CN106946848B - 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 - Google Patents
作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 Download PDFInfo
- Publication number
- CN106946848B CN106946848B CN201710190023.7A CN201710190023A CN106946848B CN 106946848 B CN106946848 B CN 106946848B CN 201710190023 A CN201710190023 A CN 201710190023A CN 106946848 B CN106946848 B CN 106946848B
- Authority
- CN
- China
- Prior art keywords
- compound
- group
- formula
- pyridine
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 title description 2
- 239000002571 phosphodiesterase inhibitor Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 334
- 238000000034 method Methods 0.000 claims abstract description 125
- 239000000203 mixture Substances 0.000 claims abstract description 102
- 230000008569 process Effects 0.000 claims abstract description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 122
- 125000000217 alkyl group Chemical group 0.000 claims description 95
- 239000000243 solution Substances 0.000 claims description 86
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 75
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 67
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 65
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 claims description 58
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 claims description 56
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 54
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 54
- 238000006467 substitution reaction Methods 0.000 claims description 52
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 50
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 48
- 150000003839 salts Chemical class 0.000 claims description 44
- 239000007787 solid Substances 0.000 claims description 36
- 239000012074 organic phase Substances 0.000 claims description 30
- 229920006395 saturated elastomer Polymers 0.000 claims description 29
- 150000001204 N-oxides Chemical class 0.000 claims description 26
- 125000001188 haloalkyl group Chemical group 0.000 claims description 25
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 24
- 238000011282 treatment Methods 0.000 claims description 21
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 20
- 239000012267 brine Substances 0.000 claims description 19
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 claims description 19
- 238000003756 stirring Methods 0.000 claims description 18
- 238000001914 filtration Methods 0.000 claims description 16
- 125000002950 monocyclic group Chemical group 0.000 claims description 15
- 238000002360 preparation method Methods 0.000 claims description 14
- 125000005843 halogen group Chemical group 0.000 claims description 13
- 150000003254 radicals Chemical class 0.000 claims description 13
- 239000002244 precipitate Substances 0.000 claims description 12
- 125000001424 substituent group Chemical group 0.000 claims description 12
- 239000003814 drug Substances 0.000 claims description 10
- 238000005406 washing Methods 0.000 claims description 10
- 201000010099 disease Diseases 0.000 claims description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 9
- 208000006673 asthma Diseases 0.000 claims description 8
- 208000023504 respiratory system disease Diseases 0.000 claims description 8
- 239000000463 material Substances 0.000 claims description 7
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 6
- 208000000884 Airway Obstruction Diseases 0.000 claims description 5
- KNQLTBDJNDODBN-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(carbamoylamino)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound NC(=O)NC1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 KNQLTBDJNDODBN-AHWVRZQESA-N 0.000 claims description 5
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 5
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 4
- 239000008346 aqueous phase Substances 0.000 claims description 4
- 229940124748 beta 2 agonist Drugs 0.000 claims description 4
- 239000003246 corticosteroid Substances 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 3
- 201000008937 atopic dermatitis Diseases 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 3
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 claims description 2
- CMLFRMDBDNHMRA-UHFFFAOYSA-N 2h-1,2-benzoxazine Chemical group C1=CC=C2C=CNOC2=C1 CMLFRMDBDNHMRA-UHFFFAOYSA-N 0.000 claims description 2
- 206010039085 Rhinitis allergic Diseases 0.000 claims description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 2
- 201000010105 allergic rhinitis Diseases 0.000 claims description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 2
- 125000002757 morpholinyl group Chemical group 0.000 claims description 2
- 125000004193 piperazinyl group Chemical group 0.000 claims description 2
- 229930192474 thiophene Natural products 0.000 claims description 2
- 238000011321 prophylaxis Methods 0.000 claims 3
- 230000001022 anti-muscarinic effect Effects 0.000 claims 1
- 125000002943 quinolinyl group Chemical class N1=C(C=CC2=CC=CC=C12)* 0.000 claims 1
- 210000002345 respiratory system Anatomy 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 16
- MSYGAHOHLUJIKV-UHFFFAOYSA-N 3,5-dimethyl-1-(3-nitrophenyl)-1h-pyrazole-4-carboxylic acid ethyl ester Chemical compound CC1=C(C(=O)OCC)C(C)=NN1C1=CC=CC([N+]([O-])=O)=C1 MSYGAHOHLUJIKV-UHFFFAOYSA-N 0.000 abstract description 10
- 102000011017 Type 4 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 abstract description 10
- 108010037584 Type 4 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 abstract description 10
- 101100296719 Caenorhabditis elegans pde-4 gene Proteins 0.000 abstract description 6
- 150000001298 alcohols Chemical class 0.000 abstract description 6
- 230000001225 therapeutic effect Effects 0.000 abstract description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 164
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 93
- 238000006243 chemical reaction Methods 0.000 description 69
- 239000002904 solvent Substances 0.000 description 66
- 229910052938 sodium sulfate Inorganic materials 0.000 description 60
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 56
- 239000007832 Na2SO4 Substances 0.000 description 55
- -1 hydrogen Chemical class 0.000 description 45
- 230000015572 biosynthetic process Effects 0.000 description 42
- 238000003786 synthesis reaction Methods 0.000 description 42
- 238000005160 1H NMR spectroscopy Methods 0.000 description 41
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 40
- 235000019439 ethyl acetate Nutrition 0.000 description 40
- 239000000047 product Substances 0.000 description 31
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 30
- 238000000746 purification Methods 0.000 description 29
- 239000011541 reaction mixture Substances 0.000 description 29
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 28
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 28
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 27
- 101100296720 Dictyostelium discoideum Pde4 gene Proteins 0.000 description 26
- 101100082610 Plasmodium falciparum (isolate 3D7) PDEdelta gene Proteins 0.000 description 26
- 238000001035 drying Methods 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 26
- 230000002829 reductive effect Effects 0.000 description 25
- 238000002953 preparative HPLC Methods 0.000 description 24
- 239000003153 chemical reaction reagent Substances 0.000 description 22
- 239000012043 crude product Substances 0.000 description 21
- 229910052739 hydrogen Inorganic materials 0.000 description 20
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 19
- 230000000694 effects Effects 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 18
- 239000001257 hydrogen Substances 0.000 description 18
- ILVXOBCQQYKLDS-UHFFFAOYSA-N pyridine N-oxide Chemical compound [O-][N+]1=CC=CC=C1 ILVXOBCQQYKLDS-UHFFFAOYSA-N 0.000 description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- 239000002243 precursor Substances 0.000 description 16
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 15
- 238000000605 extraction Methods 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- 229910052757 nitrogen Inorganic materials 0.000 description 14
- 229910000027 potassium carbonate Inorganic materials 0.000 description 14
- 238000012360 testing method Methods 0.000 description 14
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 14
- QGXYRJFTOQJWBR-HNNXBMFYSA-N (1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethanol Chemical compound C([C@H](O)C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)C1=C(Cl)C=[N+]([O-])C=C1Cl QGXYRJFTOQJWBR-HNNXBMFYSA-N 0.000 description 13
- OEKHQHPOQBNHKX-MKSBGGEFSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1,3-thiazolidine-2-carboxylate;hydrochloride Chemical compound Cl.ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@@H]1SCCN1 OEKHQHPOQBNHKX-MKSBGGEFSA-N 0.000 description 13
- 238000001704 evaporation Methods 0.000 description 13
- 229910052736 halogen Inorganic materials 0.000 description 13
- 150000002367 halogens Chemical class 0.000 description 13
- 230000005764 inhibitory process Effects 0.000 description 13
- 239000002585 base Substances 0.000 description 12
- 125000000392 cycloalkenyl group Chemical group 0.000 description 12
- 239000000543 intermediate Substances 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 11
- 239000007864 aqueous solution Substances 0.000 description 11
- 238000003818 flash chromatography Methods 0.000 description 11
- 125000004433 nitrogen atom Chemical group N* 0.000 description 11
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 11
- 239000012453 solvate Substances 0.000 description 11
- 239000003643 water by type Substances 0.000 description 11
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 10
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 10
- IBWDCEQOKMRKEX-MKSBGGEFSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-pyrrolidine-2-carboxylate;hydrochloride Chemical compound Cl.ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1NCCC1 IBWDCEQOKMRKEX-MKSBGGEFSA-N 0.000 description 10
- ZUDBENSYOYSHEH-QSAPEBAKSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(4-aminophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC(N)=CC=C1S(=O)(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 ZUDBENSYOYSHEH-QSAPEBAKSA-N 0.000 description 10
- 238000004458 analytical method Methods 0.000 description 10
- 125000000623 heterocyclic group Chemical group 0.000 description 10
- 125000006588 heterocycloalkylene group Chemical group 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- 125000003118 aryl group Chemical group 0.000 description 9
- 125000004429 atom Chemical group 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 239000007789 gas Substances 0.000 description 9
- 150000002431 hydrogen Chemical class 0.000 description 9
- 239000012047 saturated solution Substances 0.000 description 9
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- 125000005605 benzo group Chemical group 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 8
- 125000001072 heteroaryl group Chemical group 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- FUCFLINELPEUMS-LBPRGKRZSA-N (1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-methoxyphenyl]ethanol Chemical compound C1=C(OC(F)F)C(OC)=CC([C@@H](O)CC=2C(=C[N+]([O-])=CC=2Cl)Cl)=C1 FUCFLINELPEUMS-LBPRGKRZSA-N 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 7
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 7
- SFAIXXZCLPGBGM-LROBGIAVSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)C1SCCN1 SFAIXXZCLPGBGM-LROBGIAVSA-N 0.000 description 7
- 125000003342 alkenyl group Chemical group 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000000460 chlorine Substances 0.000 description 7
- 239000002158 endotoxin Substances 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 238000010438 heat treatment Methods 0.000 description 7
- 229920006008 lipopolysaccharide Polymers 0.000 description 7
- 239000000523 sample Substances 0.000 description 7
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 6
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 6
- RGUKYNXWOWSRET-UHFFFAOYSA-N 4-pyrrolidin-1-ylpyridine Chemical compound C1CCCN1C1=CC=NC=C1 RGUKYNXWOWSRET-UHFFFAOYSA-N 0.000 description 6
- BTRIVKDXFFFIEU-UHFFFAOYSA-N C(=O)O.[N+]1(=CC=CC=C1)[O-] Chemical compound C(=O)O.[N+]1(=CC=CC=C1)[O-] BTRIVKDXFFFIEU-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 6
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 6
- KOVUQUXRMIAWMX-WNJJXGMVSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 KOVUQUXRMIAWMX-WNJJXGMVSA-N 0.000 description 6
- OGAAISVRRIRGFR-BDYUSTAISA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(hydroxymethyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound OCC1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 OGAAISVRRIRGFR-BDYUSTAISA-N 0.000 description 6
- IRNFAQNJEGCMRQ-BXXZMZEQSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(4-aminobenzoyl)-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC(N)=CC=C1C(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 IRNFAQNJEGCMRQ-BXXZMZEQSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 125000000304 alkynyl group Chemical group 0.000 description 6
- 229910052801 chlorine Inorganic materials 0.000 description 6
- 238000004587 chromatography analysis Methods 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 125000006254 cycloalkyl carbonyl group Chemical group 0.000 description 6
- 230000008020 evaporation Effects 0.000 description 6
- 239000000284 extract Substances 0.000 description 6
- 239000012458 free base Substances 0.000 description 6
- 125000004043 oxo group Chemical group O=* 0.000 description 6
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 6
- 239000003208 petroleum Substances 0.000 description 6
- 239000003880 polar aprotic solvent Substances 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 230000002265 prevention Effects 0.000 description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 description 6
- QLGNIBMYQWRIBE-UHFFFAOYSA-N (5-formyl-2-methoxyphenyl) methanesulfonate Chemical compound COC1=CC=C(C=O)C=C1OS(C)(=O)=O QLGNIBMYQWRIBE-UHFFFAOYSA-N 0.000 description 5
- DYSGQMXWHGNRQV-GMAHTHKFSA-N 1-o-tert-butyl 2-o-[(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-pyrrolidine-1,2-dicarboxylate Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(=O)O[C@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)CC1=C(Cl)C=[N+]([O-])C=C1Cl DYSGQMXWHGNRQV-GMAHTHKFSA-N 0.000 description 5
- LWDFZAVDBWZJDQ-UHFFFAOYSA-N 3-(4-aminophenyl)sulfonyl-1,3-thiazolidine-2-carboxylic acid Chemical compound C1=CC(N)=CC=C1S(=O)(=O)N1C(C(O)=O)SCC1 LWDFZAVDBWZJDQ-UHFFFAOYSA-N 0.000 description 5
- KTPRNUKJCZVUHM-UHFFFAOYSA-N 3-(4-nitrobenzoyl)-1,3-thiazolidine-2-carboxylic acid Chemical compound OC(=O)C1SCCN1C(=O)C1=CC=C([N+]([O-])=O)C=C1 KTPRNUKJCZVUHM-UHFFFAOYSA-N 0.000 description 5
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 5
- OSTOSMNFNFSKHG-IGKIAQTJSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-(4-methoxy-3-methylsulfonyloxybenzoyl)pyrrolidine-2-carboxylate Chemical compound C1=C(OS(C)(=O)=O)C(OC)=CC=C1C(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)CCC1 OSTOSMNFNFSKHG-IGKIAQTJSA-N 0.000 description 5
- ZUDBENSYOYSHEH-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(4-aminophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC(N)=CC=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 ZUDBENSYOYSHEH-AHWVRZQESA-N 0.000 description 5
- IDCTVLPYJQMERN-IZEXYCQBSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[2-[3-(dimethylcarbamoyl)phenyl]-2-oxoethyl]-1,3-thiazolidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(C(=O)CN2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 IDCTVLPYJQMERN-IZEXYCQBSA-N 0.000 description 5
- DARLVEVDMUDYGO-DHIFEGFHSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[2-[3-(dimethylcarbamoyl)phenyl]acetyl]-1,3-thiazolidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(CC(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 DARLVEVDMUDYGO-DHIFEGFHSA-N 0.000 description 5
- OMFFBWNFKDJNCS-MHZLTWQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 1-[3-(dimethylcarbamoyl)phenyl]sulfonylazetidine-3-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2CC(C2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 OMFFBWNFKDJNCS-MHZLTWQESA-N 0.000 description 5
- JJMBHJAUBJEJMA-PVCWFJFTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 2-[3-(benzenesulfonyl)-1,3-thiazolidin-2-yl]acetate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)CC1N(S(=O)(=O)C=2C=CC=CC=2)CCS1 JJMBHJAUBJEJMA-PVCWFJFTSA-N 0.000 description 5
- WNUIZIFZJNGBTB-BXXZMZEQSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(4-nitrobenzoyl)-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC([N+](=O)[O-])=CC=C1C(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 WNUIZIFZJNGBTB-BXXZMZEQSA-N 0.000 description 5
- HYIJIGDLEDTUKF-OZXSUGGESA-N [(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-methoxyphenyl]ethyl] (2s)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=C(OC(F)F)C(OC)=CC([C@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)OC(=O)[C@H]2N(CCS2)S(=O)(=O)C=2C=C(C=CC=2)C(=O)N(C)C)=C1 HYIJIGDLEDTUKF-OZXSUGGESA-N 0.000 description 5
- MZHZBYJGGVOARU-MILIPEGGSA-N [1-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound COC1=CC=C(C(CC=2C(=C[N+]([O-])=CC=2Cl)Cl)OC(=O)[C@H]2N(CCS2)S(=O)(=O)C=2C=C(C=CC=2)C(=O)N(C)C)C=C1OCC1CC1 MZHZBYJGGVOARU-MILIPEGGSA-N 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 150000004675 formic acid derivatives Chemical class 0.000 description 5
- SGCUNWDQNYKLHS-UHFFFAOYSA-N methyl 3-(4-nitrobenzoyl)-1,3-thiazolidine-2-carboxylate Chemical compound COC(=O)C1SCCN1C(=O)C1=CC=C([N+]([O-])=O)C=C1 SGCUNWDQNYKLHS-UHFFFAOYSA-N 0.000 description 5
- 125000006413 ring segment Chemical group 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- DYAUJEIALQJPGW-ZDUSSCGKSA-N (1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-(3,4-dimethoxyphenyl)ethanol Chemical compound C1=C(OC)C(OC)=CC=C1[C@@H](O)CC1=C(Cl)C=[N+]([O-])C=C1Cl DYAUJEIALQJPGW-ZDUSSCGKSA-N 0.000 description 4
- JUSWZYFYLXTMLJ-JTQLQIEISA-N (2s)-1-(benzenesulfonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1S(=O)(=O)C1=CC=CC=C1 JUSWZYFYLXTMLJ-JTQLQIEISA-N 0.000 description 4
- HOINLISSDXOOHG-LBPRGKRZSA-N (2s)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylic acid Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(O)=O)=C1 HOINLISSDXOOHG-LBPRGKRZSA-N 0.000 description 4
- CFFRTSUKMZTPTJ-UHFFFAOYSA-N 1-[3-(dimethylcarbamoyl)phenyl]sulfonylazetidine-3-carboxylic acid Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2CC(C2)C(O)=O)=C1 CFFRTSUKMZTPTJ-UHFFFAOYSA-N 0.000 description 4
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- WYSLGSINIUGBBN-UHFFFAOYSA-N 2-(3,5-dichloropyridin-4-yl)-1-(3,4-dimethoxyphenyl)ethanol Chemical compound C1=C(OC)C(OC)=CC=C1C(O)CC1=C(Cl)C=NC=C1Cl WYSLGSINIUGBBN-UHFFFAOYSA-N 0.000 description 4
- YCIPGXXDKNSRSG-UHFFFAOYSA-N 2-(3-benzoyl-1,3-thiazolidin-2-yl)acetic acid Chemical compound OC(=O)CC1SCCN1C(=O)C1=CC=CC=C1 YCIPGXXDKNSRSG-UHFFFAOYSA-N 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 4
- GYOXRSRJMNDCLO-UHFFFAOYSA-N 2-[3-(benzenesulfonyl)-1,3-thiazolidin-2-yl]acetic acid Chemical compound OC(=O)CC1SCCN1S(=O)(=O)C1=CC=CC=C1 GYOXRSRJMNDCLO-UHFFFAOYSA-N 0.000 description 4
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 4
- BIAIGUIFZWVGJW-UHFFFAOYSA-N 3-(dimethylcarbamoyl)benzenesulfonyl chloride Chemical compound CN(C)C(=O)C1=CC=CC(S(Cl)(=O)=O)=C1 BIAIGUIFZWVGJW-UHFFFAOYSA-N 0.000 description 4
- ROCYKUBOYVRXFK-UHFFFAOYSA-N 3-(hydroxymethyl)benzenesulfonyl chloride Chemical compound OCC1=CC=CC(S(Cl)(=O)=O)=C1 ROCYKUBOYVRXFK-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 4
- NLUYIEYATOCTRB-GMAHTHKFSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-(1-methylimidazol-2-yl)sulfonylpyrrolidine-2-carboxylate Chemical compound CN1C=CN=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)CCC1 NLUYIEYATOCTRB-GMAHTHKFSA-N 0.000 description 4
- UXWMIEZOKBQEFF-XCZPVHLTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-[2-(benzenesulfonyl)ethyl]pyrrolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(CCS(=O)(=O)C=2C=CC=CC=2)CCC1 UXWMIEZOKBQEFF-XCZPVHLTSA-N 0.000 description 4
- RPENNGYARXJSPW-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(2-morpholin-4-ylethylsulfonyl)-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(S(=O)(=O)CCN2CCOCC2)CCS1 RPENNGYARXJSPW-AHWVRZQESA-N 0.000 description 4
- KZJKOWXMVWRLGW-FIBWVYCGSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[[3-(dimethylcarbamoyl)phenyl]methylsulfonyl]-1,3-thiazolidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(CS(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 KZJKOWXMVWRLGW-FIBWVYCGSA-N 0.000 description 4
- IHXUPARXGAPFKL-UNMCSNQZSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-ethenylsulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(S(=O)(=O)C=C)CCS1 IHXUPARXGAPFKL-UNMCSNQZSA-N 0.000 description 4
- ICVVZCURHILIGU-PVCWFJFTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 2-(3-benzoyl-1,3-thiazolidin-2-yl)acetate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)CC1N(C(=O)C=2C=CC=CC=2)CCS1 ICVVZCURHILIGU-PVCWFJFTSA-N 0.000 description 4
- BTWAZHBUYKGZMG-QSAPEBAKSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(4-nitrophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 BTWAZHBUYKGZMG-QSAPEBAKSA-N 0.000 description 4
- MMCKEEIDGGUAMJ-CHQVSRGASA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(5-formylthiophene-2-carbonyl)-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)C1N(C(=O)C=2SC(C=O)=CC=2)CCS1 MMCKEEIDGGUAMJ-CHQVSRGASA-N 0.000 description 4
- YAMBPONVOKNWJI-CEBUJLNPSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-[3-(dimethylcarbamoyl)-4-methoxyphenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=C(C(=O)N(C)C)C(OC)=CC=C1S(=O)(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 YAMBPONVOKNWJI-CEBUJLNPSA-N 0.000 description 4
- CYMZVNCZKMOJSM-GMAHTHKFSA-N [(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-methoxyphenyl]ethyl] (2s)-1-(benzenesulfonyl)pyrrolidine-2-carboxylate Chemical compound C1=C(OC(F)F)C(OC)=CC([C@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)OC(=O)[C@H]2N(CCC2)S(=O)(=O)C=2C=CC=CC=2)=C1 CYMZVNCZKMOJSM-GMAHTHKFSA-N 0.000 description 4
- DYGSHXYVDQSQFN-UHFFFAOYSA-N [3-(dimethylcarbamoyl)phenyl]methanesulfonic acid Chemical compound CN(C)C(=O)C1=CC=CC(CS(O)(=O)=O)=C1 DYGSHXYVDQSQFN-UHFFFAOYSA-N 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 239000012296 anti-solvent Substances 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 238000004807 desolvation Methods 0.000 description 4
- 230000008034 disappearance Effects 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- BHFCAIGTPIWDEH-UHFFFAOYSA-N ethyl 2-(3-benzoyl-1,3-thiazolidin-2-yl)acetate Chemical compound CCOC(=O)CC1SCCN1C(=O)C1=CC=CC=C1 BHFCAIGTPIWDEH-UHFFFAOYSA-N 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- JVTZFYYHCGSXJV-UHFFFAOYSA-N isovanillin Chemical compound COC1=CC=C(C=O)C=C1O JVTZFYYHCGSXJV-UHFFFAOYSA-N 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- DTPZZPFIILRELI-QMMMGPOBSA-N methyl (2s)-1-(1-methylimidazol-2-yl)sulfonylpyrrolidine-2-carboxylate Chemical compound COC(=O)[C@@H]1CCCN1S(=O)(=O)C1=NC=CN1C DTPZZPFIILRELI-QMMMGPOBSA-N 0.000 description 4
- GPKBSOKBDAZIEO-UHFFFAOYSA-N methyl 3-(4-aminophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate;hydrochloride Chemical compound Cl.COC(=O)C1SCCN1S(=O)(=O)C1=CC=C(N)C=C1 GPKBSOKBDAZIEO-UHFFFAOYSA-N 0.000 description 4
- GOAHJROYOQZKOP-UHFFFAOYSA-N methyl 3-(4-nitrophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound COC(=O)C1SCCN1S(=O)(=O)C1=CC=C([N+]([O-])=O)C=C1 GOAHJROYOQZKOP-UHFFFAOYSA-N 0.000 description 4
- AKRDTUGPGSGJKM-UHFFFAOYSA-N methyl 3-(cyclopropylmethoxy)-5-nitrobenzoate Chemical compound [O-][N+](=O)C1=CC(C(=O)OC)=CC(OCC2CC2)=C1 AKRDTUGPGSGJKM-UHFFFAOYSA-N 0.000 description 4
- DZJMYOWIOIADSI-UHFFFAOYSA-N methyl 3-amino-5-(cyclopropylmethoxy)benzoate Chemical compound COC(=O)C1=CC(N)=CC(OCC2CC2)=C1 DZJMYOWIOIADSI-UHFFFAOYSA-N 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- 239000011343 solid material Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 4
- 238000000825 ultraviolet detection Methods 0.000 description 4
- 239000003039 volatile agent Substances 0.000 description 4
- LCPKSEWLZFNOKC-ZETCQYMHSA-N (2s)-1-(1-methylimidazol-2-yl)sulfonylpyrrolidine-2-carboxylic acid Chemical compound CN1C=CN=C1S(=O)(=O)N1[C@H](C(O)=O)CCC1 LCPKSEWLZFNOKC-ZETCQYMHSA-N 0.000 description 3
- MTFAUTLDDYSZCF-LBPRGKRZSA-N (2s)-1-[2-(benzenesulfonyl)ethyl]pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1CCS(=O)(=O)C1=CC=CC=C1 MTFAUTLDDYSZCF-LBPRGKRZSA-N 0.000 description 3
- WHLYWVOVWPFHJJ-UHFFFAOYSA-N 1-phenyl-2H-pyridin-2-ol Chemical class C1(=CC=CC=C1)N1C(C=CC=C1)O WHLYWVOVWPFHJJ-UHFFFAOYSA-N 0.000 description 3
- PKTJKKARCIJTJR-UHFFFAOYSA-N 2,2-difluoro-1,3-dioxolane Chemical group FC1(F)OCCO1 PKTJKKARCIJTJR-UHFFFAOYSA-N 0.000 description 3
- YBHYECGRNFZJPC-UHFFFAOYSA-N 3,5-dichloro-4-methylpyridine Chemical compound CC1=C(Cl)C=NC=C1Cl YBHYECGRNFZJPC-UHFFFAOYSA-N 0.000 description 3
- NHXUHPBZPWEYCJ-UHFFFAOYSA-N 3-(1,3-dioxolan-2-ylmethyl)-n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC(CC2OCCO2)=C1 NHXUHPBZPWEYCJ-UHFFFAOYSA-N 0.000 description 3
- ZHAIUOURNNOUCX-ICSRJNTNSA-N 3-o-tert-butyl 2-o-[(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-methoxyphenyl]ethyl] (2s)-1,3-thiazolidine-2,3-dicarboxylate Chemical compound C1=C(OC(F)F)C(OC)=CC([C@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)OC(=O)[C@H]2N(CCS2)C(=O)OC(C)(C)C)=C1 ZHAIUOURNNOUCX-ICSRJNTNSA-N 0.000 description 3
- MNOMRRLCFKFAQS-UHFFFAOYSA-N 4-methoxy-3-methylsulfonyloxybenzoic acid Chemical compound COC1=CC=C(C(O)=O)C=C1OS(C)(=O)=O MNOMRRLCFKFAQS-UHFFFAOYSA-N 0.000 description 3
- POAFUYQIIJDEKA-PVCWFJFTSA-N 5-[[2-[(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethoxy]carbonyl-1,3-thiazolidin-3-yl]sulfonyl]-2-methoxybenzoic acid Chemical compound C1=C(C(O)=O)C(OC)=CC=C1S(=O)(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 POAFUYQIIJDEKA-PVCWFJFTSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 201000001320 Atherosclerosis Diseases 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 101000851058 Homo sapiens Neutrophil elastase Proteins 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical class OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 3
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- XROYEFVBNPTBTQ-BDYUSTAISA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-(2-phenylacetyl)pyrrolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(C(=O)CC=2C=CC=CC=2)CCC1 XROYEFVBNPTBTQ-BDYUSTAISA-N 0.000 description 3
- VREOXWMRPFUVDA-IZEXYCQBSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-[4-(cyclopropylmethoxy)-3-(methanesulfonamido)benzoyl]pyrrolidine-2-carboxylate Chemical compound CS(=O)(=O)NC1=CC(C(=O)N2[C@@H](CCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=CC=C1OCC1CC1 VREOXWMRPFUVDA-IZEXYCQBSA-N 0.000 description 3
- MOIOGCOQFIXYDS-UPVQGACJSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(cyclopropylmethyl)-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(CC2CC2)CCS1 MOIOGCOQFIXYDS-UPVQGACJSA-N 0.000 description 3
- IIPCABVKJXPLIM-GPNIZQGCSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 1-[(4-methoxycarbonyl-5-methylfuran-2-yl)methyl]pyrrolidine-2-carboxylate Chemical compound O1C(C)=C(C(=O)OC)C=C1CN1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)CCC1 IIPCABVKJXPLIM-GPNIZQGCSA-N 0.000 description 3
- PUCCSAIKOZJWMA-UMBBOSETSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-[5-[(dimethylamino)methyl]thiophene-2-carbonyl]-1,3-thiazolidine-2-carboxylate;formic acid Chemical compound OC=O.S1C(CN(C)C)=CC=C1C(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 PUCCSAIKOZJWMA-UMBBOSETSA-N 0.000 description 3
- QCCWLLLDMRLLRO-VXKWHMMOSA-N [(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-hydroxyphenyl]ethyl] (2s)-2-acetyloxy-2-phenylacetate Chemical compound C([C@H](OC(=O)[C@@H](OC(=O)C)C=1C=CC=CC=1)C=1C=C(O)C(OC(F)F)=CC=1)C1=C(Cl)C=[N+]([O-])C=C1Cl QCCWLLLDMRLLRO-VXKWHMMOSA-N 0.000 description 3
- NDVSCVBHNZYDQF-ZCYQVOJMSA-N [(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-hydroxyphenyl]ethyl] (2s)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(O)C(OC(F)F)=CC=2)=C1 NDVSCVBHNZYDQF-ZCYQVOJMSA-N 0.000 description 3
- CTKBJAKGAFSLTM-NBLXOJGSSA-N [(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-methoxyphenyl]ethyl] (2s)-1,3-thiazolidine-2-carboxylate;hydrochloride Chemical compound Cl.C1=C(OC(F)F)C(OC)=CC([C@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)OC(=O)[C@@H]2SCCN2)=C1 CTKBJAKGAFSLTM-NBLXOJGSSA-N 0.000 description 3
- SCTZLUXZXSYOEQ-GMAHTHKFSA-N [(1s)-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)-1-[4-(difluoromethoxy)-3-methoxyphenyl]ethyl] (2s)-2-acetyloxy-2-phenylacetate Chemical compound C1=C(OC(F)F)C(OC)=CC([C@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)OC(=O)[C@@H](OC(C)=O)C=2C=CC=CC=2)=C1 SCTZLUXZXSYOEQ-GMAHTHKFSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 3
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 3
- VSSXWIUOHKRTFW-IBGZPJMESA-N benzyl (2s)-1-[2-(benzenesulfonyl)ethyl]pyrrolidine-2-carboxylate Chemical compound O=C([C@H]1N(CCC1)CCS(=O)(=O)C=1C=CC=CC=1)OCC1=CC=CC=C1 VSSXWIUOHKRTFW-IBGZPJMESA-N 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 238000004296 chiral HPLC Methods 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- IXUDAPXXSRQPDH-AWEZNQCLSA-N ethyl (2s)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound CCOC(=O)[C@@H]1SCCN1S(=O)(=O)C1=CC=CC(C(=O)N(C)C)=C1 IXUDAPXXSRQPDH-AWEZNQCLSA-N 0.000 description 3
- INXCCJSUTXNTSE-UHFFFAOYSA-N ethyl 2-[3-(benzenesulfonyl)-1,3-thiazolidin-2-yl]acetate Chemical compound CCOC(=O)CC1SCCN1S(=O)(=O)C1=CC=CC=C1 INXCCJSUTXNTSE-UHFFFAOYSA-N 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 102000052502 human ELANE Human genes 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- YGTQIXUXZVNVKG-UHFFFAOYSA-N methyl 1,3-thiazolidin-3-ium-2-carboxylate;chloride Chemical compound Cl.COC(=O)C1NCCS1 YGTQIXUXZVNVKG-UHFFFAOYSA-N 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 3
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 239000007800 oxidant agent Substances 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000003380 propellant Substances 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- MNDBXUUTURYVHR-UHFFFAOYSA-N roflumilast Chemical compound FC(F)OC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OCC1CC1 MNDBXUUTURYVHR-UHFFFAOYSA-N 0.000 description 3
- 229960002586 roflumilast Drugs 0.000 description 3
- HJORMJIFDVBMOB-UHFFFAOYSA-N rolipram Chemical compound COC1=CC=C(C2CC(=O)NC2)C=C1OC1CCCC1 HJORMJIFDVBMOB-UHFFFAOYSA-N 0.000 description 3
- 229950005741 rolipram Drugs 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 238000001665 trituration Methods 0.000 description 3
- WYSLGSINIUGBBN-ZDUSSCGKSA-N (1s)-2-(3,5-dichloropyridin-4-yl)-1-(3,4-dimethoxyphenyl)ethanol Chemical compound C1=C(OC)C(OC)=CC=C1[C@@H](O)CC1=C(Cl)C=NC=C1Cl WYSLGSINIUGBBN-ZDUSSCGKSA-N 0.000 description 2
- OBCUSTCTKLTMBX-VIFPVBQESA-N (2s)-2-acetyloxy-2-phenylacetic acid Chemical compound CC(=O)O[C@H](C(O)=O)C1=CC=CC=C1 OBCUSTCTKLTMBX-VIFPVBQESA-N 0.000 description 2
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 2
- XPSCJSYYSCLZJV-UHFFFAOYSA-N 1-[3-(cyclopropylmethoxy)-4-methoxyphenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethanol Chemical compound COC1=CC=C(C(O)CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=C1OCC1CC1 XPSCJSYYSCLZJV-UHFFFAOYSA-N 0.000 description 2
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- CMWTZPSULFXXJA-UHFFFAOYSA-M 2-(6-methoxynaphthalen-2-yl)propanoate Chemical compound C1=C(C(C)C([O-])=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-M 0.000 description 2
- HOOOJUUXVWLJHN-UHFFFAOYSA-N 2-[3-(dimethylcarbamoyl)phenyl]acetic acid Chemical compound CN(C)C(=O)C1=CC=CC(CC(O)=O)=C1 HOOOJUUXVWLJHN-UHFFFAOYSA-N 0.000 description 2
- BBSIIQBJJLOFOR-UHFFFAOYSA-N 2-acetyl-n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC=C1C(C)=O BBSIIQBJJLOFOR-UHFFFAOYSA-N 0.000 description 2
- VHCSBTPOPKFYIU-UHFFFAOYSA-N 2-chloroethanesulfonyl chloride Chemical compound ClCCS(Cl)(=O)=O VHCSBTPOPKFYIU-UHFFFAOYSA-N 0.000 description 2
- YYOOVPWRMYDQAA-UHFFFAOYSA-N 3-(2-bromoacetyl)-n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC(C(=O)CBr)=C1 YYOOVPWRMYDQAA-UHFFFAOYSA-N 0.000 description 2
- STTCCVHGTYFYPY-UHFFFAOYSA-N 3-(cyclopropylmethoxy)-5-[methylsulfonyl(2-morpholin-4-ylethyl)amino]benzoic acid Chemical compound C=1C(OCC2CC2)=CC(C(O)=O)=CC=1N(S(=O)(=O)C)CCN1CCOCC1 STTCCVHGTYFYPY-UHFFFAOYSA-N 0.000 description 2
- ZAPMTSHEXFEPSD-UHFFFAOYSA-N 4-(2-chloroethyl)morpholine Chemical compound ClCCN1CCOCC1 ZAPMTSHEXFEPSD-UHFFFAOYSA-N 0.000 description 2
- JXRGUPLJCCDGKG-UHFFFAOYSA-N 4-nitrobenzenesulfonyl chloride Chemical compound [O-][N+](=O)C1=CC=C(S(Cl)(=O)=O)C=C1 JXRGUPLJCCDGKG-UHFFFAOYSA-N 0.000 description 2
- 125000006164 6-membered heteroaryl group Chemical group 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 208000001089 Multiple system atrophy Diseases 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 206010037211 Psychomotor hyperactivity Diseases 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 206010040070 Septic Shock Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 206010047700 Vomiting Diseases 0.000 description 2
- VLRUOABXJCTRCS-ICSRJNTNSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-pyrrolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1NCCC1 VLRUOABXJCTRCS-ICSRJNTNSA-N 0.000 description 2
- GLWNSNCHEHPUSD-ZZHFZYNASA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(2-oxo-2-thiophen-2-ylethyl)-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)C1N(CC(=O)C=2SC=CC=2)CCS1 GLWNSNCHEHPUSD-ZZHFZYNASA-N 0.000 description 2
- UXRMPSPVURBNRR-PVCWFJFTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-[4-(methanesulfonamido)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC(NS(=O)(=O)C)=CC=C1S(=O)(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 UXRMPSPVURBNRR-PVCWFJFTSA-N 0.000 description 2
- BSONSMRXCLIVHC-MOJIJOCKSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloropyridin-4-yl)ethyl] 3-[4-[methylsulfonyl(2-morpholin-4-ylethyl)amino]phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C=1C=C(S(=O)(=O)N2C(SCC2)C(=O)O[C@@H](CC=2C(=CN=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)C=CC=1N(S(=O)(=O)C)CCN1CCOCC1 BSONSMRXCLIVHC-MOJIJOCKSA-N 0.000 description 2
- ICBYHUWJYZQHDV-UHFFFAOYSA-N [3-(dimethylcarbamoyl)phenyl]methanesulfonyl chloride Chemical compound CN(C)C(=O)C1=CC=CC(CS(Cl)(=O)=O)=C1 ICBYHUWJYZQHDV-UHFFFAOYSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 208000002205 allergic conjunctivitis Diseases 0.000 description 2
- 239000000010 aprotic solvent Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 229940124630 bronchodilator Drugs 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 229960001334 corticosteroids Drugs 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 2
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 2
- ZANISHQWRNLICS-UHFFFAOYSA-N ethyl 2-(1,3-thiazolidin-2-yl)acetate Chemical compound CCOC(=O)CC1NCCS1 ZANISHQWRNLICS-UHFFFAOYSA-N 0.000 description 2
- XTULMSXFIHGYFS-VLSRWLAYSA-N fluticasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(F)[C@@]4(C)C=CC(=O)C=C4[C@@H](F)C[C@H]3[C@@H]2C[C@H]1C)C(=O)SCF)C(=O)C1=CC=CO1 XTULMSXFIHGYFS-VLSRWLAYSA-N 0.000 description 2
- 230000027119 gastric acid secretion Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 238000002664 inhalation therapy Methods 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 150000002617 leukotrienes Chemical class 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- BLWYXBNNBYXPPL-YFKPBYRVSA-N methyl (2s)-pyrrolidine-2-carboxylate Chemical compound COC(=O)[C@@H]1CCCN1 BLWYXBNNBYXPPL-YFKPBYRVSA-N 0.000 description 2
- BMAMQYNHUPUMSD-UHFFFAOYSA-N methyl 3-(cyclopropylmethoxy)-5-(methanesulfonamido)benzoate Chemical compound COC(=O)C1=CC(NS(C)(=O)=O)=CC(OCC2CC2)=C1 BMAMQYNHUPUMSD-UHFFFAOYSA-N 0.000 description 2
- ZWSMBBMMWQPBEL-UHFFFAOYSA-N methyl 3-(cyclopropylmethoxy)-5-[methylsulfonyl(2-morpholin-4-ylethyl)amino]benzoate Chemical compound C=1C(N(CCN2CCOCC2)S(C)(=O)=O)=CC(C(=O)OC)=CC=1OCC1CC1 ZWSMBBMMWQPBEL-UHFFFAOYSA-N 0.000 description 2
- HTYJYEKZUXJKKW-UHFFFAOYSA-N methyl 3-hydroxy-5-nitrobenzoate Chemical compound COC(=O)C1=CC(O)=CC([N+]([O-])=O)=C1 HTYJYEKZUXJKKW-UHFFFAOYSA-N 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 210000003097 mucus Anatomy 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 239000003149 muscarinic antagonist Substances 0.000 description 2
- JERXUPDBWDWFCF-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-2-[1-[(4-fluorophenyl)methyl]pyrrolo[2,3-b]pyridin-3-yl]-2-oxoacetamide Chemical compound C1=CC(F)=CC=C1CN1C2=NC=CC=C2C(C(=O)C(=O)NC=2C(=CN=CC=2Cl)Cl)=C1 JERXUPDBWDWFCF-UHFFFAOYSA-N 0.000 description 2
- 230000010494 opalescence Effects 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 210000001711 oxyntic cell Anatomy 0.000 description 2
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 2
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- GZNAASVAJNXPPW-UHFFFAOYSA-M tin(4+) chloride dihydrate Chemical compound O.O.[Cl-].[Sn+4] GZNAASVAJNXPPW-UHFFFAOYSA-M 0.000 description 2
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Substances O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 2
- WJUFSDZVCOTFON-UHFFFAOYSA-N veratraldehyde Chemical compound COC1=CC=C(C=O)C=C1OC WJUFSDZVCOTFON-UHFFFAOYSA-N 0.000 description 2
- 238000001851 vibrational circular dichroism spectroscopy Methods 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 1
- XWTYSIMOBUGWOL-UHFFFAOYSA-N (+-)-Terbutaline Chemical compound CC(C)(C)NCC(O)C1=CC(O)=CC(O)=C1 XWTYSIMOBUGWOL-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-SECBINFHSA-N (2R)-2-(6-methoxy-2-naphthalenyl)propanoic acid Chemical compound C1=C([C@@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-SECBINFHSA-N 0.000 description 1
- VNYLFYQAPZJKAL-IKQZNINXSA-N (2r,3r)-2,3-dihydroxybutanedioic acid;ethyl (2s)-1,3-thiazolidine-2-carboxylate Chemical compound CCOC(=O)[C@H]1NCCS1.OC(=O)[C@H](O)[C@@H](O)C(O)=O VNYLFYQAPZJKAL-IKQZNINXSA-N 0.000 description 1
- ZQEBQGAAWMOMAI-ZETCQYMHSA-N (2s)-1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCC[C@H]1C(O)=O ZQEBQGAAWMOMAI-ZETCQYMHSA-N 0.000 description 1
- XNROFTAJEGCDCT-NSHDSACASA-N (2s)-1-benzylpyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1CC1=CC=CC=C1 XNROFTAJEGCDCT-NSHDSACASA-N 0.000 description 1
- HYAXPNDMEODKHI-LURJTMIESA-N (2s)-3-[(2-methylpropan-2-yl)oxycarbonyl]-1,3-thiazolidine-2-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CCS[C@H]1C(O)=O HYAXPNDMEODKHI-LURJTMIESA-N 0.000 description 1
- NDAUXUAQIAJITI-LBPRGKRZSA-N (R)-salbutamol Chemical compound CC(C)(C)NC[C@H](O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-LBPRGKRZSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical class C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- QZMQKPGVXNSITP-UHFFFAOYSA-N 1,3-benzodioxole-4-carbaldehyde Chemical compound O=CC1=CC=CC2=C1OCO2 QZMQKPGVXNSITP-UHFFFAOYSA-N 0.000 description 1
- 150000005529 1,3-benzodioxoles Chemical class 0.000 description 1
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 1
- VIGPKJYEYGGCLK-UHFFFAOYSA-N 1-methylimidazole-2-sulfonyl chloride Chemical compound CN1C=CN=C1S(Cl)(=O)=O VIGPKJYEYGGCLK-UHFFFAOYSA-N 0.000 description 1
- DZJLJUXZISUCGU-UHFFFAOYSA-N 1-oxidopyridin-1-ium;hydrochloride Chemical compound Cl.[O-][N+]1=CC=CC=C1 DZJLJUXZISUCGU-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- RCPIMTCZGQJSGZ-HNNXBMFYSA-N 2-[2-[(3s)-3-(dimethylamino)pyrrolidin-1-yl]pyridin-3-yl]-5-ethyl-7-methoxy-3,1-benzoxazin-4-one Chemical compound O1C(=O)C=2C(CC)=CC(OC)=CC=2N=C1C1=CC=CN=C1N1CC[C@H](N(C)C)C1 RCPIMTCZGQJSGZ-HNNXBMFYSA-N 0.000 description 1
- DHQQXRRWRZFGDW-WBAXXEDZSA-N 2-[3-[2-[[(2s)-3-methyl-1-oxo-1-[(2s)-2-[[(3s)-1,1,1-trifluoro-4-methyl-2-oxopentan-3-yl]carbamoyl]pyrrolidin-1-yl]butan-2-yl]amino]-2-oxoethyl]-2-oxoimidazolidin-1-yl]acetic acid Chemical compound N([C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)C(F)(F)F)C(=O)CN1CCN(CC(O)=O)C1=O DHQQXRRWRZFGDW-WBAXXEDZSA-N 0.000 description 1
- UHWNENCHFSDZQP-UHFFFAOYSA-N 2-bromo-1-thiophen-2-ylethanone Chemical compound BrCC(=O)C1=CC=CS1 UHWNENCHFSDZQP-UHFFFAOYSA-N 0.000 description 1
- VMZCDNSFRSVYKQ-UHFFFAOYSA-N 2-phenylacetyl chloride Chemical compound ClC(=O)CC1=CC=CC=C1 VMZCDNSFRSVYKQ-UHFFFAOYSA-N 0.000 description 1
- OZMJVFGAVKZJMT-UHFFFAOYSA-N 3-(1,3-dioxolan-2-ylmethyl)benzoic acid Chemical compound OC(=O)C1=CC=CC(CC2OCCO2)=C1 OZMJVFGAVKZJMT-UHFFFAOYSA-N 0.000 description 1
- IAIVBMDMECTBOB-UHFFFAOYSA-N 3-(chloromethyl)-n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC(CCl)=C1 IAIVBMDMECTBOB-UHFFFAOYSA-N 0.000 description 1
- JMWYNUHGKFJVIB-QGFXQWJDSA-N 3-[(3R,9S,12S,15S,18S,24S,30S,33S,36S,39S,42S,45S,48S)-12-(4-aminobutyl)-15-(3-amino-3-oxopropyl)-30-[(2S)-butan-2-yl]-39-[(1R)-1-hydroxyethyl]-33-(hydroxymethyl)-9-[(4-hydroxyphenyl)methyl]-36-methyl-45-octyl-2,8,11,14,17,23,29,32,35,38,41,44,47-tridecaoxo-1,7,10,13,16,22,28,31,34,37,40,43,46-tridecazapentacyclo[46.3.0.03,7.018,22.024,28]henpentacontan-42-yl]propanoic acid Chemical compound C([C@H]1C(=O)N2CCC[C@@H]2C(=O)N2CCC[C@H]2C(=O)N[C@H](C(N[C@@H](CCC(O)=O)C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N2CCC[C@H]2C(=O)N2CCC[C@H]2C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N1)[C@@H](C)CC)[C@@H](C)O)=O)CCCCCCCC)C1=CC=C(O)C=C1 JMWYNUHGKFJVIB-QGFXQWJDSA-N 0.000 description 1
- ZAUYJHXTZBLVKG-UHFFFAOYSA-N 3-acetyl-n,n-dimethylbenzamide Chemical compound CN(C)C(=O)C1=CC=CC(C(C)=O)=C1 ZAUYJHXTZBLVKG-UHFFFAOYSA-N 0.000 description 1
- CHZPJUSFUDUEMZ-UHFFFAOYSA-N 3-acetylbenzoic acid Chemical compound CC(=O)C1=CC=CC(C(O)=O)=C1 CHZPJUSFUDUEMZ-UHFFFAOYSA-N 0.000 description 1
- LMRKXSDOAFUINK-UHFFFAOYSA-N 3-chlorosulfonylbenzoic acid Chemical compound OC(=O)C1=CC=CC(S(Cl)(=O)=O)=C1 LMRKXSDOAFUINK-UHFFFAOYSA-N 0.000 description 1
- BIAVGWDGIJKWRM-FQEVSTJZSA-N 3-hydroxy-2-phenyl-n-[(1s)-1-phenylpropyl]quinoline-4-carboxamide Chemical compound N([C@@H](CC)C=1C=CC=CC=1)C(=O)C(C1=CC=CC=C1N=1)=C(O)C=1C1=CC=CC=C1 BIAVGWDGIJKWRM-FQEVSTJZSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- UBMGTTRDNUKZMT-UHFFFAOYSA-N 4-[(1-hydroxy-3h-2,1-benzoxaborol-5-yl)oxy]benzene-1,2-dicarbonitrile Chemical compound C=1C=C2B(O)OCC2=CC=1OC1=CC=C(C#N)C(C#N)=C1 UBMGTTRDNUKZMT-UHFFFAOYSA-N 0.000 description 1
- SKDHHIUENRGTHK-UHFFFAOYSA-N 4-nitrobenzoyl chloride Chemical compound [O-][N+](=O)C1=CC=C(C(Cl)=O)C=C1 SKDHHIUENRGTHK-UHFFFAOYSA-N 0.000 description 1
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 description 1
- IHOXNOQMRZISPV-YJYMSZOUSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-methoxyphenyl)propan-2-yl]azaniumyl]ethyl]-2-oxo-1h-quinolin-8-olate Chemical compound C1=CC(OC)=CC=C1C[C@@H](C)NC[C@H](O)C1=CC=C(O)C2=C1C=CC(=O)N2 IHOXNOQMRZISPV-YJYMSZOUSA-N 0.000 description 1
- OUBZCDOQEMLMAB-UHFFFAOYSA-N 5-chlorosulfonyl-2-methoxybenzoic acid Chemical compound COC1=CC=C(S(Cl)(=O)=O)C=C1C(O)=O OUBZCDOQEMLMAB-UHFFFAOYSA-N 0.000 description 1
- VFEAMMGYJFFXKV-UHFFFAOYSA-N 5-formylthiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(C=O)S1 VFEAMMGYJFFXKV-UHFFFAOYSA-N 0.000 description 1
- GOZMBJCYMQQACI-UHFFFAOYSA-N 6,7-dimethyl-3-[[methyl-[2-[methyl-[[1-[3-(trifluoromethyl)phenyl]indol-3-yl]methyl]amino]ethyl]amino]methyl]chromen-4-one;dihydrochloride Chemical compound Cl.Cl.C=1OC2=CC(C)=C(C)C=C2C(=O)C=1CN(C)CCN(C)CC(C1=CC=CC=C11)=CN1C1=CC=CC(C(F)(F)F)=C1 GOZMBJCYMQQACI-UHFFFAOYSA-N 0.000 description 1
- XDBHURGONHZNJF-UHFFFAOYSA-N 6-[2-(3,4-diethoxyphenyl)-1,3-thiazol-4-yl]pyridine-2-carboxylic acid Chemical compound C1=C(OCC)C(OCC)=CC=C1C1=NC(C=2N=C(C=CC=2)C(O)=O)=CS1 XDBHURGONHZNJF-UHFFFAOYSA-N 0.000 description 1
- JFHROPTYMMSOLG-UHFFFAOYSA-N 6-[3-(dimethylcarbamoyl)phenyl]sulfonyl-4-(3-methoxyanilino)-8-methylquinoline-3-carboxamide Chemical compound COC1=CC=CC(NC=2C3=CC(=CC(C)=C3N=CC=2C(N)=O)S(=O)(=O)C=2C=C(C=CC=2)C(=O)N(C)C)=C1 JFHROPTYMMSOLG-UHFFFAOYSA-N 0.000 description 1
- QNQZWEGMKJBHEM-UHFFFAOYSA-N 6-methyl-5-(2-methylpyrazol-3-yl)-n-[(5-methylsulfonylpyridin-2-yl)methyl]-2-oxo-1-[3-(trifluoromethyl)phenyl]pyridine-3-carboxamide Chemical compound O=C1N(C=2C=C(C=CC=2)C(F)(F)F)C(C)=C(C=2N(N=CC=2)C)C=C1C(=O)NCC1=CC=C(S(C)(=O)=O)C=N1 QNQZWEGMKJBHEM-UHFFFAOYSA-N 0.000 description 1
- WDYVUKGVKRZQNM-UHFFFAOYSA-N 6-phosphonohexylphosphonic acid Chemical compound OP(O)(=O)CCCCCCP(O)(O)=O WDYVUKGVKRZQNM-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 206010068172 Anal pruritus Diseases 0.000 description 1
- 206010051113 Arterial restenosis Diseases 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 229910015844 BCl3 Inorganic materials 0.000 description 1
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 206010006458 Bronchitis chronic Diseases 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 208000032544 Cicatrix Diseases 0.000 description 1
- LUKZNWIVRBCLON-GXOBDPJESA-N Ciclesonide Chemical compound C1([C@H]2O[C@@]3([C@H](O2)C[C@@H]2[C@@]3(C[C@H](O)[C@@H]3[C@@]4(C)C=CC(=O)C=C4CC[C@H]32)C)C(=O)COC(=O)C(C)C)CCCCC1 LUKZNWIVRBCLON-GXOBDPJESA-N 0.000 description 1
- IHDDYKNRVLPVEV-FFKPOUSOSA-N Cl.COC1=C(OC)C(OC)=CC(C(=O)N2C[C@](CCN3CCC4(CC3)[S@](CC3=CC=CC=C34)=O)(OCC2)C=2C=C(Cl)C(Cl)=CC=2)=C1 Chemical compound Cl.COC1=C(OC)C(OC)=CC(C(=O)N2C[C@](CCN3CCC4(CC3)[S@](CC3=CC=CC=C34)=O)(OCC2)C=2C=C(Cl)C(Cl)=CC=2)=C1 IHDDYKNRVLPVEV-FFKPOUSOSA-N 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 208000006926 Discoid Lupus Erythematosus Diseases 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000002149 Elafin Human genes 0.000 description 1
- 108010015972 Elafin Proteins 0.000 description 1
- 206010014824 Endotoxic shock Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 201000011240 Frontotemporal dementia Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010018367 Glomerulonephritis chronic Diseases 0.000 description 1
- VPNYRYCIDCJBOM-UHFFFAOYSA-M Glycopyrronium bromide Chemical compound [Br-].C1[N+](C)(C)CCC1OC(=O)C(O)(C=1C=CC=CC=1)C1CCCC1 VPNYRYCIDCJBOM-UHFFFAOYSA-M 0.000 description 1
- 206010019617 Henoch-Schonlein purpura Diseases 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- 206010020649 Hyperkeratosis Diseases 0.000 description 1
- 229940122296 IKK2 inhibitor Drugs 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 208000031814 IgA Vasculitis Diseases 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100021854 Inhibitor of nuclear factor kappa-B kinase subunit beta Human genes 0.000 description 1
- 101710205525 Inhibitor of nuclear factor kappa-B kinase subunit beta Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 208000001126 Keratosis Diseases 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- 229930182821 L-proline Natural products 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 206010069698 Langerhans' cell histiocytosis Diseases 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 206010071759 Muscle oedema Diseases 0.000 description 1
- 108010014632 NF-kappa B kinase Proteins 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 201000009053 Neurodermatitis Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- RCPIMTCZGQJSGZ-UHFFFAOYSA-N O1C(=O)C=2C(CC)=CC(OC)=CC=2N=C1C1=CC=CN=C1N1CCC(N(C)C)C1 Chemical compound O1C(=O)C=2C(CC)=CC(OC)=CC=2N=C1C1=CC=CN=C1N1CCC(N(C)C)C1 RCPIMTCZGQJSGZ-UHFFFAOYSA-N 0.000 description 1
- 239000012425 OXONE® Substances 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- NMLMACJWHPHKGR-NCOIDOBVSA-N P(1),P(4)-bis(uridin-5'-yl) tetraphosphate Chemical compound N1([C@@H]2O[C@@H]([C@H]([C@H]2O)O)COP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@H]([C@@H](O2)N2C(NC(=O)C=C2)=O)O)O)C=CC(=O)NC1=O NMLMACJWHPHKGR-NCOIDOBVSA-N 0.000 description 1
- KCAJXIDMCNPGHZ-UHFFFAOYSA-N PH 797804 Chemical compound CNC(=O)C1=CC=C(C)C(N2C(C(Br)=C(OCC=3C(=CC(F)=CC=3)F)C=C2C)=O)=C1 KCAJXIDMCNPGHZ-UHFFFAOYSA-N 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 208000000609 Pick Disease of the Brain Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Chemical class OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 208000009544 Pruritus Ani Diseases 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 208000006311 Pyoderma Diseases 0.000 description 1
- 239000005869 Pyraclostrobin Substances 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- BPZSYCZIITTYBL-YJYMSZOUSA-N R-Formoterol Chemical compound C1=CC(OC)=CC=C1C[C@@H](C)NC[C@H](O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-YJYMSZOUSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 206010039793 Seborrhoeic dermatitis Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 201000002661 Spondylitis Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Chemical class OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 206010042496 Sunburn Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000024780 Urticaria Diseases 0.000 description 1
- YEEZWCHGZNKEEK-UHFFFAOYSA-N Zafirlukast Chemical compound COC1=CC(C(=O)NS(=O)(=O)C=2C(=CC=CC=2)C)=CC=C1CC(C1=C2)=CN(C)C1=CC=C2NC(=O)OC1CCCC1 YEEZWCHGZNKEEK-UHFFFAOYSA-N 0.000 description 1
- XKTXHHVUFAHYGM-IGKIAQTJSA-N [(1S)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloropyridin-4-yl)ethyl] (2S)-3-[1-methyl-5-(methylcarbamoyl)pyrrol-3-yl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound CNC(=O)c1cc(cn1C)S(=O)(=O)N1CCS[C@H]1C(=O)O[C@@H](Cc1c(Cl)cncc1Cl)c1ccc(OC(F)F)c(OCC2CC2)c1 XKTXHHVUFAHYGM-IGKIAQTJSA-N 0.000 description 1
- UGOQFUDGPPMJNZ-FIBWVYCGSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-[3-(dimethylcarbamoyl)-4-methoxyphenyl]sulfonylpiperidine-2-carboxylate Chemical compound C1=C(C(=O)N(C)C)C(OC)=CC=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)CCCC1 UGOQFUDGPPMJNZ-FIBWVYCGSA-N 0.000 description 1
- FVJAJDBFJYPVHJ-YTMVLYRLSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-[3-(dimethylcarbamoyl)phenyl]sulfonylpiperidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](CCCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 FVJAJDBFJYPVHJ-YTMVLYRLSA-N 0.000 description 1
- MTVYRGXACZGKJC-XCZPVHLTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-1-[3-(dimethylcarbamoyl)phenyl]sulfonylpyrrolidine-2-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](CCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 MTVYRGXACZGKJC-XCZPVHLTSA-N 0.000 description 1
- XRPXHTLRHVBCJL-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-2-acetyloxy-2-phenylacetate Chemical compound C([C@H](OC(=O)[C@@H](OC(=O)C)C=1C=CC=CC=1)C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)C1=C(Cl)C=[N+]([O-])C=C1Cl XRPXHTLRHVBCJL-AHWVRZQESA-N 0.000 description 1
- BGUMVCLFIHNLIQ-GMAHTHKFSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(1-methylimidazol-2-yl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound CN1C=CN=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 BGUMVCLFIHNLIQ-GMAHTHKFSA-N 0.000 description 1
- RMBMOTAIHPNVBP-LSYYVWMOSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(3,4-dimethoxyphenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=C(OC)C(OC)=CC=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 RMBMOTAIHPNVBP-LSYYVWMOSA-N 0.000 description 1
- IHKRLXUVQSEXLI-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(3-aminophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound NC1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 IHKRLXUVQSEXLI-AHWVRZQESA-N 0.000 description 1
- ZERXSOCWMFZOCF-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(3-sulfamoylphenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound NS(=O)(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 ZERXSOCWMFZOCF-AHWVRZQESA-N 0.000 description 1
- WGMKARWCFXMPMU-BDYUSTAISA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(4-cyanophenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(S(=O)(=O)C=2C=CC(=CC=2)C#N)CCS1 WGMKARWCFXMPMU-BDYUSTAISA-N 0.000 description 1
- KMXAECLWROFHEQ-BDYUSTAISA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(4-methoxyphenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 KMXAECLWROFHEQ-BDYUSTAISA-N 0.000 description 1
- LNSVVJAEOAXFJM-BDYUSTAISA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(4-methylsulfonylphenyl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=CC(S(=O)(=O)C)=CC=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 LNSVVJAEOAXFJM-BDYUSTAISA-N 0.000 description 1
- CQIOWOZNRBMYTQ-AHWVRZQESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-(benzenesulfonyl)-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(S(=O)(=O)C=2C=CC=CC=2)CCS1 CQIOWOZNRBMYTQ-AHWVRZQESA-N 0.000 description 1
- NIKMCXVBVZIDRF-WNJJXGMVSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[(1-methyl-2-oxo-3h-indol-5-yl)sulfonyl]-1,3-thiazolidine-2-carboxylate Chemical compound C([C@H](OC(=O)[C@@H]1SCCN1S(=O)(=O)C=1C=C2CC(=O)N(C2=CC=1)C)C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)C1=C(Cl)C=[N+]([O-])C=C1Cl NIKMCXVBVZIDRF-WNJJXGMVSA-N 0.000 description 1
- FTYLSYQFNDIQJM-IGKIAQTJSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[(3-methyl-[1,2]oxazolo[5,4-b]pyridin-5-yl)sulfonyl]-1,3-thiazolidine-2-carboxylate Chemical compound C([C@H](OC(=O)[C@@H]1SCCN1S(=O)(=O)C1=CN=C2ON=C(C2=C1)C)C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)C1=C(Cl)C=[N+]([O-])C=C1Cl FTYLSYQFNDIQJM-IGKIAQTJSA-N 0.000 description 1
- PDAKAOWBRGLLIX-NYDCQLBNSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(4-methylpiperazine-1-carbonyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1CN(C)CCN1C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 PDAKAOWBRGLLIX-NYDCQLBNSA-N 0.000 description 1
- YAMBPONVOKNWJI-FIBWVYCGSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(dimethylcarbamoyl)-4-methoxyphenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C1=C(C(=O)N(C)C)C(OC)=CC=C1S(=O)(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 YAMBPONVOKNWJI-FIBWVYCGSA-N 0.000 description 1
- JXGLWYARMJTRKG-XCZPVHLTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[3-(dimethylsulfamoyl)phenyl]sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound CN(C)S(=O)(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](SCC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 JXGLWYARMJTRKG-XCZPVHLTSA-N 0.000 description 1
- LDHUCIBHSSZKBK-WNJJXGMVSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-[[5-(pyrrolidine-1-carbonyl)-1h-pyrrol-3-yl]sulfonyl]-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(S(=O)(=O)C=2C=C(NC=2)C(=O)N2CCCC2)CCS1 LDHUCIBHSSZKBK-WNJJXGMVSA-N 0.000 description 1
- VGVYQKLMVAAGTL-ZCYQVOJMSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (2s)-3-pyridin-3-ylsulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)[C@H]1N(S(=O)(=O)C=2C=NC=CC=2)CCS1 VGVYQKLMVAAGTL-ZCYQVOJMSA-N 0.000 description 1
- ZNBYQFKUVLIBOS-BDYUSTAISA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] (4r)-3-[3-(dimethylcarbamoyl)phenyl]sulfonyl-1,3-thiazolidine-4-carboxylate Chemical compound CN(C)C(=O)C1=CC=CC(S(=O)(=O)N2[C@@H](CSC2)C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=C1 ZNBYQFKUVLIBOS-BDYUSTAISA-N 0.000 description 1
- OEKHQHPOQBNHKX-ISZYABMKSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 1,3-thiazolidine-2-carboxylate;hydrochloride Chemical compound Cl.ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)C1SCCN1 OEKHQHPOQBNHKX-ISZYABMKSA-N 0.000 description 1
- JEYXLDGAQMAOCR-ALLRNTDFSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(3-formylbenzoyl)-1,3-thiazolidine-2-carboxylate Chemical compound ClC1=C[N+]([O-])=CC(Cl)=C1C[C@@H](C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)OC(=O)C1N(C(=O)C=2C=C(C=O)C=CC=2)CCS1 JEYXLDGAQMAOCR-ALLRNTDFSA-N 0.000 description 1
- ADISPUZFHWISCT-ZZDYIDRTSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-[5-[(dimethylamino)methyl]thiophene-2-carbonyl]-1,3-thiazolidine-2-carboxylate Chemical compound S1C(CN(C)C)=CC=C1C(=O)N1C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)SCC1 ADISPUZFHWISCT-ZZDYIDRTSA-N 0.000 description 1
- MWZVCRDVMVPEIF-IGKIAQTJSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloropyridin-4-yl)ethyl] (2s)-1-(4-methoxy-3-methylsulfonyloxybenzoyl)pyrrolidine-2-carboxylate Chemical compound C1=C(OS(C)(=O)=O)C(OC)=CC=C1C(=O)N1[C@H](C(=O)O[C@@H](CC=2C(=CN=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)CCC1 MWZVCRDVMVPEIF-IGKIAQTJSA-N 0.000 description 1
- JFGMFYHVRLFDJF-IZEXYCQBSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloropyridin-4-yl)ethyl] (2s)-1-[4-(cyclopropylmethoxy)-3-(methanesulfonamido)benzoyl]pyrrolidine-2-carboxylate Chemical compound CS(=O)(=O)NC1=CC(C(=O)N2[C@@H](CCC2)C(=O)O[C@@H](CC=2C(=CN=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)=CC=C1OCC1CC1 JFGMFYHVRLFDJF-IZEXYCQBSA-N 0.000 description 1
- FFCPAPWIBACXPD-LSYYVWMOSA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloropyridin-4-yl)ethyl] (2s)-3-(1,3-dimethylpyrazolo[3,4-b]pyridin-5-yl)sulfonyl-1,3-thiazolidine-2-carboxylate Chemical compound C([C@H](OC(=O)[C@@H]1SCCN1S(=O)(=O)C1=CN=C2N(C)N=C(C2=C1)C)C=1C=C(OCC2CC2)C(OC(F)F)=CC=1)C1=C(Cl)C=NC=C1Cl FFCPAPWIBACXPD-LSYYVWMOSA-N 0.000 description 1
- YPFLFUJKZDAXRA-UHFFFAOYSA-N [3-(carbamoylamino)-2-(2,4-dichlorobenzoyl)-1-benzofuran-6-yl] methanesulfonate Chemical compound O1C2=CC(OS(=O)(=O)C)=CC=C2C(NC(N)=O)=C1C(=O)C1=CC=C(Cl)C=C1Cl YPFLFUJKZDAXRA-UHFFFAOYSA-N 0.000 description 1
- SFYAXIFVXBKRPK-QFIPXVFZSA-N abediterol Chemical compound C([C@H](O)C=1C=2C=CC(=O)NC=2C(O)=CC=1)NCCCCCCOCC(F)(F)C1=CC=CC=C1 SFYAXIFVXBKRPK-QFIPXVFZSA-N 0.000 description 1
- ASMXXROZKSBQIH-VITNCHFBSA-N aclidinium Chemical compound C([C@@H](C(CC1)CC2)OC(=O)C(O)(C=3SC=CC=3)C=3SC=CC=3)[N+]21CCCOC1=CC=CC=C1 ASMXXROZKSBQIH-VITNCHFBSA-N 0.000 description 1
- 229940019903 aclidinium Drugs 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 210000005091 airway smooth muscle Anatomy 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 208000004631 alopecia areata Diseases 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 229940065524 anticholinergics inhalants for obstructive airway diseases Drugs 0.000 description 1
- IMOZEMNVLZVGJZ-QGZVFWFLSA-N apremilast Chemical compound C1=C(OC)C(OCC)=CC([C@@H](CS(C)(=O)=O)N2C(C3=C(NC(C)=O)C=CC=C3C2=O)=O)=C1 IMOZEMNVLZVGJZ-QGZVFWFLSA-N 0.000 description 1
- 229960001164 apremilast Drugs 0.000 description 1
- 229960001692 arformoterol Drugs 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 208000024998 atopic conjunctivitis Diseases 0.000 description 1
- GFZWHAAOIVMHOI-UHFFFAOYSA-N azetidine-3-carboxylic acid Chemical compound OC(=O)C1CNC1 GFZWHAAOIVMHOI-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 229960003060 bambuterol Drugs 0.000 description 1
- ANZXOIAKUNOVQU-UHFFFAOYSA-N bambuterol Chemical compound CN(C)C(=O)OC1=CC(OC(=O)N(C)C)=CC(C(O)CNC(C)(C)C)=C1 ANZXOIAKUNOVQU-UHFFFAOYSA-N 0.000 description 1
- 229950000210 beclometasone dipropionate Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 102000014974 beta2-adrenergic receptor activity proteins Human genes 0.000 description 1
- 108040006828 beta2-adrenergic receptor activity proteins Proteins 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical class O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 229950010713 carmoterol Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- CFBUZOUXXHZCFB-OYOVHJISSA-N chembl511115 Chemical compound COC1=CC=C([C@@]2(CC[C@H](CC2)C(O)=O)C#N)C=C1OC1CCCC1 CFBUZOUXXHZCFB-OYOVHJISSA-N 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 208000007451 chronic bronchitis Diseases 0.000 description 1
- 229960003728 ciclesonide Drugs 0.000 description 1
- 229950001653 cilomilast Drugs 0.000 description 1
- 229960001117 clenbuterol Drugs 0.000 description 1
- STJMRWALKKWQGH-UHFFFAOYSA-N clenbuterol Chemical compound CC(C)(C)NCC(O)C1=CC(Cl)=C(N)C(Cl)=C1 STJMRWALKKWQGH-UHFFFAOYSA-N 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- USZAGAREISWJDP-UHFFFAOYSA-N crisaborole Chemical compound C=1C=C2B(O)OCC2=CC=1OC1=CC=C(C#N)C=C1 USZAGAREISWJDP-UHFFFAOYSA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- 208000004921 cutaneous lupus erythematosus Diseases 0.000 description 1
- 125000005366 cycloalkylthio group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- JMYVMOUINOAAPA-UHFFFAOYSA-N cyclopropanecarbaldehyde Chemical compound O=CC1CC1 JMYVMOUINOAAPA-UHFFFAOYSA-N 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 229950003529 diquafosol Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 201000006549 dyspepsia Diseases 0.000 description 1
- MDCUNMLZLNGCQA-HWOAGHQOSA-N elafin Chemical compound N([C@H](C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H]1C(=O)N2CCC[C@H]2C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H]2CSSC[C@H]3C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N[C@@H](C)C(=O)N[C@@H](CSSC[C@H]4C(=O)N5CCC[C@H]5C(=O)NCC(=O)N[C@H](C(N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H]5N(CCC5)C(=O)[C@H]5N(CCC5)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCSC)NC(=O)[C@H](C)NC2=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N4)C(=O)N[C@@H](CSSC1)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N3)=O)[C@@H](C)CC)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(N)=O)C(O)=O)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)C(C)C)C(C)C)C(=O)[C@@H]1CCCN1C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)N MDCUNMLZLNGCQA-HWOAGHQOSA-N 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- CSOBIBXVIYAXFM-BYNJWEBRSA-N ensifentrine Chemical compound c-12cc(OC)c(OC)cc2CCn(c(n2CCNC(N)=O)=O)c-1c\c2=N/c1c(C)cc(C)cc1C CSOBIBXVIYAXFM-BYNJWEBRSA-N 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 208000003401 eosinophilic granuloma Diseases 0.000 description 1
- UJTPZISIAWDGFF-UHFFFAOYSA-N ethenylsulfonylbenzene Chemical compound C=CS(=O)(=O)C1=CC=CC=C1 UJTPZISIAWDGFF-UHFFFAOYSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- 229960001022 fenoterol Drugs 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229960001469 fluticasone furoate Drugs 0.000 description 1
- WMWTYOKRWGGJOA-CENSZEJFSA-N fluticasone propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O WMWTYOKRWGGJOA-CENSZEJFSA-N 0.000 description 1
- 229960000289 fluticasone propionate Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960002848 formoterol Drugs 0.000 description 1
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229940015042 glycopyrrolate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 208000024798 heartburn Diseases 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 150000005828 hydrofluoroalkanes Chemical class 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 238000007866 imination reaction Methods 0.000 description 1
- 208000015446 immunoglobulin a vasculitis Diseases 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 229960004078 indacaterol Drugs 0.000 description 1
- QZZUEBNBZAPZLX-QFIPXVFZSA-N indacaterol Chemical compound N1C(=O)C=CC2=C1C(O)=CC=C2[C@@H](O)CNC1CC(C=C(C(=C2)CC)CC)=C2C1 QZZUEBNBZAPZLX-QFIPXVFZSA-N 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229940125369 inhaled corticosteroids Drugs 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000037041 intracellular level Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- OEXHQOGQTVQTAT-JRNQLAHRSA-N ipratropium Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 OEXHQOGQTVQTAT-JRNQLAHRSA-N 0.000 description 1
- 229960001888 ipratropium Drugs 0.000 description 1
- 229940039009 isoproterenol Drugs 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 229950008204 levosalbutamol Drugs 0.000 description 1
- 239000000865 liniment Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical class OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Chemical class 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- WINMHOHYPKKTEM-UHFFFAOYSA-N methyl 5-formyl-2-methylfuran-3-carboxylate Chemical compound COC(=O)C=1C=C(C=O)OC=1C WINMHOHYPKKTEM-UHFFFAOYSA-N 0.000 description 1
- 229950001768 milveterol Drugs 0.000 description 1
- 239000003595 mist Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- WOFMFGQZHJDGCX-ZULDAHANSA-N mometasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(Cl)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)C(=O)CCl)C(=O)C1=CC=CO1 WOFMFGQZHJDGCX-ZULDAHANSA-N 0.000 description 1
- 229960002744 mometasone furoate Drugs 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 230000003843 mucus production Effects 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- NPGREARFJMFTDF-UHFFFAOYSA-N n-(3,5-dichloro-1-hydroxypyridin-4-ylidene)-8-methoxy-2-(trifluoromethyl)quinoline-5-carboxamide Chemical compound C12=CC=C(C(F)(F)F)N=C2C(OC)=CC=C1C(=O)N=C1C(Cl)=CN(O)C=C1Cl NPGREARFJMFTDF-UHFFFAOYSA-N 0.000 description 1
- DPHDSIQHVGSITN-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-2-[1-[(4-fluorophenyl)methyl]-5-hydroxyindol-3-yl]-2-oxoacetamide Chemical compound C1=C(C(=O)C(=O)NC=2C(=CN=CC=2Cl)Cl)C2=CC(O)=CC=C2N1CC1=CC=C(F)C=C1 DPHDSIQHVGSITN-UHFFFAOYSA-N 0.000 description 1
- BHCJHYIMNHXLOM-WVDRJWPYSA-N n-[(e,2r)-1-(3,4-dichlorophenyl)-5-oxo-5-[[(3r)-2-oxoazepan-3-yl]amino]pent-3-en-2-yl]-n-methyl-3,5-bis(trifluoromethyl)benzamide Chemical compound C([C@@H](N(C)C(=O)C=1C=C(C=C(C=1)C(F)(F)F)C(F)(F)F)\C=C\C(=O)N[C@H]1C(NCCCC1)=O)C1=CC=C(Cl)C(Cl)=C1 BHCJHYIMNHXLOM-WVDRJWPYSA-N 0.000 description 1
- ZVULMJYRVAVKCP-UHFFFAOYSA-N n-[2-(diethylamino)ethyl]-n-[2-[2-(4-hydroxy-2-oxo-3h-1,3-benzothiazol-7-yl)ethylamino]ethyl]-3-(2-naphthalen-1-ylethoxy)propanamide Chemical compound C1=CC=C2C(CCOCCC(=O)N(CCNCCC=3C=4SC(=O)NC=4C(O)=CC=3)CCN(CC)CC)=CC=CC2=C1 ZVULMJYRVAVKCP-UHFFFAOYSA-N 0.000 description 1
- QDZOBXFRIVOQBR-LJQANCHMSA-N n-[2-[(1s)-1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-3-oxo-1h-isoindol-4-yl]cyclopropanecarboxamide Chemical compound C1=C(OC)C(OCC)=CC([C@@H](CS(C)(=O)=O)N2C(C3=C(NC(=O)C4CC4)C=CC=C3C2)=O)=C1 QDZOBXFRIVOQBR-LJQANCHMSA-N 0.000 description 1
- BMKINZUHKYLSKI-DQEYMECFSA-N n-[2-hydroxy-5-[(1r)-1-hydroxy-2-[2-[4-[[(2r)-2-hydroxy-2-phenylethyl]amino]phenyl]ethylamino]ethyl]phenyl]formamide Chemical compound C1([C@@H](O)CNC2=CC=C(C=C2)CCNC[C@H](O)C=2C=C(NC=O)C(O)=CC=2)=CC=CC=C1 BMKINZUHKYLSKI-DQEYMECFSA-N 0.000 description 1
- DRMJRHUMYBYDIX-UHFFFAOYSA-N n-[4-[5-(4-fluorophenyl)-3-methyl-2-methylsulfinylimidazol-4-yl]pyridin-2-yl]acetamide Chemical compound C1=NC(NC(=O)C)=CC(C=2N(C(=NC=2C=2C=CC(F)=CC=2)S(C)=O)C)=C1 DRMJRHUMYBYDIX-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 230000000414 obstructive effect Effects 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- KCEHVJZZIGJAAW-FERBBOLQSA-N olodaterol hydrochloride Chemical compound Cl.C1=CC(OC)=CC=C1CC(C)(C)NC[C@H](O)C1=CC(O)=CC2=C1OCC(=O)N2 KCEHVJZZIGJAAW-FERBBOLQSA-N 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- NVOYVOBDTVTBDX-PMEUIYRNSA-N oxitropium Chemical class CC[N+]1(C)[C@H]2C[C@@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)[C@H](CO)C1=CC=CC=C1 NVOYVOBDTVTBDX-PMEUIYRNSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- HJKYXKSLRZKNSI-UHFFFAOYSA-I pentapotassium;hydrogen sulfate;oxido sulfate;sulfuric acid Chemical compound [K+].[K+].[K+].[K+].[K+].OS([O-])(=O)=O.[O-]S([O-])(=O)=O.OS(=O)(=O)O[O-].OS(=O)(=O)O[O-] HJKYXKSLRZKNSI-UHFFFAOYSA-I 0.000 description 1
- XCRBXWCUXJNEFX-UHFFFAOYSA-N peroxybenzoic acid Chemical compound OOC(=O)C1=CC=CC=C1 XCRBXWCUXJNEFX-UHFFFAOYSA-N 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- ISWRGOKTTBVCFA-UHFFFAOYSA-N pirfenidone Chemical compound C1=C(C)C=CC(=O)N1C1=CC=CC=C1 ISWRGOKTTBVCFA-UHFFFAOYSA-N 0.000 description 1
- 229960003073 pirfenidone Drugs 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 206010035653 pneumoconiosis Diseases 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical compound [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- OKBMCNHOEMXPTM-UHFFFAOYSA-M potassium peroxymonosulfate Chemical compound [K+].OOS([O-])(=O)=O OKBMCNHOEMXPTM-UHFFFAOYSA-M 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229960004583 pranlukast Drugs 0.000 description 1
- UAJUXJSXCLUTNU-UHFFFAOYSA-N pranlukast Chemical compound C=1C=C(OCCCCC=2C=CC=CC=2)C=CC=1C(=O)NC(C=1)=CC=C(C(C=2)=O)C=1OC=2C=1N=NNN=1 UAJUXJSXCLUTNU-UHFFFAOYSA-N 0.000 description 1
- 229960002288 procaterol Drugs 0.000 description 1
- FKNXQNWAXFXVNW-BLLLJJGKSA-N procaterol Chemical compound N1C(=O)C=CC2=C1C(O)=CC=C2[C@@H](O)[C@@H](NC(C)C)CC FKNXQNWAXFXVNW-BLLLJJGKSA-N 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- CPNGPNLZQNNVQM-UHFFFAOYSA-N pteridine Chemical compound N1=CN=CC2=NC=CN=C21 CPNGPNLZQNNVQM-UHFFFAOYSA-N 0.000 description 1
- HZRSNVGNWUDEFX-UHFFFAOYSA-N pyraclostrobin Chemical compound COC(=O)N(OC)C1=CC=CC=C1COC1=NN(C=2C=CC(Cl)=CC=2)C=C1 HZRSNVGNWUDEFX-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000004648 relaxation of smooth muscle Effects 0.000 description 1
- 229960002720 reproterol Drugs 0.000 description 1
- WVLAAKXASPCBGT-UHFFFAOYSA-N reproterol Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CCCNCC(O)C1=CC(O)=CC(O)=C1 WVLAAKXASPCBGT-UHFFFAOYSA-N 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 230000000552 rheumatic effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 201000004700 rosacea Diseases 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 150000003902 salicylic acid esters Chemical class 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 230000037387 scars Effects 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 208000008742 seborrheic dermatitis Diseases 0.000 description 1
- 238000011894 semi-preparative HPLC Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 238000010583 slow cooling Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 1
- 229960002218 sodium chlorite Drugs 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000008279 sol Substances 0.000 description 1
- 208000020431 spinal cord injury Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 230000006103 sulfonylation Effects 0.000 description 1
- 238000005694 sulfonylation reaction Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- ZMELOYOKMZBMRB-DLBZAZTESA-N talmapimod Chemical compound C([C@@H](C)N(C[C@@H]1C)C(=O)C=2C(=CC=3N(C)C=C(C=3C=2)C(=O)C(=O)N(C)C)Cl)N1CC1=CC=C(F)C=C1 ZMELOYOKMZBMRB-DLBZAZTESA-N 0.000 description 1
- 229950008389 talmapimod Drugs 0.000 description 1
- 229950011332 talnetant Drugs 0.000 description 1
- 229960000195 terbutaline Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229950002896 tetomilast Drugs 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- PASYJVRFGUDDEW-WMUGRWSXSA-J tetrasodium;[[(2r,3s,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methoxy-oxidophosphoryl] [[[(2r,3s,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-oxidophosphoryl]oxy-oxidophosphoryl] phosphate Chemical compound [Na+].[Na+].[Na+].[Na+].O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])(=O)OP([O-])(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C(NC(=O)C=C2)=O)O)[C@@H](O)C1 PASYJVRFGUDDEW-WMUGRWSXSA-J 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- LERNTVKEWCAPOY-DZZGSBJMSA-N tiotropium Chemical compound O([C@H]1C[C@@H]2[N+]([C@H](C1)[C@@H]1[C@H]2O1)(C)C)C(=O)C(O)(C=1SC=CC=1)C1=CC=CS1 LERNTVKEWCAPOY-DZZGSBJMSA-N 0.000 description 1
- 229940110309 tiotropium Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000583 toxicological profile Toxicity 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Chemical class OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- OYYDSUSKLWTMMQ-JKHIJQBDSA-N trospium Chemical compound [N+]12([C@@H]3CC[C@H]2C[C@H](C3)OC(=O)C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CCCC1 OYYDSUSKLWTMMQ-JKHIJQBDSA-N 0.000 description 1
- 229960001491 trospium Drugs 0.000 description 1
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 201000005539 vernal conjunctivitis Diseases 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229960004764 zafirlukast Drugs 0.000 description 1
- 229940032528 zemaira Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4425—Pyridinium derivatives, e.g. pralidoxime, pyridostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/538—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
- A61K31/635—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide having a heterocyclic ring, e.g. sulfadiazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/28—Radicals substituted by singly-bound oxygen or sulphur atoms
- C07D213/30—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Abstract
本发明涉及磷酸二酯酶4(PDE4)的抑制剂。更特别地,本发明涉及化合物,其是1‑苯基‑2‑吡啶基烷基醇的衍生物,制备所述化合物的方法,含有它们的组合物及其治疗用途。
Description
发明领域
本发明涉及磷酸二酯酶4(PDE4)的抑制剂。更特别,本发明涉及化 合物,其是1-苯基-2-吡啶基烷基醇的衍生物,制备所述化合物的方法, 含有它们的组合物及其治疗用途。
发明背景
气道阻塞是许多严重呼吸系统疾病的特征,包括哮喘和慢性阻塞性 肺疾病(COPD)。导致气道阻塞的事件包括气道壁水肿,增加的粘液产生 和炎症。
目前,用于治疗呼吸系统疾病比如哮喘和COPD的药物通过吸入 给予。吸入途径相比全身性途径的优势之一是在作用位置直接递送药物 的可能性,这降低全身性副作用,从而引起更快速的临床应答和更高的 治疗比率。
吸入的皮质类固醇是目前对于哮喘特选的维持疗法,并且与支气管 扩张药β2-激动剂一起用于缓解急性症状,它们构成对于该疾病的当前 疗法的支柱。目前的COPD的治理大致是症状性的:通过使用吸入的抗 胆碱能药和吸入的β2-肾上腺素受体激动剂的支气管扩张疗法。然而, 皮质类固醇并不像它们在哮喘中那样减少COPD中的炎性反应。
鉴于治疗炎性呼吸系统疾病比如哮喘和COPD的抗炎性效果已广 泛研究的又一类治疗剂由磷酸二酯酶(PDEs)的抑制剂,尤其是磷酸二酯 酶4型(此后称为PDE4)的抑制剂所代表。
充当PDE4抑制剂的各种化合物已公开于现有技术。然而,数种第 一代PDE4抑制剂比如咯利普兰和吡拉米司特的有用性由于其不希望的 副作用而受到限制。所述不希望的副作用包括由于其对中枢神经系统中 的PDE4的作用的恶心和呕吐,以及由于对肠壁细胞中的PDE4的作用 的胃酸分泌。
所述副作用的原因已广泛研究。
已发现PDE4存在代表不同构象的两种不同形式,所述构象指定为 高亲和力咯利普兰结合位点或HPDE4,特别是存在于中枢神经系统和壁 细胞中;和低亲和力咯利普兰结合位点或LPDE4(Jacobitz,S等人Mol. Pharmacol,1996,50,891-899),存在于免疫和炎性细胞中。虽然两种 形式均显得展示催化活性,它们对于抑制剂的敏感性并不相同。尤其是,具有对LPDE4更高亲和力的化合物显得更不倾向于诱导副作用比如恶 心、呕吐和增加的胃酸分泌。
靶向LPDE4的努力已经获得第二代PDE4抑制剂比如罗氟司特的 选择性的轻度改善。虽然如此,罗氟司特在给药不足的情况下才实现了 可接受的副作用特征。
充当PDE4抑制剂的其它类别的化合物已公开于现有技术中。
例如,尤其是EP 1634606公开酮衍生物比如苯并呋喃或1,3-苯并 二氧杂环戊烯衍生物。
尤其是,WO 9402465公开下述通式的酮衍生物

其中R1是低级烷基而R2可以是烷基,烯基,环烷基,环烷基,环 烯基,环烷硫基或环烯硫基。
Celltech药业的WO 9535281涉及三取代的苯基衍生物。
WO2009/018909公开1-苯基-2-吡啶基烷基醇的衍生物,其具有下 文报告的通式

用作磷酸二酯酶4(PDE4)的抑制剂。
WO2009/077068还公开1-苯基-2-吡啶基烷基醇的衍生物,其具有 下文报告的通式

用作磷酸二酯酶4(PDE4)的抑制剂。
WO2010/089107还公开1-苯基-2-吡啶基烷基醇的衍生物,其具有 下文报告的通式

尽管上述报告迄今已经公开了数种PDE4抑制剂,仍然需要更多的 PDE4抑制剂。特别地,仍然需要更多的具有PDE4酶高亲和力的PDE4 抑制剂。还特别有利的是,鉴定其它的PDE4抑制剂,其具有PDE4酶 高亲和力并且会显示作为吸入治疗的适当的可发展性特征,例如在降低 的副作用方面的特征。
副作用的所述降低可以通过例如药物的低全身性暴露来实现;从 而,在某些药代动力学特征方面的适当特征,特别是代谢清除率,对该 目标来说可以是关键。
本发明通过提供本发明化合物来应对上述需要。
发明概要
本发明涉及化合物充当磷酸二酯酶4(PDE4)的抑制剂,制备所述化 合物的方法,含有它们的组合物及其治疗用途。
尤其是,本发明涉及通式(I)的1-苯基-2-吡啶基烷基醇的衍生物

其中:
R1选自:
-H;
-(C3-C7)环烷基羰基;
-(C1-C6)烷基,任选由选自下述的一个或多个取代基取代: (C3-C7)环烷基或(C5-C7)环烯基;
-(C1-C6)卤代烷基;
-(C3-C7)环烷基;
-(C5-C7)环烯基;
-(C2-C6)烯基;和
-(C2-C6)炔基;
R2选自:
-H;
-(C3-C7)环烷基羰基;
-(C1-C6)烷基,任选由选自下述的一个或多个取代基取代: (C3-C7)环烷基或(C5-C7)环烯基;
-(C1-C6)卤代烷基;
-(C3-C7)环烷基;
-(C5-C7)环烯基;
-(C2-C6)烯基;和
-(C2-C6)炔基;
或者,在R19不是氢的情况下,R2与R19一起形成如下定义的式(x) 基团;
或R1和R2与互相连接的原子一起形成式(q)的2,2-二氟-1,3-二氧杂 环戊烷环,其稠合至携带基团-OR1和-OR2的苯基部分,其中星号指出 与所述苯基环共享的碳原子:

R19是氢,或者,如果不是氢,其与R2一起形成式(x)基团,其中用 (1)和(2)标记的键分别指出基团(x)与携带基团R19和R2的原子的连接点

从而R2和R19与互相连接的原子一起形成式(w)的环,其稠合至携 带基团-OR2和R19的苯基环,其中星号指出与所述苯基环共享的碳原子:

R3是一个或多个独立地选自H,CN,NO2,CF3和卤素原子的取 代基;
Z是基团-(CH2)n-,其中n是0或1;
A是饱和单环(C3-C7)亚杂环烷基基团;
K选自:
--(CH2)mC(O)R4,其中m可以是0或1;
--C(O)(CH2)jR4,其中j可以是1或2;
--SO2(CH2)pR4,其中p可以是0,1或2;
--(CH2)ySO2R4,其中y可以是1或2;
--(CH2)zR4,其中z可以是1或2;和
--C(O)(CH2)2SO2R4;
R4是单环或双环环系,其可以是饱和的、部分不饱和的或完全不 饱和的,比如芳基、(C3-C8)环烷基、(C3-C7)杂环烷基或杂芳基,所述环 任选由一个或多个可以相同或不同且独立地选自下述的基团R5取代:
-(C1-C6)烷基,任选由一个或多个独立选自下述的基团取代: (C3-C7)环烷基,-OH和基团-NR18C(O)(C1-C4)烷基,其中R18是氢或 (C1-C4)烷基;
-(C3-C7)杂环烷基;
-5,6-元杂芳基,其任选由一个或两个基团(C1-C4)烷基取代;
-(C1-C6)卤代烷基;
-(C3-C7)杂环烷基(C1-C4)烷基;
-基团-OR6,其中R6选自
-H:
-(C1-C6)卤代烷基;
-基团-SO2R7,其中R7是(C1-C4)烷基;
-基团-C(O)R7,其中R7是(C1-C4)烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代或被- NR8R9如下定义的基团取代;和
-(C3-C7)环烷基;
-基团-SR20,其中R20选自
-H:
-(C1-C6)卤代烷基;
-基团-C(O)R7,其中R7是(C1-C4)烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代或被基团 -NR8R9取代;和
-(C3-C7)环烷基;
-卤素原子;
-CN;
-NO2;
-NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-(C1-C4)亚烷基-NR13R14,其中R13和R14是不同或相同的并且 独立地选自:H和(C1-C6)烷基,其任选用(C3-C7)环烷基或(C3-C7)杂环烷 基取代;或它们与其所连接的氮原子一起形成饱和的或部分饱和的 (C3-C7)杂环;
-(C1-C6)烷基,任选用(C3-C7)环烷基,(C3-C7)杂环烷基,基团 -OH或(C1-C6)烷氧基取代;
-基团-SO2R15,其中R15选自:(C1-C4)烷基,任选由(C3-C7)环 烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;和苯基,任选由一个或 多个(C1-C6)烷基,卤素或基团-OH取代;
-基团-C(O)R16,其中R16选自:(C1-C6)烷基,任选由(C3-C7) 环烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;苯基,任选由一个或 多个(C1-C6)烷基,卤素或-OH取代;和基团-NH2;
-基团-C(O)OR17,其中R17选自:(C1-C6)烷基,任选由(C3-C7) 环烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;苯基,任选由一个或 多个(C1-C6)烷基,卤素或-OH取代;和基团-NH2;
或它们与其所连接的氮原子一起形成饱和的或部分饱和的杂环,其 是任选由一个或多个(C1-C6)烷基或氧代基团取代;
-如上定义的(C1-C4)亚烷基-NR8R9;
-COR10,其中R10是苯基或(C1-C6)烷基;
-氧代;
--SO2R11,其中R11是(C1-C4)烷基,OH或NR8R9,其中R8和 R9如前文所定义;
--COOR12,其中R12是H,(C1-C4)烷基或(C1-C4)亚烷基-NR8R9, 其中R8和R9如前文所定义;和
--CONR8R9,其中R8和R9如前文所定义;
其中,如果出现在多于一个基团中,基团R6,R7,R8,R9,R10, R11,R12,R13,R14,R15,R16,R17,R18,R19和R20在每次出现时可以 具有相同或不同的含义;
其在吡啶环上的N-氧化物,及其药学上可接受的盐,或溶剂化物。
在优选的实施方式中,本发明涉及通式(IG)的1-苯基-2-吡啶基烷 基醇的衍生物

其中:
R1选自:
-H;
-(C3-C7)环烷基羰基;
-(C1-C6)烷基,任选由选自下述的一个或多个取代基取代: (C3-C7)环烷基或(C5-C7)环烯基;
-(C1-C6)卤代烷基;
-(C3-C7)环烷基;
-(C5-C7)环烯基;
-(C2-C6)烯基;和
-(C2-C6)炔基;
R2选自:
-H;
-(C3-C7)环烷基羰基;
-(C1-C6)烷基,任选由选自下述的一个或多个取代基取代: (C3-C7)环烷基或(C5-C7)环烯基;
-(C1-C6)卤代烷基;
-(C3-C7)环烷基;
-(C5-C7)环烯基;
-(C2-C6)烯基;和
-(C2-C6)炔基;
或R1和R2与互相连接的原子一起形成式(q)的2,2-二氟-1,3-二氧杂 环戊烷环,其稠合至携带基团-OR1和-OR2的苯基部分,其中星号指出 与所述苯基环共享的碳原子:

R19是氢;
R3是一个或多个独立地选自H,CN,NO2,CF3和卤素原子的取 代基;
Z是基团-(CH2)n-,其中n是0或1;
A是饱和单环(C3-C7)亚杂环烷基基团;
K选自:
--(CH2)mC(O)R4,其中m可以是0或1;
--C(O)(CH2)jR4,其中j可以是1或2;
--SO2(CH2)pR4,其中p可以是0,1或2;
--(CH2)ySO2R4,其中y可以是1或2;
--(CH2)zR4,其中z可以是1或2;和
--C(O)(CH2)2SO2R4;
R4是单环或双环环系,其可以是饱和的、部分不饱和的或完全不 饱和的,比如芳基、(C3-C8)环烷基、(C3-C7)杂环烷基或杂芳基,所述环 任选由一个或多个可以相同或不同的且独立地选自下述的基团R5取代:
-(C1-C6)烷基,任选由一个或多个独立地选自下述的基团取代: (C3-C7)环烷基,-OH和基团-NR18C(O)(C1-C4)烷基,其中R18是氢或 (C1-C4)烷基;
-(C3-C7)杂环烷基;
-5,6-元杂芳基,其任选由一个或两个基团(C1-C4)烷基取代;
-(C1-C6)卤代烷基;
-(C3-C7)杂环烷基(C1-C4)烷基;
-基团-OR6,其中R6选自
-H:
-(C1-C6)卤代烷基;
-基团-SO2R7,其中R7是(C1-C4)烷基;
-基团-C(O)R7,其中R7是(C1-C4)烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代或被基团 -NR8R9取代;和
-(C3-C7)环烷基;
-基团-SR20,其中R20选自
-H:
-(C1-C6)卤代烷基;
-基团-C(O)R7,其中R7是(C1-C4)烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代或被如下 定义的基团-NR8R9取代;和
-(C3-C7)环烷基;
-卤素原子;
-CN;
-NO2;
-NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-C1-C4)亚烷基-NR13R14,其中R13和R14是不同或相同的并且 独立地选自:H和(C1-C6)烷基,其任选用(C3-C7)环烷基或(C3-C7)杂环烷 基取代;或它们与其所连接的氮原子一起形成饱和的或部分饱和的 (C3-C7)杂环;
-(C1-C6)烷基,任选用(C3-C7)环烷基,(C3-C7)杂环烷基,基团 -OH或(C1-C6)烷氧基取代;
-基团-SO2R15,其中R15选自:(C1-C4)烷基,任选由(C3-C7)环 烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;和苯基,任选由一个或 多个(C1-C6)烷基,卤素或基团-OH取代;
-基团-C(O)R16,其中R16选自:(C1-C6)烷基,任选由(C3-C7) 环烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;苯基,任选由一个或 多个(C1-C6)烷基,卤素或-OH取代;和基团-NH2;
-基团-C(O)OR17,其中R17选自:(C1-C6)烷基,任选由(C3-C7) 环烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;苯基,任选由一个或 多个(C1-C6)烷基,卤素或-OH取代;和基团-NH2;
或者它们与其所连接的氮原子一起形成饱和的或部分饱和的杂环, 其任选由一个或多个(C1-C6)烷基或氧代基团取代;
-如上定义的(C1-C4)亚烷基-NR8R9;
-COR10,其中R10是苯基或(C1-C6)烷基;
-氧代;
--SO2R11,其中R11是(C1-C4)烷基,OH或NR8R9,其中R8和 R9如前文所定义;
--COOR12,其中R12是H或(C1-C4)烷基或(C1-C4)亚烷基 -NR8R9,其中R8和R9如前文所定义;和
--CONR8R9,其中R8和R9如前文所定义;
其中,如果出现在多于一个基团中,基团R6,R7,R8,R9,R10, R11,R12,R13,R14,R15,R16,R17,R18,R19和R20在每次出现时可以 具有相同或不同的含义;
其在吡啶环上的N-氧化物,及其药学上可接受的盐,或溶剂化物。
在又一优选实施方式中,本发明涉及通式(IL)的1-苯基-2-吡啶基烷 基醇的衍生物

其中:
R1和R2是不同或相同的并且独立地选自:
-H;
-(C3-C7)环烷基羰基;
-(C1-C6)烷基,任选由选自下述的一个或多个取代基取代: (C3-C7)环烷基或(C5-C7)环烯基;
-(C1-C6)卤代烷基;
-(C3-C7)环烷基;
-(C5-C7)环烯基;
-(C2-C6)烯基;和
-(C2-C6)炔基;
R3是一个或多个独立地选自H,CN,NO2,CF3和卤素原子的取 代基;
Z是基团-(CH2)n-,其中n是0或1;
A是饱和单环(C3-C7)亚杂环烷基基团;
K选自:
--(CH2)mC(O)R4,其中m可以是0或1;
--C(O)(CH2)R4;
--SO2(CH2)pR4,其中p可以是0或1;
--CH2SO2R4;和
--CH2R4;
R4是单环或双环环系,其可以是饱和的、部分不饱和的或完全不 饱和的,比如芳基、(C3-C8)环烷基、(C3-C7)杂环烷基或杂芳基,所述环 任选由一个或多个可以相同或不同的且独立地选自下述的基团R5取代:
-(C1-C6)烷基,任选由一个或多个(C3-C7)环烷基取代;
-(C3-C7)杂环烷基;
-(C3-C7)杂环烷基(C1-C4)烷基;
-基团-OR6,其中R6选自
-H:
-(C1-C6)卤代烷基;
-基团-SO2R7,其中R7是(C1-C4)烷基;
-基团-C(O)R7,其中R7是(C1-C4)烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代或被基团 -NR8R9取代;和
-(C3-C7)环烷基;
-基团-SR20,其中R20选自
-H:
-(C1-C6)卤代烷基;
-基团-C(O)R7,其中R7是(C1-C4)烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代或被如下 定义的基团-NR8R9取代;和
-(C3-C7)环烷基;
-卤素原子;
-CN;
-NO2;
-NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-(C1-C4)亚烷基-NR13R14,其中R13和R14是不同或相同的并且 独立地选自:H和(C1-C6)烷基,其任选用(C3-C7)环烷基或(C3-C7)杂环烷 基取代;或它们与其所连接的氮原子一起形成饱和的或部分饱和的 (C3-C7)杂环;
-(C1-C6)烷基,任选用(C3-C7)环烷基,(C3-C7)杂环烷基,基团 -OH或(C1-C6)烷氧基取代;
-基团-SO2R15,其中R15选自:(C1-C4)烷基,任选由(C3-C7)环 烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;和苯基,任选由一个或 多个(C1-C6)烷基,卤素或基团-OH取代;
-基团-C(O)R16,其中R16选自:(C1-C6)烷基,任选由(C3-C7) 环烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;苯基,任选由一个或 多个(C1-C6)烷基,卤素或-OH取代;和基团-NH2;
-基团-C(O)OR17,其中R17选自:(C1-C6)烷基,任选由(C3-C7) 环烷基或(C3-C7)杂环烷基取代;(C3-C7)杂环烷基;苯基,任选由一个或 多个(C1-C6)烷基,卤素或-OH取代;和基团-NH2;
或它们与其所连接的氮原子一起形成饱和的或部分饱和的杂环,其 是任选由一个或多个(C1-C6)烷基或氧代基团取代;
-如上定义的(C1-C4)亚烷基-NR8R9;
-COR10,其中R10是苯基或(C1-C6)烷基;
-氧代;
--SO2R11,其中R11是(C1-C4)烷基,OH或NR8R9,其中R8和 R9如前文所定义;
--COOR12,其中R12是H或(C1-C4)烷基或(C1-C4)亚烷基 -NR8R9,其中R8和R9如前文所定义;和
--CONR8R9,其中R8和R9如前文所定义;
其中,如果出现在多于一个基团中,基团R6,R7,R8,R9,R10, R11,R12,R13,R14,R15,R16,R17和R20在每次出现时可以具有相同或 不同的含义;
它们在吡啶环上的N-氧化物,及其药学上可接受的盐,或溶剂化 物。
本发明还牵涉相应的式(I)化合物的吡啶环上的N-氧化物。
本发明也涵盖其药学上可接受的盐和/或溶剂化物。
术语"药学上可接受的盐",如本文所用,是指式(I)化合物或其相应 的吡啶环上的N-氧化物的衍生物,其中通过用常规期望药学上可接受的 任意碱或酸将如果存在的任意游离酸或碱性基团转化为相应的加成盐, 来适宜地修饰母体化合物。
从而,所述盐的适宜实例可以包括碱性残基比如氨基基团,以及无 机酸或有机酸残基比如羧酸基团的无机或有机酸加成盐。
在本发明中能够适宜地用来制备盐的无机碱的阳离子包含碱金属 或碱土金属比如钾、钠、钙或镁离子。
通过将充当碱的主要化合物与无机或有机酸反应以形成盐获得的 那些,包括例如盐酸,硫酸,磷酸,甲烷磺酸,樟脑磺酸,草酸,马来 酸,丁二酸和柠檬酸的盐。
有机化学领域的技术人员将理解许多有机化合物能够与它们在其 中反应的或者自其沉淀或结晶的溶剂形成复合物。这些复合物称为"溶 剂化物"。本发明化合物的药学上可接受的溶剂化物属于本发明的范围 内。
本发明范围内还包括式(I)化合物的多晶型和晶型,其吡啶环上的 N-氧化物的多晶型和晶型,或其药学上可接受的盐或溶剂化物的多晶型 和晶型。
此后,本发明任意方面所定义的式(I),(IG),(IL)化合物,吡啶环 上相应的N-氧化物,其实施方式,对映体,非对映异构体,它们的药学 上可接受的盐和溶剂化物,和多晶型或晶形(除了描述于化学过程中的中 间体化合物)称为"本发明化合物"。
本发明还包含用于制备本发明化合物的方法。
本发明也提供本发明化合物单独或与一种或多种药学上可接受的 载体混合的组合的药物组合物。
在又一方面,本发明提供本发明化合物作为药物的用途。
在一方面,本发明提供本发明化合物用于制备药物的用途。
尤其是,本发明提供本发明化合物用于预防和/或治疗表征为磷酸 二酯酶4(PDE4)过度活性和/或其中希望抑制PDE4活性的任意疾病的用 途。
尤其是,本发明化合物单独或与其它活性成分相组合可以给予用于 预防和/或治疗表征为气道阻塞比如哮喘和COPD的呼吸道疾病。
在又一方面,本发明提供本发明化合物用于制备药物的用途,所述 药物用于预防和/或治疗表征为磷酸二酯酶4(PDE4)过度活性和/或其中 希望抑制PDE4活性的任意疾病。
此外,本发明提供用于预防和/或治疗其中希望PDE4抑制的任意 疾病的方法,所述方法包括向有此需要的患者给予治疗有效量的本发明 化合物。
定义
术语"卤素原子"如本文所用包括氟,氯,溴,和碘,优选氯。
如本文所用,术语"(C1-Cx)烷基",其中x是大于1的整数,是指 直链的和支化的烷基,其中组分碳原子数为1至x。特别的烷基是甲基, 乙基,正丙基,异丙基和叔丁基。
类似地,术语"(C1-Cx)亚烷基",是指二价(C1-Cx)烷基残基,其中 (C1-Cx)烷基是如上定义的。
术语"(C1-Cx)烷氧基",其中x是大于1的整数,是指直链的和支 化的烷氧基基团,其中组分碳原子数是1至x。特别的烷基是甲氧基, 乙氧基,正丙氧基,异丙氧基和叔丁氧基。
措辞"(C1-Cx)卤代烷基"是指上述定义的"(C1-Cx)烷基"基团,其中 一个或多个氢原子用一个或多个能够相互相同或不同的卤素原子替换。
从而所述(C1-C6)卤代烷基的实例可以包括卤化的、多卤化的和完全 卤化的烷基,其中全部氢原子用卤素原子替换,例如三氟甲基或二氟甲 基基团。
术语"(C3-Cy)环烷基",其中y是大于或等于3的整数,是指饱和 的含有3至y个环碳原子的环状烃基团。实例包括环丙基,环丁基,环 戊基,环己基和环庚基。
衍生的措辞"(C3-Cy)杂环烷基"是指单环(C3-Cy)环烷基,其中至少 一个环碳原子用杂原子(例如N,NH,S或O)替换。(C3-Cy)杂环烷基的 非限制性实例是:吡咯烷基,噻唑烷基,哌嗪基,哌啶基,吗啉基,硫 吗啉基,氮杂环丁烷基。
类似地,术语"(C3-Cy)亚杂环烷基"是指二价(C3-Cy)杂环烷基残基, 其中(C3-Cy)杂环烷基是如上定义的。
措辞"(C3-Cy)环烷基羰基"是指(C3-Cy)环烷基CO-基团,其中基团 "(C3-Cy)环烷基"具有上文定义的含义。
术语"(C2-C6)烯基"是指直链或支化的,共轭或非共轭的,具有一 个或多个顺式或反式构型的双键的碳链,其中原子数为2至6。
术语"(C5-Cz)环烯基",其中z是大于或等于5的整数,是指含有5 至z环碳原子和一个或多个双键的环状烃基团。
术语"(C2-C6)炔基"是指具有一个或多个三键的直链或支化的碳 链,其中原子数是2至6。
术语"(C3-Cy)杂环烷基(C1-Cx)烷基"是指上述"(C1-Cx)烷基"基团, 其中一个或多个氢原子用一个或多个"(C3-Cy)杂环烷基"基团替换。
如本文所用,措辞"环系"是指单环或双环环系,其可以是饱和的、 部分不饱和的或不饱和的,比如芳基、(C3-C8)环烷基、(C3-C7)杂环烷基 或杂芳基,具有5至11个环原子,其中至少一个环原子是杂原子(例如 N,S或O)。
措辞"芳基"是指单环或双环系,其具有6至10个环原子,其中至 少一个环是芳族的。
措辞"杂芳基"是指具有5至11个环原子的单环或双环系,其中至 少一个环是芳族并且其中至少一个环原子是杂原子(例如N,NH,S或 O)。
适宜的芳基或5,6-元杂芳基单环系统的实例包括例如苯基,噻吩, 苯,吡咯,吡唑,咪唑,异噁唑,噁唑,异噻唑,噻唑,吡啶,咪唑烷, 呋喃残基等。
适宜的芳基或杂芳基双环系统的实例包括萘,联苯撑,嘌呤,蝶啶, 苯并三唑,喹啉,异喹啉,吲哚,异吲哚,苯并噻吩,二氢苯并二氧杂 环己二烯,二氢苯并二氧杂环庚二烯,苯并噁嗪残基等。
发明详述
本发明涉及一类充当磷酸二酯酶4(PDE4)的抑制剂的化合物。
所述类别的化合物抑制环状核苷酸,尤其是一磷酸环状腺苷(cAMP) 转化为其无活性的5’-单核苷酸形式。
在气道中,对升高的细胞内水平的环状核苷酸尤其是cAMP的生 理学应答,导致免疫和促炎性细胞比如肥大细胞,巨噬细胞,T淋巴细 胞,嗜酸性粒细胞和中性白细胞的活性的抑制,引起炎性介导物包括细 胞因子比如IL-1、IL-3和肿瘤坏死因素-α(TNF-α)的释放的减少。
其还导致气道平滑肌松弛和水肿减少。
本发明涉及通式(I)的1-苯基-2-吡啶基烷基醇的衍生物,吡啶环上 的N-氧化物及其药学上可接受的盐或溶剂化物,

其中R1,R2,R3,R19,Z,A和K如上定义。
对本领域技术人员来说明显的是,通式(I)化合物至少含有1个立构 中心,亦即下述的星号碳原子(1),因此作为旋光立体异构体存在。

在根据本发明的化合物具有至少一个立构中心的情况下,它们可以 相应地作为对映体存在。在根据本发明的化合物具有两个或更多个立构 中心的情况下,它们可以额外地作为非对映异构体存在。应理解全部所 述异构体及其任意比例的混合物均涵盖在本发明范围内。
在优选实施方式中,本发明涉及式(I)’化合物,其是如上定义的式 (I)化合物,其中碳(1)的绝对构型如下所示:

碳(1)的绝对构型的赋予是基于Cahn-Ingold-Prelog命名,基于优 选顺序基团。
在一种优选实施方式中,对于式(I)化合物,碳(1)的绝对构型是(S)。
在式(I)化合物具有第二立构中心,亦即下述星号碳(2)的情况下, 它们作为至少四种非对映异构体存在:

所述四种非对映异构体如下所代表:

并且属于本发明范围内。
在优选实施方式中,本发明涉及式(I)”化合物,其是如上定义的式 (I)’化合物,其中碳(2)的绝对构型如下所示:

在又一优选实施方式中,本发明涉及式(I)”’化合物,其是如上定义 的式(I)’化合物,其中碳(2)的绝对构型如下所示:

对于式(I)”和(I)”’化合物,碳(1)和(2)的绝对构型的赋予是基于 Cahn-Ingold-Prelog命名,基于基团优选顺序。
应理解上下文对式(I)化合物所描述的全部优选的基团或实施方式 还都可以相互组合并适用于式(IG)化合物,(IL),(I)’,(I)”,(I)”’,(I)”” 和(I)””’,视情况修正。
在优选实施方式中,本发明提供式(IH)化合物,其是式(I)化合物的 吡啶环的N-氧化物衍生物,或其药学上可接受的盐:

在优选实施方式中,2-吡啶基环具有两个是卤素原子的R3取代基。 在又一优选实施方式中,所述R3取代基是吡啶环3位和5位的2个氯原 子。
在一种优选实施方式中,R1是(C1-C6)卤代烷基或(C1-C6)烷基。
在一种优选实施方式中,R2是(C1-C6)烷基,其任选由(C3-C7)环烷 基取代或是(C3-C7)环烷基。
在又一优选实施方式中,R1和R2与互相连接的原子一起形成式(q) 的2,2-二氟-1,3-二氧杂环戊烷环,其稠合至携带基团-OR1和-OR2的苯基 部分,其中星号指出与所述苯基环共享的碳原子:

在又一优选的实施方式中,R1是(C1-C6)卤代烷基而R2是(C1-C6) 烷基,其被(C3-C7)环烷基取代。
在又一优选实施方式中,R1是(C1-C6)烷基和R2是(C1-C6)烷基。
在优选实施方式中,R19是氢。
在又一优选实施方式中,R19是氢,R1是(C1-C6)卤代烷基而R2是 (C1-C6)烷基,其被(C3-C7)环烷基取代。
在又一优选实施方式中,R19,如果不是氢,与R2一起形成式(x) 基团,其中(1)和(2)标记的键分别指出基团(x)与携带基团R19和R2原子 的连接点

从而R2和R19与互相连接的原子一起形成式(w)的环,其稠合至携 带基团-R2和R19的苯基环,其中星号指出与所述苯基环共享的碳原子:

通式(I)化合物的优选组是,其中2-吡啶基环在3位和5位用2个氯 原子取代,具有通式(IA)

其中R1,R2,R19,K,z和A如前文对式(I)化合物所定义;及其相 应吡啶环上的N-氧化物,或药学上可接受的盐。
式(I)化合物的又一优选组是如下所示的通式(IB):

其中R3,K,Z和A如前文对式(I)化合物所定义;及其相应吡啶 环上的N-氧化物,或药学上可接受的盐。
式(I)化合物的又一优选组是如下所示的通式(IC):

其中K,Z和A如前文对式(I)化合物所定义;及其相应吡啶环上 的N-氧化物,或药学上可接受的盐。
在一种优选实施方式中,A是(C3-C7)亚杂环烷基基团,其包含代表 与基团K连接点的氮原子,如下所示:

在又一优选实施方式中,A选自下述的二价残基:

其中符号[3]和[4]指出基团A分别与基团Z和K的连接点。
在又一优选实施方式中,A选自下述的二价残基:

其中符号[3]和[4]指出基团A分别与基团Z和K的连接点。
在额外的优选实施方式中,A是基团

其中符号[3]和[4]指出基团A分别与基团Z和K的连接点。
在优选实施方式中,Z是0。
式(I)化合物的又一优选组是如下所示的通式(ID):

其中R1,R2,R3和K如前文对式(I)化合物所定义,R19是氢,Z 是键而A是上述的噻唑烷二价残基基团;及其相应吡啶环上的N-氧化 物,或药学上可接受的盐。
式(I)化合物的又一优选组是如下所示的通式(ID”’):

其中R1,R2,R3和K如前文对式(I)化合物所定义,R19是氢,Z 是键,A是噻唑烷二价残基基团并且立构中心具有上文所述的绝对构型; 及其相应吡啶环上的N-氧化物,或药学上可接受的盐。
在一种实施方式中,对于式(ID)或(ID”’)化合物,R1是(C1-C6)卤代 烷基,R2是(C1-C6)烷基,其被(C3-C7)环烷基取代,2-吡啶基环,在3和 5位被2个氯R3基团取代,并且K是基团

式(I)化合物的又一优选组是如下所示的通式(IE):

其中R1,R2,R3和K如前文对式(I)化合物所定义,Z是键,R19 是氢和A是上文所述的吡咯烷二价残基基团;及其相应吡啶环上的N- 氧化物,或药学上可接受的盐。
式(I)化合物的又一优选组是如下所示的通式(IE”’):

其中R1,R2,R3和K如前文对式(I)化合物所定义,Z是键,R19 是氢,A是吡咯烷二价残基基团和立构中心具有上文所述的绝对构型; 及其相应吡啶环上的N-氧化物,或药学上可接受的盐。
在一种实施方式中,对于式(IE)或(IE”’)化合物,R1是(C1-C6)卤代 烷基,R2是(C1-C6)烷基,其被(C3-C7)环烷基取代,2-吡啶基环,其在3 和5位被2个氯R3基团取代,而K是基团

在一种优选实施方式中,K选自下述基团:

其中符号[5]指出基团K与基团A的连接点。
在又一优选实施方式中,K选自下述基团:

其中符号[5]指出基团K与基团A的连接点。
在又一优选实施方式中,K选自下述基团:

其中符号[5]指出基团K与基团A的连接点。
在又一优选实施方式中,K是基团

其中符号[5]指出基团K与基团A的连接点。
在优选实施方式中,R4选自:基团苯基,5,6-元杂芳基基团,单环 (C3-C7)杂环烷基和双环环系;并且其各自任选由一个或多个基团R5取 代。
在一种优选实施方式中,R4是基团苯基或5,6-元杂芳基基团,其各 自任选由一个或多个基团R5取代。
在又一优选实施方式中,R4是基团苯基,其任选由一个或多个基 团R5取代。
在又一优选实施方式中,R4是5,6-元杂芳基基团,其任选由一个或 多个基团R5取代。
在又一优选实施方式中,R4是单环(C3-C7)杂环烷基,任选由一个 或多个基团R5取代。
在又一优选实施方式中,R4是双环环系,任选由一个或多个基团 R5取代。
在一种优选实施方式中,取代基R5的数量是0、1或2。在又一优 选实施方式中,该数量是1。
在一种优选实施方式中,R5独立地选自:
-(C1-C6)烷基,任选由一个或多个独立地选自下述的基团取代: (C3-C7)环烷基,-OH和基团-NR18C(O)(C1-C4)烷基,其中R18是氢或 (C1-C4)烷基-(C3-C7)杂环烷基;
-5,6-元杂芳基,其任选由一个或两个基团(C1-C4)烷基取代;
-(C1-C6)卤代烷基;
-(C3-C7)杂环烷基(C1-C4)烷基;
-基团-OR6,其中R6选自:
-(C1-C6)卤代烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代;
-基团-SO2R7,其中R7是(C1-C4)烷基;
-卤素原子;
-氰基;
-NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-(C1-C6)烷基,任选用(C3-C7)环烷基,(C3-C7)杂环烷基取代;
-基团-SO2R15,其中R15是(C1-C4)烷基;
或它们与其所连接的氮原子一起形成饱和的或部分饱和的杂环,其 是任选由一个或多个(C1-C6)烷基或氧代基团取代;
-(C1-C4)亚烷基-NR8R9;
-COR10,其中R10是苯基或(C1-C6)烷基;
-氧代;
--SO2R11,其中R11是NR8R9,其中R8和R9如前文所定义;
--COOR12,其中R12是H或(C1-C4)烷基或(C1-C4)亚烷基 -NR8R9,其中R8和R9如前文所定义;和
--CONR8R9,其中R8和R9如前文所定义。
在又一优选实施方式中,R5独立地选自:
-(C1-C6)烷基;
-(C3-C7)杂环烷基;
-(C3-C7)杂环烷基(C1-C4)烷基;
-基团-OR6,其中R6选自:
-(C1-C6)卤代烷基;
-(C1-C10)烷基,任选由一个或多个(C3-C7)环烷基取代;
-基团-SO2R7,其中R7是(C1-C4)烷基;
-卤素原子;
-NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-(C1-C6)烷基,任选用(C3-C7)环烷基,(C3-C7)杂环烷基取代;
-基团-SO2R15,其中R15是(C1-C4)烷基;
或它们与其所连接的氮原子一起形成饱和的或部分饱和的杂环,其 任选由一个或多个(C1-C6)烷基或氧代基团取代;
-(C1-C4)亚烷基-NR8R9;
-COR10,其中R10是苯基或(C1-C6)烷基;
-氧代;
--SO2R11,其中R11是NR8R9,其中R8和R9如前文所定义;
--COOR12,其中R12是H或(C1-C4)烷基或(C1-C4)亚烷基 -NR8R9,其中R8和R9如前文所定义;和
--CONR8R9,其中R8和R9如前文所定义。
在又一优选实施方式中,R5独立地选自:
-(C1-C6)烷基;
-(C3-C7)杂环烷基(C1-C4)烷基;
-基团-OR6,其中R6是(C1-C10)烷基,任选由一个或多个(C3-C7) 环烷基取代;
-卤素原子;
-NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-(C1-C6)烷基,任选用(C3-C7)环烷基,(C3-C7)杂环烷基取代;
-基团-SO2R15,其中R15是(C1-C4)烷基;
或它们与其所连接的氮原子一起形成饱和的或部分饱和的杂环,其 是任选由(C1-C6)烷基或氧代取代;
--COOR12,其中R12是H或(C1-C4)烷基或(C1-C4)亚烷基 -NR8R9,其中R8和R9如前文所定义;和
--CONR8R9,其中R8和R9如前文所定义。
在又一优选实施方式中,R5选自:
--(C1-C6)烷基;
--NR8R9,其中R8和R9是不同或相同的并且独立地选自:
-H;
-(C1-C6)烷基;和
-CONR8R9,其中R8和R9如前文所定义。
式(I)化合物的又一优选组是如下所示的通式(IF):

其中Z是键,R19是氢,A是(C3-C7)亚杂环烷基基团,其包含代表 与基团K的连接点的氮原子,K选自下述基团:

R4是基团苯基或5,6-元杂芳基基团,其各自任选由一个或多个基团 R5取代:及其相应吡啶环上的N-氧化物,或药学上可接受的盐。
根据优选实施方式,本发明提供报告如下的化合物:












或其药学上可接受的盐或溶剂化物。
在又一优选实施方式中,本发明化合物选自:
4-((S)-2-((S)-3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧基)-2-(3-(环丙基 甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-1-(3-(二甲基氨基甲酰基)苯基磺酰基)吡咯烷-2-羰氧基)乙基) 吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((R)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻唑烷-4-羰氧基)乙基) 吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-1-(3-(二甲基氨基甲酰基)苯基磺酰基)-哌啶-2-羰氧基)乙基)吡 啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-1-(3-(二甲基氨基甲酰基)-4-甲氧基苯基磺酰基)-哌啶-2-羰氧 基)乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-(4-甲基哌嗪-1-羰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(吡啶-3-基磺酰基)噻唑烷-2-羰氧基)乙基)-吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)-4-甲氧基苯基磺酰基)-噻唑烷-2-羰 氧基)乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(1-甲基-1H-咪唑-2-基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1- 氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-氨磺酰基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化 物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(4-(甲磺酰基)苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3,4-二甲氧基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-(N,N-二甲基氨磺酰基)苯基磺酰基)噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(4-甲氧基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化 物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(5-(吡咯烷-1-羰基)-1H-吡咯-3-基磺酰基)噻唑烷-2-羰氧基) 乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(1-甲基-5-(甲基氨基甲酰基)-1H-吡咯-3-基磺酰基)噻唑烷-2- 羰氧基)乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(1,3-二甲基-1H-吡唑并[3,4-b]吡啶-5-基磺酰基)噻唑烷-2-羰 氧基)乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-甲基异噁唑并[5,4-b]吡啶-5-基磺酰基)噻唑烷-2-羰氧基) 乙基)吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-(羟基甲基)苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(3-脲基苯基磺酰基)噻唑烷-2-羰氧基)乙基)-吡啶1-氧化物;
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-3-(1-甲基-2-氧代吲哚啉-5-基磺酰基)噻唑烷-2-羰氧基)乙基)吡 啶1-氧化物;
3,5-二氯-4-((S)-2-((S)-3-(4-氰基苯基磺酰基)噻唑烷-2-羰氧 基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙基)-吡啶1-氧化物;
及其药学上可接受的盐或溶剂化物。
在本发明的一方面中,提供根据下述方案1中报告的一般合成路线 用于制备本发明化合物的方法,请参照在下述段落中详述的特定合成方 案。
能够使用且描述如下且报告于各方案中的方法不应被视为限制能 用于制备本发明化合物的合成方法的范围。
方案1

在下述方案中,对于式(II)至(XIX)化合物,除非另有指定,基团 R1至R20,Z,A,和K具有上文对式(I)化合物所描述的相同含义。
式(Ia)化合物,也即式(I)化合物,其中K是基团-(CH2)R4,可以根 据下文报告的方案2a制备:将式(II)化合物,其中A’是包含基团-NH- 的(C3-C7)亚杂环烷基基团,与适当的式(III)化合物反应。
方案2(S2a)

典型反应条件包括在适宜的偶极溶剂比如THF、甲醇、乙醇或 DCM中,在适当的还原剂比如三乙酰氧基硼氢化钠、氰基硼氢化钠或 硼氢化钠,并且在适当的酸比如乙酸,甲醇或乙酸铵中的HCl存在下, 将式(II)化合物与式(III)化合物反应。有用的是在加入还原剂之前施行亚 胺化。反应在室温稳定进行1至12小时。
另选地式(Ia)化合物,也即式(I)化合物,其中K是基团-(CH2)R4, 可以根据报告如下的方案2b制备:将式(II)化合物,其中A’是包含基团 -NH-的(C3-C7)亚杂环烷基基团,与适当的式(XVI)化合物反应。
方案2b(S2b)

典型反应条件包括在适宜的极性非质子溶剂比如乙腈或DMF中, 在适当的碱比如K2CO3、碱性碳酸氢盐,TEA或DIPEA存在下,在室 温至70℃的温度下,将式(XVI)化合物,其中X是离去基团比如Cl或 Br,与式(II)化合物反应。
式(Ib)化合物,也即式(I)化合物,其中K是基团-C(O)(CH2)jR4, 可以根据报告如下的方案3a制备:将如上定义的式(II)化合物,与适当 的式(IV)化合物反应。
方案3(S3a)

典型反应条件包括在适宜的偶极非质子溶剂比如DMF、氯仿或 DCM中,在适当的缩合剂比如EDC、DCC、HOBT、HOAT或CDI, 和如果必需在适当的试剂比如DMAP、HOBT、4-吡咯烷子基吡啶(4-PPY) 或其它4-烷基氨基吡啶存在下,在室温下,将式(II)化合物与式(IV)化合 物反应。
另选地式(Ib)化合物,也即式(I)化合物,其中K是基团 -C(O)(CH2)jR4,可以根据报告如下的方案3b制备:将如上定义的式(II) 化合物,与适当的式(XVII)化合物反应
方案3b(S3b)
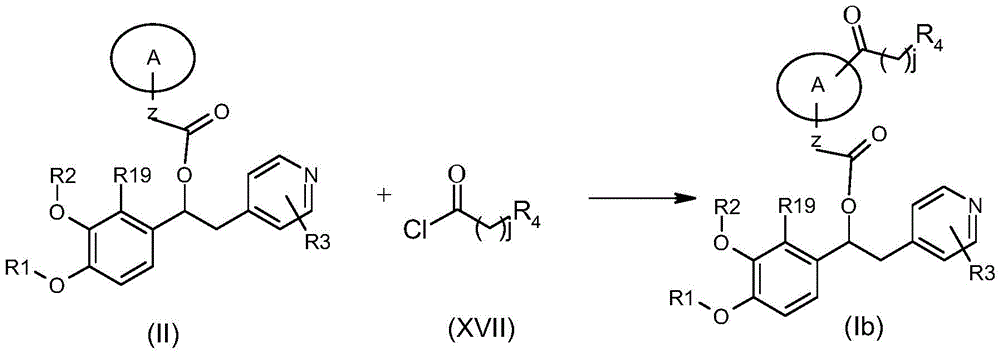
典型反应条件包括在适宜的溶剂比如吡啶或DCM中,和在如果必 需的适当的碱比如TEA、DIPEA、DBU或其它有机碱存在下,在0℃至 室温的温度下,将式(II)化合物与式(XVII)化合物反应。
式(Ic)化合物,也即式(I)化合物,其中K是基团-(CH2)mC(O)R4, 可以根据报告如下的方案4制备:将如上定义的式(II)化合物,与适当的 式(V)化合物反应,其中Hal代表适宜的卤素离去基团。
方案4(S4)

典型反应条件包括,在适宜的极性非质子溶剂比如DMF或乙腈中, 在适当的碱比如K2CO3、碱性碳酸氢盐、TEA或DIPEA存在下,在室 温至50℃的温度,将式(II)化合物与式(V)化合物反应。
式(Id)化合物,也即式(I)化合物,其中K是基团-(CH2)ySO2R4,可 以根据报告如下的方案5制备:将如上定义的式(II)化合物,与适当的式 (VI)化合物反应,其中Hal代表适宜的卤素离去基团。
方案5(S5)

典型反应条件包括在适宜的极性非质子溶剂比如DMF或乙腈中, 在适当的碱比如K2CO3、碱性碳酸氢盐、TEA或DIPEA存在下,在室 温至50℃的温度,将式(II)化合物与式(VI)化合物反应。
式(Id)化合物,也即式(I)化合物,其中K是基团-(CH2)ySO2R4和y 是1,还可以根据报告如下的方案46制备:将如上定义的式(II)化合物, 与适当的式(XVIII)化合物反应,其中Hal代表适宜的卤素离去基团
方案46(S46)

典型反应条件包括,在适宜的极性非质子溶剂比如DMF或乙腈, 在适当的碱比如K2CO3、碱性碳酸氢盐、TEA或DIPEA存在下,在室 温至50℃的温度,将式(II)化合物与式(XVIII)化合物反应。在适宜的极 性溶剂比如DCM、氯仿、EtOH或MeOH中,在室温至60℃的温度, 将这样获得的化合物(XIX)继续与适宜的氧化剂比如MCPBA或过氧化 氢反应。
式(Ie)化合物,也即式(I)化合物,其中K是基团-SO2(CH2)pR4,可 以根据报告如下的方案6制备:将如上定义的式(II)化合物,与适当的式 (VII)化合物反应。
方案6(S6)

典型反应条件包括在适宜的溶剂比如吡啶或DCM中,和如果必需 的话,在适当的碱比如TEA、DIPEA、DBU或其它有机碱存在下,在0℃ 至室温的温度,将式(II)化合物与式(VII)化合物反应。
另选地,式(I)化合物,可以根据报告如下的方案7制备:将式(VIII) 化合物,与适当的式(IX)化合物反应。
方案7(S7)

典型反应条件包括在适宜的极性非质子溶剂比如DMF、THF、氯 仿或DCM中,在适当的缩合剂比如EDC、DCC或CDI和在适当的试 剂比如DMAP、HOBT、4-吡咯烷子基吡啶(4-PPY)或其它4-烷基氨基吡 啶存在下,在室温下,将式(VIII)化合物与式(IX)化合物反应;在本领域 技术人员已知或描述于T.W.Green and P.Wutz的"protection groups in organicsynthesis"(Wiley-Interscience publication,1999)的条件下进行 可能存在的保护基团的除去。
如上定义的式(II)化合物,可以根据报告如下的方案8制备:将式 (X)化合物,其中A”是包含用适宜保护基保护的基团-N-的(C3-C7)亚杂环 烷基基团,与适当的式(XI)化合物反应,随后在适当条件下除去N-保护 基团。
方案8(S8)

典型的偶联反应条件包括,在适宜的极性非质子溶剂比如DMF、 THF、氯仿或DCM中,在适当的(偶联)缩合剂比如EDC、DCC或CDI 和在适当的试剂比如DMAP、HOBT、4-吡咯烷子基吡啶(4-PPY)或其它 4-烷基氨基吡啶存在下,在室温下,将式(VIII)化合物与式(X)化合物反 应;在本领域技术人员已知或描述于T.W.Green and P.Wutz的 "protectiongroups in organic synthesis"(Wiley-Interscience publication, 1999)的条件下进行保护基团的除去,例如在保护基团是叔丁氧基羰基的 情况下,则脱保护可以在酸性条件下方便地进行(比如二噁烷中的HCl 或AcOEt中的HCl或CH2Cl2中的TFA)。
另选地能够预先形成相应酰氯:在适宜的非质子溶剂比如DCM 中,于0度,如果必需的话,在催化量DMF的存在下,将化合物(X)与 草酰氯或亚硫酰氯或本领域技术人员熟知的其它试剂反应;且依次加入 化合物(VIII)和适当的碱比如TEA或DIPEA。
式(If)化合物,也即式(I)化合物,其中K是基团-SO2(CH2)pR4和 R2是氢,可以根据报告如下的方案42制备:在适当条件下反应如上定 义的式(Ie)化合物,其中R2是(C1-C6)烷基,任选由一个(C3-C7)环烷基取 代。
方案42(S42)

典型的反应条件包括在室温至40度的温度,将如上定义的式(Ie) 化合物与适宜的酸比如TFA或BBr3或BCl3反应。
通式(I)化合物2-吡啶基环上的N-氧化物及其实施方式可以根据于 文献中可获得的和技术人员熟知的方法来制备。例如,它们可以这样制 备:将通式(I)化合物或其实施方式溶于CH2Cl2或CHCl3,然后将氧化 剂比如间氯过氧苯甲酸(mCPBA)加至所得溶液。可以使用的其它氧化剂 是过氧化氢,过氧苯甲酸和过氧乙酸。
另选地,尤其是对于A或A’是用对氧化敏感的官能团取代的环的 那些化合物,相应的N-氧化物这样制备:在引入其它官能团之前进行氧 化步骤,例如在式(II)或(VIII)化合物上。
在优选实施方式中,用于制备式(I)化合物或其实施方式的方法起始 自式(VIII)化合物的吡啶环上的N-氧化物而进行,从而允许制备吡啶环 上的N-氧化物形式的式(I)化合物或其实施方式。
通式(III),(IV),(V),(VI),(VII),(VIII),(IX),(XVI),(XVII), (XVIII),(XIX)和(X)化合物可以是可商购的,其制备可以特别地描述于 文献中或它们可以根据于文献中可获得的和本领域技术人员已知的方法 制备。
尤其是,式(VIII)化合物和相应吡啶环上的N-氧化物还可以按照国 际专利申请WO2009/018909或WO2010/089107的描述制备。
在一种实施方式中,根据报告如下的方案43提供式(IDa)化合物, 也即式(ID)化合物的吡啶环上的N-氧化物衍生物,其中R9是氢,K是基 团-SO2(CH2)pR4且其中立构中心的绝对构型如下所示的制备的优选过 程:
方案43(S43)

描述于方案43的方法的典型反应条件包括:a)将式(VII)化合物的 吡啶(3-30体积优选8体积)溶液加至冷冻的式(XI)化合物的吡啶(3-30体 积优选8体积)溶液,在室温下搅拌所得溶液;c)将溶液倾入过量的含水 HCl;d)过滤沉淀的物质,将其用水洗涤或d’)用AcOEt萃取水相,用 含水HCl 1M、盐水洗涤,蒸发所得有机相;和任选地e)将得自步骤d) 或d’)的固体溶于AcOEt,倾至硅胶垫上,用AcOEt/MeOH[100:0至 (90:10)]洗脱,真空蒸发所得溶液;或e’)通过快速色谱法纯化产品,用 DCM/异-PrOH洗脱。
在更优选实施方式中,如上文报告根据方案43所获得的式(IDa)化 合物通过下述过程结晶,包括:f)将化合物溶于EtOH(8体积);g)在室 温下激烈地搅拌过夜;h)过滤形成的固体;和任选地,i)用EtOH(2体 积)洗涤得自步骤h)的固体和l)真空干燥固体。
在又一优选实施方式中,方案43的步骤l)这样进行:首先在室温 下减压干燥固体,随后在60℃减压干燥固体。
在一种实施方式中,对于制备如上定义的式(XI)化合物,根据报告 如下的方案44提供优选的过程:
方案44(S44)

对于描述于方案44的过程的典型的反应条件包括:a)在室温下, 在搅拌下将浓HCl(约5M;大过量)的无水AcOEt(9体积)溶液加入式 (XII)化合物的AcOEt(6体积)溶液;b)搅拌;c)过滤沉淀的固体;任选 地d)用AcOEt洗涤获得的固体;和任选地e)在室温下减压干燥获得的 固体。
在一种实施方式中,对于制备如上定义的式(XII)化合物,根据报 告如下的方案45提供优选过程:
方案45(S45)

对于描述于方案45的过程的典型反应条件包括:a)将式(XIV)化合 物、DMAP和EDC加入式(XV)化合物的DMF溶液;b)搅拌混合物,优 选过夜;c)将混合物倾入冷水;d)过滤沉淀;任选地e)将沉淀溶于DCM 中,用水洗涤溶液,干燥和蒸发溶剂;和任选地f)在搅拌下,将得自步 骤d)或e)的固体溶于沸腾的MTBE(3.5体积),加入石油醚(4体积),在 室温下搅拌,过滤获得的固体和将其在室温下减压干燥。
在优选实施方式中,按顺序进行根据方案43、44和45的过程,获 得结晶的式(IDa)化合物。
在优选实施方式中,提供制备式(IDaa)化合物,也即式(IDa)化合物 的过程,其中R1是(C1-C6)卤代烷基,R2是(C1-C6)烷基,其被下述取代: (C3-C7)环烷基,在3和5位由2个氯R3基团取代的2-吡啶基环,K是基 团

而R4是苯基基团,其任选由一个或多个基团R5取代;该过程包括 按顺序进行上述方案43、44和45中提供的反应。
在本发明的一方面中,提供式(II)化合物,它们的吡啶环上的N-氧 化物,或其盐作为制备式(I)化合物的过程的中间体

其中A’是包含基团-NH-的(C3-C7)亚杂环烷基基团。
在一种实施方式中,提供如上定义的式(XI)化合物,作为制备式 (IDa)化合物的过程的中间体。在又一优选实施方式中,对于式(XI)化合 物,R1是(C1-C6)卤代烷基而R2是(C1-C6)烷基,其被下述取代:(C3-C7) 环烷基和3和5位被2个基团R3取代的吡啶环。
在本发明的又一方面中,提供如上定义的式(II)化合物,其充当磷 酸二酯酶4(PDE4)的抑制剂,从而满足鉴定其它PDE4抑制剂的上述需 要,其具有PDE4酶高亲和力,并且作为吸入治疗可能显示适当可发展 性特征,例如在降低的副作用方面的特征。
本发明也提供含有式(II)化合物的组合物及其治疗用途。
在可行时,上述对于式(I)化合物的优选实施方式和组也适用式(II) 化合物,可作必要的修正。
所描述的过程是特别有利的,原因是其易于通过技术人员已知的任 意合适变型来合适地调节,从而获得任意所希望的本发明化合物。上述 变型属于本发明范围以内。
从全部上文,本领域技术人员应清楚任意所描述的基团可以原样存 在或以任何适当保护的形式存在。
尤其是,式(II),(III),(IV),(V),(VI),(VII),(VIII),(IX),(X), (XI),(XII),(XIII),(XIV),(XV),(XVI),(XVII),(XVIII)和(XIX) 化合物中存在的并且能产生不希望副反应和副产物的官能团,需要在烷 基化、酰化、偶联、氧化或磺酰化发生之前得到适当保护。类似地,那 些相同受保护基团的随后的脱保护可以在完成所述反应之后进行。
在本发明中,除非另有指定,术语"保护基团"指定保护性的基团, 其可以保持所结合至的基团的功能。一般地,保护性的基团用来保持氨 基,羟基,或羧基官能。从而,适当的保护基团可以包括例如苄基、苄 氧基羰基、叔丁氧基羰基、烷基或苄基酯等,其是本领域技术人员熟知 的[参见一般参考文献T.W.Green;Protective Groups in OrganicSynthesis(Wiley,N.Y.1999)]。
类似地,所述基团例如包括羰基、羟基或氨基基团中任意的选择性 保护和脱保护,可以根据有机合成化学普遍使用的很熟知的方法来实现。
式(I)化合物或其吡啶环上的N-氧化物的任选成盐可以这样进行: 将游离酸性或氨基基团中任意适当转化为相应药学上可接受的盐。在该 情况中,对于本发明化合物任选成盐所用的操作条件全部属于技术人员 的普通知识。
从全部上文,技术人员应清楚的是,上述过程,涵盖用于制备适宜 的本发明化合物的其任意变型,可以方便地进行修饰,从而使得反应条 件适应特定需要,例如选择适当的缩合剂、溶剂和保护性的基团,视情 况而定。
本发明也提供本发明化合物或式(II)化合物的药物组合物,其与一 种或多种药学上可接受的载体混合,例如描述于Remington’s Pharmaceutical Sciences Handbook,XVII Ed.,Mack Pub.,N.Y.,U.S.A 中的那些。
本发明化合物或式(II)化合物的给予可以根据患者需要来实现,例 如经口,鼻部,经肠胃外(经皮下,经静脉内,经肌肉内,胸骨内和通过 输注),吸入,直肠,阴道,局部,区域,经皮,和通过眼部给药。各种 固体口服剂型可以用于给予本发明化合物,包括固体形式比如片剂,软 胶囊,胶囊,囊片,颗粒剂,糖锭和整装粉末。本发明化合物或式(II) 化合物可以单独给予或与本领域已知的各种药学上可接受的载体,稀释 剂(比如蔗糖,甘露醇,乳糖,淀粉)和赋形剂相组合给予,包括但不限 于助悬剂,增溶剂,缓冲试剂,粘合剂,崩解剂,防腐剂,着色剂,香 料,润滑剂等。时间释放胶囊,片剂和凝胶也有利地用于给予本发明化 合物或式(II)化合物。
各种液体口服剂型还可以用于给予本发明化合物或式(II)化合物, 包括水溶液和非水溶液,乳液,悬浮液,糖浆剂和酏剂。所述剂型还能 够含有本领域已知的适宜的惰性稀释剂比如水和本领域已知的适宜的赋 形剂比如防腐剂,润湿剂,甜味剂,香料,以及用于乳化和/或悬浮本发 明化合物或式(II)化合物的试剂。本发明化合物或式(II)化合物可以注射, 例如经静脉内,以等渗无菌溶液形式。其它制剂也是可能的。
直肠给药本发明化合物或式(II)化合物的栓剂可以这样制备:将化 合物与适宜的赋形剂比如可可油、水杨酸类和聚乙二醇相混合。
用于阴道给药的配制剂可以呈霜剂,凝胶,糊剂,沫状物,或喷雾 配制剂形式,其除了活性成分之外,还含有所述本领域已知的适宜载体。
对于局部给药,药物组合物可以呈适于向皮肤、眼、耳或鼻给药的 霜剂,软膏剂,搽剂,洗剂,乳液,悬浮液,凝胶,溶液,糊剂,粉末, 喷雾剂,和滴剂形式。局部给药还可以牵涉经由装置比如透皮贴剂的经 皮给药。
对于治疗呼吸道疾病,根据本发明的化合物或式(II)化合物优选通 过吸入给予。
可吸入制剂包括可吸入粉末,含推进剂的计量气雾剂或不含推进剂 的可吸入配制剂。
对于作为干粉给药,可以使用现有技术已知的单剂量或多剂量吸入 剂。在此情况下,粉末可充入明胶、塑料或其它胶囊、药筒或泡罩包装 或储库中。
可以将一般非毒性和对本发明化合物化学惰性的稀释剂或载体,例 如乳糖或适于改善可呼吸级分的任意其它添加剂,加入粉化的本发明化 合物或式(II)化合物中。
含有推进剂气体比如氢氟烷烃的吸入气雾剂可以以溶液或分散形 式含有本发明化合物或式(II)化合物。推进剂-驱动的配制剂还可以含有 其它成分比如共溶剂,稳定剂和任选地其它赋形剂。
包含本发明化合物或式(II)化合物的不含推进剂的可吸入配制剂可 以呈含水、含醇的或含水乙醇媒介中的溶液或悬浮液,并且它们可以通 过现有技术已知的喷射或超声雾化器递送或通过软雾雾化器比如  来递送。
来递送。
本发明化合物或式(II)化合物可以作为唯一活性剂给予或与其它药 物活性成分组合给予,所述其它药物活性成分包括目前用于治疗呼吸系 统障碍中的那些,例如β2-激动剂,抗毒蕈碱药,皮质类固醇,有丝分 裂原-活化的蛋白质激酶类(P38MAP激酶)抑制剂,核因子κ-B激酶亚单 位β(IKK2)抑制剂,人类中性白细胞弹性酶(HNE)抑制剂,磷酸二酯酶4(PDE4)抑制剂,白细胞三烯调节剂,非甾族抗炎剂(NSAIDs)和粘液调节 剂。
本发明也提供本发明化合物或式(II)化合物与β2-激动剂的组合, β2-激动剂选自卡莫特罗,GSK-642444,茚达特罗,米维特罗(milveterol), 阿福特罗,福莫特罗,沙丁胺醇,左旋沙丁胺醇,特布他林,AZD-3199, BI-1744-CL,LAS-100977,班布特罗,异丙肾上腺素,丙卡特罗,克仑 特罗,瑞普特罗,非诺特罗和ASF-1020及其盐。
本发明也提供本发明化合物或式(II)化合物与皮质类固醇的组合, 皮质类固醇选自丙醋氟替卡松,糠酸氟替卡松,糠酸莫米松,丙酸倍氯 米松,环索奈德,布地奈德,GSK685698,GSK 870086。
本发明也提供本发明化合物或式(II)化合物与抗毒蕈碱药的组合, 抗毒蕈碱药选自阿地铵(aclidinium),噻托溴铵,异丙托铵,曲司铵,格 隆铵和氧托品盐。
本发明也提供本发明化合物或式(II)化合物与PDE4抑制剂的组 合,抑制剂选自AN-2728,AN-2898,CBS-3595,apremilast,ELB-353, KF-66490,K-34,LAS-37779,IBFB-211913,AWD-12-281,西潘茶碱, 西洛司特,罗氟司特,BAY19-8004和SCH-351591,AN-6415,indus-82010,TPI-PD3,ELB-353,CC-11050,GSK-256066,奥米司 特,OX-914,替托司特,MEM-1414和RPL-554。
本发明也提供本发明化合物或式(II)化合物与P38MAP激酶抑制 剂的组合,P38MAP激酶抑制剂选自塞马莫德,talmapimod,吡非尼酮, PH-797804,GSK-725,minokine和losmapimod及其盐。
在优选实施方式中,本发明提供本发明化合物或式(II)化合物与 IKK2抑制剂的组合。
本发明也提供本发明化合物或式(II)化合物与HNE抑制剂的组合, HNE抑制剂选自AAT,ADC-7828,Aeriva,TAPI,AE-3763,KRP-109, AX-9657,POL-6014,AER-002,AGTC-0106,respriva,AZD-9668, zemaira,AAT IV,PGX-100,elafin,SPHD-400,α1-蛋白酶抑制剂C 和吸入的α1-蛋白酶抑制剂。
本发明也提供本发明化合物或式(II)化合物与白细胞三烯调节剂的 组合,白细胞三烯调节剂选自孟鲁司特,扎鲁司特和普仑司特。
本发明也提供本发明化合物或式(II)化合物,与NSAID的组合, NSAID选自布洛芬和酮洛芬。
本发明也提供本发明化合物或式(II)化合物与粘液调节剂的组合, 粘液调节剂选自INS-37217,地夸磷索,西贝那德,CS-003,他奈坦, DNK-333,MSI-1956和吉非替尼。
本发明化合物的剂量取决于各种因素,包括待治疗的特定疾病,症 状的严重性,给药途径,剂量间隔频率,所用的特定化合物,化合物的 效力、毒理学特征和药代动力学特征。
有利地,本发明化合物或式(II)化合物可以以例如0.001至1000mg/ 天,优选0.1至500mg/天的剂量给予。
在通过吸入途径给予时,本发明化合物或式(II)化合物的剂量有利 地是0.01至20mg/天,优选0.1至10mg/天。
优选,本发明化合物或式(II)化合物单独或与其它活性成分相组合 可以给予用于预防和/或治疗任何梗阻性呼吸系统疾病比如哮喘,慢性支 气管炎和慢性阻塞性肺疾病(COPD)。
然而,本发明化合物或式(II)化合物可以给予用于预防和/或治疗其 中需要PDE4抑制的任何疾病。所述疾病包括:变应性疾病状态比如特 应性皮炎,荨麻疹,变应性鼻炎,变应性结膜炎,春季结膜炎,嗜曙红 性肉芽肿,牛皮癣,炎性关节炎,类风湿性关节炎,脓毒性休克,溃疡 性结肠炎,克罗恩病,心肌和脑的再灌注损伤,慢性肾小球肾炎,内毒 素性休克,囊性纤维化,动脉再狭窄,动脉粥样硬化,角化病,风湿样 脊柱炎,骨关节炎,灼热,糖尿病,尘肺,毒性和变应性接触湿疹,特 应性湿疹,脂溢性湿疹,单纯性苔藓,晒斑,肛门生殖区瘙痒症,斑秃, 肥厚性瘢痕,盘状红斑狼疮,系统性红斑狼疮,滤泡和大面积脓皮病, 内源性和外源性痤疮,痤疮酒渣鼻,Beghet’s疾病,类过敏性紫癜肾炎, 炎性肠病,白血病,多发性硬化,胃肠道疾病,自身免疫性疾病等。
它们也包括神经和精神病障碍比如阿尔茨海默病,多发性硬化,淀 粉样动脉粥样硬化(ALS),多系统萎缩(MSA),精神分裂症,帕金森病, 亨廷顿舞蹈病,匹克病,抑郁,卒中,和脊髓损伤。
本发明现通过下述非限制性实施例进一步描述。
实施例
化合物的化学名称用Cambridge Software的Structure To Name Enterprise10.0产生。
缩写
EDC=1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)盐酸盐;DMAP=4- 二甲基氨基吡啶;DMF=二甲基甲酰胺;EtOAc或AcOEt=乙酸乙酯; RT=室温;THF=四氢呋喃;DCM=二氯甲烷;Et2O=二乙醚;MeOH =甲醇;n-BuOH=正丁醇;EtOH=乙醇;IprOH或IPA=异丙醇;(Ipr)2O =二异丙基醚;MIK=甲基异丁基酮;MEK=甲基乙基酮;MTBE=甲基 叔丁基醚;AcOH=乙酸;vv=体积;v/w=体积/重量比;w/w=重量/重 量比;
一般实验细节
NMR表征:
1H-NMR谱图在400MHz Varian AS400光谱仪上记录。化学位移 报告为相对内标三甲基硅烷(TMS)的按ppm计的δ值。偶联常数(J值) 按赫兹(Hz)提供而多重性用下述缩写报告(s=单峰,d=二重峰,t=三重峰, q=四重峰,m=多重峰,br=宽,nd=未测定)。
或者
1H-NMR谱图在Bruker ARX300光谱仪上,于300.13MHz(1H), 用氘化溶剂比如氘化二甲亚砜(DMSO-d6)或氘化氯仿(CDCl3)记录。设备 配有多核反相探针和温度控制器。化学位移表达为相对四甲基硅烷(d单 元)的低磁场方向,按百万分之一(ppm)计。多重度如下指出:(s)单峰, (d)二重峰,(dd)双二重峰,(ddd)三二重峰,(t)三重峰,(dt)双三重峰, (q)四重峰,(m)多重峰,(br s)宽信号。偶联常数J以赫兹(Hz)为单位表 达。
LC/UV/MS分析方法
据估计,LC/MS保留时间受实验误差影响为±0.5分钟。
LC/UV/MS-方法1
LC设备:HPLC Alliance Waters(或等价物)
柱:Kinetex 2.6u C18 100A 100x 4.6mm(Phenomenex)
柱温(℃):50.0
流动相:HCOONH4 0.025M pH3(A);乙腈(B)
流速(ml/min):2.0(在MS中按1:10分裂)
停止时间(mins):17.0
梯度:
| 时间(min) | %A | %B |
| 0.00 | 80.0 | 20.0 |
| 10.00 | 20.0 | 80.0 |
| 12.00 | 20.0 | 80.0 |
| 14.00 | 80.0 | 20.0 |
| 17.00 | 80.0 | 20.0 |
UV检测:通道1 245nm;通道2 254nm
注射体积(ul):5.00
样品溶剂:乙腈
MS设备:Waters Quattro Micro API(或等价物)
极性ES+
毛细管(kV)3.20
锥电压(V)20.00
提取电压(V)2.00
RF透镜(V)0.3
极性ES-
毛细管(kV)3.20
锥电压(V)20.00
提取电压(V)3.00
RF透镜(V)0.3
来源温度(℃)110
去溶剂化温度(℃)210
锥气体流(L/Hr)150
去溶剂化气体流(L/Hr)650
扫描持续时间(秒):1.00
扫描间延缓(秒):0.10
质量范围:125至1000
LC/UV/MS-方法2
LC设备:Acquity Waters UPLC(或等价物)
柱:Kinetex 1.7u XB-C18 100A 100x 2.1mm(Phenomenex)
柱温(℃)50.0
流动相:HCOONH4 0.025M pH3(A);乙腈+0.1%甲酸(B)
流速(ml/min)0.65(在MS中按1:3分裂)
停止时间(mins)10.0
梯度:
| 时间(min) | %A | %B |
| 0.00 | 80.0 | 20.0 |
| 5.50 | 20.0 | 80.0 |
| 7.50 | 20.0 | 80.0 |
| 8.00 | 80.0 | 20.0 |
| 10.00 | 80.0 | 20.0 |
UV检测:波长254nm
注射体积(ul)-2.00
样品溶剂:乙腈
MS设备:Waters ZQ(或等价物)
极性ES+
毛细管(kV)3.00
锥电压(V)20.00
提取电压(V)3.00
RF透镜(V)1.0
极性ES-
毛细管(kV)3.00
锥电压(V)20.00
提取电压(V)3.00
RF透镜(V)1.0
来源温度(℃)110
去溶剂化温度(℃)210
锥气体流(L/Hr)150
去溶剂化气体流(L/Hr)650
质量范围:100至950
扫描时间(秒):0.32
LC/UV/MS-方法3
LC设备:Acquity Waters UPLC(或等价物)通过接口与2996 PDA 检测器联用
柱:Acquity UPLC BEH C18 1.7um 50x2.1mm
柱温(℃)40.0
流动相:95:5 H2O:ACN+(0.1%TFA)(A);5:95 H2O:ACN+(0.1% TFA)(B)
流速(ml/min)0.6(在MS中按1:6分裂)
停止时间(mins)8.5
梯度:
| 时间(min) | %A | %B |
| 0.00 | 95.0 | 5.0 |
| 0.50 | 95.0 | 5.0 |
| 6.00 | 0.0 | 100.0 |
| 7.00 | 0.0 | 100.0 |
| 7.10 | 95.0 | 5.0 |
| 8.50 | 95.0 | 5.0 |
UV检测:BPI检测(开始波长nm 210,终止波长nm 400,采样速 率谱图/秒=20)
注射体积(ul)-1.00
样品溶剂:DMSO:MeOH:ACN比率1:3:3
MS设备:Waters ZQ(或等价物)
极性ES
毛细管(kV)3.20
锥电压(V)25.00
提取电压(V)3.00
RF透镜(V)0.1
极性ES-
毛细管(kV)3.00
锥电压(V)20.00
提取电压(V)3.00
RF透镜(V)0.3
来源温度(℃)150
去溶剂化温度(℃)350
锥气体流(L/Hr)110
去溶剂化气体流(L/Hr)800
质量范围:60至1200
扫描时间(秒):0.4
LC/UV/MS-方法4
LC设备:Acquity Waters UPLC(或等价物)通过接口与2996 PDA 检测器联用
柱:Kinetex C18 1.7um 50x2.1mm(Phenomenex)
柱温(℃):40.0
流动相:95:5 H2O:ACN+(0.1%TFA)(A);5:95 H2O:ACN+(0.1% TFA)(B)
流速(ml/min):0.5(在MS中不裂分)
停止时间(mins):4.40
梯度:
| 时间(min) | %A | %B |
| 0.00 | 95.0 | 5.0 |
| 0.30 | 95.0 | 5.0 |
| 3.30 | 0.0 | 100.0 |
| 3.90 | 0.0 | 100.0 |
| 4.40 | 95.0 | 5.0 |
UV检测:BPI检测(开始波长nm 200,终止波长nm 400,采样速 率点/秒=20)
注射体积(ul):4.00
样品溶剂:乙腈
MS设备:ZQ(或等价物)
极性ES
毛细管(kV)3.25
锥电压(V)27.00
提取电压(V)3.00
RF透镜(V)0.4
来源温度(℃)120
去溶剂化温度(℃)400
锥气体流(L/Hr)100
去溶剂化气体流(L/Hr)800
扫描时间(秒):0.42
质量范围:100至800
制备型反相HPLC条件
制备型HPLC-方法1
Waters Micromass ZQ/样品处理器2767
光电二极管阵列检测器2996;
柱:XTerra Prep MS C18柱(5μm,19x 150mm,Waters)
流速:20ml/min,具有MS检测
UV波长:254nm。
流动相:溶剂A(水:MeCN:HCOOH 95:5:0.05);溶剂B(水: MeCN:HCOOH 5:95:0.05)
梯度:
| 时间(min) | %A | %B |
| 0.00 | 100.0 | 0.00 |
| 1.00 | 100 | 0.00 |
| 10.00 | 0.00 | 100.0 |
| 11.00 | 0.00 | 100.0 |
| 12.00 | 100.0 | 0.00 |
制备型HPLC-方法2
柱:Waters Symmetry Prep C18 17um 19x300
流速:20ml/min
流动相:90%H2O,10%乙腈,0.05%TFA(A);10%H2O,90% 乙腈,0.05%TFA(B)
梯度:
| 时间(min) | %A | %B |
| 0.00 | 95 | 5 |
| 2.5 | 95 | 5 |
| 22 | 0 | 100 |
| 30 | 0 | 100 |
制备型HPLC-方法3
Waters Micromass ZQ/样品处理器2767
光电二极管阵列检测器:2996
柱:XTERRA Prep MS C18 10um 19x300
流速:20ml/min
流动相:H2O,0.1%TFA(A);乙腈,0.1%TFA(B)
梯度:
| 时间(min) | %A | %B |
| 0.00 | 90 | 10 |
| 2 | 90 | 10 |
| 23 | 0 | 100 |
| 30 | 0 | 100 |
调理:
| 时间(min) | %A | %B |
| 30.5 | 90 | 10 |
| 32 | 90 | 10 |
手性HLPC:
对映体纯度在Hewlett Packard 1050 HPLC系统上测定,用 Chiracel OD柱(5μ4.6X250mm),用各特定实施例中指出的不同比率的 己烷和异丙醇的等度混合物洗脱。
流速=0.8ml/min
UV检测=230nm。
旋光(活性)确定
化合物的比旋光度用旋光仪Perkin Elmer型号241或341测量。
温度(℃)25
路径长度(dm)1
波长钠D-线(589nm)
需要微波加热的实验用Biotage Initiator Sixty设备来进行。
成盐程序
除非另有说明,实验部分描述的盐根据下述程序之一来获得:
甲酸盐:在描述于盐名称栏中的情况下,含有一个或多个碱性中心 和通过反相HPLC纯化的(方法1)的化合物作为甲酸盐获得:减压蒸发 自色谱法收集的清洁级分,不加任何进一步碱性处理。
三氟乙酸盐:在描述于盐名称栏中的情况下,含有一个或多个碱性 中心和通过反相HPLC纯化的(方法2或3)的化合物作为2,2,2-三氟乙酸 盐获得:减压蒸发自色谱法收集的清洁级分,不加任何进一步碱性处理。
盐酸盐:在描述于盐名称栏中的情况下,含有在酸性条件下发生 Boc脱保护而不加任何进一步碱性后处理的一个或多个碱性中心的化合 物作为盐酸盐获得。
在技术人员已知的条件下,将碱用相应的酸溶液处理,获得任意其 它盐。
如果需要,用NMR确定盐的化学计量。
在下述程序中,在各原料之后,一般提供化合物编号。其目的是帮 助有本领域化学家。原料不一定制备自标明的批次。
在提及使用"相似"或"类似"程序时,本领域技术人员能认识到, 上述程序可以牵涉次要的变化,例如反应温度,试剂/溶剂量,反应时间, 后处理条件或色谱纯化条件。
通过未经定义的线键指出的立构中心,代表作为单一非对映异构体 或对映体获得的化合物中的那些,但是其绝对构型并未确定。
下述实施例所描述的化合物中的许多已制备自立体化学纯的原料, 例如95%ee。
在指出时,赋予实施例中的化合物的立体化学的假设是原料解析立 构中心的绝对构型在整个任何后续反应条件下得到保持。
所描述的某些化合物的绝对构型已通过结晶物质的X射线或VCD (振动圆二色性)分析确认为正确。
在指出时,通过LC/UV/MS确定的非对映异构体比率,据估计受 实验误差影响为±1%。另选地非对映异构体的比率通过1H NMR确定, 据估计在单一非对映异构体用NMR分析检测的情况下是>95:5。
特定实施例的详细合成途径和程序描述于下文方案9-41和46。列 于表12的醇中间体的合成描述于所报告的专利申请中。
表12

下述程序中所用的中间体是可商购的,技术人员通过本领域熟知合 成途径可获得的,或着按照下述实施例描述的合成程序可获得的。
实施例17
(S)-3,5-二氯-4-(2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-羟基乙基)吡啶 1-氧化物164)
方案23

步骤1:4-((S)-2-((S)-2-乙酰氧基-2-苯基乙酰氧基)-2-(3-(环丙基-甲 氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(161)
将(S)-2-乙酰氧基-2-苯乙酸(0.924g,4.76mmol),(S)-3,5-二氯 -4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟基乙基)吡啶1-氧化物 (1.0g,2.380mmol),EDC(0.684g,3.57mmol)和DMAP(0.436g,3.57 mmol)在DCM(150ml)中的混合物在室温搅拌24小时。加入更多的 (S)-2-乙酰氧基-2-苯乙酸(0.350g,1.802mmol),EDC(0.456g,2.380mmol)和DMAP(0.300g,2.456mmol)并继续搅拌3小时以完成转化。 用含水1N HCl,然后用含水1M K2CO3将反应混合物洗涤两次;有机层 在Na2SO4上干燥,蒸发至干。残余物与iPrOH(30ml)研磨,过滤,提 供所希望产品(1.27g,2.129mmol,89%收率)。MS/ESI+596.18[MH]+
步骤2:4-((S)-2-((S)-2-乙酰氧基-2-苯基乙酰氧基)-2-(4-(二氟甲氧 基)-3-羟基苯基)乙基)-3,5-二氯吡啶1-氧化物(162)
将4-((S)-2-((S)-2-乙酰氧基-2-苯基乙酰氧基)-2-(3-(环丙基甲氧 基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(1.27g,2.129mmol) 用三氟乙酸(15ml,195mmol)处理,所得溶液在室温搅拌20小时。反 应混合物用DCM稀释,用水洗涤两次;有机层在Na2SO4上干燥,蒸发 至干。残余物通过色谱法在硅胶上纯化(DCM/EtOAc=3:2至1:1)。合并 混合的级分,与iPr2O/Et2O(10:1)混合物研磨。然后合并收集的固体, 自色谱法得到纯级分,提供所希望的化合物(1.08g,1.991mmol,94% 收率);MS/ESI+542.11[MH]+
1H NMR(300MHz,DMSO-d6)δppm 8.56(s,2H),7.27-7.50(m, 5H),6.98(d,1H),6.81(d,1H),7.00(t,1H),6.54(dd,1H),5.89 (dd,1H),5.84(s,1H),3.40(dd,1H),3.18(dd,1H),2.13(s,3H)
步骤3:4-((S)-2-((S)-2-乙酰氧基-2-苯基乙酰氧基)-2-(4-(二氟甲氧 基)-3-甲氧基苯基)乙基)-3,5-二氯吡啶1-氧化物(163)
将4-((S)-2-((S)-2-乙酰氧基-2-苯基乙酰氧基)-2-(4-(二氟甲氧基)-3- 羟基苯基)乙基)-3,5-二氯吡啶1-氧化物(1.080g,1.991mmol),碘甲烷 (0.162ml,2.59mmol)和碳酸钾(0.550g,3.98mmol)的CH3CN(40ml) 悬浮液在室温激烈地搅拌20小时。反应混合物在DCM与水间分配,有 机层在Na2SO4上干燥。真空除去溶剂,提供所希望的化合物(0.984g,1.769mmol,89%收率)。MS/ESI+556.17[MH]+。粗化合物不加进一步 纯化地使用。
步骤4:(S)-3,5-二氯-4-(2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-羟基乙 基)吡啶1-氧化物(164)
将4-((S)-2-((S)-2-乙酰氧基-2-苯基乙酰氧基)-2-(4-(二氟甲氧基)-3- 甲氧基苯基)乙基)-3,5-二氯吡啶1-氧化物(984mg,1.769mmol)溶于 MeOH(50ml)和DCM(10ml)的混合物。加入含水饱和NaHCO3溶液(10 ml,11.00mmol),所得悬浮液在室温搅拌2小时。反应混合物在水与 DCM间分配;有机层在Na2SO4上干燥,蒸发至干,提供所希望的化合 物(650mg,1.71mmol,97%收率)。MS/ESI+380.03[MH]。
列于表13的化合物用与方案23中的描述类似的程序,用适宜的烷 基化剂并在65℃进行步骤3制备。
表13

实施例18
合成(S)-3,5-二氯-4-(2-(3,4-二甲氧基苯基)-2-羟基乙基)吡啶1-氧化 物(170)
方案24

步骤1:合成2-(3,5-二氯吡啶-4-基)-1-(3,4-二甲氧基苯基)乙醇(166)
在氩气氛下将3,5-二氯-4-甲基吡啶(160)(54g,331mmol)溶于无水 THF(480mL),将其在-78℃于干冰/丙酮浴中冷却。滴加LHMDS 1N THF溶液(331ml,331mmol),将温度保持在-78℃。在-78℃搅拌混合物 1小时。随后,滴加将3,4-二甲氧基苯甲醛(50g,301mmol)的无水THF (120ml)溶液,保持温度在-78℃。在加入完成时,让混合物在室温温热。
将反应倾倒入冰和水(1L)中,搅拌混合物直至大量沉淀形成。过滤 固体,和溶于乙酸乙酯(500ml),在Na2SO4上干燥,真空蒸发溶剂。粗 制品在CHCl3/己烷中结晶。过滤沉淀,用己烷洗涤,在40℃真空干燥8 小时,提供55g(收率45%)。在40℃真空蒸发母液溶液,溶于乙酸乙酯 (200ml)并用200ml水萃取。有机溶液在Na2SO4上干燥,在40℃真空 蒸发溶剂。粗制品在CHCl3/己烷中结晶,获得额外15g的所希望产品(166) (总收率70%)。
步骤2:合成((R)-2-(3,5-二氯吡啶-4-基)-1-(3,4-二甲氧基苯基)乙基) 2-(6-甲氧基萘-2-基)丙酸酯(167)
将中间体166(50g,152mmol),(R)-2-(6-甲氧基萘-2-基)丙酸(38.6 g,168mmol),DMAP(20.5g,168mmol)和EDC(43.8g,229mmol) 溶于DMF(300ml),在室温搅拌反应混合物2小时。在该时间之后,加 水(500ml),发生沉淀时搅拌溶液。过滤固体,溶于DCM(500ml)。有机溶液用含水HCl 1N(2x500ml),饱和NaHCO3水溶液(500ml)洗涤, 在Na2SO4上干燥。真空蒸发溶剂,在EtOH(300ml)中超声固体残余物, 研磨1小时。过滤收集所得沉淀,在40℃真空干燥4小时,提供79g(收 率99%)化合物167,是非对映异构体的混合物。
步骤3:合成(R)-((S)-2-(3,5-二氯吡啶-4-基)-1-(3,4-二甲氧基苯基) 乙基)2-(6-甲氧基萘-2-基)丙酸酯(168)
将中间体167(79g,146mmol)溶于CHCl3(100ml),缓慢加入 MeOH(30ml)直至持续乳白色,在室温放置混合物2小时。过滤收集形 成的固体,通过CHCl3/MeOH(70ml/20ml)溶剂系统重结晶,获得35g 的化合物168(收率88%,ee 98%)。
手性HPLC分析Rt=42.33分钟(快速异构体);洗脱液:己烷:异 丙醇97:3
1H NMR(600MHz,氯仿-d)δppm 8.04(s,2H),7.67(d,J=8.79Hz, 1H),7.58(d,J=8.52Hz,1H),7.53(m,1H),7.12-7.20(m,3H), 6.95(dd,J=8.24,1.92Hz,1H),6.78-6.88(m,2H),6.14(dd,J=10.44, 4.12Hz,1H),3.95(s,3H),3.88(s,3H),3.78-3.81(m,4H),3.55 (dd,J=13.73,10.44Hz,1H),3.14(dd,J=13.60,4.26Hz,1H),1.44 (d,J=7.14Hz,3H)。
步骤4:合成(S)-2-(3,5-二氯吡啶-4-基)-1-(3,4-二甲氧基苯基)乙醇 (169)
将中间体168(30g,56mmol)溶于MeOH,缓慢加入甲苯。将叔 丁醇钾缓慢加至悬浮液。在室温搅拌混合物24小时。反应用水稀释(500 ml),含水混合物用CHCl3(500ml)萃取。有机层在Na2SO4上干燥,真 空蒸发溶剂。残余物自CHCl3(100ml)和己烷(20ml,直至持续乳白)结 晶。浓缩母液,以相同方式中重结晶,提供第二批希望的化合物。共获 得16g的化合物169(收率87%)。
手性HPLC分析Rt=58.03分钟;洗脱液:己烷:异丙醇95:5。
 (c=0.506,甲醇)
(c=0.506,甲醇)
1H NMR(400MHz,丙酮)δppm 8.47(s,2H),6.96-7.15(m,1H), 6.87(m,2H),4.93-5.21(m,1H),4.50(d,J=3.97Hz,1H),3.78(s, 6H),3.44(dd,J=12.79,8.38Hz,1H),3.22(dd,J=13.01,5.51Hz, 1H)。
MS/ESI+[MH]+:328.19
步骤5:合成(S)-3,5-二氯-4-(2-(3,4-二甲氧基苯基)-2-羟基乙基)吡啶 1-氧化物(170)
将化合物169(4g,12mmol)溶于乙酸乙酯,和将间-CPB酸加至 溶液。混合物在室温搅拌5小时。过滤收集形成的固体,用乙酸乙酯洗 涤,真空干燥,提供1.72g的标题化合物(收率41%)。手性HPLC分析 Rt=22.16分钟;洗脱液:己烷:异丙醇6:4。 (c=0.253,甲 醇/CHCl3 1:1)。MS/ESI+[MH]+:344.19
(c=0.253,甲 醇/CHCl3 1:1)。MS/ESI+[MH]+:344.19
1H NMR(400MHz,氯仿-d)δppm 8.15(s,2H),6.99(m,1H), 6.79-6.88(m,2H),5.03(dd,J=8.50,5.32Hz,1H),3.75-3.98(m, 6H),3.42(dd,J=13.57,8.56Hz,1H),3.19(dd,J=13.51,5.32Hz, 1H),2.06-2.15(m,1H)。
实施例36
合成3,5-二氯-4-(2-(2,2-二甲基苯并[d][1,3]二氧杂环戊烯-5-基)-2- 羟基乙基)吡啶1-氧化物(172)
方案25

步骤1:合成2-(3,5-二氯吡啶-4-基)-1-(2,2-二氟苯并[d][1,3]二氧杂 环戊烯-5-基)乙醇(171)
在氩气氛下将3,5-二氯-4-甲基吡啶(166)(4.37g,0.016mol)溶于无 水THF(40mL),将其在-78℃于干冰/丙酮浴中冷却。滴加LHMDS 1N THF溶液(28ml,28mmol),保持温度在-78℃。在-78℃搅拌混合物1 小时。此后,滴加2,3-二氟-3,4-苯并二氧杂环戊烯甲醛(5g,0.026mol) 的无水THF(10ml)溶液,保持温度在-78℃。在加入完成的情况下,让 混合物在室温温热。
将反应倾倒入冰和水中,水相用乙酸乙酯(3x)萃取。经合并的有机 相在Na2SO4上干燥,真空蒸发溶剂。粗制品在石油醚/己烷1/1中结晶。 过滤沉淀,用己烷洗涤,在40℃真空干燥8小时,提供6.4g(收率45%)。
MS/ESI+349.14[MH]+;1H NMR(200MHz,氯仿-d) ppm 8.71(s, 2H),7.48-7.68(m,2H),6.85-7.07(m,1H),4.61(m,1H),4.11- 4.44(m,2H)。
ppm 8.71(s, 2H),7.48-7.68(m,2H),6.85-7.07(m,1H),4.61(m,1H),4.11- 4.44(m,2H)。
步骤2:合成3,5-二氯-4-(2-(2,2-二甲基苯并[d][1,3]二氧杂环戊烯-5- 基)-2-羟基乙基)吡啶1-氧化物(172)
将2-(3,5-二氯吡啶-4-基)-1-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5- 基)乙醇(2g,5.74mmol)溶于EtOAc(40ml),将溶液冷却至0℃。加入 间-CPBA(5.29g,22.98mmol),所得混合物在室温搅拌24小时。将反 应混合物冷却至0℃,过滤白色固体沉淀,用冷DCM洗涤两次,提供 所希望产品(1.738g,4.77mmol,83%);MS/ESI+364.03[MH]+
实施例19:
合成3,5-二氯-4-(2-羟基-2-(4-甲氧基螺-[苯并[d][1,3]二氧杂环戊烯 -2,1'-环戊烷]-7-基)乙基)吡啶1-氧化物(174)
方案26

步骤1:3,5-二氯-4-(2-(4-甲氧基螺[苯并[d][1,3]二氧杂环戊烯-2,1'- 环戊烷]-7-基)-2-氧代乙基)吡啶1-氧化物(173)
向在0℃冷却的2-(3,5-二氯吡啶-4-基)-1-(4-甲氧基螺[苯并[d][1,3] 二氧杂环戊烯-2,1'-环戊烷]-7-基)乙酮(如EP1535920的描述制备,4.75g, 12.05mmol)的EtOAc(125ml)溶液,加入间-CPBA(11.09g,48.2mmol), 在室温搅拌反应混合物24小时。加入更多的间-CPBA(5.54g,24.10 mmol),继续搅拌额外的24小时。混合物用含水1M K2CO3洗涤数次,有机层在Na2SO4上干燥,蒸发至干。残余物通过色谱法在硅胶上纯化, (DCM/EtOAc=3/1至1/2),提供3,5-二氯-4-(2-(4-甲氧基螺[苯并[d][1,3] 二氧杂环戊烯-2,1'-环戊烷]-7-基)-2-氧代乙基)吡啶1-氧化物(2.14g,5.22 mmol,43.3%收率);MS/ESI+410.10[MH]+。
步骤2:3,5-二氯-4-(2-羟基-2-(4-甲氧基螺-[苯并[d][1,3]二氧杂环戊 烯-2,1'-环戊烷]-7-基)乙基)吡啶1-氧化物(174)
向3,5-二氯-4-(2-(4-甲氧基螺-[苯并[d][1,3]二氧杂环戊烯-2,1'-环戊 烷]-7-基)-2-氧代乙基)吡啶1-氧化物(2.14g,5.22mmol)的MeOH(100ml) 溶液,分批加入固体硼氢化钠(0.197g,5.22mmol),混合物在室温下搅 拌过夜。在2小时内加入额外的硼氢化钠(0.394g,10.44mmol),再继 续搅拌12小时。真空浓缩混合物,用EtOAc稀释,用含水1N NaOH洗涤两次。有机层在Na2SO4上干燥,减压除去溶剂,残余物通过色谱 法(EtOAc)在硅胶上纯化,提供3,5-二氯-4-(2-羟基-2-(4-甲氧基螺[苯并 [d][1,3]二氧杂环戊烯-2,1'-环戊烷]-7-基)乙基)吡啶1-氧化物(650mg, 1.577mmol,30.2%收率);MS/ESI+412.10[MH]+。
实施例1
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-吡咯烷-2羰氧基)乙基)吡啶1-氧化物盐酸盐(3)
方案9

步骤1:4-((S)-2-((S)-1-(叔-丁氧基羰基)吡咯烷-2-羰氧基)-2-(3-(环 丙基甲氧基)-4-(二氟甲氧基)苯基)-乙基)-3,5-二氯吡啶1-氧化物(2)
将(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟 基乙基)吡啶1-氧化物(1)(550mg,1.309mmol),(S)-1-(叔-丁氧基羰基) 吡咯烷-2-羧酸(282mg,1.309mmol),EDC(251mg,1.309mmol)和 DMAP(160mg,1.309mmol)溶于DMF(5ml)。在室温搅拌反应48小 时,实现完成。在此时间之后,反应用HCl 1M淬灭,用EtOAc萃取。 有机萃取物用HCl 1M(x3)并且用K2CO3 5%(x3)洗涤,随后在Na2SO4上干燥,真空浓缩,产生800mg的希望产品(收率99%)。MS/ESI+617.16 [MH]+
步骤2:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-吡咯烷-2-羰氧基)乙基)-吡啶1-氧化物盐酸盐(3)
将4-((S)-2-((S)-1-(叔-丁氧基羰基)吡咯烷-2-羰氧基)-2-(3-(环丙基 甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(2)(300mg, 0.486mmol)溶于二噁烷/HCl(4M,2ml),在室温搅拌8小时。在此时 间之后,在旋蒸仪上减压除去溶剂,真空炉中干燥过夜,产生希望的产 品,是盐酸盐(200mg;收率80%)。
MS/ESI+517.2[MH]+;tR/min(方法1)=3.75;非对映体比率 =>99:1;[αD]=-32.80(c=0.25;CHCl3)
1H NMR(400MHz,甲醇-d4)δppm 8.49(s,2H),7.18(d,J=7.94 Hz,1H),7.11(d,J=1.76Hz,1H),7.04(d,J=1.76Hz,1H),6.78(t, J=75.00Hz,1H),6.10-6.17(m,1H),4.38-4.51(m,1H),3.85-3.99 (m,2H),3.66(m,2H),3.39-3.51(m,1H),2.40-2.60(m,1H), 1.89-2.17(m,4H),1.15-1.36(m,1H),0.64(dd,J=7.94,1.32Hz, 2H),0.33-0.47(m,2H)。
通过用适宜的原料获得列于表1的化合物。
作为游离碱获得的化合物进行碱性后处理,代替上述的减压除去溶 剂(Ex:NaHCO3饱和溶液),随后用极性有机溶剂(Ex:AcOEt)萃取以 便除去进行步骤2(方案9)时自发发生的与HCl的成盐。
通过将其适当Boc-保护的前体与AcOEt/HCl(5M)反应,随后在室 温下过滤自发沉淀自反应混合物的盐酸盐,获得化合物6,7,181,183。
用EtOAc中的HCl进行步骤2,并不进行加热地除去溶剂,获得 化合物177,178,和179。









实施例2
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(1-((4-(甲氧羰基)-5-甲基呋喃-2-基)甲基)吡咯烷-2-羰氧基)乙基)吡 啶1-氧化物(15)
方案10
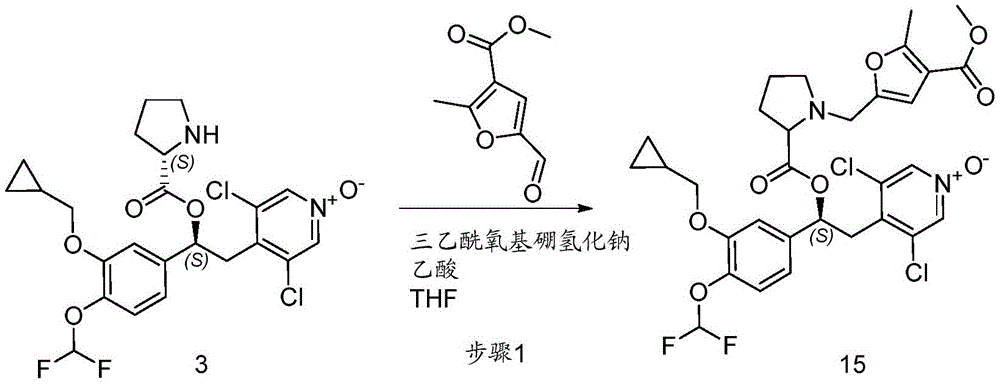
将3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-吡咯烷-2-羰氧基)乙基)吡啶1-氧化物(3)(40mg,0.077mmol) 溶于THF(1ml),向其加入5-甲酰基-2-甲基呋喃-3-羧酸甲酯(13.00mg, 0.077mmol)和乙酸(4.64mg,0.077mmol)。反应在室温搅拌30分钟, 随后加入三乙酰氧基硼氢化钠(16.39mg,0.077mmol)。在此时间之后,再搅拌反应2hrs,随后完成。除去溶剂,残余物在EtOAc与含水HCl 1M 间分配。然后有机层用K2CO3 5%水溶液洗涤,在Na2SO4上干燥。真空 除去溶剂,获得透明的油状物(35mg;收率68%),将其通过制备型HPLC 纯化,产生18mg所希望产品的非对映体的混合物(47.5:52.5),是透明 的油状物(收率36%)。MS/ESI+668.9[MH]+;tR=5.97;6.15分钟(方法 1);非对映体比率=47:53。
列于表3的化合物以与方案10步骤1中的描述类似的程序,通过 将前体(5)与适宜试剂反应制备。
自(5)获得的非对映体的混合物,将其通过制备型HPLC设备(方法 1,一般实验细节部分)分离,提供两种非对映体(16)和(17),根据它们在 一般实验细节部分(方法1)描述的色谱条件下的观察保留时间分别确认 为快速和慢速异构体。


实施例3
合成3,5-二氯-4-((S)-2-((S)-1-(4-(环丙基甲氧基)-3-(甲基磺酰氨基) 苯甲酰基)吡咯烷-2-羰氧基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基) 乙基)吡啶1-氧化物(19)
方案11
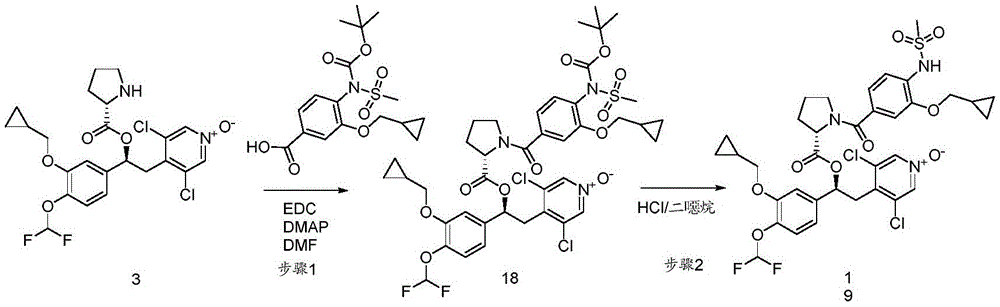
步骤1:4-((S)-2-((S)-1-(3-(N-(叔-丁氧基羰基)甲基磺酰氨基)-4-(环 丙基甲氧基)苯甲酰基)吡咯烷-2-羰氧基)-2-(3-(环丙基甲氧基)-4-(二氟甲 氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(18)
将3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-吡咯烷-2-羰氧基)乙基)吡啶1-氧化物(3)(40mg;0.077mmol) 置于50ml圆底烧瓶,溶于DMF(2ml)。将可按照WO2010/089107的 描述获得的3-(N-(叔-丁氧基羰基)甲基磺酰氨基)-4-(环丙基甲氧基)苯甲 酸(40mg,0.104mmol加入反应溶液,随后加入EDC(20.0mg,0.104mmol)和DMAP(15.0mg,0.123mmol)。反应在室温搅拌6小时,随后 完成,通过加入20ml含水HCl 1M淬灭。水层用EtOAc萃取,用HCl 1M(x3)和含水K2CO3 5%(x3)洗涤。所得有机萃取物用Na2SO4脱水, 在滤纸上过滤,在旋转式蒸发仪上减压除去溶剂。油残余物通过制备型HPLC(方法1)纯化,产生30mg的希望产品(收率44%)。MS/ESI+884.1 [MH]+
1H NMR(400MHz,丙酮) ppm 8.28(s,2H),7.33(d,J=7.94Hz, 1H),7.14-7.23(m,4H),6.68-7.13(m,2H),6.16(dd,J=9.92,4.19 Hz,1H),4.52(dd,J=7.94,6.17Hz,1H),3.88-4.12(m,6H),3.56 -3.74(m,3H),3.53(s,3H),3.34(dd,J=14.11,4.41Hz,1H),1.74-1.93(m,2H),1.47(s,9H),1.25-1.35(m,2H),0.51-0.69(m,4 H),0.27-0.48(m,4H)。
ppm 8.28(s,2H),7.33(d,J=7.94Hz, 1H),7.14-7.23(m,4H),6.68-7.13(m,2H),6.16(dd,J=9.92,4.19 Hz,1H),4.52(dd,J=7.94,6.17Hz,1H),3.88-4.12(m,6H),3.56 -3.74(m,3H),3.53(s,3H),3.34(dd,J=14.11,4.41Hz,1H),1.74-1.93(m,2H),1.47(s,9H),1.25-1.35(m,2H),0.51-0.69(m,4 H),0.27-0.48(m,4H)。
步骤2:3,5-二氯-4-((S)-2-((S)-1-(4-(环丙基甲氧基)-3-(甲基磺酰氨 基)苯甲酰基)吡咯烷-2-羰氧基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)乙基)吡啶1-氧化物(19)
将4-((S)-2-((S)-1-(3-(N-(叔-丁氧基羰基)甲基磺酰氨基)-4-(环丙基 甲氧基)苯甲酰基)吡咯烷-2-羰氧基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基) 苯基)乙基)-3,5-二氯吡啶1-氧化物(18)(30mg,0.034mmol)溶于 HCl/EOAc(4M;2ml),在室温搅拌10小时实现完成。反应通过加入 K2CO3 5%淬灭,用EtOAc萃取。所得有机萃取物在Na2SO4上干燥,在滤纸上过滤,在旋转式蒸发仪上减压除去溶剂。残余物自EtOH:己 烷(1:3)重结晶,产生白色固体(18mg;收率68%)的标题化合物。MS/ESI+ 784.1[MH]+;tR=5.72(方法1);[αD]=-48.92(c=3.7;DCM);非对映 体比率>99:1。
1H NMR(400MHz,氯仿-d)δppm 8.16(br.s.,2H),7.53(m,1H), 7.09-7.20(m,3H),7.05(m,1H),6.96(m,2H),6.41-6.63-6.84(t, 1H,CHF2),6.08-6.17(m,1H),4.53-4.66(m,1H),3.81-3.98(m, 4H),3.61(m,3H),3.22-3.39(m,1H),3.02(s,3H),2.20-2.34(m, 1H),1.78-2.02(m,3H),1.27(m,2H),0.59-0.75(m,4H),0.28- 0.42(m,4H)。
列于表4的化合物用上文方案11实施例3的描述类似的程序,将 所列适当前体与适宜试剂反应,随后进行下表指出的纯化步骤,而不是 上述的重结晶制备。


实施例4
合成4-((2S)-2-(3-(4-氨基苯甲酰基)噻唑烷-2-羰氧基)-2-(3-(环丙基 甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(26)
方案12

步骤1:3-(4-硝基苯甲酰基)噻唑烷-2-羧酸甲酯(23)
将噻唑烷-2-羧酸甲酯盐酸盐(200mg,1.089mmol)溶于DCM (2ml)。加入DMAP(173mg,1.416mmol)和4-硝基苯甲酰基氯化物(263 mg,1.416mmol),在室温搅拌反应2h,实现完成。反应混合物用DMC 稀释,用含水HCl 1M萃取。有机相用HCl 1N和盐水洗涤,在Na2SO4上干燥和真空浓缩,提供3-(4-硝基苯甲酰基)噻唑烷-2-羧酸甲酯(220mg, 0.742mmol,68%收率)。MS/ESI+297.05[MH]+
步骤2:3-(4-硝基苯甲酰基)噻唑烷-2-羧酸(24)
将3-(4-硝基苯甲酰基)噻唑烷-2-羧酸甲酯(220mg,0.742mmol)溶 于THF(2ml)。加入LiOH 1M(1ml,1.000mmol),在室温搅拌反应 6h实现完成。反应混合物用HCl 1N稀释,用EtOAc萃取。有机相在 Na2SO4上干燥和真空浓缩,提供3-(4-硝基苯甲酰基)噻唑烷-2-羧酸(180 mg,0.638mmol,86%收率)。MS/ESI+283.03[MH]+
步骤3:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(4-硝基苯甲酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(25)
将(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯基)-2-羟 基乙基)吡啶1-氧化物(100mg,0.238mmol),3-(4-硝基苯甲酰基)噻唑烷 -2-羧酸(134mg,0.476mmol),DMAP(34.9mg,0.286mmol)和EDC(137 mg,0.714mmol)溶于DMF(1.5ml)。在室温搅拌反应2h实现完成。反 应混合物用水稀释,沉淀用水洗涤,溶于EtOAc,用HCl 1N,Na2CO3饱和溶液和盐水萃取。有机相在Na2SO4上干燥和真空浓缩,提供3,5- 二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-(3-(4-硝基苯甲 酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(130mg,0.190mmol,80% 收率)。
MS/ESI+684.07[MH]+
步骤4:4-((2S)-2-(3-(4-氨基苯甲酰基)噻唑烷-2-羰氧基)-2-(3-(环丙 基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(26)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-(3-(4-硝基苯甲酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(130mg, 0.190mmol)溶于THF(3ml)。加入氯化锡(II)二水合物(257mg,1.140 mmol),在室温搅拌混合物4天。真空除去溶剂,将粗制产品溶于EtOAc, 用Na2CO3饱和溶液稀释。将硅藻土加至两相,混合物在硅藻土垫上过滤。有机相用Na2CO3饱和溶液,盐水洗涤,在Na2SO4上干燥和真空浓 缩。粗制产品在Et2O中研磨,提供4-((2S)-2-(3-(4-氨基苯甲酰基)噻唑 烷-2-羰氧基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡 啶1-氧化物(30mg,0.046mmol,24%收率)。MS/ESI+653.8[MH]+; tR=5.80;6.00(方法1);非对映体比率30:70。
实施例5
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-1-(4-甲氧基-3-(甲磺酰基氧基)-苯甲酰基)吡咯烷-2-羰氧基)乙 基)吡啶1-(30)
方案13

步骤1:5-甲酰基-2-甲氧基苯基甲磺酸酯(28)
将3-羟基-4-甲氧基苯甲醛(27)(0.5g,3.291mmol)溶于DCM,加入 甲磺酰氯(0.376g,3.29mmol)随后是三乙胺(0.499g,4.931mmol)。反 应溶液在室温搅拌2小时,在此时间之后,将其用含水HCl 1M淬灭, 用EtOAc萃取。然后,将有机萃取物用含水K2CO3 5%洗涤,在Na2SO4上干燥,在旋蒸仪上除去溶剂。获得的所希望产品,是白色粉末(0.750g, 收率99%)。MS/ESI+231.02[MH]+
步骤2:4-甲氧基-3-(甲磺酰基氧基)苯甲酸(29)
将5-甲酰基-2-甲氧基苯基甲磺酸酯(28)(0.750g,3.261mmol)和氨 基磺酸(0.316g,3.261mmol)溶于乙酸(10ml),通过冰浴冷却。将亚氯 酸钠(0.589g,6.521mmol)溶于水(4ml)并缓慢加至冷的反应溶液。让反 应混合物在室温温热,搅拌约2小时。在此时间之后,通过UPLC-MS 分析观察到完全转化。加水(15ml)引起白色固体沉淀,将其过滤,用水洗涤数次(0.700g,收率87%)。
MS/ESI+247.02[MH]+
步骤3:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-1-(4-甲氧基-3-(甲磺酰基氧基)苯甲酰基)-吡咯烷-2-羰氧基)乙 基)吡啶1-氧化物(30)
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-((S)-1-(4-甲氧基-3-(甲磺酰基氧基)苯甲酰基)吡咯烷-2-羰氧基)乙 基)吡啶1-氧化物(30)这样制备:按照与步骤1中(实施例3)的描述类似的 程序。MS/ESI+745.0[MH]+;[αD]=31.30(c=0.34;CHCl3),tR=5.57(方 法1);非对映体比率>99:1。1H NMR(400MHz,氯仿-d) ppm 8.17(s, 2H),7.56(m,2H),7.13(m,4H),6.60(t,J=75.00Hz,1H),6.04 -6.21(m,1H),4.46-4.75(m,1H),3.95(s,3H),3.85(m,2H), 3.50-3.70(m,3H),3.25-3.31(m,1H),3.22(s,3H),2.19-2.41(m, 1H),1.74-2.03(m,3H),1.14-1.34(m,1H),0.64(d,J=7.09Hz, 2H),0.34(d,J=4.16Hz,2H)。
ppm 8.17(s, 2H),7.56(m,2H),7.13(m,4H),6.60(t,J=75.00Hz,1H),6.04 -6.21(m,1H),4.46-4.75(m,1H),3.95(s,3H),3.85(m,2H), 3.50-3.70(m,3H),3.25-3.31(m,1H),3.22(s,3H),2.19-2.41(m, 1H),1.74-2.03(m,3H),1.14-1.34(m,1H),0.64(d,J=7.09Hz, 2H),0.34(d,J=4.16Hz,2H)。
列于表5的化合物根据与步骤3中(方案13)的描述类似的程序,将 所列的适当前体与适宜的商业试剂反应,随后进行如下所指的适当纯化 步骤,而不是制备型HPLC制备。








列于表14的化合物根据与步骤3中(方案13)的描述类似的程序制 备:将所列适当前体与适宜的商业试剂反应,用DCM而不是DMF作 为溶剂,随后进行下文指出的适当纯化步骤。起始自中间体3或179制 备化合物193,194和195,作为游离碱获得,随后用含水1MNaHCO3对盐酸盐进行碱性处理,随后用DCM萃取。






实施例20
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(2-(3-(二甲基氨基甲酰基)苯基)-乙酰基)-噻唑烷-2-羰氧基) 乙基)吡啶1-氧化物(198)
方案27

步骤1:3-((1,3-二氧杂环戊烷-2-基)甲基)-N,N-二甲基苯甲酰胺 (196)
将3-((1,3-二氧杂环戊烷-2-基)甲基)苯甲酸(250mg,1.201mmol), 二甲胺盐酸盐(147mg,1.801mmol),EDC(345mg,1.801mmol)和 DMAP(513mg,4.20mmol)溶于DCM(30ml),溶液在室温搅拌1小时。 反应混合物用1N HCl洗涤两次,有机层在Na2SO4上干燥。减压除去溶 剂,提供所希望产品(244mg,1.037mmol,86%收率)MS/ESI+236.18 [MH]+。
步骤2:2-(3-(二甲基氨基甲酰基)苯基)乙酸(197)
向3-((1,3-二氧杂环戊烷-2-基)甲基)-N,N-二甲基苯甲酰胺(244mg,1.037mmol)的THF(30ml)溶液加入水(20ml),过一硫酸氢钾(1913mg, 3.11mmol)和含水37%HCl(2ml,23.92mmol),混合物在室温搅拌24 小时。加入额外的过一硫酸氢钾(1.0g,1.627mmol)和含水37%HCl(1 ml,11.96mmol),继续在室温搅拌24小时。反应混合物用水稀释(100 ml),用DCM(2x 70ml)萃取两次;经合并的有机层在Na2SO4上干燥, 蒸发至干,提供所希望产品(200mg,0.965mmol,93%收率)MS/ESI+ 208.20[MH]+。
步骤3:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(2-(3-(二甲基氨基甲酰基)苯基)乙酰基)-噻唑烷-2-羰氧基)乙 基)吡啶1-氧化物(198)
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(2-(3-(二甲基氨基甲酰基)苯基)乙酰基)-噻唑烷-2-羰氧基)乙 基)吡啶1-氧化物根据与步骤3中(方案13)实施例5中的描述类似的程序 制备,用DCM作溶剂。用聚合物支持的异氰酸酯,随后制备型HPLC 处理,将其纯化(方法2)(10%收率)。
MS/ESI+724.28[MH]+,tR=3.82分钟(方法3);非对映体比率>95:5 (1H NMR);
1H NMR(300MHz,DMSO-d6)δppm 8.52(s,2H),7.23-7.48(m, 4H),7.15(d,1H),7.06(d,1H),6.91(dd,1H),6.79-7.43(m,1H), 5.93(dd,1H),5.41(s,1H),3.93(dd,2H),3.88(d,2H),3.82(s, 2H),3.41(dd,1H),3.27(dd,1H),3.00-3.22(m,2H),2.96(br.s.,3H),2.90(br.s.,3H),1.06-1.39(m,1H),0.46-0.63(m,2H),0.06 -0.41(m,2H)
列于表15的化合物根据与方案27中的描述类似的程序制备,将所 列适当前体(用含水饱和NaHCO3碱性处理盐酸盐,随后用DCM萃取, 作为游离碱获得),随后进行下述所指的适当纯化步骤。

实施例6
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(3-(环丙基甲氧基)-5-(N-(2-吗啉代乙基)甲基磺酰氨基)苯甲酰基) 噻唑烷-2-羰氧基)乙基)吡啶1-氧化物甲酸盐(49)
方案14

步骤1:3-(环丙基甲氧基)-5-硝基苯甲酸甲酯(44)
将3-羟基-5-硝基苯甲酸甲酯(43)(1.6g,8.1mmol)溶于DMF(15 ml)。加入(溴甲基)环丙烷(2.2g,16.2mmol)和K2CO3(1.7g,12.2mmol), 在80℃搅拌混合物2hrs。反应在室温冷却,用水稀释和过滤。沉淀溶于 乙酸乙酯,有机相在Na2SO4上干燥,真空蒸发,提供1.65g的所希望 产品(收率81%)。
MS/ESI+252.08[MH]+
步骤2:3-氨基-5-(环丙基甲氧基)苯甲酸甲酯(45)
将3-(环丙基甲氧基)-5-硝基苯甲酸甲酯(44)(4.9g,19.5mmol)溶于 MeOH(200ml)和加入Pd/C 5%(1.5g,0.7mmol)。在40psi的Parr设 备上,在氢气氛下振摇溶液1hr。催化剂在硅藻土垫过滤,真空蒸发溶 剂,提供3.67g的所希望产品(收率85%)。
MS/ESI+222.11[MH]+
步骤3:3-(环丙基甲氧基)-5-(甲基磺酰氨基)苯甲酸甲酯(46)
将3-氨基-5-(环丙基甲氧基)苯甲酸甲酯(45)(1.3g,5.9mmol)溶于 吡啶(4ml)。在0℃缓慢地加入甲烷磺酰氯(0.6ml,7.7mmol),在室温搅 拌混合物2.5hrs。反应用HCl 1N水溶液稀释,产品用乙酸乙酯萃取。 有机相用HCl 1N洗涤,在Na2SO4上干燥,真空蒸发,提供1.7g的所 希望产品(收率97%)。MS/ESI+300.08[MH]+
步骤4:3-(环丙基甲氧基)-5-(N-(2-吗啉代乙基)甲基磺酰氨基)苯甲 酸甲酯(47)
将3-(环丙基甲氧基)-5-(甲基磺酰氨基)苯甲酸甲酯(46)(3g,10.02 mmol)溶于DMF(25ml)。加入4-(2-氯乙基)吗啉(4.5g,30.1mmol)和 K2CO3(2.1g,15.03mmol),在60℃搅拌混合物2hrs。反应用水稀释, 用乙酸乙酯萃取。有机相用水洗涤,在Na2SO4上干燥,真空蒸发,提 供3g的所希望产品(收率73%)。
MS/ESI+413.17[MH]+
步骤5:3-(环丙基甲氧基)-5-(N-(2-吗啉代乙基)甲基磺酰氨基)苯甲 酸(48)
将3-(环丙基甲氧基)-5-(N-(2-吗啉代乙基)-甲基磺酰氨基)苯甲酸甲 酯(47)(3g,7.3mmol)溶于MeOH(45ml)。加入NaOH 1N(9ml)水溶液, 在室温下搅拌混合物过夜。反应用HCl 1N(9ml)水溶液稀释,真空除去 溶剂,产生3.7g的所希望产品(定量的收率)。
MS/ESI+399.15[MH]+
步骤6:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(3-(环丙基甲氧基)-5-(N-(2-吗啉代乙基)甲基磺酰氨基)苯甲酰基) 噻唑烷-2-羰氧基)乙基)吡啶1-氧化物甲酸盐(49)
根据与实施例3步骤1的描述类似的程序制备化合物(49),起始自 化合物5。粗制产品通过制备型HPLC纯化,获得化合物(49),是甲酸 盐。
MS/ESI+915.3[MH]+;tR=3.37;3.43(方法2);非对映体比率= 47:53。
实施例7
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(5-((二甲基氨基)甲基)噻吩-2-羰基)噻唑烷-2-羰氧基)乙基)吡啶 1-氧化物甲酸盐(51)
方案15

步骤1:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(5-甲酰基噻吩-2-羰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(50)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(5)(200mg,0.374mmol), 5-甲酰基噻吩-2-羧酸(233mg,1.494mmol),DMAP(100mg,0.822mmol) 和EDC(358mg,1.868mmol)溶于DMF(2ml)。反应在室温下搅拌过 夜,然后用水稀释,沉淀用水洗涤,溶于AcOEt和用HCl 1N水溶液, Na2CO3饱和水溶液和盐水洗涤。有机相在Na2SO4上干燥和真空浓缩, 提供3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-(3-(5- 甲酰基噻吩-2-羰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(150mg,收率 60%)。
MS/ESI+673.04[MH]+
步骤2:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(5-((二甲基氨基)甲基)噻吩-2-羰基)噻唑烷-2-羰基氧基)乙基)吡 啶1-氧化物(51)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(3-甲酰基苯甲酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(50) (50mg,0.075mmol)溶于THF(1ml)。加入乙酸(8.58μl,0.150mmol) 和二甲胺的2M THF溶液(7.95μl,0.150mmol),混合物在室温搅拌30 分钟。加入三乙酰氧基硼氢化钠(32mg,0.150mmol),混合物在室温搅 拌3小时实现完成。反应混合物用水稀释和用AcOEt萃取。有机相在 Na2SO4上干燥和真空浓缩。粗制产品通过半制备型HPLC纯化(方法1), 提供所希望的产品,是甲酸盐(16mg,收率31%)。MS/ESI+702.2[MH] +;tR(方法2)=2.54;2.68分钟;非对映体比率=21:79
实施例8
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)-苯基磺酰基)噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(52)
方案16

将3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(6)(300mg,0.525 mmol)溶于Py(3ml,37.1mmol)。加入3-(二甲基氨基甲酰基)苯-1-磺酰 氯(156mg,0.630mmol),反应在室温搅拌4h实现完成。反应混合物用 HCl 1N水溶液稀释,用乙酸乙酯萃取。有机相用HCl 1N水溶液和盐水 洗涤,在Na2SO4上干燥和真空浓缩。粗制产品通过快速色谱法 (DCM/IsoPrOH 98/2)纯化,提供3,5-二氯-4-((S)-2-(3-(环丙基甲氧 基)-4-(二氟甲氧基)-苯基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻 唑烷-2-羰氧基)乙基)吡啶1-氧化物(250mg,0.335mmol,63.8%收率)。
MS/ESI+746.2[MH]+;[αD]=-43.30(c=0.51;CHCl3),tR=3.66(方 法1);非对映体比率=>99/1;1H NMR(400MHz,DMSO-d6) ppm 8.58 (s,2H),7.88-7.97(m,1H),7.75-7.82(m,1H),7.68-7.74(m,1 H),7.14-7.21(m,1H),7.09-7.13(m,1H),7.08(t,J=75.00Hz,1 H),6.92-6.99(m,1H),5.91-6.10(m,1H),5.54(s,1H),3.79-3.94 (m,3H),3.60-3.71(m,1H),3.41-3.51(m,1H),3.26-3.32(m, 1H),3.02(s,3H),2.92-3.00(m,1H),2.89(s,3H),2.56-2.70(m, 1H),1.20-1.27(m,1H),0.53-0.60(m,1H),0.29-0.36(m,1H)。
ppm 8.58 (s,2H),7.88-7.97(m,1H),7.75-7.82(m,1H),7.68-7.74(m,1 H),7.14-7.21(m,1H),7.09-7.13(m,1H),7.08(t,J=75.00Hz,1 H),6.92-6.99(m,1H),5.91-6.10(m,1H),5.54(s,1H),3.79-3.94 (m,3H),3.60-3.71(m,1H),3.41-3.51(m,1H),3.26-3.32(m, 1H),3.02(s,3H),2.92-3.00(m,1H),2.89(s,3H),2.56-2.70(m, 1H),1.20-1.27(m,1H),0.53-0.60(m,1H),0.29-0.36(m,1H)。
列于表6的化合物根据与方案16的描述类似的程序制备:将所列 适当前体与商业适宜的试剂反应,如果需要,随后进行报告如下的适当 纯化步骤。在MW辐射下加热(50℃,30分钟)用于合成化合物224。




















































上文表6中描述的某些化合物在下表7描述的条件下进一步结晶, 以获得一种或多种晶型。
在表7提及结晶条件(A)、(B)、(C)、(D)或(E)的情况下,使用的是 下述操作条件:
温度循环实验-(A)
在所选的各溶剂系统中制备物质浆料。大约10mg物质制浆于约 200μl溶剂中(如果使用溶解物质的透明溶液)。浆料在40℃以4小时循环 进行温度循环3天的时间段(在各4小时时间段结束之后的冷却/加热速 率是约1℃/min)。分离存在的任何固体,在分析之前让其在环境条件下 干燥。
缓慢冷却实验(B)
其这样进行:将物质在各所选溶剂系统中的饱和溶液置于2℃的环 境约3天。形成饱和溶液,将其暴露于有关的实验条件。然后,回收任 何固体物质,在分析之前让其在环境条件下干燥。
快速冷却实验-(C)
其这样进行:将物质在各所选溶剂系统中的饱和溶液置于-18℃的 环境约3天。形成饱和溶液,将其暴露于有关的实验条件。然后,回收 任何固体物质,在分析之前让其在环境条件下干燥。
蒸发实验-(D)
对上文描述的饱和混合物进行蒸发实验。在环境条件下让溶剂自由 蒸发地进行。然后回收任何固体物质,在溶剂蒸发至干之后进行分析。
抗溶剂(Anti-solvent)加入实验-(E)
对物质在各自分别的溶剂系统中饱和溶液进行抗溶剂加入实验:加 入抗溶剂直至发生沉淀。然后回收任何固体物质,在分析之前让其在环 境条件下干燥。











实施例9
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基) -2-(3-(3-(二甲基氨基甲酰基)-4-甲氧基苯基磺酰基)噻唑烷-2-羰氧基)乙 基)吡啶1-氧化物(123)
方案17

步骤1:4-((2S)-2-(3-(3-羧基-4-甲氧基苯基磺酰基)-噻唑烷-2-羰氧 基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化 物,(122)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-(噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(5)(60mg,0.112mmol)溶于 吡啶(1ml),然后在0℃加入5-(氯磺酰)-2-甲氧基苯甲酸(56ml,0.224 mmol),混合物在室温搅拌2hrs。反应用HCl 1N淬灭,产品用AcOEt 萃取。有机相用HCl 1N(x2)和盐水洗涤,然后在Na2SO4上干燥。除去 溶剂,产生70mg所希望的化合物(收率83%)。MS/ESI+749.0[MH]+; tR=5.94;6.02(方法1);非对映体比率=32:68。
步骤2:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(3-(二甲基氨基甲酰基)-4-甲氧基苯基磺酰基)噻唑烷-2-羰氧基) 乙基)吡啶1-氧化物(123)
将4-((2S)-2-(3-(3-羧基-4-甲氧基苯基磺酰基)噻唑烷-2-羰氧 基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化 物(122)(70mg,0.093mmol)溶于DMF(1ml)。加入CDI(18mg,0.112 mmol),在室温搅拌混合物30分钟。然后加入二甲胺的2M THF溶液 (300μl,0.600mmol),混合物在室温搅拌2hrs。反应用水淬灭,产品用 AcOEt萃取。有机层用水(2x)和NaCl饱和溶液洗涤,在Na2SO4上干燥, 真空蒸发。粗制产品通过制备型HPLC纯化(方法1)条件,提供70mg 所希望的化合物(收率97%)。MS/ESI+776.1[MH]+;tR(方法1)=6.40; 6.50分钟;非对映体比率=36:64。
列于表8的化合物根据与方案17的描述类似的程序制备:通过将 所列适当前体与适宜的试剂反应,随后进行下文指出的适当纯化程序。





列于表9的化合物按照与方案17的描述类似的条件制备:通过将 所列适当前体与适宜试剂反应,在步骤2中用醇亲核物质代替胺,随后 进行下文指出的纯化步骤。

实施例10
合成4-((2S)-2-(3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧基)-2-(3-(环丙 基甲氧基)-4-(二氟甲氧基)苯基)-乙基)-3,5-二氯吡啶1-氧化物(135)
方案18

步骤1:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(4-硝基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(134)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-(噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(6)(1.2g,2.24mmol) 溶于吡啶(6ml),然后在0℃加入4-硝基苯-1-磺酰氯(596mg,2.69mmol), 混合物在室温搅拌2hrs。反应用HCl 1N淬灭,产品用AcOEt萃取。有 机相用HCl 1N(x2)和盐水洗涤,然后在Na2SO4上干燥。除去溶剂,产生1.5g的所希望的化合物(收率93%)。MS/ESI+720.04[MH]+
步骤2:4-((2S)-2-(3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧基)-2-(3-(环 丙基甲氧基)-4-(二氟甲氧基)苯基)-乙基)-3,5-二氯吡啶1-氧化物(135)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(4-硝基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(134) (1g,1.388mmol)溶于THF(10ml)。加入氯化锡(II)二水合物(3.13g, 13.881mmol),混合物在室温搅拌2天。真空除去溶剂,粗制产品溶于 AcOEt和用HCl 1N稀释。将硅藻土加入乳液,在硅藻土垫上过滤混合物。有机相用HCl 1N,盐水洗涤,在Na2SO4上干燥和真空浓缩,提供 1.5g的粗制品,通过快速色谱法(DCM/IPA 97:3)纯化,获得780mg的 最终化合物(1.130mmol,收率81%)。MS/ESI+689.9[MH]+;tR=6.30; 6.40(方法1);非对映体比率=38:62
列于表10的化合物根据方案18的描述类似的程序制备:将所列的 相应前体与适宜试剂反应,随后进行下文指出的纯化步骤。




实施例11
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(4-(N-(2-吗啉代乙基)-甲基磺酰氨基)苯基磺酰基)噻唑烷-2-羰氧 基)乙基)-吡啶1-氧化物甲酸盐(143)
方案19

步骤1:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(4-(甲基磺酰氨基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物(142)
将根据与实施例9类似的程序获得的4-((2S)-2-(3-(4-氨基苯基磺酰 基)噻唑烷-2-羰氧基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙 基)-3,5-二氯吡啶1-氧化物(135)(128mg,0.185mmol)溶于DCM(1.5 ml)。加入吡啶(29.3mg,0.371mmol)和甲烷磺酰氯(36.1mg,0.315 mmol),反应在室温搅拌3hrs实现完成。反应混合物用DCM稀释和用 HCl1N萃取。有机相用HCl 1N和盐水洗涤,在Na2SO4上干燥和真空 浓缩。粗制产品通过制备型HPLC纯化(方法1),提供80mg的最终化 合物(收率56%)。MS/ESI+767.9[MH]+,tR=6.25;6.35分钟(方法1); 非对映体比率:40:60。
步骤2:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(3-(4-(N-(2-吗啉代乙基)-甲基磺酰氨基)苯基磺酰基)噻唑烷-2-羰氧 基)乙基)-吡啶1-氧化物甲酸盐(143)
将3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-(3-(4-(甲基磺酰氨基)苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物,(142)(80mg,0.104mmol)溶于DMF(1.5ml)。加入4-(2-氯乙基) 吗啉(78mg,0.520mmol)和K2CO3(17.26mg,0.125mmol),反应在45℃ 搅拌6hrs实现完成。反应混合物用水稀释和用AcOEt萃取。有机相在 Na2SO4上干燥和真空浓缩。粗制产品通过制备型HPLC纯化(方法1), 提供40mg的最终化合物,是甲酸盐(收率44%)。
MS/ESI+880.9[MH]+;5tR=5.15;5.28分钟(方法1);非对映体比 率=37:63。
实施例12
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基) -2-(3-(2-氧代-2-(噻吩-2-基)乙基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物 (144)
方案20

将3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-(噻 唑烷-2-羰氧基)乙基)吡啶1-氧化物(5)(70mg,0.131mmol)溶于DMF(1 ml)。加入K2CO3(22mg,0.157mmol)和2-溴-1-(噻吩-2-基)乙酮(80mg, 0.392mmol),反应在45℃搅拌3hrs实现完成。反应混合物用水稀释和 用AcOEt萃取。有机相在Na2SO4上干燥和真空浓缩。粗制产品通过制 备型HPLC纯化(方法1),提供50mg的最终的产品,(收率58%)。 MS/ESI+658.9[MH]+;tR(方法2)=7.10;7.24min;非对映体比率=44:56
列于表16的化合物根据与方案20的描述类似的程序制备:将所列 适当前体(在NaHCO3饱和水溶液碱性处理盐酸盐之后随后用DCM萃取 作为游离碱获得的)与适宜商业试剂反应,用CH3CN而不是DMF作溶 剂,在70℃加热,随后进行下文指出的适当纯化步骤。



实施例21
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(2-(3-(二甲基氨基甲酰基)苯基)-2-氧代乙基)-噻唑烷-2-羰氧 基)乙基)吡啶1-氧化物(261)
方案28

步骤1:乙酰基-N,N-二甲基苯甲酰胺(259)
将3-乙酰基苯甲酸(400mg,2.437mmol),二甲胺盐酸盐(238mg, 2.92mmol),EDC(701mg,3.66mmol)和DMAP(447mg,3.66mmol) 溶于DCM(80ml),所得溶液在室温下搅拌过夜。反应混合物用含水1N HCl洗涤两次和用盐水洗涤;有机相在Na2SO4上干燥,过滤和蒸发,提供所希望产品(450mg,2.353mmol,97%收率);MS/ESI+192.12[MH] +。
步骤2:3-(2-溴乙酰基)-N,N-二甲基苯甲酰胺(260)
向3-乙酰基-N,N-二甲基苯甲酰胺(450mg,2.353mmol)的DCM(20 ml)溶液滴加溴(0.121ml,2.353mmol)。所得深色溶液在室温搅拌24小 时。加入更多的溴(60ml,1164mmol)继续搅拌4小时。溶液用NaHCO3饱和水溶液洗涤两次,有机层在Na2SO4上干燥。真空除去溶剂,提供 所希望产品(526mg,1.947mmol,83%收率),将其不加进一步纯化地 用于后续步骤。MS/ESI+269.96[MH]+
步骤3:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(2-(3-(二甲基氨基甲酰基)苯基)-2-氧代乙基)-噻唑烷-2-羰氧 基)乙基)吡啶1-氧化物(261)
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(2-(3-(二甲基氨基甲酰基)苯基)-2-氧代乙基)-噻唑烷-2-羰氧 基)乙基)吡啶1-氧化物根据方案20(实施例12)的描述类似的程序制备, 起始自中间体(6)(在NaHCO3饱和水溶液碱性处理盐酸盐随后用DCM 萃取作为游离碱获得的),用CH3CN作溶剂和在60℃加热。将其通过 制备型HPLC(方法2)纯化(41%收率);MS/ESI+724.24[MH]+;tR(方 法3)=3.82分钟;非对映体比率>95:5(1H NMR);[αD]=-40.6(c=0.38, DCM)。
1H NMR(B)(300MHz,DMSO-d6)δppm 8.54(s,2H),8.00(dt, 1H),7.96(t,1H),7.67(dt,1H),7.60(t,1H),7.18(d,1H),7.13 (d,1H),6.96(dd,1H),6.81-7.41(m,1H),5.98(dd,1H),4.90(s, 1H),4.13(d,1H),4.02(d,1H),3.92(d,2H),3.45-3.55(m,1H), 3.40(dd,1H),3.28(m,0H),3.13-3.23(m,1H),3.01(br.s.,3H), 2.86-2.94(m,4H),2.82-3.05(m,2H),1.07-1.39(m,1H),0.45- 0.70(m,2H),0.18-0.44(m,2H)
列于表17的化合物根据与方案28的描述类似的程序制备:反应所 列适当前体(在NaHCO3饱和水溶液碱性处理盐酸盐随后用DCM萃取作 为游离碱获得的),在步骤3中在70℃加热,随后进行下文指出的适当 纯化步骤。

实施例13
合成4-((S)-2-((S)-3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧基)-2-(3-(环 丙基甲氧基)-4-(二氟甲氧基)-苯基)乙基)-3,5-二氯吡啶1-氧化物(150)
方案21

步骤1:3-(4-硝基苯基磺酰基)噻唑烷-2-羧酸甲酯(146)
将噻唑烷-2-羧酸甲酯盐酸盐(145)(20g;109mmol)溶于吡啶(100 ml),然后在0℃加入4-硝基苯-1-磺酰氯(27g,124mmol),搅拌混合物 3小时。在此时间之后,反应用HCl 1N淬灭以沉淀固体,将其在玻璃上 过滤,用水洗涤数次。橙色固体在丙酮中研磨,用丙酮(x2)洗涤,真空 干燥,产生27g(76%)。
MS/ESI+333.01[MH]+
步骤2:3-(4-氨基苯基磺酰基)噻唑烷-2-羧酸甲酯盐酸盐(147)
于50psi氢化3-(4-硝基苯基磺酰基)噻唑烷-2-羧酸甲酯(146) (5.74g;17.27mmol)和5%Pd/C(16g;7.547mmol)在600ml MeOH和 400ml HCl 1N中的混合物10hrs。在此时间之后,反应在C盐上过滤, 用MeOH洗涤,真空浓缩溶液,产生所希望的产品(10.47g;72%)。 MS/ESI+312.04[MH]+
步骤3:3-(4-氨基苯基磺酰基)噻唑烷-2-羧酸(148)
向3-(4-氨基苯基磺酰基)噻唑烷-2-羧酸甲酯盐酸盐(147)(7.43g, 24.57mmol)的MeOH(62ml)溶液加入LiOH 1M(62ml)。在室温下搅拌 溶液1h,然后用HCl 1M调节pH至6。减压蒸发MeOH,加入HCl 1M 直至pH=3,于0度冷却混合物3hrs,直至完全沉淀。过滤固体,用水 洗涤,于45度在真空中干燥,提供6.44g的所希望产品(收率91%)。
MS/ESI+289.02[MH]+
步骤4:4-((2S)-2-(3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧基)-2-(3-(环 丙基甲氧基)-4-(二氟甲氧基)苯基)-乙基)-3,5-二氯吡啶1-氧化物(149)
向(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟 基乙基)吡啶1-氧化物(1.55g,3.69mmol)的DMF(20ml),EDC(2.83g, 14.77mmol)溶液加入3-(4-氨基苯基磺酰基)噻唑烷-2-羧酸(148)(1.81g, 6.28mmol)。于0度冷却混合物和加入DMAP(0.541g,4.43mmol)。在 -20度搅拌混合物20分钟,然后让其温热至室温,搅拌3hrs。将反应混 合物倾至水中(500ml),沉淀通过过滤分离。过滤的固体用水洗涤(150 ml),在真空中干燥于45度,提供2.49g的所希望产品(收率98%)。 MS/ESI+689.8[MH]+
步骤5:4-((S)-2-((S)-3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧 基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-乙基)-3,5-二氯吡啶1-氧 化物(150)
于50度将4-((2S)-2-(3-(4-氨基苯基磺酰基)噻唑烷-2-羰氧 基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化 物(149)(2.49g,3.61mmol)溶于EtOH(35ml),将甲磺酸的EtOH 10% w/w溶液加入(3.47g,3.61mmol)以实现成盐。滤出甲磺酸盐,再次从热 EtOH(25ml)结晶。将盐溶于CH2Cl2(40ml),加入NaHCO3饱和溶胶(30 ml)。在室温搅拌混合物30分钟,然后分离两相。有机相在Na2SO4上干 燥和减压蒸发,提供固体,其从热EtOH(25ml)再次结晶,产生800mg 的所希望产品。MS/ESI+689.8[MH]+;tR(方法1)=6.37分钟;[αD]= +38.57(c=0.49;CHCl3)。1H NMR(400MHz,DMSO-d6)δppm 8.58(s, 2H),7.42-7.52(m,2H),7.14-7.20(m,1H),7.05-7.12(m,2H), 6.90-6.97(m,1H),6.60-6.69(m,2H),6.15-6.24(s,2H),5.96-6.05 (m,1H),5.29(s,1H),3.86-3.95(m,2H),3.68-3.80(m,1H), 3.52(d,J=47.63Hz,1H),3.38-3.48(m,1H),3.29(m,1H),2.84- 2.99(m,1H),2.53-2.60(m,1H),1.22-1.36(m,1H),0.46-0.63(m, 2H),0.28-0.40(m,2H)。
列于表11的化合物根据与方案21(步骤1-4)的描述类似的程序制 备,随后通过快速色谱法用DCM/正己烷/异PrOH/EtOH=55/40/4/1洗脱 纯化化合物(131)。

实施例14
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-1-(1-甲基-1H-咪唑-2-基磺酰基)吡咯烷-2-羰氧基)乙基)吡啶1- 氧化物(155)
方案22

步骤1:(S)-1-(1-甲基-1H-咪唑-2-基磺酰基)吡咯烷-2-羧酸甲酯(153)
将(S)-吡咯烷-2-羧酸甲酯(152)(50mg;0.387mmol)溶于吡啶(1 ml),然后在0℃加入1-甲基-1H-咪唑-2-磺酰氯(70mg,0.387mmol), 搅拌混合物3小时。在此时间之后,反应用HCl 1N淬灭,用AcOEt萃 取两次。有机相在Na2SO4上干燥,真空蒸发,产生50mg(收率47%)。
MS/ESI+274.08[MH]+
步骤2:(S)-1-(1-甲基-1H-咪唑-2-基磺酰基)吡咯烷-2-羧酸(154)
在微波辐射下于100度,将(S)-1-(1-甲基-1H-咪唑-2-基磺酰基)吡咯 烷-2-羧酸甲酯(153)(50mg,0.183mmol)的HCl/二噁烷4M(1ml)溶液 反应30分钟。然后减压蒸发二噁烷,产生40mg的所希望的化合物(收 率84%)。
MS/ESI+260.06[MH]+
步骤3:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-1-(1-甲基-1H-咪唑-2-基磺酰基)吡咯烷-2-羰氧基)乙基)吡啶1- 氧化物(155)
向(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟 基乙基)吡啶1-氧化物(20mg,0.048mmol)溶于DMF(1ml)的溶液加入 EDC(9mg,0.048mmol)和3-(4-氨基苯基磺酰基)噻唑烷-2-羧酸(154)(40 mg,0.154mmol)。于0度冷却混合物,加入DMAP(6mg,0.048mmol)。 在-20度搅拌混合物20分钟,然后让其温热至室温,搅拌3hrs。将反应 混合物倾至水中,用AcOEt(x3)萃取。有机相在Na2SO4上干燥,真空 蒸发,提供30mg的所希望产品(收率95%)。MS/ESI+661.3[MH]+;tR (方法2)=3.60分钟;非对映体比率=99:1;1H NMR(400MHz, DMSO-d6) ppm 8.51(s,2H),7.47(m,1H),7.18(d,J=7.94Hz,1H), 7.13(d,J=1.76Hz,1H),7.08(d,J=4.41Hz,2H),6.98(dd,J=8.38, 1.76Hz,1H),6.00(dd,J=9.48,4.63Hz,1H),4.47(dd,J=8.82, 4.41Hz,1H),3.92(dd,J=7.06,1.76Hz,2H),3.84(s,3H),3.37- 3.55(m,3H),3.24(dd,J=14.11,4.41Hz,1H),2.22-2.37(m,1H), 1.83-1.95(m,1H),1.65-1.82(m,2H),1.12-1.30(m,1H),0.49- 0.64(m,2H),0.28-0.42(m,2H)。
ppm 8.51(s,2H),7.47(m,1H),7.18(d,J=7.94Hz,1H), 7.13(d,J=1.76Hz,1H),7.08(d,J=4.41Hz,2H),6.98(dd,J=8.38, 1.76Hz,1H),6.00(dd,J=9.48,4.63Hz,1H),4.47(dd,J=8.82, 4.41Hz,1H),3.92(dd,J=7.06,1.76Hz,2H),3.84(s,3H),3.37- 3.55(m,3H),3.24(dd,J=14.11,4.41Hz,1H),2.22-2.37(m,1H), 1.83-1.95(m,1H),1.65-1.82(m,2H),1.12-1.30(m,1H),0.49- 0.64(m,2H),0.28-0.42(m,2H)。
实施例22
合成3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-羟基苯基)-2-((S)-3-(3-(二 甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(263)
方案47

将3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(化合物52)(200mg,0.268mmol)溶于2,2,2-三氟乙酸(2 ml,0.268mmol),溶液在室温搅拌过夜。反应混合物用DCM稀释,真 空浓缩(2X),提供粗制品,将其通过制备型HPLC(方法1)纯化,提供 3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-羟基苯基)-2-((S)-3-(3-(二甲基氨基 甲酰基)-苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(90mg,0.130 mmol,49%收率)。MS/ESI+661.3[MH]+;tR(方法2)=2.49;非对映体 比率=99:1;1H NMR(400MHz,DMSO-d6)δppm10.05(s,1H),8.58(s, 2H),7.85 7.97(m,2H),7.67-7.80(m,2H),7.11(d,J=8.38Hz,1H),7.04(t,J=75.00Hz,1H),6.94(d,J=2.21Hz,1H),6.83(m,1 H),5.86-6.02(m,1H),5.47(s,1H),3.78-3.92(m,1H),3.56-3.68 (m,1H),3.37-3.49(m,1H),3.20-3.28(m,1H),2.93-3.09(m, 4H),2.89(s,3H),2.66(m 1H)。
实施例23
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(环丙基甲基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(264)
方案30

将3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(6)(150mg,0.262 mmol)溶于MeOH(10ml),加入环丙烷甲醛(19.60μl,0.262mmol),随 后加入氰基硼氢化钠(33.0mg,0.525mmol)。所得混合物在室温搅拌2 小时。减压除去挥发物,残余物通过制备型HPLC(方法2)纯化。需要 提供硅胶上的快速色谱法(DCM/MeOH=99/1)进一步纯化,提供3,5-二 氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-((S)-3-(环丙基甲 基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(25mg,0.042mmol,16.17%收 率)。MS/ESI+588.94[MH]+;tR(方法3)=3.54;非对映体比率=>95:5(1H NMR);αD]=-30.1(c=0.31,DCM);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.58(s,2H),7.19(d, 1H),7.13(d,1H),6.97(dd,1H),7.08(t,1H),5.95(dd,1H), 4.96(s,1H),3.93(d,2H),3.38-3.56(m,1H),3.40(dd,1H),3.26 (dd,1H),2.98-3.17(m,1H),2.76-2.95(m,2H),2.31(dd,1H), 2.17(dd,1H),1.14-1.35(m,1H),0.72-0.98(m,1H),0.53-0.66(m, 2H),0.43-0.53(m,2H),0.26-0.42(m,2H),-0.03-0.19(m,2H)
列于表18的化合物根据与方案30的描述类似的程序制备:将所列 适当前体与商业适宜的试剂反应,随后进行报告如下的适当纯化步骤。




实施例24
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-脲基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物 (269)
方案31

在10℃,将4-((S)-2-((S)-3-(3-氨基苯基磺酰基)噻唑烷-2-羰氧 基)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化 物140(140,如实施例10的描述制备,210mg,0.304mmol)溶于AcOH (4ml)和水(2ml)的混合物,立即加入氰酸钾(99mg,1.216mmol)。反应 在10℃搅拌1小时。加水(30ml),过滤收集沉淀,通过制备型HPLC(方 法2)纯化,提供3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-脲基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物 (136mg,0.185mmol,61.0%收率)。MS/ESI+733.05[MH]+;tR(方法 3)=5.39;非对映体比率>95:5(1H NMR);[αD]=-78.85(c=0.4,DCM);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.95(s,1H),8.58(s,2 H),8.08(t,1H),7.62(ddd,1H),7.49(t,1H),7.36(dt,1H),7.18 (d,1H),7.12(d,1H),6.96(dd,1H),7.08(t,1H),6.02(dd,1H), 6.01(br.S.,2H),5.30(s,1H),3.91(d,2H),3.61-3.82(m,2H), 3.49-3.54(m,1H),3.31(dd,1H),2.99(dt,1H),2.73(dt,1H), 1.09-1.38(m,1H),0.48-0.62(m,2H),0.21-0.45(m,2H)
实施例25
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-(羟基甲基)-苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物(271)
方案32

步骤1:3-(羟基甲基)苯-1-磺酰氯(270)
向在0℃冷却的3-(氯磺酰)苯甲酸(0.300g,1.360mmol)的无水THF (6ml)溶液,加入BH3*THF配合物的1M THF溶液(5.44ml,5.44mmol), 让所得混合物温热至室温和搅拌过夜。加入额外的BH3*THF配合物1M THF溶液(1.360ml,1.360mmol),在室温继续搅拌3天。混合物仔细地 用2M HCl淬灭,用盐水稀释,用EtOAc萃取两次。经合并的有机层用 盐水洗涤,在硫酸钠上干燥。除去溶剂,粗制品通过硅胶柱(石油醚: EtOAc=70:30)过滤纯化,提供3-(羟基甲基)苯-1-磺酰氯(0.086g,0.416 mmol,30.6%收率)。MS/ESI+不可检测的[MH]+。
步骤2:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-(羟基甲基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物(271)
向在0℃冷却的3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧 基)苯基)-2-((S)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(6)(0.183g, 0.320mmol)的DCM(5ml)、CH3CN(2ml)和吡啶(0.078ml,0.960mmol) 溶液,加入3-(羟基甲基)苯-1-磺酰氯(0.086g,0.416mmol)的DCM(2ml) 溶液,将所得混合物温热至RT并搅拌。减压除去挥发物;残余物通过 制备型HPLC纯化(方法2,在中性条件下,不加TFA),冻干收集的级 分,提供3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(3-(羟基甲基)苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧 化物(0.100g,0.142mmol,44.3%收率);MS/ESI+705.14[MH]+;tR(方 法3)=3.78;非对映体比率>95:5(1H NMR);[αD]=-65.67(c=0.42; MeOH);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.58(s,2H),7.83(t,1 H),7.73(dt,1H),7.68(dt,1H),7.61(t,1H),7.19(d,1H),7.12 (d,1H),6.97(dd,1H),7.08(t,1H),6.03(dd,1H),5.44(t,1H), 5.38(s,1H),4.62(d,2H),3.91(d,2H),3.76(dt,1H),3.67(dt, 1H),3.47(dd,1H),3.31(dd,1H),2.98(dt,1H),2.68(dt,1H), 1.03-1.38(m,1H),0.49-0.67(m,2H),0.15-0.45(m,2H)。
实施例26
合成4-((2S)-2-(2-(3-苯甲酰基噻唑烷-2-基)乙酰氧基)-2-(3-(环丙基 甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(274)
方案33

步骤1:2-(3-苯甲酰基噻唑烷-2-基)乙酸乙酯(272)
将2-(噻唑烷-2-基)乙酸乙酯(0.100g,0.571mmol)(按照描述于J.Chem.Soc.Perkin Trans.I,1987,1845-1851的合成方案制备),苯甲 酸(0.084g,0.685mmol),EDC(0.219g,1.141mmol)和DMAP(0.070g, 0.571mmol)在DCM(10ml)中的混合物在室温搅拌1小时。混合物用 DCM稀释,用含水1N HCl,1N NaHCO3和盐水洗涤;有机相在硫酸钠 上干燥,除去溶剂。残余物通过快速色谱法在硅胶柱(石油醚:EtOAc= 80:20至70:30)上纯化,产生2-(3-苯甲酰基噻唑烷-2-基)乙酸乙酯(0.085 g,0.304mmol,53%收率)。MS/ESI+280.0[MH]+。
步骤2:2-(3-苯甲酰基噻唑烷-2-基)乙酸(273)
向2-(3-苯甲酰基噻唑烷-2-基)乙酸乙酯(0.083g,0.297mmol)的二 噁烷(4ml)溶液加入含水37%HCl(4ml),混合物在室温搅拌20小时。 减压除去挥发物,产生粗制2-(3-苯甲酰基噻唑烷-2-基)乙酸(0.074g, 0.294mmol,99%收率),其不加纯化地使用。MS/ESI+251.9[MH]+。
步骤3:4-((2S)-2-(2-(3-苯甲酰基噻唑烷-2-基)乙酰氧基)-2-(3-(环丙 基甲氧基)-4-(二氟甲氧基)苯基)乙基)-3,5-二氯吡啶1-氧化物(274)
将2-(3-苯甲酰基噻唑烷-2-基)乙酸(0.074g,0.294mmol),(S)-3,5- 二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟基乙基)吡啶1-氧 化物(0.103g,0.245mmol),EDC(0.141g,0.736mmol)和DMAP(0.030 g,0.245mmol)在DCM(10ml)中的混合物在室温搅拌2小时。混合物 用DCM稀释,用1N HCl,1N NaHCO3和盐水洗涤;有机相在硫酸钠 上干燥,除去溶剂。粗制品通过制备型HPLC(方法2),随后硅胶柱上 的快速色谱法(DCM:MeOH=99:1)纯化,产生标题化合物,是非对 映体混合物(0.065g,0.099mmol,40.5%收率);MS/ESI+653.19[MH]+; tR(方法3)=3.92;非对映体比率=1:1(1H NMR)。
列于表19的化合物根据与方案33的描述类似的程序制备:使用适 宜的试剂,随后进行报告如下的适当纯化步骤。
表19

实施例27
合成3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(2-(3-(苯基磺酰基)噻唑烷-2-基)乙酰氧基)乙基)吡啶1-氧化物(278)
方案48

步骤1:2-(3-(苯基磺酰基)噻唑烷-2-基)乙酸乙酯(276)
向在0℃冷却的2-(噻唑烷-2-基)乙酸乙酯(0.100g,0.571mmol)(按 照J.Chem.Soc.Perkin Trans.I,1987,1845-1851描述的合成方案制备) 的吡啶(4ml)溶液,加入苯磺酰氯(0.088ml,0.685mmol),反应在室温 搅拌2小时。混合物在EtOAc与1N HCl间分配;有机相用1N HCl和 盐水洗涤,在硫酸钠上干燥。除去溶剂,粗制品通过快速色谱法在硅胶 上纯化(石油醚:EtOAc=90:10至80:20),提供2-(3-(苯基磺酰基)噻 唑烷-2-基)乙酸乙酯(0.096g,0.304mmol,53.3%收率)。MS/ESI+316.0 [MH]+。
步骤2:2-(3-(苯基磺酰基)噻唑烷-2-基)乙酸(277)
向2-(3-(苯基磺酰基)噻唑烷-2-基)乙酸乙酯(0.096g,0.304mmol) 的二噁烷(5ml)溶液加入含水37%HCl(5ml),混合物在室温搅拌25小 时。减压除去挥发物,提供2-(3-(苯基磺酰基)噻唑烷-2-基)乙酸(0.083g, 0.289mmol,95%收率),其不加纯化地使用。MS/ESI+310.0[Mna]+。
步骤3:3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(2-(3-(苯基磺酰基)噻唑烷-2-基)乙酰氧基)乙基)吡啶1-氧化物(278)
将2-(3-(苯基磺酰基)噻唑烷-2-基)乙酸(0.083g,0.289mmol), (S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟基乙基)吡 啶1-氧化物(0.101g,0.241mmol),EDC(0.046g,0.241mmol)和DMAP (0.029g,0.241mmol)在DCM(10ml)中的混合物在室温搅拌1小时。混 合物用DCM稀释,用1N HCl,1N NaHCO3和盐水洗涤;有机相在硫 酸钠上干燥,除去溶剂。粗制品通过制备型HPLC(方法2)纯化,提供 3,5-二氯-4-((2S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-(2-(3-(苯基 磺酰基)噻唑烷-2-基)乙酰氧基)乙基)吡啶1-氧化物(0.122g,0.177mmol, 73.5%收率);MS/ESI+689.14[MH]+;tR(方法3)=4.09;非对映体比率 =1:1(1H NMR)。
列于表20的化合物根据与方案48的描述类似的程序制备:使用适 宜的试剂,随后进行报告如下的适当纯化步骤。
表20

实施例28
(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)-苯 基)-2-(1-(3-(二甲基氨基甲酰基)苯基磺酰基)氮杂环丁烷-3-羰氧基)乙基) 吡啶1-氧化物(281)
方案34

步骤1:1-(3-(二甲基氨基甲酰基)苯基磺酰基)氮杂环丁烷-3-羧酸 (280)
向在0℃冷却的氮杂环丁烷-3-羧酸(100mg,0.989mmol)在THF(6 ml)和含水1MNa2CO3(6ml,6.00mmol)混合物中的悬浮液,加入3-(二 甲基氨基甲酰基)苯-1-磺酰氯(269mg,1.088mmol),反应在0℃搅拌1 小时。混合物用Et2O萃取,弃去有机层。水层通过加入固体KHSO4 (pH=3)仔细地酸化,用EtOAc萃取两次。经合并的有机层在Na2SO4上 干燥,蒸发至干,产生粗制1-(3-(二甲基氨基甲酰基)苯基磺酰基)氮杂环 丁烷-3-羧酸(255mg,0.816mmol,83%收率),其不加纯化地使用。
MS/ESI+313.12[MH]+
步骤2:(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-(1-(3-(二甲基氨基甲酰基)-苯基磺酰基)氮杂环丁烷-3-羰氧基)乙基) 吡啶1-氧化物(281)
将1-(3-(二甲基氨基甲酰基)苯基磺酰基)氮杂环丁烷-3-羧酸(255 mg,0.816mmol),(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基) 苯基)-2-羟基乙基)吡啶1-氧化物(175mg,0.416mmol),EDC(96mg, 0.500mmol)和DMAP(61.0mg,0.500mmol)的DCM(30ml)溶液在室 温下搅拌过夜。加入更多的EDC(80mg,0.416mmol)和DMAP(61.0 mg,0.500mmol),再继续搅拌2小时。反应混合物用含水1N HCl然后 用含水1M Na2CO3洗涤两次,有机相在Na2SO4上干燥,减压除去溶剂。 残余物通过制备型HPLC(方法2)纯化,提供(S)-3,5-二氯-4-(2-(3-(环丙 基甲氧基)-4-(二氟甲氧基)苯基)-2-(1-(3-(二甲基氨基甲酰基)苯基磺酰基) 氮杂环丁烷-3-羰氧基)乙基)吡啶1-氧化物(192mg,0.269mmol,64.5% 收率);MS/ESI+714.16[MH]+;[αD]=-14.3(c=0,37,DCM);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.56(s,2H),7.68-7.90 (m,4H),7.16(d,1H),7.02(d,1H),6.85(dd,1H),7.07(t,1H), 5.82(dd,1H),3.85-4.06(m,2H),3.89(d,2H),3.71(dd,1H), 3.56(dd,1H),3.42-3.49(m,2H),3.14(dd,1H),3.01(br.S.,3H), 2.86(br.S.,3H),1.02-1.37(m,1H),0.50-0.78(m,2H),0.07-0.50 (m,2H)
列于表21的化合物根据与方案34的描述类似的程序制备:将所列 的适当氨基酸前体与商业适宜的试剂反应,随后进行报告如下的适当纯 化步骤,如果需要。为了制备化合物283和化合物284,步骤1用水作 溶剂来完成。




实施例29
合成3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(2-吗啉代乙基磺酰基)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物 (287)
方案35

步骤1:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(乙烯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(286)
向在0℃冷却的3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧 基)苯基)-2-((S)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(6)(500mg, 0.874mmol)的DCM(10ml)溶液加入吡啶(212μl,2.62mmol)和2-氯乙 烷磺酰氯(137μl,1.312mmol),让混合物温热至室温并搅拌2小时。在 0℃加入额外的吡啶(707μl,8.74mmol)和2-氯乙烷磺酰氯(137μl,1.312 mmol),混合物在室温反应6小时。混合物用DCM稀释,用1N HCl 和盐水洗涤;有机层在Na2SO4上干燥和真空除去溶剂。所得粗制品通 过快速色谱法(DCM/MeOH=98/2)在硅胶上纯化,产生3,5-二氯 -4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-((S)-3-(乙烯基磺酰 基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(360mg,0.576mmol,66%收 率);MS/ESI+624.9[MH]+。
步骤2:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(2-吗啉代乙基磺酰基)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物 (287)
向3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(乙烯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(110 mg,0.176mmol)的EtOH(5ml)溶液加入吗啉(35.2μl,0.352mmol),混 合物在室温反应1小时。减压除去溶剂,粗制品通过制备型HPLC(方 法3)纯化,产生3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-3-(乙烯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物,是三 氟乙酸盐;MS/ESI+712.14[MH]+;tR(方法3)=3.16;非对映体比率>95:5 (1H NMR);[αD]=-126.6(c=0.23DCM);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.55(s,2H),7.19(d, 1H),7.11(d,1H),6.97(dd,1H),7.08(t,1H),6.03(dd,1H), 5.58(s,1H),3.94-4.02(m,1H),3.91(d,2H),3.61-3.74(m,1H), 3.45(dd,1H),3.31(dd,1H),3.06-3.81(m,12H),3.00-3.25(m, 2H),1.03-1.35(m,1H),0.47-0.70(m,2H),0.10-0.45(m,2H)
列于表22的化合物根据与方案35的描述类似的程序制备:将所列 适当前体与商业适宜的试剂反应,随后进行适当的纯化步骤。

实施例30:
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-1-(2-(苯基磺酰基)乙基)吡咯烷-2-羰氧基)乙基)吡啶1-氧化物 (291)
方案36

步骤1:(S)-苄基1-(2-(苯基磺酰基)乙基)吡咯烷-2-羧酸酯(289)
将(S)-苄基吡咯烷-2-羧酸酯盐酸盐(250mg,1.034mmol),乙烯基 磺酰基苯(261mg,1.551mmol)和TEA(0.216ml,1.551mmol)的EtOH (15ml)溶液在室温搅拌24小时。然后,反应混合物用DCM(50ml)稀释, 用含水饱和NH4Cl洗涤两次。有机层在Na2SO4上干燥,真空除去溶剂。 将残余物溶于DCM(15ml)和EtOH(15ml),加入PS-氨基丁三醇(游离 -NH2基团:4.7mmol/g,0.5g,0.35mmol)。悬浮液在室温搅拌3天。 滤出树脂,溶液蒸发至干,提供所希望产品(379mg,1.01mmol,98% 收率)。MS/ESI+374.10[MH]+
步骤2:(S)-1-(2-(苯基磺酰基)乙基)吡咯烷-2-羧酸(290)
将10%Pd/C(10.80mg,0.101mmol)的水(2ml)悬浮液加入(S)-苄 基1-(2-(苯基磺酰基)乙基)吡咯烷-2-羧酸酯(379mg,1.015mmol)的 MeOH(15ml)溶液。在Parr设备中于30psi,在室温将混合物氢化1 小时。滤出催化剂,将所得透明溶液蒸发至干,提供所希望产品(272mg, 0.860mmol,95%收率)。MS/ESI+284.02[MH]+。
步骤3:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-1-(2-(苯基磺酰基)乙基)吡咯烷-2-羰氧基)乙基)吡啶1-氧化物 (291)
将(S)-3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-羟 基乙基)吡啶1-氧化物(150mg,0.357mmol),(S)-1-(2-(苯基磺酰基)乙基) 吡咯烷-2-羧酸(152mg,0.535mmol),EDC(103mg,0.535mmol)和 DMAP(21.80mg,0.178mmol)的DCM(30ml)溶液在室温搅拌1小时。 反应混合物用1N HCl洗涤两次,在Na2SO4上干燥,蒸发至干。残余物通过制备型HPLC(方法2)纯化,将收集的级分蒸发至干,再溶解于 DCM,通过PL-HCO3柱(200mg,0.36毫摩尔)洗脱。将洗脱液蒸发至 干,提供标题化合物(110mg,0.160mmol,45%收率);[αD]=-33.8 (c=0.44,DCM);MS/ESI+685.42[MH]+;tR=3.15分钟(方法3);非对 映体比率>95:5(1H NMR);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.55(s,2H),7.83-7.98 (m,2H),7.69-7.82(m,1H),7.53-7.69(m,2H),7.17(d,1H), 7.06(d,1H),6.93(dd,1H),7.06(t,1H),5.92(dd,1H),3.80-4.04 (m,2H),3.43-3.58(m,1H),3.41(dd,1H),3.23(dd,1H),2.61 -3.09(m,5H),2.25-2.44(m,1H),1.84-2.08(m,1H),1.37-1.78 (m,3H),1.09-1.33(m,1H),0.48-0.73(m,2H),0.25-0.43(m, 2H)
实施例31:
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯 基)-2-((S)-1-(2-苯基乙酰基)吡咯烷-2-羰氧基)乙基)吡啶1-氧化物(292)
方案37

在0℃,逐滴向3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基) 苯基)-2-((S)-吡咯烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(6)(200mg, 0.361mmol)和吡啶(292μl,3.61mmol)的无水DCM(5ml)溶液加入苯基 -乙酰氯(84mg,0.542mmol)的无水DCM(1ml)溶液,让混合物温热至 室温并搅拌2小时。加入EtOAc(20ml),混合物用含水5%柠檬酸和盐水洗涤;有机相在Na2SO4上干燥,过滤和蒸发。残余物通过快速色谱 法(DCM/MeOH=95/5)在硅胶上纯化。需要通过制备型HPLC(方法2) 进一步纯化,提供3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基) 苯基)-2-((S)-1-(2-苯基乙酰基)吡咯烷-2-羰氧基)乙基)吡啶1-氧化物(35 mg,0.055mmol,15.25%收率);MS/ESI+635.23[MH]+;tR=3.91分 钟(方法3);非对映体比率>95:5(1H NMR);[αD]=-28.26(c=0.23, MeOH);
1H NMR(B)(300MHz,DMSO-d6 353K)δppm 8.40(s,2H),7.16 -7.39(m,5H),7.13(d,1H),7.08(d,1H),6.89-6.95(m,1H), 6.97(t,1H),5.85-6.08(m,1H),4.26-4.45(m,1H),3.91(d,2H), 3.66(br.S.,2H),3.47-3.63(m,3H),3.27(dd,1H),2.01-2.25(m, 1H),1.83-1.99(m,1H),1.49-1.83(m,2H),1.07-1.26(m,1H), 0.44-0.69(m,2H),0.19-0.40(m,2H)
实施例32:
3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苄基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(295)
方案38

步骤1:(3-(二甲基氨基甲酰基)苯基)甲磺酸(293)
向3-(氯甲基)-N,N-二甲基苯甲酰胺(1.15g,5.82mmol)的水(30ml) 悬浮液加入亚硫酸钠(1.100g,8.73mmol),混合物在100℃加热1小时。 真空除去溶剂,将残余物悬浮于MeOH(40ml)。加入HCl的4M二噁 烷(5ml)溶液,滤出不溶的无机盐。蒸发滤液至干,残余物通过与CH3CN 数次研磨纯化,提供(3-(二甲基氨基甲酰基)苯基)甲磺酸(1.04g,4.27 mmol,73.5%收率)。MS/ESI+243.95[MH]+。
步骤2:(3-(二甲基氨基甲酰基)苯基)甲烷磺酰氯(294)
向(3-(二甲基氨基甲酰基)苯基)甲磺酸(500mg,2.055mmol)的 DCM(40ml)悬浮液加入亚硫酰氯(0.900ml,12.33mmol),所得混合物 在室温搅拌20小时。将反应混合物倾至碎冰中,分离有机相,在Na2SO4上干燥。真空除去溶剂,提供希望的产品(357mg,1.364mmol,66%收 率)。MS/ESI+261.96[MH]+。
步骤3:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苄基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(295)
根据与方案16(实施例8)的描述类似的程序,获得3,5-二氯 -4-((S)-2-(3-(环丙基甲氧基)-4-(二氟-甲氧基)苯基)-2-((S)-3-(3-(二甲基氨 基甲酰基)苄基磺酰基)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物。将其用聚 合物支持的异氰酸酯捕获剂,随后用制备型HPLC(方法2)处理进行纯 化(20%收率);MS/ESI+760.17[MH]+,tR=3.78分钟(方法3);非对映 体比率>95:5(1H NMR);[αD]=-15.8(c=3.0,DCM);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.53(s,2H),7.38-7.55 (m,4H),7.17(d,1H),7.08(d,1H),6.94(dd,1H),7.07(t,1H), 6.00(dd,1H),5.27(s,1H),4.67(d,1H),4.61(d,1H),3.85-3.98 (m,2H),3.74-3.85(m,1H),3.51-3.66(m,1H),3.36-3.50(m, 2H),3.20-3.26(m,1H),3.04-3.14(m,1H),2.99(br.S.,3H), 2.92(br.S.,3H),1.04-1.42(m,1H),0.46-0.66(m,2H),0.20-0.41 (m,2H)
列于表23的化合物根据与方案38的描述类似的程序获得:使得用 饱和NaHCO3水溶液碱性处理盐酸盐之后作为游离碱获得且随后用 DCM萃取的适当前体进行反应。纯化步骤描述如下。

实施例33:
合成3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(299)
方案39

步骤1:4-((S)-2-((S)-3-(叔-丁氧基羰基)噻唑烷-2-羰氧基)-2-(4-(二 氟甲氧基)-3-甲氧基苯基)乙基)-3,5-二氯吡啶1-氧化物(297)
将(S)-3-(叔-丁氧基羰基)噻唑烷-2-羧酸(479mg,2.052mmol), (S)-3,5-二氯-4-(2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-羟基乙基)吡啶1-氧 化物(164)(根据与方案23的描述类似的程序制备),(650mg,1.710 mmol),EDC(492mg,2.56mmol)和DMAP(313mg,2.56mmol)的 DCM(60ml)溶液在室温搅拌3小时。反应混合物用DCM稀释,用含水 1N HCl洗涤两次;有机层在Na2SO4上干燥,蒸发至干,提供所希望的 化合物(定量收率)。MS/ESI+595.24[MH]+。
步骤2:3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-((S)-噻 唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(298)
向在0℃冷却的4-((S)-2-((S)-3-(叔-丁氧基羰基)噻唑烷-2-羰氧 基)-2-(4-(二氟甲氧基)-3-甲氧基苯基)乙基)-3,5-二氯吡啶1-氧化物(1.710 mmol)的EtOAc(10ml)溶液,加入HCl的4M EtOAc(10ml,40.0mmol) 溶液,所得混合物在室温搅拌2小时。加入更多的HCl的4M EtOAc(10 ml,40.0mmol)溶液,在0℃再搅拌溶液额外的2小时,使其完全转化。将溶液减压浓缩至10ml(浴温:10℃;分压:8psi),然后加入iPr2O(20 ml),产品沉淀,是粘性树胶状固体。在让固体沉降之后,通过吸取除去 溶剂。在室温真空干燥残余物,提供3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3- 甲氧基苯基)-2-((S)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(0.890g, 1.674mmol,97%收率),将其不加任意额外纯化地用于后续步骤。 MS/ESI+494.97[MH]。
步骤3:3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(299)
向在0℃冷却的3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯 基)-2-((S)-噻唑烷-2-羰氧基)乙基)吡啶1-氧化物盐酸盐(350mg,0.658 mmol)的吡啶(6ml)溶液,滴加3-(二甲基氨基甲酰基)苯-1-磺酰氯(245 mg,0.987mmol)的DCM(3ml)溶液,反应在0℃搅拌1小时。混合物 用DCM(30ml)稀释,用含水1N HCl洗涤两次;有机层在Na2SO4上干 燥,真空除去溶剂。残余物通过制备型HPLC(方法2),随后在硅胶 (DCM/MeOH=97/3)上快速色谱法纯化,提供3,5-二氯-4-((S)-2-(4-(二氟 甲氧基)-3-甲氧基苯基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻唑 烷-2-羰氧基)乙基)吡啶1-氧化物(147mg,0.208mmol,31.6%收率); MS/ESI+705.97[MH]+,tR=3.28分钟(方法3);非对映体比率=95:5(1H NMR);[αD]=-43.1(c=0.57,DCM);
1H NMR(300MHz,DMSO-d6)δppm 8.57(s,2H),7.94(dt,1H), 7.90(t,1H),7.77(dt,1H),7.71(t,1H),7.18(d,1H),7.15(d,1 H),6.97(dd,1H),7.07(t,1H),6.04(dd,1H),5.54(s,1H),3.84 (s,3H),3.75-3.94(m,1H),3.66(dt,1H),3.48(dd,1H),3.33(dd, 1H),3.02(br.S.,3H),2.98(dd,1H),2.90(br.S.,3H),2.66(dt, 1H)
列于表24的化合物根据与方案39的描述类似的程序制备:将所列 适当醇与商业适宜的试剂反应,随后进行报告如下的适当纯化步骤。化 合物303作为从非对映异构体混合物的第二洗脱的非对映异构体而获 得。



实施例34
合成3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-((S)-1-(苯 基磺酰基)吡咯烷-2-羰氧基)乙基)吡啶1-氧化物(化合物305)
方案40

步骤1:(S)-1-(苯基磺酰基)吡咯烷-2-羧酸(304)
将苯磺酰氯(4.03ml,31.3mmol)加至(S)-吡咯烷-2-羧酸(3g,26.1 mmol)在THF(50ml)和含水1M Na2CO3(60ml,60.0mmol)中的冷却悬 浮液(0℃),反应在0℃搅拌1小时。混合物用Et2O萃取两次,弃去有机 层。加入固体KHSO4仔细地酸化水相至pH=3,用EtOAc萃取两次。 经合并的有机层在Na2SO4上干燥,蒸发至干,提供(S)-1-(苯基磺酰基) 吡咯烷-2-羧酸(6.1g,23.89mmol,92%收率)。MS/ESI+256.10[MH]+
步骤2:3,5-二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯 基)-2-((S)-1-(苯基磺酰基)吡咯烷-2-羰氧基)乙基)吡啶1-氧化物(305)
将(S)-3,5-二氯-4-(2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-羟基乙基)吡 啶1-氧化物(164)(55mg,0.145mmol),(S)-1-(苯基磺酰基)吡咯烷-2-羧 酸(111mg,0.434mmol),EDC(83mg,0.434mmol)和DMAP(53.0mg, 0.434mmol)在DCM(20ml)中的混合物在室温搅拌3小时。反应混合物 用含水1N HCl然后用含水1M K2CO3洗涤两次;有机层在Na2SO4上干 燥,减压除去溶剂。残余物通过制备型HPLC(方法2)纯化,提供3,5- 二氯-4-((S)-2-(4-(二氟甲氧基)-3-甲氧基苯基)-2-((S)-1-(苯基磺酰基)吡咯 烷-2-羰氧基)乙基)吡啶1-氧化物(45mg,0.073mmol,50.4%收率); MS/ESI+617.15[MH]+,tR=3.71分钟(方法3);非对映体比率>95:5(1H NMR);[αD]=-60.3(c=0.39,DCM);
1H NMR(B)(300MHz,DMSO-d6)δppm 8.61(s,2H),7.75-7.85 (m,2H),7.69-7.76(m,1H),7.56-7.69(m,2H),7.19(d,1H), 7.18(d,1H),7.00(dd,1H),7.07(t,1H),6.05(dd,1H),4.16(dd, 1H),3.86(s,3H),3.49(dd,1H),3.39-3.44(m,1H),3.29(dd,1 H),3.09-3.23(m,1H),1.83-2.10(m,1H),1.59-1.82(m,2H), 1.44-1.59(m,1H)
列于表25的化合物根据与方案40的描述类似的程序制备:用商业 适宜试剂,并且反应所列的适当醇,随后进行报告如下的适当纯化步骤。 化合物315作为来自非对映异构体混合物的第二洗脱的非对映异构体获 得。






实施例35:
3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-甲氧基苯基)-2-((S)-3-(3-(二甲 基氨基甲酰基)苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物(320)
方案41

步骤1:(S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羧酸乙 酯(318)
将(S)-噻唑烷-2-羧酸乙酯(2R,3R)-2,3-二羟基琥珀酸盐(如Bull. KoreanChem.Soc.2010,31,2709的描述制备)(4g,12.85mmol)倾倒 入在冰浴中预先冷却至0℃的含有饱和NaHCO3(50ml,55.0mmol)水 溶液和Et2O(100.0ml)的分液漏斗。振摇混合物以溶解固体;分相,水 层用Et2O(100ml)再次萃取。经合并的有机层在Na2SO4上干燥,不加 热地蒸发至干。残余物溶于THF(50ml),将溶液冷却至0℃,加入饱和 NaHCO3水溶液(50ml,55.0mmol)。在0℃于激烈搅拌下,将3-(二甲 基氨基甲酰基)苯-1-磺酰氯(3.18g,12.85mmol)的THF(50ml)溶液加入 两相混合物,让反应置于室温4小时。混合物在EtOAc与水间分配,水 层用EtOAc萃取。经合并的有机层用含水1N HCl和盐水洗涤,在Na2SO4上干燥,蒸发至干,提供(S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻唑 烷-2-羧酸乙酯(3.79g,10.18mmol,79%收率);MS/ESI+373.04[MH]+
步骤2:(S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻唑烷-2-羧酸 (319)
将(S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻唑烷-2-羧酸乙酯 (3.79g,10.18mmol)溶于MeOH(30ml),THF(30.0ml)和水(30.0ml) 的混合物。加入LiOH(0.487g,20.35mmol),反应在室温搅拌30分钟。 混合物用含水1N HCl(pH=1)酸化,用水稀释,用EtOAc萃取两次。经 合并的有机层在Na2SO4上干燥,蒸发至干。残余物与Et2O(15ml)和石 油醚(25ml)的混合物研磨,在过滤后产生(S)-3-(3-(二甲基氨基甲酰基) 苯基磺酰基)噻唑烷-2-羧酸(1.86g,5.40mmol,53.1%收率);MS/ESI+ 344.94[MH]+;[αD]=-36.1(c=1.67,MeOH)。
为了确定对映体的纯度,将该中间体与醇1(EDC,DMAP,DCM) 偶联,提供化合物52:非对映体比率=95:5。
步骤3:3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-甲氧基苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)-噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(320)
向3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-甲氧基苯基)-2-羟基乙基)吡 啶1-氧化物(156)(160mg,0.416mmol),EDC(160mg,0.833mmol) 和DMAP(102mg,0.833mmol)的DCM(50ml)溶液加入(S)-3-(3-(二甲 基氨基甲酰基)苯基磺酰基)噻唑烷-2-羧酸(172mg,0.500mmol),所得 混合物在室温搅拌24小时。反应混合物用含水1N HCl洗涤两次,有机 层在Na2SO4上干燥。真空除去溶剂,残余物通过制备型HPLC(方法2) 纯化,提供3,5-二氯-4-(2-(3-(环丙基甲氧基)-4-甲氧基苯 基)-2-((S)-3-(3-(二甲基氨基甲酰基)苯基磺酰基)噻唑烷-2-羰氧基)乙基) 吡啶1-氧化物(122mg,0.172mmol,41%收率);MS/ESI+710.25[MH] +,tR=3.25分钟(方法3);非对映体比率1:1(1H NMR)。
列于表26的化合物根据方案41制备:用商业适宜试剂,反应所列 的适当醇,随后进行报告如下的适当纯化步骤。用苯磺酰氯所获得的中 间体的对映体纯度如上文所述在步骤2中确定(参比化合物:89;非对映 体比率=92:8)。




本发明化合物的药理学活性
实施例15
在无细胞测试中,在体外确定PDE4抑制性活性
在U937人类单核细胞的上清液细胞裂解液中,测定PDE4活性。 基本上按照TorphyTJ等人J.Pharmacol.Exp.Ther.1992; 263:1195-1205的描述,培养细胞、收获并制备上清液级分。
在含GlutaMAXTM-I培养基的RPMI 1640中,在37℃,5%CO2生长U937细胞(CellBank,Interlab Cell Line Collection,ICLC HTL94002),所述培养基补充有10%胎牛血清和100μg/ml Pen-strep (Gibco)。
收获细胞,通过离心(150x g,8分钟)在冷PBS中洗涤两次。将经 洗涤的细胞再悬浮于冷Krebs-Ringer-Henseleit缓冲剂,最终浓度为20X 106细胞/ml,并且超声。于15000xg离心20分钟之后,汇集上清液, 分为等分试样并在-80℃储存。
通过测试cAMP从温育混合物的消失,在细胞上清液中确定PDE4 活性。
试验化合物的浓度为10-12M至10-6M。通过酶热灭活(2.5分钟, 在100℃)停止反应,用PerkinElmer的‘LANCE cAMP测试’,遵照提供 商的说明,来测定残余cAMP含量。
作为本发明代表的测试化合物的结果示于下表,表示为产生50% cAMP消失抑制的试验化合物的nM浓度(IC50)的平均值±标准偏差:

在上表中,PDE4结合能力(IC50值)所指如下:>10nM‘+‘;10-1 nM‘++’;1-0.1nM‘+++’;<0.1nM‘++++’。
PDE4活性的抑制百分比这样计算:假定不存在抑制剂的cAMP消 失为100%,而热灭活样品中的cAMP消失为0%。
类似地,测试的式(II)化合物的结果(表示为产生50%cAMP消失 抑制的试验化合物的nM浓度的平均值±标准偏差)(IC50)示于下表:
| 化合物 | PDE4抑制 |
| 8,11, | +++ |
| 3,9,10,14 | ++ |
在上表中,PDE4结合能力(IC50值)所指如下:>10nM‘+‘;10-1 nM‘++’;1-0.1nM‘+++’;<0.1nM‘++++’。
实施例16
在外周血液单核细胞(PBMCs)测试中,在体外确定PDE4抑制性活 性
根据前文描述(Hatzelmann A等人J.Pharmacol.Exp.Ther.2001; 297:267-279;Draheim R等人J.Pharmacol.Exp.Ther.2004; 308:555-563)的方法进行测试,该测试基于PDE4抑制剂对脂多糖(LPS)- 诱导的肿瘤坏死因子-α(外周血液单核细胞(PBMCs)中的TNF-α释放)所 发挥的已知的抑制性活性。
在存在或不存在(50microl)试验化合物的情况下,将冷冻保存的人 类PBMCs(100μl/孔)在96-孔板(105细胞/孔)中温育30分钟,试验化合 物的浓度范围是10-12M至10-6M或10-13M至10-7M。随后加入LPS(3 ng/ml)。
在加湿的温育器中,于95%空气和5%CO2气氛下,于37℃温育 18小时之后,收集培养基,通过ELISA测量TNF-α。
作为本发明代表的测试化合物的结果示于下表:表示为产生50% LPS-诱导的TNF-α释放抑制的试验化合物摩尔浓度(IC50)的平均值 ±95%置信度边界:

在上表中,PDE4结合能力(IC50值)所指如下:>10nM‘+‘;10-1 nM‘++’;1-0.1nM‘+++’;<0.1nM‘++++’。
测试化合物的效果计算为TNF-α释放的抑制百分比:假定不存在 抑制剂化合物的情况下,LPS-诱导的TNF-α产生为100%而在不存在 LPS的情况下,PBMCs的基础TNF-α产生为0%。
类似地,对于式(II)化合物,测试化合物的结果示于下表:表示为 产生50%LPS-诱导的TNF-α释放抑制的试验化合物摩尔浓度(IC50)的 平均值±95%置信度边界:
| 化合物 | PDE4抑制 |
| 6,11 | +++ |
| 8,9, | ++ |
在上表中,PDE4结合能力(IC50值)所指如下:>10nM‘+‘;10-1 nM‘++’;1-0.1nM‘+++’;<0.1nM‘++++’。
Claims (10)
1.通式(IDa)化合物

R1选自:
-C1-C6烷基;
-C1-C6卤代烷基;
R2选自:
-C1-C6烷基,任选由C3-C7环烷基取代;
-C3-C7环烷基;
p是0或1或2;
R3取代基是吡啶环3位和5位的2个氯原子;
R4是单环或双环环系,其是饱和的、部分不饱和的或完全不饱和的,所述单环环系选自单环芳基、单环C3-C8环烷基、单环C3-C7杂环烷基或单环杂芳基,其中所述单环芳基是苯基,所述单环(C3-C8)环烷基选自环丙基,所述单环(C3-C7)杂环烷基选自吗啉基和哌嗪基,所述单环杂芳基选自噻吩、吡咯、咪唑、噻唑、吡啶、呋喃残基,且其中所述双环环系选自部分不饱和的喹啉,吲哚,异吲哚,苯并噁嗪残基,
所述环由一个或多个基团R5取代,其是相同或不同的且独立地选自:
-C1-C6烷基,任选由一个或多个独立选自下述的基团取代:-OH;
-基团-OR6,其中R6选自
-C1-C10烷基,任选由一个或多个C3-C7环烷基取代;
-卤素原子;
-CN;
-NR8R9,其中R8和R9是不同或相同的,并且独立地选自:
-H;
--COR10,其中R10是C1-C6烷基;
--SO2R11,其中R11是C1-C4烷基;和
--COOR12,其中R12是H、C1-C4烷基;
--CONR8R9,其中R8和R9如上定义,
其吡啶环上的N-氧化物衍生物,或药学上可接受的盐。
2.化合物,其选自下述化合物:
























或其药学上可接受的盐。
3.以下化合物或其药学上可接受的盐:3,5-二氯-4-((S)-2-(3-(环丙基甲氧基)-4-(二氟甲氧基)苯基)-2-((S)-3-(3-脲基苯基磺酰基)噻唑烷-2-羰氧基)乙基)吡啶1-氧化物。
4.用于制备根据权利要求1的式(IDa)化合物的方法,包括:
-进行步骤1):

所述步骤1包括:a)将式(XIV)化合物,4-二甲基氨基吡啶,和1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)盐酸盐加入式(XV)化合物的二甲基甲酰胺溶液;b)搅拌混合物;c)将混合物倾入冷水;d)过滤沉淀;
-随后进行步骤2):

所述步骤2包括:a)在搅拌下,在室温下,将浓HCl的9体积的无水乙酸乙酯溶液加入式(XII)化合物的乙酸乙酯溶液;b)搅拌;c)过滤沉淀的固体;任选地d)用乙酸乙酯洗涤获得的固体;
-然后进行步骤3):

所述步骤3包括:a)将式(VII)化合物的吡啶溶液加入冷藏的式(XI)化合物的吡啶溶液,在室温下搅拌所得溶液;c)将该溶液倾入过量的HCl水溶液;d)过滤沉淀的物质和将其用水洗涤或者d’)用乙酸乙酯萃取水相,用HCl 1M、盐水洗涤和蒸发所得有机相;f)将化合物溶于8体积的乙醇;g)在室温下激烈地搅拌过夜;h)过滤形成的固体;
其中式(XV),(XII),(XI),(VII)和(IDa)化合物中的R1、R2、R3、R4和p具有按照式(I)化合物的含义。
5.如权利要求1至3中任一项的化合物与第二药物活性组分的组合,所述第二药物活性组分选自β2-激动剂、皮质类固醇和抗毒蕈碱药类别。
6.药物组合物,包含如权利要求1至3中任一项的化合物,或根据权利要求5的组合,和一种或多种药学上可接受的载体和/或赋形剂。
7.如权利要求1至3中任一项的化合物制备用于预防和/或治疗变应性鼻炎的药物的用途。
8.如权利要求1至3中任一项的化合物制备用于预防和/或治疗特应性皮炎的药物的用途。
9.如权利要求1至3中任一项的化合物用于制备药物的用途,所述药物用于预防和/或治疗表征为气道阻塞的呼吸道疾病。
10.如权利要求9的用途,其中所述表征为气道阻塞的呼吸道疾病选自哮喘和COPD。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11168853.7 | 2011-06-06 | ||
| EP11168853 | 2011-06-06 | ||
| CN201280025202.0A CN103562185A (zh) | 2011-06-06 | 2012-06-05 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280025202.0A Division CN103562185A (zh) | 2011-06-06 | 2012-06-05 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN106946848A CN106946848A (zh) | 2017-07-14 |
| CN106946848B true CN106946848B (zh) | 2022-01-04 |
Family
ID=46458441
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201710190023.7A Active CN106946848B (zh) | 2011-06-06 | 2012-06-05 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 |
| CN201280025202.0A Pending CN103562185A (zh) | 2011-06-06 | 2012-06-05 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280025202.0A Pending CN103562185A (zh) | 2011-06-06 | 2012-06-05 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 |
Country Status (15)
| Country | Link |
|---|---|
| US (3) | US20130005716A1 (zh) |
| EP (1) | EP2718267B1 (zh) |
| JP (1) | JP2014518203A (zh) |
| KR (1) | KR20140028049A (zh) |
| CN (2) | CN106946848B (zh) |
| AR (1) | AR086676A1 (zh) |
| AU (1) | AU2012266514A1 (zh) |
| BR (1) | BR112013028959A2 (zh) |
| CA (1) | CA2838435A1 (zh) |
| MX (1) | MX2013013557A (zh) |
| RU (1) | RU2626956C2 (zh) |
| SG (1) | SG195319A1 (zh) |
| UA (1) | UA111198C2 (zh) |
| WO (1) | WO2012168226A1 (zh) |
| ZA (1) | ZA201309150B (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2022783A1 (en) | 2007-08-08 | 2009-02-11 | CHIESI FARMACEUTICI S.p.A. | "Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors" |
| EP2216327A1 (en) | 2009-02-06 | 2010-08-11 | CHIESI FARMACEUTICI S.p.A. | Benzoic acid (1-phenyl-2-pyridin-4-yl)ethyl esters as phosphodiesterase inhibitors |
| AU2011287711B2 (en) | 2010-08-03 | 2016-10-27 | Chiesi Farmaceutici S.P.A. | Dry powder formulation comprising a phosphodiesterase inhibitor |
| WO2013045280A1 (en) | 2011-09-26 | 2013-04-04 | Chiesi Farmaceutici S.P.A. | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors |
| RU2637945C2 (ru) | 2012-06-04 | 2017-12-08 | КЬЕЗИ ФАРМАЧЕУТИЧИ С.п.А. | Производные 1-фенил-2-пиридинил-алкиловых спиртов в качестве ингибиторов фосфодиэстеразы |
| RU2015121037A (ru) | 2012-12-05 | 2017-01-11 | КЬЕЗИ ФАРМАЧЕУТИЧИ С.п.А. | Производные фенилэтилпиридина в качестве ингибиторов pde-4 |
| EP2928880B1 (en) | 2012-12-05 | 2018-02-14 | Chiesi Farmaceutici S.p.A. | Phenylethylpyridine derivatives as pde4-inhibitors and muscarinic receptor antagonists |
| WO2014086849A1 (en) | 2012-12-05 | 2014-06-12 | Chiesi Farmaceutici S.P.A. | Phenylethylpyridine derivatives as pde4-inhibitors |
| CN104822659A (zh) | 2012-12-05 | 2015-08-05 | 奇斯药制品公司 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇衍生物 |
| US9326976B2 (en) * | 2014-06-05 | 2016-05-03 | Chiesi Farmaceutici S.P.A. | Carbamate derivatives |
| MA42048A (fr) * | 2015-05-07 | 2018-03-14 | Chiesi Farm Spa | Dérivés d'aminoesters |
| AR104822A1 (es) * | 2015-06-01 | 2017-08-16 | Chiesi Farm Spa | Derivado de aminoésteres |
| TW201710254A (zh) * | 2015-06-01 | 2017-03-16 | 吉斯藥品公司 | 胺基酯衍生物 |
| WO2017089347A1 (en) | 2015-11-25 | 2017-06-01 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Methods and pharmaceutical compositions for the treatment of braf inhibitor resistant melanomas |
| CN107094329B (zh) * | 2017-05-03 | 2019-01-29 | 矽力杰半导体技术(杭州)有限公司 | Led驱动电路 |
| CN115466169B (zh) * | 2021-06-10 | 2024-03-26 | 中国医学科学院药物研究所 | 取代邻苯二酚醚类化合物及其制备方法和应用 |
| WO2023178378A1 (en) * | 2022-03-21 | 2023-09-28 | DMTC Limited | Mip inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009018909A2 (en) * | 2007-08-08 | 2009-02-12 | Chiesi Farmaceutici S.P.A. | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors |
| WO2009077068A1 (en) * | 2007-12-14 | 2009-06-25 | Chiesi Farmaceutici S.P.A. | Ester derivatives as phosphodiesterase inhibitors |
| CN101490004A (zh) * | 2006-07-14 | 2009-07-22 | 奇斯药制品公司 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烯基醇的衍生物 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2140441C (en) | 1992-07-28 | 2006-11-21 | Garry Fenton | Inhibitors of c-amp phosphodiesterase and tnf |
| US5786354A (en) | 1994-06-21 | 1998-07-28 | Celltech Therapeutics, Limited | Tri-substituted phenyl derivatives and processes for their preparation |
| SE9903995D0 (sv) * | 1999-11-03 | 1999-11-03 | Astra Ab | New combination |
| AU2003252467A1 (en) | 2002-07-03 | 2004-01-23 | Kyowa Hakko Kogyo Co., Ltd. | Process for preparation of 1,3-benzodioxole-2-spiro- cycloalkane derivatives |
| JPWO2004096274A1 (ja) | 2003-03-31 | 2006-07-13 | 協和醗酵工業株式会社 | 気道内投与剤 |
| CN101123971A (zh) | 2003-07-31 | 2008-02-13 | 埃匹吉尼斯医药有限公司 | 治疗哮喘或慢性阻塞性肺病的脱氢表雄酮或硫酸脱氢表雄酮与pde-4抑制剂的组合 |
| CN101123970A (zh) * | 2003-07-31 | 2008-02-13 | 埃匹吉尼斯医药有限公司 | 治疗哮喘或慢性阻塞性肺病的脱氢表雄酮或硫酸脱氢表雄酮与抗胆碱能支气管扩张剂的组合 |
| US20090274676A1 (en) | 2003-07-31 | 2009-11-05 | Robinson Cynthia B | Combination of dehydroepiandrosterone or dehydroepiandrosterone-sulfate with a pde-4 inhibitor for treatment of asthma or chronic obstructive pulmonary disease |
| HN2005000795A (es) * | 2004-10-15 | 2010-08-19 | Aventis Pharma Inc | Pirimidinas como antagonistas del receptor de prostaglandina d2 |
| JP2008174213A (ja) * | 2006-12-20 | 2008-07-31 | Nsk Ltd | 舵角可変式ステアリング装置 |
| EP2110375A1 (en) | 2008-04-14 | 2009-10-21 | CHIESI FARMACEUTICI S.p.A. | Phosphodiesterase-4 inhibitors belonging to the tertiary amine class |
| EP2216327A1 (en) | 2009-02-06 | 2010-08-11 | CHIESI FARMACEUTICI S.p.A. | Benzoic acid (1-phenyl-2-pyridin-4-yl)ethyl esters as phosphodiesterase inhibitors |
| AU2011287711B2 (en) | 2010-08-03 | 2016-10-27 | Chiesi Farmaceutici S.P.A. | Dry powder formulation comprising a phosphodiesterase inhibitor |
| CA2807406C (en) | 2010-08-03 | 2018-12-11 | Chiesi Farmaceutici S.P.A. | Pharmaceutical formulation comprising a phosphodiesterase inhibitor |
| WO2013045280A1 (en) | 2011-09-26 | 2013-04-04 | Chiesi Farmaceutici S.P.A. | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors |
| BR112014009471A2 (pt) * | 2011-10-21 | 2017-04-18 | Chiesi Farm Spa | compostos, combinação de um composto, composição farmacêutica, uso de um composto, dispositivo e kit |
| RU2637945C2 (ru) * | 2012-06-04 | 2017-12-08 | КЬЕЗИ ФАРМАЧЕУТИЧИ С.п.А. | Производные 1-фенил-2-пиридинил-алкиловых спиртов в качестве ингибиторов фосфодиэстеразы |
| RU2015121037A (ru) * | 2012-12-05 | 2017-01-11 | КЬЕЗИ ФАРМАЧЕУТИЧИ С.п.А. | Производные фенилэтилпиридина в качестве ингибиторов pde-4 |
-
2012
- 2012-06-05 WO PCT/EP2012/060579 patent/WO2012168226A1/en active Application Filing
- 2012-06-05 EP EP12732555.3A patent/EP2718267B1/en active Active
- 2012-06-05 AR ARP120101983A patent/AR086676A1/es unknown
- 2012-06-05 CA CA2838435A patent/CA2838435A1/en not_active Abandoned
- 2012-06-05 US US13/488,818 patent/US20130005716A1/en not_active Abandoned
- 2012-06-05 JP JP2014514031A patent/JP2014518203A/ja active Pending
- 2012-06-05 CN CN201710190023.7A patent/CN106946848B/zh active Active
- 2012-06-05 UA UAA201314221A patent/UA111198C2/uk unknown
- 2012-06-05 SG SG2013090147A patent/SG195319A1/en unknown
- 2012-06-05 CN CN201280025202.0A patent/CN103562185A/zh active Pending
- 2012-06-05 KR KR1020137031957A patent/KR20140028049A/ko not_active Application Discontinuation
- 2012-06-05 RU RU2013154117A patent/RU2626956C2/ru active
- 2012-06-05 MX MX2013013557A patent/MX2013013557A/es not_active Application Discontinuation
- 2012-06-05 AU AU2012266514A patent/AU2012266514A1/en not_active Abandoned
- 2012-06-05 BR BR112013028959A patent/BR112013028959A2/pt not_active IP Right Cessation
-
2013
- 2013-10-08 US US14/048,651 patent/US20140057882A1/en not_active Abandoned
- 2013-12-05 ZA ZA2013/09150A patent/ZA201309150B/en unknown
-
2015
- 2015-09-24 US US14/863,915 patent/US9931327B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101490004A (zh) * | 2006-07-14 | 2009-07-22 | 奇斯药制品公司 | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烯基醇的衍生物 |
| WO2009018909A2 (en) * | 2007-08-08 | 2009-02-12 | Chiesi Farmaceutici S.P.A. | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors |
| WO2009077068A1 (en) * | 2007-12-14 | 2009-06-25 | Chiesi Farmaceutici S.P.A. | Ester derivatives as phosphodiesterase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2626956C2 (ru) | 2017-08-02 |
| EP2718267B1 (en) | 2016-08-10 |
| US9931327B2 (en) | 2018-04-03 |
| CA2838435A1 (en) | 2012-12-13 |
| SG195319A1 (en) | 2013-12-30 |
| CN106946848A (zh) | 2017-07-14 |
| AR086676A1 (es) | 2014-01-15 |
| MX2013013557A (es) | 2013-12-16 |
| EP2718267A1 (en) | 2014-04-16 |
| UA111198C2 (uk) | 2016-04-11 |
| KR20140028049A (ko) | 2014-03-07 |
| WO2012168226A1 (en) | 2012-12-13 |
| AU2012266514A1 (en) | 2014-01-09 |
| RU2013154117A (ru) | 2015-06-10 |
| US20160008338A1 (en) | 2016-01-14 |
| ZA201309150B (en) | 2015-03-25 |
| BR112013028959A2 (pt) | 2016-08-30 |
| CN103562185A (zh) | 2014-02-05 |
| US20130005716A1 (en) | 2013-01-03 |
| US20140057882A1 (en) | 2014-02-27 |
| JP2014518203A (ja) | 2014-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN106946848B (zh) | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇的衍生物 | |
| EP3728210A1 (en) | Isoxazole n-linked carbamoyl cyclohexyl acids as lpa antagonists | |
| EP2855450B1 (en) | Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors | |
| RU2617401C2 (ru) | Производные 1-фенил 2-пиридинилалкиловых спиртов в качестве ингибиторов фосфодиэстеразы | |
| EP2928879B1 (en) | Phenylethylpyridine derivatives as pde4-inhibitors and muscarinic receptor antagonists | |
| CN107207473B (zh) | 具有毒蕈碱受体拮抗剂和β2肾上腺素能受体激动剂活性的化合物 | |
| JP4242274B2 (ja) | 因子Xa阻害剤としてのピロリジン誘導体 | |
| CN110698459B (zh) | 作为磷酸二酯酶抑制剂的1-苯基-2-吡啶基烷基醇衍生物 | |
| CA2932486A1 (en) | Benzhydryl derivatives for the treatment of respiratory diseases | |
| EP3152204B1 (en) | Carbamate derivatives which are both phosphodiesterase 4 (pde4) enzyme inhibitors and muscarinic m3 receptor antagonists | |
| TW201302726A (zh) | 作為磷酸二酯酶抑制劑之1-苯基-2-吡啶基烷基醇衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1235785 Country of ref document: HK |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant |