CN112955147A - 用于治疗nash/nafld和相关疾病的组合 - Google Patents
用于治疗nash/nafld和相关疾病的组合 Download PDFInfo
- Publication number
- CN112955147A CN112955147A CN201980071389.XA CN201980071389A CN112955147A CN 112955147 A CN112955147 A CN 112955147A CN 201980071389 A CN201980071389 A CN 201980071389A CN 112955147 A CN112955147 A CN 112955147A
- Authority
- CN
- China
- Prior art keywords
- pharmaceutically acceptable
- inhibitors
- acceptable salt
- compound
- alcoholic steatohepatitis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images













Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/438—The ring being spiro-condensed with carbocyclic or heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Gastroenterology & Hepatology (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
Abstract
本文描述用于治疗哺乳动物的包括非酒精性脂肪性肝炎(NASH)在内的疾病的(S)‑2‑(5‑((3‑乙氧基吡啶‑2‑基)氧基)吡啶‑3‑基)‑N‑(四氢呋喃‑3‑基)嘧啶‑5‑甲酰胺或其药学上可接受的盐和4‑(4‑(1‑异丙基‑7‑氧代‑1,4,6,7‑四氢螺[吲唑‑5,4'‑哌啶]‑1'‑羰基)‑6‑甲氧基吡啶‑2‑基)苯甲酸或其药学上可接受的盐的组合。
Description
发明领域
本发明涉及用于治疗非酒精性脂肪性肝病(NAFLD)和与其相关的疾病如非酒精性脂肪性肝炎(NASH)和代谢相关疾病的新药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,且在相同或单独的组合物中包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药物上可接受的盐。
发明背景
非酒精性脂肪性肝炎(NASH)为非酒精性脂肪性肝病(NAFLD,定义为存在≥5%肝性脂肪变性)的临床和组织学亚型,与所有原因死亡率的增加、硬化和末期肝病以及心血管死亡率的增加,及肝相关和非肝相关癌症二者发生率的增加有关(Sanyal等人, Hepatology2015;61(4):1392-1405)。NAFLD为代谢性综合征的肝表现,且为涵盖脂肪变性、NASH、纤维化、硬化及最终肝细胞癌的一系列的肝病况。NAFLD和NASH被认为是主要的脂肪肝疾病,因为它们占具有肝脂质升高的个体的最大比例。NAFLD/NASH的严重程度基于脂质的存在、炎性细胞浸润、肝细胞气球样变(hepatocyte ballooning)和纤维化程度。目前,治疗选项限于相关病况的管理(EASL-EASD-EASO Clinical Practice Guidelines, J. Hepatol.2016;64(6):1388-1402)。
脂质代谢的改变已推测有助于NAFLD和NASH的分子发病机制。脂肪变性为NASH发病机制的必要但非充分的组分 (Day C, 和James O., Hepatology. 1998;27(6):1463-6)。与此一致,多项研究已证明,脂肪变性的严重程度预示着伴随脂肪性肝炎的风险以及进展为硬化的风险(Sorensen等人, Lancet. 1984;2(8397):241-4;Wanless I和Lentz J,Hepatology 1990;12(5):1106-10;Reeves H,等人, J. Hepatol. 1996;25(5):677-83)。肝性脂肪变性是TG生产/摄入肝和清除/移除不平衡的结果(Cohen JC,等人, Science.2011;332(6037):1519-1523)。据推测降低脂肪变性(支撑NAFLD/NASH发展的代谢驱动物)将导致肝炎症和纤维化的后续改善。
乙酰CoA羧化酶(ACC)和二酰基甘油酰基转移酶2 (DGAT2)为调节脂质代谢的两个关键酶。ACC催化脂肪从头合成(DNL)过程中的必要和限速步骤(Saggerson D, Annu. Rev. Nutr. 2008;28:253-72.)。另外,ACC也通过酶肉毒碱棕榈酰转移酶1 (CPT1)的变构调节来调节脂肪酸的线粒体β-氧化(Saggerson, 2008;Waite M和Wakil SJ. J. Biol. Chem.1962;237:2750-2757.)。最近数据也表明,通过ACC抑制来压制DNL可通过限制分泌炎性白细胞介素17 (IL-17)的T辅助17谱系的T细胞(Th17细胞)的形成,和有利于抗炎FoxP3(+)调节性T (Treg)细胞的发展而直接减轻炎症(Berod L等人 Nat. Med. 2014;20(11):1327-33)。
推测通过至少两种独立的机制抑制ACC活性对患有NASH的患者有益。如上所概述,患有NAFLD的人显示肝DNL显著升高且推测通过药理性肝ACC抑制使此增加的通量正常化来降低脂肪变性。此外,ACC抑制剂增加脂肪酸氧化的效应也可有助于降低肝脂肪含量。与此一致,ACC抑制剂已被证明抑制DNL。参见Griffith DA,等人 J. Med. Chem. 2014;57(24):10512-10526;Kim CW,等人 Cell Metab. 2017;26, 394-406; Stiede K,,等人Hepatology. 2017;66(2) :324-334;Lawitz EJ,等人 Clin Gastroenterol Hepatol.2018 (https://doi.org/10.1016/j.cgh.2018.04.042)。此外,预期在分泌IL-17的T细胞中抑制DNL通过限制炎性Th17细胞的形成(Berod等人, 2014),一种在NASH发病机制中可能重要的途径(Rau M,等人 J. Immunol. 2016;196(1):97-105),和有利于抗炎Treg细胞的发展来压制肝性炎症。另外,ACC抑制可降低星状细胞的活化和纤维化(Ross等人, 2019)。
甘油三酯或三酰甘油(TG)表示哺乳动物中的主要能量储存形式。TG通过甘油与三种不同的链长度及饱和度的脂肪酸依次酯化而形成(Coleman, R. A., 和Mashek, D. G.2011. Chem. Rev. 111:6359-6386)。将肠或肝中合成的TG分别包入乳糜微粒或极低密度脂蛋白(VLDL)中,且输出至外周组织,在此它们被脂蛋白脂肪酶(LPL)水解成组成它们的脂肪酸和甘油。所得非酯化脂肪酸(NEFA)可代谢而进一步产生能量或再酯化并储存。
在正常生理条件下,能量密集型TG在各种脂肪储库中维持螯合,直至需要其释放为止,随即,其水解成甘油和游离脂肪酸,此类甘油和游离脂肪酸然后释放至血流中。此过程通过胰岛素和激素(诸如儿茶酚胺)的逆向作用而紧密地调节,该逆向作用促进TG储存物在各种生理条件下蓄积和移动。在餐后设置中,胰岛素起作用而抑制脂肪分解,从而限制以NEFA形式的能量释放且确保饮食脂质适当储存于脂肪储库中。然而,在患有2型糖尿病的患者中,胰岛素抑制脂肪分解的能力不足,且来自脂肪细胞的NEFA通量不当地升高。此又导致递送至组织(诸如肌肉和肝)的脂质增加。在没有能量需求下,TG和其他的脂质代谢物(诸如二酰基甘油(DAG))会累积且造成胰岛素敏感度的丧失(Erion, D. M., 和Shulman, G. I.2010. Nat Med 16: 400-402)。肌肉中的胰岛素抗性以葡萄糖摄取和糖原储存的降低为特征,而在肝中,胰岛素信号传导的丧失引起失调的葡萄糖输出和富含TG的VLDL的生产过量(2型糖尿病的特点)(Choi, S. H., 和Ginsberg, H. N. 2011. Trends Endocrinol. Metab. 22: 353-363)。认为富含TG的VLDL (所谓的VLDL1粒子)的分泌升高会刺激小而密低密度脂蛋白(sdLDL)产生,sdLDL为与冠心病的风险升高相关联的LDL的促动脉粥样硬化亚级分(St-Pierre, A. C.et.al. 2005. Arterioscler. Thromb. Vasc. Biol. 25:553-559)。
在哺乳动物中,已表征两种二酰基甘油酰基转移酶(DGAT)酶(DGAT1和DGAT2)。虽然这些酶催化相同的酶反应,但是它们各自的氨基酸序列不相关且它们占有不同的基因家族。携有编码DGAT1的基因破坏的小鼠对饮食诱发的肥胖具有抗性且具有升高的能量消耗和活性(Smith, S. J.等人, 2000. Nat Genet 25:87-90)。Dgat1-/-小鼠表现出失调的乳糜微粒吸收后释放且在肠细胞中累积脂质(Buhman, K. K.等人2002. J. Biol. Chem.277:25474-25479)。在这些小鼠中观察到的代谢上有利的表型表明由肠中DGAT1表达的丧失而驱动(Lee, B.,等人 2010. J. Lipid Res. 51:1770-1780)。重要地,尽管在雌性Dgat1-/-小鼠中有泌乳缺陷,但是这些动物保留合成TG的能力,表明有另外的DGAT酶存在。此观察结果以及自真菌拉曼被孢霉(Mortierella rammaniana)分离第二DGAT导致对DGAT2的鉴定和表征(Yen, C. L.等人 2008. J. Lipid Res. 49:2283-2301)。
DGAT2高度表达于肝和脂肪中,且不像DGAT1,其表现出对DAG敏锐的底物特异性(Yen, C.L., 2008)。在啮齿动物中的DGAT2基因缺失导致有缺陷的子宫内生长、严重的脂血症、受损的皮肤屏障功能,以及产后早期死亡(Stone, S. J.等人 2004. J. Biol. Chem. 279:11767-11776)。由于DGAT2的丧失所引起的致命性,我们对DGAT2的生理学角色的大多数理解源自于在代谢疾病的啮齿动物模型中用反义寡核苷酸(ASO)执行的研究。在此设置中,抑制肝DGAT2导致血浆脂蛋白概况的改善(总胆固醇和TG减少)和肝脂质负荷的降低,伴随着改善的胰岛素敏感度和全身葡萄糖控制(Liu, Y.等人 2008. Biochim. Biophys. Acta 1781: 97-104; Choi, C. S.等人 2007. J. Biol. Chem. 282: 22678-22688; Yu, X. X.等人 2005. Hepatology 42: 362-371)。虽然未完全阐明构成这些观察的基础的分子机制,但显然DGAT2的抑制导致编码涉及脂肪生成的蛋白的多个基因的表达下调,包括固醇调节元件结合蛋白1c (SREBP1c)和硬脂酰CoA去饱和酶1 (SCD1)(Choi,2007; Yu, 2005)。同时,氧化途径被诱发,如增加的基因表达所证明的,例如肉毒碱棕榈酰转移酶1 (CPT1) (Choi,2007)。这些变化的最终结果为降低肝DAG和TG脂质的水平,这又导致改善的肝中胰岛素响应。此外,DGAT2抑制压制肝VLDL TG的分泌和循环胆固醇水平的降低。最终,血浆载脂蛋白B(APOB)水平被压制,可能由于用于新合成的APOB蛋白的脂质化的TG供应降低(Liu, 2008;Yu, 2005)。DGAT2抑制对血糖控制和血浆胆固醇概况二者的有益效应表明此靶标可能有治疗代谢疾病的价值(Choi,2007)。此外,DGAT2活性的压制导致肝脂质累积降低的观察表明此酶的抑制剂可能具有治疗NASH的效用。
鉴于上述,需要一种药物,例如,口服药物,其含有以下组合:(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(DGAT2抑制剂)或其药学上可接受的盐,且在相同或单独的组合物中包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸(ACCI抑制剂)或其药学上可接受的盐。本文所述的特定组合符合目前的需求。
发明概述
本发明涉及一种药物组合物,其包含与药学上可接受的赋形剂混合的各自以治疗有效量存在的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
本发明也涉及一种药物组合物,其包含与药学上可接受的赋形剂混合的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐。
本发明也涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
本发明也涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合的步骤。
本发明也涉及一种用于在人类中治疗脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎或伴有硬化且伴有肝细胞癌的非酒精性脂肪性肝炎的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的第一组合物和第二组合物,和任选的第三组合物的步骤,其中
i. 第一组合物包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,和药学上可接受的赋形剂;
ii. 第二组合物包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,和药学上可接受的赋形剂;和
iii. 第三组合物包含选自以下的药剂:抗炎剂、抗糖尿病剂、抗纤维化剂、抗脂肪变性剂、胆固醇/脂质调节剂和抗糖尿病剂,以及药学上可接受的赋形剂。
本发明也涉及一种用于在人类中治疗脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎或伴有硬化且伴有肝细胞癌的非酒精性脂肪性肝炎的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的两种单独的药物组合物的步骤,所述药物组合物包含:
i. 第一组合物,其包含与药学上可接受的赋形剂混合的以治疗有效量存在的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐;
ii. 第二组合物,其包含与药学上可接受的赋形剂混合的以治疗有效量存在的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐;以及任选地
iii. 第三组合物,其包含至少一种选自以下的另外药剂:GLP-1R激动剂、KHK抑制剂或FXR激动剂,以及药学上可接受的赋形剂。
本发明也涉及一种用于治疗心力衰竭、充血性心力衰竭、冠心病、外周血管疾病、肾血管疾病、肺动脉高压、血管炎、急性冠状动脉综合征和心血管风险改变的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
本发明也涉及一种用于治疗肥胖、I型糖尿病、II型糖尿病、特发性I型糖尿病(Ib型)、成人隐匿性自身免疫糖尿病(LADA)、早发2型糖尿病(EOD)、青少年起病非典型糖尿病(YOAD)、青少年的成人起病型糖尿病(MODY)、营养不良相关性糖尿病、妊娠糖尿病、冠心病、缺血性中风、血管成形术后再狭窄、外周血管疾病、间歇性跛行、心肌梗塞、血脂异常、餐后脂血症、糖耐量受损(IGT)的病况、空腹血糖受损的病况、代谢性酸中毒、酮病、关节炎、糖尿病视网膜病变、黄斑变性、白内障、糖尿病肾病、肾小球硬化症、慢性肾衰竭、糖尿病神经病变、代谢综合征、综合征X、高血糖症、高胰岛素血症、高甘油三脂血症、胰岛素抵抗、葡萄糖代谢受损、皮肤与结缔组织病症、足部溃疡与溃疡性结肠炎、内皮功能障碍与血管顺应性受损、高载脂蛋白B脂蛋白血症和枫糖尿症的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
本发明也涉及一种用于治疗肝细胞癌、肾脏肾透明细胞癌、头颈部鳞状细胞癌、结直肠腺癌、间皮瘤、胃腺癌、肾上腺皮质癌、肾脏乳头状细胞癌、子宫颈和子宫颈内癌、膀胱尿路上皮癌、肺腺癌的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
应理解前文的一般说明和下列详细说明均仅为示例性和解释性的,并非限制要求保护的发明。
附图说明
图1为显示实施例1的DGAT2i化合物的结晶形式1的特征x射线粉末衍射图(纵轴:强度(CPS);横轴:2θ (度))。
图2为显示实施例1的DGAT2i化合物的结晶形式2的特征x射线粉末衍射图(纵轴:强度(CPS);横轴:2θ (度))。
图3显示在配备Cu辐射源的Bruker AXS D4 Endeavor 衍射仪上进行的化合物A的形式1的说明性PXRD图。
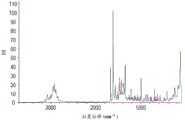
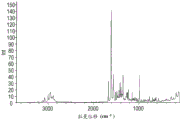
图4显示使用连接至FT-IR工作台的Nicolet NXR FT-拉曼配件收集的化合物A的形式1的说明性拉曼光谱。
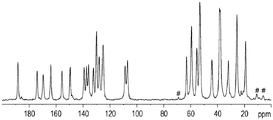
图5显示在位于Bruker-BioSpin Avance III 500 MHz (1H频率) NMR光谱仪中的Bruker-BioSpin CPMAS探针上进行的化合物A的形式1的说明性13C ssNMR图。
图6显示在配备Cu辐射源的Bruker AXS D4 Endeavor衍射仪上进行的化合物A的形式2的说明性PXRD图。
图7显示使用连接至FT-IR工作台的Nicolet NXR FT-拉曼配件收集的化合物A的形式2的说明性拉曼光谱。
图8显示在位于Bruker-BioSpin Avance III 500 MHz (1H频率) NMR光谱仪中的Bruker-BioSpin CPMAS探针上进行的化合物A的形式2的说明性13C ssNMR图。
图9显示化合物A的形式2的说明性的单晶结构。
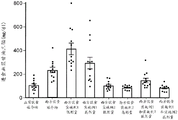
图10总结在西方饮食喂养的Sprague Dawley大鼠中在进食状态下测量的口服施用作为单一疗法和组合的化合物A和化合物D对血浆甘油三酯水平的影响。
图11总结在西方饮食喂养的Sprague Dawley大鼠中在禁食状态下测量的口服施用作为单一疗法和组合的化合物A和化合物D对血浆甘油三酯水平的影响。
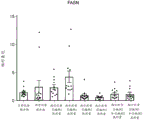
图12总结在西方饮食喂养的大鼠中施用作为单一疗法和组合的化合物A和化合物D对SREBP-1核定位的影响。
图13总结在西方饮食喂养的大鼠中施用作为单一疗法和组合的化合物A和化合物D对肝脂肪生成基因表达的影响,特别是乙酰CoA羧化酶(ACC1)。
图14总结在西方饮食喂养的大鼠中施用作为单一疗法和组合的化合物A和化合物D对肝脂肪生成基因表达的影响,特别是脂肪酸合成酶(FASN)。
图15总结在西方饮食喂养的大鼠中施用作为单一疗法和组合的化合物A和化合物D对肝脂肪生成基因表达的影响,特别是固醇CoA去饱和酶(SCD1)。
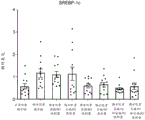
图16总结在西方饮食喂养的大鼠中施用作为单一疗法和组合的化合物A和化合物D对肝脂肪生成基因表达的影响,特别是固醇调节元件结合蛋白1c (SREBP-1c)。
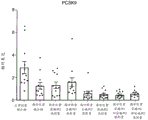
图17总结在西方饮食喂养的大鼠中施用作为单一疗法和组合的化合物A和化合物D对肝脂肪生成基因表达的影响,特别是前蛋白转化酶枯草杆菌蛋白酶/kexin9型(PCSK9)。
图18总结在西方饮食喂养的Sprague Dawley大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对肝甘油三酯水平的影响。
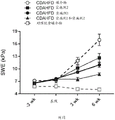
图19总结在胆碱缺乏和高脂肪饮食(CDAHFD)喂养的雄性Wistar Hann大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对肝的弹性(肝的炎症和纤维化的标志物)的影响。
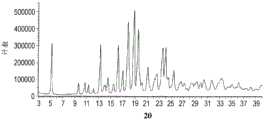
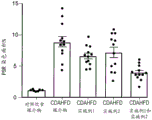
图20总结在CDAHFD喂养的雄性Wistar Hann大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对肝α平滑肌肌动蛋白(αSMA)免疫组织化学(肌纤维母细胞活化和纤维发生的标志物)的影响。
图21总结在CDAHFD喂养的雄性Wistar Hann大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对肝天狼星红染色的影响。
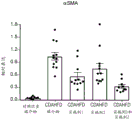
图22总结在CDAHFD喂养的雄性Wistar Hann大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对肝α平滑肌肌动蛋白(αSMA)基因表达的影响。
图23总结在CDAHFD喂养的雄性Wistar Hann大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对肝胶原蛋白1A1基因表达的影响。
图24总结在CDAHFD喂养的雄性Wistar Hann大鼠中口服施用作为单一疗法和组合的化合物A和化合物D对离子化钙结合适配分子1 (ionized calcium binding adaptermolecule 1)染色的影响。
发明详述
本发明可通过参考下列本发明的示例性实施方案的详细说明和其中包括的实施例而更容易地理解。
应理解本发明不限于特定的制备合成方法,其当然可以改变。也应理解本文中所使用的术语仅出于说明特定实施方案的目的,并不意欲为限制。在本说明书和随后的权利要求书中,将参考许多术语,这些术语应定义为具有下列意义:
如本文说明书所用,"一(a)或(an)"可意指一或多个。如本文权利要求书所用,当与词"包含"结合使用时,词"一(a)或(an)"可意指一个或多于一个。如本文所用,"另一"可意指至少两个或更多个。
术语"约"是指相对术语,表示其所指的标称值加或减10%的近似值,在一个实施方案中,加或减5%,在另一实施方案中,加或减2%。就本公开的领域而言,除非特别说明该值要求更窄的范围,否则此近似水平是适当的。
"化合物"当在本文中使用时,包括任何药学上可接受的衍生物或变体,包括构象异构体(例如顺式和反式异构体)和所有光学异构体(例如对映异构体和非对映异构体),此类异构体的外消旋、非对应异构体和其他混合物,以及溶剂合物、水合物、同晶型体、多晶型物、互变异构体、酯、盐形式和前药。表述"前药"是指作为药物前体的化合物,其在施用后经由一些化学或生理过程而体内释放药物(例如前药在达到生理pH时或通过酶作用转化成所需的药物形式)。示例性前药在裂解时释放对应的游离酸,且本发明化合物的此类可水解的酯形成残基包括但不限于具有羧基部分的那些,其中游离氢被下列替代:(C1-C4)烷基、(C2-C7)烷酰氧基甲基、具有4至9个碳原子的1-(烷酰氧基)乙基、具有5至10个碳原子的1-甲基-1-(烷酰氧基)-乙基、具有3至6个碳原子的烷氧羰基氧基甲基、具有4至7个碳原子的1-(烷氧羰基氧基)乙基、具有5至8个碳原子的1-甲基-1-(烷氧羰基氧基)乙基、具有3至9个碳原子的N-(烷氧羰基)氨基甲基、具有4至10个碳原子的1-(N-(烷氧羰基)氨基)乙基、3-酞基、4-巴豆酸内酯基(crotonolactonyl)、γ-丁内酯-4-基、二-N,N-(C1-C2)烷氨基(C2-C3)烷基(诸如β-二甲基氨基乙基)、氨基甲酰基-(C1-C2)烷基、N,N-二(C1-C2)烷基氨基甲酰基-(C1-C2)烷基和哌啶子基-、吡咯烷子基-或吗啉代(C2-C3)烷基。
如本文所用,箭头," "或波浪线,"
"或波浪线," "表示取代基与另一基团的连接点。
"表示取代基与另一基团的连接点。
"患者"是指温血动物,诸如豚鼠、小鼠、大鼠、沙鼠、猫、兔、狗、牛、山羊、绵羊、马、猴、黑猩猩和人类。"哺乳动物"为患者。
"药学上可接受的"意指物质或组合物必须与构成制剂的其他成分和/或以其治疗的哺乳动物在化学上和/或毒理学上相容。
如本文所用,下列术语具有用于施用药剂的一般意义:QD表示每天一次,和BID表示每天两次。
如本文所用,表述"反应惰性溶剂"和"惰性溶剂"是指不会以不利地影响所需的产物的产率的方式与起始材料、试剂、中间体或产物相互作用的溶剂或其混合物。
如本文所用,术语"选择性(selectivity)"或"选择性的(selective)"是指化合物在第一测定中的效应大于相同化合物在第二测定中的效应。例如,在"肠选择性的"化合物中,第一测定用于化合物在肠中的半衰期和第二测定用于化合物在肝中的半衰期。
"治疗有效量"表示治疗本文所述的特定疾病、病况或病症的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸(化合物A)的量与(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(化合物D)的量的组合,任选与另外一种或多种化合物的量组合。
4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸为选择性ACC抑制剂,且依据US 8,859,577 (其为国际申请号PCT/IB2011/054119的美国国家阶段,其全部为了所有目的特此以其整体通过引用并入本文)的实施例9制备成游离酸。化合物的结晶形式描述于国际专利申请号PCT/IB2018/058966中,其在2019年5月31日公开为WO 2019/102311。
(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺为DGAT2抑制剂且为美国公开的专利申请US 2018-0051012A1 (为了所有目的特此以其整体通过引用并入本文)的实施例1。
如本文所用,术语"治疗(treating、treat、treatment)"包含预防性(preventative)(即预防性(prophylactic));和姑息治疗,即减轻、缓和或减慢患者的疾病(或病况)进展或与疾病(或病况)相关的任何组织损害;和逆转,其中患者的疾病(或病况)不仅缓和且与疾病(或病况)相关的任何组织损伤处于比在开始治疗时更好的状态。后者可发生自例如但不限于下列中的任何一个或多个:NASH消退的证明和/或基于肝活检的纤维化评分的改善;进展为硬化、肝细胞癌和/或其他肝相关结局的发生率较低;非酒精性脂肪性肝炎活性的血清或成像基标志物水平的降低或改善;非酒精性脂肪性肝炎疾病活性的降低或改善;或非酒精性脂肪性肝炎的医疗后果的降低。
似乎ACC抑制剂的施用可对降低肝TG有正面作用且对NASH的治疗可能有其他有益作用。据报导循环TG水平升高为肝ACC抑制的机制结果(Kim等人, 2017),尽管仅部分抑制DNL的ACC抑制剂的剂量不会使循环TG升高(Bergman等人, (2018) J. of Hepatology,Volume 68, S582)。WO2016/112305提供使用单独或与一种或多种另外的治疗剂一起的ACC抑制剂治疗、稳定或减轻非酒精性脂肪性肝病的严重程度或进展的方法。
已经发现4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的施用,任选地以药学上可接受的盐施用,如在人类对象中所观察到的,在西方饮食喂养的Sprague Dawley大鼠中有可能导致循环TG升高(通常由血浆测量)。
本发明化合物可含有不对称或手性中心,且因此以不同的立体异构体形式存在。除非另有规定,意欲使本发明化合物的所有立体异构体形式以及其混合物(包括外消旋混合物)形成本发明的一部分。此外,本发明包含所有的几何和位置异构体。例如,若本发明化合物并入双键或稠合环,则顺式和反式形式二者以及混合物均包含在本发明的范围内。
本发明的手性化合物(及其手性前体)可使用色谱法(通常为高压液相色谱法(HPLC)或超临界流体色谱法(SFC))在具有不对称固定相的树脂上和由含有0至50%异丙醇(通常为2%至20%)的烃(通常为庚烷或己烷)和0至5%烷胺(通常为0.1%二乙胺(DEA)或异丙胺)所组成的流动相以富集对映异构体的形式获得。洗脱液的浓缩提供富集的混合物。
非对映异构体混合物可通过本领域技术人员已知的方法(诸如通过色谱法和/或分级结晶)根据其物理化学差异分离成它们各个的非对映异构体。对映异构体可如下分离:通过与适当的光学活性化合物(例如手性助剂诸如手性醇或Mosher酰氯)的反应而将对映异构体混合物转化成非对映异构体混合物,分离非对映异构体并将各个非对映异构体转化(例如水解)成相对应的纯对映异构体。对映异构体也可通过使用手性HPLC柱来分离。或者,特定的立体异构体可由以下方式合成:通过使用光学活性起始材料,使用光学活性试剂、底物、催化剂或溶剂通过不对称合成,或通过不对称变换而将一种立体异构体转化成另一种。
在本发明化合物具有二或多个立体中心且在名称中给出绝对或相对立体化学的情况下,名称R和S分别是指根据用于各分子的常规IUPAC编号方案而以升序编号(1、2、3等)的各立体中心。在本发明化合物具有一个或多个立体中心且未在名称或结构中给出立体化学的情况下,则应理解名称或结构意欲包含化合物的所有形式,包括外消旋形式。
本发明的中间体和化合物也可能可以不同的互变异构体形式存在,且所有此类形式均包含在本发明的范围内。术语"互变异构体"或"互变异构体形式"是指经由低能垒可相互转化的不同能量的结构异构体。例如,质子互变异构体(也称为质子转移互变异构体(prototropic tautomer))包括经由质子迁移的相互转化,诸如酮-烯醇和亚胺-烯胺异构化。
价键互变异构体包括通过重组一些键合电子进行的相互转化。
本发明所要求保护的化合物的范围内包括本发明化合物的所有立体异构体、几何异构体和互变异构体形式,包括表现出超过一种类型的异构现象的化合物及其一种或多种的混合物。也包括酸加成盐或碱盐,其中抗衡离子具有光学活性,例如D-乳酸盐或L-赖氨酸;或是外消旋的,例如DL-酒石酸盐或DL-精氨酸。
本发明包括所有药学上可接受的同位素标记的本发明化合物,其中一或多个原子被具有相同原子序数但原子质量或质量数与通常在自然中发现的原子质量或质量数不同的原子替代。
适合于包含在本发明化合物中的同位素的实例包括下列的同位素:氢(诸如2H和3H)、碳(诸如11C、13C和14C)、氯(诸如36Cl)、氟(诸如18F)、碘(诸如123I、124I和125I)、氮(诸如13N和15N)、氧(诸如15O、17O和18O)、磷(诸如32P)和硫(诸如35S)。
某些同位素标记的本发明化合物(例如,并入放射性同位素的那些)可用于药物和/或底物组织分布研究。放射性同位素氚(即3H)和碳-14(即14C)鉴于它们容易并入和现成的检测方法而特别可用于此目的。
被较重的同位素(诸如氘,即2H)替代可提供因较大代谢稳定性所产生的某些治疗优势,例如体内半衰期的增加或剂量需求的降低,且因此在一些情况下可是优选的。
被正电子放射同位素(诸如11C、18F、15O和13N)替代可用于供检查底物受体占有率的正电子发射断层扫描(PET)研究。
同位素标记的本发明化合物通常可通过本领域技术人员已知的常规技术来制备,或可通过与所附实施例和制备中所述类似的方法使用适当同位素标记的试剂置换先前所采用的未经标记的试剂来制备。
本发明化合物可被分离且以本身形式使用,或可能的话,以其药学上可接受的盐形式使用。术语"盐"是指本发明化合物的无机盐和有机盐。这些盐可在化合物的最终分离和纯化期间原位制备,或通过以适当有机或无机酸或碱单独处理化合物且分离因此形成的盐而制备。用于制备前述本发明的碱化合物的药学上可接受的酸加成盐的酸为形成无毒性酸加成盐的那些(即含有药理学上可接受的阴离子的盐,诸如盐酸盐、氢溴酸盐、氢碘酸盐、硝酸盐、硫酸盐、硫酸氢盐、磷酸盐、酸式磷酸盐、乙酸盐、乳酸盐、柠檬酸盐、酸式柠檬酸盐、酒石酸盐、酒石酸氢盐、琥珀酸盐、马来酸盐、富马酸盐、葡糖酸盐、蔗糖酸盐、苯甲酸盐、甲磺酸盐、乙磺酸盐、苯磺酸盐、萘酸盐、甲磺酸盐、葡庚糖酸盐、乳糖酸盐(lactobionate)、月桂基磺酸盐、六氟磷酸盐、苯磺酸盐、甲苯磺酸盐、甲酸盐、三氟乙酸盐、草酸盐、苯磺酸盐(besylate)、棕榈酸盐、双羟萘酸盐、丙二酸盐、硬脂酸盐、月桂酸盐、苹果酸盐、硼酸盐、对甲苯磺酸盐和双羟萘酸盐(即1,1'-亚甲基-双-(2-羟基-3-萘甲酸盐))。
本发明也涉及本发明化合物的碱加成盐。可用作制备性质上为酸性的那些本发明化合物的药学上可接受的碱盐的试剂的化学碱为与此类化合物形成无毒碱盐的那些。此类无毒碱盐包括但不限于衍生自此类药理学上可接受的阳离子(诸如碱金属阳离子(例如,锂、钾和钠)和碱土金属阳离子(例如,钙和镁))的那些、铵或水溶性胺加成盐诸如N-甲基葡糖胺(甲葡胺(meglumine))、四甲铵、四乙铵、甲胺、二甲胺、三甲胺、三乙胺、乙胺,以及低级烷醇铵(alkanolammonium)和药学上可接受的有机胺的其他碱盐。参见例如Berge等人,J. Pharm. Sci.,66,1-19 (1977)。
某些本发明化合物可以超过一种的晶型存在(一般称为"多晶型物(polymorphs)")。多晶型物可通过在各种条件下结晶而制备,例如,使用用于重结晶的不同的溶剂或不同的溶剂混合物;在不同温度下结晶;和/或各种冷却模式,在结晶期间范围从非常快的到非常慢的冷却。多晶型物也可通过加热或熔化本发明化合物,然后逐渐或快速的冷却而获得。多晶型物的存在可以通过固体探针NMR光谱法、IR光谱法、差示扫描量热法、粉末X射线衍射或此类其他技术测定。
在一个实施方案中,本发明涉及一种药物组合物,其包含与药学上可接受的赋形剂混合的各自以治疗有效量存在的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
在进一步的实施方案中,该组合物进一步包含至少一种选自以下的另外药剂:抗炎剂、抗糖尿病剂和胆固醇/脂质调节剂。
在进一步的实施方案中,该组合物进一步包含至少一种选自以下的另外药剂:[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
在进一步的实施方案中,该药物组合物含有呈下列结构的结晶固体的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺:

或其药学上可接受的盐。
在进一步的实施方案中,该结晶固体具有包含5.3±0.2、7.7±0.2和15.4±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末X射线衍射图。
在进一步的实施方案中,该结晶固体具有包含6.5±0.2、9.3±0.2和13.6±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末X射线衍射图。
在进一步的实施方案中,该药物组合物含有呈下列结构的结晶固体的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐:

或其药学上可接受的盐。
在进一步的实施方案中,该结晶固体为4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的2-氨基-2-(羟甲基)丙烷-1,3-二醇盐。
在进一步的实施方案中,该药物组合物进一步包含至少一种选自以下的另外药剂:[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
在另一实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的患者施用治疗有效量的S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
在进一步的实施方案中,该疾病或病况为脂肪肝。在另一实施方案中,该疾病或病况为非酒精性脂肪性肝病。在另一实施方案中,该疾病或病况为非酒精性脂肪性肝炎。在另一实施方案中,该疾病或病况为伴有肝纤维化的非酒精性脂肪性肝炎。在另一实施方案中,该疾病或病况为伴有硬化的非酒精性脂肪性肝炎。在另一实施方案中,该疾病或病况为伴有硬化且伴有肝细胞癌的非酒精性脂肪性肝炎。在另一实施方案中,该疾病或病况为伴有硬化且伴有代谢相关疾病的非酒精性脂肪性肝炎。
在进一步的实施方案中,所述方法包括至少一种其他药剂,其中该药剂选自:乙酰CoA羧化酶-(ACC)抑制剂,二酰基甘油O-酰基转移酶1 (DGAT-1)抑制剂,单酰基甘油O-酰基转移酶抑制剂,磷酸二酯酶(PDE)-10抑制剂,AMPK活化剂,磺酰脲,美格列奈(meglitinide),α-淀粉酶抑制剂,α-葡萄糖苷水解酶抑制剂,α-葡萄糖苷酶抑制剂,PPARγ激动剂,PPAR α/γ激动剂,双胍,胰高血糖素样肽1 (GLP-1)调节剂,利拉鲁肽(liraglutide),阿必鲁肽(albiglutide),艾塞那肽(exenatide),阿必鲁肽(albiglutide),利西拉肽(lixisenatide),杜拉鲁肽(dulaglutide),索马鲁肽(semaglutide),蛋白酪氨酸磷酸酶-1B (PTP-1B)抑制剂,SIRT-1活化剂,二肽基肽酶IV(DPP-IV)抑制剂,胰岛素促泌剂,脂肪酸氧化抑制剂,A2拮抗剂,c-jun氨基末端激酶(JNK)抑制剂,葡萄糖激酶活化剂(GKa),胰岛素,胰岛素模拟剂,糖原磷酸化酶抑制剂,VPAC2受体激动剂,SGLT2抑制剂,胰高血糖素受体调节剂,GPR119调节剂,FGF21衍生物或类似物,TGR5受体调节剂,GPBAR1受体调节剂,GPR40激动剂,GPR120调节剂,高亲和性烟酸受体(HM74A)活化剂,SGLT1抑制剂,肉毒碱棕榈酰转移酶的抑制剂或调节剂,果糖-1,6-二磷酸酶的抑制剂,醛糖还原酶的抑制剂,盐皮质激素受体抑制剂,TORC2的抑制剂,CCR2和/或CCR5的抑制剂,PKC同种型(isoform)(例如,PKCα、PKCβ、PKCγ)的抑制剂,脂肪酸合成酶的抑制剂,丝氨酸棕榈酰转移酶的抑制剂,GPR81、GPR39、GPR43、GPR41、GPR105、Kv1.3、视黄醇结合蛋白4、糖皮质激素受体、生长抑素受体的调节剂,PDHK2或PDHK4的抑制剂或调节剂,MAP4K4的抑制剂,IL1家族(包括IL1β)的调节剂,HMG-CoA还原酶抑制剂,角鲨烯合成酶抑制剂,贝特类(fibrate),胆汁酸螯合剂,ACAT抑制剂,MTP抑制剂,脂加氧酶抑制剂,胆固醇吸收抑制剂,PCSK9调节剂,胆固醇酯转移蛋白抑制剂和RXRα的调节剂。
在进一步的实施方案中,所述方法包括至少一种其他药剂,其中该药剂选自:半胱胺或其药学上可接受的盐,胱胺或其药学上可接受的盐,抗氧化剂化合物,卵磷脂,维生素B复合物,胆盐制剂,大麻素-1 (CB1)受体的拮抗剂,大麻素-1 (CB1)受体的反向激动剂,过氧化物酶体增殖物活化受体活性调节剂,苯并硫氮杂䓬或苯并硫杂䓬化合物,抑制蛋白酪氨酸磷酸酶PTPRU的RNA反义构建体,杂原子连接的经取代的哌啶及其衍生物,能够抑制硬脂酰辅酶αδ-9去饱和酶的氮杂环戊烷衍生物,具有脂联素(adiponectin)的促泌剂或诱导剂活性的酰基酰胺化合物,季铵化合物,醋酸格拉替雷(Glatiramer acetate),穿透素(pentraxin)蛋白,HMG-CoA还原酶抑制剂,n-乙酰基半胱氨酸,异黄酮化合物,大环内酯类抗生素,半乳糖凝集素(galectin)抑制剂,抗体或其任何组合。
在进一步的实施方案中,所述方法包括至少一种其他药剂,其中该药剂选自:[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
在另一实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合的步骤。
在进一步的实施方案中,所述方法包括至少一种其他药剂,其中该药剂选自:[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
在另一实施方案中,本发明涉及一种用于在人类中治疗脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎或伴有硬化且伴有肝细胞癌的非酒精性脂肪性肝炎的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的两种单独的药物组合物的步骤,所述药物组合物包含:
i. 第一组合物,其包含与药学上可接受的赋形剂混合的以治疗有效量存在的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐;
ii. 第二组合物,其包含与药学上可接受的赋形剂混合的以治疗有效量存在的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐;以及任选地
iii. 第三组合物,其包含至少一种选自以下的另外药剂:抗炎剂、抗糖尿病剂、抗纤维化剂、抗脂肪变性剂、胆固醇/脂质调节剂和抗糖尿病剂,以及药学上可接受的赋形剂。
在进一步的实施方案中,所述第一组合物和所述第二组合物同时施用。在另一实施方案中,该组合物包含第一组合物、第二组合物和第三组合物。
在进一步的实施方案中,所述方法包括第三组合物,其中该至少一种其他药剂选自[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
在本发明的另一实施方案中,与药学上可接受的赋形剂混合的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合以治疗有效量存在于药物组合物中。
在本发明的另一实施方案中,与药学上可接受的赋形剂混合的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合以治疗有效量存在于药物组合物中。
在本发明的另一实施方案中,该组合物进一步包括至少一种选自以下的另外药剂:GLP-1R激动剂、KHK抑制剂、FXR激动剂、抗炎剂、抗糖尿病剂、抗纤维化剂、抗脂肪变性剂和胆固醇/脂质调节剂。
在另一实施方案中,用于治疗代谢或代谢相关的疾病、病况或病症的方法包括向患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合的步骤。
在另一实施方案中,用于治疗选自以下的病况的方法包括施用治疗有效量的化合物A或其药学上可接受的盐和治疗有效量的化合物D或其药学上可接受的盐:高脂血症、I型糖尿病、II型糖尿病、特发性I型糖尿病(Ib型)、成人隐匿性自身免疫糖尿病(LADA)、早发2型糖尿病(EOD)、青少年起病非典型糖尿病(YOAD)、青少年的成人起病型糖尿病(MODY)、营养不良相关性糖尿病、妊娠糖尿病、冠心病、缺血性中风、血管成形术后再狭窄、外周血管疾病、间歇性跛行、心肌梗塞(例如坏死和凋亡)、血脂异常、餐后脂血症、糖耐量受损(IGT)的病况、空腹血糖受损的病况、代谢性酸中毒、酮病、关节炎、肥胖、骨质疏松症、高血压、充血性心力衰竭、左心室肥大、外周动脉疾病、糖尿病视网膜病变、黄斑变性、白内障、糖尿病肾病、肾小球硬化症、慢性肾衰竭、糖尿病神经病变、代谢综合征、综合征X、经前综合征、冠心病、心绞痛、血栓症、动脉粥样硬化、心肌梗塞、短暂性脑缺血发作、中风、血管再狭窄、高血糖症、高胰岛素血症、高脂血症、高甘油三脂血症、胰岛素抵抗、葡萄糖代谢受损、糖耐量受损的病况、空腹血糖受损的病况、肥胖、勃起功能障碍、皮肤与结缔组织病症、足部溃疡与溃疡性结肠炎、内皮功能障碍和血管顺应性受损、高载脂蛋白B脂蛋白血症、阿尔茨海默病、精神分裂症、认知受损、炎性肠病、溃疡性结肠炎、克罗恩病和肠易激综合征、非酒精性脂肪性肝炎(NASH)、非酒精性脂肪性肝病(NAFLD)。
在进一步的实施方案中,用于治疗代谢或代谢相关的疾病、病况或病症的方法包括向需要此类治疗的患者施用至少两种单独的药物组合物的步骤,所述药物组合物包含:
(i) 第一组合物,其包含与药学上可接受的赋形剂混合的以治疗有效量存在的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺,或其药学上可接受的盐;
(ii) 第二组合物,其包含与药学上可接受的赋形剂混合的以治疗有效量存在的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐;以及任选地
(iii) 第三组合物,其包含至少一种选自以下的另外药剂:GLP-1R激动剂、KHK抑制剂、FXR激动剂、抗炎剂、抗糖尿病剂、抗纤维化剂、抗脂肪变性剂和胆固醇/脂质调节剂和抗糖尿病剂,以及药学上可接受的赋形剂。
在又进一步的实施方案中,当同时施用所述第一组合物、所述第二组合物和所述第三组合物时,进行本发明的方法。
在又一实施方案中,当相继且以任何顺序施用第一组合物、所述第二组合物和所述第三组合物时,进行本发明的方法。
在一个实施方案中,当施用三种药剂时,同时施用该第一药剂和该第二药剂和相继施用该第三药剂。在另一实施方案中,相继且以任何顺序施用三种单独的药剂。
在一个实施方案中,当施用三种药剂时,该第三药剂包含GLP-1R激动剂。GLP-1R激动剂2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药物盐[诸如其2-氨基-2-(羟甲基)丙烷-1,3-二醇盐,也称为其tris盐],以及其他GLP-1R激动剂和制造这些化合物的方法描述于美国专利号10,208,019中,为了所有目的特此将其公开内容以其整体通过引用并入本文。
在某些实施方案中,该GLP-1R激动剂选自:利拉鲁肽、阿必鲁肽、艾塞那肽、阿必鲁肽、利西拉肽、杜拉鲁肽、索马鲁肽、HM15211、LY3298176、Medi-0382、NN-9924、TTP-054、TTP-273、efpeglenatide、在WO2018109607中所述的那些和DIAST-X2。
GLP-1为由L细胞在肠中响应于摄入食物而分泌的30个氨基酸长的肠促胰岛素激素。GLP-1已显示以生理和葡萄糖依赖性方式刺激胰岛素分泌、降低胰高血糖素分泌、抑制胃排空、降低食欲和刺激β细胞增殖。在非临床实验中,GLP-1通过刺激对葡萄糖依赖性胰岛素分泌重要的基因转录和通过促进β细胞新生而促进持续的β细胞活力(Meier,等人Biodrugs. 2003; 17 (2): 93-102)。
在健康的个体中,GLP-1通过刺激胰脏的葡萄糖依赖性胰岛素分泌,导致外周的葡萄糖吸收增加而在调节餐后血糖水平中起重要作用。GLP-1也抑制胰高血糖素分泌,导致肝葡萄糖输出量降低。此外,GLP-1延迟胃排空和减慢小肠运动,延迟食物吸收。在患有T2DM的人中,没有或降低正常的餐后GLP-1上升(Vilsboll T,等人 Diabetes. 2001. 50; 609-613)。
Holst (Physiol. Rev. 2007, 87, 1409)和Meier (Nat. Rev. Endocrinol.2012, 8, 728)描述GLP-1受体激动剂(诸如GLP-1、利拉鲁肽和exendin-4)具有3种通过降低空腹和餐后葡萄糖(FPG和PPG)以改善患者(诸如患有T2DM的那些)的血糖控制的主要药理活性:(i)葡萄糖依赖性胰岛素分泌增加(改善的第一和第二期),(ii)在高血糖状况下的胰高血糖素抑制活性,(iii)延迟胃排空速率,导致膳食衍生的葡萄糖的吸收推迟。
在另一实施方案中,当施用三种药剂时,第三药剂包含KHK抑制剂。
在某些实施方案中,该KHK抑制剂为[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸或药学上可接受的盐。[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸(包括其结晶游离酸形式)为己酮糖激酶(ketohexokinase)抑制剂且描述于美国专利号9,809,579的实施例4中,为了所有目的特此将其公开内容以其整体通过引用并入本文。
在某些实施方案中,该KHK抑制剂为[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸的结晶游离酸形式。
己酮糖激酶(KHK)为果糖代谢中的主要酶且催化果糖转化成果糖-1-磷酸(F1P)。KHK表达为由第三外显子的可变剪接产生的两个可变mRNA剪接变体(表示为KHKa和KHKc)。KHKc用于果糖磷酸化的亲和力和能力比KHKa大许多,如低许多的Km所证明的(Ishimoto,Lanaspa等人, PNAS 109, 4320-4325, 2012)。虽然KHKa广泛表达,但KHKc的表达在肝、肾脏和肠(体内果糖代谢的主要部位)中最高(Diggle CP,等人 (2009) J Histochem Cytochem 57:763–774; Ishimoto, Lanaspa,等人, PNAS 109, 4320-4325, 2012)。另外,已在人类中报告功能突变的缺失,并称为原发性果糖尿症(OMIM #229800),在摄入糖之后,除了尿液中出现果糖外,没有不利影响。
涉及果糖代谢的更严重病况为醛缩酶B (GENE:ALDOB)中的缺陷所引起的遗传性果糖不耐受(HFI, OMIM #229600),该醛缩酶B为负责分解F1P的酶且在途径中紧接KHK步骤的下游(Bouteldja N, 等人, J. Inherit. Metab. Dis. 2010 Apr;33(2):105-12;Tolan, DR, Hum Mutat. 1995;6(3):210-8; http://www.omim.org/entry/229600)。其为在20,000人中影响估计1人的罕见病症,且突变导致F1P的累积、ATP损耗,以及尿酸增加,这些的组合造成低血糖症、高尿酸血症和乳酸酸中毒,以及其他代谢紊乱。HFI损害身体代谢饮食果糖的能力,导致急性症状,诸如呕吐、严重低血糖症、腹泻和腹部窘迫感,导致长期生长缺陷,肝和肾脏受损和潜在地死亡(Ali M等人, J. Med. Genet. 1998 May:35(5):353-65)。患者通常在诊断之前生命的最初几年遭受痛苦,且唯一治疗过程为避免饮食中的果糖。大多数食品中都存在这种大量营养素,因而产生挑战性。除了身体症状,许多患者由于其不寻常的饮食而经历情感和社会孤立,且一直努力坚持严格的饮食限制(HFI-INFODiscussion Board, http://hfiinfo.proboards.com. 于2015年12月14日访问)。即使当它们表现出无症状时,一些患者发展NAFLD和肾脏疾病,其强调自我强加饮食限制作为唯一治疗选项的不足,和对此病况的高度未满足的医学要求。
在高血糖病况中,内源性果糖产生经由多元醇途径(使用山梨糖醇作为中间体将葡萄糖转化为果糖的途径)出现。此途径的活性随高血糖症而增加。在这些研究中,作者证实保护无KHK小鼠免受葡萄糖引起的体重增加、胰岛素抵抗和肝性脂肪变性,表明在高血糖病况下,内源性产生的果糖可促成胰岛素抵抗和肝性脂肪变性(Lanaspa, M.A.,等人,Nature Comm. 4, 2434, 2013)。因此,预期抑制KHK对其中涉及内源性或摄入的果糖中的任一个或两个的改变的许多疾病有益处。
在另一实施方案中,当施用三种药剂时,该第三药剂包含FXR激动剂。在某些实施方案中,该FXR激动剂选自:托品费索(tropifexor)(2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸)("托品费索(Tropifexor)");cilofexor (GS-9674);奥贝胆酸(obeticholic acid);LY2562175;Met409;TERN-101;和EDP-305及其药学上可接受的盐。FXR激动剂托品费索或其药学上可接受的盐描述于例如美国专利号9,150,568的实施例1-1B中,为了所有目的特此将其公开内容以其整体通过引用并入本文。托品费索(Tropifexor)的化学名称为2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸。
法尼醇X受体(FXR)为核激素受体超家族的成员且主要表达在肝、肾脏和肠中(参见,例如,Seol等人 (1995) Mol. Endocrinol. 9:72-85和Forman等人 (1995) Cell 81:687-693)。它充当与维甲酸X受体(RXR)的异二聚体和结合至靶基因启动子中的反应元件以调节基因转录。FXR-RXR异二聚体以最高亲和力结合至反向重复-1 (IR-1)反应元件,其中结合共有受体(consensus receptor)的六聚体被一个核苷酸隔开。FXR为相关过程的一部分,FXR被胆汁酸(胆固醇代谢的终产物)活化(参见,例如,Makishima等人 (1999) Science284: 1362-1365, Parks等人 (1999) Science 284:1365-1368, Wang等人 (1999) Mol.Cell. 3:543-553),其用于抑制胆固醇分解代谢。也参见Urizar等人 (2000) J. Biol.Chem. 275:39313-39317。
FXR为胆固醇稳态、甘油三酯合成以及脂肪生成的关键调节剂(Crawley, ExpertOpinion Ther. Patents (2010), 20(8): 1047-1057)。除了治疗血脂异常外,已描述FXR的多种适应证,包括治疗肝病、糖尿病、维生素D相关疾病、药物引起的副作用和肝炎。(Crawley,同上)。尽管新颖FXR激动剂的开发已有进展,但仍有很大的改进空间。
在某些其他实施方案中,当施用三种药剂时,第三药剂包含SGLT2抑制剂、二甲双胍(metformin)、肠促胰岛素类似物、肠促胰岛素受体调节剂、DPP-4抑制剂或PPAR激动剂。
在某些其他实施方案中,当施用三种药剂时,该第三药剂为选自二甲双胍、西格列汀(sitagliptin)或艾格列净(ertugliflozin)的抗糖尿剂。
在某些其他实施方案中,当施用三种药剂时,该第三药剂为选自ACE抑制剂、血管紧张素受体阻断剂、钙通道阻断剂或血管舒张剂的抗心力衰竭剂。
本发明另外包含适合用于进行上述治疗方法的试剂盒。在一个实施方案中,该试剂盒含有包含一种或多种本发明的药剂(化合物)或其药学上可接受的盐的第一剂型和用于该剂型的容器,该第一剂型的量足以进行本发明的方法。
在另一实施方案中,该试剂盒含有包含一种本发明的药剂(化合物)或其药学上可接受的盐的第一剂型和用于第一剂型的容器,和包含本发明的另一种药剂(化合物)或其药学上可接受的盐的第二剂型和用于第二剂型的容器,其中两个剂型的量都足以进行本发明的方法。
在另一实施方案中,该试剂盒含有包含一种本发明的药剂(化合物)或其药学上可接受的盐的第一剂型和用于第一剂型的容器、包含本发明的另一种药剂(化合物)或其药学上可接受的盐的第二剂型和用于第二剂型的容器,以及包含本发明的另一种药剂(化合物)或其药学上可接受的盐的第三剂型和用于第三剂型的容器,其中所有三种剂型的量都足以进行本发明的方法。
在本发明的某些实施方案中,提供一种试剂盒,其包含:包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐的第一剂型和用于第一剂型的容器;和包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的第二剂型和用于第二剂型的容器,其中两种剂型的量都足以进行本发明的方法。
在本发明的另一实施方案中,提供一种试剂盒,其包含:包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐的第一剂型和用于第一剂型的容器;包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的第二剂型和用于第二剂型的容器和包含另一种药剂(化合物)或其药学上可接受的盐的第三剂型和用于第三剂型的容器,其中所有三种剂型的量都足以进行本发明的方法。
在一些实施方案中,试剂盒的剂型可同时或按时间顺序交错施用,即在不同的时间点且以相同或不同的时间间隔施用试剂盒的任何一种剂型。
在任何上述试剂盒中,可提供单独保留各剂型的装置,诸如容器、分开的瓶或分开的箔包。此类试剂盒的实例为用于包装片剂、胶囊等的熟知的罩板包装。此类试剂盒特别适合于施用不同的剂型(例如,口服和肠胃外)、在不同的剂量间隔施用单独的药剂(化合物)或相互滴定单独的药剂(化合物)。为了协助顺应性,试剂盒可包含施用的指示且可提供所谓的记忆辅助设备。
本发明也涵盖已知用于包装、分配和施用本发明的药剂(化合物)的各种其他试剂盒。在进一步的实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,HMG-CoA还原酶抑制剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,HMG-CoA还原酶抑制剂(选自普伐他汀(pravastatin)、匹伐他汀(pitavastatin)、洛伐他汀(lovastatin)、阿托伐他汀(atorvastatin)、辛伐他汀(simvastatin)、氟伐他汀(fluvastatin)、伊伐他汀(itavastatin)、尼伐他汀(nisvastatin)、尼贝他汀(nisbastatin)、瑞舒伐他汀(rosuvastatin)、atavastatin和维沙他汀(visastatin)),以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,阿托伐他汀,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和HMG-CoA还原酶抑制剂。在某些实施方案中,该HMG-CoA还原酶抑制剂为阿托伐他汀。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和HMG-CoA还原酶抑制剂的步骤。在某些实施方案中,该HMG-CoA还原酶抑制剂为阿托伐他汀。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,贝特类药物(fibrate agent),以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,选自吉非罗齐(gemfibrozil)、非诺贝特(fenofibrate)和氯贝特(clofibrate)的贝特类药物,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,非诺贝特,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和贝特类药物。在某些实施方案中,该贝特类药物为非诺贝特。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和贝特类药物的步骤。在某些实施方案中,该贝特类药物为非诺贝特。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,胆汁酸螯合剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,选自考来烯胺(questran)、考来替泊(colestipol)和考来维仑(colesevelam)的胆汁酸螯合剂,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和胆汁酸螯合剂。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和胆汁酸螯合剂的步骤。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,胆固醇吸收抑制剂,以及药学上可接受的赋形剂。在某些实施方案中,该胆固醇吸收抑制剂为依泽替米贝(ezetimibe)。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和胆固醇吸收抑制剂。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和胆固醇吸收抑制剂的步骤。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,烟酸类药物,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,选自烟酸、niacor和slo-niacin的烟酸类药物,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和烟酸类药物。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和烟酸类药物的步骤。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,PCSK9调节剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,选自阿利库单抗(alirocumab)和依伏库单抗(evolucumab)的PCSK9调节剂,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和PCSK9调节剂。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐和PCSK9调节剂的步骤。
在任何前述实施方案中,4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸为结晶固体。在某些实施方案中,该结晶固体为4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的2-氨基-2-(羟甲基)丙烷-1,3-二醇盐。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,HMG-CoA还原酶抑制剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,选自普伐他汀、匹伐他汀、洛伐他汀、阿托伐他汀、辛伐他汀、氟伐他汀、伊伐他汀、尼伐他汀、尼贝他汀、瑞舒伐他汀、atavastatin和维沙他汀的HMG-CoA还原酶抑制剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,阿托伐他汀,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和HMG-CoA还原酶抑制剂。在某些实施方案中,该HMG-CoA还原酶抑制剂为阿托伐他汀。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和HMG-CoA还原酶抑制剂的步骤。在某些实施方案中,该HMG-CoA还原酶抑制剂为阿托伐他汀。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,贝特类药物,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,选自吉非罗齐、非诺贝特和氯贝特的贝特类药物,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,非诺贝特,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和贝特类药物。在某些实施方案中,该贝特类药物为非诺贝特。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和贝特类药物的步骤。在某些实施方案中,该贝特类药物为非诺贝特。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,胆汁酸螯合剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,选自考来烯胺、考来替泊和考来维仑的胆汁酸螯合剂,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和胆汁酸螯合剂。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和胆汁酸螯合剂的步骤。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,胆固醇吸收抑制剂,以及药学上可接受的赋形剂。在某些实施方案中,该胆固醇吸收抑制剂为依泽替米贝。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和胆固醇吸收抑制剂。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和胆固醇吸收抑制剂的步骤。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,烟酸类药物,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,选自烟酸、niacor和slo-niacin的烟酸类药物,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和烟酸类药物。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和烟酸类药物的步骤。
在进一步的实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,PCSK9调节剂,以及药学上可接受的赋形剂。
在某些实施方案中,本发明涉及一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,选自阿利库单抗和依伏库单抗的PCSK9调节剂,以及药学上可接受的赋形剂。
在进一步的实施方案中,本发明涉及一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和PCSK9调节剂。
在进一步的实施方案中,本发明涉及一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和PCSK9调节剂的步骤。
在任何前述实施方案中,(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺为结晶固体。在某些实施方案中,该结晶固体具有包含5.3±0.2、7.7±0.2和15.4±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末X射线衍射图。在某些其他实施方案中,该结晶固体具有包含6.5±0.2、9.3±0.2和13.6±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末X射线衍射图。
本发明化合物可以经由包括类似于化学领域中熟知的那些的方法的合成途径合成,特别是根据本文所含的说明。起始材料通常得自商业来源,诸如Aldrich Chemicals(Milwaukee, WI),或使用本领域技术人员熟知的方法容易地制备(例如通过在Louis F.Fieser和Mary Fieser, Reagents for Organic Synthesis, v.1-19, Wiley, New York(1967-1999 ed.),或Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed.Springer-Verlag, Berlin,包括增刊(也经由Beilstein在线数据库取得)中一般描述的方法制备)。(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺的制备呈现于US 2018-0051012A1的实施例1中,为了所有目的特此以其整体通过引用并入本文。4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的制备在US 8,859,577的实施例9中,为了所有目的特此以其整体通过引用并入本文。[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸(包括其结晶游离酸形式)的制备描述于美国专利号9,809,579的实施例4中。GLP-1R激动剂的制备描述于美国专利号10,208,019中。
组合药剂
本发明化合物可以单独施用,或者作为单独的药剂一起施用,或者以固定剂量组合施用,或者与一种或多种另外的治疗剂组合施用。"组合施用"或"组合疗法"意指化合物A和化合物D作为仅有的两种治疗剂一起施用,或与一种或多种另外的治疗剂组合施用,其同时施用于治疗的哺乳动物。当组合施用时,各组分可在相同的时间或在不同的时间点以任何顺序相继施用。因此,各组分可单独但在时间上足够接近地施用以提供所需治疗效应。因此,本文所述的治疗方法包括使用组合药剂以组合施用三种或更多种药剂。
组合药剂以治疗有效量施用于哺乳动物。"治疗有效量"意指当单独或与另外的治疗剂组合施用于哺乳动物时,本发明化合物的量有效治疗所需的疾病/病况(例如,NASH)。
用于治疗非酒精性脂肪性肝炎(NASH)和/或非酒精性脂肪性肝病(NAFLD)的优选药剂(即抗NASH和抗NAFLD剂)是乙酰CoA羧化酶(ACC)抑制剂、己酮糖激酶(KHK)抑制剂、GLP-1受体激动剂、FXR激动剂、CB1拮抗剂、ASK1抑制剂、CCR2和/或CCR5的抑制剂、PNPLA3抑制剂、羟基类固醇17-β脱氢酶(HSD17B13)抑制剂、DGAT1抑制剂、FGF21类似物、FGF19类似物、SGLT2抑制剂、PPAR激动剂、AMPK活化剂、SCD1抑制剂或MPO抑制剂。共同受让的12/01/2017提交的专利申请PCT/IB2017/057577涉及GLP-1受体激动剂。最优选的是FXR激动剂、凋亡信号调节激酶1 (ASK1)抑制剂、PPAR激动剂、GLP-1受体激动剂、SGLT抑制剂、ACC抑制剂和KHK抑制剂。
鉴于本发明化合物的NASH/NAFLD活性,它们可与治疗非酒精性脂肪性肝炎(NASH)和/或非酒精性脂肪性肝病(NAFLD)及相关疾病/病况的其他药剂共同施用,诸如奥利司他(Orlistat)、TZD和其他胰岛素敏化剂、FGF21类似物、二甲双胍,ω-3-酸乙酯(例如,Lovaza)、贝特类(Fibrates)、HMG-CoA还原酶抑制剂、依泽替米贝、普罗布考(Probucol)、熊脱氧胆酸(Ursodeoxycholic acid)、TGR5激动剂、FXR激动剂、维生素E、甜菜碱、己酮可可碱(Pentoxifylline)、CB1拮抗剂、肉碱、N-乙酰半胱氨酸、还原型谷胱甘肽、氯卡色林(lorcaserin)、纳曲酮(naltrexone)和安非他酮(buproprion)的组合、SGLT2抑制剂(包括达格列净(dapagliflozin)、卡格列净(canagliflozin)、依帕列净(empagliflozin)、托格列净(tofogliflozin)、艾格列净(ertugliflozin)、ASP-1941、THR1474、TS-071、ISIS388626和LX4211以及WO2010023594中的那些)、芬特明(Phentermine)、托吡酯(Topiramate)、GLP-1受体激动剂、GIP受体激动剂、双重GLP-1受体/胰高血糖素受体激动剂(即OPK88003、MEDI0382、JNJ-64565111、NN9277、BI 456906)、双重GLP-1受体/GIP受体激动剂(即,替西帕肽(Tirzepatide)(LY3298176)、N9423)、血管紧张素-受体阻断剂、乙酰CoA羧化酶(ACC)抑制剂、BCKDK抑制剂、己酮糖激酶(KHK)抑制剂、ASK1抑制剂、支链α酮酸脱氢酶激酶抑制剂(BCBK抑制剂)、CCR2和/或CCR5的抑制剂、PNPLA3抑制剂、DGAT1抑制剂、FGF21类似物、FGF19类似物、PPAR激动剂、FXR激动剂、AMPK活化剂、SCD1抑制剂或MPO抑制剂。
示例性ACC抑制剂包括4-(4-[(1-异丙基-7-氧代-1,4,6,7-四氢-1'H-螺[吲唑-5,4'-哌啶]-1'-基)羰基]-6-甲氧基吡啶-2-基)苯甲酸;和firsocostat (GS-0976)及其药学上可接受的盐。
示例性DGAT2抑制剂包括(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺;
2-(5-((3-乙氧基-5-氟吡啶-2-基)氧基)吡啶-3-基)-N-((3R,4S)-4-氟哌啶-3-基)嘧啶-5-甲酰胺;
2-(5-((3-乙氧基-5-氟吡啶-2-基)氧基)吡啶-3-基)-N-((3S,5S)-5-氟哌啶-3-基)嘧啶-5-甲酰胺;
2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-((3R,4S)-4-氟哌啶-3-基)嘧啶-5-甲酰胺;
2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-((3R,4R)-4-氟哌啶-3-基)嘧啶-5-甲酰胺;
2-(5-((3-乙氧基-5-氟吡啶-2-基)氧基)吡啶-3-基)-N-((3R,4R)-4-氟哌啶-3-基)嘧啶-5-甲酰胺;和
2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-((3S,5S)-5-氟哌啶-3-基)嘧啶-5-甲酰胺。
适当抗糖尿病剂的实例包括,例如,胰岛素,二甲双胍,DPPIV抑制剂,GLP-1受体激动剂、类似物和模拟物,SGLT1和SGLT2抑制剂。适当抗糖尿病剂包括乙酰CoA羧化酶-(ACC)抑制剂(诸如在WO2009144554、WO2003072197、WO2009144555和WO2008065508中描述的那些),二酰基甘油O-酰基转移酶1 (DGAT-1)抑制剂(诸如在WO09016462或WO2010086820中描述的那些、AZD7687或LCQ908),单酰基甘油O-酰基转移酶抑制剂,磷酸二酯酶(PDE)-10抑制剂,AMPK活化剂,磺酰脲(例如,乙酰苯磺酰环己脲(acetohexamide)、氯磺丙脲(chlorpropamide)、氯磺丙脲(diabinese)、格列苯脲(glibenclamide)、格列吡嗪(glipizide)、格列本脲(glyburide)、格列美脲(glimepiride)、格列齐特(gliclazide)、格列太特(glipentide)、格列喹酮(gliquidone)、格列索脲(glisolamide)、妥拉磺脲(tolazamide)和甲苯磺丁脲(tolbutamide)),美格列奈,α-淀粉酶抑制剂(例如,淀粉酶抑肽(tendamistat)、萃他汀(trestatin)和AL-3688),α-葡萄糖苷水解酶抑制剂(例如,阿卡波糖(acarbose)),α-葡萄糖苷酶抑制剂(例如,脂解素(adiposine)、卡格列波糖(camiglibose)、乙格列酯(emiglitate)、米格列醇(miglitol)、伏格列波糖(voglibose)、普拉米星-Q(pradimicin-Q)和salbostatin),PPARγ激动剂(例如,巴格列酮(balaglitazone)、环格列酮(ciglitazone)、达格列酮(darglitazone)、恩格列酮(englitazone)、伊沙列酮(isaglitazone)、吡格列酮(pioglitazone)和罗格列酮(rosiglitazone)),PPAR α/γ激动剂(例如,CLX-0940、GW-1536、GW-1929、GW-2433、KRP-297、L-796449、LR-90、MK-0767和SB-219994),双胍(例如,二甲双胍),胰高血糖素样肽1(GLP-1)调节剂(诸如激动剂(例如exendin-3和exendin-4)、利拉鲁肽、阿必鲁肽、艾塞那肽(Byetta®)、阿必鲁肽、利西拉肽、杜拉鲁肽、索马鲁肽、NN-9924、TTP-054),蛋白酪氨酸磷酸酶-1B (PTP-1B)抑制剂(例如,曲度奎明(trodusquemine)、西替欧醛(hyrtiosal)提取物和由Zhang, S.,等人,Drug Discovery Today, 12(9/10), 373-381(2007)公开的化合物),SIRT-1活化剂(例如,白藜芦醇(resveratrol)、GSK2245840或GSK184072),二肽基肽酶IV(DPP-IV)抑制剂(例如,在WO2005116014中的那些、西格列汀、维格列汀(vildagliptin)、阿格列汀(alogliptin)、度格列汀(dutogliptin)、利拉利汀(linagliptin)和沙格列汀(saxagliptin)),胰岛素促泌剂,脂肪酸氧化抑制剂,A2拮抗剂,c-jun氨基末端激酶(JNK)抑制剂,葡萄糖激酶活化剂(GKa)(诸如WO2010103437、WO2010103438、WO2010013161、WO2007122482中描述的那些、TTP-399、TTP-355、TTP-547、AZD1656、ARRY403、MK-0599、TAK-329、AZD5658或GKM-001),胰岛素,胰岛素模拟剂,糖原磷酸化酶抑制剂(例如,GSK1362885),VPAC2受体激动剂,SGLT2抑制剂(诸如在E.C. Chao等人 Nature ReviewsDrug Discovery 9, 551-559 (2010年7月)中描述的那些,包括达格列净、卡格列净、依帕列净、托格列净(CSG452)、艾格列净、ASP-1941、THR1474、TS-071、ISIS388626和LX4211以及在WO2010023594中的那些),胰高血糖素受体调节剂(诸如在Demong, D.E.等人 AnnualReports in Medicinal Chemistry 2008, 43, 119-137中所述的那些),GPR119调节剂(特别是激动剂,诸如在WO2010140092、WO2010128425、WO2010128414、WO2010106457、Jones,R.M.等人于Medicinal Chemistry 2009, 44, 149-170中描述的那些(例如MBX-2982、GSK1292263、APD597和PSN821))、FGF21衍生物或类似物(诸如在Kharitonenkov, A.等人,Current Opinion in Investigational Drugs 2009, 10 (4)359-364中描述的那些),TGR5(也称为GPBAR1)受体调节剂(特别是激动剂,诸如在Zhong, M., Current Topics inMedicinal Chemistry, 2010, 10(4), 386-396中描述的那些和INT777),GPR40激动剂(诸如Medina, J.C., Annual Reports in Medicinal Chemistry, 2008, 43, 75-85中描述的那些,包括但不限于TAK-875),GPR120调节剂(特别是激动剂),高亲和性烟酸受体(HM74A)活化剂和SGLT1抑制剂(诸如GSK1614235)。可与本发明化合物组合的抗糖尿病剂的另一代表性列表可见于例如WO2011005611的第28页第35行至第30页第19行。优选抗糖尿病剂为二甲双胍和DPP-IV抑制剂(例如,西格列汀、维格列汀、阿格列汀、度格列汀、利拉利汀和沙格列汀)。其他抗糖尿病剂可包括肉毒碱棕榈酰转移酶的抑制剂或调节剂,果糖1,6-二磷酸酶的抑制剂,醛糖还原酶的抑制剂,盐皮质激素受体抑制剂,TORC2的抑制剂,CCR2和/或CCR5的抑制剂、PKC同种型(isoform)(例如,PKCα、PKCβ、PKCγ)的抑制剂,脂肪酸合成酶的抑制剂,丝氨酸棕榈酰转移酶的抑制剂,GPR81、GPR39、GPR43、GPR41、GPR105、Kv1.3、视黄醇结合蛋白4、糖皮质激素受体、生长抑素受体(例如,SSTR1、SSTR2、SSTR3和SSTR5)的调节剂,PDHK2或PDHK4的抑制剂或调节剂,MAP4K4的抑制剂,包括IL1β在内的IL1家族的调节剂,RXRα的调节剂。此外,适当抗糖尿病剂包括由Carpino, P.A., Goodwin, B. Expert Opin.Ther. Pat, 2010, 20(12), 1627-51所列的机制。
适当抗肥胖剂包括11β-羟基类固醇脱氢酶-1 (11β-HSD型1)抑制剂、硬脂酰-CoA去饱和酶-1(SCD-1)抑制剂、MCR-4激动剂、胆囊收缩素-A (CCK-A)激动剂、单胺再摄取抑制剂(诸如西布曲明(sibutramine))、拟交感神经剂、β3肾上腺素激动剂、多巴胺激动剂(诸如溴隐亭(bromocriptine))、黑色素细胞-刺激激素类似物、5HT2c激动剂、黑色素浓缩激素拮抗剂、瘦素(OB蛋白)、瘦素类似物、瘦素激动剂、甘丙肽拮抗剂、脂肪酶抑制剂(诸如四氢利普司他汀(tetrahydrolipstatin),即奥利司他)、厌食剂(诸如铃蟾肽激动剂)、神经肽-Y拮抗剂(例如,NPY Y5拮抗剂)、PYY3-36 (包括其类似物)、拟甲状腺素剂、脱氢表雄酮或其类似物、糖皮质激素激动剂或拮抗剂、食欲肽拮抗剂、胰高血糖素样肽-1激动剂、睫状神经营养因子(诸如得自Regeneron Pharmaceuticals, Inc., Tarrytown, NY 和 Procter &Gamble Company, Cincinnati, OH的Axokine™)、人类刺鼠关联蛋白(AGRP)抑制剂、饥饿素拮抗剂、组胺3拮抗剂或反向激动剂、神经调节肽U激动剂、MTP/ApoB抑制剂(例如,肠选择性MTP抑制剂,诸如地洛他派(dirlotapide))、阿片类拮抗剂、食欲肽拮抗剂、纳曲酮和安非他酮的组合等。
用于本发明的组合的优选抗肥胖剂包括肠选择性MTP抑制剂(例如,地洛他派、米瑞他匹(mitratapide)和英普他派(implitapide)、R56918 (CAS编号403987)和CAS编号913541-47-6)、CCKa激动剂(例如,在PCT公开号WO 2005/116034或US公开号2005-0267100A1中描述的N-苄基-2-[4-(1H-吲哚-3-基甲基)-5-氧代-1-苯基-4,5-二氢-2,3,6,10b-四氮杂-苯并[e]薁-6-基]-N-异丙基乙酰胺)、5HT2c激动剂(例如,氯卡色林)、MCR4激动剂(例如,US 6,818,658中描述的化合物)、脂肪酶抑制剂(例如,赛利司他(Cetilistat))、PYY3-36(如本文所用的"PYY3-36"包括类似物,诸如聚乙二醇化PYY3-36,例如美国公开2006/0178501中描述的那些)、阿片类拮抗剂(例如,纳曲酮)、纳曲酮和安非他酮的组合、油酰雌酮(CAS编号180003-17-2)、奥尼匹肽(obinepitide) (TM30338)、普兰林肽(pramlintide) (Symlin®)、特索芬辛(tesofensine) (NS2330)、瘦素、利拉鲁肽、溴隐亭、奥利司他、艾塞那肽(Byetta®)、AOD-9604 (CAS编号221231-10-3)、芬特明和托吡酯(商标名:Qsymia)和西布曲明。优选地,本发明化合物和组合疗法与运动和合理的饮食结合施用。
本发明化合物可与下列胆固醇调节剂(包括降胆固醇剂)组合使用:诸如脂肪酶抑制剂、HMG-CoA还原酶抑制剂、HMG-CoA合成酶抑制剂、HMG-CoA还原酶基因表达抑制剂、HMG-CoA合成酶基因表达抑制剂、MTP/Apo B分泌抑制剂、CETP抑制剂、胆汁酸吸收抑制剂、胆固醇吸收抑制剂、胆固醇合成抑制剂、角鲨烯合成酶抑制剂、角鲨烯环氧酶抑制剂、角鲨烯环化酶抑制剂、组合的角鲨烯环氧酶/角鲨烯环化酶抑制剂、贝特类、烟酸、离子交换树脂、抗氧化剂、ACAT抑制剂或胆汁酸螯合剂或诸如米泊美生(mipomersen)的药剂。
适当降胆固醇/脂质剂和脂质谱疗法的实例包括:HMG-CoA还原酶抑制剂(例如,普伐他汀、匹伐他汀、洛伐他汀、阿托伐他汀、辛伐他汀、氟伐他汀、NK-104 (也称为伊伐他汀或尼伐他汀或尼贝他汀),以及ZD-4522 (也称为瑞舒伐他汀或atavastatin或维沙他汀));角鲨烯合成酶抑制剂;贝特类(例如,吉非罗齐、非诺贝特、氯贝特);胆汁酸螯合剂(诸如考来烯胺、考来替泊、考来维仑);ACAT抑制剂;MTP抑制剂;脂加氧酶抑制剂;胆固醇吸收抑制剂(例如,依泽替米贝);烟酸类药物(例如,烟酸、niacor、slo-niacin);ω-3脂肪酸;和胆固醇酯转移蛋白抑制剂。其他动脉粥样硬化剂包括PCSK9调节剂(例如,阿利库单抗和依伏库单抗)。
在另一实施方案中,本发明化合物可与治疗非酒精性脂肪性肝炎(NASH)和/或非酒精性脂肪性肝病(NAFLD)的药剂共同施用,诸如奥利司他、TZD和其他胰岛素敏化剂、FGF21类似物、二甲双胍、ω-3-酸乙酯(例如,Lovaza)、贝特类、HMG-CoA还原酶抑制剂、Ezitimbe、普罗布考、熊脱氧胆酸、TGR5激动剂、FXR激动剂、维生素E、甜菜碱、己酮可可碱、CB1拮抗剂、肉碱、N-乙酰半胱氨酸、还原型谷胱甘肽、氯卡色林、纳曲酮和安非他酮的组合、SGLT2抑制剂、芬特明、托吡酯、肠促胰岛素(GLP与GIP)类似物和血管紧张素受体阻断剂。
在另一实施方案中,另外药剂选自:半胱胺或其药学上可接受的盐,胱胺或其药学上可接受的盐,抗氧化剂化合物、卵磷脂、维生素B复合物、胆盐制剂、大麻素-1 (CB1)受体的拮抗剂、大麻素-1 (CB1)受体的反向激动剂、过氧化物酶体增殖物活化受体活性调节剂、苯并硫氮杂䓬或苯并硫杂䓬化合物、抑制蛋白酪氨酸磷酸酶PTPRU的RNA反义构建体、杂原子连接的经取代的哌啶和其衍生物、能够抑制硬脂酰辅酶αδ-9去饱和酶的氮杂环戊烷衍生物、具有脂联素的促泌剂或诱导剂活性的酰基酰胺化合物、季铵化合物、醋酸格拉替雷、穿透素蛋白、HMG-CoA还原酶抑制剂、N-乙酰基半胱氨酸、异黄酮化合物、大环内酯类抗生素、半乳糖凝集素抑制剂、抗体或其任何组合。
另外的治疗剂包括抗凝剂或凝结抑制剂、抗血小板剂或血小板抑制剂、凝血酶抑制剂、血栓溶解剂或纤维蛋白溶解剂、抗心律不齐剂、抗高血压剂、钙通道阻断剂(L型和T型)、强心苷、利尿剂、盐皮质激素受体拮抗剂、NO供给剂(诸如有机硝酸盐)、NO促进剂(诸如磷酸二酯酶抑制剂)、降胆固醇/脂质剂和脂质谱疗法、抗糖尿病剂、抗抑郁剂、抗炎剂(类固醇和非类固醇)、抗骨质疏松症剂、激素替代疗法、口服避孕药、抗肥胖剂、抗焦虑剂、抗增殖剂、抗肿瘤剂、抗溃疡和胃食管反流病剂、生长激素和/或生长激素促分泌素、甲状腺模拟物(包括甲状腺激素受体拮抗剂)、抗感染剂、抗病毒剂、抗细菌剂和抗真菌剂。
包括用于ICU设置的药剂,例如多巴酚丁胺(dobutamine)、多巴胺(dopamine)、肾上腺素(epinephrine)、硝化甘油(nitroglycerin)、硝普盐(nitroprusside)等等。
包括可用于治疗血管炎的组合药剂,例如硫唑嘌呤(azathioprine)、环磷酰胺、霉酚酸酯(mycophenolate mofetil)、利妥昔单抗(rituximab)等等。
在另一实施方案中,本发明提供一种组合,其中第三药剂为至少一种选自下列的药剂:Xa因子抑制剂、抗凝剂、抗血小板剂、凝血酶抑制剂、血栓溶解剂和纤维蛋白溶解剂。示例性Xa因子抑制剂包括阿哌沙班(apixaban)和利伐沙班(rivaroxaban)。适合与本发明化合物组合使用的抗凝剂的实例包括肝素(例如,普通肝素和低分子量肝素,诸如依诺肝素(enoxaparin)和达肝素(dalteparin))。
在另一优选实施方案中,该第三药剂为至少一种选自下列的药剂:华法林(warfarin)、达比加群(dabigatran)、普通肝素、低分子量肝素、合成五碳糖、水蛭素、阿加曲班(argatrobanas)、阿司匹林(aspirin)、布洛芬(ibuprofen)、萘普生(naproxen)、舒林酸(sulindac)、吲哚美辛(indomethacin)、甲芬那酸(mefenamate)、屈昔康(droxicam)、双氯芬酸(diclofenac)、苯磺唑酮(sulfinpyrazone)、吡罗昔康(piroxicam)、噻氯匹定(ticlopidine)、氯吡格雷(clopidogrel)、替罗非班(tirofiban)、依替巴肽(eptifibatide)、阿昔单抗(abciximab)、美拉加群(melagatran)、二硫酸根合水蛭素、组织纤溶酶原活化物、经修饰的组织纤溶酶原活化物、阿尼普酶(anistreplase)、尿激酶和链激酶。
优选第三药剂为至少一种抗血小板剂。尤其优选的抗血小板剂为阿司匹林和氯吡格雷。
如本文所用,术语抗血小板剂(或血小板抑制剂)表示抑制血小板功能(例如通过抑制血小板的聚集、粘附或颗粒分泌)的药剂。该药剂包括但不限于各种已知的非甾体类抗炎药(NSAIDS),诸如阿司匹林、布洛芬、萘普生、舒林酸、吲哚美辛、甲芬那酸、屈昔康、双氯芬酸、苯磺唑酮、吡罗昔康,以及其药学上可接受的盐或前药。在NSAIDS中,阿司匹林 (乙酰水杨酸或ASA)和COX-2抑制剂诸如CELEBREX或吡罗昔康是优选的。其他适当血小板抑制剂包括IIb/IIIa拮抗剂(例如替罗非班、依替巴肽和阿昔单抗)、凝血噁烷A2受体拮抗剂(例如伊非曲班(ifetroban))、凝血噁烷A2-合成酶抑制剂、PDE-III抑制剂(例如Pletal、双嘧达莫(dipyridamole)),以及其药学上可接受的盐或前药。
如本文所用,术语抗血小板剂(或血小板抑制剂)也意欲包括ADP (二磷酸腺苷)受体拮抗剂(优选为嘌呤受体P2Y1和P2Y12的拮抗剂,且P2Y12甚至是更优选的)。优选P2Y12受体拮抗剂包括替卡格雷(ticagrelor)、普拉格雷(prasugrel)、噻氯匹定(ticlopidine)和氯吡格雷,包括其药学上可接受的盐或前药。氯吡格雷为甚至更优选的药剂。噻氯匹定和氯吡格雷也是优选的化合物,因为已知它们在使用时对胃肠道温和。
如本文所用,术语凝血酶抑制剂(或抗凝血酶剂)表示丝氨酸蛋白酶凝血酶的抑制剂。通过抑制凝血酶,中断各种凝血酶介导的过程,诸如凝血酶介导的血小板活化(即,例如血小板聚集和/或纤溶酶原活化物抑制剂-1和/或血清素的颗粒分泌)和/或纤维蛋白形成。许多凝血酶抑制剂是本领域技术人员已知的,且预期这些抑制剂与本发明化合物组合使用。此类抑制剂包括但不限于boroarginine衍生物、boropeptides、达比加群、肝素、水蛭素、阿加曲班和美拉加群,包括其药学上可接受的盐和前药。boroarginine衍生物和boropeptides包括硼酸的N-乙酰基和肽衍生物,诸如赖氨酸、鸟氨酸、精氨酸、高精氨酸和它们对应的异硫脲鎓(isothiouronium)类似物的C末端α-氨基硼酸衍生物。如本文所用,术语水蛭素包括水蛭素的适当衍生物或类似物,在本文中称为水蛭肽(hirulog),诸如二硫酸根合水蛭素。如本文所用,术语血栓溶解剂或纤维蛋白溶解剂(或血栓溶解药或纤维蛋白溶解药)表示溶解血块(血栓)的药剂。此类药剂包括组织纤溶酶原活化物(天然或重组)及其修饰形式、阿尼普酶、尿激酶、链激酶、替奈普酶(tenecteplase;TNK)、兰替普酶(lanoteplase;nPA)、VIIa因子抑制剂、PAI-1抑制剂(即,组织纤溶酶原活化物抑制剂的去活剂)、α2-抗纤维蛋白溶酶抑制剂,以及茴香酰化(anisoylated)纤溶酶原链激酶活化物复合物,包括其药学上可接受的盐或前药。如本文所用,术语阿尼普酶是指茴香酰化纤溶酶原链激酶活化物复合物,如例如EP 028,489中所述,其公开内容特此通过引用并入本文。如本文所用,术语尿激酶意欲表示双链尿激酶和单链尿激酶二者,后者在本文中也称为尿激酶原。
适当抗心律不齐剂的实例包括:I类药剂(诸如普罗帕酮(propafenone));II类药剂(诸如美托洛尔(metoprolol)、阿替洛尔(atenolol)、卡维地洛(carvedilol)和普萘洛尔(propranolol));III类药剂(诸如索他洛尔(sotalol)、多非利特(dofetilide)、胺碘酮(amiodarone)、阿齐利特(azimilide)和伊布利特(ibutilide));IV类药剂(诸如地尔硫卓(diltiazem)和维拉帕米(verapamil));K+通道开放剂,诸如IAch抑制剂和IKur抑制剂(例如,诸如WO01/40231中所公开的那些的化合物)。
本发明化合物可与下列抗心力衰竭剂共同施用:诸如ACE抑制剂(例如,卡托普利(captopril)、依那普利(enalapril)、福辛普利(fosinopril)、赖诺普利(Lisinopril)、培哚普利(perindopril)、喹那普利(quinapril)、雷米普利(Ramipril)、群多普利(trandolapril)),血管紧张素 II受体阻断剂(例如,坎地沙坦(Candesartan)、氯沙坦(Losartan)、缬沙坦(Valsartan)),血管紧张素受体脑啡肽酶抑制剂(沙库比曲(sacubitril)/缬沙坦),If通道阻断剂伊伐布雷定(Ivabradine),β-肾上腺素阻断剂(例如,比索洛尔(bisoprolol)、琥珀酸美托洛尔、卡维地洛)、醛固酮拮抗剂(例如,螺内酯、依普利酮(eplerenone)),肼屈嗪(hydralazine)和硝酸异山梨酯,利尿剂(例如,呋塞米(furosemide)、布美他尼(bumetanide)、托拉塞米(torsemide)、氯噻嗪(chlorothiazide)、阿米洛利(amiloride)、氢氯噻嗪(hydrochlorothiazide)、吲达帕胺(Indapamide)、美托拉宗(Metolazone)、三氨蝶呤(Triamterene))或地高辛(digoxin)。
本发明化合物可与抗高血压剂组合使用,且此类抗高血压活性由本领域技术人员根据标准测定(例如,血压测量)容易地确定。适当抗高血压剂的实例包括:α肾上腺素阻断剂;β肾上腺素阻断剂;钙通道阻断剂(例如,地尔硫卓、维拉帕米、硝苯地平(nifedipine)和氨氯地平(amlodipine));血管舒张剂(例如,肼屈嗪(hydralazine));利尿剂(例如,氯噻嗪、氢氯噻嗪、氟甲噻嗪(flumethiazide)、氢氟噻嗪(hydroflumethiazide)、苄氟噻嗪(bendroflumethiazide)、甲氯噻嗪(methylchlorothiazide)、三氯噻嗪(trichloromethiazide)、多噻嗪(polythiazide)、苄噻嗪(benzthiazide)、依他尼酸三克那汾(ethacrynic acid tricrynafen)、氯噻酮(chlorthalidone)、托拉塞米、呋塞米、吲达帕胺、metozolone、姆索明(musolimine)、布美他尼、三氨蝶呤、阿米洛利、螺内酯);肾素抑制剂;ACE抑制剂(例如,卡托普利、佐芬普利(zofenopril)、福辛普利、依那普利、西那普利(ceranopril)、西拉普利(cilazopril)、地拉普利(delapril)、喷托普利(pentopril)、培哚普利、喹那普利、雷米普利、群多普利、赖诺普利);AT-1受体拮抗剂(例如氯沙坦、艾比沙坦(irbesartan)、缬沙坦);血管紧张素受体脑啡肽酶抑制剂(沙库比曲/缬沙坦);β-肾上腺素阻断剂(例如,比索洛尔、琥珀酸美托洛尔、卡维地洛);ET受体拮抗剂(例如,西他生坦(sitaxsentan)、阿曲生坦(atrsentan)和在美国专利号5,612,359和6,043,265中公开的化合物);双重ET/AII拮抗剂(例如在WO 00/01389中公开的化合物);中性肽链内切酶(NEP)抑制剂;血管肽酶抑制剂(双重NEP-ACE抑制剂)(例如,吉莫曲拉(gemopatrilat)和硝酸盐)。示例性抗心绞痛剂为伊伐布雷定(ivabradine)。
适当钙通道阻断剂(L型或T型)的实例包括地尔硫卓、维拉帕米、硝苯地平和氨氯地平和米贝地尔(mibefradil)。
适当强心苷的实例包括毛地黄(digitalis)和哇巴因(ouabain)。
在一个实施方案中,本发明化合物可与一种或多种利尿剂共同施用。适当利尿剂的实例包括(a)亨氏环利尿剂(loop diuretics),诸如呋塞米(诸如LASIX™)、托拉塞米(诸如DEMADEX™)、bemetanide (诸如BUMEXTM),以及依他尼酸(ethacrynic acid)(诸如EDECRIN™);(b)噻嗪型利尿剂,诸如氯噻嗪(诸如DIURIL™、ESIDRIX™或 HYDRODIURIL™)、氢氯噻嗪(诸如MICROZIDE™或ORETIC™)、苄噻嗪、氢氟噻嗪(诸如SALURON™)、苄氟噻嗪、甲氯噻嗪、多噻嗪、三氯噻嗪和吲达帕胺(诸如LOZOL™);(c)苄甲内酰胺型利尿剂,诸如氯噻酮(诸如HYGROTON™)和美托拉宗(诸如ZAROXOLYN™);(d)喹唑啉型利尿剂,诸如喹乙唑酮(quinethazone);和(e)保钾利尿剂诸如三氨蝶呤(诸如DYRENIUM™)和阿米洛利(诸如MIDAMOR™或MODURETIC™)。
在另一实施方案中,本发明化合物可与亨氏环利尿剂共同施用。在又另一实施方案中,该亨氏环利尿剂选自呋塞米和托拉塞米。在又另一实施方案中,本发明化合物可与呋塞米共同施用。在又另一实施方案中,本发明化合物可与托拉塞米(其可任选地为托拉塞米的受控释放或改性释放形式)共同施用。
在另一实施方案中,本发明化合物可与噻嗪型利尿剂共同施用。在又另一实施方案中,该噻嗪型利尿剂选自:氯噻嗪和氢氯噻嗪。在又另一实施方案中,本发明化合物可与氯噻嗪共同施用。在又另一实施方案中,本发明化合物可与氢氯噻嗪共同施用。
在另一实施方案中,本发明化合物可与苄甲内酰胺型利尿剂共同施用。在又另一实施方案中,该苄甲内酰胺型利尿剂为氯噻酮。适当盐皮质激素受体拮抗剂的实例包括螺内酯和依普利酮。适当磷酸二酯酶抑制剂的实例包括:PDE III抑制剂(诸如西洛他唑(cilostazol));和PDE V抑制剂(诸如西地那非(sildenafil))。
本领域技术人员应理解本发明化合物也可与其他心血管或脑血管治疗(包括PCI、支架、药物洗脱支架、干细胞疗法)和医疗装置(诸如植入起搏器、除颤器或心脏再同步化治疗)结合使用。
特别是当以单一剂量单位提供时,组合的活性成分之间可能存在化学相互作用。为此原因,当第一治疗剂和第二治疗剂组合于单一剂量单位时,将它们配制以使得虽然活性成分组合于单一剂量单位中,但活性成分间的物理接触被最小化(即降低)。例如,一种活性成分可为肠溶包衣的。通过将一种活性成分肠溶包衣,则不仅可能使组合的活性成分之间的接触最小化,也可能控制这些组分中的一种在胃肠道中释放,使得这些组分中的一种不在胃中释放而是于肠中释放。有效成分中的一种也可用在整个胃肠道中影响持续释放且也用于使组合的活性成分之间的物理接触最小化的材料包衣。再者,持续释放组分可另外肠溶包衣以使此组分的释放仅在肠内发生。又另一方法将涉及组合产物的制剂,其中一种组分用持续和/或肠溶释放聚合物包衣,且其他组分也用聚合物诸如低粘度级的羟丙基甲基纤维素(HPMC)或本领域已知的其他适当材料包衣,以进一步分开活性成分。聚合物包衣用于形成与其他组分相互作用的另外的屏障。
一旦拥有本公开,本领域技术人员将显而易见这些以及其他将本发明的组合产品的组分之间的接触最小化的方式,无论是以单一剂型施用,还是以单独形式但同时以相同的方式施用。
在组合疗法治疗中,通过常规方法将本发明化合物和其他药物疗法施用于哺乳动物(例如,人类,男性或女性)。
各治疗剂(例如化合物A、化合物D和任何另外的治疗剂)的剂量通常取决于许多因素,包括治疗对象的健康、所需的治疗程度、并行疗法(若存在)的性质和种类,以及治疗频率与所需效应的性质。通常,各治疗剂的剂量范围在每天每千克个体体重约0.001 mg至约100 mg的范围内,优选在每天每千克个体体重约0.1 mg至约10 mg的范围内。然而,也可取决于治疗对象的年龄和重量、意欲的施用途径、施用的特定抗肥胖剂等等而需要在一般剂量范围上有一些变化。对于特定患者的剂量范围和最佳剂量的确定也完全在受益于本公开的本领域普通技术人员的能力范围内。
根据本发明的治疗方法,本发明化合物或本发明化合物和至少一种另外的药剂的组合(在本文称为"组合")优选地以药物组合物的形式施用于需要此类治疗的对象。在本发明的组合方面中,本发明化合物和至少一种其他药剂(例如,另一抗肥胖剂)可单独施用或以包含两者的药物组合物施用。通常优选的是此类施用为口服。
当本发明化合物与至少一种其他药剂的组合一起施用时,此类施用可为按时间相继的或同时的。通常优选同时施用药物组合。对于相继施用,本发明化合物与另外的药剂可以任何顺序施用。通常优选的是此类施用为口服。尤其优选的是此类施用为口服且同时的。当本发明化合物与另外的药剂相继施用时,各施用可通过相同或不同的方法进行。
根据本发明的方法,本发明化合物或组合优选地以药物组合物的形式施用。因此,本发明化合物或组合可以任何常规的口服、直肠、经皮、肠胃外(例如静脉内、肌内或皮下)、脑池内、阴道内、腹膜内、局部(例如粉末、软膏、乳膏、喷雾剂或洗剂)、颊或鼻剂型(例如喷雾剂、滴剂或吸入剂)单独或一起施用于患者。
本发明化合物或组合可单独施用,但是通常与本领域已知且关于意欲的施用途径和标准药物实践而选择的一种或多种适当药物赋形剂、佐剂、稀释剂或载体混合施用。本发明化合物或组合可取决于所需的施用途径和释放曲线(release profile)的特异性(与治疗需求相称)而配制以提供立即释放、延迟释放、改性释放、持续释放、脉冲释放或控制释放剂型。
药物组合物包含本发明化合物或组合,其量通常在组合物的约1%至约75%、80%、85%、90%或甚至95%(以重量计)的范围内,通常在约1%、2%或3%至约50%、60%或70%范围内,更常在约1%、2%或3%至少于50%的范围内,诸如约25%、30%或35%。
制备具有特定量的活性化合物的各种药物组合物的方法是本领域技术人员已知的。例如,参见Remington:The Practice of Pharmacy, Lippincott Williams和Wilkins,Baltimore Md. 20.sup.th ed. 2000。
适合于肠胃外注射的组合物通常包括药学上可接受的无菌水性或非水性溶液、分散体、悬浮液或乳液,和用于重构成无菌可注射溶液或分散体的无菌粉末。适当水性和非水性载体或稀释剂(包括溶剂和媒介物)的实例包括水、乙醇、多元醇(丙二醇、聚乙二醇、甘油等等)、其适当混合物、甘油三酯(包括植物油,诸如橄榄油)和可注射有机酯(诸如油酸乙酯)。优选载体为得自Condea Vista Co., Cranford, N.J 的Miglyol®品牌的具有甘油或丙二醇的辛酸/癸酸酯(例如Miglyol.RTM. 812、Miglyol.RTM. 829、Miglyol.RTM. 840)。可例如通过使用包衣(诸如卵磷脂)、在分散体的情况中通过维持所需粒度和通过使用表面活性剂而维持适当的流动性。
用于肠胃外注射的这些组合物也可含有赋形剂,诸如防腐剂、润湿剂、乳化剂和分散剂。组合物的微生物污染的预防可以各种抗细菌剂和抗真菌剂,例如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等完成。也可能需要包括等渗剂,例如糖、氯化钠等。可注射的药物组合物的延长吸收可通过使用能够延迟吸收的试剂(例如单硬脂酸铝和明胶)达成。
用于口服施用的固体剂型包括胶囊、片剂、咀嚼片、含片、丸剂、粉末和多微粒制剂(颗粒)。在此类固体剂型中,本发明化合物或组合与至少一种惰性赋形剂、稀释剂或载体混合。适当赋形剂、稀释剂或载体包括材料诸如柠檬酸钠或磷酸二钙和/或(a)一种或多种填充剂或增量剂(例如微晶纤维素(以Avicel.TM.自FMC Corp.取得)、淀粉、乳糖、蔗糖、甘露醇、硅酸、木糖醇、山梨醇、右旋糖、磷酸氢钙、糊精、α-环糊精、β-环糊精、聚乙二醇、中链脂肪酸、氧化钛、氧化镁、氧化铝等等);(b)一种或多种粘合剂(例如羧甲基纤维素、甲基纤维素、羟丙基纤维素、羟丙基甲基纤维素、明胶、阿拉伯胶(gum arabic)、乙基纤维素、聚乙烯醇、普鲁兰糖(pullulan)、预胶化淀粉、琼脂、黄蓍胶、海藻酸盐、明胶、聚乙烯吡咯烷酮、蔗糖、阿拉伯胶(acacia)等等);(c)一种或多种保湿剂(例如甘油等等);(d)一种或多种崩解剂(例如琼脂、碳酸钙、马铃薯或木薯淀粉、海藻酸、某些复合硅酸盐、碳酸钠、月桂基硫酸钠、淀粉乙醇酸钠(以Explotab™自Edward Mendell Co.取得)、交联聚乙烯基吡咯烷酮、交联羧甲基纤维素钠A型(以Ac-di-sol.TM.取得)、波拉克林钾(离子交换树脂)等等);(e)一种或多种溶液阻滞剂(例如石蜡等等);(f)一种或多种吸收加速剂(例如季铵化合物等等);(g)一种或多种润湿剂(例如鲸蜡醇、单硬脂酸甘油酯等等);(h)一种或多种吸附剂(例如高岭土、膨润土等等);和/或(i)一种或多种润滑剂(例如滑石、硬脂酸钙、硬脂酸镁、硬脂酸、聚氧乙烯硬脂酸酯(polyoxyl stearate)、鲸蜡醇、滑石、氢化蓖麻油、脂肪酸的蔗糖酯、二甲基聚硅氧烷、微晶蜡、黄蜂蜡、白蜂蜡、固体聚乙二醇、月桂基硫酸钠等等)。在胶囊和片剂的情况中,剂型也可包含缓冲剂。
类似类型的固体组合物也可用作使用此类赋形剂如乳糖(lactose或milk sugar)以及高分子量聚乙二醇等等的软或硬填充的明胶胶囊中的填充剂。
固体剂型(诸如片剂、糖衣丸、胶囊和颗粒)可以用包衣和壳制备,诸如肠溶包衣和本领域熟知的其他包衣。它们也可含有遮光剂,且也可具有以延迟方式释放本发明化合物和/或另外的药剂的此类组合物。可使用的包埋组合物的实例为聚合物质和蜡。若适当时,药物也可呈具有上述赋形剂中的一种或多种的微包封形式。
对于片剂,活性剂通常占制剂的少于50% (以重量计),例如少于约10%,诸如5%或2.5%,以重量计。制剂的主要部分包含填充剂、稀释剂、崩解剂、润滑剂和任选的调味剂。这些赋形剂的组成为本领域所熟知。经常,填充剂/稀释剂包含下列组分中的两种或更多种的混合物:微晶纤维素、甘露醇、乳糖(所有类型)、淀粉和磷酸二钙。填充剂/稀释剂混合物通常占制剂的少于98%,且优选为少于95%,例如93.5%。优选崩解剂包括Ac-di-sol.TM.、Explotab.TM.、淀粉和月桂基硫酸钠。当存在时,崩解剂通常占制剂的少于10%,或少于5%,例如约3%。优选润滑剂为硬脂酸镁。当存在时,润滑剂通常占制剂的少于5%,或少于3%,例如约1%。
片剂可以标准的压片方法制造,例如直接压缩或湿法、干法或熔融制粒,熔融凝结法,和挤压。片剂核心可为单层或多层,且可以本领域已知适当外包衣来包衣。
用于口服施用的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆和酏剂。除了本发明化合物或组合之外,液体剂型可含有本领域常使用的惰性稀释剂,诸如水或其他溶剂、增溶剂和乳化剂,例如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苯甲酯、丙二醇、1,3-丁二醇、二甲基甲酰胺、油(例如棉籽油、花生油、玉米胚芽油、橄榄油、蓖麻油、芝麻籽油等等)、Miglyole™ (取自CONDEA Vista Co., Cranford, N.J.)、甘油、四氢糠醇、聚乙二醇和脱水山梨醇的脂肪酸酯或这些物质的混合物等等。
除了此类惰性稀释剂之外,组合物也可包括赋形剂,诸如润湿剂、乳化和悬浮剂、甜味剂、调味剂和香味剂。
本发明化合物或组合的口服液体形式包括溶液,其中活性化合物完全溶解。溶剂的实例包括适合于口服施用的所有药学上有先例的溶剂,特别是本发明化合物在其中显示良好溶解度的那些,例如聚乙二醇、聚丙二醇、食用油和基于甘油基和甘油酯的系统。基于甘油基和甘油酯的系统可包括例如下列品牌产品(和对应的一般产品):Captex™ 355 EP(三辛酸甘油酯/癸酸甘油酯,来自Abitec, Columbus Ohio)、Crodamol™ GTC/C (中链甘油三酯,来自Croda, Cowick Hall, UK) 或Labrafac™ CC (中链甘油三酯,来自Gattefosse)、Captex™ 500P (三乙酸甘油酯,即三醋精,来自Abitec)、Capmul™ MCM (中链甘油单酯和甘油二酯,来自Abitec)、Migyol™ 812 (辛酸/癸酸甘油三酯,来自Condea,Cranford N.J.)、Migyol™ 829 (辛酸/癸酸/丁二酸甘油三酯,来自Condea)、Migyol™840 (丙二醇二辛酸酯/二癸酸酯,来自Condea)、Labrafil™ M1944CS (油酰基聚乙二醇-6甘油酯,来自Gattefosse)、Peceol™ (单油酸甘油酯,来自Gattefosse)和Maisine™ 35-1(单油酸甘油酯,来自Gattefosse)。特别关注的是中链(约C8至C10)甘油三酯油。这些溶剂常构成该组合物的主要部分,即大于约50%,一般是大于约80%,例如约95%或99%。也可与溶剂一起包括佐剂与添加剂,主要作为味觉掩蔽剂、适口性与调味剂、抗氧化剂、稳定剂、质地和粘度调节剂和增溶剂。
除了本发明的化合物或组合之外,悬浮液可另外包含载体诸如悬浮剂,例如乙氧基化异硬脂醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、偏氢氧化铝、膨润土、琼脂和黄蓍胶或这些物质的混合物等。
用于直肠或阴道施用的组合物优选地包含栓剂,其可通过将本发明化合物或组合与适当无刺激性赋形剂或载体(诸如可可脂、聚乙二醇或栓剂蜡)混合而制备,该赋形剂或载体在一般室温下为固体,但在体温下为液体,且因此在直肠或阴道腔中熔化,从而释放活性组分。
用于局部施用本发明化合物或组合的剂型包括软膏、乳膏、洗剂、粉末和喷雾剂。药物与药学上可接受的赋形剂、稀释剂或载体和可能需要的任何防腐剂、缓冲剂或推进剂混合。
当化合物难溶于水(例如少于约1 μg/mL)时,溶解在非水性溶剂(诸如上文讨论的中链甘油三酯油)中的液体组合物为用于这些化合物的优选剂型。
固体无定型分散体(包括由喷雾干燥法所形成的分散体)也为用于本发明的难溶性化合物的优选剂型。"固体无定型分散体"意指其中至少一部分的难溶性化合物呈无定型形式且分散于水溶性聚合物中的固体材料。"无定型"意指难溶性化合物不为结晶的。"结晶的"意指化合物在各维度均表现出至少100个重复单位的三维度长程有序(long-rangeorder)。因此,术语无定型不仅意欲包括基本上无序的材料,并且意欲包括可具有一些小的有序度,但该有序小于三个维度和/或有序仅于短距离内的材料。无定型材料可以通过本领域已知的技术诸如粉末X射线衍射(PXRD)结晶学、固态NMR或热技术诸如差示扫描量热法(DSC)进行表征。
优选地,在该固体无定型分散体中至少大部分(即至少约60 wt %)的难溶性化合物为无定型。该化合物可存在于在该固体无定型分散体中的相对纯的无定型域或区域中,作为均匀地分布在整个聚合物内或这些状态或位于它们中间的那些状态的任何组合的化合物的固溶体。优选地,该固体无定型分散体是基本均匀的,使得该无定型化合物尽可能均匀地分布在整个聚合物内。如本文所用,"基本上均匀的"意指存在于在该固体无定型分散体中的相对纯的无定型域或区域中的化合物的部分相对小,约少于药物总量的20 wt %,且优选为少于10 wt %。
适合用于固体无定型分散体的水溶性聚合物应为惰性的,就它们不以不利的方式与难溶性化合物化学反应的意义而言,为药学上可接受的,且在生理学相关的pH (例如,1-8)下在水溶液中具有至少一些溶解度。聚合物可为中性或可离子化的,且在1-8的pH范围的至少一部分内应具有至少0.1 mg/mL的水性溶解度。
适合与本发明一起使用的水溶性聚合物可为纤维素或非纤维素的。聚合物在水溶液中可为中性或可离子化的。其中,可离子化和纤维素聚合物是优选的,可离子化的纤维素聚合物是更优选的。
示例性水溶性聚合物包括醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)、羟丙基甲基纤维素(HPMC)、羟丙基甲基纤维素酞酸酯(HPMCP)、羧甲基乙基纤维素(CMEC)、醋酸纤维素酞酸酯(CAP)、醋酸纤维素偏苯三甲酸酯(CAT)、聚乙烯吡咯烷酮(PVP)、羟丙基纤维素(HPC)、甲基纤维素(MC)、环氧乙烷与环氧丙烷的嵌段共聚物(PEO/PPO,也称为泊洛沙姆(poloxamer)),及其混合物。尤其优选的聚合物包括HPMCAS、HPMC、HPMCP、CMEC、CAP、CAT、PVP、泊洛沙姆,及其混合物。最优选的是HPMCAS。参见欧洲专利申请公开号0 901 786 A2,其公开内容通过引用并入本文。
固体无定型分散体可根据用于形成固体无定型分散体的任何方法制备,所述方法使得至少大部分(至少60%)的难溶性化合物为无定型态。此类方法包括机械法、热法和溶剂法。示例性机械法包括研磨和挤压;熔融法,包括高温融溶、溶剂改性融溶和熔融-凝结法;和溶剂法,包括非溶剂沉淀、喷涂和喷雾干燥。参见例如下列的美国专利,将其相关公开内容通过引用并入本文:第5,456,923和5,939,099号,其描述通过挤压法形成分散体;第5,340,591和4,673,564号,其描述通过研磨法形成分散体;和第5,707,646和4,894,235号,其描述通过熔融凝结法形成分散体。在一个优选方法中,固体无定型分散体通过喷雾干燥形成,如欧洲专利申请公开号0 901 786 A2中所公开。在该方法中,将化合物和聚合物溶解在溶剂(诸如丙酮或甲醇)中,然后通过喷雾干燥快速地自溶液移除溶剂以形成固体无定型分散体。可根据需要制备含有至多约99 wt %的化合物的固体无定型分散体,例如1 wt %、5wt %、10 wt %、25 wt %、50 wt %、75 wt %、95 wt %或98 wt %。
固体分散体本身可用作剂型,或其可在制备其他剂型(诸如胶囊、片剂、溶液或悬浮液)时充当制造用产品(manufacturing-use-product)(MUP)。水性悬浮液的实例为1:1(w/w)化合物/HPMCAS-HF喷雾干燥分散体的水性悬浮液,含有在2%聚山梨酯80中的2.5 mg/mL的化合物。通常将用于片剂或胶囊的固体分散体与通常在此类剂型中发现的其他赋形剂或佐剂混合。例如,用于胶囊的示例性填充剂含有2:1 (w/w)化合物/HPMCAS-MF喷雾干燥分散体(60%)、乳糖(快速流动)(15%)、微晶纤维素(例如,Avicel(R0-102)) (15.8%)、淀粉钠(7%)、月桂基硫酸钠(2%)和硬脂酸镁(1%)。
HPMCAS聚合物可分别以Aqoat(R)-LF、Aqoat(R)-MF和Aqoat(R)-HF以低级、中级和高级自Shin-Etsu Chemical Co., LTD, Tokyo, Japan获得。较高的MF和HF级别通常是优选的。
以下段落说明可用于非人类动物的示例性制剂、剂量等。化合物A或其药学上可接受的盐与化合物D或其药学上可接受的盐的组合,作为两种药剂或以与另一药剂组合的形式的施用,可以口服或非口服方式进行。 将一定量的化合物A或其药学上可接受的盐与化合物D或其药学上可接受的盐一起或与另一药剂组合施用以使接收到有效剂量。通常,口服施用于动物的每日剂量为约0.01 mg/kg至约1,000 mg/kg体重,例如约0.01 mg/kg至约300mg/kg,或约0.01 mg/kg至约100 mg/kg,或约0.01 mg/kg至约50 mg/kg体重,或约0.01 mg/kg至约25 mg/kg,或约0.01 mg/kg至约10 mg/kg,或约0.01 mg/kg至约5 mg/kg。所施用的化合物A的每日剂量可为2mg、3mg、5mg、10mg、15mg、20mg、25mg、30mg或50mg。每日剂量可分为多次剂量,诸如BID/Q12小时的给药间隔。例如,在某些情况下,化合物A的每日剂量可以15mg q12小时施用。所施用的化合物D的每日剂量可为50mg、100mg、200mg或300mg。每日剂量可分为多次剂量,诸如BID/Q12小时的给药间隔。例如,在某些情况下,化合物D的每日剂量可以300 mg q12小时施用。
方便地,本发明化合物(或组合)可携带于饮用水中,使得治疗剂量的化合物与每日供水一起摄取。化合物可直接计量至饮用水中,优选地以液体、水溶性浓缩物(诸如水溶性盐的水溶液)形式。
方便地,本发明化合物(或组合)也可以原样或以动物饲料补充物(也称为预混物或浓缩物)的形式直接加入到饲料中。化合物在赋形剂、稀释剂或载体中的预混物或浓缩物更通常用于将该药剂包括于饲料中。适当赋形剂、稀释剂或载体根据需要为液体或固体,诸如水、各种粗粉(诸如苜蓿粉、大豆粉、棉籽油粉、亚麻籽油粉、玉米穗轴粉和玉米粉、糖蜜、尿素、骨粉)和诸如常用于家禽饲料中的矿物混合物。特别有效的赋形剂、稀释剂或载体为各自的动物饲料本身;即此类饲料的一小部分。载体有助于化合物均匀分布于与预混物掺合的最终饲料中。优选地,化合物充分掺合于预混物中,且随后掺合于饲料中。在此方面,化合物可分散或溶解在适当油性媒介物(诸如大豆油、玉米油、棉籽油等等)或挥发性有机溶剂中,且然后与载体掺合。应理解化合物在浓缩物中的比例能够广泛变化,因为化合物在最终饲料中的量可通过掺合适当比例的预混物与饲料而调整,以获得所需的水平的化合物。
高效浓缩物可由饲料制造商与蛋白质载体(诸如上文所述的大豆油粉和其他粗粉)掺合以生产浓缩的补充物,其适合于直接喂食动物。在此类情况中,准许动物食用平常的饮食。或者,此类浓缩的补充物可直接加入到饲料中,以生产含有治疗有效水平的本发明化合物的营养学上平衡的最终饲料。通过标准程序,例如在双壳搅拌机中,将混合物彻底掺合以确保均匀性。
如果补充物用作饲料的顶料(top dressing),则其同样有助于确保化合物在加料饲料顶部各处的分布的均匀性。
有效增加瘦肉沉积和改善瘦肉对脂肪比的饮用水和饲料通常通过混合本发明化合物与足够量的动物饲料以在饲料或水中提供约10-3至约500 ppm的化合物而制备。
优选的含药猪、牛、绵羊和山羊饲料通常含有每吨饲料约1至约400克的本发明化合物(或组合),用于这些动物的最优量通常为每吨饲料约50克至约300克。
优选的家禽和家养宠物饲料通常含有每吨饲料约1克至约400克,且优选为约10克至约400克的本发明化合物(或组合)。
对于动物的肠胃外施用,本发明化合物(或组合)可以糊剂或小丸的形式制备且通常在动物的头或耳的皮肤下以植入物施用,其中寻求瘦肉沉积的增加和瘦肉对脂肪比的改善。
糊剂制剂可通过将药物分散于药学上可接受的油(诸如花生油、芝麻油、玉米油等)中而制备。
含有效量的化合物A或其药学上可接受的盐,组合有组合化合物D或其药学上可接受的盐(药物组合物或组合)的小丸可通过将化合物A或其药学上可接受的盐,与化合物D或其药学上可接受的盐与稀释剂(诸如卡波蜡(carbowax)、巴西棕榈蜡等等)混合而制备,且可加入润滑剂(诸如硬脂酸镁或硬脂酸钙)以改善制丸方法。
当然,应理解可以向动物施用多于一种的小丸,以实现提供增加瘦肉沉积和改善所需的瘦肉对脂肪比的所需的剂量水平。而且,也可在动物治疗期间定期进行植入,以便于在动物体内维持适当的药物水平。
本发明具有若干有利的兽医学特征。对于希望自宠物动物增加瘦和/或削减不需要的脂肪的宠物主人或兽医,本发明提供可实现此的方式。对于家禽、肉用牛和猪饲养者而言,利用本发明的方法可生产出较精瘦的动物,这些动物在肉类工业值得较高的销售价格。
实施例
除非另有规定,否则起始材料通常可从商业来源获得,诸如Aldrich ChemicalsCo.(Milwaukee, WI)、Lancaster Synthesis, Inc.(Windham, NH)、Acros Organics(Fairlawn, NJ)、Maybridge Chemical Company, Ltd. (Cornwall, England)和TygerScientific (Princeton, NJ)。已使用某些常用缩写和首字母缩略词,其可包括:AcOH (乙酸)、DBU (1,8-二氮杂双环[5.4.0]十一碳-7-烯)、CDI (1,1'-羰基二咪唑)、DCM (二氯甲烷)、DEA (二乙胺)、DIPEA (N,N-二异丙基乙胺)、DMAP (4-二甲基氨基吡啶)、DMF (N,N'-二甲基甲酰胺)、DMSO (二甲基亚砜)、EDCI (N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺)、Et2O (乙醚)、EtOAc (乙酸乙酯)、EtOH (乙醇)、G或g(克)、HATU (2-(1H-7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸甲铵)、HBTU (O-苯并三唑-1-基-N,N,N',N'-四甲基脲鎓六氟磷酸盐)、HOBT (1-羟基苯并三唑)、H或h(小时)、IPA (异丙醇)、KHMDS (六甲基二硅氮烷钾)、MeOH (甲醇)、L或l(升)、mL (毫升)、MTBE (叔丁基甲基醚)、mg (毫克)、NaBH(OAc)3(三乙酰氧基硼氢化钠)、NaHMDS (六甲基二硅氮烷钠)、NMP (N-甲基吡咯烷酮)、RH(相对湿度)、RT或rt (与环境温度相同的室温(约20至25℃))、SEM ([2-(三甲基甲硅烷基)乙氧基]甲基)、TEA (三乙胺)、TFA (三氟乙酸)、THF (四氢呋喃)和T3P (丙烷膦酸酐)。
1H核磁共振(NMR)光谱在所有情况下与所提出的结构一致。特征化学位移(δ)以相对于氘化溶剂中的残留质子信号(CHCl3于7.27 ppm;CD2HOD于3.31 ppm)的百万分率(ppm)给出,并使用指定主要峰的常规缩写报告:例如s,单峰;d,双峰;t,三重峰;q,四重峰;m,多重峰;br,宽峰。
ssNMR意指固态NMR。
PXRD意指粉末X射线衍射。
术语"基本上相同"当用于描述X射线粉末衍射图时意指包括其中峰在+/-0.2º2Ɵ的标准偏差范围内的图。
如本文所用,涉及特定结晶形式的术语"基本上纯的"意指结晶形式包括小于10%,优选小于5%,优选小于3%,优选小于1% (以重量计)的任何其他物理形式的化合物A或化合物D。
反应在空气中进行,或当使用氧或湿气敏感性试剂或中间体时,则反应在惰性气氛(氮气或氩气)下进行。在适当时,将反应设备在动态真空下使用热风枪干燥且使用无水溶剂(来自Aldrich Chemical Company, Milwaukee, Wisconsin的Sure-SealTM产品或来自EMD Chemicals, Gibbstown, NJ的DriSolvTM产品)。使用商业溶剂和试剂而无需进一步纯化。当指示时,反应使用Biotage Initiator或Personal Chemistry Emrys Optimizer微波通过微波照射加热。反应进程使用薄层色谱法(TLC)、液相色谱法-质谱法(LCMS)、高效液相色谱法(HPLC)和/或气相色谱法-质谱法(GCMS)分析监测。TLC在预涂布的硅胶板上利用荧光指示剂(254 nm激发波长)进行,且在UV光下和/或以I2、KMnO4、CoCl2、磷钼酸和/或钼酸铵铈染色剂显现。LCMS数据在Agilent 1100 Series仪器上利用Leap Technologies自动取样器、Gemini C18柱、MeCN/水梯度和TFA、甲酸或氢氧化铵改性剂获取。柱洗脱剂使用以正离子和负离子模式二者自100 Da至1200 Da扫描的Waters ZQ质谱仪分析。也使用其他类似的仪器。HPLC数据在Agilent 1100 Series仪器上使用Gemini或XBridge C18柱、MeCN/水梯度和TFA或氢氧化铵改性剂获取。GCMS数据使用Hewlett Packard 6890烘箱利用HP 6890注射器、HP-1柱(12 m×0.2 mm×0.33 µm)和氦载气获取。样品在使用电子电离自50 Da至550Da扫描的HP 5973质量选择检测器上分析。纯化使用Isco CombiFlash Companion、AnaLogix IntelliFlash 280、Biotage SP1或Biotage Isolera One仪器和预装填的IscoRediSep或Biotage Snap二氧化硅筒通过中效液相色谱法(MPLC)进行。手性纯化使用Berger或Thar仪器;ChiralPAK-AD、-AS、-IC、Chiralcel-OD或-OJ柱;和CO2与MeOH、EtOH、iPrOH或MeCN的混合物(单独或使用TFA或iPrNH2改性)通过手性超临界流体色谱法(SFC)进行。使用UV检测触发级分收集。
质谱法数据由LCMS分析报告。质谱法(MS)经由大气压化学电离(APCI)、电喷雾电离(ESI)、电子碰撞电离(EI)或电子散射(ES)电离源进行。质子核磁光谱法(1H NMR)化学位移自四甲基硅烷向低磁场以百万分率给出且记录在300、400、500或600 MHz Varian光谱仪上。化学位移以参考氘化溶剂残留峰的百万分率(ppm,δ)表示。峰形如下描述:s,单峰;d,双峰;t,三重峰;q,四重峰;quin,五重峰;m,多重峰;br s,宽单峰;app,表观。分析性SFC数据在如上文所述的Berger分析仪器上获取。旋光性数据在PerkinElmer型343旋光仪上使用1dm槽获取。硅胶色谱法主要使用中压力Biotage或ISCO系统使用由各种商业供应商(包括Biotage和ISCO)预装填的柱进行。微分析由Quantitative Technologies Inc.进行且在计算值的0.4%内。
除非另有说明,否则化学反应在室温(约23摄氏度)下进行。
下述化合物和中间体使用由ChemBioDraw Ultra,12.0版(CambridgeSoft Corp.,Cambridge, Massachusetts)提供的命名约定命名。由ChemBioDraw Ultra,12.0版提供的命名约定是本领域技术人员所熟知的,并且认为由ChemBioDraw Ultra,12.0版提供的命名约定一般与IUPAC (国际纯粹与应用化学联合会(International Union for Pure andApplied Chemistry))涉及有机化学命名法的建议和CAS索引规则相符。除非另有说明,否则所有的反应物是商业上获得的而不经进一步纯化或使用文献中已知的方法制备。
术语"浓缩的"、"蒸发的"和"在真空中浓缩的"是指以低于60℃的浴温在旋转蒸发仪上于减压下移除溶剂。缩写"min"和"h"分别代表"分钟"和"小时"。术语"TLC"是指薄层色谱法,"室温或环境温度"意指18至25℃的温度,"GCMS"是指气相色谱法-质谱法,"LCMS"是指液相色谱法-质谱法,"UPLC"是指超高效液相色谱法和"HPLC"是指高压液相色谱法,"SFC"是指超临界流体色谱法。
氢化可在帕尔震荡器(Parr Shaker)中在加压氢气下或在Thales-nano H-Cube流动氢化设备中于全氢气下和1-2 mL/min的流速下在指定的温度下进行。
HPLC、UPLC、LCMS、GCMS和SFC保留时间使用程序中说明的方法测量。
中间体和实施例的制备
实施例1 (DGAT2i 化合物/化合物D):(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺

步骤1:3-乙氧基吡啶
在15℃下将碳酸铯(12 mol,1.5 equiv)和碘乙烷(9.7 mol,1.2 equiv)加入到3-羟基吡啶(8.10 mol,1.0 equiv)在丙酮(12 L)中的溶液中。将反应混合物在室温下搅拌24小时。将反应混合物过滤并将有机层浓缩以产生粗制产物。加入乙酸乙酯(20 L)并用水(3×5 L)洗涤。将有机层经硫酸钠干燥、过滤并浓缩以产生呈油状物的3-乙氧基吡啶(620 g,62%)。1H NMR (400 MHz, CDCl3) δ 1.44 (t, 3H), 4.07 (q, 2H), 7.15-7.23 (m, 2H),8.20 (dd, 1H), 8.30 (d, 1H)。
步骤2:3-乙氧基吡啶-1-氧化物
在10℃下将间氯过氧苯甲酸(6.5 mol,1.3 equiv)加入到3-乙氧基吡啶(5.0mol,1.0 equiv)在二氯甲烷(12 L)中的溶液中。将反应混合物在室温下搅拌24小时。加入硫代硫酸钠(4 kg,在5 L的水中)。将反应混合物在15℃下搅拌2小时。加入另一部分的硫代硫酸钠(1.5 kg,在5 L的水中)。将反应混合物在15℃下搅拌1小时。将混合物用二氯甲烷(16×10 L)萃取。将合并的有机层浓缩以产生粗制产物。通过硅胶柱色谱法(二氯甲烷:甲醇;100:1-10:1)将粗制产物纯化以产生呈棕色油状物的标题化合物(680 g,97%)。通过在室温下与石油醚(4 L)一起研磨24小时将此进一步纯化以产生呈黄色固体的3-乙氧基吡啶-1-氧化物(580 g,83%)。1H NMR (400 MHz, CDCl3) δ 1.41 (t, 3H), 4.02 (q, 2H),6.84 (dd, 1H), 7.12 (dd, 1H), 7.85 (d, 1H), 7.91-7.95 (m, 1H)。
步骤3:2-((5-溴吡啶-3-基)氧基)-3-乙氧基吡啶
将此反应分五个平行批次进行。
在室温下将二异丙基乙胺(2.69 mol,3.7 equiv)和溴三吡咯烷子基鏻六氟磷酸盐(0.93 mol,1.3 equiv)加入到3-乙氧基吡啶-1-氧化物(0.72 mol,1.0 equiv)和3-溴-5-羟基吡啶(0.72 mol,1.0 equiv)在四氢呋喃(2500 mL)中的搅拌溶液中。将反应混合物在室温下搅拌2天,然后将单独的批次合并为单一批次。将得到的悬浮液浓缩至干并溶解在二氯甲烷(25 L)中。将有机层用1N氢氧化钠(15 L)、水(3×20 L)和盐水(20 L)洗涤。将有机层经硫酸钠干燥、过滤并浓缩以产生油状物。通过硅胶柱色谱法(石油醚:乙酸乙酯;10:1-1:1)将粗制油状物纯化以产生呈棕色固体的粗制产物。将此固体与甲基叔丁基醚:石油醚(1:10;11 L)一起研磨以提供呈暗黄色固体的2-((5-溴吡啶-3-基)氧基)-3-乙氧基吡啶(730 g,69%)。1H NMR (400 MHz, CDCl3) δ 1.49 (t, 3H), 4.16 (q, 2H), 7.04 (dd,1H), 7.25 (dd, 1H), 7.68-7.73 (m, 2H), 8.44 (d, 1H), 8.49 (d, 1H)。MS (ES+)297.1 (M+H)。
步骤4:2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸乙酯
将2-((5-溴吡啶-3-基)氧基)-3-乙氧基吡啶(300 mmol,1.0 equiv)在四氢呋喃(1.3 L)中的溶液用氮气脱气30分钟。在室温下以保持内部温度低于30℃的速率加入TurboGrignard (390 mmol,1.3 equiv,1.3 M,在四氢呋喃中)。使反应混合物冷却至室温并搅拌3小时。将反应冷却至10℃并以保持温度低于15℃的速率加入氯化锌(390 mmol,1.3equiv,1.9 M,在2-甲基四氢呋喃中)。使所得悬浮液温热至室温,直到所有沉淀溶解为止,并且然后冷却回至10℃。以固体加入2-氯嘧啶-5-甲酸乙酯(360 mmol,1.2 equiv)和二氯[双(2-(二苯基膦基)苯基)醚]钯(II) (6.00 mmol,0.02 equiv)。将所得悬浮液用氮气脱气30分钟,然后加热至50℃持续16小时。在水性条件下将反应进行后处理,然后相继用乙二胺四乙酸二钠盐、硫代二氧化硅(thiosilica)和木炭处理以移除金属杂质。将粗制化合物从甲醇(450 mL)重结晶以产生呈淡黄色固体的2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸乙酯(77 g,70%)。1H NMR (400 MHz, CDCl3) δ 1.44 (t, 3H), 1.50 (t,3H), 4.19 (q, 2H), 4.46 (q, 2H), 7.00-7.04 (m, 1H), 7.25 (s, 1H), 7.71 (d,1H), 8.59 (s, 1H), 8.66 (d, 1H), 9.32 (s, 2H), 9.55 (s, 1H)。
步骤5:2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸(中间体1)
将氢氧化钠(307 mmol,1.5 equiv,4M水溶液)和甲醇(50 mL)加入到2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸酯(205 mmol,1.0 equiv)在四氢呋喃(300mL)中的悬浮液中。将所得溶液在室温下搅拌3小时。将反应混合物用水(400 mL)稀释并用2:1乙醚:庚烷(2×300 mL)萃取。用4M盐酸将水层酸化至pH 4。将所得悬浮液在室温下搅拌1小时。将固体过滤,用水洗涤,并干燥以产生呈淡黄色固体的2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸(69 g,100%)。1H NMR (400 MHz, DMSO-d6) δ1.37 (t, 3H),4.18 (q, 2H), 7.19 (dd, 1H), 7.58 (dd, 1H), 7.70 (dd, 1H), 8.35-8.40 (m, 1H),8.66 (d, 1H), 9.33 (s, 2H), 9.41 (d, 1H), 13.9 (br. s, 1H)。
步骤6:(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(实施例1 (DGAT2i 化合物))
将草酰氯(13.8 mL,160 mmol,1.2 equiv)和二甲基甲酰胺(0.510 mL,6.65mmol,0.05 equiv)加入到2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸(45.0 g,133 mmol,1.0 equiv)在二氯甲烷(500 mL)中的悬浮液中。当达到溶解时,将悬浮液搅拌2小时。将反应混合物浓缩以产生呈红色固体的粗制酰氯。在0℃下将(S)-四氢呋喃-3-胺(12.2 g,140 mmol,1.05 equiv)和二异丙基乙胺(51.0 mL,293 mmol,2.2 equiv)在四氢呋喃(100 mL)中的溶液滴加到粗制酰氯在二氯甲烷(200 mL)中的溶液中。使反应温热至室温并搅拌16小时。加入水(1.0 L)和乙酸乙酯(600 mL)并将有机层分离,用饱和碳酸氢钠洗涤,经硫酸镁干燥,并过滤。将滤液用活性炭(20 g)处理,在65℃下搅拌20分钟。将悬浮液温热过滤并将滤液浓缩至淡黄色固体,将其从甲醇/乙酸乙酯(1:4,1 L)重结晶以产生呈无色固体的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(43.5 g,81%)。将标题化合物与以相同方式制备的先前批次(108.7 g,266.8mmol)合并,并在80℃下用乙酸乙酯(1.0 L)制浆4小时。使悬浮液冷却至室温并搅拌4天。将固体过滤,用乙酸乙酯(3×200 mL)洗涤并在高真空下在50℃下干燥24小时以产生呈无色固体的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(100.5 g,92%)。1H NMR (300 MHz, DMSO-d6) δ 1.38 (t, 3H), 1.89-1.98 (m, 1H),2.15-2.26 (m, 1H), 3.65 (dd, 1H), 3.70-3.78 (m, 1H), 3.85-3.92 (m, 2H), 4.18(q, 2H), 4.46-4.55 (m, 1H), 7.18 (dd, 1H), 7.58 (dd, 1H), 7.69 (dd, 1H), 8.37(dd, 1H), 8.64 (d, 1H), 8.95 (d, 1H), 9.28 (s, 2H), 9.39 (d, 1H)。MS (ES+)408.4 (M+H)。熔点177.5℃。C21H21N5O4的元素分析:计算值C, 61.91;H, 5.20;N, 17.19;实测值C, 61.86;H, 5.18;N, 17.30。
通过粉末X射线衍射(PXRD)分析表征来自此程序的固体形式,并指定为化合物D的形式1。
用于制备(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-
基)嘧啶-5-甲酰胺(实施例1 (化合物D))的替代步骤6
将乙腈(35 mL)、2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)嘧啶-5-甲酸(5.0g,15 mmol)和(S)-四氢呋喃-3-胺盐酸盐(2.2 g,18 mmol,1.2 equiv)装入100 mL反应器中。装入二异丙基乙胺(18 mL,103 mmol,7.0 equiv),同时保持温度在20℃至30℃。以保持温度低于45℃的速率装入丙烷膦酸酐(T3P)在乙腈(21 mL,30 mmol,2.0 equiv)中的溶液。将反应器加热至40±5℃持续1小时,然后取样用于反应完成。将反应冷却至20℃至25℃并加入四氢呋喃(25 mL)。装入碳酸氢钠的溶液(0.5M,40 mL)并将混合物搅拌1小时。检查并测量pH为8.5。加入乙酸乙酯(40 mL)并将混合物搅拌15分钟。使混合物沉降,并分离相。将水层转移至分液漏斗中并用乙酸乙酯(100 mL)反萃取。合并有机相并用水(40 mL)洗涤。将有机层分批转移至100 mL反应器,并在真空下浓缩至低体积。加入甲基乙基酮(100 mL)并将混合物浓缩至约60 mL的最终体积。移除真空,并将浆料加热至回流并保持直到固体从反应器壁上洗下来。将浆料经2小时冷却至15℃并粒化过夜。通过过滤分离固体,用甲基乙基酮洗涤反应器和滤饼两次(各10 mL)。将固体在真空烘箱中在50℃下干燥,以产生4.86 g(81%)的所需产物。通过PXRD分析表征来自此程序的固体形式,并指定为化合物D的形式2。
化合物D的形式2至形式1的转化
将(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(实施例1)的形式2 (10.0 g,24.6 mmol,1.00 equiv.)、甲基乙基酮(8.8 mL/g,88.0 mL)和水(1.2 mL/g,12.0 mL)装入100 mL反应器中。将反应器经30分钟加热至50℃。在约44℃下出现完全溶解。将反应器经30分钟冷却至40℃,然后装入(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(实施例1的化合物D)的晶种形式1 (0.050 g,0.123 mmol,0.0050 equiv.)。接种后,将浑浊的浆料搅拌1小时,然后经2小时冷却至5℃,并且然后在5℃下搅拌12小时。取出过程中对照样品并通过PXRD分析进行表征,以确认固体为化合物D的形式1。将该浆料过滤,并且将反应器和滤饼用0℃的甲基乙基酮(2.5 mL/g,25 mL)洗涤。将固体在真空烘箱中在50℃下干燥,以产生8.15 g(81.5%)的所需产物。所需产物的PXRD图与化合物D的形式1一致。
粉末X射线衍射:
使用配备有Cu辐射源(1.54056Å的Kα平均波长)、配备有利用gobel镜的双原色(twin primary)的Bruker AXS D8 Advance衍射仪进行粉末X射线衍射分析。通过PSD-LynxEye检测器检测衍射的辐射。原色和次色(secondary)都配备有2.5索勒狭缝(sollerslit)。将X射线管电压和安培数分别设定于40 kV和40 mA。在θ-θ测角计中使用每步6秒的扫描速度以1000步自3.0至40.0度2θ的锁定式配对扫描(locked couple scan)收集数据。通过放置在硅低背景样品架(C79298A3244B261)上准备样品。使用Bruker DIFFRAC Plus软件收集数据。以EVA diffract plus软件进行分析。
在峰检索之前不处理PXRD数据档案。使用EVA软件中的峰检索算法,使用5的临界值和0.2的宽度值选择峰。自动化分配的输出经目视检查以确保正确性且若必要时手动调整。通常选择具有≥3%的相对强度的峰。还将未解析或与噪声一致的峰丢弃。与来自USP中所述的PXRD的峰位置相关的典型误差在+/- 0.2°内(USP-941)。
表1:表征结晶材料实施例1 (化合物D)的关键PXRD峰
| 实施例1的形式1 | 实施例1的形式2 |
| 角度2Θ (°) | 角度2Θ (°) |
| 5.3, 7.7, 15.4 | 6.5, 9.3, 13.6 |
图1为显示实施例1 (化合物D)的结晶形式1的特征x射线粉末衍射图(纵轴:强度(CPS);横轴:2θ (度))。
图2为显示实施例1 (化合物D)的结晶形式2的特征x射线粉末衍射图(纵轴:强度(CPS);横轴:2θ (度))。
实施例2:4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸,化合物A (ACCi 化合物)的制备:
在化合物A的制备中,应注意本文所述的一些制备方法可能需要保护远程官能团(例如式I前体中的伯胺、仲胺、羧基)。此类保护的需求将根据远程官能团的性质和制备方法的条件而改变。此类保护的需求由本领域技术人员容易地确定。此类保护/脱保护方法的使用也在本领域技术范围内。对于保护基及其用途的一般说明,参见T.W. Greene,Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991。此外,本发明不限于本文提供的特定合成方法,其可改变。
中间体A1:1-异丙基-4,6-二氢螺[吲唑-5,4'-哌啶]-7(1H)-酮盐酸盐

步骤1. 9-氧代-3-氮杂螺[5.5]十一碳-7-烯-3-甲酸叔丁酯

在25-30℃下将4-甲酰基哌啶-1-甲酸叔丁酯(108 Kg)、环己烷(1080 L)和吡咯烷(64.8 Kg)装入干燥反应器中。将混合物搅拌5-10 min,并且然后加热至回流,持续12-16h,同时使用Dean-Stark分水器收集水。然后将反应混合物冷却至50-60℃,在该温度下施加真空以蒸馏过量吡咯烷和环己烷。然后将反应混合物冷却至25-30℃,并装入环己烷(648 L),随后装入甲基乙烯基酮(49.63 Kg)。将混合物搅拌12-16 h,然后过滤并将滤液装入干净且干燥的反应器中。将溶液冷却至10-15℃,然后缓慢加入乙酸(54.75 Kg)在水(54 L)中的溶液,保持温度低于15℃。加入结束时,将混合物温热至25-30℃并搅拌12-16 h。分离层,并将水层用乙酸乙酯(324 L)萃取。将合并的有机层用碳酸氢钠(32.34 Kg)在水(324 L)中的溶液洗涤,然后经硫酸钠干燥。将固体用乙酸乙酯(54 L)洗涤,并将合并的滤液在减压下在低于40℃浓缩。将正庚烷(216 L)装入反应器中,并在减压且低于40℃下进行蒸馏直至干燥。将混合物冷却至25-30℃并将正庚烷(216 L)装入反应器中。固体形成后,将混合物搅拌1-2h。然后将固体过滤,用正庚烷(54 L)洗涤并在40-50℃下干燥10-12 h,以产生所需的材料(90.1 Kg,67%产率)。
步骤2. (E)-10-((二甲基氨基)亚甲基)-9-氧代-3-氮杂螺[5.5]十一碳-7-烯-3-甲酸叔丁酯

在氮气氛下在25-30℃下在干净且干燥的反应器中装入9-氧代-3-氮杂螺[5.5]十一碳-7-烯-3-甲酸叔丁酯(50 Kg)、N,N-二甲基甲酰胺(500 L)和N,N-二甲基甲酰胺二甲缩醛(135 Kg)。将反应混合物搅拌5-10 min,然后加热至120-130℃,持续20 h。然后将混合物冷却至50-60℃并在高真空下在低于60℃下蒸馏溶剂。在低于45℃下装入混合二甲苯(200L)并在高真空下在低于60℃下蒸馏溶剂。用另一批混合二甲苯(200 L)重复此操作。然后将甲苯(200 L)装入反应器中并在高真空下在低于60℃下蒸馏溶剂。用第二批甲苯(200 L)重复此操作。然后在低于30℃下装入甲基叔丁基醚(100 L),并在高真空下在低于40℃下蒸馏溶剂。将混合物冷却至15-20℃并在低于20℃下装入甲基叔丁基醚(100 L)。将混合物搅拌20-30 min并将固体过滤,用甲基叔丁基醚(50 L)洗涤并在没有真空下在50-55℃下干燥10h以提供所需的化合物(52.1 Kg,87%产率)。1H NMR (400 MHz, CDCl3) δ ppm 7.48 (s,1H), 6.57 (d, J=9.97 Hz, 1H), 5.99 (d, J=10.16 Hz, 1H), 3.32-3.51 (m, 4H),3.06 (s, 6H), 2.72 (s, 2H), 1.57-1.66 (m, 2H), 1.41-1.53 (m, 11H)。
步骤3. 1-异丙基-1,4-二氢螺[吲唑-5,4'-哌啶]-1'-甲酸叔丁酯

在25-30℃下将(E)-10-((二甲基氨基)亚甲基)-9-氧代-3-氮杂螺[5.5]十一碳-7-烯-3-甲酸叔丁酯(80 Kg)、甲苯(704 L)和三甲胺(16 L)装入干净且干燥的反应器中。将反应混合物温热至70-80℃,并经4-5 h加入异丙基肼盐酸盐在甲醇中的溶液(1.25equiv.,总共141 Kg)。然后将反应混合物在70-80℃搅拌8-10 h,然后冷却至15-25℃。然后缓慢加入柠檬酸(48 Kg)在水(480 L)中的溶液,使内部温度保持低于25℃。加入乙酸乙酯(208 L)并将混合物搅拌10 min。分离层,并将有机层依次用柠檬酸(48 Kg)在水(480 L)中的溶液,然后仅用水(320 L) 洗涤。将合并的水层用乙酸乙酯(320 L)萃取。然后将合并的有机层经硫酸钠(8 Kg)干燥并在减压下并在低于40℃下将溶剂蒸发至干。将二氯甲烷(240L)装入反应器中并在25-30℃下将混合物搅拌直至澄清。在25-30℃下依次装入活性炭(1.84 Kg)、硅酸镁(1.84 Kg)和硅胶(32 Kg,100-200目)并将不均匀混合物搅拌1 h。然后将浆料在通过混合Hyflow supercell (8 Kg)和二氯甲烷(40 L)制备的Hyflow床上过滤。将滤饼用二氯甲烷洗涤(三次120 L)。将合并的滤液装回反应器中并在减压下在低于40℃下蒸发溶剂。然后装入正庚烷(160 L),并在减压下在低于40℃下蒸馏。将正庚烷(200 L)装入反应器中并将混合物冷却至0-5℃。搅拌12-15 h后,在0℃下将固体过滤,用冷却(0-5℃)正庚烷(160 L)洗涤并在真空下在40-50℃下干燥以提供标题化合物(82.4 Kg,75%)。1HNMR (400 MHz, CDCl3) δ ppm 7.25 (s, 1H), 6.42 (dd, J=10.05, 0.49 Hz, 1H) 5.84(d, J=9.95 Hz, 1H), 4.42-4.52 (m, 1H), 3.36-3.53 (m, 4H), 2.62 (s, 2H) 1.56-1.68 (m, 2H) 1.45-1.55 (m, 17H)。
步骤4. 1-异丙基-4,6-二氢螺[吲唑-5,4'-哌啶]-7(1H)-酮盐酸盐

在25-30℃下将1-异丙基-1,4-二氢螺[吲唑-5,4'-哌啶]-1'-甲酸叔丁酯(60 Kg)和甲醇(600 L)装入干净且干燥的反应器中。在25-30℃下经30-40 min分5部分加入N-溴代琥珀酰亚胺(32.4 Kg)并持续搅拌30-60 min。缓慢加入硫代硫酸钠五水合物(5.4 Kg)在水(102 L)中的溶液,保持内部温度低于30℃。将混合物搅拌20-30 min,然后在减压下在低于45℃下蒸发溶剂。将残余物冷却至25-30℃并将2-甲基四氢呋喃(420 L)与水(90 L)一起装入反应器中。将混合物搅拌15-20 min,然后分离层,将水层进一步用2-甲基四氢呋喃(120L)萃取。将合并的有机萃取物在25-30℃下用氢氧化钠(4.8 Kg)在水(120 L)中的溶液处理15-20 min。分离层并将有机层用水(120 L)洗涤,随后用氯化钠(12 Kg)在水(120 L)中的溶液洗涤,然后经硫酸钠(6 Kg)干燥。过滤后,将滤饼用2-甲基四氢呋喃(30 L)洗涤并将合并的滤液装回至反应器中。在减压下在低于45℃下将溶剂完全蒸馏并将残余物溶解在四氢呋喃(201 L)中。在25-30℃下将叔丁醇钾(60.6 Kg)和四氢呋喃(360 L)装入另一干净且干燥的反应器中。将残余物在四氢呋喃中的溶液缓慢加入到该混合物中,保持温度低于30℃。然后将反应混合物温热至60-65℃并在此温度下保持1-2 h。完成后,将混合物冷却至0-10℃,并用盐酸的溶液(1 N,196 L)缓慢淬灭,保持内温低于10℃。使反应混合物温热至25-30℃,并装入乙酸乙酯(798 L)。搅拌15-20 min后,分离层,并将水层进一步用乙酸乙酯(160L)萃取。将合并的有机层用水(160 L)洗涤,经硫酸钠(8 Kg)干燥、过滤,并将滤饼用乙酸乙酯(300 L)洗涤。在减压下在低于45℃下将溶剂完全蒸馏,并在25-30℃下将乙酸乙酯(540L),随后甲醇(156 L)装入反应器中。将混合物冷却至0-5℃,在此时缓慢加入乙酰氯(79.8Kg),保持温度在指定范围内。然后使混合物温热至20-25℃,且在该温度下在搅拌下保持4-5小时。将所得浆料过滤并将固体用乙酸乙酯(120 L)洗涤,然后在40-45℃下干燥8-10 h,以提供所需的粗制产物(33.5 Kg,65%)。
通过在干净且干燥的反应器中在25-30℃下将此粗制固体(56.8 Kg)溶解在甲醇(454.4 L)中进行最终纯化步骤。将溶液搅拌30-45 min,然后在25-30℃下通过0.2微米的筒式过滤器进入干净且干燥的反应器。在减压下在低于50℃下蒸馏甲醇,直至残余~1vol溶剂。将反应混合物冷却至25-30℃并通过0.2微米筒式过滤器装入新乙腈(113.6 L)。在减压下在低于50℃下蒸馏溶剂,直至残余~1vol溶剂。将反应混合物冷却至25-30℃并将新乙腈(190 L)通过0.2微米筒式过滤器装入反应器中。将混合物温热至65-70℃并搅拌45 min,然后冷却至25-30℃并搅拌1 h。将所得浆料过滤,并将滤饼用冷却(15℃)乙腈(56.8 L)洗涤。将固体在减压下在40-50℃下干燥8 h以提供中间体A1 (36.4 Kg,64%)。1H NMR (400 MHz,CD3OD) δ ppm 7.43 (s, 1H), 5.32-5.42 (m, 1H), 3.15-3.25 (m, 4H), 2.89 (s,2H), 2.64 (s, 2H), 1.69-1.90 (m, 4H), 1.37-1.45 (m, 6H);ESI [M+H]+=248。
中间体A2:2-(4-(叔丁氧基羰基)苯基)-6-甲氧基异烟酸

在20-25℃下将2,6-二氯异烟酸(30 Kg)和甲醇(120 L)装入干净且干燥的反应器中。将浆料搅拌5 min,然后加热至65℃(回流)。然后经由加料漏斗经至少4 h缓慢装入甲醇钠在甲醇中的溶液(30%,87.2 Kg)。用甲醇(15 L)冲洗漏斗,并在65℃下进行搅拌至少15h。然后将混合物冷却至45℃,并在减压下蒸馏,直至残余体积为~90 L。然后在40-45℃下将碳酸氢钾(28.2 Kg)和碳酸钾(21.6 Kg)在水(180 L)中的溶液装入反应器中。含有水溶液的反应器用水(21 L)冲洗,并将洗涤液装入反应混合物中。将混合物在减压下在低于80℃下蒸馏,直到残余体积为~240 L,然后冷却至20-25℃。
将4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯甲酸叔丁酯(52.3 Kg)和二噁烷(340 Kg)装入另一干净且干燥的反应器中,并在2-25℃下搅拌直至完全溶解。然后在40℃加热前反应器的内容物,以确保完全溶解并转移到此新反应器中。将反应混合物冷却至20-25℃,并经由真空/氮气循环进行脱氧步骤。将混合物进一步冷却至0-10℃并在氮气流下将乙酸钯(0.65 Kg),随后将三苯基膦(2.46 Kg)装入反应器中。将混合物温热至20-25℃,并经由真空/氮气循环进行另一脱氧步骤。然后将混合物加热至80℃,并在此温度下保持至少18 h。将混合物冷却至20-25℃,然后将甲基叔丁基醚(133.2 Kg)和水(30 L)依次装入反应器中。分离层,并用水(110 L)稀释水层,然后用甲基叔丁基醚(110 L)萃取。将合并的有机萃取物用柠檬酸(52 Kg)在水(84 L)中的溶液洗涤,并分离层。将水层进一步用甲基叔丁基醚(88.8 Kg)萃取并将有机层合并,然后用三分之一的氯化钠(43 Kg)在水(80L)中的溶液洗涤三次。最后一次层分离后,将有机层通过含有木炭筒的帕尔(pall)过滤器过滤,并将滤饼用甲基叔丁基醚(11.2 Kg)洗涤。将滤液在减压下在低于50℃下蒸馏降至~90 L,并且然后依次与庚烷(120 L)在低于50℃下共同蒸馏并降至~120 L。然后将混合物经1 h冷却至20-25℃,然后在此温度搅拌另外1 h。将浆料过滤并将滤饼用庚烷(3×18 L)洗涤三次,然后用乙腈(3×18 L)洗涤三次。将所得湿固体在真空下并在氮气流下在低于45℃下干燥至少15 h,以提供中间体A2 (44.6 Kg,87%产率)。1H NMR (400 MHz, CDCl3) δ ppm8.13 (s, 2H), 8.09 (s, 2H), 7.97 (d, J=1.17 Hz, 1H), 7.34 (d, J=0.98 Hz, 1H),4.08 (s, 3H), 1.61 (s, 9H);ESI [M+H]+=330。
中间体A3:4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸叔丁酯

将2-(4-(叔丁氧基羰基)苯基)-6-甲氧基异烟酸(中间体A2,15.2 g,46.2 mmol)和乙酸乙酯(140 mL)装入圆底烧瓶中。一次性加入1,1'-羰基二咪唑 (8.98 g,55.4 mmol)并在rt下搅拌1 h。加入1-异丙基-4,6-二氢螺[吲唑-5,4'-哌啶]-7(1H)-酮盐酸盐(中间体A1,14.8 g,52.2 mmol),随后加入N,N-二异丙基乙胺(9.1 mL,52.2 mL)并将反应在rt下搅拌18 h。加入2 M HCl水溶液(40 mL),随后加入1 M 硫酸氢钾(40 mL)和50 mL的庚烷。将所得混合物在rt下搅拌1h。将混合物转移至分液漏斗。分离有机相,依次用水(20 mL)、饱和碳酸氢钠(30 mL)、水(20 mL)、盐水(20 mL)洗涤,经20 g的硫酸镁和10 g的硅胶干燥,过滤并在真空中浓缩。浓缩快要结束时,固体开始形成。在80℃下将残余物在40 mL的乙酸乙酯中搅拌并缓慢滴加庚烷(120 mL)。将混合物在80℃下搅拌1 h,然后经1 h在搅拌下缓慢冷却至室温并在rt下搅拌18 h。经由过滤收集固体,用水和乙酸乙酯-庚烷(1:3)洗涤,并在真空下在50℃下干燥18 h以获得中间体A3 (19.64 g,76%产率)。
中间体A3的替代制备:
在20-25℃下将乙腈(219 Kg)和2-(4-(叔丁氧基羰基)苯基)-6-甲氧基异烟酸(中间体A2,34.8 Kg)装入干净且干燥的反应器中。将混合物搅拌5 min,然后1,1-羰基二咪唑(18.9 Kg)分三个连续部分装入。将浆料在20-25℃下进一步搅拌至少1 h,然后经由泵将1-异丙基-4,6-二氢螺[吲唑-5,4'-哌啶]-7(1H)-酮盐酸盐(中间体A1,33.0 Kg)装入反应器中,随后装入N,N-二异丙基乙胺(20.5 Kg)。将试剂泵以及反应器壁用乙腈(13.7 Kg)洗涤,并在20-25℃下进行搅拌至少2小时。完成后,将混合物用4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸叔丁酯(中间体A3,209g)接种并搅拌至少30 min。确认结晶开始后,经1小时装入柠檬酸单水合物(58.5 Kg)在水(257 L)中的溶液。将所得浆料在20-25℃下进一步搅拌至少2 h,然后过滤并将滤饼用乙腈(68.4 Kg)和水(87 L)的混合物洗涤。此洗涤液也用于冲洗反应器。在低于55℃下在减压下干燥固体,提供中间体A3 (43.44 Kg,73%产率)。
化合物A (呈游离酸):4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸

将4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸叔丁酯(3.7 g,6.6 mmol)和甲苯(25 mL)装入圆底烧瓶中。在搅拌下滴加85%磷酸(3.0 mL)并将反应加热至60℃,持续4小时。形成无色的稠胶状物。将反应冷却至rt并加水。观察到白色固体。丢弃甲苯有机层,保留水层和固体。加入乙酸乙酯(60 mL)和加入4N NaOH溶液以调整pH至~7。分离层,并用乙酸乙酯(50 mL)萃取水层。将合并的乙酸乙酯有机层经硫酸钠干燥、过滤,并在真空中浓缩,以提供白色固体。将这些固体在50℃下溶解于乙酸乙酯(80 mL)中并缓慢加入庚烷(90 mL)。除去热并将混合物冷却至rt并搅拌16h。经由过滤收集所得固体,用母液冲洗,并干燥,以提供呈白色固体的标题化合物(化合物A游离形式,2.15 g,65%产率)。
化合物A (呈游离酸)的替代制备:
在20-25℃下将乙腈(130.4 Kg)和4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸叔丁酯(中间体A3,20.72 Kg)装入干净的干燥反应器中。将混合物搅拌5 min,然后在温和的氮气吹扫下装入对甲苯磺酸(8.5Kg)。将反应混合物温热至70℃,并在该温度下保持至少6.5 h。一经完成,就将混合物冷却至40℃,用化合物A(104 g)接种并经至少1 h缓慢装入水(83 L)。将混合物在40℃下进一步搅拌最少4 h,然后经2 h冷却至20-25℃。进一步搅拌至少2 h,随后过滤,并将滤饼用乙腈(33 Kg)和水(41 L)的溶液冲洗。此洗涤液也用于冲洗反应器。将所得固体在减压下在低于55℃下干燥以提供化合物A (16.5 Kg,89%产率)。
化合物A的形式1 (化合物A的无水单-tris)的制备:

将4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸(151 mg,0.300 mmol)和3 mL的乙醇装入小瓶中。将混合物加热至80℃持续5分钟以溶解固体,并且然后冷却至rt。加入三(羟甲基)氨基甲烷(39 mg,0.32mmol),并将混合物在rt下搅拌过夜。滴加庚烷(2.25 mL)以产生浆料,将其加热至50℃以产生透明溶液。将混合物在搅拌下冷却至rt过夜。观察到白色固体,并将混合物搅拌另外三天。将该材料过滤并在真空烘箱中在50℃下干燥过夜以产生形式1 (151 mg,0.242 mmol,81%产率)。
化合物A的形式1 (化合物A的无水单-tris)的替代制备:
将乙醇(83 L)装入干净且干燥的反应器中,随后加入化合物A(9.43 Kg)和tris(2.55 Kg),同时将混合物保持在20-25℃的温度下。用乙醇(2 L)冲洗槽壁,并将所得混合物在65-70℃下加热,在此温度下保持至少30分钟,直到所有固体溶解,然后冷却至45-50℃。通过10 µm在线聚丙烯过滤器进行温过滤,并将反应器以及过滤器用乙醇(9 L)洗涤。通过相同的在线过滤器将正庚烷(24 L)装入温溶液中,并在45-50℃下用乙醇(0.5 L)中的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸无水tris盐(100 g)接种混合物。将温度保持至少2 h,然后经至少2 h冷却至20-25℃。搅拌进行至少5天。然后将浆料过滤,并将滤饼用乙醇(13 L)和正庚烷(6 L)的混合物洗涤。将固体在减压下在低于45℃下干燥至少12 h,提供实施例1 (11.7 Kg,77%)。
化合物A的形式2 (化合物A的单-tris盐的三水合物)的制备:

从化合物A的形式1的转化获得化合物A的形式2。将形式1 (1.7214 g,2.760mmol)、异丙醇(16.50 mL,215.8 mmol)和水(688 μL,38.190 mmol)加入到50 mL EasyMax反应器中。在25℃的反应器夹套温度下将混合物搅拌(300 rpm)约72 hr。然后将反应混合物经15 min温热至40℃并在40℃下保持约24小时,一次冷却至20℃以取出样品用于测试。通过PXRD看到形式的混合物;因此,加入另外的水(688 μL,38.190 mmol)。将搅拌速率增加到400 rpm,并使浆料搅拌6小时,并且然后冷却至15℃。在60 mL/40 M过滤器上分离固体并用96/4异丙醇/水洗涤。通过PXRD所得材料与化合物A的形式2一致。
化合物A的形式2 (化合物A的单-tris盐的三水合物)的替代制备:
将异丙醇(60.4 Kg)装入干净且干燥的反应器中,并加入化合物A (16.68 Kg)和tris (4.42 kg),同时将混合物保持在20-25℃的温度下。将混合物搅拌5 min,然后装入水(6.7 Kg)并将浆料加热至55℃。通过在线10 µm聚丙烯过滤器将现在的澄清溶液过滤至预热的干净且干燥的反应器(50-55℃)中。然后将溶液用呈三水合物的化合物A的单-tris盐(167 g)接种。确认晶种持续存在后,将混合物经至少2 h冷却至15℃,然后在15℃下保持最少16 h。过滤浆料,并用冷却的异丙醇(13.1 Kg)洗涤滤饼。然后将固体在减压下在低于25℃下干燥以仅提供化合物A的形式2 (22.1 Kg,98%产率)。
化合物A的形式1为无水的且在环境温度下在低于约0.2的水分活度(20% RH)下为热力学稳定的。化合物A的形式1具有与化合物A的图3中所示基本上相同的PXRD图。以2Ɵ±0.2º 2Ɵ表示的化合物A的形式1的特征PXRD峰在9.6、10.7和11.3。图3中的PXRD图的峰位置和强度提供于表2中。
表2 – 化合物A的形式1的PXRD峰和相对强度

化合物A的形式1具有与图4中所示基本上相同的拉曼光谱。化合物A的形式1具有以cm-1表示在568、698、989、1218、1511、1561和1615(±2 cm-1)的特征拉曼峰位移。图4中的化合物A的形式1的峰位置(±2 cm-1)和归一化强度(W=弱,M=中,S=强)列于表3中。
表3 – 化合物A的形式1的拉曼峰和归一化强度

化合物A的形式1具有与图5中所示基本上相同的13C ssNMR光谱。化合物A的形式1具有以ppm表示的在22.9、146.2、157.9、161.9和172.9 (±0.2 ppm)的特征13C ssNMR化学位移。如图5中所示的化合物A的形式1的13C化学位移(±0.2 ppm)列于表4中。
表4 – 化合物A的形式1的13C化学位移和强度

化合物A的形式2为三水合物且在环境温度和20% RH下在高于约0.2的水分活度下为热力学稳定的。化合物A的形式2具有与图6中所示基本上相同的PXRD图。以2Ɵ±0.2º 2Ɵ表示的化合物A的形式2的特征PXRD峰是在8.4、9.0、10.5、15.0和24.7。图6中的PXRD图的峰位置和强度提供于表5中。
表5 – 化合物A的形式2的PXRD峰和相对强度

化合物A的形式2具有与图7中所示基本上相同的拉曼光谱。化合物A的形式2具有以cm-1表示在562、692、984、1225、1507、1557和1610±2 cm-1的特征拉曼峰位移。图7中的化合物A的形式2的峰位置(±2 cm-1)和归一化强度(W=弱,M=中,S=强)列于表6中。
表6 – 化合物A的形式2的拉曼峰和归一化强度

化合物A的形式2具有与图8中所示基本上相同的13C ssNMR光谱。化合物A的形式2具有以ppm表示在19.2、149.5、155.6、163.8和188.3(±0.2 ppm)的特征13C ssNMR化学位移。如图8中所示的化合物A的形式2的13C化学位移(±0.2 ppm)列于表7中。
表7 – 化合物A的形式2的13C化学位移和强度

根据本文提供的公开内容,本领域普通技术人员将理解化合物A的形式1和形式2各自可通过几种不同的光谱峰或图以不同的组合来独特地识别。下述为特征峰值的示例性组合,其可用于单独识别化合物A的形式1和形式2,但绝不应将这些示例性组合视为限制本文公开的其他峰值组合。
为了确认化合物A的形式2中存在三个水分子,使用Bruker D8 Venture衍射仪在室温下收集数据。参见图9。使用单斜类空间群P21/c中的SHELX软件套件(5.1版,BrukerAXS,1997)通过固有相位解析结构。随后通过全矩阵最小二乘法精修(refine)结构。使用各向异性位移参数找到并精修所有的非氢原子。
从Fourier差分图中找到位于氮和氧上的氢原子并且在距离受限的情况下进行精修。将剩余的氢原子置于计算的位置并使其搭载于其载体原子上。
最终R指数为7.2%。最终差分Fourier显示没有丢失或错位的电子密度。
表8提供关于化合物A的形式2收集的数据:
表8

4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的结晶2-氨基-2-(羟甲基)丙烷-1,3-二醇盐。此结晶盐通常称为化合物A的tris盐。
化合物A的结晶tris盐,其中4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸和该盐的比率为1:1。
化合物A的结晶tris盐,其中该结晶盐为无水结晶盐。
化合物A的无水结晶tris盐,其中所述无水结晶盐具有包含在9.6、10.7和11.3 2Ɵ (±0.2º 2Ɵ)的衍射角的峰的PXRD图。
化合物A的无水结晶tris盐,其中所述无水结晶盐具有包含在1511、1561和1615cm-1 (±2 cm-1)的峰位移的拉曼光谱。
化合物A的无水结晶tris盐,其中所述无水结晶盐具有包含在22.9、146.2和161.9ppm (±0.2 ppm)的化学位移的13C ssNMR光谱。
化合物A的无水结晶tris盐,其中所述无水结晶盐具有选自以下的分析参数:包含在1511和1615 cm-1 (±2cm-1)的峰位移的拉曼光谱和包含至少一个在22.9、146.2或161.9ppm (±0.2 ppm)的化学位移的13C ssNMR光谱。
化合物A的无水结晶tris盐,其中所述无水结晶盐为基本上纯的。
化合物A的结晶tris盐,其中该结晶盐为三水合物结晶盐。
化合物A的三水合物结晶tris盐,其中所述三水合物结晶盐具有包含在8.4、9.0和10.5 2Ɵ (±0.2º 2Ɵ)的衍射角的峰的PXRD图。
化合物A的三水合物结晶tris盐,其中所述三水合物结晶盐具有包含在1507、1557和1610 cm-1 (±2cm-1)的峰位移的拉曼光谱。
化合物A的三水合物结晶tris盐,其中所述三水合物结晶盐具有包含在19.2、149.5和163.8 ppm (±0.2 ppm)的化学位移的13C ssNMR光谱。
化合物A的三水合物结晶tris盐,其中所述三水合物结晶盐具有选自以下的分析参数:
包含在8.4和9.0 2Ɵ (±0.2º 2Ɵ)的衍射角的峰的PXRD图,
包含在1557和1610 cm-1 (±2 cm-1)的峰位移的拉曼光谱,和
包含至少一个在19.2、149.5或163.8 ppm (±0.2 ppm)的化学位移的13C ssNMR光谱。
化合物A的三水合物结晶tris盐,其中所述三水合物结晶盐具有选自以下的分析参数:包含在8.4和9.0 2Ɵ (±0.2º 2Ɵ)的衍射角的峰的PXRD图,和包含至少一个在1507、1557或1610 cm-1 (±2cm-1)的峰位移的拉曼光谱。
化合物A的三水合物结晶tris盐,其中所述三水合物结晶盐具有选自以下的分析参数:包含在8.4和9.0 2Ɵ (±0.2º 2Ɵ)的衍射角的峰的PXRD图,和包含至少在一个在19.2、149.5或163.8 ppm (±0.2 ppm)的化学位移的13C ssNMR光谱。
药理数据
下列方案当然可由本领域技术人员改变。
在雄性Sprague-Dawley大鼠(Charles River (Boston, MA))中进行随机、媒介物对照、8平行组研究以获得循环和肝TG水平。使用标准实验室条件安置96只大鼠(~200 g);将它们成对安置并保持于12:12小时的反向明暗时刻表(在8:00 AM关灯)。在到达后将大鼠随机分为正常饮食(chow)或西方饮食组和剂量组,并在研究开始之前对标准大鼠正常饮食或西方饮食给予14天的导入期。标准实验室啮齿动物正常饮食(5053)来自LabDiet (PMI,St Louis, Missouri)。西方饮食(D12079Bi)来自Research Diets (New Brunswick, NJ)。
为了进行这些研究,制备媒介物以制造在去离子水中的0.5% (wt/体积)甲基纤维素(MC, Sigma Aldrich;274429)。用各化合物制备含有化合物A (由tris盐制备)或化合物D的储备溶液,以提供使得10 mL的溶液将递送所需的mg/kg剂量量的浓度,其中所使用的大鼠平均重量为约200 g。
从研究的第1天开始,向大鼠口服给予(10 mL/kg)的媒介物对照(在去离子水中的0.5% MC (wt/体积%)),低剂量或高剂量的化合物A (分别为1 mg/kg或10 mg/kg QD),低剂量或高剂量的化合物D (分别为5 mg/kg或30 mg/kg BID),或共同施用低剂量(化合物A为1mg/kg QD和化合物D为5 mg/kg BID),或共同施用高剂量(化合物A为10 mg/kg QD和化合物D为30 mg/kg BID)。
进食血浆分析物:在给药后2小时(进入暗循环2小时)经由侧尾静脉收集用于确定进食血浆TG浓度的血液,转移至涂有乙二胺四乙酸二钾(K2EDTA)的BD Microtainer管(PN365974),并在4℃下离心。然后在Siemens Chemistry XPT临床分析仪(Malven, PA)上使用Siemens甘油三酯_2测定试剂(ref 10335892)分析所得血浆样品。
禁食血浆分析物:在4小时禁食后给药后2小时(进入暗循环2小时)经由侧尾静脉收集用于确定禁食血浆TG的血液,转移至涂有K2EDTA的BD Microtainer管(PN365974),并在4℃下离心。然后在Siemens Chemistry XPT临床分析仪(Malven, PA)上使用Siemens甘油三酯2测定试剂(ref 10335892)分析所得血浆样品。
在研究的最后一天(第28天),在4小时禁食后给药后2小时处死大鼠以收集组织:在给药后2小时,经由侧尾静脉收集用于确定血浆分析物的血液,并且然后以CO2窒息处死动物。将血液转移至涂有K2EDTA的BD Microtainer管(PN365974),在4℃下离心并将血浆转移至96孔微量滴定板中并保存在-20℃下。迅速取出肝,以在液氮中预冷却的Wollenberg钳冷冻夹紧,单独包裹在铝箔中,并且随后保存在-80℃下。
组织粉碎:在于液氮中冷却的铝块上将冷冻的肝迅速粉碎,确保组织在整个粉碎过程中保持冷冻。将粉碎组织转移并于7 mL聚丙烯锥形管中保存在-80℃下直到分析。
肝甘油三酯的提取:将约50至100 mg的粉碎组织加入到含有800 µL冰冷的1:1CHCl3:MEOH的2 mL裂解基质D管(MP Bio)中。立即使用Qiagen组织裂解仪II (Qiagen CatNo. 85300)在4℃下以30 Hz提取样品4分钟。然后将匀浆转移至13×100 mm玻璃管中,并置于冰上。然后将裂解管用800 µL的1:1 CHCl3:MeOH冲洗,涡旋30秒,并加入到13×100 mm玻璃管中。在冰上时,将2.4 mL的100% CHCl3加入到所有玻璃小瓶中,使CHCl3:MeOH的比率达到4:2。然后将样品放入-20℃冷柜中过夜。在次日,加入1.75 mL 1M KCl H2O以使CHCl3:MeOH:H2O的比率达到4:2:1.75。然后将样品涡旋30秒,并在4℃下以1500 rpm×15 min离心。离心后,将有机相转移至新的13×100 mm提取管中,在N2下在37℃下干燥,并再悬浮于750 µL CHCl3中。将氨基丙基固相提取(SPE)筒(Waters Cat No. 054560, 6 mL, 500 mg)润湿并用5 mL己烷洗涤。洗涤后,将200 µL的在CHCl3中的样品提取物加入到该筒中并在不干燥柱的情况下真空除去。然后用5 ml的2:1 CHCl3:异丙醇/50 µM丁基化的羟基甲苯洗脱中性脂质。然后将样品在N2下在37℃下干燥,并再悬浮于1.75 ml的98:2异辛烷:异丙醇中。将样品通过0.2 µM注射器式滤器过滤,然后注射至HPLC氰基丙基柱(3.5 µM粒径,4.6×150mm柱,Agilent Zorbax Eclipse XBD-CN)上。运行方法为使用溶剂A (1000:1:2,异辛烷:异丙醇:乙酸)和溶剂B (50:50异丙醇:甲基叔丁基醚)以27分钟的运行时间进行4 µL注射。在第0-3分钟,将溶剂组成保持于100%溶剂A。在第3-8分钟,将溶剂组成从100%溶剂A改变至95%溶剂A和5%溶剂B。在第8-18分钟,将溶剂组成改变至50:50比率。在第18-19分钟,将溶剂组成改变回至100%溶剂A并在第19-27分钟保持在该组成。
使用标准方法通过超速离心从一部分粉碎肝样品中制备核和膜级分,每个处理组汇集在一起。通过Western印迹分析来自核提取物和膜级分的样品的SREBP1。钙联结蛋白(calnexin)的Western印迹用作膜级分的标志物、肌动蛋白作为总样品装载的标志物,和组蛋白(Histone) 2B作为核级分的标志物。使用相对单位来定量核SREBP1水平,并归一化至组蛋白2B,以控制核分级(fractionation)和凝胶加载过程中的样品损失。
将另一部分的粉碎肝进行处理并分析脂肪生成基因的表达。针对ACC1、FASN、SCD1、PCSK9和SREBP-1c的大鼠taqman探针全部在qPCR上使用Actb作为管家基因进行评估。
与当施用作为单一疗法的化合物A时的血浆和肝TG水平相比,(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺(化合物D)或其药学上可接受的盐与4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸(化合物A)或其药学上可接受的盐的组合的施用导致血浆(图10、11)和肝TG (图18)水平显著降低。
西方饮食喂食导致进食状态血浆TG是正常饮食喂食大鼠的2.2倍(图10)。口服施用低剂量(1 mg/kg QD)或高剂量(10 mg/kg QD)的仅化合物A (单一疗法)分别导致进食状态的血浆TG是媒介物施用的西方饮食喂食的大鼠的1.7倍和1.3倍。相反地,相对于媒介物施用的西方饮食喂食的大鼠,口服施用低剂量(5 mg/kg BID)或高剂量(30 mg/kg BID)的仅化合物D (单一疗法)分别使进食状态下的血浆TG降低55%和63%。化合物A和化合物D的共同施用导致完全阻断在进食状态下的化合物A介导的血浆TG的增加。相对于媒介物施用的西方饮食喂食的大鼠,口服共同施用均低剂量或均高剂量的化合物A和化合物D使进食状态的血浆TG水平降低37%和64%。
西方饮食喂食导致禁食血浆TG是正常饮食喂食大鼠的1.6倍(图11)。口服施用低和高剂量的作为单一疗法的化合物A分别导致禁食血浆TG是媒介物施用的西方饮食喂食大鼠的2.4倍和1.7倍。相反地,相对于媒介物施用的西方饮食喂食大鼠,口服施用低和高剂量的作为单一疗法的化合物D分别使禁食血浆TG降低20%和35%。口服共同施用各自均为低剂量或各自均为高剂量的化合物A和化合物D完全缓解当仅施用化合物A时所观察到的化合物A介导的禁食血浆TG增加。低剂量组(109 mg/dl)和高剂量组(81 mg/dl)的共同施用化合物A和化合物D的禁食血浆TG水平都类似于媒介物施用的西方饮食喂食大鼠(96 mg/dl)。
在来自施用媒介物、高剂量化合物A单一疗法、高剂量化合物D单一疗法或共同施用高剂量化合物A和高剂量化合物D的西方饮食喂食大鼠的样品中比较核SREBP-1定位(图12)。相对于媒介物处理的西方饮食喂食大鼠,施用化合物A产生SREBP-1的核定位增加,显示SREBP-1活化增加。相反地,施用化合物D降低SREBP-1核定位和SREBP-1活化。共同施用化合物A和化合物D阻断了化合物A介导的核SREBP-1定位的增加,与仅化合物A的单一疗法相比,产生50%降低。
相对于正常饮食喂食的媒介物处理的大鼠,喂食西方饮食且以媒介物处理的动物趋向于显示以下脂肪生成基因的表达增加:ACC1 (图13)、FASN (图14)、SCD1 (图15)和SREBP1 (图16),但不包括PCSK9 (图17),其在西方饮食喂食大鼠中较低。相对于西方饮食喂食和媒介物处理的动物,施用化合物A趋向于ACC1、FASN (仅化合物A高剂量)、SCD的表达的进一步增加,但不包括PCSK9和SREBP1。相反地,施用化合物D降低了所有脂肪生成基因的表达。共同施用化合物A和化合物D导致表达水平与在媒介物处理的西方饮食喂食大鼠中观察到的水平相当或更低。
媒介物施用的西方饮食喂食大鼠显示肝甘油三酯累积是正常饮食喂食大鼠的约2.7-倍(图18)。相对于媒介物施用的西方饮食喂食大鼠,口服施用低剂量和高剂量的化合物A分别产生脂肪变性降低36%和53%。同样地,相对于媒介物施用的西方饮食喂食大鼠,口服施用低剂量和高剂量的化合物D分别使脂肪变性降低25%和30%。相对于媒介物施用的西方饮食喂食大鼠,口服共同施用低或高剂量的化合物A和化合物D二者分别降低脂肪变性50%和73%。口服施用化合物A和化合物D的低剂量或高剂量组合所产生的脂肪变性降低大于施用相同剂量水平的作为单一疗法的化合物A或化合物D所观察到的。
在喂食胆碱缺乏和高脂肪饮食(CDAHFD)(研究饮食;A16092003)的雄性Wistar-Han大鼠(Charles River (Boston, MA))中进行随机、媒介物对照、5平行组研究以鉴定当以单一疗法单独施用或组合施用化合物A或化合物D时,肝炎和纤维化标志物改善的差异。使用标准实验室条件安置60只大鼠(~200 g);将它们成对安置并保持于12:12小时的反向明暗时刻表(在8:00 AM关灯)。在研究开始之前的6周开始喂食大鼠胆碱缺乏和高脂肪饮食(CDAHFD)。随机分为4个给药组(n=12/组)的大鼠每日两次接受施用媒介物、化合物A (5mg/kg)单一疗法、化合物D (30 mg/kg)单一疗法或共同施用化合物A (5 mg/kg)和化合物D(30 mg/kg),持续6周时段。使用在整个研究中保持正常饮食且每日两次施用媒介物的动物(n=12)作为对照组。在开始施用化合物之前并在施用化合物后3周和6周收集血液样品以评估循环标志物。在第-3周、第0周(第一次给药之前)、第3周和第6周进行剪切波弹性成像(Aixplorer Ultimate imager, Supersoinc imagine)测量,以评估炎症和纤维化随时间的进展。在药物施用6周后评估组织学,其相当于以CDAHFD进行12周。结果提供为各给药组的动物的平均值。
以CDAHFD进行12周后,以CO2窒息处死动物。收获肝的右侧叶、内叶和左侧叶。从每只动物的左侧叶、右内叶和右侧叶取得切片,并固定在福尔马林中并加工成石蜡块。将每只动物的左侧叶的一个切片冷冻保存在最佳切割温度(OCT)化合物中。将来自每只动物的其余肝冷冻并在液氮中冷却的铝块上快速粉碎,以确保整个粉碎过程中组织保持冷冻。转移粉碎的组织并在-80℃下保存直到分析。处理来自每只动物的一部分粉碎的肝样品并分析纤维发生的基因表达标志物。针对αSMA和COL1A1的大鼠taqman探针全部在qPCR上使用Actb作为管家基因进行评估。
下列终点由委员会认证的兽医病理学家通过定性组织学评价和定量组织形态测定术来评价:通过αSMA免疫组织化学(IHC)的肝星状细胞活化和分化为肌纤维母细胞;通过天狼星红染色的与纤维化有关的胶原蛋白。使用Visiopharm软件分析图像。统一应用具有阈值参数的Visiopharm应用以鉴定组织切片并定量各IHC (DAB (3,3'-二氨基联苯胺)阳性)或组织化学染色玻片上的靶标,以面积百分比表示:(目标染色面积/总组织ROI-空白)×100%。非参数统计数据用于分析来自此研究的数据。组值报告为平均值+/-平均值的标准误差。
在研究期间,相对于喂食正常饮食且施用媒介物的对照动物,接受CDAHFD且施用媒介物的动物显示肝硬度明显增加(使用剪切波弹性成像(SWE)评估(以千帕(kPa)测量),指示进行性肝炎和纤维化(图19)。施用作为单一疗法的化合物A或化合物D各自降低肝的硬度,表明肝炎和/或纤维化降低。化合物A和化合物D的共同施用产生的肝硬度降低大于作为单一疗法的任一药剂(图19)。
相对于喂食正常饮食且施用媒介物的对照动物,接受CDAHFD且施用媒介物的动物显示肝α平滑肌肌动蛋白(αSMA)染色显著增加,指示肌纤维母细胞活化和纤维发生(图20)。施用作为单一疗法的化合物A或化合物D各自分别使αSMA染色降低41%和23%,表明肌纤维母细胞活化和纤维发生降低。化合物A和化合物D的共同施用产生的αSMA染色降低大于作为单一疗法的任一药剂,使染色降低72%(图20)。
相对于喂食正常饮食且施用媒介物的对照动物,接受CDAHFD且施用媒介物的动物显示天狼星红(PSR)染色明显增加,指示胶原蛋白沉积和纤维化(图21)。施用作为单一疗法的化合物A或化合物D各自分别使PSR染色降低26%和20%,表明胶原蛋白沉积和纤维化降低。化合物A和化合物D的共同施用产生的PSR染色降低大于作为单一疗法的任一药剂,使染色降低56% (图21)。
相对于喂食正常饮食且施用媒介物的对照动物,接受CDAHFD且施用媒介物的动物显示离子化钙结合适配分子1 (Iba1)染色明显增加,指示肝巨噬细胞活化 (图24)。施用作为单一疗法的化合物A使Iba1染色降低15%,表明肝炎状况(tone)降低。然而施用作为单一疗法的化合物D并没有改变Iba1染色,化合物A和化合物D的共同施用产生的Iba1染色降低大于作为单一疗法施用的化合物A,使染色降低33%(图24)。
相对于喂食正常饮食且施用媒介物的对照动物,接受CDAHFD且施用媒介物的动物显示肝α平滑肌肌动蛋白(αSMA)(图22)和胶原蛋白A1A (COL1A1)(图23)基因表达明显增加,指示肌纤维母细胞活化和纤维发生。施用作为单一疗法的化合物A或化合物D各自降低肝αSMA和COL1A1基因表达,指示肝肌纤维母细胞活化和纤维发生降低。化合物A和化合物D的共同施用产生的肝αSMA (图22)和COL1A1 (图23)基因表达降低大于作为单一疗法的任一药剂。
在整个本申请中,引用了各种出版物。这些出版物的公开内容出于所有目的特此通过引用以其整体并入本申请中。
本领域技术人员将显而易见的是在本发明中可在不背离本发明的范围或精神下进行各种的修饰和变化。从考虑本文所公开的本发明的说明书和实施出发,本领域技术人员显而易见的是本发明的其他实施方案。意欲将说明书和实施例仅视为示例性的,本发明的真正范围和精神由以下权利要求书指示。
Claims (33)
1.一种药物组合物,其包含与药学上可接受的赋形剂混合的各自以治疗有效量存在的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
2.一种药物组合物,其包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐,4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐,以及药学上可接受的赋形剂。
3.权利要求1或2的组合物,其进一步包含至少一种选自以下的另外药剂:抗炎剂、抗糖尿病剂和胆固醇/脂质调节剂。
4.一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合。
5.一种用于治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐。
6.权利要求4或5的方法,其中所述疾病或病况为脂肪肝。
7.权利要求4或5的方法,其中所述疾病或病况为非酒精性脂肪性肝病。
8.权利要求4或5的方法,其中所述疾病或病况为非酒精性脂肪性肝炎。
9.权利要求4或5的方法,其中所述疾病或病况为伴有肝纤维化的非酒精性脂肪性肝炎。
10.权利要求4或5的方法,其中所述疾病或病况为伴有硬化的非酒精性脂肪性肝炎。
11.权利要求10的方法,其中所述疾病或病况为伴有硬化且伴有肝细胞癌的非酒精性脂肪性肝炎。
12.权利要求10的方法,其中所述疾病或病况为伴有硬化且伴有代谢相关疾病的非酒精性脂肪性肝炎。
13.权利要求4或5的方法,其中所述方法包括进一步施用治疗有效量的至少一种选自以下的其他药剂:乙酰CoA羧化酶-(ACC)抑制剂,二酰基甘油O-酰基转移酶1 (DGAT-1)抑制剂,单酰基甘油O-酰基转移酶抑制剂,磷酸二酯酶(PDE)-10抑制剂,AMPK活化剂,磺酰脲,美格列奈,α-淀粉酶抑制剂,α-葡萄糖苷水解酶抑制剂,α-葡萄糖苷酶抑制剂,PPARγ激动剂,PPAR α/γ激动剂,双胍,胰高血糖素样肽1 (GLP-1)调节剂,利拉鲁肽,阿必鲁肽,艾塞那肽,阿必鲁肽,利西拉肽,杜拉鲁肽,索马鲁肽,蛋白酪氨酸磷酸酶-1B (PTP-1B)抑制剂,SIRT-1活化剂,二肽基肽酶IV(DPP-IV)抑制剂,胰岛素促泌剂,脂肪酸氧化抑制剂,A2拮抗剂,c-jun氨基末端激酶(JNK)抑制剂,葡萄糖激酶活化剂(GKa),胰岛素,胰岛素模拟剂,糖原磷酸化酶抑制剂,VPAC2受体激动剂,SGLT2抑制剂,胰高血糖素受体调节剂,GPR119调节剂,FGF21衍生物或类似物,TGR5受体调节剂,GPBAR1受体调节剂,GPR40激动剂,GPR120调节剂,高亲和性烟酸受体(HM74A)活化剂,SGLT1抑制剂,肉毒碱棕榈酰转移酶的抑制剂或调节剂,果糖-1,6-二磷酸酶的抑制剂,醛糖还原酶的抑制剂,盐皮质激素受体抑制剂,TORC2的抑制剂,CCR2和/或CCR5的抑制剂,PKC同种型(例如,PKCα、PKCβ、PKCγ)的抑制剂,脂肪酸合成酶的抑制剂,丝氨酸棕榈酰转移酶的抑制剂,GPR81、GPR39、GPR43、GPR41、GPR105、Kv1.3、视黄醇结合蛋白4、糖皮质激素受体、生长抑素受体的调节剂,PDHK2或PDHK4的抑制剂或调节剂,MAP4K4的抑制剂,包括IL1β在内的IL1家族的调节剂,HMG-CoA还原酶抑制剂,角鲨烯合成酶抑制剂,贝特类,胆汁酸螯合剂,ACAT抑制剂,MTP抑制剂,脂加氧酶抑制剂,胆固醇吸收抑制剂,PCSK9调节剂,胆固醇酯转移蛋白抑制剂和RXRα的调节剂。
14.一种用于在人类中降低非酒精性脂肪性肝病或非酒精性脂肪性肝炎分级评分系统严重程度的至少一点、降低非酒精性脂肪性肝炎活性的血清标志物水平、降低非酒精性脂肪性肝炎疾病活性或降低非酒精性脂肪性肝炎的医疗后果的方法,所述方法包括向需要此类降低的患者施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐与至少治疗有效量的4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐的组合的步骤。
15.一种用于在人类中治疗脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎或伴有硬化且伴有肝细胞癌的非酒精性脂肪性肝炎的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的第一药剂和第二药剂和任选的第三药剂的步骤,其中:
(i) 所述第一药剂包含(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐;
(ii) 所述第二药剂包含4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐;和
(iii) 所述第三药剂包含选自以下的药剂:抗炎剂、抗糖尿病剂、抗纤维化剂、抗脂肪变性剂、胆固醇/脂质调节剂和抗糖尿病剂。
16.根据权利要求4、5、14或15中任一项的方法,其中(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺为下列结构的结晶固体:

或其药学上可接受的盐。
17.权利要求16的方法,其中所述结晶固体具有包含5.3±0.2、7.7±0.2和15.4±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末x射线衍射图。
18.权利要求16的方法,其中所述结晶固体具有包含6.5±0.2、9.3±0.2和13.6±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末x射线衍射图。
19.根据权利要求4、5、14或15中任一项的方法,其中4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸为下列结构的结晶固体:

或其药学上可接受的盐。
20.权利要求19的方法,其中所述结晶固体为4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的2-氨基-2-(羟甲基)丙烷-1,3-二醇盐。
21.如权利要求1或权利要求2所述的药物组合物,其中(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺为下列结构的结晶固体:

或其药学上可接受的盐。
22.如权利要求21所述的药物组合物,其中所述结晶固体具有包含5.3±0.2、7.7±0.2和15.4±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末x射线衍射图。
23.如权利要求21所述的药物组合物,其中所述结晶固体具有包含6.5±0.2、9.3±0.2和13.6±0.2的2-θ值(CuKα辐射,1.54056 Å的波长)的粉末x射线衍射图。
24.如权利要求1或权利要求2所述的药物组合物,其中4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸为下列结构的结晶固体:

或其药学上可接受的盐。
25.如权利要求24所述的药物组合物,其中所述结晶固体为4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸的2-氨基-2-(羟甲基)丙烷-1,3-二醇盐。
26.权利要求2的药物组合物,其中所述另外药剂选自:[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
27.如权利要求14-15中任一项所述的方法,其中使用至少一种其他药剂,并且所述其他药剂选自:[(1R,5S,6R)-3-{2-[(2S)-2-甲基氮杂环丁-1-基]-6-(三氟甲基)嘧啶-4-基}-3-氮杂双环[3.1.0]己-6-基]乙酸;2-[(1R,3R,5S)-3-({5-环丙基-3-[2-(三氟甲氧基)苯基]-1,2-噁唑-4-基}甲氧基)-8-氮杂双环[3.2.1]辛-8-基]-4-氟-1,3-苯并噻唑-6-甲酸;或2-[(4-{6-[(4-氰基-2-氟苄基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2S)-氧杂环丁-2-基甲基]-1H-苯并咪唑-6-甲酸或其药学上可接受的盐。
28.一种治疗心力衰竭、充血性心力衰竭、冠心病、外周血管疾病、肾血管疾病、肺动脉高压、血管炎、急性冠状动脉综合征和心血管风险改变的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐。
29.一种治疗肥胖、I型糖尿病、II型糖尿病、特发性I型糖尿病(Ib型)、成人隐匿性自身免疫糖尿病(LADA)、早发2型糖尿病(EOD)、青少年起病非典型糖尿病(YOAD)、青少年的成人起病型糖尿病(MODY)、营养不良相关性糖尿病、妊娠糖尿病、冠心病、缺血性中风、血管成形术后再狭窄、外周血管疾病、间歇性跛行、心肌梗塞、血脂异常、餐后脂血症、糖耐量受损(IGT)的病况、空腹血糖受损的病况、代谢性酸中毒、酮病、关节炎、糖尿病视网膜病变、黄斑变性、白内障、糖尿病肾病、肾小球硬化症、慢性肾衰竭、糖尿病神经病变、代谢综合征、综合征X、高血糖症、高胰岛素血症、高甘油三脂血症、胰岛素抵抗、葡萄糖代谢受损、皮肤与结缔组织病症、足部溃疡与溃疡性结肠炎、内皮功能障碍与血管顺应性受损、高载脂蛋白B脂蛋白血症和枫糖尿症的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐。
30.如权利要求29所述的方法,其中治疗II型糖尿病。
31.一种治疗肝细胞癌、肾脏肾透明细胞癌、头颈部鳞状细胞癌、结直肠腺癌、间皮瘤、胃腺癌、肾上腺皮质癌、肾脏乳头状细胞癌、子宫颈和子宫颈内癌、膀胱尿路上皮癌、肺腺癌的方法,所述方法包括向需要此类治疗的人类施用治疗有效量的(S)-2-(5-((3-乙氧基吡啶-2-基)氧基)吡啶-3-基)-N-(四氢呋喃-3-基)嘧啶-5-甲酰胺或其药学上可接受的盐和4-(4-(1-异丙基-7-氧代-1,4,6,7-四氢螺[吲唑-5,4'-哌啶]-1'-羰基)-6-甲氧基吡啶-2-基)苯甲酸或其药学上可接受的盐。
32.如权利要求31所述的方法,其中治疗肝细胞癌。
33.一种治疗选自脂肪肝、非酒精性脂肪性肝病、非酒精性脂肪性肝炎、伴有肝纤维化的非酒精性脂肪性肝炎、伴有硬化的非酒精性脂肪性肝炎,以及伴有硬化且伴有肝细胞癌或伴有代谢相关疾病的非酒精性脂肪性肝炎的疾病或病况的方法,所述方法包括向需要此类治疗的人类施用根据权利要求2的组合物。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862726172P | 2018-08-31 | 2018-08-31 | |
| US62/726172 | 2018-08-31 | ||
| US201962883860P | 2019-08-07 | 2019-08-07 | |
| US62/883860 | 2019-08-07 | ||
| PCT/IB2019/057259 WO2020044266A1 (en) | 2018-08-31 | 2019-08-28 | Combinations for treatment of nash/nafld and related diseases |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112955147A true CN112955147A (zh) | 2021-06-11 |
Family
ID=68165623
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980071389.XA Pending CN112955147A (zh) | 2018-08-31 | 2019-08-28 | 用于治疗nash/nafld和相关疾病的组合 |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US11254660B2 (zh) |
| EP (1) | EP3843740A1 (zh) |
| JP (1) | JP7161605B2 (zh) |
| KR (1) | KR102614808B1 (zh) |
| CN (1) | CN112955147A (zh) |
| AU (1) | AU2019329884B2 (zh) |
| BR (1) | BR112021003039A2 (zh) |
| CA (1) | CA3110601C (zh) |
| CL (1) | CL2021000491A1 (zh) |
| CO (1) | CO2021002230A2 (zh) |
| CR (1) | CR20210110A (zh) |
| DO (1) | DOP2021000036A (zh) |
| EC (1) | ECSP21012501A (zh) |
| IL (1) | IL281093A (zh) |
| MA (1) | MA53496A (zh) |
| MX (1) | MX2021002428A (zh) |
| PH (1) | PH12021550339A1 (zh) |
| SG (1) | SG11202101503RA (zh) |
| TW (1) | TWI718644B (zh) |
| UY (1) | UY38351A (zh) |
| WO (1) | WO2020044266A1 (zh) |
| ZA (1) | ZA202101090B (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11590161B2 (en) * | 2018-08-13 | 2023-02-28 | Viscera Labs, Inc. | Therapeutic composition and methods |
| KR20210106447A (ko) * | 2018-11-22 | 2021-08-30 | 치루 레고르 테라퓨틱스 인코포레이티드 | Glp-1r 효능제 및 그의 용도 |
| EP3906022A1 (en) * | 2018-12-31 | 2021-11-10 | Coherus Biosciences, Inc. | Compositions and methods to treat non-alcoholic fatty liver diseases (nafld) |
| US10954221B2 (en) | 2019-04-12 | 2021-03-23 | Qilu Regor Therapeutics Inc. | GLP-1R agonists and uses thereof |
| JP2022058085A (ja) * | 2020-02-24 | 2022-04-11 | ファイザー・インク | ジアシルグリセロールアシルトランスフェラーゼ2阻害剤とアセチル-CoAカルボキシラーゼ阻害剤との組合せ |
| JP2021134211A (ja) * | 2020-02-24 | 2021-09-13 | ファイザー・インク | Nafld/nashおよび関連疾患の処置のための組合せ |
| IL296100A (en) * | 2020-03-11 | 2022-11-01 | Dong A St Co Ltd | A medicinal preparation for the prevention or treatment of non-alcoholic steatohepatitis |
| WO2021211959A2 (en) * | 2020-04-18 | 2021-10-21 | Inipharm, Inc. | Methods and compositions for treating drug induced steatohepatitis |
| US11478533B2 (en) | 2020-04-27 | 2022-10-25 | Novo Nordisk A/S | Semaglutide for use in medicine |
| CN113816948B (zh) * | 2020-06-19 | 2023-08-11 | 江苏恒瑞医药股份有限公司 | 稠合咪唑类衍生物、其制备方法及其在医药上的应用 |
| CN116323599A (zh) | 2020-08-06 | 2023-06-23 | 加舒布鲁姆生物公司 | 杂环glp-1激动剂 |
| WO2022040600A1 (en) * | 2020-08-21 | 2022-02-24 | Terns Pharmaceuticals, Inc. | Compounds as glp-1r agonists |
| CN112635049A (zh) * | 2020-12-23 | 2021-04-09 | 无锡市第二人民医院 | 一种研究HSD17B13 rs72613567基因变异和肾功能损伤之间关系的方法 |
| CN118401519A (zh) | 2021-10-25 | 2024-07-26 | 拓臻制药公司 | 作为glp-1r激动剂的化合物 |
| WO2023169456A1 (en) | 2022-03-09 | 2023-09-14 | Gasherbrum Bio , Inc. | Heterocyclic glp-1 agonists |
| WO2023198140A1 (en) | 2022-04-14 | 2023-10-19 | Gasherbrum Bio, Inc. | Heterocyclic glp-1 agonists |
| WO2024125602A1 (en) | 2022-12-15 | 2024-06-20 | Gasherbrum Bio, Inc. | Salts and solid forms of a compound having glp-1 agonist activity |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012042433A1 (en) * | 2010-09-30 | 2012-04-05 | Pfizer Inc. | N1-PYRAZOLOSPIROKETONE ACETYL-CoA CARBOXYLASE INHIBITORS |
| US20180051012A1 (en) * | 2016-08-19 | 2018-02-22 | Pfizer Inc. | Diacylglycerol acyl transferase 2 inhibitors |
Family Cites Families (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1146866A (en) | 1979-07-05 | 1983-05-24 | Yamanouchi Pharmaceutical Co. Ltd. | Process for the production of sustained release pharmaceutical composition of solid medical material |
| EP0028489B1 (en) | 1979-11-05 | 1983-10-05 | Beecham Group Plc | Enzyme derivatives, and their preparation |
| DE3438830A1 (de) | 1984-10-23 | 1986-04-30 | Rentschler Arzneimittel | Nifedipin enthaltende darreichungsform und verfahren zu ihrer herstellung |
| AU1537292A (en) | 1991-04-16 | 1992-11-17 | Nippon Shinyaku Co. Ltd. | Method of manufacturing solid dispersion |
| US5340591A (en) | 1992-01-24 | 1994-08-23 | Fujisawa Pharmaceutical Co., Ltd. | Method of producing a solid dispersion of the sparingly water-soluble drug, nilvadipine |
| JP3265680B2 (ja) | 1992-03-12 | 2002-03-11 | 大正製薬株式会社 | 経口製剤用組成物 |
| US5612359A (en) | 1994-08-26 | 1997-03-18 | Bristol-Myers Squibb Company | Substituted biphenyl isoxazole sulfonamides |
| DE19504832A1 (de) | 1995-02-14 | 1996-08-22 | Basf Ag | Feste Wirkstoff-Zubereitungen |
| TW536540B (en) | 1997-01-30 | 2003-06-11 | Bristol Myers Squibb Co | Endothelin antagonists: N-[[2'-[[(4,5-dimethyl-3-isoxazolyl)amino]sulfonyl]-4-(2-oxazolyl)[1,1'-biphenyl]-2-yl]methyl]-N,3,3-trimethylbutanamide and N-(4,5-dimethyl-3-isoxazolyl)-2'-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-4'-(2-oxazolyl)[1,1'-biphe |
| EP0901786B1 (en) | 1997-08-11 | 2007-06-13 | Pfizer Products Inc. | Solid pharmaceutical dispersions with enhanced bioavailability |
| IL140622A0 (en) | 1998-07-06 | 2002-02-10 | Bristol Myers Squibb Co | Biphenyl sufonamide derivatives, pharmaceutical compositions containing the same and methods for the preparation thereof |
| MY125533A (en) | 1999-12-06 | 2006-08-30 | Bristol Myers Squibb Co | Heterocyclic dihydropyrimidine compounds |
| EP1383501B1 (en) | 2001-02-28 | 2007-04-04 | Merck & Co., Inc. | Acylated piperidine derivatives as melanocortin-4 receptor agonists |
| EP1478437B1 (en) | 2002-02-27 | 2005-08-31 | Pfizer Products Inc. | Acc inhibitors |
| GEP20084421B (en) | 2004-05-12 | 2008-07-10 | Pfizer Prod Inc | Proline derivatives and their use as dipeptidyl peptidase iv inhibitors |
| JP4069159B2 (ja) | 2004-05-25 | 2008-04-02 | ファイザー・プロダクツ・インク | テトラアザベンゾ[e]アズレン誘導体及びそれらのアナログ |
| PA8660701A1 (es) | 2005-02-04 | 2006-09-22 | Pfizer Prod Inc | Agonistas de pyy y sus usos |
| SI2463283T1 (sl) | 2006-04-20 | 2014-09-30 | Pfizer Products Inc. | Kondezirane fenil amino heterociklične spojine za preprečevanje in zdravljenje bolezni, ki jih posreduje glukokinaza |
| JP2010511035A (ja) | 2006-11-29 | 2010-04-08 | ファイザー・プロダクツ・インク | スピロケトンアセチルCoAカルボキシラーゼ阻害剤 |
| US20090036425A1 (en) | 2007-08-02 | 2009-02-05 | Pfizer Inc | Substituted bicyclolactam compounds |
| EP2297164A1 (en) | 2008-05-28 | 2011-03-23 | Pfizer Inc. | Pyrazolospiroketone acetyl-coa carboxylase inhibitors |
| ES2545231T3 (es) | 2008-05-28 | 2015-09-09 | Pfizer Inc. | Inhibidores de pirazoloespirocetona acetil-CoA carboxilasa |
| CA2729581A1 (en) | 2008-07-29 | 2010-02-04 | Pfizer Inc. | Fluorinated heteroaryls |
| EA018492B1 (ru) | 2008-08-28 | 2013-08-30 | Пфайзер Инк. | Диоксабицикло[3.2.1]октан-2,3,4-триольные производные |
| WO2010086820A1 (en) | 2009-02-02 | 2010-08-05 | Pfizer Inc. | 4-amino-5-oxo-7, 8-dihydropyrimido [5,4-f] [1,4] oxazepin-6 (5h) -yl) phenyl derivatives, pharmaceutical compositions and uses thereof |
| EP2406253B1 (en) | 2009-03-11 | 2013-07-03 | Pfizer Inc. | Benzofuranyl derivatives used as glucokinase inhibitors |
| CA2754685A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Substituted indazole amides |
| CA2754523A1 (en) | 2009-03-20 | 2010-09-23 | Pfizer Inc. | 3-oxa-7-azabicyclo[3.3.1]nonanes |
| JP2012526097A (ja) | 2009-05-08 | 2012-10-25 | ファイザー・インク | Gpr119調節因子 |
| EP2427448A1 (en) | 2009-05-08 | 2012-03-14 | Pfizer Inc. | Gpr 119 modulators |
| AU2010255422B2 (en) | 2009-06-05 | 2014-04-10 | Pfizer Inc. | 1- ( piperidin-4-yl) -pyrazole derivatives as GPR 119 modulators |
| WO2011005611A1 (en) | 2009-07-09 | 2011-01-13 | Merck Sharp & Dohme Corp. | Neuromedin u receptor agonists and uses thereof |
| CH702192A1 (de) | 2009-11-04 | 2011-05-13 | New Dent Ag | Keramisches Implantat. |
| CU24152B1 (es) | 2010-12-20 | 2016-02-29 | Irm Llc | 1,2 oxazol-8-azabiciclo[3,2,1]octano 8 il como moduladores de fxr |
| EP3597271A1 (en) | 2015-01-09 | 2020-01-22 | Gilead Apollo, LLC | Acc inhibitor combination therapy for the treatment of non-alcoholic fatty liver disease |
| SG11201804363UA (en) | 2015-12-29 | 2018-07-30 | Pfizer | Substituted 3-azabicyclo[3.1.0]hexanes as ketohexokinase inhibitors |
| CN106271089B (zh) | 2016-09-30 | 2019-01-25 | 英诺激光科技股份有限公司 | 一种激光薄膜刻蚀装置及方法 |
| PL3555064T3 (pl) | 2016-12-16 | 2023-03-06 | Pfizer Inc. | Agoności receptora GLP-1 i ich zastosowania |
| CA3082867C (en) | 2017-11-21 | 2022-05-10 | Pfizer Inc. | Crystalline 2-amino-2-(hydroxymethyl)propane-1,3-diol salt of 4-(4-(1-isopropyl-7-oxo-1,4,6,7-tetrahydrospiro[indazole-5,4'-piperidine]-1'-carbonyl)-6-methoxypyridin-2-yl)benzoic acid |
-
2019
- 2019-08-28 CR CR20210110A patent/CR20210110A/es unknown
- 2019-08-28 JP JP2021510174A patent/JP7161605B2/ja active Active
- 2019-08-28 EP EP19783644.8A patent/EP3843740A1/en active Pending
- 2019-08-28 CN CN201980071389.XA patent/CN112955147A/zh active Pending
- 2019-08-28 KR KR1020217009415A patent/KR102614808B1/ko active IP Right Grant
- 2019-08-28 US US16/553,818 patent/US11254660B2/en active Active
- 2019-08-28 AU AU2019329884A patent/AU2019329884B2/en active Active
- 2019-08-28 WO PCT/IB2019/057259 patent/WO2020044266A1/en active Application Filing
- 2019-08-28 BR BR112021003039-5A patent/BR112021003039A2/pt unknown
- 2019-08-28 TW TW108130771A patent/TWI718644B/zh active
- 2019-08-28 CA CA3110601A patent/CA3110601C/en active Active
- 2019-08-28 MX MX2021002428A patent/MX2021002428A/es unknown
- 2019-08-28 MA MA053496A patent/MA53496A/fr unknown
- 2019-08-28 SG SG11202101503RA patent/SG11202101503RA/en unknown
- 2019-08-29 UY UY0001038351A patent/UY38351A/es not_active Application Discontinuation
-
2021
- 2021-02-17 PH PH12021550339A patent/PH12021550339A1/en unknown
- 2021-02-17 ZA ZA2021/01090A patent/ZA202101090B/en unknown
- 2021-02-23 EC ECSENADI202112501A patent/ECSP21012501A/es unknown
- 2021-02-23 CO CONC2021/0002230A patent/CO2021002230A2/es unknown
- 2021-02-24 IL IL281093A patent/IL281093A/en unknown
- 2021-02-25 DO DO2021000036A patent/DOP2021000036A/es unknown
- 2021-02-26 CL CL2021000491A patent/CL2021000491A1/es unknown
- 2021-10-11 US US17/498,256 patent/US20220023299A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012042433A1 (en) * | 2010-09-30 | 2012-04-05 | Pfizer Inc. | N1-PYRAZOLOSPIROKETONE ACETYL-CoA CARBOXYLASE INHIBITORS |
| US20180051012A1 (en) * | 2016-08-19 | 2018-02-22 | Pfizer Inc. | Diacylglycerol acyl transferase 2 inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112021003039A2 (pt) | 2021-05-18 |
| EP3843740A1 (en) | 2021-07-07 |
| IL281093A (en) | 2021-04-29 |
| DOP2021000036A (es) | 2021-04-15 |
| US20200071306A1 (en) | 2020-03-05 |
| MX2021002428A (es) | 2023-01-02 |
| AU2019329884A1 (en) | 2021-03-11 |
| UY38351A (es) | 2020-03-31 |
| CO2021002230A2 (es) | 2021-03-08 |
| US20220023299A1 (en) | 2022-01-27 |
| CL2021000491A1 (es) | 2021-08-20 |
| ECSP21012501A (es) | 2021-03-31 |
| CR20210110A (es) | 2021-05-13 |
| WO2020044266A1 (en) | 2020-03-05 |
| CA3110601A1 (en) | 2020-03-05 |
| TW202026292A (zh) | 2020-07-16 |
| TWI718644B (zh) | 2021-02-11 |
| JP7161605B2 (ja) | 2022-10-26 |
| KR20210053929A (ko) | 2021-05-12 |
| SG11202101503RA (en) | 2021-03-30 |
| ZA202101090B (en) | 2022-06-29 |
| US11254660B2 (en) | 2022-02-22 |
| KR102614808B1 (ko) | 2023-12-19 |
| AU2019329884B2 (en) | 2022-01-27 |
| JP2021535126A (ja) | 2021-12-16 |
| CA3110601C (en) | 2023-09-05 |
| PH12021550339A1 (en) | 2021-10-04 |
| MA53496A (fr) | 2021-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11254660B2 (en) | Combinations for treatment of NASH/NAFLD and related diseases | |
| US11866425B2 (en) | Diacylglycerol acyl transferase 2 inhibitors | |
| US20220387402A1 (en) | Combinations comprising benzodioxol as glp-1r agonists for use in the treatment of nash/nafld and related diseases | |
| US9789110B2 (en) | Diacylglycerol acyltransferase 2 inhibitors | |
| JP2022058085A (ja) | ジアシルグリセロールアシルトランスフェラーゼ2阻害剤とアセチル-CoAカルボキシラーゼ阻害剤との組合せ | |
| RU2776369C1 (ru) | Комбинации для лечения насг/нажбп и связанных с ними заболеваний | |
| EA045908B1 (ru) | Комбинации для лечения nash/nafld и родственных заболеваний | |
| WO2023026180A1 (en) | Amorphous form of (s)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-n-(tetrahydrofuran-3- yl)pyrimidine-5-carboxamide | |
| BR112019001976B1 (pt) | Inibidores da diacilglicerol aciltransferase 2, composições que os compreendem e usos dos mesmos | |
| OA19183A (en) | Diacylglycerol Acyltransferase 2 inhibitors. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 40055015 Country of ref document: HK |