CN113166364A - 由具有超高分子量的聚乙烯纤维和交联多异氰酸酯生产复合材料的方法 - Google Patents
由具有超高分子量的聚乙烯纤维和交联多异氰酸酯生产复合材料的方法 Download PDFInfo
- Publication number
- CN113166364A CN113166364A CN201980033031.8A CN201980033031A CN113166364A CN 113166364 A CN113166364 A CN 113166364A CN 201980033031 A CN201980033031 A CN 201980033031A CN 113166364 A CN113166364 A CN 113166364A
- Authority
- CN
- China
- Prior art keywords
- polyisocyanate
- crosslinking
- fibers
- polyisocyanate composition
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/09—Processes comprising oligomerisation of isocyanates or isothiocyanates involving reaction of a part of the isocyanate or isothiocyanate groups with each other in the reaction mixture
- C08G18/092—Processes comprising oligomerisation of isocyanates or isothiocyanates involving reaction of a part of the isocyanate or isothiocyanate groups with each other in the reaction mixture oligomerisation to isocyanurate groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B33—ADDITIVE MANUFACTURING TECHNOLOGY
- B33Y—ADDITIVE MANUFACTURING, i.e. MANUFACTURING OF THREE-DIMENSIONAL [3D] OBJECTS BY ADDITIVE DEPOSITION, ADDITIVE AGGLOMERATION OR ADDITIVE LAYERING, e.g. BY 3D PRINTING, STEREOLITHOGRAPHY OR SELECTIVE LASER SINTERING
- B33Y70/00—Materials specially adapted for additive manufacturing
- B33Y70/10—Composites of different types of material, e.g. mixtures of ceramics and polymers or mixtures of metals and biomaterials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/09—Processes comprising oligomerisation of isocyanates or isothiocyanates involving reaction of a part of the isocyanate or isothiocyanate groups with each other in the reaction mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/166—Catalysts not provided for in the groups C08G18/18 - C08G18/26
- C08G18/168—Organic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/22—Catalysts containing metal compounds
- C08G18/225—Catalysts containing metal compounds of alkali or alkaline earth metals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/22—Catalysts containing metal compounds
- C08G18/24—Catalysts containing metal compounds of tin
- C08G18/244—Catalysts containing metal compounds of tin tin salts of carboxylic acids
- C08G18/246—Catalysts containing metal compounds of tin tin salts of carboxylic acids containing also tin-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/73—Polyisocyanates or polyisothiocyanates acyclic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/77—Polyisocyanates or polyisothiocyanates having heteroatoms in addition to the isocyanate or isothiocyanate nitrogen and oxygen or sulfur
- C08G18/78—Nitrogen
- C08G18/79—Nitrogen characterised by the polyisocyanates used, these having groups formed by oligomerisation of isocyanates or isothiocyanates
- C08G18/791—Nitrogen characterised by the polyisocyanates used, these having groups formed by oligomerisation of isocyanates or isothiocyanates containing isocyanurate groups
- C08G18/792—Nitrogen characterised by the polyisocyanates used, these having groups formed by oligomerisation of isocyanates or isothiocyanates containing isocyanurate groups formed by oligomerisation of aliphatic and/or cycloaliphatic isocyanates or isothiocyanates
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Ceramic Engineering (AREA)
- Civil Engineering (AREA)
- Composite Materials (AREA)
- Structural Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Polyurethanes Or Polyureas (AREA)
Abstract
本发明涉及由具有超高分子量的聚乙烯纤维和交联多异氰酸酯生产复合材料的方法,涉及可由此获得的复合材料并涉及这样的复合材料用于生产组件的用途,以及涉及由根据本发明的复合材料组成或含有根据本发明的复合材料的组件。
Description
本发明涉及由具有超高分子量的聚乙烯纤维和交联多异氰酸酯生产复合材料的方法,涉及可由此获得的复合材料并涉及这样的复合材料用于生产组件的用途,以及涉及由根据本发明的复合材料组成或含有根据本发明的复合材料的组件。
在化学工业中广泛使用基于聚合物(聚酰胺、聚乙烯等)的合成纤维。特别有意义的这一类的代表是基于聚乙烯(PE)的纤维,例如所谓的高性能聚乙烯纤维(HPPE)。这些通常由具有极高分子量(>500 kg/mol)的线性聚乙烯组成。它们因此也被称为超高分子量PE纤维(UHMWPE)。WO 2015/059268描述了UHMWPE的生产方法。EP 0 504 954还描述了具有500kg/mol以上的分子量的PE的优异材料性质。在此特别强调如耐磨性或耐化学品性之类的性质。
通常通过所谓的凝胶纺丝法获得由UHMWPE制成的纤维。EP 2 287 371和WO 2012/139934描述了通过这种方法生产UHMWPE纤维。由此可获得的纤维的特征本身在于高结晶度和优异的材料性质,如极高拉伸强度和弹性模量以及极低重量。实际上该纤维具有迄今已知的最高强度/重量比(P.K. Mallick;Fiber-Reinforced Composites – MaterialsManufacturing and Design;2008;CRC Press – Taylor & Francis Group;Boca Raton)。这样的纤维可以Dyneema或Spectra之类的商品名购得。这些纤维主要用于绳子、绳索和悬带。
原则上希望也在复合材料中利用UHMWPE纤维的所提到的材料性质,例如通过将纤维包埋在塑料树脂中。但是,这迄今只能在非常有限的程度上实现(P.K. Mallick;Fiber-Reinforced Composites – Materials Manufacturing and Design;2008;CRC Press –Taylor & Francis Group;Boca Raton)。尽管在文献中不乏这方面的尝试,但由于纤维的极差可润湿性和因此塑料树脂对纤维的相对差的附着力,UHMWPE纤维复合材料的有效生产尚未成功。
迄今没有充分解决的另一问题在于例如通过合适的非极性基质材料在较高温度下良好润湿纤维要么失败,因为尤其在升高的温度下,相容的基质材料通过溶解造成晶体结构的破坏(例如基于苯乙烯和/或丁二烯的非极性化合物),因此使强度受损,要么基质的固化过程的必要温度超过大约150℃的纤维的温度稳定性并且由于不受控熔融和重结晶过程而破坏对UHMWPE纤维的强度负责的纤维材料的显著长程有序。
已经令人惊讶地发现,当基于具有至少200的异氰酸酯基团/异氰酸酯反应性基团比的异氰酸酯制剂的塑料以液体形式且以>10重量%的异氰酸酯浓度(在此被定义为反应性树脂组分中的异氰酸酯基团的重量比例)与UHMWPE纤维接触并在<150℃的反应温度下在UHMWPE纤维存在下反应时适合作为UHMWPE纤维的包埋树脂,其中所用异氰酸酯的>50%以通过三聚的形式反应以提供对称或不对称聚异氰脲酸酯。
在第一实施方案中,本发明涉及由聚合物纤维和交联多异氰酸酯生产复合材料的方法,其包括下列步骤:
a) 提供含有多异氰酸酯的多异氰酸酯组合物A,和
b) 多异氰酸酯组合物A在至少一种聚合物纤维B和至少一种交联催化剂C存在下催化交联以提供由聚合物纤维和交联多异氰酸酯制成的复合材料。
复合材料在本申请的意义上的特征在于聚合物纤维B包埋在通过多异氰酸酯组合物中包含的多异氰酸酯的催化交联形成的聚合物基质中。该复合材料可具有用所用生产方法可实现的各种任意形状。
在一个优选实施方案中,根据本发明的方法的特征在于不需要进行聚合物纤维B的预处理以使其与多异氰酸酯组合物A相容。特别地,根据本发明的复合材料可根据上述方法生产而不在实施方法步骤b)之前对聚合物纤维B施以气体等离子体处理,用波长< 400nm的紫外线照射或进行氧化处理,特别是用过氧化物、氧化性酸或臭氧。
用有机溶剂、无机溶剂或通过机械处理清洗聚合物纤维不被理解为是本申请中的增容。
多异氰酸酯组合物A
本文所用的术语“多异氰酸酯”是在分子中含有两个或更多个异氰酸酯基团(这被本领域技术人员理解为是指通用结构–N=C=O的游离异氰酸酯基团)的化合物的集合术语。这些多异氰酸酯的最简单和最重要的代表是二异氰酸酯。这些具有通用结构O=C=N–R–N=C=O,其中R通常代表脂族、脂环族和/或芳族基团。
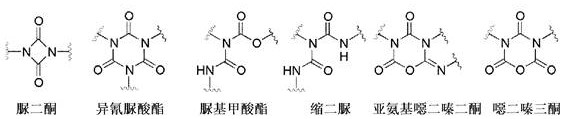
由于多官能度(至少两个异氰酸酯基团),可以由多异氰酸酯生产多种聚合物(例如聚氨酯、聚脲和聚异氰脲酸酯)和低分子量化合物(例如具有脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构的那些)。
当在本文中以通用术语提到“多异氰酸酯”时,这相同地是指单体和/或低聚多异氰酸酯。但是,为了理解本发明的许多方面,重要的是区分单体二异氰酸酯和低聚多异氰酸酯。当在本文中提到“低聚多异氰酸酯”时,这是指由至少两个单体二异氰酸酯分子形成的多异氰酸酯,即构成或含有由至少两个单体二异氰酸酯分子形成的反应产物的化合物。
由单体二异氰酸酯生产低聚多异氰酸酯在本申请中也被称为单体二异氰酸酯的改性。本文所用的这种“改性”在此是指单体二异氰酸酯的反应以产生具有脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构的低聚多异氰酸酯。
例如,己二异氰酸酯(HDI)是“单体二异氰酸酯”,因为其含有两个异氰酸酯基团并且不是至少两个多异氰酸酯分子的反应产物:
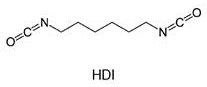
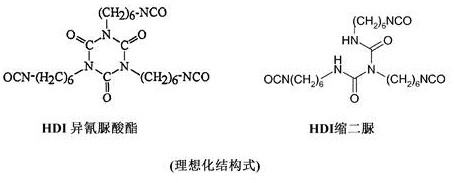
相反,仍具有至少两个异氰酸酯基团的至少两个HDI分子的反应产物是本发明的意义上的“低聚多异氰酸酯”。从单体HDI出发,这样的“低聚多异氰酸酯”的代表是例如HDI异氰脲酸酯和HDI缩二脲,它们各自由三个单体HDI单元形成:
“多异氰酸酯组合物A”在本发明的意义上是指初始反应混合物中的异氰酸酯组分。换言之,这是初始反应混合物中的所有具有异氰酸酯基团的化合物的总和。多异氰酸酯组合物A因此用作本发明的方法中的反应物。当在本文中提到“多异氰酸酯组合物A”,尤其是“提供多异氰酸酯组合物A”时,这是指多异氰酸酯组合物A作为反应物存在并用作反应物。
原则上,单体和低聚多异氰酸酯同等适用于根据本发明的多异氰酸酯组合物A。因此,多异氰酸酯组合物A可基本由单体多异氰酸酯或基本由低聚多异氰酸酯组成。或者,其可包含任意混合比的低聚和单体多异氰酸酯。
在本发明的一个优选实施方案中,用作交联中的反应物的多异氰酸酯组合物A具有低单体含量(即低单体二异氰酸酯含量)并已含有低聚多异氰酸酯。术语“具有低单体含量”和“具有低单体二异氰酸酯含量”在本文中相对于多异氰酸酯组合物A同义使用。
由于使用单体多异氰酸酯时生成比使用相应质量的低聚多异氰酸酯时更多的热,使用具有极高比例的单体多异氰酸酯的多异氰酸酯组合物A时具有形成的复合材料部件中的温度在催化交联的过程中超过所用PE纤维的熔点的风险。因此优选调节多异氰酸酯组合物中的单体多异氰酸酯的比例以使催化交联过程中的温度不超过150℃,优选140℃,特别优选130℃。导致超过上述温度限的单体多异氰酸酯的比例取决于另外的参数,特别是要生产的工件的形状,即表面积/体积比,取决于工件总重量中的纤维状填料比例以及取决于反应速率和输出反应热的可能性。后者又基本取决于所用催化剂的类型和浓度。但是,在个别情况下可以简单方式通过在催化交联的过程中在组件上的各种点使用温度传感器测量温度来测定单体多异氰酸酯的可能最大比例。因此可由本领域技术人员用常规方法实验测定临界极限。
当多异氰酸酯组合物A具有在每种情况下基于多异氰酸酯组合物A的重量计最多80重量%,尤其最多50重量%或最多20重量%的多异氰酸酯组合物A中的单体二异氰酸酯比例时,建立特别有实用意义的结果。优选的是,多异氰酸酯组合物A具有最多5重量%,尤其最多2.0重量%,更优选最多1.0重量%的单体二异氰酸酯含量,在每种情况下基于多异氰酸酯组合物A的重量计。当聚合物组合物A基本不含单体二异氰酸酯时,建立特别好的结果。“基本不含”在此是指单体二异氰酸酯的含量为基于多异氰酸酯组合物A的重量计最多0.5重量%。
在本发明的一个特别优选的实施方案中,多异氰酸酯组合物A完全或在至少80、85、90、95、98、99或99.5重量%的程度上由低聚多异氰酸酯组成,在每种情况下基于多异氰酸酯组合物A中包含的单体和低聚多异氰酸酯的重量计。在此优选的是至少99重量%的低聚多异氰酸酯含量。低聚多异氰酸酯的这种含量基于如供应时的多异氰酸酯组合物A。换言之,低聚多异氰酸酯不是在本发明的方法的过程中作为中间体形成的,而是在反应开始时已存在于用作反应物的多异氰酸酯组合物A中。
具有低单体含量或基本不含单体异氰酸酯的多异氰酸酯组合物可通过在每种情况下在实际改性反应后进行至少一个用于分离出未转化的过量单体二异氰酸酯的进一步方法步骤获得。可以通过本身已知的方法,优选通过在高真空下的薄膜蒸馏或通过用对异氰酸酯基团呈惰性的合适溶剂,例如脂族或脂环族烃,如戊烷、己烷、庚烷、环戊烷或环己烷萃取而以特别实用的方式实现这种单体分离。
在本发明的一个优选实施方案中,根据本发明的多异氰酸酯组合物A通过将单体二异氰酸酯改性和随后分离出未转化的单体获得。
但是,在本发明的一个特定实施方案中,具有低单体含量的多异氰酸酯组合物A含有外部单体二异氰酸酯。在本文中,“外部单体二异氰酸酯”是指其不同于已用于生产多异氰酸酯组合物A中包含的低聚多异氰酸酯的单体二异氰酸酯。
外部单体二异氰酸酯的加入可能有利于实现特殊技术效果,例如特定硬度。当多异氰酸酯组合物A具有在每种情况下基于多异氰酸酯组合物A的重量计最多50重量%,优选最多35重量%,更优选最多20重量%,最优选最多10重量%的多异氰酸酯组合物A中的外部单体二异氰酸酯的比例时,建立特别有实用意义的结果。优选的是,多异氰酸酯组合物A具有在每种情况下基于多异氰酸酯组合物A的重量计最多5重量%,优选最多2.0重量%,更优选最多1.0重量%的外部单体二异氰酸酯含量。
在根据本发明的方法的另一特定实施方案中,多异氰酸酯组合物A含有单体单异氰酸酯或具有大于2的异氰酸酯官能度,即具有每分子多于两个异氰酸酯基团的单体异氰酸酯。已经发现单体单异氰酸酯或具有大于2的异氰酸酯官能度的单体异氰酸酯的加入是有利的以影响该材料的网络密度。当多异氰酸酯组合物A具有在每种情况下基于多异氰酸酯组合物A的重量计最多20重量%,尤其最多15重量%或最多10重量%的多异氰酸酯组合物A中的单体单异氰酸酯或具有大于2的异氰酸酯官能度的单体异氰酸酯的比例时,建立特别有实用意义的结果。优选地,多异氰酸酯组合物A具有在每种情况下基于多异氰酸酯组合物A的重量计最多5重量%,尤其最多2.0重量%,更优选最多1.0重量%的单体单异氰酸酯或具有大于2的异氰酸酯官能度的单体异氰酸酯的含量。优选的是,在根据本发明的交联反应中不一起使用单体单异氰酸酯或具有大于2的异氰酸酯官能度的单体异氰酸酯。
根据本发明,低聚多异氰酸酯特别可具有脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构。在本发明的一个实施方案中,低聚多异氰酸酯具有下列低聚结构类型的至少一种或其混合物:
在本发明的一个优选实施方案中,使用聚合物组合物A,其中异氰脲酸酯结构比例为基于多异氰酸酯组合物A中存在的选自脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和噁二嗪三酮结构的低聚结构的总和计至少50摩尔%,优选至少60摩尔%,更优选至少70摩尔%,再更优选至少80摩尔%,再更优选至少90摩尔%,尤其优选至少95摩尔%。
在本发明的另一个优选的实施方案中,在根据本发明的方法中,使用除异氰脲酸酯结构外还含有至少一种具有脲二酮、缩二脲、脲基甲酸酯、亚氨基噁二嗪二酮和噁二嗪三酮结构及其混合物的附加低聚多异氰酸酯的多异氰酸酯组合物A。
可以例如通过NMR谱法测定多异氰酸酯A中的脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构的比例。在此可以优选使用13C NMR谱法,优选为质子解耦形式,因为所提到的低聚结构产生特征信号。
无论基础低聚结构如何(脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构),用于本发明的方法的低聚多异氰酸酯组合物A和/或其中包含的低聚多异氰酸酯优选具有2.0至5.0,优选2.3至4.5的(平均)NCO官能度。
当根据本发明使用的多异氰酸酯组合物A具有在每种情况下基于多异氰酸酯组合物A的重量计8.0重量%至28.0重量%,优选14.0重量%至25.0重量%的异氰酸酯基团含量时,建立特别有实用意义的结果。所述异氰酸酯基团可以是封闭或自由形式的。上文提到的异氰酸酯含量此时基于除去封闭剂后异氰酸酯基团的理论比例。
根据本发明用于多异氰酸酯组合物A的具有脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构的低聚多异氰酸酯的生产方法例如描述在J. Prakt. Chem. 336 (1994) 185 - 200、DE-A 1 670 666、DE-A 1 954 093、DE-A 2 414413、DE-A 2 452 532、DE-A 2 641 380、DE-A 3 700 209、DE-A 3 900 053和DE-A 3 928503或EP-A 0 336 205、EP-A 0 339 396和EP-A 0 798 299中。
在本发明的一个附加或替代性的实施方案中,根据本发明的多异氰酸酯组合物A被定义为其含有无论所用改性反应的性质如何已由单体二异氰酸酯在遵循5%至45%,优选10%至40%,更优选15%至30%的低聚度的情况下获得的低聚多异氰酸酯。“低聚度”在此被理解为是指在生产过程中为形成脲二酮、异氰脲酸酯、脲基甲酸酯、缩二脲、亚氨基噁二嗪二酮和/或噁二嗪三酮结构而消耗的最初存在于起始混合物中的异氰酸酯基团的百分比。
用于生产用于本发明的方法的多异氰酸酯组合物A和其中包含的单体和/或低聚多异氰酸酯的合适多异氰酸酯是可以各种方式获得的任意多异氰酸酯,例如通过在液相或气相中光气化或通过无光气途径,例如通过热氨基甲酸酯裂解。当该多异氰酸酯是单体二异氰酸酯时,建立特别好的结果。优选的单体二异氰酸酯是具有140至400 g/mol的分子量、具有脂族、脂环族、芳脂族和/或芳族键合的异氰酸酯基团的那些,例如1,4-丁烷二异氰酸酯(BDI)、1,5-戊烷二异氰酸酯(PDI)、1,6-己烷二异氰酸酯(HDI)、2-甲基-1,5-戊烷二异氰酸酯、1,5-二异氰酸根合-2,2-二甲基戊烷、2,2,4-或2,4,4-三甲基-1,6-己烷二异氰酸酯、1,10-癸烷二异氰酸酯、1,3-和1,4-环己烷二异氰酸酯、1,4-二异氰酸根合-3,3,5-三甲基环己烷、1,3-二异氰酸根合-2-甲基环己烷、1,3-二异氰酸根合-4-甲基环己烷、1-异氰酸根合-3,3,5-三甲基-5-异氰酸根合甲基环己烷(异佛尔酮二异氰酸酯;IPDI)、1-异氰酸根合-1-甲基-4(3)-异氰酸根合甲基环己烷、2,4'-和4,4'-二环己基甲烷二异氰酸酯(H12MDI)、1,3-和1,4-双(异氰酸根合甲基)环己烷、双(异氰酸根合甲基)降冰片烷(NBDI)、4,4'-二异氰酸根合-3,3'-二甲基二环己基甲烷、4,4'-二异氰酸根合-3,3',5,5'-四甲基二环己基甲烷、4,4'-二异氰酸根合-1,1'-二(环己基)、4,4'-二异氰酸根合-3,3'-二甲基-1,1'-二(环己基)、4,4'-二异氰酸根合-2,2',5,5'-四甲基-1,1'-二(环己基)、1,8-二异氰酸根合-对薄荷烷、1,3-金刚烷二异氰酸酯、1,3-二甲基-5,7-金刚烷二异氰酸酯、1,3-和1,4-双(异氰酸根合甲基)苯(苯二甲基二异氰酸酯;XDI)、1,3-和1,4-双(1-异氰酸根合-1-甲基乙基)苯(TMXDI)和双(4-(1-异氰酸根合-1-甲基乙基)苯基)碳酸酯、2,4-和2,6-甲苯二异氰酸酯(TDI)、2,4'-和4,4'-二苯甲烷二异氰酸酯(MDI)、1,5-萘二异氰酸酯和这些二异氰酸酯的任意混合物。同样合适的其它二异氰酸酯另外可见于例如Justus Liebigs Annalen derChemie, 第562卷(1949)第75-136页。
可任选用于多异氰酸酯组合物A的合适的单体单异氰酸酯是例如异氰酸正丁酯、异氰酸正戊酯、异氰酸正己酯、异氰酸正庚酯、异氰酸正辛酯、异氰酸十一烷基酯、异氰酸十二烷基酯、异氰酸十四烷基酯、异氰酸鲸蜡酯、异氰酸硬脂酯、异氰酸环戊酯、异氰酸环己酯、异氰酸3-或4-甲基环己酯或这些单异氰酸酯的任意混合物。可任选添加到多异氰酸酯组合物A中的具有大于2的异氰酸酯官能度的单体异氰酸酯的实例是4-异氰酸根合甲基辛烷1,8-二异氰酸酯(壬烷三异氰酸酯;TIN)。
在本发明的一个实施方案中,多异氰酸酯组合物A含有在每种情况下基于多异氰酸酯组合物A的重量计最多30重量%,尤其是最多20重量%、最多15重量%、最多10重量%、最多5重量%或最多1重量%的芳族多异氰酸酯。本文所用的“芳族多异氰酸酯”是指具有至少一个芳族键合的异氰酸酯基团的多异氰酸酯。
芳族键合的异氰酸酯基团被理解为是指键合到芳烃基上的异氰酸酯基团。
在本发明的方法的一个优选实施方案中,使用仅具有脂族和/或脂环族键合的异氰酸酯基团的多异氰酸酯组合物A。
脂族和脂环族键合的异氰酸酯基团分别被理解为是指键合到脂族和脂环族烃基上的异氰酸酯基团。在本发明的方法的另一优选实施方案中,使用由一种或多种低聚多异氰酸酯组成或包含一种或多种低聚多异氰酸酯的多异氰酸酯组合物A,其中所述一种或多种低聚多异氰酸酯仅具有脂族和/或脂环族键合的异氰酸酯基团。
在本发明的另一个实施方案中,多异氰酸酯组合物A在至少50、70、85、90、95、98或99重量%的程度上由仅具有脂族和/或脂环族键合的异氰酸酯基团的多异氰酸酯组成,在每种情况下基于多异氰酸酯组合物A的重量计。实际实验已经表明,用其中包含的低聚多异氰酸酯仅具有脂族和/或脂环族键合的异氰酸酯基团的多异氰酸酯组合物A可实现特别好的结果。
在本发明的方法的一个特别优选的实施方案中,使用由一种或多种低聚多异氰酸酯组成或包含一种或多种低聚多异氰酸酯的多异氰酸酯组合物A,其中所述一种或多种低聚多异氰酸酯基于1,4-丁烷二异氰酸酯(BDI)、1,5-戊烷二异氰酸酯(PDI)、1,6-己烷二异氰酸酯(HDI)、异佛尔酮二异氰酸酯(IPDI)或4,4'-二环己基甲烷二异氰酸酯(H12MDI)或其混合物形成。含低聚HDI的多异氰酸酯组合物A在此优选。
在本发明的一个特别优选的实施方案中,多异氰酸酯组合物A的特征进一步在于其在催化交联前具有最多45 mN/m,优选最多40 mN/m,非常特别优选最多35 mN/m的表面张力并在交联后具有最多50 mN/m,优选最多45 mN/m,非常特别优选最多40 mN/m的表面能。
在一个优选实施方案中,多异氰酸酯组合物A和根据本发明由其在多异氰酸酯组合物A交联后可获得的聚合物的表面张力之间的能量Δ为至少2 mN/m和最多20 mN/m,优选至少4 mN/m和最多15 mN/m,特别优选至少6 mN/m和最多12 mN/m。
在一个特别优选的实施方案中,多异氰酸酯组合物A的表面张力(能)比根据本发明使用的聚合物纤维的表面能小最多5 mN/m和大最多10 mN/m,并且根据本发明可获得的聚合物组合物A的交联聚合物的表面能比根据本发明使用的聚合物纤维的表面能大至少1mN/m和大最多20 mN/m。
发现根据本发明的多异氰酸酯组合物A与根据本发明可由其获得的交联聚合物的表面张力和表面能的所列比率特别有利于实现根据本发明的聚合物纤维的表面的良好润湿。
还已经令人惊讶地发现,根据本发明使用的多异氰酸酯组合物A的相对低表面张力(能),与转化成根据本发明可获得的交联聚合物后表面能的相对小变化结合,使得能够在特别具有低表面能的聚合物纤维的初始润湿中实现特别好的结果。已经进一步发现,当形成的聚合物相的表面能仅在本发明的范围内改变时,根据本发明交联的多异氰酸酯组合物A的所得聚合物的附着力特别好。
所列表面张力和表面能在每种情况下通过本领域技术人员常用的方法在23℃下测定。优选通过动态法,例如气泡压力法测量表面张力。
优选在此通过使用试验墨水的接触角法或Wilhelmy法(用于纤维的单纤维法)测定交联多异氰酸酯组合物A的聚合物表面和聚合物纤维的表面能。
在进一步优选的实施方案中,在聚合物纤维复合材料的形成过程中在交联过程中所用多异氰酸酯组合物A的收缩率在纤维方向上是垂直于纤维方向的<1/1.5。
在进一步优选的实施方案中,在聚合物纤维复合材料的形成过程中在交联过程中所用多异氰酸酯组合物A的收缩率< 10%,优选< 6%,特别优选< 5%,非常特别优选< 4%。
聚合物纤维B
各种合成纤维原则上都适合作为聚合物纤维B)。聚合物纤维B)优选选自纤维素纤维、再生蛋白质纤维、聚交酯纤维、甲壳质纤维、聚酯纤维、聚酰胺纤维、聚酰亚胺纤维、聚二酰亚胺纤维、聚丙烯酸系纤维、聚丙烯腈纤维、聚四氟乙烯纤维、聚氯化物纤维、聚氨酯纤维、聚丙烯纤维和聚乙烯纤维。
聚合物纤维更优选非极性。特别优选的非极性聚合物纤维是聚乙烯和聚丙烯纤维。非常特别优选的是,聚合物纤维B)是聚乙烯纤维。尤其优选的是下文定义的超高分子量聚乙烯纤维(UHMWPE)。
术语“聚合物纤维B”还是指至少两种上述类型的聚合物纤维的组合。但是,优选使用仅由上述类型之一的纤维组成的聚合物纤维B。
术语“超高分子量聚乙烯纤维”涉及由聚乙烯(PE)制成的纤维。该PE具有至少360kg/mol,更优选至少500 kg/mol,再更优选至少1000 kg/mol,最优选至少1600 kg/mol的数均摩尔质量。在此优选不超过11400 kg/mol的上限。数均摩尔质量特别优选在500 kg/mol至8400 kg/mol,非常特别优选1600 kg/mol至8400 kg/mol的范围内。
在遵循上述数均分子量的情况下根据本发明可用的PE纤维的多分散性(重均摩尔质量与数均摩尔质量的比率)为最多4.0;优选最多3.5;更优选最多3.0,最优选最多2.8。在此,多分散性的下限为至少1.1。
优选纤维的拉伸强度大于2500 N/mm2。聚乙烯链的平行取向优选为至少80%,更优选至少90%,特别优选至少95%。
特别合适的纤维可以“Dyneema”商标购自Koninklijke DSM N.V.和以“Spectra”商标购自Honeywell International Inc.。
根据本发明合适的纤维可通过EP 2 287 371、WO 2012/139934和WO 2014/187948中描述的方法获得。
纤维可单向排列,即互相平行。但是,根据本发明也可以使用机织物和针织物。这些可排列在一个或多个层中。单向取向纤维与机织物和/或针织物的组合根据本发明也有可能。
在本发明的一个优选实施方案中,聚异氰脲酸酯复合材料中的纤维含量基于聚异氰脲酸酯复合材料计大于3重量%,优选大于10重量%,更优选大于15重量%,优选大于20重量%,再更优选大于30重量%,尤其是50重量%、60重量%、70重量%。
原则上,聚乙烯纤维表现出与聚合物基质的差结合并且需要通过合适的预处理增容。这可例如通过如Bahramian等人, 2015, “Ultra-high-molecular-weightpolyethylene fiber reinforced dental composites: Effect of fiber surfacetreatment on mechanical properties of the composites" Dental Materials, 第31卷, 1022至1029描述的硅烷化和/或电晕处理实施。但是在本专利申请基于的研究中令人惊讶地发现,当使用根据本发明的异氰脲酸酯塑料作为基质时不需要PE纤维的预处理。在合适的溶剂,优选丙酮中清洗就足以确保PE纤维与基质材料的充分附着。
交联催化剂C
可用于交联反应的催化剂C包括基本所有在最多150℃,优选最多130℃,特别优选最多100℃的反应温度下催化异氰酸酯基团的交联以提供选自异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的至少一种结构的催化剂。
特别优选的交联催化剂C是加速异氰酸酯基团三聚成异氰脲酸酯或脲二酮结构的化合物。由于根据所用催化剂,结构的形成经常伴随着副反应,例如三聚形成亚氨基噁二嗪二酮(所谓的不对称三聚体)并且当起始多异氰酸酯中存在氨基甲酸酯基团时伴随着脲基甲酸酯化反应,术语“三聚”在本发明中应被理解为也同义地表示这些另外发生的反应。
在一个特定实施方案中,根据本发明的催化剂可优选通过脲二酮形成的中间步骤催化三聚。
用于本发明的方法的合适的催化剂C是例如简单叔胺,例如三乙基胺、三丁基胺、N,N-二甲基苯胺、N-乙基哌啶或N,N'-二甲基哌嗪。合适的催化剂还包括GB 2 221 465中描述的叔羟基烷基胺,例如三乙醇胺、N-甲基二乙醇胺、二甲基乙醇胺、N-异丙基二乙醇胺和1-(2-羟乙基)吡咯烷,或从GB 2 222 161中获知的催化剂体系,其由叔双环胺,例如DBU与低分子量的简单脂族醇的混合物组成。
适用于本发明的方法的另一些三聚催化剂C是例如从DE-A 1 667 309、EP-A 0013 880和EP-A 0 047 452中获知的季铵氢氧化物,例如四乙基氢氧化铵、三甲基苄基氢氧化铵、N,N-二甲基-N-十二烷基-N-(2-羟乙基)氢氧化铵、N-(2-羟乙基)-N,N-二甲基-N-(2,2'-二羟甲基丁基)氢氧化铵和1-(2-羟乙基)-1,4-二氮杂双环[2.2.2]辛烷氢氧化物(环氧乙烷和水加成到1,4-二氮杂双环[2.2.2]辛烷上的单加成物),从EP-A 37 65或EP-A 10589中获知的季羟基烷基铵氢氧化物,例如N,N,N-三甲基-N-(2-羟乙基)氢氧化铵,从DE-A2631733、EP-A 0 671 426、EP-A 1 599 526和US 4,789,705中获知的三烷基羟烷基铵羧酸盐,例如对叔丁基苯甲酸N,N,N-三甲基-N-2-羟丙基铵和2-乙基己酸N,N,N-三甲基-N-2-羟丙基铵,从EP-A 1 229 016中获知的季苄基铵羧酸盐,例如特戊酸N-苄基-N,N-二甲基-N-乙基铵、2-乙基己酸N-苄基-N,N-二甲基-N-乙基铵、2-乙基己酸N-苄基-N,N,N-三丁基铵、2-乙基己酸N,N-二甲基-N-乙基-N-(4-甲氧基苄基)铵或特戊酸N,N,N-三丁基-N-(4-甲氧基苄基)铵,从WO 2005/087828中获知的四取代铵α-羟基羧酸盐,例如四甲基乳酸铵,从EP-A 0 339 396、EP-A 0 379 914和EP-A 0 443 167中获知的季铵或鏻氟化物,例如具有C8-C10-烷基的N-甲基-N,N,N-三烷基铵氟化物、氟化N,N,N,N-四-正丁基铵、氟化N,N,N-三甲基-N-苄基铵、氟化四甲基鏻、氟化四乙基鏻或氟化四正丁基鏻,从EP-A 0 798 299、EP-A 0896 009和EP-A 0 962 455中获知的季铵和鏻多氟化物,例如氢多氟化苄基三甲基铵,从EP-A 0 668 271中获知并可通过叔胺与碳酸二烷基酯的反应获得的烷基碳酸四烷基铵,或甜菜碱结构化的季铵烷基碳酸盐,从WO 1999/023128中获知的季铵碳酸氢盐,例如胆碱碳酸氢盐,从EP 0 102 482中获知并可由叔胺与含磷酸的起烷基化作用的酯获得的季铵盐,例如三乙胺、DABCO或N-甲基吗啉与甲基膦酸二甲酯的反应产物,或从WO 2013/167404中获知的内酰胺的四取代铵盐,例如三辛基铵己内酰胺盐或十二烷基三甲基铵己内酰胺盐。
合适的是具有最多14个碳原子的直链或支化烷羧酸,例如丁酸、戊酸、己酸、2-乙基己酸、庚酸、辛酸、壬酸和更高级同系物的已知钠和钾盐。
同样适合作为用于本发明的方法的三聚催化剂C是多种不同的金属化合物。合适的实例是在DE-A 3 240 613中作为催化剂描述的锰、铁、钴、镍、铜、锌、锆、铈或铅的辛酸盐和环烷酸盐或其与锂、钠、钾、钙或钡的乙酸盐的混合物、DE-A 3 219 608公开的具有最多10个碳原子的直链或支化烷羧酸,如丙酸、丁酸、戊酸、己酸、庚酸、辛酸、壬酸、癸酸和十一烷酸的钠和钾盐,EP-A 0 100 129公开的具有2至20个碳原子的脂族、脂环族或芳族单羧酸和多羧酸的碱金属或碱土金属盐,如苯甲酸钠或苯甲酸钾,GB-PS 1 391 066和GB-PS 1386 399公开的碱金属酚盐,如苯酚钠或苯酚钾,GB 809 809公开的碱金属和碱土金属氧化物、氢氧化物、碳酸盐、醇盐和酚盐,可烯醇化的化合物的碱金属盐和弱脂族或脂环族羧酸的金属盐,如甲醇钠、乙酸钠、乙酸钾、乙酰乙酸钠、2-乙基己酸铅和环烷酸铅,EP-A 0 056158和EP-A 0 056 159公开的与冠醚或聚醚醇络合的碱性碱金属化合物,如络合的羧酸钠或羧酸钾,和/或EP-A 0 033 581公开的吡咯烷酮钾盐,申请EP 13196508.9公开的钛、锆和/或铪的单核或多核络合物,如四正丁氧基锆、四-2-乙基己酸锆和四-2-乙基己醇锆,和European Polymer Journal, 第16卷, 147-148 (1979)中描述的类型的锡化合物,如二氯化二丁基锡、二氯化二苯基锡、三苯基锡烷醇、乙酸三丁基锡、辛酸锡、二丁基(二甲氧基)锡烷和咪唑三丁基锡。
适用于本发明的方法的另一些交联催化剂可见于例如J. H. Saunders和K. C.Frisch, Polyurethanes Chemistry and Technology, 第94页及其后(1962)和其中引用的文献。
催化剂C可以独自或以与彼此的任意混合物的形式用于根据本发明的方法。
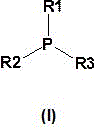
特别适用于根据本发明的方法的是通式(I)的有机膦催化剂,或通式(I)的这些叔有机膦催化剂的混合物
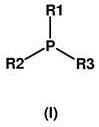
其中
R1、R2和R3是相同或不同基团并且各自是具有最多10个碳原子的烷基或环烷基,优选具有2至8个碳原子的烷基或具有3至8个碳原子的环烷基,具有7至10,优选7个碳原子的芳烷基或具有6至10,优选6个碳原子并任选被具有最多10个,优选1至6个碳原子的烷基取代的芳基,条件是这些基团的最多一个是芳基且这些基团的至少一个是烷基或环烷基,或其中
R1和R2在性质上是脂族的并与磷原子一起互相连接形成具有4至6个环成员的杂环环,其中R3是具有最多4个碳原子的烷基。
合适的叔有机膦催化剂是例如具有直链脂族取代基的叔膦,如三甲基膦、三乙基膦、三正丙基膦、三丙基膦、二丁基乙基膦、三正丁基膦、三异丁基膦、三叔丁基膦、戊基二甲基膦、戊基二乙基膦、戊基二丙基膦、戊基二丁基膦、戊基二己基膦、二戊基甲基膦、二戊基乙基膦、二戊基丙基膦、二戊基丁基膦、二戊基己基膦、二戊基辛基膦、三戊基膦、己基二甲基膦、己基二乙基膦、己基二丙基膦、己基二丁基膦、二己基甲基膦、二己基乙基膦、二己基丙基膦、二己基丁基膦、三己基膦、三辛基膦、三苄基膦、苄基二甲基膦、二甲基苯基膦或丁基磷杂环戊烷。
适用于根据本发明的方法的另一些叔有机膦催化剂是例如从EP 1 422 223 A1中获知的具有至少一个直接键合到磷上的脂环族基团的叔膦,例如环戊基二甲基膦、环戊基二乙基膦、环戊基二正丙基膦、环戊基二异丙基膦、具有任意异构丁基的环戊基二丁基膦、具有任意异构己基的环戊基二己基膦、具有任意异构辛基的环戊基二辛基膦、二环戊基甲基膦、二环戊基乙基膦、二环戊基-正丙基膦、二环戊基异丙基膦、具有任意异构丁基的二环戊基丁基膦、具有任意异构己基的二环戊基己基膦、具有任意异构辛基的二环戊基辛基膦、三环戊基膦、环己基二甲基膦、环己基二乙基膦、环己基二正丙基膦、环己基二异丙基膦、具有任意异构丁基的环己基二丁基膦、具有任意异构己基的环己基二己基膦、具有任意异构辛基的环己基二辛基膦、二环己基甲基膦、二环己基乙基膦、二环己基-正丙基膦、二环己基异丙基膦、具有任意异构丁基的二环己基丁基膦、具有任意异构己基的二环己基己基膦、具有任意异构辛基的二环己基辛基膦和三环己基膦。
用于根据本发明的方法的另一些合适的叔有机膦催化剂是例如从EP 1 982 979A1中获知并具有一个或两个直接键合到磷上的叔烷基的叔膦,例如叔丁基二甲基膦、叔丁基二乙基膦、叔丁基二正丙基膦、叔丁基二异丙基膦、具有任意异构丁基作为非叔丁基的叔丁基二丁基膦、具有任意异构己基但其中最多一个己基具有直接键合到磷上的叔碳原子的叔丁基二己基膦、具有任意异构辛基但其中最多一个辛基具有直接键合到磷上的叔碳原子的叔丁基二辛基膦、二叔丁基甲基膦、二叔丁基乙基膦、二叔丁基正丙基膦、二叔丁基异丙基膦、其中非叔丁基可以是正丁基、异丁基、2-丁基或环丁基的二叔丁基丁基膦、具有任意异构己基(其没有直接键合到磷上的叔碳原子)的二叔丁基己基膦、具有任意异构辛基(其没有直接键合到磷上的叔碳原子)的二叔丁基辛基膦、叔戊基二甲基膦、叔戊基二乙基膦、叔戊基二正丙基膦、叔戊基二异丙基膦、具有任意异构丁基但其中最多一个丁基是叔丁基的叔戊基二丁基膦、具有任意异构己基但其中最多一个己基具有直接键合到磷上的叔碳原子的叔戊基二己基膦、具有任意异构辛基但其中最多一个辛基具有直接键合到磷上的叔碳原子的叔戊基二辛基膦、二叔戊基乙基膦、二叔戊基乙基膦、二叔戊基正丙基膦、二叔戊基异丙基膦、其中丁基可以是正丁基、异丁基、2-丁基或环丁基的二叔戊基丁基膦、具有任意异构己基(其没有直接键合到磷上的叔碳原子)的二叔戊基己基膦、具有任意异构辛基(其没有直接键合到磷上的叔碳原子)的二叔戊基辛基膦、金刚烷基二甲基膦、金刚烷基二乙基膦、金刚烷基二正丙基膦、金刚烷基二异丙基膦、具有任意异构丁基但其中最多一个丁基具有直接键合到磷上的叔碳原子的金刚烷基二丁基膦、具有任意异构己基但其中最多一个己基具有直接键合到磷上的叔碳原子的金刚烷基二己基膦、具有任意异构辛基但其中最多一个辛基具有直接键合到磷上的叔碳原子的金刚烷基二辛基膦、二金刚烷基甲基膦、二金刚烷基乙基膦、二金刚烷基正丙基膦、二金刚烷基异丙基膦、其中丁基可以是正丁基、异丁基、2-丁基或环丁基的二金刚烷基丁基膦、具有任意异构己基(其没有直接键合到磷上的叔碳原子)的二金刚烷基己基膦和具有任意异构己基(其没有直接键合到磷上的叔碳原子)的二金刚烷基辛基膦。
在根据本发明的方法中,叔有机膦催化剂优选选自所提到的具有直链脂族取代基的叔膦。
非常特别优选的叔有机膦催化剂是三正丁基膦和/或三辛基膦。
在根据本发明的方法中,叔有机膦催化剂通常以基于所用多异氰酸酯组合物A的重量计的0.0005重量%至10.0重量%,优选0.01重量%至5.0重量%,更优选0.1重量%至3.0重量%,最优选0.5重量%至2.0重量%的浓度使用。用于根据本发明的方法的叔有机膦催化剂通常在引发低聚反应所需的量下在多异氰酸酯组合物A中具有足够的溶解度。在这一实施方案中,催化剂C因此优选以纯形式添加到多异氰酸酯组合物A中。
但是,任选地,叔有机膦催化剂也可溶解在合适的有机溶剂中使用以改进它们的可并入性。在此,可在极宽范围内自由地选择催化剂溶液的稀释度。这样的催化剂溶液通常在大约0.01重量%的浓度以上是催化活性的。
在一个优选实施方案中,所用含磷催化剂对氧化敏感并且在几小时至几周后通过氧化转化成不再催化活性并优选无色并优选阻燃的化合物。这样的催化剂是例如具有(环)脂族基团的膦。
同样特别合适的是具有2至20个碳原子的脂族、脂环族或芳族单羧酸和多羧酸的碱金属或碱土金属盐。上述羧酸之一的钾盐还更优选。乙酸钾特别优选。
但是,WO 2016/170057、WO 2016/170059或WO 2016/170061中提到的所有催化剂原则上也合适,只要它们在上述温度范围内催化交联反应。
特别适合作为催化剂C的是式(II)的催化剂和它们的加合物。当使用催化剂C1和C2的组合时,上述化合物优选用作催化剂C2。
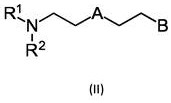
其中R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基;
A选自O、S和NR3,其中R3选自氢、甲基、乙基、丙基、异丙基、丁基和异丁基;且
B,独立于A,选自OH、SH、NHR4和NH2,其中R4选自甲基、乙基和丙基。
在一个优选实施方案中,A是NR3,其中R3选自氢、甲基、乙基、丙基、异丙基、丁基和异丁基。R3优选是甲基或乙基。R3特别优选是甲基。
在这种实施方案的第一变体中,B是OH且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在这种实施方案的第二变体中,B是SH且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在这种实施方案的第三变体中,B是NHR4且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。在这一变体中,R4选自甲基、乙基和丙基。优选的是,R4是甲基或乙基。R4特别优选是甲基。
在这种实施方案的第四变体中,B是NH2且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在另一个优选的实施方案中,A是氧。
在这种实施方案的第一变体中,B是OH且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在这种实施方案的第二变体中,B是SH且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在这种实施方案的第三变体中,B是NHR4且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。在这一变体中,R4选自甲基、乙基和丙基。优选的是,R4是甲基或乙基。R4特别优选是甲基。
在这种实施方案的第四变体中,B是NH2且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在还另一个优选的实施方案中,A是硫。
在这种实施方案的第一变体中,B是OH且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在这种实施方案的第二变体中,B是SH且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
在这种实施方案的第三变体中,B是NHR4且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。在这一变体中,R4选自甲基、乙基和丙基。优选的是,R4是甲基或乙基。R4特别优选是甲基。
在这种实施方案的第四变体中,B是NH2且R1和R2互相独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、支化C5-烷基、非支化C5-烷基、支化C6-烷基、非支化C6-烷基、支化C7-烷基和非支化C7-烷基。优选的是,R1和R2互相独立地为甲基或乙基。R1和R2特别优选是甲基。
式(II)的化合物和具有至少一个异氰酸酯基团的化合物的加合物也合适。
上位术语“加合物”被理解为是指式(II)的化合物与具有至少一个异氰酸酯基团的化合物的氨基甲酸酯加合物、硫代氨基甲酸酯加合物和脲加合物。氨基甲酸酯加合物特别优选。通过异氰酸酯与式(II)中定义的化合物的官能团B反应,形成本发明的加合物。当B是羟基时,形成氨基甲酸酯加合物。当B是硫醇基团时,形成硫代氨基甲酸酯加合物。当B是NH2或NHR4时,形成脲加合物。
合适的催化剂溶剂是例如对异氰酸酯基团呈惰性的溶剂,例如己烷、甲苯、二甲苯、氯苯、乙酸乙酯、乙酸丁酯、二乙二醇二甲基醚、二丙二醇二甲基醚、乙二醇单甲基或单乙基醚乙酸酯、二乙二醇乙基和丁基醚乙酸酯、丙二醇单甲基醚乙酸酯、乙酸1-甲氧基-2-丙基酯、乙酸3-甲氧基-正丁基酯、丙二醇二乙酸酯、丙酮、甲乙酮、甲基异丁基酮、环己酮、内酯如β-丙内酯、γ-丁内酯、ε-己内酯和ε-甲基己内酯,以及如N-甲基吡咯烷酮和N-甲基己内酰胺、碳酸1,2-亚丙酯、二氯甲烷、二甲亚砜、磷酸三乙酯之类的溶剂或这些溶剂的任意混合物。
如果在根据本发明的方法中使用催化剂溶剂,优选使用带有异氰酸酯反应性基团并可并入聚异氰脲酸酯树脂中的催化剂溶剂。这样的溶剂的实例是一元或多元简单醇,例如甲醇、乙醇、正丙醇、异丙醇、正丁醇、正己醇、2-乙基-1-己醇、乙二醇、丙二醇、异构丁二醇、2-乙基己烷-1,3-二醇或甘油;醚醇,例如1-甲氧基-2-丙醇、3-乙基-3-羟甲基氧杂环丁烷、四氢糠醇、乙二醇单甲基醚、乙二醇单乙基醚、乙二醇单丁基醚、二乙二醇单甲基醚、二乙二醇单乙基醚、二乙二醇单丁基醚、二乙二醇、二丙二醇或液体较高分子量聚乙二醇、聚丙二醇、混合聚乙二醇/聚丙二醇及其单烷基醚;酯醇,例如乙二醇单乙酸酯、丙二醇单月桂酸酯、甘油单乙酸酯和二乙酸酯、甘油单丁酸酯或2,2,4-三甲基戊烷-1,3-二醇单异丁酸酯;不饱和醇,例如烯丙醇、1,1-二甲基烯丙醇或油醇;芳脂族醇,例如苄醇;N-单取代酰胺,例如N-甲基甲酰胺、N-甲基乙酰胺、氰基乙酰胺或2-吡咯烷酮,或这些溶剂的任意混合物。
在一个特定实施方案中,使用至少一种交联催化剂C1和至少一种交联催化剂C2。
第一催化剂C1在低于100℃,优选低于80℃,更优选低于60℃,再更优选低于50℃的反应温度下催化异氰酸酯基团的交联以提供选自异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的至少一种结构。
第二催化剂C2在至少50℃,更优选至少60℃,再更优选至少80℃,最优选至少100℃的反应温度下催化至少一种上述交联反应。在此优选的是,这种催化剂C2在低于100℃,优选低于80℃,更优选低于60℃,再更优选低于50℃的温度下只有低活性。
当其在最多1小时,优选最多3小时,更优选最多24小时内催化多异氰酸酯组合物A中存在的至少15摩尔%的异氰酸酯基团的交联时,该催化剂在所示温度下具有所需活性。
术语“低活性”是指在至少1小时,更优选至少3小时,再更优选至少24小时的时期内多异氰酸酯组合物A中存在的最多10摩尔%的异氰酸酯基团交联。
所述第一交联催化剂C1优选是如上所述的式(I)的有机膦催化剂。第二催化剂C2可以是任意催化剂。优选使用WO 2016/170057、WO 2016/170059或WO/2016/170061中提到的催化剂之一。更优选的是,第二催化剂C2是具有2至20个碳原子的脂族、脂环族或芳族单羧酸和多羧酸的碱金属或碱土金属盐。再更优选的是,第二催化剂C2是上述羧酸之一的钾盐。第二催化剂特别优选是乙酸钾。
催化交联
术语“异氰酸酯组合物A的催化交联”是指其中多异氰酸酯组合物A中包含的异氰酸酯基团与彼此反应和因此使多异氰酸酯组合物A中包含的单体和/或低聚异氰酸酯与彼此交联的过程。由于通过交联催化剂C促进这种反应,其也被称为“催化交联”。优选通过形成选自异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的至少一种结构实施交联。特别通过形成异氰脲酸酯基团和至少一种其它的上述结构实施交联。
在本发明的一个优选实施方案中,通过形成异氰脲酸酯基团而在至少30摩尔%,更优选至少40摩尔%,再更优选至少50摩尔%,再更优选至少60摩尔%,特别优选至少70摩尔%,非常特别优选至少80摩尔%的程度上实施催化交联。通过将固化材料中的异氰脲酸酯基团数基于异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的总数计,确定上述值。
特别优选的是,在方法步骤b)开始时,表示为异氰酸酯指数的异氰酸酯基团与异氰酸酯反应性基团的摩尔比为至少100,优选至少150,再更优选至少200。在本文中“异氰酸酯反应性基团”应被理解为是指氨基、硫醇基和羟基,特别优选羟基。在此,这些上述基团如何引入在方法步骤b)开始时存在的混合物中并不重要。这可通过纤维B中的杂质、通过将例如催化剂溶剂添加到交联催化剂C中或通过直接添加实现。在各种情况下必须在方法步骤b)开始时遵循上述比率。
如实施例2中所示,高浓度多元醇的存在或大量氨基甲酸酯基团的形成导致聚合物纤维没有稳定包埋到基质中。因此有利的是限制反应混合物中的对异氰酸酯基团呈反应性的基团的浓度。
根据本发明在方法步骤b)的过程中,在形成的复合材料的所有部分中遵循下文定义的温度范围。这一温度也被称为“反应温度”。所述温度应区别于在形成的复合材料外部的温度,即“环境温度”。
优选在-20℃至150℃的反应温度下进行催化交联。特别优选在0℃至130℃,非常特别优选20℃至120℃的温度范围内进行固化。
当需要特别高的玻璃化转变温度时,优选在100℃至140℃的反应温度下进行催化交联。
由于异氰酸酯基团的催化交联是放热过程,在催化交联过程中的反应温度不仅取决于环境温度。其也尤其受下列参数影响:形成的复合材料的每单位重量的异氰酸酯比例、工件的尺寸和形状(即放热与经表面输出热的比率)、工件的主动冷却(或如果必要,主动加热)和催化剂的选择(在相同的热输出速率下更快反应导致更强加热)。
本领域技术人员知道他们可利用这些参数以控制在复合材料中的催化交联过程中存在的反应温度。因此例如可通过选择适当浓度的合适催化剂而降低反应速率来补偿基于所得复合材料的总重量计的异氰酸酯基团的高重量比例。
可用温度传感器监测该反应的温度分布,从而有可能在简单的初步实验中调节上述参数以遵循所需温度范围。
在一个特定实施方案中,使用对各组件的工艺模拟来优化用于基质的催化交联的温度分布和催化剂选择以提供至少50摩尔%的异氰脲酸酯结构。在此,在不同环境温度或模具温度下对所需组件/半成品,例如拉挤型材、预浸料、灌注模具、SMC模具使用不同催化剂浓度或催化剂组成和不同温度分布,其中任选在反应过程中借助热电偶或温度传感器测量基质温度。由这一参数组开发出关于温度和催化剂的理想加工策略。
优选使用上述膦作为至少一种催化剂组分进行在上文定义的温度范围内的催化三聚。但是,在这些温度范围内实现异氰酸酯基团交联的各种其它催化剂也合适。
多异氰酸酯组合物A中的异氰酸酯基团的催化交联优选在反应结束时使得多异氰酸酯组合物A中最初存在的游离异氰酸酯基团的至少70%,优选至少80%,更优选至少90%,非常特别优选至少95%已反应。换言之,通过根据本发明的方法获得的复合材料的基质中优选仅存在多异氰酸酯组合物A中最初包含的异氰酸酯基团的最多30%、最多20%,特别优选最多10%,非常特别优选最多5%。
可最初通过NCO含量的滴定测定法测定交联反应的过程,但随着反应进行,反应混合物的胶凝和固化迅速开始,因此使得湿化学分析法不可能。然后仅可以通过光谱法,例如通过红外光谱法使用在大约2270 cm-1的异氰酸酯谱带强度监测异氰酸酯基团的进一步转化,或通过DSC/DMA监测基质Tg的提高。
在本发明的一个特别优选的实施方案中,方法步骤b)中的催化交联在两个阶段中进行。
在方法步骤b1)的过程中多异氰酸酯组合物A的温度下限优选为至少-20℃,更优选为0℃,再更优选为20℃,最优选为30℃。对于方法步骤b1),温度范围特别优选为至少20℃至最多120℃。
方法步骤b1)优选进行至少30分钟。
多异氰酸酯组合物A的温度随后在方法步骤b2)中与方法步骤b1)相比提高至少20℃。多异氰酸酯组合物A的温度在这种情况下达到至少50℃,但其中优选不超过150℃的温度。在此温度下继续交联。由于较高交联温度导致固化多异氰酸酯组合物A的较高玻璃化转变温度,这使得可以获得其基质具有提高的玻璃化转变温度的复合材料。
方法步骤b2)优选进行至少5分钟。
在这一实施方案中优选使用如本申请中上文定义的至少一种交联催化剂C1和至少一种交联催化剂C2的组合。
“多异氰酸酯组合物A在至少一种聚合物纤维B存在下的催化交联”不排除除根据本发明使用的聚合物纤维B外还存在其它有机或无机填料。尤其根据本发明的是如本申请中定义的聚合物纤维B与其它纤维的混合物。
但是优选的是,基于所有有机和无机纤维状和非纤维状填料的总数计,聚合物纤维B的体积比例为至少20体积%,更优选至少40体积%,更优选至少50体积%,再更优选至少70体积%,非常特别优选至少90体积%。
在本发明的一个特别优选的实施方案中,聚合物纤维B的上文定义的体积比例为至少95体积%。
本发明进一步涉及复合材料,其特征在于所述复合材料具有根据DIN EN ISO1183-1测定的最多1.2 kg/l,优选最多1.15,特别优选最多1.1,非常特别优选最多1.05的密度。在此,弹性模量为至少3 GPa,优选至少5 GPa,更优选至少10 GPa,非常特别优选至少15 GPa。优选在根据DIN EN ISO 14125:2011-05的三点弯曲试验中测定弹性模量。所述复合材料的特征进一步在于其含有聚合物纤维,优选聚乙烯纤维,特别优选尚未增容的超高分子量聚乙烯纤维B。复合材料的基质优选由具有至少100,更优选至少150,特别优选至少200的异氰酸酯指数的催化交联的多异氰酸酯组合物A形成。
在上文定义的材料中,聚合物基质中的聚异氰脲酸酯基团的比例为基于异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的总数计的至少20摩尔%,优选至少25摩尔%,特别优选至少30摩尔%,非常特别优选至少35摩尔%。
聚合物基质中的聚异氰脲酸酯基团的比例为基于异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的总数计至少30摩尔%。
产品和用途
在另一个实施方案中,本发明涉及通过本发明的方法可获得的复合材料。
在进一步实施方案中,本发明涉及通过本发明的方法可获得的复合材料用于生产半成品或组件的用途。通过本发明的方法生产的组件优选是型材、管道、片材或任意其它成型制品。这些可用于各种行业,如汽车制造和造船、航空航天、房屋和设备建造、个人防护、电子产品、家具制造、采油、医疗技术或体育用品。在此应该具体提到飞机、火车、汽车、船舶等中的构造、防弹和/或碰撞相关组件。
优选实施方案是来自“片状模塑料法”(SMC)的任意三维成型制品,例如外壳、门、顶部模块、保险杠,来自拉挤成型法的成型制品,如型材、管道和棒材,以及由预浸料的使用形成的任意成型制品或增强元件,例如管道、机翼,以及由灌注法形成的任意成型制品,例如风电叶片、桥梁和建筑物中的结构元件,以及如通过长丝缠绕形成的旋转对称的任意元件,例如桅杆、压力容器、管道,以及如通过反应性注射成型形成的任意成型制品。
在还另一个实施方案中,本发明涉及含有或由通过本发明的方法可获得的复合材料组成的上述组件。
在另一个实施方案中,本发明涉及根据本发明的复合材料用于生产成型制品的用途和由根据本发明的复合材料组成或含有根据本发明的复合材料的成型制品。
本文所用的“成型制品”特别是在其最小尺寸方向上具有至少0.5 mm,优选至少1mm,特别优选至少2 mm的厚度的物体。本文所用的“成型制品”特别不是薄膜或膜。
下列实施例仅用于例示本发明。它们无意以任何方式限制权利要求书的保护范围。
实施例
一般信息:
除非另行指明,所有的百分比数据为重量百分比(重量%)。
在进行实验时存在的23℃的环境温度被称为RT(室温)。
测量方法:
下文详述的用于测定适当参数的方法用于进行和评估实施例并且也是通常用于测定根据本发明的相关参数的方法。
通过DSC测定相变
根据DIN EN 61006用Mettler DSC 12E(德国Giessen的Mettler Toledo GmbH)借助DSC(差示扫描量热法)测定相变。通过铟和铅的熔融起始温度实施校准。将10毫克物质称入到标准胶囊中。通过以20 K/min的加热速率从-50℃至+200℃的三次加热和随后以320K/min的冷却速率冷却,实施测量。借助液氮实施冷却。所用吹扫气体是氮气。所示的值各自基于第一加热曲线的评估,因为在研究的反应体系中,在高温下的测量过程中可能由于DSC中的热应力而发生样品变化。由在热流曲线的最大值处的温度获得熔融温度Tm。由在玻璃化转变步骤的半高处的温度获得玻璃化转变温度Tg。
红外光谱的测定
在配有ATR单元的Bruker公司的 FT-IR光谱仪上测量红外光谱。
扫描电子显微术
在FEI公司的ESEM Quanta 400型号的扫描电子显微镜(具有钨阴极)上捕集扫描电子显微照片。加速电压为10.0 kV。为了增强图像对比度,使用二次电子/反向散射检测实现检测。检测器电压为+100 V。使用10 mm的样品距离和25°的样品角度操作。
起始化合物
多异氰酸酯A1: HDI三聚物(NCO官能度 > 3),NCO含量为23.0重量%,来自Covestro AG公司。在23℃下的粘度为大约1200 mPa·s(DIN EN ISO 3219/A.3)。
催化剂K1: 三辛基膦以97重量%的纯度获自Sigma-Aldrich公司。
催化剂K2: 二月桂酸二丁基锡以95重量%的纯度获自Sigma-Aldrich公司。
聚乙二醇(PEG) 400以> 99重量%的纯度获自ACROS公司。
乙酸钾以> 99重量%的纯度获自ACROS公司。
甘油以> 99重量%的纯度获自ACROS公司。
除催化剂外的所有原材料在使用前在真空下脱气;聚乙二醇和甘油另外干燥。
PE纤维是来自DSM公司的Dyneema商标的凝胶纺丝UHMWPE纤维。
PE织物是由来自DSM公司的Dyneema商标的凝胶纺丝UHMWPE纤维制成的机织材料(0°/90°)。
所用PE纤维的热性质:
通过DSC测定PE纤维的热性质。由第一加热曲线得到151.5℃的熔融温度Tm,其中熔化热ΔHm为269.4 J/g,而由第二加热曲线得到137.7℃的熔融温度Tm,其中熔化热ΔHm为147.1 J/g。这种行为可归因于凝胶纺丝纤维的高结晶度并与通过其它手段制成的PE纤维相比允许在聚合物树脂交联过程中的更高温度。
催化剂K3的生产:
乙酸钾(5.0克)在室温下在PEG 400(95.0克)中搅拌直至其全部溶解。这获得乙酸钾在PEG 400中的5重量%溶液,其未经进一步处理即用作催化剂。
催化剂K4的生产:
乙酸钾(10.0克)在室温下在PEG 400(90.0克)中搅拌直至其全部溶解。这获得乙酸钾在PEG 400中的10重量%溶液,其未经进一步处理即用作催化剂。
反应混合物的生产
除非另行指明,通过在来自Hauschild公司的高速混合器DAC 150.1 FVZ中在23℃下以2750 min-1混合多异氰酸酯A1与相应量的催化剂K(K1-4)和任选相应量的甘油来生产反应混合物。然后未经进一步处理将其倒入合适的模具中以便交联,或添加到相应的PE纤维或PE织物中以便进一步加工。
纤维的清洗
为了从PE纤维和PE织物上脱除可能的增容剂及其残留物,在使用前通过置于丙酮中30分钟和随后冲洗和在室温下干燥来清洗纤维。
聚异氰脲酸酯复合材料的生产
通过将PE纤维与相应的反应混合物混合或将反应混合物倒在PE织物上和随后固化反应混合物,获得聚异氰脲酸酯复合材料。
通过真空灌注生产聚异氰脲酸酯复合材料
仅用PE织物进行通过真空灌注生产聚异氰脲酸酯复合材料。使用标准文献中已知的装置(例如Hammami, A.和Gebart, B. R. (2000), Analysis of the vacuum infusionmolding process. Polym Compos, 21: 28–40)。
实施例1(本发明)
如上所述,多异氰酸酯A1(84.0克)和催化剂K4(1.68克)的混合物在高速混合器中在2750转/分钟下混合6分钟。随后将反应混合物添加到最初装在铝杯中的PE纤维(4克)中并搅拌。由此获得的混合物在循环空气烘箱中在100℃下预固化2小时,随后在140℃下固化10分钟。由此获得的材料是固体、清澈和无泡的。通过压裂在试样上产生断裂面,随后在扫描电子显微镜中检查其中树脂与纤维的附着力。发现纤维-树脂附着力特别好,因为纤维在压裂时断开而非从树脂中拉出。此外,在纤维周围没有可识别的气体包层。
实施例2(对比)
如上所述,多异氰酸酯A1(83.35克)、甘油(14.02克)和催化剂K2(0.013克)的混合物在高速混合器中在2750转/分钟下混合6分钟。随后将反应混合物添加到最初装在铝杯中的PE纤维(4克)中并搅拌。由此获得的混合物在循环空气烘箱中在100℃下预固化30分钟。在预固化过程中,由于缺乏与所得聚氨酯基质的相容性,纤维沉淀/沉积在树脂表面,因此停止实验。无法进行通过扫描电子显微术测定纤维-树脂附着力。
实施例3(对比)
如上所述,多异氰酸酯A1(84.0克)和催化剂K4(1.68克)的混合物在高速混合器中在2750转/分钟下混合6分钟。随后将反应混合物添加到最初装在铝杯中的PE纤维(4克)中并搅拌。由此获得的混合物在循环空气烘箱中在140℃下固化10分钟。由于交联反应的放热性,复合材料的温度在此超过PE纤维的熔融温度。尽管由此获得的材料是固体,但其表现出棕色变色和明显起泡。无法进行通过扫描电子显微术测定纤维-树脂附着力,因为由于基质的反应焓,该纤维在固化过程中已熔融。
实施例4(本发明)
如上所述,多异氰酸酯A1(100.0克)、催化剂K1(0.5克)和催化剂K3(3.14克)的混合物在高速混合器中在2750转/分钟下混合6分钟。随后将反应混合物倒在PE织物(5 x 5cm2)上。由此获得的样品在室温下固化5天。由此获得的材料是固体、清澈和无泡的。PE织物的结构也清晰可辩。在通过相同程序获得的试样上通过压裂产生断裂面,随后在扫描电子显微镜中检查其中的树脂与织物的附着力。结果显示织物与树脂之间的良好相容性和附着力。
实施例5(本发明)
如上所述,多异氰酸酯A1(98.0克)和催化剂K1(2.0克)的混合物在高速混合器中在2750转/分钟下混合6分钟。然后在真空灌注装置中将反应混合物施加到PE织物上。层压件在真空下在室温下保持5天。由此获得的材料是固体且无泡的。红外光谱显示小于50%的残留异氰酸酯含量(在2270 cm-1的谱带)。
实施例6(本发明)
如上所述,多异氰酸酯A1(98.0克)和催化剂K1(2.0克)的混合物在高速混合器中在2750转/分钟下混合6分钟。然后在真空灌注装置中将反应混合物施加到PE织物上。层压件在真空下在室温下保持5天。试样随后在循环空气烘箱中在120℃下继续热处理2小时。由此获得的材料是固体且无泡的。红外光谱显示小于50%的残留异氰酸酯含量(在2270 cm-1的谱带)。
实施例7(本发明)
如上所述,多异氰酸酯A1(98.0克)、催化剂K1(1.0克)和催化剂K3(6.28克)的混合物在高速混合器中在2750转/分钟下混合6分钟。然后在真空灌注装置中将反应混合物施加到PE织物上。层压件在真空下在室温下保持5天。由此获得的材料是固体且无泡的。红外光谱显示小于50%的残留异氰酸酯含量(在2270 cm-1的谱带)。
实施例8(本发明)
如上所述,多异氰酸酯A1(98.0克)、催化剂K1(1.0克)和催化剂K3(6.28克)的混合物在高速混合器中在2750转/分钟下混合6分钟。然后在真空灌注装置中将反应混合物施加到PE织物上。层压件在真空下在室温下保持5天。试样随后在循环空气烘箱中在120℃下继续热处理2小时。由此获得的材料是固体且无泡的。红外光谱显示小于20%的残留异氰酸酯含量(在2270 cm-1的谱带)。
实施例9(本发明)
如上所述,多异氰酸酯A1(98.0克)、催化剂K1(1.0克)和催化剂K3(6.28克)的混合物在高速混合器中在2750转/分钟下混合6分钟。然后在真空灌注装置中将反应混合物施加到PE织物上。层压件在真空下在室温下保持1天。由此获得的材料是固体且无泡的。红外光谱显示小于50%的残留异氰酸酯含量(在2270 cm-1的谱带)。
实施例10(本发明)
如上所述,多异氰酸酯A1(98.0克)、催化剂K1(1.0克)和催化剂K3(6.28克)的混合物在高速混合器中在2750转/分钟下混合6分钟。然后在真空灌注装置中将反应混合物施加到PE织物上。层压件在真空下在室温下保持1天。试样随后在循环空气烘箱中在120℃下继续热处理24小时。由此获得的材料是固体且无泡的。红外光谱没有显示残留异氰酸酯含量(在2270 cm-1的谱带)。
论述
实施例表明只有通过使用多异氰酸酯作为反应性组分并在一种或多种三聚催化剂存在下将其催化转化以提供交联聚异氰脲酸酯才充分润湿基于聚乙烯纤维的填料,因此能够生产基于聚合物纤维的复合材料(实施例1)。对比例2表明同时使用多异氰酸酯和多元醇并将其催化转化成交联聚氨酯导致填料和基质之间的相容性问题。这导致填料分离并因此没有产生复合材料。在没有预先增容的情况下无法获得基于聚氨酯的复合材料。
此外,实施例清楚表明温度控制在复合材料的形成过程中是必要的,以使反应温度始终保持在低于填料的熔点。在实施例1中在100℃下预交联中和随后在140℃下继续交联中,通过各个方法步骤的放热性释放的热都不足以超过PE纤维的熔融温度,而对比例3表明尽管140℃的环境温度低于PE纤维的熔点,但通过各个方法步骤的放热性释放的热导致超过PE纤维的熔点,因为无法足够快地输出反应热。
除填料与基质之间的相容性和保持填料的初始性质外,对工业应用而言希望的是在尽可能短的时间内实现基质中的反应性基团的尽可能高转化率。通过在真空灌注装置中润湿PE织物和随后在低温(RT)下使用在这一温度范围内活性的催化剂的预交联与在高温(120℃)下使用又在这一温度范围内表现出提高的活性但在低温下并非如此的另一催化剂的继续交联的组合,可以实现这种高转化率(实施例10)。
Claims (16)
1.由聚合物纤维和交联多异氰酸酯生产复合材料的方法,其包括下列步骤:
a) 提供含有多异氰酸酯的多异氰酸酯组合物A,和
b) 多异氰酸酯组合物A在至少一种聚合物纤维B和至少一种交联催化剂C存在下催化交联以提供由聚合物纤维和交联多异氰酸酯制成的复合材料,
其中在方法步骤b)中的催化交联在两个分开的方法步骤b1)和b2)中运行,其特征在于方法步骤b1)中使温度保持在最多100℃并在后续方法步骤b2)中提高到大于100℃但最多200℃。
2.如权利要求1中所述的方法,其特征在于所述至少一种交联催化剂C选自通式(I)的膦催化剂
和具有2至20个碳原子的脂族、脂环族或芳族单羧酸和多羧酸的盐。
3.如权利要求1或2中所述的方法,其中使用至少两种不同的交联催化剂C1和C2,其中第一交联催化剂C1在低于50℃的反应温度下催化异氰酸酯基团的交联以提供选自异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的至少一种结构,且第二交联催化剂C2在至少80℃的反应温度下催化至少一种上述反应。
4.如权利要求1至3任一项中所述的方法,其中所述聚合物纤维由聚乙烯组成。
5.如权利要求4中所述的方法,其中所述聚合物纤维由具有至少360 kg/mol的分子量的聚乙烯组成。
6.如权利要求4或5中所述的方法,其中所述聚乙烯纤维由具有1.1至4.0的多分散性的聚乙烯组成。
7.如权利要求1至6任一项中所述的方法,其中所用聚合物纤维的拉伸强度为至少2500N/mm2。
8.如权利要求1至7任一项中所述的方法,其中在催化交联之前和之后,所述多异氰酸酯组合物具有比未处理的聚合物纤维B的表面能低不大于5 mN/m和高不大于20 mN/m的表面能。
9.如权利要求1至8任一项中所述的方法,其特征在于所述多异氰酸酯组合物A以至少50重量%的比例由1,4-丁烷二异氰酸酯、1,5-戊烷二异氰酸酯、1,6-己烷二异氰酸酯、异佛尔酮二异氰酸酯或4,4'-二环己基甲烷二异氰酸酯或其混合物的反应产物形成。
10.如权利要求1至9任一项中所述的方法,其特征在于所述多异氰酸酯组合物A)具有1.5至6.0的平均NCO官能度。
11.如权利要求1至10任一项中所述的方法,其中在方法步骤b)中在最多150℃的温度下进行异氰酸酯的催化交联以提供交联多异氰酸酯。
12.通过根据权利要求1至11任一项的方法获得或可获得的复合材料。
13.复合材料,其特征在于所述复合材料具有根据DIN EN ISO 1183-1测定的最多1.2kg/l的密度和> 3 GPa的弹性模量,含有聚合物纤维B并且其聚合物基质由具有至少100的异氰酸酯指数的多异氰酸酯组合物A形成。
14.如权利要求13中所述的复合材料,其特征在于所述聚合物基质中的聚异氰脲酸酯基团的比例为基于异氰脲酸酯、脲二酮、缩二脲、脲、亚氨基噁二嗪二酮、噁二嗪三酮和脲基甲酸酯基团的总数计的至少30摩尔%。
15.根据权利要求12至14任一项的复合材料用于生产三维制品的用途。
16.三维制品,其由如权利要求12至14任一项中所述的复合材料组成或含有如权利要求12至14任一项中所述的复合材料。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18172971.6 | 2018-05-17 | ||
| EP18172971 | 2018-05-17 | ||
| PCT/EP2019/062218 WO2019219614A1 (de) | 2018-05-17 | 2019-05-13 | Verfahren zur herstellung von verbundwerkstoffen aus polyethylenfasern mit ultrahohem molekulargewicht und vernetzten polyisocyanaten |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113166364A true CN113166364A (zh) | 2021-07-23 |
Family
ID=62200297
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980033031.8A Pending CN113166364A (zh) | 2018-05-17 | 2019-05-13 | 由具有超高分子量的聚乙烯纤维和交联多异氰酸酯生产复合材料的方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20210214487A1 (zh) |
| EP (1) | EP3794050A1 (zh) |
| CN (1) | CN113166364A (zh) |
| WO (1) | WO2019219614A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118290783A (zh) * | 2024-06-06 | 2024-07-05 | 长春三友智造科技发展有限公司 | 一种纤维增强聚氨酯复合材料及其制备方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117396532A (zh) * | 2021-07-23 | 2024-01-12 | Sika技术股份公司 | 具有高透明度的聚异氰脲酸酯塑料 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816325A (en) * | 1987-04-08 | 1989-03-28 | Mobay Corporation | Thermally flexible polyurethane coatings for substrates containing aramid fibers |
| CN106147164A (zh) * | 2015-04-23 | 2016-11-23 | 上海微创医疗器械(集团)有限公司 | 一种医用复合材料及其制备方法 |
| WO2018054776A1 (en) * | 2016-09-20 | 2018-03-29 | Covestro Deutschland Ag | Anisotropic composite materials based on polyisocyanates |
Family Cites Families (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB809809A (en) | 1956-11-16 | 1959-03-04 | Ici Ltd | Polymeric isocyanates and their manufacture |
| GB1200542A (en) | 1967-01-19 | 1970-07-29 | Takeda Chemical Industries Ltd | A method for producing isocyanate trimers |
| DE1954093C3 (de) | 1968-11-15 | 1978-12-21 | Mobay Chemical Corp., Pittsburgh, Pa. (V.St.A.) | Verfahren zur Herstellung von polymeren organischen Isocyanaten |
| GB1386399A (en) | 1971-07-16 | 1975-03-05 | Ici Ltd | Isocyanurate polymers |
| GB1391066A (en) | 1971-07-16 | 1975-04-16 | Ici Ltd | Urethane oils |
| DE2414413C3 (de) | 1974-03-26 | 1978-08-24 | Bayer Ag, 5090 Leverkusen | Verwendung von Lösungen von Polyisocyanaten mit Isocyanuratstruktur in Zweikomponenten-Polyurethan-Lacken |
| DE2452532C3 (de) | 1974-11-06 | 1978-08-24 | Bayer Ag, 5090 Leverkusen | Verfahren zur Herstellung von Polyisocyanaten mit Isocyanurat-Struktur |
| US4040992A (en) | 1975-07-29 | 1977-08-09 | Air Products And Chemicals, Inc. | Catalysis of organic isocyanate reactions |
| JPS5936574B2 (ja) * | 1975-09-22 | 1984-09-04 | 三菱化学株式会社 | 繊維強化プラスチツクフオ−ムを連続的に製造する方法 |
| DE2641380C2 (de) | 1976-09-15 | 1989-11-23 | Bayer Ag, 5090 Leverkusen | Verfahren zur Herstellung von Polyisocyanaten mit Isocyanuratstruktur |
| DE2806731A1 (de) | 1978-02-17 | 1979-08-23 | Bayer Ag | Verfahren zur herstellung von isocyanuratgruppen aufweisenden polyisocyanaten |
| US4232133A (en) * | 1978-07-27 | 1980-11-04 | Ici Americas Inc. | Polyisocyanurate containing molding compositions |
| CA1112243A (en) | 1978-09-08 | 1981-11-10 | Manfred Bock | Process for the preparation of polyisocyanates containing isocyanurate groups and the use thereof |
| DE2901479A1 (de) | 1979-01-16 | 1980-07-24 | Bayer Ag | Neue isocyanato-isocyanurate, ein verfahren zu ihrer herstellung, sowie ihre verwendung als isocyanatkomponente in polyurethan-lacken |
| CA1127644A (en) | 1980-01-28 | 1982-07-13 | Anupama Mishra | Isocyanurate products and polyurethanes therefrom |
| DE3033860A1 (de) | 1980-09-09 | 1982-04-15 | Bayer Ag, 5090 Leverkusen | Neue isocyanato-isocyanurate, ein verfahren zu ihrer herstellung, sowie ihre verwendung als isocyanatkomponente in polyurethanlacken |
| DE3100262A1 (de) | 1981-01-08 | 1982-08-05 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von isocyanuratgruppen aufweisenden polyisocyanaten, als katalysator-komponente fuer dieses verfahren geeignete loesungen, sowie die verwendung der verfahrensprodukte als isocyanat-komponente bei der herstellung von polyurethanen |
| DE3100263A1 (de) | 1981-01-08 | 1982-08-12 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von isocyanuratgruppen aufweisenden polyisocyanaten und ihre verwendung bei der herstellung von polyurethanen |
| JPS58162581A (ja) | 1982-03-19 | 1983-09-27 | Nippon Polyurethan Kogyo Kk | ポリウレタン塗料用組成物 |
| DE3227489A1 (de) | 1982-07-23 | 1984-01-26 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von isocyanuratgruppen aufweisenden polyisocyanaten und ihre verwendung als isocyanatkomponente zur herstellung von polyurethanen |
| PT77070B (en) | 1982-07-29 | 1986-01-27 | Dsm Resins Bv | Oligomerisation of polyisocyanates |
| AT375652B (de) | 1982-10-29 | 1984-08-27 | Valentina Alexandro Postnikova | Verfahren zur herstellung von arylaliphatischen polyisozyanuraten |
| JPH0678418B2 (ja) | 1986-03-10 | 1994-10-05 | 大日本インキ化学工業株式会社 | 樹脂組成物 |
| DE3700209A1 (de) | 1987-01-07 | 1988-07-21 | Bayer Ag | Verfahren zur herstellung von polyisocyanaten mit biuretstruktur |
| DE3811350A1 (de) | 1988-04-02 | 1989-10-19 | Bayer Ag | Verfahren zur herstellung von isocyanuratpolyisocyanaten, die nach diesem verfahren erhaltenen verbindungen und ihre verwendung |
| DE3814167A1 (de) | 1988-04-27 | 1989-11-09 | Bayer Ag | Verfahren zur herstellung von isocyanuratgruppen aufweisenden polyisocyanaten und ihre verwendung |
| CA1334848C (en) | 1988-08-05 | 1995-03-21 | William E. Slack | Process for the production of polyisocyanates which contain isocyanurate groups |
| CA1334849C (en) | 1988-08-24 | 1995-03-21 | Bayer Corporation | Process for the production of polyisocyanates which contain isocyanurate groups |
| DE3900053A1 (de) | 1989-01-03 | 1990-07-12 | Bayer Ag | Verfahren zur herstellung von uretdion- und isocyanuratgruppen aufweisenden polyisocyanaten, die nach diesem verfahren erhaeltlichen polyisocyanate und ihre verwendung in zweikomponenten-polyurethanlacken |
| DE3902078A1 (de) | 1989-01-25 | 1990-07-26 | Bayer Ag | Verfahren zur herstellung von modifizierten, isocyanuratgruppen aufweisenden polyisocyanaten und ihre verwendung |
| DE3928503A1 (de) | 1989-08-29 | 1991-03-07 | Bayer Ag | Verfahren zur herstellung von loesungen von isocyanuratgruppen aufweisenden polyisocyanaten in lackloesungsmitteln und ihre verwendung |
| DE4005762A1 (de) | 1990-02-23 | 1991-08-29 | Bayer Ag | Trimerisierungskatalysatoren, ein verfahren zu ihrer herstellung und ihre verwendung bei der herstellung von isocyanuratgruppen aufweisenden polyisocyanaten |
| US5073576A (en) * | 1990-06-28 | 1991-12-17 | Basf Corporation | Polyurethane-polyisocyanurate structural rim systems with enhanced processing |
| NL9100279A (nl) | 1991-02-18 | 1992-09-16 | Stamicarbon | Microporeuze folie uit polyetheen en werkwijze voor de vervaardiging daarvan. |
| DE4405055A1 (de) | 1994-02-17 | 1995-08-24 | Basf Ag | Verfahren zur Herstellung von Isocyanuratgruppen aufweisenden Polyisocyanaten und ihre Verwendung |
| DE4405054A1 (de) | 1994-02-17 | 1995-08-24 | Basf Ag | Modifizierte (cyclo)aliphatische Polyisocyanatmischungen, Verfahren zu ihrer Herstellung und ihre Verwendung |
| DE19611849A1 (de) | 1996-03-26 | 1997-10-02 | Bayer Ag | Neue Isocyanattrimerisate und Isocyanattrimerisatmischungen, deren Herstellung und Verwendung |
| DE19734048A1 (de) | 1997-08-06 | 1999-02-11 | Bayer Ag | Verfahren zur Herstellung von Polyisocyanaten, damit hergestellte Polyisocyanate und deren Verwendung |
| ZA9810038B (en) | 1997-11-04 | 2000-05-03 | Rhodia Chimie Sa | A catalyst and a method for the trimerization of isocyanates. |
| DE59903289D1 (de) | 1998-06-02 | 2002-12-12 | Bayer Ag | Verfahren zur Herstellung Iminooxadiazindiongruppen enthaltender Polyisocyanate |
| US20060160979A1 (en) * | 2000-05-25 | 2006-07-20 | Benecke Herman P | Method for forming a crosslinked polymer by temperature control |
| DE10065176A1 (de) | 2000-12-23 | 2002-06-27 | Degussa | Katalysator und Verfahren zur Herstellung von niedrigviskosen und farbreduzierten isocyanuratgruppenhaltigen Polyisocyanaten |
| DE10254878A1 (de) | 2002-11-25 | 2004-06-03 | Bayer Ag | Herstellung uretdiongruppenhaltiger Polyisocyanate |
| WO2004078820A1 (en) | 2003-02-28 | 2004-09-16 | Dow Global Technologies Inc. | Preparation of isocyanurate group containing polyisocyanate mixtures |
| US7811673B2 (en) | 2003-12-12 | 2010-10-12 | Toyo Boseki Kabushiki Kaisha | High strength polyethylene fiber |
| DE102004012571A1 (de) | 2004-03-12 | 2005-09-29 | Basf Ag | Verfahren zur Herstellung von Isocyanuratgruppen aufweisenden Polyisocyanaten und ihre Verwendung |
| US20060173128A1 (en) * | 2004-11-15 | 2006-08-03 | Huntsman International Llc | Pultrusion systems and process |
| CN101466517B (zh) * | 2006-06-14 | 2012-02-22 | 亨茨曼国际有限公司 | 复合面板 |
| DE102007018015A1 (de) | 2007-04-17 | 2008-10-23 | Bayer Materialscience Ag | Herstellung uretdiongruppenhaltiger Polyisocyanate |
| NO2697414T3 (zh) | 2011-04-13 | 2018-02-03 | ||
| WO2013167404A1 (en) | 2012-05-08 | 2013-11-14 | Basf Se | Preparation of polyisocyanates having isocyanurate groups and their use |
| BR112015029163B1 (pt) | 2013-05-23 | 2022-07-05 | Dsm Ip Assets B.V. | Fibra uhmwpe |
| KR20220056863A (ko) | 2013-10-25 | 2022-05-06 | 디에스엠 아이피 어셋츠 비.브이. | 초고분자량 폴리에틸렌의 제조 |
| JP2018513900A (ja) | 2015-04-21 | 2018-05-31 | コベストロ、ドイチュラント、アクチエンゲゼルシャフトCovestro Deutschland Ag | 断熱条件下で製造されたポリイソシアヌレートポリマーに基づく固体 |
| EP3424975A1 (en) | 2015-04-21 | 2019-01-09 | Covestro Deutschland AG | Polyisocyanurate polymers and process for the production of polyisocyanurate polymers |
| US10597484B2 (en) | 2015-04-21 | 2020-03-24 | Covestro Deutschland Ag | Polyisocyanurate plastics having high thermal stability |
| WO2017149031A1 (de) * | 2016-03-04 | 2017-09-08 | Covestro Deutschland Ag | Verfahren zur herstellung von faserverbundbauteilen |
| EP3421516A1 (de) * | 2017-06-28 | 2019-01-02 | Covestro Deutschland AG | Gefärbte kunststoffe basierend auf vernetzten polyisocyanaten |
-
2019
- 2019-05-13 CN CN201980033031.8A patent/CN113166364A/zh active Pending
- 2019-05-13 US US17/056,418 patent/US20210214487A1/en not_active Abandoned
- 2019-05-13 WO PCT/EP2019/062218 patent/WO2019219614A1/de not_active Ceased
- 2019-05-13 EP EP19722646.7A patent/EP3794050A1/de not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816325A (en) * | 1987-04-08 | 1989-03-28 | Mobay Corporation | Thermally flexible polyurethane coatings for substrates containing aramid fibers |
| CN106147164A (zh) * | 2015-04-23 | 2016-11-23 | 上海微创医疗器械(集团)有限公司 | 一种医用复合材料及其制备方法 |
| WO2018054776A1 (en) * | 2016-09-20 | 2018-03-29 | Covestro Deutschland Ag | Anisotropic composite materials based on polyisocyanates |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118290783A (zh) * | 2024-06-06 | 2024-07-05 | 长春三友智造科技发展有限公司 | 一种纤维增强聚氨酯复合材料及其制备方法 |
| CN118290783B (zh) * | 2024-06-06 | 2024-09-03 | 长春三友智造科技发展有限公司 | 一种纤维增强聚氨酯复合材料及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20210214487A1 (en) | 2021-07-15 |
| WO2019219614A1 (de) | 2019-11-21 |
| EP3794050A1 (de) | 2021-03-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11236191B2 (en) | Method for producing a polyisocyanurate composite material | |
| CN107438635B (zh) | 制造聚异氰脲酸酯塑料的方法 | |
| CN107531874B (zh) | 聚异氰脲酸酯聚合物以及制备聚异氰脲酸酯聚合物的方法 | |
| US11286332B2 (en) | Hydrophobically modified polyisocyanurate plastic and method for production thereof | |
| KR102480612B1 (ko) | 복합 폴리이소시아누레이트 재료를 제조하는 방법 | |
| CN107531876B (zh) | 具有高热稳定性的聚异氰脲酸酯塑料 | |
| EP3507321B1 (en) | Process for producing polyisocyanurate plastics by means of phosphine catalysis | |
| CN112204065B (zh) | 制备多异氰酸酯聚合物和聚异氰脲酸酯塑料的方法 | |
| CN113166364A (zh) | 由具有超高分子量的聚乙烯纤维和交联多异氰酸酯生产复合材料的方法 | |
| US12157790B2 (en) | Composite materials based on dual-cure urethane polymers and dual-cure isocyanurate polymers | |
| CN111479840A (zh) | 基于热潜催化剂的聚氨酯复合材料 | |
| JP2019536849A (ja) | ブロックトポリイソシアネートの触媒架橋によるプラスチックの製造 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| WD01 | Invention patent application deemed withdrawn after publication | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20210723 |