CN102858338A - 治疗疼痛及其它适应症的药物组合物 - Google Patents
治疗疼痛及其它适应症的药物组合物 Download PDFInfo
- Publication number
- CN102858338A CN102858338A CN2011800171616A CN201180017161A CN102858338A CN 102858338 A CN102858338 A CN 102858338A CN 2011800171616 A CN2011800171616 A CN 2011800171616A CN 201180017161 A CN201180017161 A CN 201180017161A CN 102858338 A CN102858338 A CN 102858338A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- optionally
- group
- halogeno
- hydroxyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 C*c(nc(*)[o]1)c1S*C Chemical compound C*c(nc(*)[o]1)c1S*C 0.000 description 9
- YTKBQEQZRIKXHF-UHFFFAOYSA-N C=CCC(c1cnc(cc(cc2)-c3c[o]c(-c(cc4)ccc4F)n3)c2c1)F Chemical compound C=CCC(c1cnc(cc(cc2)-c3c[o]c(-c(cc4)ccc4F)n3)c2c1)F YTKBQEQZRIKXHF-UHFFFAOYSA-N 0.000 description 1
- FRSDVKAQJOMFIF-UHFFFAOYSA-N CC(c(nc1)ccc1-c(nc(C)[o]1)c1Sc(nc1)ccc1Cl)=O Chemical compound CC(c(nc1)ccc1-c(nc(C)[o]1)c1Sc(nc1)ccc1Cl)=O FRSDVKAQJOMFIF-UHFFFAOYSA-N 0.000 description 1
- CAZDNMWZZXLBSI-UHFFFAOYSA-N CSc(nn1)ccc1Cl Chemical compound CSc(nn1)ccc1Cl CAZDNMWZZXLBSI-UHFFFAOYSA-N 0.000 description 1
- ZWLYWUZOMJSBGF-UHFFFAOYSA-N Cc(cc1)ccc1-c1c[o]c(-c2ncccc2)n1 Chemical compound Cc(cc1)ccc1-c1c[o]c(-c2ncccc2)n1 ZWLYWUZOMJSBGF-UHFFFAOYSA-N 0.000 description 1
Images
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Landscapes
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Pulmonology (AREA)
- Ophthalmology & Optometry (AREA)
- Endocrinology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Anesthesiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Addiction (AREA)
- Child & Adolescent Psychology (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Gastroenterology & Hepatology (AREA)
- Dermatology (AREA)
- Obesity (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Nutrition Science (AREA)
Abstract
本发明涉及可用于治疗FAAH介导的疾病、障碍或病症的组合物,其包含FAAH抑制剂和第二个活性剂,包括选择的咪唑或噁唑FAAH抑制剂和第二个活性剂。该组合物可用于治疗许多疾病、障碍或病症,包括骨关节炎,类风湿性关节炎,糖尿病性神经病变,带状疱疹神经痛,骨骼肌疼痛和肌纤维痛,以及急性疼痛,偏头痛,睡眠障碍,阿尔茨海默氏病和帕金森氏症。在另一个方面,本发明涉及用于治疗神经性和感受伤害性疼痛的组合物,所述组合物包含艾托考昔。
Description
本发明的背景
在一方面,本文公开的本发明涉及用于治疗疼痛及其它FAAH介导的疾病、障碍和病症的组合物。尤其是,本文公开的本发明涉及药物组合物,其包含选择的FAAH抑制剂和第二个活性剂。
在另一个方面,本文公开的本发明涉及用于治疗神经性和感受伤害性疼痛的组合物,所述组合物包含艾托考昔。
在另一个方面,本文公开的本发明涉及用于治疗神经性和感受伤害性疼痛的组合物,所述组合物包含艾托考昔和选择的FAAH抑制剂。
本文公开了抑制脂肪酸酰胺水解酶(FAAH)活性的化合物、包含该化合物的组合物和它们的使用方法。本文公开的化合物作为脂肪酸酰胺水解酶(FAAH)的抑制剂可有效用于治疗疾病、障碍或病症,这种疾病、障碍或病症可受益于抑制脂肪酸酰胺水解酶和内源性的脂肪酸酰胺的提高。
脂肪酸酰胺水解酶(FAAH)是在整个CNS中(Freund等人,Physiol. Rev. 2003;83:1017-1066)以及周围组织中丰富表达的酶,例如,在胰腺、脑、肾脏、骨骼肌、胎盘和肝脏中(Giang, D. K.等人,Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 2238-2242; Cravatt 等人Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 29, 10821-10826)。FAAH水解内源性信号类脂的脂肪酸酰胺(FAA)家族。脂肪酸酰胺的常规种类包含N-酰基乙醇酰胺(NAEs)和脂肪酸伯酰胺(FAPAs)。NAEs的例子包括花生四烯酸乙醇胺(AEA)。十六酰胺乙醇(PEA)和油酰乙醇胺(OEA)。FAPAs的例子包括9-Z-十八碳烯酰胺或油酰胺(McKinney M K 和 Cravatt B F. 2005. Annu Rev Biochem 74:411-32)。内源性信号类脂的另一种脂肪酸酰胺家族是N-酰基牛磺酸,一旦FAAH缺失或受到抑制,其显现出升高现象,并且似乎作用于瞬时受体电位(TRP)钙通道家族,虽然功能性结果仍然不清楚(Saghatelian A,等人Biochemistry. 2004, 43:14332-9, Saghatelian A,等人Biochemistry, 2006, 45:9007-9015)。除了脂肪酸酰胺之外,FAAH还可以水解某些脂肪酸酯,例如,2-花生四烯酸甘油(2-AG)(另一种内源性大麻素)(Mechoulam等人Biochem. Pharmacol. 1995; 50:83-90; Stella等人Nature, 1997; 388:773-778; Suguria等人Biochem. Biophys. Res. Commun. 1995; 215:89-97)。
人们预期抑制FAAH可导致花生四烯酸乙醇胺及其它脂肪酸酰胺水平提高。脂肪酸酰胺提高可导致伤感阈值提高。由此,FAAH的抑制剂可有效用于治疗疼痛(Cravatt, BF; Lichtman, AH Current Opinion in Chemical Biology 2003, 7, 469-475)。这种抑制剂可有效用于治疗其它障碍,这种障碍可以使用脂肪酸酰胺或大麻素受体的调节剂来治疗,例如,焦虑症,睡眠障碍,阿尔茨海默氏病和帕金森氏症,进食障碍,代谢失调,心血管障碍和炎症(Simon 等人 Archives of Gen. Psychiatry, 2006, 63, 824-830. Kunos, G等人Pharmacol Rev 2006, 58,389-462)。在一些实施方案中,FAAH抑制剂化合物可能是外围限制性化合物,并且可能不会显著地影响神经障碍,例如,抑郁症和焦虑症。最终,在动物模型中,表明大麻素受体的激动作用还可降低动脉粥样硬化的发展(参见Steffens等人Nature, 2005, 434, 782-786; 和 Steffens等人,Curr Opin. Lipid., 2006, 17, 519-526)。由此,人们预期内源性的大麻碱(cannabinergic)脂肪酸酰胺(例如,花生四烯酸乙醇胺)的水平提高可有效地治疗动脉粥样硬化或降低形成动脉粥样硬化的危险。
抑制FAAH还可导致十六酰胺乙醇的提高,人们认为其部分地通过过氧物酶体增殖子-激活的受体α(PPAR-α)的激活来进行工作,从而管理多个途径,包括,例如,神经性和炎症性病症(例如,惊厥、神经毒性、痉挛(spacticity))中的疼痛感觉,并且减轻炎症,例如,播散性神经性皮炎和关节炎(LoVerme J等人The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 2005, 67, 15-19; LoVerme J等人The search for the palmitoylethanolamide receptor. Life Sci 2005, 77: 1685-1698. Lambert DM等人The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem 2002, 9: 663-674; Eberlein B,等人Adjuvant treatment of atopic eczema: assessment of an emollient containing N-palmitoylethanolamine(ATOPA study). J Eur Acad Dermatol Venereol. 2008, 22:73-82. Re G,等人Palmitoylethanolamide, endocannabinoids and related cannabimimetic compounds in protection against tissue inflammation and pain: potential use in companion animals.Vet J. 2007 173:21-30.)。由此,抑制FAAH可有效用于治疗各种疼痛和炎症性病症,例如骨关节炎,类风湿性关节炎,糖尿病性神经病变,带状疱疹神经痛,骨骼肌疼痛和肌纤维痛。
人们还认为某些脂肪酸酰胺(例如,OEA)通过过氧物酶体增殖子-激活的受体α(PPAR-α)起作用,从而调节各种生理学过程,包括,例如,摄食和脂解。与此一致,表明人体脂组织可结合和代谢内源性大麻素,例如花生四烯酸乙醇胺和2-花生四烯酸甘油(参见Spoto等人,Biochimie 2006, 88, 1889-1897; 和 Matias等人,J. Clin. Endocrin. & Met., 2006, 91, 3171-3180)。由此,体内抑制FAAH活性导致体脂肪、体重、热量摄入和肝脏甘油三酯水平降低。然而,与通过PPAR-α起作用的其它抗脂血症的药剂(例如,贝特类)不同,FAAH抑制剂不会导致不良副作用,例如皮疹、疲劳、头痛、勃起功能障碍,以及更少的,导致贫血、白血球减少症、血管性水肿和肝炎(参见,例如,Muscari等人,Cardiology, 2002, 97: 115-121)。
许多脂肪酸酰胺根据需求而产生,并快速地被FAAH降解。结果,人们认为FAAH造成的水解是调节中枢神经系统以及周围组织和体液中脂肪酸酰胺水平的主要步骤之一。与脂肪酸酰胺的广泛排列生物效果(内源性大麻素和非内源性大麻素机理)相结合,FAAH的广泛分布说明,抑制FAAH可改变许多组织和体液中的脂肪酸酰胺的水平,并且可以用于治疗许多不同病症。FAAH抑制剂提高内源性脂肪酸酰胺的水平。FAAH抑制剂阻碍内源性大麻素的降解,并且提高这些内源性物质的组织水平。在这方面,FAAH抑制剂可以用于预防和治疗其中涉及到内源性大麻素和/或被FAAH酶代谢的任何其它基质的病变。
各种脂肪酸乙醇酰胺具有重要的和不同的生理功能。结果,选择性抑制FAAH酶活性的抑制剂分子可以相应地选择性调节FAAH基质的细胞和胞外浓度。在需要FAAH酶抑作用的情况下,当将生物学相容的FAAH抑制剂配制为针对任何临床适应症的治疗剂时,其可以是有效的药用化合物。在一些实施方案中,可以优先抑制周围组织中的FAAH活性。在一些实施方案中,显著地穿过血脑屏障的FAAH抑制剂可用于优先抑制周围组织中的FAAH活性。在一些实施方案中,优先抑制周围组织中的FAAH活性的FAAH抑制剂可以使FAAH抑制在中枢神经系统中的效果最小。在一些实施方案中,优选,抑制周围组织中的FAAH活性,并且使中枢神经系统中的FAAH抑制减到最小。
本发明概述
本发明涉及可用于治疗FAAH介导的疾病、障碍或病症的组合物,其包含FAAH抑制剂和第二个活性剂。该组合物可用于治疗许多疾病、障碍或病症,包括骨关节炎,类风湿性关节炎,糖尿病性神经病变,带状疱疹神经痛,骨骼肌疼痛和肌纤维痛,以及急性疼痛,偏头痛,睡眠障碍,阿尔茨海默氏病和帕金森氏症。
在另一个方面,本文公开的本发明涉及用于治疗神经性和感受伤害性疼痛的组合物,所述组合物包含艾托考昔。
在另一个方面,本发明涉及使用这些组合物的方法。
附图的简要说明
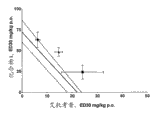
图1
该图描述了与FAAH抑制剂化合物A共同给予的艾托考昔的止痛效果的等效线图解(三个不同的剂量比例:3:1、1:1、0.1:1)。实线是与止痛效果的加和相关的预测线。图例说明:剂量比例(艾托考昔:化合物A)▲=3:1,■=1:1,●=0.3:1(Zmix与Zadd比较不是对于任何比例都是统计上显著的(P.0.05))。
本发明的详细说明
在一方面,本发明涉及药物组合物,其包含:
抑制FAAH的式I化合物:

如下文所定义
或抑制FAAH的式II化合物:

如下文所定义
和第二个活性剂,例如药剂。
在该方面中,存在上位组,其中第二个活性剂可有效用于治疗下列:疼痛(例如,急性疼痛,慢性疼痛,神经性疼痛,偏头痛;由炎症(例如,关节炎,骨关节炎,脊椎炎,类风湿性关节炎,克罗恩氏病和过敏性肠综合症)所引起的疼痛和神经性疼痛),焦虑症,进食障碍(例如,厌食,贪食症),肥胖症,眼内压升高,青光眼,心血管病症,抑郁症,炎症性病症(过敏,呼吸炎症,皮肤炎症和胃肠炎症),哮喘,克罗恩氏病和炎症性肠病,食物过敏,哮喘,皮肤炎症,呕吐,触摸痛,痛觉过敏,头痛,内脏疼痛,牙齿疼痛,与灼伤相关的疼痛,月经疼痛,痛经,原发性痛经,类风湿性关节炎,幼年型类风湿关节炎,骨关节炎,外科手术之后的疼痛(例如,与矫形外科、妇科手术、腹部外科、切口、口腔外科相关的疼痛)和背痛,癫痫和癫痫状引起的损伤,接触兴奋性神经毒素,兴奋性中毒,局部缺血性脑损伤,脑缺血,外伤性损伤(例如脑损伤),抑郁症,焦虑症,睡眠障碍,阿尔茨海默氏病,帕金森氏症,亨丁顿舞蹈症,肌萎缩侧索硬化,多发性脑硬化,妥瑞症,精神分裂症,青光眼,疼痛,成瘾,炎症,过敏性反应,进食障碍,低血压症,高血压症,呼吸问题,癌(肿瘤生长),化疗并发症,窒息,注意力缺陷病症和胃肠疾病,包括恶心和呕吐,胃溃疡,分泌型腹泻,麻痹性肠梗阻,炎症性肠病,结肠癌,胃-食道回流病症,瘙痒,脂肪肝疾病和非酒精脂肪肝炎(NASH)和过敏性肠综合症(IBS)。
本发明还涉及治疗选自下列的疾病的方法:急性疼痛,慢性疼痛,神经性疼痛,偏头痛;由炎症所引起的疼痛和神经性疼痛,焦虑症,进食障碍,肥胖症,眼内压升高,青光眼,心血管病症,抑郁症,炎症性病症,哮喘,克罗恩氏病和炎症性肠病,食物过敏,哮喘,皮肤炎症,呕吐,触摸痛,痛觉过敏,头痛,内脏疼痛,牙齿疼痛,与灼伤相关的疼痛,月经疼痛,痛经,原发性痛经,类风湿性关节炎,幼年型类风湿关节炎,骨关节炎,外科手术之后的疼痛,妇科手术、腹部外科、切口、口腔外科和背痛,癫痫和癫痫状引起的损伤,接触兴奋性神经毒素,兴奋性中毒,局部缺血性脑损伤,脑缺血,外伤性损伤,抑郁症,焦虑症,睡眠障碍,阿尔茨海默氏病,帕金森氏症,亨丁顿舞蹈症,肌萎缩侧索硬化,多发性脑硬化,妥瑞症,精神分裂症,青光眼,疼痛,成瘾,炎症,过敏性反应,进食障碍,低血压症,高血压症,呼吸问题,癌肿瘤生长,化疗并发症,窒息,注意力缺陷病症和胃肠疾病,包括恶心和呕吐,胃溃疡,分泌型腹泻,麻痹性肠梗阻,炎症性肠病,结肠癌,胃食道回流病症,瘙痒,脂肪肝疾病和非酒精脂肪肝炎和过敏性肠综合症,该方法包括:给予按照式I或II的化合物和第二个活性剂的组合物。
在该上位组之内,存在下位组,其中第二个活性剂可有效用于治疗骨关节炎,类风湿性关节炎,炎症性疼痛,神经性和感受伤害性疼痛,糖尿病性神经病变,带状疱疹神经痛,骨骼肌疼痛和肌纤维痛,以及急性疼痛,偏头痛,睡眠障碍,阿尔茨海默氏病和帕金森氏症。
在该下位组内,存在一个类,其中第二个活性剂可有效用于治疗炎症性疼痛、神经性和感受伤害性疼痛。
在该类之内,存在子类,其中第二个活性剂是依托昔布(etoricixib)。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,其可有效用于治疗许多病症。在病症之中:疼痛(例如,急性疼痛,慢性疼痛,神经性疼痛,偏头痛;由炎症(例如,关节炎,骨关节炎,脊椎炎,类风湿性关节炎,克罗恩氏病和过敏性肠综合症)所引起的疼痛,丘脑性疼痛综合症和神经性疼痛),焦虑症,进食障碍(例如,厌食,贪食症),肥胖症,眼内压升高,青光眼,心血管病症,抑郁症,炎症性病症(过敏,呼吸炎症,皮肤炎症和胃肠炎症),哮喘,克罗恩氏病和炎症性肠病。可以治疗的其它病症包括:食物过敏,哮喘,皮肤炎症,呕吐,触摸痛,痛觉过敏,头痛,内脏疼痛,牙齿疼痛,与灼伤相关的疼痛,月经疼痛,痛经,原发性痛经,类风湿性关节炎,幼年型类风湿关节炎,骨关节炎,外科手术之后的疼痛(例如,与矫形外科、妇科手术、腹部外科、切口、口腔外科相关的疼痛)和背痛,
预期本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可有效用于治疗和/或预防许多病症。预期这些FAAH抑制剂可减少一或多种这种病症的一或多种症状。
本发明的FAAH抑制剂单独或与第二个活性剂联用,可用于预防和/或治疗例如癫痫和癫痫状引起的损伤,接触兴奋性神经毒素,兴奋性中毒,局部缺血性脑损伤,脑缺血,外伤性损伤(例如脑损伤),抑郁症,焦虑症,睡眠障碍,阿尔茨海默氏病,帕金森氏症,亨丁顿舞蹈症,肌萎缩侧索硬化,多发性脑硬化,妥瑞症,精神分裂症,青光眼,疼痛,成瘾,炎症,过敏性反应,进食障碍,低血压症,高血压症,呼吸问题,癌(肿瘤生长),化疗并发症,窒息,注意力缺陷病症和胃肠疾病,包括恶心和呕吐,胃溃疡,分泌型腹泻,麻痹性肠梗阻,炎症性肠病,结肠癌,胃-食道回流病症,瘙痒,脂肪肝疾病和非酒精脂肪肝炎(NASH)。FAAH抑制剂还可以用于治疗过敏性肠综合症(IBS),通常与痉挛相关的病症,腹痛,腹胀,便秘和腹泻。IBS的三个主要类型:便秘型(IBS-C)、腹泻型(IBS-D)和便秘和腹泻两者都出现的交叉型(IBS-A)。
青光眼和眼睛病症
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可用于预防和/或治疗青光眼及其它以眼睛高血压症为特征的病症。
睡眠障碍
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可用于预防和/或治疗睡眠障碍,这种睡眠障碍影响患者入睡和/或保持熟睡的能力和/或导致不能熟睡。术语“睡眠障碍“包括失眠,夜惊,磨牙症,梦游病,睡眠呼吸暂停,多动腿综合征,不能熟睡,季节性情感障碍,生理节奏适应障碍,等等。
失眠典型地分为:入睡性失眠,在这种情况下,患者需要30分钟以上才能入睡;和睡眠维持性失眠,在这种情况下,患者在预期的睡眠期间有超过30分钟是醒着的,或例如,在目标苏醒时间之前苏醒,并且不能重新睡眠。睡眠障碍包括内源性障碍,例如睡眠呼吸暂停,和与行为或外部环境因素相关的障碍。例如,睡眠障碍包括患者难以调节到新的生理节奏,例如,由于时差综合症;晚上延长工作班次或工作班次不规则;等等。睡眠障碍还可以在患有其它障碍、疾病或损伤的患者中出现,或在用其它药物治疗的患者中出现,在这种情况下,患者难以睡着和/或难以保持熟睡,或不能熟睡。例如,本发明公开的方法可在由于进行化疗或损伤或应激反应或情绪障碍(例如抑郁症、焦虑症,等等)的结果而难以睡眠的患者中有效用于诱导催眠。
睡眠障碍包括本领域技术人员所认为是睡眠障碍的病症,例如,本领域已知的病症或预计是睡眠障碍或发现是睡眠障碍的病症。参见,例如,Thorpy. MJ International Classification of Sleep Disorders, Revised: Diagnostic and Coding Manual.American Sleep Disorders Association; Rochester, Minnesota 1997; 和 JCD CM, International Classification of Diseases, Ninth Revision, Clinical Modification, National Center for Health Statistics. Hyattsville, MD。
睡眠障碍通常可以分类为:睡眠异常,例如,内因性、外因性和生理节奏障碍;异态睡眠,例如,觉醒、睡眠-醒觉转换和快速眼运动(REM)相关的障碍,及其它异态睡眠;与精神、神经和油性医学障碍(oilier medical disorders)相关的障碍;及其它睡眠障碍。
内因性睡眠障碍包括,例如,精神生理性失眠,睡眠状态错觉,特发性失眠,嗜眠病,复发性嗜睡,特发性嗜睡,外伤后嗜睡,阻塞性睡眠无呼吸综合症,中枢性睡眠无呼吸综合症,中枢性肺泡换气过低综合症,周期性四肢运动障碍,多动腿综合症,等等。
外因性睡眠障碍包括,例如,睡眠卫生不良,环境性睡眠障碍,高空性失眠,睡眠调节障碍,睡眠不足综合症,设限性睡眠障碍,入睡相关性障碍(sleep-upsilonnset association disorder),食物过敏性失眠,夜间进食(饮酒)综合症,安眠剂依赖性睡眠障碍,兴奋剂依赖性睡眠障碍,洒精依赖性睡眠障碍,毒素诱导的睡眠障碍,等等。
生理节奏性睡眠障碍包括,例如,时区改变(时差)综合症,换班工作睡眠障碍,不规律的睡眠-醒觉模式,睡眠相位延迟综合症,睡眠相位提前综合症,非24小时睡眠-觉醒障碍,等等。
睡眠觉醒障碍包括,例如,觉醒混淆,梦游,夜惊,等等。
睡眠-醒觉转换障碍包括,例如,节奏性运动障碍,睡眠抽动,梦呓,夜间腿部痛性痉挛,等等。
REM相关的睡眠障碍包括,例如,梦魇,睡眠性麻痹,睡眠相关阴茎勃起障碍,睡眠相关痛性勃起,REM睡眠相关的房窦停止,REM睡眠行为障碍,等等。
其它异态睡眠包括,例如,睡眠磨牙症,睡眠遗尿,睡眠相关的异常吞咽综合症,夜间突发性的肌张力障碍,无法解释的夜间突然死亡综合症,原发性打鼾,婴儿睡眠呼吸暂停,先天性的中枢性换气不足综合症,婴儿猝亡综合征,良性初生儿睡眠肌阵挛,等等。“睡眠障碍”还可以在患有其它医学障碍、疾病或损伤的患者中出现,或在用其它药物治疗或医学治疗的患者中出现,在这种情况下,患者难以睡着和/或难以保持熟睡,或不能熟睡,例如,患者感受到睡眠剥夺。例如,对其它病症进行医学治疗(例如,化疗或手术)之后,或由于疼痛或其它物理性伤害的结果,一些患者难以睡眠。
本领域众所周知,某些医学障碍,例如,中枢神经系统(CNS)障碍,例如,精神或神经障碍,例如焦虑症,可能具有睡眠障碍成分,例如,睡眠剥夺。由此,治疗睡眠障碍还包括治疗其它障碍中的睡眠障碍成分,例如,CNS障碍。进一步的,治疗CNS障碍中的睡眠障碍成分还可以具有改善与该障碍有关的其它症状的有益效果。例如,在具有睡眠剥夺的一些焦虑症患者中,治疗睡眠剥夺成分还能够治疗焦虑症成分。由此,本发明还包括治疗这种医学障碍的方法。
与精神障碍有关的睡眠障碍包括精神病,情绪障碍焦虑症,恐慌障碍,成瘾,等等。具体的精神障碍包括,例如,抑郁症,强迫性的强制障碍,情感神经症/障碍,抑郁神经症/障碍,焦虑性神经症,精神抑郁障碍,行为障碍,情绪障碍,精神分裂症,躁狂性抑郁症,精神错乱,酒精中毒,等等。
与神经障碍有关的睡眠障碍包括,例如,脑褪化障碍,痴呆,震颤性麻痹,致命性家族性失眠,睡眠相关的癫痫,睡眠的癫痫性电持续状态,睡眠相关的头痛,等等。与其它医学障碍相关的睡眠障碍包括,例如,嗜眠病,夜间心脏缺血,慢性阻塞性肺病,睡眠相关的哮喘,睡眠相关的胃食管回流,消化性溃疡疾病,纤维炎综合症,等等。
在一些情况下,睡眠障碍还与疼痛有关,例如,与多动腿综合征有关的神经性疼痛;偏头痛;对疼痛的敏感性增加或放大,例如痛觉过敏、灼痛和触摸痛;急性疼痛;烧伤疼痛(bum pain);非典型性的面部疼痛:神经性疼痛;背痛;复杂区域疼痛综合症1和11;关节炎疼痛:运动创伤疼痛;与感染相关的疼痛,例如,HIV.后骨髓灰质炎综合症和疱疹后神经痛;假肢痛;阵痛;癌疼痛;化疗后疼痛;中风后疼痛,手术后疼痛;神经痛;与内脏疼痛有关的病症,包括过敏性肠综合症,偏头痛和心绞痛;等等。
其它睡眠障碍包括,例如,短睡者,长睡者,清醒不足综合症,断续性肌阵挛,睡眠多汗症,月经相关的睡眠障碍,妊娠相关的睡眠障碍,恐怖性入睡前幻觉,睡眠相关的神经性呼吸增块,睡眠相关的喉痉挛,睡眠窒息综合症,等等。
失眠典型地分为:入睡性失眠,在这种情况下,患者需要30分钟以上才能入睡;和睡眠维持性失眠,在这种情况下,患者在预期的睡眠期间有超过30分钟是醒着的,或例如,在目标觉醒时间之前觉醒,同时很难或不能重新睡眠。一些公开的化合物可有效治疗入睡性失眠和睡眠维持性失眠、由生理节奏适应障碍引起的失眠或由CNS障碍引起的失眠。在一个实施方案中,治疗患者的生理节奏适应障碍。在另一个实施方案中,治疗患者由情绪障碍引起的失眠。在其它实施方案中,治疗患者的睡眠呼吸暂停、梦游症、夜惊、多动腿综合征、入睡性失眠和睡眠维持性失眠。在其它实施方案中,治疗患者的入睡性失眠或睡眠维持性失眠。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可用于诱导、延长和/或增加睡眠。这可以包括:睡眠障碍的治疗,睡眠障碍就是由于一些内在或外界因素(例如,疼痛、应激反应或焦虑症、滥用兴奋剂或镇静剂或暂时紊乱的生活方式)而难以获得满意的睡眠,并且该治疗就使用方而言,可以包括选择性的需求以获得特别有利的睡眠时间。如此,举例来说,可能出现一种需求,即在人希望对预期到的随后几天或近期的重要事件可以充分警觉和提起精神。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其可以帮助实现下列目标中的任一个目标:开始睡眠,尤其是一期睡眠;保持熟睡;睡得很好;觉醒后精神;觉醒后警觉;更快开始一期睡眠;增加睡眠周期的持续期间;减少觉醒的次数和持续期间;增加睡眠的总时间;提高睡得很好的概率;减少失眠,尤其是长期或轻微-中度的失眠;减少睡眠期间的干扰;和提高睡眠质量。满足这些目标可以通过任何标准来测定,或通过已知的主观或客观测定方法来测定,例如,Karolinska量表、Loughborough睡眠对数表或光计量法(actimetry)。
改善睡眠可以有助于保持苏醒;保持警觉;保持精神;和更好地进行第二天。
精神恢复程度和睡眠质量可以通过Loughborough睡眠对数表的早晨对数表来测定,恢复精神或睡眠质量的最高程度表示为1,最低程度表示为5。相应地,在本文中,恢复精神或睡眠质量的提高的百分比可以通过平均恢复精神或睡眠质量的降低来测定。
极其警觉、很警觉或警觉的感觉反应可以例如通过Karolinska 9-分数量表来测定。睡眠参数的其它测定包括可以通过光计量法(actimetry)测定的睡眠干扰指数(SDI)和睡眠开始时间(TTSO)。
本发明的FAAH抑制剂可以与目前所使用的治疗睡眠障碍的治疗剂联用,例如,阿地白介素(Proleukin),金刚烷胺(Symmetrel),巴氯芬(Lioresal),苄普地尔(Vascor),卡立普多(Soma),氯硝西泮(Klonopin),地西泮(Valium),苯海拉明(Sominex,Nytoi),Doxylamipie(Unisom),艾司唑仑(ProSoni),氟西泮(Dalmane),加巴喷丁,氯羟安定(Ativan),Le vod opa-carb idopa(Sincmet),褪黑激素,利他林(Ritalin),Modanfinil(Provigil),佩默林(Cylert),培高利特,Pramipexoie,Pronietliazine(Phenergan),夸西泮(Doral),金刚烷乙胺(Flumadine),Sibutxamuie(Meridia),羟丁酸钠,合成的结合雌激素(Cenestin),替马西泮(Restoril),三唑仑(Halcion),扎来普隆(Sonata)和唑吡坦(Arnbien)。
肥胖症相关的障碍
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其可以用于治疗肥胖症和/或降低或控制体重(或脂肪)或预防和/或治疗肥胖症或与过量消耗食物、乙醇及其它促进食欲的物质相关的其它食欲相关的障碍。本发明化合物可以用于调节脂类代谢、降低体脂肪(例如,通过提高脂肪使用来降低体脂肪)或降低(或抑制)食欲(例如,通过诱导饱胀感来降低食欲)。肥胖症是存在过量体脂肪的病症。在很多情况下,如果个体具有大于或等于30 kg/nr的体重指数(BMA),或如果个体具有至少一种合并症并且BMI大于或等于27 kg/m2,则认为个体肥胖。在某些情况下,处于肥胖症危险之中的患者是具有25 kg/m2至小于30 kg/m2的BMI的另外的健康患者(otherwise healthy subject),或具有至少一种合并症同时BMI为25 kg/m2至小于27 kg/m2的患者。
人们认为,在BMI更低的亚洲人中,出现与肥胖症相关的危险提高。在一些情况下,亚洲人的肥胖症是指患者具有至少一种肥胖症诱导或肥胖症相关的合并症的病症,这种合并症需要减轻体重或通过减轻体重来改善,这种患者具有大于或等于25 kg/nr的BMI。在亚洲人中,肥胖患者有时是指具有至少一种肥胖症诱导或肥胖症相关的合并症的患者,这种合并症需要减轻体重或通过减轻体重来改善,具有大于或等于25 kg/m2的BMI。在一些情况下,处于肥胖症危险之中的亚洲人是BMI大于23 kg/m2至小于25 kg/m2的患者。
肥胖症诱导的或肥胖症相关的合并症包括但不局限于:糖尿病,非胰岛素依赖性II型糖尿病,葡糖耐量受损,空腹血糖受损,抗胰岛素性综合症,血脂异常,高血压症,血尿酸过多,痛风,冠状动脉病,心肌梗塞,狭心症,睡眠无呼吸综合症,匹克威克综合征,脂肪肝,脑梗塞,脑血栓,暂时性缺血性发作,整形外科障碍,变形性关节炎,腰痛,月经病(emmeniopatliy)和不孕。尤其是,合并症包括:高血压症,高脂质血症,血脂异常,葡萄糖耐受不良,心血管疾病,睡眠呼吸暂停,糖尿病,及其它肥胖症相关的病症。
治疗(肥胖症和肥胖症相关的障碍)是指给予本文所描述的化合物,以便减轻肥胖患者的体重或保持其体重。相对于给予本文所描述化合物之前不久的患者体重,治疗的一种结果可以减轻肥胖患者的体重。治疗的另一结果可以防止先前由于饮食、锻炼或药物治疗所失去体重的体重恢复。治疗的另一个结果可以减少肥胖症相关疾病的出现和/或降低其严重程度。治疗可以合适地导致患者减少食物或热量摄入,包括:减少总的食物摄入,或减少饮食的具体组分(例如碳水化合物或油脂)的摄入;和/或抑制营养吸收;和/或抑制代谢率的降低;和在需要其的患者中减轻体重。治疗还可以导致代谢速度的改变,例如,提高代谢速度,而不是抑制代谢速度的降低(或除了抑制代谢速度的降低之外);和/或使通常由体重减轻引起的代谢抗性最小。
预防(肥胖症和肥胖症相关障碍)是指给予本文所描述的化合物,以便减轻处于肥胖症危险之中患者的体重或保持其体重。相对于给予本文所描述化合物之前不久的患者体重,预防的一种结果可以减轻处于肥胖症危险之中的患者的体重。预防的另一结果可以防止先前由于饮食、锻炼或药物所失去体重的体重恢复。如果在处于肥胖症危险之中的患者的肥胖症发病之前给予治疗,则预防的另一个结果可以防止肥胖症出现。如果在处于肥胖症危险之中的患者的肥胖症发病之前给予治疗,则预防的另一个结果可以降低肥胖症相关障碍的出现和/或降低其严重程度。此外,如果在已经形成肥胖的患者中开始治疗,则这种治疗可以防止肥胖症相关障碍的出现、发展或严重程度,例如,但不局限于:动脉硬化,II型糖尿病,多囊卵巢疾病,心血管疾病,骨关节炎,皮肤障碍,高血压症,抗胰岛素性,高胆固醇血症,高甘油三酯血症和胆结石。
肥胖症相关障碍是与肥胖症相关、由肥胖症所引起或起因于肥胖症的障碍。肥胖症相关障碍的例子包括:进食过量和贪食症,高血压症,糖尿病,血浆胰岛素浓度升高和抗胰岛素性,血脂异常,高脂质血症,子宫内膜、乳房、前列腺和结肠癌,骨关节炎,阻塞性睡眠呼吸暂停,胆石病,胆结石,心脏病,心律异常和心律失常,心肌梗塞,充血性心力衰竭,冠心病,猝死,中风,多囊卵巢疾病,颅咽管瘤,Prader-Willi综合症,Frupsilonhhch's综合症,GH缺乏患者,正常变异矮身材,特纳氏综合症及显示出代谢活动降低或作为全部不含脂肪物质的百分比的剩余能量消耗的降低的其它病理性病症,例如,患有急性淋巴母细胞性白血病的儿童。本文所描述的化合物可以用于降低或控制体重(或脂肪)或预防和/或治疗肥胖症或其它食欲相关的障碍,这种障碍与过量消耗食物、酒类(ethapiol)及其它促进食欲的物质有关。本发明化合物可以用于调节脂类代谢、降低体脂肪(例如,通过提高脂肪使用来降低体脂肪)或降低(或抑制)食欲(例如,通过诱导饱胀感来降低食欲)。
肥胖症相关障碍的进一步例子是代谢综合症,亦称综合症X,抗胰岛素性综合症,性和生殖功能障碍,例如不孕,男性性腺机能减退和女性多毛症,胃肠活动障碍,例如肥胖症相关的胃食管回流,呼吸障碍,例如,肥胖-肺换气低下综合症(匹克威克综合征),心血管障碍,炎症,例如,血管系统的系统炎症,动脉硬化,高胆固醇血症,高尿酸血症,下背疼痛,胆囊疾病,痛风和肾癌。本文所描述的化合物还可用于降低肥胖的次生结果的危险,例如,降低左心室肥厚的危险。
本发明的FAAH抑制剂可以与抗肥胖药剂联合给予,抗肥胖药剂包括但不限于:11? HSD-I(11-beta羟基甾体脱氢酶1型)抑制剂,例如BVT 3498,BVT 2733,3-(1-金刚烷基)-4-乙基-5-(乙基硫基)-4H-1.2,4-三唑(lriazole),3-(L-金刚烷基)-5-(3,4,5-三甲氧基苯基)-4-甲基-4H-l,2,4-三唑,3-金刚烷基-4,5,6,7.8,9,10,11,12,3a-十氢-l,2,4-三唑并[4,3-a][11]轮烯和公开在WO01/90091、WO01/90090、WO01/90092和WO02/072084中的那些化合物;5HT(血清素)转运体抑制剂,例如帕罗西汀,氟西汀,芬氟拉明,氟伏沙明,舍曲林和imipramipie,和公开在WOO03/00663中的那些化合物;5HT拮抗剂,例如在WO03/037871、WO03/037887中的那些拮抗剂,等等;5HTIa调节剂,例如公开在WO03/031439中的那些调节剂,等等;5HT-2激动剂;5HT2c(血清素受体2c)激动剂,例如BVT933,DPCA37215,IK264,PNU 22394,WAY161503,R-1065和YM 348,和公开在美国专利US 3,914,250和PCT公开WO02/36596、WO02/48124、WO02/10169、WOO1/66548、WO02/44152、WO02/51844、WO02/40456和WO02/40457中的那些;5HT6受体调节剂,例如,在WO03/030901、WO03/035061、WO03/039547等等中的那些,ACC2(乙酰基-CoA羧化酶-2)抑制剂;酰基-雌激素,例如油酰基雌酮,公开在del Mar-Crasa. M.等人(Obesity Research,9:202-9(2001))和日本专利申请JP 2000256190中;α-硫辛酸(α-LA);减食欲二环化合物,例如1426(Avepitis)和 1954(Aventis),和公开在WO00/18749、 WO01/32638、 WO01/62746、 WO01/62747和 WO03/015769中的化合物;AOD9604;食欲抑制剂,例如,在WO03/40107中的那些;ATL-962(Alizymc PLC);苯佐卡因:盐酸苄非他明(bcnzphetamine)(Didrex), 墨角藻(bladderwrack)(focus vesiculosus);BKS3(韩蛙皮素受体亚型3)激动剂;安非他酮;咖啡因;CB 1(大麻素-1受体)拮抗剂/反向激动剂,例如利莫那班(Acomplia;Sanofi Synthelabo),SR-147778(Sanofi Synthelabo),BAY 65-2520(Bayer)和SLV 319(Solvay)和公开在下列中的那些:美国专利US4,973.587,5,013.837,5,081,122,5,112,820,5,292,736,5.532.237,5,624,941,6,028,084和6,509,367和WO96/33159,WO97/29079,WO98/31227,WO98/33765,WO98/37061,WO98/41519,WO98/43635,WO98/43636,WO99/02499,WO00/10967,WO00/10968,WO01/09120,WO01/58869,WO01/64632,WO01/64633,WO01/64634,WO01/70700,WO01/96330,WO02/076949,WO03/006007,WO03/007887,WO03/020217,WO03/026647,WO03/026648,WO03/027069,WO03/027076,WO03/027114,WO03/037332,WO03/040107,WO03/086940,WO03/084943和US6,509,367和EPO申请EP-658546;CCK激动剂;CCK-A(缩胆囊肽-A)激动剂,例如AR-R 15849,GI 181771,JMV-180,A-71378,A-71623和SR146131,和美国专利US 5,739,106所描述的那些;壳聚糖;铬;CNTF(睫状神经营养因子),例如Cl-181771(Glaxo-SmitliKline),SR146131(Sanofi Synthelabo),butabindide,PD170292和PD 149164(Pfizer);CNTF衍生物,例如,阿索开(Regeneron),和公开在PCT申请WO 94/09134、WO 98/22128和WO 99/43813中的那些;共轭的亚油酸;促肾上腺皮质激素-释放因子激动剂;脱氢异雄甾酮;DGATl(二脂酰甘油酰基转移酶1)抑制剂;DGAT2(二脂酰甘油酰基转移酶2)抑制剂;二羧酸酯转运体抑制剂;盐酸安非拉酮(Tenuate);二肽基肽酶IV(DP-IV)抑制剂,例如,异亮氨酸噻唑烷,缬氨酸pyrrolidide,NVP-DPP728,LLAMDAF237,P93/01,TSL 225,TMC-2A/2B/2C,FE 999011,P9310/K364,VIP 0177,SDZ 274-444,西他列汀和公开在Pratley 和 Salsali(2007)Curr Med Res Opin. 23:919-31中的DP-IV抑制剂化合物和公开在下列中的化合物:PCT公开WO02/083128,WO02/062764,WO03/000180,WO03/000181,WO03/000250,WO03/002530,WO03/002531,WO03/002553,WO03/002593,WO03/004498,WO03/004496,WO03/017936,WO03/024942,WO03/024965,WO03/033524,WO03/037327和EP1258476;麻黄;exendin-4(glp-1的抑制剂);FAS(脂肪酸合酶)抑制剂,例如浅蓝菌素和C75;脂肪再吸收抑制剂,例如,在WO03/053451等等中的那些;脂肪酸转运体抑制剂;纤维(车前草,车前草瓜尔胶纤维);甘丙肽拮抗剂;山羊豆(Goat's Rue. French Lilac);藤黄;石蚕属植物(teucrium chamaedrys);生长素释放肽拮抗剂,例如,公开在PCT申请WO 01/87335和WO 02/08250中的那些;GLP-I(胰高血糖素样肽1)激动剂(例如exendin-4);glp-1(胰高血糖素样肽-1);糖皮质激素拮抗剂;葡萄糖转运体抑制剂;生长激素促分泌受体激动剂/拮抗剂,例如NN703,海沙瑞林(hexarelin),MK-0677,SM-5 130686,CP-424.391,L-G92.429和L-163.255和例如公开在美国专利US 6,358,951、美国专利申请2002/049196和2002/022637和PCT申请WO 01/56592和WO 02/32888中的那些;生长激素促泌剂,例如,公开和具体描述在美国专利US 5,536,716中的那些;H3(组胺113)拮抗剂/反向激动剂,例如噻普酰胺(thioperamide),3-(1M-咪唑-4-gammal)丙基N-(4戊烯基)氨基甲酸酯),clobenpropit,iodophenpropit,imoproxifan,GT2394(Gliatech)和A331440,和公开在PCT公开WO02/15905中的那些和O-[3-(1H-咪唑-4-基)丙醇]氨基甲酸酯(Kiec-Kononowicz,K.等人,Pharmazie,55:349-55(2000)),含有哌啶类(pipcridine)的组胺H3受体拮抗剂(Lazewska,D.等人,Phapinazie,56:927-32(2001),二苯甲酮衍生物和相关的化合物(Sasse,15 A.等人,Arch. Pharm.(Weinheim)334:45-52(2001)),取代的N-苯基氨基甲酸酯(plienylcarbarnates)(Reidemeister,S.等人,Pharmazie,55:83-6(2000))和proxifan衍生物(Sasse,A.等人,J. Med. Chem. 43:3335-43(2000))和组胺H3受体调节剂,例如,公开在WO03/024928和WO03/024929中的那些;白介素-6(II-6)和其调节剂,例如,在WO03/057237等等中的那些;L肉毒碱;瘦素衍生物,例如,公开在美国专利US 5,552,524、5,552,523、5,552,522、5,521,283和PCT国际公开WO96/23513、WO96/23514、WO96/23515、WO96/23516、WO96/23517、WO96/23518、WO96/23519和WO96/23520中的那些;瘦素,包括重组体人瘦素(PEG-OB,Hoffman La Roche)和重组体甲硫氨酰人瘦素(Amgen);脂肪酶抑制剂,例如四氢利泼斯汀(tetrahydrolipstatin)(奥利司他/Xenical?),Triton WR 1339,RHC80267,利普司他汀(lipstatin),茶皂素(teasaponin)和didhyiumbelliferyl phosphate,FL-386,WAY-121898,Bay-N-3176,缬氨内酯(valilactone),esteracin,ebelactonc A,ebclactone B和RHC 80267和公开在PCT公开WO01/77094和美国专利US 4,598,089、4,452,813、5,512,565、5,391,571、5,602,151、4,405,644、4,189,438和4,242,453中的那些;脂类代谢调节剂,例如山楂酸,高根二醇,熊果酸,熊果醇,桦木酸,桦木醇,等等,和公开在WO03/011267中的化合物;Mc3r(黑皮质素3受体)激动剂;Mc4r(黑皮质素4受体)激动剂,例如CHIR86036(Chiron),ME-10142,ME-10145和HS-131(Melacure),和公开在下列中的那些:PCT公开WO99/64002,WO00/74679,WO0l/991752,WO01/25192,WO01/52880,WO01/74844,WO01/70708,WO01/70337,WO01/91752,WO02/059095,WO02/Q59107,WO02/059108,WO02/059117,WO02/06276,WO02/1216G,WO02/11715,WO02/12178,WO02/15909,WO02/38544,WO02/068387,WO02/068388,WO02/067869,WO02/081430,WO03/06604,WO03/007949,WO03/009847,WO03/009850,WO03/013509和WO03/031410;McSr(黑皮质素5受体)调节剂,例如,公开在WO97/19952、WO00/15826、WO00/15790、US 20030092041中的那些;MCH2R(黑色素富集激素2R)激动剂/拮抗剂;黑色素富集激素拮抗剂;黑色素富集激素1受体(MCHR)拮抗剂,例如,T-226296(Takeda),SNP-7941(Synapic),和公开在WO01/21169、WO01/82925、WO01/87834、WO02/051809、WO02/06245、WO02/076929、WO02/076947、WO02/04433、WO02/51809、WO02/083134、WO02/094799、WO03/004027、WO03/13574、WO03/15769、WO03/028641、WO03/035624、WO03/033476、WO03/033480和日本专利申请JP 13226269和JP1437059中的那些;黑皮质素激动剂,例如,Melanotan II或描述在WO 99/64002和WO 00/74679中的那些;二甲双胍(Glucopliage?);mGluRS调节剂,例如,公开在WO03/029210、WO03/047581、WO03/048137、WO03/051315、WO03/051833、WO03/053922、WO03/059904等等中的那些;一元胺再摄取抑制剂,例如西布曲明(sibutralrnine)(Meridia?/Reductil?)和其盐,和公开在美国专利US 4,746,680、4,806,570和5,436,272和美国专利公开2002/0006964和WO01/27068和WO01/62341中的那些化合物;NE(去甲肾上腺素)输送抑制剂,例如,GW 320659,despiramine,他舒普仑(talsupram)和诺米芬新;水皂角(nomame herba);非选择性的血清素/nupsilonrepinephrine输送抑制剂,例如西布曲明或芬氟拉明;NPY 1拮抗剂,例如BIBP3226,J-115814,BIBO 3304,LY-357897,CP-671906,G1-264879A,和公开在美国专利US 6,001,836和PCT专利公开WO96/14307、WO01/23387、WO99/51600、WO01/85690、WO01/85098、WO01/85173和WO01/89528中的那些;NPY5(神经肽Y Y5)拮抗剂,例如152,804,GW-569180A,GW-594884A,GW-587081X,CW-548118X,FR235208,FR226928,FR240662,FR252384,1229U91,Cl-264879A,CGP71683LAMDA,LY-377897,LY 366377,PD-160170,SR-120562LAMDA,SR-120819LAMDA,JCF-104和H409/22和公开在美国专利US 6,140,354、6,191,160、6,258,837、6,313,298、6,326,375、6,329,395、6,335,345、6,337,332、6,329,395和6,340,683、欧洲专利EP-0.010691和EP-01044970和PCT公开WO97/19682、WO97/20820、WO97/20821、WO97/20822、WO97/20823、WO98/27063、WOOO/107409、WO00/185714、WO00/185730、WO00/64880、WO00/68197、WO00/69849、WO01/09120、WO01/14376、WO01/85714、WO01/85730、WO01/07409、WO01/02379、WO01/23388、WO01/23389、WO01/44201、WOOl/62737、WO01/62738、WO01/09120、WO02/20488、WO02/22592、WO02/48152、WO02/49648、WO02/051806、WO02/094789、WO03/009845、WO03/014083、WO03/022849、WO03/028726和Norman等人J. Med. Cliern. 43:4288-4312 2000)中的那些化合物;阿片样物质拮抗剂,例如纳美芬(nalmefene)(Revex?),3-甲氧基纳曲酮,纳洛酮和纳曲酮和公开在WO00/21509中的那些;食欲素拮抗剂,例如,SB-334867-A和公开在PCT公开WO01/96302、WO01/68609、WO02/44172、WO02/51232、WO02/51838、WO02/089800、WO02/090355、WO03/023561、WO03/032991和WO03/037847中的那些;PDE(磷酸二酯酶)抑制剂,例如茶碱,已酮可可豆碱,敏喘宁,西地那非,anirinone,米利酮,cilostainide,洛利普利(rolipram)和西洛司特(cilomilast);肽YY和其片段和变体(例如YY3-36(PYY3-36)(N. Engl. J Med. 349:941, 2003;ikpeapge daspeelnry yaslrliylnl vtrqry)和PYY激动剂,例如,公开在WO03/026591中的那些;phendimetraxine:苯丁胺,磷酸酯转运体抑制剂;磷酸二酯酶-3B(PDE3B)抑制剂;phytophapin化合物57(CP 644.673);丙酮酸酯;SCD-I(硬脂酰基-CoA脱氢酶-1)抑制剂;血清素再摄取抑制剂,例如右芬氟拉明,氟西汀,和在美国专利US 6,365,633和WO01/27060和WO01/162341中的那些;T7I(Tularik;Inc;Boulder CO);甲状腺激素β激动剂,例如KB-2611(KaroBioBMS),和公开在WO02/15845和日本专利申请JP 2000256190中的那些;托吡酯(Topimax?);转录因子调节剂,例如,公开在WO03/026576中的那些;UCP-1(未耦合蛋白-1)、2或3活化剂,例如植烷酸,4-((e)-2-(5,6,7,8-四氢-5,5,8,8-四甲基-2-萘)-1-丙烯基)苯甲酸(TTNPB),视黄酸,和公开在PCT专利申请WO 99/00123中的那些;?3(β肾上腺素能受体3)激动剂,例如AD9677/TAK677(Dainlppon/Takeda),CL-316,243,SB 418790,BRL-37344,L-796568,BMS-196085,BRL-35135LAMDA,CGP12177A,BTA-243,GW 427353,曲卡君(Trecadrine),Zeneca D7114,N5984(Nisshin Kyorin),LY-377604(Lilly)和SR 59119lamda,和公开在美国专利US 5,705,515、US 5,451,677和PCT公开WO94/18161、WO95/29159、WO97/46556、WO98/04526和WO98/32753、WO01/74782、WO02/32897、WO03/014113、WO03/016276、WO03/016307、WO03/024948、WO03/024953和WO03/037881中的那些;β-羟基甾体脱氢酶-1抑制剂(β-HSD-I);β-羟基-β-甲基丁酸酯。
焦虑症相关的障碍
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可用于治疗焦虑症(包括广泛性焦虑症、恐慌障碍和社交焦虑症)和抑郁症。焦虑症是一组心理问题,其关键特征包括:过度焦虑,恐惧,烦恼,逃避和强迫性的仪式,并且产生或导致紊乱病状、过度使用保健服务和功能损害。在美国和其它大部分国家中,它们是最普遍的精神病症。在Diagnostic and Statistical Manual of Mental Disorders(第四版,Revised 1994,由American Psychiatric Association Washington,D C.出版,393-444页)中列出的焦虑症包括:恐慌障碍(有和没有广场恐怖症),没有恐慌障碍历史的广场恐怖症,特定恐惧症,社交恐惧症,强迫性-强制性障碍(OCD),创伤后精神紧张性障碍(PTSD),急性精神紧张性障碍,广泛性焦虑症(GAD),由于常规医学病症所造成的焦虑症,物质诱导的焦虑症,特定恐惧症和未另外注明的焦虑症。
强迫性的强制性障碍的特点在于:再现性和持久性想法、思想、念头或想象(偏执狂),这些是人们认为过分或不合理的自我矛盾和/或重复、有目的和有意的行为(强迫症)。偏执狂或强迫症导致明显的痛苦、耗时和/或显著地妨碍社交或职业功能。
恐慌障碍的特点在于:复发性的意想不到的恐慌发作和相关的担忧其它攻击,担心攻击暗示或结果,和/或与攻击相关的行为的显著变化。恐慌发作的定义为:强烈恐惧或不适的离散时期,其中下列症状中的四个(或更多个)症状突然形成并在10分钟之内达到峰值:(1)心悸、心怦怦跳或心率加快;(2)出汗;(3)发抖或摇动;(4)感觉呼吸急促或窒息;(5)感觉气阻;(6)胸痛或不适;(7)恶心或腹部痛苦;(8)感觉晕眩不稳、头昏眼花或晕厥;(9)现实感丧失(非真实感)或人格解体(丧失个人本体):(10)害怕失去控制;(11)害怕死亡;(12)感觉异常(麻木或发麻);和(13)冷或热潮红。恐慌障碍可能与或可能不与广场恐怖症有关,或是非理性的,并且通常丧失对远离公众的惧怕能力。
社交焦虑症,亦称社交恐惧症,其特点在于明显和持久性地惧怕一或多种社交情境或行为情境,在该情境中,使人暴露于不熟悉的人群或存在被其它人仔细检查的可能性。接触恐惧状况几乎总是引起焦虑症,其可以接近恐慌发作的强度。避开恐惧状况或承受强烈的焦虑或痛苦。在恐惧状况中,逃避、忧虑预期或痛苦显著地干扰人的正常工作、职业或学术功能或社交活动或关系,或明显地忧虑患有恐惧症。在行为焦虑症或害羞的程度较小的情况下,不需要精神药物治疗。
广泛性焦虑症的特点在于:过分焦虑和烦恼(惴惴不安地期待),其至少保持6个月,并且人发现难以控制。它必须与下列6个症状中的至少3个症状有关:不安或感觉兴奋或紧张,容易疲劳,难以集中或头脑变得茫然,烦躁,肌肉紧张和睡眠紊乱。
这种障碍的诊断标准描述在DSM-IV(本文以引证的方式将其结合)的进一步说明(American Psychiatric Association,1994)中。
DSM111-R/IV所定义的创伤后精神紧张性障碍(PTSD)需要接触创伤活动,这种活动包括实际或威胁的死亡或严重伤害,或威胁自身或其它人的身体完整性,并且涉及强烈恐惧、无助或恐怖反应。以接触创伤活动的结果形式出现的症状包括:以打扰思维、倒叙或做梦形式重新感受该活动,暗示接触该活动时产生强烈心理忧虑和生理反应;逃避回忆该创伤活动的状况,不能详细回忆该活动,和/或常规响应性的麻木,表现为对重要活动的兴趣减弱,疏远其它人,限制影响范围或感觉前途渺茫;和自动觉醒的症状,包括过度警觉、吃惊反应夸大、睡眠紊乱、集中度受损和烦躁或突然生气。PTSD诊断要求症状存在至少一个月,而且它们导致临床上显著的社交、职业或其它重要功能区域的忧虑或损害。
单独或联用的本发明化合物可有效治疗诊断患有强迫性强制性障碍的患者的偏执狂和强迫症,其基于进行合适的试验,包括但不局限于下列中的任一个:Yale-Brown强迫量表(YBOCS)(对于成人),国立精神卫生研究所综合OCD量表(NIMH GOCS)和CGl-疾病严重程度量表。进一步包括的是,本发明化合物可有效引起这些试验中测定的某些因素的好转,例如,减少YBOCS总分中的一些点。还包括的是,本发明的化合物可有效防止强迫性强制性障碍的复发。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可有效用于治疗已经诊断患有恐慌障碍的患者的恐慌障碍,这种诊断基于恐慌发作的出现频率,或借助于CGI-疾病严重程度量表。进一步包括的是,本文所描述的化合物可有效引起某些因素(在这些评价中测定)的改善,例如,降低频率或消除恐慌发作,在CGI疾病严重程度量表中有改善或CGI综合改善评分为1(非常改善)、2(很改善)或3(最低限度改善)。还包括的是,本发明的化合物可有效防止恐慌障碍的复发。
本发明的FAAH抑制剂单独或与第二个活性剂联用,可有效治疗诊断患有社交焦虑症的患者的社交焦虑症,诊断基于进行下列任何一个试验:Liebowitz社交焦虑症量表(LSAS),CGI-疾病严重程度量表,Hamilton焦虑症评级量表(HAM-A),Hamilton抑郁症评级量表(HAM-D),DSM-IV的V轴社交和职业功能评价量表,Il轴(ICD10)世界卫生组织伤残评价,附表2(DAS-2),Sheehan伤残量表,Schneier伤残图,世界卫生组织生活质量-100(WHOQOL-100),或Ballenger. JC等人1998,J Clin Psychiatry 59 Suppl 17:54-60.所描述的其它试验,本文以引证的方式将其结合。进一步包括的是,本发明的FAAH抑制剂可有效引起改善(用这些试验测定),例如,Liebowitz社交焦虑症量表(LSAS)1中的基线变化,或CGI综合改善评分为1(非常改善)、2(很改善)或3(最低限度改善)。还包括的是,本发明的FAAH抑制剂可有效防止社交焦虑症的复发。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可有效用于治疗已经诊断患有广泛性焦虑症的患者的广泛性焦虑症,这种诊断基于DSM-IV所描述的诊断标准。进一步包括的是,本文所描述化合物可有效减少这种障碍的症状,例如,下列症状:过分烦恼和焦虑,难以控制烦恼,不安,或感觉兴奋或紧张,容易疲劳,难以集中精力,或头脑变得茫然,烦躁,肌肉紧张或睡眠紊乱。还包括的是,本发明的化合物可有效防止广泛性焦虑症的复发。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可有效用于治疗已经诊断患有PTSD的患者的PTSD,这种诊断基于进行任何下列试验:临床应用的PTSD诊断量表第2部分(CAPS)和患者评价的事件影响量表(IES)。进一步包括的是,本文所描述的化合物可有效引起CAPS、IES、CCI-疾病严重程度或CGI综合改善试验的评分的改善。还包括的是,本发明的化合物可有效防止PTSD的复发。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可用于预防、控制或治疗精神分裂症、偏执狂或多巴胺传输的其它相关障碍。
本发明的FAAH抑制剂可以与抗焦虑症药剂联合给药。抗焦虑症药剂的种类包括:苯并二氮杂 (例如阿普唑仑(Xanax?),利眠宁,氯硝西泮,钾氯氮,地西泮,halazeparn,lupsilonrazepam,奥沙西泮(oxazeprarn)和环丙二氮
(例如阿普唑仑(Xanax?),利眠宁,氯硝西泮,钾氯氮,地西泮,halazeparn,lupsilonrazepam,奥沙西泮(oxazeprarn)和环丙二氮 和其可药用盐);5-HT1A激动剂或拮抗剂,尤其是5HT1A部分激动剂(例如,5-HT1A受体部分激动剂丁螺环酮,氟辛克生,吉吡隆和伊沙匹隆,和其可药用盐);促皮质素释放因子(CRF)拮抗剂(包括WO 94/13643、WO 94/13644、WO 94/13661、WO 94/13676和WO 94/13677中所描述那些);吩噻嗪类(phenotliiazines)(包括普鲁米近,氯丙嗪和三氟拉嗪);单胺氧化酶抑制剂(MAOIs,例如异唑肼(Marplan?),苯乙肼(Nardil?),反苯环丙胺(Parnate?)和司来吉兰,和其可药用盐);单胺氧化酶的可逆性抑制剂(RIMAs,例如吗氯贝胺和其可药用盐);三环抗抑郁药(TCAs,例如阿米替林(Elavil?),anioxapine,氯米帕明,地昔帕明(Norpramin?),多塞平,丙咪嗪(imiprainine)(Tofranil?),maptroline,去甲替林(Aventyl?和Pamelor?),奋乃静,普罗替林和三甲丙咪嗪(Surmentil?)和其可药用盐));非典型性的抗抑郁剂,包括安非他酮,锂,奈法唑酮,曲唑酮和乙氧苯氧甲基吗啉,和其可药用盐;和选择性的血清素再摄取抑制剂(SSRI,例如帕罗西汀(Paxil?),文拉法新,氟伏沙明(fluvoxaminc),氟西汀(Prozac?),西酞普兰(Celexa?),艾司西酞普兰(escitaloprain)和舍曲林(Zoloft?)和其可药用盐)。
和其可药用盐);5-HT1A激动剂或拮抗剂,尤其是5HT1A部分激动剂(例如,5-HT1A受体部分激动剂丁螺环酮,氟辛克生,吉吡隆和伊沙匹隆,和其可药用盐);促皮质素释放因子(CRF)拮抗剂(包括WO 94/13643、WO 94/13644、WO 94/13661、WO 94/13676和WO 94/13677中所描述那些);吩噻嗪类(phenotliiazines)(包括普鲁米近,氯丙嗪和三氟拉嗪);单胺氧化酶抑制剂(MAOIs,例如异唑肼(Marplan?),苯乙肼(Nardil?),反苯环丙胺(Parnate?)和司来吉兰,和其可药用盐);单胺氧化酶的可逆性抑制剂(RIMAs,例如吗氯贝胺和其可药用盐);三环抗抑郁药(TCAs,例如阿米替林(Elavil?),anioxapine,氯米帕明,地昔帕明(Norpramin?),多塞平,丙咪嗪(imiprainine)(Tofranil?),maptroline,去甲替林(Aventyl?和Pamelor?),奋乃静,普罗替林和三甲丙咪嗪(Surmentil?)和其可药用盐));非典型性的抗抑郁剂,包括安非他酮,锂,奈法唑酮,曲唑酮和乙氧苯氧甲基吗啉,和其可药用盐;和选择性的血清素再摄取抑制剂(SSRI,例如帕罗西汀(Paxil?),文拉法新,氟伏沙明(fluvoxaminc),氟西汀(Prozac?),西酞普兰(Celexa?),艾司西酞普兰(escitaloprain)和舍曲林(Zoloft?)和其可药用盐)。
本发明的FAAH抑制剂还可以与具有止痛活性的第二个药剂共同治疗使用。可以共同治疗使用的镇痛药包括但不局限于:NSAIDs(例如,阿西美辛,醋氨酚,乙酰水杨酸,阿氯芬酸,阿明洛芬,阿扎丙宗,阿司匹林,苯噁洛芬,bezpiperylon,布氯酸,卡洛芬,环氯茚酸,双氯芬酸,双氯芬酸,二氟尼柳,diflusinal,依托度酸,芬布芬,芬布芬,fendofenac,芬克洛酸,非诺洛芬,芬替酸,非普拉宗,氟芬那酸,氟苯柳,氟苯柳,氟洛芬,氟比洛芬,氟比洛芬,呋罗芬酸,异丁苯乙酸,布洛芬,消炎痛,吲哚美辛(indonietliacin),吲哚洛芬,伊索克酸,异噁噻酰胺,酮洛芬,keloprupsilonfen,酮咯酸,inedofenajnic acid,甲氯灭酸,niefenaniic acid,甲芬那酸,咪洛芬,rpiofebutazone,萘丁美酮奥沙普秦,萘普生,萘普生,尼氟灭酸,奥沙普秦,oxpinac,羟保泰松,非那西汀,苯丁唑酮,苯丁唑酮,吡罗昔康,吡罗昔康,吡咯洛,普拉洛芬,舒多昔康,替诺昔康,柳氮磺吡啶舒林酸,舒洛芬,泰普菲酸,硫平酸,硫恶洛芬,托芬那酸,甲苯酰吡酸,甲苯酰吡酸,齐多美辛,佐美酸和佐美酸),非麻醉止痛剂,例如反胺苯环醇,阿片样物质或麻醉止痛剂(例如,APF112,β富纳曲胺(funaltrexamine),丁丙诺啡,butorphanupsilonl,可待因,cypridime,地佐辛,双氢可待因,地芬诺酯(diphenyloxylate),脑啡肽五肽,非多托秦(fedotozinc),芬太尼,氢可酮,氢吗啡酮(hydromophihone),利富吩,洛哌丁胺,派替啶,卡波卡因,美沙酮,甲基纳洛酮(methyl nalozone),吗啡,纳布啡(nalbuphine),纳美芬(nalmefene),纳洛肼(naloxonazine),纳洛酮,纳曲酮,纳曲吲哚(naltrindole),nor-binaltorphimlne,氧可酮,羟吗啡酮(oxyrnorphone),镇痛新,达尔丰和三甲丁酯),NK1受体拮抗剂(例如,依洛匹坦(ezlopitant)和SR-14033,SSR-241585),CCK受体拮抗剂(例如,氯谷胺(loxiglumide)),NK3受体拮抗剂(例如,他奈坦,奥沙奈坦(osanetant)SR-HZSOl1,SSR-ZdISSS),去甲肾上腺素血清素再摄取抑制剂(NSRI;例如,米那普仑),香兰素受体激动剂和拮抗剂,大麻素受体激动剂(例如,arvanil),sialorphin,作为脑啡肽酶(neprilysin)的抑制剂的化合物或肽氟雷法胺(Frakefamide)(H-Tyr~D-Ala-Phe(F)-Phe-NH2;WO 01/019849 Al),Tyr-Arg(京都啡肽),CCK受体激动剂(例如,caerupsilonlein),芋螺毒素肽,胸腺肽(thyrnulin)的肽类似物,右氯谷胺(氯谷胺(loxiglumide)的R-异构体:WO 88/05774)和止痛肽(例如,endomupsilonrphin-1,内吗啡肽(endomorphin)-2,nocistatin,dalargin,luprupsilonn和物质P)。
另外,某些抗抑郁剂可以共同治疗使用,这是因为它们具有止痛或另外有益于与止痛剂联合使用。这种抗抑郁剂的例子包括:选择性的血清素再摄取抑制剂(例如,氟西汀,帕罗西汀,舍曲林),血清素-去甲肾上腺素双重摄取抑制剂,文拉法新和萘发扎酮(nefazadone)。某些抗惊厥(convulsanls)药物具有止痛活性,并且可有效用于共同治疗。这种抗惊厥剂包括:加巴喷丁,卡马西平,苯妥英,丙戊酸盐,氯硝西泮,托吡酯和乐命达。人们认为这种药剂尤其可用于治疗神经性疼痛,例如,治疗三叉神经痛、带状疱疹神经痛和糖尿病性神经病变的疼痛。在共同治疗中使用的其它化合物包括:α-2-肾上腺素能受体激动剂(例如,替扎尼定和可乐定),慢心律,皮质类甾醇,阻断NMDA(iN-甲基-天冬氨酸)受体的化合物(例如,右美沙芬,氯胺酮和金刚烷胺),甘氨酸拮抗剂,卡立普多,环苯扎林,各种阿片剂,非mu阿片样物质镇咳药(例如右美沙芬,capiniphen,卡拉美芬和喷托维林),阿片样物质镇咳药(例如可待因,氢可酮,美他沙酮(metaxolone))。本文所描述的化合物还可以与下列结合使用:吸入性气体一氧化氮(治疗肺血管收缩或呼吸道收缩),血栓烷A2受体拮抗剂,兴奋剂(即咖啡因),H2-拮抗剂(例如甲胺呋硫),抗酸剂(例如氢氧化铝或氢氧化镁),除气剂(例如二甲基硅油),减充血剂(例如苯肾上腺素,苯丙醇胺,伪麻黄碱,氧甲唑啉,肾上腺素(ephinephrine),萘唑啉,丁苄唑啉,六氢脱氧麻黄碱,或左旋脱氧麻黄碱),前列腺素(例如迷索前列醇,恩前列素,利奥前列素(rioprostil),奥诺前列素(ornoprostol)或罗沙前列醇(rosaprostol)),利尿剂,镇静或非镇静作用组胺HI受体拮抗剂/抗组织胺(anlihistamines)(即能够阻断、抑制、降低或妨碍组胺和它的受体之间相互作用的任何化合物),包括但不局限于:4 阿司米唑(asternizole),阿伐斯汀,安他唑啉,阿司咪唑,阿扎他定,氮 斯汀(azelasline),aslamizole,溴苯那敏,马来酸溴苯吡胺,卡比沙明,卡瑞斯汀,西替利嗪,扑尔敏,马来酸氯苯那敏,西眯替丁,氯马斯汀,赛克力嗪,赛庚啶,脱碳乙氧基氯雷他定(descarboethoxyloratadine),右氯苯那敏,二甲茚定,苯海拉明,二苯拉林,琥珀酸杜克西拉明,多西拉敏(doxylarnine),依巴斯汀,乙氟利嗪(efletipizine),依匹斯汀,法莫替丁,非索非那定,羟嗪,羟嗪,酮替酚,左卡巴斯汀,左西替利嗪,左西替利嗪,氯雷他定,美其敏,吡拉明,美喹他嗪,甲吡咯烷基甲吩噻嗪,米安舍林,咪唑斯汀,诺拉斯丁,诺阿司咪唑(norasternizole),iioraztoinizole,苯茚胺,抗感明,匹库马特,普鲁米近,pynlamine,吡拉明(pyrilamiiie),雷尼替丁(ianitidine),替美斯汀,特非那定,异丁嗪,曲吡那敏(tripelenarnine)和曲普利啶;5HT1激动剂,例如triptan(例如舒马曲坦或那拉曲坦(naratriplan)),腺苷Al激动剂,HP ligaml,钠通道阻断剂(例如乐命达),物质P拮抗剂(例如NK拮抗剂),大麻素,5-脂氧合酶抑制剂,白细胞三烯受体拮抗剂/白细胞三烯拮抗剂/LTD4拮抗剂(即,能够阻断、抑制、降低或妨碍白细胞三烯和Cys LTl受体之间相互作用的任何化合物),包括但不局限于:扎鲁司特,孟鲁司特,孟鲁司特钠(Singulair?),普仑司特,伊拉司特,泊比司特(pobilukast),SKB-106,203和US 5,565,473所描述的具有LTD4拮抗活性的化合物,DMARU(例如氨甲喋呤),神经元稳定抗癫痫药,单胺能摄取抑制剂(例如文拉法新),基质金属蛋白酶抑制剂,一氧化氮,合酶(NOS)抑制剂,例如iNOS或nNOS抑制剂,肿瘤坏死因子的释放或作用的抑制剂,抗体治疗,例如单克隆抗体治疗,抗病毒剂,例如核苷抑制剂(例如拉夫米定)或免疫系统调节剂(例如,干扰素),局部麻醉剂,已知的FLAMDALAMDAH抑制剂(例如,PMSF,UKB532,URB597或BMS-I,以及WO04033652、US 6,462,054、US 2003/0092734、US 2002/0188009、US 2003/0195226和WO04/033422所描述的那些),抗抑郁剂(例如,VPI-013),脂肪酸酰胺(例如花生四烯酸乙醇胺,N-棕榈酰乙醇胺,N-油酰基乙醇胺,2-花生四烯酰丙三醇或油酰胺)arvanil,US 20040122089中描述的anadamide和arvanil的类似物,和质子泵抑制剂(例如,奥美拉唑,埃索美拉唑,兰索拉唑,泮妥拉唑(pantorazole)和雷贝拉唑(rabeprazole))。
斯汀(azelasline),aslamizole,溴苯那敏,马来酸溴苯吡胺,卡比沙明,卡瑞斯汀,西替利嗪,扑尔敏,马来酸氯苯那敏,西眯替丁,氯马斯汀,赛克力嗪,赛庚啶,脱碳乙氧基氯雷他定(descarboethoxyloratadine),右氯苯那敏,二甲茚定,苯海拉明,二苯拉林,琥珀酸杜克西拉明,多西拉敏(doxylarnine),依巴斯汀,乙氟利嗪(efletipizine),依匹斯汀,法莫替丁,非索非那定,羟嗪,羟嗪,酮替酚,左卡巴斯汀,左西替利嗪,左西替利嗪,氯雷他定,美其敏,吡拉明,美喹他嗪,甲吡咯烷基甲吩噻嗪,米安舍林,咪唑斯汀,诺拉斯丁,诺阿司咪唑(norasternizole),iioraztoinizole,苯茚胺,抗感明,匹库马特,普鲁米近,pynlamine,吡拉明(pyrilamiiie),雷尼替丁(ianitidine),替美斯汀,特非那定,异丁嗪,曲吡那敏(tripelenarnine)和曲普利啶;5HT1激动剂,例如triptan(例如舒马曲坦或那拉曲坦(naratriplan)),腺苷Al激动剂,HP ligaml,钠通道阻断剂(例如乐命达),物质P拮抗剂(例如NK拮抗剂),大麻素,5-脂氧合酶抑制剂,白细胞三烯受体拮抗剂/白细胞三烯拮抗剂/LTD4拮抗剂(即,能够阻断、抑制、降低或妨碍白细胞三烯和Cys LTl受体之间相互作用的任何化合物),包括但不局限于:扎鲁司特,孟鲁司特,孟鲁司特钠(Singulair?),普仑司特,伊拉司特,泊比司特(pobilukast),SKB-106,203和US 5,565,473所描述的具有LTD4拮抗活性的化合物,DMARU(例如氨甲喋呤),神经元稳定抗癫痫药,单胺能摄取抑制剂(例如文拉法新),基质金属蛋白酶抑制剂,一氧化氮,合酶(NOS)抑制剂,例如iNOS或nNOS抑制剂,肿瘤坏死因子的释放或作用的抑制剂,抗体治疗,例如单克隆抗体治疗,抗病毒剂,例如核苷抑制剂(例如拉夫米定)或免疫系统调节剂(例如,干扰素),局部麻醉剂,已知的FLAMDALAMDAH抑制剂(例如,PMSF,UKB532,URB597或BMS-I,以及WO04033652、US 6,462,054、US 2003/0092734、US 2002/0188009、US 2003/0195226和WO04/033422所描述的那些),抗抑郁剂(例如,VPI-013),脂肪酸酰胺(例如花生四烯酸乙醇胺,N-棕榈酰乙醇胺,N-油酰基乙醇胺,2-花生四烯酰丙三醇或油酰胺)arvanil,US 20040122089中描述的anadamide和arvanil的类似物,和质子泵抑制剂(例如,奥美拉唑,埃索美拉唑,兰索拉唑,泮妥拉唑(pantorazole)和雷贝拉唑(rabeprazole))。
本发明的FAAH抑制剂还可以与作为大麻素受体拮抗剂的第二个药剂共同治疗使用,以便预防和/或治疗肥胖症及其它食欲相关的障碍。
合并病症(co-morbid)的联用药
本领域技术人员可以理解,与本文所描述化合物联合给予的治疗可以针对本文所描述化合物靶向的相同或不同的障碍靶向。
可以首先给予本文所描述的化合物,而后给予其它治疗;或首先给予其它治疗,或它们可以在两个单独的组合物中或合并在单一组合物中同时给予。其它治疗是本领域已知的任何治疗,这种治疗可以治疗、预防或减轻靶向障碍的症状,例如睡眠障碍,或其它障碍,例如,其它CNS障碍。另外,在本发明的一些实施方案中,化合物与靶向障碍的其它已知的治疗联合给予。此外,当其它治疗与所公开的FAAH抑制剂联合给予时,其包括有利于患者的任何药剂。
例如,在其它治疗是药物的一些实施方案中,其可以给予单独的制剂,或在与本文所描述化合物相同的制剂中给予。本文所描述的FAAH抑制化合物与任何一或多种商购的、柜台销售的或处方药物联合给予,这些药物包括但不局限于:抗微生物剂,抑菌剂,杀菌剂,激素,退热剂,抗糖尿病药剂,支气管扩张药,止泻药,抗心律失常药,冠状动脉膨胀药剂,糖苷,解痉药,抗高血压药,抗抑郁剂,抗焦虑药,其它精神神经疾病治疗药剂,皮质类甾醇,镇痛药,避孕用品,非甾体抗炎症药物,血糖降低药剂,胆固醇降低药剂,抗惊厥剂,其它抗癫痫药剂,imrnunoinodulatupsilonrs,抗胆碱能药,交感神经阻滞药,拟交感神经药,血管舒张药剂,抗凝血剂,抗心律失常药,具有各种药理活性的前列腺素,利尿剂,睡眠助剂,抗组胺(antihistainiriic)药剂,抗肿瘤剂,肿瘤消解药剂,抗雄激素,抗疟药,抗麻疯病药剂和各种其它类型的药物。参见Goodman 和 Oilman's The Basis of Therapeutics {Eighth Edition, Pergainopi Press, Inc, USA, 1990)和The Merck Index(Eleventh Edition, iMerck AND Co., Inc., USA, 1989)。
用于治疗糖尿病的联用药
与本发明的FAAH抑制剂联合使用的合适药剂包括抗糖尿病药,例如(1)PPARGAMMA激动剂,例如格列酮类(例如,环格列酮;达格列酮;恩格列酮;伊沙列酮(isaglitazone)(MCC-555);吡格列酮;罗格列酮;曲格列酮;BRL49653;CLX-0921;5-BTZD和GW-0207,LG-100641和LY-300512等等,和公开在PCT公开W097/10813、WO97/27857、WO97/28115、WO97/28137、WO97/27847、WO03/000685、WO03/027112、WO03/035602、WO03/048130、WO03/055867等等中的化合物;(2)双缩胍,例如丁福明;二甲双胍;和苯乙双胍,等等;(3)蛋白质酪氨酸磷酸酶-IB(PTP-IB)抑制剂,例如ISlS 11371,和公开在WO03/032916、WO03/032982、WO03/041729、WO03/055883中的那些;(4)磺酰脲,例如醋酸己脲;氨磺丁脲(carbutamidc);氯磺丙脲;氯磺丙脲:格列本脲;格列甲嗪;优降糖(格列本脲);格列美脲;格列齐特;格列太特;格列喹酮;格列索脲;甲磺吖庚脲;和甲苯磺丁脲,等等;(5)氯茴苯酸类,例如瑞格列奈和那格列萘,等等;(6)α葡糖苷水解酶抑制剂,例如阿卡波糖;脂解素;camiglihose;乙格列酯;米格列醇;伏格列醇;帕地霉素(pradimicin)-Q;柳氮定(salbostatin);CKD-711;MDL-25,637:MDL-73,945;和MOR 14,等等;(7)α-淀粉酶抑制剂,例如淀粉酶抑肽(tendamistat),萃他丁(trestatin)和A 1-3688,等等;(8)胰岛素促泌剂,例如linogliricle;和A-4166,等等;(9)脂肪酸氧化抑制剂,例如氯莫克舍和乙莫克舍,等等;(10)A2拮抗剂,例如咪格列唑(midaglizole);isagliclole;德格列哚;咪唑克生;earoxan;和氟洛克生,等等;(11)胰岛素或胰岛素模拟物,例如biota,LP-100,诺和锐(novarapid),地特胰岛素,赖脯胰岛素,甘精胰岛素,胰岛素锌混悬液(长效和超长效);Lys-Pro胰岛素,GLP-I(73-7)(胰岛素促生肽(insulintropin));和GLP-I(7-36)-NH2),等等;(12)非噻唑二酮类,例如JT-501和法格列他扎(GW-2570/G1-262579),等等;(13)PPARALPHA/γ双重激动剂,例如BVT-142、CLX-0940、GW-1536、CW-1929、GW-2433、KRP-297、L-796449、LR-90、MK-0767、SB 219994、inuraglitazar和reglitazar(JTT-501)和公开在WO99/1G758、WO99/19313、WO99/20614、WO99/38850、WO00/23415、WO00/23417、WO00/23445、WO00/50414、WO01/00579、WO01/79150、WO02/062799、WO03/004458、WO03/016265、WO03/018010、WO03/033481、WO03/033450、WO03/033453、WO03/043985、WO031053976中的那些;和(14)其它胰岛素敏化药物;(15)VPAC2受体激动剂;(16)GLK调节剂,例如,公开在WO03/015774中的那些;(17)类视黄醇调节剂,例如,公开在WO03/000249中的那些;(18)GSK 3?/GSK 3抑制剂,例如4-[2-(2-溴苯基)-4-(4-氟苯基-1H-咪唑-5-基)吡啶,和公开在WO03/024447、WO03/037869、WO03/037877、WO03/037891、WO03/068773、EP 1295884、EP 1295885等等中的化合物;(19)糖原磷酸化酶(HGLPa)抑制剂,例如,公开在WO03/037864中的那些;(20)ATP消耗促进剂,例如,公开在WO03/007990中的那些;(21)TRB3抑制剂,(22)香兰素受体配体,例如,公开在WO03/049702中的那些;(23)降血糖药,例如,公开在WO03/015781、WO03/040114中的那些,(24)糖原合酶激酶3抑制剂,例如,公开在WO03/035663中的那些,(25)和公开在WO99/51225、US 20030134890、WO01/24786、WO03/059870中的那些药剂;(26)公开在WO03/057827等等中的胰岛素响应性DNA结合蛋白-1(IRDBP-1);(27)腺苷A2拮抗剂,例如,公开在WO03/035639、WO03/035640等等中的那些。
治疗高脂质血症使用的联用药
与本发明的FAAH抑制剂联合使用的合适药剂包括脂质降低药剂,例如:
(1)胆汁酸螯合剂,例如,考来烯胺,考来维仑(coleseveleni),考来替泊,交联葡聚糖的二烷基氨基烷基衍生物;Colestid?;L Cholest?;和Questran?,等等;(2)HMG-CoA还原酶抑制剂,例如阿托伐他汀,柏伐他汀,carvastatin,西立伐他汀,克伐他汀,达伐他汀,氟伐他汀,格仑伐地汀(glenvastatin),itavaslatin,洛伐他汀,美伐他汀,匹伐他汀(pilavastatin),普伐他汀,雷伐它汀(rivastatin),罗苏伐他汀,西伐他汀,sirrivastatin和ZD-4522,等等,和公开在WO03/033481中的化合物;(3)HMG-CoA合酶抑制剂;(4)胆固醇吸收抑制剂,例如植物醇酯,β-谷甾醇,植物甾醇苷,例如替奎安(Tiqueside);和氮杂环丁酮,例如依泽替米贝,等等;(5)脂酰辅酶A-胆固醇转酰酶(ACAT)抑制剂,例如阿伐麦布(avasimibe)(Current Opinion in Investigational Drugs. 3(9):291-297(2003)),依鲁麦布(efludmibe),KY505,SMP 797,CL-277,082(Clin Pharmacol Ther. 48(2):189-94(1990)),等等;(6)CETP抑制剂,例如,在Nature,406(6792):203-7JTT 705(2000)中确定的JTT 705,托彻普(torcetrapib)(描述在US20030186952和WO20Q00171G4中的CP-529,414),CP 532,032,BAY03-2149,SC 591,SC 795,等等,包括描述在Current Opinion in Investigational Drugs. 4(3):291-297(2003)中的那些;(7)角鲨烯合成酶抑制剂,(8)抗氧化剂,例如murobucol,AG I-1067等等;(9)PPAKALPHA激动剂,例如苄氯贝特(beclofibrate),苯扎贝特(Benzafibrate),比尼贝特,环丙贝特,克利贝特,氯贝特,依托贝特,非诺贝特,gemcabene和吉非贝齐(gemfibrozil),lifibrol,GW 7647,BM 170744,LY518674;及其它纤维酸衍生物,例如Atromid、Lopid和Tricor,和公开在WO03/033456、WO03/033481、WO03/043997、WO03/048116、WO03/053974、WO03/059864、WO03/05875等等中的那些;(10)FXR受体调节剂,例如GW 4064,SR 103912,等等;(11)LXR受体调节剂,例如GW 3965,BOl 3137和XTCOl 79628,和公开在US20030125357、WO03/045382、WO03/053352、WO03/059874等等中的那些;(12)脂蛋白合成抑制剂,例如烟碱酸;(13)肾素血管紧张素系统抑制剂;(14)PPARDELTA部分激动剂,例如,公开在WO03/024395中的那些;(15)胆汁酸再吸收抑制剂,例如BARl 1453,SC435,PHA384640,S8921,AZD7706,等等;(16)PPARDELTA激动剂,例如GW 501516和GW 590735,等等,例如,公开在W097/28149、WO01/79197、WO02/14291、WO02/46154、WO02/46176、WO02/076957、WO03/016291、WO03/033493中的那些;(17)甘油三酯合成抑制剂;(18)微粒体甘油三酯转移(MTI?)抑制剂,例如inplitapide,LAB687和CP346086,等等;(19)转录调节剂;(20)鲨烯环氧酶抑制剂;(21)低密度脂蛋白(LDL)受体诱导剂;(22)血小板凝聚抑制剂;(23)5-LO或FLAP抑制剂;和(24)烟碱酸受体激动剂;(25)PPAR调节剂,例如,公开在WO99/07357、WO99/11255、WO99/12534、WO99/15520、WO99/46232、WO00/12491、WO00/23442、WO00/236331、WO00/236332、WO00/218355、WO00/238553、WO01/25181,WO01/79150、WO02/79162、WO02/100403、WO02/102780、WO02/081428、WO03/0162G5、WO03/033453、WO03/042194、WO03/043997、WO03/066581等等中的那些;(26)公开在WO03/039535中的烟碱酸结合的铬;(27)公开在WO03/040114中的取代的酸衍生物;(28)载脂蛋白B抑制剂,例如,公开在WO02/090347、WO02/28835、WO03/045921、WO03/047575中的那些;(29)因子Xa调节剂,例如,公开在WO03/047517、WO03/047520和WO03/048081中的那些。
治疗高血压症使用的联用药
与本发明的FAAH抑制剂联合使用的合适药剂包括抗高血压药,例如:(1)利尿剂,例如噻嗪,包括氯噻酮,clilorthiazide,二氯苯磺胺,双氢氟噻嗪,吲达帕胺(indapainide),多噻嗪(polytlnazide)和hydrochlorotliiazide,环利尿剂,例如布美他尼,依他尼酸,利尿磺胺和托拉塞米(torsemide);保钾药,例如阿米洛利和三氨蝶啶;和醛甾酮拮抗剂,例如螺甾内酯,依普利酮(epirenone),等等;(2)β-肾上腺素能的阻断剂,例如醋丁洛尔,阿替洛尔,倍他洛尔,贝凡洛尔,比索洛尔,波吲洛尔,卡替洛尔,卡维地洛(caivedilol),塞利洛尔,艾司洛尔,茚诺洛尔,美托洛尔,萘羟心安,奈比洛尔(nebivolol),喷布洛尔,吲哚洛尔,普萘洛尔,甲磺胺心定,特他洛尔(tertatolol),替利洛尔(tilisolol)和噻吗心安,等等;(3)钙通道阻断剂,例如氨氯地平,阿雷地平,阿折地平,巴尼地平,贝尼地平,苄普地尔(bepridtl),西尼地平(cinaldipine),氯维地平,地尔硫,依福地平,非洛地平,加洛帕米,伊拉地平,拉西地平,来米地平(lemildipine),乐卡地平,尼卡地平,硝苯地平,尼伐地平,尼莫地平,尼索地平(ntsoldipine),尼群地平,马尼地平,普拉地平(pranidipine)和维拉帕米,等等;(4)血管紧张肽转化酶(LAMDACE)抑制剂,例如贝那普利;卡托普利;施瑞普利(ceranapril);西拉普利;地拉普利;依那普利;enalopril;fosinoppil;咪达普利;赖诺普利;losinopril;莫昔普利(Moexipril);喹那普利;喹普利拉(quinaprilat);雷米普利;培哚普利(peripidopril);培哚普利(perindropril);喹那普利(quanipril);螺普利(spirapnl);替莫普利(tenocapril);群多普利和佐芬普利(zofenopril),等等;(5)中性肽链内切酶抑制剂,例如奥马趋拉(oinapatrilat),坎沙曲(cadoxatril)和依卡曲尔,法西多曲(fosidotril),山帕曲拉(sampatrilat),LAMDAVE7688,ER4030,等等;(6)内皮素拮抗剂,例如替唑生坦(tezosentan),lamda308165和YM62899,等等;(7)血管扩张剂,例如肼苯哒嗪,可乐定,米诺地尔和烟醇,等等;(8)血管紧张素II受体拮抗剂,例如aprosartan,坎地沙坦,依普罗沙坦(eprosarlan),伊贝沙坦,氯沙坦,olmesartapi,普拉沙坦(pratosartan),他索沙坦,替米沙坦(lelmisartan),缬沙坦(vaisartan)和EXP-3137,FIG828K和RNH6270,等等;(9)α/β肾上腺素能阻断剂,例如尼普洛尔,阿罗洛尔和氨磺洛尔,等等;(10)α1阻断剂,例如特拉唑嗪,呱胺甲尿啶,哌唑嗪,布那唑嗪,曲马唑嗪,多沙唑嗪,萘派地尔,uidoramin,WHP 164和XENOlO,等等;(11)α2激动剂,例如洛非西定(lofexidine),噻美尼定(tiamenidine),莫索尼定(moxonidine),利美尼定(rilmenidine)和胍那苄(guanobenz),等等;(12)醛甾酮抑制剂,等等;和(13)血管生成素(angiopoietin)-2-结合剂,例如那公开在WO03/030833中的那些。
COX-2和FAAH相关的治疗方法
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可用于治疗病症或障碍,在这种病症或障碍中,认为降低或消除COX-2活性和/或FAAH活性和/或MAGL是合乎需要的。由此,它们可以用于能够使用COX-2抑制剂或FAAH抑制剂或MAGL抑制剂的任何状况,它们也可用于其它状况。例如,化合物和相关的前体药物可用于治疗炎症性障碍,包括:认为炎症是该障碍的重要组成部分的障碍和认为炎症是该障碍的相对次要组成部分的障碍,从而治疗急性和慢性疼痛(止痛剂)和治疗发热(退热剂)。在可以治疗的炎症性障碍中有自身免疫障碍。
可以治疗的障碍包括:关节炎(包括类风湿性关节炎,脊椎关节,痛风性关节炎,褪化性关节疾病(即骨关节炎),系统性红斑狼疮,强直性脊柱炎,急性肩部疼痛,牛皮癣和幼年关节炎),哮喘,动脉粥样硬化,骨质疏松症,支气管炎,腱炎,滑囊炎,皮肤炎症(即银屑病,湿疹,灼伤,皮炎),遗尿,嗜酸性疾病,胃肠道障碍(包括炎症性肠病,消化性溃疡,局限性回肠炎,憩室炎,胃肠出血,克罗恩氏病,胃炎,过敏性肠综合症(IBS-C,IBS-A和IBS-D)和溃疡性结肠炎),和用促胃动力药剂改善的障碍(即肠梗阻,例如,手术后的肠梗阻和败血症期间的肠梗阻);胃食管反流病(GORD,或它的同义语GERD);嗜酸性的食管炎,胃轻瘫(gastroparebis),例如糖尿病性的胃轻瘫;食品不耐性和食品变态反应及其它功能性肠障碍,例如,非溃疡性消化不良(NUD)和非心因性胸痛(NCCP)。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,还可以用于治疗与下列相关的症状:流感或其它病毒感染,普通感冒,拉伤和扭伤,肌炎,神经痛,滑膜炎,伤害(例如运动伤害和手术和牙齿处理之后的那些伤害),凝血障碍,肾病(例如,肾功能受损),眼睛障碍(包括青光眼,视网膜炎,视网膜病,葡萄膜炎和眼睛组织急性损伤),肝脏疾病(即,炎症性的肝脏疾病,包括慢性乙型病毒性肝炎,慢性丙型病毒性肝炎,酒精肝损伤,夏科氏肝硬变,自身免疫肝炎,非酒精性脂肪肝炎(NASH)和肝脏移植物排斥),和肺部炎症性疾病(例如,包括哮喘,过敏性鼻炎,呼吸困难综合征,慢性支气管炎和肺气肿)。包含本文所描述的FAAH化合物和其相关前体药物的组合物还可以用于治疗例如与下列相关的炎症:血管疾病,偏头痛,紧张性头痛,结节性动脉周围炎,甲状腺炎,再生障碍性贫血,淋巴肉芽肿病,硬皮症(sclcrodoma),风湿热,I型糖尿病,重症肌无力,结节病,肾病综合症,白塞氏综合征,多肌炎,牙龈炎,超敏反应,结膜炎,多发性脑硬化和缺血(例如,心肌缺血),等等。该化合物可用于治疗与脑障碍相关的神经炎症(例如,帕金森氏症和阿尔茨海默氏病)和与颅辐射性损伤相关的慢性炎症。该化合物可用于治疗急性炎症性病症(例如,由感染引起的那些病症)和慢性炎症性病症(例如,由哮喘、关节炎和炎症性肠病引起的那些病症)。FAAH化合物还可有效用于治疗与创伤和非炎性肌痛相关的炎症。该化合物还可以在手术或摄取抗凝血剂之前给予。该化合物可以降低血栓性心血管状况的危险,这种状况定义为:已知由血小板凝聚、血栓和随后的缺血性临床情况所引起的任何类型突发状况,包括血栓形成或血栓栓塞性中风,心肌缺血,心肌梗塞,狭心症,暂时性缺血性发作(TIA;黑矇症),可逆性的缺血性神经缺陷和任何血管床(内脏、肾、主动脉、周围等等)中的任何类似的血栓形成状况。
本发明的FAAH抑制剂可以抑制由激素所引起的子宫收缩和前列腺素类引起的平滑肌收缩。该化合物可用以治疗早产、月经痛、月经不规则和痛经。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,可以抑制细胞的致瘤性转化和转移性肿瘤生长。本文所描述的化合物与降低腺瘤结肠直肠息肉的死亡数目有关。由此,该化合物和前体药物还可有效用于降低某些癌症的危险,例如,实质固态瘤癌症,例如结肠或结肠直肠癌。该化合物和前体药物还可以用于治疗、预防所有癌症,包括膀胱癌,与HER-2/neu的超表达有关的癌症/宫颈癌,皮肤癌,食道癌,头和颈癌,肺癌(包括非小细胞肺癌),肾癌,胰腺癌,前列腺癌,胆囊癌和胆管癌和子宫内膜癌,胃癌,胶质瘤,肝细胞癌,结肠癌,腺瘤,乳腺癌,卵巢癌和涎腺癌。另外,该化合物和前体药物可以治疗大肠癌和前列腺癌。该化合物还可有效用于患者处于癌症危险之中的情况,包括口腔恶变前的病变,宫颈上皮内瘤样病变,慢性肝炎,胆管增生,非典型性的肺腺瘤增生,前列腺的,上皮内瘤样病变,膀胱发育异常,皮肤的光化性角化病,结肠直肠腺瘤,胃组织变形和Barrett's食道。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其还可用于治疗认知障碍,例如痴呆,尤其是褪化性痴呆(包括老年痴呆,阿尔茨海默氏病(和其前兆),皮克氏病,遗传性舞蹈病,帕金森氏症和Creutzfeldt-Jakob疾病),血管痴呆(包括多发性脑梗塞性痴呆),以及与颅内占位性病变、创伤、感染和相关病症(包括HIV感染)、变形、毒素、缺氧症和维生素缺乏症有关的痴呆;和与年龄有关的轻微的认知损害,尤其是年龄相关的记忆损害。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其还可以通过抑制神经元自由基的形成(由此抑制氧化应激)来防止神经元损伤,并因此用于治疗中风、癫痫和癫痫性的癫痫发作(包括癫痫大发作、小发作、肌肉阵挛性癫痫和部分性发作)。该化合物可以用于控制或抑制癫痫发作(包括化学引起的那些癫痫发作)。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其可以用于治疗所有各种疼痛,包括:与咳嗽病症相关的疼痛,与癌症相关法疼痛,手术前的疼痛,关节炎疼痛及其它形式的慢性疼痛,例如,手术后的疼痛,腰尻痛,肌骨胳疼痛,头痛,偏头痛,肌肉疼痛,下背疼痛和颈疼痛,牙痛等等。该化合物还可用于治疗神经性疼痛。神经性疼痛综合症可以在神经元损伤之后形成,并且所产生的疼痛可以持续几个月或几年,甚至在已经治愈原始损伤之后仍然持续。神经元损伤可以出现在外周神经、后根、脊髓或脑的某些区域中。神经性疼痛综合症传统上按照促成它们的疾病或状况来分类。神经性疼痛综合症包括:糖尿病性神经病变;坐骨神经痛;背痛,非特异性的下背疼痛;多发性脑硬化疼痛;肌纤维痛;HIV相关的神经病;神经痛,例如,疱疹后的神经痛和三叉神经痛;与慢性酒精中毒、甲状腺机能减退、尿毒症或维生素缺乏相关的疼痛;与压迫神经相关的疼痛(例如,腕管综合征)和由创伤、截肢/假肢痛、癌症、毒素或慢性炎症性病症引起的疼痛。神经性疼痛的症状惊人地不同,并且通常被描述为自发性射击和刀割样痛或不断发展的灼痛。另外,还有与普通非疼痛感觉相关的疼痛,例如“发麻”(感觉异常和感觉迟钝),对触摸的敏感性提高(感觉过敏),无害刺激之后的疼痛感觉(动态、静态或热触摸痛),对有害刺激物的敏感性提高(热、冷、机械性痛觉过敏),除去刺激之后继续具有痛觉(痛觉过敏)或不存在选择性感觉传导路或其存在缺陷(痛觉减退)。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其还可以治疗和/或预防环加氧酶介导的增殖障碍,例如,可能出现在糖尿病性视网膜病和肿瘤血管生成中的障碍。该化合物可能用来抑制血管生成,例如,出现在湿性黄斑变性中的血管生成。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其还可以用于治疗性行为问题和/或改善性行为。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其可有效用于预防和/或治疗疼痛,尤其是急性或慢性神经性疼痛,偏头痛,神经性疼痛,包括与疱疹病毒和糖尿病相关的形式,与炎症性疾病、关节炎、类风湿性关节炎、骨关节炎、脊椎炎、痛风、血管、克罗恩氏病、过敏性肠综合症相关的急性或慢性疼痛和神经未稍周围的急性/急剧或慢性疼痛。该化合物还可以用于预防和/或治疗呕吐,眩晕,呕和恶心,尤其是化疗之后,食品性能问题/摄食障碍(即进食障碍,尤其是厌食和各种性质的恶病体质,与癌症及其它消瘦病症相关的体重减轻,或贪食症),神经病变,精神病学的震颤(例如,运动障碍,肌张力障碍,痉挛状态,强迫性的强制行为,Tourette's综合症,任何性质和起源的所有形式的抑郁症和焦虑症,情绪紊乱,精神病),急性或慢性神经变性疾病(例如,帕金森氏症,阿尔茨海默氏病,老年精神错乱,亨廷顿氏舞蹈病,与脑缺血和颅和骨髓损伤相关的病变,癫痫,睡眠障碍(睡眠呼吸暂停),心血管疾病(尤其是高血压症,心因性心律失常,动脉硬化,心力衰竭,心因性缺血,肾缺血),癌症(皮肤良性肿瘤,乳头状瘤和脑肿瘤,前列腺肿瘤,脑肿瘤(恶性胶质瘤,髓状上皮瘤,medullary blastoinas,神经母细胞瘤,肿瘤起源,星形细胞瘤,成星形细胞瘤,室管膜瘤,寡枝神经胶质细胞瘤,血管或纤维丛肿瘤,神经上皮瘤,骨骺肿瘤,ependyblastoinas,恶性脑膜瘤,肉瘤病,恶性黑色素瘤,schvvan细胞癌),免疫系统的障碍(尤其是自身免疫疾病,包括银屑病,红斑狼疮),结合或结缔组织的疾病,Sjogren's综合症,椎关节炎关节僵硬,未分化型椎关节炎,贝切特氏病,自身免疫溶血性贫血症,多发性脑硬化,肌萎缩性侧索硬化,arnyloses,移植排斥和影响胚细胞的疾病,变态反应性疾病(即,快速或迟发性过敏,过敏性鼻炎或结膜炎,接触性皮炎),病毒或细菌寄生性传染病(即AIDS,脑膜炎),炎症性疾病(尤其是关节炎疾病:关节炎,类风湿性关节炎,骨关节炎,脊椎炎,痛风,血管疾病,克罗恩氏病,过敏性肠综合症,骨质疏松症,银屑病,眼睛感染和障碍(即眼睛高血压症,青光眼,湿性黄斑变性),,肺疾病(即,呼吸道的疾病,支气管痉挛,咳嗽,哮喘,慢性支气管炎,呼吸道的慢性阻塞,肺气肿),胃肠机能紊乱(即过敏性肠综合症,肠炎症性的障碍,溃疡,腹泻,酸回流),尿失禁,膀胱炎症,运动障碍,精神运动障碍,高血压症和AIDS相关的综合症。该化合物可以用作睡眠助剂,治疗失眠或引起睡眠。该化合物可以用于降低或控制体重(或脂肪),或预防和/或治疗肥胖症或与过量消耗食品、酒精及其它促进食欲的物质相关的其它食欲相关的障碍。本发明化合物可以用于调节脂类代谢、降低体脂肪(例如,通过提高脂肪使用来降低体脂肪)或降低(或抑制)食欲(例如,通过诱导饱胀感来降低食欲)。该化合物可以用于预防、控制或治疗精神分裂症、偏执狂或其它相关的障碍或多巴胺传输的其它障碍。
本发明的FAAH抑制剂在单独或与第二个活性剂联用的情况下,还可以用于治疗焦虑症(包括广泛性焦虑症、恐慌障碍和社交焦虑症)和抑郁症。
在本发明的FAAH抑制剂单独或与第二个活性剂联用的情况下,其还可以治疗频尿,例如,治疗尿失禁、尿急或膀胱活动过度。频尿是指以排泄少量尿或使少量尿通过为特征的病症(比正常排尿更频繁)。间质性膀胱炎、慢性前列腺炎、神经病(例如,起因于神经性膀胱或脑梗塞)、下尿路前列腺肥大和衰老是与频尿相关的病症。
在一方面,本发明涉及药物组合物,其包含:
抑制FAAH的式I化合物:

或其可药用盐,其中:
X是S或SO;
n是0、1或2;
R1选自:
(1)芳基,和
(2)HET1,
其中R1任选被取代基R4和R5单或二取代;且其中R4和R5独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)单、二或三卤代OC1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-OC1-4烷基,
(e)任选被一或两个选自羟基、CN、-CHF2和-CF3的取代基取代的-C1-4烷基,
(f)任选被羟基、卤代基或CN取代的-C1-2烷基-C3-6环烷基,
(g)-S(O)nC1-4烷基,
(h)-S(O)nNR6R7,
(i)-C(O)-NH-NR8R9,
(j)-C(O)-OH,
(k)任选被卤代基或羟基取代的-C(O)-OC1-4烷基,
(l)-C(O)-NR10R11,
(m)任选被卤代基单、二或三取代的-C(O)-C1-4烷基,
(o)-C(NR12)-NR13R14,
(p)HET4,
(q)芳基,
(r)-C(O)-NH-NH-C(O)H,
(s)-CH2-C(O)-O-C1-4烷基,而CH2可以任选被C1-4烷基或OH取代
(t)-CH2-C(O)NR15R16,而CH2可以任选被C1-4烷基或OH取代,和
(u)-NR17R18,
其中选项(p)和(q)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基;
(9)-C(O)-NR19R20,
(10)-NH2
(11)氧代,
(12)=S,
条件是,选项(q)上的取代基不是氧代或=S,
其中R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、R16、R17、R18、R19和R20各自独立地选自H和C1-4烷基,
或
R6和R7或R8和R9或R10和R11或R13和R14或R15和R16或R17和R18或R19和R20与它们此处相连接的氮连接形成环,形成4至7个原子的5元杂环,所述环含有1、2、3或4个选自N、O和S的杂原子,所述环任选被独立地选自下列的取代基单或二取代:卤代基,羟基,氧代,C1-4烷基,羟基C1-4烷基,卤代基C1-4烷基,-C(O)-C1-4烷基和-S(O)nC1-4烷基;
R2选自:
(1)芳基,
(2)HET3,
(3)-CH2-芳基,
(4)-CH2-HET3,
(5)-C1-6烷基,和
(6)-C3-6环烷基,
其中R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(e)-CF3,
(f)任选被羟基或卤代基取代的-OC1-4烷基,
(g)-C(O)O-C1-3烷基,和
(h)任选被卤代基、C1-4烷基或-OC1-4烷基取代的-S-芳基;
R3选自:
(1)芳基,
(2)HET5,和
(3)C3-6环烷基,
其中R3任选被独立地选自下列的取代基单或二取代:
(a)羟基,
(b)卤代基,
(c)-C3-6环烷基,
(d)-OC3-5环烷基,
(e)-C1-4烷基,
(f)-OC1-4烷基,
(g)-C(O)CH3
(h)单、二或三卤代C1-4烷基,
(i)单、二或三卤代-OC1-4烷基,和
(j)-S(O)n-C1-4烷基;
其中芳基是单或双环芳香环系统;HET1、HET3、HET4和HET5各自独立地是5至10元芳香、部分芳香或非芳香单或双环,或其N-氧化物,所述(环)含有1至4个选自O、S和N的杂原子,并且任选被1至2个氧代取代;
或抑制FAHH的式I化合物
或其可药用盐,其中:
n=0、1或2
R1选自:
(1)苯基,和
(2)HET1,
其中选项(1)和(2)被下列取代

其中R5选自:
(a)卤代基,
(b)-CN,
(c)卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-OC1-4烷基,
(e)任选被一或两个选自羟基、CN、-CHF2和-CF3的取代基取代的-C1-4烷基,
(f)任选被羟基、卤代基或CN取代的-C1-2烷基-C3-6环烷基,
(g)-S(O)nC1-4烷基,
(h)-S(O)nNR6R7,
(i)-C(O)-OH,
(j)任选被卤代基或羟基取代的-C(O)-OC1-4烷基,
(k)-C(O)-NR10R11,
(l)任选被卤代基单、二或三取代的-C(O)-C1-4烷基,
(m)HET2,
(n)芳基,
(o)-CH2-C(O)-O-C1-4烷基,而CH2可以任选被C1-4烷基或OH取代
(t)-CH2-C(O)NR15R16,而CH2可以任选被C1-4烷基或OH取代,和
(u)-NR17R18,
其中选项(m)和(m)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)-NR19R20,
(9)-NH2
(10)氧代,
(11)=S,
其中R6、R7、R10、R11、R15、R16、R17、R18、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被卤代基单、二或三取代,
或
R6和R7或R10和R11或R15和R16或R17和R18或R19和R20与它们相连接的原子连接在一起,形成4至7个原子的5元杂环,所述环含有1、2、3或4个选自N、O和S的杂原子,所述环任选被独立地选自下列的取代基单或二取代:卤代基,羟基,氧代,C1-4烷基,羟基C1-4烷基,卤代C1-4烷基,-C(O)-C1-4烷基和-S(O)nC1-4烷基;
R2选自:
(1)氢,
(2)芳基,
(3)HET3,
(4)-CH2-芳基,
(5)-CH2-HET3,
(6)-C1-6烷基,和
(7)-C3-6环烷基,
其中选项(2)、(3)、(4)、(5)、(6)和(7)任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(e)-CF3,
(f)任选被羟基或卤代基取代的-OC1-4烷基,
(g)-C(O)O-C1-3烷基;
R3选自:
(1)芳基,
(2)HET4,和
(3)C3-6环烷基,
其中选项(1)、(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)羟基,
(b)卤代基,
(c)-C3-6环烷基,
(d)-OC3-5环烷基,
(e)-C1-4烷基,
(f)-OC1-4烷基,
(g)-C(O)CH3
(h)单、二或三卤代C1-4烷基,
(i)单、二或三卤代-OC1-4烷基,和
(j)-S(O)n-C1-4烷基;和
R4选自:
(1)-C1-4烷基,
(2)-卤代C1-4烷基,
(3)H;和
HET1、HET2、HET3和HET4各自独立地是5至10元芳香、部分芳香或非芳香单或双环,含有1-4个选自O、S和N的杂原子,并且任选被1-2个氧代取代。
在该方面,存在上位组,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,和
(9)噁唑基,
其中选项(1)至(9)被下列取代

其中R5选自:
(b)-CN,
(c)卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)任选被羟基或CN取代的-C1-4烷基,
(f)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(h)-S(O)nC1-4烷基,其中n是1或2,
(i)-S(O)2NR6R7,
(j)-C(O)-NR10R11,
(k)HET2,
(l)芳基,和
其中选项(k)和(l)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中该C1-4烷基任选被卤代基单、二或三取代;
和第二个活性剂,例如,选自下列的药剂:
COX-2抑制剂(例如艾托考昔(arcoxia)或西乐葆(CELEBREX));NSAID(例如乙酰水杨酸,水杨酸,水杨酰胺,双水杨酯,二氟尼柳,龙胆酸,消炎痛,舒林酸,甲苯酰吡酸,双氯芬酸,依托度酸,萘丁美酮,布洛芬(lbuprofen),非诺洛芬,酮洛芬,氟比洛芬,舒洛芬,卡洛芬,萘普生,酮咯酸,奥沙普秦,甲芬那酸,甲氯灭酸钠,吡罗昔康和美洛昔康);M-阿片样物质受体激动剂(例如,反胺苯环醇);GABA类似物(例如加巴喷丁),普加巴林;PPARα激动剂,CB1或CB2受体拮抗剂;醋氨酚;多巴胺D2受体拮抗剂;和黑皮质素受体调节剂。
NSAID和COX-2抑制剂是已知的有用的消炎剂、退热剂和止痛剂。
在该方面内,是式I化合物的上位组,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,
(9)噁唑基,和
(10)噁二唑基;
其中R1任选被取代基R4和R5单或二取代,其中R4和R5独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)任选被羟基或CN取代的-C1-4烷基,
(f)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(h)-S(O)nC1-4烷基,其中n是0、1或2,
(i)-S(O)nNR6R7,
(j)-C(O)-NR10R11,
(k)HET4,
(l)芳基,和
其中选项(k)和(l)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基。
在该上位组内,存在式I化合物的下位组,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,
(5)哒嗪基,
(6)1,2,4-噁二唑基,和
(7)1,3,4-噁二唑基,
任选被取代基R4和R5单或二取代,它们独立地选自:
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)nC1-4烷基,
(c)-C(O)-NR10R11,
(d)HET4,和
(e)卤代基,
其中HET4任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基。
在该方面内,存在式I化合物的上位组,其中:
R2选自:
(1)芳基,
(2)HET3,
(3)-CH2芳基,和
(4)-CH2HET3,
其中R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基。
在该上位组内,存在式I化合物的下位组,其中:
R2选自:
(1)芳基,和
(2)HET3,
其中R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3。
在该下位组内,存在式I化合物的一类,其中:
R2选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噁唑基,
(8)吡唑基,
(9)1,2,4-噁二唑基,和
(10)1,3,4-噁二唑基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代。
在该方面内,存在式I化合物的上位组,其中:
R3选自:
(1)芳基,和
(2)HET5,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-OC1-4烷基,
(d)单、二或三卤代C1-4烷基,和
(e)单、二或三卤代-OC1-4烷基。
在该上位组内,存在式I化合物的下位组,其中:
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
在该方面内,存在式I化合物的上位组,其中X是S。
在该方面内,存在下式的上位组

其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,
(9)噁唑基,和
(10)噁二唑;
其中R1任选被取代基R4和R5单或二取代,它们独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)任选被羟基或CN取代的-C1-4烷基,
(f)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(h)-S(O)nC1-4烷基,其中n是0、1或2,
(i)-S(O)nNR6R7,
(j)-C(O)-NR10R11,
(k)HET4,
(l)芳基,和
其中选项(k)和(l)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基;
R2选自:
(1)芳基,
(2)HET3,
(3)-C1-6烷基,和
(4)-C3-6环烷基,
其中选项R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基;和
R3选自:
(1)芳基,和
(2)HET5,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-C1-4烷基,
(d)-OC1-4烷基,
(e)单、二或三卤代C1-4烷基,和
(f)单、二或三卤代-OC1-4烷基。
在该上位组内,存在式Ia化合物的下位组,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,
(5)哒嗪基,
(6)1,2,4-噁二唑基,和
(7)1,3,4-噁二唑基,
任选被取代基R4和R5单或二取代,它们独立地选自:
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)nC1-4烷基,
(c)-C(O)-NR10R11,
(d)HET4,和
(e)卤代基,
其中HET4任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基。
R2选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噁唑基,
(8)吡唑基,
(9)1,2,4-噁二唑基,和
(10)1,3,4-噁二唑基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代;和
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
在该上位组内,存在下式的下位组

其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
其中R1任选被取代基R4和R5单或二取代,它们独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)-C(CH3)2-OH;
R2选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)吡唑基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代;和
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
在该下位组内,存在式Ia化合物的一类,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)吡嗪基,
其中R1任选被取代基R4和R5单或二取代,它们独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)-C(CH3)2-OH;
R2选自:
(1)苯基,
(2)吡啶基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代;和
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
实施例1至138举例说明了式I的化合物。
在该方面内,存在式II化合物的上位组,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,和
(5)哒嗪基,
其中选项(1)至(5)被下列取代

R5选自
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)2C1-4烷基,
(c)-C(O)-NR10R11,
(d)HET2,和
(e)卤代基,
其中选项(d)任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被卤代基单、二或三取代。
在该方面内,存在式II化合物的上位组,其中:
R2选自:
(1)氢,
(2)芳基,
(3)HET3,
(4)-C1-6烷基,和
(5)-C3-6环烷基,
其中选项(2)、(3)、(4)和(5)任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基。
在该上位组内,存在式II化合物的下位组,其中:
R2选自:
(1)氢,
(2)-C1-6烷基,和
(3)-C3-6环烷基,
其中选项(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3。
在该方面内,存在式II化合物的上位组,其中:
R3选自:
(1)苯基,和
(2)HET4,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-C1-4烷基,
(d)-OC1-4烷基,
(e)单、二或三卤代C1-4烷基,和
(f)单、二或三卤代-OC1-4烷基。
在该上位组内,存在式II化合物的下位组,其中:
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
(4)哒嗪基,
(5)吡嗪基,
其中选项(1)、(2)、(3)、(4)和(5)各自任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
在该方面内,存在式II化合物的上位组,其中:

或其可药用盐,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,和
(9)噁唑基,
其中选项(1)至(9)被下列取代

且R5选自
(a)-CN,
(b)卤代C1-4烷基,
(c)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(d)任选被羟基或CN取代的-C1-4烷基,
(e)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(g)-S(O)nC1-4烷基,其中n是1或2,
(h)-S(O)2NR6R7,
(i)-C(O)-NR10R11,
(j)HET2,
(k)芳基,和
其中选项(j)和(k)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被氚化或被卤代基单、二或三取代,或
R2选自:
(1)氢,
(2)芳基,
(3)HET3,
(4)-C1-6烷基,和
(5)-C3-6环烷基,
其中选项(2)、(3)、(4)和(5)任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基;和
R3选自:
(1)苯基,和
(2)HET4,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-C1-4烷基,
(d)-OC1-4烷基,
(e)单、二或三卤代C1-4烷基,和
(f)单、二或三卤代-OC1-4烷基;
R4选自:
(1)任选氚化的-C1-4烷基,和
(3)H;
在该上位组内,存在式II化合物的下位组,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,和
(5)哒嗪基,
其中选项(1)至(5)被下列取代

且R5选自
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)2C1-4烷基,
(c)-C(O)-NR10R11,和
(d)HET2,
其中选项(d)任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被氚化或被卤代基单、二或三取代,或
R2选自:
(1)氢,
(2)-C1-6烷基,和
(3)-C3-6环烷基,
其中选项(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3;
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
(4)吡嗪基,和
(5)哒嗪基,
其中选项(1)、(2)、(3)、(4)和(5)各自任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
在该下位组内,存在式II化合物的一类,其中:
R1选自:
(1)苯基,和
(2)吡啶基,
其中选项(1)和(2)被下列取代

且R5选自
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)2C1-4烷基,
(c)-C(O)-NR10R11,
(d)HET2,和
其中选项(d)任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被氚化、被卤代基单、二或三取代,或
R2选自:
(1)氢,
(2)-C1-6烷基,和
(3)-C3-6环烷基,
其中选项(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3;
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中选项(1)、(2)和(3)各自任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
实施例1B至43B举例说明了式II的化合物。
本发明的化合物可以含有一个或多个不对称中心,并可以由此以消旋体和消旋混合物、单一对映体、非对映体混合物和单一非对映体的形式出现。根据分子上各种取代基的性质,可以存在额外的不对称中心。每个这种不对称中心独立地产生两个旋光异构体,混合物中的所有合适的旋光异构体和非对映体和纯的或部分纯的化合物包括在本发明范围之内。本发明包括这些化合物的所有这种异构形式。式I显示了该类化合物的结构(没有优选的立体化学)。可以利用本文所公开方法的合适改型,按照本领域已知的方式独立地合成这些非对映体或将它们色谱分离。可以利用晶体产物或衍生的晶体中间体的X-射线晶体衍射来确定它们的绝对立体化学,如有必要,使用含有已知绝对构型的不对称中心的试剂。如果需要的话,可以分离化合物的外消旋混合物,以便分离出单一对映体。可以利用本领域众所周知的方法进行分离,例如,化合物的外消旋混合物与对映体纯化合物偶合,形成非对映体混合物,而后通过标准方法(例如分级结晶或色谱)进行单一非对映体的分离。偶合反应常常使用对映体纯的酸或碱,以形成盐。然后,通过所加入的手性残基的断裂,非对映体的衍生物可以转变为纯的对映体。还可以通过使用手性固定相的色谱方法(本领域众所周知的方法)来直接分离化合物的消旋混合物。或者,利用本领域众所周知的方法,使用已知构型的光学纯的起始原料或试剂,通过立体选择性合成,可以获得化合物的任何对映体。
本发明还包括式I化合物的所有可药用同位素变体,其中一个或多个原子被具有相同原子序数但原子量或质量数不同于自然界通常发现的原子量或质量数的原子替代。
适合于包含在本发明化合物中的同位素的例子包括:氢的同位素,例如2H和3H,碳的同位素,例如11C、13C和14C,氮的同位素,例如13N和15N,氧的同位素,例如15O、17O和18O,磷的同位素,例如32P,硫的同位素,例如35S,氟的同位素,例如18F,碘的同位素,例如23I和125I,和氯的同位素,例如36Cl。
某些同位素标记的式I化合物(例如,结合放射性同位素的那些)可有效用于药物和/或基质组织分布研究。考虑到结合的容易程度和现有的检测方法,放射性同位素氚(即3H)和碳14(即14C)尤其可用于这种目的。
用重同位素例如氘(即,2H)进行替代,因为代谢性稳定性更好,可以得到某些治疗有利结果(例如,提高体内半衰期或降低剂量要求),由此可以在一些情况下优选使用。
用正电子发射同位素(例如11C、18F、15O和13N)替代,可有效用于检验基质受体占有率的正电子发射断层扫描成像(PET)研究。
利用本领域技术人员熟知的传统方法,或利用与伴随实施例所描述方法类似的方法,使用合适的同位素标记的试剂来代替先前使用的非标记的试剂,通常可以制备同位素标记的式I化合物。
使用下列定义描述本发明,除非另有陈述。
术语“卤素”或“卤代基”包括F、Cl、Br和I。
术语“烷基”是指具有指定碳原子数目的直链或支链结构和其组合。由此,例如,C1-6烷基包括甲基,乙基,丙基,2-丙基,仲和叔丁基,丁基,戊基,己基,1,1-二甲基乙基。
术语“烷氧基”是指具有指定碳原子数目的直链、支链或环构型的烷氧基。C1-6烷氧基,例如,包括甲氧基,乙氧基,丙氧基,异丙氧基,等等。
术语“烷硫基”是指具有指定碳原子数目的直链、支链或环构型的烷硫基。C1-6烷硫基,例如,包括甲硫基,丙硫基,异丙硫基,等等。
术语“烯基”是指具有指定碳原子数目的、具有至少一个碳-碳双键的直链或支链结构和其组合,其中氢可以被额外的碳-碳双键替代。C2-6烯基,例如,包括乙烯基,丙烯基,1-甲基乙烯基,丁烯基等等。
术语“炔基”是指具有指定碳原子数目的、具有至少一个碳-碳三键的直链或支链结构和其组合。C3-6炔基,例如,包括丙炔基,1-甲基乙炔基,丁炔基等等。
术语“环烷基”是指指定碳原子数目的任选与直链或支链结构相结合的单、二或三环结构。环烷基的例子包括环丙基,环戊基,环庚基,金刚烷基,环十二烷基甲基,2-乙基-1-二环[4.4.0]癸基,等等。
术语“芳基”定义为:单或双环芳香环系统,并且包括例如苯基、萘基,等等。
术语“芳烷基”是指1至6个碳原子的上述烷基,其中一个烷基氢原子被上述芳基取代(例如,苄基,等等)。
术语“芳氧基”是指通过氧原子与分子连接的上述芳基(芳基-O),并且包括,例如,苯氧基,萘氧基等等。
术语“芳烷氧基”是指通过氧原子与分子连接的上述芳烷基(芳烷基-O),并且包括,例如,苄氧基等等。
术语“芳硫基”定义为:通过硫原子与分子连接的上述芳基(芳基-S),并且包括,例如,苯硫基,萘硫基等等。
术语“芳酰基”是指通过羰基与分子连接的上述芳基(芳基-C(O)-),并且包括,例如,苯甲酰基,萘酰基等等。
术语“芳酰基氧基”是指通过氧原子与分子连接的上述芳酰基(芳酰基-O),并且包括,例如,苯甲酰氧基或苯酰氧基,萘甲酰氧基等等。
术语“HET”,例如在“HET1”、“HET2”、“HET3”、“HET4”、”HET1”、“HET2”、“HET3”、“HET4”中,定义为5至10元芳香、部分芳香或非芳香单或双环,含有1-4个选自O、S和N的杂原子,并且任选被1-2个氧代取代。在合适的情况下,Het基团包括N-氧化物。优选,“HET”是含有1-3个选自O、S和N的杂原子的5或6元芳香或非芳香单环,例如,吡啶,嘧啶,哒嗪,呋喃,噻吩,噻唑,噁唑,异噁唑等等,或HET是含有1-3个选自O、S和N的杂原子的9或10元芳香或部分芳香双环,例如,苯并呋喃,苯并噻吩,吲哚,吡喃并吡咯,苯并吡喃,喹啉,苯并环己基,萘啶等等。“HET”还包括下列:苯并咪唑基,苯并呋喃基,苯并吡唑基,苯并三唑基,苯并噻吩基,苯并噁唑基,咔唑基,咔啉基,噌琳基,呋喃基,咪唑基,二氢吲哚基,吲哚基,吲嗪基(indolazinyl),吲唑基,异苯并呋喃基,异氮茚基,异喹啉基,异噻唑基,异噁唑基,萘啶基,噁二唑基,噁唑基,吡嗪基,吡唑基,吡啶并吡啶基,哒嗪基,吡啶基,嘧啶基,吡咯基,喹唑啉基,喹啉基,喹喔啉基,噻二唑基,噻唑基,噻吩基,三唑基,氮杂环丁烷基,1,4-二噁烷基,六氢氮杂 基,哌嗪基,哌啶基,吡咯烷基,吗啉基,硫吗啉基,二氢苯并咪唑基,二氢苯并呋喃基,二氢苯并噻吩基,二氢苯并噁唑基,二氢呋喃基,二氢咪唑基,二氢吲哚基,二氢异噁唑基,二氢异噻唑基,二氢噁二唑基,二氢噁唑基,二氢吡嗪基,二氢吡唑基,二氢吡啶基,二氢嘧啶基,二氢吡咯基,二氢喹啉基,二氢四唑基,二氢噻二唑基,二氢噻唑基,二氢噻吩基,二氢三唑基,二氢氮杂环丁烷基,亚甲基二氧基苯甲酰基,四氢呋喃基和四氢噻吩基。在一方面,“Het”选自吡啶基,哒嗪基,嘧啶基,吡嗪基,噻唑基,噻吩基,吡咯基,噁唑基和噁二唑。
基,哌嗪基,哌啶基,吡咯烷基,吗啉基,硫吗啉基,二氢苯并咪唑基,二氢苯并呋喃基,二氢苯并噻吩基,二氢苯并噁唑基,二氢呋喃基,二氢咪唑基,二氢吲哚基,二氢异噁唑基,二氢异噻唑基,二氢噁二唑基,二氢噁唑基,二氢吡嗪基,二氢吡唑基,二氢吡啶基,二氢嘧啶基,二氢吡咯基,二氢喹啉基,二氢四唑基,二氢噻二唑基,二氢噻唑基,二氢噻吩基,二氢三唑基,二氢氮杂环丁烷基,亚甲基二氧基苯甲酰基,四氢呋喃基和四氢噻吩基。在一方面,“Het”选自吡啶基,哒嗪基,嘧啶基,吡嗪基,噻唑基,噻吩基,吡咯基,噁唑基和噁二唑。
对于上述所有定义,当在说明书中提到时,每个提到的基团与所有其它提到的相同基团无关。例如,如果R1和R2 都是HET,HET的定义彼此无关,并且R1和R2可以是不同的HET基团,例如呋喃和噻吩。
式I化合物选择性地抑制FAAH的能力使得它们可用于治疗、预防或逆转各种炎症性和非炎性疾病和病症的发展。
受益于抑制FAAH酶活性的疾病、障碍、综合症和/或病症包括,例如,阿尔茨海默氏病,精神分裂症,抑郁症,酒精中毒,成瘾,自杀,帕金森氏症,亨丁顿舞蹈症,中风,呕吐,流产,胚移植,内毒素性休克,肝硬化,动脉粥样硬化,癌症,创伤性颅脑损伤,青光眼和骨水泥植入综合症。
受益于抑制FAAH活性的其它疾病、障碍、综合症和/或病症包括,例如,多发性脑硬化,视网膜炎,肌萎缩性侧索硬化,免疫缺陷性病毒引起的脑炎,注意缺陷性多动障碍,疼痛,感受伤害性疼痛,神经性疼痛,炎症性疼痛,非炎症性疼痛,出血性膀胱炎疼痛,与疱疹病毒相关的疼痛,与糖尿病相关的疼痛,周围神经性疼痛,中枢性疼痛,丘脑性疼痛综合症,传导阻滞性疼痛,慢性感受伤害性疼痛,刺激感受伤害性受体,幻想和短时急性疼痛,手术后的疼痛,癌症疼痛,与多发性脑硬化相关的疼痛和痉挛状态,蛛网膜炎,神经根病,神经痛,身体疼痛,深部疼痛,表面疼痛,内脏疼痛,急性疼痛,慢性疼痛,猛爆型疼痛,慢性背痛,腰椎手术失败综合症,肌纤维痛,中风后疼痛,三叉神经痛,坐骨神经痛,放射治疗疼痛,复杂区域疼痛综合症,灼痛,反射交感性营养不良,假肢痛和肌筋膜疼痛。
受益于抑制FAAH活性的其它疾病、障碍、综合症和/或病症包括,肥胖症,高脂质血症,代谢失调,摄食和禁食,改变食欲,应激反应,记忆,衰老,高血压症,脓毒性休克,心原性休克,肠炎和活动性,过敏性肠综合症,大肠炎,腹泻,回肠炎,缺血,脑缺血,肝缺血,心肌梗塞,脑兴奋性中毒,癫痫发作,发热性癫痫发作,神经毒性,神经病,睡眠,诱导睡眠,延长睡眠,失眠和炎症性疾病。受益于抑制FAAH活性的神经和心理障碍包括,例如,疼痛,抑郁症,焦虑症,广泛性焦虑症(GAD),强迫性的强制障碍,应激反应,应激性尿失禁,注意缺陷多动障碍,精神分裂症,精神病,帕金森氏症,肌肉痉挛状态,癫痫,运动障碍(diskenesia),癫痫发作障碍,时差综合症和失眠。
FAAH抑制剂还可以治疗各种代谢综合症、疾病、障碍和/或病症,包括但不局限于:抗胰岛素性综合症,糖尿病,高脂质血症,脂肪肝疾病,肥胖症,动脉粥样硬化和动脉硬化。FAAH抑制剂还可以治疗各种疼痛综合症、疾病、障碍和/或病症,包括但不局限于以下列为特征的那些:非炎性疼痛,炎症性疼痛,周围神经疼痛,中枢性疼痛,传导阻滞性疼痛,慢性感受伤害性疼痛,刺激感受伤害性受体,幻想和短促急性疼痛。
抑制FAAH活性还可以用于治疗各种涉及炎症的病症。这种病症包括但不局限于:关节炎(例如类风湿性关节炎,肩部腱炎或滑囊炎,痛风性关节炎和风湿性多肌痛),器官特异性炎症性疾病(例如甲状腺炎,肝炎,炎症性的肠疾病),哮喘,其它自身免疫疾病(例如多发性脑硬化),慢性阻塞性肺病(COPD),过敏性鼻炎和心血管疾病。
在某些情况下,FAAH抑制剂可有效用于预防神经变性或用于神经保护。
另外,已经表明,当FAAH活性降低或不存在时,它的基质之一(花生四烯酸乙醇胺)起到COX-2的基质的作用,其使花生四烯酸乙醇胺转变为前列腺胺(prostamides)(Weber等人J Lipid,Res. 2004;45:757)。在FAAH抑制剂的存在下,某些前列腺胺(prostamides)的浓度升高。某些前列腺胺(prostamides)与眼内压降低和眼压降低有关。由此,在一个实施方案中,FAAH抑制剂可用于治疗青光眼。
在一些实施方案中,FAAH抑制剂可用于治疗或降低EMDs的危险,包括但不局限于:肥胖症,食欲障碍,超重,皮下脂肪丰富,I型和II型糖尿病,高血糖症,血脂异常,脂肪肝炎,肝脏皮脂腺病,非酒精性脂肪肝炎,综合症X,抗胰岛素性,糖尿病性的血脂异常,厌食,贪食症,神经性厌食症,高脂质血症,高甘油三酯血症,动脉粥样硬化,动脉硬化,炎症性的障碍或病症,阿尔茨海默氏病,克罗恩氏病,血管炎症,炎症性的肠障碍,类风湿性关节炎,哮喘,血栓或恶病体质。
在其它实施方案中,FAAH抑制剂可用于治疗或降低抗胰岛素性综合症和糖尿病的危险,即,原发性实质性糖尿病,例如I型糖尿病或II型糖尿病,和继发性非实质性糖尿病。给予含有治疗有效量的体内FAAH抑制剂的组合物,可降低糖尿病症状的严重程度或降低形成糖尿病症状的危险,例如动脉粥样硬化,高血压症,高脂质血症,肝脏皮脂腺病,肾病,神经病,视网膜病,脚溃疡或白内障。
在另一个实施方案中,FAAH抑制剂可用于治疗食品滥用行为,尤其是容易导致超重的那些食品滥用行为,例如,贪食症、爱好糖或脂肪和非胰岛素依赖性糖尿病。
在一些实施方案中,FAAH抑制剂可用于治疗患有EMD以及患有抑郁症或焦虑症的患者。优选,患者在给予FAAH抑制剂组合物之前是诊断为患有抑郁或精神病症的患者。由此,对于EMD和抑郁或焦虑症两者,给予该患者治疗有效剂量的FAAH抑制剂。
优选,治疗的患者是人。然而,该方法还可以用于治疗非人类的哺乳动物。尤其使用EMDs的动物模型,例如,描述在例如美国专利US 6,946,491中的那些动物模型。
FAAH抑制剂组合物还可以在希望降低其体重(为了美容,而不一定是医学因素)的个体中用于降低体重。
应理解,当使用本文所描述的任何联用药时,本发明的FAAH化合物和另一个活性剂两者可以在合理的期间内给予患者。化合物可以在相同的可药用载体中,并因此可以同时给予。它们可以在独立的药用载体中,例如,同时摄取的常规口服剂型。术语“联用药”还是指化合物提供于独立剂型中并且顺序给予的情况。因此,例如,可以以片剂形式给予一种活性组分,而后,在合理的期间内,可以以口服剂型(例如片剂)或迅速溶解的口服剂型给予第二种活性组分。“迅速溶解的口服制剂”是指口服递送形式,当放在患者的舌头上时,其在大约10秒钟之内溶解。“合理的期间”是指不超过大约1小时的时间周期。也就是说,例如,如果以片剂形式提供第一个活性组分,则在一小时之内,应该给予第二个活性组分,可以在相同的剂型中,或在能够有效递送药物的另一个剂型中。
FAAH抑制剂组合物可以与降低循环胆固醇水平的药物(例如,他汀类(statin),烟碱酸,纤维酸衍生物或胆汁酸结合树脂)联合给予。FAAH抑制剂组合物还可以与体重减轻药物(例如,奥利司他)或食欲抑制剂(例如安非拉酮,马吲哚(mazindole),奥利司他,苯二甲吗啉,苯丁胺或西布曲明)联合使用。
术语“治疗”不但包括治疗患者,以便除去患者的疾病或病症体征和症状,而且包括预防性治疗无症状的患者,以便预防疾病或病症的发作,或预防、减缓或逆转疾病或病症的发展。术语“治疗有效量”是指引起研究人员、兽医、医生或其它临床医师所研究的组织、系统、动物或人的生物或医学响应的药物或药剂的数量。该术语还包括预防或降低研究人员、兽医、医生或其它临床医师所试图预防的组织、系统、动物或人出现生物或医学状况的危险的药物数量。
术语“治疗”不但包括治疗患者,以便除去患者的疾病或病症体征和症状,而且包括预防性治疗无症状的患者,以便预防疾病或病症的发作,或预防、减缓或逆转疾病或病症的发展。术语“治疗有效量”是指引起研究人员、兽医、医生或其它临床医师所研究的组织、系统、动物或人的生物或医学响应的药物或药剂的数量。该术语还包括预防或降低研究人员、兽医、医生或其它临床医师所试图预防的组织、系统、动物或人出现生物或医学状况的危险的药物数量。
下列缩写具有指定的含义:
AIBN=2.2'-偶氮二异丁腈
B.P.=过氧苯甲酰
Bn=苄基
CCl4=四氯化碳
D=-O(CH2)3O-
DAST=二乙胺三氟化硫
DCC=二环己基碳二亚胺
DCI=1-(3-二甲基氨基丙基)-3-乙基碳二亚胺
DEAD=偶氮二甲酸二乙基酯
DIBAL=二异丁基氢化铝
DME=乙二醇二甲醚
DMAP=4-(二甲基氨基)吡啶
DMF=N,N-二甲基甲酰胺
DMSO=二甲亚砜
Et3N=三乙胺
LDA=二异丙基胺基锂
m-CPBA=间氯过氧苯甲酸
NBS=N-溴代琥珀酰亚胺
NSAID=非甾体抗炎症药物
PCC=氯铬酸吡啶
PDC=重铬酸吡啶盐
Ph=苯基
1,2-Ph =1,2-苯二基
Pyr=吡啶二基
Qn=7-氯喹啉-2-基
Rs=-CH2SCH2CH2Ph
r.t.=室温
rac.=消旋
THF=四氢呋喃
THP=四氢吡喃-2-基
烷基缩写
Me=甲基
Et=乙基
n-Pr=正丙基
i-Pr=异丙基
n-Bu=正丁基
i-Bu=异丁基
s-Bu=仲丁基
t-Bu=叔丁基
c-Pr=环丙基
c-Bu =环丁基
c-Pen =环戊基
c-Hex=环己基。
本文所描述的一些化合物可以含有一个或多个不对称中心,并可以由此产生非对映体和旋光异构体。本发明包括这种可能的非对映体以及它们的消旋和拆分、对映体纯形式和其可药用盐。
一些本文所描述化合物含有烯双键,除非另外说明,否则,包括E和Z几何异构体。
本发明的药物组合物包含作为活性组分的式I化合物或其可药用盐,并且还可以含有可药用载体和任选的其它治疗组分。术语“可药用盐”是指由可药用无毒的碱(包括无机碱和有机碱)制备的盐。衍生自无机碱的盐包括下列的盐:铝,铵,钙,铜,铁,亚铁,锂,镁,锰盐,二价锰,钾,钠,锌,等等。尤其优选的是铵、钙、镁、钾和钠盐。衍生自可药用有机无毒碱的盐包括:伯、仲和叔胺的盐,取代的胺的盐,包括天然存在的取代的胺、环胺和碱离子交换树脂,例如精氨酸,甜菜碱,咖啡因,胆碱,N,N'-二苄基乙二胺,二乙胺,2-二乙氨基乙醇,2-二甲氨基乙醇,乙醇胺,乙二胺,N-乙基-吗啉,N-乙基哌啶,葡糖胺,氨基葡糖,组氨酸,哈胺(hydrabamine),异丙胺,赖氨酸,葡甲胺,吗啉,哌嗪,哌啶,多胺树脂,普鲁卡因,嘌呤,可可碱,三乙胺,三甲胺,三丙胺,氨基丁三醇,等等。
当本发明的化合物是碱性化合物时,盐可以由可药用无毒的酸(包括无机和有机酸)来制备。这种酸包括,例如,乙酸,苯磺酸,苯甲酸,樟脑磺酸,柠檬酸,乙磺酸,富马酸,葡糖酸,谷胺酸,氢溴酸,盐酸,羟乙磺酸,乳酸,马来酸,苹果酸,扁桃酸,甲磺酸,粘酸,硝酸,双羟萘酸,泛酸,磷酸,琥珀酸,硫酸,酒石酸,对甲苯磺酸等等。尤其优选的是柠檬酸,氢溴酸,盐酸,马来酸,磷酸,硫酸和酒石酸。
可以理解,在后面治疗方法的讨论中,提及式I的化合物还包括可药用盐。
当然,式I化合物的预防或治疗剂量的大小随着所治疗病症的性质和严重程度和使用的具体式I化合物和它的给药途径而变化。它还根据各种因素而变化,包括个体患者的年龄,重量,常规健康情况,性别,饮食,给药时间,排泄速度,药物联用和反应。通常,日剂量为每千克哺乳动物体重大约0.001 mg至大约100 mg,优选每千克0.01 mg至大约10 mg。另一方面,在某些情况下,需要使用超出这些限制的剂量。
可以与载体物质组合制备单一剂型的活性组分的数量根据所治疗宿主和具体给药模式的不同而变化。例如,为人口服给药设计的制剂可以包含大约0.5 mg至大约5 g活性剂,活性剂与合适和适当数量的载体混合,载体可以在整个组合物的大约5至大约95%之间变化。剂量单位形式通常含有大约1 mg至大约2 g活性组分,典型地含有25 mg,50 mg,100 mg,200 mg,300 mg,400 mg,500 mg,600 mg,800 mg或1000 mg。
为了治疗FAAH介导的疾病,可以口服、局部、胃肠外、吸入喷雾剂或直肠给予式I的化合物,式I的化合物在含有常规无毒的可药用载体、佐剂和赋形剂的剂量单位制剂中。本文使用的术语“肠胃外”包括皮下、静脉内、肌内、胸骨内注射或输液技术。除了治疗恒温动物之外,例如小鼠,大鼠,马,牛,羊,狗,猫等等,本发明的化合物还可有效治疗人。
含有活性组分的药物组合物可以在适合于口服使用的形式中,例如,片剂,锭剂,糖锭,溶液剂,水或油混悬剂,可分散性粉剂或颗粒剂,乳剂,硬或软胶囊,糖浆剂或酏剂。为口服使用设计的组合物可以按照制备药物组合物领域已知的任何方法来制备,并且为了提供药学精美和适口的制剂,这种组合物可以含有一或多种选自甜味剂、调味剂、着色剂和防腐剂的试剂。片剂可以含有活性组分与适合于制备片剂的无毒可药用赋形剂的混合物。这些赋形剂可以是,例如,惰性稀释剂,例如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠;造粒和崩解剂,例如,玉米淀粉或海藻酸;粘合剂,例如淀粉、明胶或阿拉伯胶,和润滑剂,例如硬脂酸镁、硬脂酸或滑石粉。片剂可以是无包衣片剂,或可以利用已知的技术将它们包衣,以便在胃肠道中延迟崩解和吸收,并由此提供长时间的持续作用。例如,可以使用时间延迟物质,例如单硬脂酸甘油酯或二硬脂酸甘油酯。还可以利用美国专利US 4,256,108、4,166,452和4,265,874所描述的技术,将它们包衣,以便形成用于控制释放的渗透治疗片剂。
还可以以硬明胶胶囊形式提供口服使用的制剂,在硬明胶胶囊中,活性组分与惰性固体稀释剂(例如,碳酸钙、磷酸钙或高岭土)混合,或以软明胶胶囊形式提供制剂,在软明胶胶囊中,活性组分与水可互溶的溶剂(例如,丙二醇、PEGs和乙醇)或油介质(例如,花生油、液体石蜡或橄榄油)混合。
水悬剂含有活性物质与适合于制备水悬剂的赋形剂的混合物。这种赋形剂是悬浮剂,例如羧甲基纤维素钠,甲基纤维素,羟丙基甲基纤维素,海藻酸钠,聚乙烯吡咯烷酮,黄芪胶和阿拉伯树胶;分散或湿润剂可以是天然存在的磷脂,例如磷脂酰胆碱,或氧化烯与脂肪酸的缩合产物,例如硬脂酸聚氧乙烯酯,或氧化乙烯与长链脂族醇的缩合产物,例如十七烷环氧丙烷鲸蜡醇(heptadecaethyleneoxycetanol),或氧化乙烯与衍生自脂肪酸和己糖醇的偏酯的缩合产物,例如聚氧乙烯山梨糖醇一油酸酯,或氧化乙烯与衍生自脂肪酸和己糖醇酐的偏酯的缩合产物,例如,聚乙烯单油酸山梨醇酐酯。水悬剂还可以含有一或多种防腐剂,例如对羟基苯甲酸乙酯或对羟基苯甲酸正丙基酯,一或多种着色剂,一或多种调味剂和一或多种甜味剂,例如蔗糖、糖精或阿斯巴甜。
油状混悬剂可以通过将活性组分悬浮在植物油(例如花生油、橄榄油、芝麻油或椰子油)或矿物油(例如液体石蜡)中来配制。油性混悬剂可以含有增稠剂,例如蜂蜡、硬石蜡或鲸蜡醇。可以加入甜味剂(例如,上面列出的那些)和调味剂,以便提供适口的口服制剂。可以通过加入抗氧化剂(例如抗环血酸)来保存这些组合物。
适合于制备水悬剂(加入水)的可分散性粉剂和颗粒剂提供活性组分与分散或湿润剂、悬浮剂和一或多种防腐剂的混合物。可以通过上述那些来举例说明合适的分散或湿润剂和悬浮剂。还可以存在额外的赋形剂,例如甜味剂、调味剂和着色剂。
本发明的药物组合物还可以是水包油乳剂形式。油相可以是植物油,例如橄榄油或花生油,或矿物油,例如液体石蜡或这些的混合物。合适的乳化剂可以是天然存在的磷脂,例如大豆、磷脂酰胆碱和衍生自脂肪酸和己糖醇酸酐的酯或偏酯,例如单油酸山梨醇酐酯,和所述偏酯与氧化乙烯的缩合产物,例如聚氧乙烯单油酸山梨醇酐酯。乳剂还可以含有甜味剂和调味剂。
糖浆剂和酏剂可以与甜味剂一起配制,例如丙三醇,丙二醇,山梨糖醇或蔗糖。这种制剂还可以含有缓和剂,防腐剂,调味剂和着色剂。药物组合物可以是无菌注射水溶液或油脂性混悬剂形式。这种混悬剂可以使用上述那些合适的分散剂或湿润剂和悬浮剂、按照已知的技术来配制。无菌注射制剂还可以是无菌注射溶液或悬浮液(在无毒的、胃肠外可接受的稀释剂或溶剂中,例如在1,3-丁二醇中的溶液)。在可以使用的可接受的赋形剂和溶剂之中,可以使用水、林格溶液和等渗氯化钠溶液。还可以使用共溶剂,例如乙醇、丙二醇或聚乙二醇。另外,无菌的不挥发油通常用作溶剂或悬浮介质。为了该目的,可以使用任何柔和的不挥发油,包括合成的甘油一酯或甘油二酯。另外,在注射制剂中可以使用脂肪酸,例如油酸。
式I的化合物还可以以直肠给药的栓剂形式给予。这些组合物可以如下制备:将药物与合适的无刺激性的赋形剂混合,该赋形剂在环境温度下是固体,但在直肠温度下是液体,并因此在直肠中融化,从而释放药物。这种物质是可可脂和聚乙二醇。
对于局部使用来说,使用含有式I化合物的乳膏剂、软膏剂、凝胶剂、溶液剂或混悬剂,等等。(对本申请来说,局部施用包括漱口剂和含漱液)。局部制剂通常可以由药用载体、共溶剂、乳化剂、渗透增强剂、防腐剂系统和软化剂组成。
试验
下列试验举例说明本发明的应用性:
对本发明的化合物进行药理学评价,以便测定它们对酶FAAH(脂肪酸酰胺水解酶)的抑制效果。
为了有助于在试验中形成针对人的稳定细胞系,形成鼠和大鼠的全长FAAH。人FAAH cDNA(登记号码:NM_001441.1)购买于Origene(Rockville,MD)。使用XbaI和EcoRI限制位点将全长FAAH亚克隆到哺乳动物表达载体(pcDEF.neo)中,并用于稳定的细胞系繁殖。

分别使用引物1和2或引物1和3(参见表),由脑cDNA(BD Biosciences,San Jose,CA)通过逆转录酶聚合酶链式反应(RT-PCR)扩增鼠(登录号NM_010173)和大鼠FAAH(登录号NM_024132)。将得到的PCR产物连接到确定的pCR4 TOPO和DNA序列上。使用EcoRI(鼠)或KpnI和EcoRI(大鼠)限制位点,将全长鼠FAAH亚克隆到哺乳动物表达载体(pcDEFneo)中。按照生产商方案(AMAXA),转染中国仓鼠卵巢细胞(CHO)。为了分离单一克隆物,转染后四十八小时,将细胞胰蛋白酶化,并转入96孔板中,板中有Iscove's DMEM培养基(补充有2mM谷氨酰胺,10%胎牛血清,1 mg/ml遗传霉素和HT Supplement(0.1 mM次黄嘌呤钠,0.016 mM胸腺嘧啶核苷))。在遗传霉素中选择之后,选择单一克隆物,并使用全细胞荧光花生四烯酸乙醇胺试验(由Ramarao等人(2005)改进),评价FAAH活性。除去组织培养基之后,取出细胞,随后加入Cellstripper(Mediatech,Inc. Manassas,VA),并转入96孔黑色透明底试验板中,在1,000rpm下离心3 min,除去介质,并用试验缓冲剂(50mM Tris pH8.0,1mM EDTA,0.1%不含脂肪酸的BSA)替换。通过加入荧光基质AMC花生四烯酰胺(Cayman Chemical,Ann Arbor,Michigan)至1μM来起始反应,并使该反应在室温下进行2小时。用CytoFluor Multiplate读数器监测荧光释放。选择表达最高数量的FAAH活性的细胞,用于研究FAAH抑制剂。
溶胞产物和微粒体的制备
表达FAAH的CHO细胞用于制备粗品细胞溶胞产物或微粒体部分。为了采集细胞,将组织培养基倾析,用不含Ca++Mg++的PBS洗涤单层三次,在不含酶的分离介质(Millipore Corp,Billerica,MA)中15分钟之后,回收细胞。通过在2000 rpm下离心15分钟来收集细胞,并用50 mM HEPES(pH7.4)(含有1mM EDTA和蛋白酶抑制剂抑肽酶(1 mg/ml)和亮肽素(100μM))将细胞球粒重新悬浮。将该悬浮液在4℃下超声处理,在4℃、在12,000xg(14,600rpm,SS34转子)下离心20分钟之后,回收细胞溶胞产物,形成粗品细胞碎片、核、过氧物酶体、溶酶体和线粒体的球粒;上清液或细胞溶胞产物用于FAAH酶试验。在某些情况下,在4℃、在27,000 rpm(100,000 x g)(SW28转子)下将细胞溶胞产物进一步离心50分钟,制备富集FAAH的微粒体部分。将含有FAAH富集的微粒体的球粒重新悬浮在50 mM HEPES、(pH7,4)1 mM EDTA中,和通过使物质经过23号针头,使任何的残留DNA被剪断,并在使用之前,将酶的等分样品保存在-80℃。
FAAH试验
一些试验已经用于证明抑制活性。在辐射酶试验中证明了酶活性,该试验基于测定花生四烯酸乙醇胺[乙醇胺1-.sup.3H](American Radiolabeled Chemicals;1mCi/ml)被FAAH(Life Sciences(1995),56,1999-2005和Journal of Pharmacology and Experimented Therapeutics(1997),283,729-734),Analytical,Biochemistry(2003),318,270-5)水解的产物(乙醇胺[3H])。另外,进行常规试验,通过7-氨基4-甲基香豆素释放时荧光增强来监测花生酰基-7-氨基-4-甲基香豆素酰胺(AAMCA)的水解(λEX=355 nm,(λEM=460 nm),Analytical,Biochemistry(2005),343,143-51。
使用荧光基质AAMCA(Cayman chemical,Ann Arbor,MI)或3H-花生四烯酸乙醇胺([乙醇胺-1-3H]American Radiolabeled Chemicals;1mCi/ml),对按照所描述方法制备的细胞溶胞产物或微粒体部分或以全细胞形式进行试验。在Costar黑色壁透明底的板中进行细胞溶胞产物或微粒体试验,向每个孔中加入FAAH_CHO(全细胞、细胞溶胞产物或微粒体)(在试验缓冲剂(50 mM磷酸盐,pH8.0,1 mM EDTA,200 mM KCl,0.2%丙三醇,0.1%不含脂肪酸的BSA)中),而后加入DMSO或化合物,并在22-25℃下培养15分钟。AAMCA基质用于获得1μM的最后浓度,并使该反应在室温下进行1-3小时。通过在CytoFluor多板读数器中读板,监测荧光释放(作为FAAH活性的量度)(Ex:360/40nM;Em:460/40nM)。用不含Ca++Mg++的PBS冲洗组织培养烧瓶三次之后,用采集的细胞进行全细胞试验,在不含酶的分离培养基中培养10分钟,并在桌面离心机中、在1,000rpm下离心5分钟。将细胞再悬浮在试验缓冲剂中,目标细胞数目为4x104个细胞/试验(96孔)、1x104个细胞/试验(384孔),并按照所描述方法进行试验。
或者,使用冷的花生四烯酸乙醇胺稀释的花生四烯酸乙醇胺[乙醇胺1-.sup.3H](比活性为10 Ci/mmol)进行试验,获得1μM花生四烯酸乙醇胺(~50,000 cpm)的最终试验浓度。在25℃下,将酶(CHO细胞溶胞产物,脑或肝脏均浆)在试验缓冲剂(50 mM磷酸盐,pH8.0,1 mM EDTA,200 mM KCl,0.2%丙三醇,0.1%脂肪酸,不含BSA)中用抑制剂培养30分钟。加入2个体积的氯仿:甲醇(1:1),并涡旋混合,从而终止该反应。在室温下进行离心步骤(2000 rpm,10分钟)之后,回收含有释放的3H-乙醇酰胺的水相,并通过液体闪烁法进行定量,作为FAAH酶活性的反映。
Ramarao M.K.,等人A fluorescence-based assay for fatty acid amide hydrolase compatible with high-throughput screening, Anal. Biochem., 343:143-51(2005)
Wilson S.J.,等人,A high-throughput-compatible assay for determining the activity of fatty acid amide hydrolase, Anal Biochem, 318:270-5(2003)。
对实施例1至29的每一个实施例进行试验并发现显示出生物活性。具体实施例的结果提供于下面。在这些试验中,发现实施例1至27中的每一个实施例具有3μM的IC50值,或更低的IC50值。
本发明化合物的制备
本发明的化合物可以如下制备:按照下面反应路线和实施例或其改型所表示的方法,使用容易得到的起始原料、试剂和合成有机化学领域普通技术人员众所周知的常规程序。反应路线中的变量的具体定义只是用于说明的目的,并不是用来限制所描述的方法。
本发明化合物的制备
本发明的化合物可以如下制备:按照下面反应路线和实施例或其改型所表示的方法,使用容易得到的起始原料、试剂和合成有机化学领域普通技术人员众所周知的常规程序。反应路线中的变量的具体定义只是用于说明的目的,并不是用来限制所描述的方法。
本发明化合物的制备
本发明的化合物可以如下制备:按照下面反应路线和实施例或其改型所表示的方法,使用容易得到的起始原料、试剂和合成有机化学领域普通技术人员众所周知的常规程序。反应路线中的变量的具体定义只是用于说明的目的,并不是用来限制所描述的方法。
一般反应路线
中间体1

2-(4-氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体2

2-(3-氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体3

2-(3,5-二氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体4

2-(3,4-二氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体5

2-(4-氯苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体6

2-(3-氯-4-氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体7

2-(4-氯-3-氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体8
2-(4-氟-2-甲基苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体9
5-[2-(4-氟苯基)-1,3-噁唑-4-基]吡嗪-2-甲酸甲酯
步骤A. 将中间体1(2.66 g,8.55 mmol)、二戊酰二硼(2.60 g,10.3 mmol)、KOAc(1.68 g,17.1 mmol)和Pd(dppf)Cl2(0.70g,0.86 mmol)的1,4-二噁烷(25 mL)溶液加热至140℃,保持30分钟。根据TLC分析判断反应完毕后,将该溶液浓缩至干,在硅胶上纯化,得到相应的硼酸中间体,将其立即使用。
步骤B. 将步骤A制备的硼酸(1.00 g,4.80 mmol)、5-氯吡嗪-2-甲酸甲酯(1.70 g,10.0 mmol)、Pd(PPh3)4(558 mg,0.48 mmol)、K2CO3(2.00g,14.5 mmol)溶于甲苯(10 mL)和水(1 mL)中,并脱气5分钟。而后,将该溶液在微波反应器中加热至120℃,保持30分钟。根据TLC分析判断反应完毕后,用蒸馏水稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(290 mg)。LC/MS: m/e 300.1(M+H)。
中间体10

2-吡嗪甲酸,5-[2-(4-氟苯基)-5-碘代-4-噁唑基]-甲基酯
将中间体9(1.40 g,4.70 mmol)、NIS(1.30 g,5.60 mmol)、TFA(0.40 mL)的CH3CN(100 mL)溶液在室温下搅拌12小时。反应完毕后,用饱和Na2S2O3水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(684 mg)。LC/MS: m/e 425.9(M+H)+. 1H NMR(500 MHz, 丙酮-d6): δ4.01(s, 3H), 7.41(t, J=8.8 Hz , 2H), 8.20-8.25(m, 2H), 9.28(s, 1H), 9.39(s, 1H)。
中间体11
2-{5-[2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇
将中间体1(60 g,0.20 mol)、二戊酰二硼(bis-pinacolatodiboron, 500g,0.25 mol)、KOAc(57.0,0.58 mol)、Pd(dppf)Cl2(7.90 g,9.60 mmol)和dppf(5.34g,9.60 mmol)的1,4-二噁烷(1.6 L)溶液加热至101℃,保持3小时。根据TLC分析判断反应完毕后,将该反应冷却至65℃。在此时,加入2-(5-溴代吡啶-2-基)丙-2-醇(62.6 g,0.30 mol)和Pd(PPh3)2Cl2(13.6 g,0.02 mol),而后加入逐滴Na2CO3水溶液(193 mL,0.40 mol,2M)。将该溶液加热至91℃,保持12小时。根据LC/MS分析判断反应完毕后,用蒸馏水稀释该溶液,并用EtOAc提取(2x)。除去合并的有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(38.50 g)。LC/MS: m/e 299.1(M+H)。
中间体12

2-{5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇
将中间体11(38.5 g,0.13 mol)和NBS(28.0 g,0.16 mol)的CH2Cl2(1340 mL)溶液在室温搅拌12小时。反应完毕后,用饱和NaS2O3水溶液稀释该溶液。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(31.97g)。LC/MS: m/e 377.0(M+H)+。
中间体13
4-[2-(4-氟苯基)-1,3-噁唑-4-基]苄腈
将中间体1(560 mg,1.80 mol)、(4-氰基苯基)硼酸(291 mg,2.00 mmol)、K2CO3(497 mg,3.60 mmol)和Pd(PPh3)4(104 mg,0.09 mmol)的1,4-二噁烷(10 mL)溶液加热至110℃,保持20分钟。根据TLC分析判断反应完毕后,将该反应浓缩至干,在硅胶上纯化,得到标题化合物(470 mg)。LC/MS: m/e 265.2(M+H)。
中间体14

4-[2-(4-氟苯基)-5-碘代-1,3-噁唑-4-基]苄腈
将中间体13(476 mg,1.80 mmol)、NIS(608 mg,2.70 mmol)、TFA(0.14 mL)的CH2Cl2(15 mL)溶液在室温搅拌12小时。反应完毕后,用饱和Na2S2O3水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(700 mg)。LC/MS: m/e 391.1(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ7.41(t, 2H), 7.94(d, 2H), 8.20(m, 2H), 8.36(d, 2H)。
中间体15
2-{5-[2-(4-氯苯基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇
利用与中间体11类似的方式制备目标化合物,只不过中间体5与2-(5-溴代吡啶-2-基)丙-2-醇偶合(XXX g)。LC/MS: m/e 315.1(M+H)。
中间体16
4-{4-[6-(1-羟基-1-甲基乙基)吡啶-3-基]-1,3-噁唑-2-基}苄腈
将中间体15(200 mg,0.60 mmol)、Pd2dba3(93 mg,0.10 mmol)、S-Phos(104 mg,0.25 mmol)和Zn(CN)2(112 mg,0.90 mmol)的10 mL 99:1 v:v DMF:H2O溶液在微波反应器中加热至180℃,保持30分钟。根据LC/MS分析判断反应完毕后,用蒸馏水稀释该溶液,并用EtOAc提取(2x)。除去合并的有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(194 mg)。LC/MS: m/e 306.1(M+H)。
中间体17

4-{5-溴-4-[6-(1-羟基-1-甲基乙基)吡啶-3-基]-1,3-噁唑-2-基}苄腈
将中间体16(476 mg,1.80 mmol)和NBS(608 mg,2.70 mmol)的CH2Cl2(15 mL)溶液在室温搅拌12小时。反应完毕后,用饱和Na2S2O3水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(43.7 mg)。LC/MS: m/e 384.0(M+H)+。
实施例1

5-[5-[(5-氯代吡啶-2-基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡嗪-2-甲酸甲酯
将溶于18 mL NMP的5-氯吡啶-2-硫醇(305 mg,2.10 mmol)溶液用NaH(84 mg,2.10 mmol)处理。将得到的溶液在室温下搅拌30分钟,而后将中间体10(684 mg,1.60 mmol)和CuI(306 mg,1.60 mmol)加入到该溶液中。将得到的黑色溶液加热至120℃,保持2小时。而后,将该溶液倒入快速搅拌的9:1 NH4Cl:NH4OH和EtOAc溶液中。一旦有机层澄清,取出有机层,而后用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(410 mg)。LC/MS: m/e 443.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ4.01(s, 3H), 7.37-7.41(m, 2H), 8.04(m, 2H), 8.70(s, 1H), 9.28(d, J=1.5 Hz, 1H), 9.44(d, J=1.0 Hz, 1H)。
使用实施例1的方法,由合适的起始原料制备表1中的化合物。
表1

实施例3

2-{5-[5-[(5-氯代吡啶-2-基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡嗪-2-基}丙-2-醇
在室温下,将5-[5-[(5-氯代吡啶-2-基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡嗪-2-甲酸甲酯(实施例1)(410 mg,0.93 mmol)的THF(20 mL)溶液用甲基溴化镁(3.1 mL,9.3 mmol,3.0 M,在THF中)处理。根据TLC分析判断反应完毕后,用饱和NH4Cl水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(90 mg)。LC/MS: m/e 442.9(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ1.58(s, 6H), 4.58(s, 1H), 7.42(m, 3H), 7,76(dd, J=2.6, 8.8 Hz, 2H), 8.32(m, 2H), 8.43(d, J=2.7 Hz, 1H), 8.96(s,1H), 9.19(s,1H)。
使用实施例3的方法,由合适的起始原料制备表2中的化合物。
表2

实施例9
2-[5-[(4-氯苯基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]-5-甲基吡嗪
在室温下,将5-[5-[(4-氯苯基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡嗪-2-甲酸甲酯(实施例1)(24 mg,0.05 mmol)的THF(5 mL)溶液用甲基溴化镁(0.2 mL,0.5 mmol,3.0 M,在THF中)处理。根据TLC分析判断反应完毕后,用饱和NH4Cl水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(6.3 mg)。LC/MS: m/e 397.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ 2.55(s, 3H), 7.46(m, 5H), 8.06(m, 2H), 8.50(s, 1H), 8.55(s, 1H), 9.10(d, J=1.1Hz, 1H) 。
使用实施例9的方法,由合适的起始原料制备表3中的化合物。
表3

实施例12
2-{5-[5-[(5-氯代吡啶-2-基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇
将溶于200 mL NMP中的5-氯吡啶-2-硫醇(27.3 g,0.20 mol)溶液用NaH(7.7 g,0.20 mol)处理。将得到的溶液在室温下搅拌30分钟,而后通过加入漏斗加入溶于200 mL NMP中的中间体12(31.9 g,0.08 mol)。最后,将CuI(16.3 g,0.08 mol)加入到该溶液中。将得到的黑色溶液加热至120℃,保持2小时。而后,将该溶液冷却至室温。一旦在室温下,将该溶液倒入快速搅拌的9:1 NH4Cl:NH4OH和EtOAc溶液中。一旦澄清,取出有机层,而后用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(31.87g)。LC/MS: m/e 442.1(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ1.76(s, 6H), 5.01(s, 1H), 7.40(m, 3H), 7.80(m, 2H), 8.25(m, 2H), 8.44(dd, J=2.3, 8.2 Hz, 1H), 8.44(d, J=2.3 Hz, 1H), 9.20(d, J=1.4 Hz, 1H)。
实施例12a

2-{5-[5-[(5-氯代吡啶-2-基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇,盐酸盐
将实施例12(138 mg,0.31 mmol)的溶液吸收在7 mL IPAC中,并加热到65℃。一旦完全溶解,逐滴加入HCl(78μl,0.31 mmol,4N,在二噁烷中)。将得到的浆液在65℃下保持2小时,而后冷却至室温。将该浆液过滤,得到白色固体(100.7 mg)。LC/MS: m/e 442.1(M+H)+ 。
使用实施例12的方法,由合适的起始原料制备表4中的化合物。
表4




实施例45

2-{6-[5-[(5-氯代吡啶-2-基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-3-基}丙-2-醇
按照实施例12所描述的方法制备标题化合物,用2-(6-溴代吡啶-3-基)丙-2-醇替代2-(5-溴代吡啶-2-基)丙-2-醇。在硅胶上纯化该油,得到标题化合物(74 mg)。LC/MS: m/e 442.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ1.59(s, 6H), 4.42(s, 1H), 7.36(m, 3H), 7.75(dd, J=2.6, 8.6 Hz, 1H), 8.06(m, 2H), 8.21(m, 2H), 8.43(d, J=2.5 Hz, 1H), 8.77(s, 1H)。
使用实施例45的方法,由合适的起始原料制备表5中的化合物。
表5
。
实施例47

4-[5-[(4-氯苯基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]苄腈
将溶于5 mL NMP中的4-氯苯硫酚(389 mg,2.70 mmol)溶液用NaH(108 mg,2.70 mmol)处理。将得到的溶液在室温下搅拌30分钟,而后将中间体14(700 mg,1.80 mmol)和CuI(342 mg,1.80 mmol)加入到该溶液中。将得到的黑色溶液加热至120℃,保持2小时。而后,将该溶液倒入快速搅拌的9:1 NH4Cl:NH4OH和EtOAc溶液中。一旦有机层澄清,取出有机层,而后用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物。LC/MS: m/e 407.8(M+H)+。
实施例48

3-{4-[5-[(4-氯苯基)硫基]-2-(4-氟苯基)-1,3-噁唑-4-基]苯基}-1,2,4-噁二唑
向在10 mL EtOH中的实施例47(100 mg,0.25 mmol)中加入1.0 mL 50 wt% NH2OH水溶液和15 mg K2CO3。通过微波辐射,将该反应加热至120℃,保持5分钟。将该反应混合物浓缩至干,并将残余物溶于5 mL原甲酸三乙酯、10 mL EtOH和1 mL TFA中。通过微波辐射,将该反应加热至100℃,保持10分钟。除去挥发物,并将残余物在硅胶上纯化,得到标题化合物(111 mg)。LC/MS: m/e 450.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ7.37-7.41(m, 6H), 8.21(m, 4H), 8.40(m, 2H), 9.41(s, 1H)。
实施例49

4-{5-[(5-氯代吡啶-2-基)硫基]-4-[6-(1-羟基-1-甲基乙基)吡啶-3-基]-1,3-噁唑-2-基}苄腈
按照实施例12所描述的方法制备标题化合物,使用中间体17(42 mg,0.10 mmol)和5-氯吡啶-2-硫醇(35.0 mg,0.24 mmol)。在硅胶上纯化该油,得到标题化合物(44.6 mg)。LC/MS: m/e 449.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ1.53(s, 6H), 4.61(s, 1H), 7.44(d, J=8.7 Hz, 1H), 7.80(m, 2H), 8.03(d, J=8.5 Hz, 2H), 8.36(d, J=8.5 Hz, 2H), 8.43(d, J=2.5 Hz, 1H), 8.45(t, J=2.5 Hz, 1H), 9.20(d, J=2.1 Hz, 1H) 。
实施例50

2-(5-{5-[(4-氯苯基)硫基]-2-吡啶-2-基-1,3-噁唑-4-基}吡啶-2-基)丙-2-醇,三氟乙酸盐
2-溴-1-(6-溴代吡啶-3-基)乙酮
步骤A. 向1-(6-溴-吡啶-3-基)-乙酮(20.3 g,101mmol)和氯化铝(200 mg,1.5mmol)的氯仿(288 mL)溶液中加入溴(5.23 mL,101 mmol)。将该混合物在室温下搅拌16小时。根据LC/MS分析判断反应完毕后,用饱和NaHCO3水溶液稀释该溶液,并用DCM提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到31 g 2-溴-1-(6-溴代吡啶-3-基)乙酮,其立即使用。LC/MS: m/e 277.9(M+H)。
2-溴-5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶
步骤B. 将步骤A的2-溴-1-(6-溴代吡啶-3-基)乙酮(2.3 g,8.25 mmol)和吡啶-2-甲酰胺(1 g,8.25 mmol)的混合物在85℃下熔融。继续加热,直到该混合物达到140℃为止,在此时,产物固化。加入冰、EtOAc和饱和NaHCO3水溶液。然后用EtOAc/THF(3:1)反萃取水层。将收集的有机物用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到250 mg(10%产率)2-溴-5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶。LC/MS: m/e 302.0(M+H)。
5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
步骤C. 将步骤B的2-溴-5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶(250 mg,0.827 mmol)、dppf(92 mg,0.166 mmol)、Pd(OAc)2(19 mg,0.0826 mmol)、TEA(0.137 mL,0.993 mmol)在MeOH(1.4 mL)和DMF(1.4 mL)中的混合物鼓入一氧化碳15分钟。然后在充满一氧化碳的球囊条件下放置该混合物,并在室温搅拌0.5小时,而后加热至75℃,保持16小时。根据LCMS分析判断反应完毕后,用蒸馏水稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,通过硅藻土垫过滤,浓缩,在硅胶上纯化,得到200 mg(86%产率)5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯。LCMS: m/e 282.1(M+H)。
2-[5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-基]丙-2-醇
步骤D. 在0℃,向步骤C的5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯(75mg,0.267mmol)的THF(1 mL)溶液中加入3M甲基溴化镁的二乙醚(0.533 mL,1.6 mmol)溶液。除去冰浴,并将该反应混合物在氮气氛围中搅拌1小时。根据LCMS分析判断反应完毕后,用饱和NH4Cl水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到2-[5-(2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-基]丙-2-醇,其立即使用。LC/MS: m/e 282.1(M+H)。

2-[5-(5-溴-2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-基]丙-2-醇
步骤E. 向步骤D的2-[5-(2-吡啶-2-基-1.3-噁唑-4-基)吡啶-2-基]丙-2-醇(75 mg,0.267 mmol)的DCM(1 mL)溶液中加入NBS(62 mg,0.347 mmol)。将该反应混合物在室温下搅拌16小时。加入水,并将该混合物用DCM提取。将该有机物干燥(MgSO4),浓缩,得到2-[5-(5-溴-2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-基]丙-2-醇,其不用进一步纯化就使用。LCMS: m/z 360.0(M+H)+。
步骤F. 向4-氯硫苯酚(38 mg,0.264 mmol)的NMP(0.5 mL)溶液中加入NaH(11 mg,0.264 mmol),并在氮气氛围中、在室温下搅拌0.5小时。向得到的钠盐中加入步骤E的2-[5-(5-溴-2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-基]丙-2-醇(38 mg,0.105 mmol)的NMP(0.5 mL)溶液,而后加入CuI(20 mg,0.105 mmol)。然后将该混合物在120℃下、在氮气氛围中加热2小时。加入饱和氯化铵水溶液(4.5 mL)和氢氧化铵(0.5 mL),并将该混合物在室温下搅拌0.5小时。将该混合物用EtOAc提取三次。将合并的有机物干燥(MgSO4),浓缩,用反相HPLC纯化,得到20 mg(35%产率,3步)标题化合物的TFA盐。LCMS: m/z 424.1(M+H)+。 1H NMR(500MHz, CO(CD3)2: δ 9.25(1H, s), 8.75(1H, m), 8.50(1H, m), 8.32(1H, d), 8.06(1H, m), 7.82(1H, m), 7.60(1H, m), 7.43(4H, br), 1.55(6H, s)。
实施例51
2-(5-{5-[(5-氯代吡啶-2-基)硫基]-2-吡啶-2-基-1,3-噁唑-4-基}吡啶-2-基)丙-2-醇,三氟乙酸盐
向5-氯吡啶-2-硫醇(38 mg,0.264 mmol)的NMP(0.5 mL)溶液中加入NaH(11 mg,0.264 mmol),并在氮气氛围中、在室温下搅拌0.5小时。向得到的钠盐中加入2-[5-(5-溴-2-吡啶-2-基-1,3-噁唑-4-基)吡啶-2-基]丙-2-醇(38 mg,0.105 mmol)的NMP(0.5 mL)溶液,而后加入CuI(20 mg,0.105 mmol)。然后将该混合物在120℃下、在氮气氛围中加热2小时。加入饱和氯化铵水溶液(4.5 mL)和氢氧化铵(0.5 mL),并将该混合物在室温下搅拌0.5小时。将该混合物用EtOAc提取三次。将合并的有机物干燥(MgSO4),浓缩,用反相HPLC纯化,得到18 mg(32%产率,3步)标题化合物的TFA盐。LCMS: m/z 425.1(M+H)+。 1H NMR(500MHz, CO(CD3)2: δ 9.22 (1H,s), 8.77(1H, s), 8.55(2H, br), 8.34(1H, m), 8.07(1H, m), 7.81(2H, br), 7.61(1H, m), 7.42(1H, d)1.54(6H, s)。
实施例52

5-[(4-氯苯基)硫基]-4-(4-氰基苯基)-2-苯基-1,3-噁唑

4-(2-苯基-1,3-噁唑-4-基)苄腈
步骤A. 将2-溴-1-(4-氰基苯基)乙酮(4 g,17.85 mmol)和苯甲酰胺(5.41g,44.6 mmol)的混合物加热至135℃,保持3小时。然后将该反应混合物冷却,并在二乙醚和水之间分配。用醚提取水层两次,并将合并的有机层用1N NaOH、1N HCl、水和盐水洗涤,用MgSO4干燥。浓缩之后,将固体残余物溶于CHCl3中。通过玻璃砂漏斗过滤未溶解的固体,并弃置。使CHCl3溶液通过二氧化硅垫过滤,蒸干,得到2.9 g(66%产率)4-(2-苯基-1,3-噁唑-4-基)苄腈。LCMS: m/z 247.1(M+H)+。

4-(5-碘代-2-苯基-1,3-噁唑-4-基)苄腈
步骤B. 将步骤A的产物(140 mg,0.57 mmol)溶于2 mL氯仿中,向其中加入NIS(282 mg,1.35 mmol)和2滴TFA。在室温下搅拌两天之后,用二氯甲烷稀释该反应,用NaHCO3水溶液、Na2S2O3水溶液、水和盐水洗涤。用MgSO4干燥有机层,过滤,浓缩,得到186 mg(88%产率)4-(5-碘代-2-苯基-1,3-噁唑-4-基)苄腈。LCMS: m/z 373.0(M+H)+。
步骤C. 将CuI(4.8 mg,0.025 mmol)、K2CO3(138 mg,1 mmol)、步骤B的产物(186 mg,0.5 mmol)和4-氯苯硫酚(72 mg,0.5 mmol)加入到烧瓶中,将烧瓶排气,并用氮气反填充(3个循环)。在室温下,通过注射器加入2-丙醇(2 mL)和乙二醇(0.056 mL,1 mmol)。将该反应混合物在80℃下加热18小时。然后用EtOAc稀释该反应,过滤,浓缩,并使残余物经过二氧化硅柱(0-20% EtOAc/己烷),得到标题化合物。1H NMR(500 MHz,(CDCl3): 8.38(d, 2H), 8.19(d, 2H), 7.78(d, 2H), 7.57(m, 3H), 7.31(d, 2H), 7.25(d, 2H)。LCMS: m/z 389.0(M+H)+。
实施例53

3-(4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯基)-1,2,4-噁二唑
向在2 mL EtOH中的3-(4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苄腈(30 mg,0.075 mmol)中加入0.25 mL 50% NH2OH水溶液和催化数量的K2CO3。通过微波辐射,将该反应在120℃加热,保持1小时。将该反应混合物浓缩至干,并将残余物溶于5 mL原甲酸三乙酯中。加入催化数量的TFA,并将该反应在130℃下加热3小时。除去挥发物,并将残余物用反相HPLC纯化,得到12mg(37%产率)标题化合物。: m/z 432.1(M+H)+。 1H NMR(500MHz, CDCl3: δ 8.8 (1H,s), 8.39(2H, d), 8.21(2H, d), 8.19(1H, m), 7.59(4H, br), 7.24(4H, br)。
实施例54

2-(4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯基)-1,3,4-噁二唑

4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯甲酸
步骤A. 将3-(4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苄腈(30 mg,0.077 mmol)的乙醇(1 mL)和2N NaOH(1 mL)溶液回流加热16小时。加入EtOAc,而后加入饱和氯化铵水溶液。干燥(MgSO4)有机物,浓缩,得到4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯甲酸,其不用进一步纯化就使用。LCMS: m/z 407.1(M+H)+。

4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯甲酸甲酯
步骤B. 将步骤A的4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯甲酸(32 mg,0.077 mmol)溶于MeOH(0.5 mL)和DCM(0.5 mL)中。在0℃慢慢地加入三甲基甲硅烷基重氮甲烷(2.0M,在醚中),直到黄色持久为止。蒸发挥发物,得到4-{5-[(4-氯苯基)硫基]-2-苯基-1,3,-噁唑-4-基}苯甲酸甲酯,其不用进一步纯化就使用。LCMS: m/z 421.1(M+H)+。

4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯甲酰肼
步骤C. 将步骤B的4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯甲酸甲酯(33 mg,0.077mmol)悬浮在1 mL EtOH和0.5 mL无水肼中,并加热至回流,保持2小时。加入EtOAc,并用水洗涤3次。干燥(MgSO4)有机物,浓缩,得到4-{5-[(4-氯苯基)硫基]-2-苯基-1,3,-噁唑-4-基}苯甲酰肼,其不用进一步纯化就使用。LCMS: m/z 421.1(M+H)+。
步骤D. 将步骤C的4-{5-[(4-氯苯基)硫基]-2-苯基-1,3,-噁唑-4-基}苯甲酰肼(33 mg,0.077mmol)溶于5 mL原甲酸三乙酯中。加入催化数量的TFA,并将该反应在130℃下加热2小时。除去挥发物,并将残余物用反相HPLC纯化,得到12 mg(36%,4步)标题化合物2-(4-{5-[(4-氯苯基)硫基]-2-苯基-1,3-噁唑-4-基}苯基)-1,3,4-噁二唑。LCMS: m/z 432.1(M+H)+。 1H NMR(500MHz, CDCl3: δ 8.55 (1H,s), 8.40(2H, d), 8.19(4H, br), 7.55(3H, br), 7.30(4H, br) 。
实施例55

5-[(4-氯苯基)硫基]-4-[4-(甲基磺酰基)苯基]-2-苯基-1,3-噁唑

4-[4-(甲基磺酰基)苯基]-2-苯基-1,3-噁唑
步骤A. 将2-溴-1-[4-(甲基磺酰基)苯基]乙酮(2 g,7.2 mmol)和苯甲酰胺(0.87 g,7.2 mmol)的混合物加热至140~180℃,保持4小时。当TLC表明该反应已经完成时,冷却该混合物,并在EtOAc和水之间分配。用EtOAc提取水层两次,并将合并的有机层用水和盐水洗涤,用MgSO4干燥。浓缩之后,用柱纯化残余物(用PE:EA=10:1洗脱),得到0.6 g(产率30%)4-[4-(甲基磺酰基)苯基]-2-苯基-1,3-噁唑。

5-溴-4-[4-(甲基磺酰基)苯基]-2-苯基-1,3-噁唑
步骤B. 在室温下,向步骤A产物(0.7 g,2.34 mmol)的乙酸(20 mL)和CHCl3(30 mL)溶液中逐滴加入Br2(0.41 g),并将该混合物搅拌2小时。将该反应混合物倒入水中,并用EtOAc提取三次。将合并的有机层用NaHCO3水溶液和盐水洗涤,用Na2SO4干燥。浓缩之后,用柱纯化残余物(PE:EA=4:1),得到0.7 g(产率80%)5-溴-4-[4-(甲基磺酰基)苯基]-2-苯基-1,3-噁唑。
步骤C. 在室温下,在氮气氛围中,向步骤B产物(0.2 g,0.53 mmol)和4-氯苯硫酚(0.076 g,0.53 mmol)的乙醇溶液中加入KOH(34 mg,0.6 mmol),然后将该混合物加热至回流过夜。冷却后,通过抽吸收集沉淀,并将滤饼用乙醇洗涤。干燥之后,获得200 mg(产率80%)标题化合物。1H-NMR(400 MHz, DMSO)δ 8.30(d, 2 H, Ar-H), 8.06(m, 4 H, Ar-H), 7.60(m, 3 H, Ar-H), 7.40(m, 4H, Ar-H), 3.26(s, 3 H, CH3)。
实施例56

2-{5-[(4-氯苯基)硫基]-4-[4-(甲基磺酰基)苯基]-1,3-噁唑-2-基}吡啶
2-{4-[4-(甲基磺酰基)苯基]-1,3-噁唑-2-基}吡啶
步骤A. 将2-溴-1-[4-(甲基磺酰基)苯基]乙酮(500 mg,1.8 mmol)和吡啶2-甲酰胺(551 mg,4.51 mmol)的混合物加热至150℃,保持1小时。然后将该反应混合物冷却,并在乙酸乙酯和水之间分配。用乙酸乙酯提取水层两次,并将合并的有机层用水和盐水洗涤,用MgSO4干燥。浓缩之后,将固体残余物溶于甲醇中,并用与质谱直接连接的HPLC纯化,得到21 mg 2-{4-[4-(甲基磺酰基)苯基]-1,3-噁唑-2-基}吡啶。LCMS: m/z 301.0(M+H)+。

2-{5-碘代-4-[4-(甲基磺酰基)苯基]-1,3-噁唑-2-基}吡啶
步骤B. 将步骤A的产物(20 mg,0.067 mmol)溶于1 mL氯仿中,向其中加入NIS(22.5 mg,0.1 mmol)和1滴TFA。在室温下搅拌2小时之后,用二氯甲烷稀释该反应,用NaHCO3水溶液、Na2S2O3水溶液、水和盐水洗涤。用MgSO4干燥有机层,过滤,浓缩,得到2-{5-碘代-4-[4-(甲基磺酰基)苯基]-1,3-噁唑-2-基}吡啶。LCMS: m/z 427.0(M+H)+。
步骤C. 将CuI(2 mg,0.01 mmol)、K2CO3(6.5 mg,0.05 mmol)、步骤B的产物(10 mg,0.023 mmol)和4-氯苯硫酚(3.4 mg,0.023 mmol)加入到烧瓶中,将烧瓶排气,并用氮气反填充(3个循环)。在室温下,通过注射器加入2-丙醇(0.5 mL)和0.01 mL乙二醇。将该反应混合物在80℃下加热18小时。然后将该反应用乙腈稀释,并通过硅藻土过滤。使滤液通过与质谱直接连接的HPLC,得到标题化合物。1H NMR(500 MHz,(CDCl3): δ8.82(宽单峰, 1H), 8.47(d, 2H), 8.23(d, 1H), 8.05(d, 2H), 7.91(t, 1H), 7.46(t, 1H), 7.23(AB quartet, 4H), 3.11(s, 3H)。 LCMS: m/z 443.0(M+H)+。
| 实施例 | 人溶胞产物IC50(nM) | 人全细胞IC50(nM) | 大鼠全细胞IC50(nM) |
| Ex 58 | 37 | 112 | 74 |
| Ex 59 | 20 | 67 | 40 |
| Ex 62 | 23 | 41 | 29 |
| Ex 65 | 27 | 29 | 21 |
| Ex 68 | 15 | 100 | 83 |
| Ex 71 | 10 | 30 | 14 |
| Ex 74 | 8 | 37 | 34 |
| Ex 78 | 28 | 69 | 39 |
| Ex 80 | 35 | 67 | 25 |
| Ex 90 | 46 | 1002 | 247 |
| Ex 96 | 17 | 133 | 63 |
| Ex 97 | 20 | NA | 10 |
| Ex 98 | 44 | 222 | 35 |
| Ex 100 | 161 | 337 | 39 |
| Ex 102 | 12 | 35 | 17 |
| Ex 107 | 24 | 91 | 11 |
| Ex 108 | 5 | 20 | 17 |
| Ex 111 | 11 | 64 | 24 |
| Ex 119 | 28 | 47 | 20 |
| Ex 122 | 161 | 474 | 146 |
| Ex 123 | 74 | 510 | 286 |
| Ex 124 | 11 | 98 | 16 |
| Ex 125 | 93 | 2291 | 680 |
| Ex 131 | 140 | 1119 | 782 |
。
中间体18
2-(2,4,5-三氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体19

2-(4-甲基苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体20

2-苯基-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体21

2-[4-(三氟甲氧基)苯基]-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体22

2-(4-甲氧基苯基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体23

2-(5-氟吡啶-3-基)-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体24

2-环丙基-1,3-噁唑-4-基三氟甲磺酸酯
使用Langille,N.F.;Dakin,L.A.;Panek,J.S. Org. Lett.,2002,4,2485所描述的方法,制备标题化合物。
中间体25

2-(4-氟苄基)-1,3-噁唑-4-基三氟甲磺酸酯
A. 向4-氟苯乙酰氯(2.0 g,12.0 mmol)的25 mLCH2Cl2搅拌溶液中加入1.7 g(12.0 mmol)氰酸银。将得到的浆液在室温下搅拌3小时。而后,通过硅藻土过滤该溶液,然后将滤液在下一步中用作原料。
将溶于DCM中的酰基异氰酸酯冷却至0℃,并用TMS重氮甲烷(6.9 mL,14.0 mmol,2.0 M溶液,在Et2O中)处理。将得到的黄色溶液升温至室温,并搅拌1小时。根据TLC分析判断反应完毕后,将该溶液浓缩至干,在硅胶上纯化,得到1.3 g噁唑啉酮中间体,将其直接三氟甲磺酸化。
在-78℃,将噁唑啉酮(1.3 g,7 mmol)用Tf2O(1.7 mL,10.0 mmol)和TEA(2.0 mL,14.0 mmol)处理。1小时之后,用饱和NaCl水溶液稀释该溶液,并升温至室温。取出有机层,用MgSO4干燥,过滤,浓缩至干,得到油。在硅胶上纯化该油,得到标题化合物(768 mg)。1H NMR(500 MHz, 丙酮-d6): δ4.21(s, 2H), 7.16(m, 2H), 7.40(m, 2H), 8.23(s, 1H)。
中间体26

4-[2-(4-氟苯基)-1,3-噁唑-4-基]苯甲酸甲酯
将中间体1(3.09 g,9.9 mmol)、4-[(甲氧羰基)苯基]硼酸(2.1 g,12.0 mmol)、Pd(dppf)Cl2、(405 mg,0.5 mmol)和CsF(3.0g,19.9 mmol)溶于二噁烷(150 mL)中,并加热到100℃,保持12小时。根据TLC分析判断反应完成后,将该溶液浓缩至干,在硅胶上纯化,得到标题化合物(2.50 g)。LC/MS: m/e 395.8(M+H) 。
中间体27

将中间体26(1.06g,3.6 mmol)和NBS(952 mg,5.4 mmol)的CH2Cl2(50 mL)溶液在室温搅拌12小时。反应完毕后,用饱和NaS2O3水溶液稀释该溶液。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(1.01g)。LC/MS: m/e 375.8(M+H)+。
中间体28

2-(4-氟苯基)-4-[4-(甲基磺酰基)苯基]-1,3-噁唑
利用与中间体26类似的方式制备目标化合物,只不过中间体1与[4-(甲基磺酰基)苯基]硼酸偶合。LC/MS: m/e 318.1(M+H) 。
中间体29

5-溴-2-(4-氟苯基)-4-[4-(甲基磺酰基)苯基]-1,3-噁唑
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 395.9(M+H)+。
中间体30
5-[2-(4-氟苯基)-1,3-噁唑-4-基]嘧啶-2-腈
利用与中间体11类似的方式制备目标化合物,只不过中间体1与5-溴嘧啶-2-腈偶合。LC/MS: m/e 267.0(M+H)+。
中间体31

5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]嘧啶-2-腈
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 345.0(M+H)+。
中间体32

5-[2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-腈
利用与中间体11类似的方式制备目标化合物,只不过中间体1与5-溴吡啶-2-腈偶合。LC/MS: m/e 266.0(M+H)。
中间体33

5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-腈
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 343.9(M+H)+。
中间体34

4-[2-(4-氟苯基)-1,3-噁唑-4-基]哌啶
将4-氟苯甲酰胺(4.54 g,32.7 mmol)和4-(溴乙酰基)-哌啶-1-甲酸叔丁基酯(5.0 g,16.3 mmol)的DMF(40 mL)溶液在145℃下加热16小时。反应完毕后,将该溶液冷却至室温,并浓缩,得到黑色油。用反相HPLC纯化该油,得到标题化合物(760 mg)。LC/MS: m/e 247.08(M+H)+。
中间体35

4-[2-(4-氟苯基)-1,3-噁唑-4-基]-1-(甲基磺酰基)哌啶
向4-[2-(4-氟苯基)-1,3-噁唑-4-基]哌啶(220 mg,0.90 mmol)的DCM(20 mL)溶液用DIEA(0.31 mL,1.8 mmol)处理,并在室温下搅拌15分钟。将甲磺酰氯(0.2 mL,2.7 mmol)慢慢地加入到该溶液中,并将得到的混合物在室温下搅拌2小时。反应完毕后,将DCM(20 mL)和水(40 mL)加入到该混合物中,并分配两个层。将有机层用MgSO4干燥,过滤,浓缩。用反相HPLC纯化残余物,得到标题化合物(100 mg)。LC/MS: m/e 325.2(M+H)+。
中间体36
2-{5-[2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}-2-甲基丙酸甲酯
利用与中间体11类似的方式制备目标化合物,只不过中间体1与2-(5-溴代吡啶-2-基)-2-甲基丙酸甲酯偶合(Kodanko, J.J.; Morys, A.J.; Lippard, S.J. Org. Lett. 2005, 7, 4585)。LC/MS: m/e 295.4(M+H)+。
中间体37

2-{5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}-2-甲基丙酸甲酯
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 373.05(M+H)+。
中间体38
5-(1-乙氧基乙烯基)吡啶-2-甲酸甲酯
向5-溴吡啶-2-甲酸甲酯(25g,116 mmol)的二噁烷(30ml)溶液中加入Pd(PPh3)4(6.7 g,5.8 mmol)和(1-乙氧基乙烯基)三丁基锡(46g,127.0 mmol)。在氮气氛围中,将得到的溶液加热至回流,并保持12小时。一旦根据LC/MS分析判断反应完毕后,用EtOAc稀释该反应,用KF溶液(10%水溶液)洗涤,通过硅藻土过滤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(20.4g)。LC/MS: m/e 208.1(M+H)+。
中间体39

5-(溴乙酰基)吡啶-2-甲酸甲酯
在室温下,向中间体38(20.3 g,98.0 mmol)的THF/水(700ml/46ml)溶液中加入一份NBS(15.0 g,98.0 mmol),并将得到的溶液在室温下搅拌30分钟。根据LC/MS分析判断反应完毕后,将该反应浓缩至干,在硅胶上纯化,得到标题化合物(19.5 g)。LC/MS: m/e 259.9(M+H)+。
中间体40

5-{[(环丙羰基)氧基]乙酰基}吡啶-2-甲酸甲酯
将环丙基甲酸(5.0 g,58.1 mmol)、中间体39(15.0 g,58.1 mmol)和K2CO3(9.63g,69.7 mmol)在DMF(50 mL)中的混合物、在室温搅拌12小时。根据LC/MS分析判断反应完毕后,用水稀释该反应,并将得到的沉淀过滤,得到标题化合物(8.54g)。LC/MS: m/e 263.9(M+H)+。
中间体41
5-(1-乙氧基乙烯基)-2-(甲基磺酰基)吡啶
利用与中间体38类似的方式制备目标化合物,只不过从5-溴-2-甲基磺酰基吡啶开始。LC/MS: m/e 228.05(M+H)+。
中间体42

2-溴-1-[6-(甲基磺酰基)吡啶-3-基]乙酮
利用与中间体39类似的方式制备标题化合物。LC/MS: m/e 279.76(M+H)+。
中间体43

环丙基甲酸2-[6-(甲基磺酰基)吡啶-3-基]-2-氧代乙基酯
利用与中间体40类似的方式制备标题化合物。LC/MS: m/e 283.9(M+H)+。
中间体44

5-(2-环丙基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
向中间体40(2.0g,7.6 mmol)的对二甲苯(130 mL)溶液中加入乙酰胺(2.24g,38.0 mmol)和BF3 ·OEt2(1.9 mL,15.2 mmol)。将得到的溶液回流加热72小时。而后,用饱和NaHCO3溶液稀释该反应,并用EtOAc提取。将有机层用盐水洗涤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(862 mg)。LC/MS: m/e 245.0(M+H)+。
中间体45
5-(5-溴-2-环丙基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
利用与中间体27类似的方式制备目标化合物,起始于中间体44。LC/MS: m/e 324.8(M+H)+。
中间体46

2-{6-[2-(4-氟苯基)-1,3-噁唑-4-基]哒嗪-3-基}丙-2-醇
利用与中间体11类似的方式制备目标化合物,只不过中间体1与2-(6-氯哒嗪-3-基)丙-2-醇偶合. LC/MS: m/e 380.0(M+H)+。
中间体47

2-{6-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]哒嗪-3-基}丙-2-醇
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 380.0(M+H)+。
中间体48
2-(4-氟苯基)-4-[(三甲基甲硅烷基)乙炔基]-1,3-噁唑
向中间体1(2.1 g,6.8 mmol)的DMF(5 mL)溶液中加入TMS乙炔(1.9 mL,13.6 mmol)、Pd(PPh3)Cl2(49 mg,0.07 mmol)、CuI(26 mg,0.14 mmol)、LiCl(433 mg,10.2 mmol)和二乙胺(9.2 mL,89 mmol)。将得到的溶液在微波反应器中、在120℃加热5分钟。而后,用饱和NH4Cl溶液稀释该反应,并用EtOAc提取。将有机层用盐水洗涤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(1.40 g)。LC/MS: m/e 262.1(M+H)+。
中间体49

4-乙炔基-2-(4-氟苯基)-1,3-噁唑
向中间体48(1.4 g,5.4 mmol)的MeOH(25 mL)溶液中加入K2CO3(746 mg,5.4 mmol)。将得到的溶液加热,搅拌12小时。而后,用水和Et2O稀释该溶液。用MgSO4干燥有机层,过滤,浓缩,得到标题化合物(1.01 g)。LC/MS: m/e 188.1(M+H)+。
中间体50
2-(4-氟苯基)-1,3-噁唑-4-腈
向中间体1(2.1 g,6.8 mmol)的DMF(15 mL)溶液中加入Pd(PPh3)4(787 mg,0.68 mmol)和Zn(CN)2(1.20 g,10.2 mmol)。将得到的溶液在微波反应器中、在120℃加热15分钟。而后,用饱和NH4Cl溶液稀释该反应,并用EtOAc提取。将有机层用盐水洗涤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(260 mg)。LC/MS: m/e 189.2(M+H)+。
中间体51

5-[2-(4-氟苯基)-1,3-噁唑-4-基]异噁唑-3-甲酸乙酯
向中间体49(1.1 g,5.9 mmol)的THF/DCM 1:1(40 mL)搅拌溶液中加入(2Z)-氯(肟基)乙酸乙酯(1.3 g,8.8 mmol)和TEA(2.4 mL,17.6 mmol)。将得到的溶液在室温下搅拌48小时。而后,将该溶液浓缩,在硅胶上纯化,得到标题化合物(469 mg)。LC/MS: m/e 303.0(M+H)+。
中间体52

5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]异噁唑-3-甲酸乙酯
利用与中间体27类似的方式制备目标化合物,起始于中间体51。LC/MS: m/e 382.9(M+H)+。
中间体53
2-[2-(4-氟苯基)-1,3-噁唑-4-基]-1,3-噻唑-4-甲酸甲酯
利用与中间体11类似的方式制备目标化合物,只不过中间体1与2-溴噻唑-4-甲酸甲酯偶合。LC/MS: m/e 04.9(M+H)+。
中间体54

2-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-1,3-噻唑-4-甲酸甲酯
利用与中间体27类似的方式制备目标化合物,起始于中间体53。LC/MS: m/e 384.9(M+H)+。
中间体55

5-[2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基磺酰基)吡啶
利用与中间体11类似的方式制备目标化合物,只不过中间体1与2-溴-5-甲基磺酰基吡啶偶合。LC/MS: m/e 318.9(M+H)+。
中间体56

5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基磺酰基)吡啶
利用与中间体27类似的方式制备目标化合物,起始于中间体55。LC/MS: m/e 398.9(M+H)+。
中间体57

5-[2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基硫基)吡啶
利用与中间体11类似的方式制备目标化合物,只不过中间体1与5-溴-2-甲硫基吡啶偶合。LC/MS: m/e 286.9(M+H)+。
中间体58

5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基硫基)吡啶
利用与中间体27类似的方式制备目标化合物,起始于中间体57。LC/MS: m/e 366.8(M+H)+。
中间体59

(R)-5-[2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基亚磺酰基)吡啶和(S)-5-[2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基亚磺酰基)吡啶
在0℃,用4小时向中间体54(1.8 g,6.3 mmol)的DCM(400 mL)溶液中逐滴加入mCPBA(1.4 g,6.3 mmol)的DCM(100ml)溶液。加入完成之后,将该溶液额外搅拌30分钟。根据LC/MS分析判断反应完毕后,用饱和NaHSO3溶液淬灭该反应,用DCM提取,用饱和Na2CO3溶液、盐水洗涤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(1.14g)。LC/MS: m/e 302.9(M+H)+。
中间体60

(R)-5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基亚磺酰基)吡啶和(S)-5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-2-(甲基亚磺酰基)吡啶
利用与中间体27类似的方式制备目标化合物,起始于中间体59。LC/MS: m/e 282.8(M+H)+。
中间体61

1H-吡唑-4-甲酰胺
将1H-吡唑-4-甲酸(2.0 g,17.8 mmol)和亚硫酰氯(20 mL,168 mmol)的混合物加热至回流。4小时之后,浓缩该反应混合物,而后减压干燥0.5小时。将得到的残余物溶于CH2Cl2(35 mL)中,冷却至0℃,并加入到氢氧化铵(46.8 mL,357 mmol)的CH2Cl2(20 mL)溶液中。将该反应混合物加热至室温,并搅拌12小时。而后,浓缩该混合物,加入CH3OH/CH2Cl2(1:10,40 ml),并搅拌10分钟。过滤该溶液,并用CH3OH/CH2Cl2(1:10)洗涤。浓缩滤液,得到标题化合物(1.5g),其不用纯化就用于下一步。LC/MS: m/e 112.0(M+H)+。
中间体62

1-乙基-1H-吡唑-4-甲酰胺
向中间体61(1.5 g,13.5 mmol)的DMF(4 mL)溶液中加入粉末K2CO3(5.6 g,40.5 mmol)。10分钟之后,加入溴乙烷(1.2 mL,16.2 mmol),并将该混合物在室温下搅拌12小时。将该反应混合物用EtOAc稀释,用水洗涤,用MgSO4干燥,浓缩,得到标题化合物白色固体(1.0 g),其不用纯化就用于下一步。LC/MS: m/e 140.1(M+H)+。
中间体63

5-[2-(1-乙基-1H-吡唑-4-基)-1,3-噁唑-4-基]吡啶-2-甲酸甲酯
在密封管中,向中间体39(650 mg,2.5 mmol)的甲苯(20 mL)溶液中加入中间体62(876 mg,6.3 mmol)。将该反应混合物加热至120℃,保持12小时。然后浓缩该反应混合物,在硅胶上纯化,得到标题化合物白色固体(100 mg)。LC/MS: m/e 299.2(M+H)+。
中间体64

5-[5-溴-2-(1-乙基-1H-吡唑-4-基)-1,3-噁唑-4-基]吡啶-2-甲酸甲酯
利用与中间体27类似的方式制备目标化合物,起始于中间体63。LC/MS: m/e 379.2(M+H)+。
中间体65

5-(2-环丙基-1,3-噁唑-4-基)-2-(甲基磺酰基)吡啶
利用与中间体44类似的方式制备目标化合物,只不过起始于中间体43。LC/MS: m/e 264.9(M+H)+。
中间体66

5-(5-溴-2-环丙基-1,3-噁唑-4-基)-2-(甲基磺酰基)吡啶
利用与中间体27类似的方式制备目标化合物,起始于中间体62。LC/MS: m/e 344.8(M+H)+。
中间体67

5-{[(环丁基羰基)氧基]乙酰基}吡啶-2-甲酸甲酯
利用与中间体40类似的方式制备目标化合物,只不过中间体39与环丁基甲酸偶合。LC/MS: m/e 278.0(M+H)+
中间体68

5-(2-环丁基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
利用与中间体44类似的方式制备目标化合物,起始于中间体64。LC/MS: m/e 259.1(M+H)+ 。
中间体69

5-(5-溴-2-环丁基-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
利用与中间体27类似的方式制备目标化合物,起始于中间体68。LC/MS: m/e 338.9(M+H)+
中间体70

5-({[(5-氯代吡啶-3-基)羰基]氧基}乙酰基)吡啶-2-甲酸甲酯
利用与中间体40类似的方式制备目标化合物,只不过中间体39与5-氯吡啶-3-甲酸偶合。LC/MS: m/e 335.0(M+H)+。
中间体71

5-[2-(5-氯代吡啶-3-基)-1,3-噁唑-4-基]吡啶-2-甲酸甲酯
利用与中间体44类似的方式制备目标化合物,起始于中间体70。LC/MS: m/e 315.9(M+H)+。
中间体72

5-[5-溴-2-(5-氯代吡啶-3-基)-1,3-噁唑-4-基]吡啶-2-甲酸甲酯
利用与中间体27类似的方式制备目标化合物,起始于中间体71。LC/MS: m/e 395.8(M+H)+。
中间体73

{5-[2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}乙腈
利用与中间体11类似的方式制备目标化合物,只不过中间体1与(5-溴代吡啶-2-基)乙腈偶合。LC/MS: m/e 280.0(M+H)+。
中间体74

{5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}乙腈
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 359.8(M+H)+。
中间体75

1-{5-[2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}-3-羟基环丁腈
在室温下,向中间体73(100mg,0.4 mmol)的DMF(8 mL)溶液中加入NaH(31.5 mg,0.8 mmol),而后加入表氯醇(39.8 mg,0.4 mmol)。将得到的溶液在室温下搅拌1小时。根据TLC分析判断反应完毕后,用水淬灭该反应,用EtOAc提取,用盐水洗涤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(16mg)。LC/MS: m/e 336.1(M+H)+。
中间体76
1-{5-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]吡啶-2-基}-3-羟基环丁腈
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 415.9(M+H)+。
中间体77

2-{5-[2-(4-氟苄基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇
利用与中间体11类似的方式制备目标化合物,只不过用中间体25代替中间体1。LC/MS: m/e 313.1(M+H)+。
中间体78

2-{5-[5-溴-2-(4-氟苄基)-1,3-噁唑-4-基]吡啶-2-基}丙-2-醇
利用与中间体27类似的方式制备目标化合物。LC/MS: m/e 393.0(M+H)+。
中间体79

2-[2-(4-氟苯基)-1,3-噁唑-4-基]嘧啶-5-甲酸甲酯
利用与中间体11类似的方式制备目标化合物,只不过中间体1与2-氯嘧啶-5-甲酸甲酯偶合。LC/MS: m/e 300.1(M+H)+。
中间体80

2-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]嘧啶-5-甲酸甲酯
利用与中间体27类似的方式制备目标化合物,起始于中间体79。LC/MS: m/e 377.9(M+H)+。
中间体81

6-[2-(4-氟苯基)-1,3-噁唑-4-基]-3,4-二氢异喹啉-1(2H)-酮
利用与中间体11类似的方式制备目标化合物,只不过中间体1与6-溴-3,4-二氢异喹啉-1(2H)-酮偶合(Bioorg. Med. Chem. Lett., 2006, 16, 2584)。LC/MS: m/e 309.3(M+H)+。
中间体82

6-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-3,4-二氢异喹啉-1-(2H)-酮
利用与中间体27类似的方式制备目标化合物,起始于中间体81。LC/MS: m/e 388.9(M+H)+。
中间体83

7-溴喹啉-3-甲醛
使用Sato,I.;Nakao,T.;Sugie,R.;Kawasaki,T.;Soai,K. Synthesis 2004,9,1419所描述的方法制备标题化合物。
中间体84

7-溴-3-(二氟甲基)喹啉
将中间体83(72 mg,0.30 mmol)溶解在CH2Cl2(1 mL)中,并加入Deoxo-Fluor(0.096 mL,0.519 mmol)的CH2Cl2(1 mL)溶液,而后加入EtOH(0.004 mL,0.069 mmol)。在室温下搅拌过夜。用CH2Cl2稀释,加入饱和NaHCO3。用CH2Cl2提取(3x),用MgSO4干燥,过滤,蒸发,在室温下高真空干燥。浅黄色油。用制备TLC纯化(SiO2,20 x 20 cm,1000微米,1个板;己烷-EtOAc,9:1),得到标题化合物(59mg)。LC/MS :[M+H]+ m/e 258, 260(M+H)+。
中间体85

3-(二氟甲基)-7-[2-(4-氟苯基)-1,3-噁唑-4-基]喹啉
利用与中间体11类似的方式制备目标化合物,只不过中间体1与7-溴-3-(二氟甲基)喹啉偶合。LC/MS: m/e 341.5。
中间体86

7-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-3-(二氟甲基)喹啉
利用与中间体27类似的方式制备目标化合物,起始于中间体85。LC/MS: m/e 421(M+H)+。
中间体87

6-溴-2-(二氟甲基)喹啉
将6-溴喹啉-2-甲醛(472 mg,2 mmol)悬浮在CH2Cl2(2 mL)中,并加入Deoxo-Fluor(0.627 mL,3.4 mmol)的CH2Cl2(2 mL)溶液,而后加入EtOH(0.023 mL,0.4 mmol)。在室温下搅拌48小时。用CH2Cl2稀释,加入饱和NaHCO3。用CH2Cl2提取(3x),用盐水洗涤提取物(1x),用MgSO4干燥,过滤,蒸发,在室温下高真空干燥。将浅棕色固体溶于少量的CH2Cl2-MeOH中,并与少量硅胶一起搅拌15分钟。过滤,蒸发,在室温下高真空干燥,得到标题化合物(491mg)。LC/MS : m/e 258, 260(M+H)+。
中间体88

2-(二氟甲基)-6-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)喹啉
在密封小瓶中,将中间体87(504 mg,1.953 mmol)、二(戊酰)二硼(506 mg,1.992 mmol)、PdCl2(dppf)(43 mg,0.059 mmol)和KOAc(575 mg,5.86 mmol)与DMSO(4.0 mL)一起混合。通过鼓入氮气进行脱气,而后用氮气包围容器,并用Teflon塞子密封。加热到80℃。加热并搅拌过夜。
16小时之后,冷却至室温。用水稀释,用EtOAc提取(3x),用盐水(1x)洗涤,用MgSO4干燥,用炭脱色,过滤,蒸发,在室温下高真空干燥,得到标题化合物(788mg)。LC/MS: m/e 306(M+H)+。
中间体89
2-(二氟甲基)-6-[2-(4-氟苯基)-1,3-噁唑-4-基]喹啉
在密封管中,将2-(4-氟苯基)-1,3-噁唑-4-基三氟甲磺酸酯(185 mg,0.593 mmol)和中间体88(263 mg,0.652 mmol)溶解在DMF(3.2 mL)中,并加入PdCl2(dppf)(13 mg,0.018 mmol),而后加入Na2CO3(314 mg,2.97 mmol)和水(0.72 mL)。将烧瓶用Teflon塞子密封,并在90℃加热。5小时之后,将该反应冷却至室温,用水稀释,并用CH2Cl2提取(3x)。用盐水洗涤提取物(1x),用MgSO4干燥,用炭脱色,通过助滤土过滤。将滤液蒸发至干,在室温下高真空干燥。用制备TLC纯化褐色固体(SiO2,20 x 20 cm,1000微米,3个板;己烷-EtOAc,3:1),得到标题化合物(109mg)。LC/MS: m/e 341(M+H)+。
中间体90

6-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-2-(二氟甲基)喹啉
利用与中间体27类似的方式制备目标化合物,起始于中间体89。LC/MS: m/e 421(M+H)+。
中间体91

6-溴-N,N-二甲基喹啉-2-甲酰胺
将6-溴喹啉-2-甲酸(1.0 g,3.93 mmol)悬浮在CH2Cl2(20 mL)中,加入DMF(0.91 mL,11.78 mmol),并在冰浴中冷却。用几分钟逐滴加入草酰氯(0.688 mL,7.86 mmol)。升温至室温,搅拌1小时,然后鼓入几分钟二甲胺气体。将暗琥珀色混合物在室温下搅拌过夜。用水稀释该溶液,用CH2Cl2(3x)提取。用盐水(1x)洗涤,用MgSO4干燥,用炭脱色,过滤,蒸发,在室温下高真空干燥,得到标题化合物(990mg)。LC/MS: m/e 279, 281(M+H)+。
中间体92
6-[2-(4-氟苯基)-1,3-噁唑-4-基]-N,N-二甲基喹啉-2-甲酰胺
利用与中间体11类似的方式制备目标化合物,只不过中间体1与中间体91偶合。LC/MS: m/e 362.4(M+H)+。
中间体 93

6-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-N,N-二甲基喹啉-2-甲酰胺
利用与中间体27类似的方式制备目标化合物,起始于中间体92。LC/MS: m/e 442.1(M+H)+。
中间体94

3-氯-6-(甲基硫基)哒嗪
将2,5-二氯哒嗪(8.7 g,58.4 mmol)溶解在DMF(30 mL)中,并用15分钟加入CH3SNa(4.1 g,58.5 mmol)的DMF(60 mL)溶液。使用冷水浴控制轻微的放热。在室温下搅拌12小时。蒸发大部分DMF(~50 mL),然后当固体沉淀时,用大量水稀释。在室温下搅拌2小时,然后过滤白色固体,并用水洗涤。将固体溶解在CH2Cl2中,分离出水,并用MgSO4干燥。过滤,蒸发,在室温下高真空干燥,得到标题化合物(5.77g)。LC/MS: m/e 161(M+H)+。
中间体95

3-[2-(4-氟苯基)-1,3-噁唑-4-基]-6-(甲基硫基)哒嗪
利用与中间体11类似的方式制备目标化合物,只不过中间体1与中间体94偶合。LC/MS: m/e 288(M+H)+。
中间体96

3-[2-(4-氟苯基)-1,3-噁唑-4-基]-6-(甲基磺酰基)哒嗪
将中间体95(135 mg,0.47 mmol)在MeOH(25.0 mL)中用过硫酸氢钾制剂(oxone,867 mg,1.41 mmol)的水(5.0 mL)溶液逐滴处理,并在室温下搅拌。然后蒸干该溶液,用CH2Cl2提取(3x)。用MgSO4干燥合并的有机提取物,过滤,蒸发,得到标题化合物(134mg)。LC/MS: m/e 320(M+H)+。
中间体97

3-[5-溴-2-(4-氟苯基)-1,3-噁唑-4-基]-6-(甲基磺酰基)哒嗪
利用与中间体27类似的方式制备目标化合物,起始于中间体96。LC/MS: m/e 399.7(M+H)+。
中间体98

(1S,2S)-2-(4-溴苯基)环丙基甲酸乙酯
向配备有磁力搅拌器的单颈1 L圆底烧瓶中加入265 mL甲基叔丁基醚。将该烧瓶排气,并用氮气吹扫三次。加入2,2'-异亚丙基二[(4R)-4-叔丁基-2-噁唑烷](2.39 g,8.03 mmol),而后加入三氟甲磺酸铜(I)-苯复合物(4.49 g,8.03 mmol)。将该绿色悬浮液在室温下搅拌大约2小时,然后过滤。将滤液加入到配备有机械搅拌器、热电偶、氮气鼓入器和加入漏斗的4颈5 L圆底烧瓶中。然后,将4-溴苯乙烯(150 g,0.803 mol)加入到此溶液中,并将该反应用冰/水浴冷却至0℃。将重氮基醋酸乙酯(167 mL,1.606 mol)溶于1675 mL MTBE中,并将该溶液排气/用氮气吹扫三次。然后将此溶液加入到加入漏斗中,并逐滴加入到该反应混合物中。观察到轻微放热。慢慢地加入重氮基醋酸乙酯,历时该周末,并将该反应慢慢地升温至室温。将该反应倒入大的提取器中,用4L MTBE稀释。将有机物用2x1L 3%氢氧化铵水溶液和2L盐水洗涤,用无水硫酸镁干燥,过滤,浓缩。将残余物溶于庚烷和少量二氯甲烷中,注射到用庚烷预装填的ISCO 1500 g柱上。该柱用1个柱体积的100%庚烷、6.5个柱体积的0-20%乙酸乙酯/庚烷洗脱,和保持在8个柱体积的20%乙酸乙酯/庚烷。收集含有产物的馏分,浓缩,得到191 g(产率88%)标题化合物。1H NMR(500 MHz,(CDCl3): δ7.42(d, 2H), 7.01(d, 2H), 4.21(q, 2H), 2.49(m, 1H), 1.88(m, 1H), 1.62(m, 2H), 1.25(t, 3H)。
使用实施例12的方法,由合适的起始原料制备表5中的化合物。
表5


实施例70

5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}吡啶-2-腈
利用与实施例12类似的方式制备标题化合物,起始于中间体33。LC/MS: m/e 409.9(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ7.39(m, 2H), 7.48(d, J=8.5 Hz, 1H), 7.83(m, 1H), 8.05(d, J=8.5 Hz, 1H), 8.24(m, 2H), 8.45(d, J=2.5 Hz, 1H), 8.72(m, 1H), 9.44(d, J=1.5 Hz, 1H) 。
实施例71

5-氯-2-({2-(4-氟苯基)-4-[6-(1,2,4-噁二唑-3-基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶
向在10 mL EtOH中的实施例70(100 mg,0.25 mmol)中加入1.0 mL 50 wt% NH2OH水溶液和15 mg K2CO3。通过微波辐射,将该反应加热至120℃,保持5分钟。将该反应混合物浓缩至干,并将残余物溶于5 mL原甲酸三乙酯、10 mL EtOH和1 mL TFA中。通过微波辐射,将该反应加热至100℃,保持10分钟。除去挥发物,并将残余物在硅胶上纯化,得到标题化合物(64 mg)。LC/MS: m/e 452.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ7.37-7.41(m, 3H), 7.82(m, 1H), 8.27(m, 2H), 8.47(d, J=2.0 Hz, 1H), 8.69(d, J=6.5 Hz, 1H), 9.47(s, 1H)。
实施例72

5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}嘧啶-2-腈
利用与实施例12类似的方式制备标题化合物,起始于中间体31。LC/MS: m/e 410.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ7.41(m, 2H), 7.53(d, J=8.5 Hz, 1H), 7.84(m, 1H), 8.26(m, 2H), 8.45(d, J=2.5 Hz, 1H), 9.61(s, 2H)。
实施例73

1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}嘧啶-2-基)乙酮
在室温下,将5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}嘧啶-2-腈(实施例72)(87 mg,0.21 mmol)的THF(5 mL)溶液用甲基溴化镁(0.7 mL,2.1 mmol,3.0 M,在THF中)处理。根据TLC分析判断反应完毕后,用饱和NH4Cl水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(13 mg)。LC/MS: m/e 427.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ 2.70(s, 3H), 7.41(m, 2H), 7.51(d, J=9.0 Hz, 1H), 7.83(m, 1H), 8.27(m, 2H), 8.45(d, J=2.5 Hz, 1H), 9.57(s, 2H)。
实施例74
2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}嘧啶-2-基)丙-2-醇
在室温下,将1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}嘧啶-2-基)乙酮(实施例73)(12 mg,0.03 mmol)的THF(5 mL)溶液用甲基溴化镁(0.09 mL,0.3 mmol,3.0M,在THF中)处理。根据TLC分析判断反应完毕后,用饱和NH4Cl水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(6.3 mg)。LC/MS: m/e 443.0(M+H)+。 1H NMR(500 MHz, 丙酮-d6): δ 1.54(s, 6H), 4.56(s, 1H), 7.39(m, 2H), 7.47(d, J=8.5Hz, 1H), 7.82(m, 1H), 8.25(m, 2H), 8.45(d, J=2.5Hz, 1H), 9.39(s, 2H)。
使用实施例12的方法,由合适的起始原料制备表6中的化合物。
表6

实施例78

5-氯-2-({2-(4-氟苯基)-4-[4-(甲基磺酰基)苯基]-1,3-噁唑-5-基}硫烷基)吡啶
将溶于60 mL NMP中的中间体29(1.30 g,3.30 mmol)、5-氯吡啶-2-硫醇(573 mg,3.90 mmol)和K2CO3(1.36 g,9.80 mmol)的搅拌溶液加热至60℃,保持1小时。而后,用蒸馏水和EtOAc稀释该溶液。取出有机层,而后用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(130 mg)。LC/MS: m/e 460.7(M+H)+。 1H NMR(500 MHz, CDCl3): δ 3.09(s, 3H), 7.05(d, J=8.5 Hz, 1H), 7.22(m, 2H), 7.56(m, 1H), 8.01(d, J=8.5 Hz, 2H), 8.19(m, 2H), 8.37(d, J=8.5 Hz, 2H), 8.41(d, J=2.5 Hz, 1H)。
实施例79

4-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}苯甲酸甲酯
将溶于20 mL NMP中的中间体27(500 mg,1.30 mmol)、5-氯吡啶-2-硫醇(290 mg,2.00 mmol)和K2CO3(551 mg,4.00 mmol)的搅拌溶液加热至80℃,保持12小时。而后,用蒸馏水和EtOAc稀释该溶液。取出有机层,而后用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(330 mg)。LC/MS: m/e 440.9(M+H)+。 1H NMR(500 MHz, CDCl3): δ 3.95(s, 3H), 7.02(d, J=8.5 Hz, 1H), 7.22(m, 2H), 7.56(m, 1H), 8.11(d, J=8.5 Hz, 2H), 8.13(m, 2H), 8.25(d, J=8.5 Hz, 2H), 8.43(d, J=2.5 Hz, 1H)。
实施例80

2-(4-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}苯基)丙-2-醇
在室温下,将4-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}苯甲酸甲酯(实施例79)(127 mg,0.29 mmol)的THF(10 mL)溶液用甲基溴化镁(0.50 mL,1.4 mmol,3.0 M,在THF中)处理。根据TLC分析判断反应完毕后,用饱和NH4Cl水溶液稀释该溶液,并用EtOAc提取。取出有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到标题化合物(100 mg)。LC/MS: m/e 441.0(M+H)+。
实施例81

4-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}-1-(甲基磺酰基)哌啶三氟乙酸盐
将中间体35(100 mg,0.925 mmol)的CH2Cl2(10 mL)溶液在室温下搅拌16小时。TLC判断反应完毕后,用CH2Cl2(20 mL)和饱和Na2S2O3水溶液(30mL)稀释该溶液。取出有机层,用MgSO4干燥,过滤,浓缩,得到相应的溴化物。该物质在下一步中直接使用。此时,向5-氯吡啶-2-硫醇(79 mg,0.564 mmol)的DME(2 mL)溶液中加入K2CO3(113 mg,0.818 mmol),并在室温下搅拌15分钟。将新制备的溴化物(110 mg,0.273 mmol)、新亚铜试剂(neocuproine,14.0 mg,0.068 mmol)和CuI(13 mg,0.068 mmol)的DME(2 mL)溶液加入到该混合物中,并加热到90℃,保持2小时。将该溶液冷却至室温,真空浓缩,并将残余物用反相HPLC纯化,得到9 mg最终化合物的TFA盐。LCMS: m/z 468.0(M+H)+。
使用实施例12的方法,由合适的起始原料制备表7中的化合物。
表7

实施例84

2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}吡啶-2-基)-2-甲基丙酸甲酯
利用与实施例12类似的方式制备标题化合物,起始于中间体37。LC/MS: m/e 484.1(M+H)+。
实施例85

3-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}吡啶-2-基)-2,3-二甲基丁-2-醇
利用与实施例80类似的方式制备标题化合物,起始于实施例84。LC/MS: m/e 484.2(M+H)+。1H NMR(500MHz, 丙酮-d6): δ 1.04(s, 6H), 1.41(s, 6H), 7.39(m, 3H),(7.36(d, J=8 Hz, 1H), 7.80(dd, J=2.5, 8.5 Hz, 1H), 8.24(m, 2H), 8.44(m, 2H), 9.23(d, J=1.5 Hz, 1H)。
实施例86

2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}吡啶-2-基)-2-甲基丙-1-醇
在-78℃,向实施例84(120mg,0.2 mmol)的THF(10 mL)溶液中加入DIBAl-H(1.0M/甲苯,0.6 mL,0.6 mmol)。将得到的溶液在-78℃下搅拌1小时。然后将该反应混合物倒入强力搅拌的罗谢尔盐溶液/EtOAc(1:1)中。一旦有机层澄清,分离各层,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(95.7mg)。
LC/MS: m/e 456.1(M+H)+。1H NMR(500MHz, 丙酮-d6): δ 1.35(s, 6H), 3.73(d, J=5.5Hz, 2H), 4.08(t, J=5.5Hz, 1H)7.40(m, 3H), 7.58(d, J=7.5Hz, 1H), 7.82(dd, J=3, 9Hz, 1H), 8.25(m, 2H), 8.39(dd, J=2.5, 8.5Hz, 1H), 8.47(d, 2.5J=2.5Hz, 1H), 9.21(s, 1H)。
实施例87
5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丙基-1,3-噁唑-4-基}吡啶-2-甲酸甲酯
在室温下,向中间体45(2.2g,6.8 mmol)的NMP(65 mL)溶液中加入5-氯吡啶-2-硫醇(1.19g,8.17 mmol)和K2CO3(2.82 g,20.4 mmol)。将得到的溶液在60℃下加热过夜。根据LC/MS分析判断该反应完毕后,用水稀释该反应,用EtOAc提取,用盐水洗涤合并的有机层,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(2.54g)。LC/MS: m/e 387.9(M+H)+。
实施例88

1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丙基-1,3-噁唑-4-基}吡啶-2-基)乙酮
在室温下,向中间体87(2.54g,6.55 mmol)的THF(260ml)溶液中加入MeMgBr(3.0M/Et2O,21.8ml,65.5mmol),并将得到的混合物在室温下搅拌2小时。根据TLC分析判断该反应完毕后,通过加入饱和NH4Cl溶液猝灭该反应,用EtOAc提取,用盐水洗涤有机层,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(188mg)。LC/MS: m/e 371.8(M+H)+。
实施例89

1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丙基-1,3-噁唑-4-基}吡啶-2-基)乙醇
在室温下,向实施例88(16 mg,0.04 mmol)的MeOH(10 mL)溶液中加入NaBH4(1.6 mg,0.04 mmol)。将得到的溶液在室温下搅拌1小时。根据TLC分析判断反应完毕后,将该反应浓缩至干,在硅胶上纯化,得到标题化合物(12mg)。LC/MS: m/e 373.9(M+H)+。
实施例90

2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丙基-1,3-噁唑-4-基}吡啶-2-基)丙-2-醇
向室温的实施例87(2.54g,6.55 mmol)的THF(260ml)溶液中加入MeMgBr(3.0M/Et2O,21.8ml,65.5mmol)。将得到的混合物在室温下搅拌2小时。根据TLC分析判断反应完毕后,通过加入饱和NH4Cl溶液来猝灭该反应,并用EtOAc提取。将有机层用盐水洗涤,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(1.77g)。LC/MS: m/e 387.9(M+H)+。1H NMR(500MHz, CDCl3): δ 1.22(m, 4H), 1.56(s, 6H), 2.19(m, 1H), 4.85(s, 1H), 6.96(d, J=8.5Hz, 1H), 7.42(d, J=8 Hz, 1H), 7.55(dd, J=2.5, 8.5Hz, 1H), 8.32(dd, J=2, 8.5Hz, 1H), 8.43(d, J=2.5 Hz, 1H), 9.14(d, J=1.5Hz, 1H)。
使用实施例12的方法,由合适的起始原料制备表8中的化合物。
表8

实施例96

2-(6-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}哒嗪-3-基)丙-2-醇
利用与实施例12类似的方式制备目标化合物,只不过用中间体47代替中间体12。LC/MS: m/e 443.2(M+H)+。1H NMR(500MHz, 丙酮-d6): δ 1.64(s, 6H), 4.70(s, 1H), 7.38(t, J=8.5 Hz, 2H), 7.43(d, J=8.5 Hz, 1H), 7.76(dd, J=2.5, 8.5Hz, 1H), 8.09(d, J=9Hz, 1H), 8.22(m, 2H), 8.27(d, J=8.5Hz, 1H), 8.43(d, J=2.5Hz, 1H)。
实施例97

5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}异噁唑-3-甲酸乙酯
利用与实施例78类似的方式制备目标化合物,只不过用中间体52代替中间体29。LC/MS: m/e 445.9(M+H)+ 。
实施例98

2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}异噁唑-3-基)丙-2-醇
利用与实施例80类似的方式制备标题化合物,起始于实施例97。LC/MS: m/e 431.9(M+H)+ 。
实施例99

2-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-Y l}-1,3-噻唑-4-甲酸甲酯
利用与实施例78类似的方式制备目标化合物,只不过用中间体54代替中间体29。LC/MS: m/e 447.9(M+H)+ 。
实施例100

2-(2-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}-1,3-噻唑-4-基)丙-2-醇
利用与实施例80类似的方式制备标题化合物,起始于实施例99。LC/MS: m/e 447.9(M+H)+。
实施例101

5-氯-2-({2-环丙基-4-[6-(甲基磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶
利用与实施例78类似的方式制备目标化合物,只不过用中间体66代替中间体29。LC/MS: m/e 407.8(M+H)+。1H NMR(500MHz, CDCl3): δ 1.22(m, 4H), 2.20(m, 1H), 3.25(s, 3H), 7.04(d, J=8.5 Hz, 1H), 7.59(dd, J=2.5, 8.5Hz, 1H), 8.12(d, J=8Hz, 1H), 8.46(d, J=2.Hz, 1H), 8.59(dd, J=2, 8.5Hz, 1H), 9.38(s, 1H)。
实施例102

5-氯-2-({2-(4-氟苯基)-4-[6-(甲基磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶
利用与实施例78类似的方式制备目标化合物,只不过用中间体56代替中间体29。LC/MS: m/e 461.8(M+H)+ NMR(500MHz, CDCl3): δ 3.28(s, 3H), 7.14(d, J=2.5 Hz, 1H), 7.24(t, J=8.5 Hz, 2H), 7.60(dd, J=2.5, 8.5Hz, 1H), 8.18(m, 3H), 8.40(d, J=2.5Hz, 1H), 8.72(dd, J=2, 8Hz, 1H), 9.49(d, J=2Hz, 1H)。
使用实施例78的方法,由合适的起始原料制备表9中的化合物。
表9
注释:实施例106是消旋的。
实施例107

5-氯-2-({2-(4-氟苯基)-4-[6-(甲基磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)嘧啶
利用与实施例78类似的方式制备目标化合物,只不过中间体56与5-氯嘧啶-2-硫醇偶合。LC/MS: m/e 461.8(M+H)+ NMR(500MHz, CDCl3): δ 3.28(s, 3H), 7.14(d, J=2.5 Hz, 1H), 7.24(t, J=8.5 Hz, 2H), 7.60(dd, J=2.5, 8.5Hz, 1H), 8.18(m, 3H), 8.40(d, J=2.5Hz, 1H), 8.72(dd, J=2, 8Hz, 1H), 9.49(d, J=2Hz, 1H)。
实施例108
(R)-5-氯-2-({2-(4-氟苯基)-4-[6-(甲基亚磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶和(S)-5-氯-2-({2-(4-氟苯基)-4-[6-(甲基亚磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶
利用与实施例78类似的方式制备目标化合物,只不过用中间体60代替中间体29。LC/MS: m/e 445.8(M+H)+ 。
实施例109

(R)-5-氟-2-({2-(4-氟苯基)-4-[6-(甲基亚磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶和(S)-5-氟-2-({2-(4-氟苯基)-4-[6-(甲基亚磺酰基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶
利用与实施例108类似的方式制备目标化合物,起始于中间体60,并用5-氟吡啶-2-硫醇替代5-氯吡啶-2-硫醇。LC/MS: m/e 445.8(M+H)+ 。
实施例110

5-(5-[(4-氯苯基)硫烷基]-2-(1-乙基-1H-吡唑-4-基)-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
利用与实施例12类似的方式制备标题化合物,起始于中间体61。LC/MS: m/e 441.2(M+H)+。
实施例111

2-(5-{5-[(4-氯苯基)硫烷基]-2-(1-乙基-1H-吡唑-4-基)-1,3-噁唑-4-基)吡啶-2-基}丙-2-醇
利用与实施例80类似的方式制备标题化合物,起始于实施例110。LC/MS: m/e 441.3(M+H)+。
实施例112

5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(1-乙基-1H-吡唑-4-基)-1,3-噁唑-4-基)吡啶-2-甲酸甲酯
利用与实施例12类似的方式制备标题化合物,起始于中间体64,并用5-氟吡啶-2-硫醇替代4-氯苯硫酚。LC/MS: m/e 441.9(M+H)+。
实施例113
2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(1-乙基-1H-吡唑-4-基)-1,3-噁唑-4-基}吡啶-2-基}丙-2-醇
利用与实施例80类似的方式制备标题化合物,起始于实施例112。LC/MS: m/e 442.1(M+H)+。
实施例114

5-氯-2-({2-(4-氟苯基)-4-[6-(甲基硫基)吡啶-3-基]-1,3-噁唑-5-基}硫烷基)吡啶
利用与实施例78类似的方式制备目标化合物,只不过用中间体58代替中间体29。LC/MS: m/e 429.8(M+H)+。NMR(500MHz, CDCl3): δ 2.62(s, 3H), 7.02(d, J=8.5 Hz, 1H), 7.22(t, J=8.5 Hz, 2H), 7.26(d, J=8.5Hz, 1H), 7.55(dd, J=2.5, 8.5Hz, 1H), 8.17(m, 2H), 8.24(dd, J=2.5, 8.5Hz, 1H), 8.42(d, J=2.5Hz, 1H), 9.20(s, 1H)。
实施例115

5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丁基-1,3-噁唑-4-基}吡啶-2-甲酸甲酯
利用与实施例87类似的方式制备目标化合物,只不过用中间体69代替中间体45。LC/MS: m/e 401.9(M+H)+。
实施例116A和实施例116B

2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丁基-1,3-噁唑-4-基}吡啶-2-基)丙-2-醇

1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丁基-1,3-噁唑-4-基}吡啶-2-基)乙酮
在室温下,向实施例115(264mg,0.6 mmol)的THF(20ml)溶液中加入MeMgBr(3.0m/Et2O,2.19ml,6.6 mmol),并将得到的混合物在室温下搅拌2小时。根据TLC分析判断该反应完毕后,通过加入饱和NH4Cl溶液猝灭该反应,用EtOAc提取,用盐水洗涤有机层,用MgSO4干燥,过滤,浓缩,在硅胶上纯化,得到标题化合物(201mg)以及副产物甲基酮。
对于116A:LC/MS: m/e 401.9(M+H)+。1H NMR(500MHz, CDCl3): δ 1.57(s, 6H), 2.12(m, 2H), 2.51(m, 4H), 3.76(m, 1H), 4.88(s, 1H), 6.97(d, J=8.5Hz, 1H), 7.43(d, J=8.5 Hz, 1H), 7.56(dd, J=3.0, 8.5Hz, 1H), 8.34(dd, J=2.5, 8.5Hz, 1H), 8.42(d, J=2.5 Hz, 1H), 9.16(d, J=1.5Hz, 1H)。
对于116B:m/e 385.9(M+H)+。1H NMR(500MHz, CDCl3): δ 2.10(m, 2H), 2.51(m, 4H), 2.74(s, 3H), 3.77(m, 1H), 4.90(s, 1H), 7.02(d, J=8.5Hz, 1H), 7.56(d, J=7.0 Hz, 1H), 8.10(d, J=8.5Hz, 1H), 8.41(s,1H), 8.50(d, J=8.0 Hz, 1H), 9.32(s, 1H)。
实施例117

(R)-1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丁基-1,3-噁唑-4-基}吡啶-2-基)乙醇和(S)-1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-环丁基-1,3-噁唑-4-基}吡啶-2-基)乙醇
利用与实施例89类似的方式制备目标化合物,只不过用实施例116B代替实施例88。LC/MS: m/e 387.9(M+H)+。1H NMR(500MHz, CDCl3): δ 1.52(d, J=6.5Hz, 3H), 2.08(m, 2H), 2.50(m,4H), 3.75(m, 1H), 4.13(br, 1H), 4.93(m, 1H), 6.95(d, J=9Hz, 1H), 7.33(d, J=8Hz, 1H), 7.54(dd, J=2.5, 8.5Hz, 1H), 8.33(dd, J=2.0, 8.0Hz, 1Hz), 8.40(d, J=2Hz, 1H), 8.18(d, J=1.5Hz, 1H)。
实施例118

5-{2-(5-氯代吡啶-3-基)-5-[(5-氯代吡啶-2-基)硫烷基]-1,3-噁唑-4-基}吡啶-2-甲酸甲酯
利用与实施例78类似的方式制备目标化合物,只不过用中间体72代替中间体29。LC/MS: m/e 458.8(M+H)+。
实施例119

2-(5-{2-(5-氯代吡啶-3-基)-5-[(5-氯代吡啶-2-基)硫烷基]-1,3-噁唑-4-基}吡啶-2-基)丙-2-醇
利用与实施例80类似的方式制备标题化合物,起始于实施例118。LC/MS: m/e 458.8(M+H)+。1H NMR(500MHz, CDCl3): δ 1.60(s, 6H), 4.81(s, 1H), 7.13(d, J=8.5Hz, 1H), 7.49(d, J=8.0 Hz, 1H), 7.60(dd, J=2.5, 8.5Hz, 1H), 8.44(m, 3H), 8.74(d, J=2.5 Hz, 1H), 9.27(dd, J=2.0, 6.5Hz, 2H)。
实施例120
1-(5-{2-(5-氯代吡啶-3-基)-5-[(5-氯代吡啶-2-基)硫烷基]-1,3-噁唑-4-基}吡啶-2-基)乙酮
利用与实施例116B类似的方式制备目标化合物,起始于实施例118。LC/MS: m/e 442.8(M+H)+。1H NMR(500MHz, CDCl3): δ 2.77(s, 3H), 7.17(d, J=8.0Hz, 1H), 7.61(d, J=7.5 Hz, 1H), 8.14(d, J=7.5Hz, 1H), 8.39(s, 1H), 8.44(s, 1H), 8.58(d, J=7.5 Hz, 1H), 8.75(s, 1H), 9.28(s, 1H), 9.45(s, 1H)。
实施例121

(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}吡啶-2-基)乙腈
利用与实施例78类似的方式制备目标化合物,只不过用中间体74代替中间体29。LC/MS: m/e 422.8(M+H)+。1H NMR(500MHz, CDCl3): δ 4.00(s, 3H), 7.08(d, J=9.0Hz, 1H), 7.23(t, J=8.5 Hz, 2H), 7.53(d, J=8.5Hz, 1H), 7.57(dd, J=2.5, 8.0Hz, 1H), 8.19(m, 2H), 8.41(d, J=2.5 Hz, 1H), 8.49(dd, J=2.0, 8.0Hz, 1H), 9.33(d, J=2.5Hz 1H)。
实施例122

1-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}吡啶-2-基)-3-羟基环丁腈
利用与实施例78类似的方式制备目标化合物,只不过用中间体76代替中间体29。LC/MS: m/e 478.9(M+H)+。1H NMR(500MHz, 丙酮-d6): δ 1.68(m, 1H), 2.01(m, 1H), 2.33(m, 1H), 3.73(m, 1H), 4.03(m, 1H), 7.40(m, 3H), 7.80(m, 2H), 8.24(m, 2H), 8.46(d, J=2.5Hz, 1H), 8.52(dd, J=2.5, 8.5Hz, 1H), 9.18(s, 1H)。
实施例123
2-(5-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苄基)-1,3-噁唑-4-基}吡啶-2-基)丙-2-醇
利用与实施例78类似的方式制备目标化合物,只不过用中间体78代替中间体29。LC/MS: m/e 456.0(M+H)+。1H NMR(500MHz, 丙酮-d6): δ 1.51(m, 1H), 4.31(s, 2H), 4.59(s, 1H), 7.15(t, J=8.5Hz, 2H), 7.23(d, J=8.5Hz, 1H), 7.47(m, 2H), 7.50(d, J=8.5Hz, 1H), 7.79(dd, J=2.5, 8.5Hz, 1H), 8.33(dd, J=2.5, 8.5Hz, 1H), 8.44(d, J=2.5Hz, 1H), 9.10(d, J=2.0Hz, 1H)。
实施例124
2-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}嘧啶-5-甲酸甲酯
利用与实施例78类似的方式制备目标化合物,只不过用中间体80代替中间体29。LC/MS: m/e 442.9(M+H)+。1H NMR(500MHz, CDCl3): δ4.02(s, 3H), 7.20(t, J=8.5Hz, 2H), 7.27(m, 1H), 7.60(dd, J=2.5, 8.5Hz, 1H), 8.19(m, 2H), 8.44(d, J=2.5Hz, 1H), 9.39(s, 2H)。
按照实施例50步骤F所描述的方法制备表10中的实施例。
表10


实施例133

6-{5-[(4-氯苯基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}-3,4-二氢异喹啉-1(2H)-酮
利用与实施例12类似的方式制备标题化合物,起始于中间体82。LC/MS: m/e 451.2(M+H)+。
实施例134

7-{5-[(4-氯苯基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}-3-(二氟甲基)喹啉
将中间体86溶解在NMP(1 mL)中的4-氯硫苯酚(23 mg,0.157 mmol)中,并加入NaH的60%油分散体(6.3 mg,0.157 mmol)。气体猛烈逸出,并且反应混合物的颜色变成暗紫色。在室温下搅拌20分钟。然后在密封小瓶中将中间体(36 mg,0.071 mmol)的NMP(1 mL)溶液、上面制备的硫醇盐溶液和CuI(13.6 mg,0.071 mmol)合并,用氮气脱气,用Teflon塞子密封,并加热到120℃。加热7小时,然后冷却至室温,并搅拌过夜。用饱和NaHCO3(9 mL)和浓NH3(1 mL)稀释,用EtOAc提取(3x)。用盐水洗涤(1x)提取物,用MgSO4干燥,过滤,蒸发,在室温下高真空干燥。用制备TLC纯化该琥珀色油(SiO2,20 x 20 cm,1000微米,3个板;己烷-EtOAc,3:1),得到标题化合物(26mg)。LC/MS : m/e 482.9(M+H)+。 1H NMR(500 MHz, CDCl3) δ 6.94(t, J=55.85 Hz, 1H), 7.24(t, J=8.55 Hz, 2H), 7.3(m, 4H), 8.01(d, J=8.5 Hz, 1H), 8.22(m, 2H), 8.36(s, 1H), 8.52(d, J=8.5 Hz, 1H), 9.06(s, 1H), 9.1(s, 1H)。
实施例135

6-{5-[(4-氯苯基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}-2-(二氟甲基)喹啉
利用与实施例12类似的方式制备标题化合物,起始于中间体90。LC/MS: m/e 483.1(M+H)+。 1H NMR(500 MHz, CDCl3) δ 6.84(t, J=55.35, 1H), 7.25(t, J=8.6 Hz, 2H), 7.3(m, 4H), 7.8(d, J=8.5 Hz, 1H), 8.22(m, 3H), 8.42(d, J=8.7 Hz, 1H), 8.69(dd, J=1.8, 8.9 Hz, 1H), 8.72(s, 1H)。
实施例136

6-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-Y l}-N,N-二甲基喹啉-2-甲酰胺
利用与实施例12类似的方式制备目标化合物,起始于中间体93。LC/MS: m/e 505.1(M+H)+。1H NMR(500 MHz, CDCl3) δ 3.21(s, 3H), 3.24(s, 3H), 7.085(d, J=8.7 Hz, 1H), 7.25(t, J=8.2 Hz, 2H), 7.56(d, J=9.1 Hz, 1H), 7.77(d, J=8.3 Hz, 1H), 8.19(d, J=8.4 Hz, 1H), 8.24(m, 2H), 8.35(d, J=8.8 Hz, 1H), 8.45(s, 1H), 8.6(d, J=9.2 Hz, 1H), 8.69(s, 1H)。
实施例137

3-{5-[(5-氯代吡啶-2-基)硫烷基]-2-(4-氟苯基)-1,3-噁唑-4-基}-6-(甲基磺酰基)哒嗪
利用与实施例12类似的方式制备目标化合物,起始于中间体97。LC/MS: m/e 462.8(M+H)+。 1H NMR(500 MHz, CDCl3) δ 3.50(s, 3H), 7.23(m, 2H), 7.33(d, J=8.7 Hz, 1H), 7.63(dd, J=2.1, 8.5 Hz, 1H), 8.15(m, 2H), 8.3(d, J=8.8 Hz, 1H), 8.42(s, 1H), 8.58(t, J=8.7 Hz, 1H)。
实施例138

(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-2-环丙基-1,3-噁唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺
步骤A. 将中间体24(478 mg,1.858 mmol)、PdCl2(dppf)-CH2Cl2加合物(68 mg,0.093 mmol)、dppf(51 mg,0.093 mmol)、KOAc(烘干)(547 mg,5.57 mmol)、二(戊酰)二硼(613 mg,2.415 mmol)的二噁烷(4.3 mL)溶液放置在氮气氛围中,并在150℃下通过微波辐射加热20分钟。向该混合物中加入中间体98(500mg,1.858 mmol)、二(三苯基膦)氯化钯(II)(130 mg,0.186 mmol)、碳酸钠(1 mL 1M水溶液)。将该混合物在150℃下通过微波辐射加热45分钟。加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩。使残余物通过二氧化硅柱(0-30% EtOAc/己烷),得到(1S,2S)-2-[4-(2-环丙基-1,3-噁唑-4-基)苯基]环丙基甲酸乙酯(239 mg,43%)。LC/MS: m/z 298.1(M+H)+。
步骤B. 将前述步骤的产物(400 mg,1.345 mmol)和NBS(311 mg,1.749 mmol)的CH2Cl2(4.5 mL)溶液在室温下搅拌3小时。反应完毕后,用饱和NaS2O3水溶液稀释该溶液。除去有机层,用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到(1S,2S)-2-[4-(5-溴-2-环丙基-1,3-噁唑-4-基)苯基]环丙基甲酸乙酯(335 mg,66%)。LC/MS: m/z 376.2(M+H)+。
步骤C. 将溶于2 mL NMP中的5-氯吡啶-2-硫醇(201 mg,1.382 mmol)溶液用NaH(55 mg,1.382 mmol)处理。将得到的溶液在室温下搅拌30分钟,而后加入前述步骤的产物(260 mg,0.691 mmol)和CuI(132 mg,0.691 mmol)。将得到的黑色溶液加热至120℃,保持16小时。而后,将该溶液倒入快速搅拌的9:1 NH4Cl:NH4OH和EtOAc溶液中。一旦有机层澄清,除去有机层,而后用MgSO4干燥,过滤,浓缩,得到油。在硅胶上纯化该油,得到(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-2-环丙基-1,3-噁唑-4-基}苯基)环丙基甲酸乙酯。LC/MS: m/z 441.1(M+H)+。
步骤D. 将前述步骤的产物(140 mg,0.318 mmol)溶于1 mL乙腈中,向其中加入1 mL水,而后加入过量KOH球粒。将该反应在80℃下搅拌3小时。将其冷却至室温之后,用浓HCl将该反应混合物的pH值调节至6。加入EtOAc,并将该混合物用水和盐水洗涤,干燥,浓缩至干,得到(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-2-环丙基-1,3-噁唑-4-基}苯基)环丙基甲酸,其不用进一步纯化就在下一步中使用。LC/MS: m/z 413.1(M+H)+。
步骤E. 将前述步骤的产物(30 mg,0.073 mmol)、HOBT(28 mg,0.182 mmol)和EDC(35 mg,0.182 mmol)溶于1 mL DMF中,向其中加入Hunig's碱(0.075 mL,0.436 mmol)和二甲胺(2M THF溶液,0.363 mL,0.727 mmol)。将该反应在75℃下加热45分钟。冷却至室温后,用EtOAc稀释该反应,并将该反应混合物用水和盐水洗涤,干燥,浓缩至干。将该标题化合物溶于热甲醇中,然后慢慢地冷却至-20℃,进行结晶。LC/MS: m/z 440.1(M+H)+。1H NMR(500 MHz, CD3OD): δ8.39(s, 1H), 7.84(d, 2H), 7.71(d, 1H), 7.20(d, 2H), 7.06(d, 1H), 3.16(s, 3H), 2.97(s, 3H), 4.22(m, 1H), 2.4-2.2(br, 2H), 1.6-1.1(br, 2H) 。
一般反应路线B
反应路线1B

在反应路线1B中,在钯介导的交叉偶合条件下,适当取代的、商购的咪唑A(其中X=Br或I)与含有R1的偶合对(coupling partner)反应,提供B。使用NIS或NCS,通过标准卤化反应,B转变为C。最后,通过铜或钯催化,在C和硫醇D之间形成硫醚,得到最终产物E。
反应路线2B

反应路线2B举例说明了实施例的合成,其中适当取代的咪唑不是商购产品。在这种情况下,在NaHCO3的存在下,将脒A和-溴酮B在THF/水中回流,得到咪唑C,用R4X将其烷基化,得到E。一旦得到取代的咪唑E,其余步骤与反应路线1所描述的那些步骤相同。
反应路线3B
在反应路线3B中,在碱(例如NaH)的存在下,用含有合适放射性核素的试剂(例如[11C]-甲基碘,[3H]-甲基碘,[18F]-氟甲基溴或[18F]-氟乙基溴)将A中的仲酰胺烷基化,分别得到叔酰胺B、C、D或E。
反应路线4B

类似地,在反应路线4B中,在碱(例如Cs2CO3或K2CO3)的存在下,用含有合适放射性核素的试剂(例如[11C]-甲基碘,[3H]-甲基碘或[18F]-氟乙基溴)将咪唑A烷基化,分别得到N-取代的咪唑B、C或D。
中间体1B
5-氯吡啶-2-硫醇
将2,5-二氯吡啶(5.0 g)和硫脲(2.57 g)悬浮在50.0 mL EtOH中,并将该混合物在95℃下加热。22小时之后,冷却该反应溶液,并慢慢地加入2.84 g KOH的5.0 mL水溶液。将该溶液在95℃下加热2小时,冷却,倒入100 mL 0.5N NaOH中,用乙酸使其呈酸性。用二氯甲烷提取产物,用水洗涤,用MgSO4干燥,过滤。浓缩有机层,得到2.3 g标题化合物。1H NMR(500 MHz,(CD3OD): 7.78(s, 1H), 7.44(d, 1H), 7.39(d, 1H), 4.39(s, 1H)。LCMS: m/z 146.0(M+H)+。
中间体2B
(1S,2S)-2-(4-溴苯基)环丙基甲酸乙酯

向配备有磁力搅拌器的单颈1 L圆底烧瓶中加入265 mL甲基叔丁基醚。将该烧瓶排气,并用氮气吹扫三次。加入2,2'-异亚丙基二[(4R)-4-叔丁基-2-噁唑烷](2.39 g,8.03 mmol),而后加入三氟甲磺酸铜(I)-苯复合物(4.49 g,8.03 mmol)。将该绿色悬浮液在室温下搅拌大约2小时,然后过滤。将滤液加入到配备有机械搅拌器、热电偶、氮气鼓入器和加入漏斗的4颈5 L圆底烧瓶中。然后,将4-溴苯乙烯(150 g,0.803 mol)加入到此溶液中,并将该反应用冰/水浴冷却至0℃。将重氮基醋酸乙酯(167 mL,1.606 mol)溶于1675 mL MTBE中,并将该溶液排气/用氮气吹扫三次。然后将此溶液加入到加入漏斗中,并逐滴加入到该反应混合物中。观察到轻微放热。慢慢地加入重氮基醋酸乙酯,历时该周末,并将该反应慢慢地升温至室温。将该反应倒入大的提取器中,用4L MTBE稀释。将有机物用2x1L 3%氢氧化铵水溶液和2L盐水洗涤,用无水硫酸镁干燥,过滤,浓缩。将残余物溶于庚烷和少量二氯甲烷中,注射到预先装入庚烷的ISCO 1500 g柱上。用1个柱体积的100%庚烷、6.5个柱体积的0-20%乙酸乙酯/庚烷洗脱该柱,并用8个柱体积的20%乙酸乙酯/庚烷保持。收集含有产物的馏分,浓缩,得到191 g(产率88%)标题化合物。1H NMR(500 MHz,(CDCl3):7.42(d, 2H), 7.01(d, 2H), 4.21(q, 2H), 2.49(m, 1H), 1.88(m, 1H), 1.62(m, 2H), 1.25(t, 3H)。
中间体3B
(1S,2S)-2-[4-(1H-咪唑-4-基)苯基]环丙基甲酸乙酯

步骤1:将3M EtMgBr/二乙醚(4.58 mL,13.75 mmol)慢慢地加入到4-碘代-1-三苯甲基-1H-咪唑(5 g,11.46 mmol)的100 mL THF溶液中,并在室温下搅拌。30分钟之后,加入ZnCl2(3.12 g,23 mmol),并将该混合物在室温下搅拌1小时。加入中间体2(3.08 g,11.46 mmol),而后加入Pd(PPh3)4(662 mg,0.573 mmol),并将该反应混合物回流加热4小时。此时,LCMS显示100%转化为产物(rt=1.19分钟)。将该反应冷却至室温,用NH4Cl水溶液(30 ml)淬灭。析出无机盐,将其过滤除去。分离水层,用水(30 mL)和盐水(30 mL)洗涤有机层。将有机层干燥(MgSO4),过滤,真空蒸发。用快速色谱纯化残余物(10-80% EA/己烷),得到4.1 g(产率71.8%)(1S,2S)-2-[4-(1-三苯甲基-1H-咪唑-4-基)苯基]环丙基甲酸乙酯。LCMS: m/z 499(M+H)+。
步骤2:将(1S,2S)-2-[4-(1-三苯甲基-1H-咪唑-4-基)苯基]环丙基甲酸乙酯(4.1 g,得自于步骤1)悬浮在30 mL甲醇和30 mL 1N HCl中。将该反应混合物回流加热2小时。真空蒸发溶剂,并将残余物与醚(100 mL)一起研磨。弃置液体有机层。固体是目标产物HCl盐。向固体中加入100 mL EtOAc和13 mL 1N NaOH,以便释放游离碱。在分离漏斗中摇动水溶液/有机物混合物。弃置水层,并将有机层用盐水洗涤,用MgSO4干燥,过滤,真空浓缩,得到标题化合物(1.4 g,66.4%)。1H NMR(500 MHz,(CD3OD): 7.98(s, 1H), 7.58(d, 2H), 7.39(s, 1H), 7.17(d, 2H), 4.18(q, 2H), 2.43(m, 1H), 1.86(m, 1H), 1.57(m, 1H), 1.37(m, 1H), 1.24(t, 3H)。 LCMS: m/z 257(M+H)+。
中间体4B
2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸

步骤1:将2-溴-1-(4-溴苯基)乙酮(8 g,28.8 mmol)的30 mL甲酰胺溶液在140℃下搅拌24小时。将该反应冷却至室温,并用EtOAc稀释。将该反应混合物用NaHCO3水溶液、水(3次)和盐水洗涤,用MgSO4干燥,浓缩,得到3.1 g粗品4-(4-溴苯基)-1H-咪唑,其不用进一步纯化就在下一步中使用。
步骤2:向步骤1产物(3.1 g,13.90 mmol)的50 mL THF溶液中加入碘甲烷(1.74 mL,27.8 mmol)和碳酸铯(5.43 g,16.68 mmol)。将反应在室温搅拌过夜。将EtOAc(150 mL)加入到该反应中,并将该混合物用水(2次)和盐水洗涤,用MgSO4干燥,并浓缩至干。用二氧化硅柱纯化残余物(10-80% EtOAc/己烷),得到2.8 g(产率85%)4-(4-溴苯基)-1-甲基-1H-咪唑。LCMS:[M+1]+=237。
步骤3:向4-(4-溴苯基)-1-甲基-1H-咪唑(步骤2产物,2.8 g,9.45 mmol)的二氯甲烷(30 mL)溶液中加入N-碘代琥珀酰亚胺(1.913 g,8.50 mmol)和六滴三氟乙酸。将该反应混合物在室温下搅拌16小时。用碳酸氢钠水溶液中和该混合物,并用二氯甲烷提取有机物。然后用硫代硫酸钠水溶液洗涤有机物,而后用水洗涤三次,然后干燥(MgSO4)。浓缩溶剂,得到4-(4-溴苯基)-5-碘代-1-甲基-1H-咪唑,其不用进一步纯化就使用。LCMS:[M+1]+=363。
步骤4:在氮气氛围中,向前述步骤产物(3.4 g,9.45 mmol)、碳酸钾(2.61 g,18.90 mmol)、碘化铜(I)(0.18 g,0.945 mmol)和中间体1(2.064 g,14.17 mmol)的31.5 mL异丙醇无水悬浮液中加入乙二醇(1.054 mL,18.90 mmol)。将该反应混合物在80℃下搅拌16小时。加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩,在100 g硅胶上纯化,用20-100%乙酸乙酯/己烷的梯度进行洗脱,得到2-{[4-(4-溴苯基)-1-甲基-1H-咪唑-5-基]硫基}-5-氯吡啶(1.9 g,5.0 mmol)。LCMS:[M+1]+=380。
步骤5:将Pd2(dba)3(0.481 g,0.525 mmol)、三叔丁基磷四氟硼酸盐(tri-tert-butylphosphonium tetrafluoroborate)(0.305 g,1.051 mmol)的DMF(15 mL)溶液在室温下搅拌10分钟。然后加入前述步骤的产物(1 g,2.63 mmol),并将得到的混合物在室温下另外搅拌10分钟,而后加入N-环己基-N-甲基环己胺(1.350 mL,6.30 mmol)、丙烯酸甲酯(2.3 mL,25.4 mmol)和DMF(50 mL)。在室温下搅拌15分钟之后,将该反应加热至120℃,保持1小时。冷却至室温后,加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩,在40 g硅胶上纯化,用50-100%乙酸乙酯/己烷的梯度进行洗脱,得到3-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)丙烯酸甲酯(0.9 g,2.3 mmol)。LCMS:[M+1]+=386。
步骤6:将氢化钠(60%,在矿物油中)、(0.233 g,5.83 mmol)和三甲基碘化亚砜(1.540 g,7.00 mmol)的DMSO(40 mL)溶液在室温下搅拌1小时。加入前述步骤的产物(0.9 g,2.3 mmol),并将得到的混合物在室温下搅拌30分钟,而后加热至50℃,保持30分钟。加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩,得到2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸甲酯(0.5 g,1.250 mmol)。LCMS:[M+1]+=400。
步骤7:向前述步骤产物(0.5 g,1.250 mmol)的乙醇(22 mL)和水(8 mL)溶液中加入过量氢氧化钾。将得到的混合物加热至回流,保持1小时,冷却,用氯化铵水溶液中和,用乙酸乙酯提取若干次,得到标题化合物粗品残余物,其不用进一步纯化就可用于下一步。或者,可以用反相HPLC纯化残余物。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.43(s, 1H), 8.00(s, 2H), 7.96(d, 2H), 7.73(d, 1H), 7.18(d, 2H), 6.95(d, 1H), 3.71(s, 3H), 2.44(m, 1H), 1.89(m, 1H), 1.50(m, 1H), 0.96(m, 1H)。LCMS:[M+1]+=385。
中间体5B
(1S,2S)-2-[4-(5-碘代-1-甲基-1H-咪唑-4-基)苯基]环丙基甲酸乙酯
步骤1:将3M EtMgBr/二乙醚(6.27 mL,18.81 mmol)慢慢地加入到4-碘代-1-甲基-1H-咪唑(3.26 g,15.67 mmol)的100 mL THF溶液中,并在室温下搅拌。30分钟之后,加入ZnCl2(4.27 g,31.3 mmol),并将该混合物在室温下搅拌1小时。加入中间体2(4.22 g,15.67 mmol),而后加入Pd(PPh3)4(906 mg,0.784 mmol),并将该反应混合物回流加热4小时。此时,LCMS显示100%转化为产物(rt=0.95分钟)。将该反应冷却至室温,用NH4Cl水溶液(30 ml)淬灭。析出无机盐,将其过滤除去。分离水层,用水(30 mL)和盐水(30 mL)洗涤有机层。将有机层干燥(MgSO4),过滤,真空蒸发。将残余物重新溶于DCM(200 mL)中,并用水(2X)和盐水洗涤有机层(除去一些含有Br的无机物种)。将DCM层干燥(MgSO4),过滤,真空蒸发。用快速色谱纯化残余物(60-90% EtOAc/己烷),得到2.9 g(产率68%)(1S,2S)-2-[4-(1-甲基-1H-咪唑-4-基)苯基]环丙基甲酸乙酯。LCMS: m/z 271(M+H)+。
步骤2:向(1S,2S)-2-[4-(1-甲基-1H-咪唑-4-基)苯基]环丙基甲酸乙酯(步骤1的产物,2.8 g,10.36 mmol)的二氯甲烷(104 mL)溶液中加入N-碘代琥珀酰亚胺(2.1 g,9.33 mmol)。将该反应混合物在室温下搅拌16小时。用二氯甲烷稀释该混合物,用硫代硫酸钠水溶液洗涤,而后用水洗涤三次,然后干燥(MgSO4)。浓缩溶剂,得到标题化合物橙色油,其不用进一步纯化就可以用于下一步。LCMS:[M+1]+=396。
中间体6B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸

按照与中间体4(步骤4和7)所描述方法相同的方法,起始于中间体5,制备标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.43(s, 1H), 8.00(s, 2H), 7.96(d, 2H), 7.73(d, 1H), 7.18(d, 2H), 6.95(d, 1H), 3.71(s, 3H), 2.44(m, 1H), 1.89(m, 1H), 1.50(m, 1H), 0.96(m, 1H)。LCMS:[M+1]+=386。
中间体7B
(1S,2S)-2-(4-{5-[(4-氯苯基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸

按照与中间体4(步骤4和7)所描述方法相同的方法,起始于4-氯硫苯酚和中间体5B,制备标题化合物。LCMS:[M+1]+=385。
中间体8B
(1R,2R)-2-[4-(5-碘代-1,2-二甲基-1H-咪唑-4-基)苯基]环丙基甲酸酯

步骤1:将2 g(7.43 mmol)(1R,2R)-2-(4-溴苯基)环丙基甲酸乙酯(以同样的方式制备的中间体2的对映体,但使用2,2'-异亚丙基二[(4S)-4-叔丁基-2-噁唑烷])、PdCl2(dppf)-CH2Cl2加合物(0.303 g,0.372 mmol)、dppf(0.206 g,0.372 mmol)、乙酸钾(烘干)(2.188 g,22.29 mmol)、二(戊酰)二硼(2.453 g,9.66 mmol)的二噁烷(17ml)溶液放置在氮气氛围中,并在150℃下通过微波辐射加热20分钟。加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩,在50 g硅胶上纯化,用0-20%乙酸乙酯/己烷的梯度进行洗脱,得到(1R,2R)-2-[4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)苯基]环丙基甲酸乙酯(2.4 g,7.59 mmol)。1H NMR(500 MHz),[(CD3)2CO]: 7.67(d, 2H), 7.20(d, 2H), 4.15(m, 1H), 2.06(m, 1H), 1.33(s, 12H), 1.24(m, 2H)。
步骤2:向前述步骤的产物乙基酯(0.5 g,1.581 mmol)、4-溴-1,2-二甲基-1H-咪唑(0.692 g,3.95 mmol)和四(0.365 g,0.316 mmol)溶液中加入碳酸钠(3.2 mL,2M水溶液)。将该混合物在150℃下通过微波辐射加热45分钟。加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩,得到(1R,2R)-2-[4-(1,2-二甲基-1H-咪唑-4-基)苯基]环丙基甲酸乙酯,其不用进一步纯化就在下一步中使用。LCMS:[M+1]+=284。
步骤3:向前述步骤的产物乙基酯(0.45 g,1.583 mmol)的二氯甲烷(5 mL)溶液中加入N-碘代琥珀酰亚胺(0.427 g,1.90 mmol)和三滴三氟乙酸。将该反应混合物在室温下搅拌1小时。用碳酸氢钠水溶液中和该混合物,并用二氯甲烷提取有机物。然后用硫代硫酸钠水溶液洗涤有机物,而后用水洗涤三次,然后干燥(MgSO4)。浓缩溶剂,得到标题化合物,其不用进一步纯化就使用。LCMS:[M+1]+=410。
中间体9B
4-(4-溴苯基)-2-环丙基-1-甲基-1H-咪唑

步骤1:在回流条件下,使用加入漏斗,向含有环丙基脒HCl盐(5.99 g,50 mmol)、NaHCO3(10 g,119 mmol)、THF(40 mL)和水(10 mL)的3颈烧瓶中加入2-溴-1-(4-溴苯基)乙酮(15.2 g,55 mmol)的30 mL THF溶液。加入完成之后,将该反应混合物回流加热过夜。蒸发除去THF,并加入EtOAc。用水和盐水洗涤该混合物。干燥有机层,浓缩,得到油。用二氧化硅柱纯化粗品,用EtOAc/DCM/己烷的1:1:1混合物洗脱,得到2.43 g(产率18%)4-(4-溴苯基)-2-环丙基-1H-咪唑。LCMS:[M+1]+=263。
步骤2:向4-(4-溴苯基)-2-环丙基-1H-咪唑(2.43 g,9.23 mmol)和碳酸铯(6.02 g,18.47 mmol)的THF(30 mL)溶液中加入碘甲烷(1.27 mL,20.31 mmol)。将该反应在室温下搅拌19小时。加入水,并将该混合物用乙酸乙酯提取。将有机物干燥(MgSO4),浓缩,得到标题化合物,其不用进一步纯化就使用。LCMS:[M+1]+=277。
中间体10B
(1S,2S)-2-{4-[1-(2-氟乙基)-5-碘代-1H-咪唑-4-基]苯基}环丙基甲酸乙酯
步骤1:向中间体3(0.5 g,1.95 mmol)的4 mL DMF溶液中加入1-氟-2-碘乙烷(0.34 g,1.95 mmol)和碳酸铯(0.7 g,2.15 mmol)。将该反应在90℃下搅拌3小时。将EtOAc(50 mL)加入到该反应中,并将该混合物用水(2次)和盐水洗涤,用MgSO4干燥,并浓缩至干。用二氧化硅柱纯化残余物(10-80% EtOAc/己烷),得到0.45 g(产率76%)(1S,2S)-2-{4-[1-(2-氟乙基)-1H-咪唑-4-基]苯基}环丙基甲酸乙酯。LCMS:[M+1]+=303。
步骤2:向(1S,2S)-2-{4-[1-(2-氟乙基)-1H-咪唑-4-基]苯基}环丙基甲酸乙酯(450 mg,1.488 mmol)的二氯甲烷(5 mL)溶液中加入N-碘代琥珀酰亚胺(352 mg,1.563 mmol)和三滴三氟乙酸。将该反应在室温下搅拌3小时。用碳酸氢钠水溶液中和该混合物,并用二氯甲烷提取有机物。然后用硫代硫酸钠水溶液洗涤有机物,而后用水洗涤三次。干燥(MgSO4)有机物,浓缩,在20 g硅胶上纯化,用35-100%乙酸乙酯/己烷的梯度进行洗脱,得到标题化合物棕色油(110 mg,0.257 mmol)。LCMS:[M+1]+=429。
中间体11B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酰肼

步骤1:起始于中间体5,按照与中间体4(步骤4)所描述方法相同的方法,制备(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸乙酯中间体。LCMS:[M+1]+=414。
步骤2:将前述步骤的产物(0.5g,1.208 mmol)悬浮在乙醇(3 mL)和水合肼(2 mL)中,并回流加热6小时。真空蒸发挥发物,得到标题化合物。LCMS:[M+1]+=400。
实施例1B
2-(4-{5-[(5-氯代吡啶-2-基-)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺
向中间体4(50 mg,0.130 mmol)、1-羟基苯并三唑水合物(24 mg,0.155 mmol)、N,N'-二异丙基碳酰亚胺(20 mg,0.155 mmol)和盐酸二甲胺(63 mg,0.777 mmol)的DMF(1 mL)溶液中加入Hunig's碱(0.226 mL,1.296 mmol)。将得到的混合物加热至80℃,保持30分钟,并对该混合物进行反相HPLC。收集含有产物的馏分,并浓缩。如果目标是三氟乙酸盐,则可以通过冷冻干燥器除去溶剂。如果目标是游离碱,则用乙酸乙酯稀释残余物,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.43(s, 1H), 7.99(s, 1H), 7.92(d, 2H), 7.71(d, 1H), 7.15(d, 2H), 6.93(d, 1H), 3.71(s, 6H), 3.15(s, 3H), 2.32(m, 1H), 2.21(m, 1H), 1.46(m, 1H), 1.21(m, 1H)。LCMS:[M+1]+=413。 人FAAH溶胞产物试验:IC50=1.4 nM。
按照实施例1所描述的方法,使用合适的胺和中间体4作为起始原料,制备表1中的实施例。
表1
*LCMS 5 min方法。
实施例5B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

向中间体6(100 mg,0.259 mmol)、1-羟基苯并三唑水合物(99 mg,0.648 mmol)、N-[3-(二甲基氨基)丙基]-N'-乙基碳酰亚胺盐酸盐(124 mg,0.648 mmol)和二甲胺(2M,在THF中)(3 mL,1.500 mmol)的二噁烷(1 mL)溶液中加入Hunig's碱(0.272 mL,1.555 mmol)。将得到的混合物加热至80℃,保持30分钟,并对该混合物进行反相HPLC。收集含有产物的馏分,并浓缩。如果目标是三氟乙酸盐,则可以通过冷冻干燥器除去溶剂。如果目标是游离碱,则用乙酸乙酯稀释残余物,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.43(s, 1H), 7.99(s, 1H), 7.92(d, 2H), 7.71(d, 1H), 7.15(d, 2H), 6.93(d, 1H), 3.71(s, 6H), 3.15(s, 3H), 2.32(m, 1H), 2.21(m, 1H), 1.46(m, 1H), 1.21(m, 1H)。LCMS:[M+1]+=413。人FAAH溶胞产物试验:IC50=1.0 nM。
按照实施例5所描述的方法,使用合适的胺和中间体6作为起始原料,制备表2B中的实施例。
表2

实施例9B
(1S,2S)-2-(4-{5-[(4-氯苯基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酰胺
按照与实施例5所描述方法相同的方法,起始于中间体7B,制备标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 7.97(br, 3H), 7.32(d, 2H), 7.16(d, 2H), 7.05(d, 2H), 3.66(s, 3H), 3.14(s, 3H), 3.02(s, 3H), 2.34(m, 1H), 2.21(m, 1H), 1.47(m 1H), 1.21(m, 1H)。LCMS:[M+1]+=412。人FAAH溶胞产物试验:IC50=0.3 nM。
实施例10B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N-(2-氟乙基)环丙基甲酰胺

按照与实施例5所描述方法相同的合成方法,起始于2-氟乙胺盐酸盐和中间体6B,而后通过用甲醇重结晶来进行纯化,制备标题化合物。1H NMR(500 MHz),[CDCl3]: 8.30(s, 1H), 7.82(br, 3H), 7.60(d, 1H), 7.19(d, 2H), 6.80(d, 1H), 4.60(m, 1H), 4.50(m, 1H), 3.90(s, 3H), 3.60(br, 2H), 2.45(m, 1H), 1.85(m, 1H), 1.65(m, 1H), 1.20(m, 1H)。LCMS:[M+1]+=430。人FAAH溶胞产物试验:IC50=1.5 nM。
实施例11B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N-(2-氟乙基)-N-甲基环丙基甲酰胺

向实施例10(10 mg,0.023 mmol)的DMF(1 mL)溶液中加入氢化钠(60%,在矿物油中)(6 mg,0.139 mmol)和碘甲烷(0.009 mL,0.139 mmol)。将该反应混合物在室温下搅拌30分钟。加入水,并将该混合物用乙酸乙酯提取。干燥(MgSO4)有机物,浓缩,在4 g硅胶上纯化,用0-5%三乙胺/乙酸乙酯的梯度进行洗脱,得到标题化合物。1H NMR(500 MHz),[CD3OD]: 8.34(s, 1H), 8.04(s, 1H), 7.73(m, 2H), 7.63(d, 1H), 7.14(m, 2H), 6.92(d, 1H), 4.58(m, 1H), 4.48(m, 1H), 3.66(s, 3H), 3.19(s, 3H), 3.00(br, 2H), 2.37(m, 1H), 2.18(m, 1H), 1.53(m, 1H), 1.31(m, 1H)。LCMS:[M+1]+=445。人FAAH溶胞产物试验:IC50=3.0 nM。
实施例12B
(1R,2R)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1,2-二甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

步骤1:按照与中间体4(步骤4和7)所描述方法相同的方法,起始于中间体8,制备(1R,2R)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1,2-二甲基-1H-咪唑-4-基}苯基)环丙基甲酸。
步骤2:按照与实施例5所描述方法相同的方法,起始于前述步骤的产物,制备标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.45(s, 1H)7.84(d, 2H), 7.82(d, 1H), 7.31(d, 1H), 7.25(d, 2H), 3.79(s, 3H), 3.15(s, 3H), 2.91(s, 3H), 2.78(s, 3H), 2.36(m, 1H), 2.28(m, 1H), 1.48(m, 1H), 1.25(m, 1H)。LCMS:[M+1]+=427。人FAAH溶胞产物试验:IC50=13.6 nM。
实施例13B
5-[(5-氯代吡啶-2-基)硫基]-2-环丙基-4-(4-{2-[(二甲基氨基)羰基]环丙基}苯基)-1-甲基-1H-咪唑-3-鎓三氟乙酸盐

按照与实施例1所描述方法相同的方法,起始于中间体9,制备标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.45(s, 1H), 7.78(br, 3H), 7.20(br, 3H), 3.87(s, 3H), 2.33(m, 1H), 2.30(m, 1H), 2.24(m, 1H)1.47(m, 1H), 1.32(m, 2H), 1.23-1.18(br, 3H)。LCMS:[M+1]+=453。人FAAH溶胞产物试验:IC50=48.3 nM。
实施例14B
5-[(5-氯代吡啶-2-基)硫基]-4-(4-{(1S,2S)-2-[(二甲基氨基)羰基]环丙基}苯基)-1-(2-氟乙基)-1H-咪唑-3-鎓三氟乙酸盐

步骤1:按照与中间体4(步骤4和7)所描述方法相同的方法,起始于中间体10B,制备(1S,2S)-2-{4-[5-[(5-氯代吡啶-2-基)硫基]-1-(2-氟乙基)-1H-咪唑-4-基]苯基}环丙基甲酸。
步骤2:按照与实施例5所描述方法相同的方法,起始于前述步骤的产物,制备标题化合物。1H NMR(500 MHz),[CD3COD]: 8.37(s, 1H), 7.76(d, 1H), 7.67(br, 2H), 7.27(br, 4H), 4.77(m, 1H), 4.68(m, 1H), 4.60(m, 1H), 4.55(m, 1H), 3.16(s, 3H), 2.96(s, 3H), 2.40(m, 1H), 2.24(m, 1H), 1.54(m, 1H), 1.34(m, 1H)。LCMS:[M+1]+=445。人FAAH溶胞产物试验:IC50=3.2 nM。
按照实施例5所描述的方法,使用合适的胺和(1S,2S)-2-{4-[5-[(5-氯代吡啶-2-基)硫基]-1-(2-氟乙基)-1H-咪唑-4-基]苯基}环丙基甲酸(实施例14,步骤1)作为起始原料,制备表3B中的实施例。
表3B

*LCMS 5 min方法。
实施例17B
5-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]-1,3,4-噁二唑-2(3H)-酮

将中间体11B(275 mg,0.688 mmol)溶于THF(0.5 mL)中,在-78℃向其中加入光气(PhMe溶液,1.375 mmol)。在-78℃下搅拌30-60分钟之后,用NaHCO3水溶液淬灭该反应,并用EtOAc提取产物。用水、盐水洗涤有机层,用MgSO4干燥,过滤,浓缩。用反相HPLC纯化残余物。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2SO]: 8.47(s, 1H), 8.09(s, 1H), 7.79(br, 3H), 7.18(d, 2H), 6.93(d, 1H), 2.44(m, 1H), 2.17(br, 4H), 1.52(m, 1H), 1.45(m, 1H)。LCMS:[M+1]+=426。人FAAH溶胞产物试验:IC50=4.5 nM。
实施例18B
5-氯-2-[(4-{4-[(1S,2S)-2-(5-甲氧基-1,3,4-噁二唑-2-基)环丙基]苯基}-1-甲基-1H-咪唑-5-基)硫基]吡啶

向中间体11B(45 mg,0.113 mmol)的四甲氧基甲烷(2 mL)溶液中加入两滴三氟乙酸。将该混合物加热至回流,保持30分钟。蒸发挥发物,并将残余物用反相HPLC纯化。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2SO]: 8.88(s, 1H), 8.47(s, 1H), 7.83(d, 2H), 7.29(br, 2H), 7.15(br, 2H), 3.15(s, 3H), 2.57-2.48(br, 1H), 2.42(s, 3H), 1.63(br, 2H), 1.27(m, 1H)。LCMS:[M+1]+=440。人FAAH溶胞产物试验:IC50=15.6 nM。
实施例19B
5-氯-2-[(1-甲基-4-{4-[(1S,2S)-2-(5-甲基-1,3,4-噁二唑-2-基)环丙基]苯基}-1H-咪唑-5-基)硫基]吡啶

按照实施例18所描述的方法,起始于中间体11B和原乙酸三甲酯,制备标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.43(s, 1H), 8.01(s 1H), 7.97(d, 2H), 7.71(s, 1H), 7.23(d, 2H), 6.95(d, 1H), 3.69(s, 3H), 3.61(m, 1H), 2.45(s, 3H), 1.69(m, 1H), 1.64(m, 1H), 0.89(m, 1H)。LCMS:[M+1]+=424。人FAAH溶胞产物试验:IC50=46 nM。
实施例20B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

步骤1:将中间体3B(860 mg,3.36 mmol)溶于二氯甲烷(15 mL)中,向其中加入NIS(679 mg,3.02 mmol)。将该反应在室温下搅拌30分钟,然后用二氯甲烷(60 mL)稀释,用NaHCO3水溶液(30 mL)淬灭。分离各层之后,用Na2S2O3水溶液、水(x2)和盐水洗涤有机层,用MgSO4干燥,过滤,真空浓缩。粗品不用进一步纯化就用于下一步。LCMS: min[M+1]=383。
步骤2:向微波管中加入CuI(2 mg,0.01 mmol)、1,10-菲咯啉(2 mg,0.11 mmol)、K2CO3(14 mg,0.11 mmol)、上述步骤1产物(20 mg,0.05 mmol)、中间体1(9 mg,0.06 mmol),排气,并用氮气反填充(三个循环)。将管密封,并在氮气氛围中加入DMSO(1 mL)。将密封管放进预热至100℃的油浴中,并将该反应混合物在此温度下搅拌4小时。将其冷却至室温之后,将该反应混合物在10 mL NaCl水溶液和20 mL EtOAc之间分配。分离有机层,并用10 mL EtOAc提取水层。将合并的有机层用水、盐水洗涤,干燥,浓缩。将残余物用二氧化硅柱纯化,用70-100% EtOAc/己烷洗脱,得到5 mg(26%产率)(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1H-咪唑-4-基}苯基)环丙基甲酸乙酯。LCMS:[M+1]=400。
步骤3:将(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1H-咪唑-4-基}苯基)环丙基甲酸乙酯(86 mg,0.215 mmol)溶于6 mL乙腈中,向其中加入2 mL水,而后加入过量KOH球粒。将该反应在80℃下搅拌30分钟。将其冷却至室温之后,用浓HCl将该反应混合物的pH值调节至6。加入EtOAc(50 mL),并将该混合物用水和盐水洗涤,干燥,浓缩至干,得到相应的酸,其不用进一步纯化就用于下一步。LCMS: m/z 372.0(M+H)+。
步骤4:将(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1H-咪唑-4-基}苯基)环丙基甲酸(步骤3产物,73 mg,0.196 mmol)、HOBT(60 mg,0.393 mmol)和EDC(113 mg,0.589 mmol)溶于3 mL DMF中,向其中加入Hunig's碱(0.206 mL,1.178 mmol)和二甲胺(2M THF溶液,0.294 mL,0.589 mmol)。将该反应在80℃下加热30-60分钟。冷却至室温,用EtOAc(80 mL)稀释该反应,并将该反应混合物用水(2-3次)和盐水洗涤,干燥,浓缩至干。将残余物用二氧化硅柱纯化(80-100% EtOAc/己烷),得到62 mg(79%)标题化合物。1H NMR(500 MHz,(CD3OD):8.38(s, 1H), 7.97(s, 1H), 7.65(d, 2H), 7.59(d, 1H), 7.18(d, 2H), 6.82(d, 1H), 3.16(s, 3H), 2.97(s, 3H), 2.38(m, 1H), 2.19(m, 1H), 1.54(m, 1H), 1.27(m, 1H)。 LCMS: m/z 399(M+H)+。 人FAAH溶胞产物试验:IC50=649.6 nM。
实施例21B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸甲酯

将中间体6B(30 mg,0.078 mmol)溶于1 mL二氯甲烷和1 mL MeOH中,在0℃向其中逐滴加入三甲基甲硅烷基重氮甲烷(2M醚溶液),直到橙-黄色持久为止。浓缩该反应混合物,并将残余物用二氧化硅柱纯化(20-80% EtOAc/己烷),得到标题化合物。LCMS: m/z 400(M+H)+。 人FAAH溶胞产物试验:IC50=0.2 nM。
实施例22B
2-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]丙-2-醇

将实施例21B的产物(15 mg,0.038 mmol)溶于1 mL THF中,在0℃,向其中加入MeMgBr(3M醚溶液,0.075 mL,0.225 mmol)。在0℃下搅拌10分钟之后,用NH4Cl水溶液淬灭该反应。加入EtOAc(20 mL),并将该混合物用水和盐水洗涤,干燥,浓缩至干。用二氧化硅柱纯化残余物(70-100% EtOAc/己烷),得到标题化合物。1H NMR(500 MHz,(CDCl3): 8.41(s, 1H), 8.39(s, 1H), 7.86(d, 2H), 7.51(d, 1H), 7.13(d, 2H), 6.86(d, 1H), 3.78(s, 3H), 1.95(m, 1H), 1.31(s, 6H), 1.29(m, 1H), 1.05(m, 1H), 0.86(m, 1H)。 LCMS: m/z 400(M+H)+。人FAAH溶胞产物试验:IC50=8.7 nM。
实施例23B
[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]甲醇

步骤1:向烧瓶中加入中间体5B(4.02 g,10.15 mmol)、中间体1(1.77 g,12.17 mmol)、CuI(97 mg,0.51 mmol)和K2CO3(2.8 g,20.29 mmol),排气,用氮气反填充(三个循环)。在氮气氛围中,加入DME(50 mL),并将该反应在80℃加热过夜。将其冷却至室温之后,用150 mL EtOAc稀释该反应混合物。用水、盐水洗涤该反应混合物,干燥,浓缩。用二氧化硅柱纯化残余物,用40-80% EtOAc/己烷洗脱,得到3.75 g(89%产率)(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基甲酸乙酯。LCMS: m/z 414(M+H)+。
步骤2:将步骤1的产物(150 mg,0.362 mmol)溶于THF(2 mL)中,在0℃,向其中加入100 mg LAH(2.63 mmol)。将该反应在0℃搅拌10分钟,用Fischer后处理方法猝灭:仔细顺序地逐滴加入0.1 mL水、0.1 mL 15% NaOH溶液和0.3 mL水,提供容易冲洗个过滤的颗粒状无机物沉淀。将粒状沉淀过滤,并用EtOAc洗涤。将合并的有机溶液浓缩,得到粗品,将其用柱纯化(95-100% EtOAc/己烷),得到标题化合物。1H NMR(500 MHz,(CDCl3): 8.39(s, 1H), 8.05(s, 1H), 7.86(d, 2H), 7.48(d, 1H), 7.06(d, 2H), 6.81(d, 1H), 3.67(s, 3H), 3.61(d, 2H), 3.02(broad s, 1H), 1.83(m, 1H), 1.45(m, 1H), 0.97(m, 2H)。 LCMS: m/z 372(M+H)+。人FAAH溶胞产物试验:IC50=88.4 nM。
实施例24B
5-氯-2-[(4-{4-[(1S,2S)-2-(甲氧基甲基)环丙基]苯基}-1-甲基-1H-咪唑-5-基)硫基]吡啶
将[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]甲醇(实施例23的产物,20 mg,0.054 mmol)溶于DMF(1 mL)中,在0℃,向其中加入NaH(0.25 mmol)和MeI(0.25 mmol)。将该反应加热至室温,并搅拌30分钟。用1 mL NH4Cl水溶液淬灭该反应,并用5 mL EtOAc稀释。用水(两次)和盐水洗涤该混合物。将有机层干燥并浓缩,得到粗品,将其用柱纯化(60-100% EtOAc/己烷),得到标题化合物。1H NMR(500 MHz,(CDCl3): 9.82(s, 1H), 8.37(s, 1H), 7.88(d, 2H), 7.58(d, 1H), 7.15(d, 2H), 7.07(d, 1H), 3.97(s, 3H), 3.42(d, 2H), 3.39(s, 3H), 1.82(m, 1H), 1.44(m, 1H), 0.99(m, 2H)。 LCMS: m/z 386(M+H)+。 人FAAH溶胞产物试验:IC50=35.5 nM。
实施例25B
5-氯-2-{[4-(4-{(1S,2S)-2-[(2-氟乙氧基)甲基]环丙基}苯基)-1-甲基-1H-咪唑-5-基]硫基}吡啶

将[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]甲醇(实施例23的产物,20 mg,0.054 mmol)溶于DMF(1 mL)中,在0℃,向其中加入NaH(0.25 mmol)和1-氟-2-碘乙烷(0.25 mmol)。将该反应加热至室温,而后在55℃下加热2小时。将其冷却至室温之后,用1 mL NH4Cl水溶液淬灭该反应,并用5 mL EtOAc稀释。用水(两次)和盐水洗涤该混合物。将有机层干燥并浓缩,得到粗品,将其用柱纯化(40-100% EtOAc/己烷),得到标题化合物。1H NMR(500 MHz,(CDCl3): 9.83(s, 1H), 8.38(s, 1H), 7.88(d, 2H), 7.63(d, 1H), 7.19(d, 1H), 7.18(d, 2H), 4.63(m, 1H), 4.58(m, 1H), 4.02(s, 3H), 3.76(m, 1H), 4.71(m, 1H), 3.57(d, 2H), 1.85(m, 1H), 1.46(m, 1H), 1.03(m, 2H)。 LCMS: m/z 418(M+H)+。 人FAAH溶胞产物试验:IC50=69.3 nM。
实施例26B
[
3
H](1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

将(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N-甲基环丙基甲酰胺(实施例6的产物,1 mg,0.0025 mmol)溶于DMF(200 ul,无水)中,并在氮气氛围中、在冰/水浴中冷却。将NaH(1M,100 ug)与50uL THF混合,并加入到该溶液中,将冷却浴换为干冰/甲醇。将该反应混合物强力搅拌20分钟,而后用注射器加入[3H]MeI/甲苯(100 mCi,80 Ci/mmol,50 uL)。用2 x 50 uL甲苯冲洗注射器,并将所有的冲洗液加入到该反应混合物中。在干冰/甲醇浴中,将该反应溶液保持搅拌1小时,而后在室温下搅拌过夜。HPLC显示了甲基化产物。用旋转蒸发器充分地干燥该反应溶液,并将残余物溶于80% ACN/水(1%TFA)中。HPLC和LC-MS显示了30%产物以及其它副产物和起始原料。用半制备HPLC纯化该混合物:Phenomenex Luna苯基-己基,4 mL/min,254 nm,70% Aq(0.1% TFA):30% ACN,等度洗脱(isocratic),得到目标产物3H-L-002311600,Tr=~12.9 min。sep-pak提取之后,将示踪物保存在脱气乙醇中(0289561-0003)。收集3.66mCi/11.5ml EtOH。SA=66.76 Ci/mmol。HPLC分析:a. Phenomenex Polar-RP 80A,1.0 mL/min,254 nm,25-45% ACN/水(0.1% TFA),20 min,Tr=16.9 min。b. Phenomenex Luna苯基-己基,1.0 mL/min,254nm,30% ACN/水(0.1%TFA),20 min,Tr=11.5 min。
实施例27B
[
3
H](1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

向具有搅拌棒的2 mL HPLC小瓶中加入(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺(实施例20B的产物,1 mg,0.0025 mmol)、Cs2CO3(2.45 mg,0.0075 mmol)和DMF(0.2 mL),而后加入一个安瓿的CT3I(用0.1 mL DMF洗涤安瓿,并也将其加入到反应中)。将该混合物搅拌2小时。用EtOH和ACN稀释粗品,过滤。真空浓缩滤液,除去挥发物。用RP HPLC纯化残余物(Synergi Polar RP 80A,4u,10 x 250 mm,5 ml/min,45% ACN/水,PDA检测器,2 x 0.2 ml注射)。通过C18 sep-pak过滤转换溶剂之后,进行第一次注射,得到33.88 mCi 75.83 Ci/mmol,并在10 mL 绝对(abs) EtOH中递送。用相同的柱检测纯度(4.6 x 250 mm,1 ml/min,254和220 nm)。还保持第二次注射(~30 mCi,在10 ml EtOH中)。
实施例28B
[
11
C](1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

步骤1:合成[11C]碘甲烷。使用Siemens RDS-111回旋加速器,制备[11C]CO2,并使用GE Medical Systems TRACERlab FCX系统,将[11C]CO2转变为[11C]MeI。
步骤2:将[11C]MeI(得自于步骤1)收集在含有16 ul NaH(0.5g/20ml DMF)的(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N-甲基环丙烷-甲酰胺(实施例6的产物,0.25mg)/DMF(0.25 mL)的RT混合物中。在65℃,将该反应混合物转入2 mL v-小瓶中,加热5分钟,用水(0.8 mL)稀释,并注射到HPLC(Gemini C18,10 X 150 mm,Phenomenex)上。用含有25% A和75% B的溶剂系统洗脱目标峰(A=MeCN,B=95:5:0.1 H2O:MeCN:TFA,5ml/min,保留时间~6.5分钟),并收集在旋转蒸发器上的热圆底烧瓶中。浓缩该溶液,并真空转移至隔膜封盖的5 mL v-小瓶中。用乙醇(0.1 mL)和盐水(1-2 mL)冲洗圆底烧瓶,并真空转移至相同的v-小瓶中,得到13.6 mCi的[11C](1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N,N-二甲基-环丙基甲酰胺。
实施例29B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-[
11
C]甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺

将[11C]MeI(利用实施例28B所公开的方法合成)收集在含有Cs2CO3的(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺(实施例20的产物,0.20mg)/DMF(0.20 mL)的RT混合物中。在65℃,将该反应混合物转入2 mL v-小瓶中,加热5分钟,用水(0.8 mL)稀释,并注射到HPLC(Gemini C18,10 X 150 mm,Phenomenex)上。用含有22% A和78% B的溶剂系统洗脱目标峰(A=MeCN,B=95:5:0.1 H2O:MeCN:TFA,5ml/min,保留时间~11.5分钟),收集在旋转蒸发器上的热圆底烧瓶中。浓缩该溶液,并真空转移至隔膜封盖的5 mL v-小瓶中。用乙醇(0.1 mL)和盐水(1-2 mL)冲洗圆底烧瓶,并真空转移至相同的v-小瓶中,得到35.4 mCi的(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-[11C]甲基-1H-咪唑-4-基}苯基)-N,N-二甲基环丙基甲酰胺。
中间体12B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙腈

在0℃,向实施例4B(300 mg,0.779 mmol)的磷酸三甲酯(1 mL,8.64 mmol)溶液中逐滴加入氯甲酸三氯甲酯(0.145 mL,1.169 mmol)。然后将该混合物加热到60℃,使该反应完成,并除去光气。用碳酸氢钠水溶液中和该混合物,并用EtOAc提取有机物。然后用水和盐水洗涤有机物,然后干燥(MgSO4)。浓缩溶剂,得到标题化合物,其不用进一步纯化就使用。LCMS:[M+1]+=367。
中间体13B
(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)-N'-羟基环丙甲脒

向中间体11B(30 mg,0.082 mmol)的2 mL EtOH溶液中加入0.25 mL 50% NH2OH水溶液和催化数量的K2CO3。通过微波辐射,将该反应加热至120℃,保持1小时。将该反应混合物浓缩至干,得到标题化合物,其不用进一步纯化就使用。LCMS:[M+1]+=400。
中间体14B
5-{(1S,2S)-2-[4-(5-碘代-1-甲基-1H-咪唑-4-基)苯基]环丙基}-1,3,4-噁二唑-2(3H)-酮

步骤1:按照与中间体11(步骤2)所描述方法相同的方法,起始于中间体5B,制备(1S,2S)-2-[4-(5-碘代-1-甲基-1H-咪唑-4-基)苯基]环丙基甲酰肼。LCMS:[M+1]+=383。
步骤2:按照实施例17B所描述方法,起始于前述步骤的产物,制备标题化合物。LCMS:[M+1]+=409。
实施例30B
5-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]-3-甲基-1,3,4-噁二唑-2(3H)-酮

向实施例17B(5 mg,0.012 mmol)和过量碳酸铯的0.5 mL DMF溶液中加入2滴碘甲烷。将该反应在室温下搅拌16小时,而后过滤,通过反相HPLC纯化。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.43(s, 1H), 8.13(s, 1H), 7.96(d, 2H), 7.75(d, 1H), 7.24(d, 2H), 7.02(d, 1H), 3.75(s, 3H), 3.30(s, 3H)2.56(m, 1H), 2.20(m, 1H), 1.60(br, 2H)。LCMS:[M+1]+=440。人FAAH溶胞产物试验:IC50=170.6 nM。
实施例31B
5-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]-1,3,4-噁二唑-2(3H)-硫酮

向中间体11B(50 mg,0.125 mmol)和1,1'-硫代羰基(carbonothioyl)二(1H-咪唑)(50 mg,0.281 mmol)的DCM(1 mL)溶液中加入TEA(0.05 mL,0.359 mmol)。将该反应在室温下搅拌1小时,而后蒸发溶剂,通过反相HPLC纯化残余物。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[(CD3)2CO]: 8.42(s, 1H), 8.00-7.93(br, 2H), 7.71(d, 1H), 7.21(d, 2H), 6.95(d, 2H)3.70(s, 3H), 2.40(m, 1H), 1.98(m, 1H), 1.55(m, 1H), 1.30(m, 2H).。LCMS:[M+1]+=442。人FAAH溶胞产物试验:IC50=677.3 nM。
实施例32B
5-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]-2,4-二氢-3H-1,2,4-三唑-3-硫酮

将中间体11B(100 mg,0.250 mmol)和过量异硫氰酸钾的乙酸(1 mL)和水(1 mL)溶液加热至60℃,保持3小时。用NaOH水溶液(5N)将pH值调节至10,并将该溶液回流2小时,而后过滤,通过反相HPLC纯化。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[CDCl3]: 8.42(s, 1H), 8.38(s, 1H), 7.73(d, 2H), 7.48(d, 1H), 6.91(d, 2H), 6.82(d, 1H), 3.68(s, 3H), 2.46(m, 1H), 1.95(m, 1H), 1.52(m, 1H), 1.28(m, 2H)。LCMS:[M+1]+=441。人FAAH溶胞产物试验:IC50=304 nM。
实施例33B
5-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]-2,4-二氢-3H-1,2,4-三唑-3-酮

将中间体11B(100 mg,0.250 mmol)和过量异氰酸钾的乙酸(1 mL)和水(1 mL)溶液在室温下搅拌3小时。用NaOH水溶液(5N)将pH值调节至10,并将该溶液回流2小时,而后过滤,通过反相HPLC纯化。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[CDCl3]: 8.38(br, 2H), 7.87(d, 2H), 7.50(d, 1H), 7.09(d, 2H), 6.83(d, 1H), 6.90(s, 3H), 1.84(br, 2H), 1.29(m, 2H)。LCMS:[M+1]+=425。人FAAH溶胞产物试验:IC50=545.3 nM。
实施例34B
5-[(1S,2S)-2-(4-{5-[(5-氯代吡啶-2-基)硫基]-1-甲基-1H-咪唑-4-基}苯基)环丙基]-4-甲基-2,4-二氢-3H-1,2,4-三唑-3-酮

按照实施例33B所描述的方法,使用异氰酸甲酯,制备标题化合物。1H NMR(500 MHz),[CDCl3]: 10.30(s, 1H), 8.40(s, 1H), 7.90(d, 2H), 7.25(s, 1H), 7.45(d, 1H), 7.15(d, 2H), 6.80(d, 1H), 3.65(s, 3H), 3.25(s, 3H), 2.40(m, 1H), 1.90(m, 1H), 1.70(m, 1H), 1.45(m, 1H)。LCMS:[M+1]+=439。人FAAH溶胞产物试验:IC50=893.5 nM。
实施例35B
5-氯-2-[(1-甲基-4-{4-[(1S,2S)-2-(1,2,4-噁二唑-3-基)环丙基]苯基}-1H-咪唑-5-基)硫基]吡啶
将中间体13B(30 mg,0.075 mmol)溶于2 mL原甲酸三乙酯中。加入催化数量的TFA,并将该反应在130℃下加热3小时。除去挥发物,并将残余物用反相HPLC纯化。收集含有产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[CDCl3]: 8.60(s, 1H), 8.39(s, 1H), 8.01(s, 1H), 7.96(d, 2H), 7.50(d, 1H), 7.17(d, 2H), 6.80(d, 1H), 3.69(s, 3H), 2.59(m, 1H), 2.47(m, 1H), 1.73(m, 1H), 1.56(m, 1H)。LCMS:[M+1]+=410。人FAAH溶胞产物试验:IC50=58.38 nM。
实施例36B
5-[(5-氯代吡啶-2-基)硫基]-1-甲基-4-{4-[(1S,2S)-2-(5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)环丙基]苯基}-1H-咪唑-3-鎓三氟乙酸盐

向中间体13B(107 mg,0.268 mmol)的吡啶(1 mL)溶液中加入氯甲酸乙酯(0.025 mL,0.268 mmol)。将该反应加热至100℃,保持2小时。蒸发挥发物,并通过反相HPLC纯化残余物。蒸发含有产物的馏分,得到标题化合物。1H NMR(500 MHz),[CD3OD]: 8.38(s, 1H), 7.77(d, 1H), 7.69(br, 3H), 7.29(br, 3H), 3.83(s, 3H), 2.54(m, 1H), 2.13(m, 1H),1.65(m, 1H), 1.60(m, 1H)。LCMS:[M+1]+=426。人FAAH溶胞产物试验:IC50=95.45 nM。
实施例37B
5-[(5-氯代吡啶-2-基)硫基]-1-甲基-4-{4-[(1S,2S)-2-(2H-四唑-5-基)环丙基]苯基}-1H-咪唑-3-鎓三氟乙酸盐

将中间体12(100 mg,0.273 mmol)和叠氮化三甲基锡(0.231 mL,1.363 mmol)的二甲苯(1 mL)无水溶液在氮气氛围中加热至140℃,保持2小时。蒸发挥发物,并通过反相HPLC纯化残余物。蒸发含有产物的馏分,得到标题化合物。1H NMR(500 MHz),[CD3OD]: 9.04(br, 1H), 8.38(s, 1H), 7.78(d, 1H), 7.67(br, 3H), 7.28(br, 3H), 3.86(s, 3H), 2.64(m, 1H), 2.53(m, 1H), 1.80-1.75(br, 2H)。LCMS:[M+1]+=410。人FAAH溶胞产物试验:IC50=167.3 nM。
实施例38B
5-氯-2-[(1-甲基-4-{4-[(1S,2S)-2-(2-甲基-2H-四唑-5-基)环丙基]苯基}-1H-咪唑-5-基)硫基]吡啶

向实施例37B(10 mg,0.024 mmol)和过量碳酸钾的DMF(0.5 mL)溶液中加入3滴碘甲烷。将该反应在室温下搅拌1小时,而后过滤,通过反相HPLC纯化。收集含有主要产物的馏分,用乙酸乙酯稀释,用碳酸氢钠水溶液、水和盐水洗涤。将有机层干燥(MgSO4),过滤,浓缩,得到标题化合物。1H NMR(500 MHz),[CD3OD]: 8.38(s, 1H), 7.78(d, 2H), 7.76(br, 3H), 7.30(br, 3H), 3.83(s, 3H), 3.66(s, 3H), 2.66(m, 1H), 2.45(m, 1H), 1.85-1.72(br, 2H)。LCMS:[M+1]+=424。人FAAH溶胞产物试验:IC50=37 nM。
按照中间体4(步骤4)所描述的方法,使用中间体14B和合适的硫醇作为起始原料,制备表4B中的实施例。
表4

MicroPET摄像机成像
将一只大鼠麻醉(氯胺酮/ace-普马嗪),放在摄像机前,其尾静脉中插入便于注射的套管。将50pe导管放置在股静脉中,用于收集血样。在注射放射性示踪剂之前2小时,口服给予另一只大鼠未标记的脂肪酸酰胺水解酶(FAAH)抑制剂,以便说明非特异性结合和剂量占有率。通过大鼠的尾静脉注射1 mCi/大鼠的11C标记的FAAH抑制剂,并用几毫升生理盐水(normal saline)冲洗导管。每次扫描一只大鼠。当注射放射性示踪剂时,开始采集图像。获得90分钟时间的成像,随后用戊巴比妥钠使大鼠安乐死。在相加图像上画出兴趣部位(ROIs),包括脑,然后用于分析随后成像中的计数率。用ROI中的计数率除以整个大鼠的计数率,然后乘以100,将计数率转变为%-剂量/ROI。
在注射的时候,从股导管处收集血液,并在第一个两分钟将两滴血液收集到每个管中,然后获取300微升血液样品,用于代谢物校正,并测定血浆和全血在5、15、30、45、60和90分钟时的放射性。刚好在注射PET示踪物之前和扫描90分钟之后,从预先注射未标记的脂肪酸酰胺水解酶抑制剂的大鼠中获取300微升血浆样品,用于血浆药物浓度测定。
在猕猴中的PET成像:
用氯胺酮I.M.(15 mpk)将禁食的猕猴(7-11 kg)麻醉,并将猴子放置在PET摄像机床中。将I.V.导管插入右侧隐静脉中。为了动脉取样,使右侧股区域无菌,放置动脉插管,并缝合固定。随后用异氟烷保持麻醉。在整个研究期间,将动物插管,并放在异氟烷(2-2.5%)上,同时通入医学级压缩空气,每分钟呼吸大约23次。调节I:E比例、呼吸体积和呼吸速度,保持CO2水平为~40mmHg,SpO2水平为95至100%。连接温度传感器、脉搏血氧仪和呼气末二氧化碳分压器。将动物放在K-模块加热板上,以便保持体温,在顶部放置另一个垫,并将动物仰卧、头向前地放在摄像机扫描架内。在整个研究中,用1 ml/min乳酸Ringer's IV保持常规输液治疗。IV注射11C标记的FAAH抑制剂的等分样品,从注射时间开始进行发射成像,并持续90分钟。
通过动脉插管将全血样品收集到肝素管中,测定全血和血浆的放射性。将样品离心,并在PET配体注射后10、20、30、45、60、90和120秒钟进行20ul全血和血浆的计数。获取血液样品(0.5 ml),用于代谢物校正,并在3、5、15、30、60和90分钟时测定血浆和全血的放射性。
在单独的实验中,在注射放射性示踪剂之前21小时,口服给予禁食猕猴未标记的FAAH抑制剂(赋形剂:Imwitor/Tween)。获取血浆样品(1 ml),测定20.5、21、22、22.5小时的血浆药物浓度。在21小时,按照与上述方案相同的方案,IV注射11C标记的FAAH抑制剂的等分样品,从注射时间开始进行发射成像,并持续90分钟。将给予FAAH抑制剂之后的脑的各个区域的示踪物结合与没有给予FAAH抑制剂情况下的脑的相同区域的示踪物结合进行比较,测定占有率。
联用研究

在大鼠的炎症性疼痛的Complete Freunds Adjuvant模型(Eid等人,2008)中,当共同给予艾托考昔时,标准等辐射分析方法(Tallarida等人1989)用于评价FAAH抑制剂化合物A是否表现出加和或协同作用。以1:1、0.3:1和3:1的PO(p.o.,口服)剂量比例来试验艾托考昔和化合物A。
总的说来,数据表明,在记录的观察结果和预测的加和线之间没有显著差异(图1)。因此,这些数据表明,艾托考昔和FAAH抑制剂化合物A在它们针对炎症性疼痛的止痛效果方面具有加和效果。
Tallarida R.J.等人,Statistical analysis of drug-drug and site-site interactions with isobolograms. Life Sci., 45(11): 947-961(1989)。
Eid, S.等人,HC-030031, a TRPA1 selective antagonist, attenuates inflammatory-and neuropathy-induced mechanical hypersensitivity. Molecular Pain, 4: 48(2008)。
Claims (25)
1.药物组合物,包含:
抑制FAAH的式I化合物:

或其可药用盐,其中:
X是S或SO;
n是0、1或2;
R1选自:
(1)芳基,和
(2)HET1,
其中R1任选被取代基R4和R5单或二取代;且其中R4和R5独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)单、二或三卤代OC1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-OC1-4烷基,
(e)任选被一或两个选自羟基、CN、-CHF2和-CF3的取代基取代的-C1-4烷基,
(f)任选被羟基、卤代基或CN取代的-C1-2烷基-C3-6环烷基,
(g)-S(O)nC1-4烷基,
(h)-S(O)nNR6R7,
(i)-C(O)-NH-NR8R9,
(j)-C(O)-OH,
(k)任选被卤代基或羟基取代的-C(O)-OC1-4烷基,
(l)-C(O)-NR10R11,
(m)任选被卤代基单、二或三取代的-C(O)-C1-4烷基,
(o)-C(NR12)-NR13R14,
(p)HET4,
(q)芳基,
(r)-C(O)-NH-NH-C(O)H,
(s)-CH2-C(O)-O-C1-4烷基,而CH2可以任选被C1-4烷基或OH取代
(t)-CH2-C(O)NR15R16,而CH2可以任选被C1-4烷基或OH取代,和
(u)-NR17R18,
其中选项(p)和(q)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基;
(9)-C(O)-NR19R20,
(10)-NH2
(11)氧代,
(12)=S,
条件是,选项(q)上的取代基不是氧代或=S,
其中R6、R7、R8、R9、R10、R11、R12、R13、R14、R15、R16、R17、R18、R19和R20各自独立地选自H和C1-4烷基,
或
R6和R7或R8和R9或R10和R11或R13和R14或R15和R16或R17和R18或R19和R20与它们此处相连接的氮连接形成环,形成4至7个原子的5元杂环,所述环含有1、2、3或4个选自N、O和S的杂原子,所述环任选被独立地选自下列的取代基单或二取代:卤代基,羟基,氧代,C1-4烷基,羟基C1-4烷基,卤代基C1-4烷基,-C(O)-C1-4烷基和-S(O)nC1-4烷基;
R2选自:
(1)芳基,
(2)HET3,
(3)-CH2-芳基,
(4)-CH2-HET3,
(5)-C1-6烷基,和
(6)-C3-6环烷基,
其中R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(e)-CF3,
(f)任选被羟基或卤代基取代的-OC1-4烷基,
(g)-C(O)O-C1-3烷基,和
(h)任选被卤代基、C1-4烷基或-OC1-4烷基取代的-S-芳基;
R3选自:
(1)芳基,
(2)HET5,和
(3)C3-6环烷基,
其中R3任选被独立地选自下列的取代基单或二取代:
(a)羟基,
(b)卤代基,
(c)-C3-6环烷基,
(d)-OC3-5环烷基,
(e)-C1-4烷基,
(f)-OC1-4烷基,
(g)-C(O)CH3
(h)单、二或三卤代C1-4烷基,
(i)单、二或三卤代-OC1-4烷基,和
(j)-S(O)n-C1-4烷基;
其中芳基是单或双环芳香环系统;HET1、HET3、HET4和HET5各自独立地是5至10元芳香、部分芳香或非芳香单或双环,或其N-氧化物,所述基团含有1至4个选自O、S和N的杂原子,并且任选被1至2个氧代基取代;
或抑制FAAH的式II化合物

或其可药用盐,其中:
n=0、1或2
R1选自:
(1)苯基,和
(2)HET1,
其中选项(1)和(2)被下列取代

其中R5选自:
(a)卤代基,
(b)-CN,
(c)卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-OC1-4烷基,
(e)任选被一或两个选自羟基、CN、-CHF2和-CF3的取代基取代的-C1-4烷基,
(f)任选被羟基、卤代基或CN取代的-C1-2烷基-C3-6环烷基,
(g)-S(O)nC1-4烷基,
(h)-S(O)nNR6R7,
(i)-C(O)-OH,
(j)任选被卤代基或羟基取代的-C(O)-OC1-4烷基,
(k)-C(O)-NR10R11,
(l)任选被卤代基单、二或三取代的-C(O)-C1-4烷基,
(m)HET2,
(n)芳基,
(o)-CH2-C(O)-O-C1-4烷基,而CH2可以任选被C1-4烷基或OH取代
(t)-CH2-C(O)NR15R16,而CH2可以任选被C1-4烷基或OH取代,和
(u)-NR17R18,
其中选项(m)和(m)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)-NR19R20,
(9)-NH2
(10)氧代,
(11)=S,
其中R6、R7、R10、R11、R15、R16、R17、R18、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被卤代基单、二或三取代,
或
R6和R7或R10和R11或R15和R16或R17和R18或R19和R20与它们此处相连接的原子连接在一起,形成4至7个原子的5元杂环,所述环含有1、2、3或4个选自N、O和S的杂原子,所述环任选被独立地选自下列的取代基单或二取代:卤代基,羟基,氧代,C1-4烷基,羟基C1-4烷基,卤代C1-4烷基,-C(O)-C1-4烷基和-S(O)nC1-4烷基;
R2选自:
(1)氢,
(2)芳基,
(3)HET3,
(4)-CH2-芳基,
(5)-CH2-HET3,
(6)-C1-6烷基,和
(7)-C3-6环烷基,
其中选项(2)、(3)、(4)、(5)、(6)和(7)任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(e)-CF3,
(f)任选被羟基或卤代基取代的-OC1-4烷基,
(g)-C(O)O-C1-3烷基;
R3选自:
(1)芳基,
(2)HET4,和
(3)C3-6环烷基,
其中选项(1)、(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)羟基,
(b)卤代基,
(c)-C3-6环烷基,
(d)-OC3-5环烷基,
(e)-C1-4烷基,
(f)-OC1-4烷基,
(g)-C(O)CH3
(h)单、二或三卤代C1-4烷基,
(i)单、二或三卤代-OC1-4烷基,和
(j)-S(O)n-C1-4烷基;和
R4选自:
(1)-C1-4烷基,
(2)-卤代C1-4烷基,
(3)H;和
HET1、HET2、HET3和HET4各自独立地是5至10元芳香、部分芳香或非芳香单或双环,含有1-4个选自O、S和N的杂原子,并且任选被1-2个氧代基取代;
在该方面,存在上位组,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,和
(9)噁唑基,
其中选项(1)至(9)被下列取代

其中R5选自:
(b)-CN,
(c)卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)任选被羟基或CN取代的-C1-4烷基,
(f)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(h)-S(O)nC1-4烷基,其中n是1或2,
(i)-S(O)2NR6R7,
(j)-C(O)-NR10R11,
(k)HET2,
(l)芳基,和
其中选项(k)和(l)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中该C1-4烷基任选被卤代基单、二或三取代;
和第二个活性剂,该药剂可有效用于治疗:
急性疼痛,慢性疼痛,神经性疼痛,偏头痛;由炎症所引起的疼痛和神经性疼痛,焦虑症,进食障碍,肥胖症,眼内压升高,青光眼,心血管病症,抑郁症,炎症性病症,哮喘,克罗恩氏病和炎症性肠病,食物过敏,哮喘,皮肤炎症,呕吐,触摸痛,痛觉过敏,头痛,内脏疼痛,牙齿疼痛,与灼伤相关的疼痛,月经疼痛,痛经,原发性痛经,类风湿性关节炎,幼年型类风湿关节炎,骨关节炎,外科手术之后的疼痛,妇科手术、腹部外科、切口、口腔外科和背痛,癫痫和癫痫状引起的损伤,接触兴奋性神经毒素,兴奋性中毒,局部缺血性脑损伤,脑缺血,外伤性损伤,抑郁症,焦虑症,睡眠障碍,阿尔茨海默氏病,帕金森氏症,亨丁顿舞蹈症,肌萎缩侧索硬化,多发性脑硬化,妥瑞症,精神分裂症,青光眼,疼痛,成瘾,炎症,过敏性反应,进食障碍,低血压症,高血压症,呼吸问题,癌肿瘤生长,化疗并发症,窒息,注意力缺陷病症和胃肠疾病,包括恶心和呕吐,胃溃疡,分泌型腹泻,麻痹性肠梗阻,炎症性肠病,结肠癌,胃食道回流病症,瘙痒,脂肪肝疾病和非酒精脂肪肝炎和过敏性肠综合症。
2.按照权利要求1的药物组合物,其中第二个活性剂可有效用于治疗急性疼痛,慢性疼痛,神经性疼痛,与偏头痛相关的疼痛,偏头痛预防,由炎症所引起的疼痛,神经性疼痛,疱疹后神经痛,由于化疗引起的周围性疼痛,神经病,由于HIV引起的周围神经病的疼痛,由于核苷逆转录酶抑制剂引起的周围神经病的疼痛,疼痛性糖尿病性神经病变,由于肌纤维痛所引起的疼痛,触摸痛,痛觉过敏,内脏疼痛,牙齿疼痛,与灼伤相关的疼痛,月经疼痛,痛经,原发性痛经,类风湿性关节炎,幼年型类风湿关节炎,骨关节炎,手术后的疼痛,妇科手术、腹部外科、切口、口腔外科和背痛,头痛,偏头痛,抑郁症,焦虑症,睡眠障碍,阿尔茨海默氏病,帕金森氏症,进食障碍和肥胖症。
3.按照权利要求2的药物组合物,其中第二个活性剂可有效用于治疗骨关节炎,类风湿性关节炎,炎症性疼痛,神经性和感受伤害性疼痛,糖尿病性神经病变,带状疱疹神经痛,骨骼肌疼痛和肌纤维痛,以及急性疼痛,偏头痛,睡眠障碍,阿尔茨海默氏病和帕金森氏症。
4.按照权利要求3的药物组合物,其中第二个活性剂可有效用于治疗炎症性疼痛、神经性和感受伤害性疼痛。
5.按照权利要求4的药物组合物,其中第二个活性剂是依托昔布(etoricixib)。
6. 药物组合物,其中权利要求1的FAAH抑制剂是式I的FAAH抑制剂,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,
(9)噁唑基,和
(10)噁二唑基;
其中R1任选被取代基R4和R5单或二取代,其中R4和R5独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)任选被羟基或CN取代的-C1-4烷基,
(f)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(h)-S(O)nC1-4烷基,其中n是0、1或2,
(i)-S(O)nNR6R7,
(j)-C(O)-NR10R11,
(k)HET4,
(l)芳基,和
其中选项(k)和(l)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基。
7.药物组合物,其中权利要求6的FAAH抑制剂是式I的FAAH抑制剂,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,
(5)哒嗪基,
(6)1,2,4-噁二唑基,和
(7)1,3,4-噁二唑基,
任选被取代基R4和R5单或二取代,它们独立地选自:
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)nC1-4烷基,
(c)-C(O)-NR10R11,
(d)HET4,和
(e)卤代基,
其中HET4任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基。
8.药物组合物,其中权利要求1的FAAH抑制剂是式I的FAAH抑制剂,其中
R2选自:
(1)芳基,
(2)HET3,
(3)-CH2芳基,和
(4)-CH2HET3,
其中R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基。
9.药物组合物,其中权利要求8的FAAH抑制剂是式I的FAAH抑制剂,其中
R2选自:
(1)芳基,和
(2)HET3,
其中R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3。
10.药物组合物,其中权利要求9的FAAH抑制剂是式I的FAAH抑制剂,其中
R2选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噁唑基,
(8)吡唑基,
(9)1,2,4-噁二唑基,和
(10)1,3,4-噁二唑基,
其中R2任选被卤代基、任选被卤代基取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代。
11.药物组合物,其中权利要求1的FAAH抑制剂是式I的FAAH抑制剂,其中
R3选自:
(1)芳基,和
(2)HET5,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-OC1-4烷基,
(d)单、二或三卤代C1-4烷基,和
(e)单、二或三卤代-OC1-4烷基。
12.药物组合物,其中权利要求11的FAAH抑制剂是式I的FAAH抑制剂,其中
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
13.药物组合物,其中权利要求1的FAAH抑制剂是式Ia的FAAH抑制剂,

其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,
(9)噁唑基,和
(10)噁二唑基;
其中R1任选被取代基R4和R5单或二取代,它们独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)任选被羟基或CN取代的-C1-4烷基,
(f)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(h)-S(O)nC1-4烷基,其中n是0、1或2,
(i)-S(O)nNR6R7,
(j)-C(O)-NR10R11,
(k)HET4,
(l)芳基,和
其中选项(k)和(l)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基;
R2选自:
(1)芳基,
(2)HET3,
(3)-C1-6烷基,和
(4)-C3-6环烷基,
其中选项R2任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基;和
R3选自:
(1)芳基,和
(2)HET5,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-C1-4烷基,
(d)-OC1-4烷基,
(e)单、二或三卤代C1-4烷基,和
(f)单、二或三卤代-OC1-4烷基。
14.药物组合物,其中权利要求13的FAAH抑制剂是式Ia的FAAH抑制剂,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,
(5)哒嗪基,
(6)1,2,4-噁二唑基,和
(7)1,3,4-噁二唑基,
任选被取代基R4和R5单或二取代,它们独立地选自:
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)nC1-4烷基,
(c)-C(O)-NR10R11,
(d)HET4,和
(e)卤代基,
其中HET4任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基。
15.R2选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噁唑基,
(8)吡唑基,
(9)1,2,4-噁二唑基,和
(10)1,3,4-噁二唑基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代;和
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
16.药物组合物,其中权利要求14的FAAH抑制剂是式Ia的FAAH抑制剂,

其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
其中R1任选被取代基R4和R5单或二取代,它们独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)-C(CH3)2-OH;
R2选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)吡唑基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代;和
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
17.药物组合物,其中权利要求15的FAAH抑制剂是式Ia的FAAH抑制剂,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)吡嗪基,
其中R1任选被取代基R4和R5单或二取代,它们独立地选自:
(a)卤代基,
(b)-CN,
(c)单、二或三卤代C1-4烷基,
(d)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(e)-C(CH3)2-OH;
R2选自:
(1)苯基,
(2)吡啶基,
其中R2任选被卤代基、任选被卤素取代的OC1-4烷基、-C1-4卤代烷基、羟基和CN单或二取代;和
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中R3任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
18.药物组合物,其中权利要求1的FAAH抑制剂是式II的FAAH抑制剂,
其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,和
(5)哒嗪基,
其中选项(1)至(5)被下列取代

R5选自
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)2C1-4烷基,
(c)-C(O)-NR10R11,
(d)HET2,和
(e)卤代基,
其中选项(d)任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被卤代基单、二或三取代。
19.药物组合物,其中权利要求1的FAAH抑制剂是式II的FAAH抑制剂,其中
R2选自:
(1)氢,
(2)芳基,
(3)HET3,
(4)-C1-6烷基,和
(5)-C3-6环烷基,
其中选项(2)、(3)、(4)和(5)任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基。
20.药物组合物,其中权利要求18的FAAH抑制剂是式II的FAAH抑制剂,其中
R2选自:
(1)氢,
(2)-C1-6烷基,和
(3)-C3-6环烷基,
其中选项(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3。
21.药物组合物,其中权利要求1的FAAH抑制剂是式II的FAAH抑制剂,其中
R3选自:
(1)苯基,和
(2)HET4,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-C1-4烷基,
(d)-OC1-4烷基,
(e)单、二或三卤代C1-4烷基,和
(f)单、二或三卤代-OC1-4烷基。
22.药物组合物,其中权利要求20的FAAH抑制剂是式II的FAAH抑制剂,其中
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
(4)哒嗪基,
(5)吡嗪基,
其中选项(1)、(2)、(3)、(4)和(5)各自任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
23.药物组合物,其中权利要求1的FAAH抑制剂是式IIa或IIb的FAAH抑制剂,
或其可药用盐,其中:
R1选自:
(1)苯基,
(2)吡啶基,
(3)哒嗪基,
(4)嘧啶基,
(5)吡嗪基,
(6)噻唑基,
(7)噻吩基,
(8)吡咯基,和
(9)噁唑基,
其中选项(1)至(9)被下列取代
R5选自
(a)-CN,
(b)卤代C1-4烷基,
(c)任选被羟基、卤代基或氨基取代的-O-C1-4烷基,
(d)任选被羟基或CN取代的-C1-4烷基,
(e)任选被羟基取代的-C1-2烷基-C3-6环烷基,
(g)-S(O)nC1-4烷基,其中n是1或2,
(h)-S(O)2NR6R7,
(i)-C(O)-NR10R11,
(j)HET2,
(k)芳基,和
其中选项(j)和(k)各自任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R6、R7、R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被氚化或被卤代基单、二或三取代,或
R2选自:
(1)氢,
(2)芳基,
(3)HET3,
(4)-C1-6烷基,和
(5)-C3-6环烷基,
其中选项(2)、(3)、(4)和(5)任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-C1-4烷基,
(f)-C1-4卤代烷基,和
(g)任选被卤代基或羟基取代的-OC1-4烷基;和
R3选自:
(1)苯基,和
(2)HET4,
其中选项(1)和(2)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-C3-6环烷基,
(c)-C1-4烷基,
(d)-OC1-4烷基,
(e)单、二或三卤代C1-4烷基,和
(f)单、二或三卤代-OC1-4烷基;
R4选自:
(1)任选氚化的-C1-4烷基,和
(3)H;
药物组合物,其中权利要求22的FAAH抑制剂是式IIa或IIb的FAAH抑制剂,其中
R1选自:
(1)苯基,
(2)吡啶基,
(3)嘧啶基,
(4)吡嗪基,和
(5)哒嗪基,
其中选项(1)至(5)被下列取代

R5选自
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)2C1-4烷基,
(c)-C(O)-NR10R11,和
(d)HET2,
其中选项(d)任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被氚化或被卤代基单、二或三取代,或
R2选自:
(1)氢,
(2)-C1-6烷基,和
(3)-C3-6环烷基,
其中选项(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3;
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
(4)吡嗪基,和
(5)哒嗪基,
其中选项(1)、(2)、(3)、(4)和(5)各自任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
24.药物组合物,其中权利要求23的FAAH抑制剂是式IIa或IIb的FAAH抑制剂,其中
R1选自:
(1)苯基,和
(2)吡啶基,
其中选项(1)和(2)被下列取代

R5选自
(a)任选被羟基取代的-C1-4烷基,
(b)-S(O)2C1-4烷基,
(c)-C(O)-NR10R11,
(d)HET2,和
其中选项(d)任选被选自下列的取代基单或二取代:
(1)卤代基,
(2)-CN,
(3)-OH,
(4)任选被羟基、卤代基或氰基取代的-C1-4烷基,
(5)-CF3,
(6)任选被羟基或卤代基取代的-OC1-4烷基,
(7)-C(O)OH,和
(8)-C(O)O-C1-3烷基,和
(9)-C(O)-NR19R20,
其中R10、R11、R19和R20各自独立地选自H和C1-4烷基,其中C1-4烷基任选被氚化被卤代基单、二或三取代,或
R2选自:
(1)氢,
(2)-C1-6烷基,和
(3)-C3-6环烷基,
其中选项(2)和(3)各自任选被独立地选自下列的取代基单或二取代:
(a)卤代基,
(b)-CN,
(c)-OH,
(d)-羟基C1-4烷基,
(e)-CH3,
(f)-CF3,和
(g)-OCH3;
R3选自:
(1)苯基,
(2)嘧啶基,
(3)吡啶基,
其中选项(1)、(2)和(3)各自任选被卤代基、卤代C1-4烷基或任选被卤代基取代的-OC1-4烷基单或二取代。
25.治疗选自下列疾病的方法:急性疼痛,慢性疼痛,神经性疼痛,偏头痛;由炎症所引起的疼痛和神经性疼痛,焦虑症,进食障碍,肥胖症,眼内压升高,青光眼,心血管病症,抑郁症,炎症性病症,哮喘,克罗恩氏病和炎症性肠病,食物过敏,哮喘,皮肤炎症,呕吐,触摸痛,痛觉过敏,头痛,内脏疼痛,牙齿疼痛,与灼伤相关的疼痛,月经疼痛,痛经,原发性痛经,类风湿性关节炎,幼年型类风湿关节炎,骨关节炎,外科手术之后的疼痛,妇科手术、腹部外科、切口、口腔外科和背痛,癫痫和癫痫状引起的损伤,接触兴奋性神经毒素,兴奋性中毒,局部缺血性脑损伤,脑缺血,外伤性损伤,抑郁症,焦虑症,睡眠障碍,阿尔茨海默氏病,帕金森氏症,亨丁顿舞蹈症,肌萎缩侧索硬化,多发性脑硬化,妥瑞症,精神分裂症,青光眼,疼痛,成瘾,炎症,过敏性反应,进食障碍,低血压症,高血压症,呼吸问题,癌肿瘤生长,化疗并发症,窒息,注意力缺陷病症和胃肠疾病,包括恶心和呕吐,胃溃疡,分泌型腹泻,麻痹性肠梗阻,炎症性肠病,结肠癌,胃食道回流病症,瘙痒,脂肪肝疾病和非酒精脂肪肝炎和过敏性肠综合症,该方法包括:给予按照权利要求1的组合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US29908710P | 2010-01-28 | 2010-01-28 | |
| US61/299,087 | 2010-01-28 | ||
| PCT/US2011/022412 WO2011094209A1 (en) | 2010-01-28 | 2011-01-25 | Pharmaceutical compositions for the treatment of pain and other indicatons |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN102858338A true CN102858338A (zh) | 2013-01-02 |
Family
ID=44319717
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2011800171616A Pending CN102858338A (zh) | 2010-01-28 | 2011-01-25 | 治疗疼痛及其它适应症的药物组合物 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20130030000A1 (zh) |
| EP (1) | EP2528603A4 (zh) |
| JP (1) | JP2013518110A (zh) |
| KR (1) | KR20120123691A (zh) |
| CN (1) | CN102858338A (zh) |
| AU (1) | AU2011209754A1 (zh) |
| BR (1) | BR112012018913A2 (zh) |
| CA (1) | CA2786888A1 (zh) |
| MX (1) | MX2012008801A (zh) |
| RU (1) | RU2012136624A (zh) |
| WO (1) | WO2011094209A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110709084A (zh) * | 2017-03-13 | 2020-01-17 | 阿比德治疗公司 | 双重magl和faah抑制剂 |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2011001313A (es) * | 2008-08-04 | 2011-03-04 | Merck Sharp & Dohme | Derivados de oxazol utiles como inhibidores de la amida hidrolasa de acidos grasos. |
| AU2011238487B2 (en) * | 2010-04-08 | 2015-10-01 | Merck Sharp & Dohme Corp. | Oxazole derivatives useful as modulators of FAAH |
| US9006433B2 (en) | 2010-04-21 | 2015-04-14 | Merck Sharp & Dohme Corp. | Substituted pyrimidines |
| KR101801864B1 (ko) | 2010-06-16 | 2017-11-27 | 인플래머토리 리스폰스 리서치, 아이엔씨. | 인플루엔자, 감기 및 염증의 치료에서 레보세티리진 및 몬테루카스트의 용도 |
| JO3407B1 (ar) | 2012-05-31 | 2019-10-20 | Eisai R&D Man Co Ltd | مركبات رباعي هيدرو بيرازولو بيريميدين |
| WO2014164299A1 (en) | 2013-03-13 | 2014-10-09 | Inflammatory Response Research, Inc. | Use of levocetirizine and montelukast in the treatment of autoimmune disorders |
| RU2672871C2 (ru) * | 2013-03-13 | 2018-11-20 | Инфламматори Респонс Ресёрч, Инк. | Применение левоцетиризина и монтелукаста при лечении травматических повреждений |
| CN105263579B (zh) | 2013-03-13 | 2020-01-10 | 炎症反应研究公司 | 左西替利嗪和孟鲁司特在治疗血管炎中的用途 |
| KR102365952B1 (ko) | 2013-10-14 | 2022-02-22 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 선택적으로 치환된 퀴놀린 화합물 |
| KR102103256B1 (ko) | 2013-10-14 | 2020-04-23 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 선택적으로 치환된 퀴놀린 화합물 |
| US20160304553A1 (en) * | 2013-12-04 | 2016-10-20 | Galmed Research & Development Ltd. | Aramchol salts |
| WO2016036588A1 (en) * | 2014-09-03 | 2016-03-10 | Merck Sharp & Dohme Corp. | Pharmaceutical suspensions containing etoricoxib |
| EP3193875B1 (en) | 2014-09-15 | 2022-02-16 | Inflammatory Response Research, Inc. | Levocetirizine and montelukast in the treatment of inflammation mediated conditions |
| EP3649128A1 (en) | 2017-07-07 | 2020-05-13 | Syngenta Participations AG | Pesticidally active heterocyclic derivatives with sulfur containing substituents |
| KR102257685B1 (ko) | 2018-09-20 | 2021-05-31 | 성균관대학교산학협력단 | Cox-2 억제제를 유효성분으로 포함하는 소염진통 예방 또는 치료용 정제 조성물 |
| CN110156710B (zh) * | 2019-04-30 | 2022-10-28 | 贵州省中国科学院天然产物化学重点实验室(贵州医科大学天然产物化学重点实验室) | 一种多取代噁唑类化合物的制备方法 |
| JP7464955B2 (ja) | 2020-02-27 | 2024-04-10 | 国立大学法人千葉大学 | ヨードオキサゾール化合物の製造方法、オキサゾール化合物の製造方法 |
| WO2021236518A1 (en) * | 2020-05-19 | 2021-11-25 | IRR, Inc. | Levocetirizine and montelukast in the treatment of sepsis and symptoms thereof |
| IL308880A (en) * | 2021-05-28 | 2024-01-01 | Ananda Scient Inc | Methods for treating post-traumatic stress syndrome and traumatic brain injuries using cannabinoids |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1703407A (zh) * | 2002-10-08 | 2005-11-30 | 斯克里普斯研究学院 | 脂肪酸酰胺水解酶抑制剂 |
| CN101146786A (zh) * | 2004-12-30 | 2008-03-19 | 詹森药业有限公司 | 作为脂肪酸酰胺水解酶调节剂的哌嗪基脲和哌啶基脲 |
| US20080275046A1 (en) * | 2005-03-31 | 2008-11-06 | Ucb Pharma, S.A. | Compounds Comprising an Oxazole or Thiazole Moiety, Processes for Making Them, and Their Uses |
| WO2009152025A1 (en) * | 2008-06-11 | 2009-12-17 | Merck & Co., Inc. | Imidazole derivatives useful as inhibitors of faah |
| CN102171196A (zh) * | 2008-08-04 | 2011-08-31 | 默沙东公司 | 用作脂肪酸酰胺水解酶的抑制剂的*唑衍生物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2152082B1 (en) * | 2007-05-25 | 2012-08-29 | The Scripps Research Institute | Tetracyclic inhibitors of fatty acid amide hydrolase |
| CA2727242A1 (en) * | 2008-06-11 | 2009-12-17 | Merck Sharp & Dohme Corp. | Pyrazole derivatives useful as inhibitors of faah |
-
2011
- 2011-01-25 MX MX2012008801A patent/MX2012008801A/es not_active Application Discontinuation
- 2011-01-25 JP JP2012551232A patent/JP2013518110A/ja not_active Withdrawn
- 2011-01-25 WO PCT/US2011/022412 patent/WO2011094209A1/en active Application Filing
- 2011-01-25 CA CA2786888A patent/CA2786888A1/en not_active Abandoned
- 2011-01-25 AU AU2011209754A patent/AU2011209754A1/en not_active Abandoned
- 2011-01-25 US US13/574,303 patent/US20130030000A1/en not_active Abandoned
- 2011-01-25 RU RU2012136624/15A patent/RU2012136624A/ru not_active Application Discontinuation
- 2011-01-25 BR BR112012018913A patent/BR112012018913A2/pt not_active IP Right Cessation
- 2011-01-25 EP EP11737514.7A patent/EP2528603A4/en not_active Withdrawn
- 2011-01-25 CN CN2011800171616A patent/CN102858338A/zh active Pending
- 2011-01-25 KR KR1020127022379A patent/KR20120123691A/ko not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1703407A (zh) * | 2002-10-08 | 2005-11-30 | 斯克里普斯研究学院 | 脂肪酸酰胺水解酶抑制剂 |
| CN101146786A (zh) * | 2004-12-30 | 2008-03-19 | 詹森药业有限公司 | 作为脂肪酸酰胺水解酶调节剂的哌嗪基脲和哌啶基脲 |
| US20080275046A1 (en) * | 2005-03-31 | 2008-11-06 | Ucb Pharma, S.A. | Compounds Comprising an Oxazole or Thiazole Moiety, Processes for Making Them, and Their Uses |
| WO2009152025A1 (en) * | 2008-06-11 | 2009-12-17 | Merck & Co., Inc. | Imidazole derivatives useful as inhibitors of faah |
| CN102171196A (zh) * | 2008-08-04 | 2011-08-31 | 默沙东公司 | 用作脂肪酸酰胺水解酶的抑制剂的*唑衍生物 |
Non-Patent Citations (2)
| Title |
|---|
| JENNIFER D. KREISBERG ET AL.: "Pummerer reaction methodology for the synthesis of 5-thiophenyl substituted oxazoles", 《TETRAHEDRON LETTERS》 * |
| S. G. PIL’O ET AL.: "Synthesis of New 5-Mercapto-1,3-oxazole Derivatives on the Basis of 2-Acylamino-3,3-dichloroacrylonitriles and Their Analogs", 《RUSSIAN JOURNAL OF GENERAL CHEMISTRY》 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110709084A (zh) * | 2017-03-13 | 2020-01-17 | 阿比德治疗公司 | 双重magl和faah抑制剂 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2528603A1 (en) | 2012-12-05 |
| AU2011209754A1 (en) | 2012-07-26 |
| BR112012018913A2 (pt) | 2017-06-20 |
| JP2013518110A (ja) | 2013-05-20 |
| MX2012008801A (es) | 2012-08-17 |
| RU2012136624A (ru) | 2014-03-10 |
| WO2011094209A1 (en) | 2011-08-04 |
| CA2786888A1 (en) | 2011-08-04 |
| KR20120123691A (ko) | 2012-11-09 |
| US20130030000A1 (en) | 2013-01-31 |
| EP2528603A4 (en) | 2013-09-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102858338A (zh) | 治疗疼痛及其它适应症的药物组合物 | |
| CN1918128B (zh) | 稠环4-氧代-嘧啶衍生物 | |
| CN1922183B (zh) | 含氮的稠合杂芳环衍生物 | |
| CN103476258B (zh) | 用作抗糖尿病药剂的新的环状氮杂苯并咪唑衍生物 | |
| CN100567270C (zh) | 磺酰胺衍生物 | |
| CN102171196B (zh) | 用作脂肪酸酰胺水解酶的抑制剂的*唑衍生物 | |
| TWI412365B (zh) | 4-苯基-6-(2,2,2-三氟-1-苯乙氧基)嘧啶系化合物與其使用方法 | |
| JP5406716B2 (ja) | インドール化合物 | |
| CA2741666C (en) | P2x3, receptor antagonists for treatment of pain | |
| AU2016372028B2 (en) | Methods of treating hyperalgesia | |
| JP4735261B2 (ja) | ヘテロアリールオキシ含窒素飽和ヘテロ環誘導体 | |
| JP2018162260A (ja) | カンナビノイド受容体モジュレーター | |
| CN107108637A (zh) | 三唑并嘧啶化合物及其用途 | |
| CN104364235B (zh) | 作为ttx‑s阻断剂的酰胺衍生物 | |
| TW200800181A (en) | Pharmaceutical composition for anticancer | |
| JP2021001215A (ja) | α−アミノ−β−カルボキシムコン酸セミアルデヒド脱炭酸酵素の阻害剤 | |
| US20060167075A1 (en) | Modulators of FAAH | |
| TW200914437A (en) | FAAH inhibitors | |
| CN103874695A (zh) | 2-吡啶基氧基-4-腈食欲素受体拮抗剂 | |
| CN101111480A (zh) | 取代的吡啶酮衍生物 | |
| TW200407138A (en) | N-substituted-2-oxodihydropyridine derivatives | |
| CN101155788A (zh) | 4(3h)-喹唑啉酮衍生物的晶体 | |
| CN105517552A (zh) | 基于咪唑并[1,2-b]哒嗪的化合物、包含它们的组合物及其使用方法 | |
| JP5319518B2 (ja) | インドールジオン誘導体 | |
| CN101917987A (zh) | 治疗精神病障碍的aldh-2抑制剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20130102 |