WO2023145941A1 - エタノールの変換方法、及びその他炭化水素の製造方法 - Google Patents
エタノールの変換方法、及びその他炭化水素の製造方法 Download PDFInfo
- Publication number
- WO2023145941A1 WO2023145941A1 PCT/JP2023/002899 JP2023002899W WO2023145941A1 WO 2023145941 A1 WO2023145941 A1 WO 2023145941A1 JP 2023002899 W JP2023002899 W JP 2023002899W WO 2023145941 A1 WO2023145941 A1 WO 2023145941A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ethanol
- fraction
- catalyst
- reaction
- carbon atoms
- Prior art date
Links
Images










Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/40—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the pentasil type, e.g. types ZSM-5, ZSM-8 or ZSM-11, as exemplified by patent documents US3702886, GB1334243 and US3709979, respectively
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C1/00—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon
- C07C1/20—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon starting from organic compounds containing only oxygen atoms as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/02—Alkenes
- C07C11/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/02—Alkenes
- C07C11/08—Alkenes with four carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/02—Alkenes
- C07C11/10—Alkenes with five carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C15/00—Cyclic hydrocarbons containing only six-membered aromatic rings as cyclic parts
- C07C15/02—Monocyclic hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2/00—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms
- C07C2/02—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by addition between unsaturated hydrocarbons
- C07C2/04—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by addition between unsaturated hydrocarbons by oligomerisation of well-defined unsaturated hydrocarbons without ring formation
- C07C2/06—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by addition between unsaturated hydrocarbons by oligomerisation of well-defined unsaturated hydrocarbons without ring formation of alkenes, i.e. acyclic hydrocarbons having only one carbon-to-carbon double bond
- C07C2/08—Catalytic processes
- C07C2/12—Catalytic processes with crystalline alumino-silicates or with catalysts comprising molecular sieves
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C7/00—Purification; Separation; Use of additives
- C07C7/04—Purification; Separation; Use of additives by distillation
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P30/00—Technologies relating to oil refining and petrochemical industry
- Y02P30/40—Ethylene production
Definitions
- the content of elemental phosphorus in the catalyst indicates a value measured using a fluorescent X-ray spectrometer.
- the content of elemental phosphorus can be measured using a commercially available fluorescent X-ray analyzer under normal conditions according to the instruction manual. Measurement conditions can be a tube voltage of 50 kV and a tube current of 50 mA using P-K ⁇ rays.
- the content of elemental silver in the catalyst indicates a value measured using a fluorescent X-ray spectrometer.
- the content of elemental silver can be measured using a commercially available fluorescent X-ray analyzer under normal conditions according to the instruction manual. Measurement conditions can be a tube voltage of 50 kV and a tube current of 50 mA using P-K ⁇ rays.
- a separation step In the separation step, the target compound is separated from the reaction gas.
- a separation step can separate ethylene, propylene and aromatic compounds from the reaction gas.
- the recycling step at least part of the fraction A is recycled to the reaction step as part of the mixed raw material.
- the fraction C obtained from the cooling step is the first distillation column 2, and the fraction A mainly containing hydrocarbons having 1 to 3 carbon atoms and the fraction mainly containing hydrocarbons having 4 to 6 carbon atoms.
- B Furthermore, by supplying the fraction A to the second distillation column 8, the fraction A is a fraction A-1 mainly containing hydrocarbons having 2 carbon atoms and a fraction A-1 mainly containing hydrocarbons having 3 carbon atoms.
- A-2 By using the fraction A-1 as a recycled raw material, the propylene concentration in the mixed raw material is lowered, and propylene can be produced efficiently.
- the method for producing hydrocarbons according to the present embodiment includes contacting a mixed raw material containing ethylene and ethanol with a catalyst in an adiabatic reactor to obtain a reaction gas containing an olefin having 3 or more carbon atoms. .
- the details of the production method are as described in the above ethanol conversion method, and the preferred embodiments thereof are also the same.
- the method for producing propylene according to this embodiment includes: a propylene separation step of separating a fraction mainly containing propylene from the reaction gas obtained by the ethanol conversion method; including.
- the method for producing an acrylonitrile-based polymer according to this embodiment includes: A step of polymerizing a polymerizable composition containing acrylonitrile obtained by the method for producing acrylonitrile described above; including.
- the polymerizable composition may contain acrylonitrile alone as a monomer, or may contain other monomers having unsaturated bonds.
- Effective raw material supply mass flow rate (kg/hr) Ethylene flow rate (kg/hr) + Ethylene conversion ethanol flow rate (kg/hr) + C4-C6 olefin flow rate (kg/hr) + Carbon numbers other than ethanol from 1 to 6 oxygenate flow rate (kg/hr)
- Effective raw material supply mass flow rate (kg/hr) Ethylene flow rate (kg/hr) + Ethylene conversion ethanol flow rate (kg/hr) + C4-C6 olefin flow rate (kg/hr) + Carbon numbers other than ethanol from 1 to 6 oxygenate flow rate (kg/hr)
- a mixed feed gas ethylene 1.0 kg/hr, ethanol 4.6 kg/hr, steam 0.7 kg/hr
- the raw material introduced into the reactor 1 has an ethylene/ethanol molar ratio of 0.80 and an olefin having 4 to 6 carbon atoms/ethylene molar ratio of 0.23.
- Table 9 shows the composition of the components (mainly hydrocarbons having 4 or more carbon atoms) recycled from the fraction B contained in the raw material.
- the average propylene yield from the start of the reaction to the end of the reaction is 24.5% by mass, and the coke yield of the zeolite-containing catalyst after 48 hours of operation is 321 mass ppm.
- the recovery yield of propylene obtained at the outlet of the separation step is 14.8% by mass, and the recovery yield of aromatic compounds is 13.4% by mass.
Abstract
(1)エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法、(2)メタノール及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法、及び(3)エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることと、前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離することと、前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすることと、を含む、エタノールの変換方法。
Description
本発明は、エタノールの変換方法、及びその他炭化水素の製造方法に関する。
低級オレフィン等の炭化水素は化学産業における重要な基幹原料であり、特に、プロピレンは需要増加が見込まれるため、種々の製造方法の開発・改良が盛んになされてきた。その中でも、一般的なプロピレンの製造方法としては、ゼオライトを活性種とした触媒にナフサやオレフィン類を接触させる方法が知られている。
例えば特許文献1ではエチレン原料からのプロピレンの製造方法が開示されている。特許文献2及び3では、それぞれペンタシル型ゼオライトと酸化亜鉛セリウム担持ゼオライトを使用したエタノールからプロピレンへの変換技術が公開されている。例えば、特許文献4では、含酸素化合物(Oxygenate)と炭素数4以上のオレフィンとを原料とした低級オレフィンの製造方法が、特許文献5では低級アルコールとナフサとを原料とした低級オレフィンの製造方法が示されている。例えば、特許文献6ではメタノールを含む原料からプロピレンを製造する方法が開示されている。
本発明は、以下の実施形態を包含する。
[1]
エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法。
[2]
前記混合原料中のエチレン/エタノールのモル比が0.20~2.5である、[1]に記載のエタノールの変換方法。
[3]
前記混合原料中のエチレン/エタノールのモル比が0.20~2.0である、[1]又は[2]に記載のエタノールの変換方法。
[4]
前記反応ガスからエチレン及びプロピレンを分離することを含む、[1]~[3]のいずれかに記載のエタノールの変換方法。
[5]
前記混合原料が炭素数4~6のオレフィンを含有する、[1]~[4]のいずれかに記載のエタノールの変換方法。
[6]
前記混合原料中の炭素数4~6のオレフィン/エチレンのモル比が3.0以下である、[5]に記載のエタノールの変換方法。
[7]
メタノール及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法。
[8]
前記混合原料が、メタノールを含む、[1]~[6]のいずれかに記載のエタノールの変換方法。
[9]
前記混合原料中のメタノール/エタノールのモル比が0.050~2.0である、[7]又は[8]に記載のエタノールの変換方法。
[10]
前記混合原料中のメタノール/エタノールのモル比が0.20~1.5である、[7]~[9]のいずれかに記載のエタノールの変換方法。
[11]
前記反応ガスからエチレン及びプロピレンを分離することを含む、[7]~[10]のいずれかに記載のエタノールの変換方法。
[12]
前記混合原料が炭素数4~6のオレフィンを含有する、[7]~[11]のいずれかに記載のエタノールの変換方法。
[13]
前記混合原料中の炭素数4~6のオレフィン/メタノールのモル比が3.0以下である、[7]~[12]のいずれかに記載のエタノールの変換方法。
[14]
前記反応ガス又は反応ガスを精製した留分の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いることを含む、[7]~[13]のいずれかに記載のエタノールの変換方法。
[15]
エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることと、
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離することと、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすることと、
を含む、エタノールの変換方法。
[16]
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離することと、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすることと、
を含む、[1]~[14]のいずれかに記載のエタノールの変換方法。
[17]
前記留分Aを、第2の蒸留塔により、炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離することを含み、
前記リサイクルすることが、前記留分A-1の少なくとも一部を、前記反応ガスを得ることにリサイクルし、前記混合原料の一部として用いることを含む、
[15]又は[16]に記載のエタノールの変換方法。
[18]
前記リサイクルすることが、前記留分Bの少なくとも一部を、前記反応ガスを得ることにリサイクルし、前記混合原料の一部として用いることを含む、
[15]~[17]のいずれかに記載のエタノールの変換方法。
[19]
前記反応ガスを冷却することにより、炭素数6以下の炭化水素を主に含む留分Cと、水及び炭素数7以上の炭化水素化合物を主に含む留分Dとに分離すること、
を含む、[15]~[18]のいずれかに記載のエタノールの変換方法。
[20]
前記第1の蒸留塔により分離することが、前記第1の蒸留塔にサイドカット段を設け、中間抜出流出液を得ることを含む、[15]~[19]のいずれかに記載のエタノールの変換方法。
[21]
前記反応ガスを、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~8の炭化水素を主に含む留分Bとに分離すること、並びに
前記留分Aからエチレン及びプロピレンを分離すること、
を含む、[1]~[20]のいずれかに記載のエタノールの変換方法。
[22]
前記留分Bの少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いることを含む、[21]に記載のエタノールの変換方法。
[23]
前記留分Bを、炭素数4~6の脂肪族炭化水素を主に含む留分B1と、芳香族化合物を主に含む留分B2とに分離すること、及び
前記留分B1の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いること、
を含む、[21]又は[22]に記載のエタノールの変換方法。
[24]
前記留分Bをスチームクラッキングに付すことにより、エチレン及びプロピレンを含有するスチームクラッキング生成物を得ること、並びに
前記スチームクラッキング生成物からエチレン及びプロピレンを分離すること、
を含む、[21]~[23]のいずれかに記載のエタノールの変換方法。
[25]
前記反応器が固定床断熱型反応器である、[1]~[24]のいずれかに記載のエタノールの変換方法。
[26]
前記反応器が固定床一段断熱型反応器である、[1]~[25]のいずれかに記載のエタノールの変換方法。
[27]
前記触媒に付着したコークを燃焼させることを含む、[1]~[26]のいずれかに記載のエタノールの変換方法。
[28]
触媒床出口温度が450℃~590℃である、[1]~[27]のいずれかに記載のエタノールの変換方法。
[29]
触媒床入口温度が450℃~590℃である、[1]~[28]のいずれかに記載のエタノールの変換方法。
[30]
触媒床出口温度と、触媒床入口温度との温度差が-80K~80Kである、[1]~[29]のいずれかに記載のエタノールの変換方法。
[31]
前記触媒がゼオライト含有触媒である、[1]~[30]のいずれかに記載のエタノールの変換方法。
[32]
前記ゼオライト含有触媒が中間細孔径ゼオライトを含む、[31]に記載のエタノールの変換方法。
[33]
前記ゼオライト含有触媒中のゼオライトのシリカ/アルミナのモル比が20~2000である、[31]又は[32]に記載のエタノールの変換方法。
[34]
前記ゼオライト含有触媒がリン元素又は銀元素を含む、[31]~[33]のいずれかに記載のエタノールの変換方法。
[35]
エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、炭化水素の製造方法。
[36]
炭素数2以上の炭化水素を分解する、クラッキング工程と、
前記クラッキング工程で得られた成分を精製する精製工程と、
を有し、
前記精製工程で、[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガス又はその精製留分を合流させる、炭化水素の製造方法。
[37]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程、
を含む、モノマーの製造方法。
[38]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからオレフィンを主に含む留分を分離するオレフィン分離工程、
を含む、オレフィンの製造方法。
[39]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程、
を含む、プロピレンの製造方法。
[40]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程、
を含む、エチレンの製造方法。
[41]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからジエンを主に含む留分を分離するジエン分離工程、
を含む、ジエンの製造方法。
[42]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程と、
前記不飽和炭化水素分離工程により得られた不飽和炭化水素からアクリルモノマーを得るアクリルモノマー製造工程と、
を含む、アクリルモノマーの製造方法。
[43]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程と、
前記プロピレン分離工程により得られたプロピレンからアクリロニトリルを得るアクリロニトリル製造工程と、
を含む、アクリロニトリルの製造方法。
[44]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程と、
前記エチレン分離工程により得られたエチレンからスチレンを得るスチレン製造工程と、
を含む、スチレンの製造方法。
[45]
[37]に記載の製造方法により得られるモノマーを重合する工程、
を含む、重合体の製造方法。
[46]
[38]に記載の製造方法により得られるオレフィンを含む重合性組成物を重合する工程、
を含む、オレフィン系重合体の製造方法。
[47]
[39]に記載の製造方法により得られるプロピレンを含む重合性組成物を重合する工程、
を含む、ポリプロピレン系重合体の製造方法。
[48]
[40]に記載の製造方法により得られるエチレンを含む重合性組成物を重合する工程、
を含む、ポリエチレン系重合体の製造方法。
[49]
[41]に記載の製造方法により得られるジエンを含む重合性組成物を重合する工程、
を含む、ジエン系重合体の製造方法。
[50]
[42]に記載の製造方法により得られるアクリルモノマーを含む重合性組成物を重合する工程、
を含む、アクリルモノマー系重合体の製造方法。
[51]
[43]に記載の製造方法により得られるアクリロニトリルを含む重合性組成物を重合する工程、
を含む、アクリロニトリル系重合体の製造方法。
[52]
[44]に記載の製造方法により得られるスチレンを含む重合性組成物を重合する工程、
を含む、スチレン系重合体の製造方法。
[53]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスから芳香族化合物を主に含む留分を分離する芳香族化合物分離工程、
を含む、芳香族化合物の製造方法。
[54]
[53]に記載の製造方法により得られた芳香族化合物から芳香族モノマーを得る芳香族モノマー製造工程、
を含む、芳香族モノマーの製造方法。
[55]
[53]に記載の製造方法により得られる芳香族モノマーを含む重合性組成物を重合する工程、
を含む、芳香族モノマー系重合体の製造方法。
[56]
エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得る反応器と、
前記反応ガスを、炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離する第1の蒸留塔と、
を備え、
前記留分Aの少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いる、エタノールの変換装置。
[57]
前記留分Aを、炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離する第2の蒸留塔を備え、
前記留分A-2の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いる、[57]に記載のエタノールの変換装置。
[58]
前記第1の蒸留塔が、中間抜出流出液を得るためのサイドカット段を有する、[58]に記載のエタノールの変換装置。
[1]
エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法。
[2]
前記混合原料中のエチレン/エタノールのモル比が0.20~2.5である、[1]に記載のエタノールの変換方法。
[3]
前記混合原料中のエチレン/エタノールのモル比が0.20~2.0である、[1]又は[2]に記載のエタノールの変換方法。
[4]
前記反応ガスからエチレン及びプロピレンを分離することを含む、[1]~[3]のいずれかに記載のエタノールの変換方法。
[5]
前記混合原料が炭素数4~6のオレフィンを含有する、[1]~[4]のいずれかに記載のエタノールの変換方法。
[6]
前記混合原料中の炭素数4~6のオレフィン/エチレンのモル比が3.0以下である、[5]に記載のエタノールの変換方法。
[7]
メタノール及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法。
[8]
前記混合原料が、メタノールを含む、[1]~[6]のいずれかに記載のエタノールの変換方法。
[9]
前記混合原料中のメタノール/エタノールのモル比が0.050~2.0である、[7]又は[8]に記載のエタノールの変換方法。
[10]
前記混合原料中のメタノール/エタノールのモル比が0.20~1.5である、[7]~[9]のいずれかに記載のエタノールの変換方法。
[11]
前記反応ガスからエチレン及びプロピレンを分離することを含む、[7]~[10]のいずれかに記載のエタノールの変換方法。
[12]
前記混合原料が炭素数4~6のオレフィンを含有する、[7]~[11]のいずれかに記載のエタノールの変換方法。
[13]
前記混合原料中の炭素数4~6のオレフィン/メタノールのモル比が3.0以下である、[7]~[12]のいずれかに記載のエタノールの変換方法。
[14]
前記反応ガス又は反応ガスを精製した留分の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いることを含む、[7]~[13]のいずれかに記載のエタノールの変換方法。
[15]
エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることと、
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離することと、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすることと、
を含む、エタノールの変換方法。
[16]
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離することと、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすることと、
を含む、[1]~[14]のいずれかに記載のエタノールの変換方法。
[17]
前記留分Aを、第2の蒸留塔により、炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離することを含み、
前記リサイクルすることが、前記留分A-1の少なくとも一部を、前記反応ガスを得ることにリサイクルし、前記混合原料の一部として用いることを含む、
[15]又は[16]に記載のエタノールの変換方法。
[18]
前記リサイクルすることが、前記留分Bの少なくとも一部を、前記反応ガスを得ることにリサイクルし、前記混合原料の一部として用いることを含む、
[15]~[17]のいずれかに記載のエタノールの変換方法。
[19]
前記反応ガスを冷却することにより、炭素数6以下の炭化水素を主に含む留分Cと、水及び炭素数7以上の炭化水素化合物を主に含む留分Dとに分離すること、
を含む、[15]~[18]のいずれかに記載のエタノールの変換方法。
[20]
前記第1の蒸留塔により分離することが、前記第1の蒸留塔にサイドカット段を設け、中間抜出流出液を得ることを含む、[15]~[19]のいずれかに記載のエタノールの変換方法。
[21]
前記反応ガスを、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~8の炭化水素を主に含む留分Bとに分離すること、並びに
前記留分Aからエチレン及びプロピレンを分離すること、
を含む、[1]~[20]のいずれかに記載のエタノールの変換方法。
[22]
前記留分Bの少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いることを含む、[21]に記載のエタノールの変換方法。
[23]
前記留分Bを、炭素数4~6の脂肪族炭化水素を主に含む留分B1と、芳香族化合物を主に含む留分B2とに分離すること、及び
前記留分B1の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いること、
を含む、[21]又は[22]に記載のエタノールの変換方法。
[24]
前記留分Bをスチームクラッキングに付すことにより、エチレン及びプロピレンを含有するスチームクラッキング生成物を得ること、並びに
前記スチームクラッキング生成物からエチレン及びプロピレンを分離すること、
を含む、[21]~[23]のいずれかに記載のエタノールの変換方法。
[25]
前記反応器が固定床断熱型反応器である、[1]~[24]のいずれかに記載のエタノールの変換方法。
[26]
前記反応器が固定床一段断熱型反応器である、[1]~[25]のいずれかに記載のエタノールの変換方法。
[27]
前記触媒に付着したコークを燃焼させることを含む、[1]~[26]のいずれかに記載のエタノールの変換方法。
[28]
触媒床出口温度が450℃~590℃である、[1]~[27]のいずれかに記載のエタノールの変換方法。
[29]
触媒床入口温度が450℃~590℃である、[1]~[28]のいずれかに記載のエタノールの変換方法。
[30]
触媒床出口温度と、触媒床入口温度との温度差が-80K~80Kである、[1]~[29]のいずれかに記載のエタノールの変換方法。
[31]
前記触媒がゼオライト含有触媒である、[1]~[30]のいずれかに記載のエタノールの変換方法。
[32]
前記ゼオライト含有触媒が中間細孔径ゼオライトを含む、[31]に記載のエタノールの変換方法。
[33]
前記ゼオライト含有触媒中のゼオライトのシリカ/アルミナのモル比が20~2000である、[31]又は[32]に記載のエタノールの変換方法。
[34]
前記ゼオライト含有触媒がリン元素又は銀元素を含む、[31]~[33]のいずれかに記載のエタノールの変換方法。
[35]
エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、炭化水素の製造方法。
[36]
炭素数2以上の炭化水素を分解する、クラッキング工程と、
前記クラッキング工程で得られた成分を精製する精製工程と、
を有し、
前記精製工程で、[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガス又はその精製留分を合流させる、炭化水素の製造方法。
[37]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程、
を含む、モノマーの製造方法。
[38]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからオレフィンを主に含む留分を分離するオレフィン分離工程、
を含む、オレフィンの製造方法。
[39]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程、
を含む、プロピレンの製造方法。
[40]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程、
を含む、エチレンの製造方法。
[41]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからジエンを主に含む留分を分離するジエン分離工程、
を含む、ジエンの製造方法。
[42]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程と、
前記不飽和炭化水素分離工程により得られた不飽和炭化水素からアクリルモノマーを得るアクリルモノマー製造工程と、
を含む、アクリルモノマーの製造方法。
[43]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程と、
前記プロピレン分離工程により得られたプロピレンからアクリロニトリルを得るアクリロニトリル製造工程と、
を含む、アクリロニトリルの製造方法。
[44]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程と、
前記エチレン分離工程により得られたエチレンからスチレンを得るスチレン製造工程と、
を含む、スチレンの製造方法。
[45]
[37]に記載の製造方法により得られるモノマーを重合する工程、
を含む、重合体の製造方法。
[46]
[38]に記載の製造方法により得られるオレフィンを含む重合性組成物を重合する工程、
を含む、オレフィン系重合体の製造方法。
[47]
[39]に記載の製造方法により得られるプロピレンを含む重合性組成物を重合する工程、
を含む、ポリプロピレン系重合体の製造方法。
[48]
[40]に記載の製造方法により得られるエチレンを含む重合性組成物を重合する工程、
を含む、ポリエチレン系重合体の製造方法。
[49]
[41]に記載の製造方法により得られるジエンを含む重合性組成物を重合する工程、
を含む、ジエン系重合体の製造方法。
[50]
[42]に記載の製造方法により得られるアクリルモノマーを含む重合性組成物を重合する工程、
を含む、アクリルモノマー系重合体の製造方法。
[51]
[43]に記載の製造方法により得られるアクリロニトリルを含む重合性組成物を重合する工程、
を含む、アクリロニトリル系重合体の製造方法。
[52]
[44]に記載の製造方法により得られるスチレンを含む重合性組成物を重合する工程、
を含む、スチレン系重合体の製造方法。
[53]
[1]~[34]のいずれかに記載のエタノールの変換方法により得られた反応ガスから芳香族化合物を主に含む留分を分離する芳香族化合物分離工程、
を含む、芳香族化合物の製造方法。
[54]
[53]に記載の製造方法により得られた芳香族化合物から芳香族モノマーを得る芳香族モノマー製造工程、
を含む、芳香族モノマーの製造方法。
[55]
[53]に記載の製造方法により得られる芳香族モノマーを含む重合性組成物を重合する工程、
を含む、芳香族モノマー系重合体の製造方法。
[56]
エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得る反応器と、
前記反応ガスを、炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離する第1の蒸留塔と、
を備え、
前記留分Aの少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いる、エタノールの変換装置。
[57]
前記留分Aを、炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離する第2の蒸留塔を備え、
前記留分A-2の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いる、[57]に記載のエタノールの変換装置。
[58]
前記第1の蒸留塔が、中間抜出流出液を得るためのサイドカット段を有する、[58]に記載のエタノールの変換装置。
本発明について、以下具体的に説明する。なお、本発明は以下の実施の形態(本実施形態)に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
本明細書において、「~」を用いて示された数値範囲は、「~」の前後に記載される数値をそれぞれ最小値及び最大値として含む範囲を示す。本明細書に段階的に記載されている数値範囲において、ある段階の数値範囲の上限値又は下限値は、他の段階の数値範囲の上限値又は下限値と任意に組み合わせることができる。
本明細書において「目的化合物」とは、オレフィン、芳香族化合物等の炭化水素である。オレフィンとしては、例えば、エチレン、プロピレン、ブテン、ブタジエン等が挙げられる。芳香族化合物としては、例えば、ベンゼン、トルエン、キシレン等が挙げられる。
目的化合物は、需要等の情勢に応じて適宜変更することも可能であるが、本実施形態のエタノールの変換方法においては、炭素数2のエタノールから、炭素数3のプロピレン、炭素数6以上の芳香族化合物を得ることもできる。
目的化合物は、需要等の情勢に応じて適宜変更することも可能であるが、本実施形態のエタノールの変換方法においては、炭素数2のエタノールから、炭素数3のプロピレン、炭素数6以上の芳香族化合物を得ることもできる。
[第1実施形態]
はじめに、第1実施形態に係るエタノールの変換方法について説明する。
はじめに、第1実施形態に係るエタノールの変換方法について説明する。
<エタノールの変換方法-第1実施形態->
第1実施形態のエタノールの変換方法は、エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ること(以下、「反応工程」ともいう。)を含む。
第1実施形態のエタノールの変換方法は、エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ること(以下、「反応工程」ともいう。)を含む。
従来、オレフィン類以外にも、近年の環境保全意識の高まりから、バイオマス原料から製造されるエタノールなどのアルコール類を原料とした化成品製造が注目を集めている。中でも、エタノールはエチレンと同じ炭素数2の化合物であることから、エタノールを変換することで、効率的なプロピレン等の炭化水素化合物の製造方法の早期開発が期待されている。
化成品製造プロセスを実用化する際、反応器及びその様式の選定は操作の容易性や環境負荷の大小に大きな影響を与える要素となる。固定床断熱型反応器は反応器の加熱又は冷却を行わず、更に、流動床などの反応器と比較して複雑な構造を有していないため、設計、建築及び操業の負荷が小さい理想的な反応器であり、例えば、固定床一段断熱型反応器を採用できればその利点は更に大きくなる。しかしながら、当該反応器は吸熱又は発熱の大きい反応系には適用できないという難点も有している。
反応熱の影響による反応器内の温度変化を抑制する方法としては、複数の化合物を混合して原料として使用することで吸熱と発熱とを示す異なる反応を引き起こし、系内の発熱と吸熱とを相殺する、つまり、熱中和状態とすることが有効である。ただし、反応系の熱中和を指向して単一の目的物を与える異なる原料を選定する際には、それぞれの変換を阻害しないように注意し原料を選定する必要があり、相手となる原料の性状を考慮した組み合わせの開発が必要となるため各素反応の反応熱だけに着目して原料を選定することは困難である。
特許文献1~3では、それぞれ各原料をゼオライト触媒で目的のオレフィンへと変換する技術が公開されているが、これらの技術は吸熱及び発熱が大きくなるため断熱型反応器へと応用することはできない。
特許文献4及び5では、アルコールなどの含酸素化合物を原料とする発熱反応を利用した熱中和技術が開示されているが、吸熱反応を誘引するエタノールを原料とする反応を断熱型反応器で実施することはできない。
そこで、本実施形態は、断熱型反応器を使用し、反応器内の温度を制御して収率良く目的化合物の得られるエタノールを変換する方法等を提供することを目的とする。
本発明者らは、上記の課題を達成するために鋭意検討した結果、エタノールから炭素数3以上のオレフィンへの変換及びエチレンから炭素数3以上のオレフィンへの変換に対して反応系全体として反応熱を制御することで、断熱型反応器を使用し、反応器内の温度を制御して収率良く目的化合物へとエタノールを変換できることを見出した。
本実施形態によれば、断熱型反応器を使用し、反応器内の温度を制御して収率良くプロピレン等の目的化合物を製造することができる。また、本実施形態によれば、断熱型反応器を使用することで操業の負荷が小さく、エネルギー効率が高い環境調和型のエタノールの変換が実施できる。また、断熱型反応器を使用した場合にも、収率良くプロピレン等の目的化合物を与え、コーキング劣化を抑制できるため、エタノールの変換方法として好適である。
本発明者は、上述の通り、エチレンやエタノールを単独で原料とし、断熱型反応器でプロピレンの製造を試みるとプロピレンの等の目的化合物の収率が低下する、又は、触媒のコーキング劣化が進行するとの知見を見出した。なお、触媒のコーキング劣化とは、触媒表面にコークが付着し触媒の活性を低下させることを意味する。
本発明者は、このような知見に基づき、エチレン及びエタノールを含有する混合原料を、断熱型反応器に充填した触媒と接触させると収率良くプロピレン等の目的化合物を与え、コーキング劣化を抑制できることを見出した。
この要因は互いを阻害しない吸熱反応と発熱反応を組み合わせることで熱中和を達成することができたことと想定される。エチレンからプロピレンへの変換反応は発熱反応である。また、エタノールからプロピレンへの変換反応は、触媒の存在下では、エタノールからエチレンへの脱水反応とエチレンからプロピレンへの変換反応の2段反応から成る吸熱反応である。本実施形態では、エチレンからプロピレンへの変換反応とエタノールからプロピレンへの変換反応が互いの反応を阻害せずに進行することを見出し、これらの発熱反応と吸熱反応とを組み合わせることで断熱型反応器においても反応条件の制御を容易に行うことができたと考えられる。ただし、要因はこれに限定されない。
(原料)
本実施形態のエタノールの変換方法は、エチレン及びエタノールを含有する混合原料を用いる。当該混合原料を用いることで、断熱型反応器を使用し、反応器内の温度を制御して収率よくプロピレンを製造することができる。
本実施形態のエタノールの変換方法は、エチレン及びエタノールを含有する混合原料を用いる。当該混合原料を用いることで、断熱型反応器を使用し、反応器内の温度を制御して収率よくプロピレンを製造することができる。
混合原料中、エチレン/エタノールのモル比は、0.20~2.5であることが好ましく、0.20~2.0であることがより好ましく、0.30~1.8であることが更に好ましく、0.30~1.5であることが特に好ましい。
エチレンとしては種々の製法で製造されたものを使用することができる。例えば、ナフサ及び/又はエタンの熱分解、エタンの直接又は酸化的脱水素反応、又は、エタノールの脱水反応により得られるものを使用することができる。エタノールも同様に種々の方法で製造されたものを使用することができる。これらの中でも、環境調和性に優れる観点から、バイオエタノールや廃棄物由来のエタノールを使用することが好ましい。これらのエチレン及びエタノールはその製造工程において、水が副生する、又は、反応器内に水が共存する工程を経ることが多い。したがって、本実施形態のエタノールの変換方法では、原料であるエチレン及びエタノールが水を含有することがある。
本実施形態のエタノールの変換方法において、混合原料は、炭素数4~6のオレフィンを更に含んでいてもよい。炭素数4~6のオレフィンは、エチレン及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物を与えることができる。炭素数4~6のオレフィンとしては、例えば、ブテン、ペンテン、ヘキセンが挙げられる。なお、本明細書において「オレフィン」という用語は、直鎖状、分岐状及び環状オレフィンに加え、シクロパラフィンを含むものとする。
混合原料中、炭素数4~6のオレフィン/エチレンのモル比は、3.0以下であることが好ましく、1.0以下であることがより好ましく、0.5以下であることが更に好ましく、0.15~0.5であることがより更に好ましい。
本実施形態のエタノールの変換方法において、混合原料は、エタノール以外の炭素数1~6の含酸素化合物を更に含んでいてもよい。炭素数1~6の含酸素化合物は、エチレン及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物を与えることができる。エタノール以外の炭素数1~6の含酸素化合物としては、例えば、メタノール、プロパノール、ジメチルエーテル、ジエチルエーテルが挙げられる。
混合原料中、エタノール以外の炭素数1~6の含酸素化合物/エタノールのモル比は、1.0以下であることが好ましく、0.5以下であることがより好ましい。
なお、エチレン、エタノール、炭素数4~6のオレフィン、エタノール以外の炭素数1~6の含酸素化合物を総称して「有効原料」ともいう。
混合原料は、上記の有効原料のほか、パラフィンなどの飽和脂肪族炭化水素、炭素数7以上のオレフィン、炭素数7以上の含酸素化合物を含んでいてもよい。これら飽和脂肪族炭化水素、炭素数7以上のオレフィン、及び炭素数7以上の含酸素化合物は、エチレン及びエタノールと同様に触媒と接触させることで、脱水素反応や脱水反応の組合せによりプロピレン等の目的化合物へと変換され得るが、上述の有効原料よりも反応性が低い。
また、混合原料には反応工程より得られた炭素数3以上のオレフィンを含有する反応ガスから炭素数4以上のオレフィンの全量又は一部を分離工程により分離したものを含むことができる。このように、所謂、リサイクル反応システムを用いることにより、オレフィン原料の有効利用を図ることができる。
混合原料は、反応工程によってプロピレン等の目的化合物へと変換され得る上述の原料の他に、窒素などの不活性ガスなどを含んでもよい。そのほか、混合原料は、希釈ガスとして、水素、メタンを含んでいてもよいが、水素希釈は行わないことが好ましい。水素は触媒のコーキング劣化を抑制するために使用されることがあるが、同時に生成プロピレン等の水素化反応が起こり、プロピレン純度(プロピレン/(プロピレン+プロパン))を低下させる悪影響がある。本実施形態の方法においては、水素希釈を行わなかった場合にも触媒のコーキング劣化速度は小さく、安定な運転が可能であるため水素希釈を行わないほうが好ましい。ただし、反応ガスから分離された留分のリサイクル等によって反応器に供給される少量の水素は上記水素希釈におけるような悪影響を生じない。
混合原料中のエチレンと炭素数4~6のオレフィンとエタノールとの合計割合が混合原料供給質量流量に対して、40質量%以上であることが好ましく、50質量%以上であることがより好ましい。混合原料供給質量流量とは、不活性成分を含む、反応器に供される全ての化合物の合計流量のことである。
混合原料中、エチレン及びエタノールの合計含有量は、有効原料供給質量に対して、好ましくは30~100質量%であり、より好ましくは40~100質量%であり、更に好ましくは50~100質量%である。ただし、エタノールは、エチレンとして換算した質量をエタノール質量及び有効原料供給質量の計算に使用する。
混合原料中、炭素数4~6のオレフィンの合計含有量は、有効原料供給質量に対して、好ましくは65質量%以下であり、より好ましくは10~55質量%である。
有効原料供給質量とは、有効原料の時間あたりの合計供給質量である。ただし、エタノールはエチレンとして換算した質量を計算に使用する。
本実施形態のエタノールの変換方法では、混合原料は水を含んでもよい。混合原料に含まれるエチレン及びエタノールは種々の製造方法によって製造されることから、「製造工程で発生した水」を含有していてもよい。ここでの「製造工程で発生した水」とは、エチレン及び/又はエタノールの製造過程において発生し、除去されていない水分のことをいう。
本実施形態のエタノールの変換方法では、「製造工程で発生した水」に追加して混合原料に水蒸気を含ませることができる。水蒸気はオレフィン分圧を低下させることでコーキング劣化を抑制し、低級オレフィンの収率を向上させる効果がある。一方で、水蒸気はゼオライトの脱アルミニウムを促進する可能性があるため、好ましくは「製造工程で発生した水」に追加して混合原料に水蒸気を含ませない。
(断熱型反応器)
本実施形態のエタノールの変換方法では断熱型反応器を使用する。断熱型反応器に関しては、Adiabatic Fixed-Bed Reactors(Elsevier,2014,Ch.1,P.4,L.5~24 ISBN:978-0-12-801306-9)の記載を参照することができる。断熱型反応器の例としては、固定床断熱型反応器、移動床断熱型反応器、流動床断熱型反応器が挙げられるが、本実施形態の方法には固定床断熱型反応器が好ましい。固定床断熱型反応器の中でも固定触媒床が一段だけの固定床一段断熱型反応器がより好ましい。反応に伴い触媒上には炭素質(コーク)が蓄積するため、反応を継続しながらこの炭素質の燃焼除去が可能な多塔切り替え式の固定床一段断熱型反応器が好ましい。
本実施形態のエタノールの変換方法では断熱型反応器を使用する。断熱型反応器に関しては、Adiabatic Fixed-Bed Reactors(Elsevier,2014,Ch.1,P.4,L.5~24 ISBN:978-0-12-801306-9)の記載を参照することができる。断熱型反応器の例としては、固定床断熱型反応器、移動床断熱型反応器、流動床断熱型反応器が挙げられるが、本実施形態の方法には固定床断熱型反応器が好ましい。固定床断熱型反応器の中でも固定触媒床が一段だけの固定床一段断熱型反応器がより好ましい。反応に伴い触媒上には炭素質(コーク)が蓄積するため、反応を継続しながらこの炭素質の燃焼除去が可能な多塔切り替え式の固定床一段断熱型反応器が好ましい。
図1は、固定床一段断熱型反応器の概略構成図である。固定床一段断熱型反応器1は、外周に断熱材121が設けられた反応筐体12と、触媒床13と、反応器入口14と、反応器出口15とを備える。反応筐体12は、外周に断熱材121が設けられることで、反応器内の熱を外部に逃がさない。本実施形態に係る製造方法においては、反応による発熱及び吸熱により反応器内の温度を制御することができる。
触媒床13には、後述する触媒が充填される。触媒床13の触媒床入口131に接する直前に第1シース熱電対161が設けられる。触媒床13の触媒床出口132を通過した直後に第2シース熱電対162が設けられる。これらの熱電対により、触媒床入口131に接触する直前の混合原料と触媒床出口132を通過した直後の反応ガスの温度を測定する。触媒床13は、多段式であってもよいが、図1に示すとおり一段型であることが好ましい。
固定床一段断熱型反応器1では、反応器入口14から混合原料を導入し、触媒床13と接触させ、反応器出口15から反応ガスを取り出す。
(反応工程の条件)
本実施形態のエタノールの変換方法では、反応温度が300℃以上であってもよい。生成するオレフィンには熱的平衡が存在しており、プロピレン収率をより向上させる観点から、反応温度は450℃以上であることが好ましい。更に、高温で促進されるコーキング劣化加速を抑制する観点から、反応温度は、600℃未満であることが好ましい。より具体的には、触媒床入口における反応ガスの温度が450℃~590℃であることが好ましく、触媒出口における反応ガスの温度が450℃~590℃であることが好ましい。
本実施形態のエタノールの変換方法では、反応温度が300℃以上であってもよい。生成するオレフィンには熱的平衡が存在しており、プロピレン収率をより向上させる観点から、反応温度は450℃以上であることが好ましい。更に、高温で促進されるコーキング劣化加速を抑制する観点から、反応温度は、600℃未満であることが好ましい。より具体的には、触媒床入口における反応ガスの温度が450℃~590℃であることが好ましく、触媒出口における反応ガスの温度が450℃~590℃であることが好ましい。
触媒床の出口温度と触媒床の入口温度との温度差が-80K~80Kであることが好ましく、-60K~60Kであることがより好ましい。
なお、触媒床の入口温度とは、断熱型反応器に充填されている触媒床に原料流体が接する直前の混合原料の温度である。触媒床の出口温度とは、反応ガスが触媒床を通過した直後の反応ガスの温度である。ここでいう混合原料及び反応ガスの温度とは、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0d~0.8dの間における温度を指す。入出平均反応温度は、図1に示すように、触媒床の入口温度と触媒床の出口温度を計測し、計算式:〔触媒床の入口温度+触媒床の出口温度〕/2により算出する値(以下、単に「反応温度」ともいう。)としている。
反応圧力は、好ましくは0.01~3.0MPaGであり、より好ましくは0.01~ 1.0MPaGである。
有効原料の供給速度は、触媒の質量基準の空間速度(WHSV)で、好ましくは0.1~1000hr-1であり、より好ましくは0.1~500hr-1であり、更に好ましくは0.5~100hr-1である。本実施形態のエタノールの変換方法では、下式のように、エタノールをエチレンとして換算した上でWHSVを算出している。また、有効原料供給質量流量は、プロピレン、芳香族化合物等の目的化合物の生産性に優れる観点から、好ましくは1kg/hr以上であり、より好ましくは10kg/hr以上であり、更に好ましくは1,000kg/hr以上である。
WHSV(hr-1)=有効原料供給質量流量(kg/hr)/触媒量(kg)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
(触媒)
本実施形態のエタノールの変換方法における触媒は、オレフィン及びエタノールをプロピレン等の目的化合物へと変換する触媒能を示す固体触媒である。このような触媒としては、触媒の熱耐久性及びプロピレン選択性に優れる観点から、ゼオライト含有触媒が好ましい。従来のゼオライトによるオレフィン製造に共通する課題として、炭化水素との反応によりゼオライト細孔内部に重質な炭素質(コーク)が蓄積し失活するコーキング劣化が挙げられる。触媒性能を再生するためには酸素分子を含有する雰囲気下でコークを燃焼除去することが好ましいが、このコーク燃焼に伴いゼオライトの構造崩壊が進行し、再生不可能な触媒の永久劣化が誘引される。本実施形態のエタノールの変換方法によればコークの生成を抑制することができるため、ゼオライト含有触媒を用いても活性を維持しやすくなる。
本実施形態のエタノールの変換方法における触媒は、オレフィン及びエタノールをプロピレン等の目的化合物へと変換する触媒能を示す固体触媒である。このような触媒としては、触媒の熱耐久性及びプロピレン選択性に優れる観点から、ゼオライト含有触媒が好ましい。従来のゼオライトによるオレフィン製造に共通する課題として、炭化水素との反応によりゼオライト細孔内部に重質な炭素質(コーク)が蓄積し失活するコーキング劣化が挙げられる。触媒性能を再生するためには酸素分子を含有する雰囲気下でコークを燃焼除去することが好ましいが、このコーク燃焼に伴いゼオライトの構造崩壊が進行し、再生不可能な触媒の永久劣化が誘引される。本実施形態のエタノールの変換方法によればコークの生成を抑制することができるため、ゼオライト含有触媒を用いても活性を維持しやすくなる。
≪ゼオライト含有触媒≫
ゼオライト含有触媒とは、ゼオライトを活性種として含有する触媒粉又は成形体のことである。本実施形態のエタノールの変換方法においては、上記のゼオライト含有触媒中のゼオライトとして、5~6Åの細孔径を有する、所謂、中間細孔径ゼオライトを用いることが好ましい。中間細孔径ゼオライトは、「細孔径の範囲が、A型ゼオライトに代表される小細孔径ゼオライトの細孔径と、モルデナイトやX型やY型ゼオライトに代表される大細孔径ゼオライトの細孔径の中間にあるゼオライト」を意味する。「中間細孔径ゼオライト」は、その結晶構造中にいわゆる酸素10員環を有する。
ゼオライト含有触媒とは、ゼオライトを活性種として含有する触媒粉又は成形体のことである。本実施形態のエタノールの変換方法においては、上記のゼオライト含有触媒中のゼオライトとして、5~6Åの細孔径を有する、所謂、中間細孔径ゼオライトを用いることが好ましい。中間細孔径ゼオライトは、「細孔径の範囲が、A型ゼオライトに代表される小細孔径ゼオライトの細孔径と、モルデナイトやX型やY型ゼオライトに代表される大細孔径ゼオライトの細孔径の中間にあるゼオライト」を意味する。「中間細孔径ゼオライト」は、その結晶構造中にいわゆる酸素10員環を有する。
中間細孔径ゼオライトの例としては、ZSM-5、ZSM-8、ZSM-11、ZSM-12、ZSM-21、ZSM-23、ZSM-35、ZSM-38等が挙げられるが、中でもZSM-5、ZSM-11、ZSM-8などのZSM-5型ゼオライトやZSM-38が好ましい。また、Stud.Surf.Sci.Catal.1987,33,167-215に記載のZSM-5、ZSM-11に類似のゼオライトを用いることができ、この中でも、触媒性能(触媒活性とコーキングに対する耐久性)に優れる観点から、MFI型ゼオライトが好ましく、ZSM-5がより好ましい。
本実施形態のゼオライト含有触媒に含まれるゼオライトのシリカ/アルミナ(SiO2/Al2O3)モル比は、適宜選択することができるが、触媒活性とプロピレン選択性に優れる観点から、好ましくは20~2000であり、触媒の耐久性を高める観点から、より好ましくは100~1500であり、更に好ましくは300~1200であり、より更に好ましくは800~1200である。ゼオライト含有触媒に含まれるゼオライトのシリカ/アルミナ(SiO2/Al2O3)モル比は、20~400であってもよく、100~300であってもよい。ゼオライトのシリ力/アルミナモル比は公知の方法により測定することができ、例えば、ゼオライトをアルカリ水溶液に完全に溶解し、得られる溶液をプラズマ発光分光分析法等により分析し、求めることができる。
本実施形態のゼオライトの合成方法に特に制限はないが、従来知られているMFI型ゼオライトの水熱合成方法の各種条件を最適化することにより製造できる。一般に、水熱合成法で効率よくMFI型ゼオライトを得る手段としては、適切な有機構造規定剤(SDA)を用いて水熱合成する方法、水熱合成されたMFIゼオライトを種結晶として添加して水熱合成する方法、又は、結晶段階にある種スラリーとして添加して水熱合成する方法がある。なお、ここで用いられる有機構造規定剤(SDA)としては、例えば、アンモニウム塩、尿素化合物、アミン、及びアルコール等が挙げられる。また、有機のSDAだけでなく、無機の陽イオンや陰イオンも構造に関わることが知られており、ゼオライト合成は各成分の複合的な働きに依存する。以上に述べたようなMFI型ゼオライトの水熱合成方法において、原材料や添加物(SDA)の種類、添加物量、pH、シリ力/アルミナモル比、媒体、陽イオン、陰イオンの存在比などの原料仕込み組成、合成温度、合成時間等の合成条件を適宜、最適化することで好適な触媒を得ることができる。
具体的には、例えば、特許5426983号公報に記載されている種スラリーを用いて合成する方法や、The Hydrothermal Synthesis of Zeolites(Chemcal Reviews,2003,103,663-702)に例示された方法が挙げられる。
また、上述した特定の物性及び組成を有するMFIゼオライトであれば、市販されているゼオライトを用いることもできる。
本実施形態におけるゼオライト含有触媒はリン元素又は銀元素を含むことが好ましい。
リン元素の形態としては、リンの重合物(例えば、ポリリン酸)、リンの酸化物(例えば、P2O5)、リンがゼオライトのアルミニウムに付加した態様の化合物等が挙げられる。また、それらの複数が含まれていてもよい。リン元素は、ゼオライトがアルミニウムを含有する場合、ゼオライトの脱アルミニウムを抑制する効果や、場合によっては、プロピレン収率を向上させる効果がある。特に、高温水蒸気雰囲気に曝される用途の場合、脱アルミニウムによりゼオライト含有触媒の特性が変化し易いので、脱アルミニウムを抑制する効果がより向上する。
ゼオライト含有触媒に含まれるリン元素の含有量は、触媒全体の質量に対して、好ましくは0.01~2.0質量%であり、脱アルミニウム抑制の効果に優れる観点から、より好ましくは0.05~2.0質量%である。
本実施形態において、触媒中のリン元素の含有量は、蛍光X線分析装置を用いて測定した値を示す。リン元素の含有量の測定は、市販の蛍光X線分析装置を使用し、取扱説明書に沿って通常の条件で測定すればよく、例えば、Rigaku製、商品名「RIX3000」を使用する場合、測定条件は、P-Kα線を用い、管球電圧50kV、管球電流50mAとすることができる。
本実施形態においては、ゼオライト含有触媒に含まれるリン元素の原料としてリン酸及び/又はリン酸塩(以下、「リン原料」ともいう。)を用いる。リン原料としてはリン酸塩がより好ましく、リン酸塩の中でも、25℃において100gの水に対して1g以上の溶解度を示す化合物がより好ましい。
リン酸としては、例えば、リン酸、ピロリン酸が挙げられ、リン酸塩としては、例えば、リン酸アンモニウム、リン酸水素二アンモニウム、リン酸二水素アンモニウム、リン酸水素アンモニウムナトリウム等のリン酸アンモニウム塩、リン酸水素カリウム、リン酸水素アルミニウム、リン酸ナトリウム、リン酸カリウム等が挙げられる。中でも、水に対する溶解度が比較的高いリン酸アンモニウム塩が好ましく、より好ましくは、リン酸アンモニウム、リン酸水素二アンモニウム、リン酸二水素アンモニウムからなる群より選択される少なくとも1種である。これらは単独で用いても2種以上を組み合わせて用いてもよい。
銀元素の形態としては、例えば銀イオンである。銀元素は、ゼオライトの酸点を制御することでゼオライトの耐水熱性を向上させる効果がある。
ゼオライト含有触媒に含まれる銀元素の含有量は、触媒全体の質量に対して、好ましくは0.01~2.0質量%であり、含有量あたりの耐水熱性の向上の効果に優れる観点から、より好ましくは0.05~2.0質量%である。
本実施形態において、触媒中の銀元素の含有量は、蛍光X線分析装置を用いて測定した値を示す。銀元素の含有量の測定は、市販の蛍光X線分析装置を使用し、取扱説明書に沿って通常の条件で測定すればよく、例えば、Rigaku製、商品名「RIX3000」を使用する場合、測定条件は、P-Kα線を用い、管球電圧50kV、管球電流50mAとすることができる。
本実施形態においては、ゼオライト含有触媒に含まれる銀元素の原料として硝酸銀が挙げられる。ナトリウムをカウンターカチオンとして含有するゼオライト含有触媒を用いて、硝酸銀とイオン交換し、焼結することで銀元素を含有するゼオライト含有触媒が得られる。ゼオライト中のカウンターカチオンであるナトリウムと硝酸銀とのイオン交換は、硝酸銀の水溶液にゼオライト、又は、ゼオライト含有触媒を浸漬した後、水洗することで実施できる。この時、浸漬と水洗とを複数回実施することでイオンの交換率を向上させることができる。
本実施形態のゼオライト含有触媒は、上述した特定の物性及び組成を有するゼオライトを用いて、例えば、以下の通りに成形して製造することができる。その成形方法は、特に限定されず、一般的な方法を用いることができる。具体的には、触媒成分を圧縮成形する方法や押出成形する方法、流動床反応方式に最適なスプレードライ成形法が挙げられる。
また、成形にはバインダーを用いることができる。バインダーとしては、特に制限されず、例えば、シリカ、アルミナ、カオリンを単独、又は、混合して使用することができる。これらのバインダーは、市販のものを使用することができる。ゼオライト/バインダーの質量比率は、好ましくは10/90~90/10の範囲であり、より好ましくは20/80~80/20の範囲である。コーキングを抑制できる観点から、シリカバインダーであることが好ましい。
本実施形態のエタノールの変換方法では、ゼオライト含有触媒を原料と接触させるのに先立って、ゼオライト含有触媒に前処理工程を実施してもよい。好ましい前処理工程としては、水蒸気の存在下、300℃以上の温度で加熱処理する工程が挙げられる。前処理を行うと、触媒の劣化抑制や選択性改善の効果がより顕著となる傾向にある。上記の方法の場合、300℃以上900℃以下の温度で、雰囲気は特に限定されないが、空気あるいは窒素等の不活性ガスとスチーム(水蒸気)との混合ガスを流通させ、水蒸気分圧0.01気圧以上の条件下で処理することが好ましい。加熱処理温度としては、400℃以上700℃以下の温度がより好ましい。また、本前処理工程はエタノールを変換する反応器を使って行うことができる。
(生成物:炭素数3以上のオレフィンを含む反応ガス)
本実施形態のエタノールの変換方法では、混合原料を触媒と接触させることで炭素数3以上のオレフィンを含む反応ガスを得る。「反応ガス」とは、混合原料を触媒と接触による反応後のガス組成物を意味する。反応ガスは、エチレンを含んでいてもよい。反応ガスは、水素、炭素数1~3の脂肪族炭化水素、炭素数4~8の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を含んでいてもよい。
本実施形態のエタノールの変換方法では、混合原料を触媒と接触させることで炭素数3以上のオレフィンを含む反応ガスを得る。「反応ガス」とは、混合原料を触媒と接触による反応後のガス組成物を意味する。反応ガスは、エチレンを含んでいてもよい。反応ガスは、水素、炭素数1~3の脂肪族炭化水素、炭素数4~8の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を含んでいてもよい。
〔再生工程〕
触媒を、長期間反応に用いるとコークが触媒上に付着しコーキング劣化を起こす場合がある。触媒がコーキング劣化を起こした場合には、例えば、酸素含有ガスと接触させ400~700℃の温度で触媒上のコークを燃焼除去することにより、コーキング劣化を起こした触媒を再生させること(以下、「再生工程」ともいう。)ができる。酸素含有ガスとしては、例えば、空気、及び、空気又は酸素と不活性ガスとの混合ガスが挙げられる。酸素含有ガスの酸素濃度は、好ましくは0.1~2.0体積%である。触媒は、反応器より抜き出し反応器外で再生処理を行う反応器外再生と反応器より抜き出さずに反応器内で再生処理を行う反応器内再生のどちらの再生方法を採用してもよい。また、切り替え式反応器を採用することで反応-再生切り替え運転を行うこともできる。
触媒を、長期間反応に用いるとコークが触媒上に付着しコーキング劣化を起こす場合がある。触媒がコーキング劣化を起こした場合には、例えば、酸素含有ガスと接触させ400~700℃の温度で触媒上のコークを燃焼除去することにより、コーキング劣化を起こした触媒を再生させること(以下、「再生工程」ともいう。)ができる。酸素含有ガスとしては、例えば、空気、及び、空気又は酸素と不活性ガスとの混合ガスが挙げられる。酸素含有ガスの酸素濃度は、好ましくは0.1~2.0体積%である。触媒は、反応器より抜き出し反応器外で再生処理を行う反応器外再生と反応器より抜き出さずに反応器内で再生処理を行う反応器内再生のどちらの再生方法を採用してもよい。また、切り替え式反応器を採用することで反応-再生切り替え運転を行うこともできる。
(反応-再生切り替え運転)
反応-再生切り替え運転とは、2塔又は多塔切り替え式の断熱型反応器を用い、反応工程と再生工程とを同時に行う運転操作である。例えば、3塔切り替え式の場合は、2塔が反応工程に使用され、同時に、残りの1塔が触媒再生に使用される。その後、反応工程に使用されていたうちの1塔の反応工程を停止して触媒再生を行い、触媒再生に使用していた1塔で反応工程を行うことで、2塔分の生産能力を維持しつつ、触媒再生を行うことができる。このような反応形式をメリーゴーランド方式とも呼び、触媒再生のために製造工程を止める必要がないため、生産効率に優れる観点から好ましい。
反応-再生切り替え運転とは、2塔又は多塔切り替え式の断熱型反応器を用い、反応工程と再生工程とを同時に行う運転操作である。例えば、3塔切り替え式の場合は、2塔が反応工程に使用され、同時に、残りの1塔が触媒再生に使用される。その後、反応工程に使用されていたうちの1塔の反応工程を停止して触媒再生を行い、触媒再生に使用していた1塔で反応工程を行うことで、2塔分の生産能力を維持しつつ、触媒再生を行うことができる。このような反応形式をメリーゴーランド方式とも呼び、触媒再生のために製造工程を止める必要がないため、生産効率に優れる観点から好ましい。
〔分離工程〕
本実施形態のエタノールの変換方法は、反応ガスからプロピレン等の目的化合物を分離すること(以下、「分離工程」ともいう。)を含むことができる。分離工程により当該反応ガスからエチレンとプロピレンを分離することができる。
本実施形態のエタノールの変換方法は、反応ガスからプロピレン等の目的化合物を分離すること(以下、「分離工程」ともいう。)を含むことができる。分離工程により当該反応ガスからエチレンとプロピレンを分離することができる。
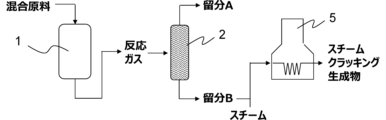
本実施形態に係る方法は、図2に示すように、反応器1と、蒸留塔2と、蒸留塔3と、蒸留塔4とを有する装置により実施することができる。反応器1で行われる反応工程により得られた反応ガスを蒸留塔2で、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~8の炭化水素を主に含む留分Bとに分離することができる。なお、蒸留塔2で蒸留する前に反応ガスから凝縮させた水を除去してもよい(図示せず)。蒸留塔3及び蒸留塔4で、留分Aからエチレン及びプロピレンを分離することによって、該反応ガスからのエチレン及びプロピレンの分離を効率的に行う。また、反応ガス及び/又は留分Aの少なくとも一部をエチレンプラントの精製系に導入し、該精製系で反応ガスからエチレン及びプロピレンを分離してもよい。分離工程により得られた留分Bをはじめとする各種留分は反応器に原料としてリサイクルすることができる。
本実施形態に係る方法は、図3に示すように、反応器1と、蒸留塔2と、スチームクラッキング装置5とを有する装置により実施することができる。分離工程により得られた留分Bをスチームクラッキングに付すことにより、エチレン及びプロピレンを含有するスチームクラッキング生成物を得、スチームクラッキング生成物からエチレン及びプロピレンを分離することでプロセス全体の効率性を高めることができる。スチームクラッキングとは、加熱蒸気と共に留分中の化合物を熱分解することを意味する。
本実施形態に係る方法は、図4に示すように、反応器1と、冷却装置6と、蒸留塔2と、油水分離機7とを有する装置により実施することができる。反応器1で行われる反応工程により得られた反応ガスを冷却装置6により冷却する、冷却工程を設けることができる。冷却工程により、反応ガスを炭素数2~6の脂肪族炭化水素を主に含む留分Cと、水、炭素数7以上の脂肪族炭化水素及び芳香族化合物を主に含む留分Dとに分離することができる。留分Cは冷却工程においてガス成分として得られ、留分Dは液成分として回収される。該留分Dから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。
冷却工程で回収された留分Dは、油水分離機7において炭化水素を主に含む留分Eと水を主に含む留分Fとに分離される。該留分Eから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。芳香族化合物の分離は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。
本実施形態に係る方法は、図5に示すように、反応器1と、蒸留塔2と、蒸留塔8とを有する装置により実施することができる。蒸留塔8により、留分Bをリサイクルする際、炭素数4~6の脂肪族炭化水素、好ましくはオレフィン、を主に含む留分B1と、芳香族化合物を主に含む留分B2とに分離し、留分B1の少なくとも一部を反応器にリサイクルすることが好ましい。留分B2を除去した留分B1をリサイクル原料とすることで、プロピレンに変換されない芳香族化合物を原料から除去することができ、効率よくプロピレンを製造することができる。このように、留分Bより留分B2として芳香族化合物を分離することもできる。芳香族化合物の更なる分離精製は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。
また、本実施形態に係る方法は、図6に示すように、反応器1と、蒸留塔2と、蒸留塔9とを有する装置により実施することができる。蒸留塔9により、留分Bを炭素数4~8の炭化水素を主に含む留分B3と炭素数9以上の炭化水素を主に含む留分B4とに分離し、留分B3の少なくとも一部を反応器にリサイクルすることが好ましい。留分B4を除去した留分B3をリサイクル原料とすることで、コーキング劣化を促進させ得る重質分を原料から除去することができ、触媒のコーキング劣化を抑制することができる。
各種留分において「主に含む」とは、「主に含む」と記載された成分の合計質量が、留分全量に対して、50質量%超であることを意味する。
なお、これらの分離工程は、蒸留、抽出など種々の公知の方法を組み合わせることで実施することができる。
[第2実施形態]
続いて、第2実施形態に係るエタノールの変換方法について説明する。
続いて、第2実施形態に係るエタノールの変換方法について説明する。
<エタノールの変換方法-第2実施形態->
第2実施形態のエタノールの変換方法は、メタノール及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ること(以下、「反応工程」とも言う。)を含む。
なお、上述の反応ガスから分離することでプロピレンを得ることができ、上述の反応ガスから分離することで芳香族化合物を得ることもできる。
第2実施形態のエタノールの変換方法は、メタノール及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ること(以下、「反応工程」とも言う。)を含む。
なお、上述の反応ガスから分離することでプロピレンを得ることができ、上述の反応ガスから分離することで芳香族化合物を得ることもできる。
従来、化成品製造プロセスを実用化する際、反応器及びその様式の選定は操作の容易性や環境負荷の大小に大きな影響を与える要素となる。固定床断熱型反応器は反応器の加熱又は冷却を行わず、更に、流動床などの反応器と比較して複雑な構造を有していないため、設計、建築及び操業の負荷が小さい理想的な反応器であり、例えば、固定床一段断熱型反応器を採用できればその利点は更に大きくなる。しかしながら、当該反応器は吸熱又は発熱の大きい反応系には適用できないという難点も有している。
特許文献2,3,6では、それぞれ各原料をゼオライト触媒で目的のオレフィンへと変換する技術が公開されているが、これらの技術は吸熱及び発熱が大きくなるため断熱型反応器へと応用することはできない。
そこで、本実施形態は、断熱型反応器を使用し、反応器内の温度を制御して、アルコールを収率良く目的化合物へと変換する方法、プロピレンの製造方法、及び芳香族化合物の製造方法を提供することを目的とする。
本発明者らは、上記の課題を達成するために鋭意検討した結果、エタノールとメタノールを混合した原料から炭素数3以上のオレフィン等の目的化合物への変換に対して反応器内において反応熱を制御することで、断熱型反応器を使用し、反応温度を制御して、アルコールを収率良く目的化合物へと変換することができることを見出した。
本実施形態によれば、断熱型反応器を使用し、反応器内の温度を制御して、アルコールを収率良く目的化合物へと変換することができる。また、本実施形態によれば、断熱型反応器を使用することで操業の負荷が小さく、エネルギー効率が高い環境調和型のアルコール変換方法が実施できる。また、断熱型反応器を使用した場合にも、アルコールを収率良く目的化合物へと変換することができ、触媒のコーキング劣化を抑制できるため、エタノールの変換方法として好適である。
本発明者は、メタノールやエタノールを単独で原料とし、断熱型反応器でアルコールの変換を試みると、プロピレン、芳香族化合物等の高付加価値化合物の収率が低下する、又は、触媒のコーキング劣化が進行するとの知見を見出した。なお、触媒のコーキング劣化とは、触媒表面にコークが付着し触媒の活性を低下させることを意味する。
本発明者は、このような知見に基づき、メタノール及びエタノールを含有する混合原料を、断熱型反応器に充填した触媒と接触させるとアルコールを収率良く目的化合物へと変換することができ、かつコーキング劣化を抑制できることを見出した。
この要因は互いを阻害しない吸熱反応と発熱反応を組み合わせることで熱中和を達成することができたことと想定される。例えば、メタノールからプロピレンへの変換反応は発熱反応であり、エタノールからプロピレンへの変換反応は吸熱反応である。本実施形態では、反応器内で吸熱反応と発熱反応とが互いの反応を阻害せずに進行することを見出し、これらの発熱反応と吸熱反応とを組み合わせることで断熱型反応器においても反応条件の制御を容易に行うことができた。ただし、要因はこれに限定されない。
この要因は互いを阻害しない吸熱反応と発熱反応を組み合わせることで熱中和を達成することができたことと想定される。例えば、メタノールからプロピレンへの変換反応は発熱反応であり、エタノールからプロピレンへの変換反応は吸熱反応である。本実施形態では、反応器内で吸熱反応と発熱反応とが互いの反応を阻害せずに進行することを見出し、これらの発熱反応と吸熱反応とを組み合わせることで断熱型反応器においても反応条件の制御を容易に行うことができた。ただし、要因はこれに限定されない。
(原料)
本実施形態のエタノールの変換方法は、メタノール及びエタノールを含有する混合原料を用いる。当該混合原料を用いることで、断熱型反応器を使用し、反応器内の温度を制御して収率よくプロピレン等の目的化合物を製造することができる。メタノール及びエタノールの少なくとも一方が、環境調和性に優れる観点から、バイオマス由来であることが好ましい。なお、バイオマスとは動植物を起源とする化石資源以外の有機性資源を指し、バイオマス由来とはバイオマスを原料として製造された化合物であることをいう。
本実施形態のエタノールの変換方法は、メタノール及びエタノールを含有する混合原料を用いる。当該混合原料を用いることで、断熱型反応器を使用し、反応器内の温度を制御して収率よくプロピレン等の目的化合物を製造することができる。メタノール及びエタノールの少なくとも一方が、環境調和性に優れる観点から、バイオマス由来であることが好ましい。なお、バイオマスとは動植物を起源とする化石資源以外の有機性資源を指し、バイオマス由来とはバイオマスを原料として製造された化合物であることをいう。
混合原料中、メタノール/エタノールのモル比は、0.050~2.0であることが好ましく、0.20~1.5であることがより好ましく、0.30~1.5であることが更に好ましく、0.30~1.0であることがより更に好ましい。
メタノールとしては種々の製法で製造されたものを使用することができる。例えば、天然ガスや石炭より得られる一酸化炭素の水素化や二酸化炭素の水素化、木酢液の蒸留により得られるものを使用することができる。エタノールも同様に種々の製法で製造されたものを使用することができる。この中でも、環境調和性に優れる観点から、バイオエタノールや廃棄物由来のエタノールを使用することが好ましい。これらのメタノール及びエタノールはその製造工程において、水が副生する、又は、反応器内に水が共存する工程を経ることが多い。したがって、本実施形態のエタノールの変換方法では、原料であるメタノール及びエタノールが水を含有することがある。
本実施形態のエタノールの変換方法において、混合原料は、炭素数4~6のオレフィンを更に含んでいてもよい。炭素数4~6のオレフィンは、メタノール及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物を与えることができる。なお、上記の「オレフィン」という用語は、直鎖状、分岐状及び環状オレフィンに加え、シクロパラフィンを含むものとする。
炭素数4~6のオレフィンとしては、例えば、ブテン、ペンテン、ヘキセンが挙げられる。混合原料中、炭素数4~6のオレフィン/メタノールのモル比は、3.0以下であることが好ましく、1.0以下であることがより好ましく、0.5以下であることが更に好ましく、0.15~0.5であることがより更に好ましい。
混合原料はエチレンを更に含んでいてもよい。エチレンは、メタノール及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物を与えることができる。混合原料中、エチレン/エタノールのモル比は、0.05以上であることが好ましく、0.1以上であることがより好ましく、0.1~2.0であることが更に好ましい。
本実施形態のエタノールの変換方法において、混合原料は、メタノール及びエタノール以外の炭素数1~6の含酸素化合物を更に含んでいてもよい。炭素数1~6の含酸素化合物は、メタノール及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物を与えることができる。メタノール及びエタノール以外の炭素数1~6の含酸素化合物としては、例えば、プロパノール、ジメチルエーテル、ジエチルエーテルが挙げられる。
混合原料中、メタノール及びエタノール以外の炭素数1~6の含酸素化合物/エタノールのモル比は、1.0以下であることが好ましく、0.5以下であることがより好ましい。
なお、エチレン、エタノール、炭素数4~6のオレフィン、エタノール以外の炭素数1~6の含酸素化合物を総称して「有効原料」ともいう。
混合原料は、上記の有効原料のほか、パラフィンなどの飽和脂肪族炭化水素、炭素数7以上のオレフィン、炭素数7以上の含酸素化合物を含むことができる。これら飽和脂肪族炭化水素、炭素数7以上のオレフィンや炭素数7以上の含酸素化合物はメタノール及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物へと変換され得るが、上述の有効原料よりも反応性が低い。
また、混合原料には反応工程より得られた炭素数4以上のオレフィンを含有する反応ガス及び反応ガスを精製した留分の全量又は一部を含むことができる。このように、所謂、リサイクル反応システムを用いることにより、原料の有効利用を図ることができる。
混合原料は、反応工程によってプロピレン等の目的化合物へと変換され得る上述の原料の他に、窒素などの不活性ガスなどを含んでもよい。そのほか、混合原料は、希釈ガスとして、水素、メタンを含んでいてもよいが、水素を含まないこと(水素希釈は行わないこと)が好ましい。水素は触媒のコーキング劣化を抑制するために使用されることがあるが、同時に生成プロピレン等の水素化反応が起こり、プロピレン純度(プロピレン/(プロピレン+プロパン))[mol/mol]を低下させる悪影響がある。本実施形態の方法においては、水素希釈を行わなかった場合にも触媒のコーキング劣化速度は小さく、安定な運転が可能であるため水素希釈を行わないほうが好ましい。ただし、反応ガスから分離された留分のリサイクル等によって反応器に供給される少量の水素は上記水素希釈におけるような悪影響を生じない。
混合原料中のメタノール、炭素数4~6のオレフィン、エタノールとエチレンとの合計割合が混合原料供給質量流量の総量に対して、40質量%以上であることが好ましく、50質量%以上であることがより好ましい。混合原料供給質量流量の総量とは、不活性ガスを含む、反応器に供給される全ての化合物の合計流量のことである。
混合原料中、メタノール及びエタノールの合計含有量は、有効原料供給質量に対して、好ましくは30~100質量%であり、より好ましくは40~100質量%であり、更に好ましくは50~100質量%である。ただし、メタノール及びエタノールは、それぞれメチレン及びエチレンとして換算した質量をメタノール質量、エタノール質量及び有効原料供給質量の計算に使用する。
混合原料中、炭素数4~6のオレフィンの合計含有量は、有効原料供給質量に対して、好ましくは65質量%以下であり、より好ましくは10~55質量%である。
有効原料供給質量とは、有効原料の時間あたりの合計供給質量である。ただし、メタノール及びエタノールはそれぞれメチレン及びエチレンとして換算した質量を計算に使用する。
本実施形態のエタノールの変換方法では、混合原料は、水を含んでいてもよい。混合原料に含まれるメタノール及びエタノールは種々の製造方法によって製造されることから、「製造工程で発生した水」を含有している。ここでの「製造工程で発生した水」とは、例えば、メタノール及び/又はエタノールの製造過程において発生し、除去されていない水分に由来する水分のことをいう。
本実施形態のエタノールの変換方法では、混合原料は、「製造工程で発生した水」に加えて、水蒸気を含んでもよい。水蒸気はオレフィン分圧を低下させることでコーキング劣化を抑制し、低級オレフィンの収率を向上させる効果がある。一方で、水蒸気はゼオライトの脱アルミニウムを促進する可能性があるため、好ましくは「製造工程で発生した水」に追加して混合原料に水蒸気を含ませない。
(断熱型反応器)
本実施形態のエタノールの変換方法では断熱型反応器を使用する。断熱型反応器に関しては、Adiabatic Fixed-Bed Reactors(Elsevier,2014,Ch.1,P.4,L.5~24 ISBN:978-0-12-801306-9)の記載を参照することができる。断熱型反応器の例としては、固定床断熱型反応器、移動床断熱型反応器、流動床断熱型反応器が挙げられるが、本実施形態の方法には固定床断熱型反応器が好ましい。固定床断熱型反応器の中でも固定触媒床が一段だけの固定床一段断熱型反応器がより好ましい。反応に伴い触媒上には炭素質(コーク)が蓄積するため、反応を継続しながらこの炭素質の燃焼除去が可能な多塔切り替え式の固定床一段断熱型反応器が好ましい。
本実施形態のエタノールの変換方法では断熱型反応器を使用する。断熱型反応器に関しては、Adiabatic Fixed-Bed Reactors(Elsevier,2014,Ch.1,P.4,L.5~24 ISBN:978-0-12-801306-9)の記載を参照することができる。断熱型反応器の例としては、固定床断熱型反応器、移動床断熱型反応器、流動床断熱型反応器が挙げられるが、本実施形態の方法には固定床断熱型反応器が好ましい。固定床断熱型反応器の中でも固定触媒床が一段だけの固定床一段断熱型反応器がより好ましい。反応に伴い触媒上には炭素質(コーク)が蓄積するため、反応を継続しながらこの炭素質の燃焼除去が可能な多塔切り替え式の固定床一段断熱型反応器が好ましい。
図1は、固定床一段断熱型反応器の概略構成図である。固定床一段断熱型反応器1は、外周に断熱材121が設けられた反応筐体12と、触媒床13と、反応器入口14と、反応器出口15とを備える。反応筐体12は、外周に断熱材121が設けられることで、反応器内の熱を外部に逃がさない。本実施形態に係る製造方法においては、反応による発熱及び吸熱により反応器内の温度を制御することができる。
触媒床13には、後述する触媒が充填される。触媒床13の触媒床入口131に接する直前に第1シース熱電対161が設けられる。触媒床3の触媒床出口132を通過した直後に第2シース熱電対162が設けられる。これらの熱電対により、触媒床入口131に接触する直前の混合原料と触媒床出口132を通過した直後の反応ガスの温度を測定する。触媒床13は、多段式であってもよいが、図1に示すとおり一段型であることが好ましい。
固定床一段断熱型反応器1では、反応器入口14から混合原料を導入し、触媒床13と接触させ、反応器出口15から反応ガスを取り出す。
(反応工程の条件)
本実施形態のエタノールの変換方法では、反応温度が300℃以上であってもよい。生成するオレフィンには熱的平衡が存在しており、プロピレン収率をより向上させる観点から、反応温度は450℃以上であることが好ましい。更に、高温で促進されるコーキング劣化加速を抑制する観点から、反応温度は、600℃未満であることが好ましい。より具体的には、触媒床入口における反応ガスの温度が450℃~590℃であることが好ましく、触媒出口における反応ガスの温度が450℃~590℃であることが好ましい。
本実施形態のエタノールの変換方法では、反応温度が300℃以上であってもよい。生成するオレフィンには熱的平衡が存在しており、プロピレン収率をより向上させる観点から、反応温度は450℃以上であることが好ましい。更に、高温で促進されるコーキング劣化加速を抑制する観点から、反応温度は、600℃未満であることが好ましい。より具体的には、触媒床入口における反応ガスの温度が450℃~590℃であることが好ましく、触媒出口における反応ガスの温度が450℃~590℃であることが好ましい。
触媒床の出口温度と触媒床の入口温度との温度差が-70K~70Kであることが好ましく、-60K~60Kであることがより好ましい。
なお、触媒床の入口温度とは、断熱型反応器に充填されている触媒床に原料流体が接する直前の混合原料の温度である。触媒床の出口温度とは、反応ガスが触媒床を通過した直後の反応ガスの温度である。ここでいう混合原料及び反応ガスの温度とは、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0d~0.8dの間における温度を指す。入出平均反応温度は、図1に示すように、触媒床の入口温度と触媒床の出口温度を計測し、計算式:〔触媒床の入口温度+触媒床の出口温度〕/2により算出する値(以下、単に「反応温度」ともいう。)としている。
反応圧力は、好ましくは0.01~3.0MPaGの範囲、より好ましくは0.01~ 1.0MPaGの範囲である。
有効原料の供給速度は、触媒の質量基準の空間速度(WHSV)で、好ましくは0.1~1000hr-1であり、より好ましくは0.1~500hr-1であり、更に好ましくは0.5~100hr-1である。本実施形態のエタノールの変換方法では、下式のように、エタノールをエチレンとして換算した上でWHSVを算出している。また、有効原料供給質量流量は、プロピレン、芳香族化合物などの目的化合物の生産性に優れる観点から、好ましくは1kg/hr以上であり、より好ましくは10kg/hr以上であり、更に好ましくは1t/hr以上である。
WHSV(hr-1)=有効原料供給質量流量(kg/hr)/触媒量(kg)
有効原料供給質量流量(kg/hr)=メチレン換算メタノール流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エチレン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
メチレン換算メタノール流量(kg/hr)=メタノール流量(kg/hr)×0.438
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×0.609
有効原料供給質量流量(kg/hr)=メチレン換算メタノール流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エチレン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
メチレン換算メタノール流量(kg/hr)=メタノール流量(kg/hr)×0.438
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×0.609
(触媒)
本実施形態のエタノールの変換方法における触媒は、オレフィン、メタノール及びエタノールをプロピレン、及び芳香族化合物等の目的化合物へと変換する触媒能を示す固体触媒である。このような触媒としては、触媒の熱耐久性及びプロピレン選択性に優れる観点から、ゼオライト含有触媒が好ましい。従来のゼオライトによるオレフィン製造に共通する課題として、炭化水素との反応によりゼオライト細孔内部に重質な炭素質(コーク)が蓄積し失活するコーキング劣化が挙げられる。触媒性能を再生するためには酸素分子を含有する雰囲気下でコークを燃焼除去する必要があるが、このコーク燃焼に伴いゼオライトの構造崩壊が進行し、再生不可能な触媒の永久劣化が誘引される。本実施形態のエタノールの変換方法によればコークの生成を抑制することができるため、ゼオライト含有触媒を用いても活性を維持しやすくなる。
本実施形態のエタノールの変換方法における触媒は、オレフィン、メタノール及びエタノールをプロピレン、及び芳香族化合物等の目的化合物へと変換する触媒能を示す固体触媒である。このような触媒としては、触媒の熱耐久性及びプロピレン選択性に優れる観点から、ゼオライト含有触媒が好ましい。従来のゼオライトによるオレフィン製造に共通する課題として、炭化水素との反応によりゼオライト細孔内部に重質な炭素質(コーク)が蓄積し失活するコーキング劣化が挙げられる。触媒性能を再生するためには酸素分子を含有する雰囲気下でコークを燃焼除去する必要があるが、このコーク燃焼に伴いゼオライトの構造崩壊が進行し、再生不可能な触媒の永久劣化が誘引される。本実施形態のエタノールの変換方法によればコークの生成を抑制することができるため、ゼオライト含有触媒を用いても活性を維持しやすくなる。
≪ゼオライト含有触媒≫
ゼオライト含有触媒とは、ゼオライトを活性種として含有する触媒粉又は成形体のことである。本実施形態のエタノールの変換方法においては、上記のゼオライト含有触媒中のゼオライトとして、5~6Åの細孔径を有する、所謂、中間細孔径ゼオライトを用いることが好ましい。中間細孔径ゼオライトは、「細孔径の範囲が、A型ゼオライトに代表される小細孔径ゼオライトの細孔径と、モルデナイトやX型やY型ゼオライトに代表される大細孔径ゼオライトの細孔径の中間にあるゼオライト」を意味する。「中間細孔径ゼオライト」は、その結晶構造中にいわゆる酸素10員環を有する。
ゼオライト含有触媒とは、ゼオライトを活性種として含有する触媒粉又は成形体のことである。本実施形態のエタノールの変換方法においては、上記のゼオライト含有触媒中のゼオライトとして、5~6Åの細孔径を有する、所謂、中間細孔径ゼオライトを用いることが好ましい。中間細孔径ゼオライトは、「細孔径の範囲が、A型ゼオライトに代表される小細孔径ゼオライトの細孔径と、モルデナイトやX型やY型ゼオライトに代表される大細孔径ゼオライトの細孔径の中間にあるゼオライト」を意味する。「中間細孔径ゼオライト」は、その結晶構造中にいわゆる酸素10員環を有する。
中間細孔径ゼオライトの例としては、ZSM-5、ZSM-8、ZSM-11、ZSM-12、ZSM-21、ZSM-23、ZSM-35、ZSM-38等が挙げられるが、中でもZSM-5、ZSM-11、ZSM-8などのZSM-5型ゼオライトやZSM-38が好ましい。また、Stud.Surf.Sci.Catal.1987,33,167-215に記載のZSM-5、ZSM-11に類似のゼオライトを用いることができ、この中でも、触媒性能(触媒活性とコーキングに対する耐久性)に優れる観点から、MFI型ゼオライトが好ましく、ZSM-5がより好ましい。
本実施形態のゼオライト含有触媒に含まれるゼオライトのシリカ/アルミナ(SiO2/Al2O3)モル比は適宜選択することができるが、触媒活性と選択性に優れる観点から、好ましくは20~2000であり、触媒の耐久性を高める観点から、より好ましくは100~1500であり、更に好ましくは300~1200であり、より更に好ましくは800~1200である。ゼオライト含有触媒に含まれるゼオライトのシリカ/アルミナ(SiO2/Al2O3)モル比は、20~400であってもよく、100~300であってもよい。ゼオライトのシリ力/アルミナモル比は公知の方法により測定することができ、例えば、ゼオライトをアルカリ水溶液に完全に溶解し、得られる溶液をプラズマ発光分光分析法等により分析し、求めることができる。
本実施形態のゼオライトの合成方法に特に制限はないが、従来知られているMFI型ゼオライトの水熱合成方法の各種条件を最適化することにより製造できる。一般に、水熱合成法で効率よくMFI型ゼオライトを得る手段としては、適切な有機構造規定剤(SDA)を用いて水熱合成する方法、水熱合成されたMFIゼオライトを種結晶として添加して水熱合成する方法、又は、結晶段階にある種スラリーとして添加して水熱合成する方法がある。
なお、ここで用いられる有機構造規定剤(SDA)としては、例えば、アンモニウム塩、尿素化合物、アミン、アルコール等が挙げられる。また、有機のSDAだけでなく、無機の陽イオンや陰イオンも構造に関わることが知られており、ゼオライト合成は各成分の複合的な働きに依存する。以上に述べたようなMFI型ゼオライトの水熱合成方法において、原材料や添加物(SDA)の種類、添加物量、pH、シリ力/アルミナモル比、媒体、陽イオン、陰イオンの存在比などの原料仕込み組成、合成温度、合成時間等の合成条件を適宜、最適化することで好適な触媒を得ることができる。
なお、ここで用いられる有機構造規定剤(SDA)としては、例えば、アンモニウム塩、尿素化合物、アミン、アルコール等が挙げられる。また、有機のSDAだけでなく、無機の陽イオンや陰イオンも構造に関わることが知られており、ゼオライト合成は各成分の複合的な働きに依存する。以上に述べたようなMFI型ゼオライトの水熱合成方法において、原材料や添加物(SDA)の種類、添加物量、pH、シリ力/アルミナモル比、媒体、陽イオン、陰イオンの存在比などの原料仕込み組成、合成温度、合成時間等の合成条件を適宜、最適化することで好適な触媒を得ることができる。
具体的には、例えば、特許5426983号公報に記載されている種スラリーを用いて合成する方法や、The Hydrothermal Synthesis of Zeolites(Chemcal Reviews,2003,103,663-702)に例示された方法が挙げられる。
また、上述した特定の物性及び組成を有するMFIゼオライトであれば、市販されているゼオライトを用いることもできる。
本実施形態におけるゼオライト含有触媒はリン元素又は銀元素を含むことが好ましい。
リン元素の形態としては、リンの重合物(例えば、ポリリン酸)、リンの酸化物(例えば、P2O5)、リンがゼオライトのアルミニウムに付加した態様の化合物等が挙げられる。また、それらの複数が含まれていてもよい。リン元素は、ゼオライトがアルミニウムを含有する場合、ゼオライトの脱アルミニウムを抑制する効果や、場合によっては、プロピレン製造反応のプロピレン収率を向上させる効果がある。本実施形態に係る方法ではアルコールの脱水反応により反応器内で水が生成し、原料であるメタノール及びエタノールが水を含有することもあるため、反応器内が高温水蒸気雰囲気となりやすく、脱アルミニウムによりゼオライト含有触媒の特性が変化し易いが、ゼオライト含有触媒がリン元素を含有することで、ゼオライトの脱アルミニウムを抑制する効果がより向上する。
ゼオライト含有触媒に含まれるリン元素の含有量は、触媒全体の質量に対して、好ましくは0.01~2.0質量%であり、脱アルミニウム抑制の効果に優れる観点から、より好ましくは0.05~2.0質量%である。
本実施形態において、触媒中のリン元素の含有量は、蛍光X線分析装置を用いて測定した値を示す。リン元素の含有量の測定は、市販の蛍光X線分析装置を使用し、取扱説明書に沿って通常の条件で測定すればよく、例えば、Rigaku製、商品名「RIX3000」を使用する場合、測定条件は、P-Kα線を用い、管球電圧50kV、管球電流50mAとすることができる。
本実施形態においては、ゼオライト含有触媒に含まれるリン元素の原料としてリン酸及び/又はリン酸塩(以下、「リン原料」ともいう。)を用いる。リン原料としてはリン酸塩がより好ましく、リン酸塩の中でも、25℃において100gの水に対して1g以上の溶解度を示す化合物がより好ましい。
リン酸としては、例えば、リン酸、ピロリン酸が挙げられ、リン酸塩としては、例えば、リン酸アンモニウム、リン酸水素二アンモニウム、リン酸二水素アンモニウム、リン酸水素アンモニウムナトリウム等のリン酸アンモニウム塩、リン酸水素カリウム、リン酸水素アルミニウム、リン酸ナトリウム、リン酸カリウム等が挙げられる。中でも、水に対する溶解度が比較的高いリン酸アンモニウム塩が好ましく、より好ましくは、リン酸アンモニウム、リン酸水素二アンモニウム、リン酸二水素アンモニウムからなる群より選択される少なくとも1種である。これらは単独で用いても2種以上を組み合わせて用いてもよい。
銀元素の形態としては、例えば銀イオンである。銀元素は、ゼオライトの酸点を制御することでゼオライトの耐水熱性を向上させる効果がある。
ゼオライト含有触媒に含まれる銀元素の含有量は、触媒全体の質量に対して、好ましくは0.01~2.0質量%であり、含有量あたりの耐水熱性の向上の効果に優れる観点から、より好ましくは0.05~2.0質量%である。
本実施形態において、触媒中の銀元素の含有量は、蛍光X線分析装置を用いて測定した値を示す。銀元素の含有量の測定は、市販の蛍光X線分析装置を使用し、取扱説明書に沿って通常の条件で測定すればよく、例えば、Rigaku製、商品名「RIX3000」を使用する場合、測定条件は、P-Kα線を用い、管球電圧50kV、管球電流50mAとすることができる。
本実施形態においては、ゼオライト含有触媒に含まれる銀元素の原料として硝酸銀が挙げられる。ナトリウムをカウンターカチオンとして含有するゼオライト含有触媒を用いて、硝酸銀とイオン交換し、焼結することで銀元素を含有するゼオライト含有触媒が得られる。ゼオライト中のカウンターカチオンであるナトリウムと硝酸銀とのイオン交換は、硝酸銀の水溶液にゼオライト、又は、ゼオライト含有触媒を浸漬した後、水洗することで実施できる。この時、浸漬と水洗とを複数回実施することでイオンの交換率を向上させることができる。
本実施形態のゼオライト含有触媒は、上述した特定の物性及び組成を有するゼオライトを用いて、例えば、以下の通りに成形して製造することができる。その成形方法は、特に限定されず、一般的な方法を用いることができる。具体的には、触媒成分を圧縮成形する方法や押出成形する方法、流動床反応方式に最適なスプレードライ成形法が挙げられる。
また、成形にはバインダーを用いることができる。バインダーとしては、特に制限されず、例えば、シリカ、アルミナ、カオリンを単独、又は、混合して使用することができる。これらのバインダーは、市販のものを使用することができる。ゼオライト/バインダーの質量比率は、好ましくは10/90~90/10の範囲であり、より好ましくは20/80~80/20の範囲である。コーキングを抑制できる観点から、シリカバインダーであることが好ましい。
本実施形態のエタノールの変換方法では、ゼオライト含有触媒を原料と接触させるのに先立って、ゼオライト含有触媒に前処理工程を実施してもよい。好ましい前処理工程としては、水蒸気の存在下、300℃以上の温度で加熱処理する工程が挙げられる。前処理を行うと、触媒の劣化抑制や選択性改善の効果がより顕著となる傾向にある。上記の方法の場合、300℃以上900℃以下の温度で、雰囲気は特に限定されないが、空気あるいは窒素等の不活性ガスとスチーム(水蒸気)との混合ガスを流通させ、水蒸気分圧0.01気圧以上の条件下で処理することが好ましい。加熱処理温度としては、400℃以上700℃以下の温度がより好ましい。また、本前処理工程はアルコールを変換する反応器を使って行うことができる。
(生成物:炭素数3以上のオレフィンを含む反応ガス)
本実施形態のエタノールの変換方法では、混合原料を触媒と接触させることで炭素数3以上のオレフィンを含む反応ガスを得る。「反応ガス」とは、混合原料を触媒と接触による反応後のガス組成物を意味する。反応ガスは、エチレンを含んでいてもよい。反応ガスは、水素、炭素数1~3の脂肪族炭化水素、炭素数4~8の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を含んでいてもよい。
本明細書では、炭素数1~3の脂肪族炭化水素、炭素数4~8の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を目的化合物と称する。
本実施形態のエタノールの変換方法では、混合原料を触媒と接触させることで炭素数3以上のオレフィンを含む反応ガスを得る。「反応ガス」とは、混合原料を触媒と接触による反応後のガス組成物を意味する。反応ガスは、エチレンを含んでいてもよい。反応ガスは、水素、炭素数1~3の脂肪族炭化水素、炭素数4~8の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を含んでいてもよい。
本明細書では、炭素数1~3の脂肪族炭化水素、炭素数4~8の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を目的化合物と称する。
〔再生工程〕
触媒を、長期間反応に用いるとコークが触媒上に付着しコーキング劣化を起こす場合がある。触媒がコーキング劣化を起こした場合には、例えば、酸素含有ガスと接触させ400~700℃の温度で触媒上のコークを燃焼除去することにより、コーキング劣化を起こした触媒を再生させる(以下、「再生工程」ともいう。)ことができる。酸素含有ガスとしては、例えば、空気、及び、空気又は酸素と不活性ガスとの混合ガスが挙げられる。酸素含有ガスの酸素濃度は、好ましくは0.1~2.0体積%である。触媒は、反応器より抜き出し反応器外で再生処理を行う反応器外再生と反応器より抜き出さずに反応器内で再生処理を行う反応器内再生のどちらの再生方法を採用してもよい。また、切り替え式反応器を採用することで反応-再生切り替え運転を行うこともできる。
触媒を、長期間反応に用いるとコークが触媒上に付着しコーキング劣化を起こす場合がある。触媒がコーキング劣化を起こした場合には、例えば、酸素含有ガスと接触させ400~700℃の温度で触媒上のコークを燃焼除去することにより、コーキング劣化を起こした触媒を再生させる(以下、「再生工程」ともいう。)ことができる。酸素含有ガスとしては、例えば、空気、及び、空気又は酸素と不活性ガスとの混合ガスが挙げられる。酸素含有ガスの酸素濃度は、好ましくは0.1~2.0体積%である。触媒は、反応器より抜き出し反応器外で再生処理を行う反応器外再生と反応器より抜き出さずに反応器内で再生処理を行う反応器内再生のどちらの再生方法を採用してもよい。また、切り替え式反応器を採用することで反応-再生切り替え運転を行うこともできる。
(反応-再生切り替え運転)
反応-再生切り替え運転とは、2塔又は多塔切り替え式の断熱型反応器を用い、反応工程と再生工程とを同時に行う運転操作である。例えば、3塔切り替え式の場合は、2塔が反応工程に使用され、同時に、残りの1塔が触媒再生に使用される。その後、反応工程に使用されていたうちの1塔の反応工程を停止して触媒再生を行い、触媒再生に使用していた1塔で反応工程を行うことで、2塔分の生産能力を維持しつつ、触媒再生を行うことができる。このような反応形式をメリーゴーランド方式とも呼び、触媒再生のために製造工程を止める必要がないため、生産効率に優れる観点から好ましい。
反応-再生切り替え運転とは、2塔又は多塔切り替え式の断熱型反応器を用い、反応工程と再生工程とを同時に行う運転操作である。例えば、3塔切り替え式の場合は、2塔が反応工程に使用され、同時に、残りの1塔が触媒再生に使用される。その後、反応工程に使用されていたうちの1塔の反応工程を停止して触媒再生を行い、触媒再生に使用していた1塔で反応工程を行うことで、2塔分の生産能力を維持しつつ、触媒再生を行うことができる。このような反応形式をメリーゴーランド方式とも呼び、触媒再生のために製造工程を止める必要がないため、生産効率に優れる観点から好ましい。
〔分離工程〕
分離工程では、反応ガスから目的化合物を分離する。分離工程により当該反応ガスからエチレン、プロピレン及び芳香族化合物を分離することができる。
分離工程では、反応ガスから目的化合物を分離する。分離工程により当該反応ガスからエチレン、プロピレン及び芳香族化合物を分離することができる。
本実施形態に係る方法は、図2に示すように、反応器1と、蒸留塔2と、蒸留塔3と、蒸留塔4とを有する装置により実施することができる。反応器1で行われる反応工程により得られた反応ガスを蒸留塔2で、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~8の炭化水素を主に含む留分Bとに分離することができる。なお、蒸留塔2で蒸留する前に反応ガスから凝縮させた水を除去してもよい(図示せず)。蒸留塔3及び蒸留塔4で、留分Aからエチレン及びプロピレンを分離することによって、該反応ガスからのエチレン及びプロピレンの分離を効率的に行う。また、反応ガス及び/又は留分Aの少なくとも一部をエチレンプラントの精製系に導入し、該精製系で反応ガスからエチレン及びプロピレンを分離してもよい。分離工程により得られた留分Bをはじめとする各種留分は反応器に原料としてリサイクルすることができる。
本実施形態に係る方法は、図3に示すように、反応器1と、蒸留塔2と、スチームクラッキング装置5とを有する装置により実施することができる。分離工程により得られた留分Bをスチームクラッキングに付すことにより、エチレン及びプロピレンを含有するスチームクラッキング生成物を得、スチームクラッキング生成物からエチレン及びプロピレンを分離することでプロセス全体の効率性を高めることができる。スチームクラッキングとは、加熱蒸気と共に留分中の化合物を熱分解することを意味する。
本実施形態に係る方法は、図4に示すように、反応器1と、冷却装置6と、蒸留塔2と、油水分離機7とを有する装置により実施することができる。反応器1で行われる反応工程により得られた反応ガスを冷却装置6により冷却する、冷却工程を設けることができる。冷却工程により、反応ガスを炭素数2~6の脂肪族炭化水素を主に含む留分Cと、水、炭素数7以上の脂肪族炭化水素及び芳香族化合物を主に含む留分Dとに分離することができる。留分Cは冷却工程においてガス成分として得られ、留分Dは液成分として回収される。該留分Dから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。
冷却工程で回収された留分Dは、油水分離機7において炭化水素を主に含む留分Eと水を主に含む留分Fとに分離される。該留分Eから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。芳香族化合物の分離は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。
本実施形態に係る方法は、図5に示すように、反応器1と、蒸留塔2と、蒸留塔8とを有する装置により実施することができる。蒸留塔8により、留分Bをリサイクルする際、炭素数4~6の脂肪族炭化水素、好ましくはオレフィン、を主に含む留分B1と、芳香族化合物を主に含む留分B2とに分離し、留分B1の少なくとも一部を反応器にリサイクルすることが好ましい。留分B2を除去した留分B1をリサイクル原料とすることで、プロピレンに変換されない芳香族化合物を原料から除去することができ、効率よくプロピレンを製造することができる。このように、留分Bより留分B2として芳香族化合物を分離することもできる。芳香族化合物の更なる分離精製は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。
また、本実施形態に係る方法は、図6に示すように、反応器1と、蒸留塔2と、蒸留塔9とを有する装置により実施することができる。蒸留塔9により、留分Bを炭素数4~8の炭化水素を主に含む留分B3と炭素数9以上の炭化水素を主に含む留分B4とに分離し、留分B3の少なくとも一部を反応器にリサイクルすることが好ましい。留分B4を除去した留分B3をリサイクル原料とすることで、コーキング劣化を促進させ得る重質分を原料から除去することができ、触媒のコーキング劣化を抑制することができる。
各種留分において「主に含む」とは、「主に含む」と記載された成分の合計質量が、各留分に対して、50質量%超であることを意味する。
なお、これらの分離工程は、蒸留、抽出など種々の公知の方法を組み合わせることで実施することができる。
[第3実施形態]
続いて、第3実施形態に係るエタノールの変換方法について説明する。
続いて、第3実施形態に係るエタノールの変換方法について説明する。
<エタノールの変換方法-第3実施形態->
本実施形態に係るエタノールの変換方法は、
エタノール及びエチレンを含有する混合原料を、断熱型反応器内で変換触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ること(以下、「反応工程」ともいう)と、
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離すること(以下、「第1分離工程」ともいう)と、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすること(以下、「リサイクル工程」ともいう)と、
を含む。
以上の本実施形態によれば、断熱型反応器を使用し、反応器内の温度を制御して、エタノールを収率良く目的化合物へと変換する方法を提供することができる。
なお、上述の反応ガスから分離することでプロピレンを得ることができ、上述の反応ガスから分離することで芳香族化合物を得ることもできる。
本実施形態に係るエタノールの変換方法は、
エタノール及びエチレンを含有する混合原料を、断熱型反応器内で変換触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ること(以下、「反応工程」ともいう)と、
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離すること(以下、「第1分離工程」ともいう)と、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすること(以下、「リサイクル工程」ともいう)と、
を含む。
以上の本実施形態によれば、断熱型反応器を使用し、反応器内の温度を制御して、エタノールを収率良く目的化合物へと変換する方法を提供することができる。
なお、上述の反応ガスから分離することでプロピレンを得ることができ、上述の反応ガスから分離することで芳香族化合物を得ることもできる。
従来、化成品製造プロセスを実用化する際、反応器及びその反応様式の選定は操作の容易性や環境負荷の大小に大きな影響を与える要素となる。断熱型反応器は反応器の加熱又は冷却が必要なく簡便な構成であり、設計、建築及び操業の負荷が小さい理想的な反応器であり、例えば、固定床一段断熱型反応器を採用できればその利点は更に大きくなる。しかしながら、断熱型反応器は、吸熱又は発熱の大きい反応系では温度制御に課題を有する。
特許文献1~3では、それぞれ各原料をゼオライト触媒で目的のオレフィンへと変換する技術が公開されているが、これらの技術は吸熱及び発熱が大きくなるため断熱型反応器へと応用することはできない。
特許文献4及び5では、アルコールなどの含酸素化合物を原料とする発熱反応を利用した熱中和技術が開示されているが、吸熱反応を誘引するエタノールを原料とする反応を断熱型反応器で実施することはできない。
特許文献4及び5では、アルコールなどの含酸素化合物を原料とする発熱反応を利用した熱中和技術が開示されているが、吸熱反応を誘引するエタノールを原料とする反応を断熱型反応器で実施することはできない。
エタノール及びエチレンを含有する混合原料を触媒により変換すると、目的とする化合物のほかにも化合物が反応ガスに含まれる。これらの化合物を原料として有効利用することで、上述の反応器の温度制御の課題を解決しながら、エタノール及びエチレンを収率よく目的化合物へと変換する方法を検討する。
そこで、本発明は、断熱型反応器を使用し、反応器内の温度を制御して、エタノール及びエチレンを収率よく目的化合物へと変換するエタノールの変換方法、プロピレンの製造方法、芳香族化合物の製造方法、並びに、エタノール及びエチレンの変換装置を提供することを目的とする。
本発明者らは、上記の課題を達成するために鋭意検討した結果、エタノール及びエチレンの変換により得られた反応ガスを分離工程に供した後、反応工程にリサイクルすることで、エタノール及びエチレンから炭素数3以上のオレフィン等への変換に対して反応熱を制御することが可能になり、断熱型反応器を使用し、当該反応器内の温度を制御しながら、エタノール及びエチレンを収率よく目的化合物へと変換できることを見出した。
本実施形態によれば、断熱型反応器を使用し、反応器内の温度を制御して、エタノールを収率良く目的化合物へと変換するエタノールの変換方法、プロピレンの製造方法、芳香族化合物の製造方法、並びに、エタノール及びエチレンの変換装置を提供することができる。
<反応工程>
本実施形態に係る反応工程では、エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得る。また、本実施形態によれば、断熱型反応器を使用することで操業の負荷が小さく、エネルギー効率が高い環境調和型の変換方法が実施できる。また、後述のリサイクル工程により、断熱型反応器を使用した場合にも、エタノール及びエチレンを収率良く目的化合物へと変換することができ、触媒のコーキング劣化を抑制できるため、エタノールの変換方法として好適である。
本実施形態に係る反応工程では、エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得る。また、本実施形態によれば、断熱型反応器を使用することで操業の負荷が小さく、エネルギー効率が高い環境調和型の変換方法が実施できる。また、後述のリサイクル工程により、断熱型反応器を使用した場合にも、エタノール及びエチレンを収率良く目的化合物へと変換することができ、触媒のコーキング劣化を抑制できるため、エタノールの変換方法として好適である。
本発明者は、エチレンやエタノールを単独で原料とし、断熱型反応器でプロピレンの製造を試みるとプロピレンの収率が低下する、又は、触媒のコーキング劣化が進行するとの知見を見出した。なお、触媒のコーキング劣化とは、触媒表面にコークが付着し触媒の活性を低下させることを意味する。
本発明者は、このような知見に基づき、エチレン及びエタノールを含有する混合原料を、断熱型反応器に充填した触媒と接触させると収率良くプロピレンを与え、コーキング劣化を抑制できることを見出した。
この要因は互いを阻害しない吸熱反応と発熱反応を組み合わせることで熱中和を達成することができたことと想定される。エチレンからプロピレンへの変換反応は発熱反応である。また、エタノールからプロピレンへの変換反応は、触媒の存在下では、エタノールからエチレンへの脱水反応とエチレンからプロピレンへの変換反応の2段反応による吸熱反応である。本実施形態では、エチレンからプロピレンへの変換反応とエタノールからプロピレンへの変換反応が互いの反応を阻害せずに進行することを見出し、これらの発熱反応と吸熱反応とを組み合わせることで断熱型反応器においても反応条件の制御を行うことができたと考えられる。ただし、要因はこれに限定されない。
(原料)
本実施形態のエタノールの変換方法では、エチレン及びエタノールを含有する混合原料を用いる。当該混合原料を用いることで、断熱型反応器を使用し、反応器内の温度を制御して収率よくプロピレン等の目的化合物を製造することができる。エタノールは、環境調和性に優れる観点から、バイオマス由来であることが好ましい。なお、バイオマスとは動植物を起源とする化石資源以外の有機性資源を指し、バイオマス由来とはバイオマスを原料として製造された化合物であることをいう。
本実施形態のエタノールの変換方法では、エチレン及びエタノールを含有する混合原料を用いる。当該混合原料を用いることで、断熱型反応器を使用し、反応器内の温度を制御して収率よくプロピレン等の目的化合物を製造することができる。エタノールは、環境調和性に優れる観点から、バイオマス由来であることが好ましい。なお、バイオマスとは動植物を起源とする化石資源以外の有機性資源を指し、バイオマス由来とはバイオマスを原料として製造された化合物であることをいう。
混合原料に含まれるエチレンは、反応ガスより分離された留分に含まれるエチレンの一部又は全量を原料としてリサイクルすることで供給される。このように、所謂、リサイクル反応システムを用いることにより、原料の有効利用を図ることができる。
混合原料は、原料比率の可変性に優れる観点から、反応ガスより分離された留分に含まれるエチレンに加えて、「追加のエチレン」を更に含むことができる。「追加のエチレン」としては種々の製法で製造されたものを使用することができる。例えば、ナフサ及び/又はエタンの熱分解、エタンの直接又は酸化的脱水素反応、又は、エタノールの脱水反応により得られるものを使用することができる。
これらのエチレン及びエタノールはその製造過程において、水が副生する、又は、反応器内に水が共存する過程を経ることが多い。したがって、本実施形態の変換方法では、原料であるエチレン及びエタノールが水を含有することがある。
混合原料中、エチレン/エタノールのモル比は、反応器内の温度を制御して、エタノールを収率良く目的化合物へと変換する観点から、0.20~2.5であることが好ましく、0.20~2.0であることがより好ましく、0.30~1.5であることが更に好ましく、0.30~1.0であることが特に好ましい。
本実施形態のエタノールの変換方法において、混合原料は、メタノールを更に含んでいてもよい。メタノールとしては種々の製法で製造されたものを使用することができる。例えば、天然ガスや石炭より得られる一酸化炭素の水素化や二酸化炭素の水素化、木酢液の蒸留により得られるものを使用することができる。
本実施形態のエタノールの変換方法において、混合原料は、炭素数4~6のオレフィンを更に含んでいてもよい。炭素数4~6のオレフィンは、エチレン及びエタノールと同様に、触媒と接触させることでプロピレンなどの目的化合物を与えることができる。炭素数4~6のオレフィンとしては、例えば、ブテン、ペンテン、ヘキセンが挙げられる。なお、明細書において「オレフィン」という用語は、直鎖状、分岐状及び環状オレフィンに加え、シクロパラフィンを含むものとする。
混合原料中、炭素数4~6のオレフィン/エチレンのモル比は、3.0以下であることが好ましく、1.0以下であることがより好ましく、0.5以下であることが更に好ましく、0.15~0.5であることがより更に好ましい。
本実施形態のエタノールの変換方法において、混合原料は、エタノール以外の炭素数1~6の含酸素化合物を更に含んでいてもよい。炭素数1~6の含酸素化合物は、エチレン及びエタノールと同様に、触媒と接触させることでプロピレン等の目的化合物を与えることができる。エタノール以外の炭素数1~6の含酸素化合物としては、例えば、メタノール、プロパノール、ジメチルエーテル、ジエチルエーテルが挙げられる。
混合原料中、エタノール以外の炭素数1~6の含酸素化合物/エタノールのモル比は、1.0以下であることが好ましく、0.5以下であることがより好ましい。
なお、エチレン、エタノール、炭素数4~6のオレフィン、エタノール以外の炭素数1~6の含酸素化合物を総称して「有効原料」ともいう。
混合原料は、上記の有効原料のほか、パラフィンなどの飽和脂肪族炭化水素、炭素数7以上のオレフィン、炭素数7以上の含酸素化合物を含んでいてもよい。飽和脂肪族炭化水素、炭素数7以上のオレフィン、及び炭素数7以上の含酸素化合物は、エチレン及びエタノールと同様に、触媒と接触させることで目的化合物へと変換され得るが、上述の有効原料よりも反応性が低い。
混合原料は、反応工程によってプロピレンへと変換され得る上述の原料の他に、窒素などの不活性ガスなどを含んでもよい。そのほか、混合原料は、希釈ガスとして、水素、メタンを含んでいてもよいが、水素希釈は行わないことが好ましい。水素は触媒のコーキング劣化を抑制するために使用されることがあるが、同時に生成プロピレン等の水素化反応が起こり、プロピレン純度(プロピレン/(プロピレン+プロパン))[mol/mol]を低下させる悪影響がある。本実施形態の方法においては、水素希釈を行わなかった場合にも触媒のコーキング劣化速度は小さく、安定な運転が可能であるため水素希釈を行わないほうが好ましい。
混合原料中のエチレンと炭素数4~6のオレフィンとエタノールとの合計割合が混合原料供給質量流量に対して、40質量%以上であることが好ましく、50質量%以上であることがより好ましい。混合原料供給質量流量とは、不活性成分を含む、反応器に供される全ての化合物の合計流量のことである。
混合原料中、エチレン及びエタノールの合計含有量は、有効原料供給質量に対して、好ましくは30~100質量%であり、より好ましくは40~100質量%であり、更に好ましくは50~100質量%である。ただし、エタノールは、エチレンとして換算した質量をエタノール質量及び有効原料供給質量の計算に使用する。
混合原料中、炭素数4~6のオレフィンの合計含有量は、有効原料供給質量に対して、好ましくは65質量%以下であり、より好ましくは10~55質量%である。
本実施形態のエタノールの変換方法では、混合原料は水を含んでもよい。混合原料に含まれるエチレン及びエタノールは種々の製造方法によって製造されることから、「製造工程で発生した水」を含有していてもよい。ここでの「製造工程で発生した水」とは、エチレン及び/又はエタノールの製造過程において発生し、除去されていない水分のことをいう。
本実施形態のエタノールの変換方法では、混合原料は、「製造過程で発生した水」に追加して混合原料に水蒸気を含んでもよい。水蒸気はオレフィン分圧を低下させることでコーキング劣化を抑制し、低級オレフィンの収率を向上させる効果がある。一方で、水蒸気はゼオライトの脱アルミニウムを促進する可能性があるため、好ましくは「製造過程で発生した水」に追加して混合原料に水蒸気を含まない。
(断熱型反応器)
本実施形態のエタノールの変換方法では断熱型反応器を使用することを特徴とする。断熱型反応器に関しては、Adiabatic Fixed-Bed Reactors(Elsevier,2014,Ch.1,P.4,L.5~24 ISBN:978-0-12-801306-9)の記載を参照することができる。断熱型反応器の例としては、固定床断熱型反応器、移動床断熱型反応器、流動床断熱型反応器が挙げられるが、本実施形態の方法には固定床断熱型反応器が好ましい。固定床断熱型反応器の中でも固定触媒床が一段だけの固定床一段断熱型反応器がより好ましい。反応に伴い触媒上には炭素質(コーク)が蓄積するため、反応を継続しながらこの炭素質の燃焼除去が可能な多塔切り替え式の固定床一段断熱型反応器が好ましい。
本実施形態のエタノールの変換方法では断熱型反応器を使用することを特徴とする。断熱型反応器に関しては、Adiabatic Fixed-Bed Reactors(Elsevier,2014,Ch.1,P.4,L.5~24 ISBN:978-0-12-801306-9)の記載を参照することができる。断熱型反応器の例としては、固定床断熱型反応器、移動床断熱型反応器、流動床断熱型反応器が挙げられるが、本実施形態の方法には固定床断熱型反応器が好ましい。固定床断熱型反応器の中でも固定触媒床が一段だけの固定床一段断熱型反応器がより好ましい。反応に伴い触媒上には炭素質(コーク)が蓄積するため、反応を継続しながらこの炭素質の燃焼除去が可能な多塔切り替え式の固定床一段断熱型反応器が好ましい。
図1は、固定床一段断熱型反応器の概略構成図である。固定床一段断熱型反応器1は、外周に断熱材121が設けられた反応筐体12と、触媒床13と、反応器入口14と、反応器出口15とを備える。反応筐体12は、外周に断熱材121が設けられることで、反応器内の熱を外部に逃がさない。本実施形態に係る製造方法においては、反応による発熱及び吸熱により反応器内の温度を制御することができる。
触媒床13には、後述する触媒が充填される。触媒床13の触媒床入口131に接する直前に第1シース熱電対161が設けられる。触媒床3の触媒床出口132を通過した直後に第2シース熱電対162が設けられる。これらの熱電対により、触媒床入口131に接触する直前の混合原料と触媒床出口132を通過した直後の反応ガスの温度を測定する。触媒床13は、多段式であってもよいが、図1に示すとおり一段型であることが好ましい。
固定床一段断熱型反応器1では、反応器入口14から混合原料を導入し、触媒床13と接触させ、反応器出口15から反応ガスを取り出す。
(反応工程の条件)
本実施形態のエタノールの変換方法では、反応温度が300℃以上であってもよい。生成するオレフィンには熱的平衡が存在しており、プロピレン収率をより向上させる観点から、反応温度は450℃以上であることが好ましい。更に、高温で促進されるコーキング劣化加速を抑制する観点から、反応温度は、600℃未満であることが好ましい。より具体的には、触媒床入口における反応ガスの温度が450℃~590℃であることが好ましく、触媒出口における反応ガスの温度が450℃~590℃であることが好ましい。
本実施形態のエタノールの変換方法では、反応温度が300℃以上であってもよい。生成するオレフィンには熱的平衡が存在しており、プロピレン収率をより向上させる観点から、反応温度は450℃以上であることが好ましい。更に、高温で促進されるコーキング劣化加速を抑制する観点から、反応温度は、600℃未満であることが好ましい。より具体的には、触媒床入口における反応ガスの温度が450℃~590℃であることが好ましく、触媒出口における反応ガスの温度が450℃~590℃であることが好ましい。
触媒床の出口温度と触媒床の入口温度との温度差が-80K~80Kであることが好ましく、-60K~60Kであることがより好ましい。
なお、触媒床の入口温度とは、断熱型反応器に充填されている触媒床に原料流体が接する直前の混合原料の温度である。触媒床の出口温度とは、反応ガスが触媒床を通過した直後の反応ガスの温度である。ここでいう混合原料及び反応ガスの温度とは、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0d~0.8dの間における温度を指す。入出平均反応温度は、図1に示すように、触媒床の入口温度と触媒床の出口温度を計測し、計算式:〔触媒床の入口温度+触媒床の出口温度〕/2により算出する値(以下、単に「反応温度」ともいう。)としている。
反応圧力は、好ましくは0.01~3.0MPaGの範囲、より好ましくは0.01~1.0MPaGの範囲である。
有効原料の供給速度は、触媒の質量基準の空間速度(WHSV)で、好ましくは0.1~1000hr-1であり、より好ましくは0.1~500hr-1であり、更に好ましくは0.5~100hr-1である。本実施形態のエタノールの変換方法では、下式のように、エタノールをエチレンとして換算した上でWHSVを算出する。また、有効原料供給質量流量は、目的化合物の生産性に優れる観点から、好ましくは1kg/hr以上であり、より好ましくは10kg/hr以上であり、更に好ましくは1t/hr以上である。
WHSV(hr-1)=有効原料供給質量流量(kg/hr)/触媒量(kg)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール質量流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール質量流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
(触媒)
本実施形態のエタノールの変換方法における触媒は、オレフィン及びエタノールをプロピレン等の目的化合物へと変換する触媒能を示す固体触媒である。このような触媒としては、触媒の熱耐久性に優れる観点から、ゼオライト含有触媒が好ましい。従来のゼオライトによるオレフィン製造に共通する課題として、炭化水素との反応によりゼオライト細孔内部に重質な炭素質(コーク)が蓄積し失活するコーキング劣化が挙げられる。触媒性能を再生するためには酸素分子を含有する雰囲気下でコークを燃焼除去する必要があるが、このコーク燃焼に伴いゼオライトの構造崩壊が進行し、再生不可能な触媒の永久劣化が誘引される。本実施形態のエタノールの変換方法によればコークの生成を抑制することができるため、ゼオライト含有触媒を用いても活性を維持しやすくなる。
本実施形態のエタノールの変換方法における触媒は、オレフィン及びエタノールをプロピレン等の目的化合物へと変換する触媒能を示す固体触媒である。このような触媒としては、触媒の熱耐久性に優れる観点から、ゼオライト含有触媒が好ましい。従来のゼオライトによるオレフィン製造に共通する課題として、炭化水素との反応によりゼオライト細孔内部に重質な炭素質(コーク)が蓄積し失活するコーキング劣化が挙げられる。触媒性能を再生するためには酸素分子を含有する雰囲気下でコークを燃焼除去する必要があるが、このコーク燃焼に伴いゼオライトの構造崩壊が進行し、再生不可能な触媒の永久劣化が誘引される。本実施形態のエタノールの変換方法によればコークの生成を抑制することができるため、ゼオライト含有触媒を用いても活性を維持しやすくなる。
≪ゼオライト含有触媒≫
ゼオライト含有触媒とは、ゼオライトを活性種として含有する触媒粉又は成形体のことである。本実施形態のエタノールの変換方法においては、上記のゼオライト含有触媒中のゼオライトとして、5~6Åの細孔径を有する、所謂、中間細孔径ゼオライトを用いることが好ましい。中間細孔径ゼオライトは、「細孔径の範囲が、A型ゼオライトに代表される小細孔径ゼオライトの細孔径と、モルデナイトやX型やY型ゼオライトに代表される大細孔径ゼオライトの細孔径の中間にあるゼオライト」を意味する。「中間細孔径ゼオライト」は、その結晶構造中にいわゆる酸素10員環を有する。
ゼオライト含有触媒とは、ゼオライトを活性種として含有する触媒粉又は成形体のことである。本実施形態のエタノールの変換方法においては、上記のゼオライト含有触媒中のゼオライトとして、5~6Åの細孔径を有する、所謂、中間細孔径ゼオライトを用いることが好ましい。中間細孔径ゼオライトは、「細孔径の範囲が、A型ゼオライトに代表される小細孔径ゼオライトの細孔径と、モルデナイトやX型やY型ゼオライトに代表される大細孔径ゼオライトの細孔径の中間にあるゼオライト」を意味する。「中間細孔径ゼオライト」は、その結晶構造中にいわゆる酸素10員環を有する。
中間細孔径ゼオライトの例としては、ZSM-5、ZSM-8、ZSM-11、ZSM-12、ZSM-21、ZSM-23、ZSM-35、ZSM-38等が挙げられるが、中でもZSM-5、ZSM-11、ZSM-8などのZSM-5型ゼオライトやZSM-38が好ましい。また、Stud.Surf.Sci.Catal.1987,33,167-215に記載のZSM-5、ZSM-11に類似のゼオライトを用いることができ、この中でも、触媒性能(触媒活性とコーキングに対する耐久性)に優れる観点から、MFI型ゼオライトが好ましく、ZSM-5がより好ましい。
本実施形態のゼオライト含有触媒に含まれるゼオライトのシリカ/アルミナ(SiO2/Al2O3)モル比は適宜選択することができるが、触媒活性と選択性に優れる観点から、好ましくは20~2000であり、触媒の耐久性を高める観点から、より好ましくは200~1500であり、更に好ましくは300~1200であり、より更に好ましくは800~1200である。ゼオライト含有触媒に含まれるゼオライトのシリカ/アルミナ(SiO2/Al2O3)モル比は、20~1200であってもよく、150~1000であってもよい。ゼオライトのシリカ/アルミナモル比は公知の方法により測定することができ、例えば、ゼオライトをアルカリ水溶液に完全に溶解し、得られる溶液をプラズマ発光分光分析法等により分析し、求めることができる。
本実施形態のゼオライトの合成方法に特に制限はないが、従来知られているMFI型ゼオライトの水熱合成方法の各種条件を最適化することにより製造できる。一般に、水熱合成法で効率よくMFI型ゼオライトを得る手段としては、適切な有機構造規定剤(SDA)を用いて水熱合成する方法、水熱合成されたMFIゼオライトを種結晶として添加して水熱合成する方法、又は、結晶段階にある種スラリーとして添加して水熱合成する方法がある。なお、ここで用いられる有機構造規定剤(SDA)としては、例えば、アンモニウム塩、尿素化合物、アミン、及びアルコール等が挙げられる。また、有機のSDAだけでなく、無機の陽イオンや陰イオンも構造に関わることが知られており、ゼオライト合成は各成分の複合的な働きに依存する。以上に述べたようなMFI型ゼオライトの水熱合成方法において、原材料や添加物(SDA)の種類、添加物量、pH、シリカ/アルミナモル比、媒体、陽イオン、陰イオンの存在比などの原料仕込み組成、合成温度、合成時間等の合成条件を適宜、最適化することで好適な触媒を得ることができる。
具体的には、例えば、特許5426983号公報に記載されている種スラリーを用いて合成する方法や、The Hydrothermal Synthesis of Zeolites(Chemcal Reviews,2003,103,663-702)に例示された方法が挙げられる。
また、上述した特定の物性及び組成を有するMFIゼオライトであれば、市販されているゼオライトを用いることもできる。
本実施形態におけるゼオライト含有触媒はリン元素又は銀元素を含むことが好ましい。
リン元素の形態としては、リンの重合物(例えば、ポリリン酸)、リンの酸化物(例えば、P2O5)、リンがゼオライトのアルミニウムに付加した態様の化合物等が挙げられる。また、それらの複数が含まれていてもよい。リン元素は、ゼオライトがアルミニウムを含有する場合、ゼオライトの脱アルミニウムを抑制する効果や、場合によって、プロピレン製造反応のプロピレン収率を向上させる効果がある。本実施形態に係る方法では反応器内で水が生成し、かつ、原料であるエタノールが水を含有することもあるため、反応器内が脱アルミニウムを引き起こす高温水蒸気雰囲気となりやすい。脱アルミニウムすることでゼオライト含有触媒の構造崩壊による活性劣化が引き起こされるが、ゼオライト含有触媒がリン元素を含有することで、ゼオライトの脱アルミニウムが抑制する効果がより向上する。
ゼオライト含有触媒に含まれるリン元素の含有量は、触媒全体の質量に対して、好ましくは0.01~2.0質量%であり、脱アルミニウム抑制の効果に優れる観点から、より好ましくは0.05~2.0質量%である。
本実施形態において、触媒中のリン元素の含有量は、蛍光X線分析装置を用いて測定した値を示す。リン元素の含有量の測定は、市販の蛍光X線分析装置を使用し、取扱説明書に沿って通常の条件で測定すればよく、例えば、Rigaku製、商品名「RIX3000」を使用する場合、測定条件は、P-Kα線を用い、管球電圧50kV、管球電流50mAとすることができる。
本実施形態においては、ゼオライト含有触媒に含まれるリン元素の原料としてリン酸及び/又はリン酸塩(以下、「リン原料」ともいう。)を用いる。リン原料としてはリン酸塩がより好ましく、リン酸塩の中でも、25℃において100gの水に対して1g以上の溶解度を示す化合物がより好ましい。
リン酸としては、例えば、リン酸、ピロリン酸が挙げられ、リン酸塩としては、例えば、リン酸アンモニウム、リン酸水素二アンモニウム、リン酸二水素アンモニウム、リン酸水素アンモニウムナトリウム等のリン酸アンモニウム塩、リン酸水素カリウム、リン酸水素アルミニウム、リン酸ナトリウム、リン酸カリウム等が挙げられる。中でも、水に対する溶解度が比較的高いリン酸アンモニウム塩が好ましく、より好ましくは、リン酸アンモニウム、リン酸水素二アンモニウム、リン酸二水素アンモニウムからなる群より選択される少なくとも1種である。これらは単独で用いても2種以上を組み合わせて用いてもよい。
銀元素の形態としては、例えば銀イオンである。銀元素は、ゼオライトの酸点を制御することでゼオライトの耐水熱性を向上させる効果がある。
ゼオライト含有触媒に含まれる銀元素の含有量は、触媒全体の質量に対して、好ましくは0.01~2.0質量%であり、含有量あたりの耐水熱性の向上の効果に優れる観点から、より好ましくは0.05~2.0質量%である。
本実施形態において、触媒中の銀元素の含有量は、蛍光X線分析装置を用いて測定した値を示す。銀元素の含有量の測定は、市販の蛍光X線分析装置を使用し、取扱説明書に沿って通常の条件で測定すればよく、例えば、Rigaku製、商品名「RIX3000」を使用する場合、測定条件は、P-Kα線を用い、管球電圧50kV、管球電流50mAとすることができる。
本実施形態においては、ゼオライト含有触媒に含まれる銀元素の原料として硝酸銀が挙げられる。ナトリウムをカウンターカチオンとして含有するゼオライト含有触媒を用いて、硝酸銀とイオン交換し、焼結することで銀元素を含有するゼオライト含有触媒が得られる。ゼオライト中のカウンターカチオンであるナトリウムと硝酸銀とのイオン交換は、硝酸銀の水溶液にゼオライト、又は、ゼオライト含有触媒を浸漬した後、水洗することで実施できる。この時、浸漬と水洗とを複数回実施することでイオンの交換率を向上させることができる。
本実施形態のゼオライト含有触媒は、上述した特定の物性及び組成を有するゼオライトを用いて、例えば、以下の通りに成形して製造することができる。その成形方法は、特に限定されず、一般的な方法を用いることができる。具体的には、触媒成分を圧縮成形する方法や押出成形する方法、流動床反応方式に最適なスプレードライ成形法が挙げられる。
また、成形にはバインダーを用いることができる。バインダーとしては、特に制限されず、例えば、シリカ、アルミナ、カオリンを単独、又は、混合して使用することができる。これらのバインダーは、市販のものを使用することができる。ゼオライト/バインダーの質量比率は、好ましくは10/90~90/10の範囲であり、より好ましくは20/80~80/20の範囲である。この中でも、コーキング耐性に優れる観点から、シリカバインダーを使用することが好ましい。
また、成形にはバインダーを用いることができる。バインダーとしては、特に制限されず、例えば、シリカ、アルミナ、カオリンを単独、又は、混合して使用することができる。これらのバインダーは、市販のものを使用することができる。ゼオライト/バインダーの質量比率は、好ましくは10/90~90/10の範囲であり、より好ましくは20/80~80/20の範囲である。この中でも、コーキング耐性に優れる観点から、シリカバインダーを使用することが好ましい。
本実施形態のエタノールの変換方法では、ゼオライト含有触媒を原料と接触させるのに先立って、ゼオライト含有触媒に前処理工程を実施してもよい。好ましい前処理工程としては、水蒸気の存在下、300℃以上の温度で加熱処理する工程が挙げられる。前処理を行うと、触媒の劣化抑制や選択性改善の効果がより顕著となる傾向にある。上記の方法の場合、300℃以上900℃以下の温度で、雰囲気は特に限定されないが、空気あるいは窒素等の不活性ガスとスチーム(水蒸気)との混合ガスを流通させ、水蒸気分圧0.01気圧以上の条件下で処理することが好ましい。加熱処理温度としては、400℃以上700℃以下の温度がより好ましい。また、本前処理工程はエタノール及びエチレンを変換する反応器を使って行うことができる。
〔再生工程〕
本実施形態のエタノールの変換方法では、触媒に付着したコークを燃焼させる再生工程(以下、「再生工程」ともいう。)を含んでいてもよい。触媒を、長期間反応に用いるとコークが触媒上に付着しコーキング劣化を起こす場合がある。触媒がコーキング劣化を起こした場合には、例えば、酸素含有ガスと接触させ400~700℃の温度で触媒上のコークを燃焼除去することにより、コーキング劣化を起こした触媒を再生させることができる。酸素含有ガスとしては、例えば、空気、及び、空気又は酸素と不活性ガスとの混合ガスが挙げられる。酸素含有ガスの酸素濃度は、好ましくは0.1~2.0体積%である。触媒は、反応器より抜き出し反応器外で再生処理を行う反応器外再生と反応器より抜き出さずに反応器内で再生処理を行う反応器内再生のどちらの再生方法を採用してもよい。また、切り替え式反応器を採用することで反応-再生切り替え運転を行うこともできる。
本実施形態のエタノールの変換方法では、触媒に付着したコークを燃焼させる再生工程(以下、「再生工程」ともいう。)を含んでいてもよい。触媒を、長期間反応に用いるとコークが触媒上に付着しコーキング劣化を起こす場合がある。触媒がコーキング劣化を起こした場合には、例えば、酸素含有ガスと接触させ400~700℃の温度で触媒上のコークを燃焼除去することにより、コーキング劣化を起こした触媒を再生させることができる。酸素含有ガスとしては、例えば、空気、及び、空気又は酸素と不活性ガスとの混合ガスが挙げられる。酸素含有ガスの酸素濃度は、好ましくは0.1~2.0体積%である。触媒は、反応器より抜き出し反応器外で再生処理を行う反応器外再生と反応器より抜き出さずに反応器内で再生処理を行う反応器内再生のどちらの再生方法を採用してもよい。また、切り替え式反応器を採用することで反応-再生切り替え運転を行うこともできる。
(反応-再生切り替え運転)
反応-再生切り替え運転とは、2塔又は多塔切り替え式の断熱型反応器を用い、反応工程と再生工程とを同時に行う運転操作である。例えば、3塔切り替え式の場合は、2塔が反応工程に使用され、同時に、残りの1塔が触媒再生に使用される。その後、反応工程に使用されていたうちの1塔の反応工程を停止して触媒再生を行い、触媒再生に使用していた1塔で反応工程を行うことで、2塔分の生産能力を維持しつつ、触媒再生を行うことができる。このような反応形式をメリーゴーランド方式とも呼び、触媒再生のために製造工程を止める必要がないため、生産効率に優れる観点から好ましい。
反応-再生切り替え運転とは、2塔又は多塔切り替え式の断熱型反応器を用い、反応工程と再生工程とを同時に行う運転操作である。例えば、3塔切り替え式の場合は、2塔が反応工程に使用され、同時に、残りの1塔が触媒再生に使用される。その後、反応工程に使用されていたうちの1塔の反応工程を停止して触媒再生を行い、触媒再生に使用していた1塔で反応工程を行うことで、2塔分の生産能力を維持しつつ、触媒再生を行うことができる。このような反応形式をメリーゴーランド方式とも呼び、触媒再生のために製造工程を止める必要がないため、生産効率に優れる観点から好ましい。
〔生成物:炭素数3以上のオレフィンを含む反応ガス〕
本実施形態のエタノールの変換方法では、混合原料を触媒と接触させることで炭素数3以上のオレフィンを含む反応ガスを得る。「反応ガス」とは、混合原料を触媒と接触による反応後のガス組成物を意味する。反応ガスは、エチレンを含んでいてもよい。反応ガスは、水素、炭素数1~3の脂肪族炭化水素、炭素数4~6の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を含んでいてもよい。
本明細書では、炭素数1~3の脂肪族炭化水素、炭素数4~6の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を目的化合物と称する。
本実施形態のエタノールの変換方法では、混合原料を触媒と接触させることで炭素数3以上のオレフィンを含む反応ガスを得る。「反応ガス」とは、混合原料を触媒と接触による反応後のガス組成物を意味する。反応ガスは、エチレンを含んでいてもよい。反応ガスは、水素、炭素数1~3の脂肪族炭化水素、炭素数4~6の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を含んでいてもよい。
本明細書では、炭素数1~3の脂肪族炭化水素、炭素数4~6の脂肪族炭化水素、芳香族化合物、及び炭素数9以上の炭化水素を目的化合物と称する。
<分離工程及びリサイクル工程>
本実施形態に係る分離工程では、上述の反応工程で得られた反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離する。
本実施形態に係る分離工程では、上述の反応工程で得られた反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離する。
そして、リサイクル工程では、前記留分Aの少なくとも一部、を反応工程に前記混合原料の一部としてリサイクルする。
リサイクルとは、反応ガスを分離工程に供することで得られる留分の全量又は一部を、再び反応工程に供することで原料として使用することをいう。この時、リサイクルされる留分をリサイクル留分という。リサイクル留分を反応工程に供することで、エタノール等に由来する炭素分を効率的に目的化合物へと変換することができる。
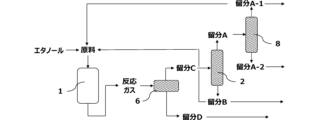
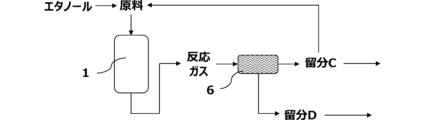
本実施形態に係る方法は、図7に示すように、固定床一段断熱型反応器1(以下、単に「反応器1」ともいう)と、第1の蒸留塔2とを有する装置により実施してもよい。反応器1で行われる反応工程により得られた反応ガスは、第1の蒸留塔2で、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離される。図示しない蒸留塔で留分Aからエチレンを分離し、さらに、図示しない蒸留塔でプロピレンを分離することによって、該反応ガスからのエチレン及びプロピレンの分離を効率的に行う。また、図示しないが、反応ガス及び/又は留分Aの少なくとも一部をエチレンプラントの精製系に導入し、該精製系で反応ガスからエチレン、プロピレン等の目的化合物を分離してもよい。留分Aの少なくとも一部は、反応器に原料としてリサイクルする。また、留分Bも反応器1に原料としてリサイクルしてもよい。
本実施形態に係る方法は、図8に示すように、反応器1と、第1の蒸留塔2と、スチームクラッキング装置5とを有する装置により実施することができる。分離工程により得られた留分Bをスチームクラッキングに付すことにより、エチレン及びプロピレンを含有するスチームクラッキング生成物を得、スチームクラッキング生成物からエチレン及びプロピレンを分離することでプロセス全体の効率性を高めることができる。スチームクラッキングとは、加熱蒸気と共に留分中の化合物を熱分解することを意味する。
本実施形態に係る方法は、図9に示すように、反応器1と、冷却装置6と、第1の蒸留塔2と、油水分離機7とを有する装置により実施することができる。反応器1で行われる反応工程により得られた反応ガスを冷却装置6により冷却する冷却工程を設けることで、反応ガスを炭素数2~6の脂肪族炭化水素を主に含む留分Cと、水、炭素数7以上の脂肪族炭化水素及び芳香族化合物を主に含む留分Dとに分離することができる。留分Cは冷却工程においてガス成分として得られ、留分Dは液成分として回収される。冷却装置としては、反応ガスと冷却剤とを直接接触させて熱交換を行わせる直接式熱交換器や、壁によって分けられた空間に反応ガスと冷却剤とを流通させ壁を通して熱交換を行わせる間接(隔壁)式熱交換器を使用することができる。
冷却工程で回収された留分Cは、第1の蒸留塔2で、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離される。留分Aの少なくとも一部を、反応器1に混合原料の一部としてリサイクルしてもよい。加えて、留分Bの少なくとも一部を、反応器1に混合原料の一部としてリサイクルしてもよい。
冷却工程で回収された留分Dは、油水分離機7において炭化水素を主に含む留分Eと水を主に含む留分Fとに分離される。該留分Eから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。芳香族化合物の分離は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。
冷却工程で回収された留分Cは、第1の蒸留塔2で、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離される。留分Aの少なくとも一部を、反応器1に混合原料の一部としてリサイクルしてもよい。加えて、留分Bの少なくとも一部を、反応器1に混合原料の一部としてリサイクルしてもよい。
冷却工程で回収された留分Dは、油水分離機7において炭化水素を主に含む留分Eと水を主に含む留分Fとに分離される。該留分Eから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。芳香族化合物の分離は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。
本実施形態に係る方法は、図10に示すように、反応器1と、冷却装置6と、第1の蒸留塔2と、第2の蒸留塔8とを有する装置により実施することができる。反応器1で行われる反応工程により得られた反応ガスを冷却装置6により冷却する、冷却工程を設けることで、反応ガスを炭素数2~6の脂肪族炭化水素を主に含む留分Cと、水、炭素数7以上の脂肪族炭化水素及び芳香族化合物を主に含む留分Dとに分離することができる。留分Cは冷却工程においてガス成分として得られ、留分Dは液成分として回収される。冷却工程で回収された留分Dから芳香族化合物を分離することによって、該反応ガスから芳香族化合物の分離を効率的に行う。芳香族化合物の分離は、例えば、蒸留、抽出蒸留、抽出、晶析及びこれらの組み合わせで実施される。冷却工程より得られた留分Cは、第1の蒸留塔2で、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離される。更に、留分Aを第2の蒸留塔8に供給することで、留分Aは炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離される。留分A-1をリサイクル原料とすることで、混合原料中のプロピレン濃度が低下し、効率よくプロピレンを製造することができる。
図11に示すように、本実施形態に係る方法の蒸留塔はサイドカット段を設けてもよい。サイドカット段より、目的の化合物が含まれる中間抜出留分を得ることで、分離効率を向上させることができる。第1の蒸留塔2の中間部より炭素数3の炭化水素を主に含む留分Eを抜き出すサイドカット段を蒸留塔に設けることで、プロピレンの回収収率が向上する。
各種留分において「主に含む」とは、「主に含む」と記載された成分の合計質量が、各留分に対して、50質量%超であることを意味する。なお、これらの分離工程は、蒸留、抽出など種々の公知の方法を組み合わせることで実施することができる。
[炭化水素等の製造方法]
第1~3の実施形態により得られる反応ガスから目的化合物を分離することで各種化学品が得られる。
つまり本実施形態に係る炭化水素の製造方法は、エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む。当該製造方法の詳細は上述のエタノールの変換方法で説明した通りであり、その好適な実施態様も同様である。
第1~3の実施形態により得られる反応ガスから目的化合物を分離することで各種化学品が得られる。
つまり本実施形態に係る炭化水素の製造方法は、エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む。当該製造方法の詳細は上述のエタノールの変換方法で説明した通りであり、その好適な実施態様も同様である。
当該製造方法により得られる炭化水素としては、例えば、プロピレン、エチレン、ブテン、ペンテン、ヘキセン、ヘプテン等のオレフィン、1,3-ブタジエン、イソプレン等のジエンなどの脂肪族不飽和炭化水素、ベンゼン、トルエンなどの芳香族炭化水素、エタン、プロパン、ブタン、ペンタンなどの脂肪族飽和炭化水素が挙げられる。なお、芳香族炭化水素は、常圧における沸点が500℃以下であることが好ましい。
得られた炭化水素をクラッカーの精製系に導入することで、効率的に目的の炭化水素化合物を精製することができる。
本実施形態に係る炭化水素の製造方法は、
炭素数2以上の炭化水素を分解する、クラッキング工程と、
前記クラッキング工程で得られた成分を精製する精製工程と、
を有し、
前記精製工程で、上述のエタノールの変換方法により得られた反応ガス又はその精製留分を合流させる。
以上の構成により、従来のクラッカーを利用し、エタノール資源から目的の炭化水素を得ることができる。例えばエタノールの変換方法における原料として、バイオエタノールを使用することで、バイオ由来の炭化水素を製造することができる。
本実施形態に係る炭化水素の製造方法は、
炭素数2以上の炭化水素を分解する、クラッキング工程と、
前記クラッキング工程で得られた成分を精製する精製工程と、
を有し、
前記精製工程で、上述のエタノールの変換方法により得られた反応ガス又はその精製留分を合流させる。
以上の構成により、従来のクラッカーを利用し、エタノール資源から目的の炭化水素を得ることができる。例えばエタノールの変換方法における原料として、バイオエタノールを使用することで、バイオ由来の炭化水素を製造することができる。
炭素数2以上の炭化水素としては、例えば、エチレン、プロピレン、ブテン、パラフィン、芳香族炭化水素が挙げられる。炭素数2以上の炭化水素として、ナフサを使用してもよい。
クラッカーとしては、エタンクラッカー、ナフサクラッカー等に用いられる熱分解炉を用いてもよい。
なお、精製工程においては、従来のエタンクラッカー又はナフサクラッカーに用いられる精製系が用いられ、例えば、蒸留塔、クエンチ塔などを備える設備により蒸留を行ってもよい。
上述のエタノールの変換方法により得られた反応ガス又はその精製留分は、上述の精製工程で合流させるが、合流させる成分に応じて合流箇所を適宜選択する。
本実施形態に係るモノマーの製造方法は、
上述のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程、
を含む。
不飽和炭化水素としては、例えば、プロピレン、エチレン、ブテン、ブタン、ペンテン、ヘキセン、ヘプテン等のオレフィン、1,3-ブタジエン、イソプレン等のジエンなどの脂肪族不飽和炭化水素が挙げられる。
上述のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程、
を含む。
不飽和炭化水素としては、例えば、プロピレン、エチレン、ブテン、ブタン、ペンテン、ヘキセン、ヘプテン等のオレフィン、1,3-ブタジエン、イソプレン等のジエンなどの脂肪族不飽和炭化水素が挙げられる。
本実施形態に係るオレフィンの製造方法は、
上述のエタノールの変換方法により得られた反応ガスからオレフィンを主に含む留分を分離するオレフィン分離工程、
を含む。
上述のエタノールの変換方法により得られた反応ガスからオレフィンを主に含む留分を分離するオレフィン分離工程、
を含む。
本実施形態に係るプロピレンの製造方法は、
上述のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程、
を含む。
上述のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程、
を含む。
本実施形態に係るエチレンの製造方法は、
上述のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程、
を含む。
上述のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程、
を含む。
本実施形態に係るジエンの製造方法は、
上述のエタノールの変換方法により得られた反応ガスからジエンを主に含む留分を分離するジエン分離工程、
を含む。
上述のエタノールの変換方法により得られた反応ガスからジエンを主に含む留分を分離するジエン分離工程、
を含む。
本実施形態に係るモノマーの製造方法は、更に不飽和炭化水素を変換する工程を有していてもよい。
本実施形態に係るアクリルモノマーの製造方法は、
上述のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程と、
前記不飽和炭化水素分離工程により得られた不飽和炭化水素からアクリルモノマーを得るアクリルモノマー製造工程と、
を含む。
アクリルモノマー製造工程は、不飽和炭化水素からアクリルモノマーを導入する公知の方法が用いられる。
上述のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程と、
前記不飽和炭化水素分離工程により得られた不飽和炭化水素からアクリルモノマーを得るアクリルモノマー製造工程と、
を含む。
アクリルモノマー製造工程は、不飽和炭化水素からアクリルモノマーを導入する公知の方法が用いられる。
本実施形態に係るアクリロニトリルの製造方法は、
上述のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程と、
前記プロピレン分離工程により得られたプロピレンからアクリロニトリルを得るアクリロニトリル製造工程と、
を含む。
アクリルモノマー製造工程は、不飽和炭化水素からアクリルモノマーを導入する公知の方法が用いられる。
上述のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程と、
前記プロピレン分離工程により得られたプロピレンからアクリロニトリルを得るアクリロニトリル製造工程と、
を含む。
アクリルモノマー製造工程は、不飽和炭化水素からアクリルモノマーを導入する公知の方法が用いられる。
本実施形態に係るスチレンの製造方法は、
上述のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程と、
前記エチレン分離工程により得られたエチレンからスチレンを得るスチレン製造工程と、
を含む。
上述のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程と、
前記エチレン分離工程により得られたエチレンからスチレンを得るスチレン製造工程と、
を含む。
上述の製造方法により得られたモノマーを更に重合してもよい。
本実施形態に係る重合体の製造方法は、
上述のモノマーの製造方法により得られるモノマーを重合する工程、
を含む。
モノマーを重合する工程は、従来の重合方法を用いて重合を行うことができ、各種開始剤、重合触媒を用いて重合することができる。
本実施形態に係る重合体の製造方法は、
上述のモノマーの製造方法により得られるモノマーを重合する工程、
を含む。
モノマーを重合する工程は、従来の重合方法を用いて重合を行うことができ、各種開始剤、重合触媒を用いて重合することができる。
本実施形態に係るオレフィン系重合体の製造方法は、
上述のオレフィンの製造方法により得られるオレフィンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてオレフィン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のオレフィンの製造方法により得られるオレフィンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてオレフィン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
本実施形態に係るポリプロピレン系重合体の製造方法は、
上述のプロピレンの製造方法により得られるプロピレンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてプロピレン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のプロピレンの製造方法により得られるプロピレンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてプロピレン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
本実施形態に係るポリエチレン系重合体の製造方法は、
上述のエチレンの製造方法により得られるエチレンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてエチレン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のエチレンの製造方法により得られるエチレンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてエチレン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
本実施形態に係るジエン系重合体の製造方法は、
上述のジエンの製造方法により得られるジエンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてジエン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のジエンの製造方法により得られるジエンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてジエン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
本実施形態に係るアクリルモノマー系重合体の製造方法は、
上述のアクリルモノマーの製造方法により得られるアクリルモノマーを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてアクリルモノマー単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のアクリルモノマーの製造方法により得られるアクリルモノマーを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてアクリルモノマー単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
本実施形態に係るアクリロニトリル系重合体の製造方法は、
上述のアクリロニトリルの製造方法により得られるアクリロニトリルを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてアクリロニトリル単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のアクリロニトリルの製造方法により得られるアクリロニトリルを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてアクリロニトリル単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
本実施形態に係るスチレン系重合体の製造方法は、
上述のスチレンの製造方法により得られるスチレンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてスチレン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のスチレンの製造方法により得られるスチレンを含む重合性組成物を重合する工程、
を含む。
ここで重合性組成物は、モノマーとしてスチレン単独であってもよく、他の不飽和結合を有するモノマーを含有していてもよい。
上述のエタノールの変換方法により得られる反応ガスから芳香族化合物を分離してもよい。
本実施形態に係る芳香族化合物の製造方法は、
上述のエタノールの変換方法により得られた反応ガスから芳香族化合物を主に含む留分を分離する芳香族化合物分離工程、
を含む。
芳香族炭化水素としては、例えば、ベンゼン、トルエン、キシレンなどが挙げられる。
本実施形態に係る芳香族化合物の製造方法は、
上述のエタノールの変換方法により得られた反応ガスから芳香族化合物を主に含む留分を分離する芳香族化合物分離工程、
を含む。
芳香族炭化水素としては、例えば、ベンゼン、トルエン、キシレンなどが挙げられる。
以下に実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
[各種物性の測定方法]
各種物性の測定方法は下記の通りである。
各種物性の測定方法は下記の通りである。
(1)ゼオライト含有触媒中のゼオライトのシリカ/アルミナのモル比
ゼオライトを水酸化ナトリウム溶液に完全に溶解した溶解液を準備した。その溶解液中に含まれるSi及びAlの量をICP(誘導結合プラズマ)発光分析装置(Rigaku製、商品名「JY138」)を用いて常法により測定し、その結果からシリカ/アルミナのモル比を導出した。測定条件は、高周波パワー:1kw、プラズマガス:13L/min、シースガス:0.15L/mim、ネブライザーガス:0.25L/min、Si測定波長:251.60nm、Al測定波長:396.152nmに設定した。
ゼオライトを水酸化ナトリウム溶液に完全に溶解した溶解液を準備した。その溶解液中に含まれるSi及びAlの量をICP(誘導結合プラズマ)発光分析装置(Rigaku製、商品名「JY138」)を用いて常法により測定し、その結果からシリカ/アルミナのモル比を導出した。測定条件は、高周波パワー:1kw、プラズマガス:13L/min、シースガス:0.15L/mim、ネブライザーガス:0.25L/min、Si測定波長:251.60nm、Al測定波長:396.152nmに設定した。
(2)ゼオライト含有触媒のリン元素含有量及び銀元素含有量
ゼオライト含有触媒中のリン元素及び銀元素の含有量は、蛍光X線分析装置(Rigaku製、商品名「RIX3000」)を用いて、常法により測定した。
ゼオライト含有触媒中のリン元素及び銀元素の含有量は、蛍光X線分析装置(Rigaku製、商品名「RIX3000」)を用いて、常法により測定した。
(3)ゼオライトの構造タイプ
ゼオライト含有触媒中のゼオライトの構造タイプは、X線分析装置(Rigaku製、商品名「RINT」)を用いて、ゼオライトのX線回折パターンを測定し、公知のゼオライトの回折パターンを参照することで同定した。測定条件は以下の通りである。
Cu陰極
管球電圧:40kV
管球電流:30mA
スキャンスピード:1deg/分
ゼオライト含有触媒中のゼオライトの構造タイプは、X線分析装置(Rigaku製、商品名「RINT」)を用いて、ゼオライトのX線回折パターンを測定し、公知のゼオライトの回折パターンを参照することで同定した。測定条件は以下の通りである。
Cu陰極
管球電圧:40kV
管球電流:30mA
スキャンスピード:1deg/分
[ゼオライト含有触媒の調製方法]
(ゼオライト含有触媒1の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比200)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径1.6mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体に対し、所定量のリン酸水素二アンモニウム水溶液を担持させてリン担持品を得た。得られたリン担持品を焼成炉を用いて、空気雰囲気下、600℃で5時間焼成した。焼成品を反応器に充填し、水蒸気を80体積%含む水蒸気-窒素混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒1を得た。ゼオライト含有触媒中に含まれるリン元素の含有量を測定したところ、0.36質量%であった。
(ゼオライト含有触媒1の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比200)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径1.6mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体に対し、所定量のリン酸水素二アンモニウム水溶液を担持させてリン担持品を得た。得られたリン担持品を焼成炉を用いて、空気雰囲気下、600℃で5時間焼成した。焼成品を反応器に充填し、水蒸気を80体積%含む水蒸気-窒素混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒1を得た。ゼオライト含有触媒中に含まれるリン元素の含有量を測定したところ、0.36質量%であった。
(ゼオライト含有触媒2の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比980)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径2.1mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体を5時間焼成してゼオライト含有触媒2を得た。
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比980)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径2.1mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体を5時間焼成してゼオライト含有触媒2を得た。
(ゼオライト含有触媒3の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比980)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径2.1mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体に対し、所定量のリン酸水素二アンモニウム水溶液を担持させてリン担持品を得た。得られたリン担持品を、空気雰囲気下、600℃で5時間焼成した。焼成品を反応器に充填し、水蒸気を80体積%含む水蒸気-窒素混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒3を得た。この時、ゼオライト含有触媒中に含まれるリン元素の含有量は0.032質量%であった。
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比980)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径2.1mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体に対し、所定量のリン酸水素二アンモニウム水溶液を担持させてリン担持品を得た。得られたリン担持品を、空気雰囲気下、600℃で5時間焼成した。焼成品を反応器に充填し、水蒸気を80体積%含む水蒸気-窒素混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒3を得た。この時、ゼオライト含有触媒中に含まれるリン元素の含有量は0.032質量%であった。
(ゼオライト含有触媒4の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比980)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径2.1mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体を600℃で5時間焼成して触媒前駆体を得た。得られた触媒前駆体を0.1N硝酸ナトリウム水溶液中で1時間攪拌した後、ろ過洗浄し、600℃で5時間焼成することでナトリウム交換体を得た。ナトリウム交換体を0.01N硝酸銀水溶液中で1時間攪拌した後、ろ過洗浄する工程を3回繰り返し、600℃で5時間焼成することで銀交換体を得た。銀交換体に、水蒸気を80体積%含む水蒸気-空気混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒2を得た。この時、ゼオライト含有触媒中に含まれる銀元素の含有量は0.16質量%であった。
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比980)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径2.1mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体を600℃で5時間焼成して触媒前駆体を得た。得られた触媒前駆体を0.1N硝酸ナトリウム水溶液中で1時間攪拌した後、ろ過洗浄し、600℃で5時間焼成することでナトリウム交換体を得た。ナトリウム交換体を0.01N硝酸銀水溶液中で1時間攪拌した後、ろ過洗浄する工程を3回繰り返し、600℃で5時間焼成することで銀交換体を得た。銀交換体に、水蒸気を80体積%含む水蒸気-空気混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒2を得た。この時、ゼオライト含有触媒中に含まれる銀元素の含有量は0.16質量%であった。
(ゼオライト含有触媒5の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比302)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径1.6mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体を空気雰囲気下、600℃で5時間焼成してゼオライト含有触媒5を得た。
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比302)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径1.6mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体を空気雰囲気下、600℃で5時間焼成してゼオライト含有触媒5を得た。
(ゼオライト含有触媒6の調製)
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比302)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径1.6mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体に対し、所定量のリン酸水素二アンモニウム水溶液を担持させてリン担持品を得た。得られたリン担持品を、空気雰囲気下、600℃で5時間焼成した。焼成品を反応器に充填し、水蒸気を80体積%含む水蒸気-窒素混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒5を得た。この時、ゼオライト含有触媒中に含まれるリン元素の含有量は0.085質量%であった。
中間細孔径ゼオライトであるプロトン型ZSM-5(シリカ/アルミナのモル比302)70質量部とシリカ30質量部(コロイダルシリカ及びヒュームドシリカを使って水分量を調整)より得られた粘土を混錬後、押出成形を実施し、直径1.6mm、長さ4~6mmに調整した押出成形体を得た。得られた成形体に対し、所定量のリン酸水素二アンモニウム水溶液を担持させてリン担持品を得た。得られたリン担持品を、空気雰囲気下、600℃で5時間焼成した。焼成品を反応器に充填し、水蒸気を80体積%含む水蒸気-窒素混合ガスを、圧力0.1MPa、温度600℃の条件下で24時間供給、流通することでゼオライト含有触媒5を得た。この時、ゼオライト含有触媒中に含まれるリン元素の含有量は0.085質量%であった。
[第1実施形態の実施例]
以下に第1実施形態の実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
以下に第1実施形態の実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
[エタノールの変換方法]
(反応装置)
以下実施例及び比較例は図1に示す固定床一段断熱型反応器1を用いて評価を行った。
(反応装置)
以下実施例及び比較例は図1に示す固定床一段断熱型反応器1を用いて評価を行った。
(原料)
実施例及び比較例におけるエチレン/エタノールのモル比及び炭素数4~6のオレフィン/エチレンのモル比は下式に倣い算出した。
実施例及び比較例におけるエチレン/エタノールのモル比及び炭素数4~6のオレフィン/エチレンのモル比は下式に倣い算出した。
エチレン/エタノールのモル比(-)=エチレンモル流量(mol/hr)/エタノールモル流量(mol/hr)
エチレン/炭素数4~6のオレフィンのモル比(-)=エチレンモル流量(mol/hr)/炭素数4~6のオレフィン流量(mol/hr)
(温度測定)
触媒床入口の温度及び触媒床出口の温度は反応器外部から挿入した熱電対によって測定を行った。具体的には、図1に示すように、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0.5d~0.6dにおける温度を測定した。なお、本熱電対挿入による放熱の影響は無視できるほど小さかった。
触媒床入口の温度及び触媒床出口の温度は反応器外部から挿入した熱電対によって測定を行った。具体的には、図1に示すように、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0.5d~0.6dにおける温度を測定した。なお、本熱電対挿入による放熱の影響は無視できるほど小さかった。
(反応評価)
以下実施例及び比較例に従って、入出平均反応温度が540℃になるように反応を実施した。反応開始から3hごとに反応器出口ガスの一部をサンプリングし、ガスクロマトグラフ(以下、単に「GC」ともいう、TCD、FID検出器)に導入して反応ガス組成の分析を行った。反応開始から48時間後に反応を停止した。反応開始から反応停止までのGC分析結果の平均値を算出した。なお、入出平均反応温度は下式に従い算出し、表中では「反応温度」と表記した。
以下実施例及び比較例に従って、入出平均反応温度が540℃になるように反応を実施した。反応開始から3hごとに反応器出口ガスの一部をサンプリングし、ガスクロマトグラフ(以下、単に「GC」ともいう、TCD、FID検出器)に導入して反応ガス組成の分析を行った。反応開始から48時間後に反応を停止した。反応開始から反応停止までのGC分析結果の平均値を算出した。なお、入出平均反応温度は下式に従い算出し、表中では「反応温度」と表記した。
入出平均反応温度(℃)=[触媒床入口温度(℃)+触媒床出口温度(℃)]/2
(コーク収率)
以下実施例及び比較例において、反応停止後、窒素を反応器に供給して炭化水素のパージを行い、触媒床を500℃に保った。次いで、酸素濃度2体積%の空気/窒素を流通させ、触媒上のコークを燃焼除去した。この際、反応器出口ガスを再生ガスとして定期的にサンプリングして、ガスクロマトグラフを用いて再生ガスの分析を行い、CO2、COの濃度を測定し、この値から触媒に付着していたカーボン量を求め、これをコーク量とした。ガスクロマトグラフを用いた再生ガス分析の方法については下記(ガスクロマトグラフの分析条件)に記載した。以下、実施例及び比較例におけるコーク収率は以下の式に従い、求めた。
以下実施例及び比較例において、反応停止後、窒素を反応器に供給して炭化水素のパージを行い、触媒床を500℃に保った。次いで、酸素濃度2体積%の空気/窒素を流通させ、触媒上のコークを燃焼除去した。この際、反応器出口ガスを再生ガスとして定期的にサンプリングして、ガスクロマトグラフを用いて再生ガスの分析を行い、CO2、COの濃度を測定し、この値から触媒に付着していたカーボン量を求め、これをコーク量とした。ガスクロマトグラフを用いた再生ガス分析の方法については下記(ガスクロマトグラフの分析条件)に記載した。以下、実施例及び比較例におけるコーク収率は以下の式に従い、求めた。
コーク収率(質量ppm)=コーク量/[有効原料供給質量流量(kg/hr)×反応時間(hr)]
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
(ガスクロマトグラフの分析条件)
[再生ガス分析]
装置:島津製作所GC-8A
カラム:以下のカラム(1)とカラム(2)を並列に連結したものを使用する。
カラム(1)80~100メッシュのモレキュラーシーブ5A(和光純薬製)を充填したSUS製カラム(内径3mm、長さ3m)
カラム(2)80~100メッシュの米国WATERS ASSOCIATES社製Porapac-Q(内径3mm、長さ2m)及びSUS製抵抗カラム(内径3mm、長さ1m)を直接に連結したもの
カラム温度:70℃
キャリアーガス(ヘリウム)流量:60mL/分
[再生ガス分析]
装置:島津製作所GC-8A
カラム:以下のカラム(1)とカラム(2)を並列に連結したものを使用する。
カラム(1)80~100メッシュのモレキュラーシーブ5A(和光純薬製)を充填したSUS製カラム(内径3mm、長さ3m)
カラム(2)80~100メッシュの米国WATERS ASSOCIATES社製Porapac-Q(内径3mm、長さ2m)及びSUS製抵抗カラム(内径3mm、長さ1m)を直接に連結したもの
カラム温度:70℃
キャリアーガス(ヘリウム)流量:60mL/分
[反応ガス分析]
装置:島津製作所製GC-2030
カラム:米国SUPELCO社製カスタムキャピラリーカラムSPB-1(内径0.25mm、長さ60m、フイルム厚3.0μm)
サンプルガス量:1mL(サンプリングラインは200℃~300℃に保温)
昇温プログラム:40℃で12分間保持し、次いで5℃/分で200℃まで昇温した後、200℃で22分間保持する。
スプリット比:200対1
キャリアーガス(窒素)流量:120mL/分
FID検出器:エアー供給圧50kPa(約500mL/分)、水素供給圧60kPa(約50mL/分)
測定方法:TCD検出器とFID検出器を直列に連結して、水素はTCD検出器で検出したデータ、炭化水素やエタノールなどの含酸素物はFID検出器で検出したデータを元に組成分析を行い、検量線法を用いて反応ガス中の目的化合物の濃度を求め、反応により生成した時間当たりの質量を求めた。なお、収率は下式で算出した。
装置:島津製作所製GC-2030
カラム:米国SUPELCO社製カスタムキャピラリーカラムSPB-1(内径0.25mm、長さ60m、フイルム厚3.0μm)
サンプルガス量:1mL(サンプリングラインは200℃~300℃に保温)
昇温プログラム:40℃で12分間保持し、次いで5℃/分で200℃まで昇温した後、200℃で22分間保持する。
スプリット比:200対1
キャリアーガス(窒素)流量:120mL/分
FID検出器:エアー供給圧50kPa(約500mL/分)、水素供給圧60kPa(約50mL/分)
測定方法:TCD検出器とFID検出器を直列に連結して、水素はTCD検出器で検出したデータ、炭化水素やエタノールなどの含酸素物はFID検出器で検出したデータを元に組成分析を行い、検量線法を用いて反応ガス中の目的化合物の濃度を求め、反応により生成した時間当たりの質量を求めた。なお、収率は下式で算出した。
プロピレン収率(%)=反応により生成した時間当たりのプロピレン質量(kg/hr)/有効原料供給質量流量(kg/hr)
芳香族収率(%)=反応により生成した時間当たりの芳香族化合物質量(kg/hr)/有効原料供給質量流量(kg/hr)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
[実施例1]
エチレン/エタノールのモル比が0.37のエチレン、エタノール及び水蒸気の混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は539℃、出口温度は542℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は23.2質量%、平均の芳香族収率は2.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。反応結果及び反応条件の詳細を表1に示す。
エチレン/エタノールのモル比が0.37のエチレン、エタノール及び水蒸気の混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は539℃、出口温度は542℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は23.2質量%、平均の芳香族収率は2.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。反応結果及び反応条件の詳細を表1に示す。
[比較例1]
エチレンと水蒸気を含まず、エタノール原料ガスのみを用いて、触媒床入口温度を588℃に調節した以外は実施例1と同様に反応を行った。この時、触媒床の出口温度は492℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.3%、平均の芳香族収率は1.5質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は507質量ppmであった。反応結果を反応条件とともに表1に示す。
エチレンと水蒸気を含まず、エタノール原料ガスのみを用いて、触媒床入口温度を588℃に調節した以外は実施例1と同様に反応を行った。この時、触媒床の出口温度は492℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.3%、平均の芳香族収率は1.5質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は507質量ppmであった。反応結果を反応条件とともに表1に示す。
[比較例2]
エタノールを含まず、エチレン原料ガスのみを用いて、触媒床入口温度を450℃とした以外は実施例1と同様に反応を行った。すると、触媒床出口の温度が650℃を超えてなお上昇し続けたため、安全性の観点より、1時間経過時点で反応を停止した。本比較例より、激しい発熱を呈する反応を断熱型反応器で効率的に実施することは困難であるとわかる。
エタノールを含まず、エチレン原料ガスのみを用いて、触媒床入口温度を450℃とした以外は実施例1と同様に反応を行った。すると、触媒床出口の温度が650℃を超えてなお上昇し続けたため、安全性の観点より、1時間経過時点で反応を停止した。本比較例より、激しい発熱を呈する反応を断熱型反応器で効率的に実施することは困難であるとわかる。
[実施例2~6]
それぞれゼオライト含有触媒2、ゼオライト含有触媒3、ゼオライト含有触媒4、ゼオライト含有触媒5及びゼオライト含有触媒6を使用した以外は実施例1と同様に反応を行った。それぞれの入出温度差や反応成績は表1に示す。
それぞれゼオライト含有触媒2、ゼオライト含有触媒3、ゼオライト含有触媒4、ゼオライト含有触媒5及びゼオライト含有触媒6を使用した以外は実施例1と同様に反応を行った。それぞれの入出温度差や反応成績は表1に示す。

実施例1及び比較例1~2の結果から、特定範囲のエチレン/エタノールのモル比の混合原料を用いることで高いプロピレン収率と触媒へのコーク堆積の抑制が両立できることが判った。
[実施例7]
エチレン/エタノールのモル比が0.25、炭素数4~6のオレフィン/エチレンのモル比が0.7であるエチレン、エタノール、表2に示す組成比の炭素数4~8の炭化水素を主に含む炭素数4以上の炭化水素、及び水蒸気の混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=4.9となるように供給し、反応を行った。この時、触媒床の入口温度は568℃、出口温度は512℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は22.5質量%、平均の芳香族収率は3.0質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は315質量ppmであった。反応結果及び反応条件の詳細を表3に示す。
エチレン/エタノールのモル比が0.25、炭素数4~6のオレフィン/エチレンのモル比が0.7であるエチレン、エタノール、表2に示す組成比の炭素数4~8の炭化水素を主に含む炭素数4以上の炭化水素、及び水蒸気の混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=4.9となるように供給し、反応を行った。この時、触媒床の入口温度は568℃、出口温度は512℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は22.5質量%、平均の芳香族収率は3.0質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は315質量ppmであった。反応結果及び反応条件の詳細を表3に示す。
[実施例8]
エチレン/エタノールのモル比が0.82、炭素数4~6のオレフィン/エチレンのモル比が0.3である混合原料ガスを用い、触媒床の入口温度を539℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は541℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は23.7質量%、平均の芳香族収率は2.6質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は273質量ppmであった。反応結果を反応条件とともに表3に示す。
エチレン/エタノールのモル比が0.82、炭素数4~6のオレフィン/エチレンのモル比が0.3である混合原料ガスを用い、触媒床の入口温度を539℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は541℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は23.7質量%、平均の芳香族収率は2.6質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は273質量ppmであった。反応結果を反応条件とともに表3に示す。
[実施例9]
エチレン/エタノールのモル比が1.5、炭素数4~6のオレフィン/エチレンのモル比が0.2である混合原料ガスを用い、触媒床の入口温度を522℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は558℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は23.2質量%、平均の芳香族収率は2.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は277質量ppmであった。反応結果を反応条件とともに表3に示す。
エチレン/エタノールのモル比が1.5、炭素数4~6のオレフィン/エチレンのモル比が0.2である混合原料ガスを用い、触媒床の入口温度を522℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は558℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は23.2質量%、平均の芳香族収率は2.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は277質量ppmであった。反応結果を反応条件とともに表3に示す。
[実施例10]
エチレン/エタノールのモル比が2.3、炭素数4~6のオレフィン/エチレンのモル比が0.2である混合原料ガスを用い、触媒床の入口温度を511℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は569℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は22.7質量%、平均の芳香族収率は2.4質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は318質量ppmであった。反応結果を反応条件とともに表3に示す。
エチレン/エタノールのモル比が2.3、炭素数4~6のオレフィン/エチレンのモル比が0.2である混合原料ガスを用い、触媒床の入口温度を511℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は569℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は22.7質量%、平均の芳香族収率は2.4質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は318質量ppmであった。反応結果を反応条件とともに表3に示す。
[実施例11]
エチレン/エタノールのモル比が0.25、炭素数4~6のオレフィン/エチレンのモル比が2.00である混合原料ガスを用い、触媒床の入口温度を572℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は508℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は20.4質量%、平均の芳香族収率は3,9質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は320質量ppmであった。反応結果を反応条件とともに表3に示す。
エチレン/エタノールのモル比が0.25、炭素数4~6のオレフィン/エチレンのモル比が2.00である混合原料ガスを用い、触媒床の入口温度を572℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は508℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は20.4質量%、平均の芳香族収率は3,9質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は320質量ppmであった。反応結果を反応条件とともに表3に示す。
[比較例3]
エチレンを用いず、エタノールと炭素数4~6のオレフィンとを原料ガスとして用い、触媒床の入口温度を590℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は490℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は17.6質量%、平均の芳香族収率は1.4質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は507質量ppmであった。反応結果を反応条件とともに表3に示す。
エチレンを用いず、エタノールと炭素数4~6のオレフィンとを原料ガスとして用い、触媒床の入口温度を590℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は490℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は17.6質量%、平均の芳香族収率は1.4質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は507質量ppmであった。反応結果を反応条件とともに表3に示す。
[比較例4]
エタノールを用いず、炭素数4~6のオレフィン/エチレンのモル比が0.1である混合原料ガスを用い、触媒床入口温度を477℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は603℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は16.5質量%、平均の芳香族収率は1.2質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は565質量ppmであった。反応結果を反応条件とともに表3に示す。
エタノールを用いず、炭素数4~6のオレフィン/エチレンのモル比が0.1である混合原料ガスを用い、触媒床入口温度を477℃に調節した以外は実施例7と同様に反応を行った。この時、触媒床の出口温度は603℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は16.5質量%、平均の芳香族収率は1.2質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は565質量ppmであった。反応結果を反応条件とともに表3に示す。


[第2実施形態の実施例]
以下に第2実施形態の実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
以下に第2実施形態の実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
[エタノールの変換方法]
(反応装置)
以下実施例及び比較例は図1に示す固定床一段断熱型反応器1を用いて評価を行った。
(反応装置)
以下実施例及び比較例は図1に示す固定床一段断熱型反応器1を用いて評価を行った。
(原料)
実施例及び比較例におけるメタノール/エタノールのモル比、炭素数4~6のオレフィン/メタノールのモル比及びエチレン/エタノールのモル比は下式に倣い算出した。
メタノール/エタノールのモル比(-)=メタノールモル流量(mol/hr)/エタノールモル流量(mol/hr)
メタノール/炭素数4~6のオレフィンのモル比(-)=メタノールモル流量(mol/hr)/炭素数4~6のオレフィン流量(mol/hr)
エチレン/エタノールのモル比(-)=エチレンモル流量(mol/hr)/エタノールモル流量(mol/hr)
実施例及び比較例におけるメタノール/エタノールのモル比、炭素数4~6のオレフィン/メタノールのモル比及びエチレン/エタノールのモル比は下式に倣い算出した。
メタノール/エタノールのモル比(-)=メタノールモル流量(mol/hr)/エタノールモル流量(mol/hr)
メタノール/炭素数4~6のオレフィンのモル比(-)=メタノールモル流量(mol/hr)/炭素数4~6のオレフィン流量(mol/hr)
エチレン/エタノールのモル比(-)=エチレンモル流量(mol/hr)/エタノールモル流量(mol/hr)
(温度測定)
触媒床入口の温度及び触媒床出口の温度は反応器外部から挿入した熱電対によって測定を行った。具体的には、図1に示すように、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0.5d~0.6dにおける温度を測定した。なお、本熱電対挿入による放熱の影響は無視できるほど小さかった。
触媒床入口の温度及び触媒床出口の温度は反応器外部から挿入した熱電対によって測定を行った。具体的には、図1に示すように、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0.5d~0.6dにおける温度を測定した。なお、本熱電対挿入による放熱の影響は無視できるほど小さかった。
(反応評価)
以下実施例及び比較例に従って、入出平均反応温度が540℃になるように反応を実施した。反応開始から3hごとに反応器出口ガスの一部をサンプリングし、ガスクロマトグラフ(TCD、FID検出器)に導入して反応ガス組成の分析を行った。反応開始から48時間後に反応を停止した。反応開始から反応停止までのGC分析結果の平均値を算出した。なお、入出平均反応温度は下式に従い算出し、表中では「反応温度」と表記した。
入出平均反応温度(℃)=[触媒床入口温度(℃)+触媒床出口温度(℃)]/2
以下実施例及び比較例に従って、入出平均反応温度が540℃になるように反応を実施した。反応開始から3hごとに反応器出口ガスの一部をサンプリングし、ガスクロマトグラフ(TCD、FID検出器)に導入して反応ガス組成の分析を行った。反応開始から48時間後に反応を停止した。反応開始から反応停止までのGC分析結果の平均値を算出した。なお、入出平均反応温度は下式に従い算出し、表中では「反応温度」と表記した。
入出平均反応温度(℃)=[触媒床入口温度(℃)+触媒床出口温度(℃)]/2
(コーク収率)
以下実施例及び比較例において、反応停止後、窒素を反応器に供給して炭化水素のパージを行い、触媒床を500℃に保った。次いで、酸素濃度2体積%の空気/窒素を流通させ、触媒上のコークを燃焼除去した。この際、反応器出口ガスを定期的にサンプリングして、ガスクロマトグラフを用いて再生ガスの分析を行い、CO2、COの濃度を測定し、この値から触媒に付着していたカーボン量を求め、これをコーク量とした。ガスクロマトグラフを用いた再生ガス分析の方法については下記(ガスクロマトグラフの分析条件)に記載した。以下、実施例及び比較例におけるコーク収率は以下の式に従い、求めた。
コーク収率(質量ppm)=コーク量/[有効原料供給質量流量(kg/hr)×反応時間(hr)]
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+メチレン換算メタノール流量+炭素数4~6のオレフィン流量(kg/hr)+メタノール及びエタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×0.609
メチレン換算メタノール流量(kg/hr)=メタノール流量(kg/hr)×0.438
以下実施例及び比較例において、反応停止後、窒素を反応器に供給して炭化水素のパージを行い、触媒床を500℃に保った。次いで、酸素濃度2体積%の空気/窒素を流通させ、触媒上のコークを燃焼除去した。この際、反応器出口ガスを定期的にサンプリングして、ガスクロマトグラフを用いて再生ガスの分析を行い、CO2、COの濃度を測定し、この値から触媒に付着していたカーボン量を求め、これをコーク量とした。ガスクロマトグラフを用いた再生ガス分析の方法については下記(ガスクロマトグラフの分析条件)に記載した。以下、実施例及び比較例におけるコーク収率は以下の式に従い、求めた。
コーク収率(質量ppm)=コーク量/[有効原料供給質量流量(kg/hr)×反応時間(hr)]
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+メチレン換算メタノール流量+炭素数4~6のオレフィン流量(kg/hr)+メタノール及びエタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×0.609
メチレン換算メタノール流量(kg/hr)=メタノール流量(kg/hr)×0.438
(ガスクロマトグラフの分析条件)
≪再生ガス分析≫
装置:島津製作所GC-8A
カラム:以下のカラム(1)とカラム(2)を並列に連結したものを使用する。
カラム(1)80~100メッシュのモレキュラーシーブ5A(和光純薬製)を充填したSUS製カラム(内径3mm、長さ3m)
カラム(2)80~100メッシュの米国WATERS ASSOCIATES社製Porapac-Q(内径3mm、長さ2m)及びSUS製抵抗カラム(内径3mm、長さ1m)を直接に連結したもの
カラム温度:70℃
キャリアーガス(ヘリウム)流量:60mL/分
≪再生ガス分析≫
装置:島津製作所GC-8A
カラム:以下のカラム(1)とカラム(2)を並列に連結したものを使用する。
カラム(1)80~100メッシュのモレキュラーシーブ5A(和光純薬製)を充填したSUS製カラム(内径3mm、長さ3m)
カラム(2)80~100メッシュの米国WATERS ASSOCIATES社製Porapac-Q(内径3mm、長さ2m)及びSUS製抵抗カラム(内径3mm、長さ1m)を直接に連結したもの
カラム温度:70℃
キャリアーガス(ヘリウム)流量:60mL/分
≪反応ガス分析≫
装置:島津製作所製GC-2030
カラム:米国SUPELCO社製カスタムキャピラリーカラムSPB-1(内径0.25mm、長さ60m、フイルム厚3.0μm)
サンプルガス量:1mL(サンプリングラインは200℃~300℃に保温)
昇温プログラム:40℃で12分間保持し、次いで5℃/分で200℃まで昇温した後、200℃で22分間保持する。
スプリット比:200対1
キャリアーガス(窒素)流量:120mL/分
FID検出器:エアー供給圧50kPa(約500mL/分)、水素供給圧60kPa(約50mL/分)
測定方法:TCD検出器とFID検出器を直列に連結して、水素はTCD検出器で検出したデータ、炭化水素やエタノールなどの含酸素物はFID検出器で検出したデータを元に組成分析を行い、検量線法を用いて反応ガス中のプロピレン濃度、ベンゼン濃度、トルエン濃度及び炭素数8の芳香族炭化水素濃度を求め、反応により生成した時間当たりのプロピレン質量及び芳香族質量を求めた。なお、反応により生成した時間当たりの芳香族質量は反応により生成した時間当たりのベンゼン、トルエン及び炭素数8の芳香族炭化水素の合計質量である。
プロピレン収率及び芳香族収率は下式で算出した。
プロピレン収率(質量%)=反応により生成した時間当たりのプロピレン質量(kg/hr)/有効原料供給質量流量(kg/hr)
芳香族収率(質量%)=反応により生成した時間当たりの芳香族質量(kg/hr)/有効原料供給質量流量(kg/hr)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+メチレン換算メタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+メタノール及びエタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×0.609
メチレン換算メタノール流量(kg/hr)=メタノール流量(kg/hr)×0.438
装置:島津製作所製GC-2030
カラム:米国SUPELCO社製カスタムキャピラリーカラムSPB-1(内径0.25mm、長さ60m、フイルム厚3.0μm)
サンプルガス量:1mL(サンプリングラインは200℃~300℃に保温)
昇温プログラム:40℃で12分間保持し、次いで5℃/分で200℃まで昇温した後、200℃で22分間保持する。
スプリット比:200対1
キャリアーガス(窒素)流量:120mL/分
FID検出器:エアー供給圧50kPa(約500mL/分)、水素供給圧60kPa(約50mL/分)
測定方法:TCD検出器とFID検出器を直列に連結して、水素はTCD検出器で検出したデータ、炭化水素やエタノールなどの含酸素物はFID検出器で検出したデータを元に組成分析を行い、検量線法を用いて反応ガス中のプロピレン濃度、ベンゼン濃度、トルエン濃度及び炭素数8の芳香族炭化水素濃度を求め、反応により生成した時間当たりのプロピレン質量及び芳香族質量を求めた。なお、反応により生成した時間当たりの芳香族質量は反応により生成した時間当たりのベンゼン、トルエン及び炭素数8の芳香族炭化水素の合計質量である。
プロピレン収率及び芳香族収率は下式で算出した。
プロピレン収率(質量%)=反応により生成した時間当たりのプロピレン質量(kg/hr)/有効原料供給質量流量(kg/hr)
芳香族収率(質量%)=反応により生成した時間当たりの芳香族質量(kg/hr)/有効原料供給質量流量(kg/hr)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+メチレン換算メタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+メタノール及びエタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×0.609
メチレン換算メタノール流量(kg/hr)=メタノール流量(kg/hr)×0.438
[実施例B1]
メタノール/エタノールのモル比が0.22のメタノール及びエタノールの混合原料ガスを加熱して、ゼオライト含有触媒1が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は563℃、出口温度は518℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は277質量ppmであった。反応結果及び反応条件の詳細を表4に示す。
メタノール/エタノールのモル比が0.22のメタノール及びエタノールの混合原料ガスを加熱して、ゼオライト含有触媒1が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は563℃、出口温度は518℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は277質量ppmであった。反応結果及び反応条件の詳細を表4に示す。
[実施例B2]
メタノール/エタノールのモル比が0.50のメタノール及びエタノールの混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は536℃、出口温度は544℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は20.1質量%、芳香族収率は7.5質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。反応結果及び反応条件の詳細を表4に示す。
メタノール/エタノールのモル比が0.50のメタノール及びエタノールの混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は536℃、出口温度は544℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は20.1質量%、芳香族収率は7.5質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。反応結果及び反応条件の詳細を表4に示す。
[実施例B3]
メタノール/エタノールのモル比が0.86のメタノール及びエタノールの混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は511℃、出口温度は569℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.3質量%、芳香族収率は8.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は357質量ppmであった。反応結果及び反応条件の詳細を表4に示す。
メタノール/エタノールのモル比が0.86のメタノール及びエタノールの混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は511℃、出口温度は569℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.3質量%、芳香族収率は8.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は357質量ppmであった。反応結果及び反応条件の詳細を表4に示す。
[比較例B1]
メタノールを含まず、エタノール原料ガスのみを用いて、触媒床入口温度を588℃に調節した以外は実施例B1と同様に反応を行った。この時、触媒床の出口温度は492℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は18.3%、芳香族収率は1.2質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は483質量ppmであった。反応結果を反応条件とともに表4に示す。
メタノールを含まず、エタノール原料ガスのみを用いて、触媒床入口温度を588℃に調節した以外は実施例B1と同様に反応を行った。この時、触媒床の出口温度は492℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は18.3%、芳香族収率は1.2質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は483質量ppmであった。反応結果を反応条件とともに表4に示す。
[比較例B2]
エタノールを含まず、メタノール原料ガスのみを用いて、触媒床入口温度を450℃とした以外は実施例B1と同様に反応を行った。すると、触媒床出口の温度が650℃を超えてなお上昇し続けたため、安全性の観点より、1時間経過時点で反応を停止した。本比較例より、激しい発熱を呈する反応を断熱型反応器で効率的に実施することは困難であるとわかる。
エタノールを含まず、メタノール原料ガスのみを用いて、触媒床入口温度を450℃とした以外は実施例B1と同様に反応を行った。すると、触媒床出口の温度が650℃を超えてなお上昇し続けたため、安全性の観点より、1時間経過時点で反応を停止した。本比較例より、激しい発熱を呈する反応を断熱型反応器で効率的に実施することは困難であるとわかる。

実施例B1~B3及び比較例B1~B2の結果から、特定範囲のメタノール/エタノールのモル比の混合原料を用いることで高いプロピレン収率及び芳香族収率と触媒へのコーク堆積の抑制が両立できることが判った。
[実施例B4]
メタノール/エタノールのモル比が0.35、炭素数4~6のオレフィン/メタノールのモル比が0.97であるメタノール、エタノール及び表5に示す組成比の炭素数4~8の炭化水素を主に含む炭素数4以上の炭化水素の混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=4.9となるように供給し、反応を行った。この時、触媒床の入口温度は566℃、出口温度は514℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は18.9質量%、芳香族収率は8.3質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は315質量ppmであった。反応結果及び反応条件の詳細を表6に示す。なお、本実施例では原料における芳香族濃度と反応ガスにおける芳香族濃度の差分より芳香族収率を算出している。
メタノール/エタノールのモル比が0.35、炭素数4~6のオレフィン/メタノールのモル比が0.97であるメタノール、エタノール及び表5に示す組成比の炭素数4~8の炭化水素を主に含む炭素数4以上の炭化水素の混合原料ガスを加熱して、ゼオライト含有触媒が充填された反応器にWHSV=4.9となるように供給し、反応を行った。この時、触媒床の入口温度は566℃、出口温度は514℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は18.9質量%、芳香族収率は8.3質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は315質量ppmであった。反応結果及び反応条件の詳細を表6に示す。なお、本実施例では原料における芳香族濃度と反応ガスにおける芳香族濃度の差分より芳香族収率を算出している。
[実施例B5]
メタノール/エタノールのモル比が0.50、炭素数4~6のオレフィン/メタノールのモル比が0.49である混合原料ガスを用い、触媒床の入口温度を558℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は522℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は297質量ppmであった。反応結果を反応条件とともに表6に示す。
メタノール/エタノールのモル比が0.50、炭素数4~6のオレフィン/メタノールのモル比が0.49である混合原料ガスを用い、触媒床の入口温度を558℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は522℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.8質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は297質量ppmであった。反応結果を反応条件とともに表6に示す。
[実施例B6]
メタノール/エタノールのモル比が0.86、炭素数4~6のオレフィン/メタノールのモル比が0.32である混合原料ガスを用い、触媒床の入口温度を541℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は539℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.9質量%、芳香族収率は7.6質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。反応結果を反応条件とともに表6に示す。
メタノール/エタノールのモル比が0.86、炭素数4~6のオレフィン/メタノールのモル比が0.32である混合原料ガスを用い、触媒床の入口温度を541℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は539℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.9質量%、芳香族収率は7.6質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。反応結果を反応条件とともに表6に示す。
[実施例B7]
メタノール/エタノールのモル比が1.33、炭素数4~6のオレフィン/メタノールのモル比が0.24である混合原料ガスを用い、触媒床の入口温度を526℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は554℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.6質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は266質量ppmであった。反応結果を反応条件とともに表6に示す。
メタノール/エタノールのモル比が1.33、炭素数4~6のオレフィン/メタノールのモル比が0.24である混合原料ガスを用い、触媒床の入口温度を526℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は554℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.6質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は266質量ppmであった。反応結果を反応条件とともに表6に示す。
[比較例B3]
エタノールを用いず、メタノールと炭素数4~6のオレフィンとを原料ガスとして用い、触媒床の入口温度を441℃に調節した以外は実施例B2と同様に反応を行った。この時、触媒床の出口温度は639℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は14.8質量%、芳香族収率は11.3質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は684質量ppmであった。反応結果を反応条件とともに表6に示す。
エタノールを用いず、メタノールと炭素数4~6のオレフィンとを原料ガスとして用い、触媒床の入口温度を441℃に調節した以外は実施例B2と同様に反応を行った。この時、触媒床の出口温度は639℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は14.8質量%、芳香族収率は11.3質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は684質量ppmであった。反応結果を反応条件とともに表6に示す。


[実施例B8]
メタノール/エタノールのモル比が0.40、炭素数4~6のオレフィン/メタノールのモル比が0.24、エチレン/エタノールのモル比が0.13である混合原料ガスを用い、触媒床の入口温度を555℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は525℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.4質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は251質量ppmであった。反応結果を反応条件とともに表7に示す。
メタノール/エタノールのモル比が0.40、炭素数4~6のオレフィン/メタノールのモル比が0.24、エチレン/エタノールのモル比が0.13である混合原料ガスを用い、触媒床の入口温度を555℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は525℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.5質量%、芳香族収率は7.4質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は251質量ppmであった。反応結果を反応条件とともに表7に示す。
[実施例B9]
メタノール/エタノールのモル比が0.46、炭素数4~6のオレフィン/メタノールのモル比が0.20、エチレン/エタノールのモル比が0.31である混合原料ガスを用い、触媒床の入口温度を544℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は536℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.8質量%、芳香族収率は7.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は280質量ppmであった。反応結果を反応条件とともに表7に示す。
メタノール/エタノールのモル比が0.46、炭素数4~6のオレフィン/メタノールのモル比が0.20、エチレン/エタノールのモル比が0.31である混合原料ガスを用い、触媒床の入口温度を544℃に調節した以外は実施例B4と同様に反応を行った。この時、触媒床の出口温度は536℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.8質量%、芳香族収率は7.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は280質量ppmであった。反応結果を反応条件とともに表7に示す。
[実施例B10]
メタノール/エタノールのモル比が0.078、エチレン/エタノールのモル比が0.26である混合原料ガスを用い、触媒床の入口温度を546℃に調節した以外は実施例B1と同様に反応を行った。この時、触媒床の出口温度は534℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.8質量%、芳香族収率は7.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は291質量ppmであった。反応結果を反応条件とともに表7に示す。
メタノール/エタノールのモル比が0.078、エチレン/エタノールのモル比が0.26である混合原料ガスを用い、触媒床の入口温度を546℃に調節した以外は実施例B1と同様に反応を行った。この時、触媒床の出口温度は534℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は19.8質量%、芳香族収率は7.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は291質量ppmであった。反応結果を反応条件とともに表7に示す。

[第3実施形態の実施例]
以下に第3実施形態の実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
以下に第3実施形態の実施例を示して、本発明をより詳細に説明するが、本発明は以下に記載の実施例によって制限されるものではない。
[エタノールの変換方法]
(反応装置)
以下実施例及び比較例は図1に示す固定床一段断熱型反応器1を用いて評価を行う。
(反応装置)
以下実施例及び比較例は図1に示す固定床一段断熱型反応器1を用いて評価を行う。
(原料)
実施例及び比較例におけるエチレン/エタノールのモル比及び炭素数4~6のオレフィン/エチレンのモル比は下式で算出する。
エチレン/エタノールのモル比(-)=エチレンモル流量(mol/hr)/エタノールモル流量(mol/hr)
炭素数4~6のオレフィン/エチレンのモル比(-)=炭素数4~6のオレフィン流量(mol/hr)/エチレンモル流量(mol/hr)
実施例及び比較例におけるエチレン/エタノールのモル比及び炭素数4~6のオレフィン/エチレンのモル比は下式で算出する。
エチレン/エタノールのモル比(-)=エチレンモル流量(mol/hr)/エタノールモル流量(mol/hr)
炭素数4~6のオレフィン/エチレンのモル比(-)=炭素数4~6のオレフィン流量(mol/hr)/エチレンモル流量(mol/hr)
(温度測定)
触媒床入口の温度及び触媒床出口の温度は反応器外部から挿入する熱電対によって測定を行う。具体的には、図1に示すように、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0.5d~0.6dにおける温度を測定する。なお、本熱電対挿入による放熱の影響は無視できるほど小さい。
触媒床入口の温度及び触媒床出口の温度は反応器外部から挿入する熱電対によって測定を行う。具体的には、図1に示すように、流体の流れ方向に垂直な平面において、反応器の中心を0とし、反応器の中心から反応器内壁面までの距離をdとすると、0.5d~0.6dにおける温度を測定する。なお、本熱電対挿入による放熱の影響は無視できるほど小さい。
(反応評価)
以下実施例及び比較例に従って、入出平均反応温度が540℃になるように反応を実施する。反応開始から3hごとに反応器出口ガスの一部をサンプリングし、ガスクロマトグラフ(TCD、FID検出器)に導入して反応ガス組成の分析を行う。反応開始から48時間後に反応を停止した。反応開始から反応停止までのGC分析結果の平均値を算出する。なお、入出平均反応温度は下式に従い算出し、表中では「反応温度」と表記する。
入出平均反応温度(℃)=[触媒床入口温度(℃)+触媒床出口温度(℃)]/2
以下実施例及び比較例に従って、入出平均反応温度が540℃になるように反応を実施する。反応開始から3hごとに反応器出口ガスの一部をサンプリングし、ガスクロマトグラフ(TCD、FID検出器)に導入して反応ガス組成の分析を行う。反応開始から48時間後に反応を停止した。反応開始から反応停止までのGC分析結果の平均値を算出する。なお、入出平均反応温度は下式に従い算出し、表中では「反応温度」と表記する。
入出平均反応温度(℃)=[触媒床入口温度(℃)+触媒床出口温度(℃)]/2
(コーク収率)
以下実施例及び比較例において、反応停止後、窒素を反応器に供給して炭化水素のパージを行い、触媒床を500℃に保つ。次いで、酸素濃度2体積%の空気/窒素を流通させ、触媒上のコークを燃焼除去する。この際、反応器出口ガスを定期的にサンプリングして、ガスクロマトグラフを用いて再生ガスの分析を行い、CO2、COの濃度を測定し、この値から触媒に付着していたカーボン量を求め、これをコーク量とする。ガスクロマトグラフを用いた再生ガス分析の方法については下記(ガスクロマトグラフの分析条件)に記載する。以下、実施例及び比較例におけるコーク収率は以下の式に従い、求める。
以下実施例及び比較例において、反応停止後、窒素を反応器に供給して炭化水素のパージを行い、触媒床を500℃に保つ。次いで、酸素濃度2体積%の空気/窒素を流通させ、触媒上のコークを燃焼除去する。この際、反応器出口ガスを定期的にサンプリングして、ガスクロマトグラフを用いて再生ガスの分析を行い、CO2、COの濃度を測定し、この値から触媒に付着していたカーボン量を求め、これをコーク量とする。ガスクロマトグラフを用いた再生ガス分析の方法については下記(ガスクロマトグラフの分析条件)に記載する。以下、実施例及び比較例におけるコーク収率は以下の式に従い、求める。
コーク収率(質量ppm)=コーク量/[有効原料供給質量流量(kg/hr)×反応時間(hr)]
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
[ガスクロマトグラフの分析条件]
(再生ガス分析)
装置:島津製作所GC-8A
カラム:以下のカラム(1)とカラム(2)を並列に連結したものを使用する。
カラム(1)80~100メッシュのモレキュラーシーブ5A(富士フイルム和光純薬製)を充填したSUS製カラム(内径3mm、長さ3m)
カラム(2)80~100メッシュの米国WATERS ASSOCIATES社製Porapac-Q(内径3mm、長さ2m)及びSUS製抵抗カラム(内径3mm、長さ1m)を直接に連結したもの
カラム温度:70℃
キャリアーガス(ヘリウム)流量:60mL/分
(再生ガス分析)
装置:島津製作所GC-8A
カラム:以下のカラム(1)とカラム(2)を並列に連結したものを使用する。
カラム(1)80~100メッシュのモレキュラーシーブ5A(富士フイルム和光純薬製)を充填したSUS製カラム(内径3mm、長さ3m)
カラム(2)80~100メッシュの米国WATERS ASSOCIATES社製Porapac-Q(内径3mm、長さ2m)及びSUS製抵抗カラム(内径3mm、長さ1m)を直接に連結したもの
カラム温度:70℃
キャリアーガス(ヘリウム)流量:60mL/分
(反応ガス分析)
装置:島津製作所製GC-2030
カラム:米国SUPELCO社製カスタムキャピラリーカラムSPB-1(内径0.25mm、長さ60m、フイルム厚3.0μm)
サンプルガス量:1mL(サンプリングラインは200℃~300℃に保温)
昇温プログラム:40℃で12分間保持し、次いで5℃/分で200℃まで昇温した後、200℃で22分間保持する。
スプリット比:200対1
キャリアーガス(窒素)流量:120mL/分
FID検出器:エアー供給圧50kPa(約500mL/分)、水素供給圧60kPa(約50mL/分)
測定方法:TCD検出器とFID検出器を直列に連結して、水素はTCD検出器で検出したデータ、炭化水素やエタノールなどの含酸素物はFID検出器で検出したデータを元に組成分析を行い、検量線法を用いて反応ガス中の目的化合物の濃度を求め、反応により生成した時間当たりの質量を求める。
装置:島津製作所製GC-2030
カラム:米国SUPELCO社製カスタムキャピラリーカラムSPB-1(内径0.25mm、長さ60m、フイルム厚3.0μm)
サンプルガス量:1mL(サンプリングラインは200℃~300℃に保温)
昇温プログラム:40℃で12分間保持し、次いで5℃/分で200℃まで昇温した後、200℃で22分間保持する。
スプリット比:200対1
キャリアーガス(窒素)流量:120mL/分
FID検出器:エアー供給圧50kPa(約500mL/分)、水素供給圧60kPa(約50mL/分)
測定方法:TCD検出器とFID検出器を直列に連結して、水素はTCD検出器で検出したデータ、炭化水素やエタノールなどの含酸素物はFID検出器で検出したデータを元に組成分析を行い、検量線法を用いて反応ガス中の目的化合物の濃度を求め、反応により生成した時間当たりの質量を求める。
(プロピレン収率)
プロピレン収率は反応におけるプロピレンへの選択性を表し、下式で算出する。
プロピレン収率(質量%)=反応により生成した時間当たりのプロピレン質量(kg/hr)/有効原料供給質量流量(kg/hr)
プロピレン収率は反応におけるプロピレンへの選択性を表し、下式で算出する。
プロピレン収率(質量%)=反応により生成した時間当たりのプロピレン質量(kg/hr)/有効原料供給質量流量(kg/hr)
有効原料供給質量流量(kg/hr)=エチレン流量(kg/hr)+エチレン換算エタノール流量(kg/hr)+炭素数4~6のオレフィン流量(kg/hr)+エタノール以外の炭素数1~6の含酸素化合物流量(kg/hr)
エチレン換算エタノール流量(kg/hr)=エタノール流量(kg/hr)×エチレン分子量(g/mol)/エタノール分子量(g/mol)
(回収収率)
プロピレン及び芳香族化合物の回収収率は下式で算出する。各成分の回収収率は、反応器に導入される原料に対する各成分の排出量を表している。排出量とは、分離工程を経て系外に排出される各成分の時間当たりの質量である。
プロピレンの回収収率(質量%)=プロピレン排出量(kg/hr)/混合原料供給質量流量(kg/hr)
芳香族化合物の回収収率(質量%)=芳香族化合物排出量(kg/hr)/混合原料供給質量流量(kg/hr)
プロピレン及び芳香族化合物の回収収率は下式で算出する。各成分の回収収率は、反応器に導入される原料に対する各成分の排出量を表している。排出量とは、分離工程を経て系外に排出される各成分の時間当たりの質量である。
プロピレンの回収収率(質量%)=プロピレン排出量(kg/hr)/混合原料供給質量流量(kg/hr)
芳香族化合物の回収収率(質量%)=芳香族化合物排出量(kg/hr)/混合原料供給質量流量(kg/hr)
[参考例1]
エチレン/エタノールのモル比が0.37である混合原料ガス(エチレン1.0kg/hr、エタノール4.6kg/hr、水蒸気0.7kg/hr)を加熱して、ゼオライト含有触媒1が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は539℃、出口温度は542℃であり、入出平均反応温度は540℃であり、反応開始から反応停止までの平均のプロピレン収率は23.2質量%、芳香族収率は2.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。
本参考例より、エチレンとエタノールのモル比率を制御することで、反応温度を制御しつつ、エチレン及びエタノールを目的化合物へと変換できることを見出した。
エチレン/エタノールのモル比が0.37である混合原料ガス(エチレン1.0kg/hr、エタノール4.6kg/hr、水蒸気0.7kg/hr)を加熱して、ゼオライト含有触媒1が充填された反応器にWHSV=3.8となるように供給し、反応を行った。この時、触媒床の入口温度は539℃、出口温度は542℃であり、入出平均反応温度は540℃であり、反応開始から反応停止までの平均のプロピレン収率は23.2質量%、芳香族収率は2.7質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は270質量ppmであった。
本参考例より、エチレンとエタノールのモル比率を制御することで、反応温度を制御しつつ、エチレン及びエタノールを目的化合物へと変換できることを見出した。
[参考例2]
エチレン/エタノールのモル比が0.82、エチレン/炭素数4~6のオレフィンのモル比が0.3である混合原料ガス(エチレン1.7kg/hr、エタノール3.4kg/hr、炭素数4~6のオレフィン1.1kg/hr、及び水蒸気1.1kg/hr)を加熱して、ゼオライト含有触媒1が充填された反応器にWHSV=4.9となるように供給し、反応を行った。この時、触媒床の入口温度は539℃、出口温度は541℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は22.5質量%、芳香族収率は13.1質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は273質量ppmであった。
エチレン/エタノールのモル比が0.82、エチレン/炭素数4~6のオレフィンのモル比が0.3である混合原料ガス(エチレン1.7kg/hr、エタノール3.4kg/hr、炭素数4~6のオレフィン1.1kg/hr、及び水蒸気1.1kg/hr)を加熱して、ゼオライト含有触媒1が充填された反応器にWHSV=4.9となるように供給し、反応を行った。この時、触媒床の入口温度は539℃、出口温度は541℃であり、入出平均反応温度は540℃であった。反応開始から反応停止までの平均のプロピレン収率は22.5質量%、芳香族収率は13.1質量%であった。また、48h運転終了後のゼオライト含有触媒のコーク収率は273質量ppmであった。
[実施例C1]
図9に示す装置により、本実施形態に係るエタノールの変換方法の効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。留分Aのリサイクル比率が0.60、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1の導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.43とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器への供給速度は、WHSV=3.8とし、触媒床の入口温度は553℃とすると、出口温度は527℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は22.1質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は251質量ppmである。分離工程の出口で得られるプロピレンの回収収率は5.4質量%、芳香族化合物の回収収率は12.3質量%である。
図9に示す装置により、本実施形態に係るエタノールの変換方法の効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。留分Aのリサイクル比率が0.60、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1の導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.43とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器への供給速度は、WHSV=3.8とし、触媒床の入口温度は553℃とすると、出口温度は527℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は22.1質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は251質量ppmである。分離工程の出口で得られるプロピレンの回収収率は5.4質量%、芳香族化合物の回収収率は12.3質量%である。
[実施例C2]
図10に示す装置により、本実施形態に係るエタノールの変換方法の効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。得られた留分Aを第2の蒸留塔8に供することで炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とに分離する。留分A-1のリサイクル比率が0.72、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.36とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は547℃、出口温度は533℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は23.5質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は235質量ppmである。分離工程の出口で得られるプロピレンの回収収率は15.5質量%、芳香族化合物の回収収率は14.0質量%である。
実施例C1と実施例C2との比較より、炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とを分離する蒸留塔を設けることでプロピレンの回収収率が向上することがわかる。
図10に示す装置により、本実施形態に係るエタノールの変換方法の効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。得られた留分Aを第2の蒸留塔8に供することで炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とに分離する。留分A-1のリサイクル比率が0.72、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.36とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は547℃、出口温度は533℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は23.5質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は235質量ppmである。分離工程の出口で得られるプロピレンの回収収率は15.5質量%、芳香族化合物の回収収率は14.0質量%である。
実施例C1と実施例C2との比較より、炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とを分離する蒸留塔を設けることでプロピレンの回収収率が向上することがわかる。
[実施例C3]
図11に示す装置により、本実施形態に係る変換方法の効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3未満の炭化水素を主に含む留分A、炭素数4以上の炭化水素を主に含む留分B、炭素数3の炭化水素を主に含む留分Eとに分離する。留分Aのリサイクル比率が0.78、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.39とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は549℃とすると、出口温度は531℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は23.8質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は240質量ppmである。分離工程の出口で得られるプロピレンの回収収率は12.1質量%、芳香族化合物の回収収率は12.0質量%である。
実施例C1と実施例C3との比較より、第1の蒸留塔2の中間部より炭素数3の炭化水素を主に含む留分Eを抜き出すサイドカット段を蒸留塔に設けることでプロピレンの回収収率が向上することがわかる。
図11に示す装置により、本実施形態に係る変換方法の効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3未満の炭化水素を主に含む留分A、炭素数4以上の炭化水素を主に含む留分B、炭素数3の炭化水素を主に含む留分Eとに分離する。留分Aのリサイクル比率が0.78、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.39とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は549℃とすると、出口温度は531℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は23.8質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は240質量ppmである。分離工程の出口で得られるプロピレンの回収収率は12.1質量%、芳香族化合物の回収収率は12.0質量%である。
実施例C1と実施例C3との比較より、第1の蒸留塔2の中間部より炭素数3の炭化水素を主に含む留分Eを抜き出すサイドカット段を蒸留塔に設けることでプロピレンの回収収率が向上することがわかる。
[比較例C1]
図12に示す、反応器1及び冷却装置6を有する装置により、変換方法を行ったときの効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cのリサイクル比率が0.67となるように、これらの留分を原料として用いる。以上のプロセスについて定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1の導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.29とする。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は534℃とすると、出口温度は546℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は23.0質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は255質量ppmである。分離工程の出口で得られるプロピレンの回収収率は4.4質量%、芳香族化合物の回収収率は12.1質量%である。
実施例C1と比較例C1との比較より、反応ガスをリサイクルする際、蒸留工程を設けることでプロピレンの回収収率が向上するとわかる。
図12に示す、反応器1及び冷却装置6を有する装置により、変換方法を行ったときの効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cのリサイクル比率が0.67となるように、これらの留分を原料として用いる。以上のプロセスについて定常運転した場合のプロピレンの収率等を表8に示す。
なお、反応器1の導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.29とする。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は534℃とすると、出口温度は546℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は23.0質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は255質量ppmである。分離工程の出口で得られるプロピレンの回収収率は4.4質量%、芳香族化合物の回収収率は12.1質量%である。
実施例C1と比較例C1との比較より、反応ガスをリサイクルする際、蒸留工程を設けることでプロピレンの回収収率が向上するとわかる。
[比較例C2]
図13に示す、反応器1を有する装置により、変換方法を行ったときの効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。反応ガスのリサイクル比率が0.65となるようにエタノールと混合することで上記原料と同様の組成の混合原料を得る。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.15とする。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は537℃とすると、出口温度は543℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は23.6質量%である。また、48h運転終了後のゼオライト含有触媒のコーク収率は310質量ppmである。分離工程の出口で得られるプロピレンの回収収率は2.8質量%、芳香族化合物の回収収率は7.4質量%である。
実施例C1、比較例C1及び比較例C2の比較より、反応ガスをリサイクルする際、蒸留工程及び冷却工程を設けることでプロピレンの回収収率が向上するとわかる。
図13に示す、反応器1を有する装置により、変換方法を行ったときの効果を検証する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。反応ガスのリサイクル比率が0.65となるようにエタノールと混合することで上記原料と同様の組成の混合原料を得る。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.40、炭素数4~6のオレフィン/エチレンのモル比が0.15とする。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は537℃とすると、出口温度は543℃であり、入出平均反応温度は540℃である。反応開始から反応停止までの平均のプロピレン収率は23.6質量%である。また、48h運転終了後のゼオライト含有触媒のコーク収率は310質量ppmである。分離工程の出口で得られるプロピレンの回収収率は2.8質量%、芳香族化合物の回収収率は7.4質量%である。
実施例C1、比較例C1及び比較例C2の比較より、反応ガスをリサイクルする際、蒸留工程及び冷却工程を設けることでプロピレンの回収収率が向上するとわかる。


[実施例C4]
反応器1に導入する原料組成を変更する以外は実施例C2と同様にして実施する。留分A-1のリサイクル比率が0.42、留分Bのリサイクル比率が0.90となるようにこれらの留分を原料として用い、上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.20、炭素数4~6のオレフィン/エチレンのモル比が0.61とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は563℃、出口温度は517℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は22.1質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は256質量ppmである。分離工程の出口で得られるプロピレンの回収収率は13.8質量%、芳香族化合物の回収収率は12.5質量%である。
反応器1に導入する原料組成を変更する以外は実施例C2と同様にして実施する。留分A-1のリサイクル比率が0.42、留分Bのリサイクル比率が0.90となるようにこれらの留分を原料として用い、上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.20、炭素数4~6のオレフィン/エチレンのモル比が0.61とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は563℃、出口温度は517℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は22.1質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は256質量ppmである。分離工程の出口で得られるプロピレンの回収収率は13.8質量%、芳香族化合物の回収収率は12.5質量%である。
[実施例C5]
反応器1に導入する原料組成を変更する以外は実施例C2と同様にして実施する。留分A-1のリサイクル比率が0.94、留分Bのリサイクル比率が0.90となるようにこれらの留分を原料として用い、上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.60、炭素数4~6のオレフィン/エチレンのモル比が0.27とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は534℃、出口温度は546℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は23.8質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は220質量ppmである。分離工程の出口で得られるプロピレンの回収収率は17.1質量%、芳香族化合物の回収収率は15.5質量%である。
反応器1に導入する原料組成を変更する以外は実施例C2と同様にして実施する。留分A-1のリサイクル比率が0.94、留分Bのリサイクル比率が0.90となるようにこれらの留分を原料として用い、上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.60、炭素数4~6のオレフィン/エチレンのモル比が0.27とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は534℃、出口温度は546℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は23.8質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は220質量ppmである。分離工程の出口で得られるプロピレンの回収収率は17.1質量%、芳香族化合物の回収収率は15.5質量%である。
[実施例C6]
留分Bのリサイクルを行わなかったこと以外は実施例C4と同様にして実施する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。得られた留分Aを第2の蒸留塔8に供することで炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とに分離する。留分A-1のリサイクル比率が0.41となるように留分A-1を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.20とする。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は563℃、出口温度は517℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は24.5質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は210質量ppmである。分離工程の出口で得られるプロピレンの回収収率は18.5質量%、芳香族化合物の回収収率は1.7質量%である。
実施例C2と実施例C6との比較より、炭素数4以上の炭化水素を主に含む留分Bをリサイクルすると、芳香族化合物の回収収率が向上することがわかる。
留分Bのリサイクルを行わなかったこと以外は実施例C4と同様にして実施する。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。得られた留分Aを第2の蒸留塔8に供することで炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とに分離する。留分A-1のリサイクル比率が0.41となるように留分A-1を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.20とする。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は563℃、出口温度は517℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は24.5質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は210質量ppmである。分離工程の出口で得られるプロピレンの回収収率は18.5質量%、芳香族化合物の回収収率は1.7質量%である。
実施例C2と実施例C6との比較より、炭素数4以上の炭化水素を主に含む留分Bをリサイクルすると、芳香族化合物の回収収率が向上することがわかる。
[実施例C7]
図10に示す装置により、原料として外部のエチレンを導入する場合、本実施形態に係る変換方法の効果を検証する。本実施例では留分A-1に含まれるエチレンに加えて、追加のエチレンを原料に外部から加える。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。得られた留分Aを第2の蒸留塔8に供することで炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とに分離する。留分A-1のリサイクル比率が0.28、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.80、炭素数4~6のオレフィン/エチレンのモル比が0.23とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は520℃、出口温度は560℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は24.5質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は321質量ppmである。分離工程の出口で得られるプロピレンの回収収率は14.8質量%、芳香族化合物の回収収率は13.4質量%である。
図10に示す装置により、原料として外部のエチレンを導入する場合、本実施形態に係る変換方法の効果を検証する。本実施例では留分A-1に含まれるエチレンに加えて、追加のエチレンを原料に外部から加える。原料をゼオライト含有触媒1が充填された反応器1に供給し反応ガスを得る。得られた反応ガスを冷却装置6に供することで炭素数6以下の炭化水素を主に含む留分Cと水及び炭素数7以上の炭化水素を主に含む留分Dとを分離する。留分Cを第1の蒸留塔2に供することで炭素数3以下の炭化水素を主に含む留分Aと炭素数4以上の炭化水素を主に含む留分Bとに分離する。得られた留分Aを第2の蒸留塔8に供することで炭素数2以下の炭化水素を主に含む留分A-1と炭素数3の炭化水素を主に含む留分A-2とに分離する。留分A-1のリサイクル比率が0.28、留分Bのリサイクル比率が0.90となるように、これらの留分を原料として用いる。以上のプロセスについて上述の参考例1及び2の結果を基に、定常運転した場合のプロピレンの収率等を表10に示す。
なお、反応器1へ導入する原料は、エチレン/エタノールモル比が0.80、炭素数4~6のオレフィン/エチレンのモル比が0.23とする。原料に含まれる留分Bからリサイクルされる成分(主に炭素数4以上の炭化水素)の組成は表9に示す。原料の反応器1への供給速度はWHSV=3.8とし、触媒床の入口温度は520℃、出口温度は560℃であり、入出平均反応温度は540℃とする。反応開始から反応停止までの平均のプロピレン収率は24.5質量%であり、48h運転終了後のゼオライト含有触媒のコーク収率は321質量ppmである。分離工程の出口で得られるプロピレンの回収収率は14.8質量%、芳香族化合物の回収収率は13.4質量%である。

本発明によれば、断熱型反応器を使用し、反応器内の温度を制御して、エタノールを収率良く目的化合物へと変換する方法を提供することができるため、プロピレン、芳香族化合物等の目的化合物を化成品の合成などに使用できるため産業上の利用可能性を有する。
1:断熱型反応器、12:反応筐体、121:断熱材、13:触媒床、131:触媒床入口、132:触媒床出口、14:反応器入口、15:反応器出口、161:第1シース熱電対、162:第2シース熱電対、2,3,4,8:蒸留塔、5:スチームクラッキング装置、6:冷却装置、7:油水分離機
Claims (56)
- エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法。
- 前記混合原料中のエチレン/エタノールのモル比が0.20~2.5である、請求項1に記載のエタノールの変換方法。
- 前記混合原料中のエチレン/エタノールのモル比が0.20~2.0である、請求項1に記載のエタノールの変換方法。
- 前記反応ガスからエチレン及びプロピレンを分離することを含む、請求項1に記載のエタノールの変換方法。
- 前記混合原料が炭素数4~6のオレフィンを含有する、請求項1に記載のエタノールの変換方法。
- 前記混合原料中の炭素数4~6のオレフィン/エチレンのモル比が3.0以下である、請求項5に記載のエタノールの変換方法。
- メタノール及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、エタノールの変換方法。
- 前記混合原料中のメタノール/エタノールのモル比が0.050~2.0である、請求項7に記載のエタノールの変換方法。
- 前記混合原料中のメタノール/エタノールのモル比が0.20~1.5である、請求項7に記載のエタノールの変換方法。
- 前記反応ガスからエチレン及びプロピレンを分離することを含む、請求項7に記載のエタノールの変換方法。
- 前記混合原料が炭素数4~6のオレフィンを含有する、請求項7に記載のエタノールの変換方法。
- 前記混合原料中の炭素数4~6のオレフィン/メタノールのモル比が3.0以下である、請求項7に記載のエタノールの変換方法。
- 前記反応ガス又は反応ガスを精製した留分の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いることを含む、請求項7に記載のエタノールの変換方法。
- エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることと、
前記反応ガスを、第1の蒸留塔により炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離することと、
前記留分Aの少なくとも一部を、前記反応工程に前記混合原料の一部としてリサイクルすることと、
を含む、エタノールの変換方法。 - 前記留分Aを、第2の蒸留塔により、炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離することを含み、
前記リサイクルすることが、前記留分A-1の少なくとも一部を、前記反応ガスを得ることにリサイクルし、前記混合原料の一部として用いることを含む、
請求項14に記載のエタノールの変換方法。 - 前記リサイクルすることが、前記留分Bの少なくとも一部を、前記反応ガスを得ることにリサイクルし、前記混合原料の一部として用いることを含む、
請求項14に記載のエタノールの変換方法。 - 前記反応ガスを冷却することにより、炭素数6以下の炭化水素を主に含む留分Cと、水及び炭素数7以上の炭化水素化合物を主に含む留分Dとに分離すること、
を含む、請求項14に記載のエタノールの変換方法。 - 前記第1の蒸留塔により分離することが、前記第1の蒸留塔にサイドカット段を設け、中間抜出流出液を得ることを含む、請求項14に記載のエタノールの変換方法。
- 前記反応ガスを、炭素数1~3の炭化水素を主に含む留分Aと、炭素数4~8の炭化水素を主に含む留分Bとに分離すること、並びに
前記留分Aからエチレン及びプロピレンを分離すること、
を含む、請求項1に記載のエタノールの変換方法。 - 前記留分Bの少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いることを含む、請求項19に記載のエタノールの変換方法。
- 前記留分Bを、炭素数4~6の脂肪族炭化水素を主に含む留分B1と、芳香族化合物を主に含む留分B2とに分離すること、及び
前記留分B1の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いること、
を含む、請求項19に記載のエタノールの変換方法。 - 前記留分Bをスチームクラッキングに付すことにより、エチレン及びプロピレンを含有するスチームクラッキング生成物を得ること、並びに
前記スチームクラッキング生成物からエチレン及びプロピレンを分離すること、
を含む、請求項19に記載のエタノールの変換方法。 - 前記反応器が固定床断熱型反応器である、請求項1に記載のエタノールの変換方法。
- 前記反応器が固定床一段断熱型反応器である、請求項1に記載のエタノールの変換方法。
- 前記触媒に付着したコークを燃焼させることを含む、請求項1に記載のエタノールの変換方法。
- 触媒床出口温度が450℃~590℃である、請求項1に記載のエタノールの変換方法。
- 触媒床入口温度が450℃~590℃である、請求項1に記載のエタノールの変換方法。
- 触媒床出口温度と、触媒床入口温度との温度差が-80K~80Kである、請求項1に記載のエタノールの変換方法。
- 前記触媒がゼオライト含有触媒である、請求項1に記載のエタノールの変換方法。
- 前記ゼオライト含有触媒が中間細孔径ゼオライトを含む、請求項29に記載のエタノールの変換方法。
- 前記ゼオライト含有触媒中のゼオライトのシリカ/アルミナのモル比が20~2000である、請求項29に記載のエタノールの変換方法。
- 前記ゼオライト含有触媒がリン元素又は銀元素を含む、請求項29に記載のエタノールの変換方法。
- エチレン及びエタノールを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得ることを含む、炭化水素の製造方法。
- 炭素数2以上の炭化水素を分解する、クラッキング工程と、
前記クラッキング工程で得られた成分を精製する精製工程と、
を有し、
前記精製工程で、請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガス又はその精製留分を合流させる、炭化水素の製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程、
を含む、モノマーの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスからオレフィンを主に含む留分を分離するオレフィン分離工程、
を含む、オレフィンの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程、
を含む、プロピレンの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程、
を含む、エチレンの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスからジエンを主に含む留分を分離するジエン分離工程、
を含む、ジエンの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスから不飽和炭化水素を主に含む留分を分離する不飽和炭化水素分離工程と、
前記不飽和炭化水素分離工程により得られた不飽和炭化水素からアクリルモノマーを得るアクリルモノマー製造工程と、
を含む、アクリルモノマーの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスからプロピレンを主に含む留分を分離するプロピレン分離工程と、
前記プロピレン分離工程により得られたプロピレンからアクリロニトリルを得るアクリロニトリル製造工程と、
を含む、アクリロニトリルの製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスからエチレンを主に含む留分を分離するエチレン分離工程と、
前記エチレン分離工程により得られたエチレンからスチレンを得るスチレン製造工程と、
を含む、スチレンの製造方法。 - 請求項35に記載の製造方法により得られるモノマーを重合する工程、
を含む、重合体の製造方法。 - 請求項36に記載の製造方法により得られるオレフィンを含む重合性組成物を重合する工程、
を含む、オレフィン系重合体の製造方法。 - 請求項37に記載の製造方法により得られるプロピレンを含む重合性組成物を重合する工程、
を含む、ポリプロピレン系重合体の製造方法。 - 請求項38に記載の製造方法により得られるエチレンを含む重合性組成物を重合する工程、
を含む、ポリエチレン系重合体の製造方法。 - 請求項39に記載の製造方法により得られるジエンを含む重合性組成物を重合する工程、
を含む、ジエン系重合体の製造方法。 - 請求項40に記載の製造方法により得られるアクリルモノマーを含む重合性組成物を重合する工程、
を含む、アクリルモノマー系重合体の製造方法。 - 請求項41に記載の製造方法により得られるアクリロニトリルを含む重合性組成物を重合する工程、
を含む、アクリロニトリル系重合体の製造方法。 - 請求項42に記載の製造方法により得られるスチレンを含む重合性組成物を重合する工程、
を含む、スチレン系重合体の製造方法。 - 請求項1~32のいずれか一項に記載のエタノールの変換方法により得られた反応ガスから芳香族化合物を主に含む留分を分離する芳香族化合物分離工程、
を含む、芳香族化合物の製造方法。 - 請求項51に記載の製造方法により得られた芳香族化合物から芳香族モノマーを得る芳香族モノマー製造工程、
を含む、芳香族モノマーの製造方法。 - 請求項52に記載の製造方法により得られる芳香族モノマーを含む重合性組成物を重合する工程、
を含む、芳香族モノマー系重合体の製造方法。 - エタノール及びエチレンを含有する混合原料を、断熱型反応器内で触媒と接触させ、炭素数3以上のオレフィンを含有する反応ガスを得る反応器と、
前記反応ガスを、炭素数2~3の炭化水素を主に含む留分Aと、炭素数4~6の炭化水素を主に含む留分Bとに分離する第1の蒸留塔と、
を備え、
前記留分Aの少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いる、エタノールの変換装置。 - 前記留分Aを、炭素数2の炭化水素を主に含む留分A-1と、炭素数3の炭化水素を主に含む留分A-2とに分離する第2の蒸留塔を備え、
前記留分A-2の少なくとも一部を前記反応器にリサイクルし、前記混合原料の一部として用いる、請求項54に記載のエタノールの変換装置。 - 前記第1の蒸留塔が、中間抜出流出液を得るためのサイドカット段を有する、請求項55に記載のエタノールの変換装置。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022-012969 | 2022-01-31 | ||
| JP2022012969 | 2022-01-31 | ||
| JP2022088596 | 2022-05-31 | ||
| JP2022-088596 | 2022-05-31 | ||
| JP2022-137041 | 2022-08-30 | ||
| JP2022137041 | 2022-08-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023145941A1 true WO2023145941A1 (ja) | 2023-08-03 |
Family
ID=87471695
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/002899 WO2023145941A1 (ja) | 2022-01-31 | 2023-01-30 | エタノールの変換方法、及びその他炭化水素の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202340124A (ja) |
| WO (1) | WO2023145941A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024070182A1 (ja) * | 2022-09-30 | 2024-04-04 | 旭化成株式会社 | エタノールの変換方法、炭化水素の製造方法、プロピレンの製造方法、芳香族化合物の製造方法、及び、エタノールの変換装置 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5436203A (en) * | 1977-08-09 | 1979-03-16 | Petroleo Brasileiro Sa | Process for preparing ethene |
| JP2007502796A (ja) * | 2003-08-19 | 2007-02-15 | トータル・ペトロケミカルズ・リサーチ・フエリユイ | オレフィンの生産 |
| JP2007290991A (ja) * | 2006-04-24 | 2007-11-08 | Idemitsu Kosan Co Ltd | 含酸素化合物からのオレフィン類の製造方法 |
| JP2007291076A (ja) * | 2006-03-30 | 2007-11-08 | Mitsubishi Chemicals Corp | プロピレンの製造方法 |
| US20110288358A1 (en) * | 2008-12-11 | 2011-11-24 | Lurgi Gmbh | Method for producing a product containing c3h6 and c2h4 |
| US20130245290A1 (en) * | 2011-09-07 | 2013-09-19 | Shell Oil Company | Process for preparing ethylene and propylene |
-
2023
- 2023-01-30 WO PCT/JP2023/002899 patent/WO2023145941A1/ja unknown
- 2023-01-31 TW TW112103226A patent/TW202340124A/zh unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5436203A (en) * | 1977-08-09 | 1979-03-16 | Petroleo Brasileiro Sa | Process for preparing ethene |
| JP2007502796A (ja) * | 2003-08-19 | 2007-02-15 | トータル・ペトロケミカルズ・リサーチ・フエリユイ | オレフィンの生産 |
| JP2007291076A (ja) * | 2006-03-30 | 2007-11-08 | Mitsubishi Chemicals Corp | プロピレンの製造方法 |
| JP2007290991A (ja) * | 2006-04-24 | 2007-11-08 | Idemitsu Kosan Co Ltd | 含酸素化合物からのオレフィン類の製造方法 |
| US20110288358A1 (en) * | 2008-12-11 | 2011-11-24 | Lurgi Gmbh | Method for producing a product containing c3h6 and c2h4 |
| US20130245290A1 (en) * | 2011-09-07 | 2013-09-19 | Shell Oil Company | Process for preparing ethylene and propylene |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024070182A1 (ja) * | 2022-09-30 | 2024-04-04 | 旭化成株式会社 | エタノールの変換方法、炭化水素の製造方法、プロピレンの製造方法、芳香族化合物の製造方法、及び、エタノールの変換装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202340124A (zh) | 2023-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DK2238094T3 (en) | DEHYDRATION OF ALCOHOLS ON CRYSTALLINIC SILICATES | |
| KR101217915B1 (ko) | 에탄올로부터의 올레핀의 제조 방법 | |
| JP3707607B2 (ja) | エチレンおよびプロピレンの製造方法 | |
| EP2244823B1 (en) | Process for the preparation of an olefinic product | |
| KR101227221B1 (ko) | 에탄올로부터의 올레핀의 제조 방법 | |
| KR20120094033A (ko) | 프로필렌 생성을 향상시키기 위한 스팀 크래커 유닛의 디보틀네킹 | |
| WO1996010548A1 (fr) | Procede de production d'hydrocarbure aromatique | |
| JP5562245B2 (ja) | オレフィンの製造方法およびその製造装置 | |
| WO2006009099A1 (ja) | エチレン及びプロピレンの製造法 | |
| JP2010533743A (ja) | オレフィンの製造方法 | |
| JPWO2009031445A1 (ja) | プロピレンの製造方法 | |
| WO2023145941A1 (ja) | エタノールの変換方法、及びその他炭化水素の製造方法 | |
| JP2008056593A (ja) | プロピレンの製造方法 | |
| JP2006008655A (ja) | プロピレンの製造方法 | |
| US20150119617A1 (en) | Process for Converting Oxygenates to Olefins | |
| WO2007019787A1 (fr) | Procede de fabrication d’olefines inferieures sous pression negative | |
| JP2023177331A (ja) | アルコールの変換方法、及び炭化水素の製造方法 | |
| WO2024070182A1 (ja) | エタノールの変換方法、炭化水素の製造方法、プロピレンの製造方法、芳香族化合物の製造方法、及び、エタノールの変換装置 | |
| JP2024052594A (ja) | エタノールの変換方法、炭化水素の製造方法、プロピレンの製造方法、芳香族化合物の製造方法、並びに、エタノールの変換装置 | |
| JP2024051654A (ja) | エタノールの変換方法、炭化水素の製造方法、プロピレンの製造方法、芳香族化合物の製造方法、並びに、エタノールの変換装置 | |
| JP2024052593A (ja) | エタノールの変換方法、炭化水素の製造方法、プロピレンの製造方法、芳香族化合物の製造方法、並びに、エタノールの変換装置 | |
| US20100130801A1 (en) | Catalytic Conversion of Alkylaromatic Compounds | |
| WO2014207134A1 (en) | An olefin cracking catalyst | |
| WO2010072725A1 (en) | Process for the preparation of an olefinic product and an oxygenate conversion catalyst | |
| US20150018591A1 (en) | Method of Converting Oxygenates to Olefins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23747151 Country of ref document: EP Kind code of ref document: A1 |