具体实施方式
本文所用的术语是依据该领域认可的含义(除非特别指出),且对本领域的技术人员而言是易于理解的。现用以下制备实施例说明本发明。
一般流程A
1-[1-亚甲基-4-(溴亚甲基)亚苯基]-4,8,11-三(二乙氧基磷酰基)-1,4,8,11-四氮杂环十四烷
将α,α′-二溴-对-二甲苯(13.2g,0.05mol)加入到搅拌的在CH3CN(150ml)中的4,8,11-三(二乙氧基磷酰基)-1,4,8,11-四-氮杂环十四烷(见Bridger等人,J.Med.Chem.1995,38,366-378)(6.1g,0.01mo)和K2CO3(1.89g,0.013mol)中,70℃搅拌反应液1小时。将溶液冷却到室温,在减压下除去溶剂。残留物分配在盐水(50ml)和CH2Cl2(100ml)间。将有机相分开,干燥(Na2SO4),浓缩到最小体积。滤去固体,在减压下蒸发溶剂,得到浅黄色油状粗产物。硅胶柱层析纯化(CH2Cl2/CH3OH,25∶1),得到浅黄色油状1-[1-亚甲基-4-(溴亚甲基)亚苯基]-4,8,11-三(二乙氧基磷酰基)-1,4,8,11-四氮杂环十四烷(4.7g,59%)。1H NMR(CDCl3)d1.21-1.37(m,18H),1.66-1.74(m,2H),1.82-1.91(m,2H),2.30-2.35(m,2H),2.58-2.63(m,2H),2.99-3.16(m,12H),3.48(s,2H),3.95-4.07(m,12H),4.48(s,2H),7.21-7.35(4H).
一般流程B
用胺将溴苄基环胺中间体第二次烷基化(例见Bridger等人,J.Med.Chem.1995,38,366-378)
80℃,在搅拌下将在CH3CN(10ml)中的1-[1-亚甲基-4-(溴亚甲基)亚苯基]-4,8,11-三(二乙氧基磷酰基)-1,4,8,11-四氮杂环十四烷(0.6mmol)溶液,历时15-20分钟,逐滴加入到悬浮有K2CO3(1.5当量)的适当胺(5.0当量)的干燥CH3CN(5ml)溶液中。80℃再搅拌1小时后,将溶液浓缩至干,残留物分配在CH2Cl2和水间。分离出有机层,用水洗涤(3x),然后干燥(MgSO4)和蒸发。粗制残留物在硅胶柱上进行层析纯化(以5-15%MeOH/CH2Cl2为洗脱液),得到粘稠的油。
一般流程C
室温用HBr/HOAc将二乙氧基磷酰氨基的去保护(例见:Bridger等人,J.Med.Chem.1995.38.366-378)
将在醋酸(Aldrich,5ml)中的30%HBr(Aldrich,5ml)加入到搅拌的在醋酸(3ml)中的流程B得到的保护的环胺衍生物(0.1-0.5mmol)的溶液中,室温搅拌14小时。过滤收集得到的沉淀,用醋酸然后用Et2O洗涤。然后将固体溶解在水(3ml)中,用活性炭(100mg)处理,然后将混合物加热到80℃,30分钟。用硅藻土过滤该热溶液,将滤液浓缩到约1ml,然后加入醋酸,迅速形成白色沉淀。过滤收集白色沉淀,真空干燥。
以下为用这些方法制备的化合物:
实施例1
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-2-(氨基-甲基)吡啶六氢溴酸盐(AMD 3465)
白色固体:Mp 200-205℃(dec);1H NMR(D2O)d 2.04(m,4H),3.20-3.40(m,8H),3.40-3.60(m,8H),4.34(s,2H),4.38(s,2H),4.51(s,2H),7.50(m,4H),7.75(t,1H,J=6.6Hz),7.82(d,1H,J=7.9Hz),8.26(t,1H,J=7.9Hz),8.63(d,1H,J=5.3Hz);13C NMR(D2O)d 18.30,18.96,37.04,37.28,37.40,40.92,41.13,41.49,44.26,47.61,48.01,51.29,58.88,127.46,127.75,130.40,131.05,131.23,131.47,132.10,132.44,144.95,145.81,146.01;FAB MS m/z 493(M+H81Br,7),491(M+H79Br,7),411(M+H,100).
分析(C24H38N6·6HBr);计算值C,32.36;H,4.98;N,9.44;Br,53.21.实验值C,32.20;H,5.00;N,9.30;Br,53.10.
实施例2
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-N-甲基-2-(氨基甲基)吡啶六氢溴酸盐水合物(AMD 3538)
白色固体:Mp 220-225℃(dec);1H NMR(D2O)d 2.06(m,4H),2.76(s,3H),3.20-3.65(m,16H),4.47(bs,4H),4.65(s,2H),7.54(bs,4H),7.80(t,1H),7.87(d,1H),8.28(t,1H),8.68(d,1H);13C NMR(D2O)d 18.14,18.75,18.89,36.74,37.04,37.15,37.62,40.38,40.72,40.91,41.28,44.05,47.50,56.98,58.88,60.28,127.60,128.86,130.78,130.96,132.16,132.64,144.91,145.04,146.12;FAB MS m/z 507(M+H81Br,27),507(M+H79Br,22),425(M+H,100).
分析(C25H40N6·6HBr·1.5H2O);计算值C,32.04;H,5.27;N,8.97;Br,51.16.实验值C,31.88;H,5.30;N,8.93;Br,51.00.
实施例3
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-4-(氨基-甲基)吡啶六氢溴酸盐(AMD 3500)
白色固体:mp 201-204℃(dec);1H NMR(D2O)d 1.91-2.12(m,4H),3.00-3.49(m,16H),4.13(s,2H),4.34(s,2H),4.53(s,2H),7.39-7.57(m,4H),8.02(d,2H,J=6.3Hz),8.74(d,2H,J=6.3Hz);13C NMR(D2O)d 18.26,18.88,36.94,37.29,37.36,40.89,41.06,41.44,44.21,47.61,49.17,51.43,59.02,127.84,130.21,131.64,132.15,132.45,142.19,151.67;FAB MS m/z 493(M+H81Br,8),491(M+H79Br,10),411(M+H,83),320(37),247(58),201(100).
分析(C24H38N6·6HBr);计算值C,32.17;H,4.95;N,9.34;Br,53.50.实验值C,32.16;H,5.03;N,9.41;Br,53.28.
实施例4
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-3-(氨基-甲基)吡啶六氢溴酸盐(AMD 3499)
白色固体:mp 198-202℃(dec);1H NMR(D2O)d 1.83-2.07(m,4H),2.96-3.47(m,16H),4.11(s,2H),4.32(s,2H),4.49(s,2H),7.38-7.56(m,4H),8.04(t,1H,J=6.4Hz),8.63(d,1H,J=8.3Hz),8.76(d,1H,J=5.6Hz),8.86(s,1H);13C NMR(D2O)d18.23,18.87,36.92,37.29(2C),40.88,41.05,41.43,44.17,47.22,47.60,51.18,59.04,128.29,130.01,131.49,132.14,132.66(2C),142.55,142.76,148.98;FAB MS m/z 493(M+H81Br,7),491(M+H79Br,6),411(M+H,100),320(33),247(24).
分析(C24H38N6·6HBr);计算值C,32.17;H,4.95;N,9.34;Br,53.50.实验值C,32.08;H,5.02;N,9.25;Br,53.28.
实施例5
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-2-(氨基-甲基-5-甲基)吡嗪五氢溴酸盐(AMD 3498)
白色固体:mp 194-197℃(dec);1H NMR(D2O)d 1.93-2.12(m,4H),2.42(s,3H),3.25(s,8H),3.48(s,8H),4.28(s,2H),4.30(s,2H),4.33(s,2H),7.44(s,4H),8.33(s,1H),8.46(s,1H);13C NMR(D2O)d 18.01,18.72,19.80,36.66,37.05,37.13,40.70,40.89,41.27,43.99,47.47,48.14,50.61,59.06,129.97,131.43,132.04,132.99,140.93,144.98,146.49,153.51;FAB MS m/z 509(M+H81Br,17),507(M+H79Br,15),426(M+H,100),320(21),247(20).
分析(C24H39N7·5.5HBr);计算值C,33.10;H,5.15;N,11.26;Br,50.47.实验值C,32.80;H,5.41;N,11.00;Br,50.58.
实施例6
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-2-(氨基-乙基)吡啶六氢溴酸盐(AMD 3497)
白色固体:mp 195-198℃(dec);1H NMR(D2O)d 1.98-2.17(m,4H),3.20-3.38(m,8H),3.38-3.63(m,12H),4.27(s,2H),4.39(s,2H),7.50(s,4H),7.80-7.89(m,2H),8.42(m,1H),8.58(d,1H,J=5.8Hz);13C NMR(D2O)d 18.51,19.14,29.85,37.56(3C),41.21,41.41,41.82,44.57,45.27,47.83,51.10,58.74,126.35,127.93,130.66,131.27,131.99,132.69,141.89,147.79,150.91;FAB MS m/z 507(M+H81Br,40),505(M+H79Br,34),425(M+H,100).
分析(C25H40N6·6HBr);计算值C,32.99;H,5.09;N,9.23;Br,52.67.实验值C,32.79;H,5.34;N,9.11;Br,52.45.
实施例7
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-2-(氨基-甲基)噻吩五氢溴酸盐(AMD 3516)
白色固体:mp 245-248℃(dec);1H NMR(D2O)d 1.87-2.12(m,4H),3.02-3.51(m,16H),4.17(s,4H),4.38(s,2H),6.97(t,1H,J=3.9Hz),7.13(d,1H,J=3.1Hz),7.41(s,5H);13C NMR(D2O)d 18.80,19.52,38.03,(3C),41.59(2C),42.21,44.89(2C),48.15,49.83,58.52,128.13,129.12,131.15,131.47,131.50,131.90,132.42,132.87;FAB MS m/z 498(M+H81Br,11),496(M+H79Br,9),416(M+H,53),218(100),201(64).
分析(C23H37N5S·5HBr);计算值C,33.68;H,5.16;N,8.54;Br,48.71.实验值C,33.85;H,5.22;N,8.50;Br,48.52.
实施例8
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-2-(氨基-乙基)硫醇五氢溴酸盐二水合物(AMD 3530)
白色固体:mp 234-236℃(dec);1H NMR(D2O)d 1.75-2.05(m,4H),2.75-3.45(m,20H),4.05(s,2H),4.15(s,2H),7.35(s,4H);FAB MS m/z 462(MH+H81Br,15),460(MH+H79Br,15),380(M+H,100),300(64),279(47),239(49).
分析(C20H37N5S·5HBr2 H2O0.5HOAc)期望值C,29.67;H,5.69;N,8.24;Br,46.99.实验值C,29.31;H,5.72;N,8.25;Br,46.64.
实施例9
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-2-氨基-苄胺五氢溴酸盐(AMD 3517)
白色固体:mp 203-206℃(dec);1H NMR(D2O)d 1.85-2.13(m,4H),3.02-3.58(m,16H),4.23(s,2H),4.31(s,4H),7.23-7.54(m,8H);13C NMR(D2O)d 18.03,19.29,37.78(3C),41.37(2C),42.00,44.82,46.25,47.96,51.16,58.68,124.04,124.40,129.40,130.75,131.21(2C),131.88,131.96,132.46,132.83;FAB MS m/z 507(M+H81Br,15),505(M+H79Br,18),425(M+H,100),320(30),201(51).
分析(C25H40N6·5.75HBr·0.5H2O).计算值C,33.42;H,5.19;N,9.35;Br,51.14.实验值C,33.69;H,5.35;N,9.00;Br,51.13.
实施例10
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-4-氨基-苄胺六氢溴酸盐(AMD 3544)
黄色固体:mp 120-125℃(dec);1H NMR(D2O)d 1.8-2.0(m,4H),2.9-3.4(m,16H),4.1(s,2H),4.18(s,4H),7.2-7.5(m,8H);13C NMR(D2O)d 18.86,19.57,38.14,41.76,43.74,45.14,48.24,50.14,50.42,51.49,58.38,124.13,131.13,131.30,131.83,131.92,131.96,132.67;FAB MS m/z 507(M+H81Br,5),505(M+H79Br,5),425(M+H,45),201(47),155(75),106(100).
分析(C25H40N6·6HBr·HOAc)期望值C,33.43;H,5.19;N,8.66;Br,49.42;O,3.30.实验值C,33.42;H,5.49;N,8.62;Br,49.23.
实施例11
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-4-(氨基-乙基)咪唑六氢溴酸盐(AMD 3543)
灰白色固体:mp 135-140℃(dec);1H NMR(D2O)d 1.75(m,2H),190(m,2H),2.70-3.27(m,20H),3.77(s,2H),4.14(s,2H),7.18(s,1H),7.25(d,2H,J=7.97Hz),7.37(d,2H,J=7.97Hz),8.48(s,1H);FAB MS m/z 496(M+H81Br,5),494(M+H79Br,5),414(M+H,17),201(15).
分析(C23H39N7·6HBr)期望值C,30.73;H,5.04;N,10.91;Br,53.32.实验值C,30.39;H,5.41;N,10.41;Br,53.66.
实施例12
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-苄胺五氢溴酸盐(AMD3529)
灰白色固体:mp 245-250℃(dec);1H NMR(D2O)d 1.9-2.1(m,4H),3.2-3.6(m,16H),4.12(s,2H),4.15(s,2H),4.36(s,2H),7.30(s,5H),7.41(d,2H,J=8.3Hz)7.46(d,2H,J=8.3Hz);13C NMR(D2O)d 18.43,19.06,37.29,37.46,37.63,41.09,41.32,41.68,44.46,47.74,50.18,51.00,58.79,129.53,129.97,130.18,130.35,130.68,131.18,131.92,133.14;FAB MS m/z 492(M+H81Br,13),490(M+H79Br,13),410(M+H,100),201(36).
分析(C25H39N5·5HBr);期望值C,36.88;H,5.45;N,8.60;Br,49.07.实验值C,36.79;H,5.56;N,8.48;Br,48.79.
用MTT方法(J.Virol.Methods 120:309-321(1988))在筛选中测试本发明的化合物。用浓度为100 CCID
50的HIV-1(HTLV-IIIB)或HIV-2(LAV-2 ROD)攻击MT-4细胞(2.5×10
4/孔),并在各种浓度的待测化合物存在下进行培养,这些化合物是在用病毒攻击后迅速加入的。37℃在CO
2培养箱培养5天后,用MTT(四唑)方法确定活细胞的数量。下表1中列出了这些化合物的抗病毒活性和细胞毒性,分别以EC
50(μg/ml)和CC
50(μg/ml)表示。用相应于CC
50对EC
50之比的计算选择性指数(SI)并确定潜在的治疗有效性。
|
表1 |
|
抗HIV活性数据 |
|
化合物 |
CC50(ug/mL) |
EC50(ug/mL) |
SI |
| | |
HIV-1(IIIB) |
HIV-2 |
HIV-1 |
|
1 AMD3465 |
>250 |
0.008 |
0.032 |
3×104 |
|
2 AMD3538 |
209 |
0.1 |
6.7 |
2.0×103 |
|
3 AMD3500 |
>250 |
0.6 |
10.3 |
417 |
|
4 AMD3499 |
>250 |
1.8 |
28.5 |
138 |
|
5 AMD3498 |
>250 |
0.2 |
7.1 |
1.2×103 |
|
6 AMD3497 |
>250 |
1.8 |
3.8 |
138 |
|
7 AMD3516 |
158 |
0.7 |
9.8 |
225 |
|
8 AMD3530 |
175 |
0.5 |
2.0 |
350 |
|
9 AMD3517 |
153 |
0.8 |
10.6 |
191 |
|
10 AMD3544 |
222 |
0.7 |
3.7 |
317 |
|
11 AMD3543 |
239 |
0.2 |
1.0 |
1×103 |
|
12 AMD3529 |
130 |
0.4 |
2.6 |
325 |
在本研究领域中,认为任何表现出选择性指数大于100的化合物具有进一步研究的潜力。HIV是最具攻击性病毒中的一种,上述结果大体上显示了对其它逆转录病毒和其它病毒的活性。
实施例13
N-[4-(1,4,7-三氮杂环十四烷基)-1,4-亚苯基二(亚甲基)-2(氨基甲基)吡啶(AMD 7049)
N,N′-二(2-硝基苯磺酰基)-1,7-庚二胺
将在CH2Cl2(40ml)中的2-硝基苯磺酰氯(18.80g,84.83mmol)加入到在CH2Cl2(70ml)中的1,7-庚二胺(5.01g,38.5mmol)和Et3N(13.5ml,96.9mmol)中。室温氮气中搅拌该混合液72小时,然后真空浓缩。在二乙醚(100ml)中搅拌残留物,过滤收集沉淀,用水(300ml)洗后用二乙醚(300ml)洗,得到灰色固体(18.5g,96%):1H NMR(DMF-d7)d 1.21(m,6H),1.49(m,4H),3.04(m,4H),7.87(m,2H),7.95(m,4H),8.04(m,2H),8.15(m,2H).
一般流程D
4-二乙氧基磷酰基-1,7-二(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷
将在80℃的在DMF(50ml)中的N-(二乙氧基磷酰基)-O,O′-二(2-甲磺酰基)二-乙醇胺(Bridger等人,J.Med.Chem.1995,38,366-378)(7.95g,20.0mmol)溶液逐滴加入到在氮气氛下维持于80℃的在DMF(500ml)中的N,N′-二(2-硝基苯磺酰基)-1,7-庚二胺(9.00g,18.0mmol)和Cs2CO3(17.8g,54.6mmol)的搅拌溶液中,历时8小时。继续加热17小时,然后冷却混合液,真空浓缩。残留物分配在CHCl3(140ml)和H2O(80ml)间,分出水层并用CHCl3(3×40ml)提取。干燥(MgSO4)混合的有机提取物,真空浓缩,硅胶(乙酸乙酯)柱层析纯化残留物,得到所需的黄色结晶固体大环(2.85g,带有DMF杂质)。
为了除去不希望的DMF杂质,将残留物溶解在EtOAc(75ml)中,依次用5%NaHCO3(2×10ml)和盐水(5×10ml)洗涤该溶液,干燥(MgSO4),蒸发,得到黄色非晶态固体(2.52g,20%):1H NMR(CDCl3)d 1.32(t,6H,J=7.1Hz),1.51(m,6H),1.61(m,4H),3.33(m,12H),4.03(m,4H),7.61(m,2H),7.71(m,4H),8.03(m,2H).
一般流程E
1,7-二(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷的合成
在搅拌下的在醋酸(5ml)中的上述大环化合物(1.88g,2.66mmol)的悬浮液中加入新鲜的在醋酸(20ml)中的饱和HBr(g)溶液,将所得匀质溶液在室温搅拌22小时。在反应液中加入二乙醚(250ml)后,得到沉淀,任其沉淀至烧瓶底部,并滗去上清液。通过倾析法用醚洗涤沉淀(重复3次),然后将残留物分配在CH2Cl2(40ml)和1NNaOH(25ml)水溶液间。用CH2Cl2(2×20ml)提取分出的水层,用盐水(20ml)洗涤混合的有机提取物,然后干燥(MgSO4),真空浓缩,得到黄色非晶态固体(1.23g,81%):1H NMR(CDCl3)d 1.46-1.67(m,10H),2.90(m,4H),3.34(m,8H),7.61(m,2H),7.70(m,4H),7.97(m,2H).
4-溴甲基苯甲醇
氮气下搅拌冷却到-78℃的在干CH2Cl2(150ml)中的4-溴甲基苯甲酸甲酯(5.73g,25mmol),在该溶液中逐滴加入DIBAL-H(82.5ml,在THF中的1.0M溶液)。-78℃继续搅拌1.5小时,然后让反应液升温到0℃,用水淬灭反应。分出有机层,用CH2Cl2(2×100ml)提取水层。干燥(MgSO4)混合的有机提取物,蒸发,得到所需的醇(5.0g,100%)(白色固体):1H NMR(CDCl3)d 1.84(br,1H),4.49(s,2H),4.67(s,2H),7.33(d,2H,J=8.2Hz),7.38(d,2H,J=8.2Hz).
N-2(硝基苯磺酰基)-2-(氨基甲基)吡啶
氮气下,通过插管将在干CH2Cl2(120ml)中的2-硝基苯磺酰氯(16.62g,0.075mol)逐滴加入到搅拌的在干CH2Cl2(150ml)中的2-(氨基甲基)吡啶(5.41g,0.05mol)和Et3N(13.9ml,0.10mol)的溶液中。室温搅拌反应混合液3小时,然后用水(20ml)淬灭反应。分出水层,用EtOAc(5×80ml)提取。干燥(MgSO4)混合的有机提取物,蒸发至小体积,得到白色沉淀,通过过滤收集,用CH2Cl2洗涤,得到所需的产物(11.37g,78%),白色固体:1H NMR(丙酮d6)d 4.46(s,2H),7.19(dd,1H,J=7.4,4.5Hz),7.25-7.35(br s,1H),7.39(d,1H,J=7.7Hz),7.68(ddd,1H,J=7.7,7.5,1.8Hz),7.76-7.88(m,2H),7.94(dd,1H,J=7.7,1.5Hz),8.04(dd,1H,J=7.5,1.8Hz),8.38(d,1H,J=4.5Hz).
N-[1-亚甲基-4(羟基亚甲基)-亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
氮气下,将在干CH3CN(150ml)中的N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(5.87g,20mmol)、4-溴甲基苯甲醇(4.02g,20mmol)和K2CO3(5.53g,40mmol)混合液搅拌并在60℃加热4小时。然后让混合液的温度降至室温,蒸发溶剂,残留物分配在水和CH2Cl2间。用CH2Cl2提取分出的水相,干燥(MgSO4)混合的有机提取物,蒸发。将残留物悬浮于乙酸乙酯/己烷(1∶1)中,过滤收集,得到所需的产物(6.87g,83%),白色固体:1H NMR(CDCl3)d 1.78(t,1H,J=5.8Hz),4.58(s,2H)4.60(s,2H),4.64(d,2H,J=5.8Hz),7.13-7.26(m,6H),7.54-7.59(m,2H),7.66-7.68(m,2H),7.98(d,1H,J=7.4Hz),8.40(d,1H,J=3.8Hz).
N-[1-亚甲基-4(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
氮气下,在冰浴中冷却的搅拌下的在CH2Cl2(20ml)中的上述醇(1.91g,4.62mmol)和Et3N(2.0ml,14mmol)溶液中,加入甲磺酰氯(0.73ml,9.4mmol),然后再回流加热该反应混合液6小时。用CH2Cl2(60ml)稀释该溶液,用10%HCl水溶液(2×20ml)、5% NaHCO3(20ml)水溶液和H2O(25ml)洗涤,然后干燥(MgSO4),真空浓缩,得到橙色油(1.95g,98%):1H NMR(CDCl3)d 4.52(s,2H),4.60(s,4H),7.12-7.26(m,6H),7.55(m,2H),7.67(d,2H,J=4.0Hz),7.94(d,1H,J=8.0Hz),8.41(d,1H,J=4.8Hz).此物无需进一步纯化即可用。
一般流程F
N-[4-[1,7-二(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷基]-1,4-亚苯基二(亚甲基)-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
氮气下,将1,7-二(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷(1.1 g,1.9mmo)、以上所得的氯化物(0.98g,2.3mmol)和K2CO3(0.85g,6.2mmol)的混合物在CH3CN(30ml)中回流加热62小时。真空蒸发溶剂,残留物分配在CH2Cl2(100ml)和盐水(70ml)间。分出水相,用CH2Cl2(40ml)提取,干燥(MgSO4)混合的有机提取物,真空浓缩。在硅胶柱上层析纯化(3%MeOH/CH2Cl2)残留物,蒸发,将含有所需产物的流分在硅胶柱上作第二次纯化(乙酸乙酯),得到浅黄色非晶态固体(940mg,49%):1H NMR(CDCl3)d 1.44(br s,6H),1.60(br s,4H),2.75(m,4H),3.23-3.33(m,8H),3.59(s,2H),4.58(s,2H),4.59(s,2H),7.08-7.20(m,6H),7.55-7.70(m,10H),7.82(dd,2H,J=7.6,1.6Hz),7.99(d,1H,J=7.8Hz),8.40(d,1H,J=4.7Hz).
N-[4-(1,4,7-三氮杂环十四烷基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶五氢溴酸盐二水合物
室温,将从上所得的中间体(870mg,0.90mmol)、K3CO3(1.15g,8.32mmol)和苯硫酚(0.33ml,3.2mmol)在DMF(12ml)中搅拌7.5小时。真空浓缩混合液,残留物分配在CH2Cl2(30ml)和H2O(15ml)间。分出有机相,用5%NaHCO3(10ml)、然后用H2O(10ml)洗涤,干燥(MgSO4),真空浓缩。通过在碱性氧化铝柱上的层析纯化黄色残留物(CH2Cl2,1%MeOH/CH2Cl2,和10%MeOH/CH2Cl2),得到游离碱,黄色油(134mg,36%):1H NMR(CDCl3)d 1.48(br s,6H),1.60(br s,4H),2.61(m,12H),3.56(s,2H),3.83(s,2H),3.92(s,2H),7.16(m,1H),7.24(m,2H),7.32(m,3H),7.79(m,1H),8.56(d,1H,J=4.7Hz).
将该游离碱(134mg,0.33mmol)溶解在EtOH(4ml)中,加入在EtOH(9ml)中新鲜制备的饱和HBr(g)溶液,得到白色沉淀。搅拌混合液5分钟,加入二乙醚(15ml)。让沉淀降至烧瓶底部,滗去上清液。然后将固体溶解在MeOH(5ml)中,用大量的醚再次沉淀,用醚倾滗冲洗(15×),在减压下(室温)通过蒸发除去最后痕量的醚。40℃真空干燥固体16小时,得到所需的产物,白色固体(178mg,63%):1H NMR(DMSO-d6)d 1.44(br s,6H),1.75(br s,4H),3.04(br s,8H),3.37(m,4H),4.06(br s,2H),4.31(s,2H),4.38(s,2H),7.52-7.68(m,6H),8.01(m,1H),8.70(d,1H,J=5.0Hz);FAB-MS m/z 492(MH+H81Br),490(MH+H79Br),410(M+H).分析计算C25H39N5·5HBr0.1Et2O·2.3H2O:C,35.35;H,5.79;N,8.11;Br,46.29.实验值:C,35.55;H,5.70;N,8.18;Br,46.17.
实施例14
N-[7-(4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD 7050)
2,6-二(2-氨基乙基)吡啶的制备见Bridger等人的美国专利No.5,698,546,该专利本文全部纳入作为参考。
2,6-二[N-(2-硝基苯磺酰基)-2-氨基乙基]吡啶
将在CH2Cl2(20ml)中的2-硝基苯磺酰氯(8.01g,36.1mmol)加入到搅拌的在CH2Cl2(35ml)中的2,6-二(2-氨基乙基)吡啶(2.7g,16mmol)和Et3N(5.7ml,41mmol)的溶液中,室温氮气下搅拌该混合液42小时。用盐水(25ml)洗涤混合液,干燥(MgSO4)有机相,真空浓缩。在硅胶柱上层析纯化褐色残留物(50%,然后60%THF/己烷),得到浅黄色固体(5.2g,59%):1H NMR(CDCl3)d 3.01(m,4H),3.52(m,4H),6.38(m,2H),6.94(d,2H,J=7.7,Hz),7.47(t,1H,J=7.7Hz),7.72(m,4H),7.82(m,2H),8.13(m,2H).
7-二乙氧基磷酰基-4,10-二(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯
用一般流程D:将2,6-二[N-(2-硝基苯磺酰基)-2-氨基乙基]吡啶(5.2g,9.7mmol)和N-(二乙氧基磷酰基)-O,O′-二(2-甲基磺酰基)二-乙醇胺(4.25g,10.7mmol)反应,硅胶柱纯化(60%,然后90%THF/己烷)反应产物,得到标题化合物,黄色非晶态固体(1.48g,21%):1H NMR(CDCl3)d 1.23(t,6H,J=7.1Hz),2.60(m,4H),2.98-3.08(m,8H),3.84-3.94(m,8H),7.11(d,2H,J=7.6Hz),7.56-7.74(m,7H),8.07(m,2H).
4,10-二(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯
用一般流程E:将7-二乙氧基磷酰基-4,10-二(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(1.04g,1.4mmol)反应,得到标题化合物,黄色非晶态固体(744mg,88%):1H NMR(CDCl3)d 2.81(m,4H),3.08(m,4H),3.33(m,4H),3.88(m,4H),7.07(d,2H,J=7.7Hz),7.54-7.71(m,7H),8.02(m,2H).
N-[7-[4,10-二(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程F:将4,10-二(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(740mg,1.2mmol)和N-[1-亚甲基-4-(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(610mg,1.4mmol)反应,硅胶柱纯化(50%,然后80%THF/己烷)反应产物,得到标题化合物,黄色非晶态固体(648mg,54%):1H NMR(CDCl3)d 2.26(m,4H),3.03(m,8H),3.37(s,2H),3.94(m,4H),4.56(s,2H),4.57(s,2H),6.95-7.17(m,8H),7.52-7.72(m,11H),7.85(m,2H),7.98(d,1H,J=7.7Hz),8.39(d,1H,J=4.8Hz).
一般流程G
N-[7-(4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶六氢溴酸盐三水合物
将苯硫酚(0.24ml,2.3mmol)加入到在含有K2CO3(806mg,5.83mmol)的DMF(9ml)中的N-[7-[4,10-双(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(640mg,0.64mmol)中,室温搅拌该混合液2小时。真空浓缩混合液,残留物分配在乙酸乙酯(30ml)和水(10ml)间。分出有机相,用5%NaHCO3(3×5ml)、然后用盐水(5ml)洗涤。用CH2Cl2(3×10ml)提取混合水相。干燥(MgSO4)混合的有机提取物,在氧化铝柱上层析纯化残留物(CH2Cl2,然后10%MeOH/CH2Cl2),得到标题化合物的游离碱,黄色油(83mg,29%):1H NMR(CDCl3)d 2.57(m,8H),3.01(s,8H),3.36(s,2H),3.78(s,2H),3.92(s,2H),6.64(d,2H,J=8.0Hz),7.07(m,4H),7.18(m,1H),7.33(d,1H,J=7.7Hz),7.67(m,2H),8.58(d,1H,J=4.8Hz).
将该游离碱(74mg,0.1 7mmol)溶解在MeOH(3ml)中,加入在MeOH(7ml)中新鲜配制的饱和HBr(g)溶液,得到白色沉淀。搅拌混合液5分钟,加入二乙醚(10ml),让沉淀降至烧瓶底部,倾滗除去上清液。用MeOH(5×5ml)、然后用醚(10×5ml)倾滗洗涤固体,真空蒸发以除去最后痕量的醚,40℃真空干燥17.5小时,得到标题化合物,白色固体(153mg,93%):1H NMR(DMSO-d6)d 2.81(br s,4H),3.28(m,8H),3.61(br s,4H),3.85(s,2H),4.27(s,2H),4.36(s,2H),7.29(d,2H,J=7.7Hz),7.36(d,2H,J=7.7Hz),7.53(m,3H),7.63(d,1H,J=7.7Hz),7.80(t,1H,J=7.7Hz),7.99(m,1H),8.69(d,1H,J=5.3Hz);FAB-MS m/z 527(MH+H81Br),525(MH+H79Br),445(M+H).分析计算C27H36N66HBr3H2O:C,32.95;H,4.92;N,8.54;Br,48.72.实验值:C,32.75;H,4.89;N,8.39;Br,48.61.
实施例15
N-[7-(4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD 7051)
1,3-亚苯基二(亚甲基)二胺
将Raney镍(约20g,先用CH3OH洗涤若干次)加入到在CH3OH(用NH3饱和的,150ml)中的1,3-亚苯基二乙腈(9.37,60mmol)的溶液中,45psi在Parr装置上将混合液氢化48小时。将反应液滤过硅藻土,蒸发滤液得到粗产物(9.45g,96%),淡绿色油:1H NMR(CDCl3)δ0.80-1.50(br s,4H),2.70-2.76(m,4H),2.94-2.99(m,4H),7.01-7.07(m,3H),7.18-7.26(m,1H).
N,N′-二(2-硝基苯磺酰基)-1,3-亚苯基二(亚甲基)二胺
氮气下,通过插管将在干CH2Cl2(70ml)中的2-硝基苯磺酰氯(19.94g,0.090mol)的溶液逐滴加入到搅拌的在CH2Cl2(80ml)中的1,3-苯氧基二(亚乙基)二胺(4.92g,0.030mol)和Et3N(16.7ml,0.12mol)的溶液中。室温搅拌反应混合液过夜,然后用水(20ml)淬灭反应。过滤收集沉淀,用H2O、CH3OH和Et2O洗涤,得到所需的产物(9.22g,58%),白色固体:1H NMR(DMSO-d6)δ2.66(t,4H,J=7.7Hz),3.08-3.18(br s,4H),6.94(d,2H,J=6.4Hz),6.98(s,1H),7.12(dd,1H,J=6.4,6.4Hz),7.78-7.84(br m,4H),7.90-7.64(br m,4H),8.16(br s,2H).
7-二乙氧基磷酰基-4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯
用一般流程D:将N,N′-二(2-硝基苯磺酰基)-1,3-亚苯基二(亚乙基)二胺(8.74g,16.4mmol)与N-(二乙氧基磷酰基)-O,O′-二(2-甲基磺酰基)二-乙醇胺(6.50g,16.4mmol)反应,然后用硅胶柱纯化(1∶15∶35 CH3OH-Et2O-CH2Cl2)反应产物,得到标题化合物,黄色泡沫状(4.03g,33%):1H NMR(CDCl3)δ1.21(t,6H,J=6.4Hz),2.39-2.46(br m,4H),2.83-2.97(br m,8H),3.68-3.72(m,4H),3.80-3.92(m,4H),7.16(d,2H,J=6.5Hz),7.18(s,1H),7.24(dd,1H,J=6.5,6.5Hz),7.60-7.74(m,6H),8.04-8.08(m,2H).
4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯
用一般流程E:将7-二乙氧基磷酰基-4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(1.27g,1.72mmol)反应,然后用硅胶柱纯化(1∶15∶25 CH3OH-EtOAc-CH2Cl2,然后用在CH2Cl2中的20%CH3OH)反应产物,得到标题化合物,浅黄色泡沫状(574mg,57%):1H NMR(CDCl3)δ1.42-1.50(br,1H),2.01(t,4H,J=5.4Hz),2.90-3.10(br m,4H),3.08(t,4H,J=5.4Hz),3.56-3.60(br m,4H),7.16(d,2H,J=6.8Hz),7.31(dd,1H,J=6.8,6.8Hz),7.36(s,1H),7.61-7.63(m,2H),7.70-7.73(m,4H),8.01-8.04(m,2H).
N-[7-[4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程F:将4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(420mg,0.7mmol)和N-[1-亚甲基-4-(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(302mg,0.7mmol)反应,然后用硅胶柱纯化(1∶3 Et2O-CH2Cl2)反应产物,得到标题化合物,苍黄色固体(491mg,70%):1H NMR(CDCl3)δ1.97-2.02(br m,4H),2.73-2.78(br m,4H),2.90-2.94(br m,4H),3.32(s,2H),3.64-3.67(br m,4H),4.55(s,2H),4.58(s,2H),6.93(d,2H,J=8.0Hz),7.04(d,2H,J=8.0Hz),7.09-7.16(br m,4H),7.23(s,1H),7.29(dd,1H,J=7.9,7.9Hz),7.51-7.72(m,10H),7.80-7.83(m,2H),7.98(d,1H,J=7.8Hz),8.39(m,1H).
N-[7-(4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶五氢溴酸盐二水合物
用一般流程G:用N-[7-[4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(380mg,0.38mmol)反应,然后用碱性氧化铝柱纯化反应产物(1∶20CH3OH-CH2Cl2),得到标题化合物的游离碱。
用在CH3OH中的饱和HBr(g)溶液将该游离碱转化成氢溴酸盐,真空干燥过夜,得到标题化合物(110mg,34%全程产率),白色固体:1H NMR(DMSO-d6)δ2.80-2.88(br s,4H),3.02-3.06(br s,4H),3.10-3.16(br s,4H),3.38-3.44(br s,4H),3.80-3.86(br s,2H),4.25-4.30(br s,2H),4.33-4.37(br s,2H),7.27-7.32(br m,4H),7.42-7.63(br m,6H),7.96(dd,1H,J=7.7,7.7Hz),8.10-8.30(br s,3H),8.69(d,1H,J=4.9Hz),9.45-9.62(br s,2H);FAB-MS m/z 526(MH+H81Br),524(MH+H79Br),444(M+H,100);分析计算C28H42N5Br5·2H2O:C,38.03;H,5.24;N,7.92;Br,45.18.实验值:C,38.37;H,5.28;N,7.76;Br,45.36.
实施例16
N-[1-(1,4,7-三氮杂环十四烷基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD 7059)
一般流程H
4-二乙氧基磷酰基-7-(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷
氮气下,将在DMF(8ml)中的苯硫酚(0.15ml,1.46mmol)溶液逐滴加入到搅拌的在DMF(11ml)中的4-二乙氧基磷酰基-1,7-二(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷(1.32g,1.87mmol)和K2CO3(654mg,4.73mmol)溶液中,历时1小时。再搅拌该混合物3小时,然后真空浓缩。残留物分配在CHCl3(50ml)和H2O(25ml)间。分出水相,用CHCl3(3×20ml)提取,干燥(MgSO4)混合的有机提取物,真空浓缩。在碱性氧化铝柱上层析纯化残留物(CHCl3,然后3%MeOH/CHCl3),得到标题化合物,黄色油(178mg,23%):1H NMR(CDCl3)δ1.31(t,6H,J=7.0Hz),1.40-1.67(m,10H),2.65(m,2H),2.78(m,2H),3.12(m,2H),3.26-3.37(m,4H),3.48(m,2H),3.97-4.09(m,4H),7.61(m,1H),7.68(m,2H),8.06(m,1H).
N-[1-[4-二乙氧基磷酰基-7-(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程F:将4-二乙氧基磷酰基-7-(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷(236mg,0.453mmol)和N-[1-亚甲基-4-(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(238mg,0.551mmol)反应,然后硅胶柱纯化(50%,然后80%THF/己烷)反应产物,得到标题化合物,黄色非晶态固体(305mg,73%):1H NMR(CDCl3)δ1.27(t,6H,J=7.1Hz),1.43(br s,8H),1.63(br s,2H),2.32(br s,2H),2.55(m,2H),3.13-3.41(m,8H),3.49(s,2H),3.85-4.02(m,4H),4.57(s,2H),4.58(s,2H),7.07-7.22(m,6H),7.51-7.71(m,7H),7.98(d,1H,J=7.4Hz),8.04(m,1H),8.41(d,1H,J=4.0Hz).
N-[1-[7-(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程E:将N-[1-[4-二乙氧基磷酰基-7-(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(300mg,0.328mmol)反应,得到标题化合物,黄色非晶态固体(214mg,84%):1H NMR(CDCl3)δ1.34-1.44(m,8H),1.69(br s,2H),2.34(m,2H),2.52(m,2H),2.62(m,2H),2.82(m,2H),3.42(m,6H),4.58(s,2H),4.59(s,2H),7.08-7.24(m,6H),7.52-7.71(m,7H),8.01(m,2H),8.42(d,1H,J=4.1Hz).
N-[1-(1,4,7-三氮杂环十四烷基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶五氢溴酸盐二水合物
将在乙腈(3ml)中的N-[1-[7-(2-硝基苯磺酰基)-1,4,7-三氮杂环十四烷基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(209mg,0.268mmol)、K2CO3(298mg,2.16mmol)和苯硫酚(0.17ml,1.7mmol)混合物加热到50℃,16.5小时。用CH2Cl2(10ml)稀释反应混合物,用盐水(10ml)洗涤。用CH2Cl2(3×5ml)提取分出的水相,干燥(MgSO4)混合的有机提取物,蒸发。碱性氧化铝柱上层析纯化残留物(CHCl3,然后10%MeOH/CHCl3),得到标题化合物的游离碱,黄色油(92mg,84%):1H NMR(CDCl3)δ1.21-1.62(m,10H),2.40-2.49(m,4H),2.60(m,6H),2.79(m,2H),3.49(s,2H),3.80(s,2H),3.91(s,2H),7.14(m,1H),7.28(m,5H),7.62(m,1H),8.54(d,1H,J=4.4Hz).
用在MeOH中的饱和HBr(g)溶液将游离碱(86mg,0.21mmol)转化成氢溴酸盐(见一般流程G),40℃真空干燥15.5小时,得到标题化合物,白色固体(128mg,70%):1H NMR(D2O)δ1.48(br s,6H),1.82(m,4H),3.22-3.36(m,10H),3.50(brs,2H),4.48(s,4H),4.64(s,2H),7.62(s,4H),7.88(m,1H),7.94(d,1H,J=8.0Hz),8.38(m,1H),8.77(d,1H,J=5.2Hz);FAB-MS m/z 492(MH+H81Br),490(M+H79Br),410(M+H).分析计算C25H39N5·5HBr·2.5H2O·0.1Et2O:C,35.20;H,5.82;N,8.08;Br,46.10.实验值:C,35.48;H,5.66;N,8.10;Br,46.05.
实施例17
N-[4-[4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD 7063)
7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯
用一般流程H:将7-二乙氧基磷酰基-4,10-二(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(1.48g,2.00mmol)与苯硫酚反应(添加后,将反应混合物再加热到50℃,1.5小时),硅胶柱纯化反应产物(8%MeOH/CHCl3),得到标题化合物,浅黄色油(423mg,52%):1H NMR(CDCl3)δ1.23(t,6H,J=7.0Hz),2.50(br s,2H),2.79(br s,2H),3.02-3.15(m,10H),3.82-3.98(m,6H),7.06(d,2H,J=7.6Hz),7.54-7.63(m,2H),7.70(m,2H),8.01(br s,1H).
N-[4-[7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程F:将7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(410mg,0.738mmol)和N-[1-亚甲基-4-(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(397mg,0.919mmol)反应,然后硅胶柱纯化(50%,80%,然后90%THF/己烷)反应产物,得到标题化合物,白色非晶态固体(441mg,63%):1H NMR(CDCl3)δ1.15(t,6H,J=7.0Hz),2.42(m,4H),2.77(m,2H),2.92-3.02(m,6H),3.10(m,2H),3.59(s,2H),3.66-3.91(m,6H),4.58(s,2H),4.59(s,2H),6.94(d,1H,J=7.6Hz),7.07-7.14(m,6H),7.22(d,1H,J=7.8Hz),7.51-7.72(m,8H),8.00(d,1H,J=7.8Hz),8.04(m,1H),8.42(d,1H,J=4.0Hz).
N-[4-[7-二乙氧基磷酰基-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-氨基甲基)吡啶
将在CH3CN(3.5ml)中的N-[4-[7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(434mg,0.456mmol)、K2CO3(508mg,3.68mmol)和苯硫酚(0.28ml,2.7mmol)混合物在氮气下加热到50℃,15小时。冷却后,反应液分配在CHCl3(15ml)和盐水(15ml)间,分出水层,用CHCl3(3×5ml)提取。干燥(MgSO4)混合的有机提取物,真空浓缩。碱性氧化铝柱上层析纯化残留物(CHCl3,然后10%MeOH/CHCl3),得到黄色油(218mg,82%):1H NMR(CDCl3)δ1.16(t,6H,J=7.1Hz),2.35(m,2H),2.55(m,2H),2.75(m,2H),2.82(m,2H),2.96-3.08(m,6H),3.16(m,2H),3.68(s,2H),3.69-3.88(m,4H),3.82(s,2H),3.93(s,2H),6.95(d,1H,J=7.6Hz),7.00(d,1H,J=7.5Hz),7.15-7.34(m,6H),7.50(m,1H),7.65(m,1H),8.56(d,1H,J=4.7Hz).
N-[4-[4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶六氢溴酸盐水合物
在搅拌下的在醋酸(0.6ml)中的N-[4-[7-二乙氧基磷酰基-4,7,10,17-四氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-氨基甲基)吡啶(211mg,0.36mmol)溶液中加入新鲜的在醋酸(6ml)中的饱和HBr(g)溶液,室温搅拌反应混合物4小时。加入二乙醚(10ml)后得到沉淀,让沉淀降至烧瓶底部,倾滗除去上清液。通过倾滗用MeOH(4×5ml)和醚(6×5ml)洗涤固体,减压下蒸发除去痕量残留的醚。40℃真空干燥产物17小时,得到标题化合物,苍黄色固体(223mg,63%):1H NMR(D2O)δ3.14-3.36(m,10H),3.55(m,4H),3.75(m,2H),4.45(s,2H),4.50(s,2H),4.64(s,2H),7.22(m,2H),7.53(s,4H),7.70(m,1H),7.95(m,1H),8.00(d,1H,J=7.9Hz),8.46(m,1H),8.79(d,1H,J=3.9Hz);FAB-MSm/z 527(MH+H81Br),525(MH+H79Br),445(M+H).分析计算C27H36N6·6HBr1.5H2O·0.2Et2O:C,34.35;H,4.87;N,8.65;Br,49.33.实验值:C,34.57;H,5.04;N,8.68;Br,49.09.
实施例18
N-[4-[4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD 7058)
7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯
用一般流程H:用7-二乙氧基磷酰基-4,10-二(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(1.11g,1.5mmol)反应,然后硅胶柱纯化反应产物(2∶5∶20 CH3OH-Et2O-CH2Cl2,然后1∶5 CH3OH-CH2Cl2),得到标题化合物,苍黄色油(300mg,54%):1H NMR(CDCl3)δ1.21(t,6H,J=7.1Hz),1.78-1.92(br s,1H),2.31-2.38(br m,2H),2.56-2.60(br m,2H),2.81-2.98(br m,10H),3.60-3.64(br m,2H),3.75-3.91(m,4H),7.05-7.12(m,2H),7.24-7.29(m,2H),7.60-7.63(m,1H),7.68-7.71(m,2H),8.02-8.06(m,1H).
N-[4-[7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程F:将7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯(290mg,0.52mmol)和N-[1-亚甲基-4-(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(271mg,0.63mmol)反应,然后硅胶柱纯化(1∶12∶12 CH3OH-Et2O-CH2Cl2)反应产物,得到标题化合物,苍黄色固体(298mg,60%):1H NMR(CDCl3)δ1.17(t,6H,J=7.0Hz),2.29-2.45(br m,4H),2.55-2.65(br m,2H),2.71-2.75(br s,4H),2.85-2.91(br m,2H),2.96-2.98(br m,2H),3.57(s,2H),3.67-3.84(br m,6H),4.57-4.61(br s,4H),7.07-7.28(br m,10H),7.55-7.71(br m,7H),7.99-8.02(m,2H),8.42-8.46(m,1H).
N-[4-[10-(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶
用一般流程E:用N-[4-[7-二乙氧基磷酰基-10-(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(290mg,0.31mmol)反应,得到标题化合物,白色固体(240mg,95%):1H NMR(CDCl3)δ1.65-1.79(br s,1H,与H2O峰一致),2.15-2.19(br m,4H),2.44-2.48(br m,2H),2.61-2.65(br m,2H),2.67-2.71(br m,2H),3.00-3.04(br m,2H),3.10-3.14(br m,2H),3.56-3.60(br s,4H),4.55(s,2H),4.61(s,2H),6.96(d,1H,J=7.8Hz),7.02-7.10(br m,6H),7.22-7.28(br m,3H),7.52-7.72(br m,7H),7.96-7.99(m,2H),8.42-8.46(m,1H).本化合物无需进一步纯化即可使用。
N-[4-[4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶五氢溴酸盐二水合物
用一般流程G:用N-[4-[10-(2-硝基苯磺酰基)-4,7,10-三氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基]-1,4-亚苯基二(亚甲基)]-N-(2-硝基苯磺酰基)-2-(氨基甲基)吡啶(236mg,0.29mmol)反应,然后用氧化铝柱纯化反应产物(1∶99 CH3OH-CH2Cl2,然后1∶10 CH3OH-CH2Cl2),得到标题化合物的游离碱,苍黄色油(111mg,86%):1H NMR(CDCl3)δ2.24-2.28(br s,3H),2.43-2.50(br m,4H),2.58-2.62(br m,2H),2.73-2.79(br m,8H),2.95-2.98(br m,2H),3.50(s,2H),3.77(s,2H),3.90(s,2H),6.83-6.87(br m,3H),7.05-7.33(br m,7H),7.63-7.67(m,1H),8.54-8.56(m,1H).
用在CH3OH中的饱和HBr(g)溶液将该游离碱转化成氢溴酸盐,真空干燥,得到标题化合物(101mg,52%),白色固体:1H NMR(D2O)δ2.90-2.94(br m,2H),2.97-3.01(br m,2H),3.12-3.16(br m,2H),3.17-3.21(br m,2H),3.24-3.28(br m,4H),3.47-3.51(br m,2H),3.57-3.61(br m,2H),4.38-4.42(m,6H),7.34-7.40(m,2H),7.46-7.60(m,8H),7.90-7.94(m,1H),8.58-8.62(m,1H);FAB-MS m/z 526(MH+H81Br),524(MH+H79Br),444(M+H,100);分析计算C28H42N5Br5·2.5H2O:C,37.65;H,5.30;N,7.84;Br,44.73.实验值:C,37.53;H,5.26;N,7.79;Br,44.75.
表2
|
化合物 |
对mAb 12G5结合的抑制IC50 a(ng/ml) |
|
AMD3100 |
27 |
|
AMD3465 |
3 |
|
AMD7049 |
52 |
|
AMD7050 |
1 |
|
AMD7051 |
7 |
|
AMD7058 |
>1000 |
|
AMD7059 |
>1000 |
|
AMD7063 |
9 |
|
SDF-1αb |
270 |
a在SUP-T1细胞中对mAb 12G5结合到CXCR-4的抑制
bCXCR-4的天然配体(Bleul等人,Nature,382:829-832(1996);Oberlin等人,Nature,382:833-835(1996))。
表3
|
化合物 |
%对Ca2+流动(浓度)或IC50 a(ng/ml)的抑制 |
|
AMD3100 |
5 |
|
AMD3465 |
1 |
|
AMD7049 |
100%(1μg/ml) |
|
AMD7050 |
100%(1μg/ml) |
|
AMD7051 |
100%(1μg/ml) |
|
AMD7058 |
44%(1μg/ml) |
|
AMD7059 |
36%(1μg/ml) |
|
AMD7063 |
100%(1μg/ml) |
a对由SDF-1α结合到SUP-T1细胞上的CXCR-4诱导的信号转导(增加细胞内Ca2+流动)的抑制
以下所有的化合物(包括AMD-3484,见图28)都是用以下方法合成的:Bridger等人,J.Med.Chem.1995,38,366-378;J.Med.Chem.1996,39,109-119和美国专利No.5,583,131、美国专利No.5,698,546和美国专利No.5,817,807,这些文献本文全部纳入作为参考。
实施例19
1-[2,6-二甲氧基吡啶-4-基(亚甲基)]-1,4,8,11-四氮杂环十四烷四氢溴酸盐(AMD 7032)
1H NMR(D2O)δ1.78(m,2H),1.88-1.92(m,2H),2.59-3.03(m,16H),3.60(s,2H),3.91(s,6H),6.44(s,2H);13C NMR(D2O)δ26.75,27.91,48.34,49.21,49.89,50.96,52.01,52.86,54.88,57.15,57.53,59.42,142.65,157.42,166.42;FAB MSm/z 352(M+H);分析(C18H33N5O2 4.1HBr0.25H2O);计算值C,31.44;H,5.51;N,10.18;Br,47.64.实验值C,31.17;H,5.61;N,9.92;Br,47.54.
实施例20
1-[2-氯吡啶-4-基(亚甲基)]-1,4,8,11-四氮杂环十四烷四盐酸盐一水合物(AMD 7048)
1H NMR(D2O)δ1.92(m,2H),2.12(m,2H),2.77-2.80(m,4H),2.96-3.39(m,12H),3.85(s,2H),7.33(d,1H,J=5.4Hz),7.44(s,1H),8.40(d,1H,J=5.4Hz);13C NMR(D2O)δ24.75,27.59,47.40,47.55,49.11,49.23,52.12,52.40,53.81,54.42,56.98,126.97,128.30,151.90,152.34,153.78;FAB MS m/z 326(M+H);分析(C16H28N5Cl.4.2HCl.0.5HOAc.1.1H2O);计算值C,38.61;H,6.94;N,13.24;Cl,34.86.实验值C,38.63;H,6.94;N,13.52;Cl,34.61.
实施例21
1-[2,6-二甲基吡啶-4-基(亚甲基)]-1,4,8,11-四氮杂环十四烷五氢溴酸盐二水合物(AMD 7060)
1H NMR(D2O)δ1.77(m,2H),1.93(m,2H),2.48(s,6H),2.61-3.00(m,16H),3.61(s,2H),7.07(s,2H);13C NMR(D2O)δ25.30,26.22,27.49,47.75,48.65,49.43,50.41,51.58,52.19,54.09,56.63,58.46;FAB MS m/z320(M+H);分析(C18H33N5.5HBr.0.5HOAc.1.7H2O);计算值C,29.08;H,5.57;N,8.92;Br,50.91.实验值C,29.04;H,5.60;N,8.73;Br,50.87.
实施例22
1-[2-甲基吡啶-4-基(亚甲基)]-1,4,8,11-四氮杂环十四烷五氢溴酸盐二水合物(AMD 7061)
1H NMR(D2O)δ2.01-2.08(m,2H),2.22(m,2H),2.70-2.72(m,2H),2.77(s,3H),2.91-2.92(m,2H),3.33-3.52(m,12H),4.00(s,2H),7.86-7.89(m,2H),8.56(d,1H,J=5.7Hz);FAB MS m/z 306(M+H);分析(C17H31N5.4.9HBr.0.3HOAc.2.1H2O);计算值C,27.9;H,5.49;N,9.24;Br,51.67.实验值C,28.08;H,5.50;N,9.56;Br,51.56.
实施例23
1-[2,6-二氯吡啶-4-基(亚甲基)]-1,4,8,11-四氮杂环十四烷三盐酸盐双水合物(AMD 3451)
1H NMR(D2O)d 1.83-1.88(m,2H),2.04-2.08(m,2H),2.58-2.62(m,2H),2.79-2.81(m,2H),3.12-3.44(m,12H),3.69(s,2H),7.30(s,2H);13C NMR(D2O)36.26,37.69,55.26,56.18,58.33,58.56,58.92,59.23,63.57,65.44,70.72,140.37,166.14,167.37;FAB MS m/z 360(M+H).分析(C16H34N5Cl5O2):计算值.C,38.00;H,6.78;N,13.85;Cl,35.05.实验值:C,38.33;H,6.42;N,13.88;Cl,35.43.
实施例24
1-[2-氯吡啶-5-基(亚甲基)]-1,4,8,11-四氮杂环十四烷四盐酸盐半水合物(AMD 3454)
1H NMR(D2O)δ1.96-2.09(br m,4H),3.02-3.17(m,4H),3.19-3.28(br m,8H),3.40(s,4H),4.10(s,2H),7.40(d,1H,J=8.2Hz),7.80(d,1H,J=8.2Hz),8.27(s,1H);13C NMR(D2O)δ19.36,19.47,38.17,38.64,39.06,41.74,41.88,42.18,45.66,48.29,54.62,125.59,126.69,142.79,150.77,151.75;FAB-MS m/z 326(M+H).分析计算C16H28N5Cl.4HCl.0.5H2O:C,39.98;H,6.92;N,14.57;Cl,36.87.实验值:C,40.36;H,7.06;N,14.56;Cl,36.92.
一般流程A、B和C用于制备以下化合物:
实施例25
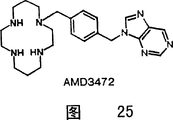
N-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-嘌呤五氢溴酸盐二水合物(AMD 3472)
1H NMR(D2O)δ1.88-2.05(br m,4H),3.06-3.22(br m,8H),3.27-3.44(brm,8H),4.22(s,2H),5.59(s,2H),7.29(s,4H),8.80(s,1H),9.11(s,1H),9.28(s,1H);13C NMR(D2O)δ18.39,19.25,37.24,37.55,37.71,41.13,41.37,41.71,44.41,47.73,54.87,129.45,131.81,132.53,136.67,140.96,147.88,152.46,154.37;FAB-MS m/z 423(M+H).分析计算C23H34N8.5HBr.2H2O.0.5CH3CO2H:C,32.27;H,5.07;N,12.54;Br,44.73.实验值:C,32.66;H,4.81;N,12.41;Br,44.58.
实施例26
1-[1,4,8,11-四氮杂环十四烷基-1,4-亚苯基二(亚甲基)]-4-苯基哌嗪五氢溴酸盐水合物(AMD 3526)
1H NMR(D2O)δ1.88-2.06(br m,4H),3.11-3.53(br m,24H),4.30(s,2H),4.32(s,2H),6.89-6.97(m,3H),7.19-7.24(m,2H),7.49(s,4H);13C NMR(D2O)δ18.74,19.37,37.34,41.47,41.76,42.03,44.31,47.45,48.26,51.16,58.48,59.29,118.18,122.34,129.99,130.37,131.53,131.85,132.62,148.47;FAB-MS m/z 547(M+H81Br),545(M+H79Br),465(M+H).分析计算C28H44N6.5HBr.H2O:C,37.90;H,5.79;N,9.47;Br,45.03.实验值:C,37.72;H,5.98;N,9.38;Br,46.78.
表4
|
化合物 |
IC50 a(μg/mL) |
|
AMD3451 |
8.9 |
|
AMD3472 |
45.4 |
|
AMD3454 |
32.3 |
|
AMD3526 |
82 |
|
AMD3100 |
>100 |
aAMD化合物对抗HIV-1 BaL感染U87.CD4.CCR5的50%抑制浓度(IC50)(μg/ml)
实施例27
对胶原诱导的关节炎的抑制
证明本发明所用的一个化合物可抑制突变型小鼠模型中胶原诱导的关节炎(CIA)。
方法
实验动物处理
如下所述将胶原注射到由十只小鼠组成的对照组。由八只小鼠组成的处理组也注射胶原,在胶原注射后的14天以5mg/ml的浓度,用渗透泵静脉注射化合物(1,1′-[1,4-亚苯基二(亚甲基)]二-1,4,8,11-四氮杂环十四烷)(AMD 3100,见图2 7)(200μl,Alga,0.5μl/小时)。
突变型小鼠
Huang S等人,Science 259:1742,(1993)已描述了编码IFN-γ受体(IFN-γRKO)α链的基因被破坏的突变型小鼠品系(129/Sv/Ev)的传代和基本特征。将这些IFN-γRKO小鼠与DBA/1野生型小鼠回交10代,得到用于本研究的DBA/1 IFN-γRKO小鼠。在Leuven大学的实验动物中心IFN-γRKO小鼠和野生型小鼠交配。实验是在8到12周龄大的小鼠上进行的,但在每个实验中,突变型小鼠和野生型小鼠年龄匹配的限制为5天。每个实验组中雄性和雌性小鼠的比例保持在0.8和1.3之间。
诱导胶原诱导的关节炎和关节炎的临床确认
用以下方法实现胶原诱导的关节炎(见Vermeire等人,Int.J.Immunol.158:5507.5513,(1997))。6℃,以2mg/ml通过搅拌将天然鸡胶原II(EPC.Owensville.MO)溶解在0.05M醋酸中过夜,然后用等体积的含有1.5mg/ml热致死乳酪分枝杆菌(Difco,Detroit,MI)的不完全弗氏佐剂(IFA)或完全弗氏佐剂(CFA)乳化。通过在尾底部单次皮内注射100μl乳剂致敏小鼠。每天检查小鼠患关节炎的征兆。对每条肢用计分系统记录病重情况。计分0:正常;计分1:一个关节红色和/或肿胀;计分2:多于一个关节红色和/或肿胀;计分3:整个爪子红色和/或肿胀;计分4:畸形或僵硬。
组织学检查
在缓冲过的盐水-B5固定剂(10%带汞的福尔马林)中固定脾和前肢及后肢。另外,在10%福尔马林或纯甲醇中固定组织(见:Vermeire等人,J.Immunol.,158,5507-5513,1997)。然后用甲酸将四肢脱钙过夜。用苏木精和曙红染色4微米厚的石蜡切片。用以下三个参数评估关节炎的严重度:单形和分叶核细胞的渗入、滑膜的增生和parmus的形成。每个参数都以0-3的计分(无;弱、中等和严重)记录。
体内抗体处理
通过对姥鲛烷致敏的无胸腺裸鼠的腹腔内接种(nu/nu NMRI背景)生长的杂交瘤产生单克隆抗体。通过在小鼠抗大鼠κ链单克隆抗体上的亲和层析纯化抗MuIFN-γ(F3,率IgG24)的中和单克隆抗体(Billiau A等人,J.Immunol.140:1506,(1988))。中和滴度(终点稀释度,相应于30单位/ml小鼠IFN-γ对由门果病毒攻击的小鼠1929细胞的抗病毒作用中和的50%)是1053U/ml(IgG含量,1.4mg/ml)。用杂交瘤C17.8(由Dr.G.Trinchieri,Wistar Institute,Philadelphia,PA,友情提供)产生IgG24抗体对鼠科IL-12的中和率。在蛋白质G(Pharmacia Uppsala,Sweden)上亲和层析抗体。从用20F3(大鼠×小鼠)杂交瘤(美国典型培养物保藏中心,MD)接种的胸腺较少的裸鼠腹水制备抗鼠科IL-6抗体。在抗大鼠κ链单克隆抗体-琼脂糖凝胶柱上亲和层析大鼠IgG抗体。中和滴度(终点稀释度,相应于10U鼠科1L6/ml对细胞生长作用中和的50%)为1055(IgG含量:2.9mg/ml)。用无关的大鼠IgG24作为同位素对照,它是从大鼠浆细胞瘤(经Dr.H.Bazin,University ofLouvain.Medical School.Brussels.Belgium允许得到的)的腹水制备的。用在HiloadQ琼脂糖凝胶上的阴离子交换层析纯化IgG,并在Superdex 200(Pharmacia)上作凝胶过滤。用显色鲎变形细胞裂解物测定法(LabiVitrum,Stockholm,Sweden)测试抗IFNγ、抗IL-12、抗IL-6和不相关IgG24的内毒素含量,发现内毒素均少于2ng/ml。用200μl无热原盐水注射。
结果
处理14天后,对照组的10只大鼠中有七只证实患了关节炎,而接受处理的动物(8只)中只有1只证明患有关节炎。见图29。处理后20天,唯一的那只被处理的动物(患病的)不发展关节炎病理。另外,接受处理的动物与对照动物相比证明没有任何明显的体重减少。见图30。另外,接受处理的动物维持了健康动物(没有注射胶原)的体重(没有显示曲线)。
实施例28
成胶质细胞瘤的治疗
可将本发明的化合物(如AMD 3100)用于治疗成胶质细胞瘤、纤维瘤、星形细胞瘤或骨髓瘤(侵袭中枢神经系统的)。这些化合物的用法可按照标准临床实践和流程,按上述实施例提供的剂量,并按照临床目的(如显影、免疫学或其它方法)。
例如,Sehgal等人,J.of Surg.Oncolo.69:99-104(1998)(″Sehgal I″)和Sehgal等人,J.of Surg.Oncolo.69:239-248(1998)(″Sehgal II″)已报道了在成胶质细胞瘤肿瘤细胞增殖中趋化因子受体结合的病原学或与其相关的内容。CXCR4与其受体的结合对成胶质细胞瘤细胞形成和/或增殖具有重要的作用。用本发明的化合物(如AMD 3100)抑制CXCR4与其天然受体配体的结合,为中枢神经系统的肿瘤(由趋化因子如CXCR4调节或与其相关)的治疗提供了一种新药。
实施例29
非小细胞肺癌的治疗
可将本发明的化合物(如AMD 3100)用于治疗非小细胞肺癌。这些化合物的用法可按照标准临床实践和流程,按上述实施例提供的剂量,并按照临床目的(如显影、免疫学或其它方法)。
例如,已发现在非小细胞肺癌中CXC趋化因子调节血管生成或与其有关(见Arengerg等人,J.of Leukocyte Biol:62:554562(1997);和Moore等人,TCM,vol8(2):51-58(1998)Elsevier Science.Inc.)。CXC趋化因子与它们各自受体的结合对非小细胞肺癌形成和/或增殖(由增加的生成血管活性促进)具有重要的作用。用本发明的化合物(如AMD 3100)抑制CXC趋化因子与它们天然受体配体的结合,为肿瘤如非小细胞肺癌(由趋化因子水平的增加调节或与其相关)的治疗提供了一种新药。
实施例30
N-[4-(1,7-二氮杂环十四烷基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶
本发明的化合物还包括分子式I的化合物,其中V带有2-6个任选地被取代的胺氮,它们彼此被2个或多个任选地被取代的碳原子所隔开。这种化合物的一个例子,包括:
N-[4-(1,7-二氮杂环十四烷基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD-Exp 1:见图34)。
AMD-Exp 1、Exp-2和Exp 3可用我们以前公开的制备连碳的(carbon-linked)氮杂环的方法(Bridger等人,J.Med.Chem.1995,38,366)改进后制得,简单总结如下:将上述中间体,N-[1-亚甲基-4-(氯亚甲基)亚苯基]-N-(2-硝基苯磺酰基)-2-氨基甲基)吡啶(或Dep保护的中间体)与丙二酸二乙酯钠盐反应,得到相应的二酯。
将二酯还原成相应的二醇,然后用甲磺酰氯衍生得到二甲磺酸酯。用氰化钠亲核取代二甲磺酸酯,得到所需的二腈,用硼烷、THF将其还原成二胺,用2-硝基苯磺酰氯衍生,用于后续的大环化反应。
用适当衍生的二醇(如1,7-庚二醇二-对-甲苯磺酸酯)大环化和进行后续的去保护(如上述实施例给出的)得到例如AMD-Exp 1.。类似地,用1,3-亚苯基二乙醇和2,6-吡啶二乙醇可分别得到AMD-Exp 2和AMD-Exp 3。
另外,AMD-Exp 1、AMD-Exp 2和AMD-Exp 3,每种都可被制成药物组合物,即在药学上可接受的载体中含有治疗有效剂量的化合物的药物组合物。
另外,AMD-Exp 1、AMD-Exp 2和AMD-Exp 3,每种都可按上述方法用于治疗大量趋化因子调节的疾病和病症,包括:HIV或FIV的感染;由内皮细胞功能调节的疾病;与肠胃道血管形成、造血、或小脑发育相关的疾病;与初级白血球流动或溢出和应答刺激剂的白血球组织侵入相关的疾病;有效结合于趋化因子受体的化合物;炎症;癌;中枢神经系统发育疾病;HIV;FIV;脉管发育疾病;心脏发育疾病;造血和其它趋化因子调节疾病或失调。
其它可用AMD-Exp 1治疗的疾病或病症的例子还包括:关节炎或多发性硬化症引起的炎症;与实体肿瘤、淋巴瘤、转移性肿瘤、成胶质细胞瘤和其它恶性肿瘤相关的癌症;非小细胞肺癌;肺癌;乳房癌;前列腺癌;和其它器官的癌症。另外,可用AMD-Exp 1治疗的失调或病症包括:用抑制或促进血管生成或诱导血管生成的停滞法治疗的失调。
实施例31
N-[7-(4,10-二氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶
本发明的化合物还包括分子式I的化合物,其中V带有2-6个任选地被取代的胺氮,它们彼此被2个或多个任选地被取代的碳原子所隔开。这种化合物的例子,包括:
N-[7-(4,10-二氮杂二环[13.3.1]十七碳-1(17),13,15-三烯基)-1,4-亚苯基二(亚甲基)]-2-(氨基甲基)吡啶(AMD-Exp 2:见图36)。
AMD Exp-2和Exp 3可用上述实施例30的方法制备。另外,AMD-Exp 2可制成药物组合物,即在药学上可接受的载体中含有治疗有效剂量的化合物的药物组合物。
另外,AMD-Exp 2可按上述方法用于治疗大量趋化因子调节的疾病和病症,包括:HIV或FIV的感染;由内皮细胞功能调节的疾病;与肠胃道血管形成、造血、或小脑发育相关的疾病;与初级白血球流动或溢出和应答刺激剂的白血球组织侵入相关的疾病;有效结合于趋化因子受体的化合物;炎症;癌;中枢神经系统发育疾病;HIV;FIV;脉管发育疾病;心脏发育疾病;造血和其它趋化因子调节的疾病或失调。
其它可用AMD-Exp 2治疗的疾病或病症的例子还包括:关节炎或多发性硬化症引起的炎症;与实体肿瘤、淋巴瘤、转移性肿瘤、成胶质细胞瘤和其它恶性肿瘤相关的癌症;非小细胞肺癌;肺癌;乳房癌;前列腺癌;和其它器官的癌症。另外,可用AMD-Exp 2治疗的失调或病症包括:用抑制或促进血管生成或诱导血管生成的停滞法治疗的失调。
按熟知的原理,将这些活性化合物以配制好的药物组合物形式给予,并将化合物(较佳地以单元剂量形式)与药学上可接受的稀释剂、载体或赋形剂组合。这些组合物可以溶液或悬浮液的形式用于注射或冲洗,或以胶囊、片剂、糖衣丸或其它固态组合物或溶液或悬浮液用于口服,或制成阴道栓或栓剂,或上述任何药物的持续缓释形式以供植入。适合的稀释剂、载体、赋形剂和其它组分是熟知的。也可期望制成局部给药的组合物,如软膏或乳膏。本发明的化合物可以组合物的形式使用或单独使用。
本发明的药物组合物可用普通的制药方法确定制成单位剂量,适宜在人体或动物中提供剂量范围为0.01-100mg/kg体重/天的活性化合物,以单剂量或若干次较少的剂量给药。优选的剂量范围为0.01-30mg/kg体重/天(静脉(iv)或腹膜腔内(ip))。其它活性化合物可用于组合物或这些活性化合物,或辅助治疗可包括在治疗疗程中。包括治疗有效剂量的新化合物(所述的化合物有效地结合于趋化因子受体)的药物组合物对治疗患者是有用的。
本发明还企图用这些组合物制成药物以治疗受HIV或FIV感染的患者,和/或治疗受内皮细胞功能调节的疾病和/或治疗与肠胃道血管生成、造血或小脑发育相关的疾病。
在治疗受HIV或FIV感染的患者的方法中,以包含在药学上可接受的载体中的治疗有效剂量的药物组合物的形式,将药物组合物给予所述的患者。在治疗患有与内皮细胞功能调节相关的疾病的患者的方法中,以包含在药学上可接受的载体中的治疗有效剂量的药物组合物的形式,将药物组合物给予所述的患者。通过给予所述的患者以包含在药学上可接受载体中的治疗有效剂量的药物组合物,本发明还尝试了治疗与肠胃道血管生成、造血或小脑发育相关的疾病。
通过给予所述的患者以包含在药学上可接受载体中的治疗有效剂量的药物组合物,本发明还尝试了治疗与初级白血球流动或溢出和应答刺激剂的白血球组织侵入相关的疾病。通过给予一位患者以包含在药学上可接受载体中的治疗有效剂量的药物组合物,本发明的方法还尝试了治疗该患者,其中所述的化合物有效地结合于趋化因子受体。
本发明还进一步尝试了用于治疗人或动物的以下疾病的药物组合物和方法:肾异体移植排斥、炎症、癌、中枢神经系统发育疾病、HIV、脉管发育疾病、造血和其它受趋化因子调节的疾病或失调。本发明还提供对以下疾病(但不限制于)的治疗:关节炎、多发性硬化症、受HIV或FIV感染的痴呆、帕金森病、阿耳茨海默氏病和炎症。本发明的药物组合物和方法还可治疗癌症(但不限制于):实体瘤、淋巴瘤、转移性肿瘤、成胶质细胞瘤和其它恶性肿瘤。本发明的药物组合物还提供对以下癌症(但不限制于)的治疗:非小细胞肺癌、肺癌、乳房癌、前列腺癌和其它器官的癌。
用本发明的药物组合物还可完成对其它疾病或失调的治疗,包括(但不限制于):用抑制或促进血管生成或诱导血管生成的停滞法治疗的失调;由趋化因子调节的发育失调。
本发明还提供了预防患者得疾病或失调的方法,通过给予一位患者以治疗有效剂量的本发明的药物组合物足够的一段时间以有效预防疾病或失调。