CN109641911B - seco-环丙吡咯并吲哚化合物和其抗体-药物缀合物以及制备和使用方法 - Google Patents
seco-环丙吡咯并吲哚化合物和其抗体-药物缀合物以及制备和使用方法 Download PDFInfo
- Publication number
- CN109641911B CN109641911B CN201780050827.5A CN201780050827A CN109641911B CN 109641911 B CN109641911 B CN 109641911B CN 201780050827 A CN201780050827 A CN 201780050827A CN 109641911 B CN109641911 B CN 109641911B
- Authority
- CN
- China
- Prior art keywords
- alkyl
- compound
- alkyl radical
- antibody
- independently
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940049595 antibody-drug conjugate Drugs 0.000 title claims abstract description 25
- 239000000611 antibody drug conjugate Substances 0.000 title claims abstract description 8
- 238000000034 method Methods 0.000 title description 34
- 125000000217 alkyl group Chemical group 0.000 claims description 102
- 150000001875 compounds Chemical class 0.000 claims description 79
- 206010028980 Neoplasm Diseases 0.000 claims description 48
- 125000003118 aryl group Chemical group 0.000 claims description 42
- 239000000562 conjugate Substances 0.000 claims description 41
- 201000011510 cancer Diseases 0.000 claims description 35
- 239000003814 drug Substances 0.000 claims description 33
- 238000006467 substitution reaction Methods 0.000 claims description 22
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims description 18
- 229910052801 chlorine Inorganic materials 0.000 claims description 16
- 229910052757 nitrogen Inorganic materials 0.000 claims description 15
- 229910052794 bromium Inorganic materials 0.000 claims description 14
- 229910052731 fluorine Inorganic materials 0.000 claims description 14
- 125000001072 heteroaryl group Chemical group 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 229910052799 carbon Inorganic materials 0.000 claims description 11
- 150000003839 salts Chemical class 0.000 claims description 11
- 235000018417 cysteine Nutrition 0.000 claims description 10
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 10
- 235000004554 glutamine Nutrition 0.000 claims description 10
- 229960002743 glutamine Drugs 0.000 claims description 10
- 239000004471 Glycine Substances 0.000 claims description 9
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 claims description 9
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 claims description 9
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims description 9
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 9
- 229910004013 NO 2 Inorganic materials 0.000 claims description 9
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 9
- 229960002433 cysteine Drugs 0.000 claims description 9
- 229960002449 glycine Drugs 0.000 claims description 9
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 8
- 239000004472 Lysine Substances 0.000 claims description 8
- 229960003646 lysine Drugs 0.000 claims description 8
- 235000018977 lysine Nutrition 0.000 claims description 8
- 206010009944 Colon cancer Diseases 0.000 claims description 6
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 6
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 6
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 claims description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims description 6
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 claims description 6
- 206010033128 Ovarian cancer Diseases 0.000 claims description 6
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 6
- 235000003704 aspartic acid Nutrition 0.000 claims description 6
- 229960005261 aspartic acid Drugs 0.000 claims description 6
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 claims description 6
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 claims description 6
- 235000013477 citrulline Nutrition 0.000 claims description 6
- 229960002173 citrulline Drugs 0.000 claims description 6
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 claims description 6
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 5
- 239000004475 Arginine Substances 0.000 claims description 5
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 5
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 claims description 5
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 5
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 5
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims description 5
- 235000009697 arginine Nutrition 0.000 claims description 5
- 229960003121 arginine Drugs 0.000 claims description 5
- 235000009582 asparagine Nutrition 0.000 claims description 5
- 229960001230 asparagine Drugs 0.000 claims description 5
- 208000029742 colonic neoplasm Diseases 0.000 claims description 5
- 235000013922 glutamic acid Nutrition 0.000 claims description 5
- 229960002989 glutamic acid Drugs 0.000 claims description 5
- 239000004220 glutamic acid Substances 0.000 claims description 5
- 239000004474 valine Substances 0.000 claims description 5
- 229960004295 valine Drugs 0.000 claims description 5
- 235000014393 valine Nutrition 0.000 claims description 5
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 4
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 4
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 4
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 4
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 4
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 claims description 4
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 claims description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 4
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 4
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 4
- 235000004279 alanine Nutrition 0.000 claims description 4
- 229960003767 alanine Drugs 0.000 claims description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 claims description 4
- UHBYWPGGCSDKFX-VKHMYHEASA-N gamma-carboxy-L-glutamic acid Chemical compound OC(=O)[C@@H](N)CC(C(O)=O)C(O)=O UHBYWPGGCSDKFX-VKHMYHEASA-N 0.000 claims description 4
- 206010017758 gastric cancer Diseases 0.000 claims description 4
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 4
- 229960000310 isoleucine Drugs 0.000 claims description 4
- 229960003136 leucine Drugs 0.000 claims description 4
- 201000005202 lung cancer Diseases 0.000 claims description 4
- 208000020816 lung neoplasm Diseases 0.000 claims description 4
- 229930182817 methionine Natural products 0.000 claims description 4
- 229960004452 methionine Drugs 0.000 claims description 4
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 4
- 229960005190 phenylalanine Drugs 0.000 claims description 4
- 201000011549 stomach cancer Diseases 0.000 claims description 4
- 125000003107 substituted aryl group Chemical group 0.000 claims description 4
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 claims description 3
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 claims description 3
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 claims description 3
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 claims description 3
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 claims description 3
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 claims description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 claims description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 3
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 claims description 3
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 claims description 3
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 claims description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 3
- 239000004473 Threonine Substances 0.000 claims description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 3
- 229940000635 beta-alanine Drugs 0.000 claims description 3
- 229960003692 gamma aminobutyric acid Drugs 0.000 claims description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims description 3
- 229960002885 histidine Drugs 0.000 claims description 3
- 229960003104 ornithine Drugs 0.000 claims description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims 1
- 239000002246 antineoplastic agent Substances 0.000 abstract description 5
- 230000003389 potentiating effect Effects 0.000 abstract description 4
- 239000011541 reaction mixture Substances 0.000 description 66
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 50
- -1 CPI compound Chemical class 0.000 description 40
- 238000006243 chemical reaction Methods 0.000 description 40
- 210000004027 cell Anatomy 0.000 description 39
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 38
- 230000021615 conjugation Effects 0.000 description 37
- 239000000243 solution Substances 0.000 description 37
- 150000001413 amino acids Chemical class 0.000 description 34
- 125000005647 linker group Chemical group 0.000 description 32
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 31
- 229940024606 amino acid Drugs 0.000 description 30
- 235000001014 amino acid Nutrition 0.000 description 30
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 29
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 29
- 239000000427 antigen Substances 0.000 description 28
- 102000036639 antigens Human genes 0.000 description 28
- 108091007433 antigens Proteins 0.000 description 28
- 230000027455 binding Effects 0.000 description 26
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 229940079593 drug Drugs 0.000 description 24
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 23
- 239000007787 solid Substances 0.000 description 22
- 125000003396 thiol group Chemical group [H]S* 0.000 description 22
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 21
- 239000000203 mixture Substances 0.000 description 19
- 108090000765 processed proteins & peptides Proteins 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 235000019439 ethyl acetate Nutrition 0.000 description 18
- 125000006850 spacer group Chemical group 0.000 description 18
- 238000003776 cleavage reaction Methods 0.000 description 17
- 230000007017 scission Effects 0.000 description 17
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 16
- 238000003786 synthesis reaction Methods 0.000 description 15
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 14
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 14
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- 108060008539 Transglutaminase Proteins 0.000 description 12
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 12
- 102000004196 processed proteins & peptides Human genes 0.000 description 12
- 102000003601 transglutaminase Human genes 0.000 description 12
- 150000001412 amines Chemical class 0.000 description 11
- 239000012267 brine Substances 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 125000000753 cycloalkyl group Chemical group 0.000 description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 11
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 11
- 230000008685 targeting Effects 0.000 description 11
- 230000037396 body weight Effects 0.000 description 10
- 239000000460 chlorine Substances 0.000 description 10
- 239000012634 fragment Substances 0.000 description 10
- 229910052736 halogen Inorganic materials 0.000 description 10
- 150000002367 halogens Chemical class 0.000 description 10
- SFJWZPGYDWHHIZ-UHFFFAOYSA-N methyl 2-isocyano-2-methylpropanoate Chemical group COC(=O)C(C)(C)[N+]#[C-] SFJWZPGYDWHHIZ-UHFFFAOYSA-N 0.000 description 10
- 239000008194 pharmaceutical composition Substances 0.000 description 10
- 102000035195 Peptidases Human genes 0.000 description 9
- 108091005804 Peptidases Proteins 0.000 description 9
- 239000004365 Protease Substances 0.000 description 9
- 229910052770 Uranium Inorganic materials 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 125000003277 amino group Chemical group 0.000 description 9
- 238000001035 drying Methods 0.000 description 9
- 229920001184 polypeptide Polymers 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 239000011734 sodium Substances 0.000 description 9
- 101000662009 Homo sapiens UDP-N-acetylglucosamine pyrophosphorylase Proteins 0.000 description 8
- 102100037921 UDP-N-acetylglucosamine pyrophosphorylase Human genes 0.000 description 8
- 125000001931 aliphatic group Chemical group 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 125000000524 functional group Chemical group 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 238000002953 preparative HPLC Methods 0.000 description 8
- 235000018102 proteins Nutrition 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 239000002002 slurry Substances 0.000 description 8
- 125000001424 substituent group Chemical group 0.000 description 8
- 125000003342 alkenyl group Chemical group 0.000 description 7
- 125000003545 alkoxy group Chemical group 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 125000004093 cyano group Chemical group *C#N 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 125000001188 haloalkyl group Chemical group 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 7
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 235000019253 formic acid Nutrition 0.000 description 6
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 6
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 6
- LDNCWSAHVWXJNF-ZEELXFFVSA-N 2-(3-aminopropyl)-1-[(e)-[(2e)-2-[[n'-(3-aminopropyl)carbamimidoyl]hydrazinylidene]ethylidene]amino]guanidine Chemical compound NCCCN=C(N)N\N=C\C=N\NC(N)=NCCCN LDNCWSAHVWXJNF-ZEELXFFVSA-N 0.000 description 5
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 238000006845 Michael addition reaction Methods 0.000 description 5
- HSRXSKHRSXRCFC-WDSKDSINSA-N Val-Ala Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](C)C(O)=O HSRXSKHRSXRCFC-WDSKDSINSA-N 0.000 description 5
- 125000000304 alkynyl group Chemical group 0.000 description 5
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 5
- 230000004071 biological effect Effects 0.000 description 5
- 238000004364 calculation method Methods 0.000 description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 230000013595 glycosylation Effects 0.000 description 5
- 238000006206 glycosylation reaction Methods 0.000 description 5
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 5
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- 150000003573 thiols Chemical class 0.000 description 5
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 4
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 4
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- ZUHQCDZJPTXVCU-UHFFFAOYSA-N C1#CCCC2=CC=CC=C2C2=CC=CC=C21 Chemical compound C1#CCCC2=CC=CC=C2C2=CC=CC=C21 ZUHQCDZJPTXVCU-UHFFFAOYSA-N 0.000 description 4
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 4
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 244000180577 Sambucus australis Species 0.000 description 4
- 235000018734 Sambucus australis Nutrition 0.000 description 4
- JKHXYJKMNSSFFL-IUCAKERBSA-N Val-Lys Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN JKHXYJKMNSSFFL-IUCAKERBSA-N 0.000 description 4
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 4
- HAXFWIACAGNFHA-UHFFFAOYSA-N aldrithiol Chemical compound C=1C=CC=NC=1SSC1=CC=CC=N1 HAXFWIACAGNFHA-UHFFFAOYSA-N 0.000 description 4
- 125000004414 alkyl thio group Chemical group 0.000 description 4
- 230000002152 alkylating effect Effects 0.000 description 4
- 125000005110 aryl thio group Chemical group 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 150000001720 carbohydrates Chemical group 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 229940126142 compound 16 Drugs 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- ZPWOOKQUDFIEIX-UHFFFAOYSA-N cyclooctyne Chemical compound C1CCCC#CCC1 ZPWOOKQUDFIEIX-UHFFFAOYSA-N 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 210000004602 germ cell Anatomy 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 229960003330 pentetic acid Drugs 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical class OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 4
- 108010073969 valyllysine Proteins 0.000 description 4
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 3
- 108090000712 Cathepsin B Proteins 0.000 description 3
- 102000004225 Cathepsin B Human genes 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 3
- 102000003735 Mesothelin Human genes 0.000 description 3
- 108090000015 Mesothelin Proteins 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 241001495137 Streptomyces mobaraensis Species 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 235000008206 alpha-amino acids Nutrition 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 229940125773 compound 10 Drugs 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 229940125846 compound 25 Drugs 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 230000012202 endocytosis Effects 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 210000003712 lysosome Anatomy 0.000 description 3
- 230000001868 lysosomic effect Effects 0.000 description 3
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 230000003595 spectral effect Effects 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000011550 stock solution Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- 238000006177 thiolation reaction Methods 0.000 description 3
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 2
- KAFZOLYKKCWUBI-HPMAGDRPSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-3-amino-2-[[(2s)-2-[[(2s)-2-(3-cyclohexylpropanoylamino)-4-methylpentanoyl]amino]-5-methylhexanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]butanediamide Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](CCC(C)C)C(=O)N[C@@H](CN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(N)=O)C(N)=O)C(=O)CCC1CCCCC1 KAFZOLYKKCWUBI-HPMAGDRPSA-N 0.000 description 2
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 2
- ZXSBHXZKWRIEIA-JTQLQIEISA-N (2s)-3-(4-acetylphenyl)-2-azaniumylpropanoate Chemical compound CC(=O)C1=CC=C(C[C@H](N)C(O)=O)C=C1 ZXSBHXZKWRIEIA-JTQLQIEISA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 2
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 2
- YCTNWTBGSOCMRO-UHFFFAOYSA-N 3,5-dinitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC(C=O)=CC([N+]([O-])=O)=C1 YCTNWTBGSOCMRO-UHFFFAOYSA-N 0.000 description 2
- UDKJQEGOILJIJN-UHFFFAOYSA-N 3-nitro-5-phenylmethoxybenzaldehyde Chemical compound O=CC1=CC([N+](=O)[O-])=CC(OCC=2C=CC=CC=2)=C1 UDKJQEGOILJIJN-UHFFFAOYSA-N 0.000 description 2
- YEBJVSLNUMZXRJ-UHFFFAOYSA-N 5-methoxyindole-2-carboxylic acid Chemical compound COC1=CC=C2NC(C(O)=O)=CC2=C1 YEBJVSLNUMZXRJ-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 2
- AAQGRPOPTAUUBM-ZLUOBGJFSA-N Ala-Ala-Asn Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(O)=O AAQGRPOPTAUUBM-ZLUOBGJFSA-N 0.000 description 2
- XWKBWZXGNXTDKY-ZKWXMUAHSA-N Asp-Val-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](N)CC(O)=O XWKBWZXGNXTDKY-ZKWXMUAHSA-N 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 2
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 229940045513 CTLA4 antagonist Drugs 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 108010084457 Cathepsins Proteins 0.000 description 2
- 102000005600 Cathepsins Human genes 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 2
- 108010008177 Fd immunoglobulins Proteins 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 102000010956 Glypican Human genes 0.000 description 2
- 108050001154 Glypican Proteins 0.000 description 2
- 108050007237 Glypican-3 Proteins 0.000 description 2
- 244000041633 Grewia tenax Species 0.000 description 2
- 235000005612 Grewia tenax Nutrition 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 102100039813 Inactive tyrosine-protein kinase 7 Human genes 0.000 description 2
- 208000018142 Leiomyosarcoma Diseases 0.000 description 2
- VWPJQIHBBOJWDN-DCAQKATOSA-N Lys-Val-Ala Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(O)=O VWPJQIHBBOJWDN-DCAQKATOSA-N 0.000 description 2
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 2
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 2
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical group ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 2
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 2
- 206010041067 Small cell lung cancer Diseases 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 102100031293 Thimet oligopeptidase Human genes 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 108700019146 Transgenes Proteins 0.000 description 2
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 2
- XXDVDTMEVBYRPK-XPUUQOCRSA-N Val-Gln Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O XXDVDTMEVBYRPK-XPUUQOCRSA-N 0.000 description 2
- IOUPEELXVYPCPG-UHFFFAOYSA-N Valylglycine Chemical compound CC(C)C(N)C(=O)NCC(O)=O IOUPEELXVYPCPG-UHFFFAOYSA-N 0.000 description 2
- DDNCQMVWWZOMLN-IRLDBZIGSA-N Vinpocetine Chemical compound C1=CC=C2C(CCN3CCC4)=C5[C@@H]3[C@]4(CC)C=C(C(=O)OCC)N5C2=C1 DDNCQMVWWZOMLN-IRLDBZIGSA-N 0.000 description 2
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 238000007792 addition Methods 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 150000001335 aliphatic alkanes Chemical class 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 125000005237 alkyleneamino group Chemical group 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 230000004154 complement system Effects 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125810 compound 20 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940126208 compound 22 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940125851 compound 27 Drugs 0.000 description 2
- 229940125877 compound 31 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 239000002619 cytotoxin Substances 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 150000002019 disulfides Chemical class 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 210000001163 endosome Anatomy 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 2
- 150000007857 hydrazones Chemical class 0.000 description 2
- 125000002349 hydroxyamino group Chemical group [H]ON([H])[*] 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 125000000879 imine group Chemical group 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 230000002132 lysosomal effect Effects 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 231100000057 systemic toxicity Toxicity 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 230000004797 therapeutic response Effects 0.000 description 2
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 108010073106 thimet oligopeptidase Proteins 0.000 description 2
- 230000034005 thiol-disulfide exchange Effects 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000007056 transamidation reaction Methods 0.000 description 2
- 238000011282 treatment Methods 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- 229960000744 vinpocetine Drugs 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- NNRZVBFMEBWXBX-QMMMGPOBSA-N (2s)-2-(2-diazohydrazinyl)-3-phenylpropanoic acid Chemical compound [N-]=[N+]=NN[C@H](C(=O)O)CC1=CC=CC=C1 NNRZVBFMEBWXBX-QMMMGPOBSA-N 0.000 description 1
- AEVBPXDFDKBGLT-YOUFYPILSA-N (2s,3s,4r,5r)-n-[2-[4-(diethoxyphosphorylmethyl)anilino]-2-oxoethyl]-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolane-2-carboxamide Chemical compound C1=CC(CP(=O)(OCC)OCC)=CC=C1NC(=O)CNC(=O)[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 AEVBPXDFDKBGLT-YOUFYPILSA-N 0.000 description 1
- GPHYIQCSMDYRGJ-UHFFFAOYSA-N (3,5-dinitrophenyl)methanol Chemical compound OCC1=CC([N+]([O-])=O)=CC([N+]([O-])=O)=C1 GPHYIQCSMDYRGJ-UHFFFAOYSA-N 0.000 description 1
- AXKGIPZJYUNAIW-UHFFFAOYSA-N (4-aminophenyl)methanol Chemical compound NC1=CC=C(CO)C=C1 AXKGIPZJYUNAIW-UHFFFAOYSA-N 0.000 description 1
- OMJKFYKNWZZKTK-POHAHGRESA-N (5z)-5-(dimethylaminohydrazinylidene)imidazole-4-carboxamide Chemical compound CN(C)N\N=C1/N=CN=C1C(N)=O OMJKFYKNWZZKTK-POHAHGRESA-N 0.000 description 1
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 description 1
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 description 1
- 125000006729 (C2-C5) alkenyl group Chemical group 0.000 description 1
- 125000006730 (C2-C5) alkynyl group Chemical group 0.000 description 1
- UOORRWUZONOOLO-OWOJBTEDSA-N (E)-1,3-dichloropropene Chemical compound ClC\C=C\Cl UOORRWUZONOOLO-OWOJBTEDSA-N 0.000 description 1
- UVNPEUJXKZFWSJ-LMTQTHQJSA-N (R)-N-[(4S)-8-[6-amino-5-[(3,3-difluoro-2-oxo-1H-pyrrolo[2,3-b]pyridin-4-yl)sulfanyl]pyrazin-2-yl]-2-oxa-8-azaspiro[4.5]decan-4-yl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H]1COCC11CCN(CC1)c1cnc(Sc2ccnc3NC(=O)C(F)(F)c23)c(N)n1 UVNPEUJXKZFWSJ-LMTQTHQJSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- VTWKXBJHBHYJBI-SOFGYWHQSA-N (ne)-n-benzylidenehydroxylamine Chemical compound O\N=C\C1=CC=CC=C1 VTWKXBJHBHYJBI-SOFGYWHQSA-N 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- IGERFAHWSHDDHX-UHFFFAOYSA-N 1,3-dioxanyl Chemical group [CH]1OCCCO1 IGERFAHWSHDDHX-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- CQMJEZQEVXQEJB-UHFFFAOYSA-N 1-hydroxy-1,3-dioxobenziodoxole Chemical compound C1=CC=C2I(O)(=O)OC(=O)C2=C1 CQMJEZQEVXQEJB-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- SYOANZBNGDEJFH-UHFFFAOYSA-N 2,5-dihydro-1h-triazole Chemical group C1NNN=C1 SYOANZBNGDEJFH-UHFFFAOYSA-N 0.000 description 1
- KFXDYZXVIHNBFD-UHFFFAOYSA-N 2-(thian-2-ylsulfonyl)thiane Chemical compound C1CCCSC1S(=O)(=O)C1CCCCS1 KFXDYZXVIHNBFD-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- DJQYYYCQOZMCRC-UHFFFAOYSA-N 2-aminopropane-1,3-dithiol Chemical group SCC(N)CS DJQYYYCQOZMCRC-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- 125000000143 2-carboxyethyl group Chemical group [H]OC(=O)C([H])([H])C([H])([H])* 0.000 description 1
- ANDCDPBTSFSYLB-UHFFFAOYSA-N 2-cyclopropylpyrrolo[2,3-e]indole Chemical compound C1CC1C(C=C1C=C2)=NC1=C1C2=NC=C1 ANDCDPBTSFSYLB-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- LQILVUYCDHSGEU-UHFFFAOYSA-N 4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexane-1-carboxylic acid Chemical compound C1CC(C(=O)O)CCC1CN1C(=O)C=CC1=O LQILVUYCDHSGEU-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- SJISCEAZUHNOMD-UHFFFAOYSA-N 4-phenylcyclohexan-1-amine Chemical class C1CC(N)CCC1C1=CC=CC=C1 SJISCEAZUHNOMD-UHFFFAOYSA-N 0.000 description 1
- KUZSBKJSGSKPJH-VXGBXAGGSA-N 5-[(9R)-6-[(3R)-3-methylmorpholin-4-yl]-11-oxa-1,3,5-triazatricyclo[7.4.0.02,7]trideca-2,4,6-trien-4-yl]pyrazin-2-amine Chemical compound C[C@@H]1COCCN1c1nc(nc2N3CCOC[C@H]3Cc12)-c1cnc(N)cn1 KUZSBKJSGSKPJH-VXGBXAGGSA-N 0.000 description 1
- TVEXGJYMHHTVKP-UHFFFAOYSA-N 6-oxabicyclo[3.2.1]oct-3-en-7-one Chemical compound C1C2C(=O)OC1C=CC2 TVEXGJYMHHTVKP-UHFFFAOYSA-N 0.000 description 1
- JTWMOWRMSZZHDR-UHFFFAOYSA-N 7-(5-hydroxy-2-methylphenyl)-6-(2-methoxyphenyl)-4-methylpurino[7,8-a]imidazole-1,3-dione Chemical compound COC1=CC=CC=C1N(C(=CN12)C=3C(=CC=C(O)C=3)C)C2=NC2=C1C(=O)NC(=O)N2C JTWMOWRMSZZHDR-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- ZIBWKCRKNFYTPT-ZKWXMUAHSA-N Ala-Asn-Val Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(O)=O ZIBWKCRKNFYTPT-ZKWXMUAHSA-N 0.000 description 1
- LIWMQSWFLXEGMA-WDSKDSINSA-N Ala-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](C)N LIWMQSWFLXEGMA-WDSKDSINSA-N 0.000 description 1
- 208000037540 Alveolar soft tissue sarcoma Diseases 0.000 description 1
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 1
- 102000030431 Asparaginyl endopeptidase Human genes 0.000 description 1
- 208000036170 B-Cell Marginal Zone Lymphoma Diseases 0.000 description 1
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 1
- 102100038078 CD276 antigen Human genes 0.000 description 1
- 102100032912 CD44 antigen Human genes 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 240000005589 Calophyllum inophyllum Species 0.000 description 1
- 102100033620 Calponin-1 Human genes 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 239000004381 Choline salt Substances 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- 108010005843 Cysteine Proteases Proteins 0.000 description 1
- 102000005927 Cysteine Proteases Human genes 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- 230000000970 DNA cross-linking effect Effects 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- 108700022150 Designed Ankyrin Repeat Proteins Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 1
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 1
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 1
- 101000884279 Homo sapiens CD276 antigen Proteins 0.000 description 1
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 1
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 1
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 1
- 101000606465 Homo sapiens Inactive tyrosine-protein kinase 7 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 1
- 101000955999 Homo sapiens V-set domain-containing T-cell activation inhibitor 1 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- 101710099452 Inactive tyrosine-protein kinase 7 Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 1
- QEFRNWWLZKMPFJ-ZXPFJRLXSA-N L-methionine (R)-S-oxide Chemical compound C[S@@](=O)CC[C@H]([NH3+])C([O-])=O QEFRNWWLZKMPFJ-ZXPFJRLXSA-N 0.000 description 1
- QEFRNWWLZKMPFJ-UHFFFAOYSA-N L-methionine sulphoxide Natural products CS(=O)CCC(N)C(O)=O QEFRNWWLZKMPFJ-UHFFFAOYSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 1
- 102000019298 Lipocalin Human genes 0.000 description 1
- 108050006654 Lipocalin Proteins 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- NVGBPTNZLWRQSY-UWVGGRQHSA-N Lys-Lys Chemical compound NCCCC[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN NVGBPTNZLWRQSY-UWVGGRQHSA-N 0.000 description 1
- 201000003791 MALT lymphoma Diseases 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108010090665 Mannosyl-Glycoprotein Endo-beta-N-Acetylglucosaminidase Proteins 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 229930126263 Maytansine Natural products 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- GHAZCVNUKKZTLG-UHFFFAOYSA-N N-ethyl-succinimide Natural products CCN1C(=O)CCC1=O GHAZCVNUKKZTLG-UHFFFAOYSA-N 0.000 description 1
- HDFGOPSGAURCEO-UHFFFAOYSA-N N-ethylmaleimide Chemical compound CCN1C(=O)C=CC1=O HDFGOPSGAURCEO-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 125000004633 N-oxo-pyridyl group Chemical group 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 108090000028 Neprilysin Proteins 0.000 description 1
- 102000003729 Neprilysin Human genes 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 102000000447 Peptide-N4-(N-acetyl-beta-glucosaminyl) Asparagine Amidase Human genes 0.000 description 1
- 108010055817 Peptide-N4-(N-acetyl-beta-glucosaminyl) Asparagine Amidase Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- JXASPPWQHFOWPL-UHFFFAOYSA-N Tamarixin Natural products C1=C(O)C(OC)=CC=C1C1=C(OC2C(C(O)C(O)C(CO)O2)O)C(=O)C2=C(O)C=C(O)C=C2O1 JXASPPWQHFOWPL-UHFFFAOYSA-N 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 1
- MNLAVFKVRUQAKW-UHFFFAOYSA-N VR nerve agent Chemical compound CCN(CC)CCSP(C)(=O)OCC(C)C MNLAVFKVRUQAKW-UHFFFAOYSA-N 0.000 description 1
- QRZVUAAKNRHEOP-GUBZILKMSA-N Val-Ala-Val Chemical compound [H]N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(O)=O QRZVUAAKNRHEOP-GUBZILKMSA-N 0.000 description 1
- AEMPCGRFEZTWIF-IHRRRGAJSA-N Val-Leu-Lys Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(O)=O AEMPCGRFEZTWIF-IHRRRGAJSA-N 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NPUXORBZRBIOMQ-RUZDIDTESA-N [(2R)-1-[[4-[[3-(benzenesulfonylmethyl)-5-methylphenoxy]methyl]phenyl]methyl]-2-pyrrolidinyl]methanol Chemical compound C=1C(OCC=2C=CC(CN3[C@H](CCC3)CO)=CC=2)=CC(C)=CC=1CS(=O)(=O)C1=CC=CC=C1 NPUXORBZRBIOMQ-RUZDIDTESA-N 0.000 description 1
- 125000004062 acenaphthenyl group Chemical group C1(CC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 150000001371 alpha-amino acids Chemical class 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 208000008524 alveolar soft part sarcoma Diseases 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000005018 aryl alkenyl group Chemical group 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000005015 aryl alkynyl group Chemical group 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 1
- 108010055066 asparaginylendopeptidase Proteins 0.000 description 1
- 108010044540 auristatin Proteins 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- ICCBZGUDUOMNOF-UHFFFAOYSA-N azidoamine Chemical compound NN=[N+]=[N-] ICCBZGUDUOMNOF-UHFFFAOYSA-N 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical class C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- CTOUWUYDDUSBQE-UHFFFAOYSA-N benzyl piperazine-1-carboxylate Chemical compound C1CNCCN1C(=O)OCC1=CC=CC=C1 CTOUWUYDDUSBQE-UHFFFAOYSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000012496 blank sample Substances 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 229960001467 bortezomib Drugs 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- XEVRDFDBXJMZFG-UHFFFAOYSA-N carbonyl dihydrazine Chemical compound NNC(=O)NN XEVRDFDBXJMZFG-UHFFFAOYSA-N 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 235000019417 choline salt Nutrition 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 108010006226 cryptophycin Proteins 0.000 description 1
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 1
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 1
- 125000002944 cyanoaryl group Chemical group 0.000 description 1
- 125000002993 cycloalkylene group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 150000001945 cysteines Chemical class 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000022811 deglycosylation Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000004988 dibenzothienyl group Chemical group C1(=CC=CC=2SC3=C(C21)C=CC=C3)* 0.000 description 1
- 150000005332 diethylamines Chemical class 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- 102000006815 folate receptor Human genes 0.000 description 1
- 108020005243 folate receptor Proteins 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 201000003444 follicular lymphoma Diseases 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229930182480 glucuronide Natural products 0.000 description 1
- 150000008134 glucuronides Chemical class 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002338 glycosides Chemical class 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 125000004970 halomethyl group Chemical group 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- 125000006588 heterocycloalkylene group Chemical group 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 150000002433 hydrophilic molecules Chemical class 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 210000003026 hypopharynx Anatomy 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 108010050180 kallistatin Proteins 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 229950007634 kitasamycin Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 210000000867 larynx Anatomy 0.000 description 1
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- XYJOGTQLTFNMQG-KJHBSLKPSA-N leucomycin V Chemical compound CO[C@H]1[C@H](O)CC(=O)O[C@H](C)C\C=C\C=C\[C@H](O)[C@H](C)C[C@H](CC=O)[C@@H]1O[C@H]1[C@H](O)[C@@H](N(C)C)[C@H](O[C@@H]2O[C@@H](C)[C@H](O)[C@](C)(O)C2)[C@@H](C)O1 XYJOGTQLTFNMQG-KJHBSLKPSA-N 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- IHLVCKWPAMTVTG-UHFFFAOYSA-N lithium;carbanide Chemical compound [Li+].[CH3-] IHLVCKWPAMTVTG-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000007422 luminescence assay Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000007919 lymphoplasmacytic lymphoma Diseases 0.000 description 1
- 108010054155 lysyllysine Proteins 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 201000009020 malignant peripheral nerve sheath tumor Diseases 0.000 description 1
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- RXYCUKSOYJWGPE-UHFFFAOYSA-N methyl 2-azidoacetate Chemical compound COC(=O)CN=[N+]=[N-] RXYCUKSOYJWGPE-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- LSDPWZHWYPCBBB-UHFFFAOYSA-O methylsulfide anion Chemical compound [SH2+]C LSDPWZHWYPCBBB-UHFFFAOYSA-O 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 210000001989 nasopharynx Anatomy 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 208000029974 neurofibrosarcoma Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 230000030147 nuclear export Effects 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 210000003300 oropharynx Anatomy 0.000 description 1
- FIYYMXYOBLWYQO-UHFFFAOYSA-N ortho-iodylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1I(=O)=O FIYYMXYOBLWYQO-UHFFFAOYSA-N 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 208000007312 paraganglioma Diseases 0.000 description 1
- 210000003695 paranasal sinus Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000007030 peptide scission Effects 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- SBMSLRMNBSMKQC-UHFFFAOYSA-N pyrrolidin-1-amine Chemical compound NN1CCCC1 SBMSLRMNBSMKQC-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- 150000003248 quinolines Chemical class 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 201000006845 reticulosarcoma Diseases 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- ATEBXHFBFRCZMA-VXTBVIBXSA-N rifabutin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC(=C2N3)C(=O)C=4C(O)=C5C)C)OC)C5=C1C=4C2=NC13CCN(CC(C)C)CC1 ATEBXHFBFRCZMA-VXTBVIBXSA-N 0.000 description 1
- 229960000885 rifabutin Drugs 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000007659 semicarbazones Chemical class 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- VUFNRPJNRFOTGK-UHFFFAOYSA-M sodium;1-[4-[(2,5-dioxopyrrol-1-yl)methyl]cyclohexanecarbonyl]oxy-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].O=C1C(S(=O)(=O)[O-])CC(=O)N1OC(=O)C1CCC(CN2C(C=CC2=O)=O)CC1 VUFNRPJNRFOTGK-UHFFFAOYSA-M 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 108090000250 sortase A Proteins 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 1
- 125000002653 sulfanylmethyl group Chemical group [H]SC([H])([H])[*] 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- UOORRWUZONOOLO-UHFFFAOYSA-N telone II Natural products ClCC=CCl UOORRWUZONOOLO-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- QKSQWQOAUQFORH-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonylimino]carbamate Chemical compound CC(C)(C)OC(=O)N=NC(=O)OC(C)(C)C QKSQWQOAUQFORH-UHFFFAOYSA-N 0.000 description 1
- 125000006092 tetrahydro-1,1-dioxothienyl group Chemical group 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- WCNFFKHKJLERFM-UHFFFAOYSA-N thiomorpholinyl sulfone group Chemical group N1(CCSCC1)S(=O)(=O)N1CCSCC1 WCNFFKHKJLERFM-UHFFFAOYSA-N 0.000 description 1
- ZCAGUOCUDGWENZ-UHFFFAOYSA-N thiomorpholinyl sulfoxide group Chemical group N1(CCSCC1)S(=O)N1CCSCC1 ZCAGUOCUDGWENZ-UHFFFAOYSA-N 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Images
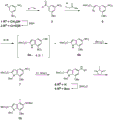
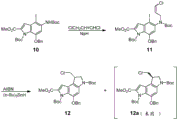
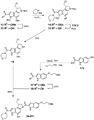
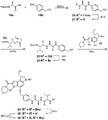
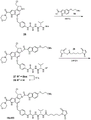
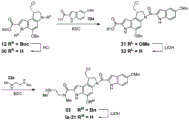
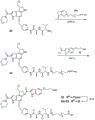

