WO2019143193A1 - 재조합 폴리펩타이드 생산용 n-말단 융합 파트너 및 이를 이용하여 재조합 폴리 펩타이드를 생산하는방법 - Google Patents
재조합 폴리펩타이드 생산용 n-말단 융합 파트너 및 이를 이용하여 재조합 폴리 펩타이드를 생산하는방법 Download PDFInfo
- Publication number
- WO2019143193A1 WO2019143193A1 PCT/KR2019/000782 KR2019000782W WO2019143193A1 WO 2019143193 A1 WO2019143193 A1 WO 2019143193A1 KR 2019000782 W KR2019000782 W KR 2019000782W WO 2019143193 A1 WO2019143193 A1 WO 2019143193A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino acid
- seq
- acid sequence
- polypeptide
- fusion
- Prior art date
Links
- 230000004927 fusion Effects 0.000 title claims abstract description 408
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 389
- 102000004196 processed proteins & peptides Human genes 0.000 title claims abstract description 365
- 229920001184 polypeptide Polymers 0.000 title claims abstract description 361
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 32
- 210000004027 cell Anatomy 0.000 claims abstract description 173
- 108091005804 Peptidases Proteins 0.000 claims abstract description 29
- 239000004365 Protease Substances 0.000 claims abstract description 23
- 235000001014 amino acid Nutrition 0.000 claims description 103
- 150000001413 amino acids Chemical group 0.000 claims description 98
- 229940024606 amino acid Drugs 0.000 claims description 93
- OGBMKVWORPGQRR-UMXFMPSGSA-N teriparatide Chemical compound C([C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](N)CO)C(C)C)[C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CNC=N1 OGBMKVWORPGQRR-UMXFMPSGSA-N 0.000 claims description 69
- 238000000034 method Methods 0.000 claims description 52
- 238000003776 cleavage reaction Methods 0.000 claims description 48
- 108010076818 TEV protease Proteins 0.000 claims description 44
- 230000007017 scission Effects 0.000 claims description 43
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 42
- 239000004475 Arginine Substances 0.000 claims description 39
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims description 39
- 229960003121 arginine Drugs 0.000 claims description 39
- 235000009697 arginine Nutrition 0.000 claims description 39
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 34
- 235000009582 asparagine Nutrition 0.000 claims description 34
- 229960001230 asparagine Drugs 0.000 claims description 34
- 235000014304 histidine Nutrition 0.000 claims description 34
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims description 34
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 34
- 229960000310 isoleucine Drugs 0.000 claims description 34
- 235000014705 isoleucine Nutrition 0.000 claims description 34
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 33
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 28
- 239000002773 nucleotide Substances 0.000 claims description 26
- 125000003729 nucleotide group Chemical group 0.000 claims description 26
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 22
- 235000004554 glutamine Nutrition 0.000 claims description 22
- 239000004471 Glycine Substances 0.000 claims description 21
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 20
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 claims description 20
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 20
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 20
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 claims description 20
- 235000004279 alanine Nutrition 0.000 claims description 20
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 claims description 19
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 19
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 19
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 19
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 19
- 229930182817 methionine Natural products 0.000 claims description 19
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 19
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 19
- 239000004474 valine Substances 0.000 claims description 19
- 102000004190 Enzymes Human genes 0.000 claims description 18
- 108090000790 Enzymes Proteins 0.000 claims description 18
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 claims description 18
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 18
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 18
- 239000004473 Threonine Substances 0.000 claims description 18
- 235000018417 cysteine Nutrition 0.000 claims description 18
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 18
- 235000004400 serine Nutrition 0.000 claims description 18
- 235000008521 threonine Nutrition 0.000 claims description 18
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 17
- 239000004472 Lysine Substances 0.000 claims description 17
- 239000013604 expression vector Substances 0.000 claims description 17
- 235000018977 lysine Nutrition 0.000 claims description 17
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 16
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 claims description 16
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 claims description 15
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 claims description 15
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 claims description 15
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims description 14
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 claims description 12
- 235000013922 glutamic acid Nutrition 0.000 claims description 12
- 239000004220 glutamic acid Substances 0.000 claims description 12
- -1 trimepodone Chemical compound 0.000 claims description 11
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 9
- 235000003704 aspartic acid Nutrition 0.000 claims description 9
- 229960005261 aspartic acid Drugs 0.000 claims description 9
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 claims description 9
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 8
- 238000012258 culturing Methods 0.000 claims description 7
- 101001135770 Homo sapiens Parathyroid hormone Proteins 0.000 claims description 6
- 101001135995 Homo sapiens Probable peptidyl-tRNA hydrolase Proteins 0.000 claims description 6
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 6
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 6
- 102000058004 human PTH Human genes 0.000 claims description 6
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 claims description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical class N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 4
- 210000004899 c-terminal region Anatomy 0.000 claims description 3
- 108010013369 Enteropeptidase Proteins 0.000 claims description 2
- 102100029727 Enteropeptidase Human genes 0.000 claims description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims description 2
- 102400000757 Ubiquitin Human genes 0.000 claims description 2
- 108090000848 Ubiquitin Proteins 0.000 claims description 2
- 230000012010 growth Effects 0.000 claims description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 claims 2
- 108090000604 Hydrolases Proteins 0.000 claims 1
- 102000004157 Hydrolases Human genes 0.000 claims 1
- 239000003292 glue Substances 0.000 claims 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 abstract description 33
- 102000035195 Peptidases Human genes 0.000 abstract description 27
- 238000005215 recombination Methods 0.000 abstract description 2
- 230000006798 recombination Effects 0.000 abstract description 2
- 210000003000 inclusion body Anatomy 0.000 abstract 1
- 125000003275 alpha amino acid group Chemical group 0.000 description 158
- 230000014509 gene expression Effects 0.000 description 118
- 229960005460 teriparatide Drugs 0.000 description 84
- 108090000623 proteins and genes Proteins 0.000 description 76
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 51
- 102000004169 proteins and genes Human genes 0.000 description 50
- 239000000523 sample Substances 0.000 description 47
- 241000588724 Escherichia coli Species 0.000 description 44
- 235000018102 proteins Nutrition 0.000 description 42
- 239000000872 buffer Substances 0.000 description 41
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 38
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 38
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 36
- 239000013613 expression plasmid Substances 0.000 description 36
- 238000004458 analytical method Methods 0.000 description 33
- 230000003287 optical effect Effects 0.000 description 32
- 239000000499 gel Substances 0.000 description 30
- 238000000746 purification Methods 0.000 description 26
- 239000002253 acid Substances 0.000 description 23
- 239000007983 Tris buffer Substances 0.000 description 21
- 239000002609 medium Substances 0.000 description 21
- 235000019419 proteases Nutrition 0.000 description 20
- 229910000162 sodium phosphate Inorganic materials 0.000 description 20
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 20
- 239000004202 carbamide Substances 0.000 description 19
- 230000006698 induction Effects 0.000 description 19
- 229930027917 kanamycin Natural products 0.000 description 19
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 19
- 229960000318 kanamycin Drugs 0.000 description 19
- 229930182823 kanamycin A Natural products 0.000 description 19
- 239000001488 sodium phosphate Substances 0.000 description 19
- 239000006228 supernatant Substances 0.000 description 19
- 239000012141 concentrate Substances 0.000 description 18
- 239000011780 sodium chloride Substances 0.000 description 18
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 18
- 239000012149 elution buffer Substances 0.000 description 17
- 239000013612 plasmid Substances 0.000 description 16
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 15
- 239000010426 asphalt Substances 0.000 description 14
- 230000003834 intracellular effect Effects 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 239000011537 solubilization buffer Substances 0.000 description 14
- 125000000539 amino acid group Chemical group 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 108091008146 restriction endonucleases Proteins 0.000 description 12
- 238000004587 chromatography analysis Methods 0.000 description 11
- 230000001965 increasing effect Effects 0.000 description 11
- 239000012722 SDS sample buffer Substances 0.000 description 10
- 239000012634 fragment Substances 0.000 description 10
- 229960002989 glutamic acid Drugs 0.000 description 10
- 239000011347 resin Substances 0.000 description 10
- 229920005989 resin Polymers 0.000 description 10
- 238000010217 densitometric analysis Methods 0.000 description 9
- 239000001963 growth medium Substances 0.000 description 9
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 9
- 239000003550 marker Substances 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 7
- YSDQQAXHVYUZIW-QCIJIYAXSA-N Liraglutide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC)C(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 YSDQQAXHVYUZIW-QCIJIYAXSA-N 0.000 description 7
- 238000002835 absorbance Methods 0.000 description 7
- 239000001110 calcium chloride Substances 0.000 description 7
- 229910001628 calcium chloride Inorganic materials 0.000 description 7
- 239000013592 cell lysate Substances 0.000 description 7
- 229960001174 ecallantide Drugs 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 210000004185 liver Anatomy 0.000 description 7
- 238000013365 molecular weight analysis method Methods 0.000 description 7
- 229960001267 nesiritide Drugs 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 108010019598 Liraglutide Proteins 0.000 description 6
- 239000007853 buffer solution Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 238000010835 comparative analysis Methods 0.000 description 6
- NKLPQNGYXWVELD-UHFFFAOYSA-M coomassie brilliant blue Chemical compound [Na+].C1=CC(OCC)=CC=C1NC1=CC=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C=CC(=CC=2)N(CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=C1 NKLPQNGYXWVELD-UHFFFAOYSA-M 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 6
- 229960002701 liraglutide Drugs 0.000 description 6
- 239000012528 membrane Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 238000010186 staining Methods 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 239000013598 vector Substances 0.000 description 6
- 108020004414 DNA Proteins 0.000 description 5
- 102400000326 Glucagon-like peptide 2 Human genes 0.000 description 5
- 101800000221 Glucagon-like peptide 2 Proteins 0.000 description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 5
- 230000015556 catabolic process Effects 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 239000013024 dilution buffer Substances 0.000 description 5
- TWSALRJGPBVBQU-PKQQPRCHSA-N glucagon-like peptide 2 Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(O)=O)[C@@H](C)CC)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=CC=C1 TWSALRJGPBVBQU-PKQQPRCHSA-N 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 150000007523 nucleic acids Chemical class 0.000 description 5
- 238000003752 polymerase chain reaction Methods 0.000 description 5
- 239000002243 precursor Substances 0.000 description 5
- 238000002741 site-directed mutagenesis Methods 0.000 description 5
- 238000005063 solubilization Methods 0.000 description 5
- 230000007928 solubilization Effects 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 241000894006 Bacteria Species 0.000 description 4
- 108020004705 Codon Proteins 0.000 description 4
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 4
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 230000003196 chaotropic effect Effects 0.000 description 4
- 238000005520 cutting process Methods 0.000 description 4
- 108010011867 ecallantide Proteins 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 239000006167 equilibration buffer Substances 0.000 description 4
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 4
- VBGWSQKGUZHFPS-VGMMZINCSA-N kalbitor Chemical compound C([C@H]1C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]2C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=3C=CC=CC=3)C(=O)N[C@H](C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)NCC(=O)NCC(=O)N[C@H]3CSSC[C@H](NC(=O)[C@@H]4CCCN4C(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CO)NC(=O)[C@H](CC=4NC=NC=4)NC(=O)[C@H](CCSC)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCC(O)=O)CSSC[C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC3=O)CSSC2)C(=O)N[C@@H]([C@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=2NC=NC=2)C(=O)N2CCC[C@H]2C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N1)[C@@H](C)CC)[C@H](C)O)=O)[C@@H](C)CC)C1=CC=CC=C1 VBGWSQKGUZHFPS-VGMMZINCSA-N 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- 239000008188 pellet Substances 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 230000001629 suppression Effects 0.000 description 4
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 3
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 3
- 108020005038 Terminator Codon Proteins 0.000 description 3
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 3
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 3
- 235000011130 ammonium sulphate Nutrition 0.000 description 3
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 3
- 239000012148 binding buffer Substances 0.000 description 3
- 239000003729 cation exchange resin Substances 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 230000007515 enzymatic degradation Effects 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 238000000855 fermentation Methods 0.000 description 3
- 230000004151 fermentation Effects 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 229960000789 guanidine hydrochloride Drugs 0.000 description 3
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 3
- 238000004255 ion exchange chromatography Methods 0.000 description 3
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 3
- 238000011218 seed culture Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 229910052709 silver Inorganic materials 0.000 description 3
- 239000004332 silver Substances 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- 101800000407 Brain natriuretic peptide 32 Proteins 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 108010011459 Exenatide Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102000007079 Peptide Fragments Human genes 0.000 description 2
- 108010033276 Peptide Fragments Proteins 0.000 description 2
- 208000035977 Rare disease Diseases 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 241000723792 Tobacco etch virus Species 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 238000005277 cation exchange chromatography Methods 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 238000012790 confirmation Methods 0.000 description 2
- 230000000593 degrading effect Effects 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 230000012202 endocytosis Effects 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- HPNRHPKXQZSDFX-OAQDCNSJSA-N nesiritide Chemical compound C([C@H]1C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)CNC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CO)C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1N=CNC=1)C(O)=O)=O)[C@@H](C)CC)C1=CC=CC=C1 HPNRHPKXQZSDFX-OAQDCNSJSA-N 0.000 description 2
- 230000011164 ossification Effects 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229920002704 polyhistidine Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 235000019833 protease Nutrition 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 102220195310 rs1057517867 Human genes 0.000 description 2
- 239000012723 sample buffer Substances 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000010421 standard material Substances 0.000 description 2
- 210000001685 thyroid gland Anatomy 0.000 description 2
- 239000013603 viral vector Substances 0.000 description 2
- 101150084750 1 gene Proteins 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- QFVHZQCOUORWEI-UHFFFAOYSA-N 4-[(4-anilino-5-sulfonaphthalen-1-yl)diazenyl]-5-hydroxynaphthalene-2,7-disulfonic acid Chemical compound C=12C(O)=CC(S(O)(=O)=O)=CC2=CC(S(O)(=O)=O)=CC=1N=NC(C1=CC=CC(=C11)S(O)(=O)=O)=CC=C1NC1=CC=CC=C1 QFVHZQCOUORWEI-UHFFFAOYSA-N 0.000 description 1
- 244000003416 Asparagus officinalis Species 0.000 description 1
- 235000005340 Asparagus officinalis Nutrition 0.000 description 1
- 206010061692 Benign muscle neoplasm Diseases 0.000 description 1
- 101710125089 Bindin Proteins 0.000 description 1
- 102400000967 Bradykinin Human genes 0.000 description 1
- 101800004538 Bradykinin Proteins 0.000 description 1
- 101000879203 Caenorhabditis elegans Small ubiquitin-related modifier Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000070928 Calligonum comosum Species 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 108010093668 Deubiquitinating Enzymes Proteins 0.000 description 1
- 102000001477 Deubiquitinating Enzymes Human genes 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010051425 Enterocutaneous fistula Diseases 0.000 description 1
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 1
- 238000012366 Fed-batch cultivation Methods 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- QXZGBUJJYSLZLT-UHFFFAOYSA-N H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH Natural products NC(N)=NCCCC(N)C(=O)N1CCCC1C(=O)N1C(C(=O)NCC(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CO)C(=O)N2C(CCC2)C(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CCCN=C(N)N)C(O)=O)CCC1 QXZGBUJJYSLZLT-UHFFFAOYSA-N 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 206010019860 Hereditary angioedema Diseases 0.000 description 1
- 208000013038 Hypocalcemia Diseases 0.000 description 1
- 208000000038 Hypoparathyroidism Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 1
- 208000008081 Intestinal Fistula Diseases 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 201000004458 Myoma Diseases 0.000 description 1
- KMAOMYOPEIRFLB-SFHVURJKSA-N N-Palmitoyl glutamic acid Chemical compound CCCCCCCCCCCCCCCC(=O)N[C@H](C(O)=O)CCC(O)=O KMAOMYOPEIRFLB-SFHVURJKSA-N 0.000 description 1
- 108020001621 Natriuretic Peptide Proteins 0.000 description 1
- 102000004571 Natriuretic peptide Human genes 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000051619 SUMO-1 Human genes 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 101000829189 Staphylococcus aureus Glutamyl endopeptidase Proteins 0.000 description 1
- 108010049264 Teriparatide Proteins 0.000 description 1
- JZRWCGZRTZMZEH-UHFFFAOYSA-N Thiamine Natural products CC1=C(CCO)SC=[N+]1CC1=CN=C(C)N=C1N JZRWCGZRTZMZEH-UHFFFAOYSA-N 0.000 description 1
- 102100036407 Thioredoxin Human genes 0.000 description 1
- 241000209140 Triticum Species 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 235000009754 Vitis X bourquina Nutrition 0.000 description 1
- 235000012333 Vitis X labruscana Nutrition 0.000 description 1
- 240000006365 Vitis vinifera Species 0.000 description 1
- 235000014787 Vitis vinifera Nutrition 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 210000004381 amniotic fluid Anatomy 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 108010062796 arginyllysine Proteins 0.000 description 1
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- QXZGBUJJYSLZLT-FDISYFBBSA-N bradykinin Chemical compound NC(=N)NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)CCC1 QXZGBUJJYSLZLT-FDISYFBBSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 230000001332 colony forming effect Effects 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000003898 enterocutaneous fistula Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 229960001519 exenatide Drugs 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- 210000003918 fraction a Anatomy 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 1
- 235000013882 gravy Nutrition 0.000 description 1
- 239000008062 guanidine hydrochloride buffer Substances 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000000705 hypocalcaemia Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000002608 insulinlike Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000007154 intracellular accumulation Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 101150011498 lad gene Proteins 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000013028 medium composition Substances 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- 239000000692 natriuretic peptide Substances 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003016 pheromone Substances 0.000 description 1
- 229920000724 poly(L-arginine) polymer Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 108010011110 polyarginine Proteins 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- GCYXWQUSHADNBF-AAEALURTSA-N preproglucagon 78-108 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 GCYXWQUSHADNBF-AAEALURTSA-N 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000012846 protein folding Effects 0.000 description 1
- 239000012521 purified sample Substances 0.000 description 1
- 238000004451 qualitative analysis Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 230000036647 reaction Effects 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012064 sodium phosphate buffer Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000007655 standard test method Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 239000013076 target substance Substances 0.000 description 1
- CILIXQOJUNDIDU-ASQIGDHWSA-N teduglutide Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(O)=O)[C@@H](C)CC)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=CC=C1 CILIXQOJUNDIDU-ASQIGDHWSA-N 0.000 description 1
- 235000019157 thiamine Nutrition 0.000 description 1
- KYMBYSLLVAOCFI-UHFFFAOYSA-N thiamine Chemical compound CC1=C(CCO)SCN1CC1=CN=C(C)N=C1N KYMBYSLLVAOCFI-UHFFFAOYSA-N 0.000 description 1
- 229960003495 thiamine Drugs 0.000 description 1
- 239000011721 thiamine Substances 0.000 description 1
- 108060008226 thioredoxin Proteins 0.000 description 1
- 229940094937 thioredoxin Drugs 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/65—Insulin-like growth factors, i.e. somatomedins, e.g. IGF-1, IGF-2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
- C07K14/4703—Inhibitors; Suppressors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/58—Atrial natriuretic factor complex; Atriopeptin; Atrial natriuretic peptide [ANP]; Cardionatrin; Cardiodilatin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/605—Glucagons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/62—Insulins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/635—Parathyroid hormone, i.e. parathormone; Parathyroid hormone-related peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/08—Linear peptides containing only normal peptide links having 12 to 20 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/20—Fusion polypeptide containing a tag with affinity for a non-protein ligand
- C07K2319/21—Fusion polypeptide containing a tag with affinity for a non-protein ligand containing a His-tag
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/35—Fusion polypeptide containing a fusion for enhanced stability/folding during expression, e.g. fusions with chaperones or thioredoxin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/50—Fusion polypeptide containing protease site
Definitions
- the present invention relates to a novel N- terminal fusion partner, a fusion polypeptide comprising said fusion partner and a desired polypeptide, and a method for producing said polypeptide by using said fusion polypeptide.
- a target protein (target polypeptide) is produced by cloning a nucleic acid of various target proteins into an expression vector to obtain a recombinant expression vector, transforming it into a suitable host cell and culturing it.
- the whole or a part of the target protein is degraded by the degrading enzyme (for example, protease or peptidase) inherent to the target protein, and the yield of the target protein is lowered.
- the degrading enzyme for example, protease or peptidase
- N- terminal fusion partner consisting of a novel amino acid sequence for producing a recombinant polypeptide.
- 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 is another object of the present invention is to provide a fusion polypeptide comprising said first terminal fusion partner with the objective polypeptide.
- Yet another object of the present invention is to provide a nucleotide encoding the fusion polypeptide, an expression vector comprising the nucleotide, and a 5 host cell comprising the expression vector.
- an aspect of the present invention is to provide a fusion partner target polypeptide comprising an amino acid sequence represented by the following general formula 1 , and a fusion polypeptide comprising a linker between the fusion partner and the target polypeptide
- the peptide is provided :
- 25 is an integer of 0 or 1 .
- Another aspect of the present invention provides a nucleotide encoding the fusion polypeptide, an expression vector comprising the nucleotide, and a host cell comprising the expression vector.
- Another aspect of the present invention is a method for culturing a host cell comprising culturing the host cell
- the novel fusion partner according to the present invention it is possible to improve the yield of the target polypeptide (recombinant polypeptide) compared to the conventional fusion partner.
- the target polypeptide having the amino terminal The fusion polypeptide comprising the fusion partner according to the present invention is induced in the host cell in the form of an intracellular form 10, and thus is useful as a protease or the like of a host cell It is possible to provide a method for producing a recombinant peptide having improved stability and yield as compared with the case of using a conventional fusion partner.
- Figure 1 shows the results of analysis of the total cell fraction of 1-34 fusion polypeptides expressed in silver colony-forming E. coli (Lane marker protein, lane 1: suppression 1-34 (strain No. 01), lane 2:? (3 ⁇ 4) 7 - 1 - 1? 1 - 34 (strain number? 0002), lane 3:? Lane 4:? 643 Inner-to-34 (Bacteria No. 20 ? 0004)).
- 3 ⁇ 4 is also produced from recombinant E. coli with the fed-batch culture? Oh, my 5 - is the result of analysis after sampling the 30 states 1-34 hourly. 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 4 is expressed in recombinant E. coli? 5 (span 2-7) my - !!! 3 TM 1-34 Fused Poly
- FIG. 5 is a graph showing the results of a 1/3-fold change in the second or third amino acid residues of the intramuscular subunit (.gamma.) Between Yak and Myungjang with isoleucine (I), asparagine (arginine 00, aspartic acid (I)
- Figure 83 is in the insoluble fraction in Example 7 through a chromatographic ⁇ -? Is a result of purifying the fusion polypeptide 1 3 ⁇ 41-34 solid line in (chromatogram, broken lines, dotted lines absorbance at one wavelength in each of 280, the conductivity ( ≪ / RTI > indicating the proportion of elution buffer).
- the ceramics were purified through chromatography (77 ⁇ -1 -1 ? 1111-34 fusion polypep
- Lanes 2 to 4 passage liquid fraction
- lanes 8 to 11 eluent fraction
- the arrows represent Examples 7-11 - 1 1? 13 ⁇ 41-34 fusion polypeptides.
- FIG. 9 ( 3) is the result of purifying the ( .15 internal-1 1? 1111-34 fusion polypeptide in the insoluble fraction through chromatography (the solid line, dashed line and dotted line in the chromatogram show absorbance at 280 and 1 wavelength, Conductivity ( «wave, representing the ratio of elution buffer).
- Bar 3 represents the fusion polypeptide of 1-34.
- the concentration of the insoluble fraction in the insoluble fraction is measured by chromatography .
- chromatography See solid line, dashed line and dotted line in the chromatogram.
- conductivity refers to the ratio of (conduct ivi y t), the elution buffer).
- FIG. 10B shows the result of SDS-PAGE analysis of the PG43-H6TEV-hPTH1 -34 fusion polypeptide purified through chromatography (lane M: marker protein, lane s: five samples before purification, lane 2 to 4: Fraction, lanes 8 to 11: eluate fraction).
- the arrow indicates the PG43-H6TEV-hPTHI-34 fusion polypeptide.
- FIG. 11 shows the result of cleavage of the fusion polypeptide of each purified sample using TEV protease and analysis of the fraction by SDS-PAGE (lane M: marker protein, lane C: sample not treated with TEY protease, lane T : Sample treated with TEV protease, 10 lane 1: PG07-H6TEV-hPTHl-34, lane 2: PG15-H6TEV-hPTHl-34, lane 3: PG43- H6TEV-hPTHl-34).
- FIG. 12 shows the result of cleavage of the purified PG15-H6TEV-hPTHl-34 fusion polypeptide using TEV protease and the fraction was analyzed by S-PAGE (lane M: marker protein, lane C: TEV protease , Lane T: a sample in which 15 TEV proteases were treated).
- FIG. 13A shows the results of separating PG15-H6TEV and hPTHI-34 from the PG15-H6TEV-hPTH1-34 fusion polypeptide using isoelectric point difference (the solid line, the broken line, and the dotted line in the chromatogram are 280 nm wavelength absorbance, conductivity (conductivit y), represents the proportion of the elution buffer) from the.
- Figure 14a shows the results of separating PG15-H6TEV and hPTHl-34 from the PG15-H6TEV-hPTHl-34 fusion polypeptide using an average hydrophobicity difference. (Chromat 25 rams show solid and dotted lines at 280 nm wavelengths And the ratio of elution buffer).
- FIG. 14B shows the result of S-PAGE analysis of the fraction of PG15-H6TEV-hPTH1-34 fusion polypeptide isolated using an average hydrophobicity difference (lane M: marker protein, lane s: pre- 5: 1 st peak fractions, lanes 1-7: 2 nd peak fractions).
- FIG. 17 is a graph 5 showing the retention time and purity of the recombinant suppression 1-34 according to the present invention in Reference Example 1 -34 of the United States Pharmacopoeia (this 1-34 identification standard test method).
- Soluble fraction lane I: insoluble fraction, lane 5:? 029 my-0-28 mycelium (strain No.? 09), lane 6:? 036 my- 043 My _____ 1 3 - 281? (Strain number (; 011)).
- FIG. 22 is 043- ⁇ 1 - My kkyo 13-1 ⁇ ( ⁇ 281? (I), asparagine) and arginine (10, asphalt acid ([)) in the second or third amino acid residue of the amino acid sequence of SEQ ID NO:
- Fig. 23 shows the results of? 643- Suppression 1 - 012-11 (281? M? 43 is substituted with isoleucine (I), asparagine) and arginine (10, aspartic acid). 3 -
- FIG. 25 shows the chromatograms of? 043? - (! -1 secreted 8 Seed 5 This is the result of purification of the fusion polypeptide (solid line in chromatogram, dotted lines indicate the absorbance at 280 ⁇ wavelength and the elution buffer ratio, respectively).
- FIG. 26 shows the chromatograms of? 643 internal (-) - 11 (281? Fused poly
- Figure 27 is purified by the TEV protease 043 I -? ⁇ 1 3 -1 ⁇ 81 ? fusion
- Figure 28 is a kkyo purified according to the invention will tanaen or the result of measuring a molecular weight of 281?.
- Figure 30 Total cell fraction of E. coli Escherichia coli separated into soluble and insoluble fractions
- Fig. 35 shows the result of the Sho-paulo analysis of the? 043 internal-0? _2 fusion polypeptide purified through chromatography (lane 1-marker protein, pre-lane retinas, liver: 1 to 5: eluate fraction). The arrow indicates the? 643 internal-0 null -2 fusion polypeptide.
- FIG. 37 shows the results of measuring the molecular weight of the purified (in-situ) 2 (according to the present invention).
- Figure 38 shows the effect of the ecarantide fusion polypeptides expressed in recombinant E. coli
- Fig. 39 shows the results of analysis of the total cell fraction of the ecartant fusion polypeptides expressed in recombinant E. coli as soluble and insoluble fractions, followed by deficient lakes
- Figure 41 shows the expression of nasiridate fusion polypeptides expressed in recombinant E. coli
- Lane I insoluble fraction
- lane 1 ⁇ 1 -1 1? 13 ⁇ 41-84 (strain No. 0027)
- lane 2 ? (77-stage liver lysate No. 28)
- lane 3 - Large 1-84 (strain number
- FIG. 45 shows the results of the analysis of (? 07 internal-1? 111-84 fusion polypeptide
- liver, liver: passage liquid fraction, lane 1: 5: effluent fraction). What is the arrow? Yes 7 My 61 -1 1? RTI ID 0.0 > 1-84 < / RTI > fusion polypeptides.
- Lane 0 sample not treated with protease
- lane I sample treated with protease 30
- 2019/143193 1 »(1 ⁇ 1 will say that shows the result of measuring the molecular weight of the purified 13 ⁇ 41-84 ⁇ 2019/000782 Figure 47 in accordance with the invention eunbon.
- Figure 48 is a schematic representation of the structure of each of the fusion polypeptides expressed in the strains (001,? 0003, PG031,? 32 and? 033). Best Mode for Carrying Out the Invention
- One embodiment of the present invention provides a fusion partner termination polypeptide comprising an amino acid sequence represented by the following general formula 1 and a fusion polypeptide comprising a linker between the fusion partner and the polypeptide of interest:
- 331 to 336 are, independently from each other, isoleucine (11 I), glycine 1 ⁇ 21, G), alanine (not show), proline (0, I 3), valine, V), leucine (1 11, or methionine ,
- amino acid sequence is 1 to 36 amino acids starting from the amino acid at position 1 of the amino acid sequence represented by SEQ ID NO: 666,
- the above 331 to 336 show that, in a state in which they are poisonously accumulated together with isoleucine (11 I), proline ( 0 ), leucine ( 11 11 ), asparagine (show II), arginine Bar 10 and asphaltic acid 3 I).
- the terminal fusion partner may be composed of 7 amino acids.
- the amino acid sequence of SEQ ID NO: 666 is 1, 2, 3, 4, 5, 6, 7 , 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 dog, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35,
- n is an integer of 1, it may be 8, 15, 22, 29 or 36 amino acids starting from the amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: 666.
- the N- terminal fusion partner according to the present invention is a peptide having 7 to 43 amino acids and having a relatively small molecular weight.
- the target polypeptide such as hPTH 1-34 is produced by using this peptide, it is possible to obtain a better yield than hPTH 1-34 in a case, for example, it has been schematically indicated in the following table 1 the rate of hPTH 1-34 in the recombinant fusion polypeptide.
- the conventional fusion partners have a ratio of? 1-34 in the fusion polypeptide of only 8% to 29%
- the fusion partner according to the present invention has a? 1-34 in the fusion fusion polypeptide Is found to be in the range of 37% to 62% .
- 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 also fusion partner in accordance with the present invention can be a fusion polypeptide can be accumulated in a high concentration into the host cell as insoluble in FOCE induces insoluble expression of the fusion polypeptide. Therefore, there is an advantage in that it is easy to obtain a five-purpose polypeptide having a high yield in a host cell such as Escherichia coli (Myoma 11 ) in which all or part of the polypeptide may be degraded or cleaved by a protease and a peptidase.
- Myoma 11 Escherichia coli
- the " end fusion partner " may include an amino acid sequence represented by the following general formula ( 2 ).
- 3 31 is an isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine, tryptophan, asparagine, serine, threonine, cysteine, glutamine, arginine, lysine, histidine,
- the ⁇ is 1 to 36 amino acids starting from 15th amino acid in position 1 of the amino acid sequence shown in SEQ ID NO: 666,
- the above-mentioned 3 may be selected from the group consisting of isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine and trimodocyte .
- isoleucine (I 6 , I) asparagine,
- the terminal fusion partner may be composed of 7 amino acids.
- the II is an integer of 1, wherein ⁇ is 1 starting from amino acid position 1 in the amino acid sequence shown in SEQ ID NO: 666 25, 2, 3, 4, 5, 6, 7 Dogs, 8, 9, 10, 11, 12,
- the formula-terminal fusion partner consisting of the amino acid sequence represented by 2 is the amino acid sequence of SEQ ID NO: 8, 30, 52, 74, 96 or 118 Lt; / RTI >
- 331 is asparagine, serine, threonine, cysteine,
- the amino acid sequence represented by the general formula (2) consisting of 10-terminal fusion partner is the number of days will be made of the amino acid sequence of SEQ ID NO: 10, 32, 54, 76, 98, or 120.
- 331 may be an asphaltic acid or glutamic acid.
- the first terminal fusion partner consisting of the amino acid sequence represented by the general formula 2 can 15 may be made of the amino acid sequence of SEQ ID NO: 11, 33, 55, 77, 99 or 121.
- terminal fusion partner may include an amino acid sequence represented by the following general formula (3).
- Table 332 shows the results of the determination of the amino acid sequence of isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine, trimepodone, asparagine, serine, threonine, cysteine, glutamine, arginine, lysine, histidine,
- amino acid sequence is 1 to 36 amino acids starting from the 25 amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: 666,
- the terminal fusion partner comprises seven 2019/143193 1 »(1 ⁇ 1 may be made of amino ⁇ 2019/000782.
- the 2 is 1, 2, 3, 4, 5, 6, 7 starting from the amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: 666 , 8, 9, 10, 11, 12,
- II is an integer of 1, it may be 8, 15, 22, 29 or 36 amino acids starting from the amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: 666.
- the third size 2 may be selected from isoleucine, glycine, alanine, proline, valine, leucine, methionine 10, phenylalanine, tyrosine, and trim the group consisting of tryptophan.
- the N-terminal fusion partner consisting of the amino acid sequence represented by the general formula 3 may be an amino acid sequence of SEQ ID NO: 9, 31, 53, 75, 97 or 119.
- 332 may be selected from the group consisting of asparagine, serine, threonine, cysteine and glutamine.
- the terminal fusion partner consisting of the amino acid sequence represented by the general formula 3 may be an amino acid sequence of SEQ ID NO: 12 , 34, 56, 78, 100 or 122 .
- the 332 can be 20 selected from the group consisting of arginine, lysine and histidine.
- the terminal fusion partner consisting of the amino acid sequence represented by the general formula 3 may be made of the number of days of the amino acid sequence of SEQ ID NO: 13, 35, 57, 79, 101 or 123.
- 332 may be an asphaltic acid or glutamic acid.
- the I-terminal fusion consisting of the amino acid sequence represented by the general formula 3 partner may be one consisting of SEQ ID NO: 25, 14, 36, 58, 80, 102 or 124 amino acid sequence.
- the I-terminal fusion partner may include an amino acid sequence represented by the following general formula ( 4 ).
- 3 33 is an isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine tyrosine, trimriptan, asparagine serine, threonine, cysteine, glutamine arginine, lysine, histidine, asphalt acid,
- ⁇ is 1 to 36 amino acids starting from amino acid position 1 in the amino acid sequence shown in SEQ ID NO: 666,
- 333 may be selected from the group consisting of isoleucine glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine,
- the terminal fusion partner may be composed of 7 amino acids.
- the above-mentioned 15 is an amino acid sequence having 1, 2, 3, 4, 5, 6, or 7 starting from amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: Eight, nine, ten, eleven, twelve, twelve, thirteen, fourteen, fifteen, sixteen, seventeen, eighteenth, nineteen, twenty, twenty- 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35,
- It can be 36 amino acids. Specifically, when II is an integer of 1, it may be 8, 15, 22, 29 or 36 amino acids starting from the amino acid at position 1 of the amino acid sequence shown in SEQ ID NO:
- the -terminal fusion partner consisting of the amino acid sequence represented by the general formula 4 may be an amino acid sequence of SEQ ID NO: 15, 37, 59, 81, 103 or 125.
- the above-mentioned 333 may be selected from the group consisting of asparagine, serine, threonine, cysteine and glutamine.
- the terminal fusion partner comprising the amino acid sequence represented by Formula 4 may be an amino acid sequence of SEQ ID NO: 16, 38, 60, 82, 104 or 126 .
- the above-mentioned 333 can be obtained from the group consisting of arginine, lysine and histidine
- the amino acid sequence represented by the general formula 4 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 consisting terminal fusion partner is the number of days will be made of the amino acid sequence of SEQ ID NO: 9, 31, 53, 75, 97 or 119.
- the above-mentioned 3 may be an asphaltic acid or a glutamic acid.
- the 1-terminal fusion partner consisting of the amino acid sequence represented by the general formula 4 may be an amino acid sequence consisting of 5, 39, 61, 83, 105 or 127 of SEQ ID NO:
- the fusion partner may include an amino acid sequence represented by the following general formula ( 5 ).
- 3 34 is an isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine, trimepodone, asparagine, serine, threonine, cysteine, glutamine, arginine, lysine, histidine,
- the ⁇ is 1 to 36 amino acids starting from amino acid position 1 in the amino acid sequence shown in SEQ ID NO: 666,
- the 334 may be selected from the 20 isoleucine, glycine, alanine, proline, valine, leucine, methionine, the group consisting of phenylalanine, tyrosine and tryptophan.
- the 331 may be selected from the group consisting of isoleucine (I 1 1), asparagine, arginine (, 10 and 31 for aspartic acid), I)).
- the I-terminal fusion partner may be composed of 7 amino acids.
- the above-mentioned II is an integer of 1
- the above-mentioned 25 is the amino acid sequence of 1, 2, 3, 4, 5, 6, 7 , 8, 9, 10, 11, 12, 13, 71], 14, 711, 15, 71], 16, 71], one hanging, 18, 71], 19 wae, 2 why, 21 7 1], 22 7 11, 23 7], 24 7 1], 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 Or 36 amino acids.
- II is an integer of 1, it means that from the amino acid at position 1 of the amino acid sequence represented by SEQ ID NO: 66 Is 1 ⁇ 1 ⁇ 8, 15, 22, 29 or 36 amino acids days starting 2019/000782: 2019/143193 1 »(.
- the terminal fusion partner consisting of the amino acid sequence represented by the general formula 5 may be an amino acid sequence of SEQ ID NO: 8, 40, 62, 84, 106 or 128.
- the above-mentioned 334 may be selected from the group consisting of asparagine, serine, threonine, cysteine and glutamine. In one embodiment,
- the chuckling 4 may be 10 selected from the group consisting of arginine, lysine and histidine.
- the -terminal fusion partner consisting of the amino acid sequence represented by the general formula 5 may be an amino acid sequence of SEQ ID NO: 20 , 42 , 64 , 86 , 108 or 130 .
- 334 may be an asphaltic acid or glutamic acid. In one embodiment,
- the 1-terminal fusion partner may include an amino acid sequence represented by the following general formula ( 6 ).
- 3 35 is an isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine, trimepodone, asparagine, serine, threonine, cysteine, glutamine, arginine, lysine, histidine,
- the ⁇ is 1 to 36 amino acids starting from amino acid position 1 in the amino acid sequence shown in SEQ ID NO: 666,
- 335 may be selected from the group consisting of isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine,
- the silver isoleucine (11 I), asparagine, 2019/143193 1 »(1 ⁇ 1 may be selected from ⁇ 2019/000782 arginine (I, 10, and aspartic acid 3 is the group consisting of.
- the -terminal fusion partner may be composed of 7 amino acids. Further, when the II is an integer of 1, wherein ⁇ is 1 starting from amino acid position 1 in the amino acid sequence shown in SEQ ID NO: 666, two, three, four, five, six, seven , 8, 9, 10, 11, 12,
- II is an integer of 1, it may be 8, 15, 22, 29 or 36 amino acids starting from the amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: 666.
- the -terminal fusion partner consisting of the amino acid sequence represented by the general formula 6 may be composed of the amino acid sequence of SEQ ID NO: 22, 44, 66, 88, 110 or 132.
- the 3- fold 5 may be selected from the group consisting of asparagine, serine, threonine, cysteine, and glutamine.
- the terminal fusion partner comprising the amino acid sequence represented by the general formula (6) may be an amino acid sequence of SEQ ID NO: 23, 45, 67, 89, 111 or 135.
- the 335 may be selected from the group consisting of arginine, lysine, and histidine.
- the terminal fusion partner consisting of the amino acid sequence represented by Formula 6 may be an amino acid sequence of SEQ ID NO: 24, 46, 68, 90, 112 or 134.
- 335 may be an asphaltic acid or glutamic acid.
- the I-terminal fusion partner consisting of the amino acid sequence represented by the general formula (6) may be one comprising the amino acid sequence of SEQ ID NO: 25, 47, 69, 91, 113 or 135.
- the terminal fusion partner may include an amino acid sequence represented by the following general formula (7).
- 3 36 is an isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine trimopeptide, asparagine serine, threonine cysteine glutamine, arginine lysine, histidine, asphalt acid or glutamic acid,
- the 336 may be selected from the 10 isoleucine, glycine, alanine, proline, valine, leucine, methionine, phenylalanine, tyrosine, and the group consisting of tryptophan.
- 331 can be selected from the group consisting of isoleucine (11 I), asparagine (show, arginine (10 and asphalt acid (Show 31 ), I)
- the terminal fusion partner may be composed of 7 amino acids.
- the above-mentioned 15 is 1, 2, 3, 4, 5, 6, 7 starting from the amino acid at position 1 of the amino acid sequence shown in SEQ ID NO: , 8, 9, 10, 11, 12,
- It can be 36 amino acids. Specifically, when the II is an integer of 1, wherein 1 is 8, 15, 22, 29 or 36 amino acids, the number of days starting from the amino acid in position 1 of the amino acid sequence shown in 20 SEQ ID NO: 666.
- the one- terminal fusion partner consisting of the amino acid sequence represented by the general formula 7 may be an amino acid sequence of SEQ ID NO: 26, 48, 70, 92, 114 or 136
- 25 , 336 may be selected from the group consisting of asparagine, serine, threonine, cysteine and glutamine. In one embodiment,
- 336 is selected from the group consisting of arginine, lysine and histidine
- the amino acid sequence represented by the general formula ( 7 ) 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 consisting terminal fusion partner is the number of days will be made of the amino acid sequence of SEQ ID NO: 28, 50, 72, 94, 116 or 138.
- the 336 may be an asphaltic acid or a glutamic acid.
- the I-terminal fusion partner comprising the amino acid sequence represented by the general formula (7) may comprise the amino acid sequence of SEQ ID NO: 29, 51, 73, 95,
- the terminal fusion partner may be composed of 7 amino acids.
- an I terminal fusion partner consisting of 7 amino acids is named as 7 " Respectively.
- 10 may be 8, 15, 22, 29 or 36 amino acids starting from the amino acid at position 1 of the amino acid sequence of SEQ ID NO: 666 when II is an integer of 1.
- the terminal fusion partner may be composed of 15, 22, 29, 36 or 43 amino acids.
- the I-terminal fusion partner is a peptide having 7 to 43 amino acids, and may be composed of 7 to 20 consecutive amino acids at the terminal of SEQ ID NO: 119.
- the fusion partner may be composed of one amino acid sequence.
- the number of amino acids in the fusion partner can be adjusted depending on the characteristics of the polypeptide of interest. For example, seven , eight ,
- the terminal fusion partner is selected from the group consisting of SEQ ID NOS: 9, 31,
- Yet another aspect of the present invention provides a fusion fusion polypeptide comprising a novel terminal fusion partner, a desired polypeptide, and a linker between the terminal fusion partner and the desired polypeptide as described above.
- the linker may include an affinity tag.
- an "affinity tag” may be used for various purposes as a peptide or nucleic acid sequence that can be introduced into a recombinant fusion polypeptide or a nucleic acid encoding the same, and may be, for example, to enhance the purification efficiency of a polypeptide of interest.
- Affinity tags that may be used can be any suitable material known in the art to be used according to the desired 5 bars.
- the affinity tag used in the present invention may be a polyhistidine tag (SEQ ID NO: 7 or 8 ), a polylysine tag (SEQ ID NO: 9 or 10 ) or a polyarginine tag (SEQ ID NO: 11 or 12 ).
- the linker may comprise a protease recognition sequence.
- the protease is a proteolytic enzyme that recognizes a specific amino acid sequence and cleaves a peptide bond between the peptide 10 binding in the recognized sequence or the first amino acid of the polypeptide fused with the last amino acid of the sequence.
- the linker having the protease recognition sequence By including the linker having the protease recognition sequence, the amino terminal (including the affinity tag when the affinity tag is used) containing the cleavage enzyme recognition sequence and the one terminal of the objective 15 polypeptide are separated during purification of the polypeptide at the final stage The desired polypeptide can be recovered.
- the protease recognition sequence may be a recognition sequence selected from the group consisting of tobacco etch virus protease recognition sequence enterokinase recognition sequence, ubiquitin hydrolase recognition sequence factor purine, and combinations thereof.
- the protease recognition sequence May include any one of the amino acid sequences of SEQ ID NOS: 146 to 20 150.
- " objective polypeptide & quot ; as used herein refers to a polypeptide to be obtained through a recombinant production system.
- the objective polypeptide can be protected from degradation by enzymes in the host cell by not only enhancing the expression level through fusion with a -terminal fusion partner according to the present invention but also accumulating 25 in the cell in the form of an insoluble intracellular form,
- the target polypeptide may include any one of the amino acid sequences of SEQ ID NOS: 18 to 27.
- the target polypeptide has an amino acid sequence of 2 to 3 and 15 to 3, , 2.51 out 3 to 14 ⁇ ), 3 to 13 ⁇ , 3.51 out 3 to 12 suppress 3, 4 ⁇ ) 3 to 11 ⁇ ) 3 .
- the target polypeptide is human parathyroid hormone 1-340? TM 1-34), human parathyroid hormone I-84 (hPTH 1-84), glucagon-like peptide-GLP- 1), lira glut ide precursor peptide, exenatide, (IGF-1), glucagon-like peptide-2 (GLP-2), tedu glut ide , ecal lantide,
- hPTH 1-34 Human Parathyroid Hormone 1_34
- hPTH 1-34 Human Parathyroid Hormone 1_34
- aa 115 amino acids secreted by the thyroid gland
- peptides secreted into the blood after the removal of the pro peptide 10 act to increase calcium concentration in the blood and stimulate bone formation.
- hPTH 1-34 is a peptide having 34 amino acids at the amino terminal portion of human parathyroid hormone and may be referred to as Teri paratide .
- the hPTH 1-34 polypeptide may be composed of the amino acid sequence of SEQ ID NO: 151, and the 15 amino acid sequence may be encoded by the nucleotide sequence of SEQ ID NO: 292.
- the human parathyroid hormone I-84 is a peptide having 84 amino acids derived from the pre-pro-peptide of 115 amino acids (aa) secreted by the thyroid gland. And it is known to stimulate bone formation.
- hPTH 1-84 are commonly used for the treatment of rare diseases 20 hypocalcemia (h ypocalcemia) or hypoparathyroidism (h ypoparathyroidi sm).
- the hPTH 1-84 polypeptide may comprise the amino acid sequence of SEQ ID NO: 628, and the amino acid sequence may be encoded by the nucleotide sequence of SEQ ID NO: 633 .
- the target polypeptide may be composed of 25 amino acid sequences of the amino acid sequence of SEQ ID NO: 151, 340, 341, 484, 485, 628, 638, 642 and 652.
- the glucagon-like peptide-1 is a polypeptide consisting of 31 amino acids.
- liraglutide is an analogue thereof, N-Palmitoyl-L-glutamic 30 (GLP-1) in which the 28th lysine of GLP-1 is replaced by arginine (K28R) and the amino group of the 20th lysine residue is composed of palmitoyl and glutamic acid acid.
- Lilaglutide is a type 2 diabetes or In general, liraglutide produces a liraglutide precursor peptide (GLP-1K28R) in which N- Palmitoyl- L-glutamic acid is not bound, and N- It can be produced by the process of binding palmitoyl-L-glutamic acid (Dunweber, Jensen et al. 2007).
- GLP-1K28R liraglutide precursor peptide
- N- Palmitoyl- L-glutamic acid N- Palmitoyl- L-glutamic acid is not bound
- N- It can be produced by the process of binding palmitoyl-L-glutamic acid (Dunweber, Jensen et al. 2007).
- the GLP-1 polypeptide may comprise the amino acid sequence of SEQ ID NO: 340 and the amino acid sequence may be encoded by the nucleotide sequence of SEQ ID NO: 475 .
- the liraglutide precursor peptide may be composed of the amino acid sequence of SEQ ID NO: 341 , and the amino acid sequence may be encoded by the nucleotide sequence of SEQ ID NO: 476 .
- the glucagon-like peptide- 2 (GLP-2) is a polypeptide consisting of 33 amino acids.
- GLP-2 is a form in which the second alanine of GLP-2 is replaced by glycine (A2G) as its analogue. It can be used as a treatment for rare bowel syndrome, chemotherapy-induced diarrhea and enterocutaneous fistula.
- the GLP-2 polypeptide may comprise the amino acid sequence of SEQ ID NO: 484 and the amino acid sequence may be the nucleotide sequence of SEQ ID NO: 619 .
- the above-mentioned edentulutide polypeptide may be composed of the amino acid sequence of SEQ ID NO: 485 , and the amino acid sequence may be encoded by the nucleotide sequence of SEQ ID NO: 620 .
- the calantide is a polypeptide consisting of 60 amino acids, which can inhibit human plasma calicrin, resulting in conversion of high molecular weight calicaine to Bradykinin It acts to inhibit.
- Calantide can be used as a treatment for hereditary angioedema, a rare disease.
- the calantide polypeptide may comprise the amino acid sequence of SEQ ID NO: 42 , and the amino acid sequence may be encoded by the nucleotide sequence of SEQ ID NO: 647 .
- the nisylideide is a polypeptide composed of 32 amino acids and is a secreted B-type sodium natriuretic peptide of human ventricular myocardium.
- Nercilideide can be used as a remedy for congestive heart failure.
- 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 polypeptide may be the amino acid sequence of SEQ ID NO: 652, the amino acid sequence can be encoded by the nucleotide sequence of SEQ ID NO: 657.
- Exenatide polypeptide may be made to the amino acid sequence of SEQ ID NO: 638 and the amino acid sequences can be coded by the nucleotide sequence of SEQ ID NO: 5 639, and the insulin-like growth factor-1 (101 ⁇ -1) a poly peptides may be achieved in the amino acid sequence of SEQ ID NO: 640, the amino acid sequence can be encoded by the nucleotide sequence of SEQ ID NO: 641.
- the fusion polypeptide according to the present invention has an isoelectric point of 10 different from that of the fusion polypeptide comprising the amino acid sequence of SEQ ID NO: 1 and the polypeptide of interest, so that the polypeptide of interest can be easily purified with high purity.
- the value of the terminal fusion partner having the amino acid sequence of SEQ ID NOS: 8 to 139 of the present invention may be 9.5 to 10.5 .
- the values of the terminal fusion partners having the amino acid sequences of SEQ ID NOS: 9, 15, 31, 53, 75, 97 or 119 may be 9.52, 11.72, 10.27, 10.27, 10.43, 10.42, respectively.
- the ? 1 values of the liraglutide precursor peptide tadglutide and ecarantid nasirilide were 8.29 , 9.10 , 5.53 , 4.17 , 5.58 , and 10.95, respectively. That is, the 1 1 values of the fusion partner and the fusion partners containing the pl value of the 20- purpose polypeptide are substantially different from each other. Therefore, the purification of the target polypeptide from the fusion partner can be easily performed by a method such as ion exchange chromatography or isoelectric point precipitation using a method such as ion exchange chromatography or isoelectric point precipitation.
- novel fusion polypeptide comprising the fusion partner, a linker and the object polypeptide of SEQ ID NO: 160 to 291, 343 to 474, 487 to 618, 630 to 632, 644 to 646 and 654 to 656 Or an amino acid sequence of any one of amino acid sequences.
- Another aspect of the invention is to provide 30 nucleotides encoding the fusion polypeptides described above, such as from 160 to 2 91 , 343 to 474, 487 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 to 618, 630 to 632, 644 to 646 and 654 to 656 may be of any of the coding amino acid sequence, wherein the oligonucleotide is SEQ ID NO: 294, 295, 478 to 483 , 621 to 627 , 635 to 637, 649 to 651, and 659 to 661 , respectively.
- vector " can be introduced into a host cell and recombined and inserted into a host cell genome, or the vector is understood as a nucleic acid means comprising a nucleotide sequence that can be spontaneously replicated as an episome.
- the vector includes a linear nucleic acid plasmid, 10 phagemid, a cosmid, a vector, a viral vector and analogues thereof. Examples of viral vectors include, but are not limited to, retroviruses, adenoviruses, and adeno-associated viruses.
- the plasmid may comprise a selectable marker, such as an antibiotic resistance gene, and the host cell harboring the plasmid may be cultured under selective conditions.
- host cell " refers to a prokaryotic or eukaryotic cell into which a recombinant expression vector may be introduced.
- transduction " as used herein, (E. G., A vector) into the cell.
- the host cell may be transformed to include a nucleotide encoding the fusion polypeptide of the present invention and used for expression and / or secretion of the desired polypeptide.
- Preferred host cells that can be used in the present invention are E. coli cells, hybridoma cells ⁇ ) myeloma cells, 293 cells, Chinese hamster ovary cells ((3 ⁇ 4 () 0 611) 3 ⁇ 41 cells, human amniotic fluid-derived cell 0 of immortality: 3 ⁇ 4 ⁇ 611) Or 0 < / RTI > cells. 25 for an example used to express the fusion polypeptides in this invention the host strain is E. coli 21 (3), wherein the genes and methods for their use are well known in the art.
- Another aspect of the invention (3) culturing the aforementioned host cell, (step of purifying the fusion polypeptide expressed in the host cell, and ((: the cutting the purified enzyme and incubated 30 fusion polypeptide To recover the desired polypeptide
- the method comprising the steps of: (a) preparing a recombinant polypeptide of interest;
- a host cell comprising an expression vector having a nucleotide encoding the fusion polypeptide of the present invention is cultured.
- the host cells can be cultured in any fermentation manner, for example, batch, fed-batch, semi-continuous and continuous fermentation methods can be used.
- the fermentation medium may be selected from complex or restricted media.
- a restriction medium is selected. Restricted media can be supplemented with low levels of amino acids, vitamins such as thiamine or other ingredients.
- the production of fusion polypeptides can be accomplished in a fermenter culture.
- a fermenter culture For example, it can be cultured at 37 ° C in a fermentor containing 2 L of restriction medium and maintained at pH 6.8 by addition of hydrochloric acid or ammonia.
- the dissolved oxygen may be maintained in excess over the injection of pure oxygen according to the stirring speed and the air flow 15 (air f low ra te) increases within the fermentor, or need.
- the injection solution containing the glucose or glycerol for cell culture to culture the cells in a high concentration in a fermentor feeding so lut ion) can be delivered to the culture medium.
- the target culture for induction fused by the addition of IPTG Expression of the polypeptide may be initiated.
- Optimum expression conditions can be determined by varying the optical density, IPTG concentration, pH, temperature, and dissolved oxygen level of each cell during induction.
- the optical density of cells at the time of induction can be varied in the range of A 600 30 to 300 .
- IPTG concentration is 1.0 mM to 0.01 mM to 25 range, the pH is 5.
- the culture from the fermenter is centrifuged to collect the cells and the cell pellet can be frozen at a temperature of -80 ° C.
- the culture sample can be analyzed by 30 methods such as SDS-PAGE for the analysis of the expression level of the recombinant fusion polypeptide. 2019/143193 1 » (: 1 ⁇ ⁇ 2019/000782
- the expression can be converted to the final concentration of 1 ⁇ ( ⁇ reulyak 0.01 to about 1.0 was added to the culture.
- the culture After addition of 5 inducing agent, the culture can be incubated for a period of time, for example, about 12 hours, during which the recombinant protein is expressed. After adding the inducing agent, the culture can be cultured for about 4 to 48 hours.
- the cell culture medium can be centrifuged to remove the medium (supernatant) in which the cells are not present and to recover the cells.
- the cell culture was 12,000 ⁇ 10 to 30 minutes (4 []), centrifuged under the conditions to remove the supernatant to obtain an insoluble fraction.
- the cell is a high pressure mechanical cell disruption device
- microfluidizer 0 0 £ 111 ⁇ 261 ⁇ ) to destroy it.
- the resuspended cells may be destroyed, for example, using sonication.
- Any of the methods well known in the art for dissolving cells can be used to recover intracellular accumulations of intracellular cells.
- chemical and / or enzymatic cytolytic reagents such as cell wall lysis enzymes ( 26 ) and may be used.
- step (k) the fusion polypeptide expressed in the host cell cultured in step (k) is purified.
- the insoluble fraction mainly contains a fusion polypeptide expressed in the form of an insoluble intracellular form.
- Solubilization of the intrinsic solids present in the insoluble fraction can be carried out under denaturing conditions involving chaotropic agents.
- the effervescent solubilization conditions can include the use of a buffer containing 25 chaotropic agents, and the effervescent solubilization buffer can comprise chaotropic agent urea or guanidine hydrochloride, sodium phosphate or tris buffer, and sodium chloride.
- the solubilization buffer comprises imidazole.
- the intracellular solubilization buffer contained 4
- 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 within FOCE solubilization buffer is from 5 to 100 of sodium phosphate or tris (1 to 7 ⁇
- 1 ⁇ (: I FOCE solubilization buffer for may include imidazole from 0 to 50.
- I FOCE solubilization buffer 81 elements, 20 5-tris 500 NaCl 50 imidazole, including the reproduction of the insoluble fraction obtained by centrifugation of the dissolved cell-suspended by fusion of water insoluble fraction polypeptide to 1 ⁇ 7.4 You can solubilize the porcine.
- polypeptides or polypeptides of interest can be isolated or purified from other proteins and cell debris by size exclusion anion or cation exchange, hydrophobic interaction, or affinity chromatography methods.
- the fusion polypeptide of the present invention may be a polyhistidine tag ( 6 -
- the proportion of the elution buffer (8 elements, 20 tris, 500 25 sodium chloride, 500 imidazole, 1 out 7.4 ) was stepwise increased to 100% to elute the fusion polypeptide bound to the column and the fraction was collected can do.
- the purified polypeptide is cultured with the cleavage enzyme by the above-mentioned method to recover the desired polypeptide.
- fusion polypeptide can be suitably cleaved by a cleavage enzyme
- fusion polypeptide can be released in a suitable form.
- Purified fusion Since 8 fragments are added to the polypeptide fractions, it is preferable to dilute the fractions so that the urea concentration is 1 M using a dilution buffer ( 20 mM Tris, pH 7.4) to prevent denaturation of the cleavage enzyme.
- the fusion polypeptide can be diluted to a urea concentration of 1 M after purification and can be present in a buffer containing 20 mM Tris, pH 7.4, 1 M urea, 62.5 mM sodium chloride, 62.5 mM 5 imidazole.
- the recombinant fusion polypeptide is reacted with a cleavage enzyme to cleave the amino terminus fusion partner and the polypeptide of interest with an affinity tag and a cleavage enzyme recognition sequence.
- a cleavage enzyme to cleave the amino terminus fusion partner and the polypeptide of interest with an affinity tag and a cleavage enzyme recognition sequence.
- Methods for protease cleavage are well known in the art and described in literature Any suitable method may be used.
- the T EV protease is added to the fusion polypeptide 10 diluted to an urea concentration of 1 after purification to a final concentration of 500, and the cleavage reaction is allowed to proceed at room temperature for 6 hours or more.
- about 60% to about 100% of the recombinant fusion polypeptide is cleaved by TEV protease.
- the yield of the recombinant fusion polypeptide or the desired polypeptide is known in the art, for example, SDS-PAGE - can be determined by the (so dium dodecyl sul fate polyacrylamide B gel electrophoresis) or Western blotting (Wes tern blot) analysis .
- a gel (gel) electricity by SDS-PAGE gel is stained (staini ng), discoloration (destaining), through a digital imaging process, it is possible to approximate quantitative and qualitative analysis of the recombinant fusion polypeptide or the desired polypeptide.
- concentration of the purified fusion polypeptide or polypeptide of interest is also important. Also, the concentration of the purified fusion polypeptide or polypeptide of interest
- Western blot analysis to determine the yield or purity of the purified fusion polypeptide or polypeptide of interest can be performed using a method known in the art for transferring isolated proteins to the nitrocellulose membrane on SDS-PAGE gels and using antibodies 25 specific for the desired polypeptide Can be carried out according to known methods.
- an enzyme- linked immuno sorbent assay ELISA may be used to determine the purity of the desired polypeptide.
- the yield of the purified fusion polypeptide or polypeptide of interest is determined by the amount of the purified fusion polypeptide or polypeptide of interest per volume of culture (e. G
- the measure of yield of the polypeptides described herein is based on the amount of the corresponding polypeptide expressed in its complete form.
- the yield is expressed as the amount of purified fusion polypeptide or target polypeptide relative to the volume of the culture, the cultured cell density or cell concentration may be considered in determining the yield.
- the yield of the desired polypeptide obtained after cleavage with the cleavage enzyme may be about 0.54 g / L to about 13.5 g / L.
- the yield of the desired polypeptide is 5
- An embodiment of the present invention is to obtain a desired polypeptide by using a fusion polypeptide having the amino acid sequence of SEQ ID NO: 1 and a recombinant fusion polypeptide to obtain a desired polypeptide by enzymatic degradation in the cell or inappropriate folding And the like can be minimized to provide 15 methods of producing the desired physical peptide at a high yield.
- An example of a specific production method will be described by way of examples.
- the hPTH 1-34 fusion polypeptide comprises one of PG07 (SEQ ID NO: 9), PG15 (SEQ ID NO: 31), PG43 (SEQ ID NO: 119) and 6 histidine tag (SEQ ID NO: 140) and TEV protease Recognition sequence (SEQ ID NO: 146) and the amino acid sequence of hPTH 1-34 (SEQ ID NO: 151).
- the hPTH 1-34 fusion polypeptide (H6TEV-hPTH 1-34) used as a control 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 amino terminal does not include the fusion partner, six histidine tag (SEQ ID NO: 14, 0), a protease recognition sequence (amino acid sequence of SEQ ID NO: 146) and for 1-34 (SEQ ID NO: 151 ).
- the gene of each fusion polypeptide contains the restriction enzyme, the recognition sequence of? 01 and ? 1, and one termination codon.
- the nucleotide sequence encoding the 11 1 1-34 fusion polypeptide corresponds to SEQ ID NOS: 294 to 296 , respectively, and the control corresponds to SEQ ID NO: 293 .
- Expression vector capable of regulating expression was ? 2613 .
- the fabricated for 1-34 fusion polypeptide expression plasmids was confirmed whether or not the correct base sequence cloned via zero check.
- Escherichia coli 21 ( ⁇ 3 ) cells were transformed by the chemical method using calcium chloride using the prepared plasmids of ISSM TM 1-34 fusion polypeptide.
- Expression 3 TM 1-34 Expression of fusion polypeptide The E. coli transfected with plasmids
- Transformed E. coli are, respectively, kanamycin is 50 / ⁇ /] concentration
- kanamycin is 50 / ⁇ /] concentration
- E. coli transformed with plasmids was dissolved at room temperature, and then 50 transformants were added to a 1 L culture medium in which kanamycin was present at a concentration of 50 cells / In a test tube containing 5
- E. coli cultured Escherichia coli 2 in was added to a flask containing 200 ml of LB liquid medium containing kanamycin at a concentration of 50 // g / m < 2 > and cultured in a shaking incubator at 37 [deg.] C.
- the optical density of the expression induction the cells are concentrated in the cell such that the 10.0, which was crushed to 50 mM 10 sodium phosphate, was resuspended in pH 7.2 buffer ultrasonic cell crusher cells using (ultrasonic p rocessor, Cole-Parmer ).
- the disrupted cells were labeled with total cell fractions.
- the cell lysate was centrifuged at 12,000 x g for 15 minutes at 4 ° C. The supernatant was collected and labeled with soluble fraction A.
- Insoluble fraction by using an ultrasonic cell crusher to 500 50 mM sodium phosphate, pH 7.2 buffer at 15 was suspended, and reproduction of the cover with the insoluble fraction.
- Example 1.4 Identification of the produced hPTH 1-34 by SDS-PAGE analysis
- the total cell, soluble and insoluble fractions of 50 were mixed in SDS sample buffer (SDS sam ple buffer 2 , concentrate, Si gma ) , each of which had been concentrated twice with 50 M , and then heated at 95 ° C for 5 minutes, Of the protein can be denatured.
- SDS sample buffer SDS sam ple buffer 2 , concentrate, Si gma
- the 20 denatured proteins in the sample were allowed to separate in the gel according to their molecular weight sizes using 16% SDS-PA lake gel and TANK buffer.
- SDS-PAGE the gel was stained with a staining buffer containing Coomassie blue R-250 and decolorized with decolorizing buffer to show only the stained protein. The results are shown in Fig. 1 and Fig.
- HPTE-hPTH 1-34 (6.9 kDa molecular weight), PG15-H6TEV-hPTH 30 1-34 (7.9 kDa), which is the fusion partner of the fusion partners PG7, PG15 and PG43 according to the present invention
- PG43-H6TEV-hPTH 1-34 (molecular weight of 10.6 kDa) Level was increased relative to the control (H6TEV-hPTH 1-34).
- Densitometric analysis revealed that the expression level of PG15-H6TEV-hPTH 1-34 by fusion of P15 among PG7, PG15 and PG43 was the highest among all the hPTH 1-34 fusion polypeptides.
- FIG. 2 it can be seen that all hPTH 1-34 Fusion 5 polypeptides, including the control group, are not observed in the soluble fraction but all appear in the insoluble fraction.
- Example 1.5 PG15-H6TEV-hPTH 1-34
- cells were cultured in a 37 ° C fermentor containing 2 L of restriction medium using the medium composition , And the pH was maintained at 6.8 by addition of hydrochloric acid and 10 ammonia.
- a feeding solution containing glucose was injected into the culture medium during cell culture. After 8 hours of incubation, 1.0 mM IPTG was added to induce the expression of PG15-H6TEV-hPTH 1-34 for 11 hours.
- Example 1.6 Improved expression level of hPTH 1-34 fusion 20 polypeptide by amino acid substitution of N-terminal fusion partner
- each amino acid residue was substituted with isoleucine, 21 mutants of hPTH 1-34 fusion polypeptide substituted with asparagine, arginine and asphalt acid were prepared, and the intracellular expression levels of PG15-H6TEV-hPTHI-34 were compared.
- Plasmid DNA for expression of variants of hPTH 1-34 fusion polypeptide was generated using site-directed mutagenesis.
- mutant template template (tem pi ate) is a PG15-H6TEV-hPTHl-34 expression plasmid was used
- pSGK477 primer is an amino acid substitution site of the nucleotide sequence is changed normal direction (forward) and backward (reverse) of each of the mutant Single strand DNA oligomers were used.
- the primers used in the experiments are shown in Table 3 below.
- E. coli ⁇ 1 21 ( ⁇ 3 ) cells were transformed by chemical methods using calcium chloride using expression plasmids of mutant polypeptides.
- TM 1-34 The expression plasmids were transformed E. coli of a fusion polypeptide are kanamycin 0 3 ⁇ 4113111: included at a concentration of 50 // / 111 I for ( ⁇ 1) Colonies were formed in the solid medium. The transformed Escherichia coli was transformed into kanamycin
- the slow TM 1-34 fusion polypeptide mutant expression plasmids were transfected after 50 dissolved in a concentrated solution of E. coli cells at room temperature in the storage at a temperature!
- the optical density of the cells (culprit 600 ) became about 1.0 after about 3 hours of incubation, the end of one liver ( ⁇ And the concentration was adjusted to 0.1 mM to induce the expression of hPTH 1-34 fusion polypeptide.
- Optical density was measured 4 hours after induction of expression to measure optical density of cells.
- the cells were concentrated to an optical density of 10.0 in the expression-induced cells, resuspended in 50 mM sodium phosphate, pH 7.2 buffer, and then disrupted using an ultrasonic processor (Cole-Parmer). The disrupted cells were labeled with total cell fractions. Cell lysate was centrifuged for 15 minutes at a temperature 12,000X g, 4 ° C. The supernatant was recovered, which was labeled with a soluble fraction insoluble fraction by using an ultrasonic cell crusher to 500 50 mM sodium phosphate, pH 7.2 buffer at 10 Resuspended and insoluble fractions.
- the total cell, soluble and insoluble fractions of 50 were mixed with SDS sample buffer (SDS sam ple buffer, 2x concentrate, Si gma ) , each of which was 50 times concentrated, and then heated at 95 ° C for 5 minutes, To be denatured.
- SDS sample buffer SDS sam ple buffer, 2x concentrate, Si gma
- the denatured proteins in the sample were allowed to separate in gels according to a molecular weight of 15 using 16% SDS-PAGE gel and TANK buffer.
- the gel was stained with a staining buffer containing Coomassie blue R-250 and decolorized with decolorizing buffer to show only the stained protein. The results are shown in Figs.
- FIG. 9A PG9-H6TEV-hPTH 1-34 of 7.9 kDa was synthesized with a N-terminal fusion partner, a 6-histidine tag and a TEV protease-recognizing sequence-specific PG15-H6TEV 30 fragment at a yield of almost 100% Peptides, hPTH 1-34.
- Example 4 Purification of hPTH 1-34
- an amino-terminal fusion partner (PG15-H6TEV) pi of hPTH 1-34 glass 15 by the cutting comprises a tag and a cleavage recognition sequence for the enzyme purification of the pi 3.
- PG15-H6TEV having a pi value of 11.72 has a positive charge
- hPTH 1-34 having a pi value of 8.29 has a positive charge. Therefore, PG15-H6TEV and hPTH 1- 34 is easy to separate.
- a sample in which PG15-H6TEV-hPTH 1-34 is cleaved and PG15- 20 H6TEV and hPTH 1-34 are mixed is mixed with cation exchange resin
- hPTH 1-34 having an anion was identified in the flow throu gh fraction without being bound to the cation exchange resin, and PG15-H6TEV having a cation was bound to the cation exchange resin and then sodium chloride And it was confirmed that it was eluted by increasing the concentration. Therefore, the N-terminal fusion partners having a relatively high value of 25 pi can be removed using ion exchange chromatography, so that the separation and purification of hPTH 1-34 can be facilitated.
- Example 4.2 Separation and purification of hFffl 1-34 by hydrophobic interaction chromatography
- the N-terminal fusion partners of the present invention having a relatively low average hydrophobicity value can be removed using hydrophobic interaction chromatography, so that the separation and purification of hPTH 1-34 can be facilitated.
- hPTH 1 -34 obtained from PG15-H6TEV-hPTH 1-34 34 was within the error range of the theoretical molecular weight. Therefore, it was confirmed that the protein was completely expressed in E. coli without partial cleavage or degradation of amino or carboxy terminal by proteolytic enzyme.
- TEV protease recognizes the ENLFQ 5 sequence, which is the recognition sequence in PG15-H6TEV-hPTH 1-34, and correctly cleaves the peptide bond between the last amino acid, Q (glutamine), and the first amino acid of hPTH 1-34, S .
- both hPTH 1-34 showed the same residence time and the purity of the recombinant hPTH 1-34 was found to be over 99.5% (FIG. 17).
- USP 15 (USP 15 Catalog # 1643962) and the recombinant hPTH 1-34 produced by the present invention were further purified using the standard peptide hPTH 1-34 (USP 15 Catalog # 1643962) in addition to the identification method of hPTH 1-34 in the United States Pharmacopoeia Equivalence of hPTH 1-34 was analyzed.
- the two hPTH 1-34 were treated with Staphylococcus aureus V8 protease and separated into five peptide fragments and analyzed by reverse phase HPLC.
- the gene for the GLP-1K28R fusion polypeptide has overlap extension
- the GLP-1K28R fusion polypeptide comprises PG07 (SEQ ID NO: 9), PG15C SEQ ID NO: 31 as the amino terminal fusion partner, PG22 (SEQ ID NO: 53), PG29 (SEQ ID NO: 75), PG36 ), The 6 histidine tag (SEQ ID NO: 140), the TEV protease recognition sequence (SEQ ID NO: 146) and the amino acid sequence of GLP-1K28R (SEQ ID NO: 341).
- 2019/143193 1 »(1 ⁇ 1 ⁇ 2019/000782 was used as a control group ( ⁇ 1 - 28 mu fusion polypeptide air - ⁇ 1 3 - 2810 does not include the amino-terminal fusion partner, six histidine tag ( SEQ ID NO: 140),
- each fusion polypeptide includes restriction enzymes, recognition sequences of? 01 and? 101 and one termination codon. 012-1281? Nucleotide sequences encoding fusion polypeptides correspond to SEQ ID NOS: 478 to 483, respectively, and control group corresponds to SEQ ID NO: 477.
- the fusion polypeptide gene fragment was digested with uva 01 restriction enzyme, and the expression vector capable of regulating expression by 1 liver (including the T7 promoter, 1 30 operator, and 1: 1 gene) Respectively.
- Each of kanamycin is 50 ⁇ / 1 present in a concentration of 11 ⁇ were cultured in a liquid medium, by the addition of 50% glycerol in the same volume of the culture medium is created and the cell concentrate, it was stored in the freezer of a temperature of _80 d.
- Example 7.2 The cultivation of transformed cells and the expression of
- the kkyo 13 kept at a temperature 8010 - 1 ⁇ (281? Expression of fusion polypeptides
- the cell concentrates of E. coli transformed with the plasmids were dissolved at room temperature and then 50 were added to a test tube containing 51 ⁇ l of LB liquid medium in which kanamycin was present at a concentration of 50 ⁇ g / M and incubated in a shaking incubator at 37 ° C for 12 Lt; / RTI > 2 ml of cultured Escherichia coli was added to a flask containing 5 200 in LB liquid medium containing kanamycin at a concentration of 50 g / ffli and cultured in a shaking incubator at 37 ° C.
- Cells were concentrated and resuspended in 50 mM sodium phosphate, pH 7.2 buffer, so that the optical density of the expression-induced cells was 10.0 .
- Cells were disrupted using an ultrasound processor ( Cole -Parmer ). The disrupted cells were labeled with the cell fraction. The cell lysate was centrifuged at 12,000 xg for 15 min at 4 ° C
- the total cell, soluble and insoluble fractions of 50 components were mixed in SDS 20 sample buffer (SDS sam ple buffer 2X concentrate, Si gma ), each of which was concentrated twice , and then heated at a temperature of 5 ° C for 5 minutes, Of the protein can be denatured.
- the denatured proteins in the sample were allowed to separate in the gel according to molecular weight size using 16% SDS-PAGE gel and TANK buffer. After SDS-PAGE, the gel was stained with a staining buffer containing Coomassie blue R-25 ° and decolorized with decolorizing 25 buffer to show only the stained protein. The results are shown in Fig. 19 and Fig.
- the band of H6TEV-GLP-1K28R ( molecular weight of 5.1 kDa ) without the fusion partner containing the amino acid sequence of SEQ ID NO: 1 as the control group was not detected in the SDS-PAGE gel, It appears to be a 30-decomposed by the protease.
- Expression of GLP- 1K28R fusion polypeptide in SDS-PAGE gels It was confirmed from PG07-H6TEV-GLP-1K28R (molecular weight of 6.1 kDa) fused with the amino terminal fusion partner PG07 having the smallest molecular weight.
- GLP-1K28R fusion polypeptide which is a fused 5- terminal fusion partner, PG15, PG22, PG29, PG36 and PG43, the molecular weight of PG22-H6TEV-GLP-1K28RC7.9 kDa ), PG29-H6TEV-GLP-1K28R (molecular weight of 8.4 kDa), PG36-H6TEV-GLP-1K28R (molecular weight of 9.1 kDa) and PG43? H6TEV_GLP-1K28R (molecular weight of 11.7 kDa) -1K28R) compared to the
- PG07, and PG15 were significantly higher than that of the fusion fusion polypeptide (FIGS. 20 and 21).
- Plasmid DNA for the expression of variants of the GLP-1K28R fusion polypeptide was constructed using site-directed mutagenesis.
- the template for the site-directed mutagenesis was PG43-H6TEV-GLP-1K28R expression
- Plasmids were identified by sequencing to confirm whether they were cloned correctly. Created 0 less - 1 other 81? A chemical method using a calcium chloride using a variant expression plasmid for a fusion polypeptide (for 3) 21 ⁇ E. coli cells were transformed. ( 1 defeat 81? E. coli expression plasmid of the fusion polypeptide are transformed are kanamycin (1 3 ⁇ 4113113 ⁇ 40:) containing 0, // 50 levels of the / 111 beam for Colonies were formed in the solid medium. The transformed Escherichia coli was transformed into kanamycin
- Creating a cell concentrate was added with the same volume of the culture medium, which-801: were stored at a temperature nyaeng bundle.
- optical density of the culture after about 3 hours the cells (( ⁇ 600) is about 1.0 when the 1 (; by addition of a final concentration was set at 0.1 kkyo 13-281? Expression of the fusion polypeptide was induced.
- Optical density was measured 4 hours after the induction of induction and the optical density of the cells was measured.
- the optical density of the expression induction the cells are concentrated in the cell such that the 10.0 and 50 sodium phosphate, was resuspended in 7.2 buffer ultrasonic cell crusher -
- the cells were disrupted by using a (111 to 33011 ⁇ ⁇ mi).
- the disrupted cells were labeled with total cell fractions.
- Cell lysate was centrifuged for 15 minutes at 12,000X 1 4 0 ° temperature. The supernatant was collected and labeled with a soluble fraction.
- Insoluble fraction was labeled with 50 0 50 sodium phosphate, reproduced by using an ultrasonic cell crusher to 7.2 buffer solution, and suspended insoluble fraction.
- a total of 50 cells, soluble and insoluble fractions of each 50/4 «twice concentrated ⁇ of £ The samples were heated at 95 ° C for 5 minutes in a mixture of SDS sam ple buffer 2 ⁇ concentrate (Sigma) to denature the protein of each sample.
- the denatured proteins in the sample were allowed to separate in the gel according to molecular weight size using 16% SDS-PAGE gel and TANK buffer. After SDS-PAGE, the gel was stained with a staining buffer containing Coomassie blue R-250 and then decolorized with decolorizing buffer to show only stained proteins.
- the frozen cell pellet of the cells expressed on the flask scale was thawed by the addition of 50 M of 50 mM sodium phosphate, pH 7.2 buffer.
- the resuspended cells were disrupted using an ultrasonic processor (Cole-Parmer). Dissolved cells were recovered 12,000 rpm (12,000Xg) conditions the insoluble fraction FOCE within 30 minutes and centrifuged to remove the supernatant liquid 20 containing the recombinant fusion polypeptide from.
- Centrifugation was carried out at 12,000 x g for 30 minutes and the supernatant was filtered through a (0.45 / 0.2) membrane filter.
- GLP-1K28R fusion in the soluble insoluble fraction For purification of the polypeptides, an AKTA pure 25 chromatographic system (GE Healthcare) equipped with an S9 sample pump (Sam ple pump S9) and a F9-C fraction collector (Fraction col- lector F9-C) was used.
- solubilized, insoluble fraction sample was injected into a HisTra p FF 1 m column (GE Healthcare) pre-equilibrated with an intracellular solubilization buffer (8 M urea, 20 mM Tris, 500 mM sodium chloride, 50 mM imidazole, pH 5 7.4) .
- the column is washed, and step by step from 100% to 5 times the elution buffer of the column volume (8 M urea, 20 mM Tris, 500 mM sodium chloride, 500 mM, pH 7.4 imidazole) with equilibration buffer of 10 times column volume of So as to elute the GLP-1K28R fusion polypeptide bound to the resin of the column.
- the results of 10 fractions obtained after elution are shown in the figures respectively (Fig. 25 and Fig. 26).
- the GLP-1K28R fusion polypeptide in the solubilized insoluble fraction sample injected into the column was mostly eluted after binding to the resin in the column, and showed a purity of 95% or more.
- dilution buffer (20 mM Tris, pH 7.4) was added to dilute the urea concentration to 1 M.
- TEV protease was added to the diluted recombinant fusion polypeptide at a final concentration of 500, and cleavage reaction was allowed to proceed for 12 hours at room temperature.
- the molecular weight of GLP-1K28R obtained from PG43-H6TEV-GLP-1K28R was 3382.59 Da and was within the error range from the theoretical molecular weight of 3383.72 Da. Therefore, proteolytic enzyme ) was expressed in complete form without partial cleavage or degradation of amino or carboxy terminal end by 5.
- TEV protease 10 determines to accurately cut the peptide bond between the PG43-H6TEV-GLP-1K28R within the recognition sequence is recognized the ENLFQ sequence and the last amino acid of Q (glutamine) and the H (histidine), the first amino acid of the GLP-1K28R do.
- Example 11 Preparation and production of the terminus glutide (GLP-2A2G) fusion polypeptide.
- GLP-2A2G fusion polypeptide was synthesized using the overla p extension 15 polymerase chain reaction (OE-PCR) method.
- the GLP-2A2G fusion polypeptide may be a fusion partner of amino terminus PG07 (SEQ ID NO: 9), PG15 SEQ ID NO: 31), PG22 (SEQ ID NO: 53), PG29 SEQ ID NO: 75, PG36 6 histidine tag one of SEQ ID NO: 119) and is (with the SEQ ID NO: 140), TEV protease recognition sequence (SEQ ID NO: 146) and amino acid sequence (SEQ ID NO: 20, 485 of the GLP-2A2G).
- the GLP-2A2G fusion polypeptide (H6TEV-GLP-2A2G) used as a control does not contain an amino terminal fusion partner and has 6 histidine tags (SEQ ID NO: 140), TEV protease recognition sequence (SEQ ID NO: 146) (SEQ ID NO: 485).
- the genes of each fusion polypeptide include restriction endonucleases Ndel, Ncol 25, and the recognition sequence of the second and one stop codon.
- the nucleotide sequences encoding the GLP-2A2G fusion polypeptides correspond to SEQ ID NOS: 622 to 627, respectively, and the control group corresponds to SEQ ID NO: 621.
- the GLP-2A2G fusion polypeptide gene fragment synthesized by 0E-PCR was amplified using NdeI and 2 expression cassettes were prepared by restriction endonuclease digestion using restriction enzymes, and expression vectors capable of regulating expression by 1 ⁇ including 1 7 promoter, 1 30 operator and 1 301 gene ? £ 2 613 .
- the expression plasmids of the present invention can be chemically synthesized using calcium chloride
- Escherichia coli transfected with the expression plasmids of the polypeptides formed colonies in kanamycin 0 and one solid medium containing 131117 ( ⁇ 11 ) at a concentration of 50 // / 111 mo. After transfection of E. coli were respectively cultured in kanamycin 50 // [ ⁇ liquid medium is present in a concentration of / I TM, 50% glycerol in the same volume of the culture medium
- Plasmids were transfected after a cell concentrate of E. coli was dissolved at room temperature for 50 ⁇ kanamycin is 50 1 ⁇ / 111 1 liquid medium present in a first concentration of was added to the test tube containing 5111 yo in a shaking incubator for 37 V Temperature 12 Lt; / RTI > Seed bacteria
- Example 11.3 Preparation of samples for comparative analysis of expression levels The cells were concentrated to an optical density of the expression-induced cells of 10.0, resuspended in 50 mM sodium phosphate, pH 7.2 buffer, and then disrupted using an ultrasonic processor (Cole-Parmer). Cells were labeled with total cell fractions. The cell lysate was centrifuged at 12,000 x g for 15 min at 4 ° C
- the insoluble fraction was resuspended in 500 ml of 50 mM sodium phosphate, pH 7.2 buffer using an ultrasonic cell crusher and labeled with an insoluble fraction.
- PG07-H6TEV-GLP-2A2G (molecular weight of 6.5 kDa), PG15-H6TEV-GLP-2A2G (7.5 kDa) which is a GLP-2A2G fusion polypeptide fused with amino terminal fusion partners PG07, PG15, PG22, PG29, PG36 and PG43 of molecular weight), PG22-H6TEV-GLP- 2A2G (7.5 kDa molecular weight), PG29-H6TEV-GLP- 2A2G (8.3 kDa molecular weight), PG36-H6TEV-GLP- 2A2GO .5 molecular weight of 25 kDa) and a PG43-H6TEV GLP-2A2G, PG36-H6TEV-GLP-2A2G and PG43-H6TEV-GLP-2A2G (12.1 kDa molecular weight) GLP-2A2G.
- a plasmid DNA for expressing variants of GLP-2A2G fusion polypeptide was produced using the positioning mutation (site-directed muta genesis) method.
- the mold (template) for positioning mutant was used as a PG43-H6TEV- GLP-2A2G expression plasmid pSGK523.
- the primers used in the experiments are shown in Table 7 below.
- the fabricated ⁇ 1 - 2 I was transformed to 20 fusion polypeptides of the mutant E. coli expression chemical methods 1 3 cells with calcium chloride using a plasmid.
- 2 shows the expression plasmid for 20 fusion polypeptide are transduced - kkyo
- the cells were disrupted by the optical density of the 10 expression induced concentrating the cell to be 10.0 using the resuspended ultrasonic cell crusher (u ltrasonic processor, Co le- Parmer) was in 50 mM sodium phosphate, pH 7.2 buffer solution
- the crushed cells were labeled with the cell fraction.
- the cell lysate was centrifuged at 12,000 ⁇ g for 15 minutes at 4 ° C.
- the supernatant was recovered and labeled with a soluble fraction
- the insoluble fraction was reconstituted using an ultrasonic cell crusher with 15 ⁇ l of 50 mM sodium phosphate, pH 7.2 buffer Labeled with a turbid and insoluble fraction.
- SDS sample buffer SDS sam ple buffer 2X concentrate, Si gma
- the 20 denatured proteins in the sample were allowed to separate in the gel according to molecular weight size using 16% SDS-PAGE gel and TANK buffer.
- the S-PA lake gel was dyed with a dye buffer containing Coomassie blue R-250, and decolorized with decolorizing buffer to show only stained proteins.
- the frozen cell pellet of the cells expressed in the size flask were thawed by the addition of 50 m e 50 mM sodium phosphate, pH 7.2 buffer.
- the resuspended cells were disrupted using an ultrasonic processor Cole-Parmer .
- the lysed cells were centrifuged at 12,000 rpm (12,000 xg) for 30 minutes, and the supernatant was removed to recover the insoluble intracellular fraction containing the recombinant fusion polypeptide.
- the recovered insoluble inner foam fraction was added 20 M of intrathecal solubilization buffer (8 M urea, 10 20 mM Tris, 500 mM sodium chloride, 50 mM imidazole, pH 7.4) and incubated at 25 ° C for 3 hours with shaking Thereby allowing the recombinant fusion polypeptide in the form of an insoluble form in the insoluble fraction to be solubilized.
- the solubilized insoluble fraction sample was centrifuged at 12,000 xg for 30 minutes and the supernatant was filtered through a (0.45 / 0.2) membrane filter.
- GLP-2A2G fusion polypeptides PG43-H6TEV-GLP-2A2G, which has the highest expression level, was purified.
- an ARTA pure 25 chromatography system 20 equipped with an S9 sample pump S9 and a F9-C fraction collector F9-C (GE Healthcare).
- the solubilized insoluble fraction sample was diluted with an intracellular solubilization buffer (8 M urea, 20 mM Tris, 500 mM sodium chloride, 50 mM imidazole, pH
- the column elution buffer wash and five times the column volume of equilibration buffer of 10 times column volume (8 M urea, 20 mM Tris-500 mM sodium chloride, 500 25 mM imidazole, pH 7.4) for step-by-step as the 100%
- the results of the fraction obtained after elution are shown in the respective figures (Fig. 34 and Fig. 35).
- the GLP-1K28R fusion polypeptide in the solubilized insoluble fraction sample injected into the column was mostly eluted after binding to the resin in the column, and showed a purity of 95% or more.
- TEV protease recognizes the ENLFQ 25 sequence, a recognition sequence in PG43-H6TEV-GLP-2A2G, and correctly cleaves peptide bonds between the last amino acid, Q (glutamine), and the first amino acid, H (histidine), of GLP-2A2G do. (The target peptide was not expressed and the solubility test was not performed).
- the gene for the calantide fusion polypeptide at 30 is overexpression polymerase chain reaction (OE-PCR). (SEQ ID NO: 9) and PG43 (SEQ ID NO: 119), the 6 histidine tag (SEQ ID NO: 140) and the TEV protease recognition sequence (SEQ ID NO: SEQ ID NO: 146) and the amino acid sequence of Ecarantide (SEQ ID NO: 638).
- OE-PCR polymerase chain reaction
- Ecallantide used as a control does not contain an amino terminal fusion partner and has a 6 histidine tag (SEQ ID NO: 140), a TEV protease recognition sequence (SEQ ID NO: 146) and an amino acid sequence of ecarantide (SEQ ID NO: 642 ).
- the gene of each fusion polypeptide contains the recognition sequence of the restriction enzymes NdeI, NcoI and I and one stop codon.
- the nucleotide sequences (PG07, PG15 and PG43) coding for the calantide fusion polypeptides correspond to SEQ ID NOS: 644 to 646, respectively, and the control group corresponds to SEQ ID NO: 643.
- Calantide fusion polypeptide expression plasmids were prepared and stored in the freezer at -8010 in the same manner as that performed in Example 1.1 .
- the culture of the transformed cells with the expression plasmids of the deposited calendared fusion polypeptides at 800 and the expression of the ecarantoide was carried out in the same manner as in Example 1.2 .
- Example 15 4. Confirmation of produced ecarantide by SDS-PAGE analysis The protein of each sample was treated under the same conditions and conditions as in Example 1.4, and the results are shown in Figs. 38 and 39.
- the band of H6TEV-Ecallantide (molecular weight of 8.8 kDa) without the fusion partner containing the amino acid sequence of SEQ ID NO: 1, which is the control group, exhibited the lowest expression compared to the taracortide fusion polypeptides Respectively.
- PG07-H6TEV-Eca11 ant i de (9.8 kDa molecular weight), PG15-H6TEV- 10 Ecal lantide (molecular weight of 10.8 kDa), which are fused to the amino terminal fusion partners PG7, PG15 and PG43, and The expression level of PG43-H6TEV-Eca 11 ant i (molecular weight of 15.4 kDa) was confirmed to be increased compared to the control (H6TEV-Ecallantide). Expression levels of PG07-H6TEV-Ecallantide by fusion of PG7 among PG7, PG15 and PG43 were found to be highest among all the E. coli-fused fusion polypeptides by densitometric analysis.
- the nasiridide fusion polypeptide used as a control (H6TEV-
- Ecallantide does not include an amino terminal fusion partner and includes a 6 histidine tag (SEQ ID NO: 140), a TEV protease recognition sequence (SEQ ID NO: 146), and an amino acid sequence of nasiridide (SEQ ID NO: 652).
- the gene of each fusion 30 polypeptide is composed of the recognition sequences of restriction enzymes NdeI, NcoI, And 1 ⁇ 1 ⁇ 2019/000782 comprises a termination codon: 0 2019/143193 1 »(.
- the nucleotide sequences encoding the nasiridate fusion polypeptides correspond to SEQ ID NOS: 654 to 656, respectively, and the control group corresponds to SEQ ID NO: 653.
- Nasiridide fusion polypeptide expression plasmids were prepared in the same manner as that performed in Example 1.1 and stored in a freezer at -80 ° C.
- the production of the nasiridide-related sample was carried out in the same manner as in Example 1.3 .
- the expression of the nasiridide fusion polypeptide in the gelatin of the yeast yomoyomi was the lowest amino-terminal fusion
- the partner PG15 was identified from the fused PG15-H6TEV-Nesiri tide (molecular weight of 7.2 kDa).
- the expression level of PG43-H6TEV-Nesiri tide (molecular weight of 11.8 kDa), which is a fusion of the amino-terminal fusion partner PG43, was confirmed to be improved compared to that of PG15-H6TEV-Nesiritide (molecular weight of 7.2 kDa).
- Densitometer 5 analysis revealed that PG43-H6TEV-1 by fusion of PG43 among PG07, PG15,
- the level of expression of Nesiritide was found to be the highest among all the nesylideide fusion polypeptides.
- hPTH 1-84 fusion polypeptide 1 5 hPTH gene for the 1-84 fusion polypeptide was synthesized using the overla p extension polymerase chain reaction (OE -PCR) method.
- OE -PCR overla p extension polymerase chain reaction
- the hPTH 1-84 fusion polypeptide has one of 6 histidine tags (SEQ ID NO: 140), TEV protease recognition sequence (SEQ ID NO: 9), PG15 (SEQ ID NO: 146) and amino acid 20 sequence of hPTH 1-84 (SEQ ID NO: 18).
- the hPTH 1-84 fusion polypeptide (H6TEV-hPTH 1-84) used as a control does not contain an amino terminal fusion partner, and the 6 histidine tag (SEQ ID NO: 140), the TEV protease recognition sequence (SEQ ID NO: 146)
- the amino acid sequence of 1-84 (SEQ ID NO: 628 ) is included.
- Gene of each fusion polypeptide comprises a recognition sequence, and one stop codon of the restriction 25 enzyme Ndel, Ncol and Xhol.
- the nucleotide sequences encoding the hPTH 1-84 fusion polypeptides correspond to SEQ ID NOS: 635 to 637, respectively, and the control group corresponds to SEQ ID NO: 654.
- hPTH 1-84 fusion polypeptide expression plasmids P SGK543, pSGK544, pSGK545 and pSGK546 shown in Table 10 below .
- 30 hPTH 1-84 fusion polypeptide gene fragments synthesized by 0 E-PCR were ligated with Ndel and Xhol restriction enzymes Using 2019/143193 1 »(? 1 ⁇ 1 ⁇ 2019/000782 cutting, 17 promoter, operator 1 and the 1301 gene vector capable of expression control by the expression 1 ⁇ include cloned in ⁇ 23 ⁇ 4.
- Escherichia coli 21 ( 3 ) cells were transformed by a chemical method using calcium chloride using polypeptide expression plasmids.
- the total cell, soluble and insoluble fractions of 50 were mixed in SDS sample buffer (SDS sample buffer 2, concentrate, Sigma), each 50 times concentrated, and then heated at 95 ° C for 5 minutes, To be denatured.
- the denatured proteins in the sample were allowed to separate in a gel according to a molecular weight of 10 using 16% SDS-PAGE gel and TANK buffer. After SDS-PAGE, the gel was stained with a staining buffer containing Coomassie blue R-250 and decolorized with decolorizing buffer to show only the stained protein. The results are shown in Figs. 42 and 43.
- the band of H6TEV-hPTHl-84 (molecular weight of 11.2 kDa) without the 15 fusion partner containing the amino acid sequence of SEQ ID NO: 1 as a control group was compared with other hPTH 1-84 fusion polypeptides Showed the lowest expression level.
- Densitometric analysis revealed that the expression level of PG15-H6TEV-hPTHl-84 by fusion of PG15 among PG07, PG15, and PG43 was the highest among all hPTH 1-84 fusion polypeptides. 43 , all the hPTH 1-84 fusion polypeptides including the control group were observed in the soluble fraction, but the larger the 25 amino terminal fusion partner was, the greater the insoluble fraction of the hPTH 1-84 fusion polypeptide was PG 43-H6TEV-hPTHl-84 fused with the larger PG 43 can be found to account for about 70% of the total protein in the insoluble fraction.
- hPTH 1-84 fusion polypeptides have a high solubility expression ratio
- the fractions were purified hPTH 1-84 fusion polypeptides.
- the frozen cell pellet of the cells expressed on the flask scale was thawed and resuspended by addition of 20 m of endogenous solubilization buffer (8 M urea, 20 mM Tris, 500 mM sodium chloride, 50 mM imidazole, pH 7.4).
- the resuspended cells were disrupted using an ultrasonic processor (5 Cole-Parmer).
- the lysed cells were centrifuged at 12,000 rpm (12,000 xg) for 30 minutes, and the supernatant was removed to remove the insoluble intracellular fraction containing the recombinant fusion polypeptide and recover the supernatant as a soluble fraction.
- the soluble fraction sample was centrifuged at 12,000 x g for 30 minutes and the supernatant was filtered through a (0.45 / 0.2 pm) membrane filter.
- the highest expression level of PG15- h6TEV-hPTHl-84 among the four hPTHl-84 fusion polypeptides was purified.
- an AKTA pure 25 chromatography system 15 GE Pure Chromatography 15 system equipped with a S9 sample pump (Sam ple pump S9) and a F9-C fraction collector ( F9- Healthcare) was used.
- the solubilized insoluble fraction sample was injected into a HisTra p FF 1 mL column (GE Healthcare) pre-equilibrated with an epiotropic solubilization buffer (8 M urea, 20 mM Tris, 500 mM sodium chloride, 50 mM imidazole, pH 7.4). After injection is complete, the column is washed, and, step by step from 100% to 5-fold column volume of elution buffer, 8 M urea, 20 mM Tris, 500 mM sodium chloride, 500 20 mM pH 7.4 imidazole) with equilibration buffer of 10 times column volume of So as to elute the hPTHl-84 fusion polypeptide bound to the resin of the column.
- an epiotropic solubilization buffer 8 M urea, 20 mM Tris, 500 mM sodium chloride, 50 mM imidazole, pH 7.4
- dilution buffer (20 mM Tris, pH 7.4) was added to dilute to a concentration of 1 M of urea.
- TEV protease was added to the diluted recombinant fusion polypeptide at a final concentration of 500 and 30 so that the cleavage reaction proceeded at room temperature for 12 hours.
- SDS-PAGE analysis was performed after cleavage in order to confirm the cleavage by TEV protease, and the results are shown in the figure (FIG. 46).
- hPTHl-84 fused polypeptide (PG15-h6TEV-hPTHl-84) by TEV protease by SDS-PAGE showed that the hPTHl-84 fusion polypeptide of 13.2 kDa had 5 amino Terminal fusion partner with 6 Histidine tag and TEV protease recognition sequence fused to PG15-H6TEV and the target polypeptide hPTH 1-84.
- the molecular weight of 15 of hPTH 1-84 obtained from PG15-H6TEV-hPTHl-84 was 9425.54 Da, which was in agreement with the theoretical molecular weight of 9424.73 Da within the error range. Therefore, ( Proteolyt ic enzyme) without partial cleavage or degradation of amino or carboxy terminus.
- TEV protease is determined to precisely cut the peptide bond between the PG15-H6TEV-hPTHl-84 within the recognition sequence of ENLFQ recognize the 20 sequence and the last amino acid of Q (glutamine) with the first amino acid, S (serine) of hPTHl-84 do.
- Example 21 Comparison of expression levels of hPIH 1-34 fusion polypeptides by fusion partner location
- Example 21.1 Additional production of hPTH 1-34 fusion polypeptide expression plasmid
- the hPTH 1-34 fusion polypeptide has the amino acid sequence of PG15 (SEQ ID NO: 31), 6 histidine tag (SEQ ID NO: 140), TEV protease recognition sequence (SEQ ID NO: 146) SEQ ID NO: 151).
- fusion genes of the fusion polypeptide are recognized by the restriction enzymes Ndel, Ncol and J. And 1 ⁇ 1 ⁇ include 2019/000782 sequence and one stop codon: 2019/143193 1 »(.
- the nucleotide sequences encoding the above viii-34 fusion polypeptides correspond to SEQ ID NOS: 294 and 295, respectively.
- Expression plasmids were prepared in the same manner as that performed in Example 1.1 and stored in a -801: temperature freezer.
- Example 21.2 Culturing and expression of transformed cells 1-34 and expression
- Each of the .alpha. And .beta. 033 cells were cultured in a flask containing 200% depleted 15 medium and 11 () was added to induce the expression of the large 1-34 fusion polypeptide.
- the structure of each fusion polypeptide is shown in FIG.
- the present invention-terminal fusion partner is 13 ⁇ 4 O 5 1-34 and fusion expression of poly be fused to the amino-terminal in the peptide according to the 1-34 TM .
- the affinity tag TM 1-34 When removed from the fusion polypeptide or the carboxyl terminal position of the I-terminal fusion partner, 1- TM 34 charges manifest a 5 fusion polypeptide can be maintained. However, the expression is maintained However, when it is located at the amino terminus of the terminal fusion partner, it can be confirmed that the expression level of TM 1-34 fusion polypeptide is remarkably reduced.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Endocrinology (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Toxicology (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
본 발명은 신규한 N-말단 융합 파트너, 상기 융합 파트너와 목적 폴리펩 타이드를 포함하는 융합 폴리펩타이드 및 이를 이용하여 목적 폴리펩타이드를 생 산하는 방법에 관한 것이다. 본 발명에 따른 신규한 융합 파트너를 사용하면, 종 래 융합 파트너 대비 목적 폴리펩타이드 (재조합 폴리펩타이드)의 수율을 향상시 킬 수 있다. 특히, 상대적으로 분자량이 작으며 분해되기 쉬운 아미노 말단을 갖 는 목적 폴리펩타이드를 유전자 재조합 기술을 이용하여 제조하는 경우에 유용하 다. 또한, 본 발명에 따른 융합 파트너를 포함하는 융합 폴리템타이드는 숙주세 포 내에서 내포체 형태로 발현이 유도됨으로써 숙주세포의 단백질 분해효소 등으 로부터 보호되어 안정적으로 목적 폴리펩타이드를 생산할 수 있는 장점이 있다. 따라서, 종래의 융합 파트너를 사용한 경우보다 안정성 및 수율이 향상된 재조합 펩타이드 생산 방법을 제공할 수 있다.
Description
명세서 재조합 폴리펩타이드 생산용 N-말단융합 파트너 및 이를 이용하여 재조합 폴리 펩타이드를생산하는방법 기술분야
본 발명은 신규한 N-말단 융합 파트너,상기 융합 파트너와 목적 폴리펩 타이드를 포함하는 융합폴리펩타이드 및 이를 이용하여 목적 폴리펩타이드를 생 산하는 방법에 관한 것이다. 배경기술
최근 유전공학 및 생명공학 기술의 발달로 대장균, 효모,동식물세포 등 을 이용하여 유용한 외래 단백질이 많이 생산되고,이들 단백질이 의약품 등으로 널리 이용되고 있다.구체적으로, 면역 조절제, 효소 저해제 및 호르몬 같은 의 약 및 연구용 단백질이나 반응 첨가 효소와 같은 산업용 단백질에 대한 생산 공 정 기술 개발 및 산업화가추진되고 있다.
이중에서도 유전자 재조합 기술은 여러 타겟 단백질들의 핵산을 발현벡터 에 클로닝하여 재조합 발현벡터를 얻고,이를 적당한 숙주세포에 형질전환시켜 배양함으로써 타겟 단백질(목적 폴리펩타이드)을 생산하는 방법이다.다만, 숙주 세포에 내재된 분해 효소(예를 들어, 프로테아제 (protease) 또는 펩티다제 (peptidase))에 의해 타겟 단백질의 전체 또는 일부가분해되어 수율이 낮아지거 나, 융합 파트너로 사용하는 펩타이드의 크기가 제조하고자 하는 타겟 단백질의 크기에 비해 너무 커서 수율이 현저히 떨어지는문제가 있다.
따라서,유전자 재조합 기술을 이용하여 타겟 단백질을 대량 생산하고자 하는 경우, 타겟 단백질를 안정적으로 발현시키면서도 생산 수율을 향상시킬 수 있는융합파트너를 개발하는 것이 중요하다. 기술적 과제
본 발명의 목적은 재조합 폴리펩타이드를 생산하기 위한 신규한 아미노산 서열로 이루어진 N-말단융합파트너를 제공하는 것이다.
2019/143193 1»(:1^1{2019/000782 본 발명의 다른 목적은 상기 1말단 융합 파트너와 목적 폴리펩타이드를 포함하는융합폴리펩타이드를 제공하는 것이다.
본 발명의 또 다른 목적은 상기 융합 폴리펩타이드를 코딩하는 뉴클레오 타이드, 상기 뉴클레오타이드를 포함하는 발현벡터 및 상기 발현벡터를 포함하는 5 숙주세포를 제공하는 것이다.
본 발명의 또 다른 목적은 상기 융합 폴리펩타이드를 이용하여 목적 폴리 펩타이드를 생산하는 방법을 제공하는 것이다. 과제 해결 수단
10 상기 목적을 달성하기 위해 본 발명의 일 측면은, 하기 일반식 1로 표시 되는 아미노산서열로 이루어진 말단융합파트너 목적 폴리펩타이드 및 상기 말단 융합 파트너와 상기 목적 폴리펩타이드 사이에 링커를 포함하는 융합 폴 리펩타이드를 제공한다:
[일반식 1]
15 1^1; -父크 -父五크요 - 3크3 - 884 -父885 -父336-(å)11
상기 일반식 1에서,

20 (쇼 , ) , 세린比아 , 幻, 트레오닌(¾!·, I) , 시스테인 , 0 , 글루타민 ½111, 이, 아르기닌( 2, 10, 리신(切 V , 히스티딘(먀 引, 아스팔트산 31), I)) 및 글루탐산(이11, 으로이루어진군으로부터 선택되고,
상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고
25 상기 은 0또는 1의 정수이다.
본 발명의 다른 측면은 상기 융합 폴리펩타이드를 코딩하는 뉴클레오타 이드, 상기 뉴클레오타이드를 포함하는 발현벡터 및 상기 발현벡터를 포함하는 숙주세포를 제공한다.
본 발명의 또 다른 측면은 상기 숙주세포를 배양하는 단계 상기 숙주세
30 포에서 발현된 융합 폴리펩타이드를 정제하는 단계 및 상기 정제된 융합 폴리펩
2019/143193 1»(:1^1{2019/000782 타이드를 절단 효소와 배양하여 목적 폴리펩타이드를 회수하는 단계를 포함하는 재조합폴리펩타이드의 생산방법을 제공한다. 발명의 효과
5 본 발명에 따른 신규한 융합 파트너를 사용하면, 종래 융합 파트너 대비 목적 폴리펩타이드(재조합 폴리펩타이드)의 수율을 향상시킬 수 있다.특히 상 대적으로 분자량이 작으며 분해되기 쉬운 아미노 말단을 갖는 목적 폴리펩타이드 를 유전자 재조합 기술을 이용하여 제조하는 경우에 유용하다.또한, 본 발명에 따른융합 파트너를 포함하는 융합폴리펩타이드는 숙주세포 내에서 내포체 형태 10 로 발현이 유도됨으로써 숙주세포의 단백질 분해효소 등으로부터 보호되어 안정 적으로 목적 폴리펩타이드를 생산할 수 있는 장점이 있다.따라서, 종래의 융합 파트너를 사용한 경우보다 안정성 및 수율이 향상된 재조합 펩타이드 생산 방법 을제공할수 있다.
15 도면의 간단한설명
도 1은재조합대장균에서 발현된 1-34융합폴리펩타이드들의 총세 포 분획을 況)구쇼湖로 분석한 결과이다(레인 마커 단백질, 레인 1: 抑 - 1-34(균주번호 ?的01), 레인 2: ?(¾)7-抑1 - 1¾1-34(균주번호 ?0002), 레 인 3: ?아5내 - 대1-34(균주 번호 (}003); 레인 4: ?643내的 - 대1-34(균 20 주번호 ?0004)) .
도 2는재조합대장균에서 발현된 ™ 1-34융합폴리펩타이드들의 총세
 , , ,
, , ,
태1-34(균주번호 ?0001), 레인 2: ?(}07내 斗 1¾1-34(균주번호 ?0002), 레 25 인 3: 야크내 -비 따 균주 번호 (}003); 레인 4: ?043내61 -111 31¾1-34(균 주번호 ?0004)) .
도 33은 ?아5-11 -出:)™ 1-34를 대량으로 생산하기 위한 유가식 배양 시 시간에 따른 광학밀도(0.1).600) 및 1 犯투여시간을나타낸 그래프이다.
도 ¾는 유가식 배양을 통해 재조합 대장균으로부터 생산된 ?아5내 - 30 태 1-34를 시간별로 샘플링한후 로 분석한 결과이다.
2019/143193 1»(:1^1{2019/000782 도 4는 재조합 대장균에서 발현된 ? 5(스2-7)내 -!!!3™ 1-34융합폴리

도 5는 야크내 -뱌江 간 내 ?(}15의 2번째 또는 3번째 아미노산 잔기 를 이소루신(I), 아스파라긴( , 아르기닌 00 , 아스팔트산(I))으로 치환한 1¾

도 7은므(}15-11 -1少1111-34 내 ?아5의 6번째 또는 7번째 아미노산 잔기 를 이소루신(I) 아스파라 \긴어) 아르기닌00 아스팔트산(I))으로 치환한 바!}!
1-34융합폴리펩타이드의 변이체를況 쇼묘로분석한결과이다.
도 83는 크로마토그래피를 통해 불용성 분획 내의 ?예7내的 - 1¾1-34 융합 폴리펩타이드를 정제한 결과이다(크로마토그램에서 실선, 파선, 점선들은 각각 280에1 파장에서의 흡광도, 전도도(«파如 , 용출 완충액의 비율을 나타냄).
도예는크로마토그래피를통해 정제한 ?(¾7내 -11?1111-34융합폴리펩

료, 레인 2~4: 통과액 분획, 레인 8~11: 용출액 분획). 화살표는 ?예7-11 - 11?1¾1-34융합폴리펩타이드를나타낸다.
도 93는 크로마토그래피를 통해 불용성 분획 내의 (}15내的 -11?1111-34 융합 폴리펩타이드를 정제한 결과이다(크로마토그램에서 실선, 파선, 점선들은 각각 280에1 파장에서의 흡광도, 전도도(«파 , 용출 완충액의 비율을 나타냄).
도 %는크로마토그래피를통해 정제한 ?아5-1161^- 1¾1-34융합폴리펩

바3재1-34융합폴리펩타이드를나타낸다.
도 10크는 크로마토크래피를 통해 불용성 분획 내의 ?643내況 -11?대1-34 융합 폴리펩타이드를 정제한 결과이다(크로마토그램에서 실선, 파선, 점선들은
각각 280 nm 파장에서의 흡광도, 전도도(conduct ivi ty), 용출 완충액의 비율을 나타냄).
도 10b는 크로마토그래피를 통해 정제한 PG43-H6TEV-hPTHl-34 융합 폴리 펩타이드를 SDS-PAGE로 분석한 결과이다(레인 M: 마커 단백질, 레인 s: 정제 전 5 시료, 레인 2~4: 통과액 분획, 레인 8~11: 용출액 분획). 화살표는 PG43-H6TEV- hPTHl-34융합폴리펩타이드를나타낸다.
도 11은 TEV프로테아제를 이용하여 정제된 각시료의 융합폴리펩타이드 를절단한후분획을 SDS-PAGE로분석한결과이다(레인 M: 마커 단백질, 레인 C: TEY프로테아제를 처리하지 않은시료, 레인 T: TEV프로테아제를 처리한시료, 10 레인 1: PG07-H6TEV-hPTHl-34, 레인 2: PG15-H6TEV-hPTHl-34, 레인 3: PG43- H6TEV-hPTHl-34) .
도 12는 TEV프로테아제를 이용하여 정제된 PG15-H6TEV-hPTHl-34융합폴 리펩타이드를 절단한후 분획을況) S-PAGE로 분석한 결과이다(레인 M: 마커 단백 질, 레인 C: TEV프로테아제를 처리하지 않은 시료, 레인 T: TEV프로테아제를 15 처리한시료).
도 13a는 등전점 차이를 이용하여 PG15-H6TEV-hPTHl-34 융합 폴리펩타이 드로부터 PG15-H6TEV와 hPTHl-34를분리한결과를나타낸 것이다(크로마토그램에 서 실선, 파선, 점선들은 각각 280 nm 파장에서의 흡광도, 전도도 (conductivity), 용출완충액의 비율을나타냄).
20 도 13b는등전점 차이를 이용하여 분리한 PG15-H6TEV-hPTHl-34융합폴리 펩타이드의 분획을 SDS-PAGE로 분석한 결과이다(레인 M: 마커 단백질, 레인 s: 정제 전시료, 레인 1~3: 통과액 분획, 레인 5~9: 용출액 분획).
도 14a는평균소수성 차이를 이용하여 PG15-H6TEV-hPTHl-34융합폴리펩 타이드로부터 PG15-H6TEV와 hPTHl-34를분리한결과를나타낸 것이다(크로마토그 25 램에서 실선, 점선들은각각 280 nm파장에서의 흡광도및 용출완충액의 비율을 나타냄).
도 14b는평균소수성 차이를 이용하여 분리한 PG15-H6TEV-hPTHl-34융합 폴리펩타이드의 분획을況 S-PAGE로 분석한 결과이다(레인 M: 마커 단백질, 레인 s: 정제 전시료, 레인 1~5: 1st peak분획, 레인 1~7: 2nd peak분획).
30 도 15는 hPTH 1-34표준물질의 분자량을측정한결과를나타낸 것이다.
2019/143193 1»(:1^1{2019/000782 도 16은 본 발명에 따라 정제된 1¾ 1-34의 분자량을 측정한 결과를 나 타낸 것이다.
도 17은 미국약전(此 의 1-34 동정 기준시험법으로 표준물질 재 1-34와본 발명에 따른 재조합 抑 1-34의 체류시간 및 순도를 확인한그래프이 5 다.
도 18은 역상 크로마토그래피 및 미국약전(此 의 대 1-34동정 기준시 험법 중 펩타이드 맵핑 방법을 통해 표준물질 대 1-34와 본 발명에 따른 재조 합 抑 1-34의 동등성을분석한결과이다.

가용성 분획, 레인 I: 불용성 분획, 레인 1: 抑 -꾜13-1他81?(균주 번호 ?的05), 레인 2: (}07-期1 -此13-11(28묘(균주 번호 ?¥06), 레인 3: ?아5내的 - (표少-1敗81?(균주번호 (¾)07), 레인 4: ? 22-抑1^ -此1)- 28묘(균주번호 ?(¾08)).
20 도 21은재조합 대장균의 총세포분획을가용성 및 불용성 분획으로분리

가용성 분획, 레인 I: 불용성 분획, 레인 5: ?029내 - 0 - 28묘(균주번호 ?예09), 레인 6: ?036내 -꾜?- 281?(균주 번호 的10), 레인 7: ?043내 _ 꾜13- 281?(균주번호 (;011)).
25 도 22는 043-抑1 -꾜13-1}(281^내 ? 43의 2번째 또는 3번째 아미노산 잔 기를 이소루신(I) , 아스파라긴어), 아르기닌(10, 아스팔트산([))으로 치환한此므 -

도 23은 ?643-抑1 - 012-11(281?내 ? 43의 4번째 또는 5번째 아미노산 잔 기를 이소루신(I), 아스파라긴어), 아르기닌(10, 아스팔트산山)으로치환한比!3-
30 1他8요융합폴리펩타이드의 변이체를 로분석한결과이다.
2019/143193 1»(:1^1{2019/000782 도 24는 ?043-11 -꾜15-1狀81?내 43의 6번째 또는 7번째 아미노산 잔 기를 이소루신(I), 아스파라긴어), 아르기닌 00 , 아스팔트산(I))으로 치환한(}1少 -

도 25는 크로마토그래피를 통해 불용성 분획 내의 ?043내 -(! -1秘 8묘 5 융합 폴리펩타이드를 정제한 결과이다(크로마토그램에서 실선 , 점선들은 각각 280 ^파장에서의 흡광도및 용출완충액의 비율을나타냄).
도 26은 크로마토그래피를 통해 정제한 ?643내的 -(^^-11(281?융합 폴리

시료, 肝: 통과액 분획, 레인 ^ 용출액 분획). 화살표는 ?643내 ^ -(}내-1他8묘 10 융합폴리펩타이드를나타낸다.
도 27은 TEV 프로테아제를 이용하여 정제된 ?043내 -此13-1他81? 융합

처리한시료).
15 도 28은 본 발명에 따라정제된 꾜?- 281?의 분자량을 측정한 결과를 나 타낸것이다.
이다(레인 ¾1: 마커 단백질, 레인 1: 11 -꾜13-2쇼2(}(균주 번호 ?的12), 레인 2: (¾)7내 ^ -(}내-2쇼2(}(균주번호 ?的13), 레인 3: ?(}15내的 -(¾ -2쇼26(균주번호 20 的14), 레인 4: ?022내 ^ -(¾ -2쇼2(}(균주 번호 ?的15), 레인 5: ?029내 -
(표 쇼요(균주번호 的16), 레인 6: ?036내 - 0 -2쇼2(;(균주번호 ?0017), 레 인 7: ?043내 -꾜?-2쇼2((균주번호 ?¥18)).
도 30은재조합대장균의 총세포분획을가용성 및 불용성 분획으로분리

25 레인 I: 불용성 분획, 레인 5: ?629내 ^ -꾜13-2요2(}(균주 번호므的16), 레인 6: ?636내附 -(내 쇼요(균주번호 (}017), 레인 7: ?643내的 -(}1 -2요2(;(균주번호 (»18)).
를 이소루신(I), 아스파라긴어), 아르기닌(10, 아스팔트산(미으로 치환한 比
30 2요20융합폴리펩타이드의 변이체를況 로분석한결과이다.
2019/143193 1»(:1^1{2019/000782

를 이소루신(I), 아스파라긴( , 아르기닌犯), 아스팔트산(⑴으로 치환한 0 - 2쇼26융합폴리펩타이드의 변이체를況 쇼湖로분석한결과이다. 5

도 34는크로마토그래피를통해 불용성 분획 내의 ?043내 -(¾ -2융합 폴리펩타이드를 정제한 결과이다(크로마토그램에서 실선, 점선들은 각각 280 ^ 파장에서의 흡광도및 용출완충액의 비율을나타냄).
10 도 35는 크로마토그래피를 통해 정제한 ?043내 - 0夕_2 융합 폴리펩타 이드를 쇼抑로 분석한 결과이다(레인 1- 마커 단백질, 레인 정제 전 시 료, 肝: 통과액 분획 , 레인 1~5: 용출액 분획). 화살표는 ?643내 - 0少-2융합 폴리펩타이드를나타낸다.

처리한시료).
도 37은본발명에 따라정제된(}내-2요2(;의 분자량을측정한결과를나타 낸것이다.
20 도 38은 재조합 대장균에서 발현된 에칼란타이드 융합 폴리펩타이드들의

묘 -此 川 균주 번호 PG019) , 레인 2: (¾)7-}1 -此311¾ 1(16(균주 번 호 ?0020), 레인 3: ?아5내 -此311 1川6(균주 번호 PG021), 레인 4: PG43 - }{61 -묘0311 1 (16(균주번호 ?(}022)).
25 도 39는 재조합 대장균에서 발현된 에칼란타이드 융합 폴리펩타이드들의 총세포 분획을 가용성 및 불용성 분획으로분리한후 敗구요湖로 분석한결과이

恥 - ^3113삵川6(균주 번호 ?的19), 레인 2: ?예7내 - ^ 13 6(균주 번 호 ?的20), 레인 3: ?아5내的 - 13 川6(균주 번호 (}021), 레인 4: ?GA3-
30 - 3113삵 6(균주번호 ?^022)).
2019/143193 1»(:1^1{2019/000782 도 40은 재조합 대장균에서 발현된 네시리타이드 융합 폴리펩타이드들의

5 !오†; (균주번호 (¾)26)).
도 41은 재조합 대장균에서 발현된 네시리타이드 융합 폴리펩타이드들의

?0029) , 레인 4: ?043내61£:’\ 11) 1-84(균주번호 ?0030)) .
15 도 43은 재조합대장균의 총세포분획을가용성 및 불용성 분획으로분리

레인 I : 불용성 분획 , 레인 1: 볘1 -11?1¾1-84(균주번호 ?0027), 레인 2: ?(¾7- 期別 肝 생 균주 번호 ?的28), 레인 3: ?供5내 - 대1-84(균주 번호
?예29), 레인 4: ?043내61 - 대1-84(균주번호므(;030)).
20 도 44는 크로마토그래피를 통해 불용성 분획 내의 ?的7-抑1 -1^}11-84 융합 폴리펩타이드를 정제한 결과이다(크로마토그램에서 실색, 점선들은 각각 280 ¥파장에서의 흡광도및용출완충액의 비율을나타냄).
도 45는크로마토그래피를통해 정제한므(}07내 - 1^111-84융합폴리펩

25 료, 肝: 통과액 분획 , 레인 1~5: 용출액 분획). 화살표는 ?예7내61 -11?대1-84 융합폴리펩타이드를나타낸다.

질, 레인 0: 프로테아제를 처리하지 않은 시료, 레인 I: 프로테아제를 30 처리한시료).
2019/143193 1»(:1^1{2019/000782 도 47은본발명에 따라정제된 1¾1-84의 분자량을측정한결과를나타 낸것이다.
도 48은균주므(}001, ?0003, PG031, ?的32및 ?(}033에서 발현시킨 각각의 융합폴리펩타이드들의 구조를도식화한것이다. 발명의 실시를위한최선의 형태
본 발명의 일 실시예는 하기 일반식 1로 표시되는 아미노산 서열로 이루어진 말단융합파트너 목적 폴리펩타이드 및 상기 말단 융합 파트너와 상기 목적 폴리펩타이드사이에 링커를포함하는융합폴리펩타이드를 제공한다:
[일반식 1]
상기 일반식 1에서,
331내지 336은, 서로독립적으로, 이소루신(11 I), 글리신 ½1 , G), 알라닌(시 쇼), 프롤린( 0 , I 3), 발린 , V), 루신(1 11, 나, 메티오닌 ,

아르기닌( 的, 리신 0^ 則, 히스티딘(바 , 아스팔트산 31), I)) 및 글루탐산 ½111, 으로이루어진군으로부터 선택되고,
상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
구체적으로, 상기 331내지 336은, 서로독집적으로, 이소루신(11 I), 프롤린( 0, ), 루신(1 11, 나, 아스파라긴(쇼 , II), 아르기닌( 당, 요), 히스티딘(바 10 및 아스팔트산 3 I))으로 이루어진 군으로부터 선택될 수 있다.
상기 II이 0의 정수인 경우, 상기 말단 융합 파트너는 7개의 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개, 13개, 14개, 15개, 16개, 17개, 18개, 19개, 20개, 21개, 22개, 23개, 24개,
25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는
36개의 아미노산일 수 있다. 구체적으로, 상기 n이 1의 정수인 경우, 상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 8개, 15개, 22개, 29개또는 36개의 아미노산일수있다.
재조합 미생물 시스템을 이용하여 여러 종류의 목적 폴리펩타이드를 생산하는 경우, 그 타겟 물질의 물성에 따라 숙주세포 내 효소에 의한 분해, 낮은 발현 수준, 부적절한 단백질 폴딩 및/또는 낮은 mRNA 안정성 등으로 인해 수율이 저하될 우려가 있다. 예를 들어, 종래 융합 파트너로 활용되는 MBP (maltose binding protein) , 글루타티온 S-전달효소 (glutathione-S- transferase), 티오레독신 (thioredoxin), SUMO, 유비퀴틴 등은 각각 397, 216, 106, 101, 76 개의 아미노산을 갖는데, 비교적 저분자량의 목적 폴리펩타이드 등을생산할때수율이 좋지 못한문제가있었다.
이에 반해 본 발명에 따른 상기 N-말단 융합 파트너는 7개 내지 43개의 아미노산으로 이루어진 상대적으로 분자량이 작은 펩타이드로서 이를 적용하여 hPTH 1-34 등의 목적 폴리펩타이드를 생산하는 경우 기존의 융합 파트너들을 활용한 경우보다 향상된 수율로 hPTH 1-34를 수득할 수 있다.예를 들어, 재조합 융합폴리펩타이드에서 hPTH 1-34의 비율을 아래 표 1에 개략적으로 나타내었다.
【표 1】

내{!11-34를기준으로링커를포함하여 계산
표 1에 나타난 바와 같이, 종래의 융합 파트너들은 융합 폴리펩타이드에서 ™ 1-34의 비율이 8% 내지 29%에 그치는 반면, 본 발명에 따른 융합 파트너가 융합된 융합 폴리펩타이드에서의 ™ 1-34의 비율은 37% 내지 62%로 확인된다.따라서 동일한 농도의 융합 폴리펩타이드로부터 얻을 수

향상될 수 있다.
2019/143193 1»(:1^1{2019/000782 또한 본 발명에 따른 융합 파트너는 융합 폴리펩타이드의 불용성 발현을 유도하여 융합 폴리펩타이드가 불용성 내포체로서 숙주세포 내로 고농도로 축적되게 할 수 있다.따라서 대장균(묘 아 11) 등의 숙주세포 내에서 프로테아제 및 펩티다제에 의해 전부 혹은 일부가 분해 혹은 절단될 우려가 있는 5 목적 폴리펩타이드를 고수율로수득하기 용이한 장점이 있다.
예를 들어, 상기 ^말단 융합파트너는 하기 일반식 2로 표시되는 아미노산서열을포함하는 것일 수 있다.
[일반식 2]
]\犯七 -父크겄 1-1 16- 용- 0-1元11-1118-(2)11
10 상기 일반식 2에서,
331은 이소루신 , 글리신 , 알라닌, 프롤린, 발린 , 루신, 메티오닌 , 페닐알라닌, 티로신, 트립토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며 ,
상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 15 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
또한, 상기 3 은 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐알라닌, 티로신 및 트림토판으로 이루어진 군으로부터 선택될 수 있다. 구체적으로, 상기 ¾ 은 이소루신(116, I), 아스파라긴 에, ,
20 아르기닌( 10 및 아스팔트산 31), £>)으로 이루어진 군으로부터 선택될 수 있다.
상기 이 0의 정수인 경우, 상기 말단 융합 파트너는 7개의 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 25 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개,
13개, 14개, 15개, 16개, 17개, 18개, 19개, 20개, 21개, 22개, 23개, 24개,
25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는
36개의 아미노산일 수 있다. 구체적으로, 상기 II이 1의 정수인 경우, 상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터
30 시작하는 8개, 15개, 22개, 29개 또는 36개의 아미노산일수 있다.
2019/143193 1»(:1^1{2019/000782 일 구체예로, 상기 일반식 2로 표시되는 아미노산 서열로 이루어진 말단 융합파트너는 서열번호 8, 30, 52, 74, 96 또는 118의 아미노산 서열로 이루어진 것일수있다.
또한, 상기 331은 아스파라긴, 세린, 트레오닌, 시스테인 및
5 글루타민으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식
2로 표시되는 아미노산 서열로 이루어진 1말단 융합파트너는 서열번호 9, 31,
53, 75, 97또는 119의 아미노산서열로이루어진 것일수있다.
선택될 수 있다. 일 구체예로, 상기 일반식 2로 표시되는 아미노산 서열로 10 이루어진 말단 융합파트너는 서열번호 10, 32, 54, 76, 98 또는 120의 아미노산서열로이루어진 것일수있다.
또한, 상기 331은 아스팔트산 또는 글루탐산일 수 있다. 일 구체예로, 상기 일반식 2로 표시되는 아미노산 서열로 이루어진 1말단 융합파트너는 서열번호 11, 33, 55, 77, 99 또는 121의 아미노산 서열로 이루어진 것일 수 15 있다.
또한, 상기 말단 융합파트너는 하기 일반식 3으로 표시되는 아미노산 서열을포함하는것일수있다.
[일반식 3]
]¾ :-요311-¾32 - / ·요-1)1· 0-[ 11내 13-(å)11
20 상기 일반식 3에서,
표332는 이소루신, 글리신 , 알라닌 , 프롤린, 발린 , 루신, 메티오닌 , 페닐알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며 ,
상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 25 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
아르기닌( 8, 10 및 아스팔트산 31), I))으로 이루어진 군으로부터 선택될 수 있다.
30 상기 II이 0의 정수인 경우, 상기 말단 융합 파트너는 7개의
2019/143193 1»(:1^1{2019/000782 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 2는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개,
13개, 14개, 15개, 16개, 17개, 18개, 19개, 20개, 21개, 22개, 23개, 24개,
5 25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는 36개의 아미노산일 수 있다. 구체적으로, 상기 II이 1의 정수인 경우, 상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 8개, 15개, 22개 , 29개 또는 36개의 아미노산일수 있다.
또한, 상기 3크2는 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 10 메티오닌, 페닐알라닌, 티로신 및 트림토판으로 이루어진 군으로부터 선택될 수 있다.
일 구체예로, 상기 일반식 3으로 표시되는 아미노산서열로 이루어진 N- 말단 융합파트너는 서열번호 9, 31, 53, 75, 97 또는 119의 아미노산 서열로 이루어진 것일수있다.
15 또한, 상기 332는 아스파라긴, 세린, 트레오닌, 시스테인 및 글루타민으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식 3으로표시되는아미노산서열로 이루어진 말단융합파트너는서열번호 12, 34, 56, 78, 100또는 122의 아미노산서열로이루어진 것일수 있다.
또한, 상기 332는 아르기닌, 리신 및 히스티딘으로 이루어진 군으로부터 20 선택될 수 있다. 일 구체예로, 상기 일반식 3으로 표시되는 아미노산 서열로 이루어진 말단 융합파트너는 서열번호 13, 35, 57, 79, 101 또는 123의 아미노산서열로이루어진 것일수 있다.
또한, 상기 332는 아스팔트산 또는 글루탐산일 수 있다. 일 구체예로, 상기 일반식 3으로 표시되는 아미노산 서열로 이루어진 I말단 융합파트너는 25 서열번호 14, 36, 58, 80, 102 또는 124의 아미노산 서열로 이루어진 것일 수 있다.
또한, 상기 I말단 융합파트너는 하기 일반식 4로 표시되는 아미노산 서열을포함하는것일수있다.
[일반식 4]
30 -요 -:! 16-¾83-!)1 0 - 1 1 - 3_(å)11
2019/143193 1»(:1^1{2019/000782 상기 일반식 4에서,
333은 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐알라닌 티로신, 트림토판, 아스파라긴 세린, 트레오닌, 시스테인, 글루타민 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며,
5 상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
또한, 상기 333은 이소루신 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐알라닌, 티로신 및 트림토판으로 이루어진 군으로부터 선택될 수

아르기닌( 10 및 아스팔트산 31) I))으로 이루어진 군으로부터 선택될 수 있다.
상기 II이 0의 정수인 경우, 상기 말단 융합 파트너는 7개의 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 15 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개, 13개, 14개, 15개, 16개, 17개, 18개, 19개, 20개, 21개, 22개, 23개, 24개, 25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는
36개의 아미노산일 수 있다. 구체적으로, 상기 II이 1의 정수인 경우, 상기 V든 20 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 8개, 15개, 22개, 29개또는 36개의 아미노산일수 있다.
일 구체예로 상기 일반식 4로 표시되는 아미노산 서열로 이루어진 - 말단 융합파트너는 서열번호 15, 37, 59, 81, 103 또는 125의 아미노산서열로 이루어진 것일수있다.
25 또한, 상기 333은 아스파라긴, 세린, 트레오닌, 시스테인 및 글루타민으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식 4로표시되는 아미노산서열로 이루어진 말단융합파트너는서열번호 16, 38, 60, 82, 104또는 126의 아미노산서열로이루어진 것일수 있다.
또한, 상기 333은 아르기닌, 리신 및 히스티딘으로 이루어진 군으로부터
30 선택될 수 있다. 일 구체예로 상기 일반식 4로 표시되는 아미노산 서열로
2019/143193 1»(:1^1{2019/000782 이루어진 말단융합파트너는서열번호 9, 31, 53, 75, 97또는 119의 아미노산 서열로이루어진 것일수 있다.
또한, 상기 에3은 아스팔트산 또는 글루탐산일 수 있다. 일 구체예로, 상기 일반식 4로 표시되는 아미노산 서열로 이루어진 1말단 융합파트너는 5 서열번호 17, 39, 61, 83, 105 또는 127의 아미노산 서열로 이루어진 것일 수 있다.
또한, 상기 -말단 융합파트너가 하기 일반식 5로 표시되는 아미노산 서열을포함하는것일수있다.
[밀반식 5]
10 1¾ -요311-1 16- 용- 384 - 1 그 -미3-(2)11
상기 일반식 5에서,
334는 이소루신, 글리신 , 알라닌, 프롤린, 발린 , 루신 , 메티오닌, 페닐알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며,
15 상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
또한, 상기 334는 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐알라닌, 티로신 및 트립토판으로 이루어진 군으로부터 선택될 수 20 있다. 구체적으로, 상기 331은 이소루신(11 I), 아스파라긴 , , 아르기닌( 용, 10 및 아스팔트산 31), I))으로 이루어진 군으로부터 선택될 수 있다.
상기 이 0의 정수인 경우, 상기 I말단 융합 파트너는 7개의 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 25 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개, 1371] , 14711 , 1571] , 1671] , 1괘, 1871] , 19ᅫ, 2왜 , 2171] , 22711 , 237]] , 2471] , 25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는 36개의 아미노산일 수 있다. 구체적으로, 상기 II이 1의 정수인 경우, 상기 는 30 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터
2019/143193 1»(:1^1{2019/000782 시작하는 8개, 15개, 22개, 29개또는 36개의 아미노산일수있다.
일 구체예로, 상기 일반식 5로 표시되는 아미노산 서열로 이루어진 말단 융합파트너는 서열번호 8, 40, 62, 84, 106 또는 128의 아미노산 서열로 이루어진 것일수있다.
5 또한, 상기 334는 아스파라긴, 세린, 트레오닌, 시스테인 및 글루타민으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식

63, 85, 107또는 129의 아미노산서열로이루어진 것일수 있다.
또한, 상기 크크4는아르기닌, 리신 및 히스티딘으로 이루어진 군으로부터 10 선택될 수 있다. 일 구체예로, 상기 일반식 5로 표시되는 아미노산 서열로 이루어진 -말단 융합파트너는 서열번호 20, 42, 64, 86, 108 또는 130의 아미노산서열로이루어진 것일수 있다.
또한, 상기 334는 아스팔트산 또는 글루탐산일 수 있다. 일 구체예로,
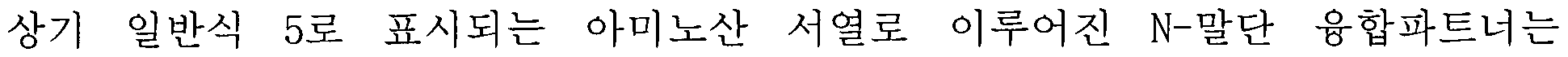
15 서열번호 21, 43, 65, 87, 190 또는 131의 아미노산 서열로 이루어진 것일 수 있다.
또한, 상기 1말단 융합파트너는 하기 일반식 6으로 표시되는 아미노산 서열을포함하는것일수있다.
[일반식 6]
20 ] 1 :-쇼311-1 16- 용_1)1,0 - 3크5 - 3_(å)11
상기 일반식 6에서,
335는 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며 ,
25 상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
또한, 상기 335는 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐알라닌, 티로신 및 트림토판으로 이루어진 군으로부터 선택될 수
30 있다. 구체적으로, 상기 은 이소루신 (11 I), 아스파라긴 , ,
2019/143193 1»(:1^1{2019/000782 아르기닌( 요, 10 및 아스팔트산 3 이으로 이루어진 군으로부터 선택될 수 있다.
상기 II이 0의 정수인 경우, 상기 -말단 융합 파트너는 7개의 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개,
13개, 14개, 15개, 16개, 17개, 18개, 19개, 20개, 21개, 22개, 23개, 24개,
25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는 36개의 아미노산일 수 있다. 구체적으로, 상기 II이 1의 정수인 경우, 상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 8개, 15개, 22개, 29개또는 36개의 아미노산일수 있다.
일 구체예로, 상기 일반식 6으로 표시되는 아미노산서열로 이루어진 - 말단 융합파트너는 서열번호 22, 44, 66, 88, 110 또는 132의 아미노산 서열로 이루어진 것일수있다.
또한, 상기 3크5는 아스파라긴, 세린, 트레오닌, 시스테인 및 글루타민으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식 6으로표시되는아미노산서열로 이루어진 말단융합파트너는서열번호 23 45, 67, 89, 111또는 135의 아미노산서열로이루어진 것일수 있다.
또한, 상기 335는아르기닌, 리신 및 히스티딘으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식 6으로 표시되는 아미노산 서열로 이루어진 말단 융합파트너는 서열번호 24, 46, 68, 90, 112 또는 134의 아미노산서열로이루어진 것일수있다.
또한, 상기 335는 아스팔트산 또는 글루탐산일 수 있다. 일 구체예로, 상기 일반식 6으로 표시되는 아미노산 서열로 이루어진 I말단 융합파트너는 서열번호 25, 47, 69, 91, 113 또는 135의 아미노산 서열로 이루어진 것일 수 있다.
또한, 상기 말단 융합파트너는 하기 일반식 7로 표시되는 아미노산 서열을포함하는것일수있다.
[일반식 7]
-쇼 - 16- 요-131 0 - 해 -
2019/143193 1»(:1^1{2019/000782 상기 일반식 7에서
336은 이소루신, 글리신, 알라닌, 프롤린 , 발린, 루신, 메티오닌, 페닐알라닌, 티로신 트림토판, 아스파라긴 세린, 트레오닌 시스테인 글루타민, 아르기닌 리신, 히스티딘, 아스팔트산또는글루탐산이며,
5 상기 V己 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
또한, 상기 336은 이소루신, 글리신, 알라닌, 프롤린, 발린 루신, 메티오닌, 페닐알라닌, 티로신 및 트립토판으로 이루어진 군으로부터 선택될 수 10 있다. 구체적으로, 상기 331은 이소루신(11 I), 아스파라긴(쇼 , , 아르기닌( 10 및 아스팔트산(쇼31), I))으로 이루어진 군으로부터 선택될 수 있다
상기 II이 0의 정수인 경우, 상기 말단 융합 파트너는 7개의 아미노산으로 이루어진 것일 수 있다. 또한, 상기 II이 1의 정수인 경우, 상기 15 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 1개, 2개, 3개, 4개, 5개, 6개, 7개, 8개, 9개, 10개, 11개, 12개,
13개, 14개, 15개, 16개, 17개, 18개, 19개, 20개, 21개, 22개, 23개, 24개, 25개, 26개, 27개, 28개, 29개, 30개, 31개, 32개, 33개, 34개, 35개 또는
36개의 아미노산일 수 있다. 구체적으로, 상기 II이 1의 정수인 경우, 상기 1는 20 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 8개, 15개, 22개, 29개또는 36개의 아미노산일수있다.
일 구체예로 상기 일반식 7로 표시되는 아미노산 서열로 이루어진 1 말단융합파트너는 서열번호 26, 48, 70, 92, 114또는 136의 아미노산 서열로 이루어진 것일수있다
25 또한, 상기 336은 아스파라긴, 세린, 트레오닌, 시스테인 및 글루타민으로 이루어진 군으로부터 선택될 수 있다. 일 구체예로, 상기 일반식

71, 93, 115또는 137의 아미노산서열로이루어진 것일수있다.
또한 상기 336은아르기닌, 리신 및 히스티딘으로 이루어진 군으로부터
30 선택될 수 있다. 일 구체예로, 상기 일반식 7로 표시되는 아미노산 서열로
2019/143193 1»(:1^1{2019/000782 이루어진 말단 융합파트너는 서열번호 28, 50, 72, 94, 116 또는 138의 아미노산서열로이루어진 것일수있다.
또한, 상기 336은 아스팔트산 또는 글루탐산일 수 있다. 일 구체예로, 상기 일반식 7로 표시되는 아미노산 서열로 이루어진 I말단 융합파트너는 5 서열번호 29, 51, 73, 95, 117 또는 139의 아미노산 서열로 이루어진 것일 수 있다.
상기 일반식 1내지 7에 있어서, 상기 II이 0의 정수인 경우, 상기 말단 융합파트너는 7개의 아미노산으로 이루어진 것일 수 있으며, 본발명에서 7개의 아미노산으로 이루어진 I말단 융합 파트너를 7的7”로 명명하였다. 또한, 10 상기 II이 1의 정수인 경우, 상기 는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노산에서부터 시작하는 8개, 15개, 22개, 29개 또는 36개의 아미노산일 수 있다. 이 경우, 상기 말단 융합 파트너는 15개, 22개, 29개, 36개 또는 43개의 아미노산으로 이루어진 것일 수 있으며, 본 발명에서 15개, 22개, 29개, 36개 또는 43개의 아미노산으로 이루어진 I말단 융합

I말단 유도체일 수 있다. 또한, 상기 I말단 융합 파트너는 7 내지 43개의 아미노산을 갖는 펩타이드로서, 서열번호 119의 말단에서 (:-말단으로 7 내지 20 43개의 연속적인아미노산으로이루어진 것일수 있다.
하나의 아미노산 서열로 이루어진 것일 수 있다. 상기 융합 파트너의 아미노산 갯수는목적 폴리펩타이드의 특성에 따라조절될수 있다. 예를들어, 7개, 8개,
9개, 10개, 13개, 15개, 17개, 22개, 25개, 27개, 29개, 30개, 33개, 38개, 40개, 25 43개 등일 수 있다. 일 구체예로서, 상기 말단융합파트너는서열번호 9, 31,
53, 75, 97또는 119로표시되는아미노산서열로이루어진 것일수있다.
본 발명의 또 다른 측면은, 전술한 바와 같은 신규한 말단 융합 파트너, 목적 폴리펩타이드 및 상기 말단 융합 파트너와 상기 목적 폴리펩타이드 사이에 링커를 포함하는융합폴리펩타이드를 제공한다.
30 상기 링커는 친화성 태그를 포함할 수 있다.본 발명에서 사용된 용어
2019/143193 1»(:1^1{2019/000782
"친화성 태그"는 재조합 융합 폴리펩타이드 또는 이를 코딩하는 핵산에 도입될 수 있는 펩타이드 또는 핵산 서열로서 다양한 목적으로 사용될 수 있으며 예를 들어 목적 폴리펩타이드의 정제 효율을 높이기 위한 것일 수 있다.본 발명에서 사용될 수 있는 친화성 태그는 당업계에 공지된 임의의 적합한 물질을 원하는 5 바에 따라 사용할 수 있다. 예를 들어, 본 발명에 사용되는 친화성 태그는 폴리히스티딘 태그(서열번호 7또는 8) 폴리라이신 태그(서열번호 9또는 10) 또는 폴리아르기닌 태그(서열번호 11또는 12)일 수 있다.
또한, 상기 링커는 프로테아제 인식 서열을 포함할 수 있다. 프로테아제란 특정 아미노산 서열을 인식하여, 인식한 서열 내의 펩타이드 10 결합이나 그 서열의 마지막 아미노산과 융합된 폴리펩타이드의 첫번째 아미노산과의 펩타이드 결합을 절단하는 단백질 분해효소이다.본 발명에 따른 융합 폴리펩타이드는 프로테아제 인식 서열을 갖는 링커를 포함함으로써 최종 단계에서 폴리 펩타이드 정제시 절단효소 인식 서열이 포함된 아미노 말단(친화성 태그를 사용한 경우에는 친화성 태그도 포함)과 목적 15 폴리펩타이드의 1말단을분리시켜 목적 폴리펩타이드를 회수할수 있다.
구체적으로, 상기 프로테아제 인식 서열은 담배 식각 바이러스 프로테아제 인식 서열 엔테로키나아제 인식 서열, 유비퀴틴 가수분해효소 인식 서열 인자 퓨린 및 이들의 조합으로 이루어진 군으로부터 선택되는 인식 서열일 수 있다.예를 들어 상기 프로테아제 인식 서열은 서열번호 146내지 20 150의 아미노산서열 중 어느 하나를포함할수 있다.
본 발명에서 사용된 용어 ’’목적 폴리펩타이드’’는 재조합 생산 시스템을 통해 수득하고자하는폴리펩타이드를 의미한다.
상기 목적 폴리펩타이드는 본 발명에 따른 -말단 융합 파트너와 융합을 통해 발현수준이 향상될 뿐만 아니라, 불용성 내포체 형태로 세포 내에 25 축적됨으로써 숙주세포 내 효소에 의한 분해로부터 보호될 수 있어, 결과적으로 더 높은 수율로 수득될 수 있다.또한, 상기 목적 폴리펩타이드는 서열번호 18 내지 27의 아미노산 서열 중 어느 하나를 포함할 수 있다.바람직하게는 상기 목적 폴리펩타이드는 2 ¾고3내지 15壯)3, 2.51出3내지 14壯)크 3壯 내지 13 壯 , 3.51出3내지 12抑3, 4壯)3내지 11壯)3의 분자량을 갖는 것일 수 있다.
30 구체적으로, 상기 목적 폴리펩타이드는 인간 부갑상선 호르몬 1-340?™
1-34), 인간부갑상선 호르몬 l-84(hPTH 1-84), 글루카곤 유사 펩타이드- GLP- 1), 리라글루타이드 (liraglut ide) 전구체 펩타이드, 엑세나타이드 (exenat ide), 인슐린 유사 성장인자- l(IGF-l), 글루카곤 유사 펩타이드- 2(GLP-2), 테두글루타이드 (teduglut ide), 에칼란타이드 (ecal lantide),
5 네시리타이드 (nesiritide) , 인슐린 및 인슐린 유사체로 이루어진 군으로부터 선택되는어느하나일수있다.
상기 인간 부갑상선 호르몬의 아미노 말단 단편인 hPTH 1-34 (Human Parathyroid Hormone 1_34)는, 갑상선에 의해 분비되는 115개 아미노산 (aa)의 프리-프로-펩타이드 (prepropept ide) 형태로 발현되어 신호서열 및 프로펩타이드 10 제거 후에 혈액으로 분비되는 펩타이드로서, 혈액 내 칼슘 농도를 증가시키는 작용을 하며 뼈 형성을 자극하는 것으로 알려져 있다. hPTH 1-34는 인간 부갑상선 호르몬의 아미노 말단 부위의 34개의 아미노산을 갖는 펩타이드로, 테리파라타이드 (Teriparat ide)로 지칭되기도 한다. 예를 들어, 상기 hPTH 1-34 폴리펩타이드는 서열번호 151의 아미노산 서열로 이루어질 수 있고, 상기 15 아미노산서열은서열번호 292의 염기서열에 의해 코딩될수 있다.
또한, 상기 인간 부갑상선 호르몬 l-84(hPTH 1-84)는, 갑상선에 의해 분비되는 115개 아미노산 (aa)의 프리-프로-펩타이드로부터 유래한 84개의 아미노산을 갖는 펩타이드로, 혈액 내 칼슘 농도를 증가시키는 작용을 하며 뼈 형성을 자극하는 것으로 알려져 있다. hPTH 1-84는 일반적으로 희귀질환인 20 저칼슘혈증 (hypocalcemia) 또는 부갑상선 기능저하증 (hypoparathyroidi sm)의 치료제로사용된다. 예를 들어, 상기 hPTH 1-84폴리펩타이드는서열번호 628의 아미노산 서열로 이루어질 수 있고, 상기 아미노산 서열은 서열번호 633의 염기서열에 의해 코딩될수 있다. 상기 목적 폴리펩타이드는서열번호 151, 340, 341, 484, 485, 628, 638, 642및 652의 아미노산서열 중어느하나의 아미노산 25 서열로이루어진 것일수있다.
또한, 상기 글루카곤 유사 펩타이드- l(GLP-l)은 31개의 아미노산으로 구성된 폴리펩타이드이다. 이와 관련하여, 리라글루타이드는 이의 유사체로서 GLP-1의 28번째 라이신이 아르기닌으로 치환 (K28R)되어 있으며, 20번째 라이신 잔기의 아미노 그룹에 팔미트산과 글루탐산으로 구성된 N-Palmitoyl-L-glutamic 30 acid가 결합되어 있는 형태를 갖는다. 리라글루타이드는 제 2형 당뇨병 또는
비만을 치료제로 사용될 수 있으며, 일반적으로 리라글루타이드는 N-Palmitoyl- L-glutamic acid가 결합되어 있지 않은 리라글루타이드 전구체 펩타이드 (GLP- 1K28R)를 생산하고, 이의 20번째 라이신 잔기에 N-Palmitoyl-L-glutamic acid를 결합시키는공정으로생산할수 있다 (Dunweber , Jensen et al . 2007) . 예를들어 , 상기 GLP-1 폴리펩타이드는서열번호 340의 아미노산서열로 이루어질 수 있고, 상기 아미노산서열은 서열번호 475의 염기서열에 의해 코딩될 수 있다. 또한, 상기 리라글루타이드 전구체 펩타이드 (GLP-1K28R)는 서열번호 341의 아미노산 서열로 이루어질 수 있고, 상기 아미노산 서열은 서열번호 476의 염기서열에 의해코딩될수 있다.
또한, 상기 글루카곤 유사 펩타이드- 2(GLP-2)는 33개의 아미노산으로 구성된 폴리펩타이드이다. 이와 관련하여 , 테두글루타이드는 이의 유사체로서 GLP-2의 2번째 알라닌이 글리신으로 치환 (A2G)되어 있는 형태이다. 테두글루타이드는 희귀질환인 단장증후군 (Short Bowel Syndrome) , 화학 요법 유발성 설사 (Chemotherapy- Induced Diarrhea) 및 장피 누공 (Enterocutaneous Fistula)의 치료제로 사용될 수 있다. 예를 들어, 상기 GLP-2 폴리펩타이드는 서열번호 484의 아미노산 서열로 이루어질 수 있고, 상기 아미노산 서열은 서열번호 619의 염기서열로 코딩될 수 있다. 또한, 상기 테두글루타이드 폴리펩타이드 (GLP-2A2G)는 서열번호 485의 아미노산 서열로 이루어질 수 있고, 상기 아미노산서열은서열번호 620의 염기서열에 의해 코딩될수 있다.
또한, 상기 에칼란타이드는 60개의 아미노산으로 구성된 폴리펩타이드로서, 인간 혈장 내 칼리크레인 (kal l ikrein)의 저해할 수 있으며, 결과적으로고분자량을갖는칼리크레인이 브래디키닌 (Bradykinin)으로 전환되는 것을 저해하는 작용을 한다. 에칼란타이드는 희귀질환인 유전성 혈관부종 (Hereditary Angioedema)의 치료제로사용될 수 있다. 예를 들어, 상기 에칼란타이드폴리펩타이드는서열번호 642의 아미노산서열로 이루어질 수 있고, 상기 아미노산서열은서열번호 647의 염기서열에 의해 코딩될수 있다.
또한, 상기 네시리타이드는 32개의 아미노산으로 구성된 폴리펩타이드로서, 인간 심실 심근 (ventricular myocardium)의 분비되는 B형 나트륨이뇨 펩타이드 (natriuretic peptide)이다. 네시리타이드는 울혈성 심부전증의 치료제로 사용될 수 있다. 예를 들어, 상기 네시리타이드
2019/143193 1»(:1^1{2019/000782 폴리펩타이드는 서열번호 652의 아미노산 서열로 이루어질 수 있고, 상기 아미노산서열은서열번호 657의 염기서열에 의해 코딩될수 있다.
또한, 상기 엑세나타이드 폴리펩타이드는 서열번호 638의 아미노산 서열로 이루어질 수 있고 상기 아미노산 서열은 서열번호 639의 염기서열로 5 코딩될 수 있고 상기 인슐린 유사성장인자- 1(101^-1) 폴리펩타이드는서열번호 640의 아미노산서열로이루어질수 있고, 상기 아미노산서열은서열번호 641의 염기서열에 의해 코딩될수있다.
한편 본 발명에 따른 융합 폴리펩타이드는, 서열번호 1의 아미노산 서열을 포함하는 융합 파트너와 목적 폴리펩타이드가 서로 상이한 등전점을 10 가짐으로써 목적 폴리펩타이드가 고순도로 용이하게 정제될 수 있다. 단백질의

이의 등전점에 따라분리될수있다.
예를 들어, 본 발명의 서열번호 8 내지 139의 아미노산서열을 갖는 말단융합파트너의 값은 9.5내지 10.5일 수 있다. 구체적으로 서열번호 9, 15 31, 53, 75, 97또는 119의 아미노산서열을 갖는 말단융합파트너의 이값은 각각 9.52, 11.72, 10.27, 10.27, 10.43, 10.42일수 있다.
리라글루타이드 전구체 펩타이드 테두글루타이드, 에칼란타이드 네시리타이드의 ?1 값은 각각 8.29, 9.10, 5.53, 4.17, 5.58, 10.95이다. 즉, 20 목적 폴리펩타이드들의 pl 값과 말단 융합 파트너 및 이를 포함하는 융합 파트너들의 1 1 값은 실질적으로 상이하다. 따라서 이온 교환 크로마토그래피, 등전점 침전법 등의 방법을 이용하여 융합 파트너로부터 목적 폴리펩타이드의 정제를 이온 교환 크로마토그래피, 등전점 침전법 등의 방법을 이용하여 용이하게할수있다.
25 또한, 상기 융합 파트너, 링커 및 목적 폴리펩타이드를 포함하는 신규한 융합폴리펩타이드는서열번호 160내지 291, 343내지 474, 487내지 618, 630 내지 632, 644 내지 646 및 654 내지 656의 아미노산 서열 중 어느 하나의 아미노산서열로이루어진 것일수있다.
본 발명의 다른 측면은 전술한 융합 폴리펩타이드를 코딩하는 30 뉴클레오타이드를제공할수있고, 예를들어 160내지 291, 343내지 474, 487
2019/143193 1»(:1^1{2019/000782 내지 618, 630 내지 632, 644 내지 646 및 654 내지 656의 아미노산 서열 중 어느하나를 코딩할수 있으며, 상기 뉴클레오타이드는서열번호 294, 295, 478 내지 483, 621 내지 627, 635 내지 637, 649 내지 651 및 659 내지 661의 염기서열중어느하나의 염기서열로이루어진 것일수있다.
5 본 발명의 또 다른 측면은, 전술한 융합 폴리펩타이드를 코딩하는 뉴클레오타이드분자를포함하는발현벡터를제공한다. 본발명에서 사용된 용어 "벡터”는숙주세포에 도입되어 숙주세포유전체 내로 재조합및 삽입될수 있다. 또는 상기 벡터는 에피좀으로서 자발적으로 복제될 수 있는 뉴클레오타이드 서열을포함하는 핵산수단으로 이해된다. 상기 벡터는 선형 핵산 플라스미드, 10 파지미드, 코스미드, ^ 벡터, 바이러스 벡터 및 이의 유사체들을 포함한다. 바이러스 벡터의 예로는 레트로바이러스, 아데노바이러스, 및 아데노-관련 바이러스를 포함하나 이에 제한되지 않는다. 또한 상기 플라스미드는 항생제 내성 유전자와 같은 선별 마커를 포함할 수 있고, 플라스미드를 유지하는 숙주세포는선택적인조건하에서 배양될수있다.
15 본 발명에서 사용된 용어 "숙주세포'’는 재조합 발현벡터가 도입될 수 있는 원핵세포 또는 진핵세포를 나타낸다. 본 발명에서 사용된 용어, "형질도입'’은, 당업계에 공지된 기술을 사용하여 세포 내로 핵산(예를 들어, 벡터)을도입하는것을의미한다.
본발명의 다른측면은 상기 발현벡터를포함하는숙주세포를 제공한다. 20 상기 숙주세포는, 본 발명의 융합 폴리펩타이드를 코딩하는 뉴클레오타이드가 포함되도록 형질전환되어 목적 폴리펩타이드의 발현 및/또는 분비에 이용될 수 있다. 본 발명에 사용될 수 있는 바람직한 숙주세포는 대장균 세포 불사의 하이브리도마 세포 八) 골수종 세포, 293 세포 중국 햄스터 난소 세포((¾() 0611) ¾1 세포, 인간 양수유래 세포 0:¾江 611) 또는 0»세포를포함한다. 25 예를 들어 본 발명에서 융합폴리펩타이드를 발현시키는데 사용된 숙주 균주는 대장균 21(에3)이며, 상기 유전자 및 이들의 사용 방법은 당업계에 공지되어 있다.
본 발명의 또 다른 측면은, (3) 전술한 숙주세포를 배양하는 단계, ( 숙주세포에서 발현된 융합 폴리펩타이드를 정제하는 단계 및 ((:) 상기 정제된 30 융합 폴리펩타이드를 절단 효소와 배양하여 목적 폴리펩타이드를 회수하는
단계를 포함하는 목적 폴리펩타이드 재조합 폴리펩타이드 의 생산방법을 제공한다.
상기 (a) 단계에서는, 본 발명의 융합 폴리펩타이드를 코딩하는 뉴클레오타이드를갖는발현벡터를포함하는숙주세포를배양한다 .
5 이때, 상기 숙주세포는 임의의 발효방식으로배양될 수 있다, 예를들어, 회분식 , 유가식, 반연속식 및 연속식 발효 방식이 이용될 수 있다. 실시예에서 , 발효배지는복합배지 혹은제한배지로부터 선택될수 있다. 특정 실시예에서, 제한 배지가 선택된다. 제한 배지는 낮은 수준의 아미노산, 티아민과 같은 비타민또는다른성분들로보충될수 있다. 본발명의 방법에 유용한배양절차 10 및 무기염 배지의 상세한 설명은 문헌 (Riesenberg, Schulz et al . 1991)에 기재되어 있다.
예를들어, 융합폴리펩타이드의 생산은발효기 배양에서 달성될수 있다. 예를들어, 2 L의 제한 배지를 함유하는 발효기에서 37°C의 온도에서 배양하고, 염산이나 암모니아 첨가를 통해 pH 6.8로 유지할 수 있다. 용존 산소는 교반 15 속도 및 발효기 내로 공기 유속 (air f low rate) 증가, 또는 필요에 따라 순수산소 주입을 통해 과량으로 유지할 수 있다. 발효기 내의 세포를 고농도로 배양하기 위해 세포 배양 동안 포도당 혹은 글리세롤이 포함된 주입 용액 (feeding solut ion)을배양액에 전달할수 있다.
또한, 상기 조건을 유지하다가, 유도를 위한 목표 배양액의 광학 20 밀도 (opt ical density) , 예를 들어, 600 nm 파장에서 특정 값의 광학 밀도 (A600)에 도달하면, IPTG를 첨가하여 융합폴리펩타이드의 발현이 개시될수 있다. 유도시 세포의 광학밀도, IPTG농도, pH, 온도, 용존산소수준, 각각을 변화시켜 최적의 발현 조건을 결정할 수 있다. 또한, 유도 시 세포의 광학 밀도는 A600 30 내지 300의 범위에서 변화시킬 수 있다. IPTG 농도는 0.01 mM 25 내지 1.0 mM범위로, pH는 5.5내지 7.5범위로, 온도는 15 °C 내지 37 °C 범위로, 그리고 공기 유속은 1 m (분당 배지 리터당 공기 리터) 내지 5 wm 범위로 변화시킬 수 있다. 유도 후 4시간 내지 48시간 후에, 발효기로부터의 배양액을 원심분리하여 세포를 수거하고, 세포 펠렛은 -80°C 온도에서 냉동할 수 있다. 배양액 시료는 재조합융합폴리펩타이드 발현 정도 분석을 위해 SDS-PAGE등의 30 방법으로분석할수있다.
2019/143193 1»(:1^1{2019/000782

때조건에서 행해질 수 있다. 1^0 계열 프로모터를 가진 발현 구조물이 사용될 때, 발현은 1肝(}를약 0.01 내지 약 1.0 의 최종농도로배양물에 첨가하여 유도할수있다.
5 유도제를 첨가한 후, 배양액은 일정 기간, 예를 들어, 약 12시간 동안 배양할 수 있는데, 이 기간에 재조합 단백질이 발현된다. 유도제를 첨가한후, 배양액은약 4시간내지 48시간동안배양할수 있다.
세포 배양액은 원심분리하여 세포가 존재하지 않는 배지(상등액)를 제거하고세포를회수할수 있다. 예를들어 세포배양액은 12,000印 조건에서 10 30분간(4〔〕) 원심분리하고 상등액을 제거하여 불용성 분획을 얻을 수 있다. 원심분리에 의해 얻은 불용성 분획에 불용성 내포체 형태로 존재하는 재조합 융합 폴리펩타이드의 가용화를 위해 불용성 분획을 요소나 염산구아니딘과 같은 카오트로픽제((±30打01)比 3요6 )가 포함된 완충액으로 재현탁할 수 있다. 실시예에서 세포는 고압 기계적 세포 파쇄 장치(예를 들어
15 마이크로플루다이저 0 0£ 111 ^261·))를 사용하여 파괴한다. 재현탁된 세포는 예를 들어, 초음파 처리를 사용하여 파괴할 수 있다. 당업계에 공지된, 세포를 용해시키는데 적합한임의의 방법을 이용하여 세포내 축적된 내포체들을회수할 수 있다. 예를 들어 실시양태에서, 화학적 및/또는 효소적 세포 용해 시약, 예를들어, 세포벽 용해 효소( 2 6) 및 쇼가사용될수있다.
20 상기 ( 단계에서는, 상기 (크)단계에서 배양한 숙주세포에서 발현된 융합폴리펩타이드를정제한다.
이때 불용성 분획에는 불용성 내포체 형태로 발현된 융합 폴리펩타이드가 주로 존재한다. 불용성 분획에 존재하는 내포체의 가용화는 카오트로픽제가포함된 변성 조건하에서 수행할수 있다. 내포체 가용화조건은, 25 카오트로픽제가 포함된 완충액의 사용을 포함할 수 있고, 내포체 가용화 완충액은 카오트로픽제로 요소 또는 염산구아니딘, 인산나트륨 또는 트리스 완충액 및 염화나트륨을 포함할 수 있다. 또한 친화성 크로마토그래피를

가용화완충액은 이미다졸을포함한다. 실시예에서, 내포체 가용화완충액은 4
30 내지 10 의 요소또는 3 내지 8 의 염산구아니딘을포함할수 있다. 또한,
2019/143193 1»(:1^1{2019/000782 내포체 가용화 완충액은 5 내지 100 인산나트륨 또는 트리스(1出 7 내지

포함할수 있다. 또한, 1齡(:을위한내포체 가용화완충액은 0 내지 50 의 이미다졸을 포함할 수 있다. 이때, 내포체 가용화 완충액은, 8 1 요소, 20 5 트리스 500 염화나트륨 50 이미다졸, 1出 7.4를포함하고 용해된 세포의 원심분리로 얻어진 불용성 분획을 재현탁하여 불용성 분획의 융합 폴리펩타이드의 내포체를가용화할수 있다.
예를 들어 내포체 가용화 완충액으로 불용성 분획의 내포체를 가용화 시키는경우, 2 0 내지 8 0 온도에서 약 1시간내지 6시간동안진탕배양한후 10 12,000 께 (12,000 용) 조건에서 30분간(41:) 원심분리하여 불용성 분획에 존재하는 파쇄된 세포 잔해를 제거하고 가용화된 융합폴리펩타이드가포함되어 있는 상등액을 얻는다. 상등액은 적층형 필터(( 아11 라) 및 막 필터로 여과하여 불용성 및 고형 성분을제거하여 정제용 컬럼에 주입될 수 있도록한다. 또한, 불용성 내포체 형태로 발현된 후 가용화된 재조합 융합
15 폴리펩타이드 또는 목적 폴리펩타이드는 크기 배제 음이온 또는 양이온 교환, 소수성 상호작용, 또는친화성 크로마토그래피 방법에 의해 다른단백질 및 세포 잔해로부터 분리하거나정제할수 있다.
예를 들어, 본 발명의 융합 폴리펩타이드는 폴리히스티딘 태그(6 -

내포체 가용화완충액으로세척 후용출완충액(8 요소, 20 트리스, 500 25 염화나트륨, 500 이미다졸, 1出 7.4)의 비율을 100%로단계적 증가시켜 컬럼에 결합된융합폴리펩타이드를용출시키고분획을수집할수 있다.
상기 ( ) 단계에서는, 전술한방법으로정제된 융합폴리펩타이드를 절단 효소와배양하여 목적 폴리펩타이드를회수한다.
이때 융합 폴리펩타이드는 절단효소에 의해 적합하게 절단될 수 있어,
30 목적 폴리펩타이드를 적합한 형태로 유리시킬 수 있다. 정제된 융합
폴리펩타이드 분획에는 8 의 요소가 첨가되어 있으므로 절단효소의 변성을 막기 위해 희석 완충액(20 mM트리스, pH 7.4)를 이용하여 요소 농도가 1 M이 되도록 희석하는 것이 바람직하다. 융합 폴리펩타이드는 정제 후 요소 농도가 1 M이 되도록 희석되어 20 mM트리스, pH 7.4, 1 M요소, 62.5 mM염화나트륨, 62.5 mM 5 이미다졸을 함유하는 완충액에 존재할 수 있다. 재조합 융합 폴리펩타이드는 절단효소와 반응하여 친화성 태그 및 절단효소 인식 서열이 포함된 아미노 말단 융합 파트너와목적 폴리펩타이드로 절단된다.프로테아제 절단 방법은 당업계에 공지되고 제조사의 지시서를 포함한문헌에 기재된 임의의 적합한 방법이 이용될 수 있다. 바람직하게는, 정제 후 요소 농도가 1 이 되도록 희석된 융합 10 폴리펩타이드에 TEV 프로테아제를 최종 농도가 500 이이 되도록 첨가하고 실온에서 6시간 이상 절단 반응이 진행될 수 있도록 한다. 예를 들어, TEV 프로테아제에 의해 재조합융합폴리펩타이드는 약 60%내지 약 100%가 절단된다. 재조합 융합 폴리펩타이드 또는 목적 폴리펩타이드의 수율은 당업자에게 공지된 방법, 예를 들어, SDS-PAGE( sodium dodecyl sul fate - polyacrylamide B gel electrophoresis) 또는 웨스턴 블롯 (Western blot ) 분석에 의해 결정할 수 있다. SDS-PAGE로 전기영동된 겔 (gel )은 염색 (staining) , 탈색 (destaining) , 디지털 영상화과정을거쳐 재조합융합폴리펩타이드또는목적 폴리펩타이드의 대략적인정량및 정성 분석이 가능하다.
또한, 정제된 융합 폴리펩타이드 또는 목적 폴리펩타이드의 농도는
20 당업계에 공지되고 문헌에 기재된 방법에 의한 흡광도 분광법으로 결정될 수 있다.
정제된 융합 폴리펩타이드 또는 목적 폴리펩타이드의 수율 또는 순도를 결정하기 위한 웨스턴 블롯 분석은 SDS-PAGE 겔 상에서 분리된 단백질을 니트로셀룰로오스 막으로 이동시키고, 목적 폴리펩타이드에 특이적인 항체를 25 이용하는 당업계에 공지된 적합한 방법에 따라 수행될 수 있다. 실시예에서, 목적 폴리펩타이드의 순도를측정하기 위한방법 중하나로 ELISA (enzyme- l inked immunosorbent assay)방법을이용할수있다.
정제된 융합 폴리펩타이드 또는 목적 폴리펩타이드의 수율은 배양액의 부피당 정제된 융합 폴리펩타이드 또는 목적 폴리펩타이드의 양 (예를 들어, g
30 또는 mg의 단백질/리터의 배양액 부피), 융합폴리펩타이드의 백분율 (예를들어,
재조합 융합 폴리펩타이드의 양/전체 세포 단백질(total cell protein)의 양), 및 건조 균체량(dry cell weight)에 대한 백분율 또는 비율을 포함한다. 본원에 기재된 폴리펩타이드의 수율의 척도는 완전한 형태로 발현된 해당 폴리펩타이드의 양을 기준으로 한다.
5 수율이 배양액 부피에 대한 정제된 융합 폴리펩타이드 또는 목적 폴리펩타이드의 양으로 표현되는 경우, 배양된 세포 밀도 혹은 세포 농도가 수율을 결정하는데 고려될 수 있다.
또한, 절단효소로 절단된 후 얻어진 목적 폴리펩타이드의 수율은 약 0.54 g/L내지 약 13.5 g/L일 수 있다.본 발명에서,목적 폴리펩타이드의 수율은 5

본 발명의 구체예는 서열번호 1의 아미노산 서열을 갖는 융합 파트너와 재조합된 융합 폴리펩타이드를 이용하여 목적 폴리펩타이드를 수득함으로써, 목적 폴리펩타이드가 세포 내에서 효소에 의해 분해되거나, 부적절한 폴딩이 발생하는 등의 문제점을 최소화하여 목적 물리펩타이드를 고수율로 생산하는 15 방법을 제공할 수 있다. 구체적인 생산 방법의 일 예시는 실시예를 통해 설명하기로 한다. 발명의 실시를위한형태
이하, 본 발명을 하기 실시예에 의하여 더욱 상세하게 설명한다. 단, 20 하기 실시예는본 발명을 예시하기 위한 것일 뿐, 본 발명의 범위가 이들만으로 한정되는것은아니다.
실시예 1. hPTH 1-34융합폴리펩타이드의 제조및 생산
실시예 1.1. hPTH 1-34융합폴리펩타이드발현플라스미드의 제작
hPTH 1-34 융합 폴리펩타이드에 대한 유전자는 overlap extension 25 polymerase chain react i on(0E-PCR) 방법을 이용하여 합성하였다. 이때, 상기 hPTH 1-34융합폴리펩타이드는아미노말단융합파트너로서 PG07(서열번호 9), PG15(서열번호 31), PG43(서열번호 119) 중 하나와 6 히스티딘 태그(서열번호 140) 및 TEV 프로테아제 인식 서열(서열번호 146) 및 hPTH 1-34의 아미노산 서열(서열번호 151)이 포함되어 있다.
30 대조군으로서 사용된 hPTH 1-34 융합 폴리펩타이드(H6TEV-hPTH 1-34)는
2019/143193 1»(:1^1{2019/000782 아미노말단융합파트너가포함되어 있지 않고, 6히스티딘 태그(서열번호 140), 프로테아제 인식 서열(서열번호 146) 및 대 1-34의 아미노산 서열(서열번호 151)이 포함되어 있다. 각 융합 폴리펩타이드의 유전자는 제한 효소 , ^01 및 ¾1이의 인식 서열과 1개의 종결코돈을포함한다. 상기 11 11 1-34 융합 폴리펩타이드를 코딩하는 뉴클레오타이드 서열은 각각 서열번호 294 내지 296에 해당하며, 대조군은서열번호 293에 해당한다.

발현조절이 가능한발현벡터 ?£ 2613에 클로닝하였다.
【표 2】

제작된 대 1-34융합 폴리펩타이드 발현 플라스미드들은 0 염기서열 확인을 통해 정확한 클로닝 여부를 확인하였다. 제작된 1正™ 1-34 융합 폴리펩타이드 발현 플라스미드들을 이용하여 염화칼슘을 이용한 화학적인 방법으로 대장균 此21(孤3) 세포를 형질전환하였다. 出3™ 1-34 융합 폴리펩타이드의 발현 플라스미드들이 형질도입된 대장균들은

형성하였다. 형질전환된 대장균들은 각각 카나마이신이 50 /保/】 의 농도로 존재하는 1고 액체 배지에 배양한 후, 50% 글리세롤을 배양액과 동일한 부피로 첨가하여 세포 농축액( 11 10을 만들고, 이를 -801: 온도의 냉동고에 보관하였다.
-80V 온도에서 보관된 느!™ 1-34 융합 폴리펩타이드의 발현 플라스미드들이 형질전환된 대장균의 세포 농축액을 실온에서 녹인 후 50 成를 카나마이신이 50能/成의 농도로 존재하는 1止 액체 배지 5 이 포함된 시험관에
첨가하여 37 °C 온도의 진탕배양기에서 12시간 동안 종균 배양하였다. 종균 배양된 대장균 2 in모는 카나마이신이 50 //g/m£의 농도로 존재하는 LB 액체 배지 200 m요이 포함된 플라스크에 첨가하여 37 °C 온도의 진탕배양기에서 배양하였다. 배양 약 3시간 후 세포의 광학밀도 (0D600)가 약 1.0이 되었을 때, IPTG의 최종 5 농도가 0.1 mM이 되도록 첨가하여 hPTH 1-34 융합 폴리펩타이드의 발현을 유도하였다. 발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
실시예 1.3. 발현수준비교분석을위한시료의 제조
발현 유도가된 세포의 광학밀도가 10.0이 되도록세포를농축하고 50 mM 10 인산나트륨, pH 7.2완충액으로 재현탁시킨 후초음파세포 파쇄기 (ultrasonic processor , Cole-Parmer )를 이용하여 세포를파쇄하였다. 파쇄된 세포는총세포 분획으로 표지하였다. 세포 파쇄액은 12,000Xg, 4°C 온도에서 15분간 원심분리하였다. 상등액을회수하고가용성 분획 A로표지하였다. 불용성 분획은 500 의 50 mM인산나트륨, pH 7.2완충액으로초음파세포파쇄기를 이용하여 15 재현탁하고불용성 분획으로표지하였다.
실시예 1.4. SDS-PAGE분석을통한생산된 hPTH 1-34의 확인
50 의 총세포, 가용성 및 불용성 분획을각각 50 M의 2배 농축된 SDS 시료 완충액 (SDS sample buffer 2公· concentrate , Sigma)에 혼합한 후, 95 °C 온도에서 5분간가열하여 각시료의 단백질이 변성될수 있도록하였다. 시료내 20 변성된 단백질들은 16% SDS-PA湖 겔 및 TANK완충액을 이용하여 분자량 크기에 따라 겔 내에서 분리될 수 있도록 하였다. SDS-PAGE 후 겔을 쿠마시 블루 (Coomassie blue) R-250이 포함된 염색 완충액으로 염색한 후, 탈색 완충액으로 탈색하여 염색된 단백질만 보일 수 있도록 하였다. 그 결과를 도 1 및도 2에 나타내었다.
25 도 1을참조하면, 대조군인 서열번호 1의 아미노산서열을포함하는융합 파트너가 포함되지 않은 H6TEV-hPTH 1-34(5.9 kDa의 분자량)의 밴드는 본원 발명에 따른 hPTH 1-34융합폴리펩타이드들과 비교하여 가장낮은 발현 수준을 나타냈다. 본발명에 따른융합파트너인 PG7, PG15및 PG43이 융합된 hPTH 1-34 융합폴리펩타이드인 PG07-H6TEV-hPTH 1-34(6.9 kDa의 분자량) , PG15-H6TEV-hPTH 30 1-34(7.9 kDa의 분자량) 및 PG43-H6TEV-hPTH 1-34(10.6 kDa의 분자량)의 발현
수준은 대조군 (H6TEV-hPTH 1-34) 대비 증가된 것이 확인할수 있다. 덴시토미터 분석을 통해 PG7, PG15 및 PG43 중 P15의 융합에 의한 PG15-H6TEV-hPTH 1-34의 발현수준이 모든 hPTH 1-34융합폴리펩타이드중가장높은 것으로 확인되었다. 또한, 도 2를 참조하면, 대조군을 포함한 모든 hPTH 1-34 융합 5 폴리펩타이드는 가용성 분획에서는 관찰되지 않고 모두 불용성 분획에서 나타나는 것을 알수 있다.
실시예 1.5. PG15-H6TEV-hPTH 1-34대량생산을위한유가식 배양 문헌 (Riesenberg, Schulz et al . 1991)에 기재되어 배지 조성을 이용하여 2 L의 제한 배지를 함유하는 37°C의 발효기에서 세포를 배양하고, 염산과 10 암모니아 첨가를 통해 pH 6.8로 유지하였다. 발효기 내의 세포를 고농도로 배양하기 위해 세포 배양 동안포도당이 포함된 주입 용액 (feeding solution)을 배양액에 주입하였다. 배양 8시간 후 1.0 mM 농도의 IPTG를 첨가하여 11시간 동안 PG15-H6TEV-hPTH 1-34의 발현을유도하였다.
그 후, SDS-PAGE 분석을 통해 PG15-H6TEV-hPTH 1-34의 발현 수준을 15 확인하였다. SDS-PAGE분석결과, IPTG에 의한발현 유도후 세포성장및 PG15- H6TEV-hPTH 1-34의 발현 수준은 지속적으로 증가하는 것을 확인할 수 있었다. 덴시토미터 분석결과 PG15-H6TEV-hPTH 1_34의 발현수준은총단백질의 약 27%로 확인되었다 (도 3) .
실시예 1.6. N-말단융합 파트너의 아미노산 치환에 의한 hPTH 1-34융합 20 폴리펩타이드의 발현수준향상
PG15-H6TEV-hPTHl-34 내 PG15의 N-말단 서열이 hPTH 1-34 융합 폴리펩타이드의 발현 수준에 미치는 영향을 알아보기 위해 PG15의 아미노산 서열의 2번부터 7번까지 총 6개의 아미노산을 결실시킨 PG15(A2-7)-H6TEV- hPTHl-34(서열번호 339)의 발현 플라스미드를 제작하여 대장균 내 발현 수준을 25 PG15-H6TEV-hPTHl-34와 비교하였다. 형질전환부터 況S-PAGE를 이용한 발현수준 분석은실시예 1.2내지 1.4와동일하게 수행하였다.
SDS-PAGE 젤의 덴시토미터 분석을 통해 PG15(A2-7)-H6TEV-hPTHl-34의 발현수준은 PG15-H6TEV-hPTHl-34와 비교하여 5배 이상 감소하는 것으로 확인되었다 (도 4) . 따라서, PG15-H6TEV-hPTHl-34 내 PG15의 2번부터 7번까지
30 서열이 hPTH 1-34 융합 폴리펩타이드의 발현 수준에 크게 영향을 주는 것을
확인하였다. ;;:
또한, PG15-H6TEV-hPTHl-34 내 PG15의 2번부터 7번까지 6개의 아미노산 잔기의 변화가 hPTH 1-34 융합 폴리펩타이드의 발현 수준에 미치는 영향을 알아보기 위해 각각의 아미노산 잔기를 이소루신, 아스파라긴, 아르기닌, 아스팔트산으로 치환한 총 21개의 hPTH 1-34 융합 폴리펩타이드의 변이체를 제작하여 PG15-H6TEV-hPTHl-34와의 세포내 발현수준을비교하였다.
hPTH 1-34 융합 폴리펩타이드의 변이체 발현을 위한 플라스미드 DNA는 위치 지정 돌연변이 (site-directed mutagenesis)방법을 이용하여 제작하였다. 위치 지정 돌연변이를 위한 주형 (tempi ate)은 PG15-H6TEV-hPTHl-34 발현 플라스미드인 pSGK477을 사용하였고 프라이머는 각각의 변이체의 아미노산 치환부위의 염기서열이 변경된 정방향 (forward) 및 역방향 (reverse)의 단일가닥 DNA올리고머 를 이용하였다. 실험에 사용된프라이머를하기 표 3에 나타내었다.
【표 3】 ·


각각의 변이체에 대한 위치 지정 돌연변이 후 얻어진 발현 플라스미드들은 0 염기서열 확인을통해 정확한클로닝 여부를확인하였다. 제작된 !¾ 1-34 융합 폴리펩타이드의 변이체 발현 플라스미드들을 이용하여 염화칼슘을 이용한 화학적인 방법으로 대장균 ^121(^3) 세포를 형질전환하였다. ™ 1-34 융합 폴리펩타이드의 발현 플라스미드들이 형질전환된 대장균들은 카나마이신 0¾113111(:^1)이 50 //용/111요의 농도로 포함된
 고체배지에서 콜로니를 형성하였다. 형질전환된 대장균들을 각각 카나마이신이
고체배지에서 콜로니를 형성하였다. 형질전환된 대장균들을 각각 카나마이신이

배양액과 동일한 부피로 첨가하여 세포 농축액을 만들고, 이를 -8010 온도의 냉동고에 보관하였다.
-80 0 온도에서 보관된 느!™ 1-34 융합 폴리펩타이드의 변이체 발현 플라스미드들이 형질전환된 대장균의 세포 농축액을 실온에서 녹인 후 50 를

배양된 대장균 2 111요은 카나마이신이 50 용/111보의 농도로 존재하는 1止 액체 배지 200 111요이 포함된 플라스크에 첨가하여 370 온도의 진탕배양기에서 배양하였다. 배양 약 3시간후 세포의 광학밀도((犯600)가 약 1.0이 되었을 때, 1肝(^의 최종
농도가 0.1 mM이 되도록 첨가하여 hPTH 1-34 융합 폴리펩타이드의 발현을 유도하였다.발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
발현 유도가 된 세포의 광학밀도가 10.0이 되도록 세포를 농축하고 50 mM 5 인산 나트륨, pH 7.2 완충액으로 재현탁시킨 후 초음파 세포 파쇄기(ultrasonic processor, Cole-Parmer)를 이용하여 세포를 파쇄하였다.파쇄된 세포는 총 세포 분획으로 표지하였다. 세포 파쇄액은 12,000Xg, 4°C 온도에서 15분간 원심분리하였다.상등액을 회수하고 가용성 분획으로 표지하였다.불용성 분획은 500 의 50 mM인산 나트륨, pH 7.2완충액으로 초음파 세포 파쇄기를 이용하여 10 재현탁하고불용성 분획으로 표지하였다.
50 의 총세포, 가용성 및 불용성 분획을각각 50成의 2배 농축된 SDS 시료 완충액 (SDS sample buffer 2x concentrate, Sigma)에 혼합한 후, 95 °C 온도에서 5분간가열하여 각시료의 단백질이 변성될 수 있도록하였다. 시료내 변성된 단백질들은 16% SDS-PAGE 겔 및 TANK 완충액을 이용하여 분자량 크기에 15 따라 겔 내에서 분리될 수 있도록 하였다. SDS-PAGE 후 겔을 쿠마시 블루 (Coomassie blue) R-250이 포함된 염색 완충액으로 염색한 후, 탈색 완충액으로 탈색하여 염색된 단백질만 보일 수 있도록 하였다. 그 결과를 도 5 내지 도 7에 나타내었다.
대조군인 PG15-H6TEV-hPTHl-34의 발현 수준과 비교하여 PG15-H6TEV- 20 hPTHl-34 내 PG15의 2번부터 7번까지 6개의 아미노산 잔기의 변화에 의해 발현 수준이 향상된 변이체와 감소된 변이체들을 확인하였다. 특히, 덴시토미터 분석을 통해 4번째 잔기가 아스팔트산으로 치환된 변이주와 7번째 잔기가 아르기닌으로 치환된 변이주는 대조군 대비 3배 이상 발현 수준이 향상되었다.
실시예 2. hPTH 1-34융합폴리펩타이드의 회수 및 정제
25 실시예 2.1.세포파쇄 및 불용성 내포체 회수
인산 나트륨, pH 7.2 완충액을 첨가하여 해동하였다. 재현탁된 세포는 초음파 세포 파쇄기(ultrasonic processor, Cole-Parmer)를 이용하여 파쇄하였다. 용해된 세포는 12,000 rpm(12,000Xg) 조건에서 30분간 원심분리하고 상등액을
30 제거하여 재조합융합폴리펩타이드가포함된 불용성 내포체 분획을회수하였다.
실시예 2.2. 불용성 내포체가용화
20 mM 트리스, 500 mM 염화나트륨, 50 mM 이미다졸, pH 7.4)을 첨가하고 25 °C 온도에서 4시간 동안 진탕배양하여 불용성 분획 내 내포체 형태의 재조합 융합 5 폴리펩타이드가 가용화될 수 있도록 하였다. 가용화된 불용성 분획 시료는 12,000Xg에서 30분간 원심분리하고 상등액을 막필터(0.45八).2 _)를 통해 여과하였다.
실시예 2.3. hPTH 1-34융합폴리펩타이드정제
가용화된 불용성 분획 내의 hPTH 1-34 융합 폴리펩타이드의 정제를 10 위하여 S9시료 펌프(Sample pump S9) 및 F9-C분획 수집기(Fraction col lector F9-C)이 장착된 AKTA pure 25 크로마토그라피 시스템(GE Healthcare)을 이용하였다. 가용화된 불용성 분획 시료는 내포체 가용화 완충액(8 M요소, 20 mM 트리스, 500 mM 염화나트륨, 50 mM 이미다졸, pH 7.4)으로 미리 평형화된 HisTrap FF 1 in^ 컬럼(GE Healthcare)에 주입하였다. 주입이 완료된 후 컬럼은 15 5배 컬럼 부피의 평형 완충액으로 세척하고, 5배 컬럼 부피의 용출 완충액(8 M 요소, 20 mM 트리스, 500 mM 염화나트륨, 500 mM 이미다졸, pH 7.4)을 단계적으로 100%가 되도록 증가시켜 컬럼의 수지에 결합되어 있는 hPTH 1-34 융합 폴리펩타이드를 용출하였다. 용출 후 얻어진 분획의 결과를 각각 도 8a 내지 도 10b에 나타내었다.
20 실시예 3. 프로테아제처리를통한링커 서열 절단
약 5 m요의 정제된 hPTH 1-34 융합 폴리펩타이드의 분획을 합친 후 140 의 희석 완충액(20 mM 트리스, pH 7.4)을 첨가하여 요소의 농도가 1 이 되도록 희석하였다. 희석된 재조합융합폴리펩타이드에 TEV프로테아제의 최종 농도가 500 nM이 되도록 첨가하고실온에서 12시간동안 절단 반응이 진행될 수 25 있도록하였다.
TEV 프로테아제에 의한 절단 여부를 확인하기 위해, 절단 후 SDS-PAGE 분석을 수행하여 그 결과를 도 11 및 도 12에 나타내었다. 한편, 도 9a를 참조하면, 7.9 kDa의 PG15-H6TEV-hPTH 1-34은 거의 100%의 수율로 N-말단 융합 파트너, 6 히스티딘 태그 및 TEV 프로테아제 인식서열 융합체인 PG15-H6TEV 30 단편과목적 폴리펩타이드인 hPTH 1-34단편으로분리되는것을확인하였다.
실시예 4. hPTH 1-34정제
실시예 4.1. 양이온교환크로마토그래피를통한 hFffl 1-34분리 및정제
PG15-H6TEV-hPTH 1-34 융합 폴리펩타이드에서, TEV 프로테아제의 절단에 의해 유리된 hPTH 1-34의 정제를위하여 S9시료 펌프 (Sample pump S9) 및 F9-C
5 분획 수집기 (Fraction col lector F9-C)이 장착된 AKTA pure 25 크로마토그라피 시스템 (GE Healthcare)을 이용하였다. TEV 프로테아제로 절단된 각각의 시료를 결합 완충액 (20 mM 아세트산 암모늄, pH 9.3)으로 완충액 교환 후 동일 완충액으로미리 평형화된 Hi Trap SP FF 1 in^컬럼 (GE Healthcare)에 주입하였다. 주입이 완료된 후 컬럼을 5배 컬럼 부피의 결합완충액으로 세척하고, 5배 컬럼 10 부피의 용출 완충액 (20 mM 아세트산 암모늄, 500 mM 염화나트륨, pH 9.3)을 점진적으로 100%가 되도록 선형으로 10배의 컬럼 부피 동안 증가시켜 컬럼의 수지에 결합되어 있는 hPTH 1-34를 용출하였다. 분획 수집기의 정제 분획은 상기에 기재된 SDS-PAGE방법으로분석하였다.
재조합 융합 폴리펩타이드 (PG15-H6TEV-hPTH 1-34)의 절단에 의해 유리된 15 hPTH 1-34의 pi는 정제용 태그 및 절단효소 인식 서열이 포함된 아미노 말단 융합 파트너 (PG15-H6TEV)의 pi보다 약 3이 낮다. 구체적으로, pH 9.3의 완충액에서 11.72의 pi 값을 갖는 PG15-H6TEV는 양전하를 갖고 8.29의 pi 값을 갖는 hPTH 1-34는양전하를가지므로, 이온교환크로마토그래피를이용한 PG15- H6TEV와 hPTH 1-34의 분리가용이하다. PG15-H6TEV-hPTH 1-34이 절단되어 PG15- 20 H6TEV와 hPTH 1-34가 혼합되어 있는 시료를 양이온 교환 수지가 충진되어 있는
HiTrap SP FF 1 mL 컬럼에 주입하였다. 도 13a 및 13b를 참조하면, 음이온을 띄고 있는 hPTH 1-34는양이온교환수지에 결합되지 않고통과액 (flow through) 분획에서 확인되었고양이온을띄고 있는 PG15-H6TEV는양이온교환수지에 결합 후염화나트륨농도증가에 의해 용출되는 것을확인하였다. 따라서 비교적 높은 25 pi 값을 갖는 본 발명에 따른 N-말단 융합 파트너들은 이온 교환 크로마토그래피를 이용하여 제거 가능하므로 hPTH 1-34의 분리 정제를 용이하게 할수있다.
실시예 4.2. 소수성 상호작용크로마토그래피를 통한 hFffl 1-34분리 및 정제
30 PG15-H6TEV-hPTH 1-34 융합 폴리펩타이드에서 , TEV 프로테아제의 절단에
의해 유리된 hPTH 1-34의 정제를 위하여 S9시료 펌프 (Sample pump S9) 및 F9-C 분획 수집기 (Fraction col lector F9-C)이 장착된 AKTA pure 25 크로마토그라피 시스템 (GE Healthcare)을 이용하였다. TEV 프로테아제로 절단된 각각의 시료를 결합 완충액 (50 mM 인산 나트륨, 1.5 M황산 암모늄, pH 7.0)으로 완충액 교환 5 후동일 완충액으로미리 평형화된 HiTrap Butyl HP 1 mL컬럼 (GE Healthcare)에 주입하였다. 주입이 완료된 후 컬럼을 5배 컬럼 부피의 결합 완충액으로 세척하고, 5배 컬럼 부피의 용출 완충액 (50 mM 인산 나트륨, pH 7.0)을 점진적으로 100%가 되도록 선형으로 30배의 컬럼 부피 동안 증가시켜 컬럼의 수지에 결합되어 있는 hPTH 1-34를 용출하였다. 분획 수집기의 정제 분획은 10 상기에 기재된 SDS-PAGE방법으로분석하였다.
재조합 융합 폴리펩타이드 (PG15-H6TEV-hPTH 1-34)의 절단에 의해 유리된 hPTH 1-34의 평균 소수성 (GRAVY) 값 (-0.671)은 정제용 태그 및 절단효소 인식 서열이 포함된 아미노 말단융합파트너 (PG15-H6TEV)의 평균 소수성 값 (-1.272) 보다 0.488만큼 높으므로 소수성 상호작용 크로마토그래피를 이용한 PG15- 15 H6TEV와 hPTH 1-34의 분리가 용이하다. 도 14a 및 14b에 나타난 바와 같이, 대부분의 PG15-H6TEV와 hPTH 1-34는 소수성 상호작용 수지에 결합하여 통과액 (f low through) 분획에서 검출되지 않았다. 황산 암모늄이 포함되어 있지 않은용출 완충액 비율이 서서히 증가됨에 따라낮은 평균소수성을 갖는 PG15- H6TEV부터 용출되었고 용출 완충액 비율이 더 올라가 황산 암모늄 농도가 20 낮아지면서 hPTH 1-34가 용출되는 결과를 확인하였다. 따라서 상대적은 낮은 평균소수성 값을갖는본발명에 따른 N-말단융합파트너들은소수성 상호작용 크로마토그래피를 이용하여 제거 가능하므로 hPTH 1-34의 분리 정제를 용이하게 할수있다.
실시예 5. 절단후의 hPTH 1-34의 분자량분석
25 완전한 형태의 PG15-H6TEV-hPTH 1-34의 발현 유무, TEV 프로테아제에 의한 정확한 절단 및 절단 후 얻어진 hPTH 1-34의 변형 (modification) 유무를 확인하기 위해 MALTI-T0F MS를 이용한 분자량 분석을 실시하였다. hPTH 1-34 표준 물질의 분자량을 측정한 결과와 본 발명에 따라 수득된 hPTH 1-34의 분자량을측정한결과를도 15및 도 16에 나타내었다.
30 도 15및 도 16를 참고하면, PG15-H6TEV-hPTH 1-34로부터 수득된 hPTH 1-
34의 분자량은 이론적인 분자량과 오차범위 이내에서 일치하였으므로 대장균 내에서 단백질 분해효소(proteolyt ic enzyme)에 의한 아미노 혹은 카르복시 말단의 부분적인 절단이나 분해 없이 완전한 형태로 발현된 것을 확인할 수 있었다. 따라서 TEV프로테아제는 PG15-H6TEV-hPTH 1-34내 인식서열인 ENLFQ 5 서열을 인식하고 마지막 아미노산인 Q(글루타민)와 hPTH 1-34의 첫번째 아미노산인 S(세린)사이의 펩타이드 결합을 정확하게 절단하는 것으로 판단된다.
실시예 6. 정제된 hPTH 1-34의 역상 HPLC분석
미국약전⑴ SP 39, Officai l Monographs, Teriparatide, 6058-6062)에 명시되어 있는 hPTH 1-34의 동정 (ident i ficat ion) 기준시험법으로 표준품 hPTH 10 1-34 (USP Catalog #1643962)와본 발명을 통해 생산된 재조합 hPTH 1-34을 역상
HPLC로 분석하였다. 그 결과, 두 hPTH 1-34 모두 동일한 체류시간을 보였고 재조합 hPTH 1-34의 순도는 99.5%이상으로확인되었다 (도 17) .
또한, 미국약전 (USP)에 hPTH 1-34의 동정 (identif ication) 방법 중 추가적으로 펩타이드 맵핑 (peptide mapping) 방법을 통해 표준품 hPTH 1-34 (USP 15 Catalog #1643962)와 본 발명을 통해 생산된 재조합 hPTH 1-34의 동등성을 분석하였다. 상기 두 hPTH 1-34에 각각 Staphylococcus aureus V8 protease를 처리하여 5개의 펩타이드 조각 (pept ide fragment)으로 분리한 후 역상 HPLC로 분석해 본결과, 두 hPTH 1-34에서 분리된 5개의 펩타이드조각들이 모두동일한 체류시간을 보여 표준품 hPTH 1-34와 재조합 hPTH 1-34의 동등성이 20 확인되었다 (도 18) .
실시예 7. 리라글루타이드전구체 펩타이드 (GLP-1X28R)융합폴리펩타이드 제조및 생산
실시예 7.1. GLP-1K28R융합폴리펩타이드발현플라스미드의 제작
GLP-1K28R 융합 폴리펩타이드에 대한 유전자는 overlap extension
25 polymerase chain react ion (0E-PCR) 방법을 이용하여 합성하였다. 이때, 상기
GLP-1K28R융합폴리펩타이드는아미노말단융합파트너로서 PG07(서열번호 9), PG15C서열번호 31) , PG22(서열번호 53) , PG29(서열번호 75) , PG36(서열번호 97) 및 PG43C서열번호 119) 중 하나와 6 히스티딘 태그 (서열번호 140), TEV 프로테아제 인식서열 (서열번호 146) 및 GLP-1K28R의 아미노산 서열 (서열번호 30 341)이 포함되어 있다.
2019/143193 1»(:1^1{2019/000782 대조군으로서 사용된 (}1少- 28묘 융합 폴리펩타이드어 -比13- 2810는 아미노말단융합파트너가포함되어 있지 않고, 6히스티딘 태그(서열번호 140),

341)이 포함되어 있다. 각융합폴리펩타이드의 유전자는 제한 효소 , ^01 및 ¾101의 인식서열과 1개의 종결 코돈을 포함한다. 상기 012-1281? 융합 폴리펩타이드를 코딩하는 뉴클레오타이드 서열은 각각서열번호 478 내지 483에 해당하며 , 대조군은서열번호 477에 해당한다.
아래 표 4에 기재한 0止- 281? 융합 폴리펩타이드 발현 플라스미드 ?3況530, ?3況495, ?3(¾496, ?3(¾500, ?3예501, ?(¾502 및 ?(¾497을 제작하기 위해서, (¾-灰고로 합성된 꾜13-11(281? 융합 폴리펩타이드 유전자 단편을 및 ¾01 제한 효소를 이용하여 절단하고, T7 프로모터, 130 작동자 및 1 (:1 유전자를 포함하여 1肝(}에 의한 발현 조절이 가능한 발현벡터 ?附261)에 클로닝하였다.
【표 4]

제작된 亂1 3-1他81?융합 폴리펩타이드 발현 플라스미드들은 0 염기서열 확인을 통해 정확한 클로닝 여부를 확인하였다. 제작된 此 1狀81? 융합 폴리펩타이드발현플라스미드들은 염화칼슘을 이용한화학적인 방법으로 대장균 此21( 3) 세포를 형질전환하였다. 0少- 281? 융합 폴리펩타이드의 발현 플라스미드들이 형질도입된 대장균들은 카나마이신(뇨 크미 !!)이 50 론/111次의

각각카나마이신이 50能/ 11 의 농도로존재하는 1止 액체 배지에 배양한후, 50% 글리세롤을배양액과동일한부피로 첨가하여 세포농축액을만들고, 이를 _80ᄃ 온도의 냉동고에 보관하였다.
실시예 7.2. 형질전환된세포의 배양및(^-1他8요의 발현
-8010 온도에서 보관된 꾜13-1}(281? 융합 폴리펩타이드의 발현
플라스미드들이 형질전환된 대장균의 세포 농축액을 실온에서 녹인 후 50 를 카나마이신이 50 //g/M의 농도로 존재하는 LB액체 배지 51就이 포함된 시험관에 첨가하여 37°C 온도의 진탕배양기에서 12시간 동안 종균 배양하였다. 종균 배양된 대장균 2 mi을 카나마이신이 50;«g/ffli의 농도로 존재하는 LB액체 배지 5 200 in보이 포함된 플라스크에 첨가하여 37°C 온도의 진탕배양기에서 배양하였다. 배양 약 3시간 후 세포의 광학밀도(OD600)가 약 1.0이 되었을 때 IPTG를 최종 농도 0.1 mM이 되도록 첨가하여 GLP-1K28R 융합 폴리펩타이드의 발현을 유도하였다. 발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
10 실시예 7.3. 발현수준비교분석을위한시료의 제조
발현 유도가 된 세포의 광학밀도가 10.0이 되도록 세포를 농축하고 50 mM 인산 나트륨, pH 7.2 완충액으로 재현탁시킨 후 초음파 세포 파쇄기(ultrasonic processor, Cole-Parmer)를 이용하여 세포를 파쇄하였다.파쇄된 세포는 종 세포 분획으로 표지하였다. 세포 파쇄액은 12,000Xg, 4°C 온도에서 15분간
15 원심분리하였다.상등액을 회수하고 가용성 분획으로 표지하였다.불용성 분획은
500 의 50 mM인산 나트륨, pH 7.2완충액으로 초음파 세포 파쇄기를 이용하여 재현탁하고불용성 분획으로 표지하였다.
실시예 7.4. SDS-PAGE분석을통한생산된 GLP-1K28R의 확인
50成의 총세포, 가용성 및 불용성 분획을각각 50成의 2배 농축된 SDS 20 시료 완충액 (SDS sample buffer 2X concentrate, Sigma)에 혼합한 후, %°C 온도에서 5분간가열하여 각시료의 단백질이 변성될수 있도록하였다. 시료내 변성된 단백질들은 16% SDS-PAGE 겔 및 TANK 완충액을 이용하여 분자량 크기에 따라 겔 내에서 분리될 수 있도록 하였다. SDS-PAGE 후 겔을 쿠마시 블루 (Coomassie blue) R-25◦이 포함된 염색 완충액으로 염색한 후, 탈색 25 완충액으로 탈색하여 염색된 단백질만보일 수 있도록 하였다. 그 결과를도 19 및도 20에 나타내었다.
도 19를 참조하면, 대조군인 서열번호 1의 아미노산 서열을 포함하는 융합 파트너가포함되지 않은 H6TEV-GLP-1K28R(5.1 kDa의 분자량)의 밴드는 SDS- PAGE 젤에서 검출되지 않아 발현 후 세포내 단백질 분해효소에 의해 분해된 30 것으로 보인다. SDS-PAGE 젤에서 GLP-1K28R 융합 폴리펩타이드의 발현은
분자량이 가장 작은 아미노 말단융합 파트너인 PG07이 융합된 PG07-H6TEV-GLP- 1K28R(6.1 kDa의 분자량)부터 확인되었다.
아미노 말단 융합 파트너인 PG15가 융합된 PG15-H6TEV-GLP-1K28R(7.1 kDa의 분자량)의 발현 수준은 PG07-H6TEV-GLP-1K28R 대비 증가하였다. 아미노
5 말단 융합 파트너인 PG15, PG22, PG29, PG36 및 PG43이 융합된 GLP-1K28R융합 폴리펩타이드인 PG15-H6TEV-GLP-1K28R(7.1 kDa의 분자량), PG22-H6TEV-GLP- 1K28RC7.9 kDa의 분자량), PG29-H6TEV-GLP- 1K28R( 8.4 kDa의 분자량), PG36- H6TEV-GLP-1K28R(9.1 kDa의 분자량) 및 PG43ᅳ H6TEV_GLP-1K28R(11.7 kDa의 분자량)의 발현 수준은 대조군(H6TEV-GLP-1K28R) 대비 매우 향상된 것이
10 확인되었다.
덴시토미터 분석을통해 PG07, PG15, PG22, PG29, PG36및 PG43중 PG22, PG29, PG36 및 PG43이 융합된 융합 폴리펩타이드들의 발현 수준은 유사하였고
PG07, PG15이 융합된융합폴리펩타이드보다월등히 높은 것으로확인되었다(도 20및 도 21).
15 또한, 도 20을 참조하면, 대조군을 포함한 모든 GLP-1K28R 융합 폴리펩타이드는 가용성 분획에서는 관찰되지 않고 모두 불용성 분획에서 나타나는것을알수 있다(레인 1: H6TEV-GLP-1K28R(균주번호 PG005) 및 레인 2: PG07-H6TEV-GLP-1K28R(균주 번호 PG006)의 경우 목적 펩타이드가 발현되지 않거나발현수준이 낮아가용성 테스트를수행하지 않았음).
20 실시예 7.5. N-말단융합파트너의 아미노산치환에 의한 GLP-1K28R융합 폴리펩타이드의 발현수준변화
PG43-H6TEV-GLP-1K28R내 PG43의 2번부터 7번까지 6개의 아미노산잔기의 변화가 GLP-1K28R 융합 폴리펩타이드의 발현 수준에 미치는 영향을 알아보기 위해 각각의 아미노산 잔기를 이소루신, 아스파라긴, 아르기닌, 아스팔트산으로
25 치환한 총 22개의 GLP-1K28R 융합 폴리펩타이드의 변이체를 제작하여 PG43- H6TEV-GLP-1K28R와의 세포내 발현수준을비교하였다.
GLP-1K28R 융합 폴리펩타이드의 변이체 발현을 위한 플라스미드 DNA는 위치 지정 돌연변이(site-directed mutagenesis)방법을 이용하여 제작하였다. 위치 지정 돌연변이를 위한 주형(template)은 PG43-H6TEV-GLP-1K28R 발현
30 플라스미드인 pSGK497을 사용하였다. 프라이머는 각각의 변이체의 아미노산
2019/143193 1»(:1/10公019/000782

쇼올리고머를이용하였다. 실험에 사용된프라이머를하기 표 5에 나타내었다.
【표 5】


플라스미드들은 염기서열 확인을통해 정확한클로닝 여부를확인하였다. 제작된 0少-1他81? 융합 폴리펩타이드의 변이체 발현 플라스미드들을 이용하여 염화칼슘을 이용한 화학적인 방법으로 대장균 此21(에3) 세포를 형질전환하였다. ( 1敗81? 융합 폴리펩타이드의 발현 플라스미드들이 형질전환된 대장균들은 카나마이신(1¾11311¾0: )0ᅵ 50 //용/111보의 농도로 포함된
 고체배지에서 콜로니를 형성하였다 . 형질전환된 대장균들을 각각 카나마이신이
고체배지에서 콜로니를 형성하였다 . 형질전환된 대장균들을 각각 카나마이신이

배양액과 동일한 부피로 첨가하여 세포 농축액을 만들고, 이를 -801: 온도의 넁동고에 보관하였다.

배양 약 3시간후 세포의 광학밀도((©600)가 약 1.0이 되었을 때, 1 (;의 최종 농도가 0.1 이 되도록 첨가하여 꾜13- 281? 융합 폴리펩타이드의 발현을 유도하였다. 발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
발현유도가된 세포의 광학밀도가 10.0이 되도록세포를농축하고 50 인산 나트륨,
 7.2완충액으로 재현탁시킨 후초음파세포 파쇄기(111打33011比 에 - ^미 )를 이용하여 세포를파쇄하였다. 파쇄된 세포는총세포 분획으로 표지하였다. 세포 파쇄액은 12,000X1 4°0 온도에서 15분간 원심분리하였다. 상등액을회수하고가용성 분획으로표지하였다. 불용성 분획은 500 의 50 인산나트륨, 7.2완충액으로초음파세포파쇄기를 이용하여 재현탁하고불용성 분획으로표지하였다.
7.2완충액으로 재현탁시킨 후초음파세포 파쇄기(111打33011比 에 - ^미 )를 이용하여 세포를파쇄하였다. 파쇄된 세포는총세포 분획으로 표지하였다. 세포 파쇄액은 12,000X1 4°0 온도에서 15분간 원심분리하였다. 상등액을회수하고가용성 분획으로표지하였다. 불용성 분획은 500 의 50 인산나트륨, 7.2완충액으로초음파세포파쇄기를 이용하여 재현탁하고불용성 분획으로표지하였다.
50 의 총세포, 가용성 및 불용성 분획을각각 50 /4 «의 2배 농축된況 £
시료 완중액 (SDS sample buffer 2x concentrate, Sigma)에 혼합한 투, 95 °C 온도에서 5분간가열하여 각시료의 단백질이 변성될수 있도록하였다. 시료내 변성된 단백질들은 16% SDS-PAGE 겔 및 TANK 완충액을 이용하여 분자량 크기에 따라 겔 내에서 분리될 수 있도록 하였다. SDS-PAGE 후 겔을 쿠마시 5 블루 (Coomassie blue) R-250이 포함된 염색 완충액으로 염색한 후, 탈색 완충액으로탈색하여 염색된단백질만보일 수있도록하였다.
도 22내지 도 24에 나타낸 바와 같이, 대조군인 PG43-H6TEV-GLP-1K28R의 발현 수준과 비교하여 PG43-H6TEV-GLP-1K28R 내 PG43의 2번부터 7번까지 6개의 아미노산 잔기의 변화에 의해 발현 수준이 향상된 변이체와 감소된 변이체들을 10 확인하였다.특히,덴시토미터 분석을 통해 2번째 잔기가 아스팔트산으로 치환된 변이주와 7번째 잔기가 아이소루신, 아스파라긴, 아르기닌, 아스팔트산으로 치환된 변이주들은 대조군 대비 2 내지 3배 이상 발현 수준이 향상되는 것으로 확인되었다.
실시예 8. GLP-1K28R융합폴리펩타이드의 회수 및 정제
15 실시예 8.1. 세포파쇄 및 불용성 내포체 회수
플라스크 규모에서 발현된 세포의 냉동된 세포 펠렛을 50 M의 50 mM 인산 나트륨, pH 7.2 완충액을 첨가하여 해동하였다. 재현탁된 세포는 초음파 세포 파쇄기 (ultrasonic processor , Cole-Parmer)를 이용하여 파쇄하였다. 용해된 세포는 12,000 rpm (12,000Xg) 조건에서 30분간 원심분리하고 상등액을 20 제거하여 재조합융합폴리펩타이드가포함된 불용성 내포체 분획을회수하였다.
실시예 8.2. 불용성 내포체 가용화
회수된 불용성 내포체 분획에 20 의 내포체 가용화 완충액 (8 M 요소, 20 mM 트리스, 500 mM 염화나트륨, 50 mM 이미다졸, pH 7.4)을 첨가하고 25 °C 온도에서 4시간 동안 진탕배양하여 불용성 분획 내 내포체 형태의 재조합 융합 25 폴리펩타이드가 가용화될 수 있도록 하였다. 가용화된 불용성 분획 시료는
12,000Xg에서 30분간 원심분리하고 상등액을 (0.45/0.2 ) 막필터를 통해 여과하였다.
실시예 8.3. GLP-1K28R융합폴리펩타이드정제
7개 GLP-1K28R융합폴리펩타이드중가장발현 수준이 높은 PG43-H6TEV- 30 GLP-1K28R를 정제하였다. 먼저, 가용화된 불용성 분획 내의 GLP-1K28R 융합
폴리펩타이드의 정제를 위하여 S9 시료 펌프 (Sample pump S9) 및 F9-C 분획 수집기 (Fraction col lector F9-C)이 장착된 AKTA pure 25 크로마토그라피 시스템 (GE Healthcare)을 이용하였다. 가용화된 불용성 분획 시료는 내포체 가용화완충액 (8 M요소, 20 mM트리스, 500 mM염화나트륨, 50 mM이미다졸, pH 5 7.4)으로미리 평형화된 HisTrap FF 1 m요컬럼 (GE Healthcare)에 주입하였다.
주입이 완료된 후 컬럼은 5배 컬럼 부피의 평형 완충액으로 세척하고 5배 컬럼 부피의 용출 완충액(8 M요소, 20 mM트리스, 500 mM염화나트륨, 500 mM 이미다졸, pH 7.4)을 단계적으로 100%가 되도록 증가시켜 컬럼의 수지에 결합되어 있는 GLP-1K28R 융합 폴리펩타이드를 용출하였다. 용출 후 얻어진 10 분획의 결과를 각각 도면에 나타내었다(도 25 및 도 26). 컬럼에 주입된 가용화된 불용성 분획 시료 내의 GLP-1K28R융합폴리펩타이드는 대부분 컬럼 내 수지에 결합후 용출되어 95%이상의 순도를보였다.
실시예 9.프로테아제 처리를통한링커 서열 절단
약 5 m요의 정제된 GLP-1K28R 융합 폴리펩타이드의 분획을 합친 후 140
15 m£의 희석 완충액 (20 mM트리스, pH 7.4)을 첨가하여 요소농도가 1 M이 되도록 희석하였다. 희석된 재조합융합폴리펩타이드에 TEV프로테아제를 최종 농도가 500이이 되도록 첨가하고실온에서 12시간동안 절단 반응이 진행될 수 있도록 하였다.
TEV 프로테아제에 의한 절단 여부를 확인하기 위해, 절단 후 SDS-PAGE 20 분석을 수행하여 그 결과를 도면에 나타내었다 (도 27). TEV프로테아제에 의한 GLP-1K28R융합폴리펩타이드 (PG43-H6TEV-GLP-1K28R)의 절단전후를 SDS-PA賊로 분석해본 결과 7.9 kDa의 GLP-1K28R 융합 폴리펩타이드는 거의 100%의 수율로 아미노 말단 융합 파트너와 6 히스티딘 태그 및 TEV 프로테아제 인식서열 융합체인 PG43-H6TEV와 목적 폴리펩타이드인 GLP-1K28R로 절단되는 것을 25 확인하였다.
실시예 10. 절단후의 GLP-1K28R의 분자량분석
완전한 형태의 GLP-1K28R 융합 폴리펩타이드 (PG43-H6TEV-GLP-1K28R)의 발현 유무, TEV프로테아제에 의한정확한 절단 및 절단후 얻어진 GLP-1K28R의 변형 (modification) 유무를확인하기 위해 MALTI-T0F MS를 이용한분자량분석을 30 실시하였다. 본발명에 따라수득된 GLP-1K28R의 분자량을측정한결과를도면에
나타내었다.
도 28을 참고하면, PG43-H6TEV-GLP-1K28R로부터 수득된 GLP-1K28R의 분자량은 3382.59 Da으로 측정되어 이론적인 분자량인 3383.72 Da과 오차범위 이내에서 일치하였으므로 대장균 내에서 단백질 분해효소(proteolyt i c enzyme)에 5 의한 아미노혹은 카르복시 말단의 부분적인 절단이나분해 없이 완전한 형태로 발현된 것을확인할수있었다.
따라서 TEV 프로테아제는 PG43-H6TEV-GLP-1K28R 내 인식서열인 ENLFQ 서열을 인식하고 마지막 아미노산인 Q(글루타민)와 GLP-1K28R의 첫번째 아미노산인 H(히스티딘) 사이의 펩타이드 결합을 정확하게 절단하는 것으로 10 판단된다. 실시예 11. 테두글루타이드(GLP-2A2G)융합폴리펩타이드제조및생산 실시예 11.1. GLP-2A2G융합폴리펩타이드발현플라스미드의 제작
GLP-2A2G 융합 폴리펩타이드에 대한 유전자는 overlap extension 15 polymerase chain reaction (OE-PCR) 방법을 이용하여 합성하였다. 이때, 상기 GLP-2A2G융합폴리펩타이드는 아미노 말단융합 파트너로서 PG07(서열번호 9), PG15서열번호 31), PG22(서열번호 53), PG29서열번호 75), PG36(서열번호 97) 및 PG43(서열번호 119) 중 하나와 6 히스티딘 태그(서열번호 140), TEV 프로테아제 인식서열(서열번호 146) 및 GLP-2A2G의 아미노산 서열(서열번호 20 485)이 포함되어 있다.
대조군으로서 사용된 GLP-2A2G 융합 폴리펩타이드(H6TEV-GLP-2A2G)는 아미노말단융합파트너가포함되어 있지 않고, 6히스티딘 태그(서열번호 140), TEV프로테아제 인식서열(서열번호 146) 및 GLP-2A2G의 아미노산서열(서열번호 485)이 포함되어 있다. 각융합폴리펩타이드의 유전자는 제한효소 Ndel , Ncol 25 및 제이의 인식서열과 1개의 종결 코돈을 포함한다. 상기 GLP-2A2G 융합 폴리펩타이드를코딩하는 뉴클레오타이드서열은 각각서열번호 622 내지 627에 해당하며, 대조군은서열번호 621에 해당한다.
아래 표 6에 기재한 GLP-2A2G 융합 폴리펩타이드 발현 플라스미드 PSGK520, pSGK521, pSGK522, pSGK547, pSGK548, pSGK549및 pSGK523을 제작하기
30 위해서, 0E-PCR로 합성된 GLP-2A2G 융합 폴리펩타이드 유전자 단편을 Ndel 및
2019/143193 1»(:1^1{2019/000782 ¾01 제한 효소를 이용하여 절단하고, 17 프로모터, 130 작동자 및 1301 유전자를 포함하여 1 ^에 의한 발현 조절이 가능한 발현벡터 ?£ 2613에 클로닝하였다.
【표 6]

확인을 통해 정확한 클로닝 여부를 확인하였다. 제작된 꾜?-2쇼20 융합 폴리펩타이드 발현 플라스미드들을 이용하여 염화칼슘을 이용한 화학적인

폴리펩타이드의 발현 플라스미드들이 형질도입된 대장균들은 카나마이신 0에131117(^11)이 50 //용/111모의 농도로 포함된 1 고체배지에서 콜로니를 형성하였다. 형질전환된 대장균들은 각각 카나마이신이 50 //은/™요의 농도로 존재하는 [出 액체 배지에 배양한 후, 50% 글리세롤을 배양액과 동일한 부피로

플라스미드들이 형질전환된 대장균의 세포 농축액을 실온에서 녹인 후 50 成를 카나마이신이 50 1塔/1111의 농도로존재하는 1 액체 배지 5 111요이 포함된 시험관에 첨가하여 37 V 온도의 진탕배양기에서 12시간 동안 종균 배양하였다. 종균

200 111보이 포함된 플라스크에 첨가하여 37 °0 온도의 진탕배양기에서 배양하였다. 배양 약 3시간 후 세포의 광학밀도((犯600)가 약 1.0이 되었을 때 1 (}를 최종 농도 0.1 이 되도록 첨가하여 꾜 쇼요 융합 폴리펩타이드의 발현을 유도하였다. 발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
실시예 11.3. 발현수준비교분석을위한시료의 제조
발현 유도가 된 세포의 광학밀도가 10.0이 되도록 세포를 농축하고 50 mM 인산 나트륨, pH 7.2 완충액으로 재현탁시킨 후 초음파 세포 파쇄기(ultrasonic processor, Cole-Parmer)를 이용하여 세포를 파쇄하였다.파쇄된 세포는 총 세포 분획으로 표지하였다. 세포 파쇄액은 12,000Xg, 4°C 온도에서 15분간
5 원심분리하였다.상등액을 회수하고 가용성 분획으로 표지하였다.불용성 분획은 500 의 50 mM인산 나트륨, pH 7.2완충액으로 초음파세포 파쇄기를 이용하여 재현탁하고불용성 분획으로표지하였다.
실시예 11.4. SDS-PAGE분석을통한생산된 GLP-2A2G의 확인
50 의 총세포, 가용성 및 불용성 분획을각각 50成의 2배 농축된 SDS 10 시료 완충액 sample buffer 2X concentrate, Sigma)에 혼합한 후, 95 °C 온도에서 5분간가열하여 각시료의 단백질이 변성될 수 있도록하였다. 시료내 변성된 단백질들은 16% SDS-PAGE 겔 및 TANK 완충액을 이용하여 분자량 크기에 따라 겔 내에서 분리될 수 있도록 하였다. SDS-PAGE 후 겔을 쿠마시 블루(Coomassie blue) R-250이 포함된 염색 완충액으로 염색한 후 탈색 15 완충액으로 탈색하여 염색된 단백질만보일 수 있도록 하였다. 그 결과를도 29 및도 30에 나타내었다.
도 29를 참조하면, 대조군인 서열번호 1의 아미노산 서열을 포함하는 융합 파트너가포함되지 않은 H6TEV-GLP-2A2G(5.5 kDa의 분자량)의 밴드는 SDS- PAGE 젤에서 검출되지 않아 발현 후 세포내 단백질 분해효소에 의해 분해된
20 것으로보인다.
아미노 말단 융합 파트너인 PG07, PG15, PG22, PG29, PG36 및 PG43이 융합된 GLP-2A2G융합 폴리펩타이드인 PG07-H6TEV-GLP-2A2G(6.5 kDa의 분자량), PG15-H6TEV-GLP-2A2G(7.5 kDa의 분자량), PG22-H6TEV-GLP-2A2G(7.5 kDa의 분자량), PG29-H6TEV-GLP-2A2G(8.3 kDa의 분자량), PG36-H6TEV-GLP-2A2GO .5 25 kDa의 분자량) 및 PG43-H6TEV-GLP-2A2G( 12.1 kDa의 분자량) 중 SDS-PAGE분석을 통해 발현 확인이 된 것은 PG22-H6TEV-GLP-2A2G, PG29-H6TEV-GLP-2A2G , PG36- H6TEV-GLP-2A2G및 PG43-H6TEV-GLP-2A2G이었다.
덴시토미터 분석을 통해 PG07, PG15, PG22, PG29, PG36 및 PG43 중 PG43의 융합에 의한 PG43-H6TEV-GLP-2A2G의 발현 수준이 모든 GLP-2A2G 융합 30 폴리펩타이드중가장높은것으로확인되었다
또한, 도 30을 참조하면, 발현이 확인된 모든 GLP-2A2G 융합 폴리펩타이드들은 가용성 분획에서는 관찰되지 않고 모두 불용성 분획에서 나타나는 것을 알 수 있다(레인 1: H6TEV-GLP-2A2G(균주 번호 PG012), 레인 2: PG07-H6TEV-GLP-2A2G(균주 번호 PG013), 레인 3: PG15-H6TEV-GLP-2A2G(균주 번호 PG014) 및 레인 4: PG22-H6TEV-GLP-2A2G(균주 번호 PG015)의 경우 목적 펩타이드가발현되지 않아가용성 테스트를수행하지 않았음).
실시예 11.5. N-말단융합파트너의 아미노산치환에 의한 GLP-2A2G융합 폴리펩타이드의 발현수준변화
PG43-H6TEV-GLP-2A2G 내 PG43의 2번부터 7번까지 6개의 아미노산 잔기의 변화가 GLP-2A2G융합폴리펩타이드의 발현수준에 미치는 영향을알아보기 위해 각각의 아미노산 잔기를 이소루신, 아스파라긴, 아르기닌, 아스팔트산으로 치환한총 22개의 GLP-2A2G융합폴리펩타이드의 변이체를제작하여 PG43-H6TEV- GLP-2A2G와의 세포내 발현수준을비교하였다.
구체적으로, GLP-2A2G 융합 폴리펩타이드의 변이체 발현을 위한 플라스미드 DNA는 위치 지정 돌연변이(site-directed mutagenesis) 방법을 이용하여 제작하였다. 위치 지정 돌연변이를위한주형(template)은 PG43-H6TEV- GLP-2A2G발현플라스미드인 pSGK523을사용하였다. 프라이머는각각의 변이체의 아미노산 치환부위의 염기서열이 변경된 정방향(forward) 및 역방향(reverse)의 단일가닥 DNA 올리고머를 이용하였다. 실험에 사용된 프라이머는 하기 표 7에 나타내었다.
【표 7】


제작된 亂1-2요20 융합 폴리펩타이드의 변이체 발현 플라스미드들을 이용하여 염화칼슘을 이용한 화학적인 방법으로 대장균 1( 3) 세포를 형질전환하였다. 꾜 -2쇼20융합폴리펩타이드의 발현 플라스미드들이 형질도입된

부피로 첨가하여 세포농축액을만들고 이를 -801: 온도의 냉동고에 보관하였다.
-801: 온도에서 보관된 융합 폴리펩타이드의 변이체 발현
플라스미드들이 형질전환된 대장균의 세포 농축액을 실온에서 녹인 후 50成를 카나마이신이 50 //g/n 의 농도로 존재하는 LB액체 배지 5 M이 포함된 시험관에 첨가하여 37°C 온도의 진탕배양기에서 12시간 동안 종균 배양하였다. 종균

5 200 m요이 포함된 플라스크에 첨가하여 37°C 온도의 진탕배양기에서 배양하였다. 배양 약 3시간 후 세포의 광학밀도(0D600)가 약 1.0이 되었을 때, IPTG의 최종 농도가 0.1 mM이 되도록 첨가하여 GLP-2A2G 융합 폴리펩타이드의 발현을 유도하였다.발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
10 발현 유도가 된 세포의 광학밀도가 10.0이 되도록 세포를 농축하고 50 mM 인산 나트륨, pH 7.2 완충액으로 재현탁시킨 후 초음파 세포 파쇄기(ultrasonic processor, Cole-Parmer)를 이용하여 세포를 파쇄하였다.파쇄된 세포는 종 세포 분획으로 표지하였다. 세포 파쇄액은 12,000Xg, 4°C 온도에서 15분간 원심분리하였다.상등액을 회수하고 가용성 분획으로 표지하였다.불용성 분획은 15 500 의 50 mM인산 나트륨, pH 7.2완충액으로 초음파세포 파쇄기를 이용하여 재현탁하고불용성 분획으로표지하였다.
50 «의 총세포, 가용성 및 불용성 분획을각각 50 의 2배 농축된 SDS 시료 완충액 (SDS sample buffer 2X concentrate, Sigma)에 혼합한 후, 95 °C 온도에서 5분간가열하여 각시료의 단백질이 변성될수 있도록하였다. 시료내 20 변성된 단백질들은 16% SDS-PAGE 겔 및 TANK 완충액을 이용하여 분자량 크기에 따라 겔 내에서 분리될 수 있도록 하였다. 況 S-PA湖 후 겔을 쿠마시 블루 (Coomassie blue) R-250이 포함된 염색 완충액으로 염색한 후, 탈색 완충액으로탈색하여 염색된단백질만보일수있도록하였다.
도 31 내지 도 33에 나타난 바와 같이 , 대조군인 PG43-H6TEV-GLP-2A2G의 25 발현 수준과 비교하여 PG43-H6TEV-GLP-2A2G 내 PG43의 2번부터 7번까지 6개의 아미노산 잔기의 변화에 의해 발현 수준이 변화된 변이체들을 SDS-PA湖 젤 및 덴시토미터 분석을 통해 확인하였다. PG43의 2번부터 7번까지 6개의 아미노산 잔기의 변화는 GLP-2A2G 융합 폴리펩타이드의 발현 수준을 크게 향상시키지 못하고 5번째 잔기가 아르기닌으로 치환된 변이주와 7번째 잔기가 아르기닌으로 30 치환된 변이주는대조군대비 50%이하로발현수준이 감소되었다.
실시예 12. GLP-2A2G융합폴리펩타이드의 회수 및 정제
실시예 12.1. 세포파쇄 및 불용성 내포체 회수
플라스크 규모에서 발현된 세포의 냉동된 세포 펠렛을 50 me의 50 mM 인산 나트륨, pH 7.2 완충액을 첨가하여 해동하였다. 재현탁된 세포는 초음파 5 세포 파쇄기(ultrasonic processor Cole-Parmer)를 이용하여 파쇄하였다. 용해된 세포는 12,000 rpm (12,000Xg) 조건에서 30분간 원심분리하고 상등액을 제거하여 재조합융합폴리펩타이드가포함된 불용성 내포체 분획을회수하였다. 실시예 12.2. 불용성 내포체 가용화
회수된 불용성 내포체 분획에 20 M의 내포체 가용화 완충액 (8 M 요소, 10 20 mM 트리스, 500 mM 염화나트륨, 50 mM 이미다졸, pH 7.4)을 첨가하고 25 °C 온도에서 4시간 동안 진탕배양하여 불용성 분획 내 내포체 형태의 재조합 융합 폴리펩타이드가 가용화될 수 있도록 하였다. 가용화된 불용성 분획 시료는 12,000Xg에서 30분간 원심분리하고 상등액을 (0.45/0.2 ) 막필터를 통해 여과하였다.
15 실시예 12.3. GLP-2A2G융합폴리팹타이드정제
7개 GLP-2A2G융합폴리펩타이드 중 가장 발현 수준이 높은 PG43-H6TEV- GLP-2A2G를 정제하였다. 먼저, 가용화된 불용성 분획 내의 GLP-2A2G 융합 폴리펩타이드의 정제를 위하여 S9 시료 펌프 (Sample pump S9) 및 F9-C 분획 수집기 (Fract ion col lector F9-C)이 장착된 ARTA pure 25 크로마토그라피 20 시스템 (GE Healthcare)을 이용하였다. 가용화된 불용성 분획 시료는 내포체 가용화완충액 (8 M요소, 20 mM트리스, 500 mM염화나트륨, 50 mM이미다졸, pH

주입이 완료된 후 컬럼은 5배 컬럼 부피의 평형 완충액으로 세척하고, 5배 컬럼 부피의 용출 완충액(8 M요소, 20 mM트리스 500 mM염화나트륨, 500 25 mM 이미다졸, pH 7.4)을 단계적으로 100%가 되도록 증가시켜 컬럼의 수지에 결합되어 있는 GLP-2A2G 융합 폴리펩타이드를 용출하였다. 용출 후 얻어진 분획의 결과를 각각 도면에 나타내었다(도 34 및 도 35). 컬럼에 주입된 가용화된 불용성 분획 시료 내의 GLP-1K28R융합폴리펩타이드는 대부분 컬럼 내 수지에 결합후용출되어 95%이상의 순도를보였다.
30 실시예 13.프로테아제 처리를통한링커 서열 절단
약 5 m요의 정제된 GLP-2A2G 융합 폴리펩타이드의 분획을 합친 후 140 m보의 희석 완충액(20 mM트리스, pH 7.4)을 첨가하여 요소농도가 1 이 되도록 희석하였다. 희석된 재조합융합폴리펩타이드에 TEV프로테아제를 최종 농도가 500 이 되도록 첨가하고실온에서 12시간동안 절단 반응이 진행될 수 있도록 5 하였다.
TEV 프로테아제에 의한 절단 여부를 확인하기 위해, 절단 후 SDS-PAGE 분석을 수행하여 그 결과를 도면에 나타내었다(도 36). TEV프로테아제에 의한 GLP-2A2G 융합 폴리펩타이드(PG43-H6TEV-GLP-2A2G)의 절단 전 후를 SDS-PAGE로 분석해본 결과 12.1 kDa의 GLP-2A2G 융합 폴리펩타이드는 거의 100%의 수율로 10 아미노 말단 융합 파트너와 6 히스티딘 태그 및 TEV 프로테아제 인식서열 융합체인 PG43-H6TEV와 목적 폴리펩타이드인 GLP-2A2G로 절단되는 것을 확인하였다.
실시예 14. 절단후의 GLP-2A2G의분자량분석
완전한 형태의 GLP-2A2G융합폴리펩타이드(PG43-H6TEV-GLP-2A2G)의 발현 15 유무, TEV 프로테아제에 의한 정확한 절단 및 절단 후 얻어진 GLP-2A2G의 변형(modification) 유무를확인하기 위해 MALTI-TOF MS를 이용한분자량분석을 실시하였다. 본 발명에 따라수득된 GLP-2A2G의 분자량을 측정한 결과를도면에 나타내었다.
도 37를 참고하면, PG43-H6TEV-GLP-2A2G로부터 수득된 GLP-2A2G의
20 분자량은 3753.10 Da으로 측정되어 이론적인 분자량인 3752.13 Da과 오차범위 이내에서 일치하였으므로 대장균 내에서 단백질 분해효소(proteolyt i c enzyme)에 의한아미노혹은카르복시 말단의 부분적인 절단이나분해 없이 완전한 형태로 발현된 것을확인할수있었다.
따라서 TEV 프로테아제는 PG43-H6TEV-GLP-2A2G 내 인식서열인 ENLFQ 25 서열을 인식하고 마지막 아미노산인 Q(글루타민)와 GLP-2A2G의 첫번째 아미노산인 H(히스티딘) 사이의 펩타이드 결합을 정확하게 절단하는 것으로 판단된다. (목적 펩타이드가발현되지 않아가용성 테스트를수행하지 않았음). 실시예 15. 에칼란타이드의 제조및 생산
실시예 15.1에칼란타이드융합폴리펩타이드발현 플라스미드의 제작
30 에칼란타이드 융합 폴리펩타이드에 대한 유전자는 over l ap extens ion
polymerase chain reaction (OE-PCR) 방법을 이용하여 합성하였다. 이때, 상기 에칼란타이드 융합폴리펩타이드는 아미노 말단융합 파트너로서 PG07서열번호 9), PG15서열번호 31), PG43(서열번호 119) 중 하나와 6 히스티딘 태그(서열번호 140) 및 TEV 프로테아제 인식 서열(서열번호 146) 및 에칼란타이드의 아미노산서열(서열번호 638)이 포함되어 있다.
대조군으로서 사용된 에칼란타이드 융합 폴리펩타이드어 - Ecallantide)는 아미노 말단 융합 파트너가 포함되어 있지 않고, 6 히스티딘 태그(서열번호 140), TEV 프로테아제 인식 서열(서열번호 146) 및 에칼란타이드의 아미노산 서열(서열번호 642)이 포함되어 있다. 각 융합 폴리펩타이드의 유전자는 제한 효소 Ndel , Ncol 및 제이의 인식 서열과 1개의 종결 코돈을 포함한다. 상기 에칼란타이드 융합 폴리펩타이드를 코딩하는 뉴클레오타이드 서열(PG07, PG15 및 PG43)은 각각 서열번호 644 내지 646에 해당하며, 대조군은서열번호 643에 해당한다.
아래 표 8에 기재한 에칼란타이드 융합 폴리펩타이드 발현 플라스미드 pSGK512 , pSGK513 , pSGK514 및 pSGK515를 제작하기 위해서, 湖- PCR로 합성된 에칼란타이드 융합 폴리펩타이드 유전자 단편을 Ndel 및 Xhol 제한 효소를 이용하여 절단하고, T7프로모터, lac작동자및 Lad 유전자를포함하여 IPTG에 의한발현조절이 가능한발현벡터 pET26b에 클로닝하였다.
【표 8]

실시예 15.2. 형질전환된 세포의 배양및 에칼란타이드의 발현
-800 에서 보관된 에칼란타이드 융합 폴리펩타이드의 발현 플라스미드들로 형질전환된 세포의 배양 및 에칼란타이드의 발현은 실시예 1.2와 동일한방법으로수행되었다.
실시예 15.3. 발현수준 비교분석을위한시료의 제조
에칼란타이드 관련 시료의 제조는 실시예 1.3과 동일한 방법으로
수행되었다.
실시예 15.4. SDS-PAGE분석을통한생산된 에칼란타이드의 확인 실시예 1.4와 동일한 방법 및 조건으로 각 시료의 단백질을 처리한 후, 그결과를도 38및 도 39에 나타내었다.
5 도 38을 참조하면, 대조군인 서열번호 1의 아미노산 서열을 포함하는 융합파트너가포함되지 않은 H6TEV-Ecallantide(8.8 kDa의 분자량)의 밴드는타 에칼란타이드 융합 폴리펩타이드들과 비교하여 가장 낮은 발현 수준을 보였다. 아미노 말단 융합 파트너인 PG7, PG15 및 PG43이 융합된 에칼란타이드 융합 폴리펩타이드인 PG07-H6TEV-Eca 11 ant i de( 9.8 kDa의 분자량), PG15-H6TEV- 10 Ecal lantide(10.8 kDa의 분자량) 및 PG43-H6TEV-Eca 11 ant i de( 15.4 kDa의 분자량)의 발현 수준은 대조군(H6TEV-Ecallantide) 대비 증가된 것이 확인되었다. 덴시토미터 분석을통해 PG7, PG15및 PG43중 PG07의 융합에 의한 PG07-H6TEV- Ecallantide의 발현 수준이 모든 에칼란타이드 융합폴리펩타이드 중 가장높은 것으로확인되었다.
15 또한 도 39를 참조하면, 대조군을 포함한 모든 에칼란타이드 융합 폴리팹타이드는 가용성 분획에서는 관찰되지 않고 모두 불용성 분획에서 나타나는 것을 알수 있다.
실시예 16.네시리타이드의 제조 및 생산
실시예 16.1네시리타이드융합폴리펩타이드발현플라스미드의 제작
20 네시리타이드 융합 폴리펩타이드에 대한 유전자는 overlap extension polymerase chain react ion(OE-PCR) 방법을 이용하여 합성하였다. 이때, 상기 네시리타이드 융합 폴리펩타이드는 아미노 말단 융합 파트너로서 PG07(서열번호 9), PG15C서열번호 31), PG43C서열번호 119) 중 하나와 6 히스티딘 태그(서열번호 140) 및 TEV 프로테아제 인식 서열(서열번호 146) 및
25 네시리타이드의 아미노산서열(서열번호 652)이 포함되어 있다.
대조군으로서 사용된 네시리타이드 융합 폴리펩타이드(H6TEV-
Ecallantide)는 아미노 말단 융합 파트너가 포함되어 있지 않고, 6 히스티딘 태그(서열번호 140), TEV 프로테아제 인식 서열(서열번호 146) 및 네시리타이드의 아미노산 서열(서열번호 652)이 포함되어 있다. 각 융합 30 폴리펩타이드의 유전자는 제한 효소 Ndel , Ncol 및 제이의 인식 서열과 1개의
0 2019/143193 1»(:1^1{2019/000782 종결 코돈을 포함한다. 상기 네시리타이드 융합 폴리펩타이드를 코딩하는 뉴클레오타이드 서열은 각각 서열번호 654 내지 656에 해당하며, 대조군은 서열번호 653에 해당한다.
아래 표 9에 기재한 네시리타이드 융합 폴리펩타이드 발현 플라스미드 ?3況516, ?3(¾517, ?3(¾518 및 ?(¾519를 제작하기 위해서, 孤-^고로 합성된 네시리타이드 융합 폴리펩타이드 유전자 단편을 (¾1 및 ¾01 제한 효소를 이용하여 절단하고, 17프로모터, 130작동자및 1301 유전자를포함하여 1 에 의한발현조절이 가능한발현벡터 ?£ 261)에 클로닝하였다.
【표 9】

실시예 16.2. 형질전환된 세포의 배양및 네시리타이드의 발현
플라스미드들이 형질전환된 세포의 배양 및 네시리타이드의 발현은 실시예 1.2와 동일한 방법으로수행되었다.
실시예 16.3. 발현 수준비교분석을위한시료의 제조
네시리타이드 관련 시료의 제조는 실시예 1.3과 동일한 방법으로 수행되었다.
실시예 1.4와 동일한 방법 및 조건으로 각 시료의 단백질을 처리한 후, 그 결과를도 40및 도 41에 나타내었다.
도 40을 참조하면, 대조군인 서열번호 1의 아미노산 서열을 포함하는 융합 파트너가 포함되지 않은 볘 -此3 ^ 6(5.2 吐)3의 분자량)와 아미노 말단 융합 파트너 ?에7 이 융합된 네시리타이드 융합 폴리펩타이드인 에

발현 후 세포내 단백질 분해효소에 의해 분해된 것으로 보인다.況요 요묘 젤에서 네시리타이드 융합폴리펩타이드의 발현은 분자량이 가장작은 아미노 말단 융합
파트너인 PG15가 융합된 PG15-H6TEV-Nesiri tide(7.2 kDa의 분자량)부터 확인되었다. 아미노 말단 융합 파트너인 PG43이 융합된 네시리타이드 융합 폴리펩타이드인 PG43-H6TEV-Nesiri tide(11.8 kDa의 분자량)의 발현 수준은 PG15- H6TEV-Nesiritide(7.2 kDa의 분자량) 대비 향상된 것이 확인되었다. 덴시토미터 5 분석을 통해 PG07, PG15, 및 PG43 중 PG43의 융합에 의한 PG43-H6TEV-
Nesiritide의 발현 수준이 모든 네시리타이드 융합 폴리펩타이드 중 가장 높은 것으로확인되었다.
또한, 도 41을 참조하면, 대조군을 포함한 모든 네시리타이드 융합 폴리펩타이드는 가용성 분획에서는 관찰되지 않고 모두 불용성 분획에서 10 나타나는 것을 알수 있다(레인 1: H6TEV-Nesiritide(균주 번호 PG023) 및 레인 2: PG07-H6TEV-Nesiritide(균주 번호 PG024)의 경우 목적 펩타이드가 발현되지 않아가용성 테스트를수행하지 않았음).
실시예 17. hPTH 1-84융합폴리펩타이드제조및 생산
실시예 17.1. hPTH 1-84융합폴리펩타이드발현플라스미드의 제작
15 hPTH 1-84 융합 폴리펩타이드에 대한 유전자는 overlap extension polymerase chain reaction (OE-PCR) 방법을 이용하여 합성하였다. 이때, 상기 hPTH 1-84융합폴리펩타이드는아미노말단융합파트너로서 PG07서열번호 9), PG15(서열번호 31), PG43서열번호 119)중 하나와 6 히스티딘 태그(서열번호 140), TEV 프로테아제 인식 서열(서열번호 146) 및 hPTH 1-84의 아미노산 20 서열(서열번호 18)이 포함되어 있다.
대조군으로서 사용된 hPTH 1-84 융합 폴리펩타이드(H6TEV-hPTH 1-84)는 아미노말단융합파트너가포함되어 있지 않고, 6히스티딘 태그(서열번호 140), TEV 프로테아제 인식 서열(서열번호 146) 및 hPTH 1-84의 아미노산 서열(서열번호 628)이 포함되어 있다. 각 융합 폴리펩타이드의 유전자는 제한 25 효소 Ndel , Ncol 및 Xhol의 인식 서열과 1개의 종결코돈을포함한다. 상기 hPTH 1-84 융합 폴리펩타이드를 코딩하는 뉴클레오타이드 서열은 각각 서열번호 635 내지 637에 해당하며, 대조군은서열번호 654에 해당한다.
아래 표 10에 기재한 hPTH 1-84 융합 폴리펩타이드 발현 플라스미드 PSGK543, pSGK544, pSGK545 및 pSGK546를 제작하기 위해서, 0E-PCR로 합성된 30 hPTH 1-84융합폴리펩타이드유전자단편을 Ndel 및 Xhol 제한효소를 이용하여
2019/143193 1»(:1^1{2019/000782 절단하고, 17 프로모터, 1 작동자 및 1301 유전자를 포함하여 1的^에 의한 발현조절이 가능한발현벡터 ?肝2¾에 클로닝하였다.
【표 10】

폴리펩타이드발현플라스미드들을 이용해 염화칼슘을 이용한화학적인 방법으로 대장균此21( 3) 세포를 형질전환하였다. ™ 1-84융합폴리펩타이드의 발현 플라스미드들이 형질도입된 대장균들은 카나마이신 ¾3113111(^11)이 50 //은/파公의

각각카나마이신이 50쌔/ 의 농도로존재하는 1고 액체 배지에 배양한후 50% 글리세롤을배양액과동일한부피로 첨가하여 세포농축액을만들고, 이를 -80亡 온도의 넁동고에 보관하였다.
실시예 17.2. 형질전환된세포의 배양및 매 1-84의 발현
-80 0 온도에서 보관된 ™ 1-84 융합 폴리펩타이드의 발현 플라스미드들이 형질전환된 대장균의 세포 농축액을 실온에서 녹인 후 50 成를 카나마이신이 50 / 111요의 농도로존재하는 18,액체 배지 5 111모이 포함된 시험관에 첨가하여 37 V 온도의 진탕배양기에서 12시간 동안 종균 배양하였다. 종균 배양된 대장균 2 111요을 카나마이신이 50;쌔/111모의 농도로 존재하는 1止 액체 배지

배양 약 3시간 후 세포의 광학밀도(孤600)가 약 1.0이 되었을 때 1的^를 최종 농도 0.1 이 되도록 첨가하여 1¾ 1-84 융합 폴리펩타이드의 발현을 유도하였다. 발현 유도 4시간 후에 광학 밀도를 측정하여 세포의 광학 밀도를 측정하였다.
실시예 17.3. 발현수준비교분석을위한시료의 제조
발현 유도가된 세포의 광학밀도가 10.0이 되도록세포를농축하고 50

근 이, 에6-?3111161·)를 이용하여 세포를파쇄하였다. 파쇄된 세포는종세포
분획으로 표지하였다. 세포 파쇄액은 12,000Xg, 4°C 온도에서 15분간 원심분리하였다.상등액을 회수하고 가용성 분획으로 표지하였다.불용성 분획은 500 의 50 mM인산 나트륨, pH 7.2완충액으로 초음파세포 파쇄기를 이용하여 재현탁하고불용성 분획으로 표지하였다.
5 실시예 17.4. SDS-PAGE분석을통한생산된 hPTH 1-84의 확인
50 의 총세포, 가용성 및 불용성 분획을각각 50成의 2배 농축된 SDS 시료 완충액(SDS sample buffer 2公、 concentrate, Sigma)에 혼합한 후, 95 °C 온도에서 5분간가열하여 각시료의 단백질이 변성될수 있도록하였다. 시료내 변성된 단백질들은 16% SDS-PAGE 겔 및 TANK 완충액을 이용하여 분자량 크기에 10 따라 겔 내에서 분리될 수 있도록 하였다. SDS-PAGE 후 겔을 쿠마시 블루(Coomassie blue) R-250이 포함된 염색 완충액으로 염색한 후 탈색 완충액으로 탈색하여 염색된 단백질만보일 수 있도록 하였다. 그 결과를도 42 및도 43에 나타내었다.
도 42를 참조하면, 대조군인 서열번호 1의 아미노산 서열을 포함하는 15 융합 파트너가포함되지 않은 H6TEV-hPTHl-84 (11.2 kDa의 분자량)의 밴드는 타 hPTH 1-84융합폴리펩타이드들과비교하여 가장낮은발현수준을보였다.
아미노 말단 융합 파트너인 PG07, PG15 및 PG43이 융합된 hPTH 1-84 융합 폴리펩타이드인 PG07-H6TEV-hPTHl-84( 12.2 kDa의 분자량), PG15-H6TEV- hPTHl-84(13.2 kDa의 분자량) 및 PG43-H6TEV-hPTHl-84( 15.9 kDa의 분자량)의 20 발현 수준은 대조군(H6TEV-hPTHl-84) 대비 향상된 것이 확인되었다. 덴시토미터 분석을통해 PG07, PG15, 및 PG43중 PG15의 융합에 의한 PG15-H6TEV-hPTHl-84의 발현수준이 모든 hPTH 1-84융합폴리펩타이드중가장높은것으로확인되었다. 또한, 도 43을 참조하면, 대조군을 포함한 모든 hPTH 1-84 융합 폴리펩타이드는 가용성 분획에서 관찰되었지만 아미노 말단 융합 파트너의 25 크기가 커질수록 hPTH 1-84융합폴리펩타이드의 불용성 분획 비율이 증가하였고 가장 크기가 큰 PG43이 융합된 PG43-H6TEV-hPTHl-84은 총 단백질의 약 70%가 불용성 분획에서 나타나는 것을 알수 있다.
실시예 18. hPTHl-84융합폴리펩타이드의 회수및 정제
실시예 18.1.세포파쇄 및 가용화
30 4개의 hPTH 1-84 융합 폴리펩타이드들은 가용성 발현비율이 높아 총세포
분획에서 hPTH 1-84융합폴리펩타이드를정제하였다. 플라스크규모에서 발현된 세포의 냉동된 세포 펠렛을 20 m보의 내포체 가용화 완충액 (8 M 요소, 20 mM 트리스, 500 mM 염화나트륨, 50 mM 이미다졸, pH 7.4)을 첨가하여 해동 및 재현탁하였다. 재현탁된 세포는 초음파 세포 파쇄기 (ultrasonic processor , 5 Cole-Parmer)를 이용하여 파쇄하였다. 용해된 세포는 12,000 rpm (12,000Xg) 조건에서 30분간 원심분리하고 상등액을 제거하여 재조합 융합 폴리펩타이드가 포함된 불용성 내포체 분획을 제거하고 가용성 분획인 상등액을 회수하였다. 가용성 분획 시료는 12,000Xg에서 30분간원심분리하고상등액을 (0.45/0.2 pm) 막필터를통해 여과하였다.
10 실시예 18.2. hPTHl-84융합폴리펩타이드정제
4개의 hPTHl-84 융합 폴리펩타이드 중 가장 발현 수준이 높은 PG15- h6TEV-hPTHl-84를 정제하였다. 먼저, 가용성 분획 내의 hPTHl-84 융합 폴리펩타이드의 정제를 위하여 S9 시료 펌프 (Sample pump S9) 및 F9-C 분획 수집기 (Fraction col lector F9-C)이 장착된 AKTA pure 25 크로마토그라피 15 시스템 (GE Healthcare)을 이용하였다. 가용화된 불용성 분획 시료는 내포체 가용화완충액 (8 M요소, 20 mM트리스, 500 mM염화나트륨, 50 mM이미다졸, pH 7.4)으로미리 평형화된 HisTrap FF 1 mL컬럼 (GE Healthcare)에 주입하였다. 주입이 완료된 후 컬럼은 5배 컬럼 부피의 평형 완충액으로 세척하고, 5배 컬럼 부피의 용출 완충액8 M요소, 20 mM트리스, 500 mM염화나트륨, 500 20 mM 이미다졸 pH 7.4)을 단계적으로 100%가 되도록 증가시켜 컬럼의 수지에 결합되어 있는 hPTHl-84 융합 폴리펩타이드를 용출하였다. 용출 후 얻어진 분획의 결과를 각각 도면에 나타내었다(도 44 및 도 45). 컬럼에 주입된 가용화된 불용성 분획 시료 내의 hPTH 1-84융합 폴리펩타이드는 대부분 컬럼 내 수지에 결합후용출되어 90%이상의 순도를보였다.
25 실시예 19. 프로테아제 처리를통한링커 서열 절단
의 희석 완충액 (20 mM트리스, pH 7.4)을 첨가하여 요소농도가 1 M이 되도록 희석하였다. 희석된 재조합융합 폴리펩타이드에 TEV프로테아제를 최종 농도가 500이이 되도록 첨가하고실온에서 12시간동안 절단 반응이 진행될 수 있도록 30 하였다.
TEV 프로테아제에 의한 절단 여부를 확인하기 위해, 절단 후 SDS-PAGE 분석을 수행하여 그 결과를 도면에 나타내었다(도 46). TEV 프로테아제에 의한 hPTH 1-84융합 폴리펩타이드(PG15-h6TEV-hPTHl-84)의 절단 전 후를 SDS-PAGE로 분석해본 결과 13.2 kDa의 hPTHl-84 융합 폴리펩타이드는 거의 100%의 수율로 5 아미노 말단 융합 파트너와 6 히스티딘 태그 및 TEV 프로테아제 인식서열 융합체인 PG15-H6TEV와 목적 폴리펩타이드인 hPTH 1-84로 절단되는 것을 확인하였다.
실시예 20. 절단후의 hPTH 1-84의분자량분석
완전한 형태의 hPTH 1-84 융합 폴리펩타이드(PG15-H6TEV-hPTHl-84)의 10 발현 유무, TEV프로테아제에 의한 정확한 절단 및 절단 후 얻어진 hPTHl-84의 변형(modification) 유무를 확인하기 위해 MALTI-TOF MS를 이용한분자량분석을 실시하였다. 본발명에 따라수득된 hPTHl-84의 분자량을측정한결과를도 47에 나타내었다.
도면 47을 참고하면, PG15-H6TEV-hPTHl-84로부터 수득된 hPTH 1-84의 15 분자량은 9425.54 Da으로 측정되어 이론적인 분자량인 9424.73 Da과분자량과 오차범위 이내에서 일치하였으므로 대장균 내에서 단백질 분해효소(proteolyt ic enzyme)에 의한 아미노 혹은 카르복시 말단의 부분적인 절단이나 분해 없이 완전한형태로발현된 것을확인할수있었다.
따라서 TEV 프로테아제는 PG15-H6TEV-hPTHl-84 내 인식서열인 ENLFQ 20 서열을 인식하고 마지막 아미노산인 Q(글루타민)와 hPTHl-84의 첫번째 아미노산인 S(세린)사이의 펩타이드 결합을정확하게 절단하는것으로판단된다.
실시예 21. 융합 파트너의 위치에 따른 hPIH 1-34융합폴리펩타이드의 발현수준비교
실시예 21.1. hPTH 1-34융합폴리펩타이드발현플라스미드의 추가제작
25 hPTH 1-34 융합 폴리펩타이드에 대한 유전자는 overlap extension polymerase chain reaction (0E-PCR) 방법을 이용하여 합성하였다. 이때, 상기 hPTH 1-34융합폴리펩타이드는아미노말단융합파트너로서 PG15(서열번호 31), 6히스티딘 태그(서열번호 140), TEV프로테아제 인식 서열(서열번호 146) 또는 hPTH 1-34의 아미노산서열(서열번호 151)이 포함되어 있다.
30 각융합폴리펩타이드의 유전자는 제한 효소 Ndel , Ncol 및 제이의 인식
2019/143193 1»(:1^1{2019/000782 서열과 1개의 종결 코돈을 포함한다. 상기 대 1-34 융합 폴리펩타이드를 코딩하는뉴클레오타이드서열은각각서열번호 294및 295에 해당한다.
아래 표 11에 기재한 1-34 융합 폴리펩타이드 발현 플라스미드 ?3(¾554, ?3部555, ?3(¾556를제작하기 위해서, 孤- ¾로합성된 대 1-34융합 폴리펩타이드 유전자 단편을 ( 1 및 ¾101 제한 효소를 이용하여 절단하고, 17

발현플라스미드들은실시예 1.1에서 수행된 것과동일한방법으로제작되어 -801: 온도의 냉동고에 보관하였다.
【표 11】

실시예 21.2. 형질전환된세포의 배양및出해 1-34와발현
?0032 및 (;033 각각을 200 減의 15 배지가 포함된 플라스크에서 배양하고 11 (}를 첨가하여 대 1-34 융합 폴리펩타이드의 발현을 유도하였다. 각각의 융합폴리펩타이드의 구조를도식화하여 도 48에 나타내었다.
분석하였다(도 48). 아미노 융합 파트너가 융합되어 있지 않는

34(5.9 抑3의 분자량)의 밴드는 검출되지 않았고 표 41的¾1-34의 아미노 말단에 ?015 태그가 융합된 ?아5내 - 1¾1-34(7.9 四3의 분자량)은 높은 수준으로 발현되는 것을 확인하였다. ?아5-抑1 - 대1-34 내 친화성 태그인 抑(6 -뱌 ½ 13 가 제거된 (}15 - 1 -!1 ¾1-34(7.1 뇨加의 분자량)는 ?015 - }½1 - 대1-34와유사한수준으로발현되는것으로확인되었다.
반면에 볘서열을아미노말단서열로융합위치를 변경시킨抑?아5 - - 1^111-34(7.9抑3의 분자량)의 발현 수준은 발현 유무만 확인할수 있을 정도로 매우 낮은 것을 알 수 있다. 또한, 11 -11?1¾1-34의 카르복시 말단에 ( 5

크기의 밴드가검출되지 않아거의 발현되지 않은것으로판단된다.
2019/143193 1»(:1^1{2019/000782 결론적으로 본 발명에 따른 말단 융합 파트너인 아5는 1¾ 1-34융합 폴리펩타이드에서 아미노 말단에 융합되어야 ™ 1-34의 고발현을 유도하는 것을 알 수 있다. 또한, 친화성 태그는 ™ 1-34 융합 폴리펩타이드에서 제거되거나 I말단 융합 파트너의 카르복시 말단에 위치하는 경우, ™ 1-34 5 융합 폴리펩타이드의 고발현성은 유지될 수 있다.그러나, 발현이 유지되지만 말단 융합 파트너의 아미노 말단에 위치하는 경우 ™ 1-34 융합 폴리펩타이드의 발현 수준이 현저히 감소되는 것을 확인할수 있다.
Claims
[일반식 1]
¾¾卜크 - 크요 - 333-¾34-¾35 - 836-(2)11
상기 일반식 1에서,
331내지 3크6은, 서로독립적으로, 이소루신(11 I), 글리신 ½1九 0,

比), 페닐알라닌 0¾ 幻, 티로신 0>1·, X), 트립토판 대, ¾0, 아스파라긴 , 비, 세린比라, 幻, 트레오닌(¾ ·, V , 시스테인 0方3, 0 , 글루타민 ½1 0), 아 르기닌( , 리신 0 10, 히스티딘( , 아스팔트산 3 미 및 글루 탐산 ½111, 으로이루어진군으로부터 선택되고,
상기 å는서열번호 666으로표시되는아미노산서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
2. 제 1항에 있어서,
상기 말단 융합파트너가 하기 일반식 2로 표시되는 아미노산서열을 포 함하는 것인, 융합폴리펩타이드:
[일반식 2]
상기 일반식 2에서,
3 은 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐 알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며,
상기 1는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고
상기 II은 0또는 1의 정수이다.
的
2019/143193 1»(:1^1{2019/000782
3. 제 1항에 있어서,
상기 I말단 융합파트너가 하기 일반식 3으로 표시되는 아미노산 서열을 포함하는 것인 융합폴리펩타이드:
[일반식 3]
상기 일반식 3에서,
332는 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐 알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며 ,
상기 å는서열번호 666으로표시되는 아미노산서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
4. 제 1항에 있어서,
상기 말단융합파트너가하기 일반식 4로표시되는아미노산서열을포 함하는것인, 융합폴리펩타이드:
[일반식 4]
1\161; -쇼 !!- 11근-父크크 !3!· 0 - 1元11 - II 1 ( å ) II
상기 일반식 4에서,
333은 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐 알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며,
상기 å는서열번호 666으로표시되는아미노산서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
5. 제 1항에 있어서,
상기 ^말단 융합파트너가 하기 일반식 5로 표시되는 아미노산 서열을 포 함하는 것인, 융합폴리펩타이드:
[일반식 5]
2019/143193 1»(:1^1{2019/000782 -요 - 1 1근- 용-父크크간-七 내 13-(2)11
상기 일반식 5에서,
3크4는 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신, 메티오닌, 페닐 알라닌, 티로신, 트립토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며,
상기 å는서열번호 666으로표시되는 아미노산서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
6. 제 1항에 있어서,
상기 -말단 융합파트너가 하기 일반식 6으로 표시되는 아미노산 서열을 포함하는 것인, 융합폴리펩타이드:
[일반식 6]
1¾61;-쇼311-1 1 - 용- 0 -父3크5 -미 -⑵!!
상기 일반식 6에서,
335는 이소루신, 글리신, 알라닌, 프롤린, 발린 , 루신 , 메티오닌, 페닐 알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민, 아르기닌, 리신, 히스티딘, 아스팔트산또는글루탐산이며 ,
상기 å는 서열번호 666으로 표시되는 아미노산 서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고
상기 II은 0또는 1의 정수이다.
7. 제 1항에 있어서,
상기 말단 융합파트너가 하기 일반식 7로 표시되는 아미노산서열을 포 함하는 것인 융합폴리펩타이드:
[일반식 7]
¾¾卜쇼511-1 16-/\ - 0-1611-¾36-(2)11
상기 일반식 7에서
336은 이소루신, 글리신, 알라닌, 프롤린, 발린, 루신 , 메티오닌 , 페닐 알라닌, 티로신, 트림토판, 아스파라긴, 세린, 트레오닌, 시스테인, 글루타민,
2019/143193 1»(:1^1{2019/000782 아르기닌, 리신, 히스티딘 , 아스팔트산또는글루탐산이며,
상기 2는서열번호 666으로표시되는아미노산서열의 1번 위치의 아미노 산에서부터 시작하는 1개 내지 36개의 아미노산이고,
상기 II은 0또는 1의 정수이다.
8. 제 1항에 있어서,
상기 말단융합파트너는서열번호 8내지 139 중 어느 하나의 아미노 산서열로이루어진 것인, 융합폴리펩타이드.
9. 제 1항에 있어서,
되는아미노산서열로이루어진 것인, 융합폴리펩타이드.
10. 제 1항에 있어서,
상기 링커는친화성 태그를포함하는, 융합폴리펩타이드.
11. 제 1항에 있어서,
상기 링커는프로테아제 인식 서열을포함하는, 융합폴리펩타이드.
12. 제 11항에 있어서,
상기 프로테아제 인식 서열은 담배 식각 바이러스 프로테아제 인식 서열 엔테로키나아제 인식 서열, 유비퀴틴 카르복시 말단 가수분해효소 인식 서열, 인

되는 융합폴리펩타이드.
13. 제 1항에 있어서,
상기 목적 폴리펩타이드는 인간 부갑상선 호르몬 1-34(11?1¾ 1-34), 인간 부갑상선 호르몬 1-8401?™ 1-34), 글루카곤 유사 펩타이드- 1(此13-1), 리라글루

성장인자- 1( -1), 글루카곤 유사 펩타이드- 2½내-2), 테두글루타이드
2019/143193 1»(:1^1{2019/000782

및 인슐린 유사체로 이루어진 군으로부터 선택되는 어느 하나인, 융합 폴리펩타 이드.
14. 제 1항에 있어서,
상기 목적 폴리펩타이드는서열번호 151, 340, 341, 484, 485, 628, 638, 642및 652의 아미노산서열중어느하나의 아미노산서열로 이루어진 것인, 융 합폴리펩타이드.
15. 제 1항에 있어서,
상기 융합폴리펩타이드는서열번호 160내지 291, 343내지 474, 487내 지 618, 630내지 632, 644내지 646 및 654내지 656의 아미노산서열 중어느 하나의 아미노산서열로이루어진 것인, 융합폴리펩타이드.
16. 제 1항 내지 제 15항중 어느 한항에 따른 융합 폴리펩타이드를 코딩하는 뉴클레오타이드.
17. 제 16항에 있어서,
상기 뉴클레오타이드는서열번호 294, 295, 478내지 483, 621내지 627, 635내지 637, 649내지 651및 659내지 661의 염기서열 중어느하나의 염기서 열로이루어진 것인, 뉴클레오타이드.
18. 제16항에 따른뉴클레오타이드를포함하는 발현벡터.
19. 제18항에 따른 발현벡터를포함하는숙주세포.
20. (a) 제19항에 따른숙주세포를 배양하는단계;
0 상기 숙주세포에서 발현된 융합폴리펩타이드를 정제하는 단계 및 상기 정제된 융합 폴리펩타이드를 절단 효소와 배양하여 목적 폴리펩 타이드를 회수하는 단계를포함하는 목적 폴리펩타이드의 생산방법.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/963,066 US11267863B2 (en) | 2018-01-19 | 2019-01-18 | N-terminal fusion partner for producing recombinant polypeptide, and method for producing recombinant polypeptide using same |
| EP19741746.2A EP3741774A4 (en) | 2018-01-19 | 2019-01-18 | N-TERMINAL FUSION PARTNER FOR THE MANUFACTURING OF A RECOMBINANT POLYPEPTID AND PROCESS FOR THE MANUFACTURING OF A RECOMBINANT POLYPEPTID USING THESE |
| JP2020540276A JP2021511785A (ja) | 2018-01-19 | 2019-01-18 | 組換えポリペプチド生産用n末端融合パートナーおよびこれを用いた組換えポリペプチドの生産方法 |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2018-0006875 | 2018-01-19 | ||
| KR20180006875 | 2018-01-19 | ||
| KR10-2018-0018255 | 2018-02-14 | ||
| KR10-2018-0018278 | 2018-02-14 | ||
| KR20180018278 | 2018-02-14 | ||
| KR10-2018-0018232 | 2018-02-14 | ||
| KR20180018255 | 2018-02-14 | ||
| KR20180018232 | 2018-02-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2019143193A1 true WO2019143193A1 (ko) | 2019-07-25 |
| WO2019143193A9 WO2019143193A9 (ko) | 2019-09-06 |
Family
ID=67302350
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2019/000782 WO2019143193A1 (ko) | 2018-01-19 | 2019-01-18 | 재조합 폴리펩타이드 생산용 n-말단 융합 파트너 및 이를 이용하여 재조합 폴리 펩타이드를 생산하는방법 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11267863B2 (ko) |
| EP (1) | EP3741774A4 (ko) |
| JP (1) | JP2021511785A (ko) |
| WO (1) | WO2019143193A1 (ko) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021048878A1 (en) * | 2019-09-13 | 2021-03-18 | Biological E Limited | N-terminal extension sequence for expression of recombinant therapeutic peptides |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111018965B (zh) * | 2019-12-30 | 2023-05-09 | 重庆艾力彼生物科技有限公司 | 一种重组甲状旁腺素pth(1-34)的纯化方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140069131A (ko) * | 2011-09-12 | 2014-06-09 | 아뮤닉스 오퍼레이팅 인코포레이티드 | 글루카곤-유사 펩티드-2 조성물 및 이의 제조 및 사용 방법 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4086196A (en) | 1975-03-28 | 1978-04-25 | Armour Pharmaceutical Company | Parathyroid hormone |
| AU736670B2 (en) * | 1996-09-26 | 2001-08-02 | Medical Research Council | Chaperone fragments |
| KR100890184B1 (ko) | 2007-09-06 | 2009-03-25 | 고려대학교 산학협력단 | SlyD를 융합파트너로 이용한 재조합 단백질의 제조방법 |
| WO2009066320A2 (en) | 2007-09-21 | 2009-05-28 | Cadila Healthcare Limited | Fusion protein systems suitable for high expression of peptides |
| KR20230113857A (ko) | 2010-04-15 | 2023-08-01 | 코디악 사이언시스 인코포레이티드 | 고분자량 쌍성이온-함유 중합체 |
| NZ630851A (en) | 2012-06-08 | 2016-07-29 | Alkermes Inc | Fusion polypeptides comprising mucin-domain polypeptide linkers |
| EP2746390A1 (en) | 2012-12-19 | 2014-06-25 | Sandoz Ag | Method for producing a recombinant protein of interest |
| CN105209598A (zh) * | 2013-02-22 | 2015-12-30 | 代尔夫特理工大学 | 用于产量增加的方法中的重组体微生物 |
| AP2016009663A0 (en) | 2014-07-30 | 2016-12-31 | Ngm Biopharmaceuticals Inc | Compositions and methods of use for treating metabolic disorders |
| US10118956B2 (en) | 2014-12-01 | 2018-11-06 | Pfenex Inc. | Fusion partners for peptide production |
| US10046058B2 (en) | 2014-12-02 | 2018-08-14 | Rezolute, Inc. | Use of hydrophobic organic acids to increase hydrophobicity of proteins and protein conjugates |
| CN105969712B (zh) | 2016-05-13 | 2019-09-20 | 江南大学 | 一种共表达分子伴侣蛋白提高重组大肠杆菌1,2,4-丁三醇产量的方法 |
| KR20190047376A (ko) | 2017-10-27 | 2019-05-08 | 주식회사 녹십자 | 개선된 면역글로불린의 정제방법 |
-
2019
- 2019-01-18 WO PCT/KR2019/000782 patent/WO2019143193A1/ko unknown
- 2019-01-18 EP EP19741746.2A patent/EP3741774A4/en not_active Withdrawn
- 2019-01-18 JP JP2020540276A patent/JP2021511785A/ja active Pending
- 2019-01-18 US US16/963,066 patent/US11267863B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140069131A (ko) * | 2011-09-12 | 2014-06-09 | 아뮤닉스 오퍼레이팅 인코포레이티드 | 글루카곤-유사 펩티드-2 조성물 및 이의 제조 및 사용 방법 |
Non-Patent Citations (6)
| Title |
|---|
| ANG, D. ET AL.: "Pseudo-T-even Bacteriophage RB49 Encodes CocO, a Cochaperonin for GroEL, Which Can Substitute for Escherichia coli's GroES and Bacteriophage T4's Gp31", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 276, no. 12, 4 December 2000 (2000-12-04), pages 8720 - 8726, XP055629775 * |
| DATABASE Nucleotide 9 February 2016 (2016-02-09), "PREDICTED: Myotis davidii parathyroid hormone-like (LOC102764295), mRNA", XP055630645, retrieved from NCBI Database accession no. XM_006773197.2 * |
| DATABASE Protein 19 March 2015 (2015-03-19), "glucagon-like protein [Camelus ferus", XP055630641, retrieved from NCBI Database accession no. EQB77340. 1 * |
| DATABASE Protein 22 March 2015 (2015-03-22), "Parathyroid hormone containing protein [Cricetulus griseus", XP055630642, retrieved from NCBI Database accession no. ERE79926.1 * |
| KYRATSOUS, C. A. ET AL.: "Chaperone-fusion expression plasmid vectors for improved solubility of recombinant proteins in Escherichia coli", GENE, vol. 440, 26 March 2009 (2009-03-26), pages 9 - 15, XP026127941, doi:10.1016/j.gene.2009.03.011 * |
| See also references of EP3741774A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021048878A1 (en) * | 2019-09-13 | 2021-03-18 | Biological E Limited | N-terminal extension sequence for expression of recombinant therapeutic peptides |
| CN114651063A (zh) * | 2019-09-13 | 2022-06-21 | 生物E有限公司 | 用于表达重组治疗性肽的n-末端延伸序列 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019143193A9 (ko) | 2019-09-06 |
| US20200347111A1 (en) | 2020-11-05 |
| EP3741774A4 (en) | 2021-06-02 |
| JP2021511785A (ja) | 2021-05-13 |
| EP3741774A1 (en) | 2020-11-25 |
| US11267863B2 (en) | 2022-03-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113767168B (zh) | 高效引入赖氨酸衍生物的氨酰基—tRNA合成酶 | |
| JPH02501112A (ja) | 免疫親和性による精製方法 | |
| JPH04505259A (ja) | 副甲状腺ホルモンの製造のための組換えdna法 | |
| US6437101B1 (en) | Methods for protein purification using aqueous two-phase extraction | |
| WO2019143193A1 (ko) | 재조합 폴리펩타이드 생산용 n-말단 융합 파트너 및 이를 이용하여 재조합 폴리 펩타이드를 생산하는방법 | |
| CN114790474A (zh) | 一种索马鲁肽的制备方法 | |
| KR100989413B1 (ko) | 새로운 융합파트너를 이용한 재조합 단백질의 제조방법 | |
| JP4088584B2 (ja) | 融合タンパク質から目的タンパク質を分離する方法。 | |
| CN113614113B (zh) | 含有荧光蛋白片段的融合蛋白及其用途 | |
| EP0437544A1 (en) | Recombinant pdgf and methods for production | |
| CN114933658B (zh) | 一种短肽元件及其应用方法 | |
| KR102064810B1 (ko) | 재조합 폴리펩타이드 생산용 n-말단 융합 파트너 및 이를 이용하여 재조합 폴리펩타이드를 생산하는 방법 | |
| CN114380903B (zh) | 一种胰岛素或其类似物前体 | |
| CN112646044B (zh) | TFF2-Fc融合蛋白及其高效表达生产方法 | |
| KR0161656B1 (ko) | 글루카곤의 생산방법 | |
| JPH07316197A (ja) | 成長ホルモン放出因子の製造方法 | |
| WO1988005816A1 (en) | Polypeptide | |
| CA1301676C (en) | Genetic expression of somatostatin as hybrid polypeptide | |
| KR102009709B1 (ko) | 융합 폴리펩타이드를 이용하여 인간 부갑상선 호르몬 1-84를 생산하는 방법 | |
| KR100535265B1 (ko) | 융합 단백질로부터 목적 단백질을 분리하는 방법 | |
| KR102345012B1 (ko) | GroES 융합을 이용한 인간부갑상선호르몬 1-34의 생산방법 | |
| KR100407792B1 (ko) | 인간 글루카곤 유사펩타이드를 융합파트너로 이용한재조합 단백질의 제조방법 | |
| KR102017542B1 (ko) | 융합 폴리펩타이드를 이용하여 글루카곤 유사 펩타이드-2 또는 이의 유사체를 생산하는 방법 | |
| JP2564536B2 (ja) | 改良されたタンパク質分泌促進能を有するシグナルペプチドをコ−ドしているdna配列 | |
| CN116083475A (zh) | 一种在毕赤酵母中表达人igf-1或其类似物lr3 igf-1的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19741746 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020540276 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019741746 Country of ref document: EP Effective date: 20200819 |