CN106460033B - 使用核酸条形码分析与单细胞缔合的核酸 - Google Patents
使用核酸条形码分析与单细胞缔合的核酸 Download PDFInfo
- Publication number
- CN106460033B CN106460033B CN201480076617.XA CN201480076617A CN106460033B CN 106460033 B CN106460033 B CN 106460033B CN 201480076617 A CN201480076617 A CN 201480076617A CN 106460033 B CN106460033 B CN 106460033B
- Authority
- CN
- China
- Prior art keywords
- sample
- barcode
- adapter
- sequence
- rna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


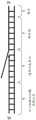




































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6804—Nucleic acid analysis using immunogens
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L3/00—Containers or dishes for laboratory use, e.g. laboratory glassware; Droppers
- B01L3/50—Containers for the purpose of retaining a material to be analysed, e.g. test tubes
- B01L3/502—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures
- B01L3/5027—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures by integrated microfluidic structures, i.e. dimensions of channels and chambers are such that surface tension forces are important, e.g. lab-on-a-chip
- B01L3/502769—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures by integrated microfluidic structures, i.e. dimensions of channels and chambers are such that surface tension forces are important, e.g. lab-on-a-chip characterised by multiphase flow arrangements
- B01L3/502784—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures by integrated microfluidic structures, i.e. dimensions of channels and chambers are such that surface tension forces are important, e.g. lab-on-a-chip characterised by multiphase flow arrangements specially adapted for droplet or plug flow, e.g. digital microfluidics
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
- C12P19/34—Polynucleotides, e.g. nucleic acids, oligoribonucleotides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6834—Enzymatic or biochemical coupling of nucleic acids to a solid phase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/10—Modifications characterised by
- C12Q2525/131—Modifications characterised by incorporating a restriction site
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/10—Modifications characterised by
- C12Q2525/155—Modifications characterised by incorporating/generating a new priming site
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/10—Modifications characterised by
- C12Q2525/191—Modifications characterised by incorporating an adaptor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2563/00—Nucleic acid detection characterized by the use of physical, structural and functional properties
- C12Q2563/149—Particles, e.g. beads
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2563/00—Nucleic acid detection characterized by the use of physical, structural and functional properties
- C12Q2563/159—Microreactors, e.g. emulsion PCR or sequencing, droplet PCR, microcapsules, i.e. non-liquid containers with a range of different permeability's for different reaction components
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2563/00—Nucleic acid detection characterized by the use of physical, structural and functional properties
- C12Q2563/185—Nucleic acid dedicated to use as a hidden marker/bar code, e.g. inclusion of nucleic acids to mark art objects or animals
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2565/00—Nucleic acid analysis characterised by mode or means of detection
- C12Q2565/60—Detection means characterised by use of a special device
- C12Q2565/629—Detection means characterised by use of a special device being a microfluidic device
Abstract
本文提供使用核酸条形码来分析与单细胞缔合的核酸的方法和组合物。根据一些实施方案,一种用于制造一种或多种所关注的聚核苷酸的方法包括:获得多种与一种或多种样品缔合的RNA,其中所述样品获自一个或多个受试者,各RNA与单一样品缔合,并且与各样品缔合的RNA存在于独立反应容积中;添加衔接分子至与各样品缔合的RNA中,其中所述衔接分子使用酶反应产生并且包含通用引发序列、条形码序列和结合位点;以及将所述条形码序列并入一种或多种与各样品缔合的聚核苷酸中,由此制造所述一种或多种所关注的聚核苷酸。
Description
相关申请的交叉引用
本申请要求名称为“Analysis of Nucleic Acids Associated with SingleCells using Nucleic Acid Barcodes”并且在2013年12月30日提交的美国临时申请号61/922,012的权益,所述临时申请的完整内容出于所有目的以引用的方式并入本文。
作为ASCII文本文件递交的“序列表”、表格或计算机程序列表附件的引用
在2014年12月30日创建(18,227,200字节,机器格式IBM-PC,MS-Windows操作系统)的文件97519-920777.txt中写出的序列表出于所有目的以引用的方式整体并入本文。在文件表18中写出的表18.[条形码_部分1].[条形码_部分1].txt,2,039,808字节;在文件表18中写出的表18.[条形码_部分2].[条形码_部分2].txt,90,112字节;在文件表22中写出的表22.[i5指数引物].[i5指数引物].txt,4,096字节;在文件表32中写出的表32.[孔-条形码].[孔-条形码].txt,4,096字节;在文件表32中写出的表32.[板-条形码].[板-条形码].txt,4,096字节(均在2014年12月24日创建,机器格式IBM-PC,MS-Windows操作系统)出于所有目的以引用的方式整体并入本文。
发明背景
如免疫球蛋白(Ig)和T细胞受体(TCR)基因的可变基因由接合点之间具有P/N核苷酸添加的V(D)J基因区段的重排形成。完全功能Ig或TCR蛋白通过两种基因(对于Ig为重链和轻链基因,对于αβTCR为α和β基因并且对于γδTCR为γ和δ基因)的缔合形成。这种组合方法产生各种各样的不同的可能序列。
这种谱系使免疫系统能够回应于生物体尚未遇到的新颖免疫损伤。免疫球蛋白基因还经历体细胞超突变,其进一步增加谱系大小。
对应地,允许天然Ig或TCR蛋白的表达以研究其功能特性的对可变基因的任何核酸分析不仅需要对个别B(对于Ig基因)或T细胞(对于TCR基因)测序,而且需要构成蛋白的两种基因的天然配对。这可通过单细胞克隆和Sanger测序进行,但为缓慢并且费力的(参看例如Wrammert等人,Nature,2008,453:667-671)。
已经开发了高通量方法来对天然配对基因进行高通量测序,并且分为两种方法。第一种方法是使独特核酸条形码识别符连接于来自细胞的核酸,并且如果基因共享所述相同条形码并且因此起源于相同细胞,那么经由使所述基因以生物信息学方式连接在一起来实现配对(PCT/US2012/000221)。第二种方法是使来自两种基因的核酸以物理方式连接在一起(参看例如美国专利号7,749,697)。
所述第一种方法是优良的,因为其允许多种基因的配对(如鉴别特异性T细胞或B细胞子集的B或T细胞共同表达基因),而所述第二种方法限于以物理方式连接一些核酸。迄今,实验数据仅关于其中已经以物理方式连接不超过两种核酸的情形存在。
使核酸明确地缔合至单细胞(所述第一种方法)而非使其经由连接彼此缔合(所述第二种方法)具有优势。当核酸彼此缔合时,可难以区别PCR和测序误差与真实生物变异。必须关于测序平台的精确性进行假设并且读数基于百分比相似性截止值任意地被指定为不同序列,即具有>95%相似性的所有读数被指定为序列并且其间的任何差异均假定是归因于测序误差。这不能区别彼此非常相似的序列(参看Zhu等人,Frontiers inMicrobiology,2012,3:315)。
此外,使用被指定为序列的读数的相对频率关于多少细胞共享所述同一序列进行假定。这是近似量度并且受到PCR扩增偏差影响,如本领域中众所周知。因此,使Ig或TCR核酸彼此缔合可仅产生近似值,而非进行测序的谱系的真实表示(参看Zhu等人,Frontiersin Microbiology,2012,3:315)。
然而,使用核酸条形码使核酸缔合至单细胞允许明确区别来自单B或T细胞的相似或甚至同一序列,因为各读数可被指定为细胞。
此外,通过用所有与细胞缔合的读数创建共有序列,可获得非常精确并且几乎完全无误差序列并且可获得进行测序的谱系的精确表示。这也可普及至细胞中所有核酸的分析。
另外,递送独特条形码至各单细胞的技术困难仍存在。连接核酸条形码至可变基因的目前最佳技术在水溶液中具有独特条形码并且各条形码甚至在连接条形码至可变基因核酸的反应之前存在于独立存储容器中(PCT/US2012/000221),否则所述核酸条形码将在使用之前混合。这产生了对数千种细胞编条形码的逻辑困难,因为需要大量容器含有个别条形码。
对于大量存储容器的需求也使这种方法与其中独特条形码无法个别地吸移至各个别反应容器(其也将含有单细胞)中的任一种方法不可相容。一个实例为纳升大小的反应容器,如纳米孔方法,其中将独特条形码个别地吸移至各纳米孔中为不切实际的,因为存在数千至数十万个纳米孔。
这在其中使用油包水乳液产生液滴的纳米液滴方法中也是不可行的,因为数十万个纳米液滴仅由一些水流产生(参看例如Dolomite Microfluidics或RaindanceTechnologies的产品),并且在递送至纳米液滴之前不可能在个别存储容器中具有独特条形码。
一种递送独特条形码至个别反应容器的方法是使用有限稀释来将独特条形码沉积于大多数反应容器中。可执行连接于可操纵物体(如珠粒)的条形码的有限稀释,所述可操纵物体各自具有所连接的一种特定条形码的多个拷贝,或可执行溶液中条形码的有限稀释。在稀释所述珠粒后,一种特定核酸条形码的多个拷贝存在于反应容器中,而在溶液中稀释条形码后,特定核酸条形码的仅单一拷贝存在于反应容器中。
此外,添加核酸条形码至存在于反应容器中的所关注的样品源性核酸中将在所引入的条形码扩增以确保其足量存在于反应腔室中的情况下更完全。例如,典型哺乳动物细胞含有mRNA的大致400,000个拷贝。为了最大化总体单细胞分析的效率,应对尽可能多的这些mRNA拷贝编条形码。因此,至少特定核酸条形码的大致相同数目的拷贝,因为mRNA拷贝需要存在于反应容器中。溶液中条形码的有限稀释导致反应容器中仅有特定条形码的单一拷贝,而具有条形码的小(例如1-2μm直径)珠粒的稀释将预期提供最大数万个拷贝。因此,在任一情形中所述条形码的扩增均为重要的以在反应容器中产生足量的特定核酸条形码,使得所述条形码成功添加至最大数目的样品源性核酸中。然而,预期珠粒提供显著更多的起始物质并且因此提供显著更佳的条形码扩增。另外,充分大的珠粒可含有数十万个核酸条形码分子。在这种情形中,核酸条形码从所述珠粒裂解可足以在反应容器中产生足量的特定核酸条形码。
此外,如果核酸连接于固体表面,那么其将不会如溶液中的核酸般自由地到处移动。对于核酸互补碱基配对,固相动力学比液相动力学慢得多,并且可使得条形码以低得多的效率添加至所关注的核酸中。优选地,核酸条形码应在参与条形码反应之前存在于液相中。
本发明比先前发明(PCT/US2012/000221)在连接独特条形码至各样品方面有所改进,其中各样品通常为单细胞,但可普及至任何类型的样品。本发明使得独特条形码能够递送至任何类型的反应容器,并且还适用于纳升大小的反应容器并且不需要使独特核酸条形码保持于独立存储容器中。其经得起但无需手动地吸移独特条形码至各反应容器中。其递送独特条形码或独特条形码集合的一个或多个拷贝至各反应容器中并且所述条形码在以快速液相动力学发生于液相中的反应中连接于所关注的核酸。由于所述反应连接条形码至细胞中的所有所关注的核酸,即细胞中所有反转录的RNA,本发明使得能够进行单细胞转录组学分析,并且不限于使免疫球蛋白可变基因缔合至特异性样品。此外,所述扩增反应可在充分低的温度下发生使得其可与中温酶(其在其它情况下在高温下失活)相容以添加条形码至所关注的核酸中。
发明概述
本文公开使用核酸条形码来分析与单细胞缔合的核酸的方法和组合物。本文公开的一种用于制造一种或多种所关注的聚核苷酸的方法包括获得多种与一种或多种样品缔合的核酸,其中所述样品获自一个或多个受试者,并且所述与样品缔合的核酸存在于独立反应容积中。所述核酸可为RNA或DNA分子(例如cDNA分子)。在一些实施方案中,衔接分子添加至与样品缔合的核酸中。在一些实施方案中,所述衔接分子使用酶反应产生并且包含通用引发序列、条形码序列和结合位点。在一些实施方案中,所述条形码序列并入至与样品缔合的一种或多种聚核苷酸中,由此制造所述一种或多种所关注的聚核苷酸。在一些实施方案中,所述方法包括添加衔接分子至与样品缔合的核酸中,其中所述衔接分子使用酶反应产生并且包含通用引发序列、条形码序列和结合位点;和将所述条形码序列并入与样品缔合的一种或多种聚核苷酸中,由此制造所述一种或多种所关注的聚核苷酸。
本文公开一种用于制造一种或多种所关注的聚核苷酸的方法。所述方法包括获得多种与一种或多种样品缔合的RNA,其中所述样品获自一个或多个受试者,并且与样品缔合的RNA存在于独立反应容积中;添加衔接分子至与样品缔合的RNA中,其中所述衔接分子使用酶反应产生并且包含通用引发序列、条形码序列和结合位点;以及将所述条形码序列并入一种或多种与样品缔合的聚核苷酸中,由此制造所述一种或多种所关注的聚核苷酸。在一些实施方案中,各RNA或所述多种RNA中的至少一者与来自所述一种或多种样品的单一样品缔合。所述方法的一些实施方案进一步包括使用所述酶反应产生所述衔接分子。
在一些实施方案中,所述衔接分子通过使模板分子与一种或多种酶接触而产生。在一些实施方案中,所述模板分子为包含RNA聚合酶(RNAP)启动子的DNA分子,并且所述一种或多种酶包括RNA聚合酶。所述RNAP启动子可选自由T7、T3和SP6组成的组。在一些实施方案中,所述模板分子为包含切口核酸内切酶限制位点的DNA分子,并且所述一种或多种酶包括切口核酸内切酶和链置换DNA聚合酶。所述切口核酸内切酶限制位点可选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组。所述链置换DNA聚合酶可选自由Klenow exo-、Bst大片段和Bst大片段的工程改造的变体组成的组。所述DNA分子可为双链分子或单链分子,所述单链分子适用作用于产生双链分子的模板。
在一些实施方案中,所述模板分子结合于固体支撑物,所述固体支撑物与水溶液接触,并且所述衔接分子在其产生时释放至所述水溶液中。在一些实施方案中,添加所述衔接分子至所述与一种样品缔合的RNA中包括组合所述水溶液与其中存在所述RNA的所述反应容积。在一些实施方案中,所述水溶液与所述与一种样品缔合的RNA存在于相同反应容积中。在一些实施方案中,所述模板分子包含核酸内切酶限制位点,所述一种或多种酶包含限制核酸内切酶,并且所述衔接分子包含所述模板分子的一部分,所述部分在所述模板分子与所述限制核酸内切酶接触后产生并且释放至所述水溶液中。在一些实施方案中,所述固体支撑物为珠粒或表面(例如微量滴定孔或管的表面)。
在所述方法的一些实施方案中,所述衔接分子在添加所述衔接分子至所述与一种样品缔合的RNA中之前以游离形式处于溶液中。在一些实施方案中,所述衔接分子在隔室中产生,并且添加所述衔接分子至所述与一种样品缔合的RNA中包括组合所述隔室与其中存在所述RNA的所述反应容积。在一些实施方案中,所述衔接分子在其中存在添加有所述衔接分子的所述RNA的所述反应容积中产生。在一些实施方案中,所述衔接分子不在其中存在添加有所述衔接分子的所述RNA的所述反应容积中产生。在一些实施方案中,所述酶反应为等温反应。在一些实施方案中,所述衔接分子进一步包含独特分子识别符(UMI)序列。在一些实施方案中,所述衔接分子为RNA分子。所述衔接分子可使用RNAP产生。
在所述方法的一些实施方案中,所述衔接分子为DNA分子。所述衔接分子可使用DNAP产生。
在一些实施方案中,制造所述一种或多种所关注的聚核苷酸包括反转录与所述样品缔合的所述RNA,由此合成多种第一链cDNA,与所述样品缔合的所述RNA中的至少一些包含与所述衔接分子的所述结合位点互补的序列区,并且所述衔接分子用作反转录的引物,使得所述条形码序列并入与所述样品缔合的第一链cDNA中。在这些实施方案中,所述结合位点可包含聚T区或随机区。所述结合位点可出现于所述衔接分子的3’端。所述衔接分子可在隔室中产生,并且反转录与样品缔合的所述RNA可在组合所述隔室与其中存在所述RNA的所述反应容积之后发生。反转录与样品缔合的所述RNA可在其中产生添加至所述RNA中的衔接分子的相同反应容积中发生。
所述方法的一些实施方案进一步包括反转录与样品缔合的所述RNA以获得多种cDNA,其中反转录RNA包括使用反转录酶和第一链引物合成cDNA的第一链。在这些实施方案中,反转录酶可为MMLV H-反转录酶。所述衔接分子可在隔室中产生,并且添加所述衔接分子至所述与一种样品缔合的RNA中包括组合所述隔室与其中存在所述RNA的所述反应容积。cDNA的第一链可在组合所述隔室与所述反应容积之前或之后合成。
在一些实施方案中,反转录与样品缔合的所述RNA在其中产生添加至所述RNA中的衔接分子的相同反应容积中发生。在这些实施方案中,所述反应容积中的缓冲液可包含在pH 8.0至pH 8.8的pH范围内的Tris、钾离子、氯离子、硫酸根离子、铵离子、乙酸离子或镁离子中的至少一者。
在一些实施方案中,所述反转录酶具有模板转换活性,与所述样品缔合的cDNA的至少一些第一链包含3’悬垂物,所述衔接分子的所述结合位点包含与所述3’悬垂物互补的3’部分,并且所述衔接分子用作所述反转录酶的模板,使得所述条形码序列并入与所述样品缔合的cDNA的第一链中。在这些实施方案中,所述3’悬垂物可包含一个或多个C核苷酸并且所述结合位点的所述3’部分可包含一个或多个G核苷酸。所述第一链引物可包含聚T区或随机序列。
在一些实施方案中,制造所关注的聚核苷酸包括使用第一(例如正向)引物和第二(例如反向)引物扩增用于各样品的cDNA的所述第一链,所述第二引物具有与所述第一链引物的至少一部分相同的序列,其中所述第一引物或所述第二引物为所述衔接分子。在这些实施方案中,所述第一引物或所述第二引物可为所述衔接分子。所述第一链引物可包含聚T区或随机序列。
在所述方法的一些实施方案中,各样品包含细胞。所述细胞可为血细胞、免疫细胞、组织细胞或肿瘤细胞。在一些实施方案中,所述细胞为B细胞或T细胞。所述B细胞可为成浆细胞、记忆B细胞或浆细胞。在一些实施方案中,与各样品缔合的所述RNA包含mRNA,例如至少1、3、10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000种mRNA。在一些实施方案中,与各样品缔合的所述RNA包含细胞的转录组或细胞的总RNA。在一些实施方案中,每种样品制造至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000种所关注的聚核苷酸。在一些实施方案中,所述一种或多种样品包含至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000个细胞。在一些实施方案中,所述一种或多种样品获自相同受试者。一些实施方案进一步包括使所述样品与溶解缓冲液接触。
一些实施方案进一步包括使所述样品与核酸标记物接触,由此允许所述核酸标记物结合于所述样品的子集;和洗涤所述样品,由此从所述核酸标记物不结合的样品去除所述核酸标记物,其中关于所述子集内的样品,添加至与所述样品缔合的所述RNA中的所述衔接分子也添加至所述核酸标记物中,并且使用所述标记的核酸标记物制造一种或多种所关注的聚核苷酸。在这些实施方案中,所述核酸标记物可包含偶联于分子标记的核酸。所述分子标记可为抗体、抗原或蛋白。所述分子标记可对一种或多种细胞表面部分具有亲和力。在一些实施方案中,所述核酸为RNA。在一些实施方案中,所述核酸为DNA并且可包含RNAP启动子。在一些实施方案中,所述样品与第一核酸标记物和第二核酸标记物接触,其中所述第一核酸标记物包含偶联于第一分子标记的第一核酸,并且所述第二核酸标记物包含偶联于第二分子标记的第二核酸。所述第一核酸和第二核酸可包含不同的序列区。在一些实施方案中,所述第一和第二分子标记为不同的(例如,不同的细胞表面抗原的两种不同抗体)。因此,所述方法允许用包含衔接分子的核酸标记物多重标记如单细胞的样品,并且制造一种或多种与所述样品缔合的所关注的聚核苷酸。
在所述方法的一些实施方案中,所述一种或多种样品获自相同受试者。在一些实施方案中,所述一种或多种样品获自至少3、10、30或100个不同受试者。
本文还公开条形码衔接构建体。一些所述条形码衔接构建体包含RNAP启动子序列、通用引发序列、条形码序列和结合位点。所述RNAP启动子可选自由T7、T3和SP6组成的组。其它条形码衔接构建体包含切口核酸内切酶限制位点、通用引发序列、条形码序列和结合位点。所述切口核酸内切酶限制位点可选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组。
本文进一步公开一种固体支撑物,其包含如上文所述的条形码衔接构建体。在一些实施方案中,所述条形码衔接构建体经由共价键结合于所述固体支撑物。在一些实施方案中,所述条形码衔接构建体的多个拷贝结合于所述固体支撑物。例如,所述条形码衔接构建体的至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000个拷贝可结合于所述固体支撑物。在一些实施方案中,所述条形码衔接构建体的各拷贝包含所述相同条形码序列。本文还公开一种衔接模板文库,其包含多种偶联于所述衔接构建体的多个拷贝的固体支撑物。在一些实施方案中,所述多种固体支撑物包含至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000种固体支撑物。在一些实施方案中,所述固体支撑物中的至少两者包含具有不同条形码序列或UMI序列的衔接构建体。在一些实施方案中,所述多种固体支撑物中的每一种固体支撑物均包含具有不同条形码序列或不同UMI序列的衔接构建体。
本文还公开一种核酸标记物,其包含偶联于分子标记的核酸。在一些实施方案中,所述分子标记为抗体、抗原或蛋白。在一些实施方案中,所述分子标记对一种或多种细胞表面部分具有亲和力。在一些实施方案中,所述核酸为RNA。在一些实施方案中,所述核酸为DNA。所述DNA可包含RNAP启动子序列。在一些实施方案中,描述多种核酸标记物,其中所述多种中的至少一者包含第一分子标记(即,第一抗体)并且所述多种中的至少一者包含第二分子标记(即,第二抗体)。在一些实施方案中,所述第一和第二分子标记为不同的,因此提供适用于用本文所述的核酸标记物多重标记不同的细胞表面部分(例如不同的细胞表面抗原)的组合物。
本文进一步公开包含本文所述的衔接构建体的试剂盒。所述试剂盒可包含多种偶联于本文所述的衔接构建体的固体支撑物。在一些实施方案中,所述试剂盒包含衔接模板文库,所述文库包含多种衔接构建体。在一些实施方案中,所述试剂盒包含衔接模板文库,所述文库包含多种偶联于多种固体支撑物的衔接构建体。所述试剂盒可进一步包含用于通过酶反应由所述衔接构建体产生本文所述的衔接分子的酶。在一些实施方案中,所述试剂盒包含本文所述的细胞悬浮缓冲液。
本文进一步公开一种包含渗透保护剂的细胞悬浮缓冲液。在一些实施方案中,所述渗透保护剂为甜菜碱或其密切结构类似物。例如,所述渗透保护剂可为甘氨酸甜菜碱。在一些实施方案中,所述渗透保护剂为糖或多元醇。例如,所述渗透保护剂可为海藻糖。在一些实施方案中,所述渗透保护剂为氨基酸。例如,所述渗透保护剂可为脯氨酸。在所述细胞悬浮缓冲液的一些实施方案中,所述缓冲液的摩尔渗透压浓度为约250-350mOsm/L。在一些实施方案中,所述渗透保护剂贡献所述缓冲液的所述摩尔渗透压浓度的多达10%、20%、30%、40%、50%、60%、70%、80%、90%或100%。在一些实施方案中,所述缓冲液包含约230-330mM甜菜碱和约10mM NaCl。
本文还公开一种使聚核苷酸连接于固体支撑物的方法,其中所述聚核苷酸含有条形码序列。所述方法包括以下步骤:a)产生反相乳液的亲水性隔室,所述亲水性隔室含有:固体支撑物、包含条形码序列的条形码寡核苷酸和经由捕获部分结合于所述固体支撑物的表面的寡核苷酸,其中所述结合的寡核苷酸包含与所述条形码寡核苷酸的3’序列互补的3’序列;和b)执行聚合酶延伸反应以将所述条形码序列并入所述固体支撑物上的所述结合的寡核苷酸中。在一些实施方案中,所述条形码寡核苷酸进一步包含与PCR反向引物序列同一或互补的5’序列。这些实施方案可进一步包括使用荧光团标记的反向引物执行PCR反应。在一些实施方案中,所述固体支撑物为珠粒。在一些实施方案中,所述捕获部分为抗生蛋白链菌素。在一些实施方案中,所述捕获部分包含羧基、环氧基或羟基。在一些实施方案中,所述捕获部分包含金以捕获硫醇化寡核苷酸。
在一些实施方案中,所述条形码寡核苷酸进一步包含通用引发序列和结合位点。所述条形码寡核苷酸可进一步包含选自由T7、T3和SP6组成的组的RNAP启动子。或者或另外,所述条形码寡核苷酸可进一步包含选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组的切口核酸内切酶限制位点。所述结合位点可为一个或多个G核苷酸。
还公开另一种使聚核苷酸连接于固体支撑物的方法,其中所述聚核苷酸含有条形码序列。所述方法包括以下步骤:a)提供:固体支撑物、包含W序列的第一条形码寡核苷酸和经由捕获部分结合于所述固体支撑物的表面的寡核苷酸,其中所述结合的寡核苷酸包含(i)S1x序列和(ii)与所述第一条形码寡核苷酸的3’序列互补的序列;b)执行聚合酶延伸反应或接合反应以将所述W序列并入所述结合的寡核苷酸中;c)提供第二条形码寡核苷酸,其包含(i)S2y序列和(ii)与由步骤b)产生的所述结合的寡核苷酸的3’端互补的3’序列;以及d)执行聚合酶延伸反应或接合反应以将所述S2y序列并入所述结合的寡核苷酸中,由此使聚核苷酸连接于所述固体支撑物,其中所述聚核苷酸含有条形码序列,并且所述条形码序列包含所述S1x、W和S2y序列。
在这种方法的一些实施方案中,所述固体支撑物为珠粒。在一些实施方案中,所述捕获部分为抗生蛋白链菌素。在一些实施方案中,所述捕获部分包含羧基、环氧基或羟基。在一些实施方案中,所述捕获部分包含金以捕获硫醇化寡核苷酸。在一些实施方案中,选择的条形码寡核苷酸进一步包含通用引发序列和结合位点,所述选择的条形码寡核苷酸为所述第一条形码寡核苷酸或所述第二条形码寡核苷酸。所述选择的条形码寡核苷酸可进一步包含选自由T7、T3和SP6组成的组的RNAP启动子。或者或另外,所述选择的条形码寡核苷酸可进一步包含选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组的切口核酸内切酶限制位点。所述结合位点可为一个或多个G核苷酸。
本文进一步公开一种通过前述方法的任何实施方案制备的固体支撑物,其中所述固体支撑物连接于聚核苷酸并且所述聚核苷酸含有条形码序列。还公开一种条形码文库,其包含多种这些固体支撑物。
另外,本文公开一种用于封装细胞、条形码衔接模板和用于制造所关注的聚核苷酸的试剂的微流体液滴器件。所述器件包括(a)三个独立控制的压力源、(b)三个微流体途径、(c)三个流量传感器、(d)两个样品环管、(e)微流体液滴芯片和(f)样品收集容器,其中:各压力源通过所述微流体途径之一耦合于并且驱动流体,所述流量传感器之一沿着各微流体途径安置于所述个别压力源的下游,第一微流体途径穿过第一样品环管,第二微流体途径穿过第二样品环管,所述第一和第二样品环管与热冷却单元接触,所述第一和第二微流体途径在第一接合点合并以形成组合途径,所述组合途径和第三微流体途径在第二接合点合并以形成样品途径,所述第二接合点出现于所述微流体液滴芯片内和所述第一接合点的下游,并且所述样品途径进入所述第二接合点的下游的所述样品收集容器中,使得(a)-(f)流体连接。
在所述器件的一些实施方案中,各压力源包括压力泵。在一些实施方案中,各压力源包括注射泵。在一些实施方案中,所述第一样品环管经过配置以计量朝向所述微流体液滴芯片的水溶液的流量,其中所述水溶液包含细胞和条形码衔接模板。在一些实施方案中,所述第二样品环管经过配置以计量朝向所述微流体液滴芯片的反应混合物的流量,其中所述反应混合物包含用于细胞溶解的试剂和用于制造所关注的聚核苷酸的试剂。在一些实施方案中,所述第三微流体途径经过配置以递送油/表面活性剂混合物至所述微流体液滴芯片。在一些实施方案中,所述热冷却单元包括珀耳帖器件。在一些实施方案中,所述热冷却单元包括冰箱。在一些实施方案中,所述第一接合点出现于所述液滴芯片内。在一些实施方案中,所述第三微流体途径分成所述微流体液滴芯片的上游的两个子途径,所述两个子途径在所述第二接合点与所述组合途径合并,并且所述第二接合点具有流动聚焦几何形状。在一些实施方案中,所述第二接合点具有t形接合几何形状。在一些实施方案中,所述第一微流体途径经过配置以容纳细胞,并且所述第二微流体途径经过配置以容纳结合于固体支撑物的条形码衔接模板。
本文公开一种用于制造一种或多种所关注的聚核苷酸的方法,所述方法包括获得cDNA文库,所述文库包含多种与一种或多种获自一个或多个受试者的样品缔合的cDNA,其中各cDNA与所述一种或多种样品中的单一样品缔合,并且其中与各样品缔合的所述cDNA存在于独立容器或隔室中。在一些实施方案中,衔接分子添加至与各样品缔合的所述cDNA中以制造所述一种或多种所关注的聚核苷酸。在一些实施方案中,所述衔接分子由包含通用引发序列、条形码和cDNA结合位点的衔接构建体产生。
在一些方面,所述衔接分子使用等温反应产生。在一些方面,所述衔接构建体进一步包含RNA聚合酶(RNAP)启动子。在一些方面,所述RNAP启动子选自由T7、T3和SP6组成的组。在一些方面,所述衔接构建体进一步包含切口核酸内切酶限制位点。在一些方面,所述切口核酸内切酶限制位点选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组。在一些方面,所述衔接子为通过RNAP产生的RNA衔接子。在一些方面,所述衔接子为通过切口核酸内切酶和链置换DNA聚合酶产生的DNA衔接子。在一些方面,所述链置换DNA聚合酶选自由Klenow exo-和Bst大片段和其工程改造的变体(如Bst 2.0)组成的组。
在一些方面,所述方法进一步包括使所述衔接分子的3’端连接于所述文库中的各cDNA的3’端以制造所述一种或多种所关注的聚核苷酸。
在一些方面,所述衔接子通过使所述衔接子退火至在反转录反应期间产生的cDNA的‘3尾来添加。在一些方面,各cDNA包含至少一个C核苷酸,其中C位于各cDNA的3’端,其中所述衔接区包含至少一个G核苷酸,其中G位于所述衔接区的3’端,并且其中所述衔接区经由所述G与C之间的结合连接于各cDNA。在一些方面,所述衔接分子为单链的,并且进一步包括通过允许酶使所述衔接分子呈双链而将所述衔接分子的互补序列并入各cDNA中。在一些方面,所述衔接分子的互补序列通过MMLV H-反转录酶并入各cDNA中以制造所关注的聚核苷酸。
在一些方面,各样品包含细胞。在一些方面,所述细胞为血细胞、免疫细胞、组织细胞或肿瘤细胞。在一些实施方案中,所述细胞为B细胞或T细胞。在一些方面,所述B细胞为成浆细胞、记忆B细胞或浆细胞。
本文还公开一种使条形码连接于固体支撑物的方法,所述方法包括以下步骤:a)产生反相乳液的亲水性隔室,所述亲水性隔室包含:其中所含的固体支撑物,其中所述固体支撑物包含经由捕获部分结合于表面的寡核苷酸,其中所述寡核苷酸包含与条形码寡核苷酸上的3’序列互补的3’序列;条形码寡核苷酸,其包含与所述结合的寡核苷酸的3’端互补的3’序列;及条形码序列;和b)执行聚合酶延伸反应以将所述条形码的序列添加至所述固体支撑物上的所述结合的寡核苷酸中。
在一些方面,所述条形码寡核苷酸进一步包含与反向PCR引物同一或互补的5’序列。在一些方面,所述方法进一步包括使用荧光团标记的反向引物执行PCR反应。
在一些方面,所述固体支撑物为珠粒或表面。在一些方面,所述捕获部分为抗生蛋白链菌素。在一些方面,所述条形码寡核苷酸进一步包含RNA聚合酶(RNAP)启动子和/或核酸内切酶限制位点、通用引发序列、cDNA结合位点。在一些方面,所述RNAP启动子选自由T7、T3和SP6组成的组。在一些方面,所述切口核酸内切酶限制位点选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组。在一些方面,所述cDNA结合位点为一个或多个G核苷酸。
本文还公开一种使条形码连接于固体支撑物的方法,所述方法包括以下步骤:a)提供固体支撑物,具有经由捕获部分结合于所述固体支撑物的寡核苷酸,其中所述寡核苷酸包含S1x序列和与第一条形码寡核苷酸上的3’序列互补的序列;第一条形码寡核苷酸,其包含与所述结合的寡核苷酸的序列互补的3’序列,和W序列;和b)执行聚合酶延伸反应或接合反应以将所述W序列添加至所述固体支撑物上的所述结合的寡核苷酸的所述S1x序列中;c)提供具有S2y序列的第二条形码寡核苷酸,其包含与在步骤b)中延伸的所述寡核苷酸的3’端互补的3’序列;d)执行聚合酶延伸反应或接合反应以将所述S2y序列添加至所述固体支撑物上的所述结合的寡核苷酸的所述S1x和W序列中,其中所述条形码序列包含所述S1x、W和S2y序列。
在一些方面,所述固体支撑物为珠粒。在一些方面,所述捕获部分为抗生蛋白链菌素。在一些方面,所述第一或第二条形码寡核苷酸进一步包含RNA聚合酶(RNAP)启动子和/或切口核酸内切酶限制位点、通用引发序列、cDNA结合位点。在一些方面,所述RNAP启动子选自由T7、T3和SP6组成的组。在一些方面,所述核酸内切酶限制位点选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组。在一些方面,所述cDNA结合位点为一个或多个G核苷酸。
本文还公开一种通过上文所公开的方法中的任一者产生的具有连接的条形码的固体支撑物。本文还公开一种珠粒条形码文库,其包含多种具有连接的条形码的所述固体支撑物。
本文还公开一种条形码衔接构建体,其包含通用引发序列、条形码和cDNA结合位点。在一些方面,所述构建体进一步包含RNAP启动子。在一些方面,所述RNAP启动子选自由T7、T3和SP6组成的组。在一些方面,所述构建体进一步包含切口核酸内切酶限制位点。在一些方面,所述切口核酸内切酶限制位点选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI组成的组。
本文还公开一种条形码衔接模板珠粒,其包含固体支撑物和经由捕获部分结合于所述固体支撑物的条形码衔接分子,其中所述条形码衔接分子包含条形码序列和cDNA结合位点。在一些方面,所述cDNA结合位点包含一个或多个G核苷酸。在一些方面,所述条形码序列包含序列S1x-W-S2y。本文还公开一种珠粒条形码文库,其包含多种如上文所公开的条形码衔接模板珠粒。
本文还公开一种聚核苷酸文库,其包含多种条形码衔接模板珠粒,所述珠粒包含固体支撑物和经由捕获部分结合于所述固体支撑物的条形码衔接分子,其中所述条形码衔接分子包含条形码序列和cDNA结合位点,其中cDNA区偶联于所述衔接子的3’端。
在一些方面,所述cDNA结合位点包含一个或多个G核苷酸。在一些方面,所述条形码序列包含序列S1x-W-S2y。
在一些方面,所述cDNA源于B细胞。在一些方面,所述B细胞为成浆细胞、记忆B细胞或浆细胞。在一些方面,所述cDNA源于B细胞源性可变免疫球蛋白区。
本文还公开如图17-19所示的微流体液滴器件。
附图简述
这些和其它特征、方面和优势关于以下描述和附图将变得更好理解,在附图中:
图1为根据本发明的一些实施方案的衔接分子或用于产生衔接分子的模板分子的图。衔接分子的序列可包括RNA聚合酶启动子和/或切口核酸内切酶位点,随后为通用引发序列(用于后续用于使引物退火的PCR步骤中),随后为条形码序列和核酸结合序列。
图2A和2B显示根据本发明的一些实施方案扩增或产生衔接分子的方法。在图2A中,RNA条形码衔接子在线性扩增反应中通过RNAP(如T7)合成,所述RNAP结合于DNA模板上的启动子序列并且合成单链条形码衔接子RNA。在图2B中,如Nt.BbvCI(NEB)的切口核酸内切酶用于在DNA模板的有义链上引入切口。DNA条形码衔接子接着在扩增反应中通过如Klenow exo-的链置换酶合成,所述链置换酶延伸切口并且置换所述单链条形码衔接子。
图3显示根据本发明的一些实施方案将条形码序列并入第一链cDNA中。此处合成RNA条形码衔接子以证明对cDNA编条形码。也可使用DNA条形码衔接子(在图2B中合成)。RNAP停止其启动子并且合成RNA条形码衔接子(图3,左上部)。在所述相同反应中,发生反转录并且产生第一链cDNA(右上部)。基于MMLV的H-反转录酶具有3’拖尾活性并且添加数个dC至所述第一链cDNA的3’端。所述条形码衔接子与所述拖尾dC(底部)碱基配对并且所述反转录酶使用所述条形码衔接子作为模板继续转录,从而将所述条形码序列并入所述第一链cDNA中。因此对所述反应中的所有mRNA编条形码。
图4显示在本发明的实施方案中RNA条形码衔接子具有比DNA条形码衔接子少的背景。在图3的编条形码反应中,寡聚(dT)和条形码衔接子均存在,并且两种寡核苷酸可引发反转录反应。当所述反应用寡聚(dT)(图4,顶部)引发时,所述反应照常进行。当所述RT反应用DNA条形码衔接子(中部)错误引发时,在PCR期间正向引物可停止有义和反义链并且引起非所需产物的扩增。当所述RT反应用RNA条形码衔接子(底部)引发时,当在PCR1中使用校正读码DNA聚合酶时生长链无法使用RNA核苷酸作为模板,并且因此错误引发的cDNA将不会在有义和反义链上含有条形码衔接序列。因此,非所需产物不应按指数规律扩增,导致显著减少背景。
图5A-C为卡通画,说明根据本发明的一些实施方案用于产生条形码衔接子和执行反转录的反应容积的分离。条形码衔接分子可在多个第一反应容积(如液滴)中酶法产生,所述第一反应容积由图5A中的垂直线表示。各第一反应容积可含有水溶液中的条形码衔接分子,所述条形码衔接分子均具有相同条形码序列。分别地,RNA分子可在多个第二反应容积中反转录,所述第二反应容积由图5B中的水平线表示。各第二反应容积可含有均源于相同样品的RNA分子。所述第一和第二反应容积可接着如通过合并液滴组合,所述液滴如由图5C中的交叉线表示。图5A和5B中的反应产物混合在一起,使得一种条形码序列被引入对应于各样品的反应容积中。所述条形码序列可并入第一链cDNA或PCR产物中。
图6A-D显示在本发明的各种实施方案中扩增条形码衔接模板以制造条形码衔接分子。图6A显示连接于如珠粒的固体表面的条形码衔接模板。图6B显示水溶液中的条形码衔接分子,所述分子由图6A中的条形码衔接模板的扩增产生。图6C显示单一条形码衔接模板分子。所述分子在水溶液中并且容纳在容器内部。图6D显示图6C的容器,其具有多种条形码衔接分子,所述分子由单一模板分子的扩增产生。
图7A-D显示由模板产生条形码衔接分子,其中所述模板连接于固体表面。在产生后,所述条形码衔接分子在水溶液中。图7A和7B显示连接于固体表面的条形码衔接模板。图7C显示由图7A中的条形码衔接模板酶法扩增的条形码衔接分子。图7D显示在图7B中的条形码衔接模板由所述固体表面化学或酶法裂解之后释放至溶液中的条形码衔接分子。
图8显示使用DNA条形码衔接子将条形码序列并入cDNA的第一链中。(顶部)所述条形码衔接子(包括3’聚T区)由条形码衔接模板使用DNA聚合酶产生。条形码衔接分子在水溶液中。(底部)所述条形码衔接子退火至mRNA的聚-A尾并且充当反转录的引物。所述条形码序列并入cDNA的第一链的5’端中。
图9显示使用DNA条形码衔接子将条形码序列并入cDNA的第一链中。(顶部)所述条形码衔接子(包括3’随机或半随机序列区)由条形码衔接模板使用DNA聚合酶产生。条形码衔接分子在水溶液中。(底部)所述条形码衔接子通过退火至至少部分地与所述3’序列区互补的RNA区而充当反转录的引物。所述条形码序列并入cDNA的第一链的5’端中。
图10为消除个别吸移步骤的编条形码工作流的示意性概述。简单地说,编条形码反应发生在油包水液滴中,其中细胞和含有条形码衔接子的珠粒通过液滴产生器件分布。条形码衔接子酶法扩增或从如珠粒的固体表面释放,并且所述条形码添加至来自细胞的所有转录物中。
图11显示使用充当RT-PCR的正向引物的DNA条形码衔接子将条形码序列并入扩增子中。所述条形码衔接子由DNA模板使用DNA聚合酶(左上部)酶法产生。条形码衔接分子在水溶液中。在独立反应容积中,或在相同反应容积中,使用mRNA模板、反转录酶、含有聚T区的引物和模板转换寡核苷酸合成cDNA的第一链(右上部)。所述模板转换寡核苷酸含有与所述条形码衔接子中的序列区互补的序列区。所述条形码序列接着在所述cDNA的PCR扩增期间并入扩增子中(底部)。所述条形码衔接子充当PCR的正向引物。
图12显示使用充当RT-PCR的反向引物的DNA条形码衔接子将条形码序列并入扩增子中。所述条形码衔接子由DNA模板使用DNA聚合酶(左上部)酶法产生。条形码衔接分子在水溶液中。在独立反应容积中,或在相同反应容积中,使用mRNA模板、反转录酶、含有聚T区的引物和模板转换寡核苷酸合成cDNA的第一链(右上部)。所述引物含有与所述条形码衔接子中的3’序列区互补的5’序列区。所述条形码序列接着在所述cDNA的PCR扩增期间并入扩增子中(底部)。所述条形码衔接子充当PCR的反向引物。
图13显示使用充当RT-PCR的反向引物的DNA条形码衔接子将条形码序列并入扩增子中。所述条形码衔接子由DNA模板使用DNA聚合酶酶法产生(左上部)。条形码衔接分子在水溶液中。在独立反应容积中,或在相同反应容积中,使用mRNA模板、反转录酶、含有3’随机序列区的引物和模板转换寡核苷酸合成cDNA的第一链(右上部)。所述引物可通过所述随机序列区退火至所述mRNA,并且还含有与所述条形码衔接子中的3’序列区互补的5’序列区。所述条形码序列接着在所述cDNA的PCR扩增期间并入扩增子中(底部)。所述条形码衔接子充当PCR的反向引物。
图14A-C说明根据本发明的实施方案使用核酸标记物查询用于所选表型的细胞群体的方法。除了对来自细胞的RNA编条形码,还可对包括来自非细胞来源的RNA的任何RNA编条形码。非细胞RNA可通过任何方式,如通过用核酸标记物标记细胞而被引入反应容积中。这种标记物可包括偶联于分子标记的核酸,所述分子标记如抗体(图14A)、抗原(图14B)或pMHC(图14C)。所述核酸标记物可结合于所述群体中的一些或所有细胞,视细胞的表型和其对所述分子标记的亲和力而定。所述群体中的所有细胞均可接着溶解并且可对各细胞中的mRNA编条形码。关于结合所述核酸标记物的细胞,也可对缔合的核酸编条形码。这种核酸可为RNA或具有RNAP启动子的dsDNA模板,所述启动子如T7、T3或SP6启动子。测序可接着使非内源性RNA序列与特异性细胞缔合,由此检测何种细胞结合于所述分子标记。不同分子标记可偶联于不同核酸序列,使得能够鉴别多种细胞表型。
图15显示根据本发明的一些实施方案在一种反应中合成条形码衔接模板珠粒。(左侧)珠粒偶联于寡核苷酸。偶联可通过将生物素标记的寡核苷酸偶联于抗生蛋白链菌素涂布的珠粒上进行,并且也可使用所属领域中已知的其它方式偶联。(右侧)偶联的珠粒、正向和反向引物以及含有条形码序列和与所述正向和反向引物互补的序列的条形码寡核苷酸均存在于反应容器中,其中所述条形码寡核苷酸优选地以仅单一拷贝存在。接着进行PCR以扩增所述条形码序列并且将其并入珠粒偶联的寡核苷酸中以形成条形码衔接模板珠粒。
图16显示根据本发明的一些实施方案在多个步骤中合成条形码衔接模板珠粒。(顶部)珠粒偶联于含有独特S1序列的寡核苷酸的(多个拷贝)。执行多种独立偶联反应,其中各偶联反应使用含有不同的独特S1序列的寡核苷酸。各自偶联于具有不同的独特S1序列的寡核苷酸的珠粒接着汇集在一起,形成具有S1x序列的珠粒的文库。(中间)这些珠粒接着用于延伸反应。在各反应中,含有独特W序列的寡核苷酸与偶联于所述珠粒的含S1x寡核苷酸互补地碱基配对,并且执行使用DNA聚合酶的延伸反应。汇集来自所有延伸反应的珠粒,并且形成含有S1x序列各自与独特W序列的组合的珠粒的文库。(底部)使来自前一步骤的双链DNA变性并且从所述珠粒洗去反义链。如先前在所述珠粒上执行额外的独立延伸反应,但与偶联于所述珠粒的含S1x和W寡核苷酸互补地碱基配对的寡核苷酸在各独立反应中含有不同的独特S2序列。汇集来自所有延伸反应的珠粒,并且获得含有条形码衔接模板的珠粒的文库,其中S1x、W和S2y序列的组合形成所述条形码序列。大量的独特条形码序列可因此在这种组合方法中获得。此外,多种独特W序列可各自与S1x和S2y序列组合,产生一般格式S1x-Wz-S2y的条形码。
图17显示根据本发明的实施方案的液滴器件。三个Dolomite P-泵配备有流量传感器。第一个P-泵经由微流体管直接连接于2-Reagent液滴芯片,所述微流体管并入T形接合点以将线路分成两个输入。这是油输入线路。另两个P-泵经由流体管连接于FEP样品环管,所述环管配合于珀耳帖器件的凹槽中,所述珀耳帖器件用于在所述器件操作时保持样品冷冻,并且这些环管各自连接于所述2-Reagent液滴芯片。各样品环管在其前端并入四通阀,使得样品可借助于注射器装载于所述环管中。第一个样品环管欲填充细胞和编条形码珠粒悬浮液,而第二个环管欲填充RT/溶解混合物。所述样品环管可水平地并且在所述液滴芯片上方或与所述液滴芯片相齐定向以便避免任何上坡区段,细胞和珠粒可能难以行进通过所述上坡区段。
图18提供图17所示的液滴器件的配置的细节。零件由IDEX H&S零件号给出:1.0.A)1528(110mm);1.0.B)P-732;1.0.C)P-232/P-248;1.0.D)1688(300mm);1.0.E)M-645;1.0.F)P-630;1.0.H)P-632;1.0.J)P-702;1.0.K)1529(50mm);1.0.L)V-101D;1.0.N)P-732;1.0.O)P-624;1.0.T)1531(900mm);1.2.A)P-630;1.2.B)1516(500mm);1.2.C)P-702;1.2.D)1529(150mm);1.2.E)P-702;1.2.G)1560(150mm);1.3.A)1528(135mm);1.5.A)1516(150mm);1.5.B)1529(300mm);1.7.A)61005;1.7.B)65020;2.0.A)1477(1254mm);2.0.B)1527(1254mm);2.0.C)1520(120mm);2.0.D)1520(600mm);2.0.E)1520(200mm);2.0.F)1520(200mm)。出口管(从所述芯片至所述样品收集管)为1562(180mm)。
图19显示本文所述的液滴器件的替代实施方案。所述样品环管与冰箱接触。
图20显示由条形码衔接模板珠粒扩增的RNA条形码衔接子,所述珠粒使用多步骤方法制造。条形码衔接模板珠粒用于体外转录反应中。存在来自分别使用S1-寡聚+W-寡聚-a+S2-寡聚-a和S1-寡聚+w-寡聚-b+S2-寡聚-b制造的珠粒的条带。
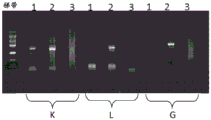
图21显示在多种缓冲液中执行的编条形码反应。1、2和3是指三种反应缓冲液,其分别为下文所述的0.5x MMLV、1x Thermopol DF和0.5x TAE缓冲液。K、L和G是指κ、λ和γ免疫球蛋白链。所有链在所用的不同反应缓冲液中扩增。
图22显示编条形码反应使用RNA条形码更好地工作。1、2和3是指三种反应条件,其为使用RNA条形码衔接子的1x MMLV和0.5x MMLV条件以及使用DNA条形码衔接子的1xMMLV。K、L和G是指κ、λ和γ免疫球蛋白链。使用DNA衔接子的反应中的条带由于高背景而模糊。
图23显示在具有条形码衔接模板的液滴反应容器中由于对单一B细胞编条形码而扩增的产物。对应于κ和λ轻链(“K/L”)和μ重链(“M”)的条带可清楚可见。
图24显示在与编条形码珠粒共封装于油包水乳液中之后轻链(κ/λ)和重链(γ)靶标的RT/PCR扩增。各样品在成对泳道中运行,一个泳道用于κ/λ轻链(左侧)并且一个泳道用于γ重链(右侧)。乳液样品包括细胞+珠粒共封装实验样品(细胞+珠粒)以及两个对照样品,所述对照样品相同地制备,除了在一种对照样品中,条形码模板衔接珠粒用水性条形码衔接模板(细胞+水性BC)置换,并且在一种对照样品中,细胞用获自AllCells的纯化的人类PBMC RNA模板置换(RNA+珠粒)。还包括整体阳性和阴性对照,其不进入乳液器件(分别为R-和R+1)。产物条带关于所述实验样品和所有阳性对照均可见,并且在阴性对照中不存在。
图25说明使用多种条形码衔接模板类型制造条形码衔接模板珠粒的方法。含条形码寡核苷酸由预期长度82bp成功地产生(左上部)。成功地获得单色条形码衔接模板珠粒(右侧)。顶部图形最初门控于AF647-珠粒上并且底部图形最初门控于FAM-Cy3-珠粒上,使得在两个图形中绘出的闸门仅显示单色珠粒。珠粒成功地用于对RNA编条形码(左下部)。此处,T细胞受体α和β链成功地编条形码并且扩增。先前产生的珠粒用作阳性对照(泳道1-2),并且单色条形码衔接模板珠粒(泳道4-7)与阴性对照(泳道3)进行比较。DNA在2%琼脂糖凝胶上进行分析,其中100bp梯带装载于左侧泳道中。
图26说明通过将条形码衔接模板珠粒和细胞封装于变化大小的液滴中来对T细胞受体α链有效编条形码。编条形码RNA在编条形码之后扩增并且在2%琼脂糖凝胶上进行分析。
图27显示TCRα和β链的文库PCR扩增产物。产物在2%琼脂糖凝胶上显现。100bp梯带装载于右侧泳道中。
图28显示IFNγ、CD8和CD4基因的文库PCR扩增产物。产物在2%琼脂糖凝胶上显现。100bp梯带装载于右侧泳道中。
图29显示转录组学文库的文库PCR扩增产物。产物在2%琼脂糖凝胶上显现。100bp梯带装载于右侧泳道中。
定义
当所述术语用于本文中时,将序列“并入”聚核苷酸中是指通过磷酸二酯键共价连接一系列核苷酸与所述聚核苷酸的剩余部分,例如在所述聚核苷酸的3’或5’端,其中所述核苷酸依所述序列所指示的顺序连接。如果聚核苷酸含有序列或其互补序列,那么所述序列已经“并入”所述聚核苷酸中,或等效地,所述聚核苷酸“并入”所述序列。序列并入聚核苷酸中可酶法(例如,通过接合或聚合)或使用化学合成(例如,通过亚磷酰胺化学)发生。
如本文所用,术语“扩增(amplify)”和“扩增(amplification)”是指完全或部分地酶法拷贝聚核苷酸的序列,以便产生更多也含有所述序列或其互补序列的聚核苷酸。正在拷贝的序列被称作模板序列。扩增的实例包括通过RNA聚合酶进行的DNA模板化RNA合成、通过反转录酶进行的RNA模板化第一链cDNA合成以及使用热稳定性DNA聚合酶进行的DNA模板化PCR扩增。扩增包括所有引物-延伸反应。
如本文所用,术语“等温”是指在恒定温度或温度范围下进行的反应,如酶反应。
术语“缔合(associated)”在本文中用于指样品与DNA分子、RNA分子或起源于或源于所述样品的其它聚核苷酸之间的关系。如果聚核苷酸为内源性聚核苷酸,那么所述聚核苷酸与样品缔合,即,当样品选自或源于内源性聚核苷酸时,所述聚核苷酸存在于所述样品中。例如,细胞的内源性mRNA与所述细胞缔合。由这些mRNA的反转录产生的cDNA和由所述cDNA的PCR扩增产生的DNA扩增子含有所述mRNA的序列并且还与所述细胞缔合。与样品缔合的聚核苷酸无需位于或合成于所述样品中,并且即使在所述样品已经受到破坏后(例如,在细胞已经溶解后)仍被认为与所述样品缔合。分子编条形码或其它技术可用于确定混合物中的何种聚核苷酸与特定样品缔合。
当所述术语用于本文中时,“反应容积”(或等效地,“容器”或“隔室”)为其中可容纳例如水溶液的液体的容积并且所述容积保持与液体的其它所述容积或周围媒介物分离(例如,隔离)的空间。反应容积与其周围环境之间的分离可由所述反应容积周围的固体屏障或由相分离引起。例如,悬浮于疏水性载液中的水性微流体液滴可构成反应容积,因为水在所述载液中不可混溶。因此,在所述载液中彼此分离的两种液滴保持分离,并且溶解于一种液滴中的核酸或其它亲水性物质无法离开所述液滴或运送至另一液滴。反应容积也可通过例如烧瓶、烧杯、离心管以及多孔板中的孔来界定。
“添加”条形码衔接子至与样品缔合的RNA中涉及将所述衔接分子引入含有这些RNA的反应容积中,使得所述RNA可参与编条形码反应。在添加后,所述条形码衔接子可例如通过与RNA杂交直接与一种或多种RNA反应,或可参与其中RNA分子充当模板的聚合反应或一系列反应(例如反转录或RT-PCR)。
在一些方面,组合物可包括聚核苷酸。术语“聚核苷酸”是指如DNA分子和RNA分子和其类似物的核酸(例如,使用核苷酸类似物或使用核酸化学产生的DNA或RNA)。如所需,聚核苷酸可例如使用公认的核酸化学以合成方式或使用例如聚合酶酶法制得并且必要时可进行修饰。典型修饰包括甲基化、生物素标记和其它所属领域已知的修饰。另外,聚核苷酸可为单链的或双链的并且必要时连接于可检测部分。在一些方面,聚核苷酸可包括杂合分子,例如包含DNA和RNA。
“G”、“C”、“A”、“T”和“U”各自一般代表分别含有鸟嘌呤、胞嘧啶、腺嘌呤、胸腺嘧啶核苷和尿嘧啶作为碱基的核苷酸。然而,应了解术语“核糖核苷酸”或“核苷酸”也可指修饰的核苷酸或代替置换部分。熟练人员充分了解鸟嘌呤、胞嘧啶、腺嘌呤和尿嘧啶可由其它部分置换而不会大致改变包含具有所述置换部分的核苷酸的寡核苷酸的碱基配对特性。例如而不限于,包含肌苷作为其碱基的核苷酸可与含有腺嘌呤、胞嘧啶或尿嘧啶的核苷酸碱基配对。因此,含有尿嘧啶、鸟嘌呤或腺嘌呤的核苷酸可在核苷酸序列中由含有例如肌苷的核苷酸置换。在另一实施例中,所述寡核苷酸中任何位置的腺嘌呤和胞嘧啶可分别用鸟嘌呤和尿嘧啶置换以形成与靶标mRNA碱基配对的G-U摇摆。含有所述置换部分的序列适用于本文所述的组合物和方法。
如本文所用,并且除非另外指示,否则术语“互补”在用于描述相对于第二核苷酸序列的第一核苷酸序列时是指包含所述第一核苷酸序列的聚核苷酸在某些条件下与包含所述第二核苷酸序列的聚核苷酸杂交并且形成双链体结构的能力,如熟练人员应了解。所述条件可例如为严格条件,其中严格条件可包括:400mM NaCl,40mM PIPES pH 6.4,1mMEDTA,50℃或70℃持续12-16小时,随后洗涤。其它条件可适用,如可在生物体内部遇到的生理学相关条件。熟练人员将能够根据杂交核苷酸的最终应用确定主要适用于两个序列的互补性的测试的条件的集合。
互补序列包括包含第一核苷酸序列的聚核苷酸区域与包含第二核苷酸序列的聚核苷酸区域在一个或两个核苷酸序列的长度或长度的一部分上的碱基配对。所述序列可在本文中称作关于彼此“互补”。然而,在第一序列在本文中称作关于第二序列“大致互补”的情况下,所述两个序列可为互补的,或其可在碱基配对的区域内包括一个或多个但一般不超过约5、4、3或2个错配碱基对。关于具有错配碱基对的两个序列,所述序列将被视为“大致互补”,只要所述两个核苷酸序列经由碱基配对彼此结合。
如本文所用,“互补”序列也可包括非Watson-Crick碱基对和/或由非天然和修饰的核苷酸形成的碱基对,或完全由所述碱基对形成,只要实现关于其杂交能力的上述实施方案。所述非Watson-Crick碱基对包括但不限于G:U摇摆或Hoogstein碱基配对。
术语百分比“同一性”在两个或更多个核酸或多肽序列的情形中是指如使用下文所述的序列比较算法之一(例如BLASTP和BLASTN或熟练人员可获得的其它算法)或通过目视检查所测量,当关于最大对应进行比较和比对时,具有规定的相同核苷酸或氨基酸残基百分率的两个或更多个序列或子序列。视应用而定,百分比“同一性”可在进行比较的序列区域内,例如在功能结构域内存在,或替代地在待比较的两个序列的全长内存在。
关于序列比较,典型地一个序列充当与测试序列进行比较的参考序列。当使用序列比较算法时,将测试和参考序列输入计算机中,必要时指定子序列座标并且指定序列算法程序参数。所述序列比较算法接着基于指定的程序参数计算所述测试序列相对于所述参考序列的百分比序列同一性。
可进行用于比较的序列的最佳比对,例如通过Smith和Waterman,Adv.Appl.Math.2:482(1981)的局部同源性算法,通过Needleman和Wunsch,J.Mol.Biol.48:443(1970)的同源性比对算法,通过Pearson和Lipman,Proc.Nat'l.Acad.Sci.USA 85:2444(1988)的相似性搜索方法,通过这些算法的计算机化执行(GAP、BESTFIT、FASTA和TFASTA,Wisconsin Genetics Software Package,Genetics ComputerGroup,575Science Dr.,Madison,Wis.)或通过目视检查(一般参看Ausubel等人,下文)。
适用于确定百分比序列同一性和序列相似性的算法的一个实例为BLAST算法,其描述于Altschul等人,J.Mol.Biol.215:403-410(1990)中。用于执行BLAST分析的软件可通过National Center for Biotechnology Information网站公开可得。BLAST算法参数W、T和X确定所述算法的灵敏性和速度。BLASTN算法(关于核苷酸序列)使用字长(W)11、期望值(E)10、M=5、N=-4和两个链的比较作为缺省值。
同一性序列包括包含第一核苷酸序列的聚核苷酸与包含第二核苷酸序列的聚核苷酸在一个或两个核苷酸序列的完整长度内的100%同一性。所述序列可在本文中称作关于彼此“完全同一”。然而,在一些方面,在第一序列在本文中称作关于第二序列“大致同一”的情况下,所述两个序列可为完全互补的,或其可在比对后具有一个或多个但一般不超过约5、4、3或2个错配核苷酸。在一些方面,在第一序列在本文中称作关于第二序列“大致同一”的情况下,所述两个序列可为完全互补的,或其可彼此至少约50、60、70、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98或99%同一。为了确定本文所述的两个核苷酸序列的百分比同一性,可使用上文所述的BLASTN的缺省设定。
在第一序列在本文中称作关于第二序列的同一性“相异”的情况下,所述两个序列在比对后具有至少一个或多个错配核苷酸。在一些方面,相异序列可在比对后具有2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个错配核苷酸。在一些方面,相异序列可彼此约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、60、70、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99或少于100%同一。在一些方面,在第一序列在本文中称作关于第二序列“相异”的情况下,所述两个序列可具有大致或完全同一序列,不过基于所述序列内的修饰的不同模式彼此不同。所述修饰是本领域中一般已知的,例如甲基化。
在一些方面,聚核苷酸可存在于聚核苷酸的文库中。在一些方面,聚核苷酸文库可包括多种聚核苷酸。在一些方面,所述多种聚核苷酸中的各聚核苷酸可源于单一样品。在一些方面,单一样品可包括单细胞,如B细胞。
本文中使用常规表示法来描述核苷酸序列:单链核苷酸序列的左手端为5'端;双链核苷酸序列的左手方向被称作5'方向。核苷酸5'至3'添加至新生RNA转录物中的方向被称作转录方向。与mRNA具有相同序列的DNA链被称作“编码链”;在与由所述DNA转录的mRNA具有相同序列的DNA链上并且位于所述RNA转录物的5'至5'端的序列被称作“上游序列”;在与所述RNA具有相同序列的DNA链上并且在所述编码RNA转录物的3'至3'端的序列被称作“下游序列”。
术语“信使RNA”或“mRNA”是指无内含子并且可翻译成多肽的RNA。
术语“cDNA”是指与mRNA互补或同一的DNA,呈单链或双链形式。
术语“扩增子”是指核酸扩增反应(例如RT-PCR)的扩增产物。
术语“杂交”是指与互补核酸特异性非共价结合相互作用的序列。杂交可针对核酸序列的全部或一部分发生。所属领域的技术人员应认识到核酸双链体或杂合物的稳定性可由Tm确定。关于杂交条件的额外指导可发现于Current Protocols in MolecularBiology,John Wiley&Sons,N.Y.,1989,6.3.1-6.3.6中和Sambrook等人,MolecularCloning,a Laboratory Manual,Cold Spring Harbor Laboratory Press,1989,第3卷中。
如本文所用,“区域”是指聚核苷酸的核苷酸序列的邻接部分。本文中描述的区域的实例包括鉴别区、样品鉴别区、板鉴别区、衔接区等。在一些方面,聚核苷酸可包括一个或多个区域。在一些方面,聚核苷酸可包括少于2、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个区域。在一些方面,区域可偶联。在一些方面,区域可操作性偶联。在一些方面,区域可物理偶联。
如本文所用,“可变区”是指由基因重组或基因转换事件产生的可变核苷酸序列,所述事件如V(D)J重组和在上游VH基因区段与重排VDJ基因之间的同源重组以制造最终表达的基因产物。实例为但不限于免疫球蛋白基因和T细胞受体基因。例如,其可包括从所关注的T细胞或B细胞分离的免疫球蛋白的V、J和/或D区或T细胞受体序列,所述细胞如活化的T细胞或活化的B细胞。
如本文所用,“B细胞可变免疫球蛋白区”是指从B细胞分离的可变免疫球蛋白核苷酸序列。例如,可变免疫球蛋白序列可包括从所关注的B细胞分离的免疫球蛋白序列的V、J和/或D区,所述细胞如记忆B细胞、活化的B细胞或成浆细胞。
如本文所用,“条形码”或“条形码序列”是指可偶联于至少一个核苷酸序列以便例如稍后鉴别所述至少一个核苷酸序列的任何独特序列标记。
如本文所用,“条形码集合”是指可偶联于来自样品的核苷酸序列的序列的任何独特集合,其中核苷酸序列偶联于所述集合中的一个条形码序列,以便例如稍后鉴别所述核苷酸序列。
术语“条形码衔接子”、“编条形码衔接子”和“条形码衔接分子”在本文中可互换使用以指包含独特条形码序列的寡核苷酸。
术语“条形码衔接模板”、“衔接模板”、“模板分子”、“条形码衔接构建体”和“衔接构建体”在本文中可互换使用以指可用作模板来扩增并且制造单链条形码衔接分子的包含条形码序列的核酸分子。
如本文所用,“条形码衔接模板珠粒”是指偶联于一种或多种条形码衔接模板的珠粒。
如本文所用,“编条形码”或“编条形码反应”是指连接条形码序列或条形码序列的互补序列与核酸的反应。所述条形码衔接子无需必定与所述核酸共价连接,而是所述条形码序列信息本身与所述核酸连接或并入所述核酸中。“对核酸编条形码”、“对细胞编条形码”、“对来自细胞的核酸编条形码”、“对来自反应容器的核酸编条形码”和“对反应容器编条形码”可互换使用。
如本文所用,“鉴别区”是指可偶联于至少一个核苷酸序列以便例如稍后鉴别所述至少一个核苷酸序列的核苷酸序列标记(例如独特条形码序列)。在一些方面,条形码序列用作样品鉴别区。在一些方面,条形码集合用作样品鉴别区。
如本文所用,“免疫球蛋白区”是指来自抗体的一个或两个链(重链和轻链)的核苷酸序列的邻接部分。
如本文所用,“衔接区”或“衔接分子”是指偶联第一核苷酸序列至第二核苷酸序列的连接子。在一些方面,衔接区可包括充当连接子的核苷酸序列的邻接部分。在一些方面,衔接区或衔接分子可包括结合位点,如cDNA结合位点。例如,结合位点可具有序列GGG并且经由GGG与CCC之间的结合偶联第一序列至第二序列。在一些方面,所述衔接区或衔接分子可包含如RNA聚合酶启动子、切口核酸内切酶限制位点、通用引发序列、条形码和cDNA结合位点的元件。
术语“样品”可包括从受试者(例如哺乳动物受试者、动物受试者、人类受试者或非人类动物受试者)取得的RNA、DNA、单细胞或多个细胞或细胞的片段或体液的等分试样。样品可由所属领域的技术人员使用现在已知或以后发现的任何方式进行选择,所述方式包括离心、静脉穿刺、抽血、排泄、擦拭、射精、按摩、活组织检查、针吸、灌洗样品、刮削、手术切口、激光捕获显微切割、梯度分离或干预或所属领域中已知的其它方式。样品也可由所属领域的技术人员使用已知与所关注的样品缔合的一种或多种标记物进行选择。样品也可使用所属领域中已知的方法进行选择,所述方法如细胞分选和FACS。
发明详述
本发明的实施方案提供一种在各反应容器中产生独特核酸编条形码衔接子的方法,使得所述核酸编条形码衔接子在水相中但产生所述衔接子的模板可连接于固体表面(如连接于珠粒)或以游离形式处于溶液中。核酸编条形码衔接子为包含独特条形码序列的任何聚核苷酸序列并且可或可不具有修饰(例如,生物素标记或含有C18间隔区)或含有修饰的聚核苷酸(如2’-O-甲基RNA碱基)。
还提供使用本文公开的方法产生的组合物。相应地,本发明提供RNA和DNA衔接子的组合物和用于其产生的构建体。尤其还提供条形码衔接模板珠粒文库、装载有RNA条形码衔接子的乳液液滴文库、含有具有细胞的条形码文库的乳液、编条形码cDNA文库和微流体液滴产生器件。
在一些实施方案中,所述编条形码衔接模板为双链DNA(dsDNA)模板,其包含以下序列:5’-T7启动子–通用引发序列–条形码序列–结合序列-3’。所述T7启动子序列允许通过T7 RNA聚合酶由模板合成RNA编条形码衔接子。所述通用引发序列用于与下游使用的PCR引物互补。所述结合序列由一个或多个鸟嘌呤碱基(G)组成并且允许所述编条形码衔接子与第一链cDNA的3’端互补碱基配对(图1)。
可使用其它启动子序列,如但不限于T3和SP6启动子序列,其允许分别通过T3和SP6 RNA聚合酶合成RNA编条形码衔接子。也可使用不具有特异性启动子序列的其它RNA聚合酶,只要在大部分情形中合成全长或接近全长编条形码衔接子(图2A)。也可使用等温扩增,典型地使用具有链置换活性的DNA聚合酶,如Bst大片段和Klenow 3’→5’exo-,只要在大部分情形中合成全长或接近全长编条形码衔接子。可使用特异性引物或切口核酸内切酶序列来代替启动子序列,视所用的等温扩增方法而定(图2B)。因此产生的编条形码衔接子将包含DNA核苷酸而非RNA核苷酸。RNA或DNA编条形码衔接子可连接于所关注的聚核苷酸。
先前已经描述连接编条形码衔接子至第一链cDNA的3’端(PCT/US2012/000221)。简单地说,H-MMLV反转录酶具有3’dC拖尾活性并且添加非模板化dC至第一链cDNA。如果以至少一个G终止的编条形码衔接子也存在,那么所述衔接子可与所述第一链cDNA的3’dC碱基配对并且所述反转录酶经历模板转换并且使用所述编条形码衔接子作为模板继续转录。所述反转录酶因此经由磷酸二酯键共价添加所述条形码序列至所述第一链cDNA的3’端(图3)。
在一些实施方案中,编条形码衔接子使用T7 RNA聚合酶由含有5’T7启动子的双链DNA(dsDNA)线性扩增。在一些实施方案中,所述编条形码衔接子在与所述反转录反应相同的反应中线性扩增。由dsDNA模板扩增编条形码衔接子至少提供以下优势:
1.编条形码衔接模板可连接于珠粒(每个珠粒独特条形码)并且存储于相同存储容器中
2.独特编条形码衔接子的多个拷贝可递送至反应容器中而不使用个别吸移步骤
3.编条形码衔接子扩增,从而克服可连接于各珠粒的聚核苷酸的有限量
4.扩增的条形码在水相中并且利用快得多的液相而非固相动力学
还存在使用RNA编条形码衔接子而非DNA编条形码衔接子时所涉及的优势:
1.RNA编条形码衔接子可在连接所述条形码序列至所关注的聚核苷酸的模板转换反应中更有效,因为反转录酶典型地使用RNA而非DNA作为模板并且模板转换由所述反转录酶体内使用以在反转录病毒的复制中转换为RNA模板。
2.当在下游PCR反应中使用校正读码DNA聚合酶时,使用完全RNA转录物作为衔接子导致较少背景。当所述条形码衔接子错误引发并且起始反转录时,背景出现,导致在第一链cDNA的5’和3’端均添加条形码衔接序列。这些可在PCR中通过与所述条形码衔接子互补的仅一种引物扩增。然而,如果校正读码DNA聚合酶在PCR期间使用,那么其将不转录所述RNA引物(图4),从而消除来自条形码衔接子错误引发的背景。
由于涉及大量的编条形码反应,NextGen测序最适合对编条形码核酸测序以利用生物信息学使来自相同反应容器的核酸彼此缔合。额外条形码可与相异于另一样品集合的样品集合缔合并且可使用具有独特条形码序列的PCR引物缔合。这些额外条形码也称作板-ID。板-ID赋予优势,如在同一测序运作中区别不同样品集合,或利用生物信息学追踪并且消除不同样品集合之间的任何潜在污染。
由于PCR和NextGen测序误差不可避免,本文所述的条形码可设计成在序列空间中以合理距离(例如汉明或编辑距离)隔开,使得任何两种条形码的序列彼此区别将在于至少数个核苷酸。因此,可正确地指定大多数条形码测序读数,具有小百分率的未指定和错误指定条形码。
在一些实施方案中,设计预定条形码序列,其中以最小汉明或编辑距离隔开。在一些实施方案中,条形码包含随机核苷酸,如(N)15,其产生415的总体可能空间,或约10亿个独特条形码序列。如果待编条形码的样品的数目比这一总体空间少得多,例如1百万个或总体条形码空间的0.1%,那么我们预期所述条形码将彼此隔开充分距离,使得应正确地指定大多数条形码。
只要错误指定率充分低,可简单地检测并且弃去错误指定的测序读数,因为连接于所述错误指定的条形码序列的核酸不同于共有序列。我们应预期针对缔合于条形码序列的各基因(例如γ重链、TCRα链)的共有序列由正确地指定的读数组装,因为所述条形码序列是设计成隔开充分距离。
反应容器中的样品可用独特条形码或独特条形码集合编条形码。独特条形码集合可通过例如每个反应容器递送两种或更多种条形码衔接模板珠粒来使用,并且样品的各核酸用所述独特条形码集合中的一种条形码编条形码。核酸接着通过使用独特条形码集合缔合至样品。
一种区别何种条形码集合用于何种样品的方法是检查来自NextGen测序的读数。预期各条形码序列与来自不同样品的组装的重叠群缔合,因为条形码序列在独特条形码集合中重新使用。但预期来自相同样品的重叠群为同一的。例如,可观察到同一的免疫球蛋白γ重链重叠群使用条形码序列a、b和c。并且可观察到条形码序列a、b和d与另一免疫球蛋白γ重链重叠群缔合。由此,我们可接着推断a、b和c构成条形码集合1,并且a、b和d构成条形码集合2。
在一些实施方案中,N个独特条形码序列的条形码衔接模板珠粒的文库充分多样以对n种样品编条形码,使得大多数样品由独特条形码或独特条形码集合编条形码。如果条形码衔接模板珠粒的数目极大地超过N,那么补替抽样法可接近,并且由独特条形码编条形码的样品的数目U遵循二项式分布并且如下给出:

其中k=1,并且p=1/N。
未用独特条形码编条形码的样品(并且因此具有两种或更多种彼此缔合的样品)的分数如下给出:
1-U/n
N、n和未用独特条形码编条形码的样品的分数之间的关系在表1中给出。
表1.未用独特条形码编条形码的样品的分数

如可见,如果N=10n,那么>90%的样品将用独特条形码编条形码。
用独特条形码集合U集合(在集合中具有x个条形码)编条形码的样品的数目也遵循二项式分布,并且可被视为具有 种独特条形码组合的条形码文库(N假定为充分大,使得组合基本上无重复),其中nx个条形码用于对n种样品编条形码并且如下给出:
种独特条形码组合的条形码文库(N假定为充分大,使得组合基本上无重复),其中nx个条形码用于对n种样品编条形码并且如下给出:

其中k=1,并且
未用独特条形码编条形码的样品(并且因此具有两种或更多种彼此缔合的样品)的分数如下给出:
1-U集合/n
N、n、x和未用独特条形码编条形码的样品的分数之间的关系在表2和3中给出。
表2.当x=2时,未用独特条形码集合编条形码的样品的分数

表3.当x=3时,未用独特条形码集合编条形码的样品的分数

如可见,当使用独特条形码集合来代替独特条形码时,需要条形码衔接文库中少得多的独特条形码来对类似数目的样品编条形码,使得大多数样品可用独特条形码集合鉴别。
I.方法
A.制造所关注的聚核苷酸
在一些方面,本发明提供用于制造一种或多种所关注的聚核苷酸的方法。所述聚核苷酸可为编条形码核酸,例如含有条形码的cDNA或DNA扩增子,其中共同条形码或条形码集合指示一组聚核苷酸源于相同样品。根据所述方法,如下文所述获得多种与一种或多种样品缔合的RNA。与各样品缔合的所述RNA存在于独立反应容积中。衔接分子接着添加至与各样品缔合的所述RNA中以将条形码序列并入一种或多种源于所述RNA的聚核苷酸中。
为了最大化编条形码反应动力学,所述条形码衔接子优选地在其添加至所述RNA中之前或同时以游离形式处于溶液中。添加所述条形码衔接子可通过吸移,通过将一种反应容积倾入另一种中,或通过合并两种或更多种反应容积来实现。例如,所述条形码衔接子可在一种反应容积中产生和/或封装,所述反应容积可接着与含有与一种样品缔合的RNA的另一反应容积组合(图5A-C)。在一些实施方案中,添加至来自样品的RNA中的条形码衔接子在其中存在所述RNA的反应容积中原位产生。
在一些实施方案中,条形码衔接子由条形码衔接模板酶法产生。条形码衔接模板可为含有条形码序列的双链DNA分子,以及其它序列区域以促进条形码衔接子的产生和核酸的后续编条形码(图1)。条形码衔接模板可使用标准分子克隆技术制备。在一些实施方案中,条形码衔接模板包括用于RNA聚合酶(RNAP)的启动子,如T7、T3或SP6启动子。RNA条形码衔接子可接着通过使模板分子与适当RNAP接触并且使体外转录发生而产生(图2A)。在一些实施方案中,条形码衔接模板包括切口核酸内切酶限制位点,如Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI或Nt.BsmAI位点。DNA条形码衔接子可由所述模板通过使所述模板与对所述限制位点具特异性的切口核酸内切酶接触并且接着使所述模板暴露于链置换DNA聚合酶而产生(图2B)。合适链置换DNA聚合酶的实例包括Klenow exo-片段、Bst大片段和其工程改造的变体。一般来说,条形码衔接子由条形码衔接模板通过使所述模板与一种或多种酶接触而产生。在一些实施方案中,所述酶反应为等温反应。
条形码衔接模板在其用于产生条形码衔接子时可以游离形式处于溶液中,或其可结合于固体支撑物。可用于本发明方法和组合物的实施方案中的固体支撑物的实例包括珠粒、色谱树脂、多孔板、微量离心管或具有固体表面的其它物体。条形码衔接模板可使用任何所需机制或捕获化学,例如生物素-抗生物素蛋白、生物素-抗生蛋白链菌素或金-硫醇相互作用结合于固体支撑物。在一些实施方案中,与条形码衔接模板连接的任何固体支撑物均与水溶液接触,并且由所述模板产生的条形码衔接分子在其产生时释放至这一溶液中(图6A、6B、7A-D)。所述水溶液可与待添加所述条形码衔接分子的样品所缔合的RNA分子处于相同反应容积中。也就是说,所述条形码衔接分子可原位产生用于所述编条形码反应。或者,与用于条形码衔接模板的固体支撑物接触的水溶液可容纳于不同于靶标RNA的反应容积中,并且由所述模板产生的条形码衔接子可在组合所述两种反应容积后添加至这些RNA中。
在一些实施方案中,条形码衔接子通过自固体支撑物裂解条形码衔接模板而产生(图7B和7D)。模板分子可含有核酸内切酶限制位点,所述位点促进所述模板分子在暴露于适当酶(例如,限制核酸内切酶)后裂解。在所述裂解后释放至溶液中的核酸分子可用作条形码衔接子并且直接参与编条形码反应,或可经受进一步酶反应(例如体外转录)以产生衔接分子。
无论条形码衔接分子如何产生,可制备这些分子的文库以对来自多种样品的核酸编条形码。衔接分子可分离至不同反应容积中,使得各反应容积含有例如平均一种衔接分子。或者,各反应容积可含有衔接分子的多个拷贝,其中各拷贝含有相同条形码序列。所述反应容积可为微流体液滴或可封入微量离心管或其它容器中。
除条形码序列外,条形码衔接分子还可包括通用引发序列或通用引发区和结合位点,如下文在“组合物”中描述。所述衔接分子还可包括独特分子识别符(UMI)序列。在一些实施方案中,UMI序列含有随机化核苷酸并且与所述条形码序列无关并入所述条形码衔接子(或产生所述衔接子的条形码衔接模板)中。因此,含有相同条形码序列的条形码衔接分子的集合可含有不同UMI序列。在其中所述含有相同条形码序列但含有不同UMI序列的条形码衔接分子的集合添加至所述与一种样品缔合的RNA中的实施方案中,每一个RNA序列均可在编条形码期间连接于不同UMI序列。制备具有UMI序列的条形码衔接模板珠粒的方法公开于下文实施例12和13中,其中各珠粒上的模板分子含有相同条形码序列和不同UMI序列的文库。
条形码衔接子可为RNA或DNA分子,或RNA-DNA杂合物。例如,衔接子可包括在共同寡核苷酸链中共价连接于DNA核苷酸的RNA核苷酸。条形码衔接子也可为单链或双链的。如果为双链的,那么所述条形码衔接子可具有一个或多个钝端或具有单链悬垂物的末端。
在一些实施方案中,所述条形码衔接子为单链DNA分子并且充当反转录的引物。所述条形码衔接子可使用DNA聚合酶(DNAP)产生。此处,所述条形码衔接子的结合位点为RNA结合位点(例如mRNA结合位点)并且含有与一种或多种RNA中的序列区互补的序列区。在一些实施方案中,所述结合位点与添加所述条形码衔接子的样品中的所有RNA所共有的序列区互补。例如,所述结合位点可为聚T区,其与真核生物mRNA的聚-A尾互补(图8)。或者或另外,所述结合位点可包括随机序列区(图9)。在添加所述条形码衔接子至与样品缔合的所述RNA中之后,可发生反转录并且可合成cDNA的第一链,使得所述条形码序列并入所述cDNA的第一链中。应认识到,反转录需要适当条件,例如适当缓冲液和反转录酶的存在,和适用于使所述条形码衔接子退火至RNA的温度和所述酶的活性。还应认识到,涉及DNA引物和RNA模板的反转录在所述引物的3’端与所述模板互补并且可直接退火至所述模板时最有效。相应地,可设计所述条形码衔接子使得所述结合位点出现于所述衔接分子的3’端。
当所述条形码衔接子用作反转录中第一链cDNA合成的引物时,并且在涉及反转录的本发明方法的其它实施方案(下文所述)中,所述反转录反应可发生在其中产生所述条形码衔接子的相同反应容积中。因此,在所述条形码衔接子产生时,所述条形码衔接子可添加至样品或与样品缔合的所述RNA中。例如,微流体液滴可含有与条形码衔接模板结合的珠粒,和细胞(图10)。如果一种或多种酶也存在于所述液滴中,如切口核酸内切酶、链置换DNA聚合酶或RNA聚合酶,那么可产生条形码衔接分子。如果溶解试剂存在于所述液滴中以从所述细胞释放RNA,并且如果存在反转录酶、引物和其它适当试剂,那么可接着发生反转录。用于产生条形码衔接子并且促进溶解和反转录的酶和试剂可例如通过合并含有所述酶和试剂的液滴与含有所述珠粒和细胞的液滴同时添加至所述液滴中,或可分步添加。
在本发明方法的一些实施方案中,与各样品缔合的所述RNA反转录,但所述条形码衔接子并未引发第一链cDNA合成。相反,使用含有聚T区、随机序列或其它RNA结合位点的标准DNA引物。在这些实施方案中,所述条形码衔接子可在其中发生第一链cDNA合成的相同隔室或反应容积中产生。在这种情况下,在反应容积中包括具有在约8.0至8.8的pH下的Tris、钾离子、氯离子、硫酸根离子、铵离子、乙酸离子和/或镁离子的缓冲液可为有益的。或者,可产生所述条形码衔接子并且可在不同隔室中发生第一链cDNA合成,在所述情况下所述隔室必要时可在第一链cDNA合成之前或之后组合。所述隔室也可在所述条形码衔接子产生之前或之后组合。关于进行酶反应并且组合隔室的不同可能性提供优化反应条件的灵活性。然而,无论所述条形码衔接子如何添加至与样品缔合的所述RNA中,所述条形码衔接子可在第一链cDNA合成期间或之后立即参与酶编条形码反应。
如上文所述,本发明方法可使用反转录酶(例如MMLV H-反转录酶),所述反转录酶在到达模板RNA的5’端后添加一个或多个非模板化核苷酸(如C)至新生cDNA链的末端。这些核苷酸在RNA/DNA双链体的一个末端形成3’DNA悬垂物。如果第二RNA分子含有与所述非模板化核苷酸互补的序列区(例如在其3’端的聚鸟嘌呤区),并且结合于所述非模板化核苷酸,那么所述反转录酶可转换模板并且继续延伸cDNA,现在是用所述第二RNA分子作为模板。所述第二RNA分子在本文中称作并且在所属领域中已知为模板转换寡核苷酸。
在本发明方法的实施方案中,所述条形码衔接子充当用于反转录的模板转换寡核苷酸(图3)。因此,所述条形码序列在模板转换后并入cDNA的第一链中,并且存在于由所述cDNA的第一链的扩增(例如,通过PCR)产生的DNA分子中。在这些实施方案中,可使用具有模板转换活性的任何反转录酶。所述条形码衔接子的结合位点为cDNA结合位点并且优选地出现于所述衔接分子的3’端。所述结合位点可包括鸟嘌呤区(包含一个或多个G核苷酸),或至少部分地与通过所述反转录酶产生的3’悬垂物的序列互补的任何其它序列。应认识到所述悬垂物序列和因此用于所述条形码衔接子的结合位点的适当序列可取决于用于所述方法的反转录酶的选择。
在其它实施方案中,与各样品缔合的所述RNA反转录,但条形码序列根本未并入cDNA的第一链中。也就是说,所述条形码衔接子并未充当用于第一链cDNA合成的引物或链转换寡核苷酸。更确切地说,所述条形码衔接子充当用于所述cDNA的第一链或其互补序列的PCR扩增的引物。在这些实施方案中,所述cDNA使用正向引物和反向引物扩增,其中所述反向引物具有与用于第一链cDNA合成的引物的至少一部分相同的序列。所述条形码衔接子可为正向引物或反向引物,并且为单链DNA寡核苷酸。当所述条形码衔接子为正向引物时,其可退火至在链转换后由所述cDNA的延伸产生的第一链cDNA(或其互补序列)的一部分(图11)。或者,所述条形码衔接子可退火至在来自样品的RNA上模板化的第一链cDNA的一部分。因此,模板转换和模板转换寡核苷酸添加至用于样品的反应容积中无需发生来进行本发明的这些实施方案。当所述条形码衔接子为反向引物时,其可结合用于第一链cDNA合成的任何引物使用,所述引物包括包括随机序列的引物(图12和13)。
本发明的方法可用任何所需样品实施。在一些实施方案中,各样品包括细胞,并且可为例如单细胞。细胞可封入如微流体液滴的反应容积中,并且必要时可溶解以释放RNA分子至所述反应容积中。为此,所述细胞可在任何便利时间与溶解缓冲液接触。所述细胞可为B细胞,例如成浆细胞、记忆B细胞或浆细胞,或任何其它种类的细胞。
本发明人已经发现细胞可在溶解之前有利地悬浮于包含渗透保护剂的细胞悬浮缓冲液中。所述渗透保护剂可保护所述细胞免于渗透性应激并且确保细胞生理学在编条形码之前保持稳定或未受扰。在一些实施方案中,细胞连同条形码衔接分子和/或条形码衔接模板悬浮于所述细胞悬浮缓冲液中。在一些实施方案中,细胞在与用于反转录、PCR和/或溶解的试剂接触之前悬浮于所述细胞悬浮缓冲液中。所述细胞悬浮缓冲液可包括于任何反应容积中并且可与本文中关于形成和组合水性反应容积所述的方法相容。
在一些实施方案中,所述细胞悬浮缓冲液中的渗透保护剂为甜菜碱或其密切结构类似物。甜菜碱和密切结构类似物的实例包括甘氨酸甜菜碱(还称为N,N,N-三甲基甘氨酸)、脯氨酸甜菜碱(还称为水苏碱)、β-丙氨酸甜菜碱、四氢嘧啶、胆碱-O-硫酸、葫芦巴碱、二甲基巯基丙酸(DMSP)和二甲基噻亭。在一些实施方案中,所述渗透保护剂为甘氨酸甜菜碱。除了充当渗透保护剂以外,甜菜碱还已经显示减少PCR中二级结构的形成并且改进扩增的特异性。甜菜碱包括于本发明方法中可因此为一般有益的。
在一些实施方案中,所述渗透保护剂为糖或多元醇,如海藻糖。其它适用的糖或多元醇包括蔗糖、果糖、棉子糖、甘露醇和肌醇。在一些实施方案中,所述渗透保护剂为氨基酸,如脯氨酸。单一渗透保护剂可包括于所述细胞悬浮缓冲液中,或可组合包括多种渗透保护剂。各渗透保护剂可以任何适用浓度存在。在一些实施方案中,所述细胞悬浮缓冲液的摩尔渗透压浓度为约250-350mOsm/L。在一些实施方案中,所述渗透保护剂贡献所述缓冲液的所述摩尔渗透压浓度的多达10%、20%、30%、40%、50%、60%、70%、80%、90%或100%。本文中使用的示例性细胞悬浮缓冲液(参看例如实施例7-9、11和14)包括约230-330mM甜菜碱和约10mM NaCl。
在其中各样品包括至少一种细胞的实施方案中,与所述样品缔合的所述RNA可包括mRNA。所述样品可包括例如至少1、3、10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000种mRNA分子,其可代表多种基因、等位基因、阅读框或相异序列。在一些实施方案中,与所述样品缔合的所述RNA包括来自所述样品的所有mRNA、所述细胞的完全或部分转录组或来自所述细胞的总RNA。
应认识到可对每种样品更多RNA编条形码并且如果更多条形码衔接分子可递送至用于各样品的反应容积,那么可制造更多所关注的聚核苷酸。然而,不受任何理论束缚,本发明方法不限制每种样品可编条形码的RNA的数目。相应地,每种样品制造的所关注的聚核苷酸的数目可为至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000。各所关注的聚核苷酸可存在于多个拷贝中。此外,可在所述方法的一种实施中编条形码的细胞或样品的数目仅受制备多种具有独特条形码序列的条形码衔接模板的挑战(上文讨论)限制。在一些实施方案中,所述一种或多种样品包括至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000个细胞。样品(例如,各自为单细胞)可获自相同受试者或不同受试者。例如,至少2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90或100个不同受试者可提供样品。
本发明方法也可用于使用核酸标记物查询用于所关注的表型的细胞群体。所述核酸标记物包括连接于结合剂的核酸,其可特异性结合于来自提供或不提供所述表型的群体的细胞的子集。例如,所述结合剂可结合于某些蛋白、糖蛋白、糖脂或存在于一些细胞的表面上的其它部分。在一些实施方案中,所述结合剂为分子标记,如抗体、抗原或蛋白(图14A-C)。在一些实施方案中,所述结合剂为肽-MHC复合物。所述核酸可使用非共价捕获部分共价连接于所述结合剂,或在其它情况下如所需。
为了查询用于所述表型的细胞,使细胞与所述核酸标记物接触并且接着洗涤。因此,所述核酸标记物仅保留于与所述结合剂结合的细胞上。所述细胞可接着封入反应容积中并且如上文所述溶解,使得可对所述细胞中的RNA编条形码。在编条形码反应期间,也对所述核酸标记物的核酸编条形码,使得所述标记物序列出现于针对保留所述标记物的细胞的RNA或扩增子测序数据中。在一些实施方案中,所述核酸标记物的核酸为具有所述群体的细胞的非内源性序列的RNA分子。在一些实施方案中,所述核酸为包含RNAP启动子的双链DNA分子。因此,所述核酸在与所述细胞(或其溶解产物)相同的反应容积中时可转录,并且所得RNA分子可连同来自所述细胞的RNA一起编条形码。
细胞可针对多种表型使用多种核酸标记物来查询,各核酸标记物包括连接于不同核酸序列的不同结合剂。例如,细胞可与第一核酸标记物和第二核酸标记物接触,其中各核酸标记物包括连接于核酸的分子标记。所述两种核酸标记物的分子标记可彼此不同(例如,为不同蛋白或对不同细胞表面部分具有亲和力)。连接于这些分子标记的核酸可含有全部或部分地彼此不同的序列。细胞可同时或依序与两种或更多种核酸标记物接触。
作为进一步实例,三个抗体可连接于不同的非内源性RNA序列,并且关于用这些抗体处理的细胞的编条形码测序数据可披露各细胞是否提供针对所述抗体中的无一者、一些或全部的靶标。编条形码扩增子的拷贝数也可逐步披露表型,例如不同细胞上的细胞表面部分的相对丰度,其中所述部分由所述核酸标记物靶向。
B.连接聚核苷酸至固体支撑物
本发明的另一方面提供用于使聚核苷酸连接于固体支撑物的方法,其中所述聚核苷酸含有条形码序列。所述聚核苷酸可为条形码衔接模板或所述模板的前体。所述聚核苷酸可因此如上文所述用于酶法产生条形码衔接子并且将所述条形码序列并入源于RNA的扩增子中。
在一些实施方案中,所述方法涉及产生反相乳液的亲水性隔室(即,水性液滴)。所述隔室可如所需例如通过将水溶液混入疏水性载液中并且任选地搅动所述混合物来产生。所述水溶液可具有固体支撑物、寡核苷酸和悬浮于其中的试剂,使得当形成所述隔室时,各隔室含有用于使所述聚核苷酸连接于所述固体支撑物的所有必需组分。在这些实施方案中,在添加所述固体支撑物至所述隔室中之前,寡核苷酸经由捕获部分结合于所述固体支撑物的表面。这种寡核苷酸在本文中称作“结合的寡核苷酸”并且含有与条形码寡核苷酸的3’序列互补的3’序列。所述聚核苷酸因此通过聚合酶延伸反应在所述固体支撑物上形成,所述反应涉及所述结合的寡核苷酸和条形码寡核苷酸,并且这种反应发生在所述隔室内。
在优选实施方案中,当形成所述亲水性隔室时,所述条形码寡核苷酸以低或限制浓度存在(例如,每个隔室一个分子)。当具有随机化序列的条形码寡核苷酸的文库用于制备多种条形码模板珠粒时,这种浓度是有利的。如果假定每一种条形码寡核苷酸具有不同条形码序列,并且各隔室中的固体支撑物需要仅具有一种条形码序列,那么每个隔室可存在一种条形码寡核苷酸(至多或平均)。一旦满足这种条件,多种固体支撑物(例如多种珠粒)可存在于隔室中,或者所述结合的寡核苷酸的多个拷贝可结合于各固体支撑物,但由所述隔室中的聚合酶延伸反应产生的所有聚核苷酸均将含有相同的条形码序列。
用于本发明方法的优选固体支撑物为珠粒,例如由金属和/或聚合物质制得并且具有在约0.1至10微米范围内的直径的球形珠粒。具有其它特征的珠粒可替代或另外使用。所述固体支撑物可用捕获部分官能化以连接所述结合的寡核苷酸至所述表面(图15,左侧)。捕获部分的实例包括抗生物素蛋白、抗生蛋白链菌素、生物素、羧基、环氧基、羟基、硫醇基和金。一些捕获部分具有与其特异性地并且非共价地结合的结合搭配物。例如,抗生蛋白链菌素采用生物素作为其结合搭配物。所述捕获部分可直接地(例如共价地)偶联于所述固体支撑物,并且所述结合搭配物可偶联于所述结合的寡核苷酸,或反之亦然,使得所述结合的寡核苷酸通过非共价相互作用结合于所述固体支撑物。其它捕获部分提供所述结合的寡核苷酸与固体支撑物之间的直接共价连接。
所述结合的寡核苷酸优选地为在其5’端结合于所述固体支撑物的单链DNA分子。因此,所述结合的寡核苷酸的3’端以游离形式处于溶液中并且当杂交至所述条形码寡核苷酸时,可通过如DNA聚合酶的酶延伸。所述延伸反应使用所述条形码寡核苷酸模板化,使得所述条形码序列并入结合于所述珠粒的DNA链中。必要时,所述结合的寡核苷酸和/或所述条形码寡核苷酸可具有设计成使分子内二级结构最小化的序列。
所述条形码寡核苷酸可含有上文讨论的序列区,如通用引发序列和/或结合位点。在用所述结合的寡核苷酸和所述条形码寡核苷酸执行引物延伸反应后,这些序列区将并入结合于所述固体支撑物的聚核苷酸中。如果所述聚核苷酸随后用作条形码衔接模板,那么所述序列区也将存在于由所述模板产生的条形码衔接分子中。如RNAP启动子和/或切口核酸内切酶限制位点的其它序列可包括于所述条形码寡核苷酸中以促进条形码衔接分子的酶制造。所述RNAP启动子可选自由T7、T3和SP6启动子组成的组。所述切口核酸内切酶限制位点可选自由Nt.BbvCI、Nt.BspQI、Nt.BsmAI、Nt.BstNBI、Nt.AlwI和Nt.BsmAI位点组成的组。所述条形码寡核苷酸内的结合位点可含有一个或多个G核苷酸。
在一些实施方案中,所述条形码序列和其它序列区使用PCR并入连接于固体支撑物的所述结合的寡核苷酸和/或所述聚核苷酸中(图15,右侧)。在这些实施方案中,所述条形码寡核苷酸充当PCR的模板,并且所述结合的寡核苷酸充当引物,其中所述结合的寡核苷酸的酶延伸从其3’端进行。所述条形码寡核苷酸还包括与PCR反向引物序列同一或互补的5’序列。因此,反向引物可退火至所述条形码寡核苷酸(或其互补序列)的5’端并且引物延伸沿与所述结合的寡核苷酸的方向相对的方向。必要时,这种反向引物可经荧光团标记,使得通过PCR产生并且连接于所述固体支撑物的聚核苷酸为荧光性的。所述标记可用于确定固体支撑物(例如珠粒)是否已经成功地连接于包括所述条形码序列的聚核苷酸。
上述方法可在单一步骤中执行。在本发明方法的其它实施方案中,含有条形码序列的聚核苷酸在多个步骤中连接于固体支撑物。在这些实施方案中,所述条形码序列由例如S1x、W和S2y区的数个序列区构成。这些序列区可作为两种或更多种条形码寡核苷酸的一部分引入所述聚核苷酸中,其中各条形码寡核苷酸用于独立步骤或酶反应中。在由所述独立步骤产生的聚核苷酸中,所述S1x、W和S2y区不必定为邻接的。各种S1x、W和S2y序列可组合于不同固体支撑物上以形成不同条形码序列或条形码序列的文库。
为了在多个步骤中连接聚核苷酸至固体支撑物(其中所述聚核苷酸含有条形码序列),如上文所述提供固体支撑物和结合于所述固体支撑物的寡核苷酸。所述固体支撑物和结合的寡核苷酸可提供于乳液的亲水性隔室中,或任何其它所需反应容积中。还提供第一条形码寡核苷酸(图16,顶部和中间)。所述结合的寡核苷酸包含S1x序列和与所述第一条形码寡核苷酸的3’序列互补的序列。所述第一条形码寡核苷酸包含W序列。在所述多步骤程序的第一步骤中,执行聚合酶延伸反应或接合反应以将所述W序列并入所述结合的寡核苷酸中。因此,在这一步骤后,所述S1x序列和W序列存在于结合于所述固体支撑物的相同核酸链中。如果使用延伸反应,那么如上文关于单一步骤程序所讨论,所述结合的寡核苷酸可充当引物并且所述第一条形码寡核苷酸可充当模板,使得所述结合的寡核苷酸从其3’端延伸。在一些实施方案中,所述第一条形码寡核苷酸中与所述结合的寡核苷酸中的S1x序列互补的部分含有肌苷区。
随后,提供第二条形码寡核苷酸以将S2y序列并入所述结合的寡核苷酸中(图16,底部)。所述第二条形码寡核苷酸包含所述S2y序列,以及与由所述多步骤程序的第一步骤产生的所述结合的寡核苷酸的3’端互补的3’序列。因此,所述第二条形码寡核苷酸可包括与所述第一条形码寡核苷酸的一部分互补或同一的序列区。所述第二条形码寡核苷酸通过聚合酶延伸反应或接合反应与所述结合的寡核苷酸(现在延伸以包括所述S1x序列和所述W序列)反应。在这一步骤后,所述S1x、W和S2y序列均存在于结合于所述固体支撑物的相同核酸链中。
如所需,相同或不同反应条件可用于多步骤程序的第一和第二步骤中以连接聚核苷酸至固体支撑物。例如,相同的酶(例如DNA聚合酶)或不同酶(例如DNA聚合酶和连接酶)可用于所述第一条形码寡核苷酸和第二条形码寡核苷酸的反应中,不过使用相同的酶可更有效。为了混合试剂和所述固体支撑物用于连续步骤,试剂可分配至反应容积中,并且反应容积可分裂、组合或以其它方式处理,均如所需。例如,所述固体支撑物和结合的寡核苷酸可分布至多个反应容积中,并且不同的第一条形码寡核苷酸可添加至各反应容积中,使得不同的W序列偶联于相同S1x序列。这些反应容积各自可又分成更多的用于添加所述第二条形码寡核苷酸的容积,使得多个S2y序列偶联于各W序列。在一些实施方案中,洗涤固体支撑物以去除未结合的寡核苷酸。在一些实施方案中,在将所述W序列并入所述结合的寡核苷酸中之后加热固体支撑物,以熔融所述结合的寡核苷酸和第一条形码寡核苷酸的双链体,并且使所述结合的寡核苷酸和第二条形码寡核苷酸退火。
可包括于条形码衔接分子和/或条形码衔接模板中的序列区(如通用引发序列、结合位点、RNAP启动子或切口核酸内切酶限制位点)可如所需分布于所述第一条形码寡核苷酸与所述第二条形码寡核苷酸之间。例如,所有所述序列可包括于一种条形码寡核苷酸中,或一些可包括于一种条形码寡核苷酸中并且一些可包括于其它条形码寡核苷酸中。在一些实施方案中,选择的条形码寡核苷酸进一步包含通用引发序列和结合位点,所述选择的条形码寡核苷酸为所述第一条形码寡核苷酸或所述第二条形码寡核苷酸。在一些实施方案中,这种选择的条形码寡核苷酸还包含RNAP启动子或切口核酸内切酶限制位点。应认识到本发明方法提供用于将不同序列区并入条形码衔接模板中的多种选项。这些模板和用于制备所述模板的寡核苷酸的最佳设计可取决于用于酶法产生条形码衔接分子和对RNA编条形码的机制。
本文关于连接聚核苷酸至固体支撑物所述的方法中的任一者均可用于制备一种或多种用于对样品、细胞或RNA编条形码的固体支撑物。连接于各固体支撑物的聚核苷酸包括条形码序列并且可充当条形码衔接模板。本发明方法还可用于制备条形码文库,所述文库包括多种固体支撑物,各固体支撑物与条形码序列缔合。任何两种固体支撑物(例如珠粒)可具有完全或部分地彼此不同的条形码序列。在一些实施方案中,所述条形码文库中的每一种固体支撑物与不同条形码序列缔合。
根据本发明方法制备的条形码衔接模板珠粒包括结合于条形码衔接模板的珠粒。所述珠粒可结合于所述模板分子的多个拷贝,例如至少10、30、100、300、1,000、3,000、10,000、30,000、100,000、300,000或1,000,000个拷贝。在一些实施方案中,结合于一种珠粒的模板分子的各拷贝包括相同条形码序列。在其中所述模板分子具有形式S1x-W-S2y的条形码序列的实施方案中,结合于一种珠粒的模板分子的各拷贝包括相同S1x、W和/或S2y序列。本发明方法还允许制备包含多种条形码衔接模板珠粒的珠粒条形码文库。所述文库中的每一种珠粒可与不同条形码序列缔合,并且各珠粒上的条形码衔接模板的拷贝可包含相同条形码序列。
在一些实施方案中,本发明方法可用于通过在条形码衔接模板珠粒上以物理方式捕获由一种或多种样品(例如细胞)制备或获得的cDNA来制备聚核苷酸文库。各珠粒包括在3’端具有cDNA结合位点的模板分子。所述珠粒可与酶接触以使所述结合位点为单链的(例如,在以游离形式处于溶液中的模板分子的末端留下3’悬垂物)。所述珠粒接着与来自样品的一种或多种cDNA接触,使得所述cDNA通过所述结合位点结合于所述模板分子的拷贝。在优选实施方案中,所述结合位点包括一个或多个G核苷酸(例如聚G区),并且与通过反转录酶添加至cDNA的末端的非模板化聚C区互补。
聚核苷酸文库中的珠粒可如所需使用,例如以对来自多种样品的cDNA测序或分离来自不同样品的cDNA。在后一种情况下,对应于不同样品的珠粒可使用离心或磁性沉淀,并且接着使用标准方法再悬浮和分离。必要时,在cDNA结合于珠粒上的模板分子后,所述模板分子可酶法延伸,由此将cDNA序列并入结合所述珠粒的DNA双链体中并且使这些序列与条形码序列缔合。如果来自样品的cDNA分子的拷贝数可相当于珠粒上的条形码衔接模板的拷贝数,那么这些cDNA分子可被捕获于少量珠粒(例如,每种样品至多约1、3、10、30、100、300或1000个珠粒)上。来自样品的RNA可使用标准方法或如上文所讨论进行反转录以产生cDNA。B细胞(例如成浆细胞、记忆B细胞和浆细胞)可用作样品,并且在一些实施方案中,所述cDNA为B细胞源性可变免疫球蛋白区。
II.组合物
A.聚核苷酸
在一些方面,聚核苷酸可包括cDNA区。在一些方面,聚核苷酸可包括样品鉴别(条形码)-衔接区。在一些方面,聚核苷酸可包括样品鉴别(条形码)区。在一些方面,聚核苷酸可包括衔接区。在一些方面,聚核苷酸可包括通用引物区。在一些方面,聚核苷酸可包括扩增子区。在一些方面,聚核苷酸可包括板鉴别区。在一些方面,聚核苷酸可包括第一板鉴别区。在一些方面,聚核苷酸可包括第二板鉴别区。在一些方面,聚核苷酸可包括限制位点区。在一些方面,聚核苷酸可包括第一限制位点区。在一些方面,聚核苷酸可包括第二限制位点区。在一些方面,聚核苷酸可包括测序区。在一些方面,聚核苷酸可包括第一测序区。在一些方面,聚核苷酸可包括第二测序区。
在一些方面,聚核苷酸可包括多种本文所述的任何区域。例如,聚核苷酸可包括第一样品鉴别(条形码)区和第二样品鉴别(条形码)区。在一些方面,所述第一样品鉴别(条形码)区和所述第二样品鉴别(条形码)区为同一或大致同一的。在一些方面,所述第一样品鉴别(条形码)区和所述第二样品(条形码)鉴别区为相异的。在一些方面,鉴别(条形码)区偶联于可变免疫球蛋白区。
在一些方面,区域的序列将至少足够长以充当针对PCR反应中的引物或探针的靶标序列。在一些方面,区域可为1个至大于5000个碱基对长。例如,区域可为1-10,000个核苷酸长,例如2-30个核苷酸长,包括其间所有子范围。作为非限制性实例,区域可为1-30个核苷酸、1-26个核苷酸、1-23个核苷酸、1-22个核苷酸、1-21个核苷酸、1-20个核苷酸、1-19个核苷酸、1-18个核苷酸、1-17个核苷酸、18-30个核苷酸、18-26个核苷酸、18-23个核苷酸、18-22个核苷酸、18-21个核苷酸、18-20个核苷酸、19-30个核苷酸、19-26个核苷酸、19-23个核苷酸、19-22个核苷酸、19-21个核苷酸、19-20个核苷酸、20-30个核苷酸、20-26个核苷酸、20-25个核苷酸、20-24个核苷酸、20-23个核苷酸、20-22个核苷酸、20-21个核苷酸、21-30个核苷酸、21-26个核苷酸、21-25个核苷酸、21-24个核苷酸、21-23个核苷酸或21-22个核苷酸。在一些方面,区域可为约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个核苷酸长。在一些方面,区域可为少于50、50-100、100-200、200-300、300-400、400-500、500-600、600-700、700-800、800-900、900-1000或大于1000个核苷酸长。在一些方面,区域可为少于1000、1000-2000、2000-3000、3000-4000、4000-5000、5000-6000、6000-7000、7000-8000、8000-9000、9000-10000或大于10000个核苷酸长。在一些方面,区域可包括本文所公开的聚核苷酸的至少两个核酸、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少15个、至少20个或更多个核苷酸。
在一些方面,聚核苷酸可源于单一样品或与单一样品缔合。在一些方面,区域可源于单一样品或与单一样品缔合。在一些方面,cDNA区可源于单一样品或与单一样品缔合。在一些方面,扩增子区可源于单一样品或与单一样品缔合。“单一样品”包括包含从单一来源取得的聚核苷酸的样品。在一些方面,单一来源包括在特定时间点或在特定位置,例如在受试者或细胞烧瓶或细胞板中取得的样品。在一些方面,第一单一样品在第一时间点从第一受试者取得并且第二单一样品在相异于所述第一时间点的第二时间点从所述第一受试者取得。在一些方面,第一单一样品在第一位置从第一受试者取得并且第二样品在相异于所述第一位置的第二位置从所述第一受试者取得。在一些方面,第一单一样品在一个时间点从第一受试者取得并且第二单一样品在一个时间点从第二受试者取得。在一些方面,第一单一样品在一个位置从第一受试者取得并且第二样品在一个位置从第二受试者取得。在一个实施方案中,样品包含包括源于一种或多种B细胞的mRNA的聚核苷酸。在另一实施方案中,样品包含包括源于一种或多种B细胞的cDNA的聚核苷酸。在另一实施方案中,单一样品包含源于一种或多种B细胞的mRNA,所述细胞分选至96孔或384孔板的单一孔中。样品一般源于原核细胞(例如细菌细胞)、真核细胞(例如哺乳动物和酵母细胞)或如病毒或噬菌体的遗传物质的其它来源。如本文所用的术语“哺乳动物(mammal)”或“哺乳动物(mammalian)”包括人类和非人类并且包括但不限于人类、非人类灵长类动物、犬科动物、猫科动物、鼠科动物、牛、马和猪。在一些方面,本发明方法适用于具有至少96个孔、至少384个孔、至少1536个孔或更多孔的板中的单一样品。在其它方面,本发明方法适用于各自具有至少96个孔的至少一个、两个、三个、四个、五个、六个、七个、八个、十个、十五个、二十个、三十个或更多个板中的单一样品。
在一些方面,5’衔接区序列和/或样品鉴别区例如在RT期间添加至来自单一样品的所有cDNA中并且不仅仅添加至Ig基因中。在一些方面,3’基因特异性引物(GSP)可用于扩增所述单一样品中的任何表达的基因。在一些方面,扩增具有5’可变区的基因,例如T细胞受体和B细胞受体而无需多重简并5’引物来扩增所关注的基因。GSP可包括对IgG、IgM、IgD、IgA、IgE、TCR链和其它所关注的基因具有特异性的引物。
在一些方面,也可例如使用巢式GSP执行多轮PCR。关于所述巢式GSP,用于第二轮PCR的GSP相对于用于第一轮PCR的GSP的杂交位置沿其靶标基因序列在位置5'与所述序列杂交。
在一些方面,cDNA区或扩增子区可包括DNA聚核苷酸。在一些方面,cDNA区或扩增子区可包括cDNA聚核苷酸。在一些方面,cDNA区或扩增子区可包括与DNA聚核苷酸杂交的RNA聚核苷酸。在一些方面,cDNA区或扩增子区可包括与cDNA聚核苷酸杂交的mRNA聚核苷酸。
在一些方面,通用引物区不完全与任何人类外显子互补。在一些方面,通用引物区不完全与任何表达的人类基因互补。在一些方面,通用引物区具有最小二级结构。
在一些方面,扩增子区包含免疫球蛋白重链扩增子序列。在一些方面,扩增子区包含免疫球蛋白轻链扩增子序列。在一些方面,扩增子区包含T细胞受体α扩增子序列。在一些方面,扩增子区包含T细胞受体β扩增子序列。
在一些方面,聚核苷酸存在于聚核苷酸文库中并且可基于所述聚核苷酸的区域与存在于所述文库中的其它聚核苷酸区别。
在一些方面,源于第一单一样品的文库中的各聚核苷酸的样品鉴别区的序列相异于源于一种或多种相异于所述第一单一样品的样品的文库中的其它聚核苷酸的样品鉴别区的序列。在一些方面,源于第一单一样品的文库中的各聚核苷酸的样品鉴别区的序列与源于一种或多种相异于所述第一单一样品的样品的文库中的其它聚核苷酸的样品鉴别区的序列的不同之处在于至少1个核苷酸。在一些方面,源于第一单一样品的文库中的各聚核苷酸的样品鉴别区的序列与源于一种或多种相异于所述第一单一样品的样品的文库中的其它聚核苷酸的样品鉴别区的序列的不同之处在于至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个核苷酸。在一些方面,源于第一单一样品的文库中的各聚核苷酸的样品鉴别区的序列可与源于一种或多种相异于所述第一单一样品的样品的文库中的其它聚核苷酸的样品鉴别区的序列约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、60、70、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99或少于100%同一。在一些方面,源于第一单一样品的文库中的各聚核苷酸的样品鉴别区的序列与源于一种或多种相异于所述第一单一样品的样品的文库中的其它聚核苷酸的样品鉴别区的序列少于100%同一。在一些方面,样品鉴别区充当由单一样品反转录的所有第一链cDNA上的数字条形码。在一些方面,所述样品鉴别区为至少1个核苷酸长。在一些方面,样品鉴别区可包含至少3个核苷酸,并且样品鉴别区的彼此不同之处可在于至少1个核苷酸。在一个实施方案中,样品鉴别区为3-15个核苷酸长并且彼此不同之处在于至少1个核苷酸。在一些方面,样品鉴别区可包含至少64种变体(使用3个核苷酸长的样品鉴别区,其中各样品-ID彼此不同之处在于至少1个核苷酸),或在一些方面更大数目的变体。在一些方面,连接3’至所述样品鉴别区的序列可为包含至少1个G的衔接区。在一个优选实施方案中,连接3’至所述样品鉴别区的序列可为包含至少2个G的衔接区。在一个实施方案中,连接至样品鉴别区的5’端的序列为通用引物序列,其可在PCR扩增期间使用以避免对后续添加5’通用引物序列(通过接合或另一种方法)的需要或多重简并5’引物来扩增具有可变5’区的基因的使用。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第一板鉴别区的序列相异于源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第一板鉴别区的序列。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第一板鉴别区的序列与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第一板鉴别区的序列的不同之处在于至少1个核苷酸。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第一板鉴别区的序列与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第一板鉴别区的序列的不同之处在于至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个核苷酸。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第一板鉴别区的序列可与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第一板鉴别区的序列约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、60、70、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99或少于100%同一。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第一板鉴别区的序列与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第一板鉴别区的序列少于100%同一。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第二板鉴别区的序列相异于源于一种或多种不同于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第二板鉴别区的序列。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第二板鉴别区的序列与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第二板鉴别区的序列的不同之处在于至少1个核苷酸。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第二板鉴别区的序列与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第二板鉴别区的序列的不同之处在于至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50个或更多个核苷酸。在一些方面,所述第二板鉴别区的序列与聚核苷酸上的所述第一板鉴别区的序列同一。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第二板鉴别区的序列可与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第二板鉴别区的序列约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、60、70、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99或少于100%同一。在一些方面,源于单一样品的第一集合的文库中的各聚核苷酸的第二板鉴别区的序列与源于一种或多种相异于所述单一样品的第一集合的单一样品集合的文库中的其它聚核苷酸的第二板鉴别区的序列少于100%同一。在一些方面,板鉴别区(例如第一板鉴别区或第二板鉴别区)可包含至少2个核苷酸,并且板鉴别区彼此不同之处在于至少1个核苷酸。在一个实施方案中,板鉴别区为2-10个核苷酸长并且彼此不同之处在于至少1个核苷酸。在一些方面,板鉴别区的使用仅在一些实施方案中发现,因为更大数目的不同样品鉴别区(每种待分析的单一样品一个)的使用可消除对板鉴别区的需要。在一些方面,板鉴别区用于减少需要合成的含有样品鉴别区的独特寡核苷酸的数目。
在一些方面,聚核苷酸包括一个或多个衔接区。在一些方面,衔接区包括一个或多个G。在一些方面,衔接区包括2、3、4、5、6、7、8、9、10个或更多个G。在一些方面,衔接区使用MMLV H-反转录酶的模板转换特性连接至cDNA的3’端。存在连接衔接区的不同方法,包括但不限于用具有5’侧接衔接区序列的引物进行PCR、粘性和钝端接合、模板转换介导的核苷酸添加或共价连接核苷酸至所述聚核苷酸的5’端、3’端或5’和3’端的其它方法。这些方法可使用分子生物学中通常使用的酶的特性。PCR可使用例如嗜热DNA聚合酶。通过用留下悬垂端的限制酶切割dsDNA或通过如TdT(末端转移酶)的酶的3’拖尾活性产生互补或大致互补的粘性端。粘性和钝端可接着使用如T4连接酶的连接酶与互补衔接区接合。模板转换利用MMLV H-反转录酶的3’拖尾活性来添加一个或多个胞嘧啶(C)至cDNA的3’端并且利用其将来自mRNA的模板转换为具有互补G的衔接区的能力。在一些方面,cDNA在其3’端包括1、2、3、4、5、6、7、8、9、10个或更多个C。
在一些方面,聚核苷酸包括一个或多个限制位点区。限制位点区包括一个或多个限制位点。限制位点可包括:NheI、XhoI、BstBI、EcoRI、SacII、BbvCI、PspXI、AgeI、ApaI、KpnI、Acc65I、XmaI、BstEII、DraIII、PacI、FseI、AsiSI和AscI。在一些方面,可使用任何稀有8-切割酶限制位点。
在一些方面,本文所述的聚核苷酸的一个或多个区域可操作性地偶联于所述聚核苷酸的一个或多个其它区域。在一些方面,单一聚核苷酸的两个或更多个相异区域可操作性地偶联。例如,通用引物区可操作性地偶联于衔接区。在一些方面,两个或更多个区域可操作性地偶联在一起,所述区域的序列大致彼此同一或描述同一。例如,第一样品鉴别区可操作性地偶联于第二样品鉴别区。在一些方面,所述第一样品鉴别区和所述第二样品鉴别区的序列为同一或大致同一的。在一些方面,所述第一样品鉴别区和所述第二样品鉴别区的序列为不同的或相异的。
在一些方面,本文所述的聚核苷酸的一个或多个区域可偶联于所述聚核苷酸的一个或多个其它区域。在一些方面,单一聚核苷酸的两个或更多个相异区域可偶联。例如,通用引物区可偶联于衔接区。在一些方面,两个或更多个区域可偶联在一起,所述区域的序列大致彼此同一或描述同一。例如,第一样品鉴别区可偶联于第二样品鉴别区。在一些方面,所述第一样品鉴别区和所述第二样品鉴别区的序列为同一或大致同一的。在一些方面,所述第一样品鉴别区和所述第二样品鉴别区的序列为不同的或相异的。
在一些方面,聚核苷酸包括序列5’-A-B-3’,其中A为样品鉴别区,并且其中B为衔接区。在一些方面,聚核苷酸包括序列5’-A-B-C-3’,其中A为通用引物区,其中B为样品鉴别区,并且其中C为衔接区。在一些方面,聚核苷酸包括序列5’-A-B-C-3’,其中A为样品鉴别区,其中B为衔接区,并且其中C为源于单一样品的扩增子区。在一些方面,聚核苷酸包括序列5’-A-B-C-D-3’,其中A为通用引物区,其中B为样品鉴别区,其中C为衔接区,并且其中D为源于单一样品的扩增子区。在一些方面,聚核苷酸包括序列5’-A-B-C-D-E-3’,其中A为板鉴别区,其中B为通用引物区,其中C为样品鉴别区,其中D为衔接区,并且其中E为源于单一样品的扩增子区。在一些方面,聚核苷酸包括序列5’-A-B-C-D-E-F-3’,其中A为第一限制位点区,其中B为通用引物区,其中C为样品鉴别区,其中D为衔接区,其中E为源于单一样品的扩增子区,并且其中F为第二限制位点区。
在一些方面,上述序列中每一者的区域可以不同顺序重排,例如5’-C-A-D-B-3’或5’-E-A-C-B-D-F-3’或5’-B-A-3’。在一些方面,上述序列的一个或多个区域可缺失,例如5’-A-D-3’或5’-B-C-3’。在一些方面,一个或多个额外区域可添加至上述序列中,例如5’-A-A2-B-3’或5’-A-B-C-D-E-F-G-3’。在所述实施例中,所述一个或多个额外区域可为本文所公开的任何区域或其等效物。在一些方面,上述序列的一个或多个区域可进行修饰,例如甲基化。
在一些方面,聚核苷酸可包括衔接分子。在一些方面,聚核苷酸衔接分子可包括通用引物区、样品鉴别区和衔接区,其中所述通用引物区的3’端偶联于所述样品鉴别区的5’端,并且其中所述样品鉴别区的3’端偶联于所述衔接区的5’端。在一些方面,衔接分子包括包含至少2个结合于通过反转录酶在第一链cDNA的3'端添加的C的核苷酸的聚核苷酸。在一些方面,衔接分子包括包含3-6个G(DNA G)的脱氧核糖聚核苷酸。在另一实施方案中,衔接分子包括包含3-6个G(RNA G)的核糖聚核苷酸。在其它实施方案中,所述衔接分子可利用核苷酸类似物,如锁核酸(LNA),例如LNA G。在其它实施方案中,核苷酸碱基也可为通用或简并碱基,如5-硝基吲哚和3-硝基吡咯,所述碱基可以任何组合与C以及其它核苷酸碱基配对。
在一些方面,聚核苷酸可包括引物或探针。在一些方面,引物可包括通用引物区和板鉴别区,并且其中所述板鉴别区的3’端偶联于所述通用引物区的5’端。
在一些方面,组合物可包括聚核苷酸组合物文库。在一些方面,聚核苷酸组合物文库包括多种聚核苷酸组合物。在一些方面,各组合物存在于独立容器中。在一些方面,容器可为试管。在一些方面,容器可为板中的孔。在一些方面,容器可为96孔板中的孔。在一些方面,容器可为384孔板中的孔。在一些方面,各组合物包含源于单一样品的cDNA区。在一些方面,各组合物包含样品鉴别-衔接区,其包含偶联于衔接区的样品鉴别区。在一些方面,文库中各样品鉴别-衔接区的样品鉴别区的序列相异于所述文库中存在于各独立容器中的其它样品鉴别-衔接区的样品鉴别区的核苷酸序列。在一些方面,所述样品鉴别-衔接区连接至cDNA区。在一些方面,所述样品鉴别-衔接区通过其3’区之间的结合连接至cDNA区。在一些方面,所述样品鉴别-衔接区通过G:C结合连接至cDNA区。在一些方面,所述cDNA区包含与DNA聚核苷酸杂交的RNA聚核苷酸。在一些方面,所述cDNA区包含与cDNA聚核苷酸杂交的mRNA聚核苷酸。
在一些方面,聚核苷酸文库中的多种聚核苷酸组合物可包含至少2个、至少3个、至少10个、至少30个、至少100个、至少300个、至少1000个、至少3000个、至少10,000个、至少30,000个、至少100,000个、至少300,000个、至少1,000,000个、至少3,000,000个、至少10,000,000个、至少30,000,000个或更多个成员。在其它方面,聚核苷酸文库中的多种聚核苷酸组合物可包含细胞样品的整个转录组的至少2种、至少3种、至少10种、至少30种、至少100种、至少300种、至少1000种、至少3000种、至少10,000种、至少30,000种或更多种基因。在其它方面,聚核苷酸文库中的多种聚核苷酸组合物包含至少1种、至少2种、至少3种、至少10种、至少30种、至少100种、至少300种、至少1000种、至少10,000种、至少100,000种、至少1,000,000种、至少10,000,000种、至少1,000,000,000种或更多种存在于个体血液中的不同抗体物质。这些抗体物质可由成浆细胞、浆细胞、记忆B细胞、长寿命浆细胞、原生B细胞、其它B谱系细胞或其组合表达。
B.载体
在一些方面,组合物可包括载体。术语“载体”用于指载体核酸分子,核酸序列可插入其中以引入至细胞中,所述核酸序列可在所述细胞中复制。载体可用于由核酸序列转化宿主细胞。在一些方面,载体可包括一种或多种本文所述的聚核苷酸。在一个实施方案中,编码靶标多肽的核酸序列的文库可引入细胞群体中,由此允许筛选文库。核酸序列可为“外源的”或“异源的”,这意指其相对于其中正引入所述载体的细胞而言是外来的,或所述序列与所述细胞中的序列同源,但在宿主细胞核酸内通常未发现所述序列的位置中。载体包括质粒、粘粒和病毒(例如噬菌体)。所属领域的技术人员可通过标准重组技术建构载体,所述技术描述于Maniatis等人,1988和Ausubel等人,1994中,所述参考文献均以引用的方式并入本文中。在一些方面,载体可为具有预先工程改造的抗体恒定区的载体。以这种方式,所属领域的技术人员可仅克隆所关注的抗体的VDJ区并且将这些区域克隆至所述预先工程改造的载体中。
术语“表达载体”是指含有编码能够被转录的基因产物的至少一部分的核酸序列的载体。在一些情况下,RNA分子接着翻译成蛋白、多肽或肽。表达载体可含有多种“控制序列”,所述控制序列是指用于特定宿主生物体中操作性地连接的编码序列的转录和可能发生的翻译的核酸序列。除了管理转录和翻译的控制序列以外,载体和表达载体还可含有也发挥其它功能的核酸序列。
在一些方面,载体可包括启动子。在一些方面,载体可包括增强子。“启动子”为控制序列,其为核酸序列中控制转录的起始和速率的区域。其可含有遗传元件,在所述遗传元件处可结合调控蛋白和分子,如RNA聚合酶和其它转录因子。措辞“操作性地定位”、“操作性地连接”、“在控制下”和“在转录控制下”意指启动子相对于核酸序列处于正确功能位置和/或定向中以控制所述序列的转录起始和/或表达。启动子可或可不结合“增强子”使用,所述增强子是指核酸序列的转录活化中涉及的顺式作用调控序列。
启动子可为与基因或序列天然缔合的启动子,如可通过分离位于编码区段和/或外显子上游的5'非编码序列来获得。所述启动子可称为“内源的”。同样,增强子可为与核酸序列天然缔合的增强子,位于所述序列的下游或上游。或者,通过在重组或异源启动子的控制下定位编码核酸区段将获得某些优势,所述重组或异源启动子是指通常不与核酸序列在其天然环境中缔合的启动子。重组或异源增强子也是指通常不与核酸序列在其天然环境中缔合的增强子。所述启动子或增强子可包括其它基因的启动子或增强子,和从任何其它原核细胞分离的启动子或增强子,和非“天然存在”的启动子或增强子,即含有不同转录调控区的不同元件,和/或改变表达的突变。除了以合成方式制造启动子和增强子的核酸序列以外,还可使用重组克隆和/或核酸扩增技术(包括PCR)结合本文所公开的组合物制造序列(参看美国专利号4,683,202、美国专利号5,928,906,各自以引用的方式并入本文中)。
在一些方面,启动子和/或增强子有效地指导选择用于表达的细胞类型中DNA区段的表达。可使用的所述启动子的一个实例为大肠杆菌阿拉伯糖或T7启动子。分子生物学领域的技术人员一般熟悉用于蛋白表达的启动子、增强子和细胞类型组合的使用,例如参看Sambrook等人(1989),以引用的方式并入本文中。所用的启动子可在指导所引入的DNA区段的高程度表达的适当条件下为组成性的、组织特异性的、诱导性的和/或适用的,如在重组蛋白和/或肽的大规模制造中为有利的。所述启动子可为异源的或内源的。
在一些方面,载体可包括起始信号和/或内部核糖体结合位点。也可包括特异性起始信号以用于编码序列的有效翻译。这些信号包括ATG起始密码子或相邻序列。可需要提供外源翻译控制信号,包括ATG起始密码子。所属领域的技术人员将轻易地能够确定这种信号并且提供必需信号。众所周知,起始密码子必须与所需编码序列的阅读框“同框”以确保整个插入物的翻译。所述外源翻译控制信号和起始密码子可为天然的或合成的。表达效率可通过包括适当转录增强子元件来增强。
在一些方面,载体可包括增加或优化编码所关注的基因的DNA区段的表达水平的序列。所述序列的实例包括在表达的mRNA中添加内含子(Brinster,R.L.等人(1988)Introns increase transcriptional efficiency in transgenicmice.Proc.Natl.Acad.Sci.USA 85,836–40;Choi,T.等人(1991)A generic intronincreases gene expression in transgenic mice.Mol.Cell.Biol.11,3070–4)。用于优化DNA区段的表达的方法的另一实例为“密码子优化”。密码子优化涉及在DNA区段中插入沉默突变以减少使用稀有密码子来优化蛋白翻译(Codon engineering for improvedantibody expression in mammalian cells.Carton JM,Sauerwald T,Hawley-Nelson P,Morse B,Peffer N,Beck H,Lu J,Cotty A,Amegadzie B,Sweet R.Protein ExprPurif.2007年10月;55(2):279-86.2007年6月16日电子出版)。
在一些方面,载体可包括多克隆位点。载体可包括多克隆位点(MCS),其为含有多个限制酶位点的核酸区,所述限制酶位点中的任一者均可结合标准重组技术使用以消化所述载体(参看Carbonelli等人,1999,Levenson等人,1998,和Cocea,1997,以引用的方式并入本文中)。“限制酶消化”是指用仅在核酸分子中的特异性位置处起作用的酶使核酸分子催化裂解。这些限制酶中的多种可在市面上购得。所述酶的使用由所属领域的技术人员理解。载体经常使用限制酶线性化或片段化,所述限制酶在所述MCS内进行切割以使得外源序列能够接合至所述载体。“接合”是指在两个核酸片段之间形成磷酸二酯键的过程,所述两个核酸片段可或可不彼此邻接。涉及限制酶和接合反应的技术为重组技术领域的技术人员众所周知的。
在一些方面,载体可包括末端信号。所述载体或构建体一般将包含至少一个末端信号。“末端信号”或“终止子”包含通过RNA聚合酶特异性终止RNA转录物所涉及的DNA序列。因此,在某些实施方案中,涵盖终止RNA转录物的制造的末端信号。终止子可为体内实现所需信使水平必需的。
预期使用的终止子包括本文所述或所属领域的技术人员已知的任何已知的转录终止子,包括但不限于例如ρ依赖性或ρ独立性终止子。在某些实施方案中,终止信号可为可转录或可翻译序列的缺乏,如归因于序列截短。
在一些方面,载体可包括复制起点。
为了在宿主细胞中繁殖载体,其可含有一个或多个复制起点位点(通常称为“ori”),所述载体为起始复制的特异性核酸序列。
在一些方面,载体可包括一种或多种可选择和/或可筛选标记物。在某些实施方案中,含有核酸构建体的细胞可通过在表达载体中包括标记物进行体外或体内鉴别。所述标记物将向细胞赋予可鉴别改变,从而允许容易鉴别含有所述表达载体的细胞。一般来说,可选择标记物为赋予允许选择的特性的标记物。阳性可选择标记物为其中所述标记物的存在允许其选择的标记物,而阴性可选择标记物为其中其存在防止其选择的标记物。阳性可选择标记物的实例为耐药性标记物。
通常,包括药物选择标记物有助于克隆和鉴别转化体,例如赋予对新霉素、嘌呤霉素、潮霉素、DHFR、GPT、博来霉素和组氨醇的抗性的基因为适用的可选择标记物。除了赋予允许基于条件的执行区别转化体的表型的标记物以外,还预期其它类型的标记物,包括可筛选标记物(如GFP),其基础为比色分析。或者,可利用可筛选酶,如氯霉素乙酰基转移酶(CAT)。所属领域的技术人员还应知晓如何使用免疫标记物,可能结合FACS分析。所用标记物未被视为重要的,只要其能够与编码基因产物的核酸同时表达即可。可选择和可筛选标记物的其它实例为所属领域的技术人员众所周知的。
一方面,所述载体可表达编码多种所关注的多肽的DNA区段。例如,编码免疫球蛋白重链和轻链的DNA区段可通过单一载体编码并且表达。一方面,两种DNA区段可包括于相同表达的RNA上并且使用内部核糖体结合位点(IRES)序列以使所述DNA区段表达为独立多肽(Pinkstaff JK,Chappell SA,Mauro VP,Edelman GM,Krushel LA.,Internalinitiation of translation of five dendritically localized neuronal mRNAs.,Proc Natl Acad Sci U S A.2001年2月27日;98(5):2770-5.2001年2月20日电子出版)。另一方面,各DNA区段具有其本身的启动子区,导致独立mRNA的表达(Andersen CR,NielsenLS,Baer A,Tolstrup AB,Weilguny D.Efficient Expression from One CMV EnhancerControlling Two Core Promoters.Mol Biotechnol.2010年11月27日.[印刷版之前的电子版])。
C.宿主细胞和表达系统
在一些方面,组合物可包括宿主细胞。在一些方面,宿主细胞可包括本文所述的聚核苷酸或载体。在一些方面,宿主细胞可包括真核细胞(例如昆虫、酵母或哺乳动物)或原核细胞(例如细菌)。在表达异源核酸序列的背景下,“宿主细胞”可指原核细胞,并且其包括能够复制载体和/或表达由载体编码的异源基因的任何可转化生物体。宿主细胞可并且已经用作载体的接受者。宿主细胞可“转染”或“转化”,其是指将外源核酸转移或引入宿主细胞中的过程。转化细胞包括原代受试者细胞和其后代。
在特定实施方案中,宿主细胞为革兰氏阴性细菌细胞。这些细菌适合使用,因为其在内膜与外膜之间具有细胞周质间隙并且具体说来,前述内膜在周质与细胞质之间,其还称作细胞质膜。因而,可能使用具有所述细胞周质间隙的任何其它细胞。革兰氏阴性细菌的实例包括但不限于大肠杆菌、绿浓假单胞菌、霍乱弧菌、鼠伤寒沙门氏菌、福氏志贺氏菌、流感嗜血杆菌、百日咳博德特氏菌、解淀粉欧文氏菌、根瘤菌属。革兰氏阴性细菌细胞可进一步作为细菌细胞定义,所述细菌细胞已经用包含能够结合所选配体的候选结合多肽的融合多肽的编码序列转化。所述多肽锚定于细胞质膜的外面,面对细胞周质间隙,并且可包含抗体编码序列或另一序列。一种用于所述多肽的表达的方式是使前导序列连接至能够引起所述引导的多肽。
多种原核细胞系和培养物可供用作宿主细胞,并且其可通过美国菌种保藏中心(ATCC)获得,美国菌种保藏中心(ATCC)为充当活培养物和遗传物质的档案馆的机构。适当宿主可由所属领域的技术人员基于载体骨架和所需结果确定。举例来说,质粒或粘粒可引入原核生物宿主细胞中用于多种载体的复制。作为用于载体复制和/或表达的宿主细胞使用的细菌细胞包括DH5-α、JM109和KC8,以及多种市面上可购得的细菌宿主,如SURETM感受态细胞和SOLOPACKTM Gold细胞(STRATAGENETM,La Jolla)。在一些方面,如大肠杆菌LE392的其它细菌细胞预期用作宿主细胞。
来自多种细胞类型和生物体的多种宿主细胞可获得并且将为所属领域的技术人员已知。同样,病毒载体可结合原核宿主细胞,尤其是允许用于所述载体的复制或表达的宿主细胞使用。一些载体可使用控制序列,所述控制序列允许其在原核和真核细胞中复制和/或表达。所属领域的技术人员将进一步了解孵育所有上述宿主细胞以维持所述细胞并且允许载体的复制的条件。还了解和知晓将允许载体的大规模制造以及由载体和其同源多肽、蛋白或肽编码的核酸的制造的技术和条件。
在一些方面,宿主细胞为哺乳动物的。实例包括CHO细胞、CHO-K1细胞或CHO-S细胞。其它哺乳动物宿主细胞包括NS0细胞,和为dhfr-的CHO细胞,例如CHO-dhfr-、DUKX-B11CHO细胞和DG44 CHO细胞。
存在多种表达系统,其可包含本文所公开的组合物的至少一部分或全部。表达系统可包括真核表达系统和原核表达系统。所述系统可能用于例如制造经鉴别为能够结合特定配体的多肽产品。基于原核生物的系统可用于制造核酸序列,或其同源多肽、蛋白和肽。多种所述系统可在市面上并且广泛购得。表达系统的其它实例包含含有如T7、Tac、Trc、BAD、λpL、四环素或Lac启动子的强原核生物启动子的载体、pET表达系统和大肠杆菌表达系统。
D.多肽
在一些方面,组合物可包括多肽。在一些方面,由本文所述的聚核苷酸编码的多肽可例如由宿主细胞表达。术语“多肽”或“蛋白”包括具有原生蛋白的氨基酸序列的大分子,也就是说由天然存在和非重组细胞制造的蛋白;或其由遗传工程改造的或重组细胞制造,并且包含具有原生蛋白的氨基酸序列的分子,或具有原生序列的一个或多个氨基酸的缺失、添加和/或取代的分子。所述术语还包括其中一种或多种氨基酸为对应的天然存在氨基酸和聚合物的化学类似物的氨基酸聚合物。术语“多肽”和“蛋白”涵盖抗原结合蛋白、抗体或具有抗原结合蛋白的一个或多个氨基酸的缺失、添加和/或取代的序列。术语“多肽片段”是指如与全长原生蛋白相比具有氨基端缺失、羧基端缺失和/或内部缺失的多肽。所述片段还可含有如与原生蛋白相比经过修饰的氨基酸。在某些实施方案中,片段为约5个至500个氨基酸长。例如,片段可为至少5、6、8、10、14、20、50、70、100、110、150、200、250、300、350、400个或450个氨基酸长。适用的多肽片段包括抗体的免疫功能片段,包括结合结构域。在结合抗体的情况下,适用的片段包括但不限于CDR区、重链和/或轻链的可变结构域、抗体链的一部分或仅其可变区(包括两个CDR)等。
术语“分离蛋白”意指主题蛋白(1)不含至少一些通常将与其一起发现的其它蛋白,(2)基本上不含来自相同来源,例如来自相同物种的其它蛋白,(3)由来自不同物种的细胞表达,(4)已经与在自然界与其缔合的聚核苷酸、脂质、碳水化合物或其它物质的至少约50%分离,(5)与在自然界未与其缔合的多肽操作性地缔合(通过共价或非共价相互作用),或(6)不在自然界存在。典型地,“分离蛋白”构成给定样品的至少约5%、至少约10%、至少约25%或至少约50%。合成起源的基因组DNA、cDNA、mRNA或其它RNA、核酸或其任何组合可编码所述分离蛋白。优选地,所述分离蛋白大致不含在其天然环境中发现的蛋白或多肽或其它污染物,所述蛋白或多肽或其它污染物将干扰其治疗、诊断、预防、研究或其它用途。
在一些方面,多肽可包括抗原结合蛋白(ABP)。如本文所用的“抗原结合蛋白”(“ABP”)意指结合规定的靶标抗原的任何蛋白。“抗原结合蛋白”包括但不限于抗体和其结合部分,如免疫功能片段。肽体为抗原结合蛋白的另一实例。如本文所用的术语抗体或免疫球蛋白链(重链或轻链)抗原结合蛋白的“免疫功能片段”(或简单地“片段”)为抗原结合蛋白物质,其包含缺乏存在于全长链中的至少一些氨基酸但仍能够特异性结合于抗原的抗体的一部分(无论所述部分如何获得或合成)。所述片段为生物学活性的,因为其结合于靶标抗原并且可与其它抗原结合蛋白(包括完整抗体)竞争结合于给定的表位。在一些实施方案中,所述片段为中和片段。这些生物学活性片段可通过重组DNA技术制造,或可通过抗原结合蛋白(包括完整抗体)的酶或化学裂解制造。免疫功能免疫球蛋白片段包括但不限于Fab、双功能抗体(与轻链可变结构域在相同多肽上的重链可变结构域,经由短肽连接子连接,所述短肽连接子过短而无法允许相同链上的两个结构域之间的配对)、Fab'、F(ab')2、Fv、结构域抗体和单链抗体,并且可源于任何哺乳动物来源,包括但不限于人类、小鼠、大鼠、骆驼科动物或兔。进一步预期,本文所公开的抗原结合蛋白的功能部分(例如一个或多个CDR)可能共价结合于第二蛋白或小分子以产生针对身体中的特定靶标的治疗剂,从而具有双功能治疗特性或具有延长血清半衰期。如所属领域的技术人员应了解,抗原结合蛋白可包括非蛋白组分。关于抗原结合蛋白和抗体的额外细节(如修饰、变体、制造方法和筛选方法)可发现于美国专利公布20110027287中,所述专利公布以引用的方式整体并入本文中来达成所有目的。
在一些方面,多肽可包括抗体。术语“抗体”是指任何同型的完整免疫球蛋白,或可与所述完整抗体竞争特异性结合于靶标抗原的其片段,并且包括例如嵌合抗体、人源化抗体、完全人抗体和双特异性抗体。“抗体”为抗原结合蛋白物质。完整抗体一般将包含至少两个全长重链和两个全长轻链,但在一些情况下可包括较少链,如天然存在于骆驼科动物中的抗体,其可仅包含重链。抗体可单独源于单一来源,或可为“嵌合的”,也就是说所述抗体的不同部分可源于两种不同抗体。所述抗原结合蛋白、抗体或结合片段可在杂交瘤中,通过重组DNA技术,或通过完整抗体的酶或化学裂解制造。除非另外指示,否则术语“抗体”除包含两个全长重链和两个全长轻链的抗体以外还包括其衍生物、变体、片段和突变蛋白。此外,除非明确地排除,否则抗体分别包括单克隆抗体、双特异性抗体、微抗体、结构域抗体、合成抗体(本文中有时称作“抗体模拟物”)、嵌合抗体、人源化抗体、人抗体、抗体融合物(本文中有时称作“抗体缀合物”)和其片段。在一些实施方案中,所述术语还涵盖肽体。
治疗有效量的ABP可施用于有需要的受试者。ABP可配制于药物组合物中。这些组合物除一种或多种ABP以外还可包含药学上可接受的赋形剂、载体、缓冲液、稳定剂或所属领域的技术人员众所周知的其它物质。所述物质应为无毒的并且不应干扰活性成分的功效。所述载体或其它物质的精确性质可取决于施用途径,例如口服、静脉内、皮肤或皮下、经鼻、肌肉内、腹膜内途径。
用于口服施用的药物组合物可呈片剂、胶囊、粉剂或液体形式。片剂可包括固体载体,如明胶或佐剂。液体药物组合物一般包括液体载体,如水、石油、动物或植物油、矿物油或合成油。可包括生理盐水溶液、右旋糖或其它糖溶液或二醇,如乙二醇、丙二醇或聚乙二醇。
关于静脉内、皮肤或皮下注射或在痛苦位点处注射,活性成分将呈肠胃外可接受的水溶液形式,所述水溶液无热原质并且具有合适的pH、等张性和稳定性。所属领域的技术人员充分能够使用例如等张媒介物制备合适溶液,所述等张媒介物如氯化钠注射液、林格氏注射液、乳酸化林格氏注射液。可如所需包括防腐剂、稳定剂、缓冲液、抗氧化剂和/或其它添加剂。
ABP施用优选地呈“治疗有效量”或“预防有效量”(视情况而定,不过预防可被视为疗法),这足以显示对个体的益处。实际施用量和施用速率和时程将取决于正在治疗的疾病的性质和严重程度。治疗处方(例如对剂量的决定等)在一般从业者和其它医师的职责内,并且典型地考虑待治疗的病症、个别患者的病况、递送位点、施用方法和从业者已知的其它因素。上文提及的技术和方案的实例可发现于Remington's Pharmaceutical Sciences,第16版,Osol,A.(编),1980中。
组合物可单独或与其它治疗组合(同时或依序,取决于待治疗的病况)施用。
III.免疫细胞
样品可包括免疫细胞。所述免疫细胞可包括T细胞和B细胞。T细胞(T淋巴细胞)包括例如表达T细胞受体的细胞。B细胞包括例如活化B细胞、胚B细胞、浆细胞、成浆细胞、记忆B细胞、B1细胞、B2细胞、边缘区B细胞和滤泡B细胞。T细胞包括活化T细胞、胚T细胞、辅助T细胞(效应T细胞或Th细胞)、细胞毒性T细胞(CTL)、记忆T细胞、中央记忆T细胞、效应记忆T细胞和调控T细胞。样品可包括单细胞(例如单一T或B细胞)或至少1,000个、至少10,000个、至少100,000个、至少250,000个、至少500,000个、至少750,000个或至少1,000,000个细胞。
A.B细胞
如本文所用,“B细胞”是指具有至少一个重排免疫球蛋白基因座的任何细胞。B细胞可包括至少一个重排免疫球蛋白重链基因座或至少一个重排免疫球蛋白轻链基因座。B细胞可包括至少一个重排免疫球蛋白重链基因座和至少一个重排免疫球蛋白轻链基因座。B细胞为淋巴细胞,其为适应性免疫系统的一部分。B细胞可包括将抗体以膜结合形式表达为细胞表面上的B细胞受体(BCR)或表达为分泌抗体的任何细胞。B细胞可表达免疫球蛋白(抗体、B细胞受体)。抗体可包括由重和轻免疫球蛋白链形成的异质二聚体。所述重链由可变、多样性和接合(VDJ)基因的基因重排形成以形成可变区,所述可变区接合至恒定区。所述轻链由可变和接合(VJ)基因的基因重排形成以形成可变区,所述可变区接着接合至恒定区。由于可能大量的接合组合,所述抗体基因(其也为BCR)的可变区具有巨大多样性,使得B细胞能够识别任何外来抗原并且对其作出反应。
B.B细胞活化和分化
当B细胞在发炎免疫反应的背景中识别抗原时,B细胞活化并且分化。其通过包括2种信号来活化,一种信号递送通过BCR(重排免疫球蛋白的膜结合形式),并且另一种信号递送通过CD40或另一共刺激分子。这一第二信号可通过与辅助T细胞的相互作用来提供,所述辅助T细胞在其表面上表达CD40的配体(CD40L)。B细胞接着增殖并且可经历体细胞超突变,其中所述抗体基因的核苷酸序列产生随机改变,并且选择其抗体对B细胞具有较高亲和力的B细胞。其也可经历“类别转换”,其中编码IgM同型的重链的恒定区转换为编码IgG、IgA或IgE同型的恒定区。分化B细胞可作为记忆B细胞终止,所述记忆B细胞通常具有较高亲和力并且进行类别转换,不过一些记忆B细胞仍具有IgM同型。记忆B细胞也可活化并且分化为成浆细胞并且最终分化为浆细胞。分化B细胞也可首先变为成浆细胞,所述成浆细胞接着分化以变为浆细胞。
C.亲和力成熟和克隆家族
克隆家族一般通过使用相关免疫球蛋白重链和/或轻链V(D)J序列由2种或更多种样品定义。相关免疫球蛋白重链V(D)J序列可通过其对在基因组中编码的V(D)J基因区段的共同使用来鉴别。在克隆家族内一般存在子家族,所述子家族基于其V(D)J区段内的共同突变而变化,可在B细胞基因重组和体细胞超突变期间产生。
活化B细胞在淋巴或其它组织内迁移并且形成胚中心,在所述组织内所述细胞经历亲和力成熟。B细胞也可在胚中心外部经历亲和力成熟。在亲和力成熟期间,B细胞经历其抗体基因的随机突变,所述随机突变集中在所述基因的互补决定区(CDR)中,所述互补决定区编码直接结合于并且识别靶标抗原的抗体的部分,所述B细胞针对所述靶标抗原活化。这由初始增殖B细胞产生子克隆,所述子克隆表达略微不同于初始克隆并且彼此不同的免疫球蛋白。克隆竞争抗原并且选择较高亲和力克隆,而较低亲和力克隆通过细胞凋亡死亡。这一过程导致B细胞的“亲和力成熟”并且因此导致表达以较高亲和力结合于所述抗原的免疫球蛋白的B细胞的产生。起源于相同‘亲本’B细胞的所有B细胞形成克隆家族,并且这些克隆家族包括识别相同或相似抗原表位的B细胞。在一些方面,我们预期以较高出现率存在的克隆代表以较高亲和力结合于抗原的克隆,因为最高亲和力克隆在亲和力成熟期间进行选择。在一些方面,具有不同V(D)J区段使用的克隆展现不同的结合特征。在一些方面,具有相同V(D)J区段使用但具有不同突变的克隆展现不同的结合特征。
D.记忆B细胞
记忆B细胞通常为亲和力成熟B细胞,并且可进行类别转换。这些细胞为可更快速地对后续抗原挑战作出反应的细胞,显著地将针对抗原的亲和力成熟抗体分泌所包括的时间从原生生物体中的约14天减少至约7天。
E.成浆细胞和浆细胞
浆细胞可为长寿命或短寿命的。长寿命浆细胞可在生物体的终生存活,而短寿命浆细胞可持续3-4天。长寿命浆细胞停留在发炎区域中、粘膜区域中(在IgA分泌浆细胞的情况下)、二级淋巴组织中(如脾或淋巴结)或骨髓中。为了到达这些发散区域,注定变成长寿命浆细胞的成浆细胞可在利用多种趋化因子梯度通行至适当区域之前首先行进通过血流。成浆细胞为亲和力成熟细胞,典型地进行类别转换,并且通常分泌抗体,不过所分泌抗体的量一般低于由浆细胞制造的抗体的量。浆细胞为专用抗体分泌者。
F.TCR和BCR基因的特征
由于鉴别重组存在于各个别适应性免疫细胞的DNA以及其相关RNA转录物中,可对RNA或DNA测序。来自T细胞或B细胞的重组序列也可称作克隆型。所述DNA或RNA可对应于编码抗体的来自T细胞受体(TCR)基因或免疫球蛋白(Ig)基因的序列。例如,所述DNA或RNA可对应于编码TCR的α、β、γ或δ链的序列。在大多数T细胞中,TCR为由α链和β链组成的异质二聚体。TCR-α链通过VJ重组产生,并且β链受体通过V(D)J重组产生。对于TCR-β链,在人类中存在48个V区段、2个D区段和13个J区段。在两个接合点中的每一者处,可缺失数个碱基并且可添加其它碱基(称为N和P核苷酸)。在少数T细胞中,TCR由γ和δ链组成。TCRγ链通过VJ重组产生,并且TCRδ链通过V(D)J重组产生(Kenneth Murphy、Paul Travers和Mark Walport,Janeway's Immunology第7版,Garland Science,2007,其以引用的方式整体并入本文中)。
在所述方法中分析的DNA和RNA可对应于编码具有恒定区(α、δ、γ、ε或μ)的重链免疫球蛋白(IgH)或具有恒定区λ或κ的轻链免疫球蛋白(IgK或IgL)的序列。各抗体可具有两个同一轻链和两个同一重链。各链由恒定(C)和可变区构成。对于重链,可变区由可变(V)、多样性(D)和接合(J)区段构成。编码这些区段中的每一种类型的数个相异序列存在于基因组中。特异性VDJ重组事件在B细胞的发育期间发生,标记所述细胞产生特异性重链。轻链中的多样性以类似方式产生,除了不存在D区,因此仅有VJ重组。体细胞突变通常接近所述重组的位点发生,引起数种核苷酸的添加或缺失,进一步增加由B细胞产生的重链和轻链的多样性。由B细胞产生的抗体的可能多样性接着为不同重链和轻链的产物。所述重链和轻链的可变区有助于形成抗原识别(或结合)区或位点。在这种多样性中添加体细胞超突变过程,其可在针对一些表位作出特异性反应之后发生。在这种过程中,突变在能够识别所述特异性表位的那些B细胞中发生,导致可能更强烈地结合所述特异性表位的抗体的更大多样性。所有这些因素均有助于由B细胞产生的抗体的极大多样性。可产生数十亿种并且可能超过一万亿种相异抗体。关于产生T细胞多样性的基础前提类似于关于由B细胞产生抗体的基础前提。T细胞和B细胞活化的要素为其结合于表位。特异性细胞的活化导致更多相同类型细胞的制造,导致克隆扩张。
互补决定区(CDR)或高变区为抗原受体(例如T细胞受体和免疫球蛋白)的可变结构域中可结合抗原的序列。各抗原受体的链含有三种CDR(CDR1、CDR2和CDR3)。产生T细胞(α和β)和免疫球蛋白(IgH和IgK或IgL)的两种多肽有助于三种CDR的形成。
CDR1和CDR2中由TCR-β编码的部分位于47个功能V区段之一内。CDR的大多数多样性发现于CDR3中,其中所述多样性由T淋巴细胞的发育期间的体细胞重组事件产生。
BCR的极大多样性存在于个体间和个体内。所述BCR由编码抗体重链和轻链的两种基因IgH和IgK(或IgL)构成。结合抗原和MHC分子的三种互补决定区(CDR)序列在IgH和IgK(或IgL)中具有大多数多样性。CDR1和CDR2中由IgH编码的部分位于44个功能V区段之一内。原生B细胞的大多数多样性在CDR3的产生中通过B淋巴细胞的发育期间的体细胞重组事件出现。所述重组可产生具有所述V、D和J区段中每样一个的分子。在人类中,存在44个V、27个D和6个J区段;因此,存在超过7,000种组合的理论可能性。在小部分BCR中(约5%),发现两个D区段。此外,在两个接合点中的每一者处,可缺失数个碱基并且可添加其它碱基(称为N和P核苷酸),从而产生极大多样性程度。在B细胞活化后,发生通过体细胞超突变实现的亲和力成熟过程。在这一过程中,活化B细胞的后代细胞在基因中聚集不同体细胞突变,其中在CDR区中具有较高突变浓度,导致产生对抗原具有较高亲和力的抗体。除体细胞超突变以外,活化B细胞还经历同型转换的过程。具有相同可变区段的抗体可具有不同形式(同型),视恒定区段而定。尽管所有原生B细胞均表达IgM(或IgD),活化B细胞主要表达IgG,以及IgM、IgA和IgE。从IgM(和/或IgD)至IgG、IgA或IgE的这一表达转换通过重组事件发生,引起一种细胞专用于制造特异性同型。对于各IgM、IgD和IgE存在一个区段,对于IgA存在两个区段,并且对于IgG存在四个区段。
IV.计算机执行
在一些方面,一种或多种本文所述的方法可在计算机上执行。在一个实施方案中,计算机包含至少一个耦合于芯片组的处理器。在一些实施方案中,所述芯片组耦合于存储器、存储器件、键盘、图形适配器、定点器件和/或网络适配器。显示器典型地耦合于所述图形适配器。在一个实施方案中,所述芯片组的功能性由存储控制器中心和/或I/O控制器中心提供。在另一实施方案中,所述存储器直接耦合于所述处理器而非所述芯片组。
所述存储器件为能够容纳数据的任何器件,如硬盘驱动器、光盘只读存储器(CD-ROM)、DVD或固态存储器件。所述存储器容纳由所述处理器使用的指令和数据。所述定点器件可为鼠标、游标控制球或其它类型的定点器件,并且与键盘组合使用以将数据输入计算机系统中。所述图形适配器在显示器上显示图像和其它信息。所述网络适配器将计算机系统耦合于局域或广域网络。
如所属领域中已知,计算机可具有与先前所述的那些不同和/或其它组件。另外,计算机可缺乏某些组件。此外,所述存储器件可为计算机局部的和/或远程的(如在存储区域网络(SAN)内具体化)。
如所属领域中已知,使计算机适应以执行用于提供本文所述的功能性的计算机程序模块。如本文所用,术语“模块”是指用于提供规定的功能性的计算机程序逻辑。因此,模块可在硬件、固件和/或软件中执行。在一个实施方案中,程序模块存储在所述存储器件上,装载于所述存储器中,并且由所述处理器执行。
本文所述的实体的实施方案可包括其它和/或与此处所述的模块不同的模块。另外,归因于所述模块的功能性可在其它实施方案中由其它或不同模块执行。此外,这一描述出于清晰并且便利的目的偶尔省略术语“模块”。
V.试剂盒
本文进一步公开包含本文所述的衔接构建体的试剂盒。试剂盒可包含多种偶联于本文所述的衔接构建体的固体支撑物。在一些实施方案中,所述试剂盒包含衔接模板文库,所述文库包含多种衔接模板。在一些实施方案中,所述试剂盒包含衔接模板文库,所述文库包含多种偶联于多种固体支撑物的衔接模板。所述试剂盒可进一步包含用于通过酶反应由所述衔接模板构建体产生本文所述的衔接分子(例如,条形码衔接分子)的酶。在一些实施方案中,所述试剂盒包含本文所述的细胞悬浮缓冲液。
试剂盒可包括本文所公开的聚核苷酸、聚核苷酸文库、载体和/或宿主细胞和使用说明书。所述试剂盒可在合适容器中包含本文所公开的聚核苷酸、聚核苷酸文库、载体和/或宿主细胞、一种或多种对照和所属领域中众所周知的多种缓冲液、试剂、酶和其它标准成分。
所述容器可包括在包含一个或多个孔的板上的至少一个孔。所述容器可包括至少一个小瓶、试管、烧瓶、瓶、注射器或其它容器构件,聚核苷酸、聚核苷酸文库、载体和/或宿主细胞可放入其中,并且在一些情况下合适地等分试样。在提供额外组件的情况下,所述试剂盒可含有额外容器,这一组件可放入所述额外容器中。所述试剂盒还可包括用于容纳所述聚核苷酸、聚核苷酸文库、载体和/或宿主细胞的构件和密切限制于商业规模的任何其它试剂容器。所述容器可包括注射或吹塑成型塑料容器,所需小瓶保留于其中。可包括贴有使用说明书和/或警告标签的容器。
VI.器件
本发明的实施方案包括用于产生和传送反应容积的器件。这些容积可以微流体规模出现并且可与载液进行相分离。可由所述器件处理的反应容积的实例包括反相乳液中的水性液滴(即,水/油乳液)。所述器件允许条形码衔接模板、条形码衔接分子、样品(例如细胞)和/或从这些样品获得的RNA分别地或一起封装于液滴中。所述器件还允许将试剂引入液滴中,使得条形码衔接分子可酶法产生并且可对来自个别样品的RNA编条形码。
如本文所用并且要求的器件的非限制性实例描绘于图17-19中。熟练技术人员应认识到这些器件的变化形式也可建构并且用于本发明方法中。器件一般包括三个微流体途径,各途径耦合于压力源和流量传感器。用于微流体途径的压力源驱动流体通过所述途径,并且出现于所述压力源的下游的所述流量传感器可用于测量通过所述途径的流动速率。在一些实施方案中,第一途径101和第二途径102在第一接合点104处合并以形成组合途径,所述组合途径接着与第三途径103在第二接合点105处合并。所述第二接合点出现于微流体液滴芯片中并且可为产生微流体液滴的位点。
如本文所述的器件可由可获自IDEX Corporation(Lake Forest,Illinois,U.S.A.)的管和射流组件并且使用可获自Dolomite Microfluidics(Charlestown,Massachusetts,U.S.A.)的微流体液滴芯片组装。所述微流体液滴芯片的一些特征描述于美国专利号7,268,167、7,375,140、7,717,615、7,772,287、8,741,192和8,883,864中,所述专利以引用的方式并入本文中。合适的压力源包括注射泵和压力泵。压力泵可获自Dolomite Microfluidics。所述压力源可独立地控制。
在一些实施方案中,所述第一和第二微流体途径传送水溶液。各途径可包括注射口和阀(例如四通阀)以使在所述注射口中引入的溶液与所述途径在同一条线上。在一些实施方案中,容纳水性载液的储槽安置在各四通阀的上游。当所述水性载液在下游被驱动或在下游推动水溶液的插塞朝向所述第一接合点时,所述载液可与所述水溶液在所述四通阀中混合。在一些实施方案中,流阻器安置在各微流体途径中。
一旦水溶液被引入所述第一或第二微流体途径中,其可传递通过样品环管,所述样品环管计量所述溶液朝向所述第一接合点的流动。计量可如所需实现,例如使用流体阻力或沿所述样品环管安置的阀。在一些实施方案中,一个样品环管与所述第一和第二微流体途径中的每一者缔合,并且所述样品环管与热冷却单元接触。可包括所述热冷却单元以防止所述水溶液中酶、核酸或其它生物组分的热变性,或针对酶反应建立最佳温度。所述热冷却单元中与用于所述第一和第二微流体途径的样品环管接触的部分可独立地或联合加以控制。任何物质或装置均可用作热冷却单元,其限制条件是其可使传递通过所述样品环管的水溶液的温度偏离环境温度。合适的热冷却器件的实例为珀耳帖器件和冰箱。
在一些实施方案中,传送通过所述第一微流体途径的水溶液含有细胞和条形码衔接模板珠粒。在一些实施方案中,传送通过所述第二微流体途径的水溶液含有用于细胞溶解的试剂和用于制造所关注的聚核苷酸的试剂(例如,用于产生条形码衔接分子的酶)。与各微流体途径缔合的注射口、阀和/或样品环管可经过配置或经过定制以容纳传递通过所述途径的水溶液的内含物。例如,与所述第一微流体途径缔合的样品环管可具有放大的内径以容纳细胞和珠粒。应认识到关于在所述第一和第二微流体途径之间分配细胞、珠粒和试剂存在多种其它选项,使得这些组分均在所述第一接合点组合。例如,细胞可传送通过所述第一微流体途径并且珠粒可传送通过所述第二微流体途径。各途径可鉴于其携带的水溶液的内含物,如所需经过配置。
由所述第一微流体途径和所述第二微流体途径的合并产生的组合途径又与所述第三微流体途径在所述微流体液滴芯片中合并。这发生在所述第二接合点,其在所述第一接合点的下游。可在所述第一接合点与第二接合点之间建立任何所需距离。在一些实施方案中,所述第一接合点也位于所述微流体液滴芯片内。在一些实施方案中,所述第一接合点恰在所述第二接合点上游,使得所述组合途径中的流体在与来自所述第三微流体途径的流体组合之前行进可忽略的距离(例如,小于10、3、1、0.3或0.1cm)。这种安排可减少所述组合途径中组分的混合。在一些实施方案中,所述器件中的所述微流体途径的尺寸(在所述微流体液滴芯片的内部和/或外部)使得流体的移动通过层流管理。
所述第三微流体途径可经过配置以递送油/表面活性剂混合物至所述微流体液滴芯片。因此,在所述器件中的第二接合点,水相和疏水相可混合并且可形成微流体液滴。可选择所述第二接合点的几何形状以确保这些液滴具有所需特征。例如,可选择几何形状以促进在所述微流体途径中在合适的流动速率下形成具有所需大小并且彼此间隔开所需距离的单分散液滴。在一些实施方案中,所述第三微流体途径分成所述微流体液滴芯片的上游的两个子途径,所述两个子途径在所述第二接合点连同所述组合(水性)途径合并在一起。所述两个子途径可以大角度(例如,大约或至少30、60、90、120、150或180度)彼此逼近,使得所述油/表面活性剂混合物在其进入所述第二接合点时形成围绕所述水性混合物的外壳。使用这种几何形状,水性液滴被从所述水性混合物‘夹断’并且当其离开所述接合点时以大约与所述水性混合物相同的方向流动。这种产生液滴的方法在所属领域中被称作流动聚焦。在其它实施方案中,所述组合水性途径以恰当的角度与所述第三微流体途径相交,因此使所述第二接合点具有t形接合几何形状。在这些实施方案中,油/表面活性剂混合物笔直流动通过所述接合点。所述水性混合物以垂直于由这种混合物形成的液滴被携带离开所述接合点的方向的方向逼近所述接合点。多种微流体几何形状中液滴形成的物理学描述于Thorsen等人,Phys.Rev.Lett.86,4163-4166,2001中和别处。
由所述含有水性混合物的组合途径和所述含有油/表面活性剂混合物的第三微流体途径的合并形成的含有液滴的流体途径构成样品途径。所述样品途径被递送至出现在所述第二接合点的下游的样品收集容器。在所述样品收集容器中,液滴可经受热循环。所述液滴也可被破开并且可收集编条形码核酸。
在操作中,本文所述的器件可用于将条形码衔接模板珠粒和细胞封装于水性微流体液滴中,使得各液滴平均含有大约一种珠粒和一种细胞。各液滴中的珠粒和细胞的数目可如所需调整,例如通过调节装载于所述器件中的溶液中珠粒或细胞的浓度,或通过调节所述三个微流体途径中的流动速率。包括于各液滴中的试剂允许条形码衔接分子由所述液滴中的所述一种珠粒酶法产生。这些试剂还允许所述一种细胞溶解并且允许来自所述细胞的RNA经历编条形码反应。因此,来自所述细胞的RNA可在所述液滴内编条形码,并且当混合来自多种细胞的核酸时,源于这些RNA的核酸(并且含有条形码序列)可稍后追溯到一种细胞。
VII.实施例
A.实施例1:在单一反应中制造条形码衔接模板珠粒文库。
使用下文所述的方法以使用乳液PCR产生条形码衔接模板珠粒文库,其中执行聚合酶链反应(PCR)来连接独特条形码衔接模板至各珠粒(参看图15)。
表4:用于在单一反应中制造条形码衔接模板珠粒文库的寡核苷酸


抗生蛋白链菌素涂布的M-270 (Life Technologies)与生物素标记的寡核苷酸(“emB_T7bridge2”)偶联:
(Life Technologies)与生物素标记的寡核苷酸(“emB_T7bridge2”)偶联:
1.珠粒通过轻微涡旋再悬浮
2. 1mL M270珠粒(大约6.7×108个珠粒)放入三个1.5mL微量离心管中的每一者中,总计3mL
3.放在磁铁上持续3分钟。
4.从各管取出上清液并且再悬浮于1mL(1x体积)结合/洗涤缓冲液(BWB;1M NaCl、5mM Tris、0.5mM EDTA)中
5.再重复步骤4两次,随后最终再悬浮于540μL体积BWB中
6. 60μL 100μM emB_T7bridge2添加至珠粒中并且在轻微旋转下孵育15分钟
7.在孵育后,珠粒用1mL BWB缓冲液洗涤3次,并且组合至单一管中
8.珠粒在4℃下与0.01%叠氮化钠一起存储
9.珠粒在使用前用10mM Tris洗涤3次
条形码寡核苷酸和正向和反向引物在基于乳液的PCR中添加至来自以上的偶联珠粒中:
1.以下PCR混合物(3mL总体积)在三个1.5mL微量离心管(VWR目录号20170-650)中制备:

2.制备油-表面活性剂混合物(1mL总体积):
a.矿物油(Sigma) 900μL
b.EM90(Evonik) 100uL
3. 800μL油-表面活性剂混合物和200μL PCR混合物组合至15个Axygen 2.0mLMaxymum Recovery锥形底微量离心管(MCT-200-L-C)中的每一者中。管密封并且振荡持续3秒
4.管放入Qiagen TissueLyzer II中,并且在14Hz下振荡持续5分钟
5.所述乳液在VWR 96孔PCR板(83007-374)的孔中分配,其中每孔添加160μL乳液
6.管使用以下程序进行热循环:

乳液被破乳并且回收珠粒:
1.将所述PCR板的内含物转移至1.5mL微量离心管(VWR20170-650)中,每个管具有不超过0.5mL乳液体积
2. 100uL 1uM emB_T7bridgefree引物添加至各管中
3.管用异丁醇终止,密封并且振荡以充分混合
4.管在14,000rpm下离心持续1min
5.管放在磁条上以将所述珠粒吸引至所述管的侧面,接着尽可能抽吸尽可能多的上清液,同时留下沉淀的珠粒
6.添加1mL异丁醇,通过上下吸移充分混合,直至剩余油/乳液体积已经分散至所述异丁醇中
7.管放在磁条上以将所述珠粒吸引至所述管的侧面,接着抽吸所述异丁醇。来自所有管的珠粒通过首先从其中将组合所述珠粒的管抽吸上清液并且接着从另一管转移全部体积,酌留在所述磁铁处收集所述珠粒的时间,接着抽吸上清液并且重复而组合至所述单一管中
8.添加1mL新鲜异丁醇,充分混合并且搁置60秒
9.抽吸异丁醇
10.添加1mL 100%乙醇,充分混合并且搁置60秒
11.抽吸乙醇
12.重复步骤10和11
13.添加1mL 70%乙醇,充分混合并且搁置60秒
14.抽吸乙醇
15.重复步骤13和14
16.添加1mL PBS,充分混合并且搁置60秒。
17.抽吸PBS
18.重复步骤16和17
并入条形码衔接模板的珠粒接着使用Becton Dickenson FACS Aria III,利用来自并入emB_Rv3反向引物中的Alexa Fluor 647染料的荧光与未编条形码珠粒分选。
珠粒在4℃下存储于0.01%叠氮化钠中以便存储。
注意这一实施例制造具有T7 RNAP启动子序列的条形码衔接模板珠粒以便由T7RNAP扩增条形码衔接子。通过用其它RNAP启动子序列置换emB-T7bridge2中的T7 RNAP启动子序列“TAA TAC GAC TCA CTA TAG G”(SEQ ID NO:6),可使用其它RNAP扩增条形码衔接子。另外,通过用切口核酸内切酶位点(如Nt.BbvCI的“CCT CAG C”)置换所述启动子序列,可使用切口核酸内切酶(例如Nt.BbvCI)和链置换DNAP(如Klenow exo-)扩增条形码衔接子。
另外,emB-BCbridge2中的“HH HTH HHH THH HHT HHH THH HH”(SEQ ID NO:3)产生约3.87亿种独特条形码。当这一条形码文库用于对甚至例如1千万种细胞编条形码时,仅使用2.5%的所述独特条形码。预期大多数所述条形码彼此相距充分距离,使得大多数来自NextGen测序的条形码序列读数容易彼此区别(其中一部分读数被弃去),而不考虑PCR和测序误差。
所述乳液可使用所属领域中已知的多种方法制得,并且在这种情况下使用振荡方法制得并且所得液滴为多分散的,其中平均液滴直径为约25μm。条形码寡核苷酸用正向和反向引物扩增并且所述反向引物用荧光标签标记,所述荧光标签在这一实施例中为AlexaFluor 647,使得并入条形码衔接模板的珠粒可与未标记珠粒区别。并入条形码衔接模板的明亮荧光珠粒接着与暗淡未标记珠粒进行FACS分选。
在这一实施例中的珠粒和条形码寡核苷酸的规定浓度下,通过泊松分布将珠粒以平均每个液滴约7个珠粒装载于液滴中,并且我们观察到大约28%的液滴含有独特条形码寡核苷酸的一种或多种拷贝,而剩余的液滴根本不含条形码寡核苷酸。在含有至少一种条形码寡核苷酸的液滴中,约70%应精确地含有一种条形码寡核苷酸,而剩余约30%应含有两种或更多种条形码。因此,所述条形码模板衔接珠粒文库的约70%为单克隆的(每个珠粒一种独特条形码序列)并且约30%为多克隆的。
下文所述的方法的最终产量为大约1200万个条形码衔接模板珠粒,其中约840万个为单克隆条形码衔接模板珠粒。并且尽管液滴填充有平均每个液滴约7个珠粒,在破乳所述乳液后,珠粒的产率为约2%。基于二项式分布,约770万种独特条形码序列存在于这一条形码衔接模板珠粒文库中。
珠粒和条形码寡核苷酸的浓度可经过调节以获得条形码衔接模板珠粒文库,其中存在不同比例的单克隆和多克隆珠粒和不同数目的独特条形码序列。这将允许对来自单细胞的核酸编条形码以实现不同比例的核酸经由独特条形码或独特条形码的集合缔合至单细胞,并且还改变从进一步分析弃去的编条形码核酸的百分率。
这种条形码衔接模板珠粒制造方法可优化以实现例如90%:10%、99%:1%或任何其它比率的单克隆:多克隆珠粒比率。这种相对于目前约70%:30%比率的改进可通过数种不同方法实现,包括进一步稀释含有所述条形码序列的寡核苷酸(在这种情况下为emB-BCbridge2),使得较少拷贝在所述乳液中的液滴中分配,导致封装于任何给定液滴中的多种条形码序列的发生率降低。
B.实施例2:在单一反应II中制造条形码衔接模板珠粒文库。
使用下文所述的方法以使用乳液PCR产生条形码衔接模板珠粒文库,其中执行聚合酶链反应(PCR)来连接独特条形码衔接模板至各珠粒(参看图15)。
表5:用于在单一反应II中制造条形码衔接模板珠粒文库的寡核苷酸


抗生蛋白链菌素涂布的M-270 (Life Technologies)与生物素标记的寡核苷酸(“emB_T7bridgeIsceI”)偶联:
(Life Technologies)与生物素标记的寡核苷酸(“emB_T7bridgeIsceI”)偶联:
1.珠粒通过轻微涡旋再悬浮
2. 1mL M270珠粒(大约6.7×108个珠粒)放入三个1.5mL微量离心管中的每一者中,总计3mL
3.放在磁铁上持续3分钟。
4.从各管取出上清液并且再悬浮于1mL(1x体积)结合/洗涤缓冲液(BWB;1M NaCl、5mM Tris、0.5mM EDTA)中
5.再重复步骤4两次,随后最终再悬浮于540μL体积BWB中
6. 60μL 100μM emB_T7bridgeIsceI添加至珠粒中并且在轻微旋转下孵育15分钟
7.在孵育后,珠粒用1mL BWB缓冲液洗涤3次,并且组合至单一管中
8.珠粒在4℃下与0.01%叠氮化钠一起存储
9.珠粒在使用前用10mM Tris洗涤3次
条形码寡核苷酸和正向和反向引物在基于乳液的PCR中添加至来自以上的偶联珠粒中:
1.以下PCR混合物(3mL总体积)在三个1.5mL微量离心管(VWR目录号20170-650)中制备:


2.制备油-表面活性剂混合物(1mL总体积):
a.矿物油(Sigma) 900μL
b.EM90(Evonik) 100μL
3. 800μL油-表面活性剂混合物和200μL PCR混合物组合至15个Axygen 2.0mLMaxymum Recovery锥形底微量离心管(MCT-200-L-C)中的每一者中。管密封并且振荡持续3秒
4.管放入Qiagen TissueLyzer II中,并且在14Hz下振荡持续5分钟
5.所述乳液在VWR 96孔PCR板(83007-374)的孔中分配,其中每孔添加160μL乳液
6.管使用以下程序进行热循环:


乳液被破乳并且回收珠粒:
1.将所述PCR板的内含物转移至1.5mL微量离心管(VWR20170-650)中,每个管具有不超过0.5mL乳液体积
2. 100uL 1uM emB_T7bridgefreeIsceI_2引物添加至各管中
3.管用异丁醇终止,密封并且振荡以充分混合
4.管在14,000rpm下离心持续1min
5.管放在磁条上以将所述珠粒吸引至所述管的侧面,接着尽可能抽吸尽可能多的上清液,同时留下沉淀的珠粒
6.添加1mL异丁醇,通过上下吸移充分混合,直至剩余油/乳液体积已经分散至所述异丁醇中
7.管放在磁条上以将所述珠粒吸引至所述管的侧面,接着抽吸所述异丁醇。来自所有管的珠粒通过首先从其中将组合所述珠粒的管抽吸上清液并且接着从另一管转移全部体积,酌留在所述磁铁处收集所述珠粒的时间,接着抽吸上清液并且重复而组合至所述单一管中
8.添加1mL新鲜异丁醇,充分混合并且搁置60秒
9.抽吸异丁醇
10.添加1mL 100%乙醇,充分混合并且搁置60秒
11.抽吸乙醇
12.重复步骤10和11
13.添加1mL 70%乙醇,充分混合并且搁置60秒
14.抽吸乙醇
15.重复步骤13和14
16.添加1mL PBS,充分混合并且搁置60秒。
17.抽吸PBS
18.重复步骤16和17
并入条形码衔接模板的珠粒接着使用Becton Dickenson FACS Aria III,利用来自并入emB_IsceI_RV反向引物中的Alexa Fluor 647染料的荧光与未编条形码珠粒分选。
珠粒在4℃下存储于0.01%叠氮化钠中以便存储。
注意这一实施例制造具有T7 RNAP启动子序列的条形码衔接模板珠粒以便由T7RNAP扩增条形码衔接子。通过用其它RNAP启动子序列置换emB-T7bridgeIsceI中的T7 RNAP启动子序列“TAA TAC GAC TCA CTA TAG G”(SEQ ID NO:6),可使用其它RNAP扩增条形码衔接子。另外,通过用切口核酸内切酶位点(如Nt.BbvCI的“CCT CAG C”)置换所述启动子序列,可使用切口核酸内切酶(例如Nt.BbvCI)和链置换DNAP(如Klenow exo-)扩增条形码衔接子。
另外,emB-BCbridgeIsceI_2中的“HH HTH HHH THH HHT HHH THH HH”(SEQ IDNO:3)产生约3.87亿种独特条形码。当这一条形码文库用于对甚至例如1千万种细胞编条形码时,仅使用2.5%的所述独特条形码。预期大多数所述条形码彼此相距充分距离,使得大多数来自NextGen测序的条形码序列读数容易彼此区别(其中一部分读数被弃去),而不考虑PCR和测序误差。
在这一实施例中的珠粒和条形码寡核苷酸的规定浓度下,通过泊松分布将珠粒以平均每个液滴约7个珠粒装载于液滴中,并且我们观察到大约25%的液滴含有独特条形码寡核苷酸的一种或多种拷贝,而剩余的液滴根本不含条形码寡核苷酸。在含有至少一种条形码寡核苷酸的液滴中,约75%精确地含有一种条形码寡核苷酸,而剩余约25%含有两种或更多种条形码。因此,所述条形码模板衔接珠粒文库的约75%为单克隆的(每个珠粒一种独特条形码序列)并且约25%为多克隆的。
下文所述的方法的最终产量为大约5千万个条形码衔接模板珠粒,其中约3750万个为单克隆珠粒。尽管液滴填充有平均每个液滴约7个珠粒,在破乳所述乳液后,珠粒的产率为约11%。基于二项式分布,存在约2800万个具有独特条形码序列的单克隆珠粒。
珠粒和条形码寡核苷酸的浓度可经过调节以获得条形码衔接模板珠粒文库,其中存在不同比例的单克隆和多克隆珠粒和不同数目的独特条形码序列。这将允许对来自单细胞的核酸编条形码以实现不同比例的核酸经由独特条形码或独特条形码的集合缔合至单细胞,并且还改变从进一步分析弃去的编条形码核酸的百分率。
C.实施例3:在多步骤中制造条形码衔接模板珠粒文库。
在这一实施例中,进行根据图16的反应,除了使用仅一种S1、一种W和一种S2条形码序列。因此,未发生偶联于不同S1序列的珠粒的汇集并且同样,在聚合酶延伸反应以添加W序列至所述S1寡核苷酸中后,珠粒未汇集。
这一实施例可容易地扩展至根据图16简单地通过具有多种具有独特条形码序列的S1-寡核苷酸、W-寡核苷酸和S2-寡核苷酸来进行。
表6:用于在单一反应中制造条形码衔接模板珠粒文库的寡核苷酸

抗生蛋白链菌素涂布的M-270 (Life Technologies)在个别反应中与生物素标记的含有S1序列的寡核苷酸偶联:
(Life Technologies)在个别反应中与生物素标记的含有S1序列的寡核苷酸偶联:
1.珠粒通过轻微涡旋再悬浮
2.M270珠粒(Life Technologies)放在磁铁上持续3分钟。
3.从各管取出上清液并且再悬浮于(1x体积)0.5x结合/洗涤缓冲液(BWB;1MNaCl、5mM Tris、0.5mM EDTA)中
4.再重复步骤4两次,随后最终再悬浮于BWB缓冲液中
5. 10μM S1-寡核苷酸添加至珠粒中并且在轻微旋转下孵育15分钟
6.在孵育后,珠粒用BWB缓冲液洗涤3次
7.珠粒在4℃下与0.01%叠氮化钠一起存储
8.珠粒在使用前用10mM Tris洗涤3次
偶联的珠粒接着汇集在一起,并且执行使用w-寡核苷酸的延伸反应。
关于w延伸反应:

在55℃下在振荡孵育器中孵育过夜,在800rpm下振荡。
珠粒汇集并且用1x BWB缓冲液洗涤三次。反义链接着在70℃熔融缓冲液(50mMNaCl、10mM Tris pH 8.0)中熔融。珠粒用磁铁沉淀并且完全去除上清液,接着珠粒在1mLTE0.1中洗涤三次并且以1mg/20uL再悬浮于TE0.1中。
关于s2延伸反应(每250μg珠粒):


S2-寡核苷酸-a与S1+w-a珠粒一起使用,并且S2-寡核苷酸-b与S1+w-b珠粒一起使用。在60℃下孵育10min,接着缓慢冷却至37℃。在37℃下孵育2小时,在800rpm下振荡。接着使反应冷却至室温。
接着添加以下:
dNTP(NEB) 1μL
大肠杆菌焦磷酸酶(NEB) 0.1μL
Klenow片段(NEB) 1μL
反应在25℃下孵育3小时,在800rpm下振荡。用1μL dNTP更新每小时反应。
珠粒汇集并且用1x BWB缓冲液洗涤三次。珠粒在4℃下与0.01%叠氮化钠一起存储并且在使用前用10mM Tris洗涤3次。
条形码衔接模板珠粒的小等分试样也用于使用T7 RNAP的体外转录反应中以确定所述珠粒的制造是否成功。如果成功,那么T7RNAP将能够从存在于s1-寡核苷酸序列中的双链T7启动子转录RNA。使用Megascript T7试剂盒(Life Technologies)并且遵循制造商的说明书。5μL反应在RNA Flashgel(Lonza)上运行。参看图20。
如由S1、W和S2序列的组合形成的独特条形码序列的数目可如所需增加或降低。例如,如表1中可见,如果独特条形码的数目如通过二项式分布所确定为待编条形码的细胞的数目的约10倍大,那么我们可预期约10%的细胞共享同一条形码并且因此在使核酸彼此生物信息学连接(这可检测为超过一种可变基因核酸,如两个免疫球蛋白重链或两个TCRα链彼此缔合)期间被弃去。因此,由所述文库,我们可预期约90%的编条形码细胞用独特序列成功地编条形码,使得核酸能够彼此适当信息学连接。
因此,所需的S1x、W和S2y序列的数目取决于待编条形码的细胞的所需数目。在表7中,预计W-延伸反应在96孔板中发生,并且使用同一数目的S1x和S2y序列。如可见,为了对1千万种细胞编条形码,需要至多323种S1x和S2y寡核苷酸和960种Wz寡核苷酸。这些为可管理数目,尤其当所述反应在96孔板中进行时,使得有必要总计仅18个96孔板执行所述反应以制造所需大小的条形码衔接模板珠粒文库。
表7.获得足够大小的条形码衔接模板文库以对来自所需数目的细胞的核酸编条形码所需的S1x、Wz和S2y序列的数目

此外,可需要将S1x、S2y和Wz中的条形码设计成最小汉明距离隔开,其中这一最小值为2。使用这一最小值,仅使用与所述条形码序列精确匹配的来自NextGen测序的条形码序列读数;弃去具有误差的条形码序列读数。如果所用的汉明距离或编辑距离增加至最小值3,那么误差校正为可能的。
注意这一实施例制造具有T7 RNAP启动子序列的条形码衔接模板珠粒以便由T7RNAP扩增条形码衔接子。通过用其它RNAP启动子序列置换emB-T7bridge2中的T7 RNAP启动子序列“TAA TAC GAC TCA CTA TAG G”(SEQ ID NO:6),可使用其它RNAP扩增条形码衔接子。另外,通过用切口核酸内切酶位点(如Nt.BbvCI的“CCT CAG C”)置换所述启动子序列,可使用切口核酸内切酶(例如Nt.BbvCI)和链置换DNAP(如Klenow exo-)扩增条形码衔接子。
D.实施例4:制造水性条形码衔接模板。
在这一实施例中,合成未偶联于珠粒的水性条形码衔接模板以证明本发明方法的广泛适用性。
如下文所述制备反应混合物:

反应混合物接着以每孔25μL等分试样至96孔PCR板中并且如下进行热循环:


所得PCR产物为所述条形码衔接模板,其接着钝化以去除A悬垂物:
NEBuffer 2 162μL
10mM dNTP 30μL
T4DNA聚合酶(New England Biolabs) 2μL
2.5μL所述钝化混合物添加至各25μL反应容积中,并且在12℃下孵育15分钟。1μL250mM EDTA接着添加至各25μL反应容积中并且加热至75℃持续20分钟以使所述酶失活。
净化所述反应并且定量:
1.反应接着使用Zymo Research RNA清洁和浓缩试剂盒遵循制造商说明书汇集并且净化
2.使用Picogreen定量试剂盒(Life Technologies)来定量所述DNA并且将条形码衔接模板浓度调节至55ng/uL
注意这一实施例制造具有T7 RNAP启动子序列的条形码衔接模板珠粒以便由T7RNAP扩增条形码衔接子。通过用其它RNAP启动子序列置换emB-T7bridge2中的T7 RNAP启动子序列“TAA TAC GAC TCA CTA TAG G”(SEQ ID NO:6),可使用其它RNAP扩增条形码衔接子。另外,通过用切口核酸内切酶位点(如Nt.BbvCI的“CCT CAG C”)置换所述启动子序列,可使用切口核酸内切酶(例如Nt.BbvCI)和链置换DNAP(如Klenow exo-)扩增条形码衔接子。
E.实施例5:添加来自条形码衔接模板的条形码至不同反应缓冲液中的mRNA中。
这一实施例显示本发明方法可在多种不同缓冲液中使用。如上文实施例4中所述制造条形码衔接模板。
表8.反应缓冲液的组成

设置以下反应:
使用0.5x MMLV缓冲液


以上加热至55℃持续3分钟,接着添加以下:

在42℃下执行来自条形码衔接模板的条形码衔接子的T7 RNAP线性扩增、反转录和条形码添加至第一链cDNA中持续2小时。
使用Thermopol缓冲液:


以上加热至55℃持续3分钟,接着添加以下:

在42℃下执行来自条形码衔接模板的条形码衔接子的T7 RNAP线性扩增、反转录和条形码添加至第一链cDNA中持续2小时。
使用TAE缓冲液:


以上加热至55℃持续3分钟,接着添加以下:

在42℃下执行来自条形码衔接模板的条形码衔接子的T7 RNAP线性扩增、反转录和条形码添加至第一链cDNA中持续2小时。
所述反应接着使用经过修改的传统苯酚/氯仿方法净化:
1. 200μL TE0.1(10mM Tris pH 8.0、0.1mM EDTA)添加至各反应混合物中
2. 200μL苯酚/氯仿/异戊醇(Sigma)添加至各反应混合物中并且在预旋转GelPhase Lock管(5Prime)中剧烈地振荡
3.Gel Phase Lock管在14,000g下离心持续3分钟并且顶部水性部分被吸移至Amicon 100kDa柱(Millipore)中并且在14,000g下旋转3分钟
4. 450μL TE(10mM Tris,pH 8.0,1mM EDTA)接着被吸移至所述Amicon柱中,并且在14,000g下旋转3分钟
5. 450μL 10mM Tris(pH8.0)接着被吸移至所述Amicon柱中并且在14,000g下旋转5分钟
6.所述Amicon柱倒置至新的收集管中并且在1000g下旋转2分钟以收集含有纯化的mRNA/第一链cDNA双链体的洗出液
接着执行两轮PCR(PCR1和PCR2):
表9.PCR1和PCR2引物序列

每个RT反应设置以下PCR1 Phusion(Thermo Scientific)反应混合物:



来自PCR1的反应物接着稀释50倍并且在3个独立PCR2反应中用作模板,一个反应用于κ轻链,一个反应用于λ轻链并且一个反应用于γ重链。
每个RT反应设置以下PCR2 Phusion(Thermo Scientific)反应混合物:



5μL产物在凝胶上运行(图21)。如可见,所述编条形码反应在多种含有多种不同离子的不同缓冲液中良好地工作,所述离子如钾离子、铵离子、氯离子、硫酸根离子和乙酸根离子。
F.实施例6:由条形码衔接模板扩增的RNA条形码衔接子比未扩增DNA条形码衔接子更好地工作。
这一实施例显示本发明方法可在多种具有不同盐浓度的不同缓冲液中使用。此外,使用由条形码衔接模板产生的扩增的RNA条形码衔接子比仅添加DNA条形码衔接子至所述反应中更好地工作(即,产生所需的扩增的反应产物),据推测因为使用RNA条形码衔接子的反应导致较低背景(参看图4)。如上文实施例4中所述制造条形码衔接模板。
表10.额外寡核苷酸序列

设置以下反应并且缓冲液组成如表8中:
使用1x MMLV缓冲液


以上加热至55℃持续3分钟,接着添加以下:

在42℃下执行来自条形码衔接模板的条形码衔接子的T7 RNAP线性扩增、反转录和条形码添加至第一链cDNA中持续2小时。
使用0.5x MMLV缓冲液


以上加热至55℃持续3分钟,接着添加以下:

在42℃下执行来自条形码衔接模板的条形码衔接子的T7 RNAP线性扩增、反转录和条形码添加至第一链cDNA中持续2小时。
使用DNA条形码衔接子

以上加热至55℃持续3分钟,接着添加以下:
Ribolock(Thermo Scientific) 0.4μL
T4gp32(NEB) 1μL
Maxima H-RTase(Thermo Scientific) 1μL
在42℃下执行反转录和条形码添加至第一链cDNA中持续2小时。
所述反应接着使用经过修改的传统苯酚/氯仿方法净化:
1.将200μL TE0.1(10mM Tris pH 8.0、0.1mM EDTA)添加至各反应混合物中
2.将200μL苯酚/氯仿/异戊醇(Sigma)添加至各反应混合物中并且在预旋转GelPhase Lock管(5Prime)中剧烈地振荡
3.Gel Phase Lock管在14,000g下离心持续3分钟并且顶部水性部分被吸移至Amicon 100kDa柱(Millipore)中并且在14,000g下旋转3分钟
4. 450μL TE(10mM Tris,pH 8.0,1mM EDTA)接着被吸移至所述Amicon柱中,并且在14,000g下旋转3分钟
5. 450μL 10mM Tris(pH8.0)接着被吸移至所述Amicon柱中并且在14,000g下旋转5分钟
6.所述Amicon柱倒置至新的收集管中并且在1000g下旋转2分钟以收集含有纯化的mRNA/第一链cDNA双链体的洗出液
接着执行两轮PCR(PCR1和PCR2):
使用条形码衔接模板的每个RT反应设置以下PCR1 Phusion(Thermo Scientific)反应混合物:


使用DNA条形码衔接子的每个RT反应设置以下PCR1 Phusion(ThermoScientific)反应混合物:



来自PCR1的反应物接着稀释50倍并且在3个独立PCR2反应中用作模板,一个反应用于κ轻链,一个反应用于λ轻链,并且一个反应用于γ重链。
每个PCR1反应设置以下PCR2 Phusion(Thermo Scientific)反应混合物:



5μL产物在凝胶上运行(图22)。如可见,所述编条形码反应在具有不同盐浓度的缓冲液中良好地工作。虽然所述反应由于所述T7RNAP的盐敏感性在低盐缓冲液(0.5x MMLV)中更好地工作,但其也在较高盐缓冲液(1x MMLV)中工作。注意由于当使用DNA条形码衔接子时在RT步骤期间的非特异性引发(参考图4),存在异常高的背景并且所需条带模糊。
G.实施例7:在使用微流体液滴器件制造的液滴中使用水性条形码衔接模板对来自细胞的核酸编条形码。
使用用于产生单分散乳液的器件来封装单细胞连同编条形码珠粒和编条形码分析所必需的其它试剂。三个Dolomite P-泵配备有流量传感器(Dolomite 3200016、3200095和3200098)。第一个P-泵经由微流体管直接连接于2-Reagent液滴芯片(Dolomite3200287),所述微流体管并入T形接合点以将线路分成两个输入。这是油输入线路。另两个P-泵经由流体管连接于PEEK样品环管,所述环管缠绕冰箱,所述冰箱用于在所述器件操作时保持样品冷冻,并且这些环管各自连接于所述2-Reagent液滴芯片。各样品环管在其前端并入四通阀,使得样品可能借助于注射器装载于所述环管中。所述第一样品环管填充有细胞,而所述第二环管填充有RT/编条形码/溶解混合物。所述器件配置的实施例如图17-19中所示。所述冰箱在使用前填充有冰。
鼠科动物B220+B细胞群体进行FACS分选并且使用具有10mM NaCl和0.5mg/ml BSA的300mM甜菜碱作为悬浮缓冲液来制备细胞悬浮液。细胞以4,500个细胞/μL的浓度使用。
如下制备RT/水性条形码混合物:


使用注射器将所述细胞悬浮液装载于一个样品环管中并且将所述RT/编条形码/溶解混合物装载于另一样品环管中。以使得单一液滴中多种细胞或条形码的出现减至最少同时使那些浓度保持足够高的方式选择细胞和条形码浓度,使得足够多的细胞与条形码一起封装。转换所述四通阀,使得所述样品环管与所述泵在同一条线上,并且所有三个泵均启动。所述两个水性输入以一定速率流动,使得其以1:2(细胞悬浮液:RT/编条形码/溶解混合物)比率混合。所述水性和油输入以一定速率流动,使得形成约50μm直径的液滴,并且在足够高的流动速率下流动,使得细胞流动通过所述器件。在Sorenson Bioscience0.2mL PCR管中收集乳液。在所述样品已经产生后,其首先进行预加热步骤(在55℃下3分钟)并且接着在42℃下孵育2小时以使所述反应继续进行。在所述反应后,使用下文所述的“破乳非珠粒乳液”方法破乳所述乳液。这制造了纯化的cDNA样品用于后续PCR扩增和测序。
如下破乳非珠粒乳液:
1. 200μL TE、400μL苯酚/氯仿/异戊醇、800μL氯仿吸移至预旋转Gel Phase Lock管中
2.各样品吸移至对应的Gel Phase Lock管中
3.管在14,000g下短暂离心持续3分钟
4.水层吸移至100kDa Amicon管(Millipore)中。
5.管在14,000g下短暂离心持续3分钟
6. 450μL TE吸移至所述Amicon管中
7.管在14,000g下短暂离心持续3分钟
8. 450μL 10mM Tris添加至所述Amicon管中
9.管在14,000g下短暂离心持续5分钟
10.Amicon管倒置放入新鲜收集管中
11.管在1,000g下短暂离心持续2分钟
接着执行两轮PCR(PCR1和PCR2),除了表9中列出的一些引物序列外还使用以下引物。
表11.用于鼠科动物免疫球蛋白基因的PCR的额外引物


使用条形码衔接模板的每个RT反应设置以下PCR1 Phusion(Thermo Scientific)反应混合物:



来自PCR1的反应物接着稀释50倍并且在3个独立PCR2反应中用作模板,一个反应用于κ和λ轻链,一个反应用于μ重链,并且一个反应用于γ重链。
每个PCR1反应设置以下PCR2 Phusion(Thermo Scientific)反应混合物:



5μL PCR产物在凝胶上运行(图23)。对应于κ和λ轻链以及对应于μ重链的条带清楚可见。仅所述μ重链扩增,因为预期大多数B220+B细胞为IgM+原生B细胞。
因此扩增的免疫球蛋白重链和轻链可纯化并且准备用于下一代测序,如但不限于454测序。由于这一实施例使用每个反应容器>1个拷贝的浓度的条形码衔接模板,将独特条形码集合而非独特条形码并入各反应容器中的核酸中。配对的免疫球蛋白重链和轻链可通过使其共享独特条形码集合而非通过独特条形码彼此缔合。
条形码衔接模板也可在一定浓度下使用,使得通过有限稀释,大多数含有条形码衔接模板的反应容器将以每个反应容器1个拷贝含有所述条形码衔接模板。在这种情况下,配对的免疫球蛋白重链和轻链可通过使其共享独特条形码序列彼此缔合。
H.实施例8:在使用微流体液滴器件制造的液滴中使用条形码衔接模板珠粒对来自细胞的核酸编条形码。
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。使用如实施例7中所述的产生液滴的微流体器件,其中唯一差异在于所述第一样品环管含有细胞和如实施例1、2或3中制得的条形码衔接模板珠粒。
鼠科动物B220+B细胞群体进行FACS分选并且使用具有10mM NaCl和0.5mg/ml BSA的300mM甜菜碱作为悬浮缓冲液来制备细胞和条形码衔接模板珠粒悬浮液。细胞以4,500个细胞/μL的浓度包括在内并且珠粒以60,000个珠粒/μL的浓度使用。
如下制备RT混合物:

使用注射器将所述细胞和编条形码珠粒悬浮液装载于一个样品环管中并且将所述RT/编条形码/溶解混合物装载于另一样品环管中。转换所述四通阀,使得所述样品环管与所述泵在同一条线上,并且所有三个泵均启动。所述两个水性输入以一定速率流动,使得其以1:2(细胞和珠粒悬浮液:RT/编条形码/溶解混合物)比率混合。所述水性和油输入以一定速率流动,使得形成约50um直径的液滴,并且在足够高的流动速率下流动,使得细胞和珠粒流动通过所述器件。在Sorenson Bioscience 0.2mL PCR管中收集乳液。在所述样品已经产生后,其首先进行加热步骤(在55℃下3分钟)并且接着在42℃下孵育2小时以使所述RT/编条形码反应继续进行。在所述编条形码反应后,使用实施例7中所述的“破乳非珠粒乳液”方法破乳所述乳液。如实施例7中执行后续PCR反应。
因此扩增的免疫球蛋白重链和轻链纯化并且准备用于下一代测序,如但不限于454测序。由于这一实施例以每个反应容器约1种珠粒使用条形码衔接模板珠粒,配对的免疫球蛋白重链和轻链通过其共同使用独特条形码序列来配对。
I.实施例9:使用用DNA聚合酶由条形码衔接模板珠粒扩增的条形码衔接子对来自细胞的核酸编条形码。
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。使用如实施例7中所述的产生液滴的微流体器件,其中唯一差异在于所述第一样品环管含有细胞和如实施例1、2或3中制得的条形码衔接模板珠粒。在这一实施例中,所述条形码衔接模板珠粒包含5’Nt.BbvCI切口核酸内切酶序列而非T7 RNAP启动子序列以允许通过DNA聚合酶扩增条形码衔接子。
鼠科动物B220+B细胞群体进行FACS分选并且使用具有10mM NaCl和0.5mg/ml BSA的300mM甜菜碱作为悬浮缓冲液来制备细胞和条形码衔接模板珠粒悬浮液。细胞以4,500个细胞/uL的浓度包括在内并且珠粒以60,000个珠粒/μL的浓度使用。
如下制备RT混合物:

使用注射器将所述细胞和编条形码珠粒悬浮液装载于一个样品环管中并且将所述RT/编条形码/溶解混合物装载于另一样品环管中。转换所述四通阀,使得所述样品环管与所述泵在同一条线上,并且所有三个泵均启动。所述两个水性输入以一定速率流动,使得其以1:2(细胞和珠粒悬浮液:RT/编条形码/溶解混合物)比率混合。所述水性和油输入以一定速率流动,使得形成约50um直径的液滴,并且在足够高的流动速率下流动,使得细胞和珠粒流动通过所述器件。在Sorenson Bioscience 0.2mL PCR管中收集乳液。在所述样品已经产生后,其首先进行加热步骤(在55℃下3分钟)并且接着在42℃下孵育2小时以使所述RT/编条形码反应继续进行。在所述编条形码反应后,使用实施例7中所述的破乳非珠粒乳液摂方法破乳所述乳液。如实施例7中执行后续PCR反应。
因此扩增的免疫球蛋白重链和轻链纯化并且准备用于下一代测序,如但不限于454测序。由于这一实施例以每个反应容器约1种珠粒使用条形码衔接模板珠粒,配对的免疫球蛋白重链和轻链通过其共同使用独特条形码序列来配对。
J.实施例10:在多孔反应容器中使用条形码衔接模板对来自细胞的核酸编条形码。
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。合成具有如图1中的组成的条形码衔接模板,作为来自如IDT的厂商的双链体寡核苷酸。各独特条形码衔接模板保持于不同存储容器中,使得条形码序列无混合或交叉污染。活化B细胞(成浆细胞)为使用FACS Aria II(Becton Dickenson)分选至10μL溶解缓冲液中进入96孔板的所有孔中的单细胞。各孔中所述缓冲液的组成为:


所述板接着在55℃下孵育3分钟,接着在42℃下孵育2小时用于使所述RT/编条形码反应发生。96孔板的所有孔中的反应接着汇集在一起并且使用经过修改的传统苯酚/氯仿方法执行净化:
1. 400μL苯酚/氯仿/异戊醇(Sigma)添加至预旋转Gel Phase Lock管(5Prime)中并且剧烈地振荡
2.Gel Phase Lock管在14,000g下离心持续3分钟并且顶部水性部分被吸移至Amicon 100kDa柱(Millipore)中并且在14,000g下旋转3分钟
3.必要时重复步骤2以使整个水性容积旋转通过所述Amicon柱
4. 450μL TE(10mM Tris,pH 8.0,1mM EDTA)接着被吸移至所述Amicon柱中,并且在14,000g下旋转3分钟
5. 450μL 10mM Tris(pH8.0)接着被吸移至所述Amicon柱中并且在14,000g下旋转5分钟
6.所述Amicon柱倒置至新的收集管中并且在1000g下旋转2分钟以收集含有纯化的mRNA/第一链cDNA双链体的洗出液
设置以下PCR1 Phusion(Thermo Scientific)反应混合物:



来自PCR1的反应物接着稀释50倍并且在3个独立PCR2反应中用作模板,一个反应用于κ轻链,一个反应用于λ轻链并且一个反应用于γ重链。
设置以下PCR2 Phusion(Thermo Scientific)反应混合物:


因此扩增的免疫球蛋白重链和轻链纯化并且准备用于下一代测序,如但不限于454测序。由于这一实施例使用单独地吸移至各反应容器(在这种情况下96孔板的孔)中的独特条形码衔接模板,配对的免疫球蛋白重链和轻链通过其共同使用独特条形码序列在生物信息学上配对。
K.实施例11:在使用微流体液滴器件制造的液滴中使用条形码衔接模板珠粒对来自细胞的核酸编条形码。
使用下文所述的方法以使用乳液PCR产生条形码衔接模板珠粒文库,其中执行聚合酶链反应(PCR)来连接独特条形码衔接模板至各珠粒(参看图15)。
表12:用于在单一反应中制造条形码衔接模板珠粒文库的寡核苷酸

抗生蛋白链菌素涂布的M-270 (Life Technologies)与生物素标记的寡核苷酸(“emB_T7bridgeIsceI”)偶联:
(Life Technologies)与生物素标记的寡核苷酸(“emB_T7bridgeIsceI”)偶联:
1.珠粒通过轻微涡旋再悬浮
2. 1mL M270珠粒(大约2×109个珠粒)放入三个1.5mL微量离心管中的每一者中,总计3mL
3.放在磁铁上持续3分钟。
4.从各管取出上清液并且再悬浮于1mL(1x体积)结合/洗涤缓冲液(BWB;1M NaCl、5mM Tris、0.5mM EDTA)中
5.再重复步骤4两次,随后最终再悬浮于540μL体积BWB中
6. 60μL 100μM emB_T7bridge2添加至珠粒中并且在轻微旋转下孵育15分钟
7.在孵育后,珠粒用1mL BWB缓冲液洗涤3次,并且组合至单一管中
8.珠粒在4℃下与0.01%叠氮化钠一起存储
9.珠粒在使用前用10mM Tris洗涤3次
条形码寡核苷酸和正向和反向引物在基于乳液的PCR中添加至来自以上的偶联珠粒中:
1.以下PCR混合物(3mL总体积)在三个1.5mL微量离心管(VWR目录号20170-650)中制备:


2.制备油-表面活性剂混合物(20mL总体积):
a.矿物油(Sigma) 18.4mL
b.EM90(Evonik) 1.6mL
3. 800μL油-表面活性剂混合物和200μL PCR混合物组合至12个Axygen 2.0mLMaxymum Recovery锥形底微量离心管(MCT-200-L-C)中的每一者中。管密封并且振荡持续3秒
4.管放入Qiagen TissueLyzer II中,并且在14Hz下振荡持续5分钟
5.所述乳液在VWR 96孔PCR板(83007-374)的孔中分配,其中每孔添加160μL乳液
6.管使用以下程序进行热循环:


乳液被破乳并且回收珠粒:
1.将所述PCR板的内含物转移至1.5mL微量离心管(VWR20170-650)中,每个管具有不超过0.5mL乳液体积
2. 100uL 1uM emB_T7bridgefreeIsceI_2引物添加至各管中
3.管用异丁醇终止,密封并且振荡以充分混合
4.管在14,000rpm下离心持续1min
5.管放在磁条上以将所述珠粒吸引至所述管的侧面,接着尽可能抽吸尽可能多的上清液,同时留下沉淀的珠粒
6.添加1mL异丁醇,通过上下吸移充分混合,直至剩余油/乳液体积已经分散至所述异丁醇中
7.管放在磁条上以将所述珠粒吸引至所述管的侧面,接着抽吸所述异丁醇。来自所有管的珠粒通过首先从其中将组合所述珠粒的单一管抽吸上清液并且接着从另一管转移全部体积,酌留在所述磁铁处收集所述珠粒的时间,接着抽吸上清液并且重复而组合至所述单一管中
8.添加1mL新鲜异丁醇,充分混合并且搁置60秒
9.抽吸异丁醇
10.添加1mL 100%乙醇,充分混合并且搁置60秒
11.抽吸乙醇
12.重复步骤10和11
13.添加1mL 70%乙醇,充分混合并且搁置60秒
14.抽吸乙醇
15.重复步骤13和14
16.添加1mL PBS,充分混合并且搁置60秒。
17.抽吸PBS
18.重复步骤16和17。
并入条形码衔接模板的珠粒接着使用Becton Dickenson FACS Aria III,利用来自并入emB_IsceI_RV反向引物中的Alexa Fluor 647染料的荧光与未编条形码珠粒分选。
珠粒在4℃下存储于0.01%叠氮化钠中以便存储。使用图17-19中所示并且实施例7中所述的微流体器件封装单细胞连同编条形码珠粒和编条形码分析所必需的其它试剂。CD19+IgG+记忆B细胞群体进行FACS分选并且在完全IMDM培养基(IMDM+10%FBS+100U/mLIL-2、50ng/mL IL-21、50ng/mL CD40L、5μg/mL抗CD40L mAb和1x Normocin)中培养6天,接着使用具有10mM NaCl和0.5mg/ml BSA的300mM甜菜碱作为悬浮缓冲液来制备细胞悬浮液。细胞以2,500个细胞/μL的浓度使用并且编条形码珠粒以100,000个珠粒/μL的浓度使用。
如下制备RT/水性条形码混合物:


使用注射器将所述细胞和珠粒悬浮液装载于一个样品环管中并且将所述RT/编条形码/溶解混合物装载于另一样品环管中。以使得单一液滴中多种细胞或条形码的出现减至最少同时使那些浓度保持足够高的方式选择细胞和珠粒浓度,使得足够多的细胞与珠粒一起封装,谨记细胞和珠粒不会以与所述悬浮流体相同的速率迁移通过所述管,从而有效地引起稀释。转换所述四通阀,使得所述样品环管与所述泵在同一条线上,并且所有三个泵均启动。所述两个水性输入以一定速率流动,使得其以1:2(细胞悬浮液:RT/编条形码/溶解混合物)比率混合。所述水性和油输入以一定速率流动,使得形成约150μm直径的液滴,尤其1μL/min(细胞/珠粒悬浮液线路)、2μL/min(RT混合物线路)、3μL/min(油线路)。在SorensonBioscience 0.2mL PCR管中收集乳液。在所述样品已经产生后,其首先进行预加热步骤(在50℃下3分钟)并且接着在42℃下孵育2小时以使所述反应继续进行。在所述反应后,使用下文所述的方案破乳所述乳液。这制造了纯化的cDNA样品用于后续PCR扩增和测序。
使用以下程序来破乳所述乳液并且回收产物:
1.Phase lock管(5Prime)短暂离心以将凝胶推动至底部。
2.样品和200μL TE缓冲液、400μL苯酚氯仿混合物、800μL氯仿添加至各锁相管中。
3.管在14,000g下短暂离心持续3分钟
4.水层转移至第二预旋转锁相管中并且添加相等体积的苯酚氯仿
5.管在14,000g下短暂离心持续3分钟
6. 450μL TE添加至所述Amicon过滤器中
7.重复步骤5和6
8. 450μL 10mM Tris添加至所述Amicon过滤器中
9.重复步骤5
10.各过滤装置接着倒置放入新的收集管中
11.管在1000g下旋转2分钟,并且净化的样品旋转至所述收集管中
接着执行两轮PCR(PCR1和PCR2),除了表13中列出的一些引物序列外还使用以下引物。
表13:

使用条形码衔接模板的每个RT反应设置以下PCR1Q5(NEB)反应混合物:



来自PCR1的反应物接着在10mM Tris-HCl(pH 8.0)中稀释25倍并且在两个独立PCR2反应中用作模板,一个反应用于κ和λ轻链并且一个反应用于γ重链。
每个PCR1反应设置以下PCR2 Q5(NEB)反应混合物:



10μL PCR产物在凝胶上运行(图24)。对应于κ和λ轻链以及对应于γ重链的条带清楚可见。
独立地对重链和轻链扩增子执行两种4循环PCR反应以添加454LibA测序衔接子。在LibPCR1中,“A”衔接子添加至所述扩增子的5’端,并且“B”衔接子添加至3’端;并且在LibPCR2中反之亦然。LibPCR细节如下,其中Lib1-FR引物混合物用于LibPCR1中并且Lib2-FR混合物用于LibPCR2中,并且所述引物列于表14中。


表14:


接着使用Ampure(Beckman Coulter)珠粒净化根据制造商的说明书使用0.68:1的珠粒:DNA比率并且使用凝胶纯化使用Flashgel Recovery gel(Lonza)根据制造商的说明书纯化扩增子。
接着使用Kapa qPCR文库定量(KAPA)根据制造商的说明书定量扩增子,并且接着将适量的重链和轻链扩增子文库用于454乳液PCR,并且破乳所述乳液并且将克隆扩增的454珠粒装载至454测序仪上用于根据制造商的说明书进行测序。由于A和B衔接子添加至所述扩增子的5’和3’端,我们能够由两种方向测序并且获得正向和反向读数。
由标准454运行产生序列,并且分析所得序列,不过也可能已经使用了其它下一代测序平台。
通过写入计算机程序来分析序列。所述计算机程序对来自454皮升滴定板的区域的序列读数执行以下步骤。区域1序列源于如上文所述产生的重链文库。区域2序列源于如上文所述产生的轻链文库。关于各读数,用计算机计算两个全局-局部比对以确定随后与来自表15的序列T2’和T1匹配的链。所述全局-局部比对对匹配计分为0、错误匹配为-1并且使用间隙开放罚分和间隙延伸罚分-1。分数需要大于-4或弃去读数。关于重链区,841×103个读数中的611×103个读数满足比对分数限制。关于轻链区,856×103个读数中的617×103个读数满足比对分数限制。基于所述全局-局部比对,从所述读数推断出所述DNA条形码的序列。关于满足所述比对分数限制的重链区读数,397×103个读数具有与预期模式一致的条形码序列并且被指定具有所观察的条形码。关于满足所述比对分数限制的轻链区读数,437×103个读数具有与预期模式一致的条形码序列并且被指定具有所观察的条形码。
具有同一DNA条形码序列的读数被分在一组用于组装。具有同一条形码的读数的分组使用newbler(454组装器)组装。关于与区域2序列具有同一条形码序列的区域1序列的组装共有序列被分组至重链和轻链对集合中。
所述重链和轻链对集合含有源于存在于乳液RT泡沫中的一个或多个B细胞的重链和轻链序列。
在所述重链和轻链读数对集合中,2,551对具有至少10个来自所述重链区的读数和至少10个来自所述轻链区的读数。在2,551个所述对中,1,820对已经组装至精确地一个重链和精确地一个轻链。发现那些对中的61对具有在所产生的序列的整个数据集中独特的重链和轻链。
由具有共有条形码“GCCGACCACGGCACAAGCGCCGAAAAT”(SEQ ID NO:124)的编条形码重链和轻链读数产生的配对的重链和轻链序列的实例为“MEFGLSWLFLVAILKGVQCGVQLLESGGGLVQPGGSLRLSCAGSQFTFSTYAMNWVRQAPGKGLEWVSGISGDGYRIQYADSVEGRFSISRDNSNNMVYLQMTSLRAEDTAVYFCAKDLFPRTIGYFDYWGQGTRVTVSS”(SEQ ID NO:125)(重链氨基酸序列)和“MEAPAQLLFLLLLWLPDTTGKIVMTQSPATLSVSPGERATLSCRASQSISINLAWYQHKPGQAPRLLIYGASTRATAIPARFSGSVSGTEFTLTISSLQSEDFAVYYCQQYDDWPRTFGQGTKVEI”(SEQ ID NO:126)(轻链氨基酸序列)。
所述分析证明缔合来自B细胞的重链序列与来自B细胞的对应的轻链序列的能力。
表15:用于鉴别来自B细胞的读数中的DNA条形码的序列


L.实施例12:在使用微流体液滴器件制造的液滴中使用条形码衔接模板珠粒对来自细胞的核酸编条形码。
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。如实施例11中制备条形码衔接模板珠粒文库,除了emB_BCbridgeISceI_2用emB_BCbridgeISceI_N置换并且emB_IsceI_RV用emB_ISceI_RV_n置换。emB_ISceI_RV_n含有独特分子识别符(UMI),使得当制备时,所述条形码衔接模板珠粒文库将包含各自具有独特样品条形码和随机H(A、C、T核苷酸)八聚体UMI的珠粒以对具有不同UMI的个别mRNA分子编条形码。
表16:用于在单一反应中制造具有样品条形码和UMI的条形码衔接模板珠粒文库的额外寡核苷酸

细胞连同珠粒被封装于液滴中用于如实施例11所述的编条形码反应,其中唯一差异在于使用PBMC而非活化记忆B细胞,并且所用的寡聚(dT)为寡聚dT_n,其中所述序列为CAC GAC CGG TGC TCG ATT TAG TTT TTT TTT TTT TTT TTT TTT TTT T(SEQ ID NO:50)。所述乳液接着如实施例11所述被破乳。
接着执行一轮PCR,使用以下引物:
表17:


每个反应使用以下PCR Q5(NEB)反应混合物,并且设置多种反应,并且各反应循环持续从15-26个循环的不同数目的循环以发现最佳循环数来使用:



5μL PCR产物在凝胶上运行并且在后续下游步骤中使用产生良好产物量但未过度循环的循环数。
接着根据Illumina的配对末端测序试剂盒制备产物并且在Illumina高通量测序仪上对正向末端测序,不过也可能已经使用其它测序平台。产生并且分析序列。接着使用样品条形码对个别细胞指定读数,并且接着使用UMI以使用所属领域中充分确立的方法执行单细胞RNA测序分析(Nat Methods.2014年2月;11(2):163-6.doi:10.1038/nmeth.2772.2013年12月22日电子出版)。
M.实施例13:使用组合产生的条形码的条形码衔接模板合成
在这一实施例中合成条形码衔接模板珠粒文库。
由两种寡核苷酸BC_部分1_有义和BC_部分2_类型(1、2或3)_反义组合产生含条形码寡核苷酸(如在图15中)。各BC_部分1_有义和BC_部分2_类型(1、2或3)_反义寡核苷酸分别含有独特序列“条形码部分1”和“条形码部分2”。这些组合的序列产生独特条形码序列。根据Generalized DNA barcode design based on Hamming codes,Bystrykh 2012 PLoSOne.2012 7:e36852的方法,“条形码部分1”和“条形码部分2”分别为(16,11)和(12,7)汉明码。因此,因此设计的条形码为误差校正的。
BC_部分2寡核苷酸还分为三种类型BC_部分2_类型1_反义、BC_部分2_类型2_反义和BC_部分2_类型3_反义。这允许用3种不同的非错误引发反向引物(Rv_类型1、Rv_类型2和Rv_类型3)扩增产生条形码衔接模板。当那些反向引物各自共价偶联于不同荧光团时,可经由在不同颜色中的荧光鉴别产生的条形码衔接模板珠粒。另外,具有超过一种类型的条形码类型的条形码衔接模板珠粒将在超过一种颜色中发荧光。由于这一实施例中的条形码衔接模板珠粒是在emPCR中制得,利用有限稀释将具有一种含条形码寡核苷酸的珠粒与所需引物一起放入液滴中。泊松统计学指示小百分率的液滴将含有超过一种含条形码寡核苷酸,有效产生非单码条形码衔接模板珠粒。通过具有不同类型的在不同颜色中发荧光的条形码衔接模板珠粒,随后对单色珠粒的FACS分选将极大地增加经由条形码衔接模板珠粒的emPCR产生获得的单码珠粒的百分率。
表18.组合产生的条形码-序列


[条形码部分1]=SEQ ID NO:127-45126;[条形码部分2]=SEQ ID NO:45127-47561。
含条形码寡核苷酸是使用表19中的条件和以下热循环条件PCR产生的:94℃持续2min,随后53℃持续2小时,94℃持续15s、53℃持续30s和68℃持续20s的7个循环,接着其后为68℃持续1min和10℃保持。所述反应使用Zymo DNA净化和浓缩试剂盒净化并且用Qubit(Life Technologies)定量浓度。
表19.用于制造含条形码寡核苷酸的反应混合物


含条形码寡核苷酸的82bp大小在凝胶上确认(图25,左上部)。
9. 8μm SuperAvidin微球珠粒(Bang’s Lab)与生物素标记的SAV-珠粒-连接子寡核苷酸偶联。1500万个珠粒与60μL 10μM寡核苷酸一起孵育1小时,并且接着用BWB缓冲液(1M NaCl于TE中)洗涤3次,随后在10mM Tris中洗涤3次以产生偶联的SAV_珠粒_连接子珠粒。
产生条形码衔接模板珠粒的emPCR如下继续进行:
表20.用于制造条形码衔接模板珠粒的反应混合物

通过振荡乳液油与表20中的反应混合物产生乳液。所述乳液油制剂为10mL AR20硅油(Sigma)、7.5mL 7225C Formulation Aid(Dow Corning)、7.5mL 0749 Resin(DowCorning)和0.1%Triton X-100(Sigma)。12mL乳液油与4mL模拟混合物(无表20中的反应混合物的寡核苷酸、引物和酶)一起在30Hz下在TissueLyser(Qiagen)中振荡持续5min,并且接着在添加4mL反应混合物后在12Hz下振荡持续5min。这产生直径在30-80um之间的较大液滴的大多数。热循环条件为:

通过用破乳混合物1洗涤,随后用破乳混合物2洗涤,随后70%乙醇洗涤和TE洗涤来破乳乳液。珠粒再悬浮于具有0.001%Tween 20的TE中。



珠粒在BD FACS Jazz上运行并且分选明亮、单色珠粒(图25,右部)。进行如实施例14中执行的编条形码反应以验证所述珠粒适用作用于对RNA编条形码的条形码衔接子,除了所述反应在开放PCR中用PCR管中的多种珠粒并且用纯化的PBMC RNA进行。如图25左下部中,获得条带,其显示珠粒确实适用作用于对RNA编条形码的条形码衔接模板。
N.实施例14:使用不同体积的液滴中的条形码衔接模板珠粒对来自T细胞的核酸编条形码
冷冻保存的PBMC解冻并且在AIM V培养基(Life Technology)中以3百万个细胞/mL的密度孵育过夜。T细胞接着使用CD3微珠粒(Miltenyi Biotec)根据制造商的说明书用磁激活细胞分选(MACS)分离。简单地说,T细胞在300g下离心持续10分钟,并且在4℃下在含有20%CD3微珠粒的MACS缓冲液(2%胎牛血清和2mM EDTA于1X PBS中)中悬浮持续15分钟。磁性标记的T细胞接着使用磁性分离柱分离,随后用1X伊屋诺霉素和1X佛波醇12-肉豆蔻酸13-乙酸(PMA)共刺激持续3小时。在去除所述含有两种刺激物的培养基后,细胞与作为抗凝集剂的1X DNA酶(Sigma)一起孵育15分钟。
使细胞离心以去除含有DNA酶的上清液,并且用含有5%1M NaCl、1.5%500mMEDTA、33.8%4M甜菜碱和7.5%20mg/ml牛血清白蛋白(BSA)的细胞悬浮缓冲液(CSB)洗涤3次。在再悬浮于1mL CSB中之后,细胞还用40μm细胞过滤器(BD Falcon)过滤以去除细胞团块。细胞悬浮液接着如实施例8中在液滴发生器器件中运行以将细胞和条形码衔接模板珠粒封装至液滴中,其中所述珠粒如实施例13中产生。在这一实施例中,细胞和珠粒封装至不同大小的液滴中:1.4、3.1和5.6nL。
含有细胞和条形码的液滴通过在以下最终反应缓冲液组合物中在50℃下孵育3分钟,随后在42℃下孵育3小时而进行反转录:
RT反应混合物

所述乳液接着用苯酚/氯仿混合物破乳并且如实施例8中在Amicon 100kDa柱(Millipore)中浓缩。cDNA经受PCR1的18个循环,随后使用下文列出的每个RT反应的反应混合物和表21中列出的热循环条件进行PCR2。所用引物在表22中。


表21.PCR1和PCR2的热循环条件

表22.PCR1和PCR2的引物序列


[i5指数引物]=SEQ ID NO:47562-47605。
如由图26可见,在所测试的3种液滴容积处,所述反应成功地完成。
O.实施例15:扩增来自编条形码核酸的TCRα和β基因并且测序
编条形码T细胞cDNA如实施例14中所述产生。简单地说,PBMC用1X伊屋诺霉素和PMA在AIM V培养基中共刺激持续3小时。表达CD3、CD4或CD8的T细胞经过磁性标记并且单独地使用MACS试剂盒(Miltenyi Biotec)分离并且穿过液体器件以封装细胞与如实施例13中产生的条形码衔接模板珠粒。含有细胞和条形码的乳液在50℃下反转录持续3分钟并且在42℃下反转录持续3小时。所述乳液接着用苯酚/氯仿混合物破乳并且使用Amicon 100kDa柱(Millipore)浓缩。
反转录和PCR1和PCR2如实施例14中执行,其中不同的指数_sID引物用于各样品,各引物具有独特指数ID条形码。这允许在相同下一代测序运行中进行样品的汇集和多路复用,其中不同样品经由所述指数ID条形码彼此区别。
PCR2产物接着用AMPure磁性珠粒(Roche)根据制造商说明书以1μl PCR 2产物:1.8μl磁性珠粒的比率浓缩。样品接着准备使用额外文库PCR用于Illumina测序以添加用于Illumina测序的衔接子。所用引物列于表23中。

用于文库PCR的热循环条件


表23.用于文库PCR扩增的引物序列

扩增产物用Pippin Prep和DNA纯化和浓缩试剂盒(Zymo Research)清洁以去除小片段并且用琼脂糖凝胶电泳(图27)分析,并且使用Illumina测序来测序。
分析来自Illumina测序的配对末端读数以确定T细胞受体(TCR)生殖系TCR CDR3并且推断全长序列。测序产生21,207,225个已过滤的配对末端读数。所述DNA条形码用于基于正向读数序列向个别T细胞内的TCR的转录物指定配对读数。所述正向读数内所述DNA条形码的鉴别使用python脚本进行。关于各正向读数,与固定序列1的编辑距离使用全局/局部比对计算。需要2或更小的编辑距离或弃去所述读数对。根据固定序列1的位置和条形码部分1(BC1)和条形码部分2(BC2)的已知长度,从所述正向读数推断出候选BC1和BC2序列。检查BC1和BC2以验证其分别满足用于汉明(16,11)或汉明(12,7)DNA条形码的汉明条件(关于指定序列的序列和彼此的相对位置参看表18)。基于BC1和BC2,向特异性T细胞指定配对读数。结果,向T细胞指定3,712,013个读数对。
向T细胞指定的配对读数接着使用程序blastn以10-5的e值截止与V、J和恒定生殖系TCR序列的已知变体进行比较。如果所述对的任一读数由blast计分为对生殖系的一次碰撞,那么所述生殖系和相关等位基因的计数关于(由BC1、BC2鉴别的细胞的)对应的TCRα或β链的增量为一。除了关于各生殖系等位基因组合和特异性细胞,还计分对其造成一次碰撞的序列的清单。
关于由BC1和BC2的独特组合鉴别的各细胞,接着基于上文指示的计数的大多数指定关于α和β链的v、j和/或恒定生殖系等位基因组成,并且关于各生殖系,选择具有与其缔合的最长HSP的序列作为所述转录物中关于所述生殖系的代表性部分。
接着,使用以下步骤确定CDR3区的组成。关于各j生殖系,可能时确定满足模式FG*G的4种氨基酸(AA)的序列的位置,并且鉴别在其序列的最后10个AA中具有CA的组合的v生殖系的清单。关于各细胞,在关于j翻译的代表性序列的所有三个框中探寻j生殖系的4种AA的模式和所述CA组合。所述CDR3被确定为在CA与所述4种AA的模式之间的AA序列。所述TCR的推定AA序列通过组合v生殖系的AA序列直至CA,随后组合所述CDR3序列,随后组合由所述4种AA的模式开始的j生殖系的AA序列来获得。使用类似方法,确定所述CDR3的核苷酸序列和所述TCR的推定全长核苷酸序列。
通过基于D生殖系与所述CDR3的核苷酸序列之间的全局-局部比对评估编辑距离来分析所述D生殖系和D等位基因。向所述TCR指定D生殖系/等位基因,限制条件是与最接近生殖系序列的编辑距离小于或等于2。
表24显示关于经过处理的样品的概述统计学,包括编条形码细胞、具有指定的TCRα或β链的细胞、具有指定的TCRα和β的细胞的估计数目,和推断的全长α或β链的数目。
表24.TCRα和β链


P.实施例16:扩增来自编条形码核酸的细胞亚型特异性基因并且测序
编条形码T细胞cDNA如实施例15中所述产生。简单地说,PBMC用1X伊屋诺霉素和PMA在AIM V培养基中共刺激持续3小时。表达CD3、CD4或CD8的T细胞经过磁性标记并且单独地使用MACS试剂盒(Miltenyi Biotec)分离并且穿过液体器件。含有细胞和条形码的乳液如实施例14中在50℃下反转录持续3分钟并且在42℃下反转录持续3小时。所述乳液接着用苯酚/氯仿混合物破乳并且使用Amicon 100kDa柱(Millipore)浓缩。接着使用表21中的热循环条件,连同如表25中列出的关于T细胞靶向子集基因(例如CD4、CD8和干扰素γ(IFNγ))的特异性引物执行在不同循环下的PCR1和PCR2。反应混合物制备如下:


表25.除了用于PCR 1和PCR 2中的序列以外用于PCR1和PCR2的T细胞靶向基因反向引物序列。


PCR2产物接着准备如实施例15中用于Illumina测序,并且所述产物在Illumina测序之前用琼脂糖凝胶电泳(图27)分析。
分析来自Illumina测序的配对末端读数以基于基因特异性标记物确定T细胞亚型。测序产生19,205,611个已过滤的配对末端读数。所述DNA条形码用于基于正向读数序列向个别T细胞内的转录物指定配对读数。所述正向读数内所述DNA条形码的鉴别使用python脚本进行。关于各正向读数,与固定序列1的编辑距离使用全局/局部比对计算。需要2或更小的编辑距离或弃去所述读数对。根据固定序列1的位置和条形码部分1(BC1)和条形码部分2(BC2)的已知长度,从所述正向读数推断出候选BC1和BC2序列。检查BC1和BC2以验证其分别满足用于汉明(16,11)或汉明(12,7)DNA条形码的汉明条件。关于满足所述汉明条件的正向读数,基于X、固定序列2和分子条形码的已知长度推断出候选分子条形码(关于指定序列的序列和彼此的相对位置参看表18)。如果所述分子条形码序列无“C”核苷酸,那么向T细胞(基于BC1和BC2)和所述T细胞内的特异性转录物(基于所述分子条形码)指定配对读数。向个别T细胞内的转录物指定3,902,569个读数对。
向T细胞转录物指定的配对读数接着使用程序blastn以10-6的e值截止并且设定百分率_同一性至98与标记物基因的已知剪切变体进行比较。如果所述对的任一读数由blast计分为一次碰撞,那么来自所述T细胞的对应的转录物(由BC1、BC2和所述分子条形码鉴别)与所述标记物基因建立联系。
关于由BC1和BC2的独特组合鉴别的各细胞,通过对由与给定标记物基因相关的读数对观察到的不同分子条形码的数目计数来确定可见来自所述给定标记物基因的转录物的不同次数。
基于所述标记物基因确定检测到的各类型的T细胞的数目。将其中确定至少一个CD4转录物和一个IFNγ转录物被指定的T细胞计数为Th1细胞。将其中确定至少一个CD4转录物被指定并且无IFNγ转录物被指定的T细胞计数为非Th1CD4样品。将其中确定至少一个CD8转录物和一个IFNγ转录物被鉴别的T细胞计数为IFNγ+细胞毒性T细胞。将其中确定至少一个CD8转录物和无IFNγ转录物被指定的T细胞计数为IFNγ-细胞毒性T细胞。
表26显示检测到的CD4 T细胞的总数,Th1 CD4 T细胞、总的细胞毒性T细胞和IFNγ+细胞毒性T细胞的数目通过使用此处所述的程序处理三种不同样品产生。
表26.子集概述。
| 对象 | CD4 | CD4 Th1 | IFNγ-CD8 | IFNγ+CD8 |
| SBJCT3 | 19 | 0 | 31 | 0 |
| SBJCT4 | 26 | 1 | 43 | 1 |
| SBJCT5 | 28 | 0 | 26 | 2 |
Q.实施例17:执行单细胞转录组学
编条形码T细胞cDNA如实施例15中所述产生。简单地说,PBMC用1X伊屋诺霉素和PMA在AIM V培养基中共刺激持续3小时。表达CD3、CD4或CD8的T细胞经过磁性标记并且单独地使用MACS试剂盒(Miltenyi Biotec)分离并且穿过液体器件。含有细胞和条形码的乳液如实施例14中在50℃下反转录持续3分钟并且在42℃下反转录持续3小时。所述乳液接着用苯酚/氯仿混合物破乳并且使用Amicon 100kDa柱(Millipore)浓缩。执行单一轮的PCR以扩增整个转录组,条件显示如下:
整个转录组PCR条件
| H<sub>2</sub>O | 28.525μL |
| 5x Q5缓冲液 | 12μL |
| Mg++ | 0.375μL |
| DMSO | 2.4μL |
| dNTP | 1.25μL |
| 指数sID(2.5μM) | 5.00μL |
| PCR1_短_n_v2(10μM) | 1.25μL |
| PCR1-RV-N-v2(10μM) | 1.25μL |
| ET-SSB | 0.625μL |
| BSA | 0.625μL |
| Tipp | 1.2μL |
| Q5酶 | 0.5μL |
| 模板 | 5μL |
| 总计 | 60μL |
热循环条件

使用PCR的5个循环,在与上文相同的PCR条件和热循环条件下,但使用FW2-n-V2作为正向引物将衔接子添加至文库中。接着汇集样品,使用Ampure珠粒清洁并且准备使用Nextera XT DNA制备试剂盒(Illumina),使用制造商的说明书进行Illumina测序,除了使用5ng DNA模板,并且在扩增步骤期间替代地使用定制内部引物,所述试剂盒将DNA片段化成较小片段。所用的内部引物Next_i5_n_v2和Next_i7_n确保将仅扩增含有所述条形码的片段化片段。运行凝胶并且显示于图29中。
表27.用于整个转录组扩增和文库制备的额外引物


使用Illumina NextSeq仪器对编条形码扩增子文库测序。分析配对末端读数以将配对读数与个别细胞联系在一起,并且鉴别在那些细胞中表达的基因。测序产生371,918,220个已过滤的配对末端读数。所述DNA条形码用于基于正向读数序列向个别细胞内的转录物指定配对读数。所述正向读数内所述DNA条形码的鉴别使用python脚本进行。关于各正向读数,与固定序列1的编辑距离使用全局/局部比对计算。需要2或更小的编辑距离或弃去所述读数对。根据固定序列1的位置和BC1和BC2的已知长度,从所述正向读数推断出候选BC1和BC2序列。检查BC1和BC2以验证其分别满足用于汉明(16,11)或汉明(12,7)DNA条形码的汉明条件。关于满足所述汉明条件的正向读数,基于X、固定序列2和分子条形码的已知长度推断出候选分子条形码。如果所述分子条形码序列无“C”核苷酸,那么向细胞(基于BC1和BC2)和所述细胞内的特异性转录物(基于所述分子条形码)指定配对读数。向个别细胞内的转录物指定37,110.172个读数对。
向细胞转录物指定的配对读数接着使用程序blastn以10-6的e值截止并且设定百分率_同一性至98与如Ensembl(www.ensembl.org)的release 78中所报告的基因的已知剪切变体进行比较。如果所述对的任一读数由blast计分为一次碰撞,那么来自所述细胞的对应的转录物(由BC1、BC2和所述分子条形码鉴别)与所述基因建立联系。如果存在超过一次blast碰撞,那么通过发现对正向和反向读数具有HSP长度的最大总和的基因来选择最佳匹配。在两种不同基因之间建立联系的情况下,将所述读数对指定至基因被视为不明确的并且不进行进一步考虑。
关于由BC1和BC2的独特组合鉴别的各细胞,通过对由与给定基因相关的读数对观察到的不同分子条形码的数目计数来确定可见来自所述给定基因的转录物的不同次数。
表33显示在使用这种程序处理四种样品后最经常检测到的基因。所述表显示Ensembl基因ID、所述基因的Ensembl描述和其中检测到所述基因的细胞的数目。
R.实施例18:将条形码衔接子并入第一链cDNA的5’端中
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。例如通过液滴发生器器件将细胞和条形码衔接模板一起放在反应容器中,由此大多数反应容器仅具有一种细胞和一种模板分子,或一种细胞和一种条形码衔接模板珠粒,并且所述反应容器为油包水液滴,如实施例14中。所述条形码衔接序列包含固定序列、条形码序列、任选地存在的UMI,和寡聚(dT)或随机或半随机序列(分别为表28中的条形码_衔接子_5c_寡聚dT和条形码_衔接子_5c_随机体)或其组合。所述模板转换寡核苷酸(TSO)包含固定序列、任选地存在的UMI和第一链cDNA互补序列(表28中的5’衔接子)。
所述反转录反应在以下反应条件下在50℃下执行3分钟,随后在42℃下执行3小时:
RT反应混合物


当所述条形码衔接子引发RT反应并且并入第一链cDNA的5’端中时,编条形码在所述RT反应期间发生。条形码衔接子由RNAP或DNAP产生(在条形码衔接模板上具有适当RNA启动子或链置换DNAP识别位点,如通过切口酶产生的切口),因为反转录能够利用DNA和RNA作为引物(图8和9)。
所述乳液如实施例14中被破乳,并且所得编条形码核酸文库接着汇集并且使用正向和反向引物扩增,所述引物包含与固定序列互补的序列,所述固定序列如实施例17中分别通过编条形码反应中的5’衔接子和条形码_衔接子_5c_寡聚dT或条形码_衔接子_5c_随机体添加。反应条件显示如下:

热循环条件


所关注的靶标基因也可通过使用包含基因特异性序列的正向引物并且使用包含与如实施例14和16中通过编条形码反应中的条形码_衔接子_5c_寡聚dT或条形码_衔接子_5c_随机体添加的固定序列互补的序列的反向引物执行扩增来扩增。关于在两个连续PCR反应中扩增TCRα和β链的反应条件显示如下,其中PCR1的产物在用于PCR2中之前稀释50倍:

PCR1和PCR2的热循环条件


所述文库接着准备用于下一代测序,如在Illumina或Ion Torrent平台上。
表28.引物序列

S.实施例19:在PCR期间将条形码衔接子并入5’端中
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。例如通过液滴发生器器件将细胞和条形码衔接模板一起放在反应容器中,由此大多数反应容器仅具有一种细胞和一种模板分子,或一种细胞和一种条形码衔接模板珠粒,并且所述反应容器为油包水液滴,如实施例14中。所述模板转换寡核苷酸(TSO)包含固定序列、任选地存在的UMI和第一链cDNA互补序列(表29中的5’衔接子)。3’衔接序列包含固定序列、任选地存在的UMI,和寡聚(dT)或随机或半随机序列(分别为表29中的3’_衔接子_寡聚dT和3’_衔接子_随机体)或其组合。
使用细胞和条形码衔接模板珠粒的反转录反应在以下反应条件下在50℃下执行3分钟,随后在42℃下执行3小时,随后在标准PCR循环条件下执行:
RT反应混合物


5’_PCR_条形码_衔接_引物由条形码_衔接_模板使用DNAP(在条形码衔接模板上具有适当链置换DNAP识别位点,如通过切口酶产生的切口)产生。此处,Klenow片段用作DNAP并且Nt.BbvCI用作切口核酸内切酶,并且所述识别位点为“CCTCAGC”。在反转录后,具有与添加至第一链cDNA的衔接序列互补的其3’端的引物用于扩增,其中正向引物为由条形码衔接模板产生的5’_PCR_条形码_衔接_引物,并且反向引物为3’_PCR_引物。
当所述条形码衔接子(5’_PCR_条形码_衔接_引物)为正向引物并且所述条形码衔接子并入第一链cDNA的5’端中时,编条形码在所述PCR反应期间发生(图11)。
所关注的靶标基因也可通过使用作为正向引物的5’_PCR_条形码_衔接_引物和包含基因特异性序列的反向引物执行扩增来扩增。
所述文库接着汇集并且准备用于下一代测序,如在Illumina或Ion Torrent平台上。
表29.引物序列


T.实施例20:在PCR期间将条形码衔接子并入3’端中
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。这一实施例类似于实施例19,除了由条形码衔接模板产生的条形码衔接子用作PCR中的反向引物。如实施例19中执行反转录,并且在PCR中,5’_PCR_引物为正向引物,并且3’_PCR_条形码_衔接_引物由条形码_衔接_模板产生并且用作反向引物(图12)。使用细胞和条形码衔接模板珠粒的反转录反应在以下反应条件下在50℃下执行3分钟,随后在42℃下执行3小时,随后在标准PCR循环条件下执行:
RT反应混合物

所关注的靶标基因也可通过使用包含基因特异性序列的正向引物和作为反向引物的3’_PCR_条形码_衔接_引物执行扩增来扩增。
所述文库接着汇集并且准备用于下一代测序,如在Illumina或Ion Torrent平台上。
表30.引物序列

U.实施例21:对来自非细胞来源的RNA编条形码
在本发明的实施方案中,对反应容器中的所有RNA均编条形码,限制条件是所述反应中所用的引物可结合于特定RNA并且起始针对特定RNA的反转录。因此,也可对外源引入的RNA编条形码。在这一实施例中,对使用体外转录产生的RNA编条形码。
SpikeIn序列根据IDT排序并且用Phusion DNA聚合酶使用作为引物的SPIKEIN-FW和SPIKEIN-RV进行PCR扩增以获得具有5’T7RNAP启动子序列和3’聚A尾的双链材料。所述产物接着用Qiagen MinElute试剂盒净化并且所述DNA产物用于由Life Technologies的T7MEGAScript试剂盒进行体外转录。因此获得的RNA接着通过使用Amicon 30kDA柱(Millipore)用10mM Tris洗涤并且浓缩来净化。
在八个96孔板的各孔中,单一记忆B细胞连同0.5ng酵母tRNA(LifeTechnologies)和0.1pg Spike-In RNA进行反转录。在每孔10μL反应中,反应为:


所述反应在55℃下孵育3分钟,并且接着在42℃下孵育2小时。96孔板中的各孔具有在孔ID-衔接子中不同的孔条形码。所述反应接着通过结合第一链cDNA与结合于生物素标记的寡聚dT的抗生蛋白链菌素顺磁C1 Dynabeads(Life Technologies),并且接着使用磁铁来拉开所述第一链cDNA,并且用BWB缓冲液(2M NaCl于TE中)将其洗涤3次,并且接着用10mM Tris洗涤3次来净化,并且再悬浮于15μL 10mM Tris中。
进行两轮PCR扩增以扩增重链和轻链免疫球蛋白基因。不同板条形码序列添加至不同板中的所有汇集的编条形码cDNA中。
关于PCR1的每孔:

来自PCR1的产物稀释50倍并且用于PCR2。关于PCR2的每孔反应:

将所得扩增材料的量标准化并且如实施例11中关于454测序来制备。用于这一实施例中的引物可发现于表13、14和32中。
获得的454读数基于板-和孔-ID条形码分类。因此,读数可分类返回特定板中的初始孔。在剪去所述条形码序列后,读数用Newbler组装。关于各重叠群,我们执行所述重叠群与所述Spike-In序列的Smith-Waterman比对,关于匹配使用计分矩阵2,关于错误匹配使用计分矩阵-1,关于间隙开口使用计分矩阵-1并且关于间隙延伸使用计分矩阵-1。具有>800的分数的任何重叠群均被视为匹配。我们对各板上观察到匹配的孔的数目计数。所述Spike-In序列在大多数孔中检测到(表31)。
表31.检测到Spike-In序列的孔


表32.所用的序列

[孔-条形码]=SEQ ID NOS:47606-47711;[板-条形码]=SEQ ID NOS:47712-47719。
V.实施例22:对来自非细胞来源的RNA编条形码以鉴别细胞群体
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。如实施例21所示,可对外源引入的RNA编条形码。在这一实施例中,使用编条形码RNA来鉴别特异性细胞群体。Spike-In DNA如实施例21中产生,除了SPIKEIN-FW具有5’NH2修饰。其使用All-in-One抗体-寡核苷酸缀合试剂盒(Solulink)缀合于抗CD4抗体。使用体外转录由Spike-In DNA产生的RNA也可替代地缀合于抗CD4抗体。
如实施例15中制备T细胞并且测序,其中额外步骤为所述T细胞与所述Spike-In缀合的抗CD4抗体一起孵育,接着在液滴发生器上运行所述T细胞并且随后对所述RNA编条形码。获得的读数基于指数-ID和通过条形码衔接子添加的条形码分类。因此,读数可分类返回初始反应容器。进行所述重叠群与所述Spike-In序列的Smith-Waterman比对,关于匹配使用计分矩阵2,关于错误匹配使用计分矩阵-1,关于间隙开口使用计分矩阵-1并且关于间隙延伸使用计分矩阵-1。具有>800的分数的任何重叠群均被视为匹配。我们接着对其中观察到匹配的反应容器计数。关于其中检测到所述Spike-In序列的反应容器,所述T细胞接着被鉴别为CD4T细胞(图14A)。可使用与不同Spike-In序列偶联的多种抗体,其中末端结果为具有不同细胞表面抗原的不同细胞可在相同实验运行中得到鉴别。
W.实施例23:对来自非细胞来源的RNA编条形码以鉴别抗原特异性B细胞
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。在这一实施例中,对外源引入的RNA编条形码并且用于鉴别抗原特异性B细胞。Spike-In DNA如实施例21中产生,除了SPIKEIN-FW具有5’NH2修饰。其使用All-in-One抗体-寡核苷酸缀合试剂盒(Solulink)缀合于流感血球凝集素抗原。使用体外转录由Spike-In DNA产生的RNA也可替代地缀合于血球凝集素。
来自流感疫苗免疫小鼠的B细胞如实施例8中制备并且进行测序,其中额外步骤为所述B细胞在对其编条形码之前与所述Spike-In缀合的抗原一起孵育。获得的读数基于指数-ID和通过条形码衔接子添加的条形码分类。因此,读数可分类返回初始反应容器。进行所述重叠群与所述Spike-In序列的Smith-Waterman比对,关于匹配使用计分矩阵2,关于错误匹配使用计分矩阵-1,关于间隙开口使用计分矩阵-1并且关于间隙延伸使用计分矩阵-1。具有>800的分数的任何重叠群均被视为匹配。我们接着对其中观察到匹配的反应容器计数。关于其中检测到所述Spike-In序列的反应容器,所述B细胞接着被鉴别为血球凝集素特异性的(图14B)。可使用与不同Spike-In序列偶联的多种抗原,其中末端结果为对不同抗原具特异性的不同B细胞可在相同实验运行中得到鉴别。
X.实施例24:对来自非细胞来源的RNA编条形码以鉴别抗原特异性T细胞
这一实施例描述了基于预测结果而非实际获得的结果的本发明的实施方案。在这一实施例中,对外源引入的RNA编条形码并且用于鉴别抗原特异性B细胞。Spike-In DNA如实施例21中产生,除了SPIKEIN-FW具有5’NH2修饰。其使用All-in-One抗体-寡核苷酸缀合试剂盒(Solulink)缀合于特定肽-MHC抗原。使用体外转录由Spike-In DNA产生的RNA也可替代地缀合于肽-MHC复合物。
如实施例15中制备T细胞并且测序,其中额外步骤为所述T细胞与所述Spike-In缀合的抗CD4抗体一起孵育,接着在液滴发生器上运行所述T细胞并且随后对所述RNA编条形码。获得的读数基于指数-ID和通过条形码衔接子添加的条形码分类。因此,读数可分类返回初始反应容器。进行所述重叠群与所述Spike-In序列的Smith-Waterman比对,关于匹配使用计分矩阵2,关于错误匹配使用计分矩阵-1,关于间隙开口使用计分矩阵-1并且关于间隙延伸使用计分矩阵-1。具有>800的分数的任何重叠群均被视为匹配。我们接着对其中观察到匹配的反应容器计数。关于其中检测到所述Spike-In序列的反应容器,所述T细胞接着被鉴别为抗原特异性的(图14C)。可使用与不同Spike-In序列偶联的多种不同肽-MHC,其中末端结果为识别不同肽-MHC的不同T细胞可在相同实验运行中得到鉴别。
表33.最经常观察到的基因。

应了解,本文所述的实施例和实施方案仅是出于说明性目的并且根据其的各种修改或变化将被推荐给本领域技术人员并且将包括在本申请的精神和范围以及随附权利要求书的范围内。本文所引用的所有公布、序列登录号、专利和专利申请针对所有目的以引用的方式整体并入本文。
Claims (136)
1.一种用于制造一种或多种所关注的聚核苷酸的方法,所述方法包括:
获得多种与一种或多种样品缔合的RNA,其中
所述样品获自一个或多个受试者,并且
与样品缔合的所述RNA存在于独立反应容积中,其中所述反应容积包含与其它的液体容积分离的液体容积;
添加衔接分子至与所述样品缔合的所述RNA中,其中所述衔接分子使用RNA聚合酶(RNAP)产生并且包含RNAP启动子、通用引发序列、条形码序列和结合位点;
其中所述衔接分子通过使模板分子与RNA聚合酶(RNAP)接触而产生,所述模板分子为包含RNA聚合酶(RNAP)启动子的DNA分子,其中所述RNAP启动子选自由T7、T3和SP6组成的组;以及
将所述条形码序列并入一种或多种与所述样品缔合的聚核苷酸中,
由此制造所述一种或多种所关注的聚核苷酸。
2.如权利要求1所述的方法,其进一步包括使用所述RNA聚合酶产生所述衔接分子。
3.如权利要求1所述的方法,其中:
所述模板分子结合于固体支撑物,
所述固体支撑物与水溶液接触,并且
所述衔接分子在其产生时释放至所述水溶液中。
4.如权利要求3所述的方法,其中添加所述衔接分子至所述与一种样品缔合的RNA中包括组合所述水溶液与其中存在所述RNA的所述反应容积。
5.如权利要求3所述的方法,其中所述水溶液与所述与一种样品缔合的RNA存在于相同反应容积中。
6.如权利要求3所述的方法,其中所述固体支撑物为珠粒或表面。
7.如权利要求1所述的方法,其中所述衔接分子在添加所述衔接分子至所述与一种样品缔合的RNA中之前以游离形式处于溶液中。
8.如权利要求1所述的方法,其中所述衔接分子在反应容积中产生,并且添加所述衔接分子至所述与一种样品缔合的RNA中包括组合所述反应容积与其中存在所述RNA的所述反应容积。
9.如权利要求1所述的方法,其中所述衔接分子在其中存在添加有所述衔接分子的所述RNA的所述反应容积中产生。
10.如权利要求1所述的方法,其中所述衔接分子未在其中存在添加有所述衔接分子的所述RNA的所述反应容积中产生。
11.如权利要求1所述的方法,其中所述衔接分子在等温反应中产生。
12.如权利要求1所述的方法,其中所述衔接分子进一步包含独特分子识别符(UMI)序列。
13.如权利要求1所述的方法,其中所述衔接分子为RNA分子。
14.如权利要求1所述的方法,其中:
制造所述一种或多种所关注的聚核苷酸包括反转录与所述样品缔合的所述RNA,由此合成多种第一链cDNA,
与所述样品缔合的所述RNA中的至少一些包含与所述衔接分子的所述结合位点互补的序列区,并且
所述衔接分子用作反转录的引物,使得所述条形码序列并入与所述样品缔合的第一链cDNA中。
15.如权利要求14所述的方法,其中所述结合位点包含聚T区。
16.如权利要求14所述的方法,其中所述结合位点包含随机区。
17.如权利要求14所述的方法,其中所述结合位点出现于所述衔接分子的3’端。
18.如权利要求14所述的方法,其中所述衔接分子在反应容积中产生,并且反转录与所述样品缔合的所述RNA发生在组合所述反应容积与其中存在所述RNA的所述反应容积后。
19.如权利要求14所述的方法,其中反转录与所述样品缔合的所述RNA发生在其中产生添加至所述RNA中的所述衔接分子的相同反应容积中。
20.如权利要求1所述的方法,其进一步包括反转录与所述样品缔合的所述RNA以获得多种cDNA,其中反转录RNA包括使用反转录酶和第一链引物合成cDNA的第一链。
21.如权利要求20所述的方法,其中所述反转录酶为MMLV H-反转录酶。
22.如权利要求20所述的方法,其中所述衔接分子在反应容积中产生,并且添加所述衔接分子至所述与一种样品缔合的RNA中包括组合所述反应容积与其中存在所述RNA的所述反应容积。
23.如权利要求22所述的方法,其中cDNA的第一链在组合所述反应容积与其中存在所述RNA的所述反应容积之前合成。
24.如权利要求22所述的方法,其中cDNA的第一链在组合所述反应容积与其中存在所述RNA的所述反应容积之后合成。
25.如权利要求20所述的方法,其中反转录与所述样品缔合的所述RNA发生在其中产生添加至所述RNA中的所述衔接分子的相同反应容积中。
26.如权利要求25所述的方法,其中所述反应容积中的缓冲液包含Tris、钾离子、氯离子、硫酸根离子、铵离子、乙酸离子或镁离子中的至少一者,其中所述缓冲液具有pH 8.0至pH 8.8的pH范围。
27.如权利要求20所述的方法,其中:
所述反转录酶具有模板转换活性,
与所述样品缔合的cDNA的至少一些第一链包含3’悬垂物,
所述衔接分子的所述结合位点包含与所述3’悬垂物互补的3’部分,并且
所述衔接分子用作所述反转录酶的模板,使得所述条形码序列并入与所述样品缔合的cDNA的第一链中。
28.如权利要求27所述的方法,其中所述3’悬垂物包含一个或多个C核苷酸并且所述结合位点的所述3’部分包含一个或多个G核苷酸。
29.如权利要求27所述的方法,其中所述第一链引物包含聚T区。
30.如权利要求27所述的方法,其中所述第一链引物包含随机序列。
31.如权利要求20所述的方法,其中制造所关注的聚核苷酸包括使用第一引物和第二引物扩增用于所述样品的cDNA的所述第一链,所述第二引物具有与所述第一链引物的至少一部分相同的序列,其中所述第一引物或所述第二引物为所述衔接分子。
32.如权利要求31所述的方法,其中所述第一引物为所述衔接分子。
33.如权利要求31所述的方法,其中所述第二引物为所述衔接分子。
34.如权利要求31所述的方法,其中所述第一链引物包含聚T区。
35.如权利要求31所述的方法,其中所述第一链引物包含随机序列。
36.如权利要求1所述的方法,其中所述样品包含细胞。
37.如权利要求36所述的方法,其中所述细胞为血细胞、免疫细胞、组织细胞或肿瘤细胞。
38.如权利要求37所述的方法,其中所述细胞为B细胞或T细胞。
39.如权利要求36所述的方法,其中与所述样品缔合的所述RNA包含mRNA。
40.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少1种mRNA。
41.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含细胞的至少部分转录组。
42.如权利要求36所述的方法,其中与所述样品缔合的所述RNA包含细胞的总RNA。
43.如权利要求36所述的方法,其中每种样品制造至少10种所关注的聚核苷酸。
44.如权利要求36所述的方法,其中所述一种或多种样品包含至少10个细胞。
45.如权利要求36所述的方法,其中所述一种或多种样品获自相同受试者。
46.如权利要求36所述的方法,其进一步包括使所述样品与溶解缓冲液接触。
47.如权利要求36所述的方法,其进一步包括:
使所述样品与核酸标记物接触,由此允许所述核酸标记物结合于所述样品的子集;和
洗涤所述样品,由此从所述核酸标记物不结合的样品去除所述核酸标记物,
其中关于所述子集内的样品,添加至与所述样品缔合的所述RNA中的所述衔接分子也添加至所述核酸标记物中,并且使用所述核酸标记物制造一种或多种所关注的聚核苷酸。
48.如权利要求47所述的方法,其中所述核酸标记物包含偶联于分子标记的核酸。
49.如权利要求48所述的方法,其中所述分子标记为蛋白。
50.如权利要求48所述的方法,其中所述分子标记对一种或多种细胞表面部分具有亲和力。
51.如权利要求48所述的方法,其中所述核酸为RNA。
52.如权利要求48所述的方法,其中所述核酸为DNA。
53.如权利要求52所述的方法,其中所述DNA包含RNAP启动子。
54.如权利要求48至53中任一项所述的方法,其中:
所述样品与第一核酸标记物和第二核酸标记物接触,
所述第一核酸标记物包含偶联于第一分子标记的第一核酸,
所述第二核酸标记物包含偶联于第二分子标记的第二核酸,
所述第一核酸和第二核酸包含不同的序列区,并且
所述第一和第二分子标记为不同的。
55.如权利要求1所述的方法,其中所述一种或多种样品获自相同受试者。
56.如权利要求1所述的方法,其中所述一种或多种样品获自至少3个不同受试者。
57.一种衔接构建体,其包含RNAP启动子、通用引发序列、条形码序列和结合位点,其中所述衔接构建体通过使模板分子与RNA聚合酶(RNAP)接触而产生,所述模板分子为包含RNA聚合酶(RNAP)启动子的DNA分子,其中所述RNAP启动子选自由T7、T3和SP6组成的组。
58.如权利要求57所述的衔接构建体,其进一步包含独特分子识别符(UMI)序列。
59.一种固体支撑物,其包含如权利要求57所述的衔接构建体。
60.如权利要求59所述的固体支撑物,其中所述衔接构建体经由共价键结合于所述固体支撑物。
61.如权利要求59所述的固体支撑物,其中所述衔接构建体的多个拷贝结合于所述固体支撑物。
62.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少10个拷贝结合于所述固体支撑物。
63.如权利要求61所述的固体支撑物,其中所述衔接构建体的各拷贝包含相同条形码序列。
64.一种衔接模板文库,其包含多种如权利要求59所述的固体支撑物。
65.如权利要求64所述的衔接模板文库,其中所述多种包含至少10种固体支撑物。
66.一种试剂盒,其包含如权利要求57所述的衔接构建体、如权利要求59所述的固体支撑物或如权利要求64所述的衔接模板文库。
67.如权利要求66所述的试剂盒,其进一步包含用于由如权利要求57所述的衔接构建体产生包含RNAP启动子、通用引发序列、条形码序列和结合位点的衔接分子的酶。
68.如权利要求66或67所述的试剂盒,其进一步包含含有渗透保护剂的细胞悬浮缓冲液。
69.如权利要求68所述的试剂盒,其中所述渗透保护剂为甜菜碱或与其结构非常类似的化合物。
70.如权利要求69所述的试剂盒,其中所述渗透保护剂为甘氨酸甜菜碱。
71.如权利要求68所述的试剂盒,其中所述渗透保护剂为糖或多元醇。
72.如权利要求71所述的试剂盒,其中所述渗透保护剂为海藻糖。
73.如权利要求68所述的试剂盒,其中所述渗透保护剂为氨基酸。
74.如权利要求73所述的试剂盒,其中所述渗透保护剂为脯氨酸。
75.如权利要求68所述的试剂盒,其中所述缓冲液的摩尔渗透压浓度为250-350mOsm/L。
76.如权利要求75所述的试剂盒,其中所述渗透保护剂贡献了多达10%、20%、30%、40%、50%、60%、70%、80%、90%或100%的所述缓冲液的所述摩尔渗透压浓度。
77.如权利要求68所述的试剂盒,其中所述缓冲液包含230-330mM甜菜碱和约10mMNaCl。
78.如权利要求64所述的衔接模板文库,其中所述固体支撑物中的至少两者包含具有不同条形码序列或UMI序列的衔接构建体。
79.如权利要求78所述的衔接模板文库,其中所述多种固体支撑物中的每一种固体支撑物均包含具有不同条形码序列或不同UMI序列的衔接构建体。
80.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少3种mRNA。
81.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少10种mRNA。
82.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少30种mRNA。
83.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少100种mRNA。
84.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少300种mRNA。
85.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少1000种mRNA。
86.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少3000种mRNA。
87.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少10,000种mRNA。
88.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少30,000种mRNA。
89.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少100,000种mRNA。
90.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少300,000种mRNA。
91.如权利要求39所述的方法,其中与所述样品缔合的所述RNA包含至少1,000,000种mRNA。
92.如权利要求36所述的方法,其中每种样品制造至少30种所关注的聚核苷酸。
93.如权利要求36所述的方法,其中每种样品制造至少100种所关注的聚核苷酸。
94.如权利要求36所述的方法,其中每种样品制造至少300种所关注的聚核苷酸。
95.如权利要求36所述的方法,其中每种样品制造至少1,000种所关注的聚核苷酸。
96.如权利要求36所述的方法,其中每种样品制造至少3,000种所关注的聚核苷酸。
97.如权利要求36所述的方法,其中每种样品制造至少10,000种所关注的聚核苷酸。
98.如权利要求36所述的方法,其中每种样品制造至少30,000种所关注的聚核苷酸。
99.如权利要求36所述的方法,其中每种样品制造至少100,000种所关注的聚核苷酸。
100.如权利要求36所述的方法,其中每种样品制造至少300,000种所关注的聚核苷酸。
101.如权利要求36所述的方法,其中每种样品制造至少1,000,000种所关注的聚核苷酸。
102.如权利要求36所述的方法,其中所述一种或多种样品包含至少30个细胞。
103.如权利要求36所述的方法,其中所述一种或多种样品包含至少100个细胞。
104.如权利要求36所述的方法,其中所述一种或多种样品包含至少300个细胞。
105.如权利要求36所述的方法,其中所述一种或多种样品包含至少1,000个细胞。
106.如权利要求36所述的方法,其中所述一种或多种样品包含至少3,000个细胞。
107.如权利要求36所述的方法,其中所述一种或多种样品包含至少10,000个细胞。
108.如权利要求36所述的方法,其中所述一种或多种样品包含至少30,000个细胞。
109.如权利要求36所述的方法,其中所述一种或多种样品包含至少100,000个细胞。
110.如权利要求36所述的方法,其中所述一种或多种样品包含至少300,000个细胞。
111.如权利要求36所述的方法,其中所述一种或多种样品包含至少1,000,000个细胞。
112.如权利要求1所述的方法,其中所述一种或多种样品获自至少10个不同受试者。
113.如权利要求1所述的方法,其中所述一种或多种样品获自至少30个不同受试者。
114.如权利要求1所述的方法,其中所述一种或多种样品获自至少100个不同受试者。
115.如权利要求49所述的方法,其中所述分子标记为抗体。
116.如权利要求49所述的方法,其中所述分子标记为抗原。
117.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少30个拷贝结合于所述固体支撑物。
118.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少100个拷贝结合于所述固体支撑物。
119.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少300个拷贝结合于所述固体支撑物。
120.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少1,000个拷贝结合于所述固体支撑物。
121.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少3,000个拷贝结合于所述固体支撑物。
122.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少10,000个拷贝结合于所述固体支撑物。
123.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少30,000个拷贝结合于所述固体支撑物。
124.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少100,000个拷贝结合于所述固体支撑物。
125.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少300,000个拷贝结合于所述固体支撑物。
126.如权利要求61所述的固体支撑物,其中所述衔接构建体的至少1,000,000个拷贝结合于所述固体支撑物。
127.如权利要求64所述的衔接模板文库,其中所述多种包含至少30种固体支撑物。
128.如权利要求64所述的衔接模板文库,其中所述多种包含至少100种固体支撑物。
129.如权利要求64所述的衔接模板文库,其中所述多种包含至少300种固体支撑物。
130.如权利要求64所述的衔接模板文库,其中所述多种包含至少1,000种固体支撑物。
131.如权利要求64所述的衔接模板文库,其中所述多种包含至少3,000种固体支撑物。
132.如权利要求64所述的衔接模板文库,其中所述多种包含至少10,000种固体支撑物。
133.如权利要求64所述的衔接模板文库,其中所述多种包含至少30,000种固体支撑物。
134.如权利要求64所述的衔接模板文库,其中所述多种包含至少100,000种固体支撑物。
135.如权利要求64所述的衔接模板文库,其中所述多种包含至少300,000种固体支撑物。
136.如权利要求64所述的衔接模板文库,其中所述多种包含至少1,000,000种固体支撑物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202111683991.4A CN114717291A (zh) | 2013-12-30 | 2014-12-30 | 使用核酸条形码分析与单细胞缔合的核酸 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361922012P | 2013-12-30 | 2013-12-30 | |
| US61/922,012 | 2013-12-30 | ||
| PCT/US2014/072898 WO2015103339A1 (en) | 2013-12-30 | 2014-12-30 | Analysis of nucleic acids associated with single cells using nucleic acid barcodes |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111683991.4A Division CN114717291A (zh) | 2013-12-30 | 2014-12-30 | 使用核酸条形码分析与单细胞缔合的核酸 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN106460033A CN106460033A (zh) | 2017-02-22 |
| CN106460033B true CN106460033B (zh) | 2021-12-24 |
Family
ID=52424112
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201480076617.XA Active CN106460033B (zh) | 2013-12-30 | 2014-12-30 | 使用核酸条形码分析与单细胞缔合的核酸 |
| CN202111683991.4A Pending CN114717291A (zh) | 2013-12-30 | 2014-12-30 | 使用核酸条形码分析与单细胞缔合的核酸 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202111683991.4A Pending CN114717291A (zh) | 2013-12-30 | 2014-12-30 | 使用核酸条形码分析与单细胞缔合的核酸 |
Country Status (13)
| Country | Link |
|---|---|
| US (6) | US9580736B2 (zh) |
| EP (2) | EP3089822B1 (zh) |
| JP (1) | JP6608368B2 (zh) |
| KR (2) | KR102433825B1 (zh) |
| CN (2) | CN106460033B (zh) |
| AU (1) | AU2014373757B2 (zh) |
| CA (1) | CA2935122C (zh) |
| DK (1) | DK3089822T3 (zh) |
| ES (1) | ES2912183T3 (zh) |
| PL (1) | PL3089822T3 (zh) |
| PT (1) | PT3089822T (zh) |
| SG (2) | SG11201605344YA (zh) |
| WO (1) | WO2015103339A1 (zh) |
Families Citing this family (150)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9424392B2 (en) | 2005-11-26 | 2016-08-23 | Natera, Inc. | System and method for cleaning noisy genetic data from target individuals using genetic data from genetically related individuals |
| US8825412B2 (en) | 2010-05-18 | 2014-09-02 | Natera, Inc. | Methods for non-invasive prenatal ploidy calling |
| US8835358B2 (en) | 2009-12-15 | 2014-09-16 | Cellular Research, Inc. | Digital counting of individual molecules by stochastic attachment of diverse labels |
| US20190010543A1 (en) | 2010-05-18 | 2019-01-10 | Natera, Inc. | Methods for simultaneous amplification of target loci |
| US11408031B2 (en) | 2010-05-18 | 2022-08-09 | Natera, Inc. | Methods for non-invasive prenatal paternity testing |
| US11939634B2 (en) | 2010-05-18 | 2024-03-26 | Natera, Inc. | Methods for simultaneous amplification of target loci |
| US11332785B2 (en) | 2010-05-18 | 2022-05-17 | Natera, Inc. | Methods for non-invasive prenatal ploidy calling |
| US11332793B2 (en) | 2010-05-18 | 2022-05-17 | Natera, Inc. | Methods for simultaneous amplification of target loci |
| US11322224B2 (en) | 2010-05-18 | 2022-05-03 | Natera, Inc. | Methods for non-invasive prenatal ploidy calling |
| US11326208B2 (en) | 2010-05-18 | 2022-05-10 | Natera, Inc. | Methods for nested PCR amplification of cell-free DNA |
| US9677118B2 (en) | 2014-04-21 | 2017-06-13 | Natera, Inc. | Methods for simultaneous amplification of target loci |
| US11339429B2 (en) | 2010-05-18 | 2022-05-24 | Natera, Inc. | Methods for non-invasive prenatal ploidy calling |
| WO2012106546A2 (en) | 2011-02-02 | 2012-08-09 | University Of Washington Through Its Center For Commercialization | Massively parallel continguity mapping |
| SG11201405274WA (en) | 2012-02-27 | 2014-10-30 | Cellular Res Inc | Compositions and kits for molecular counting |
| EP3305918B1 (en) | 2012-03-05 | 2020-06-03 | President and Fellows of Harvard College | Methods for epigenetic sequencing |
| US11591637B2 (en) | 2012-08-14 | 2023-02-28 | 10X Genomics, Inc. | Compositions and methods for sample processing |
| US9701998B2 (en) | 2012-12-14 | 2017-07-11 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| US10221442B2 (en) | 2012-08-14 | 2019-03-05 | 10X Genomics, Inc. | Compositions and methods for sample processing |
| US10323279B2 (en) | 2012-08-14 | 2019-06-18 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| US10273541B2 (en) | 2012-08-14 | 2019-04-30 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| US10400280B2 (en) | 2012-08-14 | 2019-09-03 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| US20150376609A1 (en) | 2014-06-26 | 2015-12-31 | 10X Genomics, Inc. | Methods of Analyzing Nucleic Acids from Individual Cells or Cell Populations |
| US9951386B2 (en) | 2014-06-26 | 2018-04-24 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| MX364957B (es) | 2012-08-14 | 2019-05-15 | 10X Genomics Inc | Composiciones y metodos para microcapsulas. |
| US10752949B2 (en) | 2012-08-14 | 2020-08-25 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| CA2894694C (en) | 2012-12-14 | 2023-04-25 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| US10533221B2 (en) | 2012-12-14 | 2020-01-14 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| CA2900481A1 (en) | 2013-02-08 | 2014-08-14 | 10X Genomics, Inc. | Polynucleotide barcode generation |
| DK3553175T3 (da) | 2013-03-13 | 2021-08-23 | Illumina Inc | Fremgangsmåde til fremstilling af et nukleinsyresekvenseringsbibliotek |
| GB2525568B (en) * | 2013-03-15 | 2020-10-14 | Abvitro Llc | Single cell barcoding for antibody discovery |
| SG10201806890VA (en) | 2013-08-28 | 2018-09-27 | Cellular Res Inc | Massively parallel single cell analysis |
| US9582877B2 (en) | 2013-10-07 | 2017-02-28 | Cellular Research, Inc. | Methods and systems for digitally counting features on arrays |
| ES2890078T3 (es) | 2013-12-20 | 2022-01-17 | Illumina Inc | Conservación de la información de conectividad genómica en muestras de ADN genómico fragmentado |
| ES2912183T3 (es) | 2013-12-30 | 2022-05-24 | Atreca Inc | Análisis de ácidos nucleicos asociados a células individuales utilizando códigos de barras de ácidos nucleicos |
| AU2015243445B2 (en) | 2014-04-10 | 2020-05-28 | 10X Genomics, Inc. | Fluidic devices, systems, and methods for encapsulating and partitioning reagents, and applications of same |
| CN107075543B (zh) * | 2014-04-21 | 2021-11-16 | 哈佛学院院长及董事 | 用于条形码化核酸的系统和方法 |
| US20150298091A1 (en) | 2014-04-21 | 2015-10-22 | President And Fellows Of Harvard College | Systems and methods for barcoding nucleic acids |
| RU2717641C2 (ru) | 2014-04-21 | 2020-03-24 | Натера, Инк. | Обнаружение мутаций и плоидности в хромосомных сегментах |
| CA3060708A1 (en) * | 2014-04-29 | 2015-11-05 | Illumina, Inc | Multiplexed single cell gene expression analysis using template switch and tagmentation |
| US10975371B2 (en) | 2014-04-29 | 2021-04-13 | Illumina, Inc. | Nucleic acid sequence analysis from single cells |
| AU2015318011B2 (en) * | 2014-09-15 | 2020-07-23 | Abvitro Llc | High-throughput nucleotide library sequencing |
| IL299976A (en) | 2014-10-17 | 2023-03-01 | Illumina Cambridge Ltd | Continuity-preserving transposition |
| PT3207134T (pt) * | 2014-10-17 | 2019-09-17 | Illumina Cambridge Ltd | Transposição que preserva a contiguidade |
| US20160122817A1 (en) | 2014-10-29 | 2016-05-05 | 10X Genomics, Inc. | Methods and compositions for targeted nucleic acid sequencing |
| US9975122B2 (en) | 2014-11-05 | 2018-05-22 | 10X Genomics, Inc. | Instrument systems for integrated sample processing |
| SG11201705615UA (en) | 2015-01-12 | 2017-08-30 | 10X Genomics Inc | Processes and systems for preparing nucleic acid sequencing libraries and libraries prepared using same |
| ES2824700T3 (es) | 2015-02-19 | 2021-05-13 | Becton Dickinson Co | Análisis unicelular de alto rendimiento que combina información proteómica y genómica |
| EP3262188B1 (en) | 2015-02-24 | 2021-05-05 | 10X Genomics, Inc. | Methods for targeted nucleic acid sequence coverage |
| EP3262407B1 (en) | 2015-02-24 | 2023-08-30 | 10X Genomics, Inc. | Partition processing methods and systems |
| US9727810B2 (en) | 2015-02-27 | 2017-08-08 | Cellular Research, Inc. | Spatially addressable molecular barcoding |
| EP3262189B1 (en) * | 2015-02-27 | 2021-12-08 | Becton, Dickinson and Company | Methods for barcoding nucleic acids for sequencing |
| EP3277843A2 (en) | 2015-03-30 | 2018-02-07 | Cellular Research, Inc. | Methods and compositions for combinatorial barcoding |
| JP2018511341A (ja) | 2015-04-17 | 2018-04-26 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 遺伝子配列決定および他の適用のためのバーコード化システムおよび方法 |
| CN107580632B (zh) | 2015-04-23 | 2021-12-28 | 贝克顿迪金森公司 | 用于全转录组扩增的方法和组合物 |
| US11479812B2 (en) | 2015-05-11 | 2022-10-25 | Natera, Inc. | Methods and compositions for determining ploidy |
| WO2016196229A1 (en) | 2015-06-01 | 2016-12-08 | Cellular Research, Inc. | Methods for rna quantification |
| CN115928221A (zh) * | 2015-08-28 | 2023-04-07 | Illumina公司 | 单细胞核酸序列分析 |
| WO2017044574A1 (en) | 2015-09-11 | 2017-03-16 | Cellular Research, Inc. | Methods and compositions for nucleic acid library normalization |
| US11156611B2 (en) * | 2015-09-24 | 2021-10-26 | Abvitro Llc | Single cell characterization using affinity-oligonucleotide conjugates and vessel barcoded polynucleotides |
| KR20180097536A (ko) * | 2015-11-04 | 2018-08-31 | 아트레카, 인크. | 단일 세포와 연관된 핵산의 분석을 위한 핵산 바코드의 조합 세트 |
| US20170141793A1 (en) * | 2015-11-13 | 2017-05-18 | Microsoft Technology Licensing, Llc | Error correction for nucleotide data stores |
| SG11201804086VA (en) | 2015-12-04 | 2018-06-28 | 10X Genomics Inc | Methods and compositions for nucleic acid analysis |
| US11965891B2 (en) * | 2015-12-30 | 2024-04-23 | Bio-Rad Laboratories, Inc. | Digital protein quantification |
| JP7179329B2 (ja) * | 2016-01-14 | 2022-11-29 | ヨーロピアン モレキュラー バイオロジー ラボラトリー | リガンド誘発型の細胞発現のマイクロ流体分析 |
| US11702702B2 (en) * | 2016-04-15 | 2023-07-18 | Predicine, Inc. | Systems and methods for detecting genetic alterations |
| US10822643B2 (en) | 2016-05-02 | 2020-11-03 | Cellular Research, Inc. | Accurate molecular barcoding |
| WO2017197338A1 (en) | 2016-05-13 | 2017-11-16 | 10X Genomics, Inc. | Microfluidic systems and methods of use |
| US10301677B2 (en) | 2016-05-25 | 2019-05-28 | Cellular Research, Inc. | Normalization of nucleic acid libraries |
| WO2017205344A1 (en) * | 2016-05-25 | 2017-11-30 | Bio-Rad Laboratories, Inc. | Digital proximity assay |
| CN109074430B (zh) | 2016-05-26 | 2022-03-29 | 贝克顿迪金森公司 | 分子标记计数调整方法 |
| US10202641B2 (en) | 2016-05-31 | 2019-02-12 | Cellular Research, Inc. | Error correction in amplification of samples |
| US10640763B2 (en) | 2016-05-31 | 2020-05-05 | Cellular Research, Inc. | Molecular indexing of internal sequences |
| CN105950612B (zh) * | 2016-07-08 | 2019-06-21 | 北京全式金生物技术有限公司 | 一种高效的dna接头连接方法 |
| EP3494214A4 (en) | 2016-08-05 | 2020-03-04 | Bio-Rad Laboratories, Inc. | SECOND RANK DIRECT |
| WO2018031760A1 (en) * | 2016-08-10 | 2018-02-15 | Grail, Inc. | Methods of preparing dual-indexed dna libraries for bisulfite conversion sequencing |
| WO2018058073A2 (en) | 2016-09-26 | 2018-03-29 | Cellular Research, Inc. | Measurement of protein expression using reagents with barcoded oligonucleotide sequences |
| US11485996B2 (en) | 2016-10-04 | 2022-11-01 | Natera, Inc. | Methods for characterizing copy number variation using proximity-litigation sequencing |
| EP4026905B1 (en) | 2016-10-19 | 2024-04-17 | 10X Genomics, Inc. | Methods for barcoding nucleic acid molecules from individual cells or cell populations |
| WO2018081113A1 (en) | 2016-10-24 | 2018-05-03 | Sawaya Sterling | Concealing information present within nucleic acids |
| KR20190077061A (ko) | 2016-11-08 | 2019-07-02 | 셀룰러 리서치, 인크. | 세포 표지 분류 방법 |
| EP3539035B1 (en) | 2016-11-08 | 2024-04-17 | Becton, Dickinson and Company | Methods for expression profile classification |
| WO2018089550A1 (en) * | 2016-11-10 | 2018-05-17 | Takara Bio Usa, Inc. | Methods of producing amplified double stranded deoxyribonucleic acids and compositions and kits for use therein |
| US10011870B2 (en) * | 2016-12-07 | 2018-07-03 | Natera, Inc. | Compositions and methods for identifying nucleic acid molecules |
| US10815525B2 (en) | 2016-12-22 | 2020-10-27 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| US10011872B1 (en) * | 2016-12-22 | 2018-07-03 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| EP3559262A4 (en) * | 2016-12-22 | 2020-07-22 | Illumina, Inc. | NETWORKS PRESENTING QUALITY CONTROL TRACERS |
| US10550429B2 (en) | 2016-12-22 | 2020-02-04 | 10X Genomics, Inc. | Methods and systems for processing polynucleotides |
| JP7104048B2 (ja) | 2017-01-13 | 2022-07-20 | セルラー リサーチ, インコーポレイテッド | 流体チャネルの親水性コーティング |
| EP4029939B1 (en) | 2017-01-30 | 2023-06-28 | 10X Genomics, Inc. | Methods and systems for droplet-based single cell barcoding |
| CN110382708A (zh) | 2017-02-01 | 2019-10-25 | 赛卢拉研究公司 | 使用阻断性寡核苷酸进行选择性扩增 |
| EP3375889B1 (en) | 2017-03-17 | 2019-12-11 | HiFiBiO SAS | Single cell analysis |
| JP7289792B2 (ja) * | 2017-04-21 | 2023-06-12 | メサ バイオテック,インク. | 流体検査用カセット |
| CN116121243A (zh) * | 2017-04-27 | 2023-05-16 | 深圳华大基因股份有限公司 | Dna标签及其应用 |
| WO2018203576A1 (ja) | 2017-05-02 | 2018-11-08 | 国立大学法人 東京大学 | 一細胞の非破壊的計測情報とゲノム関連情報とを一体的に検出する方法 |
| US20180320173A1 (en) * | 2017-05-05 | 2018-11-08 | Scipio Bioscience | Methods for trapping and barcoding discrete biological units in hydrogel |
| CN116064732A (zh) | 2017-05-26 | 2023-05-05 | 10X基因组学有限公司 | 转座酶可接近性染色质的单细胞分析 |
| US10400235B2 (en) | 2017-05-26 | 2019-09-03 | 10X Genomics, Inc. | Single cell analysis of transposase accessible chromatin |
| CA3059559A1 (en) | 2017-06-05 | 2018-12-13 | Becton, Dickinson And Company | Sample indexing for single cells |
| CN107502607A (zh) * | 2017-06-20 | 2017-12-22 | 浙江大学 | 一种大量组织、细胞样本mRNA的分子条形码标记、文库构建、测序的方法 |
| WO2019060716A1 (en) | 2017-09-25 | 2019-03-28 | Freenome Holdings, Inc. | SAMPLE EXTRACTION METHODS AND SYSTEMS |
| SG11201913654QA (en) | 2017-11-15 | 2020-01-30 | 10X Genomics Inc | Functionalized gel beads |
| US20190153438A1 (en) * | 2017-11-15 | 2019-05-23 | Viome, Inc. | Methods and compositions for preparing polynucleotide libraries |
| US10829815B2 (en) | 2017-11-17 | 2020-11-10 | 10X Genomics, Inc. | Methods and systems for associating physical and genetic properties of biological particles |
| EP3717661A1 (en) | 2017-11-27 | 2020-10-07 | The Trustees of Columbia University in the City of New York | Rna printing and sequencing devices, methods, and systems |
| US20210371853A1 (en) * | 2017-12-09 | 2021-12-02 | Viome, Inc. | Methods for nucleic acid library creation |
| EP3728636A1 (en) | 2017-12-19 | 2020-10-28 | Becton, Dickinson and Company | Particles associated with oligonucleotides |
| KR102014470B1 (ko) * | 2017-12-29 | 2019-08-26 | 한국과학기술원 | 핵산의 절단 및 중합연쇄반응 시스템 기반 등온 핵산증폭기술을 이용한 표적 rna 검출 방법 |
| WO2019133874A1 (en) * | 2017-12-31 | 2019-07-04 | Berkeley Lights, Inc. | General functional assay |
| WO2019147663A1 (en) * | 2018-01-24 | 2019-08-01 | Freenome Holdings, Inc. | Methods and systems for abnormality detection in the patterns of nucleic acids |
| WO2019157529A1 (en) * | 2018-02-12 | 2019-08-15 | 10X Genomics, Inc. | Methods characterizing multiple analytes from individual cells or cell populations |
| SG11202008080RA (en) * | 2018-02-22 | 2020-09-29 | 10X Genomics Inc | Ligation mediated analysis of nucleic acids |
| US11639928B2 (en) | 2018-02-22 | 2023-05-02 | 10X Genomics, Inc. | Methods and systems for characterizing analytes from individual cells or cell populations |
| CN112236518A (zh) * | 2018-02-23 | 2021-01-15 | 耶鲁大学 | 单细胞冻融裂解 |
| EP3762494B1 (en) * | 2018-03-07 | 2022-11-23 | Wisconsin Alumni Research Foundation | High throughput nucleic acid profiling of single cells |
| SG11202009889VA (en) | 2018-04-06 | 2020-11-27 | 10X Genomics Inc | Systems and methods for quality control in single cell processing |
| US11773441B2 (en) | 2018-05-03 | 2023-10-03 | Becton, Dickinson And Company | High throughput multiomics sample analysis |
| JP7358388B2 (ja) | 2018-05-03 | 2023-10-10 | ベクトン・ディキンソン・アンド・カンパニー | 反対側の転写物末端における分子バーコーディング |
| SG11202001827UA (en) * | 2018-05-17 | 2020-03-30 | Illumina Inc | High-throughput single-cell sequencing with reduced amplification bias |
| US20200032335A1 (en) | 2018-07-27 | 2020-01-30 | 10X Genomics, Inc. | Systems and methods for metabolome analysis |
| WO2020046833A1 (en) * | 2018-08-28 | 2020-03-05 | Cellular Research, Inc. | Sample multiplexing using carbohydrate-binding and membrane-permeable reagents |
| US11639517B2 (en) | 2018-10-01 | 2023-05-02 | Becton, Dickinson And Company | Determining 5′ transcript sequences |
| WO2020072907A1 (en) * | 2018-10-05 | 2020-04-09 | Board Of Regents, The University Of Texas System | Solid-phase n-terminal peptide capture and release |
| US20220010373A1 (en) | 2018-11-07 | 2022-01-13 | The University Of Tokyo | Method for detecting genome-related information of cell coexisting with at least one type of test substance |
| US11932849B2 (en) | 2018-11-08 | 2024-03-19 | Becton, Dickinson And Company | Whole transcriptome analysis of single cells using random priming |
| WO2020118200A1 (en) * | 2018-12-07 | 2020-06-11 | Qiagen Sciences, Llc | Methods for preparing cdna samples for rna sequencing, and cdna samples and uses thereof |
| US11459607B1 (en) * | 2018-12-10 | 2022-10-04 | 10X Genomics, Inc. | Systems and methods for processing-nucleic acid molecules from a single cell using sequential co-partitioning and composite barcodes |
| EP3894552A1 (en) | 2018-12-13 | 2021-10-20 | Becton, Dickinson and Company | Selective extension in single cell whole transcriptome analysis |
| CN111378557B (zh) | 2018-12-26 | 2023-06-06 | 财团法人工业技术研究院 | 用于产生液珠的管状结构及液珠产生方法 |
| US11371076B2 (en) | 2019-01-16 | 2022-06-28 | Becton, Dickinson And Company | Polymerase chain reaction normalization through primer titration |
| EP3914728B1 (en) | 2019-01-23 | 2023-04-05 | Becton, Dickinson and Company | Oligonucleotides associated with antibodies |
| US11965208B2 (en) | 2019-04-19 | 2024-04-23 | Becton, Dickinson And Company | Methods of associating phenotypical data and single cell sequencing data |
| CN110241460A (zh) * | 2019-05-31 | 2019-09-17 | 南方医科大学南方医院 | 一种甄别独立样品自身交叉反应的免疫组库方法 |
| US11939622B2 (en) | 2019-07-22 | 2024-03-26 | Becton, Dickinson And Company | Single cell chromatin immunoprecipitation sequencing assay |
| BR112022002805A2 (pt) * | 2019-08-14 | 2022-08-09 | Irepertoire Inc | Método de captura por sonda para recuperação de vdj da cadeia alfa e beta de tcr por rna transcrito de forma reversa baseado em óligo-dt |
| JP2022552194A (ja) | 2019-10-10 | 2022-12-15 | 1859,インク. | マイクロ流体スクリーニングのための方法およびシステム |
| CN114729350A (zh) | 2019-11-08 | 2022-07-08 | 贝克顿迪金森公司 | 使用随机引发获得用于免疫组库测序的全长v(d)j信息 |
| EP4090328A4 (en) | 2020-01-13 | 2024-02-14 | Fluent Biosciences Inc | EMULSION-BASED ACTIVE SCREENING |
| WO2021146207A1 (en) | 2020-01-13 | 2021-07-22 | Becton, Dickinson And Company | Methods and compositions for quantitation of proteins and rna |
| CN115768558A (zh) | 2020-01-13 | 2023-03-07 | 福路伦特生物科学公司 | 用于单细胞基因谱分析的方法和系统 |
| CN115698282A (zh) | 2020-01-13 | 2023-02-03 | 福路伦特生物科学公司 | 单细胞测序 |
| JP2023511440A (ja) | 2020-01-22 | 2023-03-17 | アトレカ インコーポレイテッド | 複数の転写産物のハイスループット連結 |
| EP4121016A1 (en) | 2020-03-16 | 2023-01-25 | Fluent Biosciences Inc. | Multi-omic analysis in monodisperse droplets |
| WO2021231779A1 (en) | 2020-05-14 | 2021-11-18 | Becton, Dickinson And Company | Primers for immune repertoire profiling |
| US11932901B2 (en) | 2020-07-13 | 2024-03-19 | Becton, Dickinson And Company | Target enrichment using nucleic acid probes for scRNAseq |
| CN116635533A (zh) | 2020-11-20 | 2023-08-22 | 贝克顿迪金森公司 | 高表达的蛋白和低表达的蛋白的谱分析 |
| MX2023007257A (es) * | 2020-12-18 | 2023-07-03 | Ionis Pharmaceuticals Inc | Compuestos y metodos para modular el factor xii. |
| JP2023554399A (ja) * | 2020-12-22 | 2023-12-27 | ハイファイバイオ エスアエス | マイクロ流体方法及びシステム |
| WO2022182682A1 (en) | 2021-02-23 | 2022-09-01 | 10X Genomics, Inc. | Probe-based analysis of nucleic acids and proteins |
| WO2023091884A1 (en) * | 2021-11-16 | 2023-05-25 | Pioneer Hi-Bred International, Inc. | Maize event das-01131-3 and methods for detection thereof |
| EP4353825A1 (en) * | 2022-10-10 | 2024-04-17 | bisy GmbH | Modified promoter sequences |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2036989A1 (en) * | 2007-09-12 | 2009-03-18 | Institut Pasteur | Single cell based reporter assay to monitor gene expression patterns with high spatio-temporal resolution |
| WO2013134261A1 (en) * | 2012-03-05 | 2013-09-12 | President And Fellows Of Harvard College | Systems and methods for epigenetic sequencing |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US5624711A (en) * | 1995-04-27 | 1997-04-29 | Affymax Technologies, N.V. | Derivatization of solid supports and methods for oligomer synthesis |
| US5928906A (en) | 1996-05-09 | 1999-07-27 | Sequenom, Inc. | Process for direct sequencing during template amplification |
| WO2002068104A1 (en) | 2001-02-23 | 2002-09-06 | Japan Science And Technology Corporation | Process for producing emulsion and microcapsules and apparatus therefor |
| US7198897B2 (en) | 2001-12-19 | 2007-04-03 | Brandeis University | Late-PCR |
| US7976779B2 (en) * | 2002-06-26 | 2011-07-12 | California Institute Of Technology | Integrated LC-ESI on a chip |
| EP1636337A4 (en) | 2003-06-20 | 2007-07-04 | Illumina Inc | METHODS AND COMPOSITIONS USEFUL FOR THE AMPLIFICATION AND GENOTYPING OF THE GENOME |
| TWI333977B (en) | 2003-09-18 | 2010-12-01 | Symphogen As | Method for linking sequences of interest |
| EP1757357B1 (en) | 2004-03-23 | 2013-04-24 | Japan Science and Technology Agency | Method and device for producing micro-droplets |
| JOP20080381B1 (ar) | 2007-08-23 | 2023-03-28 | Amgen Inc | بروتينات مرتبطة بمولدات مضادات تتفاعل مع بروبروتين كونفيرتاز سيتيليزين ككسين من النوع 9 (pcsk9) |
| US20100179075A1 (en) * | 2008-07-30 | 2010-07-15 | Life Technologies Corporation | Particles for use in supported nucleic acid ligation and detection sequencing |
| US9625454B2 (en) | 2009-09-04 | 2017-04-18 | The Research Foundation For The State University Of New York | Rapid and continuous analyte processing in droplet microfluidic devices |
| EP2420579A1 (en) * | 2010-08-17 | 2012-02-22 | QIAGEN GmbH | Helicase dependent isothermal amplification using nicking enzymes |
| US8883421B2 (en) * | 2010-09-10 | 2014-11-11 | New England Biolabs, Inc. | Method for reducing adapter-dimer formation |
| US10392726B2 (en) | 2010-10-08 | 2019-08-27 | President And Fellows Of Harvard College | High-throughput immune sequencing |
| CA2814049C (en) * | 2010-10-08 | 2021-07-13 | President And Fellows Of Harvard College | High-throughput single cell barcoding |
| EP2652155B1 (en) | 2010-12-16 | 2016-11-16 | Gigagen, Inc. | Methods for massively parallel analysis of nucleic acids in single cells |
| EP2675819B1 (en) * | 2011-02-18 | 2020-04-08 | Bio-Rad Laboratories, Inc. | Compositions and methods for molecular labeling |
| WO2012129363A2 (en) | 2011-03-24 | 2012-09-27 | President And Fellows Of Harvard College | Single cell nucleic acid detection and analysis |
| US20140141442A1 (en) * | 2011-04-05 | 2014-05-22 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Linear dna amplification |
| SI2702146T1 (sl) | 2011-04-28 | 2019-06-28 | The Board Of Trustees Of The Leland Stanford Junior University | Identifikacija polinukleotidov, povezanih z vzorcem |
| US20150125865A1 (en) | 2011-12-23 | 2015-05-07 | Gigagen, Inc. | Methods And Apparatuses For Droplet Mixing |
| EP2817418B1 (en) | 2012-02-24 | 2017-10-11 | Raindance Technologies, Inc. | Labeling and sample preparation for sequencing |
| NO2694769T3 (zh) * | 2012-03-06 | 2018-03-03 | ||
| SG11201407901PA (en) * | 2012-05-21 | 2015-01-29 | Fluidigm Corp | Single-particle analysis of particle populations |
| JP6558830B2 (ja) | 2012-06-15 | 2019-08-14 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | 複数転写産物のハイスループットシークエンシング |
| WO2014108850A2 (en) * | 2013-01-09 | 2014-07-17 | Yeda Research And Development Co. Ltd. | High throughput transcriptome analysis |
| CA2921603C (en) * | 2013-08-23 | 2019-11-05 | Ludwig Institute For Cancer Research Ltd | Methods and compositions for cdna synthesis and single-cell transcriptome profiling using template switching reaction |
| GB201317301D0 (en) * | 2013-09-30 | 2013-11-13 | Linnarsson Sten | Method for capturing and encoding nucleic acid from a plurality of single cells |
| ES2912183T3 (es) | 2013-12-30 | 2022-05-24 | Atreca Inc | Análisis de ácidos nucleicos asociados a células individuales utilizando códigos de barras de ácidos nucleicos |
| US20150298091A1 (en) * | 2014-04-21 | 2015-10-22 | President And Fellows Of Harvard College | Systems and methods for barcoding nucleic acids |
-
2014
- 2014-12-30 ES ES14830928T patent/ES2912183T3/es active Active
- 2014-12-30 DK DK14830928.9T patent/DK3089822T3/da active
- 2014-12-30 CN CN201480076617.XA patent/CN106460033B/zh active Active
- 2014-12-30 EP EP14830928.9A patent/EP3089822B1/en active Active
- 2014-12-30 CN CN202111683991.4A patent/CN114717291A/zh active Pending
- 2014-12-30 SG SG11201605344YA patent/SG11201605344YA/en unknown
- 2014-12-30 PT PT148309289T patent/PT3089822T/pt unknown
- 2014-12-30 SG SG10201807112XA patent/SG10201807112XA/en unknown
- 2014-12-30 US US14/586,857 patent/US9580736B2/en active Active
- 2014-12-30 PL PL14830928.9T patent/PL3089822T3/pl unknown
- 2014-12-30 KR KR1020167020514A patent/KR102433825B1/ko active IP Right Grant
- 2014-12-30 WO PCT/US2014/072898 patent/WO2015103339A1/en active Application Filing
- 2014-12-30 JP JP2016544526A patent/JP6608368B2/ja active Active
- 2014-12-30 AU AU2014373757A patent/AU2014373757B2/en active Active
- 2014-12-30 CA CA2935122A patent/CA2935122C/en active Active
- 2014-12-30 EP EP22165189.6A patent/EP4094834A1/en active Pending
- 2014-12-30 KR KR1020227028110A patent/KR20220119751A/ko not_active Application Discontinuation
-
2017
- 2017-02-08 US US15/428,064 patent/US10316345B2/en active Active
-
2019
- 2019-05-03 US US16/402,626 patent/US20200123582A1/en active Pending
-
2022
- 2022-04-08 US US17/716,617 patent/US20220243240A1/en active Pending
- 2022-06-17 US US17/842,966 patent/US20220389471A1/en active Pending
- 2022-06-17 US US17/842,968 patent/US20220389472A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2036989A1 (en) * | 2007-09-12 | 2009-03-18 | Institut Pasteur | Single cell based reporter assay to monitor gene expression patterns with high spatio-temporal resolution |
| WO2013134261A1 (en) * | 2012-03-05 | 2013-09-12 | President And Fellows Of Harvard College | Systems and methods for epigenetic sequencing |
Also Published As
| Publication number | Publication date |
|---|---|
| US20220243240A1 (en) | 2022-08-04 |
| US20220389472A1 (en) | 2022-12-08 |
| US20170369921A1 (en) | 2017-12-28 |
| US20200123582A1 (en) | 2020-04-23 |
| CN106460033A (zh) | 2017-02-22 |
| WO2015103339A1 (en) | 2015-07-09 |
| AU2014373757A1 (en) | 2016-07-07 |
| DK3089822T3 (da) | 2022-05-02 |
| JP6608368B2 (ja) | 2019-11-20 |
| ES2912183T3 (es) | 2022-05-24 |
| US9580736B2 (en) | 2017-02-28 |
| SG11201605344YA (en) | 2016-07-28 |
| AU2014373757B2 (en) | 2019-12-12 |
| US20150329891A1 (en) | 2015-11-19 |
| CA2935122A1 (en) | 2015-07-09 |
| EP3089822A1 (en) | 2016-11-09 |
| US10316345B2 (en) | 2019-06-11 |
| KR102433825B1 (ko) | 2022-08-31 |
| KR20160108377A (ko) | 2016-09-19 |
| PL3089822T3 (pl) | 2022-09-19 |
| SG10201807112XA (en) | 2018-09-27 |
| CA2935122C (en) | 2023-09-19 |
| CN114717291A (zh) | 2022-07-08 |
| EP3089822B1 (en) | 2022-04-06 |
| US20220389471A1 (en) | 2022-12-08 |
| KR20220119751A (ko) | 2022-08-30 |
| EP4094834A1 (en) | 2022-11-30 |
| JP2017506877A (ja) | 2017-03-16 |
| PT3089822T (pt) | 2022-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN106460033B (zh) | 使用核酸条形码分析与单细胞缔合的核酸 | |
| US11591652B2 (en) | System and methods for massively parallel analysis of nucleic acids in single cells | |
| US20220155319A1 (en) | Use of nanoexpression to interrogate antibody repertoires | |
| US20220325275A1 (en) | Methods of Barcoding Nucleic Acid for Detection and Sequencing | |
| EP3615683B1 (en) | Methods for linking polynucleotides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |