WO2020168939A1 - 杂环化合物、包含其的药物组合物及其制备方法和用途 - Google Patents
杂环化合物、包含其的药物组合物及其制备方法和用途 Download PDFInfo
- Publication number
- WO2020168939A1 WO2020168939A1 PCT/CN2020/074696 CN2020074696W WO2020168939A1 WO 2020168939 A1 WO2020168939 A1 WO 2020168939A1 CN 2020074696 W CN2020074696 W CN 2020074696W WO 2020168939 A1 WO2020168939 A1 WO 2020168939A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- group
- cycloalkyl
- alkoxy
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 152
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 26
- 150000002391 heterocyclic compounds Chemical class 0.000 title abstract description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 713
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 20
- 230000000694 effects Effects 0.000 claims abstract description 20
- 201000010099 disease Diseases 0.000 claims abstract description 17
- 238000006243 chemical reaction Methods 0.000 claims description 410
- 125000000217 alkyl group Chemical group 0.000 claims description 258
- 125000000623 heterocyclic group Chemical group 0.000 claims description 212
- 125000003545 alkoxy group Chemical group 0.000 claims description 210
- 125000001424 substituent group Chemical group 0.000 claims description 147
- 150000002367 halogens Chemical class 0.000 claims description 137
- 229910052736 halogen Inorganic materials 0.000 claims description 136
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 105
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 104
- 125000003118 aryl group Chemical group 0.000 claims description 104
- -1 4-CN-pyridin-2-yl Chemical group 0.000 claims description 97
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 94
- 125000001072 heteroaryl group Chemical group 0.000 claims description 91
- 229910052799 carbon Inorganic materials 0.000 claims description 88
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 85
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 82
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 79
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims description 77
- 125000002853 C1-C4 hydroxyalkyl group Chemical group 0.000 claims description 71
- 239000000203 mixture Substances 0.000 claims description 61
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 53
- 239000000460 chlorine Substances 0.000 claims description 45
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 34
- 229910052739 hydrogen Inorganic materials 0.000 claims description 34
- 238000000034 method Methods 0.000 claims description 32
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 claims description 31
- 206010028980 Neoplasm Diseases 0.000 claims description 30
- 125000004429 atom Chemical group 0.000 claims description 29
- 239000000651 prodrug Substances 0.000 claims description 29
- 229940002612 prodrug Drugs 0.000 claims description 29
- 239000013078 crystal Substances 0.000 claims description 27
- 239000002207 metabolite Substances 0.000 claims description 27
- 229910052801 chlorine Inorganic materials 0.000 claims description 26
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 26
- 150000001204 N-oxides Chemical class 0.000 claims description 25
- 239000012453 solvate Substances 0.000 claims description 25
- 238000006467 substitution reaction Methods 0.000 claims description 25
- 125000006716 (C1-C6) heteroalkyl group Chemical group 0.000 claims description 24
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 23
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 23
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 22
- 125000004076 pyridyl group Chemical group 0.000 claims description 22
- 150000003839 salts Chemical class 0.000 claims description 22
- 125000000335 thiazolyl group Chemical group 0.000 claims description 22
- 229910052731 fluorine Inorganic materials 0.000 claims description 21
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 20
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 19
- 125000002947 alkylene group Chemical group 0.000 claims description 19
- 125000004432 carbon atom Chemical group C* 0.000 claims description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 19
- 229910052796 boron Inorganic materials 0.000 claims description 17
- 125000005842 heteroatom Chemical group 0.000 claims description 17
- 125000002883 imidazolyl group Chemical group 0.000 claims description 16
- 229910052794 bromium Inorganic materials 0.000 claims description 15
- 125000004474 heteroalkylene group Chemical group 0.000 claims description 15
- 229910052740 iodine Inorganic materials 0.000 claims description 15
- 125000002971 oxazolyl group Chemical group 0.000 claims description 15
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 15
- 239000003153 chemical reaction reagent Substances 0.000 claims description 14
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 13
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 13
- 230000002378 acidificating effect Effects 0.000 claims description 12
- 239000003814 drug Substances 0.000 claims description 11
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 10
- 125000004122 cyclic group Chemical group 0.000 claims description 10
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 10
- 239000011737 fluorine Substances 0.000 claims description 10
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 9
- 150000001412 amines Chemical class 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- 125000002541 furyl group Chemical group 0.000 claims description 9
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims description 8
- 125000003342 alkenyl group Chemical group 0.000 claims description 8
- 125000000304 alkynyl group Chemical group 0.000 claims description 7
- 125000004419 alkynylene group Chemical group 0.000 claims description 7
- 201000011510 cancer Diseases 0.000 claims description 7
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 7
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 7
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 6
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims description 6
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 claims description 6
- 125000004193 piperazinyl group Chemical group 0.000 claims description 6
- 230000002265 prevention Effects 0.000 claims description 6
- 125000004450 alkenylene group Chemical group 0.000 claims description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 5
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 5
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 5
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 5
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 5
- 230000003449 preventive effect Effects 0.000 claims description 5
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 5
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 5
- 125000001544 thienyl group Chemical group 0.000 claims description 5
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 4
- 125000006590 (C2-C6) alkenylene group Chemical group 0.000 claims description 4
- 125000006591 (C2-C6) alkynylene group Chemical group 0.000 claims description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 4
- 125000002355 alkine group Chemical group 0.000 claims description 4
- 238000006482 condensation reaction Methods 0.000 claims description 4
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 claims description 4
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 4
- 125000001113 thiadiazolyl group Chemical group 0.000 claims description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 3
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 3
- 208000009018 Medullary thyroid cancer Diseases 0.000 claims description 3
- 206010033701 Papillary thyroid cancer Diseases 0.000 claims description 3
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 3
- 150000001335 aliphatic alkanes Chemical group 0.000 claims description 3
- 125000001352 cyclobutyloxy group Chemical group C1(CCC1)O* 0.000 claims description 3
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 3
- 238000010534 nucleophilic substitution reaction Methods 0.000 claims description 3
- 125000003386 piperidinyl group Chemical group 0.000 claims description 3
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 3
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 2
- 206010006187 Breast cancer Diseases 0.000 claims description 2
- 208000026310 Breast neoplasm Diseases 0.000 claims description 2
- 206010009944 Colon cancer Diseases 0.000 claims description 2
- 208000032612 Glial tumor Diseases 0.000 claims description 2
- 206010018338 Glioma Diseases 0.000 claims description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 2
- 206010025323 Lymphomas Diseases 0.000 claims description 2
- 208000034578 Multiple myelomas Diseases 0.000 claims description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 2
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical group C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 claims description 2
- 201000008211 brain sarcoma Diseases 0.000 claims description 2
- 208000029742 colonic neoplasm Diseases 0.000 claims description 2
- 201000010536 head and neck cancer Diseases 0.000 claims description 2
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 2
- 201000007270 liver cancer Diseases 0.000 claims description 2
- 208000014018 liver neoplasm Diseases 0.000 claims description 2
- 201000005202 lung cancer Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- 201000001441 melanoma Diseases 0.000 claims description 2
- 208000029522 neoplastic syndrome Diseases 0.000 claims description 2
- 206010038038 rectal cancer Diseases 0.000 claims description 2
- 201000001275 rectum cancer Diseases 0.000 claims description 2
- 201000002510 thyroid cancer Diseases 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims 2
- QPMLSUSACCOBDK-UHFFFAOYSA-N diazepane Chemical compound C1CCNNCC1 QPMLSUSACCOBDK-UHFFFAOYSA-N 0.000 claims 1
- SNOOUWRIMMFWNE-UHFFFAOYSA-M sodium;6-[(3,4,5-trimethoxybenzoyl)amino]hexanoate Chemical compound [Na+].COC1=CC(C(=O)NCCCCCC([O-])=O)=CC(OC)=C1OC SNOOUWRIMMFWNE-UHFFFAOYSA-M 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 269
- 239000000243 solution Substances 0.000 description 240
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 141
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 128
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 118
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 116
- 230000002829 reductive effect Effects 0.000 description 104
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 85
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 82
- 238000005481 NMR spectroscopy Methods 0.000 description 76
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 74
- 238000002953 preparative HPLC Methods 0.000 description 74
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 69
- 239000012074 organic phase Substances 0.000 description 69
- 229910052757 nitrogen Inorganic materials 0.000 description 62
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 48
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 46
- JFOZKMSJYSPYLN-QHCPKHFHSA-N lifitegrast Chemical compound CS(=O)(=O)C1=CC=CC(C[C@H](NC(=O)C=2C(=C3CCN(CC3=CC=2Cl)C(=O)C=2C=C3OC=CC3=CC=2)Cl)C(O)=O)=C1 JFOZKMSJYSPYLN-QHCPKHFHSA-N 0.000 description 45
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 43
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 42
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 41
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 41
- 238000010898 silica gel chromatography Methods 0.000 description 39
- 238000003818 flash chromatography Methods 0.000 description 35
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 34
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 33
- 239000000741 silica gel Substances 0.000 description 33
- 229910002027 silica gel Inorganic materials 0.000 description 33
- 230000005764 inhibitory process Effects 0.000 description 28
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 27
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 26
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 25
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 23
- 229920006395 saturated elastomer Polymers 0.000 description 23
- 239000002904 solvent Substances 0.000 description 23
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 22
- 238000003756 stirring Methods 0.000 description 22
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 21
- 229910052717 sulfur Inorganic materials 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 229910052760 oxygen Inorganic materials 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 17
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 17
- 239000001301 oxygen Substances 0.000 description 17
- 239000011593 sulfur Substances 0.000 description 17
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 16
- 230000002401 inhibitory effect Effects 0.000 description 16
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- 238000010791 quenching Methods 0.000 description 15
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 14
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 14
- 229910000024 caesium carbonate Inorganic materials 0.000 description 14
- 239000011734 sodium Substances 0.000 description 14
- 102000004190 Enzymes Human genes 0.000 description 13
- 108090000790 Enzymes Proteins 0.000 description 13
- 239000002253 acid Substances 0.000 description 13
- 239000007864 aqueous solution Substances 0.000 description 13
- 239000012043 crude product Substances 0.000 description 13
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 12
- 229940126657 Compound 17 Drugs 0.000 description 12
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 11
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical class [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 11
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 11
- 229940125904 compound 1 Drugs 0.000 description 11
- 239000002994 raw material Substances 0.000 description 11
- 238000006069 Suzuki reaction reaction Methods 0.000 description 10
- 238000001514 detection method Methods 0.000 description 10
- 238000005859 coupling reaction Methods 0.000 description 9
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 102200006099 rs79658334 Human genes 0.000 description 9
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 8
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 8
- GBLBJPZSROAGMF-RWYJCYHVSA-N CO[C@@]1(CC[C@@H](CC1)C1=NC(NC2=NNC(C)=C2)=CC(C)=N1)C(=O)N[C@@H](C)C1=CC=C(N=C1)N1C=C(F)C=N1 Chemical compound CO[C@@]1(CC[C@@H](CC1)C1=NC(NC2=NNC(C)=C2)=CC(C)=N1)C(=O)N[C@@H](C)C1=CC=C(N=C1)N1C=C(F)C=N1 GBLBJPZSROAGMF-RWYJCYHVSA-N 0.000 description 8
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 8
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- 238000012746 preparative thin layer chromatography Methods 0.000 description 8
- 125000006413 ring segment Chemical group 0.000 description 8
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 8
- 235000017557 sodium bicarbonate Nutrition 0.000 description 8
- 125000003003 spiro group Chemical group 0.000 description 8
- 239000003981 vehicle Substances 0.000 description 8
- WGABOZPQOOZAOI-UHFFFAOYSA-N 2-[4-[[(3,5-dimethoxy-4-methylbenzoyl)-(3-phenylpropyl)amino]methyl]phenyl]acetic acid Chemical compound COC1=C(C)C(OC)=CC(C(=O)N(CCCC=2C=CC=CC=2)CC=2C=CC(CC(O)=O)=CC=2)=C1 WGABOZPQOOZAOI-UHFFFAOYSA-N 0.000 description 7
- IPGZOYHFPGLILW-UHFFFAOYSA-N 2-[6-[6-[(6-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)OC IPGZOYHFPGLILW-UHFFFAOYSA-N 0.000 description 7
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 7
- 235000019270 ammonium chloride Nutrition 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 229940126212 compound 17a Drugs 0.000 description 7
- 239000011259 mixed solution Substances 0.000 description 7
- 239000000825 pharmaceutical preparation Substances 0.000 description 7
- CCBQGNNSKDHKIJ-UHFFFAOYSA-N 2-[6-[6-[[4-(4-fluoropyrazol-1-yl)phenyl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=C(C=C6)N7C=C(C=N7)F CCBQGNNSKDHKIJ-UHFFFAOYSA-N 0.000 description 6
- AGNVMNUXVGZSKX-UHFFFAOYSA-N 2-[6-[6-[[6-[4-(fluoromethyl)pyrazol-1-yl]pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C=N7)CF AGNVMNUXVGZSKX-UHFFFAOYSA-N 0.000 description 6
- MMPIPXGBBXBZGM-UHFFFAOYSA-N 6-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-4-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyridin-2-amine Chemical compound CC1=CC(=NC(=C1)NC2=NNC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C=N7)F MMPIPXGBBXBZGM-UHFFFAOYSA-N 0.000 description 6
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 6
- OYRRZWATULMEPF-UHFFFAOYSA-N pyrimidin-4-amine Chemical compound NC1=CC=NC=N1 OYRRZWATULMEPF-UHFFFAOYSA-N 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 238000001308 synthesis method Methods 0.000 description 6
- FPLPLWYHDFSPTD-UHFFFAOYSA-N tert-butyl 3-cyclopropyloxypyrazole-1-carboxylate Chemical compound C1(CC1)OC1=NN(C=C1)C(=O)OC(C)(C)C FPLPLWYHDFSPTD-UHFFFAOYSA-N 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- PHDIJLFSKNMCMI-ITGJKDDRSA-N (3R,4S,5R,6R)-6-(hydroxymethyl)-4-(8-quinolin-6-yloxyoctoxy)oxane-2,3,5-triol Chemical compound OC[C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)OCCCCCCCCOC=1C=C2C=CC=NC2=CC=1)O PHDIJLFSKNMCMI-ITGJKDDRSA-N 0.000 description 5
- AVOKZFCYCRLYJD-UHFFFAOYSA-N 2-[6-[6-[1-[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]ethyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5C(C)C6=CN=C(C=C6)N7C=C(C=N7)F AVOKZFCYCRLYJD-UHFFFAOYSA-N 0.000 description 5
- HUWYKCMGPFHMBA-UHFFFAOYSA-N 2-[6-[6-[[4-(3-fluoropyrazol-1-yl)phenyl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=C(C=C6)N7C=CC(=N7)F HUWYKCMGPFHMBA-UHFFFAOYSA-N 0.000 description 5
- WKMMMFKQFTYEBC-UHFFFAOYSA-N 2-[6-[6-[[6-(3-cyclopropyl-4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C(=N7)C8CC8)F WKMMMFKQFTYEBC-UHFFFAOYSA-N 0.000 description 5
- GASBTFHFDVWFRJ-UHFFFAOYSA-N 3-(5-bromopyridin-2-yl)-3,6-diazabicyclo[3.1.1]heptane Chemical compound BrC1=CC=C(N=C1)N1CC2CC(C1)N2 GASBTFHFDVWFRJ-UHFFFAOYSA-N 0.000 description 5
- GKCBDWSJLBRMJN-UHFFFAOYSA-N 6-(1H-pyrrol-2-yl)pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1C1=CC=CN1 GKCBDWSJLBRMJN-UHFFFAOYSA-N 0.000 description 5
- HDQMDNXAJHYHEW-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(1,3-oxazol-2-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)C7=NC=CO7 HDQMDNXAJHYHEW-UHFFFAOYSA-N 0.000 description 5
- MLPIIRIAXGRMTD-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(1-propan-2-ylpyrazol-4-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)C7=CN(N=C7)C(C)C MLPIIRIAXGRMTD-UHFFFAOYSA-N 0.000 description 5
- DRSHPLMAZZNXKE-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(1H-pyrrol-2-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)C7=CC=CN7 DRSHPLMAZZNXKE-UHFFFAOYSA-N 0.000 description 5
- KKEJWJYZOWWVER-UHFFFAOYSA-N [1-[5-(1,3-dioxolan-2-yl)pyridin-2-yl]pyrazol-3-yl]methanol Chemical compound OCC(C=C1)=NN1C1=NC=C(C2OCCO2)C=C1 KKEJWJYZOWWVER-UHFFFAOYSA-N 0.000 description 5
- YZQSXJNNHVXWKW-UHFFFAOYSA-N [1-[5-[[3-[5-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]pyridin-2-yl]-3,6-diazabicyclo[3.1.1]heptan-6-yl]methyl]pyridin-2-yl]pyrazol-3-yl]methanol Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=CC(=N7)CO YZQSXJNNHVXWKW-UHFFFAOYSA-N 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 5
- CJBUQMVVKYLWRD-UHFFFAOYSA-N methyl 1-[5-(1,3-dioxolan-2-yl)pyridin-2-yl]pyrazole-3-carboxylate Chemical compound COC(C(C=C1)=NN1C1=NC=C(C2OCCO2)C=C1)=O CJBUQMVVKYLWRD-UHFFFAOYSA-N 0.000 description 5
- 239000012046 mixed solvent Substances 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- CUQOHAYJWVTKDE-UHFFFAOYSA-N potassium;butan-1-olate Chemical compound [K+].CCCC[O-] CUQOHAYJWVTKDE-UHFFFAOYSA-N 0.000 description 5
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 5
- 235000018102 proteins Nutrition 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 102200006308 rs74799832 Human genes 0.000 description 5
- SYXYWTXQFUUWLP-UHFFFAOYSA-N sodium;butan-1-olate Chemical compound [Na+].CCCC[O-] SYXYWTXQFUUWLP-UHFFFAOYSA-N 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 4
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 4
- OEKBNMATJYLCTO-UHFFFAOYSA-N 1-(6-pyrazol-1-ylpyridin-3-yl)ethanone Chemical compound N1=CC(C(=O)C)=CC=C1N1N=CC=C1 OEKBNMATJYLCTO-UHFFFAOYSA-N 0.000 description 4
- IMZWSOSYNFVECD-UHFFFAOYSA-N 1-(oxan-2-yl)pyrazole Chemical compound O1CCCCC1N1N=CC=C1 IMZWSOSYNFVECD-UHFFFAOYSA-N 0.000 description 4
- BBAZVRZRDZVNRN-UHFFFAOYSA-N 1-[4-(1,3-dioxolan-2-yl)phenyl]-3-fluoropyrazole Chemical compound FC(C=C1)=NN1C1=CC=C(C2OCCO2)C=C1 BBAZVRZRDZVNRN-UHFFFAOYSA-N 0.000 description 4
- ZODFSGUXDXEVCY-UHFFFAOYSA-N 1-[4-(1,3-dioxolan-2-yl)phenyl]-4-methylpyrazole Chemical compound CC1=CN(C2=CC=C(C3OCCO3)C=C2)N=C1 ZODFSGUXDXEVCY-UHFFFAOYSA-N 0.000 description 4
- DZOQWRBXSPQTTD-UHFFFAOYSA-N 2-(3,4-dimethylpyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine Chemical compound CC1=CN(C2=NC=C(C3OCCO3)C=C2)N=C1C DZOQWRBXSPQTTD-UHFFFAOYSA-N 0.000 description 4
- PRVCVXISSVQKDG-UHFFFAOYSA-N 2-(3-cyclopropyl-4-fluoropyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine Chemical compound FC1=CN(C2=NC=C(C3OCCO3)C=C2)N=C1C1CC1 PRVCVXISSVQKDG-UHFFFAOYSA-N 0.000 description 4
- DRQRMDKLCYPDFW-UHFFFAOYSA-N 2-(5-cyclopropyloxypyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine Chemical compound C(C1)C1OC1=CC=NN1C1=NC=C(C2OCCO2)C=C1 DRQRMDKLCYPDFW-UHFFFAOYSA-N 0.000 description 4
- GPQMQIVJBRGNHA-UHFFFAOYSA-N 2-[5-(1,3-dioxolan-2-yl)pyridin-2-yl]-1,3-oxazole Chemical compound C1OC(C(C=C2)=CN=C2C2=NC=CO2)OC1 GPQMQIVJBRGNHA-UHFFFAOYSA-N 0.000 description 4
- BCSHRERPHLTPEE-NRFANRHFSA-N 2-[[5-chloro-2-[[(6s)-6-[4-(2-hydroxyethyl)piperazin-1-yl]-1-methoxy-6,7,8,9-tetrahydro-5h-benzo[7]annulen-2-yl]amino]pyrimidin-4-yl]amino]-n-methylbenzamide Chemical compound CNC(=O)C1=CC=CC=C1NC1=NC(NC=2C(=C3CCC[C@@H](CC3=CC=2)N2CCN(CCO)CC2)OC)=NC=C1Cl BCSHRERPHLTPEE-NRFANRHFSA-N 0.000 description 4
- FZZMTSNZRBFGGU-UHFFFAOYSA-N 2-chloro-7-fluoroquinazolin-4-amine Chemical compound FC1=CC=C2C(N)=NC(Cl)=NC2=C1 FZZMTSNZRBFGGU-UHFFFAOYSA-N 0.000 description 4
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 4
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 4
- OCLDTYBZCFXNBS-UHFFFAOYSA-N 4-(3-fluoropyrazol-1-yl)benzaldehyde Chemical compound C1=CC(=CC=C1C=O)N2C=CC(=N2)F OCLDTYBZCFXNBS-UHFFFAOYSA-N 0.000 description 4
- JYDZTGYCBBUXTA-UHFFFAOYSA-N 4-(4-fluoropyrazol-1-yl)benzaldehyde Chemical compound C1=CC(=CC=C1C=O)N2C=C(C=N2)F JYDZTGYCBBUXTA-UHFFFAOYSA-N 0.000 description 4
- REMCWTMSJFZBQE-UHFFFAOYSA-N 4-(4-methylpyrazol-1-yl)benzaldehyde Chemical compound C1=C(C)C=NN1C1=CC=C(C=O)C=C1 REMCWTMSJFZBQE-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- PQKWJERKABWLBF-UHFFFAOYSA-N 4-[5-(1,3-dioxolan-2-yl)pyridin-2-yl]-1,3-thiazole Chemical compound C1OC(C(C=C2)=CN=C2C2=CSC=N2)OC1 PQKWJERKABWLBF-UHFFFAOYSA-N 0.000 description 4
- FKHCIFMAVQHHRU-UHFFFAOYSA-N 4-fluoro-1-(oxan-2-yl)pyrazole Chemical compound FC=1C=NN(C=1)C1OCCCC1 FKHCIFMAVQHHRU-UHFFFAOYSA-N 0.000 description 4
- DUHAOYYLLVATHT-UHFFFAOYSA-N 4-fluoro-5-methyl-1-(oxan-2-yl)pyrazole Chemical compound CC1=C(C=NN1C2CCCCO2)F DUHAOYYLLVATHT-UHFFFAOYSA-N 0.000 description 4
- PPGRDLZPSDHBIC-UHFFFAOYSA-N 4-pyrazol-1-ylbenzaldehyde Chemical compound C1=CC(C=O)=CC=C1N1N=CC=C1 PPGRDLZPSDHBIC-UHFFFAOYSA-N 0.000 description 4
- UPSGXDRWBLHFPA-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(1-methylpyrrol-2-yl)pyridine Chemical compound CN1C(C2=NC=C(C3OCCO3)C=C2)=CC=C1 UPSGXDRWBLHFPA-UHFFFAOYSA-N 0.000 description 4
- MCFZJJZBRKANSE-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(1-propan-2-ylpyrazol-4-yl)pyridine Chemical compound CC(C)N1N=CC(C2=NC=C(C3OCCO3)C=C2)=C1 MCFZJJZBRKANSE-UHFFFAOYSA-N 0.000 description 4
- HOPXPLQMUZXJNP-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(1H-pyrazol-4-yl)pyridine Chemical compound C1OC(C(C=C2)=CN=C2C2=CNN=C2)OC1 HOPXPLQMUZXJNP-UHFFFAOYSA-N 0.000 description 4
- LFHAFDGYUJTKHQ-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(1H-pyrrol-2-yl)pyridine Chemical compound C1OC(C(C=C2)=CN=C2C2=CC=CN2)OC1 LFHAFDGYUJTKHQ-UHFFFAOYSA-N 0.000 description 4
- TWKCJPYYKCELSF-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(4-fluoro-3-iodopyrazol-1-yl)pyridine Chemical compound FC1=CN(C2=NC=C(C3OCCO3)C=C2)N=C1I TWKCJPYYKCELSF-UHFFFAOYSA-N 0.000 description 4
- CKRHQSRPESWBHR-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(4-fluoro-5-methylpyrazol-1-yl)pyridine Chemical compound CC(N(C1=NC=C(C2OCCO2)C=C1)N=C1)=C1F CKRHQSRPESWBHR-UHFFFAOYSA-N 0.000 description 4
- KONRBANLYLPIKA-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(4-fluoroimidazol-1-yl)pyridine Chemical compound FC(N=C1)=CN1C1=NC=C(C2OCCO2)C=C1 KONRBANLYLPIKA-UHFFFAOYSA-N 0.000 description 4
- MDNDXQBGRMHQSY-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(5-ethylpyrazol-1-yl)pyridine Chemical compound CCC1=CC=NN1C1=NC=C(C2OCCO2)C=C1 MDNDXQBGRMHQSY-UHFFFAOYSA-N 0.000 description 4
- PSWMKKONWQRUSN-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-[3-(fluoromethyl)pyrazol-1-yl]pyridine Chemical compound FCC(C=C1)=NN1C1=NC=C(C2OCCO2)C=C1 PSWMKKONWQRUSN-UHFFFAOYSA-N 0.000 description 4
- GWYVYHNVDKQBPE-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-[4-(fluoromethyl)pyrazol-1-yl]pyridine Chemical compound FCC1=CN(C2=NC=C(C3OCCO3)C=C2)N=C1 GWYVYHNVDKQBPE-UHFFFAOYSA-N 0.000 description 4
- LJIZCKFBPWTUJS-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-pyridin-2-ylpyridine Chemical compound O1C(OCC1)C=1C=CC(=NC=1)C1=NC=CC=C1 LJIZCKFBPWTUJS-UHFFFAOYSA-N 0.000 description 4
- LPLTWSXDWJUAJK-UHFFFAOYSA-N 6-(1,3-oxazol-2-yl)pyridine-3-carbaldehyde Chemical compound O=Cc1ccc(nc1)-c1ncco1 LPLTWSXDWJUAJK-UHFFFAOYSA-N 0.000 description 4
- JGNPURVMQPNSTH-UHFFFAOYSA-N 6-(3,4-dimethylpyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound CC1=CN(C2=NC=C(C=O)C=C2)N=C1C JGNPURVMQPNSTH-UHFFFAOYSA-N 0.000 description 4
- ZNXZYWIXVLPNOC-UHFFFAOYSA-N 6-(3-cyclopropyl-4-fluoropyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1N1N=C(C2CC2)C(F)=C1 ZNXZYWIXVLPNOC-UHFFFAOYSA-N 0.000 description 4
- XLCJUXAFHAOPFY-UHFFFAOYSA-N 6-(4-fluoroimidazol-1-yl)pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1N1C=NC(F)=C1 XLCJUXAFHAOPFY-UHFFFAOYSA-N 0.000 description 4
- QYZNWVFLBVKGEJ-UHFFFAOYSA-N 6-(5-cyclopropyloxypyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1N1N=CC=C1OC1CC1 QYZNWVFLBVKGEJ-UHFFFAOYSA-N 0.000 description 4
- QWKBNPMDWQWNBM-UHFFFAOYSA-N 6-(5-methylfuran-2-yl)pyridine-3-carbaldehyde Chemical compound CC1=CC=C(C2=NC=C(C=O)C=C2)O1 QWKBNPMDWQWNBM-UHFFFAOYSA-N 0.000 description 4
- MWDVXGQIOBRCCS-UHFFFAOYSA-N 6-[3-(2-hydroxypropan-2-yl)pyrazol-1-yl]pyridine-3-carbaldehyde Chemical compound CC(C)(C(C=C1)=NN1C1=NC=C(C=O)C=C1)O MWDVXGQIOBRCCS-UHFFFAOYSA-N 0.000 description 4
- LFHIRRZYXRDMOI-UHFFFAOYSA-N 6-[3-(hydroxymethyl)pyrazol-1-yl]pyridine-3-carbaldehyde Chemical compound OCC(C=C1)=NN1C1=NC=C(C=O)C=C1 LFHIRRZYXRDMOI-UHFFFAOYSA-N 0.000 description 4
- OZYVUWSVLRCPIL-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[4-(6-methylpyridin-3-yl)oxypiperidin-1-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=NC=C(C=C1)OC2CCN(CC2)C3=NC=C(C=C3)C4=NC(=CC(=N4)NC5=NNC(=C5)C)C OZYVUWSVLRCPIL-UHFFFAOYSA-N 0.000 description 4
- OQJJZWKSHAZGDC-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[1-(6-pyrazol-1-ylpyridin-3-yl)ethyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC(C1=CC=C(N2N=CC=C2)N=C1)N(C(C1)C2)C1CN2C(N=C1)=CC=C1C1=NC(C)=CC(NC2=NNC(C)=C2)=N1 OQJJZWKSHAZGDC-UHFFFAOYSA-N 0.000 description 4
- MDVKIUVBPCRSOO-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(5-methylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC=NN1C2=NC=C(C=C2)CN3C4CC3CN(C4)C5=NC=C(C=C5)C6=NC(=CC(=N6)NC7=NNC(=C7)C)C MDVKIUVBPCRSOO-UHFFFAOYSA-N 0.000 description 4
- LPWKFUVWWQYLOD-UHFFFAOYSA-N 9-(dimethylamino)-3-(4-methylphenyl)pyrido[2,3]thieno[2,4-d]pyrimidin-4-one Chemical compound C1=2C(N(C)C)=CN=CC=2SC(C2=O)=C1N=CN2C1=CC=C(C)C=C1 LPWKFUVWWQYLOD-UHFFFAOYSA-N 0.000 description 4
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 4
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 4
- 238000007125 Buchwald synthesis reaction Methods 0.000 description 4
- KESASNHUBYMRHU-UHFFFAOYSA-N CCc1ccnn1C1CCCCO1 Chemical compound CCc1ccnn1C1CCCCO1 KESASNHUBYMRHU-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- 229910010082 LiAlH Inorganic materials 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- 101150077555 Ret gene Proteins 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 238000006887 Ullmann reaction Methods 0.000 description 4
- 230000000259 anti-tumor effect Effects 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 229940125797 compound 12 Drugs 0.000 description 4
- 229940125846 compound 25 Drugs 0.000 description 4
- JCWIWBWXCVGEAN-UHFFFAOYSA-L cyclopentyl(diphenyl)phosphane;dichloropalladium;iron Chemical compound [Fe].Cl[Pd]Cl.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1 JCWIWBWXCVGEAN-UHFFFAOYSA-L 0.000 description 4
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 4
- AVEZVOSRCARTNH-UHFFFAOYSA-N ethyl 1-[5-(1,3-dioxolan-2-yl)pyridin-2-yl]pyrazole-4-carboxylate Chemical compound CCOC(=O)c1cnn(c1)-c1ccc(cn1)C1OCCO1 AVEZVOSRCARTNH-UHFFFAOYSA-N 0.000 description 4
- 125000001188 haloalkyl group Chemical group 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 4
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 102200006097 rs79658334 Human genes 0.000 description 4
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- SLGLGIGMDYMKQL-UHFFFAOYSA-N tert-butyl 2-(5-formylpyridin-2-yl)pyrrole-1-carboxylate Chemical compound C(C)(C)(C)OC(=O)N1C(=CC=C1)C1=NC=C(C=C1)C=O SLGLGIGMDYMKQL-UHFFFAOYSA-N 0.000 description 4
- ZMYZIFHKQIRSAP-UHFFFAOYSA-N tert-butyl 2-[5-[[3-[5-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]pyridin-2-yl]-3,6-diazabicyclo[3.1.1]heptan-6-yl]methyl]pyridin-2-yl]pyrrole-1-carboxylate Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)C7=CC=CN7C(=O)OC(C)(C)C ZMYZIFHKQIRSAP-UHFFFAOYSA-N 0.000 description 4
- LCNHANYFRQTWGE-UHFFFAOYSA-N tert-butyl 4-(6-methylpyridin-3-yl)oxypiperidine-1-carboxylate Chemical compound C1=NC(C)=CC=C1OC1CCN(C(=O)OC(C)(C)C)CC1 LCNHANYFRQTWGE-UHFFFAOYSA-N 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- LAJAFFLJAJMYLK-CVOKMOJFSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[[(7s)-4-methoxy-7-morpholin-4-yl-6,7,8,9-tetrahydro-5h-benzo[7]annulen-3-yl]amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound N1([C@H]2CCC3=CC=C(C(=C3CC2)OC)NC=2N=C(C(=CN=2)Cl)N[C@H]2[C@H]([C@@]3([H])C[C@@]2(C=C3)[H])C(N)=O)CCOCC1 LAJAFFLJAJMYLK-CVOKMOJFSA-N 0.000 description 3
- FOLCUFKJHSQMEL-BIXPGCQOSA-N (4-butylcyclohexyl) N-[(2S)-4-methyl-1-oxo-1-[[(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]pentan-2-yl]carbamate Chemical compound CCCCC1CCC(CC1)OC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C[C@@H]2CCNC2=O)C=O FOLCUFKJHSQMEL-BIXPGCQOSA-N 0.000 description 3
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 3
- PNHBRYIAJCYNDA-VQCQRNETSA-N (4r)-6-[2-[2-ethyl-4-(4-fluorophenyl)-6-phenylpyridin-3-yl]ethyl]-4-hydroxyoxan-2-one Chemical compound C([C@H](O)C1)C(=O)OC1CCC=1C(CC)=NC(C=2C=CC=CC=2)=CC=1C1=CC=C(F)C=C1 PNHBRYIAJCYNDA-VQCQRNETSA-N 0.000 description 3
- FRJJJAKBRKABFA-TYFAACHXSA-N (4r,6s)-6-[(e)-2-[6-chloro-4-(4-fluorophenyl)-2-propan-2-ylquinolin-3-yl]ethenyl]-4-hydroxyoxan-2-one Chemical compound C(\[C@H]1OC(=O)C[C@H](O)C1)=C/C=1C(C(C)C)=NC2=CC=C(Cl)C=C2C=1C1=CC=C(F)C=C1 FRJJJAKBRKABFA-TYFAACHXSA-N 0.000 description 3
- HNXASLOFQPRNLF-UHFFFAOYSA-N (6-methoxypyridin-3-yl)-[4-[5-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]pyridin-2-yl]piperazin-1-yl]methanone Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CCN(CC4)C(=O)C5=CN=C(C=C5)OC HNXASLOFQPRNLF-UHFFFAOYSA-N 0.000 description 3
- UXKLQDCALAWFIU-VKNDCNMPSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-tetradecoxy-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@H](O)[C@H](C(O2)(C(O)=O)C(O)(C(O1)C(O)=O)C(O)=O)OCCCCCCCCCCCCCC)C1=CC=CC=C1 UXKLQDCALAWFIU-VKNDCNMPSA-N 0.000 description 3
- QPAGOTNPJABYCP-FITNPZAZSA-N (E)-N-(oxan-4-yl)-N'-[[(3R)-1,2,3,4-tetrahydroisoquinolin-3-yl]methyl]-N'-[(8S)-5,6,7,8-tetrahydroquinolin-8-yl]but-2-ene-1,4-diamine Chemical compound O1CCC(CC1)NC\C=C\CN([C@H]1CCCC=2C=CC=NC1=2)C[C@@H]1NCC2=CC=CC=C2C1 QPAGOTNPJABYCP-FITNPZAZSA-N 0.000 description 3
- APJSHECCIRQQDV-ZRDIBKRKSA-N (e)-3-[4-hydroxy-3-(5,5,8,8-tetramethyl-3-pentoxy-6,7-dihydronaphthalen-2-yl)phenyl]prop-2-enoic acid Chemical compound CCCCCOC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C1=CC(\C=C\C(O)=O)=CC=C1O APJSHECCIRQQDV-ZRDIBKRKSA-N 0.000 description 3
- JNPGUXGVLNJQSQ-BGGMYYEUSA-M (e,3r,5s)-7-[4-(4-fluorophenyl)-1,2-di(propan-2-yl)pyrrol-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound CC(C)N1C(C(C)C)=C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=C1 JNPGUXGVLNJQSQ-BGGMYYEUSA-M 0.000 description 3
- VAVHMEQFYYBAPR-ITWZMISCSA-N (e,3r,5s)-7-[4-(4-fluorophenyl)-1-phenyl-2-propan-2-ylpyrrol-3-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound CC(C)C1=C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)C(C=2C=CC(F)=CC=2)=CN1C1=CC=CC=C1 VAVHMEQFYYBAPR-ITWZMISCSA-N 0.000 description 3
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 3
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 3
- YRTFLDFDKPFNCJ-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-2-oxo-4-[3-(3-pyrrolidin-1-ylpropoxy)phenyl]-1,8-naphthyridin-3-yl]urea;dihydrochloride Chemical compound Cl.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C(C=1)=CC=CC=1OCCCN1CCCC1 YRTFLDFDKPFNCJ-UHFFFAOYSA-N 0.000 description 3
- ZFNNBIMQDHBELV-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-cyclohexylpropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;dihydrochloride Chemical compound Cl.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C(C=1)=CC=CC=1OCCCC1CCCCC1 ZFNNBIMQDHBELV-UHFFFAOYSA-N 0.000 description 3
- NZTAVQFTTHHBJR-UHFFFAOYSA-N 2-[6-[6-[(2-fluoro-5-methoxyphenyl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=C(C=CC(=C6)OC)F NZTAVQFTTHHBJR-UHFFFAOYSA-N 0.000 description 3
- MIEQUKHLEPOKLG-UHFFFAOYSA-N 2-[6-[6-[(5-fluoropyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC(=CN=C6)F MIEQUKHLEPOKLG-UHFFFAOYSA-N 0.000 description 3
- PKLXSVWOENNNGM-UHFFFAOYSA-N 2-[6-[6-[[6-(3-cyclopropylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=CC(=N7)C8CC8 PKLXSVWOENNNGM-UHFFFAOYSA-N 0.000 description 3
- PAYROHWFGZADBR-UHFFFAOYSA-N 2-[[4-amino-5-(5-iodo-4-methoxy-2-propan-2-ylphenoxy)pyrimidin-2-yl]amino]propane-1,3-diol Chemical compound C1=C(I)C(OC)=CC(C(C)C)=C1OC1=CN=C(NC(CO)CO)N=C1N PAYROHWFGZADBR-UHFFFAOYSA-N 0.000 description 3
- CODBZFJPKJDNDT-UHFFFAOYSA-N 2-[[5-[3-(dimethylamino)propyl]-2-methylpyridin-3-yl]amino]-9-(trifluoromethyl)-5,7-dihydropyrimido[5,4-d][1]benzazepine-6-thione Chemical compound CN(C)CCCC1=CN=C(C)C(NC=2N=C3C4=CC=C(C=C4NC(=S)CC3=CN=2)C(F)(F)F)=C1 CODBZFJPKJDNDT-UHFFFAOYSA-N 0.000 description 3
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 3
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 3
- MWDVCHRYCKXEBY-LBPRGKRZSA-N 3-chloro-n-[2-oxo-2-[[(1s)-1-phenylethyl]amino]ethyl]benzamide Chemical compound N([C@@H](C)C=1C=CC=CC=1)C(=O)CNC(=O)C1=CC=CC(Cl)=C1 MWDVCHRYCKXEBY-LBPRGKRZSA-N 0.000 description 3
- GTHAEVGKECHJQB-UHFFFAOYSA-N 3-fluoro-5-piperidin-4-yloxypyridine Chemical compound FC1=CN=CC(OC2CCNCC2)=C1 GTHAEVGKECHJQB-UHFFFAOYSA-N 0.000 description 3
- YOHXJNOVNYMVJF-UHFFFAOYSA-N 4-[[3-[5-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]pyridin-2-yl]-3,6-diazabicyclo[3.1.1]heptan-6-yl]methyl]benzonitrile Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=C(C=C6)C#N YOHXJNOVNYMVJF-UHFFFAOYSA-N 0.000 description 3
- WGZYVHFXLPDNDL-UHFFFAOYSA-N 4-benzoyl-2-phenylpyrazole-3-carbonitrile Chemical compound C=1C=CC=CC=1C(=O)C(=C1C#N)C=NN1C1=CC=CC=C1 WGZYVHFXLPDNDL-UHFFFAOYSA-N 0.000 description 3
- GSDQYSSLIKJJOG-UHFFFAOYSA-N 4-chloro-2-(3-chloroanilino)benzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1NC1=CC=CC(Cl)=C1 GSDQYSSLIKJJOG-UHFFFAOYSA-N 0.000 description 3
- RYXJXPHWRBWEEB-UHFFFAOYSA-N 4-fluoro-1h-pyrazole Chemical group FC=1C=NNC=1 RYXJXPHWRBWEEB-UHFFFAOYSA-N 0.000 description 3
- XPNAEMIKNVCSTP-UHFFFAOYSA-N 4-fluoro-5-iodo-1h-pyrazole Chemical compound FC=1C=NNC=1I XPNAEMIKNVCSTP-UHFFFAOYSA-N 0.000 description 3
- OFPLNBAFXLMBEM-UHFFFAOYSA-N 4-fluoro-5-methyl-1h-pyrazole Chemical compound CC1=NNC=C1F OFPLNBAFXLMBEM-UHFFFAOYSA-N 0.000 description 3
- MVXAKOGJWVQPKC-UHFFFAOYSA-N 5-(3-ethynyl-5-fluorophenyl)-2-pyridin-2-yl-4,6,7,8-tetrahydro-[1,3]oxazolo[4,5-c]azepine Chemical compound FC1=CC(C#C)=CC(N2CC=3N=C(OC=3CCC2)C=2N=CC=CC=2)=C1 MVXAKOGJWVQPKC-UHFFFAOYSA-N 0.000 description 3
- KUZSBKJSGSKPJH-VXGBXAGGSA-N 5-[(9R)-6-[(3R)-3-methylmorpholin-4-yl]-11-oxa-1,3,5-triazatricyclo[7.4.0.02,7]trideca-2,4,6-trien-4-yl]pyrazin-2-amine Chemical compound C[C@@H]1COCCN1c1nc(nc2N3CCOC[C@H]3Cc12)-c1cnc(N)cn1 KUZSBKJSGSKPJH-VXGBXAGGSA-N 0.000 description 3
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 3
- PQLLSEKDCGVFFV-UHFFFAOYSA-N 5-cyclobutyloxy-1h-pyrazole Chemical compound C1CCC1OC1=CC=NN1 PQLLSEKDCGVFFV-UHFFFAOYSA-N 0.000 description 3
- HIHOEGPXVVKJPP-JTQLQIEISA-N 5-fluoro-2-[[(1s)-1-(5-fluoropyridin-2-yl)ethyl]amino]-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyridine-3-carbonitrile Chemical compound N([C@@H](C)C=1N=CC(F)=CC=1)C(C(=CC=1F)C#N)=NC=1NC=1C=C(C)NN=1 HIHOEGPXVVKJPP-JTQLQIEISA-N 0.000 description 3
- DOSHLMXHBSRCPS-UHFFFAOYSA-N 6-(1,3-thiazol-4-yl)pyridine-3-carbaldehyde Chemical compound O=Cc1ccc(nc1)-c1cscn1 DOSHLMXHBSRCPS-UHFFFAOYSA-N 0.000 description 3
- QOWHHGWCYXXTSO-UHFFFAOYSA-N 6-(1-methylpyrrol-2-yl)pyridine-3-carbaldehyde Chemical compound Cn1cccc1-c1ccc(C=O)cn1 QOWHHGWCYXXTSO-UHFFFAOYSA-N 0.000 description 3
- SPFDUPQKQYCVSL-UHFFFAOYSA-N 6-(1-propan-2-ylpyrazol-4-yl)pyridine-3-carbaldehyde Chemical compound CC(C)N1N=CC(C2=NC=C(C=O)C=C2)=C1 SPFDUPQKQYCVSL-UHFFFAOYSA-N 0.000 description 3
- QQBQITGFNUKKIW-UHFFFAOYSA-N 6-(4-fluoro-5-methylpyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound CC(N(C1=NC=C(C=O)C=C1)N=C1)=C1F QQBQITGFNUKKIW-UHFFFAOYSA-N 0.000 description 3
- YATJSCRGSVSRRP-UHFFFAOYSA-N 6-(5-fluoro-1H-pyrrol-2-yl)pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1C(N1)=CC=C1F YATJSCRGSVSRRP-UHFFFAOYSA-N 0.000 description 3
- PHZCRDCZHBTWIM-UHFFFAOYSA-N 6-[2-(oxan-2-yl)pyrazol-3-yl]pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1C1=CC=NN1C1OCCCC1 PHZCRDCZHBTWIM-UHFFFAOYSA-N 0.000 description 3
- HNQMAXYTSXZQNB-UHFFFAOYSA-N 6-[3-(fluoromethyl)pyrazol-1-yl]pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1N1N=C(CF)C=C1 HNQMAXYTSXZQNB-UHFFFAOYSA-N 0.000 description 3
- GJAWBLCUQKCCNP-UHFFFAOYSA-N 6-[4-(fluoromethyl)pyrazol-1-yl]pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1N1N=CC(CF)=C1 GJAWBLCUQKCCNP-UHFFFAOYSA-N 0.000 description 3
- NYRALBSJJJBAMN-UHFFFAOYSA-N 6-pyridin-2-ylpyridine-3-carbaldehyde Chemical compound N1=CC(C=O)=CC=C1C1=CC=CC=N1 NYRALBSJJJBAMN-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- XASOHFCUIQARJT-UHFFFAOYSA-N 8-methoxy-6-[7-(2-morpholin-4-ylethoxy)imidazo[1,2-a]pyridin-3-yl]-2-(2,2,2-trifluoroethyl)-3,4-dihydroisoquinolin-1-one Chemical compound C(N1C(=O)C2=C(OC)C=C(C=3N4C(=NC=3)C=C(C=C4)OCCN3CCOCC3)C=C2CC1)C(F)(F)F XASOHFCUIQARJT-UHFFFAOYSA-N 0.000 description 3
- IRBAWVGZNJIROV-SFHVURJKSA-N 9-(2-cyclopropylethynyl)-2-[[(2s)-1,4-dioxan-2-yl]methoxy]-6,7-dihydropyrimido[6,1-a]isoquinolin-4-one Chemical compound C1=C2C3=CC=C(C#CC4CC4)C=C3CCN2C(=O)N=C1OC[C@@H]1COCCO1 IRBAWVGZNJIROV-SFHVURJKSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 241001251200 Agelas Species 0.000 description 3
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 3
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 208000037196 Medullary thyroid carcinoma Diseases 0.000 description 3
- OHVIRCSJFFPEGC-XMMPIXPASA-N N-[3-[[4-[[(2R)-2-(hydroxymethyl)pyrrolidin-1-yl]methyl]phenyl]methoxy]phenyl]benzamide Chemical compound OC[C@H]1CCCN1Cc1ccc(COc2cccc(NC(=O)c3ccccc3)c2)cc1 OHVIRCSJFFPEGC-XMMPIXPASA-N 0.000 description 3
- 229940125907 SJ995973 Drugs 0.000 description 3
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 3
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 3
- NELWQUQCCZMRPB-UBPLGANQSA-N [(2r,3r,4r,5r)-4-acetyloxy-5-(4-amino-5-ethenyl-2-oxopyrimidin-1-yl)-2-methyloxolan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](C)O[C@H]1N1C(=O)N=C(N)C(C=C)=C1 NELWQUQCCZMRPB-UBPLGANQSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- AJRQIXBBIDPNGK-BVSLBCMMSA-N benzyl n-[(2s)-1-[[(2s)-1-(1,3-benzothiazol-2-yl)-1-oxo-3-[(3s)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]carbamate Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](C[C@H]1C(NCC1)=O)C(=O)C=1SC2=CC=CC=C2N=1)C(=O)OCC1=CC=CC=C1 AJRQIXBBIDPNGK-BVSLBCMMSA-N 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- AEULIVPVIDOLIN-UHFFFAOYSA-N cep-11981 Chemical compound C1=C2C3=C4CNC(=O)C4=C4C5=CN(C)N=C5CCC4=C3N(CC(C)C)C2=CC=C1NC1=NC=CC=N1 AEULIVPVIDOLIN-UHFFFAOYSA-N 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- 229940125773 compound 10 Drugs 0.000 description 3
- 229940125851 compound 27 Drugs 0.000 description 3
- 229940127204 compound 29 Drugs 0.000 description 3
- 229940125877 compound 31 Drugs 0.000 description 3
- 229940125844 compound 46 Drugs 0.000 description 3
- 229940127271 compound 49 Drugs 0.000 description 3
- 229940125872 compound 4d Drugs 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- NCNJHAHPOUUEQX-LDVROUIZSA-N n-[(2s,3r)-3-hydroxy-1-phenyl-4-[[(1r)-1-phenylethyl]amino]butan-2-yl]-4-(4-methylpiperazine-1-carbonyl)benzamide Chemical compound C([C@@H]([C@H](O)CN[C@H](C)C=1C=CC=CC=1)NC(=O)C=1C=CC(=CC=1)C(=O)N1CCN(C)CC1)C1=CC=CC=C1 NCNJHAHPOUUEQX-LDVROUIZSA-N 0.000 description 3
- PHVXTQIROLEEDB-UHFFFAOYSA-N n-[2-(2-chlorophenyl)ethyl]-4-[[3-(2-methylphenyl)piperidin-1-yl]methyl]-n-pyrrolidin-3-ylbenzamide Chemical compound CC1=CC=CC=C1C1CN(CC=2C=CC(=CC=2)C(=O)N(CCC=2C(=CC=CC=2)Cl)C2CNCC2)CCC1 PHVXTQIROLEEDB-UHFFFAOYSA-N 0.000 description 3
- XZMHJYWMCRQSSI-UHFFFAOYSA-N n-[5-[2-(3-acetylanilino)-1,3-thiazol-4-yl]-4-methyl-1,3-thiazol-2-yl]benzamide Chemical compound CC(=O)C1=CC=CC(NC=2SC=C(N=2)C2=C(N=C(NC(=O)C=3C=CC=CC=3)S2)C)=C1 XZMHJYWMCRQSSI-UHFFFAOYSA-N 0.000 description 3
- LVBWRNKEBGYENR-UHFFFAOYSA-N n-cyclopropyl-4-[8-(oxan-4-ylmethylamino)-6-[(1,1,1-trifluoro-2-methylpropan-2-yl)amino]imidazo[1,2-b]pyridazin-3-yl]benzamide Chemical compound C12=NC=C(C=3C=CC(=CC=3)C(=O)NC3CC3)N2N=C(NC(C)(C)C(F)(F)F)C=C1NCC1CCOCC1 LVBWRNKEBGYENR-UHFFFAOYSA-N 0.000 description 3
- 125000004043 oxo group Chemical group O=* 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 3
- 238000006268 reductive amination reaction Methods 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 210000001685 thyroid gland Anatomy 0.000 description 3
- 208000013818 thyroid gland medullary carcinoma Diseases 0.000 description 3
- 239000010936 titanium Substances 0.000 description 3
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 2
- ASQOQJYHIYYTEJ-GBESFXJTSA-N (1r,7s,9as)-7-decyl-2,3,4,6,7,8,9,9a-octahydro-1h-quinolizin-1-ol Chemical compound O[C@@H]1CCCN2C[C@@H](CCCCCCCCCC)CC[C@H]21 ASQOQJYHIYYTEJ-GBESFXJTSA-N 0.000 description 2
- KAFZOLYKKCWUBI-HPMAGDRPSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-3-amino-2-[[(2s)-2-[[(2s)-2-(3-cyclohexylpropanoylamino)-4-methylpentanoyl]amino]-5-methylhexanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]butanediamide Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](CCC(C)C)C(=O)N[C@@H](CN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(N)=O)C(N)=O)C(=O)CCC1CCCCC1 KAFZOLYKKCWUBI-HPMAGDRPSA-N 0.000 description 2
- HPJGEESDHAUUQR-SKGSPYGFSA-N (2s)-2-[[(2s)-5-(diaminomethylideneamino)-2-[[(2s)-1-[(2s)-5-(diaminomethylideneamino)-2-[[(2s)-2-[[(2s)-3-naphthalen-2-yl-2-(3-pyridin-3-ylpropanoylamino)propanoyl]amino]-3-phenylpropanoyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]amino]buta Chemical compound NC(=O)C[C@@H](C(N)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=C2C=CC=CC2=CC=1)NC(=O)CCC=1C=NC=CC=1)CC1=CC=CC=C1 HPJGEESDHAUUQR-SKGSPYGFSA-N 0.000 description 2
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 2
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 2
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 2
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- ROHGINRQAFHUPY-UHFFFAOYSA-N 1-n,1-n'-dimethylcyclohexane-1,1-diamine Chemical compound CNC1(NC)CCCCC1 ROHGINRQAFHUPY-UHFFFAOYSA-N 0.000 description 2
- HKLLHQRDMJGVBP-UHFFFAOYSA-N 2-[6-[6-[[6-(4-chloro-3-methylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C(=N7)C)Cl HKLLHQRDMJGVBP-UHFFFAOYSA-N 0.000 description 2
- MEGOGRHPDAHNGS-UHFFFAOYSA-N 2-[6-[6-[[6-(4-chloro-5-methylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C(=C(C=N7)Cl)C MEGOGRHPDAHNGS-UHFFFAOYSA-N 0.000 description 2
- DXSJNZOTJDAXNQ-UHFFFAOYSA-N 2-[6-[6-[[6-(4-fluoro-5-methylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C(=C(C=N7)F)C DXSJNZOTJDAXNQ-UHFFFAOYSA-N 0.000 description 2
- SYAIWXURFWCECS-UHFFFAOYSA-N 2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C=N7)F SYAIWXURFWCECS-UHFFFAOYSA-N 0.000 description 2
- PTBOZBOGTMGKEM-UHFFFAOYSA-N 2-[6-[6-[[6-(5-ethylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CCC1=CC=NN1C2=NC=C(C=C2)CN3C4CC3CN(C4)C5=NC=C(C=C5)C6=NC(=CC(=N6)NC7=NNC(=C7)C)C PTBOZBOGTMGKEM-UHFFFAOYSA-N 0.000 description 2
- IUYVQHKOIHBGRC-UHFFFAOYSA-N 2-methyl-5-piperidin-4-yloxypyridine Chemical compound C1=NC(C)=CC=C1OC1CCNCC1 IUYVQHKOIHBGRC-UHFFFAOYSA-N 0.000 description 2
- DFRAKBCRUYUFNT-UHFFFAOYSA-N 3,8-dicyclohexyl-2,4,7,9-tetrahydro-[1,3]oxazino[5,6-h][1,3]benzoxazine Chemical compound C1CCCCC1N1CC(C=CC2=C3OCN(C2)C2CCCCC2)=C3OC1 DFRAKBCRUYUFNT-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- WDTSGEMGTMOHMD-UHFFFAOYSA-N 5-cyclopropyloxy-1h-pyrazole Chemical compound C1CC1OC1=CC=NN1 WDTSGEMGTMOHMD-UHFFFAOYSA-N 0.000 description 2
- HWUPTWDARIVICI-UHFFFAOYSA-N 5-fluoro-1h-imidazole Chemical group FC1=CN=CN1 HWUPTWDARIVICI-UHFFFAOYSA-N 0.000 description 2
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 2
- IZLUQURPZLMHHS-UHFFFAOYSA-N 6-(5-ethylpyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound CCC1=CC=NN1C1=NC=C(C=O)C=C1 IZLUQURPZLMHHS-UHFFFAOYSA-N 0.000 description 2
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 2
- LTUZPODERZUPRD-UHFFFAOYSA-N 6-chloro-2-(1h-indol-3-yl)-4-phenylquinoline Chemical compound C12=CC(Cl)=CC=C2N=C(C=2C3=CC=CC=C3NC=2)C=C1C1=CC=CC=C1 LTUZPODERZUPRD-UHFFFAOYSA-N 0.000 description 2
- OQJJZWKSHAZGDC-QUVATAORSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[(1R)-1-(6-pyrazol-1-ylpyridin-3-yl)ethyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5[C@H](C)C6=CN=C(C=C6)N7C=CC=N7 OQJJZWKSHAZGDC-QUVATAORSA-N 0.000 description 2
- OQJJZWKSHAZGDC-XOYNAWAESA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[(1S)-1-(6-pyrazol-1-ylpyridin-3-yl)ethyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5[C@@H](C)C6=CN=C(C=C6)N7C=CC=N7 OQJJZWKSHAZGDC-XOYNAWAESA-N 0.000 description 2
- VNPFCXKYSNVPAB-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[(4-pyrazol-1-ylphenyl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=C(C=C6)N7C=CC=N7 VNPFCXKYSNVPAB-UHFFFAOYSA-N 0.000 description 2
- UUCQXOUTRDCOIF-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(1-methylpyrrol-2-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)C7=CC=CN7C UUCQXOUTRDCOIF-UHFFFAOYSA-N 0.000 description 2
- KTMHTWHJUOGILM-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(5-propan-2-ylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C(=CC=N7)C(C)C KTMHTWHJUOGILM-UHFFFAOYSA-N 0.000 description 2
- ROFVEXUMMXZLPA-UHFFFAOYSA-N Bipyridyl Chemical compound N1=CC=CC=C1C1=CC=CC=N1 ROFVEXUMMXZLPA-UHFFFAOYSA-N 0.000 description 2
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 2
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- 230000005526 G1 to G0 transition Effects 0.000 description 2
- 238000003043 HTRF KinEASE-TK Methods 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- WABIADMEUUZCJN-UHFFFAOYSA-N N-(5-cyclopropyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methylpyrimidin-4-amine Chemical compound CC1=CC(=NC(=N1)C2=CN=C(C=C2)N3CC4CC(C3)N4CC5=CN=C(C=C5)N6C=C(C=N6)F)NC7=NNC(=C7)C8CC8 WABIADMEUUZCJN-UHFFFAOYSA-N 0.000 description 2
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 2
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 2
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 2
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 2
- QBYJBZPUGVGKQQ-SJJAEHHWSA-N aldrin Chemical compound C1[C@H]2C=C[C@@H]1[C@H]1[C@@](C3(Cl)Cl)(Cl)C(Cl)=C(Cl)[C@@]3(Cl)[C@H]12 QBYJBZPUGVGKQQ-SJJAEHHWSA-N 0.000 description 2
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 2
- 125000005529 alkyleneoxy group Chemical group 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940126540 compound 41 Drugs 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- ASQQEOXYFGEFKQ-UHFFFAOYSA-N dioxirane Chemical compound C1OO1 ASQQEOXYFGEFKQ-UHFFFAOYSA-N 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 2
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 description 2
- BGXZIBSLBRKDTP-UHFFFAOYSA-N methyl 9-(4-chloroanilino)-[1,3]thiazolo[5,4-f]quinazoline-2-carboximidate Chemical compound C12=C3SC(C(=N)OC)=NC3=CC=C2N=CN=C1NC1=CC=C(Cl)C=C1 BGXZIBSLBRKDTP-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 2
- MUJNAWXXOJRNGK-UHFFFAOYSA-N n-[3-(6-methyl-1,2,3,4-tetrahydrocarbazol-9-yl)propyl]cyclohexanamine Chemical compound C1=2CCCCC=2C2=CC(C)=CC=C2N1CCCNC1CCCCC1 MUJNAWXXOJRNGK-UHFFFAOYSA-N 0.000 description 2
- YGBMCLDVRUGXOV-UHFFFAOYSA-N n-[6-[6-chloro-5-[(4-fluorophenyl)sulfonylamino]pyridin-3-yl]-1,3-benzothiazol-2-yl]acetamide Chemical compound C1=C2SC(NC(=O)C)=NC2=CC=C1C(C=1)=CN=C(Cl)C=1NS(=O)(=O)C1=CC=C(F)C=C1 YGBMCLDVRUGXOV-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 230000009822 protein phosphorylation Effects 0.000 description 2
- QJZUKDFHGGYHMC-UHFFFAOYSA-N pyridine-3-carbaldehyde Chemical compound O=CC1=CC=CN=C1 QJZUKDFHGGYHMC-UHFFFAOYSA-N 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- ORIHZIZPTZTNCU-YVMONPNESA-N salicylaldoxime Chemical compound O\N=C/C1=CC=CC=C1O ORIHZIZPTZTNCU-YVMONPNESA-N 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- PWQLFIKTGRINFF-UHFFFAOYSA-N tert-butyl 4-hydroxypiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(O)CC1 PWQLFIKTGRINFF-UHFFFAOYSA-N 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229910052722 tritium Inorganic materials 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 1
- KSOVGRCOLZZTPF-QMKUDKLTSA-N (1s,2s,3r,4r)-3-[[5-fluoro-2-[3-methyl-4-(4-methylpiperazin-1-yl)anilino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound N([C@H]1[C@H]([C@@]2([H])C[C@@]1(C=C2)[H])C(N)=O)C(C(=CN=1)F)=NC=1NC(C=C1C)=CC=C1N1CCN(C)CC1 KSOVGRCOLZZTPF-QMKUDKLTSA-N 0.000 description 1
- LJIOTBMDLVHTBO-CUYJMHBOSA-N (2s)-2-amino-n-[(1r,2r)-1-cyano-2-[4-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]phenyl]cyclopropyl]butanamide Chemical compound CC[C@H](N)C(=O)N[C@]1(C#N)C[C@@H]1C1=CC=C(C=2C=CC(=CC=2)S(=O)(=O)N2CCN(C)CC2)C=C1 LJIOTBMDLVHTBO-CUYJMHBOSA-N 0.000 description 1
- TWYYFYNJOJGNFP-CUXYNZQBSA-N (2s,4r,5s,6s)-2-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-2-carbamoyl-4-[[(e,4s,6s)-4,6-dimethyloct-2-enoyl]oxymethyl]-5-hydroxy-1,3-dioxane-4,5,6-tricarboxylic acid Chemical compound O1[C@H](C(O)=O)[C@](C(O)=O)(O)[C@](COC(=O)/C=C/[C@@H](C)C[C@@H](C)CC)(C(O)=O)O[C@]1(C(N)=O)CCC(=C)[C@@H](OC(C)=O)[C@H](C)CC1=CC=CC=C1 TWYYFYNJOJGNFP-CUXYNZQBSA-N 0.000 description 1
- VUDZSIYXZUYWSC-DBRKOABJSA-N (4r)-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 VUDZSIYXZUYWSC-DBRKOABJSA-N 0.000 description 1
- VGNCBRNRHXEODV-XXVHXNRLSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-6-dodecoxy-4,7-dihydroxy-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@H](O)[C@H](C(O2)(C(O)=O)C(O)(C(O1)C(O)=O)C(O)=O)OCCCCCCCCCCCC)C1=CC=CC=C1 VGNCBRNRHXEODV-XXVHXNRLSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000006648 (C1-C8) haloalkyl group Chemical group 0.000 description 1
- 125000006569 (C5-C6) heterocyclic group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- PPTXVXKCQZKFBN-UHFFFAOYSA-N (S)-(-)-1,1'-Bi-2-naphthol Chemical compound C1=CC=C2C(C3=C4C=CC=CC4=CC=C3O)=C(O)C=CC2=C1 PPTXVXKCQZKFBN-UHFFFAOYSA-N 0.000 description 1
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N 1,1'-Carbonyldiimidazole Substances C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- GWHTZJJRUUTEJJ-UHFFFAOYSA-N 1,2-diazabicyclo[3.1.1]heptane Chemical compound C1N2CC1CCN2 GWHTZJJRUUTEJJ-UHFFFAOYSA-N 0.000 description 1
- MUKKGHQBUKOMTD-UHFFFAOYSA-N 1-(6-bromopyridin-3-yl)ethanone Chemical group CC(=O)C1=CC=C(Br)N=C1 MUKKGHQBUKOMTD-UHFFFAOYSA-N 0.000 description 1
- SZCBDIVMCGFVPW-UHFFFAOYSA-N 1-[4-(aminomethyl)-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-(3-methoxyphenyl)-2-oxo-1,8-naphthyridin-3-yl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=C(CN)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OC)=C1 SZCBDIVMCGFVPW-UHFFFAOYSA-N 0.000 description 1
- QGRRJTQMMXJUNP-UHFFFAOYSA-N 1-pyrrolidin-1-ylpyrrolidine Chemical group C1CCCN1N1CCCC1 QGRRJTQMMXJUNP-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- ZFJADFUGRVREEC-UHFFFAOYSA-N 2-(3,5-dimethylpyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine Chemical compound CC1=CC(C)=NN1C1=NC=C(C2OCCO2)C=C1 ZFJADFUGRVREEC-UHFFFAOYSA-N 0.000 description 1
- PTWKZPXUDHDPOT-UHFFFAOYSA-N 2-(4-chloro-3-methylpyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine Chemical compound CC(C(Cl)=C1)=NN1C1=NC=C(C2OCCO2)C=C1 PTWKZPXUDHDPOT-UHFFFAOYSA-N 0.000 description 1
- CZLKZXQPFNYSQW-UHFFFAOYSA-N 2-(5-cyclobutyloxypyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine Chemical compound C(C1)CC1OC1=CC=NN1C1=NC=C(C2OCCO2)C=C1 CZLKZXQPFNYSQW-UHFFFAOYSA-N 0.000 description 1
- WNOUZCPFEJYQNW-UHFFFAOYSA-N 2-[1-[5-(1,3-dioxolan-2-yl)pyridin-2-yl]pyrazol-3-yl]propan-2-ol Chemical compound CC(C)(C(C=C1)=NN1C1=NC=C(C2OCCO2)C=C1)O WNOUZCPFEJYQNW-UHFFFAOYSA-N 0.000 description 1
- LOEXAKKLMUJLSZ-UHFFFAOYSA-N 2-[6-(6-benzyl-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=CC=C6 LOEXAKKLMUJLSZ-UHFFFAOYSA-N 0.000 description 1
- ZESOGGRVZOBGJE-UHFFFAOYSA-N 2-[6-[4-(5-fluoropyridin-3-yl)oxypiperidin-1-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CCC(CC4)OC5=CC(=CN=C5)F ZESOGGRVZOBGJE-UHFFFAOYSA-N 0.000 description 1
- FSWMBXDKKZCUTL-UHFFFAOYSA-N 2-[6-[4-[(4-methoxyphenyl)methyl]piperazin-1-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CCN(CC4)CC5=CC=C(C=C5)OC FSWMBXDKKZCUTL-UHFFFAOYSA-N 0.000 description 1
- YTPOGLBRGGQGLW-UHFFFAOYSA-N 2-[6-[4-[(6-methoxypyridin-3-yl)methyl]piperazin-1-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CCN(CC4)CC5=CN=C(C=C5)OC YTPOGLBRGGQGLW-UHFFFAOYSA-N 0.000 description 1
- AVOKZFCYCRLYJD-NNAHLOSHSA-N 2-[6-[6-[(1R)-1-[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]ethyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5[C@H](C)C6=CN=C(C=C6)N7C=C(C=N7)F AVOKZFCYCRLYJD-NNAHLOSHSA-N 0.000 description 1
- RIINABUEACCVQV-UHFFFAOYSA-N 2-[6-[6-[(4-ethoxyphenyl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CCOC1=CC=C(C=C1)CN2C3CC2CN(C3)C4=NC=C(C=C4)C5=NC(=CC(=N5)NC6=NNC(=C6)C)C RIINABUEACCVQV-UHFFFAOYSA-N 0.000 description 1
- KFBIZYDIVUUSBH-UHFFFAOYSA-N 2-[6-[6-[(4-methoxyphenyl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=C(C=C6)OC KFBIZYDIVUUSBH-UHFFFAOYSA-N 0.000 description 1
- UQBIUISAJNCIMJ-UHFFFAOYSA-N 2-[6-[6-[(5-chloropyridin-2-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=NC=C(C=C6)Cl UQBIUISAJNCIMJ-UHFFFAOYSA-N 0.000 description 1
- TXGNGPDDWVWGKB-UHFFFAOYSA-N 2-[6-[6-[(5-fluoropyridin-2-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=NC=C(C=C6)F TXGNGPDDWVWGKB-UHFFFAOYSA-N 0.000 description 1
- LKHBSROVQRORSS-UHFFFAOYSA-N 2-[6-[6-[(5-methoxypyridin-2-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=NC=C(C=C6)OC LKHBSROVQRORSS-UHFFFAOYSA-N 0.000 description 1
- DAOARHOWWAZYSR-UHFFFAOYSA-N 2-[6-[6-[(5-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC(=CN=C6)OC DAOARHOWWAZYSR-UHFFFAOYSA-N 0.000 description 1
- RJFDCGNCWOPMGS-UHFFFAOYSA-N 2-[6-[6-[(6-bromopyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)Br RJFDCGNCWOPMGS-UHFFFAOYSA-N 0.000 description 1
- CWGXYXLABFOAIO-UHFFFAOYSA-N 2-[6-[6-[(6-chloropyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)Cl CWGXYXLABFOAIO-UHFFFAOYSA-N 0.000 description 1
- FYSSUUSRURMRNE-UHFFFAOYSA-N 2-[6-[6-[1-(4-methoxyphenyl)ethyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5C(C)C6=CC=C(C=C6)OC FYSSUUSRURMRNE-UHFFFAOYSA-N 0.000 description 1
- TYXWLARPMCOJNE-UHFFFAOYSA-N 2-[6-[6-[[6-(3-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=CC(=N7)F TYXWLARPMCOJNE-UHFFFAOYSA-N 0.000 description 1
- WCHMQFKYDYKAPB-UHFFFAOYSA-N 2-[6-[6-[[6-(4-fluoro-3-methylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C(=N7)C)F WCHMQFKYDYKAPB-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- QHKASGSOHIFJSG-UHFFFAOYSA-N 2-chloro-n-(5-cyclopropyl-1h-pyrazol-3-yl)-6-methylpyrimidin-4-amine Chemical compound ClC1=NC(C)=CC(NC2=NNC(=C2)C2CC2)=N1 QHKASGSOHIFJSG-UHFFFAOYSA-N 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- ZAZHRJCWMGXICW-UHFFFAOYSA-N 3-(5-bromopyridin-2-yl)-6-[(6-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptane Chemical compound BrC=1C=CC(=NC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC ZAZHRJCWMGXICW-UHFFFAOYSA-N 0.000 description 1
- HNFMVVHMKGFCMB-UHFFFAOYSA-N 3-[3-[4-(1-aminocyclobutyl)phenyl]-5-phenylimidazo[4,5-b]pyridin-2-yl]pyridin-2-amine Chemical compound NC1=NC=CC=C1C1=NC2=CC=C(C=3C=CC=CC=3)N=C2N1C1=CC=C(C2(N)CCC2)C=C1 HNFMVVHMKGFCMB-UHFFFAOYSA-N 0.000 description 1
- HCLQARMRCPEALF-DNQXCXABSA-N 3-[[(2r)-2-[(1r)-2-[[1-(1-benzothiophen-2-yl)-2-methylpropan-2-yl]amino]-1-hydroxyethyl]pyrrolidin-1-yl]methyl]benzonitrile Chemical compound C([C@@H]1[C@H](O)CNC(C)(CC=2SC3=CC=CC=C3C=2)C)CCN1CC1=CC=CC(C#N)=C1 HCLQARMRCPEALF-DNQXCXABSA-N 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- 125000006042 4-hexenyl group Chemical group 0.000 description 1
- 125000003119 4-methyl-3-pentenyl group Chemical group [H]\C(=C(/C([H])([H])[H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- DGFUWAIXBVTTIC-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(5-propan-2-ylpyrazol-1-yl)pyridine Chemical compound CC(C)C1=CC=NN1C1=NC=C(C2OCCO2)C=C1 DGFUWAIXBVTTIC-UHFFFAOYSA-N 0.000 description 1
- BPCPVLDEKUTCMK-UHFFFAOYSA-N 5-(1,3-dioxolan-2-yl)-2-(6-methoxypyridin-3-yl)pyridine Chemical compound COC(N=C1)=CC=C1C1=NC=C(C2OCCO2)C=C1 BPCPVLDEKUTCMK-UHFFFAOYSA-N 0.000 description 1
- TXWDVWSJMDFNQY-UHFFFAOYSA-N 5-cyclopropyl-1h-pyrazole Chemical group C1CC1C1=CC=NN1 TXWDVWSJMDFNQY-UHFFFAOYSA-N 0.000 description 1
- CBNLNXLAIMQSTR-UHFFFAOYSA-N 5-ethyl-1h-pyrazole Chemical compound CCC1=CC=NN1 CBNLNXLAIMQSTR-UHFFFAOYSA-N 0.000 description 1
- WNDHCIJGEKNYNF-UHFFFAOYSA-N 5-fluoro-1h-pyrazole Chemical group FC=1C=CNN=1 WNDHCIJGEKNYNF-UHFFFAOYSA-N 0.000 description 1
- ONAFWZGKNKXRFB-UHFFFAOYSA-N 5-fluoro-2-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound CC1=NC=C(F)C=C1B1OC(C)(C)C(C)(C)O1 ONAFWZGKNKXRFB-UHFFFAOYSA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- XKVUYEYANWFIJX-UHFFFAOYSA-N 5-methyl-1h-pyrazole Chemical group CC1=CC=NN1 XKVUYEYANWFIJX-UHFFFAOYSA-N 0.000 description 1
- ICLZBFMJMMVKMB-UHFFFAOYSA-N 6-(3,5-dimethylpyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound N1=C(C)C=C(C)N1C1=CC=C(C=O)C=N1 ICLZBFMJMMVKMB-UHFFFAOYSA-N 0.000 description 1
- LEFLUBFIUIHRDD-UHFFFAOYSA-N 6-(4-chloro-3-methylpyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound C1=C(Cl)C(C)=NN1C1=CC=C(C=O)C=N1 LEFLUBFIUIHRDD-UHFFFAOYSA-N 0.000 description 1
- MJOKPSMSYSDZIT-UHFFFAOYSA-N 6-(4-fluoropyrazol-1-yl)pyridine-3-carboxylic acid Chemical compound C1=CC(=NC=C1C(=O)O)N2C=C(C=N2)F MJOKPSMSYSDZIT-UHFFFAOYSA-N 0.000 description 1
- DRJQFNYAKHMAQQ-UHFFFAOYSA-N 6-(5-cyclobutyloxypyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound O=CC(C=C1)=CN=C1N1N=CC=C1OC1CCC1 DRJQFNYAKHMAQQ-UHFFFAOYSA-N 0.000 description 1
- XYGCIUYURREPAX-UHFFFAOYSA-N 6-(5-propan-2-ylpyrazol-1-yl)pyridine-3-carbaldehyde Chemical compound CC(C)C1=CC=NN1C1=NC=C(C=O)C=C1 XYGCIUYURREPAX-UHFFFAOYSA-N 0.000 description 1
- LLQHSBBZNDXTIV-UHFFFAOYSA-N 6-[5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-4,5-dihydro-1,2-oxazol-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC1CC(=NO1)C1=CC2=C(NC(O2)=O)C=C1 LLQHSBBZNDXTIV-UHFFFAOYSA-N 0.000 description 1
- VJDQQJPRYKRDMK-UHFFFAOYSA-N 6-bromo-4-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyridin-2-amine Chemical compound CC1=CC(=NC(=C1)Br)NC2=NNC(=C2)C VJDQQJPRYKRDMK-UHFFFAOYSA-N 0.000 description 1
- PVUKGNBRJFTFNJ-UHFFFAOYSA-N 6-bromopyridine-3-carbaldehyde Chemical group BrC1=CC=C(C=O)C=N1 PVUKGNBRJFTFNJ-UHFFFAOYSA-N 0.000 description 1
- QBFBXIWWFQYZNW-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[(2-methyl-1,3-thiazol-5-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(S6)C QBFBXIWWFQYZNW-UHFFFAOYSA-N 0.000 description 1
- SPDRMXGUCPHNDP-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[(4-methylpyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=C(C=NC=C1)CN2C3CC2CN(C3)C4=NC=C(C=C4)C5=NC(=CC(=N5)NC6=NNC(=C6)C)C SPDRMXGUCPHNDP-UHFFFAOYSA-N 0.000 description 1
- CWNFJUJYLGXPEP-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[(6-methylpyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=NC=C(C=C1)CN2C3CC2CN(C3)C4=NC=C(C=C4)C5=NC(=CC(=N5)NC6=NNC(=C6)C)C CWNFJUJYLGXPEP-UHFFFAOYSA-N 0.000 description 1
- XACMBMKPXWVLJY-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[4-(trifluoromethyl)phenyl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CC=C(C=C6)C(F)(F)F XACMBMKPXWVLJY-UHFFFAOYSA-N 0.000 description 1
- ASIWIIKMPRXLQZ-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(4-methylpyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)N7C=C(C=N7)C ASIWIIKMPRXLQZ-UHFFFAOYSA-N 0.000 description 1
- LMLGJRCDHPDWEQ-UHFFFAOYSA-N 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-[6-[6-[[6-(trifluoromethyl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrimidin-4-amine Chemical compound CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3=CN=C(C=C3)N4CC5CC(C4)N5CC6=CN=C(C=C6)C(F)(F)F LMLGJRCDHPDWEQ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- MITGKKFYIJJQGL-UHFFFAOYSA-N 9-(4-chlorobenzoyl)-6-methylsulfonyl-2,3-dihydro-1H-carbazol-4-one Chemical compound ClC1=CC=C(C(=O)N2C3=CC=C(C=C3C=3C(CCCC2=3)=O)S(=O)(=O)C)C=C1 MITGKKFYIJJQGL-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- OJNFYZKTIUNLIV-UHFFFAOYSA-N BrC=1C=CC(=NC=1)N1CC2N(C(C1)C2)C(=O)OC(C)(C)C Chemical compound BrC=1C=CC(=NC=1)N1CC2N(C(C1)C2)C(=O)OC(C)(C)C OJNFYZKTIUNLIV-UHFFFAOYSA-N 0.000 description 1
- XTAVDKZAUDBHFX-UHFFFAOYSA-N C1=CC(=NC=C1C=O)N2C=C(C=N2)F Chemical compound C1=CC(=NC=C1C=O)N2C=C(C=N2)F XTAVDKZAUDBHFX-UHFFFAOYSA-N 0.000 description 1
- LYCGIEUJRSJSCX-UHFFFAOYSA-N C1=NC(OC)=CC=C1C1=CC=C(C=O)C=N1 Chemical compound C1=NC(OC)=CC=C1C1=CC=C(C=O)C=N1 LYCGIEUJRSJSCX-UHFFFAOYSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 102100031048 Coiled-coil domain-containing protein 6 Human genes 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 241000938605 Crocodylia Species 0.000 description 1
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000777370 Homo sapiens Coiled-coil domain-containing protein 6 Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- HPKJGHVHQWJOOT-ZJOUEHCJSA-N N-[(2S)-3-cyclohexyl-1-oxo-1-({(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl}amino)propan-2-yl]-1H-indole-2-carboxamide Chemical compound C1C(CCCC1)C[C@H](NC(=O)C=1NC2=CC=CC=C2C=1)C(=O)N[C@@H](C[C@H]1C(=O)NCC1)C=O HPKJGHVHQWJOOT-ZJOUEHCJSA-N 0.000 description 1
- GQLSEYOOXBRDFZ-UHFFFAOYSA-N N-formylnornicotine Natural products O=CN1CCCC1C1=CC=CN=C1 GQLSEYOOXBRDFZ-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical class CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- ZNSPHKJFQDEABI-NZQKXSOJSA-N Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 Chemical compound Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 ZNSPHKJFQDEABI-NZQKXSOJSA-N 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000052575 Proto-Oncogene Human genes 0.000 description 1
- 108700020978 Proto-Oncogene Proteins 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 206010071987 RET gene mutation Diseases 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 229940100514 Syk tyrosine kinase inhibitor Drugs 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 238000005411 Van der Waals force Methods 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 125000002009 alkene group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 210000000702 aorta abdominal Anatomy 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000003943 azolyl group Chemical group 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 150000001602 bicycloalkyls Chemical group 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 125000005569 butenylene group Chemical group 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- OSQPUMRCKZAIOZ-UHFFFAOYSA-N carbon dioxide;ethanol Chemical compound CCO.O=C=O OSQPUMRCKZAIOZ-UHFFFAOYSA-N 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 239000012295 chemical reaction liquid Substances 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940125876 compound 15a Drugs 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(I) oxide Inorganic materials [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229940112669 cuprous oxide Drugs 0.000 description 1
- KRFJLUBVMFXRPN-UHFFFAOYSA-N cuprous oxide Chemical compound [O-2].[Cu+].[Cu+] KRFJLUBVMFXRPN-UHFFFAOYSA-N 0.000 description 1
- YMHQVDAATAEZLO-UHFFFAOYSA-N cyclohexane-1,1-diamine Chemical compound NC1(N)CCCCC1 YMHQVDAATAEZLO-UHFFFAOYSA-N 0.000 description 1
- 125000005725 cyclohexenylene group Chemical group 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 230000006240 deamidation Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- FHIVAFMUCKRCQO-UHFFFAOYSA-N diazinon Chemical compound CCOP(=S)(OCC)OC1=CC(C)=NC(C(C)C)=N1 FHIVAFMUCKRCQO-UHFFFAOYSA-N 0.000 description 1
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- FFHWGQQFANVOHV-UHFFFAOYSA-N dimethyldioxirane Chemical compound CC1(C)OO1 FFHWGQQFANVOHV-UHFFFAOYSA-N 0.000 description 1
- QLBHNVFOQLIYTH-UHFFFAOYSA-L dipotassium;2-[2-[bis(carboxymethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [K+].[K+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O QLBHNVFOQLIYTH-UHFFFAOYSA-L 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000005496 eutectics Effects 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical group C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- RIKMMFOAQPJVMX-UHFFFAOYSA-N fomepizole Chemical group CC=1C=NNC=1 RIKMMFOAQPJVMX-UHFFFAOYSA-N 0.000 description 1
- 229960004285 fomepizole Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 150000008040 ionic compounds Chemical class 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- VYMYIBHEBCSLGJ-UHFFFAOYSA-N methyl 6-(4-fluoropyrazol-1-yl)pyridine-3-carboxylate Chemical compound COC(=O)C1=CN=C(C=C1)N2C=C(C=N2)F VYMYIBHEBCSLGJ-UHFFFAOYSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- XAYGBKHKBBXDAK-UHFFFAOYSA-N n-[4-[4-(cyclopropylmethyl)piperazine-1-carbonyl]phenyl]quinoline-8-sulfonamide Chemical compound C=1C=C(NS(=O)(=O)C=2C3=NC=CC=C3C=CC=2)C=CC=1C(=O)N(CC1)CCN1CC1CC1 XAYGBKHKBBXDAK-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000013392 nude mouse xenograft model Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- JKDRQYIYVJVOPF-FDGPNNRMSA-L palladium(ii) acetylacetonate Chemical compound [Pd+2].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O JKDRQYIYVJVOPF-FDGPNNRMSA-L 0.000 description 1
- 150000004965 peroxy acids Chemical class 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 229960001922 sodium perborate Drugs 0.000 description 1
- YKLJGMBLPUQQOI-UHFFFAOYSA-M sodium;oxidooxy(oxo)borane Chemical compound [Na+].[O-]OB=O YKLJGMBLPUQQOI-UHFFFAOYSA-M 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- RBPINQALCJNWMF-UHFFFAOYSA-N tert-butyl 3-[(6-bromo-4-methylpyridin-2-yl)amino]-5-methylpyrazole-1-carboxylate Chemical compound CC1=CC(=NC(=C1)Br)NC2=NN(C(=C2)C)C(=O)OC(C)(C)C RBPINQALCJNWMF-UHFFFAOYSA-N 0.000 description 1
- GJDCNROAKOCYIQ-UHFFFAOYSA-N tert-butyl 3-amino-5-methylpyrazole-1-carboxylate Chemical compound CC1=CC(N)=NN1C(=O)OC(C)(C)C GJDCNROAKOCYIQ-UHFFFAOYSA-N 0.000 description 1
- PBMFDGBXWHICAF-UHFFFAOYSA-N tert-butyl 3-cyclobutyloxypyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC(=N1)OC2CCC2 PBMFDGBXWHICAF-UHFFFAOYSA-N 0.000 description 1
- PWPXQBYACJANKI-UHFFFAOYSA-N tert-butyl 4-(5-fluoropyridin-3-yl)oxypiperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1OC1=CN=CC(F)=C1 PWPXQBYACJANKI-UHFFFAOYSA-N 0.000 description 1
- JYRWUSXRTGACLY-UHFFFAOYSA-N tert-butyl 4-[[3-(4-methylsulfonylphenyl)-[1,2]oxazolo[4,5-d]pyrimidin-7-yl]oxy]piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1OC1=NC=NC2=C1ON=C2C1=CC=C(S(C)(=O)=O)C=C1 JYRWUSXRTGACLY-UHFFFAOYSA-N 0.000 description 1
- NJBGXPJITDKQPS-UHFFFAOYSA-N tert-butyl 5-oxo-1h-pyrazole-2-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC(O)=N1 NJBGXPJITDKQPS-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- 125000005455 trithianyl group Chemical group 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 238000007492 two-way ANOVA Methods 0.000 description 1
- JQSHBVHOMNKWFT-DTORHVGOSA-N varenicline Chemical compound C12=CC3=NC=CN=C3C=C2[C@H]2C[C@@H]1CNC2 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/504—Pyridazines; Hydrogenated pyridazines forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
Definitions
- the present invention relates to a new heterocyclic compound, a pharmaceutical composition containing the same, a preparation method thereof, and its use in the prevention or treatment of diseases or conditions related to RET (Rearranged during transfection) activity.
- Protein kinases are a class of enzymes that catalyze protein phosphorylation reactions. By mediating the process of cell signal transduction, protein phosphorylation regulates the physiological activities of cells, such as cell survival, proliferation, differentiation, apoptosis and metabolism.
- the dysfunction of protein kinases is closely related to many diseases, including tumors, autoimmune diseases, inflammatory reactions, central nervous system diseases, cardiovascular diseases and diabetes.
- RET is a proto-oncogene.
- the RET protein encoded by it is a transmembrane receptor tyrosine protein kinase. It consists of a cysteine-rich cadherin-like extracellular domain (used to bind ligands), The transmembrane region and the intracellular structural region with tyrosine kinase activity are composed of three parts.
- the activated RET protein can activate multiple downstream signaling pathways, including the RAS/RAF/ERK pathway, PI3K/Akt pathway and JNK pathway, leading to cell proliferation, migration and differentiation.
- RET gene changes have a more significant effect on the downstream cascade.
- RET gene mutations are mainly related to medullary thyroid cancer and papillary thyroid cancer
- RET gene fusion is mainly related to non-small cell lung cancer and chronic myeloid leukemia. Therefore, inhibiting RET activity has great medical value (Nature Reviews Cancer, 2014, 14(3): 173-86).
- RET inhibitors have great potential to treat and prevent many diseases (such as tumors, irritable bowel syndrome, etc.). There are currently five compounds in the clinical trial stage, and compounds from many companies are in the pre-clinical research stage. However, no inhibitors with RET as the main target are currently on the market. Therefore, it is necessary to develop new, high-efficiency and low-toxic RET inhibitors to meet clinical needs.
- the present invention provides a new heterocyclic compound, which has a good inhibitory effect on RET, and has good pharmacokinetics, safety and other properties.
- One aspect of the present invention provides compounds of formula I, stereoisomers, tautomers or mixtures of said compounds, N-oxides of said compounds, pharmaceutically acceptable salts, co- Crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of said compounds:
- Ring A is selected from C 6-10 aromatic ring and 5-6 membered heteroaromatic ring;
- Ring B is selected from C 3-8 cycloalkyl and 4-11 membered heterocyclic group
- X 1 is selected from CH and N;
- R 1 is selected from H, halogen, hydroxyl, cyano, C 1-6 alkyl, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy), C 3-8 cycloalkyl, 4-10 member Heterocyclyl and -NR 20a R 20b , the alkyl, heteroalkyl (e.g. alkoxy), cycloalkyl and heterocyclyl are each optionally substituted with one or more substituents selected from the following: hydroxy , Halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy and C 1-4 heteroalkyl (e.g. C 1- 4 alkoxy);
- R 3 and R 4 are absent or are each independently selected from hydroxyl, halogen, CN, C 1-6 alkyl, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy) and C 3-6 cycloalkyl, each of said alkyl, heteroalkyl (eg alkoxy) and cycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, CN, C1-4 Alkyl, C 1-4 haloalkyl, C 1-4 alkoxy and C 1-4 haloalkoxy; when m is greater than 1, two R 3 optionally together with the atoms to which they are attached form C 3-6 Cycloalkyl or 4-10 membered heterocyclic group; and/or when n is greater than 1, two R 4 optionally together with the atoms to which they are attached form a C 3-6 cycloalkyl group or 4-10 membered heterocyclic group ;
- the alkylene, heteroalkylene, alkenylene and alkynylene groups are each optionally substituted with one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-6 alkyl , C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 haloalkoxy, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy) and C 3-8 cycloalkyl;
- L is -N(R 23a )-;
- R 20a , R 20b , R 23a , R 23b , R 23c , R 24a , R 25a and R 25b are each independently selected from H, OH, C 1-6 alkyl, C 1-6 alkoxy and C 3- 8 cycloalkyl; or R 20a and R 20b , R 23a and R 23b or R 25a and R 25b together with the atoms to which they are connected form a 3-8 membered cycloalkyl or heterocyclic group, the alkyl, alkoxy Group, cycloalkyl and heterocyclic group are each optionally substituted with one or more substituents selected from the group consisting of OH, CN, halogen, NO 2 , C 1-4 alkyl, C 1-4 alkoxy, C 1-4 hydroxyalkyl, C 1-4 haloalkyl and C 1-4 haloalkoxy;
- R 30a , R 30b , R 33a , R 33b , R 34a , R 35a and R 35b are each independently selected from H, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy and C 1-6 haloalkoxy;
- R 21 , R 22 , R 31 and R 32 are each independently selected from C 1-6 alkyl, C 1-6 alkoxy, C 3-8 cycloalkyl, 4-10 membered heterocyclyl, C 6- 12 aryl groups and 5-10 membered heteroaryl groups, each of the alkyl group, alkoxy group, cycloalkyl group, heterocyclic group, aryl group and heteroaryl group is optionally substituted by one or more substituents selected from the following Substitution: OH, halogen, CN, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 haloalkyl, C 1-4 haloalkoxy, C 3-6 cycloalkyl and 4-10 members Heterocyclic group;
- n 0, 1, 2, 3 or 4;
- n 0, 1, 2, 3 or 4;
- t 0, 1, 2, 3 or 4;
- u 0, 1, 2, 3 or 4;
- the proviso is that when ring B is a piperazine ring and X 1 is CH, R 2 is not 4-CF 3 -pyridin-2-yl or 4-CN-pyridin-2-yl.
- compositions which comprises a preventive or therapeutically effective amount of a compound of the present invention, a stereoisomer, a tautomer of the compound, or a mixture thereof, and an N-oxide of the compound , A pharmaceutically acceptable salt, co-crystal, polymorph or solvate of the compound, or a stable isotope derivative, metabolite or prodrug of the compound.
- the pharmaceutical composition further comprises one or more pharmaceutically acceptable carriers.
- Another aspect of the present invention provides a compound of the present invention, a stereoisomer, a tautomer of the compound, or a mixture thereof, an N-oxide of the compound, a pharmaceutically acceptable salt of the compound, Co-crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of the compounds, or pharmaceutical compositions as described above are being prepared for the prevention or treatment of diseases related to RET activity or Use in medicine for medical conditions.
- Another aspect of the present invention provides a compound of the present invention, a stereoisomer, a tautomer of the compound, or a mixture thereof, an N-oxide of the compound, a pharmaceutically acceptable salt of the compound, Co-crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of the compounds, or pharmaceutical compositions as described above, which are used for the prevention or treatment of diseases related to RET activity or Condition.
- Another aspect of the present invention provides a method for preventing or treating diseases or conditions associated with RET activity, the method comprising administering to an individual in need thereof an effective amount of a compound of the present invention, stereoisomers, mutual Tautomers or mixtures thereof, N-oxides of the compounds, pharmaceutically acceptable salts, co-crystals, polymorphs or solvates of the compounds, or stable isotopic derivatives of the compounds, Metabolites or prodrugs, or pharmaceutical compositions as described above.
- Another aspect of the invention provides a method of preparing the compound of the invention.
- Figure 1 shows the in vivo efficacy test results of compound 17 and the control compound BLU-667 in a subcutaneous transplantation tumor model of medullary thyroid carcinoma TT cells.
- alkyl is defined as a linear or branched saturated aliphatic hydrocarbon.
- the alkyl group has 1 to 12, for example 1 to 6 carbon atoms.
- C 1-6 alkyl and C 1-4 alkyl refer to linear or branched groups having 1-6 carbon atoms and 1-4 carbon atoms, respectively (Such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl or n-hexyl), any Optionally substituted by one or more (such as 1 to 3) suitable substituents such as halogen (in this case the group is referred to as "haloalkyl”) (e.g.
- C 1-4 alkyl refers to a linear or branched aliphatic hydrocarbon chain of 1 to 4 carbon atoms (ie methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, Sec-butyl or tert-butyl).
- alkylene means a corresponding divalent group, including, for example, “C 1-8 alkylene", “C 1-6 alkylene", “C 1-4 alkylene” and the like.
- alkylene group is optionally substituted with one or more (such as 1 to 3) substituents which are the same or different.
- heteroalkyl refers to an optionally substituted alkyl group having one or more backbone chain atoms selected from atoms other than carbon, such as oxygen, nitrogen, sulfur, phosphorus, or combinations thereof .
- Numerical ranges that can be given e.g. C1-6 heteroalkyl refer to the number of carbons in the chain, including 1-6 carbon atoms in this example.
- the -CH 2 OCH 2 CH 3 group is called a C 3 heteroalkyl group.
- the connection to the rest of the molecule can be through heteroatoms or carbon atoms in the heteroalkyl chain.
- heteroalkylene means a corresponding divalent group, including, for example, “C 1-6 heteroalkylene", “C 1-4 heteroalkylene” and the like.
- haloalkyl refers to an alkyl group substituted with one or more (such as 1 to 3) identical or different halogen atoms
- C 1-8 haloalkyl refers to haloalkyl groups having 1 to 8 carbon atoms, 1 to 6 carbon atoms and 1-4 carbon atoms, respectively, such as -CF 3 , -C 2 F 5 , -CHF 2 , -CH 2 F, -CH 2 CF 3 , -CH 2 Cl or -CH 2 CH 2 CF 3 and so on.
- hydroxyalkyl refers to a group formed by replacing the hydrogen atom in an alkyl group with one or more hydroxy groups, such as C 1-4 hydroxyalkyl or C 1-3 hydroxyalkyl, Examples thereof include, but are not limited to, hydroxymethyl, hydroxyethyl, hydroxypropyl, hydroxybutyl, -CH(OH)CH 3 , -C(CH 3 ) 2 OH and the like.
- alkoxy means a group with an oxygen atom inserted at any reasonable position of an alkyl group (as defined above), preferably a C 1-8 alkoxy group, a C 1-6 alkoxy group Group, C 1-4 alkoxy or C 1-3 alkoxy.
- C 1-6 alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy Group, tert-butoxy, pentyloxy, hexyloxy, -CH 2 -OCH 3, etc., the alkoxy is optionally substituted by one or more (such as 1 to 3) substituents which are the same or different .
- alkyleneoxy refers to a divalent alkoxy group, such as -OCH 2 -, -OCH(CH 3 )CH 2 -, -OCH 2 CH 2 O-, -CH 2 CH 2 O- etc., the alkyleneoxy group is optionally substituted with one or more (such as 1 to 3) substituents which are the same or different.
- alkenyl means a linear or branched monovalent hydrocarbon group, which contains one or more double bonds and has 2-6 carbon atoms (“C 2-6 alkenyl”).
- alkynyl means a monovalent hydrocarbon group containing one or more triple bonds, which preferably has 2, 3, 4, 5 or 6 carbon atoms, such as ethynyl, 2-propynyl, 2 -Butynyl, 1,3-butadiynyl, etc.
- the alkynyl group is optionally substituted with one or more (such as 1 to 3) substituents which are the same or different.
- alkynylene is a corresponding divalent group, including, for example, “C 2-8 alkynylene", “C 2-6 alkynylene", “C 2-4 alkynylene” and the like. Examples include but are not limited to Etc., the alkynylene group is optionally substituted with one or more (such as 1 to 3) substituents which are the same or different.
- fused ring or “fused ring” refers to a ring system formed by two or more ring structures sharing two adjacent atoms with each other.
- spirocyclic ring refers to a ring system formed by two or more ring structures sharing one ring atom with each other.
- bridged ring refers to a ring system formed by two or more ring structures sharing two atoms that are not directly connected to each other.
- cycloalkyl refers to a saturated or unsaturated non-aromatic monocyclic or polycyclic (such as bicyclic) hydrocarbon ring group, including but not limited to monocyclic alkyl (such as cyclopropyl, cyclobutyl Group, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, etc.) and bicycloalkyl, including spiro ring, fused ring (fused ring) or bridged ring system (ie, spirocycloalkyl, and Cyclo (fused ring) alkyl groups and bridged cycloalkyl groups, such as bicyclo[1.1.1]pentyl, bicyclo[2.2.1]heptyl, etc.).
- monocyclic alkyl such as cyclopropyl, cyclobutyl Group, cyclopentyl, cyclohexyl, cyclo
- the cycloalkyl group is optionally substituted with one or more (such as 1 to 3) substituents which are the same or different.
- C 3-8 cycloalkyl refers to a cycloalkyl group having 3 to 8 ring-forming carbon atoms, such as a C 3-6 cycloalkyl group, which may be a monocyclic alkyl group, such as cyclopropyl, cyclobutyl , Cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl, it can also be a bicyclic alkyl group, such as C 5-8 spirocycloalkyl, C 5-8 bridged cycloalkyl, C 5-8 fused cycloalkyl , C 5-6 spirocycloalkyl, C 5-6 bridged cycloalkyl or C 5-6 condensed cycloalkyl.
- cycloalkoxy means -O-cycloalkyl, where cycloalkyl is as defined above.
- Representative examples of cycloalkoxy include, but are not limited to, cyclopropoxy, cyclobutoxy, cyclopentyloxy, cyclohexyloxy, and the like.
- 4-11 membered heterocyclic group means a heterocyclic group containing 4-11 ring atoms, including but not limited to 4-10 membered heterocyclic group, 4-9 membered heterocyclic group, 4-8 membered heterocyclic group, 4-7 membered heterocyclic group, 5-6 membered heterocyclic group, 3-8 membered heterocyclic group, 3-7 membered heterocyclic group, 4-7 membered nitrogen-containing heterocyclic group, 4-7 membered oxygen-containing heterocyclic group, 4-7 membered sulfur-containing heterocyclic group, 5-6 membered nitrogen-containing heterocyclic group, 5-6 membered oxygen-containing heterocyclic group, 5-6 membered sulfur-containing heterocyclic group, etc.
- Each of the "nitrogen-containing heterocyclic group", “oxygen-containing heterocyclic group” and “sulfur-containing heterocyclic group” optionally further contains one or more other heteroatoms selected from oxygen, nitrogen and sulfur.
- 4-11 membered heterocyclic groups include, but are not limited to, oxirane, aziridinyl, azetidinyl, oxetanyl, tetrahydrofuryl, pyrrolidinyl, pyrrolidinone (e.g.
- imidazolidinyl imidazolidinyl, pyrazolidinyl, tetrahydropyranyl, piperidinyl, morpholinyl, dithianyl, thiomorpholinyl, piperazinyl, trithianyl.
- heterocyclic group encompasses a fused ring structure, and the point of connection between the fused ring structure and other groups can be on any ring in the fused ring structure. Therefore, the heterocyclic group of the present invention also includes but is not limited to heterocyclic group and heterocyclic group, heterocyclic group and cycloalkyl group, single heterocyclic group and single heterocyclic group, single heterocyclic group and monocyclic alkyl group, for example 3-7 membered (mono) heterocyclyl and 3-7 membered (mono) heterocyclyl, 3-7 membered (mono) heterocyclyl and (mono) cycloalkyl, 3-7 membered (mono) heterocyclyl And C 4-6 (mono)cycloalkyl, examples of which include but are not limited to pyrrolidinocyclopropyl, cyclopentylazacyclopropyl, pyrrolidinocyclobutyl, pyrrolidin
- heterocyclic group encompasses bridged heterocyclic groups and spiro heterocyclic groups.
- bridged heterocycle refers to two saturated rings that share two ring atoms that are not directly connected and contain one or more (for example, 1, 2, 3, or 4) heteroatoms.
- cyclic structure including but not limited to 7-10 membered heterocyclic ring, 8-10 membered heterocyclic ring, 7-10 membered nitrogen-containing bridged heterocyclic ring, 7- 10-membered oxygen-containing bridged heterocyclic ring, 7-10 membered sulfur-containing bridged heterocyclic ring, etc., for example Wait.
- the "nitrogen-containing heterocyclic ring”, "oxygen-containing heterocyclic ring” and “sulfur-containing heterocyclic ring” optionally further contain one or more other heteroatoms selected from oxygen, nitrogen and sulfur.
- spiro heterocyclic ring refers to two or more saturated rings that share one ring atom and contain one or more (for example, 1, 2, 3, or 4) heteroatoms.
- oxygen atom, nitrogen atom, sulfur atom cyclic structure, including but not limited to 5-10 membered spiro heterocyclic ring, 6-10 membered spiro heterocyclic ring, 6-10 membered nitrogen-containing spiro heterocyclic ring, 6-10 membered Oxygen-containing spiro heterocycles, 6-10 membered sulfur-containing spiro heterocycles, etc., for example
- the "nitrogen-containing spiro heterocyclic ring", “oxygen-containing spiro heterocyclic ring”, and “sulfur-containing spiro heterocyclic ring” optionally further contain one or more other heteroatoms selected from oxygen, nitrogen, and sulfur.
- the term "6-10 membered nitrogen-containing spiroheterocyclic group" refers to two or more saturated
- a heterocyclic group may be fused with an aryl group to form a fused ring structure.
- the fused ring structure include but are not limited to:
- aryl refers to an all-carbon monocyclic or fused polycyclic aromatic group having a conjugated ⁇ -electron system.
- C 6-12 aryl (aromatic ring) means an aryl group (aromatic ring) containing 6 to 12 carbon atoms, preferably a C 6-10 aryl group (aromatic ring), preferably It is phenyl or naphthyl.
- the aryl group is optionally substituted with one or more (such as 1 to 3) identical or different substituents (eg halogen, OH, CN, NO 2 , C 1 -C 6 alkyl, etc.).
- heteroaryl or “heteroaromatic ring” refers to a monocyclic or polycyclic aromatic group containing one or more identical or different heteroatoms, including monocyclic heteroaryl and containing at least A bicyclic or polycyclic ring system of a heteroaromatic ring (an aromatic ring system containing at least one heteroatom), which can have 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 ring atoms, For example, 5, 6, 7, 8, 9 or 10 ring atoms.
- the heteroatom may be oxygen, nitrogen or sulfur.
- the term "5-10 membered heteroaryl” or “5-10 membered heteroaromatic ring” means a heteroaryl group containing 5 to 10 (e.g. 5 to 6) ring atoms (heteroaryl ring ), including 5-10 member nitrogen-containing heteroaryl group, 5-10 member oxygen-containing heteroaryl group, 5-10 member sulfur-containing heteroaryl group, 5-6 member nitrogen-containing heteroaryl group, 5-6 member oxygen-containing heteroaryl group Aryl, 5-6 membered sulfur-containing heteroaryl, etc.
- the "nitrogen-containing heteroaryl group”, “oxygen-containing heteroaryl group” and “sulfur-containing heteroaryl group” each optionally contain one or more other heteroatoms selected from oxygen, nitrogen and sulfur.
- Examples include, but are not limited to, thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, triazolyl, tetrazolyl, oxadiazolyl , Thiadiazolyl, etc., or pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, etc., and 5-10 membered cyclic groups containing these groups.
- heteroaryl encompasses a fused ring structure, and the point of connection between the fused ring structure and other groups can be on any ring in the fused ring structure. Therefore, the heteroaryl group of the present invention also includes but is not limited to (mono)heteroaryl and (mono)heteroaryl, (mono)heteroaryl and (monocyclic)aryl, (mono)heteroaryl and (mono) ) Heterocyclyl and (mono)heteroaryl and (mono)cycloalkyl, for example 5-6 membered (mono)heteroaryl and 5-6 membered (mono)heteroaryl, 5-6 membered (mono)heteroaryl, 5-6 membered (mono)hetero Aryl phenyl, 5-6 membered (mono)heteroaryl and 5-6 membered (mono)heterocyclyl or 5-6 membered (mono)heteroaryl
- halo or halogen group is defined to include F, Cl, Br, or I.
- substituted means that one or more (eg, one, two, three, or four) hydrogens on the specified atom are replaced by a selection from the indicated group, provided that no more than the specified atom is present In the case of normal valence and the substitution forms a stable compound. Combinations of substituents and/or variables are only allowed when such combinations form stable compounds.
- substituent can be (1) unsubstituted or (2) substituted. If the carbon of a substituent is described as being optionally substituted with one or more of the list of substituents, then one or more hydrogens on the carbon (to the extent of any hydrogens present) may be independently and/or together independently Optional substitution of selected substituents. If the nitrogen of a substituent is described as being optionally substituted with one or more of the list of substituents, then one or more hydrogens on the nitrogen (to the extent of any hydrogens present) may each be independently selected optionally Substituent replacement.
- each substituent is selected independently of the other. Therefore, each substituent may be the same or different from another (other) substituent.
- one or more means 1 or more than 1, such as 2, 3, 4, 5, or 10 under reasonable conditions.
- the point of attachment of a substituent can be from any suitable position of the substituent.
- the present invention also includes all pharmaceutically acceptable isotope-labeled compounds, which are the same as the compounds of the present invention, except that one or more atoms have the same atomic number but the atomic mass or mass number is different from the predominant atomic mass in nature. Or atomic substitution of mass number.
- isotopes suitable for inclusion in the compounds of the present invention include, but are not limited to, isotopes of hydrogen (such as deuterium ( 2 H), tritium ( 3 H)); isotopes of carbon (such as 11 C, 13 C, and 14 C) ; Isotopes of chlorine (such as 36 Cl); isotopes of fluorine (such as 18 F); isotopes of iodine (such as 123 I and 125 I); isotopes of nitrogen (such as 13 N and 15 N); isotopes of oxygen (such as 15 O , 17 O and 18 O); phosphorus isotopes (such as 32 P); and sulfur isotopes (such as 35 S).
- isotopes of hydrogen such as deuterium ( 2 H), tritium ( 3 H)
- isotopes of carbon such as 11 C, 13 C, and 14 C
- Isotopes of chlorine such as 36 Cl
- isotopes of fluorine
- Certain isotope-labeled compounds of the invention can be used in drug and/or substrate tissue distribution studies (such as analysis).
- the radioisotopes tritium (i.e. 3 H) and carbon-14 (i.e. 14 C) are particularly useful for this purpose because they are easy to incorporate and easy to detect.
- Substitution with positron emission isotopes (such as 11 C, 18 F, 15 O, and 13 N) can be used to test substrate receptor occupancy in positron emission tomography (PET) studies.
- the isotopically-labeled compounds of the present invention can be prepared by methods similar to those described in the attached routes and/or examples and preparations by using appropriate isotopically-labeled reagents instead of previously used non-labeled reagents.
- the pharmaceutically acceptable solvates of the present invention include those in which the crystallization solvent can be replaced by an isotope, for example, D 2 O, acetone-d 6 or DMSO-d 6 .
- stereoisomer means an isomer due to at least one asymmetric center. In compounds with one or more (for example, one, two, three or four) asymmetric centers, it can produce exo/meso mixtures, single enantiomers, and diastereoisomer mixtures And individual diastereomers. Certain individual molecules can also exist as geometric isomers (cis/trans). Similarly, the compounds of the present invention may exist in mixtures of two or more structurally different forms (commonly referred to as tautomers) in rapid equilibrium. Representative examples of tautomers include keto-enol tautomers, phenol-ketone tautomers, nitroso-oxime tautomers, imine-enamine tautomers Wait. For example, nitroso-oximes can exist in equilibrium in the following tautomeric forms in solution:
- Solid lines can be used in this article Solid wedge Virtual wedge Depicts the chemical bonds of the compounds of the invention.
- the use of a solid line to depict the bond to an asymmetric carbon atom is intended to indicate that all possible stereoisomers at that carbon atom (e.g., specific enantiomers, racemic mixtures, etc.) are included.
- the use of real or imaginary wedges to depict bonds to asymmetric carbon atoms is intended to indicate that the stereoisomers shown exist. When present in a racemic mixture, use real and imaginary wedges to define relative stereochemistry, rather than absolute stereochemistry.
- the compounds of the present invention are intended to be stereoisomers (which include cis and trans isomers, optical isomers (such as R and S enantiomers), diastereomers, Geometric isomers, rotamers, conformational isomers, atropisomers and mixtures thereof) exist in the form of.
- the compounds of the present invention can exhibit more than one type of isomerism, and are composed of mixtures thereof (for example, racemic mixtures and diastereomeric pairs).
- the present invention covers all possible crystalline forms or polymorphs of the compounds of the present invention, which can be a single polymorph or a mixture of more than one polymorph in any ratio.
- Eutectic refers to the fact that the active molecules of the drug and other physiologically acceptable molecules of acids, bases, salts, and non-ionic compounds are connected by hydrogen bonds, ⁇ - ⁇ stacking, van der Waals forces and other non-covalent bonds to be combined in the same crystal lattice. .
- compositions of the present invention may exist in free form for treatment, or, when appropriate, in the form of pharmaceutically acceptable derivatives thereof.
- pharmaceutically acceptable derivatives include, but are not limited to, pharmaceutically acceptable salts, esters, solvates, N-oxides, metabolites, or prodrugs, which can be administered to patients in need thereof. After medication, the compound of the present invention or its metabolites or residues can be directly or indirectly provided. Therefore, when “the compound of the present invention” is referred to herein, it is also intended to cover the above-mentioned various derivative forms of the compound.
- the pharmaceutically acceptable salts of the compounds of the present invention include acid addition salts and base addition salts thereof.
- acid addition salts for example, hexafluorophosphate, meglumine salt, etc.
- suitable salts see “Handbook of Pharmaceutical Salts: Properties, Selection, and Use” by Stahl and Wermuth (Wiley-VCH, 2002).
- ester means an ester derived from each of the compounds of the general formula in this application, which includes physiologically hydrolyzable esters (which can be hydrolyzed under physiological conditions to release the free acid or alcohol form of the present invention Compound).
- the compound of the present invention may itself be an ester.
- the compound of the present invention may exist in the form of a solvate (preferably a hydrate), wherein the compound of the present invention contains a polar solvent as a structural element of the crystal lattice of the compound, particularly, for example, water, methanol or ethanol.
- a polar solvent as a structural element of the crystal lattice of the compound, particularly, for example, water, methanol or ethanol.
- the amount of polar solvent, especially water can be present in a stoichiometric or non-stoichiometric ratio.
- metabolites of the compounds of the present invention that is, substances formed in the body when the compounds of the present invention are administered. Such products can be produced by, for example, oxidation, reduction, hydrolysis, amidation, deamidation, esterification, enzymatic hydrolysis, etc. of the administered compound. Therefore, the present invention includes metabolites of the compounds of the present invention, including compounds prepared by contacting the compound of the present invention with a mammal for a time sufficient to produce its metabolites.
- the present invention further includes within its scope the prodrugs of the compounds of the present invention, which are certain derivatives of the compounds of the present invention that may themselves have less or no pharmacological activity when administered to or on the body It can be converted into the compound of the present invention having the desired activity by, for example, hydrolytic cleavage.
- prodrugs will be functional group derivatives of the compound, which are easily converted into the desired therapeutically active compound in vivo.
- prodrugs please refer to "Pro-drugs as Novel Delivery Systems", Volume 14, ACS Symposium Series (T. Higuchi and V. Stella).
- prodrugs of the present invention can be used, for example, by using certain parts known to those skilled in the art as “pro-moiety (e.g., “Design of Prodrugs", described in H. Bundgaard (Elsevier, 1985))" It is prepared by substituting appropriate functional groups present in the compounds of the present invention.
- the present invention also encompasses compounds of the present invention containing protecting groups.
- protecting groups In any process of preparing the compounds of the present invention, protection of sensitive groups or reactive groups on any relevant molecule may be necessary and/or desirable, thereby forming a chemically protected form of the compounds of the present invention. This can be achieved by conventional protecting groups, such as those described in T.W. Greene & P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991, these references are incorporated herein by reference. Using methods known in the art, the protecting group can be removed at an appropriate subsequent stage.
- One aspect of the present invention provides compounds of formula I, stereoisomers, tautomers or mixtures of said compounds, N-oxides of said compounds, pharmaceutically acceptable salts, co- Crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of said compounds:
- Ring A is selected from C 6-10 aromatic ring and 5-6 membered heteroaromatic ring;
- Ring B is selected from C 3-8 cycloalkyl and 4-11 membered heterocyclic group
- X 1 is selected from CH and N;
- R 1 is selected from H, halogen, hydroxyl, cyano, C 1-6 alkyl, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy), C 3-8 cycloalkyl, 4-10 member Heterocyclyl and -NR 20a R 20b , the alkyl, heteroalkyl (e.g. alkoxy), cycloalkyl and heterocyclyl are each optionally substituted with one or more substituents selected from the following: hydroxy , Halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy and C 1-4 heteroalkyl (e.g. C 1- 4 alkoxy);
- R 3 and R 4 are absent or are each independently selected from hydroxyl, halogen, CN, C 1-6 alkyl, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy) and C 3-6 cycloalkyl, each of said alkyl, heteroalkyl (eg alkoxy) and cycloalkyl is optionally substituted with one or more substituents selected from the group consisting of halogen, CN, C1-4 Alkyl, C 1-4 haloalkyl, C 1-4 alkoxy and C 1-4 haloalkoxy; when m is greater than 1, two R 3 optionally together with the atoms to which they are attached form C 3-6 Cycloalkyl or 4-10 membered heterocyclic group; and/or when n is greater than 1, two R 4 optionally together with the atoms to which they are attached form a C 3-6 cycloalkyl group or 4-10 membered heterocyclic group ;
- the alkylene, heteroalkylene, alkenylene and alkynylene groups are each optionally substituted with one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-6 alkyl , C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 haloalkoxy, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy) and C 3-8 cycloalkyl;
- L is -N(R 23a )-;
- R 20a , R 20b , R 23a , R 23b , R 23c , R 24a , R 25a and R 25b are each independently selected from H, OH, C 1-6 alkyl, C 1-6 alkoxy and C 3- 8 cycloalkyl; or R 20a and R 20b , R 23a and R 23b or R 25a and R 25b together with the atoms to which they are connected form a 3-8 membered cycloalkyl or heterocyclic group, the alkyl, alkoxy Group, cycloalkyl and heterocyclic group are each optionally substituted with one or more substituents selected from the group consisting of OH, CN, halogen, NO 2 , C 1-4 alkyl, C 1-4 alkoxy, C 1-4 hydroxyalkyl, C 1-4 haloalkyl and C 1-4 haloalkoxy;
- R 30a , R 30b , R 33a , R 33b , R 34a , R 35a and R 35b are each independently selected from H, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy and C 1-6 haloalkoxy;
- R 21 , R 22 , R 31 and R 32 are each independently selected from C 1-6 alkyl, C 1-6 alkoxy, C 3-8 cycloalkyl, 4-10 membered heterocyclyl, C 6- 12 aryl groups and 5-10 membered heteroaryl groups, each of the alkyl group, alkoxy group, cycloalkyl group, heterocyclic group, aryl group and heteroaryl group is optionally substituted by one or more substituents selected from the following Substitution: OH, halogen, CN, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 haloalkyl, C 1-4 haloalkoxy, C 3-6 cycloalkyl and 4-10 members Heterocyclic group;
- n 0, 1, 2, 3 or 4;
- n 0, 1, 2, 3 or 4;
- t 0, 1, 2, 3 or 4;
- u 0, 1, 2, 3 or 4;
- the proviso is that when ring B is a piperazine ring and X 1 is CH, R 2 is not 4-CF 3 -pyridin-2-yl or 4-CN-pyridin-2-yl.
- ring A is a benzene ring or a 5-6 membered heteroaromatic ring; preferably, ring A is a benzene ring, a thiazole ring, a pyridine ring, a pyrazine ring or a pyrimidine ring; more preferably, ring A is It is connected to the ring where X 1 is located through the position marked with *, and is connected to ring B through the position marked with **.
- ring B is C 3-6 cycloalkyl or 5-7 membered heterocyclyl; preferably, ring B is piperidine ring, piperazine ring, azepane bridged ring or diazide A heterocycloheptane bridged ring; more preferably, ring B is It is connected to ring A by the position marked with *, and connected to L by the position marked by **.
- X 1 is CH or N; preferably, X 1 is N.
- R 1 is selected from H, halogen, hydroxy, cyano, C 1-4 alkyl, C 1-4 heteroalkyl (e.g. C 1-4 alkoxy), C 3-6 ring Alkyl and 4-10 membered heterocyclyl, each of said alkyl, heteroalkyl (e.g. alkoxy), cycloalkyl and heterocyclyl is optionally substituted with one or more substituents selected from the following: Hydroxyl, halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy and C 1-4 heteroalkyl (e.g. C 1 -4 alkoxy).
- R 1 is selected from C 1-4 alkyl, 5-membered nitrogen-containing heterocyclic group and C 1-4 heteroalkyl (eg C 1-4 alkoxy), the alkyl, hetero
- the cyclic group and the heteroalkyl group are each optionally substituted with one or more substituents selected from the group consisting of hydroxy, halogen, CN, C 1-3 alkyl, C 1-3 haloalkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkoxy, and C 1-3 heteroalkyl (e.g. C 1-4 alkoxy).
- R 1 is selected from C 1-3 alkyl (e.g. methyl), pyrrolidinyl (e.g. pyrrolidin-1-yl), and C 1-3 alkoxy (e.g. ethoxy).
- R 2 is methyl-substituted pyrazolyl (e.g. 5-methyl-1H-pyrazol-3-yl or 1-methyl-1H-pyrazol-4-yl), cyclopropyl-substituted pyrazolyl (e.g. 5-cyclopropyl-1H-pyrazol-3-yl) or -C(O)CH 3 .
- R 3 and R 4 are absent or are independently selected from hydroxyl, halogen, CN, C 1-4 alkyl, and C 1-4 alkoxy at each occurrence, the alkyl and The alkoxy groups are each optionally substituted with one or more substituents selected from the group consisting of halogen, CN, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxy and C 1-4 Haloalkoxy; when m is greater than 1, two R 3 optionally together with the atoms to which they are attached form a C 3-6 cycloalkyl or 4-10 membered heterocyclic group; and/or when n is greater than 1, two Each R 4 optionally together with the atom to which it is attached forms a C 3-6 cycloalkyl group or a 4-10 membered heterocyclic group.
- R 3 and R 4 are absent or are independently selected from hydroxyl, halogen, CN, C 1-3 alkyl, and C 1-3 alkoxy at each occurrence, said alkyl and Each alkoxy group is optionally substituted with one or more substituents selected from the group consisting of halogen, CN and C 1-3 alkyl; when m is greater than 1, two R 3 are optionally together with the atoms to which they are attached A C 3-6 cycloalkyl group or a 4-10 membered heterocyclic group is formed; and/or when n is greater than 1, two R 4 optionally together with the atoms to which they are attached form a C 3-6 cycloalkyl group or a 4- 10-membered heterocyclic group.
- R 3 and R 4 are absent or are independently selected from: F, Cl, CN, OH, C 1-3 alkyl, and C 1-3 alkoxy at each occurrence; preferably , R 3 and R 4 do not exist.
- L is selected from -O-, -S-, -C(O)-, -N(R 23a )-C(O)-, -C(O)-N(R 23c )- , C 1-4 alkylene, C 1- 4 alkylene heteroalkyl
- the alkylene and heteroalkylene groups are each optionally substituted with one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy, C 1-4 heteroalkyl (e.g. C 1-4 alkoxy) and C 3-6 cycloalkyl.
- L is selected from -O-, -C(O)-, -NHC(O)-, -C(O)NH-, C 1-3 alkylene, C 1-3 hetero alkyl
- the alkylene and heteroalkylene groups are each optionally substituted with one or more substituents selected from the group consisting of hydroxy, halogen, CN, NO 2 , C 1-3 alkyl, C 1-3 haloalkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkoxy, C 1-3 heteroalkyl (e.g. C 1-3 alkoxy) and C 3-6 cycloalkyl, wherein R 23a and R 23b are preferably H or C 1-3 alkyl.
- L is selected from -O-, -C(O)-, -NHC(O)-, -C(O)NH-, C 1-3 alkylene,
- the alkylene group is optionally substituted with one or more substituents selected from the group consisting of hydroxy, halogen, CN, C 1-3 alkyl, and C 1-3 haloalkyl.
- L is -CH 2 -, -CH(CH 3 )-, -O-, -C(O)-, -C(O)NH-or
- R 5 is selected from C 3-6 cycloalkyl, 4-10 membered heterocyclyl, C 6-12 aryl, and 5-10 membered heteroaryl, the cycloalkyl, heterocyclic Group, aryl group, and heteroaryl group are each optionally substituted with one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 haloalkyl, C 1 -4 hydroxyalkyl, C 1-4 haloalkoxy, C 1-4 heteroalkyl (e.g.
- R 5 is selected from phenyl and 5-6 membered heteroaryl (e.g., pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyrazolyl, oxazolyl, imidazolyl or thiazolyl ), the phenyl and heteroaryl groups are each optionally substituted by one or more substituents selected from the group consisting of hydroxy, halogen, CN, C 1-3 alkyl, C 1-3 haloalkyl, C 1- 3 hydroxyalkyl, C 1-3 haloalkoxy, C 1-3 heteroalkyl (e.g., phenyl and 5-6 membered heteroaryl (e.g., pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyrazolyl, oxazolyl, imidazolyl or thiazolyl ), the phenyl and heteroaryl groups
- the heterocyclic group and the heteroaryl group are each optionally substituted with one or more substituents selected from the group consisting of hydroxy, halogen, CN, C 1-3 alkyl, C 1-3 haloalkyl, C 1-3 hydroxyalkane Group, C 1-3 haloalk
- R 5 is selected from the group consisting of phenyl, pyridyl, pyrazolyl and thiazolyl, each of which is optionally selected by one or more of the following Substituent substitution of: halogen, CN, C 1-3 alkyl, C 1-3 haloalkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkoxy, C 1-3 alkoxy, C 3- 6 cycloalkyl, C 3-6 cycloalkoxy, 4-6 membered heterocyclic group, 5-8 membered heteroaryl (e.g.
- heterocyclic group and heteroaryl group are each optionally substituted with one or more substituents selected from the following: Halogen, CN, C 1-3 alkyl, C 1-3 haloalkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkoxy, C 1-3 alkoxy, C 3-6 cycloalkyl, C 3-6 cycloalkoxy and 4-6 membered heterocyclic group.
- R 5 is optionally substituted with one or more substituents selected from halogen (e.g. fluorine or chlorine), CN, C 1-3 alkyl (e.g. methyl or ethyl), C 1- 3 haloalkyl (e.g. three Fluoromethyl), C 1-3 alkoxy (e.g. methoxy or ethoxy), C 3-6 cycloalkyl (e.g. cyclopropyl), C 3-6 cycloalkoxy (e.g. cyclopropoxy Group) and 5-6 membered heteroaryl (e.g.
- halogen e.g. fluorine or chlorine
- CN C 1-3 alkyl (e.g. methyl or ethyl), C 1- 3 haloalkyl (e.g. three Fluoromethyl), C 1-3 alkoxy (e.g. methoxy or ethoxy), C 3-6 cycloalkyl (e.g. cyclopropyl), C 3-6 cycl
- R 5 is selected from the group consisting of phenyl, pyridyl, pyrazolyl and thiazolyl, each of which is optionally selected by one or more of the following Substituent substitution of: halogen, CN, C 1-3 alkyl, C 1-3 haloalkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkoxy, C 1-3 alkoxy, C 3- 6 cycloalkyl, C 3-6 cycloalkoxy, 4-6 membered heterocyclic group, 5-6 membered heteroaryl (e.g.
- heterocyclic group and heteroaryl group are each optionally substituted with one or more substituents selected from the group consisting of halogen, CN, C 1-3 Alkyl, C 1-3 haloalkyl, C 1-3 haloalkoxy, C 1-3 alkoxy, C 3-6 cycloalkyl, and 4-6 membered heterocyclic group.
- R 5 is optionally selected from halogen (e.g. fluorine or chlorine), CN, C 1-3 alkyl (e.g.
- methyl or ethyl C 1-3 haloalkyl (e.g. tri Fluoromethyl), C 1-3 alkoxy (e.g. methoxy or ethoxy), C 3-6 cycloalkyl (e.g. cyclopropyl), C 3-6 cycloalkoxy (e.g. cyclopropoxy Phenyl, pyridyl, pyrazolyl or thiazolyl substituted with substituents of five-membered heteroaryl (e.g. pyrazolyl, imidazolyl or thiazolyl), wherein the five-membered heteroaryl is optionally further It is substituted with one or more substituents selected from halogen (e.g. fluorine or chlorine), C 1-3 alkyl (e.g. methyl), C 1-3 hydroxyalkyl (e.g. hydroxymethyl or hydroxypropyl).
- halogen e.g. fluorine or chlorine
- C 1-3 alkyl e.
- R 20a , R 20b , R 23a , R 23b , R 23c , R 24a , R 25a and R 25b are each independently selected from H, C 1-4 alkyl, C 1-4 alkoxy group and a C 3-8 cycloalkyl group; or R 20a and R 20b, R 23a and R 23b, or R 25a and R 25b together with the atoms to which they are attached form a 3-8 membered cycloalkyl or heterocyclyl, said Alkyl, alkoxy, cycloalkyl and heterocyclyl are each optionally substituted with one or more substituents selected from the group consisting of OH, CN, halogen, NO 2 , C 1-4 alkyl, C 1- 4 Alkoxy, C 1-4 hydroxyalkyl, C 1-4 haloalkyl and C 1-4 haloalkoxy.
- R 20a , R 20b , R 23a , R 23b , R 23c , R 24a , R 25a and R 25b are each independently H, C 1-4 alkyl, or C 1-4 alkoxy .
- R 23a and R 23b are each independently selected from H, C 1-3 alkyl, C 1-3 alkoxy, and C 3-6 cycloalkyl; or R 23a and R 23b together with them
- R 21 , R 22 , R 31 and R 32 are each independently selected from C 1-4 alkyl, C 1-4 alkoxy, C 3-8 cycloalkyl, and 4-10 membered Heterocyclyl, the alkyl, alkoxy, cycloalkyl and heterocyclyl are each optionally substituted with one or more substituents selected from the group consisting of OH, halogen, CN, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 haloalkyl, C 1-4 haloalkoxy, C 3-6 cycloalkyl and 4-10 membered heterocyclic group.
- R 21 , R 22 , R 31 and R 32 are each independently selected from C 1-4 alkyl.
- R 30a, R 30b, R 33a, R 33b, R 34a, R 35a and R 35b are each independently selected from H, C 1-4 alkyl, C 1- 4 haloalkyl -alkyl, C 1 -4 hydroxyalkyl, C 1-4 alkoxy and C 1-4 haloalkoxy;
- R 30a , R 30b , R 33a , R 33b , R 34a , R 35a and R 35b are each independently selected from H and C 1-4 alkyl.
- n is zero.
- n 0, 1, or 2.
- t is 0 or 1.
- u is 0 or 1.
- the compound of the invention has the structure shown in Formula I-A:
- R 1 and R 2 are as defined in formula I above;
- R 23a , R 30a , R 30b , R 31 , R 32 , R 33a , R 33b , R 34a , R 35a and R 35b are as defined in Formula I above, and R 23a is preferably H or C 1-3 alkyl.
- the 5-10 membered heteroaryl group is optionally substituted by one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 Haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy, C 1-4 heteroalkyl (e.g.
- R 23a , R 30a , R 30b , R 31 , R 32 , R 33a , R 33b , R 34a , R 35a and R 35b are as defined in Formula I above, and R 23a is preferably H or C 1-3 alkyl.
- the compound of the present invention has the structure shown in Formula I-B:
- R 1 , R 2 , R 5 and R 23a are as defined in Formula I above, and R 23a is preferably H or C 1-3 alkyl.
- the compound of the present invention has the structure shown in Formula I-C:
- R 1 , R 2 , R 5 and R 23a are as defined in Formula I above, and R 23a is preferably H or C 1-3 alkyl; and when X 1 is N, R 1 , R 2 , R 5 and R 23a are as defined in Formula IA above.
- the compound of the present invention has the structure shown in Formula I-D:
- R 1 , R 2 , R 23a , R 23b and t are as defined in formula I above;
- R 5 is as defined in Formula I above;
- R 5 is C 6-12 aryl or 5-10 membered heteroaryl, wherein
- the C 6-12 aryl group and 5-10 membered heteroaryl group are each optionally substituted with one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy, C 1-4 heteroalkyl (e.g.
- the C 6-12 aryl group is optionally substituted with one or more substituents selected from the group consisting of hydroxyl, halogen, CN, NO 2 , C 1-4 alkane Group, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy, C 1-4 heteroalkyl (e.g.
- R 30a , R 30b , R 31 , R 32 , R 33a , R 33b , R 34a , R 35a and R 35b are as defined in Formula I above.
- the compound of the present invention has the structure shown in Formula I-E:
- R 1 , R 2 , R 5 , R 23a , R 23b , X 1 and t are as defined in formula ID above.
- the compounds of the invention have the structure shown in Formula I-F:
- R 1 , R 2 , R 5 , R 23a , R 23b and X 1 are as defined in formula ID above;
- R 4 is as defined in Formula I above, and is preferably C 1-3 alkyl or C 1-3 alkoxy;
- R 23c is H, C 1-3 alkyl or C 1-3 alkoxy, each of said alkyl and alkoxy is optionally substituted by one or more substituents selected from the group consisting of OH, CN, halogen , C 1-4 alkoxy and C 1-4 hydroxyalkyl;
- u is 0 or 1
- n 0 or 1.
- the compound of the present invention has the structure shown in Formula I-G:
- X 1 is CH or N
- R 1 , R 2 and R 4 are as defined in formula I above, and R 4 is preferably C 1-3 alkyl or C 1-3 alkoxy;
- n 0 or 1
- R 30a , R 30b , R 31 , R 32 , R 33a , R 33b , R 34a , R 35a and R 35b are as defined in Formula I above.
- the compound of the present invention has the structure shown in Formula I, wherein:
- Ring A is It is connected to the ring where X 1 is located through the position marked by *, and is connected to ring B through the position marked by **;
- Ring B is It is connected to ring A through the position marked by *, and connected to L through the position marked by **;
- X 1 is N
- R 1 is selected from C 1-3 alkyl (e.g. methyl), pyrrolidinyl (e.g. pyrrolidin-1-yl) and C 1-3 alkoxy (e.g. ethoxy);
- R 2 is a methyl-substituted pyrazolyl (e.g. 5-methyl-1H-pyrazol-3-yl or 1-methyl-1H-pyrazol-4-yl), a cyclopropyl-substituted pyrazolyl ( For example, 5-cyclopropyl-1H-pyrazol-3-yl) or -C(O)CH 3 ;
- R 3 and R 4 do not exist
- L is -CH 2 -, -CH(CH 3 )-, -O-, -C(O)-, -C(O)NH-or
- R 5 is optionally substituted by one or more selected from halogen (e.g. fluorine or chlorine), CN, C 1-3 alkyl (e.g. methyl or ethyl), C 1-3 haloalkyl (e.g. trifluoromethyl) ), C 1-3 alkoxy (e.g. methoxy or ethoxy), C 3-6 cycloalkyl (e.g. cyclopropyl), C 3-6 cycloalkoxy (e.g.
- halogen e.g. fluorine or chlorine
- CN C 1-3 alkyl (e.g. methyl or ethyl), C 1-3 haloalkyl (e.g. trifluoromethyl) ), C 1-3 alkoxy (e.g. methoxy or ethoxy), C 3-6 cycloalkyl (e.g. cyclopropyl), C 3-6 cycloalkoxy (e.g.
- 5-6 membered heteroaryl for example pyridyl, pyrrolyl, pyrazolyl, furyl, oxazolyl, imidazolyl or thiazolyl
- the 5-6 membered heteroaryl group is optionally further substituted by one or more selected from halogen (e.g. fluorine or chlorine), C 1-3 alkyl (e.g. methyl, ethyl or isopropyl), C 1 -3 haloalkyl (e.g. fluoromethyl), C 1-3 hydroxyalkyl (e.g.
- the present invention covers any combination of the above embodiments.
- the compounds of the invention include, but are not limited to:
- the compound of formula I-A can be synthesized by the method shown in Route A as follows:
- Hal 1 and Hal 2 are each independently F, Cl, Br or I; preferably, Hal 1 is F, Cl, Br or I, and Hal 2 is Cl, Br or I;
- R 1 is selected from H, cyano, C 1-6 alkyl, C 1-6 heteroalkyl (e.g. C 1-6 alkoxy), C 3-8 cycloalkyl, 4-6 membered heterocyclyl and -NR 20a R 20b , the alkyl, heteroalkyl (for example, alkoxy), cycloalkyl and heterocyclic groups are each optionally substituted with one or more substituents selected from the group consisting of halogen, CN, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 haloalkoxy and C 1-4 heteroalkyl (e.g. C 1-4 alkoxy);
- R 2 is selected from the group consisting of C 1-6 alkyl, C 1-6 heteroalkyl, C 3-8 cycloalkyl, 4-6 membered heterocyclyl and 5-6 membered heteroaryl, said alkyl, heteroalkyl Group, cycloalkyl, heterocyclyl and heteroaryl are each optionally substituted with one or more substituents selected from the group consisting of hydroxy, halogen, CN, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 hydroxyalkyl, C 1-4 haloalkoxy, C 1-4 heteroalkyl and C 3-6 cycloalkyl;
- R 23a is selected from H, C 1-6 alkyl, C 1-6 alkoxy and C 3-8 cycloalkyl, each of which is optionally substituted by one or more Substitution selected from the following substituents: OH, CN, halogen, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 hydroxyalkyl, C 1-4 haloalkyl, and C 1-4 haloalkoxy base;
- R 20a and R 20b are as defined in Formula I above;
- R 5 is as defined in Formula IA above.
- the first step Compound IA-1 and R 2 -NH 2 undergo a substitution or coupling reaction (such as Buchwald reaction, Suzuki reaction or Ullmann reaction) in the presence of a base to produce compound IA-2.
- a substitution or coupling reaction such as Buchwald reaction, Suzuki reaction or Ullmann reaction
- usable bases are, for example, t BuONa, t BuOK, t BuOLi, Cs 2 CO 3 , DIPEA, LiHMDS, LDA, NaHMDS, KHMDS, K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3
- usable solvents are, for example, tert-butanol, toluene, xylene, THF, DME, 1,4-dioxane, DMF, DMSO or NMP, and the reaction temperature is 40°C to 140°C.
- usable catalysts are, for example, Pd(OAc) 2 , Pd 2 (dba) 3 , Pd(dba) 2 , PdCl 2 , Pd(PPh 3 ) 4 , Pd(dppf)Cl 2 , Pd(acac) 2 or Pd(allyl) 2
- the usable ligands are PPh 3 , XPhos, SPhos, RuPhos, XantPhos, Dppf, BINOL, BINAP or Pcy 3, etc.
- the usable bases are for example t BuONa, t BuOK, t BuOLi, Cs 2 CO 3 , LiHMDS, LDA, NaHMDS, KHMDS, K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3
- usable solvents are for example toluene, xylene, THF, DME, 1, 4-Dioxane, DMF, DMSO or NMP, and the reaction temperature is 40°C to 140°C.
- the usable catalyst is, for example, Pd(PPh 3 ) 4 or Pd(dppf)Cl 2
- the usable base is, for example, Cs 2 CO 3 , K 3 PO 4 , Na 2 CO 3 , AcOK, NaHCO 3 or K 2 CO 3
- the usable solvent is, for example, 1,4-dioxane/H 2 O, DMF/H 2 O, DMSO/H 2 O or CH 3 CN/H 2 O, and the reaction temperature is 60° C. To 120°C;
- the catalyst that can be used is, for example, CuCl, CuBr, CuI, or Cu 2 O
- the ligand that can be used is, for example, salicylaldoxime, cyclohexanediamine, N,N'-dimethylethylenediamine, TMEDA or ethyl Diamine
- usable bases are, for example, t BuONa, t BuOK, t BuOLi, Cs 2 CO 3 , LiHMDS, LDA, NaHMDS, KHMDS, K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3
- the usable solvent is, for example, toluene, xylene, THF, DME, 1,4-dioxane, DMF, DMSO or NMP, and the reaction temperature is 40°C to 140°C.
- Usable bases are, for example, t BuONa, t BuOK, t BuOLi, Cs 2 CO 3 , DIPEA, LiHMDS, LDA, NaHMDS, KHMDS, K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3 .
- Usable solvents are, for example, tert-butanol, toluene, xylene, THF, DME, 1,4-dioxane, DMF, DMSO or NMP.
- the reaction temperature is 40°C to 140°C.
- the third step Compound I-A-5 reacts with boron-containing reagent to produce compound I-A-6.
- the boron-containing reagent that can be used is, for example, B 2 (pin) 2 .
- Usable catalysts are, for example, Pd(PPh 3 ) 4 , Pd(dppf)Cl 2 or Pd(dppf) 2 Cl 2 ⁇ DCM.
- Usable bases are, for example, Cs 2 CO 3 , K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3.
- Usable solvents are, for example, 1,4-dioxane, DMF, DMSO, or CH 3 CN.
- the reaction temperature is 50°C to 120°C.
- the fourth step Compound I-A-2 and I-A-6 undergo a coupling reaction (such as Suzuki reaction) to produce compound I-A-7.
- a coupling reaction such as Suzuki reaction
- Usable catalysts are, for example, Pd(PPh 3 ) 4 , Pd(dppf)Cl 2 or Pd(dppf) 2 Cl 2 ⁇ DCM.
- Usable bases are, for example, Cs 2 CO 3 , K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3 .
- the usable solvent is, for example, 1,4-dioxane, DMF, DMSO, or CH 3 CN, or a mixture of any of the above solvents and H 2 O.
- the reaction temperature is 50°C to 120°C.
- Step 5 Compound I-A-7 is deprotected under acidic conditions to produce compound I-A-8.
- Usable acids are, for example, a 1,4-dioxane solution of HCl, an EA solution of HCl, or a DCM solution of TFA.
- the reaction temperature is 0°C to 80°C.
- Step 6 Compound I-A-8 and I-A-9 undergo reductive amination reaction to produce compound I-A.
- the usable solvent is, for example, methanol, ethanol, THF, DCM, DCE, DMA, or a mixture of them and acetic acid in any ratio.
- the reducing agent that can be used is, for example, NaBH 4 , NaBH 3 CN, or NaBH(OAc) 3 .
- the reaction temperature is 0°C to 80°C.
- the reaction may be performed in the presence of a base or an acid, such as TEA or DIPEA, and the acid, such as AcOH, HCl, or Ti(O i Pr) 4 .
- the compound of Formula I-A can be synthesized by the method shown in Route B as follows:
- Hal 2 , R 1 , R 2 , R 5 and R 23a are as defined in Route A above.
- the first step Compound I-A-5 is deprotected under acidic conditions to produce compound I-A-10.
- Usable acids are, for example, a 1,4-dioxane solution of HCl, an EA solution of HCl, or a DCM solution of TFA.
- the reaction temperature is 0°C to 80°C.
- the second step Compound I-A-10 and I-A-9 undergo reductive amination to produce compound I-A-11.
- Usable bases are, for example, DIPEA or TEA.
- the reducing agent that can be used is, for example, NaBH 3 CN or NaBH(OAc) 3 .
- Usable solvents are, for example, MeOH, EtOH or DCE.
- the reaction temperature is 0°C to 80°C.
- the usable solvent is, for example, methanol, ethanol, THF, DCM, DCE, DMA, or a mixture of them and acetic acid in any ratio.
- the reducing agent that can be used is, for example, NaBH 4 , NaBH 3 CN, or NaBH(OAc) 3 .
- the reaction temperature is 0°C to 80°C.
- the reaction may be performed in the presence of a base or an acid, such as TEA or DIPEA, and the acid, such as AcOH, HCl, or Ti(O i Pr) 4 .
- the third step Compound I-A-11 reacts with boron-containing reagent to produce compound I-A-12.
- the boron-containing reagent that can be used is, for example, B 2 (pin) 2 .
- Usable catalysts are, for example, Pd(PPh 3 ) 4 , Pd(dppf)Cl 2 or Pd(dppf) 2 Cl 2 ⁇ DCM.
- Usable bases are, for example, Cs 2 CO 3 , K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3 .
- Usable solvents are, for example, 1,4-dioxane, DMF, DMSO, or CH 3 CN.
- the reaction temperature is 50°C to 120°C.
- the fourth step Compound I-A-12 and I-A-2 undergo a coupling reaction (such as Suzuki reaction) to produce compound I-A.
- a coupling reaction such as Suzuki reaction
- Usable catalysts are, for example, Pd(PPh 3 ) 4 , Pd(dppf)Cl 2 or Pd(dppf) 2 Cl 2 ⁇ DCM.
- Usable bases are, for example, Cs 2 CO 3 , K 3 PO 4 , Na 2 CO 3 , KOAc, NaHCO 3 or K 2 CO 3 .
- the usable solvent is, for example, 1,4-dioxane, DMF, DMSO, or CH 3 CN, or a mixture of any of the above solvents and H 2 O.
- the reaction temperature is 50°C to 120°C.
- the compound of formula I-B can be synthesized by the method shown in Route C as follows:
- Hal 1 , Hal 2 , R 1 , R 2 , R 5 , and R 23a are as defined in Route A above.
- the first step Compound IB-1 and R 2 -NH 2 undergo a substitution or coupling reaction (for example, Buchwald reaction, Suzuki reaction, Ullmann reaction) in the presence of a base to produce compound IB-2.
- a substitution or coupling reaction for example, Buchwald reaction, Suzuki reaction, Ullmann reaction
- reaction conditions are as described in the first step in Route A for the preparation of compounds of formula I-A.
- Second step Compound I-B-2 and I-A-12 undergo a coupling reaction (such as Suzuki reaction) to produce compound I-B.
- a coupling reaction such as Suzuki reaction
- reaction conditions are as described in the fourth step of Route A for the preparation of compounds of formula I-A.
- compounds of formula I-C can be synthesized by the method shown in Route D as follows:
- Hal 1 , Hal 2 , R 1 , R 2 , R 5 and R 23a are as defined in Route A above;
- X 1 is selected from CH and N.
- the first step Compound IC-1 and R 2 -NH 2 undergo a substitution or coupling reaction (such as Buchwald reaction, Suzuki reaction or Ullmann reaction) in the presence of a base to produce compound IC-2.
- a substitution or coupling reaction such as Buchwald reaction, Suzuki reaction or Ullmann reaction
- reaction conditions are as described in the first step in Route A for the preparation of compounds of formula I-A.
- the second step Compound I-C-3 reacts with boron-containing reagent to produce compound I-C-4.
- reaction conditions are as described in the third step of Route A for the preparation of compounds of formula I-A.
- the third step Compound I-C-2 and I-C-4 undergo a coupling reaction (such as Suzuki reaction) to produce compound I-C-5.
- a coupling reaction such as Suzuki reaction
- reaction conditions are as described in the fourth step of Route A for the preparation of compounds of formula I-A.
- Step 4 Compound I-C-5 is deprotected under acidic conditions to produce compound I-C-6.
- reaction conditions are as described in the fifth step in Route A for the preparation of compounds of formula I-A.
- Step 5 Compound I-C-6 and I-A-9 undergo reductive amination to produce compound I-C.
- reaction conditions are as described in the sixth step of Route A for the preparation of compounds of formula I-A.
- compounds of formula I-D can be synthesized by the method shown in Route E as follows:
- R 1 and R 2 are as defined in Route A above;
- R 5 is defined as the above formula ID
- R 23a and R 23b are each independently selected from H, C 1-6 alkyl, C 1-6 alkoxy and C 3-8 cycloalkyl; or R 23a and R 23b together with the C atom to which they are attached form 3-8 membered cycloalkyl or heterocyclic group, each of said alkyl, alkoxy, cycloalkyl and heterocyclic group is optionally substituted with one or more substituents selected from the following: CN, halogen, C 1-4 alkyl, C 1-4 alkoxy, C 1-4 hydroxyalkyl, C 1-4 haloalkyl and C 1-4 haloalkoxy;
- X 1 is selected from CH and N;
- t 0 or 1.
- Compound I-C-6 and I-D-1 undergo condensation reaction to produce compound I-D.
- the condensing agent that can be used is, for example, HATU, CDI, HOBt, DMAP, DCC, DIC, EDC, HBTU, HCTU or PyBOP.
- Usable bases are, for example, TEA, DIPEA, t BuOK, t BuONa, t BuOLi, NaH, NaOH, Cs 2 CO 3 , K 3 PO 4 or Na 2 CO 3 .
- Usable solvents are, for example, THF, DCM, DCE, MeOH, EtOH, DMF, DMSO, acetone, CH 3 CN, 1,4-dioxane or toluene.
- the reaction temperature is 0°C to 120°C, such as room temperature.
- compound ID-1 first reacts with an acylating reagent to form an acid halide, and then reacts with compound IC-6 optionally in the presence of a base to form a compound of formula ID.
- the acylating agent that can be used is, for example, thionyl chloride or oxalyl chloride.
- the reaction can also be carried out under a small amount of DMF catalysis.
- the base that can be used is, for example, TEA or DIPEA.
- Usable solvents are, for example, THF, DCM, DCE, CH 3 CN, 1,4-dioxane, or toluene.
- the reaction temperature is 0°C to 100°C.
- compounds of formula I-E can be synthesized by the method shown in Route F as follows:
- R 1 , R 2 , R 5 , R 23a , R 23b and t are as defined in Route E above;
- X 1 is selected from CH and N;
- Hal 2 is F, Cl, Br or I; preferably, Hal 2 is Cl, Br or I.
- the first step Compound I-C-2 and I-A-6 undergo a coupling reaction (such as Suzuki reaction) to produce compound I-E-1.
- a coupling reaction such as Suzuki reaction
- reaction conditions are as described in the fourth step of Route A for the preparation of compounds of formula I-A.
- reaction conditions are as described in the fifth step in Route A for the preparation of compounds of formula I-A.
- the third step Compound I-E-2 and I-D-1 undergo condensation reaction to produce compound I-E.
- compounds of formula I-F can be synthesized by the method shown in Route G as follows:
- R 1 , R 2 , R 23a , R 23b and R 5 are as defined in Route E above;
- X 1 is selected from CH and N;
- R 4 is absent or selected from hydroxy, CN, C 1-6 alkyl, C 1-6 haloalkyl, and C 1-6 heteroalkyl (for example, C 1-6 alkoxy);
- R 23c is H, C 1-3 alkyl or C 1-3 alkoxy, each of said alkyl and alkoxy is optionally substituted by one or more substituents selected from the group consisting of OH, CN, halogen , C 1-4 alkoxy and C 1-4 hydroxyalkyl;
- Hal 2 is F, Cl, Br or I; preferably, Hal 2 is Cl, Br or I;
- u is 0 or 1
- n 0 or 1.
- the first step Compound I-C-2 and I-F-1 undergo a coupling reaction (such as Suzuki reaction) to produce compound I-F-2.
- a coupling reaction such as Suzuki reaction
- reaction conditions are as described in the fourth step of Route A for the preparation of compounds of formula I-A.
- the second step Compound I-F-3 and amine undergo condensation reaction to produce compound I-F-4.
- reaction conditions are as described in the fifth step in Route A for the preparation of compounds of formula I-A.
- the fourth step Compound I-F-2 and I-F-5 undergo nucleophilic substitution reaction in the presence of a base to form compound I-F.
- reaction conditions are as described in the second step of Route A for the preparation of compounds of formula I-A.
- compounds of formula I-G can be synthesized by the method shown in Route H as follows:
- R 1 and R 2 are as defined in Route A above;
- X 1 is selected from CH and N;
- R 4 is selected from H, C 1-6 alkyl, C 1-6 haloalkyl and C 1-6 heteroalkyl;
- R 5 is as defined in formula IG above.
- n 0 or 1.
- the first step compound IG-1 and R 5 -OH react with Mitsunobu to produce compound IG-2.
- the reaction reagent that can be used is, for example, PPh 3 , PMe 3 , DIAD, DEAD or DBAD.
- Solvents that can be used are aprotic solvents such as THF, ether, DCM, DMF or toluene.
- the reaction temperature is -20°C to 100°C, such as room temperature.
- the acid that can be used is, for example, a 1,4-dioxane solution of HCl, an EA solution of HCl, or a DCM solution of TFA, or a reaction between the aforementioned acid solution and any solvent selected from the group consisting of THF, MeOH, and EtOH. In a mixture.
- the reaction temperature is 0°C to 80°C.
- the third step Compound I-F-2 and I-G-3 undergo a nucleophilic substitution reaction in the presence of a base to form compound I-G.
- reaction conditions are as described in the second step of Route A for the preparation of compounds of formula I-A.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a preventive or therapeutically effective amount of a compound of the present invention, a stereoisomer, a tautomer of the compound, or a mixture thereof, and N- Oxides, pharmaceutically acceptable salts, co-crystals, polymorphs or solvates of the compounds, or stable isotopic derivatives, metabolites or prodrugs of the compounds.
- the pharmaceutical composition further comprises one or more pharmaceutically acceptable carriers.
- the present invention provides pharmaceutical preparations, which are preferably solid preparations, semi-solid preparations, liquid preparations or gaseous preparations.
- the pharmaceutical composition may also include one or more other therapeutic agents.
- the pharmaceutical composition or pharmaceutical formulation is preferably administered by oral, intravenous, intraarterial, subcutaneous, intraperitoneal, intramuscular or transdermal route.
- the present invention provides a compound of the present invention, a stereoisomer, a tautomer or a mixture of the compound, an N-oxide of the compound, and a pharmaceutically acceptable compound of the compound Salts, co-crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of said compounds, or pharmaceutical compositions as described above, or pharmaceutical preparations of the present invention are prepared for prevention Or use in drugs for treating diseases or conditions related to RET activity.
- the present invention provides a compound of the present invention, a stereoisomer, a tautomer or a mixture of the compound, an N-oxide of the compound, and a pharmaceutically acceptable compound of the compound Salts, co-crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of said compounds, or pharmaceutical compositions as described above, or pharmaceutical preparations of the present invention are prepared for regulation (For example, to reduce or inhibit) the use of RET activity in medicine.
- the present invention provides a compound of the present invention, a stereoisomer, a tautomer or a mixture of the compound, an N-oxide of the compound, and a pharmaceutically acceptable compound of the compound Salts, co-crystals, polymorphs or solvates, or stable isotope derivatives, metabolites or prodrugs of said compounds, or pharmaceutical compositions as described above, or pharmaceutical preparations of the present invention, which are used for prevention Or to treat diseases or conditions related to RET activity.
- the present invention provides a method of preventing or treating diseases or conditions associated with RET activity, the method comprising administering to an individual in need thereof an effective amount of a compound of the present invention, a stereoisomer of the compound , Tautomers or mixtures thereof, N-oxides of the compounds, pharmaceutically acceptable salts, co-crystals, polymorphs or solvates of the compounds, or stable isotope derivatives of the compounds A substance, metabolite or prodrug, or a pharmaceutical composition as described above, or a pharmaceutical preparation of the present invention.
- the disease or condition associated with RET activity is preferably cancer or tumor, or irritable bowel syndrome.
- the cancer or tumor is further preferably lung cancer (such as non-small cell lung cancer), breast cancer, head and neck cancer, rectal cancer, liver cancer, lymphoma, thyroid cancer (such as medullary thyroid cancer or papillary thyroid cancer) ), colon cancer, multiple myeloma, melanoma, glioma, brain tumor or sarcoma.
- lung cancer such as non-small cell lung cancer
- breast cancer head and neck cancer
- rectal cancer liver cancer
- lymphoma such as medullary thyroid cancer or papillary thyroid cancer
- thyroid cancer such as medullary thyroid cancer or papillary thyroid cancer
- colon cancer multiple myeloma, melanoma, glioma, brain tumor or sarcoma.
- pharmaceutically acceptable carrier refers to a diluent, adjuvant, excipient or vehicle administered with the therapeutic agent, and it is suitable for contact with humans and/or within the scope of reasonable medical judgment Tissues of other animals without excessive toxicity, irritation, allergic reactions, or other problems or complications corresponding to a reasonable benefit/risk ratio.
- the pharmaceutically acceptable carrier that can be used in the pharmaceutical composition of the present invention includes, but is not limited to, sterile liquid. Examples of suitable pharmaceutically acceptable carriers are described in Remington's Pharmaceutical Sciences (1990).
- the pharmaceutical composition of the present invention can act systemically and/or locally. For this purpose, they can be administered by a suitable route.
- the pharmaceutical composition of the present invention can be administered in a suitable dosage form.
- an effective amount refers to the amount of a compound that will relieve one or more symptoms of the condition being treated to a certain extent after being administered.
- the dosage regimen can be adjusted to provide the best desired response. For example, a single bolus can be administered, several divided doses can be administered over time, or the dose can be proportionally reduced or increased as indicated by the urgent need for the treatment situation. It should be noted that the dose value may vary with the type and severity of the condition to be alleviated, and may include single or multiple doses. It should be further understood that for any particular individual, the specific dosing regimen should be adjusted over time according to the needs of the individual and the professional judgment of the person administering the composition or supervising the administration of the composition.
- the amount of the compound of the present invention administered will depend on the individual being treated, the severity of the disorder or condition, the rate of administration, the treatment of the compound, and the judgment of the prescribing physician. Generally, the effective dose is about 0.0001 to about 50 mg per kg body weight per day. In some cases, a dose level not higher than the lower limit of the aforementioned range may be sufficient, while in other cases, a larger dose can still be used without causing any harmful side effects, provided that the larger The dose is divided into several smaller doses to be administered throughout the day.
- the content or amount of the compound of the present invention in the pharmaceutical composition may be about 0.01 mg to about 1000 mg.
- treating means reversing, alleviating, or inhibiting the disease or condition to which such term is applied or the progression of one or more symptoms of such disease or condition, or Preventing such a disorder or condition or one or more symptoms of such a disorder or condition.
- “Individual” as used herein includes human or non-human animals.
- Exemplary human individuals include human individuals (referred to as patients) or normal individuals suffering from diseases such as the diseases described herein.
- “non-human animals” include all vertebrates, such as non-mammals (such as birds, amphibians, reptiles) and mammals, such as non-human primates, livestock and/or domesticated animals (such as sheep, dogs). , Cats, cows, pigs, etc.).
- the pharmaceutical composition of the present invention may also include one or more additional therapeutic or preventive agents (for example, other drugs used to treat cancer or tumor diseases).
- the methods of the present invention may also include the administration of one or more additional therapeutic or preventive agents (e.g., other drugs used to treat cancer or tumor diseases).
- the compound of the present invention is separated and purified by preparative TLC, silica gel column chromatography, Prep-HPLC and/or flash column chromatography (Flash column chromatography), and its structure is confirmed by 1 H NMR and/or MS.
- the reaction monitoring is carried out by TLC or LC-MS.
- the 1 H NMR spectroscopy method uses a Bruker superconducting nuclear magnetic resonance spectrometer (model AVACE III HD 400MHz).
- LC/MS adopts Aglient 1260Infinity/Aglient 6120Quadrupole.
- TLC uses silica gel GF 254 as the stationary phase.
- Fast column chromatography uses Biotage fast column chromatography.
- Prep-HPLC adopts Agilent 1260, Waters 2489 and GeLai 3500 chromatographs.
- the microwave reaction was performed using a BiotageInitiator microwave reactor.
- reaction temperature is room temperature (15-30°C).
- the reagents used in this application are purchased from companies such as Acros Organics, Aldrich Chemical Company, or Terbo Chemical.
- the fifth step 2-(6-(6-benzyl-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-( Preparation of 5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 1)
- the second step 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1 .1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 17)
- the fourth step 2-(6-(4-((6-methoxypyridin-3-yl)methyl)piperazin-1-yl)pyridin-3-yl)-6-methyl-N-( Preparation of 5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 49)
- Example 18 6-methyl-2-(6-(6-((6-(4-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6- Diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 62)
- Step 1 Preparation of 1-(4-(1,3-dioxolane-2-yl)phenyl)-4-methyl-1H-pyrazole (Compound 88c)
- Example 32 2-(6-(6-((6-(3,4-Dimethyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazide Heterobicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 121)
- Step 1 Preparation of 2-(3,4-Dimethyl-1H-pyrazol-1-yl)-5-(1,3-dioxolane-2-yl)pyridine (Compound 121c)
- the third step 2-(6-(6-((6-(3,4-dimethyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazide Heterobicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 121) Preparation
- the first step Preparation of 5-(1,3-dioxolane-2-yl)-2-(4-fluoro-1H-imidazol-1-yl)pyridine (compound 64b)
- the third step 2-(6-(6-((6-(4-fluoro-1H-imidazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1. 1) Preparation of heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 64)
- Example 36 2-(6-(6-(1-(6-(4-Fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 52)
- Step 1 Preparation of tert-butyl 4-((6-methylpyridin-3-yl)oxy)piperidine-1-carboxylate (Compound 119b)
- Step 1 Preparation of 1-(4-(1,3-dioxolane-2-yl)phenyl)-3-fluoro-1H-pyrazole (compound 89b)
- the third step 2-(6-(6-(4-(3-fluoro-1H-pyrazol-1-yl)benzyl)-3,6-diazabicyclo[3.1.1]heptane-3 -Yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 89)
- Example 40 6'-(6-((6-(4-Fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1 ]Heptan-3-yl)-4-methyl-N-(5-methyl-1H-pyrazol-3-yl)-[2,3'-bipyridine]-6-amine (Compound 6)
- Step 2 Preparation of 3-((6-bromo-4-methylpyridin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylic acid tert-butyl ester (Compound 6d)
- Example 41 2-(6-(6-((6'-methoxy-[2,3'-bipyridyl]-5-yl)methyl)-3,6-diazabicyclo[3.1. 1)Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 7)
- Step 1 Preparation of 5-(1,3-dioxolane-2-yl)-6'-methoxy-2,3'-bipyridine (compound 7b)
- the third step 2-(6-(6-((6'-methoxy-[2,3'-bipyridyl]-5-yl)methyl)-3,6-diazabicyclo[3.1. 1) Preparation of heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 7)
- Example 43 6-methyl-2-(6-(6-((6-(5-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6- Diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 9)
- the first step preparation of 2-chloro-N-(5-cyclopropyl-1H-pyrazol-3-yl)-6-methylpyrimidin-4-amine (compound 10c)
- the fourth step 6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3-(5-(4,4,5,5-tetramethyl Preparation of yl-1,3,2-dioxaborolan-2-yl)pyridin-2-yl)-3,6-diazabicyclo[3.1.1]heptane (compound 10f)
- the first step preparation of 2-(3,5-dimethyl-1H-pyrazol-1-yl)-5-(1,3-dioxolane-2-yl)pyridine (compound 11b)
- the third step 2-(6-(6-((6-(3,5-dimethyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazide Heterobicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 11)
- Step 1 Preparation of 5-(1,3-dioxolane-2-yl)-2-(5-isopropyl-1H-pyrazol-1-yl)pyridine (Compound 12b)
- the third step 2-(6-(6-((6-(5-isopropyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 12)
- Instrument model Biotage Isolera Prime 2.3.1, Chromatographic Column: Agela Technologies C18 spherical 20-35um 100A, 12g; Chromatographic column temperature: 25°C; Flow rate: 15.0mL/min; Detection wavelength: 254nm; Elution gradient: (0min: 0% A, 100% B; 3.0 min: 0% A, 100% B; 20 min: 80% A, 20% B); mobile phase A: 100% acetonitrile; mobile phase B: 0.5% ammonium bicarbonate aqueous solution; compound Collection time: 10.4min-11.8min.
- Instrument model Biotage Isolera Prime 2.3.1, Chromatographic Column: Agela Technologies C18 spherical 20-35um 100A, 12g; Chromatographic column temperature: 25°C; Flow rate: 15.0mL/min; Detection wavelength: 254nm; Elution gradient: (0min: 0% A, 100% B; 3.0 min: 0% A, 100% B; 20 min: 80% A, 20% B); mobile phase A: 100% acetonitrile; mobile phase B: 0.5% ammonium bicarbonate aqueous solution; compound Collection time: 11.4min-12.8min.
- Example 48 (6-(4-Fluoro-1H-pyrazol-1-yl)pyridin-3-yl)(3-(5-(4-methyl-6-((5-methyl-1H- (Pyrazol-3-yl)amino)pyrimidin-2-yl)pyridin-2-yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methanone (Compound 26)
- the third step (6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)(3-(5-(4-methyl-6-((5-methyl-1H- (Pyrazol-3-yl)amino)pyrimidin-2-yl)pyridin-2-yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methanone (Compound 26)
- Step 4 Preparation of 2-(5-cyclobutoxy-1H-pyrazol-1-yl)-5-(1,3-dioxolane-2-yl)pyridine (compound 27f)
- Example 50 2-(6-(6-((6-(4-chloro-3-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-di Azabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine and 2-(6-(6-((6-(4-chloro-5-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[ 3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 28/compound 28')
- the first step 2-(4-chloro-3-methyl-1H-pyrazol-1-yl)-5-(1,3-dioxolane-2-yl)pyridine and 2-(4-chloro Preparation of -5-methyl-1H-pyrazol-1-yl)-5-(1,3-dioxolan-2-yl)pyridine (compound 28b/compound 28b')
- the second step 6-(4-chloro-3-methyl-1H-pyrazol-1-yl)nicotinaldehyde and 6-(4-chloro-5-methyl-1H-pyrazol-1-yl) Preparation of nicotine aldehyde (compound 28c/compound 28c')
- the third step 2-(6-(6-((6-(4-chloro-3-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-di Azabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine and 2-(6-(6-((6-(4-chloro-5-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[ 3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 28/compound 28') Preparation
- Example 52 2-(6-(6-((R)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6- Diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine And 2-(6-(6-((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6-diazepine Bicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 52 -1/Compound 52-2)
- the third step Preparation of 2-(5-cyclopropoxy-1H-pyrazol-1-yl)-5-(1,3-dioxolane-2-yl)pyridine (compound 30d)
- Example 54 2-(6-(6-((6-(3-(fluoromethyl)-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazide Heterobicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 31)
- the third step Preparation of 5-(1,3-dioxolane-2-yl)-2-(3-(fluoromethyl)-1H-pyrazol-1-yl)pyridine (compound 31d)
- the fifth step 2-(6-(6-((6-(3-(fluoromethyl)-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazide Heterobicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 31)
- Step 4 Preparation of 5-(1,3-dioxolane-2-yl)-2-(5-ethyl-1H-pyrazol-1-yl)pyridine (Compound 46d)
- the sixth step 2-(6-(6-((6-(5-ethyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[ 3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 46) preparation
- Example 56 2-(6-(6-((6-(4-Fluoro-5-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-di Azabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine ( Compound 122)
- the first step the preparation of 4-fluoro-1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazole (compound 122a)
- Step 4 Preparation of 5-(1,3-dioxolane-2-yl)-2-(4-fluoro-5-methyl-1H-pyrazol-1-yl)pyridine (Compound 122d)
- the sixth step 2-(6-(6-((6-(4-fluoro-5-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-di Azabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine ( Compound 122) Preparation
- the third step 2-(6-(6-((6-(1H-pyrrol-2-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane -3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 123)
- Example 58 2-(6-(6-((6-(3-cyclopropyl-4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6- Diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 124)
- Step 2 Preparation of 5-(1,3-dioxolan-2-yl)-2-(4-fluoro-3-iodo-1H-pyrazol-1-yl)pyridine (Compound 124b)
- Step 4 Preparation of 6-(3-cyclopropyl-4-fluoro-1H-pyrazol-1-yl)nicotinaldehyde (Compound 124d)
- the fifth step 2-(6-(6-((6-(3-cyclopropyl-4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6- Diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 124)
- Example 59 2-(6-(6-((R)-1-(6-(1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine and 2-( 6-(6-((S)-1-(6-(1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6-diazabicyclo[3.1.1]heptane -3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 125-1/Compound 125-2)
- the third step 2-(6-(6-((R)-1-(6-(1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine and 2-( 6-(6-((S)-1-(6-(1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-3,6-diazabicyclo[3.1.1]heptane -3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 125-1/Compound 125-2) Preparation
- Compound 125 (250 mg) was resolved by chiral Prep-HPLC to obtain compound 125-1 (retention time 7.647 min) and compound 125-2 (retention time 11.638 min). The two compounds were differentiated and defined according to the retention time of chiral resolution, and the stereo structure was not identified. Thus, compound 125-1 (101 mg, ee: 100%), MS m/z (ESI): 533.9 [M+H] + ; compound 125-2 (100 mg, ee: 99.93%), MS m/z ( ESI): 533.9 [M+H] + .
- Example 60 2-(6-(6-((6-(4-fluoromethyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 126) And (1-(5-((3-(5-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)pyridine-2- (Yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methyl)pyridin-2-yl)-1H-pyrazol-4-yl)methanol (Compound 127)
- the first step Preparation of 1-(5-(1,3-dioxolane-2-yl)pyridin-2-yl)-1H-pyrazole-4-carboxylic acid ethyl ester (compound 126b)
- Step 2 Preparation of 1-(5-(1,3-dioxolane-2-yl)pyridin-2-yl)-1H-pyrazol-4-yl)methanol (Compound 126c)
- the fifth step 2-(6-(6-((6-(4-fluoromethyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 126) And (1-(5-((3-(5-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)pyridine-2- (Yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methyl)pyridin-2-yl)-1H-pyrazol-4-yl)methanol (Compound 127)
- Example 61 6-Methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-(6-(6-((6-(1-methyl-1H-pyrrole-2 -Yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrimidin-4-amine (compound 128)
- the fifth step 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-(6-(6-((6-(1-methyl-1H-pyrrole-2 -Yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrimidin-4-amine (compound 128)
- Example 62 6-Methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-(6-(6-((6-(thiazol-4-yl)pyridine-3- (Yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrimidin-4-amine (compound 129)
- Step 1 Preparation of 4-(5-(1,3-dioxolane-2-yl)pyridin-2-yl)thiazole (compound 129b)
- the third step 6-methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-(6-(6-((6-(thiazol-4-yl)pyridine-3- (Yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrimidin-4-amine (compound 129)
- Step 1 Preparation of 5-(1,3-dioxolane-2-yl)-2,2'-bipyridine (compound 130b)
- Example 64 2-(6-(6-((6-(5-Fluoro-1H-pyrrol-2-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1. 1)Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 131)
- Step 1 Preparation of 6-(5-fluoro-1H-pyrrol-2-yl)nicotinaldehyde (compound 131a)
- the second step 2-(6-(6-((6-(5-fluoro-1H-pyrrol-2-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1. 1) Preparation of heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 131)
- Example 65 2-(6-(6-((6-(1H-pyrazol-5-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]hepta Alkyl-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 132)
- Step 1 Preparation of 6-(1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-5-yl)nicotinaldehyde (compound 132b)
- the third step 2-(6-(6-((6-(1H-pyrazol-5-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan Alkyl-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 132)
- Example 66 6-Methyl-N-(5-methyl-1H-pyrazol-3-yl)-2-(6-(6-((6-(5-methylfuran-2-yl) (Pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrimidin-4-amine (compound 133)
- Step 1 Preparation of 2-(5-(1,3-dioxolane-2-yl)pyridin-2-yl)oxazole (compound 134b)
- Example 68 2-(6-(6-((6-(1-isopropyl-1H-pyrazol-4-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 135)
- Step 1 Preparation of 5-(1,3-dioxolane-2-yl)-2-(1H-pyrazol-4-yl)pyridine (compound 135b)
- Step 2 Preparation of 5-(1,3-dioxolane-2-yl)-2-(1-isopropyl-1H-pyrazol-4-yl)pyridine (Compound 135d)
- the fourth step 2-(6-(6-((6-(1-isopropyl-1H-pyrazol-4-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo [3.1.1]Heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 135)
- Example 69 2-(1-(5-((3-(5-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl )Pyridin-2-yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methyl)pyridin-2-yl)-1H-pyrazol-5-yl)propan-2 -Alcohol (Compound 136)
- Step 2 Preparation of 6-(3-(2-hydroxyprop-2-yl)-1H-pyrazol-1-yl)nicotinaldehyde (Compound 136b)
- the third step 2-(1-(5-((3-(5-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl )Pyridin-2-yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methyl)pyridin-2-yl)-1H-pyrazol-5-yl)propan-2 -Preparation of alcohol (compound 136)
- Example 70 (1-(5-((3-(5-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)pyridine -2-yl)-3,6-diazabicyclo[3.1.1]heptane-6-yl)methyl)pyridin-2-yl)-1H-pyrazol-3-yl)methanol (compound 137)
- Step 1 Preparation of 6-(3-(hydroxymethyl)-1H-pyrazol-1-yl)nicotinaldehyde (compound 137a)
- Example 71 2-(6-(6-((6-(4-Fluoro-3-methyl-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-di Azabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine ( Compound 138)
- Relative inhibitory activity percentage 1-(compound groups of different concentrations-blank control) / (negative control-blank control) * 100%
- y is the relative inhibitory activity percentage
- max and min are the maximum and minimum values of the fitted curve
- x is the logarithmic concentration of the compound
- Hillslope is the slope of the curve.
- the inhibitory effect of the compound of the present invention on the enzyme activity of VEGFR2 was determined according to the instructions of HTRF KinEASE-TK kit (Cisbio). After pre-incubating the VEGFR2 enzyme with test compounds of different concentrations for 30 min at room temperature, the substrate and adenosine triphosphate (ATP) were added to initiate the reaction. After incubating at room temperature for 40 min, TK antibody-cryptate and streptavidin-XL665 were added, and the detection was performed after incubating at room temperature for 45 min. With the vehicle group (DMSO) as the negative control and the buffer group (without VEGFR2 enzyme) as the blank control, the relative inhibitory activity percentages (ie, the inhibition rate) of different concentrations of compounds were calculated according to the following formula:
- Relative inhibitory activity percentage 1-(compound groups of different concentrations-blank control) / (negative control-blank control) * 100%
- y is the relative inhibitory activity percentage
- max and min are the maximum and minimum values of the fitted curve
- x is the logarithmic concentration of the compound
- Hillslope is the slope of the curve.
- N/A means not tested.
- Compound number RET-V804M inhibition rate Compound number RET-V804M inhibition rate 6(10nM) 83% 84(100nM) 72% 7(10nM) 46% 85(100nM) 77% 8(10nM) 46% 86(10nM) 64% 15(100nM) 91% 88(10nM) 62% 16(100nM) 92% 89(10nM) 70% 18(100nM) 70% 91(10nM) 70% 21(100nM) 49% 96(100nM) 84% 23(10nM) 47% 98(100nM) 63% 26(10nM) 83% 110(100nM) 62% 27(10nM) 87% 111(100nM) 46% 28'(10nM) 34% 117(100nM) 76% 29(10nM) 52% 118(100nM) 74% 30(10nM) 88% 125-2(10nM) 82% 31(10nM) 81% 126(100n
- Compound number VEGFR2 inhibition rate Compound number VEGFR2 inhibition rate 1(100nM) 38% 70(100nM) 14% 2(100nM) 29% 80(100nM) -10% 3(100nM) 14% 82(100nM) 15% 4(1000nM) 52% 83(100nM) 18% 6(100nM) -11% 86(100nM) 52% 7(100nM) 16% 88(100nM) 32% 8(300nM) 5% 89(100nM) 16% 9(300nM) 69% 91(100nM) 26% 10(300nM) 69% 96(100nM) 4% 12(300nM) 62% 119(100nM) -7% 15(100nM) 29% 120(100nM) 28% 23(100nM) -10% 121(100nM) 37%
- BLU-667 prepared according to Example 5 of WO2017/079140A1
- compound 17 were administered to male SD rats by intragastric (PO), respectively, and the plasma concentration of BLU-667 and compound 17 in rats and the concentration of brain, lung and thyroid tissues were measured , To investigate the pharmacokinetic characteristics.
- the dose of PO administration is 5 mg/kg, and the vehicle is 0.5% MC (methyl cellulose).
- PO was administered, and blood samples were collected at different time points (0h before administration, 0.25h, 0.5h, 1h, 2h, 4h, 6h, 8h and 24h after administration).
- the blood samples were anticoagulated with dipotassium ethylenediaminetetraacetate, and the plasma samples were obtained after centrifugation and stored at -80°C.
- Rats were sacrificed by bleeding from the abdominal aorta at 0.5h, 2h, 8h and 24h after PO administration, and brain, lung and thyroid were collected. After washing, homogenized with saline in a certain proportion to obtain tissue samples, which were stored in- 80°C. Each plasma sample and tissue sample were processed with precipitated protein and then analyzed by LC-MS/MS. Using WinNonlin 6.3 software, the non-compartmental model was used to calculate the pharmacokinetic parameters. The results are shown in Table 5.
- composition Compound 17 and BLU-667 were dissolved in 0.1M H 3 PO 4 aqueous solution and 0.1M HOAc aqueous solution, respectively, to prepare a clear solution (pH about 4.0).
- the vehicle control group used an aqueous H 3 PO 4 solution with a pH of about 4.0.
- the mode of administration is PO, BID.
- Tumor measurement Measure the diameter of the tumor with a vernier caliper twice a week.
- TGI tumor growth inhibition rate
Abstract
Description































































































































| 化合物编号 | 对RET-V804M的抑制率 | 对RET-M918T的抑制率 |
| 1 | 86% | 33% |
| 2 | 93% | 51% |
| 3 | 86% | 34% |
| 4 | N/A | 69% |
| 化合物编号 | RET-V804M抑制率 | 化合物编号 | RET-V804M抑制率 |
| 6(10nM) | 83% | 84(100nM) | 72% |
| 7(10nM) | 46% | 85(100nM) | 77% |
| 8(10nM) | 46% | 86(10nM) | 64% |
| 15(100nM) | 91% | 88(10nM) | 62% |
| 16(100nM) | 92% | 89(10nM) | 70% |
| 18(100nM) | 70% | 91(10nM) | 70% |
| 21(100nM) | 49% | 96(100nM) | 84% |
| 23(10nM) | 47% | 98(100nM) | 63% |
| 26(10nM) | 83% | 110(100nM) | 62% |
| 27(10nM) | 87% | 111(100nM) | 46% |
| 28'(10nM) | 34% | 117(100nM) | 76% |
| 29(10nM) | 52% | 118(100nM) | 74% |
| 30(10nM) | 88% | 125-2(10nM) | 82% |
| 31(10nM) | 81% | 126(100nM) | 82% |
| 41(10nM) | 64% | 127(100nM) | 84% |
| 46(10nM) | 92% | 128(10nM) | 48% |
| 49(100nM) | 75% | 129(10nM) | 84% |
| 50(100nM) | 50% | 130(10nM) | 44% |
| 63(10nM) | 65% | 131(10nM) | 72% |
| 64(10nM) | 79% | 132(10nM) | 84% |
| 67(100nM) | 82% | 133(10nM) | 82% |
| 69(10nM) | 56% | 134(10nM) | 50% |
| 80(10nM) | 51% | 135(10nM) | 65% |
| 82(10nM) | 43% | 137(10nM) | 93% |
| 83(10nM) | 56% | - | - |
| 化合物编号 | 对RET-WT的抑制IC 50(nM) |
| 17 | 1.32±0.31 |
| 60 | 2.70±0.69 |
| 120 | 7.33±1.19 |
| 122 | 2.35±0.24 |
| 化合物编号 | 对RET-CCDC6的抑制IC 50(nM) |
| 17 | 2.50±0.55 |
| 60 | 3.99±0.79 |
| 69 | 10.97±9.65 |
| 120 | 18.66±7.27 |
| 122 | 2.91±0.41 |
| 化合物编号 | 对RET-V804L抑制IC 50(nM) |
| 17 | 6.54±2.43 |
| 60 | 5.07±0.40 |
| 120 | 10.09±1.05 |
| 122 | 4.79±1.68 |
| 化合物编号 | 对V804M抑制IC 50(nM) |
| 9 | 5.43±0.91 |
| 10 | 3.97±0.30 |
| 11 | 7.22±0.58 |
| 12 | 6.94±0.78 |
| 17 | 1.03±0.47 |
| 25 | 6.29±0.90 |
| 28 | 10.41±1.38 |
| 52-2 | 12.95±2.34 |
| 60 | 3.77±0.52 |
| 62 | 4.53±1.23 |
| 70 | 41.36±3.73 |
| 119 | 14.21±1.27 |
| 120 | 9.12±1.85 |
| 121 | 6.22±1.39 |
| 122 | 3.33±0.75 |
| 123 | 1.88±0.11 |
| 124 | 10.68±0.98 |
| 136 | 2.35±0.13 |
| 化合物编号 | 对RET-M918T酶的抑制IC 50(nM) |
| 17 | 1.47±0.29 |
| 60 | 1.29±0.27 |
| 69 | 8.60±1.46 |
| 120 | 15.81±3.65 |
| 122 | 4.03±0.66 |
| 化合物编号 | VEGFR2抑制率 | 化合物编号 | VEGFR2抑制率 |
| 1(100nM) | 38% | 70(100nM) | 14% |
| 2(100nM) | 29% | 80(100nM) | -10% |
| 3(100nM) | 14% | 82(100nM) | 15% |
| 4(1000nM) | 52% | 83(100nM) | 18% |
| 6(100nM) | -11% | 86(100nM) | 52% |
| 7(100nM) | 16% | 88(100nM) | 32% |
| 8(300nM) | 5% | 89(100nM) | 16% |
| 9(300nM) | 69% | 91(100nM) | 26% |
| 10(300nM) | 69% | 96(100nM) | 4% |
| 12(300nM) | 62% | 119(100nM) | -7% |
| 15(100nM) | 29% | 120(100nM) | 28% |
| 23(100nM) | -10% | 121(100nM) | 37% |
| 25(300nM) | 53% | 122(300nM) | 67% |
| 26(300nM) | 64% | 123(30nM) | 56% |
| 27(30nM) | 23% | 124(30nM) | 41% |
| 28(300nM) | 23% | 125(300nM) | 44% |
| 29(300nM) | 47% | 125-2(300nM) | 63% |
| 30(30nM) | 51% | 128(100nM) | 20% |
| 31(30nM) | 17% | 129(30nM) | 52% |
| 41(100nM) | 22% | 130(100nM) | 63% |
| 46(300nM) | 79% | 131(30nM) | 50% |
| 52(100nM) | -5% | 132(30nM) | 31% |
| 52-2(300nM) | 12% | 133(50nM) | 28% |
| 60(100nM) | 65% | 134(100nM) | 11% |
| 62(100nM) | 62% | 135(100nM) | 34% |
| 63(100nM) | 38% | 136(30nM) | 18% |
| 64(100nM) | 38% | 137(30nM) | 26% |
| 69(100nM) | 2% | - | - |

Claims (12)
- 化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中所述化合物具有式I的结构:
 其中:环A选自C 6-10芳环和5-6元杂芳环;环B选自C 3-8环烷基和4-11元杂环基;X 1选自CH和N;R 1选自H、卤素、羟基、氰基、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)、C 3-8环烷基、4-10元杂环基和-NR 20aR 20b,所述烷基、杂烷基(例如烷氧基)、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基和C 1-4杂烷基(例如C 1-4烷氧基);R 2选自C 1-6烷基、C 1-6杂烷基、C 3-8环烷基、4-10元杂环基、5-10元杂芳基和-C(=O)R 21,所述烷基、杂烷基、环烷基、杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基和C 3-6环烷基;R 3和R 4不存在或者在每次出现时各自独立地选自羟基、卤素、CN、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)和C 3-6环烷基,所述烷基、杂烷基(例如烷氧基)和环烷基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基和C 1-4卤代烷氧基;当m大于1时,两个R 3任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;和/或当n大于1时,两个R 4任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;L选自-O-、-S-、-S(O)-、-S(O) 2-、-N=CR 21-、-N(R 23a)-C(O)-、C 1-6亚烷基、C 1-6亚杂烷基、C 2-6亚烯基、C 2-6亚炔基、
其中:环A选自C 6-10芳环和5-6元杂芳环;环B选自C 3-8环烷基和4-11元杂环基;X 1选自CH和N;R 1选自H、卤素、羟基、氰基、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)、C 3-8环烷基、4-10元杂环基和-NR 20aR 20b,所述烷基、杂烷基(例如烷氧基)、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基和C 1-4杂烷基(例如C 1-4烷氧基);R 2选自C 1-6烷基、C 1-6杂烷基、C 3-8环烷基、4-10元杂环基、5-10元杂芳基和-C(=O)R 21,所述烷基、杂烷基、环烷基、杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基和C 3-6环烷基;R 3和R 4不存在或者在每次出现时各自独立地选自羟基、卤素、CN、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)和C 3-6环烷基,所述烷基、杂烷基(例如烷氧基)和环烷基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基和C 1-4卤代烷氧基;当m大于1时,两个R 3任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;和/或当n大于1时,两个R 4任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;L选自-O-、-S-、-S(O)-、-S(O) 2-、-N=CR 21-、-N(R 23a)-C(O)-、C 1-6亚烷基、C 1-6亚杂烷基、C 2-6亚烯基、C 2-6亚炔基、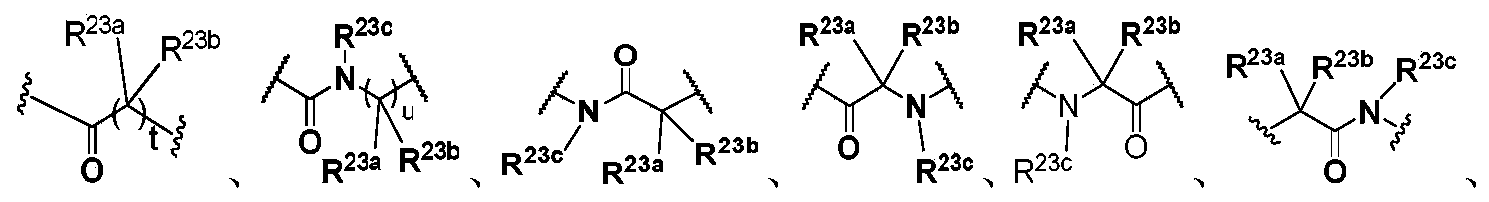
 所述亚烷基、亚杂烷基、亚烯基和亚炔基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-6烷基、C 1-6卤代烷基、C 1-6羟烷基、C 1-6卤代烷氧基、C 1-6杂烷基(例如C 1-6烷氧基)和C 3-8环烷基;或者L为-N(R 23a)-;
所述亚烷基、亚杂烷基、亚烯基和亚炔基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-6烷基、C 1-6卤代烷基、C 1-6羟烷基、C 1-6卤代烷氧基、C 1-6杂烷基(例如C 1-6烷氧基)和C 3-8环烷基;或者L为-N(R 23a)-; R 5选自羟基、卤素、CN、NO 2、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)、C 2-6烯基、C 2-6炔基、C 3-8环烷基、C 3-8环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 20aR 20b、-OR 21、-SR 21、 -S(=O)R 22、-S(=O) 2R 22、-S(=O)NR 20aR 20b、-S(=O) 2NR 20aR 20b、-NR 20aS(=O)R 20b、-NR 20aS(=O) 2R 20b、-C(=O)R 21、-C(=O)NR 23aR 23b、-NR 23aC(=O)R 23b、-OC(=O)NR 23aR 23b和-NR 24aC(=O)NR 25aR 25b,所述烷基、杂烷基(例如烷氧基)、烯基、炔基、环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基、C 3-6环烷氧基和4-10元杂环基;R 20a、R 20b、R 23a、R 23b、R 23c、R 24a、R 25a和R 25b各自独立地选自H、OH、C 1-6烷基、C 1-6烷氧基和C 3-8环烷基;或者R 20a与R 20b、R 23a与R 23b或R 25a与R 25b连同其所连接的原子一起形成3-8元环烷基或杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、NO 2、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;R 30a、R 30b、R 33a、R 33b、R 34a、R 35a和R 35b各自独立地选自H、C 1-6烷基、C 1-6卤代烷基、C 1-6羟烷基、C 1-6烷氧基和C 1-6卤代烷氧基;R 21、R 22、R 31和R 32各自独立地选自C 1-6烷基、C 1-6烷氧基、C 3-8环烷基、4-10元杂环基、C 6-12芳基和5-10元杂芳基,所述烷基、烷氧基、环烷基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:OH、卤素、CN、C 1-4烷基、C 1-4烷氧基、C 1-4卤代烷基、C 1-4卤代烷氧基、C 3-6环烷基和4-10元杂环基;m为0、1、2、3或4,优选为0;n为0、1、2、3或4,优选为0、1或2;t为0、1、2、3或4,优选为0或1;且u为0、1、2、3或4,优选为0或1;条件是,当环B为哌嗪环且X 1为CH时,R 2不是4-CF 3-吡啶-2-基或4-CN-吡啶-2-基。
R 5选自羟基、卤素、CN、NO 2、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)、C 2-6烯基、C 2-6炔基、C 3-8环烷基、C 3-8环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 20aR 20b、-OR 21、-SR 21、 -S(=O)R 22、-S(=O) 2R 22、-S(=O)NR 20aR 20b、-S(=O) 2NR 20aR 20b、-NR 20aS(=O)R 20b、-NR 20aS(=O) 2R 20b、-C(=O)R 21、-C(=O)NR 23aR 23b、-NR 23aC(=O)R 23b、-OC(=O)NR 23aR 23b和-NR 24aC(=O)NR 25aR 25b,所述烷基、杂烷基(例如烷氧基)、烯基、炔基、环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基、C 3-6环烷氧基和4-10元杂环基;R 20a、R 20b、R 23a、R 23b、R 23c、R 24a、R 25a和R 25b各自独立地选自H、OH、C 1-6烷基、C 1-6烷氧基和C 3-8环烷基;或者R 20a与R 20b、R 23a与R 23b或R 25a与R 25b连同其所连接的原子一起形成3-8元环烷基或杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、NO 2、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;R 30a、R 30b、R 33a、R 33b、R 34a、R 35a和R 35b各自独立地选自H、C 1-6烷基、C 1-6卤代烷基、C 1-6羟烷基、C 1-6烷氧基和C 1-6卤代烷氧基;R 21、R 22、R 31和R 32各自独立地选自C 1-6烷基、C 1-6烷氧基、C 3-8环烷基、4-10元杂环基、C 6-12芳基和5-10元杂芳基,所述烷基、烷氧基、环烷基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:OH、卤素、CN、C 1-4烷基、C 1-4烷氧基、C 1-4卤代烷基、C 1-4卤代烷氧基、C 3-6环烷基和4-10元杂环基;m为0、1、2、3或4,优选为0;n为0、1、2、3或4,优选为0、1或2;t为0、1、2、3或4,优选为0或1;且u为0、1、2、3或4,优选为0或1;条件是,当环B为哌嗪环且X 1为CH时,R 2不是4-CF 3-吡啶-2-基或4-CN-吡啶-2-基。 - 权利要求1所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中:环A为苯环或5-6元杂芳环;优选地,环A为苯环、噻唑环、吡啶环、吡嗪环或嘧啶环;更优选地,环A为其通过*标记的位置与X 1所在环连接,并且通过**标记的位置与环B连接;
 和/或环B为C 3-6环烷基或5-7元杂环基;优选地,环B为哌啶环、哌嗪环、氮杂环庚烷桥环或二氮杂环庚烷桥环;更优选地,环B为其通过*标记的位置与环A连接,并且通过**标记的位置与L连接;
和/或环B为C 3-6环烷基或5-7元杂环基;优选地,环B为哌啶环、哌嗪环、氮杂环庚烷桥环或二氮杂环庚烷桥环;更优选地,环B为其通过*标记的位置与环A连接,并且通过**标记的位置与L连接; 和/或X 1为CH或N,优选为N。
和/或X 1为CH或N,优选为N。 - 权利要求1或2所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中:R 1选自H、卤素、羟基、氰基、C 1-4烷基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元 杂环基,所述烷基、杂烷基(例如烷氧基)、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基和C 1-4杂烷基(例如C 1-4烷氧基);优选地,R 1选自C 1-4烷基、5元含氮杂环基和C 1-4杂烷基(例如C 1-4烷氧基),所述烷基、杂环基和杂烷基(例如烷氧基)各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基和C 1-3杂烷基(例如C 1-3烷氧基);更优选地,R 1选自C 1-3烷基(例如甲基)、吡咯烷基(例如吡咯烷-1-基)和C 1-3烷氧基(例如乙氧基);和/或R 2选自C 1-4烷基、C 1-4杂烷基、C 3-6环烷基、4-6元杂环基、5-6元杂芳基和-C(=O)R 21,所述烷基、杂烷基、环烷基、杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基和C 3-6环烷基;优选地,R 2选自C 1-3烷基、5-6元杂芳基和-C(=O)CH 3,所述烷基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3杂烷基和C 3-6环烷基;更优选地,R 2选自C 1-3烷基(例如甲基)、-C(=O)CH 3、噻吩基、吡咯基、吡唑基、咪唑基、噻唑基、噻二唑基、异噻唑基、噁唑基、噁二唑基、异噁唑基和吡啶基,所述烷基、噻吩基、吡咯基、吡唑基、咪唑基、噻唑基、噻二唑基、异噻唑基、噁唑基、噁二唑基、异噁唑基和吡啶基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基(例如甲基)、C 1-3卤代烷基、C 1-3卤代烷氧基、C 1-3杂烷基(例如C 1-3烷氧基)和C 3-6环烷基;进一步优选地,R 2为甲基取代的吡唑基(例如5-甲基-1H-吡唑-3-基或1-甲基-1H-吡唑-4-基)、环丙基取代的吡唑基(例如5-环丙基-1H-吡唑-3-基)或-C(O)CH 3。
- 权利要求1至3中任意一项所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中:R 3和R 4不存在或者在每次出现时独立地选自羟基、卤素、CN、C 1-4烷基和C 1-4烷氧基,所述烷基和烷氧基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4烷氧基和C 1-4卤代烷氧基;当m大于1时,两个R 3任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;和/或当n大于1时,两个R 4任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;优选地,R 3和R 4不存在或者在每次出现时独立地选自羟基、卤素、CN、C 1-3烷基和C 1-3烷氧基,所述烷基和烷氧基各自任选地被一个或多个选自下列的取代基取代:卤素、CN和C 1-3烷基;当m大于1时,两个R 3任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;和/或当n大于1时,两个R 4任选地与其所连接的原子一起形成C 3-6环烷基或4-10元杂环基;更优选地,R 3和R 4不存在或者在每次出现时独立地选自:F、Cl、CN、OH、C 1-3烷基和C 1-3烷氧基;进一步优选地,R 3和R 4不存在。
- 权利要求1至4中任意一项所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中:L选自-O-、-S-、-C(O)-、-N(R 23a)-C(O)-、-C(O)-N(R 23c)-、C 1-4亚烷基、C 1-4亚杂烷基、
 所述亚烷基和亚杂烷基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)和C 3-6环烷基;
所述亚烷基和亚杂烷基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)和C 3-6环烷基; 优选地,L选自-O-、-C(O)-、-NHC(O)-、-C(O)NH-、C 1-3亚烷基、C 1-3亚杂烷基、
优选地,L选自-O-、-C(O)-、-NHC(O)-、-C(O)NH-、C 1-3亚烷基、C 1-3亚杂烷基、 所述亚烷基和亚杂烷基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3杂烷基(例如C 1-3烷氧基)和C 3-6环烷基,其中R 23a和R 23b优选为H或C 1-3烷基;
所述亚烷基和亚杂烷基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3杂烷基(例如C 1-3烷氧基)和C 3-6环烷基,其中R 23a和R 23b优选为H或C 1-3烷基; 更优选地,L选自-O-、-C(O)-、-NHC(O)-、-C(O)NH-、C 1-3亚烷基、
更优选地,L选自-O-、-C(O)-、-NHC(O)-、-C(O)NH-、C 1-3亚烷基、 所述亚烷基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基和C 1-3卤代烷基;
所述亚烷基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基和C 1-3卤代烷基; 进一步优选地,L为-CH 2-、-CH(CH 3)-、-O-、-C(O)-、-C(O)NH-或
进一步优选地,L为-CH 2-、-CH(CH 3)-、-O-、-C(O)-、-C(O)NH-或

- 权利要求1至5中任意一项所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中:R 5选自羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 20aR 20b、-OR 21、-SR 21、-S(=O)R 22、-S(=O) 2R 22、-S(=O)NR 20aR 20b、-S(=O) 2NR 20aR 20b、-NR 20aS(=O)R 20b、-NR 20aS(=O) 2R 20b、-C(=O)R 21、-C(=O)NR 23aR 23b、-NR 23aC(=O)R 23b、-OC(=O)NR 23aR 23b和-NR 24aC(=O)NR 25aR 25b,所述烷基、杂烷基(例如烷氧基)、烯基、炔基、环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基、C 3-6环烷氧基和4-10元杂环基;优选地,R 5选自C 3-6环烷基、4-10元杂环基、C 6-12芳基和5-10元杂芳基,所述环烷基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1- 4烷氧基)、C 3-6环烷基、C 3-6环烷氧基和4-10元杂环基;更优选地,R 5选自C 6-10芳基和5-6元杂芳基,所述芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3杂烷基(例如C 1-3烷氧基)、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-C(=O)R 31、-C(=O)NR 33aR 33b和-NR 33aC(=O)R 33b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基、C 3-6环烷氧基和4-6元杂环基;进一步优选地,R 5选自苯基和5-6元杂芳基(例如吡啶基、嘧啶基、吡嗪基、哒嗪基、吡唑基、 噁唑基、咪唑基或噻唑基),所述苯基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3杂烷基(例如C 1-3烷氧基)、C 3-6环烷基、C 3-6环烷氧基、4-6元杂环基、5-8元杂芳基(例如吡啶基、吡咯基、吡唑基、呋喃基、噁唑基、咪唑基、噻唑基或环戊基并吡唑基)、-NR 30aR 30b、-OR 31、-C(=O)R 31、-C(=O)NR 33aR 33b和-NR 33aC(=O)R 33b,其中所述环烷基、环烷氧基、杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3杂烷基(例如C 1-3烷氧基)、C 3-6环烷基、C 3-6环烷氧基和4-6元杂环基;进一步更优选地,R 5选自苯基、吡啶基、吡唑基和噻唑基,所述苯基、吡啶基、吡唑基和噻唑基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3烷氧基、C 3-6环烷基、C 3-6环烷氧基、4-6元杂环基、5-8元杂芳基(例如吡啶基、吡咯基、吡唑基、呋喃基、噁唑基、咪唑基、噻唑基或环戊基并吡唑基)、-NR 30aR 30b和-OR 31,其中所述杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-3烷基、C 1-3卤代烷基、C 1-3羟烷基、C 1-3卤代烷氧基、C 1-3烷氧基、C 3-6环烷基、C 3-6环烷氧基和4-6元杂环基;最优选地,R 5为任选地被一个或多个选自卤素(例如氟或氯)、CN、C 1-3烷基(例如甲基或乙基)、C 1-3卤代烷基(例如三氟甲基)、C 1-3烷氧基(例如甲氧基或乙氧基)、C 3-6环烷基(例如环丙基)、C 3-6环烷氧基(例如环丙氧基)和5-6元杂芳基(例如吡啶基、吡咯基、吡唑基、呋喃基、噁唑基、咪唑基或噻唑基)的取代基取代的苯基、吡啶基、吡唑基或噻唑基,其中所述5-6元杂芳基任选地进一步被一个或多个选自卤素(例如氟或氯)、C 1-3烷基(例如甲基、乙基或异丙基)、C 1-3卤代烷基(例如氟甲基)、C 1- 3羟烷基(例如羟甲基或羟丙基)、C 1-3烷氧基(例如甲氧基)、C 3-6环烷基(例如环丙基)和C 3-6环烷氧基(例如环丙氧基或环丁氧基)的取代基取代。
- 权利要求1至6中任意一项所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中:R 20a、R 20b、R 23a、R 23b、R 23c、R 24a、R 25a和R 25b各自独立地选自H、C 1-4烷基、C 1-4烷氧基和C 3- 8环烷基;或者R 20a与R 20b、R 23a与R 23b或R 25a与R 25b连同其所连接的原子一起形成3-8元环烷基或杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、NO 2、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;优选地,R 20a、R 20b、R 23a、R 23b、R 23c、R 24a、R 25a和R 25b各自独立地为H、C 1-4烷基或C 1-4烷氧基;特别地,R 23a和R 23b各自独立地选自H、C 1-3烷基、C 1-3烷氧基和C 3-6环烷基;或者R 23a与R 23b连同其所连接的C原子一起形成C 3-6环烷基或杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:卤素、C 1-3烷基、C 1-3烷氧基、C 1-3羟烷基、C 1-3卤代烷基和C 1-3卤代烷氧基;和/或R 21、R 22、R 31和R 32各自独立地选自C 1-4烷基、C 1-4烷氧基、C 3-8环烷基和4-10元杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:OH、卤素、CN、C 1-4烷基、C 1-4烷氧基、C 1-4卤代烷基、C 1-4卤代烷氧基、C 3-6环烷基和4-10元杂环基;优选地,R 21、R 22、R 31和R 32各自独立地选自C 1-4烷基;和/或R 30a、R 30b、R 33a、R 33b、R 34a、R 35a和R 35b各自独立地选自H、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4烷氧基和C 1-4卤代烷氧基;优选地,R 30a、R 30b、R 33a、R 33b、R 34a、R 35a和R 35b各自独立地选自H和C 1-4烷基。
- 权利要求1所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药,其中所述化合物具有式I-A至式I-G之一所示的结构:
 其中:R 5选自C 6-12芳基和5-10元杂芳基,其中(1)所述C 6-12芳基任选地被一个或多个选自下列的取代基取代:C 3-6环烷氧基、C 6-12芳基、5-10元杂芳基、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷氧基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基,且(2)所述5-10元杂芳基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基C 3-6环烷氧基和4-10元杂环基;且R 1、R 2、R 23a、R 30a、R 30b、R 31、R 32、R 33a、R 33b、R 34a、R 35a和R 35b如权利要求1-7中任意一项所定义,且R 23a优选为H或C 1-3烷基;
其中:R 5选自C 6-12芳基和5-10元杂芳基,其中(1)所述C 6-12芳基任选地被一个或多个选自下列的取代基取代:C 3-6环烷氧基、C 6-12芳基、5-10元杂芳基、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷氧基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基,且(2)所述5-10元杂芳基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基C 3-6环烷氧基和4-10元杂环基;且R 1、R 2、R 23a、R 30a、R 30b、R 31、R 32、R 33a、R 33b、R 34a、R 35a和R 35b如权利要求1-7中任意一项所定义,且R 23a优选为H或C 1-3烷基; 其中:R 1、R 2、R 5和R 23a如权利要求1-7中任一项所定义,且R 23a优选为H或C 1-3烷基;
其中:R 1、R 2、R 5和R 23a如权利要求1-7中任一项所定义,且R 23a优选为H或C 1-3烷基; 其中:当X 1为CH时,R 1、R 2、R 5和R 23a如权利要求1-7中任意一项所定义,且R 23a优选为H或C 1- 3烷基;而当X 1为N时,R 1、R 2、R 5和R 23a如上文式I-A所定义;
其中:当X 1为CH时,R 1、R 2、R 5和R 23a如权利要求1-7中任意一项所定义,且R 23a优选为H或C 1- 3烷基;而当X 1为N时,R 1、R 2、R 5和R 23a如上文式I-A所定义; 其中:R 1、R 2、R 23a、R 23b和t如权利要求1-7中任意一项所定义;当X 1为CH时,R 5如权利要求1-7中任意一项所定义;且当X 1为N时,R 5为C 6-12芳基或5-10元杂芳基,其中(i)当t为0时,所述C 6-12芳基和5-10元杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基,(ii)当t为1时,(1)所述C 6-12芳基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基,且(2)所述5-10元杂芳基任选地被一个或多个选自下列的取代基取代:NO 2、C 2-6烯基、C 2-6炔基、C 3-6环烷氧基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基;且R 30a、R 30b、R 31、R 32、R 33a、R 33b、R 34a、R 35a和R 35b如权利要求1-7中任意一项所定义;
其中:R 1、R 2、R 23a、R 23b和t如权利要求1-7中任意一项所定义;当X 1为CH时,R 5如权利要求1-7中任意一项所定义;且当X 1为N时,R 5为C 6-12芳基或5-10元杂芳基,其中(i)当t为0时,所述C 6-12芳基和5-10元杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基,(ii)当t为1时,(1)所述C 6-12芳基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基,且(2)所述5-10元杂芳基任选地被一个或多个选自下列的取代基取代:NO 2、C 2-6烯基、C 2-6炔基、C 3-6环烷氧基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基;且R 30a、R 30b、R 31、R 32、R 33a、R 33b、R 34a、R 35a和R 35b如权利要求1-7中任意一项所定义; 其中:R 1、R 2、R 5、R 23a、R 23b、X 1和t如上文式I-D所定义;
其中:R 1、R 2、R 5、R 23a、R 23b、X 1和t如上文式I-D所定义; 其中:R 1、R 2、R 5、R 23a、R 23b和X 1如上文式I-D所定义;R 4如权利要求1-7中任意一项所定义,且优选为C 1-3烷基或C 1-3烷氧基;R 23c为H、C 1-3烷基或C 1-3烷氧基,所述烷基和烷氧基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、C 1-4烷氧基和C 1-4羟烷基;u为0或1;且n为0或1;
其中:R 1、R 2、R 5、R 23a、R 23b和X 1如上文式I-D所定义;R 4如权利要求1-7中任意一项所定义,且优选为C 1-3烷基或C 1-3烷氧基;R 23c为H、C 1-3烷基或C 1-3烷氧基,所述烷基和烷氧基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、C 1-4烷氧基和C 1-4羟烷基;u为0或1;且n为0或1; 其中:X 1为CH或N;R 1、R 2和R 4如权利要求1-7中任意一项所定义,且R 4优选为C 1-3烷基或C 1-3烷氧基;n为0或1;R 5选自C 6-12芳基和5-10元杂芳基,其中(1)所述C 6-12芳基任选地被一个或多个选自下列的取代基取代:C 3-6环烷氧基、C 6-12芳基、5-10元杂芳基、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷氧基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基;且(2)所述5-10元杂芳基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基;且R 30a、R 30b、R 31、R 32、R 33a、R 33b、R 34a、R 35a和R 35b如权利要求1-7中任意一项所定义。
其中:X 1为CH或N;R 1、R 2和R 4如权利要求1-7中任意一项所定义,且R 4优选为C 1-3烷基或C 1-3烷氧基;n为0或1;R 5选自C 6-12芳基和5-10元杂芳基,其中(1)所述C 6-12芳基任选地被一个或多个选自下列的取代基取代:C 3-6环烷氧基、C 6-12芳基、5-10元杂芳基、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷氧基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基;且(2)所述5-10元杂芳基任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 2-6烯基、C 2-6炔基、C 3-6环烷基、C 3-6环烷氧基、4-10元杂环基、C 6-12芳基、5-10元杂芳基、-NR 30aR 30b、-OR 31、-SR 31、-S(=O)R 32、-S(=O) 2R 32、-S(=O)NR 30aR 30b、-S(=O) 2NR 30aR 30b、-NR 30aS(=O)R 30b、-NR 30aS(=O) 2R 30b、-C(=O)R 31、-C(=O)NR 33aR 33b、-NR 33aC(=O)R 33b、-OC(=O)NR 33aR 33b和-NR 34aC(=O)NR 35aR 35b,其中所述环烷基、环烷氧基、杂环基、芳基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、NO 2、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基(例如C 1-4烷氧基)、C 3-6环烷基和4-10元杂环基;且R 30a、R 30b、R 31、R 32、R 33a、R 33b、R 34a、R 35a和R 35b如权利要求1-7中任意一项所定义。 - 权利要求1所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳 定同位素衍生物、代谢物或前药,其中所述化合物选自:




- 制备权利要求8的化合物的方法,所述方法包括以下步骤:
 其中:Hal 1和Hal 2各自独立地为F、Cl、Br或I;优选地,Hal 1为F、Cl、Br或I,且Hal 2为Cl、Br或I;R 1选自H、氰基、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)、C 3-8环烷基、4-6元杂环基和-NR 20aR 20b,所述烷基、杂烷基(例如烷氧基)、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4卤代烷氧基和C 1-4杂烷基(例如C 1-4烷氧基);R 2选自C 1-6烷基、C 1-6杂烷基、C 3-8环烷基、4-6元杂环基和5-6元杂芳基,所述烷基、杂烷基、环烷基、杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基和C 3-6环烷基;R 23a选自H、C 1-6烷基、C 1-6烷氧基和C 3-8环烷基,所述烷基、烷氧基和环烷基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;且R 20a、R 20b和R 5如权利要求8式I-A所定义;第一步:化合物I-A-1与R 2-NH 2在碱存在下反应生成化合物I-A-2;第二步:化合物I-A-3与I-A-4在碱存在下反应生成化合物I-A-5;第三步:化合物I-A-5与含硼试剂反应生成化合物I-A-6;第四步:化合物I-A-2与I-A-6反应生成化合物I-A-7;第五步:化合物I-A-7在酸性条件下脱除保护基生成化合物I-A-8;第六步:化合物I-A-8与I-A-9反应生成化合物I-A;或者,所述方法包括以下步骤:
其中:Hal 1和Hal 2各自独立地为F、Cl、Br或I;优选地,Hal 1为F、Cl、Br或I,且Hal 2为Cl、Br或I;R 1选自H、氰基、C 1-6烷基、C 1-6杂烷基(例如C 1-6烷氧基)、C 3-8环烷基、4-6元杂环基和-NR 20aR 20b,所述烷基、杂烷基(例如烷氧基)、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4卤代烷氧基和C 1-4杂烷基(例如C 1-4烷氧基);R 2选自C 1-6烷基、C 1-6杂烷基、C 3-8环烷基、4-6元杂环基和5-6元杂芳基,所述烷基、杂烷基、环烷基、杂环基和杂芳基各自任选地被一个或多个选自下列的取代基取代:羟基、卤素、CN、C 1-4烷基、C 1-4卤代烷基、C 1-4羟烷基、C 1-4卤代烷氧基、C 1-4杂烷基和C 3-6环烷基;R 23a选自H、C 1-6烷基、C 1-6烷氧基和C 3-8环烷基,所述烷基、烷氧基和环烷基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;且R 20a、R 20b和R 5如权利要求8式I-A所定义;第一步:化合物I-A-1与R 2-NH 2在碱存在下反应生成化合物I-A-2;第二步:化合物I-A-3与I-A-4在碱存在下反应生成化合物I-A-5;第三步:化合物I-A-5与含硼试剂反应生成化合物I-A-6;第四步:化合物I-A-2与I-A-6反应生成化合物I-A-7;第五步:化合物I-A-7在酸性条件下脱除保护基生成化合物I-A-8;第六步:化合物I-A-8与I-A-9反应生成化合物I-A;或者,所述方法包括以下步骤: 其中:各基团如上文路线A所定义;第一步:化合物I-A-5在酸性条件下脱除保护基生成化合物I-A-10;第二步:化合物I-A-10与I-A-9反应生成化合物I-A-11;第三步:化合物I-A-11与含硼试剂反应生成化合物I-A-12;第四步:化合物I-A-12与I-A-2反应生成化合物I-A;或者,所述方法包括以下步骤:
其中:各基团如上文路线A所定义;第一步:化合物I-A-5在酸性条件下脱除保护基生成化合物I-A-10;第二步:化合物I-A-10与I-A-9反应生成化合物I-A-11;第三步:化合物I-A-11与含硼试剂反应生成化合物I-A-12;第四步:化合物I-A-12与I-A-2反应生成化合物I-A;或者,所述方法包括以下步骤: 其中:各基团如上文路线A所定义;第一步:化合物I-B-1与R 2-NH 2在碱存在下反应生成化合物I-B-2;第二步:化合物I-B-2与I-A-12反应生成化合物I-B;或者,所述方法包括以下步骤:
其中:各基团如上文路线A所定义;第一步:化合物I-B-1与R 2-NH 2在碱存在下反应生成化合物I-B-2;第二步:化合物I-B-2与I-A-12反应生成化合物I-B;或者,所述方法包括以下步骤: 其中:各基团如上文路线A所定义;且X 1选自CH和N;第一步:化合物I-C-1与R 2-NH 2在碱存在下反应生成化合物I-C-2;第二步:化合物I-C-3与含硼试剂反应生成化合物I-C-4;第三步:化合物I-C-2与I-C-4反应生成化合物I-C-5;第四步:化合物I-C-5在酸性条件下脱除保护基生成化合物I-C-6;第五步:化合物I-C-6与I-A-9反应生成化合物I-C;或者,所述方法包括以下步骤:
其中:各基团如上文路线A所定义;且X 1选自CH和N;第一步:化合物I-C-1与R 2-NH 2在碱存在下反应生成化合物I-C-2;第二步:化合物I-C-3与含硼试剂反应生成化合物I-C-4;第三步:化合物I-C-2与I-C-4反应生成化合物I-C-5;第四步:化合物I-C-5在酸性条件下脱除保护基生成化合物I-C-6;第五步:化合物I-C-6与I-A-9反应生成化合物I-C;或者,所述方法包括以下步骤: 其中:R 1和R 2如路线A所定义;R 5如权利要求8式I-D所定义;R 23a和R 23b各自独立地选自H、C 1-6烷基、C 1-6烷氧基和C 3-8环烷基;或者R 23a与R 23b连同其所连接的C原子一起形成3-8元环烷基或杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:CN、卤素、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;X 1选自CH和N;且t为0或1;或者,所述方法包括以下步骤:
其中:R 1和R 2如路线A所定义;R 5如权利要求8式I-D所定义;R 23a和R 23b各自独立地选自H、C 1-6烷基、C 1-6烷氧基和C 3-8环烷基;或者R 23a与R 23b连同其所连接的C原子一起形成3-8元环烷基或杂环基,所述烷基、烷氧基、环烷基和杂环基各自任选地被一个或多个选自下列的取代基取代:CN、卤素、C 1-4烷基、C 1-4烷氧基、C 1-4羟烷基、C 1-4卤代烷基和C 1-4卤代烷氧基;X 1选自CH和N;且t为0或1;或者,所述方法包括以下步骤: 其中:R 1、R 2、R 5、R 23a、R 23b和t如路线E所定义;X 1选自CH和N;且Hal 2为F、Cl、Br或I;优选地,Hal 2为Cl、Br或I;第一步:化合物I-C-2与I-A-6反应生成化合物I-E-1;第二步:化合物I-E-1在酸性条件下脱除保护基生成化合物I-E-2;第三步:化合物I-E-2与I-D-1经缩合反应生成化合物I-E;或者,所述方法包括以下步骤:
其中:R 1、R 2、R 5、R 23a、R 23b和t如路线E所定义;X 1选自CH和N;且Hal 2为F、Cl、Br或I;优选地,Hal 2为Cl、Br或I;第一步:化合物I-C-2与I-A-6反应生成化合物I-E-1;第二步:化合物I-E-1在酸性条件下脱除保护基生成化合物I-E-2;第三步:化合物I-E-2与I-D-1经缩合反应生成化合物I-E;或者,所述方法包括以下步骤:
 其中:R 1、R 2、R 23a、R 23b和R 5如路线E所定义;X 1选自CH和N;R 4不存在或者选自羟基、CN、C 1-6烷基、C 1-6卤代烷基和C 1-6杂烷基(例如C 1-6烷氧基);R 23c为H、C 1-3烷基或C 1-3烷氧基,所述烷基和烷氧基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、C 1-4烷氧基和C 1-4羟烷基;Hal 2为F、Cl、Br或I;优选地,Hal 2为Cl、Br或I;u为0或1;且n为0或1;第一步:化合物I-C-2与I-F-1反应生成化合物I-F-2;第二步:化合物I-F-3与胺反应生成化合物I-F-4;第三步:化合物I-F-4在酸性条件下脱除保护基生成化合物I-F-5;第四步:化合物I-F-2与I-F-5在碱存在下反应生成化合物I-F;或者,所述方法包括以下步骤:
其中:R 1、R 2、R 23a、R 23b和R 5如路线E所定义;X 1选自CH和N;R 4不存在或者选自羟基、CN、C 1-6烷基、C 1-6卤代烷基和C 1-6杂烷基(例如C 1-6烷氧基);R 23c为H、C 1-3烷基或C 1-3烷氧基,所述烷基和烷氧基各自任选地被一个或多个选自下列的取代基取代:OH、CN、卤素、C 1-4烷氧基和C 1-4羟烷基;Hal 2为F、Cl、Br或I;优选地,Hal 2为Cl、Br或I;u为0或1;且n为0或1;第一步:化合物I-C-2与I-F-1反应生成化合物I-F-2;第二步:化合物I-F-3与胺反应生成化合物I-F-4;第三步:化合物I-F-4在酸性条件下脱除保护基生成化合物I-F-5;第四步:化合物I-F-2与I-F-5在碱存在下反应生成化合物I-F;或者,所述方法包括以下步骤: 其中:R 1和R 2如路线A所定义;X 1选自CH和N;R 4选自H、C 1-6烷基、C 1-6卤代烷基和C 1-6杂烷基;R 5如上文式I-G所定义;且n为0或1;第一步:化合物I-G-1与R 5-OH反应生成化合物I-G-2;第二步:化合物I-G-2在酸性条件下脱除保护基生成化合物I-G-3;第三步:化合物I-F-2与I-G-3在碱存在下经亲核取代反应生成化合物I-G。
其中:R 1和R 2如路线A所定义;X 1选自CH和N;R 4选自H、C 1-6烷基、C 1-6卤代烷基和C 1-6杂烷基;R 5如上文式I-G所定义;且n为0或1;第一步:化合物I-G-1与R 5-OH反应生成化合物I-G-2;第二步:化合物I-G-2在酸性条件下脱除保护基生成化合物I-G-3;第三步:化合物I-F-2与I-G-3在碱存在下经亲核取代反应生成化合物I-G。 - 药物组合物,其包含预防或治疗有效量的权利要求1-9中任意一项所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药;任选地,所述药物组合物还包含一种或多种药学上可接受的载体。
- 权利要求1-9中任意一项所述的化合物,所述化合物的立体异构体、互变异构体或其混合物,所述化合物的N-氧化物,所述化合物的药学上可接受的盐、共晶、多晶型物或溶剂合物,或者所述化合物的稳定同位素衍生物、代谢物或前药、或权利要求11所述的药物组合物在制备用于预防或治疗与RET活性相关的疾病或病况的药物中的用途;优选地,所述与RET活性相关的疾病或病况为癌症或肿瘤,或肠易激综合征;所述癌症或肿瘤进一步优选为肺癌(例如非小细胞肺癌)、乳腺癌、头颈癌、直肠癌、肝癌、淋巴瘤、甲状腺癌(例如甲状腺髓样癌或乳头状甲状腺癌)、结肠癌、多发性骨髓瘤、黑色素瘤、胶质瘤、脑瘤或肉瘤。
Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112021016299-2A BR112021016299A2 (pt) | 2019-02-19 | 2020-02-11 | Composto heterocíclico, composição farmacêutica compreendendo o mesmo, método de preparação do mesmo e uso do mesmo |
| CN202080009954.2A CN113316578B (zh) | 2019-02-19 | 2020-02-11 | 杂环化合物、包含其的药物组合物及其制备方法和用途 |
| JP2021571055A JP2022521859A (ja) | 2019-02-19 | 2020-02-11 | 複素環化合物、これを含む薬物組成物、その製造方法および使用 |
| AU2020226422A AU2020226422A1 (en) | 2019-02-19 | 2020-02-11 | Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereof |
| CA3130245A CA3130245A1 (en) | 2019-02-19 | 2020-02-11 | Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereof |
| MX2021009971A MX2021009971A (es) | 2019-02-19 | 2020-02-11 | Compuesto heterociclico, composicion farmaceutica que lo comprende, metodo para su preparacion, y uso de estos. |
| CN202311264080.7A CN117327078A (zh) | 2019-02-19 | 2020-02-11 | 杂环化合物、包含其的药物组合物及其制备方法和用途 |
| SG11202107848TA SG11202107848TA (en) | 2019-02-19 | 2020-02-11 | Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereof |
| US17/431,930 US20220144847A1 (en) | 2019-02-19 | 2020-02-11 | Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereof |
| EP20760123.8A EP3929198A4 (en) | 2019-02-19 | 2020-02-11 | HETEROCYCLIC COMPOUND, PHARMACEUTICAL COMPOSITION COMPRISING IT, METHOD FOR PREPARING IT AND ITS USE |
| CN202311221283.8A CN117263945A (zh) | 2019-02-19 | 2020-02-11 | 杂环化合物、包含其的药物组合物及其制备方法和用途 |
| KR1020217022806A KR20210131314A (ko) | 2019-02-19 | 2020-02-11 | 헤테로시클릴 화합물, 이를 포함하는 약제학적 조성물, 이의 제조 방법, 및 이의 용도 |
| IL285378A IL285378A (en) | 2019-02-19 | 2021-08-04 | A heterocyclic compound, a medical preparation containing it, a method for its preparation, and its use |
| CONC2021/0011023A CO2021011023A2 (es) | 2019-02-19 | 2021-08-20 | Compuesto heterocíclico, composición farmacéutica que lo comprende, método para su preparación, y uso de estos |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910124584 | 2019-02-19 | ||
| CN201910124584.6 | 2019-02-19 | ||
| CN201910437878 | 2019-05-24 | ||
| CN201910437878.4 | 2019-05-24 | ||
| CN201910932095.3 | 2019-09-29 | ||
| CN201910932095 | 2019-09-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020168939A1 true WO2020168939A1 (zh) | 2020-08-27 |
Family
ID=72143544
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2020/074696 WO2020168939A1 (zh) | 2019-02-19 | 2020-02-11 | 杂环化合物、包含其的药物组合物及其制备方法和用途 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20220144847A1 (zh) |
| EP (1) | EP3929198A4 (zh) |
| JP (1) | JP2022521859A (zh) |
| KR (1) | KR20210131314A (zh) |
| CN (3) | CN117327078A (zh) |
| AU (1) | AU2020226422A1 (zh) |
| BR (1) | BR112021016299A2 (zh) |
| CA (1) | CA3130245A1 (zh) |
| CL (1) | CL2021002205A1 (zh) |
| CO (1) | CO2021011023A2 (zh) |
| IL (1) | IL285378A (zh) |
| MX (1) | MX2021009971A (zh) |
| SG (1) | SG11202107848TA (zh) |
| WO (1) | WO2020168939A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022022398A1 (zh) * | 2020-07-28 | 2022-02-03 | 四川科伦博泰生物医药股份有限公司 | 一种嘧啶类化合物的盐和晶型及其制备方法 |
| CN114235976A (zh) * | 2021-11-09 | 2022-03-25 | 暨南大学 | 一种含氮杂环有机化合物中间产物的合成和分析方法 |
| WO2022199503A1 (zh) | 2021-03-24 | 2022-09-29 | 四川科伦博泰生物医药股份有限公司 | 杂环化合物治疗与激酶耐药突变相关的疾病的用途和方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101166734A (zh) * | 2005-02-28 | 2008-04-23 | 日本烟草产业株式会社 | 具有Syk抑制活性的新型的氨基吡啶化合物 |
| WO2012154518A1 (en) * | 2011-05-10 | 2012-11-15 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as syk inhibitors |
| CN103298788A (zh) * | 2010-10-28 | 2013-09-11 | 日本新药株式会社 | 吡啶衍生物及医药 |
| WO2017079140A1 (en) | 2015-11-02 | 2017-05-11 | Blueprint Medicines Corporation | Inhibitors of ret |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010208945A (ja) * | 2007-06-01 | 2010-09-24 | Mitsubishi Tanabe Pharma Corp | 複素環化合物 |
| JP2013107824A (ja) * | 2010-03-17 | 2013-06-06 | Dainippon Sumitomo Pharma Co Ltd | 新規単環ピリミジン誘導体 |
-
2020
- 2020-02-11 SG SG11202107848TA patent/SG11202107848TA/en unknown
- 2020-02-11 BR BR112021016299-2A patent/BR112021016299A2/pt unknown
- 2020-02-11 AU AU2020226422A patent/AU2020226422A1/en active Pending
- 2020-02-11 WO PCT/CN2020/074696 patent/WO2020168939A1/zh active Application Filing
- 2020-02-11 MX MX2021009971A patent/MX2021009971A/es unknown
- 2020-02-11 KR KR1020217022806A patent/KR20210131314A/ko unknown
- 2020-02-11 CN CN202311264080.7A patent/CN117327078A/zh active Pending
- 2020-02-11 CA CA3130245A patent/CA3130245A1/en active Pending
- 2020-02-11 US US17/431,930 patent/US20220144847A1/en active Pending
- 2020-02-11 EP EP20760123.8A patent/EP3929198A4/en active Pending
- 2020-02-11 CN CN202311221283.8A patent/CN117263945A/zh active Pending
- 2020-02-11 JP JP2021571055A patent/JP2022521859A/ja active Pending
- 2020-02-11 CN CN202080009954.2A patent/CN113316578B/zh active Active
-
2021
- 2021-08-04 IL IL285378A patent/IL285378A/en unknown
- 2021-08-18 CL CL2021002205A patent/CL2021002205A1/es unknown
- 2021-08-20 CO CONC2021/0011023A patent/CO2021011023A2/es unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101166734A (zh) * | 2005-02-28 | 2008-04-23 | 日本烟草产业株式会社 | 具有Syk抑制活性的新型的氨基吡啶化合物 |
| CN103298788A (zh) * | 2010-10-28 | 2013-09-11 | 日本新药株式会社 | 吡啶衍生物及医药 |
| WO2012154518A1 (en) * | 2011-05-10 | 2012-11-15 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as syk inhibitors |
| WO2017079140A1 (en) | 2015-11-02 | 2017-05-11 | Blueprint Medicines Corporation | Inhibitors of ret |
Non-Patent Citations (12)
| Title |
|---|
| DATABASE REGISTRY 2 October 2006 (2006-10-02), retrieved from STN Database accession no. 909343-99-3 * |
| DATABASE REGISTRY 2 October 2006 (2006-10-02), retrieved from STN Database accession no. 909344-03-2 * |
| DATABASE REGISTRY 2 October 2006 (2006-10-02), retrieved from STN Database accession no. 909344-04-3 * |
| DATABASE REGISTRY 2 October 2006 (2006-10-02), retrieved from STN Database accession no. 909344-08-7 * |
| DATABASE REGISTRY 22 May 2012 (2012-05-22), retrieved from STN Database accession no. 1374201-58-7 * |
| DATABASE REGISTRY 5 December 2011 (2011-12-05), retrieved from STN Database accession no. 1349069-14-2 * |
| DATABASE REGISTRY 5 December 2011 (2011-12-05), retrieved from STN Database accession no. 1349252-44-3 * |
| G. W. H. CHEESEMANE. S. G. WERSTIUK: "Pro-drugs as Novel Delivery Systems", vol. 14, 1985, ACS SYMPOSIUM SERIES, pages: 390 - 392 |
| LI, BINGKE ET AL.: "In Silico Prediction of Spleen Tyrosine Kinase Inhibitors Using Machine Learning Approaches and an Optimized Molecular Descriptor Subset Generated by Recursive Feature Elimination Method", COMPUTERS IN BIOLOGY AND MEDICINE, vol. 43, no. 4, 1 May 2013 (2013-05-01), XP055143456, ISSN: 0010-4825, DOI: 10.1016/j.compbiomed.2013.01.015 * |
| NATURE REVIEWS CANCER, vol. 14, no. 3, 2014, pages 173 - 86 |
| STAHLWERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| T. W. GREENEP. G. M. WUTS: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022022398A1 (zh) * | 2020-07-28 | 2022-02-03 | 四川科伦博泰生物医药股份有限公司 | 一种嘧啶类化合物的盐和晶型及其制备方法 |
| WO2022199503A1 (zh) | 2021-03-24 | 2022-09-29 | 四川科伦博泰生物医药股份有限公司 | 杂环化合物治疗与激酶耐药突变相关的疾病的用途和方法 |
| CN114235976A (zh) * | 2021-11-09 | 2022-03-25 | 暨南大学 | 一种含氮杂环有机化合物中间产物的合成和分析方法 |
| CN114235976B (zh) * | 2021-11-09 | 2023-11-03 | 暨南大学 | 一种含氮杂环有机化合物中间产物的合成和分析方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3929198A4 (en) | 2022-11-16 |
| CA3130245A1 (en) | 2020-08-27 |
| CO2021011023A2 (es) | 2021-09-09 |
| AU2020226422A1 (en) | 2021-09-09 |
| CN117327078A (zh) | 2024-01-02 |
| IL285378A (en) | 2021-09-30 |
| JP2022521859A (ja) | 2022-04-12 |
| CN117263945A (zh) | 2023-12-22 |
| BR112021016299A2 (pt) | 2021-10-13 |
| SG11202107848TA (en) | 2021-08-30 |
| EP3929198A1 (en) | 2021-12-29 |
| US20220144847A1 (en) | 2022-05-12 |
| KR20210131314A (ko) | 2021-11-02 |
| CL2021002205A1 (es) | 2022-04-22 |
| CN113316578B (zh) | 2023-10-31 |
| MX2021009971A (es) | 2021-10-13 |
| CN113316578A (zh) | 2021-08-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6190883B2 (ja) | 1,4−二置換ピリダジン類似体およびsmn欠損に関連する状態を処置するための方法 | |
| WO2021043077A1 (zh) | 一种取代吡嗪化合物、其制备方法和用途 | |
| WO2020168939A1 (zh) | 杂环化合物、包含其的药物组合物及其制备方法和用途 | |
| JP2017518276A (ja) | ブルトン型チロシンキナーゼ(btk)インヒビターとしての多フルオロ置換化合物 | |
| JP2020525525A (ja) | Rho−関連プロテインキナーゼ阻害剤、rho−関連プロテインキナーゼ阻害剤を含む医薬組成物、当該医薬組成物の調製方法及び使用 | |
| CN114867720A (zh) | 杂芳基类衍生物及其制备方法和用途 | |
| AU2017359023A1 (en) | 3-substituted propionic acids as alpha v integrin inhibitors | |
| CN114846005A (zh) | Shp2抑制剂及其应用 | |
| CN113950481A (zh) | 大环化合物 | |
| CN114423753A (zh) | 作为cd38抑制剂的杂双环酰胺 | |
| TW202130631A (zh) | 3—(5—甲氧基—1—側氧基異吲哚啉—2—基)哌啶—2,6—二酮衍生物及其用途 | |
| WO2022028506A1 (zh) | Sos1抑制剂、包含其的药物组合物及其用途 | |
| WO2020182018A1 (zh) | 氮杂环化合物、其制备方法及用途 | |
| WO2022237178A1 (zh) | 双环杂芳基类衍生物及其制备方法和用途 | |
| TW202214233A (zh) | 可做為tlr9抑制劑之經取代雜芳基化合物 | |
| CN111484479A (zh) | 氮杂环化合物、包含其的药物组合物及其制备方法和用途 | |
| WO2022199503A1 (zh) | 杂环化合物治疗与激酶耐药突变相关的疾病的用途和方法 | |
| CN112574201B (zh) | 芳基胺类化合物、包含其的药物组合物及其制备方法和用途 | |
| WO2022188792A1 (zh) | 具有蛋白激酶抑制活性的杂环化合物、包含其的药物组合物及其制备方法和用途 | |
| WO2024022186A1 (zh) | 甲基吡唑化合物、包含其的药物组合物及其制备方法和用途 | |
| WO2022089219A1 (zh) | 芳基酰胺化合物、包含其的药物组合物及其制备方法和用途 | |
| WO2022166796A1 (zh) | 嘧啶或吡啶并杂环类腺苷受体抑制剂及其制备方法和用途 | |
| WO2023165581A1 (zh) | 一种吡啶类衍生物及其用途 | |
| WO2024032589A1 (zh) | TGF-β抑制剂化合物及其用途 | |
| TW202104208A (zh) | 吡唑基-氨基-嘧啶基衍生物的苯醚和苯胺及其組合物和方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20760123 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3130245 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2021571055 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112021016299 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2020226422 Country of ref document: AU Date of ref document: 20200211 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2021124417 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 112021016299 Country of ref document: BR Kind code of ref document: A2 Effective date: 20210817 |
|
| ENP | Entry into the national phase |
Ref document number: 2020760123 Country of ref document: EP Effective date: 20210920 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 521430084 Country of ref document: SA |