WO2017209178A1 - パターン製造方法、半導体装置の製造方法および積層体 - Google Patents
パターン製造方法、半導体装置の製造方法および積層体 Download PDFInfo
- Publication number
- WO2017209178A1 WO2017209178A1 PCT/JP2017/020251 JP2017020251W WO2017209178A1 WO 2017209178 A1 WO2017209178 A1 WO 2017209178A1 JP 2017020251 W JP2017020251 W JP 2017020251W WO 2017209178 A1 WO2017209178 A1 WO 2017209178A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- transparent substrate
- pattern
- photosensitive resin
- acid
- Prior art date
Links
Images



Classifications
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/038—Macromolecular compounds which are rendered insoluble or differentially wettable
- G03F7/0387—Polyamides or polyimides
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/16—Coating processes; Apparatus therefor
- G03F7/168—Finishing the coated layer, e.g. drying, baking, soaking
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
- G03F7/2002—Exposure; Apparatus therefor with visible light or UV light, through an original having an opaque pattern on a transparent support, e.g. film printing, projection printing; by reflection of visible or UV light from an original such as a printed image
- G03F7/2012—Exposure; Apparatus therefor with visible light or UV light, through an original having an opaque pattern on a transparent support, e.g. film printing, projection printing; by reflection of visible or UV light from an original such as a printed image using liquid photohardening compositions, e.g. for the production of reliefs such as flexographic plates or stamps
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
- G03F7/40—Treatment after imagewise removal, e.g. baking
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L21/00—Processes or apparatus adapted for the manufacture or treatment of semiconductor or solid state devices or of parts thereof
- H01L21/02—Manufacture or treatment of semiconductor devices or of parts thereof
- H01L21/027—Making masks on semiconductor bodies for further photolithographic processing not provided for in group H01L21/18 or H01L21/34
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L23/00—Details of semiconductor or other solid state devices
- H01L23/12—Mountings, e.g. non-detachable insulating substrates
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K3/00—Apparatus or processes for manufacturing printed circuits
- H05K3/10—Apparatus or processes for manufacturing printed circuits in which conductive material is applied to the insulating support in such a manner as to form the desired conductive pattern
Definitions
- the present invention relates to a pattern manufacturing method, a semiconductor device manufacturing method, and a laminate.
- Fan-out wafer level packages are attracting attention due to the recent demand for higher integration and lower height in semiconductor devices.
- FOWLP generally, an insulating layer and a redistribution layer (RDL) are alternately stacked on a semiconductor chip.
- Photosensitive polyimide is often used for the insulating layer material
- copper is often used for the rewiring layer material.
- the number of rewiring layers tends to increase, and there are cases in which a multilayer of four layers of polyimide and three layers of rewiring layers are formed between semiconductor chips and bumps.
- FOWLP a method in which semiconductor chips are arranged on a wafer-like support (pseudo wafer) is prepared, a rewiring layer is formed after molding, and the pseudo wafer is cut (chip first).
- RDL first two methods in which a rewiring layer is first formed on the support, a semiconductor chip is arranged on the support layer, and cutting is performed after molding.
- the manufacturing yield decreases, but in the chip first method, when a defect in the redistribution layer occurs, the semiconductor chip is wasted, whereas in the RDL first method, the wiring layer is connected before the chip connection. The semiconductor chip is not wasted because it is possible to inspect and connect the chip only to a non-defective part. For this reason, the expectation for the RDL first method is increasing as a method for reducing the manufacturing cost.
- Patent Document 1 states that “a release layer mainly made of a thermoplastic polyimide resin is provided on a glass plate. A first insulating layer is formed on the release layer. By sputtering. Then, a conductor layer made of TiN, Ti and Cu is formed on the first insulating layer 50. Laser light of 308 nm is transmitted through the glass plate and irradiated to the release layer, and the release layer is softened. It is described. Further, Patent Document 2 discloses that “a method for forming an electrical structure, in which a first device wafer including a first electrical component is prepared, and the first device wafer is a first device wafer first.
- the first wafer is bonded to the first surface of the first device wafer by using a first bonding material, and the carrier wafer is bonded to the first device wafer.
- the second surface of the first device wafer is processed while being bonded, and the light from the laser passes through the carrier wafer and strikes the first bonding material.
- the first bonding material substantially releases the carrier wafer from the first device wafer by absorbing energy from the beam, the carrier wafer being substantially free of the light from the laser.
- a method comprising: removing the carrier wafer from the first device wafer and removing the remaining first bonding material from the first device wafer. .
- Patent Documents 1 and 2 were examined, and both are systems in which radiated light is irradiated from the transparent substrate side and the film is peeled off at the interface between the peeling layer and the transparent substrate. Therefore, a release layer is required in addition to the insulating layer.
- An object of the present invention is to solve such a problem, and a transparent substrate is directly formed from a laminate having a transparent substrate and a resin pattern (for example, an insulating layer) positioned on the surface of the transparent substrate. It is an object of the present invention to provide a pattern manufacturing method that can be peeled off, a semiconductor device manufacturing method, and a laminate.
- the above problem can be solved by adjusting the absorbance of the resin pattern at the wavelength to 0.5 or more and using a resin selected from polyimide and polybenzoxazole as the resin constituting the resin pattern.
- the above problem has been solved by the following means ⁇ 1>, preferably ⁇ 2> to ⁇ 13>.
- ⁇ 1> A laminate A having a transparent substrate and a resin pattern located on the surface of the transparent substrate is irradiated with radiation from the transparent substrate side of the laminate A, and the transparent substrate is peeled off from the laminate A.
- a pattern manufacturing method comprising a peeling step, wherein the resin pattern contains at least one selected from polyimide and polybenzoxazole, and the absorbance of the resin pattern at the wavelength of the emitted radiation is 0.5 or more.
- the photosensitive resin film of the laminate B having a transparent substrate and a photosensitive resin film is exposed to light, and further developed to remove the photosensitive resin film to remove the laminate A.
- the pattern manufacturing method as described in ⁇ 1> including obtaining.
- the surface area of the removed portion of the resin pattern formed by removing a part of the photosensitive resin film is 20% or less of the surface area of the entire exposed region of the photosensitive resin film. 2>.
- the laminate A includes a step of applying metal between the transparent substrate and the resin pattern on the transparent substrate and on the gap between the resin patterns or between the transparent substrate and the resin pattern. 4> The pattern manufacturing method in any one of.
- ⁇ 7> The pattern production according to any one of ⁇ 2> to ⁇ 6>, wherein the photosensitive resin film includes at least one selected from a polyimide precursor, a polyimide, a polybenzoxazole precursor, and a polybenzoxazole.
- Method. ⁇ 8> The pattern manufacturing method according to any one of ⁇ 2> to ⁇ 6>, wherein the photosensitive resin film includes at least one selected from a polyimide precursor and a polybenzoxazole precursor.
- ⁇ 9> The pattern manufacturing method according to any one of ⁇ 1> to ⁇ 8>, including a curing step of curing the resin pattern after the exposure and development step and before the peeling step.
- ⁇ 10> The pattern production method according to any one of ⁇ 2> to ⁇ 9>, wherein the development is negative development.
- ⁇ 11> A method for manufacturing a semiconductor device, comprising the pattern manufacturing method according to any one of ⁇ 1> to ⁇ 10>.
- ⁇ 12> A transparent substrate, a resin pattern located on the surface of the transparent substrate, and a metal located on the surface of the transparent substrate and located in a gap between the resin patterns, wherein the resin pattern is polyimide and polybenzo A laminate comprising at least one selected from oxazole.
- a transparent substrate a resin pattern located on the surface of the transparent substrate, a metal located on the surface of the transparent substrate and located in a gap between the resin patterns, and a semiconductor element in contact with the metal
- the laminated body in which the semiconductor element is in contact with the metal on the side opposite to the transparent substrate side, and the resin pattern includes at least one selected from polyimide and polybenzoxazole.
- a pattern manufacturing method capable of directly peeling a transparent substrate from a transparent substrate and a laminate having a resin pattern located on the surface of the transparent substrate, and a semiconductor device manufacturing method and a laminate. Now available.
- the description which does not describe substitution and unsubstituted includes the thing which has a substituent with the thing which does not have a substituent.
- the “alkyl group” includes not only an alkyl group having no substituent (unsubstituted alkyl group) but also an alkyl group having a substituent (substituted alkyl group).
- “exposure” includes not only exposure using light but also drawing using particle beams such as electron beams and ion beams.
- the light used for the exposure generally includes an active ray or radiation such as an emission line spectrum of a mercury lamp, far ultraviolet rays typified by an excimer laser, extreme ultraviolet rays (EUV light), X-rays or electron beams.
- an active ray or radiation such as an emission line spectrum of a mercury lamp, far ultraviolet rays typified by an excimer laser, extreme ultraviolet rays (EUV light), X-rays or electron beams.
- EUV light extreme ultraviolet rays
- a numerical range expressed using “to” means a range including numerical values described before and after “to” as a lower limit value and an upper limit value.
- (meth) acrylate represents both and / or “acrylate” and “methacrylate”
- (meth) allyl means both “allyl” and “methallyl”
- (Meth) acryl” represents either “acryl” and “methacryl” or any one
- “(meth) acryloyl” represents both “acryloyl” and “methacryloyl”, or Represents either.
- the term “process” is not limited to an independent process, and is included in the term if the intended action of the process is achieved even when it cannot be clearly distinguished from other processes. .
- solid content concentration is the mass percentage of the other component except a solvent with respect to the gross mass of a composition.
- a weight average molecular weight (Mw) and a number average molecular weight (Mn) are defined as polystyrene conversion values according to gel permeation chromatography (GPC measurement) unless otherwise specified.
- the weight average molecular weight (Mw) and the number average molecular weight (Mn) are, for example, HLC-8220 (manufactured by Tosoh Corporation), and guard columns HZ-L, TSKgel Super HZM-M, TSKgel.
- a transparent substrate and a laminate A having a resin pattern located on the surface of the transparent substrate (hereinafter sometimes simply referred to as “a laminate having a transparent substrate and a resin pattern”) Including a peeling step of radiating radiation from the transparent substrate side of the laminate A to peel the transparent substrate from the laminate A, wherein the resin pattern includes at least one selected from polyimide and polybenzoxazole
- the resin pattern includes at least one selected from polyimide and polybenzoxazole
- the absorbance of the resin pattern at the wavelength of the emitted radiation is 0.5 or more. The reason for this is that by using at least one resin selected from polyimide and polybenzoxazole as the resin pattern, durability against emitted light can be increased.
- the resin pattern near the transparent substrate of the resin pattern is locally heated due to the absorption of the emitted light. It is possible to ablate at the substrate interface for the time being while avoiding film quality deterioration due to overall heat generation, and it becomes possible to peel the transparent substrate directly from the laminate having the transparent substrate and the resin pattern, the resin pattern and the transparent substrate No peeling layer or the like is required between the two.
- the laminate A having a transparent substrate and a resin pattern is exposed to light having a wavelength of 500 nm or less with respect to the laminate B having a transparent substrate and a photosensitive resin film, and further developed. It is preferably used in an embodiment obtained by performing the above. That is, a laminate B having a transparent substrate and a photosensitive resin film (preferably, a laminate having a transparent substrate and a photosensitive resin film located on the surface of the transparent substrate) is exposed and developed, and the photosensitive resin film is exposed to light. Even when the resin pattern is formed by losing the property, the transparent substrate and the resin pattern are provided without damaging the resin pattern by irradiating the radiated light so as to satisfy the predetermined condition.
- the transparent substrate can be appropriately peeled from the laminate A.
- FIG. 1 is a schematic view showing a first embodiment, in which a photosensitive resin film 2 is provided on a transparent substrate 1 (photosensitive resin film manufacturing process), and exposure and development are performed to manufacture a resin pattern 3 (exposure). Development process). Thereafter, the transparent substrate 1 can be peeled off by irradiating with radiation light so as to satisfy a predetermined condition (transparent substrate peeling step). Further, after the exposure and development step, the metal 4 is applied on the transparent substrate 1 (preferably on the surface) and between the resin pattern 3 or between the transparent substrate 1 and the resin pattern 3. (Metal application process). Furthermore, the semiconductor element 5 can be applied so as to be in contact with the metal 4 (semiconductor element application process).
- a photosensitive resin film manufacturing process an exposure development process, a metal application process, a semiconductor element application process, and a transparent substrate peeling process in this order.
- a semiconductor having a semiconductor element for example, a semiconductor chip
- a metal for example, a metal wiring or an electrode
- resin patterns for example, an insulating layer
- the photosensitive resin film 2 is obtained by applying the photosensitive resin composition in layers on the transparent substrate 1, preferably on the surface. Furthermore, when the photosensitive resin composition contains a solvent, it is preferable to dry the photosensitive resin composition after applying the photosensitive resin composition in layers.
- the thickness of the photosensitive resin film is preferably 0.1 to 100 ⁇ m, more preferably 1 to 50 ⁇ m, and further preferably 3 to 20 ⁇ m.
- the type of the transparent substrate is not particularly defined as long as it is a substrate that transmits radiated light, and examples thereof include glass, quartz, liquid crystal polymer, and the like, and glass is preferable.
- the transparent substrate preferably has high heat resistance, Tg is preferably 150 ° C. or higher, and more preferably 250 ° C. or higher.
- the thickness of the transparent substrate is not particularly defined, but is usually 10 to 1000 ⁇ m.
- photosensitive resin composition As a means for applying the photosensitive resin composition in a layer form to the transparent substrate, coating is preferable. Specifically, as a means to apply, dip coating method, air knife coating method, curtain coating method, wire bar coating method, gravure coating method, extrusion coating method, spray coating method, spin coating method, slit coating method, And an inkjet method. From the viewpoint of uniformity of the thickness of the layered photosensitive resin composition (hereinafter sometimes referred to as “photosensitive resin composition layer”), the spin coating method, the slit coating method, the spray coating method, and the inkjet method are more preferable. . A desired photosensitive resin composition layer can be obtained by adjusting an appropriate solid content concentration and coating conditions according to the means to be applied.
- the coating method can be appropriately selected depending on the shape of the substrate, and a spin coat method, a spray coat method, an ink jet method or the like is preferable for a circular substrate such as a wafer, and a slit coat method, a spray coat method, an ink jet method or the like for a rectangular substrate.
- the method is preferred.
- the spin coating method for example, it can be applied at a rotational speed of 500 to 2000 rpm (revolutions per minute) for about 10 seconds to 1 minute.
- the filter pore diameter is preferably 1 ⁇ m or less, more preferably 0.5 ⁇ m or less, and even more preferably 0.1 ⁇ m or less.
- the photosensitive resin composition constituting the photosensitive resin film in this embodiment preferably contains a compound that forms a cross-linked structure upon exposure, and the composition is more preferably for negative development, and is developed with an organic solvent. More preferably, it is a negative photosensitive resin.
- the photosensitive resin composition may contain components other than these, and these components are not essential.
- the photosensitive resin composition used in the present embodiment includes a resin.
- the resin is not particularly limited, and a known resin can be used.
- the resin is preferably a high heat resistant resin.
- the photosensitive resin composition used in this embodiment preferably contains at least one resin selected from a polyimide precursor, a polyimide, a polybenzoxazole precursor, and a polybenzoxazole.
- the photosensitive resin composition includes a polyimide precursor or a polybenzoxazole precursor, and it is further preferable that the photosensitive resin composition includes a polyimide precursor.
- the photosensitive resin composition used in the present embodiment may contain only one or two or more of polyimide precursor, polyimide, polybenzoxazole precursor and polybenzoxazole.
- two or more types of resins having the same type, such as two types of polyimide precursors, and having different structures may be included.
- the resin content in the photosensitive resin composition used in the present embodiment is preferably 10 to 99% by mass, more preferably 50 to 98% by mass, and more preferably 70 to 96% by mass based on the total solid content of the photosensitive resin composition. Is more preferable.
- the resin preferably contains a polymerizable group.
- the photosensitive resin composition contains a polymerizable compound.
- a three-dimensional network is formed in the exposed area, forming a strong cross-linked film, and the photosensitive resin composition layer (cured film) is not damaged by the surface activation treatment described later, and the surface activity By the chemical treatment, the adhesion between the cured film and the metal layer or the adhesion between the cured films is more effectively improved.
- the resin includes a partial structure represented by —Ar—L—Ar—.
- Ar is each independently an aromatic group
- L is an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —CO—, —S—. , —SO 2 — or —NHCO—, and a group consisting of a combination of two or more of the above.
- Ar is preferably a phenylene group
- L is an aliphatic hydrocarbon group having 1 or 2 carbon atoms which may be substituted with a fluorine atom, —O—, —CO—, —S— or —SO 2 —.
- the aliphatic hydrocarbon group here is preferably an alkylene group.
- the polyimide precursor is not particularly defined with respect to its type and the like, and preferably includes a repeating unit represented by the following formula (2).
- a 1 and A 2 each independently represent an oxygen atom or NH
- R 111 represents a divalent organic group
- R 115 represents a tetravalent organic group
- R 113 and R 114 each independently represents a hydrogen atom or a monovalent organic group.
- a 1 and A 2 in Formula (2) are preferably an oxygen atom or NH, and more preferably an oxygen atom.
- R 111 in the formula (2) represents a divalent organic group.
- the divalent organic group include a straight chain or branched aliphatic group, a group containing a cyclic aliphatic group and an aromatic group, a straight chain or branched aliphatic group having 2 to 20 carbon atoms, a carbon number A group consisting of a cyclic aliphatic group having 6 to 20 carbon atoms, an aromatic group having 6 to 20 carbon atoms, or a combination thereof is preferable, and a group containing an aromatic group having 6 to 20 carbon atoms is more preferable.
- R 111 in the formula (2) is exemplified by a group represented by —Ar—L—Ar—.
- Ar is each independently an aromatic group
- L is an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —CO—, —S—. , —SO 2 — or —NHCO—, and a group consisting of a combination of two or more of the above.
- R 111 in formula (2) is preferably derived from a diamine.
- the diamine used in the production of the polyimide precursor include linear or branched aliphatic, cyclic aliphatic or aromatic diamine.
- One type of diamine may be used, or two or more types may be used.
- a diamine containing an aromatic group having 2 to 60 carbon atoms is more preferable.
- the following aromatic groups are mentioned as an example of an aromatic group.
- A represents a single bond or an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —C ( ⁇ O) —, —S—. , —S ( ⁇ O) 2 —, —NHCO— and a group selected from a combination thereof, a single bond, an alkylene group having 1 to 3 carbon atoms which may be substituted with a fluorine atom, It is more preferably a group selected from —O—, —C ( ⁇ O) —, —S—, —SO 2 —, —CH 2 —, —O—, —S—, —SO 2 —, More preferably, it is a divalent group selected from the group consisting of —C (CF 3 ) 2 — and —C (CH 3 ) 2 —.
- diamine examples include 1,2-diaminoethane, 1,2-diaminopropane, 1,3-diaminopropane, 1,4-diaminobutane and 1,6-diaminohexane; 1,2- or 1 , 3-diaminocyclopentane, 1,2-, 1,3- or 1,4-diaminocyclohexane, 1,2-, 1,3- or 1,4-bis (aminomethyl) cyclohexane, bis- (4- Aminocyclohexyl) methane, bis- (3-aminocyclohexyl) methane, 4,4'-diamino-3,3'-dimethylcyclohexylmethane and isophoronediamine; meta and paraphenylenediamine, diaminotoluene, 4,4'- and 3 , 3'-diaminobiphenyl, 4,4'-diaminodiphenyl ether
- diamines (DA-1) to (DA-18) shown below are also preferable.
- a diamine having at least two alkylene glycol units in the main chain is also a preferred example.
- Preferred is a diamine containing two or more ethylene glycol chains or propylene glycol chains in one molecule, and more preferred is a diamine containing no aromatic ring.
- Specific examples include Jeffermin (registered trademark) KH-511, Jeffermin (registered trademark) ED-600, Jeffermin (registered trademark) ED-900, Jeffermin (registered trademark) ED-2003, Jeffermin (registered trademark).
- EDR-148 Jeffamine (registered trademark) EDR-176, D-200, D-400, D-2000, D-4000 (above trade names, manufactured by HUNTSMAN), 1- (2- (2- (2- (2- Aminopropoxy) ethoxy) propoxy) propan-2-amine, 1- (1- (1- (2-aminopropoxy) propan-2-yl) oxy) propan-2-amine, and the like, but is not limited thereto. .
- x, y, and z are average values.
- R 111 in the formula (2) is preferably represented by —Ar—L—Ar— from the viewpoint of the flexibility of the resulting cured film.
- Ar is each independently an aromatic group
- L is an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —CO—, —S—. , —SO 2 — or —NHCO—, and a group consisting of a combination of two or more of the above.
- Ar is preferably a phenylene group
- L is an aliphatic hydrocarbon group having 1 or 2 carbon atoms which may be substituted with a fluorine atom, —O—, —CO—, —S— or —SO 2 —.
- the aliphatic hydrocarbon group here is preferably an alkylene group.
- R 111 in formula (2) is preferably a divalent organic group represented by the following formula (51) or formula (61) from the viewpoint of i-line transmittance.
- a divalent organic group represented by the formula (61) is more preferable from the viewpoint of i-line transmittance and availability.
- Formula (51) In the formula (51), R 10 to R 17 are each independently a hydrogen atom, a fluorine atom or a monovalent organic group, and at least one of R 10 to R 17 is a fluorine atom, a methyl group or a trifluoromethyl group. It is.
- Examples of the monovalent organic group represented by R 10 to R 17 include an unsubstituted alkyl group having 1 to 10 carbon atoms (preferably 1 to 6 carbon atoms) and a fluorine atom having 1 to 10 carbon atoms (preferably 1 to 6 carbon atoms). Alkyl group and the like.
- Formula (61) In formula (61), R 18 and R 19 are each independently a fluorine atom or a trifluoromethyl group.
- Diamine compounds that give the structure of formula (51) or (61) include 2,2′-dimethylbenzidine, 2,2′-bis (trifluoromethyl) -4,4′-diaminobiphenyl, 2,2′- Bis (fluoro) -4,4′-diaminobiphenyl, 4,4′-diaminooctafluorobiphenyl and the like can be mentioned. These may be used alone or in combination of two or more.
- R 115 in the formula (2) represents a tetravalent organic group.
- a tetravalent organic group containing an aromatic ring is preferable, and a group represented by the following formula (5) or formula (6) is more preferable.
- R 112 represents a single bond or an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —CO—, —S—, —SO.
- the group is selected from —S— and —SO 2 —, and —CH 2 —, —C (CF 3 ) 2 —, —C (CH 3 ) 2 —, —O—, —CO More preferred is a divalent group selected from the group consisting of —, —S— and —SO 2 —.
- R 115 in formula (2) include a tetracarboxylic acid residue remaining after removal of the anhydride group from tetracarboxylic dianhydride. Only one tetracarboxylic dianhydride may be used, or two or more tetracarboxylic dianhydrides may be used.
- the tetracarboxylic dianhydride is preferably represented by the following formula (O).
- a preferred range of R 115 has the same meaning as R 115 in formula (2), and preferred ranges are also the same.
- tetracarboxylic dianhydride examples include pyromellitic dianhydride, 3,3 ′, 4,4′-biphenyltetracarboxylic dianhydride, 3,3 ′, 4,4′-diphenyl sulfide tetra Carboxylic dianhydride, 3,3 ′, 4,4′-diphenylsulfone tetracarboxylic dianhydride, 3,3 ′, 4,4′-benzophenone tetracarboxylic dianhydride, 3,3 ′, 4 4'-diphenylmethanetetracarboxylic dianhydride, 2,2 ', 3,3'-diphenylmethanetetracarboxylic dianhydride, 2,3,3', 4'-biphenyltetracarboxylic dianhydride, 2,3 , 3 ′, 4′-benzophenonetetracarboxylic dianhydride, 4,4′-oxydi
- tetracarboxylic dianhydrides (DAA-1) to (DAA-5) shown below are also preferable examples.
- R 111 and R 115 in formula (2) has a hydroxyl group. More specifically, examples of R 111 include a residue of a bisaminophenol derivative.
- R 113 and R 114 in formula (2) each independently represent a hydrogen atom or a monovalent organic group.
- at least one of R 113 and R 114 preferably contains a polymerizable group, and more preferably both contain a polymerizable group.
- a polymerizable group is a group that can undergo a crosslinking reaction by the action of heat, radicals, and the like.
- a radical photopolymerizable group is preferable.
- the polymerizable group examples include a group having an ethylenically unsaturated bond, an alkoxymethyl group, a hydroxymethyl group, an acyloxymethyl group, an epoxy group, an oxetanyl group, a benzoxazolyl group, a blocked isocyanate group, a methylol group, and an amino group.
- a radically polymerizable group which a polyimide precursor has group which has an ethylenically unsaturated bond is preferable.
- the group having an ethylenically unsaturated bond include a vinyl group, a (meth) allyl group, a group represented by the following formula (III), and the like.
- R 200 represents a hydrogen atom or a methyl group, and a methyl group is more preferable.
- R 201 represents an alkylene group having 2 to 12 carbon atoms, —CH 2 CH (OH) CH 2 — or a polyoxyalkylene group having 4 to 30 carbon atoms.
- suitable R 201 are ethylene group, propylene group, trimethylene group, tetramethylene group, 1,2-butanediyl group, 1,3-butanediyl group, pentamethylene group, hexamethylene group, octamethylene group, dodecamethylene group.
- R 200 is a methyl group and R 201 is an ethylene group.
- R 113 or R 114 in the formula (2) may be a monovalent organic group other than the polymerizable group.
- R 113 or R 114 in Formula (2) is preferably a monovalent organic group.
- the monovalent organic group preferably contains a linear or branched alkyl group, a cyclic alkyl group, or an aromatic group.
- Examples of the monovalent organic group include an aromatic group having 1, 2 or 3 (preferably 1) acidic group bonded to carbon constituting the aryl group, and a carbon constituting the aryl group.
- Aralkyl groups having 1, 2 or 3 (preferably 1) acidic groups are particularly preferred.
- Specific examples include an aromatic group having 6 to 20 carbon atoms having an acidic group and an aralkyl group having 7 to 25 carbon atoms having an acidic group. More specifically, a phenyl group having an acidic group and a benzyl group having an acidic group can be mentioned.
- the acidic group is preferably a hydroxyl group. It is more particularly preferred that R 113 or R 114 is a hydrogen atom, 2-hydroxybenzyl, 3-hydroxybenzyl and 4-hydroxybenzyl.
- the number of carbon atoms of the alkyl group represented by R 113 or R 114 in Formula (2) is preferably 1-30.
- Examples of the linear or branched alkyl group include a methyl group, an ethyl group, a propyl group, a butyl group, a pentyl group, a hexyl group, a heptyl group, an octyl group, a nonyl group, a decyl group, a dodecyl group, a tetradecyl group, and an octadecyl group.
- the cyclic alkyl group represented by R 113 or R 114 in Formula (2) may be a monocyclic cyclic alkyl group or a polycyclic cyclic alkyl group.
- Examples of the monocyclic alkyl group include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, and a cyclooctyl group.
- Examples of the polycyclic alkyl group include an adamantyl group, a norbornyl group, a bornyl group, a camphenyl group, a decahydronaphthyl group, a tricyclodecanyl group, a tetracyclodecanyl group, a camphoroyl group, a dicyclohexyl group, and a pinenyl group. Is mentioned. Among these, a cyclohexyl group is most preferable from the viewpoint of achieving high sensitivity. Moreover, as an alkyl group substituted by the aromatic group, the linear alkyl group substituted by the aromatic group mentioned later is preferable.
- aromatic group represented by R 113 or R 114 in the formula (2) include a substituted or unsubstituted benzene ring, naphthalene ring, pentalene ring, indene ring, azulene ring, heptalene ring, indacene ring, perylene.
- the polyimide precursor forms a counter salt with a tertiary amine compound having an ethylenically unsaturated bond. You may do it.
- tertiary amine compounds having an ethylenically unsaturated bond include N, N-dimethylaminopropyl methacrylate.
- the polyimide precursor preferably has a fluorine atom in the structural unit.
- the fluorine atom content in the polyimide precursor is preferably 10% by mass or more, and more preferably 20% by mass or less.
- an aliphatic group having a siloxane structure may be copolymerized with the polyimide precursor.
- the diamine component for introducing an aliphatic group having a siloxane structure include bis (3-aminopropyl) tetramethyldisiloxane and bis (paraaminophenyl) octamethylpentasiloxane.
- the repeating unit represented by the formula (2) is preferably a repeating unit represented by the following formula (2-A). That is, at least one of the polyimide precursors is preferably a precursor having a repeating unit represented by the formula (2-A). By adopting such a structure, it becomes possible to further widen the width of the exposure latitude.
- a 1 , A 2 , R 111 , R 113 and R 114 in formula (2-A) are each independently synonymous with A 1 , A 2 , R 111 , R 113 and R 114 in formula (2).
- the preferable range is also the same.
- R 112 in formula (2-A) has the same meaning as R 112 in formula (5), and the preferred range is also the same.
- polyimide precursor one type of repeating unit represented by the formula (2) may be used, or two or more types may be used.
- the polyimide precursor may contain the structural isomer of the repeating unit represented by Formula (2).
- the polyimide precursor may also contain other types of repeating units in addition to the repeating unit represented by the above formula (2).
- a polyimide precursor in which 50 mol% or more, further 70 mol% or more, particularly 90 mol% or more of all repeating units is a repeating unit represented by the formula (2).
- the polyimide precursor is preferably obtained by reacting dicarboxylic acid or a dicarboxylic acid derivative with diamine.
- the polyimide precursor is more preferably obtained by halogenating a dicarboxylic acid or a dicarboxylic acid derivative with a halogenating agent and then reacting with a diamine.
- the polyimide precursor is, for example, a method of reacting a tetracarboxylic dianhydride and a diamine compound (partially replaced with a monoamine end-capping agent) at a low temperature, or a tetracarboxylic dianhydride (partly at a low temperature).
- a diester is obtained by tetracarboxylic dianhydride and an alcohol, and then a diamine (partially In the presence of a condensing agent and a tetracarboxylic dianhydride and an alcohol to obtain a diester, and then the remaining dicarboxylic acid is converted to an acid chloride to give a diamine (partially)
- a method such as a method of reacting with a terminal blocking agent that is a monoamine).
- an organic solvent is preferably used for the reaction.
- organic solvents may be used.
- the organic solvent can be appropriately determined according to the raw material, and examples thereof include pyridine, diethylene glycol dimethyl ether (diglyme), N-methylpyrrolidone and N-ethylpyrrolidone.
- the main chain terminal of the precursor is sealed with an end-capping agent such as an acid anhydride, monocarboxylic acid, monoacid chloride compound, or monoactive ester compound. It is preferable. Of these, it is more preferable to use a monoamine.
- the monoamine include aniline, 2-ethynylaniline, 3-ethynylaniline, 4-ethynylaniline, 5-amino-8-hydroxyquinoline, and 1-hydroxy-7.
- -Aminonaphthalene 1-hydroxy-6-aminonaphthalene, 1-hydroxy-5-aminonaphthalene, 1-hydroxy-4-aminonaphthalene, 2-hydroxy-7-aminonaphthalene, 2-hydroxy-6-aminonaphthalene, 2, -Hydroxy-5-aminonaphthalene, 1-carboxy-7-aminonaphthalene, 1-carboxy-6-aminonaphthalene, 1-carboxy-5-aminonaphthalene, 2-carboxy-7-aminonaphthalene, 2-carboxy-6- Aminonaphthalene, 2-carbo Ci-5-aminonaphthalene, 2-aminobenzoic acid, 3-aminobenzoic acid, 4-aminobenzoic acid, 4-aminosalicylic acid, 5-aminosalicylic acid, 6-aminosalicylic acid, 2-aminobenzenesulfonic acid, 3-amino Benzenesulfonic acid, 4-amino
- a step of depositing a solid may be included. Specifically, solid precipitation can be achieved by precipitating the polyimide precursor in the reaction solution in water and dissolving it in a solvent in which the polyimide precursor such as tetrahydrofuran is soluble. Then, a polyimide precursor can be dried and a powdery polyimide precursor can be obtained.
- the weight average molecular weight (Mw) of the polyimide precursor is preferably 18000 to 30000, more preferably 20000 to 27000, and further preferably 22000 to 25000.
- the number average molecular weight (Mn) is preferably 7200 to 14000, more preferably 8000 to 12000, and still more preferably 9200 to 11200.
- the dispersion degree of the polyimide precursor is preferably 2.5 or more, more preferably 2.7 or more, and further preferably 2.8 or more.
- the upper limit of the degree of dispersion of the polyimide precursor is not particularly defined, but is, for example, preferably 4.5 or less, more preferably 4.0 or less, still more preferably 3.8 or less, and still more preferably 3.2 or less, 3.1 or less is even more preferable, 3.0 or less is even more preferable, and 2.95 or less is even more preferable.
- the polyimide is not particularly limited as long as it is a polymer compound having an imide ring.
- the polyimide is preferably a compound represented by the following formula (4), more preferably a compound represented by the formula (4) and a compound having a polymerizable group.
- R 131 represents a divalent organic group
- R 132 represents a tetravalent organic group.
- the polyimide has a polymerizable group
- at least one of R 131 and R 132 may have a polymerizable group, or as shown in the following formula (4-1) or formula (4-2), You may have a polymeric group at the terminal.
- R 133 is a polymerizable group, and other groups are as defined in the formula (4).
- Formula (4-2) In formula (4-2), at least one of R 134 and R 135 is a polymerizable group, the other is an organic group, and the other groups are as defined in formula (4).
- the polymerizable group that the polyimide preferably has has the same meaning as the polymerizable group described in the polymerizable group that R 113 and R 114 in Formula (2) may contain in the polyimide precursor.
- R 131 in the formula (4) represents a divalent organic group.
- the divalent organic group include the same divalent organic groups as R 111 in formula (2), and the preferred range is also the same.
- R 131 include diamine residues remaining after removal of the amino groups of the diamine.
- the diamine include aliphatic, cycloaliphatic or aromatic diamines. Specific examples include R 111 in formula (2) of the polyimide precursor.
- R 131 in formula (4) is preferably a diamine residue having at least two alkylene glycol units in the main chain from the viewpoint of more effectively suppressing the occurrence of warpage during firing. More preferred is a diamine residue containing at least two ethylene glycol chains or propylene glycol chains in one molecule, and even more preferred is a diamine residue containing no aromatic ring.
- Examples of the diamine containing two or more of ethylene glycol chain and propylene glycol chain in one molecule include specific examples similar to the diamine capable of deriving R 111 in formula (2). It is not limited to these.
- R 132 in the formula (4) represents a tetravalent organic group.
- examples of the tetravalent organic group represented by R 132 include those similar to R 115 in formula (2), and the preferred ranges are also the same.
- four bonds of a tetravalent organic group having the following structure exemplified as R 115 in the formula (2) are bonded to four —C ( ⁇ O) — moieties in the formula (4).
- a condensed ring is formed.
- R 132 in the formula (4) examples include a tetracarboxylic acid residue remaining after the anhydride group is removed from the tetracarboxylic dianhydride.
- R 115 in the formula (2) of the polyimide precursor can be given.
- R 132 is preferably an aromatic diamine residue having 1 to 4 aromatic rings.
- R 131 and R 132 in the formula (4) has a hydroxyl group. More specifically, as R 131 in formula (4), 2,2-bis (3-hydroxy-4-aminophenyl) propane, 2,2-bis (3-hydroxy-4-aminophenyl) hexafluoropropane 2,2-bis (3-amino-4-hydroxyphenyl) propane, 2,2-bis- (3-amino-4-hydroxyphenyl) hexafluoropropane, the above (DA-1) to (DA-18) ) Is a preferred example.
- Preferred examples of R 132 in formula (4) include the above (DAA-1) to (DAA-5).
- the polyimide has a fluorine atom in the structural unit.
- the fluorine atom content in the polyimide is preferably 10% by mass or more, and preferably 20% by mass or less.
- an aliphatic group having a siloxane structure may be copolymerized with polyimide.
- the diamine component for introducing an aliphatic group having a siloxane structure include bis (3-aminopropyl) tetramethyldisiloxane and bis (paraaminophenyl) octamethylpentasiloxane.
- the main chain terminal of the polyimide is sealed with a terminal sealing agent such as monoamine, acid anhydride, monocarboxylic acid, monoacid chloride compound, monoactive ester compound, etc. It is preferable to do. Of these, it is more preferable to use a monoamine.
- a monoamine include aniline, 2-ethynylaniline, 3-ethynylaniline, 4-ethynylaniline, 5-amino-8-hydroxyquinoline, 1-hydroxy-7-aminonaphthalene and 1-hydroxy-6-aminonaphthalene.
- the polyimide preferably has an imidization ratio of 85% or more, more preferably 90% or more.
- the imidization ratio is 85% or more, film shrinkage due to ring closure that occurs when imidization is performed by heating is reduced, and the occurrence of warpage of the substrate can be suppressed.
- the polyimide may include two or more different types of repeating units of R 131 or R 132 .
- the polyimide may also contain other types of repeating units in addition to the repeating unit represented by the above formula (4).
- Polyimide may be produced by synthesizing a polyimide precursor and then cyclized by heating, or may be synthesized directly.
- a polyimide is a method of obtaining a polyimide precursor and completely imidizing it using a known imidization reaction method, or a method of stopping an imidation reaction in the middle and introducing a part of an imide structure, By blending a completely imidized polymer and its polyimide precursor, it can be synthesized utilizing a method of partially introducing an imide structure.
- Examples of commercially available polyimide products include Durimide (registered trademark) 284 (manufactured by Fujifilm) and Matrimide 5218 (manufactured by HUNTSMAN).
- the weight average molecular weight (Mw) of the polyimide is preferably 5,000 to 70,000, more preferably 8,000 to 50,000, and particularly preferably 10,000 to 30,000.
- the weight average molecular weight is more preferably 20,000 or more.
- the weight average molecular weight of at least 1 type of polyimide is the said range.
- the polybenzoxazole precursor is not particularly defined with respect to its type and the like, and preferably includes a repeating unit represented by the following formula (3).
- R 121 represents a divalent organic group
- R 122 represents a tetravalent organic group
- R 123 and R 124 each independently represents a hydrogen atom or a monovalent organic group.
- R 123 and R 124 in formula (3) have the same meaning as R 113 in formula (2), respectively, and the preferred ranges are also the same. That is, at least one of R 123 and R 124 in Formula (3) is preferably a polymerizable group.
- R 121 in the formula (3) represents a divalent organic group.
- the divalent organic group represented by R 121 is preferably a group containing at least one of an aliphatic group and an aromatic group.
- As the aliphatic group a linear aliphatic group is preferable.
- R 121 is preferably a dicarboxylic acid residue. Only one type of dicarboxylic acid residue may be used, or two or more types may be used.
- dicarboxylic acid a dicarboxylic acid containing an aliphatic group and a dicarboxylic acid containing an aromatic group are preferred, and a dicarboxylic acid containing an aromatic group is more preferred.
- dicarboxylic acid containing an aliphatic group a dicarboxylic acid containing a linear or branched (preferably linear) aliphatic group is preferable, and a linear or branched (preferably linear) aliphatic group and two COOHs are used. More preferred is a dicarboxylic acid.
- the linear or branched (preferably linear) aliphatic group preferably has 2 to 30 carbon atoms, more preferably 2 to 25 carbon atoms, still more preferably 3 to 20 carbon atoms. It is particularly preferably 15 and more preferably 5 to 10.
- the linear aliphatic group is preferably an alkylene group.
- dicarboxylic acid containing a linear aliphatic group examples include malonic acid, dimethylmalonic acid, ethylmalonic acid, isopropylmalonic acid, di-n-butylmalonic acid, succinic acid, tetrafluorosuccinic acid, methylsuccinic acid, 2, 2-dimethylsuccinic acid, 2,3-dimethylsuccinic acid, dimethylmethylsuccinic acid, glutaric acid, hexafluoroglutaric acid, 2-methylglutaric acid, 3-methylglutaric acid, 2,2-dimethylglutaric acid, 3,3-dimethylglutaric acid, 3-ethyl-3-methylglutaric acid, adipic acid, octafluoroadipic acid, 3-methyladipic acid, pimelic acid, 2,2,6,6-tetramethylpimelic acid, suberin Acid, dodecafluorosuberic acid, azelaic acid, sebacic acid, hexade
- Z is a hydrocarbon group having 1 to 6 carbon atoms, and n is an integer of 1 to 6).
- the dicarboxylic acid containing an aromatic group is preferably a dicarboxylic acid having the following aromatic group, more preferably a dicarboxylic acid comprising only the following aromatic group and two COOH.
- A represents —CH 2 —, —O—, —S—, —SO 2 —, —CO—, —NHCO—, —C (CF 3 ) 2 —, and —C (CH 3) 2 - represents a divalent radical selected from the group consisting of.
- dicarboxylic acid containing an aromatic group examples are preferably 4,4'-carbonyldibenzoic acid, 4,4'-dicarboxydiphenyl ether and terephthalic acid.
- R122 in Formula (3) represents a tetravalent organic group.
- examples of the tetravalent organic group represented by R 122 include those similar to R 115 in the above formula (2), and preferred ranges thereof are also the same.
- R 122 in formula (3) is also preferably a group derived from a bisaminophenol derivative. Examples of the group derived from a bisaminophenol derivative include 3,3′-diamino-4,4′-dihydroxybiphenyl, 4,4′-diamino-3,3′-dihydroxybiphenyl, and 3,3′-diamino-4.
- bisaminophenol derivatives having the following aromatic groups are preferred.
- X 1 represents —O—, —S—, —C (CF 3 ) 2 —, —CH 2 —, —SO 2 —, or —NHCO—.
- the bisaminophenol derivative is represented, for example, by the formula (As).
- R 1 represents a hydrogen atom, alkylene, substituted alkylene, —O—, —S—, —SO 2 —, —CO—, —NHCO—, a single bond, or the following formula (A— an organic group selected from the group of sc).
- R 2 is any one of a hydrogen atom, an alkyl group, an alkoxy group, an acyloxy group, and a cyclic alkyl group, which may be the same or different.
- R 3 is any one of a hydrogen atom, a linear or branched alkyl group, an alkoxy group, an acyloxy group, and a cyclic alkyl group, which may be the same or different.
- R 2 is an alkyl group and R 3 is an alkyl group, which means that it has high transparency to i-line and a high cyclization rate when cured at low temperature. The effect can be maintained, which is preferable.
- R 1 is more preferably alkylene or substituted alkylene.
- Specific examples of the alkylene and substituted alkylene represented by R 1 include —CH 2 —, —CH (CH 3 ) —, —C (CH 3 ) 2 —, —CH (CH 2 CH 3 ) —, —C (CH 3 ) (CH 2 CH 3 ) —, —C (CH 2 CH 3 ) (CH 2 CH 3 ) —, —CH (CH 2 CH 2 CH 3 ) —, —C (CH 3 ) (CH 2 CH 2 CH 3 ) —, —CH (CH (CH 3 ) 2 ) —, —C (CH 3 ) (CH (CH 3 ) 2 ) —, —CH (CH 2 CH 2 CH 2 CH 3 ) —, —C (CH 3 ) (CH (CH 3 ) 2 ) —, —CH (CH 2 CH 2 CH 2 CH 3 ) —, —C (CH 3 ) (CH
- the polybenzoxazole precursor may contain other types of repeating units in addition to the repeating unit represented by the above formula (3). It is preferable that the polybenzoxazole precursor contains a diamine residue represented by the following formula (SL) as another type of repeating unit in that generation of warpage of the substrate accompanying ring closure of the polybenzoxazole precursor can be suppressed. .
- SL diamine residue represented by the following formula
- Z has an a structure and a b structure
- R 1s is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms
- R 2s is a hydrocarbon group having 1 to 10 carbon atoms
- R 3s , R 4s , R 5s , R 6s are aromatic groups and the rest are hydrogen atoms or organic groups having 1 to 30 carbon atoms, which may be the same or different.
- the polymerization of the a structure and the b structure may be block polymerization or random polymerization.
- the mol% of the Z moiety is 5 to 95 mol% for the a structure, 95 to 5 mol% for the b structure, and the sum of the a and b structures is 100 mol%.
- Z include those in which R 5s and R 6s in the b structure are phenyl groups.
- the molecular weight of the diamine residue represented by the formula (SL) is preferably 400 to 4,000, more preferably 500 to 3,000.
- the diamine residue represented by the formula (SL) When the diamine residue represented by the formula (SL) is included as another type of repeating unit, it may include the remaining tetracarboxylic acid residue as a repeating unit after removing the anhydride group from the tetracarboxylic dianhydride. preferable. Examples of such tetracarboxylic acid residue, and examples of R 115 in formula (2).
- the weight average molecular weight (Mw) of the polybenzoxazole precursor is, for example, preferably 18000 to 30000, more preferably 20000 to 29000, and further preferably 22000 to 28000 when used in the composition described later.
- the number average molecular weight (Mn) is preferably 7200 to 14000, more preferably 8000 to 12000, and still more preferably 9200 to 11200.
- the degree of dispersion of the polybenzoxazole precursor is preferably 1.4 or more, more preferably 1.5 or more, and further preferably 1.6 or more.
- the upper limit value of the degree of dispersion of the polybenzoxazole precursor is not particularly defined, but is preferably 2.6 or less, more preferably 2.5 or less, further preferably 2.4 or less, and more preferably 2.3 or less. Preferably, 2.2 or less is even more preferable.
- the polybenzoxazole is not particularly limited as long as it is a polymer compound having a benzoxazole ring.
- the polybenzoxazole is preferably a compound represented by the following formula (X), more preferably a compound represented by the following formula (X) and having a polymerizable group.
- R 133 represents a divalent organic group
- R 134 represents a tetravalent organic group.
- at least one of R 133 and R 134 may have a polymerizable group, and as shown in the following formula (X-1) or (X-2), polybenzoxazole You may have a polymeric group at the terminal.
- Formula (X-1) In formula (X-1), at least one of R 135 and R 136 is a polymerizable group, the other is an organic group, and the other groups are as defined in formula (X).
- R 137 is a polymerizable group, the other is a substituent, and the other groups are as defined in the formula (X).
- the polymerizable group that polybenzoxazole preferably has has the same meaning as the polymerizable group described in the polymerizable group that the polyimide precursor has.
- R 133 represents a divalent organic group.
- the divalent organic group include an aliphatic group and an aromatic group.
- Specific examples include R 121 in the polybenzoxazole precursor (3). Preferred examples thereof are the same as those for R 121 .
- R 134 represents a tetravalent organic group.
- the tetravalent organic group include an example of R 122 in the formula (3) of the polybenzoxazole precursor. Preferred examples thereof are the same as those for R122 .
- four bonds of a tetravalent organic group exemplified as R 122 are bonded to a nitrogen atom and an oxygen atom in the above formula (X) to form a condensed ring.
- R 134 is the following organic group, the following structure is formed.
- Polybenzoxazole preferably has an oxazolation rate of 85% or more, more preferably 90% or more.
- the oxazolation rate is 85% or more, film shrinkage due to ring closure that occurs when oxazolation is performed by heating is reduced, and the occurrence of warpage can be more effectively suppressed.
- the polybenzoxazole includes the above formula (X) containing two or more different R 131 or R 132 groups. The repeating unit represented by these may be included. Polybenzoxazole may also contain other types of repeating units in addition to the repeating unit represented by the above formula (X).
- Polybenzoxazole may, for example, a bis-aminophenol derivative, a dicarboxylic acid and is reacted with a compound selected from such dicarboxylic acid dichloride and dicarboxylic acid derivatives of the dicarboxylic acids polybenzoxazole precursor including R 133, which Can be obtained by oxazolation using a known oxazolation reaction method.
- a compound selected from such dicarboxylic acid dichloride and dicarboxylic acid derivatives of the dicarboxylic acids polybenzoxazole precursor including R 133 which Can be obtained by oxazolation using a known oxazolation reaction method.
- dicarboxylic acid an active ester dicarboxylic acid derivative obtained by reacting 1-hydroxy-1,2,3-benzotriazole or the like in advance may be used in order to increase the reaction yield and the like.
- the weight average molecular weight (Mw) of polybenzoxazole is preferably 5,000 to 70,000, more preferably 8,000 to 50,000, and particularly preferably 10,000 to 30,000.
- the weight average molecular weight is more preferably 20,000 or more.
- the weight average molecular weight of at least 1 type of polybenzoxazole is the said range.
- the present embodiment can also be applied to photosensitive resins other than those described above.
- resins epoxy resins, phenol resins, and benzocyclobutene resins can be used.
- the resin has a polymerizable group or the photosensitive resin composition contains a polymerizable compound.
- the polymerizable compound is a compound having a polymerizable group, and a known compound that can be crosslinked by a radical, an acid, a base, or the like can be used.
- Examples of the polymerizable group include the polymerizable groups described in the polyimide precursor. Only 1 type of polymeric compound may be contained and 2 or more types may be contained.
- the polymerizable compound may be in any chemical form such as a monomer, a prepolymer, an oligomer or a mixture thereof, and a multimer thereof.
- the monomer type polymerizable compound (hereinafter also referred to as polymerizable monomer) is a compound different from the polymer compound.
- the polymerizable monomer is typically a low molecular compound, preferably a low molecular compound having a molecular weight of 2000 or less, more preferably a low molecular compound having a molecular weight of 1500 or less, and a low molecular compound having a molecular weight of 900 or less. More preferably it is.
- the molecular weight of the polymerizable monomer is usually 100 or more.
- the oligomer type polymerizable compound is typically a polymer having a relatively low molecular weight, and is preferably a polymer in which 10 to 100 polymerizable monomers are bonded.
- the weight average molecular weight of the oligomer type polymerizable compound is preferably 2000 to 20000, more preferably 2000 to 15000, and most preferably 2000 to 10,000.
- the number of functional groups of the polymerizable compound means the number of polymerizable groups in one molecule.
- the photosensitive resin composition preferably contains at least one bifunctional or higher polymerizable compound containing two or more polymerizable groups, and preferably contains at least one trifunctional or higher polymerizable compound. Is more preferable.
- the photosensitive resin composition preferably contains at least one trifunctional or higher functional polymerizable compound from the viewpoint that the heat resistance can be improved by forming a three-dimensional crosslinked structure. Also, a mixture of a bifunctional or lower polymerizable compound and a trifunctional or higher functional polymerizable compound may be used.
- a compound containing a group having an ethylenically unsaturated bond; a compound having a hydroxymethyl group, an alkoxymethyl group or an acyloxymethyl group; an epoxy compound; an oxetane compound; and a benzoxazine compound are preferable.
- the compound containing a group having an ethylenically unsaturated bond include unsaturated carboxylic acids (for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.), esters thereof, and amides.
- unsaturated carboxylic acids for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.
- esters thereof for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.
- esters thereof for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.
- esters thereof for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.
- esters thereof for
- a dehydration condensation reaction product with a functional carboxylic acid is also preferably used.
- an addition reaction product of an unsaturated carboxylic acid ester or amide having an electrophilic substituent such as an isocyanate group or an epoxy group with a monofunctional or polyfunctional alcohol, amine, or thiol, and a halogen group A substitution reaction product of an unsaturated carboxylic acid ester or amide having a detachable substituent such as a tosyloxy group and a monofunctional or polyfunctional alcohol, amine or thiol is also suitable.
- esters of polyhydric alcohol compounds and unsaturated carboxylic acids include acrylic acid esters such as ethylene glycol diacrylate, triethylene glycol diacrylate, 1,3-butanediol diacrylate, and tetramethylene glycol diacrylate.
- Methacrylic acid esters include tetramethylene glycol dimethacrylate, triethylene glycol dimethacrylate, neopentyl glycol dimethacrylate, trimethylolpropane trimethacrylate, trimethylolethane trimethacrylate, ethylene glycol dimethacrylate, 1,3-butanediol dimethacrylate, Hexanediol dimethacrylate, pentaerythritol dimethacrylate, pentaerythritol trimethacrylate, pentaerythritol tetramethacrylate, dipentaerythritol dimethacrylate, dipentaerythritol hexamethacrylate, sorbitol trimethacrylate, sorbitol tetramethacrylate, bis [para (3-methacryloxy-2 -Hydroxy group Epoxy) phenyl] dimethyl methane, bis - is
- Itaconic acid esters include ethylene glycol diitaconate, propylene glycol diitaconate, 1,3-butanediol diitaconate, 1,4-butanediol diitaconate, tetramethylene glycol diitaconate, pentaerythritol diitaconate And sorbitol tetritaconate.
- crotonic acid esters examples include ethylene glycol dicrotonate, tetramethylene glycol dicrotonate, pentaerythritol dicrotonate, and sorbitol tetradicrotonate.
- isocrotonic acid esters examples include ethylene glycol diisocrotonate, pentaerythritol diisocrotonate, and sorbitol tetraisocrotonate.
- maleic acid esters examples include ethylene glycol dimaleate, triethylene glycol dimaleate, pentaerythritol dimaleate, and sorbitol tetramaleate.
- esters examples include aliphatic alcohol esters described in JP-B-46-27926, JP-B-51-47334, JP-A-57-196231, and JP-A-59-5240. No. 1, JP-A-59-5241, JP-A-2-226149, those having an aromatic skeleton, those having an amino group described in JP-A-1-165613, etc. are preferably used. It is done.
- amide monomers of polyvalent amine compounds and unsaturated carboxylic acids include methylene bis-acrylamide, methylene bis-methacrylamide, 1,6-hexamethylene bis-acrylamide, 1,6-hexamethylene bis-methacrylic.
- examples include amide, diethylenetriamine trisacrylamide, xylylene bisacrylamide, and xylylene bismethacrylamide.
- Examples of other preferable amide monomers include those having a cyclohexylene structure described in JP-B No. 54-21726.
- urethane-based addition polymerizable monomers produced by using an addition reaction of isocyanate and hydroxyl group are also suitable. Specific examples thereof include, for example, one molecule described in JP-B-48-41708.
- CH 2 C (R 4) COOCH 2 CH (R 5) OH (However, R 4 and R 5 represent H or CH 3.
- urethane acrylates as described in JP-A-51-37193, JP-B-2-32293, JP-B-2-16765, JP-B-58-49860, JP-B-56- Urethane compounds having an ethylene oxide skeleton described in JP 17654, JP-B 62-39417, and JP-B 62-39418 are also suitable.
- the compound which has a boiling point of 100 degreeC or more under normal pressure is also preferable.
- monofunctional acrylates and methacrylates such as polyethylene glycol mono (meth) acrylate, polypropylene glycol mono (meth) acrylate, and phenoxyethyl (meth) acrylate; polyethylene glycol di (meth) acrylate, trimethylolethanetri (meta ) Acrylate, neopentyl glycol di (meth) acrylate, pentaerythritol tri (meth) acrylate, pentaerythritol tetra (meth) acrylate, dipentaerythritol penta (meth) acrylate, dipentaerythritol hexa (meth) acrylate, hexanediol (meth) ) Acrylate, trimethylo
- the compounds described in paragraph numbers 0254 to 0257 of JP-A-2008-292970 are also suitable.
- the polyfunctional (meth) acrylate etc. which are obtained by making the compound which has cyclic ether groups, such as glycidyl (meth) acrylate, and an ethylenically unsaturated group, react with polyfunctional carboxylic acid can also be mentioned.
- other preferable compounds containing a group having an ethylenically unsaturated bond have a fluorene ring described in JP2010-160418A, JP2010-129825A, Japanese Patent No. 4364216, and the like.
- a compound having two or more groups having an ethylenically unsaturated bond, or a cardo resin can also be used.
- Other examples include specific unsaturated compounds described in JP-B-46-43946, JP-B-1-40337, JP-B-1-40336, and JP-A-2-25493.
- vinyl phosphonic acid compounds In some cases, a structure containing a perfluoroalkyl group described in JP-A-61-22048 is preferably used.
- Journal of Japan Adhesion Association vol. 20, no. 7, pages 300 to 308 (1984), which are introduced as photopolymerizable monomers and oligomers can also be used.
- n is an integer from 0 to 14, and m is an integer from 0 to 8.
- a plurality of R and T present in the molecule may be the same or different.
- at least one of a plurality of R is —OC ( ⁇ O) CH ⁇ CH 2 or —OC
- a group represented by ( ⁇ O) C (CH 3 ) ⁇ CH 2 is represented.
- Specific examples of the compound containing a group having an ethylenically unsaturated bond represented by the above formulas (MO-1) to (MO-5) are described in paragraph numbers 0248 to 0251 of JP-A-2007-2699779. The compound that has been used can also be suitably used in this embodiment.
- Examples of the compound containing a group having an ethylenically unsaturated bond include dipentaerythritol triacrylate (as a commercially available product, KAYARAD D-330; manufactured by Nippon Kayaku Co., Ltd.), dipentaerythritol tetraacrylate (as a commercially available product, KAYARAD D).
- oligomer types can also be used.
- preferred examples include pentaerythritol derivatives and / or dipentaerythritol derivatives of the above formulas (MO-1) and (MO-2).
- Examples of commercially available polymerizable compounds include SR-494, a tetrafunctional acrylate having four ethyleneoxy chains, manufactured by Sartomer, and SR-209, a bifunctional methacrylate having four ethyleneoxy chains, DPCA-60, a 6-functional acrylate having 6 pentyleneoxy chains, TPA-330, a 3-functional acrylate having 3 isobutyleneoxy chains, urethane oligomer UAS-10, UAB-140 manufactured by Nippon Kayaku Co., Ltd.
- NK ester M-40G (Manufactured by Sanyo Kokusaku Pulp Co., Ltd.), NK ester M-40G, NK ester 4G, NK ester M-9300, NK ester A-9300, UA-7200 (manufactured by Shin-Nakamura Chemical), DPHA-40H (Nippon Kayaku Co., Ltd.) )), UA-306H, UA-306T, UA-306I, AH-600, T- 00 (manufactured by Kyoeisha Chemical Co., Ltd.), AI-600, Brenmer PME400 (manufactured by NOF Co., Ltd.), and the like.
- Examples of the compound containing a group having an ethylenically unsaturated bond are described in JP-B-48-41708, JP-A-51-37193, JP-B-2-32293, and JP-B-2-16765.
- Urethane acrylates such as those described above, and urethane compounds having an ethylene oxide skeleton described in JP-B-58-49860, JP-B-56-17654, JP-B-62-39417, and JP-B-62-39418 are also suitable.
- polymerizable compounds having an amino structure or a sulfide structure in the molecule described in JP-A-63-277653, JP-A-63-260909, and JP-A-1-105238 are described as polymerizable compounds. Monomers can also be used.
- the compound containing a group having an ethylenically unsaturated bond may be a polyfunctional monomer having an acid group such as a carboxyl group, a sulfonic acid group, or a phosphoric acid group.
- the polyfunctional monomer having an acid group is preferably an ester of an aliphatic polyhydroxy compound and an unsaturated carboxylic acid, and a non-aromatic carboxylic acid anhydride is reacted with an unreacted hydroxyl group of the aliphatic polyhydroxy compound. More preferred is a polyfunctional monomer having.
- the aliphatic polyhydroxy compound in which a non-aromatic carboxylic acid anhydride is reacted with an unreacted hydroxyl group of the aliphatic polyhydroxy compound to give an acid group, is pentaerythritol and / or diester. It is a pentaerythritol.
- examples of commercially available products include M-510 and M-520 as polybasic acid-modified acrylic oligomers manufactured by Toagosei Co., Ltd.
- the polyfunctional monomer having an acid group one kind may be used alone, or two or more kinds may be mixed and used.
- a preferable acid value of the polyfunctional monomer having an acid group is 0.1 to 40 mgKOH / g, and particularly preferably 5 to 30 mgKOH / g.
- the acid value of the polyfunctional monomer is in the above range, the production and handling properties are excellent, and further, the developability is excellent. Also, the polymerizability is good.
- the content of the compound containing a group having an ethylenically unsaturated bond is preferably 1 to 50% by mass with respect to the total solid content of the photosensitive resin composition from the viewpoint of good polymerizability and heat resistance.
- the lower limit is more preferably 5% by mass or more.
- the upper limit is more preferably 30% by mass or less.
- a compound containing a group having an ethylenically unsaturated bond may be used alone or in combination of two or more.
- the mass ratio of the resin to the compound containing a group having an ethylenically unsaturated bond (resin / polymerizable compound) is preferably 98/2 to 10/90, more preferably 95/5 to 30/70, 90 / 10 to 50/50 is most preferable. When the mass ratio of the resin and the compound containing a group having an ethylenically unsaturated bond is in the above range, a cured film that is superior in polymerizability and heat resistance can be formed.
- Formula (AM1) (Wherein t represents an integer of 1 to 20, R 4 represents a t-valent organic group having 1 to 200 carbon atoms, and R 5 represents a group represented by the following formula (AM2) or the following formula (AM3)). Is shown.)
- the compound represented by the formula (AM1) is preferably 5 parts by mass or more and 40 parts by mass or less with respect to 100 parts by mass of the polyimide precursor or the like. More preferably, it is 10 to 35 mass parts. Further, in the total polymerizable compound, the compound represented by the following formula (AM4) is contained in an amount of 10% by mass or more and 90% by mass or less, and the compound represented by the following formula (AM5) is contained in the total thermal crosslinking agent by 10% by mass or more. It is also preferable to contain 90 mass% or less.
- Formula (AM4) (Wherein R 4 represents a divalent organic group having 1 to 200 carbon atoms, and R 5 represents a group represented by the following formula (AM2) or the following formula (AM3)).
- Formula (AM5) (Wherein u represents an integer of 3 to 8, R 4 represents a u-valent organic group having 1 to 200 carbon atoms, and R 5 represents a group represented by the following formula (AM2) or the following formula (AM3)). .)
- the compound represented by the formula (AM4) include 46DMOC, 46DMOEP (trade name, manufactured by Asahi Organic Materials Co., Ltd.), DML-MBPC, DML-MBOC, DML-OCHP, DML-PCHP, DML.
- Specific examples of the compound represented by the formula (AM5) include TriML-P, TriML-35XL, TML-HQ, TML-BP, TML-pp-BPF, TML-BPA, TMOM-BP, HML-TPPHBA, HML-TPHAP, HMOM-TPPHBA, HMOM-TPPHAP (trade name, manufactured by Honshu Chemical Industry Co., Ltd.), TM-BIP-A (trade name, manufactured by Asahi Organic Materials Co., Ltd.), NIKALAC MX-280, NIKALAC MX-270, NIKALAC MW-100LM (trade name, manufactured by Sanwa Chemical Co., Ltd.).
- Epoxy compound compound having an epoxy group
- the epoxy compound is preferably a compound having two or more epoxy groups in one molecule.
- the epoxy group undergoes a cross-linking reaction at 200 ° C. or less and does not cause a dehydration reaction derived from the cross-linking, so that film shrinkage hardly occurs. For this reason, containing an epoxy compound is effective for low-temperature curing and warping of the composition.
- the epoxy compound preferably contains a polyethylene oxide group. Thereby, an elasticity modulus falls more and also curvature can be suppressed. Moreover, the film
- the polyethylene oxide group means that the number of repeating units of ethylene oxide is 2 or more, and the number of repeating units is preferably 2 to 15.
- epoxy compound examples include bisphenol A type epoxy resin; bisphenol F type epoxy resin; alkylene glycol type epoxy resin such as propylene glycol diglycidyl ether; polyalkylene glycol type epoxy resin such as polypropylene glycol diglycidyl ether; polymethyl (glycidyl Examples include, but are not limited to, epoxy group-containing silicones such as (roxypropyl) siloxane.
- Epicron (registered trademark) 850-S Epicron (registered trademark) HP-4032, Epicron (registered trademark) HP-7200, Epicron (registered trademark) HP-820, Epicron (registered trademark) HP-4700, Epicron (registered trademark) EXA-4710, Epicron (registered trademark) HP-4770, Epicron (registered trademark) EXA-859CRP, Epicron (registered trademark) EXA-1514, Epicron (registered trademark) EXA-4880, Epicron (registered trademark) EXA-4850-150, Epicron EXA-4850-1000, Epicron (registered trademark) EXA-4816, Epicron (registered trademark) EXA-4822 (trade name, manufactured by Dainippon Ink & Chemicals, Inc.), Rica Resin (registered trademark) ) BEO-60E (trade name, Shin Nippon Rika ( )), EP-4003S, EP-4000S (trade name, manufactured by
- an epoxy resin containing a polyethylene oxide group is preferable in terms of suppressing warpage and excellent heat resistance.
- Epicron (registered trademark) EXA-4880, Epicron (registered trademark) EXA-4822, and Licaredin (registered trademark) BEO-60E are preferable because they contain a polyethylene oxide group.
- the epoxy compound is preferably 5 to 50 parts by mass, more preferably 10 to 50 parts by mass, and still more preferably 10 to 40 parts by mass with respect to 100 parts by mass of the resin.
- the blending amount is 5 parts by mass or more, warpage of the cured film can be further suppressed, and when it is 50 parts by mass or less, pattern filling caused by reflow during curing can be further suppressed.
- oxetane compound compound having oxetanyl group
- examples of the oxetane compound include compounds having two or more oxetane rings in one molecule, 3-ethyl-3-hydroxymethyloxetane, 1,4-bis ⁇ [(3-ethyl-3-oxetanyl) methoxy] methyl ⁇ benzene, Examples include 3-ethyl-3- (2-ethylhexylmethyl) oxetane and 1,4-benzenedicarboxylic acid-bis [(3-ethyl-3-oxetanyl) methyl] ester.
- Aron Oxetane series (for example, OXT-121, OXT-221, OXT-191, OXT-223) manufactured by Toagosei Co., Ltd. can be preferably used. Two or more kinds may be mixed.
- the oxetane compound is preferably 5 to 50 parts by mass, more preferably 10 to 50 parts by mass, and still more preferably 10 to 40 parts by mass with respect to 100 parts by mass of the polyimide precursor or the like.
- a benzoxazine compound (compound having a benzoxazolyl group))
- a benzoxazine compound is preferable because it is a cross-linking reaction derived from a ring-opening addition reaction, so that degassing does not occur during curing, and thermal contraction is further reduced to suppress warpage.
- benzoxazine compound examples include Ba type benzoxazine, Bm type benzoxazine (trade name, manufactured by Shikoku Kasei Kogyo Co., Ltd.), benzoxazine adduct of polyhydroxystyrene resin, phenol novolac type dihydrobenzoxazine. Compounds. These may be used alone or in combination of two or more.
- the benzoxazine compound is preferably 5 to 50 parts by mass, more preferably 10 to 50 parts by mass, and still more preferably 10 to 40 parts by mass with respect to 100 parts by mass of the resin.
- the photosensitive resin composition may contain a photopolymerization initiator.
- the photosensitive resin composition contains a photo radical polymerization initiator
- the photosensitive resin composition is applied to a substrate such as a semiconductor wafer to form a photosensitive resin composition layer, and then irradiated with light. Curing due to radicals occurs, and the solubility in the light irradiation part can be reduced. Therefore, for example, by exposing the photosensitive resin composition layer through a photomask having a pattern that masks only the electrode portion, there is an advantage that regions having different solubility can be easily produced according to the electrode pattern. is there.
- the photopolymerization initiator is not particularly limited as long as it has the ability to initiate a polymerization reaction (crosslinking reaction) of the polymerizable compound, and can be appropriately selected from known photopolymerization initiators. For example, those having photosensitivity to light in the ultraviolet region to the visible region are preferable. Further, it may be an activator that generates some active radicals by generating some action with the photoexcited sensitizer.
- the photopolymerization initiator preferably contains at least one compound having a molar extinction coefficient of at least about 50 within a range of about 300 to 800 nm (preferably 330 to 500 nm). The molar extinction coefficient of the compound can be measured using a known method. Specifically, for example, it is preferable to measure with a UV-visible spectrophotometer (Cary-5 spectrophotometer manufactured by Varian) using an ethyl acetate solvent at a concentration of 0.01 g / L.
- a UV-visible spectrophotometer Cary-5
- halogenated hydrocarbon derivatives for example, those having a triazine skeleton, those having an oxadiazole skeleton, those having a trihalomethyl group
- Acylphosphine compounds such as acylphosphine oxide, oxime compounds such as hexaarylbiimidazole and oxime derivatives, organic peroxides, thio compounds, ketone compounds, aromatic onium salts, ketoxime ethers, aminoacetophenone compounds, hydroxyacetophenones, azo series
- examples thereof include compounds, azide compounds, metallocene compounds, organoboron compounds, iron arene complexes, and the like.
- halogenated hydrocarbon compounds having a triazine skeleton examples include those described in Wakabayashi et al., Bull. Chem. Soc. Japan, 42, 2924 (1969), a compound described in British Patent 1388492, a compound described in JP-A-53-133428, a compound described in German Patent 3333724, F.I. C. Schaefer et al. Org. Chem. 29, 1527 (1964), compounds described in JP-A-62-258241, compounds described in JP-A-5-281728, compounds described in JP-A-5-34920, US patents Examples thereof include compounds described in the specification of No. 42122976.
- Examples of the compounds described in US Pat. No. 4,221,976 include compounds having an oxadiazole skeleton (for example, 2-trichloromethyl-5-phenyl-1,3,4-oxadiazole, 2-trichloro Methyl-5- (4-chlorophenyl) -1,3,4-oxadiazole, 2-trichloromethyl-5- (1-naphthyl) -1,3,4-oxadiazole, 2-trichloromethyl-5 (2-naphthyl) -1,3,4-oxadiazole, 2-tribromomethyl-5-phenyl-1,3,4-oxadiazole, 2-tribromomethyl-5- (2-naphthyl)- 1,3,4-oxadiazole; 2-trichloromethyl-5-styryl-1,3,4-oxadiazole, 2-trichloromethyl-5- (4-chlorostyryl) 1,3,4-oxadiazole, 2-trichloromethyl-5-
- ketone compound examples include the compounds described in paragraph 0087 of JP-A-2015-087611, the contents of which are incorporated herein.
- Kaya Cure DETX manufactured by Nippon Kayaku Co., Ltd.
- Nippon Kayaku Co., Ltd. is also preferably used.
- hydroxyacetophenone compounds As the photopolymerization initiator, hydroxyacetophenone compounds, aminoacetophenone compounds, and acylphosphine compounds can also be suitably used. More specifically, for example, aminoacetophenone initiators described in JP-A-10-291969 and acylphosphine oxide initiators described in Japanese Patent No. 4225898 can also be used.
- hydroxyacetophenone-based initiator IRGACURE-184 (IRGACURE is a registered trademark), DAROCUR-1173, IRGACURE-500, IRGACURE-2959, and IRGACURE-127 (trade names: all manufactured by BASF) can be used.
- aminoacetophenone-based initiator commercially available products IRGACURE-907, IRGACURE-369, and IRGACURE-379 (trade names: all manufactured by BASF) can be used.
- aminoacetophenone-based initiator compounds described in JP-A-2009-191179 having a maximum absorption wavelength matched to a wavelength of 365 nm or 405 nm can also be used.
- the acylphosphine initiator include 2,4,6-trimethylbenzoyl-diphenyl-phosphine oxide.
- IRGACURE-819 and IRGACURE-TPO which are commercially available products can be used.
- the metallocene compound include IRGACURE-784 (manufactured by BASF).
- More preferred examples of the photopolymerization initiator include oxime compounds.
- the exposure latitude can be improved more effectively.
- Oxime ester compounds are particularly preferred because they have a wide exposure latitude (exposure margin) and also act as a thermal base generator.
- Specific examples of the oxime compound include compounds described in JP-A No. 2001-233842, compounds described in JP-A No. 2000-80068, and compounds described in JP-A No. 2006-342166.
- Preferable oxime compounds include, for example, compounds having the following structures, 3-benzooxyiminobutan-2-one, 3-acetoxyiminobutan-2-one, 3-propionyloxyiminobutan-2-one, 2-acetoxy Iminopentan-3-one, 2-acetoxyimino-1-phenylpropan-1-one, 2-benzoyloxyimino-1-phenylpropan-1-one, 3- (4-toluenesulfonyloxy) iminobutan-2-one And 2-ethoxycarbonyloxyimino-1-phenylpropan-1-one.
- oxime compounds include J.M. C. S. Perkin II (1979) p. 1653-1660, J.A. C. S. Perkin II (1979) pp. 156-162, Journal of Photopolymer Science and Technology (1995), pp. 156-162. 202-232 compounds, compounds described in JP-A-2000-66385, JP-A-2000-80068, JP-T 2004-534797, JP-A-2006-342166, international publication WO2015 Compound described in each publication of No. / 036910.
- IRGACURE OXE 01, IRGACURE OXE 02, IRGACURE OXE 03, IRGACURE OXE 04 (above, manufactured by BASF), Adekaoptomer N-1919 (manufactured by ADEKA Corporation, light described in JP2012-14052A) A polymerization initiator 2) is also preferably used.
- TR-PBG-304 manufactured by Changzhou Powerful Electronic New Materials Co., Ltd.
- Adeka Arkles NCI-831 and Adeka Arkles NCI-930 made by ADEKA
- DFI-091 manufactured by Daitokemix Co., Ltd.
- JP-A-2009-221114 which have an absorption maximum at 405 nm and have good sensitivity to a g-ray light source, may be used.
- the cyclic oxime compounds described in JP-A-2007-231000 and JP-A-2007-322744 can also be suitably used.
- cyclic oxime compounds in particular, cyclic oxime compounds fused to carbazole dyes described in JP2010-32985A and JP2010-185072A have high light absorptivity and high sensitivity. preferable.
- a compound described in JP-A-2009-242469 which is a compound having an unsaturated bond at a specific site of the oxime compound, can also be suitably used.
- an oxime compound having a fluorine atom examples include compounds described in JP 2010-262028 A, compounds 24, 36 to 40 described in paragraph 0345 of JP 2014-500852 A, and JP 2013. And the compound (C-3) described in paragraph 0101 of JP-A No. 164471.
- oxime compounds having a specific substituent as disclosed in JP-A-2007-267979 there are oxime compounds having a thioaryl group as disclosed in JP-A-2009-191061, and the like.
- Photopolymerization initiators are trihalomethyltriazine compounds, benzyldimethylketal compounds, ⁇ -hydroxyketone compounds, ⁇ -aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triaryls from the viewpoint of exposure sensitivity.
- Selected from the group consisting of imidazole dimers, onium salt compounds, benzothiazole compounds, benzophenone compounds, acetophenone compounds and derivatives thereof, cyclopentadiene-benzene-iron complexes and salts thereof, halomethyloxadiazole compounds, and 3-aryl substituted coumarin compounds. are preferred.
- More preferred photopolymerization initiators are trihalomethyltriazine compounds, ⁇ -aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triarylimidazole dimers, onium salt compounds, benzophenone compounds, acetophenone compounds, At least one compound selected from the group consisting of a trihalomethyltriazine compound, an ⁇ -aminoketone compound, an oxime compound, a triarylimidazole dimer, and a benzophenone compound is more preferable, and a metallocene compound or an oxime compound is more preferable, and an oxime compound. Is particularly preferred.
- Photopolymerization initiators include N, N'-tetraalkyl-4,4'-diaminobenzophenone, 2-benzyl-, such as benzophenone, N, N'-tetramethyl-4,4'-diaminobenzophenone (Michler ketone), etc.
- Aromatic ketones such as 2-dimethylamino-1- (4-morpholinophenyl) -butanone-1, 2-methyl-1- [4- (methylthio) phenyl] -2-morpholino-propanone-1, alkyl anthraquinones, etc.
- benzoin ether compounds such as benzoin alkyl ether
- benzoin compounds such as benzoin and alkylbenzoin
- benzyl derivatives such as benzyldimethyl ketal.
- a compound represented by the following formula (I) can also be used.
- R 50 represents an alkyl group having 1 to 20 carbon atoms; an alkyl group having 2 to 20 carbon atoms interrupted by one or more oxygen atoms; an alkoxy group having 1 to 12 carbon atoms; a phenyl group; An alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 12 carbon atoms, a halogen atom, a cyclopentyl group, a cyclohexyl group, an alkenyl group having 1 to 12 carbon atoms, and 2 to 2 carbon atoms interrupted by one or more oxygen atoms A phenyl group substituted with at least one of 18 alkyl groups and an alkyl group having 1 to 4 carbon atoms; or biphenylyl, and R 51 is a group represented by the formula (II) or the same as R 50 R 52 to R 54 are each independently alkyl having 1 to 12 carbons, alkoxy having 1 to 12 carbons or halogen.
- R 51 is a group represented by
- the content of the photopolymerization initiator is preferably 0.1 to 30% by mass, more preferably 0.1% by mass with respect to the total solid content of the photosensitive resin composition. -20% by mass, more preferably 0.1-10% by mass. Only one type of photopolymerization initiator may be used, or two or more types may be used. When there are two or more photopolymerization initiators, the total is preferably in the above range.
- the photosensitive resin composition further contains a migration inhibitor.
- the migration inhibitor is not particularly limited, but a heterocyclic ring (pyrrole ring, furan ring, thiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrazole ring, isoxazole ring, isothiazole ring, tetrazole ring, pyridine ring, Compounds having pyridazine ring, pyrimidine ring, pyrazine ring, piperidine ring, piperazine ring, morpholine ring, 2H-pyran ring and 6H-pyran ring, triazine ring), compounds having thioureas and mercapto groups, hindered phenol compounds , Salicylic acid derivative
- an ion trapping agent that traps anions such as halogen ions can be used.
- migration inhibitors include rust inhibitors described in paragraph 0094 of JP2013-15701, compounds described in paragraphs 0073 to 0076 of JP2009-283711, and JP2011-95956A.
- the compounds described in paragraph 0052 and the compounds described in paragraphs 0114, 0116 and 0118 of JP2012-194520A can be used.
- the migration inhibitor examples include 1H-1,2,3-triazole and 1H-tetrazole.
- the content of the migration inhibitor is preferably 0.01 to 5.0% by mass with respect to the total solid content of the photosensitive resin composition, 0.05 to 2.0% by mass is more preferable, and 0.1 to 1.0% by mass is more preferable. Only one type of migration inhibitor may be used, or two or more types may be used. When there are two or more migration inhibitors, the total is preferably within the above range.
- the photosensitive resin composition used in the present embodiment preferably contains a polymerization inhibitor.
- the polymerization inhibitor include hydroquinone, paramethoxyphenol, di-tert-butyl-paracresol, pyrogallol, para-tert-butylcatechol, parabenzoquinone, diphenyl-parabenzoquinone, 4,4′-thiobis (3-methyl).
- a polymerization inhibitor described in paragraph 0060 of JP-A-2015-127817 and compounds described in paragraphs 0031 to 0046 of international publication WO2015 / 125469 can also be used.
- the content of the polymerization inhibitor is preferably 0.01 to 5% by mass with respect to the total solid content of the photosensitive resin composition. Only one polymerization inhibitor may be used, or two or more polymerization inhibitors may be used. When two or more polymerization inhibitors are used, the total is preferably within the above range.
- the photosensitive resin composition used in this embodiment may contain a thermal base generator.
- the type of the thermal base generator is not particularly defined, but is selected from an acidic compound that generates a base when heated to 40 ° C. or higher, and an ammonium salt having an anion having an pKa1 of 0 to 4 and an ammonium cation. It is preferable to include a thermal base generator containing at least one kind.
- pKa1 represents the logarithm ( ⁇ Log 10 Ka) of the dissociation constant (Ka) of the first proton of the acid, and details will be described later.
- the cyclization reaction of the polyimide precursor and the polybenzoxazole precursor can be performed at a low temperature, and the composition can be made more stable.
- the heat base generator does not generate a base unless it is heated, cyclization of the polyimide precursor and polybenzoxazole precursor during storage is possible even if it coexists with the polyimide precursor and polybenzoxazole precursor. And is excellent in storage stability.
- the thermal base generator includes at least one selected from an acidic compound (A1) that generates a base when heated to 40 ° C. or higher, and an ammonium salt (A2) having an anion having pKa1 of 0 to 4 and an ammonium cation. It is preferable. Since the acidic compound (A1) and the ammonium salt (A2) generate a base when heated, the base generated from these compounds can promote a cyclization reaction of a polyimide precursor and a polybenzoxazole precursor, Cyclization of polyimide precursors and polybenzoxazole precursors can be performed at low temperatures.
- the solution obtained by stirring means a compound having a value measured at 20 ° C. of less than 7 using a pH (power of hydrogen) meter.
- the base generation temperature of the acidic compound (A1) and the ammonium salt (A2) is preferably 40 ° C. or higher, more preferably 120 to 200 ° C.
- the upper limit of the base generation temperature is preferably 190 ° C. or lower, more preferably 180 ° C. or lower, and further preferably 165 ° C. or lower.
- the lower limit of the base generation temperature is preferably 130 ° C or higher, and more preferably 135 ° C or higher. If the base generation temperature of the acidic compound (A1) and the ammonium salt (A2) is 120 ° C. or higher, the base is unlikely to be generated during storage.
- a photosensitive resin composition can be prepared.
- the base generation temperature of the acidic compound (A1) and the ammonium salt (A2) is 200 ° C. or lower, the cyclization temperature of the polyimide precursor, polybenzoxazole precursor, and the like can be lowered.
- the base generation temperature is measured, for example, by using differential scanning calorimetry, heating the compound to 250 ° C. at 5 ° C./min in a pressure capsule, reading the peak temperature of the lowest exothermic peak, and measuring the peak temperature as the base generation temperature. can do.
- the base generated by the thermal base generator is preferably a secondary amine or a tertiary amine, more preferably a tertiary amine. Since tertiary amine has high basicity, cyclization temperature of a polyimide precursor, a polybenzoxazole precursor, etc. can be made lower.
- the base generated by the thermal base generator preferably has a boiling point of 80 ° C. or higher, more preferably 100 ° C. or higher, and further preferably 140 ° C. or higher.
- the molecular weight of the generated base is preferably 80 to 2000.
- the lower limit is more preferably 100 or more.
- the upper limit is more preferably 500 or less.
- the molecular weight value is a theoretical value obtained from the structural formula.
- the acidic compound (A1) preferably contains one or more selected from an ammonium salt and a compound represented by the formula (101) or (102) described later.
- the ammonium salt (A2) is preferably an acidic compound.
- the ammonium salt (A2) may be a compound containing an acidic compound that generates a base when heated to 40 ° C. or higher (preferably 120 to 200 ° C.), or 40 ° C. or higher (preferably 120 to 200 ° C.). ) May be a compound excluding an acidic compound that generates a base when heated.
- the ammonium salt means a salt of an ammonium cation represented by the following formula (101) or formula (102) and an anion.
- the anion may be bonded to any part of the ammonium cation via a covalent bond, and may be outside the molecule of the ammonium cation, but may be outside the molecule of the ammonium cation. preferable.
- numerator of an ammonium cation means the case where an ammonium cation and an anion are not couple
- the anion outside the molecule of the cation moiety is also referred to as a counter anion.
- R 1 to R 6 each independently represents a hydrogen atom or a hydrocarbon group
- R 7 represents a hydrocarbon group.
- R 1 and R 2 , R 3 and R 4 , R 5 and R 6 , R 5 and R 7 in Formula (101) and Formula (102) may be bonded to each other to form a ring.
- the ammonium cation is preferably represented by any of the following formulas (Y1-1) to (Y1-5).
- R 101 represents an n-valent organic group
- R 1 and R 7 have the same meanings as formula (101) or formula (102).
- Ar 101 and Ar 102 each independently represent an aryl group
- n represents an integer of 1 or more
- m represents an integer of 0 to 5 .
- the ammonium salt preferably has an anion having an pKa1 of 0 to 4 and an ammonium cation.
- the upper limit of the anion pKa1 is more preferably 3.5 or less, and even more preferably 3.2 or less.
- the lower limit is preferably 0.5 or more, and more preferably 1.0 or more. If the pKa1 of the anion is in the above range, the polyimide precursor and the polybenzoxazole precursor can be cyclized at a low temperature, and further, the stability of the photosensitive resin composition containing the polyimide precursor and the polybenzoxazole precursor, etc. Can be improved.
- pKa1 is 4 or less, the stability of the thermal base generator is good, the generation of a base without heating can be suppressed, and the stability of the photosensitive resin composition containing a polyimide precursor and a polybenzoxazole precursor, etc. Good properties. If pKa1 is 0 or more, the generated base is not easily neutralized, and the cyclization efficiency of the polyimide precursor and polybenzoxazole precursor is good.
- the kind of anion is preferably one selected from a carboxylate anion, a phenol anion, a phosphate anion, and a sulfate anion, and a carboxylate anion is more preferable because both the stability of the salt and the thermal decomposability can be achieved.
- the ammonium salt is more preferably a salt of an ammonium cation and a carboxylate anion.
- the carboxylic acid anion is preferably a divalent or higher carboxylic acid anion having two or more carboxyl groups, and more preferably a divalent carboxylic acid anion.
- it can be set as the thermal base generator which can improve more stability, sclerosis
- an anion of a divalent carboxylic acid the stability, curability and developability of the photosensitive resin composition containing a polyimide precursor and a polybenzoxazole precursor can be further improved.
- the carboxylic acid anion is preferably a carboxylic acid anion having a pKa1 of 4 or less.
- pKa1 is more preferably 3.5 or less, and even more preferably 3.2 or less. According to this aspect, the stability of the photosensitive resin composition containing a polyimide precursor and a polybenzoxazole precursor can be further improved.
- pKa1 represents the logarithm of the reciprocal of the dissociation constant of the first proton of the acid, and the determination of Organic Structures by Physical Methods (author: Brown, HC, McDaniel, D.H., Hafliger Ed .: Braude, EA, Nachod, FC; Academic Press, New York, 1955), and Data for Biochemical Research (author: Dawson, R. M.). al; Oxford, Clarendon Press, 1959). For compounds not described in these documents, values calculated from the structural formula using software of ACD / pKa (manufactured by ACD / Labs) are used.
- the carboxylate anion is preferably represented by the following formula (X1).
- EWG represents an electron withdrawing group.
- the electron-withdrawing group means a group in which Hammett's substituent constant ⁇ m exhibits a positive value.
- ⁇ m is a review by Yusuke Tono, Journal of Synthetic Organic Chemistry, Vol. 23, No. 8 (1965) p. 631-642.
- the electron withdrawing group in this embodiment is not limited to the substituent described in the said literature.
- Me represents a methyl group
- Ac represents an acetyl group
- Ph represents a phenyl group.
- EWG is preferably a group represented by the following formulas (EWG-1) to (EWG-6).
- R x1 to R x3 each independently represent a hydrogen atom, an alkyl group, an alkenyl group, an aryl group, a hydroxyl group or a carboxyl group, and Ar represents an aromatic group Represents.
- the carboxylate anion is preferably represented by the following formula (XA).
- Formula (XA) In the formula (XA), L 10 represents a single bond or a divalent linking group selected from an alkylene group, an alkenylene group, an aromatic group, —NR X —, and a combination thereof, and R X represents a hydrogen atom Represents an alkyl group, an alkenyl group or an aryl group.
- carboxylate anion examples include a maleate anion, a phthalate anion, an N-phenyliminodiacetic acid anion, and an oxalate anion. These can be preferably used.
- thermal base generator examples include the following compounds.
- the content of the thermal base generator in the photosensitive resin composition is preferably 0.1 to 50% by mass with respect to the total solid content of the photosensitive resin composition.
- the lower limit is more preferably 0.5% by mass or more, and further preferably 1% by mass or more.
- the upper limit is more preferably 30% by mass or less, and further preferably 20% by mass or less.
- 1 type (s) or 2 or more types can be used for a thermal base generator. When using 2 or more types, it is preferable that a total amount is the said range.
- the photosensitive resin composition used in the present embodiment preferably contains a metal adhesion improver for improving the adhesion with a metal material used for electrodes, wirings and the like.
- the metal adhesion improver include sulfide compounds described in paragraphs 0046 to 0049 of JP2014-186186A and paragraphs 0032 to 0043 of JP2013-072935A.
- the metal adhesion improver also include the following compounds (N- [3- (triethoxysilyl) propyl] maleic acid monoamide and the like).
- the metal adhesion improver is preferably 0.1 to 30 parts by mass, more preferably 0.5 to 15 parts by mass with respect to 100 parts by mass of the resin.
- Adhesiveness between the cured film and the metal layer after the curing process becomes good by setting it to 0.1 parts by mass or more, and heat resistance and mechanical properties of the cured film after the curing process are good by setting it to 30 parts by mass or less. Become. Only one type of metal adhesion improver may be used, or two or more types may be used. When using 2 or more types, it is preferable that the sum total is the said range.
- solvent any known solvent can be used as long as the photosensitive resin composition can be formed into a layer.
- the solvent include compounds such as esters, ethers, ketones, aromatic hydrocarbons, and sulfoxides.
- esters include ethyl acetate, n-butyl acetate, isobutyl acetate, amyl formate, isoamyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, ⁇ -butyrolactone, and ⁇ -caprolactone , ⁇ -valerolactone, alkyl oxyacetate alkyl (eg, methyl alkyl oxyacetate, alkyl alkyl oxyacetate, butyl oxyacetate (eg, methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate, methyl ethoxyacetate, ethyl ethoxyacetate, etc.) )), 3-alkyloxypropionic acid alkyl esters (eg, methyl 3-alkyloxypropionate, ethyl
- ethers include diethylene glycol dimethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol Preferred examples include monomethyl ether acetate, propylene glycol monoethyl ether acetate, and propylene glycol monopropyl ether acetate.
- ketones include methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone, N-methyl-2-pyrrolidone and the like.
- aromatic hydrocarbons include toluene, xylene, anisole, limonene and the like.
- the solvent is preferably in the form of a mixture of two or more from the viewpoint of improving the properties of the coated surface.
- a mixed solution composed of two or more selected from dimethyl sulfoxide, ethyl carbitol acetate, butyl carbitol acetate, propylene glycol methyl ether, and propylene glycol methyl ether acetate is preferable.
- the combined use of dimethyl sulfoxide and ⁇ -butyrolactone is particularly preferred.
- the content of the solvent is preferably such that the total solid concentration of the photosensitive resin composition is 5 to 80% by mass from the viewpoint of applicability. 70 mass% is more preferable, and 10 to 60 mass% is particularly preferable.
- the solvent content may be adjusted depending on the desired thickness and coating method. For example, if the coating method is spin coating or slit coating, the content of the solvent having a solid content concentration in the above range is preferable. In the case of spray coating, the amount is preferably 0.1% by mass to 50% by mass, and more preferably 1.0% by mass to 25% by mass.
- a photosensitive resin composition layer having a desired thickness can be uniformly formed by adjusting the amount of solvent by the coating method.
- One type of solvent may be sufficient and 2 or more types may be sufficient as it. When there are two or more solvents, the total is preferably in the above range.
- the contents of N-methyl-2-pyrrolidone, N-ethyl-2-pyrrolidone, N, N-dimethylacetamide and N, N-dimethylformamide are determined based on the total mass of the photosensitive resin composition from the viewpoint of film strength. Is less than 5% by weight, more preferably less than 1% by weight, even more preferably less than 0.5% by weight, and still more preferably less than 0.1% by weight.
- the photosensitive resin composition used in the present embodiment has various additives such as a photobase generator, a thermal polymerization initiator, a thermal acid generator, and a silane as necessary, as long as the effects of the present invention are not impaired. Coupling agents, sensitizing dyes, chain transfer agents, surfactants, higher fatty acid derivatives, inorganic particles, curing agents, curing catalysts, fillers, antioxidants, ultraviolet absorbers, anti-aggregation agents, etc. can be blended. . When these additives are blended, the total blending amount is preferably 3% by mass or less of the solid content of the composition.
- the photosensitive resin composition used in the present embodiment may contain a photobase generator.
- a photobase generator generates a base upon exposure and does not exhibit activity under normal conditions of normal temperature and pressure. However, when an electromagnetic wave is irradiated and heated as an external stimulus, the base (basic substance) is generated. ) Is not particularly limited as long as it generates. Since the base generated by exposure works as a catalyst for curing the polyimide precursor, the benzoxazole precursor and the like by heating, it can be suitably used in the negative type.
- the content of the photobase generator is not particularly limited as long as a desired pattern can be formed, and can be a general content.
- the photobase generator is preferably in the range of 0.01 parts by weight or more and less than 30 parts by weight with respect to 100 parts by weight of the resin, more preferably in the range of 0.05 parts by weight to 25 parts by weight. Preferably, it is in the range of 0.1 to 20 parts by mass. Only one photobase generator may be used, or two or more photobase generators may be used. When there are two or more photobase generators, the total is preferably in the above range. In the present embodiment, known photobase generators can be used. For example, M.M. Shirai, and M.M. Tsunooka, Prog. Polym. Sci.
- An ionic compound neutralized by forming a salt with a base component, or a nonionic compound in which the base component is made latent by a urethane bond or an oxime bond such as a carbamate derivative, an oxime ester derivative, or an acyl compound Can be mentioned.
- the photobase generator that can be used in the present embodiment is not particularly limited and known ones can be used.
- oxime derivatives examples include photobase generators having a cinnamic amide structure as disclosed in JP2009-80452A and International Publication WO2009 / 123122, JP2006-1889591 and JP Photobase generator having a carbamate structure as disclosed in Japanese Patent Application Laid-Open No.
- the photosensitive resin composition used in this embodiment may contain a thermal polymerization initiator (preferably a thermal radical polymerization initiator).
- a thermal radical polymerization initiator preferably a thermal radical polymerization initiator
- the thermal radical polymerization initiator is a compound that generates radicals by heat energy and initiates or accelerates the polymerization reaction of the polymerizable compound. By adding the thermal radical polymerization initiator, the polymerization reaction of the polymerizable compound can be advanced when the cyclization reaction of the polyimide precursor and the polybenzoxazole precursor is advanced.
- the thermal radical polymerization initiator include compounds described in paragraphs 0074 to 0118 of JP-A-2008-63554.
- the content of the thermal radical polymerization initiator is preferably from 0.1 to 50% by mass, preferably from 0.1 to 50% by weight based on the total solid content of the photosensitive resin composition. 30% by mass is more preferable, and 0.1 to 20% by mass is particularly preferable. Further, the thermal radical polymerization initiator is preferably contained in an amount of 0.1 to 50 parts by mass, and more preferably 0.5 to 30 parts by mass with respect to 100 parts by mass of the polymerizable compound. According to this aspect, it is easy to form a cured film having more excellent heat resistance. Only one type of thermal radical polymerization initiator may be used, or two or more types may be used. When there are two or more thermal radical polymerization initiators, the total is preferably in the above range.
- the photosensitive resin composition used in this embodiment may contain a thermal acid generator.
- the thermal acid generator generates an acid by heating, promotes cyclization of the polyimide precursor and the polybenzoxazole precursor, and further improves the mechanical properties of the cured film. Furthermore, the thermal acid generator has an effect of promoting a crosslinking reaction of at least one compound selected from a compound having a hydroxymethyl group, an alkoxymethyl group or an acyloxymethyl group, an epoxy compound, an oxetane compound and a benzoxazine compound.
- JP2013-167742A is also preferable as the thermal acid generator.
- the content of the thermal acid generator is preferably 0.01 parts by mass or more and more preferably 0.1 parts by mass or more with respect to 100 parts by mass of the polyimide precursor and the polybenzoxazole precursor. By containing 0.01 part by mass or more, the crosslinking reaction and the cyclization of the polyimide precursor and the polybenzoxazole precursor are promoted, so that the mechanical properties and chemical resistance of the cured film can be further improved. Moreover, the content is preferably 20 parts by mass or less, more preferably 15 parts by mass or less, and particularly preferably 10 parts by mass or less from the viewpoint of electrical insulation of the cured film.
- One type of thermal acid generator may be used, or two or more types may be used. When using 2 or more types, it is preferable that a total amount becomes the said range.
- the photosensitive resin composition used in this embodiment may contain a silane coupling agent in order to improve adhesion to the substrate.
- a silane coupling agent examples include compounds described in paragraphs 0062 to 0073 of JP-A No. 2014-191002, compounds described in paragraphs 0063 to 0071 of international publication WO 2011 / 080992A1, and JP-A No. 2014-191252. Examples thereof include compounds described in paragraphs 0060 to 0061, compounds described in paragraphs 0045 to 0052 of JP 2014-41264 A, and compounds described in paragraph 0055 of international publication WO 2014/097594.
- the silane coupling agent is preferably 0.1 to 20 parts by mass, more preferably 1 to 10 parts by mass with respect to 100 parts by mass of the resin. When it is 0.1 part by mass or more, sufficient adhesion to the substrate can be imparted, and when it is 20 parts by mass or less, problems such as an increase in viscosity during storage at room temperature can be suppressed. Only one type of silane coupling agent may be used, or two or more types may be used. When using 2 or more types, it is preferable that a total amount becomes the said range.
- the photosensitive resin composition used in the present embodiment may contain a sensitizing dye.
- a sensitizing dye absorbs specific actinic radiation and enters an electronically excited state.
- the sensitizing dye in an electronically excited state comes into contact with an amine generator, a thermal radical polymerization initiator, a photopolymerization initiator, and the like, and effects such as electron transfer, energy transfer, and heat generation occur.
- the amine generator, the thermal radical polymerization initiator, and the photopolymerization initiator are decomposed by causing a chemical change to generate radicals, acids, or bases.
- preferable sensitizing dyes include those belonging to the following compounds and having an absorption wavelength in the region of 300 nm to 450 nm.
- polynuclear aromatics for example, phenanthrene, anthracene, pyrene, perylene, triphenylene, 9.10-dialkoxyanthracene
- xanthenes for example, fluorescein, eosin, erythrosine, rhodamine B, rose bengal
- thioxanthones for example, 2,4-diethylthioxanthone
- cyanines for example thiacarbocyanine, oxacarbocyanine
- merocyanines for example merocyanine, carbomerocyanine
- thiazines for example thionine, methylene blue, toluidine blue
- acridines Eg, acridine orange, chloroflavin, acriflavine
- anthrdines
- the content of the sensitizing dye is preferably 0.01 to 20% by mass, and preferably 0.1 to 15% by mass with respect to the total solid content of the photosensitive resin composition. Is more preferable, and 0.5 to 10% by mass is even more preferable.
- a sensitizing dye may be used individually by 1 type, and may use 2 or more types together.
- the photosensitive resin composition used in the present embodiment may contain a chain transfer agent.
- the chain transfer agent is defined, for example, in Polymer Dictionary 3rd Edition (edited by the Polymer Society, 2005) pages 683-684.
- As the chain transfer agent for example, a compound group having SH, PH, SiH, GeH in the molecule is used. These can generate hydrogen by donating hydrogen to a low activity radical to generate a radical, or after being oxidized and deprotonated.
- thiol compounds for example, 2-mercaptobenzimidazoles, 2-mercaptobenzthiazoles, 2-mercaptobenzoxazoles, 3-mercaptotriazoles, 5-mercaptotetrazoles, etc.
- 2-mercaptobenzimidazoles, 2-mercaptobenzthiazoles, 2-mercaptobenzoxazoles, 3-mercaptotriazoles, 5-mercaptotetrazoles, etc. can be preferably used.
- the preferable content of the chain transfer agent is preferably 0.01 to 20 parts by mass, more preferably 100 parts by mass based on the total solid content of the photosensitive resin composition. 1 to 10 parts by mass, particularly preferably 1 to 5 parts by mass. Only one type of chain transfer agent may be used, or two or more types may be used. When there are two or more chain transfer agents, the total is preferably in the above range.
- surfactant Each type of surfactant may be added to the photosensitive resin composition used in the present embodiment from the viewpoint of further improving applicability.
- the surfactant various types of surfactants such as a fluorosurfactant, a nonionic surfactant, a cationic surfactant, an anionic surfactant, and a silicone surfactant can be used.
- the following surfactants are also preferable.
- the content of the surfactant is preferably 0.001 to 2.0% by mass, more preferably 0, based on the total solid content of the photosensitive resin composition. 0.005 to 1.0 mass%. Only one surfactant may be used, or two or more surfactants may be used. When there are two or more surfactants, the total is preferably in the above range.
- a higher fatty acid derivative such as behenic acid or behenic acid amide is added to the photosensitive resin composition used in the present embodiment, and the composition is dried in the drying process after coating. It may be unevenly distributed on the surface of the object.
- the content of the higher fatty acid derivative is preferably 0.1 to 10% by mass with respect to the total solid content of the photosensitive resin composition. Only one higher fatty acid derivative may be used, or two or more higher fatty acid derivatives may be used. When two or more higher fatty acid derivatives are used, the total is preferably within the above range.
- the water content of the photosensitive resin composition used in the present embodiment is preferably less than 5% by mass, more preferably less than 1% by mass, and particularly preferably less than 0.6% by mass from the viewpoint of coating surface properties.
- the metal content of the photosensitive resin composition used in the present embodiment is preferably less than 5 ppm by weight (parts per million), more preferably less than 1 ppm by weight, and particularly preferably less than 0.5 ppm by weight from the viewpoint of insulation. preferable.
- the metal include sodium, potassium, magnesium, calcium, iron, chromium, nickel and the like. When a plurality of metals are included, the total of these metals is preferably in the above range.
- a raw material having a low metal content is selected as a raw material constituting the photosensitive resin composition.
- the raw material to be filtered may be filtered, or the inside of the apparatus may be lined with polytetrafluoroethylene or the like to perform distillation under a condition in which contamination is suppressed as much as possible.
- the halogen atom content is preferably less than 500 ppm by mass, more preferably less than 300 ppm by mass, and particularly preferably less than 200 ppm by mass from the viewpoint of wiring corrosion.
- a halogen ion is less than 5 mass ppm, More preferably, it is less than 1 mass ppm, Especially less than 0.5 mass ppm is preferable.
- the halogen atom include a chlorine atom and a bromine atom. The total of chlorine atoms and bromine atoms, or chloride ions and bromide ions is preferably in the above range.
- a step of drying the solvent may be included.
- the drying temperature is preferably 50 to 150 ° C, more preferably 70 ° C to 130 ° C, and still more preferably 90 ° C to 110 ° C.
- the drying time is preferably 30 seconds to 20 minutes, more preferably 1 minute to 10 minutes, and further preferably 3 minutes to 7 minutes.
- the resin pattern 3 is manufactured by exposing and developing the photosensitive resin film.
- the thickness of the resin pattern is the same as that of the photosensitive resin film, preferably 0.1 to 100 ⁇ m, more preferably 1 to 50 ⁇ m, and further preferably 3 to 20 ⁇ m.
- the exposure conditions are not particularly defined, and it is preferable that the solubility of the exposed portion of the photosensitive resin film in the developer is changed, and it is more preferable that the exposed portion of the photosensitive resin film can be cured.
- the exposure is preferably performed by irradiating the photosensitive resin film with light having a wavelength of 500 nm or less. By irradiating light with a wavelength of 500 nm or less, the photosensitive resin can be made difficult to peel off from the transparent substrate.
- the lower limit of the exposure wavelength is preferably 100 nm or more, more preferably 190 nm or more, and further preferably 240 nm or more.
- the upper limit of the exposure wavelength is preferably 500 nm or less, and more preferably 400 nm or less.
- the exposure dose is preferably 100 to 10000 mJ / cm 2 in terms of exposure energy at a wavelength of 365 nm, and more preferably 200 to 8000 mJ / cm 2 .
- the aperture ratio of the mask can be appropriately determined according to a desired pattern.
- the surface area of the removed portion of the resin pattern formed by removing a part of the photosensitive resin film is preferably 30% or less of the entire surface area of the photosensitive resin film, and 25% or less. Is more preferable, 20% or less is further preferable, 15% or less is further preferable, 10% or less is further preferable, 5% or less is further more preferable, and 3% or less is particularly preferable. By setting it as such a range, it exists in the tendency for the yield of the molded article obtained to improve more.
- the lower limit of the ratio of the surface area of the removed portion of the resin pattern formed by removing a part of the photosensitive resin film to the surface area of the entire region of the photosensitive resin film is not particularly defined, for example 0.5% or more.
- the size of the opening of the mask can be determined as appropriate according to the application and the like, but a mask having a maximum length of 30 ⁇ m or less (preferably, a maximum length of 3 ⁇ m or more) is exemplified.
- the gap (opening) of the resin pattern formed in the present embodiment is a part for providing a contact hole, wiring, a part for providing an electrode, or the like.
- the wiring and electrodes are formed by applying a metal described later.
- the development process may be a positive development process or a negative development process, but a negative development process is preferred.
- a negative development process By performing negative development, the unexposed part (non-exposed part) is removed.
- the development method is not particularly limited, and it is preferable that a desired pattern can be formed.
- development methods such as paddle, spray, immersion, and ultrasonic waves can be employed.
- Development is preferably performed using a developer.
- the developer can be used without particular limitation as long as the unexposed part (non-exposed part) is removed. Development using an organic solvent is preferred.
- esters include ethyl acetate, n-butyl acetate, isobutyl acetate, amyl formate, isoamyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, and lactic acid.
- Ethyl, ⁇ -butyrolactone, ⁇ -caprolactone, ⁇ -valerolactone, alkyl oxyacetate eg, alkyl oxyacetate, alkyl oxyacetate, alkyl oxyacetate (eg, methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate) , Methyl ethoxyacetate, ethyl ethoxyacetate, etc.
- alkyl esters of 3-alkyloxypropionic acid eg, methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc.
- ketones include methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone, N-methyl-2-pyrrolidone and the like.
- aromatic hydrocarbons include toluene, xylene, anisole, limonene and the like.
- Preferred examples of the sulfoxides include dimethyl sulfoxide.
- the development time is preferably 10 seconds to 5 minutes.
- the temperature at the time of development is not particularly defined, but it can usually be carried out at 20 to 40 ° C.
- rinsing may be further performed.
- the rinsing is preferably performed with a solvent different from the developer. For example, it can rinse using the solvent contained in the photosensitive resin composition.
- the rinse time is preferably 5 seconds to 1 minute.
- ⁇ Curing process> it is preferable to include a curing step after the exposure and development step and before the transparent substrate peeling step.
- a metal application process it is preferable to include the hardening process which hardens the said resin pattern before a metal application process.
- the photosensitive resin film contains at least one selected from a polyimide precursor and a polybenzoxazole precursor, the cyclization reaction of the polyimide precursor and the polybenzoxazole precursor can be advanced by the curing step. Even when the photosensitive resin film is polyimide or polybenzoxazole, it can be heated together with a crosslinking agent to form a three-dimensional network structure.
- the photosensitive resin composition contains a radically polymerizable compound or the like
- curing of the unreacted radically polymerizable compound can also proceed.
- the curing step is usually performed by raising the temperature of the photosensitive resin film to a temperature equal to or higher than the glass transition temperature of the resin constituting the photosensitive resin film.
- the photosensitive resin film contains at least one selected from a polyimide precursor and a polybenzoxazole precursor
- the final temperature reached when the temperature is raised is the cyclization temperature of the resin contained in the photosensitive resin film.
- the final ultimate temperature when the temperature is raised is preferably the highest heating temperature.
- the maximum heating temperature is preferably 100 to 500 ° C., more preferably 150 to 450 ° C., and further preferably 160 to 350 ° C.
- the temperature increase is preferably performed at a temperature increase rate of 1 to 12 ° C./min from a temperature of 20 to 150 ° C. to a maximum heating temperature, more preferably 2 to 11 ° C./min, and further preferably 3 to 10 ° C./min. .
- the temperature at the start of heating is preferably 20 to 150 ° C., more preferably 20 to 130 ° C., and further preferably 25 to 120 ° C.
- the temperature at the start of heating refers to the heating temperature at the start of the step of heating to the maximum heating temperature.
- the temperature is the temperature after drying, for example, gradually from the boiling point of the solvent contained in the photosensitive resin composition— (30 to 200) ° C. It is preferable to raise the temperature to
- Heating may be performed in stages. As an example, before raising the temperature from 25 ° C. to 180 ° C. at 3 ° C./min, placing at 180 ° C. for 60 minutes, raising the temperature from 180 ° C. to 200 ° C. at 2 ° C./min, and placing at 200 ° C. for 120 minutes. Processing steps may be performed. Heating as the pretreatment step is preferably 100 to 200 ° C, more preferably 110 to 190 ° C, and most preferably 120 to 185 ° C. In this pretreatment step, it is also preferable to carry out the treatment while irradiating UV as described in US Pat. No. 9,159,547. Such a pretreatment step can improve the properties of the cured film.
- the pretreatment step is preferably performed in a short time of about 10 seconds to 2 hours, more preferably 15 seconds to 30 minutes.
- the pretreatment process may be performed in two or more steps.
- the pretreatment process 1 may be performed in the range of 100 to 150 ° C.
- the pretreatment process 2 may be performed in the range of 150 to 200 ° C.
- the temperature raising time is preferably 20 to 200 minutes, more preferably 20 to 100 minutes.
- the cooling rate is preferably 2 ° C./min or less, and more preferably 1 ° C./min or less.
- the cooling rate in the cooling step is preferably 0.1 ° C./min or more.
- the cooling time is preferably 30 to 600 minutes, more preferably 60 to 600 minutes, and particularly preferably 120 to 600 minutes.
- the temperature after cooling is preferably 30 ° C. or lower than the glass transition temperature (Tg) of the photosensitive resin film after the curing step. If the temperature after cooling is lowered to a temperature lower by 30 ° C. or more than the Tg of the photosensitive resin film, the polyimide is sufficiently cured and the occurrence of delamination is easily suppressed.
- Tg glass transition temperature
- ⁇ Metal application process> it is preferable to have the process of applying the metal 4 to the laminated body which has the transparent substrate 1 and the resin pattern 3 located in the surface of the said transparent substrate.
- the metal 4 in the present embodiment is preferably provided in the gap between the resin patterns 3 on the surface of the transparent substrate 1.
- the method for applying the metal is not particularly defined, but includes an electroless plating method. Further, it may be provided on the transparent substrate and between the transparent substrate 1 and the resin pattern 3.
- a metal is provided between the transparent substrate 1 and the resin pattern 3, between the surface of the transparent substrate 1 and the resin pattern 3 and on the transparent substrate 1, in a direction substantially perpendicular to the transparent substrate 1, The aspect provided between the resin pattern 3 and the resin pattern 3 is illustrated.
- a metal according to the following methods (1) and (2).
- a laminate having the transparent substrate 1 and the resin pattern 3 is immersed in a plating tank containing an electroless plating solution, and a metal layer (plating film) is formed on the surface of the transparent substrate 1 and the surface of the resin pattern 3, respectively.
- the thickness of the metal layer is preferably 0.1 to 100 ⁇ m, more preferably 1 to 20 ⁇ m, and particularly preferably 1 to 10 ⁇ m.
- the metal layer may be a single layer or two or more layers.
- unnecessary metal layers such as a metal layer provided on the surface of the resin pattern 3 in the metal layer are removed by polishing or the like to expose the resin pattern.
- the metal used in this embodiment preferably includes at least one of copper, aluminum, nickel, vanadium, titanium, tantalum, chromium, cobalt, gold, silver, and tungsten.
- copper is used.
- One kind or two kinds of metals may be used.
- a plurality of resin patterns 3 and metal layers may be laminated. Specifically, after the metal application step, the step of manufacturing the resin pattern can be repeated again.
- a step of applying the semiconductor element 5 may be further included.
- the metal 4 located on the surface of the transparent substrate and in the gap of the resin pattern, and in contact with the metal.
- the gap between the resin patterns refers to a region between the resin and the resin on the side where the resin pattern is provided on the transparent substrate, and is obtained, for example, by exposing and developing a photosensitive resin. An opening.
- laminating the resin pattern it will be the lower resin pattern and / or the metal surface and the region between the resins.
- a semiconductor chip etc. are illustrated as a semiconductor element here.
- the transparent substrate 1 and the laminate 6 having a resin pattern located on the surface of the transparent substrate are irradiated with radiation from the transparent substrate side of the laminate, and the transparent substrate is peeled from the laminate.
- a peeling process is included.
- the absorbance of the resin pattern at the wavelength of the radiated light to be irradiated is set to 0.5 or more.
- the absorbance is 0.5 or more, preferably 0.8 or more, more preferably 1.0 or more, and further preferably 1.2 or more.
- the upper limit of the absorbance is not particularly high and is preferably as high as possible. However, for example, the effect of the present invention can be sufficiently achieved even with 9 or less, further 6 or less, and particularly 4 or less.
- the wavelength of the emitted light is preferably a wavelength that is transmitted through the transparent substrate and absorbed by the resin pattern, and can be appropriately determined according to the material used.
- the lower limit is preferably 170 nm or more, more preferably 220 nm or more, and further preferably 260 nm or more.
- the upper limit of the wavelength is preferably 4000 nm or less, and more preferably 3000 nm or less.
- the type of laser is not particularly defined, but YAG laser (yttrium, aluminum, garnet laser) THG (Third Harmonic Generation) (355 nm), and among excimer lasers, KrF (248 nm), XeCl (308 nm), XeF ( 351 nm) is preferred.
- the focal point is preferably set at a distance of ⁇ 10 ⁇ m distance between the transparent substrate 1 and the resin pattern 3.
- the laser light source preferably has an area of 100 to 1000000 ⁇ m 2 , a repetition frequency of 10 to 5000 Hz, and a scanning speed of 0.1 to 1000 mm / s.
- the scanning pitch is preferably 1 to 100 ⁇ m.
- the exposure amount is preferably 0.5 to 50 J / cm 2 .
- FIG. 2 is a schematic view showing a second embodiment, in which a seed layer 21 is provided on the surface of the transparent substrate 1, and a photoresist layer 22 is provided on the surface of the seed layer, and exposure and development are performed. After forming the photoresist pattern 23, the metal is applied, the photoresist pattern 23 is removed, the seed layer 21 is deleted, and the metal pattern 24 is produced (metal pattern forming step). Next, the resin pattern 3 is provided between the metal patterns 24 (resin pattern forming step).
- the transparent substrate 1 can be peeled off (transparent substrate peeling step).
- the metal pattern forming step, the resin pattern forming step, the semiconductor element application step, and the transparent substrate peeling step be performed in this order. 2 uses the same reference numerals for members common to FIG. 1 (the same applies to FIG. 3).
- a semiconductor having a semiconductor element for example, a semiconductor chip
- a metal for example, a metal wiring or an electrode
- resin patterns for example, an insulating layer
- the process of forming the metal pattern 24 is included.
- the step of forming the metal pattern preferably includes, for example, a seed layer formation step, a photoresist pattern formation step, a metal application step, a photoresist pattern and a seed layer removal step.
- the seed layer 21 is provided on the surface of the transparent substrate 1.
- the details of the transparent substrate are the same as those in the first embodiment, and the preferred range is also the same.
- the seed layer is preferably formed by sputtering, chemical vapor deposition (CVD), electroless plating, or the like. Examples of the material for the seed layer include tin, silver, and copper.
- the thickness of the seed layer is preferably 0.02 to 2 ⁇ m.
- Photoresist pattern formation process In the second embodiment, a photoresist layer 22 is provided on the seed layer 21, preferably on the surface. Next, the photoresist layer 22 is exposed and developed to form a photoresist pattern 23.
- the details of the photoresist pattern can be referred to the description of Electrology Theory, Vol. 131, No. 1, 2011, the contents of which are incorporated herein.
- the thickness of the photoresist layer is preferably 0.5 to 50 ⁇ m.
- ⁇ Metal application process it is preferable to include a step of applying a metal to the laminate having the transparent substrate 1 and the photoresist pattern 23.
- the metal in the present embodiment is applied to the pattern gap of the photoresist pattern on the transparent substrate 1 of the laminate having the transparent substrate 1 and the photoresist pattern 23.
- the method for applying the metal is not particularly defined, but includes an electroless plating method. The details of the electroless plating method and the metal are the same as those in the metal application step in the first embodiment, and the preferred range is also the same.
- a metal pattern 24 corresponding to the photoresist pattern 23 is provided.
- Photoresist pattern and seed layer removal process After the metal pattern 24 is formed, the photoresist pattern 23 is removed. The removal of the photoresist pattern can be performed by removal using a solvent or removal using a hydroxyamine-based release agent. After removing the photoresist pattern, the seed layer 21 is removed. The seed layer 21 is preferably removed by etching according to the material of the metal pattern 24.
- the resin pattern 23 is provided on the surface of the laminate having the transparent substrate 1 and the metal pattern 24.
- the manufacturing method of a resin pattern is not specifically defined, it is preferable to manufacture by applying a resin composition.
- the details of the development exposure process can be referred to the description of the first embodiment.
- the metal patterns 24 and the resin patterns 3 may be alternately stacked.
- ⁇ Semiconductor element application process> the process of applying a semiconductor element may be further included. These details are the same as those in the semiconductor element application step in the first embodiment.
- the laminate 6 having the transparent substrate 1 and the resin pattern 3 positioned on the surface of the transparent substrate is irradiated with radiation from the transparent substrate side of the laminate, and the transparent substrate is peeled off from the laminate. Including a peeling step. These details are the same as those of the transparent substrate peeling step in the first embodiment.
- FIG. 3 is a schematic view showing a third embodiment, in which a photosensitive resin film 2 is provided on the surface of the transparent substrate 1 (photosensitive resin film manufacturing process), and is exposed and developed to form a resin pattern. 3 is manufactured (exposure development step). Thereafter, the semiconductor element 5 is applied to the side of the resin pattern 3 that is not in contact with the transparent substrate 1 (semiconductor element application process). Thereafter, the transparent substrate 1 can be peeled off by irradiating the radiated light from the transparent substrate side so as to satisfy a predetermined condition (transparent substrate peeling step). After peeling the transparent substrate 1, the metal 4 can be applied from the side where the transparent substrate 1 is peeled (metal application step).
- a photosensitive resin film manufacturing process it is particularly preferable to carry out in the order of a photosensitive resin film manufacturing process, an exposure development process, a semiconductor element application process, a transparent substrate peeling process, and a metal application process.
- a semiconductor device in which the semiconductor element 5 on the surface of the resin pattern (for example, an insulating film) is bonded to the metal (for example, metal wiring or electrode) in the gap of the resin pattern 3 is obtained.
- the third embodiment will be described more specifically.
- ⁇ Curing process> it is preferable to have a hardening process after a development exposure process. It is synonymous with 1st Embodiment, and its preferable range is also the same.
- a step of applying the semiconductor element 5 is further included. Specifically, it is preferably provided on the surface of the resin pattern 3 on the surface opposite to the side in contact with the transparent substrate 1. A semiconductor chip etc. are illustrated as a semiconductor element here.
- the transparent substrate 1 and the laminate 6 having a resin pattern located on the surface of the transparent substrate are irradiated with radiation from the transparent substrate side of the laminate, and the transparent substrate is peeled from the laminate.
- a peeling process is included. These details are the same as those of the transparent substrate peeling step in the first embodiment.
- ⁇ Metal application process> it is further preferable to apply a metal to the gap between the resin patterns 3. Specifically, it is preferable to apply metal from the side where the transparent substrate 1 is peeled off.
- a method for applying metal in the present embodiment printing of metal paste is preferable.
- the thickness of the metal layer preferably corresponds to the thickness of the resin pattern. With such a configuration, the metal layer serves as an electrode.
- the metal used in this embodiment preferably includes at least one selected from copper, aluminum, nickel, vanadium, titanium, tantalum, chromium, cobalt, gold, silver, and tungsten, and printing of a metal paste is performed. In the case, silver is preferred. One kind or two kinds of metals may be used.
- the semiconductor device manufacturing method of the present invention includes the pattern manufacturing method of the present invention.
- ⁇ Laminate 1> A transparent substrate, a resin pattern located on the surface of the transparent substrate, and a metal located on the surface of the transparent substrate and located in the gap of the resin pattern, wherein the resin pattern is selected from polyimide and polybenzoxazole A laminate comprising at least one selected from the above.
- ⁇ Laminate 2> A transparent substrate, a resin pattern located on the surface of the transparent substrate, a metal located on the surface of the transparent substrate and located in a gap between the resin patterns, and a semiconductor element in contact with the metal, the semiconductor A laminate in which the element is in contact with the metal on the side opposite to the transparent substrate side, and the resin pattern includes at least one selected from polyimide and polybenzoxazole.
- the mixture was stirred at a temperature of 60 ° C. for 18 hours to produce a diester of 4,4′-oxydiphthalic acid and 2-hydroxyethyl methacrylate.
- the reaction mixture was then cooled to ⁇ 10 ° C. and 16.12 g (135.5 mmol) of SOCl 2 was added over 10 minutes while maintaining the temperature at ⁇ 10 ⁇ 4 ° C.
- the reaction mixture was diluted with 50 mL of N-methylpyrrolidone, the reaction mixture was stirred at room temperature for 2 hours.
- polyimide precursor Aa-1 The structure of polyimide precursor Aa-1 is shown below.
- ⁇ Preparation of photosensitive resin composition> The following components were mixed to prepare a photosensitive resin composition as a uniform solution.
- Photosensitive resin polyimide precursor (Aa-1) 32 parts by mass polymerizable compound B-1 6.9 parts by mass photopolymerization initiator C-1 1.0 part by mass polymerization inhibitor: parabenzoquinone (manufactured by Tokyo Chemical Industry Co., Ltd.) 0.08 parts by mass migration inhibitor: 1H-tetrazole (manufactured by Tokyo Chemical Industry Co., Ltd.) 0.12 parts by mass Metal adhesion improver: N- [3- (triethoxysilyl) propyl] maleic acid monoamide 0.70 parts by mass Solvent: ⁇ -butyrolactone 48.00 parts by mass Solvent: dimethyl sulfoxide 12.00 parts by mass
- B-1 NK ester A-9300 (manufactured by Shin-Nakamura Chemical Co., Ltd., trifunctional acrylate, the following structure)
- the photosensitive resin composition was filtered under pressure at a pressure of (0.3 MPa) through a filter having a pore width of 0.8 ⁇ m. After filtration, the photosensitive resin composition is applied to the surface of a transparent substrate (a glass substrate having a thickness of 0.6 mm) by spin coating to form a layer, dried on a hot plate at 100 ° C. for 5 minutes, and a thickness of 10 ⁇ m. A uniform photosensitive resin film was obtained.
- the photosensitive resin film side of the laminate composed of the obtained transparent substrate and the photosensitive resin film is covered with a mask (a mask having a hole having a diameter of 10 ⁇ m uniformly in the mask surface, an aperture ratio of 98%), and a stepper (Nikon NSR 2005).
- i9C was exposed at an exposure wavelength of 365 nm (i-line) and an exposure energy of 500 mJ / cm 2 .
- the resin film having the exposed portion and the unexposed portion was spin washed with cyclopentanone for 60 seconds to remove the unexposed portion (negative development). Furthermore, the residue was removed by rinsing with propylene glycol-1-monomethyl ether-2-acetate (PGMEA) to obtain a resin pattern.
- PMEA propylene glycol-1-monomethyl ether-2-acetate
- the resin pattern obtained on the surface of the transparent substrate was a resin pattern having a thickness of 6 ⁇ m and holes having a diameter of about 10 ⁇ m.
- the ratio of the surface area of the removed portion of the photosensitive resin film to the surface area of the entire region of the photosensitive resin film in the present invention is a value calculated according to a value obtained by subtracting the aperture ratio of the mask from 100%.
- an electroless copper plating film was formed by the following method.
- a water-soluble organic solvent 39 parts by mass of diethylene glycol diethyl ether (manufactured by Wako Pure Chemical Industries, Ltd.), 40.75 parts by mass of water, 20 parts by mass of nitric acid (manufactured by Wako Pure Chemical Industries, Ltd.), and palladium acetate (manufactured by Wako Pure Chemical Industries, Ltd.) 0
- a palladium catalyst solution composed of .25 parts by mass was prepared, and the laminate composed of the transparent substrate and the resin pattern obtained above was immersed in this palladium catalyst solution for 5 minutes, and then washed with water.
- electroless plating is performed for 60 minutes at an electroless plating temperature of 26 ° C. using an electroless plating solution having the following composition including Sulcup PGT (PGT-A, PGT-B, PGT-C) manufactured by Uemura Kogyo Co., Ltd. went.
- the thickness of the obtained electroless copper plating film was 3.0 ⁇ m both on the surface of the resin pattern and on the surface of the transparent substrate exposed by exposure and development (bottom of the hole).
- the electroless copper plating film on the surface was polished in order to remove the electroless copper plating film formed on the surface other than the surface (bottom of the hole) of the transparent substrate exposed by the exposure and development.
- the electroless copper plating film was removed with a cleaning liquid (acid) for polishing.
- the excess electroless copper plating film was removed by polishing, and a laminated body in which the through electrode of the electroless copper plating film was formed on the surface of the transparent substrate and in the gap (hole part) of the resin pattern was obtained.
- the laminated body was peeled off from the transparent substrate by irradiation with radiated light by the following method.
- Irradiation conditions Irradiated radiation: 355 nm pulse (YAG-THG) laser (pulse width: 5 ns)
- a fluence of 1.9 J / cm 2 was exposed.
- the absorbance of the resin pattern at the wavelength of the emitted radiation was measured by the following method. Before applying the metal (before electroless plating), select the five portions on the transparent substrate where the resin pattern is not removed, measure the absorbance at the wavelength of the emitted radiation (wavelength 355 nm), and obtain the average value. It was. The absorbance was measured using U-3900 manufactured by Hitachi High-Tech Science.
- Example 1 was performed in the same manner as Example 1 except that the mask was changed to a mask having an aperture ratio of 95%.
- Example 3 Example 1 was performed in the same manner as Example 1 except that the mask was changed to a mask with an aperture ratio of 90%.
- Example 4 Example 1 was performed in the same manner as Example 1 except that the mask was changed to a mask with an aperture ratio of 80%.
- Example 1 was performed in the same manner as Example 1 except that the mask was changed to a mask with an aperture ratio of 70%.
- Example 6 The same operation as in Example 1 was conducted except that the photosensitive resin composition in Example 1 was changed to the following photosensitive resin composition A-2.
- Photosensitive resin polyimide precursor (Aa-1) 32 parts by weight polymerizable compound B-1 6.9 parts by weight photopolymerization initiator C-1 1.0 part by weight polymerization inhibitor: 2,6-di-tert- Butyl-4-methylphenol (manufactured by Tokyo Chemical Industry Co., Ltd.) 0.1 parts by mass migration inhibitor: 1,2,4-triazole (manufactured by Tokyo Chemical Industry Co., Ltd.) 0.1 parts by mass Solvent: ⁇ -butyrolactone 48.00 parts by mass Solvent: dimethyl sulfoxide 12.00 parts by mass
- Example 7 In Example 1, the thickness of the resin pattern obtained on the surface of the transparent substrate was adjusted to adjust the absorbance to 0.55, and the others were performed in the same manner.
- Example 1 The same procedure as in Example 1 was performed to obtain a stacked body to which semiconductor chips were connected. About the obtained laminated body, although peeling of the transparent substrate was tried without irradiating radiation, it was not able to peel.
- Example 2 In Example 1, the thickness of the resin pattern obtained on the surface of the transparent substrate was adjusted to 10 ⁇ m to 3 ⁇ m, and the others were performed in the same manner. However, peeling was not possible.
- Example 3 In Example 1, the thickness of the resin pattern obtained on the surface of the transparent substrate was adjusted to adjust the absorbance to 0.45, and the others were performed in the same manner.
- the transparent substrate could be peeled from the laminate when the absorbance of the resin pattern was 0.5 or more. On the other hand, when the absorbance was less than 0.5, the transparent substrate could not be peeled from the laminate.
Landscapes
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Engineering & Computer Science (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Manufacturing & Machinery (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Computer Hardware Design (AREA)
- Power Engineering (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Exposure And Positioning Against Photoresist Photosensitive Materials (AREA)
- Materials For Photolithography (AREA)
- Photosensitive Polymer And Photoresist Processing (AREA)
Abstract
Description
近年、再配線層の数は増加する傾向にあり、半導体チップとバンプとの間をポリイミドが4層、再配線層が3層といった多層で形成するようなケースも出てきている。
一般に、再配線層の数が増えるほど製造歩留りが低下するが、チップファースト方式では再配線層の不良が発生すると半導体チップが無駄になるのに対し、RDLファースト方式ではチップ接続前に配線層を検査し、良品箇所のみにチップを接続することが可能なため半導体チップは無駄にならない。このため製造コストを低減する手法として、RDLファースト方式への期待が高まっている。
また、特許文献2には、「電気的構造を形成する方法であって、第1の電気コンポーネントを含む第1のデバイスウェハを用意し、第1のデバイスウェハは第1のデバイスウェハの第1の面に第1の電極を有し、上記第1のデバイスウェハの上記第1の面に、第1の接合材料を用いてキャリアウェハを接合し、上記キャリアウェハが上記第1のデバイスウェハに接合されたまま、上記第1のデバイスウェハの第2の面を処理し、レーザーからの光を、上記キャリアウェハを通過させて上記第1の接合材料に突き当たらせ、上記第1の接合材料がビームからのエネルギーを吸収することで上記第1の接合材料が上記キャリアウェハを上記第1のデバイスウェハから実質的に解放し、上記キャリアウェハは上記レーザーからの上記光に対して実質的に透明であり、上記第1のデバイスウェハから上記キャリアウェハを除去し、且つ上記第1のデバイスウェハから、残存する第1の接合材料を除去する、ことを有する方法。」が開示されている。
<1>透明基板と、上記透明基板の表面に位置する樹脂パターンを有する積層体Aに、上記積層体Aの透明基板側から放射光を照射して、上記積層体Aから透明基板を剥離する剥離工程を含み、上記樹脂パターンが、ポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含み、上記照射する放射光の波長における上記樹脂パターンの吸光度が0.5以上である、パターン製造方法。
<2>透明基板と感光性樹脂膜を有する積層体Bの感光性樹脂膜に対して露光し、さらに、現像処理を行って感光性樹脂膜の一部を除去することで上記積層体Aを得ることを含む、<1>に記載のパターン製造方法。
<3>上記感光性樹脂膜の一部を除去して形成された樹脂パターンの、除去された部分の表面積が、感光性樹脂膜の露光された全領域の表面積の20%以下である、<2>に記載のパターン製造方法。
<4>上記露光は、上記積層体Bの感光性樹脂膜側から行う、<2>または<3>に記載のパターン製造方法。
<5>上記積層体Aの、透明基板上であって上記樹脂パターンの間隙または透明基板の上であって透明基板と樹脂パターンの間に、金属を適用する工程を有する、<1>~<4>のいずれかに記載のパターン製造方法。
<6>上記金属が、銅、アルミニウム、ニッケル、バナジウム、チタン、タンタル、クロム、コバルト、金、銀およびタングステンから選択される少なくとも1種を含む、<5>に記載のパターン製造方法。
<7>上記感光性樹脂膜が、ポリイミド前駆体、ポリイミド、ポリベンゾオキサゾール前駆体およびポリベンゾオキサゾールから選択される少なくとも1種を含む、<2>~<6>のいずれかに記載のパターン製造方法。
<8>上記感光性樹脂膜が、ポリイミド前駆体およびポリベンゾオキサゾール前駆体から選択される少なくとも1種を含む、<2>~<6>のいずれかに記載のパターン製造方法。
<9>上記露光現像工程後、剥離工程前に、上記樹脂パターンを硬化する硬化工程を含む、<1>~<8>のいずれかに記載のパターン製造方法。
<10>上記現像がネガ型現像である、<2>~<9>のいずれかに記載のパターン製造方法。
<11><1>~<10>のいずれかに記載のパターン製造方法を含む、半導体装置の製造方法。
<12>透明基板と、上記透明基板の表面に位置する樹脂パターンと、上記透明基板の表面であって、上記樹脂パターンの間隙に位置する金属とを有し、上記樹脂パターンがポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含む積層体。
<13>透明基板と、上記透明基板の表面に位置する樹脂パターンと、上記透明基板の表面であって、上記樹脂パターンの間隙に位置する金属と、上記金属と接している半導体素子を有し、上記半導体素子が、透明基板側とは反対側で上記金属に接しており、上記樹脂パターンがポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含む積層体。
本明細書における基(原子団)の表記に於いて、置換および無置換を記していない表記は、置換基を有さないものと共に置換基を有するものをも包含するものである。例えば、「アルキル基」とは、置換基を有さないアルキル基(無置換アルキル基)のみならず、置換基を有するアルキル基(置換アルキル基)をも包含するものである。
本明細書において「露光」とは、特に断らない限り、光を用いた露光のみならず、電子線、イオンビーム等の粒子線を用いた描画も露光に含める。また、露光に用いられる光としては、一般的に、水銀灯の輝線スペクトル、エキシマレーザーに代表される遠紫外線、極紫外線(EUV光)、X線、電子線等の活性光線または放射線が挙げられる。
本明細書において、「~」を用いて表される数値範囲は、「~」の前後に記載される数値を下限値および上限値として含む範囲を意味する。
本明細書において、「(メタ)アクリレート」は、「アクリレート」および「メタクリレート」の双方、または、いずれかを表し、「(メタ)アリル」は、「アリル」および「メタリル」の双方、または、いずれかを表し、「(メタ)アクリル」は、「アクリル」および「メタクリル」の双方、または、いずれかを表し、「(メタ)アクリロイル」は、「アクリロイル」および「メタクリロイル」の双方、または、いずれかを表す。
本明細書において「工程」との語は、独立した工程だけではなく、他の工程と明確に区別できない場合であってもその工程の所期の作用が達成されれば、本用語に含まれる。
本明細書において、固形分濃度とは、組成物の総質量に対する、溶剤を除く他の成分の質量百分率である。また、固形分濃度は、特に述べない限り25℃における濃度をいう。
本明細書において、重量平均分子量(Mw)および数平均分子量(Mn)は、特に述べない限り、ゲル浸透クロマトグラフィー(GPC測定)に従い、ポリスチレン換算値として定義される。本明細書において、重量平均分子量(Mw)および数平均分子量(Mn)は、例えば、HLC-8220(東ソー(株)製)を用い、カラムとしてガードカラムHZ-L、TSKgel Super HZM-M、TSKgel Super HZ4000、TSKgel Super HZ3000、TSKgel Super HZ2000(東ソー(株)製)を用いることによって求めることができる。溶離液は特に述べない限り、THF(テトラヒドロフラン)を用いて測定したものとする。また、検出は特に述べない限り、UV線(紫外線)の波長254nm検出器を使用したものとする。
特に、本発明のパターン製造方法は、透明基板と樹脂パターンを有する積層体Aを、透明基板と感光性樹脂膜を有する積層体Bに対し、波長500nm以下の光で露光し、さらに、現像処理を行って得る態様に好ましく用いられる。すなわち、透明基板と感光性樹脂膜を有する積層体B(好ましくは、透明基板と上記透明基板の表面に位置する感光性樹脂膜を有する積層体)を露光現像して、感光性樹脂膜の感光性を喪失させることによって、樹脂パターンを形成した場合であっても、所定の条件を満たすように、放射光を照射することにより、樹脂パターンにダメージを与えることなく、透明基板と樹脂パターンを有する積層体Aから透明基板を適切に剥離できる。
さらに、上記露光現像工程の後、透明基板1上(好ましくは表面)であって樹脂パターン3の間隙または透明基板1の上であって透明基板1と樹脂パターン3の間に、金属4を適用することができる(金属適用工程)。さらにまた、金属4に接するように半導体素子5を適用することができる(半導体素子適用工程)。
本実施形態では、特に、感光性樹脂膜製造工程、露光現像工程、金属適用工程、半導体素子適用工程、透明基板剥離工程の順に行うことが好ましい。
第1の実施形態では、樹脂パターン(例えば、絶縁層)の間隙に金属(例えば、金属配線や電極)が位置し、さらに、上記金属と接している半導体素子(例えば、半導体チップ)を有する半導体装置が得られる。以下、第1の実施形態をより具体的に説明する。
感光性樹脂膜2は、透明基板1の上、好ましくは表面に、感光性樹脂組成物を層状に適用することによって得られる。さらに、感光性樹脂組成物が溶剤を含む場合、感光性樹脂組成物を層状に適用後、乾燥することが好ましい。
また、感光性樹脂膜の厚さは、0.1~100μmが好ましく、1~50μmがより好ましく、3~20μmがさらに好ましい。
透明基板は、放射光を透過する基板であれば、その種類等は特に定めるものではなく、ガラス、石英、液晶ポリマー等が例示され、ガラスが好ましい。
また、透明基板は、耐熱性が高いことが好ましく、Tgが150℃以上であることが好ましく、250℃以上であることがより好ましい。
透明基板の厚さは、特に定めるものではないが、通常、10~1000μmである。
感光性樹脂組成物を透明基板に層状に適用する手段としては、塗布が好ましい。
具体的には、適用する手段としては、ディップコート法、エアーナイフコート法、カーテンコート法、ワイヤーバーコート法、グラビアコート法、エクストルージョンコート法、スプレーコート法、スピンコート法、スリットコート法、およびインクジェット法などが例示される。層状の感光性樹脂組成物(以下、「感光性樹脂組成物層」ということがある)の厚さの均一性の観点から、スピンコート法、スリットコート法、スプレーコート法、インクジェット法がより好ましい。適用する手段に応じて適切な固形分濃度や塗布条件を調整することで、所望の感光性樹脂組成物層を得ることができる。また、基板の形状によっても塗布方法を適宜選択でき、ウェハ等の円形基板であればスピンコート法やスプレーコート法、インクジェット法等が好ましく、矩形基板であればスリットコート法やスプレーコート法、インクジェット法等が好ましい。スピンコート法の場合は、例えば、500~2000rpm(revolutions per minute)の回転数で、10秒間~1分間程度で適用することができる。
尚、感光性樹脂組成物は、その適用前に、濾過を行ってもよい。濾過は、フィルターを用いて行うことが好ましい。フィルター孔径としては、1μm以下が好ましく、0.5μm以下がより好ましく、0.1μm以下がさらに好ましい。
本実施形態における感光性樹脂膜を構成する感光性樹脂組成物は露光により架橋構造を形成する化合物を含むことが好ましく、組成物はネガ型現像用であることがより好ましく、有機溶剤で現像するネガ型感光性樹脂であることがさらに好ましい。
まず、本実施形態で用いる感光性樹脂組成物が含んでもよい成分について説明する。感光性樹脂組成物はこれら以外の成分を含んでいてもよく、また、これらの成分を必須とするわけではない。
本実施形態で用いる感光性樹脂組成物は、樹脂を含む。
本実施形態で用いる感光性樹脂組成物では、樹脂は特に制限は無く、公知の樹脂を用いることができる。樹脂は高耐熱性樹脂であることが好ましい。
本実施形態で用いる感光性樹脂組成物における、樹脂の含有量は、感光性樹脂組成物の全固形分の10~99質量%が好ましく、50~98質量%がより好ましく、70~96質量%がさらに好ましい。
また、感光性樹脂組成物が重合性化合物を含むことが好ましい。このような構成とすることにより、露光部に3次元ネットワークが形成され、強固な架橋膜となり、後述の表面活性化処理により感光性樹脂組成物層(硬化膜)がダメージを受けず、表面活性化処理により、硬化膜と金属層の密着性または硬化膜同士の密着性がより効果的に向上する。
さらにまた、樹脂が、-Ar-L-Ar-で表される部分構造を含むことが好ましい。但し、Arは、それぞれ独立に、芳香族基であり、Lは、フッ素原子で置換されていてもよい炭素数1~10の脂肪族炭化水素基、-O-、-CO-、-S-、-SO2-または-NHCO-、ならびに、上記の2つ以上の組み合わせからなる基である。このような構成とすることにより、感光性樹脂組成物層(硬化膜)が柔軟な構造となり、層間剥離の発生を抑制する効果がより効果的に発揮される。Arは、フェニレン基が好ましく、Lは、フッ素原子で置換されていてもよい炭素数1または2の脂肪族炭化水素基、-O-、-CO-、-S-または-SO2-がさらに好ましい。ここでの脂肪族炭化水素基は、アルキレン基が好ましい。
ポリイミド前駆体は、その種類等について特に定めるものではなく、下記式(2)で表される繰り返し単位を含むことが好ましい。
式(2)

式(2)におけるR111は、2価の有機基を表す。2価の有機基としては、直鎖または分岐の脂肪族基、環状の脂肪族基および芳香族基を含む基が例示され、炭素数2~20の直鎖または分岐の脂肪族基、炭素数6~20の環状の脂肪族基、炭素数6~20の芳香族基、または、これらの組み合わせからなる基が好ましく、炭素数6~20の芳香族基を含む基がより好ましい。特に好ましい実施形態として、式(2)におけるR111が-Ar-L-Ar-で表される基であることが例示される。但し、Arは、それぞれ独立に、芳香族基であり、Lは、フッ素原子で置換されていてもよい炭素数1~10の脂肪族炭化水素基、-O-、-CO-、-S-、-SO2-または-NHCO-、ならびに、上記の2つ以上の組み合わせからなる基である。これらの好ましい範囲は、上述のとおりである。
具体的には、炭素数2~20の直鎖または分岐の脂肪族基、炭素数6~20の環状の脂肪族基、炭素数6~20の芳香族基、または、これらの組み合わせからなる基を含むジアミンであることが好ましく、炭素数2~60の芳香族基からなる基を含むジアミンであることがより好ましい。芳香族基の例としては、下記の芳香族基が挙げられる。



ジェファーミン(登録商標)KH-511、ジェファーミン(登録商標)ED-600、ジェファーミン(登録商標)ED-900、ジェファーミン(登録商標)ED-2003、ジェファーミン(登録商標)EDR-148、ジェファーミン(登録商標)EDR-176の構造を以下に示す。

式(51)

R10~R17の1価の有機基として、炭素数1~10(好ましくは炭素数1~6)の無置換のアルキル基、炭素数1~10(好ましくは炭素数1~6)のフッ化アルキル基等が挙げられる。
式(61)

式(51)または(61)の構造を与えるジアミン化合物としては、2,2’-ジメチルベンジジン、2,2’-ビス(トリフルオロメチル)-4,4’-ジアミノビフェニル、2,2’-ビス(フルオロ)-4,4’-ジアミノビフェニル、4,4’-ジアミノオクタフルオロビフェニル等が挙げられる。これらは1種を用いるか、または2種以上を組み合わせて用いてもよい。
式(5)


テトラカルボン酸二無水物は、下記式(O)で表されることが好ましい。
式(O)


式(2)におけるR113およびR114の少なくとも一方が重合性基を含むことが好ましく、両方が重合性基を含むことがより好ましい。重合性基とは、熱、ラジカル等の作用により、架橋反応することが可能な基である。重合性基としては、光ラジカル重合性基が好ましい。重合性基の具体例として、エチレン性不飽和結合を有する基、アルコキシメチル基、ヒドロキシメチル基、アシルオキシメチル基、エポキシ基、オキセタニル基、ベンゾオキサゾリル基、ブロックイソシアネート基、メチロール基、アミノ基が挙げられる。ポリイミド前駆体が有するラジカル重合性基としては、エチレン性不飽和結合を有する基が好ましい。
エチレン性不飽和結合を有する基としては、ビニル基、(メタ)アリル基、下記式(III)で表される基などが挙げられる。

式(III)において、R201は、炭素数2~12のアルキレン基、-CH2CH(OH)CH2-または炭素数4~30のポリオキシアルキレン基を表す。
好適なR201の例は、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、1,2-ブタンジイル基、1,3-ブタンジイル基、ペンタメチレン基、ヘキサメチレン基、オクタメチレン基、ドデカメチレン基、-CH2CH(OH)CH2-が挙げられ、エチレン基、プロピレン基、トリメチレン基、-CH2CH(OH)CH2-がより好ましい。
特に好ましくは、R200がメチル基で、R201がエチレン基である。
有機溶剤への溶解度の観点からは、式(2)におけるR113またはR114は、1価の有機基であることが好ましい。1価の有機基としては、直鎖または分岐のアルキル基、環状アルキル基、あるいは、芳香族基を含むことが好ましい。1価の有機基としては、アリール基を構成する炭素に結合している1、2または3つ(好ましくは1つ)の酸性基を有する芳香族基、およびアリール基を構成する炭素に結合している1、2または3つ(好ましくは1つ)の酸性基を有するアラルキル基が特に好ましい。具体的には、酸性基を有する炭素数6~20の芳香族基、酸性基を有する炭素数7~25のアラルキル基が挙げられる。より具体的には、酸性基を有するフェニル基および酸性基を有するベンジル基が挙げられる。酸性基は、ヒドロキシル基が好ましい。
R113またはR114が、水素原子、2-ヒドロキシベンジル、3-ヒドロキシベンジルおよび4-ヒドロキシベンジルであることが、より特に好ましい。
式(2)におけるR113またはR114が表す環状のアルキル基は、単環の環状のアルキル基であってもよく、多環の環状のアルキル基であってもよい。単環の環状のアルキル基としては、例えば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基およびシクロオクチル基が挙げられる。多環の環状のアルキル基としては、例えば、アダマンチル基、ノルボルニル基、ボルニル基、カンフェニル基、デカヒドロナフチル基、トリシクロデカニル基、テトラシクロデカニル基、カンホロイル基、ジシクロヘキシル基およびピネニル基が挙げられる。中でも、高感度化との両立の観点から、シクロヘキシル基が最も好ましい。また、芳香族基で置換されたアルキル基としては、後述する芳香族基で置換された直鎖アルキル基が好ましい。
式(2)におけるR113またはR114が表す芳香族基としては、具体的には、置換または無置換のベンゼン環、ナフタレン環、ペンタレン環、インデン環、アズレン環、ヘプタレン環、インダセン環、ペリレン環、ペンタセン環、アセナフテン環、フェナントレン環、アントラセン環、ナフタセン環、クリセン環、トリフェニレン環、フルオレン環、ビフェニル環、ピロール環、フラン環、チオフェン環、イミダゾール環、オキサゾール環、チアゾール環、ピリジン環、ピラジン環、ピリミジン環、ピリダジン環、インドリジン環、インドール環、ベンゾフラン環、ベンゾチオフェン環、イソベンゾフラン環、キノリジン環、キノリン環、フタラジン環、ナフチリジン環、キノキサリン環、キノキサゾリン環、イソキノリン環、カルバゾール環、フェナントリジン環、アクリジン環、フェナントロリン環、チアントレン環、クロメン環、キサンテン環、フェノキサチイン環、フェノチアジン環またはフェナジン環である。ベンゼン環が最も好ましい。
式(2-A)

式(2-A)におけるR112は、式(5)におけるR112と同義であり、好ましい範囲も同様である。
ポリイミド前駆体の製造方法では、反応に際し、有機溶剤を用いることが好ましい。有機溶剤は1種でもよいし、2種以上でもよい。
有機溶剤としては、原料に応じて適宜定めることができるが、ピリジン、ジエチレングリコールジメチルエーテル(ジグリム)、N-メチルピロリドンおよびN-エチルピロリドンが例示される。
その後、ポリイミド前駆体を乾燥して、粉末状のポリイミド前駆体を得ることができる。
上記ポリイミド前駆体の分散度は、2.5以上が好ましく、2.7以上がより好ましく、2.8以上であることがさらに好ましい。ポリイミド前駆体の分散度の上限値は特に定めるものではないが、例えば、4.5以下が好ましく、4.0以下がより好ましく、3.8以下がさらに好ましく、3.2以下が一層好ましく、3.1以下がより一層好ましく、3.0以下がさらに一層好ましく、2.95以下が特に一層好ましい。
ポリイミドとしては、イミド環を有する高分子化合物であれば、特に限定されない。ポリイミドは、下記式(4)で表される化合物であることが好ましく、式(4)で表される化合物であって、重合性基を有する化合物であることがより好ましい。
式(4)

ポリイミドが重合性基を有する場合、R131およびR132の少なくとも一方に重合性基を有していてもよいし、下記式(4-1)または式(4-2)に示すようにポリイミドの末端に重合性基を有していてもよい。


また、R131としては、ジアミンのアミノ基の除去後に残存するジアミン残基が挙げられる。ジアミンとしては、脂肪族、環式脂肪族または芳香族ジアミンなどが挙げられる。具体的な例としては、ポリイミド前駆体の式(2)中のR111の例が挙げられる。
例えば、式(2)におけるR115として例示される下記構造の4価の有機基の4つの結合子が、上記式(4)中の4つの-C(=O)-の部分と結合して縮合環を形成する。

ポリイミドは、ポリイミド前駆体を得、これを、既知のイミド化反応法を用いて完全イミド化させる方法、または、途中でイミド化反応を停止し、一部イミド構造を導入する方法、さらには、完全イミド化したポリマーと、そのポリイミド前駆体をブレンドすることによって、一部イミド構造を導入する方法を利用して合成することができる。
ポリベンゾオキサゾール前駆体は、その種類等について特に定めるものではなく、下記式(3)で表される繰り返し単位を含むことが好ましい。
式(3)

脂肪族基を含むジカルボン酸としては、直鎖または分岐(好ましくは直鎖)の脂肪族基を含むジカルボン酸が好ましく、直鎖または分岐(好ましくは直鎖)の脂肪族基と2つのCOOHからなるジカルボン酸がより好ましい。直鎖または分岐(好ましくは直鎖)の脂肪族基の炭素数は、2~30であることが好ましく、2~25であることがより好ましく、3~20であることがさらに好ましく、4~15であることが特に好ましく、5~10であることが一層好ましい。直鎖の脂肪族基はアルキレン基であることが好ましい。
直鎖の脂肪族基を含むジカルボン酸としては、マロン酸、ジメチルマロン酸、エチルマロン酸、イソプロピルマロン酸、ジ-n-ブチルマロン酸、スクシン酸、テトラフルオロスクシン酸、メチルスクシン酸、2,2-ジメチルスクシン酸、2,3-ジメチルスクシン酸、ジメチルメチルスクシン酸、グルタル酸、ヘキサフルオログルタル酸、2-メチルグルタル酸、3-メチルグルタル酸、2,2-ジメチルグルタル酸、3,3-ジメチルグルタル酸、3-エチル-3-メチルグルタル酸、アジピン酸、オクタフルオロアジピン酸、3-メチルアジピン酸、ピメリン酸、2,2,6,6-テトラメチルピメリン酸、スベリン酸、ドデカフルオロスベリン酸、アゼライン酸、セバシン酸、ヘキサデカフルオロセバシン酸、1,9-ノナン二酸、ドデカン二酸、トリデカン二酸、テトラデカン二酸、ペンタデカン二酸、ヘキサデカン二酸、ヘプタデカン二酸、オクタデカン二酸、ノナデカン二酸、エイコサン二酸、ヘンエイコサン二酸、ドコサン二酸、トリコサン二酸、テトラコサン二酸、ペンタコサン二酸、ヘキサコサン二酸、ヘプタコサン二酸、オクタコサン二酸、ノナコサン二酸、トリアコンタン二酸、ヘントリアコンタン二酸、ドトリアコンタン二酸、ジグリコール酸、さらに下記式で表されるジカルボン酸等が挙げられる。


式(3)におけるR122は、また、ビスアミノフェノール誘導体由来の基であることが好ましい。ビスアミノフェノール誘導体由来の基としては、例えば、3,3’-ジアミノ-4,4’-ジヒドロキシビフェニル、4,4’-ジアミノ-3,3’-ジヒドロキシビフェニル、3,3’-ジアミノ-4,4’-ジヒドロキシジフェニルスルホン、4,4’-ジアミノ-3,3’-ジヒドロキシジフェニルスルホン、ビス-(3-アミノ-4-ヒドロキシフェニル)メタン、2,2-ビス(3-アミノ-4-ヒドロキシフェニル)プロパン、2,2-ビス-(3-アミノ-4-ヒドロキシフェニル)ヘキサフルオロプロパン、2,2-ビス-(4-アミノ-3-ヒドロキシフェニル)ヘキサフルオロプロパン、ビス-(4-アミノ-3-ヒドロキシフェニル)メタン、2,2-ビス-(4-アミノ-3-ヒドロキシフェニル)プロパン、4,4’-ジアミノ-3,3’-ジヒドロキシベンゾフェノン、3,3’-ジアミノ-4,4’-ジヒドロキシベンゾフェノン、4,4’-ジアミノ-3,3’-ジヒドロキシジフェニルエーテル、3,3’-ジアミノ-4,4’-ジヒドロキシジフェニルエーテル、1,4-ジアミノ-2,5-ジヒドロキシベンゼン、1,3-ジアミノ-2,4-ジヒドロキシベンゼン、1,3-ジアミノ-4,6-ジヒドロキシベンゼンなどが挙げられる。これらのビスアミノフェノールは単独にて、あるいは混合して使用してもよい。


式(A-s)中、R2は水素原子、アルキル基、アルコキシ基、アシルオキシ基、環状のアルキル基のいずれかであり、同一でも異なっても良い。
式(A-s)中、R3は水素原子、直鎖または分岐のアルキル基、アルコキシ基、アシルオキシ基、環状のアルキル基のいずれかであり、同一でも異なっても良い。

ポリベンゾオキサゾール前駆体の閉環に伴う基板の反りの発生を抑制できる点で、ポリベンゾオキサゾール前駆体は下記式(SL)で表されるジアミン残基を他の種類の繰り返し単位として含むことが好ましい。

また、式(SL)で表されるジアミン残基の分子量は、400~4,000であることが好ましく、500~3,000がより好ましい。式(SL)で表されるジアミン残基の分子量を上記範囲とすることで、より効果的に、ポリベンゾオキサゾール前駆体の脱水閉環後の弾性率を下げ、基板の反りを抑制できる効果と溶剤溶解性を向上させる効果を両立することができる。
上記ポリベンゾオキサゾール前駆体の分散度は、1.4以上であることが好ましく、1.5以上がより好ましく、1.6以上であることがさらに好ましい。ポリベンゾオキサゾール前駆体の分散度の上限値は特に定めるものではないが、例えば、2.6以下が好ましく、2.5以下がより好ましく、2.4以下がさらに好ましく、2.3以下が一層好ましく、2.2以下がより一層好ましい。
ポリベンゾオキサゾールとしては、ベンゾオキサゾール環を有する高分子化合物であれば、特に限定されない。ポリベンゾオキサゾールは、下記式(X)で表される化合物であることが好ましく、下記式(X)で表される化合物であって、重合性基を有する化合物であることがより好ましい。

重合性基を有する場合、R133およびR134の少なくとも一方に重合性基を有していてもよいし、下記式(X-1)または式(X-2)に示すようにポリベンゾオキサゾールの末端に重合性基を有していてもよい。


例えば、R122として例示される4価の有機基の4つの結合子が、上記式(X)中の窒素原子、酸素原子と結合して縮合環を形成する。例えば、R134が、下記有機基である場合、下記構造を形成する。

なお、ジカルボン酸の場合には反応収率等を高めるため、1-ヒドロキシ-1,2,3-ベンゾトリアゾール等を予め反応させた活性エステル型のジカルボン酸誘導体を用いてもよい。
上記以外の感光性樹脂であっても本実施形態を適用することが可能である。そのほかの樹脂としては、エポキシ樹脂やフェノール樹脂、ベンゾシクロブテン系樹脂が使用可能である。
樹脂が重合性基を有するか、感光性樹脂組成物が重合性化合物を含むことが好ましい。このような構成とすることにより、より耐熱性に優れた硬化膜を形成することができる。
重合性化合物は、重合性基を有する化合物であって、ラジカル、酸、塩基などにより架橋反応が可能な公知の化合物を用いることができる。重合性基としては、上記ポリイミド前駆体で述べた重合性基などが例示される。重合性化合物は1種のみ含まれていてもよいし、2種以上含まれていてもよい。
重合性化合物は、例えば、モノマー、プレポリマー、オリゴマーまたはそれらの混合物並びにそれらの多量体などの化学的形態のいずれであってもよい。
また、オリゴマータイプの重合性化合物は、典型的には比較的低い分子量の重合体であり、10個から100個の重合性モノマーが結合した重合体であることが好ましい。オリゴマータイプの重合性化合物の重量平均分子量は、2000~20000であることが好ましく、2000~15000がより好ましく、2000~10000であることが最も好ましい。
感光性樹脂組成物は、現像性の観点から、重合性基を2個以上含む2官能以上の重合性化合物を少なくとも1種含むことが好ましく、3官能以上の重合性化合物を少なくとも1種含むことがより好ましい。
感光性樹脂組成物は、三次元架橋構造を形成して耐熱性を向上できるという点から、3官能以上の重合性化合物を少なくとも1種含むことが好ましい。また、2官能以下の重合性化合物と3官能以上の重合性化合物との混合物であってもよい。
エチレン性不飽和結合を有する基としては、スチリル基、ビニル基、(メタ)アクリロイル基および(メタ)アリル基が好ましく、(メタ)アクリロイル基がより好ましい。
CH2=C(R4)COOCH2CH(R5)OH
(ただし、R4およびR5は、HまたはCH3を示す。)
また、特開昭51-37193号公報、特公平2-32293号公報、特公平2-16765号公報に記載されているようなウレタンアクリレート類や、特公昭58-49860号公報、特公昭56-17654号公報、特公昭62-39417号公報、特公昭62-39418号公報に記載のエチレンオキサイド系骨格を有するウレタン化合物類も好適である。
また、その他の好ましいエチレン性不飽和結合を有する基を含む化合物として、特開2010-160418号公報、特開2010-129825号公報、特許第4364216号等に記載される、フルオレン環を有し、エチレン性不飽和結合を有する基を2個以上有する化合物や、カルド樹脂も使用することが可能である。
さらに、その他の例としては、特公昭46-43946号公報、特公平1-40337号公報、特公平1-40336号公報に記載の特定の不飽和化合物や、特開平2-25493号公報に記載のビニルホスホン酸系化合物等もあげることができる。また、ある場合には、特開昭61-22048号公報に記載のペルフルオロアルキル基を含む構造が好適に使用される。さらに日本接着協会誌 vol.20、No.7、300~308ページ(1984年)に光重合性モノマーおよびオリゴマーとして紹介されているものも使用することができる。


上記式(MO-1)~(MO-5)で表される重合性化合物の各々において、複数のRの内の少なくとも1つは、-OC(=O)CH=CH2、または、-OC(=O)C(CH3)=CH2で表される基を表す。
上記式(MO-1)~(MO-5)で表される、エチレン性不飽和結合を有する基を含む化合物の具体例としては、特開2007-269779号公報の段落番号0248~0251に記載されている化合物を本実施形態においても好適に用いることができる。
また、上記式(MO-1)、式(MO-2)のペンタエリスリトール誘導体および/またはジペンタエリスリトール誘導体も好ましい例として挙げられる。
酸基を有する多官能モノマーは、1種を単独で用いてもよいが、2種以上を混合して用いてもよい。また、必要に応じて酸基を有しない多官能モノマーと酸基を有する多官能モノマーを併用してもよい。
酸基を有する多官能モノマーの好ましい酸価は、0.1~40mgKOH/gであり、特に好ましくは5~30mgKOH/gである。多官能モノマーの酸価が上記範囲であれば、製造や取扱性に優れ、さらには、現像性に優れる。また、重合性が良好である。
また、樹脂とエチレン性不飽和結合を有する基を含む化合物との質量割合(樹脂/重合性化合物)は、98/2~10/90が好ましく、95/5~30/70がより好ましく、90/10~50/50が最も好ましい。樹脂とエチレン性不飽和結合を有する基を含む化合物との質量割合が上記範囲であれば、重合性および耐熱性により優れた硬化膜を形成できる。
ヒドロキシメチル基、アルコキシメチル基またはアシルオキシメチル基を有する化合物としては、下記式(AM1)で示される化合物が好ましい。





エポキシ化合物としては、一分子中にエポキシ基を2以上有する化合物であることが好ましい。エポキシ基は、200℃以下で架橋反応し、かつ、架橋に由来する脱水反応が起こらないため膜収縮が起きにくい。このため、エポキシ化合物を含有することは、組成物の低温硬化および反りの抑制に効果的である。
オキセタン化合物としては、一分子中にオキセタン環を2つ以上有する化合物、3-エチル-3-ヒドロキシメチルオキセタン、1,4-ビス{[(3-エチル-3-オキセタニル)メトキシ]メチル}ベンゼン、3-エチル-3-(2-エチルヘキシルメチル)オキセタン、1,4-ベンゼンジカルボン酸-ビス[(3-エチル-3-オキセタニル)メチル]エステル等を挙げることができる。具体的な例としては、東亞合成株式会社製のアロンオキセタンシリーズ(例えば、OXT-121、OXT-221、OXT-191、OXT-223)が好適に使用することができ、これらは単独で、あるいは2種以上混合してもよい。
ベンゾオキサジン化合物は、開環付加反応に由来する架橋反応のため、キュア時に脱ガスが発生せず、さらに熱収縮を小さくして反りの発生が抑えられることから好ましい。
感光性樹脂組成物は、光重合開始剤を含有しても良い。特に、感光性樹脂組成物が光ラジカル重合開始剤を含むことにより、感光性樹脂組成物を半導体ウェハなどの基板に適用して感光性樹脂組成物層を形成した後、光を照射することで、ラジカルに起因する硬化が起こり、光照射部における溶解性を低下させることができる。このため、例えば、電極部のみをマスクするパターンを持つフォトマスクを介して感光性樹脂組成物層を露光することで、電極のパターンにしたがって、溶解性の異なる領域を簡便に作製できるという利点がある。
光重合開始剤は、約300~800nm(好ましくは330~500nm)の範囲内に少なくとも約50のモル吸光係数を有する化合物を、少なくとも1種含有していることが好ましい。化合物のモル吸光係数は、公知の方法を用いて測定することができる。具体的には、例えば、紫外可視分光光度計(Varian社製Cary-5 spectrophotometer)にて、酢酸エチル溶剤を用い、0.01g/Lの濃度で測定することが好ましい。
市販品では、カヤキュアーDETX(日本化薬(株)製)も好適に用いられる。
ヒドロキシアセトフェノン系開始剤としては、IRGACURE-184(IRGACUREは登録商標)、DAROCUR-1173、IRGACURE-500、IRGACURE-2959、IRGACURE-127(商品名:いずれもBASF社製)を用いることができる。
アミノアセトフェノン系開始剤としては、市販品であるIRGACURE-907、IRGACURE-369、および、IRGACURE-379(商品名:いずれもBASF社製)を用いることができる。
アミノアセトフェノン系開始剤として、365nmまたは405nm等の波長に極大吸収波長がマッチングされた特開2009-191179号公報に記載の化合物も用いることができる。
アシルホスフィン系開始剤としては、2,4,6-トリメチルベンゾイル-ジフェニル-ホスフィンオキサイドなどが挙げられる。また、市販品であるIRGACURE-819やIRGACURE-TPO(商品名:いずれもBASF社製)を用いることができる。
メタロセン化合物としては、IRGACURE-784(BASF社製)などが例示される。
オキシム化合物の具体例としては、特開2001-233842号公報に記載の化合物、特開2000-80068号公報に記載の化合物、特開2006-342166号公報に記載の化合物を用いることができる。
好ましいオキシム化合物としては、例えば、下記の構造の化合物や、3-ベンゾオキシイミノブタン-2-オン、3-アセトキシイミノブタン-2-オン、3-プロピオニルオキシイミノブタン-2-オン、2-アセトキシイミノペンタン-3-オン、2-アセトキシイミノ-1-フェニルプロパン-1-オン、2-ベンゾイルオキシイミノ-1-フェニルプロパン-1-オン、3-(4-トルエンスルホニルオキシ)イミノブタン-2-オン、および2-エトキシカルボニルオキシイミノ-1-フェニルプロパン-1-オンなどが挙げられる。

市販品ではIRGACURE OXE 01、IRGACURE OXE 02、IRGACURE OXE 03、IRGACURE OXE 04(以上、BASF社製)、アデカオプトマーN-1919((株)ADEKA製、特開2012-14052号公報に記載の光重合開始剤2)も好適に用いられる。また、TR-PBG-304(常州強力電子新材料有限公司製)、アデカアークルズNCI-831およびアデカアークルズNCI-930(ADEKA社製)も用いることができる。また、DFI-091(ダイトーケミックス株式会社製)も用いることができる。
また、特開2007-231000号公報、および、特開2007-322744号公報に記載される環状オキシム化合物も好適に用いることができる。環状オキシム化合物の中でも、特に特開2010-32985号公報、特開2010-185072号公報に記載されるカルバゾール色素に縮環した環状オキシム化合物は、高い光吸収性を有し高感度化の観点から好ましい。
また、オキシム化合物の特定部位に不飽和結合を有する化合物である、特開2009-242469号公報に記載の化合物も好適に使用することができる。
さらに、また、フッ素原子を有するオキシム化合物を用いることも可能である。そのようなオキシム化合物の具体例としては、特開2010-262028号公報に記載されている化合物、特表2014-500852号公報の0345段落に記載されている化合物24、36~40、特開2013-164471号公報の0101段落に記載されている化合物(C-3)などが挙げられる。
最も好ましいオキシム化合物としては、特開2007-269779号公報に示される特定置換基を有するオキシム化合物や、特開2009-191061号公報に示されるチオアリール基を有するオキシム化合物などが挙げられる。
さらに好ましい光重合開始剤は、トリハロメチルトリアジン化合物、α-アミノケトン化合物、アシルホスフィン化合物、フォスフィンオキサイド化合物、メタロセン化合物、オキシム化合物、トリアリールイミダゾールダイマー、オニウム塩化合物、ベンゾフェノン化合物、アセトフェノン化合物であり、トリハロメチルトリアジン化合物、α-アミノケトン化合物、オキシム化合物、トリアリールイミダゾールダイマー、ベンゾフェノン化合物からなる群より選ばれる少なくとも1種の化合物が一層好ましく、メタロセン化合物またはオキシム化合物を用いるのがより一層好ましく、オキシム化合物が特に好ましい。
また、光重合開始剤は、ベンゾフェノン、N,N′-テトラメチル-4,4′-ジアミノベンゾフェノン(ミヒラーケトン)等のN,N′-テトラアルキル-4,4′-ジアミノベンゾフェノン、2-ベンジル-2-ジメチルアミノ-1-(4-モルホリノフェニル)-ブタノン-1、2-メチル-1-[4-(メチルチオ)フェニル]-2-モルフォリノ-プロパノン-1等の芳香族ケトン、アルキルアントラキノン等の芳香環と縮環したキノン類、ベンゾインアルキルエーテル等のベンゾインエーテル化合物、ベンゾイン、アルキルベンゾイン等のベンゾイン化合物、ベンジルジメチルケタール等のベンジル誘導体などを用いることもできる。
また、下記式(I)で表される化合物を用いることもできる。


光重合開始剤は1種のみでもよいし、2種以上であってもよい。光重合開始剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
感光性樹脂組成物は、さらにマイグレーション抑制剤を含むことが好ましい。マイグレーション抑制剤を含むことにより、金属層(金属配線)由来の金属イオンが感光性樹脂組成物層内へ移動することを効果的に抑制可能となる。
マイグレーション抑制剤としては、特に制限はないが、複素環(ピロール環、フラン環、チオフェン環、イミダゾール環、オキサゾール環、チアゾール環、ピラゾール環、イソオキサゾール環、イソチアゾール環、テトラゾール環、ピリジン環、ピリダジン環、ピリミジン環、ピラジン環、ピペリジン環、ピペラジン環、モルホリン環、2H-ピラン環および6H-ピラン環、トリアジン環)を有する化合物、チオ尿素類およびメルカプト基を有する化合物、ヒンダードフェノール系化合物、サリチル酸誘導体系化合物、ヒドラジド誘導体系化合物が挙げられる。特に、トリアゾール、ベンゾトリアゾール等のトリアゾール系化合物、テトラゾール、ベンゾテトラゾール等のテトラゾール系化合物が好ましく使用できる。
マイグレーション抑制剤は1種のみでもよいし、2種以上であってもよい。マイグレーション抑制剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物は、重合禁止剤を含むことが好ましい。
重合禁止剤としては、例えば、ヒドロキノン、パラメトキシフェノール、ジ-tert-ブチル-パラクレゾール、ピロガロール、パラ-tert-ブチルカテコール、パラベンゾキノン、ジフェニル-パラベンゾキノン、4,4′-チオビス(3-メチル-6-tert-ブチルフェノール)、2,2′-メチレンビス(4-メチル-6-tert-ブチルフェノール)、N-ニトロソ-N-フェニルヒドロキシアミンアルミニウム塩、フェノチアジン、N-ニトロソジフェニルアミン、N-フェニルナフチルアミン、エチレンジアミン四酢酸、1,2-シクロヘキサンジアミン四酢酸、グリコールエーテルジアミン四酢酸、2,6-ジ-tert-ブチル-4-メチルフェノール、5-ニトロソ-8-ヒドロキシキノリン、1-ニトロソ-2-ナフトール、2-ニトロソ-1-ナフトール、2-ニトロソ-5-(N-エチル-N-スルフォプロピルアミノ)フェノール、N-ニトロソ-N-(1-ナフチル)ヒドロキシアミンアンモニウム塩、ビス(4-ヒドロキシ-3,5-tert-ブチル)フェニルメタンなどが好適に用いられる。また、特開2015-127817号公報の段落0060に記載の重合禁止剤、および、国際公開WO2015/125469号の段落0031~0046に記載の化合物を用いることもできる。
組成物が重合禁止剤を有する場合、重合禁止剤の含有量は、感光性樹脂組成物の全固形分に対して、0.01~5質量%が好ましい。
重合禁止剤は1種のみでもよいし、2種以上であってもよい。重合禁止剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物は、熱塩基発生剤を含んでいてもよい。
熱塩基発生剤としては、その種類等は特に定めるものではないが、40℃以上に加熱すると塩基を発生する酸性化合物、および、pKa1が0~4のアニオンとアンモニウムカチオンとを有するアンモニウム塩から選ばれる少なくとも1種を含む熱塩基発生剤を含むことが好ましい。ここで、pKa1とは、酸の第一のプロトンの解離定数(Ka)の対数(-Log10Ka)を表し、詳細は後述する。
このような化合物を配合することにより、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などの環化反応を低温で行うことができ、また、より安定性に優れた組成物とすることができる。また、熱塩基発生剤は、加熱しなければ塩基を発生しないので、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などと共存させても、保存中におけるポリイミド前駆体およびポリベンゾオキサゾール前駆体などの環化を抑制でき、保存安定性に優れている。
上記酸性化合物(A1)および上記アンモニウム塩(A2)は、加熱すると塩基を発生するので、これらの化合物から発生した塩基により、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などの環化反応を促進でき、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などの環化を低温で行うことができる。また、これらの化合物は、塩基により環化して硬化するポリイミド前駆体およびポリベンゾオキサゾール前駆体などと共存させても、加熱しなければポリイミド前駆体およびポリベンゾオキサゾール前駆体などの環化が殆ど進行しないので、安定性に優れたポリイミド前駆体およびポリベンゾオキサゾール前駆体などを含む感光性樹脂組成物を調製することができる。
なお、本明細書において、酸性化合物とは、化合物を容器に1g採取し、イオン交換水とテトラヒドロフランとの混合液(質量比は水/テトラヒドロフラン=1/4)を50mL加えて、室温で1時間撹拌して得られた溶液を、pH(power of hydrogen)メーターを用いて、20℃にて測定した値が7未満である化合物を意味する。
酸性化合物(A1)およびアンモニウム塩(A2)の塩基発生温度が120℃以上であれば、保存中に塩基が発生しにくいので、安定性に優れたポリイミド前駆体およびポリベンゾオキサゾール前駆体などを含む感光性樹脂組成物を調製することができる。酸性化合物(A1)およびアンモニウム塩(A2)の塩基発生温度が200℃以下であれば、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などの環化温度を低くできる。塩基発生温度は、例えば、示差走査熱量測定を用い、化合物を耐圧カプセル中5℃/分で250℃まで加熱し、最も温度が低い発熱ピークのピーク温度を読み取り、ピーク温度を塩基発生温度として測定することができる。
式(101) 式(102)


式(Y1-1)~(Y1-5)において、Ar101およびAr102は、それぞれ独立に、アリール基を表し、nは、1以上の整数を表し、mは、0~5の整数を表す。
アニオンの種類は、カルボン酸アニオン、フェノールアニオン、リン酸アニオンおよび硫酸アニオンから選ばれる1種が好ましく、塩の安定性と熱分解性を両立させられるという理由からカルボン酸アニオンがより好ましい。すなわち、アンモニウム塩は、アンモニウムカチオンとカルボン酸アニオンとの塩がより好ましい。
カルボン酸アニオンは、2個以上のカルボキシル基を持つ2価以上のカルボン酸のアニオンが好ましく、2価のカルボン酸のアニオンがより好ましい。この態様によれば、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などを含む感光性樹脂組成物の安定性、硬化性および現像性をより向上できる熱塩基発生剤とすることができる。特に、2価のカルボン酸のアニオンを用いることで、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などを含む感光性樹脂組成物の安定性、硬化性および現像性をさらに向上できる。
本実施形態において、カルボン酸アニオンは、pKa1が4以下のカルボン酸のアニオンであることが好ましい。pKa1は、3.5以下がより好ましく、3.2以下が一層好ましい。この態様によれば、ポリイミド前駆体およびポリベンゾオキサゾール前駆体などを含む感光性樹脂組成物の安定性をより向上できる。
ここでpKa1とは、酸の第一のプロトンの解離定数の逆数の対数を表し、Determination of Organic Structures by Physical Methods(著者:Brown, H. C., McDaniel, D. H., Hafliger, O., Nachod, F. C.; 編纂:Braude, E. A., Nachod, F. C.; Academic Press, New York, 1955)や、Data for Biochemical Research(著者:Dawson, R.M.C.et al; Oxford, Clarendon Press, 1959)に記載の値を参照することができる。これらの文献に記載の無い化合物については、ACD/pKa(ACD/Labs製)のソフトを用いて構造式より算出した値を用いることとする。

σmが正の値を示す置換基の例としては例えば、CF3基(σm=0.43)、CF3CO基(σm=0.63)、HC≡C基(σm=0.21)、CH2=CH基(σm=0.06)、Ac基(σm=0.38)、MeOCO基(σm=0.37)、MeCOCH=CH基(σm=0.21)、PhCO基(σm=0.34)、H2NCOCH2基(σm=0.06)などが挙げられる。なお、Meはメチル基を表し、Acはアセチル基を表し、Phはフェニル基を表す。

式(XA)




熱塩基発生剤は、1種または2種以上を用いることができる。2種以上を用いる場合は、合計量が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物は、電極や配線などに用いられる金属材料との接着性を向上させるための金属接着性改良剤を含んでいることが好ましい。金属接着性改良剤の例としては、特開2014-186186号公報の段落0046~0049や、特開2013-072935号公報の段落0032~0043に記載のスルフィド系化合物が挙げられる。金属接着性改良剤としては、また、下記化合物(N-[3-(トリエトキシシリル)プロピル]マレイン酸モノアミドなど)も例示される。

金属接着性改良剤は1種のみでもよいし、2種以上であってもよい。2種以上用いる場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物を塗布によって層状にする場合、溶剤を配合することが好ましい。溶剤は、感光性樹脂組成物を層状に形成できれば、公知のものを任意に使用できる。溶剤としては、エステル類、エーテル類、ケトン類、芳香族炭化水素類、スルホキシド類などの化合物が挙げられる。
エステル類として、例えば、酢酸エチル、酢酸-n-ブチル、酢酸イソブチル、ギ酸アミル、酢酸イソアミル、プロピオン酸ブチル、酪酸イソプロピル、酪酸エチル、酪酸ブチル、乳酸メチル、乳酸エチル、γ-ブチロラクトン、ε-カプロラクトン、δ-バレロラクトン、アルキルオキシ酢酸アルキル(例:アルキルオキシ酢酸メチル、アルキルオキシ酢酸エチルアルキル、オキシ酢酸ブチル(例えば、メトキシ酢酸メチル、メトキシ酢酸エチル、メトキシ酢酸ブチル、エトキシ酢酸メチル、エトキシ酢酸エチル等))、3-アルキルオキシプロピオン酸アルキルエステル類(例:3-アルキルオキシプロピオン酸メチル、3-アルキルオキシプロピオン酸エチル等(例えば、3-メトキシプロピオン酸メチル、3-メトキシプロピオン酸エチル、3-エトキシプロピオン酸メチル、3-エトキシプロピオン酸エチル等))、2-アルキルオキシプロピオン酸アルキルエステル類(例:2-アルキルオキシプロピオン酸メチル、2-アルキルオキシプロピオン酸エチル、2-アルキルオキシプロピオン酸プロピル等(例えば、2-メトキシプロピオン酸メチル、2-メトキシプロピオン酸エチル、2-メトキシプロピオン酸プロピル、2-エトキシプロピオン酸メチル、2-エトキシプロピオン酸エチル))、2-アルキルオキシ-2-メチルプロピオン酸メチルおよび2-オキシ-2-メチルプロピオン酸エチル(例えば、2-メトキシ-2-メチルプロピオン酸メチル、2-エトキシ-2-メチルプロピオン酸エチル等)、ピルビン酸メチル、ピルビン酸エチル、ピルビン酸プロピル、アセト酢酸メチル、アセト酢酸エチル、2-オキソブタン酸メチル、2-オキソブタン酸エチル等が好適に挙げられる。
エーテル類として、例えば、ジエチレングリコールジメチルエーテル、テトラヒドロフラン、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、メチルセロソルブアセテート、エチルセロソルブアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート等が好適に挙げられる。
ケトン類として、例えば、メチルエチルケトン、シクロヘキサノン、シクロペンタノン、2-ヘプタノン、3-ヘプタノン、N-メチル-2-ピロリドン等が好適に挙げられる。
芳香族炭化水素類として、例えば、トルエン、キシレン、アニソール、リモネン等が好適に挙げられる。
溶剤は1種のみでもよいし、2種以上であってもよい。溶剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
また、N-メチル-2-ピロリドン、N-エチル-2-ピロリドン、N,N-ジメチルアセトアミドおよびN,N-ジメチルホルムアミドの含有量は、膜強度の観点から、感光性樹脂組成物の全質量に対して5質量%未満が好ましく、1質量%未満がより好ましく、0.5質量%未満がさらに好ましく、0.1質量%未満が一層好ましい。
本実施形態で用いる感光性樹脂組成物は、本発明の効果を損なわない範囲で、必要に応じて、各種の添加物、例えば、光塩基発生剤、熱重合開始剤、熱酸発生剤、シランカップリング剤、増感色素、連鎖移動剤、界面活性剤、高級脂肪酸誘導体、無機粒子、硬化剤、硬化触媒、充填剤、酸化防止剤、紫外線吸収剤、凝集防止剤等を配合することができる。これらの添加剤を配合する場合、その合計配合量は組成物の固形分の3質量%以下とすることが好ましい。
本実施形態で用いる感光性樹脂組成物は、光塩基発生剤を含んでいてもよい。光塩基発生剤とは、露光により塩基を発生するものであり、常温常圧の通常の条件下では活性を示さないが、外部刺激として電磁波の照射と加熱が行なわれると、塩基(塩基性物質)を発生するものであれば特に限定されるものではない。露光により発生した塩基はポリイミド前駆体およびベンゾオキサゾール前駆体などを加熱により硬化させる際の触媒として働くため、ネガ型において好適に用いることができる。
光塩基発生剤は1種のみでもよいし、2種以上であってもよい。光塩基発生剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態においては、光塩基発生剤として公知のものを用いることができる。例えば、M.Shirai, and M.Tsunooka, Prog.Polym.Sci.,21,1(1996);角岡正弘,高分子加工,46,2(1997);C.Kutal,Coord.Chem.Rev.,211,353(2001);Y.Kaneko,A.Sarker, and D.Neckers,Chem.Mater.,11,170(1999);H.Tachi,M.Shirai, and M.Tsunooka,J.Photopolym.Sci.Technol.,13,153(2000);M.Winkle, and K.Graziano,J.Photopolym.Sci.Technol.,3,419(1990);M.Tsunooka,H.Tachi, and S.Yoshitaka,J.Photopolym.Sci.Technol.,9,13(1996);K.Suyama,H.Araki,M.Shirai,J.Photopolym.Sci.Technol.,19,81(2006)に記載されているように、遷移金属化合物錯体や、アンモニウム塩などの構造を有するものや、アミジン部分がカルボン酸と塩を形成することで潜在化されたもののように、塩基成分が塩を形成することにより中和されたイオン性の化合物や、カルバメート誘導体、オキシムエステル誘導体、アシル化合物などのウレタン結合やオキシム結合などにより塩基成分が潜在化された非イオン性の化合物を挙げることができる。
本実施形態に用いることができる光塩基発生剤は、特に限定されず公知のものを用いることができ、例えば、カルバメート誘導体、アミド誘導体、イミド誘導体、αコバルト錯体類、イミダゾール誘導体、桂皮酸アミド誘導体、オキシム誘導体等が挙げられる。
光塩基発生剤としては、例えば、特開2009-80452号公報および国際公開WO2009/123122号で開示されたような桂皮酸アミド構造を有する光塩基発生剤、特開2006-189591号公報および特開2008-247747号公報で開示されたようなカルバメート構造を有する光塩基発生剤、特開2007-249013号公報および特開2008-003581号公報で開示されたようなオキシム構造、カルバモイルオキシム構造を有する光塩基発生剤等が挙げられるが、これらに限定されず、その他にも公知の光塩基発生剤を用いることができる。
その他、光塩基発生剤としては、特開2012-93746号公報の段落0185~0188、0199~0200および0202に記載の化合物、特開2013-194205号公報の段落0022~0069に記載の化合物、特開2013-204019号公報の段落0026~0074に記載の化合物、ならびに国際公開WO2010/064631号の段落0052に記載の化合物が例として挙げられる。
本実施形態で用いる感光性樹脂組成物は、熱重合開始剤(好ましくは熱ラジカル重合開始剤)を含んでいてもよい。熱ラジカル重合開始剤としては、公知の熱ラジカル重合開始剤を用いることができる。
熱ラジカル重合開始剤は、熱のエネルギーによってラジカルを発生し、重合性化合物の重合反応を開始または促進させる化合物である。熱ラジカル重合開始剤を添加することによって、ポリイミド前駆体およびポリベンゾオキサゾール前駆体の環化反応を進行させる際に、重合性化合物の重合反応を進行させることができる。また、ポリイミド前駆体およびポリベンゾオキサゾール前駆体がエチレン性不飽和結合を含む場合は、ポリイミド前駆体およびポリベンゾオキサゾール前駆体の環化と共に、ポリイミド前駆体およびポリベンゾオキサゾール前駆体の重合反応を進行させることもできるので、より高度な耐熱化が達成できることとなる。
熱ラジカル重合開始剤として、具体的には、特開2008-63554号公報の段落0074~0118に記載されている化合物が挙げられる。
熱ラジカル重合開始剤は1種のみでもよいし、2種以上であってもよい。熱ラジカル重合開始剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物は、熱酸発生剤を含んでいてもよい。熱酸発生剤は、加熱により酸を発生し、ポリイミド前駆体およびポリベンゾオキサゾール前駆体の環化を促進し硬化膜の機械特性をより向上させる。さらに熱酸発生剤は、ヒドロキシメチル基、アルコキシメチル基またはアシルオキシメチル基を有する化合物、エポキシ化合物、オキセタン化合物およびベンゾオキサジン化合物から選ばれる少なくとも1種の化合物の架橋反応を促進させる効果がある。
熱酸発生剤は、1種のみ用いても、2種以上用いてもよい。2種以上用いる場合は、合計量が上記範囲となることが好ましい。
本実施形態で用いる感光性樹脂組成物は、基板との密着性を向上させるために、シランカップリング剤を含んでいてもよい。
シランカップリング剤の例としては、特開2014-191002号公報の段落0062~0073に記載の化合物、国際公開WO2011/080992A1号の段落0063~0071に記載の化合物、特開2014-191252号公報の段落0060~0061に記載の化合物、特開2014-41264号公報の段落0045~0052に記載の化合物、国際公開WO2014/097594号の段落0055に記載の化合物が挙げられる。また、特開2011-128358号公報の段落0050~0058に記載のように異なる2種以上のシランカップリング剤を用いることも好ましい。
シランカップリング剤は樹脂100質量部に対して好ましくは0.1~20質量部であり、さらに好ましくは1~10質量部の範囲である。0.1質量部以上であると、基板との充分な密着性を付与することができ、20質量部以下であると室温保存時において粘度上昇等の問題を抑制できる。
シランカップリング剤は、1種のみ用いても、2種以上用いてもよい。2種以上用いる場合は、合計量が上記範囲となることが好ましい。
本実施形態で用いる感光性樹脂組成物は、増感色素を含んでいてもよい。増感色素は、特定の活性放射線を吸収して電子励起状態となる。電子励起状態となった増感色素は、アミン発生剤、熱ラジカル重合開始剤、光重合開始剤などと接触して、電子移動、エネルギー移動、発熱などの作用が生じる。これにより、アミン発生剤、熱ラジカル重合開始剤、光重合開始剤は化学変化を起こして分解し、ラジカル、酸或いは塩基を生成する。
本実施形態で用いる感光性樹脂組成物は、連鎖移動剤を含有してもよい。連鎖移動剤は、例えば高分子辞典第三版(高分子学会編、2005年)683-684頁に定義されている。連鎖移動剤としては、例えば、分子内にSH、PH、SiH、GeHを有する化合物群が用いられる。これらは、低活性のラジカルに水素を供与して、ラジカルを生成するか、もしくは、酸化された後、脱プロトンすることによりラジカルを生成しうる。特に、チオール化合物(例えば、2-メルカプトベンズイミダゾール類、2-メルカプトベンズチアゾール類、2-メルカプトベンズオキサゾール類、3-メルカプトトリアゾール類、5-メルカプトテトラゾール類等)を好ましく用いることができる。
連鎖移動剤は1種のみでもよいし、2種以上であってもよい。連鎖移動剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物には、塗布性をより向上させる観点から、各種類の界面活性剤を添加してもよい。界面活性剤としては、フッ素系界面活性剤、ノニオン系界面活性剤、カチオン系界面活性剤、アニオン系界面活性剤、シリコーン系界面活性剤などの各種類の界面活性剤を使用できる。また、下記界面活性剤も好ましい。

界面活性剤は1種のみでもよいし、2種以上であってもよい。界面活性剤が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物には、酸素に起因する重合阻害を防止するために、ベヘン酸やベヘン酸アミドのような高級脂肪酸誘導体を添加して、塗布後の乾燥の過程で組成物の表面に偏在させてもよい。
感光性樹脂組成物が高級脂肪酸誘導体を有する場合、高級脂肪酸誘導体の含有量は、感光性樹脂組成物の全固形分に対して、0.1~10質量%が好ましい。
高級脂肪酸誘導体は1種のみでもよいし、2種以上であってもよい。高級脂肪酸誘導体が2種以上の場合は、その合計が上記範囲であることが好ましい。
本実施形態で用いる感光性樹脂組成物の水分含有量は、塗布面性状の観点から、5質量%未満が好ましく、1質量%未満がさらに好ましく、0.6質量%未満が特に好ましい。
また、感光性樹脂組成物に意図せずに含まれる金属不純物を低減する方法としては、感光性樹脂組成物を構成する原料として金属含有量が少ない原料を選択する、感光性樹脂組成物を構成する原料に対してフィルター濾過を行う、装置内をポリテトラフロロエチレン等でライニングしてコンタミネーションを可能な限り抑制した条件下で蒸留を行う等の方法を挙げることができる。
感光性樹脂組成物を層状に適用した後、溶剤を乾燥する工程を含んでいてもよい。乾燥温度は50~150℃が好ましく、70℃~130℃がより好ましく、90℃~110℃がさらに好ましい。乾燥時間としては、30秒間~20分間が好ましく、1分間~10分間がより好ましく、3分間~7分間がさらに好ましい。
本実施形態では、感光性樹脂膜を露光および現像して、樹脂パターン3を製造する。樹脂パターンの厚さは、感光性樹脂膜の厚さと同様であり、0.1~100μmが好ましく、1~50μmがより好ましく、3~20μmがさらに好ましい。
露光の条件は、特に定めるものではなく、感光性樹脂膜の露光部の現像液に対する溶解度を変化させられることが好ましく、感光性樹脂膜の露光部を硬化できることがより好ましい。例えば、露光は感光性樹脂膜に対して、波長500nm以下の光を照射して行うことが好ましい。波長が500nm以下の光を照射することにより、透明基板から感光性樹脂をはがれにくくすることができる。
露光波長の下限は、100nm以上が好ましく、190nm以上がより好ましく、240nm以上がさらに好ましい。露光波長の上限は、500nm以下が好ましく、400nm以下がより好ましい。
露光量は、波長365nmでの露光エネルギー換算で100~10000mJ/cm2であることが好ましく、200~8000mJ/cm2であることがより好ましい。
ネガ型現像の場合、感光性樹脂膜の一部を除去して形成された樹脂パターンの、除去された部分の表面積が、感光性樹脂膜全領域の表面積の30%以下が好ましく、25%以下がより好ましく、20%以下がさらに好ましく、15%以下が一層好ましく、10%以下がより一層好ましく、5%以下がさらに一層好ましく、3%以下が特に一層好ましい。このような範囲とすることにより、得られる成形品の歩留りがより向上する傾向にある。感光性樹脂膜の一部を除去して形成された樹脂パターンの、除去された部分の表面積が、感光性樹脂膜の全領域の表面積に占める割合の下限値は特に定めるものではないが、例えば、0.5%以上とすることができる。
マスクの開口部の大きさは、用途等に応じて適宜定めることができるが、最大長さが30μm以下(好ましくは、最大長さ3μm以上)の開口部となるようなマスクが例示される。
現像処理工程は、ポジ型現像処理であっても、ネガ型現像処理であってもよいが、ネガ型現像処理が好ましい。ネガ型現像を行うことにより、露光されていない部分(非露光部)が除去される。現像方法は、特に制限は無く、所望のパターンを形成できることが好ましく、例えば、パドル、スプレー、浸漬、超音波等の現像方法が採用可能である。
現像は現像液を用いて行うことが好ましい。現像液は、露光されていない部分(非露光部)が除去されるものであれば、特に制限なく使用できる。有機溶剤を用いた現像が好ましく、エステル類として、例えば、酢酸エチル、酢酸-n-ブチル、酢酸イソブチル、ギ酸アミル、酢酸イソアミル、プロピオン酸ブチル、酪酸イソプロピル、酪酸エチル、酪酸ブチル、乳酸メチル、乳酸エチル、γ-ブチロラクトン、ε-カプロラクトン、δ-バレロラクトン、アルキルオキシ酢酸アルキル(例:アルキルオキシ酢酸メチル、アルキルオキシ酢酸エチル、アルキルオキシ酢酸ブチル(例えば、メトキシ酢酸メチル、メトキシ酢酸エチル、メトキシ酢酸ブチル、エトキシ酢酸メチル、エトキシ酢酸エチル等))、3-アルキルオキシプロピオン酸アルキルエステル類(例:3-アルキルオキシプロピオン酸メチル、3-アルキルオキシプロピオン酸エチル等(例えば、3-メトキシプロピオン酸メチル、3-メトキシプロピオン酸エチル、3-エトキシプロピオン酸メチル、3-エトキシプロピオン酸エチル等))、2-アルキルオキシプロピオン酸アルキルエステル類(例:2-アルキルオキシプロピオン酸メチル、2-アルキルオキシプロピオン酸エチル、2-アルキルオキシプロピオン酸プロピル等(例えば、2-メトキシプロピオン酸メチル、2-メトキシプロピオン酸エチル、2-メトキシプロピオン酸プロピル、2-エトキシプロピオン酸メチル、2-エトキシプロピオン酸エチル))、2-アルキルオキシ-2-メチルプロピオン酸メチルおよび2-アルキルオキシ-2-メチルプロピオン酸エチル(例えば、2-メトキシ-2-メチルプロピオン酸メチル、2-エトキシ-2-メチルプロピオン酸エチル等)、ピルビン酸メチル、ピルビン酸エチル、ピルビン酸プロピル、アセト酢酸メチル、アセト酢酸エチル、2-オキソブタン酸メチル、2-オキソブタン酸エチル等、並びに、エーテル類として、例えば、ジエチレングリコールジメチルエーテル、テトラヒドロフラン、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、メチルセロソルブアセテート、エチルセロソルブアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート等が挙げられる。
ケトン類として、例えば、メチルエチルケトン、シクロヘキサノン、シクロペンタノン、2-ヘプタノン、3-ヘプタノン、N-メチル-2-ピロリドン等が挙げられる。
芳香族炭化水素類として、例えば、トルエン、キシレン、アニソール、リモネン等が挙げられる。
スルホキシド類としてジメチルスルホキシドが好適に挙げられる。
中でも3-エトキシプロピオン酸メチル、3-エトキシプロピオン酸エチル、エチルセロソルブアセテート、乳酸エチル、ジエチレングリコールジメチルエーテル、酢酸ブチル、3-メトキシプロピオン酸メチル、2-ヘプタノン、シクロヘキサノン、シクロペンタノン、γ-ブチロラクトン、ジメチルスルホキシド、エチルカルビトールアセテート、ブチルカルビトールアセテート、プロピレングリコールメチルエーテル、およびプロピレングリコールメチルエーテルアセテートが好ましく、シクロペンタノン、γ-ブチロラクトンがより好ましい。
現像時間は、10秒間~5分間が好ましい。
現像時の温度は、特に定めるものではないが、通常、20~40℃で行うことができる。
現像液を用いた処理の後、さらに、リンスを行ってもよい。リンスは、現像液とは異なる溶剤で行うことが好ましい。例えば、感光性樹脂組成物に含まれる溶剤を用いてリンスすることができる。リンス時間は、5秒間~1分間が好ましい。
本実施形態では、露光現像工程後、透明基板剥離工程前に、硬化工程を含むことが好ましい。また、金属適用工程を含む場合は、金属適用工程前に、上記樹脂パターンを硬化する硬化工程を含んでいることが好ましい。
感光性樹脂膜が、ポリイミド前駆体およびポリベンゾオキサゾール前駆体から選択される少なくとも1種を含む場合、硬化工程により、ポリイミド前駆体およびポリベンゾオキサゾール前駆体の環化反応を進行させることができる。また、感光性樹脂膜が、ポリイミドやポリベンゾオキサゾールである場合でも、架橋剤と共に加熱して、3次元ネットワーク構造を形成することができる。さらに、感光性樹脂組成物が、ラジカル重合性化合物等を含有する場合、未反応のラジカル重合性化合物の硬化なども進行させることができる。
硬化工程は、通常、感光性樹脂膜を、感光性樹脂膜を構成する樹脂のガラス転移温度以上の温度に昇温することによって行われる。また、昇温した場合の最終到達温度は、感光性樹脂膜が、ポリイミド前駆体およびポリベンゾオキサゾール前駆体から選択される少なくとも1種を含む場合、感光性樹脂膜に含まれる樹脂の環化温度以上であることが好ましい。
昇温した場合の最終到達温度は、最高加熱温度であることが好ましい。最高加熱温度としては、100~500℃が好ましく、150~450℃がより好ましく、160~350℃がさらに好ましい。
加熱開始時の温度は、20~150℃が好ましく、20℃~130℃がより好ましく、25℃~120℃がさらに好ましい。加熱開始時の温度は、最高加熱温度まで加熱する工程を開始する際の加熱温度のことをいう。例えば、感光性樹脂組成物を基板の上に適用した後、乾燥させる場合、この乾燥後の温度であり、例えば、感光性樹脂組成物に含まれる溶剤の沸点-(30~200)℃から徐々に昇温させることが好ましい。
本実施形態では、透明基板1と、上記透明基板の表面に位置する樹脂パターン3を有する積層体に金属4を適用する工程を有することが好ましい。本実施形態における金属4は、透明基板1の表面であって、樹脂パターン3の間隙に設けられることが好ましい。金属の適用方法としては、特に定めるものではないが、無電解めっき方法が挙げられる。
また、透明基板の上であって、透明基板1と樹脂パターン3の間に設けることもできる。透明基板1と樹脂パターン3の間に金属を設ける態様としては、透明基板1の表面と樹脂パターン3の間、および、透明基板1の上であって、透明基板1に概ね垂直な方向における、樹脂パターン3と樹脂パターン3の間に設ける態様が例示される。
無電解めっき方法の場合、以下の(1)および(2)の方法に従って金属を適用することが好ましい。
(1)無電解めっき液を含むめっき槽に透明基板1と樹脂パターン3を有する積層体を浸漬し、透明基板1の表面および樹脂パターン3の表面に、それぞれ、金属層(めっき膜)を形成する。このときの金属層の厚さは、0.1~100μmであることが好ましく、1~20μmであることがより好ましく、1~10μmであることが特に好ましい。金属層は1層であっても、2層以上であってもよい。
(2)次に、金属層のうち、樹脂パターン3の表面に設けられた金属層等の不要な金属層は、研磨等により削除して、樹脂パターンを露出させる。
無電解めっきの詳細は、日刊工業新聞社:プレーティング研究会編「めっき基礎のきそ」2006年6月28日、日刊工業新聞社:電気鍍金研究会編「次世代めっき技術」2004年11月30日の記載を参酌でき、これらの記載は本明細書に組み込まれる。
また、他の金属の適用方法としては、蒸着などが例示される。この場合の金属層の厚さも、上記無電解めっき方法の場合と同様の範囲が好ましい。
本実施形態では、樹脂パターン3と金属層をさらに複数層積層してもよい。具体的には、金属適用工程の後に、さらに、再度、樹脂パターンを製造する工程を繰り返すことができる。
本実施形態では、さらに、半導体素子5を適用する工程を含んでいてもよい。具体的には、透明基板1と、上記透明基板の表面に位置する樹脂パターン3と、上記透明基板の表面であって、上記樹脂パターンの間隙に位置する金属4と、上記金属と接している半導体素子5を有し、上記半導体素子5が、透明基板側とは反対側で上記金属に接するように配置することが好ましい。ここで、樹脂パターンの間隙とは、透明基板の上であって、樹脂パターンが設けられている側の、樹脂と樹脂の間の領域をいい、例えば、感光性樹脂を露光現像して得られる開口部をいう。また、樹脂パターンを積層する場合は、下層の樹脂パターンおよび/または金属の表面であって、樹脂と樹脂の間の領域となろう。
ここでの半導体素子としては、半導体チップ等が例示される。
本実施形態では、透明基板1と、上記透明基板の表面に位置する樹脂パターンを有する積層体6に、積層体の透明基板側から放射光を照射して、上記積層体から透明基板を剥離する剥離工程を含む。上記放射光の照射に際し、照射する放射光の波長における樹脂パターンの吸光度を0.5以上にする。このような構成とすることにより、樹脂パターンの透明基板側の表面付近を照射し、透明基板と樹脂パターンの剥離が可能になる。また、本実施形態では、金属なども基材表面に有するが、透明基板と金属は通常密着性が弱いため、金属も樹脂パターンと共に、積層体側に残る。
本実施形態では、上記吸光度は、0.5以上であり、0.8以上が好ましく、1.0以上がより好ましく、1.2以上がさらに好ましい。上記吸光度の上限値は、特に無く、高いほど好ましいが、例えば、9以下、さらには6以下、特には4以下でも十分に本発明の効果を達成できる。
放射光の波長は、透明基板を透過し、樹脂パターンが吸収する波長が好ましいので、使用する材料に応じて適宜さだめることができる。例えば、透過波長が広い石英の透明性が確保できる必要があり、下限が、170nm以上が好ましく、220nm以上がより好ましく、260nm以上がさらに好ましい。また、上記波長の上限は、4000nm以下が好ましく、3000nm以下がより好ましい。
レーザーの種類は特に定めるものではないが、YAGレーザー(イットリウム・アルミニウム・ガーネットレーザー)のTHG(Third Harmonic Generation)(355nm)、またエキシマレーザーのうち、KrF(248nm)、XeCl(308nm)、XeF(351nm)が好ましい。
放射光の照射の際には、焦点は、透明基板1と樹脂パターン3の界面±10μmの距離の位置にするのが好ましい。また、レーザー光源は、100~1000000μm2の面積で、繰り返し周波数10~5000Hz、走査速度0.1~1000mm/sであることが好ましい。走査ピッチは、1~100μmであることが好ましい。露光量としては、0.5~50J/cm2であることが好ましい。
図2は、第2の実施形態を示す概略図であって、透明基板1の表面にシード層21を設け、さらにシード層の表面にフォトレジスト層22を設け、露光および現像を行って、フォトレジストパターン23を形成した後、金属を適用し、フォトレジストパターン23を除去した後、シード層21を削除し、金属パターン24を作製する(金属パターン形成工程)。次に、金属パターン24の間に樹脂パターン3を設ける(樹脂パターン形成工程)。また、金属パターン24の透明基板1と反対側から、さらに、半導体素子5を適用した後(半導体素子適用工程)、所定の条件を満たすように、放射光を照射することにより、積層体6から透明基板1を剥離することができる(透明基板剥離工程)。本実施形態では、特に、金属パターン形成工程、樹脂パターン形成工程、半導体素子適用工程および透明基板剥離工程の順であることが好ましい。
尚、図2は、図1と共通する部材については、同じ符号を用いている(図3についても同じ)。
第2の実施形態では、樹脂パターン(例えば、絶縁層)の間隙に金属(例えば、金属配線や電極)が位置し、さらに、上記金属と接している半導体素子(例えば、半導体チップ)を有する半導体装置が得られる。以下、第2の実施形態をより具体的に説明する。
本実施形態では、金属パターン24を形成する工程を含む。金属パターンを形成する工程は、例えば、シード層形成工程、フォトレジストパターン形成工程、金属適用工程、フォトレジストパターンおよびシード層除去工程を含むことが好ましい。
第2の実施形態では、透明基板1の表面にシード層21を設ける。透明基板の詳細は、第1の実施形態と同義であり、好ましい範囲も同様である。
シード層は、スパッタリング、CVD(chemical vapor deposition)、無電解めっきなどで形成することが好ましい。シード層の材料としては、錫、銀、銅が例示される。シード層の厚さとしては、0.02~2μmが好ましい。
第2の実施形態では、シード層21の上、好ましくは表面に、フォトレジスト層22を設ける。次いで、フォトレジスト層22を露光現像し、フォトレジストパターン23を形成する。フォトレジストパターンの詳細としては、電学論 E,131巻1号,2011年の記載を参酌でき、これらの内容は本明細書に組み込まれる。フォトレジスト層の厚さとしては、0.5~50μmが好ましい。
本実施形態では、透明基板1と、フォトレジストパターン23を有する積層体に、金属を適用する工程を有することが好ましい。本実施形態における金属は、透明基板1とフォトレジストパターン23を有する積層体の、透明基板1の上であって、フォトレジストパターンのパターン間隙に適用される。金属の適用方法としては、特に定めるものではないが、無電解めっき方法が挙げられる。無電界めっき方法および金属の詳細は、第1の実施形態における金属適用工程と同義であり、好ましい範囲も同様である。このようしてフォトレジストパターン23に対応する金属パターン24を設ける。
金属パターン24を形成した後、フォトレジストパターン23を除去する。フォトレジストパターンの除去は、溶剤を用いた除去、ヒドロキシアミン系剥離剤を用いた除去によって行うことができる。
フォトレジストパターンを除去した後、シード層21を除去する。シード層21の除去は、金属パターン24の材質に応じて、エッチングを行って除去することが好ましい。
次いで、透明基板1と金属パターン24を有する積層体の表面に、樹脂パターン23を設ける。樹脂パターンの製造方法は、特に定めるものではないが、樹脂組成物を塗布して製造することが好ましい。好ましい実施形態としては、感光性樹脂組成物を適用し、現像露光して、金属パターン24を露出させることが好ましい。現像露光工程の詳細は、第1の実施形態の記載を参酌できる。また、樹脂組成物を塗布して硬化膜とした後、表面を削ることによって、金属パターン24を露出させてもよい。
本実施形態では、金属パターン24と樹脂パターン3をさらに交互に積層してもよい。
本実施形態では、さらに、半導体素子を適用する工程を含んでいてもよい。これらの詳細は、第1の実施形態における半導体素子適用工程と同様である。
本実施形態では、透明基板1と、上記透明基板の表面に位置する樹脂パターン3を有する積層体6に、積層体の透明基板側から放射光を照射して、上記積層体から透明基板を剥離する剥離工程を含む。これらの詳細は、第1の実施形態における透明基板剥離工程と同様である。
図3は、第3の実施形態を示す概略図であって、透明基板1の表面に感光性樹脂膜2を設け(感光性樹脂膜製造工程)、露光現像して樹脂パターン3を製造する(露光現像工程)。その後、樹脂パターン3の透明基板1と接していない側に、半導体素子5を適用する(半導体素子適用工程)。その後、所定の条件を満たすように、透明基板側から放射光を照射することにより、透明基板1を剥離することができる(透明基板剥離工程)。透明基板1を剥離した後、透明基板1を剥離した側から、金属4を適用することができる(金属適用工程)。本実施形態では、特に、感光性樹脂膜製造工程、露光現像工程、半導体素子適用工程、透明基板剥離工程および金属適用工程の順で実施することが好ましい。
第3の実施形態では、樹脂パターン(例えば、絶縁膜)の表面の半導体素子5が、樹脂パターン3の間隙の金属(例えば、金属配線や電極)と結合している半導体装置が得られる。以下、第3の実施形態をより具体的に説明する。
第1の実施形態と同義であり、好ましい範囲も同様である。
第1の実施形態と同義であり、好ましい範囲も同様である。
本実施形態では、現像露光工程の後、硬化工程を有することが好ましい。第1の実施形態と同義であり、好ましい範囲も同様である。
本実施形態では、さらに、半導体素子5を適用する工程を含んでいることが好ましい。具体的には、樹脂パターン3の表面であって、透明基板1と接している側と反対側の表面に設けることが好ましい。ここでの半導体素子としては、半導体チップ等が例示される。
本実施形態では、透明基板1と、上記透明基板の表面に位置する樹脂パターンを有する積層体6に、積層体の透明基板側から放射光を照射して、上記積層体から透明基板を剥離する剥離工程を含む。これらの詳細は、第1の実施形態における透明基板剥離工程と同様である。
本実施形態では、さらに、樹脂パターン3の間隙に金属を適用することが好ましい。具体的には、透明基板1を剥離した側から、金属を適用することが好ましい。本実施形態における金属の適用方法としては、金属ペーストの印刷が好ましい。金属が層状の場合、金属層の厚さは、樹脂パターンの厚さに相当することが好ましい。このような構成とすることにより、金属層が電極としての役割を果たす。
本実施形態で用いられる金属としては、銅、アルミニウム、ニッケル、バナジウム、チタン、タンタル、クロム、コバルト、金、銀およびタングステンから選択される少なくとも1種を含むことが好ましく、金属ペーストの印刷を行う場合、銀が好ましい。金属は、1種であっても2種であってもよい。
次に、半導体装置の製造方法について、説明する。本発明の半導体装置の製造方法は、本発明のパターン製造方法を含む。特に、以下の積層体を経て製造されることが好ましい。
<積層体1>
透明基板と、上記透明基板の表面に位置する樹脂パターンと、上記透明基板の表面であって、上記樹脂パターンの間隙に位置する金属とを有し、上記樹脂パターンがポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含む積層体。
<積層体2>
透明基板と、上記透明基板の表面に位置する樹脂パターンと、上記透明基板の表面であって、上記樹脂パターンの間隙に位置する金属と、上記金属と接している半導体素子を有し、上記半導体素子が、透明基板側とは反対側で上記金属に接しており、上記樹脂パターンがポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含む積層体。
<ポリイミド前駆体の合成>
<<4,4’-オキシジフタル酸二無水物、4,4’-オキシジアニリンおよび2-ヒドロキシエチルメタクリレートからのポリイミド前駆体Aa-1(ラジカル重合性基を有するポリイミド前駆体)の合成>>
20.0g(64.5ミリモル)の4,4’-オキシジフタル酸二無水物(4,4’-オキシジフタル酸を140℃で12時間乾燥したもの)と、18.6g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、10.7gのピリジンと、140gのダイグライム(ジエチレングリコールジメチルエーテル)を混合した。混合物を60℃の温度で18時間撹拌して、4,4’-オキシジフタル酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、反応混合物を-10℃に冷却し、温度を-10±4℃に保ちながら16.12g(135.5ミリモル)のSOCl2を10分かけて加えた。反応混合物を50mLのN-メチルピロリドンで希釈した後、反応混合物を室温で2時間撹拌した。次いで、100mLのN-メチルピロリドンに11.08g(58.7ミリモル)の4,4’-オキシジアニリンを溶解させた溶液を、20~23℃で20分かけて反応混合物に滴下した。次いで、反応混合物を室温で1晩撹拌した。次いで、5リットルの水の中に反応混合物を加えて、ポリイミド前駆体を沈殿させた。水およびポリイミド前駆体の混合物を5000rpmの速度で15分間撹拌した。水およびポリイミド前駆体の混合物を濾過して、濾液を除き、4リットルの水の中にポリイミド前駆体を含む残渣を加えた。水およびポリイミド前駆体の混合物を再度30分間撹拌し、再び濾過して、残渣としてポリイミド前駆体を得た。次いで、得られたポリイミド前駆体を減圧下で、45℃で3日間乾燥し、ポリイミド前駆体Aa-1を得た。
ポリイミド前駆体Aa-1の構造を以下に示す。

下記の成分を混合し、均一な溶液として、感光性樹脂組成物を調製した。
感光性樹脂:ポリイミド前駆体(Aa-1) 32質量部
重合性化合物B-1 6.9質量部
光重合開始剤C-1 1.0質量部
重合禁止剤:パラベンゾキノン(東京化成工業社製) 0.08質量部
マイグレーション抑制剤:1H-テトラゾール(東京化成工業社製)
0.12質量部
金属接着性改良剤:N-[3-(トリエトキシシリル)プロピル]マレイン酸モノアミド 0.70質量部
溶剤:γ-ブチロラクトン 48.00質量部
溶剤:ジメチルスルホキシド 12.00質量部
B-1:NKエステル A-9300(新中村化学工業(株)製、3官能アクリレート、下記構造)


上記感光性樹脂組成物を、細孔の幅が0.8μmのフィルターを通して、(0.3MPa)の圧力で、加圧濾過した。濾過後、感光性樹脂組成物を透明基板(厚さ0.6mmのガラス基板)の表面にスピンコート法により適用して層状にし、ホットプレート上で、100℃で5分間乾燥し、10μmの厚さの均一な感光性樹脂膜を得た。
得られた透明基板と感光性樹脂膜からなる積層体の感光性樹脂膜側をマスク(直径10μmのホールをマスク面内に均一に有するマスク、開口率98%)で覆い、ステッパー(Nikon NSR 2005 i9C)を用いて、365nm(i線)の露光波長で、500mJ/cm2の露光エネルギーで露光した。露光後に、露光部と未露光部を有する樹脂膜をシクロペンタノンで60秒間スピン洗浄して未露光部を除去した(ネガ型現像)。さらに、プロピレングリコール-1-モノメチルエーテル-2-アセタート(PGMEA)でリンスして残渣を除去して、樹脂パターンを得た。
次いで、室温から昇温して、最高加熱温度が350℃に達してから1時間加熱して樹脂パターンを硬化させ、その後室温まで冷却した。透明基板の表面に得られた樹脂パターンは、厚さ6μmで直径約10μmのホールを有する樹脂パターンであった。
尚、本発明における感光性樹脂膜の全領域の表面積に対する、感光性樹脂膜の除去された部分の表面積の割合は、100%からマスクの開口率を引いた値に従って計算した値とする。
水溶性有機溶媒として、ジエチレングリコールジエチルエーテル(和光純薬社製)39質量部、水40.75質量部、硝酸(和光純薬社製)20質量部、および酢酸パラジウム(和光純薬社製)0.25質量部からなるパラジウム触媒液を調製し、このパラジウム触媒液に、上記で得られた透明基板と樹脂パターンからなる積層体を5分間浸漬した後、水で洗浄を行った。
その後、上村工業(株)製、スルカップPGT(PGT-A、PGT-B、PGT-C)を含む下記組成の無電解めっき液を用い、無電解めっき温度26℃で、60分間無電解めっきを行った。得られた無電解銅めっき膜の厚みは、樹脂パターンの表面、および、露光現像により露出した透明基板の表面(ホールの底部)のいずれも、3.0μmであった。
・蒸留水 79.2質量%
・PGT-A 9.0質量%
・PGT-B 6.0質量%
・PGT-C 3.5質量%
・ホルマリン(和光純薬:ホルムアルデヒド液) 2.3質量%
<<照射条件>>
照射した放射光:355nmパルス(YAG-THG)レーザー(パルス幅:5ns)
・ガラスと樹脂膜の界面に焦点を合せた60μm角のレーザービーム(繰返し周波数50Hz)を1.5mm/s、走査ピッチ30μmで全面に走査した。フルエンス1.9J/cm2の露光を行った。
照射する放射光の波長における樹脂パターンの吸光度の測定は以下の方法により行った。
金属適用前(無電解めっき前)の、透明基板上の樹脂パターンの除去部が無い部分を5か所選んで、照射した放射光の波長(波長355nm)における吸光度を測定し、平均値を求めた。
吸光度の測定は、日立ハイテクサイエンス製のU-3900を用いた。
積層体からの透明基板の剥離性について、以下の通り評価した。
A:剥離できた
B:一部剥離できたが、一部が透明基板に残ってしまった
C:剥離できなかった
剥離後の積層体上の半導体チップの電極接続を全て確認し、不具合の無かった割合を評価した。単位は、%で示した。
実施例1において、マスクを、開口率が95%のマスクに変更した他は、実施例1と同様に行った。
実施例1において、マスクを、開口率が90%のマスクに変更した他は、実施例1と同様に行った。
実施例1において、マスクを、開口率が80%のマスクに変更した他は、実施例1と同様に行った。
実施例1において、マスクを、開口率が70%のマスクに変更した他は、実施例1と同様に行った。
実施例1において、感光性樹脂組成物を、下記感光性樹脂組成物A-2に変更した以外は、実施例1と同様に行った。
<<感光性樹脂組成物A-2の組成>>
感光性樹脂:ポリイミド前駆体(Aa-1) 32質量部
重合性化合物B-1 6.9質量部
光重合開始剤C-1 1.0質量部
重合禁止剤:2,6-ジ-tert-ブチル-4-メチルフェノール(東京化成工業社製) 0.1質量部
マイグレーション抑制剤:1,2,4-トリアゾール(東京化成工業社製)
0.1質量部
溶剤:γ-ブチロラクトン 48.00質量部
溶剤:ジメチルスルホキシド 12.00質量部
実施例1において、透明基板の表面に得られた樹脂パターンの厚さを調整して、吸光度が0.55になるように調整し、他は同様に行った。
実施例1と同様の手順を行って、半導体チップを接続した積層体を得た。得られた積層体について、放射光の照射をせずに、透明基板の剥離を試みたが、剥離ができなかった。
実施例1において、透明基板の表面に得られた樹脂パターンの厚さを10μmから3μmとなるように調整し、他は同様に行った。しかしながら、剥離ができなかった。
実施例1において、透明基板の表面に得られた樹脂パターンの厚さを調整して、吸光度が0.45になるように調整し、他は同様に行った。
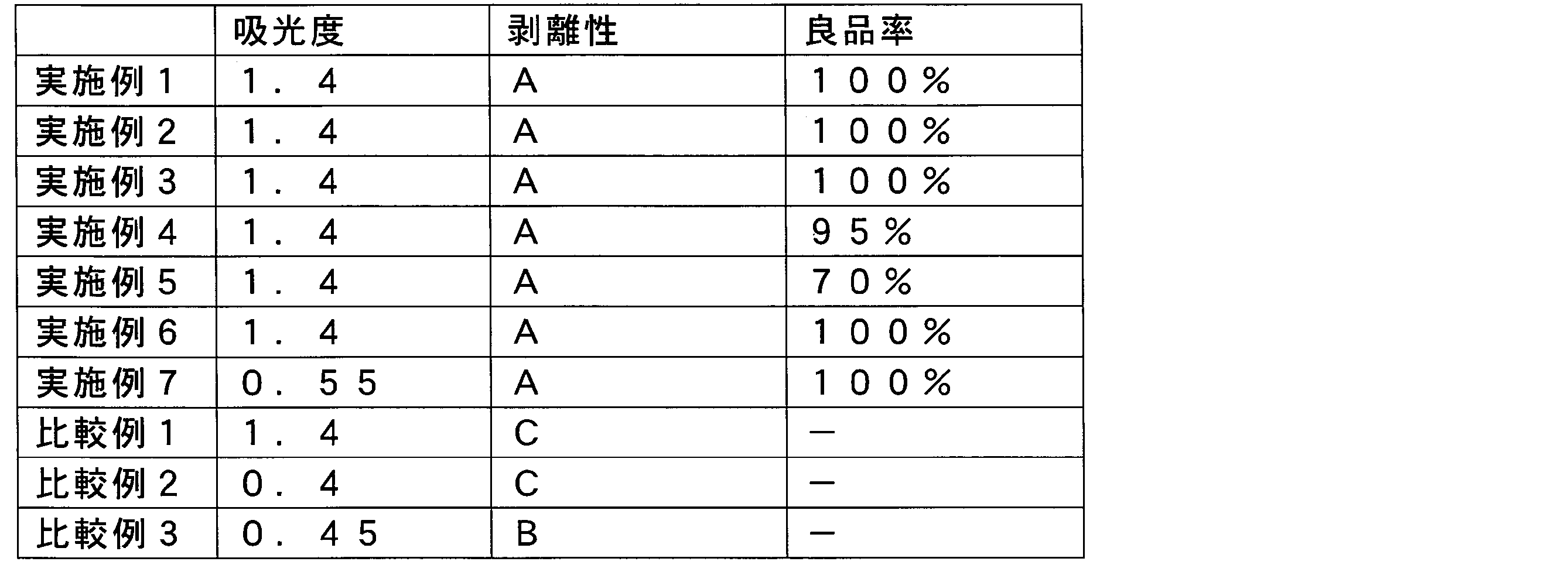
2 感光性樹脂膜
3 樹脂パターン
4 金属
5 半導体素子
6 積層体
7 マスク
21 シード層
22 フォトレジスト層
23 フォトレジストパターン
24 金属パターン
Claims (13)
- 透明基板と、前記透明基板の表面に位置する樹脂パターンを有する積層体Aに、前記積層体Aの透明基板側から放射光を照射して、前記積層体Aから透明基板を剥離する剥離工程を含み、
前記樹脂パターンが、ポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含み、
前記照射する放射光の波長における前記樹脂パターンの吸光度が0.5以上である、パターン製造方法。 - 透明基板と感光性樹脂膜を有する積層体Bの感光性樹脂膜に対して露光し、さらに、現像処理を行って感光性樹脂膜の一部を除去することで前記積層体Aを得ることを含む、請求項1に記載のパターン製造方法。
- 前記感光性樹脂膜の一部を除去して形成された樹脂パターンの、除去された部分の表面積が、前記感光性樹脂膜の露光された全領域の表面積の20%以下である、請求項2に記載のパターン製造方法。
- 前記露光は、前記積層体Bの感光性樹脂膜側から行う、請求項2または3に記載のパターン製造方法。
- 前記積層体Aの、透明基板上であって前記樹脂パターンの間隙または透明基板の上であって透明基板と樹脂パターンの間に、金属を適用する工程を有する、請求項1~4のいずれか1項に記載のパターン製造方法。
- 前記金属が、銅、アルミニウム、ニッケル、バナジウム、チタン、タンタル、クロム、コバルト、金、銀およびタングステンから選択される少なくとも1種を含む、請求項5に記載のパターン製造方法。
- 前記感光性樹脂膜が、ポリイミド前駆体、ポリイミド、ポリベンゾオキサゾール前駆体およびポリベンゾオキサゾールから選択される少なくとも1種を含む、請求項2~6のいずれか1項に記載のパターン製造方法。
- 前記感光性樹脂膜が、ポリイミド前駆体およびポリベンゾオキサゾール前駆体から選択される少なくとも1種を含む、請求項2~6のいずれか1項に記載のパターン製造方法。
- 前記露光現像工程後、剥離工程前に、前記樹脂パターンを硬化する硬化工程を含む、請求項1~8のいずれか1項に記載のパターン製造方法。
- 前記現像がネガ型現像である、請求項2~9のいずれか1項に記載のパターン製造方法。
- 請求項1~10のいずれか1項に記載のパターン製造方法を含む、半導体装置の製造方法。
- 透明基板と、前記透明基板の表面に位置する樹脂パターンと、前記透明基板の表面であって、前記樹脂パターンの間隙に位置する金属とを有し、前記樹脂パターンがポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含む積層体。
- 透明基板と、前記透明基板の表面に位置する樹脂パターンと、前記透明基板の表面であって、前記樹脂パターンの間隙に位置する金属と、前記金属と接している半導体素子を有し、前記半導体素子が、透明基板側とは反対側で前記金属に接しており、前記樹脂パターンがポリイミドおよびポリベンゾオキサゾールから選択される少なくとも1種を含む積層体。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018520957A JP6704047B2 (ja) | 2016-06-02 | 2017-05-31 | パターン製造方法、半導体装置の製造方法および積層体 |
| KR1020187034624A KR102138069B1 (ko) | 2016-06-02 | 2017-05-31 | 패턴 제조 방법, 반도체 장치의 제조 방법 및 적층체 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-111115 | 2016-06-02 | ||
| JP2016111115 | 2016-06-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017209178A1 true WO2017209178A1 (ja) | 2017-12-07 |
Family
ID=60477547
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/020251 WO2017209178A1 (ja) | 2016-06-02 | 2017-05-31 | パターン製造方法、半導体装置の製造方法および積層体 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JP6704047B2 (ja) |
| KR (1) | KR102138069B1 (ja) |
| TW (1) | TWI709817B (ja) |
| WO (1) | WO2017209178A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019109494A (ja) * | 2017-12-15 | 2019-07-04 | 旭化成株式会社 | 感光性樹脂組成物、硬化レリーフパターンの製造方法、及び半導体装置 |
| TWI696045B (zh) * | 2018-05-21 | 2020-06-11 | 日商信越化學工業股份有限公司 | 圖案形成方法 |
| JP2020106530A (ja) * | 2018-12-26 | 2020-07-09 | カムテック リミテッド | 埋め込み層の測定 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3919979A1 (en) * | 2020-06-02 | 2021-12-08 | Imec VZW | Resistless patterning mask |
| KR102600183B1 (ko) * | 2020-11-27 | 2023-11-08 | 주식회사 아큐레이저 | 반도체 소자의 전사 방법 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6298799A (ja) * | 1985-10-25 | 1987-05-08 | 株式会社リコー | 多層配線形成方法 |
| JPH01159613A (ja) * | 1987-12-16 | 1989-06-22 | Nec Kansai Ltd | 超音波半田パターンの形成方法 |
| JP2004146426A (ja) * | 2002-10-22 | 2004-05-20 | Hitachi Ltd | 電子部品を搭載した薄膜配線基板の製造方法およびマルチチップモジュール |
| JP2004207262A (ja) * | 2002-12-20 | 2004-07-22 | Fujitsu Ltd | 薄膜多層回路基板及びその製造方法 |
| JP2009043962A (ja) * | 2007-08-09 | 2009-02-26 | Sony Corp | 半導体装置の製造方法 |
| JP2012069734A (ja) * | 2010-09-24 | 2012-04-05 | Toshiba Corp | 半導体装置の製造方法 |
| JP2013074182A (ja) * | 2011-09-28 | 2013-04-22 | Nitto Denko Corp | 半導体装置の製造方法 |
| JP2015001655A (ja) * | 2013-06-17 | 2015-01-05 | 東レ株式会社 | 積層樹脂ブラックマトリックス基板 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2581427B2 (ja) * | 1993-12-16 | 1997-02-12 | 日本電気株式会社 | ポリイミド多層配線基板の製造方法 |
| JP2697661B2 (ja) * | 1995-03-17 | 1998-01-14 | 日本電気株式会社 | ポリイミド多層配線基板の製造方法 |
| JP2003231752A (ja) * | 2002-02-12 | 2003-08-19 | Toyobo Co Ltd | 感光性ポリイミド前駆体、感光性樹脂組成物、カラーフィルター、液晶駆動側基板、及び、液晶パネル |
| JP2013042052A (ja) * | 2011-08-19 | 2013-02-28 | Nec Corp | 半導体装置の製造方法 |
| JP2014011289A (ja) | 2012-06-29 | 2014-01-20 | Ibiden Co Ltd | 電子部品及び電子部品の製造方法 |
| EP2893565B1 (en) | 2012-09-05 | 2021-04-28 | Lumileds LLC | Laser de-bond of carrier wafer from device wafer |
-
2017
- 2017-05-31 KR KR1020187034624A patent/KR102138069B1/ko active IP Right Grant
- 2017-05-31 WO PCT/JP2017/020251 patent/WO2017209178A1/ja active Application Filing
- 2017-05-31 JP JP2018520957A patent/JP6704047B2/ja active Active
- 2017-06-01 TW TW106118077A patent/TWI709817B/zh active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6298799A (ja) * | 1985-10-25 | 1987-05-08 | 株式会社リコー | 多層配線形成方法 |
| JPH01159613A (ja) * | 1987-12-16 | 1989-06-22 | Nec Kansai Ltd | 超音波半田パターンの形成方法 |
| JP2004146426A (ja) * | 2002-10-22 | 2004-05-20 | Hitachi Ltd | 電子部品を搭載した薄膜配線基板の製造方法およびマルチチップモジュール |
| JP2004207262A (ja) * | 2002-12-20 | 2004-07-22 | Fujitsu Ltd | 薄膜多層回路基板及びその製造方法 |
| JP2009043962A (ja) * | 2007-08-09 | 2009-02-26 | Sony Corp | 半導体装置の製造方法 |
| JP2012069734A (ja) * | 2010-09-24 | 2012-04-05 | Toshiba Corp | 半導体装置の製造方法 |
| JP2013074182A (ja) * | 2011-09-28 | 2013-04-22 | Nitto Denko Corp | 半導体装置の製造方法 |
| JP2015001655A (ja) * | 2013-06-17 | 2015-01-05 | 東レ株式会社 | 積層樹脂ブラックマトリックス基板 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019109494A (ja) * | 2017-12-15 | 2019-07-04 | 旭化成株式会社 | 感光性樹脂組成物、硬化レリーフパターンの製造方法、及び半導体装置 |
| JP7169844B2 (ja) | 2017-12-15 | 2022-11-11 | 旭化成株式会社 | 感光性樹脂組成物、硬化レリーフパターンの製造方法、及び半導体装置 |
| TWI696045B (zh) * | 2018-05-21 | 2020-06-11 | 日商信越化學工業股份有限公司 | 圖案形成方法 |
| JP2020106530A (ja) * | 2018-12-26 | 2020-07-09 | カムテック リミテッド | 埋め込み層の測定 |
| JP7401294B2 (ja) | 2018-12-26 | 2023-12-19 | カムテック エルティーディー. | 埋め込み層の測定 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI709817B (zh) | 2020-11-11 |
| KR20190003680A (ko) | 2019-01-09 |
| TW201807495A (zh) | 2018-03-01 |
| JPWO2017209178A1 (ja) | 2019-03-22 |
| KR102138069B1 (ko) | 2020-07-27 |
| JP6704047B2 (ja) | 2020-06-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107709407B (zh) | 聚酰亚胺前体组合物、感光性树脂组合物、固化膜、固化膜的制造方法、半导体器件及聚酰亚胺前体组合物的制造方法 | |
| JP6255096B2 (ja) | 熱塩基発生剤、熱硬化性樹脂組成物、硬化膜、硬化膜の製造方法および半導体デバイス | |
| JP6167089B2 (ja) | 感光性樹脂組成物、硬化膜、硬化膜の製造方法および半導体デバイス | |
| JP6612343B2 (ja) | 前駆体組成物、感光性樹脂組成物、前駆体組成物の製造方法、硬化膜、硬化膜の製造方法および半導体デバイス | |
| JP6845848B2 (ja) | 積層体の製造方法、半導体素子の製造方法および積層体 | |
| JP6650517B2 (ja) | 硬化膜の製造方法、積層体の製造方法および半導体素子の製造方法 | |
| JP6247432B1 (ja) | 積層体、積層体の製造方法、半導体デバイス、および、半導体デバイスの製造方法 | |
| JP6511146B2 (ja) | 硬化膜の製造方法、再配線層用層間絶縁膜の製造方法、および、半導体デバイスの製造方法 | |
| WO2017110982A1 (ja) | 樹脂、組成物、硬化膜、硬化膜の製造方法および半導体デバイス | |
| JP6704047B2 (ja) | パターン製造方法、半導体装置の製造方法および積層体 | |
| JP6901596B2 (ja) | 積層体の製造方法および半導体デバイスの製造方法 | |
| JP6751159B2 (ja) | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、積層体の製造方法および半導体デバイス | |
| JP6704048B2 (ja) | ネガ型感光性樹脂組成物、硬化膜、硬化膜の製造方法、半導体デバイス、積層体の製造方法、半導体デバイスの製造方法およびポリイミド前駆体 | |
| WO2018038002A1 (ja) | 積層体の製造方法および電子デバイスの製造方法 | |
| WO2018221457A1 (ja) | 感光性樹脂組成物、ポリマー前駆体、硬化膜、積層体、硬化膜の製造方法および半導体デバイス |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018520957 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17806729 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20187034624 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17806729 Country of ref document: EP Kind code of ref document: A1 |