WO2021042911A1 - Magl抑制剂及其制备方法、用途 - Google Patents
Magl抑制剂及其制备方法、用途 Download PDFInfo
- Publication number
- WO2021042911A1 WO2021042911A1 PCT/CN2020/104989 CN2020104989W WO2021042911A1 WO 2021042911 A1 WO2021042911 A1 WO 2021042911A1 CN 2020104989 W CN2020104989 W CN 2020104989W WO 2021042911 A1 WO2021042911 A1 WO 2021042911A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aryl
- alkyl
- group
- membered heteroaryl
- maglz
- Prior art date
Links
- 0 CC(C)(C)OC(N(CC1)CCC1C(N**)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(N**)=O)=O 0.000 description 4
- JGZUDIGEMFLTCV-UHFFFAOYSA-N O=C(C(CC1)CCN1C(c(cc1)ccc1Oc1ccccc1)=O)Nc1ccccc1 Chemical compound O=C(C(CC1)CCN1C(c(cc1)ccc1Oc1ccccc1)=O)Nc1ccccc1 JGZUDIGEMFLTCV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to the field of medicine, in particular to a MAGL inhibitor and its preparation method and application.
- MAGL Monoacylglycerol Lipase
- the present invention provides a compound of formula I or a pharmaceutically acceptable salt thereof:
- R is H or a C 1-5 alkyl group, and the C 1-5 alkyl group is selected from methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, neobutyl, N-pentyl, isopentyl, neopentyl, cyclopentyl or halogen, hydroxy, carboxy, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 cycloalkyl, C 3-8 Heterocyclic group, C 3-8 cycloalkoxy group, aryl group, heteroaryl group substituted C 1-5 alkyl group; the number of R is 0, 1, 2, 3 or 4;
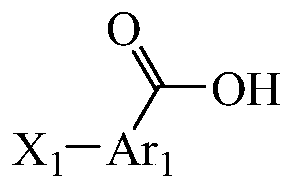
- Ar 1 and Ar 2 are each independently selected from C 5-18 aryl, heteroaryl, (C6-10 aryl)-C1-4 alkyl-, (C6-10 aryl)-C1-4 alkyl -(C6-10 aryl), (5 to 10-membered heteroaryl)-C1-4 alkyl-(C6-10 aryl), (C6-10 aryl)-(C6-10 aryl), ( C6-10 aryl)-O-(C6-10 aryl), (C6-10 aryl)-O-(5 to 10-membered heteroaryl), (5 to 10-membered heteroaryl)-O-( 5 to 10 membered heteroaryl), (5 to 10 membered heteroaryl)-(5 to 10 membered heteroaryl), any substitution position;
- X 1 and X 2 are each independently selected from H, hydroxy, carboxy, halogen, acyloxy, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 cycloalkyl, C 3-8 hetero Cyclic group, C 3-8 cycloalkoxy, aryl, heteroaryl, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 ring substituted by hydroxy, carboxy, halogen, alkyl
- One or more of an alkyl group, a C 3-8 heterocyclic group, a C 3-8 cycloalkoxy group, an aryl group, and a heteroaryl group, and the substituent positions are arbitrary.
- Ar 1 and Ar 2 are each independently selected from phenyl, C5-18 aryl, heteroaryl, (C6-10 aryl)-(C6-10 aryl), (C6-10 Aryl)-O-(C6-10 aryl);
- X 1 and X 2 are each independently selected from one or more of H, hydroxyl, halogen, C1-3 alkyl, C1-3 alkoxy, substituted
- the base position is any position.
- Ar 1 and Ar 2 are each independently selected from one or more substituents in benzene, naphthalene, diphenyl ether, indole, and biphenyl, and the substitution position is arbitrary.
- Ar 1 and Ar 2 are each independently selected from C 5-18 aryl, heteroaryl, (C6-10 aryl)-C1-4 alkyl-, (C6-10 aryl)-C1-4 Alkyl-(C6-10 aryl), (5 to 10-membered heteroaryl)-C1-4 alkyl-(C6-10 aryl), (C6-10 aryl)-(C6-10 aryl) , (C6-10 aryl)-O-(C6-10 aryl), (C6-10 aryl)-O-(5 to 10-membered heteroaryl), (5 to 10-membered heteroaryl)-O -(5 to 10 membered heteroaryl), (5 to 10 membered heteroaryl)-(5 to 10 membered heteroaryl), any substitution position;
- X 1 and X 2 are each independently selected from H, hydroxy, carboxy, halogen, acyloxy, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 cycloalkyl, C 3-8 hetero Cyclic group, C 3-8 cycloalkoxy, aryl, heteroaryl, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 ring substituted by hydroxy, carboxy, halogen, alkyl
- One or more of an alkyl group, a C 3-8 heterocyclic group, a C 3-8 cycloalkoxy group, an aryl group, and a heteroaryl group, and the substituent positions are arbitrary.
- the Ar 1 and Ar 2 are each independently selected from C 5-18 aryl, heteroaryl, (C6-10 aryl)-C1-4 alkyl, (C6-10 aryl)-C1 -4 alkyl-(C6-10 aryl), (5 to 10-membered heteroaryl)-C1-4 alkyl-(C6-10 aryl), (C6-10 aryl)-(C6-10 aryl) Group), (C6-10 aryl)-O-(C6-10 aryl), (C6-10 aryl)-O-(5 to 10-membered heteroaryl), (5 to 10-membered heteroaryl) -O-(5 to 10-membered heteroaryl), (5 to 10-membered heteroaryl)-(5 to 10-membered heteroaryl), any substitution position;
- X 1 and X 2 are each independently selected from H, hydroxy, halogen, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 heterocyclyl, C 3-8 cycloalkoxy, and One or more of hydroxy, carboxyl, halogen, alkyl-substituted C 1-3 alkyl, and C 1-3 alkoxy, and the position of the substituent is arbitrary.
- Ar 1 is one or more of benzene, naphthalene, diphenyl ether, and biphenyl, and the substitution position is arbitrary;
- X 1 is one or more of H, methoxy, hydroxyl, fluorine, and chlorine, and the position of the substituent is arbitrary;
- Ar 2 is benzene
- X 2 is one or more of H, methyl, dimethyl, methoxy, hydroxy, fluorine, and chlorine, and the position of the substituent is arbitrary.
- substitution position of Ar 1 is in the meta position.
- the Ar 1 is one of benzofuranyl, benzimidazolyl, benzoxazolyl, indolyl, benzothienyl, quinolinyl, isoquinolinyl, and purinyl.
- substitution position is arbitrary;
- X 1 is one or more of H, methyl, methoxy, hydroxy, fluorine, chlorine, and bromine, and the position of the substituent is arbitrary;
- Ar 2 is a phenyl group, the position of the substituent is arbitrary;
- X 2 is one or more of H, methyl, dimethyl, methoxy, hydroxy, fluorine, and chlorine, and the position of the substituent is arbitrary.
- the above formula 1 can be defined as formula III, as shown in the following formula
- R 1 is selected from hydroxyl, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, methoxy
- R 2 is selected from hydroxyl, fluorine, chlorine, bromine, iodine, methyl, ethyl Groups, propyl groups, methoxy groups, phenyl groups, and aromatic groups substituted by hydroxy, hydroxy, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, and methoxy groups.
- the number of R 1 , R 2 is 0 or 1 or 2 or 3 or 4; the linking mode of R 1 , R 2 and the mother core is not limited to a single single bond connection.
- the present invention provides a compound of formula b, This compound is used to synthesize a compound of formula I, wherein the Ar 2 and X 2 are those described in formula 1.
- Fragment A within the dotted line is used instead.
- the present invention provides a method for synthesizing a compound of formula I, the specific steps are as follows: salt of b or b with Reaction to obtain I; the Ar 1 and X 1 are described in formula 1.
- the condensing agent is selected from HBTU, DMC, HOBT, HOBT/EDCI, HATU, HATU/DIEPA, DCC, CDI, isopropyl Chloroformate; its structural formulas are as follows:
- the salt of b or b is obtained by deprotecting a;
- the base is an organic base, such as N,N-diisopropylethylamine (DIPEA), diethylamine (DEA) or triethylamine (TEA).
- DIPEA N,N-diisopropylethylamine
- DEA diethylamine
- TEA triethylamine
- the salt of a or a is composed of sm and Or its salt reaction
- the preparation of sm can be obtained by the prior art, or can be synthesized by those skilled in the art through a limited number of experiments on the basis of the prior art without creative labor.
- the addition of condensing agent and alkali in this step of the reaction is more conducive to the progress of the reaction.
- the condensing agent is selected from HBTU, DMC, HOBT, HOBT/EDCI, HATU, HATU/DIEPA, DCC, CDI, isopropyl chloroformate .
- the reducing agent is preferably at least one of NaBH 4 , KBH 4 , and NaBH 4 /LiCl.
- the solvents selected for the above or following reactions in the present invention are conventional solvents in the field.
- the principle of selection is to dissolve the reactants but not participate in the reaction, extract the product or make the corresponding product crystallize in it and separate the impurities.
- the numbers a, b, etc. used in the present invention are numbers that are convenient to use for describing the general formula. They can be transformed into other numbers in specific embodiments, such as 1, 2, 3, etc., all for convenience of description and do not affect the structural formula.
- the essence of its reaction equation belongs to the expression of the general formula and the general reaction equation. Those skilled in the art can judge that the substituents of the intermediates in all the above synthetic routes are determined according to the structure of the target compound.
- the substitution on the benzene ring can be one or more of the meta, meta, and para positions; for the substitution in the aromatic heterocyclic ring, it can be in the 2-, 3-, 4-, 5-position, etc. One or more of.
- substituents “one or more” and “one or more” all refer to the number of selected substituents, and the specific number is one, two, three, four...specifically,
- “Ar 1 is benzene, the position of substitution is arbitrary, X 1 is fluorine, one or more of chlorine, and the position of the substituent is arbitrary” includes the para (two), meta (two), and para positions of the phenyl group.
- (One) can be replaced by one Cl, two Cl, three Cl, or two meta positions are replaced by one Cl and one F respectively, or one Cl in the adjacent position and one F in the para position, and so on.
- the solvents selected for the above or following reactions in the present invention are conventional solvents in the field.
- the principle of selection is to dissolve the reactants but not participate in the reaction, extract the product or make the corresponding product crystallize in it and separate the impurities.
- the reaction when the reactants are in excess, the reaction can be terminated by adding a substance that can react with the excess reactants to perform a quenching reaction.
- a substance that can react with the excess reactants to perform a quenching reaction.
- water quenching or saturated ammonium chloride quenching can be used.
- the purification method of the product in each step of the reaction is selected from the group consisting of extraction, crystallization, solvent removal, and column chromatography; the operations are all conventional techniques in the field. The personnel can handle it according to the specific situation.
- the general formula and general formula synthesis method of the present invention can derive specific compounds that are not limited to these specific substances, and under the guidance of the general formula and general formula synthesis method of the present invention, those skilled in the art can obtain specific compounds without creative work. , Are all within the scope of the present invention.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising the above-mentioned compound, namely the compound covered by the general formula of I target compound and its specific substance representatives, or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable pharmaceutical excipients .
- the application provides a pharmaceutical composition, which comprises the compound described in the application or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
- the pharmaceutical composition includes, but is not limited to, an oral dosage form, a parenteral dosage form, a topical dosage form, and a rectal dosage form.
- the composition may be in liquid, solid, semi-solid, gel or aerosol form.
- the pharmaceutical composition can be oral tablets, capsules, pills, powders, sustained-release preparations, solutions and suspensions, sterile solutions, suspensions or emulsions for parenteral injection, and Ointments or creams for external use, or suppositories for rectal administration.
- the pharmaceutical composition is in a unit dosage form suitable for single administration of precise doses.
- the present invention provides all the above-mentioned compounds or their pharmaceutically acceptable salts and pharmaceutical compositions thereof, and their use as monoacylglycerol esterase inhibitors; or their use in the preparation and treatment of pathologies with monoacylglycerol esterase metabolic pathways.
- the use of drugs that characterize the disease or condition are examples of compounds or their pharmaceutically acceptable salts and pharmaceutical compositions thereof, and their use as monoacylglycerol esterase inhibitors; or their use in the preparation and treatment of pathologies with monoacylglycerol esterase metabolic pathways.
- the present invention provides a method for inhibiting monoacylglycerol esterase of all the above-mentioned compounds or their pharmaceutically acceptable salts and pharmaceutical compositions thereof.
- the method includes methods for inhibiting monoacylglycerol esterase in vivo and in vitro. Also provided are methods for treating monoacylglycerol esterase-mediated diseases or disorders with all the above-mentioned compounds or their pharmaceutically acceptable salts and pharmaceutical compositions thereof.
- the condition is selected from: metabolic disorders (such as obesity); nephropathy (such as acute inflammatory kidney injury and diabetic nephropathy); vomiting or vomiting (such as chemotherapy-induced vomiting); nausea (such as refractory nausea or Chemotherapy-induced nausea); eating disorders (e.g., anorexia or bulimia); neuropathy (e.g., diabetic neuropathy, pellagra neuropathy, alcoholic neuropathy, beriberi neuropathy); neurodegenerative disorders [Multiple Sexual sclerosis (MS), Parkinson's disease (PD), Huntington's disease, dementia, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), epilepsy, frontotemporal dementia, sleep disorders, Creutzfeldt-Jakob disease (CJD) Or prion disease]; schizophrenia; depression; bipolar disorder; tremor; dyskinesia; dystonia; spasticity; Tourette's syndrome; withdrawal syndrome [alcohol withdrawal syndrome, antidepressant withdrawal Syndrome, antipsychotic withdrawal syndrome, benz
- MS multiple sclerosis
- Devick s disease
- CNS neuropathy central pontine myelinolysis
- syphilitic myelopathy leukoencephalopathy
- Leukodystrophy Leukodystrophy
- Guillain-Barré syndrome chronic inflammatory demyelinating polyneuropathy
- anti-myelin-associated glycoprotein (MAG) peripheral neuropathy Charcot-Marie-Tooth disease
- peripheral neuropathy Myelopathy
- optic neuropathy progressive inflammatory neuropathy
- optic neuritis transverse myelitis
- cognitive impairment such as cognitive impairment related to Down’s syndrome; cognitive impairment related to Alzheimer’s disease ; Cognitive impairment related to PD; mild cognitive impairment (MCI), dementia, cognitive impairment after chemotherapy (PCCI), postoperative cognitive dysfunction (POCD)].
- MCI mild cognitive impairment
- POCD postoperative cognitive dysfunction
- the present invention provides the use of MAGL inhibitors in preparing medicines for treating diseases related to MAGL inhibitors.
- the disease associated with the MAGL inhibitor is pain, migraine, irritable bowel syndrome, or ulcerative colitis.
- the pain is traumatic pain or pathological pain.
- the pathological pain is inflammatory pain or endogenous pain.
- biogenic inflammatory pain chemical inflammatory pain, blood-derived pain, immunogenic pain, endocrine-derived pain, pain caused by metabolic disease, neurogenic pain, or cardiogenic pain.
- the pain is transient pain, acute pain or chronic pain.
- the pain is nervous system pain, cardiovascular system pain, blood system pain, respiratory system pain, digestive system pain, endocrine system pain, urinary system pain, motor system pain or immune system pain.
- the migraine is ordinary migraine, typical migraine, and cluster migraine.
- the irritable bowel syndrome is constipation-type irritable bowel syndrome, diarrhea-type irritable bowel syndrome, and constipation-diarrhea alternating-type irritable bowel syndrome.
- the MAGL inhibitor of the present invention can reduce the number of writhing times in mice caused by acetic acid, prolong the incubation time, and prolong the pain threshold of heat-stimulated mice of the mice's hot plate pain reaction. Has a significant analgesic effect.
- the MAGL inhibitor of the present invention can significantly prolong the clotting time of migraine models, increase the number of tail suspension activities, and significantly increase the 5-HT level in brain tissue. It has a significant therapeutic effect on migraine.
- the MAGL inhibitor of the present invention can significantly reduce the AWR score of the irritable bowel syndrome model and reduce the 5-HT levels in the colon and serum. It suggests that it has a significant therapeutic effect on irritable bowel syndrome. Thus, it was confirmed that the compound has the biological activity of preventing and treating irritable bowel syndrome.
- the MAGL inhibitor of the present invention can significantly increase the length of the colon in the ulcerative colitis model, reduce the total number of blood in the stool, and reduce the IL-1 content.
- the manufacturer's instructions for the use of the kit can be used, or the reaction and purification can be carried out in a manner known in the art or the instructions of this application.
- the above-mentioned techniques and methods can be implemented according to the descriptions in a number of summary and more specific documents cited and discussed in this specification according to conventional methods well-known in the art.
- groups and their substituents can be selected by those skilled in the art to provide stable structural parts and compounds.
- alkyl refers to a saturated linear or branched hydrocarbon.
- exemplary alkyl groups include, but are not limited to, linear or branched hydrocarbons of 1-6, 1-4, or 1-3 carbon atoms, referred to herein as C1-6 alkyl and C1-4 alkyl, respectively And C 1-3 alkyl.
- Exemplary alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, 2-methyl-1-butyl, 3-methyl-2-butyl, 2-methyl-1-pentyl Base, 3-methyl-1-pentyl, 4-methyl-1-pentyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl Butyl, 2,2-dimethyl-1-butyl, 3,3-dimethyl-1-butyl, 2-ethyl-1-butyl, butyl, isobutyl, tert-butyl, pentyl Base, isopentyl, neopentyl, hexyl, etc.
- the C 1-4 alkyl group mentioned in the text includes C 1-3 alkyl group, C 1-2 alkyl group, C 2-3 alkyl group, and C 2-4 alkyl group. Specific examples include but are not limited to methyl, ethyl , Propyl, isopropyl, n-butyl, isobutyl, neobutyl.
- Alkoxy refers to alkyloxy.
- the C 1-3 alkoxy group mentioned herein includes C 1-2 alkoxy group and C 2-3 alkoxy group, specific examples but not limited to methoxy group, ethoxy group, propoxy group and isopropoxy group.
- cycloalkyl refers to a saturated or partially unsaturated hydrocarbon, such as a monocyclic 5-8 membered ring system or a bicyclic 8-12 membered ring system.
- the C 3-8 cycloalkyl group described herein contains 3-8, 3-6, 4-6, 6-8, 3-7, or 5-8 carbons, and is referred to herein as a C3-8 cycloalkyl group, C3-6 cycloalkyl or C4-6 cycloalkyl is only different in form, and the meaning is equivalent.
- Exemplary cycloalkyl groups include, but are not limited to, cyclohexyl, cyclopentyl, cyclopentenyl, cyclobutyl, or cyclopropyl.
- heterocyclyl contains a monocyclic 5-8 membered ring system or a bicyclic 8-12 membered ring system containing one or more heteroatoms (e.g., one to three heteroatoms, such as nitrogen, oxygen, and sulfur).
- C 3-8 heterocyclic group means a monocyclic or polycyclic group having 3 to 8 atoms, which includes a C 3-6 heterocyclic group, a C 4-8 heterocyclic group, a C 5-8 heterocyclic group, and a C 5-8 heterocyclic group.
- heterocyclic groups include, but are not limited to, morpholinyl, thiomorpholinyl, furanyl, piperazinyl, piperidinyl, pyranyl, pyrrolidinyl, tetrahydropyranyl, tetrahydrofuranyl, and 1,3 -Dioxanyl.
- the C 3-8 cycloalkoxy group is a cycloalkyloxy group, wherein the cycloalkyl group has the same definition as the above cycloalkyl group.
- aryl refers to an aromatic mono- or multi-carbon ring group of 6 to 12 carbon atoms having at least one aromatic ring.
- the C6-10 aryl group mentioned in the text includes C6-8 aryl, C6-10 aryl, C6-9 aryl, C7-10 aryl, C8-10 aryl, C6 aryl, C7 aryl, C8 aryl , C9 aryl, C10 aryl.
- Specific examples include, but are not limited to, phenyl, mono- or poly-hydrocarbyl substituted phenyl, pentacyclopentadienyl, indenyl, naphthyl, and chamomile cyclohydrocarbyl.
- the phenyl substituted by the mono- or poly-hydrocarbyl group includes but is not limited to tolyl, ethyl phenyl, dimethyl phenyl, propyl phenyl, isopropyl phenyl, methyl ethyl phenyl; trimethyl phenyl , Butyl phenyl, methyl propyl phenyl, methyl isopropyl phenyl, diethyl phenyl, dimethyl ethyl phenyl, tetramethyl phenyl, tert-butyl phenyl, 1,2 ,3,4-Tetrahydronaphthyl, 1,2-dihydronaphthyl, indanyl and 1H-indenyl.
- the C5-18 aryl groups mentioned in the text include C6-10 aryl groups, C11-12 aryl groups, C13-15 aryl groups and C16-18 aryl groups, such as pentyl phenyl, methyl butyl phenyl, methyl iso Butyl phenyl, methyl tert-butyl phenyl, ethyl propyl phenyl, ethyl isopropyl phenyl, methyl methyl propyl phenyl, methyl methyl isopropyl phenyl, trimethyl Ethyl phenyl, pentamethyl phenyl, etc.
- heteroaryl or heteroaromatic ring or "heteroaromatic group” mentioned in the text refers to a monocyclic aromatic group containing one or more heteroatoms (for example, one to three heteroatoms, such as nitrogen, oxygen, and sulfur).
- the 5- to 10-membered heteroaryl group described herein includes 5- to 9-membered heteroaryl, 5- to 8-membered heteroaryl, 5- to 7-membered heteroaryl, 5- to 6-membered heteroaryl, and 6- to 10-membered heteroaryl.
- heteroaryl ring may be connected to adjacent groups through carbon or nitrogen.
- heteroaryl groups include, but are not limited to: benzofuranyl, isobenzofuranyl, benzo[b]thienyl, benzo[c]thienyl, isoindolyl, benzo[d]isoxazole Group, benzo[c]isoxazolyl, benzo[d]oxazolyl, benzo[d]isothiazolyl, benzo[c]isothiazolyl, benzo[d]thiazolyl, benzo[ d]imidazo
- R is selected from H and substituted or unsubstituted C 1-5 alkyl groups, the unsubstituted C 1
- the alkyl group of -5 is selected from methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, neobutyl, n-pentyl, isopentyl, neopentyl, cyclopentyl;
- the substituted C 1-5 alkyl group is halogen, hydroxy, amino, carboxyl, C 1-3 alkyl, C 1-3 alkoxy, C 3-8 cycloalkyl, C 3-8 heterocyclic group, C 3-8 cycloalkoxy, aryl, heteroaryl substituted C 1-5 alkyl.
- the R is n-propyl or
- Halogen refers to F, Cl, Br or I.
- substituent When a substituent is described by a conventional chemical formula written from left to right, the substituent also includes the chemically equivalent substituent obtained when the structural formula is written from right to left.
- CH 2 O is equivalent to OCH 2 .
- the "compounds" mentioned in this application include all stereoisomers, geometric isomers, tautomers and isotopes.
- the compounds of the present application may be asymmetric, for example, have one or more stereoisomers. Unless otherwise specified, all stereoisomers include, for example, enantiomers and diastereomers.
- the compounds of the present application containing asymmetrically substituted carbon atoms can be isolated in an optically pure form or in a racemic form. The optically active pure form can be resolved from the racemic mixture or synthesized by using chiral raw materials or chiral reagents.
- the compounds of the application also include tautomeric forms.
- the tautomeric form is derived from the exchange of a single bond with an adjacent double bond accompanied by the migration of a proton.
- the compounds of this application also include all isotopes of atoms, whether in intermediates or final compounds.
- Isotopic atoms include atoms that have the same atomic number but different mass numbers.
- isotopes of hydrogen include tritium and deuterium.
- the compounds of this application include compounds in which part of hydrogen or all hydrogen (H) is replaced by tritium (T) and/or deuterium (D); it also includes the replacement of part or all of 12 C by 13 C and/or 14 C Compounds; and other isotopes (such as N, O, P, S) between substitution compounds, such as 14 N and 15 N; 18 O and 17 O; 31 P and 32 P; 35 S and 36 S, etc.
- the compounds described herein may have one or more stereoisomeric centers, and each isomeric center may exist in the form of R or S configuration or a combination thereof.
- the compounds described herein may have one or more double bonds, and each double bond may exist in the E (trans) or Z (cis) configuration or a combination thereof.
- a specific stereoisomer, regioisomer, diastereomer, enantiomer or epimer shall be understood to include all possible isomers, such as stereoisomers Isomers, structural isomers, diastereomers, enantiomers or epimers and mixtures thereof. Therefore, the compounds described herein include all stereoisomers, structural isomers, diastereomers, enantiomers or epimeric forms that differ in configuration and their corresponding mixtures.
- treatment and other similar synonyms include alleviating, alleviating or ameliorating the symptoms of a disease or condition, inhibiting the disease or condition, for example, preventing the development of the disease or condition, alleviating the disease or condition, making the disease or condition better, and relieving the disease or condition.
- symptoms caused by a disease, or stop the symptoms of a disease or disease prevent other symptoms, ameliorate or prevent the underlying metabolic cause that causes the symptoms, in addition, the term encompasses the purpose of prevention.
- the term also includes obtaining therapeutic effects and/or preventive effects.
- the therapeutic effect refers to curing or improving the underlying disease being treated.
- the cure or improvement of one or more physiological symptoms related to the underlying disease is also a therapeutic effect.
- the composition can be administered to patients who are at risk of suffering from a specific disease, or even if a disease diagnosis has not been made, the composition can be administered to patients who have developed one or more physiological symptoms of the disease.
- an "effective amount” for treatment is the amount of the composition containing the compound disclosed herein that is required to provide significant disease relief clinically. Techniques such as dose escalation tests can be used to determine the effective amount suitable for any individual case.
- administration refers to methods capable of delivering a compound or composition to a desired site for biological action. These methods include, but are not limited to, oral route, transduodenal route, parenteral injection (including intravenous, subcutaneous, intraperitoneal, intramuscular, intraarterial injection or infusion), topical and rectal administration.
- parenteral injection including intravenous, subcutaneous, intraperitoneal, intramuscular, intraarterial injection or infusion
- topical and rectal administration topical and rectal administration.
- Those skilled in the art are familiar with the application techniques that can be used for the compounds and methods described herein, for example in Goodman and Gilman, The Pharmaceutical Basis of Therapeutics, current ed.; Pergamon; and Remington's, Pharmaceutical Science (current edition), Mack Publishing Co., Those discussed in Easton, Pa.
- pharmaceutically acceptable refers to a substance (such as a carrier or diluent) that does not affect the biological activity or properties of the compound of the present application, and is relatively non-toxic, that is, the substance can be administered to an individual without causing adverse biological reactions Or interact with any component included in the composition in an undesirable manner.
- pharmaceutical composition refers to a mixture of the compound of the present application and at least one pharmaceutically acceptable substance.
- pharmaceutically acceptable substances include, but are not limited to, carriers, stabilizers, diluents, dispersants, suspending agents, thickeners and/or excipients.
- carrier refers to a relatively non-toxic substance that facilitates the introduction of the compound of the present application into cells or tissues.
- pharmaceutically acceptable salt refers to a salt that retains the biological efficacy of the free acid and free base of the specified compound, and has no adverse effects in biology or other aspects.
- the compounds of the present application also include pharmaceutically acceptable salts.
- a pharmaceutically acceptable salt refers to a form in which the base group in the parent compound is converted into a salt form.
- Pharmaceutically acceptable salts include, but are not limited to, inorganic or organic acid salts of base groups such as amine (amino) groups.
- the pharmaceutically acceptable salt of the present application can be synthesized from the parent compound, that is, the basic group in the parent compound reacts with 1-4 equivalents of acid in a solvent system. Suitable salts are listed in Remingtong's Pharmaceutical Scicences, 17 th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 and Journal of Pharmaceutical Science, 66, 2 (1977).
- the salt in this application refers to the acid salt formed with organic acid/inorganic acid, and the basic salt formed with organic base/inorganic base.
- the basic functional group of the compound of the general formula is pyridine or imidazole (but not limited to pyridine or imidazole)
- the acidic functional group is carboxylic acid (but not limited to carboxylic acid)
- zwitterion internal salt
- N-Boc-4 piperidine carboxylic acid (458.6 mg, 2 mmol, 1 eq) was dissolved in 15 ml DCM, DIPEA (0.7 ml, 4 mmol, 2 eq) was added, HATU (912 mg, 2.4 mmol, 1.2 eq) was added, and the mixture was stirred at room temperature for 10 min.
- Aniline (0.365ml, 4mmol, 2eq) was added, stirred at room temperature for 12h, washed twice with water, washed twice with 0.1M diluted hydrochloric acid, and distilled under reduced pressure to remove the solvent to obtain Intermediate a without purification.
- Example 2-23 The preparation process of Example 2-23 is based on the general formula synthesis method, which is similar to Example 1, wherein b in Examples 22 and 23 is replaced with The MS results of each target product are equivalent to the theoretical values, and the NMR results are as follows:
- R 1 is selected from hydroxyl, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, methoxy
- R 2 is selected from hydroxyl, hydroxyl, fluorine, chlorine, bromine, iodine, methyl , Ethyl, propyl, methoxy, phenyl, and aromatic groups substituted by hydroxy, hydroxy, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, methoxy.
- the number of R 1 and R 2 is 0 or 1 or 2 or 3 or 4; the way of linking R 1 and R 2 with the mother core is not limited to a single bond.
- N-Boc-4-piperidine carboxylic acid (20) (229.3 mg, 1 mmol) was dissolved in 10 mL CH 2 Cl 2 and DIPEA (0.35 mL, 2 mmol) and HATU (456 mg, 1.2 mmol) were added.
- the mixture was stirred at room temperature for 10 min and added 3-Chloroaniline (0.21 mL, 2 mmol), stirred at room temperature for 6 h, washed three times with water, three times with 10% dilute hydrochloric acid, dried over anhydrous Na 2 SO 4 , and removed CH 2 Cl 2 by rotary evaporation to obtain intermediate 21 h without further purification.
- 100% inhibition control wells should be set up (in a 96-well plate, add 150 microliters of 1X assay buffer, 10 microliters of MAGL, and 10 microliters of DMSO, set up three replicate wells), background wells (each well Add 160 microliters of 1Xassay buffer and 10 microliters of DMSO, and set up three replicate wells). Then, graphad prism 5.0 was used to calculate and fit the IC50 curve of each inhibitor, and the specific values are shown in Table 2.
- MAGLZ-II-10 0.662 MAGLZ-II-11 0.096 MAGLZ-II-12 ND MAGLZ-II-13 0.926 MAGLZ-II-14 0.333 MAGLZ-II-15 ND MAGLZ-II-16 0.018 MAGLZ-II-17 0.015 MAGLZ-II-18 0.670 MAGLZ-II-19 2.82 MAGLZ-II-18(c) 0.0026 MAGLZ-II-18(a) 2.15 MAGLZ-II-20 9.92 MAGLZ-II-21 ND MAGLZ-II-22 1.77 MAGLZ-II-23 4.03 MAGLZ-II-24 0.275 MAGLZ-II-25 0.139 MAGLZ-II-26 12.2 MAGLZ-II-27 2.90 MAGLZ-II-29 0.252 MAGLZ-II-30 0.011 MAGLZ-II-31 ND MAGLZ-II-11(a) 2.15
- Detection reagents MAGLZ-II-11, MAGLZ-II-16, MAGLZ-II-17;
- Administration method subcutaneous injection
- Test tissue brain, heart, liver, spleen, lung, kidney, pancreas, blood;
- the three test substances were distributed in the tested brain. Among them, MAGLZ-II-16 had the highest content in the brain, followed by MAGLZ-II-17, and MAGLZ-II-11 was the lowest (see Table 3).
- Example 38 The effect of the MAGL inhibitor of the present invention on reserpine-induced depression in mice
- Group design 50 animals per batch, 3 batches; each batch of animals is randomly divided into 5 groups according to body weight: low, medium, and high-dose groups of test products, blank control group and model group. Trial product.
- MAGLZ-II-17 All indicators of Kruskal-Wallis test are statistically different (P ⁇ 0.05); Mann-Whitney U test
- the eyelids of the middle dose group had statistical differences (P ⁇ 0.05), but the average value was smaller than that of the model group, and there was no statistical difference in other indicators (P>0.05);
- MAGLZ-II-16 All indicators of Kruskal-Wallis test are statistically different (P ⁇ 0.05); Mann-Whitney U test
- the head contact in the middle dose group was statistically different from the model group (P ⁇ 0.05), the average value was larger than that of the model group, and the trend was the same as that of the blank control group, and there was no statistical difference in other indicators (P>0.05);
- the head contact and voluntary activities of the high-dose group were statistically different from the model group (P ⁇ 0.05), the average value was larger than that of the model group, and the trend was the same as that of the blank control group. There was no statistical difference in other indicators (P>0.05) .
- MAGLZ-II-16 has a certain effect on reserpine-induced depression.
- MAGLZ-II-17 may have a strengthening effect on reserpine-induced depression.
- mice Male ICR mice weighing 18-22g. Animals move under the normal 12h light and 12h dark schedule. The ambient temperature and relative humidity are maintained at 22 ⁇ 1°C and 55 ⁇ 5%, respectively.
- mice 7 groups of experimental mice, 10 in each group, set as control group (normal saline group), positive control group (fluoxetine hydrochloride capsule, 5mg/kg), 5 MAGLZ-II series administration treatment groups: MAGLZ respectively -II-06, MAGLZ-II-07, MAGLZ-II-11, MAGLZ-II-17, MAGLZ-II-18, the administration concentration is 5mg/kg.
- All drugs were administered by gavage at a dose of 20 mL/kg body weight, once a day, for 10 days. One hour after the last administration, the animal behavior test was started.
- mice forced swimming experiment was tested according to the method established by Porsolt (Porsalt R D. Behavior despair in mice: A primary screening test for antidepressants [J]. Arch. Int. Pharmacolodyn. 1977, 229).
- the mice were forced to swim alone in an open cylindrical container (10cm in diameter and 25cm in height).
- the water temperature of the container was 25 ⁇ 1°C and the water depth was 19cm.
- the total time that each animal remained immobile during 6 minutes was recorded as no Active time (in seconds).
- Each mouse was judged to be immobile when it stopped struggling, and remained suspended in the water, making only the necessary movements to keep its head above the water surface. The reduction in immobile time indicates an antidepressant effect.
- mice were hung on the tail at a distance of 10 mm from the tail of the box (250 mm ⁇ 250 mm ⁇ 300 mm) and a head distance of 5 cm from the bottom. The noise was tested in a dark room with minimal background, each mouse was hung for 6 minutes, and the immobility time was recorded at the last 4 minute interval. The criterion is that the mouse stops struggling and is completely still.
- each compound group of the MAGLZ-II series compound MAGLZ-II series can significantly shorten the cumulative immobility time of tail-suspended mice (P ⁇ 0.05) (Table 6).
- Example 40 The effect of the MAGL inhibitor of the present invention on the number of writhing times in mice caused by acetic acid
- mice 200 male ICR mice, (20 ⁇ 2)g, were randomly divided into 20 groups according to their body weight, each with 10 mice.
- the specific grouping situation is as follows: Model group: equal volume of 0.5% sodium carboxymethyl cellulose, ig
- Positive control group diclofenac sodium 10mg/kg, ig
- MAGLZ-II-11 low-dose group 7.5mg/kg, ig
- MAGLZ-II-11 middle dose group 15mg/kg, ig
- MAGLZ-II-11 high-dose group 30mg/kg, ig
- MAGLZ-II-11a low-dose group 7.5mg/kg, ig
- MAGLZ-II-11a middle dose group 15mg/kg, ig
- MAGLZ-II-11a high-dose group 30mg/kg, ig
- MAGLZ-II-18a low-dose group 7.5mg/kg, ig
- MAGLZ-II-18a middle dose group 15mg/kg, ig
- MAGLZ-II-18a high-dose group 30mg/kg, ig
- MAGLZ-II-18 low-dose group 7.5mg/kg, ig
- MAGLZ-II-18 middle dose group 15mg/kg, ig
- MAGLZ-II-18 high-dose group 30mg/kg, ig
- MAGLZ-II-18c low-dose group 7.5mg/kg, ig
- MAGLZ-II-18c middle dose group 15mg/kg, ig
- MAGLZ-II-18c high-dose group 30mg/kg, ig
- MAGLZ-II-10 low-dose group 7.5mg/kg, ig
- MAGLZ-II-10 middle dose group 15mg/kg, ig
- MAGLZ-II-10 high-dose group 30mg/kg, ig
- each compound was prepared into a suspension with 0.5% sodium carboxymethylcellulose, and then according to the dosage setting, each administration group was given by gavage; the model group was given an equal volume of 0.5% sodium carboxymethylcellulose by gavage.
- Intraperitoneal injection of acetic acid in mice caused large and long-lasting painful stimuli in the deep abdominal cavity, causing the mice to writhe.
- a 0.7% acetic acid saline solution of 0.1ml/10g was intraperitoneally injected.
- the MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, and MAGLZ-II-18c of the present invention can significantly reduce the number of writhing in mice caused by acetic acid and prolong the incubation time.
- Example 41 The effect of the MAGL inhibitor of the present invention on the painful response of the hot plate in mice
- mice Put (20 ⁇ 2)g female ICR mice on a smart hot plate instrument at 55 ⁇ 0.5°C, and record the latency (s) from the time when the sole of the mouse touches the hot plate to the hindfoot response after licking as an indicator of pain threshold, and the response is eliminated. Mice with an incubation period of ⁇ 5s or >30s or jumping.
- Example 40 The grouping and administration are the same as in Example 40.
- the intragastric administration was continued for 7 days, and the pain threshold of mice in each administration group was measured at 30, 60, 90, and 120 minutes after the last administration, and the pain threshold was calculated as 60s if the pain threshold exceeded 60s.
- the MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, and MAGLZ-II-18c of the present invention can significantly prolong the pain threshold of heat-stimulated mice.
- Example 42 Effect of the MAGL inhibitor of the present invention on the pain threshold of mice with inflammatory pain
- mice Male SD rats (180 ⁇ 20) g were injected with complete Freund's adjuvant 50 ⁇ L/mouse on the right back plantar, and the control group was injected with corresponding volume of 0.5% sodium carboxymethylcellulose to establish a foot swelling inflammatory pain model. After the model was established, the drug was administered continuously for 7 days, and the rats were given intragastrically once a day. The pain threshold (s) of mice at different time periods after administration was measured before administration, on day 1, day 3, and day 7 after administration.
- Example 40 The grouping and administration are the same as in Example 40.
- the MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, and MAGLZ-II-18c of the present invention can significantly prolong the pain threshold of mice with inflammatory pain.
- Example 43 The therapeutic effect of the MAGL inhibitor of the present invention on irritable bowel syndrome rats induced by early mother-infant separation
- Irritable bowel syndrome (Irritable Bowel Syndrome IBS): It is a common functional gastrointestinal disease, manifested by chronic abdominal pain, abdominal discomfort and changes in bowel habits, and there is no obvious intestinal pathology. This experiment mainly explores the therapeutic effect of MAGL inhibitor on irritable bowel syndrome rats induced by early mother-infant separation.
- Blank group equal volume of 0.5% sodium carboxymethyl cellulose, ig
- Model group equal volume of 0.5% sodium carboxymethyl cellulose, ig
- MAGLZ-II-11 low-dose group 7.5mg/kg, ig
- MAGLZ-II-11 middle dose group 15mg/kg, ig
- MAGLZ-II-11 high-dose group 30mg/kg, ig
- MAGLZ-II-11a low-dose group 7.5mg/kg, ig
- MAGLZ-II-11a middle dose group 15mg/kg, ig
- MAGLZ-II-11a high-dose group 30mg/kg, ig
- MAGLZ-II-18a low-dose group 7.5mg/kg, ig
- MAGLZ-II-18a middle dose group 15mg/kg, ig
- MAGLZ-II-18a high-dose group 30mg/kg, ig
- MAGLZ-II-18 low-dose group 7.5mg/kg, ig
- MAGLZ-II-18 middle dose group 15mg/kg, ig
- MAGLZ-II-18 high-dose group 30mg/kg, ig
- MAGLZ-II-18c low-dose group 7.5mg/kg, ig
- MAGLZ-II-18c middle dose group 15mg/kg, ig
- MAGLZ-II-18c high-dose group 30mg/kg, ig
- MAGLZ-II-10 low-dose group 7.5mg/kg, ig
- MAGLZ-II-10 middle dose group 15mg/kg, ig
- MAGLZ-II-10 high-dose group 30mg/kg, ig
- each compound was prepared into a suspension with 0.5% sodium carboxymethylcellulose, and then according to the dosage setting, each administration group was given by gavage; the model group was given an equal volume of 0.5% sodium carboxymethylcellulose by gavage.
- the newborn SD male rats are separated from the lactating female rats for 3 hours every day, that is, starting at 8:30 in the morning, and the lactating female rats are taken out of the cage , Then remove the newborn rat from the cage and place it in a separate cage, and then transfer this cage to the next room. After 3 hours, that is, at 11:30, return the newborn rat to the original cage. They were reunited, weaned on the 22nd day after birth, and were divided into cages on the 30th day until the 60 days were grouped and administered for a total of 20 days.
- intragastric administration was performed once a day for 20 days.
- Test index Abdominal wall withdrawal reflex score (AWR), test method: rats were fasted for 18 hours before the experiment, freely drinking, and anesthetized the rats with ether. Insert the paraffin oil-coated balloon into the rat’s colorectal 4.0cm, and use tape to The extra-anal catheter is fixed at the base of the rat's tail, and the catheter is connected to a syringe and a sphygmomanometer through a three-way connection. The rat was placed in a plexiglass observation box (20cm ⁇ 12cm ⁇ 9cm) for observation, and the experiment was started after the rat was awakened and fully adapted to the environment for 30 minutes.
- AWR Abdominal wall withdrawal reflex score
- test method rats were fasted for 18 hours before the experiment, freely drinking, and anesthetized the rats with ether. Insert the paraffin oil-coated balloon into the rat’s colorectal 4.0cm, and use tape to The extra-anal catheter is fixed
- the pressures of 20, 40, and 60mmHg were given respectively, and the expansion was continued for 20s each time, the stimulation interval was 4min, and the AWR score was repeated 3 times for each pressure.
- the AWR scoring is based on a single-blind method. The scoring standard: 0 points: no obvious behavior changes; 1 point: only simple head movement; 2 points: abdominal muscles begin to contract, but not lifted off the table; 3 points: abdominal muscles contracted significantly Flatten or lift the lower abdominal wall away from the desktop; 4 points: arched abdominal wall or bowed with the body and pelvis.
- Colon histological observation After the colon expansion experiment, take the rat's distal colon (5-6cm from the anus) and perform conventional HE staining to observe the inflammation and injury of the intestinal wall.
- MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, MAGLZ-II-18c can significantly reduce AWR score, lower colon and serum 5- HT level.
- Example 44 The effect of the MAGL inhibitor of the present invention on reserpine-induced migraine in mice
- mice 210 male ICR mice, (20 ⁇ 2) g, were randomly divided into 21 groups according to their body weight, each with 10 mice.
- the specific grouping situation is as follows: Blank group: equal volume of 0.5% sodium carboxymethyl cellulose, ig
- Model group equal volume of 0.5% sodium carboxymethyl cellulose, ig
- Positive control group Zolmitriptan 0.5mg/kg, ig
- MAGLZ-II-11 low-dose group 7.5mg/kg, ig
- MAGLZ-II-11 middle dose group 15mg/kg, ig
- MAGLZ-II-11 high-dose group 30mg/kg, ig
- MAGLZ-II-11a low-dose group 7.5mg/kg, ig
- MAGLZ-II-11a middle dose group 15mg/kg, ig
- MAGLZ-II-11a high-dose group 30mg/kg, ig
- MAGLZ-II-18a low-dose group 7.5mg/kg, ig
- MAGLZ-II-18a middle dose group 15mg/kg, ig
- MAGLZ-II-18a high-dose group 30mg/kg, ig
- MAGLZ-II-18 low-dose group 7.5mg/kg, ig
- MAGLZ-II-18 middle dose group 15mg/kg, ig
- MAGLZ-II-18 high-dose group 30mg/kg, ig
- MAGLZ-II-18c low-dose group 7.5mg/kg, ig
- MAGLZ-II-18c middle dose group 15mg/kg, ig
- MAGLZ-II-18c high-dose group 30mg/kg, ig
- MAGLZ-II-10 low-dose group 7.5mg/kg, ig
- MAGLZ-II-10 middle dose group 15mg/kg, ig
- MAGLZ-II-10 high-dose group 30mg/kg, ig
- each compound was prepared into a suspension with 0.5% sodium carboxymethylcellulose, and then according to the dosage setting, each administration group was given by gavage; the model group was given an equal volume of 0.5% sodium carboxymethylcellulose by gavage.
- mice Except for the blank control group by subcutaneous injection of normal saline, the other groups were injected subcutaneously with reserpine solution (0.2mg/kg) for a total of 10 days. After the injection of reserpine, the mice showed closed eyes, crouched and less moved, diarrhea, and eating The model is considered to be successful when the typical symptoms of reserpine are reduced, such as small amount and arched back.
- both the blank control group and the model group were given intragastric administration of the solvent, and the other groups were given the corresponding drugs and doses respectively.
- the administration was continued for 10 days.
- the 5-HT detection method is: the sample is precipitated with a 10% perchloric acid solution 1:1, and the supernatant is taken for detection after centrifugation.
- Chromatographic conditions Chromatographic column is SHIMADZU VP-ODS C 18 column (250*4.6mm, 5um), mobile phase is methanol-0.01mol/L potassium acetate buffer (10:90, V/V, use 0.2mol/L Adjust the pH to 4.00 with citric acid) isocratic elution, column temperature 25°C, flow rate 1.00ml/min, detection wavelength 275nm.
- MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, MAGLZ-II-18c can significantly prolong the clotting time.
- MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, MAGLZ-II-18c can significantly increase the number of tail suspension activities.
- MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, MAGLZ-II-18c can significantly increase reserpine-induced low 5-HT model The level of 5-HT in mouse brain tissue.
- mice were quarantined for 7 days after entering the room, and healthy male mice were selected as test animals.
- the main inspection contents during the quarantine period include, but are not limited to: whether it is consistent with the quantity and quality indicators of the animals required at the time of application; general status; weight; the above-mentioned unqualified animals are not included in this test.
- Model building After the quarantine, the mice were randomly divided into 21 groups with 10 mice in each group. The normal group was free to drink normal water, and the other groups were free to drink 1.0%-1.5% DSS solution for modeling. After 7 days of modeling, all models were free to drink normal water for 7 days. This 14 days constitutes a modeling cycle for four consecutive Cycle, modeling and administering at the same time.
- Blank group equal volume of 0.5% sodium carboxymethyl cellulose, ig
- Model group equal volume of 0.5% sodium carboxymethyl cellulose, ig
- Positive control group Mesalazine (100mg/kg), ig
- MAGLZ-II-11 low-dose group 7.5mg/kg, ig
- MAGLZ-II-11 middle dose group 15mg/kg, ig
- MAGLZ-II-11 high-dose group 30mg/kg, ig
- MAGLZ-II-11a low-dose group 7.5mg/kg, ig
- MAGLZ-II-11a middle dose group 15mg/kg, ig
- MAGLZ-II-11a high-dose group 30mg/kg, ig
- MAGLZ-II-18a low-dose group 7.5mg/kg, ig
- MAGLZ-II-18a middle dose group 15mg/kg, ig
- MAGLZ-II-18a high-dose group 30mg/kg, ig
- MAGLZ-II-18 low-dose group 7.5mg/kg, ig
- MAGLZ-II-18 middle dose group 15mg/kg, ig
- MAGLZ-II-18 high-dose group 30mg/kg, ig
- MAGLZ-II-18c low-dose group 7.5mg/kg, ig
- MAGLZ-II-18c middle dose group 15mg/kg, ig
- MAGLZ-II-18c high-dose group 30mg/kg, ig
- MAGLZ-II-10 low-dose group 7.5mg/kg, ig
- MAGLZ-II-10 middle dose group 15mg/kg, ig
- MAGLZ-II-10 high-dose group 30mg/kg, ig
- each compound was prepared into a suspension with 0.5% sodium carboxymethylcellulose, and then according to the dosage setting, each administration group was given by gavage; the model group was given an equal volume of 0.5% sodium carboxymethylcellulose by gavage.
- Experimental end point four cycles of experimental administration, anatomy on the third and fourth days of the recovery period of the fourth cycle.
- Detection indicators weigh the weight of the mice twice a week after starting the administration; observe the diarrhea and blood in the stool, and record the number of blood in the stool.
- the animals were dissected, weighed, the animals were anesthetized, blood was taken, and the serum was stored at -80°C. Take the spleen, weigh the spleen, and calculate the spleen index.
- ELISA detects the content of IL-1 ⁇ in serum. The colorectal was cut, and the length of the colorectal was measured, fixed with formaldehyde, HE stained, and pathological changes were detected.
- Table 14 The effect of the MAGL inhibitor of the present invention on the length of the colon, the number of blood in the stool, and the content of IL-1
- MAGLZ-II-11, MAGLZ-II-11a, MAGLZ-II-18a, MAGLZ-II-18, MAGLZ-II-18c can significantly increase the length of the colon, reduce the total number of blood in the stool, and reduce IL -1 content.
Abstract
Description



























| 化合物编号MAGLX | IC50(μM) |
| MAGLZ-II-01 | 0.045 |
| MAGLZ-II-02 | 0.027 |
| MAGLZ-II-03 | 0.749 |
| MAGLZ-II-04 | ND |
| MAGLZ-II-05 | 4.07 |
| MAGLZ-II-06 | 1.96 |
| MAGLZ-II-07 | 0.576 |
| MAGLZ-II-08 | 1.34 |
| MAGLZ-II-09 | 10.8 |
| MAGLZ-II-10 | 0.662 |
| MAGLZ-II-11 | 0.096 |
| MAGLZ-II-12 | ND |
| MAGLZ-II-13 | 0.926 |
| MAGLZ-II-14 | 0.333 |
| MAGLZ-II-15 | ND |
| MAGLZ-II-16 | 0.018 |
| MAGLZ-II-17 | 0.015 |
| MAGLZ-II-18 | 0.670 |
| MAGLZ-II-19 | 2.82 |
| MAGLZ-II-18(c) | 0.0026 |
| MAGLZ-II-18(a) | 2.15 |
| MAGLZ-II-20 | 9.92 |
| MAGLZ-II-21 | ND |
| MAGLZ-II-22 | 1.77 |
| MAGLZ-II-23 | 4.03 |
| MAGLZ-II-24 | 0.275 |
| MAGLZ-II-25 | 0.139 |
| MAGLZ-II-26 | 12.2 |
| MAGLZ-II-27 | 2.90 |
| MAGLZ-II-29 | 0.252 |
| MAGLZ-II-30 | 0.011 |
| MAGLZ-II-31 | ND |
| MAGLZ-II-11(a) | 2.15 |
| MAGLZ-II-11F | 0.126 |
| MAGLZ-II-11J | 1.60 |






















Claims (21)
- 式I化合物或其药学上可接受的盐:
 其中,R为H或C 1-5的烷基,所述C 1-5的烷基选自甲基,乙基,丙基,异丙基,正丁基,异丁基,新丁基,正戊基,异戊基,新戊基,环戊基,或被卤素、羟基、羧基、C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基取代的C 1-5的烷基;R的个数为0,1,2,3或4;Ar 1、Ar 2分别独立地选自C 5-18的芳基、杂芳基,(C6-10芳基)-C1-4烷基-,(C6-10芳基)-C1-4烷基-(C6-10芳基),(5至10元杂芳基)-C1-4烷基-(C6-10芳基),(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基),(C6-10芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-(5至10元杂芳基),取代位置任意;X 1、X 2分别独立地选自H,羟基,羧基,卤素,酰氧基,C 1-3烷基,C 1-3烷氧基,C 3-8环烷基,C 3-8杂环基,C 3-8环烷氧基,芳基,杂芳基,被羟基、羧基、卤素、烷基取代的C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基中的一种或多种,取代基位置任意。
其中,R为H或C 1-5的烷基,所述C 1-5的烷基选自甲基,乙基,丙基,异丙基,正丁基,异丁基,新丁基,正戊基,异戊基,新戊基,环戊基,或被卤素、羟基、羧基、C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基取代的C 1-5的烷基;R的个数为0,1,2,3或4;Ar 1、Ar 2分别独立地选自C 5-18的芳基、杂芳基,(C6-10芳基)-C1-4烷基-,(C6-10芳基)-C1-4烷基-(C6-10芳基),(5至10元杂芳基)-C1-4烷基-(C6-10芳基),(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基),(C6-10芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-(5至10元杂芳基),取代位置任意;X 1、X 2分别独立地选自H,羟基,羧基,卤素,酰氧基,C 1-3烷基,C 1-3烷氧基,C 3-8环烷基,C 3-8杂环基,C 3-8环烷氧基,芳基,杂芳基,被羟基、羧基、卤素、烷基取代的C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基中的一种或多种,取代基位置任意。 - 如权利要求1所述的化合物或其药学上可接受的盐,其特征在于,其中,R为H或C 1-5的烷基,所述C 1-5的烷基选自甲基,乙基,丙基,异丙基,正丁基,异丁基,新丁基,正戊基,异戊基,新戊基,环戊基或被卤素、羟基、羧基、C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基取代的C 1-5的烷基;R的个数为0,1,2,3或4;Ar 1、Ar 2分别独立地选自C 5-18的芳基、杂芳基,(C6-10芳基)-C1-4烷基-,(C6-10芳基)-C1-4烷基-(C6-10芳基),(5至10元杂芳基)-C1-4烷基-(C6-10芳基),(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基),(C6-10芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-(5至10元杂芳基),取代位置任意;X 1、X 2分别独立地选自H,羟基,羧基,卤素,C 1-3烷基,C 1-3烷氧基,C 3-8环烷基,C 3-8杂环基,C 3-8环烷氧基,芳基,杂芳基,被羟基、羧基、卤素、烷基取代的C 1-3烷基、C 1-3 烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基中的一种或多种,取代基位置任意。
- 如权利要求1所述的化合物或其药学上可接受的盐,其特征在于,其中,所述Ar 1、Ar 2分别独立地选自苯基,C5-18的芳基、杂芳基,(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基);X 1、X 2分别独立地选自H,羟基,卤素,酰氧基,C1-3烷基,C1-3烷氧基中的一个或多个,取代基位置为任意位置。
- 如权利要求1所述的化合物或其药学上可接受的盐,其特征在于,其中,所述Ar 1为苯,萘,二苯醚,吲哚基,联苯中的一种或多种取代基,取代位置任意。
- 如权利要求1所述的式I化合物或其药学上可接受的盐,所述的R取H,则相应的结构式为
 其中,Ar 1、Ar 2分别独立地选自C 5-18的芳基、杂芳基,(C6-10芳基)-C1-4烷基-,(C6-10芳基)-C1-4烷基-(C6-10芳基),(5至10元杂芳基)-C1-4烷基-(C6-10芳基),(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基),(C6-10芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-(5至10元杂芳基),取代位置任意;X 1、X 2分别独立地选自H,羟基,羧基,卤素,C 1-3烷基,C 1-3烷氧基,C 3-8环烷基,C 3-8杂环基,C 3-8环烷氧基,芳基,杂芳基,被羟基、羧基、卤素、烷基取代的C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基中的一种或多种,取代基位置任意。
其中,Ar 1、Ar 2分别独立地选自C 5-18的芳基、杂芳基,(C6-10芳基)-C1-4烷基-,(C6-10芳基)-C1-4烷基-(C6-10芳基),(5至10元杂芳基)-C1-4烷基-(C6-10芳基),(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基),(C6-10芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-(5至10元杂芳基),取代位置任意;X 1、X 2分别独立地选自H,羟基,羧基,卤素,C 1-3烷基,C 1-3烷氧基,C 3-8环烷基,C 3-8杂环基,C 3-8环烷氧基,芳基,杂芳基,被羟基、羧基、卤素、烷基取代的C 1-3烷基、C 1-3烷氧基、C 3-8环烷基、C 3-8杂环基、C 3-8环烷氧基、芳基、杂芳基中的一种或多种,取代基位置任意。 - 如权利要求5所述的化合物或其药学上可接受的盐,其特征在于,所述Ar 1、Ar 2分别独立地选自C5-18的芳基、杂芳基,(C6-10芳基)-C1-4烷基-,(C6-10芳基)-C1-4烷基-(C6-10芳基),(5至10元杂芳基)-C1-4烷基-(C6-10芳基),(C6-10芳基)-(C6-10芳基),(C6-10芳基)-O-(C6-10芳基),(C6-10芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-O-(5至10元杂芳基),(5至10元杂芳基)-(5至10元杂芳基),取代位置任意;X 1、X 2分别独立地选自H,羟基,卤素,C1-3烷基,C1-3烷氧基,C3-8杂环基,C3-8环烷氧基,被羟基、卤素、烷基取代的C1-3烷基、C1-3烷氧基中的一种或多种,取代基位置任意。
- 如权利要求1所述的化合物或其药学上可接受的盐,其特征在于,其中,Ar 1为苯基,萘基,二苯醚基,联苯基中的一种或多种,取代位置任意;X 1为H,甲氧基,羟基,氟,氯中的一种或多种,取代基位置任意;Ar 2为苯基,取代基位置任意;X 2为H,甲基,二甲基,甲氧基,羟基,氟,氯中的一种或多种,取代基位置任意。
- 如权利要求5所述的化合物或其药学上可接受的盐,其特征在于,所述Ar 1的取代位置在间位。
- 如权利要求1所述的化合物或其药学上可接受的盐,其特征在于,其中,所述Ar 1为苯并呋喃基、苯并咪唑基、苯并恶唑基、苯并噻吩基、吲哚基、喹啉基、异喹啉基、嘌呤基中的一种或多种,取代位置任意;X 1为H,甲基,甲氧基,羟基,氟,氯,溴中的一种或多种,取代基位置任意;Ar 2为苯基,取代基位置任意;X 2为H,甲基,二甲基,甲氧基,羟基,氟,氯中的一种或多种,取代基位置任意。
- 如权利要求1所述的式I化合物或其药学上可接受的盐,其特征在于,具体结构式选自,




- 如权利要求1的化合物,其为式III或其药学上可接受的盐;其中,所述的R 1选自羟基、氟、氯、溴、碘、甲基、乙基、丙基、甲氧基;R 2选自羟基、氟、氯、溴、碘、甲基、乙基、丙基、甲氧基、苯基、以及被羟基、羟基、氟、氯、溴、碘、甲基、乙基、丙基、甲氧基取代的芳香基。R 1、R 2的个数为0或1或2或3或4;R 1、R 2与母核的链接方式不限于单个单键连接。

- 一种用于合成如式I化合物的化合物,如式b所示,所述X 2、Ar 2如权利要求1所描述。

- 一种如式I化合物的合成方法,具体步骤如下:b或b的盐与
 反应,得到I;所述X 1、X 2、Ar 1、Ar 2为苯,萘,二苯醚,联苯中的一种或多种,取代位置任意;X 1为H,甲氧基,羟基,氟,氯中的一种或多种,取代基位置任意。
反应,得到I;所述X 1、X 2、Ar 1、Ar 2为苯,萘,二苯醚,联苯中的一种或多种,取代位置任意;X 1为H,甲氧基,羟基,氟,氯中的一种或多种,取代基位置任意。
- 如权利要求13所述的合成方法,其特征在于,所述b或b的盐与反应, 得到I;该步反应中加入缩合剂和碱,更有利于反应进行,所述的缩合剂选自HBTU、DMC、HOBT、HOBT/EDCI、HATU、HATU/DIEPA、DCC、CDI、异丙基氯甲酸酯。

- 如权利要求13所述的合成方法,其特征在于,所述b或b的盐是由a脱保护而得;

- 如权利要求13所述的合成方法,其特征在于,所述a或a的盐是由sm与或其盐反应而得;


- 药物组合物,其包含权利要求1至11中任一项的化合物或药学上可接受的盐及药学上可接受的载体。
- 如权利要求1至11中任一项的化合物或药学上可接受的盐,其用于制备治疗MAGL介导的疾病或病症的药物中的用途。
- 如权利要求18的用途,其特征在于,其中所述病症选自:代谢紊乱,肾病,呕吐或涌吐或恶心,进食障碍,神经病变,精神分裂症,抑郁症,双相障碍,震颤,运动障碍,戒断综合征,外伤性脑损伤,非外伤性脑损伤,脊髓损伤,癫痫发作,与异常细胞生长或增殖有关的病症[例如良性肿瘤或癌症,炎症性病症,免疫系统病症,肠易激综合征,溃疡性 结肠炎,急性应激障碍,物质诱导性焦虑,强迫症,焦虑症;注意缺陷障碍,注意缺陷多动障碍,疼痛,偏头痛;脱髓鞘病,及认知损害。
- 抑制MAGL的方法,其包括使所述MAGL与权利要求1至11中任一项的化合物或药学上可接受的盐接触。
- 治疗哺乳动物中MAGL介导的疾病或病症的方法,所述方法包括向所述哺乳动物给药治疗有效量的权利要求1至11中任一项的化合物或药学上可接受的盐。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/640,785 US20230090255A1 (en) | 2019-09-05 | 2020-07-27 | Magl inhibitor, preparation method therefor and use thereof |
| AU2020343737A AU2020343737A1 (en) | 2019-09-05 | 2020-07-27 | MAGL inhibitor, preparation method therefor and use thereof |
| JP2022513078A JP2022546958A (ja) | 2019-09-05 | 2020-07-27 | Magl阻害剤とその製造方法および使用 |
| EP20861878.5A EP4043436A4 (en) | 2019-09-05 | 2020-07-27 | MAGL INHIBITOR, METHOD FOR PREPARATION AND USE |
| CA3150147A CA3150147A1 (en) | 2019-09-05 | 2020-07-27 | MAGL INHIBITOR, METHOD FOR PREPARATION AND USE |
| MX2022002720A MX2022002720A (es) | 2019-09-05 | 2020-07-27 | Inhibidor de magl, procedimiento de preparacion y uso del mismo. |
| CN202080061346.6A CN114401946A (zh) | 2019-09-05 | 2020-07-27 | Magl抑制剂及其制备方法、用途 |
| KR1020227007788A KR20220058553A (ko) | 2019-09-05 | 2020-07-27 | Magl 억제제 및 이의 제조 방법, 용도 |
| BR112022004068A BR112022004068A2 (pt) | 2019-09-05 | 2020-07-27 | Inibidor da magl e seu método de preparação e uso |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910836454.5 | 2019-09-05 | ||
| CN201910836454.5A CN111518020A (zh) | 2019-02-01 | 2019-09-05 | Magl抑制剂及其制备方法、用途 |
| CN201910854235.XA CN112552228A (zh) | 2019-09-10 | 2019-09-10 | Magl抑制剂及其用途 |
| CN201910856962.X | 2019-09-10 | ||
| CN201910856962.XA CN112552229A (zh) | 2019-09-10 | 2019-09-10 | Magl抑制剂及制备方法和用途 |
| CN201910854235.X | 2019-09-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021042911A1 true WO2021042911A1 (zh) | 2021-03-11 |
Family
ID=74853072
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2020/104989 WO2021042911A1 (zh) | 2019-09-05 | 2020-07-27 | Magl抑制剂及其制备方法、用途 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20230090255A1 (zh) |
| EP (1) | EP4043436A4 (zh) |
| JP (1) | JP2022546958A (zh) |
| KR (1) | KR20220058553A (zh) |
| CN (1) | CN114401946A (zh) |
| AU (1) | AU2020343737A1 (zh) |
| BR (1) | BR112022004068A2 (zh) |
| CA (1) | CA3150147A1 (zh) |
| MX (1) | MX2022002720A (zh) |
| WO (1) | WO2021042911A1 (zh) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101128427A (zh) * | 2004-12-24 | 2008-02-20 | 阿斯利康(瑞典)有限公司 | 用作ccr2b拮抗剂的杂环化合物 |
| WO2012170098A2 (en) * | 2011-03-31 | 2012-12-13 | The Regents Of The University Of Michigan | Arbovirus inhibitors and uses thereof |
| US20160214951A1 (en) * | 2013-09-26 | 2016-07-28 | Sanford-Burnham Medical Research Institute | Ebi2 modulators |
| CN105916841A (zh) * | 2013-11-25 | 2016-08-31 | 诺沃根公司 | 作为抗癌药物的功能化的和取代的吲哚 |
| WO2017049295A1 (en) * | 2015-09-18 | 2017-03-23 | St. Jude Children's Research Hospital | Methods and compositions of inhibiting dcn1-ubc12 interaction |
| US20180079756A1 (en) * | 2015-03-30 | 2018-03-22 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| CN109311868A (zh) * | 2015-12-22 | 2019-02-05 | 尚医治疗有限责任公司 | 用于治疗癌症和炎性疾病的化合物 |
| CN109593089A (zh) * | 2019-01-24 | 2019-04-09 | 西南大学 | 二氮杂螺癸烷哌啶甲酰胺类化合物制备和应用 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR108326A1 (es) * | 2016-04-27 | 2018-08-08 | Samumed Llc | Isoquinolin-3-il carboxamidas y preparación y uso de las mismas |
| CN106279136B (zh) * | 2016-08-15 | 2019-06-21 | 中山大学 | 治疗中枢神经系统退行性疾病或脑肿瘤的化合物及其应用 |
| EP3565812B1 (en) * | 2017-01-06 | 2023-12-27 | Beyondspring Pharmaceuticals, Inc. | Tubulin binding compounds and therapeutic use thereof |
| EP3717477B1 (en) * | 2017-11-28 | 2022-07-20 | F. Hoffmann-La Roche AG | New heterocyclic compounds |
| TW201930300A (zh) * | 2017-12-15 | 2019-08-01 | 瑞士商赫孚孟拉羅股份公司 | 新雜環化合物 |
| EP3837263A1 (en) * | 2018-08-13 | 2021-06-23 | F. Hoffmann-La Roche AG | New heterocyclic compounds as monoacylglycerol lipase inhibitors |
-
2020
- 2020-07-27 EP EP20861878.5A patent/EP4043436A4/en active Pending
- 2020-07-27 WO PCT/CN2020/104989 patent/WO2021042911A1/zh unknown
- 2020-07-27 BR BR112022004068A patent/BR112022004068A2/pt unknown
- 2020-07-27 CN CN202080061346.6A patent/CN114401946A/zh active Pending
- 2020-07-27 KR KR1020227007788A patent/KR20220058553A/ko unknown
- 2020-07-27 US US17/640,785 patent/US20230090255A1/en active Pending
- 2020-07-27 AU AU2020343737A patent/AU2020343737A1/en active Pending
- 2020-07-27 MX MX2022002720A patent/MX2022002720A/es unknown
- 2020-07-27 JP JP2022513078A patent/JP2022546958A/ja active Pending
- 2020-07-27 CA CA3150147A patent/CA3150147A1/en active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101128427A (zh) * | 2004-12-24 | 2008-02-20 | 阿斯利康(瑞典)有限公司 | 用作ccr2b拮抗剂的杂环化合物 |
| WO2012170098A2 (en) * | 2011-03-31 | 2012-12-13 | The Regents Of The University Of Michigan | Arbovirus inhibitors and uses thereof |
| US20160214951A1 (en) * | 2013-09-26 | 2016-07-28 | Sanford-Burnham Medical Research Institute | Ebi2 modulators |
| CN105916841A (zh) * | 2013-11-25 | 2016-08-31 | 诺沃根公司 | 作为抗癌药物的功能化的和取代的吲哚 |
| US20180079756A1 (en) * | 2015-03-30 | 2018-03-22 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| WO2017049295A1 (en) * | 2015-09-18 | 2017-03-23 | St. Jude Children's Research Hospital | Methods and compositions of inhibiting dcn1-ubc12 interaction |
| CN109311868A (zh) * | 2015-12-22 | 2019-02-05 | 尚医治疗有限责任公司 | 用于治疗癌症和炎性疾病的化合物 |
| CN109593089A (zh) * | 2019-01-24 | 2019-04-09 | 西南大学 | 二氮杂螺癸烷哌啶甲酰胺类化合物制备和应用 |
Non-Patent Citations (40)
| Title |
|---|
| "VOGEL'S ENCYCLOPEDIA OF PRACTICAL ORGANIC CHEMISTRY", 1991, LONGMAN SCIENTIFIC AND TECHNICAL LTD., pages: 809 - 816 |
| CAREYSUNDBERG: "ADVANCED ORGANIC CHEMISTRY", vol. A, B, 2000, PLENUM PRESS |
| DATABASE REGISTRY .: "4-Piperidinecarboxamide, 1-([1,1'-biphenyl]-4-ylcarbonyl)-N-(3,5- dichlorophenyl)", XP055789075, retrieved from STN * |
| DATABASE REGISTRY .: "4-Piperidinecarboxamide, 1-(4-chlorobenzoyl)-N-(3,5-dichlorophenyl)", XP055789098, retrieved from STN Database accession no. 905171-41-7 * |
| DATABASE REGISTRY .: "4-Piperidinecarboxamide, 1-(4-chlorobenzoyl)-N-(3-methoxyphenyl)", XP055789081, retrieved from STN Database accession no. 905172-72-7 * |
| DATABASE REGISTRY .: "4-Piperidinecarboxamide, 1-(4-chlorobenzoyl)-N-(4-chlorophenyl)", XP055789111, retrieved from STN Database accession no. 864840-27-7 * |
| DATABASE REGISTRY 1 May 2003 (2003-05-01), .: "4-Piperidinecarboxamide, N-(3,5-dichlorophenyl)-1-(3-methoxybenzoyl)", XP055788552, retrieved from STN Database accession no. 508188-51-0 * |
| DATABASE REGISTRY 10 October 2005 (2005-10-10), .: "4-Piperidinecarboxamide, 1-(4-chlorobenzoyl)-N-(3-fluorophenyl)", XP055788548, retrieved from STN Database accession no. 864842-41-1 * |
| DATABASE REGISTRY 12 September 2016 (2016-09-12), .: "4-Piperidinecarboxamide, 1-(3-methoxybenzoyl)-N-(2-methoxyphenyl)", XP055788443, retrieved from STN Database accession no. 1991951-96-2 * |
| DATABASE REGISTRY 17 April 2018 (2018-04-17), .: "4-Piperidinecarboxamide, 1-([1,1'-biphenyl]-4-ylcarbonyl)-N-(3,5- dimethylphenyl)", XP055788465, retrieved from STN Database accession no. 2214031-19-1 * |
| DATABASE REGISTRY 18 June 2019 (2019-06-18), . .: "4-Piperidinecarboxamide, N-(3,5-dichlorophenyl)-1-(1H-indol-2-ylcarbonyl)", XP055788475, retrieved from STN Database accession no. 2338189-38-9 * |
| DATABASE REGISTRY 18 May 2011 (2011-05-18), .: "4-Piperidinecarboxamide, 1-(2-hydroxybenzoyl)-N-(2-methoxyphenyl)", XP055788540, retrieved from STN Database accession no. 1296587-93-3 * |
| DATABASE Registry 20 June 2019 (2019-06-20), ..: "-4-Piperidinecarboxamide, N-(3-chlorophenyl)-1-(1H-indol-2-ylcarbonyl)-", XP055788395, retrieved from STN Database accession no. 2340481-37-8 * |
| DATABASE REGISTRY 21 February 2018 (2018-02-21), "4-Piperidinecarboxamide, 1-[(5-fluoro-1H-indol-2-yl)carbonyl]-N-phenyl", XP055788462, retrieved from STN Database accession no. 2177530-54-8 * |
| DATABASE REGISTRY 22 April 2018 (2018-04-22), ..: "4-Piperidinecarboxamide, 1-([1,1'-biphenyl]-4-ylcarbonyl)-N-(3,4- dichlorophenyl)", XP055789067, retrieved from STN Database accession no. 2217251-42-6 * |
| DATABASE REGISTRY 22 April 2018 (2018-04-22), .: "4-Piperidinecarboxamide, 1-([1,1'-biphenyl]-4-ylcarbonyl)-N-(3- chlorophenyl)", XP055788467, retrieved from STN Database accession no. 2217201-40-4 * |
| DATABASE REGISTRY 22 April 2018 (2018-04-22), .: "4-Piperidinecarboxamide, 1-([1,1'-biphenyl]-4-ylcarbonyl)-N-(3- methoxyphenyl)", XP055789054, retrieved from STN Database accession no. 2217274-23-0 * |
| DATABASE REGISTRY 22 April 2018 (2018-04-22), .: "4-Piperidinecarboxamide, N-(3,4-dichlorophenyl)-1-(3-methoxybenzoyl)", XP055789045, retrieved from STN Database accession no. 2217030-90-3 * |
| DATABASE REGISTRY 22 September 2014 (2014-09-22), .: "4-Piperidinecarboxamide, 1-([1,1'-biphenyl]-4-ylcarbonyl)-N-phenyl", XP055789086, retrieved from STN Database accession no. 1624126-48-2 * |
| DATABASE REGISTRY 22 September 2014 (2014-09-22), .: "4-Piperidinecarboxamide, 1-(2-naphthalenylcarbonyl)-N-phenyl", XP055788526, retrieved from STN Database accession no. 1624146-19-5 * |
| DATABASE REGISTRY 23 April 2008 (2008-04-23), .: "4-Piperidinecarboxamide, N-(3,5-dichlorophenyl)", XP055788591, retrieved from STN Database accession no. 1016530-58-7 * |
| DATABASE REGISTRY 23 April 2008 (2008-04-23), .: "4-Piperidinecarboxamide, N-(3-chlorophenyl)-", XP055788585, retrieved from STN Database accession no. 1016748-08-5 * |
| DATABASE REGISTRY 23 April 2008 (2008-04-23), .: "4-Piperidinecarboxamide, N-(3-fluorophenyl)", XP55788587, retrieved from STN Database accession no. 1016532-14-1 * |
| DATABASE REGISTRY 23 June 2019 (2019-06-23), .: "4-Piperidinecarboxamide, N-(3,4-dichlorophenyl)-1-(1H-indol-2-ylcarbonyl)", XP055788480, retrieved from STN Database accession no. 2343019-98-5 * |
| DATABASE REGISTRY 24 June 2019 (2019-06-24), .: "4-Piperidinecarboxamide, 1-(1H-indol-2-ylcarbonyl)-N-(3-methoxyphenyl)", XP55788418, retrieved from STN Database accession no. 2344310-62-7 * |
| DATABASE REGISTRY 26 June 2002 (2002-06-26), .: "4-Piperidinecarboxamide, 1-(4-chlorobenzoyl)-N-(2-methoxyphenyl)", XP055788564, retrieved from STN Database accession no. 433952-89-7 * |
| DATABASE Registry 28 June 2019 (2019-06-28), ..: "-4-Piperidinecarboxamide, N-(3,5-dimethylphenyl)-1-(1H-indol-2-ylcarbonyl)-", XP055788407, retrieved from STN Database accession no. 2347870-36-2 * |
| DATABASE REGISTRY 29 August 2006 (2006-08-29), .: "4-Piperidinecarboxamide, 1-(4-chlorobenzoyl)-N-(3,4-dichlorophenyl)", XP055788542, retrieved from STN Database accession no. 905172-26-1 * |
| DATABASE REGISTRY 29 August 2016 (2016-08-29), .: "4-Piperidinecarboxamide, N-(3-fluorophenyl)-1-(3-methoxybenzoyl)", XP055788513, retrieved from STN Database accession no. 1981837-69-7 * |
| DATABASE REGISTRY 31 August 2011 (2011-08-31), .: "4-Piperidinecarboxamide, 1-(1H-indol-2-ylcarbonyl)-N-phenyl", XP055788530, retrieved from STN Database accession no. 1325844-20-9 * |
| DATABASE REGISTRY 31 October 2010 (2010-10-31), .: "4-Piperidinecarboxamide, N-(3-hydroxyphenyl)", XP055788569, retrieved from STN Database accession no. 1249010-19-2 * |
| DATABASE REGISTRY 7 June 2009 (2009-06-07), .: "4-Piperidinecarboxamide, N-(4-chloro-3-fluorophenyl)", XP055788580, retrieved from STN Database accession no. 1153189-42-4 * |
| DATABASE REGISTRY 8 September 2016 (2016-09-08), .: "4-Piperidinecarboxamide, N-(4-chlorophenyl)-1-(3-methoxybenzoyl)", XP055788504, retrieved from STN Database accession no. 1989422-30-1 * |
| DATABASE REGISTRY 9 February 2016 (2016-02-09), .: "4-Piperidinecarboxamide, 1-(3-methoxybenzoyl)-N-(3-methoxyphenyl)", XP055788575, retrieved from STN Database accession no. 1985280-62-3 * |
| DATABASE REGISTRY ANONYMOUS: "4-Piperidinecarboxamide, N-(3-chlorophenyl)-1-(3-methoxybenzoyl)", XP055789116, retrieved from STN Database accession no. 508188-16-7 * |
| GOODMANGILMAN: "The Pharmacological Basis of Therapeutics", 1985, MACK PUBLISHING COMPANY, pages: 1418 |
| HELLER, ACC. CHEM. RES., vol. 23, 1990, pages 128 |
| JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 2 |
| PORSOLT R DLE PICHON MJALFRE M: "Depression: a new animal model sensitive to antidepressant treatments", NATURE, vol. 266, no. 5604, 1977, pages 730 - 732 |
| ZHI ZHUOER, ZHANG WENTING, YAO JINGCHUN, SHANG YANGUO, HAO QINGJING, LIU ZHONG, REN YUSHAN, LI JIE, ZHANG GUIMIN, WANG JINXIN: "Discovery of Aryl Formyl Piperidine Derivatives as Potent, Reversible, and Selective Monoacylglycerol Lipase Inhibitors", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, vol. 63, no. 11, 11 June 2020 (2020-06-11), pages 5783 - 5796, XP055788363, ISSN: 0022-2623, DOI: 10.1021/acs.jmedchem.9b02137 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022546958A (ja) | 2022-11-10 |
| AU2020343737A1 (en) | 2022-03-31 |
| EP4043436A4 (en) | 2023-08-30 |
| CN114401946A (zh) | 2022-04-26 |
| CA3150147A1 (en) | 2021-03-11 |
| KR20220058553A (ko) | 2022-05-09 |
| MX2022002720A (es) | 2022-08-10 |
| EP4043436A1 (en) | 2022-08-17 |
| BR112022004068A2 (pt) | 2022-05-31 |
| US20230090255A1 (en) | 2023-03-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10611770B2 (en) | Condensed-ring pyrimidylamino derivative, preparation method therefor, and intermediate, pharmaceutical composition and applications thereof | |
| RU2351596C2 (ru) | Производные n-[гетероарил(пиперидин-2-ил)метил]бензамида и их применение в терапии | |
| US20070213311A1 (en) | Modulators of 11-beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same | |
| AU2019245390B2 (en) | Mono-(acid) salts of 6-aminoisoquinolines and uses thereof | |
| JP5586632B2 (ja) | 5員の複素環式化合物のアザスピラニル−アルキルカルバメートの誘導体、この調製およびこの治療的使用 | |
| CA2978458C (en) | Glucosylceramide synthase inhibitors for the treatment of diseases | |
| CA2939081A1 (en) | Cyclopropylamines as lsd1 inhibitors | |
| EA020090B1 (ru) | Способ получения и рекристаллизации кристаллического гемитартрата бензоимидазол-2-илпиримидинового производного | |
| JP6738405B2 (ja) | A2b拮抗薬としてのキサンチン置換アルキニルカルバメート/逆カルバメート | |
| WO2020115506A1 (en) | Quinolinone and benzoxazine derivatives as muscarinic m1 and/or m4 receptor agonists | |
| CN111518020A (zh) | Magl抑制剂及其制备方法、用途 | |
| CN112645869A (zh) | 一种马来酸氯苯那敏中间体的制备方法 | |
| CN108947949B (zh) | 抗焦虑氘代化合物及其医药用途 | |
| CN110577526A (zh) | 溴域结构蛋白抑制剂的盐及其制备方法和应用 | |
| CN116171153A (zh) | 用于调节剪接的组合物 | |
| EP0591027A1 (fr) | Dérivés de pipéridine, leur préparation et leur application en thérapeutique | |
| WO2021042911A1 (zh) | Magl抑制剂及其制备方法、用途 | |
| WO2021233133A1 (zh) | 用作ret激酶抑制剂的化合物及其应用 | |
| CN111518049B (zh) | Magl抑制剂、制备方法及用途 | |
| CN111518048B (zh) | Magl抑制剂、制备方法和用途 | |
| WO2020007329A1 (zh) | Hdac6选择性抑制剂的晶型及其应用 | |
| CN109535060B (zh) | 一种刺猬通路抑制剂及其制备方法和应用 | |
| CN110117301B (zh) | 用于防治神经退行性疾病的新化合物及其应用 | |
| CN112341399A (zh) | 一种1,2,4-三唑类衍生物类化合物及在治疗炎症性肠病中的应用 | |
| TW541295B (en) | A novel substituted alkylteraamine derivative for use as tachykinin antagonist |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022513078 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3150147 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112022004068 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2020343737 Country of ref document: AU Date of ref document: 20200727 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020861878 Country of ref document: EP Effective date: 20220405 |
|
| ENP | Entry into the national phase |
Ref document number: 112022004068 Country of ref document: BR Kind code of ref document: A2 Effective date: 20220304 |