CN108697700B - 氘代多潘立酮组合物和用于治疗病症的方法 - Google Patents
氘代多潘立酮组合物和用于治疗病症的方法 Download PDFInfo
- Publication number
- CN108697700B CN108697700B CN201780008640.9A CN201780008640A CN108697700B CN 108697700 B CN108697700 B CN 108697700B CN 201780008640 A CN201780008640 A CN 201780008640A CN 108697700 B CN108697700 B CN 108697700B
- Authority
- CN
- China
- Prior art keywords
- domperidone
- active agent
- release
- formulation
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000203 mixture Substances 0.000 title abstract description 241
- FGXWKSZFVQUSTL-UHFFFAOYSA-N domperidone Chemical class C12=CC=CC=C2NC(=O)N1CCCN(CC1)CCC1N1C2=CC=C(Cl)C=C2NC1=O FGXWKSZFVQUSTL-UHFFFAOYSA-N 0.000 title abstract description 197
- 238000000034 method Methods 0.000 title abstract description 54
- 229960001253 domperidone Drugs 0.000 claims abstract description 136
- 239000003814 drug Substances 0.000 claims description 90
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 50
- 206010021518 Impaired gastric emptying Diseases 0.000 claims description 32
- 150000001875 compounds Chemical class 0.000 claims description 32
- 208000001288 gastroparesis Diseases 0.000 claims description 32
- 239000008194 pharmaceutical composition Substances 0.000 claims description 29
- 150000003839 salts Chemical class 0.000 claims description 16
- 206010047700 Vomiting Diseases 0.000 claims description 9
- 206010028813 Nausea Diseases 0.000 claims description 8
- 230000008693 nausea Effects 0.000 claims description 8
- 230000008673 vomiting Effects 0.000 claims description 7
- 208000019395 Lactation disease Diseases 0.000 claims description 4
- 206010042576 Suppressed lactation Diseases 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 230000006651 lactation Effects 0.000 claims description 2
- 230000007812 deficiency Effects 0.000 claims 1
- 238000009472 formulation Methods 0.000 abstract description 138
- 230000009286 beneficial effect Effects 0.000 abstract 1
- 230000001151 other effect Effects 0.000 abstract 1
- 239000013543 active substance Substances 0.000 description 213
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 142
- 239000012730 sustained-release form Substances 0.000 description 101
- 238000013268 sustained release Methods 0.000 description 96
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 95
- 238000000576 coating method Methods 0.000 description 93
- -1 cyclic imide Chemical class 0.000 description 88
- 239000002245 particle Substances 0.000 description 88
- 229920000642 polymer Polymers 0.000 description 81
- 239000011248 coating agent Substances 0.000 description 80
- 229940079593 drug Drugs 0.000 description 77
- 239000003826 tablet Substances 0.000 description 75
- 239000002552 dosage form Substances 0.000 description 68
- 239000010410 layer Substances 0.000 description 63
- 238000002360 preparation method Methods 0.000 description 63
- 239000011324 bead Substances 0.000 description 55
- 239000002775 capsule Substances 0.000 description 54
- OAUUYDZHCOULIO-BTJKTKAUSA-N domperidone maleate Chemical compound [O-]C(=O)\C=C/C([O-])=O.O=C1[NH2+]C2=CC=CC=C2N1CCCN(CC1)CCC1N1C2=CC=C(Cl)C=C2[NH2+]C1=O OAUUYDZHCOULIO-BTJKTKAUSA-N 0.000 description 49
- 229960004268 domperidone maleate Drugs 0.000 description 49
- 239000000194 fatty acid Substances 0.000 description 49
- 239000011162 core material Substances 0.000 description 48
- 235000014113 dietary fatty acids Nutrition 0.000 description 46
- 229930195729 fatty acid Natural products 0.000 description 46
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 43
- 230000003111 delayed effect Effects 0.000 description 42
- 150000004665 fatty acids Chemical class 0.000 description 41
- 150000002632 lipids Chemical class 0.000 description 39
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 39
- 239000004094 surface-active agent Substances 0.000 description 37
- 239000011230 binding agent Substances 0.000 description 36
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 32
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 32
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 32
- 239000000463 material Substances 0.000 description 32
- 239000000243 solution Substances 0.000 description 30
- 239000002202 Polyethylene glycol Substances 0.000 description 28
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 28
- 229920001223 polyethylene glycol Polymers 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 26
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 25
- 230000001965 increasing effect Effects 0.000 description 25
- 239000002904 solvent Substances 0.000 description 25
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 24
- 239000003795 chemical substances by application Substances 0.000 description 24
- 229910052805 deuterium Inorganic materials 0.000 description 24
- 239000010408 film Substances 0.000 description 24
- 239000002609 medium Substances 0.000 description 24
- 235000010980 cellulose Nutrition 0.000 description 23
- 229920002678 cellulose Polymers 0.000 description 23
- 239000000047 product Substances 0.000 description 23
- 229920003176 water-insoluble polymer Polymers 0.000 description 23
- 229920001661 Chitosan Polymers 0.000 description 22
- 239000001913 cellulose Substances 0.000 description 22
- 238000013270 controlled release Methods 0.000 description 22
- 238000004090 dissolution Methods 0.000 description 22
- 229940057917 medium chain triglycerides Drugs 0.000 description 22
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 22
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 22
- 238000010521 absorption reaction Methods 0.000 description 21
- 239000006185 dispersion Substances 0.000 description 21
- 239000011159 matrix material Substances 0.000 description 21
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 19
- 230000004060 metabolic process Effects 0.000 description 19
- 239000000725 suspension Substances 0.000 description 19
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 18
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 18
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 18
- 239000000126 substance Substances 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 17
- 210000004027 cell Anatomy 0.000 description 17
- 230000000694 effects Effects 0.000 description 17
- 239000008187 granular material Substances 0.000 description 17
- 239000008188 pellet Substances 0.000 description 17
- 210000003491 skin Anatomy 0.000 description 17
- 235000011187 glycerol Nutrition 0.000 description 16
- 229960005150 glycerol Drugs 0.000 description 16
- 239000002207 metabolite Substances 0.000 description 16
- 239000011859 microparticle Substances 0.000 description 16
- 235000021251 pulses Nutrition 0.000 description 16
- 235000002639 sodium chloride Nutrition 0.000 description 16
- 238000011282 treatment Methods 0.000 description 16
- 239000002253 acid Substances 0.000 description 15
- 230000015572 biosynthetic process Effects 0.000 description 15
- 230000002496 gastric effect Effects 0.000 description 15
- 239000007788 liquid Substances 0.000 description 15
- 230000003232 mucoadhesive effect Effects 0.000 description 15
- 239000004014 plasticizer Substances 0.000 description 15
- 229920001577 copolymer Polymers 0.000 description 14
- 239000007903 gelatin capsule Substances 0.000 description 14
- 229920003169 water-soluble polymer Polymers 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- 239000007894 caplet Substances 0.000 description 13
- 239000012530 fluid Substances 0.000 description 13
- 229920000159 gelatin Polymers 0.000 description 13
- 235000019322 gelatine Nutrition 0.000 description 13
- 239000000843 powder Substances 0.000 description 13
- 235000000346 sugar Nutrition 0.000 description 13
- 230000032258 transport Effects 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 12
- 108010010803 Gelatin Proteins 0.000 description 12
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 12
- 238000009505 enteric coating Methods 0.000 description 12
- 239000002702 enteric coating Substances 0.000 description 12
- 239000000796 flavoring agent Substances 0.000 description 12
- 239000000499 gel Substances 0.000 description 12
- 239000008273 gelatin Substances 0.000 description 12
- 229940014259 gelatin Drugs 0.000 description 12
- 235000011852 gelatine desserts Nutrition 0.000 description 12
- 239000000314 lubricant Substances 0.000 description 12
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 12
- KSCKTBJJRVPGKM-UHFFFAOYSA-N octan-1-olate;titanium(4+) Chemical compound [Ti+4].CCCCCCCC[O-].CCCCCCCC[O-].CCCCCCCC[O-].CCCCCCCC[O-] KSCKTBJJRVPGKM-UHFFFAOYSA-N 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 235000010356 sorbitol Nutrition 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 11
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 11
- 239000001856 Ethyl cellulose Substances 0.000 description 11
- 229930195725 Mannitol Natural products 0.000 description 11
- 229920002472 Starch Polymers 0.000 description 11
- 235000019325 ethyl cellulose Nutrition 0.000 description 11
- 229920001249 ethyl cellulose Polymers 0.000 description 11
- 230000037406 food intake Effects 0.000 description 11
- 235000010355 mannitol Nutrition 0.000 description 11
- 239000000594 mannitol Substances 0.000 description 11
- 210000004877 mucosa Anatomy 0.000 description 11
- 239000002105 nanoparticle Substances 0.000 description 11
- 230000036470 plasma concentration Effects 0.000 description 11
- 229920000136 polysorbate Polymers 0.000 description 11
- 230000002829 reductive effect Effects 0.000 description 11
- 239000000600 sorbitol Substances 0.000 description 11
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 10
- 229920002261 Corn starch Polymers 0.000 description 10
- 239000000227 bioadhesive Substances 0.000 description 10
- 239000000872 buffer Substances 0.000 description 10
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 10
- 239000008120 corn starch Substances 0.000 description 10
- 239000013078 crystal Substances 0.000 description 10
- 239000000945 filler Substances 0.000 description 10
- 235000013355 food flavoring agent Nutrition 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 239000007858 starting material Substances 0.000 description 10
- 210000002784 stomach Anatomy 0.000 description 10
- 210000000434 stratum corneum Anatomy 0.000 description 10
- 230000002459 sustained effect Effects 0.000 description 10
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 9
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical class [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 9
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 9
- 229920002125 Sokalan® Polymers 0.000 description 9
- 239000000654 additive Substances 0.000 description 9
- 238000009792 diffusion process Methods 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- 230000000670 limiting effect Effects 0.000 description 9
- 229940016286 microcrystalline cellulose Drugs 0.000 description 9
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 9
- 239000008108 microcrystalline cellulose Substances 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- 210000001578 tight junction Anatomy 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 8
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 8
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 8
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 8
- 229930006000 Sucrose Natural products 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 235000010443 alginic acid Nutrition 0.000 description 8
- 229920000615 alginic acid Polymers 0.000 description 8
- 239000003963 antioxidant agent Substances 0.000 description 8
- 235000006708 antioxidants Nutrition 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- 239000004359 castor oil Substances 0.000 description 8
- 235000019438 castor oil Nutrition 0.000 description 8
- 229920002301 cellulose acetate Polymers 0.000 description 8
- 150000002148 esters Chemical group 0.000 description 8
- 235000019439 ethyl acetate Nutrition 0.000 description 8
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 8
- 230000000968 intestinal effect Effects 0.000 description 8
- 239000008101 lactose Substances 0.000 description 8
- 229960001375 lactose Drugs 0.000 description 8
- 238000002844 melting Methods 0.000 description 8
- 230000008018 melting Effects 0.000 description 8
- 210000004379 membrane Anatomy 0.000 description 8
- 239000012528 membrane Substances 0.000 description 8
- 229920000609 methyl cellulose Polymers 0.000 description 8
- 235000010981 methylcellulose Nutrition 0.000 description 8
- 239000001923 methylcellulose Substances 0.000 description 8
- 229960002900 methylcellulose Drugs 0.000 description 8
- 210000002200 mouth mucosa Anatomy 0.000 description 8
- 235000019198 oils Nutrition 0.000 description 8
- 239000007921 spray Substances 0.000 description 8
- 239000005720 sucrose Substances 0.000 description 8
- 229960004793 sucrose Drugs 0.000 description 8
- 150000003626 triacylglycerols Chemical class 0.000 description 8
- 235000015112 vegetable and seed oil Nutrition 0.000 description 8
- 239000008158 vegetable oil Substances 0.000 description 8
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 7
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 7
- 229920002307 Dextran Polymers 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 7
- 235000021355 Stearic acid Nutrition 0.000 description 7
- 125000003118 aryl group Chemical group 0.000 description 7
- 229920001400 block copolymer Polymers 0.000 description 7
- 239000011247 coating layer Substances 0.000 description 7
- 230000007423 decrease Effects 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 239000007884 disintegrant Substances 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 229920001477 hydrophilic polymer Polymers 0.000 description 7
- 230000001976 improved effect Effects 0.000 description 7
- 239000007942 layered tablet Substances 0.000 description 7
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 7
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 7
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 7
- 238000007254 oxidation reaction Methods 0.000 description 7
- 239000005017 polysaccharide Substances 0.000 description 7
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 7
- 230000000541 pulsatile effect Effects 0.000 description 7
- 239000008117 stearic acid Substances 0.000 description 7
- JOOXCMJARBKPKM-UHFFFAOYSA-N 4-oxopentanoic acid Chemical compound CC(=O)CCC(O)=O JOOXCMJARBKPKM-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 6
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 6
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 6
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 6
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 6
- 229920000148 Polycarbophil calcium Polymers 0.000 description 6
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 6
- 230000002378 acidificating effect Effects 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 6
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 6
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 6
- 229960001631 carbomer Drugs 0.000 description 6
- 239000001768 carboxy methyl cellulose Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 229940125782 compound 2 Drugs 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 238000012377 drug delivery Methods 0.000 description 6
- 238000001125 extrusion Methods 0.000 description 6
- 239000007888 film coating Substances 0.000 description 6
- 238000009501 film coating Methods 0.000 description 6
- 238000007710 freezing Methods 0.000 description 6
- 230000008014 freezing Effects 0.000 description 6
- 125000005456 glyceride group Chemical group 0.000 description 6
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 6
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 6
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 6
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 6
- 235000019359 magnesium stearate Nutrition 0.000 description 6
- 230000002503 metabolic effect Effects 0.000 description 6
- 230000007935 neutral effect Effects 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 150000007524 organic acids Chemical class 0.000 description 6
- 230000003647 oxidation Effects 0.000 description 6
- 230000035699 permeability Effects 0.000 description 6
- 229950005134 polycarbophil Drugs 0.000 description 6
- 229920001282 polysaccharide Polymers 0.000 description 6
- 150000004804 polysaccharides Chemical class 0.000 description 6
- 229950008882 polysorbate Drugs 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 6
- 238000005563 spheronization Methods 0.000 description 6
- 235000019698 starch Nutrition 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 5
- HBTAOSGHCXUEKI-UHFFFAOYSA-N 4-chloro-n,n-dimethyl-3-nitrobenzenesulfonamide Chemical compound CN(C)S(=O)(=O)C1=CC=C(Cl)C([N+]([O-])=O)=C1 HBTAOSGHCXUEKI-UHFFFAOYSA-N 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- 102000015554 Dopamine receptor Human genes 0.000 description 5
- 108050004812 Dopamine receptor Proteins 0.000 description 5
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 5
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 5
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 5
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 239000008186 active pharmaceutical agent Substances 0.000 description 5
- 150000005215 alkyl ethers Chemical class 0.000 description 5
- 239000012736 aqueous medium Substances 0.000 description 5
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 5
- 239000003833 bile salt Substances 0.000 description 5
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 5
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 5
- 229940105329 carboxymethylcellulose Drugs 0.000 description 5
- 235000010418 carrageenan Nutrition 0.000 description 5
- 229920001525 carrageenan Polymers 0.000 description 5
- 239000000679 carrageenan Substances 0.000 description 5
- 229940113118 carrageenan Drugs 0.000 description 5
- 239000007795 chemical reaction product Substances 0.000 description 5
- 239000008199 coating composition Substances 0.000 description 5
- 239000003086 colorant Substances 0.000 description 5
- 239000007891 compressed tablet Substances 0.000 description 5
- 239000007771 core particle Substances 0.000 description 5
- 229940031578 diisopropyl adipate Drugs 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 5
- 235000003599 food sweetener Nutrition 0.000 description 5
- 230000030136 gastric emptying Effects 0.000 description 5
- 210000001035 gastrointestinal tract Anatomy 0.000 description 5
- 239000008103 glucose Substances 0.000 description 5
- 229930195712 glutamate Natural products 0.000 description 5
- ACCCMOQWYVYDOT-UHFFFAOYSA-N hexane-1,1-diol Chemical compound CCCCCC(O)O ACCCMOQWYVYDOT-UHFFFAOYSA-N 0.000 description 5
- 239000012052 hydrophilic carrier Substances 0.000 description 5
- 230000002209 hydrophobic effect Effects 0.000 description 5
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000008185 minitablet Substances 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 210000003097 mucus Anatomy 0.000 description 5
- 230000003204 osmotic effect Effects 0.000 description 5
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 5
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 5
- 229920000053 polysorbate 80 Polymers 0.000 description 5
- 229940068965 polysorbates Drugs 0.000 description 5
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 5
- 239000011241 protective layer Substances 0.000 description 5
- GHBFNMLVSPCDGN-UHFFFAOYSA-N rac-1-monooctanoylglycerol Chemical compound CCCCCCCC(=O)OCC(O)CO GHBFNMLVSPCDGN-UHFFFAOYSA-N 0.000 description 5
- 239000003765 sweetening agent Substances 0.000 description 5
- 239000000454 talc Substances 0.000 description 5
- 229910052623 talc Inorganic materials 0.000 description 5
- 229940033134 talc Drugs 0.000 description 5
- 239000001993 wax Substances 0.000 description 5
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 5
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 4
- OKMWKBLSFKFYGZ-UHFFFAOYSA-N 1-behenoylglycerol Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(O)CO OKMWKBLSFKFYGZ-UHFFFAOYSA-N 0.000 description 4
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 4
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 4
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 4
- 102000007469 Actins Human genes 0.000 description 4
- 108010085238 Actins Proteins 0.000 description 4
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 4
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 4
- 239000005642 Oleic acid Substances 0.000 description 4
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 4
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 4
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 4
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 4
- DOOTYTYQINUNNV-UHFFFAOYSA-N Triethyl citrate Chemical compound CCOC(=O)CC(O)(C(=O)OCC)CC(=O)OCC DOOTYTYQINUNNV-UHFFFAOYSA-N 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 229940072056 alginate Drugs 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 229940035676 analgesics Drugs 0.000 description 4
- 239000000730 antalgic agent Substances 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 230000004888 barrier function Effects 0.000 description 4
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical class CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 4
- 230000006399 behavior Effects 0.000 description 4
- 230000036765 blood level Effects 0.000 description 4
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 4
- 229910000019 calcium carbonate Inorganic materials 0.000 description 4
- 235000010216 calcium carbonate Nutrition 0.000 description 4
- 239000001506 calcium phosphate Substances 0.000 description 4
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 4
- 239000008116 calcium stearate Substances 0.000 description 4
- 235000013539 calcium stearate Nutrition 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 235000019864 coconut oil Nutrition 0.000 description 4
- 239000003240 coconut oil Substances 0.000 description 4
- 229940099112 cornstarch Drugs 0.000 description 4
- 125000004431 deuterium atom Chemical group 0.000 description 4
- 239000012738 dissolution medium Substances 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 230000036267 drug metabolism Effects 0.000 description 4
- 230000002708 enhancing effect Effects 0.000 description 4
- 210000000981 epithelium Anatomy 0.000 description 4
- 238000010579 first pass effect Methods 0.000 description 4
- 238000004108 freeze drying Methods 0.000 description 4
- 239000001530 fumaric acid Substances 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 229940049654 glyceryl behenate Drugs 0.000 description 4
- 238000005469 granulation Methods 0.000 description 4
- 230000003179 granulation Effects 0.000 description 4
- 229960003943 hypromellose Drugs 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 238000007918 intramuscular administration Methods 0.000 description 4
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 4
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 4
- 230000005445 isotope effect Effects 0.000 description 4
- 229960001021 lactose monohydrate Drugs 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 238000007909 melt granulation Methods 0.000 description 4
- 230000037353 metabolic pathway Effects 0.000 description 4
- 210000000214 mouth Anatomy 0.000 description 4
- 210000004400 mucous membrane Anatomy 0.000 description 4
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 4
- 235000010987 pectin Nutrition 0.000 description 4
- 229920001277 pectin Polymers 0.000 description 4
- 239000001814 pectin Substances 0.000 description 4
- 150000003904 phospholipids Chemical class 0.000 description 4
- 229920001983 poloxamer Polymers 0.000 description 4
- 229920001451 polypropylene glycol Polymers 0.000 description 4
- 239000011118 polyvinyl acetate Substances 0.000 description 4
- 229920002689 polyvinyl acetate Polymers 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 229920001592 potato starch Polymers 0.000 description 4
- 125000001453 quaternary ammonium group Chemical group 0.000 description 4
- 229940100486 rice starch Drugs 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000007909 solid dosage form Substances 0.000 description 4
- 239000002047 solid lipid nanoparticle Substances 0.000 description 4
- 235000012424 soybean oil Nutrition 0.000 description 4
- 239000003549 soybean oil Substances 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 229940032147 starch Drugs 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 4
- 239000001069 triethyl citrate Substances 0.000 description 4
- VMYFZRTXGLUXMZ-UHFFFAOYSA-N triethyl citrate Natural products CCOC(=O)C(O)(C(=O)OCC)C(=O)OCC VMYFZRTXGLUXMZ-UHFFFAOYSA-N 0.000 description 4
- 235000013769 triethyl citrate Nutrition 0.000 description 4
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 4
- 210000002700 urine Anatomy 0.000 description 4
- 229940100445 wheat starch Drugs 0.000 description 4
- 229920001285 xanthan gum Polymers 0.000 description 4
- 235000010493 xanthan gum Nutrition 0.000 description 4
- 239000000230 xanthan gum Substances 0.000 description 4
- 229940082509 xanthan gum Drugs 0.000 description 4
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 3
- DOAYWDKFDPSTSV-UHFFFAOYSA-N 5-chloro-1,3-dihydro-1-(4-piperidinyl)-2h-benzimidazol-2-one Chemical compound O=C1NC2=CC(Cl)=CC=C2N1C1CCNCC1 DOAYWDKFDPSTSV-UHFFFAOYSA-N 0.000 description 3
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 3
- 208000017228 Gastrointestinal motility disease Diseases 0.000 description 3
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical class OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 3
- 102000003886 Glycoproteins Human genes 0.000 description 3
- 108090000288 Glycoproteins Proteins 0.000 description 3
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 3
- 229920002774 Maltodextrin Polymers 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 229920001800 Shellac Polymers 0.000 description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- 229920002494 Zein Polymers 0.000 description 3
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 3
- 229940035674 anesthetics Drugs 0.000 description 3
- 125000000129 anionic group Chemical group 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000008499 blood brain barrier function Effects 0.000 description 3
- 210000001218 blood-brain barrier Anatomy 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229940095672 calcium sulfate Drugs 0.000 description 3
- 235000011132 calcium sulphate Nutrition 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 230000001055 chewing effect Effects 0.000 description 3
- PUFQVTATUTYEAL-UHFFFAOYSA-N cinchocaine Chemical compound C1=CC=CC2=NC(OCCCC)=CC(C(=O)NCCN(CC)CC)=C21 PUFQVTATUTYEAL-UHFFFAOYSA-N 0.000 description 3
- 229960001747 cinchocaine Drugs 0.000 description 3
- 230000004087 circulation Effects 0.000 description 3
- 239000002131 composite material Substances 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 3
- 230000002500 effect on skin Effects 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 238000005538 encapsulation Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 235000011087 fumaric acid Nutrition 0.000 description 3
- 230000030135 gastric motility Effects 0.000 description 3
- 239000003193 general anesthetic agent Substances 0.000 description 3
- 235000001727 glucose Nutrition 0.000 description 3
- 229910021389 graphene Inorganic materials 0.000 description 3
- 239000003906 humectant Substances 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 3
- 150000002433 hydrophilic molecules Chemical class 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 3
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 239000004816 latex Substances 0.000 description 3
- 229920000126 latex Polymers 0.000 description 3
- 229940040102 levulinic acid Drugs 0.000 description 3
- 229940087305 limonene Drugs 0.000 description 3
- 235000001510 limonene Nutrition 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 description 3
- 229960004963 mesalazine Drugs 0.000 description 3
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 3
- 125000005395 methacrylic acid group Chemical group 0.000 description 3
- 239000000693 micelle Substances 0.000 description 3
- 201000006417 multiple sclerosis Diseases 0.000 description 3
- 210000003205 muscle Anatomy 0.000 description 3
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 3
- 239000002736 nonionic surfactant Substances 0.000 description 3
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 3
- 239000003605 opacifier Substances 0.000 description 3
- 239000003346 palm kernel oil Substances 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 229960000292 pectin Drugs 0.000 description 3
- 230000035515 penetration Effects 0.000 description 3
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 3
- 230000001681 protective effect Effects 0.000 description 3
- 238000007789 sealing Methods 0.000 description 3
- 235000013874 shellac Nutrition 0.000 description 3
- 229940113147 shellac Drugs 0.000 description 3
- 239000004208 shellac Substances 0.000 description 3
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 3
- 210000000813 small intestine Anatomy 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 235000015424 sodium Nutrition 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000008109 sodium starch glycolate Substances 0.000 description 3
- 239000007962 solid dispersion Substances 0.000 description 3
- 239000012798 spherical particle Substances 0.000 description 3
- 230000003068 static effect Effects 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 239000011975 tartaric acid Substances 0.000 description 3
- 235000002906 tartaric acid Nutrition 0.000 description 3
- 239000004408 titanium dioxide Substances 0.000 description 3
- 230000037317 transdermal delivery Effects 0.000 description 3
- 125000005591 trimellitate group Chemical group 0.000 description 3
- 229940088594 vitamin Drugs 0.000 description 3
- 229930003231 vitamin Natural products 0.000 description 3
- 235000013343 vitamin Nutrition 0.000 description 3
- 239000011782 vitamin Substances 0.000 description 3
- 229940093612 zein Drugs 0.000 description 3
- 239000005019 zein Substances 0.000 description 3
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- FELGMEQIXOGIFQ-CYBMUJFWSA-N (3r)-9-methyl-3-[(2-methylimidazol-1-yl)methyl]-2,3-dihydro-1h-carbazol-4-one Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FELGMEQIXOGIFQ-CYBMUJFWSA-N 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- FCGXLCNBWYIEAA-UHFFFAOYSA-N 1,3-benzothiazol-6-ylmethanamine Chemical compound NCC1=CC=C2N=CSC2=C1 FCGXLCNBWYIEAA-UHFFFAOYSA-N 0.000 description 2
- SILNNFMWIMZVEQ-UHFFFAOYSA-N 1,3-dihydrobenzimidazol-2-one Chemical compound C1=CC=C2NC(O)=NC2=C1 SILNNFMWIMZVEQ-UHFFFAOYSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Polymers CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- CTPDSKVQLSDPLC-UHFFFAOYSA-N 2-(oxolan-2-ylmethoxy)ethanol Chemical compound OCCOCC1CCCO1 CTPDSKVQLSDPLC-UHFFFAOYSA-N 0.000 description 2
- SPCKHVPPRJWQRZ-UHFFFAOYSA-N 2-benzhydryloxy-n,n-dimethylethanamine;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 SPCKHVPPRJWQRZ-UHFFFAOYSA-N 0.000 description 2
- FGXWKSZFVQUSTL-WIQKFUDUSA-N 6-chloro-3-[1-[1,1,2,2,3,3-hexadeuterio-3-(2-oxo-3H-benzimidazol-1-yl)propyl]piperidin-4-yl]-1H-benzimidazol-2-one Chemical compound C1=CC2=C(C=C1)N(C(=O)N2)C([2H])([2H])C([2H])([2H])C([2H])([2H])N1CCC(CC1)N1C(=O)NC2=C1C=CC(Cl)=C2 FGXWKSZFVQUSTL-WIQKFUDUSA-N 0.000 description 2
- QZCLKYGREBVARF-UHFFFAOYSA-N Acetyl tributyl citrate Chemical compound CCCCOC(=O)CC(C(=O)OCCCC)(OC(C)=O)CC(=O)OCCCC QZCLKYGREBVARF-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- XKJMBINCVNINCA-UHFFFAOYSA-N Alfalone Chemical compound CON(C)C(=O)NC1=CC=C(Cl)C(Cl)=C1 XKJMBINCVNINCA-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 108010037003 Buserelin Proteins 0.000 description 2
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 2
- QFOHBWFCKVYLES-UHFFFAOYSA-N Butylparaben Chemical compound CCCCOC(=O)C1=CC=C(O)C=C1 QFOHBWFCKVYLES-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 229920008347 Cellulose acetate propionate Polymers 0.000 description 2
- 239000005714 Chitosan hydrochloride Substances 0.000 description 2
- PYGXAGIECVVIOZ-UHFFFAOYSA-N Dibutyl decanedioate Chemical compound CCCCOC(=O)CCCCCCCCC(=O)OCCCC PYGXAGIECVVIOZ-UHFFFAOYSA-N 0.000 description 2
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 2
- 206010052804 Drug tolerance Diseases 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 235000010469 Glycine max Nutrition 0.000 description 2
- 229920002907 Guar gum Polymers 0.000 description 2
- 229920000569 Gum karaya Polymers 0.000 description 2
- 238000010268 HPLC based assay Methods 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 229920001479 Hydroxyethyl methyl cellulose Polymers 0.000 description 2
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N Lactic Acid Natural products CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 2
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 2
- 229920000161 Locust bean gum Polymers 0.000 description 2
- 208000002720 Malnutrition Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AOMUHOFOVNGZAN-UHFFFAOYSA-N N,N-bis(2-hydroxyethyl)dodecanamide Chemical compound CCCCCCCCCCCC(=O)N(CCO)CCO AOMUHOFOVNGZAN-UHFFFAOYSA-N 0.000 description 2
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- 229920001214 Polysorbate 60 Polymers 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 2
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 239000004147 Sorbitan trioleate Substances 0.000 description 2
- 241000934878 Sterculia Species 0.000 description 2
- 239000004376 Sucralose Substances 0.000 description 2
- ZFOZVQLOBQUTQQ-UHFFFAOYSA-N Tributyl citrate Chemical compound CCCCOC(=O)CC(O)(C(=O)OCCCC)CC(=O)OCCCC ZFOZVQLOBQUTQQ-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 2
- 238000005299 abrasion Methods 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 229920006318 anionic polymer Polymers 0.000 description 2
- 230000003474 anti-emetic effect Effects 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 239000008135 aqueous vehicle Substances 0.000 description 2
- 235000013871 bee wax Nutrition 0.000 description 2
- 239000012166 beeswax Substances 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- 229940092782 bentonite Drugs 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- BLFLLBZGZJTVJG-UHFFFAOYSA-N benzocaine Chemical compound CCOC(=O)C1=CC=C(N)C=C1 BLFLLBZGZJTVJG-UHFFFAOYSA-N 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 235000019846 buffering salt Nutrition 0.000 description 2
- CUWODFFVMXJOKD-UVLQAERKSA-N buserelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](COC(C)(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 CUWODFFVMXJOKD-UVLQAERKSA-N 0.000 description 2
- 229960002719 buserelin Drugs 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 2
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 229920006317 cationic polymer Polymers 0.000 description 2
- 239000013553 cell monolayer Substances 0.000 description 2
- 229920006217 cellulose acetate butyrate Polymers 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 235000012000 cholesterol Nutrition 0.000 description 2
- DCSUBABJRXZOMT-IRLDBZIGSA-N cisapride Chemical compound C([C@@H]([C@@H](CC1)NC(=O)C=2C(=CC(N)=C(Cl)C=2)OC)OC)N1CCCOC1=CC=C(F)C=C1 DCSUBABJRXZOMT-IRLDBZIGSA-N 0.000 description 2
- 229960005132 cisapride Drugs 0.000 description 2
- DCSUBABJRXZOMT-UHFFFAOYSA-N cisapride Natural products C1CC(NC(=O)C=2C(=CC(N)=C(Cl)C=2)OC)C(OC)CN1CCCOC1=CC=C(F)C=C1 DCSUBABJRXZOMT-UHFFFAOYSA-N 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 2
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 238000002591 computed tomography Methods 0.000 description 2
- 235000005687 corn oil Nutrition 0.000 description 2
- 239000002285 corn oil Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 2
- 230000003436 cytoskeletal effect Effects 0.000 description 2
- 230000001934 delay Effects 0.000 description 2
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 2
- 229960003964 deoxycholic acid Drugs 0.000 description 2
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 2
- 229940038472 dicalcium phosphate Drugs 0.000 description 2
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 2
- 229960001193 diclofenac sodium Drugs 0.000 description 2
- FLKPEMZONWLCSK-UHFFFAOYSA-N diethyl phthalate Chemical compound CCOC(=O)C1=CC=CC=C1C(=O)OCC FLKPEMZONWLCSK-UHFFFAOYSA-N 0.000 description 2
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 2
- 235000019621 digestibility Nutrition 0.000 description 2
- 230000001079 digestive effect Effects 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 2
- 229960000520 diphenhydramine Drugs 0.000 description 2
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 238000007922 dissolution test Methods 0.000 description 2
- QQQMUBLXDAFBRH-UHFFFAOYSA-N dodecyl 2-hydroxypropanoate Chemical compound CCCCCCCCCCCCOC(=O)C(C)O QQQMUBLXDAFBRH-UHFFFAOYSA-N 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 238000009506 drug dissolution testing Methods 0.000 description 2
- 238000009837 dry grinding Methods 0.000 description 2
- 238000002296 dynamic light scattering Methods 0.000 description 2
- 229940009662 edetate Drugs 0.000 description 2
- 230000001804 emulsifying effect Effects 0.000 description 2
- 230000002121 endocytic effect Effects 0.000 description 2
- 230000012202 endocytosis Effects 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 210000002615 epidermis Anatomy 0.000 description 2
- 229960003276 erythromycin Drugs 0.000 description 2
- 210000003238 esophagus Anatomy 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- 230000028023 exocytosis Effects 0.000 description 2
- 238000013265 extended release Methods 0.000 description 2
- 239000000835 fiber Substances 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 235000021323 fish oil Nutrition 0.000 description 2
- 235000019634 flavors Nutrition 0.000 description 2
- 238000001917 fluorescence detection Methods 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 229920001002 functional polymer Polymers 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 210000004195 gingiva Anatomy 0.000 description 2
- 229940075507 glyceryl monostearate Drugs 0.000 description 2
- 239000001087 glyceryl triacetate Substances 0.000 description 2
- 235000013773 glyceryl triacetate Nutrition 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- 235000010417 guar gum Nutrition 0.000 description 2
- 239000000665 guar gum Substances 0.000 description 2
- 229960002154 guar gum Drugs 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 210000003780 hair follicle Anatomy 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- KEMQGTRYUADPNZ-UHFFFAOYSA-N heptadecanoic acid Chemical compound CCCCCCCCCCCCCCCCC(O)=O KEMQGTRYUADPNZ-UHFFFAOYSA-N 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000009478 high shear granulation Methods 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 235000012907 honey Nutrition 0.000 description 2
- 150000004677 hydrates Chemical group 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000012051 hydrophobic carrier Substances 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 229960001680 ibuprofen Drugs 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 210000002490 intestinal epithelial cell Anatomy 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 239000007927 intramuscular injection Substances 0.000 description 2
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 2
- 235000010494 karaya gum Nutrition 0.000 description 2
- 239000000231 karaya gum Substances 0.000 description 2
- 229940039371 karaya gum Drugs 0.000 description 2
- 239000000832 lactitol Substances 0.000 description 2
- 235000010448 lactitol Nutrition 0.000 description 2
- VQHSOMBJVWLPSR-JVCRWLNRSA-N lactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-JVCRWLNRSA-N 0.000 description 2
- 229960003451 lactitol Drugs 0.000 description 2
- 229960001848 lamotrigine Drugs 0.000 description 2
- PYZRQGJRPPTADH-UHFFFAOYSA-N lamotrigine Chemical compound NC1=NC(N)=NN=C1C1=CC=CC(Cl)=C1Cl PYZRQGJRPPTADH-UHFFFAOYSA-N 0.000 description 2
- 229940031957 lauric acid diethanolamide Drugs 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 229960004194 lidocaine Drugs 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 229960004232 linoleic acid Drugs 0.000 description 2
- 150000002634 lipophilic molecules Chemical class 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 238000001972 liquid chromatography-electrospray ionisation mass spectrometry Methods 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 235000010420 locust bean gum Nutrition 0.000 description 2
- 239000000711 locust bean gum Substances 0.000 description 2
- 230000001926 lymphatic effect Effects 0.000 description 2
- 239000012931 lyophilized formulation Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- 230000001071 malnutrition Effects 0.000 description 2
- 235000000824 malnutrition Nutrition 0.000 description 2
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 2
- 235000010449 maltitol Nutrition 0.000 description 2
- 239000000845 maltitol Substances 0.000 description 2
- 229940035436 maltitol Drugs 0.000 description 2
- 235000012054 meals Nutrition 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 2
- 230000037323 metabolic rate Effects 0.000 description 2
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 229960004503 metoclopramide Drugs 0.000 description 2
- TTWJBBZEZQICBI-UHFFFAOYSA-N metoclopramide Chemical compound CCN(CC)CCNC(=O)C1=CC(Cl)=C(N)C=C1OC TTWJBBZEZQICBI-UHFFFAOYSA-N 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 150000002772 monosaccharides Chemical class 0.000 description 2
- 235000021281 monounsaturated fatty acids Nutrition 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 239000002071 nanotube Substances 0.000 description 2
- 229920005615 natural polymer Polymers 0.000 description 2
- 239000002687 nonaqueous vehicle Substances 0.000 description 2
- 229920000847 nonoxynol Polymers 0.000 description 2
- 208000015380 nutritional deficiency disease Diseases 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 229960005343 ondansetron Drugs 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 239000006191 orally-disintegrating tablet Substances 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 239000003002 pH adjusting agent Substances 0.000 description 2
- 235000019629 palatability Nutrition 0.000 description 2
- 235000019865 palm kernel oil Nutrition 0.000 description 2
- 229960005489 paracetamol Drugs 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 239000003961 penetration enhancing agent Substances 0.000 description 2
- 229960003330 pentetic acid Drugs 0.000 description 2
- 239000012466 permeate Substances 0.000 description 2
- 230000002688 persistence Effects 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- ISWRGOKTTBVCFA-UHFFFAOYSA-N pirfenidone Chemical compound C1=C(C)C=CC(=O)N1C1=CC=CC=C1 ISWRGOKTTBVCFA-UHFFFAOYSA-N 0.000 description 2
- 229920001993 poloxamer 188 Polymers 0.000 description 2
- 229940044519 poloxamer 188 Drugs 0.000 description 2
- 229920001987 poloxamine Polymers 0.000 description 2
- 229920000233 poly(alkylene oxides) Polymers 0.000 description 2
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 229920000058 polyacrylate Polymers 0.000 description 2
- 239000008389 polyethoxylated castor oil Substances 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 235000020777 polyunsaturated fatty acids Nutrition 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 229940116317 potato starch Drugs 0.000 description 2
- 229940069328 povidone Drugs 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- WIKYUJGCLQQFNW-UHFFFAOYSA-N prochlorperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 WIKYUJGCLQQFNW-UHFFFAOYSA-N 0.000 description 2
- 229960003111 prochlorperazine Drugs 0.000 description 2
- 239000000651 prodrug Chemical group 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 235000010409 propane-1,2-diol alginate Nutrition 0.000 description 2
- 239000000473 propyl gallate Substances 0.000 description 2
- 235000010388 propyl gallate Nutrition 0.000 description 2
- 229940075579 propyl gallate Drugs 0.000 description 2
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 239000002096 quantum dot Substances 0.000 description 2
- 238000011552 rat model Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical class C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 2
- 150000004671 saturated fatty acids Chemical class 0.000 description 2
- 239000000565 sealant Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 231100000245 skin permeability Toxicity 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- JGMJQSFLQWGYMQ-UHFFFAOYSA-M sodium;2,6-dichloro-n-phenylaniline;acetate Chemical compound [Na+].CC([O-])=O.ClC1=CC=CC(Cl)=C1NC1=CC=CC=C1 JGMJQSFLQWGYMQ-UHFFFAOYSA-M 0.000 description 2
- 239000007901 soft capsule Substances 0.000 description 2
- 239000012439 solid excipient Substances 0.000 description 2
- 238000007614 solvation Methods 0.000 description 2
- 235000019337 sorbitan trioleate Nutrition 0.000 description 2
- 229960000391 sorbitan trioleate Drugs 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000006190 sub-lingual tablet Substances 0.000 description 2
- 235000019408 sucralose Nutrition 0.000 description 2
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical compound O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 description 2
- 230000008093 supporting effect Effects 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 238000013269 sustained drug release Methods 0.000 description 2
- 229920001059 synthetic polymer Polymers 0.000 description 2
- 230000001839 systemic circulation Effects 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- GKCBAIGFKIBETG-UHFFFAOYSA-N tetracaine Chemical compound CCCCNC1=CC=C(C(=O)OCCN(C)C)C=C1 GKCBAIGFKIBETG-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 230000002110 toxicologic effect Effects 0.000 description 2
- 231100000027 toxicology Toxicity 0.000 description 2
- 229960002622 triacetin Drugs 0.000 description 2
- VHOCUJPBKOZGJD-UHFFFAOYSA-N triacontanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O VHOCUJPBKOZGJD-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- LADGBHLMCUINGV-UHFFFAOYSA-N tricaprin Chemical compound CCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCC)COC(=O)CCCCCCCCC LADGBHLMCUINGV-UHFFFAOYSA-N 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- 238000010977 unit operation Methods 0.000 description 2
- 210000001186 vagus nerve Anatomy 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 239000003232 water-soluble binding agent Substances 0.000 description 2
- 238000001238 wet grinding Methods 0.000 description 2
- 239000000811 xylitol Substances 0.000 description 2
- 235000010447 xylitol Nutrition 0.000 description 2
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 2
- 229960002675 xylitol Drugs 0.000 description 2
- 235000004835 α-tocopherol Nutrition 0.000 description 2
- FTLYMKDSHNWQKD-UHFFFAOYSA-N (2,4,5-trichlorophenyl)boronic acid Chemical compound OB(O)C1=CC(Cl)=C(Cl)C=C1Cl FTLYMKDSHNWQKD-UHFFFAOYSA-N 0.000 description 1
- YJRJVBRVTVDQQT-XTEKXEGTSA-N (2R,3R,4S,5R,6S)-2-(hydroxymethyl)-6-[(2R,3S,4R,5R)-4,5,6-trihydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxane-3,4,5-triol trihydrate Chemical compound O.O.O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O YJRJVBRVTVDQQT-XTEKXEGTSA-N 0.000 description 1
- LFKMOLWAKAJMHB-LFPSBFENSA-N (2R,3R,4S,5R,6S)-2-(hydroxymethyl)-6-[(2R,3S,4R,5R,6R)-4,5,6-trihydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxane-3,4,5-triol dihydrate Chemical compound O.O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O LFKMOLWAKAJMHB-LFPSBFENSA-N 0.000 description 1
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 1
- WRRSFOZOETZUPG-FFHNEAJVSA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol;hydrate Chemical compound O.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC WRRSFOZOETZUPG-FFHNEAJVSA-N 0.000 description 1
- DKSZLDSPXIWGFO-BLOJGBSASA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol;phosphoric acid;hydrate Chemical compound O.OP(O)(O)=O.OP(O)(O)=O.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC DKSZLDSPXIWGFO-BLOJGBSASA-N 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N (R)-alpha-Tocopherol Natural products OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- PPKXEPBICJTCRU-XMZRARIVSA-N (R,R)-tramadol hydrochloride Chemical compound Cl.COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 PPKXEPBICJTCRU-XMZRARIVSA-N 0.000 description 1
- ZKMNUMMKYBVTFN-HNNXBMFYSA-N (S)-ropivacaine Chemical compound CCCN1CCCC[C@H]1C(=O)NC1=C(C)C=CC=C1C ZKMNUMMKYBVTFN-HNNXBMFYSA-N 0.000 description 1
- FFJCNSLCJOQHKM-CLFAGFIQSA-N (z)-1-[(z)-octadec-9-enoxy]octadec-9-ene Chemical compound CCCCCCCC\C=C/CCCCCCCCOCCCCCCCC\C=C/CCCCCCCC FFJCNSLCJOQHKM-CLFAGFIQSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical class OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 description 1
- LEBVLXFERQHONN-UHFFFAOYSA-N 1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide Chemical compound CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-UHFFFAOYSA-N 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- RRZWKUGIZRDCPB-UHFFFAOYSA-N 2,3-dihydroxypropyl hexanoate Chemical compound CCCCCC(=O)OCC(O)CO RRZWKUGIZRDCPB-UHFFFAOYSA-N 0.000 description 1
- DAVVKEZTUOGEAK-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethyl 2-methylprop-2-enoate Chemical compound COCCOCCOC(=O)C(C)=C DAVVKEZTUOGEAK-UHFFFAOYSA-N 0.000 description 1
- FLPJVCMIKUWSDR-UHFFFAOYSA-N 2-(4-formylphenoxy)acetamide Chemical compound NC(=O)COC1=CC=C(C=O)C=C1 FLPJVCMIKUWSDR-UHFFFAOYSA-N 0.000 description 1
- FEBUJFMRSBAMES-UHFFFAOYSA-N 2-[(2-{[3,5-dihydroxy-2-(hydroxymethyl)-6-phosphanyloxan-4-yl]oxy}-3,5-dihydroxy-6-({[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}methyl)oxan-4-yl)oxy]-3,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl phosphinite Chemical compound OC1C(O)C(O)C(CO)OC1OCC1C(O)C(OC2C(C(OP)C(O)C(CO)O2)O)C(O)C(OC2C(C(CO)OC(P)C2O)O)O1 FEBUJFMRSBAMES-UHFFFAOYSA-N 0.000 description 1
- KWVPFECTOKLOBL-KTKRTIGZSA-N 2-[(z)-octadec-9-enoxy]ethanol Chemical compound CCCCCCCC\C=C/CCCCCCCCOCCO KWVPFECTOKLOBL-KTKRTIGZSA-N 0.000 description 1
- BHIZVZJETFVJMJ-UHFFFAOYSA-N 2-hydroxypropyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCC(C)O BHIZVZJETFVJMJ-UHFFFAOYSA-N 0.000 description 1
- GHHURQMJLARIDK-UHFFFAOYSA-N 2-hydroxypropyl octanoate Chemical compound CCCCCCCC(=O)OCC(C)O GHHURQMJLARIDK-UHFFFAOYSA-N 0.000 description 1
- YXYJVFYWCLAXHO-UHFFFAOYSA-N 2-methoxyethyl 2-methylprop-2-enoate Chemical compound COCCOC(=O)C(C)=C YXYJVFYWCLAXHO-UHFFFAOYSA-N 0.000 description 1
- NBUHTTJGQKIBMR-UHFFFAOYSA-N 4,6-dimethylpyrimidin-5-amine Chemical compound CC1=NC=NC(C)=C1N NBUHTTJGQKIBMR-UHFFFAOYSA-N 0.000 description 1
- ZGDLVKWIZHHWIR-UHFFFAOYSA-N 4-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl]morpholine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N2CCOCC2)N=C1 ZGDLVKWIZHHWIR-UHFFFAOYSA-N 0.000 description 1
- OSDLLIBGSJNGJE-UHFFFAOYSA-N 4-chloro-3,5-dimethylphenol Chemical compound CC1=CC(O)=CC(C)=C1Cl OSDLLIBGSJNGJE-UHFFFAOYSA-N 0.000 description 1
- SUBDBMMJDZJVOS-UHFFFAOYSA-N 5-methoxy-2-{[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl}-1H-benzimidazole Chemical compound N=1C2=CC(OC)=CC=C2NC=1S(=O)CC1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-UHFFFAOYSA-N 0.000 description 1
- USSIQXCVUWKGNF-UHFFFAOYSA-N 6-(dimethylamino)-4,4-diphenylheptan-3-one Chemical compound C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 USSIQXCVUWKGNF-UHFFFAOYSA-N 0.000 description 1
- RHAXKFFKGZJUOE-UHFFFAOYSA-N 7-acetyl-6-ethyl-3,5,8-trihydroxy-9,10-dioxoanthracene-1,2-dicarboxylic acid Chemical compound O=C1C2=CC(O)=C(C(O)=O)C(C(O)=O)=C2C(=O)C2=C1C(O)=C(CC)C(C(C)=O)=C2O RHAXKFFKGZJUOE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 208000004998 Abdominal Pain Diseases 0.000 description 1
- 206010000060 Abdominal distension Diseases 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- WBZFUFAFFUEMEI-UHFFFAOYSA-M Acesulfame k Chemical compound [K+].CC1=CC(=O)[N-]S(=O)(=O)O1 WBZFUFAFFUEMEI-UHFFFAOYSA-M 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 244000208874 Althaea officinalis Species 0.000 description 1
- 235000006576 Althaea officinalis Nutrition 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 101100248253 Arabidopsis thaliana RH40 gene Proteins 0.000 description 1
- 235000003911 Arachis Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- QTGIAADRBBLJGA-UHFFFAOYSA-N Articaine Chemical compound CCCNC(C)C(=O)NC=1C(C)=CSC=1C(=O)OC QTGIAADRBBLJGA-UHFFFAOYSA-N 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 229930003347 Atropine Natural products 0.000 description 1
- 240000005343 Azadirachta indica Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- YHFLZDPKSVADNM-UHFFFAOYSA-N C(CCCCCCCCCCCCCCCCCCCCCCCCCC)(=O)O.C(CCCCCCCCCCCCCCCCCCCCCCCCC)(=O)O Chemical compound C(CCCCCCCCCCCCCCCCCCCCCCCCCC)(=O)O.C(CCCCCCCCCCCCCCCCCCCCCCCCC)(=O)O YHFLZDPKSVADNM-UHFFFAOYSA-N 0.000 description 1
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical class C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 description 1
- HFEYMQSAJXTNIH-UHFFFAOYSA-N CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O HFEYMQSAJXTNIH-UHFFFAOYSA-N 0.000 description 1
- LKUNXBRZDFMZOK-GFCCVEGCSA-N Capric acid monoglyceride Natural products CCCCCCCCCC(=O)OC[C@H](O)CO LKUNXBRZDFMZOK-GFCCVEGCSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 239000004380 Cholic acid Substances 0.000 description 1
- 229920002567 Chondroitin Polymers 0.000 description 1
- 241000207199 Citrus Species 0.000 description 1
- QCZAWDGAVJMPTA-RNFRBKRXSA-N ClC1=CC=CC(=N1)C1=NC(=NC(=N1)N[C@@H](C(F)(F)F)C)N[C@@H](C(F)(F)F)C Chemical compound ClC1=CC=CC(=N1)C1=NC(=NC(=N1)N[C@@H](C(F)(F)F)C)N[C@@H](C(F)(F)F)C QCZAWDGAVJMPTA-RNFRBKRXSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 108050001175 Connexin Proteins 0.000 description 1
- 102000010970 Connexin Human genes 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 1
- ZAKOWWREFLAJOT-CEFNRUSXSA-N D-alpha-tocopherylacetate Chemical compound CC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C ZAKOWWREFLAJOT-CEFNRUSXSA-N 0.000 description 1
- BXZVVICBKDXVGW-NKWVEPMBSA-N Didanosine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(NC=NC2=O)=C2N=C1 BXZVVICBKDXVGW-NKWVEPMBSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 208000012661 Dyskinesia Diseases 0.000 description 1
- 229920000896 Ethulose Polymers 0.000 description 1
- 239000001859 Ethyl hydroxyethyl cellulose Substances 0.000 description 1
- VTUSIVBDOCDNHS-UHFFFAOYSA-N Etidocaine Chemical compound CCCN(CC)C(CC)C(=O)NC1=C(C)C=CC=C1C VTUSIVBDOCDNHS-UHFFFAOYSA-N 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 208000035874 Excoriation Diseases 0.000 description 1
- DJBNUMBKLMJRSA-UHFFFAOYSA-N Flecainide Chemical compound FC(F)(F)COC1=CC=C(OCC(F)(F)F)C(C(=O)NCC2NCCCC2)=C1 DJBNUMBKLMJRSA-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 241000195480 Fucus Species 0.000 description 1
- 208000012895 Gastric disease Diseases 0.000 description 1
- 102400000921 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 206010017943 Gastrointestinal conditions Diseases 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- ZTJORNVITHUQJA-UHFFFAOYSA-N Heptyl p-hydroxybenzoate Chemical compound CCCCCCCOC(=O)C1=CC=C(O)C=C1 ZTJORNVITHUQJA-UHFFFAOYSA-N 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 102220570135 Histone PARylation factor 1_L30D_mutation Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 1
- SHBUUTHKGIVMJT-UHFFFAOYSA-N Hydroxystearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OO SHBUUTHKGIVMJT-UHFFFAOYSA-N 0.000 description 1
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 206010062767 Hypophysitis Diseases 0.000 description 1
- MFESCIUQSIBMSM-UHFFFAOYSA-N I-BCP Chemical compound ClCCCBr MFESCIUQSIBMSM-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- CBIAWPMZSFFRGN-UHFFFAOYSA-N Indiplon Chemical compound CC(=O)N(C)C1=CC=CC(C=2N3N=CC(=C3N=CC=2)C(=O)C=2SC=CC=2)=C1 CBIAWPMZSFFRGN-UHFFFAOYSA-N 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- 229930192967 Laccaic acid Natural products 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 108090001060 Lipase Proteins 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 239000005913 Maltodextrin Substances 0.000 description 1
- OCJYIGYOJCODJL-UHFFFAOYSA-N Meclizine Chemical compound CC1=CC=CC(CN2CCN(CC2)C(C=2C=CC=CC=2)C=2C=CC(Cl)=CC=2)=C1 OCJYIGYOJCODJL-UHFFFAOYSA-N 0.000 description 1
- 235000013500 Melia azadirachta Nutrition 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920003094 Methocel™ K4M Polymers 0.000 description 1
- 241000408551 Meza Species 0.000 description 1
- 239000004368 Modified starch Substances 0.000 description 1
- 108050004114 Monellin Proteins 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- 241000238367 Mya arenaria Species 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 102100022365 NAD(P)H dehydrogenase [quinone] 1 Human genes 0.000 description 1
- 229930182559 Natural dye Natural products 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 102000019280 Pancreatic lipases Human genes 0.000 description 1
- 108050006759 Pancreatic lipases Proteins 0.000 description 1
- 108010019160 Pancreatin Proteins 0.000 description 1
- IQPSEEYGBUAQFF-UHFFFAOYSA-N Pantoprazole Chemical compound COC1=CC=NC(CS(=O)C=2NC3=CC=C(OC(F)F)C=C3N=2)=C1OC IQPSEEYGBUAQFF-UHFFFAOYSA-N 0.000 description 1
- 240000002834 Paulownia tomentosa Species 0.000 description 1
- 235000010678 Paulownia tomentosa Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920002507 Poloxamer 124 Polymers 0.000 description 1
- 229920002511 Poloxamer 237 Polymers 0.000 description 1
- 229920002517 Poloxamer 338 Polymers 0.000 description 1
- 229920002560 Polyethylene Glycol 3000 Polymers 0.000 description 1
- 229920001030 Polyethylene Glycol 4000 Polymers 0.000 description 1
- 108010002885 Polygeline Proteins 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 229920001219 Polysorbate 40 Polymers 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 235000016311 Primula vulgaris Nutrition 0.000 description 1
- 244000028344 Primula vulgaris Species 0.000 description 1
- 102000003946 Prolactin Human genes 0.000 description 1
- 108010057464 Prolactin Proteins 0.000 description 1
- KCLANYCVBBTKTO-UHFFFAOYSA-N Proparacaine Chemical compound CCCOC1=CC=C(C(=O)OCCN(CC)CC)C=C1N KCLANYCVBBTKTO-UHFFFAOYSA-N 0.000 description 1
- CAJIGINSTLKQMM-UHFFFAOYSA-N Propoxycaine Chemical compound CCCOC1=CC(N)=CC=C1C(=O)OCCN(CC)CC CAJIGINSTLKQMM-UHFFFAOYSA-N 0.000 description 1
- HDSBZMRLPLPFLQ-UHFFFAOYSA-N Propylene glycol alginate Chemical compound OC1C(O)C(OC)OC(C(O)=O)C1OC1C(O)C(O)C(C)C(C(=O)OCC(C)O)O1 HDSBZMRLPLPFLQ-UHFFFAOYSA-N 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 235000019484 Rapeseed oil Nutrition 0.000 description 1
- 235000019774 Rice Bran oil Nutrition 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 229920002305 Schizophyllan Polymers 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- ULUAUXLGCMPNKK-UHFFFAOYSA-N Sulfobutanedioic acid Chemical class OC(=O)CC(C(O)=O)S(O)(=O)=O ULUAUXLGCMPNKK-UHFFFAOYSA-N 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- 102000011154 Tight junction protein ZO-1 Human genes 0.000 description 1
- 108050001370 Tight junction protein ZO-1 Proteins 0.000 description 1
- AOBORMOPSGHCAX-UHFFFAOYSA-N Tocophersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-UHFFFAOYSA-N 0.000 description 1
- BAECOWNUKCLBPZ-HIUWNOOHSA-N Triolein Natural products O([C@H](OCC(=O)CCCCCCC/C=C\CCCCCCCC)COC(=O)CCCCCCC/C=C\CCCCCCCC)C(=O)CCCCCCC/C=C\CCCCCCCC BAECOWNUKCLBPZ-HIUWNOOHSA-N 0.000 description 1
- PHYFQTYBJUILEZ-UHFFFAOYSA-N Trioleoylglycerol Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCCCCCCCC)COC(=O)CCCCCCCC=CCCCCCCCC PHYFQTYBJUILEZ-UHFFFAOYSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- KGUHOFWIXKIURA-VQXBOQCVSA-N [(2r,3s,4s,5r,6r)-6-[(2s,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy-3,4,5-trihydroxyoxan-2-yl]methyl dodecanoate Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](COC(=O)CCCCCCCCCCC)O[C@@H]1O[C@@]1(CO)[C@@H](O)[C@H](O)[C@@H](CO)O1 KGUHOFWIXKIURA-VQXBOQCVSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 235000010358 acesulfame potassium Nutrition 0.000 description 1
- 229960004998 acesulfame potassium Drugs 0.000 description 1
- 239000000619 acesulfame-K Substances 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 description 1
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000005233 alkylalcohol group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- OENHQHLEOONYIE-UKMVMLAPSA-N all-trans beta-carotene Natural products CC=1CCCC(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C OENHQHLEOONYIE-UKMVMLAPSA-N 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 239000012164 animal wax Substances 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 239000000420 anogeissus latifolia wall. gum Substances 0.000 description 1
- 229940069428 antacid Drugs 0.000 description 1
- 239000003159 antacid agent Substances 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000001458 anti-acid effect Effects 0.000 description 1
- 230000001773 anti-convulsant effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000001961 anticonvulsive agent Substances 0.000 description 1
- 229960003965 antiepileptics Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 230000004596 appetite loss Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000003974 aralkylamines Chemical class 0.000 description 1
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 1
- 229960003831 articaine Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- 210000001099 axilla Anatomy 0.000 description 1
- IUKQLMGVFMDQDP-UHFFFAOYSA-N azane;piperidine Chemical compound N.C1CCNCC1 IUKQLMGVFMDQDP-UHFFFAOYSA-N 0.000 description 1
- 108010066657 azoreductase Proteins 0.000 description 1
- 210000001142 back Anatomy 0.000 description 1
- 229960005274 benzocaine Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical class C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 1
- 235000013734 beta-carotene Nutrition 0.000 description 1
- 239000011648 beta-carotene Substances 0.000 description 1
- TUPZEYHYWIEDIH-WAIFQNFQSA-N beta-carotene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2=CCCCC2(C)C TUPZEYHYWIEDIH-WAIFQNFQSA-N 0.000 description 1
- 229960002747 betacarotene Drugs 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 239000003613 bile acid Substances 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 230000002902 bimodal effect Effects 0.000 description 1
- 230000035587 bioadhesion Effects 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 239000010473 blackcurrant seed oil Substances 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 210000005178 buccal mucosa Anatomy 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 229960003150 bupivacaine Drugs 0.000 description 1
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 1
- 229960001889 buprenorphine hydrochloride Drugs 0.000 description 1
- 229940067596 butylparaben Drugs 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000000828 canola oil Substances 0.000 description 1
- 235000019519 canola oil Nutrition 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229920003123 carboxymethyl cellulose sodium Polymers 0.000 description 1
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 1
- 229940063834 carboxymethylcellulose sodium Drugs 0.000 description 1
- 230000003177 cardiotonic effect Effects 0.000 description 1
- 239000004203 carnauba wax Substances 0.000 description 1
- 235000013869 carnauba wax Nutrition 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 229940023913 cation exchange resins Drugs 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- WZNRVWBKYDHTKI-UHFFFAOYSA-N cellulose, acetate 1,2,4-benzenetricarboxylate Chemical compound OC1C(O)C(O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O.OC1C(O)C(O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O.CC(=O)OCC1OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(COC(C)=O)O1.CC(=O)OCC1OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(COC(C)=O)O1.OC(=O)C1=CC(C(=O)O)=CC=C1C(=O)OCC1C(OC2C(C(OC(=O)C=3C(=CC(=CC=3)C(O)=O)C(O)=O)C(OC(=O)C=3C(=CC(=CC=3)C(O)=O)C(O)=O)C(COC(=O)C=3C(=CC(=CC=3)C(O)=O)C(O)=O)O2)OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)C(OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)C(OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)C(OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)O1 WZNRVWBKYDHTKI-UHFFFAOYSA-N 0.000 description 1
- 229940106189 ceramide Drugs 0.000 description 1
- 150000001783 ceramides Chemical class 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 229940074979 cetyl palmitate Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- 239000013043 chemical agent Substances 0.000 description 1
- 108091008690 chemoreceptors Proteins 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 150000001805 chlorine compounds Chemical group 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960005443 chloroxylenol Drugs 0.000 description 1
- 150000001840 cholesterol esters Chemical class 0.000 description 1
- 235000019416 cholic acid Nutrition 0.000 description 1
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 1
- 229960002471 cholic acid Drugs 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- DLGJWSVWTWEWBJ-HGGSSLSASA-N chondroitin Chemical compound CC(O)=N[C@@H]1[C@H](O)O[C@H](CO)[C@H](O)[C@@H]1OC1[C@H](O)[C@H](O)C=C(C(O)=O)O1 DLGJWSVWTWEWBJ-HGGSSLSASA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 235000020971 citrus fruits Nutrition 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000000701 coagulant Substances 0.000 description 1
- 229960003920 cocaine Drugs 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Natural products C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 1
- 229960004415 codeine phosphate Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 238000009500 colour coating Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000001218 confocal laser scanning microscopy Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 229960005168 croscarmellose Drugs 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 229940095074 cyclic amp Drugs 0.000 description 1
- VXEAYBOGHINOKW-UHFFFAOYSA-N cyclobenzaprine hydrochloride Chemical compound Cl.C1=CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 VXEAYBOGHINOKW-UHFFFAOYSA-N 0.000 description 1
- 229960000500 cyclobenzaprine hydrochloride Drugs 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- ZAKOWWREFLAJOT-UHFFFAOYSA-N d-alpha-Tocopheryl acetate Natural products CC(=O)OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C ZAKOWWREFLAJOT-UHFFFAOYSA-N 0.000 description 1
- 230000020335 dealkylation Effects 0.000 description 1
- 238000006900 dealkylation reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 210000004207 dermis Anatomy 0.000 description 1
- 108700028734 des-GlyNH2(9)- argipressin Proteins 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- MJIHNNLFOKEZEW-RUZDIDTESA-N dexlansoprazole Chemical compound CC1=C(OCC(F)(F)F)C=CN=C1C[S@@](=O)C1=NC2=CC=CC=C2N1 MJIHNNLFOKEZEW-RUZDIDTESA-N 0.000 description 1
- 229960003568 dexlansoprazole Drugs 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- QMQBBUPJKANITL-MYXGOWFTSA-N dextropropoxyphene hydrochloride Chemical compound [H+].[Cl-].C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 QMQBBUPJKANITL-MYXGOWFTSA-N 0.000 description 1
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 1
- 229940099371 diacetylated monoglycerides Drugs 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 229960002656 didanosine Drugs 0.000 description 1
- 235000021045 dietary change Nutrition 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- RBOXVHNMENFORY-DNJOTXNNSA-N dihydrocodeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC RBOXVHNMENFORY-DNJOTXNNSA-N 0.000 description 1
- 229960000920 dihydrocodeine Drugs 0.000 description 1
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229940008099 dimethicone Drugs 0.000 description 1
- 239000004205 dimethyl polysiloxane Substances 0.000 description 1
- 235000019329 dioctyl sodium sulphosuccinate Nutrition 0.000 description 1
- 238000007907 direct compression Methods 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- AGDANEVFLMAYGL-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCCCC(O)=O AGDANEVFLMAYGL-UHFFFAOYSA-N 0.000 description 1
- 229960000878 docusate sodium Drugs 0.000 description 1
- WLGSIWNFEGRXDF-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O.CCCCCCCCCCCC(O)=O WLGSIWNFEGRXDF-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 239000003210 dopamine receptor blocking agent Substances 0.000 description 1
- 230000003291 dopaminomimetic effect Effects 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 229960002496 duloxetine hydrochloride Drugs 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- JWJOTENAMICLJG-QWBYCMEYSA-N dutasteride Chemical compound O=C([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)N[C@@H]4CC3)C)CC[C@@]21C)NC1=CC(C(F)(F)F)=CC=C1C(F)(F)F JWJOTENAMICLJG-QWBYCMEYSA-N 0.000 description 1
- 229960004199 dutasteride Drugs 0.000 description 1
- BZEWSEKUUPWQDQ-UHFFFAOYSA-N dyclonine Chemical compound C1=CC(OCCCC)=CC=C1C(=O)CCN1CCCCC1 BZEWSEKUUPWQDQ-UHFFFAOYSA-N 0.000 description 1
- 229960000385 dyclonine Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 201000006549 dyspepsia Diseases 0.000 description 1
- XPOQHMRABVBWPR-ZDUSSCGKSA-N efavirenz Chemical class C([C@]1(C2=CC(Cl)=CC=C2NC(=O)O1)C(F)(F)F)#CC1CC1 XPOQHMRABVBWPR-ZDUSSCGKSA-N 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000001839 endoscopy Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 210000004783 epithelial tight junction Anatomy 0.000 description 1
- 229960004770 esomeprazole Drugs 0.000 description 1
- SUBDBMMJDZJVOS-DEOSSOPVSA-N esomeprazole Chemical compound C([S@](=O)C1=NC2=CC=C(C=C2N1)OC)C1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-DEOSSOPVSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 125000005670 ethenylalkyl group Chemical group 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000007046 ethoxylation reaction Methods 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 229960001617 ethyl hydroxybenzoate Drugs 0.000 description 1
- 235000019326 ethyl hydroxyethyl cellulose Nutrition 0.000 description 1
- 235000010944 ethyl methyl cellulose Nutrition 0.000 description 1
- 239000001761 ethyl methyl cellulose Substances 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- 229940093476 ethylene glycol Drugs 0.000 description 1
- NUVBSKCKDOMJSU-UHFFFAOYSA-N ethylparaben Chemical compound CCOC(=O)C1=CC=C(O)C=C1 NUVBSKCKDOMJSU-UHFFFAOYSA-N 0.000 description 1
- 229960003976 etidocaine Drugs 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- MQOBSOSZFYZQOK-UHFFFAOYSA-N fenofibric acid Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1C(=O)C1=CC=C(Cl)C=C1 MQOBSOSZFYZQOK-UHFFFAOYSA-N 0.000 description 1
- 229960000701 fenofibric acid Drugs 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 238000000855 fermentation Methods 0.000 description 1
- 230000004151 fermentation Effects 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 206010016629 fibroma Diseases 0.000 description 1
- 206010049444 fibromatosis Diseases 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960000449 flecainide Drugs 0.000 description 1
- 210000001061 forehead Anatomy 0.000 description 1
- 210000004055 fourth ventricle Anatomy 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 235000021588 free fatty acids Nutrition 0.000 description 1
- 229960002737 fructose Drugs 0.000 description 1
- 229910003472 fullerene Inorganic materials 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 210000003976 gap junction Anatomy 0.000 description 1
- 210000004211 gastric acid Anatomy 0.000 description 1
- 230000027119 gastric acid secretion Effects 0.000 description 1
- 210000004051 gastric juice Anatomy 0.000 description 1
- 230000002640 gastrokinetic effect Effects 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 229960001031 glucose Drugs 0.000 description 1
- 230000002641 glycemic effect Effects 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- RZRNAYUHWVFMIP-HXUWFJFHSA-N glycerol monolinoleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-HXUWFJFHSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229940087068 glyceryl caprylate Drugs 0.000 description 1
- 229940074046 glyceryl laurate Drugs 0.000 description 1
- 229940075529 glyceryl stearate Drugs 0.000 description 1
- 229940074774 glycyrrhizinate Drugs 0.000 description 1
- LPLVUJXQOOQHMX-QWBHMCJMSA-N glycyrrhizinic acid Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@H](O[C@@H]1O[C@@H]1C([C@H]2[C@]([C@@H]3[C@@]([C@@]4(CC[C@@]5(C)CC[C@@](C)(C[C@H]5C4=CC3=O)C(O)=O)C)(C)CC2)(C)CC1)(C)C)C(O)=O)[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O LPLVUJXQOOQHMX-QWBHMCJMSA-N 0.000 description 1
- 235000019314 gum ghatti Nutrition 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 210000001983 hard palate Anatomy 0.000 description 1
- 201000000615 hard palate cancer Diseases 0.000 description 1
- 239000007887 hard shell capsule Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- KWLMIXQRALPRBC-UHFFFAOYSA-L hectorite Chemical compound [Li+].[OH-].[OH-].[Na+].[Mg+2].O1[Si]2([O-])O[Si]1([O-])O[Si]([O-])(O1)O[Si]1([O-])O2 KWLMIXQRALPRBC-UHFFFAOYSA-L 0.000 description 1
- 229910000271 hectorite Inorganic materials 0.000 description 1
- 230000010224 hepatic metabolism Effects 0.000 description 1
- 235000019251 heptyl p-hydroxybenzoate Nutrition 0.000 description 1
- TUFOVEWZORBKNG-UHFFFAOYSA-N hexacosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O TUFOVEWZORBKNG-UHFFFAOYSA-N 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- KYYWBEYKBLQSFW-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCC(O)=O KYYWBEYKBLQSFW-UHFFFAOYSA-N 0.000 description 1
- PXDJXZJSCPSGGI-UHFFFAOYSA-N hexadecanoic acid hexadecyl ester Natural products CCCCCCCCCCCCCCCCOC(=O)CCCCCCCCCCCCCCC PXDJXZJSCPSGGI-UHFFFAOYSA-N 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 229940072106 hydroxystearate Drugs 0.000 description 1
- 208000003532 hypothyroidism Diseases 0.000 description 1
- 230000002989 hypothyroidism Effects 0.000 description 1
- NHXTZGXYQYMODD-UHFFFAOYSA-N icosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCC(O)=O NHXTZGXYQYMODD-UHFFFAOYSA-N 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 239000012728 immediate-release (IR) tablet Substances 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- 229950003867 indiplon Drugs 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 238000009434 installation Methods 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000002563 ionic surfactant Substances 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 210000001630 jejunum Anatomy 0.000 description 1
- 230000003780 keratinization Effects 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000008463 key metabolic pathway Effects 0.000 description 1
- 229960003174 lansoprazole Drugs 0.000 description 1
- MJIHNNLFOKEZEW-UHFFFAOYSA-N lansoprazole Chemical compound CC1=C(OCC(F)(F)F)C=CN=C1CS(=O)C1=NC2=CC=CC=C2N1 MJIHNNLFOKEZEW-UHFFFAOYSA-N 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 229960004288 levobupivacaine Drugs 0.000 description 1
- LEBVLXFERQHONN-INIZCTEOSA-N levobupivacaine Chemical compound CCCCN1CCCC[C@H]1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-INIZCTEOSA-N 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229940040145 liniment Drugs 0.000 description 1
- 239000000865 liniment Substances 0.000 description 1
- 235000020778 linoleic acid Nutrition 0.000 description 1
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 235000021266 loss of appetite Nutrition 0.000 description 1
- 208000019017 loss of appetite Diseases 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 229940035034 maltodextrin Drugs 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 235000001035 marshmallow Nutrition 0.000 description 1
- 239000013563 matrix tablet Substances 0.000 description 1
- 229960001474 meclozine Drugs 0.000 description 1
- 150000002711 medium chain fatty acid esters Chemical class 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- 229940051129 meperidine hydrochloride Drugs 0.000 description 1
- 229960002409 mepivacaine Drugs 0.000 description 1
- INWLQCZOYSRPNW-UHFFFAOYSA-N mepivacaine Chemical compound CN1CCCCC1C(=O)NC1=C(C)C=CC=C1C INWLQCZOYSRPNW-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229960001797 methadone Drugs 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000012184 mineral wax Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 1
- 229960005195 morphine hydrochloride Drugs 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 230000007659 motor function Effects 0.000 description 1
- 229940051875 mucins Drugs 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- JFTURWWGPMTABQ-UHFFFAOYSA-N n,n-dimethyl-3-naphthalen-1-yloxy-3-thiophen-2-ylpropan-1-amine Chemical compound C=1C=CC2=CC=CC=C2C=1OC(CCN(C)C)C1=CC=CS1 JFTURWWGPMTABQ-UHFFFAOYSA-N 0.000 description 1
- KQCBYLOBOGMDSY-OMCTUWBCSA-N n-[(2r,3r,4r,5r,6r)-5-[(2s,3r,4r,5r,6r)-3-acetamido-5-[(2s,3r,4r,5s,6r)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4-hydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-2,4-dihydroxy-6-(hydroxymethyl)oxan-3-yl]acetamide;2-hydroxybutanedioic acid Chemical compound OC(=O)C(O)CC(O)=O.O[C@@H]1[C@@H](NC(=O)C)[C@H](O)O[C@H](CO)[C@@H]1O[C@H]1[C@H](NC(C)=O)[C@@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](CO)O1 KQCBYLOBOGMDSY-OMCTUWBCSA-N 0.000 description 1
- CDBRNDSHEYLDJV-FVGYRXGTSA-M naproxen sodium Chemical compound [Na+].C1=C([C@H](C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CDBRNDSHEYLDJV-FVGYRXGTSA-M 0.000 description 1
- 229960003940 naproxen sodium Drugs 0.000 description 1
- 239000000978 natural dye Substances 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 235000021096 natural sweeteners Nutrition 0.000 description 1
- 239000002018 neem oil Substances 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 201000004931 neurofibromatosis Diseases 0.000 description 1
- NQDJXKOVJZTUJA-UHFFFAOYSA-N nevirapine Chemical class C12=NC=CC=C2C(=O)NC=2C(C)=CC=NC=2N1C1CC1 NQDJXKOVJZTUJA-UHFFFAOYSA-N 0.000 description 1
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- SNQQPOLDUKLAAF-UHFFFAOYSA-N nonylphenol Chemical class CCCCCCCCCC1=CC=CC=C1O SNQQPOLDUKLAAF-UHFFFAOYSA-N 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- SWDYEOBSKYXKLZ-UHFFFAOYSA-N octacosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O SWDYEOBSKYXKLZ-UHFFFAOYSA-N 0.000 description 1
- RQFLGKYCYMMRMC-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O RQFLGKYCYMMRMC-UHFFFAOYSA-N 0.000 description 1
- 229960000381 omeprazole Drugs 0.000 description 1
- 229940127240 opiate Drugs 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 229960003502 oxybuprocaine Drugs 0.000 description 1
- CMHHMUWAYWTMGS-UHFFFAOYSA-N oxybuprocaine Chemical compound CCCCOC1=CC(C(=O)OCCN(CC)CC)=CC=C1N CMHHMUWAYWTMGS-UHFFFAOYSA-N 0.000 description 1
- 229960003617 oxycodone hydrochloride Drugs 0.000 description 1
- 108010014241 oxypolygelatine Proteins 0.000 description 1
- 230000008058 pain sensation Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 229940116369 pancreatic lipase Drugs 0.000 description 1
- 229940055695 pancreatin Drugs 0.000 description 1
- 229960005019 pantoprazole Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 235000011837 pasties Nutrition 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 238000005453 pelletization Methods 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical group OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000008855 peristalsis Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 239000012169 petroleum derived wax Substances 0.000 description 1
- 235000019381 petroleum wax Nutrition 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- BCIIMDOZSUCSEN-UHFFFAOYSA-N piperidin-4-amine Chemical class NC1CCNCC1 BCIIMDOZSUCSEN-UHFFFAOYSA-N 0.000 description 1
- 229960003073 pirfenidone Drugs 0.000 description 1
- 210000003635 pituitary gland Anatomy 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229960000502 poloxamer Drugs 0.000 description 1
- 229940093448 poloxamer 124 Drugs 0.000 description 1
- 229920001992 poloxamer 407 Polymers 0.000 description 1
- 229940044476 poloxamer 407 Drugs 0.000 description 1
- 229920001553 poly(ethylene glycol)-block-polylactide methyl ether Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229940113116 polyethylene glycol 1000 Drugs 0.000 description 1
- 229960004250 polygeline Drugs 0.000 description 1
- 229920001522 polyglycol ester Polymers 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 229920006254 polymer film Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 description 1
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229940068977 polysorbate 20 Drugs 0.000 description 1
- 229940101027 polysorbate 40 Drugs 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002744 polyvinyl acetate phthalate Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 239000001253 polyvinylpolypyrrolidone Substances 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 235000019814 powdered cellulose Nutrition 0.000 description 1
- 229920003124 powdered cellulose Polymers 0.000 description 1
- DQKXQSGTHWVTAD-UHFFFAOYSA-N pramocaine Chemical compound C1=CC(OCCCC)=CC=C1OCCCN1CCOCC1 DQKXQSGTHWVTAD-UHFFFAOYSA-N 0.000 description 1
- 229960001896 pramocaine Drugs 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229940126532 prescription medicine Drugs 0.000 description 1
- 229960001807 prilocaine Drugs 0.000 description 1
- MVFGUOIZUNYYSO-UHFFFAOYSA-N prilocaine Chemical compound CCCNC(C)C(=O)NC1=CC=CC=C1C MVFGUOIZUNYYSO-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 229940097325 prolactin Drugs 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 239000000770 propane-1,2-diol alginate Substances 0.000 description 1
- 229960003981 proparacaine Drugs 0.000 description 1
- 229950003255 propoxycaine Drugs 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229940026235 propylene glycol monolaurate Drugs 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- 239000011253 protective coating Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 210000004203 pyloric antrum Anatomy 0.000 description 1
- 210000001187 pylorus Anatomy 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 229960001778 rabeprazole sodium Drugs 0.000 description 1
- LKUNXBRZDFMZOK-UHFFFAOYSA-N rac-1-monodecanoylglycerol Chemical compound CCCCCCCCCC(=O)OCC(O)CO LKUNXBRZDFMZOK-UHFFFAOYSA-N 0.000 description 1
- ARIWANIATODDMH-UHFFFAOYSA-N rac-1-monolauroylglycerol Chemical compound CCCCCCCCCCCC(=O)OCC(O)CO ARIWANIATODDMH-UHFFFAOYSA-N 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000008085 renal dysfunction Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000008165 rice bran oil Substances 0.000 description 1
- XWGJFPHUCFXLBL-UHFFFAOYSA-M rongalite Chemical compound [Na+].OCS([O-])=O XWGJFPHUCFXLBL-UHFFFAOYSA-M 0.000 description 1
- 229960001549 ropivacaine Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229940085605 saccharin sodium Drugs 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 230000036186 satiety Effects 0.000 description 1
- 235000019627 satiety Nutrition 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 210000004761 scalp Anatomy 0.000 description 1
- 210000004706 scrotum Anatomy 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000012890 simulated body fluid Substances 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 230000037380 skin damage Effects 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 235000010378 sodium ascorbate Nutrition 0.000 description 1
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 1
- 229960005055 sodium ascorbate Drugs 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical compound [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- JAJWGJBVLPIOOH-IZYKLYLVSA-M sodium taurocholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 JAJWGJBVLPIOOH-IZYKLYLVSA-M 0.000 description 1
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000012453 solvate Chemical group 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 208000020431 spinal cord injury Diseases 0.000 description 1
- DKGZKTPJOSAWFA-UHFFFAOYSA-N spiperone Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCC2(C(NCN2C=2C=CC=CC=2)=O)CC1 DKGZKTPJOSAWFA-UHFFFAOYSA-N 0.000 description 1
- 229950001675 spiperone Drugs 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 229940032085 sucrose monolaurate Drugs 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000979 synthetic dye Substances 0.000 description 1
- 229920002994 synthetic fiber Polymers 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 239000003760 tallow Substances 0.000 description 1
- 210000001779 taste bud Anatomy 0.000 description 1
- BBAWEDCPNXPBQM-GDEBMMAJSA-N telaprevir Chemical class N([C@H](C(=O)N[C@H](C(=O)N1C[C@@H]2CCC[C@@H]2[C@H]1C(=O)N[C@@H](CCC)C(=O)C(=O)NC1CC1)C(C)(C)C)C1CCCCC1)C(=O)C1=CN=CC=N1 BBAWEDCPNXPBQM-GDEBMMAJSA-N 0.000 description 1
- GMUWCTKADZFKLT-UHFFFAOYSA-N tert-butyl 2-oxo-3h-benzimidazole-1-carboxylate Chemical compound C1=CC=C2NC(=O)N(C(=O)OC(C)(C)C)C2=C1 GMUWCTKADZFKLT-UHFFFAOYSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 229960002372 tetracaine Drugs 0.000 description 1
- CBYCSRICVDBHMZ-UHFFFAOYSA-N tetracosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCCCCCCCC(O)=O CBYCSRICVDBHMZ-UHFFFAOYSA-N 0.000 description 1
- ZTUXEFFFLOVXQE-UHFFFAOYSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCC(O)=O ZTUXEFFFLOVXQE-UHFFFAOYSA-N 0.000 description 1
- 239000000892 thaumatin Substances 0.000 description 1
- 235000010436 thaumatin Nutrition 0.000 description 1
- XCTYLCDETUVOIP-UHFFFAOYSA-N thiethylperazine Chemical compound C12=CC(SCC)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 XCTYLCDETUVOIP-UHFFFAOYSA-N 0.000 description 1
- 229960004869 thiethylperazine Drugs 0.000 description 1
- 229940098465 tincture Drugs 0.000 description 1
- 235000010215 titanium dioxide Nutrition 0.000 description 1
- 229960000488 tizanidine Drugs 0.000 description 1
- XFYDIVBRZNQMJC-UHFFFAOYSA-N tizanidine Chemical compound ClC=1C=CC2=NSN=C2C=1NC1=NCCN1 XFYDIVBRZNQMJC-UHFFFAOYSA-N 0.000 description 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 229940042585 tocopherol acetate Drugs 0.000 description 1
- 125000002640 tocopherol group Chemical class 0.000 description 1
- 235000019149 tocopherols Nutrition 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000723 toxicological property Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 229960003107 tramadol hydrochloride Drugs 0.000 description 1
- 229940078499 tricalcium phosphate Drugs 0.000 description 1
- 229910000391 tricalcium phosphate Inorganic materials 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- WEAPVABOECTMGR-UHFFFAOYSA-N triethyl 2-acetyloxypropane-1,2,3-tricarboxylate Chemical compound CCOC(=O)CC(C(=O)OCC)(OC(C)=O)CC(=O)OCC WEAPVABOECTMGR-UHFFFAOYSA-N 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- RRHXZLALVWBDKH-UHFFFAOYSA-M trimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azanium;chloride Chemical compound [Cl-].CC(=C)C(=O)OCC[N+](C)(C)C RRHXZLALVWBDKH-UHFFFAOYSA-M 0.000 description 1
- VLPFTAMPNXLGLX-UHFFFAOYSA-N trioctanoin Chemical compound CCCCCCCC(=O)OCC(OC(=O)CCCCCCC)COC(=O)CCCCCCC VLPFTAMPNXLGLX-UHFFFAOYSA-N 0.000 description 1
- PHYFQTYBJUILEZ-IUPFWZBJSA-N triolein Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCCCC)COC(=O)CCCCCCC\C=C/CCCCCCCC PHYFQTYBJUILEZ-IUPFWZBJSA-N 0.000 description 1
- 229940117972 triolein Drugs 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 235000014107 unsaturated dietary fats Nutrition 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 239000012178 vegetable wax Substances 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 239000000052 vinegar Substances 0.000 description 1
- 235000021419 vinegar Nutrition 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000010497 wheat germ oil Substances 0.000 description 1
- 239000009637 wintergreen oil Substances 0.000 description 1
- 229920001221 xylan Polymers 0.000 description 1
- 150000004823 xylans Chemical class 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 239000004711 α-olefin Substances 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 150000003772 α-tocopherols Chemical class 0.000 description 1
- OENHQHLEOONYIE-JLTXGRSLSA-N β-Carotene Chemical compound CC=1CCCC(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OENHQHLEOONYIE-JLTXGRSLSA-N 0.000 description 1
Images
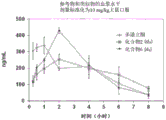

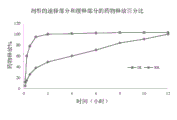
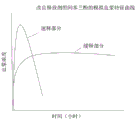

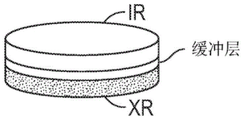

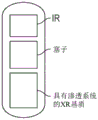

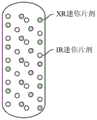






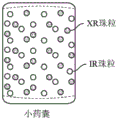




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/0056—Mouth soluble or dispersible forms; Suckable, eatable, chewable coherent forms; Forms rapidly disintegrating in the mouth; Lozenges; Lollipops; Bite capsules; Baked products; Baits or other oral forms for animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0087—Galenical forms not covered by A61K9/02 - A61K9/7023
- A61K9/0095—Drinks; Beverages; Syrups; Compositions for reconstitution thereof, e.g. powders or tablets to be dispersed in a glass of water; Veterinary drenches
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4808—Preparations in capsules, e.g. of gelatin, of chocolate characterised by the form of the capsule or the structure of the filling; Capsules containing small tablets; Capsules with outer layer for immediate drug release
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/12—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/26—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Abstract
多潘立酮组合物、方法和给药制剂通过氘代多潘立酮提供了有益的安全性和其他作用。氘代多潘立酮类似物的制备。
Description
本申请要求2016年2月4日提交的共同未决的美国第62/291,198号的优先权,其全部内容通过引用明确并入。
胃轻瘫(GP)是胃动力不起作用或不能正常发挥作用的一种症状,其可以妨碍胃部的排空并干扰消化。
GP可能是由调节消化过程的迷走神经的损伤引起的。迷走神经的损伤可能由疾病引起,如I型或II型糖尿病,或胃部或小肠手术,并可能限制神经系统向胃部肌肉发送信号的能力。病毒感染、某些药物、某些癌症治疗、硬皮病、神经系统疾病(如帕金森氏病或多发性硬化症)或甲状腺功能减退症也可引起或导致GP。
GP的诊断通常通过上消化道(GI)内窥镜检查、计算机断层扫描(CT)、肠造影、磁共振肠造影、上消化道系列检查(X射线)、胃排空研究和/或呼吸试验。GP的症状包括恶心、呕吐、血糖改变、腹痛、腹胀、少食后饱腹感、食欲不振、体重减轻以及营养不良。未经治疗的GP可导致严重的脱水、营养不良、胃中未消化的食物的硬化(胃石)以及可加剧糖尿病的血糖变化不稳定。
GP的治疗涉及标识和治疗潜在的病理。由糖尿病引起的GP可以通过饮食改变来治疗。GP可以用药物治疗以刺激胃部肌肉,例如甲氧氯普胺,红霉素以及西沙必利(cisapride)。甲氧氯普胺造成严重的副作用,例如运动障碍的出现或与其他药物的不良相互作用;红霉素易于随着患者药物耐受性的增加而丧失疗效;西沙必利的可及性有限。控制恶心和呕吐的药物包括丙氯拉嗪(prochlorperazine)、硫乙拉嗪(thiethylperazine)、苯海拉明(diphenhydramine)以及昂丹司琼(ondansetron)。
GP的症状可以通过手术治疗,如置入小肠空肠管或安装胃通气管。对于严重的情况,可以将喂食管经口或经鼻插入直接置入小肠,或经肠胃外给药。
多潘立酮为5-氯-1-(1-[3-(2-氧代-2,3-二氢-1H-苯并[d]咪唑-1-基)丙基]哌啶-4-基)-1H-苯并[d]咪唑-2(3H)-酮,其具有以下化学结构:

如本文所使用,除非另有明确说明,否则对多潘立酮的任何提及包括这些的药学上可接受的盐、酯、水合物、溶剂化物、前药形式以及衍生物,其广义地定义为经修饰或部分取代的多潘立酮化合物,实例包括但不限于加入单个原子,加入反应性基团,加入官能团,形成二聚物或多聚物,与另一分子(如抗体)结合等。
多潘立酮是一种有效的多巴胺拮抗药,其不易穿过血脑屏障;因此,多潘立酮仅表现出最小的锥体外系副作用。多潘立酮表现出胃动力和止吐活性,并通过作用于胃中的外周多巴胺受体来发挥其胃动力学作用。多潘立酮用作多巴胺D2和D3受体的外周选择性拮抗药,并起阻断记录恶心的多巴胺受体的作用。多潘立酮可以阻断幽门窦和十二指肠中的多巴胺受体,以增加上消化道中的动力。多潘立酮还可阻断脑下垂体中的多巴胺受体,这可增加催乳素的释放,导致泌乳增加,因此用于治疗泌乳不足。已评估多潘立酮用于治疗恶心和呕吐、胃轻瘫、帕金森氏病、功能性消化不良、泌乳不足、小儿反流以及其他胃肠动力障碍或病症。
发明内容
一个实施例是一种通过给予在未氯化的芳环中经4个氘氘代的多潘立酮(d4)或在连接丙基中经6个氘氘代的多潘立酮(d6)来改善胃轻瘫、恶心(作为与胃轻瘫分开或与其相关的疾病)、呕吐(作为与胃轻瘫分开或与其相关的疾病)和/或泌乳不足的治疗方法。在一个实施例中,给予多潘立酮-d4并且多潘立酮-d4优于多潘立酮-d6。
一个实施例是一种通过使具有0至4个氘的1,2-二氨基苯与反应性羰基物质反应以产生环酰亚胺,使环酰亚胺与保护基团反应以产生单保护的环酰亚胺,使单保护的环酰亚胺与具有0至6个氘的1,3-双官能丙基衍生物反应以产生中间体,使中间体与5-氯-1-(4-哌啶基)-1,3-二氢-2H-苯并咪唑-2-酮,并在中间体与5-氯-1-(4-哌啶基)-1,3-二氢-2H-苯并咪唑-2-酮反应之前或之后除去保护基团来制备氘代多潘立酮的方法。
附图说明
图1A-1B示出了口服给药的多潘立酮和氘代多潘立酮的血浆曲线(图1A)和图1A所示数据的分析(图1B)。
图2示出了速释(IR)制剂和缓释(XR)制剂的代表性药物释放曲线。
图3是来自速释部分和缓释部分的氘代多潘立酮的模拟血浆曲线。
图4示出了具有IR层和ER层的双层片剂。
图5示出了包含IR层、ER层以及缓冲层的三层片剂。
图6示出了具有ER基质和IR包衣的片剂。
图7示出了包含IR片剂、塞子以及具有渗透系统的ER片剂的胶囊。
图8示出了包含IR珠粒和ER珠粒的胶囊。
图9示出了包含IR迷你片剂和ER迷你片剂的胶囊。
图10示出了包含IR颗粒和ER颗粒的胶囊。
图11示出了包含包覆有IR层的ER珠粒的胶囊。
图12示出了包含IR颗粒和包衣ER片剂的压制片剂,IR颗粒和包衣ER片剂嵌入在压制片剂内。
图13示出了ER片剂嵌入在IR片剂内的的压制IR片剂。
图14示出了混悬在IR液体中的ER片剂。
图15示出了包含IR和ER颗粒或珠粒混合物的小药囊。
图16示出了包含泡腾IR颗粒或珠粒和包衣ER颗粒或珠粒的小药囊。
图17示出了中间层由环带分开的片剂。
图18示出了包含包衣延迟/ER药物颗粒、珠粒或颗粒的口腔崩解片;插图示出了聚合物基质中的药物。
图19示出了包含药物溶液和包衣延迟/ER药物颗粒、珠粒或颗粒的胶囊。
图20示出了包含药物溶液和包衣延迟/ER药物颗粒、珠粒或颗粒的软凝胶。
图21示出了包含包衣延迟/ER药物颗粒、珠粒或颗粒的液体载体。
具体实施方式
像所有药物一样,多潘立酮的安全性取决于其代谢。代谢降低使得药物在体内具有更长的停留时间。治疗胃轻瘫所需的多潘立酮剂量,即10mg,每天服用三次,共30mg,最多60mg,可导致心脏QT延长,这与剂量有关。由于这种影响,它在美国未被批准用于该适应症,但在欧洲和加拿大获得了批准。
多潘立酮被广泛代谢;其主要代谢途径产生5-羟基形式、N-脱烷基化形式以及羟基化形式。因此,降低多潘立酮代谢的方法将允许给予较低剂量以在患者中达到相同程度的疗效,从而降低或消除强心作用,和/或减少需要给予的剂量数,和/或提供可以提高患者耐受性或疗效的更加一致的暴露接触。
多潘立酮的总峰值血浆水平取决于其给药途径。在禁食个体中肌肉内(IM)和口服给药导致给药10分钟和30分钟后达到峰值血浆水平;栓剂给药导致给药1至2小时后达到峰值血浆水平。口服与IM给药两小时后血浆浓度更低,可能是由于肝脏首过效应和肠壁代谢。在10mg IM注射之后,在10mg片剂口服摄入之后以及在60mg片剂或滴剂口服摄入之后,峰值血浆浓度为40ng/ml,20ng/ml以及70至100ng/ml。10和100ng/ml氚代多潘立酮的人血浆蛋白结合分别为91.7%和93.0%。生物利用度,在IM注射之后相对较高,为90%,在口服之后相对较低,为13至17%,甚至由于胃酸pH值升高,因抗酸药使用而进一步降低。处方药:药物信息- Janssen-Ortho,马来酸多潘立酮。
Janssen-Ortho,马来酸多潘立酮。
与用于中枢神经系统(CNS)模型的经典配体氚代螺哌隆比较,多潘立酮选择性地和特异性地结合纹状体多巴胺受体。然而,静脉内(IV)多潘立酮给药,即使在高剂量下,由于其血脑屏障渗透性差(Reddymasu等人,《美国胃肠病学杂志(Am.J.Gastroenterology)》),在动物脑模型中不能置换标记的螺哌隆。多潘立酮对胃肠组织也有很高的亲和力;在食管、胃部和小肠中发现高浓度。多潘立酮阻断胃肠道转运的多巴胺能抑制。它很快被肝脏代谢;口服之后,32%在尿液中排泄,66%在粪便中排泄。健康个体的消除半衰期为7.5小时,肾功能障碍个体的消除半衰期为约三倍于前者。
多潘立酮的疗效是基于其能够增加食管运动功能的幅度,增强胃窦-十二指肠的收缩并更好地协调幽门的蠕动和随后的胃排空加速。多潘立酮在血脑屏障之外的脑的第四脑室中的化学感受器触发区(CTZ)具有有效的止吐活性。它没有胆碱能活性,不受阿托品的抑制。
多潘立酮调节固体和液体的胃排空,并且不改变胃酸分泌、分泌量、胃内pH值或血清胃泌素浓度。
以口服10至30mg的剂量,在餐前半小时和睡前服用,每日四次,多潘立酮显著地减少了胃肠症状和因胃轻瘫导致的入院治疗,对呕吐和恶心的中枢控制具有积极影响,并且加速了固体膳食的排空(Buckels等人,《医景一般内科(Medscape General Medicine.)》2003;5(4)www.medscape.com)。
一种降低多潘立酮代谢的方法是给予氘代形式的多潘立酮。多潘立酮被广泛代谢。氘代减缓了关键位点和代谢途径的代谢,从而以较低剂量产生较高的疗效。氘代形式的小分子增加其存留并因此降低其代谢,从而允许给予较低剂量,同时实现与较高剂量相同的疗效而没有心脏症状。
氘代多潘立酮可以通过几种途径合成或制备。通常,多潘立酮的化学结构中的任何氢可以是氢或氘。有利地,多潘立酮代谢的位点,即芳环上的所有非取代位点,可以通过加入市售氘代烷基卤化物来阻断,并且代谢物可以通过针对丙基连接基团加入市售氘代烷基卤化物来阻断。这可以显著降低多潘立酮的代谢并改善其生物利用度。
药物的氘代增加了药物半衰期,从而允许较低频率的给药以及改善的药代动力学,即吸收、分布和代谢。使用动力学同位素效应(KIE)和氘动力学同位素效应(DKIE)将氘引入药物中。Gant,《药物化学杂质(J.Med.Chem.)》(2014)57,3595-3611。
相较于氢,氘与碳形成更稳定的键。在一些情况下,氘取代可能会改变药物代谢。改变的药物代谢可以采取许多形式,例如代谢物稳定性改善,毒性代谢物形成减少和/或活性代谢物形成增加。与相应的非氘代形式相比,氘代化合物可能具有增加的半衰期和增加的全身性暴露接触。增加的半衰期和降低的代谢可以提供增强的疗效、耐受性、安全性和便利性,使得较低剂量的氘代形式可以产生与较高剂量的非氘代形式相似的结果。氘代化合物通常保留与相应的非氘代形式相同的生物化学效力和选择性。氘取代对代谢参数的任何影响高度依赖于氘取代氢的特定分子位置。
即使在具有相似化学结构的化合物中,氘取代的代谢效应不明显,也不可预测。例如,美国公开第2009/0076010号公开了富含氘的拉莫三嗪(一种抗惊厥药)。氘代率为14至100%,取决于氢被氘取代的位置。富集方法可以是通过与氘进行质子交换,或通过与富含氘的起始材料的分子合成。美国公开第2009/0131485号公开了富含氘的吡非尼酮(一种响应于细胞因子阻断成纤维细胞增殖和刺激的胶原产生抑制剂),用于治疗神经纤维瘤病、多发性硬化症和其他纤维瘤相关疾病。它公开了合成方法和同位素,以及用于增强生物利用度和给药的方法。氘代吡非尼酮的半衰期为110至140%或更高,取决于氘代程度。与非氘代形式相比,有效量为80至40%或更低。美国公开第2011/0160253号公开了富含氘的替扎尼定(一种中枢作用性α2-肾上腺素受体激动剂的苯并噻唑,用于控制与多发性硬化症、脊髓损伤等相关的肌张力过度和肌肉痉挛)。它公开了氘代方法;还公开了52.5至99.5%的富集范围和药物组合物、有效量和剂量。Harbeson和Tung,《药物化学消息2号(MECHEM NEWSNo.2)》2014年5月8-22公开了氘取代可以提高治疗剂的安全性、疗效和/或耐受性。
氘代药物已用于非临床环境中,并用作代谢和药代动力学探针,但均未被批准用作人类治疗剂。取决于期望的氘代位点,D2O可以直接交换入成品药物化合物或用于合成药物的试剂中。氘具有较低的全身毒性。氘气可以用作掺入氘的起始材料。烯键和炔键的催化氘代可以快速掺入氘。在氘气存在下,金属催化剂(如Pd、Pt和Rh)可用于在含烃的官能团中直接将氢交换为氘。氘代试剂和合成结构单元可以在市场上买到。分子的形状和大小在氘代和非氘代形式中非常相似。部分或完全氘代的化合物的微小物理性质变化是疏水性降低、羧酸和酚的酸性下降以及胺的碱性增加,但大多数这些微小差异对生物化学效力或靶选择性的影响可以忽略不计。结合同位素效应是众所周知的,并且可以对测量的氘动力学同位素效应产生正面或负面的贡献。代谢率和代谢转换也有所减少,其中代谢物的比例发生变化。个体与母体药物的暴露接触以及代谢物的变化可能会影响氘代药物的药代动力学、耐受性和疗效。氘代减少不需要的或有毒的代谢物的形成,以及增强所需代谢物的形成。代谢分流的积极作用的一个实例是氘代奈韦拉平,其使皮疹发病率和严重程度降低,并且氘代依法韦仑、茚地普隆和奥当卡替中的每一者在大鼠模型中都导致较低的副作用且具有增强的疗效。氘代罗非考昔(也称为BDD-11602)与非氘代罗非考昔相比,在大鼠模型中具有改善的药代动力学特征。氘代特拉匹韦(一种丙型肝炎病毒NS3-4A蛋白酶抑制剂)差向异构化率增加13%,但对抗病毒活性的影响可以忽略不计。对任何特定药物的代谢特征的氘代影响都是不可预测的,尽管有可能改善安全性、耐受性、疗效和给药。
一种降低多潘立酮代谢的方法是给予氘代形式的多潘立酮。氘代形式的小分子将增加其存留并因此降低其代谢,从而允许给予较低剂量但达到与较高剂量相同的疗效而没有心脏症状。多潘立酮被广泛代谢,因此氘代减缓了关键位点和关键代谢途径的代谢,从而以较低的剂量产生较高的疗效。这在随后描述的图1A和1B中示出,其中氘代化合物的曲线下面积更高。随后描述的表1显示了60分钟时间内化合物2的代谢物形成的减少。
下面的化合物1显示了一般氘代形式的多潘立酮,其中R=H或D,独立地允许存在1至10个氘。可以没有限制地使用氘代位点的任何和所有排列。最重要的氘代位点是所示的芳族化合物以及哌啶氮的烷基连接基团α的亚甲基。主要的羟基化代谢物存在于这个芳环中,氘的存在将会降低这些代谢物形成的速度。脱烷基化代谢途径通过N-氧化物的消除机理涉及那些α质子;氘的消除速度较慢,从而减缓了这种代谢途径。在一个实施例中,如本文所述使用和/或制备化合物1的氘代化合物。
多潘立酮可能在任何氢位点被氘代。在化合物2中,未取代芳环的芳香H原子的氘代产生了多潘立酮-d4。额外的连接丙基中一个或多个亚甲基位点的H的氘代产生了化合物3多潘立酮-d6或多潘立酮-d8(未示出)或化合物5多潘立酮-d10。也可以氘代一个或多个亚甲基位点的H,同时保留芳香位点的H。例如,一个亚甲基基团的H的氘代产生了化合物4多潘立酮-d2,两个亚甲基基团的H的氘代产生了多潘立酮-d4(未示出),并且所有三个亚甲基基团的H的氘代产生了化合物6多潘立酮-d6。
在一个实施例中,给予化合物2、3和/或4中的任一种。化合物2、3和4中的任一种都会影响代谢速度的降低,并且化合物2、3和4中的任一种是优选的。
仅在α亚甲基处具有氘的原料可能比丙基完全氘代的原料更昂贵且不易获得。
在一个实施例中,使用化合物2,并且化合物2是优选的。
在一个实施例中,使用化合物5,并且化合物5是优选的。
在一个实施例中,使用化合物6,并且化合物6是优选的。
在一个实施例中,使用化合物2、5和6,并且化合物2、5和6是优选的。

在一个实施例中,使用环己基胺中含1至8个氘的化合物,例如化合物13。

方案1显示了制备通常被示为化合物1的各种氘代多潘立酮化合物的一般合成。合成基于Vandenberk的美国专利第4,066,772号中的非氘代类似物。该工艺始于在芳环上用0至4个氘取代1,2-二氨基苯。在合适的溶剂中使用氯甲酸乙酯或其他类似的反应性羰基物质封闭酰亚胺,该合适的溶剂例如但不限于醚(如四氢呋喃(THF))、卤代烃(如二氯甲烷)、酮(如丙酮)、烃(如庚烷)以及酰胺(如N,N-二甲基甲酰胺(DMF))。然后用合适的保护基团(例如但不限于氨基甲酸酯、磺酰胺和乙烯基烷基)对化合物8进行单保护。然后第二个氮与包含0至6个氘的1,3-双官能丙基衍生物反应。官能团可以独立地为卤素(Br、Cl、I)、羟基或合适的离去基团(如甲苯磺酸酯或甲磺酸酯)。在一个优选实施例中,两个基团是不同的。化合物10的剩余离去基团X可以任选地交换为更具反应性的物质,例如,如果X是氯化物,则氯化物可以交换为碘化物,或者如果X是甲苯磺酸酯,可以将甲苯磺酸酯交换为三氟甲基磺酸酯(三氟甲磺酸酯)。保护基团P也可以在此步骤中被除去。
然后使用碱或其他合适的偶联方法使中间体10或12与化合物14反应。使用本领域已知的方法来制备化合物14。除去保护基产生了所需的氘代多潘立酮衍生物1。或者,可以在与14烷基化之前除去保护基团P。所有化合物根据该一般方案使用适当取代的氘代起始材料来制备。
化合物13的合成遵循针对非氘代材料的已知方法,但是以氘代4-氨基哌啶衍生物开始。
方案1:氘代多潘立酮的一般合成

图1示出了这种降低的代谢,其中相对于未氘代多潘立酮,氘代多潘立酮化合物2和6的口服多潘立酮的大鼠血浆水平的曲线下面积更高。下表示出了相对于未氘代多潘立酮,氘代多潘立酮化合物2和6在60分钟时间内在人肝细胞中的代谢物形成的减少。
表1:人肝细胞中代谢物形成的百分比


另一种降低多潘立酮代谢的方法是在舌下给药,以使活性剂在循环系统中立即可用,并绕过代谢发生的消化系统。配制成片剂、薄膜或其他合适制剂的舌下形式的多潘立酮或氘代多潘立酮可以以较低剂量给予,但具有与口服形式相当的疗效。多潘立酮的药代动力学,特别是其t1/2、pKa、log P以及Kd,使其有利于舌下给药。
另一种降低多潘立酮代谢的方法是以提供增加的表面积的颗粒形式给予活性剂。例如,可以将多潘立酮或氘代多潘立酮配制成微粒或纳米颗粒。使用生物药剂学分类系统(BCS),已知II类药物具有高渗透性和低溶解度,使得它们的生物利用度受溶剂化速率限制。在该实施例中,提供增加的表面积的微粒或纳米颗粒或其他制剂通过增加溶剂化速率来增加生物利用度,并且可以以较低剂量给予,但具有与口服形式相当的疗效。
应理解的是,其他制剂可以获得相似或相同的结果,例如使用喷雾制剂、粉末、薄膜等,并使用多潘立酮或氘代多潘立酮。
应该理解的是,这些实施例可以组合使用。作为一个实例,可以将微粒或纳米颗粒形式的多潘立酮应用于或掺入薄膜中并舌下给予。作为另一个实例,可将氘代形式的多潘立酮配制为微粒或纳米颗粒,并且在一些实施例中可以舌下给予,例如在薄膜中或薄膜上,作为喷雾剂等。在这些组合实例中,多潘立酮的剂量可能由于其增加的生物利用度和降低的代谢而进一步降低。
以下配剂和制剂可用于氘代或非氘代形式的多潘立酮。
纳米颗粒可以使用干磨或湿磨来制备。干磨工艺的实例包括美国专利公开第2013/0209569号、第2010/0092563号、第2014/0287039号、第2014/0200276号、第2014/0194487号、第2014/0255494号、第2013/0243854号、第2014/0248359号、第2014/0256781、第2014/0302127号、第2014/019395号、第2014/0220121号、第2012/0135048号、第2014/0326812号、第2009/0028948以及美国专利第9,089,471号;第9,095,496号;第9,180,096号;第9,173,854号;第9,017,721号;第8,679,544号;第8,999,387号;第8,734,847号;第8,992,982号;第9,180,096号;第9,186,328号;第8,735,450号和第8,808,751号。美国专利第9,107,827号公开了一种示例性湿磨工艺。这些制剂中的任何一种,包括但不限于薄膜、片剂、喷雾剂、溶液等,都包括舌下剂型。
多潘立酮,无论是氘代形式还是非氘代形式,都可通过皮肤给药,即透皮给药。通过皮肤的吸收(也称为经皮递送、透皮递送或皮肤递送)将多潘立酮从外表皮表面转运进入皮肤并进入全身循环。表皮表面是透皮递送的主要吸收途径,尽管少量的试剂也可以通过毛囊或腺体转移。从表皮起始表面开始,试剂在进入真皮之前通过七个表皮层,试剂从该表皮进入循环系统和/或淋巴系统。角质层是最外面或表面暴露接触的皮肤层,并且是外部试剂进入的限速障碍,因此穿过角质层的速率决定了整体吸收。主要的角质层组分是亲脂性化合物胆固醇、胆固醇酯和神经酰胺。因此,与具有较少脂质溶解度的试剂相比,具有较高脂质溶解度的试剂更快地穿透角质层并实现全身性暴露接触,但大部分试剂都可以一定程度穿透角质层。多潘立酮的溶解度与其pH值有关;多潘立酮是一种弱碱,pKa 7.89,在脂质与水之比为3.90的水中溶解度有限。
角质层的健康和完整性影响试剂渗透。例如,损坏或破坏角质层组成的诸如强酸之类的试剂被快速吸收。由于烧伤、擦伤、伤口和疾病引起的皮肤损伤也会影响吸收。一些溶剂,例如二甲亚砜(DMSO),增加了角质层的渗透性,从而充当载体并因此用作渗透增强剂或促进剂。一些表面活性剂,例如十二烷基硫酸钠,增加了水溶性试剂的皮肤渗透性,从而可能增加了皮肤对水的渗透性。
透皮递送可以通过局部给予、环境暴露接触和/或注射来实现。通过皮肤的吸收取决于试剂因素,包括但不限于浓度、分子量和亲脂/亲水性质或溶解度,还有暴露接触持续时间、皮肤的物理状态、暴露的表面以及暴露的表面上是否存在毛囊。例如,由于具有防止下层组织水分流失作用的角质层的角化分层鳞状细胞层,根据以下方案从各种皮肤表面发生试剂吸收,从最快到最慢依次为:阴囊>额头>腋下>头皮>背部>腹部>手掌/足底。
多潘立酮的皮肤施用可以允许更多的局部治疗,并且可以避免或最小化首过肝脏代谢。因此,皮肤给药可以达到较高的全身浓度。皮肤给药制剂包括贴剂、洗剂、擦剂、软膏剂、酊剂、乳膏剂、粉剂、气雾剂、凝胶等。在一个实施例中,贴剂可以控释并且可以允许多潘立酮释放7天。贴剂可以包含渗透增强剂,其可以促进递送或在一些情况下对于递送很重要。在透皮贴剂中,多潘立酮或前药形式的多潘立酮以游离碱和/或盐形式存在。
通过透皮施用吸收的多潘立酮的量可以使用本领域已知的方法和如后面所述的方法直接或间接测量。即使在体内研究中,物质差异也可能是显著的。而且,之前描述的不同制剂可影响所递送的浓度。直接浓度测量可以使用体内方法进行,即将多潘立酮直接施用于皮肤并在设定的时间测量血液和尿液中的多潘立酮,然后将结果绘制在图上并测量曲线下面积(AUC)。可以使用体外方法,因为当小心地除去皮肤时,角质层的渗透性没有显著改变。已知室内研究,使用任何多潘立酮制剂(例如薄膜、贴剂、洗剂等,参见例如Basu,《国际药物研究杂志(IRJP)》3(2012)134-45;Madishetti等人《Daru》18(2010)221-29;Khan等人《药物研究公报(Bull.Pharm.Res.)》2(2012)15-21),施用于孤立的皮肤样品的一侧表面并测量在该样品的另一侧表面上测量其浓度,例如,孤立的灌注猪皮瓣(Riviere,《基础与应用毒物学(Fundam Appl Toxicol)》7(1986)444-53)。体外方法包括静态和流通式扩散池,其实例分别是Franz扩散池和Bronaugh扩散池(Bronaugh和Stewart,《药物科学杂志(J.Pharm.Sci)》,74(1985)64-67)。静态Franz扩散池设备具有上部供应室和下部接收室,该下部接收室包含流体,其中上部室和下部室被作为膜的皮肤样品分隔。下部室中的接收流体通常是缓冲盐水,包含已知量的蛋白质,例如牛血清白蛋白或生物流体,并且与皮肤膜接触。在使用中,将载体中已知体积和浓度的多潘立酮施用到上部室并渗透穿过皮肤膜、扩散或以其他方式进入下部室中的接收流体。通常通过取样口(该取样口还取代了除去的流体体积)对该接收流体取样,并定期分析以确定透过皮肤膜的多潘立酮的量。流通型Bronaugh扩散池设备与Franz扩散池设备类似,只是在下部室中使用了流通系统,从下部室中获得样品并连续分析而不是在设定的时间点进行分析。
美国公开第2010/0255096号中公开了多潘立酮透粘膜递送的方法。粘膜粘附递送技术提供了试剂的(如多潘立酮)安全有效的递送。粘膜粘附递送技术包括在口腔粘膜中扩散的所有方法:包括跨细胞(通过细胞)和细胞旁(通过细胞周围的富含脂质的结构域)的被动扩散,载体介导的转运以及活性细胞摄取的内吞作用/胞吐作用并通过内吞途径的排泄。
黏膜、粘膜、线体腔或者外部暴露接触于环境中或者是内部器官。口腔粘膜是被覆于口腔内部的黏膜,由层状鳞状上皮(口腔上皮)和下面的结缔组织(固有层)组成。根据功能和组织学可将其进一步分为三大类:在舌背、硬腭和附着龈上发现的角化分层鳞状上皮的咀嚼粘膜;几乎遍布口腔内部发现的非角化鳞状上皮的被覆粘膜,包括被覆面颊的颊粘膜、嘴唇内侧的唇粘膜以及牙龈之间的粘膜(即牙槽粘膜)和颊/唇粘膜;以及在舌背侧的舌乳头上的味蕾区域中的特殊粘膜。生物粘附聚合物可粘附到任何潮湿的表面,因此粘膜粘附/生物粘附制剂粘附到唾液润湿的角化和非角化粘膜。
下面提供示例性透皮制剂,所有百分数均为重量/重量。虽然以下制剂列出了多潘立酮,但本领域普通技术人员知道,多潘立酮可能是指多潘立酮或氘代多潘立酮。
制剂1
多潘立酮 1%
二甲亚砜 足量至100%
制剂2
多潘立酮 1%
癸二酸二乙酯 15%
二甲亚砜 足量至100%
制剂3
多潘立酮 1%
己二酸二异丙酯20%
二甲亚砜 足量至100%
制剂4
多潘立酮 1%
异山梨糖醇二甲醚15%
二甲亚砜 足量至100%
制剂5
多潘立酮 1%
二丙二醇 10%
二甲亚砜 足量至100%
制剂6
多潘立酮 1%
己二醇 12%
二甲亚砜 足量至100%
制剂7
多潘立酮 1%
碳酸丙烯酯 5%
二甲亚砜 足量至100%
制剂8
多潘立酮 1%

二甲亚砜 足量至100%
制剂9
多潘立酮 1%
月桂酸二乙醇酰胺15%
二甲亚砜 足量至100%
制剂10
多潘立酮 1%
PEG 400 20%
二甲亚砜 足量至100%
制剂11
多潘立酮 1%
椰子酰胺 5%
二甲亚砜 足量至100%
制剂12
多潘立酮 1%
油酸 20%
二甲亚砜 足量至100%
制剂13
多潘立酮 1%
PEG-7甲醚 20%
二甲亚砜 足量至100%
制剂14
多潘立酮 1%
聚山梨酯80 15%
二甲亚砜 足量至100%
制剂15
多潘立酮 1%

二甲亚砜 足量至100%
制剂16
多潘立酮 1%

二甲亚砜 足量至100%
制剂17
多潘立酮 1%

二甲亚砜 足量至100%
制剂18
多潘立酮 1%
棕榈酸异丙酯 8%
二甲亚砜 足量至100%
制剂19
多潘立酮 1%
乙酰丙酸 5%
二甲亚砜 足量至100%
制剂20
多潘立酮 1%
月桂醇乳酸酯 5%
二甲亚砜 足量至100%
制剂21
马来酸多潘立酮1%
二甲亚砜 足量至100%
制剂22
马来酸多潘立酮1%
癸二酸二乙酯 15%
二甲亚砜 足量至100%
制剂23
马来酸多潘立酮1%
己二酸二异丙酯20%
二甲亚砜 足量至100%
制剂24
马来酸多潘立酮 1%
异山梨糖醇二甲醚15%
二甲亚砜 足量至100%
制剂25
马来酸多潘立酮1%
二丙二醇 10%
二甲亚砜 足量至100%
制剂26
马来酸多潘立酮1%
己二醇 12%
二甲亚砜 足量至100%
制剂27
马来酸多潘立酮1%
碳酸丙烯酯 5%
二甲亚砜 足量至100%
制剂28
马来酸多潘立酮1%

二甲亚砜 足量至100%
制剂29
马来酸多潘立酮 1%
月桂酸二乙醇酰胺15%
二甲亚砜 足量至100%
制剂30
马来酸多潘立酮1%
PEG 400 20%
二甲亚砜 足量至100%
制剂31
马来酸多潘立酮1%
椰子酰胺 5%
二甲亚砜 足量至100%
制剂32
马来酸多潘立酮1%
油酸 20%
二甲亚砜 足量至100%
制剂33
马来酸多潘立酮 1%
PEG-7methyl ether 20%
二甲亚砜 足量至100%
制剂34
马来酸多潘立酮1%
聚山梨酯80 15%
二甲亚砜 足量至100%
制剂35
马来酸多潘立酮 1%

二甲亚砜 足量至100%
制剂36
马来酸多潘立酮 1%

二甲亚砜 足量至100%
制剂37
马来酸多潘立酮1%

二甲亚砜 足量至100%
制剂38
马来酸多潘立酮1%
棕榈酸异丙酯 8%
二甲亚砜 足量至100%
制剂39
马来酸多潘立酮1%
乙酰丙酸 5%
二甲亚砜 足量至100%
制剂40
马来酸多潘立酮1%
月桂醇乳酸酯 5%
二甲亚砜 足量至100%
制剂41
马来酸多潘立酮 1%

二甲亚砜 足量至100%
制剂42
马来酸多潘立酮 1%


二甲亚砜 足量至100%
制剂43
马来酸多潘立酮 1%

己二醇 12%
二甲亚砜 足量至100%
制剂44
马来酸多潘立酮 1%


二甲亚砜 足量至100%
制剂45
马来酸多潘立酮 1%


二甲亚砜 足量至100%
制剂46
马来酸多潘立酮 1%

癸二酸二乙酯 20%
二甲亚砜 足量至100%
制剂47
马来酸多潘立酮 1%

己二酸二异丙酯 20%
二甲亚砜 足量至100%
制剂48
马来酸多潘立酮 1%

乙酰丙酸 10%
二甲亚砜 足量至100%
制剂49
马来酸多潘立酮 1%

月桂醇乳酸酯 10%
二甲亚砜 足量至100%
制剂50
马来酸多潘立酮 1%

聚山梨酯80 20%
二甲亚砜 足量至100%
制剂51
马来酸多潘立酮 1%



二甲亚砜 足量至100%
制剂52
马来酸多潘立酮 1%


己二醇 12%
二甲亚砜 足量至100%
制剂53
马来酸多潘立酮 1%



二甲亚砜 足量至100%
制剂54
马来酸多潘立酮 1%


己二酸二异丙酯 20%
二甲亚砜 足量至100%
制剂55
马来酸多潘立酮 1%


月桂醇乳酸酯 10%
二甲亚砜 足量至100%
制剂56
马来酸多潘立酮 1%



二甲亚砜 足量至100%
制剂57
马来酸多潘立酮 1%

己二醇 12%

二甲亚砜 足量至100%
制剂58
马来酸多潘立酮 1%

癸二酸二乙酯 20%

二甲亚砜 足量至100%
制剂59
马来酸多潘立酮 1%

己二酸二异丙酯 20%

二甲亚砜 足量至100%
制剂60
马来酸多潘立酮 1%

月桂醇乳酸酯 10%

二甲亚砜 足量至100%
制剂61
马来酸多潘立酮 1%



癸二酸二乙酯 10%
二甲亚砜 足量至100%
制剂62
马来酸多潘立酮 1%


癸二酸二乙酯 20%
二甲亚砜 足量至100%
制剂63
马来酸多潘立酮 5%


癸二酸二乙酯 20%
二甲亚砜 足量至100%
制剂64
马来酸多潘立酮 10%


癸二酸二乙酯 20%
二甲亚砜 足量至100%
制剂65
马来酸多潘立酮 1%



癸二酸二乙酯 5%
柠檬烯 5%
二甲亚砜 足量至100%
制剂66
马来酸多潘立酮 1%

柠檬烯 5%
二甲亚砜 足量至100%
制剂67
马来酸多潘立酮 1%



癸二酸二乙酯 5%
二甲亚砜 足量至100%
制剂68
多潘立酮 1%



癸二酸二乙酯 5%
二甲亚砜 足量至100%
制剂69
马来酸多潘立酮 1%



癸二酸二乙酯 5%
柠檬烯 5%
二甲亚砜 足量至100
一个实施例是一种以舌下剂型提供的用多潘立酮或氘代多潘立酮或其药学上可接受的盐治疗胃轻瘫的组合物和方法。舌下剂型的实例包括舌下片剂、生物相容性薄膜和舌下喷雾剂。例如,舌下片剂可以制备成快速崩解片(RDT)。RDT是包含药物的固体剂型,当置于舌上或舌下时,即在与唾液接触时,药物快速(≤30秒)崩解。在RDT中配制多潘立酮或氘代多潘立酮可以实现口服多潘立酮,不用水并且不用咀嚼。市售的RDT技术是冻干片、压制片、模制片、喷雾干燥粉末、薄膜和棉花糖体系(McLaughlin等人,《制药技术(Pharmaceutical Technology)》,2009年9月号副刊)。
一个实施例是一种在生物相容性纳米颗粒制剂中用多潘立酮或氘代多潘立酮或其药学上可接受的盐治疗胃轻瘫的组合物和方法。在一个实施例中,将多潘立酮或氘代多潘立酮配制用于局部施用。在一个实施例中,多潘立酮或氘代多潘立酮直接或间接配制在薄膜中和/或薄膜上。例如,可以将多潘立酮或氘代多潘立酮配制成包含在薄膜基质中,或者可以配制成薄膜的一层,或者可以配制在施加到薄膜上的载体中。该载体可以是混悬液、泡沫、乳液等。在一个实施例中,将多潘立酮或氘代多潘立酮配制成局部施用的泡沫。在一个实施例中,将多潘立酮或氘代多潘立酮配制在纳米颗粒之中、之上或与纳米颗粒缔合。在其他实施例中,将多潘立酮或氘代多潘立酮配制成固体片剂,或者配制成液体(例如糖浆、混悬液、溶液或乳液),或配制成注射剂。
薄膜制剂包括但不限于美国专利公开第2014/0271788号、第2014/0272220号、第2014/0271787号、第2014/0163060号、第2014/0070440号、第2014/0017299号、第2013/0333831号和第2013/0220526号。
纳米颗粒制剂包含任何纳米尺寸的结构,包括但不限于量子点(包括石墨烯量子点、氧化石墨烯量子点和石墨烯氧化锌量子点)、纳米管(包括石墨烯纳米管和/或碳纳米管)、富勒烯,巴基球、树状聚合物、脂质体、适体、胶束等。
该制剂提供了即用的患者依从性和最佳剂量递送以治疗胃轻瘫的症状。应该理解的是,其他的胃动力障碍也可以用所公开的组合物和方法治疗。“药学上可接受的”是指从药理学/毒理学优势方面对于患者可接受的性质和/或物质,并且对于制造药物化学家指从组合物、制剂、稳定性、患者接受度和生物利用度相关的物理/化学优势方面对于患者可接受的性质和/或物质。
药学上可接受的盐包括与药学上可接受的酸或碱(例如无机酸(例如,盐酸、硫酸、磷酸、二磷酸、氢溴酸、氢碘酸和硝酸)和有机酸(例如,柠檬酸、富马酸、马来酸、苹果酸、扁桃酸、抗坏血酸、草酸、琥珀酸、酒石酸、苯甲酸、乙酸、甲磺酸、乙磺酸、苯磺酸、环己氨磺酸(环拉酸)或对甲苯磺酸))的盐。药学上可接受的碱包括碱金属(例如,钠或钾)、碱土金属(例如,钙或镁)、氢氧化物以及有机碱(例如,烷基胺、芳烷基胺和杂环胺)。
制剂可以以固体形式口服给药,例如片剂,胶囊、锭剂或胶剂,或以液体形式口服给药,例如水性或非水性载体中的糖浆、乳液、溶液或混悬液。在固体形式中,制剂可以控释或快速溶解以快速起效。制剂也可以通过注射给药,其可以是皮下、皮内、肌内、静脉内或其他注射方法。通过注射给药的制剂可以包括在水性或非水性载体中的混悬液、溶液或乳液。其他制剂可以鼻内、阴道、直肠或透皮递送。制剂也可以透粘膜递送。该配剂可以每天一次至每天四次给药。
在一个实施例中,将多潘立酮或氘代多潘立酮溶液或混悬液放入泡罩包装中并冻干以制备单位剂量。冻干混悬液可以包含规则颗粒、微粉化颗粒或纳米颗粒。以下冻干技术平台包括 (Catalent)、LYOCTM(Cephalon)、
(Catalent)、LYOCTM(Cephalon)、 (SPIPharmaceuticals)和
(SPIPharmaceuticals)和 (Janssen)。
(Janssen)。
对于用于舌下给药的冻干RDT产品,将作为活性药物成分(API)的多潘立酮或氘代多潘立酮分散在溶于水中的聚合物结构形成物(例如明胶)和糖类(通常为甘露糖醇)的基质中。在成品中,聚合物组分的玻璃状无定形结构赋予了强度和弹性,同时保持了一定的柔性。特定的明胶等级及其相关的溶解特性确保口腔中的顺利且快速的融化。甘露糖醇在冷冻过程中结晶,从而提供优雅的外观和刚性,并确保产品在搬运和输送方面的稳固性。甘露糖醇容易溶解,因此也改善了质地、味道和口感。多潘立酮或氘代多潘立酮可以溶解在基质中或分散以形成用于给药的均匀混悬液。液体给药确保良好的剂量均匀性并且适应极低的剂量强度,例如微克。可以适应高达400mg的混悬液剂量强度,并且多潘立酮或氘代多潘立酮通常被微粉化。颗粒尺寸是一个考虑因素,因为>50μm的颗粒可能会有砂砾感。由于可溶性API降低了凝固点,溶液产品可以适应高达60mg的剂量强度。基于溶液和混悬液的产品都使用干燥单元的精细分散的多潘立酮或氘代多潘立酮,从而有助于快速分散和口感平滑。可以包含如随后所述的其他赋形剂。多潘立酮或氘代多潘立酮分散到预先形成的泡罩包装中并通过液氮快速冷却以快速冷冻。
冷冻导致在冻干期间升华的冰晶网络产生高度多孔结构。基质组分保持干燥单元的结构,但是在与水分接触时,高孔隙率导致快速渗水。基质快速溶解,导致迅速崩解(<10秒)。
冷冻之后,将多潘立酮或氘代多潘立酮冻干、定量并在密封的泡罩包装中干燥,从而提供物理和环境保护。产品稍微粘附在包装上,导致产品在泡罩袋内的最小移动,以确保输送期间的稳固性。该产品是一种晶片状结构,但具有最小的脆性和足够的强度,可从包装中取出而不会破碎。
冻干的RDT制剂的晶片状结构和高孔隙率反映出水通常是定量给药制剂的主要组分,因此干燥产品的重量显著降低并且通常主要由多潘立酮或氘代多潘立酮的剂量决定。RDT的推荐500mg重量限制可能接近冻干制剂中最高的多潘立酮或氘代多潘立酮剂量,由其快速崩解所抵消。
在舌上或舌下给药和快速分散之后,冻干制剂有效地恢复为原始的多潘立酮或氘代多潘立酮溶液或混悬液。因此,冻干的RDT片提供了固体口服剂型的便利性,具有溶液/混悬液产品的益处,适用于口腔和舌下摄入以直接提高生物利用度到全身循环中并且避免首过代谢以最小化不需要的代谢物,还具有物理和化学稳定性,保质期与常规片剂相当,即2至5年。

LYOCTM以水包油乳液起始,直接放入泡罩腔中,然后冷冻干燥。为了在冷冻过程中保持均匀性,必须包含聚合物以将基质粘度提高至几乎糊状稠度以防止沉降。增加的基质粘度降低了产品的孔隙率,从而增加了冷冻干燥时间并对崩解有负面影响。
如果本文没有指定,百分比是指重量/体积。
在一个实施例中,相对较低的速释是初始剂量,然后是缓释制剂的给予。在一个实施例中,10mg是低速释剂量,然后是24小时内的20至30mg缓释。
除非另外指出,否则制剂是剂型。片剂是剂型的非限制性实例。制剂的分散和崩解被同义使用。如本文所使用,缩写为“活性剂”的活性试剂包括多潘立酮、多潘立酮衍生物、多潘立酮类似物、氘代多潘立酮等。
活性剂包括所有形式的多潘立酮活性剂,包括氘代形式的多潘立酮,并且还包括但不限于中间体、代谢物、对映异构体、多晶型物、晶体结构、水合物、立体异构体、盐、碱、配合物、载体、类似物、衍生物和缀合物。如本文所使用,缓释和持续释放通常同义地使用。
珠粒和丸粒中的每一者是剂型的任何分立组分,例如,胶囊壳可以填充有多个珠粒和/或多个丸粒。
改良释放(MR)剂型包括但不限于以下:
速释制剂和延迟释放制剂表示与给药相关的活性剂释放的开始。速释制剂表示在给药之后相对较短的时间(例如,几分钟或更少)内开始从制剂中释放活性剂。延迟释放制剂表示不是在给药之后相对较短的时间内开始从制剂中释放活性剂,而是延迟了活性剂的释放并在给药之后相对较长的时间(例如,多于一小时)后开始或触发从制剂中释放活性剂。
快速释放制剂和缓慢释放制剂表示开始给药之后的释放速率。一旦活性剂的递送开始,活性剂可以相对快速或相对缓慢地释放。快速释放表示,在开始给药之后,在相对较短的时间内达到最大或峰值剂量。缓慢释放表示,在开始给药之后,在相对较长的时间内达到最大或峰值剂量。一旦达到,最大剂量可以任何速率(例如,快速、缓慢或恒速)下降。
持续释放制剂和连续释放制剂表示持续释放的时间,并且意味着在初始给药之后连续递送活性剂或将活性剂的递送持续很长一段时间(通常多于一小时),无论剂量释放曲线的形状如何。例如,在一段相对较长的时间内,将活性剂的释放持续在大于零的最大值和最小值之间。该释放可以是恒定剂量,或者随着时间的推移而减少的剂量。
恒定释放制剂表示正在释放的剂量。恒定释放意味着活性剂在中等长度时间或很长一段时间段内以相对恒定的剂量递送。这可以通过剂量释放曲线来表示,所述剂量释放曲线在初始给药之后相对平坦或仅缓慢倾斜,即没有非常明显的峰和谷。因此,恒定释放通常是持续的或连续的,但持续释放或连续释放可能不是恒定的。
脉冲释放制剂表示活性剂以一个或多个剂量递送,该剂量在一段时间内在最大剂量和最小剂量之间波动。这可以由具有一个或多个不同的峰或谷的剂量释放曲线表示。然而,两次或多次脉冲释放可能会产生重叠的、整体的或复合的释放曲线,其看起来是恒定的或者实际上即是恒定的。当发生两次或多次脉冲释放时,脉冲之间可能会存在或不存在一段无释放时间。通常,脉冲释放导致在约60分钟或更短的时间内释放基本上所有的活性剂。
缓释制剂在一段相对较长的时间内提供在目标剂量范围内的活性剂的释放,或在一段相对较长的时间内提供在目标剂量范围内的药物血浆水平,而不考虑释放的特定机制或性质,如持续、脉冲或恒定。
口服药物的释放曲线表示制剂在给药之后释放或递送活性剂到胃、肠等的方式和时间。已知各种方法来评估药物释放并产生释放曲线,包括模拟制剂的体内行为的体外试验,和包括速释和控释固体剂型的USP溶出测试的体外试验。
药物释放曲线与血浆曲线不同。血浆曲线指示哺乳动物(例如,接受药物制剂的患者)血流中的活性剂的剂量或水平。当从制剂释放活性剂,例如进入肠道时,可以确定随着时间流逝而变化的血流中的活性剂的存在量。
药物释放曲线可以被设计成产生期望或者目标血浆曲线,并且血浆曲线可以模拟释放曲线。例如,尽管预计活性剂的持续释放会在血浆中产生持续剂量,并且预计脉冲释放会产生脉冲(峰和谷)血浆曲线,但情况并非必然如此。血流中的活性剂的半衰期(t1/2)(其衰变速率)可能是这样的,即持续或连续的血浆曲线可能来自脉冲递送曲线。其他因素也可能起作用,如生物吸收、生物利用度和首过效应。由特定活性剂释放曲线产生的血浆曲线也可因患者而异。
生物利用度的测量是本领域已知的,包括血浆浓度-时间曲线下面积(AUC)、浓度最大值(Cmax)和Cmax达到时间(Tmax)。
AUC测量血浆浓度-时间曲线下面积,表示在给予单次剂量药物之后的药物吸收量(《雷明顿:药学的科学与实践(Remington:The Science and Practice of Pharmacy)》,编辑Gennaro 2000,第999页)。
Cmax是口服给药之后达到的最大血浆浓度(《雷明顿(Remington)》,第999页)。口服给药导致一个Cmax,但可导致多于一个峰值血浆浓度,例如在给予脉冲剂量制剂之后。
Tmax是口服给药之后达到Cmax所需的时间量,与活性剂的吸收速度有关(《雷明顿(Remington)》,第999页)。
“溶解度增强聚合物”或“结晶抑制聚合物”是指能够以适当浓度形成如本文所定义的固体分散体的弱碱性氯苯甲嗪在溶解度增强聚合物中的水溶性聚合物,例如通过首先将药物和聚合物溶解在相同的溶剂体系中,然后在合适的条件下除去溶剂。在包含溶解度增强聚合物和弱碱性药物的固体分散体的组合物的储存、运输和商业销售期间,弱碱性药物基本上被保持为分子分散体或无定形形式。
“控释”包衣包括延迟释放、持续释放、阻止释放和/或以其他方式延长药物从包覆有控释包衣的颗粒中释放。术语“控释”包括“持续释放”、“延迟释放”和“定时脉冲释放”,因此“控释包衣”包括持续释放包衣、定时释放包衣或“滞后时间“包衣。
“肠溶聚合物”是指对耐胃液的pH敏感聚合物(即,在胃中发现的低pH水平下相对不溶),并且其在肠道中发现的较高pH水平下溶解。
关于可以是剂型或剂型组分的药物组合物的“速释”是指在给予剂型之后约1小时内释放大于或等于约50%的活性剂的药物组合物,在另一个实施例中是指在给予剂型之后约1小时内释放大于约75%的活性剂的药物组合物,在另一个实施例中是指在给予剂型之后约1小时内释放大于约90%的活性剂的药物组合物,并且在其他实施例中是指在给予剂型之后约1小时内释放大于约95%的活性剂的药物组合物。该术语也可以指药物组合物,其中在滞后时间之后发生活性剂的相对快速的释放,活性剂在滞后时间内不释放或几乎不释放。
“速释(IR)珠粒”或“速释颗粒”广泛地指包含珠粒或颗粒的活性剂,其表现出相对于活性剂的“速释”性质。
“缓释(SR)珠粒”或“缓释颗粒”广泛地指包含置于含活性剂芯上的SR包衣的珠粒或颗粒。
“滞后包衣”或“定时脉冲释放包衣”(TRP)是指组合了非水溶性聚合物和肠溶聚合物的控释包衣;TPR包衣本身在预定滞后时间之后提供活性剂的速释脉冲。具有置于屏障包衣(SR包衣)上的TPR包衣的定时缓释(TSR)珠粒在预定滞后时间之后提供持续活性剂释放曲线。
“延迟释放(DR)珠粒”或“延迟释放颗粒”广泛地指含活性剂芯。DR包衣是指包含肠溶聚合物的控释包衣,任选地与增塑剂组合。
“控释(CR)珠粒”或“控释颗粒”广泛地指含活性剂芯,其具有内部SR包衣,任选地接着外部DR或TPR包衣,或内部TPR包衣,接着外部DR包衣。
“滞后时间”是指在摄入组合物或包含组合物的剂型之后,或者在组合物或包含组合物的剂型暴露接触于模拟体液之后,小于约10%的活性剂从药物组合物中释放的时间,例如利用USP设备,使用两阶段溶出介质(首先在37℃下在700mL的0.1N HCl中2小时,然后在通过加入200mL的pH调节剂获得的pH6.8条件下溶出测试)评估。
“置于……之上”,例如关于在底物上的包衣,指的是例如包衣关于底物的相对位置,但不要求包衣与底物直接接触。例如,“置于底物上”的第一包衣可以与底物直接接触,或者可以在第一包衣和底物之间插入的一种或多种介入材料或包衣。例如,置于含活性剂芯上的SR包衣可以指直接沉积在含活性剂芯或酸性晶体或含酸芯上的SR包衣,或者可以指沉积在保护密封包衣上的SR包衣,该保护密封包衣沉积在含活性剂芯上。
“密封剂层”或“保护密封包衣”是指置于含活性剂芯颗粒或功能聚合物包衣之上的保护膜,其在搬运期间保护颗粒免于磨损和摩擦,和/或使处理期间的静电最小化。
“口腔崩解片”或“ODT”是指在给药之后在不咀嚼的情况下在口腔中快速崩解的片剂。其崩解速率可以变化,但快于旨在给药之后立即吞服的常规固体剂型(例如片剂或胶囊)的崩解速率,或当被试验(例如USP<701>试验方法)时,快于可咀嚼固体剂型的崩解速率。
术语“基本崩解”是指崩解水平相当于至少约50%崩解、至少约60%崩解、至少约70%崩解、至少约80%崩解、至少约90%崩解或约100%解体。“崩解”与“溶解”不同。“崩解”是指例如包含片剂的组分颗粒的结构内聚力的分解或损失,而“溶解”是指固体在液体中的溶解,例如药物在溶剂或胃液中的溶解。
“非水溶性聚合物”是不依赖于pH或在宽pH范围(例如,pH 0至pH 14)内不溶或非常微溶于水介质的聚合物。膨胀但不溶于水介质的聚合物可以是“非水溶性的”。
“水溶性聚合物”是一种可溶解的聚合物,即不依赖于pH,大量溶解于水介质中。
“肠溶聚合物”是在肠道条件下可溶,即显大量溶解的聚合物;即在中性至碱性条件下可溶于水介质中,在酸性条件(即低pH)下不溶。
“反向肠溶聚合物”或“胃溶聚合物”是指在酸性条件下可溶且在中性和碱性条件下不溶的聚合物。
除非另有说明,否则本文所述的各种包衣或层的量(“包衣重量”)表示为干燥包衣提供的颗粒或珠粒相对于颗粒或珠粒在包衣之前的初始重量的百分比重量增加。因此,10%包衣重量是指将颗粒重量增加10%的干燥包衣。
生物等效性是指在类似条件下以相同剂量给药时,活性成分的可用性没有明显不同的吸收率和吸收程度。生物等效性可以通过药代动力学参数来测量,例如AUC和Cmax。
一个实施例是包含改良释放制剂(MR)的口服制剂。在该实施例中,单个剂型包含速释(IR)剂型和缓释(XR)剂型。如本文所使用,速释剂型在给药之后快速释放活性剂。如本文所使用,缓释剂型包括延迟释放、时间释放、控释或持续释放形式。如本文所使用,缓释剂型随时间以预定速率释放活性剂以在特定的时间内维持恒定的药物浓度并具有最小的副作用。缓释制剂可以通过随后用说明性但非限制性实例描述的各种制剂来实现,包括具有活性剂和活性剂的脂质体制剂的聚合缀合物。
递送系统可包括可以负载或不负载活性剂的芯、种或基质,以及包含活性剂和/或包含具有释放特性的层的一个或多个包衣层,其控制活性剂的起效和释放特性。芯、种或基质可以商业制备或获得。仅作为一个实例,可以存在糖或微晶纤维素芯,其具有由例如羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、聚(环氧乙烷)、聚(乙烯醇)、黄原胶、卡波姆、角叉菜胶、菌胶团等制成的亲水基质。
包衣层可以提供速释、延迟脉冲释放或持续释放。可以从速释层快速释放活性剂,通过例如使用胃液可以快速渗透的非常薄的层或包衣,从而促进活性剂的快速浸出;或将活性剂掺入混合物中,该混合物包含容易溶解并在胃液中释放活性剂的支持性粘合剂或其他惰性材料;或使用在与胃液接触时快速崩解的支持性粘合剂或其他惰性材料,其中材料和活性剂都以小颗粒快速分散到胃液中。这种快速崩解和分散材料包括例如乳糖和微晶纤维素。羟丙基甲基纤维素是混悬剂和粘合剂的一个实例。
延迟脉冲释放组分的肠溶包衣可以是pH依赖性或非pH依赖性的。持续释放组分的肠溶包衣是pH依赖性的。活化pH依赖性包衣以在已知pH范围内释放药物,该已知pH范围通常与期望延迟释放的环境的局部pH相匹配。示例性pH依赖性包衣包括邻苯二甲酸乙酸纤维素、偏苯三酸乙酸纤维素、邻苯二甲酸羟丙基甲基纤维素、邻苯二甲酸聚乙酸乙烯酯、羧甲基乙基纤维素、共聚甲基丙烯酸/甲基丙烯酸甲酯,例如商品名为 L12.5,L100或
L12.5,L100或 S12.5,S100的材料或用于获得肠溶包衣的类似化合物。也可以使用水性胶体聚合物分散体或再分散体,例如,
S12.5,S100的材料或用于获得肠溶包衣的类似化合物。也可以使用水性胶体聚合物分散体或再分散体,例如, L 30D-55、
L 30D-55、 L100-55、
L100-55、 S100、
S100、 配剂4110D(Rohm Pharma);
配剂4110D(Rohm Pharma); CPD 30(FMC);KOLLICOAT
CPD 30(FMC);KOLLICOAT 30D及30DP(BASF);
30D及30DP(BASF); 30D(Eastman Chemical)。
30D(Eastman Chemical)。
以包衣或层的形式使用肠溶系统以防止活性剂在胃中溶解的市售药物制剂的实例包括 (盐酸度洛西汀,Lilly Indianapolis IN)、
(盐酸度洛西汀,Lilly Indianapolis IN)、 (埃索美拉唑,AstraZeneca LP)、
(埃索美拉唑,AstraZeneca LP)、 (雷贝拉唑钠,Eisai Inc.和Ortho-Mc-Neil-Janssen Pharmaceuticals,Inc.)、
(雷贝拉唑钠,Eisai Inc.和Ortho-Mc-Neil-Janssen Pharmaceuticals,Inc.)、 HD(美沙拉嗪,Procter&GamblePharmaceuticals,Inc.)、
HD(美沙拉嗪,Procter&GamblePharmaceuticals,Inc.)、 (美沙拉嗪,Shire US)、
(美沙拉嗪,Shire US)、 (美沙拉嗪,Shire US)、
(美沙拉嗪,Shire US)、 EC(布地奈德胶囊,AstraZeneca)、
EC(布地奈德胶囊,AstraZeneca)、 XR(拉莫三嗪片,GlaxoSmithKline)、
XR(拉莫三嗪片,GlaxoSmithKline)、 (右兰索拉唑,Takeda Pharmaceuticals NorthAmerica,Inc.)、
(右兰索拉唑,Takeda Pharmaceuticals NorthAmerica,Inc.)、 (胰酶胶囊,Solvay S.A)、
(胰酶胶囊,Solvay S.A)、 (胰脂肪酶胶囊,AxcanPharma US)、
(胰脂肪酶胶囊,AxcanPharma US)、 (泮托拉唑,Pfizer)、
(泮托拉唑,Pfizer)、 (双丙戊酸钠,AbbottLaboratories)、
(双丙戊酸钠,AbbottLaboratories)、 (奥美拉唑,AstraZeneca)、
(奥美拉唑,AstraZeneca)、 (兰索拉唑,Novartis Consumer Health)、
(兰索拉唑,Novartis Consumer Health)、 (双氯芬酸钠,Pfizer)、
(双氯芬酸钠,Pfizer)、 (丙戊酸,Noven Therapeutics)、TRILIPIXI 4(非诺贝酸延迟释放胶囊,Abbott Laboratories)和
(丙戊酸,Noven Therapeutics)、TRILIPIXI 4(非诺贝酸延迟释放胶囊,Abbott Laboratories)和 EC(地达诺新,Bristol-Myers Squibb)。
EC(地达诺新,Bristol-Myers Squibb)。
耐醇药物组合物使用“醇保护剂”防止或延迟乙醇诱导的活性试剂从剂型中“倾卸”,这种“倾卸”可能导致过量的多潘立酮或氘代多潘立酮释放到患者体内,这可能会产生更高的Cmax,可能导致QT延长。使用耐醇组合物,可避免剂量倾卸,从而减轻这种安全隐患。醇保护剂可以是单一材料,例如,赋形剂溶液中的聚合物或材料的组合,例如聚合物的组合。在替代实施例中,醇保护剂沉积在层或包衣中,或者其为基质的形式。醇保护剂材料包括但不限于有机基邻苯二甲酸乙酸纤维素、甲基丙烯酸铵共聚物、甲基丙烯酸酯共聚物、甲基丙烯酸共聚物、天然和合成淀粉、聚环氧烷、天然和合成纤维素(包括改性纤维素,如羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、羟甲基纤维素(HMC)、甲基纤维素(MC)、羟乙基纤维素(HEC)和羧甲基纤维素(CMC))、蜡(例如,昆虫和动物蜡、植物蜡、矿物蜡、石油蜡和合成蜡)。在一个实施例中,醇保护剂是有机基邻苯二甲酸乙酸纤维素Eastman 或纤维醋法酯NF(Eastman Chemical Company,Kingsport TN USA)。醇保护剂可以以足以在给定乙醇浓度下赋予醇耐受性的量存在于制剂中,例如以重量增加20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%、150%、200%、250%、300%、350%、400%、450%和500%的量加入。
或纤维醋法酯NF(Eastman Chemical Company,Kingsport TN USA)。醇保护剂可以以足以在给定乙醇浓度下赋予醇耐受性的量存在于制剂中,例如以重量增加20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%、150%、200%、250%、300%、350%、400%、450%和500%的量加入。
在一个实施例中,活性组合物包含多个CR和IR颗粒,其中CR颗粒各自包含芯,该芯包覆有非水溶性聚合物层,接着是包含肠溶聚合物(任选地组合有水溶性聚合物)的包衣层,其中该芯包含多潘立酮和药学上可接受的聚合物粘合剂,随后是包含非水溶性聚合物(任选地组合有水溶性聚合物)的第一包衣层以及任选的肠溶聚合物的第二包衣(任选地组合有非水溶性聚合物)。
一个实施例具有多个CR和IR颗粒。CR颗粒包含芯,该芯包覆有非水溶性聚合物层,接着是包含肠溶聚合物(任选地组合有非水溶性聚合物)的包衣层;该芯包含至少通过SR层彼此分离的活性剂和药学上可接受的有机酸(例如,富马酸)。IR颗粒各自包含组合有合适的赋形剂的活性剂。在某些实施例中,组合物包含活性剂和至少一种溶解度增强有机酸,该溶解度增强有机酸能够在包衣珠粒中创建酸性pH微环境以在活性剂释放到肠道的敌对pH环境中之前溶解活性剂,药物在肠道中几乎不溶。
在一个实施例中,CR颗粒包含依次包覆有药学上可接受的有机酸(例如,琥珀酸)和药学上可接受的粘合剂(例如,羟丙基纤维素)的惰性芯(例如,糖球,纤维素球等);SR层(例如包含药学上可接受的非水溶性聚合物,如乙基纤维素,任选地用药学上可接受的增塑剂,如柠檬酸三乙酯或聚乙二醇,增塑);活性剂层和药学上可接受的粘合剂(例如,聚维酮);任选的密封层(例如包水性溶性聚合物,如羟丙基甲基纤维素);和包含非水溶性聚合物(例如,乙基纤维素(EC-10))、肠溶聚合物(例如,邻苯二甲酸羟丙甲纤维素,HP-55)以及任选的药学上可接受的增塑剂(例如,柠檬酸三乙酯(TEC))的SR层。
非pH依赖性包衣包含易于由肠道细菌中的偶氮还原酶(即偶氮聚合物)酶促活化的材料或易于由结肠中的多糖酶(天然多糖)降解的材料。偶氮聚合物的非限制性实例包括甲基丙烯酸2-羟乙酯(HEMA)和甲基丙烯酸甲酯(MMA)的共聚物。天然多糖的非限制性实例包括直链淀粉、壳聚糖、软骨素、葡聚糖和木聚糖。
“肠溶聚合物”是聚苯乙烯当量重均分子量(MW)为约50,000至150,000且包含羧基的聚合物,羧基在低于约pH 4(胃pH值范围)的pH下保持不溶,但是其电离并因此导致聚合物在高于约5.0(肠pH值范围)的pH下溶解。肠溶聚合物可以是成膜的,例如邻苯二甲酸乙酸纤维素(C-A-P)、偏苯三酸乙酸纤维素(C-A-T)、邻苯二甲酸羟丙基甲基纤维素(HPMCP)、甲基丙烯酸和丙烯酸乙酯的共聚物、琥珀酸乙酸羟丙基甲基纤维素(HPMCAS)和邻苯二甲酸聚乙酸乙烯酯(PVAP)。HPMCP的MW可以在约80,000和110,000之间,或在95,000和100,000之间。C-A-P的MW可以在约55,000和75,000之间,或在68,000和80,000之间。
持续释放组分可以包括持续释放包衣、持续释放基质和持续释放渗透系统。可以使用非水溶性聚合物、非水溶性聚合物的组合或非水溶性和水溶性聚合物的组合来制备持续释放包衣。本领域普通技术人员已知的常规持续释放聚合物可用于持续释放基质。
示例性持续释放包衣包括聚乙酸乙烯酯、乙酸纤维素、乙酸丁酸纤维素、乙酸丙酸纤维素、乙基纤维素、脂肪酸及其酯、烷基醇、蜡、玉米醇溶蛋白(醇溶玉米蛋白)和水性聚合物分散体,如 RS和RL30D、
RS和RL30D、 NE30D、
NE30D、
 SR30D和乙酸纤维素胶乳。
SR30D和乙酸纤维素胶乳。
丸粒或珠粒可以基于与活性剂的相容性和丸粒或珠粒的物理化学性质由任何药学上可接受的材料制成。
粘合剂包括纤维素衍生物,如甲基纤维素、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、聚乙烯吡咯烷酮、聚乙烯吡咯烷酮/乙酸乙烯酯共聚物等。
崩解剂包括玉米淀粉、预胶化淀粉、交联羧甲基纤维素 羟基乙酸淀粉钠
羟基乙酸淀粉钠 交联聚乙烯吡咯烷酮
交联聚乙烯吡咯烷酮 等。
等。
填充剂包括乳糖、碳酸钙、磷酸钙、硫酸钙、微晶纤维素、葡聚糖、淀粉、蔗糖、木糖醇、乳糖醇、甘露糖醇、山梨糖醇、氯化钠、聚乙二醇等。
表面活性剂包括月桂基硫酸钠、脱水山梨糖醇单油酸酯、聚氧乙烯脱水山梨糖醇单油酸酯、胆汁盐、单硬脂酸甘油酯、 系列(BASF)等。
系列(BASF)等。
增溶剂包括柠檬酸、琥珀酸、富马酸、苹果酸、酒石酸、马来酸、戊二酸、碳酸氢钠、碳酸钠等。
稳定剂包括抗氧化剂、缓冲剂、酸等。
以下信息说明了示例性但非限制性制造方法。
芯可以通过挤出滚圆、高剪切造粒、溶解或混悬分层来制备,
在挤出滚圆中,通过加入粘合剂溶液将活性剂和其他添加剂造粒。湿物质通过装备有一定尺寸筛网的挤出机。挤出物在球磨机中滚圆。将得到的丸粒干燥并过筛。
在高剪切造粒中,将活性剂和其他添加剂干混,然后通过在高剪切造粒机/混合机中加入粘合剂溶液使混合物润湿。在通过混合和研磨的组合作用润湿之后,将颗粒捏合。将得到的颗粒或丸粒干燥并过筛。
在溶解或混悬分层中,在流化床处理器或其他合适的设备中将具有或不具有粘合剂的药物溶液或分散液喷洒到具有一定粒度的起始种上,因此将活性剂包在起始种的表面上。将活性剂负载丸粒干燥。
芯颗粒具有约50微米至1500微米,优选100微米至800微米的直径。芯颗粒可以在流化床设备中包覆有交替顺序的包衣层。芯可以直接包覆有一层或多层活性剂,和/或活性剂可以掺入到芯材料中。分离层或保护层可以加入到含活性剂层的顶部,和/或活性剂层之间。分离层或保护层可以被加入到活性剂负载芯的表面上,然后肠溶延迟脉冲或持续释放层可以被包覆于其上。另一个活性剂层也可以加入到肠溶延迟脉冲或持续层以传递初始剂量。通过本领域中使用的常规包衣技术,例如锅包衣或流化床包衣,使用聚合物水溶液或合适的有机溶剂或水性聚合物分散体,可以将保护包衣层紧紧涂覆在含活性剂芯或活性剂层芯之外。用于保护层的合适材料包括纤维素衍生物,例如羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、聚乙烯吡咯烷酮、聚乙烯吡咯烷酮/乙酸乙烯酯共聚物、乙基纤维素水性分散体
 RL 30D、
RL 30D、 乙酸纤维素、乙酸丁酸纤维素、乙酸丙酸纤维素、乙基纤维素、脂肪酸及其酯、蜡、玉米醇溶蛋白和水性聚合物分散体(例如,
乙酸纤维素、乙酸丁酸纤维素、乙酸丙酸纤维素、乙基纤维素、脂肪酸及其酯、蜡、玉米醇溶蛋白和水性聚合物分散体(例如, RS和RL 30D、
RS和RL 30D、 NE 30D、
NE 30D、 和/或乙酸纤维素胶乳),单独使用或与亲水性聚合物(如羟乙基纤维素、羟丙基纤维素(
和/或乙酸纤维素胶乳),单独使用或与亲水性聚合物(如羟乙基纤维素、羟丙基纤维素( Hercules Corp.)、羟丙基甲基纤维素(
Hercules Corp.)、羟丙基甲基纤维素( Dow Chemical Corp.)、聚乙烯吡咯烷酮等)组合使用。包衣水平范围为约1%w/w至约6%w/w,优选约2%w/w至约4%w/w。
Dow Chemical Corp.)、聚乙烯吡咯烷酮等)组合使用。包衣水平范围为约1%w/w至约6%w/w,优选约2%w/w至约4%w/w。
通过本领域已知的常规包衣技术,例如锅包衣或流化床包衣,使用聚合物水溶液或合适的有机溶剂,或使用水性聚合物分散体,在有或没有密封包衣的条件下,将肠溶延迟脉冲释放或持续释放包衣层涂覆于芯。合适的包衣机在本领域中是已知的,例如市售的pH敏感聚合物,使得活性剂在酸性胃环境(pH<4.5)中不释放,但是当在延迟一段时间后pH敏感层于较高的pH下溶解时,或在单位通过胃之后,活性剂被释放并变得可用。
延迟脉冲释放组分和持续释放组分的肠溶聚合物包括例如邻苯二甲酸乙酸纤维素、偏苯三酸乙酸纤维素、邻苯二甲酸羟丙基甲基纤维素、邻苯二甲酸聚乙酸乙烯酯、羧甲基乙基纤维素、共聚甲基丙烯酸/甲基丙烯酸甲酯,例如称为 L12.5,L100或
L12.5,L100或 S12.5,S100的材料或用于获得肠溶包衣的类似化合物。也可以使用水性胶体聚合物分散体或再分散体,例如,
S12.5,S100的材料或用于获得肠溶包衣的类似化合物。也可以使用水性胶体聚合物分散体或再分散体,例如, L 30D-55、
L 30D-55、 L100-55、
L100-55、 S 100、
S 100、 配剂4110D(Rohm Pharma);
配剂4110D(Rohm Pharma); CPD 30(FMC);KOLLICOAT
CPD 30(FMC);KOLLICOAT 30D及30DP(BASF);
30D及30DP(BASF); 30D(Eastman Chemical)。
30D(Eastman Chemical)。
肠溶延迟脉冲释放和持续释放聚合物可以通过与其他已知的pH不敏感的包衣产品来修饰,该包衣产品例如带有少量甲基丙烯酰氧乙基三甲基氯化铵的中性甲基丙烯酸酯(市售为 RS和
RS和 RL);没有任何官能团的中性酯分散体(市售为
RL);没有任何官能团的中性酯分散体(市售为 NE30D);和其他非pH依赖性包衣产品。
NE30D);和其他非pH依赖性包衣产品。
在肠溶延迟脉冲释放或持续释放包衣上使用的保护层的修饰组分可以包括水渗透阻挡层(半透性聚合物),其可以在肠溶包衣之后继续包覆以降低通过肠溶包衣层的水渗透速率,并从而增加活性剂释放的滞后时间。如前所述进行包衣。
可任选地涂覆保护层或着色剂外覆层。 OPADRY
OPADRY
 和
和 的相应彩色和无色等级可防止丸粒发粘并为产品提供颜色。在一个实施例中,保护剂或彩色包衣的范围为1%w/w至6%w/w,优选约2%w/w至约3%w/w。也可以使用滑石粉。
的相应彩色和无色等级可防止丸粒发粘并为产品提供颜色。在一个实施例中,保护剂或彩色包衣的范围为1%w/w至6%w/w,优选约2%w/w至约3%w/w。也可以使用滑石粉。
可以将组分掺入到外覆配方中,例如为了促进和提供更加快速的释放。这些组分包括例如增塑剂,包括乙酰柠檬酸三乙酯、柠檬酸三乙酯、乙酰柠檬酸三丁酯、癸二酸二丁酯、三醋精、聚乙二醇、丙二醇等;润滑剂,包括滑石粉、胶体二氧化硅、硬脂酸镁、硬脂酸钙、二氧化钛、硅酸镁等。
可将组合物单独或与其他赋形剂一起掺入硬明胶胶囊中。可以将组合物掺入片剂中,例如通过掺入片剂基质中,片剂基质在摄入后快速分散颗粒。为了防止压片过程中的颗粒破坏,需要填充剂/粘合剂,例如微晶纤维素 大豆多糖
大豆多糖 预胶化淀粉(
预胶化淀粉( 1500,
1500, 1551)和聚乙二醇
1551)和聚乙二醇 其存在量的范围为约5%w/w至约75%w/w,优选范围为约25%w/w至约50%w/w。
其存在量的范围为约5%w/w至约75%w/w,优选范围为约25%w/w至约50%w/w。
赋形剂通常包括但不限于一种或多种惰性填充剂,包括微晶纤维素、大豆多糖、二水磷酸钙、硫酸钙、乳糖、蔗糖、山梨糖醇等;一种或多种赋予粉末流动性的材料,包括气相二氧化硅、硅胶、硬脂酸镁、硬脂酸钙等;一种或多种确保合适压片的润滑剂,包括聚乙二醇、亮氨酸、山嵛酸甘油酯、硬脂酸镁、硬脂酸钙、硬脂酸、氢化植物油等,其存在量的范围为约0.1%w/w至约10%w/w,优选范围为约0.3%w/w至约3.0%w/w。
摄入片剂之后,加入崩解剂以分散珠粒。崩解剂包括但不限于交联羧甲基纤维素钠 羟基乙酸淀粉钠
羟基乙酸淀粉钠
 交联聚乙烯聚吡咯烷酮
交联聚乙烯聚吡咯烷酮 等,其存在量的范围为约3%w/w至约15%w/w,优选范围为约5%w/w至约10%w/w。
等,其存在量的范围为约3%w/w至约15%w/w,优选范围为约5%w/w至约10%w/w。
在一个实施例中,片剂由颗粒形成,颗粒与 崩解剂和润滑剂一起引入混合机中,混合达规定时间(分钟)以获得均匀混合物,然后将混合物置于压片机的料斗,用压片机压制片剂。所使用的压制力足以形成片剂,但不会使珠粒或包衣破裂。
崩解剂和润滑剂一起引入混合机中,混合达规定时间(分钟)以获得均匀混合物,然后将混合物置于压片机的料斗,用压片机压制片剂。所使用的压制力足以形成片剂,但不会使珠粒或包衣破裂。
片剂可以被构建成三层,其中速释组分被干混,并且延迟脉冲释放和持续释放组分被湿法造粒。然后将片剂形成一层或三层压片。在层溶解之后,各组分被释放并且起到配制时的作用:例如速释颗粒提供快速释放,延迟脉冲释放颗粒提供延迟脉冲释放,并且持续释放颗粒在滞后时间之后提供持续释放。
本发明的一个实施例是口服多潘立酮或氘代多潘立酮制剂,其以单个剂型包含速释形式和缓释形式。本发明的一个实施例是口服多潘立酮或氘代多潘立酮制剂,其以单个剂型包含速释形式和缓释形式。
将10mg速释制剂(5mg至20mg)和20mg缓释制剂(10mg至80mg)组合的多潘立酮或氘代多潘立酮的剂型向患者递送试剂持续约12小时。这样的剂量制剂为12小时的治疗提供了单次患者剂量,从而提供患者便利性和延长的治疗,例如,患者可以在没有症状的情况下有益地体验整夜睡眠、完整的工作日、完整的休闲日等。
在实施例中,本发明的制剂包含组合物的速释(IR)部分或组分,以及缓释(XR)部分或组分或其组合。速释部分在小于约一小时的时间内递送100%速释剂量,并且缓释部分在12小时内递送缓释剂量。
在图2中示出了多潘立酮的典型溶出曲线,也被称为释放曲线。在递送系统的速释部分中,药物释放百分比在小于一小时的时间内或在一小时内接近100%;在递送系统的缓释部分中,药物释放百分比在12小时内或小于12小时的时间内接近约100%。图3是多潘立酮的模拟血浆浓度的示意图,其中速释部分的血浆药物浓度达到峰值(约为浓度的两倍),同时缓释部分的药物达到稳定状态(约为速释部分的药物的一半)。
在一个实施例中,可以直肠给予活性剂。活性剂的直肠给药可以是10mg至20mg,每天三次。直肠给药可以通过栓剂。在一个实施例中,该制剂直肠给药,例如通过栓剂给药。
该组合物可以采取多种递送形式或系统。可以使用以下制剂,这些仅是示例性的而非限制性的。口服制剂包括片剂、胶囊、小药囊、软凝胶、液体、凝胶、条状物、薄膜、粉末、颗粒、凝胶、脉冲释放物、包衣芯、延迟缓释形式、环带型药物形式、持续释放形式、片剂胶囊、粒化囊片、分层片剂等,包括这些的组合,例如片剂胶囊、粒化囊片、分层片剂等,以及活性剂和至少一种药学上可接受的赋形剂。
本领域技术人员已知片剂制剂。该片剂可以是适于口服给药的任何形状或大小,例如圆形、椭圆形等。在一个实施例中,片剂可以是速释(IR)、缓释(XR)或其组合。片剂可以是包含彼此相邻的IR层和XR层的双层片剂(图4);包含由药学上可接受的缓冲层分离的IR层和XR层的三层片剂(图5);或在基质层中包含活性剂并包覆有活性剂IR层的XR片剂(图6)。
所述组合物还可以其他递送形式提供,例如包含IR片剂、塞子和渗透药物递送系统内的XR片剂的胶囊,用于在12小时的持续时间内控制组合物的递送(图7);包含以适当比例混合的IR珠粒和XR珠粒的胶囊(图8);包含与XR迷你片剂混合的IR迷你片剂的胶囊(图9);包含包覆有缓释聚合物的IR颗粒和XR颗粒的胶囊(图10);包含包覆有IR层的XR珠粒的胶囊(图11)等。活性剂的其他递送形式可以是包含IR颗粒和包衣XR珠粒的压制片剂,IR颗粒和包衣XR珠粒嵌入在片剂内(图12);包含嵌入IR片剂内的XR片剂的压制片剂(图13);或混悬在胶囊内的速释液体药物溶液中的XR片剂(图14)。
另一种递送形式是小药囊。小药囊可以包含IR和XR颗粒或珠粒的混合物(图15),或者其可以包含泡腾IR颗粒和包衣XR颗粒的混合物(图16)。
在以下参考文献中的每一个中描述了其他速释、缓释的或持续释放、改良释放和延迟脉冲释放系统:美国公开第2005/0095295号、第2005/0106247号和第2007/0264323号;和美国专利第6,126,969号和第8,211,465号。作为一个实例,美国公开第2005/0106247号描述了具有包含非水溶性聚合物和/或水溶性聚合物的缓释包衣的缓释颗粒(例如珠粒、丸粒、颗粒等)中的药物(盐酸环苯扎林),并且一些颗粒被包含在明胶胶囊中。作为另一实例,美国公开第2007/0264323号描述了药物 (例如珠粒)的递送系统,该药物置于包括包衣层的胶囊、片剂、或小药囊,延迟脉冲释放组分,速释制剂,中间释放制剂,持续释放制剂和控释胶囊中。美国专利第6,126,969号描述了药物(对乙酰氨基酚)的递送系统,该药物例如速释/持续释放剂型的包衣和未包衣药物颗粒的组合。美国专利第8,211,465号描述了用于药物(NSAID,如布洛芬)的初始释放和该药物的第二次持续释放的剂型。在Patra等人的《渗透药物递送系统:基础和设计方法,关于药物递送和制剂的近期专利》,7(2013)1-12中描述了渗透递送系统。
(例如珠粒)的递送系统,该药物置于包括包衣层的胶囊、片剂、或小药囊,延迟脉冲释放组分,速释制剂,中间释放制剂,持续释放制剂和控释胶囊中。美国专利第6,126,969号描述了药物(对乙酰氨基酚)的递送系统,该药物例如速释/持续释放剂型的包衣和未包衣药物颗粒的组合。美国专利第8,211,465号描述了用于药物(NSAID,如布洛芬)的初始释放和该药物的第二次持续释放的剂型。在Patra等人的《渗透药物递送系统:基础和设计方法,关于药物递送和制剂的近期专利》,7(2013)1-12中描述了渗透递送系统。
剂型的活性芯可以是惰性颗粒或酸性或碱性缓冲剂晶体,其包覆有含药物成膜制剂。在一个实施例中,水溶性成膜组合物形成水溶性/分散性颗粒。或者,可以通过造粒和研磨和/或通过包含活性剂的聚合物组合物的挤出和滚圆来制备活性剂。芯中的活性剂的量取决于所需的剂量,并且通常在约5重量%至60重量%之间变化。取决于所需的释放曲线的类型和/或所选的聚合物和包衣溶剂,活性芯上的聚合物包衣通常为约4%至20%(以包衣颗粒的重量计)。本领域技术人员将能够选择适当量的活性剂包覆于芯上或掺入到芯中以实现期望的剂量。在一个实施例中,非活性芯可以是糖球或缓冲晶体或包封的缓冲晶体,例如碳酸钙、碳酸氢钠、富马酸、酒石酸等,其改变活性剂的微环境以促进其释放。
含药物颗粒可用包含非水溶性聚合物或非水溶性聚合物与水溶性聚合物的组合的缓释(XR)包衣包覆以提供XR珠粒。在实施例中,非水溶性聚合物和水溶性聚合物可以以100:0至65:35,或约95:5至70:30,或约85:15至75:25的重量比存在。缓释包衣以提供所需释放曲线需要的量施用。在实施例中,缓释包衣为包衣珠粒的约1重量%至15重量%,或为包衣珠粒的约7重量%至12重量%。
包含两个珠粒群的混合物的改良释放剂型可以如下制备。通过用活性剂和聚合物粘合剂包覆惰性颗粒(例如,糖丸种(non-pareil seed)、酸性缓冲剂晶体或碱性缓冲剂晶体)或通过造粒和研磨或通过挤出/滚圆以形成IR珠粒来制备含药物芯。IR珠粒单独以增塑非水溶性聚合物(例如,乙基纤维素)包覆或与水溶性聚合物(例如,羟丙基甲基纤维素)组合包覆以形成XR珠粒。将硬明胶胶囊XR珠粒单独或与IR珠粒组合以所需比例填充以产生提供所需释放曲线的改良释放(MR)胶囊。
据报道,使用以下溶解程序的IR珠粒在30分钟内释放至少约70%,更特别是至少约90%的活性剂。
USP设备2(桨速50rpm)与以下溶解介质一起使用:900mL 0.1N HCl(或合适的溶解介质),在37℃下,通过HPLC测定活性剂释放。
水性或药学上可接受的溶剂可用于制备含活性剂芯颗粒。用于将药物与惰性糖球粘合的成膜粘合剂的类型并不重要,但通常使用水溶性、醇溶性或丙酮/水溶性粘合剂。可以以约0.5重量%至约5重量%的浓度使用粘合剂(例如,聚乙烯吡咯烷酮(PVP)、聚环氧乙烷、羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、诸如葡聚糖的多糖、玉米淀粉),也可以使用其他浓度。活性剂可以以溶液形式存在于该包衣制剂中,或者可以以高达约35重量%的固体含量分散,这取决于包衣制剂的粘度。
将活性剂、任选的粘合剂(如PVP)、溶解速率控制聚合物(如果使用的话)以及任选的其他药学上可接受的赋形剂在行星式混合机或高剪切造粒机(如 )中混合并通过加入/喷洒造粒流体(如水或醇)来造粒。可以使用挤出机/球磨机将湿块挤出并滚圆以产生球形颗粒(珠粒)。在这些实施例中,以挤出/滚圆化芯的总重量计,有效负载可以高达90重量%。
)中混合并通过加入/喷洒造粒流体(如水或醇)来造粒。可以使用挤出机/球磨机将湿块挤出并滚圆以产生球形颗粒(珠粒)。在这些实施例中,以挤出/滚圆化芯的总重量计,有效负载可以高达90重量%。
用于XR包衣的非水溶性聚合物的说明性但非限制性实例包括乙基纤维素粉末或水性分散体(例如, ECD-30)、乙酸纤维素、聚乙酸乙烯酯(
ECD-30)、乙酸纤维素、聚乙酸乙烯酯( SR 30D,BASF),基于丙烯酸乙酯和甲基丙烯酸甲酯的中性共聚物、丙烯酸酯和甲基丙烯酸酯与季铵基团的共聚物(例如
SR 30D,BASF),基于丙烯酸乙酯和甲基丙烯酸甲酯的中性共聚物、丙烯酸酯和甲基丙烯酸酯与季铵基团的共聚物(例如 NE,RS和RS30D,RL或RL30D等)。说明性但非限制性的水溶性聚合物包括低分子量羟丙基甲基纤维素(HPMC)、甲基纤维素、羟丙基纤维素、聚乙烯吡咯烷酮和/或分子量>3000的聚乙二醇(PEG)。取决于活性剂在水中的溶解度和所使用的溶剂或胶乳混悬体基包衣制剂,通常以约1重量%至15重量%的范围内的厚度施用缓释包衣。
NE,RS和RS30D,RL或RL30D等)。说明性但非限制性的水溶性聚合物包括低分子量羟丙基甲基纤维素(HPMC)、甲基纤维素、羟丙基纤维素、聚乙烯吡咯烷酮和/或分子量>3000的聚乙二醇(PEG)。取决于活性剂在水中的溶解度和所使用的溶剂或胶乳混悬体基包衣制剂,通常以约1重量%至15重量%的范围内的厚度施用缓释包衣。
用于形成膜的包衣组合物通常被塑化。说明性但非限制性的增塑剂包括三醋精、柠檬酸三丁酯、柠檬酸三乙酯、乙酰柠檬酸三正丁酯、邻苯二甲酸二乙酯、聚乙二醇、聚丙二醇、蓖麻油、癸二酸二丁酯和/或乙酰化甘油单酯等。以聚合物计,增塑剂可包含约3重量%至约30重量%,更通常约10重量%至约25重量%。增塑剂的类型及其含量取决于聚合物(一种或多种)和包衣体系的性质(例如,基于水性或溶剂的,基于溶液或分散体,以及总固体)。
在施用缓释膜包衣以分离不同的膜层之前,可以通过施用薄羟丙基甲基纤维素(HPMC)( Clear)膜来引发颗粒。通常使用HPMC,但也可以使用其他引物,如羟丙基纤维素(HPC)。
Clear)膜来引发颗粒。通常使用HPMC,但也可以使用其他引物,如羟丙基纤维素(HPC)。
可以使用制药工业中使用的任何包衣技术将膜包衣施用于芯。在一个实施例中,使用流化床包衣。
可以使用多剂量形式,即多颗粒剂型形式(丸粒、珠粒、颗粒、迷你片剂等)或适于口服给药的其他形式的产品。如本文所使用,这些术语可互换使用以指代多颗粒剂型。
包含两种或多种珠粒群的混合物的缓释剂型可以如下制备。惰性颗粒(例如,糖丸种、酸性缓冲剂晶体或碱性缓冲剂晶体)用活性剂和聚合物粘合剂包覆以形成活性颗粒,即速释(IR)珠粒,其可以是单位剂量形式以作为单次剂量。活性颗粒用非水溶性聚合物的溶液或混悬液或水溶性聚合物和非水溶性聚合物的混合物包覆以形成缓释包衣活性颗粒,即缓释(XR)。将硬明胶胶囊XR珠粒单独并且任选地与IR珠粒组合以95:5至70:30(ER珠粒:IR珠粒)范围内的比率填充以产生呈现靶活性剂释放曲线的改良释放(MR)胶囊。
在一个实施例中,剂型具有分散在油性或脂质体系中的活性剂的速释部分和配制在疏水性载体包覆的活性剂的蜡质基质或颗粒中的另一部分。至少15%至50%的活性剂是速释部分,并且是适合快速释放的剂型。片剂胶囊的其余部分,按重量计,可包括活性剂的持续释放制剂或活性剂的持续释放制剂的一部分。
活性多潘立酮或氘代多潘立酮可以配制在脂质基递送系统中。将活性剂包封或溶解于脂质赋形剂中可导致溶解和吸收的增加,从而导致生物利用度增加。
脂质赋形剂是市售的。由于脂质会影响吸收,因此有必要了解脂质赋形剂的特性。决定脂质基制剂赋形剂的选择的因素包括混溶性、溶解能力、自分散性和促进制剂自分散的能力、消化产物的消化性和结果、刺激性、毒性、纯度、化学稳定性、胶囊相容性、熔点、成本等。
由中链甘油三酯和长链甘油三酯组成的膳食油以及各种溶剂和表面活性剂经常用于制备脂质基制剂。许多脂质是两亲的,即它们具有亲脂性部分(脂肪酸)和亲水性部分。随着脂肪酸链长的增加,熔点增加,但熔点随着脂肪酸不饱和度的增加而降低,这也增加了易氧化性。下表中提供了用于脂质基制剂中的增溶剂:
市售脂质基口服制剂中使用的溶解赋形剂

甘油三酯植物油是最常见的脂质赋形剂。它们被完全消化吸收,从而消除了安全问题。甘油三酯是长链甘油三酯(LCT)、中链甘油三酯(MCT)和短链甘油三酯(SCT)。它们对活性剂的溶解能力主要归因于酯基团的有效浓度。MCT具有比LCT更高的溶解能力并且不易氧化。来自不同植物来源的油的每种脂肪酸都具有不同比例。下面显示了各种脂质赋形剂中的脂肪酸组成。
脂质基赋形剂中发现的脂肪酸的组成:

D-α-生育酚聚乙二醇1000琥珀酸酯(维生素E TPGS)源自植物生育酚。它是水溶性的,用作水难溶性药物的吸收促进剂。纯甘油三酯存在于精炼植物油中。
混合甘油酯通过植物油的部分水解获得。甘油三酯起始原料和水解程度决定了所产生的混合甘油酯的化学组成。中链混合甘油酯不易氧化,溶解能力更强,并促进乳化。这些极性油性赋形剂还可改善溶解能力和制剂的分散性。极性油的实例包括脱水山梨糖醇三油酸酯( 85)和油酸。
85)和油酸。
共溶剂,例如乙醇、甘油、丙二醇、聚乙二醇(PEG)-400等,增加了制剂对活性剂的溶解能力并有助于分散包含高比例水溶性表面活性剂的体系。与共溶剂有关的实际限制包括可溶性活性剂由于稀释后溶解能力的丧失而析出,一些共溶剂与油的不混溶性,以及低分子量溶剂与胶囊壳的不相容性。
非水溶性表面活性剂是吸附在油-水界面的脂质赋形剂,具有中等亲水亲油平衡(HLB 8-12)。根据乙氧基化程度,它们在水中具有有限的溶解度。如果经受剪切,它们可以形成乳液,并且可以被称为在水中“可分散”。它们可以形成胶束,但由于其亲水性不足而不能自乳化。油酸酯,如聚氧乙烯(20)失水山梨糖醇三油酸酯( -85)和聚氧乙烯(20)三油酸甘油酯(
-85)和聚氧乙烯(20)三油酸甘油酯( -TO)例示了具有HLB 11-11.5的非水溶性表面活性剂。但是,平均HLB为11的
-TO)例示了具有HLB 11-11.5的非水溶性表面活性剂。但是,平均HLB为11的 -80和
-80和 -80的混合物在功能上与
-80的混合物在功能上与 -85不相似。
-85不相似。 -80和
-80和 -80的混合物具有水溶性和非水溶性分子,但
-80的混合物具有水溶性和非水溶性分子,但 -85主要具有非水溶性分子。
-85主要具有非水溶性分子。
水溶性表面活性剂是用于配制自乳化药物递送系统的最常见的表面活性剂。HLB>12的材料可通过在超过其临界胶束浓度(CMC)的纯水中溶解形成低浓度的胶束溶液。水溶性表面活性剂通过PEG与水解植物油合成,或者可以使醇与环氧乙烷反应以产生烷基醚乙氧基化物,通常使用的表面活性剂(例如,鲸蜡硬脂醇乙氧基化物“CETOMACROGOLTM”)。脱水山梨糖醇酯与环氧乙烷的反应产生聚山梨酯,主要是醚乙氧基化物。 RH40和RH60(乙氧基化氢化蓖麻油)就是这种类型的实例,它是源自植物油的材料氢化得到的。未氢化的
RH40和RH60(乙氧基化氢化蓖麻油)就是这种类型的实例,它是源自植物油的材料氢化得到的。未氢化的 EL(乙氧基化蓖麻油)也被广泛使用。
EL(乙氧基化蓖麻油)也被广泛使用。 通过抑制外排泵来增强吸收;虽然抑制机理未被确定,但它可能是由于表面活性剂分子渗入膜,吸附到外排泵表面或分子与外排泵的胞内结构域的相互作用而导致的非特异性构象变化。
通过抑制外排泵来增强吸收;虽然抑制机理未被确定,但它可能是由于表面活性剂分子渗入膜,吸附到外排泵表面或分子与外排泵的胞内结构域的相互作用而导致的非特异性构象变化。
可以加入添加剂以保护制剂免于氧化。实例包括脂溶性抗氧化剂如α-生育酚、β-胡萝卜素、没食子酸丙酯、丁基化羟基甲苯(BHT)、丁基化羟基茴香醚(BHA)等。
评估制备过程中的脂质行为,因为脂质赋形剂具有不同的化学组成,导致宽的熔化范围。使用差示扫描量热法(DSC)评估脂质的热性质,例如结晶温度、熔点、玻璃化转变温度和相对于温度的赋形剂的固体脂肪含量的测定。加热或冷却过程中的脂质组织通过热台显微镜评估。通过X射线衍射(XRD)确认脂质赋形剂的结晶度。
高效液相色谱(HPLC)和气相色谱(GC)可以确定醚、酯和脂肪酸分布的确切组成。其他化学指标包括由其皂化值确定的脂肪酸的分子量,通过碘基测定确定的烃链的饱和度,通过测量过氧化物确定的氧化变化,由酸含量测量的游离脂肪酸和通过测量游离羟基含量确定的羟基。
FDA要求的溶出度测试与脂质基制剂的体内行为不相关。胃肠道中的脂质在存在脂肪酶(胃和胰腺)的情况下经历消化过程,脂肪酶也影响脂质赋形剂的乳化和分散性质,从而导致体内溶解能力改变。因此,在选择脂质基制剂时必须考虑脂质赋形剂的消化性。生物溶解介质中的溶出度测试可以评估这种影响并预测体内行为。自乳化制剂的有效性可以通过分散测试(乳化能力和粒度)来测定。光子相关光谱(PCS)或激光衍射可用于测量粒径,目视观察可帮助预测乳化能力。
脂质基赋形剂通过影响各种生理过程,例如刺激胆汁流动和胰液分泌,延长胃排空,增加膜流动性,打开紧密连接,促进药物的淋巴转运从而避免首过代谢,以及抑制外排转运蛋白,来增强药物的口服吸收。为了评估这些影响,可以使用各种体外模型,包括肠微粒体、Caco-2细胞、使用腔室的外翻肠囊和原位灌注测定。
可以使用脂质体;这些球形双层结构在其排列方式上类似于细胞膜,并且主要是两亲磷脂(亲水头和疏水脂肪酸尾)。当水合时,这些磷脂形成球形双层结构,其定向为疏水尾朝向结构内部,亲水头朝向结构外部。亲水性物质可嵌入小球的水性内部空间中,而疏水性活性剂可嵌入内部脂肪酸层内。
可以使用固体脂质纳米颗粒(SLN)。SLN可以提高生物利用度以及受控和位点特异性药物递送,因此是口服肠道淋巴递送的潜在载体。SLN通常是球形颗粒,范围从10nm至1000nm,具有可溶解亲脂性分子的固体脂质芯基质(通过表面活性剂稳定)。主要使用的脂质包括甘油单酯(如单硬脂酸甘油酯)、甘油二酯(如甘油山嵛酸酯)、甘油三酯(如三硬脂精)、脂肪酸(如硬脂酸)、类固醇(如胆固醇)和蜡(如鲸蜡醇棕榈酸酯)。一种药物的口服生物利用度通过使用甘油单酯脂质和大豆卵磷脂以及泊洛沙姆188表面活性剂来配制包覆载药SLN的N-羧甲基壳聚糖聚合物而得到改善(Venishetty等人)。
在喷雾凝结(也称为喷雾冷却)中,将熔化的脂质喷入冷却室,并在空气接触时凝结成球形固体颗粒。从腔室底部收集固体颗粒并填充到硬明胶胶囊中或压制成片剂。超声雾化器在喷雾冷却过程中产生固体颗粒。要考虑的参数是赋形剂的熔点、制剂的粘度和腔室内允许液滴立即凝固的冷却空气温度。据报道,使用PEG4000或泊洛沙姆188作为可熔性粘合剂并使用一水乳糖作为填充剂,通过熔体造粒来制备药物颗粒。当使用硬脂酰聚氧甘油酯( 50/13)作为赋形剂时,报道了具有窄粒径分布的微粒,并显著提高了水难溶性药物的溶解度(Cavallari等人)。
50/13)作为赋形剂时,报道了具有窄粒径分布的微粒,并显著提高了水难溶性药物的溶解度(Cavallari等人)。
熔体造粒,也称为丸粒化,将活性剂粉末混合物转化成颗粒或丸粒。在高剪切混合(“泵开(pump on)”技术)的情况下将可熔性粘合剂(熔融状态)喷到粉末混合物上,或者在高剪切混合过程中,由于颗粒(固体/半固体)的摩擦,可熔性粘合剂与粉末混合物混合并熔化。熔化的粘合剂在粉末颗粒之间形成液体桥,并在受控条件下形成转变为滚圆丸粒的小颗粒。根据粉末的细度,可以使用15%至25%的脂质基粘合剂。在该过程期间要考虑的参数是粘合剂粒度、混合时间、推进速度和粘合剂在熔化时的粘度。通过配制包含药物固体分散体的熔融附聚物来改善药物的溶解速率(Seo等人)。一水乳糖与可熔性粘合剂(例如 50/13的PEG 3000)在高剪切混合机中熔融附聚。聚氧甘油酯,偏甘油酯或聚山梨酯和卵磷脂是用于形成自微乳化体系的熔体造粒技术中的示例性脂质赋形剂。
50/13的PEG 3000)在高剪切混合机中熔融附聚。聚氧甘油酯,偏甘油酯或聚山梨酯和卵磷脂是用于形成自微乳化体系的熔体造粒技术中的示例性脂质赋形剂。
在实施例中,持续释放骨架片可以使用疏水性载体或可熔性粘合剂(如硬脂酸,巴西棕榈蜡和蜂蜡)通过熔体造粒技术配制,从而使载体疏水以持续递送。
在一个实施例中,使用脉冲释放形式。脉冲释放形式包括具有一个或多个包衣的活性芯,称为“包衣芯”制剂。包衣芯还可以与适合速释的一定量的活性剂组合使用。
在一个实施例中,配制用于快速释放的一定量的活性剂与配制的至少第二量的活性剂组合,使得第二量在起效之前有一定延迟,并且第二部分的释放随时间延长或可以随时间延长,称为“延迟缓释”制剂。除非另有说明,否则均以重量百分比进一步描述这些脉冲释放剂量制剂中的每一种。
“包衣芯”制剂是剂量的活性芯,其包含惰性颗粒,例如市售的糖丸球。芯中的活性剂的量根据所要递送的剂量而变化。在一个实施例中,芯包含约5%活性剂至约90%活性剂。在一个实施例中,芯包含约5%活性剂至约60%活性剂。活动剂的量以芯的总重量计。本领域技术人员将能够选择适当量的活性剂用于包覆芯或掺入芯中以获得所需的剂型。通常,包衣芯可包含约80mg、160mg、至多约480mg活性剂。可以使用水性或药学上可接受的溶剂介质来包覆芯颗粒。可以使用任何类型的药学上可接受的惰性粘合剂来将活性剂与惰性颗粒粘合。可以使用水溶性粘合剂。可以使用醇溶性粘合剂。可以通过将粘合剂以约0.5重量%至5重量%的浓度分散在水中来使用它们,粘合剂例如聚乙烯吡咯烷酮(PVP)、羧烷基纤维素、聚环氧乙烷、多糖(如葡聚糖)、玉米淀粉、羟丙基甲基纤维素(HPMC(曾用)或羟丙甲纤维素(现用))、羟丙基纤维素等。活性剂可以以溶液形式或混悬液形式存在于该包衣制剂中。取决于包衣制剂的粘度,活性剂的浓度可以在约0.1重量%至约20重量%之间变化。
在一个实施例中,活性芯通过造粒或通过挤出和滚圆来制备。将活性剂、粘合剂(如PVP)、任选的溶解速率控制聚合物(如高粘度HPMC(羟丙甲纤维素))以及任选的其他药学上可接受的赋形剂在高剪切造粒机(例如, 造粒机)或流化床造粒机(例如,
造粒机)或流化床造粒机(例如, GPCG造粒机)中混合,通过加入/喷洒造粒流体(如水或醇)而造粒以形成附聚物,并干燥。使用挤出机将湿块挤出并滚圆以产生球形颗粒(珠粒)。在这些实施例中,以挤出或粒化芯的总重量计,药物负载量可以是90重量%。
GPCG造粒机)中混合,通过加入/喷洒造粒流体(如水或醇)而造粒以形成附聚物,并干燥。使用挤出机将湿块挤出并滚圆以产生球形颗粒(珠粒)。在这些实施例中,以挤出或粒化芯的总重量计,药物负载量可以是90重量%。
在一个实施例中,包含活性剂的颗粒上的一层膜包衣包含增塑的肠溶聚合物,另一层包含非水溶性聚合物和增塑的水分散性/肠溶聚合物的混合物。非水溶性聚合物和水分散性聚合物以约10:1至1:1,或约4:1至1:1的重量比存在。以多颗粒剂型的总重量计,包衣的总重量为约15重量%至80重量%,或约20重量%至约60重量%。
中间含酸膜是任选的。如果包括,中间含酸膜可以包括有机酸,例如富马酸、柠檬酸、琥珀酸、酒石酸、苹果酸、马来酸等;和粘合剂,例如PVP。通常使用水溶性聚合物或醇溶性聚合物。以包衣珠粒的总重量计,该含酸膜的重量为约5%至约20%。含酸膜中的酸延迟了肠溶聚合物在内层中的溶解,从而增加了滞后时间,并降低了活性剂从包衣珠粒上释放的速率。基于预测的体外/体内相关性,进一步优化聚合物膜外层的组成以及内膜层、中间膜层和外膜层的个体重量以实现活性剂的脉冲释放曲线。因此,脉冲释放剂量制剂被优化以在预定时间之后和/或在给予制剂的个体的消化道中的特定点释放一定量的活性剂。
肠溶聚合物的实例包括但不限于以下单独或组合的化合物或组合物:纤维素及其衍生物的酯(邻苯二甲酸乙酸纤维素、邻苯二甲酸羟丙基甲基纤维素、琥珀酸乙酸羟丙基甲基纤维素酯)、邻苯二甲酸聚乙酸乙烯酯、pH敏感甲基丙烯酸-甲基丙烯酸甲酯共聚物和虫胶。这些聚合物可以用作干粉或水性分散体。可使用甲基丙烯酸共聚物 L100,S100,L30D(Rohm Pharma)、邻苯二甲酸乙酸纤维素
L100,S100,L30D(Rohm Pharma)、邻苯二甲酸乙酸纤维素 (EastmanChemical Co.)、邻苯二甲酸乙酸纤维素水性分散体
(EastmanChemical Co.)、邻苯二甲酸乙酸纤维素水性分散体 (FMC Corp.)和琥珀酸乙酸羟丙基甲基纤维素水性分散体
(FMC Corp.)和琥珀酸乙酸羟丙基甲基纤维素水性分散体 (Shin Etsu K.K.)。
(Shin Etsu K.K.)。
非水溶性聚合物的实例包括但不限于以下单独或组合的化合物或组合物:纤维素衍生物(例如,乙基纤维素)、聚乙酸乙烯酯( SR 30D,BASF)、基于丙烯酸乙酯和甲基丙烯酸甲酯的中性共聚物、具有季铵基团的丙烯酸酯和甲基丙烯酸酯的共聚物,例如
SR 30D,BASF)、基于丙烯酸乙酯和甲基丙烯酸甲酯的中性共聚物、具有季铵基团的丙烯酸酯和甲基丙烯酸酯的共聚物,例如 NE,RS或RS30D,RL或RL30D等。
NE,RS或RS30D,RL或RL30D等。
可以使用本领域已知的任何药物包衣方法将膜包衣施用到芯上。例如,可以使用流化床包衣。
脉冲释放剂量制剂可以通过以下步骤制备:(i)用活性和聚合物粘合剂包覆惰性颗粒,例如糖丸种(糖球),或者通过造粒和/或挤出/滚圆制备包含活性剂的颗粒以形成活性颗粒;(ii)用塑化的肠溶包衣包覆活性颗粒,从而形成塑化的肠溶包衣活性颗粒;和(iii)用非水溶性聚合物和肠溶聚合物的混合物包覆增塑的肠溶包衣活性颗粒。释放特性可以通过互换部分(ii)和(iii)来调节。可将如前所述的有机酸加入到部分(ii)和(iii)之间的膜上以进一步调节颗粒的滞后时间和活性释放曲线。
在一个实施例中,制剂可以使用单一形式的颗粒以在口服给药数小时之后提供活性剂的时控脉冲释放,或靶向特定吸收部位。在一个实施例中,掺入包含颗粒的多包衣活性剂的剂型被组合在具有一定量的速释活性剂的复合剂量制剂中,例如组合在明胶(硬明胶或软明胶)胶囊中。该实施例提供了具有活性剂的速释部分和时控脉冲释放部分的复合剂型。
任选的速释部分和包衣芯的活性剂可各自包含约10mg、20mg等的活性剂,本发明的包衣芯剂型可包含约10至80mg活性剂。
在一个实施例中,使用延迟缓释形式。
在一个实施例中,剂型可以提供活性剂的至少双峰血液曲线,例如图2中所示的曲线。在该实施例中,剂型包含至少第一量的速释活性剂和第二量的延迟缓释活性剂。例如,在本发明的剂型给药后的第一个小时内,第一部分活性剂快速释放。存在一段实质上无活性剂释放和/或无活性剂能够进入循环和/或给予的活性剂的第二部分生物不具有生物利用度时间。然后,在另一段时间(例如几个小时)之后,释放额外的活性剂,并且该第二部分的释放在一段延长的时间内发生,例如在最初给药后长达12小时或甚至更长。该第二部分的释放通常发生在无活性剂释放的滞后时间之后,因此可能在开始释放一定量活性剂之前表现出延迟的这种剂型被称为“延迟缓释”剂型。这种剂型可以单独给药,或者可以与其他剂型组合给药。
活性剂的血液水平增加是理想的,其中血液浓度对应于给药后第一个小时内的速释之后具有生物利用度的活性剂的量。一段时间后,活性剂的血液水平下降至低于理想水平或治疗水平。在活性剂的速释部分被释放之后,活性剂的第二部分可以进入循环。在实施例中,在活性剂的血液水平开始降低之后,制剂理想般地增加和/或维持血液水平处于或高于约所需浓度,而不需要给予第二剂量的活性剂。
以下实例说明了一个实施例。活性剂的第一速释部分具有初始药代动力学特征。填充剂、赋形剂等可占最终重量百分比。
延迟持续释放或延迟缓释制剂如下。每种持续释放组合物包含一定量的活性剂,其被配制为在4小时至12小时(通常6小时至12小时)的时间内释放活性剂。
加入多元醇(如甘露糖醇)、凝结剂(如 )、凝结兼润滑剂(如硬脂酸),以产生可提供延迟释放和缓释活性制剂的颗粒。延迟释放制剂的囊片、片剂或其他剂型使用本领域已知的方法制备,包括包封方法。这样的剂型通常表现出“持续释放”血液特征,仅此而已,即剂型通常在摄入后快速释放活性剂并随着时间的推移继续释放活性剂。这些组合物也可以配制成剂型,并且可以表现出缓释特征,在摄入后12小时内释放活性剂达几小时。
)、凝结兼润滑剂(如硬脂酸),以产生可提供延迟释放和缓释活性制剂的颗粒。延迟释放制剂的囊片、片剂或其他剂型使用本领域已知的方法制备,包括包封方法。这样的剂型通常表现出“持续释放”血液特征,仅此而已,即剂型通常在摄入后快速释放活性剂并随着时间的推移继续释放活性剂。这些组合物也可以配制成剂型,并且可以表现出缓释特征,在摄入后12小时内释放活性剂达几小时。
在一个实施例中,由所述组合物形成的剂型可以任选地进行基础包衣以密封片剂用于后续加工。密封剂包括例如HPMC、(聚)乙二醇(PEG)等。
在一个实施例中,将剂型与一个或多个由一种或多种聚合物材料制成的环带结合在一起,如随后描述和图17所示。使用一个或多个周向型或其他类型的聚合物材料环带,例如相对不溶的聚合物材料,其在配药期间仅仅最小限度地消蚀或降解或不消蚀或降解。典型的不溶性聚合物包括前述的非水溶性聚合物。环带的数量,环带之间的位置或间隔以及环带的厚度可以控制活性剂的释放速率。例如,如果使用多个环带,则环带之间可以存在0.5mm、1.0mm、1.5mm、2.0mm、2.5mm或3.0mm的空间。例如,每个环带可以是0.5mm、1.0mm、1.5mm或2.0mm宽,并且具有约0.1微米至100微米、或0.1微米至50微米、或0.1微米至20微米的厚度。如图17所示,在一个实施例中,囊片具有两个周向聚合物环带,每个环带20和30具有约1mm的宽度和约2mm的间隔40。环带型制剂减缓了活性剂的释放并延长了活性剂可以释放和/或进入循环的时间,即具有生物利用度的时间。在实施例中,环带延迟了活性剂释放的开始,使得存在滞后时间,也称为期间无活性剂释放的起效延迟(delay of onset)或延迟释放。起效延迟可以是给药后0小时至4小时,或者可以是0小时至3小时,或者可以是0.5小时至4小时,或者可以是1小时至2小时。
肠溶包衣还可以包括其他赋形剂或填充剂,例如滑石粉、乳糖、磷酸二钙、润滑剂(如硬脂酸镁)等。
环带型剂型可以以约2μg/cm2至10μg/cm2,通常约7μg/cm2的水平包覆有肠溶包衣。肠溶包衣延迟了活性剂的起效,使得在剂型给药后有一段时间无活性剂释放。通常,在肠溶包衣之后,由包衣环带型剂型(例如肠溶包衣环带型囊片)造成的活性剂的起效延迟可以为0.5小时至4小时,通常为1小时至2小时。
在一个实施例中,使用本领域已知的方法将先前描述的速释剂量的活性剂与肠溶包衣环带型囊片组合,以产生单个复合剂型,例如制成单个明胶胶囊。该制剂可以定制以提供特定的所需血液特征。
在实施例中,组合物包括至少速释制剂和随后在下文中描述的持续释放制剂。持续释放制剂通常不表现出活性剂的延迟起效。剂型中的药物可能会在给药之后不具有生物利用度,持续释放制剂通常不会表现出明显的这样一段时间。
在一个实施例中,片剂胶囊是包含片剂形式的活性剂的第一部分的胶囊(其被配制用于在摄入或给药后快速释放),以及片剂形式的活性剂的至少第二部分,其被配制用于持续释放,即第二部分在摄入后继续释放一定量的活性剂达6至12小时。至少15%至50%的活性剂是速释制剂,呈片剂形式并且适合快速释放。片剂胶囊的其余部分按重量计可包括活性剂的持续释放制剂或活性剂的持续释放制剂的一部分。包含活性剂的速释制剂的片剂和包含活性剂的持续释放制剂的片剂可以使用本领域已知的方法组合在单一剂型(例如,明胶胶囊)中。
在一个实施例中,粒化囊片是胶囊或囊片,其包含配制用于快速释放的活性剂颗粒的第一部分和配制用于持续释放的片剂形式的活性剂的至少第二部分。至少15%至50%的活性剂是速释制剂,并且可以相对于片剂以颗粒形式存在。在一个实施例中,至少约80%的粒化胶囊包含通常包含在单独的囊片中的颗粒形式的速释活性剂组合物。粒化囊片剂的其余部分按重量计可以包括活性剂的持续释放制剂,或者粒化囊片可以包含活性剂的持续释放制剂的一部分。包含活性剂的速释制剂的囊片和包含活性剂的持续释放制剂的囊片可以使用本领域已知的方法组合在单一剂型(例如,明胶胶囊)中。
在一个实施例中,分层片剂包含具有两层或多层的片剂(具有配制用于快速释放的活性剂)和配制用于持续释放的活性剂层。分层片剂包含一定量的摄入后快速释放的活性剂,以及可在分层片剂摄入后快速提供一定量的活性剂达6小时至12小时的活性剂的至少第二部分。至少15%至50%的活性剂是速释制剂。在一个实施例中,至少约80%的层状片剂包含速释活性剂组合物。层状片剂的其余部分按重量计可以包括活性剂的持续释放制剂,或者可以包括活性剂的持续释放制剂的一部分。制剂可以例如在压片机中以常规方式组合,使得在加工之后,最终的片剂剂型具有两层或多层,其中至少第一层包含活性剂的速释制剂和第二层包含活性剂的持续释放制剂。
在一个实施例中,活性剂为持续释放组合物的至少20重量%至30重量%,30重量%至60重量%或70重量%,其中组合物的剩余重量为赋形剂,例如填充剂、润滑剂、聚合物等。在一个实施例中,聚合物可以以持续释放组合物的5重量%至20重量%存在,在一个实施例中以持续释放组合物的7重量%至10重量%存在,并且在一个实施例中以持续释放组合物的10重量%至16.5重量%存在。在一个实施例中,聚合物是纤维素聚合物,例如Methocel K4M并且以约10重量%存在。持续释放制剂可以通过直接压制或湿法造粒来制备。
该制剂可以压制成片剂,或者可以直接与食物混合。这种组合物应包含至少0.1%的活性化合物。组合物和配剂的百分比可以变化,例如单位重量的约2%至约60%。
赋形剂包括但不限于一种或多种药学上可接受的惰性稀释剂;可吸收的食用载体;促进崩解的崩解剂,例如改性纤维素衍生物、改性淀粉衍生物等,应注意的是,本领域技术人员认识到包括粘合剂和润滑剂在内的其他成分也会影响剂型的溶出曲线;硬或软壳明胶胶囊;磷酸二钙;粘合剂,如黄蓍胶、阿拉伯胶、玉米淀粉或明胶;崩解剂,如玉米淀粉、马铃薯淀粉、海藻酸等;润滑剂,如硬脂酸镁;甜味剂,如蔗糖、乳糖或糖精;调味剂,如薄荷、冬青油、樱桃香精;一种或多种表面活性剂,如离子表面活性剂、非离子表面活性剂和/或胆汁盐表面活性剂,其中阴离子表面活性剂包括烷基硫酸钠(十二烷基硫酸钠)和磺基琥珀酸酯衍生物(如多库酯钠),非离子表面活性剂包括聚氧乙烯脱水山梨糖醇脂肪酸酯(聚山梨酯)(如 20、
20、 80、
80、 40、
40、 20)、聚乙二醇脂肪酸酯(如
20)、聚乙二醇脂肪酸酯(如 44/14、
44/14、 50/13)、饱和聚乙二醇化甘油酯(包括甘油单酯、甘油二酯或甘油三酯)、中链甘油单酯(6至10个碳)(例如单辛酸甘油酯(
50/13)、饱和聚乙二醇化甘油酯(包括甘油单酯、甘油二酯或甘油三酯)、中链甘油单酯(6至10个碳)(例如单辛酸甘油酯( 308)、单己酸甘油酯(
308)、单己酸甘油酯( MCM C-8)、辛酸/癸酸甘油酯(
MCM C-8)、辛酸/癸酸甘油酯( MCM)、聚氧乙烯甘油基辛酸酯和聚氧乙烯甘油基己酸酯
MCM)、聚氧乙烯甘油基辛酸酯和聚氧乙烯甘油基己酸酯 )、中链脂肪酸酯(如三癸酸甘油酯和glyceryltricarilate(
)、中链脂肪酸酯(如三癸酸甘油酯和glyceryltricarilate( 612))、环氧乙烷和环氧丙烷的嵌段聚合物、聚氧乙烯-聚氧丙烯嵌段共聚物(如泊洛沙姆188(
612))、环氧乙烷和环氧丙烷的嵌段聚合物、聚氧乙烯-聚氧丙烯嵌段共聚物(如泊洛沙姆188( F-68)、泊洛沙姆237(
F-68)、泊洛沙姆237( F-87)、泊洛沙姆338(
F-87)、泊洛沙姆338( F-108)、泊洛沙姆407(
F-108)、泊洛沙姆407( F-127)、泊洛沙姆124(
F-127)、泊洛沙姆124( L-44))、聚乙二醇硬脂酸酯-聚乙氧基化(40)硬脂酸(
L-44))、聚乙二醇硬脂酸酯-聚乙氧基化(40)硬脂酸( 52)、乙氧基化蓖麻油-聚乙氧基化(60)氢化蓖麻油(
52)、乙氧基化蓖麻油-聚乙氧基化(60)氢化蓖麻油( EL)、乙氧基化氢硬脂酸聚乙二醇660羟基硬脂酸酯(
EL)、乙氧基化氢硬脂酸聚乙二醇660羟基硬脂酸酯( HS 15)、聚氧乙烯烷基醚(12至18个碳)(如聚氧乙烯20十六十八烷基醚(
HS 15)、聚氧乙烯烷基醚(12至18个碳)(如聚氧乙烯20十六十八烷基醚( G-3713)、聚氧乙烯10油基醚(
G-3713)、聚氧乙烯10油基醚( 96,
96, 97,Oleth 10)、聚乙二醇醚(TritonTM X-100、TritonTM X-114、TritonTM X-405、TritonTM N-101))、以及卵磷脂(如磷脂(二肉豆蔻酰DL-α-磷脂酰胆碱)),胆汁盐表面活性剂包括脱氧胆酸、脱氧胆酸钠、胆酸、牛磺胆酸钠;等等。胶囊剂型还可以包含液体载体。其他材料可作为包衣存在或以其他方式改变剂型的物理形式,例如片剂、丸剂,或胶囊可包覆有虫胶和/或糖。糖浆或酏剂可包含活性剂、作为甜味剂的蔗糖、作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯、染料和调味剂。
97,Oleth 10)、聚乙二醇醚(TritonTM X-100、TritonTM X-114、TritonTM X-405、TritonTM N-101))、以及卵磷脂(如磷脂(二肉豆蔻酰DL-α-磷脂酰胆碱)),胆汁盐表面活性剂包括脱氧胆酸、脱氧胆酸钠、胆酸、牛磺胆酸钠;等等。胶囊剂型还可以包含液体载体。其他材料可作为包衣存在或以其他方式改变剂型的物理形式,例如片剂、丸剂,或胶囊可包覆有虫胶和/或糖。糖浆或酏剂可包含活性剂、作为甜味剂的蔗糖、作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯、染料和调味剂。
在实施例中,制剂中可以包含其他活性剂。
在一个实施例中,剂型是液体填充的软凝胶胶囊,其包含具有脂质、表面活性剂和溶剂的赋形剂。胶囊可以包含速释、延迟释放、持续释放或控释的制剂。
该制剂可以包含赋形剂,例如一种或多种脂肪酸。该方法涉及将难溶于水的活性试剂溶解、熔化或混悬于一种或多种脂肪酸、缀合脂肪酸、具有高HLB值的(半)固体表面活性剂和/或亲水性聚合物中。合适的脂肪酸包括C10-C18脂肪酸,优选C16-C18脂肪酸。合适的缀合脂肪酸包括与甘油(例如甘油单酯)、单糖和/或聚乙二醇(PEG)缀合的C10-C18脂肪酸,优选C16-C18脂肪酸。合适的亲水性聚合物包括泊洛沙姆和泊洛沙胺。
合适的脂肪酸包括C10-C18脂肪酸,更优选C16-C18脂肪酸。示例性脂肪酸包括但不限于十二酸(月桂酸)、十四酸(肉豆蔻酸)、十六酸(棕榈酸)、十七酸(珠光脂酸)、十八酸(硬脂酸)、二十酸(花生酸)、二十二酸(山嵛酸)、二十四酸(木蜡酸)、二十六酸(蜡酸)、二十七碳酸(碳铈酸)、二十八酸(褐煤酸)、三十酸(蜂花酸)、三十二酸(虫漆醋酸)、三十三酸(蜡蜜酸)、三十四酸(格地酸)和三十五酸(蜡塑酸)。脂肪酸可以是饱和脂肪酸、单不饱和脂肪酸、多不饱和脂肪酸或其组合。
油,例如植物油(如大豆油),可以单独使用或与上面列出的包衣材料组合使用。大豆油包含14.4%饱和脂肪酸、23.3%单不饱和脂肪酸(如油酸)和57.9%多不饱和脂肪酸(如亚油酸和α亚油酸)。
在一个实施例中,脂肪酸与甘油、单糖(如山梨糖醇或脱水山梨糖醇)、聚环氧烷(如聚乙二醇和聚丙二醇)或其组合共价偶联。这些材料被称为缀合脂肪酸。合适的缀合脂肪酸包括但不限于脂肪酸聚乙二醇酯(例如,以商品名 市售的那些)、脂肪酸脱水山梨糖醇酯(例如,脱水山梨糖醇单硬脂酸酯)、上面列出的脂肪酸的甘油脂肪酸酯(例如,山嵛酸甘油酯和单硬脂酸甘油酯)以及它们的组合。
市售的那些)、脂肪酸脱水山梨糖醇酯(例如,脱水山梨糖醇单硬脂酸酯)、上面列出的脂肪酸的甘油脂肪酸酯(例如,山嵛酸甘油酯和单硬脂酸甘油酯)以及它们的组合。
脂肪酸的浓度范围为组合物(微粒和载体)的约1重量%至约20重量%,优选约5重量%至约15重量%。
非水溶性活性剂可以用一种或多种表面活性剂单独包覆,或与一种或多种脂肪酸或缀合脂肪酸和/或一种或多种亲水性聚合物组合包覆。在一个实施例中,表面活性剂具有大于约10、大于约12、大于约14或大于约16(在1-18的范围内)的HLB值。具有所期望HLB的表面活性剂在本领域中是已知的。表面活性剂可以是阴离子型、阳离子型或非离子型。在一个实施例中,表面活性剂是非离子表面活性剂。
这种表面活性剂的实例包括但不限于聚山梨酯20、40和80(以名称 销售),聚氧乙烯单硬脂酸酯、一些糖酯(如蔗糖单月桂酸酯)、乙氧基化壬基酚、α-烯烃磺酸酯、乙氧基化牛脂胺、环氧乙烷/环氧丙烷嵌段共聚物、乙氧基化大豆胺、脂肪酸和醇、聚乙氧基化蓖麻油、聚山梨酯、聚氧乙烯烷基醚和聚氧乙烯硬脂酸酯。
销售),聚氧乙烯单硬脂酸酯、一些糖酯(如蔗糖单月桂酸酯)、乙氧基化壬基酚、α-烯烃磺酸酯、乙氧基化牛脂胺、环氧乙烷/环氧丙烷嵌段共聚物、乙氧基化大豆胺、脂肪酸和醇、聚乙氧基化蓖麻油、聚山梨酯、聚氧乙烯烷基醚和聚氧乙烯硬脂酸酯。
在一个实施例中,表面活性剂是包含脂肪酸链的高HLB表面活性剂。合适的表面活性剂包括但不限于聚乙氧基化蓖麻油、聚山梨酯、聚氧乙烯烷基醚和聚氧乙烯硬脂酸酯。
聚氧乙烯蓖麻油衍生物主要包含用30至50个环氧乙烷分子的乙氧基化的蓖麻油基甘油。聚山梨酯或聚氧乙烯脱水山梨糖醇脂肪酸酯是一系列山梨糖醇及其酸酐的部分脂肪酸酯,每摩尔山梨糖醇及其酸酐与约20、5或4摩尔环氧乙烷共聚。所得产物是具有宽分子量范围的分子的混合物。聚氧乙烯烷基醚是一系列的直链脂肪醇(正醇)(如月桂醇、肉豆蔻醇、鲸蜡醇和硬脂醇)的聚氧乙烯乙二醇醚。聚氧乙烯硬脂酸酯是通过硬脂酸的聚乙氧基化产生的。
不希望受任何理论束缚,据信表面活性剂的亲水部分增强了活性试剂与水性溶解介质在体外或体内的相容性,并且脂肪酸侧链通过脂肪酸氧化增强了吸收。在脂肪酸氧化过程中,细胞内Ca2+被消耗,导致间隙连接变宽,从而允许活性试剂在细胞之间通过。此外,例如通过防止活性试剂的氧化,这种包衣颗粒可以比单独的药物更稳定。
表面活性剂的浓度为组合物(微粒和载体)的约1重量%至约50重量%,优选约5重量%至约15重量%。
合适的亲水性聚合物包括但不限于泊洛沙姆、泊洛沙胺、聚乙二醇、聚乙烯醇、聚乙烯吡咯烷酮、聚(乙烯醇)、纤维素材料(如羟丙基纤维素、羟甲基纤维素、羟丙基甲基纤维素)、明胶、羧甲基纤维素和多肽。
亲水性聚合物的浓度为组合物的约1重量%至约50重量%,更优选约5重量%至约重量15%。如果亲水性聚合物是聚乙二醇,则浓度为组合物的约1重量%至约80重量%,约30重量%至约60重量%,约35重量%至约60重量%或组合物(微粒和载体)的约40重量%至约60重量%。
在一个实施例中,通过将药物和包衣材料的混合物加入至药学上可接受的载体来形成微粒。在一个实施例中,载体是亲水性或亲脂性载体。所得到的颗粒混悬在载体中。载体可以是单一组分或多种组分的混合物。载体可以包括溶剂、表面活性剂或其他赋形剂。载体材料可以改变或更改药物从微粒释放的速率和/或药物的溶解速率。由于微粒的控释特性和载体的控释特性,组合物可以表现出双相释放曲线。改变载体材料的定性和定量组成可以允许调节活性试剂的释放曲线。载体可以包含一种或多种调节活性试剂的释放的速率控制赋形剂。示例性速率控制赋形剂包括但不限于山嵛酸甘油酯、
 氢化植物油、蜂蜡、纤维素聚合物(如羟丙甲纤维素)、海藻酸盐、
氢化植物油、蜂蜡、纤维素聚合物(如羟丙甲纤维素)、海藻酸盐、 及其组合。
及其组合。
在一个实施例中,载体是亲水性载体,其包含HLB值大于约10、大于约12、大于约14或大于约16和/或水溶性的表面活性剂。示例性亲水性载体包括但不限于聚乙二醇、聚氧乙烯32月桂酸甘油酯(可购自Abitech,商品名为 M-44)、聚氧乙烯8辛酸甘油酯(可购自Abitech,商品名为
M-44)、聚氧乙烯8辛酸甘油酯(可购自Abitech,商品名为 MC-8)和四氢呋喃聚乙二醇醚。亲水性载体可以进一步包含一种或多种可混溶的溶剂,例如甘油、乙醇、四氢呋喃聚乙二醇醚和辛酰己酰聚乙二醇-8(可购自Gattefosse S.A.,Saint Priest,France,商品名为
MC-8)和四氢呋喃聚乙二醇醚。亲水性载体可以进一步包含一种或多种可混溶的溶剂,例如甘油、乙醇、四氢呋喃聚乙二醇醚和辛酰己酰聚乙二醇-8(可购自Gattefosse S.A.,Saint Priest,France,商品名为 )。
)。
在一个实施例中,亲水性载体是水或醇。在另一个实施例中,载体是包含聚乙二醇和任选的一种或多种表面活性剂和/或水的亲水性载体混合物。在一个具体的实施例中,亲水性载体为PEG 400(例如,组合物的57重量%)、水(例如,组合物的8重量%)和 20(例如,组合物的10重量%)的混合物。亲水性载体还可以包含
20(例如,组合物的10重量%)的混合物。亲水性载体还可以包含 RH 40。亲水性载体的浓度通常为组合物(微粒和载体)的约50重量%至约85重量%,优选组合物的约70重量%至约80重量%。
RH 40。亲水性载体的浓度通常为组合物(微粒和载体)的约50重量%至约85重量%,优选组合物的约70重量%至约80重量%。
在另一个实施例中,载体是亲脂性载体。在一个优选实施例中,亲脂性载体具有小于约10的HLB值和/或油溶性。示例性亲脂性油性载体包括但不限于植物油、中链甘油单酯/甘油二酯/甘油三酯、硬脂酸甘油酯(可购自Sasol,商品名为 )、聚氧乙烯化油酸甘油酯(可购自Gattefosse,SA.,Saint Priest,France,商品名为
)、聚氧乙烯化油酸甘油酯(可购自Gattefosse,SA.,Saint Priest,France,商品名为 )、矿物油、甘油单酯和甘油二酯乳化剂(例如,单油酸甘油酯、单癸酸甘油酯、单辛酸甘油酯、单辛酸丙二醇酯和单月桂酸丙二醇酯(可购自Abitec Corp.,Columbus,Ohio,商品名为
)、矿物油、甘油单酯和甘油二酯乳化剂(例如,单油酸甘油酯、单癸酸甘油酯、单辛酸甘油酯、单辛酸丙二醇酯和单月桂酸丙二醇酯(可购自Abitec Corp.,Columbus,Ohio,商品名为 ))和二甲基聚硅氧烷(例如,二甲基硅油)。
))和二甲基聚硅氧烷(例如,二甲基硅油)。
亲脂性载体的浓度通常为组合物(微粒和载体)的约10重量%至约50重量%,优选约5重量%至约35重量%。
所述组合物可包含一种或多种药学上可接受的赋形剂,其被认为是安全和有效的,并且可被给予个体而不引起不希望的生物副作用或不希望的相互作用。示例性添加剂包括但不限于溶剂、混悬剂、分散剂、缓冲剂、pH调节剂、等渗性改性剂、防腐剂、抗微生物剂及其组合。
用于包含在本文所述组合物中的合适添加剂包括但不限于抗氧化剂(例如α生育酚,如维生素E乙酸酯、抗坏血酸、丁基化羟基茴香醚和丁基化羟基甲苯);极性溶剂(例如水、丙二醇和甘油);疏水性溶剂(例如玉米油、蓖麻油、大豆油、橄榄油、鱼油、花生油、薄荷油、红花油、芝麻油、源自椰子油或棕榈籽油的中链甘油三酯、辛酸甘油三酯、癸酸甘油三酯);和增粘剂(例如明胶、甘油、角叉菜胶、胶体二氧化硅、氢化植物油;聚维酮和海藻酸丙二醇酯)。
通常将本文所述的微粒组合物配制用于口服或肠胃外给药。合适的口服剂型包括胶囊,例如硬或软的明胶或非明胶胶囊,或口服混悬液或糖浆(例如图21)。合适的胃肠外制剂包括混悬液。
在一个实施例中,微粒组合物(混悬于亲水性或亲脂性载体中的微粒)被包封在胶囊,例如硬胶囊或软胶囊中。胶囊可以由天然和/或合成成膜聚合物制备。合适的天然成膜材料包括但不限于明胶。非明胶胶囊包括但不限于由角叉菜胶、虫胶、海藻酸盐、果胶和玉米醇溶蛋白制成的胶囊。合适的合成成膜聚合物包括但不限于甲基纤维素、琥珀酸乙酸羟丙基甲基纤维素、邻苯二甲酸羟丙基甲基纤维素、邻苯二甲酸乙酸纤维素和丙烯酸酯(如聚(甲基)丙烯酸酯)。
组合物也可以包封在肠溶胶囊中,其中胶囊包覆有肠溶包衣或胶囊壳包含肠溶聚合物,如Banner Pharmacaps,Inc.的WO 2004/030658中所述。
硬壳胶囊通常通过形成两个胶囊半部,用填充溶液填充其中一个半部,然后将两个胶囊半部密封在一起以形成成品胶囊来制备。软明胶胶囊通常使用旋转模具封装工艺制备。这样的工艺在本领域中是已知的。
胶囊壳可以包含一种或多种添加剂。合适的壳添加剂包括增塑剂、遮光剂、着色剂、湿润剂、防腐剂、调味剂和缓冲盐和酸及其组合。
增塑剂是加入到明胶中的化学试剂,使材料更柔软、更柔韧。合适的增塑剂包括但不限于甘油、山梨糖醇溶液(山梨糖醇和脱水山梨糖醇的混合物)、以及其他多元醇(如丙二醇和麦芽糖醇或其组合)。
遮光剂用于当封装的活性试剂对光敏感时遮蔽胶囊壳。合适的遮光剂包括二氧化钛、氧化锌、碳酸钙及其组合。
着色剂可用于销售和产品标识/区分目的。合适的着色剂包括合成和天然染料及其组合。
湿润剂可用于抑制软凝胶的水分活性。合适的湿润剂包括甘油和山梨糖醇,其通常是增塑剂组合物的组分。由于干燥的适当储存的软胶囊的水分活性低,微生物的最大风险来自霉菌和发酵物。为此,可以将防腐剂掺入胶囊壳中。合适的防腐剂包括对羟基苯甲酸烷基酯,如对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯和对羟基苯甲酸庚酯(统称为“对羟基苯甲酸酯”)或其组合。
调味剂可以用来掩盖填充制剂的令人不愉快的气味和味道。合适的调味剂包括合成和天然调味剂。由于可以交联明胶的醛的存在,调味剂的使用可能会有问题。因此,缓冲盐和酸可以与包含醛的调味剂一起使用,以抑制明胶的交联。
也可以使用中链甘油三酯。如本文所使用,“中链甘油三酯”意指通过甘油与三种脂肪酸的酯化形成的C6-C12酯链。中链甘油三酯有多种来源,例如椰子油、棕榈仁油等。分馏椰子油是最常用的中链甘油三酯来源。市售中链甘油三酯的实例可以包括由SasolGermany GMBH生产的 810,812或881、由Abitec Corporation生产的
810,812或881、由Abitec Corporation生产的 300,355,或810D、由Stepan Company生产的
300,355,或810D、由Stepan Company生产的 M5、由Croda Inc生产的
M5、由Croda Inc生产的 GTC/C和由Gattesfosse Group生产的
GTC/C和由Gattesfosse Group生产的 Lipophile WL 1349。在一个示例性实施例中,中链甘油三酯可以包含
Lipophile WL 1349。在一个示例性实施例中,中链甘油三酯可以包含 355,其为辛酸(C8)/癸酸(C10)甘油三酯。
355,其为辛酸(C8)/癸酸(C10)甘油三酯。
药物制剂中可以包含不同量的中链甘油三酯。在一个或多个实施例中,药物制剂可以包含约50重量%至约95重量%的中链甘油三酯,或约85重量%至约95重量%的中链甘油三酯。此外,在示例性实施例中,药物制剂可包含约100mg至约300mg或约200mg至300mg重量的中等链甘油三酯,或约225mg至275mg重量的中等链甘油三酯,或约250mg重量的中链甘油三酯。
与中链甘油三酯类似,“中链甘油单酯”和“中链甘油二酯”分别是通过甘油与一种脂肪酸或两种脂肪酸的酯化形成的C6-C12酯链。市售中链甘油单酯/甘油二酯的实例可以包括由Abitec生产的 产品。也可考虑使用还包含中链甘油三酯的中链甘油单酯/甘油二酯化合物,例如由Sasol生产的市售
产品。也可考虑使用还包含中链甘油三酯的中链甘油单酯/甘油二酯化合物,例如由Sasol生产的市售 组合物。
组合物。
在示例性实施例中,中链甘油单酯/甘油二酯可以包含 MCM,其包括辛酸(C8)/癸酸(C10)中链甘油单酯/甘油二酯。虽然
MCM,其包括辛酸(C8)/癸酸(C10)中链甘油单酯/甘油二酯。虽然 MCM产品系列的所有级别适用于本发明,例如国家处方(NF)级或
MCM产品系列的所有级别适用于本发明,例如国家处方(NF)级或 MCM EP,但可能需要使用EP级,因为它包含3%甘油,而NF级包括7%甘油。
MCM EP,但可能需要使用EP级,因为它包含3%甘油,而NF级包括7%甘油。
根据一个或多个实施例,药物制剂可以包含约5重量%至约25重量%的中链甘油单酯/甘油二酯,或约5重量%至约15重量%的中链甘油单酯/甘油二酯。此外,在示例性实施例中,药物制剂可以包含约20mg至50mg重量的中链甘油单酯/甘油二酯,或约25mg至30mg重量的中链甘油单酯/甘油二酯,或约25mg重量的中链甘油单酯/甘油二酯。
不受理论束缚,中链甘油三酯和中链甘油单酯/甘油二酯的混合物对于液体填充的硬凝胶胶囊制剂内的活性成分的生物利用度是重要的。尽管软凝胶胶囊可能仅包含中链甘油单酯/甘油二酯,但仅具有中链甘油单酯/甘油二酯的硬明胶胶囊可能不能提供成品剂型的必需的物理稳定性。但是,硬明胶胶囊内的中链甘油三酯和中链甘油单酯/甘油二酯的混合物可以实现活性药物成分的所需产品稳定性、溶解性和生物利用度。因此,根据本发明,中链甘油三酯与中链甘油单酯/甘油二酯的重量比有助于在将混合物加入胶囊中之前和之后非乳化混合物中的活性药物成分(例如度他雄胺)的溶解度和稳定性。中链甘油三酯和中链甘油单酯/甘油二酯可以以约10:1至约5:1,或约10:1至约7:1的重量比存在。
除了上述组分之外,可以使用本领域技术人员已知的其他赋形剂,例如口服组合物中使用的赋形剂可以是稀释剂、粘合剂、润滑剂、崩解剂、调味剂、着色剂、稳定剂、助流剂、增塑剂、防腐剂和甜味剂。
稀释剂可以包括液体稀释剂,例如任何长链甘油三酯(落花生油、杏仁油、花生油、棕榈油、棕榈仁油、黑加仑籽油、米糠油、大豆油、芥花籽油、玉米油、椰子油、棉籽油、蓖麻油、橄榄油、楝树油(苦楝)、芝麻油、樱草油、植物油、Lipex 108(Abitec)、小麦胚芽油、鱼油、菜籽油、葵花籽油和saffola油。在替代实施例中,预期可以使用其他稀释剂,例如选自硅酸铝钙( 106PQ)、碳酸钙、磷酸氢钙、磷酸三钙、硫酸钙、微晶纤维素、微晶硅化纤维素、粉状纤维素、葡萄糖结合剂、葡萄糖、果糖、乳糖醇、无水乳糖、一水乳糖、二水乳糖、三水乳糖、甘露糖醇山梨糖醇、淀粉、预胶化淀粉、蔗糖、滑石粉、木糖醇、麦芽糖麦芽糖糊精、麦芽糖醇、二氧化硅、HPMC及其组合。
106PQ)、碳酸钙、磷酸氢钙、磷酸三钙、硫酸钙、微晶纤维素、微晶硅化纤维素、粉状纤维素、葡萄糖结合剂、葡萄糖、果糖、乳糖醇、无水乳糖、一水乳糖、二水乳糖、三水乳糖、甘露糖醇山梨糖醇、淀粉、预胶化淀粉、蔗糖、滑石粉、木糖醇、麦芽糖麦芽糖糊精、麦芽糖醇、二氧化硅、HPMC及其组合。
该制剂包括给药途径、制备类型、活性剂的非活性成分的释放、稳定性、放大规模、新活性剂制备工艺、新制剂工艺。
体内性能评估包括药代动力学数据,如pK/pD,如Tmax、Cmax、血浆浓度曲线、疗效、副作用等。
其他释放曲线包括但不限于受控、肠溶、持续、快速、多阶段等。
本发明包括本发明的多潘立酮和氘代多潘立酮制剂的其他已知和待定用途。
先前引用的以及下面列出的每个参考文献通过引用整体并入本文:
Chang和Robinson,《第4章:片剂和颗粒通过包衣的持续药物释放,药物剂型:片剂(chapter 4:Sustained Drug Release from Tablets and Particles Through Coating,Pharmaceutical Dosage Forms:Tablets)》,第3卷,编辑Lieberman,Lachman和Schwartz,Marcel Dekker,Inc.,1991。
Campbell和Sackett,《第3章:薄膜包衣,药物单位操作:包衣(Chapter 3:Filmcoating,Pharmaceutical Unit Operations:Coating)》,编辑Avis,Shukla和Chang,Interpharm Press,Inc.,1999。
Youssef等人,《通过LC-ESI-MS/MS鉴定胃轻瘫患者血浆和尿液中的多潘立酮代谢物,外来物(Identification of Domperidone Metabolites in Plasma and Urine ofGastroparesis Patients with LC-ESI-MS/MS,Xenobiotica)》43(2013)1073-1083。
Michaud等人,《通过荧光检测测定多潘立酮和三种用于体外药物代谢研究的主要代谢产物的改进型HPLC测定法,色谱杂志(An Improved HPLC Assay with FluorescenceDetection for the Determination of Domperidone and Three Major Metabolitesfor Application to in vitro Drug Metabolism Studies,J.Chromatogr.)》B,852(2007)611-616。
所公开的组合物包含治疗量的多潘立酮或氘代多潘立酮或其药学上可接受的盐和至少一种赋形剂。赋形剂可以例如促进活性试剂的递送。如前所述,可以包括其他活性试剂,例如止痛剂、麻醉剂、抗氧化剂、抗微生物剂、抗真菌剂、维生素等。
药物组合物中可包含一种或多种止痛剂,以缓解由胃轻瘫引起的疼痛。止痛剂的实例包括但不限于单纯的止痛剂,例如扑热息痛或阿司匹林;非类固醇消炎药(NSAIDS),如布洛芬、双氯芬酸钠或萘普生钠;和/或鸦片类药物,如可待因、双氢可待因、磷酸可待因、芬太尼、美沙酮、盐酸曲马多、盐酸右丙氧芬、吗啡、羟考酮、丁丙诺啡或盐酸哌替啶。
一种或多种麻醉剂可以包含在药物组合物中以诱导由胃轻瘫引起的疼痛感的暂时和可逆的缺失。麻醉剂的实例包括但不限于利多卡因、苯佐卡因、布比卡因、阿替卡因、可卡因、依替卡因、氟卡同胺、甲哌卡因、普莫卡因、丙胺卡因、普鲁卡因、氯普鲁卡因、奥布卡因、丙美卡因、罗哌卡因、丁卡因、达克罗宁、地步卡因、氯二甲苯酚、辛可卡因、地昔伐卡因、二胺卡因、海克卡因、左布比卡因、丙氧卡因、吡咯卡因、利索卡因、罗杜卡因及其药学上可接受的衍生物。
药物组合物中可以包含一种或多种抗氧化剂。抗氧化剂的实例包括但不限于棕榈酸抗坏血酸酯、丁基化羟基茴香醚、丁基化羟基甲苯、二亚乙基三胺五乙酸(DTPA)、依地酸酯(EDTA)、单硫代甘油、抗坏血酸钠、甲醛次硫酸钠、偏亚硫酸氢钠、丁基化羟基甲苯(BHT)、丁基化羟基茴香醚(BHA)、亚硫酸氢钠、氨三乙酸酯、维生素E或其衍生物、以及没食子酸丙酯。
在一个实施例中,将多潘立酮或氘代多潘立酮以及任选的任何其他试剂掺入颗粒(包括纳米颗粒)之中或之上。多潘立酮或氘代多潘立酮颗粒可以混悬或分散在水性介质中。因此,粒度可以为微粒(μm)至纳米颗粒(nm)。
患有胃轻瘫或其他胃肠运动障碍的人或其他哺乳动物可以通过每天一次或多次定期给予本发明公开的药物组合物来治疗。包含多潘立酮或氘代多潘立酮和任何补充治疗剂的药剂以组成治疗有效剂量的量存在于组合物中。治疗有效剂量是在治疗之后相对于这些症状的给药前状态实现了一定程度的症状减轻的药剂的量。
药物组合物可以每天给予一至四次。
在一个实施例中,多潘立酮或氘代多潘立酮以0.5mg至100mg,或0.05%至10.0%,或0.07mg/kg至1.43mg/kg的范围给予。在一个实施例中,多潘立酮或氘代多潘立酮以1mg至60mg,或0.1%至6.0%,或0.014mg/kg至0.86mg/kg的范围给予。在一个实施例中,多潘立酮或氘代多潘立酮以2.0mg至30mg,或0.2%至3.0%,或0.028mg/kg至0.43mg/kg的范围给予。
该制剂还可以包含赋形剂。示例性赋形剂包括但不限于粘合剂、填充剂、溶剂、润滑剂、抗氧化剂、缓冲剂、盐、表面活性剂、维生素、颜料、调味剂、崩解剂和/或增塑剂。
可将固体赋形剂加入药物组合物中,然后研磨并形成片剂。示例性固体赋形剂包括但不限于糖,包括乳糖、蔗糖、三氯蔗糖、甘露糖醇或山梨糖醇;纤维素基材料,例如玉米淀粉、小麦淀粉、大米淀粉、马铃薯淀粉、黄蓍胶、明胶、甲基纤维素、聚乙烯吡咯烷酮、羟丙基甲基纤维素和/或羧甲基纤维素钠。促进摄入时片剂崩解的赋形剂包括但不限于琼脂、海藻酸和/或其盐、甘露糖醇、微晶纤维素、玉米淀粉、柑橘果肉、月桂基硫酸钠、膨润土、羟基乙酸淀粉钠、羧甲基纤维素钙、粘土、对准物(align)、树胶、木质纤维素、粉状天然海绵和/或阳离子交换树脂。
该组合物可以包括其他赋形剂和添加剂以改变一种或多种组合物特性,例如包衣能力、粘度、适口性等。改善适口性的赋形剂可以包括但不限于糖,如乳糖、蔗糖、三氯蔗糖、葡萄糖、甘露糖醇或山梨糖醇;天然甜味剂,如蜂蜜;纤维素基添加剂,如玉米淀粉、小麦淀粉、大米淀粉;其他甜味剂,如阿斯巴甜、环拉酸及其盐、二氢查尔酮、甘草酸盐、莫内林、糖精钠、索马甜或乙酰磺胺酸钾;和/或其他甜味剂或调味剂。
任选的粘度调节剂赋形剂可以加入组合物的液体制剂中以改变组合物的流动特性。可以修改流动特性、以结合到特定的装置或应用机构中以将组合物施用到治疗部位。示例性粘度调节赋形剂包括但不限于甘油、卡波姆均聚物、卡波姆共聚物、阿拉伯胶(阿拉伯树胶)、琼脂、硅酸铝镁、海藻酸钠、硬脂酸钠、墨角藻、膨润土、卡波姆、角叉菜胶、角豆胶、角叉菜、葡萄糖、红藻胶、明胶、印度树胶、瓜尔豆胶、梧桐胶、黄蓍胶、黄原胶、锂蒙脱石、乳糖、麦芽糖糊精、甘露糖醇、蔗糖、山梨糖醇、蜂蜜、玉米淀粉、小麦淀粉、大米淀粉、马铃薯淀粉、乙二醇、纤维素、乙基纤维素、乙基羟乙基纤维素、乙基甲基纤维素、甲基纤维素、羟乙基纤维素、羟乙基甲基纤维素、羟丙基纤维素、聚(甲基丙烯酸羟乙酯)、氧代聚明胶、果胶、聚明胶肽、碳酸丙烯酯、甲基乙烯基醚/马来酸酐共聚物(PVM/MA)、聚(甲基丙烯酸甲氧基乙酯)、聚(甲基丙烯酸甲氧基乙氧基乙酯)、羟丙基纤维素、羟丙基甲基纤维素、羧甲基-纤维素(CMC)(包括其盐)、二氧化硅、聚乙烯吡咯烷酮(PVP)和/或
药物组合物还可以包含一种或多种粘合剂、填充剂、溶剂、润滑剂、抗氧化剂、缓冲剂、盐、表面活性剂、维生素、颜料、调味剂、崩解剂和/或增塑剂。示例性粘合剂包括但不限于任何先前公开的淀粉(如玉米淀粉、小麦淀粉、大米淀粉和/或马铃薯淀粉)、纤维素衍生物(如甲基纤维素、羧甲基纤维素、羟乙基纤维素、羟乙基甲基纤维素等)、任何分子量或等级的POLYOXTM聚环氧乙烷聚合物(辐射或不辐射)、聚乙烯吡咯烷酮(PVP)、 微晶纤维素粉末等。示例性填充剂包括但不限于任何先前公开的糖和淀粉、纤维素、钙盐、硅藻土和/或二氧化钛。示例性缓冲液包括但不限于乙酸盐缓冲液、柠檬酸盐缓冲液和/或磷酸盐缓冲液。
微晶纤维素粉末等。示例性填充剂包括但不限于任何先前公开的糖和淀粉、纤维素、钙盐、硅藻土和/或二氧化钛。示例性缓冲液包括但不限于乙酸盐缓冲液、柠檬酸盐缓冲液和/或磷酸盐缓冲液。
加入到药物组合物中的表面活性剂可以是阴离子型、阳离子型、非离子型或两性离子型。示例性表面活性剂包括但不限于烷基硫酸钠(例如,十二烷基硫酸钠(SDS))、季铵和吡啶阳离子表面活性剂、聚山梨酯、脱水山梨糖醇酯、胆汁酸、胆汁酸盐、壬苯醇醚或聚氧乙烯二醇脂肪酸酯和/或泊洛沙姆。示例性润滑剂包括但不限于滑石粉、氢化脂肪油、硬脂酸镁、硬脂酸钙和/或硬脂酸。调味剂可以包括天然或合成香精。增塑剂包括但不限于甘油和山梨糖醇。
用多潘立酮或氘代多潘立酮治疗胃轻瘫和其他胃肠运动障碍的药物组合物可以配制成用于粘膜表面的局部口服给药。粘膜粘附递送技术通过口腔粘膜提供安全有效的多潘立酮或氘代多潘立酮的递送。这些粘膜粘附递送技术包括在口腔粘膜中扩散的所有方法:(i)被动扩散,包括跨细胞(通过细胞)和细胞旁(其中物质通过细胞周围的富含脂质的结构域),(ii)载体介导的转运,和(iii)内吞作用/胞吐作用,其中材料通过内吞途径被细胞主动摄取和排泄。生物粘附,也被称为粘膜粘附,定义了生物或合成材料粘附或“粘贴”到粘膜上的能力,导致材料长时间粘附在组织上。这种能力提供了在药物递送中的应用以及由于生物粘附剂剂型与吸收组织接触的时间延长而导致的药物生物利用度的增加(相对于标准剂型)。对于需要生物粘附的材料,它必须与粘液相互作用。粘液是保护粘膜的高度水合的粘稠的阴离子水凝胶层。粘液素由柔性糖蛋白链组成。
在该实施例中,所述药物组合物包含治疗量的多潘立酮或氘代多潘立酮和任何任选的其他活性试剂(如果存在的话)以及至少一种赋形剂,赋形剂可包含粘膜粘附剂或生物粘附剂以增加药物活性试剂与口腔粘膜之间的接触,并增加活性试剂的粘膜吸收。吸收表面是拟施用药物活性试剂的口腔粘膜下面的组织表面。药物组合物可以以软膏、乳膏、洗剂、凝胶、粉末或糊剂的形式施用,并且可以在有或没有闭塞的情况下通过薄膜或胶带或通过特定的粘贴绷带施用于治疗部位,或者通过膜或带阻塞或不通过。组合物还可以包含促进将组合物给予口腔粘膜的载体。
示例性粘膜粘附或生物粘附赋形剂包括但不限于天然、合成或生物聚合物;脂质、磷脂等。天然和/或合成聚合物的实例包括纤维素衍生物,如甲基纤维素、羧甲基纤维素、羟乙基纤维素、羟乙基甲基微晶纤维素等;天然树胶,如瓜尔豆胶、黄原胶、刺槐豆胶、刺梧桐树胶、硅酸镁铝等;聚丙烯酸酯,如 聚合物、聚卡波菲等;海藻酸盐、含硫醇聚合物、聚氧乙烯、分子量优选为1000至40,000Da的聚乙二醇(PEG)(无论直链或支链)、任何来源的分子量优选为1000至40,000Da的葡聚糖、嵌段共聚物(例如,各种粘度、分子量和乳酸/乙醇酸比例的乳酸和乙醇酸(如PLA、PGA、PLGA)的组合);任何数量和组合的重复单元的聚乙二醇-聚丙二醇嵌段共聚物(如
聚合物、聚卡波菲等;海藻酸盐、含硫醇聚合物、聚氧乙烯、分子量优选为1000至40,000Da的聚乙二醇(PEG)(无论直链或支链)、任何来源的分子量优选为1000至40,000Da的葡聚糖、嵌段共聚物(例如,各种粘度、分子量和乳酸/乙醇酸比例的乳酸和乙醇酸(如PLA、PGA、PLGA)的组合);任何数量和组合的重复单元的聚乙二醇-聚丙二醇嵌段共聚物(如 嵌段共聚物、
嵌段共聚物、 嵌段共聚物或
嵌段共聚物或 嵌段共聚物)、上述共聚物物理或化学连接单元的组合(例如,PEG-PLA或PEG-PLGA共聚物混合物)。生物粘附材料可以选自聚乙二醇、聚氧乙烯、聚丙烯酸聚合物(如
嵌段共聚物)、上述共聚物物理或化学连接单元的组合(例如,PEG-PLA或PEG-PLGA共聚物混合物)。生物粘附材料可以选自聚乙二醇、聚氧乙烯、聚丙烯酸聚合物(如 聚合物(如
聚合物(如 71G,934P,971P 974P)和聚卡波菲(如
71G,934P,971P 974P)和聚卡波菲(如 AA-1,CA-1,和CA-2聚卡波菲))、纤维素及其衍生物、聚乙二醇、
AA-1,CA-1,和CA-2聚卡波菲))、纤维素及其衍生物、聚乙二醇、 聚合物和/或纤维素衍生物或组合;可溶性聚乙烯吡咯烷酮聚合物(PVP)、卡波姆均聚物、卡波姆共聚物、一种或多种麦芽糖糊精、海藻酸盐、交联海藻酸盐胶凝胶、吸水膨胀但非水溶性纤维交联羧基官能聚合物、亲水性多糖胶、巯基聚合物(例如,巯基化壳聚糖、巯基化聚卡波菲、巯基化海藻酸盐、巯基化纤维素衍生物、巯基化羧甲基纤维素、巯基化聚丙烯酸或巯基化聚丙烯酸酯);凝集素、羟丙基甲基纤维素(HPMC)、纤维素衍生物、HPMA共聚物、水分散性聚羧化乙烯基聚合物、阳离子聚合物、非离子聚合物或阴离子聚合物。阳离子聚合物包括但不限于壳聚糖(Wella“低粘度”)、壳聚糖(Wella“高粘度”)、壳聚糖(Dr.Knapczyk)、代壳聚糖(daichitosan)H、代壳聚糖VH、Sea Cure 240、Sea Cure 210+、壳聚糖(Sigma)、聚卡波菲/代壳聚糖VH混合物、DEAE-葡聚糖和氨基葡聚糖。非离子聚合物包括但不限于硬葡聚糖、He-淀粉和HPC。阴离子聚合物包括但不限于羧甲基纤维素(CMC)(低、中或高粘度)、果胶、黄原胶和/或聚卡波菲。在一个实施例中,粘膜粘附剂是
聚合物和/或纤维素衍生物或组合;可溶性聚乙烯吡咯烷酮聚合物(PVP)、卡波姆均聚物、卡波姆共聚物、一种或多种麦芽糖糊精、海藻酸盐、交联海藻酸盐胶凝胶、吸水膨胀但非水溶性纤维交联羧基官能聚合物、亲水性多糖胶、巯基聚合物(例如,巯基化壳聚糖、巯基化聚卡波菲、巯基化海藻酸盐、巯基化纤维素衍生物、巯基化羧甲基纤维素、巯基化聚丙烯酸或巯基化聚丙烯酸酯);凝集素、羟丙基甲基纤维素(HPMC)、纤维素衍生物、HPMA共聚物、水分散性聚羧化乙烯基聚合物、阳离子聚合物、非离子聚合物或阴离子聚合物。阳离子聚合物包括但不限于壳聚糖(Wella“低粘度”)、壳聚糖(Wella“高粘度”)、壳聚糖(Dr.Knapczyk)、代壳聚糖(daichitosan)H、代壳聚糖VH、Sea Cure 240、Sea Cure 210+、壳聚糖(Sigma)、聚卡波菲/代壳聚糖VH混合物、DEAE-葡聚糖和氨基葡聚糖。非离子聚合物包括但不限于硬葡聚糖、He-淀粉和HPC。阴离子聚合物包括但不限于羧甲基纤维素(CMC)(低、中或高粘度)、果胶、黄原胶和/或聚卡波菲。在一个实施例中,粘膜粘附剂是 聚合物和/或纤维素衍生物。
聚合物和/或纤维素衍生物。
由于其粘膜粘附特性(Lehr等人,1992)和有利的毒理学性质,壳聚糖是跨肠上皮细胞的吸收促进剂。壳聚糖谷氨酸盐降低了培养的肠上皮细胞系(Caco-2)的体外跨上皮细胞电阻(TEER)(Borchard等人,1996)并增加了亲水性分子(如[14C]甘露糖醇,分子量(MW)182.2和明显在Caco-2细胞单层中的荧光素-葡聚糖(MW 4400))的转运(Artursson等人,1994;Borchard等人,1996;Schipper等人,1996)。类似地,与Caco-2细胞单层中的壳聚糖谷氨酸盐共同给药之后,肽类药物9-脱甘氨酰胺、8-精氨酸加压素(DGAVP,MW1412)的转运显著增加(Luessen等人,1997)。壳聚糖盐,如壳聚糖谷氨酸盐和壳聚糖盐酸盐,在体内用作肽药物的吸收促进剂。通过壳聚糖谷氨酸盐的胰岛素鼻腔给药显著降低了大鼠和绵羊的血糖水平(Illum等人,1994),并且在凝胶制剂中布舍瑞林(MW1299.5)和壳聚糖盐酸盐的十二指肠内施用将布舍瑞林的绝对生物利用度从0.1±0.1%增加至5.1±1.5%(Luessen等人,1996a)。这些吸收的增加可归因于壳聚糖对上皮紧密连接完整性的影响。紧密连接在保持细胞膜的选择性屏障功能方面和将细胞密封在一起以形成小分子也无法穿透的连续的细胞层方面起着关键作用。然而,紧密连接可透过水、电解质和一定尺寸的其他带电荷或不带电荷的分子(Madara 1989;Wilson和Washington 1989)。已知紧密连接对钙浓度、环AMP(cAMP)、渗透度、pH和细胞骨架状态的变化作出反应(Cereijido等人,1993)。
提出壳聚糖盐以浓度依赖性和pH依赖性方式打开紧密连接以允许大亲水性化合物的细胞旁转运。这些化合物的转运的增加可归因于壳聚糖的C-2位上的带正电荷的氨基与细胞膜上的负电荷位点和粘膜上皮细胞的紧密连接的相互作用,以允许打开紧密连接。已知壳聚糖谷氨酸盐诱导F肌动蛋白分布的变化(Artursson等人,1994)。还已知与细胞骨架F肌动蛋白相互作用的药物试剂同时增加细胞旁渗透性(Meza等人,1982)。这与F肌动蛋白直接或间接与紧密连接中的蛋白质(如ZO-1)相关联的假设(Madara1987)一致。Schipper等人(1997)已经表明,壳聚糖诱导细胞骨架F肌动蛋白和紧密连接蛋白ZO-1的重新分布。共聚焦激光扫描显微术已证实,壳聚糖能够打开紧密连接以允许大亲水性化合物的细胞旁转运(Borchard等人,1996;Schipper等人,1997)。通过增加药物在细胞表面的停留时间,粘膜粘附可能在此过程中起到另外的作用。
在实施例中,粘膜粘附/生物粘附赋形剂通常以约1%至约50%w/w的范围,或约1%至约40%w/w的范围,或约2%至约30%w/w的范围存在。可以使用单个粘膜粘附剂或生物粘附剂或组合。生物粘附增加了剂型在吸收部位的停留时间,从而可能导致药物生物利用度的增加。使用粘膜粘附剂有利于延长药物组合物与口腔粘膜之间的接触。当药物组合物与粘膜接触时,粘液中的水分塑化粘膜粘附剂,粘膜粘附剂随后通过与粘液中的糖蛋白形成弱键和/或与粘液中的糖蛋白和脂质机械互锁而与粘膜合并。粘膜粘附剂可以增加药物活性试剂与吸收表面接触的停留时间,并且可以促进吸收表面对药物活性试剂的吸收。
以下实例不是限制性的。
实例1
2,3-二氢(4,5,6,7-D4)-1H-1,3-苯并二唑-2-酮的制备:
将(D4)苯-1,2-二胺(1当量,2g,17.83mmol)和30ml无水DMF加入设有搅拌棒和氮气进出口的100ml圆底烧瓶中,然后在氮气下搅拌溶解,随后加入1-(1H-咪唑-1-羰基)-1H-咪唑(1当量,2.89g,17.83mmol),并在室温下搅拌22小时。真空蒸发溶剂,得到黄色浓稠油状物,用最少量的二氯甲烷(DCM)稀释结晶。通过真空过滤收集所需固体,用DCM洗涤并在真空下干燥,得到2.09g(15.13mmol,85%)所需产物。
实例2
2-氧代-2,3-二氢(4,5,6,7-D4)-1H-1,3-苯并二唑-1-羧酸叔丁酯的制备:
向设有搅拌棒和氮气进出口的100ml三颈圆底烧瓶中加入2,3-二氢(4,5,6,7-D4)-1H-1,3-苯并二唑-2-酮(1当量,2.09g,15.13mmol)和40ml无水DMF。向该搅拌溶液中分批加入氢化钠(1.1当量,197mg,8.200mmol),并将反应产物在相同条件下放置1.5小时。此后,滴加溶解于8ml无水DMF中的二碳酸二叔丁酯(1当量,3.30g,15.13mmol),并放置使其反应3小时。反应完成,用NH4Cl饱和溶液处理,然后用H2O稀释并用4×50ml EtOAc萃取。将有机部分合并,用Na2SO4干燥,过滤并真空浓缩至干。将由此获得的原材料通过硅胶色谱(Biotage ISOLERATM,KP-Sil 50g色谱柱,以Cy:EtOAc/90:10至纯AcOE的梯度洗脱)纯化,得到3.134g(13.15mmol,87%)所需化合物。
实例3
3-(3-氯丙基)-2-氧代-2,3-二氢(D4)-1H-1,3-苯并二唑-1-羧酸叔丁酯的制备:
向三颈圆底烧瓶中加入2-氧代-2,3-二氢-1H-1,3-苯并二唑-1-羧酸叔丁酯(1当量,3.134g,13.15mmol)的60毫升无水DMF溶液,并在室温下搅拌。向该溶液中分批加入碳酸钾(3当量,5.452g,39.45mmol)并在相同条件下放置30分钟。此后,向该溶液中加入1-溴-3-氯丙烷(1当量,1.300ml,13.15mmol)并在室温下搅拌过夜。然后通过用乙酸乙酯(EtOAc)和H2O稀释将反应产物淬灭。分层,水相用3×25ml EtOAc萃取,合并有机层,用盐水洗涤,用Na2SO4干燥,过滤并真空浓缩至干。将由此获得的原材料使用硅胶色谱(BiotageISOLERATM,KP-Sil 50g色谱柱,以Cy:EtOAc/90:10至Cy:EtOAc/1:1的梯度洗脱)纯化,得到所需化合物(3.929g,12.48mmol,95%)。
实例4
1-(3-碘丙基)-2,3-二氢(4,5,6,7-D4)-1H-1,3-苯并二唑-2-酮的制备:
向250mL圆底烧瓶中加入3-(3-氯丙基)-2-氧代-2,3-二氢(D4)-1H-1,3-苯并二唑-1-羧酸叔丁酯(1当量,3.929g,12.48mmol),溶于100ml乙腈中,并在室温下搅拌。分批加入碘化钠(4.5当量,8.417g,56.16mmol),并将反应产物回流过夜。冷却至室温之后,过滤反应产物并在真空下除去溶剂。将由此获得的原材料用硅胶色谱(Biotage ISOLERATM,KP-Sil100g色谱柱,以纯DCM至纯DCM:MeOH/1:1的梯度洗脱)纯化,得到所需化合物(3.631g,11.86mmol,产率=95%)。
实例5
1-{3-[4-(5-氯-2-氧代-2,3-二氢-1H-1,3-苯并二唑-1-基)哌啶-1-基]丙基}-2,3-二氢(4,5,6,7-D4)-1H-1,3-苯并二唑-2-酮(化合物2)的制备:
向500mL圆底烧瓶中加入5-氯-1-(哌啶-4-基)-2,3-二氢-1H-1,3-苯并二唑-2-酮(1.2当量,3.582g,14.23mmol),然后溶于250ml无水THF和25ml无水DMF中。该溶液在室温下在氮气下搅拌,并在10分钟内逐滴加入1-(3-碘丙基)-2,3-二氢(4,5,6,7-D4)-1H-1,3-苯并二唑-2-酮(1当量,3.631g,11.86mmol)的120ml无水THF溶液。将所得黄色溶液搅拌2小时,然后加入碳酸钾(1.5当量,2.458g,17.79mmol)并在室温下搅拌48小时直至黄色消失。过滤反应产物,将固体用EtOAc洗涤,并将滤液真空浓缩至干。将由此获得的原材料通过硅胶色谱(Biotage ISOLERATM,KP-Sil 340g色谱柱,以DCM:MeOH/98:2至DCM:MeOH/1:1的梯度洗脱)。在纯化过程结束时,得到3.245g(7.45mmol,64%)所需化合物2,其为白色结晶固体。
说明书中显示和描述的实施例仅是本领域技术人员的具体实施例,并且不以任何方式进行限制。因此,在不脱离以下权利要求书范围内的本发明的精神的情况下,可以对这些实施例进行各种更改、修改或改变。所引用的每篇参考文献的全部内容通过引用明确地并入本文。
Claims (12)
1.化合物,其是d4-多潘立酮2或其药学上可接受的盐:

2.一种药物组合物,其包含权利要求1所述的化合物和至少一种赋形剂。
3.治疗有效量的权利要求1所述的化合物在制备用于改善以下病症的药物中的用途:胃轻瘫、与胃轻瘫分开的恶心、与胃轻瘫分开的呕吐、与胃轻瘫相关的恶心、与胃轻瘫相关的呕吐、泌乳不足或及其组合。
4.根据权利要求3所述的用途,其中治疗有效量在0.5mg到100mg的范围。
5.根据权利要求3或4所述的用途,其中治疗有效量在1mg到60mg的范围。
6.根据权利要求3或4所述的用途,其中治疗有效量在2.0mg到30mg的范围。
7.根据权利要求3所述的用途,其中治疗有效量在0.07mg/kg到1.43mg/kg的范围。
8.根据权利要求3或7所述的用途,其中治疗有效量在0.014mg/kg到0.86mg/kg的范围。
9.根据权利要求3或4所述的用途,其中治疗有效量在0.028mg/kg到0.43mg/kg的范围。
10.根据权利要求3或4所述的用途,其中所述给药改善胃轻瘫。
11.根据权利要求3或4所述的用途,其中所述给药改善患者的作为与由胃轻瘫引起的恶心或呕吐分开的单独的病症的恶心或呕吐中的至少一种。
12.根据权利要求3或4所述的用途,其中所述给药改善泌乳不足。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111078948.5A CN113995755A (zh) | 2016-02-04 | 2017-02-03 | 氘代多潘立酮组合物和用于治疗病症的方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662291198P | 2016-02-04 | 2016-02-04 | |
| US62/291,198 | 2016-02-04 | ||
| PCT/US2017/016334 WO2017136617A1 (en) | 2016-02-04 | 2017-02-03 | Deuterated domperidone compositions and methods for therapy of disorders |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111078948.5A Division CN113995755A (zh) | 2016-02-04 | 2017-02-03 | 氘代多潘立酮组合物和用于治疗病症的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN108697700A CN108697700A (zh) | 2018-10-23 |
| CN108697700B true CN108697700B (zh) | 2021-08-17 |
Family
ID=59501077
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111078948.5A Pending CN113995755A (zh) | 2016-02-04 | 2017-02-03 | 氘代多潘立酮组合物和用于治疗病症的方法 |
| CN201780008640.9A Active CN108697700B (zh) | 2016-02-04 | 2017-02-03 | 氘代多潘立酮组合物和用于治疗病症的方法 |
| CN201880043378.6A Active CN111093653B (zh) | 2016-02-04 | 2018-06-28 | 氘代多潘立酮组合物、方法和制备物 |
| CN202310648870.9A Pending CN116712432A (zh) | 2016-02-04 | 2018-06-28 | 氘代多潘立酮组合物、方法和制备物 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111078948.5A Pending CN113995755A (zh) | 2016-02-04 | 2017-02-03 | 氘代多潘立酮组合物和用于治疗病症的方法 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201880043378.6A Active CN111093653B (zh) | 2016-02-04 | 2018-06-28 | 氘代多潘立酮组合物、方法和制备物 |
| CN202310648870.9A Pending CN116712432A (zh) | 2016-02-04 | 2018-06-28 | 氘代多潘立酮组合物、方法和制备物 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US10266516B2 (zh) |
| EP (2) | EP3411031A4 (zh) |
| JP (3) | JP7296185B2 (zh) |
| KR (2) | KR20180104662A (zh) |
| CN (4) | CN113995755A (zh) |
| AU (2) | AU2017213852B2 (zh) |
| BR (2) | BR112018016032B1 (zh) |
| CA (2) | CA3013123A1 (zh) |
| EA (2) | EA035515B1 (zh) |
| WO (2) | WO2017136617A1 (zh) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3288556A4 (en) | 2015-04-29 | 2018-09-19 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
| CN113995755A (zh) | 2016-02-04 | 2022-02-01 | 辛多美制药有限公司 | 氘代多潘立酮组合物和用于治疗病症的方法 |
| US10076494B2 (en) | 2016-06-16 | 2018-09-18 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| US11364226B2 (en) | 2017-06-30 | 2022-06-21 | Cinrx Pharma, Llc | Deuterated domperidone compositions, methods, and preparation |
| JP2021527634A (ja) * | 2018-06-21 | 2021-10-14 | デルマバント サイエンシズ ゲーエムベーハー | Dgat1阻害剤の局所製剤およびそれらの使用の方法 |
| CA3117682A1 (en) * | 2018-10-25 | 2020-04-30 | Cindome Pharma, Inc. | Formulations containing deuterated domperidone |
| KR20210082480A (ko) * | 2018-10-25 | 2021-07-05 | 신돔 파마, 인크. | 돔페리돈을 함유하는 제제 |
| US20220307004A1 (en) * | 2021-03-29 | 2022-09-29 | The Government Of The United States Of America, As Represented By The Secretary Of The Navy | Multi-Enzyme Nanoparticle-Assisted Stable Isotope Incorporation Into Small Molecules by Channeling |
| WO2023205368A1 (en) * | 2022-04-20 | 2023-10-26 | Cindome Pharma, Inc. | Deuterated domperidone for treating gastroparesis |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4066772A (en) | 1975-07-21 | 1978-01-03 | Janssen Pharmaceutica N.V. | 1,3-Dihydro-1-[3-(1-piperidinyl)propyl]-2H-benzimidazol-2-ones and related compounds |
| NZ181256A (en) * | 1975-07-21 | 1978-04-28 | Janssen Pharmaceutica Nv | 1-(w-benzazol-11-ylalkyl)-piperidine derivatives and pharmaceutical compositions containing certain of these derivatives |
| TW466119B (en) * | 1994-02-28 | 2001-12-01 | Janssen Pharmaceutica Nv | Film coated tablet of paracetamol and domperidone |
| FR2725986B1 (fr) * | 1994-10-21 | 1996-11-29 | Adir | Nouveaux derives de piperidine, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| US5773031A (en) | 1996-02-27 | 1998-06-30 | L. Perrigo Company | Acetaminophen sustained-release formulation |
| US20160206639A9 (en) | 2001-10-12 | 2016-07-21 | Monosol Rx, Llc | Films and Drug Delivery Systems Made Therefrom |
| US20140070440A1 (en) | 2001-10-12 | 2014-03-13 | Monosol Rx, Llc | Films and Drug Delivery Systems Made Therefrom |
| US20130220526A1 (en) | 2001-10-12 | 2013-08-29 | Monosol Rx, Llc | Films and Drug Delivery Systems Made Therefrom |
| US20140163060A1 (en) | 2001-10-12 | 2014-06-12 | Monosol Rx, Llc | Films and Drug Delivery Systems Made Therefrom |
| US20140271788A1 (en) | 2013-03-15 | 2014-09-18 | Monosol Rx, Llc | Sublingual and buccal film compositions |
| ITMI20020514A1 (it) | 2002-03-12 | 2003-09-12 | Jagotec Ag | Sistema terapeutico per il rilascio controllato di uno o piu' principi attivi |
| US9101540B2 (en) | 2002-04-12 | 2015-08-11 | Alkermes Pharma Ireland Limited | Nanoparticulate megestrol formulations |
| SI1545475T1 (sl) | 2002-10-01 | 2014-10-30 | Banner Pharmacaps, Inc. | Enteričen sestavek za pripravo ovoja mehke kapsule |
| US7387793B2 (en) | 2003-11-14 | 2008-06-17 | Eurand, Inc. | Modified release dosage forms of skeletal muscle relaxants |
| BRPI0518101A (pt) | 2004-12-31 | 2008-10-28 | Iceutica Pty Ltd | métodos de produção e sìntese de composições nanoparticuladas, composições obtidas e seu uso |
| ITMI20050477A1 (it) | 2005-03-23 | 2006-09-24 | Bouty S P A | Cerotto transdermico |
| CN100386323C (zh) * | 2006-02-27 | 2008-05-07 | 南京长澳医药科技有限公司 | 马来酸多潘立酮的合成方法 |
| US8846100B2 (en) | 2006-05-12 | 2014-09-30 | Shire Llc | Controlled dose drug delivery system |
| US8808751B2 (en) | 2006-06-30 | 2014-08-19 | Iceutica Pty Ltd. | Methods for the preparation of biologically active compounds in nanoparticulate form |
| AU2008245580A1 (en) | 2007-04-26 | 2008-11-06 | Craig A. Aronchick | Compositions and methods for transmucosal delivery of domperidone |
| AU2007357647A1 (en) | 2007-08-15 | 2009-02-19 | Mcneil-Ppc, Inc. | Immediate release and sustained release ibuprofen dosing regimen |
| WO2009035598A1 (en) | 2007-09-10 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Deuterated pirfenidone |
| US20090076010A1 (en) | 2007-09-13 | 2009-03-19 | Protia, Llc | Deuterium-enriched lamotrigine |
| WO2009146310A1 (en) | 2008-05-28 | 2009-12-03 | Concert Pharmaceuticals Inc. | Deuterated tizanidine |
| WO2010054003A2 (en) * | 2008-11-06 | 2010-05-14 | Auspex Pharmaceutical, Inc. | Methylindazole modulators of 5-ht3 receptors |
| US20100120733A1 (en) * | 2008-11-07 | 2010-05-13 | Auspex Pharmaceuticals, Inc. | Steroid modulators of glucocorticoid receptor |
| NZ620887A (en) | 2009-04-24 | 2015-08-28 | Iceutica Pty Ltd | A novel formulation of diclofenac |
| JP5906181B2 (ja) | 2009-04-24 | 2016-04-20 | イシューティカ ピーティーワイ リミテッド | 商業的ナノ粒子及びマイクロ粒子粉末の生産方法 |
| NZ595903A (en) | 2009-04-24 | 2014-03-28 | Iceutica Pty Ltd | A novel formulation of metaxalone |
| AP3530A (en) | 2009-04-24 | 2016-01-13 | Iceutica Pty Ltd | A novel formulation of naproxen. |
| KR20140077945A (ko) | 2009-04-24 | 2014-06-24 | 아이슈티카 피티와이 리미티드 | 신규 인도메타신 제제 |
| US20140017299A1 (en) | 2011-08-18 | 2014-01-16 | Monosol Rx, Llc | Steroid hormone delivery systems and methods of preparing the same |
| WO2013128562A1 (ja) | 2012-02-28 | 2013-09-06 | ニチバン株式会社 | 貼付剤 |
| US20150197525A1 (en) | 2012-06-15 | 2015-07-16 | Concert Pharmaceuticals, Inc. | Deuterated derivatives of ruxolitinib |
| AU2013277429B2 (en) * | 2012-06-19 | 2016-01-14 | Intercept Pharmaceuticals, Inc. | Preparation, uses and solid forms of obeticholic acid |
| US20140019395A1 (en) | 2012-07-12 | 2014-01-16 | Craig Charles Bauer | Method and application of Fear, Love and Methodology as protocols within and of a programming group, as a means of creating an artificial individual, software system or other independent entity capable of independent self directive creation of choice and/or self directive creation of purpose within a virtual and/or any environment |
| US9290475B2 (en) | 2013-03-14 | 2016-03-22 | Deuterx, Llc | 3-(substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| SG10201709400VA (en) | 2013-03-15 | 2018-01-30 | Iceutica Inc | Abiraterone acetate formulation |
| US20140271787A1 (en) | 2013-03-15 | 2014-09-18 | Monosol Rx, Llc | Continuous single layer film structure including discrete domains |
| US20140272220A1 (en) | 2013-03-15 | 2014-09-18 | Monosol Rx, Llc | Reduction in stress cracking of films |
| CN113995755A (zh) | 2016-02-04 | 2022-02-01 | 辛多美制药有限公司 | 氘代多潘立酮组合物和用于治疗病症的方法 |
-
2017
- 2017-02-03 CN CN202111078948.5A patent/CN113995755A/zh active Pending
- 2017-02-03 EA EA201891727A patent/EA035515B1/ru not_active IP Right Cessation
- 2017-02-03 JP JP2018535888A patent/JP7296185B2/ja active Active
- 2017-02-03 KR KR1020187023566A patent/KR20180104662A/ko active Search and Examination
- 2017-02-03 WO PCT/US2017/016334 patent/WO2017136617A1/en active Application Filing
- 2017-02-03 CN CN201780008640.9A patent/CN108697700B/zh active Active
- 2017-02-03 AU AU2017213852A patent/AU2017213852B2/en active Active
- 2017-02-03 EP EP17748198.3A patent/EP3411031A4/en active Pending
- 2017-02-03 BR BR112018016032-6A patent/BR112018016032B1/pt active IP Right Grant
- 2017-02-03 CA CA3013123A patent/CA3013123A1/en active Pending
- 2017-06-30 US US15/639,431 patent/US10266516B2/en active Active
-
2018
- 2018-06-28 EP EP18743261.2A patent/EP3644995A1/en active Pending
- 2018-06-28 CN CN201880043378.6A patent/CN111093653B/zh active Active
- 2018-06-28 JP JP2019571517A patent/JP2020534246A/ja active Pending
- 2018-06-28 WO PCT/US2018/039928 patent/WO2019006078A1/en unknown
- 2018-06-28 AU AU2018290905A patent/AU2018290905B2/en active Active
- 2018-06-28 EA EA202090183A patent/EA202090183A1/ru unknown
- 2018-06-28 CN CN202310648870.9A patent/CN116712432A/zh active Pending
- 2018-06-28 KR KR1020207002879A patent/KR20200044790A/ko not_active Application Discontinuation
- 2018-06-28 CA CA3055777A patent/CA3055777A1/en active Pending
- 2018-06-28 BR BR112019028185A patent/BR112019028185A8/pt unknown
-
2019
- 2019-04-16 US US16/385,599 patent/US10590110B2/en active Active
-
2022
- 2022-12-22 JP JP2022205876A patent/JP2023052045A/ja active Pending
Non-Patent Citations (1)
| Title |
|---|
| STN CAS:1329614-18-7;STN-REGISTRY;《STN-REGISTRY》;20110907 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108697700B (zh) | 氘代多潘立酮组合物和用于治疗病症的方法 | |
| JP7323451B2 (ja) | フロルグルシノールおよびトリメチルフロログルシノールの医薬製剤 | |
| KR101752014B1 (ko) | 고용량 및 저용량 약물들의 조합을 포함하는 구강붕해정 조성물 | |
| BRPI0615014A2 (pt) | composição farmacêutica sólida compreendendo 1-(4-cloroanilino)-4-(4-piridilmetil)ftalazina e um modificador de ph e uso da mesma | |
| US11364226B2 (en) | Deuterated domperidone compositions, methods, and preparation | |
| US20240091199A1 (en) | Deuterated Domperidone Compositions, Methods, and Preparation | |
| EA045546B1 (ru) | Способ облегчения заболевания или состояния, связанного с моторикой желудочно-кишечного тракта, с использованием дейтерированного домперидона | |
| EA046180B1 (ru) | Фармацевтический состав флороглюцинола и триметилфлороглюцинола |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1259508 Country of ref document: HK |
|
| TA01 | Transfer of patent application right |
Effective date of registration: 20200512 Address after: Ohio, USA Applicant after: Xinduomei Pharmaceutical Co., Ltd Address before: Ohio, USA Applicant before: CINRX PHARMA, LLC |
|
| TA01 | Transfer of patent application right | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |