WO2023027198A1 - トリアジン誘導体を含有する経口投与する製剤 - Google Patents
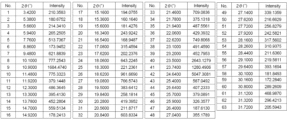
トリアジン誘導体を含有する経口投与する製剤 Download PDFInfo
- Publication number
- WO2023027198A1 WO2023027198A1 PCT/JP2022/043092 JP2022043092W WO2023027198A1 WO 2023027198 A1 WO2023027198 A1 WO 2023027198A1 JP 2022043092 W JP2022043092 W JP 2022043092W WO 2023027198 A1 WO2023027198 A1 WO 2023027198A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- vii
- acid
- compound represented
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images

























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/194—Carboxylic acids, e.g. valproic acid having two or more carboxyl groups, e.g. succinic, maleic or phthalic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Definitions
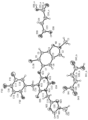
- the present invention relates to an orally administered formulation containing a triazine derivative. Specifically, it relates to an orally administered preparation containing, as an active ingredient, a triazine derivative exhibiting coronavirus 3CL protease inhibitory activity, a pharmaceutically acceptable salt thereof, or a complex thereof.
- the coronavirus which belongs to the subfamily Orthocoronavirus subfamily, Coronaviridae, order of the Nidoviridae, has a genome size of about 30 kilobases, and is the largest single-stranded + stranded RNA virus among known RNA viruses.
- Coronaviruses are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus, and there are two types of coronaviruses that infect humans: Alphacoronavirus (HCoV-229E, HCoV-229E, HCoV -NL63) and five members of the genus Betacoronavirus (HCoV-HKU1, HCoV-OC43, SARS-CoV, MERS-CoV, SARS-CoV-2).
- HCoV-229E HCoV-NL63, HCoV-HKU1, HCoV-OC43
- SARS severe acute respiratory syndrome
- MERS Middle East respiratory syndrome coronavirus
- SARS-CoV novel coronavirus
- Non-Patent Document 1 The novel coronavirus disease (COVID-19) that broke out in Wuhan, China in December 2019 spread rapidly throughout the international community, and was declared a pandemic by the WHO on March 11, 2020. As of September 21, 2022, the confirmed number of infected people reached 610 million or more, and the number of deaths reached 6.5 million or more (Non-Patent Document 1). Droplet infection, contact infection and aerosol infection have been reported as the main infection routes of SARS-CoV-2. (Non-Patent Document 2). The incubation period is about 2 to 14 days, and cold-like symptoms such as fever (87.9%), dry cough (67.7%), malaise (38.1%), and phlegm (33.4%) are typical. (Non-Patent Document 3). In severe cases, respiratory failure due to acute respiratory distress syndrome, acute lung injury, interstitial pneumonia, etc. occurs. Multiple organ failure such as renal failure and liver failure has also been reported.
- Coronaviruses synthesize two polyproteins when they infect cells. These two polyproteins contain the replication complexes that make up the viral genome, two proteases. Protease plays an essential role in cleaving polyproteins synthesized from viruses and allowing each protein to function. Of the two proteases, 3CL protease (main protease) is responsible for most of the polyprotein cleavage (Non-Patent Document 4).
- 3CL protease main protease
- PF-07304814 a prodrug of PF-00835231, at ClinicalTrials. gov
- PF-00835231 Lufotrelvir
- PF-07321332 PF-07321332:
- ClinicalTrials.com announced that a Phase 2/3 trial of PF-07321332 in combination with ritonavir will begin in COVID-19 patients with high-risk factors. gov (NCT04960202).
- PAXLOVIDTM PF-07321332; ritonavir
- Pfizer's website showed that PAXLOVIDTM (PF-07321332; ritonavir) reduced the risk of hospitalization or death by 89% compared to placebo in high-risk adults. was reported (Non-Patent Document 14).
- PAXLOVIDTM was approved for Emergency Use Authorization in the United States, and on February 10, 2022, Paxlovid® Pack was granted special approval in Japan.
- Non-Patent Documents 5 to 8 Although compounds having 3CL protease inhibitory activity are disclosed in Non-Patent Documents 5 to 8, none of the documents describes or suggests compounds, production methods, and synthetic intermediates related to the present invention. Triazine and uracil derivatives with P2X3 and/or P2X2/ 3 receptor antagonism are disclosed in US Pat. Neither description nor suggestion is made about the effect. Moreover, the production method and synthetic intermediates according to the present invention are neither described nor suggested. Non-Patent Documents 9 to 11 disclose triazine derivatives having antitumor effects, but none of these documents describe coronavirus 3CL protease inhibitory activity and antiviral effects. Relevant compounds, methods of preparation and synthetic intermediates are neither described nor suggested.
- Patent document 5 discloses a triazine derivative having galanin receptor modulating activity, but none of the documents describes or suggests 3CL protease inhibitory activity or antiviral effect. Moreover, the production method and synthetic intermediates according to the present invention are neither described nor suggested.
- the present invention relates to the following.
- the compound represented by formula (III) is represented by formula (III-1): The production method according to (1) or (2) above.
- formula (VI) (The symbols in the formula have the same meanings as above.) A method for producing a compound represented by or a salt thereof or a solvate thereof. (5) The production method according to (4) above, wherein the acid is acetic acid. (6) a compound represented by formula (VI), Formula (VII): The production method according to (4) or (5) above. (7) Formula (III): (VII), comprising obtaining a compound of formula (VII) or a salt thereof: A method for producing a compound represented by, or a salt or solvate thereof. (8) Formula (VII): or a salt thereof in the presence of fumaric acid, acetone and water.
- Formula (VII) obtained by using the production method described in any one of (1) to (7) above: The production method according to (8) above, wherein the compound represented by or a salt thereof is crystallized. (10) The production method according to (8) or (9) above, wherein the crystallization temperature is 40 to 60° C. and the crystallization time is 120 minutes or more. (11) Formula (VIII): A compound represented by or a salt thereof. (12) Formula (IX): A compound represented by or a salt thereof. (13) Formula (X): A compound represented by or a salt thereof. (14) Formula (XI): A compound represented by or a salt thereof. (15) Formula (VII): Toluene solute of the compound represented by.
- Substantially Formula (VII) Fumaric acid co-crystal form I of the compound of formula (VII), which does not contain the free form of the compound of formula (VII).
- the formulation (pharmaceutical composition) according to any one of (28) The formulation (pharmaceutical composition) according to any one of (22) to (27) above for treating and/or preventing coronavirus infection.
- (30) The formulation according to (22) above, which contains 125.0 mg of the compound represented by formula (VII) as an active ingredient.
- (32) The formulation according to (22) above, which contains 250.0 mg of the compound represented by formula (VII) as an active ingredient.
- the compound produced by the production method according to the present invention has inhibitory activity against coronavirus 3CL protease and is useful as a therapeutic and/or preventive agent for coronavirus infection. Moreover, the compound produced by the production method according to the present invention is useful as a drug substance. Furthermore, a pharmaceutical composition containing the fumaric acid co-crystal of compound (I-0005) produced by the production method according to the present invention is very useful as a therapeutic agent for novel coronavirus infection (COVID-19). .
- the production method according to the present invention is a method capable of producing the compound according to the present invention in good yield.
- the orally administered formulation (pharmaceutical composition) according to the present invention has an inhibitory activity against coronavirus 3CL protease and is useful as a therapeutic and/or preventive agent for coronavirus infection.
- FIG. 1 shows a powder X-ray diffraction pattern of fumaric acid co-crystal Form I of the compound represented by formula (VII) in Example a.
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- FIG. 2 shows a peak list of the powder X-ray analysis pattern of FIG. 1.
- FIG. Figure 2 shows the structure in the asymmetric unit of fumaric acid co-crystal Form I of the compound of formula (VII).
- FIG. 2 shows the DSC analysis results of the fumaric acid co-crystal Form I of the compound represented by formula (VII) showing the powder X-ray analysis pattern of FIG. 1.
- FIG. The horizontal axis represents temperature (°C) and the vertical axis represents heat quantity (W/g).
- FIG. 2 shows the TG/DTA analysis results of the fumaric acid co-crystal Form I of the compound represented by formula (VII) showing the powder X-ray analysis pattern of FIG. 1.
- FIG. The vertical axis indicates heat quantity ( ⁇ V) or weight change (%), and the horizontal axis indicates temperature (° C.). Cel in the figure means degrees Celsius (°C).
- FIG. 2 shows the DVS analysis results of the fumaric acid co-crystal Form I of the compound represented by formula (VII) showing the powder X-ray analysis pattern of FIG. 1.
- FIG. The crystals were stable against moisture, with no substantial weight change observed with changes in humidity. 2 shows the HPLC measurement results of compound I-005 obtained in step 4 of Example 1a.
- the pa% (peak area %) of the compound represented by formula (VII) (Compound I-005) was about 95 pa%. HPLC measurement results of compound I-005 obtained in step 4' are shown. The pa% of the compound represented by formula (VII) (Compound I-005) was about 99 pa%.
- 1 shows a powder X-ray diffraction pattern of fumaric acid co-crystal Form I of the compound represented by formula (VII) in Example b. The horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- FIG. 10 shows a peak table of the powder X-ray analysis pattern of FIG. 9; FIG. FIG.
- FIG. 2 shows a structural diagram in the asymmetric unit of fumaric acid co-crystal Form I of the compound represented by formula (VII).
- Fig. 2 shows the results of HPLC of undried crystals of the compound of formula (VII) obtained in step 5-1 of Example 1b.
- the pa% of the compound represented by formula (VII) was about 99 pa%.
- the pa% of the compound represented by formula (VII) was about 99.7 pa%, and each impurity was about 0.1 pa% or less.
- FIG. 3 shows the DSC analysis results of the fumaric acid co-crystal Form I of the compound represented by formula (VII) obtained in step 5-2 of Example 1b.
- the weight loss up to 150°C was 0.28%.
- Fig. 2 shows the HPLC results of the fumaric acid co-crystal Form I of the compound of formula (VII) obtained in step 5-2 of Example 1b.
- Fig. 2 shows the particle size distribution of the fumaric acid co-crystal Form I of the compound of formula (VII) obtained in step 5-2 of Example 1b.
- FIG. 18 shows a comparison of powder X-ray analysis patterns before and after DVS measurement. Before and after the DVS measurement, the crystal form did not change and was stable.
- 1 shows a powder X-ray diffraction pattern of the toluene solute of the compound represented by formula (VII). The horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count). 2 shows the dissolution behavior of Example 6.
- the horizontal axis represents time (minutes), and the vertical axis represents dissolution rate (%).
- 1 shows the particle size distribution of the active ingredient (fumaric acid co-crystal form I crystal of the compound of formula (VII)) used in the formulations of Examples 6A and 6B.
- Fig. 3 shows the particle size distribution of the active ingredient (fumaric acid co-crystal form I crystal of the compound of formula (VII)) used in the formulations of Examples 6C and 6D.
- the vertical axis indicates the amount of change in virus titer from baseline (log 10 (TCID 50 /mL)), and the horizontal axis indicates the evaluation time point.
- the vertical axis indicates the percentage of SARS-CoV-2 virus titer-negative persons (unit: %).
- the horizontal axis indicates the time (unit: hour) from the start of treatment. Shown is the change from baseline in the COVID-19 12-symptom total score at each time point. The vertical axis shows the amount of change from baseline in the COVID-19 12-symptom total score. The horizontal axis indicates the evaluation time points. 2 shows the dissolution behavior of Example 10.
- FIG. The horizontal axis represents time (minutes), and the vertical axis represents content-corrected dissolution rate (%).
- Halogen includes a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom. Fluorine and chlorine atoms are particularly preferred.
- Alkyl includes a linear or branched hydrocarbon group having 1 to 15 carbon atoms, preferably 1 to 10 carbon atoms, more preferably 1 to 6 carbon atoms, still more preferably 1 to 4 carbon atoms. do. For example, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl, n-heptyl, isoheptyl, n-octyl , isooctyl, n-nonyl, n-decyl and the like.
- alkyl examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl and n-pentyl. More preferred embodiments include methyl, ethyl, n-propyl, isopropyl and tert-butyl.
- C1-C4 alkyl includes a straight chain or branched hydrocarbon group having 1 to 4 carbon atoms. Examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl and the like.
- Aromatic carbocyclic group means a monocyclic or bicyclic or more cyclic aromatic hydrocarbon group. Examples include phenyl, naphthyl, anthryl, phenanthryl and the like. Six-membered aromatic carbocyclic groups include, for example, phenyl. Examples of 10-membered aromatic carbocyclic groups include naphthyl and the like. Examples of 14-membered aromatic carbocyclic groups include anthryl, phenanthryl and the like. A preferred embodiment of the "aromatic carbocyclic group” is phenyl.
- Aromatic carbocyclic ring means a ring derived from the above “aromatic carbocyclic group”.
- “Aromatic heterocyclic group” means a monocyclic or bicyclic or more aromatic cyclic group having one or more heteroatoms which are the same or different and are arbitrarily selected from O, S and N in the ring. do.
- An aromatic heterocyclic group with two or more rings includes a monocyclic or an aromatic heterocyclic group with two or more rings condensed with the ring in the above "aromatic carbocyclic group", and the bond is You may have it in any ring.
- the monocyclic aromatic heterocyclic group is preferably 5- to 8-membered, more preferably 5- or 6-membered.
- Five-membered aromatic heterocyclic groups include, for example, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, furyl, thienyl, isoxazolyl, oxazolyl, oxadiazolyl, isothiazolyl, thiazolyl, thiadiazolyl and the like.
- 6-membered aromatic heterocyclic groups include pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl and the like.
- the bicyclic aromatic heterocyclic group is preferably 8- to 10-membered, more preferably 9- or 10-membered.
- indolyl isoindolyl, indazolyl, indolizinyl, quinolinyl, isoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, naphthyridinyl, quinoxalinyl, purinyl, pteridinyl, benzimidazolyl, benzisoxazolyl, benzoxazolyl, benzoxadiazolyl, benzisothiazolyl.
- Ryl benzothiazolyl, benzothiadiazolyl, benzofuryl, isobenzofuryl, benzothienyl, benzotriazolyl, imidazopyridyl, triazolopyridyl, imidazothiazolyl, pyrazinopyridazinyl, oxazolopyridyl, thiazolopyridyl, etc. are mentioned.
- 9-membered aromatic heterocyclic groups include indolyl, isoindolyl, indazolyl, indolizinyl, purinyl, benzimidazolyl, benzisoxazolyl, benzoxazolyl, benzoxadiazolyl, benzisothiazolyl, benzothiazolyl, benzothiadiazo lyl, benzotriazolyl, benzofuranyl, imidazopyridyl, triazolopyridyl, oxazolopyridyl, thiazolopyridyl and the like.