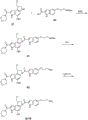
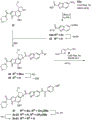
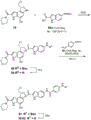
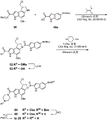

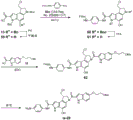
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/44—Antibodies bound to carriers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6857—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from lung cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6863—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from stomach or intestines cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6869—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of the reproductive system: ovaria, uterus, testes, prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Cell Biology (AREA)
- Oncology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pulmonology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Reproductive Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
式(I)的seco‑环丙吡咯并吲哚化合物,其中Hal,R1,R2和R3如本申请中所定义,是可用于抗体‑药物缀合物的有效抗癌剂。
Description
相关申请的交叉引用
本申请按35U.S.C.§119(e)规定,要求2016年8月19日提交的美国临时申请序列No.62/377,052的权益;其公开内容通过引用并入本文。
发明背景
本发明涉及seco-环丙吡咯并吲哚化合物,其缀合物,以及制备和使用这些化合物和缀合物的方法。
双螺旋DNA有两个沿其外部延伸的纵向螺旋沟,很像理发店标杆上的条纹。两个沟不相同:一个称为大沟,比称为小沟的另一个沟宽得多。
小沟的宽度约等于苯环的厚度。许多具有生物学活性的DNA结合分子是具有弓形足迹的基本上平面的多芳族分子,这种形状使得它们能够紧贴地在小沟中滑动。这些分子中的一类不仅与DNA结合,而且还使其烷基化,并称为DNA小沟结合剂-烷基化剂(“MGBA”)。
MGBA亚类由天然产物CC-1065、多米卡新(duocarmycin)SA和谷田霉素(yatakemycin)(Boger and Johnson 1995;Tichenor等人2007)代表。(本说明书末尾通过第一作者或发明人和年份列出了在本文引用的文献的完整引用)。它们包含烷基化亚单位和一个或多个结合亚单位,后者有助于与DNA结合但对其为化学非反应性的。在CC-1065和多米卡新SA中,烷基化亚单位位于分子的一端,而结合亚单位位于另一端。在谷田霉素中,烷基化亚单位的两侧是结合亚单位。与整体MGBA结构一致,烷基化和结合亚单位本身是多芳族的并且基本上是平面的。由于烷基化亚单位具有环丙吡咯并吲哚(“CPI”)核心结构,该亚类中的MGBA由此命名为CPI化合物。

在与DNA结合后,CPI环丙基环被活化并在腺嘌呤N3氮处烷基化DNA(Hurley等人1984)。提出用于解释活化的一种理论是结合将进一步的构象应变引入已经应变的环丙基环中,增加其反应性(Boger 2001;Boger等人1997;Tichenor等人1997)。

seco-CPI化合物是CPI化合物的变体,其中环丙基环已被打开并被卤甲基取代。虽然seco-CPI化合物本身不使DNA烷基化,但它们在体外或体内易于转化为CPI化合物,并且它们的生物学活性与后者基本相同(Li等人2012)。因此,seco-CPI化合物作为CPI化合物的合成方便的功能等价物或作为其合成的中间体受到关注(Boger等人2000)。

seco-CPI化合物的优点是它可以被前药化以控制向CPI形式的转化。将前药化基团PD连接到酚羟基上防止转化成CPI形式,除非首先裂解掉PD。可以选择PD使得其被在预期生物学作用位点(例如肿瘤)处或附近发现的药剂裂解,以降低全身毒性的风险。PD优选是酶促可裂解的基团,例如氨基甲酸酯,磷酸酯,糖苷或葡糖苷酸,它们分别可被羧基酯酶,磷酸酯酶,糖苷酶或葡糖苷酸酶裂解。见例如Kobayashi等人1994;Lajiness等人2010;Sufi等人2013;Tietze等人2001;Zhang等人2014。

对CPI化合物类似物的研究导致另一个MGBA亚类的开发,其中CPI吡咯基团被苯环替代。由于烷基化亚单位的环丙苯并吲哚(“CBI”)核心,这种化合物被称为CBI化合物。与CPI化合物一样,CBI化合物可以以seco和前药化的seco形式存在。更简单的CBI结构更易于合成,并且CBI化合物已被显示具有稳定性和生物学效力(Lajiness等人2010;Boger等人1990和1999)。

CPI和CBI化合物都是有效的细胞毒素,使得它们成为抗癌剂的有吸引力的候选物。大量的研究工作致力于合成和评估这些化合物及其seco变体用于此类用途。见例如Boger 2003;Kobayashi等人1994;Li等人1992;Nagamura and Saito 1998;Nagamura等人1996;Tichenor等人2007;Tietze等人2008。
一种产生强烈兴趣的抗癌剂是缀合物,其中药物与靶向剂连接,所述靶向剂结合癌细胞上的配体。因此,靶向剂将药物引导至癌细胞,其在癌细胞处通过几种机制中的一种释放以作用于癌细胞。
常见类型的缀合物是抗体-药物缀合物(ADC,也称为免疫缀合物)。在ADC中,药物(也称为治疗剂,细胞毒素,有效负载或弹头)与抗体共价连接,抗体的抗原是肿瘤相关抗原-即由癌细胞表达的抗原。
将抗体和药物共价连接的部分称为连接基。当每种抗体具有与其连接的一种药物时,ADC的结构可表示为:
[抗体]-[连接基]-[药物]
抗体在与抗原结合后将ADC递送至癌部位。在那里,连接基的裂解或抗体的降解释放药物。通常,ADC通过内吞作用内化入靶细胞并且药物释放在其内部发生。当ADC在血液中循环时,药物由于与抗体连接,保持无活性。因此,ADC中的药物可比普通的化疗药物明显更有效(细胞毒性的),因为其局部释放降低了全身毒性。有关ADC的综述,见Schrama等人2006。
CPI和CBI化合物以及它们的seco对应物已被提议作为ADC中的药物。见例如Boyd等人2008;Chari等人1995;Chen等人2014;Ducry等人2010;Gangwar等人2011;Ng等人2006,2009和2011;Sufi等人2013;Zhang等人2014;Zhao等人2010。
发明简单概述
本发明提供了新的和高效的seco-CPI化合物,其适合以ADC或其他缀合物形式用作药物。这些化合物具有与seco-CPI亚单位的吡咯基团连接的酰胺基团和与其另一端连接的一个或多个结合亚单位。
一方面,本发明提供具有式(I)所示结构的seco-CPI化合物:

其中
Hal是Cl或Br;
R1是
R2是H,C1-C3烷基,CO2H,CO2(C1-C3烷基),C(=O)NH2,C(=O)NH(C1-C3烷基)或C(=O)N(C1-C3烷基)2;
R3是


R4,R4’,R5,R6或R7独立地是H,OMe,OH,6元芳基(优选苯基)基团,5或6元杂芳基,NH2,NHMe,NMe2,NH(C2-C4烷基),N(C2-C4烷基)2,NHC(=O)X1,O(C2-C4烷基),O(CH2)0-2(C3-C6环烷基),O(CH2)0-2X1或

其中C2-C4烷基可以未取代或被OCH2CH2OH,OCH2CH2NH2,NHCH2CH2OH,NHCH2CH2NH2,OH或NH2取代,芳基或杂芳基可以被C1-C2烷基,OH,NH2,NH(C1-C2烷基),N(C1-C2烷基)2,F,Cl,Br,NO2或CN取代;
条件是R4,R4’,R5,R6和R7至少一者不是H;
R8和R8’独立地是H,OH,O(C1-C3烷基),Cl,Br,F,O(CH2)2-4NH2或O(CH2)2-4OH;
R9是H,C(=O)(C1-C3烷基),C(=O)NH2,C(=O)NH(C1-C3烷基),C(=O)(C1-C3烷基)2,(CH2)2-4OH,(CH2)2-4O(C1-C3烷基),(CH2)2-4NH2,(CH2)2-4NH(C1-C3烷基)或(CH2)2-4N(C1-C3烷基)2;
每个X1独立地是6元芳基(优选苯基)或5至6元杂芳基,其是未取代的或被C1-C3烷基,OH,O(C1-C3烷基),NH2,NH(C1-C3烷基),N(C1-C3烷基)2,F,Cl,Br,NO2或CN取代;
每个X2独立地是H,Me或C2-C4烷基,其可以未取代或被OCH2CH2OH,OCH2CH2NH2,NHCH2CH2OH,NHCH2CH2NH2,OH或NH2取代;且
每个X3独立地是O,NH,N(C1-C3烷基)或S;
或其药学上可接受的盐。
另一方面,本发明提供了与连接基连接的本发明的seco-CPI化合物,其适于与靶向部分,尤其是抗体缀合。
另一方面,本发明提供了通过连接基与靶向部分,尤其是抗体缀合的本发明的seco-CPI化合物的缀合物。
另一方面,本发明提供治疗患有这种癌症的受试者的癌症的方法,包括给予受试者治疗有效量的本发明化合物或其与抗体的缀合物。在优选的实施方案中,所治疗的癌症是肺癌,胃癌,卵巢癌或结肠癌。
另一方面,本发明提供了本发明的seco-CPI化合物或其与抗体的缀合物,用于治疗患有癌症的受试者。癌症可以是肺癌,胃癌,卵巢癌或结肠癌。
附图的简要说明
图1A和1B显示合成可用于制备本发明化合物的seco-CPI核心结构的方案。
图2,4,6,7,8,9,10,11和12显示了用于合成本发明的seco-CPI化合物的示例性方案。本领域技术人员将理解,在一些情况下,方案的不同主要在于组装化合物的各个区段的顺序或保护基团被除去的顺序,并且这些步骤的执行顺序可以是个人偏好的。
图3A,3B和5显示了合成本发明的seco-CPI连接基化合物的示例性方案。同样,本领域技术人员将理解,某些步骤的执行顺序的变化可能受个人偏好的影响。
发明的详细说明
定义
“抗体”意指完整抗体和任何抗原结合片段(即“抗原结合部分”)或其单链变体。完整抗体是包含通过二硫键相互连接的至少两条重(H)链和两条轻(L)链的蛋白质。每条重链包含重链可变区(VH)和包含三个结构域CH1,CH2和CH3的重链恒定区。每条轻链包含轻链可变区(VL或Vk)和包含一个单结构域CL的轻链恒定区。VH和VL区可以进一步细分为高变区,称为互补决定区(CDR),散布着更保守的框架区(FR)。每个VH和VL包含三个CDR和四个FR,以下列顺序从氨基端到羧基端排列:FR1,CDR1,FR2,CDR2,FR3,CDR3和FR4。可变区含有与抗原相互作用的结合域。恒定区可以介导抗体与宿主组织或因子的结合,包括免疫系统的各种细胞(例如效应细胞)和经典补体系统的第一组分(Clq)。如果抗体以5x 10-8M或更低,更优选1x10-8M或更低,更优选6x 10-9M或更低,更优选3x10-9M或更低,甚至更优选2x 10-9M或更低的KD与抗原X结合,则称抗体与抗原X“特异性结合”。抗体可以是嵌合的,人源化的,或优选人的。可以改造重链恒定区以影响糖基化类型或程度,延长抗体半衰期,增强或减少与效应细胞或补体系统的相互作用,或调节一些其他性质。可以通过替换,添加或缺失一个或多个氨基酸或通过用来自另一种免疫球蛋白类型的结构域替换结构域或前述的组合来完成改造。
抗体的“抗原结合片段”和“抗原结合部分”(或简称“抗体部分”或“抗体片段”)是指保留与抗原的特异性结合能力的抗体的一个或多个片段。已经显示抗体的抗原结合功能可以通过全长抗体的片段实现,例如(i)Fab片段,由VL、VH、CL和CH1结构域组成的单价片段;(ii)F(ab')2片段,包含两个Fab片段的二价片段,所述Fab片段在铰链区通过二硫键连接;(iii)Fab'片段,其基本上是具有部分铰链区的Fab(见例如Abbas等人,CellularandMolecularImmunology,第6版,Saunders Elsevier 2007);(iv)由VH和CH1结构域组成的Fd片段;(v)由抗体单臂的VL和VH结构域组成的Fv片段,(vi)dAb片段(Ward等人,(1989)Nature 341:544-546),它由VH结构域组成;(vii)孤立的互补决定区(CDR);和(viii)纳米抗体,包含单个可变结构域和两个恒定结构域的重链可变区。优选的抗原结合片段是Fab,F(ab')2,Fab',Fv和Fd片段。此外,尽管Fv片段的两个结构域VL和VH由不同的基因编码,但它们可以通过合成连接基使用重组方法连接,该合成连接基使得它们能够制成单个蛋白链,其中VL和VH区配对形成单价分子(称为单链Fv或scFv);见例如Bird等人(1988)Science242:423-426;和Huston等人(1988)Proc.Natl.Acad.Sci.USA85:5879-5883)。这种单链抗体也包括在抗体的术语“抗原结合部分”内。
“分离的抗体”是指基本上不含具有不同抗原特异性的其他抗体的抗体(例如,特异性结合抗原X的分离的抗体基本上不含特异性结合抗原X以外的抗原的抗体)。然而,特异性结合抗原X的分离的抗体可能与其他抗原(例如来自其他物种的抗原X分子)具有交叉反应性。在某些实施方案中,分离的抗体特异性结合人抗原X并且不与其他(非人)抗原X抗原交叉反应。此外,分离的抗体可以基本上不含其他细胞物质和/或化学物质。
“单克隆抗体”或“单克隆抗体组合物”是指单分子组合物的抗体分子的制剂,其显示对特定表位的单一结合特异性和亲和力。
“人抗体”是指具有可变区的抗体,其中框架区和CDR区(和恒定区,如果存在的话)均源自人种系免疫球蛋白序列。人抗体可包括后来的修饰,包括天然或合成修饰。人抗体可包括不由人种系免疫球蛋白序列编码的氨基酸残基(例如,通过体外随机或位点特异性诱变或通过体内体细胞突变引入的突变)。然而,“人抗体”不包括其中衍生自另一种哺乳动物物种(例如小鼠)的种系的CDR序列已经移植到人框架序列上的抗体。
“人单克隆抗体”是指显示单一结合特异性的抗体,其具有可变区,其中框架区和CDR区均源自人种系免疫球蛋白序列。在一个实施方案中,人单克隆抗体由杂交瘤产生,所述杂交瘤包括获自转基因非人动物例如转基因小鼠的B细胞,具有与无限增殖化细胞融合的包含人重链转基因和轻链转基因的基因组。
“脂族”是指直链或支链,饱和或不饱和的非芳族烃部分,具有指定碳原子数(例如,如“C3脂族”、“C1-5脂族”、“C1-C5脂族”或“C1至C5脂族”,后三个短语是同义词,指具有1至5个碳原子的脂族部分”),或者,当没有明确指定碳原子数时,具有1至4个碳原子(在不饱和脂族部分的情况下具有2至4个碳原子)。类似的理解适用于其他类型中的碳的数量,如C2-4烯烃,C4-C7环脂族等。类似地,诸如“(CH2)1-3”的术语应理解为下标是1、2或3的简写,因此该术语表示CH2,CH2CH2和CH2CH2CH2。
“烷基”是指饱和脂族部分,适用指定碳原子数的相同惯例。举例来说,C1-C4烷基部分包括但不限于甲基,乙基,丙基,异丙基,异丁基,叔丁基,1-丁基,2-丁基等。“亚烷基”是指烷基的二价对应物,例如CH2CH2,CH2CH2CH2和CH2CH2CH2CH2。
“烯基”是指具有至少一个碳-碳双键的脂族部分,适用指定碳原子数的相同惯例。举例来说,C2-C4烯基部分包括但不限于乙烯基(乙烯基),2-丙烯基(烯丙基或丙-2-烯基),顺式-1-丙烯基,反式-1-丙烯基,E-(或Z-)2-丁烯基,3-丁烯基,1,3-丁二烯基(丁-1,3-二烯基)等。
“炔基”是指具有至少一个碳-碳三键的脂族部分,适用指定碳原子数的相同惯例。举例来说,C2-C4炔基包括乙炔基(乙炔基),炔丙基(丙-2-炔基),1-丙炔基,丁-2-炔基等。
“环脂族”是指具有1至3个环的饱和或不饱和的非芳族烃部分,每个环具有3至8个(优选3至6个)碳原子。“环烷基”是指其中每个环饱和的环脂族部分。“环烯基”是指环脂族部分,其中至少一个环具有至少一个碳-碳双键。“环炔基”是指环脂族部分,其中至少一个环具有至少一个碳-碳三键。举例来说,环脂族部分包括但不限于环丙基,环丁基,环戊基,环戊烯基,环己基,环己烯基,环庚基,环辛基和金刚烷基。优选的环脂族部分是环烷基部分,尤其是环丙基,环丁基,环戊基和环己基。“亚环烷基”是指环烷基的二价对应物。
“杂环脂族”是指环脂族部分,其中在其至少一个环中,至多三个(优选1至2个)碳被独立地选自N,O或S的杂原子代替,其中N和S任选地可以被氧化并且N任选地可以被季铵化。优选的环脂族部分由一个大小为5-6元的环组成。类似地,“杂环烷基”,“杂环烯基”和“杂环炔基”分别指环烷基,环烯基或环炔基部分,其中其至少一个环被如此修饰。示例性的杂环脂族部分包括氮杂环丙烷基,氮杂环丁烷基,1,3-二噁烷基,氧杂环丁烷基,四氢呋喃基,吡咯烷基,哌啶基,哌嗪基,四氢吡喃基,四氢噻喃基,四氢噻喃基砜,吗啉基,硫代吗啉基,硫代吗啉基亚砜,硫代吗啉基砜,1,3-二氧杂环戊烷基,四氢-1,1-二氧代噻吩基,1,4-二噁烷基,硫杂环丁烷基等。“亚杂环烷基”是指杂环烷基的二价对应物。
“烷氧基”,“芳氧基”,“烷硫基”和“芳硫基”分别表示-O(烷基),-O(芳基),-S(烷基)和-S(芳基)。实例分别是甲氧基,苯氧基,甲硫基和苯硫基。
“卤素”或“卤”表示氟,氯,溴或碘,除非指出更窄的含义。
“芳基”是指具有单环,双环或三环环系(优选单环)的烃部分,其中每个环具有3至7个碳原子且至少一个环是芳族的。环系统中的环可以彼此稠合(如在萘基中)或彼此键合(如在联苯中)并且可以稠合或键合到非芳族环(如在茚满基或环己基苯基中)。作为进一步说明,芳基部分包括但不限于苯基,萘基,四氢萘基,茚满基,联苯基,菲基,蒽基和苊基。“亚芳基”是指芳基的二价对应物,例如1,2-亚苯基,1,3-亚苯基或1,4-亚苯基。
“杂芳基”是指具有单环,双环或三环环系(优选5至7元单环)的部分,其中每个环具有3-7个碳原子,并且至少一个环是含有1至4个杂原子的芳族环,所述杂原子独立地选自N,O或S,其中N和S任选地可以被氧化并且N任选地可以被季铵化。这种含至少一个杂原子的芳族环可以与其他类型的环稠合(如苯并呋喃基或四氢异喹啉基)或直接键合到其他类型的环(如苯基吡啶基或2-环戊基吡啶基)。作为进一步说明,杂芳基部分包括吡咯基,呋喃基,噻吩基(噻吩基),咪唑基,吡唑基,噁唑基,异噁唑基,噻唑基,异噻唑基,三唑基,四唑基,吡啶基,N-氧代吡啶基,哒嗪基,嘧啶基,吡嗪基,喹啉基,异喹啉基,喹唑啉基,噌啉基,喹唑啉基,萘啶基,苯并呋喃基,吲哚基,苯并噻吩基,噁二唑基,噻二唑基,吩噻唑基,苯并咪唑基,苯并三唑基,二苯并呋喃基,咔唑基,二苯并噻吩基,吖啶基等。“亚杂芳基”是指杂芳基的二价对应物。
当表明部分可以被取代时,例如通过使用“未取代的或取代的”或“任选取代的”表述,如“未取代或取代的C1-C5烷基”或“任选取代的杂芳基”,此类部分可具有一个或多个独立选择的取代基,数量优选1至5个,数量更优选1或2个。考虑取代基连接的部分,本领域普通技术人员可以选择取代基和取代模式,以提供化学稳定的并且可以通过本领域已知的技术以及本文所述的方法合成的化合物。当部分被确定为“未取代的或取代的”或“任选取代的”时,在优选的实施方案中,这种部分是未取代的。
“芳基烷基”,“(杂环脂族)烷基”,“芳基烯基”,“芳基炔基”,“联芳基烷基”等是指烷基、烯基或炔基部分,视情况而定,可被芳基、杂环脂族、联芳基等部分取代,视情况而定,在烷基、烯基或炔基部分具有开放(未满足)价,例如苄基,苯乙基,N-咪唑基乙基,N-吗啉代乙基等。相反,“烷基芳基”、“烯基环烷基”等是指芳基、环烷基等部分,视情况而定,可被烷基、烯基等部分取代,视情况而定,例如甲基苯基(甲苯基)或烯丙基环己基。“羟烷基”、“卤代烷基”、“烷基芳基”、“氰基芳基”等是指烷基、芳基等部分,视情况而定,被一个或多个所确定的取代基(羟基、卤素等,视情况而定)取代。
例如,允许的取代基包括但不限于烷基(尤其是甲基或乙基),烯基(尤其是烯丙基),炔基,芳基,杂芳基,环脂族,杂环脂族,卤素(尤其是氟),卤代烷基(尤其是三氟甲基),羟基,羟烷基(特别是羟乙基),氰基,硝基,烷氧基,-O(羟烷基),-O(卤代烷基)(特别是-OCF3),-O(环烷基),-O(杂环烷基),-O(芳基),烷硫基,芳硫基,=O,=NH,=N(烷基),=NOH,=NO(烷基),-C(=O)(烷基),-C(=O)H,-CO2H,-C(=O)NHOH,-C(=O)O(烷基),-C(=O)O(羟烷基),-C(=O)NH2,-C(=O)NH(烷基),-C(=O)N(烷基)2,-OC(=O)(烷基),-OC(=O)(羟烷基),-OC(=O)O(烷基),-OC(=O)O(羟烷基),-OC(=O)NH2,-OC(=O)NH(烷基),-OC(=O)N(烷基)2,叠氮基,-NH2,-NH(烷基),-N(烷基)2,-NH(芳基),-NH(羟烷基),-NHC(=O)(烷基),-NHC(=O)H,-NHC(=O)NH2,-NHC(=O)NH(烷基),-NHC(=O)N(烷基)2,-NHC(=NH)NH2,-OSO2(烷基),-SH,-S(烷基),-S(芳基),-S(环烷基),-S(=O)烷基,-SO2(烷基),-SO2NH2,-SO2NH(烷基),-SO2N(烷基)2等。
当被取代的部分是脂族部分时,优选的取代基是芳基,杂芳基,环脂族,杂环脂族,卤素,羟基,氰基,硝基,烷氧基,-O(羟烷基),-O(卤代烷基),-O(环烷基),-O(杂环烷基),-O(芳基),烷硫基,芳硫基,=O,=NH,=N(烷基),=NOH,=NO(烷基),-CO2H,-C(=O)NHOH,-C(=O)O(烷基),-C(=O)O(羟烷基),-C(=O)NH2,-C(=O)NH(烷基),-C(=O)N(烷基)2,-OC(=O)(烷基),-OC(=O)(羟烷基),-OC(=O)O(烷基),-OC(=O)O(羟烷基),-OC(=O)NH2,-OC(=O)NH(烷基),-OC(=O)N(烷基)2,叠氮基,-NH2,-NH(烷基),-N(烷基)2,-NH(芳基),-NH(羟烷基),-NHC(=O)(烷基),-NHC(=O)H,-NHC(=O)NH2,-NHC(=O)NH(烷基),-NHC(=O)N(烷基)2,-NHC(=NH)NH2,-OSO2(烷基),-SH,-S(烷基),-S(芳基),-S(=O)烷基,-S(环烷基),-SO2(烷基),-SO2NH2,-SO2NH(烷基)和-SO2N(烷基)2。更优选的取代基是卤素,羟基,氰基,硝基,烷氧基,-O(芳基),=O,=NOH,=NO(烷基),-OC(=O)(烷基),-OC(=O)O(烷基),-OC(=O)NH2,-OC(=O)NH(烷基),-OC(=O)N(烷基)2,叠氮基,-NH2,-NH(烷基),-N(烷基)2,-NH(芳基),-NHC(=O)(烷基),-NHC(=O)H,-NHC(=O)NH2,-NHC(=O)NH(烷基),-NHC(=O)N(烷基)2和-NHC(=NH)NH2。特别优选的是苯基,氰基,卤素,羟基,硝基,C1-C4烷氧基,O(C2-C4亚烷基)OH和O(C2-C4亚烷基)卤素。
当被取代的部分是环脂族,杂环脂族,芳基或杂芳基部分时,优选的取代基是烷基,烯基,炔基,卤素,卤代烷基,羟基,羟烷基,氰基,硝基,烷氧基,-O(羟烷基),-O(卤代烷基),-O(芳基),-O(环烷基),-O(杂环烷基),烷硫基,芳硫基,-C(=O)(烷基),-C(=O)H,-CO2H,-C(=O)NHOH,-C(=O)O(烷基),-C(=O)O(羟烷基),-C(=O)NH2,-C(=O)NH(烷基),-C(=O)N(烷基)2,-OC(=O)(烷基),-OC(=O)(羟烷基),-OC(=O)O(烷基),-OC(=O)O(羟烷基),-OC(=O)NH2,-OC(=O)NH(烷基),-OC(=O)N(烷基)2,叠氮基,-NH2,-NH(烷基),-N(烷基)2,-NH(芳基),-NH(羟烷基),-NHC(=O)(烷基),-NHC(=O)H,-NHC(=O)NH2,-NHC(=O)NH(烷基),-NHC(=O)N(烷基)2,-NHC(=NH)NH2,-OSO2(烷基),-SH,-S(烷基),-S(芳基),-S(环烷基),-S(=O)烷基,-SO2(烷基),-SO2NH2,-SO2NH(烷基)和-SO2N(烷基)2。更优选的取代基是烷基,烯基,卤素,卤代烷基,羟基,羟烷基,氰基,硝基,烷氧基,-O(羟烷基),-C(=O)(烷基),-C(=O)H,-CO2H,-C(=O)NHOH,-C(=O)O(烷基),-C(=O)O(羟烷基),-C(=O)NH2,-C(=O)NH(烷基),-C(=O)N(烷基)2,-OC(=O)(烷基),-OC(=O)(羟烷基),-OC(=O)O(烷基),-OC(=O)O(羟烷基),-OC(=O)NH2,-OC(=O)NH(烷基),-OC(=O)N(烷基)2,-NH2,-NH(烷基),-N(烷基)2,-NH(芳基),-NHC(=O)(烷基),-NHC(=O)H,-NHC(=O)NH2,-NHC(=O)NH(烷基),-NHC(=O)N(烷基)2和-NHC(=NH)NH2。特别优选的是C1-C4烷基,氰基,硝基,卤素和C1-C4烷氧基。
在陈述范围的情况下,如“C1-C5烷基”或“5-10%”,这样的范围包括该范围的终点,如在第一种情况下的C1和C5以及在第二种情况下的5%和10%。
除非特别指出特定的立体异构体(例如,通过结构式中相关立构中心的粗体或虚线键,通过将结构式中的双键描述为具有E或Z构型,或通过使用立体化学指定命名法),所有立体异构体都包括在本发明的范围内,作为纯化合物及其混合物。除非另有说明,否则单独的对映异构体,非对映异构体,几何异构体及其组合和混合物都被本发明包括。
本领域技术人员将理解,化合物可具有互变异构形式(例如,酮和烯醇形式)、共振形式和两性离子形式,其等同于本文使用的结构式中所描述的那些,并且所述结构式包括这种互变异构、共振或两性离子形式。
“药学上可接受的酯”是指在体内(例如在人体内)水解以产生母体化合物或其盐或本身具有与母体化合物类似的活性的酯。合适的酯包括C1-C5烷基,C2-C5烯基或C2-C5炔基酯,尤其是甲基,乙基或正丙基酯。
“药学上可接受的盐”是指适用于药物制剂的化合物的盐。当化合物具有一个或多个碱性基团时,盐可以是酸加成盐,例如硫酸盐,氢溴酸盐,酒石酸盐,甲磺酸盐,马来酸盐,柠檬酸盐,磷酸盐,乙酸盐,双羟萘酸盐(双羟萘酸盐),氢碘酸盐,硝酸盐,盐酸盐,乳酸盐,甲基硫酸盐,富马酸盐,苯甲酸盐,琥珀酸盐,甲磺酸盐,乳糖酸盐,辛二酸盐,甲苯磺酸盐等。当化合物具有一个或多个酸性基团时,盐可以是例如以下的盐:钙盐,钾盐,镁盐,葡甲胺盐,铵盐,锌盐,哌嗪盐,氨丁三醇盐,锂盐,胆碱盐,二乙胺盐,4-苯基环己胺盐,苄星盐,钠盐,四甲基铵盐等。多晶型晶体形式和溶剂化物也包括在本发明的范围内。
在本说明书的式中,横穿键的波浪线 或键末端的星号(*)表示共价连接位点。例如,陈述式
或键末端的星号(*)表示共价连接位点。例如,陈述式 中的R是
中的R是 或R是
或R是 是指
是指
在本说明书的式中,在芳族环的两个碳之间横穿芳族环的键意指连接至键的基团可位于芳族环的任何可用位置。作为说明,式 表示
表示
SECO-CPI化合物
在式(I)和存在这些变量的其他式中,以下优选实施方案单独或与其他优选实施方案组合适用,除非上下文指出不同的优选实施方案或优选实施方案的组合适用:
(i)Hal是Cl。
(ii)R2是H。
(iii)R3是

(iv)R5是OMe,OH,苯基,NH2,NHMe,NMe2,NH(C2-C4烷基),N(C2-C4烷基)2,NHC(=O)X1,O(C2-C4烷基),O(CH2)0-2(C3-C6环烷基),O(CH2)0-2X1,或
其中C2-C4烷基可以是未取代的或被OCH2CH2OH,OCH2CH2NH2,NHCH2CH2OH,NHCH2CH2NH2,OH或NH2取代,并且苯基可以被C1-C2烷基,OH,NH2,NH(C1-C2烷基),N(C1-C2烷基)2,F,Cl,Br,NO2或CN取代;(即R5为非H的R4,R4’,R5,R6或R7之一)。
(v)R1是 更优选为
更优选为 最优选为
最优选为
(vi)R4,R4’,R5,R6和R7中不超过3个,更优选不超过2个不是H。
(vii)X1中的芳基或杂芳基是苯基,吡咯基,呋喃基,噻吩基,咪唑基,吡唑基,噁唑基,异噁唑基,噻唑基,异噻唑基,三唑基,四唑基,吡啶基,哒嗪基或吡嗪基;更优选苯基,尤其是被NH2或OH取代的苯基。
(viii)当R1是 时,至少一个X2不是H。
时,至少一个X2不是H。
关于上述优选(iii),R3更优选为 甚至更优选地,其中R4,R6和R7中的每一个是H。
甚至更优选地,其中R4,R6和R7中的每一个是H。
在本发明的优选方面,根据式(I)的化合物具有由式(Ia)表示的结构,其中R1,R4,R5,R6和R7如上文发明简单概述部分中关于式(I)所定义:

根据式(Ia)的化合物的实例列于表I中,其中R4,R6和R7各自为H,除非另有说明。



优选的seco-CPI化合物是(Ia-01),其完整结构如下所示:

优选的seco-CPI化合物是(Ia-02),其完整结构如下所示:

优选的seco-CPI化合物是(Ia-03),其完整结构如下所示:

优选的seco-CPI化合物是(Ia-09),其完整结构如下所示:

优选的seco-CPI化合物是(Ia-16),其完整结构如下所示:

优选的seco-CPI化合物是(Ia-18),其完整结构如下所示:

优选的seco-CPI化合物是(Ia-30),其完整结构如下所示:

根据式(I)的化合物的另一个优选实施方案具有由式(Ib)表示的结构,其中R1,R4,R5,R6和R7如上文发明简单概述部分中关于式(I)所定义:

表II中列出了根据式(Ib)的化合物的实例(R4,R4’,R6和R7各自为H)。

根据式(I)的化合物的另一个优选实施方案具有由式(Ic)表示的结构,其中R1,R4,R5,R6和R7如上文发明简单概述部分中关于式(I)所定义:

化合物(Ic)的实例是化合物(Ic-01):

化合物(I)的另一个实施方案具有由式(Id)表示的结构,其中R1,R4,R4’,R5,R6和R7如上文发明简单概述部分中关于式(I)所定义的:

化合物(Id)的实例是(Id-01)和(Id-02):


R1的具体优选实施方案是

R5的具体优选实施方案是

不受理论束缚,据信本发明的R1基团是亲水的,有利地改善了它们的性质。具有低CLogP R1基团可能是有利的,因为它增加了seco-CPI化合物及其连接基构建体的水溶性。如下文实施例1和2中所述,与抗体的缀合在基本上水性的介质中进行,并且更可溶的seco-CPI化合物-连接基化合物可以更有效地缀合。此外,在ADC意外过早裂解释放seco-CPI化合物到血液的情况下,增加的亲水性降低了化合物渗透细胞膜并引起脱靶毒性的可能性。(ADC进入细胞是通过不同的过程:由抗体与其抗原结合引发的内吞作用。)
每个基团R1中的开放价—见例如表I—位于氮处,使得它与seco-CPI核的羧基形成酰胺键。它们的亲水性可以通过相应的胺R1H的亲水性来估计。
化合物的亲水性(或相反地,疏水性)可以从其CLogP估计,CLogP是可从其结构计算的值:较低的值表示更亲水的分子,而较高的值表示更疏水的分子。对应于表I中的R1基团的胺R1H的CLogP值如下所示。CLogP值均小于0.300,范围为-1.484至0.248。(CLogP值使用来自PerkinElmer的 Ultra软件(Version 14.0.0.126)计算。(使用相同的软件计算本说明书中其他地方引用的CLogP值。)
Ultra软件(Version 14.0.0.126)计算。(使用相同的软件计算本说明书中其他地方引用的CLogP值。)

相比而言,Tichenor等人2007公开了化合物15a和15b,其具有与seco-CPI核连接的吡咯烷基酰胺,但相应的胺(吡咯啉)的CLogP值显著更高,为0.376。

因此,在一个实施方案中,基团R1是其相应的胺R1H具有小于0.300的CLogP值的那些,如使用前述CHEMBIODRAWTM软件计算。
缀合物
概述
本发明的seco-CPI化合物本身可用作治疗剂,但优选以缀合物形式使用。更优选地,缀合物中的靶向部分是抗体或其抗原结合部分,并且其抗原是肿瘤相关抗原,即由癌细胞表达的抗原。优选地,与正常细胞相比,肿瘤相关抗原被癌细胞独特地表达或过表达。肿瘤相关抗原可位于癌细胞表面或由癌细胞分泌到其周围环境中。
因此,本发明的另一个实施方案是包含本发明的seco-CPI化合物和配体的缀合物,由式(II)表示
[D(XD)a(C)c(XZ)b]mZ(II)
其中Z是靶向部分,D是本发明的seco-CPI化合物,-(XD)aC(XZ)b-统称为“连接基部分”或“连接基”,因为它们连接Z和D。在连接基内,C是可裂解的基团,设计为在D的预期生物学作用位点处或附近裂解;XD和XZ分别是间隔D和C以及C和Z的间隔基部分(或“间隔基”);下标a,b和c独立地是0或1(即,XD、XZ和C的存在是任选的)。下标m是1、2、3、4、5、6、7、8、9或10(优选1、2、3或4)。下面更全面地描述D,XD,C,XZ和Z。
通过与其抗原或受体所在的靶组织或细胞结合,Z将缀合物引向那里。靶组织或细胞处C基团的裂解释放D以局部发挥其细胞毒性作用。在一些情况下,缀合物通过胞吞作用内化到靶细胞中,并且在靶细胞内发生裂解。以这种方式,在预期作用的位置实现D的精确递送,减少了所需的剂量。而且,D在其缀合状态下通常是生物学上无活性的(或显著较低活性的),从而减少对非靶组织或细胞的不期望的毒性。
如下标m所反映的,每个Z可以与多于一个D缀合,这取决于Z具有的可用于缀合的位点数和所用的实验条件。本领域技术人员将理解,虽然每个单独的Z与整数个D缀合,缀合物的制备可以针对D与Z的非整数比来分析,反映统计平均值。该比率称为取代比(“SR”)或药物-抗体比(“DAR”)。
靶向部分Z
优选地,靶向部分Z是抗体。为了方便和简洁而不是为了限制,本说明书中关于Z及其缀合物的详细讨论是在其为抗体的背景下书写的,但是本领域技术人员将理解其他类型的Z加以必要的变更也可以缀合。例如,具有叶酸作为靶向部分的缀合物可以靶向在其表面上具有叶酸受体的细胞(Leamon等人,Cancer Res.2008,68(23),9839)。出于同样的原因,本说明书中的详细讨论主要是以1:1的Z与D的比率(m=1)来书写的。
优选地,Z是针对肿瘤相关抗原的抗体,允许选择性靶向癌细胞。此类抗原的实例包括:间皮素,前列腺特异性膜抗原(PSMA),CD19,CD22,CD30,CD70,B7H3,B7H4(也称为O8E),蛋白酪氨酸激酶7(PTK7),磷脂酰肌醇蛋白聚糖-3,RG1,岩藻糖基-GM1,CTLA-4和CD44。抗体可以是动物(例如鼠)、嵌合、人源化的,或优选人抗体。抗体优选是单克隆抗体,尤其是单克隆人抗体。针对一些上述抗原的人单克隆抗体的制备公开于Korman等人,US 8,609,816B2(2013;B7H4,也称为08E;特别是抗体2A7,1G11和2F9);Rao-Naik等人,8,097,703B2(2012;CD19;特别是抗体5G7,13F1,46E8,21D4,21D4a,47G4,27F3和3C10);King等人,US 8,481,683B2(2013;CD22;特别是抗体12C5,19A3,16F7和23C6);Keler等人,US 7,387,776B2(2008;CD30;特别是抗体5F11,2H9和17G1);Terrett等人,US 8,124,738B2(2012;CD70;特别是抗体2H5,10B4,8B5,18E7和69A7);Korman等人,US 6,984,720B1(2006;CTLA-4;特别是抗体10D1,4B6和1E2);Vistica等人,US 8,383,118B2(2013,岩藻糖基-GM1,特别是抗体5B1,5B1a,7D4,7E4,13B8和18D5);Korman等人,US 8,008,449B2(2011;PD-1;特别是抗体17D8,2D3,4H1,5C4,4A11,7D3和5F4);Huang等人,US 2009/0297438A1(2009;PSMA.特别是抗体1C3,2A10,2F5,2C6);Cardarelli等人,US 7,875,278B2(2011;PSMA;特别是抗体4A3,7F12,8C12,8A11,16F9,2A10,2C6,2F5和1C3);Terrett等人,US 8,222,375B2(2012;PTK7;特别是抗体3G8,4D5,12C6,12C6a和7C8);Terrett等人,US 8,680,247B2(2014;磷脂酰肌醇蛋白聚糖-3;特别是抗体4A6,11E7和16D10);Harkins等人,US 7,335,748B2(2008;RG1;特别是抗体A,B,C和D);Terrett等人,US 8,268,970B2(2012;间皮素;特别是抗体3C10,6A4和7B1);Xu等人,US 2010/0092484A1(2010;CD44;特别是抗体14G9.B8.B4,2D1.A3.D12和1A9.A6.B9);Deshpande等人,US 8,258,266 B2(2012;IP10;特别是抗体1D4,1E1,2G1,3C4,6A5,6A8,7C10,8F6,10A12,10A12S和13C4);Kuhne等人,US8,450,464 B2(2013;CXCR4;特别是抗体F7,F9,D1和E2);以及Korman等人,US 7,943,743 B2(2011;PD-L1;特别是抗体3G10,12A4,10A5,5F8,10H10,1B12,7H1,11E6,12B7和13G4);其公开内容通过引用并入本文。
除了作为抗体之外,Z还可以是抗体片段(例如Fab,Fab’,F(ab’)2,Fd或Fv)或抗体模拟物,例如亲和体(affibody),结构域抗体(dAb),纳米抗体,unibody,DARPin,anticalin,versabody,duocalin,lipocalin或avimer。
Z上的几个不同反应基团中的任何一个可以是缀合位点,包括赖氨酸残基中的ε-氨基,侧链碳水化合物部分,天冬氨酸或谷氨酸侧链上的羧酸基团,半胱氨酸-半胱氨酸二硫基团和半胱氨酸巯基。对于适用于缀合的抗体反应性基团的综述,见例如Garnett,Adv.DrugDeliveryRev.2001,53,171-216和Dubowchik and Walker,Pharmacology&Therapeutics 1999,83,67-123,其公开内容通过引用并入本文。
大多数抗体具有多个赖氨酸残基,其可以通过其ε-氨基通过酰胺,脲,硫脲或氨基甲酸酯键缀合。
在另一个实施方案中,Z可以通过碳水化合物侧链缀合,因为许多抗体是糖基化的。碳水化合物侧链可以用高碘酸盐氧化以产生醛基,醛基又可以与胺反应形成亚胺基,例如在缩氨基脲,肟或腙中。如果需要,通过用氰基硼氢化钠还原,可以将亚胺基团转化为更稳定的胺基。关于通过碳水化合物侧链缀合的其他公开内容,见例如Rodwell等人,Proc.Nat’lAcad.Sci.USA 83,2632-2636(1986);其公开内容通过引用并入本文。
在另一个实施方案中,Z可以通过天冬氨酸或谷氨酸侧链羧酸缀合,例如通过转化成碳酰肼,然后与含有醛的连接基反应。见Fisch等人,Bioconjugate Chemistry 1992,3,147-153。
半胱氨酸侧链中的硫醇(-SH)基团可以通过几种方法用于形成缀合物。它可用于在其与连接基上的巯基之间形成二硫键。另一种方法是通过其迈克尔加成到连接基上的马来酰亚胺基团。
通常,尽管抗体具有半胱氨酸残基,但它们缺乏游离巯基,因为它们的所有半胱氨酸都参与链内或链间二硫键。为了产生游离巯基,可以减少天然二硫基团。见例如Packard等人,Biochemistry 1986,25,3548;King等人,CancerRes.1994,54,6176;和Doronina等人,NatureBiotechnol.2003,21,778。或者,可以通过使抗体突变,用半胱氨酸取代另一个氨基酸或将一个半胱氨酸插入多肽链来引入具有游离-SH基团的半胱氨酸。见例如Eigenbrot等人,US 7,521,541B2(2009);Chilkoti等人,Bioconjugate Chem.1994,5,504;Urnovitz等人,US 4,698,420(1987);Stimmel等人,J.Biol.Chem.2000,275,30445;Bam等人,US 7,311,902B2(2007);Kuan等人,J.Biol.Chem.1994,269,7610;Poon等人,J.Biol.Chem.1995,270,8571;Junutula等人,NatureBiotechnology 2008,26,925和Rajpal等人,2015年12月21日提交的美国临时申请No.62/270245。在另一种方法中,将半胱氨酸添加到重链或轻链的C末端。见例如Liu等人,US 8,865,875B2(2014);Cumber等人,J.Immunol.1992,149,120;King et al,CancerRes.1994,54,6176;Li等人,BioconjugateChem.2002,13,985;Yang等人,Protein Engineering2003,16,761;和Olafson等人,Protein Engineering Design&Selection 2004,17,21。本段引用的文献的公开内容通过引用并入本文。
连接基及其组分
如上所述,连接基包含最多三个元件:可裂解的基团C和任选的间隔基XZ和XD。
基团C在生理条件下是可裂解的。优选地,当缀合物在血液中循环时,它是相对稳定的,但是一旦缀合物到达靶细胞附近、靶细胞处或靶细胞内的其预期作用的位点时,它就容易被裂解。优选地,当抗体Z与靶细胞表面上展示的抗原结合时,缀合物被靶细胞内化。随后,C的裂解发生在靶细胞的囊泡体(早期内涵体,晚期内涵体,或尤其是溶酶体)中。
在一个实施方案中,C是pH敏感基团。血浆中的pH略高于中性,而溶酶体内的pH为酸性,约为5。因此,酸敏感的C将在溶酶体内以比血液中快几个数量级的速率裂解。酸敏感基团的实例是顺式-乌头酰基酰胺和腙,如Shen等人,US 4,631,190(1986);Shen等人,US5,144,011(1992);Shen等人,Biochem.Biophys.Res.Commun.1981,102,1048;和Yang等人,Proc.NatlAcad.Sci(USA)1988,85,1189中所述,其公开内容通过引用并入本文。
在另一个实施方案中,C是二硫化物。二硫化物可以通过硫醇-二硫化物交换机制裂解,其速率取决于环境硫醇浓度。由于谷胱甘肽和其他硫醇的细胞内浓度高于其血清浓度,二硫化物的裂解速率在细胞内更高。此外,硫醇-二硫化物交换的速率可以通过调节二硫化物的空间和电子特性来调节(例如,烷基-芳基二硫化物vs.烷基-烷基二硫化物;芳基环上的取代等),使得能够设计具有增强的血清稳定性或特定裂解速率的二硫键。见例如Thorpe等人,CancerRes.2008,48,6396-6403;Santi等人,US7,541,530B2(2009);Ng等人,US 6,989,452B2(2006);Ng等人,WO2002/096910A1;Boyd等人,US 7,691,962B2(2010);和Sufi等人,US8,461,117B2(2013);其公开内容通过引用并入本文。
优选的基团C是通过靶细胞内的蛋白酶而不是通过血清中的蛋白酶选择性裂解的肽。通常,肽包含1至20个氨基酸,优选1至6个氨基酸,更优选2至3个氨基酸。氨基酸可以是天然的和/或非天然的α-氨基酸。天然氨基酸是由遗传密码编码的氨基酸,以及由其衍生的氨基酸,例如,羟脯氨酸,γ-羧基谷氨酸,瓜氨酸和O-磷酸丝氨酸。在本说明书中,术语“氨基酸”还包括氨基酸类似物和模拟物。类似物是具有天然氨基酸的相同的一般H2N(R)CHCO2H结构的化合物,除了R基团不是天然氨基酸中发现的一种。类似物的实例包括高丝氨酸,正亮氨酸,甲硫氨酸-亚砜和甲硫氨酸甲基锍。氨基酸模拟物是具有与α-氨基酸的一般化学结构不同的结构但以与一种α-氨基酸类似的方式起作用的化合物。氨基酸可以具有遗传编码的氨基酸的“L”立体化学以及具有对映体“D”立体化学。
优选地,C含有氨基酸序列,该氨基酸序列是蛋白酶的裂解识别序列。许多裂解识别序列是本领域已知的。见例如Matayoshi等人Science247:954(1990);Dunn等人Meth.Enzymol.241:254(1994);Seidah等人Meth.Enzymol.244:175(1994);Thornberry,Meth.Enzymol.244:615(1994);Weber等人Meth.Enzymol.244:595(1994);Smith等人Meth.Enzymol.244:412(1994);和Bouvier等人Meth.Enzymol.248:614(1995);其公开内容通过引用并入本文。
对于不预期被细胞内化的缀合物,可以选择基团C使得它被存在于癌症附近的细胞外基质中的蛋白酶裂解,所述蛋白酶例如附近垂死的癌细胞释放的蛋白酶或癌细胞分泌的肿瘤相关蛋白酶。示例性细胞外肿瘤相关蛋白酶是纤溶酶,基质金属蛋白酶(MMP),thimet寡肽酶(TOP)和CD10。见例如Trouet等人,US 7,402,556B2(2008);Dubois等人,US7,425,541B2(2008);和Bebbington等人,US 6,897,034B2(2005)。
对于设计为被细胞内化的缀合物,C优选包含选择用于被内涵体或溶酶体蛋白酶,尤其是后者裂解的氨基酸序列。此类蛋白酶的非限制性实例包括组织蛋白酶B,C,D,H,L和S,尤其是组织蛋白酶B。示例性组织蛋白酶B可裂解肽包括Val-Ala,Val-Cit,Val-Lys,Lys-Val-Ala,Asp-Val-Ala,Val-Ala,Lys-Val-Cit,Ala-Val-Cit,Val-Gly,Val-Gln和Asp-Val-Cit。(此处,氨基酸序列以N至C方向书写,如在H2N-AA2-AA1-CO2H中,除非上下文另有明确说明。)见Dubowchik等人,Biorg.Med.Chem.Lett.1998,8,3341;Dubowchik等人,Bioorg.Med.Chem.Lett.1998,8,3347;和Dubowchik等人,Bioconjugate Chem.2002,13,855;其公开内容通过引用并入。
另一种可用于裂解肽基连接基的酶是豆荚蛋白,一种优先在Ala-Ala-Asn处裂解的溶酶体半胱氨酸蛋白酶。
在一个实施方案中,基团C是包含双氨基酸序列-AA2-AA1-的肽,其中AA1是赖氨酸,精氨酸或瓜氨酸,AA2是苯丙氨酸,缬氨酸,丙氨酸,亮氨酸或异亮氨酸。在另一个实施方案中,C由一至三个氨基酸的序列组成,选自Val-Cit,Ala-Val,Val-Ala-Val,Lys-Lys,Ala-Asn-Val,Val-Leu-Lys,Cit-Cit,Val-Lys,Ala-Ala-Asn,Lys,Cit,Ser和Glu。更优选地,它是来自前述组的2-3个氨基酸肽。
由单个氨基酸组成的可裂解基团C的制备和设计公开于Chen等人,US 8,664,407B2(2014),其公开内容通过引用并入本文。
基团C可以直接键合到Z或D;即间隔基XZ或XD视情况而定可以不存在。
当存在时,间隔基XZ提供C和Z之间的空间分离,以免前者在空间上干扰后者的抗原结合,或后者在空间上干扰前者的裂解。此外,间隔基XZ可用于赋予缀合物增加的溶解度或降低的聚集性质。间隔基XZ可包括一个或多个模块区段,其可以以任何数量的组合组装。用于间隔基XZ的合适区段的实例是:


间隔基XD如果存在,提供C和D之间的空间分离,以免后者在空间上或电子上干扰前者的裂解。间隔基XD也可用于将额外的分子质量和化学官能团引入缀合物中。通常,额外的质量和官能团将影响缀合物的血清半衰期和其他性质。因此,通过明智地选择间隔基团,可以调节缀合物的血清半衰期。间隔基XD也可以由模块化区段组装,类似于上面对于间隔基XZ的描述。
间隔基XZ和/或XD(如果存在)优选分别在Z和C或D和C之间提供4至25个原子,更优选4至20个原子的线性间隔。
除了共价连接抗体和药物之外,连接基还可以执行其他功能。例如,连接基可含有聚(乙二醇)(“PEG”)基团。由于缀合步骤通常涉及将药物-连接基与抗体在水性介质中偶联,因此PEG基团可增强药物-连接基的水溶性。而且,PEG基团可以增强溶解度或减少所得ADC中的聚集。当存在PEG基团时,它可以加入间隔基XZ/XD或两者中。PEG基团中重复单元的数量可以是2至20,优选4和10之间。
间隔基XZ或XD或两者可包含自牺牲部分。自牺牲部分是以下的部分:(1)与C键合并且与Z或D键合,且(2)具有这样的结构,使得从C基团裂解引发反应序列,导致在这种情况下自牺牲部分自身脱离Z或D。换句话说,在远离Z或D的位置处的反应(从C基团裂解)导致XZ-Z或XD-D键也破裂。在间隔基XD的情况下,期望存在自牺牲部分,因为如果在缀合物裂解后,间隔基XD或其一部分保持与D连接,则D的生物学活性可能会受损。在可裂解基团C是多肽的情况下,特别期望使用自牺牲部分,在该情况下,自牺牲部分通常位于其附近,以防止D在空间或电子上干扰肽裂解。
与D的羟基或氨基键合的示例性自牺牲部分(i)-(v)如下所示:


自牺牲部分是虚线a和b(或虚线b和c)之间的结构,显示相邻的结构特征以提供所处环境。自牺牲部分(i)和(v)与D-NH2键合(即,通过氨基缀合),而自牺牲部分(ii)、(iii)和(iv)键合到D-OH(即,通过羟基或羧基缀合)。通过酶裂解虚线b处的键—在结构(i)-(v)的情况下为肽酶和在结构(vi)的情况下为β-葡糖苷酸酶—引发自牺牲反应序列,其导致在虚线a处裂解键并随后释放D-OH或D-NH2,视情况而定。举例来说,结构(i)和(iv)的自牺牲机制如下所示:

换句话说,在自牺牲基团的一个部分裂解第一个化学键引发了步骤序列,该序列导致在自牺牲基团的不同部分的第二个化学键—将自牺牲基团与药物连接的化学键—的裂解,从而释放药物。
在某些情况下,自牺牲基团可以串联使用,如结构(vii)所示。在这种情况下,虚线c处的裂解通过1,6-消除反应触发虚线b和c之间的部分的自牺牲,然后通过环化-消除反应在虚线a和b之间的部分自牺牲。有关自牺牲部分的其他公开内容,见Carl等人,J.Med.Chem.1981,24,479;Carl等人,WO 81/01145(1981);Dubowchik等人,Pharmacology& Therapeutics 1999,83,67;Firestone等人,US 6,214,345B1(2001);Toki等人,J.Org.Chem.2002,67,1866;Doronina等人,NatureBiotechnology2003,21,778(erratum,p.941);Boyd等人,US 7,691,962B2;Boyd等人,US 2008/0279868A1;Sufi等人,WO 2008/083312A2;Feng,US 7,375,078B2;Jeffrey等人,US 8,039,273;和Senter等人,US 2003/0096743A1;其公开内容通过引用并入。
在另一个实施方案中,Z和D通过不可裂解的连接基连接,即C不存在。D的代谢最终将连接基减小为不会干扰D的生物学活性的小的附加部分。
缀合技术
本发明的缀合物优选通过以下方法制备:首先制备包含D和连接基(XD)a(C)c(XZ)b的化合物(其中XD,C,XZ,a,b和c如对式(II)所定义),以形成由式(III)表示的药物-连接基化合物:
D-(XD)a(C)c(XZ)b-R31(III)
其中R31是适合与Z上的互补官能团反应形成缀合物的官能团。合适的基团R31的实例包括氨基,叠氮化物,硫醇,环辛炔,

其中R32是Cl,Br,F,甲磺酸根或甲苯磺酸根,R33是Cl,Br,I,F,OH,-O-N-琥珀酰亚胺基,-O-(4-硝基苯基),-O-五氟苯基,或-O-四氟苯基。通常可用于制备合适的部分D-(XD)aC(XZ)b-R31的化学方法公开于Ng等人,US 7,087,600 B2(2006);Ng等人,US 6,989,452 B2(2006);Ng等人,US 7,129,261 B2(2006);Ng等人,WO 02/096910 A1;Boyd等人,US 7,691,962 B2;Chen等人,US 7,517,903 B2(2009);Gangwar等人,US 7,714,016 B2(2010);Boyd等人,US 2008/0279868 A1;Gangwar等人,US 7,847,105 B2(2010);Gangwar等人,US 7,968,586 B2(2011);Sufi等人,US 8,461,117 B2(2013);和Chen等人,US 8,664,407 B2(2014);其公开内容通过引用并入本文。
优选反应性官能团-R31是-NH2,-OH,-CO2H,-SH,马来酰亚胺基,环辛炔,叠氮基(-N3),羟基氨基(-ONH2)或N-羟基琥珀酰亚胺基。特别优选的官能团-R31是:

-OH基团可以用抗体上,例如天冬氨酸或谷氨酸侧链上的羧基酯化。
-CO2H基团可以用-OH基团酯化,或者用抗体上的氨基(例如赖氨酸侧链上的氨基)酰胺化。
N-羟基琥珀酰亚胺基团在功能上是活化的羧基,并且可以通过与氨基(例如来自赖氨酸)反应方便地酰胺化。
马来酰亚胺基团可以在迈克尔加成反应中与抗体上的-SH基团(例如,来自半胱氨酸或来自抗体的化学修饰以引入巯基官能团)缀合。
当抗体不具有可用于缀合的半胱氨酸-SH时,赖氨酸残基侧链中的ε-氨基可与2-亚氨基四氢噻吩或N-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯(“SPDP”)反应引入游离巯基(-SH)-,产生好像半胱氨酸替代物。巯基可以与马来酰亚胺或其他亲核受体基团反应以实现缀合。如下用2-亚氨基四氢噻吩说明该机理。

通常,实现每抗体2至3个巯基的硫醇化水平。关于代表性方法,见Cong等人,US 8,980,824B2(2015),其公开内容通过引用并入本文。
在相反的布置中,抗体Z可以用N-琥珀酰亚胺基4-(马来酰亚胺甲基)-环己烷甲酸酯(“SMCC”)或其磺化变体磺基-SMCC(两者都可从Sigma-Aldrich获得)修饰以向其引入马来酰亚胺基团。然后,可以用连接基上具有-SH基团的药物-连接基化合物进行缀合。
另一种缀合方法采用无铜“点击化学”,其中叠氮基团加成在应变的环辛炔上,形成1,2,3-三唑环。见例如Agard等人,J.Amer.Chem.Soc.2004,126,15046;Best,Biochemistry 2009,48,6571,其公开内容通过引用并入本文。叠氮化物可位于抗体上,而环辛炔可位于药物-连接基部分上,反之亦然。优选的环辛炔基是二苯并环辛炔(DIBO)。具有DIBO基团的各种试剂可从Invitrogen/Molecular Probes,Eugene,Oregon获得。下面的反应说明了其中DIBO基团与抗体(Ab)连接的情况下的点击化学缀合:

另一种缀合技术涉及将非天然氨基酸引入抗体,其中非天然氨基酸提供与药物部分中的反应性官能团缀合的官能团。例如,非天然氨基酸对乙酰基苯丙氨酸可以加入抗体或其他多肽中,如Tian等人,WO2008/030612 A2(2008)中所教导的。通过与连接基-药物部分上的羟基氨基形成肟,对乙酰基苯丙氨酸中的酮基可以是缀合位点。或者,可以将非天然氨基酸对叠氮基苯丙氨酸加入抗体中,以提供叠氮化物官能团用于通过点击化学缀合,如上所述。非天然氨基酸也可以使用无细胞方法加入抗体或其他多肽中,如Goerke等人,US2010/0093024 A1(2010)和Goerke等人,Biotechnol.Bioeng.2009,102(2),400-416中所教导的。上述公开内容通过引用并入本文。因此,在一个实施方案中,用于与本发明的药物制备缀合物的抗体具有被非天然氨基酸替代的一个或多个氨基酸,所述非天然氨基酸优选为对乙酰基苯丙氨酸或对叠氮基苯丙氨酸,更优选为对乙酰基苯丙氨酸。
另一种缀合技术使用酶谷氨酰胺转胺酶(优选来自Streptomyces mobaraensis或BTG的细菌性谷氨酰胺转胺酶),根据Jeger等人,Angew.Chem.Int.Ed.2010,49,9995。BTG在谷氨酰胺(胺受体)的侧链甲酰胺和亚烷基氨基(胺供体)(可以是例如赖氨酸或5-氨基-正戊基的ε-氨基)之间形成酰胺键。在典型的缀合反应中,谷氨酰胺残基位于抗体上,而亚烷基氨基位于连接基-药物部分上,如下所示:

谷氨酰胺残基在多肽链上的定位对其对BTG介导的转酰胺化的易感性具有很大影响。抗体上的谷氨酰胺残基通常都不是BTG底物。然而,如果抗体是去糖基化的-糖基化位点是重链的天冬酰胺297(N297;编号按照Kabat等人,"Sequences ofproteinsofimmunological interest,第5版,Pub.No.91-3242,U.S.Dept.Health&Human Services,NIH,Bethesda,Md.,1991(以下称为“Kabat”)中提出的EU索引)-邻近的谷氨酰胺295(Q295)对BTG敏感。通过用PNGase F(肽-N-糖苷酶F)处理,可以使抗体酶促去糖基化。或者,可以通过在恒定区中引入N297A突变来合成无糖苷抗体,以消除N297糖基化位点。此外,已经显示N297Q取代不仅消除了糖基化,而且还引入了第二个谷氨酰胺残基(在297位),其也是胺受体。因此,在一个实施方案中,与本发明药物缀合的抗体是去糖基化的。在另一个实施方案中,抗体具有N297Q取代。本领域技术人员将理解,通过合成后修饰或通过引入N297A突变的去糖基化产生每个抗体两个BTG-反应性谷氨酰胺残基(每个重链一个,在295位),而具有N297Q取代的抗体将具有四个BTG反应性谷氨酰胺残基(每个重链两个,在295和297位)。
通过向其中引入含有谷氨酰胺的肽或“标签”,也可以使抗体对BTG介导的缀合敏感,例如在Pons等人,US 2013/0230543 A1(2013)和Rao-Naik等人,PCT申请No.PCT/US2016/020192(2016年3月1日提交)中所教导。
在互补方法中,BTG的底物特异性可以通过改变其氨基酸序列来改变,使得其变得能够与未修饰的抗体中的谷氨酰胺295反应,如2015年10月2日提交的Rao-Naik等人,美国临时申请No.62/236274中所教导。
最后,尽管最常用的细菌性谷氨酰胺转胺酶来自S.mobaraensis,但可以考虑具有稍微不同的底物特异性的来自其他细菌的谷氨酰胺转胺酶,例如来自Streptoverticilliumladakanum的谷氨酰胺转胺酶(Hu等人,US 2009/0318349 A1(2009),US 2010/0099610 A1(2010)和US2010/0087371 A1(2010))。
也可以使用酶分选酶A实现缀合,如Levary等人,PLoS One 2011,6(4),e18342;Proft,Biotechnol.Lett.2010,32,1-10;Ploegh等人,WO2010/087994 A2(2010);和Mao等人,WO 2005/051976 A2(2005)所教导的。分选酶A识别基序(通常为LPXTG,其中X为任何天然氨基酸)可位于配体Z上,亲核受体基序(通常为GGG)可为式(III)中的基团R31,反之亦然。
Seco-CPI-连接基化合物
本发明的seco-CPI化合物的ADC包括与seco-CPI化合物上的官能团连接的连接基,该连接基与抗体连接。反映本领域已知的缀合技术的多样性,本发明的seco-CPI化合物可以被精心制成适于缀合抗体的许多不同的seco-CPI化合物-连接基化合物。
通常,有三个不同的用于连接基与本发明的seco-CPI化合物连接的位点,如下所示:

在(a)型seco-CPI-连接基化合物中,连接基与结合亚单位的酚羟基连接,并且在连接时,充当前药化基团,防止环化成CPI结构。在(b)型seco-CPI-连接基化合物中,连接基通过R1基团中的合适官能团例如氨基、羧基或羟基连接。在(c)型seco-CPI-连接基化合物中,连接基通过R3基团中的合适官能团例如氨基、羧基或羟基连接。在(b)和(c)型中,酚羟基任选地可以被前药化,如上所述。
在一个实施方案中,(a)型seco-CPI-连接基化合物可由式(IIIa)表示:

其中,
T是自牺牲基团;
t是0或1;
AAa和每个AAb独立地选自丙氨酸,β-丙氨酸,γ-氨基丁酸,精氨酸,天冬酰胺,天冬氨酸,γ-羧基谷氨酸,瓜氨酸,半胱氨酸,谷氨酸,谷氨酰胺,甘氨酸,组氨酸,异亮氨酸,亮氨酸,赖氨酸,蛋氨酸,正亮氨酸,正缬氨酸,鸟氨酸,苯丙氨酸,脯氨酸,丝氨酸,苏氨酸,色氨酸,酪氨酸和缬氨酸;
u是0或1;
p是1、2、3或4;
q是1、2、3、4、5、6、7、8、9、10、11或12(优选2、3、4或8);
r是1、2、3、4或5;
s是0或1;
v是0或1;
R31是H,

条件是,只有当s是1且v为0时,R31才能为H,并且只有当s是1且R31为H时,v才能为0;
且
R1,R2和R3如上文发明简单概述部分中关于式(I)所定义。
在根据式(IIIa)的seco-CPI连接基的一个实施方案中,R3是

在一个实施方案中,式(IIIa)中u是1。
部分-AAa-[AAb]p-表示长度由p的值确定的多肽(如果p是1,则为二肽,如果p为3,则为四肽等)。AAa位于多肽的羧基末端,其羧基与seco-CPI化合物的胺氮形成肽(酰胺)键。相反,最后的AAb位于多肽的氨基末端,其α-氨基与 形成肽键,分别取决于s是1还是0。优选的多肽-AAa-[AAb]p-是Val-Ala,Val-Cit,Val-Lys,Lys-Val-Ala,Asp-Val-Ala,Val-Ala,Lys-Val-Cit,Ala-Val-Cit,Val-Gly,Val-Gln和Asp-Val-Cit,以常规的N至C方向书写,如H2N-Val-Cit-CO2H中)。更优选地,多肽是Val-Cit,Val-Lys或Val-Ala。优选地,多肽-AAa-[AAb]p-可被靶(癌)细胞内发现的酶裂解,所述酶例如组织蛋白酶,尤其是组织蛋白酶B。
形成肽键,分别取决于s是1还是0。优选的多肽-AAa-[AAb]p-是Val-Ala,Val-Cit,Val-Lys,Lys-Val-Ala,Asp-Val-Ala,Val-Ala,Lys-Val-Cit,Ala-Val-Cit,Val-Gly,Val-Gln和Asp-Val-Cit,以常规的N至C方向书写,如H2N-Val-Cit-CO2H中)。更优选地,多肽是Val-Cit,Val-Lys或Val-Ala。优选地,多肽-AAa-[AAb]p-可被靶(癌)细胞内发现的酶裂解,所述酶例如组织蛋白酶,尤其是组织蛋白酶B。
如下标所示,t等于0或1,任选存在自牺牲基团T。优选的自牺牲基团T是对氨基苄醇(PABA)基团,其结构如下所示(酚氧未显示),星号(*)表示与seco-CPI化合物的酚氧键合的末端,波浪线 表示与多肽-AAa-[AAb]p-键合的末端。
表示与多肽-AAa-[AAb]p-键合的末端。

另一种优选的自牺牲基团T是对氨基苄基氧基羰基(PABC)基团,其结构如下所示:

在一些情况下,PABA基团可优于PABC基团,因为其更具水解稳定性。
在一个优选的实施方案中,基团R31是

在另一个优选的实施方案中,基团R31是

或者R31是H而v是0并且s是1。在每种情况下,结果分别是以下结构的具有氨基末端基团的seco-CPI-连接基化合物

其适合作为谷氨酰胺转胺酶介导的缀合的胺供体。
优选的(a)型seco-CPI-连接基化合物根据式(IIIa')

其中
R4,R5,R6和R7如上文发明简单概述部分中关于式(I)所定义;
R10是
R11,R12和R13独立地为H,CH3,CH(CH3)2,CH2CO2H,CH2CH2CO2H,CH2C(=O)NH2,CH2CH2C(=O)NH2,(CH2)4NH2,(CH2)3NHC(=NH)NH2或(CH2)3NHC(=O)NH2(即,R11,R12和R13对应于氨基酸甘氨酸,缬氨酸,天冬氨酸,谷氨酸,天冬酰胺,谷氨酰胺,赖氨酸,精氨酸和瓜氨酸的侧链残基);和
R14是

优选地,R10中的肽是二肽—即与肽中的氨基酸HO2CCH(R13)NH2相关的下标为零—对应于下式

根据式(IIIa')的具体化合物示于表III中,其中R4,R6和R7为H,除非另有说明,且R10为A或B:


优选的seco-CPI-连接基化合物是(IIIa-06),其完整结构如下所示:

优选的seco-CPI-连接基化合物是(IIIa-09),其完整结构如下所示:

优选的seco-CPI-连接基化合物是(IIIa-12),其完整结构如下所示:

如上所述,具有A型连接基的seco-CPI-连接基化合物被设计用于通过与抗体上的巯基的迈克尔加成反应进行缀合。具有B型连接基的那些被设计用于谷氨酰胺转胺酶介导的缀合,同样如上所述。
更优选地,在式(IIIa')中,R4,R6和R7各自为H;并且R5是


缀合物
在一个实施方案中,本发明的缀合物衍生自(a)型seco-CPI-连接基化合物,并且可以由式(IVa)表示:

其中,
Ab是抗体;
R40是键,
其中与Ab键合的R40的开放价用星号(*)表示,与(CH2)r键合的R40的开放价用波浪线 表示;
表示;
m是1、2、3或4;
v是0或1,条件是只有当s是1且R40为键时v才能为0,并且只有当v为0且s是1时R40才能为键;
T,t,AAa,AAb,u,p,q,s,r和XA如关于式(IIIa)所定义;和
R1,R2和R3如上文发明简单概述部分中所定义。
当R40是键时, 中的氮根据下式直接与抗体Ab键合:
中的氮根据下式直接与抗体Ab键合:

在根据式(IVa)的缀合物的一个实施方案中,R3是

在一个优选的实施方案中,式(IVa)中u是1。
在式(IVa)中,如果下标t和u都是0,则连接基是不可裂解的类型并且依赖于抗体Ab的降解以释放药物。如果聚乙二醇组分的存在是有益的,例如通过在缀合期间增加药物-连接基化合物的溶解度而有益,并且不干扰药物的生物学活性,则任选地可以存在聚乙二醇组分(即,s是1)。
药物组合物
另一方面,本公开提供药物组合物,其包含与药学上可接受的载体或赋形剂一起配制的本发明化合物或其缀合物。它可任选地含有一种或多种另外的药物活性成分,例如抗体或另一种药物。药物组合物可以在与另一种治疗剂,尤其是另一种抗癌剂的组合疗法中给药。
药物组合物可包含一种或多种赋形剂。可以使用的赋形剂包括载体,表面活性剂,增稠剂或乳化剂,固体粘合剂,分散或悬浮助剂,增溶剂,着色剂,调味剂,包衣剂,崩解剂,润滑剂,甜味剂,防腐剂,等渗剂及其组合。Gennaro编辑,Remington:The ScienceandPractice ofPharmacy,第20版,(Lippincott Williams & Wilkins 2003)教导了合适的赋形剂的选择和使用,其公开内容通过引用并入本文。
优选地,药物组合物适用于静脉内,肌肉内,皮下,肠胃外,脊柱或表皮施用(例如,通过注射或输注)。取决于施用途径,活性化合物可以被包覆在材料中以保护其免受可能使其失活的酸和其他天然条件的作用。短语“肠胃外给药”是指除肠内和局部给药以外的给药方式,通常通过注射给药,包括但不限于静脉内,肌肉内,动脉内,鞘内,囊内,眼内,心内,皮内,腹膜内,经气管,皮下,表皮下,关节内,囊下,蛛网膜下,脊柱内,硬膜外和胸骨内注射和输注。或者,药物组合物可以通过非肠胃外途径给药,例如局部,表皮或粘膜给药途径,例如,鼻内,口服,阴道,直肠,舌下或局部给药。
药物组合物可以是无菌水溶液或分散液的形式。它们还可以配制成微乳液,脂质体或适于实现高药物浓度的其他有序结构。该组合物还可以以冻干物的形式提供,用于在给药前在水中重构。
可以与载体材料组合以产生单一剂型的活性成分的量将根据所治疗的受试者和特定的给药方式而变化,并且通常是产生治疗效果的组合物的量。通常,基于百分之百,该量为约0.01%至约99%的活性成分,优选约0.1%至约70%,最优选约1%至约30%的活性成分与药学上可接受的载体组合。
调整剂量方案以提供治疗反应。例如,可以施用单次推注,可以随时间施用几个分开的剂量,或者可以根据情况的紧急性按比例减少或增加剂量。以剂量单位形式配制肠胃外组合物以便于给药和使剂量均匀是特别有利的。“剂量单位形式”是指适合作为待治疗受试者的单位剂量的物理上离散的单位;每个单位含有经计算产生所需的治疗反应的预定量的活性化合物,以及所需的药物载体。
剂量范围为宿主体重的约0.0001至100mg/kg,更通常为0.01至5mg/kg。例如,剂量可以是0.3mg/kg体重,1mg/kg体重,3mg/kg体重,5mg/kg体重或10mg/kg体重或在1-10mg/kg范围内或者0.1至5mg/kg范围内。示例性治疗方案是每周一次,每两周一次,每三周一次,每四周一次,每月一次,每三个月一次,或每三至六个月一次给药。优选的剂量方案包括1mg/kg体重或3mg/kg体重,通过静脉内给药,使用以下给药方案之一:(i)每四周一次,给予六次剂量,然后每三个月一次;(ii)每三周一次;(iii)3mg/kg体重一次,然后每三周1mg/kg体重。在一些方法中,调节剂量以达到约1-1000μg/mL的血浆抗体浓度,并且在一些方法中达到约25-300μg/mL。
本发明化合物的“治疗有效量”优选导致疾病症状的严重程度降低,疾病无症状期的频率和持续时间增加,或预防由患有疾病引起的损伤或残疾。例如,对于携带肿瘤的受试者的治疗,“治疗有效量”优选抑制肿瘤生长至少约20%,更优选至少约40%,甚至更优选至少约60%,并且进一步更优选至少约80%,相对于未治疗的受试者而言。治疗有效量的治疗化合物可以降低肿瘤大小,或以其他方式改善受试者的症状,所述受试者通常是人,但可以是另一种哺乳动物。
药物组合物可以是控释或持续释放制剂,包括植入物,透皮贴剂和微囊化递送系统。可以使用可生物降解的、生物相容的聚合物,例如乙烯乙酸乙烯酯,聚酐,聚乙醇酸,胶原,聚原酸酯和聚乳酸。见例如Sustainedand ControlledReleaseDrugDelivery Systems,J.R.Robinson编辑,Marcel Dekker,Inc.,New York,1978。
可以通过医疗装置给药,例如(1)无针皮下注射装置(例如,US5,399,163;5,383,851;5,312,335;5,064,413;4,941,880;4,790,824;和4,596,556);(2)微输液泵(US 4,487,603);(3)透皮装置(US 4,486,194);(4)输液装置(US 4,447,233和4,447,224);和(5)渗透装置(US 4,439,196和4,475,196);其公开内容通过引用并入本文。
在某些实施方案中,可以配制药物组合物以确保体内适当的分布。例如,为了确保本发明的治疗化合物穿过血脑屏障,可以将它们配制成脂质体,其可以另外包含靶向部分以增强向特定细胞或器官的选择性转运。见例如US 4,522,811;5,374,548;5,416,016;和5,399,331;Ranade,J.Clin.Pharmacol.1989,29,685;Umezawa等人,Biochem.Biophys.Res.Commun.1988,153,1038;Bloeman等人,FEBSLett.1995,357,140;Briscoe等人,Am.J.Physiol.1995,1233,134;Schreier等人,J.Biol.Chem.1994,269,9090;Keinanen and Laukkanen,FEBSLett.1994,346,123;和Killion and Fidler,Immunomethods 1994,4,273。
用途
本发明化合物或其缀合物可用于治疗疾病,例如但不限于过度增殖性疾病,包括:头颈部癌症,包括头部,颈部,鼻腔,鼻旁窦,鼻咽,口腔,口咽,喉,下咽,唾液腺和副神经节瘤的肿瘤;肝脏和胆管癌,特别是肝细胞癌;肠癌,特别是结肠直肠癌;卵巢癌;小细胞和非小细胞肺癌(SCLC和NSCLC);乳腺癌肉瘤,如纤维肉瘤,恶性纤维组织细胞瘤,胚胎性横纹肌肉瘤,平滑肌肉瘤(leiomysosarcoma),神经纤维肉瘤,骨肉瘤,滑膜肉瘤,脂肪肉瘤和肺泡软部分肉瘤;白血病,如急性早幼粒细胞白血病(APL),急性髓性白血病(AML),急性淋巴细胞白血病(ALL)和慢性髓性白血病(CML);中枢神经系统的肿瘤,特别是脑癌;多发性骨髓瘤(MM),淋巴瘤如霍奇金淋巴瘤,淋巴浆细胞样淋巴瘤,滤泡性淋巴瘤,粘膜相关淋巴组织淋巴瘤,套细胞淋巴瘤,B细胞系大细胞淋巴瘤,伯基特淋巴瘤和T细胞间变性大细胞淋巴瘤。临床上,本文所述组合物的方法和用途的实施将导致癌性生长的大小或数量的减少和/或相关症状的减少(如果适用)。病理学上,本文所述组合物的方法和用途的实施将产生病理相关的反应,例如:抑制癌细胞增殖,减少癌症或肿瘤的大小,预防进一步的转移,以及抑制肿瘤血管生成。治疗此类疾病的方法包括向受试者施用治疗有效量的本发明组合。必要时可以重复该方法。
本发明的化合物或其缀合物可以与其他治疗剂组合给药,所述其他治疗剂包括抗体,烷基化剂,血管生成抑制剂,抗代谢物,DNA裂解剂,DNA交联剂,DNA嵌入剂,DNA小沟结合剂,烯二炔,热休克蛋白90抑制剂,组蛋白去乙酰化酶抑制剂,免疫调节剂,微管稳定剂,核苷(嘌呤或嘧啶)类似物,核输出抑制剂,蛋白酶体抑制剂,拓扑异构酶(I或II)抑制剂,酪氨酸激酶抑制剂和丝氨酸/苏氨酸激酶抑制剂。具体治疗剂包括阿达木单抗,安沙霉素P3,奥瑞他汀,苯达莫司汀,贝伐单抗,比卡鲁胺,博来霉素,硼替佐米,白消安,人源性激肽释放酶结合蛋白(callistatin)A,喜树碱,卡培他滨,卡铂,卡莫司汀,西妥昔单抗,顺铂,克拉屈滨,阿糖胞苷,cryptophycin,达卡巴嗪,达沙替尼,柔红霉素,多西紫杉醇,多柔比星,多米卡新,dynemycin A,埃博霉素,依托泊苷,氟尿嘧啶,氟达拉滨,5-氟尿嘧啶,吉非替尼,吉西他滨,伊匹单抗,羟基脲,伊马替尼,英夫利昔单抗,干扰素,白细胞介素,β-拉帕醌,来那度胺,伊立替康,美登素,氮芥,美法仑,6-巯基嘌呤,甲氨蝶呤,丝裂霉素C,尼罗替尼,纳武单抗,奥沙利铂,紫杉醇,丙卡巴肼,辛二酰苯胺异羟肟酸(SAHA),6-硫鸟嘌呤,塞替派,替尼泊苷,拓扑替康,曲妥珠单抗,曲古抑菌素A,长春碱,长春新碱和长春地辛。
实施例
通过参考以下实施例可以进一步理解本发明的实践,这些实施例是以说明而非限制的方式提供的。
实施例1-通过迈克尔加成的缀合
该一般方法基于通过赖氨酸ε-氨基与2-亚氨基四氢噻吩的反应将游离巯基引入抗体,然后与含马来酰亚胺的药物-连接基部分反应,如上所述。最初,将抗体缓冲液交换到含有50mMNaCl和2mM二亚乙基三胺五乙酸(DTPA)的0.1M磷酸盐缓冲液(pH8.0)中并浓缩至5-10mg/mL。通过向抗体中添加2-亚氨基四氢噻吩来实现硫醇化。待加入的2-亚氨基四氢噻吩的量可以通过初步实验确定,并且因抗体而异。在初步实验中,向抗体中滴定加入增加量的2-亚氨基四氢噻吩,并在室温下(“RT”,约25℃)与抗体培养1小时后,使用SEPHADEXTMG-25柱将抗体脱盐至50mM HEPES,5mM甘氨酸,2mM DTPA,pH 5.5,通过与二硫代二吡啶(DTDP)反应快速测定引入的巯基数。巯基与DTDP的反应导致硫代吡啶的释放,其可以在324nm下通过光谱法监测。通常使用蛋白质浓度为0.5-1.0mg/mL的样品。280nm处的吸光度可用于精确测定样品中蛋白质的浓度,然后将各样品的等分试样(0.9mL)与0.1mLDTDP(5mM乙醇储备溶液)在室温下培养10分钟。单独缓冲液加DTDP的空白样品也同时培养。10分钟后,测量324nm处的吸光度,并使用19,800M-1的硫代吡啶消光系数定量巯基的数量。
通常需要每个抗体约2-3个巯基的硫醇化水平。例如,对于一些抗体,这可以通过添加15倍摩尔过量的2-亚氨基四氢噻吩然后在室温下培养1小时来实现。然后将抗体与2-亚氨基四氢噻吩以所需的摩尔比培养,然后脱盐至缀合缓冲液(50mM HEPES,5mM甘氨酸,2mM DTPA,pH5.5))。将硫醇化物质保持在冰上,同时如上所述定量引入的巯基数。
在验证引入的巯基数之后,以每个巯基2.5倍摩尔过量添加seco-CPI-连接基部分。使缀合反应在含有终浓度25%丙二醇和5%海藻糖的缀合缓冲液中进行。通常,将药物-连接基储备溶液溶解在100%DMSO中。将储备溶液直接添加到硫醇化抗体中。
将缀合反应混合物在室温下培养2小时,同时温和搅拌。然后将10倍摩尔过量的N-乙基马来酰亚胺(在DMSO中的100mM储备液)加入到缀合混合物中并再搅拌1小时以封闭任何未反应的巯基。然后通过0.2μ过滤器过滤样品。通过TFF VivaFlow 50Sartorius 30MWCOPES膜将该物质缓冲液交换至10mg/mL甘氨酸,20mg/mL山梨糖醇,15%乙腈(“ACN”)pH 5.0(5X TFF缓冲液交换体积),以去除任何未反应的药物。最终制剂通过TFF进行至20mg/mL山梨糖醇,10mg/mL甘氨酸,pH 5.0。
实施例2-谷氨酰胺转胺酶介导的缀合
以下方法可用于谷氨酰胺转胺酶介导的seco-CPI-连接基化合物的缀合,其中连接基具有可用作胺供体的胺基。抗体可以是具有谷氨酰胺转胺酶反应性谷氨酰胺的抗体,例如具有N297A或N297Q取代的抗体。通过重组细菌性谷氨酰胺转胺酶以5:1的抗体:酶的摩尔比进行缀合。使用标准方案在50mM Tris缓冲液(pH8.0)中进行缀合,在37℃培养过夜。将得到的缀合物在用50mM Tris,pH8.0预平衡的蛋白A柱上纯化。用0.1M柠檬酸钠缓冲液(pH3.5)洗脱缀合物。洗脱的级分用1M Tris pH9.0中和。缀合物可以配制于20mg/mL山梨糖醇,10mg/mL甘氨酸,pH5.0。
本领域技术人员将理解,这两个实施例中的条件和方法是说明性的而非限制性的,并且其变体或其他缀合方法是本领域已知的并且可用于本发明。
实施例3-seco-CPI化合物的性质
表IV显示了本发明化合物的性质,包括对各种人癌细胞系的增殖抑制效力和CLogP。使用72小时ATP发光测定法测量增殖抑制(Cheng等人,US 8,394,922B2(2013))。
测量针对以下的增殖抑制:(a)H226(人间皮瘤(肺)癌细胞系);(b)N87(人胃(胃)癌细胞系);(c)OVCAR3(人卵巢癌细胞系);(d)HCT116(人结肠癌细胞系);(e)HCT116/VM46(HCT116的多药和紫杉醇抗性亚系)。


实施例4-化合物10
该实施例和图1A涉及化合物10的合成。
将3,5-二硝基苄醇1(5g,25.2mmol)的EtOAc(160mL)溶液加入到2-碘氧基苯甲酸(“SIBX”,可得自Sigma-Aldrich,17.67g,63.1mmol)中。将反应混合物在90℃下回流20小时,冷却至室温并过滤。弃去固体,真空浓缩滤液,得到粗产物2,为黄色固体,将其溶于100mL EtOAc中,用饱和NaHCO3溶液洗涤,然后用水洗涤。然后将其用无水Na2SO4干燥。将溶液真空浓缩,得到4.2g(81%)3,5-二硝基苯甲醛2,为黄色固体。1H NMR(500MHz,DMSO-d6)δ10.24(s,1H),9.08-9.06(m,1H),9.04(d,J=2.0Hz,2H);C7H4N2O5分析计算值:196.01;实测值194.99[M-H]+。
将syn-苯甲醛肟(6.18g,51.0mmol)在N,N-二甲基甲酰胺(“DMF,”50mL)中的搅拌溶液用K2CO3(14.38g,102mmol)处理并搅拌10分钟。加入3,5-二硝基苯甲醛2(5g,25.5mmol)的DMF(50mL)溶液。将反应混合物在90℃下搅拌2小时并冷却至室温。将(溴甲基)苯(7.12mL,58.6mmol)加入到反应混合物中,然后搅拌18小时。将反应混合物用Et2O(200mL)稀释。在搅拌下缓慢加入HCl水溶液(1N,100mL)。收集乙醚层,水层用Et2O(80mL×2)和EtOAc(80mL)萃取。将合并的有机层用饱和NaHCO3溶液洗涤,然后用盐水洗涤,经MgSO4干燥,过滤,并在真空下浓缩,得到16g棕色油状物,将其在330g来自Biotage的柱上纯化(45至55%二氯甲烷/己烷),得到3.41g(52.1%)3-(苄氧基)-5-硝基苯甲醛3,为白色固体。1HNMR(500MHz,氯仿-d)δ10.07(s,1H),8.37-8.29(m,1H),8.10(t,J=2.2Hz,1H),7.82(dd,J=2.4,1.2Hz,1H),7.53-7.36(m,5H),5.24(s,2H);C14H11N1O4分析计算值:257.0;实测值279.3[M+Na]+。
在-25℃下,经20分钟,将甲醇钠(25%)(1.313mL,5.91mmol)滴加到3-(苄氧基)-5-硝基苯甲醛3(400mg,1.555mmol)和2-叠氮基乙酸甲酯4(可得自Sigma-Aldrich,591mg,5.13mmol)在四氢呋喃(“THF”,3mL)和MeOH(3mL)的混合物中的搅拌的溶液中。将反应混合物在-20℃下搅拌64小时,然后在0℃下搅拌1小时。将反应混合物倒入冰水混合物中,用二氯甲烷(“DCM”,15mL×3)萃取,用水洗涤,然后用盐水洗涤。将有机层经MgSO4干燥,得到粗产物,将其在330g来自Biotage的柱上纯化(5%至9%EtOAc/Hex),得到323mg(58.6%)化合物5:1HNMR(500MHz,氯仿-d)δ8.22(s,1H),7.84(s,1H),7.80(t,J=2.1Hz,1H),7.53-7.33(m,5H),6.86(s,1H),5.20(s,2H),3.96(s,3H);13C NMR(126MHz,氯仿-d)δ163.4,159.1,149.2,135.6,135.3,128.8,128.5,128.2,127.6,122.8,122.0,118.0,109.8,70.9,53.3;C17H14N4O5分析计算值:354.09;实测值377.30[M+Na]+。
将化合物5(313mg,0.883mmol)加入到二甲苯(80mL)中并声处理5分钟。形成均匀溶液,将其脱气两次。将反应混合物在160℃下回流20小时并冷却至室温。真空蒸发二甲苯,得到黄色固体,将其在来自Biotage的24g柱上纯化(5%至15%EtOAc/己烷),得到47.3mg(16.41%)化合物6a和160.3mg(55.6%)化合物6b。
化合物6a:1HNMR(500MHz,氯仿-d)δ10.19(br.s.,1H),8.06(d,J=2.2Hz,1H),7.63(d,J=1.9Hz,1H),7.52-7.47(m,2H),7.44(t,J=7.4Hz,2H),7.41-7.35(m,1H),5.20(s,2H),4.01(s,3H);13C NMR(126MHz,氯仿-d)δ161.2,152.7,136.2,133.2,131.2,130.5,128.8,128.3,127.6,125.6,115.2,112.0,108.9,71.5,52.4;C17H14N2O5分析计算值:327.1;实测值327.3[M+H]+。
化合物6b:1HNMR(500MHz,氯仿-d)δ9.38(br.s.,1H),8.38(s,1H),7.75(d,J=1.3Hz,1H),7.57-7.51(m,2H),7.51-7.42(m,3H),7.38(d,J=2.0Hz,1H),5.32(s,2H),3.99(s,3H);13C NMR(126MHz,氯仿-d)δ161.4,145.2,143.3,135.3,130.9,129.7,128.9,128.8,128.2,126.7,113.2,111.0,100.5,71.1,52.4;C17H14N2O5分析计算值:327.1;实测值327.3[M+H]+。
向搅拌的化合物6b(150mg,0.460mmol)的THF(4mL)溶液中加入Boc-酐(0.192mL,0.827mmol)和4-二甲基氨基吡啶(“DMAP,”11.23mg,0.092mmol)。将反应混合物在室温下搅拌18小时。加入盐水(5mL)和EtOAc(5mL)并继续搅拌5分钟。收集有机层。用10mL EtOAc萃取水层。将合并的有机层经MgSO4干燥,过滤,并在真空下浓缩,得到黄色固体,将其在来自Biotage的24g柱上纯化(5%至40%EtOAc/己烷),得到184.2mg(94%)化合物7:1HNMR(500MHz,氯仿-d)δ8.29(d,J=1.7Hz,1H),7.70(d,J=1.6Hz,1H),7.50(d,J=7.3Hz,2H),7.45-7.39(m,2H),7.39-7.33(m,2H),5.35(s,2H),3.97(s,3H),1.50(s,9H);13C NMR(126MHz,氯仿-d)δ160.4,149.2,145.5,143.5,135.1,130.1,130.0,128.8,128.6,128.1,126.3,112.6,112.4,102.1,86.4,71.2,52.4,27.2;C22H22N2O7分析计算值:426.1;实测值449.4[M+Na]+。
将锌粉(130mg,1.982mmol)和NH4Cl(212mg,3.96mmol)加入到化合物7(169mg,0.396mmol)在丙酮(4mL)/水(0.800mL)中的搅拌溶液中。将反应混合物在室温下搅拌18小时。将反应混合物在真空下浓缩,得到180mg粗化合物8,将其再溶解于EtOAc(8mL)中,用水(8mL)再用盐水(8mL)洗涤,并经MgSO4干燥。将其在真空下浓缩,得到160mg粗化合物8(棕色),将其在来自Biotage的12g柱上纯化(15%至35%EtOAc/己烷),得到138.1mg(88%)化合物8:1HNMR(500MHz,氯仿-d)δ7.46(d,J=7.3Hz,2H),7.39(t,J=7.4Hz,2H),7.35-7.30(m,1H),7.03(s,1H),6.50(d,J=1.4Hz,1H),6.29(d,J=1.4Hz,1H),5.22(s,2H),3.91(s,3H),3.53(br.s.,2H),1.49(s,9H);C22H24N2O5分析计算值:397.17;实测值397.13[M+H]+。
将Boc-酐(0.112mL,0.484mmol)加入到化合物8(128mg,0.323mmol)在THF(3.5mL)中的搅拌溶液中。将反应混合物在室温下搅拌20小时,并在真空下浓缩,得到178mg棕色蜡状粗化合物9,将其在来自Biotage的12g柱上纯化(5%至15%EtOAc/己烷),得到156mg(97%)化合物9:1H NMR(500MHz,氯仿-d)δ7.48(d,J=7.3Hz,2H),7.39(t,J=7.4Hz,2H),7.36-7.30(m,2H),7.13(s,1H),6.86(s,1H),6.41(br.s.,1H),5.24(s,2H),3.92(s,3H),1.54(s,9H),1.47(s,9H)。C27H32N2O7分析计算值:496.2;实测值497.5[M+H]+。
将N-碘代琥珀酰亚胺(127mg,0.564mmol)和乙酸(0.065mL,1.128mmol)加入到化合物9(140mg,0.282mmol)在甲苯(8mL)中的搅拌溶液中。用铝箔覆盖反应烧瓶并在室温下搅拌18小时。将反应混合物倒入水(20mL)中。加入EtOAc(10mL)并将混合物搅拌2分钟。收集有机层。用EtOAc(10mL)萃取水层。将合并的有机层用MgSO4干燥,过滤,并在真空下浓缩,得到深紫色蜡状物,将其在来自Biotage的12g柱上纯化(5%至12%EtOAc/己烷),得到110.6mg(63.0%)化合物10,为黄色蜡状物:1HNMR(500MHz,氯仿-d)δ7.81(br.s.,1H),7.50(d,J=7.1Hz,2H),7.43-7.36(m,2H),7.35-7.30(m,1H),7.12(s,1H),6.79(br.s.,1H),5.27(s,2H),3.94(s,3H),1.56(s,9H),1.43(s,9H);C27H31IN2O7分析计算值:622.1;实测值623.4[M+H]+。
实施例5-酯12
该实施例和图3B涉及酯12的制备。
在0℃下,将NaH(7.71mg,0.193mmol)和1,3-二氯丙烯(23.50μl,0.241mmol)加入到化合物10(100mg,0.161mmol)在DMF中的搅拌溶液中。将反应混合物搅拌3小时。将反应混合物倒入饱和NH4Cl(100mL)溶液中并用Et2O(2×80mL)萃取。萃取物经MgSO4干燥,过滤,并在真空下浓缩,得到棕色蜡状物,将其在来自Biotage的12g柱上纯化(5%至12%EtOAc/己烷),得到88.7mg(79%)化合物11:1HNMR(500MHz,氯仿-d)δ7.47-7.31(m,5H),7.21(s,1H),6.53(d,J=18.3Hz,1H),6.03-5.83(m,3H),5.34-5.21(m,2H),3.96(s,2H),4.57-3.70(m,2H),1.58(s,9H),1.30(d,J=9.8Hz,9H)。C30H34ClIN2O7分析计算值:696.1;实测值719.5[M+Na]+。
将氢化三正丁基锡(1.793mL,6.59mmol)和2,2'-偶氮双(2-甲基丙腈)(“AIBN,”0.085g,0.507mmol)加入到化合物11(3.5305g,5.07mmol)在苯(100mL)中的搅拌溶液中。将反应混合物脱气一次并在80℃下搅拌4小时。将反应混合物在真空下冷却至室温,得到棕色蜡状物,将其在120g来自Biotage的柱上纯化(5%至10%EtOAc/己烷),得到2.7552g化合物12和12a的混合物。通过SFC手性分离分离混合物:Chiralpak:AD-H制备柱,30×250mm,5μm;流动相:35%MeOH/CO2,130巴;温度:35℃;流速:70.0mL/min,持续13min;在220nm处UV监测。该方法得到0.9562g(33.1%)化合物12。1HNMR(500MHz,氯仿-d)δ7.52-7.30(m,6H),7.11(s,1H),5.27(s,2H),4.20-4.13(m,1H),4.01-3.82(m,6H),3.53(t,J=9.9Hz,1H),1.57(s,9H),1.43(br.s.,9H);分析计算值:C30H35ClN2O7570.2;实测值571.5[M+H]+;[α]20D-17.06(c 2.45,CHCl3)和0.9252g(32.0%)化合物12b。1HNMR(500MHz,氯仿-d)δ7.53-7.30(m,6H),7.11(s,1H),5.27(s,2H),4.22-4.13(m,1H),4.12-3.83(m,6H),3.53(t,J=10.1Hz,1H),1.57(s,9H),1.43(br.s.,9H)。C30H35ClN2O7分析计算值:570.2;实测值571.5[M+H]+;[α]20D+17.06(c 2.45,CHCl3)。
实施例6-化合物16
该实施例和图2涉及化合物16的制备。
制备酯12(1g,1.751mmol)在ACN(Aldrich,40mL)中的溶液。加入LiOH(0.125g,5.22mmol)/蒸馏水(10mL)。将反应混合物在50℃下搅拌2小时。LCMS分析显示反应完成。将反应混合物用6NHCl酸化至pH4,并在真空下浓缩以除去ACN。用EtOAC(2×50mL)萃取反应混合物。将有机相用水和盐水洗涤,用无水Na2SO4干燥,浓缩并在高真空下干燥过夜,得到粗酸13(0.98g)。ESI-LCMS(M+H):557.0。
将N,N-二异丙基乙胺(“DIPEA,”Aldrich,0.612mL,3.50mmol)加入到酸13(0.98g)、吗啉13a(Aldrich,0.458g,5.25mmol)和N,N,N',N'-四甲基-O-(7-氮杂苯并三唑-1-基)脲鎓六氟磷酸盐(“HATU”,CAS Reg.No.148893-10-1,Oakwood Chemical,0.999g,2.63mmol)的无水DMF(Acros,5mL)溶液中。将反应混合物在室温下搅拌45分钟。LCMS分析显示反应完成。将反应混合物用EtOAc,水和盐水后处理。合并有机相,用2g硅胶浓缩成浆液。将浆液在24g的COMBIFLASHTM柱上纯化,用0-35%EtOAc/己烷梯度洗脱。将预期含有产物的级分合并,浓缩并在高真空下干燥,得到化合物14,为固体(0.872g,两步产率80%)。ESI-LCMS(M+H):626.1。
制备化合物14(396mg,0.633mmol)的无水THF(Acros,5mL)溶液。加入甲酸铵(Aldrich,1.104g,8.76mmol)/蒸馏水(1mL)。将氮气鼓泡通过反应混合物5分钟,然后加入Pd/碳(Aldrich-Sigma,10%,60mg)。将反应混合物在室温下搅拌0.5小时。LCMS分析显示反应完成。将反应混合物用EtOAC(50mL)稀释并过滤。将滤液用水和盐水洗涤,经无水Na2SO4干燥,并真空浓缩,得到残余物,将其用ACN和水冷冻干燥,得到化合物15(白色固体,225mg,66%收率)。ESI-LCMS(M+H):536.2。
将三氟乙酸(“TFA”,Aldrich,1mL)加入到化合物15(78mg,0.146mmol)的无水DCM(Acros,1mL)溶液中。将反应混合物在室温下搅拌30分钟。LCMS分析显示反应完成。将反应混合物真空浓缩,并用ACN和水冷冻干燥,得到化合物16,为绿色固体(81mg,双TFA盐,98%)。ESI-LCMS(M+H):336.0。
实施例7-化合物18
该实施例涉及化合物18的制备,如图2所示。
制备乙酯17a(AlfaAesar,300mg,1.46mmol)和三苯基膦(“TPP,”Aldrich-Sigma,767mg,2.92mmol)在无水THF(Acros Organics,10mL)中的溶液。在10分钟内滴加偶氮二甲酸二叔丁酯(“DBAD,”Aldrich,673mg,2.92mmol)/无水THF(Acros,无水,2mL),然后加入2-甲氧基乙醇17b(Aldrich,445mg,5.85mmol)。将反应混合物在室温下搅拌过夜。LCMS分析显示反应几乎完成。将反应混合物用EtOAc,水和盐水后处理。合并有机相,并在真空下用硅胶(1g)浓缩,得到浆液。将浆液在24g的COMBIFLASHTM柱上纯化,用0-30%EtOAc/己烷梯度洗脱。将预期含有产物的级分合并,浓缩并在高真空下干燥,得到化合物17,为固体(314mg,1.19mmol,82%)。ESI-LCMS(M+H):264.1。
将LiOH(Aldrich,50mg)/蒸馏水(1mL)加入到化合物17(314mg,1.19mmol)的ACN(Acros,10mL)溶液中。将反应混合物在40℃下搅拌3小时。LCMS分析显示反应完成。将反应混合物用乙酸(0.5mL)中和,并在真空下浓缩至2mL。将得到的混合物加载到COMBIFLASHTMAQGold50g柱上,用0-60%ACN/水梯度(均含有0.05%甲酸,50mL/min)洗脱。将预期含有产物的级分合并并冷冻干燥,得到化合物18,为白色粉末(226g,0.961mmol,81%)。ESI-LCMS(M+H):236.0。
实施例8-Seco-CPI化合物Ia-01
该实施例和图2涉及seco-CPI化合物Ia-01的制备。
制备化合物16(5mg,0.015mmol)和18(7.0mg,0.030mmol)在无水DMF(Acros,1mL)中的溶液。加入N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺盐酸盐(“EDC,”Aldrich,5.71mg,0.030mmol)。将反应混合物在室温下搅拌2小时。LCMS分析显示反应完成。通过制备型HPLC纯化反应混合物,在KINETEXTM5μmEVO C18 150x 21.2mm柱上用45-60%ACN/水(具有0.1%TFA,20mL/min)洗脱。将预期含有产物的级分合并并冷冻干燥,得到Ia-01,为灰白色固体(4.1mg,7.4μmol,50%产率)。ESI-LCMS(M+H):553.1。1HNMR(DMSO-d6,400MHz)11.54(1H,s),11.29(1H,s),9.75(1H,s),7.75(1H,s),7.39(1H,d),7.15(1H,d),7.02(1H,d),6.91(2H,d),4.74(1H,t),4.45(1H,d),4.14(4H,m),3.89(2H,m),3.40-3.80(12H,m)。
大致遵循该实施例和图2的方法,但使用替代的前体材料,制备另外的seco-CPI化合物,如下表V中所列。

实施例9-化合物26
该实施例和图3A涉及化合物26的合成。
制备化合物19a(Bachem,7.5g,24.09mmol)和(4-氨基苯基)-甲醇19b(Aldrich,3.86g,31.3mmol)在无水DCM(Acros,50mL)和无水甲醇(Acros,50mL)中的溶液。加入2-乙氧基-1-乙氧基羰基-1,2-二氢喹啉(“EEDQ,”Bachem,8.94g,36.1mmol)。将反应混合物在室温下搅拌过夜。LCMS分析显示反应几乎完成。将反应混合物用EtOAc(400mL)稀释,并将所得溶液用水和盐水洗涤。有机相经无水Na2SO4干燥,并在真空下浓缩至100mL体积。过滤收集沉淀的白色固体,并空气干燥一个周末,得到化合物20(白色固体,8.9g,21.37mmol,89%)。ESI-LCMS(M+H):417.1。
将哌啶(Aldrich,1mL)加入到化合物20(2.2g,5.28mmol)的DMF(Acros,无水,10mL)溶液中。将反应混合物在室温下搅拌1小时。LCMS分析显示反应完成。将反应混合物用ACN和水冷冻干燥,得到粗化合物21,具有一些Fmoc残余,为白色固体(2.12g)。ESI-LCMS(M+H):195.0。
制备化合物21(2.12g,粗品)和化合物22a(Aldrich,1.610g,5.28mmol)在DMF(Acros,无水,10mL)中的溶液。加入DIPEA(Aldrich)以将反应混合物的pH调节至大于8。将反应混合物在室温下搅拌2小时。LCMS分析显示反应完成。将反应混合物用EtOAc,水和盐水后处理。合并有机相并浓缩,得到残余物。将残余物用ACN(Aldrich,5mL)和蒸馏水(10mL)再溶解。过滤悬浮液。将滤液加载到130g COMBIFLASHTMAQ Gold柱上,以75mL/min用25-40%ACN(含0.05%甲酸)和水(也含有0.05%甲酸)洗脱。将预期含有产物的级分合并并冷冻干燥,得到化合物22,为白色固体(0.89g,2.262mmol,42.8%)。ESI-LCMS(M+H):394.1。
制备化合物22(147mg,0.374mmol)和全溴甲烷(Aldrich,217mg,0.653mmol)在无水DCM(Acros,4mL)中的溶液。加入TPP(Aldrich,171mg,0.653mmol)。将反应混合物在室温下搅拌5分钟,之后形成澄清溶液。LCMS分析显示反应完成。将反应混合物加载到12gCOMBIFLASHTMGold柱上,用50-100%EtOAc/己烷(25mL/min)洗脱。将预期含有产物的级分合并,浓缩并在高真空下干燥15分钟,得到化合物23,为固体(92mg,54%)。ESI-LCMS(M+H):456.1。
将甲基锂/乙醚(Aldrich,1.6M,0.233mL,0.373mmol)加入到化合物15(图2,100mg,0.187mmol)的无水THF(Acros,1mL)溶液中。将反应混合物在室温下搅拌5分钟。加入化合物23(92mg,0.202mmol)。将反应混合物在室温下搅拌10分钟。LCMS显示反应完成。将反应混合物用EtOAc,水和盐水后处理。将有机相用无水Na2SO4干燥,浓缩并在高真空下干燥,得到化合物24(153mg,粗品)。ESI-LCMS(M+H):911.2。
将TFA(Aldrich,2mL)加入到粗化合物24(153mg)的无水DCM(Acros,0.5mL)溶液中。将反应混合物在室温下搅拌30分钟。LCMS分析显示反应完成。将反应混合物浓缩并在COMBIFLASHTMAQ Gold 30g柱上纯化,用15-30%ACN(含0.05%甲酸)/去离子水(含0.05%甲酸)以35mL/min洗脱。将预期含有产物的级分合并并冷冻干燥,得到化合物25,为白色固体(61mg,53.5%)。ESI-LCMS(M+H):611.2。
制备化合物25(61mg,0.100mmol)和BOC-酸酐(Oakwood,0.030mL,0.130mmol)在无水DMF(Acros,1mL)中的溶液。加入DIPEA(Aldrich,0.060mL,0.348mmol)。将反应混合物在室温下搅拌30分钟。LCMS分析显示反应完成。在KINETEXTMC18EVO 5μ150x22.1mm柱上,通过制备型HPLC用30-50%ACN/水(含有0.1%TFA,20mL/min)纯化反应混合物。将预期含有产物的级分合并并冷冻干燥,得到化合物26,为白色固体(60mg,84.4%)。ESI-LCMS(M+H):711.2。
实施例10-Seco-CPI-连接基化合物IIIa-03
该实施例和图3B涉及化合物IIIa-03的制备。
将DIPEA(Aldrich,0.074mL,0.422mmol)加入到化合物18(图2,29.8mg,0.127mmol)和HATU(Oakwood,48.1mg,0.127mmol)的无水DMF(Acros,0.7mL)溶液中。将反应混合物在室温下搅拌5分钟。加入化合物26(图3B,30mg,0.042mmol)。将反应混合物在室温下搅拌过夜。LCMS分析显示反应完成70%。通过制备型HPLC纯化反应,在KINETEXTMC18EVO 5μ150x22.1mm柱上用50-65%ACN/水(含有0.1%TFA,20mL/min)洗脱。将预期含有产物的级分合并并冷冻干燥,得到化合物27,为粉末(27mg,68.8%)。ESI-LCMS(M+H):928.3。
将TFA(Sigma,1mL)加入到化合物27(27mg,0.029mmol)的无水DCM(Acros,1mL)溶液中。将反应混合物在室温下搅拌15分钟。LCMS分析显示反应完成。通过制备型HPLC纯化反应,在KINETEXTMC18EVO 5μ150x22.1mm柱上用40-50%ACN/水(含有0.1%TFA,20mL/min)洗脱。将预期含有产物的级分合并并冷冻干燥,得到化合物28,为粉末(20mg,83.0%)。ESI-LCMS(M+H):828.2。
将DIPEA(Sigma,20μL,0.114mmol)加入到化合物28(20mg,0.024mmol)和6-马来酰亚胺基己酸N-羟基琥珀酰亚胺酯29(得自SIGMA-Aldrich,11.17mg,0.036mmol)的无水DMF(Acros,0.7mL)溶液中。将反应混合物在室温下搅拌30分钟。LCMS分析显示反应完成。通过制备型HPLC纯化反应混合物,在KINETEXTMC18EVO 5μ150x22.1mm柱上用50-65%乙腈/水(含有0.1%TFA,20mL/min)洗脱。将预期含有产物的级分合并并冷冻干燥,得到seco-CPI-连接基化合物IIIa-03,为粉末(12.8mg,51.9%)。ESI-LCMS(M+H):1021.5.1HNMR(DMSO-d6):δ11.98(s,1H),11.77(s,1H),9.79(s,1H),8.11(d,1H,J=6.9Hz),7.79(d,1H,J=8.6Hz),7.42(d,1H,J=8.8Hz),7.29(d,3H,J=8.3Hz),7.19(s,1H),6.98-7.03(m,4H),6.61(d,1H,J=7.19Hz),6.53(d,2H,J=8.5Hz),5.05(t,2H,J=4.7Hz),4.45(d,1H,J=10.7Hz),4.34(d,1H,J=7.1Hz),4.11-4.17(m,3H),3.52-3.72(m,10H),3.09-3.38(m,9H),2.15(m,2H),1.95(m,1H),1.48(m,4H),1.27(m,3H),1.17(m,2H),0.84(m,6H)。
大致遵循该实施例的方法和图3A-3B的方案,但使用替代的前体材料制备另外的seco-CPI-连接基化合物,如下表VI中所列出。

化合物66的制备

实施例11-Seco-CPI化合物Ia-21
该实施例和图4涉及seco-CPI化合物Ia-21的制备。
将HCl/二噁烷(4N,15mL)加入到酯12(0.25g,0.4388mmol)中。将反应混合物在50℃下搅拌5小时。LCMS分析显示反应完成。将反应混合物在氮气下吹干,得到深蓝色残余物,为粗酸30,不经进一步纯化用于下一步骤。ESI-LCMS(M+H):371.1。
将N,N-二甲基乙酰胺(“DMA”,5mL)加入到粗酸30(盐酸盐,178mg,0.437mmol),5-甲氧基-1H-吲哚-2-甲酸30a(CAS Reg.No.4382-54-1,得自Sigma-Aldrich,125mg,0.656mmol),EDC(251mg,1.311mmol),1-羟基苯并三唑(“HOBT,”201mg,1.311mmol),和NaHCO3(184mg,2.185mmol)。将黑色溶液在室温下搅拌4.5小时。LCMS分析显示产物峰的出现。将反应混合物用水(15mL)稀释,在室温下搅拌15分钟并过滤。收集粗产物,为棕色固体,并重新溶解在DCM/甲醇中。将所得溶液用硅胶(1.3g)浓缩至干,形成浆液。将浆液在15g硅胶柱上纯化,用0-3%乙酸乙酯/DCM洗脱液梯度洗脱。将含有预期产物的级分合并,浓缩并在真空下干燥,得到粗产物(206mg)。将粗产物再溶于20%甲醇/氯仿中,并用1.3g硅胶浓缩成浆液。将浆液在硅胶柱上纯化,用100%DCM和1-5%乙酸乙酯/己烷洗脱液。将含有预期产物的级分合并,浓缩并在高真空下干燥,得到略微不纯的化合物31(0.108g,0.199mmol,45.4%)。ESI-LCMS(M+H):544.4。
将THF(3mL),水(1.000mL)和MeOH(2.000mL)加入到化合物31(104mg,0.191mmol)中,并加入氢氧化锂水合物(138mg,3.29mmol)。将黄色非均相混合物搅拌过夜。LC/MS显示反应完成。将反应混合物用1NHCl酸化,然后过滤。收集固体并干燥,得到粗化合物32(0.168g,88%)。ESI-LCMS(M+H):529.9。
将DMA(0.5mL)加入到化合物32(15mg,0.028mmol),N,N'-二甲基-乙烷-1,2-二胺32a(CAS Reg.No.110-70-3,得自Sigma-Aldrich,1.247mg,0.014mmol),EDC(10.85mg,0.057mmol),HOBT(8.67mg,0.057mmol)和NaHCO3(9.51mg,0.113mmol)。LC/MS显示反应完成50%。将反应混合物用MeOH稀释,并通过制备型LC纯化,用15-100%ACN/水(均含有0.1%TFA,40mL/min)梯度洗脱。将含有预期产物的级分合并并冷冻干燥,得到化合物33(5.8mg,7.98μmol,28.2%)。ESI-LCMS(M+H):600.10。
将MeOH(0.5mL)加入到化合物33(TFA盐,5.7mg,7.98μmol),甲酸铵(15.10mg,0.239mmol)和Pd-C(1.699mg,1.596μmol)中。将反应混合物加热至回流30秒。10分钟后,LC/MS显示反应完成。过滤反应混合物,滤液通过制备型HPLC在Waters-YMC-OBD S520x100mm柱上用0-75%ACN/水(均含有0.1%TFA,25ml/min)纯化。将含有预期产物的级分合并并冷冻干燥,得到seco-CPI化合物Ia-21,为琥珀色固体(1.5mg,2.12μmol,26.5%)。ESI-LCMS(M+H):510.15.1HNMR(MeOD,500MHz)7.92(1H,s),7.41(1H,d,J=5Hz),7.18(1H,d,J=1Hz),7.14(1H,s),7.07(1H,s),6.96(1H,dd,J=10Hz),4.76(1H,t),4.61(1H,dd),4.09(2H,m),3.86(3H,s),3.80(3H,m),3.40(2H,t,J=5Hz),3.03(6H,s)。
大致遵循该实施例和图4的方法,但使用替代的前体材料,制备另外的seco-CPI化合物,如下表VII中所列。


实施例12-Seco-CPI-连接基化合物IIIa-05
该实施例和图5涉及seco-CPI-连接基化合物IIIa-05的制备
将DIPEA(Sigma,0.052mL,0.299mmol)加入到化合物25(61mg,0.100mmol)和羟基琥珀酰亚胺酯25a(QuiantaBiodesign,76mg,0.130mmol)在DMF(Acros,无水,1mL)中的溶液中。将反应混合物在室温下搅拌60分钟。LCMS分析显示反应完成。将反应混合物用乙酸(40uL)中和,加载到30g COMBIFLASHTMAQ Gold柱上,并用50-75%ACN/水(均含有0.05%甲酸,35mL/min)纯化。合并含有预期产物的级分并冷冻干燥,得到化合物34(52mg,0.048mmol,48.2%产率)。ESI-LCMS(M+H):1080.3。
将DIPEA(Sigma,0.042mL,0.241mmol)加入到酸18(34.0mg,0.144mmol),化合物34(52mg,0.048mmol)和HATU(Oakwood,54.9mg,0.144mmol)在DMF(Acros,无水,0.7mL)中的溶液中。将反应混合物在室温下搅拌过夜。LCMS分析显示反应完成70%。在KINETEXTM5μC18EVO150x21.2mm柱上,通过制备型HPLC用45-65%ACN/水(含0.1%TFA,20mL/min)纯化反应混合物。合并含有产物的级分并冷冻干燥,得到化合物35(31mg,49.8%)。ESI-LCMS(M+H):1297.3。
将哌啶(Aldrich,50μL)加入到化合物35(31mg)的DMF(Acros,无水,0.5mL)溶液中。将反应混合物在室温下搅拌30分钟。LCMS分析显示反应完成。将反应混合物用AcOH(50μL)中和,并通过制备型HPLC在KINETEXTMC185μ150x21.2mm柱上用30-40%ACN/水梯度(含0.1%TFA,20mL/min)纯化。将含有预期产物的级分合并并冷冻干燥,得到seco-CPI-连接基化合物IIIa-05(22mg,64.2%),为TFA盐。ESI-LCMS(M+H):1075.6。1HNMR(DMSO-d6):δ11.97(s,1H),11.77(s,1H),9.81(s,1H),8.15(d,1H,J=3.1Hz),7.88(d,1H,J=8.8Hz),7.79(s,3H),7.42(d,1H,J=9.1Hz),7.30(d,3H,J=8.6Hz),7.19(s,1H),6.96-7.02(m,2H),6.63(d,1H,J=1.9Hz),6.53(d,2H,J=8.6Hz),5.06(t,2H,J=4.9Hz),4.45(d,1H,J=10.7Hz),4.34(d,1H,J=7.1Hz),4.21(m,1H),4.12(s,2H),2.96-3.72(m,32H),2.36-2.48(m,4H),1.97(m,2H),1.28(d,3H,J=7.1Hz),0.85(m,6H)。
大致遵循该实施例的方法和图5的方案,但使用替代的前体材料制备另外的seco-CPI-连接基化合物,如下表VIII中所列出。


实施例13-Seco-CPI化合物Ia-15
图6显示了合成seco-CPI化合物Ia-15的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。表IX显示化合物Ia-15和类似制备的其他化合物的分析数据。



实施例14-Seco-CPI化合物Ia-18
图7显示了合成seco-CPI化合物Ia-18的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。表X显示化合物Ia-18和类似制备的另一化合物的分析数据。

实施例15-Seco-CPI化合物Ib-01和Ib-02
图8显示了合成seco-CPI化合物Ib-01和Ib-02的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。Ib-01[M+H]+=615.0;Ib-02[M+H]+=659.4。
实施例16-Seco-CPI化合物Id-02
图9显示了合成seco-CPI化合物Id-02的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。表XI显示了化合物Id-02和类似制备的其他化合物的分析数据。

实施例17-Seco-CPI化合物Ia-25
图10显示了合成seco-CPI化合物Ia-25的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。
使用1-甲基哌嗪代替1-Cbz-哌嗪类似地制备化合物Ia-26。
两种化合物的质谱数据为:Ia-25[M+H]+=493.2;Ia-26[M+H]+=507.3。
实施例18-Seco-CPI化合物Ia-27
图11显示了合成seco-CPI化合物Ia-27的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。化合物Ia-27的质谱数据:[M+H]+=588.1。
实施例19-Seco-CPI化合物Ia-29
图12显示了合成seco-CPI化合物Ia-29的方案。所用试剂及其使用条件是本领域已知的和/或已在前述实施例中举例说明。化合物Ia-29的质谱数据:[M+H]+=574.1。
实施例20-缀合物的生物学活性
大致遵循上面的实施例1的迈克尔加成方法,将Seco-CPI-连接基化合物IIIa-01,IIIa-02,IIIa-03和IIIa-04与抗间皮素抗体6A4(Terrett等人,US 8,268,970B2(2012))缀合。得到的抗体-药物缀合物分别命名为6A4/IIIa-01ADC,6A4/IIIa-02ADC,6A4/IIIa-03ADC和6A4/IIIa-04ADC。
另外,大致遵循实施例2的谷氨酰胺转胺酶介导的缀合方法,将seco-CPI-连接基化合物IIIa-06和IIIa-12与抗间皮素抗体6A4缀合,所述抗间皮素抗体6A4经改造以在重链中具有N297A(EU,如Kabat)取代。N297A取代消除了抗体的糖基化,并使附近的谷氨酰胺295(Q295)可用作转谷氨酰胺催化的转酰胺化反应的胺受体(Jeger等人,Angew.Chem.Int.Ed.2010,49,9995)。得到的抗体-药物缀合物分别命名为6A4(N297A)/IIIa-06ADC和6A4(N297A)/IIIa-12ADC。
使用3H胸苷结合测定法(Cheng等人,US 8,394,922B2(2013))测量这些ADC对抗H226和N87癌细胞的活性,两者均表达间皮素。结果列于表XII中。

前面对本发明的详细描述包括主要或专门涉及本发明的特定部分或方面的段落。应当理解,这是为了清楚和方便,特定特征可能不仅仅与其被公开的段落相关,并且本文的公开内容包括在不同段落中发现的信息的所有适当的组合。类似地,尽管这里的各附图和描述涉及本发明的具体实施方案,但是应该理解,在特定附图或实施方案的上下文中公开具体特征的情况下,在适当的程度上在另一附图或实施方案的上下文中这样的特征也可以另一个特征组合或者总体上在本发明中使用。
此外,虽然已经就某些优选实施方案具体描述了本发明,但是本发明不限于这些优选实施方案。相反,本发明的范围由所附权利要求限定。
参考
以下提供了本说明书前面由第一作者(或发明人)以及日期以缩略方式引用的以下参考文献的完整引用。出于所有目的,将这些参考文献中的每一个通过引用并入本文。
Boger,US 6,281,354B1(2001).
Boger,US 6,548,530B1(2003).
Boger和Johnson,Proc.Nat.Acad.Sci.(USA)1995,92,3642.
Boger等人,J.Org.Chem.1990,55,5823.
Boger等人,J.Am.Chem.Soc.1997,119,4987.
Boger等人,Synthesis 1999,SI,1505.
Boger等人,J.Org.Chem.2000,65,4101.
Boyd等人,US 2008/0279868A1(2008).
Chari等人,CancerRes.1995,55,4079.
Chen等人,US 8,664,407B2(2014).
Ducry等人,Bioconjug.Chem.2010,21,5.
Gangwar等人,US 7,968,586B2(2011).
Hurley等人,Science 1984,226,843.
Kobayashi等人,CancerRes.1994,54,2404.
Lajiness等人,J.Med.Chem.2010,53,7731.
Li等人,CancerRes.1992,52,4904.
Nagamura and Saito,Chem.Heterocyclic Compounds 1998,34(12),1386.
Nagamura等人,Chem.Pharm.Bull.1996,44(9),1723.
Ng等人,US 7,129,261B2(2006).
Ng等人,US 7,507,420B2(2009).
Ng等人,US 8,034,959B2(2011).
Schrama等人,NatureRev.DrugDisc.2006,5,147-159.
Sufi等人,US 8461,117B2(2013).
Tichenor等人,J.Am.Chem.Soc.2007,129,10858.
Tietze等人,ChemBioChem 2001,2,758.
Tietze等人,Bioorg.Med.Chem.2008,16,6312.
Zhang等人,US 8,852,599B2(2014).
Zhao等人,US 7,655,660B2(2010)。
Claims (19)
1.具有由式(I)表示的结构的化合物

其中
Hal是Cl或Br;
R1是 ;
;
R2是H, C1-C3烷基, CO2H, CO2(C1-C3烷基), C(=O)NH2, C(=O)NH(C1-C3烷基)或C(=O)N(C1-C3烷基)2;
R3是


R4, R4’, R5, R6或R7独立地是H, OMe, OH, 6元芳基, 5或6元杂芳基, NH2, NHMe,NMe2, NH(C2-C4烷基), N(C2-C4烷基)2, NHC(=O)X1, O(C2-C4烷基), O(CH2)0-2(C3-C6环烷基), O(CH2)0-2X1或

其中C2-C4烷基可以未取代或被OCH2CH2OH, OCH2CH2NH2, NHCH2CH2OH, NHCH2CH2NH2, OH或NH2取代,芳基或杂芳基可以被C1-C2烷基, OH, NH2, NH(C1-C2烷基), N(C1-C2烷基)2, F,Cl, Br, NO2或CN取代;
条件是R4, R4’, R5, R6和R7至少一者不是H;
R8和R8’独立地是H, OH, O(C1-C3烷基), Cl, Br, F, O(CH2)2-4NH2或O(CH2)2-4OH;
R9是H, C(=O)(C1-C3烷基), C(=O)NH2, C(=O)NH(C1-C3烷基), C(=O)( C1-C3烷基)2,(CH2)2-4OH, (CH2)2-4O(C1-C3烷基), (CH2)2-4NH2, (CH2)2-4NH(C1-C3烷基)或(CH2)2-4N(C1-C3烷基)2;
每个X1独立地是6元芳基或5至6元杂芳基,其被C1-C3烷基, OH, O(C1-C3烷基), NH2,NH(C1-C3烷基), N(C1-C3烷基)2, F, Cl, Br, NO2或CN取代;
每个X2独立地是H, Me或C2-C4烷基,其被OCH2CH2OH, OCH2CH2NH2, NHCH2CH2OH,NHCH2CH2NH2, OH或NH2取代;且
每个X3独立地是O, NH, N(C1-C3烷基)或S;
或其药学上可接受的盐。
2.根据权利要求1的化合物,其中R2是H且Hal是Cl。
3.根据权利要求1的化合物,其中R3是

4.根据权利要求1的化合物,具有由式(Ia)表示的结构:

5.具有由式(Ia)表示的结构的化合物:

其中R4,R6和R7各自为H,R1为

并且R5是

6.根据权利要求1的化合物,其中R1是相应的化合物R1H具有小于0.300的CLogP值的基团。
7.根据权利要求4的化合物,其中R1是

8.根据权利要求1的化合物,具有由式(Ib)表示的结构:

9.根据权利要求1的化合物,具有由式(Ic)表示的结构:

10.根据权利要求1的化合物,具有由式(Id)表示的结构:

11.seco-CPI-连接基化合物,具有根据式(IIIa)的结构:

其中,
T是自牺牲基团;
t是0或1;
AAa和每个AAb独立地选自丙氨酸,β-丙氨酸,γ-氨基丁酸,精氨酸,天冬酰胺,天冬氨酸,γ-羧基谷氨酸,瓜氨酸,半胱氨酸,谷氨酸,谷氨酰胺,甘氨酸,组氨酸,异亮氨酸,亮氨酸,赖氨酸,蛋氨酸,正亮氨酸,正缬氨酸,鸟氨酸,苯丙氨酸,脯氨酸,丝氨酸,苏氨酸,色氨酸,酪氨酸和缬氨酸;
u是0或1;
p是1、2、3或4;
q是1、2、3、4、5、6、7、8、9、10、11或12;
r是1、2、3、4或5;
s是0或1;
v是0或1;
R31是H, ;
;
条件是,只有当s是1且v为0时,R31才能为H,并且只有当s是1且R31为H时,v才能为0;
Hal是Cl或Br;
R1是 ;
;
R2是H, C1-C3烷基, CO2H, CO2(C1-C3烷基), C(=O)NH2, C(=O)NH(C1-C3烷基)或C(=O)N(C1-C3烷基)2;
R3是


R4, R4’, R5, R6或R7独立地是H, OMe, OH, 6元芳基, 5或6元杂芳基, NH2, NHMe,NMe2, NH(C2-C4烷基), N(C2-C4烷基)2, NHC(=O)X1, O(C2-C4烷基), O(CH2)0-2(C3-C6环烷基), O(CH2)0-2X1或

其中C2-C4烷基可以未取代或被OCH2CH2OH, OCH2CH2NH2, NHCH2CH2OH, NHCH2CH2NH2, OH或NH2取代,芳基或杂芳基可以被C1-C2烷基, OH, NH2, NH(C1-C2烷基), N(C1-C2烷基)2, F,Cl, Br, NO2或CN取代;
条件是R4, R4’, R5, R6和R7至少一者不是H;
R8和R8’独立地是H, OH, O(C1-C3烷基), Cl, Br, F, O(CH2)2-4NH2或O(CH2)2-4OH;
R9是H, C(=O)(C1-C3烷基), C(=O)NH2, C(=O)NH(C1-C3烷基), C(=O)( C1-C3烷基)2,(CH2)2-4OH, (CH2)2-4O(C1-C3烷基), (CH2)2-4NH2, (CH2)2-4NH(C1-C3烷基)或(CH2)2-4N(C1-C3烷基)2;
每个X1独立地是6元芳基或5至6元杂芳基,其被C1-C3烷基, OH, O(C1-C3烷基), NH2,NH(C1-C3烷基), N(C1-C3烷基)2, F, Cl, Br, NO2或CN取代;
每个X2独立地是H, Me或C2-C4烷基,其被OCH2CH2OH, OCH2CH2NH2, NHCH2CH2OH,NHCH2CH2NH2, OH或NH2取代;且
每个X3独立地是O, NH, N(C1-C3烷基)或S;
或其药学上可接受的盐。
12.根据权利要求11的seco-CPI-连接基化合物,其中q是2、3、4或8。
13.具有根据式(IIIa')的结构的seco-CPI-连接基化合物:

其中
R4,R6和R7各自为H;并且
R5是


R10是

R11,R12和R13独立地为H, CH3, CH(CH3)2, CH2CO2H, CH2CH2CO2H, CH2C(=O)NH2, CH2CH2C(=O)NH2, (CH2)4NH2, (CH2)3NHC(=NH)NH2或(CH2)3NHC(=O)NH2;且
R14为

14.抗体-药物缀合物,具有根据式(IVa)的结构:

其中
Ab是抗体;
R40是键,

其中与Ab键合的R40的开放价用星号*表示,与(CH2)r键合的R40的开放价用波浪线 表示;
表示;
m是1、2、3或4;
v是0或1,条件是只有当s是1且R40为键时v才能为0,并且只有当v为0且s是1时R40才能为键;
T是自牺牲基团;
t是0或1;
AAa和每个AAb独立地选自丙氨酸,β-丙氨酸,γ-氨基丁酸,精氨酸,天冬酰胺,天冬氨酸,γ-羧基谷氨酸,瓜氨酸,半胱氨酸,谷氨酸,谷氨酰胺,甘氨酸,组氨酸,异亮氨酸,亮氨酸,赖氨酸,蛋氨酸,正亮氨酸,正缬氨酸,鸟氨酸,苯丙氨酸,脯氨酸,丝氨酸,苏氨酸,色氨酸,酪氨酸和缬氨酸;
u是0或1;
p是1、2、3或4;
q是1、2、3、4、5、6、7、8、9、10、11或12;
r是1、2、3、4或5;
s是0或1;
Hal是Cl或Br;
R1是 ;
;
R2是H, C1-C3烷基, CO2H, CO2(C1-C3烷基), C(=O)NH2, C(=O)NH(C1-C3烷基)或C(=O)N(C1-C3烷基)2;
R3是

R4, R4’, R5, R6或R7独立地是H, OMe, OH, 6元芳基, 5或6元杂芳基, NH2, NHMe,NMe2, NH(C2-C4烷基), N(C2-C4烷基)2, NHC(=O)X1, O(C2-C4烷基), O(CH2)0-2(C3-C6环烷基), O(CH2)0-2X1或 ;
;
其中C2-C4烷基可以未取代或被OCH2CH2OH, OCH2CH2NH2, NHCH2CH2OH, NHCH2CH2NH2, OH或NH2取代,芳基或杂芳基可以被C1-C2烷基, OH, NH2, NH(C1-C2烷基), N(C1-C2烷基)2, F,Cl, Br, NO2或CN取代;
条件是R4, R4’, R5, R6和R7至少一者不是H;
R8和R8’独立地是H, OH, O(C1-C3烷基), Cl, Br, F, O(CH2)2-4NH2或O(CH2)2-4OH;
R9是H, C(=O)(C1-C3烷基), C(=O)NH2, C(=O)NH(C1-C3烷基), C(=O)( C1-C3烷基)2,(CH2)2-4OH, (CH2)2-4O(C1-C3烷基), (CH2)2-4NH2, (CH2)2-4NH(C1-C3烷基)或(CH2)2-4N(C1-C3烷基)2;
每个X1独立地是6元芳基或5至6元杂芳基,其被C1-C3烷基, OH, O(C1-C3烷基), NH2,NH(C1-C3烷基), N(C1-C3烷基)2, F, Cl, Br, NO2或CN取代;
每个X2独立地是H, Me或C2-C4烷基,其被OCH2CH2OH, OCH2CH2NH2, NHCH2CH2OH,NHCH2CH2NH2, OH或NH2取代;且
每个X3独立地是O, NH, N(C1-C3烷基)或S;
或其药学上可接受的盐。
15.根据权利要求14的抗体-药物缀合物,其中q是2、3、4或8。
16.根据权利要求1的化合物或其与抗体的缀合物在制备用于治疗受试者的癌症的药物中的用途,该受试者患有这种癌症。
17.根据权利要求16的用途,其中所述癌症是肺癌,胃癌,卵巢癌或结肠癌。
18.根据权利要求16的用途,其中所述化合物与抗体缀合。
19.根据权利要求18的用途,其中所述癌症是肺癌,胃癌,卵巢癌或结肠癌。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662377052P | 2016-08-19 | 2016-08-19 | |
| US62/377052 | 2016-08-19 | ||
| PCT/US2017/047465 WO2018035391A1 (en) | 2016-08-19 | 2017-08-18 | Seco-cyclopropapyrroloindole compounds, antibody-drug conjugates thereof, and methods of making and use |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109641911A CN109641911A (zh) | 2019-04-16 |
| CN109641911B true CN109641911B (zh) | 2023-02-21 |
Family
ID=59714170
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201780050827.5A Active CN109641911B (zh) | 2016-08-19 | 2017-08-18 | seco-环丙吡咯并吲哚化合物和其抗体-药物缀合物以及制备和使用方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10287291B2 (zh) |
| EP (1) | EP3500574B1 (zh) |
| JP (1) | JP7023933B2 (zh) |
| KR (1) | KR102493853B1 (zh) |
| CN (1) | CN109641911B (zh) |
| ES (1) | ES2902179T3 (zh) |
| WO (1) | WO2018035391A1 (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020112588A1 (en) | 2018-11-30 | 2020-06-04 | Bristol-Myers Squibb Company | Antibody comprising a glutamine-containing light chain c-terminal extension, conjugates thereof, and methods and uses |
| KR20210102334A (ko) | 2018-12-12 | 2021-08-19 | 브리스톨-마이어스 스큅 컴퍼니 | 트랜스글루타미나제 접합을 위해 변형된 항체, 그의 접합체, 및 방법 및 용도 |
| WO2021055306A1 (en) | 2019-09-16 | 2021-03-25 | Bristol-Myers Squibb Company | Dual capture method for analysis of antibody-drug conjugates |
| AU2022357933A1 (en) * | 2021-09-30 | 2024-04-11 | Ajinomoto Co., Inc. | Conjugate of antibody and functional substance or salt thereof, and antibody derivative and compound or salts thereof to be used in producing conjugate or salt thereof |
| WO2023151679A1 (en) * | 2022-02-11 | 2023-08-17 | Shenzhen Enduring Biotech , Ltd. | Pegylated antibody hydroxyl-bearing drug conjugate |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5091541A (en) * | 1990-02-01 | 1992-02-25 | Hoechst-Roussel Pharmaceuticals Inc. | Hexahydropyrrolo(2,3-B)indole carbamates, ureas, amides and related compounds |
| CN101616667A (zh) * | 2006-10-27 | 2009-12-30 | 百时美施贵宝公司 | 可用作激酶抑制剂的杂环酰胺化合物 |
| CN101652067A (zh) * | 2007-03-30 | 2010-02-17 | 王者制药研究发展有限公司 | 腺苷a2a受体拮抗剂 |
| CN102520153A (zh) * | 2011-12-15 | 2012-06-27 | 安徽师范大学 | 一种快速检测食品中的邻苯二甲酸二丁酯的方法 |
| CN104220442A (zh) * | 2012-04-05 | 2014-12-17 | 内尔维阿诺医学科学有限公司 | 新的烷化剂 |
Family Cites Families (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1981001145A1 (en) | 1979-10-18 | 1981-04-30 | Univ Illinois | Hydrolytic enzyme-activatible pro-drugs |
| US4475196A (en) | 1981-03-06 | 1984-10-02 | Zor Clair G | Instrument for locating faults in aircraft passenger reading light and attendant call control system |
| US4447233A (en) | 1981-04-10 | 1984-05-08 | Parker-Hannifin Corporation | Medication infusion pump |
| US5144011A (en) | 1981-06-26 | 1992-09-01 | Boston University | Acidity-sensitive spacer molecule to control the release of pharmaceuticals from molecular carriers |
| US4631190A (en) | 1981-06-26 | 1986-12-23 | Shen Wei C | Acidity-sensitive spacer molecule to control the release of pharmaceuticals from molecular carriers |
| US4439196A (en) | 1982-03-18 | 1984-03-27 | Merck & Co., Inc. | Osmotic drug delivery system |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US4447224A (en) | 1982-09-20 | 1984-05-08 | Infusaid Corporation | Variable flow implantable infusion apparatus |
| US4487603A (en) | 1982-11-26 | 1984-12-11 | Cordis Corporation | Implantable microinfusion pump system |
| US4486194A (en) | 1983-06-08 | 1984-12-04 | James Ferrara | Therapeutic device for administering medicaments through the skin |
| US4698420A (en) | 1985-02-25 | 1987-10-06 | Xoma Corporation | Antibody hybrid molecules and process for their preparation |
| US4596556A (en) | 1985-03-25 | 1986-06-24 | Bioject, Inc. | Hypodermic injection apparatus |
| US5374548A (en) | 1986-05-02 | 1994-12-20 | Genentech, Inc. | Methods and compositions for the attachment of proteins to liposomes using a glycophospholipid anchor |
| MX9203291A (es) | 1985-06-26 | 1992-08-01 | Liposome Co Inc | Metodo para acoplamiento de liposomas. |
| US4941880A (en) | 1987-06-19 | 1990-07-17 | Bioject, Inc. | Pre-filled ampule and non-invasive hypodermic injection device assembly |
| US4790824A (en) | 1987-06-19 | 1988-12-13 | Bioject, Inc. | Non-invasive hypodermic injection device |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| US5312335A (en) | 1989-11-09 | 1994-05-17 | Bioject Inc. | Needleless hypodermic injection device |
| US5064413A (en) | 1989-11-09 | 1991-11-12 | Bioject, Inc. | Needleless hypodermic injection device |
| US5383851A (en) | 1992-07-24 | 1995-01-24 | Bioject Inc. | Needleless hypodermic injection device |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| AU727608B2 (en) | 1995-10-03 | 2000-12-14 | Scripps Research Institute, The | CBI analogs of CC-1065 and the duocarmycins |
| ATE271041T1 (de) | 1997-05-22 | 2004-07-15 | Scripps Research Inst | Analoga von duocarmycin and cc-1065 |
| US7425541B2 (en) | 1998-12-11 | 2008-09-16 | Medarex, Inc. | Enzyme-cleavable prodrug compounds |
| US6984720B1 (en) | 1999-08-24 | 2006-01-10 | Medarex, Inc. | Human CTLA-4 antibodies |
| AU2001286727A1 (en) | 2000-08-24 | 2002-03-04 | Coulter Pharmaceutical, Inc. | Prodrugs activated by plasmin and their use in cancer chemotherapy |
| MXPA03011094A (es) | 2001-05-31 | 2004-12-06 | Medarex Inc | Citotoxinas, profarmacos, ligadores, y estabilizadores utiles para ello. |
| CA2450316A1 (en) | 2001-06-11 | 2002-12-19 | Medarex, Inc. | Cd10-activated prodrug compounds |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| JP4562395B2 (ja) | 2002-01-09 | 2010-10-13 | メダレックス, インク. | Cd30に対するヒトモノクローナル抗体 |
| US6756397B2 (en) | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| JP4753867B2 (ja) | 2003-04-15 | 2011-08-24 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | ヒトil−18を含むコンジュゲートおよびその置換変異体 |
| TWI353992B (en) | 2003-07-22 | 2011-12-11 | Schering Ag | Rg1 antibodies and uses thereof |
| WO2005051976A2 (en) | 2003-11-20 | 2005-06-09 | Ansata Therapeutics, Inc. | Protein and peptide ligation processes and one-step purification processes |
| EP2865687A1 (en) | 2003-12-10 | 2015-04-29 | E. R. Squibb & Sons, L.L.C. | IP-10 antibodies and their uses |
| JP5064037B2 (ja) | 2004-02-23 | 2012-10-31 | ジェネンテック, インコーポレイテッド | 複素環式自壊的リンカーおよび結合体 |
| RU2402548C2 (ru) | 2004-05-19 | 2010-10-27 | Медарекс, Инк. | Химические линкеры и их конъюгаты |
| EP1747021B1 (en) | 2004-05-19 | 2015-09-09 | E. R. Squibb & Sons, L.L.C. | Self-immolative linkers and drug conjugates |
| NZ553500A (en) | 2004-09-23 | 2009-11-27 | Genentech Inc Genentech Inc | Cysteine engineered antibodies and conjugates withCysteine engineered antibodies and conjugates with a free cysteine amino acid in the heavy chain a free cysteine amino acid in the heavy chain |
| US7875278B2 (en) | 2005-02-18 | 2011-01-25 | Medarex, Inc. | Monoclonal antibodies against prostate specific membrane antigen (PSMA) lacking in fucosyl residues |
| MX2007009878A (es) | 2005-02-18 | 2007-10-03 | Medarex Inc | Anticuerpos monoclonales para antigeno de membrana especifica para prostata (amep). |
| US7714016B2 (en) | 2005-04-08 | 2010-05-11 | Medarex, Inc. | Cytotoxic compounds and conjugates with cleavable substrates |
| CN117534755A (zh) | 2005-05-09 | 2024-02-09 | 小野药品工业株式会社 | 程序性死亡-1(pd-1)的人单克隆抗体及使用抗pd-1抗体来治疗癌症的方法 |
| KR101888321B1 (ko) | 2005-07-01 | 2018-08-13 | 이. 알. 스퀴부 앤드 선즈, 엘.엘.씨. | 예정 사멸 리간드 1 (피디-엘1)에 대한 인간 모노클로날 항체 |
| US8039273B2 (en) | 2005-07-18 | 2011-10-18 | Seattle Genetics, Inc. | β-glucuronide-linker drug conjugates |
| BRPI0617546A2 (pt) | 2005-09-26 | 2011-07-26 | Medarex Inc | conjugado de fÁrmaco-anticorpo, formulaÇço farmacÊutica, mÉtodo para matar uma cÉlula de tumor, mÉtodo para retardar ou interromper o crescimento de um tumor em um sujeito mamÍfero e composto |
| NZ566395A (en) | 2005-09-26 | 2012-03-30 | Medarex Inc | Human monoclonal antibodies to CD70 |
| BRPI0619331A2 (pt) | 2005-10-26 | 2011-09-27 | Medarex Inc | método para fazer um composto para preparar um análogo de cbi cc-1065, método para preparar um análogo de cbi cc-1065 e composto |
| WO2007059404A2 (en) | 2005-11-10 | 2007-05-24 | Medarex, Inc. | Duocarmycin derivatives as novel cytotoxic compounds and conjugates |
| PL1960434T3 (pl) | 2005-12-08 | 2012-12-31 | Squibb & Sons Llc | Ludzkie przeciwciała monoklonalne przeciw fozylowi-gm1 i sposoby zastosowania antyfukozylu-gm1 |
| EP1963371A2 (en) | 2005-12-08 | 2008-09-03 | Medarex Inc. | Human monoclonal antibodies to o8e |
| CN101360761B (zh) | 2005-12-08 | 2012-09-12 | 米德列斯公司 | 抗蛋白质酪氨酸激酶7(ptk7)的人单克隆抗体及其用途 |
| KR100869414B1 (ko) | 2005-12-13 | 2008-11-21 | 야마하 가부시키가이샤 | 건반식 음판 타악기용의 음판 및 그 제조방법, 음판타악기의 음원 유닛 및 건반식 타악기 |
| WO2007103288A2 (en) * | 2006-03-02 | 2007-09-13 | Seattle Genetics, Inc. | Engineered antibody drug conjugates |
| US8715958B2 (en) | 2006-06-29 | 2014-05-06 | The Board Of Trustees Of The Leland Stanford Junior University | Cell-free synthesis of proteins containing unnatural amino acids |
| WO2008020075A1 (en) | 2006-08-18 | 2008-02-21 | Novo Nordisk Health Care Ag | Transglutaminase variants with improved specificity |
| CA2662752C (en) | 2006-09-08 | 2016-04-12 | Ambrx, Inc. | Site specific incorporation of non-natural amino acids by vertebrate cells |
| US8450464B2 (en) | 2006-10-02 | 2013-05-28 | Medarex, Inc. | Human monoclonal antibodies that bind CXCR4 |
| MX2009005776A (es) | 2006-12-01 | 2009-06-10 | Medarex Inc | Anticuerpos humanos que se enlazan al cd 22 y sus usos. |
| UY30776A1 (es) | 2006-12-21 | 2008-07-03 | Medarex Inc | Anticuerpos cd44 |
| TWI412367B (zh) | 2006-12-28 | 2013-10-21 | Medarex Llc | 化學鏈接劑與可裂解基質以及其之綴合物 |
| JP2010519310A (ja) | 2007-02-21 | 2010-06-03 | メダレックス インコーポレイテッド | 単一のアミノ酸を有する化学リンカーおよびその複合体 |
| BRPI0808014A2 (pt) | 2007-02-22 | 2014-06-17 | Novo Nordisk Healthcare Ag | Variantes de transglutaminase com especificidade melhorada |
| SI2178921T1 (sl) | 2007-07-17 | 2016-05-31 | E.R. Squibb & Sons, L.L.C. | Monoklonska protitelesa proti glipikan-3 |
| WO2009026274A1 (en) | 2007-08-22 | 2009-02-26 | Medarex, Inc. | Site-specific attachment of drugs or other agents to engineered antibodies with c-terminal extensions |
| AU2008308956B2 (en) | 2007-10-01 | 2013-03-14 | Bristol-Myers Squibb Company | Human antibodies that bind mesothelin, and uses thereof |
| EP2391714B2 (en) | 2009-01-30 | 2019-07-24 | Whitehead Institute for Biomedical Research | Methods for ligation and uses thereof |
| US8394922B2 (en) | 2009-08-03 | 2013-03-12 | Medarex, Inc. | Antiproliferative compounds, conjugates thereof, methods therefor, and uses thereof |
| WO2011103557A1 (en) * | 2010-02-22 | 2011-08-25 | Advanced Cancer Therapeutics, Llc | Small molecule inhibitors of pfkfb3 and glycolytic flux and their methods of use as anti-cancer therapeutics |
| DK3056203T3 (da) * | 2010-04-21 | 2018-01-29 | Syntarga Bv | Konjugater af cc-1065-analoger og bifunktionelle linkere. |
| JP6105477B2 (ja) | 2010-11-05 | 2017-03-29 | ライナット ニューロサイエンス コーポレイション | 操作されたポリペプチドコンジュゲートおよびトランスグルタミナーゼを用いてそれを作製する方法 |
| US8852599B2 (en) | 2011-05-26 | 2014-10-07 | Bristol-Myers Squibb Company | Immunoconjugates, compositions for making them, and methods of making and use |
| US9056887B2 (en) * | 2011-07-26 | 2015-06-16 | Elitech Holding B.V. | Minor groove binder phosphoramidites and methods of use |
| DK2956173T3 (en) | 2013-02-14 | 2017-07-17 | Bristol Myers Squibb Co | TUBULYSIS RELATIONSHIPS, METHODS OF PRODUCING AND USING THEREOF |
| EP2976362B1 (en) * | 2013-03-19 | 2019-10-23 | Beijing Shenogen Pharma Group Ltd. | Antibodies and methods for treating estrogen receptor-associated diseases |
| US9765077B2 (en) * | 2015-05-29 | 2017-09-19 | University Of East Anglia | Synthesis of duocarmycin analogues |
-
2017
- 2017-08-18 ES ES17758392T patent/ES2902179T3/es active Active
- 2017-08-18 WO PCT/US2017/047465 patent/WO2018035391A1/en unknown
- 2017-08-18 US US15/680,424 patent/US10287291B2/en active Active
- 2017-08-18 EP EP17758392.9A patent/EP3500574B1/en active Active
- 2017-08-18 JP JP2019509530A patent/JP7023933B2/ja active Active
- 2017-08-18 CN CN201780050827.5A patent/CN109641911B/zh active Active
- 2017-08-18 KR KR1020197007464A patent/KR102493853B1/ko active IP Right Grant
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5091541A (en) * | 1990-02-01 | 1992-02-25 | Hoechst-Roussel Pharmaceuticals Inc. | Hexahydropyrrolo(2,3-B)indole carbamates, ureas, amides and related compounds |
| CN101616667A (zh) * | 2006-10-27 | 2009-12-30 | 百时美施贵宝公司 | 可用作激酶抑制剂的杂环酰胺化合物 |
| CN101652067A (zh) * | 2007-03-30 | 2010-02-17 | 王者制药研究发展有限公司 | 腺苷a2a受体拮抗剂 |
| CN102520153A (zh) * | 2011-12-15 | 2012-06-27 | 安徽师范大学 | 一种快速检测食品中的邻苯二甲酸二丁酯的方法 |
| CN104220442A (zh) * | 2012-04-05 | 2014-12-17 | 内尔维阿诺医学科学有限公司 | 新的烷化剂 |
Non-Patent Citations (2)
| Title |
|---|
| "Solid-Phase Synthesis of Duocarmycin Analogues and the Effect of C‑Terminal Substitution on Biological Activity";Michael J. Stephenson等,;《J. Org. Chem.》;20150910;第80卷;第9454-9467页 * |
| "Systematic Exploration of the Structural Features of Yatakemycin Impacting DNA Alkylation and Biological Activity";Mark S. Tichenor等,;《J. AM. CHEM. SOC.》;20070811;第129卷(第5期);第10858-10869页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2902179T3 (es) | 2022-03-25 |
| CN109641911A (zh) | 2019-04-16 |
| KR102493853B1 (ko) | 2023-01-30 |
| JP7023933B2 (ja) | 2022-02-22 |
| EP3500574B1 (en) | 2021-11-24 |
| WO2018035391A1 (en) | 2018-02-22 |
| US10287291B2 (en) | 2019-05-14 |
| KR20190039570A (ko) | 2019-04-12 |
| JP2019524845A (ja) | 2019-09-05 |
| EP3500574A1 (en) | 2019-06-26 |
| US20180051031A1 (en) | 2018-02-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9676775B2 (en) | Heteroarylene-bridged benzodiazepine dimers, conjugates thereof, and methods of making and using | |
| CN109641911B (zh) | seco-环丙吡咯并吲哚化合物和其抗体-药物缀合物以及制备和使用方法 | |
| EP3245213B1 (en) | Benzodiazepine dimers, conjugates thereof, and methods of making and using | |
| US9156850B2 (en) | Enediyne compounds, conjugates thereof, and uses and methods therefor | |
| US10869934B2 (en) | Antiproliferative compounds and conjugates made therefrom | |
| CN112188902A (zh) | 用于前药和缀合物的经修饰的自消灭部分以及使用和制造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |