CN1058503C - 一种α-烯烃聚合物的制备方法 - Google Patents
一种α-烯烃聚合物的制备方法 Download PDFInfo
- Publication number
- CN1058503C CN1058503C CN 90103481 CN90103481A CN1058503C CN 1058503 C CN1058503 C CN 1058503C CN 90103481 CN90103481 CN 90103481 CN 90103481 A CN90103481 A CN 90103481A CN 1058503 C CN1058503 C CN 1058503C
- Authority
- CN
- China
- Prior art keywords
- olefin
- alpha
- generated
- peak
- main
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images








Landscapes
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
一种具有新颖结构的聚α-烯烃,它是一种含有4个或更多个碳原子的α-烯烃的加成聚合物,其中:(a)在1,2,4-三氯苯中测量而得到的碳-13核磁共振谱中,对由直接键合到主链上的侧链亚甲基团生成的波蜂进行分析可以看出(i)一个不是由其等规立构结构生成的新的主波峰位于由其等规立构结构生成的波峰的较高磁场一侧,和(ii)所说的主波峰的强度相对于由直接键合到主链上的侧链亚甲基团生成的所有波峰强度总和之比值为等于或大于0.3,以及(b)由凝胶渗透色谱分析法测量的、并按聚丙烯计算的分子量、不低于1000。
Description
本发明涉及一种新颖的立体结构的聚α-烯烃。
迄今为止,已经知道聚α-烯烃的立体构形有等规立构结构和无规立构结构。然而,就那些具有间规立构结构的聚合物来说,已经披露的仅有立构规正度较低的间规聚丙烯和间规聚苯乙烯(日本公开特许第187708/87号等)、这些间规聚合物的加氢产品(日本公开特许第131263/89号)等等。另一方面,J.A.Ewen等人已经透露了一种使用包含过渡金属化合物(带有一个不对称配位体和一个铝氧烷)的催化剂制备聚丙烯的方法,该方法的特点在于每单位量过渡金属的高活性以及所生成的聚合物具有高的间同规正度(J.Am.Chem.Soc,1988,110,6255-6256)。
迄今尚不知道有4个或更多碳原子的α-烯烃聚合物不是等规立构或无规立构结构而是具有新的立体规整性的。但是,如果可以得到这些具有新的立体规整性的聚合物的话,人们可以期望将它们用于各种基于其立构有规性的应用中。
本发明人对新颖的有规立构的聚α-烯烃作了深入细致的研究导致完成本发明。
本发明提供一种新颖结构的聚α-烯烃,它是4个或更多碳原子的α-烯烃的加成聚合物,其中:(a)在碳-13核磁共振谱(由在1,2,4-三氯苯中测量而得到的)中,对由直接键合到主链上的侧链亚甲基团生成的波峰进行分析,可以看出(I)一个新的不是由其等规立构结构生成的主波峰,它处于由等规立构结构所生成的波峰的较高磁场的一侧,和(ii)所说主波峰的强度为相对于直接键合到主链上的侧链亚甲基团所生成的所有波峰强度总和的0.3或更大些;和(b)通过凝胶渗透色谱法测定并按聚丙烯计算的分子量不低于1000。
碳-13核磁共振谱是通过量测聚合物的1,2,4-三氯苯溶液,以四甲基硅烷为原点而获得的。在由直接键合到主链上的侧链亚甲基团生成的波峰中,上述的主波峰指出了间规键合的吸收峰,因而,主波峰的强度相对于直接键合到主链上的侧链亚甲基团所生成的所有波峰强度总和之比值,可以作为代表本发明聚α-烯烃的间同规正度的量度。谱中波峰的分布是根据C.P.Lindeman等人的文献:Anal.Chem.,Vol.43,1245(1971)。
虽然本发明的聚α-烯烃可以单独使用,但是它特别适用于作为其他热塑性树脂的改性剂。
由于它们的新颖的立体规整性,本发明的聚α-烯烃可以期望用于基于它们的立构有规性的新应用中,因而极有工业价值。
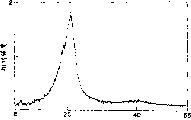
图1是间规聚丁烯-1的红外光谱。在图2中,(a)是间规聚戊烯-1的碳-13核磁共振谱,而(b)是等规聚戊烯-1的碳-13核磁共振谱。图3是间规聚戊烯-1的X-射线衍射谱的例子。图4(a)是间规聚已烯-1的碳-13核磁共振谱,而(b)是表示等规聚已烯-1的碳-13核磁共振谱。图5表示间规已烯-1的红外吸收光谱,而图6所给出的是等规聚已烯-1的红外吸收光谱。在图7中,(a)表示间规聚辛烯-1的碳-13核磁共振谱,而(b)是等规聚辛烯-1的碳-13核磁共振谱。图8是间规聚辛烯-1的红外吸收光谱,而图9表示等规聚辛烯-1的红外吸收光谱。在图10中,(a)表示间规聚十六碳烯-1的碳-13核磁共振谱。而(b)给出等规聚十六碳烯-1的核磁共振谱。在图11中,(a)是间规聚十六碳烯-1的X-射线衍射谱,而(b)给出的是等规聚十六碳烯-1的X-射线衍射谱。图12(a)是间规聚十八碳烯-1的碳-13核磁共振谱,而(b)表示等规聚十八碳烯-1的碳-13核磁共振谱。在图13中,(a)给出的是间规聚十八碳烯-1的X-射线衍射谱,而(b)是等规聚十八碳烯-1的X-射线衍射谱。
本发明的聚α-烯烃被假定为主要是间规立构结构。对其在碳-13核磁区振谱中由键合到主链上的侧链亚甲基团所生成的波峰的研究,特征性地表明一个不是由等规立构结构生成的主波峰位于由等规立构结构所生成的波峰的较高磁场的一侧,而且间规的立体规整性是如此之高,以致此主波峰的强度相对于由直接键合到侧链亚甲基团而生成的所有波峰的强度总和之比值为0.3或0.3以上,最好为0.5或0.5以上。此强度比值少于0.3的低立体规整性的聚α-烯烃不属于本发明的范围。
为了使本发明的聚合物能显示出它的特性,其分子量必须为1000或1000以上(由凝胶渗透色谱法测量并按聚丙烯计算的)。低于该分子量值的聚合物是没有用的。分子量的测量,例如是通过凝胶渗透色谱法在135℃的1,2,4-三氯苯溶液中进行的,并按聚丙烯计算出其数均分子量。
以下将阐述并举例说明本发明的聚α-烯烃的制备方法。
用于制备本发明的新颖的有规立构的聚α-烯烃的α-烯烃聚合反应催化剂是一种包含有一种过渡金属化合物的催化剂,它带有一个不对称和相互键合的配位体(一种终端相互键合的不对称的配位体)。带有一个不对称和相互键合的配位体的过渡化合物是一种带有如通式A-B-C所示的配位体的化合物,其中A和C各自为一个不同的芳族或环不饱和基团,而B是一个含有1-20个碳原子的亚烷基或亚环烷基,该过渡金属化合物可用下面的通式表示: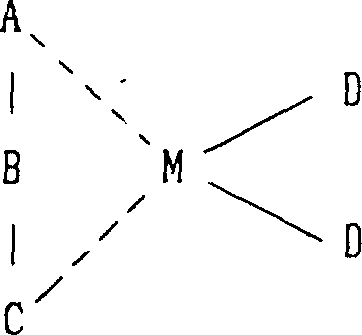 其中A、B和C如上所述,M表示钛、锆或铪,而D为含1-20个碳原子的烷基或卤原子。芳族或环不饱和基团的优选例子可包括:环戊二烯基、茚基和芴基,以及这些基团的环取代产物的基团,而含有一侧为环戊二烯基团、而另一侧为茚基或芴基或它们的环取代产物基团的配位体可被引以为例。特别是在J.A.Ewen等人写的文献(J.Am.Chem.Soc.1988,110,6255-6256)中所例举的化合物,如异丙基(环戊二烯基-1-芴基)铪二氯化物和异丙基(环戊二烯基-1-芴基)锆二氯化物可被优选作为过渡金属化合物。通常在聚合过程中结合使用一种铝氧烷。铝氧烷是通式为:
其中A、B和C如上所述,M表示钛、锆或铪,而D为含1-20个碳原子的烷基或卤原子。芳族或环不饱和基团的优选例子可包括:环戊二烯基、茚基和芴基,以及这些基团的环取代产物的基团,而含有一侧为环戊二烯基团、而另一侧为茚基或芴基或它们的环取代产物基团的配位体可被引以为例。特别是在J.A.Ewen等人写的文献(J.Am.Chem.Soc.1988,110,6255-6256)中所例举的化合物,如异丙基(环戊二烯基-1-芴基)铪二氯化物和异丙基(环戊二烯基-1-芴基)锆二氯化物可被优选作为过渡金属化合物。通常在聚合过程中结合使用一种铝氧烷。铝氧烷是通式为: 的化合物,其中R是含1-3个碳原子的烃残基。其中R为甲基(例如甲基铝氧烷)和n为5或5以上、最好n为10-100的化合物特别被优先使用。所使用的铝氧烷的比例,相对于上述的过渡金属化合物,为10-1000000摩尔倍,通常为50-5000摩尔倍。除该催化剂体系外也可以使用如下的催化剂体系,即它含有上述的过渡金属化合物(其中至少有一个D基团为烷基)并与如日本公开特许第501950/89号和第502036/89号中所示的硼化合物结合使用。
的化合物,其中R是含1-3个碳原子的烃残基。其中R为甲基(例如甲基铝氧烷)和n为5或5以上、最好n为10-100的化合物特别被优先使用。所使用的铝氧烷的比例,相对于上述的过渡金属化合物,为10-1000000摩尔倍,通常为50-5000摩尔倍。除该催化剂体系外也可以使用如下的催化剂体系,即它含有上述的过渡金属化合物(其中至少有一个D基团为烷基)并与如日本公开特许第501950/89号和第502036/89号中所示的硼化合物结合使用。
在制备本发明聚合物中使用的α-烯烃为含有4-20个碳原子的烯烃,其通式为CH2=CH-CH2-CH2-R′,其中R′为氢原子或含1-16个碳原子的烷基。所用的α-烯烃的特例有丁烯-1、戊烯-1、已烯-1、庚烯-1、辛烯-1、壬烯-1、癸烯-1、十一碳烯-1、十二碳烯-1、十三碳烯-1、十五碳烯-1、十六碳烯-1、十七碳烯-1,十八碳烯-1和5-甲基己烯-1,等等。
对于由α-烯烃形成其聚合物的条件未提出特别的限制。因此,各种各样的聚合工艺均可使用,包括使用惰性溶剂的溶液聚合工艺,在基本上无惰性生溶剂存在下使用α-烯烃作聚合介质的本体聚合工艺,以及气相聚合工艺。聚合过程通常是在-100°到200℃的温度和在常压到20公斤/厘米2表压的压力下进行的。优选的温度为-100°到100℃、压力为从常压到10公斤/厘米2表压。
本发明的聚α-烯烃不仅可以包括上述α-烯烃的均聚物,而且也可包括其与少量(例如,10%摩尔或更少的)其它的α-烯烃的共聚物,就其立体构形而言,在碳-13核磁共振谱中显示出特殊的测定结果,正如附加的权利要求书中所述的。
本发明将通过参考下列各实施例而被进一步加以说明。实施例1
在内容积为300毫升的高压釜中装有20克的丁烯-1,加入5毫克的异丙基(环戊二烯基-1-芴基)锆二氯化物和0.34克的甲基铝氧烷(其聚合度约为15)。异丙基(环戊二烯基-1-芴基)锆二氯化物是按下述方法预先制得的,即将锂加入到异丙基环戊二烯基-1-芴中按通常的方法进行合成,然后将所生成的化合物与四氯化锆进行反应;而甲基铝氧烷是由硫酸铜六水合物与三甲基铝在甲苯中进行反应而预先制得的。所得的混合物在25℃下进行聚合反应5个小时,尔后将未反应的丁烯-1通过蒸发除去。将蒸发的残留物溶于50毫升的已烷中,向该溶液加入500毫升的甲醇得到7.5克作为不溶性沉淀物的聚丁烯-1。用GPC-150C凝胶渗透色谱仪(由WatersLtd.制造)在135℃用其1,2,4-三氯苯溶液进行测量,并按聚丙烯计算,其数均分子量和重均分子量分别为14500和30000。在135℃下,在1,2,3,4-四氢化萘溶液中测得的特性粘度为0.17。用其1,2,4-三氯苯溶液测量其碳-13核磁共振谱(以0PPm四甲基硅烷为基准),作为其结果,在40.7,35.0,26.9和10.7PPm处发现4个吸收波峰。
在由直接键合到主链上的测链CH2基团生成的吸收波峰中,由间规五价键在26.9PPm处生成的吸收波峰的强度相对于在整个侧链中由直接键合到主链上的CH2基团生成的吸收波峰强度总和之比值为0.89。红外吸收光谱的测量结果如图1所示。
在根据A.Zammbelli等人的方法(Macromolecules 1987,Vol.20,1015)而合成的等规聚丁烯-1的碳-13核磁共振谱中,各波峰是以上述相同的方法在40.3、35.1、27.7和10.7PPm处而被观察到的。碳-13核磁共振谱的指定是以T.Asakura等人的文献为基础的[(Macromolecules Vol.16,786(1983)]。实施例2
在容量为300毫升的烧瓶中装有20克戊烯-1,加入5毫克实施例1中所用的异丙基(环戊二烯基-1-芴基)锆二氯化物和0.34克甲基铝氧烷(其聚合度约为16,由Toso Akuzo K.K.制造)。该混合物在5℃下进行聚合反应2个小时,然后加入大量的甲醇,获得8.2克聚戊烯-1(以甲醇不溶物形式出现)。以实施例1相同的方法对其进行分析,得出其数均分子量为25000、重均分子量为50000(按聚丙烯计算)。在135℃下,在1,2,3,4-四氢化萘溶液中测得的特性粘度为0.52。在以同样方式获得的碳-13核磁共振谱中,在大约14.9、19.9、33.5、37.5和42.0PPm处观察到5个主波峰。
在由直接键合到主链上的侧链CH2基生成的吸收蜂中,由间规键在37.5PPm处生成的吸收峰的强度相对于由直接键合到侧链CH2基团生成的吸收波峰强度总和之比值为0.72。碳-13核磁共振吸收波谱的测量结果在图2(a)所示,而其X-射线衍射谱(Cu-Kα)的测量结果如图3所示。
按照同样的步骤,通过1,2-亚乙基二茚基锆二氯化物的催化聚合可以得到6.8克的等规聚戊烯-1。以同样方式进行其碳-13核磁共振谱测定,其结果为在大约14.6、19.9、33.5、38.1和41.4PPm处发现5个吸收峰,这些值与P.Localelli等人所示的,因不同的基质而修正其偏移值后所得值是相一致的(Makromol.Chem.RapidCommun,VOl.5,495-499)。碳-13核磁共振吸收谱的测量结果如图2(b)所示。实施例3
在容量为300毫升的烧瓶中装有100克已烯-1,加入与实施例1所用的同样数量的同种催化剂。在25℃下,搅拌该混合物,使之聚合5个小时。用与实施例1相同的方法处理所生成的混合物而得到聚合物。以同样的方法对由此获得的聚合物进行测量,发现其数均分子量为35000、重均分子量为64000(按聚丙烯计算)。在碳-13核磁共振法测量结果中,在大约14.1、23.5、29.2、33.8、34.7和42.1PPm处观察到六个主吸收峰。
在由直接键合到主链上的侧链CH2基团生成的吸收波峰中,间规键在34.7PPm处吸收波峰的强度相对于由直接键合到主链上的侧链CH2基团生成的所用吸收波峰强度总和之比值为0.65。碳-13核磁共振吸收波谱的测量结果如图4(a)所示,而其红外吸收光谱的测量结果如图5所示。
按照同样的步骤,由1,2-亚乙基二茚基锆二氯化物经催化聚合可得到等规聚已烯-1。用同样的方法,测量其碳-13核磁共振吸收谱,在大约14.0、23.5、29.2、33.6、35.3和41.4PPm处观察到6个吸收峰。碳-13核磁共振法的测量结果如图4(b)所示,而其红外吸收光谱的测量结果如图6所示。实施例4
用与实施例2相同的方法,只是用100克辛烯-1替代戊烯-1,并在25℃下聚合4个小时,获得47.7克的聚合物。用同样的方法对聚合物进行分析,发现其数均分子量为29600、重均分子量为62100(按聚丙烯计算)。根据碳-13核磁共振谱分析,在大约13.9、22.9、27.0、30.2、32.2、33.8、35.0和42.2PPm处观察到8个吸收峰。
在由直接键合到主链上的侧链CH2基团生成的吸收波峰中,间规键在35.0PPm处吸收波峰强度相对于由所有侧链中的直接键合到主链上的CH2基生成的所有吸收波峰强度总和之比值为0.67。碳-13核磁共振法测量结果如图7(a)所示,而其红外吸收光谱的测量结果如图8所示。
按照同样步骤,由1,2-亚乙基二茚基锆二氯化合物的催化聚合得到33.2克的等规聚辛烯-1。同样的方法测量其碳-13核磁共振谱,在大约13.9、22.8、27.0、30.2、32.2、33.6、35.7和42.4PPm处观察到8个吸收峰。
碳-13核磁共振谱的测量结果和红外吸收光谱的测量结果分别示于图7(b)和图9中。实施例5
用实施例2同样的方法,只是用40克十六碳烯-1代替代戊烯-1,并在30℃下聚合7个小时,得到4.7克聚十六碳烯-1。用同样方法对聚合物进行分析,可知其数均分子量为15000、其重均分子量为30000(按聚丙烯计算)。在碳-13核磁共振谱中,在大约14.0、22.8、27.1、30.7、32.1、33.8、35.1和42.3PPm处发现8个很好区分开的波峰,但在29.5到30.2PPm处所看到的波峰很难区分开。根据差示扫描热法分析,该聚合物在140℃下熔化,然后温度以10℃/分的速度降低,在这种情况下,所测的结晶温度按其峰值温度为12.7℃、而在升温过程中的熔点为19.7℃和40.0℃。
在由直接键合到主链上的侧链CH2基团生成的吸收波峰中,间规键在35.1PPm处吸收波峰的强度相对于由直接键合到主链上的侧链CH2基生成的所有吸收波峰强度总和之比值为0.66。碳-13核磁共振吸收谱的测量结果和X-射线衍射谱(Cu-Kα)的测量结果分别于图10(a)和图11(a)中给出。
按照同样的步骤,通过1,2-亚乙基二茚基锆二氯化物的催化聚合得到32.8克的等规聚十六碳烯-1。用同样的方法测量其碳-13核磁共振谱,其结果在大约14.0、22.8、27.1、30.7、32.1、33.8、35.7和41.4PPm处观察到清楚区分开的8个波峰,但在29.5到30.0PPm处所看到的波峰很难区分开。用同样的方法测得结晶温度为30.2℃、熔点为52.0℃和57.3℃。碳-13核磁共振谱的测量结果如图10(b)所示,而X-射线衍射谱(Cu-Kα)的测量结果在图11(b)中给出。实施例6
按照实施例2同样的方法,只是用40克十八碳烯-1替代戊烯一1,并在30℃下聚合7个小时,得到3.6克聚十八碳烯-1。用相同方法对聚合物进行分析,得出其数均分子量为16000、其重均分子量为32000(按聚丙烯计算)。在碳-13核磁共振谱中,在大约13.9、22.8、27.0、30.7、32.4、33.8、35.1和42.3PPm处观察到很好分开的8个波峰,但在29.2到30.2PPm处所看到的波峰很难区分开。根据差示扫描热法分析,该聚合物在140℃熔化,然后以10℃/分的速率降低温度,在这种情况下,所测得的结晶温度(其峰值温度)为22.3℃和12.0℃、而在升温过程度中的熔点为15.9℃和31.9℃。
在由直接键合到主链上的侧链CH2基生成的吸收波峰中,间规键在35.1PPm处的波峰强度相对于由直接键合到主链上的所有侧链CH2基团生成的吸收波峰强度总和之比值为0.68。碳-13核磁共振吸收谱的测量结果如图12(a)所示;而其X-射线衍射谱(Cu-Kα)的测量结果在图13(a)中给出。
按照同样的步骤,通过1,2-亚乙基二茚基锆二氯化物的催化聚合得到33.9克等规聚十八碳烯-1。用同样的方法测量其碳-13核磁共振谱,其结果在大约14.0、22.8、27.1、30.8、32.1、33.5、35.7和41.4PPm观察到很好分开的8个波峰,但在29.5到30.0PPm处所看到的波蜂很难区分开。用相同的方法测得结晶温度约为36.3℃、而熔点为26.7℃和62.0℃。碳-13核磁共振谱的测量结果和X-射线衍射谱(Cu-Kα)的测量结果分别示于图12(b)和图13(b)中。实施例7
将锂加入到异丙基环戊二烯基-1-芴中,并使生成的化合物与四氯化铪(一种由四氯化锆和四氯化铪以1.5锆/98.5铪的重量比而组成的混合物,它替代了四氯化锆)反应,而合成异丙基(环戊二烯基-1-芴基)铪二氯化物。然后,除了使用异丙基(环戊二烯基-1-芴基)铪二氯化物替代丙基(环戊二烯基-1-芴基)锆二氯化物外,按照实施例1的步骤进行,生成了0.8克聚丁烯-1。用同样的方法进行分析,发现其数均分子量为29000、其重均分子量为114000(按聚丙烯计算)。在135℃下,在1,2,3,4-四氢化萘溶液中测得的特性粘度为0.47。根据碳-13核磁共振法分析,间规五价键吸收波峰的强度相对于由直接键合到主链上的所有侧链CH2基团生成的吸收波峰强度总和之比值为0.72。
Claims (9)
1.一种α-烯烃聚合物的制备方法,所述α-烯烃聚合物是一种含有4个或更多个碳原子的α-烯烃的加成聚合物,其中(a)在1,2,4-三氯苯中测量而得的碳-13核磁共振谱中,对由直接键合到主链上的侧链亚甲基团生成的波峰进行分析可以看出(i)一个不是由其等规立构结构生成的主波峰位于由其等规立构结构生成的波峰的较高磁场的一侧,和(ii)所说的主波峰的强度相对于由直接键合到主链上的侧链亚甲基团生成的所有波峰强度总和之比值为等于或大于0.3,以及(b)由凝胶渗透色谱分析法测量的、并按聚丙烯折算的分子量不低于1000,所述α-烯烃聚合物的制备是在一种催化剂存在下于-100℃至200℃的温度和常压至20kg/cm2表压的压力下通过聚合所述α-烯烃而成,所述催化剂包含具有不对称和相互键接的配位体的过渡金属化合物,该过渡金属化合物具有不对称的且相互键合的配位体,是一种具有通式A-B-C代表的配位体的化合物,其中A和C各自为不同的芳族或环状不饱和基团,B是含有1-20个碳原子的亚烷基或亚环烷基,该过渡金属化合物由下面的通式表示: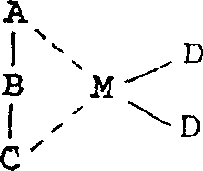 式中A、B和C的定义同上,M代表钛、锆或铪,D为具有1-20个碳原子的烷基或卤原子。
式中A、B和C的定义同上,M代表钛、锆或铪,D为具有1-20个碳原子的烷基或卤原子。
2.根据权利要求1的方法,其中α-烯烃为丁烯-1、戊烯-1、已烯-1、庚烯-1、辛烯-1、壬烯-1、癸烯-1、十一碳烯-1、十二碳烯-1、十三碳烯-1、十五碳烯-1、十六碳烯-1、十七碳烯-1或十八碳烯-1。
3.根据权利要求2的方法,其中在碳-13核磁共振谱中由直接键合到主链上的侧链亚甲基生成的各波峰中,所述的主波峰强度的相对于由直接键合到主链上的侧链亚甲基团生成的所有波峰强度总和之比值等于或大于0.5。
4.根据权利要求2的方法,其中的α-烯烃为十八碳烯-1。
5.根据权利要求3的方法,其中α-烯烃为丁烯-1。
6.根据权利要求3的方法,其中α-烯烃为己烯-1。
7.根据权利要求3的方法,其中α-烯烃为辛烯-1。
8.根据权利要求3的方法,其中α-烯烃为戊烯-1。
9.根据权利要求3的方法,其中α-烯烃为十六碳烯-1。
Applications Claiming Priority (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP141168/1989 | 1989-06-05 | ||
| JP14116889 | 1989-06-05 | ||
| JP141168/89 | 1989-06-05 | ||
| JP156707/89 | 1989-06-21 | ||
| JP156707/1989 | 1989-06-21 | ||
| JP15670789 | 1989-06-21 | ||
| JP228369/1989 | 1989-09-05 | ||
| JP228369/89 | 1989-09-05 | ||
| JP22836989 | 1989-09-05 | ||
| JP262966/1989 | 1989-10-11 | ||
| JP26296689 | 1989-10-11 | ||
| JP262966/89 | 1989-10-11 | ||
| JP278617/1989 | 1989-10-27 | ||
| JP278617/89 | 1989-10-27 | ||
| JP27861789 | 1989-10-27 | ||
| JP145090A JP2944695B2 (ja) | 1989-06-05 | 1990-01-10 | 新規なポリα―オレフィン |
| JP001450/1990 | 1990-01-10 | ||
| JP001450/90 | 1990-01-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1057056A CN1057056A (zh) | 1991-12-18 |
| CN1058503C true CN1058503C (zh) | 2000-11-15 |
Family
ID=27547610
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 90103481 Expired - Fee Related CN1058503C (zh) | 1989-06-05 | 1990-06-05 | 一种α-烯烃聚合物的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN1058503C (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106590853B (zh) * | 2016-10-20 | 2019-07-16 | 广州吉盛润滑科技有限公司 | 一种合成增压甲醇发动机油及其制备方法和应用 |
-
1990
- 1990-06-05 CN CN 90103481 patent/CN1058503C/zh not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN1057056A (zh) | 1991-12-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3307704B2 (ja) | α‐オレフィン重合体の製造法 | |
| Resconi et al. | High-molecular-weight atactic polypropylene from metallocene catalysts. 1. Me2Si (9-Flu) 2ZrX2 (X= Cl, Me) | |
| KR100414759B1 (ko) | 메탈로센 화합물, 이것의 제조 방법, 및 이것의 올레핀중합용 촉매로서의 용도 | |
| DE69127811T3 (de) | Verfahren zur herstellung von olefinpolymeren | |
| US5187250A (en) | Poly-α-olefins | |
| Chien et al. | Homogeneous binary zirconocenium catalyst systems for propylene polymerization. 1. Isotactic/atactic interfacial compatibilized polymers having thermoplastic elastomeric properties | |
| EP0611773B1 (en) | Catalyst component for use in the polymerization of alpha-olefins and process for producing alpha-olefin polymers using the same | |
| KR100746676B1 (ko) | 메탈로센 화합물, 메탈로센 화합물의 제조 방법, 올레핀 중합 촉매, 및 폴리올레핀의 제조 방법 | |
| DE69431222T2 (de) | Übergangsmetallverbindung, und diese enthaltender Polymerisationkatalysator | |
| EP0729978A1 (en) | Polyethylene and method for production thereof | |
| EP0403866A1 (en) | Novel Poly-alfa-olefins | |
| JP3323347B2 (ja) | α‐オレフィンの重合 | |
| JPH10180964A (ja) | 二軸延伸複層フィルム | |
| JP2001163924A (ja) | 分岐を有するプロピレン重合体及びその製造方法 | |
| CN1058503C (zh) | 一种α-烯烃聚合物的制备方法 | |
| JPH11349650A (ja) | ブロック共重合体 | |
| JP2001525801A (ja) | 立体剛性メタロセン化合物 | |
| Sadrtdinova et al. | Heterocene Catalysts for Copolymerization of Hex-1-ene and Polar Vinyl Monomers | |
| JP2000095791A (ja) | 遷移金属化合物、オレフィン重合用触媒成分およびα―オレフィン重合体の製造方法 | |
| JPH11349649A (ja) | プロピレン系ブロック共重合体 | |
| JP2004124044A (ja) | オレフィン重合体の製造方法 | |
| JPH11349634A (ja) | プロピレン重合体 | |
| US5334684A (en) | α-olefin-alkenylsilane copolymer and method for preparing same | |
| JP3197314B2 (ja) | 分子量分布の広いポリオレフィンの製造方法 | |
| KR930002702B1 (ko) | 신규한 폴리 α-올레핀 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |