CN103619902A - 半导体聚合物 - Google Patents
半导体聚合物 Download PDFInfo
- Publication number
- CN103619902A CN103619902A CN201280028860.5A CN201280028860A CN103619902A CN 103619902 A CN103619902 A CN 103619902A CN 201280028860 A CN201280028860 A CN 201280028860A CN 103619902 A CN103619902 A CN 103619902A
- Authority
- CN
- China
- Prior art keywords
- semi
- conducting polymer
- polymer described
- alkyl
- independently
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images










Classifications
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/20—Carbon compounds, e.g. carbon nanotubes or fullerenes
- H10K85/211—Fullerenes, e.g. C60
- H10K85/215—Fullerenes, e.g. C60 comprising substituents, e.g. PCBM
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
- C08G61/126—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds with a five-membered ring containing one sulfur atom in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L65/00—Compositions of macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain; Compositions of derivatives of such polymers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K10/00—Organic devices specially adapted for rectifying, amplifying, oscillating or switching; Organic capacitors or resistors having a potential-jump barrier or a surface barrier
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/111—Organic polymers or oligomers comprising aromatic, heteroaromatic, or aryl chains, e.g. polyaniline, polyphenylene or polyphenylene vinylene
- H10K85/113—Heteroaromatic compounds comprising sulfur or selene, e.g. polythiophene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/151—Copolymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/14—Side-groups
- C08G2261/141—Side-chains having aliphatic units
- C08G2261/1412—Saturated aliphatic units
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/14—Side-groups
- C08G2261/142—Side-chains containing oxygen
- C08G2261/1426—Side-chains containing oxygen containing carboxy groups (COOH) and/or -C(=O)O-moieties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/14—Side-groups
- C08G2261/146—Side-chains containing halogens
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/32—Monomer units or repeat units incorporating structural elements in the main chain incorporating heteroaromatic structural elements in the main chain
- C08G2261/324—Monomer units or repeat units incorporating structural elements in the main chain incorporating heteroaromatic structural elements in the main chain condensed
- C08G2261/3243—Monomer units or repeat units incorporating structural elements in the main chain incorporating heteroaromatic structural elements in the main chain condensed containing one or more sulfur atoms as the only heteroatom, e.g. benzothiophene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/90—Applications
- C08G2261/91—Photovoltaic applications
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K30/00—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation
- H10K30/30—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation comprising bulk heterojunctions, e.g. interpenetrating networks of donor and acceptor material domains
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Nanotechnology (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
- Photovoltaic Devices (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
本发明公开了可用于电光学器件和电子器件中的具有共轭单元的新型半导体光伏聚合物,所述聚合物提供改善的太阳能转换效率。所述聚合物在太阳能器件中表现出增强的太阳能转换效率。
Description
交叉引用相关专利申请并且以引用的方式并入
本专利申请要求提交于2011年4月15日的美国临时专利申请No.61/517,205的优先权,特此将该临时专利申请的全文以引用的方式并入。
政府许可权利
本发明根据美国国家科学基金会(National Science Foundation)和美国空军科学研究处(Air Force Office of Scientific Research)授予的第DMR-1004195和FA9550-09-1-0220号资助在政府支持下完成。政府可享有本发明的某些权利。
技术领域
本发明涉及基于半导体共轭聚合物的半导体聚合物。本发明还涉及它们在电光学器件和电子器件中的用途。
背景技术
本体异质结(bulk heterojunction,BHJ)聚合物太阳能电池与其无机对应物相比具有许多优点,例如低制造成本、易加工性、柔性和制备大面积器件的可能性(Braga等人,Sol.Energy Mater.Sol.Cells(《《太阳能材料与太阳能电池》》),2008,92,418)。然而,主要缺点是它们的功率转换效率(PCE)较低,这严重阻碍了将聚合物太阳能电池进一步推向实际应用。近年来,经过多个方面巨大努力,PCE已得到改善。例如,将镍氧化物和石墨烯氧化物引入作为空穴传输层的候选物以及防止电子从BHJ受体泄漏到阳极,从而取代常规的聚(3,4-乙烯二氧噻吩)(PEDOT)层。将光反射材料沉积在电极与光活性层之间,以接近定量地驱动内量子效率。采用缓慢生长、热退火和混合溶剂来优化双连续互穿网络。一般来讲,聚合物的物理特性决定开路电压(Voc)和短路电流密度(Jsc),并且PCE被定义为Pout/Pin=VocJscFF/Pin。因此,针对开发新型半导体聚合物的研究致力于:1)降低能带隙以增强Jsc;以及2)降低最高占据分子轨道(HOMO)的能级以改善Voc。
最近本体异质结(BHJ)有机光伏材料(OPV)因为它们在通过易行、低成本溶液加工技术来制造柔性、轻质太阳能电池方面的潜能引发了人们高涨的热情。(Thompson,B.C.等人,Angew.Chem.,Int.Ed.(《应用化学国际版》)2008,47,58–77;Gunes,S.等人,Chem.Rev.(《化学评论》)2007,107,1324-1338;Spangaard,H.等人,Sol.Energy Mater.Sol.Cells.(《太阳能材料与太阳能电池》)2004,83,125-146;Hoppe,H.等人,J.Mater.Res.(《材料研究杂志》)2004,19,1924-1945;Brabec,C.J.等人,Adv.Funct.Mater.(《先进功能材料》)2001,11,15-26)。为了使OPV从研发阶段完全成熟为高性价比的产品,新材料是至关重要的。大面积OPV太阳能电池的功率转换效率(PCE)应不断地改善。(G.Dennler,G.等人,J.Adv.Mater.(《先进材料杂志》)2009,21,1323-1338;Scharber,M.等人,Adv.Mater.(《先进材料》)2006,18,789-794;Coakley,K.M.等人,Chem.Mater.(《化学材料》)2004,16,4533-4542)。设计新型聚合物的主要挑战之一是对诸如形态和供体-受体能级匹配之类的物理特性的同时优化。(Cheng,J.-Y.等人,Chem.Rev.(《化学评论》)2009,109,5868-5923;Brabec,C.J.等人,J.Chem.Soc.Rev.(《化学学会评论杂志》)2011,40,1185-1199;
Bundgaard,E.等人,Sol.Energy Mater.Sol.Cells.(《太阳能材料与太阳能电池》)2007,91,954-985)。
除了这些参数之外,当对能带隙、能级和形态均进行优化时,结合大的局部偶极矩也可在电荷分离中发挥重要作用,从而增强OPV电池的性能。(Carsten,B.等人,J.Am.Chem.Soc.(《美国化学会会志》)2011,133,20468-20475)。
对表现出增加的太阳能转换效率的聚合物太阳能电池,本领域存在需要。
发明内容
本文描述的是半导体光伏衍生物,包括聚硒吩衍生物,它们在用于电光学器件和电子器件中时表现出高太阳能转换效率。还存在在电光学器件和电子器件中用作空穴传输材料且以富勒烯衍生物作为受体的半导体聚合物。该聚合物被设计成针对太阳光谱中的宽吸收实现低能带隙。
在一个方面,半导体聚合物具有选自如下的化学式:

其中X1、X2、X3和X4独立地选自O、S、Se、NH和CH2。R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基。Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基。Z1和Z2独立地选自CH和N。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。n为大于0的整数。
在另一方面,半导体聚合物具有含上文定义的取代基的式I-VI中的任一者。
附图说明
图1a示出了CHCl3溶液中的PSeB1的紫外-可见光谱(实线)和PSeB2的紫外-可见光谱(虚线)。图1b示出了PSeB1:PC61BM共混膜的紫外-可见光谱(虚线)、PSeB1:PC71BM共混膜的紫外-可见光谱(点划线)和PSeB2的紫外-可见光谱(实线)。
图2示出了(a)聚合物太阳能电池在AM1.5G条件(100mW cm-2)下的电流-电压特性。(b)聚合物太阳能电池的外部量子效率(EQE)(PTB9,深色方形;PSeB1,方形;PSeB2,三角形;PSeB3,星形)。
图3示出了PSeB1的循环伏安法曲线图。
图4示出了阐释电荷分离动力学中的偶极效应的模型。
图5示出了PTDBD1-PTDBD3的结构。
图6示出了DBD单体和PTDBD聚合物的合成路线,a)Pd(PPh3)4,K2CO3(或NaOH),甲苯/乙醇/H2O,110℃,24小时;b)CF3SO3H,P2O5,3天;c)吡啶,回流,12小时;d)Br2,CHCl3,6小时;e)n-BuLi,THF,-78℃,25分钟;f)Me3SnCl,-78℃,12小时;g)Pd(PPh3)4,甲苯/DMF,120℃,12小时。
图7示出了原始聚合物膜的吸收光谱。
图8示出了由聚合物/PC71BM构成的太阳能电池的电流-电压图(a),以及由聚合物/PC61BM构成的太阳能电池的电流-电压图(b)。
图9示出了PTDBD2的循环伏安图(A),以及PTDBD3的循环伏安图(B)。
图10示出了PTDBD2/PC71BM太阳能电池器件的EQE曲线。
图11示出了在具有一层PEDOT:PSS的硅基板上的净PTDBD2聚合物的2D GIWAXS图(a-b)和净PTDBD3聚合物的2D GIWAXS图(c-d)的扣除背景的qy和qz线切(linecut),它们在图4中以文字呈现。在这里,实线表示使用Pseudo-Voigt1型峰函数的峰的最佳拟合。
具体实施方式
除非另外定义,否则本文使用的所有技术和科学术语均具有本发明所属领域的普通技术人员通常理解的相同含义。当描述本发明的化合物、组合物、方法和工艺时,除非另外指明,否则如下术语具有如下含义。
“烷基”自身或作为另一取代基的一部分是指可以为具有指定碳原子数(即C1-8意指一个至八个碳原子)的直链、环状或支链或它们的组合的烃基。烷基的例子包括甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、仲丁基、环己基、环戊基、(环己基)甲基、环丙基甲基、二环[2.2.1]庚烷、二环[2.2.2]辛烷等。除非另外指明,否则烷基可以为取代的或未取代的。取代的烷基的例子包括卤代烷基、多卤代烷基(如多氟烷基和多氯烷基)、氨基烷基等。烷基还包括直链和支链烷基。
“烷氧基”是指-O-烷基。烷氧基的例子包括甲氧基、乙氧基、正丙氧基等。
“芳基”是指具有单个环(单环)或可稠合在一起或共价连接的多个环(二环)的多不饱和芳族烃基。具有6-10个碳原子的芳基是优选的,其中该碳原子数可通过例如C6-10指定。芳基的例子包括苯基和萘-1-基、萘-2-基、联苯基等。除非另外指明,芳基可以是取代的或未取代的。“芳氧基”是指-O-芳基;而“杂芳氧基”是指-O-杂芳基。
术语“氨基”是指-NRR’,其中R和R’独立地选自氢、烷基、芳基、芳烷基和脂环烃,它们中除氢之外所有均任选被取代。R和R’均可形成环状环系。环系可为5-7元,并且可任选与另一环基(包括环烷基、芳基和杂芳基)稠合。
“氰基”是指-CN。
“酯”是指R’C(=O)O-,其中R’为氢原子、烷基、芳基和芳基杂环,如本文所定义的。“芳基杂环”是指由如本文所定义的芳环构成的二环或三环,所述芳环通过芳环的两个相邻碳原子附接到杂环。示例性芳基杂环包括二氢吲哚、1,2,3,4-四氢喹啉等。
“酮”是指R’C(=O)-,其中R’为氢原子、烷基、芳基和芳基杂环。仅出于定义的目的,本文所述的酮包括醛。
“卤代”或“卤素”自身或作为取代基的一部分是指氯、溴、碘或氟原子。
如本文所用,术语“杂芳基”是指具有五至十个环原子的单环、二环或三环芳基或芳环,在所述环原子之中,至少一个环原子选自S、Se、O和N;零、一或两个环原子为独立地选自S、Se、O和N的另外的杂原子;并且其余的环原子为碳,其中环内所含的任何N或S可任选被氧化。杂芳基包括但不限于吡啶基、吡嗪基、嘧啶基、吡咯基、吡唑基、咪唑基、噻唑基、 唑基、异
唑基、异 唑基、噻二唑基、
唑基、噻二唑基、 二唑基、苯硫基、呋喃基、喹啉基、异喹啉基、苯并咪唑基、苯并
二唑基、苯硫基、呋喃基、喹啉基、异喹啉基、苯并咪唑基、苯并 唑基、喹
唑基、喹 啉基等。杂芳环可通过碳或杂原子键合至化学结构上。
啉基等。杂芳环可通过碳或杂原子键合至化学结构上。
“杂原子”旨在包括氧(O)、氮(N)、硫(S)、硒(Se)和硅(Si)。
作为取代的烷基的“卤代烷基”是指单卤代烷基或多卤代烷基,最通常的是被1-3个卤素原子取代。例子包括1-氯乙基、3-溴丙基、三氟甲基等。
在聚合物太阳能电池中研究了本文所述的聚合物的光伏特性。将简单的太阳能电池与具有本文所述化学式的半导体聚合物一起使用。在一些实施例中,功率转换效率(PCE)大于6%。在可能优选的实施例中,半导体聚合物产生大于5%的PCE。在一些优选的实施例中,PCE达到6.8%。
本文所述的聚合物表现出的高转换效率可能是高效的光捕获的结果。使用所述聚合物的电池吸收几乎整个可见光谱中的光。聚合物形态也有利于电荷分离和电荷输送。这可以获得高填充因子。
不希望受理论的束缚,本文所述的硒吩聚合物基于两大概念。首先,含有扩展的π共轭的供体/受体型聚合物的供体部分(苯并二硒吩)由于大Se电子轨道而将增强聚合物/富勒烯共混膜中的电荷转移,并有利于异质结太阳能电池器件中的电荷输送。
其次,受体部分(在某一实施例中为硒吩并硒吩)可支持醌式结构并导致窄聚合物能带隙,这可能对捕获太阳能有影响。由于硒吩并硒吩部分富含电子,因此可以引入诸如酯基或酮之类的吸电子基团以稳定所得的聚合物。
在另一方面,本公开还包括作为聚合物的基本单元的梯式杂并苯。梯式杂并苯具有较大的有效共轭长度,并且在理论上预测将相对于具有相同芳环数的聚杂芳族化合物显示出较窄的能带隙(Anthony,J.E.Angew.Chem.Int.Ed.(《应用化学国际版》)2008,47,452-483;Morales,Y.R.J.Phys.Chem.A(《物理化学杂志A辑》)2002,106,11283-11308;Anthony,J.E.Chem.Rev.(《化学评论》)2006,106,5028-5048)。
不希望受理论的束缚,如模型中所示(图4),在光激发时,聚合物中较大的基态局部偶极矩导致聚合物中出现较大的局部偶极变化(Δμge(D)),因此,激发态被大大极化。负电荷在缺电子噻吩并噻吩部分上集中,在这里将发生电荷向PCBM的转移(弯曲箭头),而正电荷则留在苯并[1,2-b:4,5-b’]二噻吩单元上(+)。正负组分之间的高度分离降低了激子的库仑结合能,从而使得进一步的电荷分离/电荷载流子传输变得更容易。这导致太阳能电池效率提高。
该模型指出了两种增强新型供体聚合物中的功率转换效率的方法:1.进一步增加局部偶极矩,2.扩展BDT单元中的共轭体系以增强正电荷的离域作用,从而降低电荷密度。新聚合物的设计基于增大电荷离域程度的第二种方法。选择了梯式杂并苯作为聚合物的基本单元。梯式杂并苯具有较大的有效共轭长度,并且预计将相对于具有相同芳环数的聚杂芳族化合物显示出较窄的能带隙。(Anthony,J.E.Angew.Chem.Int.Ed.(《应用化学国际版》)2008,47,452-483;Morales,Y.R.J.Phys.Chem.A(《物理化学杂志A辑》)2002,106,11283-11308;Anthony,J.E.Chem.Rev.(《化学评论》)2006,106,5028-5048)。梯式杂并苯和3-氟噻吩并[3,4-b]噻吩-2-羧酸酯的共轭共聚物可同时产生共聚物的低带隙并满足上述设计标准,从而导致太阳能电池效率增强。
不希望受任何理论的束缚,已假设了若干机理。梯式杂并苯和3-氟噻吩并[3,4-b]噻吩-2-羧酸酯的共轭共聚物可同时产生共聚物的低带隙,这将导致太阳能电池效率增强。
本文所述的半导体聚合物可由式(I)、(II)、(III)、(IV)、(V)和(VI)表示:

其中X1、X2、X3和X4独立地选自O、S、Se、NH和CH2。优选地,X1、X2、X3和X4独立地选自O、S和NH。R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基。Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基。Z1和Z2独立地选自CH和N。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。n为大于0的整数。
在一些实施例中,聚合物的数均分子量在约15至约25kDa之间。在一些实施例中,n为约1至约200。在一些实施例中,多分散指数在约1.5至约3之间。
在一些实施例中,半导体聚合物具有式(V)。X1和X2独立地选自O、S和NH。R1和R2独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3和Y4独立地选自O、S、Se和氨基。Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。n为大于0的整数。
在一些实施例中,聚合物的数均分子量在约15至约25kDa之间。在一些实施例中,n为约1至约200。在一些实施例中,多分散指数在约1.5至约3之间。
在一些实施例中,X1和/或X2为O。
在一些实施例中,R1和R2独立地为烷基。在一些实施例中,R1和R2独立地为C1-30烷基。在一些实施例中,R1和R2相同。在一些实施例中,R1和R2为2-乙基己基。
在一些实施例中,Y1和Y2为S。在一些实施例中,Y1和Y2为Se。在一些实施例中,Y3和Y4为Se。
在一些实施例中,Z为酯。在一些实施例中,Z为C1-30烷基酯。在一些实施例中,Z为-C(O)OCH2CH(C2H5)C4H9。
在一些实施例中,W为H或卤素。在一些实施例中,W为卤素。在一些实施例中,W为F。
在一些实施例中,聚合物具有式(I):

优选地,X1、X2、X3和X4独立地选自O、S和NH。R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。可交联基团可以是将一个基团通过共价化学键连接至另一个基团的任何基团。Y1、Y2、Y3和Y4独立地选自O、S、Se和氨基。n为大于0的整数。
在一些实施例中,聚合物具有式(II):

优选地,X1和X2独立地选自O、S和NH。R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3和Y4独立地选自O、S、Se和氨基。Z1和Z2独立地选自CH和N。n为大于0的整数。
在一些实施例中,聚合物具有式(III):

优选地,X1、X2、X3和X4独立地选自O、S和NH。R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基。Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。n为大于0的整数。
在一些实施例中,聚合物具有式(IV):

R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基。Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。n为大于0的整数。
在一些实施例中,聚合物具有式(V):
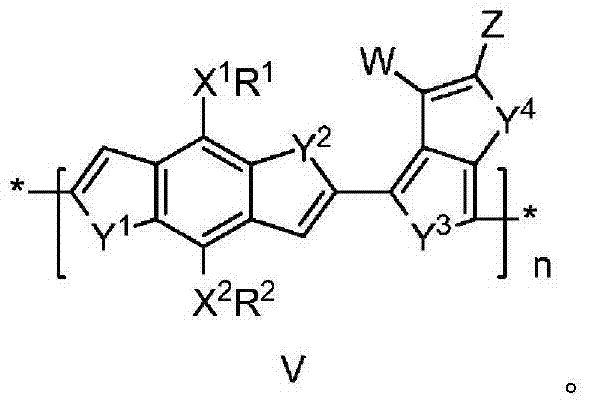
优选地,X1和X2独立地选自O、S和NH。R1和R2独立地选自H、烷基、芳基、杂芳基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。n为大于0的整数。
本公开的聚合物的具体实施例在下文示出。R1、R2和R3独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。
在一些实施例中,R1、R2和R3独立地为烷基。在一些实施例中,R1、R2和R3独立地为C1-30烷基。在一些实施例中,R1、R2和R3相同。在一些实施例中,R1、R2和R3为2-乙基己基。
在某些实施例中,富电子的Se杂原子可具有减小聚合物的能带隙的作用,从而允许更高效的光捕获。它们还可以增大电荷迁移率。因此合成了具有不同侧链的多种单体,以便获得可加工的聚合物。同时,还合成了另一种噻吩PTB家族聚合物PTB-8,其具有与PTAT-3上的那些相似的大体积侧链,且在苯并二噻吩部分上具有两个2-丁基辛氧基。(He,Feng;等人J.Am.Chem.Soc.(《美国化学学会杂志》)2011,133(10),3284-3287)。
合成了梯式大型并苯:3,7-二烷基-二噻吩并[2,3-d:2’,3’-d’]苯并[1,2-b:4,5-b’]二噻吩(DBD),并将其掺入到共轭聚合物中。
聚合物具有式(VI):

R1和R2独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇)。Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基。W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基。Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基。n为大于0的整数。
在一些实施例中,聚合物的数均分子量在约15至约25kDa之间。在一些实施例中,n为约1至约200。在一些实施例中,多分散指数在约1.5至约3之间。
在一些实施例中,R1和R2独立地为C1-30烷基。在一些实施例中,R1和R2独立地为2-乙基己基或2-丁基己基。
在一些实施例中,Y1、Y2、Y3、Y4、Y5和Y6独立地为S。
在一些实施例中,W为卤素。优选地,W为F。
在一些实施例中,Z为酯。在一些实施例中,Z为C1-30烷基酯。在一些实施例中,Z为-C(O)OCH2CH(C2H5)C4H9。在一些实施例中,Z为-C(O)OCH2CH(C4H9)C6H13。
本公开还包括本文所述的聚合物在太阳能电池、光学器件、电致发光器件、光伏电池、半导体电池或光电二极管中的用途。在一些实施例中,聚合物具有选自式(I)-(VI)的化学式。
在一些实施例中,组合物包含本文所述的半导体聚合物和吸电子富勒烯衍生物。在一些实施例中,聚合物具有选自式(I)-(VI)的化学式。在一些实施例中,吸电子富勒烯衍生物可以为[6,6]-苯基-C61-丁酸甲酯(PC61BM)或[6,6]-苯基-C71-丁酸甲酯(PC71BM)。该组合物可用在太阳能电池、光学器件、电致发光器件、光伏电池、半导体电池或光电二极管中。
成功地合成了新的梯式大型并苯:3,7-二烷基-二噻吩并[2,3-d:2',3'-d']苯并[1,2-b:4,5-b']二噻吩(DBD)并将其掺入到共轭聚合物中,该聚合物表现出低于1.7eV的能带隙(图5)。在优化聚合物的溶解性和与PC71BM的形态相容性后,聚合物BHJ太阳能电池器件实现了7.71%的PCE,这是针对太阳能电池聚合物所报道的最佳PCE。
DBD单体的合成路线在图6中示出。化合物22a-b和26的合成如本文所述。将DBD单体与4,6-二溴-3-氟噻吩并[3,4-b]噻吩-2-羧酸烷基酯(FTT)共聚。(Carsten,B.等人,P.Chem.Rev.(《化学评论》)2011,111,1493-1528)。
初始聚合物PTDBD1(图5)显示了低溶解性并且难以在有机溶剂中加工。为了提高聚合物的溶解性,通过将体积更大的烷基侧链掺入到FTT和/或DBD单元中而对聚合物的结构进行调整。所得的聚合物PTDBD2和PTDBD3(图5)显示出108.6和105.5KDa的相似重均分子量,且多分散指数分别为2.84和2.32。热重量分析(TGA)表明两种聚合物在最高至约250℃的条件下均是稳定的。
PTDBD2和PTDBD3在聚合物膜中表现出450nm至800nm的吸收,但是PTDBD3的吸收更宽并且更无特征(图7)。从吸收光谱的起始处(λ起 始)估计的聚合物的光学能带隙为1.67~1.68eV。通过在薄膜中进行的循环伏安法(CV)测量,PTDBD3的HOMO能级(-5.30eV)略低于PTDBD2的HOMO能级(-5.24eV)(图9)。(Pommerehne,J.等人,J.Adv.Mate.(《先进材料杂志》)1995,7,551-554)。PTDBD2显示出比PTDBD3高的电荷载流子迁移率,其中前者的该值为2.70×10-4cm2V-1s-1,后者的该值为1.09×10-4cm2V-1s-1,如通过对原始聚合物膜的空间电荷限制电流(SCLC)进行测量所确定。不受理论的约束,这可归因于在PTDBD3的FTT中的大非导电性双(2-乙基己基)甲基,其可能干扰聚合物膜中的有效电荷载流子传输,但与富勒烯共混时通常改变聚合物堆积形态。
为了表征PTDBD2和PTDBD3的光伏特性,以(ITO/PEDOT:PSS/聚合物:PCBM/Ca/Al)的结构制备了太阳能电池器件。用于形成聚合物太阳能电池的活性层的PTDBD2和PTDBD3的优化聚合物/PC71BM比率分别为1:1.2和1:1.3。相同的比率用于聚合物/PC61BM太阳能电池。聚合物活性层由氯苯(CB)/3%(v/v)1,8-二碘辛烷(DIO)助溶剂中的溶液旋涂。J-V曲线在图8中报告,器件性能在表1中汇总。基于PTDBD2的太阳能电池在将PC71BM用作受体材料时表现出7.71%的最高PCE。图10中的外量子效率(EQE)谱表明PTDBD2/PC71BM器件的EQE在450nm至700nm的范围内接近60%。在器件的J-V曲线中,短路电流密度(Jsc)达到最高13.7mA/cm2,而开路电压(Voc)为869mV并且填充因子(FF)为64.8%。使用相同的制造条件(除了PTDBD3/PCBM比率不同外)制备的基于PTDBD3/PC71BM的太阳能电池器件表现出882mV的改善Voc,不受理论的束缚,这可能是由于其HOMO值较低而引起的。然而,PTDBD3实现了10mA/cm2的较低Jsc,这与其52.1%的FF值相结合,导致4.92%的PCE值。当使用PC61BM制造器件时,PTDBD2还显示出更佳的太阳能电池性能,具有较高的Jsc和FF值。不受理论的束缚,PTDBD2相对于PTDBD3的光电流改善归因于其较高的电荷载流子迁移率。
表1:PTDBD2和PTDBD3


透射电子显微镜(TEM)研究表明,PTDBD2相对于PTDBD3的器件性能增强可能与由聚合物FTT单元中的不同烷基侧链引起的聚合物/PCBM共混膜的形态变化相关。与PC71BM或PC61BM共混的PTDBD3膜表现出更大且更暗的具有约100nm尺寸的特征,该特征可指示聚集的PCBM簇,而PTDBD2的共混膜显示出大小为约10~15nm的均匀精细特征。假设器件中较小的畴允许较高的聚合物-PCBM界面面积,并因此允许更有效地生成电荷载流子,从而导致在PTDBD2太阳能电池中产生更高的光电流。引入共轭聚合物的大体积侧链往往减小聚合物与PCBM分子的互混性,从而导致聚合物与PCBM分子之间出现较大的相分离。(He,F.等人,J.Am.Chem.Soc.(《美国化学会会志》)2011,133,3284-3287)。
为了更深入地了解共混膜中的纳米级形态和分子排序,应用了掠入射广角X射线衍射(GIWAXS)来研究这些体系。净PTDBD2和PTDBD3聚合物的2D GIWAXS图在q~0.35–0.45 -1表现出环状散射,对应于聚合物主链的周期性层片,且在约q~1.6 -1表现出出平面散射,对应于聚合物的π堆叠。对于PTDBD2,层片间距为15.4
-1表现出环状散射,对应于聚合物主链的周期性层片,且在约q~1.6 -1表现出出平面散射,对应于聚合物的π堆叠。对于PTDBD2,层片间距为15.4 (qy=0.408
(qy=0.408 -1),而π堆叠距离为4.0
-1),而π堆叠距离为4.0 (qz=1.55
(qz=1.55 -1)。对于PTDBD3,其层片间距为17.3(qy=0.364
-1)。对于PTDBD3,其层片间距为17.3(qy=0.364 -1),而其π堆叠距离为3.9
-1),而其π堆叠距离为3.9 (qz=1.62
(qz=1.62 -1)(图11)。仅出平面观察到的π堆叠散射表明PTDBD2和PTDBD3聚合物链优先地出膜平面堆叠,但具有更随机的取向。
-1)(图11)。仅出平面观察到的π堆叠散射表明PTDBD2和PTDBD3聚合物链优先地出膜平面堆叠,但具有更随机的取向。
在与PC71BM共混时,PTDBD2和PTDBD3均保持了其结构特性,但是与主链层片相关的峰的半峰全宽(FWHM)分别从0.132 -1变窄到0.110
-1变窄到0.110 -1(对于PTDBD2)以及从0.061
-1(对于PTDBD2)以及从0.061 -1变窄到0.042
-1变窄到0.042 -1(对于PTDBD3)。这表明因与PC71BM混合而在两种聚合物中产生了更高的结晶度,这有益于OPV器件中的电荷输送。除了从聚合物或PC71BM的漫散射外,还在聚合物/PC71BM共混膜的2D GIWAXS图中检测到了归因于界限明确的、纯的PC71BM晶体的一对清晰的散射斑点,从而表明在聚合物/PC71BM共混相中存在漂浮的PC71BM晶体。由于这些散射斑点在聚合物/PC61BM共混物中很少被观察到,因此不受理论的束缚,可能的是这些漂浮的PC71BM晶体可将光激发的电子从聚合物/PC71BM共混相中排出并将它们传输至电极,从而促进电荷分离和传输。(Clarke,T.M.等人,Chem.Rev.(《化学评论》)2010,110,6736;Deibel,C.等人,J.IEEE J.Sel.Top.QuantumElectron.(《IEEE量子电子学选题杂志》)2010,16,1517)。此外,由于PTDBD2/PC71BM膜的聚合物主链峰的FWHM是PTDBD3/PC71BM膜的两倍大,故表明PTDBD2的结晶度低得多。PTDBD2的这种低结晶度能够导致在聚合物/PC71BM共混相内在分子水平上的更佳互混性,这通过TEM观察得到证实。综合起来,不受理论的束缚,这些结构和形态特征(PTDBD2与PCBM的更佳互混性、PCBM引起的结晶度和以及漂浮的界限明确的纯PCBM晶体)是PTDBD2聚合物在与PC71BM共混时具备优异的OPV性能的原因。
-1(对于PTDBD3)。这表明因与PC71BM混合而在两种聚合物中产生了更高的结晶度,这有益于OPV器件中的电荷输送。除了从聚合物或PC71BM的漫散射外,还在聚合物/PC71BM共混膜的2D GIWAXS图中检测到了归因于界限明确的、纯的PC71BM晶体的一对清晰的散射斑点,从而表明在聚合物/PC71BM共混相中存在漂浮的PC71BM晶体。由于这些散射斑点在聚合物/PC61BM共混物中很少被观察到,因此不受理论的束缚,可能的是这些漂浮的PC71BM晶体可将光激发的电子从聚合物/PC71BM共混相中排出并将它们传输至电极,从而促进电荷分离和传输。(Clarke,T.M.等人,Chem.Rev.(《化学评论》)2010,110,6736;Deibel,C.等人,J.IEEE J.Sel.Top.QuantumElectron.(《IEEE量子电子学选题杂志》)2010,16,1517)。此外,由于PTDBD2/PC71BM膜的聚合物主链峰的FWHM是PTDBD3/PC71BM膜的两倍大,故表明PTDBD2的结晶度低得多。PTDBD2的这种低结晶度能够导致在聚合物/PC71BM共混相内在分子水平上的更佳互混性,这通过TEM观察得到证实。综合起来,不受理论的束缚,这些结构和形态特征(PTDBD2与PCBM的更佳互混性、PCBM引起的结晶度和以及漂浮的界限明确的纯PCBM晶体)是PTDBD2聚合物在与PC71BM共混时具备优异的OPV性能的原因。
基于将π共轭体系扩展至较低正电荷密度和结合能这一想法,合成了含有梯式DBD衍生物的一系列新型半导体聚合物。聚合物中的烷基侧链的最佳结构可改善聚合物链与PCBM分子的相容性,从而改善聚合物/PCBM共混膜的形态,同时增加共混膜中聚合物链的结晶度。因此,太阳能电池效率增大高达7.71%。该结果表明含有梯式DBD的共聚物可构成高性能有机太阳能电池。
除非另行指出,否则所有化学品均购自奥德里奇公司(Aldrich),并且不进一步纯化而使用。除非另外指明,否则所有反应均在氮气气氛下进行。将四氢呋喃(THF)和二乙醚(Et2O)在钠和二苯甲酮上蒸馏。4,6-二溴-3-氟噻吩并[3,4-b]噻吩-2-羧酸烷基酯根据文献中报道的程序进行制备。(Liang,Y.Y.等人,J.Am.Chem.Soc.(《美国化学会会志》)2009,131,7792-7799)。分别在Bruker DRX-400或DRX-500光谱仪上记录了400或500MHz下的1H NMR和13C NMR谱。化学位移从低频到高频以份每一百万份(ppm)报告,并参考残余溶剂共振。指示多重性的标准缩写如下使用:s=单峰,d=二重峰,t=三重峰,m=多重峰,q=四重峰以及b=宽峰。使用337nm氮激光器以四氰基对醌二甲烷(TCNQ)作为基质,在Bruker ReflexII-TOF质谱仪上记录MALDI-TOF质谱。EI质谱分析使用Varian Saturn2000GC/MS质谱仪进行。聚合物的分子量和分布通过配备Waters510HPLC泵、Waters410差示折光计和Waters486可调吸收检测器的WatersAssociates液相色谱仪使用GPC进行测定。将THF用作洗脱剂,将聚苯乙烯用作标准品。使用Shimadzu UV-2401PC分光光度计获得光学吸收谱。聚合物的薄膜由其氯苯溶液旋涂,并测量了膜吸收谱。
用于合成4,6-二溴硒吩并[3,4-b]噻吩羧酸乙基己酯单体7的方法是基于改良的报道程序(合成路线1)。通过将硼氢化钠缓慢添加至硒粉在碱性水溶液中的混合物而制备硒化钠。将所得的无色Na2Se水溶液经30分钟滴加至2,3-双氯甲基-5-甲氧羰基硒吩1的醇溶液以产生二聚体2作为唯一产物,其收率为73%。通过1H NMR确认结构,且质谱显示出592处的确切分子离子和296处的基峰。未观察到3的形成,因为Na2Se溶液以这些类型的环化典型的较快速率添加。遵循报道的程序将导致形成主要产物2(~70%)和副产物3(~10%)。由于化合物3是关键的原料,因此需要提高其收率。因此,使用了多种不同的实验条件,包括使用极性溶剂(乙醇、水和THF)中的NaHSe或Na2Se、改变添加的模式和添加试剂时的温度,结果是形成了作为唯一产物或作为主要产物的2以及作为副产物的3。据报道,二聚体2的苯类似物可按定量的收率以热或光化学的方式转变为相应的二氢苯并硒吩。这可通过双自由基中间体有利地分子内环化成五元环中的两个而形成。因此,二聚体2经减压闪速热解以良好的收率(56%)转变成了二氢硒吩并硒吩3。前两个步骤的总收率为41%,该收率是此前报道的方法的收率(10-12%)的约四倍。所有的光谱数据均符合报道的4,6-二氢硒吩并[3,4-b]硒吩-2-羧酸甲酯(3)的那些数据。通过使用H2O2进行氧化而使3芳化得到N-硒亚砜,其在用冷乙酸酐小心处理时得到相应的硒吩并[3,4-b]噻吩羧酸甲酯4。添加未冷却的乙酸酐将导致一些原料由于发生的瞬间自发放热反应而分解。使用NBS进行溴化得到了4,6-二溴硒吩并[3,4-b]噻吩羧酸甲酯5,其在碱性条件下水解时产生4,6-二溴硒吩并[3,4-b]噻吩羧酸6。为了增强聚合物的溶解性,将羧酸6通过支链2-乙基-1-己醇使用DCC和DMAP进行酯化,从而以高收率(88%)得到了相应的4,6-二溴硒吩并[3,4-b]噻吩羧酸乙基己酯7单体。
合成路线1
R=2-乙基己基
合成4,8-双(烷氧基)苯并[1,2-b:4,5-b’]二噻吩(BDT)的便利方法需要噻吩衍生物作为关键原料(Yu等人,Science(《科学》)1995,270,1789);Brabec等人,Adv.Funct.Mater.(《先进功能材料》)2001,11,15;Shaheen等人,Appl.Phys.Lett.(《应用物理学快报》)2001,78,84;Li等人,Nat.Mater.(《自然-材料学》)2005,4,864;Thompson等人,Angew.Chem.Int.Edit.(《应用化学国际版》)2008,47,58;Yu,G.;Heeger,A.J.J.Appl.Phys.(《应用物理学杂志》)1995,78,4510)。由于硒吩衍生物的可用性受到限制,因此采用了其中中心苯为原料并通过分子内环化构建硒吩环的合成方法。(Wang等人,Appl.Phys.Lett.(《应用物理学快报》)2008,92,33307;Peet等人,Nat.Mater.(《自然-材料学》)2007,6,497;Muhlbacher等人,Adv.Mater.(《先进材料》)2006,18,2884;Zou等人,J.Am.Chem.Soc.(《美国化学会会志》)2010,132,5330;Piliego等人,J.Am.Chem.Soc.(《美国化学会会志》)2010,132,7595)。
为了制备4,8-双(烷氧基)苯并[1,2-b:4,5-b’]二硒吩(BDSe),中心苯应在如合成路线2所示的合成方法的早期阶段具有烷氧基。因此,合成4,8-双(2-乙基己基)苯并[1,2-b:4,5-b’]二硒吩的原料从1,4-二溴-3,6-双(2-乙基己基)苯9开始。在存在FeCl3作为催化剂的情况下使用N-氯代琥珀酰胺(NCS)将化合物9氯化,从而以良好的收率得到了1,4-二溴-2,5-二氯-3,6-双(2-乙基己基)苯10(Liang等人,J.Am.Chem.Soc.(《美国化学会会志》)2009,131,56;Liang等人,J.Am.Chem.Soc.(《美国化学会会志》)2009,131,7792;Chen等人,Nat.Photonic.(《自然光子学》)2009,3,649;Liang等人,Adv.Mater.(先进材料》)2010,22,E135;Liang等人,Acc.Chem.Res.(《化学研究评述》)2010,43,1227)。通过钯催化的Sonogashira偶联使10与三甲基甲硅烷基乙炔反应而以中等收率(45%)得到1,4-二溴-2,5-双(2-三甲基甲硅烷基乙炔)-3,6-双(2-乙基己基)苯11。在用叔丁基锂对化合物11进行处理、接着添加元素硒再通过乙醇淬灭后,以中等收率(40%)得到所需的BDSe核12。该环化通过乙炔与硒化物阴离子中间体之间的分子内环化反应而发生(Wang等人,Appl.Phys.Lett.2008,92,33307(Wang等人,《应用物理学快报》,2008年,第92卷,第33307页);Peet等人,Nat.Mater.(《自然材料学》)2007,6,497;Muhlbacher等人,Adv.Mater.(《先进材料》)2006,18,2884;Zou等人,J.Am.Chem.Soc.(《美国化学会会志》)2010,132,5330;Piliego等人,J.Am.Chem.Soc.(《美国化学会会志》)2010,132,7595)。12的1H NMR显示在芳族区域中出现了作为唯一单峰的在7.88pm处共振的硒吩质子。通过用四丁基氟化铵处理使化合物12去甲硅烷基化而以极佳的收率(92%)得到13。用正丁基锂或叔丁基锂处理13即便进行很长的时间也未发生明显的反应。为了获得所需的单体,将13优选首先用NBS进行溴化而得到14,14将易于与正丁基锂发生锂-卤素交换。用SnMe3Cl淬灭得到目标15。经由Pd催化的Stille缩聚反应使单体聚合以良好的收率获得了聚合物PSeB1和PSeB2。
合成路线2

R=2-乙基己基
数均分子量和重均分子量通过凝胶渗透色谱法分别表征为9.2kg/mol和15.4kg/mol,且多分散度分别为1.68和2.68。热重量分析(TGA)指示聚合物在高达200℃时是稳定的。通过电子吸收光谱确认了关于聚合物能带隙的假定。氯仿溶液中PSeB1和PSeB2的紫外-可见光谱显示出分别在703和707nm处(PTB1~692nm)的最大吸收。对于两种聚合物,固体聚合物膜的吸收略微红移至713nm。从光谱起始处估计,获得1.60eV的光学能带隙。通过使用循环伏安法研究了PSeB1薄膜对玻璃碳电极的电化学行为。如图1b所示,观察到了两种材料的还原和氧化的准可逆特性。前线分子轨道的能级可使用公式ELUMO=-(4.71+Ered)、EHOMO=-(4.71+Eox)由起点推导得出。PSeB1的LUMO和HOMO能级计算值分别为-3.27eV和-5.05eV,而PSeB2的相应值分别为-3.26eV和-5.04eV。由其得到两种聚合物的能带隙为1.78eV。
用ITO/PEDOT:PSS/聚合物:富勒烯/Ca/Al的器件结构制备BHJ聚合物太阳能电池。通过分别使用1:1的聚合物与PC61BM的重量比以及1:1.2的聚合物与PC71BM的重量比旋转浇注出活性层。膜厚度在100~110nm的范围内。
图2示出了在PSeB1的不同溶剂加工条件下在1.5G照射下记录的光电流密度与电压的关系图。相应的物理参数收集在表2中。研究了由氯苯(CB)和邻二氯苯(DCB)溶剂制备的PSeB1/PC61BM的共混膜。使用CB得到的Voc和Jsc均比用DCB得到的高得多,这表明CB是用于该体系的更佳的溶剂。将1,8-二碘辛烷(DIO,2%v/v)加入CB和DCB有效地将填充因子(FF)从约41%增至约54%并导致了5.33%的高PCE值(条目4)。DIO对改变Voc无明显效果(条目3、4:626至624mV),且对改变Jsc亦无明显效果(15.21至15.89mA/cm2)。值得注意的是,15.89mA/cm2的Jsc显然属于聚合物太阳能电池的最佳值。由于PC71BM在可见光区具有更佳的吸收并同时保持与PC61BM相似的电子特性,因此通过使用1:1.2的PSeB1:PC71BM重量比制备出活性层。膜厚度也在约100~110nm。令人惊讶的是,得到了16.77mA/cm2的高Jsc。器件具有618mV的Voc、55%的FF和5.70%的PCE。
表2:具有PSeB1和PSeB2的器件的太阳能电池参数。

a1:1PSeB1/PC61BM。b1:1.2PSeB1/PC71BM。c1:1.2PSeB2/PC71BM。
与PSeB1相似,PSeB2在CB中浇铸的BHJ太阳能电池与使用DCB的那些相比具有更优异的性能。使用DCB而以DIO作为添加剂导致了628mV的Voc、13.3mA/cm2的Jsc、58%的FF以及4.82%的PCE。当用CB替代DCB时,Jsc增至15.7mA/cm2,这与PSeB1中发现的增加相似。然而,FF增至67%,而Voc保持相对不变。这与PSeB1形成鲜明的对比,对于PSeB1,在由DCB换成CB时表现出Voc和Jsc的增加而FF不变。在这些最佳条件下,PSeB2提供了6.56%的PCE,这明显高于PSeB1表现出的5.70%。
测量了在优化的条件下PSeB1和PSeB2二者的EQE,发现它们几乎相同(图2b)。二者均跨越了宽波长范围(350–800nm),且最大值在645nm处。两种聚合物的最大EQE对于PSeB1和PSeB2而言分别为63%和61%。PSeB2的曲线与PSeB1的相比略微红移,这是由于PSeB2的吸光度发生了红移(图1)。这使得PSeB2与PSeB1相比能够捕集更宽跨度的能量。
进行了透射电子显微镜检查(TEM)来研究共混膜。由CD而无DIO添加剂浇铸的PSeB1/PC71BM膜表现出粗糙的外观,从而表明形成了大的单独的聚集聚合物和富勒烯畴。由CB/DIO浇铸的PSeB1/PC71BM膜表现出了均匀精细的特征,从而表明形成了纳米级相分离,该现象是由有效的供体-受体相互作用所导致的。该特征性特征有利于电荷分离。PSeB1的空穴迁移率通过使用空间电荷限制电流(SCLC)方法测定,据发现其为约2.9×10-4cm2/V·s。该值落在此前所述PTB聚合物的同一水平中,并且足以在共混物中形成平衡的载流子迁移率并导致高填充因子。由CB/DIO浇铸的PSeB2/PC71BM膜显示出令人惊讶的粗糙外观,尽管其在结构上与PSeB1相似。实际上,其形态更接近由无DIO添加剂的CB浇铸的PSeB1/PC71BM膜的形态。然而,由CB/DIO浇铸的PSeB2/PC71BM膜表现出极高的PCE,这与聚合物的噻吩PTB系列的观察结果形成鲜明的对比。
在本发明中,合成了半导体聚合物聚硒吩并[3,4-b]硒吩-co-苯并二噻吩(PSeB1)和聚硒吩并[3,4-b]硒吩-co-苯并二硒吩(PSeB2)。光伏研究表明PSeB2是用于如BHJ太阳能电池的应用的非常有前景的材料。当与PC71BM共混时实现了6.56%的优异PCE。然而,AFM研究反映了较差的膜形态,从而表明使用该聚合物可能获得甚至更高的PCE。
在本公开中,对硒吩并硒吩单体进行了选择性氟化以增强PCE。该体系使用氟取代以增强PCE。已显示噻吩并噻吩的氟化对PCE有影响。氟化硒吩聚合物的合成在合成路线3中示出。
合成路线3

本发明还包括半导体聚合物和电子接受材料的组合物,所述电子接受材料包括但不限于巴克敏斯特富勒烯(“富勒烯”)衍生物。任何电子接受富勒烯均可与本文所述的半导体聚合物一起使用。此类富勒烯可为C60至C90。在优选的实施例中,富勒烯可以为C61、C71、C81或C91。富勒烯可以是具有下式的[6,6]-苯基-C61-丁酸甲酯(PC61BM):

聚合物和富勒烯可共混为混合物。在一个实施例中,可将共轭物与富勒烯如下所示进行连接。
本公开的一个实施例包括本文所述的半导体聚合物和共轭物在诸如太阳能电池、光学器件、电致发光器件、光伏电池、半导体电池或光电二极管之类的器件中的应用。本公开的另一个实施例包括本文所述的半导体聚合物的共轭物在太阳能电池、光学器件、电致发光器件、光伏电池、半导体电池、光电二极管或聚合物场效应晶体管中的应用。
将本文所述的半导体聚合物掺入到基板上的方法包括将一种或多种本文所述的聚合物与富勒烯衍生物溶解于溶剂中,然后将所得的复合材料涂布到基板上。
一种或多种聚合物可以与富勒烯衍生物共溶在例如1,2-二氯苯、氯苯、氯仿或甲苯中。聚合物的浓度可以为约5至约20mg/mL,或者为其中的任意组合或子组合。
然后可将聚合物/富勒烯复合材料涂布到基板上。可以使用合适的基板,如本领域中已知的铟锡氧化物(ITO)涂布的玻璃。可以使用适于层涂布或浇注的任何种类的工艺来将该复合材料涂布到基板上。可以利用旋转浇注将复合物共混物的薄层或膜涂布到基板上。复合物共混物的层可以为约80nm至约150nm厚。层厚将根据复合材料共混物的涂布和基板而不同。因此,本文提供的是包含如本文所述的半导体聚合物和富勒烯衍生物的光伏膜。
本文所述的半导体聚合物可以具有交替的四噻吩并蒽和噻吩并[3,4-b]噻吩单元。可针对光伏应用而精细调整这些聚合物的物理性质。聚合物的HOMO能级可以通过如下方式降低:将烷氧基侧链替换为供电子性较低的烷基侧链,或者向聚合物主链中引入吸电子的氟,从而引起聚合物太阳能电池的Voc提高。侧链和取代基也影响聚合物的吸收性和空穴迁移率以及与富勒烯的混溶性,这些都会影响聚合物太阳能电池性能。由混合溶剂制备的膜表现出精细分布的聚合物/富勒烯相互贯穿的网络和显著提高的太阳能电池转换效率。在基于由混合溶剂制备的氟化PTAT-3/PC61BM复合膜的太阳能电池中,可以实现超过5.6%的功率转换效率。
在整个本说明书中,对本发明的优选实施例和替代实施例给出了各种指示。然而,应当理解,本发明不限于其中的任何一种。因此,应该认为上述详细描述是说明性而非限制性的,并且还应理解所附权利要求(包括所有等同物)旨在限定本发明的精神和范围。
实例
二聚体(2).将硒金属(1.65g,21.0mmol)悬浮进氢氧化钠(1.93g,48.3mmol)在60mL水中的溶液中。将硼氢化钠(1.79g,48.3mmol)溶解于20mL水中,然后在氮气氛下将其缓慢添加至硒悬浮液中。该反应为放热反应,剧烈地放出氢气。将所得的混合物在室温下进一步搅拌4小时。将无色的硒化钠溶液滴加至2,3-双氯甲基硒吩-5-羧酸甲酯1(5.00g,17.4mmol)在200mL脱气乙醇中的溶液。在添加完成后,将所得的混合物进一步搅拌2小时。移除溶剂,将固体残余物与100mL水一起搅拌30分钟。然后,将混合物过滤而得到纯度足以用于下一步的2(3.76g,73%)。通过从吡啶/水中重结晶而获得纯样品。1H NMR(CDCl3):δ3.27(4H,s),3.78(4H,s),3.85(6H,s),8.05(2H,s)。MS(EI):计算值591.8;实测值[M+1]+592.8。
4,6-二氢硒吩并[3,4-b]硒吩-2-羧酸甲酯(3):将一端封闭的Pyrex管(长40cm,直径2cm)用于热解。将2的样品(0.25g,0.425mmol)置于管的封闭端中。将开口端连接至真空系统。将具有样品的封闭端置于电炉中(在约600℃下预热)。热解立即(约2分钟)发生,化合物3冷凝在玻璃管的冷部分(位于炉外)上。将有机产物溶解于CH2Cl2中,浓缩,使用CH2Cl2/己烷=1:1在硅胶上进行层析,得到作为黄色固体的0.14g(56%)纯的3。1HNMR(CDCl3):δ3.85(3H,s),4.08(2H,m),4.29(2H,m),7.73(1H,s)。MS(EI):计算值295.9;实测值[M+l]+296.7。
硒吩并[3,4-b]硒吩-2-羧酸甲酯(4):在搅拌下向在-20℃下冷却的0.90g(3.06mmol)3在7mL无水THF中的溶液滴加0.62g(5.3mmol)30%H2O2在3mL无水THF中的溶液。在-20℃下搅拌过夜后,将无色固体滤除,从而得到了0.66g硒亚砜。将滤液真空浓缩,过滤得到了另外0.12g硒亚砜(81%)。将硒亚砜用冷Ac2O(15mL)处理,于是发生自发放热反应,溶液颜色变得非常深。在室温下搅拌1小时后,将Ac2O用水水解,然后将混合物用CH2Cl2提取,将有机提取物用冷的饱和NaHCO3和水仔细洗涤,再用MgSO4干燥并浓缩。使用CH2Cl2在硅胶上进行柱层析,得到0.55g4(62%)。1H NMR(CDCl3):δ3.90(3H,s),7.83(1H,s),7.95(1H,dd,J=2.3,0.85Hz),8.44(1H,d,J=2.4Hz)。MS(EI):计算值293.9;实测值[M+l]294.9。
4,6-二溴硒吩并[3,4-b]硒吩-2-羧酸甲酯(5):在氮气保护下在暗处向0.34g(6.0mmol)4在3mL DMF中的溶液滴加NBS(0.52g,15.0mmol)在3mLDMF中的溶液,搅拌24小时。将反应混合物倾注进处于冰水浴中的饱和亚硫酸钠溶液,随后用二氯甲烷提取。收集有机相,并通过硫酸钠干燥。移除溶剂,使用二氯甲烷/己烷(1:1)在硅胶上进行柱纯化,得到作为浅黄色固体的0.42g(80%)标题化合物5。1H NMR(CDCl3):δ3.90(3H,s),7.75(1H,s)。MS(EI):计算值449.7;实测值[M+l]+450.7。
4,6-二溴硒吩并[3,4-b]硒吩-2-羧酸(6):向0.41g(0.91mmol)5在15mLTHF中的溶液加入LiOH·H2O(0.12g,2.86mmol)在3mL H2O中的溶液。将所得的混合物在室温和N2下搅拌4小时,然后倾注进冰水中,再通过1NHCl溶液酸化。将黄色固体过滤,用冷水洗涤,然后干燥得到0.34g6(85%)。1H NMR(DMSO):δ7.65(1H,s),13.80(1H,s)。MS(EI):计算值435.7;实测值[M-l]+434.7。
4,6-二溴硒吩并[3,4-b]硒吩-2-羧酸-2-乙基己酯(7):将0.34g(0.78mmol)6、0.21g(1.02mmol)DCC、20mg(0.16mmol)DMAP添加至具有5mLCH2Cl2的10mL圆底烧瓶中。将0.75g(10.0mmol)2-乙基己-1-醇添加至烧瓶中,然后在N2保护下保持搅拌20小时。将反应混合物倾注进30mL水中,用CH2Cl2提取。使用己烷/CH2Cl2=5:1将反应混合物直接在硅胶上层析,得到作为浅黄色固体的0.37g(88%)纯标题化合物。1H NMR(CDCl3):δ0.90-0.96(6H,m),1.32-1.46(8H,m),1.66-1.75(1H,m),4.22(2H,m),7.75(1H,s)。MS(EI):计算值547.8;实测值[M+2]+549.8。
PSeB1的合成:
称取4,6-二溴硒吩并[3,4-b]硒吩-2-羧酸2-乙基己酯(7)(0.238mg,0.432mmol)加到25mL圆底烧瓶中。加入2,6-双(三甲基锡)-4,8-双(2-乙基己氧基)苯并[1,2-b:4,5-b′]二噻吩(8)(334mg,0.432mmol)和Pd(PPh3)4(30mg)。使烧瓶经受真空随后是再充注氩气的三个连续循环。然后,将无水DMF(2mL)和无水甲苯(8mL)通过注射器加入。在氮气保护下于120℃进行聚合反应12小时。将粗产物沉淀进甲醇中,并通过过滤收集。将沉淀溶于氯仿,然后用硅藻土过滤以除去金属催化剂。通过在己烷中沉淀并在真空中干燥12小时而获得聚合物,产生325mg(92%)。1H NMR(CDCl3):δ0.80-2.40(45H,br),3.90-4.70(6H,br),7.00-7.90(2H,br)。GPC:Mw(15×103g/mol),PDI(1.68)。
2,5-二溴-3,6-二氯-1,4-二乙基己氧基苯(10):
向2,5-二溴-1,4-二乙基己氧基苯(8.60g,17.4mmol)在CH3CN(10mL)中的溶液添加NCS(7.00g,52.2mmol)和无水FeCl3(0.85g,5.22mmol)。(Z.N.Bao等人,1995,J.Am.Chem.Soc.(《美国化学会会志》),117,12426)。将所得的混合物在55℃下加热并搅拌过夜。将反应混合物用H2O(100mL)处理,然后用己烷(2×100mL)提取。将合并的有机提取物用无水MgSO4干燥,然后真空浓缩得到油状残余物,通过柱层析(己烷:CH2Cl2,95:5)将其纯化得到无色油(7.8g,80%)。1H NMR(CDCl3):δ3.87(m,4H),1.81(m,2H),1.31-1.64(m,12H),0.97(t,J=7.5Hz,6H),0.92(t,J=7.5Hz,6H)。MS(EI):计算值561.2;实测值[M+l]+562.2。
2,5-二氯-3,6-双(2-三甲基甲硅烷基乙炔基)-1,4-二乙基己氧基苯(11)。将三甲基甲硅烷基乙炔(3.1mL,21.43mmol)、PdCl2(PPh3)2(400mg,0.6mmol)和CuI(235mg,0.79mmol)相继添加至2,5-二溴-3,6-二氯-1,4-二乙基己氧基苯(10)(4.00g,7.14mmol)在二异丙胺(25mL)和甲苯(55mL)中的脱气溶液。将所得混合物回流48小时,然后用水(150mL)稀释,并用己烷(2×80mL)提取。将提取物用水(2×80mL)洗涤并干燥(MgSO4)。蒸发溶剂得到油状残余物,通过柱层析(己烷:CH2Cl2,98:2)将其纯化得到橙红色固体(1.90g,45%)。1H-NMR(CDCl3):δ3.87(m,4H),1.81(m,2H),1.31-1.64(m,12H),0.97(t,J=7.5Hz,6H),0.92(t,J=7.5Hz,6H)。MS(EI):计算值595.8;实测值[M+l]+596.8。
2,6-双(三甲基甲硅烷基)-4,8-双(2-乙基己氧基)苯并[1,2-b:4,5-b′]二硒吩(12)。
在-78℃下向11(1.0g,1.67mmol)在乙醚(20mL)中的溶液添加tBuLi(1.7M,4.0mL,7.5mmol)的戊烷溶液。将所得的混合物在相同的温度下搅拌30分钟,然后逐渐升至室温。然后将硒粉末(0.27g,3.34mmol)一次性加入,将所得的混合物搅拌30分钟。在添加乙醇(30mL)后,将混合物进一步搅拌2小时,并添加水(50mL),然后用氯仿(3×30mL)提取。将提取物用MgSO4(无水)干燥,然后真空浓缩。将残余物通过柱层析在硅胶(己烷:CH2Cl2,98:2)上纯化,得到无色微晶(0.45g,40%):1H-NMR(CDCl3):δ7.88(s,2H),4.12(d,,J=5.3Hz,4H),1.78(m,2H),1.35-1.70(m,12H),1.03(t,J=7.4Hz,6H),0.94(t,J=6.9Hz,6H),0.38(s,18H)。MS(EI):计算值684.9;实测值[M+1]+685.9。
4,8-双(2-乙基己氧基)苯并[1,2-b:4,5-b′]二硒吩(13)。
向2,6-双(三甲基甲硅烷基)-4,8-双(2-乙基己氧基)苯并[1,2-b:4,5-b′]二硒吩(11)(0.29g,0.423mmol)在THF(10mL)中的溶液添加1.3mL四丁基氟化铵(THF中的1M溶液)。将所得的溶液在室温下搅拌3小时,加入水(40mL),然后用氯仿(2×40mL)提取。将提取物用MgSO4(无水)干燥,然后真空浓缩。将残余物通过柱层析在硅胶(己烷:CH2Cl2,98:2)上纯化,得到黄色油(0.21g,92%)。1H-NMR(CDCl3):δ7.90(d,J=5.9Hz,2H),7.73(d,J=5.9Hz,2H),4.12(d,J=5.5Hz,4H),1.80(m,2H),1.35-1.70(m,12H),1.03(t,J=4.9Hz,6H),0.94(t,J=4.8Hz,6H)。MS(EI):计算值540.5;实测值[M+1]+541.5。
2,6-二溴-4,8-双(2-乙基己氧基)苯并[1,2-b:4,5-b′]二硒吩(14):
将化合物13(1.08g,2.0mmol)在N2和避光的条件下溶于无水CH2Cl2(35mL)中。将NBS(0.906g,5.09mmol)一次性加入,然后将混合物搅拌一小时或直至TLC表明反应完成。将混合物用己烷稀释,然后用H2O和盐水洗涤。干燥有机层并蒸发溶剂。将残余物用己烷通过柱洗脱,得到作为橙色油的纯5(1.05g,75%)。1H-NMR(CDCl3):δ7.68(s,2H),4.07,(d,J=5.5Hz,4H),1.80(m,2H),1.35-1.70(m,12H),1.03(t,J=4.9Hz,6H),0.94(t,J=4.8Hz,6H)。MS(EI):计算值698.3;实测值[M+1]+699.3。
2,6-双(三甲基锡)-4,8-双(2-乙基己氧基)苯并[1,2-b:4,5-b′]二硒吩(15):
将化合物14(0.08g,1.14mmol)在N2和避光的条件下溶于新蒸馏的THF(30mL)中。将溶液冷却至-78℃,然后滴加2.5M n-BuLi(1.14mL)的己烷溶液。搅拌15分钟后,让混合物缓慢升至室温,在此期间,溶液变得浑浊。向其中滴加1M SnMe3Cl(1.075mL)的己烷溶液,然后在室温下搅拌12小时。将混合物倾注进H2O中,然后用己烷提取。将有机层用H2O提取三次,干燥,得到缓慢结晶的橙色油。将残余物用异丙醇再次结晶,得到略带橙色的针状目标单体(0.74g,75%)。1H-NMR(CDCl3):δ7.86(s,2H),4.12(d,J=5.5Hz,4H),1.80(m,2H),1.35-1.70(m,12H),1.03(t,J=4.9Hz,6H),0.94(t,J=4.8Hz,6H),0.44(s,12H)。MS(EI):计算值866.1;实测值[M+1]+866.2)
PSeB2的合成。
称取化合物7(0.095g,0.173mmol)加到干燥的10mL圆底烧瓶中。添加化合物15(0.15g,0.173mmol)和Pd(PPh3)4(0.012g,0.0103mmol)。使烧瓶经受真空随后是再充注氩气的三个连续循环。然后,经由注射器加入无水DMF(1mL)和无水甲苯(3mL)。聚合反应在120℃和氩气保护下进行12小时。将粗产物沉淀进甲醇中,并通过过滤收集。将沉淀溶于氯仿,然后用硅藻土过滤以除去金属催化剂。通过在己烷中沉淀并在真空中干燥12小时,得到最终聚合物,产量为0.11g(45%)。1H-NMR(CDCl3):δ7.90-7.00(2H,br),4.70-3.90(6H,br),2.40-0.80(45H,br)。GPC:Mw(41×103g/mol),PDI(2.68)。
合成路线4:化合物20a-b和26的合成路线。

2-丁基己酸乙酯(16)
在氮气保护下将二异丙胺(45.5g,450mmol)溶于无水THF(150mL)中,然后在乙腈/干冰浴中冷却至-40℃。在搅拌下添加2.5M正丁基锂(n-BuLi)/己烷溶液(126mL,315mmol)。加入后,将混合物在冰水浴中保持30分钟,在丙酮/干冰浴中冷却至-78℃,并滴加己酸乙酯(43.3g,300mmol)。将反应混合物在相同温度下搅拌1小时,然后滴加碘丁烷(60.7g,330mmol)在1,3-二甲基-3,4,5,6-四氢-2(1H)-嘧啶酮(DMPU)(11.5g,90mmol)中的溶液。将所得的溶液升温至-40℃,搅拌2小时,保持在室温下过夜。将反应混合物用饱和的NH4Cl水溶液淬灭,然后通过旋转蒸发浓缩。将200mL Et2O和200mL水添加进烧瓶中。在用2N HCl水溶液酸化该混合物后,分离有机层,然后用10%(v/v)HCl水溶液(150mL×2)洗涤,并通过旋转蒸发浓缩。将所得的粗黄色油通过真空蒸馏纯化。收集作为无色油的化合物16(45.0g,75%)。1H NMR(CDCl3):δ0.86-0.90(6H,m),1.07-1.62(12H,m),1.25(3H,t),2.27-2.32(1H,m),4.12(2H,q)。MS(EI):计算值200.3;实测值M+H+201.2。
2-丁基己醇(17)
在氮气保护下将化合物16(54.1g,270mmol)溶于无水THF(450mL)中并在冰/水浴中冷却。将氢化铝锂(LAH)(15.3g,404mmol)分成小份添加进反应烧瓶中。将反应溶液的温度升至室温,然后回流过夜。将反应混合物冷却至室温,然后非常缓慢地倾注进冰/水溶液中。将溶液用2N HCl水溶液酸化,用Et2O(150mL×3)提取,然后通过旋转蒸发浓缩。将所得的粗制油通过真空蒸馏纯化。收集作为无色油的化合物17(32.6g,76.2%)。1H NMR(CDCl3):δ0.87-0.92(6H,m),1.17-1.42(12H,m),1.40-1.50(1H,m),3.53(2H,d)。MS(EI):计算值158.3;实测值M+H+159.2。
2-丁基己基溴(18)
在氩气保护下于室温下将溴(61.4g,384mmol)加入三苯基膦(100.7g,384mmol)和450mL二氯甲烷的溶液中。然后,经由另外的漏斗用时50分钟滴加2-丁基己醇(60.8g,384mmol),将反应溶液保持在室温下搅拌过夜。蒸发掉二氯甲烷,通过用戊烷洗涤而对浓缩物进行过滤。将滤液通过旋转蒸发浓缩,将所得的粗制黄色油通过真空蒸馏纯化。收集作为无色油的标题化合物(70.8g,83.3%)。1H NMR(CDCl3):δ0.89-0.92(6H,m),1.21-1.45(12H,m),1.54-1.67(1H,m),3.45(2H,d)。
1,4-二-2-乙基己基苯(20a)
将配有附接至氮气入口的回流冷凝器的500ml双颈烧瓶装入10.1g(0.422mol)镁屑。将反应混合物在真空下加热3分钟并将其冷却至室温后,充入200mL无水Et2O,然后滴加54.3g(0.281mol)2-乙基己基溴在50mL干乙醚中的溶液。将溶液回流3小时,然后冷却至室温。将制得的2-乙基己基溴化镁溶液用时一小时滴加至1,4-二溴苯(24.5g,0.104mol)与Ni(dppp)Cl2(0.564g,1.04mmol)在干乙醚(300mL)中的冰冷搅拌混合物中。移除冷却浴,将温度升至室温。然后将反应溶液回流过夜,冷却至0℃,再用水(50mL)接着用2N HCl(300mL)小心淬灭。在分离有机层后,将有机层用水(300mL)洗涤两次,用无水硫酸钠干燥,然后真空蒸发。柱层析(硅胶,己烷)得到纯化合物20a(18.2g,58%)。1H NMR(CDCl3):δ0.84-0.92(12H,m),1.25-1.35(16H,m),1.50-1.58(2H,m),2.50(4H,d),7.04(4H,s)。MS(EI):计算值302.5;实测值M+302.3。
1,4-二-2-丁基己基苯(20b)根据与20a相同的程序用1,4-二溴苯(24g,0.102mol)和2-丁基己基溴(54.1g,0.245mol)合成。(18.3g,50%)1H NMR(CDCl3):δ0.84-0.92(12H,m),1.23-1.34(24H,m),1.54-1.65(2H,m),2.49(4H,d),7.03(4H,S)。MS(EI):计算值358.6;实测值M+358.2。
1,4-二溴-2,5-双(2-乙基己基)苯(21a)
将催化碘添加至20a(17.9g,59.3mmol)的浓CH2Cl2溶液。在用铝箔包裹整个烧瓶以避光后,滴加溴(47.4g,0.297mol)。将反应物搅拌1天,然后添加200mL CH2Cl2。用亚硫酸氢钠水溶液除去过量的溴,然后分离有机部分。将有机部分用碳酸氢钠水溶液洗涤,并用无水硫酸钠干燥。移除溶剂,然后使用己烷在硅胶上进行柱纯化,得到目标产物(26.2g,96%)。1HNMR(CDCl3):δ0.81-0.91(12H,m),1.23-1.35(16H,m),1.64-1.75(2H,m),2.57(4H,d),7.31(4H,s)。MS(EI):计算值460.3;实测值M+460.0。
1,4-二溴-2,5-双(2-丁基己基)苯(21b)根据与21a相同的程序使用20b(16.6g,46.3mmol)合成。(22.7g,95%)1H NMR(CDCl3):δ0.85-0.91(12H,m),1.21-1.31(24H,m),1.65-1.75(2H,m),2.57(4H,d),7.31(4H,s)。MS(EI):计算值516.4;实测值M+516.2。
2,2’-(2,5-双(2-乙基己基)-1,4-亚苯基)双(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷)(22a)。
将装有在250mL DMSO中的21a(26.1g,56.8mmol)、双(频那醇合)二硼(34.5g,0.136mol)、PdCl2(dppf)(2.5g,6mol%)和乙酸钾(33.4g,0.341mol)的烧瓶在氮气氛下于80℃搅拌过夜。冷却后,将溶液真空浓缩,用150mLCH2Cl2稀释,用水和盐水洗涤,干燥(无水Na2SO4),然后蒸发掉溶剂。将粗产物通过柱层析(硅胶,己烷/EtOAc=20/1)纯化。(9.4g,30%)。1HNMR(CDCl3):δ0.86-0.90(12H,m),1.15-1.31(16H,m),1.33(24H,s),1.49-1.57(2H,m),2.77(4H,d),7.48(4H,s)。MS(EI):计算值554.5;实测值M+554.9。
2,2’-(2,5-双(2-丁基己基)-1,4-亚苯基)双(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷)(22b)根据与22a相同的程序使用21b(10.4g,20.2mmol)合成。(3.1g,25%)1H NMR(CDCl3):δ0.83-0.90(12H,m),1.17-1.33(24H,m),1.34(24H,s),1.67-1.78(2H,m),2.78(4H,d),7.48(4H,S)。MS(EI):计算值610.6;实测值M+,610.9。
3-甲硫基噻吩(24)。
将3-溴噻吩(8.7g,53.4mmol)在氩气氛下溶于干乙醚(200mL)中,然后将溶液冷却至–78℃。滴加2.5M n-BuLi的己烷溶液(21.5mL,53.8mmol),将反应混合物在–78℃下再搅拌20分钟。然后滴加二甲基二硫醚(12.5g,0.133mol),将反应混合物搅拌30分钟,然后升至室温。然后将混合物搅拌过夜。将反应溶液使用饱和氯化铵溶液淬灭。将分离的有机相用水(2×150mL)洗涤并干燥。通过旋转蒸发除去溶剂,将所得的粗制黄色油通过真空蒸馏纯化。(3.5g,50%)1H NMR(CDCl3):δ2.52(3H,S),6.97(1H,q),7.0(1H,q),7.32(1H,q)。MS(EI):计算值130.2;实测值M+129.8。
2-溴-3-甲硫基噻吩(25)
在氮气氛下于0℃向化合物24(6.69g,51.34mmol)在乙酸(50mL)和CH2Cl2(50mL)中的搅拌溶液分批加入N-溴代琥珀酰亚胺(NBS)(9.14g,51.34mmol)。让所得的反应混合物升至室温并搅拌过夜。将溶剂在真空下蒸发,然后添加CH2Cl2(150mL)和水(150mL)。分离有机相,然后用水(2×150mL)洗涤。将有机提取物用无水硫酸钠干燥,然后真空蒸发。柱层析(硅胶,己烷)得到纯的化合物25。(5.37g,50%)1H NMR(CDCl3):δ2.43(3H,s),6.89(1H,d),7.25(1H,d)。MS(EI):计算值209.1;实测值M+209.9。
2-溴-3-甲基亚磺酰基噻吩(26)
在-40℃下向化合物25(3.64g,17.4mmol)在CH2Cl2(100mL)中的溶液滴加3-氯过苯甲酸(3g,17.4mmol)在CH2Cl2(50mL)中的溶液,然后使所得的溶液在室温下搅拌过夜。将反应混合物用Na2CO3水溶液洗涤,用无水硫酸钠干燥并真空蒸发。使用柱层析(硅胶,己烷/EtOAc(1/2))得到纯化合物26(2.74g,70%)。1H NMR(CDCl3):δ2.82(3H,s),7.35(1H,d),7.43(1H,d)。MS(EI):计算值225.1;实测值M+210.1=产物-16。
27a:
在氩气氛下将化合物22a(3.09g,5.57mmol)、化合物26(3.01g,13.4mmol)、2M K2CO3水溶液(11g,40mL)和Pd(PPh3)4(644mg,10mol%)在甲苯/乙醇(60/15mL)中混合。将混合物在110℃下加热24小时,然后冷却。将溶液真空浓缩,然后添加CH2Cl2(150mL)和水(150mL)。将有机层用水和盐水洗涤,干燥(无水Na2SO4),然后蒸发掉溶剂。将粗产物通过柱层析(硅胶,己烷/THF=10/1)纯化。(2.4g,73%)。1H NMR(CDCl3):δ0.69-0.83(12H,m),1.11-1.26(16H,m),1.30-1.45(2H,m),2.40-2.64(4H,m),2.71(2H,s),2.81(4H,s),7.29(2H,d),7.50-7.60(4H,m)。MS(MALDI-TOF):计算值591.0;实测值M+587.3。
27b:
在氩气氛下将化合物22b(1.75g,2.87mmol)、化合物26(1.55g,6.88mmol)、2M NaOH水溶液(1.68g,21mL)和Pd(PPh3)4(332mg,10mol%)在甲苯/乙醇(34/10mL)中混合。将混合物在110℃下加热24小时,然后冷却。将溶液真空浓缩并添加CH2Cl2(100mL)和水(100mL)。将有机层用水和盐水洗涤,干燥(无水Na2SO4),然后蒸发掉溶剂。将粗产物通过柱层析(硅胶,己烷/THF=20/1)纯化。(1.2g,65%)。1H NMR(CDCl3):δ0.71-0.85(12H,m),1.13-1.28(24H,m),1.40-1.53(2H,m),2.38-2.61(4H,m),2.69(2H,s),2.78(4H,s),7.31(2H,d),7.50-7.60(4H,m)。MS(MALDI-TOF):计算值647.1;实测值M+646.3。
29a:
将烧瓶装上化合物27a(6.9g,11.7mmol)、五氧化二磷(627mg,4.42mmol)和三氟甲磺酸(150mL)。将混合物在室温下搅拌3天,然后倾注入冰水(1L)中。通过抽滤收集棕色沉淀并真空干燥。将粗化合物溶于吡啶(200mL)中并在140℃加热过夜。将反应溶液冷却至室温,真空浓缩,然后添加150mLCH2Cl2和150mL2N HCl水溶液。将提取的有机相用2N HCl水溶液(3×150mL)洗涤,用无水硫酸钠干燥并真空蒸发。柱层析(硅胶,己烷)得到纯化合物29a(2.47g,40%)。1H NMR(CDCl3):δ0.81-0.92(12H,m),1.20-1.59(16H,m),2.17-2.21(2H,m),3.21-3.31(4H,m),7.36(2H,d),7.56(2H,d)。MS(MALDI-TOF):计算值526.9;实测值M+527.0。
29b根据与29a相同的程序使用27b(1.9g,2.93mmol)合成。(0.34g,20%)1H NMR(CDCl3):δ0.82-0.94(12H,m),1.20-1.61(24H,m),2.15-2.22(2H,m),3.19-3.30(4H,m),7.34(2H,d),7.53(2H,d)。MS(MALDI-TOF):计算值583.0;实测值M+583.2。
30a
将化合物29a(0.266g,0.505mmol)溶于4mL CHCl3。将0.5mL CHCl3中的溴(0.052mL,1.01mmol)溶液滴加至该反应溶液。将所得的混合物在室温下搅拌6小时。将该溶液倾注进处于冰水浴中的饱和亚硫酸钠溶液并用二氯甲烷提取。将有机提取物用无水硫酸钠干燥,然后真空蒸发。使用己烷和二氯甲烷混合洗脱液在硅胶上进行柱层析,得到化合物30a(0.286g,83%)。1H NMR(CDCl3):δ0.82(6H,t),0.82-0.89(6H,t)1.21-1.55(16H,m),2.10-2.13(2H,m),3.13(4H,q),7.35(2H,s)。MS(MALDI-TOF):计算值684.7;实测值M+685.6。
30b根据与30a相同的程序使用29b(0.33g,0.566mmol)合成。(0.31g,74%)1H NMR(CDCl3):δ0.79(12H,t),1.18-1.51(24H,m),2.12-2.22(2H,m),3.13(4H,d),7.36(2H,s)。MS(MALDI-TOF):计算值740.8;实测值M+741.6。
31a
在氮气保护下将化合物30a(0.3g,0.43mmol)溶于10mL无水THF中,并在丙酮/干冰浴中冷却至-78℃。伴随搅拌添加2.5M n-BuLi/己烷溶液(0.35mL,0.88mmol)。添加后,将混合物保持在干冰浴中25分钟,滴加三甲基氯化锡溶液(1.12mL,1.12mmol,1M溶液),然后将该混合物在室温下搅拌过夜。将混合物用20mL水淬灭并用二乙醚提取。将有机相用无水硫酸钠干燥并真空蒸发。将残余物从CH2Cl2和异丙醇再结晶,得到化合物31a(0.22g,60%)。1H NMR(CDCl3):δ0.49(18H,S),0.86-0.94(12H,m),1.25-1.61(16H,m),2.15-2.29(2H,m),3.23-3.32(4H,m),7.41(2H,s)。
31b根据与31a相同的程序使用30b(0.31g,0.42mmol)合成。(336mg,88%)1H NMR(CDCl3):δ0.46(18H,S),0.80(12H,t),1.18-1.50(24H,m),2.20-2.28(2H,m),3.26(4H,d),7.38(2H,s)。
聚合物的合成
PTDBD2
称取4,6-二溴噻吩并[3,4-b]噻吩-2-羧酸-2-丁基辛酯(32.7mg,0.062mmol)加到10mL圆底烧瓶中。添加31b(56.3mg,0.062mmol)和Pd(PPh3)4(3mg)。使烧瓶经受真空随后是再充注氩气的五个连续循环。然后,经由注射器添加无水DMF(0.4mL)和无水甲苯(1.6mL)。在氮气保护下于120℃进行聚合反应12小时。将粗产物沉淀进甲醇中,并通过过滤收集。使用己烷、甲醇、氯仿和氯苯通过索氏提取而将聚合物纯化。真空干燥过夜后,获得来自氯苯级分的所得固体(30mg,50%)。1H NMR(CDCl2CDCl2):δ0.50-2.10(61H,br),2.50-2.90(4H,br),4.0-4.50(2H,br),7.20-7.70(2H,br)。GPC:Mw(108.6×103g/mol),PDI(2.82)。
PTDBD3根据与PTDBD2相似的程序使用相应的单体合成。聚合物的1H NMR和凝胶渗透色谱(GPC)数据在下面列出。
1H NMR(CDCl2CDCl2):δ0.40-2.20(72H,br),2.50-2.90(4H,br),4.0-4.40(1H,br),7.20-7.70(2H,br)。GPC:Mw(105.5×103g/mol),PDI(2.34)。
器件制造。
在80℃下将聚合物PSeB1和PSeB2以及富勒烯受体以10mg/ml的PTB1浓度共同溶于溶剂中5小时。将涂布有ITO的玻璃基板在超声下逐步于水、丙酮和异丙醇中清洁,各进行10至30分钟,然后在烘箱中干燥5小时。随后将ITO用臭氧清洁。将PEDOT(聚(乙烯二氧噻吩)):PSS(聚(苯乙烯磺酸))(Baytron P VP A14083)的薄层(~30nm)旋涂于ITO表面上。在120℃下烘焙约20分钟后,将基板转移进充满氮气的手套箱(<0.1ppm O2&H2O)中。然后由共混溶液以800-1000rpm在ITO/PEDOT:PSS基板上旋转浇注约100nm厚的聚合物/PCBM复合层,不作进一步特殊处理。然后,将膜转移进位于同一手套箱中的热蒸发仪中。在2×10-6托的真空下依次沉积25nm的Ca层和80nm的Al层。测得有效区域的面积为0.095cm2。
仪器。
紫外-可见光吸收和循环伏安法
通过Shimadzu UV-2401PC分光光度计采集光学吸收光谱。使用循环伏安法(CV)来研究聚合物的电化学特性。在0.10M四丁基六氟磷酸铵(Bu4NPF6)的乙腈溶液中以100mVs-1的扫描速率研究涂布在玻璃碳电极上的聚合物薄膜。为了校准,在相同的条件下测量二茂铁/二茂铁 (Fc/Fc+)的氧化还原电位,相对于Ag/Ag+电极其位于0.1V。假设Fc/Fc+的氧化还原电位相对于真空的绝对能级为-4.80eV。(G.Dennler,G.等人,Adv.Mater.(《先进材料》)2009,21,1323–1338;Scharber,M.等人,Adv.Mater.(《先进材料》)2006,18,789-794;Coakley,K.M.等人,Chem.Mater.(《材料化学》)2004,16,4533-4542)。
(Fc/Fc+)的氧化还原电位,相对于Ag/Ag+电极其位于0.1V。假设Fc/Fc+的氧化还原电位相对于真空的绝对能级为-4.80eV。(G.Dennler,G.等人,Adv.Mater.(《先进材料》)2009,21,1323–1338;Scharber,M.等人,Adv.Mater.(《先进材料》)2006,18,789-794;Coakley,K.M.等人,Chem.Mater.(《材料化学》)2004,16,4533-4542)。
然后根据如下公式计算最高(HOMO)和最低未占分子轨道(LUMO)的能级:
EHOMO=-(φox+4.74)eV;ELUMO=-(φred+4.74)eV
其中φox是来自CV数据的第二次扫描的起始氧化电位。
空穴迁移率。
空穴迁移率通过采集0-6V范围内的电流-电压电流并将结果拟合为空间电荷限制形式,使用ITO/PEDOT:PSS/聚合物/Al的二极管构造根据文献中所述的相似方法进行测量,其中空间电荷限制电流(SCLC)由J=9ε0εμV2/8L3表述,其中ε0是真空电容率,εr是聚合物的介电常数,μ是空穴迁移率,V是横跨器件的电压降(V=Vappl–Vr–Vbi,Vappl:向器件施加的电压;Vr:由于整个电极的接触电阻和串联电阻而导致的电压降;Vbi:由于两个电极的逸出功差异导致的内建电压),并且L为聚合物膜厚度。器件的电阻使用空白构造ITO/PEDOT:PSS/Al进行测量,并测得为约10-20Ω。Vbi由J0.5与Vappl的关系图在2.5V以上电压处的最佳拟合推导出,并测得为约1.5V。介电常数εr在分析中假定为3,该值是共轭聚合物的典型值。聚合物膜的厚度通过使用AFM进行测量。
太阳能电池制造和电流-电压测量
以优化的重量比将聚合物与PCBM共同溶解于含有或不含3%(v/v)1,8-二碘辛烷的氯苯(CB)中。聚合物浓度为10mg/mL。将涂布有ITO的玻璃基板(15Ω/sq)在超声下逐步于清洁剂、水、丙酮和异丙醇中清洁,各进行15分钟,然后在烘箱中在80℃和真空下干燥1分钟。然后在用紫外线臭氧处理20分钟后,以4000rpm将PEDOT:PSS的薄层旋涂至ITO表面上。在80℃和真空下烘焙45分钟后,将基板转移进充满氮气的手套箱(<0.1ppmO2和H2O)中,以形成活性涂层和电极。然后由共混溶液将聚合物/PCBM复合层旋转浇注于该基板上。旋涂速度为1200rpm。然后,将膜转移至位于同一手套箱中的热蒸发仪中。在2×10-6托的真空压力下依次沉积20nmCa层和60nm Al层。膜的有效面积被测量为0.0314cm2。
使用Keithley2420源测量单元在充满氮气的手套箱中测量电流密度-电压(J-V)曲线。在Newport Thermal Oriel Sol3A AAA级450W太阳模拟器(型号:94023A,2英寸×2英寸光束尺寸)下,以100mW/cm2于AM1.5G照度下测量光电流。重复用于器件制造的条件以确保可重复性。外部量子效率(EQE)测量使用Newport Oriel IQE200系统进行。
掠入射广角X射线衍射(GIWAXS)
GIWAXS测量在高级光子源阿贡国家实验室(Advanced Photon Source(APS),Argonne National Laboratory)使用波长λ=1.6868 而光束尺寸为约100μm(h)和50μm(v)的X射线在8ID-E光束线下进行。为了使结果与OPV器件的那些结果相当,在与用于制造太阳能电池器件的那些相同的条件下在PEDOT:PSS改性的Si基板上制备用于测量的样品。使用2-D PILATUS1M-F检测器捕获衍射图,该检测器位于距离样品208.7mm处。典型的GISAXS图以0.20°的入射角采集,其高于PTDBD聚合物或PTDBD:PCBM共混物的临界角并低于硅基板的临界角。因此,可检测薄膜的整个结构。此外,通过穿过反射光束中心的线切得到了qy线切,而qz线切则使用作为零的反射光束中心通过qy=0A-1处的线切实现。这些线切的背景通过拟合指数函数进行估计,并且衍射峰的参数通过使用Pseudo-Voigt1型峰函数通过最佳拟合获得。
而光束尺寸为约100μm(h)和50μm(v)的X射线在8ID-E光束线下进行。为了使结果与OPV器件的那些结果相当,在与用于制造太阳能电池器件的那些相同的条件下在PEDOT:PSS改性的Si基板上制备用于测量的样品。使用2-D PILATUS1M-F检测器捕获衍射图,该检测器位于距离样品208.7mm处。典型的GISAXS图以0.20°的入射角采集,其高于PTDBD聚合物或PTDBD:PCBM共混物的临界角并低于硅基板的临界角。因此,可检测薄膜的整个结构。此外,通过穿过反射光束中心的线切得到了qy线切,而qz线切则使用作为零的反射光束中心通过qy=0A-1处的线切实现。这些线切的背景通过拟合指数函数进行估计,并且衍射峰的参数通过使用Pseudo-Voigt1型峰函数通过最佳拟合获得。
透射电子显微镜(TEM)测量
TEM测量在EI Tecnai F30(300KV)的Gatan CCD数码显微照片上进行。用于TEM的聚合物/PCBM膜的制备条件与用于在涂布有PEDOT:PSS的ITO基板上制造器件的那些条件相同。将样品浸入水中,然后将漂浮到水面上的活性层转移至TEM网格。
受益于以上说明中提供的教导,本公开所属领域的技术人员将想到本公开的许多修改形式和其他实施例。对本领域技术人员将显而易见的是,可在不脱离本公开的范围或精神的情况下得到本公开的变型形式和修改形式。因此,应当理解,本发明不限于所公开的具体实施例,并且修改形式和其他实施例旨在包括在所附权利要求书的范围内。虽然本文采用了特定的术语,但这些术语仅用于一般性和描述性目的,并不旨在进行任何限制。
Claims (39)
1.一种具有选自式(I)-(VI)的化学式的半导体聚合物:

其中X1、X2、X3和X4独立地选自O、S、Se、NH和CH2;
R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇);
Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基;
Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基;
Z1和Z2独立地选自CH和N;
W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基;并且n为大于0的整数。
2.一种具有选自式(I)-(V)的化学式的半导体聚合物:
其中X1、X2、X3和X4独立地选自O、S、Se、NH和CH2;
R1、R2、R3和R4独立地选自H、烷基、烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、可交联部分和低聚(乙二醇);
Y1、Y2、Y3、Y4、Y5和Y6独立地选自O、S、Se和氨基;
Z选自酯、酮、酰胺、氰基、烷基、多氟烷基、多氯烷基、芳基和杂芳基;
Z1和Z2独立地选自CH和N;
W选自H、卤素、氰基、二氰基乙烯基和三氰基乙烯基;并且n为大于0的整数。
3.根据权利要求1-2中任一项所述的半导体聚合物,其具有式(I):

4.根据权利要求1-2中任一项所述的半导体聚合物,其具有式(II):

5.根据权利要求1-2中任一项所述的半导体聚合物,其具有式(III):

6.根据权利要求1-2中任一项所述的半导体聚合物,其具有式(IV):

7.根据权利要求1-2中任一项所述的半导体聚合物,其具有式(V):

8.根据权利要求7所述的半导体聚合物,其中X1和X2独立地选自O、S、NH和CH2。
9.根据权利要求7-8中任一项所述的半导体聚合物,其中X1和X2独立地为O。
10.根据权利要求7-9中任一项所述的半导体聚合物,其中R1和R2独立地为烷基。
11.根据权利要求7-10中任一项所述的半导体聚合物,其中R1和R2独立地为C1-30烷基。
12.根据权利要求7-11中任一项所述的半导体聚合物,其中R1和R2相同。
13.根据权利要求7-11中任一项所述的半导体聚合物,其中R1和R2中的任一者为2-乙基己基。
14.根据权利要求7-13中任一项所述的半导体聚合物,其中Y1和Y2为S。
15.根据权利要求7-13中任一项所述的半导体聚合物,其中Y1和Y2为Se。
16.根据权利要求7-15中任一项所述的半导体聚合物,其中Y3和Y4为Se。
17.根据权利要求7-16中任一项所述的半导体聚合物,其中Z为酯。
18.根据权利要求7-17中任一项所述的半导体聚合物,其中Z为-C(O)OCH2CH(C2H5)C4H9。
19.根据权利要求7-18中任一项所述的半导体聚合物,其中W为H或卤素。
20.根据权利要求7-19中任一项所述的半导体聚合物,其中W为F。
21.根据权利要求7所述的半导体聚合物,其具有选自

22.根据权利要求21所述的半导体聚合物,其中R1、R2和R3独立地为烷基。
23.根据权利要求21和22中任一项所述的半导体聚合物,其中R1、R2和R3独立地为2-乙基己基。
24.根据权利要求1-2中任一项所述的半导体聚合物,其具有式(VI):

25.根据权利要求24所述的半导体聚合物,其中R1和R2独立地为C1-30烷基。
26.根据权利要求24和25中任一项所述的半导体聚合物,其中R1和R2独立地为2-乙基己基或2-丁基己基。
27.根据权利要求24-26中任一项所述的半导体聚合物,其中Y1、Y2、Y3、Y4、Y5和Y6独立地为S。
28.根据权利要求24-27中任一项所述的半导体聚合物,其中W为卤素。
29.根据权利要求24-28中任一项所述的半导体聚合物,其中W为F。
30.根据权利要求24-29中任一项所述的半导体聚合物,其中Z为酯。
31.根据权利要求24-30中任一项所述的半导体聚合物,其中Z选自-C(O)OCH2CH(C2H5)C4H9和-C(O)OCH2CH(C4H9)C6H13。
32.根据权利要求24所述的半导体聚合物,其选自式PTDBD1、PTDBD2和PTDBD3:
33.根据权利要求1-32中任一项所述的半导体聚合物,其中所述聚合物的数均分子量在约15至约25kDa之间。
34.根据权利要求1-32中任一项所述的半导体聚合物,其中n为约1至约200。
35.根据权利要求1-32中任一项所述的半导体聚合物,其中多分散指数在约1.5至约3之间。
36.一种包含权利要求1-35中任一项所述的半导体聚合物和吸电子的富勒烯衍生物的组合物。
37.根据权利要求36所述的组合物,其中所述吸电子的富勒烯衍生物选自[6,6]-苯基-C61-丁酸甲酯和[6,6]-苯基-C71-丁酸甲酯。
38.权利要求1-35中任一项所述的半导体聚合物在太阳能电池、光学器件、电致发光器件、光伏电池、半导体电池或光电二极管中的用途。
39.权利要求36和37中任一项所述的组合物在太阳能电池、光学器件、电致发光器件、光伏电池、半导体电池或光电二极管中的用途。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161517205P | 2011-04-15 | 2011-04-15 | |
| US61/517,205 | 2011-04-15 | ||
| PCT/US2012/033601 WO2012142469A1 (en) | 2011-04-15 | 2012-04-13 | Semiconducting polymers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103619902A true CN103619902A (zh) | 2014-03-05 |
Family
ID=47009715
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280028860.5A Pending CN103619902A (zh) | 2011-04-15 | 2012-04-13 | 半导体聚合物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9153785B2 (zh) |
| EP (1) | EP2697283A4 (zh) |
| JP (1) | JP2014518562A (zh) |
| KR (1) | KR101545429B1 (zh) |
| CN (1) | CN103619902A (zh) |
| WO (1) | WO2012142469A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110317210A (zh) * | 2018-03-30 | 2019-10-11 | 中国科学院化学研究所 | 平面茚并茚—二噻吩类光伏受体材料、其制备方法及应用 |
| CN111269083A (zh) * | 2020-02-24 | 2020-06-12 | 珠海市柏瑞医药科技有限公司 | 一种格尔伯特酸的合成方法 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103687891A (zh) * | 2011-09-23 | 2014-03-26 | 海洋王照明科技股份有限公司 | 二氟代苯并三唑基太阳能电池聚合材料及其制备方法和应用 |
| US9006568B2 (en) | 2012-02-15 | 2015-04-14 | Phillips 66 Company | Synthesis of photovoltaic conjugated polymers |
| US9691986B2 (en) | 2012-12-03 | 2017-06-27 | Solarmer Energy, Inc. | Furan and selenophene derivatized benzo [1,2-b:4,5-b′] dithiophene-thienothiophene based conjugated polymers for high-efficiency organic solar cells |
| KR101563048B1 (ko) | 2013-05-10 | 2015-10-30 | 주식회사 엘지화학 | 광활성층, 이를 포함하는 유기 태양 전지 및 이의 제조 방법 |
| CN103626976B (zh) * | 2013-11-22 | 2016-08-17 | 武汉理工大学 | 苯并[2,1-b:3,4-b′]二硒吩类聚合物半导体材料及其应用 |
| US10818849B2 (en) * | 2015-12-29 | 2020-10-27 | The University Of Chicago | Electron acceptors based on alpha-position substituted PDI for OPV solar cells |
| JP2017149689A (ja) * | 2016-02-25 | 2017-08-31 | 株式会社トクヤマ | クロロプロパン類の製造方法 |
| CN111349003B (zh) * | 2018-12-20 | 2023-10-03 | 四川科瑞德制药股份有限公司 | 一种丙戊酸钠的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101170161A (zh) * | 2006-10-25 | 2008-04-30 | 施乐公司 | 电子器件 |
| CN101343351A (zh) * | 2007-07-13 | 2009-01-14 | 气体产品与化学公司 | 含有硒的导电性聚合物以及制备导电性聚合物的方法 |
| WO2010008672A1 (en) * | 2008-07-18 | 2010-01-21 | University Of Chicago | Semiconducting polymers |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7432340B2 (en) | 2002-07-11 | 2008-10-07 | Air Products And Chemicals, Inc. | Fluorinated alkyl substituted-thieno[3,4-]thiophene monomers and polymers therefrom |
| US7071289B2 (en) | 2002-07-11 | 2006-07-04 | The University Of Connecticut | Polymers comprising thieno [3,4-b]thiophene and methods of making and using the same |
| ATE413423T1 (de) | 2003-08-28 | 2008-11-15 | Merck Patent Gmbh | Mono-, oligo- und polythieno(2,3-b)thiophene |
| DE602004024629D1 (de) | 2003-10-15 | 2010-01-28 | Merck Patent Gmbh | Polybenzodithiophene |
| EP2044082A1 (en) | 2006-07-26 | 2009-04-08 | Merck Patent GmbH | Substituted benzodithiophenes and benzodiselenophenes |
| US7834132B2 (en) * | 2006-10-25 | 2010-11-16 | Xerox Corporation | Electronic devices |
| US7982055B2 (en) * | 2007-07-13 | 2011-07-19 | Konarka Technologies, Inc. | Heterocyclic fused selenophene monomers |
| US20090140219A1 (en) * | 2007-07-13 | 2009-06-04 | Air Products And Chemicals, Inc. | Selenium Containing Electrically Conductive Polymers and Method of Making Electrically Conductive Polymers |
| DE112009001505T5 (de) * | 2008-08-18 | 2011-07-14 | Merck Patent GmbH, 64293 | Indacenodithiophen- und Indacenodiselenophen-Polymere und ihre Verwendung als organische Halbleiter |
| US8372945B2 (en) * | 2009-07-24 | 2013-02-12 | Solarmer Energy, Inc. | Conjugated polymers with carbonyl substituted thieno[3,4-B]thiophene units for polymer solar cell active layer materials |
| KR101780083B1 (ko) | 2009-12-02 | 2017-10-10 | 바스프 에스이 | 디티에노벤조-티에노[3,2-b]티오펜 공중합체 및 고성능 용액 공정 가능한 반도체 중합체로서 이의 용도 |
| WO2011156478A2 (en) | 2010-06-08 | 2011-12-15 | The University Of North Carolina At Chapel Hill | Polymers with tunable band gaps for photonic and electronic applications |
-
2012
- 2012-04-13 US US14/111,473 patent/US9153785B2/en active Active
- 2012-04-13 CN CN201280028860.5A patent/CN103619902A/zh active Pending
- 2012-04-13 JP JP2014505356A patent/JP2014518562A/ja active Pending
- 2012-04-13 KR KR1020137030162A patent/KR101545429B1/ko not_active IP Right Cessation
- 2012-04-13 EP EP12771160.4A patent/EP2697283A4/en not_active Withdrawn
- 2012-04-13 WO PCT/US2012/033601 patent/WO2012142469A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101170161A (zh) * | 2006-10-25 | 2008-04-30 | 施乐公司 | 电子器件 |
| CN101343351A (zh) * | 2007-07-13 | 2009-01-14 | 气体产品与化学公司 | 含有硒的导电性聚合物以及制备导电性聚合物的方法 |
| WO2010008672A1 (en) * | 2008-07-18 | 2010-01-21 | University Of Chicago | Semiconducting polymers |
Non-Patent Citations (1)
| Title |
|---|
| ASIT PATRA, ET AL.: ""Tuning the Band Gap of Low-Band-Gap Polyselenophenes and Polythiophenes: The Effect of the Heteroatom"", 《CHEM. MATER.》, vol. 23, no. 3, 11 January 2011 (2011-01-11), pages 896 - 906 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110317210A (zh) * | 2018-03-30 | 2019-10-11 | 中国科学院化学研究所 | 平面茚并茚—二噻吩类光伏受体材料、其制备方法及应用 |
| CN111269083A (zh) * | 2020-02-24 | 2020-06-12 | 珠海市柏瑞医药科技有限公司 | 一种格尔伯特酸的合成方法 |
| CN111269083B (zh) * | 2020-02-24 | 2023-08-25 | 珠海市柏瑞医药科技有限公司 | 一种格尔伯特酸的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2697283A1 (en) | 2014-02-19 |
| EP2697283A4 (en) | 2015-12-16 |
| US20140145119A1 (en) | 2014-05-29 |
| KR20140010156A (ko) | 2014-01-23 |
| KR101545429B1 (ko) | 2015-08-18 |
| WO2012142469A1 (en) | 2012-10-18 |
| JP2014518562A (ja) | 2014-07-31 |
| US9153785B2 (en) | 2015-10-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103619902A (zh) | 半导体聚合物 | |
| CN102439059B (zh) | 共轭聚合物及其在光电子器件中的用途 | |
| CN102149750B (zh) | 半导体聚合物 | |
| CN102460758B (zh) | 半导体化合物和包含该半导体化合物的设备 | |
| CN108864137B (zh) | 一种受体化合物、制备方法、用途以及含有其的光伏电池 | |
| CN102762631B (zh) | 含芘导电聚合物及包括含芘导电聚合物的有机太阳能电池 | |
| CN103547582B (zh) | 用于电子应用的含稠合噻吩环的化合物及其聚合物 | |
| Tao et al. | Wide bandgap copolymers with vertical benzodithiophene dicarboxylate for high-performance polymer solar cells with an efficiency up to 7.49% | |
| Qiu et al. | The crystallinity control of polymer donor materials for high-performance organic solar cells | |
| CN103288848A (zh) | 苯并三噻吩类化合物及其制备方法和用途 | |
| CA2863606A1 (en) | Semiconducting polymers | |
| EP2530084B1 (en) | Copolymer containing fluorenylporphyrin-anthracene, preparation method and use thereof | |
| CN109912621B (zh) | 一种不对称的萘核小分子受体材料及其制备方法和应用 | |
| CN114014873B (zh) | 一类基于C2v对称氮杂稠环的化合物及其制备方法与应用 | |
| CN113527641B (zh) | 一类基于酯基侧链取代喹喔啉衍生物的聚合物材料及应用 | |
| CN109265656B (zh) | 双烷硫链取代的共轭聚合物及其制备和应用 | |
| CN108192083B (zh) | 含三氟甲基的共轭聚合物及其制备方法和应用 | |
| CN111039961A (zh) | 基于5, 6-双氟苯并噻二唑单元的齐聚物光伏供体材料的制备及应用 | |
| Kim et al. | 2, 2-dimethyl-2 H-benzimidazole based small molecules for organic solar cells | |
| Ming et al. | Fused-ring acceptor with a spiro-bridged ladder-type core for organic solar cells | |
| JP5667693B2 (ja) | キノキサリン単位含有ポルフィリン共重合体及びその製造方法、並びにその応用 | |
| Li et al. | Novel benzodithiophene unit with an alkylthiobiphenyl side chain for constructing high-efficiency polymer solar cells | |
| Wang et al. | D-(π-A) 3 type low bandgap star-shaped fused-ring electron acceptor with alkoxy-substituted thiophene as π-bridge | |
| KR101702306B1 (ko) | 신규한 유기반도체 화합물 및 이를 이용한 유기 전자 소자 | |
| CN109776568B (zh) | 一种轴对称的六元桥环萘核小分子受体材料及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20140305 |