DE60209484T2 - Verfahren zum aktivieren eines getragenen katalysators der eine kobalthaltigen verbindung enthält - Google Patents
Verfahren zum aktivieren eines getragenen katalysators der eine kobalthaltigen verbindung enthält Download PDFInfo
- Publication number
- DE60209484T2 DE60209484T2 DE60209484T DE60209484T DE60209484T2 DE 60209484 T2 DE60209484 T2 DE 60209484T2 DE 60209484 T DE60209484 T DE 60209484T DE 60209484 T DE60209484 T DE 60209484T DE 60209484 T2 DE60209484 T2 DE 60209484T2
- Authority
- DE
- Germany
- Prior art keywords
- catalyst
- hydrocarbon
- gas
- catalyst precursor
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 92
- 238000000034 method Methods 0.000 title claims abstract description 47
- 230000003213 activating effect Effects 0.000 title abstract description 7
- 150000001875 compounds Chemical class 0.000 title description 5
- 229930195733 hydrocarbon Natural products 0.000 claims abstract description 48
- 150000002430 hydrocarbons Chemical class 0.000 claims abstract description 48
- 239000012018 catalyst precursor Substances 0.000 claims abstract description 45
- 239000004215 Carbon black (E152) Substances 0.000 claims abstract description 35
- 230000008569 process Effects 0.000 claims abstract description 21
- 150000001869 cobalt compounds Chemical class 0.000 claims abstract description 12
- 238000002360 preparation method Methods 0.000 claims abstract description 5
- 238000006243 chemical reaction Methods 0.000 claims description 61
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 claims description 40
- 239000000203 mixture Substances 0.000 claims description 38
- 229910052760 oxygen Inorganic materials 0.000 claims description 29
- 229910052799 carbon Inorganic materials 0.000 claims description 22
- 239000007789 gas Substances 0.000 claims description 22
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 20
- 239000001301 oxygen Substances 0.000 claims description 20
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 19
- 229910017052 cobalt Inorganic materials 0.000 claims description 17
- 239000010941 cobalt Substances 0.000 claims description 17
- 229910052739 hydrogen Inorganic materials 0.000 claims description 17
- 239000001257 hydrogen Substances 0.000 claims description 17
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 16
- 230000004913 activation Effects 0.000 claims description 14
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims description 14
- 229910002091 carbon monoxide Inorganic materials 0.000 claims description 11
- 230000003647 oxidation Effects 0.000 claims description 11
- 238000007254 oxidation reaction Methods 0.000 claims description 11
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 claims description 8
- 150000001868 cobalt Chemical class 0.000 claims description 8
- 238000003786 synthesis reaction Methods 0.000 claims description 8
- -1 β-aluminate Chemical compound 0.000 claims description 8
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 claims description 7
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 claims description 7
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 claims description 6
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 claims description 6
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 claims description 6
- 239000012298 atmosphere Substances 0.000 claims description 6
- 230000015572 biosynthetic process Effects 0.000 claims description 6
- 229910052786 argon Inorganic materials 0.000 claims description 5
- UFMZWBIQTDUYBN-UHFFFAOYSA-N cobalt dinitrate Chemical compound [Co+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O UFMZWBIQTDUYBN-UHFFFAOYSA-N 0.000 claims description 5
- 238000005470 impregnation Methods 0.000 claims description 5
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 4
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 4
- 229910001981 cobalt nitrate Inorganic materials 0.000 claims description 4
- 239000011261 inert gas Substances 0.000 claims description 4
- 239000007858 starting material Substances 0.000 claims description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 3
- 229910021536 Zeolite Inorganic materials 0.000 claims description 3
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 3
- 239000001273 butane Substances 0.000 claims description 3
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 claims description 3
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 3
- 229910052734 helium Inorganic materials 0.000 claims description 3
- 238000005984 hydrogenation reaction Methods 0.000 claims description 3
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 claims description 3
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 claims description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 claims description 3
- 239000001294 propane Substances 0.000 claims description 3
- 239000000377 silicon dioxide Substances 0.000 claims description 3
- 239000011029 spinel Substances 0.000 claims description 3
- 229910052596 spinel Inorganic materials 0.000 claims description 3
- 239000010457 zeolite Substances 0.000 claims description 3
- 239000000395 magnesium oxide Substances 0.000 claims description 2
- 238000003980 solgel method Methods 0.000 claims description 2
- 239000001307 helium Substances 0.000 claims 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 claims 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 claims 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 claims 1
- 239000004408 titanium dioxide Substances 0.000 claims 1
- 229910001928 zirconium oxide Inorganic materials 0.000 claims 1
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 18
- 239000000047 product Substances 0.000 description 13
- 238000009826 distribution Methods 0.000 description 11
- 239000003607 modifier Substances 0.000 description 11
- 239000007800 oxidant agent Substances 0.000 description 10
- 238000001354 calcination Methods 0.000 description 8
- 239000002243 precursor Substances 0.000 description 8
- 230000000694 effects Effects 0.000 description 7
- 239000010453 quartz Substances 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- MWUXSHHQAYIFBG-UHFFFAOYSA-N nitrogen oxide Inorganic materials O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 230000008901 benefit Effects 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 230000008021 deposition Effects 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000001590 oxidative effect Effects 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 3
- 229910021193 La 2 O 3 Inorganic materials 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000012876 carrier material Substances 0.000 description 3
- 238000006555 catalytic reaction Methods 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 229910052723 transition metal Inorganic materials 0.000 description 3
- 150000003624 transition metals Chemical class 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- 229910000873 Beta-alumina solid electrolyte Inorganic materials 0.000 description 2
- 229910020599 Co 3 O 4 Inorganic materials 0.000 description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 2
- 229910004298 SiO 2 Inorganic materials 0.000 description 2
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 230000002745 absorbent Effects 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- HSJPMRKMPBAUAU-UHFFFAOYSA-N cerium(3+);trinitrate Chemical compound [Ce+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O HSJPMRKMPBAUAU-UHFFFAOYSA-N 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 229910052761 rare earth metal Inorganic materials 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- DHEQXMRUPNDRPG-UHFFFAOYSA-N strontium nitrate Chemical compound [Sr+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O DHEQXMRUPNDRPG-UHFFFAOYSA-N 0.000 description 2
- XTQHKBHJIVJGKJ-UHFFFAOYSA-N sulfur monoxide Chemical class S=O XTQHKBHJIVJGKJ-UHFFFAOYSA-N 0.000 description 2
- 229910052815 sulfur oxide Inorganic materials 0.000 description 2
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- 229910013553 LiNO Inorganic materials 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- IWOUKMZUPDVPGQ-UHFFFAOYSA-N barium nitrate Inorganic materials [Ba+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O IWOUKMZUPDVPGQ-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- ZCCIPPOKBCJFDN-UHFFFAOYSA-N calcium nitrate Inorganic materials [Ca+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O ZCCIPPOKBCJFDN-UHFFFAOYSA-N 0.000 description 1
- 229910021386 carbon form Inorganic materials 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229910000428 cobalt oxide Inorganic materials 0.000 description 1
- IVMYJDGYRUAWML-UHFFFAOYSA-N cobalt(ii) oxide Chemical compound [Co]=O IVMYJDGYRUAWML-UHFFFAOYSA-N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229910052746 lanthanum Inorganic materials 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- YIXJRHPUWRPCBB-UHFFFAOYSA-N magnesium nitrate Inorganic materials [Mg+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O YIXJRHPUWRPCBB-UHFFFAOYSA-N 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 150000001247 metal acetylides Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003345 natural gas Substances 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229910001392 phosphorus oxide Inorganic materials 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical class [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 150000002910 rare earth metals Chemical class 0.000 description 1
- 238000002407 reforming Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 238000000629 steam reforming Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- WOZZOSDBXABUFO-UHFFFAOYSA-N tri(butan-2-yloxy)alumane Chemical compound [Al+3].CCC(C)[O-].CCC(C)[O-].CCC(C)[O-] WOZZOSDBXABUFO-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 238000013022 venting Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2/00—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon
- C10G2/30—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen
- C10G2/32—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen with the use of catalysts
- C10G2/33—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen with the use of catalysts characterised by the catalyst used
- C10G2/331—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen with the use of catalysts characterised by the catalyst used containing group VIII-metals
- C10G2/332—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen with the use of catalysts characterised by the catalyst used containing group VIII-metals of the iron-group
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/75—Cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/78—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with alkali- or alkaline earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/83—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/12—Oxidising
- B01J37/14—Oxidising with gases containing free oxygen
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
- B01J37/18—Reducing with gases containing free hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen
- C01B3/02—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen
- C01B3/32—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air
- C01B3/34—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air by reaction of hydrocarbons with gasifying agents
- C01B3/38—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air by reaction of hydrocarbons with gasifying agents using catalysts
- C01B3/386—Catalytic partial combustion
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen
- C01B3/02—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen
- C01B3/32—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air
- C01B3/34—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air by reaction of hydrocarbons with gasifying agents
- C01B3/38—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air by reaction of hydrocarbons with gasifying agents using catalysts
- C01B3/40—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide, air by reaction of hydrocarbons with gasifying agents using catalysts characterised by the catalyst
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/02—Processes for making hydrogen or synthesis gas
- C01B2203/025—Processes for making hydrogen or synthesis gas containing a partial oxidation step
- C01B2203/0261—Processes for making hydrogen or synthesis gas containing a partial oxidation step containing a catalytic partial oxidation step [CPO]
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1052—Nickel or cobalt catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1082—Composition of support materials
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1094—Promotors or activators
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1235—Hydrocarbons
- C01B2203/1241—Natural gas or methane
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Combustion & Propulsion (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Inorganic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
- Die vorliegende Erfindung betrifft einen Katalysator und ein Verfahren zur Herstellung eines Katalysators, insbesondere eines Katalysators zur Verwendung bei der partiellen Oxidation eines Kohlenwasserstoffs wie beispielsweise Methan zu Synthesegas oder zur Verwendung in einer Fischer-Tropsch-Synthesereaktion oder anderen Reaktionen.
- Katalysatoren sind dafür bekannt, bei bestimmten Reaktionen nützlich zu sein. Insbesondere können sie verwendet werden, um bestimmte Reaktionen zu unterstützen, welche normalerweise in Abwesenheit des Katalysators nicht stattfinden würden. Ein Beispiel für eine Reaktion, bei der ein Katalysator nützlich ist, ist die Herstellung von Syngas, wobei es sich um ein Gemisch aus Kohlenmonoxid und Wasserstoff in veränderbaren Anteilen handelt. Es ist bekannt, Syngas durch eine Dampfreformierungsreaktion herzustellen, wobei Dampf und Methan über einen Katalysator geleitet werden. Eine derartige Reaktion ist endotherm.
- Kürzlich wurde Syngas auch durch die partielle Oxidation von Methan (POM) hergestellt. In diesem Verfahren wird Methan in der folgenden Reaktion teilweise mit Sauerstoff oxidiert:
- Die POM-Reaktion hat den großen Vorteil, dass sie exotherm ist und deshalb keiner großen Energiezufuhr bedarf. Das Verfahren wird mit weiteren Einzelheiten beispielsweise in Choudhary et al., Fuel, Bd. 77, Nr. 15, S. 1803–1807, 1998, Slagtern et al., Catalysis Today, 46, 107–115, 1998 und WO 01/36,323 beschrieben. In einer Abwandlung dieses Verfahrens können auch andere Kohlenwasserstoffe oder anstelle von Methan verwendet werden. So wie er in dieser Beschreibung Verwendung findet, soll der Begriff "POM" nicht nur die partielle Oxidation von Methan sondern auch die partielle Oxidation eines beliebigen Kohlenwasserstoffs abdecken.
- Die POM-Reaktion verwendet im Allgemeinen einen Nickel oder Kobalt enthaltenden Katalysator oder einen Edelmetallkatalysator. Beispielsweise offenbaren Choudhary et al. die Verwendung von verschiedenen derartigen Katalysatoren. Beispielsweise kann ein Kobalt enthaltender Katalysator durch Zumischen von Kobaltnitrat zu einem Träger wie beispielsweise Silicagel und entmineralisiertem Wasser zur Bildung einer dicken Paste, Mischen der Paste und Trocknen und Zersetzen der Paste an Luft bei 600 °C für 4 h vor dem Kalzinieren des Katalysators bei 900 °C für 4 h hergestellt werden. Dieser Literaturverweis zeigt auch, dass die Katalysatoren unter Verwendung von Wasserstoff reduziert werden können bevor sie verwendet werden, aber dass die Leistungsfähigkeit reduzierter und nicht reduzierter Katalysatoren vergleichbar ist. Es wurde ebenso herausgefunden, dass die besten Katalysatoren Nickel enthaltende Katalysatoren sind wie beispielsweise NiO-Tho2, Ni-ZrO2 und Ni-UO2.
- Ein anderes Beispiel für ein Verfahren, bei welchem ein Katalysator nützlich ist, ist eine Fischer-Tropsch-Synthesereaktion in, welcher ein Gemisch von Kohlenwasserstoffen aus Kohlenstoffmonoxid und N2 hergestellt wird.
- Wir haben nun einen weiteren Katalysator gefunden, der in einer Vielzahl von Reaktionen wie beispielsweise einer POM- oder einer Fischer-Tropsch-Reaktion verwendet werden kann. Wir haben überraschenderweise herausgefunden, dass dieser Katalysator einen oder mehrere Vorteile gegenüber den bekannten Katalysatoren aufweisen kann.
- Die vorliegende Erfindung stellt ein Verfahren zur Herstellung eines aktivierten Katalysators bereit umfassend 0,02 Gew.% bis 10 Gew.% Kohlenstoff, wobei das Verfahren die Aktivierung eines Katalysatorvorläufers umfasst, der eine Kobaltverbindung und einen Träger umfasst mit einem Gas, das wenigstens 5 mol% eines aus Methan, Ethan, Acetylen, Propan, Propen und Butan ausgewählten Kohlenwasserstoffs umfasst bei einer Temperatur von 300 °C bis 1000 °C, wobei der Katalysatorvorläufer ein POM; ein Fischer-Tropsch-, ein Hydroisomerisierungs- oder ein Hydrierungskatalysatorvorläufer ist und 0,05 bis 30 Gew.% Kobalt umfasst.
- Die vorliegende Erfindung stellt ferner ein Verfahren zur Herstellung eines Gemischs aus Kohlenwasserstoffen durch eine Fischer-Tropsch-Reaktion bereit, welches das Leiten eines Gemischs aus Kohlenmonoxid und H2 über einen Katalysator umfasst, wobei der Katalysator durch ein wie oben definiertes Verfahren erhalten wurde.
- Die vorliegende Erfindung stellt ferner ein Verfahren zur partiellen Oxidation eines Kohlenwasserstoffs bereit, welches das Durchleiten eines derartigen Kohlenwasserstoffs und von Sauerstoff über einen Katalysator umfasst, welcher durch ein Verfahren wie oben definiert oder einen Katalysator wie oben definiert erhalten wurde. Wie hierin verwendet, deckt die Bezeichnung "Katalysator" sowohl den Katalysator in aktiver Form als auch den Katalysator in Vorläuferform ab, da er sich in der Reaktionsumgebung verändern kann. Der Begriff "Katalysatorvorläufer" sollte breit ausgelegt werden, so dass er nicht nur einen frisch hergestellten Katalysatorvorläufer oder einen reduzierten Katalysatorvorläufer oder einen Katalysatorvorläufer umfasst, welcher nicht in einer Reaktion verwendet worden ist, welche er katalysiert, sondern auch jeden beliebigen Vorläufer, der nach Aktivierung als Katalysator verwendet werden kann, wie beispielsweise ein Katalysator, der bereits in einer Reaktion, welche er katalysiert, verwendet worden ist. In ähnlicher Weise soll der Begriff "Aktivierung" so verstanden werden, dass er nicht nur eine Aktivierung eines unbenutzten oder unreduzierten Katalysatorvorläufers umfasst, sondern auch eine Aktivierung eines benutzten oder reduzierten Katalysators. Daher umfasst der Begriff innerhalb seines Bedeutungsbereichs jede beliebige Aktivierung, einschließlich Regenerierung eines benutzten Katalysators.
- Das Verfahren der vorliegenden Erfindung verwendet eher einen Aktivierungsschritt mit einem Kohlenwasserstoff als mit Wasserstoff wie es beispielsweise in Choudhary et al. offenbart ist. Es wurde überraschenderweise herausgefunden, dass ein solcher Katalysator viele vorteilhafte Charakteristika wie beispielsweise eine bessere Aktivität und weniger Anfälligkeit gegenüber einer Deaktivierung mit der Zeit aufweisen kann.
- Der Katalysator wird durch Aktivieren eines Katalysatorvorläufers mit einem Kohlenwasserstoff hergestellt. Der Katalysatorvorläufer enthält eine Kobaltverbindung und einen Träger. Derartige Katalysatorvorläufer sind im Stand der Technik bekannt und offenbart, beispielsweise in Choudhary et al.
- Der Träger kann jeder beliebige Träger sein, welcher den Katalysator in der gewünschten Reaktion tragen kann. Derartige Träger sind im Fachbereich bekannt. Der Träger kann ein inerter Träger sein oder er kann ein aktiver Träger sein. Beispiele geeigneter Träger sind Aluminiumoxid, modifiziertes Aluminiumoxid, Spinelloxide, Silica, modifiziertes Silica, Magnesiumoxid, Titanoxid, Zirkonoxid, Zeolith, β-Aluminat und Kohlenstoffformen. Die Aluminiumoxide oder modifizierten Aluminiumoxide können beispielsweise α-Aluminiumoxid, β-Aluminiumoxid oder γ-Aluminiumoxid sein. Für β-Aluminiumoxid und Spinelloxide wie beispielsweise Bariumhexaaluminat, wurde herausgefunden, dass sie im Hinblick auf ihre Stabilität besonders nützlich sind. Der Kohlenstoff kann beispielsweise in Form von Aktivkohle oder Kohlenstoffnanoröhren vorliegen. Ein Zeolith kann abhängig vom gewünschten Endprodukt ausgewählt werden. Daher kann er beispielsweise Poren oder Kanäle umfassen. Vorzugsweise ist der Träger porös. Die Partikelgröße reicht in Abhängigkeit von der Verwendung vorzugsweise von 0,2 µm bis 0,4 mm. Der Oberflächenbereich ist wünschenswerterweise größer als 5 m2/g. Es kann ein Träger oder ein Gemisch aus zwei oder mehr Trägern verwendet werden.
- Der Katalysatorvorläufer umfasst auch eine Kobaltverbindung. Jede beliebige Kobaltverbindung kann verwendet werden, aber vorzugsweise liegt sie in Form eines Salzes vor, insbesondere eines wässrig löslichen Salzes oder Oxids. Beispiele geeigneter Kobaltsalze sind Kobaltnitrat, -acetat, -benzoat, -oxalat und -acetylacetonat. Es ist bevorzugt, die Verwendung eines Kobalthalogenids zu vermeiden, da das Halogenid mit dem Träger wechselwirken kann. Ein Beispiel eines geeigneten Kobaltoxids ist Co3O4. Es kann ein Kobaltsalz oder ein Gemisch aus zwei oder mehreren Kobaltsalzen und/oder Oxiden verwendet werden.
- Der Katalysatorvorläufer ist auf dem Träger trägergestützt. Abhängig von der Art der zu katalysierenden Reaktion kann der Katalysatorvorläufer auf jede gewünschte Art auf oder im Träger verteilt sein. Daher kann er beispielsweise im Wesentlichen durch den ganzen Träger hindurch oder nur auf der äußeren Oberfläche des Trägers verteilt sein.
- Beispielsweise kann der Katalysatorvorläufer durch Imprägnieren des Trägers mit einer Lösung eines Kobaltsalzes wie beispielsweise Kobaltnitrat, -acetat oder -oxalat hergestellt werden.
- Der Katalysatorvorläufer kann in einer beliebigen bekannten Weise trägergetützt sein. Daher kann er beispielsweise in Lösung in einem Lösungsmittel wie beispielsweise Wasser oder einem organischen Lösungsmittel wie beispielsweise einem Alkohol, welcher beispielsweise 1 bis 4 Kohlenstoffatome enthält, wie beispielsweise Methanol oder Ethanol, hinzugefügt werden und das Lösungsmittel anschließend entfernt werden. Das Lösungsmittel kann beispielsweise durch Trocknen bei Raumtemperatur (20 °C) oder darüber, beispielsweise von 50 °C bis 250 °C, für 1 bis 24 h entfernt werden. Es kann eine Kombination aus Trocknungsschritten verwendet werden. Daher kann der trägergestützte Katalysatorvorläufer beispielsweise bei Raumtemperatur für 2 bis 10 h getrocknet werden und anschließend kann er bei einer erhöhten Temperatur, beispielsweise von 100 °C bis 200 °C, insbesondere etwa 120 °C, für 2 bis 8 h getrocknet werden.
- Die die den Katalysatorvorläufer umfassende Lösung kann, falls erwünscht, weitere Komponenten umfassen. Daher kann sie beispielsweise ein Hilfsmittel oder ein Modifizierungsmittel umfassen. Geeignete Hilfsmittel sind Erdalkalisalze wie beispielsweise Magnesium-, Calcium-, Barium- und/oder Strontiumnitrat.
- Geeignete Hilfsmittel umfassen die Oxide von Alkalimetallen, Erdalkalimetallen oder Übergangsmetallen, welche aus deren löslichen Verbindungen ableitbar sind, wie beispielsweise deren Salze, z.B. LiNO3, KNO3, RbNO3, Ba(NO3)2, Mg(NO3)2, Ca(NO3)2, Sr(NO3)2, Zr(NO3)2·xH2O, Ce(NO3)3·xH2O und UO(NO3)2. Die Hilfsmittel können auf dem Träger in einer beliebigen Weise aufgebracht werden, wie beispielsweise durch Imprägnierung, insbesondere sequenzielle Imprägnierung oder Co-Imprägnierung mit der Kobaltverbindung.
- Geeignete Modifizierungsmittel sind seltene Erden-Modifizierungsmittel wie beispielsweise Übergangsmetalle oder seltene Erdensalze oder Oxide wie beispielsweise Lanthan- und/oder Cernitrat oder -acetat oder Oxide der d-Block-Übergangsmetalle wie beispielsweise Mn, W, Nb und Vn. Die Modifizierungsmittel werden im Allgemeinen aus ihren wässrigen löslichen Verbindungen wie beispielsweise Salzen abgeleitet und können in den Katalysatorträger imprägniert werden, wobei sich Kalzinierung anschließt, beispielsweise von 300 °C bis 1000 °C für 1 bis 24 h an Luft. Die Hilfsmittel und Modifizierungsmittel können einzeln oder in einer Kombination aus zweien oder mehreren verwendet werden. Vorzugsweise enthalten die trägergestützten Katalysatorvorläufer keine Katalysatorgifte wie beispielsweise Phosphoroxide, Stickstoffoxide oder Schwefeloxide oder Verbindungen und enthalten keine Komponenten mit zusätzlicher Funktionalität wie beispielsweise Absorptionsmittel, insbesondere Absorptionsmittel für Stickstoffoxide und/oder Schwefeloxide. Die trägergestützten Katalysatorvorläufer umfassen jedoch wünschenswerterweise ein Hilfsmittel und/oder ein Modifizierungsmittel.
- Falls erwünscht, kann der trägergestützte Katalysatorvorläufer kalziniert werden. Kalzinierung ist ein Schritt, der im Verfahren der vorliegenden Erfindung nicht notwendig ist. Kalzinierung kann an Luft, einem anderen Sauerstoff umfassenden Gas oder in einer inerten Atmosphäre stattfinden. Eine geeignete Kalzinierungstemperatur reicht beispielsweise von 300 °C bis 1000 °C beispielsweise von 350 °C bis 800 °C und insbesondere von 400 °C bis 600 °C für 1 bis 10 h. Es wird postuliert, obwohl wir nicht durch diese Theorie gebunden sind, dass der Kalzinierungsschritt das Kobaltsalz zu einer Oxidform oder einem Gemisch aus Oxidformen hin verändert. Der Kalzinierungsschritt kann auch das Hilfsmittel und/oder das Modifizierungsmittel zu Oxidformen umwandeln, falls diese vorliegen.
- Der trägergestützte Katalysatorvorläufer umfasst von 0,05 bis 30 Gew.% Kobalt, insbesondere 0,5 bis 15 Gew.%. Beispielsweise umfasst der trägergestützte Katalysatorvorläufer im Allgemeinen von 0,5 bis 50 Gew.% eine Kobaltverbindung, 0 bis 10 Gew.% ein Hilfsmittel und 0 bis 20 Gew.% ein Modifizierungsmittel, insbesondere 0,01 bis 5 Gew.% Modifizierungsmittel, bezogen auf das Gesamtgewicht des trägergestützten Katalysatorvorläufers. Für eine POM-Reaktion umfasst der trägergestützte Katalysatorvorläufer vorzugsweise von 0,5 bis 10 Gew.% eine Kobaltverbindung, 0 bis 5 Gew.% Hilfsmittel und 0 bis 3 Gew.% Modifizierungsmittel. Für eine Fischer-Tropsch-Reaktion umfasst der trägergestützte Katalysatorvorläufer vorzugsweise von 5 bis 40 Gew.% eine Kobaltverbindung, 0 bis 3 Gew.% Hilfsmittel und 0 bis 3 Gew.% Modifizierungsmittel. Die oben beschriebenen Prozentangaben für die Kobaltverbindungen sind entweder auf die Verbindung oder das Kobaltmetall in der Verbindung bezogen.
- Der trägergestützte Katalysatorvorläufer wird anschließend mit einem Gas umfassend Kohlenwasserstoff ausgewählt aus Methan, Ethan, Acetylen, Propan, Propen und Butan aktiviert. Es kann ein Kohlenwasserstoff oder ein Gemisch aus zwei oder mehreren Kohlenwasserstoffen verwendet werden.
- Das Gas umfasst wenigstens 5 mol% des Kohlenwasserstoffs, vorzugsweise wenigstens 10 mol%, stärker bevorzugt wenigstens 20 mol% und nochmals stärker bevorzugt wenigstens 40 mol%. Der Kohlenwasserstoff liegt in gasförmiger Form vor.
- Das Gas umfassend den Kohlenwasserstoff kann nur aus Kohlenwasserstoff bestehen oder kann beispielsweise in einer Menge von bis zu 100 mol%, von bis zu 20 mol% oder von bis zu 40 mol% aus Kohlenwasserstoff bestehen. Falls gewünscht, kann es ein Inertgas wie beispielsweise Stickstoff und/oder Argon umfassen. Es kann auch eine reaktive Komponente umfassen wie beispielsweise eine andere Komponente, welche ebenfalls den Katalysatorvorläufer aktiviert. Deshalb kann das Gas beispielsweise auch Wasserstoff umfassen. Besonders nützliche Kombinationen sind eine Kombination aus Methan und/oder Ethan mit Wasserstoff. Wenn Wasserstoff verwendet wird, kann ein beliebiges Verhältnis von Kohlenwasserstoff zu Wasserstoff verwendet werden, aber das Verhältnis ist vorzugsweise 0,04 oder 0,05 bis 100:1 auf molarer Basis, stärker bevorzugt 0,1 oder 0,5 bis 10:1.
- Die Aktivierung wird durch Einbringen des trägergestützten Katalysatorvorläufers in eine Atmosphäre aus dem aktivierendem Gas durchgeführt. Wünschenswerterweise wird das aktivierende Gas über den trägergestützten Katalysator geleitet. Die Aktivierungstemperatur reicht beispielsweise von 400 °C bis 1000 °C, insbesondere von 400 °C bis 800 °C. Die Dauer der Aktivierung beträgt im Allgemeinen wenigstens 30 min, vorzugsweise wenigstens 1 h, beispielsweise von 1 h bis zu 20 h, insbesondere 2 bis 5 h. Die Aktivierungstemperatur kann in Abhängigkeit von der Art des Katalysatorvorläufers und/oder des Kohlenwasserstoffs variieren. Für den Aktivierungsschritt wird üblicherweise Atmosphärendruck verwendet, obwohl, falls erwünscht, reduzierter oder erhöhter Druck ebenfalls verwendet werden können.
- Der Katalysatorvorläufer kann in dem Reaktionsgefäß aktiviert werden, in welchem die gewünschte Reaktion unter Verwendung des aktivierten Katalysators durchgeführt werden soll, oder er kann in einem anderen Gefäß aktiviert werden. Der aktivierte Katalysator geht eine substantielle Oxidation ein, wenn er gegenüber Luft ausgesetzt wird. Zur Stabilisierung des Katalysators kann er in einer Atmosphäre behandelt werden, welche eine kleine Menge Sauerstoff enthält, beispielsweise etwa 1 % Sauerstoff in einem Inertgas wie beispielsweise Stickstoff oder Argon. Alternativ kann der Katalysator einfach im Aktivierungsreaktor belassen werden, während eine kleine Menge Sauerstoff durch Entlüften zugeführt wird. Daher kann der aktivierte Katalysator durch Behandlung in einer reduzierten Sauerstoffatmosphäre beruhigt werden, beispielsweise umfassend weniger als 20 mol% Sauerstoff, weniger als 10 mol% Sauerstoff, weniger als 5 mol% Sauerstoff oder weniger als 2 mol% Sauerstoff für wenigstens 30 min, insbesondere für wenigstens 1 h. Der sich ergebende passivierte Katalysator kann nun für kurze Zeiträume ohne weitere substantielle Oxidation an Luft gehandhabt oder gelagert werden.
- Der trägergestützte Katalysatorvorläufer kann auch durch ein Solgelverfahren gebildet werden. Ein derartiges Verfahren ist beispielsweise in Gonzalez et al., Catalysis Today, 35 (1997), 293–317 und J. Livage, Catalysis Today, 41 (1998), 3–19 beschrieben. Beispielsweise werden in einem "vorgelierendem" Schritt zu Beginn ein Alkoxid oder ein Alkohol und ein Metallvorläufer hydrolysiert und anschließend unter Bildung eines Gels kondensiert, beispielsweise in Gegenwart von Wasser. Anschließend wird eine Kobaltverbindung in einem nachfolgenden "Nachgelierungs"-Schritt zugegeben und das Gel wird getrocknet und kalziniert.
- Beispielsweise wird Aluminium-tri-sec-butylat (ASB) in 2-Butanol mit 1,3-Butandiol behandelt. Es findet eine Hydrolysereaktion statt. Anschließend wird Co(H2O)6(NO3)2 (hydriertes Kobaltnitrat) zugegeben und das sich ergebende Gel wird für 1 h bei 85 °C gerührt. Das Lösungsmittel wird im Luftstrom oder Stickstoffstrom bei 40 °C bis 100 °C für 2 bis 3 h entfernt. Das Feststoffprodukt wird von 400 °C bis 1000 °C für 2 bis 5 h kalziniert, um einen trägergestützten Katalysatorvorläufer zu ergeben.
- Es wird postuliert, obwohl wir durch diese Theorie nicht gebunden sind, dass die Aktivierung des Katalysatorvorläufers ein Gemisch aus metallischem Kobalt und Kobaltcarbiden bildet wie beispielsweise Co2C und/oder Co3C auf dem Träger. XRD-Analyse zeigt, dass, wenn N2 verwendet wird, um den gleichen Katalysatorvorläufer zu aktivieren, die Co-Metallpeakintensität sehr viel intensiver ist, als wenn ein Kohlenwasserstoff verwendet wird, wobei dies nahelegt, dass der Kohlenwasserstoff zu kleineren Partikeln aus Kobaltmetall führt, als wenn Wasserstoff verwendet wird. Dies erhöht in vorteilhafter Weise den Oberflächenbereich des Kobaltmetalls, was wiederum die Wirksamkeit des Katalysators erhöht. Deshalb umfasst der aktivierte Katalysator Kohlenstoff in einer beliebigen Form einschließlich elementarem Kohlenstoff und Carbiden wie beispielsweise Kobaltcarbiden in einer Menge von 0,02 bis 10 Gew.%.
- Der Katalysator, welcher durch das Verfahren der vorliegenden Erfindung hergestellt wurde, kann in einem beliebigen Verfahren verwendet werden, in welchem ein Kobaltkatalysator verwendet werden kann, insbesondere wenn ein Festbettreaktor oder ein Aufschlämmungsbettreaktor verwendet wird. Beispielsweise kann er in einer POM-Reaktion, in einer Fischer-Tropsch-Reaktion, in einer Hydroisomerisierungsreaktion oder in einer Hydrierungsreaktion verwendet werden.
- Zur Erzeugung von Syngas wird in einer POM-Reaktion ein Gemisch aus einem Kohlenwasserstoff und Sauerstoff über den Katalysator geleitet. Der Kohlenwasserstoff enthält vorzugsweise 1 bis 16 Kohlenstoffatome und stärker bevorzugt 1 bis 5 Kohlenstoffatome. Am stärksten bevorzugt ist Methan oder Erdgas. Der Kohlenwasserstoff kann gesättigt oder ungesättigt sein, beispielsweise enthaltend 1, 2, 3 oder mehr Doppel- und/oder Dreifachbindungen. Er kann linear, zyklisch oder verzweigt sein. Der Kohlenwasserstoff kann auch aliphatisch oder arylisch sein oder kann aliphatische und Arylgruppen enthalten. Es kann ein Kohlenwasserstoff oder ein Gemisch aus Kohlenwasserstoffen verwendet werden.
- In der POM-Reaktion ist das Oxidationsmittel normalerweise O2. Es kann reines O2 sein. Jedoch kann es mit H2O (Dampf) oder CO2 angereichert sein, beispielsweise durch Zugabe zum Ausgangsmaterial. Daher können O2 und H2O; O2 und CO2; oder O2, H2O und CO2 verwendet werden. Dies führt zur Oxy-Dampf- bzw. Oxy-Trockenreformierung von Methan. Durch dieses Mittel kann die Exothermizität und das Produktverhältnis wie gewünscht kontrolliert werden. Das O2 und ggf. H2O und CO2 können rein verwendet werden oder können mit einem Inertgas wie beispielsweise Luft, N2, Ar oder He verdünnt verwendet werden.
- Wünschenswerterweise findet die Reaktion bei einer Temperatur von wenigstens 500 °C statt, beispielsweise von 700 °C bis 1000 °C.
- Wünschenswerterweise ist der Druck Atmosphärendruck (101 kPa) oder darüber, beispielsweise von 1 bis zu 60 Atmosphären (101 kPa bis 6080 kPa), insbesondere von 1 bis zu 30 Atmosphären (101 kPa bis 3060 kPa). Die Raumgeschwindigkeit (space velocity) der Reaktanden kann beispielsweise von 1000 h–1 bis zu 1.000.000 h–1, vorzugsweise 10.000 h–1 bis 600.000 h–1 reichen. Das Molverhältnis des oxidierten Kohlenwasserstoffs und des Sauerstoffs wird wünschenswerter so gewählt, dass ein Gemisch aus Kohlenmonoxid und Wasserstoff in einem stöichiometrischen Verhältnis erhalten wird. Daher ist beispielsweise das atomare Verhältnis von Kohlenstoff, wie beispielsweise Methan zu Sauerstoff, im Ausgangsmaterial vorzugsweise 0,9 bis 5:1 oder auch höher, insbesondere 1,0 oder 1,8 bis 3,5:1, stärker bevorzugt 1 bis 3:1, insbesondere 1 bis 2:1, insbesondere etwa 1:1, obwohl auch, falls erwünscht, höhere oder niedrigere Verhältnisse verwendet werden können.
- Es wurde herausgefunden, dass die durch das Verfahren der vorliegenden Erfindung hergestellten Katalysatoren üblicherweise eine Reihe von Vorteilen gegenüber Kobaltkatalysatoren aufweisen, welche unter Verwendung von Wasserstoff aktiviert worden sind. Daher sind die Katalysatoren beispielsweise über die Zeit stabil und leiden nicht unter einer Abnahme hinsichtlich ihrer Aktivität. Darüber hinaus weisen die Katalysatoren größere Aktivität auf, beispielsweise eine Aktivität, welche die Aktivität von Rutheniumkatalysatoren erreicht, wobei sie aber den bemerkenswerten Vorteil aufweisen, dass sie günstiger als Rutheniumkatalysatoren sind. Darüber hinaus wurde herausgefunden, dass die durch das Verfahren der vorliegenden Erfindung hergestellten Katalysatoren die Kohlenstoffablagerung nicht unterstützen, welche in industriellen Verfahren äußerst unerwünscht ist. Kobaltkatalysatoren, welche mit Wasserstoff aktiviert worden sind, leiden üblicherweise nach etwa 200 h Verwendung unter Kohlenstoffablagerung. Die Katalysatoren, welche durch das Verfahren der vorliegenden Erfindung hergestellt worden sind, leiden im Allgemeinen auch nach etwa 1000 h Verwendung nicht unter Kohlenstoffablagerung.
- Die Katalysatoren, welche durch das Verfahren der vorliegenden Erfindung hergestellt worden sind, können beispielsweise auch in einer Fischer-Tropsch-Synthesereaktion verwendet werden. Eine derartige Reaktion erzeugt ein Gemisch aus Kohlenwasserstoffen und/oder oxygenierten Kohlenwasserstoffen, z.B. gasförmigen flüssigen und/oder festen Kohlenwasserstoffen und/oder oxygenierten Kohlenwasserstoffen wie beispielsweise Alkoholen aus einem Gemisch aus Wasserstoff und Kohlenstoffmonoxid. Daher kann die Reaktion beispielsweise unverzüglich unter Verwendung von Syngas, welches durch das POM-Verfahren hergestellt wurde, in direkt verbundenen Reaktoren durchgeführt werden, wie es beispielsweise in WO 01/36,323 beschrieben ist.
- In der Fischer-Tropsch-Reaktion werden Wasserstoff und Kohlenstoffmonoxid über dem aktivierten Kobaltkatalysator bei einer Temperatur von beispielsweise 150 bis 300 °C bei Atmosphärendruck (101 kPa) oder darüber beispielsweise von 1 Atmosphäre (101 kPa) bis 20 Atmosphären (2030 kPa) umgesetzt. Es können ähnliche Vorteile für die Katalysatoren, welche durch das Verfahren der vorliegenden Erfindung hergestellt wurden, gegenüber denjenigen beobachtet werden, welche unter Verwendung von reinem Wasserstoff als einem aktivierenden Gas hergestellt wurden, wie sie für die POM-Reaktion oben diskutiert wurden. Daher kann der Katalysator beispielsweise stabiler und/oder aktiver sein. In ähnlicher Weise unterstützt er nicht die Kohlenstoffablagerung und er kann für das gewünschte Gemisch aus Kohlenwasserstoffen, insbesondere für Kohlenwasserstoffe, welche wenigstens 5 Kohlenstoffatome enthalten, selektiver sein. Er kann auch ein Gemisch aus Kohlenwasserstoffen mit einem hohen Maß an Ungesättigtheit aufweisen.
- Die vorliegende Erfindung wird nun durch die im Folgenden beschriebenen Beispiele weiter verdeutlicht.
- Beispiel 1
- 1,0 g Al2O3-Trägermaterial (Partikelgröße > 250 µm, getrocknet bei 120 °C für 2 h) wurde mit 1,0 ml 0,2 M Ba(NO3)2-Lösung imprägniert. Das Gemisch wurde bei 120 °C für 4 h getrocknet und im Anschluss durch Kalzinierung bei 600 °C für 4 h behandelt, um einen BaO-modifizierten Träger zu erhalten. Dieser modifizierte Träger (1,05 g) wurde anschließend mit 1 ml 2,4 M Co(NO3)2-Lösung für 2 h imprägniert. Der sich ergebende Feststoff wurde zu 600 °C kalziniert, um die Oxidvorstufe des Kobaltkatalysators zu ergeben. Diese wurde anschließend mit 30 ml/min 50% CH4/H2 bei 800 °C für 2 h behandelt und anschließend im Strom von 50% CH4/H2 auf Raumtemperatur abgekühlt. Der aktivierte Katalysator wurde bei Raumtemperatur mit 1,0% O2/N2 für 6 h behandelt; 0,1 g des aktivierten Katalysators wurden in einer Quarzröhre platziert; in N2 auf die ausgewählten Reaktionstemperaturen erhitzt und anschließend wurde ein Gemisch aus CH4 und Luft in den Reaktor bei 100 kPa (1 bar) eingeführt. Die Reaktionsbedingungen und Produkte sind in Tabelle 1 aufgelistet.
- Tabelle 1
- CH4-Umwandlung und Produktverteilung aus partieller Methanoxidation (POM) über 12,5 Gew.% Co/Al2O3-BaO-Katalysatoren, welche mit 50% CH4/H2 zu 800 °C für 2 h aktiviert wurden
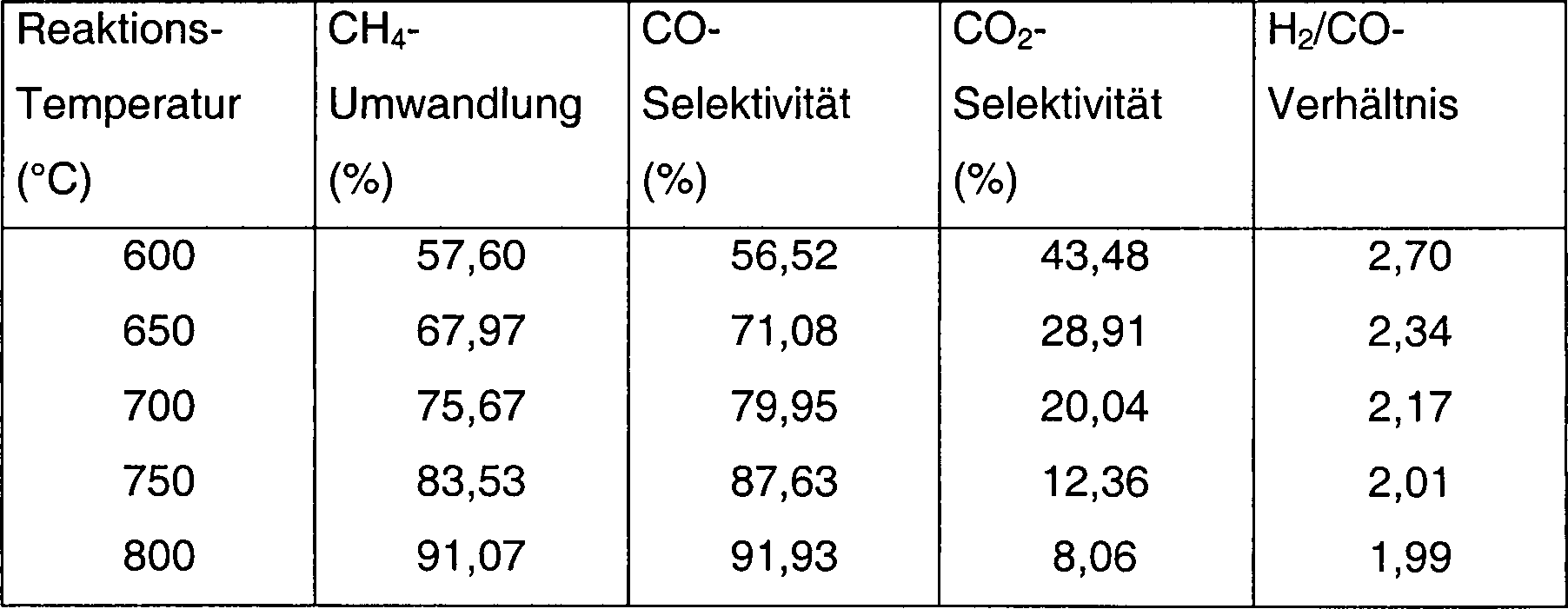
- Reaktionsbedingungen: P: 100 kPa (1 bar), GHSV: 36,000 h–1. Luft wurde als Oxidationsmittel verwendet.
- Beispiel 2
- 1,0 g Al2O3-Trägermaterial (Partikelgröße > 250 µm) wurde mit 1,0 ml 0,2 M Ba(NO3)2-Lösung imprägniert. Der Feststoff wurde bei 120 °C für 4 h getrocknet und anschließend bei 600 °C für 4 h kalziniert. Der sich ergebende BaO-modifizierte Träger (Al2O3-BaO) (1,05 g) wurde mit 1 ml 2,4 M Co(NO3)2-Lösung für 2 h imprägniert. Das Gemisch wurde anschließend bei 600 °C kalziniert, um die Oxidvorstufe des Katalysators zu ergeben. Diese wurde mit bei 30 ml/min strömendem 20% C2H6/H2 bei 630 °C behandelt und bei 630 °C für 2 h gehalten. Der Gasstrom wurde gestoppt und der Reaktor wurde ohne Exposition gegenüber Luft auf Raumtemperatur abgekühlt. Der Katalysator wurde anschließend mit 1,0% O2/N2 für 3 h behandelt. 0,1 g des zur Verwendung bereiten Kobaltkatalysators wurden in die Quarzröhre eingebracht und in N2 auf die ausgewählte Reaktionstemperatur erhitzt. Ein Reaktandengemisch aus CH4 und Luft (CH4/O2-Verhältnis, 2,01) wurde bei 100 kPa (1 bar) in den Reaktor geleitet. Die Reaktionsbedingungen und Ergebnisse sind in Tabelle 2 aufgelistet.
- Tabelle 2
- CH4-Umwandlung und Produktverteilung aus POM über 12,5 Gew.% Co/Al2O3-BaO-Katalysatoren, welche mit 20% C2H6/H2 für 2 h zu 630 °C aktiviert worden waren
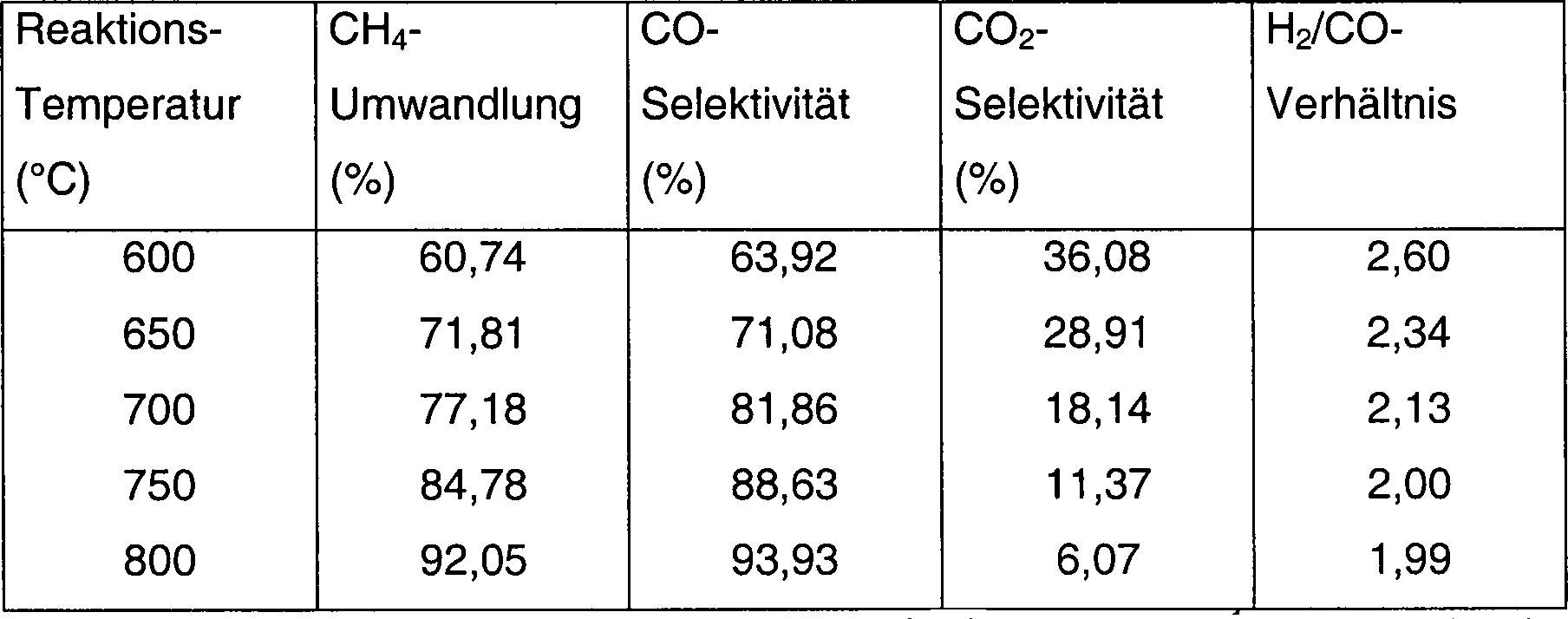
- Reaktionsbedingungen: P: 100 kPa (1 bar), GHSV: 36,200 h–1. Luft wurde als Oxidationsmittel verwendet.
- Beispiel 3
- 1,0 g Al2O3-Trägermaterial (Partikelgröße > 250 µm) wurden mit 1,0 ml 0,15 M La(NO3)s-Lösung imprägniert. Das Gemisch wurde bei 120 °C für 4 h getrocknet und anschließend bei 700 °C für 2 h kalziniert. Der sich ergebende La2O3-modifizierte Träger (Al2O3-La2O3) (1,05 g) wurde mit 1 ml 1,0 M Co(NO3)2-Lösung für 5 h imprägniert und zu 700 °C kalziniert. Er wurde anschließend mit strömendem CH4 bei 700 °C behandelt und für 1 h bei 700 °C gehalten und anschließend ohne Exposition gegenüber Luft auf Raumtemperatur abgekühlt. Anschließend wurde er mit 1,0% O2/N2 bei Raumtemperatur für 10 h behandelt. Der zur Verwendung fertige Katalysator (0,1 g) wurde in eine Quarzröhre eingebracht und auf die ausgewählten Reaktionstemperaturen im 10 ml/min CH4 erwärmt. Ein Gemisch aus CH4 und reinem Sauerstoff (CH4/O2-Verhältnis, 2,01) wurde in den Reaktor bei 100 kPa (1 bar) eingeleitet. Die Reaktionsbedingungen und die Produktverteilung sind in Tabelle 3 aufgelistet.
- Tabelle 3
- CH4-Umwandlung und Produktverteilung aus POM über 5,6 Gew.% Co/Al2O3-La2O3-Katalysatoren, welche mit reinem CH4 zu 700 °C für 2 h aktiviert worden waren
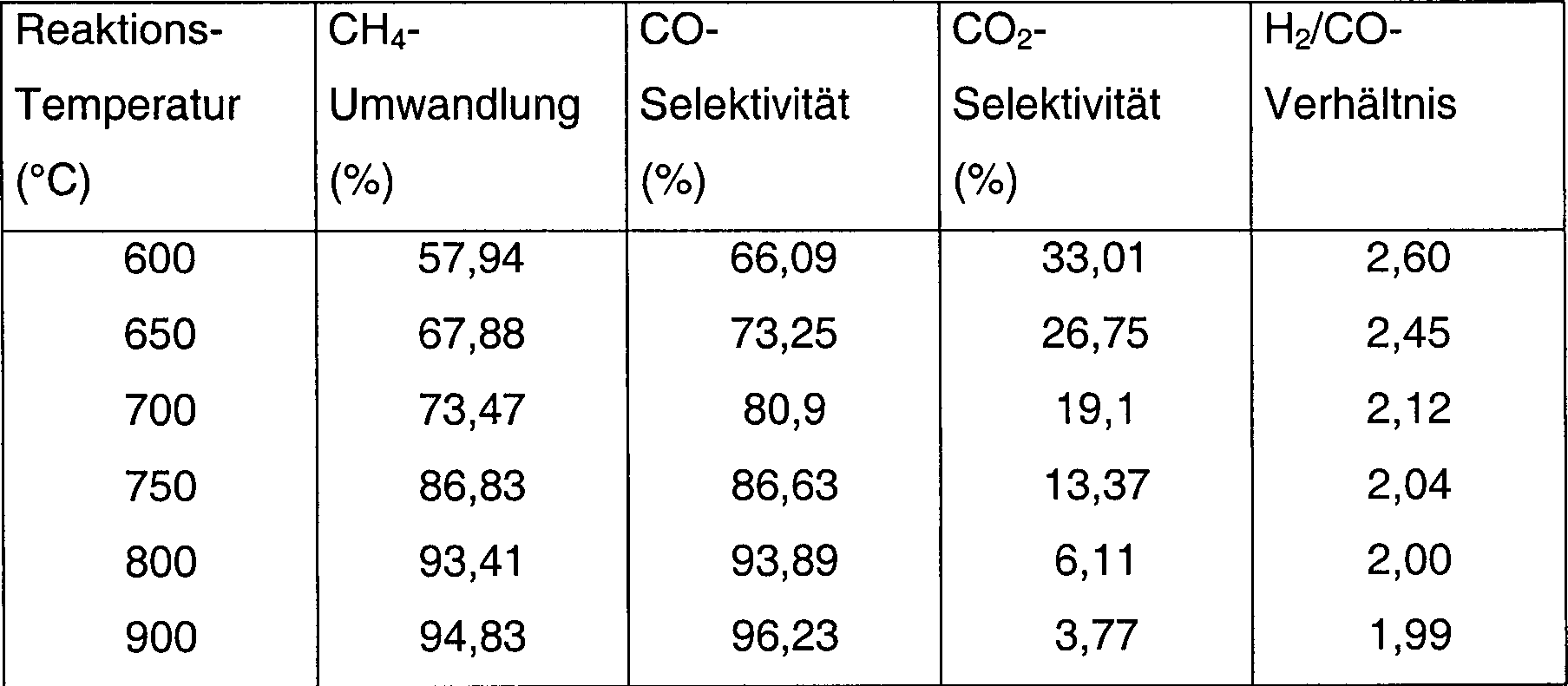
- Reaktionsbedingungen: P: 100 kPa (1 bar), GHSV: 18,600 h–1. Reiner Sauerstoff wurde als Oxidationsmittel verwendet.
- Beispiel 4
- 1,0 g α-Al2O3-Trägermaterial (Partikelgröße > 250 µm) wurden mit 1,0 ml 0,12 M Pr(NO3)3-Lösung für 10 h unter Rühren imprägniert. Es wurde anschließend bei 650 °C für 2 h kalziniert. Der sich ergebende Pr2O3-modifizierte Träger (Al2O3-Pr2O3) (1,05 g) wurde mit 1 ml 1,0 M Co(NO3)2-Lösung für 5 h imprägniert. Der Feststoff wurde zu 700 °C kalziniert und dort für 2 h gehalten, anschließend auf Raumtemperatur abgekühlt. Der sich ergebende Oxidvorläufer wurde anschließend mit 30 ml/min CH4 bei 700 °C für 5 h behandelt und anschließend auf Raumtemperatur abgekühlt. Der aktivierte Katalysator wurde mit 1,0% O2/N2 für 10 h behandelt. Der zur Verwendung fertige Katalysator (0,1 g) wurde in eine Quarzröhre eingebracht, in N2 zu 860 °C erhitzt und anschließend wurde ein Gemisch aus CH4 und Luft (CH4/O2-Verhältnis, 2,01) in den Reaktor bei einigen verschiedenen Strömungsraten und bei 100 kPa (1 bar) eingeführt. Die Reaktionsbedingungen und die Produktverteilung sind in Tabelle 4 aufgelistet.
- Tabelle 4
- Einfluß von Raumgeschwindigkeit (space velocity) auf CH4-Umwandlung und Produktverteilung aus POM über 5,6 Gew.% CO/Al2O3-Pr2O3-Katalysatoren, welche mit reinem CH4 zu 700 °C für 1 h aktiviert wurden
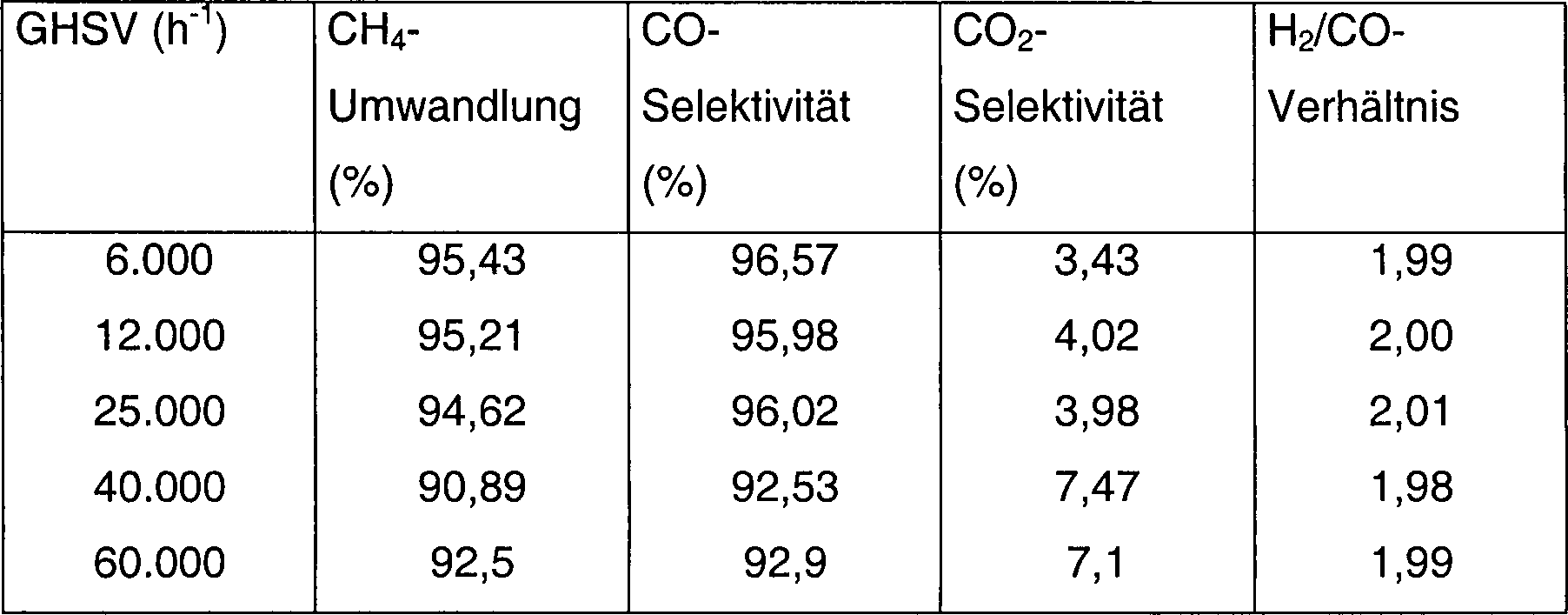
- Reaktionsbedingungen: P: 100 kPa (1 bar), Reaktionstemperatur: 860 °C, Luft wurde als Oxidationsmittel verwendet.
- Beispiel 5
- 1,0 g des BaO-modifizierten Trägermaterials (Al2O3-BaO) wurde mit 1 ml 0,75 M Co(NO3)2-Lösung für 5 h imprägniert. Der Feststoff wurde zu 750 °C kalziniert und anschließend auf Raumtemperatur abgekühlt. Er wurde anschließend mit 14 ml/min C2H6 bei 600 °C behandelt und anschließend auf Raumtemperatur abgekühlt. Der aktivierte Katalysator wurde mit 1,0 O2/N2 für 3 h behandelt. Der zur Verwendung fertige Katalysator (0,1 g) wurde in eine Quarzröhre eingebracht und auf die ausgewählten Reaktionstemperaturen in N2 erhitzt. Ein Gemisch aus C2H6 und Luft (C/O-Verhältnis, 1,0) wurde in den Reaktor bei 1000 kPa (1 bar) eingeleitet. Die Produktverteilung ist in Tabelle 5 gezeigt.
- Tabelle 5
- C2H6-Umwandlung und Produktverteilung aus POM über 4,0 Gew.% Co/Al2O3-BaO-Katalysatoren, welche mit reinem Ethan zu 600 °C für 2 h aktiviert worden waren

- Reaktionsbedingungen: P: 100 kPa (1 bar), GHSV: 25,000 h–1, Luft wurde als Oxidationsmittel verwendet.
- Beispiel 6
- 1,0 g des BaO-modifizierten Trägermaterials (Al2O3-BaO) wurde mit 1 ml 1,0 M CoC2O4-Lösung für 4 h imprägniert. Der Feststoff wurde bei 100 °C für 3 h getrocknet, zu 700 °C kalziniert und dort für 2 h gehalten, anschließend auf Raumtemperatur abgekühlt. Der sich ergebende Oxidvorläufer wurde mit 30 ml/min eines Gemischs aus 20% C2H6/H2 zu 750 °C behandelt und dort für 2 h gehalten. Er wurde anschließend auf Raumtemperatur abgekühlt und mit 1,0% O2/N2 für 2 h behandelt. Der zur Verwendung fertige Katalysator (0,1 g) wurde in eine Quarzröhre eingebracht, auf die ausgewählten Reaktionstemperaturen in stationärem 20% C2N6/H2 erhitzt und dort für 0,5 h gehalten. Ein Gemisch aus CH4 und Luft (enthaltend Dampf)(C/O-Verhältnis 1,0; H2O/CH4-Verhältnis 1,0) wurde in den Reaktor bei 100 kPa (1 bar) eingeführt. Die Reaktionsbedingungen und die Produktverteilung sind in Tabelle 6 aufgelistet.
- Tabelle 6
- CH4-Umwandlung und Produktverteilung aus einer Oxy-Dampfkombination reformiert aus Methan über 5,6 Gew.% Co/Al2O3-BaO-Katalysatoren, welche mit einem Gemisch aus Ethan und Wasserstoff zu 750 °C für 2 h aktiviert worden waren

- Reaktionsbedingung: P: 100 kPa (6 bar), GHSV: 25,000 h–1, Luft wurde als Oxidationsmittel verwendet.
- Beispiel 7
- 1,0 g des BaO-modifizierten Trägermaterials (Al2O3-BaO) wurde mit 1 ml 1,0 M Co(NO3)2-Lösung für 5 h imprägniert. Das Feststoffgemisch wurde bei 100 °C für 3 h getrocknet und bei 650 °C für 2 h kalziniert. Der sich ergebende Oxidvorläufer des Katalysators (0,1 g) wurde in eine Quarzröhre eingebracht und mit 20 ml/min eines Gemischs aus 10% CH4/H2 oder H2 zu 800 °C für 2 h behandelt. Das Reaktantengas wurde anschließend auf ein Gemisch aus CH4 und Luft (C/O-Verhältnis 1,0) umgestellt und die Temperatur wurde auf 850 °C erhöht. Die Kohlenstoffablagerung wurde nach einem 200 h-Lauf gemessen und die Reaktion wurde fortgesetzt, bis die CH4-Umwandlung auf weniger als 90% abnahm. Die Reaktionsbedingungen und Ergebnisse sind in Tabelle 7 aufgelistet.
- Menge an Kohlenstoffablagerung und Lebensdauer von 5,6 Gew.% Co/Al2O3-BaO-Katalysatoren, welche mit CH4/H2 und reinem H2 zu 800 °C für 2 h aktiviert worden waren

- Reaktionsbedingungen: P: 10 kPa (1 bar), Temperatur: 850°C, GHSV: 25,000 h–1, Luft wurde als Oxidationsmittel verwendet.
- *. Die Menge an Kohlenstoff wurde nach 200 h Zeit im Strom gemessen.
- **. Lebensdauer ist, wenn die Aktivität des Katalysators auf weniger als 90% verringert ist.
- Beispiel 8
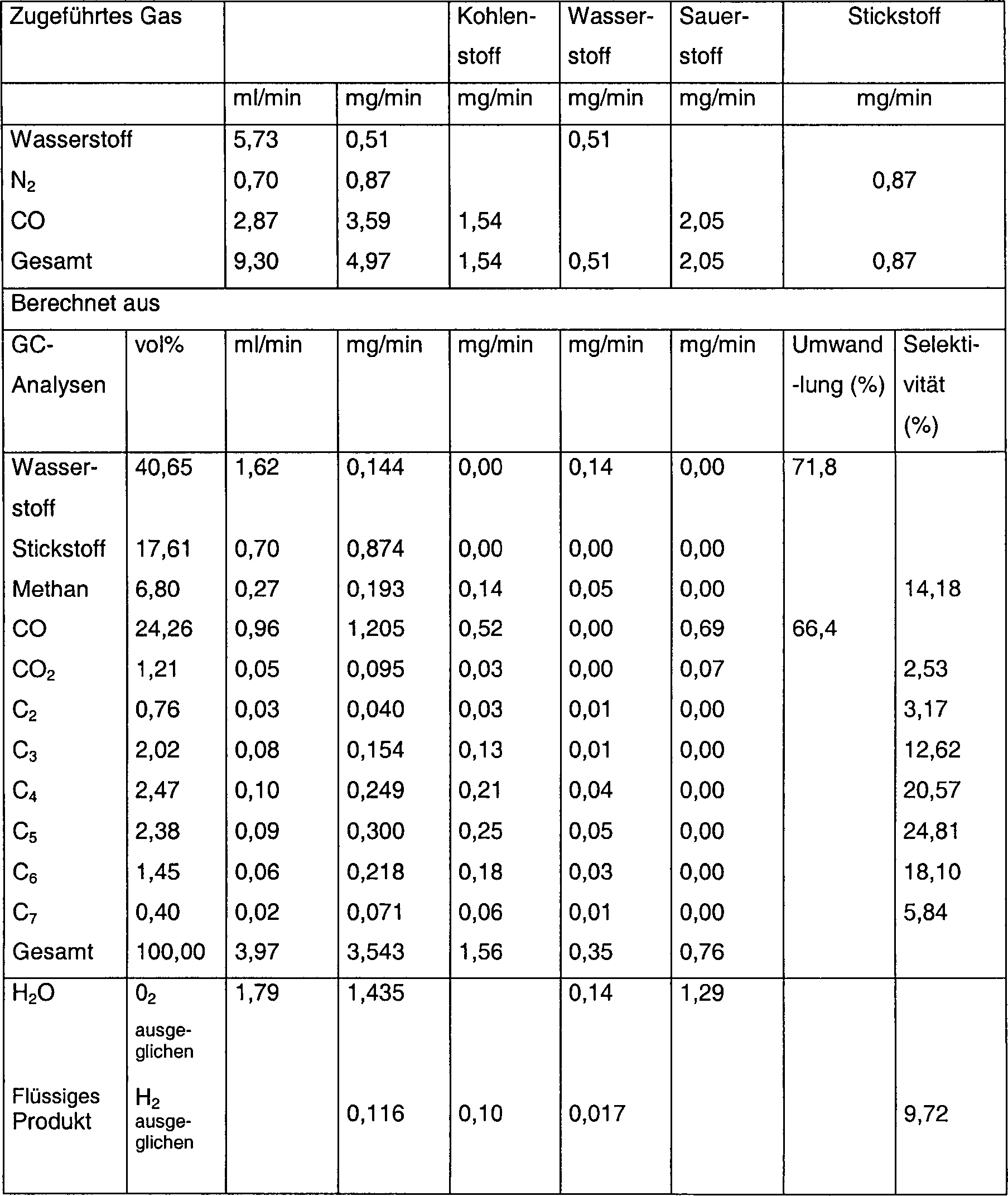
- 0,2 g SiO2 (> 250 µm, getrocknet bei 200 °C für 4 h) wurde mit 0,4 ml Wasserlösung (enthaltend 2,5 M Co(NO3)2 und 0,2 M ZrO(NO3)2) bei Raumtemperatur für 10 h imprägniert. Das System wurde bei 450 °C für 4 h an der Luft kalziniert, auf Raumtemperatur abgekühlt und anschließend mit 10% CH4/H2 bei einem Temperaturgradienten mit 2 K/min zu 500 °C aktiviert und dort für 1 h gehalten. Anschließend wurde es auf 230 °C abgekühlt und das Gas wurde mit einem Gemisch aus 9 ml/min Syngas (2H2+CO) und 1 ml/min N2 ausgetauscht, wobei der Druck auf 600 kPa (1 bar) angehoben wurde. Der Massenausgleich und Produktverteilung sind in Tabelle 8 gezeigt.
- Tabelle 8
- Massenausgleich für F-T-Synthese nach 48 h Zeit im Strom Reaktionsbedingungen: 230 °C, 600 kPa (6 bar), 0,2 g CoZr/SiO2-Katalysator, welcher mit 10% CH4/H2 aktiviert wurde

- Beispiel 9
- 1,0 g γ-Aluminiumoxid (getrocknet bei 120 °C für 4 h) wurde mit 0,8 ml 0,5 La(NO3)3-Lösung bei Raumtemperatur für 2 h unter Rühren imprägniert. Man ließ das Gemisch für 20 h an Luft stehen, anschließend wurde es für 4 h bei 600 °C kalziniert und anschließend auf Raumtemperatur abgekühlt. Das Lamodifizierte Aluminiumoxid wurde anschließend mit 1 ml 0,8 M Co(NO3)3·6H2O-Lösung imprägniert, das Gemisch wurde bei Raumtemperatur für 4 h gerührt und man ließ es für 6 h an der Luft stehen. Es wurde anschließend bei 600 °C für 4 h kalziniert, um einen Co3O4/Al2O3-La-Katalysatorvorläufer für partielle Methanoxidation zu Synthesegas bereitzustellen.
- 0,01 g des oben beschriebenen Katalysatorvorläufers wurden in einen mit einer 6 mm (o.d)-Silicaröhre ausgekleideten Edelstahlreaktor eingebracht und mit 4 ml/min reinem CH4 zu 900 °C behandelt und für 30 min bei 900 °C gehalten. Anschließend wurde ein Gemischstrom aus (6,1 ml/min CH4+2,5 ml/min O2 (rein)) in das Katalysatorbett eingeleitet und der Druck wurde auf 800 kPa (8 bar) erhöht. Die Leistungsfähigkeit des Katalysators ist in Tabelle 9 gezeigt. Tabelle 9

- Reaktionstemperatur: 950 °C; Druck 800 kPa (8 bar)
- Zusammensetzung des Ausgangsmaterials: 2,5 CH4/O2, reiner O2 wurde als Oxidationsmittel verwendet; Methan wurde im Überschuss zugeführt, um thermodynamische Effekte auszuschalten und um die Ausbeute und Selektivität für die CO2- und H2·CH4-Umwandlung zu erhöhen und die CO2-Selektivität war unter Verwendung von Luft als Oxidationsmittel besser. GHSV: 516,000 h–1.
- Dies macht es möglich, eine direkte Fischer-Tropsch-Synthese zu konfigurieren.
Claims (21)
- Verfahren zur Herstellung eines aktivierten Katalysators, der 0.02 Gew.-% bis 10 Gew.-% Kohlenstoff umfasst, welches das Aktivieren eines Katalysatorvorläufers, der eine Kobaltverbindung und einen Träger umfasst, mit einem Gas, das wenigstens 5 mol% eines aus Methan, Ethan, Acetylen, Propan, Propen und Butan ausgewählten Kohlenwasserstoffs umfasst, bei einer Temperatur von 300 °C bis 1000 °C umfasst, wobei der Katalysatorvorläufer ein POM, Fischer-Tropsch, Hydroisomerisierungs- oder Hydrierungs-Katalysatorvorläufer ist und 0.05 bis 30 Gew.-% Kobalt umfasst.
- Verfahren gemäß Anspruch 1, worin der Träger Aluminiumoxid, modifiziertes Aluminiumoxid, Siliciumdioxid, modifiziertes Siliciumdioxid, β-Aluminat, Magnesiumoxid, Titandioxid, ein Spineloxid, Zirkonoxid, ein Zeolith oder Kohlenstoff ist.
- Verfahren gemäß Anspruch 1 oder 2, worin der Kohlenwasserstoff Methan oder Ethan ist.
- Verfahren gemäß einem der vorhergehenden Ansprüche, worin das Gas wenigstens 10 mol% des Kohlenwasserstoffs umfasst.
- Verfahren gemäß Anspruch 4, worin das Gas wenigstens 20 mol% des Kohlenwasserstoffs umfasst.
- Verfahren gemäß einem der vorhergehenden Ansprüche, worin das Gas auch H2, N2, Argon, Helium oder ein Gemisch davon umfasst.
- Verfahren gemäß einem der vorhergehenden Ansprüche, worin das Gas im Wesentlichen aus dem Kohlenwasserstoff und gegebenenfalls aus einem inerten Gas und/oder Wasserstoff besteht.
- Verfahren gemäß Anspruch 6, worin das Gas N2 umfasst und das Verhältnis von Kohlenwasserstoff zu Wasserstoff auf molarer Basis von 0.04 bis 10:1 reicht.
- Verfahren gemäß einem der Ansprüche 1 bis 5, worin das Gas nur aus dem Kohlenwasserstoff besteht.
- Verfahren gemäß einem der vorhergehenden Ansprüche, worin der Katalysatorvorläufer durch Imprägnieren des Trägers mit einer Lösung eines Kobaltsalzes hergestellt worden ist.
- Verfahren gemäß Anspruch 10, worin das Kobaltsalz, Kobaltnitrat, -acetat oder -oxalat ist.
- Verfahren gemäß einem der Ansprüche 1 bis 9, worin der Katalysatorvorläufer durch ein Sol-Gel-Verfahren hergestellt worden ist.
- Verfahren gemäß einem der vorhergehenden Ansprüche, worin der Katalysatorvorläufer vor der Aktivierung kalziniert worden ist.
- Verfahren gemäß Anspruch 13, worin der Katalysatorvorläufer bei einer Temperatur von 300 bis 1000 °C kalziniert worden ist.
- Verfahren gemäß einem der vorhergehenden Ansprüche, worin der aktivierte Katalysator durch Behandlung in einer reduzierten Sauerstoffatmosphäre für wenigstens 30 Minuten beruhigt worden ist.
- Verfahren zur partiellen Oxidation eine Kohlenwasserstoffs, welches ein Leiten des Kohlenwasserstoffs und von Sauerstoff über einen Katalysator, der gemäß einem Verfahren, wie es in einem der Ansprüche 1 bis 15 bestimmt ist, erhalten worden ist, umfasst.
- Verfahren gemäß Anspruch 16, worin der Kohlenwasserstoff Methan ist und die partielle Oxidation Synthesegas erzeugt.
- Verfahren gemäß einem der Ansprüche 16 oder 17, worin das Atomverhältnis von Kohlenstoff zu Sauerstoff im Ausgangsmaterial 0.9 bis 5:1 beträgt.
- Verfahren gemäß einem der Ansprüche 16 bis 18, worin Sauerstoff in Form eines Gemisches von 02 und H2O; 02 und CO2; oder 02, N2O und CO2 vorliegt.
- Verfahren gemäß einem der Ansprüche 16 bis 19, worin der Sauerstoff mit N2, Ar oder He verdünnt ist.
- Verfahren zur Herstellung eines Gemisches von Kohlenwasserstoffen durch Fischer-Tropsch-Reaktion, welches ein Leiten eines Gemisches aus Kohlenmonoxid und H2 über einen Katalysator, der durch ein Verfahren, wie es in einem der Ansprüche 1 bis 15 bestimmt ist, erhalten worden ist, umfasst.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0115850 | 2001-06-28 | ||
| GBGB0115850.0A GB0115850D0 (en) | 2001-06-28 | 2001-06-28 | Catalyst |
| PCT/GB2002/002883 WO2003002252A1 (en) | 2001-06-28 | 2002-06-21 | A process for the activation of a catalyst comprising a cobalt compound and a support |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60209484D1 DE60209484D1 (de) | 2006-04-27 |
| DE60209484T2 true DE60209484T2 (de) | 2006-11-23 |
Family
ID=9917553
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60209484T Expired - Lifetime DE60209484T2 (de) | 2001-06-28 | 2002-06-21 | Verfahren zum aktivieren eines getragenen katalysators der eine kobalthaltigen verbindung enthält |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US7183329B2 (de) |
| EP (1) | EP1399256B1 (de) |
| CN (1) | CN100500283C (de) |
| AT (1) | ATE318651T1 (de) |
| AU (1) | AU2002304467B2 (de) |
| DE (1) | DE60209484T2 (de) |
| DK (1) | DK1399256T3 (de) |
| GB (1) | GB0115850D0 (de) |
| WO (1) | WO2003002252A1 (de) |
Families Citing this family (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2410449B (en) * | 2004-01-28 | 2008-05-21 | Statoil Asa | Fischer-Tropsch catalysts |
| US7973086B1 (en) | 2010-10-28 | 2011-07-05 | Chevron U.S.A. Inc. | Process of synthesis gas conversion to liquid hydrocarbon mixtures using alternating layers of synthesis gas conversion catalyst and hydrocracking catalyst |
| US7309479B2 (en) * | 2005-06-29 | 2007-12-18 | Samsung Engineering Co., Ltd. | Cobalt oxide catalysts |
| CN101384356A (zh) | 2006-02-14 | 2009-03-11 | 巴斯夫欧洲公司 | 吸附组合物和从物流中去除co的方法 |
| JP5198441B2 (ja) | 2006-06-21 | 2013-05-15 | ビーエーエスエフ ソシエタス・ヨーロピア | 液体プロピレン流からcoを除去するための方法 |
| JP2010510879A (ja) | 2006-12-01 | 2010-04-08 | ビーエーエスエフ ソシエタス・ヨーロピア | 物質流からcoを除去するための吸着体及びその除去方法 |
| WO2009028113A1 (ja) * | 2007-08-29 | 2009-03-05 | National University Corporation Oita University | 低温水素製造用触媒及びその製造方法と水素製造方法 |
| GB0704003D0 (en) * | 2007-03-01 | 2007-04-11 | Oxford Catalysts | Promoted carbide-based fischer-tropsch catalyst, method for its preparation and uses thereof |
| CN101688125B (zh) * | 2007-05-11 | 2015-07-01 | 沙索技术有限公司 | 催化剂 |
| US8614158B2 (en) * | 2008-02-29 | 2013-12-24 | Schlumberger Technology Corporation | Fischer-trospch and oxygenate synthesis catalyst activation/regeneration in a micro scale process |
| US8100996B2 (en) * | 2008-04-09 | 2012-01-24 | Velocys, Inc. | Process for upgrading a carbonaceous material using microchannel process technology |
| WO2009126765A2 (en) * | 2008-04-09 | 2009-10-15 | Velocys Inc. | Process for converting a carbonaceous material to methane, methanol and/or dimethyl ether using microchannel process technology |
| US7939953B2 (en) * | 2008-04-16 | 2011-05-10 | Schlumberger Technology Corporation | Micro scale fischer-tropsch and oxygenate synthesis process startup unit |
| US8293805B2 (en) * | 2008-05-29 | 2012-10-23 | Schlumberger Technology Corporation | Tracking feedstock production with micro scale gas-to-liquid units |
| US8148292B2 (en) * | 2008-07-25 | 2012-04-03 | Exxonmobil Research And Engineering Company | Preparation of high activity cobalt catalysts, the catalysts and their use |
| US8318986B2 (en) * | 2008-09-25 | 2012-11-27 | Albemarle Corporation | Methods for improving syngas-to-alcohol catalyst activity and selectivity |
| WO2010042794A2 (en) | 2008-10-10 | 2010-04-15 | Velocys Inc. | Process and apparatus employing microchannel process technology |
| US20100160464A1 (en) * | 2008-12-24 | 2010-06-24 | Chevron U.S.A. Inc. | Zeolite Supported Cobalt Hybrid Fischer-Tropsch Catalyst |
| GB2473071B (en) | 2009-09-01 | 2013-09-11 | Gtl F1 Ag | Fischer-tropsch catalysts |
| WO2011044549A1 (en) * | 2009-10-09 | 2011-04-14 | Velocys Inc. | Process for treating heavy oil |
| GB2475492B (en) | 2009-11-18 | 2014-12-31 | Gtl F1 Ag | Fischer-Tropsch synthesis |
| US7943674B1 (en) | 2009-11-20 | 2011-05-17 | Chevron U.S.A. Inc. | Zeolite supported cobalt hybrid fischer-tropsch catalyst |
| US20130102693A1 (en) * | 2010-07-02 | 2013-04-25 | Chevron U.S. Inc. | Modified fischer-tropsch catalyst and method for conversion of syngas |
| EP2603316B1 (de) | 2010-08-09 | 2017-04-19 | Gtl. F1 Ag | Fischer-tropsch-katalysatoren |
| KR20130114133A (ko) * | 2010-09-09 | 2013-10-16 | 바스프 에스이 | 구리, 아연 및 지르코늄 옥사이드 함유 흡착 조성물의 활성화 방법 |
| US8637724B2 (en) | 2010-09-09 | 2014-01-28 | Basf Se | Process for the regeneration of a copper, zinc and zirconium oxide-comprising adsorption composition |
| US8637723B2 (en) | 2010-09-09 | 2014-01-28 | Guido Henze | Process for the activation of a copper-, zinc- and zirconium oxide-comprising adsorption composition |
| US8519011B2 (en) | 2010-10-28 | 2013-08-27 | Chevron U.S.A. Inc. | Process of synthesis gas conversion to liquid hydrocarbon mixtures using alternating layers of synthesis gas conversion catalyst, hydrocracking and hydroisomerization catalyst |
| US8445550B2 (en) | 2010-11-23 | 2013-05-21 | Chevron U.S.A. Inc. | Ruthenium hybrid fischer-tropsch catalyst, and methods for preparation and use thereof |
| GB2502030C (en) * | 2011-02-07 | 2018-08-15 | Velocys Tech Limited | Catalysts |
| US9050588B2 (en) | 2011-05-27 | 2015-06-09 | Gi—Gasification International, S.A. | Fischer-tropsch catalyst activation procedure |
| CN102909099B (zh) * | 2011-08-01 | 2014-10-22 | 中国石油化工股份有限公司 | 以烃/氢混合气还原活化脱氢催化剂的方法 |
| CN102909015B (zh) * | 2011-08-01 | 2016-02-10 | 中国石油化工股份有限公司 | 以含co气体还原活化脱氢催化剂的方法 |
| CN102909100B (zh) * | 2011-08-01 | 2015-01-14 | 中国石油化工股份有限公司 | 以烃/氢混合气和合成气分段活化脱氢催化剂的方法 |
| US9566571B2 (en) | 2012-02-10 | 2017-02-14 | Basf Se | Hexaaluminate-comprising catalyst for the reforming of hydrocarbons and a reforming process |
| KR20140126366A (ko) * | 2012-02-10 | 2014-10-30 | 바스프 에스이 | 탄화수소 개질용 헥사알루미네이트-포함 촉매 및 개질 방법 |
| KR102061235B1 (ko) | 2012-03-07 | 2020-01-02 | 한국화학연구원 | 피셔-트롭시 합성용 촉매의 활성화 방법 |
| RU2662221C2 (ru) * | 2013-03-07 | 2018-07-25 | Басф Се | Катализатор риформинга углеводородов в присутствии диоксида углерода, содержащий гексаалюминат никеля |
| US9676623B2 (en) | 2013-03-14 | 2017-06-13 | Velocys, Inc. | Process and apparatus for conducting simultaneous endothermic and exothermic reactions |
| KR101816787B1 (ko) | 2013-11-28 | 2018-01-11 | 한국화학연구원 | 활성화된 피셔-트롭시 합성용 촉매의 저장방법 |
| CN107433199A (zh) * | 2016-12-12 | 2017-12-05 | 陕西学前师范学院 | 一种用于费‑托合成的C‑Si复合物载体的制备方法 |
| WO2019123251A1 (en) * | 2017-12-18 | 2019-06-27 | King Abdullah University Of Science And Technology | Catalysts for co2 hydrogenation |
| BR102018068334B1 (pt) | 2018-09-11 | 2021-12-07 | Petróleo Brasileiro S.A. - Petrobras | Processo para a preparação de hidrocarbonetos líquidos por processo de fischer- tropsch integrado a unidades de refino |
| US10434506B1 (en) | 2018-12-18 | 2019-10-08 | Emerging Fuels Technology, Inc. | Method for activation or regeneration of a catalyst |
| CN113171790B (zh) * | 2021-05-14 | 2022-08-26 | 内蒙古工业大学 | 一种ch4-co2重整催化剂及其制备方法 |
| KR20250144446A (ko) * | 2023-02-17 | 2025-10-10 | 캐즘 어드밴스드 머티리얼스, 인크. | 촉매, 촉매 전구체, 제조 공정 및 그 결과 생성된 고순도 및 제어된 형태 탄소 나노튜브 |
| IL303685B2 (en) * | 2023-06-13 | 2025-02-01 | Electriq Global Energy Solutions Ltd | Ceramic catalyst, method for its preparation and uses |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1116129A (en) * | 1967-03-02 | 1968-06-06 | Shell Int Research | Catalytic reduction of sulphur dioxide to hydrogen sulphide |
| SU774584A1 (ru) * | 1978-01-05 | 1980-10-30 | Предприятие П/Я А-7531 | Способ активации катализатора |
| CA2003394A1 (en) * | 1988-11-22 | 1990-05-22 | Sandra Bessell | Conversion of synthesis gas into hydrocarbons |
| CA2020151C (en) * | 1989-06-30 | 1997-12-09 | Malcolm Leslie Hodder Green | Catalytic conversion of methane |
| AU638741B2 (en) * | 1989-09-11 | 1993-07-08 | Broken Hill Proprietary Company Limited, The | Catalyst for conversion of synthesis gas into hydrocarbons |
| US5168091A (en) * | 1990-10-15 | 1992-12-01 | Exxon Research And Engineering Company | Activation conditions to maximize the HSC activity of supported cobalt catalysts |
| GB9114314D0 (en) * | 1991-07-02 | 1991-08-21 | British Petroleum Co Plc | Catalyst treatment for fisher-tropsch process |
| US5292705A (en) * | 1992-09-24 | 1994-03-08 | Exxon Research & Engineering Co. | Activation of hydrocarbon synthesis catalyst |
| FR2739038B1 (fr) | 1995-09-25 | 1997-11-21 | Inst Francais Du Petrole | Procede de conversion du gaz de synthese en presence d'un catalyseur comprenant du cobalt et des elements additionnels |
| US6475943B1 (en) * | 1995-11-08 | 2002-11-05 | Shell Oil Company | Catalyst activation process |
| FR2747054B1 (fr) * | 1996-04-09 | 1998-05-22 | Inst Francais Du Petrole | Procede de conversion du gaz de synthese en presence d'un catalyseur a base de cobalt et de titane |
| US6723230B1 (en) * | 1996-08-23 | 2004-04-20 | Exxonmobil Research & Engineering Company | Regeneration of iron-based hydrogen sulfide sorbents |
| US5953911A (en) * | 1998-02-04 | 1999-09-21 | Goal Line Environmental Technologies Llc | Regeneration of catalyst/absorber |
| US6579510B2 (en) * | 1999-07-30 | 2003-06-17 | Alfred E. Keller | SPOX-enhanced process for production of synthesis gas |
| JP2003519011A (ja) * | 2000-01-04 | 2003-06-17 | エクソン リサーチ アンド エンジニアリング カンパニー | 水素およびアンモニアによる炭化水素合成触媒の増強 |
| US6337353B1 (en) * | 2000-01-04 | 2002-01-08 | Exxonmobil Research And Engineering Company | Activation of hydrocarbon synthesis catalysts with hydrogen and ammonia |
| CA2340822C (en) * | 2000-03-17 | 2010-08-03 | Snamprogetti S.P.A. | Process for the production of hydrogen |
| WO2002008361A1 (en) * | 2000-07-21 | 2002-01-31 | Exxonmobil Research And Engineering Company | The use of hydrogen to regenerate metal oxide hydrogen sulfide sorbents |
-
2001
- 2001-06-28 GB GBGB0115850.0A patent/GB0115850D0/en not_active Ceased
-
2002
- 2002-06-21 DK DK02732977T patent/DK1399256T3/da active
- 2002-06-21 WO PCT/GB2002/002883 patent/WO2003002252A1/en not_active Ceased
- 2002-06-21 DE DE60209484T patent/DE60209484T2/de not_active Expired - Lifetime
- 2002-06-21 US US10/481,986 patent/US7183329B2/en not_active Expired - Fee Related
- 2002-06-21 AT AT02732977T patent/ATE318651T1/de not_active IP Right Cessation
- 2002-06-21 EP EP02732977A patent/EP1399256B1/de not_active Expired - Lifetime
- 2002-06-21 AU AU2002304467A patent/AU2002304467B2/en not_active Ceased
- 2002-06-21 CN CNB028129636A patent/CN100500283C/zh not_active Expired - Fee Related
-
2007
- 2007-01-04 US US11/649,655 patent/US7511080B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| EP1399256A1 (de) | 2004-03-24 |
| CN100500283C (zh) | 2009-06-17 |
| DE60209484D1 (de) | 2006-04-27 |
| US7183329B2 (en) | 2007-02-27 |
| US20070112080A1 (en) | 2007-05-17 |
| US7511080B2 (en) | 2009-03-31 |
| CN1541139A (zh) | 2004-10-27 |
| EP1399256B1 (de) | 2006-03-01 |
| DK1399256T3 (da) | 2006-07-10 |
| ATE318651T1 (de) | 2006-03-15 |
| US20040242941A1 (en) | 2004-12-02 |
| GB0115850D0 (en) | 2001-08-22 |
| AU2002304467B2 (en) | 2007-12-06 |
| WO2003002252A1 (en) | 2003-01-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60209484T2 (de) | Verfahren zum aktivieren eines getragenen katalysators der eine kobalthaltigen verbindung enthält | |
| DE69927032T2 (de) | Verfahren für autotherme reformierung einer kohlenwasserstoffeinspeisung | |
| DE60120692T2 (de) | Katalysatoren mit hoher kobaltoberfläche | |
| DE69905467T2 (de) | Katalytische teiloxidation mit einem rhodium-iridium legierungskatalysator | |
| US5447705A (en) | Oxidation catalyst and process for the partial oxidation of methane | |
| DE60224925T2 (de) | Katalytisches verfahren zum reformieren von kohlenwasserstoff unter verwendung des katalysators | |
| DE4102185C2 (de) | Katalysator zur Herstellung von Synthesegas durch Reformieren von leichten Kohlenwasserstoffen mit CO¶2¶ | |
| DE3882886T2 (de) | Verfahren und Katalysator für die Umwandlung von Synthesegas zu Kohlenwasserstoffen. | |
| DE69312598T2 (de) | Verfahren zur Umwandlung von Synthesegas in Kohlenwasserstoffe auf Kobalt basierender Katalysator | |
| DE69825577T2 (de) | Verfahren zur herstellung von synthesegas | |
| DE69606810T2 (de) | Verfahren zur Herstellung eines Katalysators zur Umwandlung von Synthesegas | |
| AU2002304467A1 (en) | A process for the activation of a catalyst comprising a cobalt compound and a support | |
| DE68903397T2 (de) | Traegerkatalysator fuer die nichtselektive oxydation von organischen verbindungen, verfahren fuer die nichtselektive oxydation, insbesondere von organischen verbindungen. | |
| DE10013895A1 (de) | Verfahren zur katalytischen Umsetzung von Kohlenmonoxid in einem Wasserstoff enthaltenden Gasgemisch | |
| DE69920379T2 (de) | Palladium-Ceroxid-Trägerkatalysator und Verfahren zur Herstellung von Methanol | |
| DE3103171A1 (de) | Katalysator zur methanbildung und verfahren zu seiner herstellung | |
| DE10393935T5 (de) | Fischer-Tropsch-Katalysatoren auf Eisen-Basis und Herstellungs- und Anwendungsverfahren | |
| EP2866930A1 (de) | Hochdruckverfahren zur kohlendioxid-reformierung von kohlenwasserstoffen in gegenwart von iridiumhaltigen aktivmassen | |
| DE112021000826T5 (de) | Katalysator für CO2-Methanisierungsreaktion mit hoher Aktivität und Langzeitstabilität sowie Verfahren dafür | |
| EP0695279B1 (de) | Verwendung eines katalysators zur herstellung von synthesegas | |
| DE112011105502T5 (de) | Redox-Material zum thermochemischen Wasserspalten und Verfahren zum Herstellen von Wasserstoff | |
| DE3686235T2 (de) | Hitzebestaendige zusammensetzung und verfahren zu deren herstellung. | |
| DE3586630T2 (de) | Ruthenium-rhenium-titaniumoxyd-katalysatoren und ihre verwendung zur fischer-tropsch-synthese. | |
| DE69717290T2 (de) | Fischer-tropsch katalysator und verfahren zur herstellung von kohlenwasserstoffen | |
| DE69128148T2 (de) | Katalysator basierend auf eisen/zink sowie korversion von synthesegas zu alpha-olefinen unter benutzung dieses katalysators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |