WO2020095811A1 - 吸水性樹脂 - Google Patents
吸水性樹脂 Download PDFInfo
- Publication number
- WO2020095811A1 WO2020095811A1 PCT/JP2019/042766 JP2019042766W WO2020095811A1 WO 2020095811 A1 WO2020095811 A1 WO 2020095811A1 JP 2019042766 W JP2019042766 W JP 2019042766W WO 2020095811 A1 WO2020095811 A1 WO 2020095811A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- absorbent resin
- mass
- polymerization
- aqueous solution
- Prior art date
Links
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
- B01J20/267—Cross-linked polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/15203—Properties of the article, e.g. stiffness or absorbency
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/53—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/22—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing macromolecular materials
- A61L15/24—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/24—Naturally occurring macromolecular compounds, e.g. humic acids or their derivatives
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/261—Synthetic macromolecular compounds obtained by reactions only involving carbon to carbon unsaturated bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28002—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their physical properties
- B01J20/28011—Other properties, e.g. density, crush strength
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/02—Monocarboxylic acids having less than ten carbon atoms, Derivatives thereof
- C08F20/04—Acids, Metal salts or ammonium salts thereof
- C08F20/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
- C08J3/03—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques in aqueous media
- C08J3/075—Macromolecular gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/53—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium
- A61F2013/530481—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/53—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium
- A61F2013/530481—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials
- A61F2013/530583—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials characterized by the form
- A61F2013/530635—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials characterized by the form in thin film
- A61F2013/530642—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials characterized by the form in thin film being cross-linked or polymerised in situ
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61F—FILTERS IMPLANTABLE INTO BLOOD VESSELS; PROSTHESES; DEVICES PROVIDING PATENCY TO, OR PREVENTING COLLAPSING OF, TUBULAR STRUCTURES OF THE BODY, e.g. STENTS; ORTHOPAEDIC, NURSING OR CONTRACEPTIVE DEVICES; FOMENTATION; TREATMENT OR PROTECTION OF EYES OR EARS; BANDAGES, DRESSINGS OR ABSORBENT PADS; FIRST-AID KITS
- A61F13/00—Bandages or dressings; Absorbent pads
- A61F13/15—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators
- A61F13/53—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium
- A61F2013/530481—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials
- A61F2013/530708—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials characterized by the absorbency properties
- A61F2013/530737—Absorbent pads, e.g. sanitary towels, swabs or tampons for external or internal application to the body; Supporting or fastening means therefor; Tampon applicators characterised by the absorbing medium having superabsorbent materials, i.e. highly absorbent polymer gel materials characterized by the absorbency properties by the absorbent capacity
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2220/00—Aspects relating to sorbent materials
- B01J2220/50—Aspects relating to the use of sorbent or filter aid materials
- B01J2220/68—Superabsorbents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
Definitions
- the present disclosure relates to a water absorbent resin, a method for manufacturing the same, and the like. More specifically, the present invention relates to a water-absorbent resin that constitutes an absorber preferably used for absorbent articles for sanitary materials such as sanitary items and disposable diapers, and a method for producing the same.
- Absorbent articles for sanitary materials such as sanitary products and disposable diapers are usually composed of an absorbent body containing hydrophilic fibers and a water-absorbent resin as main constituent units.
- the water-absorbent resin include starch-acrylonitrile graft copolymers.
- hydrolyzed products of polymers neutralized products of starch-acrylic acid graft copolymers, saponified products of vinyl acetate-acrylic acid ester copolymers, partially neutralized products of polyacrylic acid, and the like.
- the texture (softness) of the absorber is excellent, but the absorption performance represented by the amount of reversion is not satisfactory. Therefore, if the content of the water-absorbent resin is increased, the absorption performance after absorption of body fluid and the like tends to be improved, but the texture of the absorbent body is impaired. Further, it is difficult to uniformly disperse a large amount of the water absorbent resin in the absorbent body, and the performance improvement due to the increased amount of the water absorbent resin is not necessarily guaranteed. As described above, it is difficult to satisfy both the absorption performance and the texture in the absorber. Therefore, even if the content in the absorber is small, a water absorbent resin having a property of enhancing the absorption performance in the absorber is desired.
- the water-absorbent resin In order to improve the absorption performance of absorbent articles for hygiene materials, the water-absorbent resin is required to have high water retention capacity and high water absorption capacity under load. In order to obtain a water absorbent resin having such characteristics, research has been conducted so far. For example, the following proposals have been made. That is, by performing reverse phase suspension polymerization of water-soluble ethylenically unsaturated monomers in multiple stages, in addition to excellent water absorption, the resulting water-absorbent resin has a large particle size, a small amount of fine powder, and a sharp distribution.
- Patent Document 1 a method for producing a water-absorbent resin having high wettability with water (see Patent Document 1), reverse-phase suspension polymerization of a water-soluble ethylenically unsaturated monomer to obtain primary particles having a specific median particle size.
- a water-soluble ethylenically unsaturated monomer in the second stage is added to carry out a reverse phase suspension polymerization reaction to obtain secondary particles having a specific median particle diameter.
- Patent Document 2 primary particles obtained by polymerizing a water-soluble ethylenically unsaturated monomer by a reverse phase suspension polymerization method are further aggregated by a reverse phase suspension polymerization method.
- a water-absorbent resin having a median particle size of the primary particles of 100 to 250 ⁇ m Absorbent resin (see Patent Document 3) it is known, wherein the saline water retention capacity of the water-absorbent resin is not more than 30 g / g.
- the main purpose of the present disclosure is to provide a water absorbent resin that can reduce the amount of reversion of the liquid to be absorbed from the absorbent body even in an absorbent body having a low water absorbent resin content.
- the present inventors have found that when a water-absorbent resin satisfying a specific index is used, even in an absorber having a low content of the water-absorbent resin, the amount of reversion of the liquid to be absorbed from the absorber can be reduced. We made further improvements.
- a water-absorbent resin which is a polymer cross-linked product of a water-soluble ethylenically unsaturated monomer and has a dry-up index of 1.85 or more represented by the following formula (1).
- the total absorption capacity term ⁇ and the water absorption rate term ⁇ are obtained by the following equations (2) and (3).
- Item 2 The water absorbent resin according to Item 1, wherein the total absorption capacity term ⁇ is 0.95 or more.
- Item 3 The water absorbent resin according to Item 1 or 2, wherein the water absorption rate term ⁇ is 1.56 or more.
- Item 4. The water absorbent resin according to any one of Items 1 to 3, wherein the difference between the physiological saline absorption capacity and the physiological saline retention capacity is 18 or less.
- Item 5 The water absorbent resin according to any one of Items 1 to 4, wherein the difference between the physiological saline water absorption capacity under a load of 2.007 kPa and the physiological saline water absorption capacity under a load of 4.82 kPa is 17 to 36. ..
- Item 6 An absorbent body containing 5 to 50% by mass of the water absorbent resin according to any one of the items 1 to 5 or the following items AF.
- Item A A water-absorbent resin which is a polymer cross-linked product of a water-soluble ethylenically unsaturated monomer, which is prepared by a polymerization method satisfying at least two conditions of the following conditions (i), (ii), and (iii).
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the first stage polymerization to the internal cross-linking agent (water-soluble ethylenically unsaturated monomer / internal cross-linking agent) is 10 ⁇ 10 3 to 15 ⁇ .
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the second stage polymerization to the internal crosslinking agent (water-soluble ethylenically unsaturated monomer / internal crosslinking agent) is 15 ⁇ 10 3 to 25 ⁇ . It is 10 3 .
- the molar ratio of the total amount of the water-soluble ethylenically unsaturated monomer used in the polymerization for preparing the resin used for the post-crosslinking reaction and the post-crosslinking agent (water-soluble ethylenically unsaturated monomer / post-crosslinking agent) ) Is 2.5 ⁇ 10 3 to 4.5 ⁇ 10 3 .
- Item B The water absorbent resin according to any one of Items 1 to 5, which is prepared by a polymerization method satisfying at least two conditions of the following conditions (i), (ii), and (iii).
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the first stage polymerization to the internal cross-linking agent (water-soluble ethylenically unsaturated monomer / internal cross-linking agent) is 10 ⁇ 10 3 to 15 ⁇ . It is 10 3 .
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the second stage polymerization to the internal crosslinking agent (water-soluble ethylenically unsaturated monomer / internal crosslinking agent) is 15 ⁇ 10 3 to 25 ⁇ . It is 10 3 .
- the molar ratio of the total amount of the water-soluble ethylenically unsaturated monomer used in the polymerization to prepare the resin used for the post-crosslinking reaction and the post-crosslinking agent (water-soluble ethylenically unsaturated monomer / post-crosslinking agent) ) Is 2.5 ⁇ 10 3 to 4.5 ⁇ 10 3 .
- Term D At least one of (i) and (ii) is satisfied, and the internal crosslinking agent is selected from the group consisting of (poly) ethylene glycol diglycidyl ether, (poly) propylene glycol diglycidyl ether, and (poly) glycerin diglycidyl ether.
- the post-crosslinking agent satisfying at least (iii) is at least one selected from the group consisting of (poly) ethylene glycol diglycidyl ether, (poly) propylene glycol diglycidyl ether, and (poly) glycerin diglycidyl ether.
- the polymerization method satisfying at least two conditions of (i), (ii), and (iii) is a reverse phase suspension polymerization (preferably one or two-step reverse phase suspension polymerization, more preferably two-step).
- Term G Item 7.
- a water-absorbent resin that can reduce the amount of reversion, and a method for producing the same, even when the water-absorbent resin is used in a small amount in the absorber.
- the present disclosure preferably includes, but is not limited to, a specific water absorbent resin and a method for producing the same, and the present disclosure includes all disclosed in the present specification and recognizable by a person skilled in the art.
- a water-absorbent resin included in the present disclosure is a water-absorbent resin composed of a polymer of a water-soluble ethylenically unsaturated monomer, and is represented by the following formula (1). Is a water-absorbent resin having a dry-up index of 1.85 or more, which is represented by the product of the water absorption rate term ⁇ .
- the water absorbent resin may be referred to as the “water absorbent resin of the present disclosure”.
- the water-absorbent resin of the present disclosure preferably has a dry-up index of 1.9 to 5.0.
- the lower limit of the numerical range is, for example, 2.0, 2.1, 2.2, 2.3, 2.4, 2.5, 2.6, 2.7, 2.8, or 2.9. It may be.
- the upper limit of the numerical range is, for example, 4.9, 4.8, 4.7, 4.6, 4.5, 4.4, 4.3, 4.2, 4.1, 4.0. It may be 3.9, 3.8, 3.7, 3.6, 3.5, 3.4, 3.3, 3.2, 3.1, or 3.0.
- the numerical range is, for example, more preferably 2.0 to 4.0, further preferably 2.1 to 3.0.
- the overall absorption capacity term ⁇ of the water absorbent resin is a value calculated by the following equation (2).
- physiological saline water absorption capacity means a standard sieve having an opening of 75 ⁇ m after stirring 2.0 g of the water absorbent resin for 60 minutes while stirring 500 g of physiological saline at 600 r / min. Is a value obtained by measuring the mass of the swollen gel after standing still for 30 minutes in a state in which the sieve was tilted at an inclination angle of about 30 degrees with respect to the horizontal.
- the “physiological saline water retention capacity” means that while stirring 500 g of physiological saline at 600 r / min, 2.0 g of the water-absorbent resin is stirred for 30 minutes, and then stored in a cotton bag (Membroad No. 60). It is a value obtained by pouring and dehydrating a cotton bag for 1 minute using a dehydrator set to have a centrifugal force of 167 G, and measuring the mass of the swollen gel after dehydration.
- physiological saline water absorption capacity under a load of 2.07 kPa refers to 0.1 g of the water-absorbent resin evenly sprinkled on a cylinder having an inner diameter of 2.0 cm with a 200-mesh nylon mesh. It is determined by allowing the water-absorbent resin to absorb water under the condition that a load of 2.07 kPa is uniformly applied by the weight and measuring the amount of physiological saline 60 minutes after the start of water absorption. For the measurement, the measuring device X whose schematic configuration is shown in FIG. 1 can be preferably used.
- the “physiological saline water absorption capacity under a load of 4.82 kPa” is 4% by weight with respect to 0.9 g of the water-absorbent resin charged in a supporting cylinder having an inner diameter of 60 mm with a 400-mesh wire mesh. It is determined by allowing the water-absorbent resin to absorb water with a load of 0.82 kPa being uniformly applied, and measuring the amount of physiological saline after 60 minutes have elapsed from the start of water absorption. For the measurement, the measuring device Y whose schematic configuration is shown in FIG. 2 can be preferably used.
- the water-absorbent resin of the present disclosure preferably has a total absorption capacity term ⁇ of 0.95 or more, more preferably 1.00 to 4.00, and even more preferably 1.05 to 2.00.
- the upper limit may be 4.00, 3.00, 2.00, 1.80, 1.65, or 1.55.
- the water-absorbent resin of the present disclosure preferably has a difference between the physiological saline absorption capacity and the physiological saline retention capacity of 18 or less, more preferably 17 or less, and even more preferably 16 or less. Further, the difference between the physiological saline absorption capacity and the physiological saline retention capacity is preferably 5 or more, more preferably 8 or more, and further preferably 10 or more.
- the difference between the water absorption capacity of the water absorbent resin according to the present disclosure under a load of 2.07 kPa and the water absorption capacity of a saline solution under a load of 4.82 kPa is preferably 17 to 36, and 17 to 36 is preferable. 33 is more preferable, and 17 to 30 is further preferable.
- the water-absorption rate term ⁇ of the water-absorbent resin is represented by the ratio of the dynamic water-absorption rate to the static water-absorption rate, and is calculated by the following equation (3).
- the dynamic water absorption rate was as follows. 50 g of physiological saline solution at a temperature of 25 ° C. was stirred with a magnetic stirrer bar of 8 mm ⁇ ⁇ 30 mm, and a vortex was generated at a rotation speed of 600 r / min to obtain 2.0 g of the water absorbent resin. It can be obtained by adding all at once and measuring the time from the addition of the water absorbent resin to the point when the vortex of the liquid surface converges. More specifically, it is a value measured by the method described in Examples described later.
- the static water absorption rate is as follows: 0.1g of water absorbent resin uniformly sprinkled on a cylinder with an inner diameter of 2.0cm and a nylon mesh of 200mesh It is determined by measuring the amount of physiological saline that has absorbed water and measuring the time (seconds) required for the water-absorbent resin to absorb 25 g of physiological saline per gram. More specifically, it is a value measured by the method described in Examples described later. For the measurement, the measuring device X whose schematic configuration is shown in FIG. 1 can be preferably used.
- the water-absorbent resin of the present disclosure preferably has a water-absorption rate term ⁇ of 1.56 or more, more preferably 1.60 or more, still more preferably 1.65 or more.
- the upper limit is not particularly limited, but is preferably 3 or less, and may be 2.95, 2.9, 2.85, 2.8, 2.75, or 2.7 or less.
- the water-absorbent resin according to the present disclosure preferably has a median particle size of 200 to 600 ⁇ m, and 250 to 600 ⁇ m from the viewpoint of reducing the feeling of foreign matter when worn when used as sanitary materials such as sanitary products and disposable diapers. It is more preferably 550 ⁇ m, further preferably 300 to 500 ⁇ m.
- the water-absorbent resin obtained may be mixed with additives depending on the purpose.
- additives include inorganic powders, surfactants, oxidizing agents, reducing agents, metal chelating agents, radical chain inhibitors, antioxidants, antibacterial agents, deodorants and the like.
- inorganic powders surfactants, oxidizing agents, reducing agents, metal chelating agents, radical chain inhibitors, antioxidants, antibacterial agents, deodorants and the like.
- amorphous silica as an inorganic powder to 100 parts by mass of the water absorbent resin, the fluidity of the water absorbent resin can be improved.
- Method for producing water-absorbent resin examples include a reverse phase suspension polymerization method, an aqueous solution polymerization method, and the like.
- the reverse phase suspension polymerization method will be described in more detail below as an example of the method for producing the water absorbent resin of the present disclosure.
- the polymerization can be performed by a method known in the field of water absorbent resin preparation or a method conceivable from a known method. Among them, reverse phase suspension polymerization is preferable.
- a water-soluble ethylenically unsaturated monomer aqueous solution containing a radical polymerization initiator and optionally a crosslinking agent (internal crosslinking agent) in a hydrocarbon dispersion medium in the presence of a dispersion stabilizer is prepared. Polymerization is carried out by mixing with stirring and heating.
- the polymerization reaction may be carried out in one stage or in multiple stages of two or more stages. In the case of multiple stages, the number of stages is preferably 2 or 3 from the viewpoint of improving productivity.
- a water-soluble ethylenically unsaturated monomer aqueous solution is added to the reaction mixture obtained in the first-stage polymerization reaction and mixed.
- the second step polymerization may be carried out in the same manner as in the first step.
- multistage polymerization can also be performed by repeating the same operation. When two or more steps of polymerization are performed, the same polymerization method or different polymerization methods may be used, and the same polymerization method is more preferable. It is more preferable to use reverse phase suspension polymerization in any polymerization.
- a radical polymerization initiator in addition to the water-soluble ethylenically unsaturated monomer, a radical polymerization initiator, an internal cross-linking agent, etc. are added in the polymerization in the second and subsequent stages.
- Polymerization can be carried out by adding within the range of the molar ratio of each component to the water-soluble ethylenically unsaturated monomer described below, based on the amount of the ethylenically unsaturated monomer.
- water-soluble ethylenically unsaturated monomer examples include (meth) acrylic acid (in the present specification, “acry” and “methacryl” are collectively referred to as “(meth) acry”.
- water-soluble ethylenically unsaturated monomers may be used alone or in combination of two or more.
- (meth) acrylic acid and salts thereof, (meth) acrylamide and N, N-dimethylacrylamide are preferable, and (meth) acrylic acid and salts thereof are more preferable, because they are industrially easily available.
- acrylic acid and its salts are widely used as raw materials for water-absorbent resins, and these acrylic acid and its salts are copolymerized with the other water-soluble ethylenically unsaturated monomers described above. It can also be used.
- acrylic acid and its salt are preferably used as the main water-soluble ethylenically unsaturated monomer in an amount of 70 to 100 mol% based on the total water-soluble ethylenically unsaturated monomer.
- the above water-soluble ethylenically unsaturated monomer may be used as an aqueous solution in order to improve the dispersibility in a hydrocarbon dispersion medium during reverse phase suspension polymerization.
- the concentration of the water-soluble ethylenically unsaturated monomer in such an aqueous solution may be usually 20% by mass to a saturated concentration or less, but the water absorption performance of the resulting water-absorbent resin can be ensured while ensuring the productivity.
- the concentration of the water-soluble ethylenically unsaturated monomer is preferably 20 to 50% by mass, more preferably 22 to 45% by mass, and further preferably 24 to 36% by mass.
- the acid group may be preliminarily alkaline if necessary. You may use what was neutralized with the solvating agent.
- alkaline neutralizing agent include alkali metal salts such as sodium hydroxide, sodium carbonate, sodium hydrogen carbonate, potassium hydroxide and potassium carbonate; ammonia and the like.
- these alkaline neutralizing agents may be used in the form of an aqueous solution in order to simplify the neutralizing operation.
- the above alkaline neutralizing agents may be used alone or in combination of two or more.
- the degree of neutralization of the water-soluble ethylenically unsaturated monomer by the alkaline neutralizing agent the water absorbing performance is enhanced by increasing the osmotic pressure of the resulting water absorbent resin, and the presence of excess alkaline neutralizing agent results.
- the degree of neutralization with respect to all acid groups of the water-soluble ethylenically unsaturated monomer is preferably 40 to 90 mol%, more preferably 70 to 88 mol%.
- 75 to 85 mol% is more preferable, and 77 to 80 mol% is even more preferable.
- hydrocarbon dispersion medium examples include n-hexane, n-heptane, 2-methylhexane, 3-methylhexane, 2,3-dimethylpentane, 3-ethylpentane, n-octane and the like having 6 to 8 carbon atoms.
- Aliphatic hydrocarbons such as cyclohexane, methylcyclohexane, cyclopentane, methylcyclopentane, trans-1,2-dimethylcyclopentane, cis-1,3-dimethylcyclopentane, trans-1,3-dimethylcyclopentane
- aromatic hydrocarbons such as benzene, toluene, xylene and the like can be mentioned.
- n-hexane, n-heptane and cyclohexane are preferably used because they are industrially easily available, have stable quality, and are inexpensive.
- commercially available exol heptane manufactured by Exxon Mobil Corp .: heptane and its isomer hydrocarbon 75 to 85 mass% contained
- Goods can also be used.
- the amount of the hydrocarbon dispersion medium used is usually the first-stage water-soluble ethylenically unsaturated monomer in order to uniformly disperse the water-soluble ethylenically unsaturated monomer aqueous solution and facilitate control of the polymerization temperature. 80 to 1500 parts by mass is preferable, and 120 to 1200 parts by mass is more preferable, relative to 100 parts by mass of the body.
- a surfactant can be used as the dispersion stabilizer, and examples thereof include sucrose fatty acid ester, polyglycerin fatty acid ester, sorbitan fatty acid ester, polyoxyethylene sorbitan fatty acid ester, polyoxyethylene glycerin fatty acid ester, sorbitol fatty acid ester, and polyoxy.
- Ethylene sorbitol fatty acid ester polyoxyethylene alkyl ether, polyoxyethylene alkyl phenyl ether, polyoxyethylene castor oil, polyoxyethylene hydrogenated castor oil, alkylallyl formaldehyde condensed polyoxyethylene ether, polyoxyethylene polyoxypropylene block copolymer, poly Oxyethylene polyoxypropyl alkyl ether, polyethylene glycol fatty acid ester, alkyl glucoside N- alkyl gluconamide, polyoxyethylene fatty acid amides, it can be used polyoxyethylene alkyl amines.
- sorbitan fatty acid ester polyglycerin fatty acid ester, sucrose fatty acid ester and the like are preferable from the viewpoint of dispersion stability of the monomer aqueous solution.
- surfactants may be used alone or in combination of two or more.
- the amount of the surfactant used is a water-soluble ethylenic unsaturated used for the polymerization, from the viewpoint of maintaining a good dispersion state of the aqueous monomer solution in the hydrocarbon dispersion medium and obtaining a dispersion effect commensurate with the amount used.
- the amount is preferably 0.05 to 30 parts by mass, more preferably 0.1 to 20 parts by mass with respect to 100 parts by mass of the monomer.
- a polymer-based dispersant may be used together with the surfactant.
- Polymeric dispersants that can be used include maleic anhydride-modified polyethylene, maleic anhydride-modified polypropylene, maleic anhydride-modified ethylene / propylene copolymer, maleic anhydride-modified EPDM (ethylene / propylene / diene / terpolymer), and anhydrous.
- the amount of the polymeric dispersant used is such that the water-soluble ethylenic agent used for the polymerization is used in order to keep the dispersion state of the monomer aqueous solution in the hydrocarbon dispersion medium good and to obtain a dispersion effect commensurate with the amount used.
- the amount is preferably 0.05 to 30 parts by mass, more preferably 0.1 to 20 parts by mass with respect to 100 parts by mass of the unsaturated monomer.
- the surfactant used as the dispersion stabilizer may be added before or after the polymerization reaction is started, either before or after the addition of the aqueous monomer solution.
- the addition timing of the polymeric dispersant used in combination with the surfactant as a dispersion stabilizer may be before or after the addition of the aqueous monomer solution, but the dispersion stability and water absorption of the aqueous monomer solution may be added.
- the aqueous monomer solution is dispersed. That is, it is more preferable to disperse the aqueous monomer solution in the hydrocarbon dispersion medium in which the polymer dispersant is dispersed, and then further disperse the surfactant before the polymerization.
- radical polymerization initiator examples include persulfates such as potassium persulfate, ammonium persulfate, and sodium persulfate; methyl ethyl ketone peroxide, methyl isobutyl ketone peroxide, di-t-butyl peroxide, t-butyl cumyl peroxide.
- radical polymerization initiators potassium persulfate, ammonium persulfate, sodium persulfate and 2,2′-azobis (2-amidinopropane) dihydrochloride are preferable from the viewpoints of easy availability and easy handling.
- These radical polymerization initiators may be used alone or in combination of two or more.
- the radical polymerization initiator is usually used in an amount of 0.001 with respect to 100 mol of the water-soluble ethylenically unsaturated monomer used for the polymerization. 005 to 1 mol is preferred, 0.01 to 0.5 mol is more preferred, 0.0125 to 0.1 mol is more preferred, and 0.015 to 0.05 mol is even more preferred.
- reaction temperature of the polymerization reaction varies depending on the radical polymerization initiator used, it is usually preferably 20 to 110 ° C. from the viewpoint of rapidly advancing the polymerization to enhance the productivity and more smoothly removing the heat of polymerization. More preferably, the temperature is up to 90 ° C.
- the reaction time is usually preferably about 0.1 to 4 hours.
- Internal cross-linking agent When polymerizing the water-soluble ethylenically unsaturated monomer, a cross-linking agent may be used if necessary. When the polymerization is carried out in multiple stages, a crosslinking agent may or may not be used in all stages. In the case of multiple stages, the type of crosslinking agent used in each stage may be the same or different, and the same type is preferable.
- Such a cross-linking agent hereinafter referred to as “internal cross-linking agent” means, for example, (poly) ethylene glycol [“(poly)” means with or without the prefix “poly”].
- diols such as (poly) propylene glycol, 1,4-butanediol, trimethylolpropane, (poly) glycerin, and polyols such as triol, and unsaturated compounds such as (meth) acrylic acid, maleic acid, and fumaric acid.
- Unsaturated polyesters obtained by reacting with an acid Bisacrylamides such as N, N-methylenebisacrylamide; Di or tri (meth) acrylic acid esters obtained by reacting a polyepoxide with (meth) acrylic acid
- a polyglycidyl compound is preferable, a diglycidyl ether compound is more preferable, and a (poly) ethylene glycol diglycidyl ether is particularly preferable.
- These internal cross-linking agents may be used alone or in combination of two or more.
- the internal cross-linking agent is preferably used by adding it to the above-mentioned aqueous monomer solution.
- the amount used is 0.00001 to 1 mol with respect to 100 mol of the water-soluble ethylenically unsaturated monomer in order to sufficiently enhance the water absorption performance of the resulting water-absorbent resin. It is preferable that the amount be 0.0001 to 0.5 mol.
- a thickener When carrying out the polymerization reaction, a thickener may be added to the aqueous solution of the water-soluble ethylenically unsaturated monomer. By adjusting the viscosity of the aqueous solution by adding a thickener in this way, it is possible to control the median particle size of the resulting water-absorbent resin.
- thickener examples include hydroxyethyl cellulose, hydroxypropyl cellulose, methyl cellulose, carboxymethyl cellulose, polyacrylic acid, polyacrylic acid (partial) neutralized product, polyethylene glycol, polyacrylamide, polyethyleneimine, dextrin, sodium alginate, polyvinyl alcohol. , Polyvinylpyrrolidone, polyethylene oxide and the like can be used. If the stirring speed during polymerization is the same, the higher the viscosity of the water-soluble ethylenically unsaturated monomer aqueous solution, the larger the median particle size of the obtained particles.
- Post-crosslinking process After the polymerization step, a post-crosslinking reaction is performed on a hydrogel (a water absorbent resin obtained by polymerization and containing water) to increase the crosslink density in the vicinity of the surface of the water absorbent resin. It is possible to improve various performances such as water absorption capacity under load. In the production of the water absorbent resin of the present disclosure, post-crosslinking may be performed with a post-crosslinking agent.
- the post-crosslinking agent may be one that can react with the carboxyl group of the water absorbent resin.
- Typical examples of the post-crosslinking agent include polyols such as (poly) ethylene glycol, (poly) propylene glycol, 1,4-butanediol, trimethylolpropane and (poly) glycerin; (poly) ethylene glycol diglycidyl ether, Diglycidyl ether compounds such as (poly) propylene glycol diglycidyl ether and (poly) glycerin diglycidyl ether; epihalohydrin compounds such as epichlorohydrin, epibromhydrin, ⁇ -methylepichlorohydrin; 2,4-tolylene diisocyanate, hexamethylene diisocyanate Examples thereof include compounds having two or more reactive functional groups such as isocyanate compounds. Of these, (poly) ethylene glycol diglycidyl ether is preferable. These may be used
- the amount of the post-crosslinking agent cannot be unconditionally determined because it depends on the type of the post-crosslinking agent, but when the amount of the post-crosslinking agent used is small, the crosslink density of the surface layer of the water absorbent resin becomes insufficient. The water absorption capacity under load tends to decrease, while on the other hand, when the amount of the post-crosslinking agent used is large, the water retention capacity of the water absorbent resin tends to decrease. Therefore, the amount of the post-crosslinking agent used is usually 0.00001 to 0.01 mol, preferably 0.00005 to 0 mol, based on 1 mol of the total amount of the water-soluble ethylenically unsaturated monomer used for the polymerization. The amount may be 0.005 mol, and more preferably 0.0001 to 0.002 mol.
- the post-crosslinking agent is preferably added to a system in which 1 to 400 parts by weight of water is present per 100 parts by weight of the total amount of the water-soluble ethylenically unsaturated monomer, and 5 to 200 parts by weight of water is added. Is more preferably added to the system in which 10 to 100 parts by weight of water are present.
- the amount of water means the total amount of the water contained in the reaction system and the water used as necessary when adding the post-crosslinking agent.
- the reaction temperature in the post-crosslinking reaction is preferably 50 to 250 ° C, more preferably 60 to 180 ° C.
- the reaction time of the post-crosslinking cannot be unconditionally determined because it depends on the reaction temperature, the type and amount of the post-crosslinking agent, etc., but it is usually 1 to 300 minutes, preferably 5 to 200 minutes.
- a drying step of removing water, a hydrocarbon dispersion medium, and the like by distillation by externally applying energy such as heat to the hydrous gel-like material may be included.
- the drying process may be performed under normal pressure or under reduced pressure. Further, from the viewpoint of enhancing the drying efficiency, the drying may be performed under a stream of nitrogen or the like.
- the drying temperature is preferably 70 to 250 ° C, more preferably 80 to 180 ° C.
- the drying temperature is preferably 40 to 160 ° C, more preferably 50 to 110 ° C.
- One particularly preferable embodiment of the water-absorbent resin of the present disclosure is a water-absorbent resin produced through a polymerization reaction of a water-soluble ethylenically unsaturated monomer, and the water-absorbent resin according to the following (3) to (3)
- a water-absorbent resin satisfying at least two of the conditions can be mentioned.
- a water-absorbent resin that satisfies all three conditions is more preferable.
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the first stage polymerization to the internal cross-linking agent is 10 ⁇ 10 3 to 15 ⁇ . It is 10 3 .
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the second stage polymerization to the internal crosslinking agent is 15 ⁇ 10 3 to 25 ⁇ . It is 10 3 .
- a water-absorbent resin By preparing a water-absorbent resin so as to satisfy at least two or three of these conditions, a water-absorbent resin exhibiting the above-mentioned preferable overall absorption capacity term ⁇ and water-absorption rate term ⁇ , and thus a preferable dry-up index, is prepared. It may be possible. In other words, the water absorbent resin of the present disclosure can be preferably prepared using these conditions (i) to (iii) as an index.
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the first-stage polymerization and the internal crosslinking agent is more preferably Is 10 ⁇ 10 3 to 14 ⁇ 10 3 , more preferably 10 ⁇ 10 3 to 13 ⁇ 10 3 , and even more preferably 11 ⁇ 10 3 to 12 ⁇ 10 3 .
- the molar ratio of the water-soluble ethylenically unsaturated monomer used in the second-stage polymerization to the internal crosslinking agent is more preferably Is 17.5 ⁇ 10 3 to 24 ⁇ 10 3 , more preferably 20 ⁇ 10 3 to 23 ⁇ 10 3 , and even more preferably 21 ⁇ 10 3 to 22 ⁇ 10 3 .
- the molar ratio of the total amount of the water-soluble ethylenically unsaturated monomer used in the polymerization for preparing the resin used for the post-crosslinking reaction and the post-crosslinking agent is more preferably 3 ⁇ 10 3 to 4 ⁇ 10 3 , and further preferably 3 ⁇ 10 3 to 3.5 ⁇ 10 3 .
- the present disclosure also preferably includes a method for producing a water absorbent resin so as to satisfy these conditions.
- Absorbent body, absorbent article constitutes an absorbent body together with hydrophilic fibers, for example.
- Such an absorber is suitably used for absorbent articles used for sanitary products, sanitary materials such as disposable diapers.
- the absorber is composed of, for example, a water absorbent resin and hydrophilic fibers.
- a mixed dispersion obtained by mixing the water-absorbent resin and the hydrophilic fiber so as to have a uniform composition a sandwich in which the water-absorbent resin is sandwiched between the layered hydrophilic fibers
- the absorbent body may be blended with other components, for example, an adhesive binder such as a heat-fusible synthetic fiber for enhancing the shape retention of the absorbent body, a hot melt adhesive, or an adhesive emulsion. ..
- the content of the water absorbent resin of the present disclosure in the absorber is preferably 5 to 50% by mass, more preferably 10 to 45% by mass, and 15 to 40% by mass based on the total mass of the absorbent. % Is more preferable.
- hydrophilic fibers cotton-like pulp obtained from wood, cellulose fibers such as mechanical pulp, chemical pulp, and semi-chemical pulp, artificial cellulose fibers such as rayon and acetate, synthetic polyamide such as hydrophilized polyamide, polyester, and polyolefin. Fibers made of resin can be used.
- An absorbent article is obtained by holding the absorber between a liquid-permeable sheet (top sheet) through which liquid can pass and a liquid-impermeable sheet (back sheet) through which liquid cannot pass.
- the liquid-permeable sheet is disposed on the side that contacts the body, and the liquid-impermeable sheet is disposed on the opposite side that contacts the body.
- the liquid permeable sheet includes air-through type, spun bond type, chemical bond type, needle punch type non-woven fabrics made of fibers such as polyethylene, polypropylene and polyester, and porous synthetic resin sheets.
- Examples of the liquid impermeable sheet include a synthetic resin film made of a resin such as polyethylene, polypropylene and polyvinyl chloride.
- the cotton bag was dehydrated for 1 minute using (product of Japan Centrifuge Co., Ltd., product number: H-122), and the mass Wc (g) of the cotton bag containing the swollen gel after dehydration was measured. The same operation was performed without adding the water absorbent resin, the empty mass Wd (g) of the cotton bag when wet was measured, and the water retention capacity of the physiological saline was determined by the following formula.
- the measuring device X shown in FIG. 1 includes a buret unit 1, a conduit 2, a measuring table 3, and a measuring unit 4 placed on the measuring table 3.
- a rubber stopper 14 is connected to the upper part of the buret 10
- an air introducing pipe 11 and a cock 12 are connected to the lower part thereof
- a cock 13 is provided on the upper part of the air introducing pipe 11.
- a conduit 2 is attached from the burette portion 1 to the measuring table 3, and the diameter of the conduit 2 is 6 mm.
- a hole having a diameter of 2 mm is formed in the central portion of the measuring table 3 to which the conduit 2 is connected.
- the measuring unit 4 includes a cylinder 40, a nylon mesh 41 attached to the bottom of the cylinder 40, and a weight 42.
- the inner diameter of the cylinder 40 is 2.0 cm.
- the nylon mesh 41 is formed to have 200 mesh (opening 75 ⁇ m). Then, a predetermined amount of the water absorbing resin 5 is evenly spread on the nylon mesh 41.
- the weight 42 has a diameter of 1.9 cm and a mass of 59.8 g. The weight 42 is placed on the water absorbent resin 5, and a load of 2.07 kPa can be uniformly applied to the water absorbent resin 5.
- the cock 12 and the cock 13 of the buret part 1 are closed, and a 0.9 mass% sodium chloride aqueous solution (physiological saline solution) adjusted to 25 ° C. is put into the buret 10 from the upper part thereof, and the rubber is After plugging the upper part of the buret with the plug 14, the cock 12 and the cock 13 of the buret part 1 are opened.
- the height of the measuring table 3 is adjusted so that the tip of the conduit 2 at the center of the measuring table 3 and the air introduction port of the air introducing pipe 11 have the same height.
- the water absorbent resin 5 is evenly spread over the nylon mesh 41 of the cylinder 40, and the weight 42 is placed on the water absorbent resin 5.
- the measuring unit 4 is placed so that its central portion coincides with the conduit port at the central portion of the measuring table 3.
- the physiological saline absorption capacity under load of the water-absorbent resin 5 after 60 minutes from the start of water absorption was determined by the following formula using the specific gravity of physiological saline of 1.0 (g / mL).
- the measuring device Y shown in FIG. 2 is mainly composed of a weight 90, a support cylinder 91, a measuring unit including a piston 92, and a liquid supply unit mainly including a petri dish 6 and a glass filter 7.
- a 400-mesh (mesh opening 38 ⁇ m) stainless steel wire mesh 93 is adhered to one side (bottom surface) of a cylindrical plastic support cylinder 91 having an inner diameter of 60 mm, and the diameter inside the cylinder is slightly smaller than 60 mm.
- the piston 92 and the weight 90 are provided above the piston 92 that does not cause a gap between the wall surface of the support cylinder 91 and the vertical movement of the piston 92, and the weight of the piston 92 and the weight 90 is 4.82 kPa relative to the water absorbent resin 5.
- the load can be applied uniformly. Using such a measuring unit, 0.90 g of the water-absorbent resin 5 is uniformly sprayed on the wire net of the support cylinder 91, and then the piston 92 and the weight 90 are placed, and the mass Wf (g) of the measuring unit is set. It was measured.
- the mass Wf (g) is the sum of the masses of the support cylinder 91, the water absorbent resin 5, the piston 92, and the weight 90.
- a glass filter 7 having a diameter of 90 mm and a thickness of 5 mm was placed inside a Petri dish 6 having a diameter of 150 mm, and a 0.9 mass% sodium chloride aqueous solution (physiological saline) adjusted to 25 ⁇ 1 ° C. ) Is added to the Petri dish 6 so as to be at the same height as the upper surface of the glass filter 7, and then a piece of filter paper 8 (made by ADVANTEC, No. 2) having a diameter of 9 cm is placed on the upper portion of the glass filter 7. A liquid supply part was prepared. It was confirmed that the entire surface of the filter paper 8 was wet, and if there was excess liquid, it was blotted appropriately with a tissue.
- the measurement unit was placed on the liquid supply unit to allow the water absorbent resin 5 to absorb physiological saline under a load of 4.82 kPa.
- physiological saline was appropriately added to keep the liquid surface level constant. 60 minutes after the measurement part was placed on the liquid supply part, the measurement part was removed from the liquid supply part, and the mass Wg (g) was measured.
- the physiological saline absorption capacity (g / g) under a load of 4.82 kPa was calculated by the following formula.
- Dynamic Water Absorption Rate The measurement of the dynamic water absorption rate was performed in a room controlled at 25 ° C ⁇ 1 ° C. 50 ⁇ 0.1 g of physiological saline weighed in a 100 mL beaker was adjusted to a temperature of 25 ⁇ 0.2 ° C. in a constant temperature water bath, and then stirred with a magnetic stirrer bar (8 mm ⁇ ⁇ 30 mm ring was not used). Then, a vortex was generated at a rotation speed of 600 r / min.
- the dynamic water absorption rate (1 / min) was calculated as follows.
- Dynamic water absorption rate (1 / min) (Amount of physiological saline / Amount of water absorbent resin) / (Dynamic water absorption time (min))
- the amount of physiological saline in the burette 10 is continuously reduced from the time when the water-absorbent resin 5 under no load starts to absorb water (the amount of physiological saline absorbed by the water-absorbent resin 5) Wi (mL) was read, and the amount of physiological saline absorbed by the water absorbent resin 5 by a specific time was calculated by the following formula.
- the specific gravity of physiological saline was 1.0 g / mL.
- Amount of physiological saline absorbed by the water absorbent resin 5 Wi (mL) ⁇ 1.0 (g / mL) / mass of the water absorbent resin (g)
- the time (second) required for the water-absorbent resin 5 to absorb 25 g of physiological saline solution per 1 g The time elapsed from the time when the water-soluble resin 5 started to absorb water was measured by a stopwatch and converted into minutes, which was defined as the static water absorption time (minutes).
- the static water absorption rate (1 / min) was calculated by the following formula.
- Static water absorption rate (1 / min) 25 (g / g) ⁇ static water absorption time (min)
- a polyethylene air-through type porous liquid permeable sheet having the same size as the absorbent body and a basis weight of 22 g / m 2 is arranged on the upper surface of the absorbent body, and a polyethylene liquid impermeable sheet of the same size and the same basis weight is provided.
- the absorbent article for study was prepared by disposing the sheet on the lower surface of the absorbent body and sandwiching the absorbent body.
- Example 1 As a reflux condenser, a dropping funnel, a nitrogen gas introducing pipe, and a stirrer, a round bottom cylindrical separable flask having an inner diameter of 110 mm and a volume of 2 L equipped with a stirrer having two inclined paddle blades with a blade diameter of 50 mm in two stages was used. Got ready. To this flask, 300 g of n-heptane as a hydrocarbon dispersion medium was added, and 0.62 g of a maleic anhydride-modified ethylene / propylene copolymer (Mitsui Chemicals, Inc., Hiwax 1105A) was added as a polymer dispersant and stirred. After heating and dissolving, the mixture was cooled to 50 ° C.
- n-heptane as a hydrocarbon dispersion medium
- 0.62 g of a maleic anhydride-modified ethylene / propylene copolymer Mitsubishi Chemicals, Inc., Hiwax 1105
- a monomer aqueous solution having a monomer concentration of 30% by mass.
- the monomer concentration is the mass ratio of the water-soluble ethylenically unsaturated monomer and its salt to the total amount of the aqueous monomer solution, and will be referred to hereinafter in the same manner in the present specification.
- the monomer aqueous solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then sucrose stearate ester of HLB3 as a surfactant (5.62 g of n-heptane (Mitsubishi Chemical Foods Co., Ltd., After further adding 6.2 g of a surfactant solution prepared by heating and dissolving 0.62 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 155.3 g of water was extracted from the system while refluxing n-heptane by azeotropic distillation of n-heptane and water. Then, 3.90 g (0.45 mmol) of a 2% by mass aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to give a spherical medium particle size. 82.1 g of 350 ⁇ m water absorbent resin was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- Example 2 As a reflux condenser, a dropping funnel, a nitrogen gas introducing pipe, and a stirrer, a round bottom cylindrical separable flask having an inner diameter of 110 mm and a volume of 2 L equipped with a stirrer having two inclined paddle blades with a blade diameter of 50 mm in two stages was used. Got ready. To this flask, 300 g of n-heptane as a hydrocarbon dispersion medium was added, and 0.52 g of a maleic anhydride-modified ethylene / propylene copolymer (Mitsui Chemicals, Inc., Hiwax 1105A) was added as a polymer dispersant and stirred. After heating and dissolving, the mixture was cooled to 50 ° C.
- n-heptane as a hydrocarbon dispersion medium
- 0.52 g of a maleic anhydride-modified ethylene / propylene copolymer Mitsubishi Chemicals, Inc., Hiwax 1105
- aqueous monomer solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then 4.68 g of n-heptane was added to sucrose stearate ester of HLB3 as a surfactant (Mitsubishi Chemical Foods Co., Ltd., After further adding 5.2 g of a surfactant solution prepared by heating and dissolving 0.52 g of Ryoto Sugar Ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- the entire amount of the second-stage monomer aqueous solution was added to the first-stage polymerization slurry liquid, and the system was sufficiently replaced with nitrogen, and then, again, The flask was immersed in a 70 ° C. water bath to raise the temperature, and the second stage polymerization was carried out for 30 minutes.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 259.3 g of water was extracted from the system while refluxing n-heptane by azeotropic distillation of n-heptane and water. Then, 4.78 g (0.55 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to aggregate spherical particles. 173.3 g of a water absorbent resin having a medium particle size of 332 ⁇ m was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- Example 3 As a reflux condenser, a dropping funnel, a nitrogen gas introducing pipe, and a stirrer, a round bottom cylindrical separable flask having an inner diameter of 110 mm and a volume of 2 L equipped with a stirrer having two inclined paddle blades with a blade diameter of 50 mm in two stages was used. Got ready. To this flask, 300 g of n-heptane as a hydrocarbon dispersion medium was added, and 0.62 g of a maleic anhydride-modified ethylene / propylene copolymer (Mitsui Chemicals, Inc., Hiwax 1105A) was added as a polymer dispersant and stirred. After heating and dissolving, the mixture was cooled to 50 ° C.
- n-heptane as a hydrocarbon dispersion medium
- 0.62 g of a maleic anhydride-modified ethylene / propylene copolymer Mitsubishi Chemicals, Inc., Hiwax 1105
- the monomer aqueous solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then sucrose stearate ester of HLB3 as a surfactant (5.62 g of n-heptane (Mitsubishi Chemical Foods Co., Ltd., After further adding 6.2 g of a surfactant solution prepared by heating and dissolving 0.62 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 263.8 g of water was taken out of the system while refluxing n-heptane by azeotropic distillation of n-heptane and water. Then, 5.66 g (0.65 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to aggregate spherical particles. 205.5 g of a water absorbent resin having a medium particle diameter of 350 ⁇ m was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- Example 4 As a reflux condenser, a dropping funnel, a nitrogen gas introducing pipe, and a stirrer, a round bottom cylindrical separable flask having an inner diameter of 110 mm and a volume of 2 L equipped with a stirrer having two inclined paddle blades with a blade diameter of 50 mm in two stages was used. Got ready. To this flask, 300 g of n-heptane as a hydrocarbon dispersion medium was added, and 0.74 g of a maleic anhydride-modified ethylene / propylene copolymer (Mitsui Chemicals, Inc., Hiwax 1105A) was added as a polymer dispersant and stirred. After heating and dissolving, the mixture was cooled to 50 ° C.
- aqueous monomer solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then 6.62 g of n-heptane was added to sucrose stearate ester of HLB3 as a surfactant (Mitsubishi Chemical Foods Co., Ltd., After further adding 7.3 g of a surfactant solution prepared by heating and dissolving 0.74 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- the entire amount of the second-stage monomer aqueous solution was added to the first-stage polymerization slurry liquid, and the system was sufficiently replaced with nitrogen, and then, again, The flask was immersed in a 70 ° C. water bath to raise the temperature, and the second stage polymerization was carried out for 30 minutes.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and azeotropic distillation of n-heptane and water was carried out, and 283.6 g of water was extracted from the system while refluxing the n-heptane. Then, 6.62 g (0.76 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to aggregate spherical particles. 230.7 g of a water absorbent resin having a medium particle diameter of 361 ⁇ m was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- aqueous monomer solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then 6.62 g of n-heptane was added to sucrose stearate ester of HLB3 as a surfactant (Mitsubishi Chemical Foods Co., Ltd., After further adding 7.4 g of a surfactant solution obtained by heating and dissolving 0.74 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- the whole amount of the second-stage monomer aqueous solution was added to the first-stage polymerization slurry liquid, and the system was sufficiently replaced with nitrogen.
- the flask was again immersed in a 70 ° C. water bath to raise the temperature, and the second stage polymerization was carried out for 30 minutes.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 266.2 g of water was extracted from the system while azeotropically distilling n-heptane and water while refluxing the n-heptane. Then, 4.42 g (0.51 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to form spherical particles in an aggregated form. 229.7 g of a water absorbent resin having a medium particle diameter of 345 ⁇ m was obtained.
- the water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- aqueous monomer solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then 6.66 g of n-heptane was added to sucrose stearate ester of HLB3 as a surfactant (Mitsubishi Chemical Foods Co., Ltd., After further adding 7.4 g of a surfactant solution obtained by heating and dissolving 0.74 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- the whole amount of the second-stage monomer aqueous solution was added to the first-stage polymerization slurry liquid, and the system was sufficiently replaced with nitrogen.
- the flask was again immersed in a 70 ° C. water bath to raise the temperature, and the second stage polymerization was carried out for 30 minutes.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 261.8 g of water was taken out of the system while refluxing n-heptane by azeotropic distillation of n-heptane and water. Then, 4.42 g (0.51 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to form spherical particles in an aggregated form. 234.2 g of a water absorbent resin having a medium particle diameter of 380 ⁇ m was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- the monomer aqueous solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then 6.66 g of n-heptane was added to sucrose stearate ester of HLB3 as a surfactant (Mitsubishi Chemical Foods Co., Ltd., After further adding 7.4 g of a surfactant solution obtained by heating and dissolving 0.74 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- a surfactant solution obtained by heating and dissolving 0.74 g of lyoto sugar ester S-370
- the whole amount of the second-stage monomer aqueous solution was added to the first-stage polymerization slurry liquid, and the system was sufficiently replaced with nitrogen.
- the flask was again immersed in a 70 ° C. water bath to raise the temperature, and the second stage polymerization was carried out for 30 minutes.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 259.7 g of water was taken out of the system while refluxing n-heptane by azeotropic distillation of n-heptane and water. Then, 4.42 g (0.51 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to form spherical particles in an aggregated form. 236.0 g of a water absorbent resin having a medium particle size of 356 ⁇ m was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- the monomer aqueous solution prepared as described above was added to a separable flask and stirred for 10 minutes, and then 6.66 g of n-heptane was added to sucrose stearate ester of HLB3 as a surfactant (Mitsubishi Chemical Foods Co., Ltd., After further adding 7.4 g of a surfactant solution obtained by heating and dissolving 0.74 g of lyoto sugar ester S-370) and thoroughly replacing the inside of the system with nitrogen while stirring, the flask was immersed in a water bath at 70 ° C. Then, the temperature was raised and the polymerization was carried out for 60 minutes to obtain a first stage polymerization slurry liquid.
- a surfactant solution obtained by heating and dissolving 0.74 g of lyoto sugar ester S-370
- the whole amount of the second-stage monomer aqueous solution was added to the first-stage polymerization slurry liquid, and the system was sufficiently replaced with nitrogen.
- the flask was again immersed in a 70 ° C. water bath to raise the temperature, and the second stage polymerization was carried out for 30 minutes.
- the temperature of the reaction solution was raised in an oil bath at 125 ° C., and 260.8 g of water was extracted from the system while azeotropically distilling n-heptane and water while refluxing the n-heptane. Then, 6.62 g (0.76 mmol) of a 2 mass% aqueous solution of ethylene glycol diglycidyl ether was added as a post-crosslinking agent, and the mixture was kept at 80 ° C. for 2 hours. Then, n-heptane was evaporated to dryness to obtain a dried product.
- amorphous silica (Evonik Degussa Japan Ltd., Carplex # 80) was mixed with this dried product, and the mixture was passed through a sieve with an opening of 1000 ⁇ m to form spherical particles in an aggregated form. 235.1 g of a water absorbent resin having a medium particle diameter of 343 ⁇ m was obtained. The water absorbent resin and the absorber using the water absorbent resin were evaluated according to the various test methods described above.
- Table 1 shows the evaluation test results of the water absorbent resin.
- Table 2 shows the dry-up index of the water-absorbent resin, the overall absorption capacity term ⁇ , the water-absorption rate term ⁇ , and the evaluation result (reverse amount) of the absorbent body using the water-absorbent resin.
- the total absorption capacity term ⁇ and the water absorption rate term ⁇ are obtained by the following equations (2) and (3).
Abstract
吸収体において吸水性樹脂が少ない含有量で用いられた際でも、被吸収液の逆戻り量を低減できる吸水性樹脂及びその吸収性樹脂を含む吸収体を用いた吸収性物品が提供される。 当該吸水性樹脂は、水溶性エチレン性不飽和単量体の重合体架橋物である吸水性樹脂であって、以下式(1)で示されるドライアップ指数が1.85以上である吸水性樹脂である。式(1) ここで、総括吸収容量項αと吸水速度項βは、以下式(2)および(3)により求められる。
Description
本開示は、吸水性樹脂及びその製造方法等に関する。より詳しくは、生理用品、紙おむつ等の衛生材料用途の吸収性物品に好適に用いられる吸収体を構成する吸水性樹脂及びその製造方法等に関する。
生理用品、紙おむつ等の衛生材料用途の吸収性物品は、通常、親水性繊維と吸水性樹脂とを主な構成単位とする吸収体からなり、吸水性樹脂としては、例えば、澱粉-アクリロニトリルグラフト共重合体の加水分解物、澱粉-アクリル酸グラフト共重合体の中和物、酢酸ビニル-アクリル酸エステル共重合体のけん化物、ポリアクリル酸部分中和物等が知られている。
一般的に、吸収体において吸水性樹脂の含有量が少ない場合、吸収体の風合い(柔らかさ)が優れる一方で、逆戻り量に代表される吸収性能は満足できるものではない。そこで、吸水性樹脂の含有量を増加させれば、体液等の吸収後における吸収性能は向上する傾向にあるが、吸収体の風合いは損なわれる。また、多量の吸水性樹脂を吸収体中に均一に分散させることは難しく、吸水性樹脂が増量された分の性能向上は必ずしも保証されない。このように、吸収体において吸収性能と風合いの両方を満たすことは、困難とされている。そのため、吸収体における含有量が少量であっても、吸収体における吸収性能を高められる特性を有する吸水性樹脂が望まれている。
衛生材料用途の吸収性物品における吸収性能を高めるために、吸水性樹脂に望まれる特性としては、高い保水能や荷重下での高い吸水能等が挙げられる。このような特性を備えた吸水性樹脂を得るため、これまでにも研究が進められてきている。例えば、次のような提案がなされている。すなわち、水溶性エチレン性不飽和単量体の逆相懸濁重合を多段に行なうことにより、優れた吸水性に加えて、得られる吸水性樹脂の粒径が大きく、微粉が少なく、分布がシャープで、水に対する濡れ性の高い吸水性樹脂の製造方法(特許文献1参照)、水溶性エチレン性不飽和単量体を逆相懸濁重合して、特定の中位粒子径の1次粒子を得た後、第2段目の水溶性エチレン性不飽和単量体を添加して逆相懸濁重合反応を行ない、特定の中位粒子径の2次粒子を得る吸水性樹脂粒子の製造方法(特許文献2参照)、並びに、水溶性エチレン性不飽和単量体を逆相懸濁重合法によって重合して得られた1次粒子を、さらに逆相懸濁重合法によって凝集させて形成される吸水性樹脂であって、該1次粒子の中位粒子径が100~250μmであり、該吸水性樹脂の生理食塩水保水能が30g/g以下であることを特徴とする吸水性樹脂(特許文献3参照)等が知られている。
本開示は、吸水性樹脂の含有量が少ない吸収体においても、吸収体からの被吸収液の逆戻り量の低減を可能とする吸水性樹脂の提供を主な目的とする。
本発明者らは、特定の指数を満たす吸水性樹脂を用いると、吸水性樹脂の含有量が少ない吸収体においても、吸収体からの被吸収液の逆戻り量が低減し得る可能性を見出し、さらに改良を重ねた。
本開示は例えば以下の項に記載の主題を包含する。
項1.水溶性エチレン性不飽和単量体の重合体架橋物である吸水性樹脂であって、以下式(1)で示されるドライアップ指数が1.85以上である吸水性樹脂。
ここで、総括吸収容量項αと吸水速度項βは、以下式(2)および(3)により求められる。
項2.総括吸収容量項αが0.95以上である、項1に記載の吸水性樹脂。
項3.吸水速度項βが1.56以上である、項1又は2に記載の吸水性樹脂。
項4.生理食塩水吸水能と生理食塩水保水能の差が18以下である、項1~3のいずれかに記載の吸水性樹脂。
項5.2.07kPa荷重下での生理食塩水吸水能と4.82kPa荷重下での生理食塩水吸水能の差が17~36である、項1~4のいずれかに記載の吸水性樹脂。
項6.項1~5あるいは以下の項A~Fのいずれかに記載の吸水性樹脂を5~50質量%含む吸収体。
項A.
以下の条件(i)、(ii)、及び(iii)の少なくとも2つの条件を満たす重合方法により調製され、水溶性エチレン性不飽和単量体の重合体架橋物である吸水性樹脂。
(i)1段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が10×103~15×103である。
(ii)2段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が15×103~25×103である。
(iii)後架橋反応に用いる樹脂を調製した重合で用いた水溶性エチレン性不飽和単量体の合計量と後架橋剤とのモル比(水溶性エチレン性不飽和単量体/後架橋剤)が2.5×103~4.5×103である。
項B.
以下の条件(i)、(ii)、及び(iii)の少なくとも2つの条件を満たす重合方法により調製される、項1~5のいずれかに記載の吸水性樹脂。
(i)1段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が10×103~15×103である。
(ii)2段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が15×103~25×103である。
(iii)後架橋反応に用いる樹脂を調製した重合で用いた水溶性エチレン性不飽和単量体の合計量と後架橋剤とのモル比(水溶性エチレン性不飽和単量体/後架橋剤)が2.5×103~4.5×103である。
項C.
水溶性エチレン性不飽和単量体が、(メタ)アクリル酸である、項A又はBに記載の吸水性樹脂。
項D.
少なくとも(i)及び(ii)のいずれかを満たし、内部架橋剤が、(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、及び(ポリ)グリセリンジグリシジルエーテルからなる群より選択される少なくとも1種である、項A~Cのいずれかに記載の吸水性樹脂。
項E.
少なくとも(iii)を満たし、後架橋剤が、(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、及び(ポリ)グリセリンジグリシジルエーテルからなる群より選択される少なくとも1種である、項A~Cのいずれかに記載の吸水性樹脂。
項F.
条件(i)、(ii)、及び(iii)の少なくとも2つの条件を満たす重合方法が、逆相懸濁重合(好ましくは1段または2段の逆相懸濁重合、より好ましくは2段の逆相懸濁重合)である、項A~Eのいずれかに記載の吸水性樹脂。
項G.
前記重合方法が、条件(i)、(ii)、及び(iii)の全ての条件を満たす重合方法である、項A~Fのいずれかに記載の吸水性樹脂。
項A.
以下の条件(i)、(ii)、及び(iii)の少なくとも2つの条件を満たす重合方法により調製され、水溶性エチレン性不飽和単量体の重合体架橋物である吸水性樹脂。
(i)1段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が10×103~15×103である。
(ii)2段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が15×103~25×103である。
(iii)後架橋反応に用いる樹脂を調製した重合で用いた水溶性エチレン性不飽和単量体の合計量と後架橋剤とのモル比(水溶性エチレン性不飽和単量体/後架橋剤)が2.5×103~4.5×103である。
項B.
以下の条件(i)、(ii)、及び(iii)の少なくとも2つの条件を満たす重合方法により調製される、項1~5のいずれかに記載の吸水性樹脂。
(i)1段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が10×103~15×103である。
(ii)2段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が15×103~25×103である。
(iii)後架橋反応に用いる樹脂を調製した重合で用いた水溶性エチレン性不飽和単量体の合計量と後架橋剤とのモル比(水溶性エチレン性不飽和単量体/後架橋剤)が2.5×103~4.5×103である。
項C.
水溶性エチレン性不飽和単量体が、(メタ)アクリル酸である、項A又はBに記載の吸水性樹脂。
項D.
少なくとも(i)及び(ii)のいずれかを満たし、内部架橋剤が、(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、及び(ポリ)グリセリンジグリシジルエーテルからなる群より選択される少なくとも1種である、項A~Cのいずれかに記載の吸水性樹脂。
項E.
少なくとも(iii)を満たし、後架橋剤が、(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、及び(ポリ)グリセリンジグリシジルエーテルからなる群より選択される少なくとも1種である、項A~Cのいずれかに記載の吸水性樹脂。
項F.
条件(i)、(ii)、及び(iii)の少なくとも2つの条件を満たす重合方法が、逆相懸濁重合(好ましくは1段または2段の逆相懸濁重合、より好ましくは2段の逆相懸濁重合)である、項A~Eのいずれかに記載の吸水性樹脂。
項G.
前記重合方法が、条件(i)、(ii)、及び(iii)の全ての条件を満たす重合方法である、項A~Fのいずれかに記載の吸水性樹脂。
吸収体において吸水性樹脂が少ない含有量で用いられた際でも、逆戻り量を低減できる吸水性樹脂、及びその製造方法等が提供される。
以下、本開示に包含される各実施形態について、さらに詳細に説明する。本開示は、特定の吸水性樹脂及びその製造方法等を好ましく包含するが、これらに限定されるわけではなく、本開示は本明細書に開示され当業者が認識できる全てを包含する。
1.吸水性樹脂
本開示に包含される吸水性樹脂は、水溶性エチレン性不飽和単量体の重合体により構成される吸水性樹脂であって、以下式(1)に示す、総括吸収容量項αと吸水速度項βの積で表されるドライアップ指数が、1.85以上である吸水性樹脂である。当該吸水性樹脂を「本開示の吸水性樹脂」と表記することがある。
本開示に包含される吸水性樹脂は、水溶性エチレン性不飽和単量体の重合体により構成される吸水性樹脂であって、以下式(1)に示す、総括吸収容量項αと吸水速度項βの積で表されるドライアップ指数が、1.85以上である吸水性樹脂である。当該吸水性樹脂を「本開示の吸水性樹脂」と表記することがある。
本開示の吸水性樹脂は、ドライアップ指数が1.9~5.0であることが好ましい。当該数値範囲の下限は、例えば、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、又は2.9であってもよい。また、当該数値範囲の上限は、例えば、4.9、4.8、4.7、4.6、4.5、4.4、4.3、4.2、4.1、4.0、3.9、3.8、3.7、3.6、3.5、3.4、3.3、3.2、3.1、又は3.0であってもよい。当該数値範囲は、例えば、2.0~4.0がより好ましく、2.1~3.0がさらに好ましい。
吸水性樹脂の総括吸収容量項αは、以下式(2)により算出される値である。
ここで、本明細書において、「生理食塩水吸水能」は、生理食塩水500gを600r/minで撹拌させながら、吸水性樹脂2.0gを60分間撹拌した後、目開きが75μmの標準篩を用いてろ過し、篩を水平に対して約30度の傾斜角となるように傾けた状態で30分間静置した後、膨潤ゲルの質量を測定することにより求めた値である。
本明細書において、「生理食塩水保水能」は、生理食塩水500gを600r/minで撹拌させながら、吸水性樹脂2.0gを30分間撹拌した後、綿袋(メンブロード60番)中に注ぎ込み、遠心力が167Gとなるよう設定した脱水機を用いて綿袋を1分間脱水し、脱水後の膨潤ゲルの質量を測定することにより求めた値である。
本明細書において、「2.07kPa荷重下での生理食塩水吸水能」は、200メッシュのナイロンメッシュを付した内径2.0cmの円筒に均一に撒布された吸水性樹脂0.1gに対して、重りにより2.07kPaの荷重を均一に加えられた状態で、吸水性樹脂を吸水させ、吸水開始から60分間経過後における生理食塩水の量を測定することにより求められる。当該測定には、図1に概略構成を示した測定装置Xを好適に用いることができる。
なお、上記の通り、本明細書では、単に、生理食塩水吸水能、と表記した場合と、2.07kPa荷重下での生理食塩水吸水能、と断って表記した場合とは、明確に区別される。
本明細書において、「4.82kPa荷重下での生理食塩水吸水能」は、400メッシュの金網を付した内径60mmの支持円筒に投入された吸水性樹脂0.9gに対して、重りにより4.82kPaの荷重を均一に加えられた状態で、吸水性樹脂を吸水させ、吸水開始から60分間経過後における生理食塩水の量を測定することにより求められる。当該測定には、図2に概略構成を示した測定装置Yを好適に用いることができる。
なお、上記の通り、本明細書では、単に、生理食塩水吸水能、と表記した場合と、4.82kPa荷重下での生理食塩水吸水能、と断って表記した場合とは、明確に区別される。
本開示の吸水性樹脂は、総括吸収容量項αが0.95以上であることが好ましく、1.00~4.00がより好ましく、1.05~2.00がさらに好ましい。当該上限は、4.00、3.00、2.00、1.80、1.65、又は1.55であってもよい。
本開示の吸水性樹脂は、生理食塩水吸水能と生理食塩水保水能の差が18以下であることが好ましく、17以下がより好ましく、16以下がさらに好ましい。また、生理食塩水吸水能と生理食塩水保水能の差は、5以上であることが好ましく、8以上がより好ましく、10以上がさらに好ましい。
また、本開示に係る吸水性樹脂の2.07kPa荷重下での生理食塩水吸水能と4.82kPa荷重下での生理食塩水吸水能との差は17~36であることが好ましく、17~33がより好ましく、17~30がさらに好ましい。
吸水性樹脂の吸水速度項βは、静的吸水速度に対する動的吸水速度の比で表され、以下式(3)により算出される。
ここで、動的吸水速度は、25℃の温度の生理食塩水50gを8mmφ×30mmのマグネチックスターラーバーで攪拌して、回転数600r/minで渦を発生させ、吸水性樹脂2.0gを一度に添加し、吸水性樹脂の添加後から液面の渦が収束する時点までの時間を測定することにより求められる。より具体的には、後述される実施例に記載の方法により測定した値である。
静的吸水速度は、200メッシュのナイロンメッシュを付した内径2.0cmの円筒に均一に撒布された吸水性樹脂0.1gに対して、重りによる荷重を加えることない状態で、吸水性樹脂が吸水した生理食塩水の量を測定し、吸水性樹脂が1gあたり25gの生理食塩水を吸収するまでに要した時間(秒)を測定することにより求められる。より具体的には、後述される実施例に記載の方法により測定した値である。当該測定には、図1に概略構成を示した測定装置Xを好適に用いることができる。
本開示の吸水性樹脂は、吸水速度項βが1.56以上であることが好ましく、1.60以上がより好ましく、1.65以上がさらに好ましい。上限は特に制限されないが、3以下が好ましく、2.95、2.9、2.85、2.8、2.75、又は2.7以下であってもよい。
また、生理用品、紙おむつ等の衛生材料に用いられた場合に、装着時の異物感を少なくする観点から、本開示に係る吸水性樹脂は、中位粒子径が200~600μmが好ましく、250~550μmがより好ましく、300~500μmがさらに好ましい。
なお、得られた吸水性樹脂に、目的に応じた添加剤を配合してもよい。このような添加剤としては、無機粉末、界面活性剤、酸化剤、還元剤、金属キレート剤、ラジカル連鎖禁止剤、酸化防止剤、抗菌剤、消臭剤等が挙げられる。例えば、吸水性樹脂100質量部に対し、無機粉末として0.05~5質量部の非晶質シリカを添加することで、吸水性樹脂の流動性を向上させることができる。
2.吸水性樹脂の製造方法
本開示の吸水性樹脂を得るための方法としては、例えば、逆相懸濁重合法、水溶液重合法等が挙げられる。以下に、本開示の吸水性樹脂に関して、その製造方法の一例として、逆相懸濁重合法についてより詳しく説明する。
本開示の吸水性樹脂を得るための方法としては、例えば、逆相懸濁重合法、水溶液重合法等が挙げられる。以下に、本開示の吸水性樹脂に関して、その製造方法の一例として、逆相懸濁重合法についてより詳しく説明する。
<2-1.重合工程>
重合は、吸水性樹脂調製分野で公知の方法又は公知の方法から想到できる方法により行うことができる。中でも、逆相懸濁重合が好ましい。逆相懸濁重合法では、分散安定剤の存在下、炭化水素分散媒中、ラジカル重合開始剤および必要に応じて架橋剤(内部架橋剤)を含む水溶性エチレン性不飽和単量体水溶液を撹拌混合し、加熱することにより重合が行われる。
重合は、吸水性樹脂調製分野で公知の方法又は公知の方法から想到できる方法により行うことができる。中でも、逆相懸濁重合が好ましい。逆相懸濁重合法では、分散安定剤の存在下、炭化水素分散媒中、ラジカル重合開始剤および必要に応じて架橋剤(内部架橋剤)を含む水溶性エチレン性不飽和単量体水溶液を撹拌混合し、加熱することにより重合が行われる。
重合反応は、1段で行ってもよく、或いは、2段以上の多段で行ってもよい。多段の場合、段数は生産性を高める観点から、2又は3段であることが好ましい。多段の重合を行う場合には、後述する方法で1段目の重合を行った後、1段目の重合反応で得られた反応混合物に水溶性エチレン性不飽和単量体水溶液を添加し混合して、1段目と同様の方法で2段目の重合を行ってもよい。さらに多段の重合は、同様の操作を繰り返すことで行うこともできる。また、2段以上の重合を行う場合、重合法としては同じ重合法を用いても異なった重合法を用いてもよく、同じ重合法を用いることがより好ましい。いずれの重合においても逆相懸濁重合を用いることがさらに好ましい。
2段目以降の各段における重合では、水溶性エチレン性不飽和単量体の他に、ラジカル重合開始剤、内部架橋剤などを、2段目以降の各段における重合の際に添加する水溶性エチレン性不飽和単量体の量を基準として、後述する水溶性エチレン性不飽和単量体に対する各成分のモル比の範囲内で添加して重合を行うことができる。
[水溶性エチレン性不飽和単量体]
水溶性エチレン性不飽和単量体としては、例えば、(メタ)アクリル酸(本明細書においては「アクリ」及び「メタクリ」を合わせて「(メタ)アクリ」と表記する。以下同様。)及びその塩;2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸及びその塩;(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド、2-ヒドロキシエチル(メタ)アクリレート、N-メチロール(メタ)アクリルアミド、ポリエチレングリコールモノ(メタ)アクリレート等の非イオン性単量体;N,N-ジエチルアミノエチル(メタ)アクリレート、N,N-ジエチルアミノプロピル(メタ)アクリレート、ジエチルアミノプロピル(メタ)アクリルアミド等のアミノ基含有不飽和単量体及びその4級化物等が挙げられる。これらの水溶性エチレン性不飽和単量体は単独で用いてもよいし、2種以上を組み合わせて用いてもよい。中でも、工業的に入手が容易である点から、(メタ)アクリル酸及びその塩、(メタ)アクリルアミド、N,N-ジメチルアクリルアミドが好ましく、(メタ)アクリル酸及びその塩がより好ましい。これらの中でも、例えば、アクリル酸及びその塩が吸水性樹脂の原材料として広く用いられており、これらアクリル酸及びその塩に、前述の他の水溶性エチレン性不飽和単量体を共重合させて用いることもできる。この場合、アクリル酸及びその塩は、主となる水溶性エチレン性不飽和単量体として、総水溶性エチレン性不飽和単量体に対して70~100モル%用いられることが好ましい。
水溶性エチレン性不飽和単量体としては、例えば、(メタ)アクリル酸(本明細書においては「アクリ」及び「メタクリ」を合わせて「(メタ)アクリ」と表記する。以下同様。)及びその塩;2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸及びその塩;(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド、2-ヒドロキシエチル(メタ)アクリレート、N-メチロール(メタ)アクリルアミド、ポリエチレングリコールモノ(メタ)アクリレート等の非イオン性単量体;N,N-ジエチルアミノエチル(メタ)アクリレート、N,N-ジエチルアミノプロピル(メタ)アクリレート、ジエチルアミノプロピル(メタ)アクリルアミド等のアミノ基含有不飽和単量体及びその4級化物等が挙げられる。これらの水溶性エチレン性不飽和単量体は単独で用いてもよいし、2種以上を組み合わせて用いてもよい。中でも、工業的に入手が容易である点から、(メタ)アクリル酸及びその塩、(メタ)アクリルアミド、N,N-ジメチルアクリルアミドが好ましく、(メタ)アクリル酸及びその塩がより好ましい。これらの中でも、例えば、アクリル酸及びその塩が吸水性樹脂の原材料として広く用いられており、これらアクリル酸及びその塩に、前述の他の水溶性エチレン性不飽和単量体を共重合させて用いることもできる。この場合、アクリル酸及びその塩は、主となる水溶性エチレン性不飽和単量体として、総水溶性エチレン性不飽和単量体に対して70~100モル%用いられることが好ましい。
なお、上述の水溶性エチレン性不飽和単量体は、逆相懸濁重合する際に、炭化水素分散媒中での分散性を向上させるために水溶液にして用いてもよい。このような水溶液中における水溶性エチレン性不飽和単量体の濃度は、通常、20質量%~飽和濃度以下とすればよいが、生産性を確保しつつ、得られる吸水性樹脂の吸水性能を高める観点から、水溶性エチレン性不飽和単量体の濃度は、20~50質量%が好ましく、22~45質量%がより好ましく、24~36質量%であることがさらに好ましい。
水溶性エチレン性不飽和単量体が(メタ)アクリル酸、2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸等のように酸基を有する場合、必要に応じてその酸基が予めアルカリ性中和剤により中和されたものを用いてもよい。このようなアルカリ性中和剤としては、例えば水酸化ナトリウム、炭酸ナトリウム、炭酸水素ナトリウム、水酸化カリウム、炭酸カリウム等のアルカリ金属塩;アンモニア等が挙げられる。特にこれらのアルカリ性中和剤は、中和操作を簡便にするために水溶液の状態にして用いてもよい。
上述のアルカリ性中和剤は単独で用いてもよいし、2種以上を組み合わせて用いてもよい。アルカリ性中和剤による水溶性エチレン性不飽和単量体の中和度については、得られる吸水性樹脂の浸透圧を高めることで吸水性能を高め、かつ余剰のアルカリ性中和剤の存在に起因する安全性等に問題が生じないようにする観点から、水溶性エチレン性不飽和単量体が有する全ての酸基に対する中和度として、40~90モル%が好ましく、70~88モル%がより好ましく、75~85モル%がさらに好ましく、77~80モル%がよりさらに好ましい。
[炭化水素分散媒]
炭化水素分散媒としては、例えば、n-ヘキサン、n-ヘプタン、2-メチルヘキサン、3-メチルヘキサン、2,3-ジメチルペンタン、3-エチルペンタン、n-オクタン等の炭素数6~8の脂肪族炭化水素;シクロヘキサン、メチルシクロヘキサン、シクロペンタン、メチルシクロペンタン、trans-1,2-ジメチルシクロペンタン、cis-1,3-ジメチルシクロペンタン、trans-1,3-ジメチルシクロペンタン等の脂環族炭化水素;ベンゼン、トルエン、キシレン等の芳香族炭化水素等が挙げられる。これらの炭化水素分散媒は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。これらの炭化水素分散媒の中でも、工業的に入手が容易であり、品質が安定しており、かつ安価である点で、n-ヘキサン、n-ヘプタン及びシクロヘキサンが好適に用いられる。また、上述炭化水素分散媒の混合物の例として、市販されているエクソールヘプタン(エクソンモービル社製:ヘプタン及びその異性体の炭化水素75~85質量%含有)等が挙げられ、このような市販品を用いることもできる。
炭化水素分散媒としては、例えば、n-ヘキサン、n-ヘプタン、2-メチルヘキサン、3-メチルヘキサン、2,3-ジメチルペンタン、3-エチルペンタン、n-オクタン等の炭素数6~8の脂肪族炭化水素;シクロヘキサン、メチルシクロヘキサン、シクロペンタン、メチルシクロペンタン、trans-1,2-ジメチルシクロペンタン、cis-1,3-ジメチルシクロペンタン、trans-1,3-ジメチルシクロペンタン等の脂環族炭化水素;ベンゼン、トルエン、キシレン等の芳香族炭化水素等が挙げられる。これらの炭化水素分散媒は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。これらの炭化水素分散媒の中でも、工業的に入手が容易であり、品質が安定しており、かつ安価である点で、n-ヘキサン、n-ヘプタン及びシクロヘキサンが好適に用いられる。また、上述炭化水素分散媒の混合物の例として、市販されているエクソールヘプタン(エクソンモービル社製:ヘプタン及びその異性体の炭化水素75~85質量%含有)等が挙げられ、このような市販品を用いることもできる。
炭化水素分散媒の使用量は、水溶性エチレン性不飽和単量体水溶液を均一に分散し、重合温度の制御を容易にする観点から、通常、1段目の水溶性エチレン性不飽和単量体100質量部に対して、80~1500質量部が好ましく、120~1200質量部がより好ましい。
[分散安定剤]
分散安定剤としては界面活性剤を用いることができ、例えば、ショ糖脂肪酸エステル、ポリグリセリン脂肪酸エステル、ソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレングリセリン脂肪酸エステル、ソルビトール脂肪酸エステル、ポリオキシエチレンソルビトール脂肪酸エステル、ポリオキシエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル、ポリオキシエチレンヒマシ油、ポリオキシエチレン硬化ヒマシ油、アルキルアリルホルムアルデヒド縮合ポリオキシエチレンエーテル、ポリオキシエチレンポリオキシプロピレンブロックコポリマー、ポリオキシエチレンポリオキシプロピルアルキルエーテル、ポリエチレングリコール脂肪酸エステル、アルキルグルコシド、N-アルキルグルコンアミド、ポリオキシエチレン脂肪酸アミド、ポリオキシエチレンアルキルアミン等を用いることができる。中でも、単量体水溶液の分散安定性の面から、ソルビタン脂肪酸エステル、ポリグリセリン脂肪酸エステル、ショ糖脂肪酸エステル等が好ましい。これらの界面活性剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
分散安定剤としては界面活性剤を用いることができ、例えば、ショ糖脂肪酸エステル、ポリグリセリン脂肪酸エステル、ソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレングリセリン脂肪酸エステル、ソルビトール脂肪酸エステル、ポリオキシエチレンソルビトール脂肪酸エステル、ポリオキシエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル、ポリオキシエチレンヒマシ油、ポリオキシエチレン硬化ヒマシ油、アルキルアリルホルムアルデヒド縮合ポリオキシエチレンエーテル、ポリオキシエチレンポリオキシプロピレンブロックコポリマー、ポリオキシエチレンポリオキシプロピルアルキルエーテル、ポリエチレングリコール脂肪酸エステル、アルキルグルコシド、N-アルキルグルコンアミド、ポリオキシエチレン脂肪酸アミド、ポリオキシエチレンアルキルアミン等を用いることができる。中でも、単量体水溶液の分散安定性の面から、ソルビタン脂肪酸エステル、ポリグリセリン脂肪酸エステル、ショ糖脂肪酸エステル等が好ましい。これらの界面活性剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
界面活性剤の使用量は、炭化水素分散媒中における、単量体水溶液の分散状態を良好に保ち、かつ使用量に見合う分散効果を得る観点から、重合のために用いる水溶性エチレン性不飽和単量体100質量部に対して、好ましくは0.05~30質量部、より好ましくは0.1~20質量部である。
また、分散安定剤として、界面活性剤とともに高分子系分散剤を併用してもよい。使用できる高分子系分散剤としては、無水マレイン酸変性ポリエチレン、無水マレイン酸変性ポリプロピレン、無水マレイン酸変性エチレン・プロピレン共重合体、無水マレイン酸変性EPDM(エチレン・プロピレン・ジエン・ターポリマー)、無水マレイン酸変性ポリブタジエン、無水マレイン酸・エチレン共重合体、無水マレイン酸・プロピレン共重合体、無水マレイン酸・エチレン・プロピレン共重合体、無水マレイン酸・ブタジエン共重合体、ポリエチレン、ポリプロピレン、エチレン・プロピレン共重合体、酸化型ポリエチレン、酸化型ポリプロピレン、酸化型エチレン・プロピレン共重合体、エチレン・アクリル酸共重合体、エチルセルロース、エチルヒドロキシエチルセルロース等が挙げられる。これらの高分子系分散剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
高分子系分散剤の使用量は、炭化水素分散媒中における、単量体水溶液の分散状態を良好に保ち、かつ使用量に見合う分散効果を得る観点から、重合のために用いる水溶性エチレン性不飽和単量体100質量部に対して、好ましくは0.05~30質量部、より好ましくは0.1~20質量部である。
分散安定剤として用いられる界面活性剤の添加時期は、重合反応を開始する前であれば、単量体水溶液添加の前後のどちらであってもよい。中でも、得られる吸水性樹脂に残存する炭化水素分散媒量を低減できる観点から、単量体水溶液を分散させた後に、さらに界面活性剤を分散させてから重合を行うことが好ましい。また、分散安定剤として界面活性剤と併用される高分子系分散剤の添加時期は、単量体水溶液添加の前後のどちらであってもよいが、単量体水溶液の分散安定性と吸水性樹脂に残存する炭化水素分散媒量低減の観点から、単量体水溶液を分散させる前に添加することが好ましい。すなわち、高分子系分散剤を分散させた炭化水素分散媒に、単量体水溶液を分散させた後に、さらに界面活性剤を分散させてから重合を行うことがより好ましい。
[ラジカル重合開始剤]
ラジカル重合開始剤としては、例えば、過硫酸カリウム、過硫酸アンモニウム、及び過硫酸ナトリウム等の過硫酸塩類;メチルエチルケトンパーオキシド、メチルイソブチルケトンパーオキシド、ジ-t-ブチルパーオキシド、t-ブチルクミルパーオキシド、t-ブチルパーオキシアセテート、t-ブチルパーオキシイソブチレート、t-ブチルパーオキシピバレート、及び過酸化水素等の過酸化物類;2,2’-アゾビス(2-アミジノプロパン)二塩酸塩、2,2’-アゾビス〔2-(N-フェニルアミジノ)プロパン〕二塩酸塩、2,2’-アゾビス〔2-(N-アリルアミジノ)プロパン〕二塩酸塩、2,2’-アゾビス{2-〔1-(2-ヒドロキシエチル)-2-イミダゾリン-2-イル〕プロパン}二塩酸塩、2,2’-アゾビス{2-メチル-N-〔1,1-ビス(ヒドロキシメチル)-2-ヒドロキシエチル〕プロピオンアミド}、2,2’-アゾビス〔2-メチル-N-(2-ヒドロキシエチル)-プロピオンアミド〕、及び4,4’-アゾビス(4-シアノ吉草酸)等のアゾ化合物等を挙げることができる。これらのラジカル重合開始剤のなかでは、入手が容易で取り扱いやすいという観点から、過硫酸カリウム、過硫酸アンモニウム、過硫酸ナトリウム及び2,2’-アゾビス(2-アミジノプロパン)二塩酸塩が好ましい。これらのラジカル重合開始剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
ラジカル重合開始剤としては、例えば、過硫酸カリウム、過硫酸アンモニウム、及び過硫酸ナトリウム等の過硫酸塩類;メチルエチルケトンパーオキシド、メチルイソブチルケトンパーオキシド、ジ-t-ブチルパーオキシド、t-ブチルクミルパーオキシド、t-ブチルパーオキシアセテート、t-ブチルパーオキシイソブチレート、t-ブチルパーオキシピバレート、及び過酸化水素等の過酸化物類;2,2’-アゾビス(2-アミジノプロパン)二塩酸塩、2,2’-アゾビス〔2-(N-フェニルアミジノ)プロパン〕二塩酸塩、2,2’-アゾビス〔2-(N-アリルアミジノ)プロパン〕二塩酸塩、2,2’-アゾビス{2-〔1-(2-ヒドロキシエチル)-2-イミダゾリン-2-イル〕プロパン}二塩酸塩、2,2’-アゾビス{2-メチル-N-〔1,1-ビス(ヒドロキシメチル)-2-ヒドロキシエチル〕プロピオンアミド}、2,2’-アゾビス〔2-メチル-N-(2-ヒドロキシエチル)-プロピオンアミド〕、及び4,4’-アゾビス(4-シアノ吉草酸)等のアゾ化合物等を挙げることができる。これらのラジカル重合開始剤のなかでは、入手が容易で取り扱いやすいという観点から、過硫酸カリウム、過硫酸アンモニウム、過硫酸ナトリウム及び2,2’-アゾビス(2-アミジノプロパン)二塩酸塩が好ましい。これらのラジカル重合開始剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
ラジカル重合開始剤の使用量は、急激な重合反応を回避し、かつ、重合反応時間を短縮する観点から、通常、重合に用いられる水溶性エチレン性不飽和単量体100モルに対して0.005~1モルが好ましく、0.01~0.5モルがより好ましく、0.0125~0.1モルがさらに好ましく、0.015~0.05モルがよりさらに好ましい。
重合反応の反応温度は、使用するラジカル重合開始剤によって異なるが、重合を迅速に進行させて生産性を高めるとともに、重合熱をより円滑に除去する観点から、通常20~110℃が好ましく、40~90℃がより好ましい。反応時間は、通常、0.1時間~4時間程度が好ましい。
[内部架橋剤]
水溶性エチレン性不飽和単量体を重合する際に、必要に応じて架橋剤を使用してもよい。重合が多段である場合には、全ての段階において架橋剤を用いてもよいし、用いない段階が存在してもよい。また、多段の場合、各段階で用いる架橋剤の種類は同じ又は異なってよく、同じものが好ましい。このような架橋剤(以下、「内部架橋剤」という)としては、例えば、(ポリ)エチレングリコール〔「(ポリ)」とは「ポリ」の接頭語がある場合とない場合を意味する。以下同じ〕、(ポリ)プロピレングリコール、1,4-ブタンジオール、トリメチロールプロパン、(ポリ)グリセリン等のジオール、トリオール等のポリオール類と(メタ)アクリル酸、マレイン酸、フマル酸等の不飽和酸とを反応させて得られる不飽和ポリエステル類;N,N-メチレンビスアクリルアミド等のビスアクリルアミド類;ポリエポキシドと(メタ)アクリル酸とを反応させて得られるジまたはトリ(メタ)アクリル酸エステル類;トリレンジイソシアネート、ヘキサメチレンジイソシアネート等のポリイソシアネートと(メタ)アクリル酸ヒドロキシエチルとを反応させて得られるジ(メタ)アクリル酸カルバミルエステル類;アリル化澱粉、アリル化セルロース、ジアリルフタレート、N,N’,N’’-トリアリルイソシアヌレート、ジビニルベンゼン等の重合性不飽和基を2個以上有する化合物;(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、(ポリ)グリセリンジグリシジルエーテル等のジグリシジル化合物、トリグリシジル化合物等のポリグリシジル化合物;エピクロルヒドリン、エピブロムヒドリン、α-メチルエピクロルヒドリン等のエピハロヒドリン化合物;2,4-トリレンジイソシアネート、ヘキサメチレンジイソシアネート等のイソシアネート化合物等の反応性官能基を2個以上有する化合物等が挙げられる。これらの内部架橋剤の中でも、好ましくはポリグリシジル化合物、さらに好ましくはジグリシジルエーテル化合物、特に好ましくは(ポリ)エチレングリコールジグリシジルエーテルが挙げられる。これらの内部架橋剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。内部架橋剤は、上述の単量体水溶液に添加して用いることが好ましい。
水溶性エチレン性不飽和単量体を重合する際に、必要に応じて架橋剤を使用してもよい。重合が多段である場合には、全ての段階において架橋剤を用いてもよいし、用いない段階が存在してもよい。また、多段の場合、各段階で用いる架橋剤の種類は同じ又は異なってよく、同じものが好ましい。このような架橋剤(以下、「内部架橋剤」という)としては、例えば、(ポリ)エチレングリコール〔「(ポリ)」とは「ポリ」の接頭語がある場合とない場合を意味する。以下同じ〕、(ポリ)プロピレングリコール、1,4-ブタンジオール、トリメチロールプロパン、(ポリ)グリセリン等のジオール、トリオール等のポリオール類と(メタ)アクリル酸、マレイン酸、フマル酸等の不飽和酸とを反応させて得られる不飽和ポリエステル類;N,N-メチレンビスアクリルアミド等のビスアクリルアミド類;ポリエポキシドと(メタ)アクリル酸とを反応させて得られるジまたはトリ(メタ)アクリル酸エステル類;トリレンジイソシアネート、ヘキサメチレンジイソシアネート等のポリイソシアネートと(メタ)アクリル酸ヒドロキシエチルとを反応させて得られるジ(メタ)アクリル酸カルバミルエステル類;アリル化澱粉、アリル化セルロース、ジアリルフタレート、N,N’,N’’-トリアリルイソシアヌレート、ジビニルベンゼン等の重合性不飽和基を2個以上有する化合物;(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、(ポリ)グリセリンジグリシジルエーテル等のジグリシジル化合物、トリグリシジル化合物等のポリグリシジル化合物;エピクロルヒドリン、エピブロムヒドリン、α-メチルエピクロルヒドリン等のエピハロヒドリン化合物;2,4-トリレンジイソシアネート、ヘキサメチレンジイソシアネート等のイソシアネート化合物等の反応性官能基を2個以上有する化合物等が挙げられる。これらの内部架橋剤の中でも、好ましくはポリグリシジル化合物、さらに好ましくはジグリシジルエーテル化合物、特に好ましくは(ポリ)エチレングリコールジグリシジルエーテルが挙げられる。これらの内部架橋剤は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。内部架橋剤は、上述の単量体水溶液に添加して用いることが好ましい。
内部架橋剤を使用する場合、その使用量は、得られる吸水性樹脂の吸水性能を十分に高めるために、水溶性エチレン性不飽和単量体100モルに対して、0.00001~1モルとすることが好ましく、0.0001~0.5モルとすることがより好ましい。
[その他の成分]
この吸水性樹脂の製造方法の一例では、逆相懸濁重合を行うに際し、その他の成分を、水溶性エチレン性不飽和単量体水溶液に添加してもよい。その他の成分としては、増粘剤や連鎖移動剤等の各種の添加剤を添加することができる。
この吸水性樹脂の製造方法の一例では、逆相懸濁重合を行うに際し、その他の成分を、水溶性エチレン性不飽和単量体水溶液に添加してもよい。その他の成分としては、増粘剤や連鎖移動剤等の各種の添加剤を添加することができる。
(増粘剤)
重合反応を行うに際し、水溶性エチレン性不飽和単量体水溶液に増粘剤を添加してもよい。このように増粘剤を添加して水溶液粘度を調整することによって、得られる吸水性樹脂の中位粒子径を制御することが可能である。
重合反応を行うに際し、水溶性エチレン性不飽和単量体水溶液に増粘剤を添加してもよい。このように増粘剤を添加して水溶液粘度を調整することによって、得られる吸水性樹脂の中位粒子径を制御することが可能である。
増粘剤としては、例えば、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、メチルセルロース、カルボキシメチルセルロース、ポリアクリル酸、ポリアクリル酸(部分)中和物、ポリエチレングリコール、ポリアクリルアミド、ポリエチレンイミン、デキストリン、アルギン酸ナトリウム、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキサイド等を用いることができる。なお、重合時の攪拌速度が同じであれば、水溶性エチレン性不飽和単量体水溶液の粘度が高いほど得られる粒子の中位粒子径は大きくなる傾向にある。
<2-2.後架橋工程>
重合工程以降に、含水ゲル状物(重合により得られた吸水性樹脂であって、水を含むもの)に対して後架橋反応を施し、吸水性樹脂の表面近傍の架橋密度を高めることにより、荷重下での吸水能等の諸性能を高めることができる。本開示の吸水性樹脂製造において、後架橋剤により後架橋を施してもよい。
重合工程以降に、含水ゲル状物(重合により得られた吸水性樹脂であって、水を含むもの)に対して後架橋反応を施し、吸水性樹脂の表面近傍の架橋密度を高めることにより、荷重下での吸水能等の諸性能を高めることができる。本開示の吸水性樹脂製造において、後架橋剤により後架橋を施してもよい。
後架橋剤としては、吸水性樹脂のカルボキシル基と反応し得るものが挙げられる。後架橋剤の代表例としては、(ポリ)エチレングリコール、(ポリ)プロピレングリコール、1,4-ブタンジオール、トリメチロールプロパン、(ポリ)グリセリン等のポリオール類;(ポリ)エチレングリコールジグリシジルエーテル、(ポリ)プロピレングリコールジグリシジルエーテル、(ポリ)グリセリンジグリシジルエーテル等のジグリシジルエーテル化合物;エピクロルヒドリン、エピブロムヒドリン、α-メチルエピクロルヒドリン等のエピハロヒドリン化合物;2,4-トリレンジイソシアネート、ヘキサメチレンジイソシアネート等のイソシアネート化合物等の反応性官能基を2個以上有する化合物等が挙げられる。中でも(ポリ)エチレングリコールジグリシジルエーテルが好ましい。これらは、それぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。また、後架橋剤を水や有機溶媒等に溶解して使用してもよい。
後架橋剤の量は、後架橋剤の種類により異なるので一概には決定することができないが、後架橋剤の使用量が少ないと、吸水性樹脂の表面層の架橋密度が不十分となって荷重下での吸水能が低くなる傾向があり、一方、後架橋剤の使用量が多いと、吸水性樹脂の保水能が低下する傾向がある。このため、後架橋剤の使用量は、通常、重合に使用した水溶性エチレン性不飽和単量体の総量1モルに対して、0.00001~0.01モル、好ましくは0.00005~0.005モル、さらに好ましくは、0.0001~0.002モルとすればよい。
後架橋剤の添加時期は、水溶性エチレン性不飽和単量体の総量100質量部に対し、1~400質量部の水分が存在する系に添加することが好ましく、5~200質量部の水分が存在する系に添加することがより好ましく、10~100質量部の水分が存在する系に添加することがよりさらに好ましい。なお、水分の量は、反応系に含まれる水分と後架橋剤を添加する際に必要に応じて用いられる水分との合計量を意味する。
後架橋反応における反応温度は、50~250℃が好ましく、60~180℃がより好ましい。また、後架橋の反応時間は、反応温度、後架橋剤の種類及び量等によって異なるので一概には決定することができないが、通常、1~300分間、好ましくは5~200分間である。
<2-3.乾燥工程>
上述の含水ゲル状物に、熱等のエネルギーを外部から加えることで、水、炭化水素分散媒等を蒸留により除去する乾燥工程を含んでいてもよい。例えば、逆相懸濁重合後の含水ゲル状物から脱水を行う場合、炭化水素分散媒中に含水ゲル状物が分散している系を加熱し、水と炭化水素分散媒を共沸蒸留により系外に一旦留去することにより行われる。このとき、留去した炭化水素分散媒のみを系内へリサイクルすることにより、連続的な共沸蒸留が可能であり、かかる手法を採用することにより、系内の温度を共沸温度以下に維持することができ、樹脂が劣化しにくい等の観点から好ましい。次いで、水及び炭化水素分散媒を留去することにより、吸水性樹脂の粒子が得られる。
上述の含水ゲル状物に、熱等のエネルギーを外部から加えることで、水、炭化水素分散媒等を蒸留により除去する乾燥工程を含んでいてもよい。例えば、逆相懸濁重合後の含水ゲル状物から脱水を行う場合、炭化水素分散媒中に含水ゲル状物が分散している系を加熱し、水と炭化水素分散媒を共沸蒸留により系外に一旦留去することにより行われる。このとき、留去した炭化水素分散媒のみを系内へリサイクルすることにより、連続的な共沸蒸留が可能であり、かかる手法を採用することにより、系内の温度を共沸温度以下に維持することができ、樹脂が劣化しにくい等の観点から好ましい。次いで、水及び炭化水素分散媒を留去することにより、吸水性樹脂の粒子が得られる。
乾燥工程では、当該乾燥処理を常圧下で行ってもよく、減圧下で行ってもよい。また、乾燥効率を高める観点から、窒素等の気流下で行ってもよい。乾燥処理を常圧下で行う場合においては、乾燥温度としては、70~250℃であることが好ましく、80~180℃であることがより好ましい。また、当該乾燥処理を減圧下で行う場合においては、乾燥温度としては、40~160℃であることが好ましく、50~110℃であることがより好ましい。
本開示の吸水性樹脂の、特に好ましい一態様としては、水溶性エチレン性不飽和単量体の重合反応を経て製造される吸水性樹脂であって、次の(i)~(iii)の3条件のうち少なくとも2つの条件を満たす吸水性樹脂が挙げられる。より好ましくは3条件全てを満たす吸水性樹脂である。
(i)1段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が10×103~15×103である。
(ii)2段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)が15×103~25×103である。
(iii)後架橋反応に用いる樹脂を調製した重合で用いた水溶性エチレン性不飽和単量体の合計量と後架橋剤とのモル比(水溶性エチレン性不飽和単量体/後架橋剤)が2.5×103~4.5×103である。
これらの条件の少なくとも2つ若しくは3つを満たすように吸水性樹脂を調製することにより、上記の好ましい総括吸収容量項α及び吸水速度項β、ひいては好ましいドライアップ指数を示す吸水性樹脂を調製することが可能となり得る。言い換えれば、これら(i)~(iii)の条件を指標として、本開示の吸水性樹脂を好ましく調製し得る。
(i)の条件において、1段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)は、より好ましくは10×103~14×103であり、さらに好ましくは10×103~13×103であり、よりさらに好ましくは11×103~12×103である。
(ii)の条件において、2段目の重合に用いる水溶性エチレン性不飽和単量体と内部架橋剤とのモル比(水溶性エチレン性不飽和単量体/内部架橋剤)は、より好ましくは17.5×103~24×103であり、さらに好ましくは20×103~23×103であり、よりさらに好ましくは21×103~22×103である。
(iii)の条件において、後架橋反応に用いる樹脂を調製した重合で用いた水溶性エチレン性不飽和単量体の合計量と後架橋剤とのモル比(水溶性エチレン性不飽和単量体/後架橋剤)は、より好ましくは3×103~4×103であり、さらに好ましくは3×103~3.5×103である。
本開示は、これらの条件を満たすように吸水性樹脂を製造する方法についても好ましく包含する。
3.吸収体、吸収性物品
本開示の吸水性樹脂は、例えば、親水性繊維とともに吸収体を構成する。そのような吸収体は、生理用品、紙オムツ等の衛生材料に用いられる吸収性物品に好適に用いられる。
本開示の吸水性樹脂は、例えば、親水性繊維とともに吸収体を構成する。そのような吸収体は、生理用品、紙オムツ等の衛生材料に用いられる吸収性物品に好適に用いられる。
吸収体は、例えば、吸水性樹脂と親水性繊維より構成される。吸収体の構成としては、吸水性樹脂と親水性繊維とを均一な組成となるように混合することによって得られた混合分散体、層状の親水性繊維の間に吸水性樹脂が挟まれたサンドイッチ構造体、吸水性樹脂と親水性繊維とをティッシュ等で包んだ形態の構造体等が挙げられる。なお、吸収体には、他の成分、例えば、吸収体の形態保持性を高めるための熱融着性合成繊維、ホットメルト接着剤、接着性エマルジョン等の接着性バインダーが配合されていてもよい。
本開示の吸水性樹脂の吸収体における含有量としては、吸収体の総質量に対して5~50質量%であることが好ましく、10~45質量%であることがより好ましく、15~40質量%であることがさらに好ましい。
親水性繊維としては、木材から得られる綿状パルプ、メカニカルパルプ、ケミカルパルプ、セミケミカルパルプ等のセルロース繊維、レーヨン、アセテート等の人工セルロース繊維、親水化処理されたポリアミド、ポリエステル、ポリオレフィン等の合成樹脂からなる繊維等が挙げられる。
前記吸収体を、液体が通過し得る液体透過性シート(トップシート)と、液体が通過し得ない液体不透過性シート(バックシート)との間に保持することによって、吸収性物品とすることができる。液体透過性シートは、身体と接触する側に配され、液体不透過性シートは、身体と接する反対側に配される。
液体透過性シートとしては、ポリエチレン、ポリプロピレン、ポリエステル等の繊維からなる、エアスルー型、スパンボンド型、ケミカルボンド型、ニードルパンチ型等の不織布及び多孔質の合成樹脂シート等が挙げられる。また、液体不透過性シートとしては、ポリエチレン、ポリプロピレン、ポリ塩化ビニル等の樹脂からなる合成樹脂フィルム等が挙げられる。
なお、本明細書において「含む」とは、「本質的にからなる」と、「からなる」をも包含する(The term "comprising" includes "consisting essentially of” and "consisting of.")。また、本開示は、本明細書に説明した構成要件を任意の組み合わせを全て包含する。
また、上述した本開示の各実施形態について説明した各種特性(性質、構造、機能等)は、本開示に包含される主題を特定するにあたり、どのように組み合わせられてもよい。すなわち、本開示には、本明細書に記載される組み合わせ可能な各特性のあらゆる組み合わせからなる主題が全て包含される。
以下、例を示して本開示をより詳細に説明するが、本開示は以下の例に限定されるものではない。
<評価試験方法について>
[吸水性樹脂の評価試験]
下記の実施例及び比較例にて得られた吸水性樹脂については、下記に示す各種試験に供して評価した。以下、各試験方法について説明する。
[吸水性樹脂の評価試験]
下記の実施例及び比較例にて得られた吸水性樹脂については、下記に示す各種試験に供して評価した。以下、各試験方法について説明する。
1) 生理食塩水吸水能
500mL容のビーカーに、0.9質量%塩化ナトリウム水溶液(生理食塩水)500gを量り取り、600r/minで撹拌させながら、吸水性樹脂2.0gを、ママコが発生しないように分散させた。60分間撹拌を継続し、吸水性樹脂を十分に膨潤させた。その後、あらかじめ目開きが75μmの200mmφ標準篩の質量Wa(g)を測定しておき、これを用いて、前記ビーカーの内容物をろ過し、篩を水平に対して約30度の傾斜角となるように傾けた状態で、30分間静置することにより余剰の水分をろ別した。膨潤ゲルの入った篩の質量Wb(g)を測定し、以下式により生理食塩水吸水能を求めた。
500mL容のビーカーに、0.9質量%塩化ナトリウム水溶液(生理食塩水)500gを量り取り、600r/minで撹拌させながら、吸水性樹脂2.0gを、ママコが発生しないように分散させた。60分間撹拌を継続し、吸水性樹脂を十分に膨潤させた。その後、あらかじめ目開きが75μmの200mmφ標準篩の質量Wa(g)を測定しておき、これを用いて、前記ビーカーの内容物をろ過し、篩を水平に対して約30度の傾斜角となるように傾けた状態で、30分間静置することにより余剰の水分をろ別した。膨潤ゲルの入った篩の質量Wb(g)を測定し、以下式により生理食塩水吸水能を求めた。
生理食塩水吸水能(g/g)=[Wb-Wa](g)/吸水性樹脂の質量(g)
2) 生理食塩水保水能
500mL容のビーカーに、0.9質量%塩化ナトリウム水溶液(生理食塩水)500gを量り取り、600r/minで撹拌させながら、吸水性樹脂2.0gを、ママコが発生しないように分散させた。30分間撹拌を継続し、吸水性樹脂を十分に膨潤させた。その後、ビーカー内の膨潤ゲルと生理食塩水を綿袋(メンブロード60番、横100mm×縦200mm)中に注ぎ込み、綿袋の上部を輪ゴムで縛り、遠心力が167Gとなるよう設定した脱水機(国産遠心機株式会社製、品番:H-122)を用いて綿袋を1分間脱水し、脱水後の膨潤ゲルを含んだ綿袋の質量Wc(g)を測定した。吸水性樹脂を添加せずに同様の操作を行ない、綿袋の湿潤時の空質量Wd(g)を測定し、以下式により生理食塩水保水能を求めた。
500mL容のビーカーに、0.9質量%塩化ナトリウム水溶液(生理食塩水)500gを量り取り、600r/minで撹拌させながら、吸水性樹脂2.0gを、ママコが発生しないように分散させた。30分間撹拌を継続し、吸水性樹脂を十分に膨潤させた。その後、ビーカー内の膨潤ゲルと生理食塩水を綿袋(メンブロード60番、横100mm×縦200mm)中に注ぎ込み、綿袋の上部を輪ゴムで縛り、遠心力が167Gとなるよう設定した脱水機(国産遠心機株式会社製、品番:H-122)を用いて綿袋を1分間脱水し、脱水後の膨潤ゲルを含んだ綿袋の質量Wc(g)を測定した。吸水性樹脂を添加せずに同様の操作を行ない、綿袋の湿潤時の空質量Wd(g)を測定し、以下式により生理食塩水保水能を求めた。
生理食塩水保水能(g/g)=[Wc-Wd](g)/吸水性樹脂の質量(g)
3) 2.07kPa荷重下での生理食塩水吸水能
図1に概略構成を示した測定装置Xを用いて、吸水性樹脂の2.07kPa荷重下での生理食塩水吸水能を測定した。
図1に概略構成を示した測定装置Xを用いて、吸水性樹脂の2.07kPa荷重下での生理食塩水吸水能を測定した。
図1に示した測定装置Xは、ビュレット部1、導管2、測定台3、測定台3上に置かれた測定部4からなっている。ビュレット部1は、ビュレット10の上部にゴム栓14、下部に空気導入管11とコック12が連結されており、さらに、空気導入管11の上部はコック13がある。ビュレット部1から測定台3までは、導管2が取り付けらており、導管2の直径は6mmである。測定台3の中央部には、直径2mmの穴があいており、導管2が連結されている。測定部4は、円筒40と、この円筒40の底部に貼着されたナイロンメッシュ41と、重り42とを有している。円筒40の内径は、2.0cmである。ナイロンメッシュ41は、200メッシュ(目開き75μm)に形成されている。そして、ナイロンメッシュ41上に所定量の吸水性樹脂5が均一に撒布されるようになっている。重り42は、直径1.9cm、質量59.8gである。この重り42は、吸水性樹脂5上に置かれ、吸水性樹脂5に対して2.07kPaの荷重を均一に加えることができるようになっている。
このような構成の測定装置Xでは、まずビュレット部1のコック12とコック13を閉め、25℃に調節された0.9質量%塩化ナトリウム水溶液(生理食塩水)をビュレット10上部から入れ、ゴム栓14でビュレット上部の栓をした後、ビュレット部1のコック12、コック13を開ける。次に、測定台3中心部における導管2の先端と空気導入管11の空気導入口とが同じ高さになるように測定台3の高さの調整を行う。
一方、円筒40のナイロンメッシュ41上に0.10gの吸水性樹脂5を均一に撒布して、この吸水性樹脂5上に重り42を置く。測定部4は、その中心部が測定台3中心部の導管口に一致するようにして置く。
吸水性樹脂5が吸水し始めた時点から継続的に、ビュレット10内の生理食塩水の減少量(吸水性樹脂5が吸水した生理食塩水量)We(mL)を読み取った。吸水開始から60分間経過後における、吸水性樹脂5の荷重下での生理食塩水吸水能は、生理食塩水の比重1.0(g/mL)を用いて、以下式により求めた。
2.07kPa荷重下での生理食塩水吸水能(g/g)=We(mL)×1.0(g/mL)/吸水性樹脂の質量(g)
4) 4.82kPa荷重下での生理食塩水吸水能
図2に概略構成を示した測定装置Yを用いて、吸水性樹脂の4.82kPa荷重下での生理食塩水吸水能を測定した。
図2に概略構成を示した測定装置Yを用いて、吸水性樹脂の4.82kPa荷重下での生理食塩水吸水能を測定した。
図2に示した測定装置Yは、主に重り90、支持円筒91、ピストン92からなる測定部と、主にペトリ皿6、ガラスフィルター7からなる液供給部とからなっている。当該測定部は、内径60mmの円筒プラスチック製支持円筒91の片側(底面)に、400メッシュ(目開き38μm)のステンレス製金網93が接着されており、円筒内部には直径が60mmよりわずかに小さく支持円筒91との壁面に隙間が生じずかつ上下の動きは妨げられないピストン92とその上部に重り90を有しており、ピストン92と重り90により、吸水性樹脂5に対して4.82kPaの荷重を均一に加えることができるようになっている。このような測定部を用いて、支持円筒91の金網上に0.90gの吸水性樹脂5を均一に散布したのち、ピストン92と重り90を載置し、測定部の質量Wf(g)を測定した。質量Wf(g)は、支持円筒91、吸水性樹脂5、ピストン92、重り90それぞれの質量を合計したものである。
別途、直径150mmのペトリ皿6の内側に直径90mm、厚さ5mmのガラスフィルター7(柴田科学株式会社製)を置き、25±1℃に調整した0.9質量%塩化ナトリウム水溶液(生理食塩水)をガラスフィルター7の上面と同じ高さになるようにペトリ皿6に加えたのち、ガラスフィルター7の上部に直径9cmの濾紙8(ADVANTEC製、No.2)を1枚載置して、液供給部を調製した。濾紙8は表面全体が濡れていることを確認し、余剰の液がある場合には、ティッシュで適宜吸い取った。
上記測定部を上記液供給部に載置して、吸水性樹脂5に生理食塩水を4.82kPaの荷重下で吸収させた。液供給部の液面がガラスフィルター7の上面部分よりも低くなった際には生理食塩水を適宜追加し、液面レベルを一定に保った。上記測定部を上記液供給部に載置してから60分後に測定部を液供給部から取り外し、質量Wg(g)を測定した。
以下式により4.82kPa荷重下での生理食塩水吸水能(g/g)を求めた。
4.82kPa荷重下での生理食塩水吸水能(g/g)=[Wg-Wf](g)/吸水性樹脂の質量(g)
5) 動的吸水速度
動的吸水速度の測定は、25℃±1℃に調節された室内で行われた。100mL容のビーカー中に秤量した生理食塩水50±0.1gを、恒温水槽にて25±0.2℃の温度に調整したのち、マグネチックスターラーバー(8mmφ×30mmのリング無し)で攪拌して、回転数600r/minで渦を発生させた。吸水性樹脂2.0±0.002gを、上記生理食塩水中に一度に添加し、吸水性樹脂の添加後から液面の渦が収束する時点までの時間(秒)を測定し、分単位に換算したものを動的な吸水時間(分)とした。
動的吸水速度の測定は、25℃±1℃に調節された室内で行われた。100mL容のビーカー中に秤量した生理食塩水50±0.1gを、恒温水槽にて25±0.2℃の温度に調整したのち、マグネチックスターラーバー(8mmφ×30mmのリング無し)で攪拌して、回転数600r/minで渦を発生させた。吸水性樹脂2.0±0.002gを、上記生理食塩水中に一度に添加し、吸水性樹脂の添加後から液面の渦が収束する時点までの時間(秒)を測定し、分単位に換算したものを動的な吸水時間(分)とした。
以下により動的吸水速度(1/分)を算出した。
動的吸水速度(1/分)=(生理食塩水量÷吸水性樹脂量)÷(動的な吸水時間(分))
6) 静的吸水速度
図1に概略構成を示した測定装置Xを用いて、重り42を使用せず無荷重下での測定を行うこと以外は、前述した2.07kPa荷重下での生理食塩水吸水能と同様にして、吸水性樹脂の静的吸水速度を測定した。本測定方法においては、前述の通り、重り42を用いないため、荷重がかからない状態(言い換えれば、無荷重下)での吸水性樹脂5の吸水速度を測定した。
図1に概略構成を示した測定装置Xを用いて、重り42を使用せず無荷重下での測定を行うこと以外は、前述した2.07kPa荷重下での生理食塩水吸水能と同様にして、吸水性樹脂の静的吸水速度を測定した。本測定方法においては、前述の通り、重り42を用いないため、荷重がかからない状態(言い換えれば、無荷重下)での吸水性樹脂5の吸水速度を測定した。
上記と同様に、無荷重下の吸水性樹脂5が吸水し始めた時点から継続的に、ビュレット10内の生理食塩水の減少量(吸水性樹脂5が吸水した生理食塩水量)Wi(mL)を読み取り、特定の時間までに吸水性樹脂5の吸収した生理食塩水量を以下式により算出した。なお、生理食塩水の比重は1.0g/mLとした。
吸水性樹脂5の吸収した生理食塩水量(g/g)=Wi(mL)×1.0(g/mL)/吸水性樹脂の質量(g)
このようにして吸水性樹脂5の吸収した生理食塩水量(g/g)を測定する時、吸水性樹脂5が1gあたり25gの生理食塩水を吸収するまでに要した時間(秒)を、吸水性樹脂5が吸水し始めた時点からの経過時間としてストップウォッチにより測定し、分単位に換算したものを静的な吸水時間(分)とした。以下式により静的吸水速度(1/分)を算出した。
静的吸水速度(1/分)=25(g/g)÷静的な吸水時間(分)
7) 中位粒子径(粒度分布)
JIS標準篩を上から、目開き850μmの篩、目開き600μmの篩、目開き500μmの篩、目開き425μmの篩、目開き300μmの篩、目開き250μmの篩、目開き150μmの篩、及び受け皿の順に組み合わせた。
JIS標準篩を上から、目開き850μmの篩、目開き600μmの篩、目開き500μmの篩、目開き425μmの篩、目開き300μmの篩、目開き250μmの篩、目開き150μmの篩、及び受け皿の順に組み合わせた。
組み合わせた最上の篩に、吸水性樹脂50gを入れ、ロータップ式振とう器(飯田製作所社製)を用いて20分間振とうさせて分級した。分級後、各篩上に残った吸水性樹脂の質量を全量に対する質量百分率として算出し、粒度分布を求めた。この粒度分布に関して粒子径の大きい方から順に篩上を積算することにより、篩の目開きと篩上に残った吸水性樹脂の質量百分率の積算値との関係を対数確率紙にプロットした。確率紙上のプロットを直線で結ぶことにより、積算質量百分率50質量%に相当する粒子径を中位粒子径とした。
[吸水性樹脂を使用した吸収体の評価試験]
8) 逆戻り量
(a)試験液の調整
10L容の容器に、塩化カルシウムニ水和物2.5g、塩化マグネシウム六水和物5.0g、塩化カリウム20g、硫酸ナトリウム20g、リン酸二水素アンモニウム8.5g、リン酸水素二アンモニウム1.5g及び適量の蒸留水を入れ、完全に溶解させた。残りの蒸留水を全量追加して希釈し、さらに、少量の青色1号で着色して、試験液を調製した。
8) 逆戻り量
(a)試験液の調整
10L容の容器に、塩化カルシウムニ水和物2.5g、塩化マグネシウム六水和物5.0g、塩化カリウム20g、硫酸ナトリウム20g、リン酸二水素アンモニウム8.5g、リン酸水素二アンモニウム1.5g及び適量の蒸留水を入れ、完全に溶解させた。残りの蒸留水を全量追加して希釈し、さらに、少量の青色1号で着色して、試験液を調製した。
(b)吸収体及び吸収性物品の作製
吸水性樹脂6.6gと解砕パルプ(レオニア社製レイフロック)10gを用い、空気抄造によって均一混合することにより、40cm×12cmの大きさの吸収体コアを作製した。次に、吸収体コアと同じ大きさで、坪量16g/m2の2枚のティッシュッペーパーを吸収体コアの上下に配置した状態で、全体に196kPaの荷重を30秒間加えてプレスすることにより、吸水性樹脂の含有量が40質量%の吸収体を作製した。さらに吸収体の上面に、吸収体と同じ大きさで、坪量22g/m2のポリエチレン製エアスルー型多孔質液体透過性シートを配置し、同じ大きさ、同じ坪量のポリエチレン製液体不透過性シートを吸収体の下面に配置して、吸収体を挟みつけることにより、検討用の吸収性物品とした。
吸水性樹脂6.6gと解砕パルプ(レオニア社製レイフロック)10gを用い、空気抄造によって均一混合することにより、40cm×12cmの大きさの吸収体コアを作製した。次に、吸収体コアと同じ大きさで、坪量16g/m2の2枚のティッシュッペーパーを吸収体コアの上下に配置した状態で、全体に196kPaの荷重を30秒間加えてプレスすることにより、吸水性樹脂の含有量が40質量%の吸収体を作製した。さらに吸収体の上面に、吸収体と同じ大きさで、坪量22g/m2のポリエチレン製エアスルー型多孔質液体透過性シートを配置し、同じ大きさ、同じ坪量のポリエチレン製液体不透過性シートを吸収体の下面に配置して、吸収体を挟みつけることにより、検討用の吸収性物品とした。
(c)逆戻り量測定
まず、水平の台上に吸収性物品を置いた。吸収性物品の中心部に、内径3cmの開口部を有する液投入用シリンダーを置き、50mLの試験液をそのシリンダー内に一度に投入した。試験液が浸透したのち、前記シリンダーをはずし、吸収性物品をそのままの状態で保存し、1回目の試験液投入開始から30分後及び60分後にも、1回目と同じ位置にシリンダーを用いて同様の操作を行った。前記3回目の試験液投入から60分経過後、吸収性物品の試験液投入位置に、あらかじめ質量(Wk(g)、約70g)を測定しておいた10cm四方の濾紙を、吸収性物品の中心部に合わさるよう置き、その上に底面が10cm×10cmの質量5kgの重りを載せた。5分間の荷重後、試験後の濾紙の質量(Wl(g))を測定し、増加した質量を以下式により算出し、逆戻り量(g)とした。
まず、水平の台上に吸収性物品を置いた。吸収性物品の中心部に、内径3cmの開口部を有する液投入用シリンダーを置き、50mLの試験液をそのシリンダー内に一度に投入した。試験液が浸透したのち、前記シリンダーをはずし、吸収性物品をそのままの状態で保存し、1回目の試験液投入開始から30分後及び60分後にも、1回目と同じ位置にシリンダーを用いて同様の操作を行った。前記3回目の試験液投入から60分経過後、吸収性物品の試験液投入位置に、あらかじめ質量(Wk(g)、約70g)を測定しておいた10cm四方の濾紙を、吸収性物品の中心部に合わさるよう置き、その上に底面が10cm×10cmの質量5kgの重りを載せた。5分間の荷重後、試験後の濾紙の質量(Wl(g))を測定し、増加した質量を以下式により算出し、逆戻り量(g)とした。
逆戻り量(g)=Wl-Wk
<各実施例及び比較例>
[実施例1]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.62gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
[実施例1]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.62gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液78g(0.87モル)をとり、外部より冷却しつつ、22.4質量%の水酸化ナトリウム水溶液120.6gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース1.170g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.055g(0.203ミリモル)とイオン交換水59.0gを加えて溶解し、モノマー濃度が30質量%のモノマー水溶液を調製した。(なお、モノマー濃度とは、モノマー水溶液総量に対する、水溶性エチレン性不飽和単量体及びその塩の質量比率であり、本明細書においては、以下同様に表記する。)
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン5.62gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.62gを加熱溶解した界面活性剤溶液6.2gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
1段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら155.3gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液3.90g(0.45ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.2質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状形態で中位粒子径350μmの吸水性樹脂82.1gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[実施例2]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.52gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.52gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液65g(0.72モル)をとり、外部より冷却しつつ、22.4質量%の水酸化ナトリウム水溶液100.5gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.065g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.046g(0.170ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.011g(0.063ミリモル)とイオン交換水92.0gを加えて溶解し、モノマー濃度が25質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン4.68gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.52gを加熱溶解した界面活性剤溶液5.2gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方、別の500mL容の三角フラスコに80質量%のアクリル酸水溶液94.3g(1.05モル)をとり、外部より冷却しつつ、28.1質量%の水酸化ナトリウム水溶液116.2gを滴下して中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.066g(0.244ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.009g(0.052ミリモル)とイオン交換水1.9gを加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。
前述のセパラブルフラスコ系内を27℃に冷却した後、2段目のモノマー水溶液の全量を、1段目の重合スラリー液に添加して、系内を窒素で十分に置換した後、再度、フラスコを70℃の水浴に浸漬して昇温し、2段目の重合を30分間行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら259.3gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液4.78g(0.55ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.2質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径332μmの吸水性樹脂173.3gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[実施例3]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.62gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.62gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液78g(0.87モル)をとり、外部より冷却しつつ、22.4質量%の水酸化ナトリウム水溶液120.6gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.078g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.055g(0.203ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.013g(0.075ミリモル)とイオン交換水59.0gを加えて溶解し、モノマー濃度が30質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン5.62gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.62gを加熱溶解した界面活性剤溶液6.2gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方で、500mL容の三角フラスコに80質量%のアクリル酸水溶液110.8g(1.23モル)をとり、外部より冷却しつつ、28.1質量%の水酸化ナトリウム水溶液136.5gを滴下して中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.078g(0.289ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.010g(0.057ミリモル)を加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。2段目のモノマー水溶液を調製後は、実施例2と同様に2段目の重合を行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら263.8gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液5.66g(0.65ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.2質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径350μmの吸水性樹脂205.5gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[実施例4]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備した。このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液92g(1.02モル)をとり、外部より冷却しつつ、22.4質量%の水酸化ナトリウム水溶液142.3gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.092g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.064g(0.237ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.016g(0.092ミリモル)とイオン交換水26.0gを加えて溶解し、モノマー濃度が35質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン6.62gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.74gを加熱溶解した界面活性剤溶液7.3gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方、別の500mL容の三角フラスコに80質量%のアクリル酸水溶液128.8g(1.43モル)をとり、外部より冷却しつつ、28.1質量%の水酸化ナトリウム水溶液158.8gを滴下して中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.090g(0.333ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.012g(0.069ミリモル)を加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。
前述のセパラブルフラスコ系内を27℃に冷却した後、2段目のモノマー水溶液の全量を、1段目の重合スラリー液に添加して、系内を窒素で十分に置換した後、再度、フラスコを70℃の水浴に浸漬して昇温し、2段目の重合を30分間行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら283.6gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液6.62g(0.76ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.2質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径361μmの吸水性樹脂230.7gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[比較例1]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液92g(1.02モル)をとり、外部より冷却しつつ、21.5質量%の水酸化ナトリウム水溶液142.5gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.092g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.064g(0.237ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.010g(0.057ミリモル)とイオン交換水10.0gを加えて溶解し、モノマー濃度が37質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン6.62gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.74gを加熱溶解した界面活性剤溶液7.4gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方、別の500mL容の三角フラスコに80質量%のアクリル酸水溶液128.8g(1.43モル)をとり、外部より冷却しつつ、27.0質量%の水酸化ナトリウム水溶液142.5gを滴下して75モル%の中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.090g(0.333ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.012g(0.069ミリモル)を加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。
前述のセパラブルフラスコ系内を27℃に冷却した後、2段目の単量体水溶液の全量を、1段目の重合スラリー液に添加して、系内を窒素で十分に置換した後、再度、フラスコを70℃の水浴に浸漬して昇温し、2段目の重合を30分間行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら266.2gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液4.42g(0.51ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.2質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径345μmの吸水性樹脂229.7gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[比較例2]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液92g(1.02モル)をとり、外部より冷却しつつ、21.5質量%の水酸化ナトリウム水溶液142.5gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.092g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.064g(0.237ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.010g(0.057ミリモル)とイオン交換水10.0gを加えて溶解し、モノマー濃度が37質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン6.66gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.74gを加熱溶解した界面活性剤溶液7.4gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方、別の500mL容の三角フラスコに80質量%のアクリル酸水溶液128.8g(1.43モル)をとり、外部より冷却しつつ、27.0質量%の水酸化ナトリウム水溶液142.5gを滴下して75モル%の中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.090g(0.333ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.012g(0.069ミリモル)を加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。
前述のセパラブルフラスコ系内を27℃に冷却した後、2段目の単量体水溶液の全量を、1段目の重合スラリー液に添加して、系内を窒素で十分に置換した後、再度、フラスコを70℃の水浴に浸漬して昇温し、2段目の重合を30分間行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら261.8gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液4.42g(0.51ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.5質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径380μmの吸水性樹脂234.2gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[比較例3]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液92g(1.02モル)をとり、外部より冷却しつつ、21.5質量%の水酸化ナトリウム水溶液142.5gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.092g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.064g(0.237ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.010g(0.057ミリモル)とイオン交換水10.0gを加えて溶解し、モノマー濃度が37質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン6.66gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.74gを加熱溶解した界面活性剤溶液7.4gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方、別の500mL容の三角フラスコに80質量%のアクリル酸水溶液128.8g(1.43モル)をとり、外部より冷却しつつ、27.0質量%の水酸化ナトリウム水溶液142.5gを滴下して75モル%の中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.090g(0.333ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.012g(0.069ミリモル)を加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。
前述のセパラブルフラスコ系内を27℃に冷却した後、2段目の単量体水溶液の全量を、1段目の重合スラリー液に添加して、系内を窒素で十分に置換した後、再度、フラスコを70℃の水浴に浸漬して昇温し、2段目の重合を30分間行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら259.7gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液4.42g(0.51ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.5質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径356μmの吸水性樹脂236.0gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
[比較例4]
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
還流冷却器、滴下ロート、窒素ガス導入管、並びに、攪拌機として、翼径50mmの4枚傾斜パドル翼を2段で有する攪拌翼を備えた内径110mm、2L容の丸底円筒型セパラブルフラスコを準備し、このフラスコに、炭化水素分散媒としてn-ヘプタン300gをとり、高分子系分散剤として無水マレイン酸変性エチレン・プロピレン共重合体(三井化学株式会社、ハイワックス1105A)0.74gを添加し、攪拌しつつ加温溶解した後、50℃まで冷却した。
一方、500mL容の三角フラスコに80質量%のアクリル酸水溶液92g(1.02モル)をとり、外部より冷却しつつ、21.5質量%の水酸化ナトリウム水溶液142.5gを滴下して中和を行った後、増粘剤としてヒドロキシルエチルセルロース0.092g(住友精化株式会社、HEC AW-15F)、ラジカル重合開始剤として過硫酸カリウム0.064g(0.237ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.018g(0.103ミリモル)とイオン交換水10.0gを加えて溶解し、モノマー濃度が37質量%のモノマー水溶液を調製した。
そして、上述のように調製したモノマー水溶液をセパラブルフラスコに添加して、10分間攪拌した後、n-ヘプタン6.66gに界面活性剤としてHLB3のショ糖ステアリン酸エステル(三菱化学フーズ株式会社、リョートーシュガーエステルS-370)0.74gを加熱溶解した界面活性剤溶液7.4gをさらに添加して、攪拌しながら系内を窒素で十分に置換した後、フラスコを70℃の水浴に浸漬して昇温し、重合を60分間行うことで1段目の重合スラリー液を得た。
一方、別の500mL容の三角フラスコに80質量%のアクリル酸水溶液128.8g(1.43モル)をとり、外部より冷却しつつ、27.0質量%の水酸化ナトリウム水溶液142.5gを滴下して75モル%の中和を行った後、ラジカル重合開始剤として過硫酸カリウム0.090g(0.333ミリモル)、内部架橋剤としてエチレングリコールジグリシジルエーテル0.039g(0.224ミリモル)を加えて溶解し、モノマー濃度が44質量%の2段目のモノマー水溶液を調製した。
前述のセパラブルフラスコ系内を27℃に冷却した後、2段目の単量体水溶液の全量を、1段目の重合スラリー液に添加して、系内を窒素で十分に置換した後、再度、フラスコを70℃の水浴に浸漬して昇温し、2段目の重合を30分間行った。
2段目の重合後、125℃の油浴で反応液を昇温し、n-ヘプタンと水との共沸蒸留によりn-ヘプタンを還流しながら260.8gの水を系外へ抜き出した後、後架橋剤としてエチレングリコールジグリシジルエーテルの2質量%水溶液6.62g(0.76ミリモル)を添加し、80℃で2時間保持した。その後、n-ヘプタンを蒸発させて乾燥することによって、乾燥品を得た。この乾燥品に対して0.5質量%の非晶質シリカ(エボニックデグサジャパン株式会社、カープレックス#80)を混合し、それを目開き1000μmの篩を通過させ、球状粒子が凝集した形態で中位粒子径343μmの吸水性樹脂235.1gを得た。この吸水性樹脂およびそれを用いた吸収体を、前述の各種試験方法に従って評価した。
<評価結果について>
[吸水性樹脂および吸収体の評価結果]
下記表1に吸水性樹脂の評価試験結果を示した。また、下記表2には吸水性樹脂のドライアップ指数、総括吸収容量項α、吸水速度項β、及びその吸水性樹脂を用いた吸収体の評価結果(逆戻り量)を示した。
[吸水性樹脂および吸収体の評価結果]
下記表1に吸水性樹脂の評価試験結果を示した。また、下記表2には吸水性樹脂のドライアップ指数、総括吸収容量項α、吸水速度項β、及びその吸水性樹脂を用いた吸収体の評価結果(逆戻り量)を示した。
ここで、総括吸収容量項αと吸水速度項βは、以下式(2)および(3)により求められる。

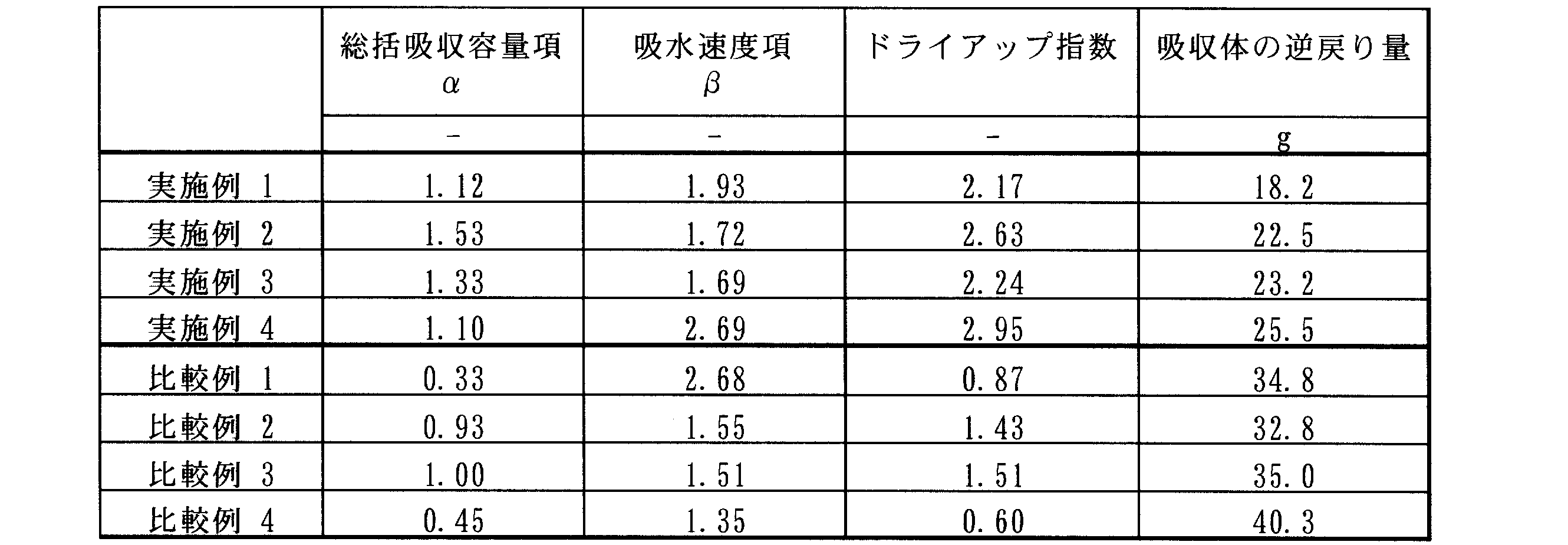
1 ビュレット部
10 ビュレット
11 空気導入管
12 コック
13 コック
14 ゴム栓
2 導管
3 測定台
4 測定部
40 円筒
41 ナイロンメッシュ
42 重り
5 吸水性樹脂粒子
6 ペトリ皿
7 ガラスフィルター
8 濾紙
90 重り
91 プラスチック製支持円筒
92 ピストン
93 ステンレス製金網
X 測定装置
Y 測定装置
10 ビュレット
11 空気導入管
12 コック
13 コック
14 ゴム栓
2 導管
3 測定台
4 測定部
40 円筒
41 ナイロンメッシュ
42 重り
5 吸水性樹脂粒子
6 ペトリ皿
7 ガラスフィルター
8 濾紙
90 重り
91 プラスチック製支持円筒
92 ピストン
93 ステンレス製金網
X 測定装置
Y 測定装置
Claims (6)
- 水溶性エチレン性不飽和単量体の重合体架橋物である吸水性樹脂であって、以下式(1)で示されるドライアップ指数が1.85以上である吸水性樹脂。



- 総括吸収容量項αが0.95以上である、請求項1に記載の吸水性樹脂。
- 吸水速度項βが1.56以上である、請求項1又は2に記載の吸水性樹脂。
- 生理食塩水吸水能と生理食塩水保水能の差が18以下である、請求項1~3のいずれかに記載の吸水性樹脂。
- 2.07kPa荷重下での生理食塩水吸水能と4.82kPa荷重下での生理食塩水吸水能の差が17~36である、請求項1~4のいずれかに記載の吸水性樹脂。
- 請求項1~5のいずれかに記載の吸水性樹脂を5~50質量%含んでなる吸収体。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/291,149 US20220126271A1 (en) | 2018-11-05 | 2019-10-31 | Water-absorbing resin |
| EP19881530.0A EP3878874A4 (en) | 2018-11-05 | 2019-10-31 | WATER-ABSORBENT RESIN |
| SG11202104506VA SG11202104506VA (en) | 2018-11-05 | 2019-10-31 | Water-absorbing resin |
| CN201980072201.3A CN113039219B (zh) | 2018-11-05 | 2019-10-31 | 吸水性树脂 |
| JP2020556019A JPWO2020095811A1 (ja) | 2018-11-05 | 2019-10-31 | 吸水性樹脂 |
| KR1020217012665A KR20210090617A (ko) | 2018-11-05 | 2019-10-31 | 흡수성 수지 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018208212 | 2018-11-05 | ||
| JP2018-208212 | 2018-11-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020095811A1 true WO2020095811A1 (ja) | 2020-05-14 |
Family
ID=70611877
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/042766 WO2020095811A1 (ja) | 2018-11-05 | 2019-10-31 | 吸水性樹脂 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20220126271A1 (ja) |
| EP (1) | EP3878874A4 (ja) |
| JP (1) | JPWO2020095811A1 (ja) |
| KR (1) | KR20210090617A (ja) |
| CN (1) | CN113039219B (ja) |
| SG (1) | SG11202104506VA (ja) |
| WO (1) | WO2020095811A1 (ja) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001523733A (ja) * | 1997-11-19 | 2001-11-27 | アンコル インターナショナル コーポレイション | 多成分超吸収性ゲル粒子 |
| WO2007123188A1 (ja) | 2006-04-24 | 2007-11-01 | Sumitomo Seika Chemicals Co., Ltd. | 吸水性樹脂粒子の製造方法、およびそれにより得られる吸水性樹脂粒子 |
| WO2012023433A1 (ja) | 2010-08-19 | 2012-02-23 | 住友精化株式会社 | 吸水性樹脂 |
| WO2016006132A1 (ja) * | 2014-07-11 | 2016-01-14 | 住友精化株式会社 | 吸水性樹脂及び吸収性物品 |
| JP2016028117A (ja) * | 2014-07-11 | 2016-02-25 | 住友精化株式会社 | 吸水性樹脂の製造方法、吸水性樹脂、吸水剤、吸収性物品 |
| JP2016028115A (ja) * | 2014-07-11 | 2016-02-25 | 住友精化株式会社 | 吸水性樹脂及び吸収性物品 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2938920B2 (ja) | 1990-01-31 | 1999-08-25 | 住友精化株式会社 | 吸水性樹脂の製造方法 |
| JP2007123188A (ja) | 2005-10-31 | 2007-05-17 | Canon Inc | 携帯型電子機器及び撮像装置 |
| EP2692744B1 (en) * | 2011-03-28 | 2017-08-23 | Sumitomo Seika Chemicals Co., Ltd. | Process for producing water-absorbing resin |
| JP5893117B2 (ja) * | 2014-07-11 | 2016-03-23 | 住友精化株式会社 | 吸水性樹脂及び吸収性物品 |
| JP5719078B1 (ja) * | 2014-07-11 | 2015-05-13 | 住友精化株式会社 | 吸水性樹脂の製造方法 |
-
2019
- 2019-10-31 KR KR1020217012665A patent/KR20210090617A/ko not_active Application Discontinuation
- 2019-10-31 CN CN201980072201.3A patent/CN113039219B/zh active Active
- 2019-10-31 EP EP19881530.0A patent/EP3878874A4/en active Pending
- 2019-10-31 SG SG11202104506VA patent/SG11202104506VA/en unknown
- 2019-10-31 JP JP2020556019A patent/JPWO2020095811A1/ja active Pending
- 2019-10-31 WO PCT/JP2019/042766 patent/WO2020095811A1/ja unknown
- 2019-10-31 US US17/291,149 patent/US20220126271A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001523733A (ja) * | 1997-11-19 | 2001-11-27 | アンコル インターナショナル コーポレイション | 多成分超吸収性ゲル粒子 |
| WO2007123188A1 (ja) | 2006-04-24 | 2007-11-01 | Sumitomo Seika Chemicals Co., Ltd. | 吸水性樹脂粒子の製造方法、およびそれにより得られる吸水性樹脂粒子 |
| WO2012023433A1 (ja) | 2010-08-19 | 2012-02-23 | 住友精化株式会社 | 吸水性樹脂 |
| WO2016006132A1 (ja) * | 2014-07-11 | 2016-01-14 | 住友精化株式会社 | 吸水性樹脂及び吸収性物品 |
| JP2016028117A (ja) * | 2014-07-11 | 2016-02-25 | 住友精化株式会社 | 吸水性樹脂の製造方法、吸水性樹脂、吸水剤、吸収性物品 |
| JP2016028115A (ja) * | 2014-07-11 | 2016-02-25 | 住友精化株式会社 | 吸水性樹脂及び吸収性物品 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3878874A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2020095811A1 (ja) | 2021-10-07 |
| SG11202104506VA (en) | 2021-05-28 |
| EP3878874A1 (en) | 2021-09-15 |
| KR20210090617A (ko) | 2021-07-20 |
| CN113039219B (zh) | 2023-04-14 |
| EP3878874A4 (en) | 2022-08-10 |
| CN113039219A (zh) | 2021-06-25 |
| US20220126271A1 (en) | 2022-04-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10265226B2 (en) | Water-absorbent resin, water-absorbent material, and water-absorbent article | |
| JP5893117B2 (ja) | 吸水性樹脂及び吸収性物品 | |
| JP7194101B2 (ja) | 吸水性樹脂 | |
| WO2016006132A1 (ja) | 吸水性樹脂及び吸収性物品 | |
| JP2016028113A (ja) | 吸水性樹脂及び吸水性樹脂の製造方法 | |
| WO2016104374A1 (ja) | 吸水性樹脂組成物 | |
| WO2018159800A1 (ja) | 吸水性樹脂及び吸収性物品 | |
| WO2018159802A1 (ja) | 吸水性樹脂及び吸収性物品 | |
| JP2016028131A (ja) | 吸水性樹脂及び吸水性樹脂の製造方法 | |
| WO2019142872A1 (ja) | 吸水性樹脂 | |
| WO2019189485A1 (ja) | 吸水性樹脂粒子 | |
| JP2021058771A (ja) | 吸水性樹脂粒子、吸収性物品、吸水性樹脂粒子を製造する方法、及び吸収体の加圧下での吸収量を高める方法 | |
| WO2020184395A1 (ja) | 吸水性樹脂粒子及びその製造方法、吸収体、吸収性物品、並びに、浸透速度の調整方法 | |
| WO2020184398A1 (ja) | 吸水性樹脂粒子及びその製造方法、吸収体、並びに、吸収性物品 | |
| WO2020184387A1 (ja) | 吸水性樹脂粒子及びその製造方法、吸収体、並びに吸収性物品 | |
| JP2016027846A (ja) | 吸水性樹脂及び吸収性物品 | |
| WO2020184391A1 (ja) | 吸収体、吸収性物品、及び、浸透速度の調整方法 | |
| JP6780047B2 (ja) | 吸水性樹脂粒子、吸収体及び吸収性物品 | |
| WO2020184392A1 (ja) | 吸水性樹脂粒子及びその製造方法 | |
| WO2020095811A1 (ja) | 吸水性樹脂 | |
| WO2020184393A1 (ja) | 吸水性樹脂粒子、吸収体及び吸収性物品 | |
| WO2020129594A1 (ja) | 吸水性樹脂、吸収体、吸収性物品、及び吸水性樹脂の製造方法 | |
| JP2024059766A (ja) | 吸水性樹脂 | |
| WO2022255300A1 (ja) | 吸水性樹脂の製造方法、吸水性樹脂、吸収体及び吸収性物品 | |
| JP7091556B2 (ja) | 吸水性樹脂粒子、及び吸水シート |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19881530 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020556019 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019881530 Country of ref document: EP Effective date: 20210607 |