de 1,4-conjugado utilizando los compuestos de vinilboro. Los ejemplos de disolventes adecuados incluyen, pero no se limitan a, agua, metanol, etanol, alcohol isopropílico, alcohol isobutílico, 1,4-dioxano, tolueno, tetrahidrofurano (THF), 2-metil-tetrahidrofurano (2-Me-THF), diglima, acetonitrilo, N-metilpirrolidona (NMP), N,N-dimetilformamida (Dm F), N,N-dimetilacetamida (DMAC), etilenglicol y combinaciones de los mismos. La etapa de reacción de adición de 1,4-conjugado que utiliza compuestos de vinilboro puede realizarse en solventes próticos o apróticos. En algunas realizaciones, la reacción se lleva a cabo en un disolvente prótico. En algunas realizaciones, el disolvente prótico es un disolvente de alcohol. En algunas realizaciones, el disolvente de alcohol es metanol. El metanol no solo proporciona un buen medio para realizar la reacción de adición de 1,4-conjugado de esta invención, sino que también es barato, está disponible y es fácil de manejar y desechar en las escalas de fabricación.
Cualquier compuesto de boro adecuado puede usarse en el procedimiento de la invención. Los ejemplos de compuestos de vinilboro adecuados incluyen, pero no se limitan a, ácidos vinilboroicos, vinil boroxinas, ésteres vinilboroicos (por ejemplo, ésteres pinacolborónicos y ésteres catecolborónicos), trivinilboranos, dialquilvinilboranos, boronatos de ácido vinílico N-imino diacético, B-vinil-9-BBN compuestos, boratos de viniltriol, sales de trifluorovinilborato y sales de tetravinilborato. Preferiblemente, el compuesto de vinilboro es un ácido vinilboroico o una sal de viniltrifluoroborato. Es decir, el grupo X que contiene boro del compuesto de fórmula III se selecciona del grupo que consiste en B(OH)2 (un ácido vinilboroico); B(OR)2 (un éster vinilboroico) en el que R es un grupo alquilo o un grupo arilo; BR2 (un dialquilvinilborano) en el que R es un grupo alquilo; BR2 (un trivinilborano) en el que R es un grupo vinilo; un grupo 9-borabiciclo [3.3.1]nonano (9-BBN); BR2 en el que R es un grupo carboxilato; BR en el que R es un grupo carboxilato bidentado; BR2 en el que R es un grupo ariloxi; BR en el que R es un grupo ariloxi bidentado, BF3M (una sal de viniltrifluoroborato) en el que M es un ión metálico, BF3M en el que M es un ión amonio o fosfonio, BR3M (una sal de trialquilvinilborato) en el que R es un grupo alquilo y M es un ion metálico o un ion amonio o fosfonio, y BR3M (una sal de tetravinilborato) en el que R es un grupo vinilo y en el que M es un ion metálico o es un ion amonio o fosfonio.
La Tabla 2, entradas 1-5 muestra que el mismo radical 2-fenilvinilo puede administrarse a4-oxi-ciclopent-2-en-1-onas sustituido en 2 2 en la reacción de adición del 1,4-conjugado mediante el ácido vinilboroico 3.1 (entrada 1), viniltrifluoroborato de potasio 3.1a (entrada 2), vinilboronato de catecol 3.1b (entrada 3), vinilboronato de pinacol 3.1c (entrada 4) y vinilboronato de ácido N-metilimino diacético 3.1d (entrada 5), para proporcionar el análogo protegido de de Prostaglandina E210.1.


Los compuestos de vinilboro III mencionados anteriormente son fáciles de preparar utilizando los procedimientos descritos en la literatura y a menudo se pueden aislar como sólidos, que a veces son cristalinos. Además, los compuestos de vinilboro III generalmente son estables al aire y la humedad (es decir, tolerantes al agua) y tienen una larga vida útil, a diferencia de los compuestos de vinilcuprato correspondientes utilizados en la metodología convencional de dos componentes que a menudo se deben usar inmediatamente o poco después de la preparación. Los compuestos de vinilboro III pueden manipularse fácilmente en el laboratorio o en la planta de fabricación, y pueden adquirirse en fuentes comerciales o prepararse internamente y luego almacenarse bajo condiciones ambientales hasta que sea necesario. Además, estos compuestos de vinilboro generalmente se consideran "no tóxicos", lo que es adecuado para aplicaciones industriales y farmacéuticas. El uso de los compuestos de vinilboro III en la presente invención es una mejora significativa con respecto al uso de compuestos de vinilcobre (vinilcupratos) del procedimiento convencional de dos componentes.
Sorprendentemente, la reacción de adición de 1,4-conjugado de esta invención puede funcionar durante un intervalo estructuralmente diverso de compuestos de vinilboro de fórmula III. Como se resume en la Tabla 3, entradas 1-8, se obtuvieron altos rendimientos de 4-oxi-ciclopentan-1-ona 2,3-disustituido 10 (análogos protegidos de prostaglandina E2) utilizando el compuesto de 4-sililoxi-ciclopent-1-ona sustituido en 2 de fórmula 2 y ácidos vinilboroicos 3, en la que R4 y R5 son H y R3 es fenilo (3.1, entrada 1), 4-metoxifenilo (3.2, entrada 2), 3-metoxifenilo (3.12, entrada 3), 4-metilfenilo (3.3, entrada 4), 3-metilfenilo (3.4, entrada 5), 2-metilfenilo (3.11, entrada 6), 4-trifluorometilfenilo (3.14, entrada 7) o 4-fluorofenilo (3.13, entrada 8). Además, cuando R4 es H y R3 y R5 forman un anillo carbocíclico (es decir, 3.20; entrada 9) se obtuvo el correspondiente compuesto de 4-oxi-ciclopentan-1-ona 2,3-disustituido de fórmula 10.20. Adicionalmente, los ácidos vinilboroicos no aromáticos 3.6 (entrada 10), 3.5 (entrada 11), 3.16 (entrada 12) y 3.15 (entrada 13) generalmente proporcionaron rendimientos de buenos a altos de los correspondientes compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituidos de fórmula 10.6, 10.5, 10.16 y 10.15 (análogos de la prostaglandina E2).



La aplicabilidad del procedimiento de la invención a la síntesis de precursores de prostaglandinas y análogos de prostaglandinas de importancia farmacéutica se demuestra mediante los ejemplos no limitantes resumidos en la Tabla 4. La reacción de adición de 1,4-conjugado del compueto 4-oxiciclopent-2-en-1-ona sustituido en 2 de fórmula 2 y ácidos vinilboroicos 3.9a (entrada 1), 3.17 (entrada 3), 3.18 (entrada 4) o 3.7 (entrada 5) proporcionaron compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituidos de fórmula 10.9a, 10.17, 10.18 y 10.7 (análogos de prostaglandina E2), respectivamente. Tales compuestos son conocidos e informados como precursores sintéticos de dinoprost, bimatoprost, travoprost y tafluprost, respectivamente, que son en sí mismos análogos de la prostaglandina F2a que se utilizan para inducir el parto o en el tratamiento de la enfermedad. Véase, por ejemplo, la patente '795 para el uso de amálogos protegidos de prostaglandina E2 10.17 y 10.18 para la síntesis de los análogos de prostaglandinas F2a bimatoprost y travoprost, respectivamente. De manera similar, el vinil pinacolboronato 2.9a (entrada 2), el trifluoroborato de vinil potasio 3.7a (entrada 6) o el pinacolboronato de vinilo 2.19 (entrada 7) se pueden usar en la reacción de adición del 1,4-conjugado para proporcionar compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituidos de fórmula 10.9a, 10.7 y 10.19, que son conocidos y reportados como precursores sintéticos de los análogos de la prostaglandina F2a dinoprost y tafluprost, y el derivado de la prostaglandina E1 lubiprostona, respectivamente. Véase, por ejemplo, la patente '958 para el uso del análogo protegido de prostaglandina E210.19 para la síntesis de lubiprostona.



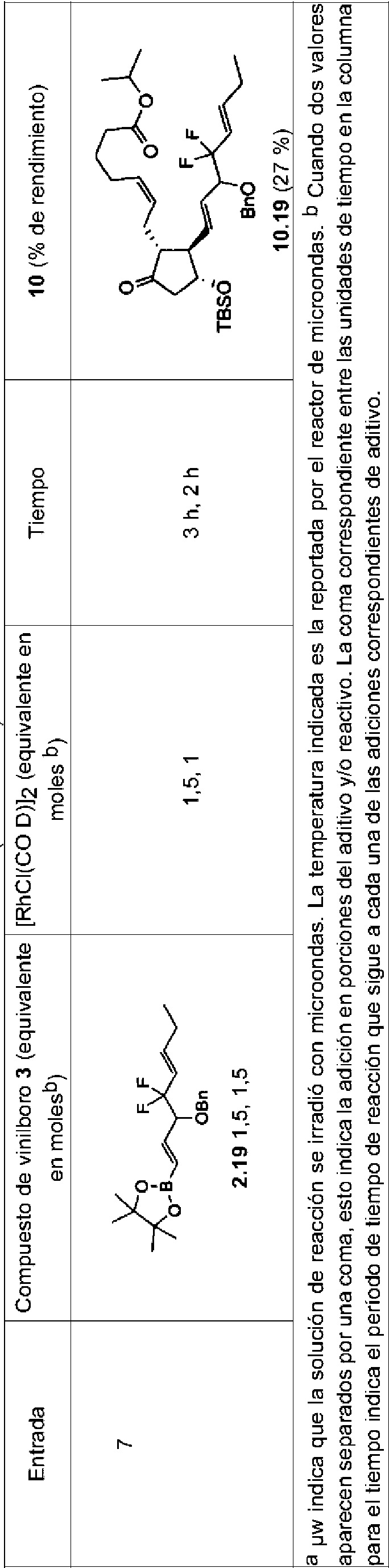
Es notable que, mientras que fallaron múltiples intentos para preparar el compuesto de 4-oxiciclopentan-1-ona 2,3-disustituido de fórmula 10.7, útil para preparar tafluprost, utilizando la metodología convencional de dos componentes utilizando el correspondiente vinilcuprato VII y el compuesto de 4-sililoxi-ciclopent-2-en-1-ona sustituido en 2 compuesto 2 (véase Tabla 5), tanto el ácido vinilboroico 3.7 (Tabla 4, entrada 5) como el trifluoroborato de vinil potasio 3.7a (entrada 6) podrían usarse para producir el compuesto de 4-oxi-ciclopentan-1-ona 2,3-disustituido de fórmula 10.7, útil en la síntesis de tafluprost como se describe en la Figura 6, con buen rendimiento. Los compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituidos de fórmula 10.9a y 10.7 se convirtieron adicionalmente (como se describe en la sección de ejemplos en este documento) en los análogos de la prostaglandina F2a dinoprost (véase, Figura 5) o tafluprost (véase, Figura 6), respectivamente, que demuestran la aplicabilidad y utilidad de la presente invención a la preparación de precursores de fármacos de prostaglandina.

La reacción de los compuestos de vinilboro III con los compuestos de 4-oxiciclopent-2-en-1-ona sustituidos en 2 de fórmula II en la etapa de reacción de adición de 1,4-conjugado solo requiere cantidades catalíticas (es decir, subestequiométricas) del aditivo metálico (por ejemplo, un compuesto de rodio, un compuesto de cobalto, un compuesto de níquel o combinaciones de los mismos). Por ejemplo, típicamente 3 por ciento en moles del dímero de rodio, [RhCl(1,5-ciclooctadieno)]2, con respecto a la cantidad en moles de compuestos de 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 de la fórmula II es suficiente para dar como resultado la conversión de alto rendimiento de los compuestos de 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 de fórmula II y los compuestos de vinilboro III a compuestos de 4-oxiciclopentan-1-ona 2,3-disustituido de fórmula I. Por el contrario, la química organocobre convencional de la metodología de dos componentes requiere cantidades estequiométricas del metal de transición, Cu(I) y, en algunos casos, Zr(IV) y los metales Li(I), y comúnmente Sn(IV), todo en cantidades estequiométricas. En la invención aquí descrita, el boro convenientemente ocupa el lugar de los metales Cu(I), Sn(IV) o Zr(IV) y/o Li(I) de la metodología convencional de dos componentes y, por lo tanto, reduce la cantidad de metales de transición y otros metales necesarios para hacer compuestos precursores de prostaglandinas de fórmula I.
En algunas realizaciones de la presente invención, el aditivo de compuesto de rodio usado en la etapa de reacción de adición de 1,4-conjugado de los compuestos de vinilboro III al compuesto de fórmula II es un precatalizador o catalizador que se usa en cantidades subestequiométricas e incluye, pero no se limita a compuestos de rodio (I) seleccionados del grupo [RhCl(1,5-ciclooctadieno)]2 (es decir, dímero de cloro(1,5-ciclooctadieno)rodio (I));
[RhCl(C2H4)2]2i [RhCl(C2H4)2]2 con un aditivo de ligando dieno; [RhCl(norbornadieno)]2; y [Rh(OH)(1,5-ciclooctadieno)]2 (es decir, dímero de hidroxi(1,5-ciclooctadieno)rodio (I)). El 1,5-ciclooctadieno a menudo se abrevia como COD, y por lo tanto [RhCl(1,5-ciclooctadieno)]2 se abrevia como [RhCl(COD)]2. En ciertas realizaciones, el aditivo de rodio es el dímero de rodio [RhCl(1,5-ciclooctadieno)]2 o el dímero de rodio [Rh(OH)(1,5-ciclooctadieno)]2.
La cantidad del aditivo metálico es preferiblemente típicamente menor que 50 por ciento en moles con respecto a la cantidad en moles del compuesto de fórmula II, tal como menor o igual a 12 por ciento en moles con respecto a la cantidad en moles del compuesto de fórmula II. o menos de 10 por ciento en moles con respecto a la cantidad en moles del compuesto de fórmula II, o menor o igual a 6 por ciento en moles con respecto a la cantidad en moles del compuesto de fórmula II, o menor o igual a 5 por ciento en moles con respecto a la cantidad molar del compuesto de fórmula II.
Cualquier aditivo básico adecuado puede usarse en el procedimiento de la invención. Ejemplos de aditivos básicos adecuados utilizados en la etapa de reacción de adición del 1,4-conjugado incluyen, pero no se limitan a, KHF2, t-BuOLi, t-BuONa, t-BuOK, K3PO4, K2CO3 , Cs2CO3, LiOH, NaOH, KOH, CsOH, KF, CsF, NaHCO3, KH2PO4, 1,3-diaminopropano, t-BuNH2, /-Pr2NH, piperidina, Et3N, 2,6-lutidina y combinaciones de los mismos. En ciertas realizaciones, el aditivo básico es un hidróxido. En realizaciones particulares, el hidróxido es hidróxido de potasio. Sin pretender estar limitado por la teoría, se cree que el aditivo básico actúa sobre el aditivo metálico para convertirlo de un precatalizador en un catalizador activo, aunque el aditivo básico también puede desempeñar otras funciones en la reacción. Por ejemplo, se ha informado que las sales de cloruro de rodio (I) son convertidas por compuesto de hidróxido en hidróxidos de rodio (I) en hidróxidos, y que estos últimos compuestos son catalíticamente activos y pueden catalizar las reacciones de adición de compuestos de vinilboro a compuestos olefínicos activados, tales como compuestos carbonílicos a,p-insaturados (J. Am. Chem. Soc. 2002, 124, 5052-5058). La cantidad del aditivo básico es preferiblemente equimolar con respecto al aditivo metálico, o mayor que equimolar con respecto al aditivo metálico.
Diversas combinaciones de diferentes solventes, aditivos básicos y temperaturas de reacción pueden dar como resultado rendimientos significativamente diferentes de compuestos de 4-oxiclopentan-1-ona 2,3-disustituidos de fórmula I y niveles de los productos de degradación 2,3,4-trisustituido-ciclopentan-1 ona de fórmula VIII. Una persona experta en la técnica entenderá que para cada compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II y compuesto de vinilboro III, la optimización de la cantidad relativa y el tipo de aditivo básico, la cantidad relativa y el tipo de aditivo metálico, la cantidad relativa y el tipo de solvente y la temperatura de reacción podrían requerirse para identificar las condiciones que proporcionan los mejores rendimientos de los compuestos de 4-oxiciclopentan-1-ona 2,3-disustituidos de fórmula I.
En algunas realizaciones, el rendimiento de la reacción de los compuestos de 4-oxi-ciclopentan-1-ona 2,3-disustituidos de fórmula I se puede aumentar mediante la adición en porciones de los compuestos de vinilboro de fórmula III y/o la adición en porciones del aditivo metálico. Por ejemplo, la Tabla 1, entradas 4-7 muestra que diferentes permutaciones de la cantidad total y el número y las cantidades de cada adición en porciones de ácido borónico 3.1 y [RhCl(1,5-ciclooctadieno)]2 dieron como resultado diferentes rendimientos del compuesto de 4-oxiciclopentan-1-ona 2,3-disustituido 10.1.
En algunas realizaciones de la presente invención, el R2 del compuesto de fórmula I y el compuesto de fórmula II es un grupo protector de hidroxilo. Ejemplos de grupos protectores de hidroxilo incluyen, pero no se limitan a, tetrahidropiranilo (THP), metoximetilo (MOM), [2-(trimetilsilil)etoxi]metilo (SEM), trialquilsililo, triarilsililo, diarilalquilsililo, bencilo, p-metoxibencilo (PMB), alquilcarbonilo, arilcarbonilo y alilo. En ciertas realizaciones, el grupo
protector de hidroxilo es un grupo trialquilsililo. En algunas realizaciones, el trialquilsililo es terc-butildimetilsililo (TBS).
En algunas realizaciones de la presente invención, el compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II es IIa o IIb en donde R2 es hidrógeno o un grupo protector de hidroxilo definido como anteriormente y R6 es un radical seleccionado del grupo que incluye H, metilo, etilo, propilo, isopropilo, butilo, isobutilo, terc-butilo, trimetilsililo, trietilsililo, triisopropilsililo, terc-butildimetilsililo y bencilo.

En realizaciones adicionales de la presente invención, el compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II es IIaa en donde R2 es hidrógeno y R6 es isopropilo (véase Tabla 6, entrada 4). En otras realizaciones de la presente invención (véase Tabla 6), el compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II se selecciona del grupo IIba donde R2 es terc-butildimetilsililo (TBS) y R6 es metilo (entrada 1), IIbb donde R2 es tetrahidropiranilo (THP) y R6 es metilo (entrada 2), y IIbc donde R2 es alilo y R6 es metilo (entrada 3).


En algunas realizaciones de la presente invención, los radicales R3, R4 y/o R5 del compuesto de fórmula I y el compuesto de fórmula III poseen uno o más grupos hidroxilo. En algunas realizaciones, los grupos hidroxilo mencionados anteriormente pueden estar protegidos. El grupo protector de hidroxilo adecuado incluye, pero no se limita a, tetrahidropiranilo (THP), metoximetilo (MOM), [2-(trimetilsilil)etoxi]metilo (SEM), trialquilsililo, triarilsililo, diarilalquilsililo, bencilo, 4-metoxibencilo (PMB) , alquilcarbonilo, arilcarbonilo y alilo. En ciertas realizaciones, el grupo protector es un grupo trialquilsililo. En algunas realizaciones, el trialquilsililo es terc-butildimetilsililo (TBS). La Tabla 7, entradas 1-8, muestra cuatro derivados diferentes del ácido vinilboroico protegido con hidroxilo 3.8a (tercbutildimetilsililo protegido; entrada 1), 3.8c (bencilo protegido; entrada 2), 3.8e (tetrahidropiranilo protegido; entrada 3), y 3.8b (acetilo protegido; entrada 4), y cuatro derivados diferentes de pinacol vinilboronato protegidos con hidroxilo 2.8a (terc-butildimetilsililo protegido; entrada 8), 2.8c (bencilo protegido; entrada 7), 2.8e (tetrahidropiranilo protegido; entrada 5) y 2.8b (acetilo protegido; entrada 6). En algunas realizaciones, el grupo hidroxilo mencionado anteriormente puede permanecer desprotegido, como en el ejemplo de la Tabla 7, entrada 9 (como en el ácido vinilboroico 2.8).



Los compuestos de vinilboro de fórmula III que son útiles en esta invención se pueden preparar por procedimientos descritos en la literatura (J. Am. Chem. Soc. 2005, 127, 5766-5767; Angew. Chem. 2003, 42, 3399-3404; J. Org. Chem. 2001, 66, 5359-5365; Angew. Chem. 2012, 51, 9385-9388; Org. Lett. 2012, 14, 6104-6107; Org. Lett. 2010, 12, 2004-2007).
Por ejemplo, cuando R4 es H, un procedimiento comprende
a) poner en contacto el compuesto alquino de fórmula IV

con un reactivo de organoboro; y
b) convertir el producto de la etapa a) para obtener III. Alternativamente, los compuestos de vinilboro de fórmula III pueden prepararse usando haluros de vinilo como materiales de partida, y los procedimientos para esta transformación son conocidos en la técnica.
El reactivo de organoborón mencionado anteriormente es un compuesto hidroborano y se selecciona del grupo que consiste en un complejo de éter de borano, tal como BH3THF o BH3OEt2; un complejo de sulfuro de borano, tal como BH3 SMe2; 9-borabiciclo[3.3.1]nonano; dímero de 9-borabiciclo [3.3.1]nonano; pinacolborano; catecolborano; un dialquilborano tal como diciclohexilborano, disiamilborano, diisopinocanfoilborano o di(isopropilprenil)borano; y complejos de bases de Lewis de mono- y dicloroborano o mono- y dibromoborano tales como BHCl2 dioxano, BH2ClOEt2, BH2ClSMe2, BHBr2 dioxano, BH2BrOEt2 o BH2BrSMe2. Los compuestos de vinilboro de fórmula III que son útiles en esta invención incluyen ácidos vinilboroicos, ésteres vinilboroicos, boronatos de ácido vinil N-imino diacético, vinil triorganoboranos (por ejemplo, vinil trialquilboranos), compuestos de B-órgano-9-BBN de vinilo (por ejemplo, compuestos de B-alquil-9-BBN de vinilo), sales de viniltrifluoroborato y sales de tetravinilborato. En ciertas realizaciones, el compuesto de vinilboro es un ácido vinilboroico o una sal de viniltrifluoroborato.
Los compuestos de 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 de fórmula II útiles en el procedimiento de la invención pueden prepararse usando procedimientos conocidos en la técnica, incluidos los descritos en la Patente U.S. N° 7.897.795, Org. Process Res. Dev. 2012, 16, 1905-1916 y Tetrahedron Lett. 2003, 44, 7411-7415.
Si bien se proporciona una guía en los ejemplos a continuación, un experto en la técnica apreciará que el procedimiento descrito en este documento funcionará bajo una variedad de condiciones relacionadas con la temperatura y el solvente o solventes utilizados (discutidos anteriormente), así como el tiempo de reacción, y la concentración de reactivos en la mezcla de reacción. Los siguientes ejemplos son ilustrativos de ciertas realizaciones de la invención y no deben considerarse como limitantes de ninguna manera.
IV. Ejemplos
Ejemplo 1 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butN-dimetil-sNamloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)

Una solución de (Z)-7-[(3R)-3-(fe/f-butil-dimetil-silaniloxi)-5-oxociclopent-1-enil]-hept-5-enoato de isopropilo (2) (57 mg, 0,15 mmol) ), ácido trans-2-fenilvinilboroico (3.1) (33 mg, 0,22 mmol), [RhCl(1,5-ciclooctadieno)]2 (1,1 mg, 2,2 pmol) y KOH acuoso (9,5 pl, KOH acuoso 3,0 M, 29 pmmol) en MeOH (1,0 ml) se agitó bajo irradiación de microondas (CEM, Discover S; Milestone, Startsynth; la temperatura se fijó a 30 °C). Después de 5 horas, se agregaron ácido trans-2-fenilvinilboroico (3.1) (11 mg, 74 pmol) y [RhCl(1,5-ciclooctadieno)]2 (1,1 mg, 2,4 pmol) y la mezcla de reacción se agitó durante otra hora. y se concentró directamente al vacío. El residuo se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando 10.1 (70 mg, 96 %).
[a]27D -57,4 (c 1,00, CHCl3); 1H RMN (400 MHz, CDCl3): 5 0,01 (s, 3H), 0,03 (s, 3H), 0,86 (s, 9H), 1,20 (d, J= 6,2 Hz, 6H), 1,59-1,69 (m, 2H), 2,05 (dd, J= 7,1, 14,1 Hz, 2H), 2,16-2,26 (m, 4H), 2,38-2,42 (m, 2H), 2,58-2,73 (m, 2H), 4,13 (dd, J= 8,4, 16,0 Hz, 1H), 4,97 (septeto, J= 6,2 Hz, 1H), 5,32-5,46 (m, 2H), 6,05 (dd, J= 8,6, 15,8 Hz, 1H), 6,50 (d,
J=15,8 Hz, 1H), 7,22-7,26 (m, 1H), 7,27-7,37 (m, 4H); 13C RMN (125 MHz, CDCla): 5 -4,8 (CH3), -4,7 (CH3), 18,1 (C), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,7 (CH), 26,7 (CH2), 34,1 (CH2), 47,6 (CH2), 54,1 (CH), 54,4 (CH), 67,4 (CH), 73,0 (CH), 126,1 (CH), 126,5 (CH), 127,4 (CH), 128,6 (CH), 129,8 (CH), 131,0 (CH), 132,9 (CH), 137,1 (C), 173,1 (C), 214,7 (C); HRMS (ESI-QTOF) calculado para [C29H44O4SÍ H]+= 485,3081, encontrado 485,3064; FTIR (KBr, puro) 2935, 2861, 1736, 1593, 1459, 1372, 1250, 1107, 838, 745 cm-1.
Ejemplo 2 - Síntesis de (Z)-7-[(1R,2R)-5-oxo-2,3-diestiril-ciclopent-3-enil]-hept-5-enoato de isopropilo (6.1)

El estándar de producto de degradación 6.1 se puede preparar utilizando el siguiente procedimiento: Una solución de 10.1 (16 mg, 33 pmol), ácido trans-2-fenilvinilboroico (3.1) (9,2 mg, 62 pmol), [RhCl(1,5-ciclooctadieno )]2 (1,1 mg, 2,25 pmol, 5,6 % en moles) y KOH acuoso (9,5 pl, KOH acuoso 3,0 M, 28,5 pmol) en MeOH (1 ml) se agitó a 60 °C. Después de 12 horas, la mezcla se concentró directamente al vacío. El residuo se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título 6,1 (13 mg, 86 %).
[a]24D -149,5 (c 0,46, CHCh)); 1H RMN (400 MHz, CDCh): 5 1,20 (d, J= 6,4, 6H), 1,58-1,72 (m, 2H), 1,98-2,12 (m, 2H), 2,13-2,34 (m, 4H), 2,36-2,58 (m, 3H), 2,64-2,85 (m, 2H), 4,97 (septeto, J= 6,4 Hz, 1H), 5,31-5,50 (m, 2H), 6,06 6,22 (m, 2H), 6,43 (dd, J= 11,6, 16,0 Hz, 2H), 7,17-7,36 (m, 10H); 13C RMN (100 MHz, CDCh): 5 21,8 (CH3), 24,8 (CH2), 25,0 (CH2), 26,7 (CH2), 30,9 (CH2), 34,1 (CH2), 44,4 (CH), 51,7 (CH), 55,5 (CH), 67,4 (CH), 77,2 (CH), 126,18 (CH), 126,22 (CH), 126,5 (CH), 127,4 (CH), 127,5 (CH), 128,5 (CH), 128,6 (CH), 130,3 (CH), 131,0 (CH), 131,2 (CH), 132,5 (CH), 137,0 (C), 173,1 (C), 216,7 (C); HRMS (ESI-TOF) calculado para [C31H36O3 H]+= 457,2737, encontrado 457,2738; FTIR (KBr, puro) 3446, 2975, 1730, 1640, 1443, 1375, 1217, 1164, 1102, 963 cm-1
Ejemplo 3 - Síntesis de (Z)-7-{(2R,3R),-3-(ferí-butil-dimetil-silamloxi)-2-[2-(4-metoxi-feml)-viml]-5-oxociclopentil}-hept-5-enoato de isopropilo (10.2)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando ácido trans-2-(4-metoxi-fenil)-vinilboroico (3.2). La mezcla de producto crduo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) produciendo (Z}-7-{(2R,3R),-3-(fe/f-butil-dimetil-silaniloxi)-2-[2-(4-metoxifenil)-vinil]-5-oxo-ciclopentil}-hept-5-enoato de isopropilo (10.2, 72 mg, 93 %).
[a]22D -50,6 (c 1,00, CHCh); 1H RMN (400 MHz, CDCh): 5 0,00 (s, 3H), 0,02 (s, 3H), 0,85 (s, 9H), 1,20 (d, J= 6,3 Hz, 6H), 1,60-1,68 (m, 2H), 2,05 (dd, J= 7,0, 14,1 Hz, 2H), 2,14-2,25 (m, 4H), 2,36-2,39 (m, 2H), 2,55-2,71 (m, 2H), 3,81 (s, 3H), 4,09 (dd, J= 8,5, 15,8 Hz, 1H), 4,97 (septeto, J= 6,3 Hz, 1H), 5,32-5,45 (m, 2H), 5,89 (dd, J= 8,4, 15,6 Hz, 1H), 6,44 (d, J= 15,6 Hz, 1H), 6,85 (d, J= 8,6 Hz, 2H), 7,28 (d, J= 8,6 Hz, 2H); 13C RMN (100 MHz, CDCh): 5 -4,8 (CH3), -4,6 (CH3), 18,0 (C), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,7 (CH3), 26,6 (CH2), 34,1 (CH2), 47,6 (CH2), 54,1 (CH), 54,4 (CH), 55,2 (CH3), 67,4 (CH), 73,1 (CH), 114,0 (CH), 126,5 (CH), 127,2 (CH), 127,5 (CH), 129,9 (C), 130,9 (CH), 132,3 (CH), 159,0 (C), 173,1 (C), 214,9 (C); HRMS (ESI-QTOF) calculado para ^ c ^ O a S i H]+= 515,3187, encontrado 515,3182; FTIR (KBr, puro) 2939, 2863, 1737, 1604, 1512, 1463, 1371, 1246, 1166, 1109 cm-1.
Ejemplo 4 Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silamloxi)-5-oxo-2-(2-p-toluil-viml)-ciclopentil]-hept-5-enoato de isopropilo (10.3)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando ácido trans-2-ptoluil-vinilboroico (3.3). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando (Z)-7-[(2R,3R)-3-(fe/f-butil-dimetil-silaniloxi)-5-oxo-2-(2-p-toluil-vinil)-ciclopentil]-hept-5-enoato de isopropilo (10.3, 71 mg, 95 %).
[a]23D-40,2 (c 1,00, CHCla); 1H RMN (400 MHz, CDCls): 5 0,00 (s, 3H), 0,02 (s, 3H), 0,85 (s, 9H), 1,20 (d, J = 6,2 Hz, 6H), 1,64 (quintet, J = 7,6 Hz, 2H), 2,04 (dd, J = 7,3, 14,2 Hz, 2H), 2,13-2,26 (m, 4H), 2,34 (s, 3H), 2,38-2,40 (m, 2H), 2,57-2,72 (m, 2H), 4,11 (dd, J = 8,0, 15,6 Hz, 1H), 4,97 (septeto, J = 6,2 Hz, 1H), 5,31-5,45 (m, 2H), 5,98 (dd, J = 8,7, 15,7 Hz, 1H), 6,46 (d, J = 15,7 Hz, 1H), 7,13 (d, J = 8,0 Hz, 2H), 7,25 (d, J = 8,0 Hz, 2H); 13C RMN (100 MHz, CDCls): 5 -4,8 (CH3), -4,7 (CH3), 18,0 (C), 21,1 (CH3), 21,8 (CH3), 24,8 (CH2), 25,0 (CH2), 25,7 (CH3), 26,6 (CH2), 34,1 (CH2), 47,6 (CH2), 54,1 (CH), 54,4 (CH), 67,3 (CH), 73,0 (CH), 126,0 (CH), 126,5 (CH), 128,7 (CH), 129,3 (C), 130,9 (CH), 132,7 (CH), 134,3 (C), 137,1 (C), 173,1 (C), 214,8 (C); HRMS (ESI-QTOF) calculado para [C30H46O4S¡ H]+= 499,3238, encontrado 499,3244; FTIR (KBr, puro) 2938, 2863, 1737, 1509, 1463, 1372, 1250, 1110, 964, 841 cm-1.
Ejemplo 5 Síntesis de (Z)-7-[(2R,3R)-3-(fert-butil-dimetil-silamloxi)-5-oxo-2-(2-m-toluil-viml)-ciclopentil]-hept-5-enoato de isopropilo (10.4)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando ácido trans-2-mtoluil-vinilboroico (3.4). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando (Z)-7-[(2R,3R)-3-(fe/f-butil-dimetil-silaniloxi)-5-oxo-2-(2-m-toluil-vinil)-ciclopentil]-hept-5-enoato de isopropilo (10.4, 73 mg, 98 %).
[a]23D-51,1 (c 1,00, CHCh); 1H RMN (400 MHz, CDCh): 5 0,01 (s, 3H), 0,03 (s, 3H), 0,86 (s, 9H), 1,20 (d, J = 6,4 Hz, 6H), 1,65 (quintet, J = 7,6 Hz, 2H), 2,05 (dd, J = 7,0, 14,2 Hz, 2H), 2,15-2,26 (m, 4H), 2,35 (s, 3H), 2,36-2,41 (m, 2H), 2,58-2,73 (m, 2H), 4,12 (dd, J = 8,4, 16,0 Hz, 1H), 4,97 (septeto, J = 6,4 Hz, 1H), 5,32-5,46 (m, 2H), 6,03 (dd, J = 8,6, 15,8 Hz, 1H), 6,47 (d, J = 15,8, 1H), 7,06 (d, J = 7,3 Hz, 1H), 7,15-7,23 (m, 3H); 13C RMN (100 MHz, CDCfe): 5 -4,8 (CH3), -4,6 (CH3), 18,0 (C), 21,4 (CH3), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,7 (CH3), 26,7 (CH2), 34,1 (CH2), 47,6 (CH2), 54,1 (CH), 54,3 (CH), 67,4 (CH), 73,1 (CH), 123,2 (CH), 126,5 (CH), 126,9 (CH), 128,2 (CH), 128,5 (CH), 129,5 (CH), 131,0 (CH), 133,0 (CH), 137,0 (C), 138,1 (C), 173,1 (C), 214,8 (C); HRMS (ESI-QTOF) calculado para [C30H46O4S¡ H]+= 499,3238, encontrado 499,3242; FTIR (KBr, puro) 2939, 2863, 1737, 1600, 1463, 1375, 1253, 1112, 971,884 cm-1.
Ejemplo 6 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silamloxi)-2-(2-metil-prop-1-eml)-5-oxo-ciclopentil]-hept-5-enoato de isopropilo (10.5)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando ácido 2-metilpropenilborónico (3.5). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando (Z)-7-[(2R,3R)-3-(fe/f-butil-dimetil-silaniloxi)-2-(2-metil-prop-1-enil)-5-oxo-ciclopentil]-hept-5-enoato de isopropilo (10.5, 65 mg, 99 %).
[a]23D -68,2 (c 1,00, CHCla); 1H RMN (400 MHz, CDCls): 5 0,00 (s, 3H), 0,01 (s, 3H), 0,86 (s, 9H), 1,22 (d, J = 6,4 Hz, 6H), 1,63-1,68 (m, 5H), 1,74 (s, 3H), 1,92-1,99 (m, 1H), 2,01-2,07 (m, 2H), 2,13-2,21 (m, 1H), 2,22-2,27 (m, 2H), 2,27-2,33 (m, 2H), 2,58-2,65 (m, 1H), 2,67-2,76 (m, 1H), 3,95 (dd, J = 8,0, 15,6 Hz, 1H), 4,92 (d, J = 9,6 Hz, 1H), 5,00 (septeto, J = 6,4 Hz, 1H), 5,30-5,43 (m, 2H); 13C RMN (100 MHz, CDCls): 5 -5,0 (CH3), 18,0 (C), 18,7 (CH3), 21,8 (CH3), 24,8 (CH2), 25,0 (CH2), 25,6 (CH3), 25,8 (CH3), 26,5 (CH2), 34,1 (CH2), 47,6 (CH2), 49,4 (CH), 55,2 (CH), 67,4 (CH), 73,6 (CH), 125,4 (CH), 127,0 (CH), 130,4 (CH), 135,4 (C), 173,1 (C), 215,6 (C); HRMS (ESI-QTOF) calculado para [C2aH44O4S¡ H]+= 437,3082, encontrado 437,3096; FTIR (KBr, puro) 2937, 2855, 1738, 1463, 1373, 1248, 1150, 1108, 880, 834 cm-1.
Ejemplo 7 - Síntesis de (Z)-7-[(2ft,3ft)-3-(ferí-butil-dimetil-silamloxi)-2-(trans-oct-1-eml)-5-oxo-ciclopentil]-hept-5-enoato de isopropilo (10.6)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando ácido trans-oct-1-enilborónico (3.6). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando (Z)-7-[(2R,3R)-3-(fe/f-butil-dimetil-silaniloxi)-2-(frans-oct-1-enil)-5-oxociclopentil]-hept-5-enoato de isopropilo (10.6, 69 mg, 93 %).
[a]23D-54,5 (c 1,00, CHCh); 1H RMN (400 MHz, CDCh): 5 0,02 (s, 3H), 0,03 (s, 3H), 0,86 (s, 9H), 1,21 (d, J = 6,4 Hz, 6H), 1,26-1,36 (m, 10H), 1,62-1,69 (m, 3H), 1,97-2,07 (m, 5H), 2,09-2,16 (m, 1H), 2,24 (t, J= 7,6 Hz, 2H), 2,26-2,43 (m, 3H), 2,61 (ddd, J = 1,1, 7,14, 18,3 Hz, 1H), 3,98 (dd, J = 8,0, 15,6 Hz, 1H), 4,99 (septeto, J = 6,4 Hz, 1H), 5,26 (dd, J= 8,4, 15,2 Hz, 1H), 5,30-5,42 (m, 2H), 5,50-5,58 (m, 1H); 13C RMN (100 MHz, CDCh): 5 -4,7 (CH3), 14,1 (CH3), 18,1 (C), 21,8 (CH3), 22,6 (CH2), 24,8 (CH2), 24,9 (CH2), 25,7 (CH3), 26,6 (CH2), 28,9 (CH2), 29,3 (CH2), 31,7 (CH2), 32,6 (CH2), 34,1 (CH2), 47,6 (CH2), 53,5 (CH), 54,2 (CH), 67,3 (CH), 73,1 (CH), 126,8 (CH), 129,6 (CH), 130,6 (CH), 134,0 (CH), 173,1 (C), 215,3 (C); HRMS (ESI-QTOF) calculado para [C2gH52O4S¡ H]+= 493,3708, encontrado 493,3702; FTIR (KBr, puro) 2930, 2860, 1739, 1462, 1371, 1250, 1111,968, 839, 775 cm-1.
Ejemplo 8 - Síntesis de (Z)-7-[(2ft,3ft)-3-(ferí-butil-dimetil-silamloxi)-2-(frans-3,3-difluoro4-fenoxi-but-1-eml)-5-oxo-ciclopentil]-hept-5-enoato de isopropilo (10.7)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando trans-3,3-difluoro-4-fenoxi-but-1-eniltrifluoroborato de potasio (3.7a). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando (Z)-7-[(2R,3R)-3-(fe/f-butil-dimetil-silaniloxi)-2-(frans-3,3-d¡fluoro-4-fenoxi-but-1-en¡l)-5-oxo-c¡clopent¡l]-hept-5-enoato de isopropilo (10.7, 52 mg, 61 %).
[a]24D -45,0 (c 1,00, CHCh); 1H RMN (600 MHz, CDCh): 5 0,03 (s, 3H), 0,04 (s, 3H), 0,86 (s, 9H), 1,21 (d, J = 6,3 Hz, 6H), 1,58-1,63 (m, 2H), 2,01 (dd, J = 7,2, 14,4 Hz, 2H), 2,11-2,22 (m, 4H), 2,35-2,38 (m, 2H), 2,57 (dd, J = 8,7, 20,2 Hz, 1H), 2,66 (dd, J = 1,0, 7,2 Hz, 1H), 4,09 (dd, J = 8,4, 16,2 Hz, 1H), 4,16-4,21 (m, 2H), 4,98 (septeto, J = 6,3 Hz, 1H), 5,28-5,32 (m, 1H), 5,37-5,41 (m, 1H), 5,85-5,91 (m, 1H), 6,11 (m, 1H), 6,91 (d, J = 8,0 Hz, 2H), 7,00 (t, J = 7,4 Hz, 1H), 7,30 (dd, J = 7,4, 8,0 Hz, 2H); 13C RMN (150 MHz, CDCh): 5 -4,8 (CH3), -4,7 (CH3), 18,0 (C), 21,8 (CH3),
24,7 (CH2), 25,0 (CH2), 25,7 (CH3), 26,6 (CH2), 34,0 (CH2), 47,4 (CH2), 53,0 (CH), 53,8 (CH), 67,4 (CH), 69,4 (t, J = 35,0 Hz, CH2), 72,3 (CH), 114,7 (CH), 117,9 (t, J = 238,5 Hz, C), 121,8 (CH), 125,5 (t, J = 24,8 Hz, CH), 126,0 (CH), 129,6 (CH), 131,4 (CH), 136,9 (t, J = 9,0 Hz, CH), 157,9 (C), 173,0 (C), 213,6 (C); HRMS (ESI-QTOF): calculado para [Cs1H46F2O5Si NH4]+= 586,3421, encontrado 582,3406; FTIR (KBr, puro) 2939, 2863, 1737, 1592, 1490, 1463, 1375, 1307, 1250, 1158 cm-1.
Ejemplo 9 - Síntesis de 7-((1R,2R,3R)-3-((fert-butildimetilsilil)oxi)-2-((E)-2-metilestiril)-5-oxociclopentil)hept-5-enoato de (Z)-isopropilo (10.11)
Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido trans-2-o-toluil-vinilboroico (3.11), la mezcla cruda de reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título 10.11 (66 mg, 88 %).
[a]25D -53,4 (c 1,00, CHCla); 1H RMN (400 MHz): 50,03 (s, 3H), 0,04 (s, 3H), 0,87 (s, 9H), 1,20 (d, J = 6,2 Hz, 6H), 1,65 (quintet, J = 7,6 Hz, 2H), 2,07 (dd, J= 6,4, 14,0 Hz, 2H), 2,13-2,30 (m, 4H), 2,34 (s, 3H), 2,35-2,45 (m, 2H), 2,60 2,72 (m, 2H), 4,15 (dd, J = 8,6, 15,8 Hz, 1H), 4,97 (septeto, J = 6,2 Hz, 1H), 5,32-5,47 (m, 2H), 5,93 (dd, J = 8,6, 15,6 Hz, 1H), 6,72 (d, J = 15,6 Hz, 1H), 7,12-7,20 (m, 3H), 7,38-7,44 (m, 1H); 13C RMN (100 MHz): 5 -4,72 (CH3), -4,65 (CH3), 18,0 (C), 19,7 (CH3), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,7 (CH3), 26,7 (CH2), 34,0 (CH2), 47,6 (CH2), 54,4 (CH), 54,5 (CH), 67,3 (CH), 73,0 (CH), 125,5 (CH), 126,1 (CH), 126,6 (CH), 127,3 (CH), 130,2 (CH), 130,7 (CH), 130,9 (CH), 131,2 (CH), 135,1 (C), 136,2 (C), 173,0 (C), 214,7 (C); HRMS (ESI-QTOF) calculado para [CscH46O4Si H]+= 499,3238, encontrado 499,3234; FTIR (KBr, puro) 3454, 2948, 1737, 1647, 1462, 1375, 1253, 1107, 969, 880 cm-1.
Ejemplo 10 Síntesis de 7-((1R,2R,3R)-3-((ferí-butildimetilsilil)oxi)-2-((E)-3-metoxiestiril)-5-oxociclopentil)hept-5-enoato de (Z)-isopropilo (10.12)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido trans-2-(3-metoxifenil) -vinilboroico (3.12), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando El compuesto del título 10.12 (72 mg, 93 %).
[a]25D -65,1 (c 1,00, CHCla); 1H RMN (400 MHz): 50,01 (s, 3H), 0,02 (s, 3H), 0,85 (s, 9H), 1,20 (d, J = 6,2 Hz, 6H), 1,69-1,70 (m, 2H), 2,05 (dd, J = 7,1, 14,1 Hz, 2H), 2,13-2,27 (m, 4H), 2,33-2,44 (m, 2H), 2,53-2,80 (m, 2H), 3,81 (s, 3H), 4,12 (dd, J = 8,6, 15,8 Hz, 1H), 4,97 (septeto, J = 6,2 Hz, 1H), 5,29-5,48 (m, 2H), 6,04 (dd, J = 8,6, 15,7 Hz, 1H), 6,47 (d, J = 15,7 Hz, 1H), 6,79 (dd, J = 2,2, 8,0 Hz, 1H), 6,89 (s, 1H), 6,94 (d, J = 7,6 Hz, 1H), 7,19-7,27 (m, 1H); 13C RMN (100 MHz): 5 -4,8 (CH3),-4,7 (CH3), 18,0 (C), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,7 (CH3), 26,6 (CH2), 34,0 (CH2), 47,5 (CH2), 54,0 (CH), 54,3 (CH), 55,1 (CH3), 67,3 (CH), 73,0 (CH), 111,6 (CH), 112,8 (CH), 118,8 (CH), 126,4 (CH), 129,5 (CH), 130,1 (CH), 131,0 (CH), 132,7 (CH), 138,5 (C), 159,8 (C), 173,0 (C), 214,6 (C); HRMS (ESI-QTOF) calculado para [C30H46O5Si H]+= 515,3187, encontrado 515,3172; FTIR (KBr, puro) 2945, 2862, 1737, 1666, 1590, 1463, 1375, 1257, 1156, 1108 cm-1.
Ejemplo 11 - Síntesis de 7-((1R,2R,3R)-3-((ferí-butildimetilsilil)oxi)-2-((E)-4-fluoroestiril)-5-oxociclopentil)hept-5-enoato de (Z)-isopropilo (10.13)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido trans-2-(4-fluorometil-fenil) -vinilboroico (3.13), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) que proporciona el compuesto del título 10.13 (69 mg, 92 %).
[a]30D -44,7 (c 1,00, CHCla); 1H RMN (400 MHz): 5 -0,01 (s, 3H), 0,01 (s, 3H), 0,85 (s, 9H), 1,19 (d, J = 6,2 Hz, 6H), 1,64 (quintet, J = 7,5 Hz, 2H), 2,04 (dd, J = 7,1, 14,2 Hz, 2H), 2,12-2,28 (m, 4H), 2,32-2,45 (m, 2H), 2,54-2,74 (m, 2H), 4,11 (dd, J = 8,6, 15,8 Hz, 1H), 4,96 (septeto, J = 6,2 Hz, 1H), 5,27-5,46 (m, 2H), 5,95 (dd, J = 8,6, 15,7 Hz, 1H), 6,46 (d, J= 15,7 Hz, 1H), 7,00 (dd, J = 8,6, 8,7 Hz, 2H), 7,26-7,34 (m, 2H); 13C RMN (100 MHz): 5 -4,8 (CH3), -4,7 (CH3), 18,0 (C), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,6 (CH3), 26,6 (CH2), 34,1 (CH2), 47,5 (CH2), 54,1 (CH), 54,3 (CH), 67,4 (CH), 72,9 (CH), 115,5 (d, J = 21,0 Hz, CH), 126,5 (CH), 127,5 (d, J = 8,0 Hz, CH), 129,5 (CH), 131,0 (CH), 131,7 (CH), 133,2 (d, J = 3,0 Hz, C), 162,2 (d, J = 245,0 Hz, C), 173,0 (C), 214,5 (C); HRMS (ESI-QTOF) calculado para [C2gH4sFO4Si H]+= 503,2987, encontrado 503,2970; FTIR (KBr, puro) 2939, 1737, 1603, 1463, 1373, 1232, 1154, 1111,845, 777 cm-1.
Ejemplo 12 - Síntesis de 7-((1ft,2ft,3ft)-3-((ferí-butildimetilsilil)oxi)-5-oxo-2-((E)-4-(trifluorometil)estiril)cidopentil)hept-5-enoato de (Z)-isopropilo (10.14)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se utilizó ácido trans-2-(4-trifluorometil-fenil) -vinilboroico (3.14), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) que proporciona el compuesto del título 10.14 (66 mg, 80 %).
[apD-40,4 (c 1,00, CHCls); 1H RMN (400 MHz): 5 -0,01 (s, 3H), 0,02 (s, 3H), 0,85 (s, 9H), 1,18 (d, J = 6,2 Hz, 6H), 1,64 (quintet, J = 7,4 Hz, 2H), 2,04 (dd, J = 7,2, 14,5 Hz, 2H), 2,14-2,28 (m, 4H), 2,35-2,43 (m, 2H), 2,59-2,75 (m, 2H), 4,13 (dd, J = 8,7, 15,9 Hz, 1H), 4,96 (septeto, J = 6,2 Hz, 1H), 5,25-5,52 (m, 2H), 6,16 (dd, J = 8,6, 15,7 Hz, 1H), 6,54 (d, J= 15,7 Hz, 1H), 7,44 (d, J = 8,2 Hz, 2H), 7,57 (d, J = 8,2 Hz, 2H); 13C RMN (100 MHz): 5 -4,7 (CH3), 18,0 (C), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,6 (CH3), 26,6 (CH2), 34,0 (CH2), 47,5 (CH2), 54,2 (CH), 54,3 (CH), 67,4 (CH), 72,8 (CH), 124,2 (q, J = 270,2 Hz, C), 125,6 (q, J = 4,0 Hz, CH), 126,2 (CH), 126,4 (CH), 129,2 (q, J = 33,0 Hz, C), 131,1 (CH), 131,7 (CH), 132,6 (CH), 140,4 (C), 173,0 (C), 214,1 (C); HRMS (ESI-QTOF) calculado para [CsoH43Fa04Si H]+= 553,2955, encontrado 553,2936; FTIR (KBr, puro) 2941, 2866, 1739, 1617, 1464, 1373, 1252, 1163, 1118, 840, 777 cm-1.
Ejemplo 13 - Síntesis de 7-((1ft,2S,3ft)-3-((ferí-butildimetilsilil)oxi)-5-oxo-2-(prop-1-en-2-il)ciclopentil)hept-5-enoato de (Z)-isopropilo (10.15)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido prop-1-en-2-ilborónico (3.15), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título 10.15 (48 mg, 76 %).
[a]25D -85,4 (c 1,00, CHCla); 1H RMN (400 MHz): 5 0,016 (s, 3H), 0,024 (s, 3H), 0,86 (s, 9H), 1,22 (d, J = 6,2 Hz, 6H), 1,58-1,69 (m, 3H), 1,73 (s, 3H), 2,04 (q, J = 7,0 Hz, 2H), 2,10-2,27 (m, 3H), 2,27-2,34 (m, 2H), 2,50 (dd, J = 8,3, 12,0 Hz, 1H), 2,66 (dd, J = 7,1, 18,4 Hz, 1H), 4,13 (dd, J = 8,2, 15,8 Hz, 1H), 4,85 (s, 1H), 4,93 (s, 1H), 4,99 (septeto, J = 6,2 Hz, 1H), 5,28-5,44 (m, 2H); 13C RMN (100 MHz): 5 -4,9 (CH3), -4,8 (CH3), 18,0 (C), 19,6 (CH3), 21,8 (CH3), 24,8 (CH2), 25,3 (CH2), 25,7 (CH3), 26,6 (CH2), 34,1 (CH2), 47,7 (CH2), 52,2 (CH), 57,7 (CH), 67,4 (CH), 71,3 (CH), 114,2 (CH2), 126,5 (CH), 130,8 (CH), 142,4 (C), 173,1 (C), 215,2 (C); HRMS (ESI-QTOF) calculado para [C24H42O4Si H]+= 423,2925, encontrado 423,2927; FTIR (KBr, puro) 3421, 2950, 2862, 1738, 1636, 1376, 1253, 1107, 888, 837, 786 cm-1.
Ejemplo 14 Síntesis de 7-((1R,2S,3R)-2-((£)-but-2-en-2-il)-3-((ferfbutildimetilsilil)oxi)-5-oxociclopentil)hept-5-enoato de (Z)-isopropilo (10.16)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se utilizó ácido (Z) -but-2-en-2-ilborónico (3.16), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título 10.16 (24 mg, 37 %).
[a]25D -43,6 (c 1,00, CHCh); 1H RMN (400 MHz): 5 -0,01 (s, 3H), 0,01 (s, 3H), 0,85 (s, 9H), 1,22 (d, J = 6,0 Hz, 6H), 1,54-1,71 (m, 8H), 1,99-2,08 (m, 2H), 2,10-2,31 (m, 6H), 2,59-2,72 (m, 1H), 3,04 (dd, J=8,8, 12,4 Hz, 1H), 4,15 (q, J = 8,8 Hz, 1H), 4,99 (septeto, J= 6,0 Hz, 1H), 5,29-5,42 (m, 2H), 5,48-5,55 (m, 1H); 13C RMN (100 MHz): 5 -5,0 (CH3), -4,9 (CH3), 13,4 (CH3), 17,9 (C), 18,7 (CH3), 21,8 (CH3), 24,8 (CH2), 24,9 (CH2), 25,7 (CH3), 26,4 (CH2), 34,1 (CH2), 47,7 (CH2), 50,7 (CH), 51,7 (CH), 67,4 (CH), 70,0 (CH), 125,0 (CH), 126,6 (CH), 130,6 (CH), 131,4 (C), 173,1 (C), 215,4 (C); HRMS (ESI-QTOF) calculado para [C2aH44O4S¡ H]+= 437,3081, encontrado 437,3087; FTIR (KBr, puro) 3452, 2938, 1736, 1667, 1461, 1373, 1250, 1108, 972, 838 cm-1.
Ejemplo 15 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silamloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)

Una solución de 2 (57 mg, 0,15 mmol), ácido trans-2-fenilvinilboroico (3.1) (33 mg, 0,22 mmol), [RhCl(1,5-ciclooctadieno)]2 (1,1 mg, 2,2 pmol) y t-BuOK acuoso (20 pl, t-BuOK acuoso 1,5 M, 30 pmol) en dioxano (1,0 ml) a 60 °C durante 4 días usando calentamiento convencional (manta de calentamiento) y luego la mezcla de reacción se concentró directamente al vacío. El residuo se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando 10.1 (39 mg, 54 %).
Ejemplo 16 Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silamloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)

Una solución de 2 (57 mg, 0,15 mmol), ácido trans-2-fenilvinilboroico (3.1) (33 mg, 0,22 mmol), [RhCl(1,5-ciclooctadieno)]2 (1,1 mg, 2,2 pmol) y t-BuOK acuoso (20 pl, t-BuOK acuoso 1,5 M, 30 pmol) en isopropanol (1,0 ml) se agitó a 60 °C durante 3 días usando calentamiento convencional (manta de calentamiento) y luego la mezcla de reacción se concentró directamente al vacío. El residuo se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando 10.1 (43 mg, 59 %).
Ejemplo 17 - Síntesis de (Z)-7-[(2R,3R)-3-(fert-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Una solución de 2 (57 mg, 0,15 mmol), ácido trans-2-fenilvinilboroico (3.1) (33 mg, 0,22 mmol), [RhCl(1,5-ciclooctadieno)]2 (1,1 mg, 2,2 pmol) y KOH acuoso (9,5 pl, KOH acuoso 3,0 M, 29 pmmol) en isopropanol (1,0 ml) a 25 °C durante 3 días y luego la mezcla de reacción se concentró directamente al vacío. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 70 % de 10.1 y una recuperación del 22 % de 2.
Ejemplo 18 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó usando KHF2 acuoso (0,3 ml, KHF2 acuoso 3,0 M, 0,9 mmol) como el aditivo básico. La mezcla de producto crudo se analizó luego por espectroscopia de 1H RMN que mostró un rendimiento del 74 % de 10.1 y una recuperación del 18 % de 2.
Ejemplo 19 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando NaOH acuoso (9,5 pl, NaOH acuoso 3,0 M, 29 pmmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 35 % de 10.1 y una recuperación del 70 % de 2.
Ejemplo 20 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando LiOH acuoso (9,5 pl, LiOH acuoso 3,0 M, 29 pmmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H r Mn que mostró un rendimiento del 25 % de 10.1 y una recuperación del 65 % de 2.
Ejemplo 21 Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando CsOH acuoso (9.5 pl, CsOH acuoso 3.0 M, 29 pmmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 22 % de 10.1 y una recuperación del 77 % de 2.
Ejemplo 22 - Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando K2CO3 acuoso (0,3 ml, K2CO3 acuoso 3,0 M, 0,9 mmol) como el aditivo básico. La mezcla de producto crudo se analizó luego por espectroscopia de 1H RMN que mostró un rendimiento del 13 % de 10.1 y una recuperación del 60 % de 2.
Ejemplo 23 Síntesis (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando K3PO4 acuoso (0,6 ml, K3PO4 acuoso 1,5 M, 0,9 mmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 54 % de 10.1 y una recuperación del 26 % de 2.
Ejemplo 24 Síntesis de (Z)-7-[(2R,3R)-3-(ferí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato
de iso p ro p ilo
(10.1)
Siguiendo el procedimiento descrito en el Ejemplo 18, el compuesto del título se sintetizó utilizando didorobis(norbomadieno)dirodio (1,0 mg, 2,2 emoles) como el aditivo metálico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 35 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 25 - Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butN-dimetN-sNanMoxi)-5-oxo-2-estinl-ciclopentN]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 18, el compuesto del título se sintetizó utilizando [RhCl(C2H4)2]2 (0,9 mg, 2,3 pmoles) como aditivo metálico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 29 % de 10.1 y una recuperación del 69 % de 2.
Ejemplo 26 - Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butN-dimetN-sNanMoxi)-5-oxo-2-estinl-ciclopentN]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando t-BuOH (0,8 ml) como disolvente. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 58 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 27 - Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butN-dimetN-sNanMoxi)-5-oxo-2-estinl-ciclopentN]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando EtOH (0,8 ml) como disolvente. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 75 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 28 - Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butN-dimetN-sNanMoxi)-5-oxo-2-estinl-ciclopentN]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando MeOH (0,8 ml) como disolvente. La mezcla de producto crudo se analizó luego mediante espectroscopía de 1H RMN que mostró un rendimiento del 80 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 29 - Síntesis de (Z)-7-[(2R,3R)-3-(íerí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando MeOH-H2O (0,8 ml, 1: 1 v/v) como disolvente. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 61 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 30 Síntesis de (Z)-7-[(2R,3R)-3-(íerí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 18, el compuesto del título se sintetizó utilizando dioxano (0,8 ml) como disolvente. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 48 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 31 Síntesis de (Z)-7-[(2R,3R)-3-(íerí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 28, el compuesto del título se sintetizó utilizando K3PO4 (0,6 ml, K3PO4 acuoso 1,5 M 0,9 mmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 60 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 32 Síntesis de (Z)-7-[(2R,3R)-3-(íerí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 28, el compuesto del título se sintetizó utilizando t-BuNH2 (0,95 ml, 9,0 mmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 50 % de 10.1 y una recuperación del 50 % de 2.
Ejemplo 33 Síntesis de (Z)-7-[(2R,3R)-3-(íerí-butil-dimetil-silaniloxi)-5-oxo-2-estiril-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 28, el compuesto del título se sintetizó utilizando piperidina (0,89 ml, 9,0 mmol) como el aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 28 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 34 Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butil-dimetil-silamloxi)-5-oxo-2-estinl-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Siguiendo el procedimiento descrito en el Ejemplo 17, el compuesto del título se sintetizó utilizando 1,3-diaminopropano (0,8 ml, 9,6 mml) como aditivo básico. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 42 % de 10.1 y una recuperación del 38 % de 2.
Ejemplo 35 Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butil-dimetil-silamloxi)-5-oxo-2-estinl-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Una solución de 2 (57 mg, 0,15 mmol), ácido trans-2-fenilvinilboroico (3.1) (33 mg, 0,22 mmol) y [RhOH(1,5-ciclooctadieno)]2 (1,0 mg, 2,2 pmol) en MeOH (1,0 ml) se agitó durante 6 horas bajo irradiación de microondas mientras la temperatura se ajustó jhasta 30 °C, y luego la mezcla de reacción se concentró directamente al vacío. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 29 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 36 - Síntesis de (Z)-7-[(2R,3R)-3-(ferf-butil-dimetil-silamloxi)-5-oxo-2-estinl-ciclopentil]-hept-5-enoato de isopropilo (10.1)
Una solución de 2 (57 mg, 0.15 mmol), ácido trans-2-fenilvinilboroico (3.1) (33 mg, 0,22 mmol) y [RhOH(1,5-ciclooctadieno)]2 (1,0 mg, 2,2 pmol) en MeOH (1 ml) se agitó a 50 °C durante 3 días usando calentamiento convencional (manta de calentamiento) y luego la mezcla de reacción se concentró directamente al vacío. La mezcla de producto crudo se analizó luego mediante espectroscopia de 1H RMN que mostró un rendimiento del 47 % de 10.1 y una recuperación del 2 % de 2.
Ejemplo 37 Síntesis de terc-butil-(1-etinil-hexiloxi) -dimetil-silano (1.8a)

A una solución de oct-1-in-3-ol (11,574 ml, 76,1 mmol), imidazol (10,4 g, 153 mmol) y DMAP (0,929 g, 7,60 mmol) en CH2Cl2 (100 ml) a 0-5 °C se añadió gota a gota una solución de TBSCl (17,2 g, 114 mmol) en CH2Cl2 (50 ml). La mezcla de reacción (suspensión blanca) se dejó calentar hasta temperatura ambiente. La mezcla se agitó a temperatura ambiente durante 20 h y se diluyó con CH2Ch (150 ml) y NH4Cl saturado acuoso (300 ml). La capa orgánica se separó y la capa acuosa se extrajo con CH2Cl2 (200 ml x 2). La capa orgánica combinada se lavó con salmuera, se secó sobre MgSO4 y se concentró. La cromatografía de columna (eluyendo con 1:20 (v/v) de EtOAc-nheptano) proporcionó el compuesto del título 1.8a en forma de un líquido incoloro (18,11 g, 99 %).
1H RMN (400 MHz, CDCla): 5 4,35 (td, J= 6,4, 2,0 Hz, 1H), 2,38 (d, ,1H), 1,72-1,67 (m, 2H), 1,62-1,45 (m, 2H), 1,44 1,30 (m, 4H), 0,91-0,86 (m, 12H), 0,04 (s, 3H), 0,02 (s, 3H); 13C RMN (100 MHz, CDCla): 585,8, 71,8, 62,8, 38,5, 31,4, 25,8 (CH2 x3), 24,8, 22,6, 18,2, 14,0, -4,6, -5,1; FTIR (KBr, puro) 3312, 2956, 2930, 2858, 1716, 1642, 1471, 1463, 1362, 1341, 1253, 1144, 1119, 1005, 969 cm'1.
Ejemplo 38 - Síntesis de 2-[3-(fert-butil-dimetil-silamloxi)-oct-1-eml]-4,4,5,5-tetrametil-[1,3,2]dioxaborolano (2.8a)

A n-heptano (10.0 ml) se le añadió terc-butil-( 1 -etinilhexoxi) -dimetil-silano (2,48 g, 10,3 mmol), 5 % en moles de ácido 4-(dimetilamino) benzoico (83 mg, 0,50 mmol) y pinacolborano (4,5 ml, 30 mmol; aceite puro) bajo atmósfera de N2 a temperatura ambiente. La solución se calentó en un baño a 100 °C durante 5,5 horas y se enfrió hasta temperatura ambiente. La mezcla se diluyó con EtOAc (30 ml) y solución saturada acuosa de NH4Cl (40 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (30 ml) y las capas orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre MgSO4 anhidro (2,5 g), se filtraron y se concentraron bajo presión reducida para proporcionar la mezcla cruda que se purificó. por cromatografía de columna (50 g de SiO2; eluyendo con n-heptano (200 ml) y luego 1:50 (v/v) MTBE-n-heptano (750 ml)) para dar el compuesto del título (2,85 g, 7,74 mmol, 75 % rendimiento).
1H RMN (400 MHz, CDCh): 5 6,37 (1H, dd, d=18,0, 5,6), 5,43 (1H, dd, d=18,0, 1,2), 4,11 (1H, m), 1,48 (2H, m), 1,29 (6H, m), 1,28 (12H, s), 0,90 (9H, s), 0,88 (3H,m), 0,04 (3H, s), 0,03 (3H, s); 13C RMN (100 MHz, CDCla): 5 156,2, 116,2, 83,0 (C x2), 74,1, 37,5, 31,9, 25,9 (CH2 *3), 24,8 (CH *2), 24,7 (CH *3), 22,6, 18,2, 14,0, -4,4, -4,9; FTIR (KBr, puro) 2956, 2929, 2857, 1715, 1642, 1463, 1471, 1362, 1339, 1255, 1165, 1087 cm'1; HRMS (ESI-QTOF) calculado para [C20H4-iBO3Si OH]-= 385,2951, encontrado 385,2961.
Ejemplo 39 - Síntesis del ácido (E)-(3 -((terc-butildimetilsilil)oxi)oct-1-en-1-il)borónico (3.8a)

A una solución de -2-[3-(terc-butil-dimetil-silaniloxi) -oct-1-enil]-4,4,5,5 tetrametil-[1,3,2]dioxaborolano (2.8a, 0,99 g , 2,69 mmol) en acetona y agua (150 ml, 2: 1 v/v) se añadió NaIO4 (1,80 g, 8,42 mmol; 3,1 equiv.) y NH4OAc (0,63 g, 8,17 mmol; 3,0 equiv.). La solución turbia resultante se agitó a temperatura ambiente durante 20 horas y se concentró bajo presión reducida para eliminar la acetona. El residuo se diluyó con EtOAc (50 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (50 ml) y las capas orgánicas combinadas se lavaron con salmuera (100 ml), se secaron sobre MgSO4 (2,0 g), se filtraron y se concentraron bajo presión reducida para proporcionar el ácido borónico del título 3.8a (820 mg, cuant.).
1H RMN (400 MHz, DMSO-d6): 5 7,57 (2H, s), 6,58 (1H, dd, d=17,8, 5,0), 5,58 (1H, dd, d=18,0, 1,2), 4,16 (1H, m), 1,39 (2H, m), 1,37 (6H, m), 0,84 (9H, s), 0,83 (3H,m), 0,01 (3H, s), -0,01 (3H, s); 13C RMN (100 MHz, DMSO-d6): 5 152,1, 122,4, 74,0, 37,2, 31,2, 25,7 (CH *3), 24,2, 22,0, 17,9, 13,8, -4,4, -4,9; FTIR (KBr, puro) 2956, 2929, 2857, 1636, 1472, 1463, 1348, 1286, 1255, 1118, 1088, 1004, 966 cm-1; HRMS (ESI-QTOF) calculado para [C-mHs-iBOsSí -H]-= 285,2063, encontrado 285,2082.
Ejemplo 40 - Síntesis de 1-(4,4,5,5-tetrametil-[1,3,2]dioxaborolan-2-il)-oct-1-en-3-ol (2.8)

A n-heptano (10,0 ml) se le añadió octi-1 -in-3-ol (1,5 g, 11,0 mmol), 5 % en moles de ácido 4-(dimetilamino) benzoico (94 mg, 0,55 mmol) y pinacolborano (6,8 ml , 45 mmol; aceite puro) bajo atmósfera de N2 a temperatura ambiente. La solución se calentó en un baño a 100 °C durante 24 horas y se enfrió hasta temperatura ambiente. La mezcla se diluyó con EtOAc (50 ml) y solución saturada acuosa de NH4Cl (60 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (50 ml) y las capas orgánicas combinadas se lavaron con salmuera (60 ml), se secaron sobre MgSO4 anhidro (3,0 g), se filtraron y se concentraron bajo presión reducida para proporcionar la mezcla cruda que se purificó por cromatografía de columna (80 g de SiO2; eluyendo con 1:20 (v/v) de MTBE-n-heptano (250 ml) y luego 1:10 (750 ml) para dar el compuesto del título (2,8, 1,06 g, 7,74 mmol, rendimiento del 38 %).
1H RMN (400 MHz, CDCla): 5 6,60 (1H, dd, J=18,0, 5,2), 5,60 (1H, dd, J=18,4, 1,2), 4,12 (1H, m), 1,93 (1H, s), 1,52 (2H, m), 1,30 (6H, m), 1,28 (12H, s), 0,88 (3H,m); 13C RMN (100 MHz, CDCl3): 5 155,4, 117,1, 83,2 (CH *2), 73,6, 36,5, 31,7, 24,9 (CH *2), 24,7 (CH *3), 22,5, 14,0; HRMS (ESI-QTOF) calculado para [C14H27BO3 OH]+= 271,2086, encontrado 271,2106.
Ejemplo 41 - Síntesis del ácido (E) -(3-hidroxioct-1-en-1-il)borónico (3.8)

A una solución de 2.8 (1,17 g, 4,6 mmol) en acetona y agua (180 ml, 2:1 v/v) se añadió NaIO4 (3,05 g, 14,3 mmol) y NH4OAc (1,06 g, 13,8 mmol). La solución turbia resultante se agitó a 40 °C durante 20 horas y se concentró bajo presión reducida para eliminar la acetona. El residuo se diluyó con EtOAc (60 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (60 ml) y las capas orgánicas combinadas se lavaron con salmuera (120 ml), se secaron sobre MgSO4 (2,5 g), se filtraron y se concentraron bajo presión reducida para proporcionar el compuesto del título (3.8).443 mg, 56 %).
1H RMN (400 MHz, DMSO-d6): 5 7,55 (s, 2H), 6,39 (dd, J= 18,0, 5,2 Hz, 1H), 5,42 (dd, J= 18,0, 1,2 Hz, 1H), 4,66 (d, J= 4,4 Hz, 1H), 3,90 (m, 1H), 1,34 (m, 2H), 1,27 (m, 6H), 0,85 (m, 3H); 13C RMN (100 MHz, DMSO-d6): 5 153,8, 122,4, 72,5, 37,2, 31,8, 25,1, 22,6, 14,4; HRMS (ESI-QTOF) calculado para [CaH-iyBOs - H]-= 171,1198, encontrado 171,1210.
Ejemplo 42 - Síntesis de acetato de oct-1-in-3-ilo (1.8b)

Un matraz se cargó con 4-dimetilaminopiridina (610 mg, 4,9930 mmol), oct-1-in-3-ol (7,3 ml, 50 mmol) y diclorometano (100 ml). La solución se enfrió hasta 0 °C y se añadió trietilamina (20,9 ml, 150 mmol), seguido de la adición gota a gota de anhídrido acético (7,09 ml, 75 mmol) en 3 ml de diclorometano. La solución se calentó gradualmente hasta temperatura ambiente. Una vez completada la reacción, la mezcla de reacción se concentró al vacío. La mezcla de reacción cruda se purificó en gel de sílice (eluyendo con 1:25 (v/v) de MTBE-hexanos) para proporcionar el compuesto del título (1.8b, 8,21 g, 97,6 % de rendimiento) como un aceite de color amarillo pálido.
1H RMN (400 MHz, CDCb): 6 5,30 (t, J= 5,2 Hz, 1H), 2,43 (d, ,1H), 2,04 (s, 3H), 1,75-1,70 (m, 2H), 1,41-1,39 (m, 2H), 1,28-1,27 (m, 4H), 0,86-0,84 (m, 3H); 13C RMN (100 MHz, CDCb): 6 169,7, 81,2, 73,4, 63,7, 34,5, 31,2, 24,5, 22,4, 20,9, 13,9.
Ejemplo 43 - Síntesis del acetato de (E)-1-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)oct-1-en-3-ilo (2.8b)

Para secar el n-heptano (15 ml) se añadió acetato de oct-1-in-3-ilo (2,5 g, 15 mmol), ácido 4-dimetilaminobenzoico (120 mg, 0,74 mmol) y pinacolborano (6,47 ml, 43,3 mmol) bajo N2. La solución se calentó hasta 90-100 °C. Una vez que se completó la reacción y se enfrió hasta temperatura ambiente, la mezcla de reacción se concentró bajo vacío y se sometió a partición entre EtOAc (50 ml) y NH4Cl acuoso al 10 % (50 ml). La fase acuosa se extrajo con EtOAc (50 ml x 2) y las fases orgánicas combinadas se lavaron con salmuera (100 ml) y se secaron sobre MgSO4. La mezcla de reacción se concentró al vacío y la mezcla de reacción cruda se purificó en gel de sílice (eluyendo con 1:25 (v/v) de MTBE-n-heptano con Et3N al 1 %) para proporcionar el compuesto del título (2.8b, 1,23 g, rendimiento del 27,9 %) como un aceite de color amarillo pálido.
1H RMN (400 MHz, CDCh): 6 6,38 (dd, J= 18,4, 5,2 Hz, 1H), 5,57 (dd, J= 17,2, 1,2 Hz, 1H), 5,32-5,27 (m, 1H), 2,07 (s, 3H), 1,64-1,59 (m, 2H), 1,34-1,24 (m, 17H), 0,90-0,86 (m, 4H); 13C RMN (100 MHz, CDCh): 6 170,3, 150,3, 83,3 (c x2), 75,0, 33,8, 31,6, 24,8, 24,7 (CH3 x4), 22,5, 21,1, 14,0 (la señal de ipso carbono (unida a B) no se observó debido a la baja intensidad) .
Ejemplo 44 - Síntesis del ácido (I) -(3-acetoxioct-1-en-1-il) borónico (3.8b)
A una solución de 2.8b (950 mg, 3,2 mmol) en acetona/H2O (3/1, v/v) (142 mL) se le añadió peryodato de sodio (2.13 g, 9.96 mmol) y acetato de amonio (742 mg, 9,63 mmol). La solución turbia resultante se agitó a 40 °C. Una vez completada la reacción, la mezcla de reacción se colocó bajo presión reducida para eliminar la acetona. El residuo se diluyó con EtOAc (100 ml) y las fases se separaron. La capa acuosa se extrajo con EtOAc (100 ml) y las capas orgánicas combinadas se lavaron con salmuera (50 ml) y se secaron sobre MgSO4. La mezcla de reacción se concentró al vacío y la mezcla de reacción cruda se purificó en gel de sílice (eluyendo con 1: 4 (v/v) acetona-nheptano) para proporcionar el compuesto del título (3.8b, 507 mg, 74 % de rendimiento) como un aceite de color amarillo pálido.
1H RMN (400 MHz, DMSO-cfe): 5 6,34 (dd, J= 18, 5,2 Hz, 1H), 5,44 (dd, J= 18,4, 1,6 Hz, 1H), 5,17-5,13 (m, 1H), 2,04 (s, 3H), 1,56-1,52 (m, 2H), 1,3-1,26 (m, 6 H), 0,88-0,84 (m, 3H); 13C RMN (100 MHz, DMSO-cfe): 5 170,1, 147,2, 75,2, 33,9, 31,4, 24,7, 22,4, 21,3, 14,3 (la señal de ipso carbono (unida a B) no se observó debido a la baja intensidad); h Rm S (Es I-QTOF) calculado para [C10H19BO4 - H]-= 213,1304, encontrado 213,1316.
Ejemplo 45 - Síntesis de {[(1-pentilprop-2-inil) oxi] metil}benceno (1.8c)

Un matraz se cargó con oct-1 -in-3-ol (6 g, 47.544 mmol) y THF (60 ml). La solución se enfrió a 0-5 °C y luego se añadió NaH al 60 % (2,9 g, 73 mmol). A la mezcla se le añadió BnBr (6 ml, 50,45 mmol) gota a gota y luego se agitó a temperatura ambiente. Una vez completada la reacción, la mezcla se inactivó mediante la adición de 30 ml de H2O a 0-15 °C. La mezcla de reacción se extrajo con EtOAc (50 ml x 2) y las fases orgánicas combinadas se lavaron con salmuera (30 ml) y se secaron sobre MgSO4. La mezcla de reacción se concentró al vacío, y la mezcla de reacción cruda se purificó sobre gel de sílice (eluyendo con EtOAc-n-heptano al 0-20 % (v/v)) para proporcionar el compuesto del título en forma de un aceite de color amarillo pálido (1.8c). 10,27 g, 47,5 mmol, rendimiento del 99,9 %).
1H RMN (400 MHz, CDCls): 5 7,40-7,28 (m, 5H), 4,84 (d, J= 12 Hz, 1H), 4,54 (d, J= 12 Hz, 1H), 4,12-4,08 (m, 1H), 2,49 (d, J= 2 Hz, 1H), 1,83-1,74 (m, 2H), 1,53-1,50 (m, 2H), 1,37-1,28 (m, 4H), 0,93-0,92 (m, 3H); 13C RMN (100 MHz, CDCls): 5 137,9, 128,4 (CH x2), 128,0 (CH x2), 127,7, 83,1,73,7, 70,5, 68,5, 35,6, 31,5, 24,9, 22,5, 14,0.
Ejemplo 46 - Síntesis de (E)-2-[3-(benciloxi)oct-1-en-1-il] -4,4,5,5-tetrametil-1,3,2-dioxaborolano (2.8c)
Para secar el n-heptano (15 ml) se agregaron 1.8c (2,18 g, 10.1 mmol), ácido 4-dimetilaminobenzoico (0,0906 g, 0,548 mmol) y pinacolborano (5 ml, 33,4 mmol) bajo N2. La solución se calentó hasta 90-100 °C. Una vez que se completó la reacción y se enfrió hasta temperatura ambiente, la mezcla de reacción se concentró bajo vacío y se sometió a partición entre EtOAc (50 ml) y NH4Cl acuoso al 10 % (50 ml). La fase acuosa se extrajo con EtOAc (50 ml x 2) y las fases orgánicas combinadas se lavaron con salmuera (100 ml) y se secaron sobre MgSO4. La mezcla de reacción se concentró al vacío y la mezcla de reacción cruda se purificó en gel de sílice (eluyendo con 1:30 (v/v) de EtOAc-n-heptano) para proporcionar el compuesto del título en forma de un aceite de color amarillo pálido (2.8c, 2,75). g, 79,2 % de rendimiento).1
1H RMN (400 MHz, CDCh): 5 7,36-7,28 (m, 5H), 6,53 (dd, J= 18, 6,8 Hz, 1H), 5,65 (dd, J= 18,4, 0,8 Hz, 1H), 4,62 (d, J= 12 Hz, 1H), 4,36 (d, J= 12 Hz, 1H), 3,82-3,80 (m, 1H), 1,65-1,26 (m, 20H), 0,90-0,97 (m, 3H); 13C RMN (100 MHz, CDCla): 5 153,6, 138,8, 128,3 (CH x2), 127,7 (CH x2), 127,4, 83,3 (C x2), 81,3, 70,5, 35,1, 31,8, 25,0, 24,8 (CH3 x4 ), 22,6, 14,1 (la señal de ipso carbono (unida a B) no se observó debido a la baja intensidad).
Ejemplo 47 - Síntesis del ácido (E)-[3-(benciloxi) oct-1-en-1-il]borónico (3.8c)

A una solución de 2.8c (1,2 g, 3,5 mmol) en acetona/H2O (3/1, v/v) (150 ml) se le añadió peryodato de sodio (1,8 g, 8,4 mmol) y acetato de amonio (0,67 g, 8,7 mmol). La solución turbia resultante se agitó a 40 °C. Una vez completada la reacción, la mezcla de reacción se colocó bajo presión reducida para eliminar la acetona. El residuo se diluyó con EtOAc (100 ml) y las fases se separaron. La capa acuosa se extrajo con EtOAc (100 ml) y las capas orgánicas combinadas se lavaron con salmuera (50 ml) y se secaron sobre MgSO4. La mezcla de reacción se concentró al vacío y la mezcla de reacción cruda se purificó sobre gel de sílice (eluyendo con 35 % de acetona-nheptano) para proporcionar el compuesto del título en forma de un aceite de color amarillo pálido (3.8c, 0,7 g, 80 % de rendimiento).
1H RMN (400 MHz, DMSO-da): 6 7,68 (s, 2H), 7,37-7,26 (m, 5H), 6,33 (dd, J= 18, 6,8 Hz, 1H), 5,52 (d, J= 18 Hz, 1H), 4,49 (d, J= 12 Hz, 1H), 4,29 (d, J= 12 Hz, 1H), 3,79-3,75 (m, 1H), 1,55-1,20 (m, 8H), 0,86-0,83 (m, 3H); 13C RMN (100 MHz, DMSO-cfe): 6 150,0, 139,3, 128,6, 127,9 (CH *2), 127,7 (CH *2), 81,1, 69,9, 35,1, 31,7, 24,9, 22,5, 14,4 (la señal de ipso carbono (unida a B) no se observó debido a la baja intensidad); HRMS (ESI-QTOF) calculado para [Ci5H23BO3 - H]-= 261,1667, encontrado 261,1681.
Ejemplo 48 - Síntesis de (E)-2-[3-(aliloxi)oct-1-en-1-il] -4,4,5,5-tetrametil-1,3,2-dioxaborolano (2.8d)

A una solución de 2,8 (1,3 g, 5,1 mmol) en THF (13 ml) se agregó una suspensión de NaH prelavado (0,8 g, 20 mmol) en THF (10 ml). La suspensión resultante se calentó a reflujo durante 30 minutos y se añadió bromuro de alilo (2,0 ml, 23 mmol). La suspensión resultante se calentó a reflujo durante 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y se vertió en hielo. La mezcla se extrajo con EtOAc (30 ml x 2) y los extractos orgánicos combinados se secaron sobre MgSO4 (2,0 g). Los compuestos volátiles se eliminaron al vacío y el residuo se purificó por cromatografía de columna (40 g de SiO2; eluyendo con 1:20 (v/v) de MTBE-n-heptano (500 ml) para dar el compuesto del título (2.8d, 861 mg, 2,93 mmol, 57 % de rendimiento).
1H RMN (400 MHz, CDCh): 6 6,45 (dd, J= 18,0, 6,4 Hz, 1H), 5,90 (m, 1H), 5,59 (dd, J= 18,0, 0,8 Hz, 1H), 5,26 (dd, J= 17,2, 3,6 Hz, 1H), 5,15 (dd, J= 17,2, 3,6 Hz, 1H), 4,05 (m, 1H), 3,79 (m, 2H), 1,48 (m, 2H), 1,38 (m, 6H), 1,28 (s, 12H), 0,88 (m, 3H); 13C RMN (100 MHz, CDCh): 6153,5, 135,1, 119,5, 116,5, 83,2 (CH *2), 81,3, 69,6, 35,0, 31,8, 25,0, 24,8 (CH *4), 22,5, 14,0.
Ejemplo 49 - Síntesis del ácido (E)-[3-(aliloxi)oct-1-en-1-il]borónico (3.8d)

A una solución de 2.8d (1,1 g, 3,7 mmol) en acetona y agua (150 ml, 2: 1 v/v) se agregó NaIO4 (2,6 g, 12,0 mmol) y NH4OAC (0,9 g, 11,1 mmol). La solución turbia resultante se agitó a 40 °C durante 24 horas y se concentró bajo presión reducida para eliminar la acetona. El residuo se diluyó con EtOAc (60 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (60 ml) y las capas orgánicas combinadas se lavaron con salmuera (100 ml), se secaron sobre MgSO4 (2,0 g), se filtraron y se concentraron bajo presión reducida para proporcionar la mezcla cruda que se purificó. por cromatografía de columna (30 g de SiO2; eluyendo con 1:10 (v/v) MTBE-n-heptano (100 ml) luego 1: 3 (400 ml) para dar el compuesto del título (3.8d, 679 mg, 3,2 mmol, 86 % de rendimiento).
1H RMN (400 MHz, DMSO-cfe): 5 7,64 (s, 2H), 6,24 (dd, J=18,0, 7,2 Hz, 1H), 5,85 (m, 1H), 5,46 (d, J= 18,0, 1H), 5,21 (dd, J= 17,2, 3,6 Hz, 1H), 5,10 (dd, J= 10,4, 1,6 Hz, 1H), 3,93 (m, 1H), 3,77 (m, 1H), 3,71 (m, 1H), 1,37 (m, 2H), 1,24 (m, 6 H), 0,84 (m, 3H); 13C RMN (100 MHz, DMSO-cfe): 5 149,4, 135,5, 125,9, 115,6, 80,7, 68,4, 34,6, 31,2, 24,4, 22,0, 13,8; HRMS (ESI-QTOF) calculado para [C11H21BO3 - H]-= 211,1511, encontrado 211,1527.
Ejemplo 50 - Síntesis de 2-(oct-1-in-3-iloxi) tetrahidro-2H-pirano (1.8e)

A una solución de oct-1-in-3-ol (11,6 ml, 76,2 mmol), DHP (13,9 ml, 152 mmol) en CH2Cl2 (100 ml) a 0-5 °C se añadió PPTS (950 mg, 3,78 mmol ) en CH2Cl2 (2,0 ml). La mezcla de reacción se dejó calentar hasta temperatura ambiente y se agitó durante 4 horas. Después de diluir con CH2Cl2 (100 ml), se añadió NH4Cl saturado acuoso (200 ml). Las capas se separaron y la capa acuosa se extrajo de nuevo con CH2O 2 (100 ml x 2). Las capas orgánicas se combinaron, se lavaron con salmuera, se secaron sobre MgSO4 y se concentraron. La cromatografía de columna (eluyendo con 1:20 (v/v) de EtOAc-n-heptano) proporcionó el compuesto del título como una mezcla de cuatro diastereoisómeros como un líquido incoloro (1.8e, 14,22 g, 89 % como una mezcla diastereomérica).
1H RMN (400 MHz, CDCla): 5 4,98-4,74 (m, 1H), 4,43-4,25 (m, 1H), 4,05-3,77 (m, 1H), 3,56-3,50 (m, 1H), 2,43-2,37 (m, 1H), 1,79-1,60 (m, 4H), 1,59-1,42 (m, 6 H), 1,35-1,28 (m, 4H), 0,91-0,84 (m, 3H); 13C RMN (100 MHz, CDCla): 5 98,2, 95,5, 84,0, 83,0, 73,1, 72,5, 67,1, 64,7, 62,3, 62,2, 35,6, 35,5, 31,5 (*2), 30,5 (*2), 25,5, 25,4, 25,0, 24,7, 22,5 (*2), 19,3, 19,1, 14,0 (*2).
Ejemplo 51 - Síntesis de (E) -2-[3-(tetrahidro-2H-piranoxi) oct-1-en-1-il] -4,4,5,5-tetrametil-1,3,2-dioxaborolano (2.8e)

Siguiendo el procedimiento descrito en el Ejemplo 38, pero utilizando el compuesto 1.8e (2,02 g, 9,59 mmol) y ácido benzoico (79 mg, 0.49 mmol), el producto crudo se purificó por cromatografía de columna (eluyendo con 1:20 (v/v) MTBE -n-heptano) que proporcionó 2.8e (2,31 g, 71 % como una mezcla diastereomérica).
1H RMN (400 MHz, CDCh): 8 6,63 (dd, J= 18,2, 5,4 Hz, 0,5 H), 6,42 (dd, J= 18,0, 6,4 Hz, 0,5H), 5,69 (dd, J= 18,0, 1,6 Hz, 0,5H), 5,57 (dd, J= 18,0, 0,8 Hz, 0,5H), 4,73 (t, J= 3,6 Hz, 0,5H), 4,61 (t, J= 3,4 Hz, 0,5H), 4,19-4,11 (m, 1H), 3,88-3,84 (m, 1H), 3,51-3,42 (m, 1H), 1,88-1,82 (m, 1H), 1,75-1,68 (m, 1H), 1,65-1,27 (m, 24H), 0,90-0,86 (m, 3H); 13C RMN (100 MHz, CDCla): 5 153,9, 153,2, 119,9 (br), 96,8, 95,7, 83,2, 83,1, 77,3, 77,1, 62,3, 62,1, 35,3, 33,8, 31,9, 31,8, 30,7, 25,5, 25,4, 25,1, 24,79, 24,75, 24,71, 24,67, 24,5, 22,5, 19,6, 19,3, 14,0.
Ejemplo 52 - Síntesis del ácido (E)-[3-(tetrahidro-2H-piranoxi)oct-1-en-1-il]borónico (3.8e)

Siguiendo el procedimiento descrito en el Ejemplo 39, pero utilizando el compuesto 2.8e (950 mg, 2,80 mmol), el producto crudo se purificó por cromatografía de columna (eluyendo con 1:10 (v/v) EtOAc-n-heptano) proporcionando 3.8e (495). mg, 69 % como mezcla diastereomérica).
1H RMN (400 MHz, DMSO-d6): 5 7,65 (s, 1H), 7,59 (s, 1H), 6,40 (dd, J= 18,0, 5,6 Hz, 0,5H), 6,21 (dd, J= 18,0, 6,8 Hz, 0,5H), 6,45 (t, J= 18,4 Hz, 1H), 4,65 (d, J= 3,2 Hz, 0,5H), 4,51 (d, J= 3,6 Hz, 0,5H), 4,01-3,98 (m, 1H), 3,78-3,71 (m, 1H), 3,43-3,39 (m, 1H), 1,74-1,70 (m, 1H), 1,62-1,58 (m, 1H), 1,51-1,40 (m, 6H), 1,26-1,24 (m, 6H), 0,87-0,84 (m, 3H); 13C RMN (100 MHz, DMSO-d6): 5 150,74, 149,5, 126,7, 123,8, 97,1, 95,1, 78,0, 77,1, 61,9, 61,7, 35,4, 34,2, 31,81, 31,74, 31,68, 30,98, 30,84, 28,9, 25,62, 25,57, 25,1, 24,5, 22,5, 19,7, 19,6, 14,4; HRMS (ESI-QTOF) calculado para [C-I3H25BO4 - H]- = 255,1773, encontrado 255,1785.
Ejemplo 53 - Síntesis de (S) -tert-butildimetil (oct-1-in-3-iloxi)silano (1.9a)

A una solución de (S)-oct-1-in-3-ol (5,08 g, 40,3 mmol), 2,6-lutidina (8,49 g, 79,2 mmol) y DMAP (0,506 g, 4,14 mmol) en CH2Cl2 (70 ml) a 0-5 °C se añadió gota a gota una solución de TBSOTf (15,8 g, 59,8 mmol) en CH2Cl2 (20 ml). La mezcla de reacción (solución de color amarillo claro) se dejó calentar hasta temperatura ambiente y se agitó
durante 16 horas. Después de añadir NH4CI saturado acuoso (50 ml), se recolectó la capa orgánica y la capa acuosa se extrajo con CH2Ch (50 ml x 2). Las capas orgánicas se combinaron, se lavaron con salmuera (100 ml), se secaron sobre MgSO4 (3,0 g), se filtraron y se concentraron. La cromatografía de columna (eluyendo con n-heptano y luego 1:20 (v/v) EtOAc-n-heptano) produjo 1.9a como un líquido incoloro (8,52 g, 88,0 %).
Ejemplo 54 - Síntesis de (S, E)-tertbutildimetil {[1-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il) oct-1-en-3-il]oxi} silano (2.9a)

A n-heptano (20.0 ml) se le agregaron 1.9a (4,9 g, 20,0 mmol), 5 % en moles de ácido 4-(dimetilamino)benzoico (170 mg, 1,0 mmol) y pinacolborano (9,2 ml, 62 mmoles; aceite puro) bajo atmósfera de N2 a temperatura ambiente. La solución se calentó en un baño a 100 °C durante 5 horas y se enfrió hasta temperatura ambiente. La mezcla se diluyó con EtOAc (40 ml) y solución saturada acuossa de NH4Cl (50 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (40 ml) y las capas orgánicas combinadas se lavaron con salmuera (80 ml), se secaron sobre MgSO4 anhidro (3,0 g), se filtraron y se concentraron bajo presión reducida para proporcionar el 2.9a crudo que fue purificado por cromatografía de columna (100 g de SO2; eluyendo con n-heptano (400 ml) y luego 1:30 (v/v) MTBE-n-heptano (900 ml)) para dar el compuesto del título (2.9a, 6,83 g, 18,5 mmol, 91 % de rendimiento).
Ejemplo 55 - Síntesis del ácido (S, E)-{3-[(terc-butildimetilsilil)oxi]oct-1-en-1-il}borónico (3.9)

A una solución de 2.9a (9,29 g, 25,2 mmol) en acetona y agua (540 ml, 2: 1 v/v) se agregó NaIO4 (15,0 g, 70,13 mmol) y NH4OAc (5,15 g, 66,80 mmol). La solución turbia resultante se agitó a 40 °C durante 20 horas y se concentró bajo presión reducida para eliminar la acetona. El residuo se diluyó con EtOAc (200 ml) y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (200 ml) y las capas orgánicas combinadas se lavaron con salmuera (200 ml), se secaron sobre MgSO4 (5,0 g), se filtraron y se concentraron bajo presión reducida para proporcionar el compuesto del título (3.9, 3,86 g, 13,5 mmol, 53,5 %).
[a]20 *25D= -1,26 (c 1.0, MeOH); HRMS (ESI-QTOF) calculado para [C^Ha-iBOaSi - H ]- 285,2063, encontrado 285,2078.
Ejemplo 56 - Síntesis de 7-((1R,2R,3R)-3-[(ferf-butMdimetMsiMl)oxi]-2-{(E)-3-[(ferf-butMdimetNsNN)oxi]-5-fenilpent-1-en-1-il)}-5-oxociclopentil)hept-5-enoato de (Z)-isopropilo (10.17)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido (S,E)-{3-[(terc-butildimetilsilil) oxi]-5-fenilpent-1-en-1-il}borónico (3.17, 72,0 mg, 0,225 mmol), La mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título 10.17 (63 mg, 64 %).
[a]29D -35,9 (c 1,00, CHCla); 1H RMN (400 MHz): 5 0,03 (s, 3H), 0,05 (s, 3H), 0,06 (s, 3H), 0,07 (s, 3H), 0,87 (s, 9H), 0,92 (s, 9H), 1,22 (d, J= 6,4 Hz, 6H), 1,57-1,72 (m, 3H), 1,74-1,89 (m, 2H), 1,92-2,11 (m, 3H), 2,11-2,43 (m, 4H), 2,52 (dt, J = 7,6, 10,8 Hz, 1H), 2,56-2,74 (m, 3H), 4,08 (q, J = 7,6 Hz, 1H), 4,20 (q, J = 5,5 Hz, 1H), 4,99 (septeto, J = 6,4 Hz, 1H), 5,28-5,47 (m, 2H), 5,50-5,68 (m, 2H), 7,10-7,22 (m, 3H), 7,24-7,32 (m, 2H); 13C RMN (100 MHz): 5 -4,7 (CH3), -4,63 (CH3), -4,59 (CH3), -4,2 (CH3), 18,0 (C), 18,2 (C), 21,8 (CH3), 24,8 (CH2), 25,3 (CH2), 25,8 (CH3), 25,9 (CH3), 26,7 (CH2), 31,6 (CH2), 34,1 (CH2), 40,2 (CH2), 47,7 (CH2), 52,7 (CH), 53,9 (CH), 67,4 (CH), 72,1 (CH), 73,3 (CH), 125,7 (CH), 126,6 (CH), 128,27 (CH), 128,33 (CH), 129,2 (CH), 130,9 (CH), 135,9 (CH), 142,3 (C), 173,1 (C), 215,3 (C); HRMS (ESI): calculado para [C3sH64O5Si2 NH4]+ = 674,4631, encontrado 674,4611; FTIR (KBr, puro) 3465, 2950, 2859, 1736, 1630, 1462, 1376, 1252, 1103, 1020, 834 cm-1.
Ejemplo 57 Síntesis de 7-((1R,2R,3R)-3-((ferí-butildimetilsilil)oxi)-2-((E)-3-((ferí-butildimetilsilil)oxi)-4-(3-(trifluorometil)fenoxi)but-1-en-1-il)-5-oxocidopentil)hept-5-enoato de (Z)-isopropilo (10.18)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó el ácido (R,E)-{3-[(tercbutildimetilsilil)oxi]-4-[3-(trifluorometil)fenoxi]but-1-en-1-il} borónico (3.17 , 88 mg, 0,225 mmol), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título 10.18 (71 mg, 65 %).
[a]29D -35,6 (c 1,00, CHCla); 1H RMN (400 MHz): 5 0,04 (s, 3H), 0,05 (s, 3H), 0,100 (s, 3H), 0,104 (s, 3H), 0,87 (s, 9H), 0,91 (s, 9H), 1,21 (d, J = 6,2 Hz, 6H), 1,58-1,72 (m, 3H), 1,92-2,11 (m, 2H), 2,12-2,45 (m, 4H), 2,52 (dt, J = 7,6, 11,2 Hz, 1H), 2,55-2,70 (m, 1H), 3,80-3,92 (m, 2H), 4,07 (q, J = 8,4 Hz, 1H), 4,50-4,62 (m, 1H), 4,98 (septeto, J = 6,4 Hz, 1H), 5,25-5,45 (m, 2H), 5,65-5,82 (m, 2H), 7,03 (dd, J = 2,0, 8,0 Hz, 1H), 7,08 (s, 1H), 7,20 (d, J = 7,6 Hz, 2H), 7,38 (dd, J = 8,0, 8,0 Hz, 1H); 13C RMN (100 MHz): 5 -4,5 (CH3), -4,6 (CH3), -4,7 (CH3), 18,0 (C), 18,3 (C), 21,8 (CH3), 24,8 (CH2), 25,1 (CH2), 25,7 (CH3), 25,8 (CH3), 26,7 (CH2), 30,9 (CH3), 34,1 (CH2), 47,6 (CH2), 53,1 (CH), 54,1 (CH), 67,4 (CH), 70,9 (CH), 72,6 (CH2), 72,9 (CH), 111,0 (q, J = 4,0 Hz, CH), 117,5 (q, J = 4,0 Hz, CH), 123,9 (q, J = 271.0 Hz, C), 126,5 (CH), 130,0 (CH), 130,9 (CH), 131,5 (CH), 131,9 (q, J = 32,2 Hz, C), 132,3 (CH), 158,9 (C), 173.0 (C), 214,7 (C); HRMS (ESI): calculado para [C3sH61F3O6Si2 NH4]+= 744,4297, encontrado 744,4291; FTIR (KBr, puro) 2945, 2862, 1737, 1602, 1457, 1330, 1249, 1167, 1123, 839 cm-1.
Ejemplo 58 - Síntesis de (Z)-7-{(1R,2R,3R)-2-[(1E,5E)-3-(benciloxi)-4,4-difluoroocta-1,5-dien-1-il]-3-[(feríbutildimetilsilil)oxi]-5-oxociclopentil}hept-5-enoato de isopropilo (10.19)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó 2-((1E, 5E) -3-(benciloxi) -4,4-difluoroocta-1,5-dien-1-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (2,19, 85 mg, 0,225 mmol), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando (Z)-7-{(1R,2R,3R)-2-[(1E,5E)-3-(benciloxi)-4,4-difluoroocta-1,5-dien-1-il]-3-[(te/f-butildimetilsilil)oxi]-5-oxociclopentil}hept-5-enoato de isopropilo (10.19, 27 mg, 28 % como un par de diastereoisómeros).
[a]22D -51,8 (c 0,33, CHCla); 1H RMN (400 MHz): 5 0,035 (s, 1,5 H), 0,044 (s, 1,5H), 0,052 (s, 1,5H), 0,065 (s, 1,5H), 0,88 (s, 9H), 1,03 (t, J = 7,4 Hz, 3H), 1,21 (d, J = 6,4 Hz, 6 H), 1,62-1,70 (m, 1H), 2,01-2,16 (m, 6 H), 2,16-2,30 (m, 2H), 2,28-2,43 (m, 2H), 2,53-2,60 (m, 1H), 2,62-2,69 (m, 1H), 3,93-4,15 (m, 2H), 4,51 (d, J = 12,0 Hz, 0,5H), 4,53 (d, J = 12,0 Hz, 0,5H), 4,67 (d, J = 12,0 Hz, 0,5H), 4,68 (d, J = 12,0 Hz, 0,5H), 4,98 (septeto, J = 6,4 Hz, 1H), 5,26-5,46 (m, 2H), 5,52-5,68 (m, 2H), 5,69-5,82 (m, 1H), 6,11-6,23 (m, 1H), 7,29-7,32 (m, 6 H); 13C RMN (100 MHz): 5 -4,87 (CH3), -4,74 (CH3), -4,67 (CH3), -4,52 (CH3), 12,7 (CH3), 18,0 (C), 21,8 (CH3), 24,79 (CH2), 24,81 (CH2), 25,0 (CH2), 25.2 (CH2), 25,72 (CH3), 25,74 (CH3), 26,63 (CH2), 26,66 (CH2), 34,05 (CH2), 34,06 (CH2), 47,7 (CH2), 53,1 (CH), 53.2 (CH), 53,75 (CH), 53,82 (CH), 67,4 (CH), 71,3 (CH2), 71,5 (CH2), 72,8 (CH), 73,0 (CH), 81,0 (q, J = 31,0 Hz, CH), 121,5 (q, J = 25,3 Hz, CH), 126,2 (CH), 126,3 (CH), 126,4 (CH), 127,7 (CH), 127,80 (CH), 127,84 (CH), 128,37 (CH), 128,41 (CH), 131,2 (CH), 131,3 (CH), 136,8 (CH), 137,2 (CH), 137,5 (C), 137,6 (C), 138,9 (q, J = 8,7 Hz, CH), 173,09 (C), 173,12 (C), 214,5 (C); HRMS (ESI-QTOF) calculado para [Cs6H54F2O5Si Na]+ = 655,3601, encontrado 655,3620; FTIR (KBr, puro) 3460, 2938, 2867, 1738, 1630, 1461, 1377, 1253, 1214, 1109, 974, 873 cm-1.
Ejemplo 59 - Síntesis de 7-{(1ft,2ft,3ft,5S)-3-[(ferí-butildimetilsilil)oxi]-2-((£)-3,3-difluoro-4-fenoxibut-1-en-1-il)-5-hidroxicid opentil}hept-5-enoato de (Z)-isopropilo (11.7)

A una solución de THF (15 ml) del compuesto 10.7 obtenido en el Ejemplo 8 (1,42 g, 2,51 mmol) se le añadió una solución de L-Selectride® (3,45 ml, 1,0 M en THF, 3,45 mmol) a -78 °C. La mezcla se agitó a -78 °C durante 40 minutos y luego se añadió NH4Cl(ac) saturado acuso (5 ml). La capa acuosa se separó y se extrajo con EtOAc (15 ml x 3). Las capas orgánicas se combinaron, se secaron sobre Na2SO4 anhidro, se filtraron y se evaporaron para proporcionar un producto crudo aceitoso, que se purificó mediante cromatografía de columna (eluyendo con 1 : 8 (v/v) EtOAc-hexanos) para proporcionar el compuesto del título 11.7 (1,34 g, 94 %)
[a]26D -9,4 (c 0,11, CHCla); 1H RMN (400 MHz): 5 0,027 (s, 3H), 0,034 (s, 3H), 0,86 (s, 9H), 1,22 (d, J = 6,2 Hz, 6 H), 1,52-1,61 (m, 1H), 1,65 (quintet, J = 7,5 Hz, 2H), 1,73-1,82 (m, 1H), 2,03-2,18 (m, 4H), 2,20-2,46 (m, 5H), 4,02 (quintet, J = 3,5 Hz, 1H), 4,06-4,32 (m, 3H), 4,99 (septeto, J = 6,2 Hz, 1H), 5,28-5,59 (m, 2H), 5,76 (dt, J = 11,1, 15,6 Hz, 1H), 6,14 (ddt, J = 2,4, 9,3, 15,7 Hz, 1H), 6,89-6,92 (m, 2H), 7,01 (t, J = 7,4 Hz, 1H), 7,26-7,31 (m, 2H); 13C RMN (100 MHz): 5 -4,9 (CH3), -4,7 (CH3), 17,9 (C), 21,8 (CH3), 24,9 (CH2), 25,7 (CH3), 26,1 (CH2), 26,6 (CH2), 34,1 (CH2), 43,4 (CH2), 50,4 (CH), 55,7 (CH), 67,4 (CH), 69,5 (t, J = 35,0 Hz, C), 73,5 (CH), 78,5 (CH), 114,7 (CH), 118,1 (t, J = 239.0 Hz, C), 121,7 (CH), 123,6 (t, J = 25,0 Hz, CH), 128,8 (CH), 129,5 (CH), 129,8 (CH), 138,6 (t, J = 9,0 Hz, CH), 158.0 (C), 173,2 (C); HRMS (ESI-QTOF) calculado para [C31H4sF2O5S¡ H]+= 567,3312, encontrado 567,3316; FTIR (KBr, puro) 3451,2954, 2879, 1724, 1595, 1498, 1463, 1375, 1301, 1246, 1101 cm-1.
Ejemplo 60 - Síntesis de tafluprost (7-[(1ft,2ft,3ft,5S)-2-((E)-3,3-difluoro-4-fenoxibut-1-en-1-il)-3,5-dihidroxiciclopentil]hept-5-enoato de (Z)-isopropilo; 12.7)
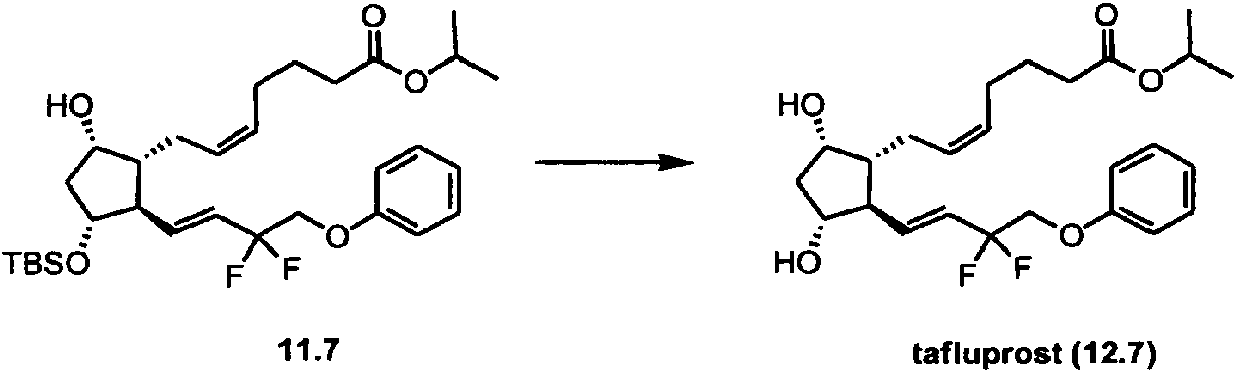
A una solución de THF (7 ml) del compuesto 11.7 obtenido en el Ejemplo 59 (2,11 g, 3,73 mmol) se añadió n-Bu4NF (4,5 ml, 1,0 M en THF, 4,5 mmol) a 0 °C y la mezcla se calentó hasta temperatura ambiente en 30 min y se agitó durante otras 3 h. Se añadió NH4Cl(ac) saturado acuoso para inactivar la reacción y las capas se separaron. La fase acuosa se extrajo con EtOAc (20 ml x 3) y las capas orgánicas se combinaron, se secaron sobre Na2SO4 anhidro, se filtraron y se evaporaron para proporcionar el producto crudo, que se purificó por cromatografía de columna (hexano/EtOAc = 1: 5) para proporcionar el compuesto del título tafluprost (12.7, 1,52 g, 90 %).
[a]23D 21,6 (c 1,00, CHCla); 1H RMN (600 MHz): 5 1,22 (d, J = 6,3 Hz, 6H), 1,58-1,69 (m, 3H), 1,85 (dt, J = 1,2, 14,4 Hz, 1H), 2,03-2,14 (m, 6H), 2,26 (dt, J = 3,0, 7,4 Hz, 2H), 2,28-2,38 (m, 1H), 2,47 (dt, J = 4,2, 9,6 Hz, 1H), 4,00-4,04 (m, 1H), 4,15-4,25 (m, 3H), 5,00 (septeto, J = 6,3 Hz, 1H), 5,33-5,43 (m, 2H), 5,80 (dt, J = 11,1, 15,7 Hz, 1H), 6,10 (ddt, J = 2,3, 9,1, 15,7 Hz, 1H), 6,91-6,92 (m, 2H), 6,97-7,02 (m, 1H), 7,28-7,31 (m, 2H); 13C RMN (150 MHz): 5 21,80 (CH3), 21,82 (CH3), 24,8 (CH2), 25,7 (CH2), 26,6 (CH2), 34,0 (CH2), 43,0 (CH2), 50,5 (CH), 55,8 (CH), 67,7 (CH), 69,3 (t, J = 34,5 Hz, CH2), 73,3 (CH), 78,0 (CH), 114,8 (CH), 118,2 (t, J = 240,0 Hz, C), 121,8 (CH), 123,6 (t, J = 24,8 Hz, CH), 128,6 (CH), 129,6 (CH), 130,1 (CH), 138,6 (t, J = 8,3 Hz, CH), 158,0 (C), 173,4 (C); 19F RMN (564 MHz): 5 -103,4 (d, J = 255,2 Hz), -104,1 (d, J = 255,5 Hz); HRMS (ESI-QTOF): calculado para [C25H34F2O5+ H]+= 453,2447, encontrado 453,2449; FTIR (KBr, puro) 3410, 2929, 1720, 1593, 1494, 1376, 1247, 1156, 1103, 1052 cm-1.
Ejemplo 61 - Síntesis de (Z)-7-((1ft,2ft,3ft)-2-{(E)-3-acetoxioct-1-en-1-il)-3-[(ferí-butildimetilsilil)oxi)]-5-oxociclopentil}hept-5-enoato de isopropilo (10.8b)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se utilizó ácido (E)-(3-acetoxioct-1-en-1-il)borónico (3.8b, 48 mg, 0,225 mmol), la mezcla cruda de reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) que proporciona (Z}-7-((1R,2R,3R)-2-{(É)-3-acetoxioct-l-en-1-il)-3-[(ferfbutildimetilsilil)oxi]-5-oxociclopentil}hept-5-enoato de isopropilo (10.8b, 20 mg, 24 % como un par de diastereoisómeros).
[a]24D -94,2 (c 0,23, CHCla); 1H RMN (400 MHz): 5 0,046 (s, 3H), 0,054 (s, 3H), 0,82-0,92 (m, 12H), 1,24 (d, J = 6,2 Hz, 6H), 1,26-1,36 (m, 7H), 1,60-1,70 (m, 3H), 1,98-2,09 (m, 6H), 2,14 (qd, J = 2,1, 9,1 Hz, 1H), 2,25 (t, J = 7,6 Hz, 2H), 2,29-2,52 (m, 3H), 2,63 (dd, J = 7,2, 18,3 Hz, 1H), 4,015 (q, J = 8,4 Hz, 0,5H), 4,023 (q, J = 8,4 Hz, 0,5H), 4,99 (septeto, J = 6,2 Hz, 1H), 5,21-5,34 (m, 2H), 5,35-5,45 (m, 1H), 5,47-5,65 (m, 2H); 13C RMN (100 MHz): 5 -4,81 (CH3), -4,80 (CH3), -4,7 (CH3), -4,6 (CH3), 13,99 (CH3), 18,0 (C), 21,3(CH3), 21,9(CH3), 22,50 (CH2), 22,52 (CH2), 24,8 (CH2), 25,0 (CH2), 25,1 (CH2), 26,6 (CH2), 26,7 (CH2), 30,9 (CH2), 31,5 (CH2), 31,6 (CH2), 34,06 (CH2), 34,08 (CH2), 34,5 (CH2), 34,6 (CH2), 47,6 (CH2), 53,3 (CH), 53,3 (CH), 53,94 (CH), 53,3,96 (CH), 67,4 (CH), 72,72 (CH), 72,74 (CH), 74,2 (CH), 74,3 (CH), 126,47 (CH), 126,53 (CH), 130,9 (CH), 131,0 (CH), 131,77 (CH), 131,84 (CH), 133,17 (CH), 133,24 (CH), 170,15 (C), 170,19 (C), 173,1 (C), 214,6 (C); HRMS (ESI-QTOF) calculado para [C31H54O6Si NH4]+= 568,4028, encontrado 568,4047; FTIR (KBr, puro): 3442, 2932, 2861, 1737, 1641, 1378, 1237, 1107, 963, 838 cm-1.
Ejemplo 62 Síntesis de (Z)-7-((1ft,2ft,3ft)-3-[(ferí-butildimetilsilil)oxi]-5-oxo-2-{(E)-3-[(tetrahidro-2H-piran-2-il)oxiloct-1-en-1-il}ciclopentil)hept-5-enoato de isopropilo (10.8e)
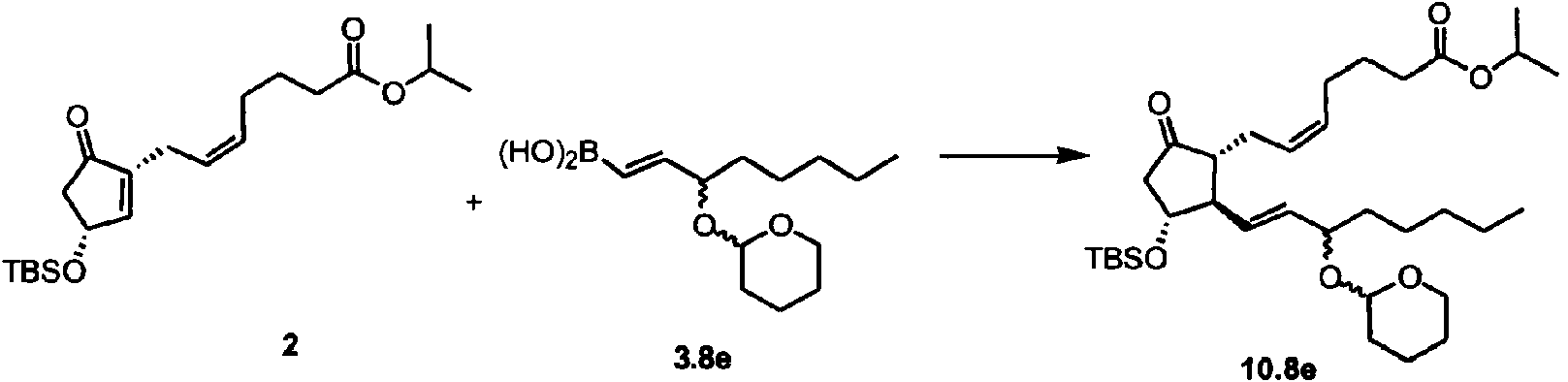
Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido (E)-(3-((tetrahidro-2H-piran-2-il)oxi) oct-1-en-1-il)borónico (3.8e, 58 mg, 0,225 mmol) ), la mezcla cruda de reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) para proporcionar (Z)-7-((1R,2R,3R)-3-[(terfbutildimetilsilil)oxi]-5-oxo-2-{(E)-3-[(tetrahidro-2H-piran-2-il)oxi]oct-1-en-1-il}ciclopentil)hept-5-enoato de isopropilo (10.8e) (56 mg, 63 % como mezcla diastereomérica).
[a]24D -62,9 (c 0,36, CHCla); 1H RMN (400 MHz): 5 0,03 (s, 1,5H), 0,04 (s, 1,5H), 0,05 (s, 1,5H), 0,06 (s, 1,5H), 0,79 0,94 (m, 12H), 1,21 (d, J= 6,3 Hz, 6H), 1,28-1,34 (m, 5H), 1,36-1,72 (m, 10H), 1,73-1,83 (m, 1H), 2,03-2,19 (m, 4H), 2,21-2,40 (m, 4H), 2,45-2,71 (m, 2H), 3,33-3,53 (m, 1H), 3,78-3,82 (m, 1H), 3,92-4,12 (m, 2H), 4,68-4,72 (m, 1H), 4,99 (septeto, J = 6,3 Hz, 1H), 5,28-5,43 (m, 2H), 5,48-5,66 (m, 2H); 13C RMN (100 MHz): 5 -4,8 (CH3), -4,7 (CH3), -4,6 (CH3), -4,5 (CH3), 14,1 (CH3), 17,95 (C), 18,0 (C), 19,42 (CH2), 19,49 (CH2), 19,51 (CH2), 21,9 (CH3), 22,6 (CH2), 24,7 (CH2), 24,8 (CH2), 24,9 (CH2), 25,1 (CH2), 25,2 (CH2), 25,3 (CH2), 25,4 (CH2), 25,5 (CH2), 25,6 (CH2), 25,75 (CH3), 25,77 (CH3), 26,7 (CH2), 30,79 (CH2), 30,82 (CH2), 31,8 (CH2), 31,9 (CH2), 34,09 (CH2), 34,11 (CH2), 34,6 (CH2), 34,8 (CH2), 36,1 (CH2), 47,7 (CH2), 52,7 (CH), 52,9 (CH), 53,1 (CH), 53,8 (CH), 53,9 (CH), 62,0 (CH2), 62,3 (CH2), 67,38 (CH), 67,41 (CH), 73,09 (CH), 73,13 (CH), 73,3 (CH), 75,7 (CH), 76,5 (CH), 77,1 (CH), 94,8 (CH), 97,0 (CH), 97,3 (CH), 126,56 (CH), 126,62 (CH), 126,7 (CH), 130,2 (CH), 130,3 (CH), 130,85 (CH), 130,91 (CH), 132,8 (CH), 133,8 (CH), 134,2 (CH), 134,8 (C), 173,0 (C), 173,09 (C), 173,12 (C), 214,8 (C), 215,3 (C), 215,4 (C); HRMS (ESI): calculado para ^ ^ c A s S i Na]+= 615,4051, encontrado 615,4063; FTIR (KBr, puro) 3424, 2937, 2862, 1737, 1638, 1463, 1375, 1248, 1113, 1020 cm-1.
Ejemplo 63 - Síntesis de (Z)-7-{(1ft,2ft,3ft)-3-[(ferí-butNdimetNsilN)oxi]-2-((E)-3-bencNoxi-1-en-1-M)-5-oxociclopentil}hept-5-enoato de isopropilo (10.8c)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido (E)-(3-(benciloxi) oct-1 -en-1-il) borónico (3.8c, 59 mg, 0,225 mmol), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con acetona-hexanos = 1:80) que proporcionó (Z)-7-((1R,2R,3R)-2-((E)-3-(benciloxi)oct-1-en-1-il)-3-((tertbutildimetilsilil)oxi)-5-oxociclopentil)hept-5-enoato de isopropilo (10.8c) (44 mg, 50 % como un par de diastereoisómeros).
Diastereómero 1: [a]24D-60,4 (c 0,63, CHCla); 1H RMN (400 MHz): 5 0,06 (s, 3H), 0,07 (s, 3H), 0,83-0,96 (m, 12H), 1.21 (d, J = 6,2 Hz, 6H), 1,26-1,53 (m, 7H), 1,60-1,73 (m, 3H), 2,02-2,11 (m, 3H), 2,12-2,27 (m, 3H), 2,28-2,45 (m, 2H), 2,54 (dt, J = 7,6, 11,4 Hz, 1H), 2,63 (dd, J = 6,9, 18,2 Hz, 1H), 3,76 (q, J = 6,5 Hz, 1H), 4,08 (q, J = 8,3 Hz, 1H), 4,35 (d, J = 11,9 Hz, 1H), 4,60 (d, J = 11,9 Hz, 1H), 4,99 (septeto, J = 6,2 Hz, 1H), 5,29-5,48 (m, 2H), 5,49-5,64 (m, 2H), 7,27-7,37 (m, 5H); 13C RMN (100 MHz): 5 -4,7 (CH3), -4,5 (CH3), 14,1 (CH3), 18,0 (C), 21,9 (CH3), 22,6 (CH2), 24.8 (CH2), 25,1 (CH2), 25,3 (CH2), 25,8 (CH3), 26,7 (CH2), 31,8 (CH2), 34,1 (CH2), 36,0 (CH2), 47,7 (CH2), 53,0 (CH), 53.9 (CH), 67,4 (CH), 70,3 (CH2), 73,2 (CH), 79,9 (CH), 126,6 (CH), 127,4 (CH), 127,6 (CH), 128,3 (CH), 131,0 (CH), 132,3 (CH), 134,2 (CH), 138,9 (C), 173,0 (C), 214,9 (C); HRMS (ESI-QTOF) calculado para ^HasOaSi Na]+= 621,3946, encontrado 621,3961; FTIR (KBr, puro) 3448, 2932, 2862, 1736, 1461, 1370, 1246, 1105, 974, 837 cm-1.
Diastereómero 2: [a]24D-37,2 (c 0,9, CHCh); 1H RMN (400 MHz): 5 0,04 (s, 3H), 0,05 (s, 3H), 0,81-0,91 (m, 12H), 1.21 (d, J = 6,3 Hz, 6H), 1,24-1,58 (m, 7H), 1,62-1,72 (m, 3H), 2,04-2,26 (m, 6H), 2,32-2,60 (m, 3H), 2,65 (dd, J = 7,0, 18,3 Hz, 1H), 3,69-3,80 (m, 1H), 4,08 (q, J = 8,0 Hz, 1H), 4,35 (d, J = 12,0 Hz, 1H), 4,59 (d, J = 12,0 Hz, 1H), 4,98 (septeto, J = 6,3 Hz, 1H), 5,30-5,46 (m, 2H), 5,49-5,62 (m, 2H), 7,23-7,37 (m, 5H); 13C RMN (100 MHz): 5 -4,7 (CH3), -4,5 (CH3), 14,1 (CH3), 18,0 (C), 21,9 (CH3), 22,6 (CH2), 24,8 (CH2), 25,1 (CH2), 25,3 (CH2), 25,8 (CH3), 26,7 (CH2), 31,8 (CH2), 34,1 (CH2), 35,9 (CH2), 47,7 (CH2), 53,2 (CH), 54,0 (CH), 67,4 (CH), 70,1 (CH2), 73,0 (CH), 79,7
(CH), 126,5 (CH), 127,4 (CH), 127,6 (CH), 128,3 (CH), 131,1 (CH), 132,6 (CH), 134,2 (CH), 138,8 (C), 173,1 (C), 214,8 (C); FTIR (KBr, puro) 3443, 2931,2853, 1736, 1459, 1373, 1246, 1106, 972, 836 cm-1.
Ejemplo 64 - Síntesis de (Z)-7-{(1ft,2ft,3ft)-3-[(ferí-butildimetilsilil)oxi]-2-((E)-3-hidroxioct-1-en-1-il)-5-oxociclopentil}hept-5-enoato de isopropilo (10.8)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto que se usó ácido (E)-(3-hidroxioct-1-en-1-il) borónico (3.8, 39 mg, 0,225 mmol), la mezcla cruda de reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) que proporcionó (Z)-7-{(1R,2R,3R)-3-[(fe/f-butildimetilsilil)oxi]-2-((£)-3-hidroxioct-1-en-1-il)-5-oxociclopentil}hept-5-enoato de isopropilo (10.8, 31 mg, 40 % como un par de diastereoisómeros).
[a]24D -60,7 (c 0,6, CHCla); 1H RMN (400 MHz): 5 0,03-0,05 (m, 6 H), 0,83-0,91 (m, 12H), 1,22 (d, J = 6,2 Hz, 6 H), 1,25-1,34 (m, 4H), 1,34-1,58 (m, 3H), 1,58-1,72 (m, 3H), 1,98-2,20 (m, 5H), 2,22-2,52 (m, 5H), 2,58-2,68 (m, 1H), 4,02 (q, J = 8,1 Hz, 1H), 4,05-4,17 (m, 1H), 4,99 (septeto, J = 6,2 Hz, 1H), 5,28-5,44 (m, 2H), 5,45-5,68 (m, 2H); 13C RMN (100 MHz): 5 -4,72 (CH3), -4,66 (CH3), -4,58 (CH3), 14,0 (CH3), 18,0 (C), 18,1 (C), 21,8 (CH3), 22,6 (CH2), 24,8 (CH2), 24,9 (CH2), 25,0 (CH2), 25,1 (CH2), 25,3 (CH2), 25,7 (CH3), 26,7 (CH2), 31,77 (CH2), 31,82 (CH2), 34,04 (CH2), 34.09 (CH2), 37,3 (CH2), 37,4 (CH2), 47,62 (CH2), 47,64 (CH2), 52,8 (CH), 53,1 (CH), 53,8 (CH), 54,1 (CH), 67,55 (CH), 67,60 (CH), 72,56 (CH), 72,62 (CH), 72,92 (CH), 73,13 (CH), 126,6 (CH), 126,7 (CH), 130,2 (CH), 130,4 (CH), 130.9 (CH), 136,3 (CH), 137,0 (CH), 173,3 (C), 173,4 (C), 214,9 (C); HRMS (ESI-QTOF) calculado para C ^ O a S i NH4]+= 526,3922, encontrado 526,3924; FTIR (KBr, puro) 3446, 2934, 2861, 1736, 1461, 1374, 1250, 1110, 968, 838 cm-1.
Ejemplo 65 - Síntesis de (Z)-7-((1ft,2ft,3ft)-3-[(ferí-butildimetilsilil)oxi]-2-{(S,£)-3-[(ferí-butildimetilsilil)]oxioct-1-en-1-il}-5-oxociclopentil)hept-5-enoato de isopropilo (10.9a)

Siguiendo los procedimientos descritos en el Ejemplo 1, excepto en una escala mayor utilizando 2 (0,30 g, 0,79 mmol) y ácido (S,E){3-[(terc-butildimetilsilil)oxi]oct-1-en-1-il}borónico (3.9, 0,34 g, 1,19 mmol), la mezcla cruda de la reacción se purificó por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título (10.9a, 0,48 g, 98 %).
[a]26D -29,6 (c 1,00, CHCla); 1H RMN (400 MHz): 5 0,02 (s, 3H), 0,043 (s, 3H), 0,049 (s, 3H), 0,052 (s, 3H), 0,87 (s, 9H), 0,89 (s, 9H), 1,22 (d, J = 6,4 Hz, 6 H), 1,23-1,35 (m, 8H), 1,35-1,52 (m, 3H), 1,66 (quintet, J = 7,5 Hz, 2H), 1,98 2,10 (m, 3H), 2,15 (dd, J = 8,4, 18,2 Hz, 1H), 2,25 (t, J = 7,6 Hz, 2H), 2,27-2,53 (m, 3H), 2,63 (ddd, J = 1,1, 7,0, 18,2 Hz, 1H), 3,98-4,13 (m, 2H), 4,99 (septeto, J = 6,4 Hz, 1H), 5,27-5,44 (m, 2H), 5,45-5,63 (m, 2H); 13C RMN (100 MHz): 5 -4,71 (CH3), -4,65 (CH3), -4,59 (CH3), -4,3 (CH3), 14,0 (CH3), 18,0 (C), 18,2 (C), 21,8 (CH3), 22,6 (CH2), 24,8 (CH2), 25,1 (CH2), 25,2 (CH2), 25,8 (CH3), 25,9 (CH3), 26,7 (CH2), 31,9 (CH2), 34,1 (CH2), 38,6 (CH2), 47,7 (CH2), 52,7 (CH), 53,9 (CH), 67,4 (CH), 72,6 (CH), 73,3 (CH), 126,6 (CH), 128,6 (CH), 130,8 (CH), 136,5 (CH), 173,1 (C), 215,4 (C); HRMS (ESI-QTOF) calculado para [C35H66O5S¡2 NH4]+= 640,4787, encontrado 640,4775; FTIR (KBr, puro) 2937, 2861, 1738, 1577, 1467, 1371, 1250, 1107, 835, 774 cm-1.
Ejemplo 66 - Síntesis de (Z)-7-((1ft,2ft,3ft,5Sj-3-[(ferí-butildimetilsilil)oxi]-2-{(S,E)-3-[(tertbutildimetilsilil)oxi]oct-1-en-1-il}-5-hidroxiciclopentil)hept-5-enoato de isopropilo (11.9a)

A una solución de THF (6,4 ml) de 10.9a (80 mg, 0,13 mmol) se agregó una solución de L-Selectride® (0,15 ml, 1,0 M en THF, 0,15 mmol) a -78 °C. La mezcla se agitó a -78 °C durante 20 minutos y se concentró directamente a temperatura ambiente para dar un residuo que se purificó por cromatografía de columna (eluyendo con 1: 8 (v/v) EtOAc-hexanos) para proporcionar el compuesto del título (11.9a %, 61 mg, 76 %).
[a]23D-6,3 (c 1,00, CHCla); 1H RMN (400 MHz): 5 0,01 (s, 3H), 0,04 (s, 3H), 0,046 (s, 3H), 0,049 (s, 3H), 0,87 (s, 9H), 0,88 (s, 9H), 1,22 (d, J = 6,2 Hz, 6H), 1,24-1,52 (m, 10H), 1,67 (quintet, J = 7,5 Hz, 2H), 1,72-1,92 (m, 2H), 2,04-2,21 (m, 3H), 2,22-2,39 (m, 4H), 2,68 (d, J = 9,2 Hz, 1H), 3,96-4,15 (m, 3H), 4,99 (septeto, J = 6,2 Hz, 1H), 5,26-5,52 (m, 4H); 13C RMN (100 MHz): 5 -4,9 (CH3), -4,8 (CH3), -4,6 (CH3), -4,3 (CH3), 14,0 (CH3), 17,8 (C), 18,2 (C), 21,8 (CH3), 22,6 (CH2), 25,0 (CH2), 25,8 (CH3), 25,9 (CH3), 26,6 (CH2), 26,7 (CH2), 31,8 (CH2), 34,1 (CH2), 38,6 (CH2), 42,9 (CH2), 51,8 (CH), 56,4 (CH), 67,3 (CH), 73,2 (CH), 74,7 (CH), 80,0 (CH), 129,2 (CH), 129,5 (CH), 130,8 (CH), 134,4 (CH), 173,2 (C); LRMS (ESI): calculado para [C35H6sO5Si2= 624,5, encontrado 624,5; HRMS (ESI-QTOF) calculado para [C35H6sO5Si2 H]+= 625,4678, encontrado 625,4676; FTIR (KBr, puro) 3433, 2936, 1722, 1636, 1457, 1250, 1088, 832, 659, 503 cm-1.
Ejemplo 67 - Síntesis ácido (Z)-7-((1ft,2ft,3ft,5S)-3-[(ferí-butNdimetNsNN)oxi]-2-{(S,E)-3-[(feríbutildimetilsilil)oxi]oct-1-en-1-il}-5-hidroxiciclopentil)hept-5-enoico (12.9a)

A una solución del compuesto 11.9a (1,70 g, 2,7 mmol) en MeOH (20 ml) se le añadió NaOMe-MeOH al 25 % (9,3 ml, 41 mmol) a temperatura ambiente y la mezcla se agitó hasta que se alcanzó la conversión completa como se indica por t Lc (1: 4 EtOAc-n-heptano). Se añadió NaOH acuoso al 10 % (10 ml) y la mezcla se calentó a 50 °C durante 2 horas. La solución se enfrió hasta temperatura ambiente y se añadió un 10 % de ácido cítrico acuoso (60 ml). Las capas se separaron y la fase acuosa se extrajo con EtOAc (60 ml x 3). Las capas orgánicas se combinaron, se lavaron con salmuera (150 ml), se secaron sobre MgSO4 (4,0 g), se filtraron y se concentraron bajo presión reducida. El residuo resultante se purificó mediante una purificación en columna sobre gel de sílice (51 g de gel de sílice; eluyendo con 1:2 (v/v) de EtOAc-n-heptano (800 ml)) para dar el compuesto del título 12.9a (1,42 g, 2,44 mmol, 90 % de rendimiento).
[a]20D+12,09 (c 1,0, CH2Cl2); 1H RMN (400 MHz, CDCls): 5 5,52-5,44 (m, 2H), 5,40-5,32 (m, 2H), 4,14 (s, 1H, br), 4,06 (m, 2H), 2,37-2,28 (m, 4H), 2,27-2,19 (m, 3H) 1,93-1,83 (m, 2H), 1,73 (m, 2H), 1,50-1,10 (m, 3H), 1,27 (m, 7H), 0,89 (m, 21H), 0,07-0,03 (m, 12H); 13C RMN (100 MHz, CDCls): 5 179,1, 134,4, 130,7, 129,6, 129,0, 80,0, 74,7, 73,2, 56,4, 51,8, 42,8, 38,5, 33,4, 31,8, 26,6, 26,5, 25,9 (x3), 25,8 (x3), 25,0, 24,6, 22,6, 18,2, 17,8, 14,0, -4,3, -4,6, -4,8, -4,9; HRMS (ESI-QTOF) calculado para [C32H62OaSi2 Na]+= 605,4028, encontrado 605,4018; FTIR (KBr, puro) 2955, 2928, 2856, 1710, 1472, 1463, 1361, 1251, 1082, 870, 835 cm-1.
Ejemplo 68 - Síntesis de dinoprost (PGF2a; 12.9)

Se añadió HCl acuoso (3 N, 6,3 ml, 19 mmol) a una solución agitada del compuesto 12.9a (1,1 g, 1,9 mmol) en THF (19 ml) a temperatura ambiente. Después de 6 horas, la solución de reacción se neutralizó con una solución sat. aq. NaHCO3 (30 ml) y se extrajo con EtOAc (40 ml x 2). Las capas orgánicas se combinaron, se lavaron con NaCl saturado acuoso (50 ml), se secó sobre MgSO4, se filtró y se concentró bajo presión reducida. El producto se purificó por cromatografía de columna sobre gel de sílice (33 g de gel de sílice; se eluyó con 1:20 (v/v) MeOH- CH2Cl2 (300 ml) y luego 1:5 (500 ml) para dar dinoprost como un aceite incoloro (12.9, 595 mg, 89 %).
[a]20D 23,7 (c 0,50, THF); 1H RMN (400 MHz, MeOD): 5 5,55-5,43 (m, 3H), 5,38-5,31 (m, 1H), 4,10 (td, J = 5,6, 2,0 Hz, 1H), 4,01 (dd, J= 12,8, 6,4 Hz, 1H), 3,83 (ddd, J = 7,6, 5,6, 5,2 Hz, 1H), 2,36-2,26 (m, 1H), 2,26-2,20 (m, 3H), 2,18-2,15 (m, 1H), 2,14-2,07 (m, 3H), 1,69-1,58 (m, 4H), 1,57-1,53 (m, 2H), 1,51-1,29 (m, 6H), 0,91 (t, J= 6,8 Hz, 3H); 13C RMN (100 MHz, MeOD): 5 178,5, 136,6, 134,3, 130,5, 130,4, 77,9, 74,1,72,3, 56,2, 50,9, 44,4, 38,5, 35,0, 33,1, 27,8, 26,5, 26,3 (x2), 23,9, 14,6; HRMS (ESI-QTOF) calculado para [C20H34O5 Na]+= 377,2298, encontrado 377,2298; FTIR (KBr, puro) 3346, 3006, 2954, 2930, 2858, 1708, 1550, 1456, 1409, 1237, 1081, 1053, 1025, 969 cm-1.
Ejemplo 69 - Síntesis de (Z)-7-{(1ft,2S,3ft)-3-[(ferí-butildimetilsilil)oxi]-2-(ciclohex-1-en-1-il)-5-oxociclopentil}hept-5-enoato de isopropilo (10.20)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando ácido ciclohex-1-en-1-ilborónico (28 mg, 0,225 mmol). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) de acetona-hexanos) produciendo el compuesto del título (10.20, 26 mg, 38 %).
1H RMN (400 MHz, CDCls): 5 0,02 (s, 3H), 0,03 (s, 3H), 0,87 (s, 9H), 1,22 (d, J = 6,2 Hz, 6H), 1,58-1,70 (m, 6H), 1,85-1,95 (m, 2H), 1,98-2,08 (m, 4H), 2,09-2,31 (m, 6H), 2,38 (dd, J = 8,5, 12,4 Hz, 1H), 2,64 (dd, J = 7,2, 18,5 Hz, 1H), 4,11 (q, J = 8,4 Hz, 1H), 5,00 (septeto, J = 6,2 Hz, 1H), 5,29-5,52 (m, 2H), 5,45-5,60 (m, 1H); HRMS (ESI-TOF) calculado para [C27H46O4Si h]+= 463,3238, encontrado 463,3240.
Ejemplo 70 - Síntesis de (£)-7-{3-[(fert-butildimetilsilil)oxi]-5-oxo-2-estirilciclopentil}heptanoato de metilo (Iba)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando 7-{3-[(tercbutildimetilsilil)oxi]-5-oxociclopent-1-en-1-il}heptanoato de metilo (IIba, 53 mg, 0,15 mmol) . La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) de acetona-hexanos) proporcionando el compuesto del título (Iba, 36 mg, 53 %).
1H RMN (400 MHz, CDCls): 5 0,01 (s, 3H), 0,02 (s, 3H), 0,86 (s, 9H), 1,22-1,33 (m, 6H), 1,35-1,48 (m, 1H), 1,50-1,70 (m, 3H), 2,02-2,12 (m, 1H), 2,19-2,30 (m, 3H), 2,54-2,73 (m, 2H), 3,64 (s, 3H), 4,11 (q, J = 8,5 Hz, 1H), 6,05 (dd, J =
8 ,6 , 15,8 Hz, 1H), 6,52 (d, J = 15,8 Hz, 1H), 7,22-7,26 (m, 1H), 7,30-7,38 (m, 4H); HRMS (ESI-TOF) calculado para [C27H42O4SÍ H]+= 459,2925, encontrado 459,2930.
Ejemplo 71 - Síntesis de (E)-7-{5-oxo-2-estiril-3-[(tetrahidro-2H-piran-2-il)oxi]cidopentil}heptanoato de metilo (Ibb)

Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando 7- {5-oxo-3-[(tetrahidro-2H-piran-2-il)oxi]ciclopent-1-en-1-il}heptanoato de metilo (IIbb , 49 mg, 0,15 mmol). La mezcla de producto crudo se purificó luego por cromatografía de columna (eluyendo con 1:80 (v/v) acetona-hexanos) proporcionando el compuesto del título (Ibb, 20 mg, 31 %) como una mezcla 1:1 de isómeros diastereoisómeros en el centro de carbono acetal.
1H RMN (400 MHz, CDCls): 5 1,21-1,36 (m, 6H), 1,38-1,82 (m, 9H), 2,03-2,12 (m, 1H), 2,17 (dd, J = 8,8, 18,4 Hz, 1H), 2,26 (td, J = 1,5, 7,5 Hz, 2H), 2,36 (dd, J = 9,0, 18,7 Hz, 1H), 2,64-2,87 (m, 2H), 3,37-3,52 (m, 1H), 3,64 (s, 3H), 3,77-3,88 (m, 1H), 4,11 (q, J = 8,6 Hz, 0,5 H), 4,27 (q, J = 8,7 Hz, 0,5 H), 4,68-4,77 (m, 1H), 6,09-6,23 (m, 1H), 6,50 6,63 (m, 1H), 7,20-7,25 (m, 1H), 7,29-7,42 (m, 4H); HRMS (ESI-TOF) calculado para [C26H36O5 Na]+= 451,2455, encontrado 451,2459.
Ejemplo 72 - Síntesis de (Z)-7-[(1ft,2ft,3ft)-3-hidroxi-5-oxo-2-((E)-estiril)ciclopentil]hept-5-enoato de isopropilo (Iaa)

Una solución de (R,Z)-7-(3-hidroxi-5-oxociclopent-1-en-1-il) hept-5-enoato de isopropilo (IIaa, 50 mg, 0,19 mmol), 3.1 (45 mg, 0,22) mmol), [RhCl(1,5-ciclooctadieno)]2 (1,4 mg, 2,8 pmol) y KOH acuoso (11,9 pl, KOH acuoso 3,0 M, 36 pmmol) en MeOH (1,0 ml) se agitó bajo irradiación de microondas (CEM, Discover S; Milestone, Startsynth; la temperatura se fijó a 30 °C). El residuo se purificó por cromatografía de columna (eluyendo con 1:20 (v/v) acetonahexanos) proporcionando el compuesto del título (Iaa, 27 mg, 39 %).
1H RMN (400 MHz, CDCls): 5 1,19 (d, J = 6,2 Hz, 6H), 1,58-1,73 (m, 2H), 1,94 (br, 1H), 2,04 (q, J = 7,2 Hz, 2H), 2,20 (t, J = 7,6 Hz, 2H), 2,32-2,54 (m, 4H), 2,56-2,65 (m, 1H), 2,72-2,81 (m, 1H), 4,48 (t, J = 4,2 Hz, 1H), 4,96 (septeto, J = 6,2 Hz, 1H), 5,25-5,45 (m, 2H), 6,34 (dd, J = 7,9, 16,1 Hz, 1H), 6,57 (d, J = 16,1 Hz, 1H), 7,23-7,28 (m, 1H), 7,31 7,41 (m, 4H); HRMS (EI) calculado para [C23H30O4]+= 370,2139, encontrado 370,2141.
Ejemplo 73 - Síntesis de (E)-7-[3-(aliloxi)-5-oxo-2-estirilciclopentil]heptanoato de metilo (Ibc)
Siguiendo el procedimiento descrito en el Ejemplo 1, el compuesto del título se sintetizó utilizando 7- [3-(aliloxi)-5-oxociclopent-1-en-1-il]heptanoato de metilo (IIbc, 42 mg, 0,15 mmol). La mezcla de producto crudo se filtró a través de una columna de gel de sílice y luego se analizó mediante espectroscopia de 1H RMN que mostró un rendimiento del 2 % del compuesto del título Ibc.
HRMS (ESI-TOF) calculado para [C24H32O4 H]+= 385.2373, encontrado 385,2375.
Claims (13)
1. Un procedimiento de preparación de un compuesto de 4-oxi-ciclopentan-l-ona 2,3-disustituido de fórmula I

el procedimiento comprende poner en contacto un compuesto4-oxi-ciclopent-2-en-1-onas sustituido en 2 de fórmula II

con un compuesto de fórmula III

en un disolvente con un aditivo metálico, opcionalmente en presencia de un aditivo básico,
para dar el compuesto de fórmula I;
en la que
R1, R3, R4 y R5 representan independientemente hidrógeno, arilo, heteroarilo, alquilo, arilalquilo, ariloxialquilo, alquenilo o alquinilo; o R3 y R5 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, que tiene opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S; o R3 y R4 se toman juntos para formar un anillo carbocíclico de 5 a 7 miembros, teniendo opcionalmente uno o dos heteroátomos como vértices de anillo, en la que los heteroátomos se seleccionan del grupo que consiste en O, N y S, y en la que cada uno de R1, R3, R4 y R5 están opcionalmente sustituidos con uno a tres miembros seleccionados del grupo que consiste en halógeno, hidroxi, alquilo C1-4, haloalquilo C1-4, alcoxi C1-4, sililoxi, ariloxi, aciloxi, un anillo heterocíclico de 5 a 7 miembros, oxo, COOH, CONH2, CONH alquilo C1-4, C(O)o Ch2 arilo C6-10, C(O)O arilo C6-10 y C(O)O alquilo C1-4;
R2 representa hidrógeno o un grupo protector de hidroxilo;
X representa un grupo que contiene boro.
2. El procedimiento de la reivindicación 1, en el que el compuesto de fórmula I es

3. El procedimiento de la reivindicación 1, en el que R3 es alquilo, arilo, arilalquilo o ariloxialquilo, cada uno de los cuales está opcionalmente sustituido con uno a tres miembros seleccionados del grupo que consiste en alquilo C1-4, alcoxi C1-4, sililoxi, ariloxi aciloxi, tetrahidropiranilo (THP), trifluorometilo y flúor.
4. El procedimiento de la reivindicación 1, en el que el disolvente se selecciona del grupo que consiste en agua, metanol, etanol, alcohol isopropílico, alcohol isobutílico, 1,4-dioxano, tolueno, tetrahidrofurano (THF), 2-metiltetrahidrofurano (2-Me-THF), diglima, acetonitrilo, N-metilpirrolidona, N,N-dimetilformamida (DMF), N,N-dimetilacetamida (DmAC), etilenglicol y combinaciones de los mismos, preferiblemente metanol.
5. El procedimiento de la reivindicación 1, en el que el aditivo metálico se selecciona del grupo que consiste en compuestos de rodio, compuestos de cobalto, compuestos de níquel y combinaciones de los mismos,
preferiblemente, en el que el compuesto de rodio es un compuesto de rodio (I) seleccionado del grupo que consiste en [RhCl(1,5-ciclooctadiene)]2, [RhCl(C2H4)2]2, [RhCl(C2H4)2]2 con un aditivo ligando dieno, [RhCl(norbornadieno)]2, [RhOH(1,5-ciclooctadieno)]2 y combinaciones de los mismos, en particular en el que el compuesto de rodio (I) se selecciona del grupo que consiste en [RhCl(1,5-ciclooctadieno)]2 y [RhOH(1,5-ciclooctadieno)]2.
6. El procedimiento de la reivindicación 1, en el que el aditivo básico se selecciona del grupo que consiste en K KHF2, #-BuOLi, t-BuONa, f-BuOK, K3PO4, K2CO3, Cs2COs, LiOH, NaOH, KOH, CsOH, KF, CsF, NaHCOs, KH2PO4, 1,3-diaminopropano, t-BuNH2, /-Pr2NH, piperidina, Et3N, 2,6-lutidina y combinaciones de los mismos, preferiblemente KOH.
7. El procedimiento de la reivindicación 1, en el que el grupo protector de hidroxilo se selecciona del grupo que consiste en tetrahidropiranilo (THP), metoximetilo (MOM), [2-(trimetilsilil) etoxi]metilo (SEM), trialquilsililo, triarilsilsilo, diarilalquilsililo, bencilo, 4-metoxibencilo (PMB), alquilcarbonilo, arilcarbonilo y alilo, preferiblemente tercbutildimetilsililo (TBS).
8. El procedimiento de la reivindicación 1, en el que el grupo X que contiene boro se selecciona del grupo que consiste en B(OH)2, B(OR)2 en el que R es un grupo alquilo o un grupo arilo, BR2 en el que R es un grupo alquilo, BR2 en el que R es un grupo vinilo, BR2 en el que R es un grupo carboxilato, BR en el que R es un grupo carboxilato bidentado, BR2 en el que R es un grupo ariloxi, BR en el que R es un grupo ariloxi bidentado, un grupo 9-borabiciclo (3.3.1) nonano (9-BBN), BF3M en el que M es un ion metálico, BF3M en el que M es un ion amonio o fosfonio, y BR3M en el que R es un grupo vinilo y en el que M es un ion metálico o es un ión amonio o fosfonio, preferiblemente en en el que el grupo X que contiene boro se selecciona del grupo que consiste en B(OH)2 y BF3M en el que M es un ion metálico.
9. El procedimiento de la reivindicación 1 se realiza a una temperatura de 0 a 80 °C.
10. El procedimiento de la reivindicación 1, en el que el aditivo metálico se usa en cantidades subestequiométricas
11. El procedimiento de la reivindicación 1, en el que el aditivo básico se usa en cantidades subestequiométricas.
12. El procedimiento de la reivindicación 1, que comprende además convertir el compuesto de 4-oxi-ciclopentan-1-ona 2,3-disustituido de fórmula I en travoprost, bimatoprost, lubiprostona, dinoprost, dinoprostona, tafluprost, carboprost, alprostadil, latanoprost o isopropilo de unoprostona.
13. El procedimiento de la reivindicación 1, en el que el compuesto de 4-oxiciclopentan-1-ona 2,3-disustituido de fórmula I se selecciona del grupo que consiste en el compuesto de fórmula 10.9a.
el compuesto de fórmula 10.17

el compuesto de fórmula 10.18.

el compuesto de fórmula 10.7
Ċ

y el compuesto de fórmula 10.19

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462022797P | 2014-07-10 | 2014-07-10 | |
| PCT/IB2015/055212 WO2016005943A1 (en) | 2014-07-10 | 2015-07-09 | Metal-catalyzed asymmetric 1,4-conjugate addition of vinylboron compounds to 2-substituted-4-oxy- cyclopent-2-en-l-ones yielding prostaglandins and prostaglandin analogs |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2724112T3 true ES2724112T3 (es) | 2019-09-06 |
Family
ID=55063668
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15819728T Active ES2724112T3 (es) | 2014-07-10 | 2015-07-09 | Adición del 1,4-conjugado asimétrico catalizado por metal de compuestos de vinilboro a 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 que producen prostaglandinas y análogos de prostaglandinas |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9670234B2 (es) |
| EP (1) | EP3166918B1 (es) |
| JP (1) | JP6643308B2 (es) |
| KR (1) | KR20170028990A (es) |
| CN (1) | CN105254657B (es) |
| AU (1) | AU2015287220B2 (es) |
| CA (1) | CA2952927C (es) |
| ES (1) | ES2724112T3 (es) |
| IL (1) | IL249632B (es) |
| NZ (1) | NZ727773A (es) |
| TW (1) | TWI541227B (es) |
| WO (1) | WO2016005943A1 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU231185B1 (hu) | 2017-07-11 | 2021-07-28 | CHINOIN Gyógyszer és Vegyészeti Termékek Gyára Zrt. | Eljárás Misoprostol előállítására és tisztítására |
| US10457623B1 (en) * | 2018-07-13 | 2019-10-29 | Chirogate International Inc. | Process for the preparation of Lubiprostone and intermediates thereof |
| US10253011B1 (en) * | 2018-07-13 | 2019-04-09 | Chirogate International Inc. | Lubiprostone crystals and methods for preparing the same |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008021029A2 (en) * | 2006-08-09 | 2008-02-21 | Merck & Co., Inc. | Process for making lactam tachykinin receptor antagonists |
| AR071312A1 (es) | 2008-04-09 | 2010-06-09 | Scinopharm Taiwan Ltd | Proceso para la preparacion de analogos de prostaglandina y sus intermediarios |
| KR101045935B1 (ko) * | 2009-03-11 | 2011-07-01 | 연성정밀화학(주) | 프로스타글란딘 유도체의 제조방법 |
| IN2013MN00733A (es) * | 2010-10-15 | 2015-06-12 | Scinopharm Kunshan Biochemical Technology Co Ltd |
-
2015
- 2015-07-03 CN CN201510388413.6A patent/CN105254657B/zh not_active Expired - Fee Related
- 2015-07-09 EP EP15819728.5A patent/EP3166918B1/en not_active Not-in-force
- 2015-07-09 WO PCT/IB2015/055212 patent/WO2016005943A1/en active Application Filing
- 2015-07-09 JP JP2017500379A patent/JP6643308B2/ja not_active Expired - Fee Related
- 2015-07-09 AU AU2015287220A patent/AU2015287220B2/en not_active Ceased
- 2015-07-09 US US14/795,506 patent/US9670234B2/en active Active
- 2015-07-09 TW TW104122398A patent/TWI541227B/zh not_active IP Right Cessation
- 2015-07-09 KR KR1020177003580A patent/KR20170028990A/ko not_active Application Discontinuation
- 2015-07-09 CA CA2952927A patent/CA2952927C/en not_active Expired - Fee Related
- 2015-07-09 ES ES15819728T patent/ES2724112T3/es active Active
- 2015-07-09 NZ NZ727773A patent/NZ727773A/en not_active IP Right Cessation
-
2016
- 2016-12-18 IL IL249632A patent/IL249632B/en active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| WO2016005943A1 (en) | 2016-01-14 |
| EP3166918B1 (en) | 2019-02-20 |
| TWI541227B (zh) | 2016-07-11 |
| CA2952927A1 (en) | 2016-01-14 |
| CN105254657B (zh) | 2018-06-15 |
| US9670234B2 (en) | 2017-06-06 |
| CA2952927C (en) | 2018-12-18 |
| AU2015287220A1 (en) | 2017-01-12 |
| JP2017520587A (ja) | 2017-07-27 |
| AU2015287220B2 (en) | 2019-08-01 |
| IL249632B (en) | 2020-04-30 |
| EP3166918A1 (en) | 2017-05-17 |
| JP6643308B2 (ja) | 2020-02-12 |
| IL249632A0 (en) | 2017-02-28 |
| US20160009740A1 (en) | 2016-01-14 |
| WO2016005943A8 (en) | 2017-02-09 |
| TW201602074A (zh) | 2016-01-16 |
| EP3166918A4 (en) | 2018-02-21 |
| NZ727773A (en) | 2020-07-31 |
| KR20170028990A (ko) | 2017-03-14 |
| CN105254657A (zh) | 2016-01-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2724112T3 (es) | Adición del 1,4-conjugado asimétrico catalizado por metal de compuestos de vinilboro a 4-oxi-ciclopent-2-en-1-onas sustituidos en 2 que producen prostaglandinas y análogos de prostaglandinas | |
| KR20160070772A (ko) | 복분해를 사용하는 프로스타글란딘 및 프로스타글란딘 중간체의 합성 경로 | |
| CA2750487A1 (en) | Methods of making lubiprostone and intermediates thereof | |
| US7897793B2 (en) | Process for preparation of 13,14-dihydro-PGF2 alpha derivatives | |
| ES2746749T3 (es) | (6R,10R)-6,10,14-Trimetilpentadecan-2-ona preparada a partir de 6,10-dimetilundec-5-en-2-ona o 6,10-dimetilundeca-5,9-dien-2-ona | |
| ES2875551T3 (es) | Método para preparar (R)-4-n-propil-dihidrofuran-2(3H)-ona ópticamente pura | |
| EP1082299B1 (en) | C11 oxymyl and hydroxylamino prostaglandins useful as medicaments | |
| JP6392749B2 (ja) | 化合物と方法 | |
| US5504107A (en) | Optically pure 4-alkenyl- or 4-alkanyl-2-hydroxytetronic acids and pharmaceutical use thereof | |
| CA2324590C (en) | C11 oxymyl and hydroxylamino prostaglandins useful as fp agonists | |
| US9242950B2 (en) | Method for preparing a fatty acid derivative | |
| US20140114086A1 (en) | Clinprost, Isocarbacyclin And Analogs Thereof And Methods Of Making The Same | |
| CN111263750B (zh) | 前列腺素衍生物的制造方法 | |
| CA2327332A1 (en) | Novel process | |
| US20150031898A1 (en) | Process for preparation of prostaglandin f2 alpha analogues | |
| WO2021070658A1 (ja) | シクロペンタン化合物の製造方法、ラクトン化合物の製造方法、ジオール化合物の製造方法、および化合物 | |
| EP2380871B1 (en) | A process for the preparation of isoserine derivatives | |
| PL224738B1 (pl) | Sposób wytwarzania analogów prostaglandyny F2α o strukturze 13,14-en-15-olu | |
| WO2013164729A1 (en) | An improved and scalable process for preparation of prostaglandin derivatives and intermediates thereof | |
| JPH0649669B2 (ja) | 光学活性アルコールの製法 | |
| JP2010138078A (ja) | 2,3−ジヒドロ−チエノ[3,4−b]フラン誘導体の製造方法、及びそれに用いられる新規化合物 |