CN111315744B - 杂芳基并四氢吡啶类化合物、其制备方法、药物组合物及应用 - Google Patents
杂芳基并四氢吡啶类化合物、其制备方法、药物组合物及应用 Download PDFInfo
- Publication number
- CN111315744B CN111315744B CN201880072563.8A CN201880072563A CN111315744B CN 111315744 B CN111315744 B CN 111315744B CN 201880072563 A CN201880072563 A CN 201880072563A CN 111315744 B CN111315744 B CN 111315744B
- Authority
- CN
- China
- Prior art keywords
- compound
- alkyl
- stereoisomer
- pharmaceutically acceptable
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明公开了作为乙肝表面抗原(HBsAg)分泌抑制剂的式I的杂芳基并四氢吡啶类化合物、其制备方法、药物组合物及应用。
Description
技术领域
本发明涉及作为乙肝表面抗原HBsAg分泌抑制剂的杂芳基并四氢吡啶类化合物、包含其的药物组合物、其制备方法及其用途。
背景技术
乙型肝炎(Hepatitis B)是一种由乙型肝炎病毒(Hepatitis B virus,HBV)感染引起的常见传染性肝病,可以进一步发展为肝硬化和肝癌等疾病。据世界卫生组织(WHO)2017年《全球肝炎报告》估计,2015年全世界有2.57亿慢性乙肝病毒感染患者(乙肝表面抗原(HBsAg)呈阳性),导致88.7万人死亡,大多死于乙肝并发症(包括肝硬化和肝癌),而且由肝炎导致的死亡率从2000年到2015年期间一直在增加。尽管预防性乙肝疫苗问世多年来HBV发病率下降,但截止2015年包括中国在内的西太平洋地区HBsAg阳性患者仍占总人口的6.2%(Global hepatitis report 2017,WHO),仅中国就有近9000万慢性乙肝患者需要抗病毒治疗。
目前FDA共批准了8个抗乙肝药物上市,可以分为两大类:干扰素和核苷(酸)类似物。干扰素类包括干扰素α-2b和聚乙二醇(PEG)化的干扰素α-2a。干扰素通过与宿主细胞表面受体作用使细胞产生抗病毒蛋白,从而抑制乙肝病毒的复制,它的缺点是有效应答率较低和严重的副作用并且需长期注射给药。口服核苷(酸)类似物包括拉米夫定、阿德福韦酯、恩替卡韦、替比夫定、替诺福韦酯和替诺福韦艾拉酚胺。核苷(酸)类似物主要通过抑制病毒多聚酶(逆转录酶)的复制来发挥作用,该类药物的缺点是长期服用易使病毒变异而产生耐药性(Jia et al.Future Med.Chem.,2015,7,587-607)。并且这些药物对HBsAg的清除(乙肝功能性治愈的标志之一(Revill et al.Nat.Rev.Gastroenterol.Hepatol.,2016,13,239-248))都收效甚微(Janssen et al.Lancet,2005,365,123-129;Marcellin etal.N.Engl.J.Med.,2004,351,1206-1217;Buster et al.Hepatology,2007,46,388-394)。因此乙肝患者迫切需要更为有效、更为安全的新药。
HBsAg分为大(L)、中(M)、小(S)三种亚型,它们是感染性病毒颗粒(Daneparticles)和亚病毒颗粒(Subviral particles,SVPs)胞膜的重要组成部分,不仅在病毒进入细胞和分泌新的病毒颗粒中起重要作用,而且在慢性乙肝患者血清中,HBsAg组装成的SVPs数可达1013/mL,含量为感染性颗粒的10万倍,如此大量且持续的SVPs表达,可能通过中和乙肝表面抗体等机制抑制机体对HBV特异性免疫反应(Cornberg et al.J.Hepatol.,2017,66,398-411)。因此HBsAg分泌抑制剂有良好的治疗HBV的应用前景。
最近Replicor公布的临床数据显示HBsAg分泌抑制剂Rep-2139和Rep-2165(核酸多聚物(NAPs))与干扰素和替诺福韦酯(TDF)联用可显著降低血清HBsAg水平,并伴随表面抗体Anti-HBs的增加,实现了对乙肝病毒的功能性控制(AASLD:The LiverMeeting.Washington,DC,October 20-24,2017.AbstractLB-24.)。但是因为需要长期注射给药必将限制患者的顺从性。
其它报道的HBsAg分泌抑制剂包含体外活性在微摩尔级别的四氮唑并嘧啶类小分子化合物HBF-0259(Dougherty et al.Antimicrob.Agents Ch.,2007,51,4427-4437)、优化HBF-0259得到的化学稳定性更好的三氮唑并嘧啶衍生物PBHBV-001和PBHBV-2-15(Yu etal.J.Med.Chem.,2011,54,5660-5670)及苯并咪唑衍生物BM-601(Xu etal.Antivir.Res.,2014,107,6-15),体外活性高至纳摩尔级别的罗氏专利中的化合物(WO2016107832)。
发明内容
本发明提供新的、高活性的HBsAg分泌抑制剂,其可以单独或与其它药物联用治疗乙型肝炎。
本发明的一个方面提供式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药:

其中:
R1选自C6-C14芳基和5-14元杂芳基,所述C6-C14芳基和5-14元杂芳基可任选地被取代基取代;
R2和R3各自独立地选自氢和未取代的C1-C6烷基,或者R2和R3和其所连接的碳原子一起形成3-7元环烷基;
R4选自-C(O)R5、-CO2R6、-C(O)NR7SO2R8、-C(O)NR9R10、可任选地被取代基取代的5元杂芳基,以及

其中:R5和R8各自独立地选自C1-C6烷基和C3-C7环烷基,所述C1-C6烷基和C3-C7环烷基可任选地被取代基取代;
R6选自氢、C1-C6烷基和C3-C7环烷基,所述C1-C6烷基和C3-C7环烷基可任选地被取代基取代;
R7、R9和R10各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基和4-7元杂环基,或者R9和R10和其所连接的氮原子一起形成4-7元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基和4-7元杂环基可任选地被取代基取代;
R11和R16各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基、C6-C14芳基、5-14元杂芳基和4-10元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基、C6-C14芳基、5-14元杂芳基和4-10元杂环基可任选地被取代基取代;
R12、R13、R14和R15各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷氧基、C1-C6烷基-OC1-C6烷基和4-7元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷氧基、C1-C6烷基-OC1-C6烷基和4-7元杂环基可任选地被取代基取代;
X和Y中之一为N,另一个选自CH和N;
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、C3-C7环烷基、氰基、硝基、-C(O)R5、-C(O)OR6、-NR7SO2R8、-SO2R8、-C(O)NR9R10、-SO2NR9R10、-NR9R10、-NR9C(O)R10、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基、羟基烷基、C1-C6烷基-NR9R10、C1-C6烷基-C(O)NR9R10、C1-C6烷基-NR9C(O)R10、芳基、杂芳基和杂环基;
条件是:当R4为任选地被取代基取代的5元杂芳基时,X和Y不同时为N。
本发明的另一方面提供药物组合物,其包含本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,以及一种或多种药学上可接受的载体。
本发明的另一方面提供本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药在制备用于治疗与HBsAg分泌过度相关的疾病的药物中的用途。
本发明的另一方面提供本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药或者本发明的药物组合物,其用于治疗与HBsAg分泌过度相关的疾病。
本发明的另一方面提供治疗与HBsAg分泌过度相关的疾病的方法,所述方法包括向需要其的个体给药有效量的本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药或者本发明的药物组合物,并任选地包括联合给药其他治疗与HBsAg分泌过度相关的疾病或病症的药剂。
本发明的另一方面提供本发明的化合物的制备方法。
化合物和制备方法
本发明的第一方面提供式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药:

其中:
R1选自C6-C14芳基和5-14元杂芳基,所述C6-C14芳基和5-14元杂芳基可任选地被取代基取代;
R2和R3各自独立地选自氢和未取代的C1-C6烷基,或者R2和R3和其所连接的碳原子一起形成3-7元环烷基;
R4选自-C(O)R5、-CO2R6、-C(O)NR7SO2R8、-C(O)NR9R10、可任选地被取代基取代的5元杂芳基,以及

其中:R5和R8各自独立地选自C1-C6烷基和C3-C7环烷基,所述C1-C6烷基和C3-C7环烷基可任选地被取代基取代;
R6选自氢、C1-C6烷基和C3-C7环烷基,所述C1-C6烷基和C3-C7环烷基可任选地被取代基取代;
R7、R9和R10各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基和4-7元杂环基,或者R9和R10和其所连接的氮原子一起形成4-7元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基和4-7元杂环基可任选地被取代基取代;
R11和R16各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基、C6-C14芳基、5-14元杂芳基和4-10元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基、C6-C14芳基、5-14元杂芳基和4-10元杂环基可任选地被取代基取代;
R12、R13、R14和R15各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷氧基、C1-C6烷基-OC1-C6烷基和4-7元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷氧基、C1-C6烷基-OC1-C6烷基和4-7元杂环基可任选地被取代基取代;
X和Y中之一为N,另一个选自CH和N;
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、C3-C7环烷基、氰基、硝基、-C(O)R5、-C(O)OR6、-NR7SO2R8、-SO2R8、-C(O)NR9R10、-SO2NR9R10、-NR9R10、-NR9C(O)R10、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基、羟基烷基、C1-C6烷基-NR9R10、C1-C6烷基-C(O)NR9R10、C1-C6烷基-NR9C(O)R10、芳基、杂芳基和杂环基。
在一些实施方案中,当R4为任选地被取代基取代的5元杂芳基时,X和Y不同时为N。
在一些实施方案中,本发明提供式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药:

其中:
R1选自C6-C14芳基和5-14元杂芳基,所述C6-C14芳基和5-14元杂芳基可任选地被取代基取代;
R2和R3各自独立地选自氢和未取代的C1-C6烷基,或R2和R3和其所连接的碳原子一起形成3-7元环烷基;
R4选自-C(O)R5、-CO2R6、-C(O)NR7SO2R8、-C(O)NR9R10、任选地被取代基取代的5元杂芳基,以及

其中:R5和R8各自独立地选自C1-C6烷基和C3-C7环烷基,所述C1-C6烷基和C3-C7环烷基可任选地被取代基取代;
R6选自氢、C1-C6烷基和C3-C7环烷基,所述C1-C6烷基和C3-C7环烷基可任选地被取代基取代;
R7、R9和R10各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基和4-7元杂环基,或者R9和R10和其所连接的氮原子一起形成4-7元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基和4-7元杂环基可任选地被取代基取代;
R11和R16各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基、C6-C14芳基、5-14元杂芳基和4-10元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷基-OC1-C6烷基、C6-C14芳基、5-14元杂芳基和4-10元杂环基可任选地被取代基取代;
R12、R13、R14和R15各自独立地选自氢、C1-C6烷基、C3-C7环烷基、C1-C6烷氧基、C1-C6烷基-OC1-C6烷基和4-7元杂环基,所述C1-C6烷基、C3-C7环烷基、C1-C6烷氧基、C1-C6烷基-OC1-C6烷基和4-7元杂环基可任选地被取代基取代;
X和Y中之一为N,另一个选自CH和N;
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C3-C7环烷基、氰基、硝基、-C(O)R5、-C(O)OR6、-NR7SO2R8、-SO2R8、-C(O)NR9R10、-SO2NR9R10、-NR9R10、-NR9C(O)R10、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基、羟基烷基、C1-C6烷基-NR9R10、C1-C6烷基-C(O)NR9R10、C1-C6烷基-NR9C(O)R10、芳基、杂芳基和杂环基。
在一些实施方案中,当R4为任选地被取代基取代的5元杂芳基时,X和Y不同时为N。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R1选自C6-C10芳基和5-10元杂芳基,所述C6-C10芳基和5-10元杂芳基可任选地被一个或多个(例如2个或3个)选自下列基团的取代基取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、卤代C1-C3烷氧基、-SO2R8、-C(O)NR9R10、-OC1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OH、-NR9R10和杂环基;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、卤代C1-C3烷氧基、-C(O)NH2、-SO2CH3、-OC1-C3烷基-OC1-C3烷基、-OC1-C3烷基-OH、-NR9R10和杂环基;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代:氟、氯、溴、甲氧基、乙氧基、丙氧基、异丙氧基、氟甲氧基、二氟甲氧基、三氟甲氧基、甲基、乙基、丙基、异丙基、氰基、氟甲基、二氟甲基、三氟甲基、羟基甲氧基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2、-SO2CH3、-NR9R10和5至10元螺杂环基;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个独立地选自下列基团组的取代基取代:
氟、氯、溴、甲氧基、乙氧基、异丙氧基、氟甲氧基、二氟甲氧基、三氟甲氧基、甲基、氰基、三氟甲基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2和-SO2CH3;
-NR9R10,其中R9和R10和其所连接的氮原子一起形成可任选地被取代基取代的4-6元杂环基,其中:
优选地,所述4-6元杂环基选自可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代的 羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基和羟基C1-C10烷基;
羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基和羟基C1-C10烷基;
更优选地,所述4-6元杂环基选自可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代的 羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和羟基C1-C3烷基-;
羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和羟基C1-C3烷基-;
更优选地,所述4-6元杂环基选自可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代的 羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基;
羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基;
更优选地,所述4-6元杂环基选自

 以及
以及
9至10元含氮螺杂环基,优选 更优选/>
更优选/>
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R1选自C6-C10芳基和5-10元杂芳基,所述C6-C10芳基和5-10元杂芳基可任选地被一个或多个下列基团取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、-SO2R8、-C(O)NR9R10、-OC1-C6烷基-OC1-C6烷基或-OC1-C6烷基-OH;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个下列基团取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、-C(O)NH2、-SO2CH3、-OC1-C3烷基-OC1-C3烷基或-OC1-C3烷基-OH;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个下列基团取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、-C(O)NH2、-SO2CH3、-OC1-C3烷基-OC1-C3烷基或-OC1-C3烷基-OH;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个下列基团取代:氟、氯、溴、甲氧基、乙氧基、异丙氧基、甲基、氰基、三氟甲基、一氟甲氧基、二氟甲氧基、三氟甲氧基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2或-SO2CH3;
优选地,R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被一个或多个下列基团取代:氟、甲氧基、甲基、氰基、三氟甲基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2或-SO2CH3。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R2和R3各自独立地选自氢和C1-C3烷基,或者R2和R3和其所连接的碳原子一起形成3-7元环烷基;
优选地,R2和R3各自独立地选自氢、甲基和乙基,或者R2和R3和其所连接的碳原子一起形成环丙基;
更优选地,R2和R3各自独立地选自氢和甲基,或者R2和R3和其所连接的碳原子一起形成环丙基;
特别优选地,R2和R3各自独立地选自氢和甲基。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R4选自-C(O)C1-C6烷基、-C(O)OC1-C6烷基、-C(O)OH、-C(O)NH2、-C(O)NH(C1-C6烷基)、-C(O)NH-SO2-C1-C6烷基、-C(O)N(C1-C6烷基)-SO2-C1-C6烷基,任选地被一个或多个独立地选自C1-C6烷基的取代基取代的5元杂芳基(例如四唑基、噻唑基、咪唑基、三唑基和噁唑基),以及
在优选的实施方案中,R4选自-C(O)C1-C3烷基、-C(O)OC1-C3烷基、-C(O)OH、-C(O)NH2、-C(O)NH-SO2-C1-C3烷基、

在优选的实施方案中,R4选自-C(O)OCH3、-C(O)OEt、-C(O)OH、-C(O)NH2、-C(O)CH3、-C(O)Et、-C(O)NH-SO2-CH3、-C(O)NH-SO2-Et、

在优选的实施方案中,R4选自-C(O)OEt、-C(O)OH、-C(O)NH2、-C(O)CH3、-C(O)NH-SO2-CH3、

在优选的实施方案中,R4选自-C(O)OEt、-C(O)OH、-C(O)NH2、-C(O)CH3和-C(O)NH-SO2-CH3。
在优选的实施方案中,R4选自-C(O)OEt和-C(O)OH,更优选地,R4为-C(O)OH。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R5和R8选自C1-C3烷基和C3-C5环烷基,所述C1-C3烷基和C3-C5环烷基可任选地被取代基取代;优选地,R5和R8为甲基。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R6选自氢、C1-C3烷基和C3-C5环烷基,所述C1-C3烷基和C3-C5环烷基可任选地被取代基取代;
优选地,R6为氢、甲基或乙基;更优选地,R6为氢或乙基;特别优选地,R6为氢。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R7、R9和R10各自独立地选自氢、C1-C3烷基、C3-C5环烷基、C1-C3烷基-OC1-C3烷基和4-6元杂环基,或者R9和R10和其所连接的氮原子一起形成4-6元杂环基,所述C1-C3烷基、C3-C5环烷基、C1-C3烷基-OC1-C3烷基或4-6元杂环基可任选地被取代基取代;
优选地,R7、R9和R10为氢;
优选地,R9和R10和其所连接的氮原子一起形成可任选地被取代基取代的4-6元杂环基,
更优选地,所述4-6元杂环基选自可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代的 羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基和羟基C1-C10烷基;
羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基和羟基C1-C10烷基;
更优选地,所述4-6元杂环基选自可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代的 羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和OH-C1-C3烷基-;
羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和OH-C1-C3烷基-;
更优选地,所述4-6元杂环基选自可任选地被一个或多个(例如2个或3个)独立地选自下列基团的取代基取代的 羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基;
羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基;
更优选地,所述4-6元杂环基选自

在一些实施方案中,本发明提供如上文所述的式I的化合物或其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药,其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被选自下列基团的取代基取代:
5至10元含氮螺杂环基,优选 和/>
和/> 更优选/>
更优选/>
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R11和R16各自独立地选自氢、C1-C3烷基、C3-C5环烷基、C1-C3烷基-OC1-C3烷基、C6-C14芳基、5-14元杂芳基和4-6元杂环基,所述C1-C3烷基、C3-C5环烷基、C1-C3烷基-OC1-C3烷基、C6-C14芳基、5-14元杂芳基和4-6元杂环基可任选地被取代基取代;
优选地,R11和R16各自独立地选自氢、C1-C3烷基、5-10元杂芳基和4-6元杂环基;
优选地,R11和R16各自独立地选自氢和甲基。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
R12、R13、R14和R15各自独立地选自氢、C1-C3烷基、C3-C5环烷基、C1-C3烷氧基、C1-C3烷基-OC1-C3烷基和4-6元杂环基,所述C1-C3烷基、C3-C5环烷基、C1-C3烷氧基、C1-C3烷基-OC1-C3烷基和4-6元杂环基可任选地被取代基取代;
优选地,R12、R13、R14和R15均为氢。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
X为N,Y为CH;或者
X为CH,Y为N;或者
X为N,Y为N。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中:
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、C3-C7环烷基、氰基、硝基、-C(O)C1-C6烷基、-C(O)OH、-C(O)OC1-C6烷基、-NHSO2C1-C6烷基、-N(C1-C6烷基)SO2C1-C6烷基、-SO2C1-C6烷基、-C(O)NH2、-C(O)NH(C1-C6烷基)、-SO2NH2、-SO2NH(C1-C6烷基)、-NH2、-NH(C1-C6烷基)、如上文关于R9和R10所定义的由二者与其所连接的N原子一起形成的4-7元杂环基、-NHC(O)C1-C6烷基、-N(C1-C6烷基)C(O)C1-C6烷基、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、被羟基取代的-OC1-C6烷基-OC1-C6烷基、羟基C1-C6烷基、C1-C6烷基-NH2、C1-C6烷基-NH(C1-C6烷基)、C1-C6烷基-C(O)NH2、C1-C6烷基-C(O)NH(C1-C6烷基)、C1-C6烷基-NHC(O)C1-C6烷基、C1-C6烷基-N(C1-C6烷基)C(O)C1-C6烷基、芳基、杂芳基和5-10元螺杂环基;
优选地,所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、-OC1-C3烷基-OH、卤代C1-C3烷氧基、C3-C6环烷基、氰基、硝基、-C(O)C1-C3烷基、-C(O)OH、-C(O)OC1-C3烷基、-NHSO2C1-C3烷基、-N(C1-C3烷基)SO2C1-C3烷基、-SO2C1-C3烷基、-C(O)NH2、-C(O)NH(C1-C3烷基)、-SO2NH2、-SO2NH(C1-C3烷基)、-NH2、-NH(C1-C3烷基)、如上文关于R9和R10所定义的由二者与其所连接的N原子一起形成的4-6元杂环基、-NHC(O)C1-C3烷基、-N(C1-C3烷基)C(O)C1-C3烷基、-C1-C3烷基-OC1-C3烷基、-OC1-C3烷基-OC1-C3烷基、被羟基取代的-OC1-C3烷基-OC1-C3烷基、羟基C1-C3烷基、C1-C3烷基-NH2、C1-C3烷基-NH(C1-C3烷基)、C1-C3烷基-C(O)NH2、C1-C3烷基-C(O)NH(C1-C3烷基)、C1-C3烷基-NHC(O)C1-C3烷基、C1-C3烷基-N(C1-C3烷基)C(O)C1-C3烷基和9至10元含氮螺杂环基;
优选地,所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基;氟、氯、溴;甲基、乙基、异丙基;氟甲基、二氟甲基、三氟甲基;甲氧基、乙氧基、异丙氧基;-OCH2OH、-OCH2CH3OH;氟甲氧基、二氟甲氧基、三氟甲氧基;环丙基、环丁基、环戊基、环己基;氰基;硝基;-C(O)CH3、-C(O)CH2CH3;-C(O)OH、-C(O)OCH3、-C(O)OCH2CH3;-NHSO2CH3、-NHSO2CH2CH3、-N(CH3)SO2CH3、-N(CH3)SO2CH2CH3;-SO2CH3、-SO2CH2CH3;-C(O)NH2、-C(O)NHCH3、-C(O)NHCH2CH3;-SO2NH2、-SO2NHCH3、-SO2NHCH2CH3;-NH2、-NHCH3、-NHCH2CH3;如上文关于R9和R10所定义的由二者与其所连接的N原子一起形成的4-6元杂环基;-NHC(O)CH3、-NHC(O)CH2CH3、-N(CH3)C(O)CH3、-N(CH3)C(O)CH2CH3;-CH2-OCH3、-CH2CH2-OCH3、-CH2-OCH2CH3、-CH2CH2-OCH2CH3;-OCH2-OCH3、-OCH2-OCH2CH3、-OCH2CH2-OCH3;被羟基取代的-OCH2-OCH3、-OCH2-OCH2CH3或-OCH2CH2-OCH3;-CH2OH、-CH2CH2OH;-CH2NH2、-CH2CH2NH2、-CH2-NH(CH3)、-CH2-NH(CH2CH3)、-CH2CH2-NH(CH3)、-CH2CH2-NH(CH2CH3);-CH2C(O)NH2、-CH2C(O)NH(CH3)、-CH2C(O)NH(CH2CH3)、-CH2CH2C(O)NH(CH3)、-CH2CH2C(O)NH(CH2CH3);-CH2-NHC(O)CH3、-CH2-N(CH3)C(O)CH3、-CH2-N(CH3)C(O)CH2CH3;和9至10元含氮螺杂环基;
优选地,所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:氟、氯、溴;甲基、乙基、异丙基;氟甲基、二氟甲基、三氟甲基;甲氧基、乙氧基、异丙氧基;-OCH2OH、-OCH2CH3OH;氟甲氧基、二氟甲氧基、三氟甲氧基;氰基;-SO2CH3、-SO2CH2CH3;-C(O)NH2、-C(O)NHCH3、-C(O)NHCH2CH3;如上文关于R9和R10所定义的由二者与其所连接的N原子一起形成的4-6元杂环基;-CH2-OCH3、-CH2CH2-OCH3、-CH2-OCH2CH3、-CH2CH2-OCH2CH3;-OCH2-OCH3、-OCH2-OCH2CH3、-OCH2CH2-OCH3;-CH2OH、-CH2CH2OH;以及
优选地,所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:氟、氯、溴;甲基;三氟甲基;甲氧基、乙氧基、异丙氧基;-OCH2CH3OH;氟甲氧基、二氟甲氧基、三氟甲氧基;氰基;-SO2CH3;-C(O)NH2;如上文关于R9和R10所定义的由二者与其所连接的N原子一起形成的4-6元杂环基;-CH2-OCH3;-OCH2CH2-OCH3;-CH2OH;以及
本发明涵盖对上述优选基团进行任意组合所得的式I的化合物。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中所述化合物为式II的化合物:

