WO2021052402A1 - 一种喜树碱衍生物及其偶联物 - Google Patents
一种喜树碱衍生物及其偶联物 Download PDFInfo
- Publication number
- WO2021052402A1 WO2021052402A1 PCT/CN2020/115807 CN2020115807W WO2021052402A1 WO 2021052402 A1 WO2021052402 A1 WO 2021052402A1 CN 2020115807 W CN2020115807 W CN 2020115807W WO 2021052402 A1 WO2021052402 A1 WO 2021052402A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- antibody
- alkyl
- cycloalkyl
- compound
- Prior art date
Links
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical class C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 title claims abstract description 29
- 239000000611 antibody drug conjugate Substances 0.000 claims abstract description 90
- 229940049595 antibody-drug conjugate Drugs 0.000 claims abstract description 90
- 239000003814 drug Substances 0.000 claims abstract description 74
- 229940079593 drug Drugs 0.000 claims abstract description 70
- 125000000217 alkyl group Chemical group 0.000 claims description 232
- 150000001875 compounds Chemical class 0.000 claims description 169
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 108
- 125000000623 heterocyclic group Chemical group 0.000 claims description 98
- 229910052805 deuterium Inorganic materials 0.000 claims description 91
- 150000003839 salts Chemical class 0.000 claims description 91
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 89
- 125000003118 aryl group Chemical group 0.000 claims description 87
- 229910052736 halogen Inorganic materials 0.000 claims description 86
- 150000002367 halogens Chemical class 0.000 claims description 85
- 125000001072 heteroaryl group Chemical group 0.000 claims description 84
- -1 cyano, amino Chemical group 0.000 claims description 77
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 77
- 125000003107 substituted aryl group Chemical group 0.000 claims description 68
- 239000012453 solvate Substances 0.000 claims description 61
- 239000000427 antigen Substances 0.000 claims description 48
- 108091007433 antigens Proteins 0.000 claims description 48
- 102000036639 antigens Human genes 0.000 claims description 48
- 239000000203 mixture Substances 0.000 claims description 48
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 47
- 206010028980 Neoplasm Diseases 0.000 claims description 45
- 125000004432 carbon atom Chemical group C* 0.000 claims description 45
- 125000004431 deuterium atom Chemical group 0.000 claims description 42
- 125000001188 haloalkyl group Chemical group 0.000 claims description 40
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 36
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 35
- 235000001014 amino acid Nutrition 0.000 claims description 33
- 150000001413 amino acids Chemical class 0.000 claims description 33
- 108090000623 proteins and genes Proteins 0.000 claims description 33
- 239000012634 fragment Substances 0.000 claims description 31
- 125000003545 alkoxy group Chemical group 0.000 claims description 30
- 125000001424 substituent group Chemical group 0.000 claims description 29
- 125000004429 atom Chemical group 0.000 claims description 28
- 230000027455 binding Effects 0.000 claims description 25
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 25
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 25
- 235000018102 proteins Nutrition 0.000 claims description 24
- 102000004169 proteins and genes Human genes 0.000 claims description 24
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 23
- 239000000126 substance Substances 0.000 claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 claims description 20
- 238000002360 preparation method Methods 0.000 claims description 20
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 18
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 18
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 17
- 239000002904 solvent Substances 0.000 claims description 17
- 229910052717 sulfur Inorganic materials 0.000 claims description 17
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims description 16
- 201000011510 cancer Diseases 0.000 claims description 16
- 125000003277 amino group Chemical group 0.000 claims description 15
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 14
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 14
- 241001529936 Murinae Species 0.000 claims description 11
- 229910052760 oxygen Inorganic materials 0.000 claims description 11
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 11
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 8
- 150000001335 aliphatic alkanes Chemical class 0.000 claims description 8
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 claims description 8
- 239000003085 diluting agent Substances 0.000 claims description 8
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 8
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 125000005842 heteroatom Chemical group 0.000 claims description 7
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 7
- 206010006187 Breast cancer Diseases 0.000 claims description 6
- 208000032612 Glial tumor Diseases 0.000 claims description 6
- 206010018338 Glioma Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 6
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims description 6
- 208000026310 Breast neoplasm Diseases 0.000 claims description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 5
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 5
- 206010060862 Prostate cancer Diseases 0.000 claims description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 5
- 125000000539 amino acid group Chemical group 0.000 claims description 5
- 239000000562 conjugate Substances 0.000 claims description 5
- 238000005859 coupling reaction Methods 0.000 claims description 5
- 201000005202 lung cancer Diseases 0.000 claims description 5
- 208000020816 lung neoplasm Diseases 0.000 claims description 5
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 5
- 201000002528 pancreatic cancer Diseases 0.000 claims description 5
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 5
- 229920001184 polypeptide Polymers 0.000 claims description 5
- 206010005003 Bladder cancer Diseases 0.000 claims description 4
- 206010005949 Bone cancer Diseases 0.000 claims description 4
- 208000018084 Bone neoplasm Diseases 0.000 claims description 4
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 4
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 4
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 4
- 206010033128 Ovarian cancer Diseases 0.000 claims description 4
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 4
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 4
- 206010038389 Renal cancer Diseases 0.000 claims description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 4
- 206010046431 Urethral cancer Diseases 0.000 claims description 4
- 206010046458 Urethral neoplasms Diseases 0.000 claims description 4
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 4
- 208000029742 colonic neoplasm Diseases 0.000 claims description 4
- 201000004101 esophageal cancer Diseases 0.000 claims description 4
- 206010017758 gastric cancer Diseases 0.000 claims description 4
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 4
- 201000010982 kidney cancer Diseases 0.000 claims description 4
- 201000007270 liver cancer Diseases 0.000 claims description 4
- 208000014018 liver neoplasm Diseases 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 206010038038 rectal cancer Diseases 0.000 claims description 4
- 201000001275 rectum cancer Diseases 0.000 claims description 4
- 201000011549 stomach cancer Diseases 0.000 claims description 4
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 4
- VNEOHNUYPRAJMX-UHFFFAOYSA-N 3-[[2-[[2-amino-3-(1h-indol-3-yl)propanoyl]amino]-4-methylpentanoyl]amino]-4-[[1-(butoxycarbonylamino)-1-oxo-3-phenylpropan-2-yl]amino]-4-oxobutanoic acid Chemical compound C=1NC2=CC=CC=C2C=1CC(N)C(=O)NC(CC(C)C)C(=O)NC(CC(O)=O)C(=O)NC(C(=O)NC(=O)OCCCC)CC1=CC=CC=C1 VNEOHNUYPRAJMX-UHFFFAOYSA-N 0.000 claims description 3
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 3
- 206010014733 Endometrial cancer Diseases 0.000 claims description 3
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 3
- 239000004471 Glycine Substances 0.000 claims description 3
- 101000606465 Homo sapiens Inactive tyrosine-protein kinase 7 Proteins 0.000 claims description 3
- 102100039813 Inactive tyrosine-protein kinase 7 Human genes 0.000 claims description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 3
- 206010025323 Lymphomas Diseases 0.000 claims description 3
- 206010029260 Neuroblastoma Diseases 0.000 claims description 3
- 108091006576 SLC34A2 Proteins 0.000 claims description 3
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 claims description 3
- 206010061934 Salivary gland cancer Diseases 0.000 claims description 3
- 206010039491 Sarcoma Diseases 0.000 claims description 3
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 3
- 102100038437 Sodium-dependent phosphate transport protein 2B Human genes 0.000 claims description 3
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 3
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 3
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 3
- 201000010881 cervical cancer Diseases 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 claims description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 3
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 claims description 3
- 201000000849 skin cancer Diseases 0.000 claims description 3
- 201000002510 thyroid cancer Diseases 0.000 claims description 3
- 206010046766 uterine cancer Diseases 0.000 claims description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 2
- OBVVKJDJORDBCH-CZDIJEQGSA-N 2-aminoacetic acid (2S)-2-amino-3-phenylpropanoic acid Chemical compound NCC(O)=O.NCC(O)=O.NCC(O)=O.OC(=O)[C@@H](N)CC1=CC=CC=C1 OBVVKJDJORDBCH-CZDIJEQGSA-N 0.000 claims description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 2
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 claims description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 2
- 239000004472 Lysine Substances 0.000 claims description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 2
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 claims description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 2
- 235000003704 aspartic acid Nutrition 0.000 claims description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 claims description 2
- 235000013477 citrulline Nutrition 0.000 claims description 2
- 229960002173 citrulline Drugs 0.000 claims description 2
- 235000013922 glutamic acid Nutrition 0.000 claims description 2
- 239000004220 glutamic acid Substances 0.000 claims description 2
- 201000005787 hematologic cancer Diseases 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 239000004474 valine Substances 0.000 claims description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 8
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical group N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 claims 1
- 102100033942 Ephrin-A4 Human genes 0.000 claims 1
- 101000925259 Homo sapiens Ephrin-A4 Proteins 0.000 claims 1
- 230000000259 anti-tumor effect Effects 0.000 abstract description 9
- 239000002246 antineoplastic agent Substances 0.000 abstract description 5
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 abstract description 3
- 229940041181 antineoplastic drug Drugs 0.000 abstract description 3
- 229940127093 camptothecin Drugs 0.000 abstract description 3
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 abstract description 3
- 230000004048 modification Effects 0.000 abstract description 2
- 238000012986 modification Methods 0.000 abstract description 2
- 230000021615 conjugation Effects 0.000 abstract 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 171
- 230000015572 biosynthetic process Effects 0.000 description 143
- 238000003786 synthesis reaction Methods 0.000 description 142
- 238000006243 chemical reaction Methods 0.000 description 94
- 239000000243 solution Substances 0.000 description 74
- 210000004027 cell Anatomy 0.000 description 51
- 150000001975 deuterium Chemical group 0.000 description 50
- 230000002829 reductive effect Effects 0.000 description 46
- 239000007787 solid Substances 0.000 description 43
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 37
- 125000005647 linker group Chemical group 0.000 description 32
- 238000000034 method Methods 0.000 description 32
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 28
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 26
- 238000002953 preparative HPLC Methods 0.000 description 24
- 239000003446 ligand Substances 0.000 description 23
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 21
- 238000010168 coupling process Methods 0.000 description 21
- 239000005457 ice water Substances 0.000 description 20
- 229960000575 trastuzumab Drugs 0.000 description 17
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 239000003053 toxin Substances 0.000 description 15
- 231100000765 toxin Toxicity 0.000 description 15
- 108700012359 toxins Proteins 0.000 description 15
- 229910052739 hydrogen Inorganic materials 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 238000001514 detection method Methods 0.000 description 11
- 239000001257 hydrogen Substances 0.000 description 11
- 238000002347 injection Methods 0.000 description 11
- 239000007924 injection Substances 0.000 description 11
- 239000002609 medium Substances 0.000 description 11
- BMTZEAOGFDXDAD-UHFFFAOYSA-M 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium;chloride Chemical compound [Cl-].COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 BMTZEAOGFDXDAD-UHFFFAOYSA-M 0.000 description 10
- VCUXVXLUOHDHKK-UHFFFAOYSA-N 2-(2-aminopyrimidin-4-yl)-4-(2-chloro-4-methoxyphenyl)-1,3-thiazole-5-carboxamide Chemical compound ClC1=CC(OC)=CC=C1C1=C(C(N)=O)SC(C=2N=C(N)N=CC=2)=N1 VCUXVXLUOHDHKK-UHFFFAOYSA-N 0.000 description 9
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 9
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 9
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 229940125846 compound 25 Drugs 0.000 description 9
- 229940125851 compound 27 Drugs 0.000 description 9
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 9
- GWNFQAKCJYEJEW-UHFFFAOYSA-N ethyl 3-[8-[[4-methyl-5-[(3-methyl-4-oxophthalazin-1-yl)methyl]-1,2,4-triazol-3-yl]sulfanyl]octanoylamino]benzoate Chemical compound CCOC(=O)C1=CC(NC(=O)CCCCCCCSC2=NN=C(CC3=NN(C)C(=O)C4=CC=CC=C34)N2C)=CC=C1 GWNFQAKCJYEJEW-UHFFFAOYSA-N 0.000 description 9
- 239000004530 micro-emulsion Substances 0.000 description 9
- 150000007523 nucleic acids Chemical class 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- 231100000331 toxic Toxicity 0.000 description 9
- 230000002588 toxic effect Effects 0.000 description 9
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 8
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 8
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 239000004480 active ingredient Substances 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 239000012298 atmosphere Substances 0.000 description 8
- 238000007796 conventional method Methods 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 102000039446 nucleic acids Human genes 0.000 description 8
- 108020004707 nucleic acids Proteins 0.000 description 8
- 125000004434 sulfur atom Chemical group 0.000 description 8
- 239000013598 vector Substances 0.000 description 8
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 7
- 108060003951 Immunoglobulin Proteins 0.000 description 7
- 239000007900 aqueous suspension Substances 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 102000018358 immunoglobulin Human genes 0.000 description 7
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- 125000003342 alkenyl group Chemical group 0.000 description 6
- 125000003282 alkyl amino group Chemical group 0.000 description 6
- 125000000304 alkynyl group Chemical group 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 125000005366 cycloalkylthio group Chemical group 0.000 description 6
- 239000013604 expression vector Substances 0.000 description 6
- 210000004602 germ cell Anatomy 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- VMFMUJZRXZXYAH-UHFFFAOYSA-N n-[5-[[5-chloro-4-[2-[2-(dimethylamino)-2-oxoacetyl]anilino]pyrimidin-2-yl]amino]-4-methoxy-2-(4-methylpiperazin-1-yl)phenyl]prop-2-enamide Chemical compound C=CC(=O)NC=1C=C(NC=2N=C(NC=3C(=CC=CC=3)C(=O)C(=O)N(C)C)C(Cl)=CN=2)C(OC)=CC=1N1CCN(C)CC1 VMFMUJZRXZXYAH-UHFFFAOYSA-N 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 230000008685 targeting Effects 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 5
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- 229940127007 Compound 39 Drugs 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 229910020008 S(O) Inorganic materials 0.000 description 5
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- 125000000000 cycloalkoxy group Chemical group 0.000 description 5
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 5
- 229940127089 cytotoxic agent Drugs 0.000 description 5
- 239000000796 flavoring agent Substances 0.000 description 5
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000012053 oil suspension Substances 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 125000006413 ring segment Chemical group 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- 239000000080 wetting agent Substances 0.000 description 5
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 4
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 4
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 0 CCC(CC(C=C1N2Cc3c([C@](CCc4c(*)c(*)c5)N(*)*)c4c5nc13)=C(CO1)C2=O)C1=* Chemical compound CCC(CC(C=C1N2Cc3c([C@](CCc4c(*)c(*)c5)N(*)*)c4c5nc13)=C(CO1)C2=O)C1=* 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 125000003275 alpha amino acid group Chemical group 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 229940125773 compound 10 Drugs 0.000 description 4
- 229940127113 compound 57 Drugs 0.000 description 4
- 239000002254 cytotoxic agent Substances 0.000 description 4
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 238000011068 loading method Methods 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 239000003765 sweetening agent Substances 0.000 description 4
- 238000010189 synthetic method Methods 0.000 description 4
- GCTFTMWXZFLTRR-GFCCVEGCSA-N (2r)-2-amino-n-[3-(difluoromethoxy)-4-(1,3-oxazol-5-yl)phenyl]-4-methylpentanamide Chemical compound FC(F)OC1=CC(NC(=O)[C@H](N)CC(C)C)=CC=C1C1=CN=CO1 GCTFTMWXZFLTRR-GFCCVEGCSA-N 0.000 description 3
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 3
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 3
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 3
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 3
- OMBVEVHRIQULKW-DNQXCXABSA-M (3r,5r)-7-[3-(4-fluorophenyl)-8-oxo-7-phenyl-1-propan-2-yl-5,6-dihydro-4h-pyrrolo[2,3-c]azepin-2-yl]-3,5-dihydroxyheptanoate Chemical compound O=C1C=2N(C(C)C)C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C=3C=CC(F)=CC=3)C=2CCCN1C1=CC=CC=C1 OMBVEVHRIQULKW-DNQXCXABSA-M 0.000 description 3
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 3
- MHSLDASSAFCCDO-UHFFFAOYSA-N 1-(5-tert-butyl-2-methylpyrazol-3-yl)-3-(4-pyridin-4-yloxyphenyl)urea Chemical compound CN1N=C(C(C)(C)C)C=C1NC(=O)NC(C=C1)=CC=C1OC1=CC=NC=C1 MHSLDASSAFCCDO-UHFFFAOYSA-N 0.000 description 3
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 3
- SCVJRXQHFJXZFZ-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound C1=2NC(N)=NC(=S)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)O1 SCVJRXQHFJXZFZ-KVQBGUIXSA-N 0.000 description 3
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 3
- QISAUDWTBBNJIR-UHFFFAOYSA-N 2-phenylmethoxyacetyl chloride Chemical compound ClC(=O)COCC1=CC=CC=C1 QISAUDWTBBNJIR-UHFFFAOYSA-N 0.000 description 3
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 3
- 101710117290 Aldo-keto reductase family 1 member C4 Proteins 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 3
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 125000004414 alkyl thio group Chemical group 0.000 description 3
- 235000010323 ascorbic acid Nutrition 0.000 description 3
- 239000011668 ascorbic acid Substances 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 229940125877 compound 31 Drugs 0.000 description 3
- 229940126540 compound 41 Drugs 0.000 description 3
- 229940125844 compound 46 Drugs 0.000 description 3
- 229940126545 compound 53 Drugs 0.000 description 3
- 229940125900 compound 59 Drugs 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 239000007884 disintegrant Substances 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 229920002521 macromolecule Polymers 0.000 description 3
- ZBELDPMWYXDLNY-UHFFFAOYSA-N methyl 9-(4-bromo-2-fluoroanilino)-[1,3]thiazolo[5,4-f]quinazoline-2-carboximidate Chemical compound C12=C3SC(C(=N)OC)=NC3=CC=C2N=CN=C1NC1=CC=C(Br)C=C1F ZBELDPMWYXDLNY-UHFFFAOYSA-N 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 3
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 3
- 239000000346 nonvolatile oil Substances 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- 238000002823 phage display Methods 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- FDBYIYFVSAHJLY-UHFFFAOYSA-N resmetirom Chemical compound N1C(=O)C(C(C)C)=CC(OC=2C(=CC(=CC=2Cl)N2C(NC(=O)C(C#N)=N2)=O)Cl)=N1 FDBYIYFVSAHJLY-UHFFFAOYSA-N 0.000 description 3
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 3
- 229960001860 salicylate Drugs 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 229960001612 trastuzumab emtansine Drugs 0.000 description 3
- 238000000108 ultra-filtration Methods 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 2
- LGNCNVVZCUVPOT-FUVGGWJZSA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-methoxy-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 LGNCNVVZCUVPOT-FUVGGWJZSA-N 0.000 description 2
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 2
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 2
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 2
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 2
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 2
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 2
- 125000005916 2-methylpentyl group Chemical group 0.000 description 2
- DFRAKBCRUYUFNT-UHFFFAOYSA-N 3,8-dicyclohexyl-2,4,7,9-tetrahydro-[1,3]oxazino[5,6-h][1,3]benzoxazine Chemical compound C1CCCCC1N1CC(C=CC2=C3OCN(C2)C2CCCCC2)=C3OC1 DFRAKBCRUYUFNT-UHFFFAOYSA-N 0.000 description 2
- WFOVEDJTASPCIR-UHFFFAOYSA-N 3-[(4-methyl-5-pyridin-4-yl-1,2,4-triazol-3-yl)methylamino]-n-[[2-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound N=1N=C(C=2C=CN=CC=2)N(C)C=1CNC(C=1)=CC=CC=1C(=O)NCC1=CC=CC=C1C(F)(F)F WFOVEDJTASPCIR-UHFFFAOYSA-N 0.000 description 2
- BGAJNPLDJJBRHK-UHFFFAOYSA-N 3-[2-[5-(3-chloro-4-propan-2-yloxyphenyl)-1,3,4-thiadiazol-2-yl]-3-methyl-6,7-dihydro-4h-pyrazolo[4,3-c]pyridin-5-yl]propanoic acid Chemical compound C1=C(Cl)C(OC(C)C)=CC=C1C1=NN=C(N2C(=C3CN(CCC(O)=O)CCC3=N2)C)S1 BGAJNPLDJJBRHK-UHFFFAOYSA-N 0.000 description 2
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000005917 3-methylpentyl group Chemical group 0.000 description 2
- APRZHQXAAWPYHS-UHFFFAOYSA-N 4-[5-[3-(carboxymethoxy)phenyl]-3-(4,5-dimethyl-1,3-thiazol-2-yl)tetrazol-3-ium-2-yl]benzenesulfonate Chemical compound S1C(C)=C(C)N=C1[N+]1=NC(C=2C=C(OCC(O)=O)C=CC=2)=NN1C1=CC=C(S([O-])(=O)=O)C=C1 APRZHQXAAWPYHS-UHFFFAOYSA-N 0.000 description 2
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 2
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 2
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 2
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 2
- XASOHFCUIQARJT-UHFFFAOYSA-N 8-methoxy-6-[7-(2-morpholin-4-ylethoxy)imidazo[1,2-a]pyridin-3-yl]-2-(2,2,2-trifluoroethyl)-3,4-dihydroisoquinolin-1-one Chemical compound C(N1C(=O)C2=C(OC)C=C(C=3N4C(=NC=3)C=C(C=C4)OCCN3CCOCC3)C=C2CC1)C(F)(F)F XASOHFCUIQARJT-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108091026890 Coding region Proteins 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 2
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 102000008072 Lymphokines Human genes 0.000 description 2
- 108010074338 Lymphokines Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- AVYVHIKSFXVDBG-UHFFFAOYSA-N N-benzyl-N-hydroxy-2,2-dimethylbutanamide Chemical compound C(C1=CC=CC=C1)N(C(C(CC)(C)C)=O)O AVYVHIKSFXVDBG-UHFFFAOYSA-N 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 2
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 229940125644 antibody drug Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 229940072107 ascorbate Drugs 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229940044683 chemotherapy drug Drugs 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940127204 compound 29 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125878 compound 36 Drugs 0.000 description 2
- 229940125807 compound 37 Drugs 0.000 description 2
- 229940125936 compound 42 Drugs 0.000 description 2
- 229940127271 compound 49 Drugs 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 239000003168 generic drug Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 2
- 229940072221 immunoglobulins Drugs 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical class CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229960005558 mertansine Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- YGBMCLDVRUGXOV-UHFFFAOYSA-N n-[6-[6-chloro-5-[(4-fluorophenyl)sulfonylamino]pyridin-3-yl]-1,3-benzothiazol-2-yl]acetamide Chemical compound C1=C2SC(NC(=O)C)=NC2=CC=C1C(C=1)=CN=C(Cl)C=1NS(=O)(=O)C1=CC=C(F)C=C1 YGBMCLDVRUGXOV-UHFFFAOYSA-N 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 229940126586 small molecule drug Drugs 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 235000012424 soybean oil Nutrition 0.000 description 2
- 239000003549 soybean oil Substances 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 1
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- UVNPEUJXKZFWSJ-LMTQTHQJSA-N (R)-N-[(4S)-8-[6-amino-5-[(3,3-difluoro-2-oxo-1H-pyrrolo[2,3-b]pyridin-4-yl)sulfanyl]pyrazin-2-yl]-2-oxa-8-azaspiro[4.5]decan-4-yl]-2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@@](=O)N[C@@H]1COCC11CCN(CC1)c1cnc(Sc2ccnc3NC(=O)C(F)(F)c23)c(N)n1 UVNPEUJXKZFWSJ-LMTQTHQJSA-N 0.000 description 1
- IWYDHOAUDWTVEP-ZETCQYMHSA-N (S)-mandelic acid Chemical compound OC(=O)[C@@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-ZETCQYMHSA-N 0.000 description 1
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 1
- GQXURJDNDYACGE-UHFFFAOYSA-N 1-hydroxycyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(O)CC1 GQXURJDNDYACGE-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003764 2,4-dimethylpentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QEBYEVQKHRUYPE-UHFFFAOYSA-N 2-(2-chlorophenyl)-5-[(1-methylpyrazol-3-yl)methyl]-4-[[methyl(pyridin-3-ylmethyl)amino]methyl]-1h-pyrazolo[4,3-c]pyridine-3,6-dione Chemical compound C1=CN(C)N=C1CN1C(=O)C=C2NN(C=3C(=CC=CC=3)Cl)C(=O)C2=C1CN(C)CC1=CC=CN=C1 QEBYEVQKHRUYPE-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-L 2-(carboxymethyl)-2-hydroxysuccinate Chemical compound [O-]C(=O)CC(O)(C(=O)O)CC([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-L 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- BVKGUTLIPHZYCX-UHFFFAOYSA-N 3,3,3-trifluoro-2-hydroxypropanoic acid Chemical compound OC(=O)C(O)C(F)(F)F BVKGUTLIPHZYCX-UHFFFAOYSA-N 0.000 description 1
- 125000004336 3,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- LRNUWMFPJHMALD-UHFFFAOYSA-N 3-chloro-4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium Chemical class COC1=NC(OC)=NC([N+]2(C)C(COCC2)Cl)=N1 LRNUWMFPJHMALD-UHFFFAOYSA-N 0.000 description 1
- 125000004337 3-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 description 1
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- UIADWSXPNFQQCZ-UHFFFAOYSA-N 4-[4-(3,4-dichlorophenyl)-5-phenyl-1,3-oxazol-2-yl]butanoic acid Chemical compound ClC=1C=C(C=CC=1Cl)C=1N=C(OC=1C1=CC=CC=C1)CCCC(=O)O UIADWSXPNFQQCZ-UHFFFAOYSA-N 0.000 description 1
- MPMKMQHJHDHPBE-RUZDIDTESA-N 4-[[(2r)-1-(1-benzothiophene-3-carbonyl)-2-methylazetidine-2-carbonyl]-[(3-chlorophenyl)methyl]amino]butanoic acid Chemical compound O=C([C@@]1(N(CC1)C(=O)C=1C2=CC=CC=C2SC=1)C)N(CCCC(O)=O)CC1=CC=CC(Cl)=C1 MPMKMQHJHDHPBE-RUZDIDTESA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 108010066676 Abrin Proteins 0.000 description 1
- 101710092462 Alpha-hemolysin Proteins 0.000 description 1
- 101710197219 Alpha-toxin Proteins 0.000 description 1
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 101000669426 Aspergillus restrictus Ribonuclease mitogillin Proteins 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 238000011729 BALB/c nude mouse Methods 0.000 description 1
- 241001112741 Bacillaceae Species 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- IWTBEZMQSMGQCJ-UHFFFAOYSA-N CC(COCc1ccccc1)NCC=C Chemical compound CC(COCc1ccccc1)NCC=C IWTBEZMQSMGQCJ-UHFFFAOYSA-N 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical class [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 101710158575 Cap-specific mRNA (nucleoside-2'-O-)-methyltransferase Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 244000168525 Croton tiglium Species 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 102000003915 DNA Topoisomerases Human genes 0.000 description 1
- 108090000323 DNA Topoisomerases Proteins 0.000 description 1
- SBJKKFFYIZUCET-JLAZNSOCSA-N Dehydro-L-ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(=O)C1=O SBJKKFFYIZUCET-JLAZNSOCSA-N 0.000 description 1
- KRHAHEQEKNJCSD-UHFFFAOYSA-N Dihydroasparagusic acid Natural products OC(=O)C(CS)CS KRHAHEQEKNJCSD-UHFFFAOYSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000588921 Enterobacteriaceae Species 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 1
- 241000606768 Haemophilus influenzae Species 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000690301 Homo sapiens Aldo-keto reductase family 1 member C4 Proteins 0.000 description 1
- 101001116548 Homo sapiens Protein CBFA2T1 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 1
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 241000221089 Jatropha Species 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 229920000168 Microcrystalline cellulose Chemical class 0.000 description 1
- 244000302512 Momordica charantia Species 0.000 description 1
- 235000009811 Momordica charantia Nutrition 0.000 description 1
- NUGPIZCTELGDOS-QHCPKHFHSA-N N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclopentanecarboxamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CC[C@@H](C=1C=NC=CC=1)NC(=O)C1CCCC1)C NUGPIZCTELGDOS-QHCPKHFHSA-N 0.000 description 1
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- 108091005461 Nucleic proteins Chemical group 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 102000016979 Other receptors Human genes 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108010019160 Pancreatin Proteins 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 101710124951 Phospholipase C Proteins 0.000 description 1
- 101100413173 Phytolacca americana PAP2 gene Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 239000002776 alpha toxin Substances 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000003452 antibody preparation method Methods 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- UORVGPXVDQYIDP-BJUDXGSMSA-N borane Chemical class [10BH3] UORVGPXVDQYIDP-BJUDXGSMSA-N 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 229960000455 brentuximab vedotin Drugs 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 230000000981 bystander Effects 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- 238000000748 compression moulding Methods 0.000 description 1
- 108091036078 conserved sequence Proteins 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- CHVJITGCYZJHLR-UHFFFAOYSA-N cyclohepta-1,3,5-triene Chemical group C1C=CC=CC=C1 CHVJITGCYZJHLR-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 229930191339 dianthin Natural products 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- JYGAZEJXUVDYHI-UHFFFAOYSA-N dihydroartemisininic acid Natural products C1CC(C)=CC2C(C(C)C(O)=O)CCC(C)C21 JYGAZEJXUVDYHI-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 229940047650 haemophilus influenzae Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000009931 harmful effect Effects 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 102000054751 human RUNX1T1 Human genes 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000005918 in vitro anti-tumor Effects 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical class IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 239000008108 microcrystalline cellulose Chemical class 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 108010010621 modeccin Proteins 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 230000001293 nucleolytic effect Effects 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 229940055695 pancreatin Drugs 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 108010076042 phenomycin Proteins 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000009465 prokaryotic expression Effects 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 235000004252 protein component Nutrition 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000022983 regulation of cell cycle Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- KSAVQLQVUXSOCR-UHFFFAOYSA-M sodium lauroyl sarcosinate Chemical compound [Na+].CCCCCCCCCCCC(=O)N(C)CC([O-])=O KSAVQLQVUXSOCR-UHFFFAOYSA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000012085 test solution Substances 0.000 description 1
- OFVLGDICTFRJMM-WESIUVDSSA-N tetracycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O OFVLGDICTFRJMM-WESIUVDSSA-N 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000008467 tissue growth Effects 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 229940049679 trastuzumab deruxtecan Drugs 0.000 description 1
- 229930013292 trichothecene Natural products 0.000 description 1
- 150000003327 trichothecene derivatives Chemical class 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68037—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a camptothecin [CPT] or derivatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1005—Tetrapeptides with the first amino acid being neutral and aliphatic
- C07K5/1008—Tetrapeptides with the first amino acid being neutral and aliphatic the side chain containing 0 or 1 carbon atoms, i.e. Gly, Ala
Definitions
- the present invention relates to camptothecin derivatives and their antibody-drug conjugates used as anti-tumor drugs. Specifically, the present invention relates to obtaining a better camptothecin anti-tumor drug through a series of molecular structure modification, so as to be more suitable as a drug for antibody coupling.
- Antibody-drug conjugate as a new type of targeted drug, generally consists of three parts: antibody or antibody-like ligand, small molecule drug base, and linker that couples the ligand to the drug.
- the antibody-drug conjugate uses the specific recognition of the antibody to the antigen, transports the drug molecule to the vicinity of the target cell and effectively releases the drug molecule to achieve the purpose of treatment.
- antibody-drug conjugates can accurately bind to tumor cells and reduce the impact on normal cells [Mullard A, (2013) Nature Reviews Drug Discovery, 12: 329-332; DiJoseph JF, Armellino DC , (2004) Blood, 103: 1807-1814].
- Kadcyla ado-trastezumab emtansine, T-DM1
- T-DM1 trastuzumab
- Herceptin Herceptin
- paclitaxel resistant advanced or metastatic breast cancer patients WO2005037992; US8088387
- ADC commonly used toxins include MMAE, T-DM1, PBD and other highly toxic toxins. Such toxins are highly toxic, and even after the formation of ADC drugs, the therapeutic window is narrow.
- Camptothecin derivatives that have anti-tumor effects by inhibiting topoisomerase I are a class of small molecules used in antibody-drug conjugates.
- Isinotecan was developed by Daiichi Sankyo. The drug was used as a single chemotherapeutic agent to advance to phase III clinical. The main indications are bone cancer, prostate cancer, breast cancer, pancreatic cancer, etc., because of side effects. The treatment window is large and the treatment window is narrow. In the end, the direct administration of exenotecan failed to be marketed.
- Daiichi Sankyo has developed exenotecan as an ADC toxin and has been successfully marketed.
- the reduction in the number of conjugated drugs will reduce the therapeutic index of ADC drugs.
- the technical problem to be solved by the present invention is to explore and find better anti-tumor camptothecin derivatives, reduce the toxicity of the compound, improve its safety and effectiveness in the application of ADC drugs, and obtain an anti-tumor ADC with excellent curative effect. drug.
- the present invention designs and synthesizes a series of camptothecin derivatives and antibody conjugates with significant anti-tumor activity, and the resulting camptothecin derivatives The toxicity of the base derivatives is significantly reduced, and finally the anti-tumor activity in vivo is tested to prove its good anti-tumor activity.
- the present invention aims to provide a kind of camptothecin derivatives and their conjugates or tautomers, mesosomes, racemates, enantiomers, non- Enantiomers or mixtures thereof, or pharmaceutically acceptable salts or solvates thereof, wherein the antibody-drug conjugate comprises an anti-tumor camptothecin compound represented by formula D.
- R is selected from deuterium atom, C 1-6 alkyl, deuterated alkyl, substituted alkyl, aryl, substituted aryl or heteroaryl, preferably C 1-6 alkyl or substituted alkyl;
- R 1 is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a haloalkyl group, a cycloalkyl group, a cycloalkylalkyl group, an alkoxyalkyl group, a carboxyl group, a carboxyl group, a heterocyclic group, an aryl group, Substituted aryl or heteroaryl;
- R 2 is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, carboxyl, carboxyl, heterocyclic, aryl, Substituted aryl or heteroaryl;
- X is selected from -C(O)-CR a R b -O-(CR 3 R 4 ) n -, -C(O)-CR a R b -NH-(CR 3 R 4 ) n -or -C( O)-CR a R b -(CR 3 R 4 ) n -S-;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 3 and R 4 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a halogenated alkyl group, a deuterated alkyl group, an alkoxy group, a hydroxyl group, an amino group, a cyano group, a nitro group, a hydroxyalkyl group , Cycloalkyl or heterocyclic group;
- R 3 and R 4 together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n is selected from an integer from 0 to 4.
- camptothecin derivatives or tautomers mesoisomers, racemates, enantiomers, diastereomers or mixtures thereof are provided , Or a pharmaceutically acceptable salt or solvate, including a compound having the general formula D 1 or a pharmaceutically acceptable salt or solvate thereof;
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- X is selected from -C(O)-CR a R b -(CR 3 R 4 ) n -O-, -C(O)-CR a R b -(CR 3 R 4 ) n -NH- or -C( O)-CR a R b -(CR 3 R 4 ) n -S-;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 3 and R 4 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a hydroxyl group, an amino group, a cyano group, a nitro group, a halogen, an alkyl group, a halogenated alkyl group, a deuterated alkyl group, an alkoxy group, and a hydroxyalkyl group. , Cycloalkyl or heterocyclic group;
- R 3 and R 4 together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n is selected from an integer from 0 to 4.
- the wavy line in formula D 1 represents a hydrogen atom, or is covalently connected to a linker unit or an antibody that binds to an antigen expressed by a tumor target cell.
- the structural unit -X- is -C(O)-CR a R b -(CR 3 R 4 ) n -O- or the structural unit -X- is -C(O)-CR a R b -(CH 2 ) n -O-;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 3 and R 4 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a halogenated alkyl group, a deuterated alkyl group, an alkoxy group, a hydroxyl group, an amino group, a cyano group, a nitro group, a hydroxyalkyl group , Cycloalkyl or heterocyclic group;
- R 3 and R 4 together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n 0 or 1.
- a camptothecin derivative or a pharmaceutically acceptable salt or solvate thereof is provided, wherein the O end of the structural unit -X- is connected to the linking unit L, and the structural unit -X- is selected from , But not limited to:
- One aspect of the present invention provides a compound represented by the general formula (D 2 ) or its tautomer, meso, racemate, enantiomer, diastereomer Or a mixture thereof, or a pharmaceutically acceptable salt or solvate thereof,
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl, preferably C 1-6 alkyl or substituted alkyl;
- R a is selected from cycloalkyl or cycloalkylalkyl, preferably C 3-6 cycloalkylalkyl or C 3-6 cycloalkyl;
- R b is selected from a hydrogen atom, a deuterium atom, an alkyl group, a haloalkyl group, a cycloalkyl group or a cycloalkylalkyl group, preferably a hydrogen atom or a deuterium atom;
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n 0 or 1
- the wavy line in formula D 2 represents a hydrogen atom, or is covalently connected to a linker unit or an antibody that binds to an antigen expressed by a target cell.
- a preferred solution in one aspect of the present invention provides a compound represented by general formula (D) or its tautomer, meso, racemate, enantiomer, diastereomer Or its mixture form, or its pharmaceutically acceptable salt or solvate, including the compound represented by the general formula (D 3 ) or its tautomer, meso, racemate, enantiomer Forms, diastereomers or mixtures thereof, or pharmaceutically acceptable salts or solvates thereof:
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl; preferably C 3-6 cycloalkylalkyl or C 3-6 cycloalkyl;
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl; preferably C 3-6 cycloalkylalkyl or C 3-6 cycloalkyl;
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n 0 or 1
- the compounds shown in the general formula (D) shown in the present invention include but are not limited to:
- Camptothecin derivatives of the compound are used in the preparation of lung cancer, kidney cancer, urethral cancer, colon cancer, rectal cancer, prostate cancer, multiform glioblastoma, ovarian cancer, pancreatic cancer, and breast cancer without limitation. Cancer, melanoma, liver cancer, bladder cancer, stomach cancer, lung cancer or esophageal cancer and other solid tumors or blood tumor drugs.
- the antibody-drug conjugate or pharmaceutically acceptable salt or solvent compound thereof according to the present invention, wherein the linker unit -L- is -L 1 -L 2- L 3 -L 4 -, the L 1 end is connected to the antibody, and the L 4 end is connected to X;
- L 1 is selected from, but not limited to: -YC(O)-, -CH 2 -C(O)-NR 5 -YC(O)- or -C(O)-YC(O)-, wherein Y is non-limitingly selected from a C 1-8 alkyl group, a C 1-8 alkyl-cycloalkyl group, or a linear or linear-cyclic heteroalkyl group of 1-8 atoms, and the heteroalkyl group includes the group consisting of: From 1-3 atoms of N, O or S, the C 1-8 alkyl, cycloalkyl, linear or linear cyclic heteroalkyl are each independently selected from deuterated atoms, halogens, and hydroxyl groups. , Cyano, nitro, amino, alkyl, heteroalkyl, substituted alkyl, carboxy, alkoxy or cycloalkyl substituted by one or more substituents;
- the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof wherein the linking unit L 1 is selected from, but not limited to: -(succinimid-3-yl -N-)-(CH 2 ) o -C(O)-, -(succinimide-3-yl-N)-CH 2 -cyclohexyl-C(O)-, -(succinimide- 3-base-N-)-(CH 2 CH 2 O) o -C(O)-, -CH 2 -C(O)-NR 5 -(CH 2 ) o -C(O)- or -C( O)-(CH 2 ) o -C(O)-, wherein o is selected from an integer of 2-8, preferably 5;
- L 2 is selected from -NR 6 (CH 2 CH 2 O) p CH 2 CH 2 C(O)-, -NR 6 (CH 2 CH 2 O) p CH 2 C(O)-, -S(CH 2 ) p C(O)- or chemical bond, where p is selected from an integer of 0-20;
- the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof is provided, wherein the linking unit L 2 is selected from -NR 6 (CH 2 CH 2 O) p CH 2 C( O)-or a chemical bond, where p is selected from an integer of 0-12.
- L 3 is non-limitingly selected from peptide residues composed of 2-7 amino acids, wherein the optional amino acid is further selected from deuterium atom, halogen, hydroxyl, cyano, amino, nitro, alkyl, substituted alkyl, alkane One or more substituents in oxy and cycloalkyl or substituted cycloalkyl;
- the antibody-drug conjugate or the pharmaceutically acceptable salt or solvent compound thereof is provided, wherein the peptide residue of L 3 is selected from one, two or more Amino acid (F), glycine (G), valine (V), lysine (K), citrulline, serine (S), glutamic acid (E) or aspartic acid (D) Polypeptide residues formed by amino acids.
- they are amino acid residues formed by one, two or more amino acids selected from phenylalanine and glycine; more preferably are tetrapeptide residues; most preferably are GGFG (glycine-glycine-phenylalanine- Glycine) composed of tetrapeptide residues.
- L 4 is selected from -NR 7 (CR 8 R 9 ) q -, -C(O)NR 7 , -C(O)NR 7 (CH 2 ) q -or a chemical bond, wherein q is selected from an integer of 0-6, Preferably it is -NR 7 (CR 8 R 9 ) q -;
- R 5 , R 6 and R 7 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, and an alkoxyalkyl group.
- R 8 and R 9 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, an alkoxyalkyl group, a hetero Cyclic, aryl, substituted aryl or heteroaryl.
- the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof is provided, wherein L 4 is selected from -NR 7 (CR 8 R 9 ) q -, and R 7 is selected from A hydrogen atom, a deuterium atom or an alkyl group, R 8 and R 9 are the same or different, and each independently has a hydrogen atom, a deuterium atom or an alkyl group, and q is 1 or 2; L 4 is preferably -NR 7 CR 8 R 9 -; L 4 is more preferably -NHCH 2 -.
- a preferred solution of one aspect of the present invention provides a drug-linker compound represented by the general formula (LXD 2 ) or its tautomer, meso, racemate, enantiomer, non- The enantiomer or its mixture form, or its pharmaceutically acceptable salt or solvate, which is a compound represented by the general formula (La-XD 2 ) or its pharmaceutically acceptable salt or solvate:
- Z is selected from, but not limited to -YC(O)-, -CH 2 -C(O)-NR 5 -YC(O)- or -C(O)-YC(O)-, wherein Y is selected from C 1-8 alkyl, C 1-8 alkyl-cycloalkyl, or linear or linear-cyclic heteroalkyl of 1-8 atoms, the heteroalkyl includes a group selected from N, O or S 1-3 atoms, the C 1-8 alkyl, cycloalkyl, linear or linear cyclic heteroalkyl are each independently selected from deuterated atoms, halogen, hydroxyl, carboxyl, cyano, nitro Substituted by one or more substituents of a group, an amino group, an alkyl group, a heteroalkyl group, a substituted alkyl group, a carboxy group, an alkoxy group or a cycloalkyl group;
- L 2 is selected from -NR 6 (CH 2 CH 2 O) p CH 2 CH 2 C(O)-, -NR 6 (CH 2 CH 2 O) p CH 2 C(O)-, -S(CH 2 ) p C(O)- or chemical bond, where p is preferably an integer of 0-20, preferably a chemical bond;
- L 3 is selected from a peptide residue consisting of 2-7 amino acids, wherein the optional amino acid is further selected from deuterium atom, halogen, hydroxyl, cyano, amino, nitro, alkyl, substituted alkyl, alkoxy and cyclic Alkyl group or substituted cycloalkyl group is substituted by one or more substituents;
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 5 , R 6 and R 7 are the same or different, and are each independently selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, and an alkoxy group Alkyl, heterocyclyl, aryl, substituted aryl or heteroaryl;
- R 8 and R 9 are the same or different, and are each independently selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, an alkoxyalkyl group, Heterocyclic group, aryl group, substituted aryl group or heteroaryl group.
- n is selected from an integer of 0 to 4, preferably 0 or 1, more preferably 0.
- a preferred solution of one aspect of the present invention provides a drug-linker compound represented by the general formula (LXD 2 ) or its tautomer, meso, racemate, enantiomer, non- Enantiomers or their mixtures, or their pharmaceutically acceptable salts or solvates, which are compounds represented by the general formula (Lb-XD 2 ) or their pharmaceutically acceptable salts or solvates:
- Z is selected from, but not limited to -YC(O)-, -CH 2 -C(O)-NR 5 -YC(O)- or -C(O)-YC(O)-, wherein Y is selected from C 1-8 alkyl, C 1-8 alkyl-cycloalkyl, or linear or linear-cyclic heteroalkyl of 1-8 atoms, the heteroalkyl includes a group selected from N, O or S 1-3 atoms, the C 1-8 alkyl, cycloalkyl, linear or linear cyclic heteroalkyl are each independently selected from deuterated atoms, halogen, hydroxyl, carboxyl, cyano, nitro Substituted by one or more substituents of a group, an amino group, an alkyl group, a heteroalkyl group, a substituted alkyl group, a carboxy group, an alkoxy group or a cycloalkyl group;
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 7 is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a haloalkyl group, a cycloalkyl group, a cycloalkylalkyl group, an alkoxyalkyl group, a heterocyclic group, an aryl group, a substituted aryl group or Heteroaryl
- R 8 and R 9 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, an alkoxyalkyl group, a heterocyclic ring Group, aryl, substituted aryl, or heteroaryl.
- n is selected from an integer of 0 to 4, preferably 0 or 1, more preferably 0.
- the antibody-drug conjugates or tautomers, mesoisomers, racemates, enantiomers, diastereomers or mixtures thereof are provided , Or a pharmaceutically acceptable salt or solvate thereof, which is an antibody-drug conjugate represented by the general formula (LX-Dr) or a pharmaceutically acceptable salt or solvate thereof, which is selected from, but not limited to in:
- the antibody-drug conjugates or tautomers, mesoisomers, racemates, enantiomers, diastereomers or mixtures thereof are provided , Or its pharmaceutically acceptable salt or solvate, which is the antibody-drug conjugate represented by the general formula (Ab-LXD r ) or its pharmaceutically acceptable salt or solvate:
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- X is selected from -C(O)-CR a R b -O-(CR 3 R 4 ) n -, -C(O)-CR a R b -NH-(CR 3 R 4 ) n -or -C( O)-CR a R b -(CR 3 R 4 ) n -S-;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 3 and R 4 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a halogenated alkyl group, a deuterated alkyl group, an alkoxy group, a hydroxyl group, an amino group, a cyano group, a nitro group, a hydroxyalkyl group , Cycloalkyl or heterocyclic group;
- R 3 and R 4 together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n is selected from an integer from 0 to 4.
- n is selected from an integer or decimal of 1-10;
- Ab is an antibody, antibody fragment or protein
- L is the connecting unit.
- the provided antibody-drug conjugate or its pharmaceutically acceptable salt or solvate is the antibody-drug conjugate represented by the general formula (Ab-LXD 2) or its A pharmaceutically acceptable salt or solvate;
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl; preferably C 3-6 cycloalkylalkyl or C 3-6 cycloalkyl;
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl; preferably C 3-6 cycloalkylalkyl or C 3-6 cycloalkyl;
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- n 0 or 1
- n is selected from an integer or a decimal number from 1 to 10; preferably n is 2 to 8, which can be an integer or a decimal; more preferably n is 3 to 8, which can be an integer or a decimal;
- the antibody-drug conjugate represented by the general formula (Ab-LX-Dr) or a pharmaceutically acceptable salt or solvate thereof is provided, which is of the general formula (Ab-L a -The antibody-drug conjugate shown in XD 2 ) or a pharmaceutically acceptable salt or solvate thereof;
- Z is selected from but not limited to -YC(O)-, -CH 2 -C(O)-NR 5 -YC(O)- or -C(O)-YC(O)-, where Y, Selected from C 1-8 alkyl, C 1-8 alkyl-cycloalkyl or linear or linear-cyclic heteroalkyl of 1-8 atoms, the heteroalkyl contains selected from N, O or 1-3 atoms of S, the C 1-8 alkyl, cycloalkyl, linear or linear cyclic heteroalkyl are each independently selected from deuterated atoms, halogen, hydroxyl, carboxyl, cyano , Nitro, amino, alkyl, heteroalkyl, substituted alkyl, carboxy, alkoxy or cycloalkyl substituted by one or more substituents;
- L 2 is selected from without limitation -NR 6 (CH 2 CH 2 O) p CH 2 CH 2 C(O)-, -NR 6 (CH 2 CH 2 O) p CH 2 C(O)-, -S (CH 2 ) p C(O)- or a chemical bond, where p is preferably an integer from 0 to 20, preferably a chemical bond;
- L 3 is non-limitingly selected from peptide residues composed of 2-7 amino acids, wherein the optional amino acid is further selected from deuterium atom, halogen, hydroxyl, cyano, amino, nitro, alkyl, substituted alkyl, alkane One or more substituents in oxy and cycloalkyl or substituted cycloalkyl;
- R is selected from deuterium atom, C 1-6 alkyl or substituted alkyl, aryl, substituted aryl or heteroaryl;
- R a is selected from a hydrogen atom, a deuterium atom, a halogen, an alkyl, deuterated alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, an aryl group, a substituted aryl group or Heteroaryl
- R b is selected from hydrogen atom, deuterium atom, halogen, alkyl, haloalkyl, deuterated alkyl, cycloalkyl, cycloalkylalkyl, alkoxyalkyl, heterocyclyl, aryl, substituted aryl or Heteroaryl
- R a , R b together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl, cycloalkylalkyl or heterocyclic group;
- R 3 and R 4 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a halogenated alkyl group, a deuterated alkyl group, an alkoxy group, a hydroxyl group, an amino group, a cyano group, a nitro group, a hydroxyalkyl group , Cycloalkyl or heterocyclic group;
- R 3 and R 4 together with the carbon atoms to which they are connected form a C 3-6 cycloalkyl group or a heterocyclic group;
- R 5 , R 6 and R 7 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, and an alkoxyalkyl group , Heterocyclic group, aryl group, substituted aryl group or heteroaryl group;
- R 8 and R 9 are the same or different, and are each independently a hydrogen atom, a deuterium atom, a halogen, an alkyl group, a deuterated alkyl group, a halogenated alkyl group, a cycloalkyl group, a cycloalkylalkyl group, an alkoxyalkyl group, a heterocyclic ring Group, aryl, substituted aryl, or heteroaryl.
- n is selected from an integer of 0 to 4, preferably 0 or 1, more preferably 0;
- n is selected from 1-10, and can be an integer or a decimal
- Ab is an antibody, antibody fragment or protein.
- the antibody-drug conjugate represented by the general formula (Ab-LXD r ) or a pharmaceutically acceptable salt or solvate thereof is provided, which is of the general formula (Ab-L b- XD 2 ) the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof;
- the antibody-drug conjugate represented by the general formula (Ab-L-X-Dr) of the present invention is selected from the following structures without limitation:
- the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof wherein the Ab is an antibody, an antibody fragment or an antigen-binding fragment thereof, the antibody It is selected from a chimeric antibody, a humanized antibody or a fully human antibody; preferably a monoclonal antibody.
- the antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof wherein the antibody, antibody fragment or antigen-binding fragment thereof is non-limitingly selected from: Anti-EGFRvIII antibody, anti-DLL-3 antibody, anti-PSMA antibody, anti-CD70 antibody, anti-MUC16 antibody, anti-ENPP3 antibody, anti-TDGF1 antibody, anti-ETBR antibody, anti-MSLN antibody, anti-TIM-1 antibody, anti-LRRC15 antibody, anti-LIV -1 antibody, anti-CanAg/AFP antibody, anti-cladin 18.2 antibody, anti-Mesothelin antibody, anti-HER2 (ErbB2) antibody, anti-EGFR antibody, anti-c-MET antibody, anti-SLITRK6 antibody, anti-KIT/CD117 antibody, anti-STEAP1 antibody, Anti-SLAMF7/CS1 antibody, anti-NaPi2B/SLC34A2 antibody, anti-GPNMB antibody, anti-HER3 (ErbB3) antibody
- a method for preparing the antibody-drug conjugate represented by the general formula (Ab-L a -XD 2 ) or a pharmaceutically acceptable salt or solvate thereof which includes the following step:
- Ab is an antibody, an antibody fragment or an antigen-binding fragment thereof
- R, R a , R b , R 7 , R 8 , R 9 , L 2 , L 3 , Z and n are as defined in the above general formula (Ab-La-XD 2 ).
- composition which contains the compound represented by the general formula (D) according to the present invention or its tautomers, mesosomes, racemates, Enantiomers, diastereomers, or mixtures thereof, or pharmaceutically acceptable salts thereof, and one or more pharmaceutically acceptable carriers, diluents or excipients.
- Another aspect of the present invention further relates to an antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof, which comprises a ligand and a drug linked to the ligand, wherein the drug is selected from the group of the present invention
- the compound represented by the general formula (D), the compound represented by the general formula (LX-Dr), or its tautomers, mesosomes, racemates, enantiomers, and non-pairs Enantiomers, or mixtures thereof, or pharmaceutically acceptable salts thereof preferably the drug is linked to the ligand via a linker, and the ligand is preferably a monoclonal antibody.
- an antibody-drug conjugate or a pharmaceutically acceptable salt or solvate thereof which comprises combining the compound represented by the general formula (D) of the present invention, the general formula The compound represented by the formula (LX-Dr), or its tautomer, meso, racemate, enantiomer, diastereomer, or a mixture thereof, or a mixture thereof.
- the step of connecting the pharmaceutically acceptable salt with the ligand is preferably connected via a linker, and the ligand is preferably a monoclonal antibody.
- Another aspect of the present invention further relates to the antibody-drug conjugate or compound of the present invention, or a pharmaceutically acceptable salt or solvate thereof, or a pharmaceutical composition thereof, for use as a medicine.
- Another aspect of the present invention further relates to the antibody-drug conjugate or compound of the present invention, or a pharmaceutically acceptable salt or solvate thereof, or a pharmaceutical composition thereof in the preparation of a medicament for the treatment or prevention of tumors.
- the antibody-drug conjugate or compound of the present invention or a pharmaceutically acceptable salt or solvate thereof, or a pharmaceutical composition thereof in the preparation of a medicament for the treatment or prevention of tumors.
- Another aspect of the present invention further relates to the antibody-drug conjugates or compounds of the present invention, or pharmaceutically acceptable salts or solvates thereof, or pharmaceutical compositions used in the preparation of drugs for the treatment and/or prevention of cancer.
- the cancer is selected from without limitation: breast cancer, ovarian cancer, cervical cancer, uterine cancer, prostate cancer, kidney cancer, urethral cancer, bladder cancer, liver cancer, stomach cancer, endometrial cancer, salivary gland cancer, esophageal cancer , Lung cancer, colon cancer, rectal cancer, colorectal cancer, bone cancer, skin cancer, thyroid cancer, pancreatic cancer, melanoma, glioma, neuroblastoma, glioma multiforme, sarcoma, lymphoma And leukemia and other solid tumors or blood tumors.
- Another aspect of the present invention further relates to a method for treating and/or preventing tumors, the method comprising administering to a patient in need thereof a therapeutically effective dose of the antibody-drug conjugate or compound of the present invention, or A pharmaceutically acceptable salt or solvate thereof or a pharmaceutical composition containing the same.
- Another aspect of the present invention further relates to a method for treating or preventing cancer, the method comprising administering to a patient in need thereof a therapeutically effective dose of the antibody-drug conjugate or compound of the present invention, or a pharmacological agent thereof.
- the cancer is selected from, without limitation, breast cancer, ovarian cancer, cervical cancer, uterine cancer, prostate cancer, kidney cancer, urethral cancer, bladder cancer , Liver cancer, stomach cancer, endometrial cancer, salivary gland cancer, esophageal cancer, lung cancer, colon cancer, rectal cancer, colorectal cancer, bone cancer, skin cancer, thyroid cancer, pancreatic cancer, melanoma, glioma, neuroblastoma Solid tumors or hematological tumors such as cell tumors, glioma multiforme, sarcomas, lymphomas and leukemias.
- the active compound can be formulated into a form suitable for administration by any appropriate route, and the active compound is preferably in a unit dose form, or in a form in which the patient can self-administer in a single dose.
- the expression of the unit dose of the compound or composition of the present invention can be tablet, capsule, cachet, bottled syrup, powder, granule, lozenge, suppository, regenerating powder or liquid preparation.
- the dosage of the compound or composition used in the treatment method of the present invention will generally vary with the severity of the disease, the weight of the patient, and the relative efficacy of the compound.
- a suitable unit dose may be 0.1-1000 mg.
- the pharmaceutical composition of the present invention may contain one or more excipients, which are non-limitingly selected from the following ingredients: fillers (diluents), binders, wetting agents, disintegrants Or excipients and so on.
- the composition may contain 0.1 to 99% by weight of the active compound.
- the pharmaceutical composition containing the active ingredient may be in a form suitable for oral administration, such as tablets, dragees, lozenges, water or oil suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or Elixirs.
- the oral composition can be prepared according to any method known in the art for preparing pharmaceutical compositions, and such compositions can have binders, fillers, lubricants, disintegrants or pharmaceutically acceptable wetting agents, etc.
- the similar composition may also contain one or more ingredients selected from the group consisting of sweeteners, flavoring agents, coloring agents and preservatives to provide pleasing and palatable pharmaceutical preparations.
- Aqueous suspensions contain the active substance and excipients suitable for the preparation of aqueous suspensions for mixing.
- the aqueous suspension may also contain one or more preservatives, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents.
- Oil suspensions can be formulated by suspending the active ingredients in vegetable oils.
- the oil suspension may contain thickeners.
- the above-mentioned sweeteners and flavoring agents can be added to provide a palatable preparation.
- the pharmaceutical composition can also be dispersible powders and granules used to prepare aqueous suspensions to provide active ingredients by adding one or more of a water-mixed dispersant, wetting agent, suspending agent or preservative. Other excipients such as sweeteners, flavoring agents and coloring agents may also be added. These compositions are preserved by adding antioxidants such as ascorbic acid.
- the pharmaceutical composition of the present invention may also be in the form of an oil-in-water emulsion.
- the pharmaceutical composition may be in the form of a sterile injectable aqueous solution.
- Acceptable solvents or solvents that can be used include water, Ringer's solution, and isotonic sodium chloride solution.
- the sterile injectable preparation may be a sterile injectable oil-in-water microemulsion in which the active ingredient is dissolved in an oil phase.
- the active ingredient is dissolved in a mixture of soybean oil and lecithin.
- the oil solution is added to the mixture of water and glycerin to form a microemulsion.
- the injection or microemulsion can be injected into the patient's bloodstream by local large-scale injection.
- a continuous intravenous delivery device can be used.
- An example of such a device is the Deltec CADD-PLUS.TM. 5400 intravenous pump.
- the pharmaceutical composition may be in the form of a sterile injection water or oil suspension for intramuscular and subcutaneous administration.
- the suspension can be formulated according to known techniques using those suitable dispersing or wetting agents and suspending agents mentioned above.
- the sterile injection preparation may also be a sterile injection solution or suspension prepared in a parenterally acceptable non-toxic diluent or solvent.
- sterile fixed oil can be conveniently used as a solvent or suspending medium.
- the compounds of the present invention can be administered in the form of suppositories for rectal administration.
- These pharmaceutical compositions can be prepared by mixing the drug with a suitable non-irritating excipient that is solid at ordinary temperatures but liquid in the rectum and thus will melt in the rectum to release the drug.
- suitable non-irritating excipient include cocoa butter, glycerinated gelatin, hydrogenated vegetable oils, polyethylene glycols of various molecular weights and mixtures of fatty acid esters of polyethylene glycol.
- the dosage of the drug depends on many factors, including but not limited to the following factors: the activity of the specific compound used, the age of the patient, the weight of the patient, the health of the patient, and the behavior of the patient. , The patient’s diet, time of administration, mode of administration, rate of excretion, combination of drugs, etc.; in addition, the best treatment mode, such as the mode of treatment, the daily dosage of the compound of the general formula, or the type of pharmaceutically acceptable salt can be based on Traditional treatment plan to verify.
- Figure 1 shows the in vivo efficacy of ADC on A431 tumor-bearing mouse model
- Figure 2 is a graph showing the in vitro activity data of compounds 6, 12, 14 and 16 of the present invention on Fadu cells;
- Figure 3 is a graph showing the in vitro activity data of compounds 6, 12, 14 and 16 of the present invention on A431 cells;
- Figure 4 is a graph showing the in vitro activity data of compounds 6, 12, 14 and 16 of the present invention on Bxpc-3 cells;
- Figure 5 is a graph showing the in vitro activity data of compounds 6, 12, 14 and 16 of the present invention on SW620 cells.
- the applicant intends to include the formulation of the trade name product, the generic drug and the active drug part of the trade name product.
- antibody is a macromolecular compound that can recognize and bind to an antigen or receptor associated with a target cell.
- the role of antibodies is to present drugs to target cell populations that bind to antibodies. These antibodies include but are not limited to protein hormones, lectins, growth factors, antibodies or other molecules that can bind to cells.
- the antibody is referred to as Ab.
- the antibody can form a link with the linking unit through its heteroatoms, preferably an antibody or an antigen-binding fragment thereof, and the antibody is selected from a chimeric antibody, a humanized antibody, and a fully human Antibody or murine antibody; preferably a monoclonal antibody.
- drug refers to a cytotoxic drug.
- the drug is denoted as D, which can have chemical molecules in tumor cells that strongly disrupt their normal growth.
- cytotoxic drugs can kill tumor cells at a sufficiently high concentration, but due to lack of specificity, while killing tumor cells, it will also cause normal cell apoptosis, leading to serious side effects.
- the term includes toxins, such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, radioisotopes (e.g.
- linker unit or “linker fragment” or “linker unit” refers to a chemical structure fragment or bond with one end connected to a ligand and the other end to a drug, and other linkers can also be connected before being connected to the drug.
- the preferred scheme of the present invention is expressed as L and L 1 -L 4 , where the L 1 end is connected to the ligand, and the L 4 end is connected to the structural unit X and then connected to the drug (D).
- the linker including extensions, spacers and amino acid units, can be synthesized by methods known in the art, such as those described in US2005-0238649A1.
- the linker may be a "breakable linker" that facilitates the release of the drug in the cell.
- breakable linker For example, acid-labile linkers (such as hydrazone), protease-sensitive (such as peptidase-sensitive) linkers, light-labile linkers, dimethyl linkers, or disulfide-containing linkers (Chari et al. Cancer Research 52:127-131, 1992); U.S. Patent No. 5,208,020).
- antibody-drug conjugate refers to that the antibody is connected to a biologically active drug through a stable linking unit.
- ADC antibody-drug conjugate
- antibody refers to an immunoglobulin, which is a tetrapeptide chain structure composed of two identical heavy chains and two identical light chains connected by interchain disulfide bonds.
- the amino acid composition and sequence of the constant region of the immunoglobulin heavy chain are different, so their antigenicity is also different.
- immunoglobulins can be divided into five categories, or isotypes of immunoglobulins, namely IgM, IgD, IgG, IgA, and IgE.
- the corresponding heavy chains are ⁇ chain, ⁇ chain, and ⁇ chain. , ⁇ chain and ⁇ chain.
- IgG can be divided into IgG1, IgG2, IgG3, and IgG4.
- the light chain is divided into a kappa chain or a lambda chain by the difference of the constant region.
- Each of the five types of Ig can have a kappa chain or a lambda chain.
- the antibodies of the present invention are preferably specific antibodies against cell surface antigens on target cells.
- Non-limiting examples are the following antibodies: anti-EGFRvIII antibody, anti-DLL-3 antibody, anti-PSMA antibody, anti-CD70 antibody, and anti-MUC16 antibody , Anti-ENPP3 antibody, anti-TDGF1 antibody, anti-ETBR antibody, anti-MSLN antibody, anti-TIM-1 antibody, anti-LRRC15 antibody, anti-LIV-1 antibody, anti-CanAg/AFP antibody, anti-cladin 18.2 antibody, anti-Mesothelin antibody, anti-HER2 (ErbB2) antibody, anti-EGFR antibody, anti-c-MET antibody, anti-SLITRK6 antibody, anti-KIT/CD117 antibody, anti-STEAP1 antibody, anti-SLAMF7/CS1 antibody, anti-NaPi2B/SLC34A2 antibody, anti-GPNMB antibody, anti-HER3 (ErbB3) Antibody, anti-MUC1/CD227 antibody, anti-AXL antibody, anti-CD166 antibody, anti-B7-H3 (CD276) antibody, anti-PT
- variable region The sequence of about 110 amino acids near the N-terminus of the antibody heavy and light chains varies greatly and is the variable region (Fv region); the remaining amino acid sequences near the C-terminus are relatively stable and are the constant region.
- the variable region includes 3 hypervariable regions (HVR) and 4 framework regions (FR) with relatively conserved sequences. Three hypervariable regions determine the specificity of the antibody, also known as complementarity determining regions (CDR).
- Each light chain variable region (LCVR) and heavy chain variable region (HCVR) consists of 3 CDR regions and 4 FR regions.
- the sequence from the amino terminal to the carboxy terminal is: FRI, CDRI, FR2, CDR2, FR3, CDR3, FR4.
- the 3 CDR regions of the light chain refer to LCDR1, LCDR2, and LCDR3; the 3 CDR regions of the heavy chain refer to HCDR1, HCDR2, and HCDR3.
- the antibodies of the present invention include murine antibodies, chimeric antibodies, humanized antibodies and fully human antibodies, preferably humanized antibodies and fully human antibodies.
- murine-derived antibody in the present disclosure refers to the preparation of antibodies with mice based on knowledge and skills in the art. During preparation, the test object is injected with a specific antigen, and then the hybridization of the antibody with the desired sequence or functional characteristics is separated and expressed
- chimeric antibody is an antibody formed by fusing the variable region of a murine antibody with the constant region of a human antibody, which can alleviate the immune response induced by the murine antibody.
- To establish a chimeric antibody it is necessary to first establish a hybridoma secreting murine-derived specific monoclonal antibodies, then clone the variable region genes from the murine hybridoma cells, and then clone the constant region genes of the human antibody as needed, and replace the murine variable region genes. It is connected with the human constant region gene to form a chimeric gene and inserted into an expression vector, and finally the chimeric antibody molecule is expressed in a eukaryotic system or a prokaryotic system.
- humanized antibody is also called CDR-grafted antibody (CDR-grafted antibody), refers to the mouse CDR sequence grafted into the human antibody variable region framework, that is, different types of human germline antibody framework sequences It can overcome the heterologous reaction induced by chimeric antibodies due to the large amount of murine protein components.
- CDR-grafted antibody refers to the mouse CDR sequence grafted into the human antibody variable region framework, that is, different types of human germline antibody framework sequences It can overcome the heterologous reaction induced by chimeric antibodies due to the large amount of murine protein components.
- Such framework sequences can be obtained from public DNA databases including germline antibody gene sequences or public references. Such as humans.
- the germline DNA sequences of the heavy and light chain variable region genes can be found in the "VBase" human germline sequence database (available on the Internet www.mrccpe.com/ac.uk/vbase ), as well as in Kabat, EA, etc., Found in 1991 Sequences of Proteins of Immunological Interest, 5th edition.
- the human antibody variable region framework sequence can be subjected to minimal reverse mutations or back mutations. Maintain activity.
- the humanized antibodies of the present invention also include humanized antibodies that are further subjected to affinity maturation of the CDRs by phage display.
- the development of monoclonal antibodies has gone through four stages, namely: murine monoclonal antibodies, chimeric monoclonal antibodies, humanized monoclonal antibodies and fully human monoclonal antibodies.
- the present invention is a fully human monoclonal antibody.
- the related technologies of fully human antibody preparation mainly include: human hybridoma technology, EBV transformed B lymphocyte technology, phage display technology (phage display), transgenic mouse antibody preparation technology (transgenic mouse) and single B cell antibody preparation technology.
- antigen-binding fragment refers to one or more fragments of an antibody that retain the ability to specifically bind to an antigen. It has been shown that fragments of full-length antibodies can be used to perform the antigen-binding function of antibodies.
- binding fragments contained in "antigen-binding fragments” include (i) Fab fragments, monovalent fragments consisting of VL, VH, CL and CH1 domains; (ii) F(ab') 2 fragments, including through the hinge region A bivalent fragment of two Fab fragments connected by a disulfide bridge, (iii) Fd fragment composed of VH and CH1 domains; (iv) Fv fragment composed of VH and VL domains of one arm of an antibody; (v ) A single domain or dAb fragment (Ward et al., (1989) Nature 341:544-546), which consists of a VH domain; and (vi) an isolated complementarity determining region (CDR) or (vii) can optionally be passed A combination
- the two domains VL and VH of the Fv fragment are encoded by separate genes, recombination methods can be used to connect them through a synthetic linker so that it can be produced as a single protein in which the VL and VH regions are paired to form a monovalent molecule.
- Chain referred to as single-chain Fv (scFv); see, for example, Bird et al. (1988) Science 242: 423-426 and Huston et al. (1 988) Proc. Nat L. Acad. Sci. USA 85: 5879-5883).
- Such single chain antibodies are also intended to be included in the "antigen-binding fragment" of the term antibody.
- the antigen-binding portion can be produced by recombinant DNA technology or by enzymatic or chemical fragmentation of the intact immunoglobulin.
- the antibodies may be antibodies of different isotypes, for example, IgG (eg, IgG1, IgG2, IgG3 or IgG4 subtype), IgA1, IgA2, IgD, IgE or lgM antibodies.
- Fab is an antibody fragment that has a molecular weight of about 50,000 and has antigen-binding activity among fragments obtained by treating IgG antibody molecules with the protease papain (cleaving the amino acid residue at position 224 of the H chain), wherein the H chain is N-terminal About half of the side and the entire L chain are joined together by disulfide bonds.
- F(ab')2 is an antibody with a molecular weight of about 100,000 obtained by digesting the lower part of the two disulfide bonds in the hinge region of IgG with the enzyme pepsin and has antigen-binding activity and contains two Fab regions connected at the hinge position. Fragment.
- Fab' is an antibody fragment with a molecular weight of about 50,000 and antigen-binding activity obtained by cleaving the disulfide bond in the hinge region of F(ab')2 described above.
- the Fab' can be produced by inserting DNA encoding the Fab' fragment of the antibody into a prokaryotic expression vector or eukaryotic expression vector and introducing the vector into a prokaryotic organism or eukaryotic organism to express Fab'.
- single-chain antibody means an antibody heavy chain variable domain (or region; (H) and antibody light chain variable domain (or region; VL) connected by a linker.
- Such scFv molecules may have the general structure: NH 2 -VL-linker-VH-COOH or NH 2 -VH-linker-VL-COOH.
- Suitable prior art linkers consist of repeated GGGGS amino acid sequences or variations thereof Body composition, for example, using 1-4 repeated variants (Holliger et al. (1993), proc. Natl. Acad. Sci. USA 90: 6444-6448).
- linkers that can be used in the present invention are described by Alfthan et al. (1995) , Protein Eng. 8: 725-731, Choi et al. (2001), Eur. J. Immuno 1.31: 94-106, Hu et al. (1996), Cancer Res. 56: 3055-3061, Kipriyanov et al. (1999) , J. Mol. Biol. 293:41-56 and Roovers et al. (2001), Cancer Immunol.
- CDR refers to one of the six hypervariable regions in the variable domain of an antibody that mainly contribute to antigen binding.
- One of the most commonly used definitions of the 6 CDRs is provided by Kabat E.A. et al. (1991) Sequences of Proteins of Immunological Interest. NIH Publication 91-3242).
- the Kabat definition of CDR only applies to the CDR1, CDR2, and CDR3 of the light chain variable domain (CDR L1, CDRL2, CDRL3 or L1, L2, L3), and the CDR2 and CDR3 of the heavy chain variable domain.
- CDR3 CDR H2, CDR H3 or H2, H3
- antibody framework refers to a part of the variable domain VL or VH, which serves as a scaffold for the antigen binding loop (CDR) of the variable domain. Essentially, it is a variable structure without CDR.
- epitopes refers to the site on an antigen where an immunoglobulin or antibody specifically binds.
- Epitopes usually include at least 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 consecutive or non-contiguous amino acids in a unique spatial conformation. See, for example, Epitope Mapping Protocols in Methods in Molecular Biology, Volume 66, G.E. Morris, et al. (1996).
- antibodies bind with an affinity (KD) of approximately less than 10 -7 M, such as approximately less than 10 -8 M, 10 -9 M, or 10 -10 M or less.
- nucleic acid molecule refers to DNA molecules and RNA molecules.
- the nucleic acid molecule may be single-stranded or double-stranded, but is preferably double-stranded DNA.
- the nucleic acid is "operably linked.” For example, if a promoter or enhancer affects the transcription of a coding sequence, the promoter or enhancer is effectively linked to the coding sequence.
- vector refers to a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked.
- the vector is a "plasmid”, which refers to a circular double-stranded DNA loop into which additional DNA segments can be ligated.
- the vector is a viral vector in which additional DNA segments can be ligated into the viral genome.
- the vectors invented herein can replicate autonomously in the host cell into which they have been introduced (for example, bacterial vectors with a bacterial origin of replication and episomal mammalian vectors) or can be integrated into the genome of the host cell after being introduced into the host cell, so as to follow The host genome replicates together (e.g., a non-episomal mammalian vector).
- Antigen-binding fragments can also be prepared by conventional methods.
- the antibody or antigen-binding fragment of the invention is genetically engineered to add one or more human FR regions to the non-human CDR region.
- the human FR germline sequence can be obtained from the 1mMunoGeneTics (MGT) website http://imgt.cines.fr by comparing the IMGT human antibody variable region germline gene database and MOE software, or from the Journal of Immunoglobulin, 2001ISBN012441351 obtain.
- host cell refers to a cell into which an expression vector has been introduced.
- Host cells may include bacteria, microorganisms, plant or animal cells.
- Bacteria that are easily transformed include members of the enterobacteriaceae, such as Escherichia coli or Salmonella strains; Bacillaceae such as Bacillus subtilis; Pneumococcus; Streptococcus and Haemophilus influenzae.
- Suitable microorganisms include Saccharomyces cerevisiae and Pichia pastoris.
- Suitable animal host cell lines include CHO (Chinese Hamster Ovary cell line) and NS0 cells.
- the engineered antibody or antigen-binding fragment of the present invention can be prepared and purified by conventional methods.
- the cDNA sequences encoding the heavy and light chains can be cloned and recombined into a GS expression vector.
- the recombinant immunoglobulin expression vector can be stably transfected into CHO cells.
- mammalian expression systems can lead to glycosylation of antibodies, especially in the highly conserved N-terminal sites of the Fc region. Positive clones are expanded in the serum-free medium of the bioreactor to produce antibodies.
- the culture medium from which the antibody is secreted can be purified by conventional techniques. For example, use A or G Sepharose FF column with adjusted buffer for purification.
- the bound antibody was eluted by the PH gradient method, and the antibody fragments were detected by SDS-PAGE and collected.
- the antibody can be filtered and concentrated by conventional methods. Soluble mixtures and polymers can also be removed by conventional methods, such as molecular sieves and ion exchange. The resulting product needs to be frozen immediately, such as -70°C, or lyophilized.
- peptide refers to a fragment of a compound between amino acids and proteins. It is composed of two or more amino acid molecules connected to each other by peptide bonds. They are structural and functional fragments of proteins, such as hormones, enzymes, etc. in essence. All are peptides.
- sucrose refers to a biological macromolecule composed of three elements: C, H, and O, which can be divided into monosaccharides, disaccharides and polybran.
- fluorescent probe refers to the characteristic fluorescence in the ultraviolet-visible-near-infrared region, and its fluorescent properties (excitation and emission wavelength, intensity, lifetime, polarization, etc.) can vary with the properties of the environment, such as polarity, refractive index A type of fluorescent molecules that change sensitively due to changes in viscosity, viscosity, etc., which non-covalently interact with nucleic acids (DNA or RNA), proteins or other macromolecular structures to change one or several fluorescent properties, which can be used for research The nature and behavior of macromolecular substances.
- toxic drug refers to a substance that inhibits or prevents the function of cells and/or causes cell death or destruction. Including toxins and other compounds that can be used in tumor treatment.
- toxin refers to any substance that can have a harmful effect on the growth or proliferation of cells, and can be small molecule toxins and their derivatives from bacteria, fungi, plants or animals, including camptothecin derivatives such as ixati Health, maytansinoid and its derivatives (CN101573384) such as DM1, DM3, DM4, auristatin F (AF) and its derivatives, such as MMAF, MMAE, 3024 (WO 2016/127790 A1), diphtheria toxin, external Toxin, ricin A chain, abrin A chain, modeccin, ⁇ -toxin (sarcin), Aleutitesfordii toxic protein, dianthin toxic protein , Phytolaca americana toxic protein (PAPI, PAPII and PAP-S), momordica charantia inhibitor, jatropha curcin, croton, sapaonaria officinalis Inhibitors, gelonin, mitogellin, restrictoc
- alkyl refers to a saturated aliphatic hydrocarbon group, which is a straight or branched chain group containing 1 to 20 carbon atoms, preferably an alkyl group containing 1 to 12 carbon atoms, more preferably containing 1 to 10 carbons The most preferred is an alkyl group containing 1 to 6 carbon atoms.
- Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, n-pentyl, 1,1-dimethylpropyl, 1 ,2-Dimethylpropyl, 2,2-Dimethylpropyl, 1-ethylpropyl, 2-methylbutyl, 3-methylbutyl, n-hexyl, 1-ethyl-2- Methylpropyl, 1,1,2-trimethylpropyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 2,2-dimethylbutyl, 1,3 -Dimethylbutyl, 2-ethylbutyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 2,3-dimethylbutyl, n-heptyl, 2 -Methylhexyl, 3-methylhexyl, 4-methylhe
- lower alkyl groups containing 1 to 6 carbon atoms More preferred are lower alkyl groups containing 1 to 6 carbon atoms.
- Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, and sec-butyl.
- Alkyl groups may be substituted or unsubstituted.
- substituents When substituted, substituents may be substituted at any available attachment point.
- the substituents are preferably one or more of the following groups, which are independently selected from alkanes Group, alkenyl, alkynyl, alkoxy, alkylthio, alkylamino, halogen, mercapto, hydroxyl, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkane Oxy, heterocycloalkoxy, cycloalkylthio, heterocycloalkylthio, oxo.
- heteroalkyl refers to an alkyl group containing one or more heteroatoms selected from N, O or S, wherein the alkyl group is as defined above.
- alkylene refers to a saturated linear or branched aliphatic hydrocarbon group, which has two residues derived from the removal of two hydrogen atoms from the same carbon atom or two different carbon atoms of the parent alkane, which is A straight or branched chain group containing 1 to 20 carbon atoms, preferably containing 1 to 12 carbon atoms, more preferably an alkylene group containing 1 to 6 carbon atoms.
- Non-limiting examples of alkylene include, but are not limited to, methylene (-CH 2 -), 1,1-ethylene [-CH(CH 3 )-], 1,2-ethylene (-CH 2 -) CH 2 )-, 1,1-propylene [-CH(CH 2 CH 3 )-], 1,2-propylene [-CH 2 CH(CH 3 )-], 1,3-propylene (-CH 2 CH 2 CH 2 -), 1,4-butylene (-CH 2 CH 2 CH 2 CH 2 -) and 1,5-butylene (-CH 2 CH 2 CH 2 CH 2 CH 2 -) Wait.
- the alkylene group may be substituted or unsubstituted. When substituted, the substituent may be substituted at any available point of attachment.
- the substituent is preferably independently optionally selected from alkyl, alkenyl, alkynyl , Alkoxy, alkylthio, alkylamino, halogen, mercapto, hydroxy, nitro, cyano, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy , Cycloalkylthio, heterocycloalkylthio and oxo groups are substituted by one or more substituents.
- alkoxy refers to -O- (alkyl) and -O- (unsubstituted cycloalkyl), where the definition of alkyl or cycloalkyl is as described above.
- alkoxy groups include: methoxy, ethoxy, propoxy, butoxy, cyclopropoxy, cyclobutoxy, cyclopentyloxy, cyclohexyloxy.
- the alkoxy group may be optionally substituted or unsubstituted.
- the substituent is preferably one or more of the following groups, which are independently selected from alkyl, alkenyl, alkynyl, alkoxy, alkane Thio, alkylamino, halogen, mercapto, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio , Heterocycloalkylthio.
- cycloalkyl refers to a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent.
- the cycloalkyl ring contains 3 to 20 carbon atoms, preferably 3 to 12 carbon atoms, more preferably 3 to 10 Carbon atoms, most preferably 3 to 8 carbon atoms.
- Non-limiting examples of monocyclic cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, cycloheptyl, cycloheptatriene Groups, cyclooctyl, etc.; polycyclic cycloalkyls include spiro, fused, and bridged cycloalkyls.
- heterocyclyl refers to a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent containing 3 to 20 ring atoms, one or more of which is selected from nitrogen, oxygen or S(O) u (where u is an integer of 0 to 2) heteroatoms, but does not include the ring part of -OO-, -OS- or -SS-, and the remaining ring atoms are carbon. It preferably contains 3 to 12 ring atoms, of which 1 to 4 are heteroatoms; more preferably, the cycloalkyl ring contains 3 to 10 ring atoms.
- Non-limiting examples of monocyclic heterocyclic groups include pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, homopiperazinyl, and the like.
- Polycyclic heterocyclic groups include spirocyclic, condensed, and bridged heterocyclic groups.
- the heterocyclyl ring may be fused to an aryl, heteroaryl or cycloalkyl ring, wherein the ring connected to the parent structure is a heterocyclic group, non-limiting examples thereof include:
- the heterocyclic group may be optionally substituted or unsubstituted.
- the substituent is preferably one or more of the following groups, which are independently selected from alkyl, alkenyl, alkynyl, alkoxy, alkane Thio, alkylamino, halogen, mercapto, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio , Heterocycloalkylthio, oxo.
- aryl refers to a 6 to 14-membered all-carbon monocyclic or fused polycyclic (that is, rings sharing adjacent pairs of carbon atoms) group with a conjugated electron system, preferably 6 to 10 members, such as benzene And naphthyl, preferably phenyl.
- the aryl ring may be fused to a heteroaryl, heterocyclic or cycloalkyl ring, wherein the ring connected to the parent structure is an aryl ring, and non-limiting examples thereof include:
- the aryl group may be substituted or unsubstituted.
- the substituent is preferably one or more of the following groups, which are independently selected from alkyl, alkenyl, alkynyl, alkoxy, alkylthio, Alkylamino, halogen, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio, heterocycloalkylthio base.
- heteroaryl refers to a heteroaromatic system containing 1 to 4 heteroatoms and 5 to 14 ring atoms, where the heteroatoms are selected from oxygen, sulfur, and nitrogen.
- Heteroaryl groups are preferably 5 to 10 members, more preferably 5 or 6 members, such as furyl, thienyl, pyridyl, pyrrolyl, N-alkylpyrrolyl, pyrimidinyl, pyrazinyl, imidazolyl, tetrakis Azole and so on.
- the heteroaryl ring may be fused to an aryl, heterocyclic or cycloalkyl ring, wherein the ring connected to the parent structure is a heteroaryl ring, non-limiting examples of which include:
- Heteroaryl groups may be optionally substituted or unsubstituted.
- the substituents are preferably one or more of the following groups, which are independently selected from alkyl, alkenyl, alkynyl, alkoxy, alkane Thio, alkylamino, halogen, mercapto, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio , Heterocycloalkylthio.
- amino protecting group is to keep the amino group unchanged when other parts of the molecule react, and to protect the amino group with a group that is easy to remove.
- Non-limiting examples include 9-fluorenylmethyloxycarbonyl, tert-butoxycarbonyl, acetyl, benzyl, allyl, p-methoxybenzyl, and the like. These groups may be optionally substituted with 1-3 substituents selected from halogen, alkoxy or nitro.
- the amino protecting group is preferably 9-fluorenylmethyloxycarbonyl.
- cycloalkylalkyl refers to an alkyl group substituted with one or more cycloalkyl groups, preferably one cycloalkyl group, where alkyl is as defined above and where cycloalkyl is as defined above.
- haloalkyl refers to an alkyl group substituted with one or more halogens, where the alkyl group is as defined above.
- deuterated alkyl refers to an alkyl group substituted with one or more deuterium atoms, where the alkyl group is as defined above.
- hydroxy refers to the -OH group.
- halogen refers to fluorine, chlorine, bromine or iodine.
- amino refers to -NH 2 .
- nitro refers to -NO 2 .
- amido refers to -C(O)N-(alkyl) or (cycloalkyl), where alkyl and cycloalkyl are as defined above.
- carboxylate group refers to -C(O)O-(alkyl) or (cycloalkyl), where alkyl and cycloalkyl are as defined above.
- the present invention also includes compounds of formula (D) in various deuterated forms.
- Each available hydrogen atom connected to a carbon atom can be independently replaced by a deuterium atom.
- Those skilled in the art can synthesize the compound of formula (D) in the deuterated form with reference to relevant literature.
- Commercially available deuterated starting materials can be used when preparing the deuterated form of the compound of formula (D), or they can be synthesized using conventional techniques using deuterated reagents.
- Deuterated reagents include, but are not limited to, deuterated borane and tri-deuterated. Borane tetrahydrofuran solution, deuterated lithium aluminum hydride, deuterated ethyl iodide and deuterated methyl iodide, etc.
- heterocyclic group optionally substituted by an alkyl group means that an alkyl group may but need not be present, and the description includes the case where the heterocyclic group is substituted by an alkyl group and the case where the heterocyclic group is not substituted by an alkyl group.
- Substituted refers to one or more hydrogen atoms in the group, preferably up to 5, more preferably 1-3 hydrogen atoms independently of each other, substituted with a corresponding number of substituents. It goes without saying that the substituents are only in their possible chemical positions, and those skilled in the art can determine (through experiment or theory) possible or impossible substitutions without too much effort. For example, an amino group or a hydroxyl group having free hydrogen may be unstable when combined with a carbon atom having an unsaturated (e.g., olefinic) bond.
- pharmaceutical composition means a mixture containing one or more of the compounds described herein or their physiologically/pharmaceutically acceptable salts or prodrugs and other chemical components, as well as other components such as physiologically/pharmaceutically acceptable Carriers and excipients.
- the purpose of the pharmaceutical composition is to promote the administration to the organism, which is beneficial to the absorption of the active ingredient and thus the biological activity.
- pharmaceutically acceptable salt refers to the salt of the antibody-drug conjugate of the present invention, or the salt of the compound described in the present invention. Such salts have It is safe and effective, and has due biological activity.
- the antibody-drug conjugate compound of the present invention contains at least one amino group, so it can form a salt with an acid.
- Non-limiting examples of pharmaceutically acceptable salts include: hydrochloride , Hydrobromide, hydroiodide, sulfate, bisulfate, citrate, acetate, succinate, ascorbate, oxalate, nitrate, pearate, hydrogen phosphate, diphosphate Hydrogen salt, salicylate, hydrogen citrate, tartrate, maleate, fumarate, formate, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate, P-toluenesulfonate.
- solvate or “solvent compound” means that the antibody-drug conjugate compound of the present invention forms a pharmaceutically acceptable solvate with one or more solvent molecules.
- solvent molecules include water, ethanol, acetonitrile, Isopropanol, DMSO, ethyl acetate.
- drug loading refers to the average number of cytotoxic drugs loaded on each antibody in the molecule. It can also be expressed as the ratio of the amount of drug to the amount of antibody. The range of drug loading can be 0-12 per antibody (Ab). One, preferably 1-10 cytotoxic drugs (D). In the embodiment of the present invention, the drug loading is expressed as m, which may be an average value of 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 as an example. Conventional methods such as UV/visible light spectroscopy, mass spectrometry, ELISA test and HPLC characteristics can be used to identify the average number of drug products per ADC molecule after the coupling reaction.
- the cytotoxic drug is coupled to the N-terminal amino group of the ligand and/or the epsilon-amino group of the lysine residue through the linking unit.
- the cytotoxic drug can be coupled to the antibody in the coupling reaction.
- the number of drug molecules will be less than the theoretical maximum.
- ligand cytotoxic drug conjugates including:
- carrier used in the drug of the present invention refers to a system that can change the way the drug enters the body and its distribution in the body, control the release rate of the drug, and deliver the drug to the targeted organ.
- the drug carrier release and targeting system can reduce drug degradation and loss, reduce side effects, and improve bioavailability.
- polymer surfactants that can be used as carriers can self-assemble to form various forms of aggregates due to their unique amphiphilic structure.
- Preferred examples are micelles, microemulsions, gels, liquid crystals, vesicles, etc. . These aggregates have the ability to contain drug molecules, and at the same time have good permeability to the membrane, and can be used as excellent drug carriers.
- excipient is an addition to the main drug in a pharmaceutical preparation, and can also be referred to as an adjuvant.
- adjuvant such as binders, fillers, disintegrants, lubricants in tablets; base parts in semi-solid preparations ointments and creams; preservatives, antioxidants, flavors, fragrances, and auxiliary agents in liquid preparations
- Solvents, emulsifiers, solubilizers, osmotic pressure regulators, colorants, etc. can all be called excipients.
- diluent is also called a filler, and its main purpose is to increase the weight and volume of the tablet.
- the addition of diluent not only guarantees a certain volume, but also reduces the deviation of the dosage of the main components, and improves the compression molding of the drug.
- an absorbent should be added to absorb the oily substance to keep it in a “dry” state, so as to facilitate the manufacture of tablets.
- the pharmaceutical composition may be in the form of a sterile injectable aqueous solution.
- the acceptable solvents and solvents that can be used are water, Ringer's solution and isotonic sodium chloride solution.
- the sterile injectable preparation may be a sterile injectable oil-in-water microemulsion in which the active ingredient is dissolved in an oil phase.
- the active ingredient is dissolved in a mixture of soybean oil and lecithin.
- the oil solution is added to the mixture of water and glycerin to form a microemulsion.
- the injection or microemulsion can be injected into the patient's bloodstream by local large-scale injection.
- a continuous intravenous delivery device can be used.
- An example of such a device is the Deltec CADD-PLUS.TM. 5400 intravenous pump.
- the pharmaceutical composition may be in the form of a sterile injection water or oil suspension for intramuscular and subcutaneous administration.
- the suspension can be formulated according to known techniques using those suitable dispersing or wetting agents and suspending agents mentioned above.
- the sterile injection preparation may also be a sterile injection solution or suspension prepared in a non-toxic parenterally acceptable diluent or solvent, for example, a solution prepared in 1,3-butanediol.
- sterile fixed oil can be conveniently used as a solvent or suspending medium. For this purpose, any blended fixed oil including synthetic mono- or diglycerides can be used.
- fatty acids such as oleic acid can also be used to prepare injections.
- the present invention relates to a cleavable linker with a specific structure and a drug with a specific structure, and an antibody drug conjugate (ADC) composed of a linker, a drug and an antibody.
- ADC antibody drug conjugate
- This type of ADC is a complex formed by linking a toxic substance to an antibody via a spacer.
- the antibody-conjugated drug (ADC) is degraded in the body to release active molecules, which play a corresponding role.
- the following terms and phrases as used herein are intended to have the following meanings.
- the brand name includes the product formula, generic drugs, and active pharmaceutical ingredients of the brand name product.
- alkyl refers to a saturated hydrocarbon group having 1-20 carbon atoms.
- alkylene groups include, but are not limited to, methyl (-CH 3 ), ethyl (-CH 2 -CH 3 ), n-propyl, n-butyl, n-pentyl, and n-hexyl.
- R', R" and R"' each independently refers to hydrogen, unsubstituted C 1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, unsubstituted C 1-8 alkyl, C 1- 8 alkoxy or C 1-8 thioalkoxy, or unsubstituted aryl -C 1-4 alkyl .
- -NR'R" includes 1-pyrrolidinyl and 4-morpholinyl.
- Acid amino acid refers to an amino acid whose isoelectric point is less than 7. Acidic amino acid molecules often contain one or more acidic groups such as carboxyl groups, which can be effectively ionized into negative ions in the structure to increase hydrophilicity. Acidic amino acids can be natural or unnatural amino acids.
- Natural amino acids refer to amino acids synthesized by biology. Natural amino acids are generally L-type, but there are a few exceptions, such as glycine, including natural and biosynthetic.
- Unnatural amino acid refers to an amino acid obtained by synthetic means.
- the ligand unit is a targeting agent that specifically binds to the target part.
- the ligand can specifically bind to cellular components or to cellular components or to other target molecules of interest.
- the target moiety or target is usually on the surface of the cell.
- the role of the ligand unit is to deliver the drug unit to a specific target cell population with which the ligand unit interacts.
- Ligands include but are not limited to proteins, polypeptides and peptides, as well as non-proteins such as sugars. Suitable ligand units include, for example, antibodies, such as full-length (complete) antibodies and antigen-binding fragments thereof.
- the ligand unit is a non-antibody targeting agent
- it can be a peptide or polypeptide, or a non-protein molecule.
- targeting agents include interferons, lymphokines, hormones, growth factors and colony stimulating factors, vitamins, nutrient transport molecules, or any other cell binding molecules or substances.
- the linker is covalently attached to the sulfur atom of the ligand.
- the sulfur atom is the sulfur atom of a cysteine residue, which forms an interchain disulfide bond of the antibody.
- the sulfur atom is a sulfur atom of a cysteine residue that has been introduced into the ligand unit, which forms an interchain disulfide bond of the antibody.
- the sulfur atom is the sulfur atom of the cysteine residue that has been introduced into the ligand unit (for example, by site-directed mutagenesis or chemical reaction).
- the sulfur atom to which the linker binds is selected from the cysteine residues that form the interchain disulfide bond of the antibody or the frontal cysteine residues that have been introduced into the ligand unit (for example, by site-directed mutagenesis or chemical reaction).
- the EU index in Kabat Kabat (Kabat EA et al., (1991)) "Sequences of proteins of Immunological Interest" (Sequences of proteins of Immunological Interest), fifth edition, NIH publication 91-3242) Numbering system.
- antibody or “antibody unit” includes any part of the structure of an antibody within the scope to which it belongs. This unit can bind, reactively associate, or complex a receptor, antigen, or target other receptor unit possessed by the cell population.
- the antibody can be any protein or protein molecule that can bind, complex, or react with a part of the cell population to be treated or biologically modified.
- the antibody constituting the antibody-drug conjugate of the present invention preferably maintains its original antigen binding ability in the wild state. Therefore, the antibody of the present invention can, preferably, specifically bind to the antigen.
- the antigens involved include, for example, tumor-associated antigens (TAA), cell surface receptor proteins and other cell surface molecules, cell survival regulators, cell proliferation regulators, and molecules related to tissue growth and differentiation (as known or predicted Functional), lymphokines, cytokines, molecules involved in cell cycle regulation, molecules involved in angiogenesis, and molecules related to angiogenesis (as known or predicted to be functional).
- TAA tumor-associated antigens
- the tumor-related factor may be a cluster differentiation factor (such as CD protein).
- Antibodies used in antibody-drug conjugates include, but are not limited to, antibodies directed against cell surface receptors and tumor-associated antigens.
- tumor-associated antigens are well-known in the industry and can be prepared by antibody preparation methods and information well-known in the industry.
- researchers are trying to find transmembrane or other tumor-related peptides. These targets can be specifically expressed on the surface of one or more cancer cells, but little or no expression on the surface of one or more non-cancer cells.
- tumor-associated polypeptides are more overexpressed on the surface of cancer cells than on the surface of non-cancer cells. Confirming that such tumor-related factors can greatly improve the specific targeting properties of antibody-based treatment of cancer.
- Tumor-associated antigens include, but are not limited to the following tumor-associated antigens (1)-(36).
- the antigen-related information well-known in the industry is marked as follows, including name, other names, and gene bank accession number.
- Nucleic acid and protein sequences corresponding to tumor-associated antigens can be found in public databases, such as Genbank.
- Antibodies targeting corresponding tumor-associated antigens include all amino acid sequence variants and homologs, which have at least 70%, 80%, 85%, 90%, or 95% homology with the sequence confirmed in the reference, or have the same
- the tumor-associated antigen sequences in the cited literature have completely identical biological properties and characteristics.
- inhibitor refers to reducing the detectable amount, or preventing it completely.
- cancer refers to a physiological condition or disease characterized by unregulated cell growth.
- Tumor includes cancer cells.
- autoimmune disease is a disease or disorder that originates from the tissues or proteins of an individual.
- pharmaceutically acceptable salt refers to a pharmaceutically acceptable organic or inorganic salt of a compound (eg, drug, drug-linker or ligand-linker-drug conjugate).
- the compound may contain at least one amino or carboxyl group, and therefore may form an addition salt with the corresponding acid or base.
- Exemplary salts include, but are not limited to: sulfate, trifluoroacetate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid Phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, salicylate, Formate, this format, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, potassium salt, sodium salt, etc.
- pharmaceutically acceptable salts have more than one point atom in the structure. Examples in which multiple charged atoms are part of a pharmaceutically acceptable salt can have multiple counter-examples.
- a pharmaceutically acceptable salt has one or more charged atoms and/or one or more counter atoms.
- linkers or “linkers of antibody-drug conjugates” can be divided into two categories: non-cleavable linkers and cleavable linkers.
- the drug release mechanism is: After the conjugate binds to the antigen and is endocytosed by the cell, the antibody is enzymatically digested in the lysosome, and the small molecule drug is released.
- An active molecule composed of amino acid residues of an antibody. The resulting changes in the structure of the drug molecule do not reduce its cytotoxicity, but because the active molecule is charged (amino acid residues), it cannot penetrate neighboring cells. Therefore, such active drugs cannot kill adjacent tumor cells that do not express the targeted antigen (antigen-negative cells) (bystander effect) (Ducry et al., 2010, Bioconjugate Chem. 21:5-13).
- reaction solution was directly concentrated under reduced pressure and purified by preparative high performance liquid chromatography (acetonitrile/pure water system).
- the target peak was collected and reduced After the acetonitrile was removed by pressure, it was lyophilized to obtain about 2 mg of compound 16 as a yellow solid with a yield of about 60%.
- reaction solution is directly concentrated under reduced pressure and purified by preparative high performance liquid chromatography (acetonitrile/pure water system).
- the target peak is collected and the acetonitrile is removed under reduced pressure.
- compound 4 (4mg, 8.9umol) was added to a 10mL bottle, 1mL absolute ethanol, 0.2mL DMF and 0.15mL NMM were added to dissolve, cooled to 0°C in an ice water bath, and compound 21 (4.1mg, 4eq is prepared by the method disclosed in the patent application "WO2013106717”), 1-hydroxybenzotriazole HOBt (3.7mg, 4eq) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide salt Salt EDCI (5.1mg, 4eq), naturally warmed to room temperature and reacted for 2 hours, TLC monitored the end of the reaction, the reaction solution was directly concentrated under reduced pressure, purified by preparative high performance liquid chromatography (acetonitrile/pure water system), and collected After removing the acetonitrile under reduced pressure, the target peak was lyophilized to obtain about 1.5 mg of compound 22A, yellow solid, about 1.5 mg of compound 22B, yellow solid
- Patent No: WO 2020063673 It is prepared by referring to the synthetic method of the known method "Patent No: WO 2020063673".
- Patent No: WO 2020063673 It is prepared by referring to the synthetic method of the known method "Patent No: WO 2020063673".
- compound 82 was fed (28 mg, 100 umol) to obtain 19 mg of target compound, yield 25%, MS m/z: [M+H] + 762.4.
- compound 88 was fed (34 mg, 70 umol) to obtain 45 mg of target compound, yield 86%, MS m/z: [M+H] + 750.9.
- compound 110 was fed (measured according to compound 109) to obtain 1.1 mg of target compound, with a two-step yield of about 13%, MS m/z: [M+H] + 1259.8.
- the antibody molecules whose monomer ratio is greater than 95% are exchanged into a phosphate buffer containing EDTA with an ultrafiltration centrifuge tube at a concentration of 10 mg/mL.
- use an ultrafiltration centrifuge tube with a molecular weight cut-off of 30KDa to exchange the liquid into PBS, and remove the uncoupled payload.
- the sample was centrifuged at 14000 rpm for 5 minutes, and the supernatant was taken for analysis.
- Mobile phase A: 50mM PB, 300mM NaCl, 200mM Arg, 5% IPA, pH6.5;
- the mobile phase A was eluted isocratically for 30 min, flow rate: 0.714 mL/min, column temperature 25°C, detection wavelength: 280 nm.
- the sample was centrifuged at 14000 rpm for 5 minutes, and the supernatant was taken for analysis.
- Mobile phase A: 1.5M ammonium sulfate, 0.025M anhydrous sodium phosphate, pH 7.0;
- the mobile phase A equilibrates the chromatographic column, the mobile phase A and B are gradient eluted, and the flow rate is 0.8mL/min;
- Example 25 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC: Tras-6
- Example 26 Prepare according to the general coupling method, and use Tr000 as the antibody to obtain ADC: Tr000-6
- Example 27 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC:Tras-9
- Example 28 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC:Tras-12
- Example 29 Prepare according to the general coupling method, and use Tr000 as the antibody to obtain ADC: Tr000-12
- Example 30 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC: Tras-14
- Example 31 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC: Tras-16
- Example 32 Prepare according to the general coupling method, and use Tr000 as the antibody to obtain ADC: Tr000-16
- Example 33 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC: Tras-18
- Example 34 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-20A
- Example 35 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-22B
- Example 36 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-PEG-6
- Example 37 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC: Tras-PEG-9
- Example 38 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-PEG-12
- Example 39 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-PEG-14
- Example 40 Prepare according to the general coupling method, and use Trastuzumab as the antibody to obtain ADC: Tras-PEG-16
- Example 41 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-PEG-18
- Example 42 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-PEG-20A
- Example 43 Prepared according to the general coupling method, using Trastuzumab as the antibody to obtain ADC: Tras-PEG-22B
- UV lamp in the biological safety cabinet 30 minutes in advance, and then turn on the ventilation for 3 minutes.
- the dilution of antibody solution prepare the test solution with the initial concentration of 5uM and 300uL in the first column of the V-type 96-well plate with the detection medium, and then add 240uL of the detection medium in the second column to the 10th column. Take 60uL from the first row of uniformity and add it to the second row, mix up and down 10 times with a row gun, discard the pipette tip, and operate the next 7 concentrations in sequence.
- MTS reagent After 4 days, take out the MTS reagent. After thawing at room temperature and avoiding light, vortex and mix thoroughly. In a biological safety cabinet, add 20 ⁇ L CellTiter One Solution Reagen MTS reagent to each 100 ⁇ L cell culture volume along the side wall of the well, and gently tap the plate surface. , After mixing the MTS solution evenly, place it in a cell culture incubator and incubate for 2 hours in the dark. After the reaction, the 96-well plate was taken out, the OD490nm absorbance value was detected in the microplate reader, and the data was recorded, sorted, analyzed and stored.
- camptothecin derivatives of the present invention have obvious in vitro anti-tumor activity (nanomolar level), which is significantly less toxic than the toxin used by Daiichi Sankyo (control compound 16).
- the designed and synthesized camptothecin derivatives are expected to become safer anti-tumor ADC drugs.
- an A431 tumor-bearing mouse model was established to evaluate the in vivo efficacy of the toxin ADC coupled drug. That is, 1 ⁇ 10 6 A431 cells were injected subcutaneously into the right side of BALB/c nude mice aged 4-6 weeks. After the average tumor size of the mice grew to 180-200mm3, the mice were randomly divided into groups, 5 in each group, at 0 , 7, 14, and 21 days were given blank control (buffer solution blank), antibody drug conjugate Tr000-6, both were administered intravenously at a dose of 10 mg/kg. The tumor volume measurement data is displayed as the mean tumor volume ⁇ SE at the time of measurement.
Abstract
Description

































































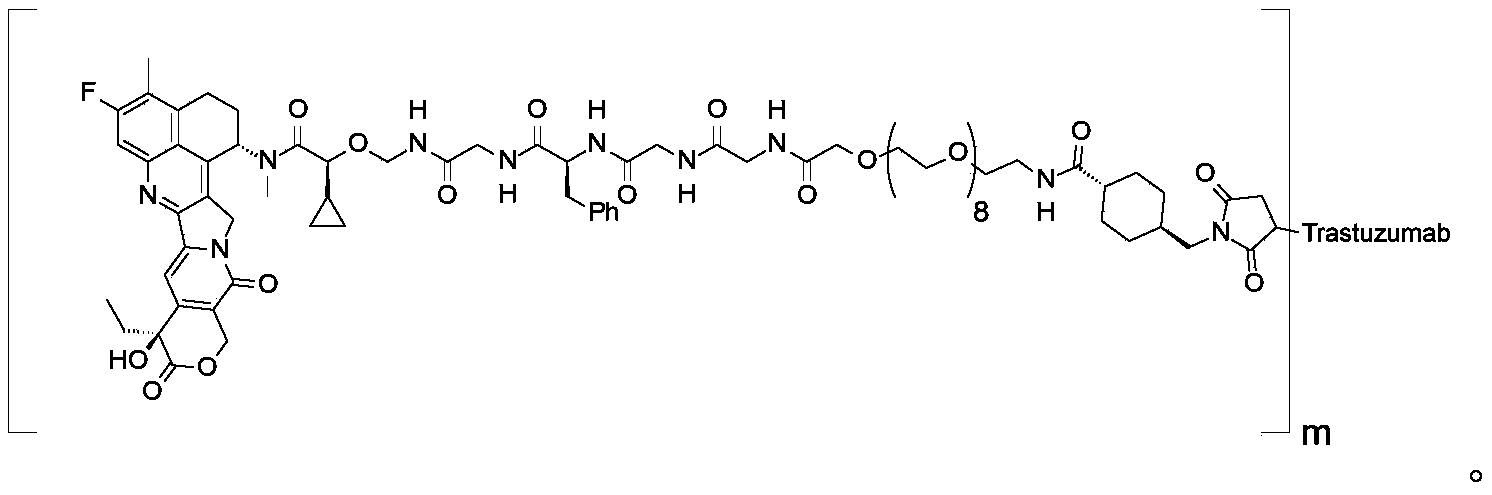

Claims (22)
- 一种喜树碱类衍生物,提供了一种通式D所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式或其药学上可接受的盐或溶剂化物:
 其中:R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R 1选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、羧基、杂环基、芳基、取代芳基或杂芳基;R 2选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、羧基、杂环基、芳基、取代芳基或杂芳基;X选自-C(O)-CR aR b-(CR 3R 4) n-O-、-C(O)-CR aR b-(CR 3R 4) n-NH-或-C(O)-CR aR b-(CR 3R 4) n-S-;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 3、R 4相同或者不同,且分别独立地为氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、烷氧基、羟基、氨基、氰基、硝基、羟烷基、环烷基或杂环基;或者,R 3、R 4连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n选自0至4的整数。
其中:R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R 1选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、羧基、杂环基、芳基、取代芳基或杂芳基;R 2选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、羧基、杂环基、芳基、取代芳基或杂芳基;X选自-C(O)-CR aR b-(CR 3R 4) n-O-、-C(O)-CR aR b-(CR 3R 4) n-NH-或-C(O)-CR aR b-(CR 3R 4) n-S-;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 3、R 4相同或者不同,且分别独立地为氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、烷氧基、羟基、氨基、氰基、硝基、羟烷基、环烷基或杂环基;或者,R 3、R 4连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n选自0至4的整数。 - 根据权利要求1所述的喜树碱类衍生物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式或其药学上可接受的盐或溶剂化物,其特征在于:通式D 1所示的化合物或其药学上可接受的盐或溶剂化物:
 其中:R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;X选自-C(O)-CR aR b-(CR 3R 4) n-O-、-C(O)-CR aR b-(CR 3R 4) n-NH-或-C(O)-CR aR b-(CR 3R 4) n-S-;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 3、R 4相同或者不同,且分别独立地为氢原子、氘原子、羟基、氨基、氰基、硝基、卤素、烷基、卤代烷基、氘代烷基、烷氧基、羟烷基、环烷基或杂环基;或者,R 3、R 4连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n选自0至4的整数;其中,式D 1中的波浪线或表示氢原子,或与接头单元或与结合靶细胞所表达抗原的抗体共价连接。
其中:R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;X选自-C(O)-CR aR b-(CR 3R 4) n-O-、-C(O)-CR aR b-(CR 3R 4) n-NH-或-C(O)-CR aR b-(CR 3R 4) n-S-;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 3、R 4相同或者不同,且分别独立地为氢原子、氘原子、羟基、氨基、氰基、硝基、卤素、烷基、卤代烷基、氘代烷基、烷氧基、羟烷基、环烷基或杂环基;或者,R 3、R 4连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n选自0至4的整数;其中,式D 1中的波浪线或表示氢原子,或与接头单元或与结合靶细胞所表达抗原的抗体共价连接。 - 根据权利要求1或2中所述的喜树碱类衍生物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其药学上可接受的盐或溶剂化物,其特征在于:X为-C(O)-CR aR b-(CR 3R 4) n-O-;R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R 3和R 4相同或不同,各自独立地选自氢原子、氘原子、卤素或烷基;R a为C 3-6环烷基烷基或C 3-6环烷基;R b选自氢原子、卤代烷基或C 3-6环烷基;优选氢原子;或者,R a和R b与其相连接的碳原子一起形成C 3-6环烷基;n为0或者1。
- 根据权利要求1或2中所述的喜树碱类衍生物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其药学上可接受的盐或溶剂化物,其特征在于:所述喜树碱类衍生物包含式D 2所示的结构:
 其中:R选自氘原子、C 1-6烷基、氚代烷基、取代烷基、芳基、取代的芳基或杂芳基;R a选自环烷基或者环烷基烷基;R b选自氢原子、氘原子、烷基、卤代烷基、环烷基或者环烷基烷基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n为0或者1;其中,式D 2中的波浪线或表示氢原子,或与接头单元或与结合靶细胞所表达抗原的抗体共价连接。
其中:R选自氘原子、C 1-6烷基、氚代烷基、取代烷基、芳基、取代的芳基或杂芳基;R a选自环烷基或者环烷基烷基;R b选自氢原子、氘原子、烷基、卤代烷基、环烷基或者环烷基烷基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n为0或者1;其中,式D 2中的波浪线或表示氢原子,或与接头单元或与结合靶细胞所表达抗原的抗体共价连接。 - 根据权利要求1或2所述的喜树碱类衍生物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其药学上可接受的盐或溶剂化物,其特征在于:X非限制性地选自:


- 根据权利要求1或2中所述的喜树碱类衍生物或其药学上可接受的盐或溶剂化物,其特征在于:其为通式D 3所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式、或其药学上可接受的盐或溶剂化物:
 其中:R选自氘原子、C 1-6烷基、氚代烷基、取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;优选C 3-6环烷基或者环烷基烷基;R b选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n为0或者1;
其中:R选自氘原子、C 1-6烷基、氚代烷基、取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;优选C 3-6环烷基或者环烷基烷基;R b选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n为0或者1; - 根据权利要求1、2或6中所述的喜树碱类衍生物或其药学上可接受的盐或溶剂化物,其特征在于:其非限制性地选自以下所示的化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式、或其药学上接受的盐或溶剂化物:

- 一种通式如(-L-X-D 2)所示的药物-连接子化合物或者其药学上可接受的盐或溶剂化物;
 其中:R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代的芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;优选C 3-6环烷基或者环烷基烷基;R b选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n为0或者1;L为连接单元;其中,式-L-X-D 2中的波浪线或表示氢原子,或与接头单元或与结合靶细胞所表达抗原的抗体共价连接。
其中:R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代的芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;优选C 3-6环烷基或者环烷基烷基;R b选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;n为0或者1;L为连接单元;其中,式-L-X-D 2中的波浪线或表示氢原子,或与接头单元或与结合靶细胞所表达抗原的抗体共价连接。 - 根据权利要求8中所述的药物-连接子化合物或者其药学上可接受的盐或溶剂化物, 其特征在于:其中X的O端与连接单元L相连。
- 根据权利要求8或9中所述的药物-连接子化合物或其药学上可接受的盐或者溶剂化物,其特征在于:其中连接单元-L-为-L 1-L 2-L 3-L 4-,,其L 1端与抗体相连,L 4端与X相连;其中:L 1选自:-(琥珀酰亚胺-3-基-N)-Y-C(O)-、-CH 2-C(O)-NR 5-Y-C(O)-或-C(O)-Y-C(O)-,其中,Y选自C 1-8烷基、C 1-8烷基-环烷基或者1-8个原子的直链或者直链-环状杂烷基;所述杂烷基包含选自N、O或者S的1-3个原子,所述的C 1-8烷基、环烷基、直链或者直链环状杂烷基各自独立地被选自氘代原子、卤素、羟基、氰基、硝基、氨基、烷基、羧基、杂烷基、取代烷基、烷氧基或者环烷基的一个或者多个取代基所取代;L 2选自:-NR 6(CH 2CH 2O) pCH 2CH 2C(O)-、-NR 6(CH 2CH 2O) pCH 2C(O)-、-S(CH 2) pC(O)-或者化学键,其中p选自0-20的整数;L 3选自:由2-7个氨基酸构成的肽残基,其中任选氨基酸进一步被选自氘原子、卤素、羟基、氰基、氨基、硝基、烷基、取代烷基、烷氧基和环烷基或者取代环烷基中的一个或多个取代基所取代;L 4选自:-NR 7(CR 8R 9) q-、-C(O)NR 7、-C(O)NR 7(CH 2) q-或者化学键,其中q选自0-6的整数;R 5、R 6和R 7相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R 8和R 9相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基。
- 根据权利要求8-10中任一所述的药物-连接子化合物或其药学上可接受的盐或溶剂化合物,其特征在于:L 1选自:-(琥珀酰亚胺-3-基-N)-Y-C(O)-、-CH 2-C(O)-NR 5-Y-C(O)-或者-C(O)-Y-C(O)-,其中Y选自C 1-8烷基、C 1-8烷基-环烷基或者1-8个原子的直链或者直链-环状杂烷基;所述杂烷基包含选自N、O或者S的1-3个原子,所述的C 1-8烷基、环烷基、直链或者直链环状杂烷基各自独立地选自氘代原子、卤素、羟基、氰基、硝基、氨基、烷基、羧基、杂烷基、取代烷基、烷氧基或者环烷基的一个或者多个取代基所取代;L 2选自:-NR 6(CH 2CH 2O) pCH 2CH 2C(O)-、-NR 6(CH 2CH 2O) pCH 2C(O)-、-S(CH 2) pC(O)-或者化学键,其中p选自0-20的整数。L 3选自:由2-7个氨基酸构成的肽残基,其中任选氨基酸进一步被选自氘原子、卤素、羟基、氰基、氨基、硝基、烷基、取代烷基、羧基、烷氧基和环烷基或者取代环烷基中的一个或多个取代基所取代;优选为苯丙氨酸(F)、甘氨酸(G)、缬氨酸(V)、赖氨酸(K)、瓜氨酸、丝氨酸(S)、谷氨酸(E)或者天冬氨酸(D)中的氨基酸形成的多肽残基;优选为由一个、两个或者多个选自苯丙氨酸和甘氨酸的氨基酸形成的氨基酸残基;最优选的为GGFG(甘氨酸-甘氨酸-苯丙氨酸-甘氨酸)组成的四肽残基;L 4选自:-NR 7(CR 8R 9) q-、-C(O)NR 7、-C(O)NR 7(CH 2) q-或者化学键,其中q选自0-6的整数,L 4优选为-NR 7CR 8R 9-;L 4更优选为-NHCH 2-。R 5、R 6和R 7相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R 8和R 9相同或者不同,且各自独立地氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基。
- 一种通式(L-X-D 2)所示的药物-连接子化合物或者其药学上可接受的盐或溶剂化物;
 其中:Z选自:-Y-C(O)-、-CH 2-C(O)-NR 5-Y-C(O)-或者-C(O)-Y-C(O)-,其中Y非限制性地选自C 1-8烷基、C 1-8烷基-环烷基或1-8个原子的直链或直链-环状杂烷基,所述杂烷基包含选自N、O或者S的1-3个原子,所述的C 1-8烷基、环烷基、直链或直链环状杂烷基各自独立地被选自氘代原子、卤素、羟基、氰基、硝基、氨基、烷基、杂烷基、取代烷基、烷氧基、羧基或环烷基的一个或者多个取代基所取代;L 2选自:-NR 6(CH 2CH 2O) pCH 2CH 2C(O)-、-NR 6(CH 2CH 2O) pCH 2C(O)-、-S(CH 2) pC(O)-或化学键,其中p选自0-20的整数;L 3选自:由2-7个氨基酸构成的肽残基,其中任选氨基酸进一步被选自氘原子、卤素、羟基、氰基、氨基、硝基、烷基、取代烷基、烷氧基和环烷基或者取代环烷基中的一个或多个取代基所取代;R选自氘原子、C 1-6烷基或取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 5、R 6和R 7相同或者不同,且各自独立地氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R 8和R 9相同或者不同,且各自独立地氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基。n选自0至4的整数。
其中:Z选自:-Y-C(O)-、-CH 2-C(O)-NR 5-Y-C(O)-或者-C(O)-Y-C(O)-,其中Y非限制性地选自C 1-8烷基、C 1-8烷基-环烷基或1-8个原子的直链或直链-环状杂烷基,所述杂烷基包含选自N、O或者S的1-3个原子,所述的C 1-8烷基、环烷基、直链或直链环状杂烷基各自独立地被选自氘代原子、卤素、羟基、氰基、硝基、氨基、烷基、杂烷基、取代烷基、烷氧基、羧基或环烷基的一个或者多个取代基所取代;L 2选自:-NR 6(CH 2CH 2O) pCH 2CH 2C(O)-、-NR 6(CH 2CH 2O) pCH 2C(O)-、-S(CH 2) pC(O)-或化学键,其中p选自0-20的整数;L 3选自:由2-7个氨基酸构成的肽残基,其中任选氨基酸进一步被选自氘原子、卤素、羟基、氰基、氨基、硝基、烷基、取代烷基、烷氧基和环烷基或者取代环烷基中的一个或多个取代基所取代;R选自氘原子、C 1-6烷基或取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 5、R 6和R 7相同或者不同,且各自独立地氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R 8和R 9相同或者不同,且各自独立地氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基。n选自0至4的整数。 - 根据权利要求12所述的通式(L-X-D 2)所示的化合物或者其药学上可接受的盐或溶剂化物,其特征在于:其为通式(L b-X-D 2)所示的化合物,或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式;
 其中:Z选自:-Y-C(O)-、-CH 2-C(O)-NR 5-Y-C(O)-或者-C(O)-Y-C(O)-,其中Y非限制性地选自C 1-8烷基、C 1-8烷基-环烷基或1-8个原子的直链或直链-环状杂烷基,所述杂烷基包含选自N、O或者S的1-3个原子,所述的C 1-8烷基、环烷基、直链或直链环状杂烷基各自独立地被选自氘代原子、卤素、羟基、氰基、硝基、氨基、烷基、杂烷基、取代烷基、烷氧基、羧基或环烷基的一个或者多个取代基所取代;R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 5和R 7相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R 8和R 9相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;n选自0至4的整数。
其中:Z选自:-Y-C(O)-、-CH 2-C(O)-NR 5-Y-C(O)-或者-C(O)-Y-C(O)-,其中Y非限制性地选自C 1-8烷基、C 1-8烷基-环烷基或1-8个原子的直链或直链-环状杂烷基,所述杂烷基包含选自N、O或者S的1-3个原子,所述的C 1-8烷基、环烷基、直链或直链环状杂烷基各自独立地被选自氘代原子、卤素、羟基、氰基、硝基、氨基、烷基、杂烷基、取代烷基、烷氧基、羧基或环烷基的一个或者多个取代基所取代;R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 5和R 7相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R 8和R 9相同或者不同,且各自独立地选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;n选自0至4的整数。 - 根据权利要求1或13中所述的通式(L-X-D 2)所示的药物-连接子化合物或者其药学上可接受的盐或溶剂化物,其特征在于:选自以下结构:





- 一种包括权利要求1-14任一所述的喜树碱衍生物或喜树碱-连接子化合物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或混合物形式,或其药学上可接受的盐或溶剂化物,与抗体连接形成如通式(Ab-L-X-Dr)所示的抗体-药物偶联物或其药学上可接受的盐或溶剂化物:
 其中:X选自-C(O)-CR aR b-(CR 3R 4) n-O-、-C(O)-CR aR b-(CR 3R 4) n-NH-或-C(O)-CR aR b-(CR 3R 4) n-S-;R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 3、R 4相同或者不同,且分别独立地选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、烷氧基、羟基、氨基、氰基、硝基、羟烷基、环烷基或杂环基;或者,R 3、R 4连同其所连接碳原子构成C 3-6环烷基或杂环基;n选自0至4的整数;m选自1-10的整数或小数;Ab为抗体、抗体片段或蛋白;L为连接单元。
其中:X选自-C(O)-CR aR b-(CR 3R 4) n-O-、-C(O)-CR aR b-(CR 3R 4) n-NH-或-C(O)-CR aR b-(CR 3R 4) n-S-;R选自氘原子、C 1-6烷基、氘代烷基、取代烷基、芳基、取代芳基或杂芳基;R a选自氢原子、氘原子、卤素、烷基、氘代烷基、卤代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;R b选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、环烷基、环烷基烷基、烷氧基烷基、杂环基、芳基、取代芳基或杂芳基;或者,R a、R b连同其所连接碳原子构成C 3-6环烷基、环烷基烷基或杂环基;R 3、R 4相同或者不同,且分别独立地选自氢原子、氘原子、卤素、烷基、卤代烷基、氘代烷基、烷氧基、羟基、氨基、氰基、硝基、羟烷基、环烷基或杂环基;或者,R 3、R 4连同其所连接碳原子构成C 3-6环烷基或杂环基;n选自0至4的整数;m选自1-10的整数或小数;Ab为抗体、抗体片段或蛋白;L为连接单元。 - 根据权利要求15中所述的抗体-药物偶联物或其药学上可接受的盐或溶剂化物,其特征在于:Ab为抗体,可通过其杂原子与连接单元形成连接键,所述抗体选自嵌合抗体、人源化抗体、全人源抗体或鼠源抗体。
- 根据权利要求15或16所述的抗体-药物偶联物或其药学上可接受的盐或溶剂化物,其特征在于:所述抗体为单克隆抗体,其选自:抗EGFRvIII抗体、抗DLL-3抗体、抗PSMA抗体、抗CD70抗体、抗MUC16抗体、抗ENPP3抗体、抗TDGF1抗体、抗ETBR抗体、抗MSLN抗体、抗TIM-1抗体、抗LRRC15抗体、抗LIV-1抗体、抗CanAg/AFP抗体、抗cladin 18.2抗体、抗Mesothelin抗体、抗HER2(ErbB2)抗体、抗EGFR抗体、抗c-MET抗体、抗SLITRK6抗体、抗KIT/CD117抗体、抗STEAP1抗体、抗SLAMF7/CS1抗体、抗NaPi2B/SLC34A2抗体、抗GPNMB抗体、抗HER3(ErbB3)抗体、抗MUC1/CD227抗体、抗AXL抗体、抗CD166抗体、抗B7-H3(CD276)抗体、抗PTK7/CCK4抗体、抗PRLR抗体、抗EFNA4抗体、抗5T4抗体、抗NOTCH3抗体、抗Nectin 4抗体、抗TROP-2抗体、抗CD142抗体、抗CA6抗体、抗GPR20抗体、抗CD174抗体、抗CD71抗体、抗EphA2抗体、抗LYPD3抗体、抗FGFR2抗体、抗FGFR3抗体、抗FRα抗体、抗CEACAMs抗体、抗GCC抗体、抗Integrin Av抗体、抗CAIX抗体、抗P-cadherin抗体、抗GD3抗体、抗Cadherin 6抗体、抗LAMP1抗体、抗FLT3抗体、抗BCMA抗体、抗CD79b抗体、抗CD19抗体、抗CD33抗体、抗CD56抗体、抗CD74抗体、抗CD22抗体、抗CD30抗体、抗CD37抗体、抗CD138抗体、抗CD352抗体、抗CD25抗体或抗CD123抗体。
- 根据权利要求15或16中所述的抗体-药物偶联物或其药学上可接受的盐或溶剂化物,其特征在于,非限制性地选自以下结构:



 其中:m选自1-10的整数或小数;Ab为抗体、抗体片段或者与抗原连接的片段。
其中:m选自1-10的整数或小数;Ab为抗体、抗体片段或者与抗原连接的片段。 - 一种制备如权利要求15-18所述通式(Ab-L a-X-Dr)所示抗体-药物偶联物或其药学上可接受的盐或溶剂化物的方法,其特征在于,包括如下步骤:
 通过抗体、抗体片段或者其抗原结合片段与通式(La-X-D 2)偶联反应,得到通式(Ab-L a-X-D 2)。
通过抗体、抗体片段或者其抗原结合片段与通式(La-X-D 2)偶联反应,得到通式(Ab-L a-X-D 2)。 - 一种药物组合物,其含有治疗有效量的根据权利要求1-19中任一项所述的抗体-药物偶联物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或其药学上可接受的盐或溶剂化物,以及药学上可接受的载体、稀释剂或赋形剂。
- 一种包括权利要求1-19中任一项所述的喜树碱类衍生物或其抗体-药物偶联物或其互变异构体、内消旋体、外消旋体、对映异构体、非对映异构体或其混合物形式,或 其药学上可接受的盐或溶剂化合物,以及药学上可接受的载体、稀释剂或者赋形剂,在制备用于治疗或预防肿瘤的药物中的用途。
- 根据权利要求21中所述的用途,其特征在于:所述的癌症优非限制性地选自:乳腺癌、卵巢癌、宫颈癌、子宫癌、前列腺癌、肾癌、尿道癌、膀胱癌、肝癌、胃癌、子宫内膜癌、唾液腺癌、食道癌、肺癌、结肠癌、直肠癌、结直肠癌、骨癌、皮肤癌、甲状腺癌、胰腺癌、黑色素瘤、神经胶质瘤、神经母细胞瘤、多形性胶质细胞瘤、肉瘤、淋巴瘤和白血病等实体瘤或血液肿瘤。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20864446.8A EP4032892A4 (en) | 2019-09-18 | 2020-09-17 | CAMPTOTHECIN DERIVATIVE AND CONJUGATE THEREOF |
| JP2022517395A JP7467610B2 (ja) | 2019-09-18 | 2020-09-17 | カンプトテシン誘導体及びその複合体 |
| BR112022004913A BR112022004913A2 (pt) | 2019-09-18 | 2020-09-17 | Derivado de camptotecina e conjugado do mesmo |
| US17/761,417 US20220411436A1 (en) | 2019-09-18 | 2020-09-17 | Camptothecin derivative and conjugate thereof |
| AU2020347715A AU2020347715A1 (en) | 2019-09-18 | 2020-09-17 | Camptothecin derivative and conjugate thereof |
| IL291337A IL291337A (en) | 2019-09-18 | 2022-03-13 | Camptothecin derivative and its conjugate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910885306 | 2019-09-18 | ||
| CN201910885306.2 | 2019-09-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021052402A1 true WO2021052402A1 (zh) | 2021-03-25 |
Family
ID=73845152
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2020/115807 WO2021052402A1 (zh) | 2019-09-18 | 2020-09-17 | 一种喜树碱衍生物及其偶联物 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20220411436A1 (zh) |
| EP (1) | EP4032892A4 (zh) |
| CN (1) | CN112125915A (zh) |
| AU (1) | AU2020347715A1 (zh) |
| BR (1) | BR112022004913A2 (zh) |
| IL (1) | IL291337A (zh) |
| WO (1) | WO2021052402A1 (zh) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022078259A1 (zh) * | 2020-10-12 | 2022-04-21 | 四川百利药业有限责任公司 | 一种氘代的喜树碱衍生物及其抗体药物偶联物 |
| WO2022236136A1 (en) * | 2021-05-07 | 2022-11-10 | ALX Oncology Inc. | Exatecan derivatives and antibody-drug conjugates thereof |
| WO2022262789A1 (zh) * | 2021-06-17 | 2022-12-22 | 明慧医药(杭州)有限公司 | 一种抗肿瘤化合物及其应用 |
| US11607459B1 (en) | 2020-09-30 | 2023-03-21 | Duality Biologics (Suzhou) Co., Ltd. | Anti-tumor compound and preparation method and use thereof |
| WO2023057814A1 (en) * | 2021-10-04 | 2023-04-13 | Vincerx Pharma Gmbh | Compounds, pharmaceutical compositions, and methods for the treatment, prevention, or management of hyperproliferative disorders |
| CN115990269A (zh) * | 2021-11-16 | 2023-04-21 | 启德医药科技(苏州)有限公司 | 依沙替康衍生物及其连接子-负载物和缀合物 |
| WO2023173026A1 (en) * | 2022-03-10 | 2023-09-14 | Sorrento Therapeutics, Inc. | Antibody-drug conjugates and uses thereof |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023201268A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating tumor antigen expressing cancers |
| US11806405B1 (en) | 2021-07-19 | 2023-11-07 | Zeno Management, Inc. | Immunoconjugates and methods |
| WO2024067811A1 (en) * | 2022-09-30 | 2024-04-04 | Beigene, Ltd. | Ligand-drug conjugate of exatecan analogue, and medical use thereof |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115298220A (zh) * | 2020-03-24 | 2022-11-04 | 上海翰森生物医药科技有限公司 | 抗体-药物偶联物及其医药用途 |
| CA3177279A1 (en) * | 2020-03-25 | 2021-09-30 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Anti-psma antibody-exatecan analogue conjugate and medical use thereof |
| EP4289851A1 (en) * | 2021-02-05 | 2023-12-13 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Camptothecin compound, preparation method therefor, and application thereof |
| WO2022166719A1 (zh) * | 2021-02-08 | 2022-08-11 | 四川百利药业有限责任公司 | 一种ca4衍生物及其配体-药物偶联物 |
| WO2022171115A1 (zh) * | 2021-02-09 | 2022-08-18 | 微境生物医药科技(上海)有限公司 | 用于adc制备的喜树碱衍生物 |
| CN116867789A (zh) * | 2021-03-31 | 2023-10-10 | 上海复旦张江生物医药股份有限公司 | 一种伊喜替康衍生物的制备方法及其中间体 |
| CN113816969B (zh) * | 2021-04-30 | 2024-01-16 | 联宁(苏州)生物制药有限公司 | 依喜替康类化合物、其抗体药物偶联物及其应用 |
| CN115850291A (zh) * | 2021-09-24 | 2023-03-28 | 石药集团巨石生物制药有限公司 | 喜树碱衍生物及其用途 |
| WO2023083381A1 (zh) * | 2021-11-15 | 2023-05-19 | 成都百利多特生物药业有限责任公司 | 双特异性抗体-喜树碱类药物偶联物及其医药用途 |
| WO2023088382A1 (zh) * | 2021-11-17 | 2023-05-25 | 石药集团巨石生物制药有限公司 | 抗体-药物偶联物及其用途 |
| CN116212044A (zh) * | 2021-12-03 | 2023-06-06 | 成都百利多特生物药业有限责任公司 | 抗人Trop2抗体-喜树碱类药物偶联物及其医药用途 |
| WO2023207710A1 (zh) * | 2022-04-29 | 2023-11-02 | 四川科伦博泰生物医药股份有限公司 | 一类抗体药物偶联物、其药物组合物及应用 |
| WO2023217064A1 (zh) * | 2022-05-09 | 2023-11-16 | 同宜医药(苏州)有限公司 | 一种喜树碱衍生物,基于其的抗体-药物偶联物和药物组合物,及其应用 |
| CN117164601A (zh) * | 2022-06-02 | 2023-12-05 | 华东师范大学 | 一种高喜树碱类小分子及其应用 |
| CN116789733A (zh) * | 2022-07-05 | 2023-09-22 | 上海药明合联生物技术有限公司 | 偶联连接子 |
| WO2024049931A1 (en) * | 2022-09-02 | 2024-03-07 | Merck Sharp & Dohme Llc | Exatecan-derived topoisomerase-1 inhibitors pharmaceutical compositions, and uses thereof |
| CN117752813A (zh) * | 2022-09-26 | 2024-03-26 | 成都百利多特生物药业有限责任公司 | 抗cd33抗体和抗cd33抗体-药物偶联物及其用途 |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| EP0737683A1 (en) | 1991-02-27 | 1996-10-16 | Pfizer Inc. | N-2-(Phthalimido)ethyl trans-piperidine-2,5-dicarboxylates as intermediates in the preparation of perhydro-1H-pyrido[1,2-a]pyrazines |
| WO2004010957A2 (en) | 2002-07-31 | 2004-02-05 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| WO2005001038A2 (en) | 2003-05-28 | 2005-01-06 | Seattle Genetics, Inc. | Recombinant anti-cd30 antibodies and uses thereof |
| WO2005037992A2 (en) | 2003-10-10 | 2005-04-28 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| US20050238649A1 (en) | 2003-11-06 | 2005-10-27 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| WO2008025020A2 (en) | 2006-08-25 | 2008-02-28 | Seattle Genetics, Inc. | Cd30 binding agents and uses thereof |
| CN101573384A (zh) | 2006-10-27 | 2009-11-04 | 健泰科生物技术公司 | 抗体和免疫偶联物及其用途 |
| WO2013106717A1 (en) | 2012-01-13 | 2013-07-18 | The General Hospital Corporation | Anesthetic compounds and related methods of use |
| CN104755494A (zh) * | 2012-10-11 | 2015-07-01 | 第一三共株式会社 | 抗体-药物偶联物 |
| CN105829346A (zh) * | 2014-01-31 | 2016-08-03 | 第三共株式会社 | 抗her2抗体-药物偶联物 |
| WO2016127790A1 (zh) | 2015-02-15 | 2016-08-18 | 江苏恒瑞医药股份有限公司 | 配体-细胞毒性药物偶联物、其制备方法及其应用 |
| CN107922477A (zh) * | 2015-06-29 | 2018-04-17 | 第三共株式会社 | 用于选择性制造抗体‑药物缀合物的方法 |
| WO2019034176A1 (zh) * | 2017-08-18 | 2019-02-21 | 四川百利药业有限责任公司 | 一种喜树碱-抗体偶联物 |
| WO2020063673A1 (zh) | 2018-09-30 | 2020-04-02 | 江苏恒瑞医药股份有限公司 | 抗b7h3抗体-依喜替康类似物偶联物及其医药用途 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3359955B2 (ja) * | 1992-07-16 | 2002-12-24 | 第一製薬株式会社 | 抗腫瘍剤 |
| ES2782248T3 (es) * | 2012-10-19 | 2020-09-11 | Daiichi Sankyo Co Ltd | Conjugado de anticuerpo y fármaco producido por la unión a través de un enlazador que tiene estructura hidrófila |
| MA52669A (fr) * | 2018-04-06 | 2021-02-17 | Seagen Inc | Conjugués peptidiques de camptothécine |
| TW202015740A (zh) * | 2018-06-07 | 2020-05-01 | 美商西雅圖遺傳學公司 | 喜樹鹼結合物 |
-
2020
- 2020-09-17 CN CN202010978407.7A patent/CN112125915A/zh active Pending
- 2020-09-17 EP EP20864446.8A patent/EP4032892A4/en active Pending
- 2020-09-17 BR BR112022004913A patent/BR112022004913A2/pt unknown
- 2020-09-17 WO PCT/CN2020/115807 patent/WO2021052402A1/zh unknown
- 2020-09-17 US US17/761,417 patent/US20220411436A1/en active Pending
- 2020-09-17 AU AU2020347715A patent/AU2020347715A1/en active Pending
-
2022
- 2022-03-13 IL IL291337A patent/IL291337A/en unknown
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| EP0737683A1 (en) | 1991-02-27 | 1996-10-16 | Pfizer Inc. | N-2-(Phthalimido)ethyl trans-piperidine-2,5-dicarboxylates as intermediates in the preparation of perhydro-1H-pyrido[1,2-a]pyrazines |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| WO2004010957A2 (en) | 2002-07-31 | 2004-02-05 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| US7659241B2 (en) | 2002-07-31 | 2010-02-09 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| WO2005001038A2 (en) | 2003-05-28 | 2005-01-06 | Seattle Genetics, Inc. | Recombinant anti-cd30 antibodies and uses thereof |
| US8088387B2 (en) | 2003-10-10 | 2012-01-03 | Immunogen Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| WO2005037992A2 (en) | 2003-10-10 | 2005-04-28 | Immunogen, Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| US20050238649A1 (en) | 2003-11-06 | 2005-10-27 | Seattle Genetics, Inc. | Monomethylvaline compounds capable of conjugation to ligands |
| WO2008025020A2 (en) | 2006-08-25 | 2008-02-28 | Seattle Genetics, Inc. | Cd30 binding agents and uses thereof |
| CN101573384A (zh) | 2006-10-27 | 2009-11-04 | 健泰科生物技术公司 | 抗体和免疫偶联物及其用途 |
| WO2013106717A1 (en) | 2012-01-13 | 2013-07-18 | The General Hospital Corporation | Anesthetic compounds and related methods of use |
| CN104755494A (zh) * | 2012-10-11 | 2015-07-01 | 第一三共株式会社 | 抗体-药物偶联物 |
| CN105829346A (zh) * | 2014-01-31 | 2016-08-03 | 第三共株式会社 | 抗her2抗体-药物偶联物 |
| WO2016127790A1 (zh) | 2015-02-15 | 2016-08-18 | 江苏恒瑞医药股份有限公司 | 配体-细胞毒性药物偶联物、其制备方法及其应用 |
| CN107922477A (zh) * | 2015-06-29 | 2018-04-17 | 第三共株式会社 | 用于选择性制造抗体‑药物缀合物的方法 |
| WO2019034176A1 (zh) * | 2017-08-18 | 2019-02-21 | 四川百利药业有限责任公司 | 一种喜树碱-抗体偶联物 |
| WO2020063673A1 (zh) | 2018-09-30 | 2020-04-02 | 江苏恒瑞医药股份有限公司 | 抗b7h3抗体-依喜替康类似物偶联物及其医药用途 |
Non-Patent Citations (21)
| Title |
|---|
| ALFTHAN ET AL., PROTEIN ENG, vol. 8, 1995, pages 725 - 731 |
| CHARI ET AL., CANCER RESEARCH, vol. 52, 1992, pages 127 - 131 |
| CHOI ET AL., EUR. J. IMMUNO, vol. 1, no. 31, 2001, pages 94 - 106 |
| DIJOSEPH JFARMELLINO DC, BLOOD, vol. 103, 2004, pages 1807 - 1814 |
| DUCRY ET AL., BIOCONJUGATE CHEM, vol. 21, 2010, pages 5 - 13 |
| G. E. MORRIS ET AL., EPITOPE MAPPING PROTOCOLS IN METHODS IN MOLECULAR BIOLOGY, vol. 66, 1996 |
| HOLLIGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HU ET AL., CANCER RES, vol. 56, pages 3055 - 3061 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 5879 - 5883 |
| J. BIOL. CHEM, vol. 243, 1968, pages 3558 |
| JONES, NATURE, vol. 321, 1986, pages 522 |
| KABAT E. A. ET AL.: "Sequences of proteins of Immunological Interest", 1991, NIH |
| KIPRIYANOV ET AL., J. MOL. BIOL., vol. 293, 1999, pages 41 - 56 |
| MULLARD A, NATURE REVIEWS DRUG DISCOVERY, vol. 12, 2013, pages 329 - 332 |
| NAT. BIOTECHNOL, vol. 21, no. 7, 2003, pages 778 - 784 |
| ORGANIC LETTERS, vol. 14, no. 18, 2012, pages 4910 - 4913 |
| QUEEN ET AL., PROC., NATL. ACAD. SCI. USA, vol. 88, 1991, pages 2869 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 327 |
| ROOVERS ET AL., CANCER IMMUNOL, 2001 |
| VERHOEYEN ET AL., SCIENCE, vol. 242, 1988, pages 1534 - 426 |
| WARD ET AL., NATURE, vol. 341, 1989, pages 544 - 546 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11607459B1 (en) | 2020-09-30 | 2023-03-21 | Duality Biologics (Suzhou) Co., Ltd. | Anti-tumor compound and preparation method and use thereof |
| US11952384B2 (en) | 2020-09-30 | 2024-04-09 | Duality Biologics (Suzhou) Co., Ltd. | Anti-tumor compound and preparation method and use thereof |
| US11685742B2 (en) | 2020-09-30 | 2023-06-27 | Duality Biologics (Suzhou) Co., Ltd. | Anti-tumor compound and preparation method and use thereof |
| WO2022078259A1 (zh) * | 2020-10-12 | 2022-04-21 | 四川百利药业有限责任公司 | 一种氘代的喜树碱衍生物及其抗体药物偶联物 |
| WO2022236136A1 (en) * | 2021-05-07 | 2022-11-10 | ALX Oncology Inc. | Exatecan derivatives and antibody-drug conjugates thereof |
| WO2022262789A1 (zh) * | 2021-06-17 | 2022-12-22 | 明慧医药(杭州)有限公司 | 一种抗肿瘤化合物及其应用 |
| US11806405B1 (en) | 2021-07-19 | 2023-11-07 | Zeno Management, Inc. | Immunoconjugates and methods |
| WO2023057814A1 (en) * | 2021-10-04 | 2023-04-13 | Vincerx Pharma Gmbh | Compounds, pharmaceutical compositions, and methods for the treatment, prevention, or management of hyperproliferative disorders |
| US11814394B2 (en) | 2021-11-16 | 2023-11-14 | Genequantum Healthcare (Suzhou) Co., Ltd. | Exatecan derivatives, linker-payloads, and conjugates and thereof |
| WO2023088235A1 (en) * | 2021-11-16 | 2023-05-25 | Genequantum Healthcare (Suzhou) Co., Ltd. | Exatecan derivatives, linker-payloads, and conjugates and thereof |
| CN115990269B (zh) * | 2021-11-16 | 2023-11-14 | 启德医药科技(苏州)有限公司 | 依沙替康衍生物及其连接子-负载物和缀合物 |
| CN115990269A (zh) * | 2021-11-16 | 2023-04-21 | 启德医药科技(苏州)有限公司 | 依沙替康衍生物及其连接子-负载物和缀合物 |
| WO2023173026A1 (en) * | 2022-03-10 | 2023-09-14 | Sorrento Therapeutics, Inc. | Antibody-drug conjugates and uses thereof |
| WO2023201267A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023201268A1 (en) | 2022-04-13 | 2023-10-19 | Gilead Sciences, Inc. | Combination therapy for treating tumor antigen expressing cancers |
| WO2024067811A1 (en) * | 2022-09-30 | 2024-04-04 | Beigene, Ltd. | Ligand-drug conjugate of exatecan analogue, and medical use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20220411436A1 (en) | 2022-12-29 |
| BR112022004913A2 (pt) | 2022-06-07 |
| EP4032892A1 (en) | 2022-07-27 |
| CN112125915A (zh) | 2020-12-25 |
| AU2020347715A1 (en) | 2022-04-07 |
| JP2022548908A (ja) | 2022-11-22 |
| EP4032892A4 (en) | 2023-10-18 |
| IL291337A (en) | 2022-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021052402A1 (zh) | 一种喜树碱衍生物及其偶联物 | |
| TWI826543B (zh) | 依喜替康類似物的配體-藥物偶聯物、其製備方法及其應用 | |
| JP6475277B2 (ja) | Trop−2に対して特異的な抗体およびこれらの使用 | |
| US20210347894A1 (en) | Anti-b7h3 antibody-exatecan analog conjugate and medicinal use thereof | |
| US20230101735A1 (en) | Anti-trop-2 antidody-exatecan analog conjugate and medical use thereof | |
| US20230144203A1 (en) | Drug conjugate of eribulin derivative, preparation method therefor and application thereof in medicine | |
| WO2021115426A1 (zh) | 抗密蛋白抗体药物偶联物及其医药用途 | |
| US20230405138A1 (en) | Anti-cd79b antibody-drug conjugate, and preparation method therefor and pharmaceutical use thereof | |
| WO2021058027A1 (zh) | 吡咯并杂芳基衍生物或其偶联物、其制备方法及其应用 | |
| CN113121639A (zh) | 澳瑞他汀类似物及其偶联物、其制备方法及其应用 | |
| US20230055408A1 (en) | Anti-cea antibody-exatecan analog conjugate and pharmaceutical use thereof | |
| US20230140397A1 (en) | Anti-psma antibody-exatecan analogue conjugate and medical use thereof | |
| JP7467610B2 (ja) | カンプトテシン誘導体及びその複合体 | |
| WO2023001300A1 (zh) | 艾日布林衍生物的药物偶联物 | |
| WO2023124963A1 (zh) | 一种可逆反应减少的抗体-药物偶联物,其制备方法及应用 | |
| CN115845080A (zh) | 艾日布林衍生物-抗叶酸受体抗体偶联物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20864446 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022517395 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020347715 Country of ref document: AU Date of ref document: 20200917 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020864446 Country of ref document: EP Effective date: 20220419 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112022004913 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112022004913 Country of ref document: BR Kind code of ref document: A2 Effective date: 20220316 |