WO2019156200A1 - 多能性幹細胞から腸管上皮細胞への分化誘導方法 - Google Patents
多能性幹細胞から腸管上皮細胞への分化誘導方法 Download PDFInfo
- Publication number
- WO2019156200A1 WO2019156200A1 PCT/JP2019/004553 JP2019004553W WO2019156200A1 WO 2019156200 A1 WO2019156200 A1 WO 2019156200A1 JP 2019004553 W JP2019004553 W JP 2019004553W WO 2019156200 A1 WO2019156200 A1 WO 2019156200A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- inhibitor
- cell
- days
- culturing
- Prior art date
Links
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0679—Cells of the gastro-intestinal tract
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/37—Digestive system
- A61K35/38—Stomach; Intestine; Goblet cells; Oral mucosa; Saliva
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5014—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing toxicity
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5073—Stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/01—Modulators of cAMP or cGMP, e.g. non-hydrolysable analogs, phosphodiesterase inhibitors, cholera toxin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/11—Epidermal growth factor [EGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/15—Transforming growth factor beta (TGF-β)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases (EC 2.)
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Definitions
- the present invention relates to a method for inducing differentiation of pluripotent stem cells into intestinal epithelial cells and use thereof.
- iPS induced pluripotent stem
- Patent Document 1 In order to provide intestinal epithelial cells used for drug absorption tests and the like, a method for selectively obtaining intestinal stem / progenitor cells from cells derived from the intestinal tract has been reported (Patent Document 1). In addition, a method for producing or maintaining pluripotent cells using an ALK5 inhibitor has been proposed (Patent Document 2).
- JP 2008-206510 A Special table 2012-511935 gazette International Publication No. 2014/132933 Pamphlet International Publication No. 2017/154795 pamphlet
- Non-Patent Documents 1 to 6 There are several reports on the induction of differentiation from iPS cells to intestinal epithelial cells (see, for example, Non-Patent Documents 1 to 6), but the differentiation induction methods in these reports are complicated and the differentiation efficiency is sufficient. In addition, pharmacokinetic analysis has not been performed in detail. Furthermore, the differentiation induction method induces differentiation using a large amount of extremely expensive growth factors and cytokines and is not suitable for practical use. The present inventors are also studying the differentiation of human iPS cells into intestinal epithelial cells, and have reported that the prepared intestinal epithelial cell-like cells have various pharmacokinetic functions (Patent Document 3). Non-Patent Documents 7 and 8). Moreover, low molecular weight compounds and conditions useful for promoting differentiation from human iPS cells to intestinal epithelial cells and acquiring functions have been found (Patent Documents 3 and 4 and Non-Patent Document 8).
- the present inventors conducted detailed studies with the aim of developing a more efficient differentiation induction method.
- intestinal stem cell-like cells obtained from iPS cells are induced to differentiate into intestinal epithelial cells, culturing the cells in the presence of a cAMP activator to actively increase cAMP levels in the cells, It was found to be extremely effective for efficient differentiation induction and maturation (acquisition of function). It also provided useful information on the combination and timing of addition of low molecular weight compounds used for differentiation induction.
- a method for inducing differentiation of pluripotent stem cells into intestinal epithelial cells comprising the following steps (1) and (2): (1) a step of differentiating pluripotent stem cells into intestinal stem cell-like cells; (2) Differentiation of intestinal stem cell-like cells obtained in step (1) into intestinal epithelial cell-like cells using MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, EGF and cAMP activator Process.
- step (1) comprises the following steps (1-1) and (1-2): (1-1) a step of differentiating pluripotent stem cells into endoderm-like cells; (1-2) A step of differentiating the endoderm-like cells obtained in step (1-1) into intestinal stem cell-like cells.
- step (2) comprises the following steps (1-1) and (1-2): (1-1) a step of differentiating pluripotent stem cells into endoderm-like cells; (1-2) A step of differentiating the endoderm-like cells obtained in step (1-1) into intestinal stem cell-like cells.
- the culture period in step (2) is 7 to 40 days.
- step (2) includes any one of the following culture steps A to D: Culturing step A: (a-1) culturing in the presence of EGF and cAMP activator and (a-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor performed after the culturing And culturing in the presence of EGF, Culturing step B: (b-1) culturing in the presence of EGF and (b-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, EGF and cAMP activity performed after the culturing Including culturing in the presence of chemicals, Culturing step C: (c-1) culturing in the presence of EGF and cAMP activator and (c-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor performed after the culturing Culturing in the presence of EGF and cAMP activators, Culturing step D: (d-1) includes
- the culture period of (a-1) is 2 to 10 days, the culture period of (a-2) is 9 to 29 days, The culture period of (b-1) is 2 days to 10 days, the culture period of (b-2) is 9 days to 19 days, The culture period of (c-1) is 2 days to 10 days, the culture period of (c-2) is 9 days to 19 days, The method according to [4], wherein the culture period of (d-1) is 15 to 25 days. [6] The method according to any one of [1] to [5], wherein the cAMP activator is forskolin.
- the culture step B includes (b-3) culture in the presence of a MEK1 inhibitor, a DNA methylation inhibitor, a TGF ⁇ receptor inhibitor, and EGF, which is performed after the culture of (b-2).
- Culturing step C is carried out after culturing of (c-2), including (c-3) culturing in the presence of a MEK1 inhibitor, a DNA methylation inhibitor, a TGF ⁇ receptor inhibitor and EGF
- Culturing step D comprises culturing in the presence of (d-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor and EGF, which is carried out after culturing of (d-1) [4 ] To [9].
- the MEK1 inhibitor is PD98059
- the DNA methylation inhibitor is 5-aza-2′-deoxycytidine
- the TGF ⁇ receptor inhibitor is A-83-01.
- [1] to [11] The method as described in any one of. [13] The method according to any one of [2] to [12], wherein activin A is used as a differentiation-inducing factor in the step (1-1).
- the pluripotent stem cell is an induced pluripotent stem cell.
- [19] The method according to [18], wherein the pharmacokinetics is metabolism, absorption, excretion, drug interaction, induction of a drug metabolizing enzyme, or induction of a drug transporter.
- Test group 1 in which 8-bromo-3 ', 5'-cyclic adenosine monophosphate (8-Br-cAMP) was added to the medium in the first half (day 8 to day 13), second half (day 13 to day 26)
- Test group 2 in which 8-Br-cAMP was added to the medium in the first half (test day 8 to day 13), and test group 3 in which 3-isobutyl-1-methylxanthine (IBMX) was added to the medium in the first half (day 8 to 13)
- Test group 4 with IBMX added to the medium on day 1 to day 26)
- Test group 5 with forskolin added to the medium in the first half (day 8 to day 13)
- Test group 6 with Forskolin added to the medium Effect of cAMP activator (forskolin) on differentiation from human iPS cells to intestinal epithelial cell-like cells (result of Experiment 1).
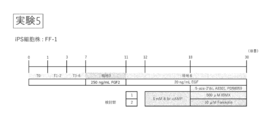
- Protocol of experiment 3 using human iPS cells (FF-1). 18 days after induction of differentiation into intestinal stem cells by culturing for 5 days in the presence of Activin (A (Day 0 to Day 5) and culturing for 4 days in the presence of FGF2 (Days 5 to 9) Differentiation into intestinal epithelial cells was induced by culturing from day 10 to day 28).
- the following test groups 1 and 2 having different medium-added components in inducing differentiation into intestinal epithelial cells were set, and the effects on differentiation were compared. Test group 1 where 8-Br-cAMP was added to the medium from day 10 to day 16 and IBMX was added to the medium from day 16 to day 28.
- Test group 2 with Forskolin added to the medium from day 18 to 30; Test group 3 with Forskolin added to the medium from day 12 to 30 Effect of cAMP activator (forskolin) on drug-metabolizing enzyme activity of human iPS cell-derived intestinal epithelial cell-like cells (result of experiment 4).
- Human primary small intestinal cell pair test group 1 is **** P ⁇ 0.0001, human primary small intestinal cell pair test group 2 is ns P> 0.05, and test group 1 vs. test group 2 is *** P ⁇ 0.001.
- IVAL's human primary small intestinal cells Lot No. HE3007 was used. Composition of medium used for experiments 1-5.
- F a value and P app drug 16 kinds of relationship F a value and P app drug.
- Differentiated enterocytes (A) and Caco-2 cells (B) were incubated for 60 minutes at 37 ° C. in transport buffer containing 16 drugs.
- the present invention relates to a method for inducing differentiation of pluripotent stem cells into the intestinal epithelial cell lineage (hereinafter also referred to as “differentiation induction method of the present invention”).
- differentiation induction method of the present invention cells exhibiting characteristics similar to those of intestinal epithelial cells constituting intestinal tissue of a living body, that is, intestinal epithelial cell-like cells can be obtained.
- pluripotency refers to the ability to differentiate into all the cells that make up a living body (differentiation pluripotency) and the ability to generate daughter cells that have the same differentiation potential as self through cell division (self-replication ability) ). Pluripotency can be evaluated by transplanting cells to be evaluated into nude mice and testing for the presence or absence of teratoma containing each of the three germ layers (ectodermal, mesoderm, and endoderm). it can.
- pluripotent stem cells examples include embryonic stem cells (ES cells), embryonic germ cells (EG cells), and induced pluripotent stem cells (iPS cells), which have both differentiation pluripotency and self-renewal ability. As long as it is a cell, it is not limited to this. Preferably, ES cells or iPS cells are used. More preferably iPS cells are used.
- the pluripotent stem cells are preferably mammalian cells (for example, primates such as humans and chimpanzees, rodents such as mice and rats), particularly preferably human cells. Therefore, in the most preferred embodiment of the present invention, human iPS cells are used as pluripotent stem cells.
- ES cells can be established, for example, by culturing an early embryo before implantation, an inner cell mass constituting the early embryo, a single blastomere, etc. (Manipulating the Mouse Embryo A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press (1994); Thomson, J. A. et al., Science, 282, 1145-1147 (1998)).
- an early embryo an early embryo produced by nuclear transfer of a nucleus of a somatic cell may be used (Wilmut et al. (Nature, 385, 810 (1997)), Cibelli et al. (Science, 280, 1256). (1998)), Akira Iriya et al.
- ES cells are available from preserving institutions or are commercially available.
- human ES cells can be obtained from the Institute of Regenerative Medicine, Kyoto University (for example, KhES-1, KhES-2 and KhES-3), WiCell Research Institute, ESI BIO, and the like.
- EG cells can be established by culturing primordial germ cells in the presence of LIF, bFGF, SCF, etc. (Matsui et al., Cell, 70, 841-847 (1992), Shamblott et al., Proc. Natl. Acad. Sci. USA, 95 (23), 13726-13731 (1998), Turnpenny et al., Stem Cells, 21 (5), 598-609, (2003)).
- iPS cells are cells having pluripotency (pluripotency) and proliferative ability produced by reprogramming somatic cells by introduction of reprogramming factors. Artificial pluripotent stem cells exhibit properties similar to ES cells. Somatic cells used for the production of iPS cells are not particularly limited, and may be differentiated somatic cells or undifferentiated stem cells. Although the origin thereof is not particularly limited, preferably, somatic cells of mammals (eg, primates such as humans and chimpanzees, rodents such as mice and rats), particularly preferably human somatic cells are used. iPS cells can be prepared by various methods reported so far. In addition, it is naturally assumed that an iPS cell production method developed in the future will be applied.
- mammals eg, primates such as humans and chimpanzees, rodents such as mice and rats
- iPS cell production The most basic method of iPS cell production is to introduce four factors, transcription factors Oct3 / 4, Sox2, Klf4 and c-Myc, into cells using viruses (Takahashi K, Yamanaka S : Cell 126 (4), 663-676, 2006; Takahashi, K, et al: Cell 131 (5), 861-72, 2007).
- Human iPS cells have been reported to be established by introducing four factors, Oct4, Sox2, Lin28 and Nonog (Yu J, et al: Science 318 (5858), 1917-1920, 2007).
- Three factors excluding c-Myc (Nakagawa M, et al: Nat. Biotechnol.
- lentiviruses (Yu J, et al: Science 318 (5858), 1917-1920, 2007), adenoviruses (Stadtfeld M, et al: Science 322 (5903 ), 945-949, 2008), plasmid (Okita K, et al: Science 322 (5903), 949-953, 2008), transposon vectors (Woltjen K, Michael IP, Mohseni P, et al: Nature 458, 766- 770, 2009; Kaji K, Norrby K, Pac a A, et al: Nature 458, 771-775, 2009; Yusa K, Rad R, Takeda J, et al: Nat Methods 6, 363-369, 2009), or Techniques using episomal vectors (Yu J, Hu K, Smuga-Otto K, Tian S, et al: Science 324, 797-801, 2009) have been developed.
- pluripotent stem cell markers such as Fbxo15, Nanog, Oct / 4, Fgf-4, Esg-1, and Cript Etc. can be selected as an index.
- the selected cells are collected as iPS cells.
- IPS cells can also be provided from, for example, Kyoto University or RIKEN BioResource Center.
- inducing differentiation means acting to differentiate along a specific cell lineage.
- iPS cells are induced to differentiate into intestinal epithelial cells.
- the differentiation inducing method of the present invention is roughly divided into two stages of induction processes, ie, a process of differentiating iPS cells into intestinal stem cell-like cells (step (1)), and the obtained intestinal stem cell-like cells are converted into intestinal epithelial cell-like cells.
- pluripotent stem cells are cultured and differentiated into intestinal stem cell-like cells.
- pluripotent stem cells are cultured under conditions that induce differentiation into intestinal stem cell-like cells.
- Culture conditions are not particularly limited as long as pluripotent stem cells differentiate into intestinal stem cell-like cells.
- the two-stage differentiation induction described below ie, to endoderm-like cells of pluripotent stem cells. Differentiation (step (1-1)) and differentiation of endoderm-like cells into intestinal stem cell-like cells (step (1-2)).
- Step (1-1) Differentiation into Endoderm-like Cells pluripotent stem cells are cultured and differentiated into endoderm-like cells.
- pluripotent stem cells are cultured under conditions that induce differentiation into endoderm.
- the culture conditions are not particularly limited as long as the pluripotent stem cells are differentiated into endoderm-like cells.
- it is cultured in a medium supplemented with activin A according to a conventional method.
- the concentration of activin A in the medium is, for example, 10 ng / mL to 200 ng / mL, preferably 20 ng / mL to 150 ng / mL.
- Serum is not limited to fetal bovine serum, and human serum, sheep serum, and the like can also be used.
- the amount of serum or serum replacement added is, for example, 0.1% (v / v) to 10% (v / v).
- An inhibitor of the Wnt / ⁇ -catenin signaling pathway eg, hexachlorophene, quercetin, Wnt3a, which is a Wnt ligand
- Wnt3a which is a Wnt ligand
- BMP4, VEGF, and FGF2 may be added to the medium to promote differentiation into endoderm-like cells.
- the concentration of BMP4 in the medium is, for example, 0.1 ng / mL to 10 ng / mL, preferably 1 ng / mL to 5 ng / mL
- the concentration of VEGF in the medium is, for example, 0.5 ng / mL to 100 ng / ng, preferably 1 ng / mL to 20 ng / mL
- the concentration of FGF2 in the medium is, for example, 0.2 ng / mL to 50 ng / mL, preferably 0.5 ng / mL to 10 ng / mL.
- This step can also be performed by the method described in International Publication No. 2014/165663 pamphlet or a method analogous thereto.
- two-stage culture is performed as step (1-1).
- the first stage culture is performed in a medium supplemented with a relatively low concentration of serum (eg, 0.1% (v / v) to 1% (v / v)), and the second stage culture is the first stage culture.
- a medium with a higher serum concentration for example, the serum concentration is 1% (v / v) to 10% (v / v)).
- Adopting the two-stage culture in this way is preferable in that the growth of undifferentiated cells is suppressed by the first-stage culture and the differentiated cells are grown in the subsequent second stage.
- the period of the step (1-1) (culture period) is, for example, 1 day to 10 days, preferably 2 days to 7 days.
- the first stage culture period is, for example, 1 to 7 days, preferably 2 to 5 days
- the second stage culture period is, for example, 1 day. Up to 6 days, preferably 1 to 4 days.
- the endoderm-like cells obtained in step (1-1) are cultured and differentiated into intestinal stem cell-like cells.
- endoderm-like cells are cultured under conditions that induce differentiation into intestinal stem cells.
- the culture conditions are not particularly limited as long as the endoderm-like cells are differentiated into intestinal stem cell-like cells.
- the culture is performed in the presence of FGF2 (fibroblast growth factor 2) or in the presence of a GSK-3 ⁇ inhibitor.
- FGF2 fibroblast growth factor 2
- human FGF2 for example, human recombinant FGF2
- the cell population obtained through the step (1-1) or a part thereof is subjected to the step (1-2) without sorting.
- the step (1-2) may be performed after selecting endoderm-like cells from the cell population obtained through the step (1-1).
- the endoderm-like cells may be selected with a flow cytometer (cell sorter) using a cell surface marker as an index.
- FGF2 In the presence of FGF2 is synonymous with the condition in which FGF2 is added to the medium. Therefore, in order to perform culture in the presence of FGF2, a medium supplemented with FGF2 may be used.
- An example of the concentration of FGF2 added is 100 ng / mL to 500 ng / mL.
- GSK-3 ⁇ inhibitor in the presence of a GSK-3 ⁇ inhibitor, is synonymous with the condition in which the GSK-3 ⁇ inhibitor is added to the medium. Therefore, in order to perform culture in the presence of a GSK-3 ⁇ inhibitor, a medium supplemented with FGF2 may be used.
- GSK-3 ⁇ inhibitors include CHIR 99021, SB216763, CHIR 98014, TWS119, Tideglusib, SB415286, BIO, AZD2858, AZD1080, AR-A014418, TDZD-8, LY2090314, IM-12, Indirubin, Bikinin, 1-Azakenpaullone be able to.
- concentration of GSK-3 ⁇ inhibitor added in the case of CHIR 99021 is 1 ⁇ M to 100 ⁇ M, preferably 3 ⁇ M to 30 ⁇ M.
- the period of the step (1-2) (culture period) is, for example, 2 days to 10 days, preferably 3 days to 7 days. If the culture period is too short, the expected effects (increased differentiation efficiency, promotion of acquisition of functions as intestinal stem cells) cannot be sufficiently obtained. On the other hand, if the culture period is too long, the differentiation efficiency is lowered.
- intestinal stem cell-like cells Differentiation into intestinal stem cell-like cells can be determined or evaluated using, for example, the expression of intestinal stem cell marker as an index.
- intestinal stem cell markers are G protein-coupled receptor 5 (LGR5) and ephrin B2 receptor (EphB2) containing leucine-rich repeats.
- Step (2) Differentiation into intestinal epithelial cell-like cells>
- MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, EGF and cAMP activator are used in combination, and the intestinal stem cell-like cells obtained in step (1) are converted into intestinal epithelial cell-like cells.
- the cAMP level is positively increased by using a cAMP activator during this differentiation induction.
- “Combination of MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, EGF and cAMP activator” means that one or more of the cultures constituting step (2) are performed. It does not require that the compounds are required, that all these compounds are used at the same time, that is, that the culture using the medium to which all these compounds are added is performed as an essential condition.
- the cell population obtained through the step (1) or a part thereof is subjected to the step (2) without sorting.
- the step (2) may be performed. Selection of intestinal stem cell-like cells may be performed, for example, with a flow cytometer (cell sorter) using a cell surface marker as an index.
- Step (2) includes one or more cultures (details will be described later).
- a medium supplemented with EGF and cAMP activator as essential components MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor and EGF are essential components
- MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, etc. are essential components
- MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, etc. Used.
- Examples of MEK1 inhibitors include PD98059, PD184352, PD184161, PD0325901, U0126, MEK inhibitor I, MEK inhibitor II, MEK1 / 2 inhibitor II, and SL327.
- examples of DNA methylation inhibitors include 5-aza-2'-deoxycytidine, 5-azacytidine, RG108, and zebralin.
- A-83-01 used in the examples described later exhibits inhibitory activity on TGF- ⁇ receptors ALK4, ALK5, and ALK7. What shows inhibitory activity with respect to one or more of ALK4, ALK5, and ALK7 may be used.
- A-83-01, SB431542, SB-505124, SB525334, D4476, ALK5 inhibitor, LY2157299, LY364947, GW788388, and RepSox satisfy the condition.
- the cAMP activator forskolin, indomethacin, NKH477 (colforsin daropate), cell-derived toxin protein (pertussis toxin, cholera toxin), PACAP-27, PACAP-38, SKF83822 and the like can be used.
- Forskolin exhibits an adenylate cyclase activation action and promotes the synthesis of intracellular cAMP.
- concentration of MEK1 inhibitor added is 4 ⁇ M to 100 ⁇ M, preferably 10 to 40 ⁇ M.
- an example of the addition concentration of a DNA methylation inhibitor in the case of 5-aza-2′-deoxycytidine is 1 ⁇ M to 25 ⁇ M, preferably 2.5 ⁇ M to 10 ⁇ M
- the addition concentration of a TGF ⁇ receptor inhibitor is 0.1 ⁇ M to 2.5 ⁇ M, preferably 0.2 ⁇ M to 1 ⁇ M.
- concentration of EGF added are 5 ng / mL to 100 ng / mL, preferably 10 ng / mL to 50 ng / mL.
- concentration of cAMP activating substance added is 1 ⁇ M to 200 ⁇ M, preferably 5 ⁇ M to 100 ⁇ M.
- concentration when using the compound different from the exemplified compound, ie, PD98059, 5-aza-2'-deoxycytidine, A-83-01 and forskolin is 1 ⁇ M to 200 ⁇ M, preferably 5 ⁇ M to 100 ⁇ M.
- concentration when using the compound different from the exemplified compound, ie, PD98059, 5-aza-2'-deoxycytidine, A-83-01 and forskolin In view of the differences in properties (especially differences in activity) of the compounds (PD98059, 5-aza-2'-deoxycytidine, A-83-01, forskolin), those skilled in the art will follow the above concentration range. Can be set. Further, whether or not the set concentration range is appropriate can be confirmed by a preliminary experiment according to an example described later.
- Step (2) is added to the above conditions, cAMP is supplied to the cells (referred to as “additional condition 1”) and cAMP degrading enzyme inhibitor is present (referred to as “additional condition 2”), or these You may decide to carry out under either of these conditions.
- the additional condition 1 condition in which cAMP is supplied to the cell
- the additional condition 1 when the additional condition 1 is employed, it can be expected that the decrease in intracellular cAMP concentration is suppressed and the differentiation induction into the intestinal epithelium, particularly the acquisition of the function as the intestinal epithelial cell is promoted. That is, the conditions can allow for the preparation of more functional intestinal epithelial cell-like cells.
- PAMP activators e.g., 8-Br-cAMP (8-Bromoadenosine-3 ′, 5′-cyclic monophosphate sodium salt, CAS Number: 76939-46-3), 6-Bnz-cAMP (N6-Benzoyladenosine- 3 ', 5'-cyclic monophosphate sodium salt salt, CAS Number: 1135306-29-4), cAMPS-Rp ((R) -Adenosine, cyclic 3', 5 '-(hydrogenphosphorothioate) triethylammonium salt, CAS Number: 151837- 09-1), cAMPS-Sp ((S) -Adenosine, cyclic 3 ', 5'-(hydrogenphosphorothioate) triethylammonium salt, CAS Number: 93602-66-5), Dibutyryl-cAMP (N6, O2'-Dibutyryl adenosine 3 ', 5'-cyclic monophosphate
- 8-CPT-cAMP 8 -(4-Chlorophenylthio) adenosine 3 ', 5'-cyclic monophosphate, CAS Number: 93882-12-3
- 8-pCPT-2'-OM e-cAMP e.g., 8- (4-Chlorophenylthio) -2′-O-methyladenosine 3 ′, 5′-cyclic monophosphate monosodium, CAS No Number: 634634207-53-7) can be used.
- concentration of cAMP derivative added is 0.1 mM to 10 mM, preferably 0.2 mM to 5 mM, more preferably 0.5 mM to 2 mM.
- addition concentration in the case of using the exemplified compound, that is, a compound different from 8-Br-cAMP, the difference between the characteristics of the compound used and the characteristics of the exemplified compound (8-Br-cAMP) (particularly In view of the difference in activity), those skilled in the art can set the concentration according to the above concentration range. Further, whether or not the set concentration range is appropriate can be confirmed by a preliminary experiment according to an example described later.
- Additional condition 2 is synonymous with the condition in which the cAMP-degrading enzyme inhibitor is added to the medium.
- the additional condition 2 it can be expected that the decrease of intracellular cAMP concentration is suppressed by inhibiting the degradation of cAMP, and the induction of differentiation into the intestinal epithelium, particularly the acquisition of the function as an intestinal epithelial cell can be expected. That is, the conditions can allow for the preparation of more functional intestinal epithelial cell-like cells.
- the additional condition 1 and the additional condition 2 are used together, it is possible to suppress a decrease in intracellular cAMP concentration while supplying cAMP to the cells. Therefore, it is an effective condition for maintaining intracellular cAMP at a high level, and it can be expected that efficient differentiation induction into intestinal epithelial cells is promoted.
- cAMP-degrading enzyme inhibitors include IBMX® (3-isobutyl-1-methylxanthine) ® (MIX), Theophylline, Papaverine, Pentoxifylline® (Trental), KS-505, 8-Methoxymethyl-IBMX, Vinpocetine® (TCV-3B), EHNA, Trequinsin (HL-725), Lixazinone (RS-82856), (LY-186126), Cilostamide (OPC3689), Bemorradan (RWJ-22867), Anergrelide (BL4162A), Indolidan (LY195115), Cilostazol (OPC-13013), Milrinone ( WIN47203), Siguazodan (SKF-94836), 5-Methyl-imazodan (CI 930), SKF-95654, Pirilobendan (UD-CG 115 BS), Enoximone (MDL 17043), Imazodan (CL 914), SKF-94120, Vesnarin
- concentration of cAMP-degrading enzyme inhibitor added is 0.05 mM to 5 mM, preferably 0.1 mM to 3 mM, more preferably 0.2 mM to 1 mM.
- addition concentration when using a compound different from the exemplified compound that is, a compound different from IBMX, considering the difference between the characteristics of the compound used and the characteristics of the exemplified compound (IBMX) (particularly the difference in activity)
- concentration range considering the difference between the characteristics of the compound used and the characteristics of the exemplified compound (IBMX) (particularly the difference in activity)
- concentration range considering the difference between the characteristics of the compound used and the characteristics of the exemplified compound (IBMX) (particularly the difference in activity)
- concentration range that is, a compound different from IBMX
- the period of step (2) (culture period) is, for example, 7 to 40 days, preferably 10 to 30 days. If the culture period is too short, the expected effects (increased differentiation efficiency, promotion of acquisition of functions as intestinal epithelial cells) cannot be sufficiently obtained. On the other hand, if the culture period is too long, the differentiation efficiency is lowered.
- intestinal epithelial cell markers include ATP binding cassette transporter B1 / multidrug resistance protein 1 (ABCB1 / MDR1), ATP binding cassette transporter G2 / breast cancer resistance protein (ABCG2 / BCRP), cytochrome P450P3A4 (CYP3A4) , Fatty acid binding protein 2 (FABP), pregnane X receptor (PXR), SLC (solute carrier) family member 5A1 / sodium conjugated glucose transporter 1 (SLC5A1 / SGLT1), SLC (solute carrier) family member 15A1 / peptide trans Porter 1 (SLC15A1 / PEPT1), SLC (solute carrier) organic anion transporter 2B1 (SLCO2B1 / OATP2B1), sucrase-i
- sucrase-isomaltase which is highly specific to the intestinal epithelium, and villin 1, CYP3A4, the main drug-metabolizing enzyme in the small intestine, SLC15A1 / PEPT1, which is involved in peptide absorption in the small intestine, and the apical membrane side of the small intestine SLC5A1 / SGLT1, which is a glucose transporter expressed in the lung, SLCO2B1 / OATP2B1, which is involved in the absorption of organic anions in the small intestine, and CES2A1, which is a hydrolase with high expression in the small intestine, are particularly effective markers.
- a cell population consisting only of the target cells (intestinal epithelial cell-like cells) or a cell population that contains the target cells in a high ratio (high purity)
- select a cell surface marker that is characteristic for the target cells may be selected and sorted as an index.
- any one of the following culture steps A to D is performed.
- ⁇ Culture process A> In the culture step A, (a-1) culturing in the presence of EGF and intracellular cAMP synthesis promoter, and (a-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor performed after the culturing. Culture in the presence of a body inhibitor and EGF. If two-stage culture is performed in this way, the effects of promoting differentiation into intestinal epithelial cells, maturation, and function acquisition can be expected.
- the culture period of (a-1) is, for example, 2 days to 10 days, preferably 4 days to 8 days
- the culture period of (a-2) is, for example, 9 days to 29 days, preferably 7 days to 27 days. Days.
- concentration of each compound, etc. said corresponding description is used.
- (A-1) culture where cAMP is supplied to the cells (referred to as “additional condition 1”) and / or in the presence of a cAMP degrading enzyme inhibitor (referred to as “additional condition 2”), or any of these You may decide to carry out on condition of this.
- additional condition 1 a culture where cAMP is supplied to the cells
- additional condition 2 a cAMP degrading enzyme inhibitor
- ⁇ Culture process B> In the culture step B, (b-1) culture in the presence of EGF, and (b-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor inhibitor, EGF and cells performed after the culture Culture is performed in the presence of an internal cAMP synthesis promoter. If two-stage culture is performed in this way, the effects of promoting differentiation into intestinal epithelial cells, maturation, and function acquisition can be expected.
- the culture period of (b-1) is, for example, 2 days to 10 days, preferably 4 days to 8 days, and the culture period of (b-2) is, for example, 9 days to 19 days, preferably 7 days to 17 days. Days.
- concentration of each compound, etc. said corresponding description is used.
- the culture in (b-1) may be performed under the conditions for supplying cAMP to cells (additional condition 1) and / or in the presence of a cAMP degrading enzyme inhibitor (additional condition 2), or any of these conditions. Good.
- additional condition 1 additional condition 1
- additional condition 2 additional condition 2
- the details of the additional condition 1 and the additional condition 2 are as described above.
- culture in (b-2) After the culture in (b-2), culture in the presence of a MEK1 inhibitor, a DNA methylation inhibitor, a TGF ⁇ receptor inhibitor and EGF (culture in (b-3)) may be performed.
- the culture period is, for example, 1 day to 10 days. If this culture is performed, the effect of promoting differentiation into intestinal epithelial cells, maturation, and function acquisition can be expected.
- ⁇ Culture process C> In the culture step C, (c-1) culturing in the presence of EGF and intracellular cAMP synthesis promoter, and (c-2) MEK1 inhibitor, DNA methylation inhibitor, TGF ⁇ receptor carried out after the culturing. Culture in the presence of a body inhibitor, EGF and an intracellular cAMP synthesis promoter. If two-stage culture is performed in this way, the effects of promoting differentiation into intestinal epithelial cells, maturation, and function acquisition can be expected.
- the culture period of (c-1) is, for example, 2 days to 10 days, preferably 4 days to 8 days, and the culture period of (c-2) is, for example, 9 days to 19 days, preferably 7 days to 17 days. Days.
- concentration of each compound, etc. said corresponding description is used.
- the culture in (c-1) may be carried out under the condition that cAMP is supplied to the cells (additional condition 1) and / or in the presence of the cAMP-degrading enzyme inhibitor (additional condition 2). Good.
- additional condition 1 the condition that cAMP is supplied to the cells
- additional condition 2 the condition that cAMP-degrading enzyme inhibitor
- culture in (c-2) After the culture in (c-2), culture in the presence of a MEK1 inhibitor, a DNA methylation inhibitor, a TGF ⁇ receptor inhibitor and EGF (culture in (c-3)) may be performed.
- the culture period is, for example, 1 day to 10 days. If this culture is performed, the effect of promoting differentiation into intestinal epithelial cells, maturation, and function acquisition can be expected.
- (d-1) culture is performed in the presence of a MEK1 inhibitor, a DNA methylation inhibitor, a TGF ⁇ receptor inhibitor, EGF and an intracellular cAMP synthesis promoter.
- This culturing step is particularly advantageous in that the culturing operation is simple, it is more effective for differentiation into intestinal epithelial cells, and a stable effect can be expected because it is a compound.
- the culture period of (d-1) is, for example, 15 days to 25 days, preferably 17 days to 23 days. In addition, about the matter which is not demonstrated in particular (The compound which can be used for each culture
- the culture in (d-1) may be performed under the conditions for supplying cAMP to the cells (additional condition 1) and / or in the presence of a cAMP-degrading enzyme inhibitor (additional condition 2), or any of these conditions. Good.
- additional condition 1 and the additional condition 2 are as described above.
- culture in the presence of a MEK1 inhibitor, a DNA methylation inhibitor, a TGF ⁇ receptor inhibitor and EGF may be performed.
- the culture period is, for example, 1 day to 10 days. If this culture is performed, the effect of promoting differentiation into intestinal epithelial cells, maturation, and function acquisition can be expected.
- each step ((1), (1-1), (1-2), (2), (a-1), (a-2), (b-1), (b- 2), (b-3), (c-1), (c-2), (c-3), (d-1), (d-2)) Good.
- the cells when the cells become confluent or subconfluent, a part of the cells is collected and transferred to another culture vessel, and the culture is continued.
- the cells may be seeded at a cell density of about 1 ⁇ 10 4 cells / cm 2 to 1 ⁇ 10 6 cells / cm 2 .
- ROCK inhibitor Rho-associated coiled-coil forming kinase / Rho-binding kinase
- culture conditions in each step constituting the present invention may be those generally adopted in animal cell culture. That is, for example, it may be cultured in an environment of 37 ° C. and 5% CO 2 .
- IMDM Iskov modified Dulbecco medium
- Ham F12 medium HamF12
- D-MEM Dulbecco modified Eagle medium
- Gibco, etc. Glasgow basic medium
- RPMI1640 medium etc.
- Two or more basic media may be used in combination.
- a basic medium for example, D- It is preferable to use a mixed medium of MEM and Ham F12 medium, D-MEM.
- components that can be added to the medium include bovine serum albumin (BSA), antibiotics, 2-mercaptoethanol, PVA, non-essential amino acids (NEAA), insulin, transferrin, and selenium.
- BSA bovine serum albumin
- NEAA non-essential amino acids
- insulin transferrin
- selenium selenium
- cells are cultured two-dimensionally using a culture dish or the like.
- intestinal epithelial cell-like cells can be obtained from pluripotent stem cells by two-dimensional culture.
- three-dimensional culture using a gel culture substrate or a three-dimensional culture plate may be performed.
- the second aspect of the present invention relates to the use of intestinal epithelial cell-like cells prepared by the differentiation induction method of the present invention.
- Various assays are provided as a first use.
- the intestinal epithelial cell-like cell of the present invention can be used in a model system of the intestine, particularly the small intestine, and is useful for evaluating pharmacokinetics (absorption, metabolism, etc.) and toxicity in the intestine, particularly the small intestine.
- the intestinal epithelial cell-like cell of the present invention can be used for evaluating the pharmacokinetics and toxicity of the compound.
- the intestinal epithelial cell-like cells of the present invention can be used to test the absorbability or membrane permeability of a test substance, drug interaction, induction of drug metabolizing enzymes, induction of drug transporters, toxicity, etc. it can. That is, the present invention evaluates the absorbability or membrane permeability of a test substance, drug interaction, induction of a drug metabolizing enzyme, induction of a drug transporter, toxicity, etc. as one of the uses of intestinal epithelial cell-like cells.
- a method (first aspect) is provided.
- a step of preparing a cell layer composed of intestinal epithelial cell-like cells obtained by the differentiation induction method of the present invention and (ii) a step of bringing a test substance into contact with the cell layer; (Iii) quantifying a test substance that has permeated through the cell layer, and evaluating the absorbability or membrane permeability of the test substance, drug interaction, induction of a drug metabolizing enzyme, induction of a drug transporter, or toxicity Do.
- the absorbability of the test substance can also be evaluated by the method described later (second aspect).
- intestinal epithelial cell-like cells are typically cultured on a semipermeable membrane (porous membrane) to form a cell layer.
- a semipermeable membrane for example, Transwell (registered trademark) provided by Corning
- a culture insert for example, Transwell (registered trademark) provided by Corning
- “Contact” in step (ii) is typically performed by adding a test substance to the medium.
- the timing of adding the test substance is not particularly limited. Therefore, after culturing is started in a medium not containing the test substance, the test substance may be added at a certain point in time, or the culture may be started in advance in a medium containing the test substance.
- organic compounds or inorganic compounds having various molecular sizes can be used as the test substance.
- organic compounds include nucleic acids, peptides, proteins, lipids (simple lipids, complex lipids (phosphoglycerides, sphingolipids, glycosylglycerides, cerebrosides, etc.), prostaglandins, isoprenoids, terpenes, steroids, polyphenols, catechins, vitamins (B1, B2, B3, B5, B6, B7, B9, B12, C, A, D, E, etc.) Existing or candidate ingredients such as pharmaceuticals, nutritional foods, food additives, agricultural chemicals, cosmetics (cosmetics)
- One of the preferable test substances is a plant extract, cell extract, culture supernatant, etc.
- the test substance can be used by simultaneously adding two or more kinds of test substances.
- the test substance may be derived from a natural product or synthesized, for example, in the latter case It is possible to build an efficient assay systems using techniques
- the period for contacting the test substance can be set arbitrarily.
- the contact period is, for example, 10 minutes to 3 days, preferably 1 hour to 1 day.
- the contact may be performed in a plurality of times.
- the test substance that has permeated the cell layer is quantified.
- a culture vessel equipped with a culture insert such as Transwell a test substance that has passed through the culture insert, that is, a sample that has moved into the upper or lower vessel through the cell layer.
- the test substance is quantified by a measurement method such as mass spectrometry, liquid chromatography, or immunological technique (for example, fluorescence immunoassay (FIA method), enzyme immunoassay (EIA method)) according to the test substance.
- FIA method fluorescence immunoassay
- EIA method enzyme immunoassay
- the absorbability or membrane permeability of the test substance Based on the quantification result (the amount of the test substance that has permeated the cell layer) and the amount of the test substance used (typically the amount added to the medium), the absorbability or membrane permeability of the test substance, drug interaction, Judge and evaluate drug-metabolizing enzyme induction, drug transporter induction, or toxicity.
- the present invention also provides a method for evaluating metabolism or absorption of a test substance as another aspect (second aspect).
- this method (I) a step of bringing a test substance into contact with the intestinal epithelial cell-like cell obtained by the differentiation induction method of the present invention, and (II) metabolism or absorption of the test substance, drug interaction, drug metabolizing enzyme Induction, drug transporter induction, or a step of measuring and evaluating toxicity.
- Step (I) that is, the contact between the intestinal epithelial cell-like cells and the test substance can be carried out in the same manner as in the above step (ii). However, it is not essential to form a cell layer in advance.
- step (II) the metabolism or absorption of the test substance, drug interaction, induction of drug metabolizing enzyme, induction of drug transporter, or toxicity is measured and evaluated (step (II)).
- step (I) that is, after contact with the test substance, metabolism or the like can be measured and evaluated without a substantial time interval, or for a certain time (for example, 10 minutes to 5 hours).
- Metabolism etc. may be measured and evaluated after the passage. Metabolism can be measured, for example, by detecting a metabolite. In this case, the expected metabolite is usually measured qualitatively or quantitatively using the culture solution after step (I) as a sample.
- An appropriate measurement method may be selected according to the metabolite. For example, mass spectrometry, liquid chromatography, immunological method (eg, fluorescence immunoassay (FIA method), enzyme immunoassay (EIA method)) ) Etc. can be adopted.
- FFA method fluorescence immunoassay
- EIA method enzyme
- the metabolic amount of the test substance can be evaluated according to the amount of the metabolite.
- the metabolic efficiency of the test substance may be calculated based on the detection result of the metabolite and the amount of the test substance used (typically, the amount added to the medium).
- Test substance using the expression of drug metabolizing enzymes (cytochrome P450 (especially CYP3A4), uridine diphosphate-glucuronyltransferase (especially UGT1A8, UGT1A10), sulfate transferase (especially SULT1A3, etc.)) in intestinal epithelial cell-like cells It is also possible to measure the metabolism.
- the expression of drug metabolizing enzymes can be assessed at the mRNA level or protein level. For example, when an increase in the mRNA level of the drug-metabolizing enzyme is observed, it can be determined that “the test substance has been metabolized”. Similarly, when an increase in the activity of the drug-metabolizing enzyme is observed, it can be determined that “the test substance has been metabolized”. As in the case of determining a metabolite as an index, quantitative determination / evaluation may be performed based on the expression level of a drug metabolizing enzyme.
- the remaining amount of the test substance in the culture solution is measured.
- the test substance is quantified using the culture solution after step (I) as a sample.
- An appropriate measuring method may be selected according to the test substance. For example, mass spectrometry, liquid chromatography, immunological techniques (for example, fluorescence immunoassay (FIA method), enzyme immunoassay (EIA method)) and the like can be employed.
- FFA method fluorescence immunoassay
- EIA method enzyme immunoassay
- the amount of absorption or absorption efficiency of the test substance can be determined and evaluated according to the degree of decrease.
- the absorption can also be evaluated by measuring the amount of the test substance taken up into the cells.
- the measurement / evaluation of metabolism and the measurement / evaluation of absorption may be performed simultaneously or in parallel.
- a cell preparation containing intestinal epithelial cell-like cells is provided as a second use of the intestinal epithelial cell-like cells prepared by the differentiation induction method of the present invention.
- the cell preparation of the present invention can be applied to the treatment of various intestinal diseases. In particular, it is expected to be used as a material for regeneration / reconstruction of intestinal epithelial tissue that is damaged (including dysfunction). That is, contribution to regenerative medicine can be expected.
- intestinal epithelial cell-like cells obtained by the method of the present invention are suspended in physiological saline or a buffer solution (for example, a phosphate buffer solution), or the cells are used. It can be prepared by producing a three-dimensional tissue (organoid or spheroid).
- 1 ⁇ 10 5 to 1 ⁇ 10 10 cells may be contained as a single dose so that a therapeutically effective amount of cells can be administered.
- the content of the cells can be appropriately adjusted in consideration of the purpose of use, the target disease, the sex of the application target (recipient), age, weight, diseased state, cell state, and the like.
- DMSO Dimethyl sulfoxide
- serum albumin for the purpose of cell protection, antibiotics for the purpose of blocking bacterial contamination, and various components (vitamins for the purpose of cell activation, proliferation or differentiation induction, etc. , Cytokines, growth factors, steroids, etc.) may be included in the cell preparation of the present invention.
- other pharmaceutically acceptable ingredients for example, carriers, excipients, disintegrants, buffers, emulsifiers, suspensions, soothing agents, stabilizers, preservatives, preservatives, physiological saline, etc. You may make it contain in the cell formulation of this invention.
- Example 1 Cells As the human iPS cells, Windy (iPS-51) and FF-1 strains were used. Windy is expressed in human fetal lung fibroblast MRC-5, octamer binding protein 3/4 (OCT3 / 4), sex determining region Y-box 2 (SOX2), kruppel-like factor 4 (KLF4), v-myc myelocytomatosis viral Oncogene homolog (avian) (c-MYC) was introduced using a pantropic retrovirus vector, and a human ES cell-like colony was cloned and was provided by Dr. Akihiro Umezawa, National Center for Child Health and Development. Mouse fetal fibroblasts (MEF) were used as feeder cells. FF-1 stock was provided by FUJIFILM Corporation.
- MEF fetal fibroblasts
- FBS fetal bovine serum
- L-Glu 2 mmol / L L-glutamine
- NEAA non-essential amino acid
- 100 units / mL penicillin G 100 ⁇ g / mL Dulbecco's modified Eagle medium (DMEM) containing streptomycin was used.
- EDTA trypsin-ethylenediaminetetraacetic acid
- Cell Banker 1 was used as the MEF stock solution.
- Windy maintenance culture includes 20% knockout serum replacement (KSR), 0.8% NEAA, 2 mmol / L L-Glu, 0.1 mmol / L 2-mercaptoethanol (2-MeE), 5 ng / mL fibroblast proliferation DMEM Ham's F-12 (DMEM / F12) containing factor (FGF) 2 was used.
- the stripping solution is Dulbecco's phosphate buffered saline (PBS) containing 1 mg / mL collagenase IV, 0.25% trypsin, 20% KSR, 1 mmol / L calcium chloride, and the storage solution is for primate ES / iPS cells.
- PBS Dulbecco's phosphate buffered saline
- trypsin 20% KSR
- MTesR1 was used for maintenance culture of the FF-1 strain.
- Induction of differentiation of human iPS cells into intestinal epithelial cells is a state where the proportion of undifferentiated colonies in human iPS cells is about 70% of the culture dish. Started with.
- Windy is 2% FBS, 100 ng / mL Activin A for 2 days in Roswell Park Memorial Laboratory (RPMI) + Glutamax medium containing 0.5% FBS, 100 ng / mL Activin A, 100 Units / mL Penicillin G, 100 ⁇ g / mL Streptomycin
- RPMI + Glutamax medium containing 100 units / mL penicillin G and 100 ⁇ g / mL streptomycin for 1 day. Thereafter, differentiation was induced to intestinal stem cells by culturing in DMEM / F12 containing 2% FBS, 1% Glutamax, 250 ng / mL FGF2 for 4 days.
- Y-27632 Rho binding kinase inhibitor
- PD98059 (20 ⁇ mol / L), 5-aza-2′-deoxycytidine (5 ⁇ mol / L), A-83-01 (low molecular compounds previously discovered by the present inventors during differentiation induction, 0.5 ⁇ mol / L), 1 mmol / L 8-bromo-3 ', 5'-cyclic adenosine monophosphate (8-Br-cAMP), 0.1 or 0.5 mmol / L 3-isobutyl-1-methylxanthine (IBMX), 10 or 30 ⁇ mol / L forskolin was added, and the effect on differentiation induction into intestinal stem cells and intestinal epithelial cells was examined.
- IBMX isobutyl-1-methylxanthine
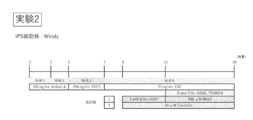
- Experiment 1 (FIG. 1) and Experiment 2 (FIG. 4) using Windy and Experiment 3 (FIG. 6), Experiment 4 (FIG. 9) and Experiment 5 (FIG. 11) using the FF-1 strain were set.
- the FF-1 strain in Experiment 3, it was cultured for 5 days (days 0 to 5) to induce differentiation into endoderm, and 4 days (days 5 to 9) to induce differentiation into intestinal stem cells. ) And 18 days (10th to 28th days) to induce differentiation into intestinal epithelial cells.
- RNA extraction Total ribonucleic acid (RNA) extraction
- RNA was extracted according to the attached manual of RNeasy (registered trademark) Mini Kit (Qiagen) after completion of induction of differentiation of human iPS cells.
- Real-Time RT-PCR Real-time reverse transcription polymerase chain reaction (Real-Time RT-PCR) Real-Time RT-PCR was performed using cDNA as a template using KAPA SYBR Fast qPCR Kit (Nippon Genetics). The operation followed the attached manual. As an endogenous control, hypoxanthine-guanine phosphoribosyltransferase (HPRT) was used to correct the measurement results.
- HPRT hypoxanthine-guanine phosphoribosyltransferase
- ABCB1 / MDR1 ATP-binding cassette transporter B1 / multidrug resistance protein 1
- CYP3A4 cytochrome P450 3A4
- FABP fatty acid binding protein 2
- PXR pregnane X receptor
- SLC5A1 / SGLT1 SLC (solute carrier) family member 5A1 / sodium conjugated glucose transporter 1: A glucose transporter expressed on the apical membrane side of the small intestine.
- SLC15A1 / PEPT1 (SLC (solute carrier) family member 15A1 / peptide transporter 1): expressed on the apical membrane side of the small intestine. Villin 1 is the main component of microvilli.
- CES2A1 Carboxylesterase 2A1.
- CES a hydrolase, has 1A and 2A1 isoforms. CES1A is expressed in the liver and CES2A1 is highly expressed in the small intestine.
- CYP3A4 and SGLT1 that are particularly important as intestinal epithelial cell markers by differentiating with forskolin alone compared to the differentiation induction method using 8-Br-cAMP and IBMX combined so far developed by the inventors Expression level was significantly increased (FIG. 5). Therefore, it was suggested that forskolin is extremely effective in promoting differentiation from human iPS cells to intestinal epithelial cells.
- CYP3A4 activity similar to that of human primary small intestinal cells was observed in the group differentiated into intestinal stem cell-like cells using FGF2 and differentiated into intestinal epithelial cells using 8-Br-cAMP and forskolin. (FIGS. 11 and 12). This suggests that forskolin may contribute not only to gene expression of intestinal markers but also to improvement of pharmacokinetic functions such as drug metabolizing enzyme activity.
- Example 2 Differentiation from human iPS cells to small intestinal epithelial cells Human iPS cells (FF-1) were treated with serum-free medium containing 100 ng / mL activin A for 24 hours to initiate differentiation. Thereafter, the cells were treated in a serum-free medium containing 100 ng / mL activin A, 2.5 ng / mL BMP4, 10 ng / mL VEGF and 5 ng / mL FGF2 for 144 hours to differentiate into endoderm.
- FF-1 Human iPS cells
- FF-1 Human iPS cells (FF-1) were treated with serum-free medium containing 100 ng / mL activin A for 24 hours to initiate differentiation. Thereafter, the cells were treated in a serum-free medium containing 100 ng / mL activin A, 2.5 ng / mL BMP4, 10 ng / mL VEGF and 5 ng / mL FGF2 for 144 hours to differentiate into endoderm.
- small intestinal stem cells by culturing in Advanced DMEM / F-12 containing 2% FBS, 1% GlutaMax, 100 units / mL penicillin G, 100 ⁇ g / mL streptomycin sulfate, 250 ng / mL FGF2 for 96 hours.
- Small intestinal stem cell-like cells treated with activin A and FGF2 were detached with Accutase and seeded on GFR Matrigel previously diluted 30-fold with human iPS medium.
- the cells were cultured for 19-23 days in Advanced DMEM / F-12 containing mL EGF and 30 ⁇ M forskolin. From 12-16 days before the end of differentiation, add PD98059 to 20 ⁇ M, 5-aza-2'-deoxycytidine (5-aza-2'-dC) to 5 ⁇ M, and A-83-01 to 0.5 ⁇ M. Differentiated into small intestinal epithelial cells.
- acebutolol, metformin, hydrochlorothiazide, sulpiride and lucifer yellow which are permeabilized by paracellular, cephalexin, lisinopril, ribavirin and enalapril, which are permeated by transporters, antipyrine and caffeine, CYP3A4, which are permeabilized by transcellular HBSS (pH 6.5) containing erythromycin, indinavir, midazolam, tacrolimus and verapamil, which are the substrates of the above, was added to the apical chamber and incubated at 37 ° C. for 60 minutes.
- each compound was 50 ⁇ M for lisinopril and caffeine, 50 ⁇ g / mL for lucifer yellow, and 10 ⁇ M for other compounds.
- HBSS pH 7.4 was added to the basal chamber. Samples were collected from the receiver chamber side every 15 minutes. The unchanged form was measured using UPLC-MS / MS. F a ⁇ F g in humans referred from an existing report (following References 1-6).
- Reference 1 Takenaka T, Harada N, Kuze J, Chiba M, Iwao T, Matsunaga T. Human small intestinal epithelial cells differentiated from adult intestinal stem cells as a novel system for predicting oral drug absorption in humans. Drug Metab Dispos. 42: 1947-54 (2014).
- Reference 2 Takenaka T, Harada N, Kuze J, Chiba M, Iwao T, Matsunaga T. Application of a Human Intestinal Epithelial Cell Monolayer to the Prediction of Oral Drug Absorption in Humans as a Superior Alternative to the Caco-2 Cell Monolayer. J Pharm Sci. 105: 915-924 (2016).
- Reference 3 Tachibana T, Kato M, Sugiyama Y.
- P (1) is a scaling factor.
- WinNonlin (Certara, USA) was used for nonlinear regression analysis.
- a correlation coefficient (R value) was calculated in order to evaluate the correlation between P app in human iPS cell-derived small intestinal epithelial cells and Caco-2 cells, and human F a ⁇ F g .
- the scaling factors in human iPS cell-derived small intestinal epithelial cells and Caco-2 cells were 0.531 ⁇ 0.083 and 3.243 ⁇ 0.992, respectively.
- P app of compounds that are transported by the compounds and transporters through the paracellular pathway as the value of F a ⁇ F g is increased in human iPS cell-derived small intestinal epithelial cells, became gradually larger value.
- P app showed almost the same value even for compounds having different F a and F g (FIG. 14).
- Correlation coefficient P app and F a ⁇ F g in the compound human iPS cell-derived small intestinal epithelial cells, except for the substrate of CYP3A4 is 0.9, and Caco-2 cells 0.56, better human iPS cell-derived small intestinal epithelial cells It was highly correlated with human F a and F g .
- Intestinal epithelial cell-like cells are useful as a model system for the small intestine, and can be used for absorption / metabolism / membrane permeability, induction of drug-metabolizing enzymes, induction of drug transporters, evaluation of toxicity, and the like.
- it is expected to be used as an active ingredient of cell preparations for treating various intestinal diseases or as a material for regenerative medicine.
Abstract
Description
[1]以下の工程(1)及び(2)を含む、多能性幹細胞を腸管上皮細胞へ分化誘導する方法:
(1)多能性幹細胞を腸管幹細胞様細胞へと分化させる工程;
(2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質を併用し、工程(1)で得られた腸管幹細胞様細胞を腸管上皮細胞様細胞へと分化させる工程。
[2]工程(1)が以下の工程(1-1)及び(1-2)からなる、[1]に記載の方法:
(1-1)多能性幹細胞を内胚葉様細胞へと分化させる工程;
(1-2)工程(1-1)で得られた内胚葉様細胞を腸管幹細胞様細胞へと分化させる工程。
[3]工程(2)における培養期間が7日間~40日間である、[1]又は[2]に記載の方法。
[4]工程(2)が、以下のA~Dのいずれかの培養工程を含む、[1]~[3]のいずれか一に記載の方法、
培養工程A:(a-1)EGF及びcAMP活性化物質の存在下での培養と、該培養の後に行われる、(a-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含む、
培養工程B:(b-1)EGFの存在下での培養と、該培養の後に行われる、(b-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質の存在下での培養を含む、
培養工程C:(c-1)EGF及びcAMP活性化物質の存在下での培養と、該培養の後に行われる、(c-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質の存在下での培養を含む、
培養工程D:(d-1)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質の存在下での培養を含む。
[5](a-1)の培養の期間は2日間~10日間であり、(a-2)の培養の期間は9日間~29日間であり、
(b-1)の培養の期間は2日間~10日間であり、(b-2)の培養の期間は9日間~19日間であり、
(c-1)の培養の期間は2日間~10日間であり、(c-2)の培養の期間は9日間~19日間であり、
(d-1)の培養の期間は15日間~25日間である、[4]に記載の方法。
[6]cAMP活性化物質がフォルスコリンである、[1]~[5]のいずれか一に記載の方法。
[7]工程(2)が、cAMPが細胞へ供給される条件及び/又はcAMP分解酵素阻害剤が存在する条件で行われる、[1]~[6]のいずれか一に記載の方法。
[8]cAMPが細胞へ供給される条件が、培地中に8-Br-cAMPが存在することである、[7]に記載の方法。
[9]cAMP分解酵素阻害剤がIBMXである、[7]又は[8]に記載の方法。
[10]培養工程Bが、(b-2)の培養の後に行われる、(b-3)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含み、
培養工程Cが、(c-2)の培養の後に行われる、(c-3)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含み、
培養工程Dが、(d-1)の培養の後に行われる、(d-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含む、[4]~[9]のいずれか一に記載の方法。
[11](b-3)の培養、(c-3)の培養、及び(d-2)の培養の期間は1日間~10日間である、[10]に記載の方法。
[12]MEK1阻害剤がPD98059であり、DNAメチル化阻害剤が5-アザ-2’-デオキシシチジンであり、TGFβ受容体阻害剤がA-83-01である、[1]~[11]のいずれか一に記載の方法。
[13]工程(1-1)における分化誘導因子としてアクチビンAを用いる、[2]~[12]のいずれか一に記載の方法。
[14]工程(1-2)における分化誘導因子としてFGF2又はGSK-3β阻害剤を用いる、[2]~[13]のいずれか一に記載の方法。
[15]多能性幹細胞が人工多能性幹細胞である、[1]~[14]のいずれか一に記載の方法。
[16]人工多能性幹細胞がヒト人工多能性幹細胞である、[15]に記載の方法。
[17][1]~[16]のいずれか一に記載の方法で得られた腸管上皮細胞様細胞。
[18][17]に記載の腸管上皮細胞様細胞を用いた、被検物質の体内動態又は毒性を評価する方法。
[19]前記体内動態が、代謝、吸収、排泄、薬物相互作用、薬物代謝酵素の誘導、又は薬物トランスポーターの誘導である、[18]に記載の方法。
[20]以下の工程(i)~(iii)を含む、[18]又は[19]に記載の方法:
(i)[17]に記載の腸管上皮細胞様細胞で構成された細胞層を用意する工程;
(ii)前記細胞層に被検物質を接触させる工程;
(iii)前記細胞層を透過した被検物質を定量し、被検物質の吸収性ないし膜透過性、薬物相互作用、薬物代謝酵素の誘導、薬物トランスポーターの誘導、又は毒性を評価する工程。
[21]以下の工程(I)及び(II)を含む、[18]又は[19]に記載の方法:
(I)[17]に記載の腸管上皮細胞様細胞に被検物質を接触させる工程;
(II)被検物質の代謝若しくは吸収、薬物相互作用、薬物代謝酵素の誘導、薬物トランスポーターの誘導、又は毒性を測定・評価する工程。
[22][17]に記載の腸管上皮細胞様細胞を含む、細胞製剤。
この工程では多能性幹細胞を培養し、腸管幹細胞様細胞へと分化させる。換言すれば、腸管幹細胞様細胞への分化を誘導する条件下で多能性幹細胞を培養する。多能性幹細胞が腸管幹細胞様細胞へ分化する限り、培養条件は特に限定されない。典型的には、多能性幹細胞が内胚葉様細胞を介して腸管幹細胞様細胞へと分化するように、以下で説明する2段階の分化誘導、即ち、多能性幹細胞の内胚葉様細胞への分化(工程(1-1))と、内胚葉様細胞の腸管幹細胞様細胞への分化(工程(1-2))を行う。
この工程では多能性幹細胞を培養し、内胚葉様細胞へと分化させる。換言すれば、内胚葉への分化を誘導する条件下で多能性幹細胞を培養する。多能性幹細胞が内胚葉様細胞に分化する限り、培養条件は特に限定されない。例えば、常法に従い、アクチビンAを添加した培地で培養する。この場合、培地中のアクチビンAの濃度を例えば10 ng/mL~200 ng/mL、好ましくは20 ng/mL~150 ng/mLとする。細胞の増殖率や維持等の観点から、培地に血清又は血清代替物(KnockOutTM Serum Replacement(KSR)など)を添加することが好ましい。血清はウシ胎仔血清に限られるものではなく、ヒト血清や羊血清等を用いることもできる。血清又は血清代替物の添加量は例えば0.1%(v/v)~10%(v/v)である。
BMP4、VEGF、およびFGF2の一以上を培地に添加し、内胚葉様細胞への分化の促進を図ってもよい。この場合、培地中のBMP4の濃度は例えば0.1 ng/mL~10 ng/mL、好ましくは1 ng/mL~5 ng/mLであり、培地中のVEGF の濃度は例えば0.5 ng/mL~100 ng/mL、好ましくは1 ng/mL~20 ng/mLであり、培地中のFGF2の濃度は例えば0.2 ng/mL~50 ng/mL、好ましくは0.5 ng/mL~10 ng/mLである 。
この工程では、工程(1-1)で得られた内胚葉様細胞を培養し、腸管幹細胞様細胞へと分化させる。換言すれば、腸管幹細胞への分化を誘導する条件下で内胚葉様細胞を培養する。内胚葉様細胞が腸管幹細胞様細胞へと分化する限り、培養条件は特に限定されない。好ましくは、FGF2(線維芽細胞増殖因子2)の存在下、又はGSK-3β阻害剤の存在下で培養を行う。FGF2としては好ましくはヒトFGF2(例えばヒト組換えFGF2)を用いる。
この工程では、MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質を併用し、工程(1)で得られた腸管幹細胞様細胞を腸管上皮細胞様細胞へと分化させる。本発明では、この分化誘導の際、cAMP活性化物質を使用することで、細胞内のcAMPレベルを積極的に上昇させる。「MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質を併用する」とは、工程(2)を構成する1又は2以上の培養を行うため、これら全ての化合物が必要とされることいい、これら全ての化合物が同時に使用されること、即ち、これら全ての化合物が添加された培地を用いた培養が行われることを必須の条件として要求するものではない。
<培養工程A>
培養工程Aでは、(a-1)EGF及び細胞内cAMP合成促進剤の存在下での培養と、当該培養の後に行われる、(a-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を行う。このように2段階の培養を行えば、腸管上皮細胞への分化促進、成熟化、機能獲得の効果が期待できる。(a-1)の培養の期間は例えば2日間~10日間、好ましくは4日間~8日間であり、(a-2)の培養の期間は例えば9日間~29日間、好ましくは7日間~27日間である。尚、特に説明しない事項(各培養に使用可能な化合物、各化合物の添加濃度等)については、上記の対応する説明が援用される。
培養工程Bでは、(b-1)EGFの存在下での培養と、当該培養の後に行われる、(b-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及び細胞内cAMP合成促進剤の存在下での培養を行う。このように2段階の培養を行えば、腸管上皮細胞への分化促進、成熟化、機能獲得の効果が期待できる。(b-1)の培養の期間は例えば2日間~10日間、好ましくは4日間~8日間であり、(b-2)の培養の期間は例えば9日間~19日間、好ましくは7日間~17日間である。尚、特に説明しない事項(各培養に使用可能な化合物、各化合物の添加濃度等)については、上記の対応する説明が援用される。
培養工程Cでは、(c-1)EGF及び細胞内cAMP合成促進剤の存在下での培養と、当該培養の後に行われる、(c-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及び細胞内cAMP合成促進剤の存在下での培養を行う。このように2段階の培養を行えば、腸管上皮細胞への分化促進、成熟化、機能獲得の効果が期待できる。(c-1)の培養の期間は例えば2日間~10日間、好ましくは4日間~8日間であり、(c-2)の培養の期間は例えば9日間~19日間、好ましくは7日間~17日間である。尚、特に説明しない事項(各培養に使用可能な化合物、各化合物の添加濃度等)については、上記の対応する説明が援用される。
培養工程Dでは、(d-1)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及び細胞内cAMP合成促進剤の存在下での培養を行う。この培養工程は、培養操作が簡便であること、腸管上皮細胞への分化に対してより効果的であること、化合物であるため安定した効果が期待できること等の点で特に有利である。(d-1)の培養の期間は例えば15日間~25日間、好ましくは17日間~23日間である。尚、特に説明しない事項(各培養に使用可能な化合物、各化合物の添加濃度等)については、上記の対応する説明が援用される。
生体の腸管上皮細胞により近い機能を示す細胞(腸管上皮細胞様細胞)を簡便且つ効率的に調製する方法の確立を目指し、以下の検討を行った。
1.方法
(1)細胞
ヒトiPS細胞はWindy(iPS-51)及びFF-1株を用いた。Windyは、ヒト胎児肺線維芽細胞MRC-5にoctamer binding protein 3/4(OCT3/4)、sex determining region Y-box 2(SOX2)、kruppel-like factor 4(KLF4)、v-myc myelocytomatosis viral oncogene homolog(avian)(c-MYC)を、パントロピックレトロウイルスベクターを用いて導入後、ヒトES細胞様コロニーをクローン化したものであり、国立成育医療研究センター梅澤明弘博士よりご供与いただいた。フィーダー細胞はマウス胎仔線維芽細胞(MEF)を使用した。FF-1株は富士フイルム株式会社より供与された。
MEFの培養には10%ウシ胎仔血清(FBS)、2 mmol/L L-グルタミン(L-Glu)、1%非必須アミノ酸(NEAA)、100 ユニット/mLペニシリンG、100μg/mLストレプトマイシンを含むダルベッコ改変イーグル培地(DMEM)を用いた。MEFの剥離液には0.05%トリプシン-エチレンジアミン四酢酸(EDTA)を、MEFの保存液にはセルバンカー1を用いた。Windyの維持培養には20%ノックアウト血清代替物(KSR)、0.8% NEAA、2 mmol/L L-Glu、0.1 mmol/L 2-メルカプトエタノール(2-MeE)、5 ng/mL線維芽細胞増殖因子(FGF)2を含むDMEM Ham’s F-12(DMEM/F12)を用いた。剥離液には1 mg/mLコラゲナーゼIV、0.25%トリプシン、20% KSR、1 mmol/L塩化カルシウムを含むダルベッコリン酸緩衝生理食塩水(PBS)を、保存液には霊長類ES/iPS細胞用凍結保存液を用いた。FF-1株の維持培養にはmTesR1を用いた。
WindyはマイトマイシンC処理を施したMEF(6×105 cells/100 mmディッシュ)上に播種し、5% CO2/95% air条件下CO2インキュベーター中37℃にて培養した。FF-1株はマトリゲルコートしたディッシュ上で培養を行った。ヒトiPS細胞の継代は、3~5日培養後、1:2~1:3のスプリット比で行った。ヒトiPS細胞は解凍48時間後に培地を交換し、それ以降は毎日交換した。
ヒトiPS細胞の腸管上皮細胞への分化誘導は、ヒトiPS細胞が培養ディッシュに対し、未分化コロニーの占める割合が約70%になった状態で開始した。Windyは0.5% FBS、100 ng/mLアクチビンA、100ユニット/mLペニシリンG、100μg/mLストレプトマイシンを含むロズウェルパーク記念研究所(RPMI)+Glutamax培地で2日間、2% FBS、100 ng/mLアクチビンA、100ユニット/mLペニシリンG、100μg/mLストレプトマイシンを含むRPMI+Glutamax培地で1日間培養することで内胚葉に分化誘導した。その後、2% FBS、1% Glutamax、250 ng/mL FGF2を含むDMEM/F12で4日間培養することで腸管幹細胞へ分化誘導した。この処理後、Y-27632(Rho結合キナーゼ阻害剤)を10 μmol/Lとなるように添加し、5% CO2/95% air条件下CO2インキュベーター中37℃にて60分間処理した細胞をアクターゼにて剥離し、あらかじめヒトiPS細胞用培地にて30倍に希釈した、成長因子を除去したマトリゲルにてコートした細胞培養用24ウェルプレートに播種した。その後、2% FBS、1% Glutamax、1% NEAA、2% B27 supplement、1% N2 supplement、100ユニット/mLペニシリンG、100μg/mLストレプトマイシン、20 ng/mL上皮細胞増殖因子(EGF)、10 μmol/L Y-27632を含むDMEM/F12で1日間、2% FBS、1% Glutamax、1% NEAA、2% B27 supplement、1% N2 supplement、100ユニット/mLペニシリンG、100μg/mLストレプトマイシン、20 ng/mL上皮細胞増殖因子(EGF)を含むDMEM/F12で18日間培養することで腸管上皮細胞へ分化誘導した。また、分化誘導の際に以前本発明者らが見出した低分子化合物であるPD98059(20 μmol/L)、5-アザ-2’-デオキシシチジン(5 μmol/L)、A-83-01(0.5 μmol/L)に加え、1 mmol/L 8-ブロモ-3’,5’-サイクリックアデノシン一リン酸(8-Br-cAMP)、0.1もしくは0.5 mmol/L 3-イソブチル-1-メチルキサンチン(IBMX)、10もしくは30 μmol/Lフォルスコリンを添加し、腸管幹細胞および腸管上皮細胞への分化誘導に及ぼす影響について検討した。
総RNAはヒトiPS細胞の分化誘導終了後、RNeasy(登録商標) Mini Kit(Qiagen)の添付マニュアルに従い抽出した。
相補的DNA(cDNA)の合成は、ReverTra Ace(登録商標) qPCR RT Kit(東洋紡株式会社)を使用した。操作は添付マニュアルに従った。
KAPA SYBR Fast qPCR Kit(日本ジェネティクス株式会社)を用い、cDNAを鋳型にしてReal-Time RT-PCRを行った。操作は添付マニュアルに従った。内在性コントロールとしてヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ(HPRT)を用い、測定結果を補正した。
分化誘導終了後、5 μmol/Lミダゾラムおよび10 μmol/L 7-ヒドロキシクマリンを含む培地(1% Glutamax、1% NEAA、1% N2 supplement、100 ユニット/mLペニシリンG、100 μg/mLストレプトマイシン、20 ng/mL EGFを含むDMEM/F12)で37℃にてインキュベーションし、24時間(実験3及び4)または2時間(実験5)経過後、培地をサンプリングした。代謝活性は、液体クロマトグラフィー-マススペクトロメーター(LC-MS/MS)を用いて測定した培地中の1-水酸化ミダゾラムもしくは7-ヒドロキシクマリングルクロニドの量より算出した。代謝実験終了後、タンパク定量を行い、代謝活性をタンパク量で補正した。
ABCB1/MDR1(ATP結合カセットトランスポーターB1/多剤耐性タンパク1):P糖タンパク質であり、排出トランスポーターとして機能する。
CYP3A4(シトクロムP450 3A4):小腸において主要な薬物代謝酵素である。
FABP(脂肪酸結合タンパク2):さまざまなサブタイプがあり、FABP2は腸型。
PXR(プレグナンX受容体):CYP3A4の発現や誘導に関与する。
SLC5A1/SGLT1(SLC(solute carrier)ファミリーメンバー5A1/ナトリウム共役型グルコーストランスポーター1):小腸の頂側膜側に発現しているグルコーストランスポーター。
SLC15A1/PEPT1(SLC(solute carrier)ファミリーメンバー15A1/ペプチドトランスポーター1):小腸の頂側膜側に発現している。
Villin 1(ビリン 1):微絨毛の主要な構成成分である。
CES2A1:カルボキシルエステラーゼ2A1。加水分解酵素であるCESには1Aと2A1のアイソフォームがあり、肝臓ではCES1Aの発現が、小腸ではCES2A1の発現が高い。
(1)腸管上皮細胞への分化誘導に対する効果の検討
分化開始後8日目以降にフォルスコリンを添加することで、各種腸管マーカーの遺伝子発現レベルの有意な上昇が認められた(図2、3)。フォルスコリンの腸管マーカー発現に対する効果は、これまでに発明者らが見出した8-Br-cAMPと類似した傾向を示したが、主要な薬物代謝酵素であるCYP3A4や、排出トランスポーターとして重要なABCB1/MDR1の発現に対するフォルスコリンの効果は8-Br-cAMPの効果を遙かに凌駕した。また、これまでに発明者らが開発した8-Br-cAMPとIBMXを併用した分化誘導法と比較して、フォルスコリン単独で分化することで、腸管上皮細胞マーカーとして特に重要であるCYP3A4およびSGLT1の発現レベルが有意に上昇した(図5)。したがって、フォルスコリンはヒトiPS細胞から腸管上皮細胞への分化促進に極めて有効であることが示唆された。
8-Br-cAMPおよびIBMXを用いて分化させた場合と比較して、8-Br-cAMPおよびフォルスコリンを用いて分化させることで、各種腸管マーカーの遺伝子発現レベルが有意に上昇するとともに(図7)、CYP3A4およびUGT活性が有意に上昇した(図8)。また、8-Br-cAMPに代え、フォルスコリンを分化誘導開始後12日目から長期間用いることで、さらに有意なCYP3A4活性の上昇が認められた(図10)。UGT活性においては、8-Br-cAMPおよびフォルスコリンを用いて分化させた群で最も高い代謝活性を示した(図10)。さらに、FGF2を用いて腸管幹細胞様細胞へ分化させ、8-Br-cAMPおよびフォルスコリンを用いて腸管上皮様細胞に分化させた群で、ヒト初代小腸細胞と同程度のCYP3A4活性が観察された(図11、図12)。このことから、フォルスコリンは腸管マーカーの遺伝子発現だけでなく、薬物代謝酵素活性などの薬物動態学的機能の向上にも寄与している可能性が考えられた。
以上の結果より、これまで以上に、より成熟した腸管上皮細胞様細胞をヒトiPS細胞から作製する方法を確立した。また、この細胞はCYP3A4などによる薬物代謝活性を十分に有しており、ヒト初代小腸細胞と同程度のCYP3A4活性を有しているものも見出された。薬物の消化管吸収試験系として現在汎用されているCaco-2細胞は薬物代謝活性の低さが問題となっていることから、本発明によって、よりヒト小腸に近い機能を有する細胞の作製が可能になるものと考えられる。
1.方法
(1)ヒトiPS細胞から小腸上皮細胞への分化
ヒトiPS細胞(FF-1)に 100 ng/mL アクチビンAを含む無血清培地を24時間処理し分化を開始した。その後、100 ng/mL アクチビンA、2.5 ng/mL BMP4、10 ng/mL VEGFおよび5 ng/mL FGF2を含む無血清培地中で144時間処理し内胚葉に分化させた。その後2% FBS、1% GlutaMax、100 units/mL ペニシリンG、100 μg/mL ストレプトマイシン硫酸塩、250 ng/mL FGF2を含むAdvanced DMEM/F-12で96時間培養することで小腸幹細胞に分化させた。アクチビン AおよびFGF2処理後の小腸幹細胞様細胞をAccutaseにて剥離し、あらかじめヒトiPS培地で30倍に希釈したGFR Matrigel上に播種した。播種後は2% FBS、0.1 mM NEAA、2 mM L-Glu、100 units/mL ペニシリン G、100 μg/mL ストレプトマイシン硫酸塩、2% B27 supplement、1% N2 supplement、1% HepExtend supplement、20 ng/mL EGFおよび30 μM フォルスコリンを含むAdvanced DMEM/F-12で19-23日間培養した。分化終了12-16日前からPD98059を20 μM、5-アザ-2’-デオキシシチジン (5-aza-2'-dC)を5 μM、A-83-01を0.5 μMになるように添加して小腸上皮細胞に分化させた。
Cell culture insert上に播種したヒトiPS細胞由来小腸上皮細胞およびCaco-2細胞のapical側のチャンバーにHBSS(pH 6.5)を、basal側のチャンバーにHBSS(pH 7.4)を添加し、37℃にて60分間プレインキュベートした。その後、パラセルラーで透過する化合物であるacebutolol、metformin、hydrochlorothiazide、sulpirideおよびlucifer yellow、トランスポーターで透過する化合物であるcephalexin、lisinopril、ribavirinおよびenalapril、トランスセルラーで透過する化合物であるantipyrineおよびcaffeine、CYP3A4の基質であるerythromycin、indinavir、midazolam、tacrolimusおよびverapamilを含むHBSS(pH 6.5)をapical側のチャンバーに添加し、37℃で60分間インキュベートした。各化合物の終濃度はlisinopril およびcaffeineは50 μM、lucifer yellowは50 μg/mL、その他の化合物は10 μMになるように添加した。またbasal側のチャンバーにはHBSS(pH 7.4)を添加した。15分毎にサンプルをレシーバーチャンバー側から回収した。未変化体を、UPLC-MS/MSを用いて測定した。ヒトにおけるFa・Fgは既存の報告から参照した(下記文献1~6)。
文献2:Takenaka T, Harada N, Kuze J, Chiba M, Iwao T, Matsunaga T. Application of a Human Intestinal Epithelial Cell Monolayer to the Prediction of Oral Drug Absorption in Humans as a Superior Alternative to the Caco-2 Cell Monolayer. J Pharm Sci. 105: 915-924 (2016).
文献3:Tachibana T, Kato M, Sugiyama Y. Prediction of nonlinear intestinal absorption of CYP3A4 and P-glycoprotein substrates from their in vitro Km values. Pharm Res. 29: 651-68 (2012).
文献4:Chong S, Dando SA, Soucek KM, Morrison RA. In vitro permeability through caco-2 cells is not quantitatively predictive of in vivo absorption for peptide-like drugs absorbed via the dipeptide transporter system. Pharm Res. 13: 120-3 (1996).
文献5:Zhu C, Jiang L, Chen TM, Hwang KK. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur J Med Chem. 37: 399-407 (2002).
文献6:Cheng KC, Li C, Uss AS. Prediction of oral drug absorption in humans--from cultured cell lines and experimental animals. Expert Opin Drug Metab Toxicol. 4: 581-90 (2008).

(1)ヒトiPS細胞株由来小腸上皮細胞の膜透過特性
ヒトにおけるFa・FgがヒトiPS細胞株由来小腸上皮細胞の膜透過試験結果のPappから予測可能かどうか検証するため、16個の化合物のPappとFa・Fgを比較した(表1)。ヒトiPS細胞由来小腸上皮細胞およびCaco-2細胞におけるCYP3A4の基質を除いた11化合物のPappとFa・Fgとの相関を非線形最小二乗法にて解析した結果、図14に示す回帰曲線が得られた。ヒトiPS細胞由来小腸上皮細胞およびCaco-2細胞におけるスケーリングファクターは、それぞれ0.531 ± 0.083、3.243 ± 0.992となった。細胞間隙経路を通る化合物やトランスポーターで輸送される化合物のPappは、ヒトiPS細胞由来小腸上皮細胞ではFa・Fgの値が大きくなるにつれ、徐々に大きな値になった。しかしながら、Caco-2細胞では異なるFa・Fgを持つ化合物でもPappはほぼ同じ値を示した(図14)。CYP3A4の基質を除いた11化合物でのPappとFa・Fgの相関係数はヒトiPS細胞由来小腸上皮細胞が0.9、Caco-2細胞が0.56と、ヒトiPS細胞由来小腸上皮細胞のほうがヒトのFa・Fgと高い相関を有していた。

膜透過試験の結果からCaco-2細胞では細胞間隙経路やトランスポーターで輸送されるFa・Fgが異なる様々な化合物が同程度の速度で透過されているが、ヒトiPS細胞由来小腸上皮細胞は化合物のFa・Fgの大きさに伴って異なる速度で透過していた(図14)。そのため、Caco-2細胞よりもヒトiPS細胞由来小腸上皮細胞でのPappがFa・Fgと高い相関を示した。この理由として、Caco-2細胞は強固なタイトジャンクションを有するため、細胞間隙経路を通る薬物の透過性が低いことや、トランスポーターの発現が低いことが原因であると考えられる。これらの結果から、ヒトiPS細胞由来小腸上皮細胞は、Caco-2細胞よりも細胞間隙経路を通過する基質およびトランスポーターに輸送される基質の膜透過の予測が優れていることが示唆された。
Claims (22)
- 以下の工程(1)及び(2)を含む、多能性幹細胞を腸管上皮細胞へ分化誘導する方法:
(1)多能性幹細胞を腸管幹細胞様細胞へと分化させる工程;
(2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質を併用し、工程(1)で得られた腸管幹細胞様細胞を腸管上皮細胞様細胞へと分化させる工程。 - 工程(1)が以下の工程(1-1)及び(1-2)からなる、請求項1に記載の方法:
(1-1)人工多能性幹細胞を内胚葉様細胞へと分化させる工程;
(1-2)工程(1-1)で得られた内胚葉様細胞を腸管幹細胞様細胞へと分化させる工程。 - 工程(2)における培養期間が7日間~40日間である、請求項1又は2に記載の方法。
- 工程(2)が、以下のA~Dのいずれかの培養工程を含む、請求項1~3のいずれか一項に記載の方法、
培養工程A:(a-1)EGF及びcAMP活性化物質の存在下での培養と、該培養の後に行われる、(a-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含む、
培養工程B:(b-1)EGFの存在下での培養と、該培養の後に行われる、(b-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質の存在下での培養を含む、
培養工程C:(c-1)EGF及びcAMP活性化物質の存在下での培養と、該培養の後に行われる、(c-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質の存在下での培養を含む、
培養工程D:(d-1)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤、EGF及びcAMP活性化物質の存在下での培養を含む。 - (a-1)の培養の期間は2日間~10日間であり、(a-2)の培養の期間は9日間~29日間であり、
(b-1)の培養の期間は2日間~10日間であり、(b-2)の培養の期間は9日間~19日間であり、
(c-1)の培養の期間は2日間~10日間であり、(c-2)の培養の期間は9日間~19日間であり、
(d-1)の培養の期間は15日間~25日間である、請求項4に記載の方法。 - cAMP活性化物質がフォルスコリンである、請求項1~5のいずれか一項に記載の方法。
- 工程(2)が、cAMPが細胞へ供給される条件及び/又はcAMP分解酵素阻害剤が存在する条件で行われる、請求項1~6のいずれか一項に記載の方法。
- cAMPが細胞へ供給される条件が、培地中に8-Br-cAMPが存在することである、請求項7に記載の方法。
- cAMP分解酵素阻害剤がIBMXである、請求項7又は8に記載の方法。
- 培養工程Bが、(b-2)の培養の後に行われる、(b-3)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含み、
培養工程Cが、(c-2)の培養の後に行われる、(c-3)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含み、
培養工程Dが、(d-1)の培養の後に行われる、(d-2)MEK1阻害剤、DNAメチル化阻害剤、TGFβ受容体阻害剤及びEGFの存在下での培養を含む、請求項4~9のいずれか一項に記載の方法。 - (b-3)の培養、(c-3)の培養、及び(d-2)の培養の期間は1日間~10日間である、請求項10に記載の方法。
- MEK1阻害剤がPD98059であり、DNAメチル化阻害剤が5-アザ-2’-デオキシシチジンであり、TGFβ受容体阻害剤がA-83-01である、請求項1~11のいずれか一項に記載の方法。
- 工程(1-1)における分化誘導因子としてアクチビンAを用いる、請求項2~12のいずれか一項に記載の方法。
- 工程(1-2)における分化誘導因子としてFGF2又はGSK-3β阻害剤を用いる、請求項2~13のいずれか一項に記載の方法。
- 多能性幹細胞が人工多能性幹細胞である、請求項1~14のいずれか一に記載の方法。
- 人工多能性幹細胞がヒト人工多能性幹細胞である、請求項15に記載の方法。
- 請求項1~16のいずれか一項に記載の方法で得られた腸管上皮細胞様細胞。
- 請求項17に記載の腸管上皮細胞様細胞を用いた、被検物質の体内動態又は毒性を評価する方法。
- 前記体内動態が、代謝、吸収、排泄、薬物相互作用、薬物代謝酵素の誘導、又は薬物トランスポーターの誘導である、請求項18に記載の方法。
- 以下の工程(i)~(iii)を含む、請求項18又は19に記載の方法:
(i)請求項17に記載の腸管上皮細胞様細胞で構成された細胞層を用意する工程;
(ii)前記細胞層に被検物質を接触させる工程;
(iii)前記細胞層を透過した被検物質を定量し、被検物質の吸収性ないし膜透過性、薬物相互作用、薬物代謝酵素の誘導、薬物トランスポーターの誘導、又は毒性を評価する工程。 - 以下の工程(I)及び(II)を含む、請求項18又は19に記載の方法:
(I)請求項17に記載の腸管上皮細胞様細胞に被検物質を接触させる工程;
(II)被検物質の代謝若しくは吸収、薬物相互作用、薬物代謝酵素の誘導、薬物トランスポーターの誘導、又は毒性を測定・評価する工程。 - 請求項17に記載の腸管上皮細胞様細胞を含む、細胞製剤。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980012236.8A CN111684058B (zh) | 2018-02-09 | 2019-02-08 | 从多能干细胞向肠上皮细胞的分化诱导方法 |
| EP19751317.9A EP3750985A4 (en) | 2018-02-09 | 2019-02-08 | METHOD FOR INDUCING THE DIFFERENTIATION OF PLURIPOTENT STEM CELLS IN INTESTINAL EPITHELIAL CELLS |
| CA3090473A CA3090473C (en) | 2018-02-09 | 2019-02-08 | Method for inducing differentiation of pluripotent stem cells into intestinal epithelial cells |
| KR1020207022847A KR102446763B1 (ko) | 2018-02-09 | 2019-02-08 | 다능성 줄기 세포로부터 장관 상피 세포로의 분화 유도 방법 |
| JP2019571161A JP7317317B2 (ja) | 2018-02-09 | 2019-02-08 | 多能性幹細胞から腸管上皮細胞への分化誘導方法 |
| US16/988,145 US20200370020A1 (en) | 2018-02-09 | 2020-08-07 | Method for inducing differentiation of pluripotent stem cells into intestinal epithelial cells |
| JP2022046424A JP7317323B2 (ja) | 2018-02-09 | 2022-03-23 | 多能性幹細胞から腸管上皮細胞への分化誘導方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-021545 | 2018-02-09 | ||
| JP2018021545 | 2018-02-09 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/988,145 Continuation US20200370020A1 (en) | 2018-02-09 | 2020-08-07 | Method for inducing differentiation of pluripotent stem cells into intestinal epithelial cells |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019156200A1 true WO2019156200A1 (ja) | 2019-08-15 |
Family
ID=67548287
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/004553 WO2019156200A1 (ja) | 2018-02-09 | 2019-02-08 | 多能性幹細胞から腸管上皮細胞への分化誘導方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20200370020A1 (ja) |
| EP (1) | EP3750985A4 (ja) |
| JP (2) | JP7317317B2 (ja) |
| KR (1) | KR102446763B1 (ja) |
| CN (1) | CN111684058B (ja) |
| CA (1) | CA3090473C (ja) |
| WO (1) | WO2019156200A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020095879A1 (ja) * | 2018-11-08 | 2020-05-14 | 公立大学法人名古屋市立大学 | 多能性幹細胞の腸管上皮細胞への分化誘導 |
| WO2020203712A1 (ja) | 2019-03-29 | 2020-10-08 | 富士フイルム株式会社 | 特定細胞に分化する能力を有する多能性幹細胞の製造方法およびその応用 |
| WO2021095848A1 (ja) | 2019-11-14 | 2021-05-20 | 富士フイルム株式会社 | 細胞の保存又は輸送器具及び細胞の輸送方法 |
| WO2021261540A1 (ja) | 2020-06-25 | 2021-12-30 | 富士フイルム株式会社 | 腸管上皮細胞の製造方法、及びその利用 |
| WO2023074648A1 (ja) * | 2021-10-25 | 2023-05-04 | 富士フイルム株式会社 | 腸内分泌細胞を製造する方法、多能性幹細胞由来の腸内分泌細胞、培地又は培地キット、およびそれらの利用 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111849860B (zh) * | 2020-06-01 | 2021-12-21 | 浙江大学 | 一种利用铁元素调控肠道干细胞分化的方法及应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008206510A (ja) | 2007-02-02 | 2008-09-11 | Institute Of Physical & Chemical Research | 腸管幹/前駆細胞の取得方法 |
| JP2012511935A (ja) | 2008-12-17 | 2012-05-31 | ザ スクリプス リサーチ インスティチュート | 幹細胞の作製と維持 |
| WO2014132933A1 (ja) | 2013-02-26 | 2014-09-04 | 公立大学法人名古屋市立大学 | 人工多能性幹細胞を腸管上皮細胞へ分化誘導する方法 |
| WO2014165663A1 (en) | 2013-04-03 | 2014-10-09 | Cellular Dynamics International, Inc. | Methods and compositions for culturing endoderm progenitor cells in suspension |
| WO2017154795A1 (ja) | 2016-03-08 | 2017-09-14 | 公立大学法人名古屋市立大学 | 人工多能性幹細胞の腸管上皮細胞への分化誘導 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3400286A1 (en) * | 2016-01-08 | 2018-11-14 | Massachusetts Institute Of Technology | Production of differentiated enteroendocrine cells and insulin producing cells |
-
2019
- 2019-02-08 CN CN201980012236.8A patent/CN111684058B/zh active Active
- 2019-02-08 CA CA3090473A patent/CA3090473C/en active Active
- 2019-02-08 EP EP19751317.9A patent/EP3750985A4/en active Pending
- 2019-02-08 WO PCT/JP2019/004553 patent/WO2019156200A1/ja unknown
- 2019-02-08 JP JP2019571161A patent/JP7317317B2/ja active Active
- 2019-02-08 KR KR1020207022847A patent/KR102446763B1/ko active IP Right Grant
-
2020
- 2020-08-07 US US16/988,145 patent/US20200370020A1/en active Pending
-
2022
- 2022-03-23 JP JP2022046424A patent/JP7317323B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008206510A (ja) | 2007-02-02 | 2008-09-11 | Institute Of Physical & Chemical Research | 腸管幹/前駆細胞の取得方法 |
| JP2012511935A (ja) | 2008-12-17 | 2012-05-31 | ザ スクリプス リサーチ インスティチュート | 幹細胞の作製と維持 |
| WO2014132933A1 (ja) | 2013-02-26 | 2014-09-04 | 公立大学法人名古屋市立大学 | 人工多能性幹細胞を腸管上皮細胞へ分化誘導する方法 |
| WO2014165663A1 (en) | 2013-04-03 | 2014-10-09 | Cellular Dynamics International, Inc. | Methods and compositions for culturing endoderm progenitor cells in suspension |
| WO2017154795A1 (ja) | 2016-03-08 | 2017-09-14 | 公立大学法人名古屋市立大学 | 人工多能性幹細胞の腸管上皮細胞への分化誘導 |
Non-Patent Citations (52)
| Title |
|---|
| "Mouse Embryo A Laboratory Manual", 1994, COLD SPRING HARBOR LABORATORY PRESS |
| BAGUISI ET AL., NATURE BIOTECHNOLOGY, vol. 17, 1999, pages 456 |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 1135306-29-4 |
| CHENG KCLI CUSS AS: "Prediction of oral drug absorption in humans-from cultured cell lines and experimental animals", EXPERT OPIN DRUG METAB TOXICOL., vol. 4, 2008, pages 581 - 90 |
| CHONG SDANDO SASOUCEK KMMORRISON RA: "In vitro permeability through caco-2 cells is not quantitatively predictive of in vivo absorption for peptide-like drugs absorbed via the dipeptide transporter system", PHARM RES., vol. 13, 1996, pages 120 - 3 |
| CHUNG Y., CELL STEM CELL, vol. 2, 2008, pages 113 - 117 |
| CIBELLI ET AL., SCIENCE, vol. 280, 1998, pages 1256 - 1147 |
| IRIYA ET AL., PROTEIN NUCLEIC ACID ENZYME, vol. 44, 1999, pages 892 |
| IWAO T., DRUG METAB DISPOS, vol. 43, no. 6, 2015, pages 603 - 610 |
| IWAO T., DRUG METAB PHARMACOKINET, vol. 29, no. 1, 2014, pages 44 - 51 |
| KABEYA T. ET AL.: "Cyclic AMP Signaling Promotes the Differentiation of Human Induced Pluripotent Stem Cells into Intestinal Epithelial Cells", DRUG METABOLISM AND DISPOSITION, vol. 46, no. 10, October 2018 (2018-10-01), pages 1411 - 1419, XP055629232, DOI: 10.1124/dmd.118.082123 * |
| KIM DKIM C HMOON J I ET AL., CELL STEM CELL, vol. 4, 2009, pages 472 - 476 |
| KIM ET AL., CELL STEM CELL, vol. 1, 2007, pages 346 - 352 |
| KIM J B ET AL., CELL, vol. 136, no. 3, 2009, pages 411 - 419 |
| KIM J B ET AL., NATURE, vol. 454, no. 7204, 2008, pages 646 - 650 |
| KLIMANSKAYAI. ET AL., NATURE, vol. 444, 2006, pages 481 - 485 |
| KODAMA N. ET AL.: "Characteristic Analysis of Intestinal Transport in Enterocyte-Like Cells Differentiated from Human Induced Pluripotent Stem Cells", DRUG METABOLISM AND DISPOSITION, vol. 44, no. 10, 2016, pages 1662 - 1667, XP055629227, DOI: 10.1124/dmd.116.069336 * |
| KODAMA N. ET AL.: "Inhibition of mitogen-activated protein kinase kinase, DNA methyltransferase, and transforming growth factor-beta promotes differentiation of human induced pluripotent stem cells into enterocytes", DRUG METABOLISM AND PHARMACOKINETICS, vol. 31, 2016, pages 193 - 200, XP055563526, doi:10.1016/j.dmpk.2016.02.002 * |
| MATSUI ET AL., CELL, vol. 70, 1992, pages 841 - 847 |
| MCCRACKEN K. W., NAT PROTOC., vol. 6, no. 12, 10 November 2011 (2011-11-10), pages 1920 - 8 |
| NAKAGAWA M ET AL., NAT. BIOTECHNOL., vol. 26, no. 11, 2008, pages 1269 - 1275 |
| NAKAJIMA ET AL., STEM CELLS, vol. 25, 2007, pages 983 - 985 |
| NATURE GENETICS, vol. 22, 1999, pages 127 |
| OGAKI S., SCI REP., vol. 5, 30 November 2015 (2015-11-30), pages 17297 |
| OGAKI S., STEM CELLS, vol. 31, no. 6, June 2013 (2013-06-01), pages 1086 - 1096 |
| OZAWA T., SCI REP., vol. 5, 12 November 2015 (2015-11-12), pages 16479 |
| PROC. NATL. ACAD. SCI. USA, vol. 96, 1999, pages 14984 |
| REVAZOVA ET AL., CLONING STEM CELLS, vol. 10, 2008, pages 11 - 24 |
| REVAZOVA ET AL., CLONING STEM CELLS, vol. 9, 2007, pages 432 - 449 |
| RIDEOUTIII ET AL., NATURE GENETICS, vol. 24, 2000, pages 109 |
| SHAMBLOTT ET AL., PROC. NATL. ACAD. SCI. USA, vol. 95, no. 23, 1998, pages 13726 - 13731 |
| SILVA J ET AL., PLOS. BIOL., vol. 6, no. 10, 2008, pages e253 |
| SPENCE J. R., NATURE, vol. 470, no. 7332, 3 February 2011 (2011-02-03), pages 105 - 109 |
| STADTFELD M ET AL., SCIENCE, vol. 322, no. 5903, 2008, pages 949 - 953 |
| STRELCHENKO N. ET AL., REPROD BIOMED ONLINE, vol. 9, 2004, pages 623 - 629 |
| TACHIBANA ET AL.: "Human Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer", CELL, 2013 |
| TACHIBANA TKATO MSUGIYAMA Y: "Prediction of nonlinear intestinal absorption of CYP3A4 and P-glycoprotein substrates from their in vitro Km values", PHARM RES., vol. 29, 2012, pages 651 - 68 |
| TAKAHASHI KYAMANAKA S, CELL, vol. 126, no. 4, 2006, pages 663 - 676 |
| TAKAHASHI, K ET AL., CELL, vol. 131, no. 5, 2007, pages 861 - 72 |
| TAKENAKA THARADA NKUZE JCHIBA MIWAO TMATSUNAGA T: "Application of a Human Intestinal Epithelial Cell Monolayer to the Prediction of Oral Drug Absorption in Humans as a Superior Alternative to the Caco-2 Cell Monolayer", J PHARM SCI., vol. 105, 2016, pages 915 - 924 |
| TAKENAKA THARADA NKUZE JCHIBA MIWAO TMATSUNAGA T: "Human small intestinal epithelial cells differentiated from adult intestinal stem cells as a novel system for predicting oral drug absorption in humans", DRUG METAB DISPOS, vol. 42, 2014, pages 1947 - 54 |
| TURNPENNY ET AL., STEM CELLS, vol. 21, no. 5, 2003, pages 598 - 609 |
| UEDA T., BIOCHEM BIOPHYS RES COMMUN., vol. 391, no. 1, 1 January 2010 (2010-01-01), pages 38 - 42 |
| WAKAYAMA ET AL., NATURE, vol. 394, 1998, pages 369 |
| WASSARMAN,P. M. ET AL., METHODS IN ENZYMOLOGY, vol. 365, 2003 |
| WILMUT ET AL., NATURE, vol. 385, 1997, pages 810 |
| WOLTJEN KMICHAEL I PMOHSENI P ET AL., NATURE, vol. 458, 2009, pages 771 - 775 |
| YU J ET AL., SCIENCE, vol. 318, no. 5858, 2007, pages 1917 - 1920 |
| YU JHU KSMUGA-OTTO KTIAN S ET AL., SCIENCE, vol. 324, 2009, pages 797 - 801 |
| YUSA KRAD RTAKEDA J ET AL., NAT METHODS, vol. 6, 2009, pages 363 - 369 |
| ZHANG X. ET AL., STEM CELLS, vol. 24, 2006, pages 2669 - 2676 |
| ZHU CJIANG LCHEN TMHWANG KK: "A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential", EUR J MED CHEM., vol. 37, 2002, pages 399 - 407 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020095879A1 (ja) * | 2018-11-08 | 2020-05-14 | 公立大学法人名古屋市立大学 | 多能性幹細胞の腸管上皮細胞への分化誘導 |
| JPWO2020095879A1 (ja) * | 2018-11-08 | 2021-09-30 | 公立大学法人名古屋市立大学 | 多能性幹細胞の腸管上皮細胞への分化誘導 |
| WO2020203712A1 (ja) | 2019-03-29 | 2020-10-08 | 富士フイルム株式会社 | 特定細胞に分化する能力を有する多能性幹細胞の製造方法およびその応用 |
| WO2021095848A1 (ja) | 2019-11-14 | 2021-05-20 | 富士フイルム株式会社 | 細胞の保存又は輸送器具及び細胞の輸送方法 |
| WO2021261540A1 (ja) | 2020-06-25 | 2021-12-30 | 富士フイルム株式会社 | 腸管上皮細胞の製造方法、及びその利用 |
| WO2023074648A1 (ja) * | 2021-10-25 | 2023-05-04 | 富士フイルム株式会社 | 腸内分泌細胞を製造する方法、多能性幹細胞由来の腸内分泌細胞、培地又は培地キット、およびそれらの利用 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20200106930A (ko) | 2020-09-15 |
| JP7317323B2 (ja) | 2023-07-31 |
| JPWO2019156200A1 (ja) | 2021-05-13 |
| KR102446763B1 (ko) | 2022-09-22 |
| CN111684058A (zh) | 2020-09-18 |
| EP3750985A1 (en) | 2020-12-16 |
| JP7317317B2 (ja) | 2023-07-31 |
| EP3750985A4 (en) | 2021-04-07 |
| JP2022075898A (ja) | 2022-05-18 |
| CA3090473A1 (en) | 2019-08-15 |
| US20200370020A1 (en) | 2020-11-26 |
| CA3090473C (en) | 2024-04-02 |
| CN111684058B (zh) | 2023-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7317323B2 (ja) | 多能性幹細胞から腸管上皮細胞への分化誘導方法 | |
| JP6949336B2 (ja) | 人工多能性幹細胞由来腸管幹細胞の維持培養 | |
| JP5938726B2 (ja) | 多能性幹細胞の分化誘導効率を改善するための方法及び培地 | |
| JP6296399B2 (ja) | 人工多能性幹細胞を腸管上皮細胞へ分化誘導する方法 | |
| JP6937036B2 (ja) | 人工多能性幹細胞の腸管上皮細胞への分化誘導 | |
| WO2022168908A1 (ja) | 多能性幹細胞由来の陰窩-絨毛様構造を有する腸管細胞の製造方法及びその用途 | |
| JP2022019411A (ja) | 人工多能性幹細胞由来の内胚葉細胞を製造する方法及びその利用 | |
| WO2021261540A1 (ja) | 腸管上皮細胞の製造方法、及びその利用 | |
| US20210147808A1 (en) | Method for producing intestinal epithelial cell and intestinal epithelial cell | |
| WO2023074648A1 (ja) | 腸内分泌細胞を製造する方法、多能性幹細胞由来の腸内分泌細胞、培地又は培地キット、およびそれらの利用 | |
| WO2020095879A1 (ja) | 多能性幹細胞の腸管上皮細胞への分化誘導 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19751317 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019571161 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3090473 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20207022847 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019751317 Country of ref document: EP Effective date: 20200909 |