WO2018061868A1 - 積層体の製造方法 - Google Patents
積層体の製造方法 Download PDFInfo
- Publication number
- WO2018061868A1 WO2018061868A1 PCT/JP2017/033714 JP2017033714W WO2018061868A1 WO 2018061868 A1 WO2018061868 A1 WO 2018061868A1 JP 2017033714 W JP2017033714 W JP 2017033714W WO 2018061868 A1 WO2018061868 A1 WO 2018061868A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- polymer layer
- weight
- laminate
- base material
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 43
- 229920000642 polymer Polymers 0.000 claims abstract description 239
- 239000000835 fiber Substances 0.000 claims abstract description 136
- 239000004816 latex Substances 0.000 claims abstract description 107
- 229920000126 latex Polymers 0.000 claims abstract description 107
- 239000000701 coagulant Substances 0.000 claims abstract description 98
- 229910052751 metal Inorganic materials 0.000 claims abstract description 34
- 239000002184 metal Substances 0.000 claims abstract description 34
- 239000000758 substrate Substances 0.000 claims abstract description 34
- 150000003839 salts Chemical class 0.000 claims abstract description 30
- 150000007524 organic acids Chemical class 0.000 claims abstract description 29
- 239000002904 solvent Substances 0.000 claims abstract description 19
- 229920000459 Nitrile rubber Polymers 0.000 claims abstract description 11
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 8
- 238000005345 coagulation Methods 0.000 claims abstract description 6
- 230000015271 coagulation Effects 0.000 claims abstract description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 4
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 claims abstract description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 claims abstract description 4
- 239000000463 material Substances 0.000 claims description 87
- 230000001681 protective effect Effects 0.000 claims description 29
- 230000000149 penetrating effect Effects 0.000 claims description 9
- 238000007711 solidification Methods 0.000 claims description 6
- 230000008023 solidification Effects 0.000 claims description 6
- 230000001112 coagulating effect Effects 0.000 abstract description 7
- 239000010410 layer Substances 0.000 description 110
- 239000002585 base Substances 0.000 description 82
- 239000000178 monomer Substances 0.000 description 81
- 239000000243 solution Substances 0.000 description 74
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 51
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- ZCCIPPOKBCJFDN-UHFFFAOYSA-N calcium nitrate Chemical compound [Ca+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O ZCCIPPOKBCJFDN-UHFFFAOYSA-N 0.000 description 26
- 239000000203 mixture Substances 0.000 description 24
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 23
- 239000003431 cross linking reagent Substances 0.000 description 23
- 238000000034 method Methods 0.000 description 23
- 238000006116 polymerization reaction Methods 0.000 description 22
- 238000000465 moulding Methods 0.000 description 21
- 229910052717 sulfur Inorganic materials 0.000 description 19
- 239000011593 sulfur Substances 0.000 description 19
- 235000011054 acetic acid Nutrition 0.000 description 17
- -1 wool Substances 0.000 description 16
- 230000000052 comparative effect Effects 0.000 description 15
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 14
- 239000002253 acid Substances 0.000 description 14
- 238000004132 cross linking Methods 0.000 description 14
- 230000003204 osmotic effect Effects 0.000 description 14
- 150000001993 dienes Chemical class 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 150000002825 nitriles Chemical class 0.000 description 11
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 10
- 239000006185 dispersion Substances 0.000 description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 238000007720 emulsion polymerization reaction Methods 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 7
- 238000010438 heat treatment Methods 0.000 description 7
- 239000011787 zinc oxide Substances 0.000 description 7
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 6
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- 238000005299 abrasion Methods 0.000 description 6
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 239000004034 viscosity adjusting agent Substances 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 5
- 229920001971 elastomer Polymers 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- YXIWHUQXZSMYRE-UHFFFAOYSA-N 1,3-benzothiazole-2-thiol Chemical compound C1=CC=C2SC(S)=NC2=C1 YXIWHUQXZSMYRE-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- 239000000919 ceramic Substances 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 239000003995 emulsifying agent Substances 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 150000002978 peroxides Chemical class 0.000 description 4
- 239000003505 polymerization initiator Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000005060 rubber Substances 0.000 description 4
- 239000002562 thickening agent Substances 0.000 description 4
- 229920002554 vinyl polymer Polymers 0.000 description 4
- 238000004073 vulcanization Methods 0.000 description 4
- YAJYJWXEWKRTPO-UHFFFAOYSA-N 2,3,3,4,4,5-hexamethylhexane-2-thiol Chemical compound CC(C)C(C)(C)C(C)(C)C(C)(C)S YAJYJWXEWKRTPO-UHFFFAOYSA-N 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 3
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 3
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 3
- 239000004677 Nylon Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 238000013329 compounding Methods 0.000 description 3
- 229920003244 diene elastomer Polymers 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 239000012535 impurity Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 229910001507 metal halide Inorganic materials 0.000 description 3
- 239000003607 modifier Substances 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 229920001778 nylon Polymers 0.000 description 3
- 235000019260 propionic acid Nutrition 0.000 description 3
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- SDJHPPZKZZWAKF-UHFFFAOYSA-N 2,3-dimethylbuta-1,3-diene Chemical compound CC(=C)C(C)=C SDJHPPZKZZWAKF-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 2
- 229940054266 2-mercaptobenzothiazole Drugs 0.000 description 2
- GIUBHMDTOCBOPA-UHFFFAOYSA-N 3h-1,3-benzothiazole-2-thione;zinc Chemical compound [Zn].C1=CC=C2SC(S)=NC2=C1 GIUBHMDTOCBOPA-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical class S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000032683 aging Effects 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 2
- 239000003945 anionic surfactant Substances 0.000 description 2
- IWOUKMZUPDVPGQ-UHFFFAOYSA-N barium nitrate Chemical compound [Ba+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O IWOUKMZUPDVPGQ-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 2
- ZQMIGQNCOMNODD-UHFFFAOYSA-N diacetyl peroxide Chemical compound CC(=O)OOC(C)=O ZQMIGQNCOMNODD-UHFFFAOYSA-N 0.000 description 2
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 2
- 230000002431 foraging effect Effects 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 235000011087 fumaric acid Nutrition 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 2
- 150000002763 monocarboxylic acids Chemical class 0.000 description 2
- 150000002823 nitrates Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 239000003002 pH adjusting agent Substances 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 230000000379 polymerizing effect Effects 0.000 description 2
- 239000005077 polysulfide Substances 0.000 description 2
- 229920001021 polysulfide Polymers 0.000 description 2
- 150000008117 polysulfides Polymers 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 125000001273 sulfonato group Chemical class [O-]S(*)(=O)=O 0.000 description 2
- 125000000542 sulfonic acid group Chemical group 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 239000012209 synthetic fiber Substances 0.000 description 2
- 229920002994 synthetic fiber Polymers 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 210000002268 wool Anatomy 0.000 description 2
- 239000002759 woven fabric Substances 0.000 description 2
- 210000000707 wrist Anatomy 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- BOXSVZNGTQTENJ-UHFFFAOYSA-L zinc dibutyldithiocarbamate Chemical compound [Zn+2].CCCCN(C([S-])=S)CCCC.CCCCN(C([S-])=S)CCCC BOXSVZNGTQTENJ-UHFFFAOYSA-L 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- ONDPHDOFVYQSGI-UHFFFAOYSA-N zinc nitrate Chemical compound [Zn+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O ONDPHDOFVYQSGI-UHFFFAOYSA-N 0.000 description 2
- PMJHHCWVYXUKFD-SNAWJCMRSA-N (E)-1,3-pentadiene Chemical compound C\C=C\C=C PMJHHCWVYXUKFD-SNAWJCMRSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- SVHAMPNLOLKSFU-UHFFFAOYSA-N 1,2,2-trichloroethenylbenzene Chemical compound ClC(Cl)=C(Cl)C1=CC=CC=C1 SVHAMPNLOLKSFU-UHFFFAOYSA-N 0.000 description 1
- 150000005206 1,2-dihydroxybenzenes Chemical class 0.000 description 1
- WGJCBBASTRWVJL-UHFFFAOYSA-N 1,3-thiazolidine-2-thione Chemical compound SC1=NCCS1 WGJCBBASTRWVJL-UHFFFAOYSA-N 0.000 description 1
- 150000005208 1,4-dihydroxybenzenes Chemical class 0.000 description 1
- AYMDJPGTQFHDSA-UHFFFAOYSA-N 1-(2-ethenoxyethoxy)-2-ethoxyethane Chemical compound CCOCCOCCOC=C AYMDJPGTQFHDSA-UHFFFAOYSA-N 0.000 description 1
- XSZYESUNPWGWFQ-UHFFFAOYSA-N 1-(2-hydroperoxypropan-2-yl)-4-methylcyclohexane Chemical compound CC1CCC(C(C)(C)OO)CC1 XSZYESUNPWGWFQ-UHFFFAOYSA-N 0.000 description 1
- LGBYJXBCVZKJBL-UHFFFAOYSA-N 1-[(2-oxoazepan-1-yl)disulfanyl]azepan-2-one Chemical compound O=C1CCCCCN1SSN1C(=O)CCCCC1 LGBYJXBCVZKJBL-UHFFFAOYSA-N 0.000 description 1
- LGNQGTFARHLQFB-UHFFFAOYSA-N 1-dodecyl-2-phenoxybenzene Chemical compound CCCCCCCCCCCCC1=CC=CC=C1OC1=CC=CC=C1 LGNQGTFARHLQFB-UHFFFAOYSA-N 0.000 description 1
- OZCMOJQQLBXBKI-UHFFFAOYSA-N 1-ethenoxy-2-methylpropane Chemical compound CC(C)COC=C OZCMOJQQLBXBKI-UHFFFAOYSA-N 0.000 description 1
- LAYAKLSFVAPMEL-UHFFFAOYSA-N 1-ethenoxydodecane Chemical compound CCCCCCCCCCCCOC=C LAYAKLSFVAPMEL-UHFFFAOYSA-N 0.000 description 1
- OVGRCEFMXPHEBL-UHFFFAOYSA-N 1-ethenoxypropane Chemical compound CCCOC=C OVGRCEFMXPHEBL-UHFFFAOYSA-N 0.000 description 1
- CISIJYCKDJSTMX-UHFFFAOYSA-N 2,2-dichloroethenylbenzene Chemical compound ClC(Cl)=CC1=CC=CC=C1 CISIJYCKDJSTMX-UHFFFAOYSA-N 0.000 description 1
- WQMSFSSGCZUEPE-UHFFFAOYSA-N 2,3-dibutyl-4-hydroxybenzenecarbodithioic acid Chemical compound CCCCc1c(O)ccc(C(S)=S)c1CCCC WQMSFSSGCZUEPE-UHFFFAOYSA-N 0.000 description 1
- RWGJDHDOFUBENT-UHFFFAOYSA-N 2,3-diethyl-4-hydroxybenzenecarbodithioic acid Chemical compound CCc1c(O)ccc(C(S)=S)c1CC RWGJDHDOFUBENT-UHFFFAOYSA-N 0.000 description 1
- GSFSVEDCYBDIGW-UHFFFAOYSA-N 2-(1,3-benzothiazol-2-yl)-6-chlorophenol Chemical compound OC1=C(Cl)C=CC=C1C1=NC2=CC=CC=C2S1 GSFSVEDCYBDIGW-UHFFFAOYSA-N 0.000 description 1
- LCPVQAHEFVXVKT-UHFFFAOYSA-N 2-(2,4-difluorophenoxy)pyridin-3-amine Chemical compound NC1=CC=CN=C1OC1=CC=C(F)C=C1F LCPVQAHEFVXVKT-UHFFFAOYSA-N 0.000 description 1
- RCPUUVXIUIWMEE-UHFFFAOYSA-N 2-(2,4-dinitrophenyl)sulfanyl-1,3-benzothiazole Chemical compound [O-][N+](=O)C1=CC([N+](=O)[O-])=CC=C1SC1=NC2=CC=CC=C2S1 RCPUUVXIUIWMEE-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- GOXQRTZXKQZDDN-UHFFFAOYSA-N 2-Ethylhexyl acrylate Chemical compound CCCCC(CC)COC(=O)C=C GOXQRTZXKQZDDN-UHFFFAOYSA-N 0.000 description 1
- SBYMUDUGTIKLCR-UHFFFAOYSA-N 2-chloroethenylbenzene Chemical compound ClC=CC1=CC=CC=C1 SBYMUDUGTIKLCR-UHFFFAOYSA-N 0.000 description 1
- NRSMWHGLCNBZSO-UHFFFAOYSA-N 2-ethylidenebutanedioic acid Chemical compound CC=C(C(O)=O)CC(O)=O NRSMWHGLCNBZSO-UHFFFAOYSA-N 0.000 description 1
- NCNAZAGHUIATKV-UHFFFAOYSA-N 2-hydroxy-2-(prop-2-enoylamino)propane-1-sulfonic acid Chemical compound OS(=O)(=O)CC(O)(C)NC(=O)C=C NCNAZAGHUIATKV-UHFFFAOYSA-N 0.000 description 1
- BTOVVHWKPVSLBI-UHFFFAOYSA-N 2-methylprop-1-enylbenzene Chemical compound CC(C)=CC1=CC=CC=C1 BTOVVHWKPVSLBI-UHFFFAOYSA-N 0.000 description 1
- RPBWMJBZQXCSFW-UHFFFAOYSA-N 2-methylpropanoyl 2-methylpropaneperoxoate Chemical compound CC(C)C(=O)OOC(=O)C(C)C RPBWMJBZQXCSFW-UHFFFAOYSA-N 0.000 description 1
- AGBXYHCHUYARJY-UHFFFAOYSA-N 2-phenylethenesulfonic acid Chemical compound OS(=O)(=O)C=CC1=CC=CC=C1 AGBXYHCHUYARJY-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- BIISIZOQPWZPPS-UHFFFAOYSA-N 2-tert-butylperoxypropan-2-ylbenzene Chemical compound CC(C)(C)OOC(C)(C)C1=CC=CC=C1 BIISIZOQPWZPPS-UHFFFAOYSA-N 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- FRIBMENBGGCKPD-UHFFFAOYSA-N 3-(2,3-dimethoxyphenyl)prop-2-enal Chemical compound COC1=CC=CC(C=CC=O)=C1OC FRIBMENBGGCKPD-UHFFFAOYSA-N 0.000 description 1
- OHXAOPZTJOUYKM-UHFFFAOYSA-N 3-Chloro-2-methylpropene Chemical compound CC(=C)CCl OHXAOPZTJOUYKM-UHFFFAOYSA-N 0.000 description 1
- ZTHJQCDAHYOPIK-UHFFFAOYSA-N 3-methylbut-2-en-2-ylbenzene Chemical compound CC(C)=C(C)C1=CC=CC=C1 ZTHJQCDAHYOPIK-UHFFFAOYSA-N 0.000 description 1
- HLBZWYXLQJQBKU-UHFFFAOYSA-N 4-(morpholin-4-yldisulfanyl)morpholine Chemical compound C1COCCN1SSN1CCOCC1 HLBZWYXLQJQBKU-UHFFFAOYSA-N 0.000 description 1
- RJMXTPCJDQRHQG-UHFFFAOYSA-N 4-hydroxy-2,3-dimethylbenzenecarbothioic s-acid Chemical compound CC1=C(C)C(C(S)=O)=CC=C1O RJMXTPCJDQRHQG-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- OSDWBNJEKMUWAV-UHFFFAOYSA-N Allyl chloride Chemical compound ClCC=C OSDWBNJEKMUWAV-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- 244000025254 Cannabis sativa Species 0.000 description 1
- 235000012766 Cannabis sativa ssp. sativa var. sativa Nutrition 0.000 description 1
- 235000012765 Cannabis sativa ssp. sativa var. spontanea Nutrition 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- HXKCUQDTMDYZJD-UHFFFAOYSA-N Methyl selenac Chemical compound CN(C)C(=S)S[Se](SC(=S)N(C)C)(SC(=S)N(C)C)SC(=S)N(C)C HXKCUQDTMDYZJD-UHFFFAOYSA-N 0.000 description 1
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 1
- CNCOEDDPFOAUMB-UHFFFAOYSA-N N-Methylolacrylamide Chemical compound OCNC(=O)C=C CNCOEDDPFOAUMB-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- BDJKDKKHLSWMEX-UHFFFAOYSA-N P(=O)(OC(C)CCl)(O)O.C(C=C)(=O)O Chemical compound P(=O)(OC(C)CCl)(O)O.C(C=C)(=O)O BDJKDKKHLSWMEX-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 1
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical compound C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- KYIKRXIYLAGAKQ-UHFFFAOYSA-N abcn Chemical compound C1CCCCC1(C#N)N=NC1(C#N)CCCCC1 KYIKRXIYLAGAKQ-UHFFFAOYSA-N 0.000 description 1
- ZCHPKWUIAASXPV-UHFFFAOYSA-N acetic acid;methanol Chemical compound OC.CC(O)=O ZCHPKWUIAASXPV-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical class CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 150000008360 acrylonitriles Chemical class 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000004996 alkyl benzenes Chemical class 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- DIZPMCHEQGEION-UHFFFAOYSA-H aluminium sulfate (anhydrous) Chemical compound [Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O DIZPMCHEQGEION-UHFFFAOYSA-H 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000002280 amphoteric surfactant Substances 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003712 anti-aging effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- ITHZDDVSAWDQPZ-UHFFFAOYSA-L barium acetate Chemical compound [Ba+2].CC([O-])=O.CC([O-])=O ITHZDDVSAWDQPZ-UHFFFAOYSA-L 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- WDIHJSXYQDMJHN-UHFFFAOYSA-L barium chloride Chemical compound [Cl-].[Cl-].[Ba+2] WDIHJSXYQDMJHN-UHFFFAOYSA-L 0.000 description 1
- 229910001626 barium chloride Inorganic materials 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 description 1
- 239000001639 calcium acetate Substances 0.000 description 1
- 235000011092 calcium acetate Nutrition 0.000 description 1
- 229960005147 calcium acetate Drugs 0.000 description 1
- 235000009120 camo Nutrition 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 235000005607 chanvre indien Nutrition 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- YACLQRRMGMJLJV-UHFFFAOYSA-N chloroprene Chemical compound ClC(=C)C=C YACLQRRMGMJLJV-UHFFFAOYSA-N 0.000 description 1
- OOCCDEMITAIZTP-UHFFFAOYSA-N cinnamyl alcohol Chemical compound OCC=CC1=CC=CC=C1 OOCCDEMITAIZTP-UHFFFAOYSA-N 0.000 description 1
- HNEGQIOMVPPMNR-IHWYPQMZSA-N citraconic acid Chemical compound OC(=O)C(/C)=C\C(O)=O HNEGQIOMVPPMNR-IHWYPQMZSA-N 0.000 description 1
- 238000004581 coalescence Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- AFZSMODLJJCVPP-UHFFFAOYSA-N dibenzothiazol-2-yl disulfide Chemical compound C1=CC=C2SC(SSC=3SC4=CC=CC=C4N=3)=NC2=C1 AFZSMODLJJCVPP-UHFFFAOYSA-N 0.000 description 1
- PCERBVBQNKZCFS-UHFFFAOYSA-N dibenzylcarbamodithioic acid Chemical compound C=1C=CC=CC=1CN(C(=S)S)CC1=CC=CC=C1 PCERBVBQNKZCFS-UHFFFAOYSA-N 0.000 description 1
- FJBFPHVGVWTDIP-UHFFFAOYSA-N dibromomethane Chemical compound BrCBr FJBFPHVGVWTDIP-UHFFFAOYSA-N 0.000 description 1
- SZRLKIKBPASKQH-UHFFFAOYSA-N dibutyldithiocarbamic acid Chemical compound CCCCN(C(S)=S)CCCC SZRLKIKBPASKQH-UHFFFAOYSA-N 0.000 description 1
- PEEFBAYDEKJSPE-UHFFFAOYSA-N dicyclohexylcarbamodithioic acid Chemical compound C1CCCCC1N(C(=S)S)C1CCCCC1 PEEFBAYDEKJSPE-UHFFFAOYSA-N 0.000 description 1
- LMBWSYZSUOEYSN-UHFFFAOYSA-N diethyldithiocarbamic acid Chemical compound CCN(CC)C(S)=S LMBWSYZSUOEYSN-UHFFFAOYSA-N 0.000 description 1
- FVCOIAYSJZGECG-UHFFFAOYSA-N diethylhydroxylamine Chemical compound CCN(O)CC FVCOIAYSJZGECG-UHFFFAOYSA-N 0.000 description 1
- 229940096818 dipentamethylenethiuram disulfide Drugs 0.000 description 1
- DHNCYZNCPMWMHP-UHFFFAOYSA-N diphenylcarbamodithioic acid Chemical compound C=1C=CC=CC=1N(C(=S)S)C1=CC=CC=C1 DHNCYZNCPMWMHP-UHFFFAOYSA-N 0.000 description 1
- PXJJSXABGXMUSU-UHFFFAOYSA-N disulfur dichloride Chemical compound ClSSCl PXJJSXABGXMUSU-UHFFFAOYSA-N 0.000 description 1
- WNAHIZMDSQCWRP-UHFFFAOYSA-N dodecane-1-thiol Chemical compound CCCCCCCCCCCCS WNAHIZMDSQCWRP-UHFFFAOYSA-N 0.000 description 1
- GVGUFUZHNYFZLC-UHFFFAOYSA-N dodecyl benzenesulfonate;sodium Chemical compound [Na].CCCCCCCCCCCCOS(=O)(=O)C1=CC=CC=C1 GVGUFUZHNYFZLC-UHFFFAOYSA-N 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- UIWXSTHGICQLQT-UHFFFAOYSA-N ethenyl propanoate Chemical compound CCC(=O)OC=C UIWXSTHGICQLQT-UHFFFAOYSA-N 0.000 description 1
- NKSJNEHGWDZZQF-UHFFFAOYSA-N ethenyl(trimethoxy)silane Chemical compound CO[Si](OC)(OC)C=C NKSJNEHGWDZZQF-UHFFFAOYSA-N 0.000 description 1
- 125000002573 ethenylidene group Chemical group [*]=C=C([H])[H] 0.000 description 1
- PARDSSZKOJRLBK-UHFFFAOYSA-N ethyl dihydrogen phosphate prop-2-enoic acid Chemical compound C(C=C)(=O)O.C(C)OP(O)(=O)O PARDSSZKOJRLBK-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- XUCNUKMRBVNAPB-UHFFFAOYSA-N fluoroethene Chemical compound FC=C XUCNUKMRBVNAPB-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N formaldehyde Natural products O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 239000011487 hemp Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- NXPHCVPFHOVZBC-UHFFFAOYSA-N hydroxylamine;sulfuric acid Chemical compound ON.OS(O)(=O)=O NXPHCVPFHOVZBC-UHFFFAOYSA-N 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 229920003049 isoprene rubber Polymers 0.000 description 1
- 238000009940 knitting Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 150000005309 metal halides Chemical class 0.000 description 1
- 229910001960 metal nitrate Inorganic materials 0.000 description 1
- FQPSGWSUVKBHSU-UHFFFAOYSA-N methacrylamide Chemical compound CC(=C)C(N)=O FQPSGWSUVKBHSU-UHFFFAOYSA-N 0.000 description 1
- FJQXCDYVZAHXNS-UHFFFAOYSA-N methadone hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 FJQXCDYVZAHXNS-UHFFFAOYSA-N 0.000 description 1
- 125000005394 methallyl group Chemical group 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- 229940088644 n,n-dimethylacrylamide Drugs 0.000 description 1
- YLGYACDQVQQZSW-UHFFFAOYSA-N n,n-dimethylprop-2-enamide Chemical compound CN(C)C(=O)C=C YLGYACDQVQQZSW-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 125000002560 nitrile group Chemical group 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- ZWWQICJTBOCQLA-UHFFFAOYSA-N o-propan-2-yl (propan-2-yloxycarbothioyldisulfanyl)methanethioate Chemical compound CC(C)OC(=S)SSC(=S)OC(C)C ZWWQICJTBOCQLA-UHFFFAOYSA-N 0.000 description 1
- KZCOBXFFBQJQHH-UHFFFAOYSA-N octane-1-thiol Chemical compound CCCCCCCCS KZCOBXFFBQJQHH-UHFFFAOYSA-N 0.000 description 1
- SRSFOMHQIATOFV-UHFFFAOYSA-N octanoyl octaneperoxoate Chemical compound CCCCCCCC(=O)OOC(=O)CCCCCCC SRSFOMHQIATOFV-UHFFFAOYSA-N 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 238000010525 oxidative degradation reaction Methods 0.000 description 1
- HVAMZGADVCBITI-UHFFFAOYSA-M pent-4-enoate Chemical compound [O-]C(=O)CCC=C HVAMZGADVCBITI-UHFFFAOYSA-M 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- KNBRWWCHBRQLNY-UHFFFAOYSA-N piperidine-1-carbothioylsulfanyl piperidine-1-carbodithioate Chemical compound C1CCCCN1C(=S)SSC(=S)N1CCCCC1 KNBRWWCHBRQLNY-UHFFFAOYSA-N 0.000 description 1
- PMJHHCWVYXUKFD-UHFFFAOYSA-N piperylene Natural products CC=CC=C PMJHHCWVYXUKFD-UHFFFAOYSA-N 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920001084 poly(chloroprene) Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229910052573 porcelain Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- FZYCEURIEDTWNS-UHFFFAOYSA-N prop-1-en-2-ylbenzene Chemical compound CC(=C)C1=CC=CC=C1.CC(=C)C1=CC=CC=C1 FZYCEURIEDTWNS-UHFFFAOYSA-N 0.000 description 1
- RAJUSMULYYBNSJ-UHFFFAOYSA-N prop-1-ene-1-sulfonic acid Chemical compound CC=CS(O)(=O)=O RAJUSMULYYBNSJ-UHFFFAOYSA-N 0.000 description 1
- QROGIFZRVHSFLM-UHFFFAOYSA-N prop-1-enylbenzene Chemical compound CC=CC1=CC=CC=C1 QROGIFZRVHSFLM-UHFFFAOYSA-N 0.000 description 1
- UIIIBRHUICCMAI-UHFFFAOYSA-N prop-2-ene-1-sulfonic acid Chemical compound OS(=O)(=O)CC=C UIIIBRHUICCMAI-UHFFFAOYSA-N 0.000 description 1
- 238000010298 pulverizing process Methods 0.000 description 1
- 239000012779 reinforcing material Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229940080264 sodium dodecylbenzenesulfonate Drugs 0.000 description 1
- CHQMHPLRPQMAMX-UHFFFAOYSA-L sodium persulfate Substances [Na+].[Na+].[O-]S(=O)(=O)OOS([O-])(=O)=O CHQMHPLRPQMAMX-UHFFFAOYSA-L 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229920003048 styrene butadiene rubber Polymers 0.000 description 1
- FWMUJAIKEJWSSY-UHFFFAOYSA-N sulfur dichloride Chemical compound ClSCl FWMUJAIKEJWSSY-UHFFFAOYSA-N 0.000 description 1
- 229940052367 sulfur,colloidal Drugs 0.000 description 1
- 238000004381 surface treatment Methods 0.000 description 1
- 239000002335 surface treatment layer Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical class ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- KUAZQDVKQLNFPE-UHFFFAOYSA-N thiram Chemical compound CN(C)C(=S)SSC(=S)N(C)C KUAZQDVKQLNFPE-UHFFFAOYSA-N 0.000 description 1
- 229960002447 thiram Drugs 0.000 description 1
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- NLVXSWCKKBEXTG-UHFFFAOYSA-N vinylsulfonic acid Chemical compound OS(=O)(=O)C=C NLVXSWCKKBEXTG-UHFFFAOYSA-N 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- RKQOSDAEEGPRER-UHFFFAOYSA-L zinc diethyldithiocarbamate Chemical compound [Zn+2].CCN(CC)C([S-])=S.CCN(CC)C([S-])=S RKQOSDAEEGPRER-UHFFFAOYSA-L 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
- 239000004711 α-olefin Substances 0.000 description 1
Images
Classifications
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/07—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with halogens; with halogen acids or salts thereof; with oxides or oxyacids of halogens or salts thereof
- D06M11/11—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with halogens; with halogen acids or salts thereof; with oxides or oxyacids of halogens or salts thereof with halogen acids or salts thereof
- D06M11/17—Halides of elements of Groups 3 or 13 of the Periodic Table
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/693—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with natural or synthetic rubber, or derivatives thereof
-
- A—HUMAN NECESSITIES
- A41—WEARING APPAREL
- A41D—OUTERWEAR; PROTECTIVE GARMENTS; ACCESSORIES
- A41D19/00—Gloves
- A41D19/0055—Plastic or rubber gloves
-
- A—HUMAN NECESSITIES
- A41—WEARING APPAREL
- A41D—OUTERWEAR; PROTECTIVE GARMENTS; ACCESSORIES
- A41D19/00—Gloves
- A41D19/04—Appliances for making gloves; Measuring devices for glove-making
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D5/00—Processes for applying liquids or other fluent materials to surfaces to obtain special surface effects, finishes or structures
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B25/00—Layered products comprising a layer of natural or synthetic rubber
- B32B25/10—Layered products comprising a layer of natural or synthetic rubber next to a fibrous or filamentary layer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/24—Acids; Salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/12—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity
- C08L101/14—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity the macromolecular compounds being water soluble or water swellable, e.g. aqueous gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
- C08L9/02—Copolymers with acrylonitrile
- C08L9/04—Latex
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/07—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with halogens; with halogen acids or salts thereof; with oxides or oxyacids of halogens or salts thereof
- D06M11/11—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with halogens; with halogen acids or salts thereof; with oxides or oxyacids of halogens or salts thereof with halogen acids or salts thereof
- D06M11/155—Halides of elements of Groups 2 or 12 of the Periodic Table
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/07—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with halogens; with halogen acids or salts thereof; with oxides or oxyacids of halogens or salts thereof
- D06M11/11—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with halogens; with halogen acids or salts thereof; with oxides or oxyacids of halogens or salts thereof with halogen acids or salts thereof
- D06M11/20—Halides of elements of Groups 4 or 14 of the Periodic Table, e.g. zirconyl chloride
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/51—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with sulfur, selenium, tellurium, polonium or compounds thereof
- D06M11/55—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with sulfur, selenium, tellurium, polonium or compounds thereof with sulfur trioxide; with sulfuric acid or thiosulfuric acid or their salts
- D06M11/56—Sulfates or thiosulfates other than of elements of Groups 3 or 13 of the Periodic Table
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/51—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with sulfur, selenium, tellurium, polonium or compounds thereof
- D06M11/55—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with sulfur, selenium, tellurium, polonium or compounds thereof with sulfur trioxide; with sulfuric acid or thiosulfuric acid or their salts
- D06M11/57—Sulfates or thiosulfates of elements of Groups 3 or 13 of the Periodic Table, e.g. alums
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/58—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with nitrogen or compounds thereof, e.g. with nitrides
- D06M11/64—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with nitrogen or compounds thereof, e.g. with nitrides with nitrogen oxides; with oxyacids of nitrogen or their salts
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M11/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising
- D06M11/58—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with nitrogen or compounds thereof, e.g. with nitrides
- D06M11/64—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with inorganic substances or complexes thereof; Such treatment combined with mechanical treatment, e.g. mercerising with nitrogen or compounds thereof, e.g. with nitrides with nitrogen oxides; with oxyacids of nitrogen or their salts
- D06M11/65—Salts of oxyacids of nitrogen
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/184—Carboxylic acids; Anhydrides, halides or salts thereof
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/184—Carboxylic acids; Anhydrides, halides or salts thereof
- D06M13/1845—Aromatic mono- or polycarboxylic acids
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/184—Carboxylic acids; Anhydrides, halides or salts thereof
- D06M13/188—Monocarboxylic acids; Anhydrides, halides or salts thereof
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/184—Carboxylic acids; Anhydrides, halides or salts thereof
- D06M13/192—Polycarboxylic acids; Anhydrides, halides or salts thereof
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/184—Carboxylic acids; Anhydrides, halides or salts thereof
- D06M13/203—Unsaturated carboxylic acids; Anhydrides, halides or salts thereof
- D06M13/2035—Aromatic acids
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/21—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/31—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds of unsaturated nitriles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D1/00—Processes for applying liquids or other fluent materials
- B05D1/36—Successively applying liquids or other fluent materials, e.g. without intermediate treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/02—Composition of the impregnated, bonded or embedded layer
- B32B2260/021—Fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/04—Impregnation, embedding, or binder material
- B32B2260/048—Natural or synthetic rubber
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2319/00—Synthetic rubber
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2377/00—Polyamides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/28—Nitrogen-containing compounds
- C08K2003/287—Calcium, strontium or barium nitrates
Definitions
- the present invention relates to a method for producing a laminate formed by forming a polymer layer on a fiber base material. Moreover, this invention relates to the manufacturing method of the protective glove using the said laminated body.
- Patent Document 1 discloses a protective glove in which a stretchable yarn is knitted at least on the wrist and rubber or resin is coated on the stretchable yarn.
- the protective glove obtained by the technique of Patent Document 1 has a problem that the flexibility when used as a work glove is not sufficient due to the influence of rubber or resin coated on the elastic yarn.
- the present invention has been made in view of such a situation, and an object thereof is to provide a method for producing a laminate having excellent flexibility and wear resistance. Moreover, this invention also aims at providing the manufacturing method of the protective glove using the laminated body obtained by such a manufacturing method.
- the present inventors have made a polymer layer formed by bringing a polymer latex into contact with a fiber substrate to form a polymer layer.
- a coagulant solution for coagulating the polymer of the polymer latex is obtained by dissolving or dispersing a metal salt and an organic acid as a coagulant in a predetermined ratio in a solvent. The inventors have found that the object can be achieved and have completed the present invention.
- a coagulant solution adhesion step for adhering a coagulant solution to a fiber substrate, and a polymer latex is brought into contact with the fiber substrate to which the coagulant solution is adhered to coagulate the polymer.
- the said metal salt is a polyvalent metal salt.
- the said organic acid is an organic acid which has at least 1 type of group of a carboxyl group, a sulfo group, a hydroxyl group, and a thiol group.
- the polymer constituting the polymer latex is preferably nitrile rubber.
- the present invention it is possible to provide a method for producing a laminate having excellent flexibility and wear resistance. Moreover, according to this invention, the manufacturing method of the protective glove using the laminated body obtained by such a manufacturing method can be provided.
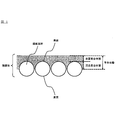
- FIG. 1 is a cross-sectional view of a laminate in which a polymer layer is formed on a fiber base material.
- the method for producing a laminate of the present invention comprises a coagulant solution adhesion step for adhering a coagulant solution to a fiber base material, and a polymer latex brought into contact with the fiber base material to which the coagulant solution is adhered.
- Coagulating step of forming a polymer layer on the fiber base material by coagulating a metal salt 0.2 to 7.0 wt% as a coagulant in the solvent as the coagulant solution.
- an organic acid 0.1 to 7.0% by weight dissolved or dispersed therein is used.
- the fiber base material used in the present invention is not particularly limited as long as it is made of fiber, but natural fibers such as cotton, wool, hemp, and wool, synthetic fibers such as polyester, polyurethane, acrylic, and nylon are used as materials. Among these, nylon is preferable.
- the fiber base material may be knitted or sewn, and may be a woven fabric or a non-woven fabric.
- the thickness of the fiber base material is not particularly limited, but is preferably 0.1 to 2.0 mm.
- the linear density of the fiber base material is not particularly limited, but is preferably 50 to 500 denier.
- the gauge number of the fiber base material is not particularly limited, but is preferably 7 to 18 gauge. Here, the number of gauges refers to the number of knitting machine needles between 1 inch.
- the fiber base material is composed of a plurality of fibers, particularly when the fiber base material is a woven fabric, the portion in which the fibers are usually folded and the overlapping degree of the fibers in the thickness direction is dense.
- the thickness may be different between the portion where the fiber overlap is dense and the portion where the fiber overlap is sparse, The thickness of the fiber base material is determined as an average value when the thickness of the portion where the fiber overlap degree is dense is defined as the thickness of the fiber base material.
- the coagulant solution used in the present invention is obtained by dissolving or dispersing 0.2 to 7.0% by weight of a metal salt as a coagulant and 0.1 to 7.0% by weight of an organic acid in a solvent. is there.
- the metal salt as a coagulant constituting the coagulant solution may be any metal salt that can coagulate the polymer in the polymer latex, and is not particularly limited.
- the metal species include lithium, sodium, and potassium.
- Monovalent metals such as magnesium, calcium, zinc, iron, barium, zirconium, copper, and the like; and trivalent metals such as aluminum.
- the salt species include inorganic acids such as nitric acid, sulfuric acid and hydrochloric acid; organic acids such as acetic acid.
- the metal species is preferably a polyvalent metal, more preferably a divalent metal, and particularly preferably calcium.
- the salt species nitric acid or hydrochloric acid is preferable, and nitric acid is particularly preferable. That is, the metal salt is preferably a polyvalent metal salt, more preferably a divalent metal nitrate or halide.
- these metal salts include nitrates such as calcium nitrate, barium nitrate and zinc nitrate; metal halide salts such as barium chloride, calcium chloride, magnesium chloride, zinc chloride and aluminum chloride; barium acetate, calcium acetate and acetic acid Acetates such as zinc; sulfates such as calcium sulfate, magnesium sulfate, and aluminum sulfate; among these, nitrates and metal halides are preferable, calcium nitrate and calcium chloride are more preferable, and calcium nitrate is particularly preferable preferable. These metal salts can be used alone or in combination of two or more.
- the organic acid constituting the coagulant solution is not particularly limited, but an organic acid having at least one kind of carboxyl group, sulfo group, hydroxy group, and thiol group is preferable.
- an organic acid having at least one kind of carboxyl group, sulfo group, hydroxy group, and thiol group is preferable.
- acetic acid, formic acid, propionic acid, citric acid, oxalic acid, ascorbic acid, malic acid, tartaric acid, propionic acid, benzoic acid, lactic acid, gluconic acid, succinic acid, fumaric acid, alkylbenzene sulfonic acid, aliphatic sulfone An acid, dodecyl diphenyl ether sulfonic acid, etc. are mentioned, Among these, organic acids having a carboxyl group such as acetic acid, formic acid, propionic acid, citric acid and oxalic acid are preferable, and acetic
- the solvent for dissolving or dispersing the metal salt and the organic acid as the coagulant is not particularly limited, but water, alcohol such as methanol and ethanol, or a mixture thereof can be used. Alcohol is preferred, and methanol is particularly preferred.
- the concentration of the metal salt as the coagulant in the coagulant solution is 0.1 to 7.0% by weight, preferably 0.1 to 6.0% by weight, more preferably 0.1 to 4.0% by weight. %, More preferably 0.1 to 2.0% by weight.
- the concentration of the organic acid in the coagulant solution is 0.2 to 7.0% by weight, preferably 0.2 to 5.0% by weight, more preferably 0.2 to 3.0% by weight. is there.
- such a coagulant solution is made to adhere to a fiber base material
- a polymer latex is made to contact the fiber base material to which the coagulant solution was made to adhere, and a polymer is coagulated.
- the polymer layer is formed on the fiber base material.
- the above-mentioned coagulant solution is attached to a fiber base material, and a polymer latex is brought into contact with the fiber base material to which the coagulant solution thus obtained is attached.
- the polymer layer is formed by the solidification of the polymer in the polymer latex progressing while a part of the polymer latex penetrates into the inside of the fiber base material. Therefore, as shown in FIG. 1, in the laminate obtained by the production method of the present invention, a polymer layer is formed on the surface of the fiber base, and a part of the polymer layer constitutes the fiber base. It penetrates to the gap between the fibers.
- FIG. 1 in the laminate obtained by the production method of the present invention, a polymer layer is formed on the surface of the fiber base, and a part of the polymer layer constitutes the fiber base. It penetrates to the gap between the fibers.
- FIG. 1 is a figure which shows sectional drawing of the laminated body obtained by the manufacturing method of this invention
- permeated the clearance gap between fiber base materials among polymer layers is a penetration polymer layer.
- the part formed on the surface of the fiber base material among polymer layers is shown as a surface polymer layer.
- the cross section of the fiber base material is simply shown for easy understanding, but the actual cross section of the fiber base material is not limited to such a shape.
- the fibers constituting the fiber base material are illustrated as being arranged in a single layer in a direction perpendicular to the paper surface. , And may be lined up so that there are two or more layers.
- the fibers constituting the fiber substrate may be single fibers (one by one extracted from the above-mentioned natural fibers or synthetic fibers), or may be twisted yarns composed of a plurality of single fibers.
- the polymer layer is described as appropriately composed of a osmotic polymer layer and a surface polymer layer. Usually, the osmotic polymer layer and the surface polymer layer are integrally formed. It will be.
- the coagulant solution for coagulating the polymer latex polymer contains a metal salt and an organic acid as a coagulant, and the metal as the coagulant.
- a salt and an organic acid having a predetermined content ratio are used, whereby a polymer layer having a certain thickness is formed on the surface of the fiber substrate, and a part of the polymer layer is formed.
- the fiber base material can be appropriately permeated, and the flexibility and abrasion resistance of the resulting laminate can be balanced to a high degree, thereby making it suitable as a protective glove such as a work glove. It can be.
- the polymer latex when the polymer latex is brought into contact with the fiber substrate to which the coagulant solution is adhered, the polymer in the polymer latex due to the action of the metal salt as the coagulant in the coagulant solution.
- the coagulation rate of the polymer can be adjusted appropriately by the action of the organic acid in the coagulant solution, so that the polymer coagulation can proceed while the polymer latex is properly infiltrated into the fiber substrate. Is. And thereby, as shown in FIG. 1, while being able to form a polymer layer of a certain amount of thickness on the surface of a fiber base material, a part of the said polymer layer is made to osmose
- the content ratio of the metal salt as the coagulant in the coagulant solution is increased (for example, the content ratio of the metal salt as the coagulant in the coagulant solution is 0.5 wt. %),
- the organic acid in the coagulant solution can properly penetrate a part of the polymer layer into the fiber base material.
- the polymer layer can be effectively prevented from being peeled off by the action of the portion of the coalesced layer that has penetrated into the fiber base material, whereby the wear resistance of the laminate can be particularly improved.
- the coagulation rate of the polymer in the polymer latex will increase, and the polymer latex will not be sufficiently penetrated before it penetrates into the fiber substrate. Since the solidification of the coalescence proceeds, the thickness of the surface polymer layer formed on the surface of the fiber base material becomes too thick among the formed polymer layers, and the flexibility of the resulting laminate is reduced. In addition, among the polymer layers to be formed, the thickness of the osmotic polymer layer formed by penetrating into the fiber substrate becomes too thin, and the polymer layer is easily peeled off from the fiber substrate. As a result, the wear resistance of the laminate is reduced.
- the content ratio of the metal salt as the coagulant in the coagulant solution is too small, the coagulation rate of the polymer in the polymer latex decreases, and the polymer latex penetrates too much into the fiber substrate.
- the polymer layer reaches the back side of the fiber base material, causing a through-through, which reduces the productivity of the laminate or inferior to the feeling of use when the resulting laminate is used as a protective glove. It becomes.
- the content ratio of the organic acid in the coagulant solution is too small, the solidification rate of the polymer in the polymer latex decreases, and the polymer latex penetrates too much into the fiber base, so that the polymer layer is a fiber group. Through-holes that reach the back surface of the material may occur, which may reduce the productivity of the laminated body, or may deteriorate the feeling of use when the obtained laminated body is used as a protective glove.
- the content of the organic acid in the coagulant solution is too high, the solidification rate of the polymer in the polymer latex is increased, and the polymer is solidified before the polymer latex sufficiently penetrates the fiber substrate.
- the thickness of the surface polymer layer formed on the surface of the fiber substrate becomes too thick, and the flexibility of the resulting laminate is reduced.
- the thickness of the penetrating polymer layer formed by penetrating into the fiber base material becomes too thin, and the polymer layer is easily peeled off from the fiber base material. The wear resistance of the laminate is reduced.
- the rubbery polymer includes natural rubber; conjugated diene rubber obtained by polymerizing or copolymerizing conjugated dienes such as butadiene and isoprene; among these, conjugated diene rubber is preferable.
- conjugated diene rubber examples include so-called nitrile rubber, isoprene rubber, styrene-butadiene rubber, chloroprene rubber and the like obtained by copolymerizing nitrile, and among these, nitrile rubber is particularly preferable.
- the nitrile rubber is not particularly limited, and a nitrile rubber obtained by copolymerizing an ⁇ , ⁇ -ethylenically unsaturated nitrile monomer and other copolymerizable monomers used as necessary can be used.
- the ⁇ , ⁇ -ethylenically unsaturated nitrile monomer is not particularly limited, and an ethylenically unsaturated compound having a nitrile group and preferably having 3 to 18 carbon atoms can be used.
- Examples of such ⁇ , ⁇ -ethylenically unsaturated nitrile monomers include acrylonitrile, methacrylonitrile, halogen-substituted acrylonitrile and the like, and among these, acrylonitrile is particularly preferable.
- These ⁇ , ⁇ -ethylenically unsaturated nitrile monomers may be used alone or in combination of two or more.
- the content ratio of the ⁇ , ⁇ -ethylenically unsaturated nitrile monomer unit in the nitrile rubber is preferably 10 to 45% by weight, more preferably 20 to 40% by weight, based on the total monomer units.
- the nitrile rubber preferably contains a conjugated diene monomer unit from the viewpoint of imparting rubber elasticity to the resulting polymer layer.
- conjugated diene monomer forming the conjugated diene monomer unit examples include 1,3-butadiene, isoprene, 2,3-dimethyl-1,3-butadiene, 1,3-pentadiene, chloroprene and the like having 4 to 4 carbon atoms. 6 conjugated diene monomers are preferred, 1,3-butadiene and isoprene are more preferred, and 1,3-butadiene is particularly preferred. In addition, these conjugated diene monomers may be used individually by 1 type, and may be used in combination of 2 or more type.
- the content ratio of the conjugated diene monomer unit is preferably 40 to 80% by weight, more preferably 52 to 78% by weight, based on all monomer units constituting the nitrile rubber.
- Nitrile rubber is also a monomer that forms ⁇ , ⁇ -ethylenically unsaturated nitrile monomer units and other ethylenically unsaturated monomers that are copolymerizable with monomers that form conjugated diene monomer units.
- An acid monomer may be included.
- Such other copolymerizable ethylenically unsaturated acid monomer is not particularly limited, and examples thereof include a carboxyl group-containing ethylenically unsaturated monomer, a sulfonic acid group-containing ethylenically unsaturated monomer, Examples thereof include phosphoric acid group-containing ethylenically unsaturated monomers.
- the carboxyl group-containing ethylenically unsaturated monomer is not particularly limited, but ethylenically unsaturated monocarboxylic acids such as acrylic acid, methacrylic acid, and crotonic acid; fumaric acid, maleic acid, itaconic acid, maleic anhydride, anhydrous And ethylenically unsaturated polyvalent carboxylic acids such as itaconic acid and anhydrides thereof; partially esterified products of ethylenically unsaturated polyvalent carboxylic acids such as methyl maleate and methyl itaconic acid; and the like.
- monocarboxylic acids such as acrylic acid, methacrylic acid, and crotonic acid
- fumaric acid, maleic acid, itaconic acid, maleic anhydride anhydrous And ethylenically unsaturated polyvalent carboxylic acids such as itaconic acid and anhydrides thereof
- the sulfonic acid group-containing ethylenically unsaturated monomer is not particularly limited, but vinyl sulfonic acid, methyl vinyl sulfonic acid, styrene sulfonic acid, (meth) allyl sulfonic acid, (meth) acrylic acid-2-sulfonic acid ethyl And 2-acrylamido-2-hydroxypropanesulfonic acid.
- the phosphoric acid group-containing ethylenically unsaturated monomer is not particularly limited, but includes (meth) acrylic acid-3-chloro-2-propyl phosphate, (meth) acrylic acid-2-ethyl phosphate, 3-allyloxy. Examples include -2-hydroxypropane phosphoric acid.
- copolymerizable ethylenically unsaturated acid monomers can be used as alkali metal salts or ammonium salts, and can be used singly or in combination of two or more. Good.
- carboxyl group-containing ethylenically unsaturated monomers are preferable, ethylenically unsaturated monocarboxylic acids are more preferable, and methacrylic acid is particularly preferable.
- the content of the other copolymerizable ethylenically unsaturated acid monomer in the polymer constituting the polymer latex used in the present invention is preferably 2 with respect to all monomer units in the polymer. ⁇ 8% by weight.
- the polymer constituting the polymer latex may contain other monomer units.
- monomers that form other monomer units include conjugated diene monomers, ethylenically unsaturated nitrile monomer units, and monomers copolymerizable with ethylenically unsaturated acid monomers. There is no particular limitation, and examples thereof include the following monomers.
- aromatic vinyl monomers such as styrene, ⁇ -methyl styrene, monochloro styrene, dichloro styrene, trichloro styrene, monomethyl styrene, dimethyl styrene, trimethyl styrene, hydroxymethyl styrene; acrylamide, Ethylenically unsaturated carboxylic acid amide monomers such as methacrylamide, N, N-dimethylacrylamide, N-methylolacrylamide; methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, (meth ) Ethylenically unsaturated carboxylic acid alkyl ester monomers such as 2-ethylhexyl acrylate; vinyl acetate monomers such as vinyl acetate, vinyl propionate and vinyl versatate; vinyl chloride, vinylidene chloride
- the content of other monomer units in the polymer constituting the polymer latex is a point of view that prevents the polymer layer from peeling off from the fiber substrate, and when used as a work glove. From the viewpoint of suppressing fatigue, and from the viewpoint of suppressing permeation of solvent gas when worn as a work glove, it is preferably 26% by weight or less, more preferably 10% by weight, based on the total monomer units in the polymer. % Or less, more preferably 7% by weight or less, and particularly preferably 5% by weight or less.
- the polymer latex used in the present invention is not particularly limited, and may be, for example, a polymer latex obtained by polymerizing a monomer mixture containing the above monomer.
- a latex obtained by emulsion polymerization, a latex obtained by phase inversion emulsification of a polymer solution obtained by solution polymerization of the monomer mixture, and the like can be used.
- the composition of the resulting copolymer can be easily adjusted by adjusting the composition of the monomer mixture used for the emulsion polymerization.
- a method of emulsion polymerization a conventionally known method can be employed.
- polymerization auxiliary materials such as an emulsifier, a polymerization initiator, and a molecular weight modifier can be used.
- the method for adding these polymerization auxiliary materials is not particularly limited, and any method such as an initial batch addition method, a divided addition method, or a continuous addition method may be used.
- the emulsifier examples include, but are not limited to, anionic surfactants, nonionic surfactants, cationic surfactants, and amphoteric surfactants. Among these, alkylbenzene sulfonates, aliphatics Anionic surfactants such as sulfonates, sulfates of higher alcohols, ⁇ -olefin sulfonates, and alkyl ether sulfates are preferred.
- the amount of the emulsifier is preferably 0.5 to 10 parts by weight, more preferably 1 to 8 parts by weight with respect to 100 parts by weight of the total monomers used.
- a radical initiator is preferable.
- the radical initiator is not particularly limited, and examples thereof include inorganic peroxides such as sodium persulfate, potassium persulfate, ammonium persulfate, potassium perphosphate, and hydrogen peroxide; t-butyl peroxide, cumene hydroperoxide, p-menthane hydroperoxide, di-t-butyl peroxide, t-butylcumyl peroxide, acetyl peroxide, isobutyryl peroxide, octanoyl peroxide, dibenzoyl peroxide, 3,5,5-trimethylhexanoyl Organic peroxides such as peroxide and t-butylperoxyisobutyrate; azo compounds such as azobisisobutyronitrile, azobis-2,4-dimethylvaleronitrile, azobiscyclohexanecarbonitrile, methyl
- inorganic peroxides or organic peroxides are preferred, inorganic peroxides are more preferable, persulfate are particularly preferred.
- These polymerization initiators may be used individually by 1 type, and may be used in combination of 2 or more type. The amount of the polymerization initiator used is preferably 0.01 to 2 parts by weight, more preferably 0.05 to 1.5 parts by weight with respect to 100 parts by weight of the total monomers used.
- the molecular weight modifier is not particularly limited.
- ⁇ -methylstyrene dimer mercaptans such as t-dodecyl mercaptan, n-dodecyl mercaptan, octyl mercaptan; halogenated carbon tetrachloride, methylene chloride, methylene bromide, etc. Hydrocarbons; sulfur-containing compounds such as tetraethylthiuram disulfide, dipentamethylene thiuram disulfide, diisopropylxanthogen disulfide; and the like.
- molecular weight regulators may be used alone or in combination of two or more.
- the amount of the molecular weight modifier used varies depending on the type, but is preferably 0.1 to 1.5 parts by weight, more preferably 0.2 to 1.0 parts by weight, based on 100 parts by weight of all monomers used. Part.
- Emulsion polymerization is usually performed in water.
- the amount of water used is preferably 80 to 500 parts by weight, more preferably 100 to 200 parts by weight, based on 100 parts by weight of the total monomers used.
- polymerization auxiliary materials other than the above may be further used as necessary.
- the polymerization auxiliary material include a chelating agent, a dispersing agent, a pH adjusting agent, a deoxidizing agent, a particle size adjusting agent and the like, and the type and amount of use thereof are not particularly limited.
- Examples of the monomer addition method include a method of adding monomers to be used in a reaction vessel all at once, a method of adding continuously or intermittently as the polymerization proceeds, and a part of the monomer is added. And a method in which the remaining monomer is continuously or intermittently added and polymerized, and any method may be employed.
- the composition of the mixture may be constant or may be changed.
- Each monomer may be added to the reaction vessel after previously mixing various monomers to be used, or may be added separately to the reaction vessel.
- the polymerization temperature at the time of emulsion polymerization is not particularly limited, but is usually 0 to 95 ° C., preferably 5 to 70 ° C.
- the polymerization time is not particularly limited, but is usually about 5 to 40 hours.
- the monomer is emulsion-polymerized, and when the predetermined polymerization conversion rate is reached, the polymerization reaction is stopped by cooling the polymerization system or adding a polymerization terminator.
- the polymerization conversion rate when stopping the polymerization reaction is usually 80% by weight or more, preferably 90% by weight or more.
- the polymerization terminator is not particularly limited as long as it is usually used in emulsion polymerization. Specific examples thereof include hydroxylamine, hydroxyamine sulfate, diethylhydroxyamine, hydroxyaminesulfonic acid and alkali metals thereof.
- Hydroxyamine compounds such as salts; sodium dimethyldithiocarbamate; hydroquinone derivatives; catechol derivatives; aromatic hydroxydithiocarboxylic acids such as hydroxydimethylbenzenethiocarboxylic acid, hydroxydiethylbenzenedithiocarboxylic acid, hydroxydibutylbenzenedithiocarboxylic acid, and alkali metal salts thereof
- Aromatic hydroxydithiocarboxylic acid compounds such as;
- the amount of the polymerization terminator used is not particularly limited, but is usually 0.05 to 2 parts by weight with respect to 100 parts by weight of the total monomers used.
- unreacted monomers may be removed to adjust the solid content concentration and pH.
- the weight average particle diameter of the polymer particles constituting the polymer latex is usually 30 to 1000 nm, preferably 50 to 500 nm, more preferably 70 to 200 nm.
- the solid content concentration of the polymer latex is usually 20 to 65% by weight, preferably 30 to 60% by weight, more preferably 35 to 55% by weight.
- the pH of the polymer latex is usually 5 to 13, preferably 7 to 10, and more preferably 7.5 to 9.
- the polymer latex used in the present invention may be a composition (latex composition) to which these are added.
- latex composition a composition (latex composition) to which these are added.
- the following explanation about the polymer latex is the same when the latex composition is used as the polymer latex.
- the crosslinking agent it is preferable to use a sulfur-based crosslinking agent.
- a sulfur type crosslinking agent Sulfur, such as powder sulfur, sulfur white, precipitation sulfur, colloidal sulfur, surface treatment sulfur, insoluble sulfur; sulfur chloride, sulfur dichloride, morpholine disulfide, alkylphenol disulfide, dibenzothia Sulfur-containing compounds such as zildisulfide, caprolactam disulfide, phosphorus-containing polysulfide, and polymer polysulfide; sulfur-donating compounds such as tetramethylthiuram disulfide, selenium dimethyldithiocarbamate, and 2- (4′-morpholinodithio) benzothiazole; Is mentioned.
- These crosslinking agents may be used alone or in combination of two or more.
- the addition amount of the sulfur-based crosslinking agent is preferably 0.01 to 5 parts by weight, more preferably 0.05 to 3 parts by weight, particularly preferably 0. 0 part by weight with respect to 100 parts by weight of the total solid content in the polymer latex. 1 to 2 parts by weight.
- the sulfur-based crosslinking agent is preferably added as a dispersion in which the sulfur-based crosslinking agent is dispersed in a solvent.
- a dispersion By adding it to the polymer latex as a dispersion, a laminate having few defects such as cracks, pinholes, and adhesion of aggregates in the resulting polymer layer can be obtained.
- the method for preparing the dispersion of the sulfur-based crosslinking agent is not particularly limited, but a method of adding a solvent to the sulfur-based crosslinking agent and pulverizing and stirring with a wet pulverizer such as a ball mill or a bead mill is preferable.
- sulfur-based crosslinking agent When sulfur is used as the sulfur-based crosslinking agent, it is preferable to use a crosslinking accelerator (vulcanization accelerator) or zinc oxide in combination.
- vulcanization accelerator vulcanization accelerator
- zinc oxide zinc oxide
- the crosslinking accelerator is not particularly limited.
- dithiocarbamine such as diethyldithiocarbamic acid, dibutyldithiocarbamic acid, di-2-ethylhexyldithiocarbamic acid, dicyclohexyldithiocarbamic acid, diphenyldithiocarbamic acid, and dibenzyldithiocarbamic acid.
- crosslinking accelerators may be used alone or in combination of two or more.
- the amount of the crosslinking accelerator used is preferably 0.1 to 10 parts by weight, more preferably 0.5 to 5 parts by weight with respect to 100 parts by weight of the total solid content in the polymer latex.
- the amount of zinc oxide used is preferably 5 parts by weight or less, more preferably 0.1 to 3 parts by weight, and still more preferably 0.5 to 2 parts by weight with respect to 100 parts by weight of the total solid content in the polymer latex. Part.
- a viscosity modifier to the polymer latex.
- group thickener, etc. are mentioned.
- the viscosity of the polymer latex is preferably 500 to 8,000 mPa ⁇ s, more preferably 2,500 to 7,000 mPa ⁇ s.
- the polymer latex when used in the state of a latex composition (for example, the above-described crosslinking agent, crosslinking accelerator, zinc oxide, viscosity modifier and the like are added to the polymer latex),
- the viscosity of the latex composition is preferably within the above range.
- a crosslinking agent As described above, a crosslinking agent, a crosslinking accelerator, zinc oxide, a viscosity modifier, and the like can be added to the polymer latex, and further, an anti-aging agent, an antioxidant, a preservative, as necessary.
- various additives such as an antibacterial agent, a wetting agent, a thickening agent, a dispersing agent, a pigment, a dye, a filler, a reinforcing material, and a pH adjusting agent can be added in a predetermined amount.
- the solid content concentration of the polymer latex in a state where no cross-linking agent is added is preferably controlled within the above-mentioned range, but the solid content of the polymer latex added with the cross-linking agent (latex composition obtained by adding the cross-linking agent).
- the partial concentration is preferably 5 to 40% by weight, more preferably 10 to 25% by weight.
- the surface tension of the polymer latex to which a crosslinking agent is added (latex composition to which a crosslinking agent is added) is preferably 25 to 40 mN / m.
- the above-described coagulant solution is attached to the above-described fiber base, and then the polymer latex is brought into contact with the fiber base to which the coagulant solution is attached to coagulate the polymer.
- the polymer latex is brought into contact with the fiber base to which the coagulant solution is attached to coagulate the polymer.
- the method for adhering the coagulant solution to the fiber substrate is not particularly limited, and examples thereof include a method of immersing the fiber substrate in the coagulant solution.
- the immersion time when the fiber base material is immersed in the coagulant solution is not particularly limited, but is preferably 30 to 1 second, more preferably 10 to 1 second.
- the fiber base material when adhered to the coagulant solution, it is preferable to immerse the fiber base material in the coagulant solution in a state where the fiber base material is put on a molding die having a desired shape.
- the molding die for covering the fiber substrate is not particularly limited, but various materials such as porcelain, glass, metal, and plastic can be used.
- the shape of the molding die may be a desired shape according to the shape of the final product.
- various molds for gloves such as a mold having a shape from the wrist to the fingertip as the mold for covering the fiber base material. .
- the drying temperature at this time is not particularly limited and may be selected according to the solvent to be used, but is preferably 10 to 80 ° C., more preferably 15 to 70 ° C.
- the drying time is not particularly limited, but is preferably 600 to 1 second, more preferably 300 to 5 seconds.
- the polymer latex is brought into contact with the fiber base material to which the coagulant solution is adhered in this manner, so that the polymer in the polymer latex is coagulated to form a polymer layer on the fiber base material. .
- the method of bringing the polymer latex into contact with the fiber base material to which the coagulant solution is attached is not particularly limited, and examples thereof include a method in which the fiber base material to which the coagulant solution is attached is immersed in the polymer latex.
- the fiber base material to which the coagulant solution is attached is immersed in the polymer latex
- the fiber base material to which the coagulant solution is attached is put on the polymer latex in a state where the fiber base material is put on a molding die having a desired shape. It is preferable to immerse.
- the fiber base material is coated with the coagulant solution as described above in a state where the fiber base material is previously covered with a molding die having a desired shape, and then the coagulant solution is adhered to the fiber base material. Is preferably immersed in a polymer latex while being covered with a molding die.
- the drying temperature at this time is not particularly limited, but is preferably 10 to 80 ° C., more preferably 15 to 80 ° C.
- the drying time is not particularly limited, but is preferably 120 minutes to 5 seconds, more preferably 60 minutes to 10 seconds.
- the temperature conditions for aging are not particularly limited, but are preferably 20 to 50 ° C.
- the time for aging is the viewpoint of preventing separation between the fiber base material and the polymer layer, the viewpoint of improving the wear resistance when the obtained laminate is used as a protective glove, and working the protective glove. From the viewpoint of suppressing the permeation of solvent gas when used as a protective glove, it is preferably 4 hours or longer and 120 hours or shorter, more preferably 24 hours or longer and 72 hours or shorter.
- the aging time in the above range, in the obtained laminate, the polymer layer appropriately penetrates into the fiber base material, so that the fiber base material and the polymer layer are prevented from peeling off. While abrasion resistance improves, permeation
- the crosslinking agent as polymer latex
- the polymer latex adhering to the fiber base material is heated.
- the heating temperature for crosslinking is preferably 60 to 160 ° C, more preferably 80 to 150 ° C. By setting the heating temperature within the above range, the time required for the crosslinking reaction can be shortened to improve the productivity of the laminate, and the obtained laminate can suppress the oxidative degradation of the polymer due to excessive heating. The physical properties of can be improved.
- the heating time for crosslinking may be appropriately selected according to the heating temperature, but is usually 5 to 120 minutes.
- the polymer latex is brought into contact with the fiber base material to which the coagulant solution is adhered as described above, so that a part of the polymer latex penetrates the fiber base material while The polymer in the combined latex coagulates, whereby a polymer layer is formed on the fiber substrate, and a laminate is obtained. Therefore, as shown in FIG. 1, the obtained laminate is solidified in a state where a part of the polymer of the polymer latex penetrates into the fiber base material to form a polymer layer, thereby improving flexibility and wear resistance. It will be excellent.
- the polymer layer is immersed in warm water at 20 to 80 ° C. for about 0.5 to 60 minutes to remove the polymer layer from the polymer layer. It is preferable to remove water-soluble impurities (emulsifier, water-soluble polymer, coagulant, etc.).
- water-soluble impurities emulsifier, water-soluble polymer, coagulant, etc.
- the treatment of immersing such a polymer layer in warm water is performed by Although it may be performed after crosslinking, it is preferably performed before crosslinking the polymer of the polymer layer from the viewpoint that water-soluble impurities can be more efficiently removed.
- a laminate can be obtained by removing the fiber base material on which the polymer layer is formed from the molding die. It can.
- a method of removing from the molding die a method of peeling from the molding die by hand, or peeling by water pressure or compressed air pressure can be employed.
- a heat treatment (post-crosslinking step) may be performed at a temperature of 60 to 120 ° C. for 10 to 120 minutes. Further, a surface treatment layer by chlorination treatment or coating treatment may be formed on the inner and / or outer surface of the laminate.
- the thickness of the portion of the polymer layer constituting the laminated body that has penetrated into the fiber substrate that is, the thickness of the osmotic polymer layer shown in FIG.
- the thickness of the osmotic polymer layer shown in FIG. is preferably 0.6 to 0.05 mm, more preferably 0.55 to 0.1 mm, and still more preferably 0.5 to 0.2 mm.
- the upper limit of thickness becomes the thickness at the time of osmosis
- the upper limit of the thickness of the osmotic polymer layer Is 0.6 mm.
- permeated the fiber base material among polymer layers ie, the thickness of the surface polymer layer shown in FIG. 1
- the thickness of the surface polymer layer shown in FIG. 1 is not specifically limited. However, it is preferably 0.6 to 0.05 mm, more preferably 0.5 to 0.1 mm, and still more preferably 0.45 to 0.12 mm.
- the ratio of the thickness of the osmotic polymer layer to the surface polymer layer in the polymer layer is not particularly limited, but the ratio of the thickness of the osmotic polymer layer to the thickness of the surface polymer layer (the thickness of the osmotic polymer layer / the surface weight).
- the thickness of the combined layer is preferably 5 to 0.2, more preferably 2 to 0.3.
- the total thickness of the polymer layer that is, the total thickness of the osmotic polymer layer and the surface polymer layer is not particularly limited, but is preferably 1.0 to 0.05 mm.
- the present invention as described above, as a coagulant solution for coagulating the polymer of the polymer latex, 0.2 to 7.0% by weight of the coagulant and 0.1 to By using 7.0% by weight dissolved or dispersed, a polymer layer having a certain thickness is formed on the surface of the fiber substrate as shown in FIG. A laminate in which a part of the fiber base material has permeated appropriately can be obtained. Therefore, according to the present invention, the obtained laminate is excellent in flexibility and wear resistance, and is suitably used as a work glove, particularly a protective glove for home use, agriculture use, fishing use, industrial use, and the like. be able to.
- the thickness of the surface polymer layer is determined by measuring the distance from the surface of the fiber substrate to the surface of the polymer layer at 10 points, and the number average of the measurement results It was obtained by calculating the value.
- the thickness of the penetrating polymer layer was determined by measuring the distance from the surface of the fiber base material to the deepest part of the penetrating polymer at 10 locations and calculating the number average value of the measurement results. Furthermore, the thickness of the whole laminated body was calculated
- the gloves manufactured in the flexible example and the comparative example were each worn by 10 people and evaluated according to the following evaluation criteria. 5: Very soft 4: Soft 3: Slightly soft 2: Hard 1: Very hard
- the abrasion resistance abrasion test was evaluated using a Martindale abrasion tester (STM633: manufactured by SATRA) according to the method described in EN388. Specifically, for the laminates produced in the examples and comparative examples, friction was repeated while applying a predetermined load, and the number of frictions until breakage was obtained. According to the number of frictions until breakage, it is divided into levels from level 0 to level 4, and the higher the level, the better the wear resistance. LEVEL 4: 8,000 rotations LEVEL 3: 2,000 or more rotations and less than 8,000 rotations LEVEL 2: 500 or more rotations and less than 2,000 rotations LEVEL 1: 100 or more rotations and 500 rotations Less than LEVEL 0: Less than 100 revolutions
- Example 1 Preparation of dispersion of compounding agent 1.0 part of colloidal sulfur (manufactured by Hosoi Chemical Co., Ltd.), 0.5 part of dispersing agent (trade name “Demol N”, manufactured by Kao Corporation), 5% potassium hydroxide aqueous solution (Wako Pure Chemical Industries Kogyo Co., Ltd. (0.0015 parts) and water (1.0 parts) were pulverized and stirred in a ball mill for 48 hours to obtain a dispersion having a solid content concentration of 50% by weight.
- colloidal sulfur manufactured by Hosoi Chemical Co., Ltd.
- dispersing agent trade name “Demol N”, manufactured by Kao Corporation
- 5% potassium hydroxide aqueous solution (Wako Pure Chemical Industries Kogyo Co., Ltd. (0.0015 parts) and water (1.0 parts) were pulverized and stirred in a ball mill for 48 hours to obtain a dispersion having a solid content concentration of 50% by weight.
- ZnDBC zinc dibutyldithiocarbamate
- BD 1,3-butadiene
- AN acrylonitrile as an ⁇ , ⁇ -ethylenically unsaturated nitrile monomer
- MAA methacrylic acid
- MAA methacrylic acid
- t-dodecyl mercaptan 4 parts 132 parts of ion-exchanged water
- sodium dodecylbenzenesulfonate 0.5 parts of ⁇ -naphthalenesulfonic acid formalin condensate sodium salt
- potassium persulfate 0.05 part of sodium ethylenediaminetetraacetate
- the pH and solid content concentration of the polymer latex are adjusted to obtain a dip-molding latex according to Example 1 having a solid content concentration of 40% and a pH of 8. It was.
- Aron manufactured by Toa Gosei Co., Ltd.
- Aron was added as a viscosity modifier to adjust the viscosity of the composition to 3,000 mPa ⁇ s to obtain a latex composition for dip molding. .
- Coagulant Solution A methanol solution prepared by dissolving 1.0% by weight of calcium nitrate as a coagulant and 3.0% by weight of acetic acid as an organic acid in methanol was prepared as a coagulant solution.
- Laminate Protective Gloves
- aging also referred to as pre-vulcanization
- a ceramic glove mold covered with a glove-shaped fiber base material material: nylon, linear density: 300 denier, gauge number: 13 gauge, thickness: 0.8 mm
- material: nylon, linear density: 300 denier, gauge number: 13 gauge, thickness: 0.8 mm was immersed in the above coagulant solution for 5 seconds, After lifting from the coagulant solution, it was dried at a temperature of 30 ° C. for 1 minute. Thereafter, the ceramic glove mold was immersed in the dip molding latex composition for 5 seconds, pulled up from the dip molding latex composition, dried at a temperature of 30 ° C.
- the polymer layer was formed on the fiber base material by making it dry on the conditions for minutes. Thereafter, the ceramic glove mold in which the polymer layer is formed is immersed in warm water at 60 ° C. for 90 seconds to elute water-soluble impurities from the polymer layer, and then dried under conditions of a temperature of 30 ° C. for 10 minutes, Furthermore, the polymer in the polymer layer was subjected to a crosslinking treatment by performing a heat treatment under conditions of a temperature of 125 ° C. for 30 minutes. Subsequently, the laminated body (protective glove) was obtained by peeling the fiber base material in which the polymer layer was formed from the ceramic glove mold.
- flexibility, abrasion resistance, penetration, and acetic acid odor were evaluated.
- the results are shown in Table 1.
- the total thickness of the surface polymer layer and the penetrating polymer layer is shown as the thickness of the entire polymer layer.
- Examples 2-5 Instead of the coagulant solution used in Example 1, 0.2% by weight of calcium nitrate and 3.0% by weight of acetic acid in methanol (Example 2), 7.0% by weight of calcium nitrate and 3.0% by weight of acetic acid Methanol solution (Example 3), 1.0 wt% calcium nitrate and 0.1 wt% acetic acid in methanol (Example 4), and 1.0 wt% calcium nitrate and 7.0 wt% acetic acid in methanol A laminate (protective glove) was obtained and evaluated in the same manner as in Example 1 except that (Example 5) was prepared and used as a coagulant solution. The results are shown in Table 1.
- Example 6 instead of the coagulant solution used in Example 1, a methanol solution containing 4.8% by weight calcium nitrate and 3.0% by weight acetic acid was prepared and used as a coagulant solution. A laminate (protective glove) was obtained and evaluated in the same manner. The results are shown in Table 1.
- Comparative Examples 1-5 Instead of the coagulant solution used in Example 1, a methanol solution in which only 1.0% by weight of calcium nitrate was dissolved (Comparative Example 1) and a methanol solution in which only 3.0% by weight of acetic acid was dissolved (Comparative Example 2) ), A methanol solution in which only 0.1% by weight of calcium nitrate is dissolved (Comparative Example 3), a methanol solution in which only 10.0% by weight of calcium nitrate is dissolved (Comparative Example 4), and 10.0% by weight of calcium nitrate A laminate (protective glove) was obtained in the same manner as in Example 1 except that 8.0% by weight of acetic acid and a methanol solution of 8.0% by weight (Comparative Example 5) were prepared and used as a coagulant solution. Evaluation was performed. The results are shown in Table 1.
- Example 2 Even when the content ratio of the organic acid is in the above range, the laminate obtained using the coagulant solution containing no coagulant was inferior in flexibility, and the inside out occurred (comparison).
- Example 3 The laminate obtained by using a coagulant solution containing a coagulant with an excessively small content and not containing an organic acid was inferior in flexibility and could cause a strike through (comparison).
- Example 4 The laminate obtained using a coagulant solution containing a coagulant with an excessively large content and not containing an organic acid was inferior in flexibility and wear resistance (Comparative Example 4).
- the laminate obtained by using the coagulant solution in which the content ratio of the coagulant and the content ratio of the organic acid are both too large is inferior in wear resistance and the acetic acid odor is confirmed (Comparative Example). 5).
Landscapes
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Gloves (AREA)
- Chemical Or Physical Treatment Of Fibers (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
Abstract
Description
本発明の積層体の製造方法において、前記有機酸がカルボキシル基、スルホ基、ヒドロキシ基、チオール基の少なくとも一種類の基を有する有機酸であることが好ましい。
本発明の積層体の製造方法において、前記重合体ラテックスを構成する重合体がニトリルゴムであることが好ましい。
なお、繊維基材は、複数の繊維により構成されるため、特に、繊維基材が織布である場合には、通常、繊維が折り重なって厚み方向における繊維の重なり度合いが密になっている部分(重なり合っている繊維の数が多い部分)と、厚み方向における繊維の重なり度合いが疎になっている部分(重なり合っている繊維の数が少ない部分)とが存在し、これらの部分を含む層(基材層)によって、構成されることとなる。そのため、繊維基材は、そのミクロ構造においては、繊維の重なり度合いが密になっている部分と、繊維の重なり度合いが疎になっている部分とで、その厚みが異なる場合があるが、上記の繊維基材の厚みは、繊維基材について、繊維の重なり度合いが密になっている部分の厚みを、その厚みとした際の平均値として、求めることとする。
これらの金属塩の具体例としては、硝酸カルシウム、硝酸バリウム、硝酸亜鉛等の硝酸塩;塩化バリウム、塩化カルシウム、塩化マグネシウム、塩化亜鉛、塩化アルミニウム等のハロゲン化金属塩;酢酸バリウム、酢酸カルシウム、酢酸亜鉛等の酢酸塩;硫酸カルシウム、硫酸マグネシウム、硫酸アルミニウム等の硫酸塩;等が挙げられ、これらの中でも、硝酸塩およびハロゲン化金属塩が好ましく、硝酸カルシウムおよび塩化カルシウムがより好ましく、硝酸カルシウムが特に好ましい。
これらの金属塩は、単独または2種以上組み合わせて用いることができる。
乳化重合して得られるラテックスを用いる場合には、乳化重合に用いる単量体混合物の組成を調節することにより、得られる共重合体の組成を容易に調節することができるようになる。乳化重合の方法としては、従来公知の方法を採用することができる。
乳化剤の使用量は、使用する全単量体100重量部に対して、好ましくは0.5~10重量部、より好ましくは1~8重量部である。
重合開始剤の使用量は、使用する全単量体100重量部に対して、好ましくは0.01~2重量部、より好ましくは0.05~1.5重量部である。
分子量調整剤の使用量は、その種類によって異なるが、使用する全単量体100重量部に対して、好ましくは0.1~1.5重量部、より好ましくは0.2~1.0重量部である。
また、各単量体は、使用する各種単量体を予め混合してから反応容器に添加しても、あるいは別々に反応容器に添加してもよい。
重合停止剤の使用量は、特に限定されないが、通常、使用する全単量体100重量部に対して、0.05~2重量部である。
架橋促進剤の使用量は、重合体ラテックス中の全固形分100重量部に対して、好ましくは0.1~10重量部、より好ましくは0.5~5重量部である。
実施例及び比較例で製造した積層体について、中指の先から12cmの掌部分の重合体層が積層された断面を、光学顕微鏡(キーエンス社製VHX-200)を用いて観察して、表面重合体層の厚み、浸透重合体層の厚み、および積層体全体の厚みを測定した。具体的な測定方法について図1を参照して説明すると、表面重合体層の厚みは、繊維基材の表面から、重合体層の表面までの距離を、10カ所測定し、測定結果の数平均値を算出することにより求めた。また、浸透重合体層の厚みは、繊維基材の表面から、浸透した重合体の最深部までの距離を、10カ所測定し、測定結果の数平均値を算出することにより求めた。さらに、積層体全体の厚みは、重合体層の表面から、繊維基材の裏面までの距離を、10カ所測定し、測定結果の数平均値を算出することにより求めた。
実施例および比較例で製造した手袋を10人にそれぞれ着用してもらい下記の評価基準で評価した。
5:非常に柔らかい
4:柔らかい
3:やや柔らかい
2:硬い
1:非常に硬い
摩耗試験はEN388に記載の方法に則って、マーチンデール式摩耗試験機(STM633:SATRA社製)を用いて評価を実施した。具体的には、実施例および比較例で製造した積層体について、所定の加重をかけながら摩擦を繰り返し、破損までの摩擦回数を得た。破損に至るまでの摩擦回数に従い、レベル0からレベル4までのレベルに分けられ、レベルが高いほど耐摩耗性に優れる。
LEVEL 4:回転数8,000回転
LEVEL 3:回転数2,000回転以上、8,000回転未満
LEVEL 2:回転数500回転以上、2,000回転未満
LEVEL 1:回転数100回転以上、500回転未満
LEVEL 0:回転数100回転未満
実施例及び比較例で製造した積層体について、繊維基材に浸透した重合体層が繊維基材の裏面まで到達しているか否かを目視にて確認し、以下の基準で評価した。
無し:重合体層が繊維基材の裏面まで到達していなかった。
有り:重合体層が繊維基材の裏面まで到達していた。
実施例及び比較例で製造した積層体を被験者10人が着用し、キーボード入力操作の軽作業を1時間行い、1時間の作業後に酢酸の臭気による不快感を覚えた人の人数をカウントし、以下の基準により評価した。
無し:不快感を覚えた人数が0人であった。
有り:不快感を覚えた人数が1人以上であった。
配合剤の分散液の調製
コロイド硫黄(細井化学工業社製)1.0部、分散剤(花王社製、商品名「デモールN」)0.5部、5%水酸化カリウム水溶液(和光純薬工業社製)0.0015部、水1.0部を、ボールミル中で48時間粉砕攪拌し、固形分濃度50重量%の分散液を得た。
重合反応器に、共役ジエン単量体として1,3-ブタジエン(以下、「BD」ということがある。)65部、α,β-エチレン性不飽和ニトリル単量体としてアクリロニトリル(以下、「AN」ということがある。)30部、エチレン性不飽和モノカルボン酸系単量体としてメタクリル酸(以下、「MAA」ということがある。)5部、t-ドデシルメルカプタン0.4部、イオン交換水132部、ドデシルベンゼンスルホン酸ナトリウム3部、β-ナフタリンスルホン酸ホルマリン縮合物ナトリウム塩0.5部、過硫酸カリウム0.3部およびエチレンジアミン四酢酸ナトリウム塩0.05部を仕込み、重合温度を30~40℃に保持して重合を行い、重合転化率が94%に達するまで反応させて、重合体ラテックスを得た。
上記のディップ成形用ラテックスに、10%アンモニア水溶液を添加して、pHを9.5に調整するとともに、ディップ成形用ラテックス中の共重合体100部に対して、それぞれ固形分換算で、コロイド硫黄1.0部(上記硫黄分散液として添加)、ZnDBC0.5部、酸化亜鉛2.0部となるように、各配合剤の水分散液を添加した。なお、各配合剤の水分散液の添加の際には、ディップ成形用ラテックスを撹拌した状態で、所定の量をゆっくり添加した。添加物が均一に混合された後に、粘度調整剤としてアロン(東亜合成(株)製)を添加して組成物の粘度を3,000mPa・sに調整し、ディップ成形用ラテックス組成物を得た。
凝固剤としての硝酸カルシウム1.0重量%、および有機酸としての酢酸3.0重量%をそれぞれメタノールに溶解させてなるメタノール溶液を、凝固剤溶液として調製した。
まず、上述したディップ成形用ラテックス組成物に対して、温度30℃、48時間の条件で、熟成(前加硫ともいう。)を施した。次いで、手袋形状の繊維基材(材質:ナイロン、線密度:300デニール、ゲージ数:13ゲージ、厚み:0.8mm)を被せたセラミックス手袋型を、上記の凝固剤溶液に5秒間浸漬し、凝固剤溶液から引き上げた後、温度30℃、1分間の条件で乾燥させた。その後、セラミックス手袋型を、上記のディップ成形用ラテックス組成物に5秒間浸漬し、ディップ成形用ラテックス組成物から引き上げた後、温度30℃、30分間の条件で乾燥させ、次いで温度70℃、10分間の条件で乾燥させることで、繊維基材上に重合体層を形成した。その後、重合体層を形成したセラミックス手袋型を、60℃の温水に90秒間浸漬して、重合体層から水溶性の不純物を溶出させた後、温度30℃、10分間の条件で乾燥させ、さらに温度125℃、30分間の条件で熱処理を行う事で、重合体層中の重合体に架橋処理を施した。次いで、重合体層が形成された繊維基材をセラミックス手袋型から剥がすことで、積層体(保護手袋)を得た。得られた積層体について、上述した方法に従い、表面重合体層の厚み、浸透重合体層の厚みおよび積層体全体の厚み、柔軟性、耐摩耗性、裏抜けならびに酢酸臭の評価を行った。結果を表1に示す。なお、表1においては、表面重合体層の厚み、および浸透重合体層の厚みの合計値を、重合体層全体の厚みとして示した。
実施例1で用いた凝固剤溶液に代えて、硝酸カルシウム0.2重量%および酢酸3.0重量%のメタノール溶液(実施例2)、硝酸カルシウム7.0重量%および酢酸3.0重量%のメタノール溶液(実施例3)、硝酸カルシウム1.0重量%および酢酸0.1重量%のメタノール溶液(実施例4)、ならびに硝酸カルシウム1.0重量%および酢酸7.0重量%のメタノール溶液(実施例5)を、それぞれ調製して凝固剤溶液として使用した以外は、実施例1と同様にして、積層体(保護手袋)を得て、同様に評価を行った。結果を表1に示す。
実施例1で用いた凝固剤溶液に代えて、硝酸カルシウム4.8重量%および酢酸3.0重量%のメタノール溶液を調製して凝固剤溶液として使用した以外は、実施例1と同様にして、積層体(保護手袋)を得て、同様に評価を行った。結果を表1に示す。
実施例1で用いた凝固剤溶液に代えて、硝酸カルシウム1.0重量%のみを溶解させたメタノール溶液(比較例1)、酢酸3.0重量%のみを溶解させたメタノール溶液(比較例2)、硝酸カルシウム0.1重量%のみを溶解させたメタノール溶液(比較例3)、硝酸カルシウム10.0重量%のみを溶解させたメタノール溶液(比較例4)、ならびに硝酸カルシウム10.0重量%および酢酸8.0重量%のメタノール溶液(比較例5)を、それぞれ調製して凝固剤溶液として使用した以外は、実施例1と同様にして、積層体(保護手袋)を得て、同様に評価を行った。結果を表1に示す。

一方、凝固剤の含有割合が上記範囲であっても、有機酸を含有しない凝固剤溶液を用いて得られた積層体は、柔軟性に劣るものであった(比較例1)。
有機酸の含有割合が上記範囲であっても、凝固剤を含有しない凝固剤溶液を用いて得られた積層体は、柔軟性に劣り、しかも裏抜けが発生してしまうものであった(比較例2)。
凝固剤の含有割合が小さすぎであり、かつ、有機酸を含有しない凝固剤溶液を用いて得られた積層体は、柔軟性に劣り、しかも裏抜けが発生してしまうものであった(比較例3)。
凝固剤の含有割合が大きすぎであり、かつ、有機酸を含有しない凝固剤溶液を用いて得られた積層体は、柔軟性および耐摩耗性に劣るものであった(比較例4)。
凝固剤の含有割合および有機酸の含有割合がいずれも大きすぎる凝固剤溶液を用いて得られた積層体は、耐摩耗性に劣り、しかも酢酸臭が確認されてしまうものであった(比較例5)。
Claims (7)
- 繊維基材に凝固剤溶液を付着させる凝固剤溶液付着工程と、
前記凝固剤溶液を付着させた前記繊維基材に、重合体ラテックスを接触させて重合体を凝固させることで、前記繊維基材上に重合体層を形成する凝固工程と、を備え、
前記凝固剤溶液として、溶媒中に、凝固剤としての金属塩0.2~7.0重量%、および有機酸0.1~7.0重量%を溶解または分散させてなるものを用いる積層体の製造方法。 - 前記金属塩が多価金属塩である請求項1に記載の積層体の製造方法。
- 前記有機酸がカルボキシル基、スルホ基、ヒドロキシ基、チオール基の少なくとも一種類の基を有する有機酸である請求項1または2に記載の積層体の製造方法。
- 前記重合体ラテックスを構成する重合体がニトリルゴムである請求項1~3のいずれかに記載の積層体の製造方法。
- 前記凝固工程において、前記重合体層のうち前記繊維基材に浸透した部分である浸透重合体層の厚みが、0.05~0.6mmとなるように、前記重合体層を形成する請求項1~4のいずれかに記載の積層体の製造方法。
- 前記凝固工程において、前記重合体層のうち前記繊維基材に浸透していない部分である表面重合体層の厚みが、0.05~0.6mmとなるように、前記重合体層を形成する請求項1~5のいずれかに記載の積層体の製造方法。
- 請求項1~6のいずれかに記載の製造方法により得られる積層体を用いる保護手袋の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780051746.7A CN109642389B (zh) | 2016-09-30 | 2017-09-19 | 层叠体的制造方法 |
| JP2018542424A JP6922923B2 (ja) | 2016-09-30 | 2017-09-19 | 積層体の製造方法 |
| US16/337,104 US11325356B2 (en) | 2016-09-30 | 2017-09-19 | Method for producing laminate |
| EP17855823.5A EP3521506A4 (en) | 2016-09-30 | 2017-09-19 | PROCESS FOR PRODUCING LAMINATE |
| KR1020197012461A KR20190053962A (ko) | 2016-09-30 | 2017-09-19 | 적층체의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-193427 | 2016-09-30 | ||
| JP2016193427 | 2016-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018061868A1 true WO2018061868A1 (ja) | 2018-04-05 |
Family
ID=61759671
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/033714 WO2018061868A1 (ja) | 2016-09-30 | 2017-09-19 | 積層体の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11325356B2 (ja) |
| EP (1) | EP3521506A4 (ja) |
| JP (1) | JP6922923B2 (ja) |
| KR (1) | KR20190053962A (ja) |
| CN (1) | CN109642389B (ja) |
| WO (1) | WO2018061868A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020045476A (ja) * | 2018-09-21 | 2020-03-26 | 住友理工株式会社 | ゴム成形体の製造方法 |
| WO2020111097A1 (ja) | 2018-11-30 | 2020-06-04 | 日本ゼオン株式会社 | ディップ成形用ラテックス組成物およびディップ成形体 |
| WO2021166835A1 (ja) | 2020-02-18 | 2021-08-26 | 日本ゼオン株式会社 | ラテックス組成物およびディップ成形体 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7180667B2 (ja) * | 2018-03-02 | 2022-11-30 | 三菱瓦斯化学株式会社 | アルミナの保護液、保護方法及びこれを用いたアルミナ層を有する半導体基板の製造方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH105304A (ja) * | 1996-06-21 | 1998-01-13 | Koki Bussan Kk | 水不透過性手袋 |
| JP2011001662A (ja) * | 2009-06-19 | 2011-01-06 | Towa Corp:Kk | 手袋及びその製造方法 |
| US20150135403A1 (en) * | 2013-11-19 | 2015-05-21 | Ansell Limited | Polymer blends of nitrile rubber and polychloroprene |
| US20160262469A1 (en) * | 2015-03-10 | 2016-09-15 | Ansell Limited | Supported glove having an abrasion resistant nitrile coating |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9061453B2 (en) * | 2009-11-02 | 2015-06-23 | Atg Ceylon (Private) Limited | Protective garments and materials therefor |
| US8883657B2 (en) * | 2011-12-16 | 2014-11-11 | Ansell Healthcare Products Llc | Latex coated high performance polyethylene fabrics |
| JP6460363B2 (ja) | 2012-10-31 | 2019-01-30 | ショーワグローブ株式会社 | 手袋 |
| US10350848B2 (en) * | 2013-11-26 | 2019-07-16 | Ansell Limited | Nitrile/polyurethane polymer blends |
-
2017
- 2017-09-19 JP JP2018542424A patent/JP6922923B2/ja active Active
- 2017-09-19 KR KR1020197012461A patent/KR20190053962A/ko not_active Application Discontinuation
- 2017-09-19 CN CN201780051746.7A patent/CN109642389B/zh active Active
- 2017-09-19 WO PCT/JP2017/033714 patent/WO2018061868A1/ja unknown
- 2017-09-19 US US16/337,104 patent/US11325356B2/en active Active
- 2017-09-19 EP EP17855823.5A patent/EP3521506A4/en not_active Withdrawn
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH105304A (ja) * | 1996-06-21 | 1998-01-13 | Koki Bussan Kk | 水不透過性手袋 |
| JP2011001662A (ja) * | 2009-06-19 | 2011-01-06 | Towa Corp:Kk | 手袋及びその製造方法 |
| US20150135403A1 (en) * | 2013-11-19 | 2015-05-21 | Ansell Limited | Polymer blends of nitrile rubber and polychloroprene |
| US20160262469A1 (en) * | 2015-03-10 | 2016-09-15 | Ansell Limited | Supported glove having an abrasion resistant nitrile coating |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3521506A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020045476A (ja) * | 2018-09-21 | 2020-03-26 | 住友理工株式会社 | ゴム成形体の製造方法 |
| JP7217114B2 (ja) | 2018-09-21 | 2023-02-02 | 住友理工株式会社 | ゴム成形体の製造方法 |
| WO2020111097A1 (ja) | 2018-11-30 | 2020-06-04 | 日本ゼオン株式会社 | ディップ成形用ラテックス組成物およびディップ成形体 |
| WO2021166835A1 (ja) | 2020-02-18 | 2021-08-26 | 日本ゼオン株式会社 | ラテックス組成物およびディップ成形体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3521506A4 (en) | 2020-05-13 |
| CN109642389B (zh) | 2021-07-09 |
| JPWO2018061868A1 (ja) | 2019-07-11 |
| EP3521506A1 (en) | 2019-08-07 |
| JP6922923B2 (ja) | 2021-08-18 |
| CN109642389A (zh) | 2019-04-16 |
| KR20190053962A (ko) | 2019-05-20 |
| US20200031100A1 (en) | 2020-01-30 |
| US11325356B2 (en) | 2022-05-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10280291B2 (en) | Dip-forming latex composition and dip-formed article | |
| WO2018061868A1 (ja) | 積層体の製造方法 | |
| WO2017014029A1 (ja) | ゴム成形品および保護手袋 | |
| US20170342243A1 (en) | Dip-formed article | |
| JP2023116582A (ja) | ラテックス組成物 | |
| JP7167917B2 (ja) | 積層体の製造方法 | |
| JP7234940B2 (ja) | ラテックス組成物 | |
| JP7095601B2 (ja) | 積層体 | |
| EP3323613B1 (en) | Dip-molded article and protective glove | |
| CN112739761B (zh) | 聚合物胶乳及层叠体 | |
| JP2020050810A (ja) | 重合体ラテックスおよび積層体 | |
| JP7163919B2 (ja) | 積層体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17855823 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018542424 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20197012461 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017855823 Country of ref document: EP Effective date: 20190430 |