CAMPO DA INVENÇÃO
[001] A presente invenção refere-se a novos compostos com atividade inibitória de fosfodiesterase, assim como a seu uso como agentes terapêuticos no tratamento de doenças e de condições inflamatórias.
FUNDAMENTOS DA INVENÇÃO
[002] As fosfodiesterases são enzimas, que catalisam a hidrólise de MP cíclico e/ ou de GMP cíclico em células par 5-AMP e 5- GMP, respectivamente, e como tais eles são críticos para a regulação celular dos níveis de cAMP ou cGMP. Das 11 fosfodiesterases até o momento identificadas, a fosfodiesterase (PDE) 4, PDE7 e PDE8 são seletivas para cAMP. PDE4 é o modulador de cAMP mais importante, expresso nas células imunes e inflamatórias, tais que neutrófilos, macrófagos e T-linfócitos (Z. Hunag e J. A. Mancini, Current Med. Chem. 134, 2006, págs. 3253- 3262). Como cAMP é um segundo mensageiro chave na modulação de respostas inflamatórias, PDE 4 foi verificado como capaz de regular as respostas inflamatórias de células inflamatórias pela modulação de citoquinas proinflamatórias, tais que TNFα, IL-2, IFN-% GM - CSF e LTB4. A inibição de PDE4 tornou-se portanto, um alvo atrativo par a terapia de doenças inflamatórias, tais que asma, doença pulmonar obstrutiva crônica (COPD), artrite reumatóide, dermatite tópica, mal de Crohn, etc. (M. D. Houslay et al., Drug Discovery Today 10 (220, 2005, págs. 1503 - 1519). Como os pacientes de dermatite atópica (AD) apresentam uma atividade de PDE aumentada, a inibição de PDE4 seria também considerada como um tratamento viável de AD (Journal of Investigative Dermatology (1986), 87(3), 372-6).
[003] A família do gene PDE4 consiste de pelo menos quatro genes, A, B, C e D, que apresentam um alto grau de homologia (V. Boswell Smith and D. Spina, Curr. Opinion Investig. Drugs 6 (11), 2006, págs. 1136- 1141). As quatro isoformas de PDE4 são expressadas de modo diferente em diferentes tecidos e tipos de célula. Deste modo, o PDE4B é expresso, de um modo predominante, em monócitos e neutrófilos, mas não no córtex e nas células epiteliais, embora o PDE4 seja expresso no pulmão, córtex, cerebelo e células -T (C. Kroegel e M. Foerster, Exp. Opinion Investig. Drugs 16 (1), 2007, págs.109- 124). Foi especulado que a inibição de PDE4D no cérebro está associada com os efeitos adversos encontrados quando da administração de inibidores de PDE4 clinicamente, primariamente náusea e emese, enquanto que a inibição de PDE4 está associada os com efeitos antiinflamatórios (B. Lipworth, Lancet 365, 2005, págs.167- 175). No entanto, os inibidores de PDE desenvolvidos até o momento não são tidos como sendo específicos para cada um das quatro isoformas de PDE4.
[004] Numerosos inibidores de PDE4 foram estudados quanto seu efeito terapêutico sobre doenças inflamatórias, primariamente a asma, a doença intestinal inflamatória e COPD. O primeiro destes, teofilina, é um inibidor de fosfodiesterase não seletivo, fraco, usado no tratamento de doenças respiratórias, tais que a asma e a COPD. O tratamento com teofilina pode, no entanto, dar origem tanto a efeitos adversos brandos como severos, por exemplo arritmia e convulsões, restringindo a utilidade clínica de teofilina (Krogel and Foerster, supra). Como a fosfodiesterase permaneceu um alvo atrativo para a terapia antiinflamatória, vários outros inibidores de PDE4 mais seletivos foram desenvolvidos e investigados em um ambiente clínico. O desenvolvimento clínico de muitos dos inibidores de PDE4 de primeira geração, tais que o rolipram, foi descontinuado devido a efeitos colaterais que limitam a dose, primariamente a náusea e emese. Os inibidores de PDE4 de segunda geração com efeitos adversos aparentemente menos pronunciados estão correntemente em ensaios clínicos (Houslay, supra).
[005] Os inibidores de PDE-4 recentemente desenvolvidos estão, por exemplo, expostos na EP 0771794 e na EP 0943613. A WO 96/31476 expõe 3,5-dicloropiridinas 4-substituídas, que são inibidores de AMP fosfodiesterase cíclica.
[006] Existe uma necessidade continuada quanto a novos inibidores de PDE4, que possuem uma janela terapêutica mais favorável, isto é, menos efeitos adversos, ao mesmo tempo em que o seu efeito antiinflamatório terapêutico é mantido. Uma revisão dos ensaios clínicos e pré- clínicos com inibidores de PDE4 seletivos, incluindo aqueles inibidores destinados ao tratamento de dermatite atópica e psoríase, foi recentemente fornecida em Inflammation & Allergy: Drug Targets, 2007, 6 (1), 17- 26.
SUMÁRIO DA INVENÇÃO
[007] Os inventores verificaram, de um modo surpreendente, que os novos compostos da presente invenção exibem uma atividade inibitória de PDE4 e podem ser úteis como agentes terapêuticos para doenças alérgicas inflamatórias, tais que a asma brônquica, rinite alérgica, e nefrite; doenças autoimuines, tais que a artrite reumatóide, esclerose múltipla, mal de Crohn, e lupus sistêmico eritematoso; doenças do sistema nervoso central, tais que a depressão, amnésia e demência, a organopatia associada com o refluxo isquêmico causado por falha cardíaca, choque, e doenças cerebrovasculares, e os similares; diabetes resistente a insulina; feridas; AIDS, e os similares.
[008] Os compostos da presente invenção podem ser também benéficos na prevenção, tratamento ou melhora de uma variedade de doenças, tais que as doenças ou condições dérmicas, tais que os distúrbios d pele inflamatórios e proliferativos e em particular, psoríase, inflamação epidérmica, alopecia, atrofia da pele, atrofia da pele induzida por esteróide, envelhecimento da pele, fotoenvelhecimento da pele, acne, dermatite, dermatite atópica, dermatite seborréica, dermatite de contato, urticária, prurido e eczema.
[009] Deste modo, a presente invenção refere-se a um composto de acordo com a fórmula I,

A I em que m e n representam independentemente 0, 1, 2, 3, 4, 5, 6 ou 7; em que G e E representam, independentemente enxofre, oxigênio, -N=, -N (R5)-, ou -N(R5)C(O)-, e R1 e R2, junto com o átomo de carbono ao qual eles estão ligados, formam um anel carboxílico insaturado ou um anel heterocíclico, que compreende um ou dois heteroátomos, selecionados a partir de oxigênio, enxofre, -S(O)-, -S(O)2-, -N(R5)-, um ou mais átomos de carbono no referido anel carbocíclico insaturado ou anel heterocíclico sendo opcionalmente substituído por um ou mais, os mesmos substituintes ou substituintes diferentes, selecionados a partir de R4; ou em que G e E representam independentemente enxofre, oxigênio, -N=, -N (R5)-, ou — N(R5)C(O)-, e R1 e R2, junto com o átomo de carbono ao qual eles estão ligados, formam um anel carbocíclico saturado, um ou mais átomos de carbono no referido anel carbocíclico saturado sendo opcionalmente substituídos por um ou mais substituintes, os mesmos ou diferentes, selecionados partir de R4, contanto que quando G é oxigênio, m e n não sejam ambos zero, e com a condição ainda de que, quando Ge E forem ambos oxigênio, a soma de m e n seja seis ou acima; R3 é halogênio, hidróxi, alquila, alquenila, alquinila, haloalquil, alcóxi, haloalcóxi, alquiltio, formila, alcoxicarbonila, alquilcarbonila ou aminocarbonila; R4 é hidrogênio, amino, tioxo, alquila, haloalquiola, hidroxialquila, alcóxi, halogênio, oxo, tia ou hidróxi; R5 é hidrogênio, alquila, haloalquila, alquilcarbonila, hidroxilquila, lcoxicarbonila, alquilsulfonila, alquilaminosulfonila ou aminosulfonila; X é uma ligação, -CH2- ou -NH-; A é arila, cicloalquila, cicloalquenila, arilalquila, heteroarila, heteroarilalauqila, heterocicloalquila ou heterocicloalquenila, opcionalmente substituído por um ou mais, os mesmos substituintes ou substituintes diferentes, selecionados partir de R4; e sais farmaceuticamente aceitáveis, hidratos, N-óxidos ou solvatos dos mesmos.
[010] Em um outro aspecto, a invenção refere-se a uma composição farmacêutica, que compreende um composto da fórmula I, como acima definido, junto com um veículo ou excipiente farmaceuticamente aceitável ou veículo(s) farmaceuticamente aceitáveis, opcionalmente junto com um ou mais outro(s) composto(s) terapeuticamente ativos.
[011] Em ainda um outro aspecto, a invenção refere-se a um composto de acordo com a fórmula I, como acima definido, e a sais farmaceuticamente aceitáveis, hidratos, N-óxidos ou solvatos do mesmo, para o uso na profilaxia, tratamento ou melhora de doenças dérmicas ou condições, ou de distúrbios de ferida cutânea agudos ou crônicos.
[012] Em um outro aspecto adicional, a invenção refere-se a um método de prevenção, tratamento ou melhora de doenças dérmicas ou condições, ou de distúrbios de ferida cutânea agudos ou crônicos, o método compreendendo administrar a uma pessoa, que sofra de pelo menos uma das referidas doenças, uma quantidade eficaz de um ou mais compostos da fórmula I, como acima definido, e sais farmaceuticamente aceitáveis, hidratos, N- óxidos ou solvatos dos mesmos; de um modo opcional junto com um veículo frmaceuticamente aceitável ou um ou mais excipientes, e de um modo opcional em combinação com outros compostos terapeuticamente ativos.
DESCRIÇÃO DETALHADA DA INVENÇÃO
[013] O termo “radical hidrocarboneto” tem a intenção de indicar um radical contendo apenas hidrogênio e átomos de carbono, podendo conter uma ou mais ligações duplas e/ ou triplas carbono- carbono, e podendo compreender porções cíclicas em combinação com porções lineares ou ramificadas. O referido hidrocarboneto compreende de 1- 20 átomos de carbono, e compreende, de um modo preferido, de 1-12, por exemplo, de 1-6, por exemplo, de 1-4, por exemplo de 1-3, por exemplo 1-2 átomos de carbono. O termo inclui alquila, alquenila, cicloalquila, cicloalquenila, alquinila e arila, arilalquila, como abaixo indicado.
[014] O termo “arila” tem a intenção de indicar um radical ou anéis carbocíclicos aromáticos, que compreendem de 6- 20 átomos de carbono, tal que de 6-14 átomos de carbono, de um modo preferido de 6-10 átomos de carbono, em particular anéis de 5-6 membros, de um modo opcional anéis carbocíclicos opcionalmente fundidos com pelo menos um anel aromático, tal que fenila, naftila, indenila e indanila.
[015] O termo “heteroarila” tem a intenção de indicar radicais de anéis aromáticos heterocíclicos, que compreendem de 1-6 heteroátomos (selecionados a partir de O, S e N) e de 1-20 átomos de carbono, tais que 1-5 heteroátomos e 1-10 átomos de carbono, tais que 1-5 heteroátomos e 1-6 átomos de carbono, tal que 1-5 heteroátomos e 1-3 átomos, em particular anéis de 5 ou 6 membros com 1- 4 heteroátomos, selecionados a partir de O, S e N, opcionalmente fundidos com anéis bicíclicos com 1-4 heteroátomos, selecionados a partir de O, S e N, ou anéis bicíclicos opcionalmente fundidos com 1-4 heteroátomos, e em que pelo menos um anel é aromático, por exemplo, piridila, quinolila, isoquinolila, indolila, tetrazolila, tiazolila, imidazolila, pirazolila, oxazolila, isoxazolila, tienila, pirazinila, isotizolila, benzimidazolila e benzofuranila.
[016] No presente contexto, o termo “alquila” tem a intenção de indicar o radical obtido quando um átomo de hidrogênio é removido a partir de um hidrocarboneto. O referido alquila compreende de 1-20, de um modo preferido de 1-12, tal que de 1-6, tal que de 1-4 átomos de carbono. O termo inclui as subclasses alquila normal (n- alquila), alquila secundária e terciária, tais que metila, etil, n- propila, isopropila, n-butila, isobutila, sec-butila, terc- butila, pentila, isopentila, hexila e isoexila.
[017] O termo “cicloalquila” tem a intenção de indicar um radical cicloalcano saturado, que compreende de 3-20 átomos de carbono, de um modo preferido de 3-10 átomos de carbono, em particular de 3-8 átomos de carbono, tal que de 3-6 átomos de carbono, incluindo anéis bicíclicos fundidos, por exemplo, ciclopropila, ciclobutil, ciclopentila, cicloexila, ou cicloeptila.
[018] O termo “heterocicloalquila” tem a intenção de indicar um radical cicloalcano, tal que acima descrito, em que um ou mais átomos de carbono são substituídos por heteroátomos, compreendendo de 1-19 átomos de carbono, por exemplo, 2-4 átomos de carbono, além disso compreendendo 1-6 heteroátomos, de um modo preferido 1, 2, ou 3 heteroátomos, selecionados a partir de O, N ou S, que podem ser opcionalmente oxidados uma ou duas vezes, por exemplo, [1,3]dioxol, oxetano, [1,3]dioxolano, [1,3] dioxano, tetraidrotiopirano, tetraidrotiopiran-1,1-dióoxido, tetraidrotiopiran-1- óxido, piperidina, tetraidrotiofeno, [1,3]-ditiano, tietano, [1,3]-ditiano-1,3- dióxido, ou tietano-1-óxido, ou incluindo anéis bicíclicos fundidos com 1- 4 heteroátomos, em que pelo menos um anel compreende um heteroátomo, e em que o outro anel pode, por exemplo, ser um anel carbocíclico, por exemplo, isoindolila.
[019] O termo “alquenila” tem a intenção de indicar um radical mono-, di-, tri-, tetra- ou pentainsaturado compreendendo de 2-10 átomos de carbono, em particular de 2-6 átomos de carbono, tal que de 2-4 átomos de carbono, por exemplo, etenila, propenila, butenila, pentenila ou hexenila.
[020] O termo “cicloalquenila” tem a intenção de indicar radicais hidrocarboneto cíclicos não- aromáticos mono-, di-, tri- ou tetrainsaturados, compreendendo de 3- 20 átomos de carbono, incluindo anéis bicíclicos fundidos, compreendendo, de um modo típico, de 3-10 átomos de crbono, tal que 3, 4 ou 6 átomos de carbono, por exemplo, ciclopropenila, ciclobutenila, ciclopentenila, cicloexenila, cicloeptenila.
[021] O termo “heterocicloalquenila” tem a intenção de indicar um radical cicloalqueno, como acima descrito, em que um ou mais átomos de carbono são substituídos por heteroátomos, compreendendo de 1-19 átomos de carbono, por exemplo 2-4 átomos de carbono, compreendendo ainda 1-6 heteroáotmos, de um modo preferido 1, 2 ou 3 heteroátomos, selecionados a partir de O, N ou S, incluindo anéis bicíclicos fundidos com de 1-4 heteroátomos, em que pelo menos um anel compreende um heteroátomo e em que o outro anel pode, por exemplo, ser um anel carbocíclico, por exemplo, diidrofuranila ou 2,5- dididro-1H-pirrolila.
[022] O termo “arilalquila” tem a intenção de indicar um radical arila, tal como acima definido, covalentemente ligado a um grupo alquila, por exemplo, benzila.
[023] O termo “heteroarilalquila” tem a intenção de indicar um radical heteroarila, como acima definido, covalentemente ligado a um grupo alquila.
[024] O termo “alquinila” tem a intenção de indicar um radical hidrocarboneto, que compreende de 1-5 ligações C-C triplas e 2-20 átomos de carbono, compreendendo, de um modo típico, de 2-10 átomos de carbono, em particular de 2-6 átomos de carbono, tal que 2-4 átomos de carbono, por exemplo, etinila, propinila, butinila, pentinila ou hexinila.
[025] O termo “halogênio” tem a intenção de indicar um substituinte a partir do 7° grupo principal da tabela periódica, tal que flúor, cloro, bromo e iodo.
[026] O termo “haloalquila” tem a intenção de indicar um grupo alquila, tal como acima definido, substituído por um ou mais átomos de halogênio, como acima definido, por exemplo, difluorometila.
[027] O termo “hidroxialquila” tem a intenção de indicar um grupo alquila, tal como acima definido, substituído por um ou mais hidróxi, por exemplo, hidroximetila, hidroxietila, hidroxipropila.
[028] O termo “alcóxi” tem a intenção de indicar um radical d fórmula -OR’, em que R’ é alquila, tal como acima indicado, por exemplo metóxi, etóxi, n-propóxi, isopropóxi, butóxi, etc.
[029] O termo “alcoxicarbonila” tem a intenção de indicar um radical da fórmula -C(O)-O-R‘, em que R’ é alquila, tal como acima indicado, por exemplo, metoxicarbonila, etoxicarbonila, n- propoxicarbonila, isopropoxicarbonila, etc.
[030] O termo “alquilcarbonila” tem a intenção de indicar um radical da fórmula -C(O)-R‘, em que R’ é alquila, tal como acima indicado, por exemplo, etanoíla, acetila.
[031] O termo “aminossulfonila” tem a intenção de indicar um radical da fórmula -S(O)2-NR”, em que R’ é como acima indicado, por exemplo, -SO2Me.
[032] O termo “anel heterocíclico” tem a intenção de incluir as definições heteroarila, heterocicloalquila e heterocicloalquenila, como acima definido, além disso incluindo sistemas de anel anelados um como o outro ou com hidrocarbonetos cíclicos, por exemplo, 2,5-diidrobenzo (b) dioxocina, 2, 3, 5, 8-tetraidro-[1,4]-dioxocina, 5, 8-diidro-[1,4]dioxocina.
[033] O termo “sal farmaceuticamente aceitável” tem a intenção de indicar os sais preparados através da reação de um composto da fórmula I com um ácido orgânico ou inorgânico adequado, tais que os ácidos clorídrico, bromídrico, iodídrico, sulfúrico, nítrico, fosfórico, fórmico, acético, 2,2- dicloroacético, adípico, ascórbico, L-aspártico, L-glutâmico, galactárico, láctico, maléico, L-málico, ftálico, cítrico, propiônico, benzóico, glutárico, glucônico, D-glucurônico, metanossulfônico, salicílico, succínico, malônico, tartárico, benzenossulfônico, etano-1,2-dissulfônico, 2-hidróxi etanossulfônico, toluenossulfônico, sulfâmico ou fumárico. Sais farmaceuticamente aceitáveis dos compostos da fórmula I podem ser também preparados através de reação com uma base adequada, tal que hidróxido de sódio, hidróxido de potássio, hidróxido de magnésio, hidróxido de cálcio, hidróxido de prata, amônia ou os similares, ou aminas não- tóxicos adequadas, tais que alquilaminas inferiores, por exemplo trietilamina, hidróxi- alquilaminas inferiores, por exemplo, 2-hidroxietilamina, bis-(2-hidroxietil)- amina, cicloalquilaminas, por exemplo dicicloexilamina, ou benzilaminas, por exemplo N,N’-dibenziletilenodiamina, e dibenzilamina, ou L- arginina ou L- lisina. Os sais obtidos través de reação com uma base adequada incluem, mas não estão limitados a, sais de sódio, sais de colina, sais de 2-(dimetilamino)- etanol, sais de 4-(2-hidroxietil)-morfolina, sais de L- lisina, sais de N-(2- hidroxietil)-pirrolidina, sais de etanolamina, sais de potássio, sais de tetrabutilamônio, sais de benziltrimetilamônio, sais de cetiltrimetilamônio, sais de tetrametilamônio, sais de tetrapropilamônio, sais de tris (hidroximetil) aminometano, sais de N-metil- D-glucamina, sais de prata, sais de benzetônio, e sais de trietanolamina.
[034] O termo “solvato” tem a intenção de indicar uma espécie formada através da interação entre um composto, por exemplo um composto da fórmula I, e um solvente, por exemplo um álcool, glicerol ou água, em que as referidas espécies estão em uma forma sólida. Quando a água é o solvente, a referida espécie é referida como a um hidrato. Modalidades da presente invenção
[035] Em uma ou mais modalidades da presente invenção, E e G são ambos oxigênio.
[036] Em uma ou mais modalidades da presente invenção, m e n são ambos um.
[037] Em uma ou mais modalidades da presente invenção, m e n são ambos zero.
[038] Em uma ou mais modalidades da presente invenção, R1 e R2, junto com o átomo de carbono ao qual eles estão ligados, formam um anel heterocíclico, que compreende um ou dois heteroátomos selecionados a partir do grupo que consiste de -O-, -S-, -S(O)-, -S(O2)-, -N=, e - N(R5)-; um ou mais átomos de carbono no anel heterocíclico sendo opcionalmente substituídos por um ou mais substituintes, os mesmos ou diferentes, selecionados a partir de R4.
[039] Em uma ou mais modalidades da presente invenção, R1 e R2, junto com o átomo de carbono ao qual eles estão ligados, formam um anel heterocicloalquila, que compreende um ou dois heteroátomos, selecionados partir do grupo, que consiste de -O-, -S-, -S(O)-, -S(O2)-, e -N (R5);; um ou mais átomos de carbono no anel heterocicloalquila sendo opcionalmente substituídos por um ou mais substituintes, os mesmos ou diferentes, selecionados a partir de R4.
[040] Em um ou mais modalidades da presente invenção, R1 e R2, junto com o átomo de carbono, ao qual eles estão ligados, formam um anel heterocíclico de 4, 5 ou 6 membros, em particular um anel heterocíclico de 6 membros.
[041] Em uma ou mais modalidades da presente invenção, o anel heterocíclico é tetraidropirano, oxetano, [1,3] dioxolano, [1,3]dioxano, tetraidrotiopirano, tetraidrotiopirano-1,1-dióxido, tetraidrotiopiran-1-óxido, piperidina, tetraidrotiofeno, [1,3]-ditiano, tietano, [1,3]-ditiano-1,3-dióxido, tietano-1-óxido, ou tietano-1,1-dióxido.
[042] Em uma ou mais modalidades da presente invenção, o anel heterocíclico formado de R1 e R2, junto com o átomo de carbono, ao qual eles estão ligados, compreende um heteroátomo ou dois heteroátomos no referido anel.
[043] Em uma ou mais modalidades da presente invenção, o(s) heteroátomo (s) é/ são oxigênio, enxofre, -S(O)-, ou -S(O)2-.
[044] Em uma ou mais modalidades da presente invenção, A representa heteroarila ou heteroarilalquila.
[045] Em uma ou mais modalidades da presente invenção, A representa piridila, pirazinila ou quinolila.
[046] Em outras modalidades, A pode representar fenila.
[047] Em uma ou mais modalidades da presente invenção, R3 representa metóxi ou etóxi.
[048] Em uma ou mais modalidades da presente invenção, X é -CH2- ou -NH-.
[049] Em uma ou mais modalidades da presente invenção, A é 4- (3,5-dicloropiridila).
[050] Em uma ou mais modalidades da presente invenção, o composto da fórmula I é representado pela fórmula Ia ou Ib:
em que X, A,G, E, R1, R2, R3, R4, R5, m e n são como acima definidos.
[051] Em uma modalidade particular da presente invenção, X = NH-, em que R3 representa alcóxi C1-6.
[052] A presente invenção inclui todas as modalidades, em que X, A, G, E, R1, R2, R3, R4 e R5 são combinados em qualquer modo aqui descrito.
[053] De um modo particular, os compostos da fórmula I podem ser selecionados a partir de um dos compostos que se seguem: 2-(3,5-Dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-piran]-4-il) etanona (Composto 101), N-(3,5-Dicloropiridina-4-il)-7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-piran]-4- carboxamida (Composto 102), 2-(3,5-Dicloro-1-óxido-piridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol-2,4’-(4H)- piran]-4-il)etanona (Composto 103), 2- (3,5-Dicloropiridina-4-il)-1-(7-metóxi- 4’,5’-diidro-espiro [1,3- benzodioxol-2,3’-(2H)tiofen]-4-il)etanona (Composto 104), 2-(3,5-Dicloropiridina-4-il)-1- (7-metóxi- espiro [1,3-benzodioxol-2,4’- piperidin]-4-il) etanona (Composto 105), 2- (3,5-Dicloropiridin-4-il)-1-(7-metóxi-1’-[metoxicarbonil]-espiro[1,3- benzodioxol-2,4’-piperidin] -4-il) etanona (Composto 106), 2-(3,5- Dicloropiridina-4-il)-1-(7- metóxi-1’-[metilsulfonil]-espiro[1,3- benzodioxol-2,4’-piperidina]-4-il) etanona (Composto 107), 2-(3,5- Dicloropiridin-4-il)-1- (7-metóxi-1’-acetil-espiro[1,3- benzodioxol- 2,4’-piperidina]-4-il) etanona (Composto 108), 2-(3,5- Dicloropiridin-4-il)-1- (7- metóxi-1’-metil-espiro[1,5-benzodioxol- 2,4’-piperidina]-4-il)etanona (Composto 109), 2-(3,5-Dicloropiridina-4-il)-1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’- (4H) - tiopiran]-4-il) etanona (Composto 110), 2-(3,5-Dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran-1’-óxido]-4-il) etanona (Composto 111), 2- (3,5-Dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol- 2,4’-(4H)-tiopiran- 1’,1’-dióxido]-4-il)etanona (Composto 112), ou 2-(3,5-Dicloro-1- óxido-piridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’- tetraidroespiro [1,3-benzodioxol-2,4’- (4H)-tiopiran-1’,1’-dióxido]-4-il) etanona (Composto 113); 2-(3-bromopiridina-4-il) -1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (Composto 114); 2-(3-Bromo- pirazin-2-il)-1-(7-metóxi-2’,3’, 5’, 6’-tetraidro-espiro[1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (Composto 115); 2-(-pirazin-2-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro[1,3- benzodioxol- 2,4’-(4H)-tiopiran]-4-il) etanona (Composto 116); 2-(-piridin-4-il)-1- (7-metóxi- 2’, 3’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol- 2,4’- (4H)- tiopiran]-4-il) etanona (Composto 117); 2-(quinolin-4-il)-1-(7-metóxi- 2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’- (4H)-tiopiran] -4-il) etanona (Composto 118); 2-(2,6-Dicloro-fenil)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (Composto 119); 2-(2-Cloro-fenil)-1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro[1,3- benzodioxol- 2,4’- (4H)-tiopiran]-4-il) etanona (Composto 120); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2H-1,5-benzodioxepin-3- (4H), 3’-oxetano]-6-il} etanona (Composto 121); 2-(3,5-Dicloro-1-óxido-piridin-4-il)-1-{9-metóxi-espiro [2H-1,5- benzodioxepin-3(4H), 3’-oxetano]-6-il} etanona (Composto 122); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro[2H-1,5-benzodioxepin- 3 (4H), 3’-tietano]-6-il}etanona (Composto 123); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro[2H-1,5-benzodioxepin-3 (4H), 3’- tietano-1’,1’-dióxido ] -6-il} etanona (Composto 124); 2-(3,5-Dicloropiridin-1-óxido-4-il)-1-{ 9-metóxi-espiro [2 H-1,5- benzodioxepin 3(4H), 3’-tietano-1’,1’-dióxido]-6-il} etnona (Composto 125); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2 H-1,5-benzodioxepin- 3(4H), 2’- (1,3-dioxolano)]-6-il} etanona (Composto 126); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [ 2H-1,5- benzodioxepin-3- (4H), 4’-tetraidropiran]-6-il} etanona (Composto 127); 2-(3,5-Dicloro-1- óxido-piridin-4-il)-1-{ 9-metóxi- espiro [2 H-1,5- benzodioxepin-3(4H), 4’-tetraidropiran]-6- il} etanona (Composto 128); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-2’,2’-dimetil-espiro [2H-1,5- benzodioxepin-3 (4H), 5’- [1,3] dioxano]-6-il}etanona (Composto 129); 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2 H-1,5-benzodioxepin- 3(4H), 5’-[1,3] dioxano]-6-il} etanona (Composto 130); 2-(3,5-Dicloro-1-óxido-piridina-4-il)-1-{ 9-metoxiespiro [2H-1,5- benzodioxepin-3(4H), 5’-[1,3]dioxano]-6- il} etanona (Composto 131); e 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2H-1,5-benzodioxepin-3(4H), 5’-[1,3] ditiano]-6-il}etanona (Composto 132). e sais farmaceuticamente aceitáveis, hidratos, N- óxidos ou solvatos dos mesmos.
[054] Em uma ou mais modalidades da presente invenção, os compostos da fórmula I possuem um peso molecular abaixo de 800 Daltons, tal que abaixo de 700 Daltons, ou abaixo de 650, 600, 550 ou 500 Daltons.
[055] Em uma ou mais modalidades da presente invenção, os compostos da fórmula I, tal como acima definidos, são úteis em terapia.
[056] Os compostos da fórmula I podem ser obtidos em forma cristalina, seja diretamente através de concentração partir de um solvente orgânico ou através de cristalização ou de recristalização a partir de um solvente orgânico ou mistura do referido solvente e cossolvente, que pode ser orgânico ou inorgânico, tal que a água. Os cristais podem ser isolados em uma forma essencialmente livre de solvente, ou como um solvato, tal que um hidrato. A invenção também abrange todas as modificações e formas cristalinas, e também misturas das mesmas.
[057] Os compostos da fórmula I podem ou não compreender átomos de carbono (quirais) assimetricamente substituídos, que dão origem à existência de formas isoméricas, por exemplo enanciômeros e possivelmente diastereômeros. A presente invenção refere-se a todos tais isômeros, seja em forma pura ou como misturas dos mesmos (por exemplo, racematos). As formas estreoméricas puras dos compostos e intermediários desta invenção podem ser obtidos através da aplicação de procedimentos conhecidos na arte. As várias formas isoméricas podem ser separadas através de métodos de separação física, tais que a cristalização seletiva e técnicas cromatográficas, por exemplo, cromatografia líquida usando fases estacionárias quirais. Os ennciômeros podem ser seprados, um a partir do outro, através da cristalização seletiva de seus sais diastereoméricos com aminas opticamente ativas, tais que I- efedrina. De um modo alternativo, os enanciômeros podem ser separados através de técnicas cromatográficas usando fases estacionárias quirais. As referidas formas estereoisoméricas podem ser também derivadas a partir das formas estereosioméricas puras dos materiais de partida apropriados, contanto que a reação ocorra de um modo estereosseletivo ou estereoespecífico. De um modo preferido, se um estereoisômero específico for desejado, o referido composto será sintetizado através de métodos de preparação estreosseletivos ou estereoespecíficos. Estes métodos irão empregar, de um modo vantajoso, materiais de partida puros quirais.
[058] Os compostos da invenção, opcionalmente em combinação com outros compostos ativos, podem ser úteis para o tratamento de doenças dérmicas ou condições, ou distúrbios de ferida cutânea aguda ou crônica, psoríase, câncer, inflamação epidérmica, alopecia, atrofia da pele, atrofia da pela induzida por esteróide, envelhecimento da pele, fotoenvelhecimento da pele, acne, dermatite, dermatite atópica, dermatite seborréica, dermatite de contato, urticária, prurido, e eczema.
[059] Além de serem úteis para o tratamento humano, os compostos da presente invenção podem ser também úteis para o tratamento veterinário de animais, que incluem mamíferos, tais que cavalos, gado, ovelhas, porcos, cachorros e gatos.
[060] Para o uso na terapia, os compostos da presente invenção estão, de um modo típico, sob a forma de uma composição farmacêutica. A invenção refere-se ainda a uma composição farmacêutica compreendendo um composto da fórmula I, opcionalmente junto com um ou mais outro(s) composto(s) terapeuticamente ativo(s), junto com um excipiente ou veículo farmaceuticamente aceitável. O excipiente precisa ser “aceitável” no sentido de ser compatível com os outros ingredientes da composição e não ser deletério para aquele que o recebe.
[061] De um modo conveniente, o ingrediente ativo compreende de 0,05 - 99,9%, em peso, da formulação.
[062] Sob a forma de uma unidade de dosagem, o composto pode ser administrado uma ou mais vezes ao dia, em intervalos apropriados, sempre dependendo, no entanto, d condição do paciente e de acordo com a prescrição feita pelo médico assistente. De um modo conveniente, uma unidade de dosagem de uma formulação contém entre 0,1 mg e 1000 mg, de um modo preferido entre 1 mg e 100 mg, tl que de 5- 50 mg de um composto da fórmula I.
[063] Uma dosagem adequada do composto da invenção irá depender, inter alia, da idade e da condição do paciente, d severidade da doença a ser tratada e de outros fatores bem conhecidos do médico assistente. O composto pode ser administrado ou por via oral, parenteral ou tópica, de acordo com programas de dosagem diferentes, por exemplo, em intervalos diários ou semanais. De um modo geral, uma dose única deverá estar em uma faixa de 0,01 a 400 mg/ kg de peso corpóreo. O composto pode ser administrado como um bolo (isto é, a dose diária total é administrada de um única vez) ou em doses divididas, duas ou mais vezes ao dia.
[064] No contexto de um tratamento tópico, pode ser apropriado fazer referência a uma “unidade de uso”, que denota uma única dose, que é capaz de ser administrada a um paciente, e que pode ser prontamente manipulada e compactada, permanecendo como uma dose unitária fisicamente e quimicamente estável, compreendendo ou o material ativo como tal, ou uma mistura deste com diluentes ou veículos farmacêuticos sólidos ou líquidos.
[065] O termo “unidade de uso”, em conexão com o uso tópico compreende um dose unitária isto é, uma dose única, capaz de ser administrada, de um modo tópico, a um paciente, em uma aplicação por centímetro quadrado da área infectada de a partir de 0,1 mg a 10 mg, e de um modo preferido de 0,2 mg a 1 mg, do ingrediente ativo em questão.
[066] É também considerado que, em certos regimes de tratamento, a administração com intervalos mais longos, por exemplo, em dias alternados, a cada semana, ou em intervalos ainda mais longos, pode ser benéfica.
[067] Se o tratamento envolver administração de um outro composto terapeuticamente ativo, é recomendado consultar Goodman & Gillman’s The Pharmacological Basis of Therapeutics, 9 a Ed., J. G. Hardman e L. E. Limbird (Eds.), McGraw- Hill, 1995, quanto a dosagens úteis de tais compostos.
[068] A administração de um composto da presente invenção com um ou mais outros compostos ativos pode ser efetuada ou de um modo concomitante ou sequencial.
[069] As formulações incluem, por exemplo, aquelas sob uma forma adequada para a administração oral (incluindo a liberação sustentada ou retardada), retal, parenteral (incluindo subcutânea, intraperitonial, intramuscular, intraarticular e intravenosa), transdérmica, oftálmica, tópica, dérmica, nasal ou bucal. A administração tópica da formulação reivindicada é particularmente adequada.
[070] As formulações podem, de um modo conveniente, ser apresentadas em uma forma unitária de dosagem e podem ser preparadas através de qualquer um dos métodos bem conhecidos na arte da farmácia, por exemplo, como exposto em Remington, The Science and Practice of Pharmacy, 20a Ed., 2000. Todos os métodos incluem o estágio e colocar o ingrediente ativo em associação com o veículo, que é constituído por um ou mis ingredientes acessórios. De um modo geral, as formulações são preparadas através da colocação uniforme e íntima do ingrediente ativo em associação com um veículo líquido ou com um veículo sólido finamente dividido, ou ambos, e então, se necessário, modelagem do produto sob a forma da formulação desejada.
[071] As formulações da presente invenção, adequadas para administração oral, podem estar sob forma de unidades distintas, tais que cápsulas, sachês, comprimidos ou pastilhas, cada qual contendo uma quantidade predeterminada do ingrediente ativo; sob a forma de um pó ou de grânulos; sob a forma de um solução ou de uma suspensão em um líquido aquoso ou em um líquido não- aquoso, tal que etanol ou glicerol; ou sob a forma de uma emulsão óleo- em- água ou de uma emulsão água- em- óleo. Tais óleos podem ser óleos comestíveis, tais que óleo de caroço de algodão, óleo de sésamo, óleo de coco, ou óleo de amendoim. Os agentes de dispersão ou de suspensão adequados para as suspensões aquosas incluem as gomas naturais ou sintéticas, tais que tragacanto, alginato, acácia, dextrano, carboximetilcelulose sódica, gelatina, metil celulose, hidroxipropilmetil celulose, hidroxipropil celulose, carbômeros e polivinil pirrolidona. Os ingredientes ativos podem ser administrados sob a forma de um bolo, de um pessário ou de uma pasta.
[072] Um comprimido pode ser produzido através da compressão ou moldagem do ingrediente ativo, de um modo opcional com um ou mis ingredientes acessórios. Os comprimidos prensados podem ser preparados através de compressão, em uma máquina adequada, do(s) ingrediente (s) ativo (s) em uma forma de fluxo livre, tal que como um pó ou grânulos, opcionalmente misturado por um aglutinante, tal que, por exemplo, lactose, glicose, amido, gelatina, goma acácia, goma tragacanto, alginato de sódio, carboximetil celulose, metil cleulose, hidroxipropilmetil celulose, polietileno glicol, ceras ou os similares; um lubrificante, tal que, por exemplo, oleato de sódio, estearato de sódio, estearato de magnésio, benzoato de sódio, acetato de sódio, cloreto de sódio, ou os similares; um agente de desintegração, tal que, por exemplo, amido, metil celulose, ágar, bentonita, croscarmelose sódica, glicolato de amido sódico, metil celulose, ágar, bentonita, croscarmelose sódica, glicolato de amido sódico, crospovidona ou os similares, ou um agente de dispersão, tal que o polisorbato 80. Os comprimidos moldados podem ser produzidos através de moldagem, em uma máquina adequada, de uma mistura do ingrediente ativo em pó e de um veículo adequado, umedecido com um diluente líquido inerte.
[073] As formulações para administração retal podem estar sob a forma de supositórios, nos quais o composto da presente invenção é misturado com sólidos solúveis ou insolúveis em água com baixo ponto de fusão, tais que manteiga de cacau, óleos vegetais hidrogenados, polietileno glicol ou ésteres de ácidos graxos de polietileno glicóis, enquanto que os elixires podem ser preparados usando palmitato de miristila.
[074] As formulações adequadas para administração parenteral compreendem, de um modo conveniente, uma preparação oleosa ou aquosa estéril dos ingredientes ativos, que é, de um modo preferido, isotônica com o sangue daquele que a recebe, por exemplo, solução salina isotônica, solução de glicose isotônica, ou solução de tampão. A formulação pode ser esterilizada, de um modo conveniente, através, por exemplo, de filtração através de um filtro retentor de bactérias, adição de um agente de esterilização à formulação, irradiação da formulação ou aquecimento da formulação. As formulações lipossômicas são expostas, por exemplo, em Encyclopedia of Pharmaceutical Technology, vol.9, 1994, e são também adequadas para administração parenteral.
[075] De um modo alternativo, os compostos da fórmula I podem ser apresentados como uma preparação sólida, estéril, por exemplo, um pó secado por congelamento, que é prontamente dissolvido em um solvente estéril, imediatamente antes do uso.
[076] As formulações transdérmicas podem estar sob a forma de um esparadrapo ou de um adesivo.
[077] As formulações adequadas para a administração oftálmica podem estar sob a forma de uma preparação aquosa estéril dos ingredientes ativos, que podem estar sob a forma microcristalina, por exemplo, sob a forma de uma suspensão microcristalina aquosa. As formulações lipossômicas ou os sistemas de polímero biodegradáveis, por exemplo, como exposto na Encyclopedia of Pharmaceutical Technology, vol. 2, 1989, podem ser também usadas para apresentar o ingrediente ativo para a administração oftálmica.
[078] As formulações adequadas para a administração tópica ou oftálmica incluem as preparações líquidas ou semilíquidas, tais que linimentos, loções, géis, pulverizações, espumas, emulsões óleo- em- água ou água- em- óleo, tais que cremes, unguentos ou pastas; ou soluções ou suspensões, tais que gotas. As composições para o tratamento oftálmico podem, de um modo preferido, também conter uma ciclodextrina.
[079] Para a administração tópica, o composto da fórmula I, pode, de um modo típico, estar presente em uma quantidade de a partir de 0,01 a 20%, em peso, da composição, tal que de 0,1 % a cerca de 10%, mas pode também estar presente em uma quantidade de até cerca de 50% da composição.
[080] As formulações adequadas para administração nasal ou bucal, incluem formulações em pó, auto- propelentes e pulverizações, tais que os aerossóis e os atomizadores. Tais formulações são expostas em maiores detalhes em, por exemplo, Modern Pharmaceutics, 2a Ed., G. S. Banker e C. T. Rhodes (Eds.), paginas 427- 432, Marcel Dekker, New York; Modern Pharmaceutics, 3 a Ed., G. S. Banker and C.; T. Rhodes (Eds.), páginas 618- 619, e 718- 721, Marcel Dekker, New York and Encyclopedia of Pharmaceutical Technology, vol. 10, J. Swarbrick and J. C. Boylan (Eds.), páginas 191- 221, Marcel Dekker, New York.
[081] Em adição os ingredientes antes mencionados, as formulações de um composto da fórmula I podem incluir um ou mais ingredientes adicionais, tais que diluentes, tampões, agentes aromatizantes, colorantes, agentes tensoativos, agentes de espessamento, conservantes, por exemplo, hidroxibenzoato de metila (incluindo antioxidantes), agentes de emulsificação, e os similares.
[082] Quando o ingrediente ativo é administrado sob a forma de sais com ácidos ou bases não- tóxicos farmaceuticamente aceitáveis, os sais preferidos são, por exemplo, facilmente solúveis em água ou ligeiramente solúveis em água, de um modo a que seja obtida uma taxa de absorção particular e apropriada.
[083] A composição farmacêutica pode, de um modo adicional, compreender um ou mais outros componentes ativos usados, de um modo convencional, no tratamento de doença dérmica ou de condições, por exemplo, selecionadas a partir do grupo, que consiste de glicocorticóides, vitamina D e análogos da vitamina D, anti- histamínicos, fator de ativação de plaqueta (PAF), antgonistas, agentes anticolinérgicos, metil xantinas, agentes e-adrenérgicos, inibidores de COX- 2, salicilatos, indometacina, flufenamato, naproxeno, timegadina, sais de ouro, penicilina, agentes de redução do colesterol no soro, agentes retinóides, sais de zinco, salicilazo sulfapiridina e inibidores de calcineurina.
[084] A invenção é descrita em maiores detalhes nos exemplos que se seguem, que não se destinam, de nenhum modo, a limitar o escopo da invenção, tal como reivindicado.
MÉTODOS DE PREPARAÇÃO
[085] Os compostos da presente invenção podem ser preparados em uma diversidade de modos, bem conhecidos daqueles versados na arte da síntese. Os compostos da fórmula I podem, por exemplo, ser preparados usando as reações e as técnicas descritas abaixo, junto com os métodos conhecidos na arte da química orgânica sintética, ou variações dos mesmos, conforme apreciado por aqueles versados n arte. Os métodos preferidos incluem, mas não estão limitados, àqueles abaixo descritos. As reações podem ser executadas em solventes apropriados para os reagentes e materiais empregados e adequados para as transformações que estão sendo efetuadas. Além disso, nos métodos sintéticos descritos abaixo, deve ser entendido que todas as condições de reação propostas, incluindo a escolha de solvente, a atmosfera de reação, a temperatura da reação, a duração do experimento e os procedimentos de elaboração, são selecionados, de um modo a que sejam condições de padrão para a reação, o que deverá ser prontamente reconhecido por aquele versado na arte. Nem todos os compostos, que recaem em uma determinada classe, podem ser compatíveis com algumas das condições de reação requeridas em alguns dos métodos descritos. Tais restrições quanto aos substituintes, que são compatíveis com as condições de reação, serão prontamente evidentes para aqueles versados na arte, e métodos alternativos podem ser usados.
[086] Os materiais de partida ou são os compostos em si conhecidos, que estão comercialmente disponíveis, ou eles podem ser preparados através de métodos sintéticos de rotina, bem conhecidos de uma pessoa versada na arte.
[087] Os compostos da presente invenção ou quaisquer intermediários podem ser purificados, se requerido, usando métodos convencionais, bem conhecidos de um químico orgânico sintético, por exemplo, os métodos descritos em “Purification of Laboratory Chemicals”, 5a Ed., 2003. Os materiais de partida ou são os compostos em si conhecidos, comercialmente disponíveis, ou eles podem ser preparados através de métodos sintéticos de rotina, bem conhecidos daqueles versados na arte.
PROCEDIMENTOS GERAIS, PREPARAÇÕES E EXEMPLOS
[088] Os espectros de ressonância magnética nuclear 1H (RMN) foram registrados em espectros de RMN de 300 MHz e 13C, em 75, 6 MHz. Os valores de desvio químico (δ, em ppm) são cotados no solvente especificado em relação a padrões internos de tetrametil silano (δ = 0,00) ou clorofórmio (δ = 7, 25), ou deutério clorofórmio (δ = 76, 81 para RMN nC). O valor de um multípleto, seja definido (dubleto (d), tripleto (t), quarteto (q)) ou não (m) no ponto médio aproximado é fornecido, a não ser que uma faixa seja citada. (bs) indica um singleto amplo. Os solventes orgânicos usados foram usualmente anidros. A cromatogrfia foi efetuada em sílica gel Merck 60 (0,040 - 0,063 mm). As rzões de solvente indicaram referir-se a v: v, a não ser que mencionado de um outro modo.
[089] As abreviações que se seguem foram usadas ao longo de todo o texto: DCM DMF DMSO Et EtOAc h L LDA LiHMDS diclorometano N,N’- Dimetilformamida Sulfóxido de dimetila etila acetato de etila hora litro lítio diisopropilamida lítio Hexametildissilazida M mili Me metila MeOH metanol RMN ressonância magnética nuclear ppt precipitado rt temperatura ambiente TsCl cloreto de p-tolueno sulfonila THF tetraidrofurano V volume
HPLC/ MS Preparativo
[090] HPLC/ MS preparativo foi executado em um sistema APS Dionex com duas bombas prep. Shimadzu PP150 e um espectrômetro de massa Thermo MSQ Plus. Coluna: Waters Xterra C-18, 150 mm x 19 mm, 5 μm; sistema de solvente: A = água (0,1% de ácido fórmico) e B= acetonitrila (0,1 % de ácido fórmico); taxa de fluxo = 18 ml/ min; método (10 min.): gradiente linear indo de 10% de B a 00% de B em 6 minutos e permanecendo em 100% de B durante mais 2 minutos. As frações foram coletadas com base em traços de íon de íons relevantes e sinal de PDA (240 - 400 nm).
HPLC/ MS Analítico
[091] Método A: O HPLC/ MS analítico foi executado em um sistema APS Dionex com uma bomba analítica P680A e um espectrômetro de massa Thermo MSQ Plus. Coluna: Waters Xterra C- 18, 150 mm x 4, 6 mm, 5 μm; sistema de solvente: A = água (0,1 % de ácido fórmico) e B= acetonitrila (0,1 % de ácido fórmico); taxa de fluxo = 1,0 ml/ min.; método (10 min): Método de gradiente linear indo de 10% de B a 100% de B em 6,6 minutos, e permanecendo em 100% durante mais 1,5 minutos.
[092] Método B: HPLC/ MS foi executado em um sistema consistindo de HPLC Waters 2795, espectrômetro de massa Micromass ZQ, Waters 996 PDA. Coluna: Waters Xterra C- 18, 50 mm x 3,0 mm, 5 μm; sistema de solvente: A = água: acetonitrila a 95: 5 (0,05% de ácido fórmico) e B= acetonitrila (0,05% de ácido fórmico); taxa de fluxo = 1,0 ml/ min.; método (8 min.: Método de gradiente linear indo de 10% de B para 100% de B em 6,0 minutos e permanecendo em 100% de B durante 1 minuto.
Procedimento geral de preparação:
[093] Os compostos da invenção podem, por exemplo, ser preparados através dos métodos gerais que se seguem:
[094] Os compostos da fórmula geral Ia, em que R1, R2 e R3 são como acima definidos, podem ser preparados como se segue:
I A
[095] Os materiais de partida da fórmula 1a são preparados de acordo com procedimentos convencionais, conhecidos para o químico versado na arte. O ácido 2,3,4-trimetóxi benzóico é di- desmetilado seletivamente nas posições 2 e 3, usando BCl3 de acordo com Kaisalo et al., Synth. Commun., (1996), 16, 645- 48.
[096] A reação subsequente do composto desprotegido em cetonas puras, éteres enólicos, cetais ou uma mistura destes com ou sem catalisadores adicionais, tais que o ácido para- tolueno sulfônico ou um ácido de Lewis, em uma temperatura a partir da temperatura ambiente até 180°C, usando o aquecimento em microondas ou convencional, resulta nos compostos 2a.
[097] A reação dos compostos com a fórmula 2a com MeI (ou sulfato de dimetila), na presença de uma base adequada, tal que K2CO3, KHCO3 ou Et3N, em um solvente adequado, tal que DMF, acetona, THF ou DCM, em temperaturas a partir da temperatura ambiente a até 100°C, fornece os compostos da fórmula 3a.
[098] O éster 3a pode ser também preparado através de métodos de esterificaçõ clássicos, usando o álcool e um ácido adequado, por exemplo, H2SO4.
[099] Os compostos da fórmula I (X = CH2) foram obtidos através da condensação do éster metílico gerado com lítio carbânions gerados a partir de A- Metila, em que A é definido como acima descrito, e uma base adequada, tal que LDA ou LiHMDS, em um solvente adequado, tal que THF, em temperaturas de menos 78°C ou em temperatura ambiente. De um modo alternativo aos lítio carbânions, um reagente de Grignard pode ser usado.
[0100] Os compostos da fórmula Ia (X= NH) foram obtidos através da reação de compostos com a fórmula 2a com (COCl)2, SOCl2 ou PCl5, em um solvente adequado, tal que DCM ou tolueno, com ou sem a quantidade catalítica de DMF, em temperaturas de 0°C a 70°C, de um modo a fornecer o cloreto ácido correspondente. Após a evaporação do solvente in vacuo, é efetuada a condensação subsequente do cloreto ácido gerado com ânions de nitrogênio, gerados pela adição de uma base adequada, tal que NaH, LDA ou LiHMDS, em um solvente adequado, tal que THF, em temperaturas de menos 78°C à temperatura ambiente para A-NH2, em que A é definido como acima descrito.
[0101] Os compostos da fórmula geral Ib, em que R1, R2 e R3 são como acima definidos, podem ser preparados como se segue:
[0102] A esterificação de 1b usando os procedimentos convencionais, por exemplo MeOH e H2SO4, resulta no éster 2b.
[0103] A alquilação de 2b usando 3b (X = Br, I, Ots) na presença de uma base adequada, tal que K2CO3, em um solvente adequado, tal que DMSO, em temperaturas a partir da temperatura ambiente a 120°C, fornecem os compostos da fórmula 4b.
[0104] Os compostos da fórmula Ib (X = CH2) foram obtidos através da condensação do éster metílico gerado com lítio carbânions gerados a partir de A- metila, em que A é definido como acima descrito, e uma base adequada, tal que LDA ou LiHMDS em um solvente adequado, tal que THF, em temperaturas de menos 78° à temperatura ambiente. De um modo alternativo aos lítio carbânions, um reagente de Grignard pode ser usado.
[0105] É esperado que a hidrólise de éster usando as condições convencionais (ácidas ou básicas) resulte no ácido carboxílico 7b, que pode ser convertido ao cloreto do ácido carboxílico e subsequentemente reagido com ânions de nitrogênio (gerados a partir de A-NH2), como descrito para a síntese de Ia (X =NH). Preparação 1: Ácido 7- metóxi-2’,3’, 5’, 6’-tetraidro-espiro [1,3-benzodioxol-2,4’-(4H)- piran]-4-carboxílico (Composto 501)
[0106] Uma suspensão do ácido 2,3- diidróxi-4-metoxibenzóico (6,04 g, 32,8 mmol) em 5,6-diidro-4-metóxi-2H-piran (20 ml, 152 mmol) foi mantida a 140°C durante três dias. Em temperatura ambiente, foi adicionado acetato de etila (200 ml) e a fase orgânica foi extraída com NaHCO 3 (2 x 50 ml). A fase aquosa foi lavada com Et2O (2 x 40 ml), acidificada ao pH = 1 com HCl concentrado e extraída com diclorometano (2 x 50 ml). A fase orgânica foi secada com MgSO4. A evaporação sob pressão reduzida forneceu traços do ácido 2,3-diidróxi-4-metoxibenzóico, junto com o ácido 7-metóxi- 2’, 3’, 5’, 6’ - tetraidro-espiro [1,3-benzodioxol-2,4’-(4H)-piran]-4- carboxílico (1,23 g, 14%). RMN 13C (DMSO) δ 164, 9, 148,2, 146, 6, 134, 5, 123, 7, 117,0, 107, 1, 106, 8, 64, 4, 56, 0, 35, 3. Preparação 2: 7- Metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3-benzodioxol-2,4’-(4H)-piran-4- carboxilto de metila (Composto 502)
[0107] Uma suspensão do ácido 7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol-2, 4’-(4H)- piran]-4-carboxílico (2,17 g, 8,15 mmol), KHCO3 (2, 58 g, 26, 0 mmol) e sulfato de dimetila (1, 58 ml, 16,7 mmol) em acetona (62 ml) foi agitada em temperatura ambiente, durante dois dias, antes que ela fosse evaporada até a secura, sob pressão reduzida. Acetato de etila (100 ml) foi então adicionado. A fase orgânica foi lavada com NaOH aquoso 0,5 M (6 x 30 ml) e evaporado té a secura, sob pressão reduzida. O produto bruto foi novamente dissolvido em diclorometano (75 ml), secado com MgSO4, e evaporado até a secura, sob pressão reduzida. A cromatografia de coluna em sílica gel convencional forneceu 7- metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol-2,4’- (4H)- piran-4-carboxilato (1,87 g, 79%). RMN 13C (CDC13) δ 164,9, 149,1, 147,2, 135,2, 124,0, 117,5, 107,1, 106,5, 65,2, 56,4, 51,8, 35,9. Procedimento Convencional A: Exemplo 1: 2-(3,5-Dic1oropiridina-4-i1)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2, 4’- (4H)- piran] -4-il) etanona (Composto 101). OMe

[0108] Uma so1ução de 7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodixol-2,4’- (4H)- piran]-4- carboxilato de metila (1,80 g, 6, 42 mmol) e 3,5- dicloro-4-picolina (1,46 g, 8,99 mmol) em tetraidrofurano (33 ml) foi resfriado a 0°C. Uma solução 1,0 M de lítio bis (trimetilsilil)amida em tetraidrofurano (19,3 ml, 19,3 mmol) foi adicionada e a mistura da reação foi deixda alcançar a temperatura ambiente durante a noite. NH4Cl (70 ml) aquoso saturado foi adicionado. A fase aquos foi extraída com diclorometano (3 x 100 ml). A fase orgânica combinada foi lavada com água (50 ml), secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A cromatografia de coluna em sílica gel, seguida pela recristalização a partir de isopropanol forneceu 2-(3,5-dicloropiridina-4-il)-1- (7-metóxi-2’, 3’, 5’, 6’- tetraidro- espiro [1,3-benzodioxol-2,4’-(4H)-piran]-4-il) etanona (1,90 g, 71%). RMN i3C (DMSO) δ 189,1, 148,2, 147,7, 147,0, 141, 2, 134, 5, 132, 8, 122,0, 118,0, 113,0, 107, 8, 64,4, 56,3, 43,5, 35,2. Exemplo 2 N-(3,5-Dicloropiridina-4-il)-7- metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’- (4H)- piran]-4- carboxamida (Composto 102) □ME

[0109] Cloreto de oxalila (92 μl, 1,1 mmol) e uma quantidade catalítica de N,N-dimetilformamida foram adicionados a uma suspensão do ácido 7- metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’- (4H)- piran]-4-carboxílico (48 mg, 0,18 mmol) em diclorometano (2 ml). Após a agitação durante uma hora em temperatura ambiente, o solvente foi removido sob pressão reduzida e o cloreto ácido bruto foi novamente dissolvido em tetraidrofurano (2 ml). Uma suspensão de 3,5-dicloropiridin-4-amina (67 mg, 0,40 mmol) e NaH (uma dispersão a 60% em óleo mineral, 16 mg, 0, 40 mmol) em tetraidrofurano (1 ml) foi agitada durante três horas, em temperatura ambiente, antes que ela fosse adicionada, em gotas, em temperatura ambiente, à solução de tetraidrofurano contendo o cloreto ácido bruto. Após ter sido agitada durante a noite em temperatura ambiente, a mistura da reação foi diluída com éter dietílico (30 ml) e a fase orgânica foi lavada com NaOH aquoso 0, 5 M (3 x 10 ml). A fase orgânica foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação através de HPLC convencional forneceu N- (3,5- dicloropiridina-2,4’-(4H)-piran]-4- carboxamida (14 mg, 19%). RMN 13C (DMSO) δ 160,8, 148,0, 146,5, 146,2, 141,1, 134,1, 130,5, 122,5, 118,2, 108,3, 107,6, 64,2, 56,2, 35,2. Exemplo 3: 2-(3,5-Dicloro-1-oxido-piridina-4-il)-1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2, 4’- (4H)- piran]-4-il)etanona (Composto 103) I. o

[0110] A uma solução de 2- (3,5-dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-piran]-4-il)etanona (41 mg, 0,99 mmol) em diclorometano (0, 5 ml) foram adicionados 30 % de H2O2 (25 μl) e metiltrioxorrênio (VII) (3 mg). A mistura foi agitada, em temperatura ambiente durante a noite, MnO2 (3 mg) foi adicionado e foi então agitado durante mais uma hora. Água (10 ml) foi adicionada e fase aquosa foi extraída com diclorometano (3 x 10 ml). A fase orgânica combinada foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação através de HPLC convencional forneceu o composto 103 (8 mg, 19%). LC/ MS (MÉTODO B): (m/z) 426, 1; 428, 1 (MH+); Temp. Amb. = 2,98 min.; pureza (UV) = 100%. Preparação 3: Tetraidro-3,3-dimetoxitiofeno (Composto 503)
[0111] Uma solução de tetraidrotiofen-3-ona (10,0 g, 97, 9 mmol), ortoformato de metila (21,4 ml, 196 mmol) e monoidrato do ácido para- toluenossulfônico (50 mg, 0, 29 mmol) em metanol seco (25 ml) foi então refluído durante uma hora. Então, NaOMe metanólico 1,0 M (0,30 ml, 0,30 mmol) foi adicionado e excesso de metanol e ortoformato de trimetila foram removidos através de destilação (pressão atmosférica). A destilação adicional sob pressão reduzida forneceu uma mistura de tetraidrotiofen-3-ona (~ 0,67 g, 7%) e tetraidro-3,3-dimetoxitiofeno (~9,8 g, 67%). RMN 13C (MeOH) δ 113,01, 50,11, 36,90, 36,11, 27,72. Preparação 4: Ácido 7-metóxi-4’,5’-diidro-espiro carboxílico (Composto 504)
[0112] Ácido p-toluenossulfônico (54 mg, 0,28 mmol) foi adicionado à mistura de tetraidro-3,3- dimetoxitiofen (~ 9,8 g, 66 mmol) e tetraidrotiofen- 3-ona (~ 0,67 g, 6,6 mmol). O banho de óleo foi aquecido a 145°C e aproximadamente um equivalente de metanol (2,7 ml, 67 mmol) foi destilado. A temperatura foi reduzida e a destilação sob pressão reduzida forneceu 7,04 g de um óleo, ao qual foi adicionado o ácido 2,3-diidróxi-4-metoxibenzóico (1,00 g, 5,43 mmol). A suspensão foi exposta ao aquecimento em microondas (180 °C, uma hora) em um vaso de reação vedado. A filtração e a purificação por HPLC convencional subsequente forneceu o composto 504 (164 mg, 11%). LC/MS (MÉTODO B): (m/z) 267, 2 (M-1); RT = 2,79 minutos; pureza (UV)= 100%. Preparação 5: 7-metóxi-4’,5’-diidro-espiro[1,3-benzodioxol-2,3’-(2H)-tiofen]-4-carboxilato de metila (Composto 505)
[0113] Uma suspensão do ácido 7-metóxi-4’,5’-diidro-espiro[1,3- benzodioxol-2,3’- (2H)-tiofen]-4- carboxílico (161 mg, 0, 600 mmol), K2CO3 (166 g, 1,20 mmol) e sulfato de dimetila (74μl, 0,78 mmol) em acetona (1 ml) foi mantida a 50°C, durante a noite. Água em temperatura ambiente (15 ml) foi adicionada e a fase aquosa foi extraída com acetato de etila (2 x 20 ml). A fase orgânica foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto 505 (24 mg, 14%). RMN »H (CDCI3) δ 7,44 (d, 1 H), 6, 56 (d, 1 H), 3, 94 (s, 3 H), 3,88 (s, 3 H), 3,32 (d, 1 H), 3,24 (d, 1 H), 3,05 (t, 2 H), 2, 49 (td, 2 H). Exemplo 4: 2-(3,5-Dicloropiridina-4-il)-1-(7-metóxi-4’,5’-diidro-espiro [1,3-benzodioxol- 2, 3’-(2H)-tiofen]-4-il) etanona (Composto 104)
[0114] Uma solução de 7-metóxi-4’,5’-diidro-espiro [1,3- benzodioxol-2,3’-(2H)-tiofen]-4-carboxilato de metila (24 mg, 85 μmol) e 3,5-dicloro-4-picolina (21 mg, 0,13 mmol) em tetraidrofurano (1 ml) foi resfriada a 0°C. Uma solução 1,0 M de lítio bis (trimetilsilil) amida em tetraidrofurano (0,26 ml, 0,26 mmol) foi adicionada e a mistura da reação foi deixada alcançar a temperatura ambiente, durante a noite. NH4Cl (10 ml) aquoso saturado foi adicionado. A fase aquosa foi extraída com diclorometano (3 x 10 ml). A fase orgânica combinada foi lavada com água (20 ml), secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto do título (12 mg, 34%). RMN 13C (DMSO) δ 189,02, 148,09, 147,51, 147,05, 140,98, 134,48, 132,69, 127,57, 122,31, 112,81, 107, 81, 56,35, 43,26, 37,54, 36,54, 25,70. Preparação 6: 1-Acetil-4,4-dimetóxi-piperidina (Composto 506)
[0115] Uma solução de 1-acetil-4-piperidona (17,9 g, 121 mmol), ortoformato de trimetila (26, 4 ml, 241 mmol) e monoidrato do ácido para- tolueno sulfônico (80 mg, 0,42 mmol) em metanol seco (34 ml) foi refluída durante 1 hora. Então, NaOMe metanólico 1,0 M (0, 42 ml, 0, 42 mmol) e excesso de metanol e ortoformato de trimetila foram removidos através de destilação (pressão atmosférica). Além disso, a destilação sob pressão reduzida forneceu 1-acetil-4,4-dimetóxi-piperidina (20, 2 g, 89%). RMN iH (DMSO) δ 3, 45 - 3,32 (m, 4 H), 3,10 (s, 6H), 1,99 (s, 3 H), 1, 72 - 1, 62 (m, 2 H), 1,61 - 1, 52 (m, 2 H). Preparação 7: 1-Acetil-1, 2, 3, 6-tetraidro-4-metóxi-piridina (Composto 507)
[0116] A 1-acetil-4,4-dimetóxi-piperidina (20,2 g, 108 mmol) foi adicionado o monoidrato do ácido para- tolueno sulfônico (80 mg, 0,42 mmol). A mistura foi então aquecida a 160°C e aproximadamente um equivalente de metanol (4,38 ml, 108 mmol) foi destilado. A temperatura foi reduzida e a destilação sob pressão reduzida forneceu uma mistura de 1- acetil-4,4-dimetóxi-piperidina (1,4 g, 7%) e 1-acetil-1,2,3,6-tetraidro-4- metóxi-piridina (14,2 g, 85%). RMN i H (DMSO) δ 4, 68 - 4,62 (m, 1 H), 4,00 - 3,88 (m, 2 H), 3, 59 - 3, 49 (m, 2 H), 3, 49 - 3,45 (m, 3 H), 2, 19 - 2,12 (m, 1 H), 2,09 - 2,03 (m, 1 H), 2,03 - 1,96 (m, 3 H). Preparação 8: Ácido 7-metóxi-1’-acetil-espiro [1,3-benzodiol-2,4’-piperidina]-4-carboxílico (Composto 508)
[0117] Uma mistura de ácido 2, 3-diidróxi-4-metoxibenzóico (1,23 g, 6,67 mmol), 1-acetil-4,4-dimetóxi- piperidina (1,4 g, 7,6 mmol) e 1-acetil- 1,2,3,6- tetraidro-4-metóxi-piridina (14,2 g, 91,5 mmol) foi exposta ao aquecimento em microondas (180°C, uma hora), em um vaso de reação vedado. A filtração e a purificação por HPLC convencional subsequente forneceu o composto 508 (0,54 g, 26%). LC/ MS (MÉTODO B): (m/ z) 308, 2 (MH+); RT = 2, 27 minutos; pureza (UV) = 95%. Preparação 9 7-Metóxi-espiro[1,3-benzodioxol-2,4’-piperidina]-4-carboxilato de metila (Composto 509)
[0118] Uma solução do ácido 7-metóxi-1’-acetil-espiro [1,3- benzodioxol-2,4’-piperidina]-4-carboxílico (143 mg, 0, 467 mmol) e LiOH (224 mg, 9,34 mmol) em água (3 ml) e MeOH (3 ml) foi aquecida a 75°C durante cinco horas. Em temperatura ambiente, a mistura foi neutralizada com HCl 2 M e evaporada até a secura, sob pressão reduzida. O ácido 7-metóxi- espiro [1,3-benzodioxol-2,4’-piperidina]-4-carboxílico bruto [LC/MS (MÉTODO B): (m/ z) 266, 2 (MH+); RT = 1, 57 minutos; pureza (UV) = 82%] foi refluída durante a noite em HCl metanólico 1, 7 M (5 ml). Água em temperatura ambiente (20 ml) foi adicionada. A fase aquosa foi lavada com Et2O (10 ml), tornada básica pela adição de Na2CO3 e extraída com diclorometano (3 x 10 ml). A fase orgânica foi secada com MgSO4 e a evaporação sob pressão reduzida forneceu 7-metóxi-espiro [1,3-benzodioxol- 2,4’-piperidina]-4-carboxilato de metila (75 mg, 57%). RMN ÃH (DMSO) δ 7,31 (d, 1 H), 6,72 (d, 1 H), 3,87 (s, 3 H), 3,78 (s, 3 H), 2, 96 - 2,77 (m, 4 H), 1, 94 - 1,83 (m, 4 H). Exemplo 5: 2-(3,5-Dicloropiridina-4-il)-1-(7-metóxi- espiro [1,3-benzodioxol-2,4’- piperidina]-4-il) etanona (Composto 105)
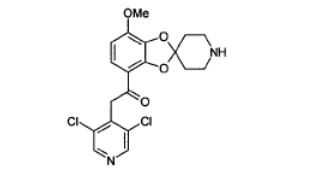
[0119] Uma solução de 7-metóxi- espiro {1,3-benzodioxol-2,4’- piperidina]-4-carboxilato de metila (75 mg, 0, 268 mol) e 3,5-dicloro-4- picolina (65 mg, 0,40 mmol) em tetraidrofurano (2, 5 ml) foi resfriada a 0°C. Uma solução 1,0 M de lítio bis (trimetilsilil) amida em tetraidrofurano (0,80 ml, 0, 80 mmol) foi adicionada e a mistura da reação foi deixada alcançar a temperatura ambiente durante a noite. NH4Cl aq1uoso saturado (10 ml) foi adicionado. A fase aquosa foi extraída com diclorometano (3 x 10 ml). A fase orgânica combinada foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto 105 (58 mg, 53%). RMN »H (DMSO) δ 8, 66 (s, 2 H), 7,38 - 7,36 (m, 1 H), 6,83 - 6, 80 (m, 1 H), 4, 62 (s, 2 H), 3, 91 (s, 3 H), 3,02 - 2, 91 (m, 4 H), 2,12 - 1,93 (m, 4 H). Exemplo 6 2-(3,5-Dicloropiridina-4-il)-1- (7-metóxi-1’-[metoxicarbonil]-espiro [1,3- benzodioxol-2,4’-piperidina]-4-il) etanona (Composto 106).
[0120] Uma solução de 2-(3,5-dicloropiridina-4-il)-1-(7-metóxi- espiro[1,3-benzodioxol-2,4‘- piperidina]-4-il) etanona (10 mg, 24 μmol), trietilamina (24 μl, 171 μl) e cloroformato de metila (10 μl, 122 μmol) em diclorometano (200 μl) foi mantida em temperatura ambiente, durante a noite. Água (500 μl) foi adicionada e a fase aquosa foi extraída com diclorometano (3 x 500 μl). A fase orgânica foi secada com MGSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto 106 (2,5 mg, 22%). RMN »H (DMSO) δ 8, 65 (s, 2 H), 7, 39 (d, 1 H), 6,84 (d, 1 H), 4,62 (s, 2 H), 3, 91 (s, 3 H), 3, 80 - 3, 66 (m, 2H), 3, 62 (s, 3 H), 3, 58 - 3, 46 (m, 2 H), 2, 18 - 1, 97 (m, 4 H). Exemplo 7: 2- (3,5-Dicloropiridina-4-il)-1-(7-metóxi-1’-[metilsulfonil] espiro [1,3- benzodioxol-2,4’- piperidina]—4-il) etanona (Composto 107). □Me
[0121] Uma solução de 2-(3,5-dicloropiridina-4-il)-1-(7-metóxi- espiro [1,3-benzodioxol-2,4’-piperidina]-4-il)etanona (10 mg, 24 μmol), trietilamina (24 μl, 171 μmol) e cloreto de mesila (10 μl, 122 μmol) em diclorometano (200 μl) foi mantida em temperatura ambiente, durante a noite. Água (500 μl) foi adicionada e a fase aquosa foi extraída com diclorometano (3 x 500 μl). A fase orgânica foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto 107 (1,8 mg, 15%). RMN (DMSO) δ 8, 66 (s, 2 H), 7,45 - 7, 38 (m, 1 H), 6,88 - 6,81 (m, 1 H), 4, 62 (s, 2 H), 3, 92 (s, 3 H), 3, 50 - 3, 36 (m, 4 H), 2, 98 (s, 3H), 2, 29 - 2,11 (m, 4 H). Exemplo 8: 2-(3,5-Dicloropiridina-4-il)-1- (7-metóxi-1’-acetil-espiro [1,3-benzodioxol- 2,4’-piperidina]-4-il) etanona (Composto 108)
[0122] Uma solução de 2-(3,5-dicloropiridina-4-il)-1-(7-metóxi- espiro[1,3- benzodioxol-2,4’-piperidina]-4-il) etanona (10 mg, 24 μmol), trietilamina (24 μl) foi mantida em temperatura ambiente, durante a noite. Água (500 μl) foi adicionada e a fase aquosa foi extraída com diclorometano (3 x 500 μl). A fase orgânica foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto 108 (7,2 mg, 65%). RMN >H (DMSO) δ 8, 66 (s, 2 H), 7, 40 (d, 1 H), 6, 84 (d, 1 H), 4, 63 (s, 2 H), 3,92 (s, 3 H), 3,90 - 3,84 (m, 1 H), 3,75 - 3,68 (m, 1 H), 3,64 - 3,58 (m, 1 H), 3,55 - 3,49 (m, 1 H), 2,21 - 2,15 (m, 1 H), 2,15 - 2, 05 (m, 5 H), 2,01 - 1, 94 (m, 1 H). Preparação 10: 4,4-Dimetoxitetraidro-(4H)-tiopirano (Composto 510)
[0123] Uma mistura de tetraidro(4H)-tiopiran-4-ona (15:0 g, 129 mmol), ortoformato de trimetila (28,3 ml, 258 mmol) e monoidrato do ácido para- tolueno sulfônico (67 mg, 0, 35 mmol) em metanol (40 ml) foi refluída durante 1 hora. A mistura da reação foi resfriada à temperatura ambiente, NaOMe 1 M (0, 35 ml, 0, 35 mmol) foi adicionado e metanol em excesso e ortoformato de trimetila foram removidos através de destilação (pressão atmosférica). A destilação adicional sob pressão reduzida forneceu 4,4- dimetoxitretraidro-(4H)-tiopirano (20,7 g, 99%). RMN iR (DMSO) δ 3, 07 (s, 6H), 2, 56 (m, 4 H), 1, 84 (m, 4H). Preparação 11: Ácido 7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol-2,4’-(4H)- tiopiran]-4-carboxílico (Composto 511).
[0124] Ácido p-tolueno sulfônico (97 mg, 0,51 mmol) foi adicionado a 4,4-dimetoxitetraidro-(4H)- tiopiran (20,7 g, 128 mmol) e a mistura foi aquecida a 145°C e mantida nesta temperatura até que aproximadamente um equivalente de metanol (5,17 ml, 128 mmol) fosse destilado. A mistura foi então resfriada a 130°C e a destilação sob pressão reduzida forneceu 10,1 g de uma mistura a 5:3 de 5, 6-diidro-4-metóxi-(2H)-tiopiran PH RMN (DMSO) δ 4, 87 (m, 1 H), 3,44 (s, 3 H), 3, 15 (dt, 2 H), 2, 72 (t, 2 H), 2,22 (m, 2 H)] e 4,4-dimetoxitetraidro-(4H)-tiopirano. Sem purificação adicional, a mistura foi adicionada ao ácido 2,3-diidróxi-4-metoxibenzóico (2,00 g,10,9 mmol) e a suspensão foi exposta a aquecimento em microondas (180°C, uma hora) em um vaso de reação vedado. Acetato de etila (100 ml) foi adicionado e a fase orgânica foi primeiramente lavada com HCl 0,5 M (40 ml) e então extraída com NaHCO3 aquoso saturado (2 x 30 ml). A fase aquosa foi então lavada com Et2O (2 x 40 ml), acidificada a um pH = 1 com HCl concentrado e extraída com diclorometano (2 x 30 ml). A fase orgânica foi secada com MgSO4. A evaporação sob pressão reduzida forneceu 7- metóxi-2’, 3’, 5’, 6’- tetraidro-espiro [1,3- benzodioxol-2,4’- (4H)-tiopiran]-4-carboxílico (1,86 g, 61%). RMN 13C (DMSO) δ 164,9, 148, 2, 146, 6, 134, 5, 123, 8, 118,0, 107, 2, 106,9, 56,1, 35,9, 25,4. Preparação 12: 7-Metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3-benzodioxol-2,4’-(4H)-tiopiran]- 4-carboxilato de metila (Composto 512).

[0125] Uma suspensão do ácido 7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol-2, 4’-(4H)- tiopiran]-4-carboxílico (570 mg, 2,02 mmol), K2CO3 (558 mg, 4,04 mmol) e sulfato de dimetila (0,25 ml, 2, 62 mmol) em acetona (14 ml) foi agitado a 50°C, durante a noite. Água em temperatura ambiente (30 ml) foi adicionada. A fase aquosa foi extraída com diclorometano (3 x 15 ml). A fase orgânica combinada foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A cromatografia de coluna em sílica gel convencional forneceu 7- metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’- (4H)-tiopiran]-4-carboxilato de metila (407 mg, 68%). RMN 13C (DMSO) δ 163, 8, 148, 1, 146, 9, 134, 6, 123,4, 118,3, 107,1, 105,9, 56,1, 35,9, 25,4. Exemplo 9: 2-(3,5-Dicloropiridina-4-il)-1-(7-metóxi- 2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (Composto 110)
[0126] Uma solução de 7-metóxi-2’,3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4- carboxilato de metila (40 mg, 0,145 mmol) e 3,5-dicloro-4-picolina (33 mg, 0, 20 mmol) em tetraidrofurano (1, 1 ml) foi resfriada a 0°C. Uma solução 1,0 M de lítio bis (trimetilsilil) amida em tetraidrofurano (0,41 ml, 0,41 mmol) foi adicionada e a mistura da reação foi deixada alcançar a temperatura ambiente, durante a noite. NH4Cl aquoso saturado (20 ml) foi adicionado. A fase aquosa foi extraída com diclorometano (3 x 15 ml). A fase orgânica combinada foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 2- (3,5- dicloropiridina-4-il)-1-(7-metóxi- 2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’- (4H)- tiopiran]-4-il) etanona (38 mg, 67%). RMN 13C (DMSO) δ 189, 1, 148, 2, 147,7, 147,0, 141, 3, 134, 5, 132, 8, 122,0, 119, 1, 113,0, 107, 9, 56, 3, 43, 6, 35,9, 25,5. Exemplo 10 2-(3,5-Dicloropiridina-4-il)-1-(7-metóxi-2,’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’-(4H) tiopiran-1’-óxido]-4-il)etanona (Composto 111).

[0127] A uma solução de 2-(3,5-dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’ - tetraidro- espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (17 mg, 40 μmol) em diclorometano (0,5 ml) foi adicionado primeiramente H2O2 0, 25 M em etanol (128 μl, 32 μmol) e então, em segundo lugar, metiltrioxorrênio (VII) (1 mg, 4 μmol). A mistura foi agitada, em temperatura ambiente, durante dois dias e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 2-(3, 5- dicloropiridina-4-il)-1-(7-metóxi-2, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4 H) tiopiran-1’-óxido]-4-il) etanona (7 mg, 40%). RMN 1H (DMSO) δ 8,66 (s, 2 H), 7,42 (d, 1 H), 6,85 (d, 1 H), 4,63 (s, 2 H), 3,93 (s, 3 H), 3, 17- 2, 94 (m, 4 H), 2,69 - 2,55 (m, 2 H), 2,36 - 2,24 (m, 2 H). Exemplo 11: 2- (3, 5-Dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2, 4’-(4H)-tiopiran-1’,1’-dióxido]-4-il)etanona (Composto 112).
[0128] A uma solução de 2-(3,5-dicloropiridina-4-il)-1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3] benzodioxol-2,4’-(4H)-tiopiran]-4-il)etanona (11 mg, 26 μmol) em diclorometano (0,25 ml) foi adicionado o ácido meta- cloroperbenzóico (10 mg, 58 μmol) e a reação foi agitada em temperatura ambiente, durante a noite. NaHCO3 aquoso saturado (1 ml) foi adicionado, e a fase aquosa foi extraída com diclorometano (2 x 2 ml). A fase orgânica combinada foi secada com MgSO4, e evaporada até a secura, sob pressão reduzida. A puririficação por HPLC convencional forneceu 2-(3,5- dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3]benzodioxol-2, 4’-(4H)- tiopiran-1’,1’-dióxido]-4-il) etanona (5 mg, 42%). RMN iR (CDCl3) δ 8, 52 (s, 2 H), 7,55 (d, 1 H), 6,68 (d, 1 H), 4,55 (s, 2 H), 3,99 (s, 3 H), 3,45 - 3,37 (m, 2 H), 3,33- 3,25 (m, 2 H), 2,79 -2,66 (m, 4 H). Exemplo 12: 2- (3,5-Dicloro-1-[óxido-piridina-4-il)-1- (7-metóxi-2’,3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’-(4H) - tiopiran-1’,1’-dióxido]-4-il) etanona (Composto 113)

[0129] A uma solução de 2-(3,5-dicloropiridina-4-il)-1-(7-metóxi-2’, 3’, 5’, 6-tetraidro - espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (10 mg, 23 μmol) em etanol absoluto (2 ml) foi adicionado primeiramente H2O2 (100 μl, 0, 97 mmol) e então, em segundo lugar, metiltrioxorrênio (VII) (2 mg, 8 μmol). A mistura foi então agitada a 40°C durante a noite, antes que fossem adicionados 5% P/ V de NaHSO3 (10 ml).
[0130] A fase aquosa foi extraída com diclorometano (3 x 20 ml). A fase orgânica combinada foi secada com MgSO4, e evaporada até a secura, sob pressão reduzida. A purificação por PPLC convencional forneceu 2-(3,5- dicloro-1-óxido- piridina-4-il) -1- (7-metóxi- 2’, 3’’, 5’, 6’-tetraidro- espiro [1,3-benzodioxol-2, 4’-(4H)-tiopiran-1’, 1’-dióxido ]-4-il) etanona (2,5 mg, 23 %). RMN iH (DMSO) iH δ 8, 64 (s, 2 H), 7,42 (d, 1H), 6,85 (d, 1 H), 4,59 (s, 2 H), 3, 92 (s, 3 H), 3,51 (m, 2 H), 3,31 (m, 2 H), 2,59 (m, 45 H). Procedimento Geral A:
[0131] LiHMDS (1 M em THF, 3, 0 eq.) foi adicionado, em gotas, a uma solução gelada do éster 512 (1 eq.) e A-metila (1,3 eq.) em THF anidro. A mistura da reação foi agitada, em temperatura ambiente, durante 12 horas, H2O (10 ml) e NH4Cl aquoso saturado (20 ml) foram então extraídos com EtOAc (3 x 50 ml). As fases orgânicas combinadas foram secadas (Na2SO4), filtradas e concentradas sob pressão reduzida. O resíduo obtido foi purificado através de cromatografia de coluna de cintilação, de um modo a fornecer cetona.
Exemplo 13: 2-(3-bromopiridina-4-il)-1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2, 4’-(4H)-tiopiran]-4-il) etanona (Composto 114).
[0132] Preparado de acordo com o Procedimento Geral A usando 18 mg de 4- bromo-3-metilpiridina (rendimento: 40%)
[0133] LC/ MS (MÉTODO B): (m/z) 436, 2 (MH+); RT = 4, 17 min.; pureza (UV)= 100% Exemplo 14: 2-(3-Bromo- pirazin-2-il)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4-il) etanona (Composto 115).
[0134] Preparado de acordo com o Procedimento Geral, usando 17 mg de 2-bromo-3-metilpirazina (rendimento: 9%).
[0135] LC/ MS (MÉTODO B): (m/ z) 437, 2; 439, 22 (MH+); RT = 4,22 min.; pureza (UV) = 100% Exemplo 15: 2-(-pirazin-2-il)-1-(7-metóxi-2’,3’,5’, 6’-tetraidro-espiro[1,3-benzodioxol- 2,4’-(4H)-tiopiran]-4-il) etanona (Composto 116).
[0136] Preparado de acordo com o Procedimento Geral A usando 6 mg de 2-metilpirazina (rendimento: 52%)
[0137] LC/ MS (MÉTODO B): (m/ z) 359, 3 (MH+); RT = 3,33 min.; pureza (UV) = 100% Exemplo 16: 2-(-piridin-4-il-)-1-(7-metóxi-2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol- 2,4’- (4H)- tiopiran]---4-il) etanona (Composto 117).
[0138] Preparado de acordo com o Procedimento geral A, usando 18 mg de 4-metil- piridina (rendimento: 13%).
[0139] LC/ MS (MÉTODO B): (M/ Z) 358, 3 (MH+); RT = 2, 50 min; pureza (UV) = 100% Exemplo 17: 2-(quinolin-4-il)-1- (7-metóxi- 2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’-(4H)- tiopiran]-4-il) etanona (Composto 118)
[0140] Preparado de acordo com o Procedimento geral A, usando 16 mg de 4- metilquinolina (rendimento: 12 %).
[0141] LC/ MS (MÉTODO b): (M/ Z) 408, 3 (MH +); RT = 33, 33 min; pureza (UV) = 100% Exemplo 18: 2-(2,6-Dicloro-fenil)-1-(7-metóxi- 2’, 3’, 5’, 6’-tetraidro- espiro [1,3- benzodioxol-2,4’- (4H)-tiopiran]-4-il) etanona (Composto 119).
[0142] Preparado de acordo com o Procedimento Geral A, usando 15 mg de 2,6-diclorotolueno (rendimento: 8%).
[0143] LC / MS (MÉTODO B): (m/ z) 425, 24 (MH+); RT = 5, 28 min; pureza (UV) = 100%. Exemplo 19: 2-(2-cloro-fenil)-1- (7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3-benzodioxol- 2,4’-(4H)- tiopiran]-4-il) etanona (Composto 120).
[0144] Uma solução de 7-metóxi-2’, 3’, 5’, 6’-tetraidro-espiro [1,3- benzodioxol-2,4’-(4H)-tiopiran]-4- carboxilato de metila (14 mg) em THF seco (500 ml) foi resfriada a 0°C, sob Ar. Cloreto de 2- clorobenzil magnésio em éter dietílico (solução 0, 25 M, 189 μl) foi adicionada e o resfriamento foi removido. Após 2 horas em temperatura ambiente, uma porção adicional de cloreto de 2- clorobenzil magnésio em éter dietílico (solução 0, 25 M, 189 μl) foi adicionada. A mistura foi então agitada durante 18 horas, água foi adicionada e extraída com acetato de etila. As fases orgânicas combinadas foram secadas com MgSO4, e evaporadas até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu o composto 120 (9,2 %).
[0145] LC/ MS (MÉTODO B): (m/ Z) 391, 22 (MH +); RT = 4, 90 minutos; pureza (UV) = 100% Preparação 13: 2,3-Diidróxi-4-metóxi benzoato de metila (Composto 513)
[0146] Uma solução do ácido 2,3-diidróxi-4-metoxibenzóico disponível (11,6 g, 63 mmol) em MeOH anidro (150 ml) foi resfriada em um banho de gelo, e H2SO4 (8 ml) foi adicionado, em gotas. A mistura da reação foi refluída durante 12 horas, e então resfriada à temperatura ambiente, e o solvente foi removido sob pressão reduzida. H2O (100 ml) e NaHCO3 aquoso saturado (50 ml) foram adicionados e extraídos com EtOAc (3 x 100 ml). A fase orgânica combinada foi secada (Na2SO4), filtrada e concentrada in vacuo, de um modo a fornecer o composto 513 como um sólido amarelo, que foi usado no próximo estágio, sem purificação adicional. LC- MS: RT = 2,31 min.; m/z 197,3 (M - H)-.
[0147] RMN iR (CDCI3): δ 10, 83 (1 H, s), 7,41 (1 H, d, J 9,0), 6, 50 (1 H, d, J 8,9), 5,45 (1 H, s), 3, 94 (3 H, s), 3, 93 (3 H, s). Procedimento Geral B:
[0148] A uma solução agitada do Composto 513 e 3 b (1,1 eq.) em DMSO anidro, foi adicionado K2CO3 (2,5 eq.) e a mistura foi agitada a 100°C durante de 4- 12 horas, sob atmosfera inerte. Após o resfriamento à temperatura ambiente, a mistura de água gelada foi adicionada, agitada durante 15 minutos, e então extraída com EtOAc (3 x 50 ml). As fases orgânicas combinadas foram secadas (Na2SO4), filtradas e concentradas sob pressão reduzida. O resíduo obtido foi purificado através de cromatografia de coluna de cintilação.
[0149] Usando o procedimento geral B, foram obtidos os compostos que se seguem: Preparação 14: Éster metílico do ácido 9-metóxi-espiro [ 2 H- 1,5-benzodioxepin-3(4 H), 3’- oxetano]-6- carboxílico (Composto 514).
[0150] Seguindo o procedimento geral, a dialquilação de 513 (198 mg, 1 mmol) com 3,3- bis (iodometil) oxetano comercial (372 mg, 1,1 mmol) em DMSO (5 ml), na presença de K2CO3 (345 mg, 2,5 mmol) forneceu o composto 514 como um material sólido branco após a purificação através de cromatografia de coluna (50-65% de EtOAc em petróleo leve). LC- MS: RT = 2,40 min.; m/ z 281, 26 (M + H)+. RMN ÍH (CDCl3): δ 7, 49 (1 H, d, J 8,8), 6,62 (1 H, d, J 8,8), 4, 61 (2 H, d, J 6,8), 4,58 (2 H, J 6,8), 4,48 (4 H, s), 3,90 (3 H, s), 3,87 (3 H, s). Preparação 15: Éster metílico do ácido 9-metóxi-espiro [2 H-1,5-benzodioxepin-3 (4H), 3’- tietano]-6- carboxílico (Composto 516) Estágio A:
[0151] 3,3-Bis (bromometil) tietano 515 foi obtido em um processo de dois estágios de 1,3- dibromo-2,2- bis (bromometil) propano, seguido o procedimento da literatura (Petrukhina, M. a; Henck, C.; Li, B.; Block, E.; Jin, J.; Zhang, S-Z.; Clerac, R. Inorg. Chem. 2005, 44, 77-84). Deste modo, uma mistura de 1,3-dibromo-2,2-bis(bromometil) propano (7,76 g, 20 mmol) e KDAc (2,28 g, 20 mmol) em THF anidro (30 ml), foi refluído durante 30 horas. O ppt foi filtrado e o filtrado foi concentrado e o resíduo obtido foi purificado através de cromatografia de coluna de cintilação (10-25% de EtOAc em petróleo leve) de um modo a fornecer o éster (2,2- (bisbromometil)-3-bromo propil) do ácido tiocético como um material sólido amarelo pálido. Uma mistura do éster (2,2-(bisbromometil)-3-bromopropil do ácido tioacético (1,53 g, 4 mmol) e NaOMe (324 mg, 6 mmol) em MeOH anidro (10 ml) foi agitado a 0°C, durante 2 horas. O MeOH foi removido in vacuo, co- evaporado com tolueno (2 x 2 ml) e o resíduo obtido foi filtrado através de um tampão curto de sílica gel, de um modo a fornecer 3,3- bis (bromometil) tietano 515 como um óleo espesso, que foi usado sem purificação adicional. Estágio B:

[0152] Seguindo o procedimento geral, a dialquilação de 513 (665 mg, 3,36 mmol) com 515 (962 mg, 3,7 mmol) em DMSO (15 ml) na presença de K2CO3 (1,16 g, 8,4 mmol) forneceu 516 como um material sólido branco, após a purificação através de cromatografia de coluna (40- 60% de ETOAc em petróleo leve). LC- MS: RT = 3, 17 minutos; m/ z 297, 19 (M + H)+. RMN >H (CDCI3): δ 7,49 (1 H, d, J 8,8), 6,63 (1 H, d, J 8,8), 4, 30 (2 H, s), 4,28 (2 H, s), 3, 90 (3 H, s), 3,87 (3 H, s), 3,11 (4 H, s). Preparação 16: Éster metílico do ácido 9-metóxi-espiro [2 H-1,5-benzodioxepin-3(4H), 2’- (1,3-dioxolano)]-6-carboxílico (Composto 518). Estágio A:
[0153] 2,2-Bis(bromometil)-1,3-dioxolano 517 foi obtido a partir de dibromoeacetona seguindo o procedimento da literatura (Valentin, M- L.; Bolte, J. Bull. Soc. Chim. Fr., 1995, 132, 1167 - 71). Deste modo, uma solução de dibromoacetona (4, 04 g, 18,7 mmol), etileno glicol (2,32 g, 37,4 mmol) e p- TsOH (25 mg) em benzeno (70 ml) foi refluída durante 12 horas, com remoção azeotrópica da água. A mistura da reação foi concentrada sob pressão reduzida, Et2O (50 ml) foi adicionado e a camada orgânica foi lavada com H2O (2 x 50 ml), secada (Na2SO4), filtrada e concentrada sob pressão reduzida. O resíduo obtido foi purificado através de cromatografia de coluna de cintilação (7- 10% de EtOAc em petróleo leve), de um modo a fornecer 2,2-bis (bromometil)-1,3-dioxolano (517) como um líquido incolor. Estágio B:
[0154] Seguindo o procedimento geral, a dialquilação de 513 (396 mg, 2 mmol) com 517 (572 mg, 2,2 mmol) em DMSO (10 ml) na presença de K2CO3 (690 mg, 5 mmol) forneceu 518 como um material sólido branco após a purificação através de cromatografia de coluna (45- 60 % de EtOAc em petróleo leve). LC- MS: RT = 2,70 minutos; m/ z 297, 18 (M + H)+, 319,16 (M + Na)+.
[0155] RMN 1H (DMSO- d6): δ 7,34 (1 H, d, J 8,8), 6,76 (1 H, d, J 8,8), 4,11 (2 H, s), 4,09 (2 H, s), 3,94 (4 H, s), 3,80 (3 H, s), 3,75 (3 H, s). Preparação 17: Éster metílico do ácido 9-metóxi- espiro [2 H-1,5-benzodioxepin-3 (4H), 4’- tetraidropiran]-6-carboxílico (Composto 520). Estágio A:
[0156] 4,4-Bis (p-toluenossulfoniloximetil) tetraidropirano (519) foi obtido em um processo de dois estágios, a partir do éster dimetílico do ácido tetraidropiran-4,4-dicarboxílico comercial. Deste modo, a uma solução gelada do diéster (3,03 g, 15 mmol) em tolueno anidro (45 ml) foi adicionado, em gotas, anidrido (70% em tolueno, 19,5 ml, 66 mmol) e a mistura foi agitada a 120°C, durante 3 horas. A mistura foi resfriada à temperatura ambiente, H2O (50 ml) foi adicionado lentamente e concentrado com sílica gel (35 g). A cromatografia em coluna rápida, com um gradiente linear de MeOH em CH2Cl2 forneceu 4,4-(bisidroximetil) tetraidropirano como um material sólido. Uma mistura de diol (1,46 g, 10 mmol) e TsCl (4,77 g, 25 mmol) em piridina anidra (25 ml) foi agitada, em temperatura ambiente, durante 48 horas. O solvente foi removido in vacuo e co- evaporado com tolueno (3 x 10 ml). CH2Cl2 (100 ml) e NaHCO3 aquoso saturado (100 ml) foram adicionados, as fases foram separadas, e fase orgânica foi secada (Na2SO4), filtrada e concentrada sob pressão reduzida. O resíduo obtido foi submetido à cromatografia de cintilação (35 - 45% de EtOAc em petróleo leve) de um modo a fornecer 4,4 - bis (p- toluenossulfoniloximetil) tetraidropirano (519) como um material sólido branco. Estágio B:

[0157] Seguindo o procedimento geral, a dialquilação do composto 513 (95 mg, 0,48 mmol) com o composto 519 (240 mg, 0,53 mmol) em DMSO (3 ml) na presença de K2CO3 (166 mg, 1,2 mmol) forneceu o composto 520 como um material sólido branco, após a purificação através de cromatografia de coluna (45-65% de EtOAc em petróleo leve). LC- MS: RT = 2,40 minutos; RMN iR (DMSO- d6): δ 7, 34 (1 H, d, J 8, 8), 6,78 (1 H, d, J 8,8), 3, 99 (4 H, s), 3, 80 (3 H, s), 3,76 (3 H, s), 3, 61 (4 H, t, J 5,5), 1, 54 (4 H, t, J 5,5). Preparação 18 Éster metílico do ácido 9-metóxi-2’, 2’-dimetil-espiro [2 H-1,5- benzodioxepin-3 (4H), 5’-[1,3]dioxano]-6-carboxílico (Composto 522). Estágio A:
[0158] Uma solução de 2,2- bis (bromometil)-1,3-propanodiol (5,0 g, 19,1 mmol), acetona anidra (20 ml) e p-TsOH (100 mg) em benzeno (745 ml) foi refluída durante 12 horas, com a remoção azeotrópica de água. A mistura da reação foi concentrada sob pressão reduzida, EtOAc (100 ml) foi adicionado e a camada orgânica foi lavada sucessivamente com H2O (3 x 30 ml) e salmoura (30 ml), secada (Na2SO4), filtrada e concentrada sob pressão reduzida. O sólido branco obtido foi triturado com n- pentano (20 ml), de um modo a fornecer 5,5-bis(bromometil)-2,2-dimetil-[1,3] dioxano (521) como cristais incolores. Estágio B:
[0159] Seguindo o procedimento geral, a dialquilação do composto 513 (1,19 g, 6 mmol) com o composto 521 (2,0 g, 6,62 mmol) em DMSO (30 ml), na presença de K2CO3 (2, 07 g, 15 mmol) forneceu o composto 522 como um material sólido branco, após a purificação através de cromatografia de coluna (30- 40% de EtOAc em petróleo leve). LC-MS: RT = 2,99 minutos; m/ z 339, 31 (M + H)+, 361, 25 (M + Na)+. RMN iR (CDCl3): δ 7,45 (1 H, d, J 8,8), 6,59 (1 H, d, J 8,8), 4,22 (2 H, s), 4,18 (2 H, s), 3, 89 (3 H, s), 3,86 (7 H, s), 1,43 (6 H, s). Preparação 19: Éster metílico do ácido 9-metóxi-espiro [2H-1,5-benzodioxepin-3(4 H), 5’- [1,3]diocano ]—6- carboxílico (Composto 524). Estágio A:
[0160] 5,5-Bis (bromometil)-[1,3] dioxano (523) foi obtido a partir de 2,2-bis(bromometil)-1,3- propanodiol seguindo o procedimento da literatura (Bitha, P.; Carvajal, S. G.; Citarella, R. V.; Delos Santos, E. F.; Durr, F. E.; Hlavka, J. J.; Lang, S. A., Jr.; Lindsay, H. L.; Thomas, J. P.; Wallace, R. E.; Yang- I, L. J. Med. Chem. 1989, 32 (9), 2063 - 7 e Mitkin, O. D.;. Wan, Y.; Kurchan A. N./ Kutateladze, A. G. Synthesis, 2001, (8), 1133-42). Deste modo, uma solução de 2,2- bis (bromometil)-1,3- propanodiol (2,5 g, 9,55 mmol), formaldeído (solução aquosa a 37%, 3,5 ml) e HCl conc. (2,0 ml) foi refluída durante 12 horas. Após ter sido resfriado à temperatura ambiente, H2O (25 ml) foi adicionado à mistura da reação, que foi então extraída com CH2Cl2 (2 x 25 ml). A camada orgânica combinada foi lavada sucessivamente com Na2CO3 aquoso saturado (25 ml) e H2O (25 ml), secada (Na2SO4), filtrada e concentrada sob pressão reduzida. O líquido incolor obtido foi então verificado como sendo > 95 % puro através de RMN ÃH, e foi usado sem purificação adicional. Estágio B:

[0161] Seguindo o procedimento geral, a dialquilação do composto 513 (198 mg, 1 mmol) com o composto 523 (301 mg, 1,1 mmol) em DMSO (5 ml) na presença de K2CO3 (345 mg, 2,5 mmol) forneceu o composto 524 como um material sólido branco, após a purificação por cromatografia de coluna (50- 65% de EtOAc em petróleo leve). LC- MS: RT = 2,65 minutos; m/ z 311,23 (M + H)+. RMN iR (DMSO d6): δ 7,37 (1 H, d, J 8,8), 6,81 (1 H, d, J 8,8), 4,80 (1H, d, J 6,4), 4,78 (1 H, d, J 6,4), 4, 07 (2 H, s), 4,01 (2 H, s), 3,81 (7 H, s), 3,76 (3 H, s). Preparação 20 Éster metílico do ácido 9-metóxi- espiro[2H-1,5-benzodioxepin-3(4H), 5’ - [1,3] ditiano]-6- carboxílico (Composto 526). Estágio A:
[0162] 4,4-Bis(hidroximetil)- [1,3] ditiano foi obtido em um processo de dois estágios, a partir do composto 523, seguindo o procedimento da literatura (Mitkin, O. D.; Wan, Y.; Kurchan, A. N.; Kutaleladze, A. G. Synthesis, 2001, (8), 1133- 42). Deste modo, uma mistura do composto 523 (1,36 g, 5 mmol) e KSAc (1,71 g, 15 mmol) em FMF anidro (10 ml) foi agitada durante 30 horas. O solvente foi removido sob pressão reduzida e co- evaporado com tolueno (3 x 5 ml). A mistura com água gelada foi adicionada e então a extração foi executada com éter diisopropílico (2 x 25 ml). As fases orgânicas combinadas foram secadas (Na2SO4), filtradas e concentradas, de um modo a fornecer 5,5- bis (acetiltiometil)-[1,3]-dioxano (1,32 g, quantitativo) como um óleo viscoso amarelo pálido. Estes foi verificado como sendo > 95% puro através de RMN ÃH e este foi usado sem purificação adicional.
[0163] Uma solução de 5,5-bis (acetiltiometil)-[1,3]dioxano (1,32 g, 5 mmol) em HCl aquoso (2 N, 25 ml) foi refluída durante 16 horas e foi então resfriada à temperatura ambiente. A mistura foi tornada alcalina através da adição, em gotas, de Na2CO3 aquoso (2 M) e então extraída com CH2Cl 2 (3 x 40 ml). A camada orgânica combinada foi secada (Na2SO4), filtrada e concentrada sob pressão reduzida, de um modo a fornecer um material sólido branco, que foi triturado com n-hexano-éter diisopropílico (2:1, 15 ml). O sólido branco foi filtrado de um modo a prover 4,4-bis (hidroximetil)-[1,3] ditiano, que foi usado sem purificação adicional.
[0164] Uma mistura de 4,4-bis(hidroximetil)-[1,3]ditiano (405 mg, 2,25 mmol) e TsCl (1,29 g, 6,75 mmol) em piridina anidra (4 ml) foi agitada, em temperatura ambiente, durante 48 horas. O solvente foi removido in vacuo e co- evaporado com tolueno (3 x 3 ml). CH2Cl2 (40 ml) e NaHCO3 aquoso saturado (40 ml) foram adicionados, as fases foram separadas e a fase orgânica foi secada (Na2SO4), filtrada e concentrada sob pressão reduzida. O resíduo obtido foi submetido à cromatografia de cintilação (35- 55% de EtOAc em petróleo leve) de um modo a prover 4,4- bis (p- toluenossulfonilóximetil) - [1,3] ditiano (525) como um material sólido branco. Estágio B:
[0165] Seguindo o procedimento geral, a dialquilação do composto 513 (297 mg, 1,5 mmol) com o composto 525 (806 mg, 1,65 mmol) em DMSO (7,5 ml), na presença de K2CO3(518 mg, 3,75 mmol) forneceu o composto 526 como um material sólido branco após a purificação através de cromatografia de coluna (40-50% de EtOAc em petróleo leve). LC- MS: RT = 3,53 minutos; m/ z 343,14 (M + H)+, 365,12 (M + Na)+, 327,28 (M- CH3)-. RMN iH (DMSO- d6): δ 7,36 (1H, d, J, 8,8), 6,81 (1 H, d, J 8,8), 4,20 (2 H, s), 4,14 (2 H, s), 3,83 (2 H, s), 3,81 (3 H, s), 3,77 (3H, s), 2,85 (4 H, s).
Procedimento Geral C:
[0166] LiHMDS (1 M em THF, 3,0 eq.) foi adicionado, em gotas, a uma solução gelada do éster 4 b e de 3,5- dicloro-4-metilpiridina comercialmente disponível (1,3 eq.) em THF anidro. A mistura da reação foi agitada em temperatura ambiente, durante 12 horas, H2O (10 ml) e NH4Cl aquoso saturado (20 ml) foram adicionados e a mistura foi extraída com EtOAc (3 x 50 ml). As fases orgânicas foram secadas (Na2SO4), filtradas e concentradas sob pressão reduzida. O resíduo obtido foi purificado através de cromatografia de coluna de cintilação, de um modo a fornecer a cetona Ib.
[0167] Usando o procedimento geral C foram obtidos os compostos que se seguem: Exemplo 20: 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2 H-1,5-benzodioxepin-3 (4H), 3’-oxetano] - 6- il} etanona (Composto 121).
[0168] Seguindo o procedimento geral, a condensação de 3,5-dicloro- 4-metilpiridina (169 mg, 1, 04 mmol) com o composto 514 (224 mg, 0,8 mmol) em THF (4 ml), na presença de LiHMDS (2,4 ml, 2,4 mmol) forneceu o composto 121 como um material sólido branco, após a purificação através de cromatografia de coluna (65- 80%) de EtOAc em petróleo leve). LC-MS: RT = 3,24; m/ z 410,07 (M + H)+, 408,19, 410,22 (M-H)-; RMN iH (DMSO- d6): δ 8,66 (2H, s), 7,45 (1 H, d, J 8,9), 6,91 (1 H, d, J 9,0), 4,66 (2 H, s) 4,57 (2 H, s), 4,50 (2 H, d, J 6,5), 4,46 (2 H, d, J 6,6), 4,36 (2 H, s), 3,85 (3 H, s). Exemplo 21 2-(3,5-Dicloro-1-óxido-piridin-4-il)-1-(9-metóxi-espiro [2 H-1, 5- benzodioxepin-3 (4H), 3’-oxetano]-6-il) etanona (Composto 122).
[0169] A uma solução de 2-(3, 5-Dicloropiridin-4-il)-1- (9-metóxi- espiro [2H-1,5-benzodioxepin-3(4H), 3’-oxetano]-6-il}etanona [121] (20,5 mg, 50 μmol) em CH2CI2 (1 ml) foi adicionado 30% de H2O2 (15 μl) e metiltrioxorrênio (VII) (5 mg). A mistura foi agitada durante 18 horas, foi adicionado MnO2 (5mg) e foi agitada durante mais uma hora. CH2Cl2 (10 ml) foi adicionado e a fase orgânica foi lavada com água. A fase orgânica combinada foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 14 mg do produto.
[0170] LC/ MS (MÉTODO B): (m/ z) 426,18; 428,20 (MH+); RT = 2,42 minutos; pureza (UV) = 100%. Exemplo 22 2-(3,5-Dicloropiridin-4-il)-1-(9-metóxi-espiro [2 H-1,5-benzodioxepin-3 (4H), 3’-tietano] -6- il} etanona (Composto 123).
[0171] Seguindo o procedimento geral, a condensação de 3,5- dicloro- 4-metilpiridina (366 mg, 2,26 mmol) com o composto 516 (516 mg, 1,74 mmol) em THF (10 ml) na presença de LiHMDS (5,2 ml, 5,2 mmol) forneceu o composto 123 como um material sólido branco, após a purificação através de cromatografia de coluna (55-65% de EtOAc em petróleo leve). LC- MS: RT = 4,23 minutos; m/ z 426,24, 428,25 (M + H)+, 424,23 (M-H)-. RMN *H (CDCl3): δ 8,51 (2H, s), 7,57 (1 H, d, J 9,2), 6,71 (1 H, d, J 9,2), 4,64 (2 H, s), 4,46 (2 H, s), 4,33 (2 H, s), 3,93 (3 H, s), 3,20 (2 H, d, J 9,9), 3,12 (2 H, d, J 9,9). Exemplo 23: 2-(3,5-Dicloropiriridin-4-il)-1-{9-metóxi-espiro [2H-1,5-benzodioxepin-3(4 H), 3’-tietano-1’, 1’-dióxido]-6- il}etanona (Composto 124).
[0172] A uma solução de 2-(3,5-dicloropiridin-4-il)-1-(9-metóxi- espiro [2H-1,5- benzodioxepin-3- (4H), 3’-tietano]-6-il}etanona [123] (42,6 mg) em CH2C12 (1 ml) foram adicionados 30% de H2O2 (38 μl) e metiltrioxorrênio (VII) (5 mg). A mistura foi agitada durante 18 horas e subsequentemente lavada com água. A fase orgânica foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 6 mg do produto.
[0173] LC/ MS (MÉTODO B): (m/z) 458,13, 460,10, 462,14 (MH+); RT = 3,30 minutos; pureza (uv) = 100%. Exemplo 24: 2-(3, 5-Dicloropiridin-1-óxido-4-il)-1-{9-metóxi-espiro [2 H-1,5- benzodioxepin-3(4H), 3’-tietano-1’,1’- dióxido]-6-il}etanona (Composto 125)
[0174] A uma solução de 2-(3,5-dicloropiridin-4-il)-1- (9-metóxi- espiro [2 H-1,5-benzodioxepin-3(4H), 3’- tietano]-6-il}etanona [123] (39,6 mg) em CH2Cl2 (1 ml) foram adicionados 30% de H2O2 (76 μl) e metiltrioxorrênio (VII) (5 mg). A mistura foi agitada durante 18 horas, e água foi subsequentemente adicionada. A fase orgânica foi secada com MgSO4, e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 8,4 mg do produto.
[0175] LC/ MS (MÉTODO B): (m/z) 474,17, 476,16, 478,18 (MH+); RT = 2,39 minutos; pureza (UV) = 100%. Exemplo 25: 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi- espiro [2 H-1,5-benzodioxepin-3 (4 H),2’-(1,3-dioxolano)]-6-il} etanona (Composto 126)
[0176] Seguindo o procedimento geral, a condensação de 3,5-dicloro- 4-metilpiridina (53 mg, 0,33 mmol) com o composto 518 (74 mg, 0,25 mmol) em THF (1,5 ml), na presença de LiHMDS (0, 75 ml, 0,75 mmol) forneceu o composto 127 como um material sólido branco, após a purificação através de cromatografia de coluna (60- 80% de EtOAc em petróleo leve). LC- MS: RT = 3,63 minutos; m/z 426,18, 428,16 (M + H)+. RMN *H (DMSO- dô) δ 8,64 (2 H, s), 7,39 (1 H, d, J 8,8), 6,86 (1 H, d, J 8,9), 4,60 (2 H, s), 4,34 (2 H, s), 4,17 (2 H, s), 3,97 (4 H, s), 3,84 (3 H, s). Exemplo 26: 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2H-1,5-benzodioxepin-2(4H), 4’-tetraidropiran]-6-il} etanona (Composto 127)
[0177] Seguindo o procedimento geral, a condensação de 3, 5-dicloro- 4-metilpiridina (59 mg, 0,36 mmol) com o composto 520 (85 mg, 0,28 mmol) em THF (1,5 ml), na presença de LiHMDS (0,85 ml, 0,85 mmol) forneceu o composto 127 como um material sólido branco após a purificação através de cromatografia de coluna (60-70% de EtOAc em petróleo leve). LC- MS: RT = 3,70 minutos; m/z 438,21, 440,21 (M + H)+, 436,30, 438,27 (M-H)-. RMN ÍH (DMSO-d6): δ 8,65 (2 H, s), 7,42 (1 H, d, J 8,9), 6,87 (1 H, d, J 9,0), 4,63 (2 H, s), 4, 25 (2 H, s), 4,07 (2 H, s), 3,84 (3 H, s), 3,64 (4 H, t, J 5,3), 1, 60 (4 H, t, J 5,3). Exemplo 27: 2-(3,5-Dicloro-1-óxido- piridin-4-il)-1-{9-metóxi-espiro [2H-1,5- benzodioxepín-3 (4H), 4’-tetraidropiran]-6-il}etanona (Composto 128)
[0178] A uma solução de 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi- espiro [2 H-1,5-benzodioxepin-3(4H), 4’-tetraidropiran]-6-il}etanona [127] (66 mg, 150 μmol) em CH2C12 (2 ml) foram adicionados 30% de H2O2 (45 μl) e metiltrioxorrênio (VII) (10 mg). A mistura foi agitada durante 18 horas, foi adicionado MnO2 (10 mg) e a mistura foi agitada durante mais uma hora. CH2Cl2 (10 ml) foram adicionados e a fase orgânica foi lavada com água. A fase orgânica combinada foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 5, 6 mg do produto.
[0179] LC/ MS (MÉTODO B): (m/ z) 454,32; 456, 32 (MH+); RT = 2, 80 minutos; pureza (UV) = 100%. Exemplo 28: 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-2’,2’-dimetil-espiro [2 H-1, 5- benzodioxepin-3(4H), 5’-[1,3]dioxano] -6-il}etanona (Composto 129).
[0180] Seguindo o procedimento geral, a condensação de 3,5- dicloro- 4-metilpiridina (93 mg, 0,58 mmol) com o composto 522 (150 mg, 0,44 mmol) em THF (2 ml), na presença de LiHMDS (1,3 ml, 1,3 mmol) forneceu o composto 129 como um material sólido branco, após a purificação através de cromatografia de coluna (55- 60% de EtOAc em petróleo leve). LC- MS: RT = 3,95 minutos; m/ z 468, 19, 470,23 (M + H)+, 466,36, 468,33 (M-H)-. RMN iH (DMSO- d6): δ 8, 65 (2H, s),7,42 (1 H, d, J 8,8), 6, 89 (1 H, d, J 9,1), 4,63 (2 H, s), 4,33 (2 H, s), 4,08 (2 H, s), 3,84 (3 H, s), 3,82 (4 H, s), 1,39 (3 H, s), 1,37 (3 H, s). Exemplo 29: 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro [2H-1,5-benzodioxepin-3(4H), 5’-[1,3] dioxano]-6-il}etanona (Composto 130).
[0181] Seguindo o procedimento geral, a condensação de 3,5-dicloro- 4-metilpiridina (211 mg, 1,3 mmol) com o composto 524 (310 mg, 1,0 mmol) em THF (6 ml), na presença de LiHMDS (3 ml, 3,0 mmol) forneceu o composto 130 como um material sólido branco, após a purificação através de cromatografia de coluna (60-80% de EtOAc em petróleo leve). LC- MS: RT = 3, 64 minutos; m/ z 440, 16, 441, 17 (M + H)+, 438, 27, 440,29 (M-H)-. RMN iH (DMSO- d6): δ 8,65 (2 H, s), 7,43 (1 H, d, J 8,9), 6, 89 (1 H, d, J 8,9), 4,84 (1 H, d, J 6, 1), 4,79 (1 H, d, J 6,1), 4,64 (2 H, s), 4,34 (2 H, s), 4, 08 (2 H, s), 3,90 (2 H, d, J 11, 4), 3,85 (3 H, s), 3,82 (2 H, d, J 11, 2). Exemplo 30: 2- (3,5-Dicloro-1-óxido-piridina-4-il)-1-{9-metóxi-espiro [2 H-1,5- benzodioxepin- 3(4 H), 5’-[1,3] dioxano ]-6-il}etanona (Composto 131).
[0182] A uma solução de 2-(3,5-dicloropiridin-4-il)-1-{9-metóxi- espiro [2 H-1,5-benzodioxepin-3(4H), 5’- [1,3]dioxano]-6-il}etanona [130] (100 mg) em CH2C12 (2 ml) foram adicionados 30% de H2O2 (120 μl) e metiltrioxorrênio (VII) (5 mg). A mistura foi agitada durante 18 horas e subsequentemente lavada com água. A fase orgânica foi secada com MgSO4 e evaporada até a secura, sob pressão reduzida. A purificação por HPLC convencional forneceu 85 mg do produto.
[0183] LC/MS (MÉTODO B): (m/z)456,23 (MH+); RT= 2,55 min; pureza (UV)=95% Exemplo 31 2-(3,5-Dicloropiridin-4-il)-1-{9-metóxi-espiro[2H-1,5-benzodioxepin- 3(4H),5’-[1,3]ditiano]-6-il}etanona (composto 132)
[0184] Seguindo o procedimento geral, condensação de 3,5-dicloro-4- metilpiridina (154 mg, 0,95mmol) com composto 256 (250 mg, 0,73 mmol) em THF (4ml) na presença de LiHMDS (2,2 ml, 2,2 mmol) composto produzido 132 como uma material sólido branco após purificação por cromatografia em coluna (45-55 % EtAc em petróleo leve). LC-MS: RT 4,39 min.; m/ z 472,15, 474,14 (M + H)+. RMN iR (DMSO- d6): δ 8,65 (2 H, s), 7,42 (1 H, d, J 8,9), 6,88 (1 H, d, J 8,9), 4,64 (2 H, s), 4,43 (2 H, s), 4,24 (2 H, s), 3,85 (3 H, s), 3,84 (2 H, s), 2,91 (4 H, s). Exemplo 32: Ensaio de PDE4
[0185] PDE4 recombinante humano (acesso Genbank n° NM - 006203) foi incubado durante 1 hora, com o composto de teste em concentrações de até 10 μM, com cAMP (1 x 10 -5M), e com uma baixa quantidade (0,021 MBq) de cAMP rotulado rdioativamente. No final da incubação, a clivagem do substrato foi avaliada pela ligação do produto de AMP a contas de SPA, que geram quimioluminescência quando ligadas ao traçador radioativo. O produto de AMP inibiu a ligação do traçador radioativo às contas, e o sinal luminescente foi comparado. Os resultados foram calculados como as concentrações molares, que resultam em 50% de inibição da clivagem do substrato comparadas às amostras de controles, e são expressas como IC50 (M).
[0186] Os resultados são apresentados na Tabela 1 abaixo. Tabela 1
Exemplo 33: Liberação de TNF-α
[0187] Células mononucleares sanguíneas periféricas humanas (PBMC) foram isoladas a partir de revestimentos acastanhados. O sangue é misturado com uma solução salina, em uma razão de 1:1, e os PBMC foram isolados usando tubos Lymphoprep TM (Nycomed, Noruega). Os PBMC foram suspensos em RPMI 1640 com 2 % de soro de novilho fetal (FCS), pen./ estrep. E L- glutamina 2 mM, em uma concentração de 5 x 105 c/ ml. As células foram previamente incubadas durante 30 minutos com os compostos de teste em placas de cultura de tecido de 96 reservatórios, e estimuladas durante 18 horas com 1 mg/ ml de lipopolissacarídeo (Sigma). O nível de TNF-α foi medido no sobrenadante da cultura por imunoensaios de enzima, usando anticorpos biotinilados primários e secundários de sistemas de R&D. Os resultados são expressos como valores de IC50, calculados a partir de curvas de inibição usando como controles positivos a secreção em reservatórios estimulados com LPS e como controles negativos a secreção em células não- estimuladas.
[0188] Os resultados são apresentados na Tabela 2 abaixo. Tabela 2:























