- Ten-membered aromatic heterocyclic groups include quinolinyl, isoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, naphthyridinyl, quinoxalinyl, pteridinyl, pyrazinopyridazinyl, and the like.
- the aromatic heterocyclic group having 3 or more rings is preferably 13- to 15-membered. Examples include carbazolyl, acridinyl, xanthenyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, dibenzofuryl and the like.
- a preferred embodiment of the "aromatic heterocyclic group" is triazolyl.
- Aromatic heterocyclic ring means a ring derived from the above “aromatic heterocyclic group”.
- Substituents of "substituted alkyl” include the following Substituent Group A. A carbon atom at any position may be bonded to one or more groups selected from Substituent Group A below. Substituent group A: halogen, cyano and nitro. Substituents of the “substituted C1-C4 alkyl” include the following Substituent Group B. A carbon atom at any position may be bonded to one or more groups selected from Substituent Group B below. Substituent group B: halogen, cyano and nitro.
- substituents on the ring of "aromatic carbocyclic ring” and “aromatic heterocyclic ring” such as “substituted aromatic carbocyclic group” and “substituted aromatic heterocyclic group”, the following substituent group C mentioned. Any atom on the ring may be bonded to one or more groups selected from Substituent Group B below. Substituent group C: halogen, cyano, nitro and alkyl.
- the compounds of formula (VI) and formula (VII) are not limited to any particular isomer, but all possible isomers (e.g. keto-enol isomers, imine-enamine isomers, diastereoisomers isomers, optical isomers, rotational isomers, etc.), racemates or mixtures thereof.
- the compound represented by formula (VII) and compound I-005 include the following tautomers.
- the fumaric acid co-crystal form I of the compound represented by formula (VII), which does not contain the free form of the compound represented by, is a measuring instrument such as powder X-ray diffraction measurement of the compound represented by formula (VII). It means the fumaric acid co-crystal Form I of the compound represented by the formula (VII) in which no peak derived from the free form is detected (below the detection limit).
- formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI), formula (VII), formula (VIII), formula (IX), formula (X) ) and one or more hydrogen, carbon and/or other atoms of the compound represented by formula (XI) (hereinafter referred to as formula (VII), etc.) are each the isotopes of hydrogen, carbon and/or other atoms body can be replaced.
- isotopes examples include 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 O , 31 P, 32 P, 35 S, 18 F , 123 I and Hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, iodine and chlorine are included, as is 36 Cl.
- Compounds represented by formula (VII) and the like also include compounds substituted with such isotopes.
- the isotope-substituted compounds are also useful as pharmaceuticals, and include all radiolabeled compounds of formula (VII) and the like.
- a “radiolabeling method” for producing the “radiolabel” is also encompassed by the present invention, and the “radiolabel” is useful as a research and/or diagnostic tool in metabolic pharmacokinetic studies, binding assays.
- the crystal of the present invention may be a deuterium converter.
- the crystals of the present invention may be labeled with isotopes (eg, 3 H, 14 C, 35 S, 125 I, etc.).
- a radiolabeled compound of formula (VII) or the like can be prepared by a method well known in the art.
- a tritium-labeled compound represented by formula (VII) or the like can be prepared by introducing tritium into a specific compound represented by formula (VII) or the like through a catalytic dehalogenation reaction using tritium.
- a suitable catalyst such as Pd/C
- a compound represented by formula (VII) or the like is reacted with tritium gas with an appropriately halogenated precursor.
- Pharmaceutically acceptable salts of the compounds represented by formula (VII) and the like can be used in the preparations of the present invention.
- Pharmaceutically acceptable salts of the compound represented by formula (VII) include, for example, compounds represented by formula (VII) and the like, alkali metals (e.g., lithium, sodium, potassium, etc.), alkaline earth metals ( calcium, barium, etc.), magnesium, transition metals (e.g., zinc, iron, etc.), ammonia, organic bases (e.g., trimethylamine, triethylamine, dicyclohexylamine, ethanolamine, diethanolamine, triethanolamine, meglumine, ethylenediamine, pyridine, picoline, quinoline, etc.) and salts with amino acids, or inorganic acids (e.g., hydrochloric acid, sulfuric acid, nitric acid, carbonic acid, hydrobromic acid, phosphoric acid, hydroiodic acid, etc.), and organic acids (e.g., formic
- a pharmaceutically acceptable salt of a compound of formula (VII) is, for example, a compound of formula (VII) and a counter molecule or counter ion, containing any number of counter molecules or counter ions. Also good.
- a pharmaceutically acceptable salt of a compound of formula (VII) refers to an ionic bond via proton transfer between the compound and a counter-molecule or counter-atom.
- a complex of the compound represented by formula (VII) or a pharmaceutically acceptable salt thereof can be used in the formulation of the present invention.
- Compounds of formula (VII) or pharmaceutically acceptable salts thereof may form solvates (e.g., hydrates, etc.), co-crystals and/or clathrates, and are herein They are described as "complexes”.
- solvate used herein may be coordinated with any number of solvent molecules (eg, water molecules, etc.) with respect to the compound represented by, for example, formula (VII).
- solvent molecules eg, water molecules, etc.
- the compound represented by formula (VII) or the like or a pharmaceutically acceptable salt thereof When the compound represented by formula (VII) or the like or a pharmaceutically acceptable salt thereof is left in the atmosphere, it may absorb water, attach adsorbed water, or form a hydrate.
- Solvent molecules include, for example, acetonitrile, chlorobenzene, chloroform, cyclohexane, 1,2-dichloroethene, dichloromethane, 1,2-dimethoxyethane, N,N-dimethylacetamide, N,N-dimethylformamide, 1,4-dioxane , 2-ethoxyethanol, ethylene glycol, formamide, hexane, methanol, 2-methoxyethanol, methylbutylketone, methylcyclohexane, N-methylpyrrolidone, nitromethane, pyridine, sulfolane, tetralin, toluene, 1,1,2-trichloroethene , xylene, acetic acid, anisole, 1-butanol, 2-butanol, n-butyl acetate, t-butyl methyl ether, cumene, dimethyl sulfoxide, ethyl
- co-crystal means a regular arrangement of counter-molecules within the same crystal lattice, and may contain any number of counter-molecules.
- a co-crystal refers to an intermolecular interaction between a compound and a counter molecule through non-covalent and non-ionic chemical interactions such as hydrogen bonding and van der Waals forces.
- a co-crystal of the compound represented by formula (VII) is composed of the compound represented by formula (VII) and a counter molecule, and may contain any number of counter molecules.
- it consists of the compound of formula (VII) and fumaric acid, and may contain any number of fumaric acids.
- the compound of formula (VII) and fumaric acid are co-crystals in a 1:1 molar ratio.
- Co-crystals are distinguished from salts in that the compounds remain essentially uncharged or neutral.
- Co-crystals are distinguished from hydrates or solvates in that the counter-molecule is not water or solvent.
- Formula (I), Formula (II), Formula (III), Formula (IV), Formula (V), Formula (VI), Formula (VII), Formula (VIII), Formula (IX), Formula (IX) of the present invention X) and the compound represented by formula (XI) or a salt thereof may form solvates (e.g., hydrates, etc.), co-crystals and/or crystal polymorphs, and the present invention provides such Various solvates, co-crystals and polymorphs are also included.
- “Solvate” refers to formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI), formula (VII), formula (VIII), formula (IX) ), formula (X), and formula (XI) may be coordinated with any number of solvent molecules (eg, water molecules, etc.). Further, formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI), formula (VII), formula (VIII), formula (IX), formula (X) ) and the compound of formula (XI) or a salt thereof may form polymorphs upon recrystallization.
- crystal means a solid in which constituent atoms, ions, molecules, etc. are arranged regularly in three dimensions, and is distinguished from amorphous solids that do not have such a regular internal structure. be. Crystals of the present invention may be single crystals, twin crystals, polycrystals, and the like. Furthermore, “crystals” may have “crystal polymorphs” that have the same composition but different arrangements in the crystal, and these are collectively referred to as “crystal forms”.
- formula (I), formula (II), formula (III), formula (IV), formula (V), formula (VI), formula (VII), formula (VIII), formula (IX), formula ( X) and compounds of formula (XI) may be converted into salts or pharmaceutically acceptable solvates thereof.
- the crystals of the present invention may be salts, hydrates, solvates, or crystal polymorphs thereof, and mixtures of two or more of them are intended to be included within the scope of the invention. be done.
- Crystalline morphology and crystallinity can be measured by a number of techniques including, for example, powder X-ray diffraction measurements, Raman spectroscopy, infrared spectroscopy, moisture adsorption-desorption measurements, differential scanning calorimetry, dissolution properties. can.
- a "polymorph” may be formed by recrystallization of a compound represented by formula (VII) or the like, a pharmaceutically acceptable salt thereof, or a complex thereof.
- Various salts, complexes (hydrates, solvates, co-crystals, clathrates), crystal polymorphs, and mixtures of two or more thereof can be used in the preparations of the present invention. can also be used.
- X-ray powder diffraction X-ray powder diffraction
- XRPD X-ray powder diffraction
- a crystalline form of a compound represented by formula (VII) or the like can be identified by powder X-ray diffraction patterns and characteristic diffraction peaks.
- the crystalline form of the compound represented by formula (VII) etc. can be distinguished from other crystalline forms by the presence of characteristic diffraction peaks.
- a characteristic diffraction peak is a peak selected from an observed diffraction pattern.
- the characteristic diffraction peaks are preferably selected from about 10, more preferably about 5, even more preferably about 3 in the diffraction pattern.
- the peak that is confirmed in the crystal and not confirmed in other crystals is a characteristic peak that is preferable for identifying the crystal.
- One or even two such characteristic peaks can characterize the crystal.
- the diffraction angle (2 ⁇ ) in powder X-ray diffraction can have an error within the range of ⁇ 0.2°, so the value of the diffraction angle in powder X-ray diffraction is within the range of about ⁇ 0.2°.
- the present invention includes not only crystals in which the diffraction angles of peaks in powder X-ray diffraction completely match, but also crystals in which the diffraction angles of peaks match with an error of about ⁇ 0.2°.
- Single crystal structure analysis It is one of the methods for identifying a crystal, and it is possible to obtain crystallographic parameters of the crystal, as well as atomic coordinates (values indicating the spatial positional relationship of each atom) and a three-dimensional structure model. See Toshio Sakurai, "A Guide to X-ray Structural Analysis,” published by Shokabo (1983), Stout & Jensen, X-Ray Structure Determination: A Practical Guide, Macmillan Co., New York (1968). Single crystal structure analysis is useful for identifying the crystal structures of complexes, salts, optical isomers, tautomers, and geometric isomers of the present invention.
- a compound represented by formula (VII) or the like has coronavirus 3CL protease inhibitory activity, and is therefore useful as a therapeutic and/or prophylactic agent for diseases associated with coronavirus 3CL protease.
- the term "therapeutic agent and/or prophylactic agent” also includes symptom improving agents.
- Diseases involving coronavirus 3CL protease include viral infections, preferably coronavirus infections.
- coronaviruses include coronaviruses that infect humans.
- Coronaviruses that infect humans include HCoV-229E, HCoV-NL63, HCoV-HKU1, HCoV-OC43, SARS-CoV, MERS-CoV, and/or SARS-CoV-2.
- coronaviruses include alphacoronaviruses and/or betacoronaviruses, more preferably betacoronaviruses, and even more preferably sarvecoviruses.
- alphacoronaviruses include HCoV-229E and HCoV-NL63. Particularly preferred is HCoV-229E.
- betacoronaviruses include HCoV-HKU1, HCoV-OC43, SARS-CoV, MERS-CoV, and/or SARS-CoV-2. HCoV-OC43 or SARS-CoV-2 is preferred, and SARS-CoV-2 is particularly preferred.
- the betacoronavirus includes betacoronavirus A strain ( ⁇ -coronavirus lineage A), betacoronavirus B strain ( ⁇ -coronavirus lineage B), and betacoronavirus C strain ( ⁇ -coronavirus lineage C). is mentioned. More preferred are ⁇ -coronavirus lineage A and ⁇ -coronavirus lineage B, particularly preferably ⁇ -coronavirus lineage B.
- Betacoronavirus lineage A includes, for example, HCoV-HKU1 and HCoV-OC43, preferably HCoV-OC43.
- Betacoronavirus lineage B includes, for example, SARS-CoV and SARS-CoV-2, preferably SARS-CoV-2.
- the beta-coronavirus lineage B preferably includes MERS-CoV.
- coronaviruses include HCoV-229E, HCoV-OC43 and/or SARS-CoV-2, particularly preferably SARS-CoV-2.
- Coronavirus infections include infections by HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1, SARS-CoV, MERS-CoV, and/or SARS-CoV-2.
- infections caused by HCoV-229E, HCoV-OC43 and/or SARS-CoV-2 particularly preferably infections caused by SARS-CoV-2.
- a novel coronavirus infection (COVID-19) is particularly preferred as the coronavirus infection.
- Step 1 Method for producing a compound represented by formula (III)
- This step is a method for producing a compound represented by formula (III), characterized by reacting a compound represented by formula (I) with a compound represented by formula (II) in the presence of an acid.
- the compound represented by formula (II) can be used usually in an amount of 1.0 to 5.0 equivalents, for example 1.0 to 3.0 equivalents, relative to the compound represented by formula (I).
- the solvent is not particularly limited as long as it allows the above steps to proceed efficiently, and an acid may be used as the solvent.
- Acids include protonic acids and Lewis acids, preferably trifluoroacetic acid.
- the amount of the acid to be used is generally 1.0 equivalents to a large excess, for example 5.0 equivalents to a large excess, relative to the compound represented by formula (I).
- the reaction temperature is not particularly limited, it can be generally carried out at about 0°C to about 50°C, preferably room temperature to 40°C.
- the reaction time is not particularly limited, it is usually 0.1 to 12 hours, preferably 0.1 to 5 hours.
- Step 2 Method for producing a compound represented by formula (VI)
- This step is a method for producing a compound represented by formula (VI), characterized by reacting a compound represented by formula (IV) with a compound represented by formula (V) in the presence of an acid.
- the compound represented by formula (V) can be used in an amount of usually 1.0 to 5.0 equivalents, for example 1.0 to 1.5 equivalents, relative to the compound represented by formula (IV).
- the solvent is not particularly limited as long as it allows the above steps to proceed efficiently, and an acid may be used as the solvent. Toluene, t-butanol, t-amyl alcohol and the like can be mentioned, and can be used alone or in combination. Toluene is preferred.
- Acids include acetic acid, 2,2-dimethylbutanoic acid and the like. Acetic acid is preferred.
- the amount of acid to be used is generally 1.0 to 10 equivalents, for example, 3.0 to 10 equivalents, relative to the compound represented by formula (IV).
- the reaction temperature is not particularly limited, but usually room temperature to about 150° C. or under microwave irradiation, preferably 50 to 150° C. or under microwave irradiation.
- the reaction time is not particularly limited, it is usually 0.1 to 12 hours, preferably 3 to 10 hours.
- Step 3 Method for preparing fumaric acid co-crystal form I of the compound of formula (VII) This step is characterized by crystallizing the compound of formula (VII) in the presence of fumaric acid, acetone and water. , a method for preparing the fumaric acid co-crystal Form I of the compound of formula (VII).
- the amount of fumaric acid to be used is generally 1.0 equivalent to 3.0 equivalents, for example, 1.0 equivalent to 1.5 equivalents, relative to the compound represented by formula (VII).
- the crystallization temperature is not particularly limited, it is usually 40 to 80°C, preferably 40 to 60°C.
- the crystallization time is not particularly limited, it is usually 1 hour or longer, preferably 2 hours or longer, and more preferably 2 to 12 hours.
- the reaction may be carried out in the presence of acetone and water, preferably at a ratio of acetone and water of 85:15 to 50:50.
- a compound represented by formula (VII), a pharmaceutically acceptable salt thereof or a complex thereof (hereinafter referred to as a compound represented by formula (VII), etc.), and a compound produced by the production method according to the present invention Since (compounds represented by formula (VII), etc.) have coronavirus 3CL protease inhibitory activity, they are useful as therapeutic and/or prophylactic agents for viral infections. Furthermore, the compound produced by the production method according to the present invention is useful as a medicine, and preferably has one or more of the following excellent characteristics. a) It has a weak inhibitory effect on CYP enzymes (eg, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, etc.).
- CYP enzymes eg, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, etc.
- b) show good pharmacokinetics such as high bioavailability and moderate clearance; c) high metabolic stability; d) Does not exhibit irreversible inhibitory action on CYP enzymes (eg, CYP3A4) within the concentration range of the measurement conditions described herein. e) not mutagenic; f) low cardiovascular risk; g) exhibit high solubility; h) High protein non-binding rate (fu value). i) have high coronavirus 3CL protease selectivity; j) It has high coronavirus growth inhibitory activity. For example, it has high coronavirus growth inhibitory activity under the addition of human serum (HS) or human serum albumin (HSA).
- HS human serum
- HSA human serum albumin
- coronavirus growth inhibitors include embodiments in which EC 50 is 10 ⁇ M or less, preferably 1 ⁇ M or less, and more preferably 100 nM or less in the CPE suppression effect confirmation test (SARS-CoV-2) described later.
- the salt, crystal, complex, or co-crystal of the compound according to the present invention is useful as a medicine, and preferably has one or more of the following excellent characteristics.
- bb) It exhibits good pharmacokinetics, such as high bioavailability, moderate clearance, high AUC, and high peak blood concentration.
- gg exhibit high solubility, high chemical stability and low hygroscopicity;
- a pharmaceutical composition containing a compound represented by formula (VII) can be administered either orally or parenterally.
- parenteral administration methods include transdermal, subcutaneous, intravenous, intraarterial, intramuscular, intraperitoneal, transmucosal, inhalation, nasal, ocular, ear and intravaginal administration.
- internal solid preparations e.g., tablets, powders, granules, capsules, pills, films, etc.
- internal liquid preparations e.g., suspensions, emulsions, elixirs, syrups, etc.
- Tablets may be sugar-coated tablets, film-coated tablets, enteric-coated tablets, sustained-release tablets, troches, sublingual tablets, buccal tablets, chewable tablets or orally disintegrating tablets, and powders and granules may be dry syrups.
- the capsules may be soft capsules, microcapsules or sustained release capsules.
- injections In the case of parenteral administration, injections, drops, external preparations (e.g., eye drops, nasal drops, ear drops, aerosols, inhalants, lotions, injections, coatings, gargles, enemas, Any commonly used dosage form such as ointments, plasters, jellies, creams, patches, poultices, powders for external use, suppositories, etc.) can be suitably administered. Injections may be emulsions such as O/W, W/O, O/W/O and W/O/W types.
- excipients suitable for the dosage form in an effective amount of the compound represented by formula (VII) (for example, the compound produced by the production method according to the present invention)
- Pharmaceutical excipients can be mixed as necessary to form a pharmaceutical composition.
- the pharmaceutical composition can be used as a pharmaceutical composition for children, the elderly, critically ill patients, or for surgery.
- a pediatric pharmaceutical composition can be used for neonates (less than 4 weeks after birth), infants (4 weeks after birth to less than 1 year old) infants (1 to 7 years old), children (7 to 15 years old) or 15 Patients between the ages of 18 and 18 can be administered.
- geriatric pharmaceutical compositions may be administered to patients 65 years of age or older.
- a pharmaceutical composition containing a compound represented by formula (VII), etc. e.g., a compound produced by the production method according to the present invention
- the dosage of the pharmaceutical composition is preferably set in consideration of the patient's age, body weight, type and degree of disease, administration route, etc., but when administered orally, it is usually 0.05 to 200 mg / kg / day, preferably within the range of 0.1 to 100 mg/kg/day. In the case of parenteral administration, it is generally 0.005 to 200 mg/kg/day, preferably 0.01 to 100 mg/kg/day, although it varies greatly depending on the route of administration. It may be administered once to several times a day.
- the compound represented by formula (VII) and the like are used for the purpose of enhancing the action of the compound or reducing the dosage of the compound, for example, other Used in combination with a therapeutic drug for novel coronavirus infection (COVID-19) (the therapeutic drug includes an approved drug and a drug under development or to be developed in the future) (hereinafter referred to as a concomitant drug) may
- a therapeutic drug for novel coronavirus infection COVID-19
- the therapeutic drug includes an approved drug and a drug under development or to be developed in the future
- a concomitant drug used in combination with a therapeutic drug for novel coronavirus infection (COVID-19)
- the therapeutic drug includes an approved drug and a drug under development or to be developed in the future
- a concomitant drug used in combination with a therapeutic drug for novel coronavirus infection (COVID-19)
- the therapeutic drug includes an approved drug and a drug under development or to be developed in the future
- the dosage of the concomitant drug can be appropriately selected based on the clinically used dosage.
- the compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination, and the like. For example, when the subject of administration is a human, 0.01 to 100 parts by weight of the concomitant drug may be used per 1 part by weight of the compound of the present invention.
- the formulation of the present invention may be a formulation for oral administration.
- internal solid preparations e.g., tablets, powders, granules, dry syrups, capsules, pills, films, etc.
- internal liquid preparations e.g., suspensions, emulsions, elixirs, etc.
- Tablets may be sugar-coated tablets, film-coated tablets, enteric-coated tablets, sustained-release tablets, troches, sublingual tablets, buccal tablets, chewable tablets or orally disintegrating tablets, and powders and granules may be dry syrups.
- the capsules may be soft capsules, microcapsules or sustained release capsules.
- Orally administered solid preparations or suspensions are preferred, orally administered solid preparations are more preferred, and tablets and granules are particularly preferred.
- any shape can be adopted as the shape of the tablet, and specifically, it can be a round, oval, spherical, rod-shaped, or doughnut-shaped tablet. Moreover, a laminated tablet, a dry-coated tablet, or the like may be used, but a single-layer tablet, which is easy to produce, is preferable. In addition, markings such as marks and letters may be added to improve distinguishability, and dividing lines may be added.
- the formulation of the present invention can be prepared for children, the elderly, severely ill patients, or for surgery by appropriately changing the effective amount, dosage form, and/or various pharmaceutical additives of the compound represented by formula (VII). It can also be a formulation of
- a pediatric pharmaceutical composition can be used for neonates (less than 4 weeks after birth), infants (4 weeks after birth to less than 1 year old) infants (1 to 7 years old), children (7 to 15 years old) or 15 Patients between the ages of 18 and 18 can be administered.
- geriatric formulations may be administered to patients 65 years of age or older.
- the dosage of the formulation of the present invention is determined in consideration of the patient's age, body weight, type and degree of disease, administration route, etc. It is desirable to set the dosage, but in the case of oral administration, it is usually 0.05-200 mg/kg/day, preferably 0.1-100 mg/kg/day. In the case of parenteral administration, it is generally 0.005 to 200 mg/kg/day, preferably 0.01 to 100 mg/kg/day, although it varies greatly depending on the route of administration. It may be administered once to several times a day.
- the weight of the compound represented by formula (VII) contained in the formulation of the present invention is not particularly limited as long as it is easy for patients to take and can be manufactured into a formulation, but is 1 to 450 mg, preferably 5 ⁇ 350 mg, more preferably 25-250 mg.
- the weight of the compound represented by formula (VII) contained in one tablet, one capsule or one package is 25 mg, 50 mg, 75 mg, 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg or 250 mg. is.
- 25 mg indicates a range of 22.5 mg to 27.5 mg, preferably 23.7 mg to 26.3 mg
- 50 mg indicates a range of 45.0 mg to 55.0 mg, preferably 47.5 mg to 52 mg.
- 75 mg indicates a range of 67.5 mg to 82.5 mg, preferably 71.2 mg to 78.8 mg
- 100 mg indicates a range of 90.0 mg to 110.0 mg, preferably represents a range of 95.0 mg to 105.0 mg, 125 mg represents a range of 112.5 mg to 137.5 mg, preferably 118.7 mg to 131.3 mg
- 150 mg represents a range of 135.0 mg to 165.0 mg , preferably 142.5 mg to 157.5 mg, 175 mg is 157.5 mg to 192.5 mg, preferably 166.2 to 183.8 mg, .0 mg, preferably 190.0 mg to 210.0 mg
- 225 mg is 202.5 mg to 247.5 mg, preferably 213.7 mg to 236.3 mg
- 250 mg is 225.0 mg -275.0 mg, preferably from 237.5 mg to 262.5 mg.
- the compound represented by formula (VII) is used for the purpose of enhancing the action of the compound or reducing the dosage of the compound, for example, other therapeutic agents for novel coronavirus infection (COVID-19) (the treatment
- the drug may be used in combination with an approved drug and a drug under development or to be developed in the future (hereinafter referred to as concomitant drug).
- the timing of administration of the compound represented by formula (VII) and the concomitant drug are not limited, and they may be administered to the subject at the same time or at different times.
- the compound represented by formula (VII) and the concomitant drug may be administered as two or more formulations containing each active ingredient, or administered as a single formulation containing those active ingredients. good too.
- the dosage of the concomitant drug can be appropriately selected based on the clinically used dosage.
- the compounding ratio of the compound represented by the formula (VII) or the like and the concomitant drug can be appropriately selected according to the subject of administration, administration route, target disease, symptom, combination, and the like. For example, when the subject of administration is a human, 0.01 to 100 parts by weight of the concomitant drug may be used per 1 part by weight of the compound represented by the formula (VII) or the like.
- the formulation of the present invention may contain a polymer, and as the polymer, a polymer listed in the Japanese Pharmacopoeia, Japanese Non-Pharmacopoeia Pharmaceutical Standards, Pharmaceutical Additives Standards, Food Additives Standards, etc. is used. be able to.
- a formulation containing a compound of formula (VII), a pharmaceutically acceptable salt thereof or a complex thereof, and a polymer can improve the solubility of the compound of formula (VII) in the formulation .
- Polymers include, for example, cellulose-based polymers, acrylic-based polymers, vinyl-based polymers, polysaccharides, and the like.
- Cellulosic polymers include hydroxypropylcellulose (HPC), hypromellose (hydroxypropylmethylcellulose) (HPMC), hydroxyethylcellulose, hydroxyethylmethylcellulose, hydroxypropylmethylcellulose acetate succinate, hydroxypropylmethylcellulose, phthalate, methylcellulose (MC), methyl Hydroxyethyl cellulose carboxymethyl ethyl cellulose, ethyl cellulose, crystalline cellulose, microcrystalline cellulose, crystalline cellulose, carmellose sodium, carmellose, carmellose sodium, carmellose calcium, powdered cellulose, low-substituted hydroxypropyl cellulose, fumaric acid, stearic acid, polyvinyl acetal A mixture of diethylaminoacetate/hydroxypropylmethylcellulose and the like can be mentioned.
- Acrylic polymers include aminoalkyl acrylate copolymer E, polyvinyl acetal diethylaminoacetate, ethyl acrylate/methyl methacrylate copolymer dispersion, aminoalkyl methacrylate copolymer, methacrylic acid copolymer, 2-methyl-5-vinylpyridine methyl acrylate/methacrylic Acid copolymers, dry methacrylic acid copolymers, dimethylaminoethyl methacrylate-methyl methacrylate copolymers, and the like.
- Vinyl polymers include polyvinylpyrrolidone, polyvinyl alcohol, polyvinyl alcohol/methyl methacrylate/acrylic acid copolymer, crospovidone, carboxyvinyl polymer, polyvinyl acetal diethylaminoacetate, and polyvinyl alcohol copolymer. Moreover, pullulan etc. are mentioned as a polysaccharide. Preferred are cellulose-based polymers, acrylic-based polymers and/or vinyl-based polymers, more preferred are cellulose-based polymers, and even more preferred is hypromellose (hydroxypropylmethylcellulose).
- an orally administered tablet containing a compound represented by formula (VII), a pharmaceutically acceptable salt thereof, or a complex thereof as an active ingredient, and a cellulosic polymer is preferred.
- one or more additives selected from the group consisting of excipients, binders, disintegrants and lubricants may be used.
- excipient also referred to as a filler
- excipients listed in the Japanese Pharmacopoeia, Japanese Non-Pharmacopoeia Pharmaceuticals Standards, Pharmaceutical Additives Standards, Food Additives Standards, or the like can be used.
- excipients include sugar derivatives, starch derivatives, cellulose derivatives, inorganic excipients, ⁇ -cyclodextrin, magnesium stearate, calcium stearate, sucrose fatty acid ester, crospovidone, soybean lecithin, tragacanth powder, arabic gum, dextran, pullulan and the like.
- sugar derivatives include sugars and sugar alcohols.
- sugars include lactose, sucrose, glucose, fructose, and sucrose.
- sugar alcohols include mannitol, sorbitol, erythritol, xylitol, powdered maltose syrup, maltitol and the like.
- starch derivatives include starch, potato starch, corn starch (corn starch), rice starch, partially pregelatinized starch, pregelatinized starch, perforated starch, sodium carboxystarch, hydroxypropyl starch, low-substituted sodium carboxymethylstarch, and the like. be done.
- Cellulose derivatives include crystalline cellulose, powdered cellulose, carmellose sodium, carmellose, croscarmellose sodium, carmellose calcium, carboxymethylethylcellulose, low-substituted hydroxypropylcellulose and the like.
- Inorganic excipients include silicate derivatives, phosphates, carbonates, sulfates, magnesium oxide, titanium oxide, calcium lactate, synthetic hydrotalcite, talc, kaolin, dried aluminum hydroxide, magnesium oxide, bentonite. etc.
- Examples of silicate derivatives include hydrous silicon dioxide, silicon dioxide such as light anhydrous silicic acid, magnesium aluminometasilicate, synthetic aluminum silicate, and calcium silicate.
- Phosphates include anhydrous calcium hydrogen phosphate, calcium monohydrogen phosphate, calcium hydrogen phosphate, sodium hydrogen phosphate, dipotassium phosphate, potassium dihydrogen phosphate, calcium dihydrogen phosphate, and sodium dihydrogen phosphate. etc.
- Carbonates include precipitated calcium carbonate, calcium carbonate, magnesium carbonate, and the like. Calcium sulfate etc. are mentioned as a sulfate. These excipients may be used by mixing two or more kinds in an appropriate ratio. Excipients in the formulations of the present invention are preferably mannitol and/or croscarmellose sodium.
- a binder may be used in the formulation of the present invention, and a binder listed in the Japanese Pharmacopoeia, the Japanese Pharmacopoeia Standards for Pharmaceutical Products, the Pharmaceutical Additives Standards, the Food Additives Codex, or the like can be used.
- binders include cellulose-based binders, starch-based binders, vinyl-based binders, polyethers, gum arabic, gum arabic powder, gum arabic powder, alginic acid, sodium alginate, sucrose, gelatin, dextrin, pullulan, Tragacanth, tragacanth powder, xanthan gum, pectin, sodium polyacrylate, agar, oak powder, guar gum, light anhydrous silicic acid, hydrogenated oil and the like.
- Cellulosic binders include carboxymethylcellulose (carmellose, CMC), carboxymethylcellulose sodium (carmellose sodium), hydroxyethylcellulose (HEC), hydroxypropylcellulose (HPC), hydroxypropylmethylcellulose (hypromellose) (HPMC), methylcellulose (MC ), crystalline cellulose, microcrystalline cellulose, ethyl cellulose, crystalline cellulose/carmellose sodium, carmellose calcium, powdered cellulose, low-substituted hydroxypropyl cellulose, and the like.
- Starch binders include starch, pregelatinized starch, partially pregelatinized starch, potato starch, wheat starch, rice starch, perforated starch, corn starch, hydroxypropyl starch, sodium starch glycolate (sodium carboxymethyl starch), etc. mentioned.
- vinyl-based binders examples include polyvinyl alcohol (PVA), polyvinylpyrrolidone (povidone) (PVP), carboxyvinyl polymer, and copolyvidone.
- Polyethers include macrogol (polyethylene glycol) 200, macrogol 300, macrogol 400, macrogol 600, macrogol 1000, macrogol 1500, macrogol 1540, macrogol 4000, macrogol 6000, macrogol 20000, glycerin. , polyoxyethylene [105] polyoxypropylene [5] glycol, propylene glycol, and the like.
- the binder in the formulations of the invention is preferably hydroxypropylcellulose (HPC).
- a disintegrant may be used in the formulation of the present invention, and a disintegrant listed in the Japanese Pharmacopoeia, Japanese Pharmaceutical Standards Outside the Japanese Pharmacopoeia, Pharmaceutical Excipients Standards, Food Additives Codex, etc. can be used.
- disintegrants include cellulose-based disintegrants, starch-based disintegrants, vinyl-based disintegrants, and magnesium aluminometasilicate.
- Cellulose-based disintegrants include carmellose, carmellose calcium, carmellose sodium, hydroxypropylcellulose, low-substituted hydroxypropylcellulose, croscarmellose sodium (Ac-Di-Sol), crystalline cellulose, powdered cellulose, and the like.
- Starch-based disintegrants include partially pregelatinized starch, potato starch, corn starch, hydroxypropyl starch, carboxymethyl starch sodium, low-substituted carboxymethyl starch sodium, sodium starch glycolate, pregelatinized starch, starch, and the like.
- vinyl-based disintegrants examples include crospovidone and polyvinyl alcohol.
- Two or more of these disintegrants may be mixed in an appropriate ratio and used.
- Super disintegrants include carmellose calcium, low-substituted hydroxypropylcellulose, croscarmellose sodium, crospovidone, sodium starch glycolate and the like.
- super disintegrants may be used by mixing two or more kinds in an appropriate ratio. Also, disintegrants and superdisintegrants may be combined.
- the disintegrant in the formulation of the present invention is preferably croscarmellose sodium.
- the formulation of the present invention may contain a lubricant, and a lubricant listed in the Japanese Pharmacopoeia, Japanese Non-Pharmacopoeia Pharmaceutical Standards, Pharmaceutical Additives Standards, Food Additives Codex, etc. should be used. can be done.
- lubricants include stearic acid and stearic acid metal salts, inorganic lubricants, hydrophobic lubricants, hydrophilic lubricants, sodium stearyl fumarate, and the like.
- stearic acid and stearic acid metal salts examples include magnesium stearate, calcium stearate, stearic acid, stearyl alcohol, polyoxyl 40 stearate, and the like.
- Inorganic lubricants include talc, light anhydrous silicic acid, hydrated silicon dioxide, magnesium carbonate, precipitated calcium carbonate, dried aluminum hydroxide gel, magnesium aluminometasilicate, magnesium silicate, synthetic aluminum silicate, magnesium oxide, Magnesium sulfate etc. are mentioned.
- Hydrophobic lubricants include cacao butter, carnauba wax, glycerin fatty acid ester, hydrogenated oil, bleached beeswax, hydrogenated soybean oil, beeswax, cetanol, sodium laurate, and the like.
- hydrophilic lubricants examples include sucrose fatty acid esters and polyethylene glycol (macrogol).
- the binder in the formulations of the invention is preferably magnesium stearate.
- the formulation of the present invention may contain a fluidizing agent, and the fluidizing agent listed in the Japanese Pharmacopoeia, Japanese Non-Pharmacopoeia Pharmaceutical Standards, Pharmaceutical Excipients Standards, Food Additives Codex, etc. should be used. can be done.
- fluidizing agents include silicon dioxide, stearic acid and its metal salts, crystalline cellulose, synthetic aluminum silicate, titanium oxide, heavy silicic anhydride, magnesium aluminate hydroxide, tribasic calcium phosphate, talc, corn starch, metasilicic acid.
- silicon dioxide examples include hydrous silicon dioxide and light anhydrous silicic acid.
- Stearic acid and metal salts thereof include stearic acid, calcium stearate, magnesium stearate and the like.
- the binder in the formulation of the present invention is preferably crystalline cellulose.
- the method for producing granules among the formulations of the present invention is not particularly limited. Specifically, additives such as active ingredients, disintegrants, and excipients are mixed to produce a mixed powder, and then the mixed powder is A granulation method, preferably a wet granulation method in which granulation is performed by adding water, water containing a binder, or a solvent, a dry granulation method in which compression molding is performed without using water, or a melt granulation method. .
- the method for producing tablets is not particularly limited. Specifically, granules are produced by the above method, and a disintegrant and a lubricant are mixed with the granules, This is a tableting method in which granules are tableted using a tableting machine.
- the formulation of the present invention may be coated with a coating layer.
- a fluidized bed granulation coating machine, a fluidized bed tumbling coating machine, or the like can be used.
- a pan coating machine, a permeable coating machine, or the like can be used. In the coating machine, while the granules and tablets are fluidized, the coating liquid is sprayed onto the granules and tablets and dried to form a coating layer.
- Boc tert-butoxycarbonyl
- CDI carbonyldiimidazole
- DBU 1,8-diazabicyclo[5.4.0]-7-undecene
- DIEA N,N-diisopropylethylamine
- DMA N,N-dimethylacetamide
- DMF N,N -dimethylformamide
- DMSO dimethylsulfoxide
- DTT dithiothreitol
- EDC 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- EDT 1,2-ethanedithiol
- EDTA ethylenediaminetetraacetic acid
- FBS fetal bovine serum
- HOBT 1- Hydroxybenzotriazole
- LHMDS lithium bis(trimethylsilyl)amide
- MEM Eagle's minimum essential medium
- NMP N-methylpyrrolidone
- Pd(OAc) 2 palladium acetate
- DSC differential scanning calorimetry
- Step 2 Synthesis of Compound 19
- Compound 18 (1.51 g, 4.04 mmol) and TFA (3.02 mL) were mixed. The reaction solution was stirred at room temperature for 4 hours and allowed to stand overnight. TFA was distilled off under reduced pressure, and toluene was added to the residue for azeotropic distillation. Isopropyl ether was added to the residue to suspend it, followed by filtration to obtain compound 19 (1.22 g, 3.84 mmol, yield 95%).
- Step 3 Synthesis of compound 20 Compound 19 (200 mg, 0.63 mmol), DMF (1.8 mL), potassium carbonate (261 mg, 1.89 mmol) and 3-(chloromethyl)-1-methyl-1H-1,2, 4-triazole hydrochloride (159 mg, 0.946 mmol) was mixed. The reaction solution was stirred at 60° C. for 2 hours and saturated aqueous ammonium chloride solution was added. The aqueous layer was extracted with ethyl acetate, and the organic layer was washed with saturated brine. The organic layer was dried over magnesium sulfate, filtered and concentrated.
- the residue was suspended in a mixed solvent of isopropyl ether, hexane, ethyl acetate and chloroform and collected by filtration. Mix the residue, DMF (1.8 mL), potassium carbonate (261 mg, 1.89 mmol) and 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride (159 mg, 0.946 mmol) bottom.
- the reaction solution was stirred at 60° C. for 6 hours and saturated aqueous ammonium chloride solution was added.
- the aqueous layer was extracted with ethyl acetate, and the organic layer was washed with saturated brine.
- the organic layer was dried over magnesium sulfate, filtered and concentrated.
- Step 4 Synthesis of compound (I-005) Compound 20 (115 mg, 0.279 mmol), THF (2.30 mL) and 6-chloro-2-methyl-2H-indazol-5-amine (60.8 mg, 0.335 mmol) ) were mixed. LHMDS (558 ⁇ L, 0.558 mmol) was added dropwise to the reaction solution at 0°C. The reaction solution was stirred at 0° C. for 2.5 hours and at room temperature for 40 minutes, and saturated aqueous ammonium chloride solution was added. After extraction with chloroform, the organic layer was concentrated.
- FIG. 7 shows the HPLC measurement results of the obtained compound I-005.
- FIG. 1 shows the powder X-ray diffraction pattern of fumaric acid co-crystal Form I of the compound represented by formula (VII).
- FIG. 2 shows the peak table of the powder X-ray diffraction pattern of FIG. In powder X-ray diffraction pattern, diffraction angle (2 ⁇ ): 7.8 ⁇ 0.2 °, 9.5 ⁇ 0.2 °, 10.1 ⁇ 0.2 °, 10.9 ⁇ 0.2 °, 13 .8 ⁇ 0.2°, 14.7 ⁇ 0.2°, 18.6 ⁇ 0.2°, 22.6 ⁇ 0.2°, 23.5 ⁇ 0.2° and 24.6 ⁇ 0. A peak was observed at 2°.
- Fig. 4 shows the DSC analysis results of the fumaric acid co-crystal form I crystal of the compound represented by formula (VII), which shows the powder X-ray analysis pattern of Fig. 1.
- the onset temperature of the endothermic peak was about 272°C.
- Fig. 5 shows the TG/DTA analysis results of the fumaric acid co-crystal Form I of the compound represented by formula (VII), which shows the powder X-ray analysis pattern of Fig. 1.
- FIG. 6 shows the DVS analysis results of the fumaric acid co-crystal Form I of the compound represented by formula (VII), which shows the powder X-ray analysis pattern of FIG.
- the compound (I-005) represented by formula (VII) can also be synthesized as follows. Step 4' To compound 20 (300 mg, 0.727 mmol), 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL), LHMDS (1 M in THF; 1.46 mL, 1.46 mmol) was added dropwise at 0°C. The reaction solution was stirred at 0° C. for 2.5 hours and at room temperature for 40 minutes, and saturated aqueous ammonium chloride solution was added. The aqueous layer was extracted with EtOAc. The organic layer was washed with saturated brine, dried over magnesium sulfate and concentrated under reduced pressure.
- BINAP 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- CPME cyclopentyl methyl ether
- CbzCl benzyl chloroformate
- DME dimethyl ether
- MEK methyl ethyl ketone
- Phase determination was performed using the direct method program ShelXT (Sheldrick, G.M., 2015), and refinement was performed using ShelXL (Sheldrick, G.M., 2015) using the full-matrix least-squares method. All temperature factors of non-hydrogen atoms were anisotropically refined. Hydrogen atoms were introduced by calculation using the default parameters of ShelXL and treated as riding atoms. All hydrogen atoms were refined with isotropic parameters. PLATON (Spek, 1991)/ORTEP (Johnson, 1976) was used for drawing.
- DSC differential scanning calorimetry
- Step 1 Synthesis of Compound 3
- Compound 1 (35.0 kg, 238.8 mol, hydrochloride), N,N-dimethylacetamide (273 L), 1,8-diazabicyclo[5,4,0]-7-undecene (87 .2 kg, 573.1 mol) and compound 2 (26.0 kg, 262.7 mol) were mixed and stirred at 25° C. for 10 minutes.
- N,N'-Carbonyldiimidazole (50.3 kg, 310.4 mol) and N,N-dimethylacetamide (7 L) were mixed with the reaction solution and stirred at 50°C for 90 minutes.
- Step 3 Synthesis of compound 7 Compound 5 (29.0 kg, 76.4 mol), trifluoroacetic acid (72.5 L) and compound 6 (16.5 kg, 152.9 mol) were mixed and stirred at 35°C for 180 minutes. . The reaction solution was cooled, ethyl acetate (348 L) was added, and the mixture was washed with 38% aqueous tripotassium phosphate solution, 2.3% brine and water. The ethyl acetate solution was concentrated to 203 L and heptane (261 L) was added.
- Step 4 Synthesis of compound 9 Compound 7 (23.3 kg, 64.1 mol), compound 8 (14.0 kg, 83.4 mol, hydrochloride), potassium iodide (6.4 kg, 38.5 mol), cesium carbonate ( 31.3 kg, 96.2 mol) and N,N-dimethylacetamide (139.8 L) were mixed and stirred at 40° C. for 360 minutes. The reaction solution was cooled to 25° C. and acetic acid (34.6 kg, 577.2 mol) was added. Insoluble matter was filtered off, and acetonitrile (93.2 L) and water (326.2 L) were added to the filtrate.
- Step 5-2 Acetone (613.5 L) and water (109.2 L) were added to half the undried crystals of the compound of formula (VII) obtained and dissolved at 50°C.
- the obtained solution was treated with activated carbon, acetone (150.2 L) and water (5.9 L) were added to the treated solution, and the mixture was concentrated to 702 L.
- the temperature of the concentrate was adjusted to 50° C., fumaric acid (4.6 kg, 72.6 mol), acetone (150.2 L) and water (5.9 L) were added and concentrated to 464 L.
- Acetone (78 L) was added to the concentrate, concentrated to 265 L, and acetone (19.5 L) was added. The slurry was thermostatted to 55° C. and stirred for 120 minutes longer.
- Synthesis step 1 of toluene solute of compound represented by formula (VII) Compound 9 (150 mg, 0.327 mmol) and compound 10 (65.4 mg, 0.360 mmol) were mixed with toluene (1.5 mL) and acetic acid (0.187 ml, 3.27 mmol) and stirred at 100°C for 9 hours. . After cooling to room temperature, heptane (1.5 mL, 10 V) was added and filtered, and the obtained crystals were washed with heptane (0.7 mL) three times. Drying under reduced pressure gave crystals of the compound represented by formula (VII) (168 mg, yield 87%).
- the obtained crystals contained 0.5 to 0.6 molecules of toluene as a solvate, and the toluene was not removed under reduced pressure drying in the normal operating range. It was confirmed that the toluene solute of the compound represented by formula (VII) was obtained in good quality.
- Step 1 Synthesis of Compound S-2
- Compound S-1 (5.50 kg, 29.5 mol), acetonitrile (21.7 kg) and glacial acetic acid (115.00 kg) were mixed and cooled to 5°C.
- a 17% sodium nitrite aqueous solution (13.03 kg) was added, and the mixture was stirred for 1 hour, heated to 25°C, and stirred for 1.5 hours.
- the insoluble matter was filtered off and washed with acetonitrile (21.7 kg) and tetrahydrofuran (49.0 kg). Water (460 L) was added to the collected filtrate.
- Step 2 Synthesis of Compound S-3
- Compound S-2 (3.25 kg, 16.4 mol) and ethyl acetate (58.7 kg) were mixed, and trimethyloxonium tetrafluoroborate (2.09 kg, 14.1 mol) was added. and stirred at 25° C. for 7 hours.
- the organic layer was washed twice with 5% aqueous sodium chloride solution (65.8 kg), treated with activated carbon, and concentrated to 42 kg.
- Step 3 Synthesis of compound S-4 Compound S-3 (1040 g, 4.9 mol), 10% palladium carbon (PE type, hydrous) (523 g, 0.25 mol) and ethyl acetate (8.99 kg) are mixed, Hydrazine monohydrate (504 g, 10.1 mol) was added and stirred at 35° C. for 3 hours. The 10% palladium on carbon was filtered off and washed with water (1560 g) and ethyl acetate (9.00 kg). 2 mol/L hydrochloric acid (750 g) was added to the collected filtrate to separate into an organic layer and an aqueous layer. The resulting aqueous layer was extracted with ethyl acetate (4.69 kg).
- Step 1 Synthesis of Dichloromethane Solution of Compound A-2
- Compound A-1 (9.2 kg, 65.1 mol) and tetrahydrofuran (64 L) were mixed and cooled to 0° C. to form slurry.
- a Red-AL/tetrahydrofuran solution obtained by mixing sodium bis(2-methoxy)aluminum hydride (Red-AL)/toluene solution (65 wt%) (26.4 kg, 84.9 mol) and tetrahydrofuran (28 L) was contained therein. It was added dropwise over 60 minutes while maintaining the temperature at 8°C or lower. After that, the mixture was stirred at 0°C to 5°C for 30 minutes.
- Acetone (4.9 kg, 84.3 mol) was added dropwise to this reaction solution over 30 minutes, and the temperature was raised to 25°C.
- Step 2 Synthesis of Compound 8 Dichloromethane (44 L) was added to the A-2/dichloromethane solution (49.8 kg of a dichloromethane solution containing 5.53 kg of A-2 (48.8 mol)) prepared in Step 1, and the mixture was heated to 25°C. temperature adjusted. A mixed solution of thionyl chloride (7.8 kg, 65.5 mol) and dichloromethane (27 L) was added dropwise over 30 minutes, and the line was washed with dichloromethane (8.2 L). bottom. Separately, a 20% sodium acetate aqueous solution (179 kg) was prepared from sodium acetate (36.2 kg, 436 mol) and tap water (143 L).
- Step 1 Synthesis of compound B-2
- Compound B-1 (79.1 kg, 499 mol) was added in portions to 98% sulfuric acid (395.7 L) cooled to 0°C to 5°C under a nitrogen atmosphere.
- Potassium nitrate (55.5 kg) was kept at an internal temperature of 0° C. to 12° C. and charged in 15 portions (at intervals of 20 minutes or longer).
- the mixture was stirred at an internal temperature of 0°C to 5°C for 5 hours.
- the above reaction solution was slowly poured into water (791 L) cooled to 0°C to 5°C while maintaining the internal temperature at 0°C to 5°C, and washed with 98% sulfuric acid (39.6L).
- Step 2 Synthesis of compound S-2 Ethanol (697 L), water (697 L) and hydrazine monohydrate (73.5 kg, 1468 mol) were mixed and heated to 45°C. A mixed solution of compound B-2 (99.6 kg, 489 mol) and ethanol (299 L) was added dropwise thereto over 60 minutes, followed by stirring for 8 hours at 45° C. to 50° C. for 9 hours. An aqueous solution prepared from potassium hydrogen carbonate (53.9 kg, 538 mol) and water (1295 L) was added dropwise over 30 minutes while maintaining the internal temperature at 40°C to 50°C. After cooling to 0° C. to 5° C. and stirring for 1 hour, filtration was carried out.
- Step 3 Synthesis of compound S-3
- Compound S-2 (84 kg, 430 mol) and ethyl acetate (1596 L) were mixed and stirred at 20°C to 30°C.
- Trimethyloxonium tetrafluoroborate (77.6 kg, 525 mol) was charged in several portions, ethyl acetate (84 L) was added, and the mixture was stirred at 25°C for 6 hours.
- a mixed solution of methanol (252 L) and ethyl acetate (420 L) was added dropwise to the above reaction solution over 2 hours to quench excess trimethyloxonium tetrafluoroborate.
- Step 4 Synthesis of Compound 10
- Compound S-3 (65.7 kg, 310 mol) and ethyl acetate (657 L) were mixed, stirred at room temperature, cooled to around 10° C., and purged with nitrogen.
- 5% platinum-carbon (57.7 kg, 53% moisture content) was added.
- the mixture was stirred for 4 hours while adjusting the internal temperature to around 25°C.
- nitrogen replacement and filtration were performed to remove the platinum-carbon catalyst.
- the organic layer was concentrated, and heptane was added dropwise to the ethyl acetate solution to form a crystallization slurry.
- Step 1 Synthesis of compound C-2
- Compound C-1 (10.00 g, 48.0 mmol, mesylate), N,N'-carbonyldiimidazole (8.18 g, 50.4 mmol), acetonitrile (60.00 mL ), and diisopropylethylamine (6.83 g, 52.8 mmol) were mixed and stirred at 10° C. for 60 minutes.
- Compound 1 (8.09 g, 55.2 mmol, hydrochloride) and diisopropylethylamine (7.14 g, 55.2 mmol) were mixed with the reaction solution and stirred at 50° C. for 210 minutes. The reaction was cooled and concentrated to 45 g.
- Step 2 Synthesis of Compound C-3
- Compound C-2 (8.00 g, 32.2 mmol), N,N'-carbonyldiimidazole (6.79 g, 41.9 mmol), tetrahydrofuran (80.0 mL), and 1 ,8-diazabicyclo[5,4,0]-7-undecene (5.40 g, 35.4 mmol) was mixed and stirred at 25° C. for 120 minutes.
- Tetrahydrofuran (80.0 mL) was added dropwise and the reaction was cooled to 0° C. to form a crystallization slurry.
- Step 3 Synthesis of Compound 9
- Compound C-3 (1.00 g, 2.3 mmol, 1,8-diazabicyclo[5.4.0]-7-undecene salt), N,N-dimethylacetamide (5.0 mL) , and compound 4 (579.2 mg, 2.6 mmol) were mixed and stirred at 70° C. for 300 minutes.
- the operation of cooling the reaction solution, adding acetonitrile (10 mL), and concentrating to 9.4 g was repeated twice.
- Compound 6 (461 mg, 4.7 mmol) and diisopropylethylamine (456 mg, 3.5 mmol) were added to the concentrate and stirred at 60°C for 160 minutes.
- Step 1 Synthesis of Compound S-3
- Compound B-2 was obtained in the same manner as in Step 1 of Reference Example 3. Subsequently, compound B-2 (30 g, 147 mmol) and NMP (120 mL) were mixed, Boc-carboxylate (56 g, 383 mmol) was added under ice cooling, and the mixture was stirred at room temperature for 30 minutes. Diisopropylethylamine (38.6 mL, 221 mmol) was added to the reaction solution, and the mixture was stirred at 90° C. for 20 hours. After the reaction solution was brought to 80° C. and water (240 mL) was added, the solution was cooled to room temperature and the precipitated insoluble matter was separated by filtration.
- the resulting solid was suspended in isopropyl acetate (100 mL).
- the resulting suspension was added to a mixture of mesylic acid (96 mL, 1474 mmol) and isopropyl acetate (100 mL) at 55°C, washed with isopropyl acetate (60 mL), and stirred at the same temperature for 25 minutes.
- Water (240 mL), aqueous sodium hydroxide solution (239 mL, 1916 mmol) and isopropyl acetate (150 mL) were added to the reaction solution under ice-cooling, and the mixture was stirred at 40°C. Isopropyl acetate (150 mL) was added to the resulting reaction solution.
- Step 1 Synthesis of Compounds T-2 and T-3 Under ice-cooling, compound T-1 (40 g, 182 mmol), concentrated sulfuric acid (200 mL, 3677 mmol) and 69% nitric acid (23.3 g, 255 mmol) were mixed and mixed with ice. The mixture was stirred from cold to room temperature for 3 hours and then allowed to stand overnight. The mixed liquid was poured into 520 mL of ice water, and dichloromethane (200 mL) was added to perform a liquid separation operation. The resulting dichloromethane solution was washed twice with 5% aqueous sodium bicarbonate solution (400 mL) and concentrated to dryness.
- Step 3 Synthesis of Compound T-5
- Compound T-4 500 mg, 1.80 mmol
- 2-propanol 2.5 mL
- tributylphosphine 802 mg, 3.96 mmol
- solvent replacement was performed three times with toluene (3 mL), and concentration was performed until the residue after concentration was 5 g.
- the reaction solution was ice-cooled to 4° C., 4 mol/L hydrochloric acid-ethyl acetate solution (1.5 mL) was added, and the mixture was stirred for 20 minutes.
- Step 4 Synthesis of Compound 10 Compound T-5 (2.015 g, 8.21 mmol), DME (20 mL), sodium tert-butoxide (1.104 g, 11.49 mmol), benzophenone imine (1.645 mL) under a nitrogen stream. , 9.80 mmol), BINAP (0.153 g, 0.246 mmol), and diacetoxypalladium (0.036 g, 0.160 mmol) were mixed and stirred at 80° C. for 9 hours and allowed to stand overnight. Ethanol (10 mL) was added to the resulting suspension and cooled to 5°C. 30% sulfuric acid (20 mL) was added little by little to the suspension, and the mixture was stirred overnight at room temperature.
- Ethyl acetate (40 mL) and water (20 mL) were added to the obtained reaction solution to carry out a liquid separation operation.
- the resulting aqueous layer was washed with ethyl acetate (10 mL), and the organic layer was washed with 10% sulfuric acid (10 mL).
- the resulting aqueous layers were combined, cooled with ice, and then neutralized to pH 8 using a 48% sodium hydroxide aqueous solution.
- Ethyl acetate (20 mL) was added, the precipitated sodium sulfate was filtered off, and a liquid separation operation was performed.
- Ethyl acetate (20 mL) was added to the resulting aqueous layer to carry out a liquid separation operation.
- Step 1 Synthesis of Compound U-2
- Compound U-1 (2.09 g, 22.6 mmol, hydrochloride) was mixed with CPME (12.04 g) and water (7 g), and potassium carbonate (4.25 g, 30.0 g) was mixed. 8 mmol) dissolved in water (7 g) was added slowly so that the temperature of the reaction solution reached 20-30°C.
- the resulting mixed solution was vigorously stirred, CbzCl (3.50 g, 20.5 mmol) was added slowly so that the temperature of the reaction solution reached 20 to 30° C., and the mixture was stirred at room temperature for 1 hour.
- the resulting solution was subjected to liquid separation, and the organic layer was washed with water (14 g) and then concentrated.
- Step 2 Synthesis of compound U-3 Methanol (31.66 g) was added to compound U-2 (8.00 g, 42.1 mmol), and after cooling to 0°C, a 28% methanol solution of sodium methoxide (2.43 g , 12.6 mmol) was added, and the mixture was stirred at the same temperature for 4 hours. To the resulting solution was added a solution of N-methylformohydrazide (3.74 g, 50.5 mmol) in methanol (19 g) at 0-5°C, and acetic acid (2.53 g, 42.1 mmol) was added to the same. added at room temperature and stirred at 0° C. for 2 hours. The obtained solution was heated to 60° C.
- Step 3 Synthesis of Compound C-1
- Compound U-3 (10 g, 29.2 mol, mesylate) and methanol (79.15 g) were mixed, stirred at room temperature, and then purged with nitrogen.
- Palladium-carbon (10% palladium) (0.5 g, 5% by weight) was added, and after purging with hydrogen, the mixture was stirred at room temperature for 7 hours. After purging with nitrogen, the palladium-carbon catalyst was removed by Celite (registered trademark) filtration. The obtained filtrate was concentrated to 50 g.
- the operation of adding MEK (40.25 g) and concentrating to 40 g was repeated twice.
- Tables 3 and 4 show atomic coordinates of non-hydrogen atoms.
- U(eq) means an equivalent isotropic temperature factor.
- Table 5 shows atomic coordinates of hydrogen atoms.
- U(iso) means an isotropic temperature factor.
- the numbers of the hydrogen atoms in Table 5 are associated with the numbers of the non-hydrogen atoms to which they are attached.
- Table 6 shows the interatomic bond distance (unit: angstrom).
- the bond distance of N10-C9 was about 1.26 ⁇ , and the bond distance of N16-C9 was about 1.37 ⁇ . Since the N10-C9 bond distance (approximately 1.26 ⁇ ) is shorter than the N16-C9 bond distance (approximately 1.37 ⁇ ), the compound of formula (VII) in fumaric acid cocrystal Form I has an imino structure : identified as
- the counter molecule of the salt or complex may be Depending on the type, it may have an imino structure or an amino structure, and even the same counter molecule may have an imino structure or an amino structure depending on the crystallization conditions and the like. It may also be a mixture of a compound having an imino structure, a salt thereof, or a complex thereof and a compound having an amino structure, a salt thereof, or a complex thereof.
- FIG. 1 shows the results of X-ray powder diffraction of fumaric acid co-crystal form I of the compound represented by formula (VII) obtained by the same production method as in step 5-2 of Example 1b.
- diffraction angle (2 ⁇ ) 7.7 ⁇ 0.2 °, 9.5 ⁇ 0.2 °, 10.0 ⁇ 0.2 °, 10.9 ⁇ 0.2 °, 13 .8 ⁇ 0.2°, 14.6 ⁇ 0.2°, 18.6 ⁇ 0.2°, 22.6 ⁇ 0.2°, 23.4 ⁇ 0.2° and 24.6 ⁇ 0.
- a peak was observed at 2°.
- FIG. 9 shows the powder X-ray diffraction pattern of fumaric acid co-crystal Form I of the compound of formula (VII). The horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- FIG. 10 shows a peak table in the powder X-ray diffraction pattern of FIG.
- FIG. 20 shows the powder X-ray diffraction pattern of the toluene solute of the compound represented by formula (VII).
- the horizontal axis represents 2 ⁇ (°), and the vertical axis represents intensity (Count).
- the molecular structure (amino body/imino body) of the toluene solute of the compound represented by formula (VII) has not been identified.
- the compound represented by the formula (VII) according to the present invention has a coronavirus 3CL protease inhibitory action and may inhibit coronavirus 3CL protease.
- the IC50 is preferably 50 ⁇ M or less, more preferably 1 ⁇ M or less, and even more preferably 100 nM or less.
- Test Example 1 Cytopathic effect (CPE) suppression effect confirmation test using human TMPRSS2-expressing Vero E6 cells (Vero E6/TMPRSS2 cells) ⁇ Operating procedure> ⁇ Dilution and dispensing of test sample Dilute the test sample with DMSO in advance to an appropriate concentration, prepare a 2- to 5-fold serial dilution series, and then dispense into a 384-well plate.
- CPE Cytopathic effect
- Test Example 2 SARS-CoV-2 3CL protease inhibitory activity test ⁇ Material> ⁇ Commercially available Recombinant SARS-CoV-2 3CL Protease - Commercially available substrate peptide Dabcyl-Lys-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-Met-Glu(Edans)-NH2 (SEQ ID NO: 1) - Internal Standard peptide Dabcyl-Lys-Thr-Ser-Ala-Val-Leu(13C6,15N)-Gln (SEQ ID NO: 2) Dabcyl-Lys-Thr-Ser-Ala-Val-Leu(13C6,15N)-Gln is described in the literature (Atherton, E.; Sheppard, R.C., "In Solid Phase Peptide Synthesis, A Practical Approach", IRL Press at Oxford University).
- an assay buffer consisting of 20 mM Tris-HCl, 1 mM EDTA, 10 mM DTT, 0.01% BSA is used.
- ⁇ Dilution and dispensing of test sample Dilute the test sample with DMSO in advance to an appropriate concentration, prepare a 2- to 5-fold serial dilution series, and then dispense into a 384-well plate. Addition of Enzyme and Substrate, Enzyme Reaction Add 8 ⁇ M substrate and 6 or 0.6 nM enzyme solution to the prepared compound plate and incubate at room temperature for 3-5 hours.
- reaction stop solution (0.067 ⁇ M Internal Standard, 0.1% formic acid, 10 or 25% acetonitrile) is added to stop the enzymatic reaction.
- Measurement of Reaction Products Reaction completed plates are measured using a Rapid Fire System 360 and mass spectrometer (Agilent, 6550 iFunnel Q-TOF) or a Rapid Fire System 365 and mass spectrometer (Agilent, 6495C Triple Quadrupole).
- a solution (75% isopropanol, 15% acetonitrile, 5 mM ammonium formate
- B solution 0.01% trifluoroacetic acid, 0.09% formic acid
- the reaction product detected by the mass spectrometer is calculated using RapidFire Integrator or a program capable of equivalent analysis and taken as a product area value.
- the internal standard detected at the same time is also calculated and used as the internal standard area value.
- ⁇ Calculation of each measurement item value> ⁇ P/IS is calculated by calculating the area value obtained in the item before calculating P/IS using the following formula.
- P / IS Product area value / Internal Standard area value 50% SARS-CoV-2 3CL protease inhibitory concentration (IC 50 ) calculation
- x is the logarithmic value of the compound concentration and y is % Inhibition
- y is % Inhibition
- Test Example 1-2 Cytopathic effect (CPE) suppression effect confirmation test using human TMPRSS2-expressing Vero E6 cells (Vero E6/TMPRSS2 cells) ⁇ Operating procedure> - Dilution and dispensing of test sample A test sample is diluted with DMSO in advance to an appropriate concentration, and after preparing a 3-fold serial dilution series, it is dispensed into a 96-well plate.
- CPE Cytopathic effect
- Test Example 2-2 SARS-CoV-2 3CL protease inhibitory activity test ⁇ Material> ⁇ Commercially available Recombinant SARS-CoV-2 3CL Protease - Commercially available substrate peptide Dabcyl-Lys-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-Met-Glu(Edans)-NH2 (SEQ ID NO: 1) - Internal Standard peptide Dabcyl-Lys-Thr-Ser-Ala-Val-Leu(13C6,15N)-Gln (SEQ ID NO: 2) Dabcyl-Lys-Thr-Ser-Ala-Val-Leu(13C6,15N)-Gln is described in the literature (Atherton, E.; Sheppard, R.C., "In Solid Phase Peptide Synthesis, A Practical Approach", IRL Press at Oxford University Press, 1989.
- ⁇ Rapid Fire Cartridge C4 type A ⁇ Operation procedure> -Preparation of assay buffer In this test, an assay buffer consisting of 20 mM Tris-HCl, 1 mM EDTA, 10 mM DTT and 0.01% BSA is used.
- test sample is diluted in advance with DMSO to an appropriate concentration, and after preparing a 3-fold serial dilution series, it is dispensed into a 384-well plate.
- Addition of Enzyme and Substrate, Enzyme Reaction Add 8 ⁇ M substrate and 6 nM enzyme solution to the prepared compound plate and incubate at room temperature for 3 hours. Thereafter, a reaction stop solution (0.072 ⁇ M Internal Standard, 0.1% formic acid, 10% acetonitrile) is added to stop the enzymatic reaction.
- Reaction completed plates are measured using a RapidFire System 360 and a mass spectrometer (Agilent, 6550 iFunnel Q-TOF).
- a solution (75% isopropanol, 15% acetonitrile, 5 mM ammonium formate) and B solution (0.01% trifluoroacetic acid, 0.09% formic acid) are used as mobile phases for measurement.
- the reaction product detected by the mass spectrometer is calculated using RapidFire Integrator and taken as a Product area value.
- the internal standard detected at the same time is also calculated and used as the internal standard area value.
- ⁇ Calculation of each measurement item value> ⁇ P/IS is calculated by calculating the area value obtained in the item before calculating P/IS using the following formula.
- P / IS Product area value / Internal Standard area value 50% SARS-CoV-2 3CL protease inhibitory concentration (IC 50 ) calculation
- IC 50 SARS-CoV-2 3CL protease inhibitory concentration
- Fumaric acid co-crystal Form I crystals of the compound of formula (VII) are weighed so that the amount of the compound of formula (VII) is about 1.8 (w/v)%, and 0.3 (w/v) /v) % polymer was added to the dissolved aqueous solution and dispersed.
- Additives include aminoalkyl methacrylate copolymer E (manufactured by Evonik), hydroxypropylcellulose (manufactured by Nippon Soda), hypromellose (manufactured by Shin-Etsu Chemical Co., Ltd.), polyvinyl alcohol (manufactured by Merck), polyvinylpyrrolidone (manufactured by BASF). , polyvinyl alcohol/methyl methacrylate/acrylic acid copolymer (manufactured by Nisshin Kasei Co., Ltd.) was used.
- Test Example 3 Solubility Evaluation 1 mL of each dispersion in Example 5 was added to 14 mL of simulated fasting simulated intestinal fluid (FaSSIF) and stirred at 37° C. and 400 rpm for 1 hour with a constant temperature stirrer. After stirring for 1 hour, the sample was filtered through a 0.45 ⁇ m filter, and the concentration of the compound represented by formula (VII) in the filtrate was measured by liquid chromatography.
- FaSSIF simulated fasting simulated intestinal fluid
- microcrystalline cellulose manufactured by Asahi Kasei
- magnesium stearate manufactured by Mallinckrodt
- light anhydrous silicic acid manufactured by Cabot
- Test Example 4 Tablet dissolution test (dissolution test method) The dissolution test was performed according to Method 2 of the Japanese Pharmacopoeia Dissolution Test Method 18th Edition (dissolution test liquid 1 containing surfactant, paddle method, paddle rotation speed: 50 rpm, result: average value of 2 tablets).
- FIG. 22 shows the particle size distribution of the active ingredient (fumaric acid co-crystal form I crystal of the compound represented by formula (VII)) used in the preparations of Examples 6A and 6B used in Test Example 4.
- the 10% particle size was 0.69 ⁇ m, the 50% particle size was 4.00 ⁇ m, and the 90% particle size was 10.80 ⁇ m.
- FIG. 23 shows the particle size distribution of the active ingredient (fumaric acid co-crystal Form I crystal of the compound represented by formula (VII)) used in the preparations of Examples 6C and 6D used in Test Example 4.
- the 10% particle size was 0.67 ⁇ m
- the 50% particle size was 3.63 ⁇ m
- the 90% particle size was 10.98 ⁇ m.
- the measurement conditions for particle size distribution are shown below.
- Test Example 5 Effect of Stabilizer in Formulation in Stability Test Over Time The results of confirming the stability over time of the formulation of Example 6A of the same lot as that used in Test Example 4 are shown.
- a Stability test method The formulation of Example 6A was stored 1) at 60°C in closed amber glass bottles for two weeks, or 2) in open amber glass bottles at 40°C and 75% relative humidity for one month. Thereafter, the amounts of analogues of the compound represented by formula (VII) in the formulation of the present invention were measured. (Sample solution preparation method) (Measuring method) The amounts of analogues of the compound represented by formula (VII) in the formulation of the present invention were measured by liquid chromatography according to the following method and conditions.
- Detector Ultraviolet absorption photometer (measurement wavelength: 222 nm)
- Column C18 column
- Column temperature constant temperature around 40° C.
- Mobile phase A 0.01 mol/L ammonium formate solution
- mobile phase B acetonitrile
- the mixing ratio of mobile phase A and mobile phase B is 9: The mixing ratio of mobile phase B was gradually increased from 1 to 1:9, and measurement was performed for 32 minutes.
- - Flow rate 0.6 mL/class
- the amount of related substances was calculated as a ratio (%) to that amount, with the total peak area of the chromatogram of the HPLC chart being 100%.
- Example 7 The total amount of related substances in the formulation of Example 6A increased after 1) storage at 60° C. for 2 weeks in closed amber glass bottles and 2) storage at 40° C. in open amber glass bottles at 75% relative humidity for 1 month. was stable. As described above, the formulation of the present invention has high stability against humidity and temperature. (Example 7)
- Test Example 6 Clinical trial (Ph2a) When repeated oral administration of the test drug (active ingredient: fumaric acid co-crystal form I crystal of the compound represented by formula (VII)) to SARS-CoV-2 infected persons with mild/moderate and asymptomatic/mild symptoms only A randomized, double-blind controlled study with placebo as a control was performed to evaluate the efficacy and safety of The primary endpoint of the Phase 2a Part is the change from baseline in SARS-CoV-2 viral titer at each time point, common to mild/moderate and asymptomatic SARS-CoV-2-infected persons. We confirmed the antiviral effect of the drug test.
- active ingredient fumaric acid co-crystal form I crystal of the compound represented by formula (VII)
- Patients with mild/moderate SARS-CoV-2 infection were selected if they met all of the following criteria: (a) Male or female patients aged 12 to 70 years. (b) Subjects diagnosed as positive for SARS-CoV-2 within 120 hours prior to enrollment. (c) Those who have been within 120 hours from the onset of COVID-19 to registration.
- Asymptomatic SARS-CoV-2 infected patients were selected if they met all of the following criteria: (a) Male or female patients aged 12 to 70 years. (b) Subjects diagnosed as positive for SARS-CoV-2 within 120 hours prior to enrollment. (c) Asymptomatic: Persons who have not had the following COVID-19 symptoms (excluding symptoms preexisting before SARS-CoV-2 infection) within 2 weeks prior to enrollment.
- Test drug 250 mg tablet The tablet contains fumaric acid co-crystal Form I of the compound of formula (VII) and 250 mg of the compound of formula (VII).
- the 250 mg tablets are tablets made by doubling each composition as in Example 6C.
- 125 mg tablet The tablet contains fumaric acid co-crystal Form I of the compound of formula (VII) and contains 125 mg of the compound of formula (VII). This 125 mg tablet is of the same composition as in Example 6C.
- placebo Placebo-D tablet A tablet indistinguishable in appearance from the 250 mg tablet described above and containing no fumaric acid co-crystal form I of the compound of formula (VII).
- Placebo-B tablet A tablet indistinguishable in appearance from the 125 mg tablet described above and containing no fumaric acid co-crystal Form I of the compound of formula (VII).
- Investigational drug 375/125 mg group by treatment group Administer 3 125 mg tablets and 3 Placebo-D tablets each on Day 1, and 1 125 mg tablet and 1 Placebo-D tablet each per day on Days 2-5.
- 750/250 mg group Administer 3 250 mg tablets and 3 Placebo-B tablets on Day 1, and 1 250 mg tablet and 1 Placebo-B tablet each on Days 2-5.
- Placebo group Administer 3 Placebo-D and 3 Placebo-B tablets on Day 1, and 1 Placebo-D and 1 Placebo-B tablet each on Days 2-5.
- Day 1 represents the first day of administration
- Day 2-Day 5" represents days 2-5 counting from the first day of administration
- Phase 2a Part The primary efficacy endpoint of the Phase 2a Part is the change from baseline in SARS-CoV-2 viral titer at each time point in mild/moderate and asymptomatic SARS-CoV-2-infected persons. be. Defined as the absolute change from baseline in the observed SARS-CoV-2 viral titer at each time point.
- Results of the primary endpoint (1) Change from baseline in SARS-CoV-2 viral titer at each time point (Phase 2a Part) Sixty-nine patients were randomized to the Phase 2a Part, 22 to the 375/125 mg group (including 1 naive), 23 to the 750/250 mg group, and 24 to the placebo group. rice field. Of the 69 cases, 44 had a positive baseline RT-PCR, of which 40 had a detectable baseline viral titer. The composition of the 40 patients was 14 in the 375/125 mg group, 13 in the 750/250 mg group, and 13 in the placebo group. The number of cases in these groups and their breakdown were calculated based on the RT-PCR measurement results and virus titer measurement results obtained by January 17, 2022.
- the final results of the Phase 2a Part were 47 with positive baseline RT-PCR and 43 with detectable baseline viral titers. rice field.
- the 43 patients consisted of 15 in the 375/125 mg group, 14 in the 750/250 mg group, and 14 in the placebo group.
- Visit The visit date specified in the clinical trial protocol is indicated by Visit, and the correspondence with the administration date (Day) and the allowable range are as follows.
- Op V means Optional Visit and indicates an optional visit date.
- SARS-CoV-2 viral titers from baseline to FIG. 24 shows the transition of the average value for each group of the amount of change in .
- This analysis included mild/moderate SARS-CoV-2-infected persons and asymptomatic SARS-CoV-2-infected persons in the analysis, and only visited visits marked as mandatory visits were displayed.
- the virus titer was below the detection limit (0.8 log 10 (TCID 50 /mL)
- the virus titer value was treated as 0.8 log 10 (TCID 50 /mL).
- the percentage of virus titer-positive patients at each time point is shown below.
- the percentage of positive persons with a virus titer of 0.8 or higher in the placebo group decreased by 80% in the 750/250 mg group and decreased by 63% in the 375/125 mg group.
- the percentage of positive persons with a virus titer of 0.8 or higher in the placebo group decreased by 54% in the 750/250 mg group and decreased by 100% in the 375/125 mg group.
- both the 750/250 mg and 375/125 mg groups tended to have a lower percentage of patients with positive virus titers compared to the placebo group. Therefore, it was suggested that administration of the pharmaceutical composition of the present invention can rapidly reduce the number of patients who shed infectious viruses.
- Results of secondary endpoints (1) Time to first confirmation of negative virus titer (Phase 2a Part) The time to first confirmed negative virus titer for SARS-CoV-2 is shown in Table 13 below and in Figure 25. Of the 69 subjects in Phase 2a Part, the results for 15 subjects in the 375/125 mg group, 13 subjects in the 750/250 mg group, and 14 subjects in the placebo group are shown.
- the subject stratum (mild/moderate, asymptomatic/mild only) is used as the stratification factor]
- the time to negative virus titers for 50% of patients was approximately 4.6 days after the start of treatment.
- the time to negative virus titers in 50% of patients was approximately 2.6 days after initiation of treatment.
- a reduction in the time to first negative viral titers in 50% of patients was confirmed by approximately 2 days.
- Figure 6 shows the change from baseline in the COVID-19 12-symptom total score at each time point.
- results are shown for 13 subjects in the 375/125 mg group, 12 subjects in the 750/250 mg group, and 14 subjects in the placebo group in subjects with mild/moderate disease.
- the 375/125 mg and 750/250 mg groups had numerical COVID-19 12-symptom total scores compared to the placebo group at all time points after Day 2 (after one dose). A trend of improvement was observed.
- Phase 2a Part Severe suppression effect
- subjects with mild/moderate symptoms were evaluated for the first time at any time after the start of administration, ordinal scale (an ordinal scale that classifies clinical severity into 8 levels, Table 14)
- Table 15 shows the percentage of subjects with a worsening of 3 or more.
- Table 15 shows the percentage of subjects with a worsening of 3 or more.
- microcrystalline cellulose manufactured by Asahi Kasei
- magnesium stearate manufactured by Mallinckrodt
- light anhydrous silicic acid manufactured by Cabot
- Test Example 7 Tablet dissolution test (dissolution test method)
- the 18th revision Japanese Pharmacopoeia dissolution test method 2nd method dissolution test liquid 1 containing surfactant, paddle method, paddle rotation speed: 50 rpm, result: 2 tablets Mean value
- 2nd method dissolution test liquid 1 containing surfactant, paddle method, paddle rotation speed: 50 rpm, result: 2 tablets Mean value
- microcrystalline cellulose manufactured by Asahi Kasei
- magnesium stearate manufactured by Mallinckrodt
- light anhydrous silicic acid manufactured by Cabot
- Test Example 8 Tablet dissolution test (dissolution test method) For 2 formulations of Example 9, 18th revision Japanese Pharmacopoeia dissolution test method 2nd method (dissolution test liquid 1 containing surfactant, paddle method, paddle rotation speed: 50 rpm, result: average value of 2 tablets ) was used to perform an elution test.
- Granules containing fumaric acid co-crystal Form I crystals of the compound of formula (VII) are prepared.
- the table below shows the formulation per granule.
- Fumaric acid co-crystal form I crystal of the compound represented by formula (VII) D-mannitol (manufactured by Roquette), powdered reduced maltose starch syrup (maltitol, manufactured by Roquette), croscarmellose sodium (manufactured by DuPont), Hydroxypropyl cellulose (manufactured by Nippon Soda Co., Ltd.), light anhydrous silicic acid (manufactured by Cabot Co.), magnesium stearate (manufactured by Mallinckrodt Co.) and sucralose (manufactured by San-Eigen FFI Co., Ltd.) were sieved through a 30-mesh sieve.
- Granulate After mixing, granulate. Granulated and sized granules, light silicic anhydride (manufactured by Cabot) and sucralose (manufactured by Saneigen FFI) are added and mixed to obtain granules having the following composition.
- Example 9 Dissolution test of granules (dissolution test method) For 2 formulations out of Example 10, 18th revision Japanese Pharmacopoeia dissolution test method 2nd method (dissolution test liquid 1 containing surfactant, paddle method, paddle rotation speed: 50 rpm, result: average of 2 granules value).
- Test Example 10 Test of stability over time of granules The results of confirming the stability over time of two formulations out of Example 10 are shown.
- a. Stability test method The formulation of Example 10 was 1) stored in a closed DUMA bottle (plastic container, GERRESHEIMER) at 40°C and 75% relative humidity for 3 weeks or 2) in a closed DUMA bottle containing silica gel for 40 days. 3) DUMA bottle closed at 40°C and 75% relative humidity for 3 weeks, or 4) DUMA bottle closed with silica gel at 40°C and 75% Stored under relative humidity for one month. Thereafter, the amounts of analogues of the compound represented by formula (VII) in the formulation of the present invention were measured.
- sample solution preparation method (Measuring method)
- the amounts of analogues of the compound represented by formula (VII) in the formulation of the present invention were measured by liquid chromatography according to the following method and conditions.
- ⁇ Detector Ultraviolet absorption photometer (measurement wavelength: 222 nm)
- ⁇ Column C18 column
- Column temperature constant temperature around 40° C.
- Mobile phase A 0.01 mol/L ammonium formate solution
- mobile phase B acetonitrile
- the mixing ratio of mobile phase A and mobile phase B is 9: The mixing ratio of mobile phase B was gradually increased from 1 to 1:9, and measurement was performed for 32 minutes.
- the formulation examples shown below are merely illustrative and are not intended to limit the scope of the invention in any way.
- the compounds of the invention can be administered by any conventional route, in particular enterally, e.g. orally, e.g. in the form of tablets, granules or capsules, or parenterally, e.g. in the form of injection solutions or suspensions. , topically, for example, in the form of a lotion, gel, ointment or cream, or in nasal or suppository form as a pharmaceutical composition.
- a pharmaceutical composition comprising a compound of the invention in free form or in pharmaceutically acceptable salt form together with at least one pharmaceutically acceptable carrier or diluent can be prepared by mixing, mixing, It can be manufactured by a granulation or coating method.
- oral compositions can be tablets, granules, capsules containing excipients, disintegrants, binders, lubricants, etc. and active ingredients.
- injectable compositions may be in the form of solutions or suspensions, may be sterilized, and may contain preservatives, stabilizers, buffers and the like.
- Suspension A suspension was prepared by adding, for example, water for injection to the drug substance of the compound represented by formula (VII).
- the compound produced by the production method according to the present invention has an inhibitory effect on coronavirus 3CL protease, and is thought to be useful as a therapeutic and/or prophylactic agent for diseases or conditions involving coronavirus 3CL protease.
- the novel synthetic intermediates or their salts according to the present invention and the production method according to the present invention are useful for pharmaceutical production.
- the formulation of the present invention has an inhibitory effect on coronavirus 3CL protease, and is considered useful as a therapeutic and/or prophylactic agent for diseases or conditions involving coronavirus 3CL protease.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Virology (AREA)
- Biophysics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Plural Heterocyclic Compounds (AREA)
- Biochemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022574839A JP7253866B1 (ja) | 2021-11-24 | 2022-11-22 | トリアジン誘導体を含有する経口投与する製剤 |
| AU2022333823A AU2022333823A1 (en) | 2021-11-24 | 2022-11-22 | Preparation for oral administration containing triazine derivative |
| EP22861484.8A EP4289432A4 (en) | 2021-11-24 | 2022-11-22 | PREPARATION FOR ORAL ADMINISTRATION CONTAINING TRIAZINE DERIVATIVES |
| KR1020237025438A KR20230119022A (ko) | 2021-11-24 | 2022-11-22 | 트라이아진 유도체를 함유하는 경구 투여하는 제제 |
| CN202280007992.3A CN117255680A (zh) | 2021-11-24 | 2022-11-22 | 含有三嗪衍生物的口服给与的制剂 |
| US18/277,445 US20240148741A1 (en) | 2021-11-24 | 2022-11-22 | Formulation for oral administration, comprising triazine derivative |
| JP2023048827A JP7466731B2 (ja) | 2021-11-24 | 2023-03-24 | トリアジン誘導体を含有する経口投与する製剤 |
Applications Claiming Priority (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-189932 | 2021-11-24 | ||
| JP2021189932 | 2021-11-24 | ||
| JP2021-191635 | 2021-11-26 | ||
| JP2021191635 | 2021-11-26 | ||
| JP2022-006725 | 2022-01-19 | ||
| JP2022006725 | 2022-01-19 | ||
| JP2022012386 | 2022-01-28 | ||
| JP2022-012386 | 2022-01-28 | ||
| JP2022017132 | 2022-02-07 | ||
| JP2022-017132 | 2022-02-07 | ||
| JP2022027629 | 2022-02-25 | ||
| JP2022-027629 | 2022-02-25 | ||
| JP2022046304 | 2022-03-23 | ||
| JP2022-046304 | 2022-03-23 | ||
| JP2022142767 | 2022-09-08 | ||
| JP2022-142767 | 2022-09-08 | ||
| CN202211151791.9A CN116514734A (zh) | 2021-11-24 | 2022-09-21 | 三嗪衍生物的制造方法和含有三嗪衍生物的口服给与的制剂 |
| CN202211151791.9 | 2022-09-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023027198A1 true WO2023027198A1 (ja) | 2023-03-02 |
Family
ID=85321709
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/043092 Ceased WO2023027198A1 (ja) | 2021-11-24 | 2022-11-22 | トリアジン誘導体を含有する経口投与する製剤 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20240148741A1 (https=) |
| EP (1) | EP4289432A4 (https=) |
| JP (2) | JP7253866B1 (https=) |
| KR (1) | KR20230119022A (https=) |
| AU (1) | AU2022333823A1 (https=) |
| WO (1) | WO2023027198A1 (https=) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116554106A (zh) * | 2023-04-28 | 2023-08-08 | 扬州市普林斯医药科技有限公司 | 一种6-氯-2-甲基-2h-吲唑-5-胺的制备方法 |
| CN116589410A (zh) * | 2023-05-29 | 2023-08-15 | 济南国鼎医药科技有限公司 | 一种5-氯-2-甲基-2h-吲唑-6-胺的合成方法 |
| CN116621817A (zh) * | 2023-07-20 | 2023-08-22 | 爱斯特(成都)生物制药股份有限公司 | 一种富马酸恩赛特韦的晶型及其制备方法、药物组合物和用途 |
| EP4122926A4 (en) * | 2021-04-14 | 2023-08-30 | Shionogi & Co., Ltd | TRIAZINE DERIVATIVE WITH VIRUS REPRODUCTION INHIBITING ACTIVITY AND PHARMACEUTICAL COMPOSITION THEREOF |
| WO2023193818A1 (zh) * | 2022-04-08 | 2023-10-12 | 湖北九康通生物医药有限公司 | 一种多取代三嗪烷类化合物的合成方法 |
| CN117357531A (zh) * | 2023-09-08 | 2024-01-09 | 湖北九康通生物医药有限公司 | 一种药物组合物、其制备方法和其应用 |
| JP2024045113A (ja) * | 2022-05-10 | 2024-04-02 | 北京遠大九和薬業有限公司 | 化合物及びフマル酸の結晶形態、医薬組成物及びコロナウイルス誘発性疾患の治療方法 |
| US11963967B2 (en) | 2020-10-16 | 2024-04-23 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US12030904B2 (en) | 2020-08-24 | 2024-07-09 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US12091420B2 (en) | 2022-08-05 | 2024-09-17 | Gilead Sciences, Inc. | SARS-COV2 main protease inhibitors |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4289432A4 (en) * | 2021-11-24 | 2025-01-15 | Shionogi & Co., Ltd | PREPARATION FOR ORAL ADMINISTRATION CONTAINING TRIAZINE DERIVATIVES |
| WO2023054732A2 (ja) * | 2022-01-19 | 2023-04-06 | 塩野義製薬株式会社 | 新型コロナウイルス感染症治療用医薬 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010092966A1 (ja) | 2009-02-13 | 2010-08-19 | 塩野義製薬株式会社 | 新規トリアジン誘導体およびそれを含有する医薬組成物 |
| WO2012009258A2 (en) | 2010-07-13 | 2012-01-19 | Edward Roberts | Peptidomimetic galanin receptor modulators |
| WO2012020749A1 (ja) | 2010-08-10 | 2012-02-16 | 塩野義製薬株式会社 | トリアジン誘導体およびそれを含有する鎮痛作用を有する医薬組成物 |
| WO2013089212A1 (ja) | 2011-12-15 | 2013-06-20 | 塩野義製薬株式会社 | 置換トリアジン誘導体およびそれらを含有する医薬組成物 |
| WO2014200078A1 (ja) | 2013-06-14 | 2014-12-18 | 塩野義製薬株式会社 | アミノトリアジン誘導体およびそれらを含有する医薬組成物 |
| WO2021205298A1 (en) | 2020-04-05 | 2021-10-14 | Pfizer Inc. | Compounds and methods for the treatment of covid-19 |
| CN113620888A (zh) | 2021-09-27 | 2021-11-09 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物及其制备方法和应用 |
| CN113666914A (zh) | 2021-09-27 | 2021-11-19 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物及其制备方法和用途 |
| CN113735838A (zh) | 2021-09-27 | 2021-12-03 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物、其制备方法及用途 |
| CN113773300A (zh) | 2021-09-27 | 2021-12-10 | 成都施贝康生物医药科技有限公司 | 磺酰胺类化合物、其制备方法及用途 |
| WO2021250648A1 (en) | 2020-09-03 | 2021-12-16 | Pfizer Inc. | Nitrile-containing antiviral compounds |
| CN113801097A (zh) | 2021-09-27 | 2021-12-17 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物、其制备方法及用途 |
| WO2022138988A1 (ja) * | 2021-04-14 | 2022-06-30 | 塩野義製薬株式会社 | ウイルス増殖阻害作用を有するトリアジン誘導体およびそれらを含有する医薬組成物 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999036410A1 (en) * | 1998-01-13 | 1999-07-22 | Scriptgen Pharmaceuticals, Inc. | Triazine antiviral compounds |
| WO2019087884A1 (ja) * | 2017-10-31 | 2019-05-09 | 富士フイルム株式会社 | 組成物、抗菌組成物、抗ウイルス用組成物、抗ノロウイルス用組成物、スプレー、ワイパー |
| EP4282420A4 (en) * | 2021-01-20 | 2024-11-27 | National University Corporation Hokkaido University | ANTIVIRAL AGENT |
| EP4313097A4 (en) * | 2021-03-29 | 2025-03-19 | Shireen Nature Company for General Trading, Ltd. | Willow extract and its use in treating coronavirus infection, inflammation, and associated medical conditions |
| EP4289432A4 (en) | 2021-11-24 | 2025-01-15 | Shionogi & Co., Ltd | PREPARATION FOR ORAL ADMINISTRATION CONTAINING TRIAZINE DERIVATIVES |
-
2022
- 2022-11-22 EP EP22861484.8A patent/EP4289432A4/en active Pending
- 2022-11-22 WO PCT/JP2022/043092 patent/WO2023027198A1/ja not_active Ceased
- 2022-11-22 US US18/277,445 patent/US20240148741A1/en active Pending
- 2022-11-22 JP JP2022574839A patent/JP7253866B1/ja active Active
- 2022-11-22 KR KR1020237025438A patent/KR20230119022A/ko active Pending
- 2022-11-22 AU AU2022333823A patent/AU2022333823A1/en active Pending
-
2023
- 2023-03-24 JP JP2023048827A patent/JP7466731B2/ja active Active
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010092966A1 (ja) | 2009-02-13 | 2010-08-19 | 塩野義製薬株式会社 | 新規トリアジン誘導体およびそれを含有する医薬組成物 |
| WO2012009258A2 (en) | 2010-07-13 | 2012-01-19 | Edward Roberts | Peptidomimetic galanin receptor modulators |
| WO2012020749A1 (ja) | 2010-08-10 | 2012-02-16 | 塩野義製薬株式会社 | トリアジン誘導体およびそれを含有する鎮痛作用を有する医薬組成物 |
| WO2013089212A1 (ja) | 2011-12-15 | 2013-06-20 | 塩野義製薬株式会社 | 置換トリアジン誘導体およびそれらを含有する医薬組成物 |
| WO2014200078A1 (ja) | 2013-06-14 | 2014-12-18 | 塩野義製薬株式会社 | アミノトリアジン誘導体およびそれらを含有する医薬組成物 |
| WO2021205298A1 (en) | 2020-04-05 | 2021-10-14 | Pfizer Inc. | Compounds and methods for the treatment of covid-19 |
| WO2021250648A1 (en) | 2020-09-03 | 2021-12-16 | Pfizer Inc. | Nitrile-containing antiviral compounds |
| WO2022138988A1 (ja) * | 2021-04-14 | 2022-06-30 | 塩野義製薬株式会社 | ウイルス増殖阻害作用を有するトリアジン誘導体およびそれらを含有する医薬組成物 |
| CN113620888A (zh) | 2021-09-27 | 2021-11-09 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物及其制备方法和应用 |
| CN113773300A (zh) | 2021-09-27 | 2021-12-10 | 成都施贝康生物医药科技有限公司 | 磺酰胺类化合物、其制备方法及用途 |
| CN113735838A (zh) | 2021-09-27 | 2021-12-03 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物、其制备方法及用途 |
| CN113801097A (zh) | 2021-09-27 | 2021-12-17 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物、其制备方法及用途 |
| CN113666914A (zh) | 2021-09-27 | 2021-11-19 | 成都施贝康生物医药科技有限公司 | 二氢嘧啶类化合物及其制备方法和用途 |
Non-Patent Citations (28)
| Title |
|---|
| "A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF'-00835231 as a potential new treatment for COVID-l_9", JOURNAL OF VIROLOGY, 26 April 2021 (2021-04-26), Retrieved from the Internet <URL:https://journals.asm.org/doi/10.1128/JVI.01819-20><doi:10.1128/JVI.01819-20> |
| "COVID-19 Dashboard by the Center for Systems Science and Engineering at Johns Hopkins University", 21 September 2022, JOHNS HOPKINS UNIVERSITY |
| "Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19", BIORXIV, Retrieved from the Internet <URL:https://doi.org/10.1101/2022.01.26.477782> |
| "Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19", J. MED. CHEM., vol. 65, 2022, pages 6499 - 6512 |
| "Isotopes in the Physical and Biomedical Sciences", vol. 1, 1987, article "Labeled Compounds" |
| "Japanese Pharmacopoeia" |
| "Pfizer's Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk Of Hospitalization Or Death By 89°,% In Interim Analysis Of Phase 2/3 EPIC-HR Study", 5 November 2021, PFIZER PRESS RELEASE |
| "Report of the WHO-China Joint Mission on Coronavirus disease 2019 (COVID-19", 28 February 2020, WHO |
| 261ST AM CHEM SOC (ACS) NATL MEET, 5 April 2021 (2021-04-05) |
| A(ΗS CENTRAL SCIENCE, vol. 7, no. 3, 2021 |
| AIMECS 2021 (AFMC INTERNATIONAL MEDICINAL CHEMISTRY SYMPOSIUM 2021, 2 December 2021 (2021-12-02) |
| AKAJI KENICHI, KONNO HIROYUKI: "Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease", MOLECULES, vol. 25, no. 17, pages 3920, XP055826867, DOI: 10.3390/molecules25173920 * |
| ARZNEIMITTEL-FORSCLMNG, vol. 6, 1984, pages 663 - 668 |
| ATHERTON, E.SHEPPARD, R. C.: "In Solid Phase Peptide Synthesis, A Practical Approach", 1989, IRL PRESS AT OXFORD UNIVERSITY PRES |
| BIOORG. MED. CHEM., vol. 5, 1997, pages 1883 - 1891 |
| CANCER TREATMENT REVIEWS, vol. 11, 1984, pages 99 - 110 |
| CELL RESEARCH, vol. 30, 2020, pages 678 - 692 |
| CONTRIBUTIONS TO ONCOLOGY, vol. 18, 1984 |
| LIU YUZHI; LIANG CHENGYUAN; XIN LIANG; REN XIAODONG; TIAN LEI; JU XINGKE; LI HAN; WANG YONGBO; ZHAO QIANQIAN; LIU HONG; CAO WENQIA: "The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, ELSEVIER, AMSTERDAM, NL, vol. 206, 6 August 2020 (2020-08-06), AMSTERDAM, NL , XP086299093, ISSN: 0223-5234, DOI: 10.1016/j.ejmech.2020.112711 * |
| OLUBIYI OLUJIDE O., OLAGUNJU MARYAM, KEUTMANN MONIKA, LOSCHWITZ JENNIFER, STRODEL BIRGIT: "High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2", MOLECULES, vol. 25, no. 14, pages 3193, XP055944494, DOI: 10.3390/molecules25143193 * |
| SAKURAI TOSHIO: "Manual of X-ray structural analysis", 1983, SHOKABO CO., LTD. |
| SCIENCE, vol. 300, 2003, pages 1763 - 1767 |
| SCIENCE, vol. 368, 2020, pages 409 - 412 |
| SCIENCE, vol. 374, 2021, pages 1586 - 1593 |
| See also references of EP4289432A4 |
| STOUTJENSEN: "X-Ray Structure Determination: A Practical Guide", 1968, MACMILLAN CO. |
| THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 382, 2020, pages 1564 - 1567 |
| UNOH YUTO, UNOH YUTO, UEHARA SHOTA, NAKAHARA KENJI, NOBORI HARUAKI, YAMATSU YUKIKO, YAMAMOTO SHIHO, MARUYAMA YUKI, TAODA YOSHIYUKI: "Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 65, no. 9, 12 May 2022 (2022-05-12), US , pages 6499 - 6512, XP093007183, ISSN: 0022-2623, DOI: 10.1021/acs.jmedchem.2c00117 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12473314B2 (en) | 2020-08-24 | 2025-11-18 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US12030904B2 (en) | 2020-08-24 | 2024-07-09 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US11963967B2 (en) | 2020-10-16 | 2024-04-23 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| US12208110B2 (en) | 2020-10-16 | 2025-01-28 | Gilead Sciences, Inc. | Phospholipid compounds and uses thereof |
| EP4400502A3 (en) * | 2021-04-14 | 2025-01-01 | Shionogi & Co., Ltd | Triazine derivatives having virus replication inhibitory activity and pharmaceutical composition comprising the same |
| EP4122926A4 (en) * | 2021-04-14 | 2023-08-30 | Shionogi & Co., Ltd | TRIAZINE DERIVATIVE WITH VIRUS REPRODUCTION INHIBITING ACTIVITY AND PHARMACEUTICAL COMPOSITION THEREOF |
| US12559474B2 (en) | 2021-04-14 | 2026-02-24 | Shionogi & Co., Ltd. | Triazine derivatives having virus replication inhibitory activity and pharmaceutical composition comprising the same |
| WO2023193818A1 (zh) * | 2022-04-08 | 2023-10-12 | 湖北九康通生物医药有限公司 | 一种多取代三嗪烷类化合物的合成方法 |
| JP2024045113A (ja) * | 2022-05-10 | 2024-04-02 | 北京遠大九和薬業有限公司 | 化合物及びフマル酸の結晶形態、医薬組成物及びコロナウイルス誘発性疾患の治療方法 |
| JP7669325B2 (ja) | 2022-05-10 | 2025-04-28 | 北京遠大九和薬業有限公司 | 化合物及びフマル酸の結晶形態、医薬組成物及びコロナウイルス誘発性疾患の治療方法 |
| US12091420B2 (en) | 2022-08-05 | 2024-09-17 | Gilead Sciences, Inc. | SARS-COV2 main protease inhibitors |
| US12410183B2 (en) | 2022-08-05 | 2025-09-09 | Gilead Sciences, Inc. | Sars-cov2 main protease inhibitors |
| CN116554106A (zh) * | 2023-04-28 | 2023-08-08 | 扬州市普林斯医药科技有限公司 | 一种6-氯-2-甲基-2h-吲唑-5-胺的制备方法 |
| CN116589410B (zh) * | 2023-05-29 | 2024-05-03 | 济南国鼎医药科技有限公司 | 一种6-氯-2-甲基-2h-吲唑-5-胺的合成方法 |
| CN116589410A (zh) * | 2023-05-29 | 2023-08-15 | 济南国鼎医药科技有限公司 | 一种5-氯-2-甲基-2h-吲唑-6-胺的合成方法 |
| CN116621817A (zh) * | 2023-07-20 | 2023-08-22 | 爱斯特(成都)生物制药股份有限公司 | 一种富马酸恩赛特韦的晶型及其制备方法、药物组合物和用途 |
| CN116621817B (zh) * | 2023-07-20 | 2023-09-29 | 爱斯特(成都)生物制药股份有限公司 | 一种富马酸恩赛特韦的晶型及其制备方法、药物组合物和用途 |
| CN117357531A (zh) * | 2023-09-08 | 2024-01-09 | 湖北九康通生物医药有限公司 | 一种药物组合物、其制备方法和其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20230119022A (ko) | 2023-08-14 |
| JP7253866B1 (ja) | 2023-04-07 |
| AU2022333823A1 (en) | 2024-05-02 |
| JP7466731B2 (ja) | 2024-04-12 |
| US20240148741A1 (en) | 2024-05-09 |
| EP4289432A4 (en) | 2025-01-15 |
| EP4289432A1 (en) | 2023-12-13 |
| JPWO2023027198A1 (https=) | 2023-03-02 |
| JP2023078413A (ja) | 2023-06-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7253866B1 (ja) | トリアジン誘導体を含有する経口投与する製剤 | |
| JP7105430B1 (ja) | ウイルス増殖阻害作用を有するトリアジン誘導体およびそれらを含有する医薬組成物 | |
| WO2023054292A1 (ja) | トリアジン誘導体を含有する医薬組成物 | |
| CN116514734A (zh) | 三嗪衍生物的制造方法和含有三嗪衍生物的口服给与的制剂 | |
| JP2021073241A (ja) | 癌を処置する方法 | |
| AU2017214589A1 (en) | Sulfonamide derivative and pharmaceutical composition containing same | |
| JP7236065B1 (ja) | トリアジン誘導体を含有する医薬組成物 | |
| KR20250004704A (ko) | 바이러스 증식 저해 활성을 갖는 유라실 유도체 및 그들을 함유하는 의약 조성물 | |
| US20250025459A1 (en) | Covid-19 treatment medicine characterized by combining 3cl protease inhibitor and covid-19 treatment drug | |
| JP2024178473A (ja) | ウイルス増殖阻害活性を有する二環性含窒素複素環誘導体およびそれらを含有する医薬組成物 | |
| JP7261529B1 (ja) | ウイルス増殖阻害作用を有するトリアジン誘導体の製造方法 | |
| CN108929329B (zh) | 2-氮杂环-5-三氟甲基-8-硝基苯并(硫代)吡喃-4-酮类化合物 | |
| CN116782904A (zh) | 含有三嗪衍生物的医药组合物 | |
| JP7701585B2 (ja) | ウラシル誘導体を含有する製剤 | |
| RU2806042C1 (ru) | Триазиновые производные, имеющие ингибиторную активность в отношении репликации вируса, и содержащая их фармацевтическая композиция | |
| TWI916636B (zh) | 新型冠狀病毒感染症治療用醫藥 | |
| HK40114521A (en) | Pharmaceutical composition containing triazine derivative | |
| AU2024307335A1 (en) | Pharmaceutical composition containing uracil derivative | |
| BR122023002208A2 (pt) | Composições farmacêuticas compreendendo derivados de triazina tendo efeito inibidor de propagação de vírus, inibidores e usos | |
| BR122023002208B1 (pt) | Composições farmacêuticas compreendendo derivados de triazina para o tratamento e/ou prevenção de covid-19 | |
| WO2025074997A1 (ja) | ウイルス増殖阻害活性を有する縮合複素環誘導体およびそれらを含有する医薬組成物 | |
| BR112022023272B1 (pt) | Derivados de triazina tendo efeito inibidor de propagação de vírus e cocristal do mesmo com ácido fumárico | |
| JPWO2020045475A1 (ja) | ピラゾロ[3,4−d]ピリミジンの結晶 | |
| HK1167644A1 (en) | 2-[[[2-[(hydroxyacetyl)amino]-4-pyridinyl]methyl]thio]-n-[4-(trifluoromethoxy)phenyl]-3-pyridinecarboxamide benzene- sulfonate, crystals of same, polymorphs thereof, and processes for production thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022574839 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22861484 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280007992.3 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 20237025438 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18277445 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022861484 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022861484 Country of ref document: EP Effective date: 20230907 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2022333823 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2022333823 Country of ref document: AU Date of ref document: 20221122 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11202402544W Country of ref document: SG |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |