WO2020050127A1 - ベンゾニトリル誘導体、発光材料およびそれを用いた発光素子 - Google Patents
ベンゾニトリル誘導体、発光材料およびそれを用いた発光素子 Download PDFInfo
- Publication number
- WO2020050127A1 WO2020050127A1 PCT/JP2019/033913 JP2019033913W WO2020050127A1 WO 2020050127 A1 WO2020050127 A1 WO 2020050127A1 JP 2019033913 W JP2019033913 W JP 2019033913W WO 2020050127 A1 WO2020050127 A1 WO 2020050127A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- substituted
- unsubstituted
- mmol
- carbazol
- Prior art date
Links
- 0 *c1c(*)c(I)c(*=C)c(*)c1[Zn] Chemical compound *c1c(*)c(I)c(*=C)c(*)c1[Zn] 0.000 description 6
- KIVHKTXAVHIDEJ-UHFFFAOYSA-N CC(C)(C)C(C1)C=Cc2c1c(CC(C)(C(C)(C)C)C=C1)c1[n]2-c(c(-[n]1c(ccc(C(C)(C)C)c2)c2c2c1ccc(C(C)(C)C)c2)c(-c(cc1)ccc1C#N)c(-[n]1c2ccc(C(C)(C)C)cc2c2c1ccc(C(C)(C)C)c2)c1-[n]2c(C=CC(C3)C(C)(C)C)c3c3c2ccc(C(C)(C)C)c3)c1C#N Chemical compound CC(C)(C)C(C1)C=Cc2c1c(CC(C)(C(C)(C)C)C=C1)c1[n]2-c(c(-[n]1c(ccc(C(C)(C)C)c2)c2c2c1ccc(C(C)(C)C)c2)c(-c(cc1)ccc1C#N)c(-[n]1c2ccc(C(C)(C)C)cc2c2c1ccc(C(C)(C)C)c2)c1-[n]2c(C=CC(C3)C(C)(C)C)c3c3c2ccc(C(C)(C)C)c3)c1C#N KIVHKTXAVHIDEJ-UHFFFAOYSA-N 0.000 description 1
- MLFFHLJMJHROIS-UHFFFAOYSA-N CC(C)(C)c(cc1)cc(c2cc(C(C)(C)C)ccc22)c1[n]2-c(c(-c(cc1)ccc1OC)c1-[n]2c3ccc(C(C)(C)C)cc3c3c2ccc(C(C)(C)C)c3)c(-c(cc2)ccc2OC)c(-[n]2c3ccc(C(C)(C)C)cc3c3c2ccc(C(C)(C)C)c3)c1C#N Chemical compound CC(C)(C)c(cc1)cc(c2cc(C(C)(C)C)ccc22)c1[n]2-c(c(-c(cc1)ccc1OC)c1-[n]2c3ccc(C(C)(C)C)cc3c3c2ccc(C(C)(C)C)c3)c(-c(cc2)ccc2OC)c(-[n]2c3ccc(C(C)(C)C)cc3c3c2ccc(C(C)(C)C)c3)c1C#N MLFFHLJMJHROIS-UHFFFAOYSA-N 0.000 description 1
- GCZGLIZRLRWAQY-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1-c(c(F)c(c(C#N)c1F)F)c1F Chemical compound CC(C)(C)c(cc1)ccc1-c(c(F)c(c(C#N)c1F)F)c1F GCZGLIZRLRWAQY-UHFFFAOYSA-N 0.000 description 1
- KGHFYQZXBHTJGH-UHFFFAOYSA-N CC(C1)C(OC)=CC=C1c(c(F)c(c(F)c1-c(cc2)ccc2OC)C#N)c1F Chemical compound CC(C1)C(OC)=CC=C1c(c(F)c(c(F)c1-c(cc2)ccc2OC)C#N)c1F KGHFYQZXBHTJGH-UHFFFAOYSA-N 0.000 description 1
- QREVRHSWIRRCGI-UHFFFAOYSA-N CC(CC(C(C)(C)C)=C1)c2c1c(cc(C(C)(C)C)cc1)c1[n]2-c(c(C#N)c(c(-[n]1c(ccc(C(C)(C)C)c2)c2c2cc(C(C)(C)C)ccc12)c1-c2ccccn2)-[n]2c(ccc(C(C)(C)C)c3)c3c3c2ccc(C(C)(C)C)c3)c1-[n]1c2ccc(C(C)(C)C)cc2c2c1ccc(C(C)(C)C)c2 Chemical compound CC(CC(C(C)(C)C)=C1)c2c1c(cc(C(C)(C)C)cc1)c1[n]2-c(c(C#N)c(c(-[n]1c(ccc(C(C)(C)C)c2)c2c2cc(C(C)(C)C)ccc12)c1-c2ccccn2)-[n]2c(ccc(C(C)(C)C)c3)c3c3c2ccc(C(C)(C)C)c3)c1-[n]1c2ccc(C(C)(C)C)cc2c2c1ccc(C(C)(C)C)c2 QREVRHSWIRRCGI-UHFFFAOYSA-N 0.000 description 1
- CNSIWXLCFOXSIQ-FXBPXSCXSA-N CC1(C=CC=CC1C=C1)c(c2ccc(cccc3)c3c22)c1[n]2-c(cc1)ccc1N(c(cc1)ccc1-[n]1c2c(/C=C\C)c(C=C)ccc2c(-c2c(C)cccc2)c1C)c(cc1)ccc1-[n]1c(c(cccc2)c2cc2)c2c2c1ccc1ccccc21 Chemical compound CC1(C=CC=CC1C=C1)c(c2ccc(cccc3)c3c22)c1[n]2-c(cc1)ccc1N(c(cc1)ccc1-[n]1c2c(/C=C\C)c(C=C)ccc2c(-c2c(C)cccc2)c1C)c(cc1)ccc1-[n]1c(c(cccc2)c2cc2)c2c2c1ccc1ccccc21 CNSIWXLCFOXSIQ-FXBPXSCXSA-N 0.000 description 1
- FMVXUJNSNQGFIN-UHFFFAOYSA-N COc(cc1)ccc1-c(c(C#N)c(c(F)c1-c(cc2)ccc2OC)F)c1F Chemical compound COc(cc1)ccc1-c(c(C#N)c(c(F)c1-c(cc2)ccc2OC)F)c1F FMVXUJNSNQGFIN-UHFFFAOYSA-N 0.000 description 1
- QJPJQTDYNZXKQF-UHFFFAOYSA-N COc(cc1)ccc1Br Chemical compound COc(cc1)ccc1Br QJPJQTDYNZXKQF-UHFFFAOYSA-N 0.000 description 1
- FKXTVUUVCNDRGT-UHFFFAOYSA-N N#Cc(c(F)c(c(-c1ncccc1)c1F)F)c1F Chemical compound N#Cc(c(F)c(c(-c1ncccc1)c1F)F)c1F FKXTVUUVCNDRGT-UHFFFAOYSA-N 0.000 description 1
- GTAZMAYDIJQPST-UHFFFAOYSA-N N#Cc(cc1)ccc1-c(c(F)c(c(C#N)c1F)F)c1F Chemical compound N#Cc(cc1)ccc1-c(c(F)c(c(C#N)c1F)F)c1F GTAZMAYDIJQPST-UHFFFAOYSA-N 0.000 description 1
- MMPKVFQMDMBXSG-UHFFFAOYSA-N N#Cc1cc(F)cc(F)c1F Chemical compound N#Cc1cc(F)cc(F)c1F MMPKVFQMDMBXSG-UHFFFAOYSA-N 0.000 description 1
- DVNOWTJCOPZGQA-UHFFFAOYSA-N c(cc1)cc(c2c3cccc2)c1[n]3-c1cc(-[n]2c3ccccc3c3c2cccc3)cc(-[n]2c3ccccc3c3c2cccc3)c1 Chemical compound c(cc1)cc(c2c3cccc2)c1[n]3-c1cc(-[n]2c3ccccc3c3c2cccc3)cc(-[n]2c3ccccc3c3c2cccc3)c1 DVNOWTJCOPZGQA-UHFFFAOYSA-N 0.000 description 1
- MMNNWKCYXNXWBG-UHFFFAOYSA-N c(cc1)ccc1-c1cc(-c2nc(-c3cccc(-c4ccccc4)c3)nc(-c3cc(-c4ccccc4)ccc3)n2)ccc1 Chemical compound c(cc1)ccc1-c1cc(-c2nc(-c3cccc(-c4ccccc4)c3)nc(-c3cc(-c4ccccc4)ccc3)n2)ccc1 MMNNWKCYXNXWBG-UHFFFAOYSA-N 0.000 description 1
Images




















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/86—Carbazoles; Hydrogenated carbazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/654—Aromatic compounds comprising a hetero atom comprising only nitrogen as heteroatom
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
- H10K85/6572—Polycyclic condensed heteroaromatic hydrocarbons comprising only nitrogen in the heteroaromatic polycondensed ring system, e.g. phenanthroline or carbazole
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
Definitions
- the present invention relates to a 2,3,4,5,6-penta-substituted benzonitrile compound, a light-emitting material, and a light-emitting element using the same, which have excellent light-emitting characteristics.
- Patent Document 1 discloses 3,5-di (3,6-diphenyl-9H-carbazol-9-yl) -2,4,6-tri (4-cyanophenyl) -benzonitrile and the like.
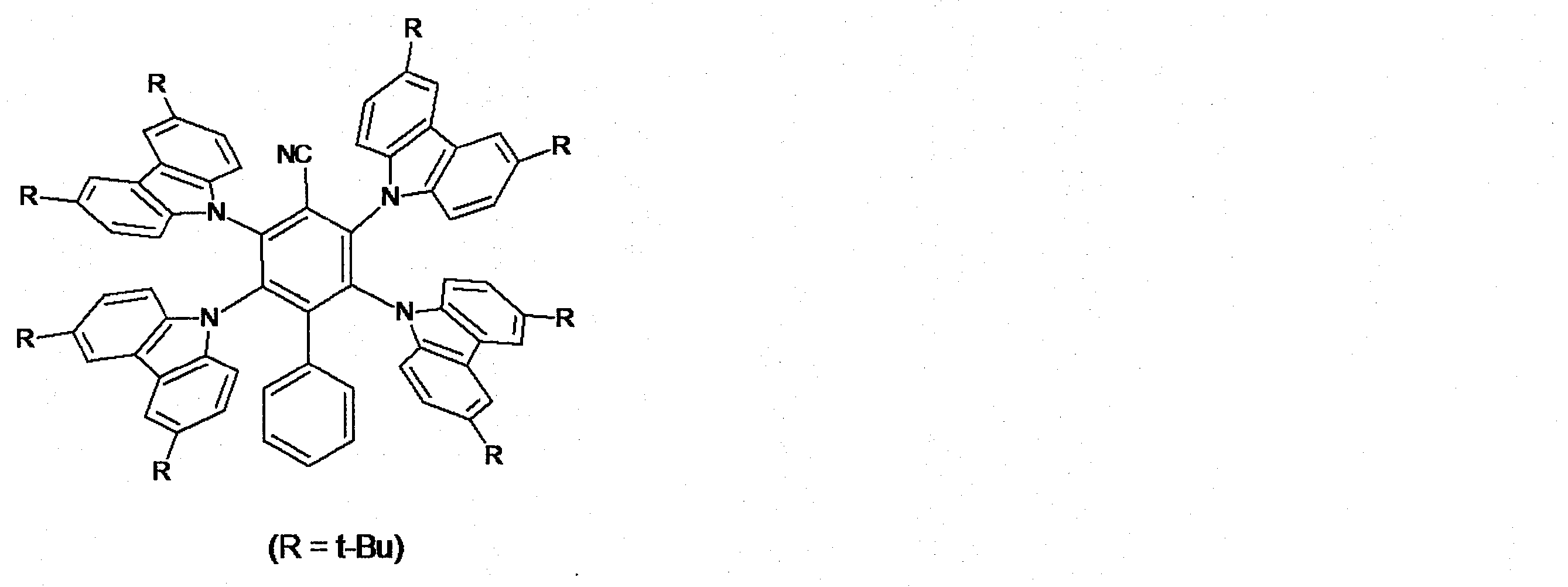
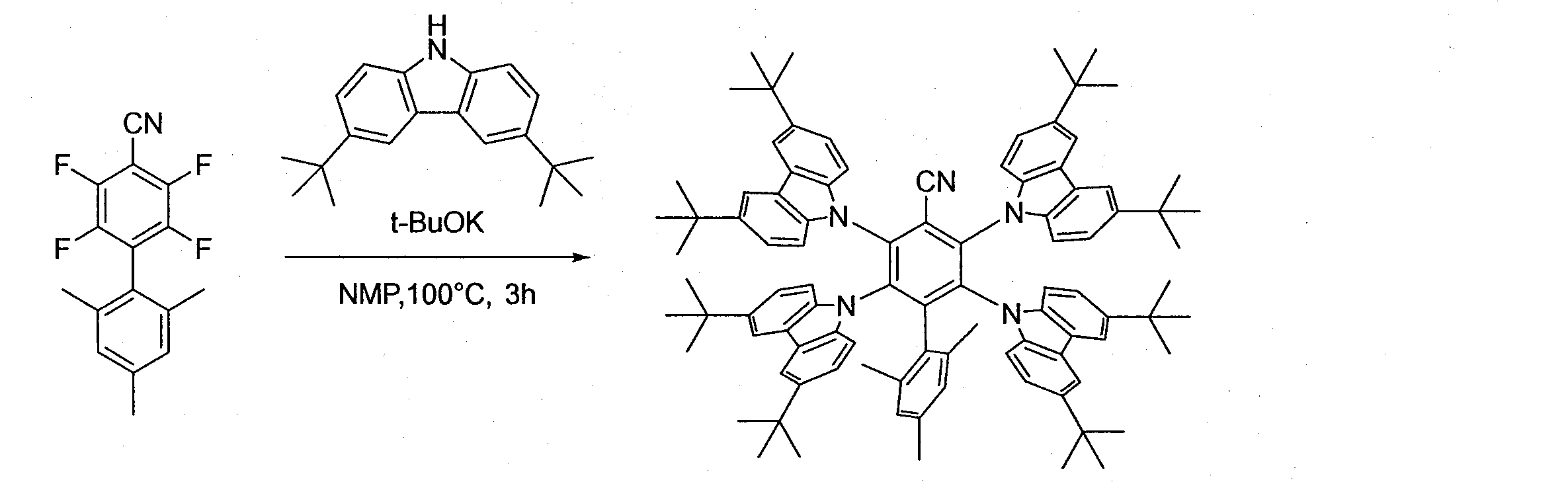
- Patent Document 2 discloses 2,3,5,6-tetra (3,6-diphenyl-9H-carbazol-9-yl) -4- (4-cyanophenyl) -benzonitrile and the like.
- Patent Document 3 discloses 2,3,5,6-tetra (9H-carbazol-9-yl) -4-phenyl-benzonitrile and the like.
- An object of the present invention is to provide a 2,3,4,5,6-pentasubstituted benzonitrile compound (hereinafter, may be referred to as “the compound of the present invention”), a light-emitting material, and a light-emitting material using the same, which have excellent light-emitting characteristics. It is to provide an element.
- L is each independently a substituted or unsubstituted aryl group or a substituted or unsubstituted heteroaryl group; n represents the number of L, is 1 or 2, Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazol-9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group Or a substituted or unsubstituted 3-phenyl-6-t-butyl-9H-carbazol-9-yl group, and m represents the number of Q and is 5-n.
- L is each independently a substituted or unsubstituted aryl group or a substituted or unsubstituted heteroaryl group
- Q is each independently a substituted or unsubstituted 3,6- It is a di-t-butyl-9H-carbazol-9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group or a substituted or unsubstituted heteroaryl group
- Q is each independently a substituted or unsubstituted 3,6- It is a di-t-butyl-9H-carbazol-9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group or a substituted or unsubstituted heteroaryl group
- Q is each independently a substituted or unsubstituted 3,6- It is a di-t-butyl-9H-carbazol-9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group or a substituted or unsubstituted heteroaryl group
- Q is each independently a substituted or unsubstituted 3,6- It is a di-t-butyl-9H-carbazol-9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is a substituted or unsubstituted nitrogen- or oxygen-containing 5- or 6-membered heteroaryl.
- L is a substituted or unsubstituted phenyl group, a substituted or unsubstituted biphenyl group, a substituted or unsubstituted naphthyl group, a substituted or unsubstituted anthryl group, a substituted or unsubstituted phenanthryl group, a substituted or unsubstituted group Pyridinyl group, substituted or unsubstituted pyrimidinyl group, substituted or unsubstituted furyl group, substituted or unsubstituted thienyl group, substituted or unsubstituted oxazolyl group, substituted or unsubstituted thiazolyl group, substituted or unsubstituted imidazolyl Group,
- a luminescent material comprising the compound according to any one of [1] to [11].
- a light-emitting element containing the light-emitting material according to [12].
- the compound of the present invention is useful as a light emitting material. Some luminescent materials according to the present invention emit delayed fluorescence. The light-emitting element containing the light-emitting material according to the present invention can realize excellent light-emitting efficiency.
- FIG. 3 is a diagram showing characteristics of voltage-current density-luminance of 3Cz-2PBN-A, 3BuCz-2PBN-A, and 3PCz-2PBN-A. It is a figure which shows the brightness-external quantum efficiency of 3Cz-2PBN-A, 3BuCz-2PBN-A, and 3PCz-2PBN-A ⁇ .
- FIG. 3 is a diagram showing characteristics of voltage-current density-luminance of 3Cz-2PBN-B, 3BuCz-2PBN-B, and 3PCz-2PBN-B. It is a figure which shows the brightness-external quantum efficiency of 3Cz-2PBN-B, 3BuCz-2PBN-B, and 3PCz-2PBN-B.
- FIG. 3 is a diagram showing characteristics of voltage-current density-luminance of 3Cz-2PBN-A, 3BuCz-2PBN-A, and 3PCz-2PBN-A.
- FIG. 4 is a diagram showing voltage-current density-luminance characteristics of 4Cz-1PBN-A and 4BuCz-1PBN-A. It is a figure which shows the brightness-external quantum efficiency of 4Cz-1PBN-A and 4BuCz-1PBN-ABN.
- FIG. 3 is a diagram showing characteristics of voltage-current density-luminance of 3BuCz-2PBN-C, 3PCz-2PBN-C, 3BuCz-2PBN-D, and 3PCz-2PBN-D. It is a figure which shows the brightness-external quantum efficiency of 3BuCz-2PBN-C, 3PCz-2PBN-C, 3BuCz-2PBN-D, and 3PCz-2PBN-D.
- FIG. 3 is a diagram showing voltage-current density-luminance characteristics of 4Cz-1PBN-A and 4BuCz-1PBN-A. It is a figure which shows the brightness-external quantum efficiency of 4Cz-1PBN-A and 4BuCz-1
- FIG. 4 is a diagram showing characteristics of voltage-current density-luminance of 4BuCz-1PBN-A, 4BuCz-1PBN-B, and 4BuCz-1PBN-C.
- FIG. 4 is a diagram showing luminance-external quantum efficiency of 4BuCz-1PBN-A, 4BuCz-1PBN-B, and 4BuCz-1PBN-C. It is a figure which shows the characteristics of voltage-current density-luminance of 4X-BCz-PBN-Bu, 4X-BCz-PBN-OMe, 4X-BCz-PBN-SMe, and 4X-BCz-PBN-CN.
- FIG. 4 is a diagram showing characteristics of voltage-current density-luminance of 4X-BCz-PBN-CO2Me, 4X-BCz-PBN-MesBN, and 4X-BCz-PBN-IPN. It is a figure which shows the brightness-external quantum efficiency of 4X-BCz-PBN-CO2Me, 4X-BCz-PBN-MesBN, and 4X-BCz-PBN-IPN.
- FIG. 3 is a diagram showing characteristics of voltage-current density-luminance of 3Y-BCz-PBN-tBu, 3Y-BCz-PBN-OMe, and 3Y-BCz-PBN-SMe.
- FIG. 3 is a diagram showing characteristics of voltage-current density-luminance of 3F-BCz-PBN-tBu, 3F-BCz-PBN-OMe, and 3F-BCz-PBN-SMe. It is a figure which shows the brightness-external quantum efficiency of 3F-BCz-PBN-tBu, 3F-BCz-PBN-OMe, and 3F-BCz-PBN-SMe.
- the 2,3,4,5,6-penta-substituted benzonitrile compound of the present invention is a compound represented by the formula (I).
- the 2,3,4,5,6-penta-substituted benzonitrile compound of the present invention is preferably a compound represented by the formula (IIa), (IIb), (IIc), (IIIa), (IIIb), (IIIc) ) Or a compound represented by the formula (IVa), more preferably a compound represented by the formula (IIa).
- the 2,3,4,5,6-penta-substituted benzonitrile compound of the present invention may be a compound represented by the formula (IIId) or (IIIe).
- L is a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazol-9-yl group It is.
- L is a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazol-9-yl group It is.
- L is a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazol-9-yl group It is.
- L is each independently a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazole- It is a 9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazole- It is a 9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazole- It is a 9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazole- It is a 9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazole- It is a 9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- L is each independently a substituted or unsubstituted aryl group
- Q is each independently a substituted or unsubstituted 3,6-di-t-butyl-9H-carbazole- It is a 9-yl group or a substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group.
- the substituted or unsubstituted 3,6-di-t-butyl-9H-carbazol-9-yl group is preferably a group represented by the formula (A).
- the substituted or unsubstituted 3,6-diphenyl-9H-carbazol-9-yl group is preferably a group represented by the formula (B).
- the substituted or unsubstituted 3-phenyl-6-t-butyl-9H-carbazol-9-yl group is preferably a group represented by the formula (C).
- R 1 , R 2 , R 3 , R 4 , R 5 and R 6 are each independently a hydrogen atom or a substituent, and * is a bonding moiety. is there.
- the term “unsubstituted” means that there is only a parent nucleus. When only the name of a group to be a mother nucleus is described, it means “unsubstituted” unless otherwise specified.
- the term “substituted” means that any hydrogen atom of the parent nucleus is replaced by a group having the same or different structure as the parent nucleus. Thus, a "substituent” is another group attached to a parent nucleus.
- the number of substituents may be one, or two or more. Two or more substituents may be the same or different.
- the “substituent” is chemically acceptable and is not particularly limited as long as it has the effects of the present invention.
- Specific examples of the group that can be a “substituent” include the following groups.
- a halogeno group such as a fluoro group, a chloro group, a bromo group, and an iodo group;
- C1-20 such as methyl group, ethyl group, n-propyl group, i-propyl group, n-butyl group, s-butyl group, i-butyl group, t-butyl group, n-pentyl group and n-hexyl group
- An alkyl group (preferably a C1-6 alkyl group); Vinyl group, 1-propenyl group, 2-propenyl group, 1-butenyl group, 2-butenyl group, 3-butenyl group, 1-methyl-2-propenyl group, 2-methyl-2-propenyl group, 1-penten
- Ethynyl 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-methyl-2-propynyl, 2-methyl-3-butynyl, 1-pentynyl , 2-pentynyl, 3-pentynyl, 4-pentynyl, 1-methyl-2-butynyl, 2-methyl-3-pentynyl, 1-hexynyl, 1,1-dimethyl-2-butynyl, etc.
- a C2-10 alkynyl group (C2-6 alkynyl group); A C3-8 cycloalkyl group such as a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, a cubanyl group; C3-8 cycloalkenyl groups such as 2-cyclopropenyl group, 2-cyclopentenyl group, 3-cyclohexenyl group and 4-cyclooctenyl group; A C6-40 aryl group such as a phenyl group and a naphthyl group (preferably a C6-10 aryl group); 5-membered heteroaryl groups such as pyrrolyl, furyl, thienyl, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, ox
- a C1-6 haloalkyl group such as a chloromethyl group, a chloroethyl group, a trifluoromethyl group, a 1,2-dichloro-n-propyl group, a 1-fluoro-n-butyl group, a perfluoro-n-pentyl group; C2-6 haloalkenyl groups such as 2-chloro-1-propenyl group and 2-fluoro-1-butenyl group; C2-6 haloalkynyl groups such as 4,4-dichloro-1-butynyl group, 4-fluoro-1-pentynyl group and 5-bromo-2-pentynyl group; A C3-6 halocycloalkyl group such as a 3,3-difluorocyclobutyl group; C1-6 haloalkoxy group such as 2-chloro-n-propoxy group, 2,3-dichlorobutoxy group, trifluoromethoxy group, 2,2,2-tri
- a C1-6 alkylaminocarbonyl group such as a methylaminocarbonyl group, a dimethylaminocarbonyl group, an ethylaminocarbonyl group, an i-propylaminocarbonyl group
- Imino C1-6 alkyl groups such as iminomethyl group, (1-imino) ethyl group, (1-imino) -n-propyl group
- Hydroxyimino C1-6 alkyl groups such as hydroxyiminomethyl group, (1-hydroxyimino) ethyl group, (1-hydroxyimino) propyl group
- a C1-6 alkoxyimino C1-6 alkyl group such as a methoxyiminomethyl group and a (1-methoxyimino) ethyl group
- a mercapto group C1-20 alkylthio groups (preferably C1-6 alkylthio groups) such as methylthio group, ethylthio group, n-propylthio group, i-propylthio group, n-butylthio group, i-butylthio group, s-butylthio group and t-butylthio group );
- a C1-6 haloalkylthio group such as a trifluoromethylthio group and a 2,2,2-trifluoroethylthio group;
- a C2-6 alkenylthio group such as a vinylthio group and an allylthio group;
- a C2-6 alkynylthio group such as an ethynylthio group and a propargylthio group;
- a C1-6 alkylsulfinyl group such as a methylsulfinyl group, an ethylsulf
- C6-20 arylamide groups such as phenylamide group, naphthylamide group, phenylacetamide group, naphthylacetamide group;
- Tri-C1-10 alkylsilyl groups such as trimethylsilyl group, triethylsilyl group and t-butyldimethylsilyl group (preferably tri-C1-6 alkylsilyl group); Tri-C6-10 arylsilyl groups such as triphenylsilyl group; In these “substituents”, any of the hydrogen atoms in the substituents may be substituted with a group having a different structure.
- C1-6 indicates that the group serving as a mother nucleus has 1-6 carbon atoms.
- the number of carbon atoms does not include the number of carbon atoms present in the substituent.
- an ethoxybutyl group is classified as a C2 alkoxy C4 alkyl group because a group serving as a mother nucleus is a butyl group and a substituent is an ethoxy group.
- Preferred substituents include a hydroxy group, a halogeno group, a cyano group, an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, an alkylthio group having 1 to 20 carbon atoms, and an alkyl substitution having 1 to 20 carbon atoms.
- Substituted carbazolyl group alkenyl group having 2 to 10 carbon atoms, alkynyl group having 2 to 10 carbon atoms, alkoxycarbonyl group having 2 to 10 carbon atoms, alkylsulfonyl group having 1 to 10 carbon atoms, haloalkyl having 1 to 10 carbon atoms Group, amide group, alkylamide group having 2 to 10 carbon atoms, trialkylsilyl group having 3 to 20 carbon atoms, trialkylsilylalkyl group having 4 to 20 carbon atoms, Trialkylsilyl alkenyl group prime 5-20 include trialkylsilyl alkynyl group and a nitro group having 5 to 20 carbon atoms.
- More preferred substituents include a halogeno group, a cyano group, an alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, an aryl group having 6 to 40 carbon atoms, a heteroaryl group having 3 to 40 carbon atoms, Examples thereof include a diarylamino group having 12 to 40 carbon atoms and a carbazolyl group having 12 to 40 carbon atoms.
- substituents include a fluoro group, a chloro group, a cyano group, an alkyl group having 1 to 10 carbon atoms, an alkoxy group having 1 to 10 carbon atoms, a dialkylamino group having 1 to 10 carbon atoms, and an aryl having 6 to 15 carbon atoms. And heteroaryl groups having 3 to 12 carbon atoms.
- substituents those which can be further substituted may be substituted by the substituent.
- the substituted or unsubstituted aryl group for L is preferably a substituted or unsubstituted phenyl group, a substituted or unsubstituted biphenyl group, a substituted or unsubstituted naphthyl group, a substituted or unsubstituted anthryl group, or a substituted or unsubstituted anthryl group.
- Substituted phenanthryl groups can be mentioned. Of these, a substituted or unsubstituted phenyl group is more preferred.
- the compound of the present invention is not particularly limited by its production method.
- the compound described in Patent Document 1 or Patent Document 2 is applied to a compound corresponding to a corresponding substituent as a starting material or described in Examples. Can be obtained.
- the synthesized compound of the present invention can be purified by column chromatography, adsorption and purification using silica gel, activated carbon, activated clay, or the like, recrystallization or crystallization using a solvent, or the like.
- the compound can be identified by NMR analysis or the like.
- the compound of the present invention can be used as a light emitting material.
- the light-emitting material of the present invention can provide a light-emitting element such as an organic photoluminescence element and an organic electroluminescence element. Since the compound of the present invention has a function of assisting light emission of another light emitting material (host material), it can be used by doping another light emitting material.
- the organic photoluminescence element which is one of the light emitting elements of the present invention, is provided with a light emitting layer containing the light emitting material of the present invention on a substrate.
- the light-emitting layer can be obtained by a coating method such as spin coating, a printing method such as an inkjet printing method, or a vapor deposition method.
- the organic electroluminescence device of the present invention has an organic layer provided between an anode and a cathode.
- the ⁇ organic layer '' in the present invention means a layer substantially located between an anode and a cathode and substantially composed of an organic substance, and these layers contain an inorganic substance within a range that does not impair the performance of the light emitting device of the present invention. May be.
- the substrate in order, an anode, a hole injection layer, a hole transport layer, an electron blocking layer, a light emitting layer, a hole blocking layer, an electron transport layer
- examples include those comprising a cathode and those further having an electron injection layer between the electron transport layer and the cathode.
- the light emitting material of the present invention may be doped not only in the light emitting layer but also in the hole injection layer, the hole transport layer, the electron blocking layer, the hole blocking layer, the electron transport layer, or the electron injection layer.
- the substrate is a support for the light emitting element, and a silicon plate, a quartz plate, a glass plate, a metal plate, a metal foil, a resin film, a resin sheet, or the like is used. Particularly, a glass plate or a plate of a transparent synthetic resin such as polyester, polymethacrylate, polycarbonate, and polysulfone is preferable.
- a synthetic resin substrate it is necessary to pay attention to gas barrier properties. If the gas barrier property of the substrate is too low, the light emitting element may be deteriorated by the outside air passing through the substrate. Therefore, it is preferable to provide a dense silicon oxide film or the like on one or both sides of the synthetic resin substrate to ensure gas barrier properties.
- An anode is provided on the substrate.
- a material having a large work function is generally used for the anode.
- the anode material include metals such as aluminum, gold, silver, nickel, palladium, and platinum; metal oxides such as indium oxide, tin oxide, ITO, zinc oxide, In 2 O 3 —ZnO, and IGZO; Examples thereof include metal halides such as copper halide, carbon black, and conductive polymers such as poly (3-methylthiophene), polypyrrole, and polyaniline.
- the formation of the anode is usually performed by a sputtering method, a vacuum evaporation method, or the like in many cases.
- fine particles of metal such as silver, fine particles of copper iodide or the like, carbon black, conductive metal oxide fine particles, conductive polymer fine powder, etc.
- they are dispersed in an appropriate binder resin solution and placed on a substrate.
- An anode can be formed by coating.
- a conductive polymer a thin film can be directly formed on a substrate by electrolytic polymerization, or an anode can be formed by applying a conductive polymer on the substrate.
- the anode can be formed by laminating two or more different substances.
- the thickness of the anode depends on the required transparency. When transparency is required, it is desirable that the visible light transmittance is usually 60% or more, preferably 80% or more. In this case, the thickness is usually 10 to 1000 nm, preferably 10 to 1000 nm. 200 nm. If opaque, the anode may be as thick as the substrate.
- the sheet resistance of the anode is preferably several hundred ⁇ / ⁇ or more.
- a hole injection layer provided as needed, in addition to a porphyrin compound represented by copper phthalocyanine, a naphthalenediamine derivative, a star burst type triphenylamine derivative, three or more triphenylamine structures in the molecule, a single bond Or acceptor heterocyclic compounds such as triphenylamine trimers and tetramers such as arylamine compounds having a structure linked by a divalent group containing no hetero atom, hexacyanoazatriphenylene, and coating type polymer materials Can be used. These materials can be formed into a thin film by a known method such as a spin coating method or an ink jet method, in addition to a vapor deposition method.
- the hole transporting material used for the hole transporting layer provided as necessary has a high hole injection efficiency from the anode and can efficiently transport the injected holes.
- the ionization potential is small, the transparency to visible light is high, the hole mobility is large, the stability is high, and impurities serving as traps are unlikely to be generated during production or use.
- the element has higher heat resistance. Therefore, a material having a Tg of 70 ° C. or more is desirable.
- triazole derivatives As a hole transport layer provided as needed, triazole derivatives, oxadiazole derivatives, imidazole derivatives, carbazole derivatives, indolocarbazole derivatives, polyarylalkane derivatives, pyrazoline derivatives, pyrazolone derivatives, phenylenediamine derivatives, arylamine derivatives, Examples include amino-substituted chalcone derivatives, oxazole derivatives, styryl anthracene derivatives, fluorenone derivatives, hydrazone derivatives, stilbene derivatives, silazane derivatives, aniline copolymers, and conductive polymer oligomers.
- NPD N, N′-diphenyl-N, N′-di (m-tolyl) -benzidine
- TPD N, N ′ Benzidine derivatives such as -diphenyl-N, N'-di ( ⁇ -naphthyl) -benzidine (hereinafter abbreviated as NPD), N, N, N ', N'-tetrabiphenylylbenzidine, 1,1-bis [ (Di-4-tolylamino) phenyl] cyclohexane (hereinafter abbreviated as TAPC), various triphenylamine trimers and tetramers, and carbazole derivatives.
- NPD -diphenyl-N, N'-di ( ⁇ -naphthyl) -benzidine
- TAPC 1,1-bis [ (Di-4-tolylamino) phenyl] cyclohexane
- TAPC 1,1-bis [ (Di-4-
- the hole transport layer may be a film having a single-layer structure or a film having a laminated structure.
- a coating type such as poly (3,4-ethylenedioxythiophene) (hereinafter abbreviated as PEDOT) / poly (styrenesulfonate) (hereinafter abbreviated as PSS) is used.
- PEDOT poly (3,4-ethylenedioxythiophene)
- PSS poly (styrenesulfonate)
- PSS poly (styrenesulfonate)
- trisbromophenylamine hexachloroantimony is further P-doped with respect to a material usually used for the layer, or a polymer having a PD structure in its partial structure.
- a material usually used for the layer or a polymer having a PD structure in its partial structure.
- Compounds and the like can be used.
- a host material having a hole-injecting / transporting property a carbazole derivative such as PPF, PPT, CBP, TCTA, or mCP can be used.
- TCTA N-carbazolyl triphenylamine
- mCP 1,3-bis (carbazol-9-yl) benzene
- Ad-Cz 2,2-bis (4-carbazol-9-ylphenyl) adamantane
- Carbazole derivatives such as 9- [4- (carbazol-9-yl) phenyl] -9- [4- (triphenylsilyl) phenyl] -9H-fluorene and triphenylsilyl groups.
- the electron blocking layer may be a film having a single layer structure or a film having a laminated structure, and these materials may be formed by a known method such as a spin coating method and an ink jet method in addition to a vapor deposition method.
- a thin film can be formed by the method described above.
- the light-emitting layer is a layer having a function of emitting light by generating excitons by recombination of holes and electrons injected from the anode and the cathode, respectively.
- the light-emitting layer may be formed using the light-emitting material of the present invention alone, or may be formed by doping a host material with the light-emitting material of the present invention.
- the host material include metal complexes of quinolinol derivatives such as PPF, PPT, and tris (8-hydroxyquinoline) aluminum (hereinafter abbreviated as Alq3), anthracene derivatives, bisstyrylbenzene derivatives, pyrene derivatives, oxazole derivatives, polyoxins, and the like.
- Examples thereof include a paraphenylene vinylene derivative, a compound having a bipyridyl group and an orthoterphenyl structure, mCP, a thiazole derivative, a benzimidazole derivative, and a polydialkylfluorene derivative.
- the light emitting layer may contain a known dopant. Examples of the dopant include quinacridone, coumarin, rubrene, anthracene, perylene and derivatives thereof, benzopyran derivatives, rhodamine derivatives, aminostyryl derivatives and the like.
- a phosphorescent material such as a green phosphorescent material such as Ir (ppy) 3, a blue phosphorescent material such as FIrpic and FIr6, and a red phosphorescent material such as Btp2Ir (acac) may be used. These can be used alone or in combination of two or more.
- the light-emitting layer may have a single-layer structure or a stacked-layer structure. These materials can be formed into a thin film by a known method such as a spin coating method or an ink jet method, in addition to a vapor deposition method.
- the lower limit of the amount of the light emitting material of the present invention that can be contained in the light emitting layer is preferably 0.1% by mass, more preferably 1% by mass, and the upper limit is preferably 50% by mass. %, More preferably 20% by mass, even more preferably 10% by mass.
- a compound having a bipyridyl group and an orthoterphenyl structure such as bathocuproin (hereinafter abbreviated as BCP), an aluminum (III) bis (2-methyl-8- Metal complexes of quinolinol derivatives such as (quinolinato) -4-phenylphenolate (hereinafter abbreviated as BAlq), various rare earth complexes, oxazole derivatives, triazole derivatives, triazine derivatives, and other compounds having a hole-blocking action may be mentioned. it can. These materials may also serve as the material of the electron transport layer. These can be used alone or in combination of two or more.
- the hole blocking layer may be a film having a single-layer structure or a film having a laminated structure. These materials can be formed into a thin film by a known method such as a spin coating method or an ink jet method, in addition to a vapor deposition method.
- an electron transport layer As an electron transport layer provided as needed, in addition to metal complexes of quinolinol derivatives such as Alq3 and BAlq, various metal complexes, triazole derivatives, triazine derivatives, oxadiazole derivatives, thiadiazole derivatives, carbodiimide derivatives, quinoxaline derivatives, A phenanthroline derivative, a silole derivative, or the like can be used. These can be used alone or in combination of two or more.
- the electron transport layer may have a single-layer structure or a multilayer structure. These materials can be formed into a thin film by a known method such as a spin coating method or an ink jet method, in addition to a vapor deposition method.
- alkali metal salts such as lithium fluoride and cesium fluoride
- alkaline earth metal salts such as magnesium fluoride
- ____ metal oxides such as aluminum oxide (merely “metal oxide ) May be used, but this can be omitted in the preferred selection of the electron transport layer and the cathode.
- a material obtained by further doping a metal such as cesium by N with respect to a material usually used for the layer can be used.
- Cathode materials include, for example, sodium, sodium-potassium alloy, lithium, tin, magnesium, magnesium / copper mixture, magnesium / aluminum mixture, magnesium / indium mixture, aluminum / aluminum oxide mixture, indium, calcium, aluminum, silver, lithium / Aluminum mixture, magnesium silver alloy, magnesium indium alloy, aluminum magnesium alloy and the like.
- a transparent conductive material By using a transparent conductive material, a transparent or translucent cathode can be obtained.
- the thickness of the cathode is usually 10 to 5000 nm, preferably 50 to 200 nm.
- the sheet resistance of the cathode is preferably several hundred ⁇ / ⁇ or more.
- a metal layer having a high work function and stable against the atmosphere such as aluminum, silver, nickel, chromium, gold, and platinum, is further laminated thereon. It is preferable to increase stability.
- a cathode interface layer may be provided between the two.
- Examples of the material used for the cathode interface layer include an aromatic diamine compound, a quinacridone compound, a naphthacene derivative, an organic silicon compound, an organic phosphorus compound, a compound having an N-phenylcarbazole skeleton, and an N-vinylcarbazole polymer. .
- the light-emitting element of the present invention can be applied to any of a single element, an element having a structure arranged in an array, and a structure in which an anode and a cathode are arranged in an XY matrix.
- a light emitting layer having a thickness of 20 nm was laminated in this order by a vacuum evaporation method (5.0.times.10@-4 Pa or less).
- PPF was used as a host material of the light emitting layer
- 2,4,6-tri (9H-carbazol-9-yl) -3,5-diphenyl-benzonitrile (3Cz-2PBN-A) was used as a dope material.
- the dough material concentration was set to 12.0% by weight.
- a cathode was formed by stacking a 10-nm thick PPF layer, a 40-nm thick B3PyPB layer, a 1-nm thick 8-hydroxyquinolitho lithium film, and a 100-nm thick aluminum film in this order by a vacuum evaporation method.
- An organic light emitting diode (OLED) was obtained. The results are shown in FIGS. This organic light-emitting diode had a maximum external quantum efficiency (EQEMax) of 13.1%.
- Example 1 except that the doping material was changed to 2,4,6-tri (3,6-diphenyl-9H-carbazol-9-yl) -3,5-diphenyl-benzonitrile (3PCz-2PBN-A) Emission evaluation was performed in the same manner. The results are shown in FIGS. EQEMax was 28.1%.
- Example 2 Comparing Example 1, Example 2, and Example 3, which are the same 2,3-phenyl-substituted skeleton, the compound of the present invention (Example 2 and Example 3) has a relatively high EQEMax and is used as a luminescent material. It turns out to be useful.
- the temperature was returned to room temperature, water and ethyl acetate were added, and the organic layer was separated.
- the aqueous layer was further extracted twice with ethyl acetate, and the combined organic layers were washed three times with water and then twice with saturated saline.
- the organic layer was dried over magnesium sulfate, filtered, and the filtrate was concentrated to obtain a crude product.
- the crude product was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate) to obtain a crude product. Further, acetone / n-hexane was added to the crude product, which was irradiated with ultrasonic waves.
- Emission evaluation was performed in the same manner as in Example 1 except that the dope material was changed to 2,3,5-tri (9H-carbazol-9-yl) -4,6-diphenyl-benzonitrile (3Cz-2PBN-B). went. The results are shown in FIGS. EQEMax was 16.7%.
- the temperature was returned to room temperature, water and ethyl acetate were added, and the organic layer was separated.
- the aqueous layer was further extracted twice with ethyl acetate, and the combined organic layers were washed three times with water and then twice with saturated saline.
- the organic layer was dried over magnesium sulfate, filtered, and the filtrate was concentrated to obtain a crude product.
- the crude product was purified by silica gel column chromatography (eluent: n-hexane / dichloromethane) to obtain a crude product. Further, dichloromethane / diethyl ether / n-hexane was added to the crude product, and the mixture was irradiated with ultrasonic waves.
- Example 1 was repeated except that the dope material was changed to 2,3,5-tri (3,6-diphenyl-9H-carbazol-9-yl) -4,6-diphenyl-benzonitrile (3PCz-2PBN-B). Emission evaluation was performed in the same manner. The results are shown in FIGS. EQEMax was 35.1%.
- the temperature was returned to room temperature, water and ethyl acetate were added, and the organic layer was separated.
- the aqueous layer was further extracted twice with ethyl acetate, and the combined organic layers were washed three times with water and then twice with saturated saline.
- the organic layer was dried over magnesium sulfate, filtered, and the filtrate was concentrated to obtain a crude product.
- the crude product was purified by silica gel column chromatography (eluent: n-hexane / dichloromethane) to obtain a crude product. Further, n-hexane was added to the crude purified product, and the mixture was irradiated with ultrasonic waves.
- N, N-dimethylformamide (5 mL) to which potassium tert-butoxide (0.73 g, 6.52 mmol) was added at room temperature was added, and the mixture was stirred at room temperature for 30 minutes. Thereafter, N, N-dimethylformamide (10 mL) to which compound 1 (0.4 g, 1.59 mmol) was added was added dropwise over 10 minutes. Thereafter, the mixture was stirred at 80 ° C. for 10 hours. Next, the temperature was returned to room temperature, water (20 mL) was added, and after 30 minutes, chloroform was added to perform extraction.
- Emission evaluation was performed in the same manner as in Example 1 except that the dope material was changed to 2,3,5,6-tetra (9H-carbazol-9-yl) -4-phenyl-benzonitrile (4Cz-1PBN-A). went. The results are shown in FIGS. EQEMax was 26.1%.
- Example 7 Comparing Example 7 and Example 8, which are the same 4-phenyl-substituted skeleton, it can be seen that the compound of the present invention (Example 7) has a relatively high EQEMax and is useful as a light emitting material.
- Example 1 was repeated except that the doping material was changed to 3,4,5-tri (3,6-diphenyl-9H-carbazol-9-yl) -2,6-diphenyl-benzonitrile (3PCz-2PBN-C). Emission evaluation was performed in the same manner. The results are shown in FIGS. EQEMax was 21.6%.
- 3,6-Diphenylcarbazole (1.75 g, 5.5 mmol) was added to a 100 mL two-necked flask purged with nitrogen, and dissolved in 10 mL of N-methyl-2-pyrrolidone. Thereto was added potassium t-butoxide (0.65 g, 5.8 mmol), and the mixture was stirred at room temperature for 1 hour. Then, 3 ′, 5 ′, 6′-trifluoro- [1,1 ′: 4 ′, 1 ′′ -terphenyl] -2′-carbonitrile (0.42 g, 1.4 mmol) was added to N- under a nitrogen stream.
- the suspension was added to 10 mL of methyl-2-pyrrolidone, and the mixture was stirred for 20 hours at 120 ° C. After returning to room temperature, water and ethyl acetate were added, the organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed three times with water and then twice with saturated saline, and the organic layer was dried over magnesium sulfate, filtered and concentrated to obtain a crude product.
- the crude product was purified by silica gel column chromatography (eluent: n-hexane / dichloromethane), and the crude product was purified again by silica gel column chromatography (eluent: n-hexane / toluene). (3PCz-2PBN-D) to 0. 93 g was obtained in a yield of 56.7%.
- Example 1 was repeated except that the doping material was changed to 3,4,6-tri (3,6-diphenyl-9H-carbazol-9-yl) -2,5-diphenyl-benzonitrile (3PCz-2PBN-D). Emission evaluation was performed in the same manner. The results are shown in FIGS. EQEMax was 20.7%.
- 3,6-Di-t-butylcarbazole (3.62 g, 13.0 mmol) was added to a 300 mL four-necked flask purged with nitrogen, and dissolved in 65 mL of N-methyl-2-pyrrolidone. To this was added potassium t-butoxide (1.58 g, 14.1 mmol), and the mixture was stirred at room temperature for 1 hour.
- 3 ′, 5 ′, 6′-Trifluoro- [1,1 ′: 4 ′, 1 ′′ -terphenyl] -2′-carbonitrile (1.00 g, 3.2 mmol) was added thereto under a nitrogen stream. The mixture was stirred for 6 hours at 120 ° C.
- 1,3-Dibromotetrafluorobenzene (6.50 g, 21.1 mmol), phenylboronic acid (2.73 g, 22.4 mmol), potassium carbonate (8.74 g, 63.3 mmol), 26 ml of water, And 65 ml of tetrahydrofuran. Degassed and then purged with argon. Thereafter, Pd (PPh 3 ) 4 (0.56 g, 0.63 mmol) was added, and the mixture was stirred under reflux with heating for 17 hours. The obtained liquid was returned to room temperature, and extracted with 100 ml of diethyl ether.
- the doped material was 2,4,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl)-[1,1′-biphenyl] -3-carbonitrile (4BuCz-1PBN-
- the luminescence was evaluated in the same manner as in Example 1 except that C) was changed.
- the results are shown in FIGS. EQEMax was 26.0%.
- the doped material was 3,4,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl)-[1,1′-biphenyl] -2-carbonitrile (4BuCz-1PBN-
- the luminescence was evaluated in the same manner as in Example 1 except that B) was used.
- the results are shown in FIGS. EQEMax was 22.3%.
- the doped material was 4 '-(tert-butyl) -2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl)-[1,1'-biphenyl]-
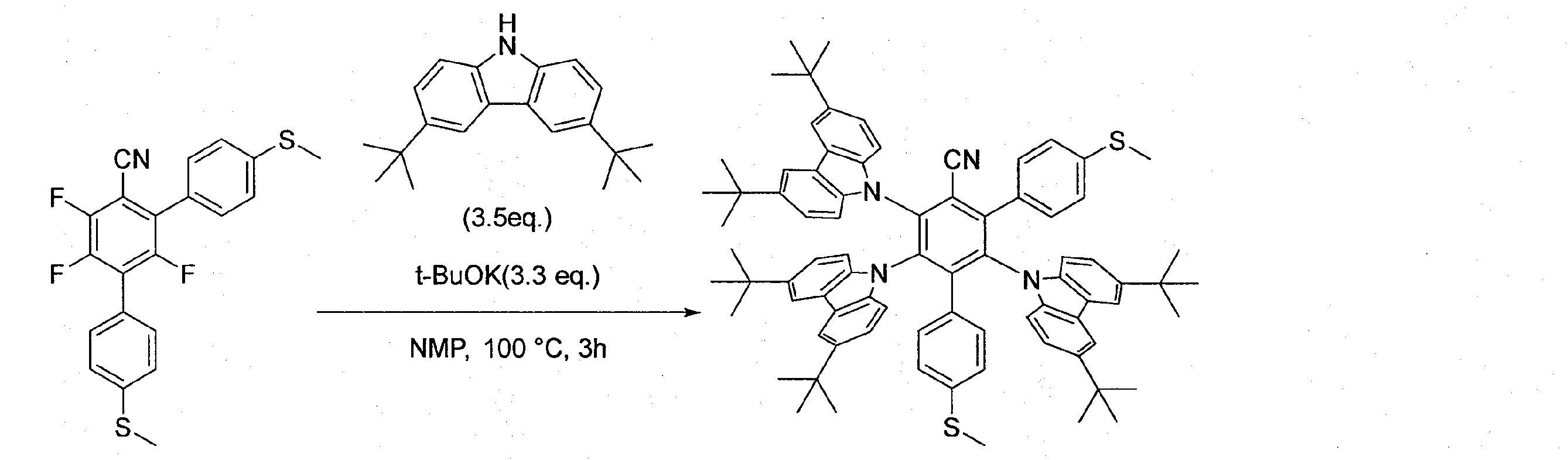
- the luminescence was evaluated in the same manner as in Example 1 except that 4-carbonitrile (4X-BCz-PBN-Bu) was used. The results are shown in FIGS. EQEMax was 26.5%.
- Dope material is 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl) -4'-methoxy- [1,1'-biphenyl] -4-carbonitrile
- the luminescence was evaluated in the same manner as in Example 1 except that (4X-BCz-PBN-OMe) was used. The results are shown in FIGS. EQEMax was 28.5%.
- Dope material is 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl) -4'-methylthio- [1,1'-biphenyl] -4-carbonitrile (4X-BCz-PBN-SMe), except that the luminescence was evaluated in the same manner as in Example 1. The results are shown in FIGS. EQEMax was 29.0%.
- the doped material was 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl)-[1,1'-biphenyl] -4,4'-dicarbonitrile ( The luminescence was evaluated in the same manner as in Example 1 except that 4X-BCz-PBN-CN) was used. The results are shown in FIGS. EQEMax was 31.6%.
- the doped material was 4'-cyano-2 ', 3', 5 ', 6'-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl)-[1,1'-biphenyl]-
- the luminescence was evaluated in the same manner as in Example 1 except that 4-carboxylic acid methyl ester (4X-BCz-PBN-CO 2 Me) was used.
- the results are shown in FIGS. EQEMax was 30.4%.
- 3,6-Di-t-butylcarbazole (2.12 g, 7.6 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 30 mL of dehydrated N-methyl-2-pyrrolidone. 82 g, 7.3 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, and the precursor (0.50 g, 1.70 mmol) dissolved in 5 mL of dehydrated N-methyl-2-pyrrolidone was added under a nitrogen stream, followed by stirring at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration.
- Emission evaluation was performed in the same manner as in Example 1 except that the dope material was changed to 4X-BCz-PBN-MesBN. The results are shown in FIGS. EQEMax was 29.5%.
- 3,6-Di-t-butylcarbazole (1.84 g, 6.6 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 27 mL of dehydrated N-methyl-2-pyrrolidone. (71 g, 6.3 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, and the precursor (0.45 g, 1.49 mmol) dissolved in 5 mL of dehydrated N-methyl-2-pyrrolidone was added under a nitrogen stream, followed by stirring at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration.
- the solid was dissolved in dichloromethane, dried over magnesium sulfate and concentrated. The residue was purified by silica gel column chromatography (n-hexane / benzene) to obtain a crude product. The crude product was added with n-hexane and irradiated with ultrasonic waves. The precipitated solid was collected by filtration and the solvent was distilled off, thereby obtaining 1.24 g of the desired product as a yellow solid (yield 62.0%).
- the doped material was 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl) -4- (pyridin-2-yl) benzonitrile (4X-BCz-PBN-
- the luminescence was evaluated in the same manner as in Example 1 except that 2-Py) was used.
- the results are shown in FIGS. EQEMax was 24.4%.
- the doped material was 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl) -4- (pyridin-3-yl) benzonitrile (4X-BCz-PBN-
- the luminescence was evaluated in the same manner as in Example 1 except that 3-Py) was used.
- the results are shown in FIGS. EQEMax was 33.6%.
- the doped material was 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl) -4- (pyridin-4-yl) benzonitrile (4X-BCz-PBN-
- the luminescence was evaluated in the same manner as in Example 1 except that 4-Py) was used.
- the results are shown in FIGS. EQEMax was 30.9%.
- the doped material was 2,3,5,6-tetrakis (3,6-di-tert-butyl-9H-carbazol-9-yl) -4- (pyrimidin-5-yl) benzonitrile (4X-BCz-PBN-
- the luminescence was evaluated in the same manner as in Example 1 except that 5-Pm) was used.
- the results are shown in FIGS. EQEMax was 30.3%.
- 3,6-Di-t-butylcarbazole (1.16 g, 4.2 mmol) was added to a 200-mL three-necked flask purged with nitrogen, dissolved in 24 mL of dehydrated N-methyl-2-pyrrolidone, and potassium t-butoxide (0.1 mL) was added. (44 g, 3.9 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, the precursor (0.50 g, 1.19 mmol) was added under a nitrogen stream, and the mixture was stirred at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration.
- Emission evaluation was performed in the same manner as in Example 1, except that the doping material was changed to 3Y-BCz-PBN-tBu. The results are shown in FIGS. EQEMax was 24.6%.
- 3,6-Di-t-butylcarbazole (1.33 g, 4.8 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 20 mL of dehydrated N-methyl-2-pyrrolidone. (44 g, 3.9 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, and the precursor (0.51 g, 1.35 mmol) dissolved in 7 mL of dehydrated N-methyl-2-pyrrolidone was added thereto under a nitrogen stream, followed by stirring at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration.
- the solid was dissolved in dichloromethane, dried over magnesium sulfate and concentrated. The residue was purified by silica gel column chromatography (n-hexane / benzene) to obtain 1.63 g of a crude target product. To 3.33 g of the crude product obtained by the same method, n-hexane / ethyl acetate was added, and the crystals precipitated by ultrasonic irradiation were collected by filtration, and the solvent was distilled off to give the target compound as a pale yellow-white solid. 2.37 g was obtained (74.1% yield).
- 3,6-Di-t-butylcarbazole (1.95 g, 7.0 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 32 mL of dehydrated N-methyl-2-pyrrolidone. 76 g, 6.8 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, a precursor (0.80 g, 2.00 mmol) was added under a nitrogen stream, and the mixture was stirred at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration. The solid was dissolved in dichloromethane, dried over magnesium sulfate and concentrated.
- Emission evaluation was performed in the same manner as in Example 1, except that the doping material was changed to 3Y-BCz-PBN-SMe. The results are shown in FIGS. EQEMax was 20.9%.
- 3,6-Di-t-butylcarbazole (1.16 g, 4.2 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 24 mL of dehydrated N-methyl-2-pyrrolidone. (44 g, 3.9 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, the precursor (0.50 g, 1.19 mmol) was added under a nitrogen stream, and the mixture was stirred at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration. The solid was dissolved in dichloromethane, dried over magnesium sulfate and concentrated.
- Emission evaluation was performed in the same manner as in Example 1, except that the doping material was changed to 3F-BCz-PBN-tBu. The results are shown in FIGS. EQEMax was 26.5%.
- 3,6-Di-t-butylcarbazole (1.51 g, 5.4 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 25 mL of dehydrated N-methyl-2-pyrrolidone. (58 g, 5.2 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, and the precursor (0.57 g, 1.54 mmol) dissolved in 6 mL of dehydrated N-methyl-2-pyrrolidone was added under a nitrogen stream, followed by stirring at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration.
- Emission evaluation was performed in the same manner as in Example 1, except that the doping material was changed to 3F-BCz-PBN-OMe. The results are shown in FIGS. EQEMax was 25.2%.
- 3,6-Di-t-butylcarbazole (1.95 g, 7.0 mmol) was added to a 200-mL three-necked flask purged with nitrogen, and dissolved in 32 mL of dehydrated N-methyl-2-pyrrolidone. 76 g, 6.8 mmol) and stirred at room temperature for 1 hour. The mixture was cooled with ice water, a precursor (0.80 g, 2.00 mmol) was added under a nitrogen stream, and the mixture was stirred at 100 ° C. for 3 hours. The reaction solution was ice-cooled, cold water was added, and the precipitated solid was collected by filtration. The solid was dissolved in dichloromethane, dried over magnesium sulfate and concentrated.
- the residue was purified by silica gel column chromatography (n-hexane / dichloromethane) to obtain a crude product.
- the crude product was added with n-hexane / diethyl ether and irradiated with ultrasonic waves.
- the precipitated crystals were collected by filtration and the solvent was distilled off to obtain 1.85 g of the desired product as a pale yellow solid (yield 78.7%). ).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Plural Heterocyclic Compounds (AREA)
- Electroluminescent Light Sources (AREA)
- Indole Compounds (AREA)
Abstract
Description
本願は、2018年9月5日に、日本に出願された特願2018-165955号、及び、2019年2月1日に、日本に出願された特願2019-017156に基づき優先権を主張し、その内容をここに援用する。
例えば、特許文献1は、3,5-ジ(3,6-ジフェニル-9H-カルバゾール-9-イル)-2,4,6-トリ(4-シアノフェニル)-ベンゾニトリルなどを開示している。特許文献2は、2,3,5,6-テトラ(3,6-ジフェニル-9H-カルバゾール-9-イル)-4-(4-シアノフェニル)-ベンゾニトリルなどを開示している。特許文献3は、2,3,5,6-テトラ(9H-カルバゾール-9-イル)-4-フェニル-ベンゾニトリルなどを開示している。

Lは、それぞれ独立に、置換若しくは無置換のアリール基、または置換若しくは無置換のヘテロアリール基であり、
nは、Lの数を表し、1または2であり、
Qは、それぞれ独立に、置換若しくは無置換の3,6-ジ-t-ブチル-9H-カルバゾール-9-イル基、置換若しくは無置換の3,6-ジフェニル-9H-カルバゾール-9-イル基、または置換若しくは無置換の3-フェニル-6-t-ブチル-9H-カルバゾール-9-イル基であり、且つ
mは、Qの数を表し、5-nである。







〔10〕 Lが、置換若しくは無置換のフェニル基、置換若しくは無置換のビフェニル基、置換若しくは無置換のナフチル基、置換若しくは無置換のアントリル基、置換若しくは無置換のフェナントリル基、置換若しくは無置換のピリジニル基、置換若しくは無置換のピリミジニル基、置換若しくは無置換のフリル基、置換若しくは無置換のチエニル基、置換若しくは無置換のオキサゾリル基、置換若しくは無置換のチアゾリル基、置換若しくは無置換のイミダゾリル基、置換若しくは無置換のインドリル基、置換若しくは無置換のキノリニル基、置換若しくは無置換のベンゾフラニル基、置換若しくは無置換のベンゾチエニル基、置換若しくは無置換のベンゾオキサゾリル基、置換若しくは無置換のベンゾチアゾリル基、または置換若しくは無置換のベンゾイミダゾリル基である前記〔1〕~〔8〕のいずれかひとつに記載の化合物。
〔11〕 Lが、置換若しくは無置換のフェニル基、置換若しくは無置換のピリジニル基、または置換若しくは無置換のピリミジニル基である、前記〔1〕~〔8〕のいずれかひとつに記載の化合物。
〔13〕 前記〔12〕に記載の発光材料を含有する発光素子。

Lは、それぞれ独立に、置換若しくは無置換のアリール基であり、
nは、Lの数を表し、1または2であり、
Qは、それぞれ独立に、置換若しくは無置換の3,6-ジ-t-ブチル-9H-カルバゾール-9-イル基、置換若しくは無置換の3,6-ジフェニル-9H-カルバゾール-9-イル基、または置換若しくは無置換の3-フェニル-6-t-ブチル-9H-カルバゾール-9-イル基であり、且つ
mは、Qの数を表し、5-nである。












一方、用語「置換(substituted)」は、母核となる基のいずれかの水素原子が、母核と同一または異なる構造の基で置換されていることを意味する。従って、「置換基」は、母核となる基に結合した他の基である。置換基は1個であってもよいし、2個以上であってもよい。2個以上の置換基は同一であってもよいし、異なるものであってもよい。
「置換基」となり得る基の具体例としては、以下の基を挙げることができる。
フルオロ基、クロロ基、ブロモ基、イオド基などのハロゲノ基;
メチル基、エチル基、n-プロピル基、i-プロピル基、n-ブチル基、s-ブチル基、i-ブチル基、t-ブチル基、n-ペンチル基、n-ヘキシル基などのC1~20アルキル基(好ましくはC1~6アルキル基);
ビニル基、1-プロペニル基、2-プロペニル基、1-ブテニル基、2-ブテニル基、3-ブテニル基、1-メチル-2-プロペニル基、2-メチル-2-プロペニル基、1-ペンテニル基、2-ペンテニル基、3-ペンテニル基、4-ペンテニル基、1-メチル-2-ブテニル基、2-メチル-2-ブテニル基、1-ヘキセニル基、2-ヘキセニル基、3-ヘキセニル基、4-ヘキセニル基、5-ヘキセニル基などのC2~10アルケニル基(好ましくはC2~6アルケニル基);
シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、キュバニル基などのC3~8シクロアルキル基;
2-シクロプロペニル基、2-シクロペンテニル基、3-シクロヘキセニル基、4-シクロオクテニル基などのC3~8シクロアルケニル基;
フェニル基、ナフチル基などのC6~40アリール基(好ましくはC6~10アリール基);
ピロリル基、フリル基、チエニル基、イミダゾリル基、ピラゾリル基、オキサゾリル基、イソオキサゾリル基、チアゾリル基、イソチアゾリル基、トリアゾリル基、オキサジアゾリル基、チアジアゾリル基、テトラゾリル基などの5員環のヘテロアリール基;
ピリジル基、ピラジニル基、ピリミジニル基、ピリダジニル基、トリアジニル基などの6員環のヘテロアリール基;
インドリル基、ベンゾフリル基、ベンゾチエニル基、ベンゾイミダゾリル基、ベンゾオキサゾリル基、ベンゾチアゾリル基、キノリル基、イソキノリル基、キノキサリニル基などの縮合環のヘテロアリール基;
オキシラニル基、テトラヒドロフリル基、ジオキソラニル基、ジオキラニル基などの環状エーテル基;
アジリジニル基、ピロリジニル基、ピペリジル基、ピペラジニル基、モルホリニル基などの環状アミノ基;
メトキシ基、エトキシ基、n-プロポキシ基、i-プロポキシ基、n-ブトキシ基、s-ブトキシ基、i-ブトキシ基、t-ブトキシ基などのC1~20アルコキシ基(好ましくはC1~6アルコキシ基);
ビニルオキシ基、アリルオキシ基、プロペニルオキシ基、ブテニルオキシ基などのC2~6アルケニルオキシ基;
エチニルオキシ基、プロパルギルオキシ基などのC2~6アルキニルオキシ基;
フェノキシ基、ナフトキシ基などのC6~10アリールオキシ基;
チアゾリルオキシ基、ピリジルオキシ基などの5~6員環のヘテロアリールオキシ基;
ホルミル基; アセチル基、プロピオニル基などのC1~6アルキルカルボニル基;
ホルミルオキシ基; アセチルオキシ基、プロピオニルオキシ基などのC1~6アルキルカルボニルオキシ基;
メトキシカルボニル基、エトキシカルボニル基、n-プロポキシカルボニル基、i-プロポキシカルボニル基、n-ブトキシカルボニル基、t-ブトキシカルボニル基などのC1~6アルコキシカルボニル基;
2-クロロ-1-プロペニル基、2-フルオロ-1-ブテニル基などのC2~6ハロアルケニル基;
4,4-ジクロロ-1-ブチニル基、4-フルオロ-1-ペンチニル基、5-ブロモ-2-ペンチニル基などのC2~6ハロアルキニル基;
3,3-ジフルオロシクロブチル基などのC3~6ハロシクロアルキル基;
2-クロロ-n-プロポキシ基、2,3-ジクロロブトキシ基、トリフルオロメトキシ基、2,2,2-トリフルオロエトキシ基などのC1~6ハロアルコキシ基;
2-クロロプロペニルオキシ基、3-ブロモブテニルオキシ基などのC2~6ハロアルケニルオキシ基;
クロロアセチル基、トリフルオロアセチル基、トリクロロアセチル基などのC1~6ハロアルキルカルボニル基;
メチルアミノ基、ジメチルアミノ基、ジエチルアミノ基などのC1~20アルキルアミノ基(好ましくはC1~6アルキルアミノ基);
アニリノ基、ナフチルアミノ基などのC6~40アリールアミノ基(好ましくはC6~10アリールアミノ基);
ホルミルアミノ基; アセチルアミノ基、プロパノイルアミノ基、ブチリルアミノ基、i-プロピルカルボニルアミノ基などのC1~6アルキルカルボニルアミノ基;
メトキシカルボニルアミノ基、エトキシカルボニルアミノ基、n-プロポキシカルボニルアミノ基、i-プロポキシカルボニルアミノ基などのC1~6アルコキシカルボニルアミノ基;
S,S-ジメチルスルホキシイミノ基などのC1~6アルキルスルホキシイミノ基;
メチルアミノカルボニル基、ジメチルアミノカルボニル基、エチルアミノカルボニル基、i-プロピルアミノカルボニル基などのC1~6アルキルアミノカルボニル基;
イミノメチル基、(1-イミノ)エチル基、(1-イミノ)-n-プロピル基などのイミノC1~6アルキル基;
ヒドロキシイミノメチル基、(1-ヒドロキシイミノ)エチル基、(1-ヒドロキシイミノ)プロピル基などのヒドロキシイミノC1~6アルキル基;
メトキシイミノメチル基、(1-メトキシイミノ)エチル基などのC1~6アルコキシイミノC1~6アルキル基;
メチルチオ基、エチルチオ基、n-プロピルチオ基、i-プロピルチオ基、n-ブチルチオ基、i-ブチルチオ基、s-ブチルチオ基、t-ブチルチオ基などのC1~20アルキルチオ基(好ましくはC1~6アルキルチオ基);
トリフルオロメチルチオ基、2,2,2-トリフルオロエチルチオ基などのC1~6ハロアルキルチオ基;
ビニルチオ基、アリルチオ基などのC2~6アルケニルチオ基;
エチニルチオ基、プロパルギルチオ基などのC2~6アルキニルチオ基;
メチルスルフィニル基、エチルスルフィニル基、t-ブチルスルフィニル基などのC1~6アルキルスルフィニル基;
トリフルオロメチルスルフィニル基、2,2,2-トリフルオロエチルスルフィニル基などのC1~6ハロアルキルスルフィニル基;
アリルスルフィニル基などのC2~6アルケニルスルフィニル基;
プロパルギルスルフィニル基などのC2~6アルキニルスルフィニル基;
メチルスルホニル基、エチルスルホニル基、t-ブチルスルホニル基などのC1~6アルキルスルホニル基;
トリフルオロメチルスルホニル基、2,2,2-トリフルオロエチルスルホニル基などのC1~6ハロアルキルスルホニル基;
アリルスルホニル基などのC2~6アルケニルスルホニル基;
プロパルギルスルホニル基などのC2~6アルキニルスルホニル基;
、N,N-ジイソブチルアセトアミド基等のC2~20アルキルアミド基;
トリフェニルシリル基などのトリC6~10アリールシリル基;
また、これらの「置換基」は、前記置換基中のいずれかの水素原子が、異なる構造の基で置換されていてもよい。
数5~20のトリアルキルシリルアルキニル基およびニトロ基が挙げられる。
より好ましい置換基としては、ハロゲノ基、シアノ基、炭素数1~20のアルキル基、炭素数1~20のアルコキシ基、炭素数6~40のアリール基、炭素数3~40のヘテロアリール基、炭素数12~40のジアリールアミノ基、炭素数12~40のカルバゾリル基を挙げることができる。
さらに好ましい置換基は、フルオロ基、クロロ基、シアノ基、炭素数1~10のアルキル基、炭素数1~10のアルコキシ基、炭素数1~10のジアルキルアミノ基、炭素数6~15のアリール基、炭素数3~12のヘテロアリール基を挙げることができる。これら置換基のうち、さらに置換可能なものは、前記置換基によって置換されていてもよい。
本発明の有機エレクトロルミネッセンス素子の一実施形態における構造としては、基板上に順次に、陽極、正孔注入層、正孔輸送層、電子阻止層、発光層、正孔阻止層、電子輸送層、陰極からなるもの、また、電子輸送層と陰極の間にさらに電子注入層を有するものを挙げることができる。これらの多層構造においては有機層を何層か省略することが可能であり、例えば基板上に順次に、陽極、正孔輸送層、発光層、電子輸送層、電子注入層、陰極とすることや、陽極、正孔輸送層、発光層、電子輸送層、陰極とすることもできる。
本発明の発光材料は、発光層のみならず、正孔注入層、正孔輸送層、電子阻止層、正孔阻止層、電子輸送層、または電子注入層にドープさせてもよい。
場合は電解重合により直接基板上に薄膜を形成したり、基板上に導電性高分子を塗布して陽極を形成したりすることもできる。
必要に応じて設けられる正孔輸送層として、トリアゾール誘導体、オキサジアゾール誘導体、イミダゾール誘導体、カルバゾール誘導体、インドロカルバゾール誘導体、ポリアリールアルカン誘導体、ピラゾリン誘導体、ピラゾロン誘導体、フェニレンジアミン誘導体、アリールアミン誘導体、アミノ置換カルコン誘導体、オキサゾール誘導体、スチリルアントラセン誘導体、フルオレノン誘導体、ヒドラゾン誘導体、スチルベン誘導体、シラザン誘導体、アニリン系共重合体、導電性高分子オリゴマーなどを挙げることができる。
より具体的に、m-カルバゾリルフェニル基を含有する化合物、N,N’-ジフェニル-N,N’-ジ(m-トリル)-ベンジジン(以後、TPDと略称する)、N,N’-ジフェニル-N,N’-ジ(α-ナフチル)-ベンジジン(以後、NPDと略称する)、N,N,N’,N’-テトラビフェニリルベンジジンなどのベンジジン誘導体、1,1-ビス[(ジ-4-トリルアミノ)フェニル]シクロヘキサン(以後、TAPCと略称する)、種々のトリフェニルアミン3量体および4量体やカルバゾール誘導体などを挙げることができる。これらは、1種単独でまたは2種以上を組み合わせて用いることができる。正孔輸送層は、単層構造の膜であってもよいし、積層構造の膜であってもよい。また、正孔の注入・輸送層として、ポリ(3,4-エチレンジオキシチオフェン)(以後、PEDOTと
略称する)/ポリ(スチレンスルフォネート)(以後、PSSと略称する)などの塗布型の高分子材料を用いることができる。これらの材料は蒸着法の他、スピンコート法やインクジェット法などの公知の方法によって薄膜形成を行うことができる。

















































ホスト材料を用いた場合、発光層に含有させることができる本発明の発光材料の量は、下限が、好ましくは0.1質量%、より好ましくは1質量%であり、上限が、好ましくは50質量%、より好ましくは20質量%、さらに好ましくは10質量%である。




























































































〔2,4,6-トリ(9H-カルバゾール-9-イル)-3,5-ジフェニル-ベンゾニトリル(3Cz-2PBN-A)の合成〕

1H-NMR(400MHz,DMSO-d6,δ) : 8.07(d,J=8.0Hz,4H), 7.81-7.78(m,8H), 7.49(td,J=8.0Hz,0.8Hz,4H), 7.37(td,J=7.2Hz,0.8Hz,2H), 7.23(t,J=7.6Hz,4H), 7.06(t,J=8.0Hz,2H), 6.81(dd,J=7.2Hz,1.2Hz,4H), 6.55(tt,J=7.6Hz,1.6Hz,2H), 6.44(t,J=7.6Hz,4H)
膜厚50nmのインジウム・スズ酸化物(ITO)からなる陽極が形成されたガラス基板上に10nm厚のHAT-CN層、40nm厚のTAPC層、10nm厚のCCP層、10nm厚のmCP層、および20nm厚の発光層をこの順で真空蒸着法(5.0×10-4Pa以下)によって積層させた。
発光層のホスト材料としてPPFを用い、ドープ材料として2,4,6-トリ(9H-カルバゾール-9-イル)-3,5-ジフェニル-ベンゾニトリル(3Cz-2PBN-A)を用いた。ドーブ材料濃度を12.0重量%に設定した。
次いで、10nm厚のPPF層、40nm厚のB3PyPB層、1nm厚の8-ヒドロキシキノリトリチウム膜、および100nm厚のアルミニウム膜をこの順で真空蒸着法にて積層させることにより陰極を形成させて、有機発光ダイオード(OLED)を得た。結果を図1および2に示す。この有機発光ダイオードは、外部量子効率の最大値(EQEMax)が13.1%であった。
〔2,4,6-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-3,5-ジフェニル-ベンゾニトリル(3PCz-2PBN-A)の合成〕

1H-NMR(400MHz,DMSO-d6,δ): 8.59(d,J=1.6Hz,4H), 8.37(d,J=2.0Hz,2H), 8.02-7.99(m,6H), 7.87-7.75(m,18H), 7.52-7.45(m,12H), 7.37-7.30(m,6H), 7.03(dd,J=7.0Hz,1.2Hz,4H), 6.66-6.58(m,6H)
ドープ材料を2,4,6-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-3,5-ジフェニル-ベンゾニトリル(3PCz-2PBN-A)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図1および2に示す。EQEMaxは28.1%であった。
〔2,4,6-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-3,5-ジフェニル-ベンゾニトリル(3BuCz-2PBN-A)の合成〕

1H-NMR(400MHz,CDCl3,δ) : 7.94(d,J=2.0Hz,4H), 7.73(d,J=1.6Hz,2H), 7.42(dd,J=8.8Hz,1.6Hz,4H), 7.27(dd,J=8.6Hz,2.0Hz,4H), 7.13(d,J=8Hz,4H), 6.98(d,J=8.8Hz,2H), 6.63(d,J=7.8Hz,4H), 6.50(t,J=6.4Hz,2H), 6.38(t,J=7.6Hz,4H), 1.39(s,36H), 1.31(s,18H)
ドープ材料を2,4,6-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-3,5-ジフェニル-ベンゾニトリル(3BuCz-2PBN-A)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図1および2に示す。EQEMaxは22.6%であった。
〔2,3,5-トリ(9H-カルバゾール-9-イル)-4,6-ジフェニル-ベンゾニトリル(3Cz-2PBN-B)の合成〕

更に水層を酢酸エチルで2回抽出し、混合した有機層を水で3回洗浄し、次いで飽和食塩水で2回洗浄した。有機層を硫酸マグネシウムで脱水し、ろ過して、ろ液を濃縮することで粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液:n-ヘキサン/酢酸エチル)で精製することで粗精製物を得た。さらに、粗精製物にアセトン/n-ヘキサンを加え、超音波照射した。その後、ろ過してn-ヘキサンで洗浄することで目的物(3Cz-2PBN-B)の淡黄色固体を2.46g、収率90.5%で得た。
1H-NMR(400MHz,DMSO-d6,δ): 7.88(d,J=8.0Hz,2H), 7.85(d,J=8.0Hz,2H), 7.82(d,J=8.0Hz,2H), 7.80(d,J=8.0Hz,2H), 7.64(d,J=8.4Hz,2H), 7.60(d,J=7.6Hz,2H), 7.41-7.37(m,2H), 7.35-7.32(m,2H), 7.25-7.21(m,2H), 7.12-7.03(m,9H), 6.89(t,J=7.6Hz,2H), 6.68(d,J=7.2Hz,2H), 6.42(t,J=7.6Hz,1H), 6.27(t,J=7.6Hz,2H)
ドープ材料を2,3,5-トリ(9H-カルバゾール-9-イル)-4,6-ジフェニル-ベンゾニトリル(3Cz-2PBN-B)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図3および4に示す。EQEMaxは16.7%であった。
〔2,3,5-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-4,6-ジフェニル-ベンゾニトリル(3PCz-2PBN-B)の合成〕

とで粗精製物を得た。さらに、粗精製物にジクロロメタン/ジエチルエーテル/n-ヘキサンを加え、超音波照射した。その後、ろ過してn-ヘキサンで洗浄することで目的物(3PCz-2PBN-B)の黄色固体を0.90g、収率96.1%で得た。
1H-NMR(400MHz,DMSO-d6,δ): 8.44(d,J=2.0Hz,2H), 8.27(d,J=1.6Hz,2H), 8.05(d,J=1.6Hz,2H), 8.00(d,J=8.8Hz,2H), 7.89(d,J=8.8Hz,2H), 7.82-7.79(m,6H), 7.73(d,J=8.8Hz,2H), 7.63(d,J=7.6Hz,4H,), 7.56(d,J=7.2Hz,4H), 7.53-7.47(m,8H), 7.43-7.26(m,16H),
7.19-7.15(m,3H), 6.97(d,J=7.2Hz,2H), 6.58-6.48(m,3H)
ドープ材料を2,3,5-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-4,6-ジフェニル-ベンゾニトリル(3PCz-2PBN-B)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図3および4に示す。EQEMaxは35.1%であった。
〔2,3,5-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-4,6-ジフェニル-ベンゾニトリル(3BuCz-2PBN-B)の合成〕

その後、ろ過してn-ヘキサンで洗浄することで目的物(3BuCz-2PBN-B)の淡黄白色固体を2.79g、収率79.3%で得た。
1H-NMR(400MHz,DMSO-d6,δ): 7.90(d,J=2.0Hz,2H), 7.74(d,J=2.0Hz,2H), 7.58(d,J=8.8Hz,2H), 7.50(d,J=1.6Hz,2H), 7.39(td,J=8.8Hz,2.0Hz,4H), 7.27(d,J=8.4Hz,2H), 7.18(d,J=8.8Hz,2H), 7.11-7.05(m,5H), 6.97(dd,J=8.8Hz,2.0Hz,2H), 6.78(d,7.2Hz,2H), 6.47(t,J=8.0Hz,1H), 6.36(t,J=8.0Hz,2H), 1.35(s,18H), 1.31(s,18H), 1.22(s,18H)
ドープ材料を2,3,5-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-4,6-ジフェニル-ベンゾニトリル(3BuCz-2PBN-B)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図3および4に示す。EQEMaxは24.4%であった。
〔2,3,5,6-テトラ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-4-フェニル-ベンゾニトリル(4BuCz-1PBN-A)の合成〕

1H NMR (400 MHz, CDCL3): δ 7.56-7.52 (m, 3H), 7.48-7.45 (m, 2H).
1H NMR (400 MHz, DMSO-d6): δ 7.76 (d, J = 1.2Hz, 4H), 7.56 (d, J = 1.2Hz, 4H), 7.46 (d, J = 8.4Hz, 4H), 7.42 (d, J = 8.8Hz, 4H), 7.09 (dd,J =8.8,1.2Hz, 6H), 7.02(dd,J = 8.4, 1.6Hz, 4H), 6.60-6.57(m, 3H), 1.32(s, 36H), 1.26(s, 36H)
ドープ材料を2,3,5,6-テトラ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-4-フェニル-ベンゾニトリル(4BuCz-1PBN-A)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図5および6に示す。EQEMaxは40.1%であった。
〔2,3,5,6-テトラ(9H-カルバゾール-9-イル)-4-フェニル-ベンゾニトリル(4Cz-1PBN-A)の合成〕

窒素置換したフラスコにカルバゾール(1.09g, 6.52mmol)、およびN,N-ジメチルホルムアミド(10mL)を加えた。その後、室温でカリウムtert-ブトキシド(0.73g,6.52mmol)を加えたN,N-ジメチルホルムアミド(5mL)を加えて、室温で30分間攪拌した。その後、化合物1(0.4g,1.59mmol)を加えたN,N-ジメチルホルムアミド(10mL)を10分間かけて滴下した。その後、80℃で10時間攪拌した。次いで、室温に戻し、水(20mL)を加え、30分間経過した後にクロロホルムを加え抽出を行った。有機層に硫酸ナトリウムを加え乾燥させて、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:9)により精製し、黄色い固体の化合物2(4Cz-1PBN-A)(1.18g,88%)を得た。
1H NMR(400MHz, DMSO-d6): δ 7.92(d,J =8.4Hz,4H),7.89-7.87(m,8H), 7.65 (d,J = 7.2Hz,4H), 7.24(td,J = 7.2,1.2Hz,4H), 7.15-7.08(m,8H), 6.94(td,J =7.4,0.8Hz,4H), 6.72(dd,J =8.4, 1.2Hz,2H), 6.44(tt,J =8.0,1.2Hz,1H), 6.31(t,J = 7.8Hz, 2H).
ドープ材料を2,3,5,6-テトラ(9H-カルバゾール-9-イル)-4-フェニル-ベンゾニトリル(4Cz-1PBN-A)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図5および6に示す。EQEMaxは26.1%であった。
〔4’,5’,6’-トリフルオロ-[1,1’:3’,1”-テルフェニル]-2’-カルボニトリル(2PBN-C)の合成〕

濃縮物をn-ヘキサン/酢酸エチル=9/1で洗浄し、目的物(2PBN-C)の白色結晶を4.43g得た。(収率75.1%)
1H-NMR(400MHz,CDCl3,δ):7.55~7.47(m,10H)
〔3,4,5-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-2,6-ジフェニル-ベンゾニトリル(3BuCz-2PBN-C)の合成〕

1H-NMR(400MHz,CDCl 3,δ): 7.45(d,4H), 7.30(d,2H), 7.28(d,2H), 7.13(d,2H), 7.04(m,6H), 6.88(d,4H), 6.87(d,4H), 6.67(d,2H), 6.49(dd,2H), 1.24(s,36H), 1.17(s,18H)
ドープ材料を3,4,5-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-2,6-ジフェニル-ベンゾニトリル(3BuCz-2PBN-C)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図7および8に示す。EQEMaxは12.0%であった。

1H-NMR(400MHz,CDCl 3,δ): 7.80(s,4H), 7.47-7.41(m,14H), 7.36-7.32(m,8H), 7.28-7.21(m,22H), 7.16-7.14(m,6H), 7.09(d,2H), 6.86(dd,2H)
ドープ材料を3,4,5-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-2,6-ジフェニル-ベンゾニトリル(3PCz-2PBN-C)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図7および8に示す。EQEMaxは21.6%であった。
〔3’,5’,6’-トリフルオロ-[1,1’:4’,1”-テルフェニル]-2’-カルボニトリル(2PBN-D)の合成〕

濃縮物にクロロホルム150mlを加えて加熱溶解した。それにn-ヘキサン300mlを加えて冷却した。析出した白色固体をろ過し、目的物(2PBN-D)を4.40g(収率74.6%)得た。
1H-NMR(400MHz,CDCl3,δ):7.54~7.47(m,10H)
〔3,4,6-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-2,5-ジフェニル-ベンゾニトリル(3PCz-2PBN-D)の合成〕

1H-NMR(400MHz,CDCl3,δ): 8.32(d,J=2.0Hz,2H), 7.33-7.70(m,8H), 7.66(d,J=2.0Hz,2H), 7.53-7.26(m,30H), 7.26-7.21(m,5H), 7.13(dd,J=8.4Hz,2.0Hz,2H), 7.05(d,J=8.4Hz,2H), 6.99(d,J=8.8Hz,2H,), 6.91-6.89(m,2H), 6.67-6.57(m,3H)
ドープ材料を3,4,6-トリ(3,6-ジフェニル-9H-カルバゾール-9-イル)-2,5-ジフェニル-ベンゾニトリル(3PCz-2PBN-D)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図7および8に示す。EQEMaxは20.7%であった。
〔3,4,6-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-2,5-ジフェニル-ベンゾニトリル(3BuCz-2PBN-D)の合成〕

それにカリウムt-ブトキシド(1.58g,14.1mmol)を加えて室温で1時間撹拌した。それに窒素気流下で3’,5’,6’-トリフルオロ-[1,1’:4’,1”-テルフェニル]-2’-カルボニトリル(1.00g,3.2mmol)を加え、120℃で6時間撹拌した。その後、130℃で14時間撹拌した。次いで、室温に戻し、水と酢酸エチルを加え、有機層を分液した。更に水層を酢酸エチルで2回抽出し、混合した有機層を水で3回洗浄し、次いで飽和食塩水で2回洗浄した。有機層を硫酸マグネシウムで脱水し、ろ過・濃縮することで粗生成物を得た。粗生成物をシリカゲルカラムクロマトグラフィー(溶出液:n-ヘキサン/ジクロロメタン、n-ヘキサン/トルエン)で精製することで目的物(3BuCz-2PBN-D)を1.73g、収率49.2%で得た。
1H-NMR(400MHz,CDCl3,δ): 8.03(d,J=1.2Hz,2H), 7.44-7.40(m,6H), 7.35(d,J=1.6Hz,2H), 7.17(d,J=8.4Hz,2H), 7.10-7.06(m,3H), 6.83-6.76(m,6H), 6.66(d,J=8.4Hz,2H), 6.62(d,J=8.8Hz,2H), 6.56-6.46(m,3H), 1.43(s,18H), 1.29(s,18H), 1.25(s,18H)
ドープ材料を3,4,6-トリ(3,6-ジ-t-ブチル-9H-カルバゾール-9-イル)-2,5-ジフェニル-ベンゾニトリル(3BuCz-2PBN-D)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図7および8に示す。EQEMaxは15.6%であった。
〔2,4,5,6-テトラフルオロ-[1,1’-ビフェニル]-3-カルボニトリル(1PBN-C)の合成〕

1H-NMR(400MHz,CDCl3,δ):7.50~7.38(m,5H)
これにジエチルエーテル50mlを加え、10%アンモニア水で洗浄し、次いで水で洗浄した。その後、硫酸マグネシウムを加えて乾燥させ、ロータリーエバポレーターで濃縮した。濃縮物をシリカゲルカラムクロマトグラフィー(n-ヘキサン/ベンゼン=2/1)にて分離精製し、目的物2,4,5,6-テトラフルオロ-[1,1’-ビフェニル]-3-カルボニトリルを2.05g(収率83.3%)得た。
1H-NMR(400MHz,CDCl3,δ):7.51-7.48(m,3H),7.40-7.37(m,2H)
〔2,4,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1’-ビフェニル]-3-カルボニトリル(4BuCz-1PBN-C)の合成〕

1H-NMR(400MHz,CDCl 3,δ):8.01(d,2H), 7.59(s,2H), 7.46~7.44(m,4H), 7.18(dd, 4H), 6.98(d,4H), 6.91(dd,4H), 6.67~6.64(m,4H), 6.55~6.52(3H,m), 6.43(dd,2H), 1.43(s,18H), 1.30(s,18H), 1.22(s,18H), 1.21(s,18H)
ドープ材料を2,4,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1’-ビフェニル]-3-カルボニトリル(4BuCz-1PBN-C)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図9および10に示す。
EQEMaxは26.0%であった。
〔3,4,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1’-ビフェニル]-2-カルボニトリル(4BuCz-1PBN-B)の合成〕

1H NMR (400 MHz, CDCl3): δ 7.58-7.53 (m, 3H), 7.51-7.45 (m, 3H)
その後、化合物1(0.30g, 1.19mmol)とN,N-ジメチルホルムアミド(10mL)との混合物を10分間かけて滴下した。その後、80℃で10時間攪拌した。その後、室温に戻し、水(20 mL)を加え、30分後にクロロホルムを加え抽出を行った。有機層に硫酸ナトリウムを加え乾燥させ、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:9)により精製し、黄色い固体の化合物2(4BuCz-1PBN-B)を(1.24g, 81%)得た。
1H NMR (400 MHz, DMSO-d6): δ 7.73 (d, J = 1.0Hz, 2H), 7.52 (d, J = 6.8Hz, 4H), 7.32-7.27 (m, 4H), 7.21-7.11 (m, 11H), 6.99 (d, J =8.4Hz, 2H), 6.87 (d,J = 8.8Hz, 2H), 6.63-6.59(m, 4H), 1.33 (s, 18H), 1.27 (s, 18H), 1.16 (d, J = 8.0Hz, 36H)
ドープ材料を3,4,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1’-ビフェニル]-2-カルボニトリル(4BuCz-1PBN-B)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図9および10に示す。EQEMaxは22.3%であった。
〔2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1’-ビフェニル]-4-カルボニトリル(4BuCz-1PBN-A)の合成〕
実施例7と同様の方法にて目的物(4BuCz-1PBN-A)を得た。
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1’-ビフェニル]-4-カルボニトリル(4BuCz-1PBN-A)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図9および10に示す。

1H-NMR(400MHz, CDCl3, δ) : 7.53(d,2H)、7.40(d,2H)、1.36(s,9H)

1H-NMR(400MHz, CDCl3, δ) : 7.55(d,4H)、7.42(d,4H)、6.94(dd,4H)、6.88(d, 4H)、6.78(dd,6H)、6.62(d,4H)、6.48(d,2H)、1.35(s,36H)、1.39(s,36H)、0.71(s,9H)
ドープ材料を4'-(tert-ブチル)-2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1'-ビフェニル]-4-カルボニトリル (4X-BCz-PBN-Bu)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図11および12に示す。EQEMaxは26.5%であった。

1H-NMR(400MHz, CDCl3, δ) : 7.41(d,2H)、7.03(d,2H)、3.87(s,3H)

1H-NMR(400MHz, CDCl3, δ) : 7.54(d,4H)、7.43(d,4H)、6.93(dd,4H)、 6.86(d, 4H)、6.81(dd,6H)、6.63(d,4H)、6.06(d,2H)、3.23(s,3H)、1.34(s,36H)、1.30(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-4'-メトキシ-[1,1'-ビフェニル]-4-カルボニトリル (4X-BCz-PBN-OMe)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図11および12に示す。EQEMaxは28.5%であった。

1H-NMR(400MHz, CDCl3, δ) : 7.37(d,2H)、7.34(d,2H)、2.53(s,3H)

1H-NMR(400MHz, CDCl3, δ) : 7.56(d,4H)、 7.44(d,4H)、 6.95(dd,4H)、6.85(d, 4H)、6.83(dd,6H)、6.64(d,4H)、6.42(d,2H)、1.96(s,3H)、1.36(s,36H)、1.32(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-4'-メチルチオ-[1,1'-ビフェニル]-4-カルボニトリル (4X-BCz-PBN-SMe)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図11および12に示す。EQEMaxは29.0%であった。

1H-NMR(400MHz, CDCl3, δ) : 7.83(d,2H)、7.58(d,2H)

1H-NMR(400MHz, CDCl3, δ) : 7.57(d,4H)、 7.46(d,4H)、7.03 (d,2H)、 6.96(dd, 4H)、6.87(d,4H)、6.84(dd,6H)、6.59(d,4H)、1.35(s,36H)、1.31(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1'-ビフェニル]-4,4'-ジカルボニトリル (4X-BCz-PBN-CN)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図11および12に示す。EQEMaxは31.6%であった。

1H-NMR(400MHz, CDCl3, δ) : 8.18(d,2H)、7.54(d,2H)、3.96(s,3H)

1H-NMR(400MHz, CDCl3, δ) : 7.54(d,4H)、7.43(d,4H)、6.93(dd,4H)、6.86(d, 4H)、6.81(dd,6H)、6.63(d,4H)、6.06(d,2H)、3.23(s,3H)、1.34(s,36H)、1.30(s,36H)
ドープ材料を4'-シアノ-2',3',5',6'-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-[1,1'-ビフェニル]-4-カルボン酸メチルエステル (4X-BCz-PBN-CO2Me)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図13および14に示す。EQEMaxは30.4%であった。

1H-NMR(400MHz,CDCl3,δ):7.01(s,2H),2.35(s,3H),2.05(s,6H)
1H-NMR(400MHz,CDCl3,δ):7.52(d,J=1.6Hz,4H),7.38(s,4H),6.89(dd,J=8.4Hz,2.0Hz,4H),6.81(d,J=8.8Hz,4H),6.73-6.68(m,8H),6.26(s,2H),2.17(s,6H),1.70(s,3H),1.35(s,36H),1.30(s,36H)
ドープ材料を4X-BCz-PBN-MesBNに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図13および14に示す。EQEMaxは29.5%であった。

1H-NMR(400MHz,CDCl3,δ):8.12(t,J=1.2Hz,1H),8.00(d,J=1.2Hz,2H)

1H-NMR(400MHz,CDCl3,δ):7.64(d,J=1.6Hz,4H),7.50(d,J=1.6Hz,4H),7.12(d,J=1.2Hz,2H),7.07(dd,J=8.8Hz,2.0Hz,4H),7.02-6.96(m,9H),6.67(d,J=8.4Hz,4H),1.36(s,36H),1.32(s,36H)
ドープ材料を4X-BCz-PBN-IPNに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図13および14に示す。EQEMaxは31.0%であった。

1H-NMR(400MHz, CDCl3, δ) : 8.80(d,1H)、7.88(m,1H)、7.51(d,1H)、7.45(m,1H)

1H-NMR(400MHz, CDCl3, δ) : 7.84(d,1H)、7.56(d,4H)、7.42(d,4H)、6.98-6.95(m, 5H)、6.92(d,4H)、6.86-6.83(m,5H)、6.77(d,4H)、6.43(dd,1H)、1.36(s,36H)、 1.30(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-4-(ピリジン-2-イル)ベンゾニトリル (4X-BCz-PBN-2-Py)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図15および16に示す。EQEMaxは24.4%であった。

1H-NMR(400MHz, CDCl3, δ) : 8.76-8.73(m,2H)、7.80(d,1H)、7.50-7.47(t,1H)

1H-NMR(400MHz, CDCl3, δ) : 8.22(d,1H)、7.81(d,1H)、7.57(d,4H)、7.45(d, 4H)、7.23(d,1H)、6.96(dd,4H)、6.89(d,4H)、6.84(dd,4H)、6.62(d,4H)、6.49(dd,1H)、 1.36(s,36H)、1.31(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-4-(ピリジン-3-イル)ベンゾニトリル (4X-BCz-PBN-3-Py)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図15および16に示す。EQEMaxは33.6%であった。

1H-NMR(400MHz, CDCl3, δ) : 8.81(d,2H)、7.38(d,2H)

1H-NMR(400MHz, CDCl3, δ) : 7.82(d,2H)、7.56(d,4H)、7.45(d,4H)、6.95(dd, 4H)、6.87-6.82(m,10H)、6.62(d,4H)、1.35(s,36H)、1.31(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-4-(ピリジン-4-イル)ベンゾニトリル (4X-BCz-PBN-4-Py)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図15および16に示す。EQEMaxは30.9%であった。

1H-NMR(400MHz, CDCl3, δ) : 9.35(s,1H)、8.91(s,2H)

1H-NMR(400MHz, CDCl3, δ) : 8.39(s,1H)、 8.27(s,2H)、7.57(d,4H)、7.47(d, 4H)、6.97(dd,4H)、6.90(d,4H)、6.86(dd,4H)、6.59(d,4H)、1.36(s,36H), 1.31(s,36H)
ドープ材料を2,3,5,6-テトラキス(3,6-ジ-tert-ブチル-9H-カルバゾール-9-イル)-4-(ピリミジン-5-イル)ベンゾニトリル (4X-BCz-PBN-5-Pm)に換えた以外は実施例1と同じ方法で発光評価を行った。結果を図15および16に示す。EQEMaxは30.3%であった。

1H-NMR(400MHz,CDCl3,δ):7.51(d,J=8.0Hz,4H),7.37(d,J=8.4Hz,4H),1.37(s,18H)

1H-NMR(400MHz,CDCl3,δ):7.94(d,J=1.2Hz,4H),7.71(d,J=2.0Hz,2H),7.38(dd,J=8.8Hz,2.0Hz,4H),7.21(dd,J=8.8Hz,2.0Hz,2H),7.10(d,J=8.8Hz,4H),6.93(d,J=8.4Hz,2H),6.43(d,J=8.0Hz,4H),6.33(d,J=8.8Hz,4H),1.39(s,36H),1.31(s,18H),0.77(s,18H)
ドープ材料を3Y-BCz-PBN-tBuに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図17および18に示す。EQEMaxは24.6%であった。

1H-NMR(400MHz,CDCl3,δ):7.37(d,J=8.8Hz,4H),7.03-7.00(m,4H),3.87(s,6H)

1H-NMR(400MHz,CDCl3,δ):7.96(d,J=2.0Hz,4H),7.76(d,J=2.0Hz,2H),7.41(dd、J=8.8Hz,2.0Hz,4H),7.27(dd,2H),7.11(d,J=8.0Hz,4H),6.96(d,J=8.8Hz,2H),6.52(d,J=8.8Hz,4H),5.92(d,J=9.2Hz,4H),3.29(s,6H),1.40(s,36H),1.33(s,18H)
ドープ材料を3Y-BCz-PBN-OMeに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図17および18に示す。EQEMaxは21.5%であった。

1H-NMR(400MHz,CDCl3,δ):7.35(s,8H),2.53(s,6H)

1H-NMR(400MHz,CDCl3,δ):7.96(d,J=2.0Hz,4H),7.77(d,J=2.0Hz,2H),7.41(dd,J=8.4Hz,2.0Hz,4H),7.26(dd,J=8.4Hz,2.0Hz,2H),6.93(d,J=8.0Hz,2H),6.50(d,J=8.4Hz,4H),6.28(d,J=8.4Hz,4H),1.99(s,6H),1.40(s,36H),1.34(s,18H)
ドープ材料を3Y-BCz-PBN-SMeに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図17および18に示す。EQEMaxは20.9%であった。

1H-NMR(400MHz,CDCl3,δ):7.53(d,J=10.4Hz,4H),7.45-7.43(m,4H),1.37(s,18H)

1H-NMR(400MHz,CDCl3,δ):7.80(s,2H),7.54(s,2H),7.38(s,2H),7.26(d,J=8.8Hz,2H),7.15(d,J=8.4Hz,2H),6.99(t,J=8.0Hz,4H),6.91(d,J=8.8Hz,4H),6.85(d,J=8.4Hz,2H),6.76(d,J=8.0Hz,2H),6.59(dd,J=10.4Hz,8.8Hz,4H),6.38(d,J=8.0Hz,2H),1.37(s,18H),1.34(s,18H),1.27(s,18H),1.09(s,9H),0.73(s,9H)
ドープ材料を3F-BCz-PBN-tBuに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図19および20に示す。EQEMaxは26.5%であった。

1H-NMR(400MHz,CDCl3,δ):7.46-7.42(m,4H),7.05-7.02(m,4H),3.87(s,6H)

1H-NMR(400MHz,CDCl3,δ):7.85(d,J=1.6Hz,2H),7.53(d,J=2.0Hz,2H),7.38(d,J=2.0Hz,2H),7.31(dd,J=8.8Hz,2.0Hz,2H),7.19(d,J=8.4Hz,2H),7.01(d,J=8.8Hz,2H),6.90(dd,J=8.0Hz,1.6Hz,2H),6.81-6.77(m,4H),6.64-6.55(m,6H),5.94(d,J=8.8Hz,2H),3.62(s,3H),3.25(s,3H),1.39(s,18H),1.34(s,18H),1.28(s,18H)
ドープ材料を3F-BCz-PBN-OMeに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図19および20に示す。EQEMaxは25.2%であった。

1H-NMR(400MHz,CDCl3,δ):7.42-7.34(m,8H),2.54(s,6H)
1H-NMR(400MHz,CDCl3,δ):7.85(d,J=1.6Hz,2H),7.53(d,J=2.0Hz,2H),7.38(d,J=1.2Hz,2H),7.31(dd,J=8.8Hz,2.0Hz,2H),7.18(d,J=8.4Hz,2H),6.99(d,J=8.8Hz,2H),6.92-6.89(m,4H),6.80-6.77(m,4H),6.60(d,J=8.4Hz,2H),6.57(d,J=8.4Hz,2H),6.29(d,J=8.4Hz,2H),2.30(s,3H),1.95(s,3H),1.39(s,18H),1.34(s,18H),1.28(s,18H)
ドープ材料を3F-BCz-PBN-SMeに換えた以外は実施例1と同じ方法で発光評価を行った。結果を図19および20に示す。EQEMaxは22.0%であった。
Claims (13)
- 式(I)で表される化合物。

nは、Lの数を表し、1または2であり、
Qは、それぞれ独立に、置換若しくは無置換の3,6-ジ-t-ブチル-9H-カルバゾール-9-イル基、置換若しくは無置換の3,6-ジフェニル-9H-カルバゾール-9-イル基、または置換若しくは無置換の3-フェニル-6-t-ブチル-9H-カルバゾール-9-イル基であり、且つ
mは、Qの数を表し、5-nである。 - 式(IIa)で表される請求項1に記載の化合物。

- 式(IIb)で表される請求項1に記載の化合物。

- 式(IIc)で表される請求項1に記載の化合物。

- 式(IIIa)で表される請求項1に記載の化合物。

- 式(IIIb)で表される請求項1に記載の化合物。

- 式(IIIc)で表される請求項1に記載の化合物。

- 式(IVa)で表される請求項1に記載の化合物。

- Lが、置換若しくは無置換の含窒素または含酸素の5員環または6員環ヘテロアリールである請求項1~8のいずれかひとつに記載の化合物。
- Lが、置換若しくは無置換のフェニル基、置換若しくは無置換のビフェニル基、置換若しくは無置換のナフチル基、置換若しくは無置換のアントリル基、置換若しくは無置換のフェナントリル基、置換若しくは無置換のピリジニル基、置換若しくは無置換のピリミジニル基、置換若しくは無置換のフリル基、置換若しくは無置換のチエニル基、置換若しくは無置換のオキサゾリル基、置換若しくは無置換のチアゾリル基、置換若しくは無置換のイミダゾリル基、置換若しくは無置換のインドリル基、置換若しくは無置換のキノリニル基、置換若しくは無置換のベンゾフラニル基、置換若しくは無置換のベンゾチエニル基、置換若しくは無置換のベンゾオキサゾリル基、置換若しくは無置換のベンゾチアゾリル基、または置換若しくは無置換のベンゾイミダゾリル基である請求項1~8のいずれかひとつに記載の化合物。
- Lが置換若しくは無置換のフェニル基、置換若しくは無置換のピリジニル基、または置換若しくは無置換のピリミジニル基である、請求項1~8のいずれかひとつに記載の化合物。
- 請求項1~11のいずれかひとつに記載の化合物を含む発光材料。
- 請求項12に記載の発光材料を含有する発光素子。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/272,550 US20210332032A1 (en) | 2018-09-05 | 2019-08-29 | Benzonitrile derivative, light-emitting material, and light-emitting element using same |
| KR1020217005954A KR102533313B1 (ko) | 2018-09-05 | 2019-08-29 | 벤조니트릴 유도체, 발광 재료 및 그것을 사용한 발광 소자 |
| CN201980057047.2A CN112638874A (zh) | 2018-09-05 | 2019-08-29 | 苯甲腈衍生物、发光材料和使用该发光材料的发光元件 |
| JP2020541162A JP7184263B2 (ja) | 2018-09-05 | 2019-08-29 | ベンゾニトリル誘導体、発光材料およびそれを用いた発光素子 |
| EP19857043.4A EP3848352A4 (en) | 2018-09-05 | 2019-08-29 | BENZONITRILE DERIVATIVE, LIGHT EMITTING MATERIAL AND LIGHT EMITTING ELEMENT THEREOF |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-165955 | 2018-09-05 | ||
| JP2018165955 | 2018-09-05 | ||
| JP2019-017156 | 2019-02-01 | ||
| JP2019017156 | 2019-02-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020050127A1 true WO2020050127A1 (ja) | 2020-03-12 |
Family
ID=69723167
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/033913 WO2020050127A1 (ja) | 2018-09-05 | 2019-08-29 | ベンゾニトリル誘導体、発光材料およびそれを用いた発光素子 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20210332032A1 (ja) |
| EP (1) | EP3848352A4 (ja) |
| JP (1) | JP7184263B2 (ja) |
| KR (1) | KR102533313B1 (ja) |
| CN (1) | CN112638874A (ja) |
| TW (1) | TWI719618B (ja) |
| WO (1) | WO2020050127A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112409241A (zh) * | 2020-11-27 | 2021-02-26 | 清华大学 | 一种有机化合物及其应用及采用该化合物的有机电致发光器 |
| CN112409240A (zh) * | 2020-11-20 | 2021-02-26 | 清华大学 | 一种有机化合物及其应用及采用该化合物的有机电致发光器 |
| CN114685353A (zh) * | 2020-12-28 | 2022-07-01 | 北京鼎材科技有限公司 | 用于有机发光器件的有机化合物、有机电致发光器件 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7226806B2 (ja) * | 2017-06-23 | 2023-02-21 | 株式会社Kyulux | 有機発光ダイオードに用いられる組成物 |
| CN115745869A (zh) * | 2022-10-28 | 2023-03-07 | 清华大学 | 一种有机化合物及采用该化合物的有机电致发光器 |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014183080A1 (en) | 2013-05-09 | 2014-11-13 | Nitto Denko Corporation | Emissive compounds for light emitting devices |
| WO2016138077A1 (en) | 2015-02-24 | 2016-09-01 | Nitto Denko Corporation | Gas sensor element |
| WO2016152605A1 (ja) * | 2015-03-23 | 2016-09-29 | 保土谷化学工業株式会社 | 有機エレクトロルミネッセンス素子用材料、発光材料および有機エレクトロルミネッセンス素子 |
| JP2016539182A (ja) * | 2013-10-30 | 2016-12-15 | 日東電工株式会社 | 発光デバイス用発光化合物 |
| WO2017115834A1 (ja) * | 2015-12-28 | 2017-07-06 | 株式会社Kyulux | 化合物、発光材料および有機発光素子 |
| WO2018047948A1 (ja) * | 2016-09-09 | 2018-03-15 | 東洋紡株式会社 | 有機発光素子ならびにそれに用いる発光材料および化合物 |
| JP2018519663A (ja) * | 2015-06-16 | 2018-07-19 | 昆山国顕光電有限公司Kunshan Go−Visionox Opto−Electronics Co., Ltd. | 有機エレクトロルミネッセンス装置及びその製造方法 |
| WO2018155642A1 (ja) * | 2017-02-24 | 2018-08-30 | 国立大学法人九州大学 | 化合物、発光材料および発光素子 |
| JP2018165955A (ja) | 2017-03-28 | 2018-10-25 | 大阪瓦斯株式会社 | 火災警報器 |
| WO2018237389A1 (en) * | 2017-06-23 | 2018-12-27 | Kyulux Inc. | MATERIAL COMPOSITION FOR USE IN ORGANIC ELECTROLUMINESCENT DIODE |
| JP2019017156A (ja) | 2017-07-05 | 2019-01-31 | Solネットワーク株式会社 | 太陽電池パネル配置 |
| US20190241549A1 (en) * | 2018-02-02 | 2019-08-08 | Kyulux, Inc. | Composition of matter for use in organic light-emitting diodes |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5624270B2 (ja) * | 2007-09-18 | 2014-11-12 | ユー・ディー・シー アイルランド リミテッド | 有機電界発光素子 |
| JP2014135466A (ja) * | 2012-04-09 | 2014-07-24 | Kyushu Univ | 有機発光素子ならびにそれに用いる発光材料および化合物 |
| TWI637944B (zh) * | 2013-11-28 | 2018-10-11 | 九州有機光材股份有限公司 | 發光材料、有機發光元件及化合物 |
| CN106316924B (zh) * | 2015-06-16 | 2020-01-24 | 清华大学 | 一种热活化延迟荧光材料 |
| JP6668152B2 (ja) * | 2015-12-28 | 2020-03-18 | 株式会社Kyulux | 化合物、発光材料および有機発光素子 |
| US11069860B2 (en) * | 2017-08-21 | 2021-07-20 | Kyulux, Inc. | Composition of matter for use in organic light-emitting diodes |
| CN109422674B (zh) * | 2017-08-25 | 2024-03-22 | 三星显示有限公司 | 有机分子,特别是用于光电子器件的有机分子 |
| CN109994628B (zh) * | 2017-12-29 | 2021-05-04 | 昆山国显光电有限公司 | 有机电致发光器件及有机电致发光器件的制备方法 |
| JP7325731B2 (ja) * | 2018-08-23 | 2023-08-15 | 国立大学法人九州大学 | 有機エレクトロルミネッセンス素子 |
| US11903308B2 (en) * | 2018-09-07 | 2024-02-13 | Idemitsu Kosan Co., Ltd. | Compound, material for organic electroluminescent element, organic electroluminescent element, and electronic device |
| TWI741639B (zh) * | 2019-06-14 | 2021-10-01 | 國立大學法人九州大學 | 間二氰苯化合物、發光材料及使用其之發光元件 |
-
2019
- 2019-08-29 JP JP2020541162A patent/JP7184263B2/ja active Active
- 2019-08-29 KR KR1020217005954A patent/KR102533313B1/ko active IP Right Grant
- 2019-08-29 WO PCT/JP2019/033913 patent/WO2020050127A1/ja unknown
- 2019-08-29 EP EP19857043.4A patent/EP3848352A4/en active Pending
- 2019-08-29 CN CN201980057047.2A patent/CN112638874A/zh active Pending
- 2019-08-29 US US17/272,550 patent/US20210332032A1/en active Pending
- 2019-09-03 TW TW108131600A patent/TWI719618B/zh active
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016526025A (ja) * | 2013-05-09 | 2016-09-01 | 日東電工株式会社 | 発光装置用の発光性化合物 |
| WO2014183080A1 (en) | 2013-05-09 | 2014-11-13 | Nitto Denko Corporation | Emissive compounds for light emitting devices |
| JP2016539182A (ja) * | 2013-10-30 | 2016-12-15 | 日東電工株式会社 | 発光デバイス用発光化合物 |
| WO2016138077A1 (en) | 2015-02-24 | 2016-09-01 | Nitto Denko Corporation | Gas sensor element |
| WO2016152605A1 (ja) * | 2015-03-23 | 2016-09-29 | 保土谷化学工業株式会社 | 有機エレクトロルミネッセンス素子用材料、発光材料および有機エレクトロルミネッセンス素子 |
| JP2018519663A (ja) * | 2015-06-16 | 2018-07-19 | 昆山国顕光電有限公司Kunshan Go−Visionox Opto−Electronics Co., Ltd. | 有機エレクトロルミネッセンス装置及びその製造方法 |
| WO2017115834A1 (ja) * | 2015-12-28 | 2017-07-06 | 株式会社Kyulux | 化合物、発光材料および有機発光素子 |
| WO2018047948A1 (ja) * | 2016-09-09 | 2018-03-15 | 東洋紡株式会社 | 有機発光素子ならびにそれに用いる発光材料および化合物 |
| WO2018155642A1 (ja) * | 2017-02-24 | 2018-08-30 | 国立大学法人九州大学 | 化合物、発光材料および発光素子 |
| JP2018165955A (ja) | 2017-03-28 | 2018-10-25 | 大阪瓦斯株式会社 | 火災警報器 |
| WO2018237389A1 (en) * | 2017-06-23 | 2018-12-27 | Kyulux Inc. | MATERIAL COMPOSITION FOR USE IN ORGANIC ELECTROLUMINESCENT DIODE |
| JP2019017156A (ja) | 2017-07-05 | 2019-01-31 | Solネットワーク株式会社 | 太陽電池パネル配置 |
| US20190241549A1 (en) * | 2018-02-02 | 2019-08-08 | Kyulux, Inc. | Composition of matter for use in organic light-emitting diodes |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3848352A4 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112409240A (zh) * | 2020-11-20 | 2021-02-26 | 清华大学 | 一种有机化合物及其应用及采用该化合物的有机电致发光器 |
| CN112409241A (zh) * | 2020-11-27 | 2021-02-26 | 清华大学 | 一种有机化合物及其应用及采用该化合物的有机电致发光器 |
| CN114685353A (zh) * | 2020-12-28 | 2022-07-01 | 北京鼎材科技有限公司 | 用于有机发光器件的有机化合物、有机电致发光器件 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI719618B (zh) | 2021-02-21 |
| EP3848352A4 (en) | 2022-06-01 |
| EP3848352A1 (en) | 2021-07-14 |
| US20210332032A1 (en) | 2021-10-28 |
| CN112638874A (zh) | 2021-04-09 |
| KR102533313B1 (ko) | 2023-05-16 |
| TW202033502A (zh) | 2020-09-16 |
| JPWO2020050127A1 (ja) | 2021-08-26 |
| JP7184263B2 (ja) | 2022-12-06 |
| KR20210039418A (ko) | 2021-04-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7184263B2 (ja) | ベンゾニトリル誘導体、発光材料およびそれを用いた発光素子 | |
| JP6994724B2 (ja) | ジシアノピラジン化合物、発光材料、およびそれを用いた発光素子 | |
| JP6958967B2 (ja) | ヘテロ環化合物およびこれを含む有機発光素子 | |
| JP6760616B2 (ja) | ジシアノn−ヘテロ環化合物、発光材料およびそれを用いた発光素子 | |
| JP6948644B2 (ja) | ジシアノペンタヘリセン化合物、発光材料およびそれを用いた発光素子 | |
| JP7386486B2 (ja) | イソフタロニトリル化合物、発光材料およびそれを用いた発光素子 | |
| WO2021210501A1 (ja) | ホウ素含有化合物、発光材料およびそれを用いた発光素子 | |
| KR102671951B1 (ko) | 이소프탈로니트릴 화합물, 발광 재료 및 그것을 사용한 발광 소자 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19857043 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020541162 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20217005954 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019857043 Country of ref document: EP Effective date: 20210406 |