其中X、Y、R1和R4如上文对于式I所定义。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中所述化合物为式III的化合物:

其中R1和R4如上文对于式I所定义;
条件是:R4不为任选地被取代基取代的5元杂芳基。
在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中所述化合物为式IV的化合物:

其中:
R1和R4如上文对于式I所定义;
R2为未取代的C1-C6烷基,优选未取代的C1-C3烷基,更优选甲基或乙基,更优选甲基;且
R3为氢或未取代的C1-C6烷基(优选未取代的C1-C3烷基,更优选甲基或乙基);优选为H或甲基;
或者
R2和R3和其所连接的碳原子一起形成3-7元环烷基,优选3-6元环烷基,例如环丙基;
特别地,所述化合物是式V或VI的化合物:

在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中所述化合物为式VII的化合物:

其中:
R4如上文对于式I所定义;
W1、W2和W3之中一个为N,另外两个为CR17;
m为0、1、2、3或4;
R17各自独立地选自下列基团:
(1)氢、卤素(例如氟、氯和溴)、C1-C6烷基和C1-C6烷氧基;
(2)-NR9R10,其中R9和R10和其所连接的氮原子一起形成任选地被取代基取代的4-6元杂环基,如上文对于式I的R9和R10所定义;以及
(3)5至10元螺杂环基,优选9至10元含氮螺杂环基,更优选 特别优选/>
特别优选/>
在优选的式VII的化合物的实施方案中,R4选自-C(O)OEt、-C(O)OH、-C(O)NH2、-C(O)CH3和-C(O)NH-SO2-CH3;
R17各自独立地选自下列基团:
(1)氢、卤素(例如氟、氯和溴)、C1-C3烷基(例如甲基、乙基和丙基)和C1-C3烷氧基(例如甲氧基和乙氧基);
(2)-NR9R10,其中R9和R10和其所连接的氮原子一起形成任选地被取代基取代的4-6元杂环基,如上文对于式I的R9和R10所定义;以及
(3) 优选/>
优选/>
在优选的式VII的化合物的实施方案中,R4为-C(O)OH;
R17各自独立地选自氢、甲基、甲氧基、氟、


在一些实施方案中,本发明提供如上文所述的式I的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,其中所述化合物为式VIII的化合物:

其中:
R3为氢或未取代的C1-C6烷基(优选未取代的C1-C3烷基,更优选甲基或乙基);优选为H或甲基;
R4如上文对于式I所定义;
R18、R19、R20和R21各自独立地选自H、卤素、氰基、卤代C1-C6烷基、C1-C6烷基、-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、-C(O)R5、-C(O)OR6、-NR7SO2R8、-SO2R8、-C(O)NR9R10、-SO2NR9R10、-NR9R10和-NR9C(O)R10;并且
R5、R6、R7、R8、R9和R10各自如上文对于式I所定义。
在优选的式VIII的化合物的实施方案中,R4选自-C(O)OEt、-C(O)OH、-C(O)NH2、-C(O)CH3和-C(O)NH-SO2-CH3;优选地,R4选自-C(O)OEt和-C(O)OH。
在优选的式VIII的化合物的实施方案中,R18、R19、R20和R21各自独立地选自H、卤素、氰基、卤代C1-C3烷基、C1-C3烷基、-OC1-C3烷基、-OC1-C3烷基-OC1-C3烷基、-OC1-C3烷基-OH、卤代C1-C3烷氧基和-S(O)2-C1-C3烷基。
在优选的式VIII的化合物的实施方案中,R18、R19、R20和R21各自独立地选自H、氟、氯、溴、氰基、三氟甲基、甲基、甲氧基、乙氧基、异丙氧基、-O-CH2-CH2-OMe、-O-CH2CH2-OH、三氟甲氧基、氟甲氧基、二氟甲氧基和-S(O)2CH3。
在优选的式VIII的化合物的实施方案中,R4选自-C(O)OEt和-C(O)OH;
R18选自H、氟、氯、甲基、氰基和三氟甲基;
R19选自H、氟、氯、甲氧基和甲基;
R20选自H、氟、氯、三氟甲基、甲基、氰基和-S(O)2CH3;
R21选自H、氟、氯、溴、甲氧基、乙氧基、-O-CH2-CH2-OMe、三氟甲基、-O-CH2-CH2-OH、氰基、-C(O)NH2、三氟甲氧基、异丙氧基、二氟甲氧基、氟甲氧基和甲基。
在一些实施方案中,本发明提供如上文所述的化合物或其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药,其中所述化合物选自:




本发明第二方面提供制备本发明的化合物的方法。
在一些实施方案中,本发明提供制备式II的化合物的方法:

其中:
R4选自-C(O)R5、-CO2R6、-C(O)NR7SO2R8和-C(O)NR9R10;
X、Y、R1、R5、R6、R7、R8、R9和R10如上文对于式I所定义;
所述方法包括以下步骤:

第一步:化合物II-1经还原反应生成化合物II-2,其中Ra为C1-C4烷基,例如甲基、乙基或异丙基,优选乙基;
例如,化合物II-1在还原剂(例如,氢气、硼氢化钠或硼氢化锂)和催化剂(例如Pd/C等)存在下,于极性质子溶剂(例如甲醇或乙醇等)中,在室温至90℃下经还原反应生成化合物II-2。
第二步:化合物II-2与R1-L经偶联或亲核取代反应生成化合物II-3,其中,其中L为离去基团,例如卤素(例如F、Cl、Br或I)或三氟甲磺酰氧基(OTf)等;
例如,化合物II-2在碱(例如碳酸钠、碳酸钾、碳酸铯或乙酸钾等)和配体(例如RuPhos、BINAP或SPhos等)的存在下,在钯催化剂(例如Pd(PPh3)4、Pd(dppf)Cl2、Pd(OAc)2或Pd2(dba)3)催化下,于惰性气体(例如N2)保护和室温至100℃下,于非极性溶剂(例如甲苯或二甲苯等)中,与R1-L经偶联反应生成化合物II-3;或者,化合物II-2在碱(例如碳酸钾、N,N-二异丙基乙胺(DIPEA)或叔丁醇钾等)的存在下,于高沸点溶剂(例如N,N-二甲基甲酰胺(DMF)或N-甲基吡咯烷酮(NMP)等)中,在100℃至160℃下,与R1-L经亲核取代反应生成化合物II-3。
第三步:化合物II-3经合适的反应转化成所述式II的目标化合物。
在一些实施方案中,本发明提供制备式II的化合物的方法:

其中:
R4选自任选地被取代基取代的5元杂芳基以及
X、Y、R1、R11、R12、R13、R14、R15和R16如上文对于式I所定义;
所述方法包括以下步骤:

第一步:化合物II-4经还原反应生成化合物II-5,其中PG1为合适的羟基保护基(例如C1-C3烷基,例如甲基、乙基或异丙基,优选甲基);
例如,化合物II-4在还原剂(例如氢气、硼氢化钠或硼氢化锂)和催化剂(例如Pd/C等)存在下,于极性质子溶剂(例如甲醇或乙醇等)中,在室温至90℃下经还原反应生成化合物II-5。
第二步:化合物II-5与R1-L经偶联或亲核取代反应生成化合物II-6,其中L为离去基团,例如卤素(例如F、Cl、Br或I)或OTf;
化合物II-5在碱(例如碳酸钠、碳酸钾、碳酸铯或乙酸钾等)和配体(例如RuPhos、BINAP或SPhos等)的存在下,在催化剂(例如Pd(PPh3)4、Pd(dppf)Cl2、Pd(OAc)2或Pd2(dba)3)催化下,于惰性气体(例如N2)保护和室温至100℃下,于非极性溶剂(例如甲苯或二甲苯等)中,与R1-L经偶联反应生成化合物II-6;或者,化合物II-5在碱(例如碳酸钾、DIPEA或叔丁醇钾等)的存在下,于高沸点溶剂(例如DMF或NMP等)中,在100℃至160℃下,与R1-L经亲核取代反应生成化合物II-6。
第三步:化合物II-6经脱保护反应生成化合物II-7;
例如,化合物II-6在质子酸(例如盐酸、氢溴酸等)或路易斯酸(例如三溴化硼乙醚溶液或三氯化铝等)存在下,在0℃至90℃下经脱保护反应(例如脱甲基化)生成化合物II-7。
第四步:使化合物II-7转化成化合物II-8,其中PG2为适合的羟基保护基(例如三氟甲磺酰基(Tf));
例如,化合物II-7在室温下与Tf2O反应生成化合物其中PG2为Tf的化合物II-8。
第五步:化合物II-8与R4-硼酸或R4-硼酸酯经偶联反应生成式II的化合物;
例如,化合物II-8在碱(例如碳酸钠、碳酸钾、碳酸铯或乙酸钾)的存在下,在钯催化剂(例如Pd(PPh3)4、Pd(dppf)Cl2或Pd2(dba)3等)催化下,在惰性气体(例如N2)保护和室温至120℃下,于非质子溶剂(例如1,4二氧六环、甲苯或二甲苯等)中与R4-硼酸或R4-硼酸酯偶联反应生成式II的化合物。
在一些实施方案中,本发明提供制备式V的化合物的方法:

其中:
R4选自-C(O)R5、-CO2R6、-C(O)NR7SO2R8和-C(O)NR9R10;
R1、R5、R6、R7、R8、R9和R10如上文对于式I所定义;
所述方法包括以下步骤:

第一步:化合物V-1与适合的有机锡化合物(如三丁基乙烯基锡)经偶联反应生成化合物V-2,其中Ra在每次出现时独立地选自C1-C4烷基,例如甲基、乙基或异丙基,优选乙基;Rb为离去基团,例如卤素(例如Cl、Br或I)或OTf;
例如,化合物V-1在配体(例如RuPhos、BINAP或SPHos等)的存在下,在钯催化剂(例如Pd(PPh3)4、Pd(dppf)Cl2、Pd(OAc)2或Pd2(dba)3)催化下,于惰性气体(例如N2)保护和室温至100℃下,于极性非质子溶剂(例如DMF等)中,与三丁基乙烯基锡经偶联反应生成化合物V-2。
第二步:化合物V-2经环化反应生成化合物V-3;
例如,化合物V-2与氯化铵(NH4Cl)于醋酸中在回流条件下反应生成化合物V-3。
第三步:使化合物V-3转化成化合物V-4,其中PG3为适合的氨基保护基,例如苄基、对甲苯磺酰基(Ts)、苯甲酰基、苄氧羰基(Cbz)、烯丙氧羰基(Alloc)、甲氧羰基、乙氧羰基或叔丁氧羰基(Boc)等;
例如,化合物V-3中NH基团经保护基PG3(例如苄基、Boc等)保护从而生成化合物V-4。
第四步:化合物V-4经Kulinkovich反应生成化合物V-5;
例如,该反应中使用的烷氧基肽催化剂可以是Ti(OiPr)4,所用的格式试剂可以是EtMgBr。
第五步:化合物V-5经脱保护反应生成化合物V-6;
第六步:化合物V-6与R1-L经偶联或亲核取代反应生成化合物V-7,其中L为离去基团,例如卤素(例如F、Cl、Br或I)或OTf等;
例如,化合物V-6在碱(例如碳酸钠、碳酸钾、碳酸铯或乙酸钾等)和配体(例如RuPhos、BINAP或SPHos等)的存在下,在钯催化剂(例如Pd(PPh3)4、Pd(dppf)Cl2、Pd(OAc)2或Pd2(dba)3)催化下,在惰性气体(例如N2)保护和室温至100℃下,于非极性溶剂(例如甲苯或二甲苯等)中,与R1-L经偶联反应生成化合物V-7;或者,化合物V-6在碱(例如碳酸钾、DIPEA或叔丁醇钾等)的存在下,于高沸点溶剂(例如DMF或NMP等)中,在100℃至160℃下,与R1-L经亲核取代反应生成化合物V-7。
第七步:化合物V-7经合适的反应转化成所述式V的目标化合物。
在一些实施方案中,本发明提供制备式VI的化合物的方法:

其中:
R4选自-C(O)R5、-CO2R6、-C(O)NR7SO2R8和-C(O)NR9R10;
R1、R5、R6、R7、R8、R9和R10如上文对于式I所定义;
所述方法包括以下步骤:

第一步:化合物VI-1与NC-C(O)ORa经[2+2+2]环加成反应生成化合物VI-2,其中PG3为氨基保护基,例如Ts、苯甲酰基、Cbz、Alloc、甲氧羰基、乙氧羰基或Boc,特别是Ts;Ra为C1-C4烷基,例如甲基、乙基或异丙基,优选甲基或乙基;
例如,化合物VI-1与氰基甲酸乙酯在适合的铑催化剂(例如Rh(COD)2BF4等)和配体(例如RuPhos、BINAP或SPHos等)存在下,于适合的溶剂(例如二氯甲烷(DCM)、1,2-二氯乙烷、或DCM与水的化合物等)中,在室温至80℃下发生[2+2+2]环加成反应生成化合物VI-2。
第二步:化合物VI-2经脱保护反应生成化合物VI-3;
例如,化合物VI-2在适合的酸(例如氢溴酸)的存在下,在80℃至120℃下经脱保护反应生成化合物VI-3。
第三步:化合物VI-3与R1-L发生偶联反应或亲核取代反应生成化合物VI-4,其中L为离去基团,例如卤素(例如F、Cl、Br或I)或OTf;
例如,化合物VI-3在适合的碱(例如碳酸钠、碳酸钾、碳酸铯或乙酸钾等)和配体(例如RuPhos、BINAP或SPHos等)的存在下,在适合的钯催化剂(例如Pd(PPh3)4、Pd2(dba)3、Pd(dppf)Cl2或Pd(OAc)2等)催化下,于惰性气体(例如N2)保护和室温至100℃下,于适合的非极性溶剂(例如甲苯或二甲苯等)中,与R1-L发生偶联反应生成化合物VI-4;或者,化合物VI-3在适合的碱(例如碳酸钾、DIPEA或叔丁醇钾等)的存在下,于适合的高沸点有机溶剂(例如DMF或NMP等)中,在100℃至160℃下,与R1-L发生亲核取代反应生成化合物VI-4。
第四步:化合物VI-4经合适的反应转化成所述式VI的目标化合物。
在上文所述的制备式II的化合物的方法的第三步、制备式V的化合物的方法的第七步以及制备式VI的化合物的方法的第四步中,所述合适的反应选自:
(1)经水解生成其中R4为-CO2H的目标化合物;
(2)与醇R6OH发生酯交换生成其中R4为-CO2R6且R6不为H的目标化合物;
(3)与HN(OMe)Me反应生成Weinreb酰胺,然后所得的Weinreb酰胺与格氏试剂R5MgBr反应生成其中R4为-C(O)R5的目标化合物;
(4)水解成酸,然后所得的酸与HNR7SO2R8发生缩合反应生成其中R4为-C(O)NR7SO2R8的目标化合物;和
(5)与HNR9R10发生胺解反应生成其中R4为-C(O)NR9R10的目标化合物,或者发生水解反应形成酸,然后所得的酸与胺HNR9R10缩合生成其中R4为-C(O)NR9R10的目标化合物。
药物组合物、制备方法和治疗方法
本发明的第三方面提供药物组合物,其包含本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,以及一种或多种药学上可接受的载体。
本发明的第四方面提供制备本发明的药物组合物的方法,所述方法包括将本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,以及一种或多种药学上可接受的载体组合。
本发明的第五方面提供药物制剂,其包含本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药或者本发明的药物组合物。
本发明的第六方面提供本发明的化合物或者其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药,本发明的药物组合物或者本发明的药物制剂在制备用于治疗与HBsAg分泌过度相关的疾病的药物中的用途。优选地,所述疾病为乙型肝炎。
本发明的第七方面提供本发明的化合物或其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物、前药,本发明的药物组合物或者本发明的药物制剂,其用于治疗与HBsAg分泌过度相关的疾病。优选地,所述疾病为乙型肝炎。
本发明的第八方面提供治疗与HBsAg分泌过度相关的疾病的方法,所述方法包括向需要其的个体给药有效量的本发明的化合物或其立体异构体、互变异构体、药学上可接受的盐、多晶型物、共晶物、溶剂合物、代谢物或前药,本发明的药物组合物或者本发明的药物制剂,并任选地包括联合给药其他与治疗与HBsAg分泌过度相关的疾病或病症的药剂。优选地,所述疾病为乙型肝炎。
定义
除非在下文中另有定义,本文中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。提及本文中使用的技术意图指在本领域中通常所理解的技术,包括那些对本领域技术人员显而易见的技术的变化或等效技术的替换。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。
如本文中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”及其在本文中的其它变体形式为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。
如本文中所使用,术语“烷基”定义为直链或支链的饱和脂肪族烃基。在一些实施方案中,烷基具有1至10个碳原子,例如1至8个碳原子(C1-C8烷基)、1至6个碳原子(C1-C6烷基)、1至4个碳原子(C1-C4烷基)、1至3个碳原子(C1-C3烷基)、2至6个碳原子(C2-C6烷基)、2至4个碳原子(C2-C4烷基)或3至4个碳原子(C3-C4烷基)。例如,如本文中所使用,术语“C1-C6烷基”指具有1至6个碳原子的直链或支链的基团(例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基或正己基)。在一些实施方案中,所述烷基任选地被一个或多个(诸如1至3个)适合的取代基如卤素取代(此时该基团被称作“卤代烷基”,例如-CF3、-C2F5、-CHF2、-CH2F、-CH2CF3、-CH2Cl或-CH2CH2CF3等)。
如本文中所使用,术语“环烷基”指饱和或不饱和的非芳族单环或多环(诸如双环)烃环(例如单环,诸如环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基,或双环,包括螺环、稠合或桥连系统(诸如双环[1.1.1]戊基、双环[2.2.1]庚基等),其任选地被一个或多个(诸如1至3个)适合的取代基取代。所述环烷基具有3至15个,例如3至10个碳原子、3至7个碳原子、3至6个碳原子、3至5个碳原子、5至7个碳原子、4至6个碳原子或5至6个碳原子等。例如,如本文中所使用,术语“C3-C7环烷基”指具有3至7个成环碳原子的饱和或不饱和的非芳族单环或多环(诸如双环)烃环(例如环丙基、环丁基、环戊基、环己基、环庚基),其任选地被一个或多个(诸如1至3个)适合的取代基取代,例如甲基取代的环丙基。术语“3-7元环烷基”指具有3至7个成环原子的饱和或不饱和的非芳族单环或多环(诸如双环)烃环(例如环丙基、环丁基、环戊基、环己基、环庚基),其任选地被一个或多个(诸如1至3个)适合的取代基取代,例如甲基取代的环丙基。
如本文中所使用,术语“芳基”指具有共轭π电子系统的全碳单环或稠合环多环芳族基团。例如,如本文中所使用,术语“C6-C14芳基”意指含有6至14个碳原子的芳族基团,术语“C6-C10芳基”意指含有6至10个碳原子的芳族基团,诸如苯基或萘基。芳基任选地被一个或多个(诸如1至3个)适合的取代基(例如卤素、-OH、-CN、-NO2、C1-C6烷基等)取代。
如本文中所使用,术语“杂芳基”指单环、双环或三环芳族环系,其具有5-14个环原子,特别是具有5、6、7、8、9、10、11、12、13或14个环原子,特别是1、2、3、4、5、6、7、8、9或10个碳原子,且其包含至少一个可以相同或不同的杂原子(例如氧、氮或硫),并且,另外在每一种情况下可为苯并稠合的。例如,如本文中所使用,术语“5-14元杂芳基”意指含有5至14个环原子的杂芳基。杂芳基的具体实例包括但不限于噻吩基、呋喃基、吡咯基、噁唑基、噻唑基、咪唑基、吡唑基、异噁唑基、异噻唑基、三唑基、四唑基、噁二唑基、噻二唑基等,或吡啶基、哒嗪基、嘧啶基、吡嗪基、三嗪基等,或氮杂吲哚,以及它们的苯并衍生物例如吲哚、苯并咪唑、喹啉、异喹啉等。
如本文中所使用,术语“卤代”或“卤素”基团定义为包括F、Cl、Br或I。
如本文中所使用,术语“烷氧基”意指通过氧原子连接至母体分子部分的如上文所定义的烷基。C1-C6烷氧基的代表性实例包括但不限于甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、戊氧基、己氧基等。
如本文中所使用,术语“羟基烷基”意指上文所定义的烷基(例如,具有1个至约10个碳原子的直链或支链烷基基团)中的氢原子中的任何一个或多个被一个或一个以上羟基取代。例如,如本文中所使用,术语“羟基C1-C10烷基”意指C1-C10烷基中的任何一个或多个氢原子被一个或多个羟基取代。这样的基团的例子包括羟基甲基、羟基乙基、羟基丙基、羟基丁基和羟基己基。
如本文中所使用,术语“杂环基”指单环或多环基团,其在环中具有2、3、4、5、6、7、8、9个碳原子和一个或多个(例如1个、2个、3个或4个)选自C(=O)、O、S、S(=O)、S(=O)2和NR(R表示氢原子或取代基,例如但不限于烷基或环烷基)的基团。特别地,3-10元杂环基为在环中具有3-10个碳原子及杂原子的基团,例如,其具有4至10、4至7、4至6、5至6个、5至7、5至9以及5至10碳原子及杂原子(分别称作4至10元、4至7元、4至6元、5至6元、5至7元、5至9元以及5至10元杂环基),例如但不限于环氧乙烷基、氮丙啶基、氮杂环丁基、氧杂环丁基、四氢呋喃基、吡咯烷基、吡咯烷酮基、咪唑烷基、吡唑烷基、四氢吡喃基、哌啶基、吗啉基、二噻烷基(dithianyl)、硫吗啉基、哌嗪基、三噻烷基(trithianyl)等;以及它们的并环衍生物,例如但不限于吡咯烷基并环丙基、环戊基并氮杂环丙基、吡咯烷基并环丁基、吡咯烷基并吡咯烷基、吡咯烷基并哌啶基、吡咯烷基并哌嗪基、吡咯烷基并吗啉基、哌啶基并吗啉基;或螺环衍生物;或苯并衍生物或杂芳基并衍生物,例如但不限于

术语“取代”指所指定的原子上的一个或多个(例如1个、2个、3个或4个)氢被从所指出的基团的选择代替,条件是未超过所指定的原子在当前情况下的正常原子价并且所述取代形成稳定的化合物。取代基和/或变量的组合仅仅当这种组合形成稳定的化合物时才是允许的。
如果基团被描述为“任选地被取代基取代”,则基团可(1)未被取代或(2)被一个或多个取代基取代。如果基团中的碳被描述为任选地被取代基列表中的一个或多个取代,则碳上的一个或多个氢(至存在的任何氢的程度)可单独和/或一起被独立地选择的任选的取代基替代。如果基团中的氮被描述为任选地被取代基列表中的一个或多个取代,则氮上的一个或多个氢(至存在的任何氢的程度)可各自被独立地选择的任选的取代基替代。
如果取代基被描述为“独立地选自”一组,则各取代基独立于另一者被选择。因此,各取代基可与另一(其他)取代基相同或不同。
如果变量或取代基可选自不同的变化方案,并且该变量或取代基不止一次地出现,那么各变化方案可以相同或不同。
如本文中所使用,术语“一个或多个”意指在合理条件下的1个或超过1个,例如2个、3个、4个、5个或10个。
除非指明,否则如本文中所使用,取代基的连接点可来自取代基的任意适宜位置。
本发明还包括所有药学上可接受的同位素化合物,其与本发明的化合物相同,除了一个或多个原子被具有相同原子序数但原子质量或质量数不同于在自然界中占优势的原子质量或质量数的原子替代。适合包含入本发明的化合物中的同位素的实例包括(但不限于)氢的同位素(例如2H、3H);碳的同位素(例如11C、13C及14C);氯的同位素(例如36Cl);氟的同位素(例如18F);碘的同位素(例如123I及125I);氮的同位素(例如13N及15N);氧的同位素(例如15O、17O及18O);磷的同位素(例如32P);及硫的同位素(例如35S)。
术语“立体异构体”表示由于至少一个不对称中心形成的异构体。在具有一个或多个(例如1个、2个、3个或4个)不对称中心的化合物中,其可产生外消旋混合物、单一对映异构体、非对映异构体混合物和单独的非对映异构体。特定个别分子也可以几何异构体(顺式/反式)存在。类似地,本发明的化合物可以两种或更多种处于快速平衡的结构不同的形式的混合物(通常称作互变异构体)存在。互变异构体的代表性实例包括酮-烯醇互变异构体、苯酚-酮互变异构体、亚硝基-肟互变异构体、亚胺-烯胺互变异构体等。例如,二氢嘧啶基团、2(1H)-吡啶酮基等在溶液中可以下列互变异构形式平衡存在。要理解,本申请的范围涵盖所有这样的以任意比例(例如60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%)的异构体或其混合物。
除非另外指明,否则本发明的化合物意欲可以立体异构体(其包括顺式及反式异构体、光学异构体(例如R及S对映异构体)、非对映异构体、几何异构体、旋转异构体、构象异构体、阻转异构体及其混合物)的形式存在。本发明的化合物可表现一种以上类型的异构现象,且由其混合物(例如外消旋混合物及非对映异构体对)组成。
例如在本发明中,式I化合物或其盐以立体异构的形式(例如,其含有一个或多个不对称碳原子)存在时,单独的立体异构体(对映异构体和非对映异构体)以及它们的混合物包括在本发明的范围内。本发明还包括式I表示的化合物或盐的单独异构体,以及与其中一个或多个手心中心反转的异构体的混合物。本发明的范围包括:立体异构体的混合物,以及纯化的对映异构体或对映异构体/非对映异构体富集的混合物。本发明包括所有对映异构体及非对映异构体所有可能的不同组合的立体异构体的混合物。本发明包括上文定义的所有具体基团的立体异构体的全部组合和子集。本发明还包括式I化合物或其盐的几何异构体,所述几何异构体包括顺反异构体。
本文用波浪线“~~”表示的结构图中的键意在表示,该结构表示顺式或反式异构体,或任意比例的顺式和反式异构体的混合物。
本发明涵盖本发明的化合物的所有可能的结晶形式或多晶型物,其可为单一多晶型物或多于一种多晶型物的任意比例的混合物。还应当理解,本发明的某些化合物可以游离形式存在用于治疗,或适当时,以其药学上可接受的衍生物形式存在。在本发明中,药学上可接受的衍生物包括但不限于:药学上可接受的盐、溶剂合物、代谢物或前药,在将它们向需要其的患者给药后,能够直接或间接提供本发明的化合物或其代谢物或残余物。因此,当在本文中提及“本发明的化合物”时,也意在涵盖化合物的上述各种衍生物形式。
本发明的化合物的药学上可接受的盐包括其酸加成盐及碱加成盐。例如六氟磷酸盐、葡甲胺盐等。适合的盐的综述参见Stahl及Wermuth的“Handbook of PharmaceuticalSalts:Properties,Selection,and Use”(Wiley-VCH,2002)。用于制备本发明的化合物的药学上可接受的盐的方法为本领域技术人员已知的。
本发明的化合物可以溶剂合物(优选水合物)的形式存在,其中本发明的化合物包含作为所述化合物晶格的结构要素的极性溶剂,特别是例如水、甲醇或乙醇。极性溶剂特别是水的量可以化学计量比或非化学计量比存在。
在本发明的范围内还包括本发明的化合物的代谢物,即在给药本发明的化合物时体内形成的物质。这样的产物可由例如被给药的化合物的氧化、还原、水解、酰胺化、脱酰胺化、酯化、脱脂化、酶解等产生。因此,本发明包括本发明的化合物的代谢物,包括通过使本发明的化合物与哺乳动物接触足以产生其代谢产物的时间的方法制得的化合物。
本发明在其范围内进一步包括本发明的化合物的前药,其为自身可具有较小药理学活性或无药理学活性的本发明的化合物的某些衍生物当被给药至身体中或其上时可通过例如水解裂解转化成具有期望活性的本发明的化合物。通常这样的前药会是所述化合物的官能团衍生物,其易于在体内转化成期望的治疗活性化合物。关于前药的使用的其他信息可参见“Pro-drugs as Novel DeliverySystems”,第14卷,ACS Symposium Series(T.Higuchi及V.Stella)及“Bioreversible Carriers in Drug Design,”PergamonPress,1987(E.B.Roche编辑,American Pharmaceutical Association)。本发明的前药可例如通过用本领域技术人员已知作为“前-部分(pro-moiety)(例如“Design ofProdrugs”,H.Bundgaard(Elsevier,1985)中所述)”的某些部分替代本发明的化合物中存在的适当官能团来制备。
本发明还涵盖含有保护基的本发明的化合物。在制备本发明的化合物的任何过程中,保护在任何有关分子上的敏感基团或反应基团可能是必需的和/或期望的,由此形成本发明的化合物的化学保护的形式。这可以通过常规的保护基实现,例如,在ProtectiveGroups in Organic Chemistry,ed.J.F.W.McOmie,Plenum Press,1973;和T.W.Greene&P.G.M.Wuts,Protective Groups in Organic Synthesis,John Wiley&Sons,1991中所述的那些保护基,这些参考文献通过援引加入本文。使用本领域已知的方法,在适当的后续阶段可以移除保护基。
本发明中“药学上可接受的载体”是指与治疗剂一同给药的稀释剂、辅剂、赋形剂或媒介物,并且其在合理的医学判断的范围内适于接触人类和/或其它动物的组织而没有过度的毒性、刺激、过敏反应或与合理的益处/风险比相应的其它问题或并发症。
在本发明的药物组合物中可使用的药学上可接受的载体包括但不限于无菌液体,例如水和油,包括那些石油、动物、植物或合成来源的油,例如花生油、大豆油、矿物油、芝麻油等。当所述药物组合物通过静脉内给药时,水是示例性载体。还可以使用生理盐水和葡萄糖及甘油水溶液作为液体载体,特别是用于注射液。适合的药物赋形剂包括淀粉、葡萄糖、乳糖、蔗糖、明胶、麦芽糖、白垩、硅胶、硬脂酸钠、单硬脂酸甘油酯、滑石、氯化钠、脱脂奶粉、甘油、丙二醇、水、乙醇等。所述组合物还可以视需要包含少量的湿润剂、乳化剂或pH缓冲剂。口服制剂可以包含标准载体,如药物级的甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、纤维素、碳酸镁等。适合的药学上可接受的载体的实例如在Remington’s PharmaceuticalSciences(1990)中所述。
本发明的组合物可以系统地作用和/或局部地作用。为此目的,它们可以适合的途径给药,例如通过注射、静脉内、动脉内、皮下、腹膜内、肌内或经皮给药;或通过口服、含服、经鼻、透粘膜、局部、以眼用制剂的形式或通过吸入给药。
对于这些给药途径,可以适合的剂型给药本发明的组合物。
所述剂型包括但不限于片剂、胶囊剂、锭剂、硬糖剂、散剂、喷雾剂、乳膏剂、软膏剂、栓剂、凝胶剂、糊剂、洗剂、软膏剂、水性混悬剂、可注射溶液剂、酏剂、糖浆剂。
如本文中所使用的术语“有效量”指被给药后会在一定程度上缓解所治疗病症的一或多种症状的化合物的量。
可调整给药方案以提供最佳所需响应。例如,可给药单次推注,可随时间给药数个分剂量,或可如治疗情况的急需所表明而按比例减少或增加剂量。要注意,剂量值可随要减轻的病况的类型及严重性而变化,且可包括单次或多次剂量。要进一步理解,对于任何特定个体,具体的给药方案应根据个体需要及给药组合物或监督组合物的给药的人员的专业判断来随时间调整。
所给药的本发明的化合物的量会取决于所治疗的个体、病症或病况的严重性、给药的速率、化合物的处置及处方医师的判断。一般而言,有效剂量在每日每kg体重约0.0001至约50mg,例如约0.01至约10mg/kg/日(单次或分次给药)。对70kg的人而言,这会合计为约0.007mg/日至约3500mg/日,例如约0.7mg/日至约700mg/日。在一些情况下,不高于前述范围的下限的剂量水平可以是足够的,而在其它情况下,仍可在不引起任何有害副作用的情况下采用较大剂量,条件是首先将所述较大剂量分成数个较小剂量以在一整天中给药。
本发明的化合物在药物组合物中的含量或用量可以是约0.01mg至约1000mg。
除非另外说明,否则如本文中所使用,术语“治疗(treating)”意指逆转、减轻、抑制这样的术语所应用的病症或病况或者这样的病症或病况的一或多种症状的进展,或预防这样的病症或病况或者这样的病症或病况的一或多种症状。
如本文所使用的“个体”包括人或非人动物。示例性人个体包括患有疾病(例如本文所述的疾病)的人个体(称为患者)或正常个体。本发明中“非人动物”包括所有脊椎动物,例如非哺乳动物(例如鸟类、两栖动物、爬行动物)和哺乳动物,例如非人灵长类、家畜和/或驯化动物(例如绵羊、犬、猫、奶牛、猪等)。
发明的有益效果
本发明的化合物对HBsAg分泌具有很强的的抑制活性,具有良好的物理化学性质(例如溶解度、物理和/或化学稳定性)、良好的药物代谢动力学性质(例如良好的生物利用度、合适的血药浓度、半衰期)、良好的安全性(较低的毒性(例如较低的心脏、肝脏毒性)和/或较少的副作用,较宽的治疗窗)等优异的性质。
具体实施方式
实施例
以下结合实施例进一步描述本发明,但提供这些实施例并非意图限制本发明的范围。
本发明中的缩写具有以下含义:


本发明化合物的结构通过核磁共振波谱(1H NMR)和/或质谱(MS)来确证。
反应的监测采用薄层色谱法(TLC)或LC-MS进行。
1H NMR光谱仪:Bruker超导核磁共振波谱仪(型号AVACE III HD 400MHz)。
LC/MS质谱仪:Aglient 1260Infinity/Aglient 6120Quadrupole。
薄层色谱法采用为硅胶GF 254为固定相。
本发明化合物可通过制备型硅胶板、硅胶柱层析、制备高效液相色谱仪(Prep-HPLC)、快速柱层析(Flash柱层析)分离纯化。
柱层析一般使用200~300目硅胶(青岛海洋)为固定相。
Flash柱层析使用Biotage快速柱色谱仪。
Prep-HPLC采用Agilent 1260色谱仪。
微波反应使用BiotageInitiator微波反应器进行。
洗脱剂的体系包括:A:二氯甲烷和甲醇;B:石油醚和乙酸乙酯,溶剂的体积比根据化合物的极性不同而进行调节。
在以下实施例中,如无特殊说明,反应的温度为室温(20℃~30℃)。
本申请中所使用的试剂购自Acros Organics、Aldrich Chemical Company或特伯化学等公司。
实施例1:7-(3,4-二氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(1)

第一步:5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(1b)
将化合物1a(2.02g,10mmol,参照文献“Heterocycl.Commun.,2000,6,25”制备而得)溶于40mL乙醇中,N2保护下加入100mg 10%Pd/C,再用H2置换N2后,于室温下反应24h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系A)得到化合物1b(0.96g)。
MS(ESI,m/z):207.2[M+H]+.
第二步:7-(3,4-二氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(1)
将化合物1b(220mg,1.07mmol)、5-溴-1,2-二氟-3-甲氧基苯(357mg,1.60mmol)、Pd(OAc)2(20mg,0.085mmol)、BINAP(106mg,0.17mmol)和Cs2CO3(869mg,2.67mmol)溶于20mL甲苯中,N2保护下加热至90℃反应过夜。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系A)得到化合物1(240mg)。
MS(ESI,m/z):349.1[M+H]+.
1H NMR(CDCl3,400MHz)δ8.53(s,1H),7.94(s,1H),6.36-6.31(m,2H),4.45(q,J=7.2Hz,2H),4.36(s,2H),3.88(s,3H),3.48(t,J=5.6Hz,2H),3.03(t,J=5.6Hz,2H),1.42(t,J=7.2Hz,3H).
实施例2:7-(3,4-二氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(2)

将化合物1(35mg,0.1mmol)溶于1mL四氢呋喃和0.5mL水中,再加入LiOH·H2O(21mg,0.5mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,用乙酸乙酯萃取,无水硫酸钠干燥旋干后,经Prep-HPLC分离得化合物2(20mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):321.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.91(s,1H),6.64-6.60(m,2H),4.50(s,2H),3.88(s,3H),3.56(t,J=5.6Hz,2H),3.00(t,J=5.6Hz,2H).
实施例3:7-(3,4-二氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-甲酰胺(3)

将化合物1(28mg,0.08mmol)溶于5mL 7N的氨甲醇溶液中,于50℃下反应过夜,过滤得到化合物3(10mg)。
MS(ESI,m/z):320.2[M+H]+.
1H NMR(CDCl3,400MHz)δ8.37(s,1H),8.03(s,1H),7.81(br,1H),6.39-6.34(m,2H),5.57(br,1H),4.38(s,2H),3.92(s,3H),3.50(t,J=5.6Hz,2H),3.06(t,J=5.6Hz,2H).
实施例4:1-(7-(3,4-二氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-基)乙酮(4)

第一步:7-(3,4-二氟-5-甲氧基苯基)-N-甲氧基-N-甲基-5,6,7,8-四氢-2,7-萘啶-3-甲酰胺(4a)
将化合物2(161mg,0.5mmol)、甲氧基甲基胺盐酸盐(74mg,0.75mmol)、HATU(229mg,0.6mmol)和DIPEA(226mg,1.75mmol)溶于3mL DMF中,于室温下搅拌2h。反应结束后用乙酸乙酯萃取,无水硫酸钠干燥旋干后,经快速柱层析(洗脱剂体系A)得到化合物4a(160mg)。
MS(ESI,m/z):364.2[M+H]+.
第二步:1-(7-(3,4-二氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-基)乙酮(4)
将化合物4a(81mg,0.22mmol)溶于2mL无水四氢呋喃中,N2保护下、冰浴下滴加入3N的甲基溴化镁的四氢呋喃溶液(0.144mL,0.433mmol),于冰浴下反应2h。反应结束后用乙酸乙酯萃取,无水硫酸钠干燥,旋干后经Prep-HPLC分离纯化得到化合物4(37mg)(Prep-HPLC条件参照实施例2)。
MS(ESI,m/z):319.1[M+H]+.
1H NMR(CDCl3,400MHz)δ6.49(s,1H),7.87(s,1H),6.39-6.35(m,2H),4.40(s,2H),3.92(s,3H),3.50(t,J=5.6Hz,2H),3.06(t,J=5.6Hz,2H),2.72(s,3H).
实施例5:7-(3,4-二氟-5-甲氧基苯基)-N-(甲磺酰基)-5,6,7,8-四氢-2,7-萘啶-3-甲酰胺(5)

将化合物2(56mg,0.174mmol)、甲基磺酰胺(34mg,0.348mmol)、DCC(72mg,0.348mmol)、DMAP(5mg,0.04mmol)溶于5mL二氯甲烷中,于室温下反应2h,然后经硅藻土抽滤,滤液旋干后经Prep-HPLC分离得到化合物5(3mg)(Prep-HPLC条件参照实施例2)。
MS(ESI,m/z):398.1[M+H]+.
1H NMR(CDCl3,400MHz)δ8.39(s,1H),8.02(s,1H),6.38-6.30(m,3H),4.39(s,2H),3.92(s,3H),3.50(t,J=5.2Hz,2H),3.40(s,3H),3.08(t,J=5.2Hz,2H).
实施例6:7-(3,4-二氟-5-(2-甲氧基乙氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(6)

第一步:7-(3,4-二氟-5-(2-甲氧基乙氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(6a)
将化合物1b(63mg,0.305mmol)、5-溴-1,2-二氟-3-(2-甲氧基乙氧基)苯(122mg,0.458mmol)、Pd(OAc)2(3mg,0.015mmol)、BINAP(19mg,0.03mmol)和Cs2CO3(253mg,0.610mmol)溶于5mL甲苯中,N2保护下加热至90℃反应过夜。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系A)得到化合物6a(30mg)。
MS(ESI,m/z):393.2[M+H]+.
第二步:7-(3,4-二氟-5-(2-甲氧基乙氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(6)
将化合物6a(30mg,0.076mmol)溶于5mL四氢呋喃和1mL水中,再加入LiOH·H2O(10mg,0.229mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物6(5mg)(Prep-HPLC条件参照实施例2)。
MS(ESI,m/z):365.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.90(s,1H),6.71-6.56(m,2H),4.49(s,2H),4.28-4.18(m,2H),3.71-3.66(m,2H),3.55(t,J=5.7Hz,2H),3.32(s,3H),2.99(t,J=5.5Hz,2H).
实施例7:7-(3-氟-4-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(7)

第一步:7-(3-氟-4-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(7a)
将化合物1b(50mg,0.242mmol)、4-溴-2-氟-1-甲基苯(69mg,0.364mmol)、Pd(OAc)2(4mg,0.017mmol)、BINAP(22mg,0.034mmol)和Cs2CO3(198mg,0.606mmol)溶于5mL甲苯中,N2保护下加热至90℃反应过夜。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系A)得到化合物7a(20mg)。
MS(ESI,m/z):393.2[M+H]+.
第二步:7-(3-氟-4-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(7)
将化合物7a(20mg,0.063mmol)溶于5mL四氢呋喃和1mL水中,再加入LiOH·H2O(10mg,0.229mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物7(2mg)(Prep-HPLC条件参照实施例2)。
MS(ESI,m/z):287.2[M+H]+.
1H NMR(CD3OD,400MHz)δ8.49(s,1H),8.01(s,1H),6.95-6.89(m,3H),4.40(s,2H),3.51(t,J=5.6Hz,2H),3.10(t,J=5.6Hz,2H),2.24(s,3H).
实施例8:7-(6-甲基吡啶-3-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(8)

第一步:7-(6-甲基吡啶-3-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(8a)
将化合物1b(50mg,0.242mmol)、5-溴-2-甲基吡啶(83mg,0.484mmol)、Pd(OAc)2(4mg,0.018mmol)、BINAP(23mg,0.036mmol)和Cs2CO3(236mg,0.726mmol)溶于10mL甲苯中,N2保护下加热至90℃反应过夜。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系A)得到化合物8a(40mg)。
MS(ESI,m/z):298.2[M+H]+.
第二步:7-(6-甲基吡啶-3-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(8)
将化合物8a(40mg,0.134mmol)溶于2mL四氢呋喃和1mL水中,再加入LiOH·H2O(28mg,0.67mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,在Prep-HPLC分离后(Prep-HPLC条件参照实施例2)得到的溶液中加入1N HCl并搅拌5min,冻干得化合物8的盐酸盐(10mg)。
MS(ESI,m/z):270.1[M+H]+.
1H NMR(D2O,400MHz)δ8.53(s,1H),8.15(d,J=2.4Hz,1H),8.01(m,2H),7.62(d,J=9.2Hz,1H),4.69(s,2H),3.88(t,J=5.6Hz,2H),3.20(m,2H),2.58(s,3H).
实施例9:7-(3,4-二氟-5-(2-羟基乙氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(10)

第一步:7-(3-(2–((叔丁基二甲基甲硅烷基)氧基)乙氧基)-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(10a)
将化合物1b(73mg,0.354mmol)、(2-(5-溴-2,3-二氟苯氧基)乙氧基)(叔丁基)二甲基硅烷(195mg,0.531mmol)、Pd(OAc)2(4.0mg,0.018mmol)、BINAP(22mg,0.035mmol)和Cs2CO3(231mg,0.708mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物10a(123mg)。
MS(ESI,m/z):493.3[M+H]+.
第二步:7-(3,4-二氟-5-(2-羟基乙氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(10)
将化合物10a(123mg,0.250mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(31mg,0.750mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物10(18mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):351.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.90(s,1H),6.71–6.57(m,2H),4.95(s,1H),4.49(s,2H),4.12(t,J=4.9Hz,2H),3.74(s,2H),3.55(t,J=5.7Hz,2H),2.99(t,J=5.5Hz,2H).
实施例10:7-(3-氟-5-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(14)

第一步:7-(3-氟-5-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(14a)
将化合物1b(50mg,242.43μmol)、1-溴-3-氟-5-甲基苯(68.74mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物14a(38mg)。
MS(ESI,m/z):315.3[M+H]+.
第二步:7-(3-氟-5-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(14)
将化合物14a(36mg,114.52μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(24.05mg,572.60μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物14(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):287.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.90(s,1H),6.70(s,1H),6.68–6.63(m,1H),6.39(d,J=9.6Hz,1H),4.52(s,2H),3.58(t,J=6.0Hz,2H),2.99(t,J=5.7Hz,2H),2.27(s,3H).
实施例11:7-(3-氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(15)

第一步:7-(3-氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(15a)
将化合物1b(50mg,242.44μmol)、1-溴-3-氟-5-甲氧基苯(75mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.48mg,606.09μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物15a(35mg)。
MS(ESI,m/z):331.3[M+H]+.
第二步:7-(3-氟-5-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(15)
将化合物15a(35mg,105.95μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(22.25mg,529.74μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物15(24mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;5.0min:70%A,30%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):303.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.89(s,1H),6.48(dt,J=12.6,2.0Hz,1H),6.38(s,1H),6.21(dt,J=10.8,2.0Hz,1H),4.52(s,2H),3.74(s,3H),3.58(t,J=5.8Hz,2H),2.98(t,J=5.8Hz,2H).
实施例12:7-(4-氟-3-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(16)

第一步:7-(4-氟-3-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(16a)
将化合物1b(50.0mg,0.242mmol)、4-溴-1-氟-2-甲氧基苯(89.0mg,0.434mmol)、Pd(OAc)2(1.6mg,0.007mmol)、BINAP(9mg,0.015mmol)和Cs2CO3(95mg,0.292mmol)溶于5mL甲苯中,N2保护下加热至90℃反应过夜。反应结束后用硅藻土过滤,滤液旋干后经制备型硅胶板分离纯化(洗脱剂体系B)得到16a(40mg)。
MS(ESI,m/z):331.1[M+H]+.
第二步:7-(4-氟-3-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(16)
将化合物16a(40mg,0.121mmol)溶于5mL四氢呋喃和1mL水中,再加入LiOH·H2O(12.8mg,0.305mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物16(2mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:40%A,60%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%三氟乙酸水溶液。
MS(ESI,m/z):303.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),7.79(dd,J=12.0,8.0Hz,1H),6.83(s,1H),6.54(d,J=8.0Hz,1H),4.46(s,2H),3.85(s,3H),3.53(t,J=4.0Hz,2H),3.02(t,J=4.0Hz,2H).
实施例13:7-(3-氰基-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(17)

第一步:7-(3-氰基-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(17a)
将化合物1b(50mg,0.24mmol)、3-溴-5-氟苄腈(63.04mg,0.32mmol)、Pd(OAc)2(5.43mg,24.24μmol)、BINAP(30.19mg,48.49μmol)和Cs2CO3(197.58mg,0.61mmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物17a(40mg)。
MS(ESI,m/z):226.1[M+H]+.
第二步:7-(3-氰基-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(17)
将化合物17a(40mg,0.12mmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(25.82mg,0.61mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,浓缩至干,经Prep-HPLC分离得化合物17(6mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):298.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.91(s,1H),7.33(s,1H),7.24–7.21(m,1H),7.08–7.06(m,1H),4.63s,2H),3.69–3.66(m,2H),3.01–2.99(m,2H).
实施例14:7-(4-氰基-3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(35)

第一步:7-(4-氰基-3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(35a)
将化合物1b(50mg,242.43μmol)、4-溴-2-氟-苄腈(72.73mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物35a(38mg)。
MS(ESI,m/z):326.3[M+H]+.
第二步:7-(4-氰基-3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(35)
将化合物35a(35mg,107.58μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(22.59mg,537.90μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物35(22mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):298.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.94(s,1H),7.66(t,J=8.8Hz,1H),7.05(dd,J=14.0,2.4Hz,1H),6.95(dd,J=8.8,2.4Hz,1H),4.71(s,2H),3.73(t,J=6.0Hz,2H),3.02(t,J=6.0Hz,2H).
实施例15:7-(4-氯-3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(36)

第一步:7-(4-氯-3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(36a)
将化合物1b(40mg,0.194mmol)、4-溴-1-氯-2-氟苯(61mg,0.291mmol)、Pd(OAc)2(2.2mg,0.010mmol)、BINAP(12mg,0.019mmol)和Cs2CO3(126mg,0.388mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物36a(65mg)。
MS(ESI,m/z):335.1[M+H]+.
第二步:7-(4-氯-3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(36)
将化合物36a(65mg,0.194mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(24mg,0.582mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物36(15mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:0%A,100%B;25.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%三氟乙酸水溶液。
MS(ESI,m/z):307.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.90(s,1H),7.37(t,J=8.9Hz,1H),7.08(dd,J=13.2,2.8Hz,1H),6.89(dd,J=9.0,2.5Hz,1H),4.55(s,2H),3.61(t,J=5.8Hz,2H),2.99(t,J=5.7Hz,2H).
实施例16:7-(3,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(37)

第一步:7-(3,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(37a)
将化合物1b(40mg,0.194mmol)、1-溴-3,5-二氟苯(56mg,0.291mmol)、Pd(OAc)2(2.2mg,0.010mmol)、BINAP(12mg,0.019mmol)和Cs2CO3(126mg,0.388mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物37a(62mg)。
MS(ESI,m/z):319.2[M+H]+.
第二步:7-(3,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(37)
将化合物37a(62mg,0.194mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(24mg,0.582mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物37(8mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;25.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%三氟乙酸水溶液。
MS(ESI,m/z):291.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.91(s,1H),6.73(d,J=9.4Hz,2H),6.48(t,J=9.2Hz,1H),4.57(s,2H),3.62(t,J=5.8Hz,2H),2.99(t,J=5.7Hz,2H).
实施例17:7-(3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(38)

第一步:7-(3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(38a)
将化合物1b(50mg,0.24mmol)、1-溴-3-氟苯(55.68mg,0.32mmol)、Pd(OAc)2(5.43mg,24.24μmol)、BINAP(30.19mg,48.49μmol)和Cs2CO3(197.58mg,0.61mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物38a(40mg)。
MS(ESI,m/z):301.1[M+H]+.
第二步:7-(3-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(38)
将化合物38a(40mg,0.13mmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(27.98mg,0.67mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物38(6mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):298.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.63-8.10(m,1H),7.86-8.45(m,1H),7.22–7.23(m,1H),6.89–6.87(m,2H),6.53–6.51(m,1H)4.51–4.45(m,2H),3.58–3.57(m,2H),2.99–2.98(m,2H).
实施例18:7-(5-氯-2-氰基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(39)

第一步:7-(5-氯-2-氰基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(39a)
将化合物1b(50.0mg,0.242mmol)、2-溴-4-氯苄腈(105.0mg,0.484mmol)、Pd(OAc)2(1.6mg,0.007mmol)、BINAP(9mg,0.015mmol)和Cs2CO3(95mg,0.292mmol)溶于5mL甲苯中,N2保护下加热至90℃反应过夜。反应结束后用硅藻土过滤,滤液旋干后经制备型硅胶板分离纯化(洗脱剂体系B)得到39a(70mg)。
MS(ESI,m/z):342.1[M+H]+.
第二步:7-(5-氯-2-氰基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(39)
将化合物39a(70mg,0.204mmol)溶于5mL四氢呋喃和1mL水中,再加入LiOH·H2O(12.8mg,0.305mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物39(9mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:40%A,60%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%三氟乙酸水溶液。
MS(ESI,m/z):314.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.53(s,1H),7.95(s,1H),7.79(d,J=8.0Hz,1H),7.33(s,1H),7.18(d,J=8.0Hz,1H),4.54(s,2H),3.62(t,J=4.0Hz,2H),3.10(t,J=4.0Hz,2H).
实施例19:7-(4-氯-3-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(40)

第一步:7-(4-氯-3-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(40a)
将化合物1b(50mg,242.43μmol)、4-溴-1-氯-2-甲氧基苯(80.54mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物40a(42mg)。
MS(ESI,m/z):347.8[M+H]+.
第二步:7-(4-氯-3-甲氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(40)
将化合物40a(26mg,74.97μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(15.74mg,374.85μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物40(8mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):319.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.58(s,1H),7.92(s,1H),7.23(d,J=8.8Hz,1H),6.78(s,1H),6.62(d,J=8.8Hz,1H),4.54(s,2H),3.88(s,3H),3.63–3.58(m,2H),3.03(d,J=5.3Hz,2H).
实施例20:7-(3-氯-4-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(41)

第一步:7-(3-氯-4-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(41a)
将化合物1b(50mg,242.43μmol)、4-溴-2-氯-1-氟苯(76.16mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物41a(48mg)。
MS(ESI,m/z):335.7[M+H]+.
第二步:7-(3-氯-4-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(41)
将化合物41a(36mg,107.54μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(22.58mg,537.68μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物41(23mg)。
Prep-HPLC条件:仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):307.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.89(s,1H),7.29(t,J=9.2Hz,1H),7.21(dd,J=6.4,3.0Hz,1H),7.09–7.01(m,1H),4.49(s,2H),3.55(t,J=5.6Hz,2H),2.99(t,J=5.6Hz,2H).
实施例21:7-(3-氯-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(42)

第一步:7-(3-氯-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(42a)
将化合物1b(50mg,242.43μmol)、1-溴-3-氯-5-氟苯(76.16mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物42a(46mg)。
MS(ESI,m/z):335.7[M+H]+.
第二步:7-(3-氯-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(42)
将化合物42a(46mg,137.41μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(28.86mg,687.03μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物42(32mg)。
Prep-HPLC条件:仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):307.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.90(s,1H),6.92(s,1H),6.87(dd,J=12.8,2.0Hz,1H),6.69(dd,J=8.4,1.6Hz,1H),4.58(s,2H),3.62(t,J=5.8Hz,2H),2.99(t,J=5.7Hz,2H).
实施例22:7-(4-氯-3,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(43)
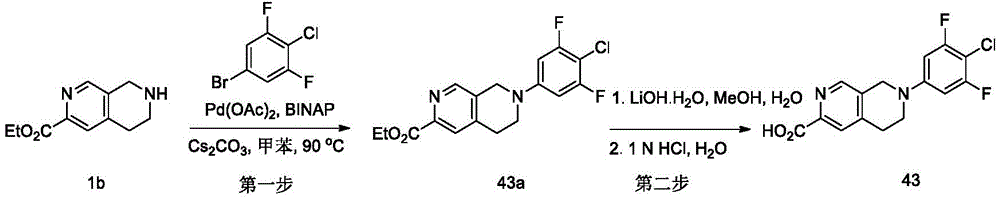
第一步:7-(4-氯-3,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(43a)
将化合物1b(50mg,0.24mmol)、5-溴-2-氯-1,3-二氟苯(71.68mg,0.32mmol)、Pd(OAc)2(5.43mg,24.24μmol)、BINAP(30.19mg,48.49μmol)和Cs2CO3(197.58mg,0.61mmol)溶于5mL甲苯中,N2保护下加热至90℃反应6h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物43a(40mg)。
MS(ESI,m/z):352.1[M+H]+.
第二步:7-(4-氯-3,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(43)
将化合物43a(40mg,0.11mmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(23.81mg,0.57mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,固体干燥得化合物43(15mg)。
MS(ESI,m/z):325.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.91(s,1H),6.99–6.96(m,2H),4.59(s,2H),3.65–3.62(m,2H),2.97–2.98(m,2H).
实施例23:7-(3-(三氟甲氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(44)

第一步:7-(3-(三氟甲氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(44a)
将化合物1b(50mg,0.242mmol)、1-溴-3-(三氟甲氧基)苯(88mg,0.364mmol)、Pd(OAc)2(2.7mg,0.012mmol)、BINAP(15mg,0.024mmol)和Cs2CO3(158mg,0.485mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物44a(73mg)。
MS(ESI,m/z):367.2[M+H]+.
第二步:7-(3-(三氟甲氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(44)
将化合物44a(73mg,0.194mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(25mg,0.600mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物44(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;25.0min:45%A,55%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):339.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.58(s,1H),7.90(s,1H),7.34(t,J=8.2Hz,1H),7.06(d,J=8.4Hz,1H),6.97(s,1H),6.71(d,J=7.7Hz,1H),4.56(s,2H),3.62(t,J=5.6Hz,2H),3.01(t,J=5.2Hz,2H).
实施例24:7-(2-氟-3-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(45)

第一步:7-(2-氟-3-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(45a)
将化合物1b(54mg,0.262mmol)、1-溴-2-氟-3-甲基苯(74mg,0.393mmol)、Pd(OAc)2(2.9mg,0.013mmol)、BINAP(16mg,0.026mmol)和Cs2CO3(171mg,0.524mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物45a(63mg)。
MS(ESI,m/z):315.2[M+H]+.
第二步:7-(2-氟-3-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(45)
将化合物45a(63mg,0.200mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(25mg,0.600mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物45(17mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:20%A,80%B;20.0min:45%A,55%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):287.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.52(s,1H),7.89(s,1H),7.06–6.86(m,3H),4.32(s,2H),3.50–3.25(m,2H),3.01(t,J=5.4Hz,2H),2.23(d,J=1.6Hz,3H).
实施例25:7-(3-氟-4-(三氟甲基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(46)

第一步:7-(3-氟-4-(三氟甲基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(46a)
将化合物1b(50mg,242.43μmol)、4-溴-2-氟-1-(三氟甲基)苯(88.37mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物46a(25mg)。
MS(ESI,m/z):369.3[M+H]+.
第二步:7-(3-氟-4-(三氟甲基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(46)
将化合物46a(25mg,137.41μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(14.25mg,339.37μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物46(7mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):341.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.59(s,1H),7.93(s,1H),7.54(t,J=8.8Hz,1H),7.06(d,J=15.2Hz,1H),6.95(d,J=8.6Hz,1H),4.68(s,2H),3.71(t,J=5.6Hz,2H),3.03(t,J=5.2Hz,2H).
实施例26:7-(3-溴-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(47)

第一步:7-(3-溴-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(47a)
将化合物1b(52mg,0.252mmol)、1,3-二溴-5-氟苯(96mg,0.378mmol)、Pd(OAc)2(2.8mg,0.013mmol)、BINAP(16mg,0.025mmol)和Cs2CO3(164mg,0.504mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物47a(57mg)。
MS(ESI,m/z):382.2[M+H]+.
第二步:7-(3-溴-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(47)
将化合物47a(57mg,0.150mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(19mg,0.450mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物47(45mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;20.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):352.0[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),7.04(s,1H),6.90(d,J=12.9Hz,1H),6.81(d,J=8.1Hz,1H),4.57(s,2H),3.62(t,J=5.8Hz,2H),2.99(t,J=5.6Hz,2H).
实施例27:7-(4-氯苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(48)

第一步:7-(4-氯苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(48a)
将化合物1b(50mg,242.43μmol)、1-溴-4-氯苯(69.62mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物48a(42mg)。
MS(ESI,m/z):317.7[M+H]+.
第二步:7-(4-氯苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(48)
将化合物48a(39mg,123.11μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(25.85mg,615.57μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物48(23mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;5min:30%A,70%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):289.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.89(s,1H),7.27(d,J=8.8Hz,2H),7.06(d,J=8.8Hz,2H),4.50(s,2H),3.57(t,J=6.0Hz,2H),2.99(t,J=5.6Hz,2H).
实施例28:7-(4-氟-2-(三氟甲基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(49)

第一步:7-(4-氟-2-(三氟甲基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(49a)
将化合物1b(50mg,242.43μmol)、1-溴-4-氟-2-(三氟甲基)苯(88.37mg,363.65μmol)、Pd(OAc)2(3.81mg,16.97μmol)、BINAP(21.13mg,33.94μmol)和Cs2CO3(197.47mg,606.07μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物49a(55mg)。
MS(ESI,m/z):369.3[M+H]+.
第二步:7-(4-氟-2-(三氟甲基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(49)
将化合物49a(55mg,149.32μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH.H2O(31.36mg,746.62μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物49(36mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):341.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.91(s,1H),7.43–7.36(m,2H),7.29(d,J=6.4Hz,1H),4.54(s,2H),3.60(t,J=5.8Hz,2H),3.02(t,J=5.6Hz,2H).
实施例29:7-(3-氯-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(50)

第一步:7-(3-氯-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(50a)
将化合物1b(42mg,0.202mmol)、5-溴-1-氯-2,3-二氟苯(55mg,0.242mmol)、Pd(OAc)2(2.3mg,0.010mmol)、BINAP(13mg,0.020mmol)和Cs2CO3(131mg,0.403mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物50a(25mg)。
MS(ESI,m/z):353.2[M+H]+.
第二步:7-(3-氯-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧(50)
将化合物50a(25mg,0.071mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(9mg,0.213mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物50(10mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;20.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):324.9[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.90(s,1H),7.15(ddd,J=14.0,6.3,3.0Hz,1H),7.03(dd,J=5.2,2.2Hz,1H),4.53(s,2H),3.59(t,J=5.9Hz,2H),2.99(t,J=5.7Hz,2H).
实施例30:7-(3,4,5-三氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(51)

第一步:7-(3,4,5-三氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(51a)
将化合物1b(50mg,0.24mmol)、5-溴-1,2,3-三氟苯(76.72mg,0.36mmol)、Pd(OAc)2(5.43mg,24.24μmol)、BINAP(30.19mg,48.49μmol)和Cs2CO3(197.58mg,0.61mmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物51a(20mg)。
MS(ESI,m/z):337.1[M+H]+.
第二步:7-(3,4,5-三氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(51)
将化合物51a(20mg,59.47μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(12.6mg,0.30mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,干燥固体得化合物51(10mg)。
MS(ESI,m/z):308.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.53(s,1H),7.90(s,1H),6.98–6.94(m,2H),4.53(s,2H),3.59–3.57(m,2H),3.00–2.97(m,2H).
实施例31:7-(3,4-二氟-5-异丙氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(52)

第一步:7-(3,4-二氟-5-异丙氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(52a)
将化合物1b(250mg,1.21mmol)、5-溴-1,2-二氟-3-异丙氧基苯(456.51mg,1.82mmol)、Pd(OAc)2(19.05mg,84.85μmol)、BINAP(105.67mg,169.70μmol)和Cs2CO3(987.38mg,3.03mmol)溶于30mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物52a(320mg)。
MS(ESI,m/z):377.4[M+H]+.
第二步:7-(3,4-二氟-5-异丙氧基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(52)
将化合物52a(320mg,850.17μmol)溶于6mL四氢呋喃和2mL水中,再加入LiOH·H2O(357.07mg,78.50mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物52(55mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。化合物保留时间Rt=6.2min。
MS(ESI,m/z):349.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.90(s,1H),6.69–6.59(m,2H),4.77–4.70(m,1H),4.48(s,2H),3.54(t,J=5.8Hz,2H),2.99(t,J=6.0Hz,2H),1.29(d,J=6.0Hz,6H).
实施例32:7-(3,4-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(53)

第一步:7-(3,4-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(53a)
将化合物1b(50mg,242.43μmol)、4-溴-1,2-二氟苯(94mg,484.87μmol)、Pd2(dba)3(22.20mg,24.24μmol)、Ru-phos(22.63mg,48.49μmol)和Cs2CO3(197.48mg,606.09μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物53a(27mg)。
MS(ESI,m/z):319.1[M+H]+.
第二步:7-(3,4-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(53)
将化合物53a(27mg,84.82μmol)溶于5mL THF和2mL水中,再加入LiOH·H2O(35.62mg,848.2μmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物53(7mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):291.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.45(s,1H),7.90(s,1H),7.13(dd,J=19.5,9.2Hz,1H),6.97(m,1H),6.85–6.78(m,1H),4.42(s,2H),3.55(t,J=5.8Hz,2H),3.06(t,J=5.7Hz,2H).
实施例33:7-(3,4-二氯-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(54)

第一步:7-(3,4-二氯-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(54a)
将化合物1b(0.1g,0.48mmol)、5-溴-1,2-二氯-3-氟苯(0.18g,0.73mmol)、Pd(OAc)2(10.86mg,48.49μmol)、BINAP(60.38mg,96.97μmol)和Cs2CO3(0.39mg,1.21mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物54a(40mg)。
MS(ESI,m/z):369.1[M+H]+.
第二步:7-(3,4-二氯-5-氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(54)
将化合物54a(40mg,0.11mmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(22.76mg,0.55mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,干燥固体得化合物54(30mg)。
MS(ESI,m/z):340.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.90(s,1H),6.98–6.94(m,2H),4.53(s,2H),3.59–3.57(m,2H),3.00–2.97(m,2H).
实施例34:7-(3,5-二氟-4-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(55)

第一步:7-(3,5-二氟-4-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(55a)
将化合物1b(0.1g,0.48mmol)、5-溴-1,3-二氟-2-甲基苯(150.56mg,0.73mmol)、Pd(OAc)2(10.86mg,48.49μmol)、BINAP(60.38mg,96.97μmol)和Cs2CO3(0.39mg,1.21mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物55a(30mg)。
MS(ESI,m/z):333.1[M+H]+.
第二步:7-(3,5-二氟-4-甲基苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(55)
将化合物55a(30mg,90.27μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(18.9mg,0.45mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,干燥固体得化合物55(18mg)。
MS(ESI,m/z):305.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.89(s,1H),6.77–6.70(m,2H),4.52(s,2H),3.59–3.56(m,2H),2.97–2.96(m,2H),2.02(s,3H).
实施例35:7-(3-(二氟甲氧基)-4-5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(56)

第一步:7-(3-(二氟甲氧基)-4-5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(56a)
将化合物1b(60mg,0.29mmol)、5-溴-1-(二氟甲氧基)-2,3-二氟苯(90.42mg,0.35mmol)、Pd(OAc)2(6.52mg,29.09μmol)、BINAP(36.23mg,58.18μmol)和Cs2CO3(235.65mg,0.73mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物56a(10mg)。
MS(ESI,m/z):385.1[M+H]+.
第二步:7-(3-(二氟甲氧基)-4-5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(56)
将化合物56a(10mg,26.02μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(5.5mg,0.13mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,干燥固体得到化合物56(4mg)。
MS(ESI,m/z):357.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.54(s,1H),7.90(s,1H),7.31–7.04(m,1H),7.07–7.02(m,1H),6.82–6.83(m,1H),4.52(s,2H),3.59–3.56(m,2H),3.01–2.98(m,2H).
实施例36:7-(3-乙氧基-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(57)

第一步:7-(3-乙氧基-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(57a)
将化合物1b(60mg,290.92μmol)、5-溴-1-乙氧基-2,3-二氟苯(124.13mg,523.66μmol)、Pd(OAc)2(13.03mg,58.18μmol)、BINAP(36.23mg,58.18μmol)和Cs2CO3(236.97mg,727.31μmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物57a(45mg)。
MS(ESI,m/z):363.1[M+H]+.
第二步:7-(3-乙氧基-4,5-二氟苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(57)
将化合物57a(45mg,124.18μmol)溶于5mL THF和2mL水中,再加入LiOH·H2O(52.16mg,1214.8μmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物57(15mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):335.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.38(s,1H),7.74(s,1H),6.64–6.55(m,2H),4.42(s,2H),4.16(m,2H),3.51(d,J=5.8Hz,2H),2.92(s,2H),1.35(t,J=7.0Hz,3H).
实施例37:7-(3,4-二氟-5-(氟甲氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(58)

第一步:7-(3,4-二氟-5-(氟甲氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(58a)
将化合物1b(89mg,0.432mmol)、5-溴-1,2-二氟-3-(氟甲氧基)苯(125mg,0.519mmol)、Pd(OAc)2(4.9mg,0.022mmol)、BINAP(27mg,0.043mmol)和Cs2CO3(282mg,0.864mmol)溶于5mL甲苯中,N2保护下加热至90℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物58a(80mg)。
MS(ESI,m/z):367.1[M+H]+.
第二步:7-(3,4-二氟-5-(氟甲氧基)苯基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(58)
将化合物58a(80mg,0.218mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(27mg,0.655mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物58(20mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16.0min:50%A,50%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):339.0[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.90(s,1H),6.93–6.82(m,1H),6.81–6.71(m,1H),6.00(d,J=7.6Hz,1H),5.88(s,1H),4.51(s,2H),3.57(t,J=5.8Hz,2H),3.00(t,J=5.5Hz,2H).
实施例38:7-(6-氟-4-甲氧基吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(59)

第一步:7-(6-氟-4-甲氧基吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯59a)
将化合物1b(60mg,290.92μmol)、2,6-二氟-4-甲氧基吡啶(84.43mmol,581.84μmol)、DIPEA(112.80mg,872.77μmol)在NMP中于微波中于150℃下反应2h,反应结束后,用乙酸乙酯萃取,旋干后经制备型硅胶板分离纯化(洗脱剂体系B)得到化合物59a(96.39mg)。
MS(ESI,m/z):332.3[M+H]+
第二步:7-(6-氟-4-甲氧基吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(59)
将化合物59a(85mg,256.53μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(107.74mg,2.57mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物59(46mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):304.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.59(s,1H),7.90(s,1H),6.29(s,1H),6.00(d,J=1.2Hz,1H),4.76(s,2H),3.84(s,3H),3.80(t,J=5.8Hz,2H),2.96(t,J=5.6Hz,2H).
实施例39:7-(2-氟-6-(3-甲氧基吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(60)

第一步:7-(2,6-二氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(60a)和7-(4,6-二氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(60b)
将化合物1b(4.5g,21.8mmol)和2,4,6-三氟吡啶(3.48g,26.18mmol)、DIPEA(8.46g,65.43mmol)溶于30mL NMP中,然后于微波中于150℃反应1h,反应结束后,用乙酸乙酯萃取,有机相旋干后经快速柱层析(洗脱剂体系B)得到化合物60a(4.5g)和化合物60b(1.6g)。
MS(ESI,m/z):320.3[M+H]+.
第二步:7-(2-氟-6-(3-甲氧基吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(60c)
将化合物60a(60mg,187.91μmol)、3-甲氧基吡咯烷盐酸盐(77.57mg,563.72μmol)、K2CO3(77.79mg,563.72μmol)溶于1mL NMP中,然后微波加热至150℃反应3h,反应结束后,用乙酸乙酯萃取,有机相旋干后经制备型硅胶板分离纯化(洗脱剂体系B)得到化合物60c(62mg)。
MS(ESI,m/z):401.4[M+H]+.
第三步:7-(2-氟-6-(3-甲氧基吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(60)
将化合物60c(60mg,149.83μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(62.93mg,1.5mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物60(26mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):373.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),5.91(s,1H),5.67(s,1H),4.63(s,2H),4.07–4.00(m,1H),3.65(t,J=5.6Hz,2H),3.47–3.39(m,2H),3.38–3.27(m,2H),3.26(s,3H),2.98(t,J=5.6Hz,2H),2.06–1.98(m,2H).
实施例40:7-(6-氟-4-(3-甲氧基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(61)

第一步:7-(6-氟-4-(3-甲氧基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(61a)和7-(4-氟-6-(3-甲氧基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(61b)
将化合物60b(400mg,1.25mmol)、3-甲氧基吡咯烷盐酸盐(517.15mg,3.76mmol)和DIPEA(809.51mg,6.26mmol)溶于5mL NMP中,微波加热至150℃反应2h。反应结束后向体系中加入50mL水,用EA萃取,有机相用无水硫酸钠干燥后过滤,有机相旋干后经制备型硅胶板分离纯化(洗脱剂体系B)得到化合物61a(370mg)和61b(150mg)。
MS(ESI,m/z):401.2[M+H]+.
第二步:7-(6-氟-4-(3-甲氧基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(61)
将化合物61a(370mg,923.97μmol)溶于10mL THF和4mL水中,再加入LiOH·H2O(387.70mg,9.24mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物61(105mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):373.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.58(s,1H),7.89(s,1H),5.72(s,1H),5.55(d,J=1.1Hz,1H),4.71(s,2H),4.07(d,J=2.7Hz,1H),3.77(dd,J=8.7,6.2Hz,2H),3.44(m,2H),3.32(m,2H),3.27(s,3H),2.95(t,J=5.7Hz,2H),2.05(m,2H).
实施例41:7-(4-氟-6-(3-甲氧基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(62)

将化合物61b(150mg,374.58μmol)溶于10mL THF和4mL水中,再加入LiOH·H2O(157.17mg,3.75mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物62(50mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):373.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.43(s,1H),7.77(s,1H),5.94(dd,J=12.5,1.5Hz,1H),5.56(dd,J=11.6,1.5Hz,1H),4.71(s,2H),4.06–4.00(m,1H),3.79(t,J=5.9Hz,2H),3.45(m,2H),3.43(m,1H),3.32(d,J=9.2Hz,1H),3.25(s,3H),2.89(t,J=5.6Hz,2H),2.02(m,2H).
实施例42:7-(2-氟-6-(2-甲基吗啉代)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(63)

第一步:7-(2-氟-6-(2-甲基吗啉代)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(63a)
将化合物60a(60mg,187.91μmol)、2-甲基吗啉(57.02mg,563.72μmol)、K2CO3(77.79mg,563.72μmol)溶于1mL NMP中,然后微波加热至150℃反应2h。反应结束后,用乙酸乙酯萃取,旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到63a(53mg)。
MS(ESI,m/z):401.4[M+H]+.
第二步:7-(2-氟-6-(2-甲基吗啉代)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(63)
将化合物63a(53mg,132.35μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(55.59mg,1.32mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物63(10mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):373.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.91(s,1H),6.05(s,1H),6.00(s,1H),4.65(s,2H),4.06–3.86(m,3H),3.67(t,J=6.0Hz,2H),3.57–3.48(m,2H),2.98(t,J=5.4Hz,2H),2.77–2.70(m,1H),2.42(t,J=12.4Hz,1H),1.16(d,J=6.4Hz,3H).
实施例43:7-(6-氟-4-(2-甲基吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(64)

第一步:7-(6-氟-4-(2-甲基吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(64a)和7-(4-氟-6-(2-甲基吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(64b)
将化合物60b(100mg,313.18μmol)、2-甲基吗啉(95.03mg,939.54μmol)、DIPEA(121.43mg,939.54μmol)溶于2mL NMP中,然后微波加热至150℃反应2h。反应结束后,用乙酸乙酯萃取,旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到64a(66mg)和64b(43mg)。
MS(ESI,m/z):401.4[M+H]+.
第二步:7-(6-氟-4-(2-甲基吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(64)
将化合物64a(66mg,164.82μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(69.22mg,1.65mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物64(50mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):373.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.89(s,1H),6.09(s,1H),5.92(s,1H),4.72(s,2H),3.91–3.72(m,5H),3.58–3.52(m,2H),2.95(t,J=5.6Hz,2H),2.83–2.73(m,1H),2.46(d,J=12.4Hz,1H),1.16(d,J=6.2Hz,3H).
实施例44:7-(4-氟-6-(2-甲基吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(65)

将化合物64b(43mg,107.38μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(45.10mg,1.07mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物65(20mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):373.3[M+H]+.
1H NMR(DMSO-d6,400MHz)8.56(s,1H),7.88(s,1H),6.07(d,J=12.0Hz,1H),5.96(d,J=12.2Hz,1H),4.74(s,2H),4.03(d,J=12.8Hz,2H),3.88(dd,J=11.4,2.4Hz,1H),3.79(t,J=5.6Hz,2H),3.57–3.49(m,2H),2.93(t,J=5.2Hz,2H),2.79–2.72(m,1H),2.45(dd,J=12.6Hz,1H),1.15(d,J=6.2Hz,3H).
实施例45:7-(4-(3,3-二氟吡咯烷-1-基)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(66)

第一步:7-(4-(3,3-二氟吡咯烷-1-基)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(66a)和7-(6-(3,3-二氟吡咯烷-1-基)-4-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(66b)
将化合物60b(100mg,0.313mmol)、3,3-二氟吡咯烷盐酸盐(135mg,0.940mmol)和DIPEA(121mg,0.940mmol)溶于2mL NMP中,N2保护下加热至150℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物66a(70mg)和66b(25mg)。
MS(ESI,m/z):407.1[M+H]+.
第二步:7-(4-(3,3-二氟吡咯烷-1-基)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(66)
将化合物66a(70mg,0.172mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(22mg,0.517mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物66(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):379.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.89(s,1H),5.79(s,1H),5.64(d,J=1.2Hz,1H),4.73(s,2H),3.85–3.74(m,4H),3.55(t,J=7.3Hz,2H),2.95(t,J=5.7Hz,2H),2.62–2.52(m,2H).
实施例46:7-(6-(3,3-二氟吡咯烷-1-基)-4-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(67)

将化合物66b(25mg,0.062mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(8mg,0.185mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物67(12mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):379.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.88(s,1H),6.08(dd,J=12.4,1.3Hz,1H),5.69(dd,J=11.3,1.3Hz,1H),4.78(s,2H),3.89–3.77(m,4H),3.58(t,J=7.3Hz,2H),2.94(t,J=5.7Hz,2H),2.60–2.51(m,2H).
实施例47:7-(2-(3,3-二氟吡咯烷-1-基)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(68)

第一步:7-(2-(3,3-二氟吡咯烷-1-基)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(68a)
将化合物60a(100mg,0.313mmol)、3,3-二氟吡咯烷盐酸盐(135mg,0.940mmol)和DIPEA(130mg,0.940mmol)溶于2mL NMP中,N2保护下加热至150℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物68a(60mg)。
MS(ESI,m/z):407.1[M+H]+.
第二步:7-(2-(3,3-二氟吡咯烷-1-基)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(68)
将化合物68a(60mg,0.148mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(19mg,0.443mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物68(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:20%A,80%B;6.0min:42.5%A,57.5%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):379.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.92(s,1H),6.01(s,1H),5.77(s,1H),4.66(s,2H),3.78(t,J=13.3Hz,2H),3.67(t,J=5.8Hz,2H),3.58(t,J=7.3Hz,2H),2.99(t,J=5.7Hz,2H),2.58–2.45(m,2H).
实施例48:7-(6-氟-4-(3-甲基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(69)

第一步:7-(6-氟-4-(3-甲基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(69a)和7-(4-氟-6-(3-甲基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(69b)
将化合物60b(97mg,0.304mmol)、3-甲基吡咯烷盐酸盐(110mg,0.911mmol)和DIPEA(118mg,0.911mmol)溶于2mL NMP中,N2保护下加热至150℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物69a(60mg)和69b(20mg)。
MS(ESI,m/z):385.2[M+H]+.
第二步:7-(6-氟-4-(3-甲基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(69)
将化合物69a(60mg,0.156mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(20mg,0.468mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物69(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16.0min:70%A,30%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):357.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.58(s,1H),7.89(s,1H),5.68(s,1H),5.51(s,1H),4.70(s,2H),3.76(s,2H),3.56–3.20(m,3H),2.95(s,2H),2.90–2.77(m,1H),2.33(td,J=14.5,7.3Hz,1H),2.08(dt,J=11.6,6.9Hz,1H),1.57(td,J=17.2,8.4Hz,1H),1.07(d,J=6.5Hz,3H).
实施例49:7-(4-氟-6-(3-甲基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(70)

将化合物69b(20mg,0.052mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(7mg,0.156mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物70(13mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):357.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.87(s,1H),5.94(d,J=12.5Hz,1H),5.53(d,J=11.7Hz,1H),4.75(s,2H),3.81(t,J=5.8Hz,2H),3.56–3.20(m,3H),2.99–2.78(m,3H),2.37–2.23(m,1H),2.05(dd,J=10.9,4.4Hz,1H),1.54(td,J=16.8,8.3Hz,1H),1.07(d,J=6.6Hz,3H).
实施例50:7-(2-氟-6-(3-甲基吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(71)

第一步:7-(2-氟-6-(3-甲基吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(71a)
将化合物60a(75mg,0.235mmol)、3-甲基吡咯烷盐酸盐(86mg,0.705mmol)和DIPEA(130mg,0.940mmol)溶于2mL NMP中,N2保护下加热至150℃反应4h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物71a(70mg)。
MS(ESI,m/z):385.2[M+H]+.
第二步:7-(2-氟-6-(3-甲基吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(71)
将化合物71a(70mg,0.148mmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(23mg,0.546mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物71(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:24.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;6.0min:70%A,30%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):357.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.91(s,1H),5.88(s,1H),5.64(s,1H),4.63(s,2H),3.64(t,J=5.8Hz,2H),3.57–3.50(m,2H),2.98(t,J=5.7Hz,2H),2.91–2.84(m,1H),2.30(dd,J=14.7,6.7Hz,2H),2.12–1.99(m,1H),1.53(dt,J=20.6,8.3Hz,1H),1.07(d,J=6.6Hz,3H).
实施例51:7-(4-(2,2-二甲基吗啉代)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(72)

第一步:7-(4-(2,2-二甲基吗啉代)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(72a)
将化合物60b(100mg,0.31mmol)、2,2-二甲基吗啉(107.53mg,0.93mmol)和DIPEA(120.66mg,0.93mmol)溶于5mL NMP中,微波加热至150℃反应2h。反应结束后,乙酸乙酯萃取,有机相浓缩干后经快速柱层析(洗脱剂体系B)得到化合物72a(30mg)。
MS(ESI,m/z):415.2[M+H]+.
第二步:7-(4-(2,2-二甲基吗啉代)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(72)
将化合物72a(30mg,72.38μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(15.2mg,0.36mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,固体干燥得化合物72(27mg)。
MS(ESI,m/z):387.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.63(s,1H),7.95(s,1H),6.06(s,1H),5.91(s,1H),4.74(s,2H),3.80–3.77(m,2H),3.71–3.68(m,2H),3.33-3.30(m,2H),3.21(s,2H),2.98–2.96(m,2H),1.18(s,6H).
实施例52:7-(2-(2,2-二甲基吗啉代)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(73)

第一步:7-(2-(2,2-二甲基吗啉代)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(73a)
将化合物60a(100mg,0.31mmol)、2,2-二甲基吗啉(107.53mg,0.93mmol)和DIPEA(120.66mg,0.93mmol)溶于2mL NMP中,微波加热至150℃反应2h。反应结束后,乙酸乙酯萃取,有机相浓缩干后经快速柱层析(洗脱剂体系B)得到化合物73a(50mg)。
MS(ESI,m/z):415.2[M+H]+.
第二步:7-(2-(2,2-二甲基吗啉代)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(73)
将化合物73a(50mg,0.12mmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(25.2mg,0.60mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,有黄色固体析出,过滤,固体干燥得化合物73(27mg)。
MS(ESI,m/z):387.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.94(s,1H),6.04(s,1H),5.97(s,1H),4.65(s,2H),3.68–3.65(m,4H),3.40–3.39(m,2H),3.28(s,2H),3.00–2.99(m,2H),1.18(s,6H).
实施例53:7-(6-氟-4-(3-甲氧基氮杂环丁烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(74)

第一步:7-(6-氟-4-(3-甲氧基氮杂环丁烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(74a)
将化合物60b(100mg,0.31mmol)、3-甲氧基氮杂环丁烷盐酸盐(116.11mg,0.93mmol)和DIPEA(120.66mg,0.93mmol)溶于2mL NMP中,微波加热至150℃反应3h。反应结束后,乙酸乙酯萃取,有机相浓缩干后经快速柱层析(洗脱剂体系B)得到化合物74a(35mg)。
MS(ESI,m/z):387.2[M+H]+.
第二步:7-(6-氟-4-(3-甲氧基氮杂环丁烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(74)
将化合物74a(30mg,77.64μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(16.4mg,0.39mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物74(9mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):359.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.46(s,1H),7.79(s,1H),5.61(s,1H),5.41–5.40(m,1H),4.65(s,2H),4.33–4.30(m,1H),4.13–4.09(m,2H),3.75–3.70(m,4H),3.24(s,3H),2.91–2.88(m,2H).
实施例54:7-(6-氟-4-(2-(羟甲基)吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(75)

第一步:7-(6-氟-4-(2-(羟甲基)吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(75a)和7-(4-氟-6-(2-(羟甲基)吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(75b)
将化合物60b(100mg,313.18μmol)、2-(羟甲基)吡咯烷盐酸盐(95.03mg,939.54μmol)和DIPEA(202.38mg,1570μmol)溶于2mL NMP中,微波加热至150℃反应2h。反应结束后向体系中加入30ml水,用EA萃取,有机相用无水硫酸钠干燥后过滤,滤液减压浓缩后柱层析(洗脱剂体系B)得到化合物75a(45mg)和75b(23mg)。
MS(ESI,m/z):401.2[M+H]+.
第二步:7-(6-氟-4-(2-(羟甲基)吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(75)
将化合物75a(45mg,112.37μmol)溶于5mL THF和2mL水中,再加入LiOH·H2O(47.15mg,1.12mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物75(7mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters XBridge Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):373.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.47(s,1H),7.92(s,1H),5.77(s,1H),5.62(s,1H),4.69(s,2H),3.81(t,J=5.8Hz,3H),3.62(dd,J=11.2,3.5Hz,1H),3.50–3.39(m,2H),3.22(dd,J=12.4,6.0Hz,1H),3.01(s,2H),2.21–2.06(m,2H),2.05–1.96(m,2H).
实施例55:7-(2-氟-6-(2-(羟甲基)吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(76)

第一步:7-(2-氟-6-(2-(羟甲基)吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(76a)
将化合物60a(60mg,187.91μmol)、2-(羟甲基)吡咯烷盐酸盐(77.57mg,563.72μmol)和DIPEA(121.43mg,939.54μmol)溶于2mL NMP中,微波加热至150℃反应2h。反应结束后向体系中加入30mL水,用乙酸乙酯萃取后将有机相旋干,经快速柱层析(洗脱剂体系B)得到化合物76a(40mg)。
MS(ESI,m/z):401.2[M+H]+.
第二步:7-(2-氟-6-(2-(羟甲基)吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(76)
将化合物76a(40mg,99.89μmol)溶于5mL THF和2mL水中,再加入LiOH·H2O(41.95mg,999.8μmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物76(15mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):373.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.90(s,1H),5.91(s,1H),5.69(s,1H),4.62(s,2H),3.95(dd,J=11.3,7.7Hz,1H),3.64(t,J=5.8Hz,2H),3.59–3.55(m,1H),3.40(s,2H),3.22–3.16(m,2H),2.98(t,J=5.7Hz,2H),2.02–1.94(m,2H),1.91–1.79(m,2H).
实施例56:7-(6-氟-4-(3-羟基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(77)

第一步:7-(6-氟-4-(3-羟基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(77a)
将化合物60b(100mg,0.31mmol)、3-羟基吡咯烷盐酸盐(116.11mg,0.94mmol)和DIPEA(120.66mg,0.93mmol)溶于2mL NMP中,微波加热至150℃反应3h。反应结束后,用乙酸乙酯萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物77a(30mg)。
MS(ESI,m/z):387.2[M+H]+.
第二步:7-(6-氟-4-(3-羟基吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(77)
将化合物77a(30mg,77.64μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(16.4mg,0.39mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得到化合物77(20mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):359.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.58(s,1H),7.88(s,1H),5.69(s,1H),5.23(m,1H),4.70(s,2H),4.38–4.37(m,1H),3.78–3.75(m,2H),3.44-3.35(m,2H),3.17–3.14(m,2H),2.96–2.93(m,2H),2.02–1.96(m,1H),1.91–1.86(m,1H).
实施例57:7-(4-氟-6-(2-(羟甲基)吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(78)

第一步:7-(4-氟-6-(2-(羟甲基)吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(78a)
将化合物60b(100mg,0.31mmol)、吗啉-2-基甲醇(110.06mg,0.94mmol)和DIPEA(120.66mg,0.93mmol)溶于2mL NMP中,微波加热至150℃反应3h。反应结束后,将反应液倒入20mL水中,用乙酸乙酯萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物78a(20mg)。
MS(ESI,m/z):417.2[M+H]+.
第二步:7-(4-氟-6-(2-(羟甲基)吗啉代)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(78)
将化合物78a(20mg,48.03μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(10.10mg,0.24mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得黄色固体化合物78(11mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):389.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.88(s,1H),6.09–6.07(m,1H),5.94–5.92(m,1H),4.70(s,2H),4.11–4.00(m,2H),3.92–3.90(m,1H),3.82–3.79(m,2H),3.54–3.50(m,4H),2.95–2.93(m,2H),2.85–2.77(m,1H),2.58–2.53(m,1H).
实施例58:7-(2-(2-乙基吗啉代)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(79)

第一步:7-(2-(2-乙基吗啉代)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(79a)
将化合物60a(100mg,313.18μmol)、2-乙基吗啉(108.21mg,939.54μmol)和DIPEA(121.43mg,939.54μmol)溶于1.5mL NMP中,然后微波加热至150℃反应2h,反应结束后,用乙酸乙酯萃取,将有机相旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到79a(105mg)。
MS(ESI,m/z):415.4[M+H]+.
第二步:7-(2-(2-乙基吗啉代)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(79)
将化合物79a(100mg,241.27μmol)溶于8mL四氢呋喃和2mL水中,再加入LiOH·H2O(101.24mg,2.41mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物79(80mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):387.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.68(s,1H),8.05(s,1H),6.17(s,1H),6.12(s,1H),4.77(s,2H),4.16(d,J=12.3Hz,1H),4.12–4.00(m,2H),3.79(t,J=5.8Hz,2H),3.62(d,J=2.7Hz,1H),3.45–3.41(m,1H),3.11(t,J=5.7Hz,2H),2.97–2.82(m,1H),2.56(dd,J=12.6,10.5Hz,1H),1.67–1.54(m,2H),1.06(t,J=7.5Hz,3H).
实施例59:7-(2-氟-6-(2-氧杂-7-氮杂螺[4.4]壬烷-7-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(80)

第一步:7-(2-氟-6-(2-氧杂-7-氮杂螺[4.4]壬烷-7-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(80a)
将化合物60a(100mg,313.18μmol)、2-氧杂-7-氮杂螺[4.4]壬烷(119.49mg,939.54μmol)和DIPEA(121.43mg,939.54μmol)溶于1.5mL NMP中,然后微波加热至150℃反应2h,反应结束后,用乙酸乙酯萃取,将有机相旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到80a(65mg)。
MS(ESI,m/z):427.1[M+H]+.
第二步:7-(2-氟-6-(2-氧杂-7-氮杂螺[4.4]壬烷-7-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(80)
将化合物80a(60mg,140.69μmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(59.03mg,1.41mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物80(30mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:26.0mL/min;检测波长:254nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):399.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),5.90(s,1H),5.67(s,1H),4.63(s,2H),3.81(t,J=7.0Hz,2H),3.64(t,J=5.8Hz,2H),3.57(s,2H),3.43(s,4H),2.98(t,J=5.7Hz,2H),1.97–1.84(m,4H).
实施例60:7-(2-氟-6-(2-(甲氧基甲基)吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(81)

第一步:7-(2-氟-6-(2-(甲氧基甲基)吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(81a)
将化合物60a(100mg,313.18μmol)、2-(甲氧基甲基)吡咯烷盐酸盐(142.47mg,939.54μmol)和DIPEA(202.38mg,1.57mmol)溶于2mL NMP中,微波加热至150℃反应2h。反应结束后向体系中加入30mL水,用乙酸乙酯萃取,将有机相用无水硫酸钠干燥后过滤,滤液减压浓缩后得到化合物81a粗品(130mg)。
MS(ESI,m/z):415.2[M+H]+.
第二步:7-(2-氟-6-(2-(甲氧基甲基)吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(81)
将化合物81a粗品(130mg,324.64μmol)溶于5mL THF和2mL水中,再加入LiOH·H2O(68.11mg,1.62mmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,减压浓缩后经Prep-HPLC分离得化合物81(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):387.2[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),5.92(s,1H),5.70(s,1H),4.63(s,2H),4.10(s,1H),3.64(t,J=5.8Hz,2H),3.48–3.45(m,1H),3.40(s,1H),3.26(s,3H),3.24–3.16(m,2H),2.98(t,J=5.7Hz,2H),2.04–1.82(m,4H).
实施例61:7-(4-(3,3-二甲基氮杂环丁烷-1-基)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(82)

第一步:7-(4-(3,3-二甲基氮杂环丁烷-1-基)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(82a)
将化合物60b(100mg,313.18μmol)、3,3-二甲基氮杂环丁烷(76.17mg,626.36μmol)和DIPEA(161.91mg,1.25mmol)溶于1mL NMP中,然后于微波下在110℃反应3h。反应结束后,用乙酸乙酯萃取,将有机相旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到82a(65mg)。
MS(ESI,m/z):385.4[M+H]+.
第二步:7-(4-(3,3-二甲基氮杂环丁烷-1-基)-6-氟吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(82)
将化合物82a(65mg,169.07μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(71.01mg,1.65mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物82(38mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):357.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.88(s,1H),5.58(s,1H),5.38(d,J=0.9Hz,1H),4.69(s,2H),3.74(t,J=5.8Hz,2H),3.61(s,4H),2.94(t,J=5.7Hz,2H),1.28(s,6H).
实施例62:7-(2-(3,3-二甲基氮杂环丁烷-1-基)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(83)

第一步:7-(2-(3,3-二甲基氮杂环丁烷-1-基)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(83a)
将化合物60a(100mg,313.18μmol)、3,3-二甲基氮杂环丁烷(76.17mg,626.36μmol)和DIPEA(161.91mg,1.25mmol)溶于1mL NMP中,然后于微波下在110℃反应2h。反应结束后,用乙酸乙酯萃取,将有机相旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到83a(70mg)。
MS(ESI,m/z):385.4[M+H]+.
第二步:7-(2-(3,3-二甲基氮杂环丁烷-1-基)-6-氟吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(83)
将化合物83a(65mg,169.07μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(71.01mg,1.65mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物83(35mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:20%A,80%B;16min:80%A,20%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):357.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.91(s,1H),5.93(s,1H),5.59(s,1H),4.61(s,2H),3.62(t,J=5.8Hz,2H),3.58(s,4H),2.97(t,J=5.7Hz,2H),1.26(s,6H).
实施例63:(S)-7-(2-氟-6-(3-氟吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(84)

第一步:(S)-7-(2-氟-6-(3-氟吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(84a)
将化合物60a(100mg,313.18μmol)、(S)-3-氟吡咯烷盐酸盐(78.65mg,626.36μmol)和DIPEA(121.43mg,939.54μmol)溶于1.5mL NMP中,然后在微波下在150℃反应2h。反应结束后,用乙酸乙酯萃取,将有机相旋干后经制备硅胶板分离纯化(洗脱剂体系B)得到84a(100mg)。
MS(ESI,m/z):389.2[M+H]+.
第二步:(S)-7-(2-氟-6-(3-氟吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(84)
将化合物84a(90mg,231.71μmol)溶于6mL四氢呋喃和1.5mL水中,再加入LiOH·H2O(97.23mg,2.32mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物84(30mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:254nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):361.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),5.94(s,1H),5.72(s,1H),5.48–5.35(m,1H),4.65(s,2H),3.68–3.63(m,3H),3.59(dd,J=14.5,5.5Hz,3H),2.99(t,J=5.7Hz,2H),2.27–2.17(m,2H).
实施例64:(R)-7-(2-氟-6-(3-氟吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(85)
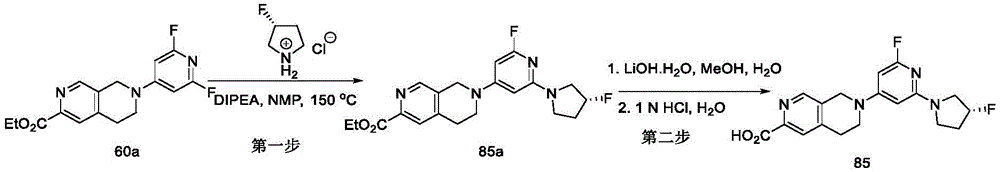
第一步:(R)-7-(2-氟-6-(3-氟吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(85a)
将化合物60a(100mg,0.31mmol)、(R)-3-氟吡咯烷盐酸盐(47mg,0.38mmol)和DIPEA(120.66mg,0.93mmol)溶于2mL NMP中,于微波下在150℃反应3h。反应结束后,将反应液倒入20mL水中,用乙酸乙酯萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物85a(25mg)。
MS(ESI,m/z):388.2[M+H]+.
第二步:(R)-7-(2-氟-6-(3-氟吡咯烷-1-基)吡啶-4-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(85)
将化合物85a(25mg,64.36μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(13.52mg,0.32mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得到化合物85(14mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):361.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.46(s,1H),8.71(s,1H),5.93(s,1H),5.71(s,1H),5.48–5.35(m,1H),4.64–4.56(m,2H),3.67–3.46(m,4H),3.41–3.34(m 2H),2.93–2.95(m,2H),2.24–2.08(m,2H).
实施例65:(R)-7-(6-氟-4-(3-氟吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(86)

第一步:(R)-7-(6-氟-4-(3-氟吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(86a)
将化合物60b(100mg,0.31mmol)、(R)-3-氟吡咯烷盐酸盐(47mg,0.38mmol)和DIPEA(120.66mg,0.93mmol)溶于2mL NMP中,于微波下在150℃反应3h。反应结束后,将反应液倒入20mL水中,用乙酸乙酯萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物86a(15mg)。
MS(ESI,m/z):388.2[M+H]+.
第二步:(R)-7-(6-氟-4-(3-氟吡咯烷-1-基)吡啶-2-基)-5,6,7,8-四氢-2,7-萘啶-3-羧酸(86)
将化合物86a(15mg,38.62μmol)溶于1mL甲醇和2mL水中,再加入LiOH·H2O(8.11mg,0.19mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得到化合物1(9mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:20.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16.0min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%碳酸氢铵水溶液。
MS(ESI,m/z):361.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.50(s,1H),7.82(s,1H),5.74(s,1H),5.59(s,1H),5.52–5.38(m,1H),4.74–4.64(m,2H),3.78–3.34(m,2H),3.61–3.34(m 4H),2.93–2.92(m,2H),2.26–2.11(m,2H).
实施例66:7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(87)

第一步:丁-3-炔-2-基(甲苯磺酰基)氨基甲酸叔丁酯(87b)
将87a(50g,184.28mmol)、丁-3-炔-2-醇(19.37g,276.41mmol)和PPh3(72.42g,276.41mmol)溶于500mL THF中,冰浴下滴入DIAD(55.84g,276.41mmol),滴加完毕后于25℃反应3h。反应结束后将反应液倒入1000mL水中,用乙酸乙酯萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物87b(56g)。
MS(ESI,m/z):324.1[M+H]+.
第二步:N-(丁-3-炔-2-基)-4-甲基苯磺酰胺(87c)
将87b(56g,173.16mmol)溶于300mL DCM中,冰浴下加入200mL TFA,于25℃反应2h。反应结束后直接将反应液减压浓缩以除去DCM和大部分的TFA,向减压浓缩后的残留液中加入300mL EA和200mL水,用碳酸氢钠溶液调至pH约为7~8。用乙酸乙酯萃取后将有机相浓缩干,得到化合物87c(40.5g)。
MS(ESI,m/z):224.1[M+H]+.
第三步:N-(丁-3-炔-1-基)-N-(丁-3-炔-2-基)-4-甲基苯磺酰胺(87d)
将87c(40g,179.14mmol,FR)、丁-3-炔-1-醇(18.83g,268.66mmol)和PPh3(70.40g,268.71mmol)溶于400mL THF中,冰浴下滴入DIAD(54.28g,268.71mmol),滴加完毕后于25℃反应3h。反应结束后将反应液倒入1000mL水中,用乙酸乙酯萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物87d(30g)。
MS(ESI,m/z):276.1[M+H]+.
第四步:8-甲基-7-甲苯磺酰基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(87e)
将87d(30g,108.95mmol)、Rh(COD)2BF4(2.21g,5.45mmol)、BINAP(3.39g,5.45mmol)溶于300mL DCM和30mL水的混合体系中,N2置换后室温搅拌10min,冰浴下滴加氰基甲酸乙酯(16.19g,163.42mmol),于25℃反应16h。反应结束后将反应液倒入100mL水中,用DCM萃取后将有机相浓缩干,经快速柱层析(洗脱剂体系B)得到化合物87e。
MS(ESI,m/z):375.1[M+H]+.
第五步:8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(87f)
将87e(13g,34.72mmol)、苯酚(8.17g,86.79mmol)溶于130mL 33%HBr的AcOH溶液中,90℃反应2h。反应结束后将反应液直接浓缩干,然后向体系中加入100mL EA,搅拌30min,过滤,将固体用EA洗涤,然后溶于100mL水中,用EA萃取2次,水相用碳酸氢钠溶液调节pH约为7~8,用异丙醇/DCM(1/3)萃取4次(100ml*4)。合并有机相,将有机相浓缩干得到化合物87f(6.65g)。
MS(ESI,m/z):221[M+H]+.
第六步:7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(87g)
将87f(3.78g,17.16mmol)、1-溴-3,5-二氟苯(6.62g,34.32mmol)、Pd(OAc)2(384.41mg,1.72mmol)、Cs2CO3(13.98g,42.90mmol)和BINAP(2.14g,3.43mmol)溶于50mL甲苯中,N2保护下加热至90℃反应16h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物得到87g(5.5g)。
MS(ESI,m/z):333.1[M+H]+.
第七步:7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(87)
将87g(2g,6.02mmol)溶于25mL四氢呋喃和10mL水中,再加入LiOH·H2O(1.26g,30.09mmol),于室温下反应3h。反应结束后用1N HCl调体系pH约为3,旋蒸除去大部分溶剂后,析出固体,抽滤,滤饼用少量水洗涤,减压干燥得化合物87(955mg)。
MS(ESI,m/z):305.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.90(s,1H),6.75–6.63(m,2H),6.42(tt,J=9.2,2.1Hz,1H),5.28(q,J=6.5Hz,1H),3.83(dt,J=13.2,4.6Hz,1H),3.37(dd,J=9.1,4.6Hz,1H),2.98(dd,J=9.4,5.5Hz,2H),1.41(d,J=6.7Hz,3H).
实施例67和68:(S)-7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸

第一步:7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(87g)的手性分离
将87g(1.175g,3.54mmol)通过手性柱分离得到峰1——87g-1(500mg,1.5mmol)和峰2——87g-2(550mg,1.65mmol)。
手性分离方法:仪器:YMC-K制备系统,色谱柱:CHIRALPAK IC 2.5cm I.D.×25cmL×5μm;色谱柱温:35℃;流速:30.0mL/min;检测波长:214nm;流动相:MeOH=100%。峰1:7.5-8.5min;峰2:11.0-12.5min。
第二步:(S)-7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸
将87g-1(500mg,1.5mmol)溶于15mL四氢呋喃和3mL水中,加入NaOH(180mg,4.5mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为5-6。旋蒸除去大部分溶剂后,析出固体,抽滤,滤饼用少量水洗涤,减压干燥得到88(420mg)。
MS(ESI,m/z):305.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.89(s,1H),6.70–6.67(m,2H),6.42–6.39(m,1H),5.30–5.26(m,1H),3.85–3.80(m,1H),3.37–3.33(m,1H),2.99–2.96(m,2H),1.41–1.39(m,3H).
将87g-2(550mg,1.65mmol)溶于12mL四氢呋喃和3mL水中,再加入LiOH·H2O(347.20mg,8.27mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋蒸除去大部分溶剂后,析出固体,抽滤,滤饼用少量水洗涤,减压干燥得到89(420mg)。
MS(ESI,m/z):305.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.90(s,1H),6.69(d,J=10.1Hz,2H),6.42(t,J=9.1Hz,1H),5.29(d,J=6.3Hz,1H),3.83(dd,J=8.8,4.4Hz,1H),3.00–2.95(m,3H),1.41(d,J=6.6Hz,3H).
实施例69:7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(90)

第一步:7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(90a)
将化合物87f(60mg,272.40μmol)、1-溴-3-氯-5-氟苯(85.60mg,408.60μmol)、Pd(OAc)2(6.11mg,27.24μmol)、BINAP(33.90mg,54.48μmol)和Cs2CO3(221.88mg,680.99μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物90a(50mg)。
MS(ESI,m/z):349.8[M+H]+.
第二步:7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(90)
将化合物90a(40mg,114.68μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(48.10mg,1.15mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物90(4mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:70%A,30%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):321.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.89(s,1H),6.88(s,1H),6.82(dd,J=13.1,2.1Hz,1H),6.63(dt,J=8.4,1.8Hz,1H),5.30(q,J=6.6Hz,1H),3.84(dt,J=13.3,4.7Hz,1H),3.04–2.91(m,3H),1.40(d,J=6.6Hz,3H).
实施例70和71:(S)-7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸

第一步:7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(90a)的手性分离
将90a(1.241g,3.56mmol)通过手性分离得到峰1——90a-1(530mg,1.52mmol)和峰2——90a-2(280mg,0.80mmol)。
手性分离方法:仪器:YMC-K制备系统,色谱柱:CHIRALPAK AY 5.0cm I.D.×25cmL×10μm;色谱柱温:35℃;流速:60.0mL/min;检测波长:254nm;流动相:EtOH=100%。峰1:8.5-10.0min;峰2:11.2-13.2min。
第二步:(S)-7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3-氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸
将90a-1(530mg,1.52mmol)溶于10mL四氢呋喃和3mL水中,再加入LiOH·H2O(318.79mg,7.60mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼用少量水洗涤,减压干燥得化合物91(355mg)。
MS(ESI,m/z):321.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.60(s,1H),7.96(s,1H),6.88(s,1H),6.83(d,J=13.0Hz,1H),6.63(d,J=8.2Hz,1H),5.34(d,J=6.5Hz,1H),3.90–3.80(m,1H),3.43–3.33(m,1H),3.00–2.95(m,2H),1.41(d,J=6.5Hz,3H).
将90a-2(280mg,0.80mmol)溶于6mL四氢呋喃和2mL水中,再加入LiOH·H2O(168.58mg,4.01mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼用少量水洗涤,减压干燥得化合物92(190mg)。
MS(ESI,m/z):321.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.90(s,1H),6.88(s,1H),6.82(dt,J=13.1,2.1Hz,1H),6.63(dt,J=8.4,1.8Hz,1H),5.31(q,J=6.5Hz,1H),3.84(dt,J=13.2,4.6Hz,1H),3.42–3.35(m,1H),2.97(d,J=4.0Hz,2H),1.40(d,J=6.6Hz,3H).
实施例72:7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(93)

第一步:7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(93a)
将化合物87f(70mg,317.80μmol)、5-溴-2-氯-1,3-二氟苯(108.42mg,476.69μmol)、Pd(OAc)2(7.10mg,31.78μmol)、BINAP(39.61mg,63.56μmol)和Cs2CO3(258.90mg,794.49μmol)溶于5mL甲苯中,N2保护下加热至90℃反应15h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物93a(52mg)。
MS(ESI,m/z):367.1[M+H]+.
第二步:7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(93)
将化合物93a(50mg,136.32μmol)溶于4mL四氢呋喃和1mL水中,再加入LiOH·H2O(57.20mg,1.36mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物93(35mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:70%A,30%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):339.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.91(s,1H),6.95(d,J=11.4Hz,2H),5.31(q,J=6.5Hz,1H),4.04(dd,J=14.2,7.1Hz,1H),3.86(dt,J=13.2,4.6Hz,1H),2.99(dd,J=9.0,5.2Hz,2H),1.42(d,J=6.6Hz,3H).
实施例73和74:(S)-7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸

第一步:7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(93a)的手性分离
将93a(0.843g,2.30mmol)通过手性分离得到峰1——93a-1(380mg,1.04mmol)和峰2——93a-2(400mg,1.09mmol)。
手性分离方法:仪器:YMC-K制备系统,色谱柱:CHIRALPAK IC 5.0cm I.D.×25cmL×10μm;色谱柱温:35℃;流速:30.0mL/min;检测波长:214nm;流动相:MeOH=100%。峰1:8.2-9.5min;峰2:12.3-14.2min。
第二步:(S)-7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(4-氯-3,5-二氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸
将93a-1(380mg,1.04mmol)溶于5mL四氢呋喃和2mL水中,再加入LiOH·H2O(217.41mg,5.18mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物94(275mg)。
MS(ESI,m/z):339.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.55(s,1H),7.90(s,1H),6.95(t,J=8.3Hz,2H),5.30(q,J=6.5Hz,1H),3.85(dt,J=13.2,4.6Hz,1H),3.36(dd,J=8.3,5.3Hz,1H),2.98(dd,J=9.0,5.2Hz,2H),1.41(d,J=6.6Hz,3H).
将93a-2(400mg,1.09mmol)溶于6mL四氢呋喃和2mL水中,再加入LiOH·H2O(229.01mg,5.45mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物95(350mg)。
MS(ESI,m/z):339.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.58(s,1H),7.97(s,1H),6.96(s,1H),6.93(s,1H),5.33(q,J=6.5Hz,1H),3.87(dt,J=13.2,4.5Hz,1H),3.45–3.33(m,1H),3.00(d,J=5.0Hz,2H),1.42(d,J=6.6Hz,3H)。
实施例75:7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(96)

第一步:7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(96a)
将化合物87f(80mg,363.20μmol)、4-溴-1-氯-2-氟苯(114.10mg,544.79μmol)、Pd(OAc)2(5.71mg,25.42μmol)、BINAP(31.66mg,50.85μmol)和Cs2CO3(259.84mg,907.99μmol)溶于5mL甲苯中,N2保护下加热至90℃反应5h。反应结束后用硅藻土过滤,将滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物96a(50mg)。
MS(ESI,m/z):349.8[M+H]+.
第二步:7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(96)
将化合物96a(50mg,143.35μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(60.21mg,1.43mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物96(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:214nm;洗脱梯度:(0min:30%A,70%B;16min:70%A,30%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):321.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.88(s,1H),7.35(t,J=9.0Hz,1H),7.04(dd,J=13.5,2.7Hz,1H),6.85(dd,J=9.1,2.5Hz,1H),5.26(q,J=6.5Hz,1H),3.80(dt,J=13.0,4.6Hz,1H),3.40–3.35(m,1H),3.00–2.92(m,2H),1.39(d,J=6.6Hz,3H).
实施例76和77:(S)-7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸

第一步:7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(96a)的手性分离
将96a(1.835g,5.26mmol)通过手性分离得到峰1——96a-1(790mg,2.26mmol)和峰2——96a-2(730mg,2.09mmol)。
手性分离方法:仪器:YMC-K制备系统,色谱柱:CHIRALPAK IC 5.0cm I.D.×25cmL×10μm;色谱柱温:35℃;流速:30.0mL/min;检测波长:214nm;流动相:MeOH=100%。峰1:8.0-9.1min;峰2:11.0-13.0min。
第二步:(S)-7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(4-氯-3-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸将96a-1(790mg,2.26mmol)溶于10mL四氢呋喃和5mL水中,再加入LiOH·H2O(475.18mg,11.32mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物97(425mg)。
MS(ESI,m/z):321.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.90(s,1H),7.35(t,J=9.0Hz,1H),7.03(dd,J=13.5,2.8Hz,1H),6.85(dd,J=9.0,2.6Hz,1H),5.27(q,J=6.6Hz,1H),3.81(dt,J=13.1,4.7Hz,1H),3.39–3.32(m,1H),2.97(dd,J=10.3,6.2Hz,2H),1.39(d,J=6.6Hz,3H).
将96a-2(730mg,2.09mmol)溶于6mL四氢呋喃和2mL水中,再加入LiOH·H2O(439.51mg,10.46mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物98(660mg)。
MS(ESI,m/z):321.7[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.64(s,1H),8.02(s,1H),7.37(t,J=8.9Hz,1H),7.04(d,J=13.4Hz,1H),6.85(d,J=8.8Hz,1H),5.33(d,J=6.5Hz,1H),3.84(d,J=13.1Hz,1H),3.43–3.32(m,1H),3.04(s,2H),1.40(d,J=6.5Hz,3H).
实施例78:7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(99)

第一步:7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(99a)
将化合物87f(1.2g,5.45mmol)、1-溴-3-乙氧基-5-氟苯(2.15g,9.81mmol)、Pd(OAc)2(122.03mg,544.79μmol)、BINAP(678.45mg,1.09mmol)和Cs2CO3(4.44g,13.62mmol)溶于30mL甲苯中,N2保护下加热至90℃反应16h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物99a(820mg)。
MS(ESI,m/z):359.2[M+H]+.
第二步:7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(99)
将化合物99a(36mg,100.44μmol)溶于3mL四氢呋喃和1mL水中,再加入LiOH·H2O(21.05mg,500μmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,经Prep-HPLC分离得化合物99(25mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:214nm;洗脱梯度:(0min:10%A,90%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):331.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.88(s,1H),6.41(d,J=12.9Hz,1H),6.32(s,1H),6.14(dd,J=10.9,2.0Hz,1H),5.24(q,J=6.4Hz,1H),4.00(q,J=7.0Hz,2H),3.83–3.72(m,1H),3.00–2.92(m,2H),1.38(d,J=6.6Hz,3H),1.31(t,J=7.0Hz,3H).
实施例79和80:(S)-7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸

第一步:7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(99a)的手性分离
将99a(0.862g,2.41mmol)通过手性分离得到峰1——99a-1(320mg,892.9mmol)和峰2——99a-2(380mg,1.06mmol)。
手性分离方法:仪器:YMC-K制备系统,色谱柱:CHIRALPAK IC 5.0cm I.D.×25cmL×10μm;色谱柱温:35℃;流速:30.0mL/min;检测波长:254nm;流动相:MeOH=100%。峰1:8.0-9.5min;峰2:11.0-13.0min。
第二步:(S)-7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3-乙氧基-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸
将99a-1(320mg,892.9mmol)溶于8mL四氢呋喃和2mL水中,再加入LiOH·H2O(188.8mg,4.5mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物100(195mg)。
MS(ESI,m/z):331.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.88(s,1H),6.43–6.39(m,1H),6.32(s,1H),6.16–6.12(m,1H),5.26–5.22(m,1H),4.03–3.98(m,2H),3.80–3.75(m,1H),3.36–3.29(m,1H),2.98–2.93(m,2H),1.84–1.37(m,3H),1.32–1.29(m,3H).
将99a-2(380mg,1.06mmol)溶于8mL四氢呋喃和2mL水中,再加入LiOH·H2O(222.44mg,5.30mmol),于室温下反应2h。反应结束后用1N HCl调体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物101(195mg)。
MS(ESI,m/z):331.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.56(s,1H),7.88(s,1H),6.42(d,J=12.8Hz,1H),6.32(s,1H),6.14(d,J=10.9Hz,1H),5.24(dd,J=13.2,6.5Hz,1H),4.00(q,J=7.0Hz,2H),3.82–3.73(m,1H),3.02–2.89(m,3H),1.37(d,J=6.6Hz,3H),1.30(t,J=7.0Hz,3H).
实施例81:7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(102)

第一步:7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(102a)
将化合物87f(2g,9.08mmol)、1-溴-3-氟-5-甲基苯(4.3g,22.7mmol)、Pd2(dba)3(830mg,0.91mmol)、BINAP(1.1g,1.8mmol)和Cs2CO3(8.8g,27.2mmol)加入60mL甲苯中,N2保护下加热至80℃反应24h。反应结束后用硅藻土过滤,滤液旋干后经快速柱层析(洗脱剂体系B)得到化合物102a(1.3g)。
MS(ESI,m/z):329.2[M+H]+.
第二步:7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(102)
将化合物102a(15mg,45.68μmol)加入1mL乙醇和2mL水中,再加入NaOH(9mg,228.4μmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,析出固体,过滤,固体干燥,得化合物102(10mg)。
MS(ESI,m/z):301.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.64(s,1H),8.00(s,1H),7.33(s,1H),6.66–6.60(m,2H),6.36–6.34(m,1H),5.31–5.29(m,1H),3.39–3.32(m,2H),3.02–3.01(m,2H),2.26(s,3H),1.39–1.38(m,3H).
实施例82和83:(S)-7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸

第一步:7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(102a)的手性分离
将102a(1.280g,3.90mmol)通过手性分离得到峰1——102a-1(600mg,1.83mmol)和峰2——102a-2(620mg,1.89mmol)。
手性分离方法:仪器:YMC-K制备系统,色谱柱:CHIRALPAK AY 5.0cm I.D.×25cmL×10μm;色谱柱温:35℃;流速:60.0mL/min;检测波长:254nm;流动相:EtOH/ACN=90%/10%。峰1:12.5-13.5min;峰2:16.5-17.5min。
第二步:(S)-7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸和(R)-7-(3-氟-5-甲基苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸
将102a-1(600mg,1.83mmol)溶于20mL四氢呋喃和4mL水中,再加入LiOH·H2O(383.3mg,9.14mmol),于室温下反应2h。反应结束后用1N HCl调节体系pH约为3,旋去大部分溶剂后,析出固体,抽滤,滤饼经少量水洗涤后,减压干燥后得化合物103(450mg)。
MS(ESI,m/z):301.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.88(s,1H),6.65(s,1H),6.61(d,J=13.1Hz,1H),6.33(d,J=9.4Hz,1H),5.24(q,J=6.1Hz,1H),3.84–3.73(m,1H),3.41–3.28(m,1H),3.04–2.89(m,2H),2.26(s,3H),1.38(d,J=6.6Hz,3H).
将102a-2(620mg,1.89mmol)溶于8mL四氢呋喃和2mL水中,再加入LiOH·H2O(396.4mg,9.44mmol),于室温下反应2h。反应结束后用1N HCl调节体系pH约为3,旋去大部分溶剂后得粗品,粗品经Prep-HPLC分离得化合物104(428mg)。
Prep-HPLC条件:
仪器型号:Agilent 1260,色谱柱:Waters SunFire Prep C18OBD(19mm×150mm×5.0μm);色谱柱温:25℃;流速:28.0mL/min;检测波长:254nm;保留时间:3.6-5.1min,洗脱梯度:(0min:30%A,70%B;16min:90%A,10%B);流动相A:100%乙腈;流动相B:0.05%甲酸水溶液。
MS(ESI,m/z):301.3[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.57(s,1H),7.88(s,1H),6.65(s,1H),6.61(d,J=13.0Hz,1H),6.33(d,J=9.4Hz,1H),5.24(q,J=6.6Hz,1H),3.89–3.73(m,1H),3.37–3.31(m,1H),3.01–2.84(m,2H),2.26(s,3H),1.38(d,J=6.7Hz,3H).
实施例84:7-(3,4-二氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸(105)

第一步:7-(3,4-二氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(105a)
将化合物87f(100mg,454μmol)、5-溴-1,2-二氯-3-氟苯(221.5mg,908μmol)、Pd2(dba)3(10mg,45μmol)、SPHos(37mg,91μmol)和Cs2CO3(370mg,101μmol)溶于5mL二甲苯中,N2保护下加热至150℃反应6h。反应结束后用硅藻土过滤,将滤液旋干后经快速柱层析(洗脱剂体系B)得化合物105a(20mg)。
MS(ESI,m/z):383.1[M+H]+.
第二步:7-(3,4-二氯-5-氟苯基)-8-甲基-5,6,7,8-四氢-2,7-萘啶-3-羧酸乙酯(105)
将化合物105a(20mg,52.2μmol)加入1mL乙醇和2mL水中,再加入NaOH(10.5mg,261μmol),于室温下反应1h。反应结束后用1N HCl调体系pH约为3,析出固体,过滤,固体干燥,得化合物105(6mg)。
MS(ESI,m/z):355.1[M+H]+.
1H NMR(DMSO-d6,400MHz)δ8.49(s,1H),7.83(s,1H),7.10–7.05(m,2H),5.31–5.27(m,1H),3.86–3.81(m,1H),3.35–3.33(m,1H),2.97–2.95(m,2H),1.41–1.38(m,3H).
生物学评价
以下实验进一步描述和解释本发明,但这些实验并非意在限制本发明的范围。
1.HBsAg分泌抑制及细胞毒性检测
实验目的:
本试验的目的是测试本发明化合物对乙肝病毒表面抗原HBsAg分泌的影响和对细胞的毒性。
实验原理:
HepG2.2.15细胞能够向培养基中分泌成熟的乙肝病毒颗粒、HBsAg和HBeAg。细胞分泌的HBsAg可以通过ELISA的方法来定量,并由此检测化合物对病毒HBsAg分泌的影响,同时检测化合物的细胞毒性。
实验方法:
试剂
HepG2.2.15细胞生长培养基:DMEM(Invitrogen 11960-044)+10%FBS(BioseraFB-1280);
HepG2.2.15检测培养基:2%FBS+DMEM
HBV HBsAg diagnostic ELISA kit(乙型肝炎病毒表面抗原检测ELISA试剂盒)(上海科华生物工程股份有限公司,S10910113);
CellTiter-Glo Luminescent Cell Viability Assay kit(发光法细胞活力检测试剂盒)(Promega G7572)
实验步骤和结果:
96孔细胞培养板接种HepG2.2.15细胞15,000个/孔,在37℃,5%CO2细胞培养箱中培养3天至细胞长至满孔,更换含不同浓度药物的培养基,继续培养7天,隔天换液。在第7天取上清液按照乙型肝炎病毒表面抗原检测ELISA试剂盒说明测定细胞上清液中的HBsAg含量,计算对HBsAg的半数抑制浓度(IC50),同时用上述细胞株,用CellTiter方法测定化合物的细胞毒性(CC50),测定次数为2次。实验结果如表1中所示。
表1.本发明化合物对HBsAg分泌抑制的IC50或抑制率及细胞毒性CC50


注:NA表示未测定。
结果表明本发明的化合物对HBsAg分泌具有很高的抑制活性,且具有很低的细胞毒性。
2.安全性评估
(1)hERG实验
采用PredictorTM hERG Fluorescence Polarization Assay Kit(ThermoFisher),按照试剂盒说明测试化合物对hERG钾离子通道的抑制作用,测试浓度为10μM,试验结果见表2。
表2本发明化合物对hERG的抑制
| 化合物 | hERG IC50(μM) |
| 1 | >10 |
| 2 | >10 |
| 3 | >10 |
| 14 | >10 |
| 15 | >10 |
| 36 | >10 |
| 37 | >10 |
| 57 | >10 |
| 60 | >10 |
| 89 | >10 |
| 92 | >10 |
| 101 | >10 |
| 104 | >10 |
结果表明本发明的化合物无hERG抑制作用,导致心脏QT间期延长的可能性小。
(2)CYP酶抑制试验
CYP450是药物代谢中最重要的酶系统,其中最主要的为CYP1A2、CYP2D6和CYP3A4。在对CYP450酶的抑制测试中,采用P450-GloTM CYP1A2Screening System、 CYP2D6CyanScreeningKit和/>
CYP2D6CyanScreeningKit和/> CYP3A4Red Screening Kit,按照试剂盒说明分别测定化合物对CYP1A2、CYP2D6和CYP3A4的抑制活性。测试结果见表3。
CYP3A4Red Screening Kit,按照试剂盒说明分别测定化合物对CYP1A2、CYP2D6和CYP3A4的抑制活性。测试结果见表3。
表3.本发明化合物对CYP酶的抑制试验结果


结果表明本发明的化合物对CYP1A2、CYP2D6、CYP3A4酶无明显抑制作用。
3.大鼠体内的药代动力学(PK)研究
药物及试剂:对于静脉注射(IV),待测化合物用5%DMSO:5%solutol:90%生理盐水配成溶液;对于口服(PO),待测化合物用0.5%MC配成混悬液。
测试用动物:雄性SPF级别SD大鼠(IV、PO各3只),购于成都达硕实验动物有限公司,所有动物给药前禁食10-14小时,给药后4小时恢复给食。
给药剂量:IV为0.5mg/kg或1mg/kg;PO为5mg/kg。
药代动力学测试:将待测化合物分别通过IV和PO给予大鼠,血样经尾静脉采血,每个样品采集约0.30mL,EDTA-K2抗凝。IV采血时间点如下:给药前0min,给药后5min、0.25h、0.5h、1h、2h、4h、6h、8h、24h;PO采血时间点如下:给药前0min,给药后0.25h、0.5h、1h、2h、4h、6h、8h、24h,并于30min内离心(4000转/min,10min,4℃)取血浆,血浆样品分析前存放于-80℃,血浆样品经沉淀蛋白处理后进行LC-MS/MS分析。质谱使用API 5500,液相色谱使用Waters ACQUITY I CLASS系统;色谱柱使用Agela HILIC column(2.1×50mm,3.0μm);流动相:B相为水+0.5%甲酸+5mM乙酸铵,A相为乙腈;流速为0.5mL/min,柱温为40℃;离子源为ESI源正离子模式,扫描方式为多重反应监测(MRM)。使用药代动力学软件WinNonlin6.3非房室模型分别计算化合物的药代动力学参数,结果如下表4所示:
表4本发明化合物在大鼠体内的药代动力学参数

*表示IV和PO实验在不同日期进行,未计算生物利用度。
结果表明,本发明的化合物在SD大鼠体内具有良好的PK性质。
4.Beagle犬体内的药代动力学(PK)研究
药物及试剂:对于静脉注射(IV),待测化合物用5%DMSO:5%solutol:90%生理盐水配成溶液;对于口服(PO),待测化合物用0.5%MC配成混悬液。
测试用动物:雄性Beagle犬(IV、PO各3只),购于北京玛斯生物技术有限公司,所有动物给药前禁食10-14小时,给药后4小时恢复给食。
给药剂量:IV为0.5mg/kg;PO为2.5mg/kg。
药代动力学测试:将待测化合物分别通过IV和PO给予犬,血样经四肢静脉采血,每个样品采集约0.50mL,EDTA-K2抗凝。IV采血时间点如下:给药前0min,给药后5min、0.25h、0.5h、1h、2h、4h、6h、8h、24h;PO采血时间点如下:给药前0min,给药后0.25h、0.5h、1h、2h、4h、6h、8h、24h,并于30min内离心(4000转/min,10min,4℃)取血浆,血浆样品分析前存放于-80℃,血浆样品经沉淀蛋白处理后进行LC-MS/MS分析。质谱使用API 5500,液相色谱使用Waters ACQUITY I CLASS系统;色谱柱使用Agela HILIC column(2.1×50mm,3.0μm);流动相:B相为水+0.5%甲酸+5mM乙酸铵,A相为乙腈;流速为0.5mL/min,柱温为40℃;离子源为ESI源正离子模式,扫描方式为多重反应监测(MRM)。使用药代动力学软件WinNonlin6.3非房室模型分别计算化合物的药代动力学参数,结果如下表5所示:
表5本发明化合物在Beagle犬体内的药代动力学参数
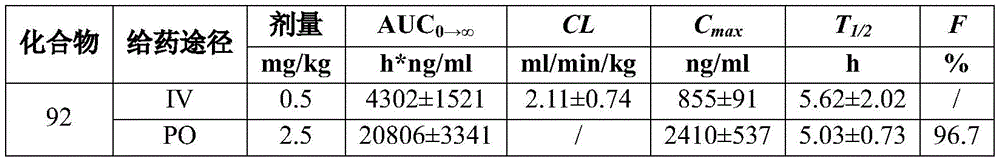
结果表明,本发明的化合物(例如化合物92)在Beagle犬体内具有优秀的PK性质。
5.食蟹猴体内的药代动力学(PK)研究
药物及试剂:对于静脉注射(IV),待测化合物用5%DMSO:5%solutol:90%生理盐水配成溶液;对于口服(PO),待测化合物用0.5%MC配成混悬液。
测试用动物:雌性食蟹猴(IV、PO各3只),购于海南金港生物技术股份有限公司,所有动物给药前禁食10-14小时,给药后4小时恢复给食。
给药剂量:IV为0.5mg/kg;PO为2.5mg/kg。
药代动力学测试:将待测化合物分别通过IV和PO给予猴,血样经四肢静脉采血,每个样品采集约0.50mL,EDTA-K2抗凝。IV采血时间点如下:给药前0min,给药后5min、0.25h、0.5h、1h、2h、4h、6h、8h、24h;PO采血时间点如下:给药前0min,给药后0.25h、0.5h、1h、2h、4h、6h、8h、24h,并于30min内离心(4000转/min,10min,4℃)取血浆,血浆样品分析前存放于-80℃,血浆样品经沉淀蛋白处理后进行LC-MS/MS分析。质谱使用API 5500,液相色谱使用Waters ACQUITY I CLASS系统;色谱柱使用Agela HILIC column(2.1×50mm,3.0μm);流动相:B相为水+0.5%甲酸+5mM乙酸铵,A相为乙腈;流速为0.5mL/min,柱温为40℃;离子源为ESI源正离子模式,扫描方式为多重反应监测(MRM)。使用药代动力学软件WinNonlin6.3非房室模型分别计算化合物的药代动力学参数,结果如下表6所示:
表6本发明化合物在食蟹猴体内的药代动力学参数

结果表明,本发明的化合物(例如化合物92)在食蟹猴体内具有优秀的PK性质。
除本文中描述的那些外,根据前述描述,本发明的多种修改也意图落入所附权利要求书的范围内。本申请中所引用的各参考文献(包括所有专利、专利申请、期刊文章、书籍及任何其它公开)均以其整体援引加入本文。
Claims (72)
1.式I的化合物或者其立体异构体、药学上可接受的盐:

其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被1、2或3个独立地选自下列基团的取代基取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、卤代C1-C3烷氧基、-C(O)NH2、-SO2CH3、-OC1-C3烷基-OC1-C3烷基、-OC1-C3烷基-OH、-NR9R10和5至10元螺杂环基;
R2和R3各自独立地选自氢和未取代的C1-C6烷基,或者R2和R3和其所连接的碳原子一起形成3-6元环烷基;
R4选自-C(O)R5、-CO2R6、-C(O)NR9R10;
其中:R5选自C1-C6烷基;
R6选自氢、C1-C6烷基和C3-C7环烷基;
R9和R10各自独立地选自氢、C1-C6烷基,或者R9和R10和其所连接的氮原子一起形成4-7元杂环基,所述4-7元杂环基可任选地被取代基取代;
X为N,Y为CH;
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、NH2、-NH(C1-C6烷基)、-C1-C6烷基-OC1-C6烷基、OC1-C6烷基-OC1-C6烷基、羟基C1-C6烷基。
2.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被1、2或3个独立地选自独立地选自下列基团的取代基取代:氟、氯、溴、甲氧基、乙氧基、丙氧基、异丙氧基、氟甲氧基、二氟甲氧基、三氟甲氧基、甲基、乙基、丙基、异丙基、氰基、氟甲基、二氟甲基、三氟甲基、羟基甲氧基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2、-SO2CH3、-NR9R10和5至10元螺杂环基。
3.权利要求2的化合物或者其立体异构体、药学上可接受的盐,其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被1、2或3个独立地选自下列基团组的取代基取代:
氟、氯、溴、甲氧基、乙氧基、异丙氧基、氟甲氧基、二氟甲氧基、三氟甲氧基、甲基、氰基、三氟甲基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2和-SO2CH3;
-NR9R10,其中R9和R10和其所连接的氮原子一起形成可任选地被取代基取代的4-6元杂环基;和
9至10元含氮螺杂环基。
4.权利要求3的化合物或者其立体异构体、药学上可接受的盐,其中:
所述4-6元杂环基选自任选地被1、2或3个独立地选自下列基团的取代基取代的
 羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基和羟基C1-C6烷基。
羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基和羟基C1-C6烷基。
5.权利要求4的化合物或者其立体异构体、药学上可接受的盐,其中:所述4-6元杂环基选自任选地被1、2或3个独立地选自下列基团的取代基取代的 羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和羟基C1-C3烷基-。
羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和羟基C1-C3烷基-。
6.权利要求5的化合物或者其立体异构体、药学上可接受的盐,其中:
所述4-6元杂环基选自任选地被1、2或3个独立地选自下列基团的取代基取代的
 羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基。
羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基。
7.权利要求6的化合物或者其立体异构体、药学上可接受的盐,其中:
所述4-6元杂环基选自

 以及所述9至10元含氮螺杂环基选自/>
以及所述9至10元含氮螺杂环基选自/>
8.权利要求7的化合物或者其立体异构体、药学上可接受的盐,其中:
所述9至10元含氮螺杂环基选自
9.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被1、2或3个下列基团取代:氰基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、-C(O)NH2、-SO2CH3、-OC1-C3烷基-OC1-C3烷基或-OC1-C3烷基-OH。
10.权利要求9的化合物或者其立体异构体、药学上可接受的盐,其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被1、2或3个下列基团取代:氟、氯、溴、甲氧基、乙氧基、异丙氧基、甲基、氰基、三氟甲基、一氟甲氧基、二氟甲氧基、三氟甲氧基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2或-SO2CH3。
11.权利要求10的化合物或者其立体异构体、药学上可接受的盐,其中:
R1选自苯基和吡啶基,所述苯基和吡啶基可任选地被1、2或3个下列基团取代:氟、甲氧基、甲基、氰基、三氟甲基、2-羟基乙氧基、2-甲氧基乙氧基、-C(O)NH2或-SO2CH3。
12.权利要求1-11中任一项的化合物或者其立体异构体、药学上可接受的盐,其中:
R2和R3各自独立地选自氢和C1-C3烷基,或者R2和R3和其所连接的碳原子一起形成3-6元环烷基。
13.权利要求12的化合物或者其立体异构体、药学上可接受的盐,其中:
R2和R3各自独立地选自氢、甲基和乙基,或者R2和R3和其所连接的碳原子一起形成环丙基。
14.权利要求13的化合物或者其立体异构体、药学上可接受的盐,其中:
R2和R3各自独立地选自氢和甲基,或者R2和R3和其所连接的碳原子一起形成环丙基。
15.权利要求14的化合物或者其立体异构体、药学上可接受的盐,其中:
R2和R3各自独立地选自氢和甲基。
16.权利要求1-11、13-15中任一项的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)C1-C6烷基、-C(O)OC1-C6烷基、-C(O)OH、-C(O)NH2和-C(O)NH(C1-C6烷基)。
17.权利要求16的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)C1-C3烷基、-C(O)OC1-C3烷基、-C(O)OH和-C(O)NH2。
18.权利要求17的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)OCH3、-C(O)OEt、-C(O)OH、-C(O)NH2、-C(O)CH3和-C(O)Et。
19.权利要求18的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)OEt、-C(O)OH、-C(O)NH2和-C(O)CH3。
20.权利要求19的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)OEt和-C(O)OH。
21.权利要求20的化合物或者其立体异构体、药学上可接受的盐,其中:
R4为-C(O)OH。
22.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
R5选自C1-C3烷基。
23.权利要求22的化合物或者其立体异构体、药学上可接受的盐,其中:
R5为甲基。
24.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
R6选自氢、C1-C3烷基和C3-C5环烷基。
25.权利要求24的化合物或者其立体异构体、药学上可接受的盐,其中:
R6为氢、甲基或乙基。
26.权利要求25的化合物或者其立体异构体、药学上可接受的盐,其中:
R6为氢或乙基。
27.权利要求26的化合物或者其立体异构体、药学上可接受的盐,其中:
R6为氢。
28.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
R9和R10各自独立地选自氢、C1-C3烷基,或者R9和R10和其所连接的氮原子一起形成4-6元杂环基,所述4-6元杂环基可任选地被取代基取代。
29.权利要求28的化合物或者其立体异构体、药学上可接受的盐,其中:
R9和R10为氢。
30.权利要求28的化合物或者其立体异构体、药学上可接受的盐,其中:
R9和R10和其所连接的氮原子一起形成可任选地被取代基取代的4-6元杂环基,所述4-6元杂环基选自任选地被1、2或3个独立地选自下列基团的取代基取代的
 羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基和羟基C1-C6烷基。
羟基、卤素、C1-C6烷基、卤代C1-C6烷基、C1-C6烷氧基、-OC1-C6烷基-OH、卤代C1-C6烷氧基、氰基、硝基、-NH2、-C1-C6烷基-OC1-C6烷基、-OC1-C6烷基-OC1-C6烷基和羟基C1-C6烷基。
31.权利要求30的化合物或者其立体异构体、药学上可接受的盐,其中:
所述4-6元杂环基选自任选地被1、2或3个独立地选自下列基团的取代基取代的
 羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和HO-C1-C3烷基-。
羟基、氟、氯、溴、C1-C3烷基、C1-C3烷氧基、-C1-C3烷基-OC1-C3烷基和HO-C1-C3烷基-。
32.权利要求31的化合物或者其立体异构体、药学上可接受的盐,其中:
所述4-6元杂环基选自任选地被1、2或3个独立地选自下列基团的取代基取代的
 羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基。
羟基、氟、氯、溴、甲基、乙基、甲氧基、乙氧基、甲氧基甲基、甲氧基乙基、羟甲基和羟乙基。
33.权利要求32的化合物或者其立体异构体、药学上可接受的盐,其中:
所述4-6元杂环基选自 
34.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基、卤素、C1-C3烷基、卤代C1-C3烷基、C1-C3烷氧基、-OC1-C3烷基-OH、卤代C1-C3烷氧基、-NH2、-NH(C1-C3烷基)、-C1-C3烷基-OC1-C3烷基、羟基C1-C3烷基。
35.权利要求34的化合物或者其立体异构体、药学上可接受的盐,其中:
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:羟基;氟、氯、溴;甲基、乙基、异丙基;氟甲基、二氟甲基、三氟甲基;甲氧基、乙氧基、异丙氧基;-OCH2OH、-OCH2CH3OH;氟甲氧基、二氟甲氧基、三氟甲氧基;-NH2、-NHCH3、-NHCH2CH3;-CH2-OCH3、-CH2CH2-OCH3、-CH2-OCH2CH3、-CH2CH2-OCH2CH3;-OCH2-OCH3、-OCH2-OCH2CH3、-OCH2CH2-OCH3;-CH2OH、-CH2CH2OH。
36.权利要求35的化合物或者其立体异构体、药学上可接受的盐,其中:
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:氟、氯、溴;甲基、乙基、异丙基;氟甲基、二氟甲基、三氟甲基;甲氧基、乙氧基、异丙氧基;-OCH2OH、-OCH2CH3OH;氟甲氧基、二氟甲氧基、三氟甲氧基;-CH2-OCH3、-CH2CH2-OCH3、-CH2-OCH2CH3、-CH2CH2-OCH2CH3;-OCH2-OCH3、-OCH2-OCH2CH3、-OCH2CH2-OCH3;-CH2OH、-CH2CH2OH。
37.权利要求36的化合物或者其立体异构体、药学上可接受的盐,其中:
所述“被取代基取代”是指任选被一个或多个独立地选自下列基团的取代基所取代:氟、氯、溴;甲基;三氟甲基;甲氧基、乙氧基、异丙氧基;-OCH2CH3OH;氟甲氧基、二氟甲氧基、三氟甲氧基;-CH2-OCH3;-OCH2CH2-OCH3;-CH2OH。
38.权利要求1-11、17-37中任一项的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物为式II的化合物:

39.权利要求1-11、17-37中任一项的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物为式IV的化合物:


其中:
R2为未取代的C1-C6烷基;且
R3为氢或未取代的C1-C6烷基;
或者
R2和R3和其所连接的碳原子一起形3-6元环烷基。
40.权利要求39的化合物或者其立体异构体、药学上可接受的盐,其中:
R2为未取代的C1-C3烷基;且
R3为氢或未取代的C1-C3烷基;
或者
R2和R3和其所连接的碳原子一起形成环丙基。
41.权利要求40的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物为式IV的化合物,其中:
R2为甲基或乙基;且
R3为氢、甲基或乙基。
42.权利要求41的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物为式IV的化合物,其中:
R2为甲基;且
R3为氢或甲基。
43.权利要求39的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物是式V或VI的化合物:

44.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物为式VII的化合物:

其中:
W1、W2和W3之中一个为N,另外两个为CR17;
m为0、1、2、3;
R17各自独立地选自下列基团:
(1)氢、卤素、C1-C3烷基和C1-C3烷氧基;
(2)-NR9R10,其中R9和R10如权利要求30所定义;以及
(3)5至10元螺杂环基。
45.权利要求44的化合物或者其立体异构体、药学上可接受的盐,其中:
R17定义中:
卤素选自氟、氯和溴;以及
5至10元螺杂环基为9至10元含氮螺杂环基。
46.权利要求45的化合物或者其立体异构体、药学上可接受的盐,其中:
R17定义中的9至10元含氮螺杂环基为
47.权利要求46的化合物或者其立体异构体、药学上可接受的盐,其中:
R17定义中的9至10元含氮螺杂环基为
48.权利要求44的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)OEt、-C(O)OH、-C(O)NH2和-C(O)CH3;
R17各自独立地选自下列基团:
(1)氢、卤素、C1-C3烷基和C1-C3烷氧基;
(2)-NR9R10,其中R9和R10如权利要求30所定义;以及
(3)
49.权利要求48的化合物或者其立体异构体、药学上可接受的盐,其中:
R17选自氢、氟、氯、溴、甲基、乙基、丙基、甲氧基、乙氧基和
50.权利要求44或48的化合物或者其立体异构体、药学上可接受的盐,其中:
R4为-C(O)OH;
R17各自独立地选自氢、甲基、甲氧基、氟、


51.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物为式VIII的化合物:

其中:
R3为氢或未取代的C1-C6烷基;
R4如权利要求1中所定义;
R18、R19、R20和R21各自独立地选自H、卤素、氰基、卤代C1-C3烷基、C1-C3烷基、-OC1-C3烷基、-OC1-C3烷基-OC1-C3烷基、-OC1-C3烷基-OH、卤代C1-C3烷氧基、-NR9R10,并且R18、R19、R20和R21中至少1个为氢;并且
R9和R10各自如权利要求1中所定义。
52.权利要求51的化合物或者其立体异构体、药学上可接受的盐,其中:
R3为氢或未取代的C1-C3烷基。
53.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中:
R3为氢、甲基或乙基。
54.权利要求53的化合物或者其立体异构体、药学上可接受的盐,其中:
R3为H或甲基。
55.权利要求51-54任一项的化合物或者其立体异构体、药学上可接受的盐,其中:
R18、R19、R20和R21各自独立地选自H、氟、氯、溴、氰基、三氟甲基、甲基、甲氧基、乙氧基、异丙氧基、-O-CH2-CH2-OMe、-O-CH2CH2-OH、三氟甲氧基、氟甲氧基和二氟甲氧基。
56.权利要求55的化合物或者其立体异构体、药学上可接受的盐,其中:
R4选自-C(O)OEt和-C(O)OH;
R18选自H、氟、氯、甲基、氰基和三氟甲基;
R19选自H、氟、氯、甲氧基和甲基;
R20选自H、氟、氯、三氟甲基、甲基和氰基;
R21选自H、氟、氯、溴、甲氧基、乙氧基、-O-CH2-CH2-OMe、三氟甲基、-O-CH2-CH2-OH、氰基、-C(O)NH2、三氟甲氧基、异丙氧基、二氟甲氧基、氟甲氧基和甲基。
57.权利要求1的化合物或者其立体异构体、药学上可接受的盐,其中所述化合物选自:




58.药物组合物,其包含权利要求1-57中任一项的化合物或者其立体异构体、药学上可接受的盐,以及一种或多种药学上可接受的载体。
59.药物制剂,其包含权利要求1-57中任一项的化合物或者其立体异构体、药学上可接受的盐,或者权利要求58的药物组合物。
60.权利要求1-57中任一项的化合物或者其立体异构体、药学上可接受的盐,权利要求58的药物组合物或者权利要求59的药物制剂在制备用于治疗与HBsAg分泌过度相关的疾病的药物中的用途。
61.权利要求60的用途,其中所述疾病为乙型肝炎。
62.权利要求60或61的用途,其中所述药物还包含其他治疗与HBsAg分泌过度相关的疾病或病症的药剂。
63.制备式II的化合物的方法:

其中:
R4选自-C(O)R5、-CO2R6和-C(O)NR9R10;
X、Y、R1、R5、R6、R9和R10如权利要求38中所定义;
所述方法包括以下步骤:

第一步:化合物II-1经还原反应生成化合物II-2,其中Ra为C1-C4烷基;
第二步:化合物II-2与R1-L经偶联或亲核取代反应生成化合物II-3,其中L为离去基团;
第三步:化合物II-3经合适的反应转化成所述式II的目标化合物。
64.权利要求63的制备方法,其中:
Ra为甲基、乙基或异丙基;和/或
L为卤素或三氟甲磺酰氧基。
65.权利要求64的制备方法,其中:
Ra为乙基;和/或
L为F、Cl、Br、I或三氟甲磺酰氧基。
66.制备式V的化合物的方法:
其中:
R4选自-C(O)R5、-CO2R6、和-C(O)NR9R10;
R1、R5、R6、R9和R10如权利要求43中所定义;
所述方法包括以下步骤:

第一步:化合物V-1与适合的有机锡化合物经偶联反应生成化合物V-2,其中Ra在每次出现时独立地选自C1-C4烷基;Rb为离去基团;
第二步:化合物V-2经环化反应生成化合物V-3;
第三步:使化合物V-3转化成化合物V-4,其中PG3为适合的氨基保护基;
第四步:化合物V-4经Kulinkovich反应生成化合物V-5;
第五步:化合物V-5经脱保护反应生成化合物V-6;
第六步:化合物V-6与R1-L经偶联或亲核取代反应生成化合物V-7,其中L为离去基团;
第七步:化合物V-7经合适的反应转化成所述式V的目标化合物。
67.权利要求66的制备方法,其中:
Ra在每次出现时独立地选自甲基、乙基或异丙基;和/或
Rb选自卤素或三氟甲磺酰氧基;和/或
PG3为苄基、对甲苯磺酰基、苯甲酰基、苄氧羰基、烯丙氧羰基、甲氧羰基、乙氧羰基或叔丁氧羰基;和/或
L为卤素或三氟甲磺酰氧基。
68.权利要求67的制备方法,其中:
Ra在每次出现时独立地选自乙基;和/或
Rb选自Cl、Br、I或三氟甲磺酰氧基;和/或
L为F、Cl、Br、I或三氟甲磺酰氧基。
69.制备式VI的化合物的方法:

其中:
R4选自-C(O)R5、-CO2R6和-C(O)NR9R10;
R1、R5、R6、R9和R10如权利要求43中所定义;
所述方法包括以下步骤:

第一步:化合物VI-1与NC-C(O)ORa经[2+2+2]环加成反应生成化合物VI-2,其中PG3为氨基保护基;Ra为C1-C4烷基;
第二步:化合物VI-2经脱保护反应生成化合物VI-3;
第三步:化合物VI-3与R1-L发生偶联反应或亲核取代反应生成化合物VI-4,其中L为离去基团;
第四步:化合物VI-4经合适的反应转化成所述式VI的目标化合物。
70.权利要求69的制备方法,其中:
PG3为苄基、对甲苯磺酰基、苯甲酰基、苄氧羰基、烯丙氧羰基、甲氧羰基、乙氧羰基或叔丁氧羰基;和/或
Ra为甲基、乙基或异丙基;和/或
L为卤素或OTf。
71.权利要求70的制备方法,其中:
PG3为对甲苯磺酰基;和/或
Ra为甲基或乙基;和/或
L为F、Cl、Br、I或OTf。
72.权利要求63-71中任一项的方法,其中所述合适的反应选自:
(1)经水解生成其中R4为-CO2H的目标化合物;
(2)与醇R6OH发生酯交换生成其中R4为-CO2R6且R6不为H的目标化合物;
(3)与HN(OMe)Me反应生成Weinreb酰胺,然后所得的Weinreb酰胺与格氏试剂R5MgBr反应生成其中R4为-C(O)R5的目标化合物;
和
(5)与HNR9R10发生胺解反应生成其中R4为-C(O)NR9R10的目标化合物,或者发生水解反应形成酸,然后所得的酸与胺HNR9R10缩合生成其中R4为-C(O)NR9R10的目标化合物。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810017350 | 2018-01-09 | ||
| CN2018100173507 | 2018-01-09 | ||
| CN201811329216 | 2018-11-09 | ||
| CN2018113292167 | 2018-11-09 | ||
| PCT/CN2018/123415 WO2019137201A1 (zh) | 2018-01-09 | 2018-12-25 | 杂芳基并四氢吡啶类化合物、其制备方法、药物组合物及应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111315744A CN111315744A (zh) | 2020-06-19 |
| CN111315744B true CN111315744B (zh) | 2023-07-14 |
Family
ID=67219307
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201880072563.8A Active CN111315744B (zh) | 2018-01-09 | 2018-12-25 | 杂芳基并四氢吡啶类化合物、其制备方法、药物组合物及应用 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN111315744B (zh) |
| WO (1) | WO2019137201A1 (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110857298B (zh) * | 2018-08-22 | 2022-05-10 | 四川科伦博泰生物医药股份有限公司 | 杂芳基并四氢吡啶类化合物、包含其的药物组合物、其制备方法及其用途 |
| CN112142730A (zh) * | 2019-06-28 | 2020-12-29 | 四川科伦博泰生物医药股份有限公司 | 杂芳基并四氢吡啶类化合物的固体形式及其制备方法 |
| KR20220124176A (ko) | 2019-12-06 | 2022-09-13 | 버텍스 파마슈티칼스 인코포레이티드 | 나트륨 채널의 조절제로서의 치환된 테트라하이드로푸란 |
| WO2021130270A1 (en) | 2019-12-24 | 2021-07-01 | F. Hoffmann-La Roche Ag | Pharmaceutical combination of antiviral agents targeting hbv and/or an immune modulator for treatment of hbv |
| AU2022284886A1 (en) | 2021-06-04 | 2023-11-30 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016177655A1 (en) * | 2015-05-04 | 2016-11-10 | F. Hoffmann-La Roche Ag | Tetrahydropyridopyrimidines and tetrahydropyridopyridines as inhibitors of hbsag (hbv surface antigen) and hbv dna production for the treatment of hepatitis b virus infections |
| CN107108610A (zh) * | 2014-12-30 | 2017-08-29 | 豪夫迈·罗氏有限公司 | 用于治疗和预防肝炎b病毒感染的新的四氢吡啶并嘧啶和四氢吡啶并吡啶化合物 |
| WO2018083081A1 (en) * | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydropyridopyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
-
2018
- 2018-12-25 WO PCT/CN2018/123415 patent/WO2019137201A1/zh active Application Filing
- 2018-12-25 CN CN201880072563.8A patent/CN111315744B/zh active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107108610A (zh) * | 2014-12-30 | 2017-08-29 | 豪夫迈·罗氏有限公司 | 用于治疗和预防肝炎b病毒感染的新的四氢吡啶并嘧啶和四氢吡啶并吡啶化合物 |
| WO2016177655A1 (en) * | 2015-05-04 | 2016-11-10 | F. Hoffmann-La Roche Ag | Tetrahydropyridopyrimidines and tetrahydropyridopyridines as inhibitors of hbsag (hbv surface antigen) and hbv dna production for the treatment of hepatitis b virus infections |
| WO2018083081A1 (en) * | 2016-11-03 | 2018-05-11 | F. Hoffmann-La Roche Ag | Novel tetrahydropyridopyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111315744A (zh) | 2020-06-19 |
| WO2019137201A1 (zh) | 2019-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111315744B (zh) | 杂芳基并四氢吡啶类化合物、其制备方法、药物组合物及应用 | |
| CN113563323B (zh) | 一类苯并噻唑基联芳基类化合物、制备方法和用途 | |
| EP3240537B1 (en) | Novel tetrahydropyridopyrimidines and tetrahydropyridopyridines for the treatment and prophylaxis of hepatitis b virus infection | |
| JP6285440B2 (ja) | 融合二環式スルファモイル誘導体並びにb型肝炎の治療のための薬剤としてのその使用 | |
| TWI715593B (zh) | 新穎羥基酸衍生物,其製備方法及含彼等之醫藥組合物 | |
| KR20210093951A (ko) | 항바이러스제로서의 작용화된 헤테로사이클 | |
| KR20220101083A (ko) | 항바이러스 헤테로사이클릭 화합물 | |
| CN112424185B (zh) | 含苯环的化合物、其制备方法及应用 | |
| JP2020526569A (ja) | 環内チアミジノアミド−アリールアミド系化合物及びb型肝炎を治療するためのその用途 | |
| EP3816163A1 (en) | Cell necrosis inhibitor, preparation method therefor and use thereof | |
| JP2009507896A (ja) | ヒスタミンh4受容体活性の調節剤としての2−アミノピリミジン誘導体 | |
| CN112368281B (zh) | 作为pde3/pde4双重抑制剂的三并环类化合物 | |
| CN113330009B (zh) | 氮杂环化合物、其制备方法及用途 | |
| CN111683662A (zh) | 取代嘧啶类化合物及其药物组合物和治疗方法 | |
| KR102600391B1 (ko) | 삼중고리형 화합물 | |
| CN113045569B (zh) | 用作ret激酶抑制剂的化合物及其应用 | |
| JP5781537B2 (ja) | ヒスタミンh4受容体アンタゴニストとしてのアミノアルキルピリミジン誘導体 | |
| CN113880865A (zh) | 吡唑[1,5-a]吡啶类化合物及其制备方法与应用 | |
| JP2022529466A (ja) | 二環式及び三環式化合物 | |
| EP4206197A1 (en) | Preparation method for novel rho-related protein kinase inhibitor and intermediate in preparation method | |
| US11773121B2 (en) | Antiviral compounds | |
| CN115368382A (zh) | Kras g12d抑制剂及其在医药上的应用 | |
| CN113166109B (zh) | 氨基吡啶类化合物及其制备方法和用途 | |
| CN111377925B (zh) | 嘌呤类衍生物、其制备方法及其在医药上的应用 | |
| CN112876419A (zh) | 烯丙胺衍生物及其制备方法和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |