WO2016159211A1 - 試験管内淘汰及び分子間相互作用解析用高速光架橋型共用リンカー、そのリンカーを用いた試験管内淘汰方法 - Google Patents
試験管内淘汰及び分子間相互作用解析用高速光架橋型共用リンカー、そのリンカーを用いた試験管内淘汰方法 Download PDFInfo
- Publication number
- WO2016159211A1 WO2016159211A1 PCT/JP2016/060611 JP2016060611W WO2016159211A1 WO 2016159211 A1 WO2016159211 A1 WO 2016159211A1 JP 2016060611 W JP2016060611 W JP 2016060611W WO 2016159211 A1 WO2016159211 A1 WO 2016159211A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- linker
- mrna
- site
- photocrosslinking
- protein
- Prior art date
Links
Images



























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1096—Processes for the isolation, preparation or purification of DNA or RNA cDNA Synthesis; Subtracted cDNA library construction, e.g. RT, RT-PCR
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B30/00—Methods of screening libraries
- C40B30/04—Methods of screening libraries by measuring the ability to specifically bind a target molecule, e.g. antibody-antigen binding, receptor-ligand binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/001—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof by chemical synthesis
- C07K14/003—Peptide-nucleic acids (PNAs)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1003—Extracting or separating nucleic acids from biological samples, e.g. pure separation or isolation methods; Conditions, buffers or apparatuses therefor
- C12N15/1006—Extracting or separating nucleic acids from biological samples, e.g. pure separation or isolation methods; Conditions, buffers or apparatuses therefor by means of a solid support carrier, e.g. particles, polymers
- C12N15/101—Extracting or separating nucleic acids from biological samples, e.g. pure separation or isolation methods; Conditions, buffers or apparatuses therefor by means of a solid support carrier, e.g. particles, polymers by chromatography, e.g. electrophoresis, ion-exchange, reverse phase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/93—Ligases (6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y605/00—Ligases forming phosphoric ester bonds (6.5)
- C12Y605/01—Ligases forming phosphoric ester bonds (6.5) forming phosphoric ester bonds (6.5.1)
- C12Y605/01001—DNA ligase (ATP) (6.5.1.1)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6845—Methods of identifying protein-protein interactions in protein mixtures
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
- C12N2310/3513—Protein; Peptide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2529/00—Culture process characterised by the use of electromagnetic stimulation
- C12N2529/10—Stimulation by light
Definitions
- the present invention relates to a linker for use in a cDNA display method, which is a genotype-phenotype association technique, and a method for using the linker. More specifically, the present invention relates to a novel puromycin linker that can be used for both in vitro test and affinity measurement, and an in vitro test method using the linker.
- antigen recognition sites and peptide aptamers of antibodies can be artificially synthesized due to their low molecular weight.
- Such a peptide aptamer can be synthesized and purified using a genotype-phenotype association technique (hereinafter, also simply referred to as “association technique”) after preparing mRNA.
- association technique includes a phage display method, a ribosome display method, an mRNA display method, and the like.
- the cDNA display method is most suitable for synthesizing peptide aptamers in consideration of the demands for automation and high throughput.
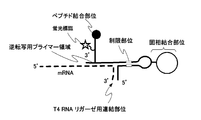
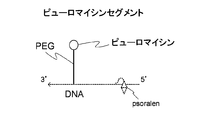
- FIG. 1A shows a solid phase binding site, a main chain comprising a T4 RNA ligase linking site for linking mRNA and the main chain of this linker with T4 RNA ligase, a reverse transcription primer region, and a peptide bond.
- a linker having a site and a fluorescent label and comprising a side chain linked to the main chain is shown (see Patent Document 1, hereinafter referred to as “Conventional Example 1”).
- FIG. 1B has almost the same structure as the linker of Conventional Example 1 except that the main chain is partially composed of double strands.
- a linker incorporating a restriction site for separating the linker from the solid phase is shown (see Patent Document 1, hereinafter referred to as “Conventional Example 2”).
- FIG. 1C shows a linker having first and second cleavage sites for separating the solid phase and the linker in addition to Conventional Example 1 (see Patent Document 1, hereinafter “Conventional Example”). 3 ”).
- FIG. 1D shows a linker in which two main chains are connected by psoralen (see Patent Document 2, hereinafter referred to as “Conventional Example 4”).
- cancer is the number one cause of death for Japanese people at present, but the proportion of the cause of death is increasing year by year.
- tumor Marker To observe whether or not a tumor is present and the main post-treatment course, once the tumor is present in the body, certain levels of certain substances in the blood or urine (tumor Marker) is detected. Then, by measuring the types of tumor markers and their blood or urine levels, the presence or absence of the tumor and its progress can be known.
- tumor markers are sugar chain antigens.
- the chain length of sugar chains on the cell surface is changed by glycosyltransferases, resulting in cancer cell-specific sugar chains, so these sugar chains are tumor markers for identifying cancer.
- glycan tumor marker the antigenic determinant (hereinafter sometimes referred to as “epitope”) of the glycan tumor marker has a general structure as schematically shown in FIG. 2 (A). As shown in Table 1, it is divided into type 1 sugar chains, type 2 sugar chains, mother nucleus sugar chains, and core proteins.
- the type 1 sugar chain and the type 2 sugar chain include those shown in Table 1 above, and these sugar chains are both glycosyltransferases on the cell surface. Is longer than that of normal cells.
- the core protein refers to a protein present around a viral nucleic acid.
- FIG. 2 (B) schematically shows the structure of CA19-9 (Carbohydrate Antigen-919-9), which is one type of sugar chain antigen against the basic structure, as an example of such a sugar chain antigen.
- CA19-9 is a molecule with a molecular weight of 874 having a structure consisting of five monosaccharides, and each monosaccharide is connected by a glycosidic bond.
- NeuNac represents N-acetylneuraminic acid
- Gal represents galactose
- Fuc represents fucose
- GlcNAc represents N-acetyl-D-glucosamine.
- GlcNAc contained in CA19-9 shown in FIG. 2 (B) is also contained in many sugar chain antigens other than CA19-9 (see Non-Patent Document 5, Hereinafter, it is referred to as “Conventional Example 6”.
- an antibody is generally composed of a light chain and a heavy chain
- the molecular weight of immunoglobulin (IgG) is known to be as large as 170 KDa.
- camelid antibodies do not have a light chain, are composed only of heavy chains, are known to have a low molecular weight of 12 KDa, high conformational plasticity, and excellent thermal stability.
- a molecule having such a region (domain) may be referred to as “VHH”.
- the above-mentioned conventional techniques are all excellent as linkers used in the cDNA display method.
- the cDNA display technology is based on the use of mRNA, and an enzyme called T4 RNA ligase is used to link the main chain to the mRNA.
- T4 RNA ligase an enzyme used to link the main chain to the mRNA.
- RNase in general molecular biology laboratories using E. coli, RNase cannot be completely removed from the sample used, so the mRNA used for peptide aptamer synthesis is degraded in vitro, which In all of the linkers of Examples 1 to 4, there was a problem that peptide aptamers were not synthesized well.
- each of the conventional linkers has a problem that it takes 30 minutes or more to connect the main chain and mRNA using an enzyme called T4 RNA ligase.
- a ligation in which mRNA, cDNA, and peptide are displayed on the linker is formed, and such a ligation is collected, and the displayed cDNA or peptide is generated and the sequence is identified.
- a cDNA library or a peptide library can be constructed.
- the linker used in the soot process cannot be used for intermolecular interaction analysis. That is, the in vitro fistula experiment (screening) for obtaining candidate clones and the evaluation of the binding of candidate clones obtained by screening could not be performed using the same linker. For this reason, separate screening must be used for screening candidate clones and the screening test for the binding of candidate clones that have been screened, and this has been a problem in terms of work efficiency and cost.
- an antibody such as an immunoglobulin.
- an antibody composed of a light chain and a heavy chain has a large molecular weight and is irreversible with heat of 70 ° C or higher. In other words, there is a problem that chemical synthesis cannot be performed.
- sugar chains have many kinds of sugars and the structure of the sugar chain itself is complicated, there is a problem that it is very difficult to obtain an antibody capable of recognizing a sugar chain antigen.
- camelid antibody such as VHH
- denaturation is reversible even when heat-treated at 90 ° C., and when it is returned to room temperature, it shows the same antigenic activity as before heat-treatment.
- the antibody production rate in camelids is low, and it takes time and effort to purify, and there is a problem that the target antibody cannot always be obtained.
- a molecule capable of specifically recognizing a carbohydrate antigen can be obtained, it can be used as a diagnostic agent to quickly and accurately determine cancer prevention and therapeutic effects. And if such a determination becomes possible, even if it suffers from cancer, it becomes possible to receive an appropriate treatment at an early stage, thereby making it possible to improve the cure rate.
- one embodiment of the present invention is a linker having a main chain and a side chain, wherein the main chain has a predetermined base sequence, is located at the 5 ′ end of the main chain, and binds to a solid phase A solid phase binding site for forming the solid phase; a solid phase cleavage site for separating the solid phase together with the solid phase binding site; a side chain linking site for linking the side chains; and the side chain linking site; A high-speed photocrosslinking site located between the solid-phase cleavage site and linking mRNA having a sequence complementary to the main chain by photocrosslinking; adjacent to the side chain linking site; A reverse transcription initiation region located at the 3 ′ end of the main chain, wherein the side chain is linked to a fluorescent label, a protein binding site located at the free end, and a side chain linking site of the main chain A formation site, and the side chain is connected to the side chain connection site at the connection
- the solid-phase cleavage site is preferably composed of a base selected from the group consisting of deoxyinosine, riboG or ribopyrimidine.
- the high-speed photocrosslinking site is preferably composed of a cyanovinylcarbazole compound, and the cyanovinylcarbazole compound is preferably 3-cyanovinylcarbazole.
- the solid phase binding site is any compound selected from the group consisting of biotin, streptavidin, alkyne, azide by click chemistry, amino group, N-hydroxysuccinimide ester (NHS), SH group, and Au, It is preferably composed of poly A bound to a compound, and the protein binding site is preferably composed of puromycin or an analogous compound thereof.
- PANS-amino acid 3'-N-aminoacyl adenosine amino acid nucleoside (AANS-amino acid), etc.
- PANS-Gly which is glycine, PANS-Val, which is valine, PANS-Ala, which is alanine, a mixture of PANS amino acids, AANS-Gly, in which the amino acid part of AANS is glycine, AANS-Val, which is valine, AANS that is alanine More preferably, it is any compound selected from the group consisting of a mixture of -Ala and AANS amino acids.
- Another aspect of the present invention includes a complementary bond forming step of complementary bonding of the main chain of the high-speed photocrosslinking shared linker for analyzing in vitro and intermolecular interactions with the desired mRNA;
- a test tube
- the solid phase is preferably magnetic particles coated with streptavidin or avidin.
- the cleavage of the conjugate in the selection step is preferably performed with any enzyme selected from the group consisting of endonuclease V, RNase T1, and RNase A. It is preferable that the main chain of the high-speed photocrosslinking shared linker has a sequence that recognizes a sugar chain antigen.
- Still another embodiment of the present invention includes a complementary bond forming step of complementary bonding of the main chain of the high-speed photocrosslinking shared linker for analyzing in vitro and intermolecular interactions with a desired mRNA;
- a photocrosslinking step of irradiating the formed main chain and mRNA with light having a wavelength of 300 to 400 nm for 0.5 to 5 minutes to form a photocrosslink; translating the mRNA bound to the linker in a cell-free translation system;
- a linker-protein fusion forming step a solid phase binding step of binding the linker-protein fusion to a solid phase; and elution of the linker-protein fusion from the solid phase under predetermined conditions; Seisuru purification step and;
- Affinity measurement linker
- the solid phase is preferably magnetic particles coated with streptavidin or avidin.
- the purification step is preferably performed at room temperature in an aqueous solution containing 0 to 100 mM NaCl.
- Still another embodiment of the present invention is an affinity measurement linker-protein produced by the above method.
- a linker for high-speed photocrosslinking cDNA display that can significantly reduce the time required for linking the linker and mRNA. Moreover, by using the linker for high-speed photocrosslinking type cDNA display, there is provided an in vitro scissor method capable of efficiently selecting candidate clones.
- linker for high-speed photocrosslinking cDNA display there is provided a method for producing a linker-protein for measuring affinity, which can be used for evaluating the binding of candidate clones obtained by the above method. Is done.
- an affinity measurement linker-protein produced by the above method is provided.
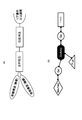
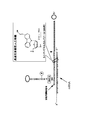
- FIG. 1A is a schematic diagram showing a linker (SBP) of Conventional Example 1 that does not have a cleavage site with a solid phase.
- FIG. 1B is a schematic diagram showing a linker of Conventional Example 2 having a double-stranded cleavage site (restriction site) with a solid phase.
- FIG. 1C is a schematic diagram showing a linker of Conventional Example 3 having a plurality of single-stranded cleavage sites with a solid phase.
- the first and second cleavage sites of the linker shown in FIG. 1C are composed of ribosylguanosine (rG) or deoxyinosine (I).
- FIG. 1D is a schematic diagram showing a linker of Conventional Example 4 in which two main chains are crosslinked with psoralen.
- FIG. 2 (A) is a diagram schematically showing the structure of an antigenic determinant of a sugar chain tumor marker.
- FIG. 2 (B) is a diagram schematically showing the structure of CA19-9, which is an example of an antigenic determinant of a sugar chain tumor marker.
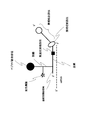
- FIG. 3A is a schematic diagram showing a high-speed photocrosslinking shared linker for analyzing in vitro test tubes and intermolecular interactions according to the present invention.
- FIG. 3B is a diagram showing the structure of the linker of the present invention shown in FIG. 3A.
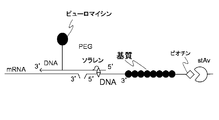
- FIG. 4 is a schematic diagram showing that the linker of the present invention can be used for both in vitro scavenging and intermolecular interaction analysis assays.
- FIG. 5 is a diagram showing the structure of the VHH library construct used in the present invention.
- FIG. 6 is a graph showing the theoretical frequency of random amino acids indicated by the mixed base codon in SEQ ID NO: 6 in the sequence listing.
- (A) shows the case where the codon is nbn
- (B) shows the case where the codon is drb
- (C) shows the case where the codon is yhm.
- FIG. 7A is a gel electrophoresis photograph showing a crosslinked state by ultraviolet irradiation when the linker of the present invention is used (+) and when it is not used ( ⁇ ).
- FIG. 7B is a gel electrophoresis photograph showing the result of examining the degradation of the mRNA-linker fusion (conjugate) by the ultraviolet irradiation time (detection by FITC).
- FIG. 7C is a gel electrophoresis photograph showing the result of examining the degradation of the mRNA-linker fusion (conjugate) by the ultraviolet irradiation time (detection by SYBR Gold).
- FIG. 7D is a gel electrophoresis photograph showing the result of examining degradation of mRNA-linker fusion (conjugate) by ultraviolet irradiation time when the linker of Conventional Example 1 is used (detection by FITC and SYBRSYGold).
- FIG. 7E is a gel electrophoresis photograph showing the result of examining the degradation of the mRNA-linker fusion (conjugate) by the ultraviolet irradiation time when the linker of the present invention is used (detection by SYBR Gold).
- FIG. 7F is a gel electrophoresis photograph showing the result of examining the degradation of mRNA-linker fusion (conjugate) by the amount of ultraviolet irradiation when the linker of the present invention is used (detection by SYBR Gold).
- FIG. 7G is a gel electrophoresis photograph showing the result of examining the influence of ultraviolet irradiation time on cDNA synthesis when the linker of the present invention is used (detection by SYBR Gold).
- FIG. 8 is a chart showing the distribution of the initial library (upper (A)) and the library (lower (B)) after three rounds in the in vitro test method.
- FIG. 9 is a chart showing the binding confirmation result by competitive elution with a low concentration of GlcNAc non-binding protein.
- FIG. 10 is a chart showing the binding confirmation results by competitive elution with a high concentration of GlcNAc non-binding protein.
- FIG. 11 is a diagram showing gel electrophoresis results showing differences in peptides contained in the washing solution and the eluate.
- FIG. 12 is a gel electrophoresis photograph showing the first round product in the preparation of the cross-linked peptide aptamer.
- FIG. 13 is a gel electrophoresis photograph showing the product of the sixth round in the preparation of the cross-linked peptide aptamer.
- FIG. 14 is a gel electrophoresis photograph showing formation of an mRNA-peptide conjugate in which a peptide is further linked to an mRNA-linker conjugate.
- FIG. 15 is a gel electrophoresis photograph showing the results of confirming the degree of progression of in-vitro fistula in the first to third rounds.
- FIG. 16 is a gel electrophoresis photograph showing the results of confirming the degree of progression of the in-vitro fistula in the fourth round.
- FIG. 17 is a gel electrophoresis photograph showing the result of confirming the degree of progression of the in-vitro fistula in the fifth round.
- FIG. 18 is a gel electrophoresis photograph showing the result of confirming the degree of progression of the in-vitro fistula in the sixth round.
- FIG. 19 is a gel electrophoresis photograph in which the difference in the product depending on the presence or absence of the cDNA display of the VHH peptide was confirmed.
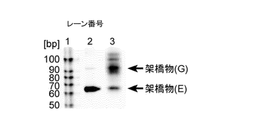
- FIG. 20 is a gel electrophoresis photograph showing that the mRNA and the linker of the present invention were crosslinked by photocrosslinking.
- FIG. 21A is a diagram showing the base sequence and restriction sites of the main chain of the linker of Conventional Example 2.
- FIG. 21B is a diagram showing that the linker of Conventional Example 2 is not cleaved by endonuclease V.
- FIG. 22 is a diagram showing that the linker of Conventional Example 3 is decomposed by RNase but the linker of Conventional Example 4 is not decomposed.
- FIG. 23A is a diagram showing segments when the linker of Conventional Example 5 is synthesized (1).
- FIG. 23B is a diagram showing segments when the linker of Conventional Example 5 is synthesized (2).
- FIG. 23C is a gel electrophoresis photograph showing the product of the synthesis process of the linker of Conventional Example 5 (1).
- FIG. 23D is a gel electrophoresis photograph showing the product of the synthesis process of the linker of Conventional Example 5 (2).
- FIG. 23E is a gel electrophoresis photograph showing the product of the synthesis process of the linker of Conventional Example 5 (3).
- the present invention is a linker for high-speed photocrosslinking cDNA display comprising (a) a main chain and (b) a side chain, wherein the main chain (a) comprises (a1) a predetermined base.
- a solid phase binding site having a sequence and located at the 5 'end of the main chain to form a bond with the solid phase; (a2) solid phase cleavage for separating the solid phase together with the solid phase binding site (A3) a side chain linking site for linking the side chains; (a4) located between the side chain linking site and the solid phase cleavage site and having a sequence complementary to the main chain a high-speed photocrosslinking site for linking mRNA to the main chain by photocrosslinking; (a5) a reverse transcription initiation region located adjacent to the side chain linking site and located at the 3 ′ end of the main chain; .
- the side chain (b) is (b1) a fluorescent label, (b2) a protein binding site located at the free end, and (b3) a linkage formation site linked to the side chain linkage site of the main chain, It has.
- the side chain (b) is connected to the side chain connection site of the main chain (a) at the connection forming site.
- the solid phase binding site (a1) is selected from the group consisting of biotin, streptavidin alkyne, azide by click chemistry, amino group, N-hydroxysuccinimide ester (NHS), SH group, and Au. It is preferably composed of any selected compound and poly A bonded to the compound. It is preferable that at least 10 or more adenines are bonded to the poly A because it can maintain an appropriate distance from the solid phase and can be easily separated from the solid phase described later, and about 20 are bonded. More preferably.
- the solid-phase cleavage site (a2) is preferably composed of a base selected from the group consisting of deoxyinosine, riboG or ribopyrimidine for the following reasons.
- the two conjugates described later ie, the linker of the present invention, the conjugate of mRNA and cDNA, or the linker of the present invention, the conjugate of mRNA, cDNA and peptide (hereinafter, both are simply fused together.
- any enzyme selected from the group consisting of endonuclease V, RNase T1, and RNase A is used. This is because if the solid-phase cleavage site is composed of any of the above bases, the solid phase can be specifically separated from the linker together with the solid-phase binding site.
- the linker of the present invention reacts with a fusion (conjugate) between the linker and the linker without affecting the mRNA, cDNA, and / or protein corresponding to the mRNA, even if they are bound. It can be recovered in the supernatant of the liquid.
- the main chain of the present invention is provided with a side chain linking site (a3) for linking side chains described later.
- a high-speed photocrosslinking site (a4) for linking mRNA having a sequence complementary to the main chain by photocrosslinking is located between the side chain linking site and the solid-phase cleavage site. Is provided.
- the high-speed photocrosslinking site is preferably composed of a cyanovinylcarbazole compound because it does not cause degradation of mRNA.
- cyanovinyl carbazole compounds examples include 3-cyanovinyl carbazole and the like, and when 3-cyanovinyl carbazole (hereinafter, sometimes referred to as “cnvK”) is used, it can be submerged in water in a very short time.
- the main chain of the linker can be bound to the mRNA to be screened using ultraviolet light having a long wavelength. This means that it is not necessary to use an enzyme such as T4 RNA ligase and the crosslinking reaction can be carried out in water.
- the enzyme reaction for ligation requires a buffer containing metal ions such as Zn 2+ , but RNase that degrades RNA cannot be completely removed from such a buffer.
- RNase When T4 RNA ligase is used for binding between mRNA and the main chain, RNase is also activated under conditions that activate T4 RNA ligase. For this reason, it often occurred that mRNA was degraded by RNase and cDNA was not synthesized.
- cnvK by constructing the high-speed photocrosslinking site with cnvK, it is possible to perform the cross-linking reaction under the condition of water in which the enzyme cannot act without using the enzyme, thus preventing the degradation of mRNA by RNase. It becomes possible.
- the wavelength of light used for crosslinking of the above high-speed photocrosslinking site is a considerably long wavelength of 300 to 400 nm as described later, and the irradiation time is short. For this reason, there is an advantage that a desired peptide corresponding to the used mRNA can be obtained without causing a problem that a thymine dimer is formed in the synthesized cDNA.
- the reverse transcription initiation region (a5) for synthesizing cDNA corresponding to the mRNA linked to this linker is formed adjacent to this side chain linking site and located at the 3 ′ end of the main chain. Has been.
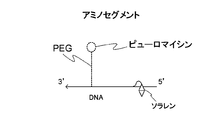
- the (b) side chain contained in the linker of the present invention includes (b1) a fluorescent label, (b2) a protein binding site located at the free end, and (b3) a side chain linking site of the main chain. And a connection forming portion to be connected.
- the (b1) fluorescent label include fluorescein, rhodamine, Cy dye, AlexaR Fluor, and the like. More specifically, the use of FITC is preferable from the viewpoint of cost.
- the protein binding site located at the free end of the side chain is preferably composed of puromycin or an analogous compound thereof.
- puromycin-related compounds include 3′-N-aminoacylpuromycin (PANS-amino acid) and 3′-N-aminoacyladenosine amino acid nucleoside (AANS-amino acid).
- PANS-Gly in which the amino acid part of PANS is glycine, PANS-Val, which is valine, PANS-Ala, which is alanine, a mixture of PANS amino acids
- AANS-Gly in which the amino acid part of AANS is glycine, and valine AANS-Val that is AANS, AANS-Ala that is alanine, a mixture of AANS amino acids, and the like.
- the (b3) linkage forming site of the side chain is preferably composed of a heteroreactive divalent reagent capable of crosslinking reaction between an amino group and an SH group, for example, N- (6-Maleimidocaproyloxy) oxysuccinimide (Hereinafter sometimes referred to as "EMCS").
- EMCS N- (6-Maleimidocaproyloxy) oxysuccinimide
- the side chain (b) is connected to the side chain connection site (a3) of the main chain at the connection formation site (b3).
- the cDNA-display linker of the present invention forms an mRNA having a desired sequence and a peptide conjugate corresponding to the mRNA, and the conjugate is immobilized on a solid phase.
- the cDNA is further ligated by reverse transcription, and then the solid phase is separated from the solid phase binding site using any enzyme selected from the group consisting of endonuclease V, RNase T1, and RNase A.
- a cDNA display can be obtained. That is, it can be used as a linker for cDNA display (FIG. 5).
- RNA of the mRNA-peptide-linker conjugate is digested with an enzyme, and then, for example, bound to a solid phase such as oligo dT magnetic beads at the solid phase binding site and eluted with an elution buffer, thereby solid phase binding.
- a protein-linker conjugate with a site can be obtained, and this conjugate can be used as it is in a binding assay using SPR (surface plasmon resonance device), QCM (quartz crystal microbalance), etc. (FIG. 4).
- the linker for cDNA display of the present invention as described above can be prepared as follows. First, the main chain of the linker of the present invention (hereinafter sometimes referred to as “poly A + cnvK segment”) is designed so that cnvK is at a desired position between the solid phase binding site and the side chain binding site. Then, chemical synthesis of DNA is performed according to a conventional method. Such chemical synthesis of DNA strands can be outsourced to a company that performs the synthesis. Such a main chain can be designed to include, for example, a reverse transcription initiation region as shown in FIG. 3B, a side chain linking site, a high-speed photocrosslinking site, and a solid phase binding site. Of the main chain shown in FIG.
- the base sequence of the main chain excluding the modified site is shown in the following sequence (SEQ ID NO: 1 in the sequence listing).
- the following main chain has BioTEG added to the 5 ′ end.
- N represents inosine
- X represents amino C6-dT.
- the side chain (puromycin-segment) of the linker of the present invention is also designed to have a desired sequence, and the chemical synthesis of DNA is performed according to a conventional method in the same manner as the Poly A + cnvK segment.
- Such chemical synthesis of DNA strands can be outsourced to a company that performs the synthesis.
- Such side chains can be designed to include, for example, a linking site as shown in FIG. 3B, a fluorescent molecule, and a protein binding site.
- the base sequence excluding the modified site can be exemplified as follows.
- the solid-phase cleavage site is preferably composed of a base selected from the group consisting of deoxyinosine, riboG or ribopyrimidine, in order to specifically cleave later from the solid phase.
- P which is the free end, is puromycin as a protein binding site.
- (5S) represents 5′Thiol C6, F represents FITC-dT, and Z represents Spacer18.
- the main chain having the above sequence is added to 0.1 to 0.3 M sodium phosphate (pH 7.0 to 7.4) containing 10 to 20 nmol (final concentration 100 to 200 ⁇ M), and EMCS (manufactured by Dojindo Laboratories Co., Ltd.) ) Is added to a final concentration of 15-18 mM and incubated at about 37 ° C. for 20-40 minutes, followed by ethanol precipitation.
- EMCS is added to a final concentration of about 16.7 mM in about 0.2 M sodium phosphate solution (about pH 7.2) containing about 15 nmol of the main chain (final concentration is about 150 ⁇ M), and about Incubate at 37 ° C. for about 30 minutes, and then ethanol precipitate using, for example, Quick-Precip Plus Solution (manufactured by Edge BioSystems).
- nmol of side chains are dissolved in 0.8-1.5 M sodium hydrogenphosphate aqueous solution containing 40-60 mM DTT so that the final concentration is 400-430 ⁇ M, and the mixture is used at room temperature using a shaker. Stir for 0.75 to 1.5 hours.
- buffer exchange is performed on this solution.
- about 37.5 nmol of the side chain is dissolved in about 1 M sodium hydrogen phosphate aqueous solution containing about 50 mM DTT so that the final concentration is about 417 ⁇ M, and about 1 hour at room temperature using a shaker.
- the buffer is exchanged with about 0.1 M sodium phosphate (about pH 7.0) containing about 0.15 M NaCl using, for example, a NAP5 column.
- the solution containing the reduced side chain subjected to the buffer exchange as described above is mixed with the above-described EMCS-modified ethanol precipitation product of the main chain and left at 2 to 6 ° C. overnight.
- DTT is added to the reaction solution so as to be 40 to 60 mM and stirred at room temperature for 15 to 60 minutes.
- ethanol precipitation is performed, and the obtained ethanol precipitate is dissolved in 50 to 200 ⁇ L of Nuclease-free water to purify.
- the solution containing the reduced side chain buffer-exchanged with the above buffer is mixed with the above-mentioned ethanol precipitation product of the EMCS-modified main chain and left at about 4 ° C. overnight.
- the eluent used for gradient elution is, for example, 0.05 to 0.2M trimethylammonium acetate (ultra pure water) for solution A, 75 to 85% acetonitrile for solution B, and the ratio of solution A during the elution period at the start. It may be reduced by about 20% over 40 to 50 minutes.
- the flow rate can be 0.5-1.5 ml / min and the fraction can be 0.5-1.5 mL.
- liquid A is about 0.1M trimethylammonium acetate (ultra pure water)
- liquid B is about 80% acetonitrile
- the ratio of liquid A during the starting elution period (about 85%) is 40 to 50 minutes. And reduce it to about 65%.
- the flow rate is about 1.0 ml / min and the fraction is about 1.0 mL.
- the components in the above fractions are confirmed by fluorescence and ultraviolet absorption (for example, 280 nm), the fractions in which peaks are observed by both detection means are collected, the solvent is evaporated using a vacuum evaporator, and then ethanol precipitation is performed.
- the linker of the present invention can be produced by dissolving in Nuclease-free water.
- the resulting linker of the present invention is stored at about ⁇ 20 ° C.
- the fraction from 30 to 32 minutes is collected, and the solvent is evaporated using a vacuum evaporator. Thereafter, for example, ethanol precipitation is performed using Quick-Precip Plus Solution, dissolved in Nuclease-free water, and stored at about -20 ° C.
- a DNA mixture is prepared by mixing a library DNA having a desired sequence and a DNA encoding a FLAG sequence at a desired ratio, for example, a molar ratio of 25,000 to 100,000: 1, and a portion thereof is transcribed. For example, 250 to 1,000 ng is transcribed on a 10 to 20 ⁇ L scale using a kit such as RiboMAX Large Scale RNA Production Systems-T7.
- a sequence used for transcription for example, the following sequence (SEQ ID NO: 2 in the sequence listing) can be used.
- N arbitrarily represents A, T, G, and C
- K represents G or T.
- DNase is added in a desired amount. Thereafter, it is further incubated at the desired temperature for a desired time, for example, at about 37 ° C. for 5-15 minutes, and the resulting mRNA is then purified.
- a desired temperature for a desired time for example, at about 37 ° C. for 3 to 5 hours
- DNase® for example, RQ1Dnase, Promega
- the obtained mRNA is purified using, for example, After Tri-Reagent RNA Clean-Up Kit (Favogen Biotech Corp.).
- biotin-cnvK linker-mRNA conjugate (hereinafter sometimes referred to as “mRNA-linker conjugate”) is obtained at the desired temperature using the cell-free translation system at the desired scale in the same manner as described above.
- Translate time For example, using 10-20 ⁇ L of the mRNA-linker conjugate, translate at a scale of 100-150 ⁇ L, for example, at about 30 ° C. for about 15 minutes using a cell-free translation system such as rabbit reticulocyte lysate.
- MgCl 2 and KCl can be added at desired concentrations to obtain an mRNA-peptide conjugate in which a peptide is further linked to the mRNA-linker conjugate.
- MgCl 2 and KCl are added to 75 mM and 900 mM, respectively, and incubated at about 37 ° C. for about 1 hour to obtain a translation reaction solution containing an mRNA-peptide conjugate.
- 0.25 to 1M EDTA (pH 7.8 to 8.2) is added at a desired concentration and incubated at a desired temperature, and ribosomes bound to the mRNA-peptide conjugate are removed. Remove. For example, about 0.5 M EDTA (pH ⁇ about 8.0) is added to a final concentration of about 83 mM and incubated at room temperature for about 5 minutes.
- the above magnetic beads are washed a desired number of times with a desired amount of 1 ⁇ binding buffer for SA, and then reverse transcription is performed. For example, it is washed 2 to 4 times with about 200 ⁇ L of 1 ⁇ binding buffer for SA, and about 100 ⁇ L of the reverse transcription reaction solution is added according to the protocol attached to ReverTra Ase (registered trademark), for example, at about 42 ° C. Reverse transcription is performed with stirring for 15 minutes to prepare an mRNA / cDNA-peptide conjugate (hereinafter sometimes referred to as “cDNA display”).
- ReverTra Ase registered trademark
- the magnetic beads are washed with a desired amount of 1 ⁇ NE buffer, and then a desired amount of 1 ⁇ NE buffer containing any enzyme selected from the group consisting of endonuclease V, RNase T1, and RNase A is added. For the desired time.
- the desired amount of 2xHis-tag wash buffer is then added, after which the supernatant is collected.
- Dynabeads MyOne C1 streptavidin is washed with about 150 ⁇ L of 1 ⁇ NE buffer, and then about 75 ⁇ L of 1 ⁇ NE buffer containing about 10 U of Endonuclease IV is added and stirred at about 37 ° C. for about 1 hour.
- About 75 ⁇ L of 2 ⁇ His-tag wash buffer (about 40 mM sodium phosphate (about pH 7.4) containing about 1 M NaCl, about 0.1% Tween-20) is then added, after which the supernatant is collected.
- the collected supernatant and magnetic beads such as His-Mag-Sepharose-Ni washed with, for example, 1xHis-tag washing buffer are mixed and stirred at a desired temperature for a desired time using a mixer.
- the beads are washed the desired number of times with 1xHis-tag wash buffer, then the desired selection buffer is added and stirred for the desired time at the desired temperature using a mixer, after which the supernatant is collected.
- about 150 ⁇ L of the collected supernatant and about 20 ⁇ L of HisHiMag Sepharose Ni (GE Healthcare, washed with 1xHis-tag washing buffer) are mixed, and Intellimixer RM-2M (Toho) Or the like, and stir at room temperature for about 1 hour.
- a desired amount from the obtained linker for example, fill a desired column with a suspension of anti-FLAG M2 affinity gel and wash with a desired amount of selection buffer. Then, a desired amount of the above supernatant is loaded onto the column and stirred at room temperature for about 1 hour using a rotator or the like. Wash the column multiple times with the desired amount of selection buffer, then add, for example, the desired concentration of FLAG peptide (manufactured by Sigma-Aldrich Japan LLC), and stir at room temperature for about 15 minutes using the above rotator, etc. The mRNA / cDNA-peptide conjugate bound to the anti-FLAG M2 affinity gel is competitively eluted.
- anti-FLAG® M2 affinity gel about 40-60% suspension
- a suitable column such as MicroSpin® Empty® Columns (GE Healthcare) and about 150-250 ⁇ L of selection buffer. Wash 2-4 times with. Thereafter, about 50 to 150 ⁇ L of the supernatant is taken and loaded onto the column, and stirred at room temperature for about 1 hour using a rotator. Wash the column 3-5 times with about 150-250 ⁇ L of selection buffer, then add about 50-150 ⁇ L of about 50-150 ng / ⁇ L of 3xFLAG peptide (manufactured by Sigma-Aldrich Japan GK) at room temperature using the above rotator. By stirring for about 15 minutes, the mRNA / cDNA-peptide conjugate bound to the anti-FLAG M2 affinity gel can be competitively eluted.
- the eluate in the column is collected by centrifugation, ethanol precipitated, and then dissolved in a desired amount of nuclease-free water.
- the ethanol precipitation product is added to about 150 to 250 ⁇ L of the PCR reaction solution, and PCR is performed.
- the eluate in the column recovered by centrifugation is ethanol precipitated using Quick-Precip Plus Solution or the like and dissolved in about 15 ⁇ L of Nuclease-free water.
- PCR is carried out as follows: The two exemplified NewYtag sequences for T7 ⁇ new and cnvK (SEQ ID NOs: 3 and 4 in the sequence listing) are shown below.
- PCR is performed by, for example, (a1) 98 ° C for 1 minute, (b1) 98 ° C for 15 seconds, (c1) 68 ° C for 30 seconds, (d1) 68 ° C for 1 minute, (b1) and ( c1) can be 25 cycles.
- the obtained PCR product is subjected to SDS gel electrophoresis, the full construct DNA is excised and purified according to a conventional method. Double-stranded DNA can be obtained by adding the purified full-construct DNA to a desired amount of PCR reaction solution, dispensing the desired amount, and performing PCR.
- the PCR product obtained by performing PCR as described above is electrophoresed on, for example, 8M urea-denatured 6% PAGE, and the full construct DNA (260 to 300 mer) is excised and purified according to a conventional method.
- About 150 to 250 ⁇ L of PCR reaction solution about 0.2 mM dNTPs, about 0.4 ⁇ M Newleft, about 0.4 ⁇ M cnvK NewYtag, and about 0.02 U / ⁇ L PrimeSTAR HS DNA polymerase
- 1xPrimeSTAR buffer containing Mg 2+
- double-stranded DNA can be obtained by dispensing about 25 to 75 ⁇ L each and performing PCR.
- the sequence of Newleft (SEQ ID NO: 5 in the sequence listing) exemplified as the peptide used here is shown below.
- PCR is performed, for example, by (a2) 98 ° C for 1 minute, (b2) 98 ° C for 10 seconds, (c2) 68 ° C for 30 seconds, (d2) 68 ° C for 1 minute, steps (b2) and (c2) 5 cycles can be performed.
- the PCR reaction solution after performing PCR as described above is collectively purified and used in the next round.
- the above procedure is performed from the transcription of the library DNA to the affinity selection in a desired round, for example, a total of 3 rounds, and the resulting full construct DNA sequence is directly sequenced to form a unit.
- the linker of the present invention displaying FLAG-DNA of the sequence described above can be obtained.
- an mRNA-protein-linker conjugate corresponding to the base sequence of the known protein can be obtained according to the same procedure as described above.
- a protein whose sequence is known for example, a B domain of A protein (hereinafter referred to as “BDA”, SEQ ID NO: 6 in the sequence listing) and the like can be used.
- a desired buffer and RNase H are added to the translation product containing the mRNA-protein-linker conjugate described above, and incubated at a desired temperature for a desired time to degrade the mRNA.
- the above magnetic beads are washed with a desired amount of a desired buffer, and then a desired amount of ultrapure water is added and incubated at a desired temperature for a desired time to elute biotin adhesion protein.
- An equal amount of the desired washing buffer is added to the biotin adhesion protein solution thus obtained.
- it is mixed with a desired amount of magnetic particles such as His Mag Sepharose Ni, and stirred at a desired temperature for a desired time using a rotator or the like.
- Dynabeads® Oligo® (dT) 25 is used as magnetic beads, and this is washed several times with about 150 to 250 ⁇ L of 1 ⁇ binding buffer for oligo dT. Thereafter, 200 to 250 ⁇ L of ultrapure water is added and incubated at about 37 ° C. for 5 to 15 minutes to elute the linker bound to the desired protein such as biotin-adhered BDA. For example, add an equal volume of 2xHis-tag washing buffer to the biotin-adhered BDA solution, and then add about 15-50 ⁇ L of magnetic beads such as His Mag Sepharose Ni (washed with 1xHis-tag washing buffer) And stir at room temperature for about 30-50 minutes using a rotator.
- the shared linker of the present invention can be used for both in vitro and intermolecular binding studies as described above.
- the cross-linked peptide aptamer for wrinkling using the cnvK linker described above can be prepared as follows.
- camelid antibodies, such as VHH are preferable because of excellent refolding and relatively long CDR3 sequences.
- VHH and a desired sequence can be linked to construct a VHH library construct, which can be used as a template DNA.
- the desired sequence include a T7 region, a Kozak region, a 5′cap region, an ⁇ region, a His-Tag, a Y-tag, a NewY tag, a region that hybridizes with a linker, and the like.
- a construct containing such a sequence for example, a construct having a DNA sequence as shown in SEQ ID NO: 7 in the sequence listing can be preferably used.
- n, b, d, r, y, h, and m each represent a Mix base
- the ratio is as follows, and the frequency of occurrence of the base is as shown in Table 2 below. is there. 6A to 6C show the theoretical frequency of each amino acid in each codon represented by nbn, drb, and yhm.
- PCR can be performed under desired conditions to amplify the construct.
- the above-mentioned VHH construct can be added to each desired amount of 5X Prime STAR Buffer, dNTP mixture, primer, Prime STAR HS HS DNA polymerase, and a desired amount, for example, a PCR solution can be prepared with ultrapure water.
- T7 ⁇ ⁇ ⁇ ⁇ ⁇ omega new (60 mer) (SEQ ID NO: 3), NewYtag_for_PolyA & cnvK-Lin (22 mer) (SEQ ID NO: 4), ⁇ RT-Lnew (32 mer) (sequence list) SEQ ID NO: 8), New left (33 mer) (SEQ ID NO: 5 in the sequence listing), NewYtag (22 mer) (SEQ ID NO: 9 in the sequence listing), and the like.
- a PCR solution having the above composition is prepared, for example, at about 98 ° C. for 1.5 to 2.5 minutes, (about 98 ° C. for 5 to 15 seconds, about 98 ° C. for about 1 second, about 65 to 70 ° C. for about 30 to about 50 seconds) can be amplified with a PCR program of 4 to 6 cycles, about 70 to 75 ° C. and 1.5 to 2.5 minutes.
- the diversity of the initial solution (stock solution) containing the VHH construct library is preferably about 2.5 to 5 ⁇ 10 11 molecules / ⁇ L because various VHH constructs contained in this library can be used as materials.
- PCR Clean-Up MiniKit Frutegen
- a PCR product having a desired chain length can be obtained by performing PCR while changing primers as necessary. For example, when New left is used among the above primers, PCR is performed instead of T7 omega new, and a desired PCR product can be obtained by, for example, a PCR program similar to the above.
- RiboMAX TM Large Scale RNA production may be a commercially available product such as Systems (Promega Corp.), it may be transcribed using the desired transcription reaction.
- a transcription reaction solution for example, a template DNA dissolved in RNase-free water can be added to T7 Transcription 5X Buffer, rNTPs, and Enzyme Mix.
- the above transcription reaction solution is incubated at, for example, about 36 to 38 ° C. for a desired time, for example, 2.5 to 3.5 hours, and Rnase-Free DNAse is added, for example, at about 36 to 38 ° C. for a desired time, for example, By incubating for 10 to 20 minutes, mRNA can be obtained.
- Rnase-Free DNAse for example, a commercially available product such as RQ1 Rnase-Free DNAse (manufactured by PROMEGA) can be used.
- RNA is purified.
- RNA purification for example, After Tri-Reagent RNA Clean-up Kit (manufactured by Favogen Biotech Corp.) can be used. It is preferable to obtain an mRNA capable of photocrosslinking with a linker by adjusting the amount of DNA used as a template so that a desired RNA can be obtained.
- the obtained purified mRNA and the Biotin-cnvK linker prepared as described above are linked by photocrosslinking.
- the photocrosslinking reaction solution containing the purified mRNA, biotin-cnvK linker, about 0.75 to 1.5 M NaCl, and about 0.2 to 0.3 M Tris-HCl is heated at about 88 to 92 ° C. for about 1 to 3 minutes, for example. Then, the temperature is lowered to about 65 to 75 ° C. in about 1 minute. It can then be annealed by heating at about 68-72 ° C. for about 0.5-2 minutes, followed by a temperature drop to about 20-30 ° C. in 15 minutes.
- the ratio of mRNA and Biotin-cnvK linker in the photocrosslinking reaction solution is preferably approximately equimolar from the viewpoint of crosslinking efficiency.
- a long wavelength ultraviolet light is irradiated in a desired amount to carry out photocrosslinking.
- a commercially available device such as CL-1000 Ultraviolet Crosslinker (manufactured by UVP)
- ultraviolet light with a wavelength of about 360 to 370 nm is irradiated under conditions of 350 to 450 mJ / cm 2 to perform photocrosslinking, and mRNA -Linker conjugates can be obtained.
- the mRNA-linker conjugate obtained as described above is translated in a cell-free translation system to form an mRNA-peptide conjugate.
- a reaction solution used for such translation for example, a solution containing Translation Mix, the above mRNA-linker conjugate, Retic Lysate, and RNase inhibitor is prepared, and finally adjusted to a desired volume with ultrapure water.
- Cell-free translation can be performed by treating the tube containing the reaction solution for translation as follows. For example, incubate at about 25-35 ° C. for about 15-25 minutes, to which about 10-15 ⁇ L of about 2.5-3.5 M KCl and about 2-4 ⁇ L of 0.5-1.5 M MgCl 2 are added and about 36 Incubate at ⁇ 38 ° C for an additional approximately 45-75 minutes. Then add about 5-15 ⁇ L of ethylenediaminetetraacetic acid (about pH 7.5-8.5), incubate at about 36-38 ° C. for about 5-15 minutes, and then add about 40-80 ⁇ L of binding buffer.
- An mRNA-peptide conjugate in which a peptide synthesized by a fine translation system is bound to the mRNA conjugate can be obtained.
- the mRNA-peptide conjugate obtained as described above is bound on the magnetic beads.
- magnetic beads include Streptavidin (SA) Magnetic beads: Dynabeads MyOne Streptavidin C1. Place 8 to 15 times the volume of the magnetic beads of the mRNA-peptide conjugate used for this binding in a Protein LoBind tube (Eppendorf) of the desired size and leave it on a magnetic stand. Discard the clear. Here, 1x binding buffer is added and resuspended, left on a magnetic stand, and the supernatant is discarded, whereby the magnetic beads can be washed and rendered RNase-free.
- the translation product (mRNA-peptide conjugate) obtained as described above is added and incubated at about 20-30 ° C. for about 75-135 minutes with stirring with a rotator. To join.
- the mRNA-peptide conjugate is immobilized on the magnetic beads in the tube, the tube is left on a magnetic stand, and the supernatant is discarded.
- the operation of adding a desired amount, for example, about 50 to 150 ⁇ L of 1 ⁇ binding buffer to the tube, resuspending the magnetic beads, and discarding the supernatant is repeated a desired number of times.
- cDNA is synthesized using a desired solution to obtain a cDNA display in which cDNA is further bound to the above mRNA-peptide conjugate.
- a desired solution for example, commercially available products such as ReverTra Ace (manufactured by Toyobo Co., Ltd.) can be used.
- ReverTra Ace manufactured by Toyobo Co., Ltd.
- a reaction solution for cDNA synthesis containing ReverTra Ace attached buffer, dNTP mixed solution, Rever Tra Ace (100 U / ⁇ L), and ultrapure water is used as a magnetic bead on which the washed mRNA-peptide conjugate is immobilized.
- a cDNA display further bound with cDNA can be obtained as immobilized on magnetic beads. .
- an enzyme for separating the cDNA display from the magnetic beads and a tag for use in purification of the separated cDNA display are provided.
- incubate ribopyrimidines such as deoxyinosine, riboG, riboT, riboC, and riboU can be used at a desired concentration, for example, 500 to 1,500 U / ⁇ L.
- the tag His-tag, Y-tag, or the like can be used. Incubation is preferably performed at about 36 to 38 ° C. for 15 to 45 minutes with stirring using a rotator or the like.
- the cDNA display obtained as described above is classified into those in which the peptide encoding the VHH sequence is expressed and those in which the peptide is not expressed.
- His Mag sepharose Ni is added to the tube containing the cDNA display and incubated at the desired temperature for the desired time, for example, at about 20-30 ° C for 0.5-1.5 hours with shaking, after which the tube is Place on a magnetic stand and remove the supernatant. Wash with tag buffer, add elution buffer and incubate with shaking at desired temperature for a desired time, eg 15-60 minutes at room temperature. By collecting the supernatant after incubation, only the cDNA display in which the VHH peptide is expressed can be obtained.
- the buffer of the solution containing the cDNA display in which the VHH peptide is expressed is changed.
- a Micro Bio-Spin TM 6 column manufactured by BIO-RAD
- BIO-RAD BIO-RAD
- gel electrophoresis is performed according to the following procedure. Prepare 10x SDS running buffer, 1.5M Tris-HCl buffer (pH 8.8), 0.5M Tris-HCl buffer (pH 6.8), 2x SDS sample buffer, 40% acrylamide solution according to the conventional method. These are used to prepare the desired concentration of stacking gel and separating gel, for example, 4% stacking gel and 15% separating gel.
- electrophoresis is performed at a desired current for a desired time, for example, 20 mA, for a time suitable for separation of the electrophoresis sample.
- a staining solution such as FITC or SYBER-gold according to a conventional method, and use an imager, for example, Typhoon FLA 9500 (GE Healthcare Japan Co., Ltd.) Analyze the results of electrophoresis.
- VHH peptide has a binding property to a desired sugar, for example, a cancer-related monosaccharide N-acetyl-D-glucosamine (GlcNAc) is confirmed as follows.
- a magnetic bead and biotinylated GlcNAc are incubated to prepare a magnetic bead on which biotinylated GlcNAc is immobilized, and a magnetic bead on which nothing is immobilized is prepared.
- an equal amount of biotin-DNA is added and incubated to check whether biotinylated GlcNAc is immobilized on the magnetic beads.
- biotinylated GlcNAc is immobilized on the magnetic beads
- PDO is a DNA fragment constructed using a desired sugar recognition peptide, for example, the POU domain of Oct1 protein.
- PDO includes the sequences T7, 5′cap, ⁇ , Kozak, GGGS, His-Tag, GGS and Y-tag, and is a peptide having the base sequence shown below ( Sequence number 10 of a sequence table).
- the GlcNAc-binding VHH peptide was expressed by repeating in vitro testing.
- a cDNA display can be obtained.
- Example 1 Preparation and characterization of the linker of the present invention (Puromycine-linker (polyA + cnvK)) (1) Preparation of the linker of the present invention (Puromycine-linker (polyA + cnvK)) Puromycine-linker (polyA + cnvK) ) Is shown in FIGS. 3A and 3B.
- the biotin fragment (poly A + cnvK: main chain) has the sequence described in SEQ ID NO: 1 in the sequence listing.
- BioTEG is bound to the 5 ′ end of the main chain, and N in the base sequence represents inosine and X represents AminoC6-dT.
- the puromycin-segment (side chain) has the following sequence.
- P as the free end of the side chain is puromycin as a protein binding site.
- (5S) represents 5′Thiol C6
- F represents FITC-dT
- Z represents Spacer18. The chemical synthesis of the main chain and side chain was outsourced to Tsukuba Oligo Service Co., Ltd.
- the buffer-reduced reduced puromycin-segment solution was mixed with the ethanol-precipitated product of the above-mentioned EMCS-modified biotin-fragment (polyA + cnvK) and allowed to stand at 4 ° C. overnight. Subsequently, DTT was added to the reaction solution to a final concentration of 50 mM and stirred at room temperature for 30 minutes. Thereafter, ethanol precipitation was performed using Quick-Precip Plus Solution (Edge BioSystems). The ethanol precipitation product was dissolved in 100 ⁇ L of Nuclease-free water (manufactured by Nacalai Tesque) and subjected to HPLC purification using a C18 column under the following conditions.
- Liquid A 0.1M trimethylammonium acetate (ultra pure water)
- Liquid B 80% acetonitrile Program: Composition ratio of liquid A and liquid B is such that 85% of liquid A at the start is 65% over 45 minutes Flow rate: 1 mL / fraction Fraction: 1 mL
- mRNA-peptide conjugates were formed was confirmed by SDS-PAGE of 4% stacking gel containing 6M urea and 6% separation gel by staining the gel with SYBR® Gold (see FIG. 7A).
- the UV irradiation time was 0 minutes, 10 minutes, and 20 minutes.
- FIG. 7A in the preparation that was not subjected to UV irradiation, no mRNA-peptide conjugate was formed at any irradiation time.
- formation of mRNA-peptide conjugate was confirmed at any irradiation time.
- Example 2 Examination of irradiation dose in photocrosslinking reaction As a model, DNA encoding the B domain of A protein (SEQ ID NO: 6 in the sequence listing) was used. Transcription was performed using RiboMAX Large Scale RNA Production Systems-T7. BDA mRNA obtained by transcription and puromycin-linker (poly A + cnvK) were added to 25 mM Tris-HCl buffer (pH 7.5) containing 100 mM NaCl so that the final concentration was 1 ⁇ M.
- UV irradiation time As shown in FIG. 7B, it was confirmed that a UV irradiation time of 30 seconds or more is sufficient for linking mRNA and linker. In order to form a stable photocrosslink while minimizing damage to the linker, the UV irradiation time was 2 minutes.
- Example 3 Model selection of FLAG epitope (1) Transcription of library 8-amino acid random library DNA (SEQ ID NO: 2 in the sequence listing) having the following sequence and FLAG sequence (SEQ ID NO: 11 in the sequence listing) were encoded.
- a DNA mixture was prepared by mixing DNA in a molar ratio of 50,000: 1, and 500 ng of the mixture was prepared using RiboMAX Large Scale RNA Production Systems-T7 (hereinafter sometimes simply referred to as “kit”). Transcribed on scale.
- N arbitrarily represents A, T, G, and C, and K represents G or T.
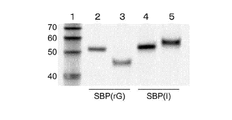
- RNA degradation was examined using the conjugate of the cnvK linker of the present invention and mRNA and the linker (SBP) of Conventional Example 1 using T4 RNA ligase.
- SBP linker
- the ligation reaction between mRNA and the linker of Conventional Example 1 was performed in a buffer at 25 ° C. for 30 minutes.
- this buffer was subjected to electrophoresis under the conditions of 200 V, 15 mA, 8 M urea-denatured 4% acrylamide gel, it was shown by SYBR staining that mRNA was degraded (FIG. 7D).
- cnvK linker of the present invention was used, mRNA degradation was not observed at all (FIG. 7E).
- a ligation reaction can be performed in water that does not contain a metal ion such as zinc instead of a buffer, so that RNase is not activated as in the case of reaction in a buffer. It was thought to be due to this.
- UV was irradiated so that the irradiation dose was 81 mJ to 810 mJ, and damage to the mRNA due to photocrosslinking was confirmed (FIG. 7F).
- FIG. 7F degradation of the mRNA-cnvK linker fusion was not observed.
- the change in the amount of cDNA was not observed as in the case where no damage was observed on the mRNA (FIG. 7G).
- 2x binding buffer for SA containing 20 mM Tris-HCl (pH 7.5), 2 M NaCl, 2 mM EDTA, 0.2% Tween-20
- 2x binding buffer for SA containing 20 mM Tris-HCl (pH 7.5), 2 M NaCl, 2 mM EDTA, 0.2% Tween-20
- 2x binding buffer for SA containing 20 mM Tris-HCl (pH 7.5), 2 M NaCl, 2 mM EDTA, 0.2% Tween-20
- the purified full construct DNA was added to a 200 ⁇ L PCR reaction solution (0.2 mM dNTPs, 0.4 ⁇ M Newleft, 0.4 ⁇ M NewYtag for cnvK, and 1xPrimeSTAR buffer (containing Mg 2+) containing 0.02 U / ⁇ L PrimeSTAR HS DNA polymerase. In addition to)), 50 ⁇ L was dispensed and the following PCR program was performed to obtain double-stranded DNA.
- the sequence of Newleft (SEQ ID NO: 5 in the sequence listing) is shown below.
- the PCR program was (a2) 98 ° C for 1 minute, (b2) 98 ° C for 10 seconds, (c2) 68 ° C for 30 seconds, (d2) 68 ° C for 1 minute, and steps (b2) and (c2) were Five cycles were performed.
- the four PCR reaction solutions were combined and purified into a column for use in the next round.
- a total of 3 rounds from (1) library DNA transcription to (7) affinity selection were performed, and the resulting full construct DNA sequence was directly sequenced. This direct sequencing was commissioned to Eurofin Genomics. As a result of direct sequencing, it was confirmed that the FLGA DNA had converged (FIGS. 8A and 8B).
- Example 4 Production of biotin adhesion protein and application to SPR device Also in this example, the B domain (BDA) of A protein was used as a model protein in the same manner as described above. Prepare mRNA-peptide conjugate for 30 pmol as mRNA from DNA encoding BDA by the same procedure as (1) to (7) of FLAG sequence model selection by cDNA display method performed in Example 3 above. did.
- the above Dynabeads Oligo (dT) 25 was washed twice with 200 ⁇ L of 1 ⁇ binding buffer for oligo dT, and then 232 ⁇ L of ultrapure water was added and incubated at 37 ° C. for 10 minutes, followed by biotin adhesion protein (hereinafter referred to as “biotin adhesion BDA” Was sometimes eluted.).
- biotin adhesion BDA biotin adhesion protein
- Example 5 Affinity measurement using Biotin-attached protein A sensor chip SA was set on Biacore X100 (manufactured by GE Healthcare). The biotin-adhered BDA solution obtained as described above was diluted 2-fold with 1 ⁇ His-tag washing buffer. This diluted biotin-adhered BDA solution was allowed to flow for 900 seconds at a flow rate of 5 ⁇ L to immobilize biotin-adhered BDA on the sensor chip.
- IgG obtained from rabbit serum was used to make 2-fold serial dilutions of 160 nM, 80 nM, 40 nM, 20 nM, and 10 nM using 1 ⁇ HBP-EP + buffer (manufactured by GE Healthcare). . These dilution series liquids were injected into the Fc1 and Fc2 channels at a flow rate of 5 ⁇ L / min, and the affinity was measured.
- EMCS crosslinking agent, manufactured by Wako Pure Chemical Industries, Ltd.
- N, N-dimethylformamide 100 mM EMCS.
- 200 ⁇ L of 0.2 ⁇ M sodium phosphate buffer (manufactured by Wako Pure Chemical Industries, Ltd., for molecular biology, pH ⁇ 7.2) was added with 10 nmol of Biotin-cnvK segment and 40 ⁇ L of 100 mM EMCS, and 30 ° C. Incubated for minutes. Thereafter, ethanol precipitation was performed to concentrate the product.
- the PCR program was 98 ° C for 2 minutes (98 ° C for 10 seconds, 98 ° C for 1 second, 68 ° C for 40 seconds) with 5 cycles at 72 ° C for 2 minutes.
- the diversity of VHH in the stock solution was 2.7 ⁇ 10 11 molecules / ⁇ L.
- the PCR product 1 obtained by the PCR 1 was purified using PCR Clean-Up MiniKit (manufactured by Favorite) to remove short-chain DNA.
- PCR solution 2 having the same composition was prepared except that 20 ⁇ M Primer 2 (New left) was replaced with 20 ⁇ M Primer 2 (T7 omega new).
- the obtained purified PCR product 1 was subjected to PCR 2 to obtain PCR product 2.
- the PCR program was 98 ° C for 2 minutes (98 ° C for 10 seconds, 98 ° C for 1 second, 68 ° C for 40 seconds) with 23 cycles at 72 ° C for 2 minutes.
- the above transcription reaction solution was incubated at 37 ° C. for 3 hours, 1 ⁇ L of RQ1 Rnase-Free DNAse (Promega) was added, and further incubated at 37 ° C. for 15 minutes. Thereafter, mRNA was purified using After Tri-Reagent RNA Clean-up Kit (manufactured by Favorgen) according to the attached protocol. The above operation was repeated up to the sixth round, and mRNA was obtained in each round.
- the amount of template DNA used in each round was as shown in Table 5 below, and the amount of x in the above table was changed according to the amount of DNA used. At that time, x was adjusted so that a sufficient amount of mRNA could be purified for subsequent photocrosslinking formation.
- the tube containing the translation reaction solution was incubated at 30 ° C. for 20 minutes. To this, 12 ⁇ L of 3M KCl and 3 ⁇ L of 1M MgCl 2 were added, and further incubated at 37 ° C. for 60 minutes. Thereafter, 10 ⁇ L of ethylenediaminetetraacetic acid (pH 8.0) was added, incubated at 37 ° C. for 10 minutes, then 50 ⁇ L of 2 ⁇ binding buffer was added, and the peptide synthesized in the non-translated system was bound to the mRNA conjugate. An mRNA-peptide conjugate was obtained.
- ethylenediaminetetraacetic acid pH 8.0
- Magnetic beads Washed to make Dynabeads MyOne Streptavidin C1 (magnetic beads) RNase-Free. Magnetic beads having a volume of 10 times the concentration of the mRNA-peptide conjugate to be introduced were placed in a 1.5 mL Protein LoBind tube (manufactured by Eppendorf), left on a magnetic stand, and the supernatant was discarded. The suspension was resuspended in 1 ⁇ binding buffer, left on a magnetic stand, the supernatant was discarded, and the magnetic beads were washed.
- the translation product (mRNA-peptide conjugate) obtained in (2-4) above was added, and incubated at 25 ° C. for 90 to 120 minutes with stirring with a rotator.
- the mRNA-peptide conjugate was applied to the magnetic beads. Combined.
- the amount of mRNA-peptide conjugate used and the incubation time are shown in Table 8 below. It was as shown in.
- centrifugation was performed at 1,000 ⁇ g for 2 minutes with the column lid opened, and 500 ⁇ L of binding buffer for glucosamine was added thereto, followed by centrifugation at 1,000 ⁇ g for 1 minute.
- the operation of adding a binding buffer for glucosamine and centrifuging at 1,000 ⁇ g for 1 minute was repeated 4 times, and the final centrifugation was performed for 3 minutes.
- the cDNA display obtained as described above was added thereto, followed by centrifugation at 1,000 ⁇ g for 4 minutes to collect the buffer-exchanged cDNA display.
- cDNA displays that bind non-specifically to biotin were excluded.
- the magnetic beads were washed with glucosamine washing buffer and incubated with biotin at 25 ° C. for 30 minutes in a tube. Thereafter, the mixture was stirred with a shaker to obtain a cDNA display that specifically binds to GlcNAc.
- Table 10 below shows the relationship between the amount of cDNA display in each round and the incubation time on the shaker.
- test tube sputum (1) Examination of test tube sputum conditions Next, using the obtained GlcNAc-specific cDNA display, test tube sputum of VHH against the target molecule GlcNAc was performed. First, the magnetic beads were washed in a test tube using a glucosamine washing buffer, biotinylated GlcNAc was added thereto, and incubated at room temperature for 30 minutes with shaking on a shaker. Then, place the tube on a magnetic stand, discard the supernatant, add glucosamine washing buffer to wash the magnetic beads again, add biotin, and shake on a shaker at room temperature for 30 minutes. Incubated while.
- the tube was allowed to stand on a magnetic stand, the supernatant was discarded, the magnetic beads were washed with a glucosamine washing buffer, and incubated with a cDNA display at 25 ° C. for 30 minutes while stirring with a rotator. Thereafter, this tube was allowed to stand on a magnetic stand and the supernatant was discarded.
- the cDNA display of GlcNAc-specific VHH was immobilized on the magnetic beads via GlcNAc.
- the conditions in each round in the test tube were as shown in Table 11 below.
- the cDNA display was immobilized on the magnetic beads via GlcNAc in the tube. Subsequently, the washing buffer for glucosamine was added and resuspended by tapping for 20 seconds. Thereafter, this tube was allowed to stand on a magnetic stand for 1 minute, and the supernatant was collected. This operation was repeated 3 to 5 times, and the collected supernatant was stored individually as a washing solution (hereinafter sometimes referred to as “Wash”). Wash was subjected to gel electrophoresis to confirm the contents. As a result, the number of times the cDNA display was washed in each round was as shown in Table 12 below.
- GlcNAc at a sufficient concentration relative to the amount of cDNA display immobilized on the magnetic beads was added, and elution was carried out with stirring with a rotator at 4 ° C. for 3 days except for the sixth round.
- the sixth round was eluted overnight.
- the cDNA display of VHH that non-specifically binds to GlcNAc was removed, followed by competitive elution, and the cDNA display of VHH that specifically binds to the target GlcNAc was recovered.
- gel electrophoresis was performed according to the following procedure.
- a 10xSDS running buffer, 1.5M Tris-HCl buffer (pH 8.8), 0.5M Tris-HCl buffer (pH 6.8), 2xSDS sample buffer, and 40% acrylamide solution were prepared as follows.
- the 10xSDS running buffer was made up to 1,000 mL by adding ultrapure water to 30.3 g of tris (hydroxymethyl) aminomethane, 144 g of glycine, and 10 g of SDS.
- 1.5M Tris-HCl buffer pH 8.8 was prepared by dissolving 36.3 g of Tris (hydroxymethyl) aminomethane in ultrapure water, adjusting the pH to 8.8 with hydrochloric acid, and then measuring up to 200 mL with ultrapure water. .
- Tris-HCl buffer pH 6.8
- 12.1 g of tris (hydroxymethyl) aminomethane was dissolved in ultrapure water, adjusted to pH 6.8 with hydrochloric acid, and then diluted to 200 mL with ultrapure water.
- 2x SDS sample buffer is 12.5mL 0.5M Tris-HCl buffer (pH 6.8), 5mL 2-mercaptoethanol, 2g SDS, 24g urea, 3g sucrose dissolved in a small amount of ultrapure water, and an appropriate amount of bromophenol.
- BPB blue
- the 40% acrylamide solution was obtained by dissolving SDS-PAGE grade 194.8 g acrylamide and 5.2 g bisacrylamide in an appropriate amount of ultrapure water and making up to 500 mL.
- the 15% separation gel (for 1 sheet) was mixed with 2.5 mL of 1.5 M Tris-HCl buffer (pH 8.8), 3.75 mL of 40% acrylamide solution, and 100 ⁇ L of 10% SDS to make 10 mL with ultrapure water. . To this, 25 ⁇ L of 20% APS and 5 ⁇ L of TEMED were added.
- the 10 ⁇ SDS running buffer was diluted 10-fold to fill the electrophoresis tank, and the gel electrophoresis was set in the electrophoresis tank and electrophoresed at 20 mA for a time suitable for separation of the electrophoresis sample. Staining was performed according to a conventional method using FITC and SYBER-gold, and the results of gel electrophoresis were analyzed using an imager (Typhoon FLA 9500, manufactured by GE Healthcare Japan Co., Ltd.).
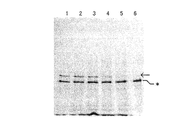
- VHH cDNA display and BDA cDNA display in a 1: 1 ratio, respectively, and incubate at room temperature for 30 minutes while stirring with a rotator. Then, it left still on a magnetic stand and the supernatant liquid was thrown away. The washing buffer for glucosamine was added and resuspended by tapping, and left on a magnetic stand for 1 minute to collect the supernatant. Subsequently, competitive elution was performed using GlcNAc to obtain an eluate. These were gel-electrophoresed with markers at 4% PAGE, 200 V for 30 minutes, and stained with SYBER-Gold. The results are shown in FIG.
- the intensity ratio of the VHH: BDA band was 9:16, and the BDA cDNA display concentration was high. Thereafter, the intensity ratio of the bands in the cleaning solution was 1: 3, and the BDA concentration was high. In contrast, in the eluate, the band intensity ratio was reversed to 4: 1, and the VHH concentration increased.
- the VHH concentration in the control was 0.6 times that of BDA, but in the eluate, the VHH concentration was 6 times higher than the BDA concentration. This indicates that the obtained VHH cDNA display has been eluted while the BDA cDNA display was not eluted. From this result, it was confirmed that the cDNA display of VHH obtained by the sixth round in vitro fistula was capable of binding to GlcNAc.
- Crosslinked peptide aptamer (BDA and VHH cDNA display) was prepared in the same manner as in Example 6 (2) above. After preparing each of these cDNA displays, both cDNA displays were placed in a tube containing magnetic beads and incubated, and in vitro was performed in the same manner as when only the VHH cDNA display was added. Washing was performed 5 times in each round, and competitive elution with GlcNAc was performed overnight at 4 ° C.
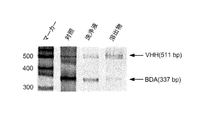
- the length of the obtained VHH DNA was the same as that of 541 bp from the first round to the sixth round, and the length of mRNA was the same as that of 541mer. As shown in FIG. 13 (B), it was confirmed that a sufficient amount of mRNA-linker conjugate was formed.
- the mRNA-peptide conjugate band was shifted upward, confirming the formation of the mRNA-peptide conjugate.
- the band detected on the mRNA-linker conjugate band was considerably thin.
- the concentration ratio of the bands was 7: 3.
- after purification indicates a purified cDNA display.
- Incubation supernatant indicates the supernatant after incubation of magnetic beads (1 st is a column) and cDNA display in each round.
- Wash liquid indicates the supernatant collected after washing by tapping.
- eluate indicates a liquid eluted by competitive elution with GlcNAc.
- the results in the first to third rounds are shown in FIGS. 15 (A) to (C).
- the first round 108 pmol of mRNA-linker conjugate was used, and the amount of biotinylated GlcNAc immobilized was 25 nmol.
- 36 pmol of mRNA-linker conjugate was used, and the amount of biotinylated GlcNAc immobilized was 1 nmol.
- 18 pmol of mRNA-linker conjugate was used, and the amount of biotinylated GlcNAc immobilized was 500 pmol.
- Gel electrophoresis was performed at 4% PAGE, 200 V for 30 minutes, and stained with SYBER Gold.
- the results of the fifth round are shown in FIGS. 17 (A) and (B).
- 8 pmol of mRNA-linker conjugate was used, and the amount of biotinylated GlcNAc immobilized was 50 nmol. It was observed that the eluate lane band was thinner than the negative wash lane band. In addition, a dark band was observed in the positive eluate lane, indicating that the glaze of GlcNAc-binding VHH was progressing.
- FIGS. 18 (A) and (B) The results of the sixth round are shown in FIGS. 18 (A) and (B).
- 2.8 pmol of mRNA-linker conjugate was used, and the amount of biotinylated GlcNAc immobilized was 50 nmol.
- the darkness of each negative and positive wash lane band was approximately the same as the darkness of each eluate lane band. Therefore, negative and positive washing solutions 5 and their eluates were subjected to gel electrophoresis at 4% PAGE and 200 V for 30 minutes and stained with SYBER Gold. The results are shown in FIG.
- the positive eluate was 22 times darker than the negative eluate, and as a result, it was confirmed that the positive eluate contains GlcNAc-binding ability VHH.
- the sequence analysis showed that CDR3 contributes to GlcNAc recognition.
- (S) is Thiol-Modifier C6 SS (compound name: o- (dimethoxytrityloxy-hexyl-dithiohexyl) -o '-(2-cyanoethyl) -N, N-diisopropyl-phosphoramidite)
- (PL) is PC Linker Phosphoramidite (Compound name: 3- (4,4'-Dimethoxytrityl) -1- (2-nitrophenyl) -1-propanyl-[(2-cyanoethyl)-(N, N-diisopropyl)]-phosphoramidite ).
- Fluorescein-dT (compound name: (5'-Dimethoxytrityloxy-5- [N-((3 ', 6'-dipivaloylfluoresceinyl) -aminohexyl) -3-acryimido] -2'-deoxyUridine-3 '-succinoyl-long chain alkylamino) and (Puro) stands for puromycin.
- Spacer 18 is Spacer Phosphoramidite 18 (compound name: 18-0-Dimethoxytritylhexaethyleneglycol, 1-[(2-cyanoethyl)-(N, N-diisopropyl)]-phosphoramidite). (All are made by Glenn Research).
- the SBP linker was obtained by cross-linking and purifying the (A) Puro-F-S segment and (B2) Hybri segment synthesized in Example 1 (1) according to the following method. Dissolve 10 nmol of Puro-FS in 100 ⁇ L of 50 mM phosphate buffer (pH 7.0), add 1 ⁇ L of 100 mM Tris [2-carboxyethyl] phosphine (TCEP, manufactured by Pierce) (final concentration: 1 mM), leave at room temperature for 6 hours, The trisylated mercapto group of -FS was reduced to a thiol group. Immediately before the cross-linking reaction, TCEP was removed and desalted using NAP-5 ⁇ Columns equilibrated with 50 mM phosphate buffer (pH ⁇ 7.0).
- mRNA BDA B domain of Protein A
- T7 promoter sequence and translation promoting sequence 5 'upstream of BDA gene BDA gene; 192 bases long), spacer region, histidine tag (His tag) and puromycin linker complementary strand region 3' downstream
- BDA whole; SEQ ID NO: 6 in the sequence listing; 367 bases in length The DNA to which the sequence was added (BDA whole; SEQ ID NO: 6 in the sequence listing; 367 bases in length) was synthesized and purified by PCR, and then attached protocol using T7 RiboMAX Express Large Scale RNA Production System (Promega). According to the procedure, 5-30 pmol / ⁇ L mRNA (BDA mRNA) was synthesized.
- T4 polynucleotide kinase 10 U / ⁇ L
- 1 ⁇ L of T4 RNA ligase 40 U / ⁇ L (both manufactured by Takara Bio Inc.) were added and held at 25 ° C. for 15 minutes.
- reaction system R1 In order to divide the generated reaction solution in half and compare it in a later experimental step, for the sample in which 10 pmol of mRNA was reacted with 10 pmol of linker, all the substances were doubled and reacted in a 40 ⁇ L system. (Reaction system R1).
- the LBP linker was obtained by crosslinking and purifying the (A) Puro-F-S segment (side chain) and the (B) Biotin-loop segment (main chain) according to the following method. First, dissolve 10 nmol of Puro-FS in 22.5 ⁇ L of 1M phosphate buffer (pH 9.0), add 2.5 ⁇ L of 1M DTT, and let stand for 1 hour at room temperature, using the tritylated mercapto group of Puro-FS as the thiol group. Reduced. Immediately before the cross-linking reaction, DTT was removed and desalted using NAP-5 Columns (manufactured by GE Healthcare) equilibrated with 20 mM phosphate buffer (pH 7.2).
- the reaction product was quickly dissolved in Puro-F-S ( ⁇ 10 nmol) reduced as described above and left at 4 ° C. overnight to prepare a sample.
- DTT was added to a final concentration of 50 mM, and the mixture was allowed to stand at 37 ° C. for 30 minutes to stop the thiol group crosslinking reaction.
- the synthesized linker was precipitated at room temperature by an ethanol precipitation method to remove unreacted Puro-F-S. Further, in order to remove unreacted Biotin-loops and their EMCS cross-linked products, purification was performed by a gradient method using HPLC under the following conditions.
- the product fractionated by HPLC was detected by electrophoresis using 16% acrylamide gel (8M urea, 60 ° C.), and the target fraction was dried under reduced pressure. Thereafter, it was dissolved in DEPC (diethylpyrocarbonate) -treated water so as to be 10 pmol / ⁇ L.
- DEPC diethylpyrocarbonate
- (B1) I represents deoxyinosine
- symbols representing (T), (TB) and the modified base are the same as those in Comparative Examples 1 and 2.
- (rG) represents riboG.
- the SBP (I) linker was obtained by cross-linking and purifying the (A) Puro-FS segment and (B) I-Hybri segment synthesized in (1) of Example 1 according to the following method.
- the SBP (rG) linker was obtained by crosslinking (A) Puro-FS and (C) rG-hybri.
- the reaction product was quickly dissolved in the Puro-F-S solution ( ⁇ 5 nmol) reduced as described above and left at 4 ° C. overnight to prepare a sample.
- DTT was added so that the final concentration was 50 mM, and the mixture was left at 37 ° C. for 30 minutes to stop the crosslinking reaction of thiol groups.
- the linker synthesized by ethanol precipitation at room temperature remove unreacted Puro-FS, and further remove unreacted I-Hybri or rG-Hybri and their EMCS cross-linked products, the same as in Example 1 HPLC purification was performed under the conditions.
- the product fractionated by HPLC was fractionated by electrophoresis on a 12% acrylamide gel containing 8 M urea under conditions of 200 V and 60 ° C. for 30 minutes.
- the desired fraction was dried under reduced pressure. Then, it was dissolved in DEPC (Diethylpyrocarbonate) -treated water to make 10 pmol / ⁇ L.
- DEPC Diethylpyrocarbonate
- linker RNase resistance test The linker SBP (rG) and SBP (I) synthesized as described above were tested for RNase resistance using RNase ONE (Promega) derived from the E. coli periplasm. I went.
- RNase ONE is an RNase having an activity of cleaving a phosphodiester bond at the 3 ′ end of each of A, C, G, and U RNAs.
- FIG. 21B 1 ⁇ L of 100 bp DNA ladder (Promega) was applied to the first lane as a size marker.
- the second lane contains untreated SBP (rG) at 0.5 pmol
- the third lane contains 5 ⁇ L of SBP (rG) treated with RNase ONE (shown as “puroFS” in the figure)
- Untreated SBP (I) was applied at 0.5 pmol
- 5 ⁇ L of SBP (I) treated with RNase ONE was applied to the fifth lane.
- (psoralen) includes Psoralen C6 Phosphoramidite ((6- [4 '-(Hydroxymethyl) -4,5', 8-trimethylpsoralen] -hexyl-1-O- (2-cyanoethyl)-(N, N -diisopropyl) -phosphoramidite)) was used.
- T-NH2 includes Amino-Modifier-C6 dT (5'-Dimethoxytrityl-5- [N- (trifluoro-acetylaminohexyl) -3-acrylimido] -2'-deoxyUridine, 3 '-[(2-cyanoethyl) -(N, N-diisopropyl)]-phosphoramidite). All of the above reagents were purchased from Glen Research. Using these reagents, the reaction was performed according to the phosphoramidite method using an automatic nucleic acid synthesizer (FIG. 23B).
- alkyne-peptide-biotin segment (SEQ ID NO: 18 in the sequence listing) having the following structure was synthesized. The following sequences are shown as structures from N-terminal to C-terminal. Here, Fmoc-Gly (Propargyl) -OH was used for Gly (Propargyl). (K-biotin) -NH 2 is modified by amidating the C-terminus of a lysine residue and binding biotin to the side chain of the lysine residue.
- the resulting cross-linked reaction product was desalted using a microbiospin column 6 (manufactured by Bio-rad) equilibrated with phosphate buffer (pH 7.2), and the desalted cross-linked reaction product was separated by polyacrylamide gel electrophoresis. DNA was stained with SybrGold (Invitrogen) and analyzed.
- Lane 1 shows a 10 bp DNA step ladder (Promega)
- Lane 2 shows an azide-segment
- Lane 3 shows a cross-linked reaction product. From this result, it was confirmed that the target crosslinked product (F) was obtained with a yield of about 70% (FIG. 23C).
- the obtained cross-linked products were separated by urea-denaturing polyacrylamide gel electrophoresis (12% acrylamide gel, 8M urea, 60 ° C.), and the DNA was stained and analyzed by SybrGold (manufactured by Invitrogen).
- Lane 1 is a 10 bp DNA step ladder (manufactured by Promega)
- lane 2 is the result of electrophoresis of the crosslinking reaction product.
- lane 1 is a 10 bp DNA step ladder (manufactured by Promega)
- lane 2 is the result of electrophoresis of the crosslinking reaction product.
- a band comparable to the sum of these molecular weights was detected, confirming that the desired cross-linked product (E) was obtained.
- the result is shown in FIG. 23D.
- this crosslinked product (E) was precipitated with ethanol in the same manner as described above to remove the unreacted puromycin segment, and further to remove the unreacted psoralen-amino segment and its EMCS crosslinked product, the following: Purification was performed by HPLC under conditions.
- the product fractionated by HPLC was again detected by electrophoresis on a 12% acrylamide gel (8 M urea, 60 ° C.), and the desired fraction was concentrated under reduced pressure, and then ethanol was used in the same manner as above. Precipitated. Thereafter, the precipitate was dissolved in water to 50 ⁇ M.
- Photocrosslinking of cross-linked product (E) and cross-linked product (F) A solution containing 1 pmol cross-linked product (E) and 1.2 pmol cross-linked product (F) containing 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl Mixed in, then heated to 60 ° C. and cooled to 25 ° C. over 10 minutes. Next, the sample was irradiated with 365 nm (2 W / cm 2 ) light using a xenon lamp as a light source for 20 minutes, and the sample was exposed to obtain an exposed sample.
- the present invention is useful in technical fields such as molecular targeted peptide drugs, drugs using low molecular weight antibodies and antibody-like proteins.
- SEQ ID NO: 1 nucleotide sequence of main chain of cnvK linker of the present invention
- SEQ ID NO: 2 nucleotide sequence of primer (T7 ⁇ new)
- SEQ ID NO: 3 nucleotide sequence of primer (NewYtag)
- SEQ ID NO: 4 nucleotide sequence sequence number of primer (Newleft) 5: Base sequence of B domain of A protein
- SEQ ID NO: 6 VHH library construct
- SEQ ID NO: 7 Random library DNA sequence used in the in vitro test method
- SEQ ID NO: 8 ⁇ RT-Lnew Sequence number 9: NewYtag (22mer)
- SEQ ID NO: 10 DNA sequence of PDO
- SEQ ID NO: 11 Base sequence of FLAG
- SEQ ID NO: 12 Base sequence of Hybri segment of conventional linker
- SEQ ID NO: 13 Base sequence of the main chain of the conventional linker
- SEQ ID NO: 14 Base sequence of the I-hybri segment of the conventional linker
- SEQ ID NO: 15 Base sequence of the rG-Hybri segment of the conventional linker
- SEQ ID NO: 16 Base of the psoralen-amino segment of the conventional linker
- SEQ ID NO: 17 Base sequence of azide segment of conventional linker
- SEQ ID NO: 18 Base sequence of biotin segment of side chain of conventional linker
Abstract
本発明は、酵素を利用することなく、候補クローンのスクリーニング及び得られた候補クローンの結合性の評価の双方に使用可能なリンカーの提供、前記リンカーを用いる試験管内淘汰方法を提供することを目的とする。本発明では、主鎖の5'末端に位置し固相との結合を形成するための固相結合部位と;前記固相結合部位ごと前記固相を切り離すための固相切断部位と;側鎖を連結するための側鎖連結部位と;主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位と;前記側鎖連結部位に隣接し、前記主鎖の3'末端に位置する逆転写開始領域と;を備える主鎖と、蛍光標識と、遊離末端に位置するタンパク質結合部位と、前記主鎖の側鎖連結部位に連結される連結形成部位と、を備える側鎖とを有し、試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーを提供する。
Description
本発明は、遺伝子型-表現型対応付け技術である、cDNAディスプレイ法に使用するためのリンカー及びその使用方法等に関する。より詳細には、試験管内淘汰及び親和性測定の双方に利用可能な新規ピューロマイシン・リンカー、そのリンカーを用いた試験管内淘汰方法に関する。
これまで、各製薬企業による抗体医薬の開発が進められてきた。抗体は、抗原分子を一定の期間を置いて複数回動物に投与し、その後、採血して得られた血液から精製する必要があるため、in vivoでの製造には時間と手間がかかる。また、最も分子量の小さいIgGでさえ分子量が150 KDaと大きいため、in vitroでの完全合成も難しい。
一方で、抗体の抗原認識部位やペプチドアプタマーは、分子量が小さいことから人工的に合成することができる。
医薬品として使用する場合にはGMPレベルの有機合成が求められるが、上記のようなペプチドアプタマーの場合にはこうしたレベルの合成も可能である。また、分子量が小さいことから化学修飾も容易であり、チップなどへの固定化も自由に行うことができ、さらに、抗体に比べると安定性が高いため、チップ等へ固定した後に、常温で保存することもできるといったメリットがある。
こうしたペプチドアプタマーは、mRNAを作成した後に、遺伝子型-表現型対応付け技術(以下、単に「対応付け技術」ということがある。)を用いて合成及び淘汰することができる。そして、こうした対応付け技術としては、cDNAディスプレイ法の外、ファージディスプレイ法、リボソームディスプレイ法、mRNAディスプレイ法等が存在する。これらのうち、自動化、ハイスループット化という要請を考慮すると、cDNAディスプレイ法がペプチドアプタマーを合成するのに最も適している。
従来、cDNAディスプレイ法で使用するために、図1A~1Dに示すようなリンカーが提案され、使用されてきた。図1Aには、固相結合部位と、mRNAとこのリンカーの主鎖とをT4 RNAリガーゼで連結するためのT4 RNAリガーゼ用連結部位と、逆転写用プライマー領域とを備える主鎖と、ペプチド結合部位及び蛍光標識を備え、前記主鎖に連結された側鎖とで構成されたリンカーが示されている(特許文献1参照、以下、「従来例1」という。)。
また、図1Bには、主鎖が一部二本鎖で構成されている点を除けば従来例1のリンカーとほぼ同様の構造を有しており、この二本鎖で構成された部分に、固相からリンカーを切り離すための制限部位が組み込まれたリンカーが示されている(特許文献1参照、以下、「従来例2」という)。
また、図1Cには、前記従来例1に加えて、固相とリンカーとを切り離すための第1及び第2切断部位を有するリンカーが示されている(特許文献1参照、以下、「従来例3」という。)。図1Dには、2本の主鎖がソラレンで連結されたリンカーが示されている(特許文献2参照、以下、「従来例4」という。)。
また、図1Cには、前記従来例1に加えて、固相とリンカーとを切り離すための第1及び第2切断部位を有するリンカーが示されている(特許文献1参照、以下、「従来例3」という。)。図1Dには、2本の主鎖がソラレンで連結されたリンカーが示されている(特許文献2参照、以下、「従来例4」という。)。
以上のようなリンカー以外に、リンカーの主鎖にcnvKを組み込んでおき、mRNAと主鎖とを光で架橋するというリンカーが提案されている(特許文献3参照、以下、「従来例5」という。)。
ところで、現在の日本人の死因の第一位は癌(腫瘍)であるが、その死因中に占める割合は、年々増加している。腫瘍に罹患しているか否か、及び主要の治療後の経過を観察するためには、腫瘍が体内に存在するようになると、血中や尿の中に、あるレベル以上の特定の物質(腫瘍マーカー)が検出されるようになる。そして、腫瘍マーカーの種類及びそれらの血中又は尿中レベルを測定することによって、腫瘍の有無やその進行度を知ることができる。
ここで、腫瘍マーカーは、ほとんどが糖鎖抗原である。正常な細胞が癌化すると、細胞表面上の糖鎖の鎖長が糖転移酵素によって変化し、癌細胞特異的な糖鎖となるため、このような糖鎖が癌を識別するための腫瘍マーカー(以下、「糖鎖性腫瘍マーカー」という。)とされている(非特許文献5参照)。ここで、糖鎖性腫瘍マーカーの抗原決定基(以下、「エピトープ」ということがある。)は、図2(A)に模式的に表すような一般的構造を有し、その構造から、下記表1に示すように、1型糖鎖、2型糖鎖、母核糖鎖、コア蛋白に分けられている。

ここで、上記1型糖鎖及び2型糖鎖には、上記表1に示すようなものがあることが知られているが、これらはいずれも、細胞表面に存在する糖鎖が糖転移酵素によって伸長されて、正常細胞の場合よりも長くなっている。また、コア蛋白はウイルスの核酸の周囲に存在するタンパク質をいう。
図2(B)に、基幹構造に対する糖鎖抗原の1種であるCA19-9(Carbohydrate Antigen 19-9)の構造を、こうした糖鎖抗原の1例として模式的に示す。CA19-9は、5つの単糖からなる構造を有する分子量874の分子であり、各単糖はグリコシド結合によってつながっている。図中、NeuNacはN-アセチルノイラミン酸、Galはガラクトース、Fucはフコース、GlcNAcはN-アセチル-D-グルコサミンをそれぞれ表す。そして、図2(B)に示したCA19-9に含まれているGlcNAcは、CA19-9以外の多くの糖鎖抗原にも含まれていることが知られている(非特許文献5参照、以下、「従来例6」という。)。
また、抗体は、一般的には軽鎖と重鎖とで構成されているため、免疫グロブリン(IgG)の分子量は、170KDaと大きいことが知られている。これに対し、ラクダ科の抗体は軽鎖を持たず、重鎖のみで構成されており、分子量が12KDaと小さく、立体構造の可塑性が高く、熱安定性に優れることが知られている。以下、このような領域(ドメイン)を有する分子を「VHH」ということがある。
Nucleic Acid Research, 2009, 1-13dot:10.1093/nar/gkp514
Ueno, S., J Biotechnol. 162, 299-302(2012)
Nemoto N, et al., Anal. Chem. 86, 8535-8540(2014)
Small T, et al., Methods in Molecular Biology 911, 2012, pp 3-13
池辺詠美、生物工学会誌、188巻、92~94頁、2014年
上述した従来技術は、cDNAディスプレイ法で使用するリンカーとしては、いずれも優れたものである。しかしながら、cDNA display 技術はmRNAを用いることが前提となっており、主鎖とmRNAとの連結にT4 RNAリガーゼという酵素を使用する。一方で、大腸菌を用いる一般の分子生物学の実験室では、使用する試料中からRNaseを完全に除去することはできないため、ペプチドアプタマーの合成に使用するmRNAが試験管内で分解されてしまい、従来例1~4のリンカーでは、いずれもペプチドアプタマーがうまく合成されないという問題点があった。
例えば、従来例3のリンカーの場合には、ディスプレイされたペプチドの回収を容易にするために、固相とリンカーとを切り離すべく、主鎖に2つの切断部位を設けている(図1C参照)。この場合、rGを組み込んだ場合にはRNaseIで切り離すこととしているが、この酵素を使用するとmRNAの分解が起こるという問題があった。このため、RNaseI以外の酵素で固相からの切り離しを可能とするために、デオキシイノシン(dI)を上記切断部位に組み込み、エンドヌクレアーゼVで切り離すように改変が行われ、mRNAの分解は減少したという点では優れた発明である。
しかし、従来例のリンカーはいずれもT4 RNAリガーゼという酵素を利用して、主鎖とmRNAとを連結させるため、その連結に30分以上はかかるという問題があった。
主鎖とmRNAとの連結時間を短縮するための方法としては、ソラレンを用いて15分程度、290~300nmの波長の紫外線(以下、「UV」ということがある。)照射を行う光架橋法が提案されている。しかし、この光架橋法では、チミジンダイマーが形成され、突然変異が起こりやすくなるという問題がある。また、mRNAにダメージを与えるため、逆転写がうまく行われず、cDNAディスプレイの効率が悪いという問題もある。さらに、ソラレンを主鎖に組み込める位置がリンカーの主鎖の5'末端に限定されており(図1D参照)、リンカーの設計上、大きな制約となっていた。
したがって、酵素を利用することなく、かつmRNAにダメージを与えずに短時間でリンカーとmRNAとを連結することができ、リンカーの設計上も制約とならない方法についての強い社会的要請があった。
また、cDNAディスプレイ法では、リンカーにmRNA、cDNA、ペプチドがディスプレイされた連結体が形成され、こうした連結体を回収し、ディスプレイされたcDNA又はペプチドを生成して配列を特定するという淘汰プロセスを繰り返すことによって、cDNAライブラリ又はペプチドライブラリを構築することができる。しかし、淘汰プロセスに使用したリンカーは、分子間相互作用解析には使用することができない。すなわち、候補クローンを得るための試験管内淘汰実験(スクリーニング)と、スクリーニングによって得られた候補クローンの結合性の評価とを、同じリンカーを用いて行うことができなかった。このため、候補クローンのスクリーニングと、スクリーニングされ前記候補クローンの結合性の評価試験とでは、それぞれ別のリンカーを使用せざるを得ず、作業効率及びコストの面で問題となっていた。
したがって、候補クローンのスクリーニングと得られた候補クローンの結合性の評価との双方に使用することができるリンカーの作製について、強い社会的要請があった。
さらに、標的タンパク質を特異的に検出するためには、免疫グロブリン等の抗体を使用することが望ましいが、軽鎖と重鎖とで構成される抗体は分子量が大きく、70℃以上の熱で不可逆的に失活するため、化学合成ができないという問題があった。また、糖鎖は、それを構成する糖の種類が多く、糖鎖の構造自体も複雑であることから、糖鎖抗原を認識できる抗体を得ることは非常に難しいという問題があった。
一方で、ラクダ科動物の抗体、例えば、VHHの場合には、90℃で熱処理をした場合でも変性は可逆的であり、室温に戻すと熱処理前と同程度の抗原活性を示す。しかし、ラクダ科動物での抗体の産生率は低く、精製にも手間がかかる上、必ずしも、目的とする抗体を得ることができる訳ではないという問題があった。
糖鎖性抗原を特異的に認識し得る分子を得ることができれば、それらを診断薬に利用することによって、癌の予防や治療効果の判定を、迅速かつ的確に行うことができる。そして、このような判定が可能となれば、例え、癌に罹患したとしても、早期に適切な治療を受けることができるようになり、それによって、治癒率を向上させることも可能となる。
このため、目的とする抗体、又は標的分子に結合し得る抗原決定基を有する分子を、迅速かつ簡便に取得することについては、強い社会的な要請があった。
本願発明の発明者らは、以上のような状況の下で鋭意研究を進め、本願発明を完成したものである。
すなわち、本発明の一態様は、主鎖と側鎖とを有するリンカーであって、前記主鎖は、所定の塩基配列を有し、前記主鎖の5'末端に位置し固相との結合を形成するための固相結合部位と;前記固相結合部位ごと前記固相を切り離すための固相切断部位と;前記側鎖を連結するための側鎖連結部位と;前記側鎖連結部位と前記固相切断部位との間に位置し、前記主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位と;前記側鎖連結部位に隣接し、前記主鎖の3'末端に位置する逆転写開始領域と;を備え、前記側鎖は、蛍光標識と、遊離末端に位置するタンパク質結合部位と、前記主鎖の側鎖連結部位に連結される連結形成部位とを備え、前記側鎖は前記連結形成部位で前記側鎖連結部位に連結されている、試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーである。
すなわち、本発明の一態様は、主鎖と側鎖とを有するリンカーであって、前記主鎖は、所定の塩基配列を有し、前記主鎖の5'末端に位置し固相との結合を形成するための固相結合部位と;前記固相結合部位ごと前記固相を切り離すための固相切断部位と;前記側鎖を連結するための側鎖連結部位と;前記側鎖連結部位と前記固相切断部位との間に位置し、前記主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位と;前記側鎖連結部位に隣接し、前記主鎖の3'末端に位置する逆転写開始領域と;を備え、前記側鎖は、蛍光標識と、遊離末端に位置するタンパク質結合部位と、前記主鎖の側鎖連結部位に連結される連結形成部位とを備え、前記側鎖は前記連結形成部位で前記側鎖連結部位に連結されている、試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーである。
ここで、前記固相切断部位は、デオキシイノシン、リボG又はリボピリミジンからなる群から選ばれる塩基で構成されることが好ましい。また、前記高速光架橋部位はシアノビニルカルバゾール化合物で構成されることが好ましく、前記シアノビニルカルバゾール化合物は、3-シアノビニルカルバゾールであることが好ましい。
前記固相結合部位は、ビオチン、ストレプトアビジン、アルキン、クリックケミストリーによるアジ化物、アミノ基、N-ヒドロキシスクシンイミドエステル(NHS)、SH基、及びAuからなる群から選ばれるいずれかの化合物と、前記化合物に結合したポリAとで構成されることが好ましく、前記タンパク質結合部位はピューロマイシン又はその類縁化合物で構成されていることが好ましい。前記ピューロマイシンの類縁化合物としては、3'-N-アミノアシルピューロマイシン(PANS-アミノ酸)及び3'-N-アミノアシルアデノシンアミノ酸のヌクレオシド(AANS-アミノ酸)等を使用することができ、PANSのアミノ酸部分がグリシンであるPANS-Gly、バリンであるPANS-Val、アラニンであるPANS-Ala、PANSアミノ酸の混合物、AANSのアミノ酸部分がグリシンであるAANS-Gly、バリンであるAANS-Val、アラニンであるAANS-Ala、AANSアミノ酸の混合物からなる群から選ばれるいずれかの化合物であることがさらに好ましい。
本発明の別の態様は、上記試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーの主鎖と所望のmRNAとを相補結合させる相補結合形成工程と;前記相補結合が形成された主鎖とmRNAとに、300~400nmの波長の光を0.5~5分間照射して光架橋を形成させる光架橋工程と;前記リンカーに結合されたmRNAを無細胞翻訳系で翻訳し、前記mRNAに対応するタンパク質を前記リンカーのタンパク質結合部位に結合させ、mRNA-タンパク質の融合体を形成する融合体形成工程と;前記融合体を固相に結合させる固相結合工程と;前記融合体に含まれるmRNAを逆転写し、前記融合体と逆転写されたcDNAとの結合体を形成する逆転写工程と;前記結合体を固相切断部位から切断し、所望のcDNAディスプレイ法を選択する選択工程と;を備える、試験管内淘汰方法である。
ここで、前記固相は、ストレプトアビジン又はアビジンでコーティングされた磁性粒子であることが好ましい。また、前記選択工程における結合体の切断は、エンドヌクレアーゼV、RNase T1、及びRNase Aからなる群から選ばれるいずれかの酵素で行われることが好ましい。前記高速光架橋型共用リンカーの主鎖は、糖鎖抗原を認識する配列を有するものであることが好ましい。
本発明のさらに別の態様は、上記試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーの主鎖と所望のmRNAとを相補結合させる相補結合形成工程と;前記相補結合が形成された主鎖とmRNAとに、300~400nmの波長の光を0.5~5分間照射して光架橋を形成させる光架橋工程と;前記リンカーに結合されたmRNAを無細胞翻訳系で翻訳し、前記mRNAに対応するタンパク質を前記リンカーのタンパク質結合部位に結合させ、mRNA-タンパク質の融合体を形成する融合体形成工程と;前記融合体をRNAで消化してリンカー-タンパク質融合体とする、リンカー-タンパク質融合体形成工程と;前記リンカー-タンパク質融合体を固相に結合させる固相結合工程と;前記リンカー-タンパク質融合体を所定の条件下で前記固相から溶出させ、精製する精製工程と;を備える親和性測定用リンカー-タンパク質の作製方法である。
ここで、前記固相は、ストレプトアビジン又はアビジンでコーティングされた磁性粒子であることが好ましい。また、前記精製工程は、0~100mM NaClを含む水溶液中、室温にて行われることが好ましい。
本発明のさらに別の態様は、上記の方法で作製された親和性測定用リンカー-タンパク質である。
本発明によれば、リンカーとmRNAとの連結に要する時間を大幅に短縮することができる、高速光架橋型cDNAディスプレイ用リンカーが提供される。また、前記高速光架橋型cDNAディスプレイ用リンカーを用いることにより、効率よく候補クローンを選択することができる、試験管内淘汰方法が提供される。
さらにまた、前記高速光架橋型cDNAディスプレイ用リンカーを用いることにより、上記の方法で得られた候補クローンの結合性の評価に使用することができる、親和性測定用リンカー-タンパク質の作製方法が提供される。加えて、上記の方法で作製された親和性測定用リンカー-タンパク質が提供される。
以下に、本発明を、図3A~5を参照しつつ、さらに詳細に説明する。
図3Aに示すように、本発明は、(a)主鎖と(b)側鎖とを備える高速光架橋型cDNAディスプレイ用リンカーであり、上記主鎖(a)は、(a1)所定の塩基配列を有し、前記主鎖の5'末端に位置し固相との結合を形成するための固相結合部位と;(a2)前記固相結合部位ごと前記固相を切り離すための固相切断部位と;(a3)前記側鎖を連結するための側鎖連結部位と;(a4)前記側鎖連結部位と前記固相切断部位との間に位置し、主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位と;(a5)前記側鎖連結部位に隣接し、前記主鎖の3'末端に位置する逆転写開始領域と;を備えている。また、上記側鎖(b)は、(b1)蛍光標識と、(b2)遊離末端に位置するタンパク質結合部位と、(b3)前記主鎖の側鎖連結部位に連結される連結形成部位と、を備えている。そして、前記側鎖(b)は、前記連結形成部位で前記主鎖(a)の側鎖連結部位に連結されている。
図3Aに示すように、本発明は、(a)主鎖と(b)側鎖とを備える高速光架橋型cDNAディスプレイ用リンカーであり、上記主鎖(a)は、(a1)所定の塩基配列を有し、前記主鎖の5'末端に位置し固相との結合を形成するための固相結合部位と;(a2)前記固相結合部位ごと前記固相を切り離すための固相切断部位と;(a3)前記側鎖を連結するための側鎖連結部位と;(a4)前記側鎖連結部位と前記固相切断部位との間に位置し、主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位と;(a5)前記側鎖連結部位に隣接し、前記主鎖の3'末端に位置する逆転写開始領域と;を備えている。また、上記側鎖(b)は、(b1)蛍光標識と、(b2)遊離末端に位置するタンパク質結合部位と、(b3)前記主鎖の側鎖連結部位に連結される連結形成部位と、を備えている。そして、前記側鎖(b)は、前記連結形成部位で前記主鎖(a)の側鎖連結部位に連結されている。
上記固相結合部位(a1)は、図3Aに示すように、ビオチン、ストレプトアビジンアルキン、クリックケミストリーによるアジ化物、アミノ基、N-ヒドロキシスクシンイミドエステル(NHS)、SH基、及びAuからなる群から選ばれるいずれかの化合物と、前記化合物に結合したポリAとで構成されていることが好ましい。上記ポリAは、少なくとも10個以上のアデニンが結合していることが、固相と適度な間隔を維持することができ、後述する固相からの切り離しがうまくできることから好ましく、約20個が結合していることがさらに好ましい。
また、上記固相切断部位(a2)はデオキシイノシン、リボG又はリボピリミジンからなる群から選ばれる塩基で構成されることが、以下の理由から好ましい。固相から、後述する2つの連結体、すなわち、本発明のリンカーとmRNAとcDNAとの連結体又は本発明のリンカーとmRNAとcDNAとペプチドとの連結体(以下、双方を合わせて単に「融合体」ということがある。)を、固相から切り離すには、エンドヌクレアーゼV、RNase T1、及びRNase Aからなる群から選ばれるいずれかの酵素を使用する。その際に固相切断部位を上記のいずれかの塩基で構成しておくと、前記リンカーから、固相結合部位ごと固相を特異的に切り離すことができるからである。
これによって、本発明のリンカーは、mRNA、cDNA及び/又はmRNAに対応するタンパク質が結合している場合でも、それらには全く影響がないまま、これらとリンカーとの融合体(連結体)を反応液の上清中に回収することができる。
また、本発明の主鎖には、後述する側鎖を連結するための側鎖連結部位(a3)が設けられている。そして、主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位(a4)が、前記側鎖連結部位と前記固相切断部位との間に位置するように設けられている。この高速光架橋部位は、シアノビニルカルバゾール化合物で構成されることが、mRNAの分解を生じさせないことから好ましい。
こうしたシアノビニルカルバゾール化合物としては、3-シアノビニルカルバゾール等を挙げることができ、3-シアノビニルカルバゾール(以下、「cnvK」ということがある。)を使用すると、水中で、きわめて短時間のうちに、リンカーの主鎖とスクリーニングしようとするmRNAとを長波長の紫外線を用いて結合させることができる。このことは、T4 RNAリガーゼ等の酵素を使用することを必要とせず、水中で架橋反応を行うことができることを意味する。
ところで、ライゲーション用の酵素反応にはZn2+等の金属イオンを含むバッファーが必要であるが、こうしたバッファー中から、RNAを分解するRNaseを完全に除去することはできない。そして、T4 RNAリガーゼをmRNAと主鎖との結合に使用すると、T4 RNAリガーゼが活性化される条件の下では、RNaseも活性化される。このため、RNaseによってmRNAが分解されてしまい、cDNAが合成されないことがしばしば起きていた。しかし、高速光架橋部位をcnvKで構成しておくことにより、酵素を使用することなく、酵素が作用できない水中という条件の下で架橋反応を行なうことができることから、RNaseによるmRNAの分解を防止することが可能となる。
また、上記の高速光架橋部位の架橋に使用する光の波長は、後述するように、300~400nmとかなり長波長であり、しかも照射時間も短い。このため、合成されたcDNA中でチミンダイマーが形成されるといった障害が発生することもなく、使用したmRNAに対応する所望のペプチドを得ることができるという利点がある。
次に、このリンカーに連結されたmRNAに対応するcDNAを合成するための逆転写開始領域(a5)は、この側鎖連結部位に隣接し、前記主鎖の3'末端に位置するように形成されている。
本発明のリンカーに含まれる(b)側鎖は、上述した通り、(b1)蛍光標識と、(b2)遊離末端に位置するタンパク質結合部位と、(b3)前記主鎖の側鎖連結部位に連結される連結形成部位とを備えている。ここで、(b1)蛍光標識としては、例えば、fluorescein、rhodamine、Cy dye、AlexaR Fluorなどを挙げることができる。より具体的には、FITCを使用することがコストの面から好ましい。
(b2)上記側鎖の遊離末端に位置するタンパク質結合部位は、ピューロマイシン又はその類縁化合物で構成されていることが好ましい。前記ピューロマイシンの類縁化合物としては、例えば、3'-N-アミノアシルピューロマイシン(PANS-アミノ酸)及び3'-N-アミノアシルアデノシンアミノ酸のヌクレオシド(AANS-アミノ酸)等を挙げることができる。より具体的には、PANSのアミノ酸部分がグリシンであるPANS-Gly、バリンであるPANS-Val、アラニンであるPANS-Ala、PANSアミノ酸の混合物、AANSのアミノ酸部分がグリシンであるAANS-Gly、バリンであるAANS-Val、アラニンであるAANS-Ala、AANSアミノ酸の混合物等を例示することができる。
ピューロマイシンを使用することが、リンカーの合成が容易であり、また、合成されたリンカーの取り扱いが容易であること、及びコストが安いということから好ましい。
上記側鎖の(b3)連結形成部位は、アミノ基とSH基との架橋反応が可能な異反応性二価性試薬で構成されていることが好ましく、例えば、N-(6-Maleimidocaproyloxy) succinimide(以下、「EMCS」ということがある。)等を使用することができる。低価格で取扱いが容易なことから、EMCSを使用することが好ましい。そして、上記側鎖(b)は、この連結形成部位(b3)で上記主鎖の側鎖連結部位(a3)に連結されている。
以上のような構成とすることによって、本発明のcDNA-ディスプレイ用リンカーに、所望の配列を有するmRNA、このmRNAに対応するペプチドの連結体を形成させ、この連結体を固相に固定化させて、逆転写によりcDNAをさらに連結させた連結体とし、その後にエンドヌクレアーゼV、RNase T1、及びRNase Aからなる群から選ばれるいずれかの酵素を用いて、固相結合部位ごと固相を切り離すことによって、cDNAディスプレイを得ることができる。すなわち、cDNAディスプレイ用のリンカーとして使用することができる(図5)。
また、mRNA-ペプチド-リンカー連結体のRNAを酵素で消化し、その後に、例えば、オリゴdT磁性ビーズ等の固相に固相結合部位で結合させ、溶出バッファーによって溶出させることによって、固相結合部位を備えたタンパク質-リンカー連結体を得ることができ、この連結体は、そのままでSPR(表面プラズモン共鳴装置)、QCM(水晶発振子マイクロバランス)等を用いた結合アッセイに使用することができる(図4)。
以上のような本発明のcDNAディスプレイ用リンカーは、以下のようにして作製することができる。まず、cnvKが固相結合部位と側鎖結合部位との間の所望の位置になるように本発明のリンカーの主鎖(以下、「poly A+cnvKセグメント」ということがある。)を設計し、DNAの化学合成を常法に従って行う。このようなDNA鎖の化学合成は、合成を行う会社に委託することもできる。
このような主鎖としては、例えば、図3Bに示すような逆転写開始領域と、側鎖連結部位と、高速光架橋部位と、固相結合部位とを含むように設計することができる。図3Bに示す主鎖のうち、修飾された部位を除く主鎖の塩基配列を下記の配列(配列表の配列番号1)に示す。下記の主鎖は、5'末端にBioTEGが付加されている。また、下記の塩基配列中、Nはイノシンを表し、XはアミノC6-dTを表す。
このような主鎖としては、例えば、図3Bに示すような逆転写開始領域と、側鎖連結部位と、高速光架橋部位と、固相結合部位とを含むように設計することができる。図3Bに示す主鎖のうち、修飾された部位を除く主鎖の塩基配列を下記の配列(配列表の配列番号1)に示す。下記の主鎖は、5'末端にBioTEGが付加されている。また、下記の塩基配列中、Nはイノシンを表し、XはアミノC6-dTを表す。
[配列1]
5'AAAAAAAAAAAAAAAAAAAASTTCCAGCCGCCCCCCGVCCT 3'
5'AAAAAAAAAAAAAAAAAAAASTTCCAGCCGCCCCCCGVCCT 3'
本発明のリンカーの側鎖(ピューロマイシン-セグメント)も、所望の配列となるように設計し、Poly A+cnvKセグメントと同様に、DNAの化学合成を常法に従って行う。このようなDNA鎖の化学合成は、合成を行う会社に委託することもできる。
このような側鎖としては、例えば、図3Bに示すような連結部位と、蛍光分子と、タンパク質結合部位とを含むように設計することができる。図3Bに示す側鎖のうち、修飾された部位を除く塩基配列は、下記のものを例示することができる。ここで、前記固相切断部位は、デオキシイノシン、リボG又はリボピリミジンからなる群から選ばれる塩基で構成されていることが、固相から後に特異的に切り離すために好ましい。下記の側鎖は、遊離末端となるPはタンパク質結合部位としてのピューロマイシンである。また、下記の塩基配列中、(5S)は5'Thiol C6を、FはFITC-dTを、そしてZは Spacer18をそれぞれ表す。
5' (5S)TCTFZZCCP
例えば、上記のような配列を有する主鎖を10~20nmol(終濃度100~200μM)を含む0.1~0.3Mのリン酸ナトリウム(pH 7.0~7.4)に、EMCS((株)同仁化学研究所製)を終濃度が15~18mMとなるように加えて、約37℃で20~40分インキュベートし、その後、エタノール沈殿をさせる。好ましくは、約15nmolの上記主鎖(終濃度が約150μM)を含む約0.2Mのリン酸ナトリウム溶液(約pH7.2)に、EMCSを終濃度が約16.7mMとなるようにを加え、約37℃で約30分インキュベートし、その後、例えば、Quick-Precip Plus Solution(Edge BioSystems社製)等を用いてエタノール沈殿させる。
次に、30~45nmol分の側鎖を終濃度が400~430μMとなるように、40~60mMのDTTを含む0.8~1.5Mのリン酸水素ナトリウム水溶液に溶解し、シェーカーを用いて、室温で0.75~1.5時間撹拌する。引き続き、この溶液についてバッファー交換を行う。好ましくは、約37.5nmol分の側鎖を終濃度が約417μMとなるように、約50mMのDTTを含む約1Mのリン酸水素ナトリウム水溶液に溶解し、シェーカーを用いて、室温にて約1時間撹拌する。引き続き、例えば、NAP5カラム等を用いて、約0.15MのNaClを含む約0.1Mのリン酸ナトリウム(約pH7.0)にバッファー交換を行う。
次いで、上記のようにバッファー交換を行った還元側鎖を含む溶液を、上記のEMCS修飾済みの主鎖のエタノール沈殿産物と混合し、2~6℃で一晩放置し、その後、終濃度が40~60mMとなるようにDTTを上記反応液に投入し、室温で15~60分間撹拌する。その後、エタノール沈殿を行い、得られたエタノール沈殿産物を50~200μLのNuclease-free waterに溶解して精製を行う。好ましくは、上記のバッファーにバッファー交換した還元側鎖を含む溶液を、上記のEMCS修飾済み主鎖のエタノール沈殿産物と混合し、約4℃で一晩放置する。
その後、終濃度が約50mMとなるようにDTTを上記の反応液中に加え、室温で約30分間撹拌し、その後、例えば、Quick-Precip Plus Solution(Edge BioSystems社製)を用いて、エタノール沈殿を行なう。得られたエタノール沈殿産物を、約100μLのNuclease-free waterに溶解し、例えば、以下の条件でC18カラムを用いてグラジエント溶出により、HPLC精製を行う。これによって、本発明のリンカーを得ることができる。
グラジエント溶出に使用する溶出液は、例えば、A液を0.05~0.2Mの酢酸トリメチルアンモニウム(超純水中)、B液を75~85%アセトニトリルとし、開始時の溶出期中のA液の割合を、40~50分かけて、20%ほど低下させるようにしてもよい。流速は0.5~1.5ml/分とし、画分は0.5~1.5mLとすることができる。好ましくは、A液を約0.1Mの酢酸トリメチルアンモニウム(超純水中)、B液を約80%アセトニトリルとし、開始時の溶出期中のA液の割合(約85%)を、40~50分かけて、約65%に低下させるようにする。流速は約1.0ml/分、画分は約1.0mLとする。
上記の画分中の成分を蛍光及び紫外吸収(例えば、280nm)で確認し、双方の検出手段でピークが見られる画分を集めて、真空エバポレターを用いて溶媒を蒸発させ、その後、エタノール沈殿を行ない、Nuclease-free waterに溶解することによって、本発明のリンカーを製造することができる。得られた本発明のリンカーは、約-20℃で保存する。例えば、30~32分までの画分で、蛍光とUVの両方でピークが見られる場合には、30~32分までの画分を集めて、真空エバポレターを用いて溶媒を蒸発させる。その後、例えば、Quick-Precip Plus Solutionを用いてエタノール沈殿を行ない、Nuclease-free waterに溶解させて約-20℃で保存すればよい。
(試験管内淘汰方法)
所望の配列を有するライブラリDNAとFLAG配列とがコードされたDNAを、所望の比率、例えば、25,000~100,000:1のモル比で混合したDNA混合物を調製し、一部を取って転写する。例えば、250~1,000ngを、RiboMAX Large Scale RNA Production Systems-T7等のキットを使用して、10~20μLのスケールで転写を行う。転写に使用する配列としては、例えば、下記のような配列(配列表の配列番号2)を使用することができる。下記の配列中、Nは、任意にA, T, G, 及びCを表し、KはG又はTを表す。
所望の配列を有するライブラリDNAとFLAG配列とがコードされたDNAを、所望の比率、例えば、25,000~100,000:1のモル比で混合したDNA混合物を調製し、一部を取って転写する。例えば、250~1,000ngを、RiboMAX Large Scale RNA Production Systems-T7等のキットを使用して、10~20μLのスケールで転写を行う。転写に使用する配列としては、例えば、下記のような配列(配列表の配列番号2)を使用することができる。下記の配列中、Nは、任意にA, T, G, 及びCを表し、KはG又はTを表す。
[配列2:アミノ酸ランダムライブラリDNA配列]
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCAGCNNKNNKNNKNNKNNKNNKNNKNNKGGAGGTGGAATTAAAAACATGTGCAATTTGAACCCACTTTTAAAAAAGTGGCTAAATGATGCAAAGGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCAGCNNKNNKNNKNNKNNKNNKNNKNNKGGAGGTGGAATTAAAAACATGTGCAATTTGAACCCACTTTTAAAAAAGTGGCTAAATGATGCAAAGGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
上記のDNA混合物を所望の温度で所望の時間、例えば、約37℃で3~5時間インキュベートした後、DNaseを所望の量で加える。その後、さらに所望の温度で所望の時間、例えば、約37℃で5~15分間インキュベートし、得られたmRNAをその後精製する。好ましくは、上記のDNA混合物を約37℃で約4時間インキュベートした後、上記のキット付属のDNase (例えば、RQ1 Dnase、プロメガ社製)を約0.5μL加え、さらに約37℃で約10分間インキュベートしてmRNAを得る。得られたmRNAは、例えば、After Tri-Reagent RNA Clean-Up Kit(Favogen Biotech Corp.製)を使用して精製する。
上記と同様にして、mRNAと本発明のリンカー(poly A+cnvK)の光架橋を、長波長のUVを0.5~5分間照射して行う。得られたビオチン-cnvKリンカー-mRNA連結体(以下、「mRNA-リンカー連結体」ということがある。)を、上記と同様に所望のスケールで、無細胞翻訳系を用いて所望の温度で所望の時間翻訳する。例えば、10~20μLのmRNA-リンカー連結体を用いて、100~150μLのスケールで、例えば、ウサギ網内赤血球ライセート等の無細胞翻訳系を使用して、約30℃で約15分間翻訳する。その後、MgCl2及びKClをそれぞれ所望の濃度で加えて、上記のmRNA-リンカー連結体にさらにペプチドが連結したmRNA-ペプチド連結体を得ることができる。例えば、それぞれ75mM及び900mMとなるようにMgCl2及びKClをそれぞれ加えて、約37℃で約1時間インキュベートし、mRNA-ペプチド連結体を含む翻訳反応液を得る。
上記のようにして得られた翻訳反応液に、0.25~1MのEDTA(pH 7.8~8.2)を所望の濃度で加えて所望の温度でインキュベートし、mRNA-ペプチド連結体に結合しているリボソームを除去する。例えば、約0.5MのEDTA(pH 約8.0)を、終濃度が約83mMとなるように加えて、室温で約5分間インキュベートする。
引き続き、上記のように処理した翻訳反応液に、所望の結合バッファーを等量加え、所望の結合バッファーで洗浄した所望の量のストレプトアビジン付きの磁性ビーズと混合し、所望の温度で所望の時間撹拌する。例えば、SA用2x結合バッファー(約20mMのTris-HCl(pH 約7.5), 約2MのNaCl、約2mMのEDTA、約0.2%のTween-20を含む)を等量加え、SA用の1x結合バッファーで洗浄済みの磁性ビーズ、例えば、Dynabeads MyOne C1 ストレプトアビジン100~200μLと混合し、約20~30℃で約20~40分撹拌する。
上記の磁性ビーズを、所望の量のSA用1x結合バッファーで所望の回数洗浄し、その後逆転写を行う。例えば、約200μLのSA用の1x結合バッファーで2~4回洗浄し、例えば、ReverTra Ase(登録商標)に付属のプロトコルに従って、約100μLの逆転写用反応液を投入し、約42℃で約15分間撹拌して逆転写を行い、mRNA/cDNA-ペプチド連結体(以下、「cDNAディスプレイ」ということがある。)を調製する。
所望の量の1xNEバッファーで、上記の磁性ビーズを洗浄し、その後所望の量のエンドヌクレアーゼV、RNase T1、及びRNase Aからなる群から選ばれるいずれかの酵素を含む1xNEバッファーを加えて、所望の温度で所望の時間撹拌する。次いで、所望量の2xHis-tag洗浄バッファーを加え、その後上清を回収する。例えば、約150μLの1xNEバッファーで、Dynabeads MyOne C1 ストレプトアビジンを洗浄し、その後、約10UのEndonuclease Vを含む、約75μLの1xNEバッファーを加えて、約37℃で約1時間撹拌する。次いで、約75μLの2xHis-tag洗浄バッファー(約1MのNaCl、約0.1%のTween-20を含む約40mMのリン酸ナトリウム(pH 約7.4))を加え、その後上清を回収する。
次いで、回収した上清と、例えば、1xHis-tag洗浄バッファーで洗浄したHis Mag Sepharose Ni等の磁性ビーズとを混合し、ミキサーを用いて、所望の温度で所望の時間撹拌する。このビーズを1xHis-tag洗浄バッファーで所望の回数洗浄し、その後、所望のセレクションバッファーを加え、ミキサーを用いて、所望の温度で所望の時間撹拌し、その後に上清を回収する。例えば、回収した上清約150μLと、約20μLのHis Mag Sepharose Ni (GEヘルスケア社製、1xHis-tag洗浄バッファーにより洗浄済み)とを混合し、インテリミキサーRM-2M((株)トーホー製)等のミキサーを用いて、室温で約1時間撹拌する。
約100μLの1xHis-tag洗浄バッファーで1~3回洗浄し、その後、EDTA濃度を高めた約30μLのセレクションバッファー(約1MのNaCl、約10mMのイミダゾール、約5mMのEDTA及び約0.1%のTween-20を含む約50mMのTris-HClバッファー(pH 約7.4))を加え、上記のミキサーを用いて、室温で約10分間撹拌した後に上清を回収する。以上のようにして、本発明のリンカーを製造することができる。
得られたリンカーから、所望量をとり、例えば、抗FLAG M2アフィニティゲルの懸濁液を、所望のカラムに充填し、所望量のセレクションバッファーで洗浄する。その後、所望量の上記の上清をカラムにロードし、ローテーター等を用いて室温で約1時間撹拌する。所望量のセレクションバッファーでカラムを複数回洗浄し、次いで、例えば、所望の濃度のFLAGペプチド(シグマアルドリッチジャパン合同会社製)を加え、上記のローテーター等を用いて室温で約15分間撹拌することにより、抗FLAG M2アフィニティゲルに結合しているmRNA/cDNA-ペプチド連結体を競合溶出させる。
例えば、約25~100μLの抗FLAG M2アフィニティゲル(約40~60%懸濁液)を、MicroSpin Empty Columns(GEヘルスケア社製)等の適当なカラムに充填し、約150~250μLのセレクションバッファーで2~4回洗浄する。その後、上記の上清から、約50~150μLをとってカラムにロードし、ローテーターを用いて室温で約1時間撹拌する。約150~250μLのセレクションバッファーで3~5回カラムを洗浄し、次いで約50~150ng/μLの3xFLAGペプチド(シグマアルドリッチジャパン合同会社製)を約50~150μL加え、上記のローテーターを用いて室温で約15分間撹拌することにより、抗FLAG M2アフィニティゲルに結合しているmRNA/cDNA-ペプチド連結体を競合溶出させることができる。
遠心操作によってカラム内の溶出液を回収し、エタノール沈殿を行なった後に、所望量のヌクレアーゼフリー水(Nuclease-free water)に溶解する。上記のエタノール沈殿産物を、約150~250μLのPCR反応液に加え、PCRを行なう。例えば、遠心操作によって回収したカラム内の溶出液を、Quick-Precip Plus Solution等を使用してエタノール沈殿させ、約15μLのNuclease-free waterに溶解する。上記のエタノール沈殿産物を、約150~250μLのPCR反応液(約0.2mMのdNTPs、約0.4μMのT7Ωnew、約4μMのcnvK用NewYtag、約0.02U/μLのPrimeSTAR HS DNAポリメラーゼを含む1xPrimeSTARバッファー(Mg2+含有)に加え、以下のようにPCRを行なう。ここで使用するペプチドとして例示した2つ、T7Ωnew及びcnvK用NewYtagの配列(配列表の配列番号3及び4)を以下に示す。
[配列3:T7Ωnewの配列]
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACA-3'
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACA-3'
[配列4:cnvK用NewYtagの配列]
5'‐TTTCCACGCCGCCCCCCGTCCTGCTTCCGCCGTGATGAT‐3'
5'‐TTTCCACGCCGCCCCCCGTCCTGCTTCCGCCGTGATGAT‐3'
次に、PCRは、例えば、(a1)98℃で1分、(b1)98℃で15秒、(c1)68℃で30秒、(d1)68℃で1分、上記(b1)及び(c1)を25サイクルとすることができる。得られたPCR産物を、SDSゲル電気泳動にかけ、フルコンストラクトDNAを切出し、常法に従って精製する。精製した上記フルコンストラクトDNAを、所望量のPCR反応液に加えて、所望量で分注し、PCRを行なうと、二本鎖DNAを得ることができる。
上記のようにPCRを行なって得られたPCR産物を、例えば、8M尿素変性6%PAGEにて泳動し、フルコンストラクトDNA(260~300mer)を切出して、常法に従って精製する。精製した上記フルコンストラクトDNAを、約150~250μLのPCR反応液(約0.2mMのdNTPs、約0.4μMのNewleft、約0.4μMのcnvK用NewYtag、及び約0.02U/μLのPrimeSTAR HS DNA polymeraseを含む1xPrimeSTARバッファー(Mg2+含有))に加え、約25~75μLずつ分注して、PCRを行なうことにより、二本鎖DNAを得ることができる。ここで使用するペプチドとして例示した、Newleftの配列(配列表の配列番号5)を以下に示す。
[配列5:Newleftの配列]
GATCCCGCGAAATTAATACGACTCACTATAGGG
GATCCCGCGAAATTAATACGACTCACTATAGGG
PCRは、例えば、(a2)98℃で1分、(b2)98℃で10秒、(c2)68℃で30秒、(d2)68℃で1分とし、ステップ(b2)及び(c2)を5サイクル行うようにすることができる。上記のようにPCRを行なった後のPCR反応液をまとめてカラム精製し、次のラウンドに使用する。以上のような手順を、ライブラリDNAの転写からアフィニティセレクションまでを、所望のラウンド、例えば、合計で3ラウンド行い、その結果得られたフルコンストラクトDNAの配列をダイレクトシーケンシングすることにより、一体となった配列のFLAG-DNAをディスプレイした本発明のリンカーを得ることができる。
次に、結合アッセイ等に使用するための本発明のリンカーについて説明する。配列が公知となっている所望のタンパク質DNAを使用し、上記と同様の手順に従って、上記公知タンパク質の塩基配列に対応するmRNA-タンパク質-リンカー連結体を得ることができる。ここで使用する配列が公知のタンパク質としては、例えば、Aタンパク質のBドメイン((以下、「BDA」という。配列表の配列番号6)等を使用することができる。
[配列6:BDAのDNA配列]
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGATAACAAATTCAACAAAGAACAACAAAATGCTTTCTATGAAATCTTACATTTACCTAACTTAAACGAAGAACAACGCAATGGTTTCATCCAAAGCCTAAAAGATGACCCAAGCCAAAGCGCTAACCTTTTAGCAGAAGCTAAAAAGCTAAATGATGCTCAAGCACCAAAAGCTGACAACAAATTCAACGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGATAACAAATTCAACAAAGAACAACAAAATGCTTTCTATGAAATCTTACATTTACCTAACTTAAACGAAGAACAACGCAATGGTTTCATCCAAAGCCTAAAAGATGACCCAAGCCAAAGCGCTAACCTTTTAGCAGAAGCTAAAAAGCTAAATGATGCTCAAGCACCAAAAGCTGACAACAAATTCAACGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
次いで、上記のmRNA-タンパク質-リンカー連結体を含む翻訳産物に、所望のバッファーとRNase Hとを加え、所望の温度で所望の時間インキュベーションして、mRNAを分解する。この反応液と等量の所望の結合バッファーを加え、上記の結合バッファーで洗浄した所望の磁性ビーズと混合し、ローテーターを用いて室温で約15~45分撹拌する。
例えば、上記のmRNA-ペプチド連結体を含む翻訳産物に、約1/9倍容の10xNEバッファー2と、約20UのRNase H(タカラバイオ(株)製)を加え、約37℃で45~75分間インキュベーションして、mRNAを分解する。次いで、例えば、この反応液と等量のオリゴdT用の2x結合バッファー(約1MのLiCl, 約2mMのEDTA、及び約0.1%のTween-20を含む約20mMのTris-HCl(pH 約7.5))を加え、約50~150μLの磁性ビーズ、例えば、Dynabeads Oligo (dT)25(サーモ・フィッシャー。サイエンティフィック(株)製、オリゴdT用の2x結合バッファーにて洗浄済み)と混合し、ローテーター等を用いて室温で約30分撹拌する。
上記の磁性ビーズを所望の量の所望のバッファーで洗浄し、その後、所望量の超純水を加えて所望の温度で所望の時間インキュベートし、ビオチン接着タンパク質を溶出する。このようにして得られたビオチン接着タンパク質溶液に対して、等量の所望の洗浄バッファーを加える。その後、所望量のHis Mag Sepharose Ni等の磁性粒子と混合し、ローテーター等を用いて所望の温度で所望の時間撹拌する。
例えば、磁性ビーズとして、Dynabeads Oligo (dT)25を使用し、これを約150~250μLのオリゴdT用1x結合バッファーで複数回洗浄する。その後、200~250μLの超純水を加えて約37℃で5~15分間インキュベートし、ビオチン接着BDA等の所望のタンパク質と結合したリンカーを溶出する。得られたビオチン接着BDA溶液に対して、例えば、等量の2xHis-tag洗浄バッファーを加え、その後、約15~50μL分のHis Mag Sepharose Ni等の磁性ビーズ(1xHis-tag洗浄バッファーで洗浄済み)と混合し、ローテーター等を用いて室温で約30~50分撹拌する。
次いで、所望の量の1xHis-tag洗浄バッファーで洗浄し、その後、所望の溶出バッファーを加えて、ミキサー等を用いて所望の温度で所望の時間撹拌し、ビオチン接着タンパク質を溶出させ、得られたビオチン接着タンパク質のDNA配列を決定する。
例えば、約50~150μLの1xHis-tag洗浄バッファーで複数回洗浄し、その後、50~60μLのHis-tag溶出バッファー(約0.5MのNaCl, 約250mMのイミダゾール、約0.05%のTween-20を含む約20mMのSリン酸ナトリウムリムバッファー(pH 約7.4))を加えて、例えば、インテリミキサーRM-2Mを用いて室温で10~20分間撹拌し、ビオチン接着タンパク質を溶出させ、得られたビオチン接着BDAのDNA配列を決定する。
この後、Biacore X100(GEヘルスケア社製)等を使用して親和性を測定し、例えば、1:1 ラングミュア結合モデル(A + B = AB)を用いたビアコアJソフトウェアを用いて、結合時(ka) 及び解離時(kd)の速度、及び親和性(KD = kd/ka)を求めることができる。
本発明の共用リンカーは、以上のように、試験管内淘汰及び分子間結合研究の双方に使用することができる。
上述したcnvKリンカーを用いて淘汰を行なうための架橋ペプチドアプタマーは、以下のようにして調製することができる。こうしたペプチドアプタマーの調製には、ラクダ科の動物の抗体、例えば、VHHを使用することが、リフォールディング性に優れ、CDR3の配列が相対的に長いことから好ましい。
例えば、常法に従って、VHHと所望の配列とを連結させて、VHHライブラリコンストラクトを構築し、鋳型DNAとして使用することができる。所望の配列としては、T7領域、Kozak領域、5'cap領域、Ω領域、His-Tag、Y-tag、NewY tag、リンカーとハイブリダイゼーションする領域等を挙げることができる。こうした配列を含むコンストラクトとしては、例えば、配列表の配列番号7に示すようなDNA配列を有するものを好適に使用することができる。下記の配列番号7中、n、b、d、r、y、h、mはそれぞれMix塩基を示しており、その比率は以下のとおりであり、塩基の出現頻度は下記表2に示す通りである。また、nbn、drb、yhmで表されるそれぞれのコドン中の理論上の各アミノ酸の出現頻度を図6(A)~(C)に示す。
(配列番号7)
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCGAGGTGCAGCTGGTGGAGAGCGGAGGAGGATCCGTGCAGGCTGGAGGAAGCCTGCGCCTGAGCTGCGCTGCTAGCGGAnbnnbndrbyhmyhmyhmdrbnbnnbnTGGTTCCGCCAGGCTCCTGGAAAGGAGCGCGAGGGAGTGnbnnbndrbyhmyhmyhmyhmdrbACCTACTACGCTGACAGCGTGAAGGGACGCTTCACCATCAGCCAGGACAACGCCAAGAACACCGTGTACCTGCAGATGAACAGCCTGAAGCCTGAGGACACCGCTATCTACTACTGCGCTGCTdrbdrbyhmyhmyhmyhmyhmyhmyhmyhmyhmyhmdrbTACTGGGGACAGGGAACCCAGGTGACCGTGGGAGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGGGGAAA-3'
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCGAGGTGCAGCTGGTGGAGAGCGGAGGAGGATCCGTGCAGGCTGGAGGAAGCCTGCGCCTGAGCTGCGCTGCTAGCGGAnbnnbndrbyhmyhmyhmdrbnbnnbnTGGTTCCGCCAGGCTCCTGGAAAGGAGCGCGAGGGAGTGnbnnbndrbyhmyhmyhmyhmdrbACCTACTACGCTGACAGCGTGAAGGGACGCTTCACCATCAGCCAGGACAACGCCAAGAACACCGTGTACCTGCAGATGAACAGCCTGAAGCCTGAGGACACCGCTATCTACTACTGCGCTGCTdrbdrbyhmyhmyhmyhmyhmyhmyhmyhmyhmyhmdrbTACTGGGGACAGGGAACCCAGGTGACCGTGGGAGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGGGGAAA-3'
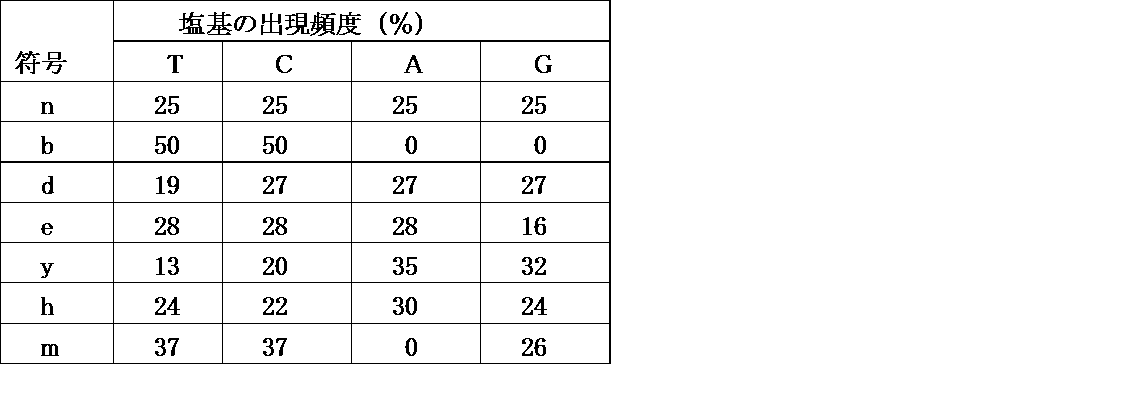
これらの配列を含むコンストラクトを作製した後に、所望の条件でPCRを行ない、コンストラクトを増幅させることができる。例えば、それぞれ所望量の5X Prime STAR Buffer、dNTP 混合物、プライマー、Prime STAR HS DNA polymeraseに、上記のVHHコンストラクトを加え、超純水で所望量、例えば、PCR溶液を調製することができる。
ここで、プライマーとしては、T7 omega new (60 mer)(配列表の配列番号3)、NewYtag_for_PolyA&cnvK-Lin (22 mer)(配列表の配列番号4)、ΩRT-Lnew (32 mer)(配列表の配列番号8)、New left (33 mer)(配列表の配列番号5)、及びNewYtag (22 mer)(配列表の配列番号9)等を挙げることができる。
上記のような組成のPCR溶液を調整し、例えば、約98℃で1.5~2.5分、(約98℃で5~15秒、約98℃で約1秒、約65~70℃で約30~50秒)を4~6サイクル、約70~75℃で1.5~2.5分というPCRプログラムで増幅させることができる。
(配列番号4)NewYtag_for_PolyA&cnvK-Lin (22 mer)
5'-TTTCCACGCCGCCCCCCGTCCT-3'
(配列番号8)ΩRT-Lnew (32 mer)
5'‐GGGGAAGTATTTTTACAACAATTACCAACAAC‐3'
5'-TTTCCACGCCGCCCCCCGTCCT-3'
(配列番号8)ΩRT-Lnew (32 mer)
5'‐GGGGAAGTATTTTTACAACAATTACCAACAAC‐3'
ここで、VHHコンストラクトライブラリを含む最初の溶液(原液)のダイバーシチは、約2.5~5x1011分子/μLとすることが、このライブラリに含まれる多様なVHHコンストラクトを材料として使用できることから好ましい。
上記のようなPCRで得られたPCR産物を、所望の鎖長のPCR産物を選択するために、常法に従って精製し、短鎖長のDNAを除去することが好ましい。こうした精製には、PCR Clean-Up MiniKit (Favorgen社製)等のキットを使用することができる。次いで、必要に応じてプライマーを変更してPCRを行なうことにより、所望の鎖長のPCR産物を得ることができる。例えば、上記のプライマーのうち、New leftを使用する場合には、これをT7 omega newに代えてPCRを行ない、例えば、上記と同様のPCRプログラムによって所望のPCR産物を得ることができる。
次いで、得られたPCR産物をmRNAに転写する。こうした転写には、例えば、RiboMAXTM Large Scale RNA production Systems(Promega社製)等の市販品を使用してもよく、所望の転写反応液を使用して転写させてもよい。こうした転写反応液としては、例えば、T7 Transcription 5X Buffer、rNTPs、Enzyme Mixに、RNase-Free水に溶解した鋳型DNAを加えたものとすることができる。
上記の転写反応溶液を、例えば、約36~38℃で所望の時間、例えば、2.5~3.5時間、インキュベートし、Rnase-Free DNAseを加えて、例えば、約36~38℃で所望の時間、例えば、10~20分間インキュベートすることにより、mRNAを得ることができる。Rnase-Free DNAseとしては、例えば、RQ1 Rnase-Free DNAse (PROMEGA社製)等の市販品を使用することができる。
その後、RNAを精製する。RNAの精製には、例えば、After Tri-Reagent RNA Clean-up Kit(Favogen Biotech Corp.製)等を使用することができる。所望のRNAを得ることができるように、鋳型となるDNAの量を調整して、リンカーとの光架橋を行なうことができるmRNAを得るようにすることが好ましい。
次いで、得られた精製mRNAと、上記のようにして作成したBiotin-cnvKリンカーとを、光架橋により連結させる。ここで、上記の精製mRNA、ビオチン-cnvKリンカー、約0.75~1.5M NaCl及び約0.2~0.3M Tris-HClを含む光架橋反応液を、例えば、約88~92℃で約1~3分加熱し、その後約1分間で約65~75℃まで降温させる。次いで、約68~72℃で約0.5~2分加熱し、その後15分間で約20~30℃まで降温させて、アニールすることができる。ここで、上記光架橋反応液中のmRNAとBiotin-cnvKリンカーとの比率は、ほぼ等モルとすることが架橋効率の点から好ましい。
アニーリング終了後、長波長の紫外光を所望の量で照射し、光架橋を行なう。例えば、CL-1000 Ultraviolet Crosslinker(UVP社製)等の市販の装置を用いて、波長約360~370nmの紫外光を、350~450mJ/cm2の条件で照射し、光架橋を行って、mRNA-リンカー連結体を得ることができる。
引き続き、以上のようにして得られたmRNA-リンカー連結体を、無細胞翻訳系で翻訳し、mRNA-ペプチド連結体を形成させる。こうした翻訳に使用する反応溶液としては、例えば、Translation Mix、上記mRNA-リンカー連結体、Retic Lysate、及びRNaseインヒビターを含む溶液を調製し、最後に、超純水で所望の液量に調整する。
無細胞系翻訳は、上記の翻訳用反応液入れたチューブを、以下のように処理して行うことができる。例えば、約25~35℃で、約15~25分間インキュベートし、ここに、約10~15μLの約2.5~3.5M KClと、約2~4μLの0.5~1.5M MgCl2とを加え、約36~38℃で、さらに約45~75分間インキュベートする。その後、約5~15μLのエチレンジアミン四酢酸(約pH7.5~8.5)を加え、約36~38℃で、約5~15分間インキュベートし、次いで約40~80μLの結合バッファーを加えることにより、無細翻訳系で合成されたペプチドが上記mRNA連結体に結合した、mRNA-ペプチド連結体を得ることができる。
次いで、上記のようにして得られたmRNA-ペプチド連結体を、磁性体ビーズ上に結合させる。こうした磁性体ビーズとしては、例えば、Streptavidin(SA) Magnetic beads:Dynabeads MyOne Streptavidin C1等を挙げることができる。この結合に使用するmRNA-ペプチド連結体の濃度の約8~15倍容の磁性体ビーズを、所望の大きさのProtein LoBindチューブ(Eppendorf社製)に入れ、磁気スタンド上に静置して上澄を捨てる。ここに、1x結合バッファーを加えて再懸濁し、磁気スタンド上に静置して上澄を捨てることにより、磁性体ビーズを洗浄し、RNaseフリーとすることができる。
次いで、上記のようにして得た翻訳産物(mRNA-ペプチド連結体)を加え、約20~30℃で約75~135分間、ローテーターで攪拌しながらインキュベートし、mRNA-ペプチド連結体を磁性体ビーズに結合させる。
以上のようにしてチューブ内で磁性体ビーズ上にmRNA-ペプチド連結体を固定化し、このチューブを磁気スタンド上に静置して上澄を捨てる。このチューブに、所望量、例えば、約50~150μLの1x結合バッファーを加えて磁性体ビーズを再懸濁させ、上澄を捨てるという操作を所望の回数繰り返す。
その後、所望の溶液を用いてcDNAを合成し、上記のmRNA-ペプチド連結体に、さらにcDNAが結合したcDNAディスプレイを得る。こうしたcDNA合成には、例えば、ReverTra Ace(東洋紡(株)製)等の市販品を使用することができる。例えば、ReverTra Ace付属バッファー、dNTP混合液、Rever Tra Ace (100U/μL)、及び超純水を含むcDNA合成用反応溶液を、上記の洗浄済のmRNA-ペプチド連結体を固定化した磁性体ビーズ入りのチューブに加え、約40~45℃で約60~120分間ローテーターで攪拌しながらインキュベートすることにより、cDNAがさらに結合したcDNAディスプレイを、磁性体ビーズ上に固定されたものとして得ることができる。
以上のようにして得られたcDNAディスプレイが結合した磁性体ビーズが入っているチューブに、cDNAディスプレイを磁性体ビーズから切り離すための酵素と、切り離されたcDNAディスプレイの精製に使用するためのtagを加えてインキュベートする。ここで、上記酵素としては、デオキシイノシン、リボG、リボT、リボC、リボU等のリボピリミジンを、所望の濃度、例えば、500~1,500U/μLで使用することができる。また、tagとしては、His-tag、Y-tag等を使用することができる。インキュベートは、約36~38℃で、15~45分間、ローテーター等を使用して撹拌しながら行うことが好ましい。
以上のようにして得られたcDNAディスプレイを、tagを使用して、VHHの配列をコードしたペプチドが発現しているものと、発現していないものとに分別する。例えば、上記cDNAディスプレイが入ったチューブに、His Mag sepharose Niを加え、所望の温度で所望の時間、例えば、約20~30℃で0.5~1.5時間、振盪しながらインキュベートし、その後、このチューブを磁気スタンド上に静置して上澄を除く。tag用のバッファーで洗浄し、溶出バッファーを加えて、所望の温度で所望の時間、例えば、室温にて15~60分間、振盪しながらインキュベートする。インキュベート後に上澄を回収することにより、VHHペプチドが発現したcDNAディスプレイだけを取得することができる。
引き続き、VHHペプチドが発現したcDNAディスプレイを含む溶液のバッファーを交換する。このバッファー交換には、例えば、Micro Bio-SpinTM 6 カラム(BIO-RAD社製)等を使用することができる。
また、ゲル電気泳動は以下の手順で行う。常法に従い、10xSDSランニングバッファー、1.5M トリス塩酸バッファー(pH 8.8)、0.5M トリス塩酸バッファー(pH 6.8)、2xSDSサンプルバッファー、40%アクリルアミド溶液を調製する。これらを用いて、所望の濃度のスタッキングゲル及び分離ゲル、例えば、4%スタッキングゲル及び15%分離ゲルを調製する。
これらのゲル電気泳動を用いて、所望の電流で所望の時間、例えば、20mAで、泳動サンプルの分離に適した時間、電気泳動を行なう。泳動終了後、FITC、SYBER-gold等の染色液を使用して、常法に従って染色を行ない、イメージャー、例えば、Typhoon FLA 9500(GEヘルスケア・ジャパン(株)製)等を用いて、ゲル電気泳動の結果を解析する。
その後、発現しているVHHペプチドが、所望の糖、例えば、癌関連単糖N-アセチル-D-グルコサミン(GlcNAc)に対する結合性を持っているか否かを以下のようにして確認する。まず、磁性体ビーズとビオチン化GlcNAcとをインキュベートしてビオチン化GlcNAcを固定化した磁性体ビーズを準備し、何も固定化していない磁性体ビーズとを準備する。まず、ビオチン-DNAを等量ずつ加えてインキュベートし、ビオチン化GlcNAcが磁性体ビーズに固定化されているかどうかを確認する。
磁性体ビーズをグルコサミン洗浄用バッファーで洗浄し、ビオチンと共に、所望の温度で所望の時間、例えば、約20~30℃にて約15~45分間、インキュベートし、その後、シェーカーで振盪しながら攪拌することによって、GlcNAcに特異的に結合するcDNAディスプレイを得ることができる。
磁性体ビーズにビオチン化GlcNAcが固定化されているか否かは、所望の糖認識ペプチド、例えば、Oct1タンパク質のPOUドメインを用いて構築したDNA断片である、PDOを使用して行うことができる。PDOは、図5に示すように、T7、5'cap、Ω、Kozak、GGGS、His-Tag、GGS及びY-tagという配列を含んでおり、以下に示すような塩基配列のペプチドである(配列表の配列番号10)。
(配列番号10)PDOのDNA配列
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACA
ATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTAC
AACTACAAGCCACCATGGATAACAAATTCAACAAAGAACAACAAAATGCT
TTCTATGAAATCTTACATTTACCTAACTTAAACGAAGAACAACGCAATGG
TTTCATCCAAAGCCTAAAAGATGACCCAAGCCAAAGCGCTAACCTTTTAG
CAGAAGCTAAAAAGCTAAATGATGCTCAAGCACCAAAAGCTGACAACAAA
TTCAACGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGAC
GGGGGGCGGCGGGGAAA -3'
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACA
ATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTAC
AACTACAAGCCACCATGGATAACAAATTCAACAAAGAACAACAAAATGCT
TTCTATGAAATCTTACATTTACCTAACTTAAACGAAGAACAACGCAATGG
TTTCATCCAAAGCCTAAAAGATGACCCAAGCCAAAGCGCTAACCTTTTAG
CAGAAGCTAAAAAGCTAAATGATGCTCAAGCACCAAAAGCTGACAACAAA
TTCAACGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGAC
GGGGGGCGGCGGGGAAA -3'
常法に従ってビオチンと結合させたPDOを使用することによって、磁性体ビーズにGlcNAcが結合しているか否かを判断することができる。磁性体ビーズを含むチューブに投入する前のPDO濃度と、インキュベーション後の上澄中のDNA濃度とを比較したときに、インキュベーション後のPDO濃度が大きく低下していれば、PDOが磁性体粒子に結合したことになるからである。
以上のようにして得られた、VHHペプチドを発現しているcDNAディスプレイと、GlcNAcを固定化した磁性体ビーズとを用いて、試験管内淘汰を繰り返すことにより、GlcNAc結合性のVHHペプチドを発現したcDNAディスプレイを得ることができる。
(実施例1)本発明のリンカー(Puromycine-linker(polyA+cnvK))の作製及び特性評価
(1)本発明のリンカー(Puromycine-linker(polyA+cnvK))の作製
Puromycine-linker(polyA+cnvK)の構造を図3A及び3Bに示す。ビオチンフラグメント(poly A+cnvK:主鎖)は、配列表の配列番号1に記載された配列を有している。ここで、上記主鎖の5'末端にはBioTEGが結合しており、また、塩基配列中のNはイノシンを、XはAminoC6-dTをそれぞれ表す。また、ピューロマイシン-セグメント(側鎖)は、下記の配列を有する。
(1)本発明のリンカー(Puromycine-linker(polyA+cnvK))の作製
Puromycine-linker(polyA+cnvK)の構造を図3A及び3Bに示す。ビオチンフラグメント(poly A+cnvK:主鎖)は、配列表の配列番号1に記載された配列を有している。ここで、上記主鎖の5'末端にはBioTEGが結合しており、また、塩基配列中のNはイノシンを、XはAminoC6-dTをそれぞれ表す。また、ピューロマイシン-セグメント(側鎖)は、下記の配列を有する。
5' (5S)TCTFZZCCP
上記の側鎖の遊離末端となるPは、タンパク質結合部位としてのピューロマイシンである。また、下記の塩基配列中、(5S)は5'Thiol C6を、FはFITC-dTを、そしてZは Spacer18をそれぞれ表す。上記の主鎖及び側鎖の化学合成は、つくばオリゴサービス(株)に委託した。
まず、0.2Mのリン酸ナトリウム(pH7.2)に、15nmolのビオチンフラグメント(poly A+cnvK) (終濃度150μM)及びEMCS((株)同仁化学研究所製、終濃度16.7mM)を加え、37℃で30分インキュベートした。その後、Quick-Precip Plus Solution(Edge BioSystems社製)を用いてエタノール沈殿させた。
次に37.5nmol分のピューロマイシン-セグメントを終濃度417μMとなるように、50mMのDTTを含む1Mのリン酸水素ナトリウム水溶液に溶解し、シェーカーを用いて室温で1時間撹拌した。次いで、NAP5カラムを用いて、0.15MのNaClを含む0.1Mのリン酸ナトリウム(pH7.0)にバッファーを交換した。
上記バッファー交換済みの還元ピューロマイシン-セグメント溶液を、上記のEMCS修飾済みビオチン-フラグメント(poly A+cnvK)のエタノール沈殿産物と混合し、4℃で一晩放置した。続いて、DTTを終濃度50mMとなるように上記反応液に投入し、室温で30分間撹拌した。その後、Quick-Precip Plus Solution(Edge BioSystems社製)を用いて、エタノール沈殿を行なった。エタノール沈殿産物を、100μLのNuclease-free water(ナカライテスク社製)に溶解し、以下の条件でC18カラムによるHPLC精製を行った。
A液:0.1Mの酢酸トリメチルアンモニウム(超純水中)
B液:80%アセトニトリル
プログラム:A液とB液との組成比は、開始時のA液85%を45分かけて65%とするグラディエント
流速:1mL/分
分画:1mL
B液:80%アセトニトリル
プログラム:A液とB液との組成比は、開始時のA液85%を45分かけて65%とするグラディエント
流速:1mL/分
分画:1mL
画分中の成分は、蛍光及び紫外吸収(280nm)で確認した。30~32分までの画分では、蛍光とUVの両方でピークが見られた。30~32分までの画分を集め、溶媒を真空エバポレターを用いて蒸発させた。その後、Quick-Precip Plus Solutionを用いてエタノール沈殿を行ない、Nuclease-free waterに溶解して-20℃で保存した。
(2)BDA mRNAとピューロマイシン-リンカー(poly A+cnvK)とのハイブリダイズ調製物の作製
RiboMAX Large Scale RNA Production Systems-T7(Promega Corp.製)を使用して転写を行なった。この転写により得られたBDA mRNA とピューロマイシン-リンカー(poly A+cnvK)とを、各々終濃度が1μMになるように100mMのNaClを含む25mMのTris-HClバッファー(pH7.5)に加えた。90℃で1分間インキュベートした後に、70℃で1分間インキュベートし、0.08℃/秒の速度で25℃まで降温させて、ピューロマイシン-リンカー(poly A+cnvK)をmRNAの3'末端側にハイブリダイズさせた。続いてCL-1000 UV Crosslinkerを用いてUV(366nm)を、約1分間照射した。以上の操作によって、BDA mRNAとピューロマイシン-リンカー(poly A+cnvK)とのハイブリダイズ調製物を得た。
RiboMAX Large Scale RNA Production Systems-T7(Promega Corp.製)を使用して転写を行なった。この転写により得られたBDA mRNA とピューロマイシン-リンカー(poly A+cnvK)とを、各々終濃度が1μMになるように100mMのNaClを含む25mMのTris-HClバッファー(pH7.5)に加えた。90℃で1分間インキュベートした後に、70℃で1分間インキュベートし、0.08℃/秒の速度で25℃まで降温させて、ピューロマイシン-リンカー(poly A+cnvK)をmRNAの3'末端側にハイブリダイズさせた。続いてCL-1000 UV Crosslinkerを用いてUV(366nm)を、約1分間照射した。以上の操作によって、BDA mRNAとピューロマイシン-リンカー(poly A+cnvK)とのハイブリダイズ調製物を得た。
次いで、上記の調製物(溶液)を半分に分け、一方に対しては、CL-1000 UV Crosslinkerを用いて2分間UV(366nm)を照射した。他方に対しては、UV照射は行わなかった。3pmolの各サンプルを、25μLスケールの無細胞翻訳系(Rabbit reticulocyte Lysate (nuclease-treated)、Promega Inc.製)を用いて、30℃で、10分間又は20分間、インキュベートした。その後、MgCl2及びKClをそれぞれ最終濃度75mM、900mMとなるように加えて37℃で1時間インキュベートし、mRNA-ペプチド連結体を形成させた。
8Mの尿素を含む4%スタッキングゲル-6%分離ゲルのSDS-PAGEにより、mRNA-ペプチド連結体が形成されるか否かを、SYBR Goldでゲルを染色して確認した(図7A参照)。UV照射時間は、0分、10分及び20分とした。図7Aに示すように、UV照射を行わなかった調製物では、いずれの照射時間でもmRNA-ペプチド連結体の形成は見られなかった。これに対し、UV照射を行った調製物では、いずれの照射時間でもmRNA-ペプチド連結体の形成が確認された。
(実施例2)光架橋反応における照射量の検討
モデルとして、Aタンパク質のBドメインがコードされたDNA(配列表の配列番号6)を使用した。RiboMAX Large Scale RNA Production Systems-T7を使用して転写を行なった。転写によって得られたBDA mRNAと、ピューロマイシン-リンカー(poly A+cnvK)を、各々終濃度が1μMとなるように、100mMのNaClを含む25mMのTris-HClバッファー(pH 7.5)に加えた。90℃で1分間インキュベートし、その後、70℃で1分間インキュベートして、0.08℃/秒の速さで25℃まで降温してピューロマイシン-リンカー(poly A+cnvK)をmRNAの3'末端側にハイブリダイズさせた。その後、CL-1000 UV Crosslinkerを用いて、366nmのUVを30秒間~150秒間UVを照射し、架橋に要する時間を検討した(図7B及び7C)。
モデルとして、Aタンパク質のBドメインがコードされたDNA(配列表の配列番号6)を使用した。RiboMAX Large Scale RNA Production Systems-T7を使用して転写を行なった。転写によって得られたBDA mRNAと、ピューロマイシン-リンカー(poly A+cnvK)を、各々終濃度が1μMとなるように、100mMのNaClを含む25mMのTris-HClバッファー(pH 7.5)に加えた。90℃で1分間インキュベートし、その後、70℃で1分間インキュベートして、0.08℃/秒の速さで25℃まで降温してピューロマイシン-リンカー(poly A+cnvK)をmRNAの3'末端側にハイブリダイズさせた。その後、CL-1000 UV Crosslinkerを用いて、366nmのUVを30秒間~150秒間UVを照射し、架橋に要する時間を検討した(図7B及び7C)。
8M尿素変性4%スタッキングゲル-6%分離ゲルを用いたSDS-PAGEを行ない、FITC及びSYBR Goldで染色して、mRNA-リンカー連結体の形成を確認した(図7B)。いずれの結果からも、mRNA-リンカーの連結体の形成が確認された。
図7Bに示すように、mRNAとリンカーとの連結には、30秒以上のUV照射時間があれば十分であることが確認された。リンカーへのダメージを最小限に抑えつつ、安定な光架橋を形成させるために、UV照射時間を2分間とした。
(実施例3)FLAGエピトープのモデルセレクション
(1)ライブラリの転写
下記の配列を有する8アミノ酸ランダムライブラリDNA(配列表の配列番号2)とFLAG配列(配列表の配列番号11)とがコードされたDNAを、50,000:1のモル比で混合したDNA混合物を調製し、その500 ngをRiboMAX Large Scale RNA Production Systems-T7(以下、単に「キット」ということがある。)を使用して、15μLのスケールで転写した。下記配列中、Nは、任意にA, T, G, 及びCを表し、KはG又はTを表す。
(1)ライブラリの転写
下記の配列を有する8アミノ酸ランダムライブラリDNA(配列表の配列番号2)とFLAG配列(配列表の配列番号11)とがコードされたDNAを、50,000:1のモル比で混合したDNA混合物を調製し、その500 ngをRiboMAX Large Scale RNA Production Systems-T7(以下、単に「キット」ということがある。)を使用して、15μLのスケールで転写した。下記配列中、Nは、任意にA, T, G, 及びCを表し、KはG又はTを表す。
[配列2:8アミノ酸ランダムライブラリDNA]
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCAGCNNKNNKNNKNNKNNKNNKNNKNNKGGAGGTGGAATTAAAAACATGTGCAATTTGAACCCACTTTTAAAAAAGTGGCTAAATGATGCAAAGGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCAGCNNKNNKNNKNNKNNKNNKNNKNNKGGAGGTGGAATTAAAAACATGTGCAATTTGAACCCACTTTTAAAAAAGTGGCTAAATGATGCAAAGGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
[配列11:FLAG DNA配列]
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCAGCGATTATAAGGACGATGACGATAAGGGGAGGTGGAATTAAAAACATGTGCAATTTGAACCCACTTTTAAAAAAGTGGCTAAATGATGCAAAGGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCAGCGATTATAAGGACGATGACGATAAGGGGAGGTGGAATTAAAAACATGTGCAATTTGAACCCACTTTTAAAAAAGTGGCTAAATGATGCAAAGGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
上記の混合物を37℃で4時間インキュベートした後、キット付属のDNase(RQ1 DNase)を0.5μL加え、さらに37℃で10分間インキュベートした。合成されたmRNAは、After Tri-Reagent RNA Clean-Up Kitを使用して精製した。
(2)光架橋
上記実施例2と同様にして、mRNAとピューロマイシン-リンカー(poly A+cnvK)の光架橋を行ない、20pmolのライブラリmRNAにピューロマイシン-リンカー(poly A+cnvK)を光架橋した。このときのUV照射時間は2分とした。
上記実施例2と同様にして、mRNAとピューロマイシン-リンカー(poly A+cnvK)の光架橋を行ない、20pmolのライブラリmRNAにピューロマイシン-リンカー(poly A+cnvK)を光架橋した。このときのUV照射時間は2分とした。
また、本発明のcnvKリンカーとmRNAとの結合体と、従来例1のリンカー(SBP)をT4 RNAリガーゼを用いた場合とのRNAの分解を検討した。まず、25℃で30分、バッファー中でmRNAと従来例1のリンカーとの連結反応を行なった。このバッファーを200V, 15mA, 8M尿素変性4%アクリルアミドゲルという条件の下で電気泳動にかけたところ、SYBR染色で、mRNAが分解していることが示された(図7D)。これに対し、本発明のcnvKリンカーを使用した場合には、mRNAの分解はまったく見られなかった(図7E)。これは、cnvKリンカーの場合には、バッファーではなく亜鉛等の金属イオンを含まない水中で連結反応をさせることができるため、バッファー中で反応させるときのようにRNaseが活性化されることがないことによるものと考えられた。
また、81mJ~810mJの照射量となるようにUVを照射し、光架橋によるmRNAへのダメージを確認した(図7F)。図7Fに示すように、mRNA-cnvKリンカー融合体の分解は見られなかった。さらに、cDNAの合成に対するUVの照射量の影響を確認したところ、mRNAに対するダメージが見られなかったのと同様に、cDNA量の変化は見られなかった(図7G)。
(3)無細胞翻訳系による翻訳
15pmolのmRNA-リンカー連結体を、上記実施例2と同様に、125μLスケールで、上記無細胞翻訳系を用いて30℃で15分間翻訳した。その後、MgCl2及びKClをそれぞれ最終濃度が75mM及び900mMとなるように加えて、37℃で1時間インキュベートし、mRNA-ペプチド連結体を含む翻訳反応液を得た。
15pmolのmRNA-リンカー連結体を、上記実施例2と同様に、125μLスケールで、上記無細胞翻訳系を用いて30℃で15分間翻訳した。その後、MgCl2及びKClをそれぞれ最終濃度が75mM及び900mMとなるように加えて、37℃で1時間インキュベートし、mRNA-ペプチド連結体を含む翻訳反応液を得た。
(4)精製及び逆転写
上記のようにして得られた翻訳反応液に、0.5MのEDTA(pH 8.0)を終濃度が83mMとなるように加えて、室温で5分間インキュベートし、mRNA-ペプチド連結体に結合しているリボソームを除去した。
上記のようにして得られた翻訳反応液に、0.5MのEDTA(pH 8.0)を終濃度が83mMとなるように加えて、室温で5分間インキュベートし、mRNA-ペプチド連結体に結合しているリボソームを除去した。
引き続き、上記のように処理した翻訳反応液に、SA用2x結合バッファー(20mMのTris-HCl(pH 7.5), 2MのNaCl、2mMのEDTA、0.2%のTween-20を含む)を等量加え、SA用の1x結合バッファーで洗浄済みのDynabeads MyOne C1 ストレプトアビジン(サーモフィッシャーサイエンティフィック(株)製) 150μLと混合し、冷却サーモブロックローテーター((株)日伸理化製、SNP-24B)を用いて25℃で30分撹拌した。
上記のDynabeads MyOne C1ストレプトアビジンを、200μLのSA用の1x結合バッファーで3回洗浄し、その後ReverTra Ase(登録商標)に付属のプロトコルに従って、100μLの逆転写用反応液を投入した。冷却サーモブロックローテーターを用いて、42℃で15分間撹拌して逆転写を行い、mRNA/cDNA-ペプチド連結体を調製した。
(5)磁性体ビーズ上からの逆転写産物の回収
150μLの1xNEバッファーで、上記(4)で反応させたDynabeads MyOne C1 ストレプトアビジンを洗浄し、その後10UのEndonuclease V(ニュー・イングランド・バイオラボ・ジャパン(株)製)を含む、75μLの1xNEバッファーを加えて、冷却サーモブロックローテーターを用いて37℃で1時間撹拌した。次いで、75μLの2xHis-tag洗浄バッファー(1MのNaCl、0.1%のTween-20を含む40mMのリン酸ナトリウムリム(pH 7.4))を加え、その後上清を回収した。
150μLの1xNEバッファーで、上記(4)で反応させたDynabeads MyOne C1 ストレプトアビジンを洗浄し、その後10UのEndonuclease V(ニュー・イングランド・バイオラボ・ジャパン(株)製)を含む、75μLの1xNEバッファーを加えて、冷却サーモブロックローテーターを用いて37℃で1時間撹拌した。次いで、75μLの2xHis-tag洗浄バッファー(1MのNaCl、0.1%のTween-20を含む40mMのリン酸ナトリウムリム(pH 7.4))を加え、その後上清を回収した。
(6)His-Tagによる精製
回収した上清150μLと、20μLのHis Mag Sepharose Ni(GEヘルスケア社製、1xHis-tag洗浄バッファーにより洗浄済み)とを混合し、インテリミキサーRM-2M((株)トーホー製)を用いて、室温で1時間撹拌した。100μLの1xHis-tag洗浄バッファーで2回洗浄し、その後、EDTA濃度を高めた30μLのセレクションバッファー(1MのNaCl、10mMのイミダゾール、5mMのEDTA及び0.1%のTween-20を含む50mMのTris-HClバッファー(pH 7.4))を加え、上記のインテリミキサーRM-2Mを使用して、室温で10分間撹拌し、その後に上清を回収した。
回収した上清150μLと、20μLのHis Mag Sepharose Ni(GEヘルスケア社製、1xHis-tag洗浄バッファーにより洗浄済み)とを混合し、インテリミキサーRM-2M((株)トーホー製)を用いて、室温で1時間撹拌した。100μLの1xHis-tag洗浄バッファーで2回洗浄し、その後、EDTA濃度を高めた30μLのセレクションバッファー(1MのNaCl、10mMのイミダゾール、5mMのEDTA及び0.1%のTween-20を含む50mMのTris-HClバッファー(pH 7.4))を加え、上記のインテリミキサーRM-2Mを使用して、室温で10分間撹拌し、その後に上清を回収した。
(7)アフィニティセレクション
50μLの抗FLAG M2アフィニティゲル(50%懸濁液)を、MicroSpin Empty Columns(GEヘルスケア社製)に充填し、200μLのセレクションバッファーで3回洗浄した。その後、上記の上清100μLをカラムにロードし、ローテーターを用いて室温で1時間撹拌した。200μLのセレクションバッファーで4回カラムを洗浄し、次いで100ng/μLの3xFLAGペプチド(シグマアルドリッチジャパン合同会社製)を100μL加え、上記のローテーターを用いて室温で15分間撹拌し、抗FLAG M2アフィニティゲルに結合しているmRNA/cDNA-ペプチド連結体を競合溶出させた。
50μLの抗FLAG M2アフィニティゲル(50%懸濁液)を、MicroSpin Empty Columns(GEヘルスケア社製)に充填し、200μLのセレクションバッファーで3回洗浄した。その後、上記の上清100μLをカラムにロードし、ローテーターを用いて室温で1時間撹拌した。200μLのセレクションバッファーで4回カラムを洗浄し、次いで100ng/μLの3xFLAGペプチド(シグマアルドリッチジャパン合同会社製)を100μL加え、上記のローテーターを用いて室温で15分間撹拌し、抗FLAG M2アフィニティゲルに結合しているmRNA/cDNA-ペプチド連結体を競合溶出させた。
遠心操作を行なってカラム内の溶出液を回収し、Quick-Precip Plus Solutionを使用したエタノール沈殿を行なった後に、15μLのNuclease-free waterに溶解した。上記のエタノール沈殿産物を、200μLのPCR反応液(0.2 mMのdNTPs、0.4μMのT7Ωnew、4μMのcnvK用NewYtag、0.02U/μLのPrimeSTAR HS DNAポリメラーゼを含む1xPrimeSTARバッファー(Mg2+含有)に加え、下記のPCRプログラムを行った。T7Ωnew及びcnvK用NewYtagの配列(配列表の配列番号3及び4)を以下に示す。
[配列3:T7Ωnewの配列]
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACA-3'
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACA-3'
[配列4:cnvK用NewYtagの配列]
5'‐TTTCCACGCCGCCCCCCGTCCTGCTTCCGCCGTGATGAT‐3'
5'‐TTTCCACGCCGCCCCCCGTCCTGCTTCCGCCGTGATGAT‐3'
次に、ここで行ったPCRプログラムを示す。(a1)98℃で1分、(b1)98℃で15秒、(c1)68℃で30秒、(d1)68℃で1分、上記(b1)及び(c1)を25サイクル行ってPCR産物を得た。上記のようにして得られたPCR産物を、8M尿素変性6%PAGEにて泳動し、フルコンストラクトDNA(274mer)を切出して、常法に従って精製した。精製した上記フルコンストラクトDNAを、200μLのPCR反応液(0.2mMのdNTPs、0.4μMのNewleft、0.4μMのcnvK用NewYtag、及び0.02U/μLのPrimeSTAR HS DNA polymeraseを含む1xPrimeSTARバッファー(Mg2+含有))に加え、50μLずつ分注して、以下のPCRプログラムを行い、二本鎖化DNAを得た。以下に、Newleftの配列(配列表の配列番号5)を示す。
[配列5:Newleftの配列]
GATCCCGCGAAATTAATACGACTCACTATAGGG
GATCCCGCGAAATTAATACGACTCACTATAGGG
PCRプログラムは、(a2)98℃で1分、(b2)98℃で10秒、(c2)68℃で30秒、(d2)68℃で1分とし、ステップ(b2)及び(c2)を5サイクル行った。
上記PCR反応液4本分をまとめてカラム精製し次ラウンドに使用した。上記(1)ライブラリDNAの転写から(7)アフィニティセレクションまでを、合計で3ラウンド行い、その結果得られたフルコンストラクトDNAの配列をダイレクトシーケンシングした。このダイレクトシーケンシングは、ユーロフィンジェノミクス(株)に委託した。ダイレクトシーケンシングの結果、FLGA DNAが収束していることを確認した(図8(A)及び8(B))。
上記PCR反応液4本分をまとめてカラム精製し次ラウンドに使用した。上記(1)ライブラリDNAの転写から(7)アフィニティセレクションまでを、合計で3ラウンド行い、その結果得られたフルコンストラクトDNAの配列をダイレクトシーケンシングした。このダイレクトシーケンシングは、ユーロフィンジェノミクス(株)に委託した。ダイレクトシーケンシングの結果、FLGA DNAが収束していることを確認した(図8(A)及び8(B))。
(実施例4)ビオチン接着タンパク質の作製およびSPR装置への応用
本実施例でも、モデルタンパク質として上記と同様にAタンパク質のBドメイン(BDA)を使用した。上記実施例3で行ったcDNAディスプレイ法によるFLAG配列のモデルセレクションの(1)~(7)までと同様の手順により、BDAをコードしたDNAから、mRNAとして30pmol分のmRNA-ペプチド連結体を調製した。
本実施例でも、モデルタンパク質として上記と同様にAタンパク質のBドメイン(BDA)を使用した。上記実施例3で行ったcDNAディスプレイ法によるFLAG配列のモデルセレクションの(1)~(7)までと同様の手順により、BDAをコードしたDNAから、mRNAとして30pmol分のmRNA-ペプチド連結体を調製した。
次いで、上記のmRNA-ペプチド連結体を含む翻訳産物に、1/9倍容の10xNEバッファー2と、20UのRNase H(タカラバイオ(株)製)を加え、37℃で1時間インキュベーションしてmRNAを分解した。この反応液と等量のオリゴdT用の2x結合バッファー(1MのLiCl, 2mMのEDTA、及び0.1%のTween-20を含む20mMのTris-HCl(pH 7.5))を加え、100μLのDynabeads Oligo (dT)25(サーモ・フィッシャー。サイエンティフィック(株)製、オリゴdT用の2x結合バッファーにて洗浄済み)と混合し、ローテーターを用いて室温で30分撹拌した。
上記Dynabeads Oligo (dT)25を200μLのオリゴdT用1x結合バッファーで2回洗浄し、その後、232μLの超純水を加えて37℃で10分間インキュベートし、ビオチン接着タンパク質(以下、「ビオチン接着BDA」をいうことがある。)を溶出した。このようにして得られたビオチン接着BDA溶液に対して、等量の2xHis-tag洗浄バッファーを加え、その後、30μL分のHis Mag Sepharose Ni(1xHis-tag洗浄バッファーで洗浄済み)と混合し、ローテーターを用いて室温で40分撹拌した。
100μLの1xHis-tag洗浄バッファーで2回洗浄し、その後、56μLのHis-tag溶出バッファー(0.5MのNaCl, 250mMのイミダゾール、0.05%のTween-20を含む20mMのSリン酸ナトリウムリムバッファー(pH7.4))を加えて、インテリミキサーRM-2Mを用いて室温で15分間撹拌し、ビオチン接着BDAを溶出させた。得られたビオチン接着BDAのDNA配列(配列表の配列番号6)は以下の通りであった。
[配列6:BDA DNA配列]
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGATAACAAATTCAACAAAGAACAACAAAATGCTTTCTATGAAATCTTACATTTACCTAACTTAAACGAAGAACAACGCAATGGTTTCATCCAAAGCCTAAAAGATGACCCAAGCCAAAGCGCTAACCTTTTAGCAGAAGCTAAAAAGCTAAATGATGCTCAAGCACCAAAAGCTGACAACAAATTCAACGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGATAACAAATTCAACAAAGAACAACAAAATGCTTTCTATGAAATCTTACATTTACCTAACTTAAACGAAGAACAACGCAATGGTTTCATCCAAAGCCTAAAAGATGACCCAAGCCAAAGCGCTAACCTTTTAGCAGAAGCTAAAAAGCTAAATGATGCTCAAGCACCAAAAGCTGACAACAAATTCAACGGGGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA
(実施例5)Biotin-attached proteinを用いた親和性測定
Biacore X100(GEヘルスケア社製)に、センサーチップSAをセットした。上記のようにして得たビオチン接着BDA溶液を、1xHis-tag洗浄バッファーで2倍希釈した。この希釈ビオチン接着BDA溶液を、流速5μLで900秒流し、センサーチップ上にビオチン接着BDAを固定化した。
Biacore X100(GEヘルスケア社製)に、センサーチップSAをセットした。上記のようにして得たビオチン接着BDA溶液を、1xHis-tag洗浄バッファーで2倍希釈した。この希釈ビオチン接着BDA溶液を、流速5μLで900秒流し、センサーチップ上にビオチン接着BDAを固定化した。
次いで、ウサギ血清から得たIgG(シグマアルドリッチ合同会社製)を、1xHBP-EP+ バッファー(GEヘルスケア社製)を用いて、160nM、80nM、40nM、20nM、及び10nMという2倍系列希釈を作製した。5μL/分の流速でFc1及びFc2チャネルに、これらの希釈系列の液体を注入し、親和性を測定した。アッセイチャネル(Fc2)のセンサーグラムは、バッファーコントロール及びFc1(コントロール)から求め、キネティックフィッティング用にオーバーレイして、結合時(ka)及び解離時(kd)の速度、及び親和性(KD=kd/ka)を求めた。このキネティックフィッティングは、1:1ラングミュア結合モデル(A+B=AB)を用いたビアコアJソフトウェアで行った。全ての実験を3回繰り返し、標準偏差つきの平均KDを示した。
グリシン-塩酸(pH 2.0)を再生溶液としたマルチサイクル法によるセンサーグラム測定を行った。得られたセンサーグラムを元に、キネティックス解析、アフィニティー解析をそれぞれ行った(図8A及び8B)。その結果、得られたKDは、1.923x10-8であり、文献値をほぼ同じであった。このことは、本発明のcnvKリンカーを用いた場合に、このリンカーと結合する分子が、それらが本来保有しているアフィニティーを発揮していることを示すものである。
(実施例6)ビオチン-cnvKリンカーの合成及び架橋ペプチドアプタマーの調製
(1)ビオチン-cnvKリンカーの合成
cnvKを含むセグメント(主鎖)と、ピューロマイシンとFITCとを含むPuro-F-S(側鎖)で構成されている本実施例で使用するcnvKリンカー(図3B参照)を、以下の方法によりビオチンとさらに結合させて、Biotin-cnvKリンカーを作成した
(1)ビオチン-cnvKリンカーの合成
cnvKを含むセグメント(主鎖)と、ピューロマイシンとFITCとを含むPuro-F-S(側鎖)で構成されている本実施例で使用するcnvKリンカー(図3B参照)を、以下の方法によりビオチンとさらに結合させて、Biotin-cnvKリンカーを作成した
9.4mgのEMCS(架橋剤、和光純薬工業(株)製)を305.5μLのN,N-ジメチルホルムアミドに溶解させ、100mMのEMCSを調製した。ついで、200μLの0.2 Mのリン酸ナトリウムバッファー(和光純薬工業(株)製、分子生物学用、pH 7.2)に、10nmolのBiotin-cnvKセグメントと40μLの100mM EMCSとを加え、37℃で30分間インキュベートした。その後、エタノール沈殿を行なって、生成物を濃縮した。
2mMのPuro-F-Sに対して、80μLの1M Na2PO4 (pH 9.0)及び10μLの1M DTTを加えて1時間、室温で振とう攪拌した。ついで、20mMのリン酸バッファーを満たしたNAP5 カラムでバッファー交換を行い、蛍光を発している画分のみを分取した。分取したPuro-F-Sに、エタノール沈殿させた後のEMCSの溶液を加え、4℃で終夜反応させた。
1M DTTを全量の1/20量だけこの反応液に加え、室温で30分間振とう攪拌し、エタノール沈殿させて、生成物を濃縮した。濃縮後、超純水を加えて100μLにメスアップし、次いで、実施例1と同様の条件にてHPLCを行ない、ビオチン-cnvKリンカーを精製した。
(2)架橋ペプチドアプタマーの調製
(2-1)VHHライブライブラリを鋳型とするPCRによるDNAの増幅
CDR1、2、3にランダム配列を有するVHHライブラリ(配列表の配列番号7)を鋳型DNAとして以下の条件でPCR1を行い、DNAを増幅させた。配列表7中のn、b、d、r、y、h、y、及びmは上記と同様である。
(2-1)VHHライブライブラリを鋳型とするPCRによるDNAの増幅
CDR1、2、3にランダム配列を有するVHHライブラリ(配列表の配列番号7)を鋳型DNAとして以下の条件でPCR1を行い、DNAを増幅させた。配列表7中のn、b、d、r、y、h、y、及びmは上記と同様である。
[配列番号7:VHHライブラリのコンストラクトの配列]
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCGAGGTGCAGCTGGTGGAGAGCGGAGGAGGATCCGTGCAGGCTGGAGGAAGCCTGCGCCTGAGCTGCGCTGCTAGCGGAnbnnbndrbyhmyhmyhmdrbnbnnbnTGGTTCCGCCAGGCTCCTGGAAAGGAGCGCGAGGGAGTGnbnnbndrbyhmyhmyhmyhmdrbACCTACTACGCTGACAGCGTGAAGGGACGCTTCACCATCAGCCAGGACAACGCCAAGAACACCGTGTACCTGCAGATGAACAGCCTGAAGCCTGAGGACACCGCTATCTACTACTGCGCTGCTdrbdrbyhmyhmyhmyhmyhmyhmyhmyhmyhmyhmdrbTACTGGGGACAGGGAACCCAGGTGACCGTGGGAGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGGGGAAA-3'
5'-GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACAACTACAAGCCACCATGGGCGAGGTGCAGCTGGTGGAGAGCGGAGGAGGATCCGTGCAGGCTGGAGGAAGCCTGCGCCTGAGCTGCGCTGCTAGCGGAnbnnbndrbyhmyhmyhmdrbnbnnbnTGGTTCCGCCAGGCTCCTGGAAAGGAGCGCGAGGGAGTGnbnnbndrbyhmyhmyhmyhmdrbACCTACTACGCTGACAGCGTGAAGGGACGCTTCACCATCAGCCAGGACAACGCCAAGAACACCGTGTACCTGCAGATGAACAGCCTGAAGCCTGAGGACACCGCTATCTACTACTGCGCTGCTdrbdrbyhmyhmyhmyhmyhmyhmyhmyhmyhmyhmdrbTACTGGGGACAGGGAACCCAGGTGACCGTGGGAGGAGGCAGCCATCATCATCATCATCACGGCGGAAGCAGGACGGGGGGCGGCGGGGAAA-3'

PCRプログラムは、98℃で2分、(98℃で10秒、98℃で1秒、68℃で40秒)を5サイクル、72℃で2分とした。また、原液のVHHのダイバーシチは、2.7x1011分子/μLであった。
上記のPCR1で得られたPCR産物1を、PCR Clean-Up MiniKit(Favorgen社製)を用いて精製し、鎖長の短いDNAを除去した。次いで、上記表3に示すPCR1用溶液のうち、20μM Primer 2 (New left)を20μM Primer 2 (T7 omega new)に代えた以外は、同じ組成のPCR溶液2を調製し、上記のようにして得られた精製PCR産物1をPCR2に供して、PCR産物2を得た。PCRプログラムは、98℃で2分、(98℃で10秒、98℃で1秒、68℃で40秒)を23サイクル、72℃で2分とした。
(2-2)mRNAへの転写
RiboMAXTM Large Scale RNA production Systems(Promega社製)を用いて、上記のPCR2で得られたDNAをmRNAに転写した。この転写反応に使用した反応溶液の組成と手順は下記の表4の通りとした。
RiboMAXTM Large Scale RNA production Systems(Promega社製)を用いて、上記のPCR2で得られたDNAをmRNAに転写した。この転写反応に使用した反応溶液の組成と手順は下記の表4の通りとした。

上記の転写反応溶液を、37℃で3時間インキュベートし、RQ1 Rnase-Free DNAse (Promega社製)を1μL加え、37℃で15分、さらにインキュベートした。その後、After Tri-Reagent RNA Clean-up Kit(Favorgen社製)を用いて、付属のプロトコルに従い、mRNAを精製した。以上の操作を第6ラウンドまで繰り返し、各ラウンドでmRNAを得た。ここで、各ラウンドで使用した鋳型DNAの量は下記表5に示す通りとし、使用したDNAの量に応じて、上記表中のxの量を変更した。その際、その後の光架橋形成のために十分な量のmRNAが精製できるように、xを調整した。
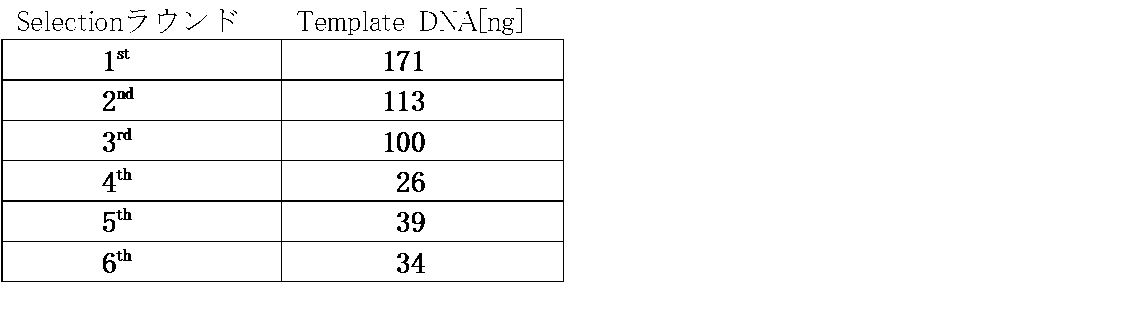
(2-3)mRNAとBiotin-cnvKリンカーとの結合
以上のようにして各セレクションラウンドで得られたmRNAを精製し、精製mRNAを得た。各セレクションラウンドで得られた精製mRNAと、Biotin-cnvKリンカーとを、以下条件で光架橋により連結させた。光架橋用反応溶液の組成を下記の表6に示す。光架橋反応溶液中のmRNAとBiotin-cnvKリンカーとの比率は、いずれの場合も1:1(モル比)とした。また、アニーリングは、90℃で2分、1分間で70℃まで降温、70℃で1分、その後15分間で25℃まで降温させることによって行った。
以上のようにして各セレクションラウンドで得られたmRNAを精製し、精製mRNAを得た。各セレクションラウンドで得られた精製mRNAと、Biotin-cnvKリンカーとを、以下条件で光架橋により連結させた。光架橋用反応溶液の組成を下記の表6に示す。光架橋反応溶液中のmRNAとBiotin-cnvKリンカーとの比率は、いずれの場合も1:1(モル比)とした。また、アニーリングは、90℃で2分、1分間で70℃まで降温、70℃で1分、その後15分間で25℃まで降温させることによって行った。

アニーリング後、CL-1000 Ultraviolet Crosslinkerを用いて、波長365nmの紫外光を405 mJ/cm2の条件で照射して光架橋を行い、各ラウンドで得られたmRNAとBiotin-cnvKリンカーとの連結体(mRNA-リンカー連結体)を得た。
(2-4)無細胞翻訳系によるmRNA-ペプチド連結体の形成
以上のようにして得られたmRNA-リンカー連結体を、無細胞翻訳系で翻訳し、mRNA-ペプチド連結体を形成させた。無細胞翻訳に用いた反応液の組成を下記表7に示す。
以上のようにして得られたmRNA-リンカー連結体を、無細胞翻訳系で翻訳し、mRNA-ペプチド連結体を形成させた。無細胞翻訳に用いた反応液の組成を下記表7に示す。

上記の翻訳用反応液入れたチューブを30℃で20分間インキュベートし、ここに、12μLの3M KClと3μLの1M MgCl2とを加え、37℃でさらに60分間インキュベートした。その後、10μLのエチレンジアミン四酢酸(pH 8.0)を加え、37℃で10分間インキュベートし、次いで50μLの2x結合バッファーを加えて、無細翻訳系で合成されたペプチドが上記mRNA連結体に結合した、mRNA-ペプチド連結体を得た。
(2-5)ストレプトアビジン磁性体ビーズへの固定
Streptavidin(SA) Magnetic beads:Dynabeads MyOne Streptavidin C1(磁性体ビーズ)をRNase-Freeとするために、洗浄を行った。投入するmRNA-ペプチド連結体の濃度の10倍容の磁性体ビーズを、1.5mL容量のProtein LoBindチューブ(Eppendorf社製)に入れ、磁気スタンド上に静置して上澄を捨てた。1x結合バッファーで再懸濁し、磁気スタンド上に静置して上澄を捨て、磁性体ビーズを洗浄した。ここに、上記(2-4)で得た翻訳産物(mRNA-ペプチド連結体)を加え、25℃で90~120分間、ローテーターで攪拌しながらインキュベートし、mRNA-ペプチド連結体を磁性体ビーズに結合させた。使用したmRNA-ペプチド連結体の量と、インキュベート時間とは下記表8
に示す通りとした。
Streptavidin(SA) Magnetic beads:Dynabeads MyOne Streptavidin C1(磁性体ビーズ)をRNase-Freeとするために、洗浄を行った。投入するmRNA-ペプチド連結体の濃度の10倍容の磁性体ビーズを、1.5mL容量のProtein LoBindチューブ(Eppendorf社製)に入れ、磁気スタンド上に静置して上澄を捨てた。1x結合バッファーで再懸濁し、磁気スタンド上に静置して上澄を捨て、磁性体ビーズを洗浄した。ここに、上記(2-4)で得た翻訳産物(mRNA-ペプチド連結体)を加え、25℃で90~120分間、ローテーターで攪拌しながらインキュベートし、mRNA-ペプチド連結体を磁性体ビーズに結合させた。使用したmRNA-ペプチド連結体の量と、インキュベート時間とは下記表8
に示す通りとした。

(2-6)cDNAへの逆転写及び磁性体ビーズからの切り離し
以上のようにしてmRNA-ペプチド連結体を磁性体ビーズ上に固定化し、磁性体ビーズが入ったチューブを磁気スタンド上に静置して上澄を捨てた。このチューブに100μLの1x結合バッファーを加えて磁性体ビーズを再懸濁させ、再度上澄を捨てて、磁性体ビーズを洗浄した。この操作を2回繰り返した。
以上のようにしてmRNA-ペプチド連結体を磁性体ビーズ上に固定化し、磁性体ビーズが入ったチューブを磁気スタンド上に静置して上澄を捨てた。このチューブに100μLの1x結合バッファーを加えて磁性体ビーズを再懸濁させ、再度上澄を捨てて、磁性体ビーズを洗浄した。この操作を2回繰り返した。
その後、ReverTra Ace(東洋紡(株)製)に付属している5xバッファーを希釈して1xバッファーとし、その100μLを用いて同様の操作を行ない、さらに磁性体ビーズを洗浄した。次いで、下記の表9に示す組成のcDNA合成用反応液を加え、42℃で90分間ローテーターで攪拌しながらインキュベートし、cDNAがさらに結合したcDNAディスプレイを得た。

以上のようにして得られたcDNAディスプレイが結合した磁性体ビーズ入りのチューブに、(x-1)μLの1x His-tag洗浄バッファーと1μLのRNase T1(1,000U/μL)とを加えて、37℃にて30分間、ローテーターで撹拌しながらインキュベートし、磁性体ビーズからcDNAディスプレイを切り離した。
(2-7)His-Tag精製及び生成物の回収
以上のようにしてcDNAディスプレイを調製し、VHHの配列をコードしたペプチドが発現しているものと、発現していないものとをHis-tag精製により分別した。先ず、上記のcDNAディスプレイが入ったチューブにHis Mag sepharose Niを加え、25℃で1時間、シェーカーで振盪しながらインキュベートした。その後、このチューブを磁気スタンド上に静置して上澄を除き、100μLの1x His tagバッファーで1回洗浄した。
以上のようにしてcDNAディスプレイを調製し、VHHの配列をコードしたペプチドが発現しているものと、発現していないものとをHis-tag精製により分別した。先ず、上記のcDNAディスプレイが入ったチューブにHis Mag sepharose Niを加え、25℃で1時間、シェーカーで振盪しながらインキュベートした。その後、このチューブを磁気スタンド上に静置して上澄を除き、100μLの1x His tagバッファーで1回洗浄した。
次いで、50μLのHis tag溶出バッファー(高濃度のイミダゾールを含む)を加えて、室温にて30分間、シェーカーを用いて振盪しながらインキュベートした。その後、磁気スタンド上に静置して上澄を回収し、VHHペプチドが発現したcDNAディスプレイだけを取得した。
His-tag精製によってVHHペプチドが発現したcDNAディスプレイを回収した後、Micro Bio-SpinTM 6 カラム(BIO-RAD社製)を用いて、試験管内淘汰に使用するためにバッファー交換を以下のように行った。
まず、カラムの蓋を開けた状態で1,000xgで2分間遠心し、ここに、500μLのグルコサミン用結合バッファーを加えて1,000xgで1分間遠心した。グルコサミン用結合バッファーを加えて1,000xgで1分間遠心するという操作を4回繰り返し、最後の遠心は3分間行った。ここに、以上のようにして得られたcDNAディスプレイを加えて、1,000xgで4分間遠心し、バッファー交換したcDNAディスプレイを回収した。
(3)癌関連単糖N-アセチル-D-グルコサミン(GlcNAc)の固定
磁性体ビーズとビオチン化GlcNAcとをインキュベートしてビオチン化GlcNAcを固定化した磁性体ビーズと、何も固定化していない磁性体ビーズとを用意し、そこにビオチン-DNAを等量ずつ加えてインキュベートし、ビオチン化GlcNAcが磁性体ビーズに固定化されているかどうかの確認を行った。
磁性体ビーズとビオチン化GlcNAcとをインキュベートしてビオチン化GlcNAcを固定化した磁性体ビーズと、何も固定化していない磁性体ビーズとを用意し、そこにビオチン-DNAを等量ずつ加えてインキュベートし、ビオチン化GlcNAcが磁性体ビーズに固定化されているかどうかの確認を行った。
セレクションの際に、ビオチンに非特異に結合するcDNAディスプレイを排除した。磁性体ビーズをグルコサミン洗浄用バッファーで洗浄し、ビオチンと共に25℃にて30分間、チューブ内でインキュベートした。その後、シェーカーで振盪しながら攪拌し、GlcNAcに特異的に結合するcDNAディスプレイを得た。下記表10に、各ラウンドにおけるcDNAディスプレイの量と、シェーカーでのインキュベート時間との関係を示す。

図9及び図10に示したように、ビオチン化GlcNAcを固定化した磁性体ビーズと反応させた場合と、固定化していない磁性体ビーズと反応させた場合とでは、上澄中のDNAの量が、固定化していないビーズを使用した方が有意に低くなっていた。このため、ビオチン化GlcNAcが磁性体ビーズ上に固定化されていることが確認された。
(実施例7)試験管内淘汰
(1)試験管内淘汰条件の検討
次いで、得られたGlcNAc特異的cDNAディスプレイを用いて、標的分子であるGlcNAcに対するVHHの試験管内淘汰を行った。まず、磁性体ビーズを試験管内でグルコサミン洗浄バッファーを用いて洗浄し、ビオチン化GlcNAcをここに加えて、室温にて30分間、シェーカーで振盪しながらインキュベートした。その後、チューブを磁気スタンド上に静置して上澄を捨て、グルコサミン洗浄用バッファーを加えて再び磁性体ビーズを洗浄し、次いで、ビオチンを加えて、室温にて30分間、シェーカーにて振盪しながらインキュベートした。
(1)試験管内淘汰条件の検討
次いで、得られたGlcNAc特異的cDNAディスプレイを用いて、標的分子であるGlcNAcに対するVHHの試験管内淘汰を行った。まず、磁性体ビーズを試験管内でグルコサミン洗浄バッファーを用いて洗浄し、ビオチン化GlcNAcをここに加えて、室温にて30分間、シェーカーで振盪しながらインキュベートした。その後、チューブを磁気スタンド上に静置して上澄を捨て、グルコサミン洗浄用バッファーを加えて再び磁性体ビーズを洗浄し、次いで、ビオチンを加えて、室温にて30分間、シェーカーにて振盪しながらインキュベートした。
インキュベート終了後、磁気スタンド上にチューブを静置して上澄を捨て、グルコサミン洗浄用バッファーで磁性体ビーズを洗浄し、cDNAディスプレイと共に、25℃にて30分間、ローテーターで撹拌しながらインキュベートした。その後、このチューブを磁気スタンド上に静置して上澄を捨てた。以上の操作によって、GlcNAc特異的VHHのcDNAディスプレイがGlcNAcを介して磁性体ビーズに固定化された。試験管内淘汰における各ラウンドでの条件は、下記表11の通りとした。
以上のようにして、チューブ内でGlcNAcを介してcDNAディスプレイを磁性体ビーズ上に固定化した。次いで、グルコサミン用洗浄バッファーを加えて、20秒間のタッピングにより再懸濁した。その後、このチューブを磁気スタンド上で1分間静置し、上澄を回収した。この操作を3~5回繰り返し、回収した上澄は洗浄液(以下、「Wash」ということがある。)として個別に保存した。Washをゲル電気泳動に供し、含有物を確認した。その結果、上記の各ラウンドにおけるcDNAディスプレイの洗浄の回数を、下記表12に示す通りとした。ゲル電気泳動の結果、第4ラウンド以降のcDNAディスプレイについては、洗浄が不十分と判断された、このため、第4ラウンド以降のcDNAディスプレイについては洗浄回数を増やし、非特異的に吸着するcDNAを除去した。
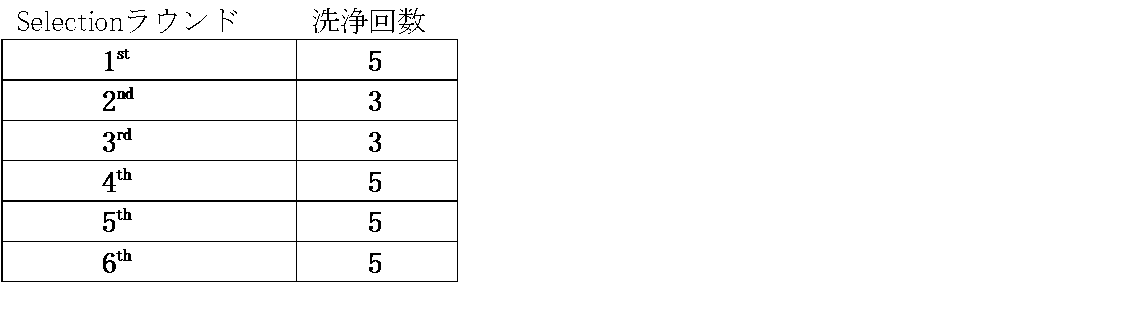
すなわち、磁性体ビーズに固定化したcDNAディスプレイ量に対して十分な濃度のGlcNAcを加え、第6ラウンドを除いて、4℃で3日間、ローテーターで攪拌しながら溶出させた。第6ラウンドは、終夜で溶出させた。この洗浄操作によってGlcNAcに非特異的に結合するVHHのcDNAディスプレイを除去し、その後に競合溶出を行なって、目的とするGlcNAcに特異的に結合するVHHのcDNAディスプレイを回収した。
また、ゲル電気泳動は以下の手順で行った。10xSDSランニングバッファー、1.5M トリス塩酸バッファー(pH 8.8)、0.5M トリス塩酸バッファー(pH 6.8)、2xSDSサンプルバッファー、40%アクリルアミド溶液を以下のようにして調製した。
10xSDSランニングバッファーは、30.3gのトリス(ヒドロキシメチル)アミノメタン、144gのグリシン、10gのSDSに超純水を加えて1,000mLにメスアップした。また、1.5M トリス塩酸バッファー(pH 8.8)は、36.3gのトリス(ヒドロキシメチル)アミノメタンを超純水に溶解し、塩酸でpHを8.8に調整した後に、超純水で200mLにメスアップした。0.5M トリス塩酸バッファー(pH 6.8)は、12.1gのトリス(ヒドロキシメチル)アミノメタンを超純水に溶解し、塩酸でpHを6.8に調整した後に、超純水で200mLにメスアップした。
2xSDSサンプルバッファーは、12.5mLの0.5M トリス塩酸バッファー(pH 6.8)、5mLの2-メルカプトエタノール、2gのSDS、24gの尿素、3gのスクロースを少量の超純水に溶解し、適量のブロモフェノールブルー(以下、「BPB」ということがある。)を加えた後に、超純水で50mLにメスアップした。40%アクリルアミド溶液は、SDS-PAGEグレードの194.8gのアクリルアミド及び5.2gのビスアクリルアミドを適量の超純水に溶解し、500mLにメスアップした。
4%スタッキングゲル(1枚分)は、1.25mLの0.5M トリス塩酸バッファー(pH 6.8)、0.5mLの40%のアクリルアミド溶液、50μLの10%SDSを混合し、超純水で5mLとした。ここに、12.5μLの20%APS(Ammonium peroxodisulfate:ペルオキソ二硫酸アンモニウム)及び6μLのTEMED(N,N,N,N-Tetramethy-ethylenediamine:N,N,N,N-テトラメチルエチルレンジアミン)を加えて調製した。
15%分離ゲル(1枚分)は、2.5mLの1.5M トリス塩酸バッファー(pH 8.8)、3.75mLの40%のアクリルアミド溶液、及び100μLの10%SDSを混合し、超純水で10mLとした。ここに、25μLの20%APS及び5μLのTEMEDを加えて調製した。
10xSDSランニングバッファーを10倍に希釈して泳動槽を満たし、ゲル電気泳動を泳動槽にセットして20mAで、泳動サンプルの分離に適した時間、泳動した。FITC及びSYBER-goldを用いて、常法に従って染色を行ない、イメージャー(Typhoon FLA 9500、GEヘルスケア・ジャパン(株)製)を用いてゲル電気泳動の結果を解析した。
(2)GlcNAc非結合性cDNAディスプレイ分子との競合アッセイ
GlcNAcを標的分子として、GlcNAcに結合しないAタンパク質のBドメイン(以下、「BDA」ということがある。)と、第6ラウンド後に回収したVHHのcDNAディスプレイとを一緒に試験管内淘汰に供し、そのGlcNAcに対する結合能を確認した。
GlcNAcを標的分子として、GlcNAcに結合しないAタンパク質のBドメイン(以下、「BDA」ということがある。)と、第6ラウンド後に回収したVHHのcDNAディスプレイとを一緒に試験管内淘汰に供し、そのGlcNAcに対する結合能を確認した。
GlcNAcを固定化した磁性ビーズを入れたチューブ内に、VHHのcDNAディスプレイとBDAのcDNAディスプレイとを、それぞれ1:1の比で加えて、室温にて30分間、ローテーターで撹拌しながらインキュベートし、その後、磁気スタンド上に静置して上澄を捨てた。グルコサミン用洗浄バッファーを加えてタッピングにより再懸濁し、磁気スタンド上に1分間静置して上澄を回収した。次いで、GlcNAcを用いて競合溶出させ、溶出物を得た。これらを、マーカーとともに、4%PAGE、200Vで30分間ゲル電気泳動し、SYBER-Goldで染色した。結果を図11に示す。
インキュベート前の対照では、VHH:BDAのバンドの強度比は9:16であり、BDAのcDNAディスプレイ濃度が高かった。その後、洗浄液でもバンドの強度比は、1:3となっており、BDA濃度が高かった。これに対し、溶出物では、バンド強度比が逆転して4:1となり、VHH濃度が高くなった。対照中のVHH濃度はBDAの0.6倍であったが、溶出物ではVHH濃度がBDA濃度よりも6倍高くなっていた。
これは、BDAのcDNAディスプレイが溶出されなかったのに対して、取得したVHHのcDNAディスプレイは溶出されてきたことを示す。この結果から、第6ラウンドの試験管内淘汰によって得られたVHHのcDNAディスプレイはGlcNAcに対して結合能があることが確認された。
これは、BDAのcDNAディスプレイが溶出されなかったのに対して、取得したVHHのcDNAディスプレイは溶出されてきたことを示す。この結果から、第6ラウンドの試験管内淘汰によって得られたVHHのcDNAディスプレイはGlcNAcに対して結合能があることが確認された。
架橋ペプチドアプタマー(BDA及びVHHのcDNAディスプレイ)は、上記実施例6(2)と同様に調製した。これらのcDNAディスプレイをそれぞれ調製した後に、磁性体ビーズを入れたチューブ中に両方のcDNAディスプレイを入れてインキュベートし、VHHのcDNAディスプレイのみを加えたときと同様にして、試験管内淘汰を行った。洗浄回数は各ラウンドで5回とし、4℃にて終夜、GlcNAcによる競合溶出を行った。
(3)架橋ペプチドアプタマーの調製
上記実施例6(2)と同様の操作で得られたVHHライブラリコンストラクトのPCR産物、それらのmRNA、それらのmRNA-リンカー連結体を、4%PAGE、200Vで30分間、ゲル電気泳動し、SYBER Gold又はFITCで染色して、PCRによる増幅及びmRNAとリンカーとの光架橋を確認した。第1及び第6ラウンドの結果を、それぞれ図12(A)及び(B)、並びに図13(A)及び(B)に示す。
上記実施例6(2)と同様の操作で得られたVHHライブラリコンストラクトのPCR産物、それらのmRNA、それらのmRNA-リンカー連結体を、4%PAGE、200Vで30分間、ゲル電気泳動し、SYBER Gold又はFITCで染色して、PCRによる増幅及びmRNAとリンカーとの光架橋を確認した。第1及び第6ラウンドの結果を、それぞれ図12(A)及び(B)、並びに図13(A)及び(B)に示す。
得られたVHH DNAの長さは、第1ラウンド~第6ラウンドまでいずれも541bpと同じであり、mRNAの長さも541merと同じであった。図13(B)に示すように、十分な量のmRNA-リンカー連結体が形成されていることが確認された。
(4)mRNA-ペプチド連結体の形成確認
mRNA-リンカー連結体、mRNA-ペプチド連結体、及び磁性体ビーズとを試験管内で共にインキュベートし、インキュベート終了後の上澄を、4%スタッキングゲル、6%ランニングゲル、200Vにて120分間、電気泳動し、FITCで染色してmRNA-ペプチド連結体の形成を確認した。mRNA-リンカー結合体は対照として使用した。結果を図14に示す。
mRNA-リンカー連結体、mRNA-ペプチド連結体、及び磁性体ビーズとを試験管内で共にインキュベートし、インキュベート終了後の上澄を、4%スタッキングゲル、6%ランニングゲル、200Vにて120分間、電気泳動し、FITCで染色してmRNA-ペプチド連結体の形成を確認した。mRNA-リンカー結合体は対照として使用した。結果を図14に示す。
mRNA-リンカー連結体と比べて、mRNA-ペプチド連結体のバンドが上にシフトしていることから、mRNA-ペプチド連結体が形成されていることが確認された。また、mRNA-ペプチド連結体と磁性体ビーズとをチューブ内で共にインキュベートした後の上澄では、mRNA-リンカー連結体のバンドの上に検出されたバンドがかなり薄くなっていた。このため、Quantity Oneを用いてインキュベート前のmRNA-ペプチド連結体(中央のカラムの濃く検出されたバンド)と比較したところ、バンドの濃度比は7:3となった。この結果、上澄中で検出されたmRNA-ペプチド連結体の量から、使用したmRNA-連結体の約57%が磁性体ビーズに固定化されたことが明らかになった。
(5)試験管内淘汰の確認
各ラウンドでGlcNAcにより競合溶出したVHH、洗浄液中に含まれるペプチドを、上記と同様の条件でPCRによって増幅させ、4%PAGE、200Vにて30分間ゲル電気泳動し、試験管内淘汰の進行度を確認した。結果を図15(A)~(C)に示す。
各ラウンドでGlcNAcにより競合溶出したVHH、洗浄液中に含まれるペプチドを、上記と同様の条件でPCRによって増幅させ、4%PAGE、200Vにて30分間ゲル電気泳動し、試験管内淘汰の進行度を確認した。結果を図15(A)~(C)に示す。
図中、「精製後」は、精製したcDNAディスプレイを示す。また、「インキュベーション上澄」は、各ラウンドで磁性体ビーズ(1st はカラム)とcDNAディスプレイとをインキュベートした後の上澄を示す。「洗浄液」は、タッピングで洗浄した後に回収した上澄を示す。さらに、「溶出物」は、GlcNAcによる競合溶出で溶出された液体を示す。
第1ラウンド~第3ラウンドでの結果を図15(A)~(C)に示す。第1ラウンドでは、mRNA-リンカー連結体を108pmol使用し、ビオチン化GlcNAc固定化量を25nmolとした。2ラウンドではmRNA-リンカー連結体を36pmol使用し、ビオチン化GlcNAc固定化量を1nmolとした。第3ラウンドでは、mRNA-リンカー連結体を18pmol使用し、ビオチン化GlcNAc固定化量を500pmolとした。ゲル電気泳動は、いずれも4%PAGE、200Vで30分間行い、SYBER Goldで染色した。
洗浄液のレーンのバンドの濃さが、ラウンドが進むについて薄くなったこと、溶出物のレーンのバンドの濃さに大きな低下がみられないことから、試験管内淘汰が進行しているものと推定された。このため、第4ラウンドからは、GlcNAc未固定磁性体ビーズと上記で得られたcDNAディスプレイとをインキュベートしたものを、ネガティブとして使用した。第4~第6ラウンドのゲル電気泳動も、4%PAGE、200Vで30分間行い、SYBER Goldで染色した。
第4ラウンドでは、mRNA-リンカー連結体を9pmol使用し、ビオチン化GlcNAc固定化量を100nmolとした。また、GlcNAc固定化磁性体ビーズとcDNAディスプレイとをインキュベートしたものをポジティブとして使用した。結果を図16(A)及び(B)に示す。ネガティブ及びポジティブの双方の溶出物のレーンにバンドが観察されたことから、淘汰は不十分と判定した。
第5ラウンドの結果を図17(A)及び(B)に示す。第5ラウンドでは、mRNA-リンカー連結体を8pmol使用し、ビオチン化GlcNAc固定化量を50nmolとした。ネガティブの洗浄液のレーンのバンドより溶出物のレーンのバンドが薄くなってきていることが観察された。また、ポジティブの溶出物のレーンでは濃いバンドが観察され、GlcNAc結合性VHHの淘汰が進んでいることが示された。
第6ラウンドの結果を図18(A)及び(B)に示す。第6ラウンドでは、mRNA-リンカー連結体を2.8pmol使用し、ビオチン化GlcNAc固定化量を50nmolとした。ネガティブ及びポジティブの各洗浄液のレーンのバンドの濃さと、各溶出物のレーンのバンドとの濃さとはほぼ同じであった。このため、ネガティブ及びポジティブの各洗浄液5、及びそれらの溶出物を、4%PAGE、200Vで30分間ゲル電気泳動し、SYBER Goldで染色した。結果を図19に示す。
また、上記マーカーの600のバンドをB1、ネガティブの溶出物をU1、ポジティブの溶出をU2として、Quantity One 1次元ゲル電気泳動解析ソフトウェア(Bio-Rad Laboratories社製)の計算ソフトを用いて、これらのバンドの濃さの比を求めた。結果を下記表13に示す。

表13に示したように、ポジティブの溶出物はネガティブのそれよりも22倍濃く、この結果、ポジティブの溶出物には、GlcNAc結合能性VHHが含まれていることが確認された。また、シーケンス解析の結果から、GlcNAcの認識には、CDR3が寄与していることが示された。
(比較例)従来リンカーの作製
従来例1~5のリンカー(以下、「従来リンカー1」のようにいう。)の構造を模式的に図1A~1Dに示す。
(比較例1)従来リンカー1(SBPリンカー)の合成及び特性評価
(1)従来リンカー1の合成
Short-ビオチン-ピューロマイシン・リンカー(SBPリンカー)を合成した。まず、下記の(A)及び(B1)合成をジーンワールド(株)(東京)及び(株)BEX(東京)に委託した。
従来例1~5のリンカー(以下、「従来リンカー1」のようにいう。)の構造を模式的に図1A~1Dに示す。
(比較例1)従来リンカー1(SBPリンカー)の合成及び特性評価
(1)従来リンカー1の合成
Short-ビオチン-ピューロマイシン・リンカー(SBPリンカー)を合成した。まず、下記の(A)及び(B1)合成をジーンワールド(株)(東京)及び(株)BEX(東京)に委託した。
(A)Puro-F-Sセグメントの合成[配列:5'-(S)-(PL)C(F)-(Spacer18)-(Spacer18)-(Spacer18)-(Spacer18)-CC-(Puro)-3']
ここで、(S)は、Thiol-Modifier C6 S-Sである(化合物名:o-(dimethoxytrityloxy-hexyl-dithiohexyl)-o'-(2-cyanoethyl)-N,N-diisopropyl-phosphoramidite)(PL)は、PC Linker Phosphoramiditeである(化合物名:3-(4,4'-Dimethoxytrityl)-1-(2-nitrophenyl)-1-propanyl-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)。(F)は、Fluorescein-dTである(化合物名:(5'-Dimethoxytrityloxy-5-[N-((3',6'-dipivaloylfluoresceinyl)-aminohexyl)-3-acryimido]-2'-deoxyUridine-3'-succinoyl-long chain alkylamino)。また、(Puro)は、ピューロマイシンを表わす。
ここで、(S)は、Thiol-Modifier C6 S-Sである(化合物名:o-(dimethoxytrityloxy-hexyl-dithiohexyl)-o'-(2-cyanoethyl)-N,N-diisopropyl-phosphoramidite)(PL)は、PC Linker Phosphoramiditeである(化合物名:3-(4,4'-Dimethoxytrityl)-1-(2-nitrophenyl)-1-propanyl-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)。(F)は、Fluorescein-dTである(化合物名:(5'-Dimethoxytrityloxy-5-[N-((3',6'-dipivaloylfluoresceinyl)-aminohexyl)-3-acryimido]-2'-deoxyUridine-3'-succinoyl-long chain alkylamino)。また、(Puro)は、ピューロマイシンを表わす。
(Spacer18)は、Spacer Phosphoramidite 18である(化合物名:18-0-Dimethoxytritylhexaethyleneglycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)。)(いずれも、Glen Research社製)。
(B1)Hybriセグメント[(26mer) 配列12:5'-CCGCBCRCCC CGCCG CCCCC CGDCC T-3']
ここで、Dはアミノ-モディファイヤーC6 dT(5'-Dimethoxytrityl-5-[N-(trifluoro acetylaminohexyl)-3-acrylimido]-2'-deoxyUridine,3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite);BはBiotin-dT(5'-Dimethoxytrityloxy-5-[N-((4-t-butylbenzoyl)-biotinyl)-aminohexyl)-3-acrylimido]-2'-deoxyUridine-3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)、RはriboGである(すべてGlen Research Search社製)。
ここで、Dはアミノ-モディファイヤーC6 dT(5'-Dimethoxytrityl-5-[N-(trifluoro acetylaminohexyl)-3-acrylimido]-2'-deoxyUridine,3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite);BはBiotin-dT(5'-Dimethoxytrityloxy-5-[N-((4-t-butylbenzoyl)-biotinyl)-aminohexyl)-3-acrylimido]-2'-deoxyUridine-3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)、RはriboGである(すべてGlen Research Search社製)。
上記SBPリンカーは、上記実施例1(1)で合成した(A)Puro-F-Sセグメントと(B2)Hybriセグメントとを以下の方法に従って架橋し精製して得た。Puro-F-S 10nmolを100μLの50mMリン酸バッファー(pH 7.0)に溶かし、100mM Tris[2-carboxyethyl]phosphine(TCEP、Pierce社製)を1μL加え(終濃度 1mM)、室温で6時間放置し、Puro-F-Sのトリチル化メルカプト基をチオール基に還元した。架橋反応を行う直前に、50mMのリン酸バッファー(pH 7.0)で平衡化したNAP-5 Columnsを用いてTCEPの除去及び脱塩を行った。
100μLの0.2 Mリン酸バッファー(pH 7.0)に、500pmol/μLのHybriを20μL及び20μLの100mM架橋剤(EMCS)を加えて良く攪拌し、37℃で30分反応させた。その後、4℃でエタノール沈殿を行って反応産物を沈殿させ、未反応のEMCSを除去した。沈殿を500μLの冷70%エタノールにて洗浄し、減圧下で乾燥させた後、0.2Mリン酸バッファー(pH 7.0)10μLに溶解した。
この反応産物に、上記還元したPuro-F-S(~10nmol)を速やかに加えて、4℃で一晩放置した。サンプルに終濃度が4mMになるようにTCEPを加え、37℃で15分間放置し、チオール基の架橋反応を停止させた。室温にてエタノール沈殿を行い、合成したリンカーを沈殿させ、未反応のPuro-F-Sを取り除き、さらに未反応のHybriセグメント、及びそれらのEMCS架橋物を取り除くために、以下の条件でHPLC精製を行った。
(HPLC条件)
カラム:COSMOSIL(登録商標)10x250mm C18-AR-300(ナカライテスク(株)製)
バッファーA:0.1M TEAA(triethylammonium acetate)
バッファーB:80%アセトニトリル(超純水で希釈したもの)
流速:0.5ml/分
濃度勾配:A及びBの混合バッファー濃度は85:15→65:15(A:B)で変化させた(33分間)。
カラム:COSMOSIL(登録商標)10x250mm C18-AR-300(ナカライテスク(株)製)
バッファーA:0.1M TEAA(triethylammonium acetate)
バッファーB:80%アセトニトリル(超純水で希釈したもの)
流速:0.5ml/分
濃度勾配:A及びBの混合バッファー濃度は85:15→65:15(A:B)で変化させた(33分間)。
HPLCにより分画された生成物を18%アクリルアミドゲル(8M 尿素、62℃)の電気泳動にて検出し、目的の画分を減圧下で乾燥させた。この後、DEPC(Diethylpyrocarbonate)処理水で溶かして、10pmol/μLとした。得られたリンカー(以下、「従来リンカー1」という。)を、図1Aに模式的に示す。
(2)mRNAの合成
モデルmRNAとしてBDA(B domain of Protein A)を用いることとした。BDA遺伝子(BDA gene; 192塩基長)の5'側上流にT7プロモーター配列と翻訳促進配列、3'側下流にスペーサー領域、ヒスチジンタグ(Hisタグ)及びピューロマイシン・リンカーとの相補鎖領域を有する配列を付加したDNA(BDA whole;配列表の配列番号6;367塩基長)をPCRによって合成、精製した後、T7 RiboMAX Express Large Scale RNA Production System(プロメガ社製))を用いて、添付のプロトコルに従って5~30 pmol/μLのmRNA(BDA mRNA)を合成した。
モデルmRNAとしてBDA(B domain of Protein A)を用いることとした。BDA遺伝子(BDA gene; 192塩基長)の5'側上流にT7プロモーター配列と翻訳促進配列、3'側下流にスペーサー領域、ヒスチジンタグ(Hisタグ)及びピューロマイシン・リンカーとの相補鎖領域を有する配列を付加したDNA(BDA whole;配列表の配列番号6;367塩基長)をPCRによって合成、精製した後、T7 RiboMAX Express Large Scale RNA Production System(プロメガ社製))を用いて、添付のプロトコルに従って5~30 pmol/μLのmRNA(BDA mRNA)を合成した。
(3)従来リンカー1の特性評価
(3-1)mRNAとの連結体の形成
T4 RNAリガーゼを用いたライゲーション反応を以下のように行った。従来リンカー2を1としたときに1~6倍のmRNAを加え、20μLのT4 RNAリガーゼバッファー(10mM 塩化マグネシウム、10mM DTT及び1mM ATPを含む50mM Tris-HCl(pH 7.5))中にて行なった。酵素を加える前にアニーリングするため、90℃で5分間アルミブロック上にて温め、次に70℃で5分間アルミブロック上にて温め、最後に室温で10分間放置した。その後、氷上に置いた。ここに、1μLのT4ポリヌクレオチドキナーゼ(10U/μL)及び1μLのT4 RNAリガーゼ(40U/μL)(いずれもタカラバイオ(株)製)を加え、25℃で15分間保持した。
(3-1)mRNAとの連結体の形成
T4 RNAリガーゼを用いたライゲーション反応を以下のように行った。従来リンカー2を1としたときに1~6倍のmRNAを加え、20μLのT4 RNAリガーゼバッファー(10mM 塩化マグネシウム、10mM DTT及び1mM ATPを含む50mM Tris-HCl(pH 7.5))中にて行なった。酵素を加える前にアニーリングするため、90℃で5分間アルミブロック上にて温め、次に70℃で5分間アルミブロック上にて温め、最後に室温で10分間放置した。その後、氷上に置いた。ここに、1μLのT4ポリヌクレオチドキナーゼ(10U/μL)及び1μLのT4 RNAリガーゼ(40U/μL)(いずれもタカラバイオ(株)製)を加え、25℃で15分間保持した。
生成反応液を半分に分割して、後の実験工程にて比較するため、リンカー10pmolに対しmRNA 10pmolを反応させたサンプルについては、全ての物質を倍に増量して40μLの系にて反応させた(反応系R1)。
(3-2)ライゲーション反応の結果
以下に示す混合比で、mRNAとSBPリンカーとの連結効率を比較した。比較は、65℃、8M尿素変性5%アクリルアミドゲルを用い、200Vにて25分間の電気泳動にて行った。サンプルを各々1μLずつ分注し、3μLのローディングバッファーと2μLのDEPC水と混合して、ゲルにロードした。結果を図20に示す。バンドの検出はリンカーに結合したFITCの蛍光により行い、未反応のリンカー又は上記連結体Aを検出した。
以下に示す混合比で、mRNAとSBPリンカーとの連結効率を比較した。比較は、65℃、8M尿素変性5%アクリルアミドゲルを用い、200Vにて25分間の電気泳動にて行った。サンプルを各々1μLずつ分注し、3μLのローディングバッファーと2μLのDEPC水と混合して、ゲルにロードした。結果を図20に示す。バンドの検出はリンカーに結合したFITCの蛍光により行い、未反応のリンカー又は上記連結体Aを検出した。
10pmolのmRNAに対して、10pmolの従来リンカー2を加え(前記40μLの反応系にて反応させたもの)(レーン1、反応系R1)、6.6pmol(レーン2、反応系R2)、3.3pmol(レーン3、反応系R3)、1.66pmol(レーン4、反応系R4)、上記の通りライゲーション反応させたものについて電気泳動を行なって観察した(図20)。図20中、矢印は未反応のリンカー、*はリンカーが連結された反応産物を示す。その結果、混合比1:1の場合を除き、連結体Aの合成量に差はなく、いずれにおいてもほぼ等量のライゲーション反応の起きたことが観察された。
(比較例2)従来リンカー2(LBPリンカー)の合成及び特性の評価
(1)従来リンカー2(LBPリンカー)の合成
Long-Biotin-ピューロマイシン・リンカー(LBPリンカー)を以下のように合成した。まず、上記(1-1)の(A)及び(B2)の合成をジーンワールド(株)及び(株)BEXに委託した。
(1)従来リンカー2(LBPリンカー)の合成
Long-Biotin-ピューロマイシン・リンカー(LBPリンカー)を以下のように合成した。まず、上記(1-1)の(A)及び(B2)の合成をジーンワールド(株)及び(株)BEXに委託した。
ビオチン-ループセグメント(56 mer)(配列表の配列番号13)中の制限部位を図21Aに示した。また、チオール-モディファイヤーC6 S-Sの化学式を下記式(IX)に示す。
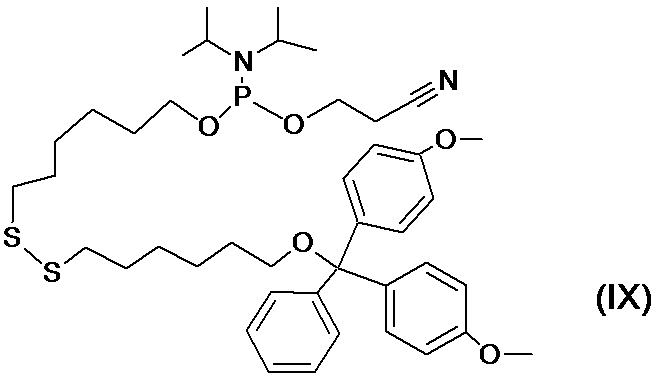
上記LBPリンカーは、前記(A)Puro-F-Sセグメント(側鎖)と(B)Biotin-loopセグメント(主鎖)を以下の方法に従って架橋し精製して得た。まず、10nmolのPuro-F-Sを22.5μLの1Mのリン酸バッファー(pH 9.0)に溶かし、2.5μLの1M DTTを加え、室温で1時間放置し、Puro-F-Sのトリチル化メルカプト基をチオール基に還元した。架橋反応を行う直前に、20mMリン酸バッファー(pH 7.2)で平衡化したNAP-5 Columns(GEヘルスケア社製)を用いてDTTの除去及び脱塩を行った。
50μLの0.2Mリン酸バッファー(pH 7.2)に、10μLの500pmol/μLのBiotin-loop及び10μLの100mMのEMCS(6-Maleimidehexanoic acid N-hydroxysuccinide ester:架橋剤、(株)同人化学研究所製)を加えて良く攪拌し、37℃で30分反応させた。その後、4℃で反応産物をエタノール沈殿させ、未反応のEMCSを除去した。沈殿物を500μLの70%エタノールにて洗浄し、減圧下で乾燥させた。
この反応産物を、上記のように還元したPuro-F-S(~10nmol)に速やかに溶解し、4℃で一晩放置してサンプルとした。このサンプルに、終濃度が50mMになるようにDTTを加え、37℃で30分間放置し、チオール基の架橋反応を停止させた。エタノール沈殿法により、室温で、合成したリンカーを沈殿させ、未反応のPuro-F-Sを除去した。さらに未反応のBiotin-loop及びそれらのEMCS架橋物を取り除くために、以下の条件でHPLCを用いたグラジエント法で精製を行った。
(HPLC)条件
カラム:Symmetry 300 C18, 5μm, 内径4.6mmx250mm (Waters Corporation製)
溶離液:下記の溶液Aと溶液Bとを混合して使用した。
溶液A:0.1M TEAA (酢酸トリエチルアンモニウム)
溶液B:80%アセトニトリル(超純水で希釈したもの)
流速:0.5mL/分
溶離液の濃度勾配:溶液A:溶液Bを、30分間で85:15→65:35に変化させた。
カラム:Symmetry 300 C18, 5μm, 内径4.6mmx250mm (Waters Corporation製)
溶離液:下記の溶液Aと溶液Bとを混合して使用した。
溶液A:0.1M TEAA (酢酸トリエチルアンモニウム)
溶液B:80%アセトニトリル(超純水で希釈したもの)
流速:0.5mL/分
溶離液の濃度勾配:溶液A:溶液Bを、30分間で85:15→65:35に変化させた。
HPLCにより分画された生成物を、16%アクリルアミドゲル(8M尿素、60℃)を使用した電気泳動にて検出し、目的の画分を減圧下に乾燥させた。この後、DEPC(二炭酸ジエチル、Diethylpyrocarbonate)処理水に10pmol/μLとなるように溶解した。
(2)従来リンカー2の特性評価
従来リンカー2がエンドヌクレアーゼV(以下、「Endo V」ということがある。M0305S、New England Biolabs, Inc.(以下、「NEB社」と略すことがある。)製)で切断されるか否かを評価した。
従来リンカー2がエンドヌクレアーゼV(以下、「Endo V」ということがある。M0305S、New England Biolabs, Inc.(以下、「NEB社」と略すことがある。)製)で切断されるか否かを評価した。
10pmolの従来リンカー1、0.5μLのEndo V(10U/μL)、1μLの10xNEバッファー及び蒸留水を含む10μLの反応液中にて、37℃で、30分間反応させた。反応終了後、P6カラム(Bio-Rad Laboratories, Inc.製)で脱塩した。従来リンカー1として5pmol相当の溶液を取り、SDS-PAGE法にて分析した。8M尿素を含有する12%ポリアクリルアミドゲルにて、60℃で、200Vの条件の下、30分間電気泳動を行った。電気泳動終了後、SYBR(登録商標) Goldにて染色し、泳動像を蛍光観察した。Endo Vによって切断された産物は検出されないことが確認された(図21B参照)。
(比較例3)従来リンカー3の合成及び特性評価
(1)従来リンカー3の合成
本発明で使用するイノシン-Short-Biotin-ピューロマイシン・リンカー(SBP(I)リンカー)及びrG-Short-Biotin-ピューロマイシン・リンカー(SBP(rG)リンカー)を以下のように合成した。まず、比較例1に記載した(A)及び(B1)に加えて、(C1)I-hybriセグメント((28mer)、配列表の配列番号14)、及び(C2)rG-Hybriセグメント((26mer) 配列表の配列番号15)という特殊DNAの合成をジーンワールド(株)(東京)に委託した。
(1)従来リンカー3の合成
本発明で使用するイノシン-Short-Biotin-ピューロマイシン・リンカー(SBP(I)リンカー)及びrG-Short-Biotin-ピューロマイシン・リンカー(SBP(rG)リンカー)を以下のように合成した。まず、比較例1に記載した(A)及び(B1)に加えて、(C1)I-hybriセグメント((28mer)、配列表の配列番号14)、及び(C2)rG-Hybriセグメント((26mer) 配列表の配列番号15)という特殊DNAの合成をジーンワールド(株)(東京)に委託した。
ここで、(B1)中、Iはデオキシイノシンを表わし、(T)、(T-B)及び修飾塩基を表す記号は、上記比較例1及び2と同様である。また、(C1)中、(rG)はリボGを表わす。
上記SBP(I)リンカーは、上記実施例1の(1)で合成した(A)Puro-F-Sセグメントと(B)I-Hybriセグメントとを、以下の方法に従って架橋し精製して得た。また、上記SBP(rG)リンカーは(A)Puro-F-Sと(C)rG-hybriを架橋して得た。
上記SBP(I)リンカーは、上記実施例1の(1)で合成した(A)Puro-F-Sセグメントと(B)I-Hybriセグメントとを、以下の方法に従って架橋し精製して得た。また、上記SBP(rG)リンカーは(A)Puro-F-Sと(C)rG-hybriを架橋して得た。
1.7μLの3mM Puro-F-Sを、22.5μLの1Mリン酸バッファー(pH 9.0)と混合し、2.5μLの1M DTTを加え、室温で1時間放置し、Puro-F-Sのトリチル化メルカプト基をチオール基に還元した。架橋反応を行う直前に、20mMリン酸バッファー(pH 7.2)で平衡化したNAP-5 Columns(GEヘルスケア社製)を用いてDTTの除去及び脱塩を行った。
50μLの0.2Mリン酸バッファー(pH 7.2)に、2.5μLの1mM I-HybriまたはrG-Hybri、及び10μLの100mMのEMCSを加えて良く攪拌し、37℃で30分反応させた。その後、反応産物を4℃でエタノール沈殿させて未反応のEMCSを除去した。沈殿物を200μLの70%エタノールで洗浄し、減圧下で乾燥させた。
この反応産物を、上記のように還元したPuro-F-S溶液(~5nmol)に速やかに溶解し、4℃で一晩放置してサンプルとした。このサンプルに、終濃度が50mMとなるようにDTTを加え、37℃で30分間放置し、チオール基の架橋反応を停止させた。室温でエタノール沈殿により合成したリンカーを沈殿させ、未反応のPuro-F-Sを取り除き、さらに未反応のI-HybriまたはrG-Hybri、及びそれらのEMCS架橋物を取り除くために、実施例1と同様の条件でHPLC精製を行った。
HPLCにより分画された生成物を、8Mの尿素を含有する12%アクリルアミドゲルにて、200V、60℃の条件下で30分間電気泳動を行い分画した。目的の画分を減圧下で乾燥させた。この後、DEPC(Diethylpyrocarbonate)処理水で溶かして、10pmol/μLとした。
(2)リンカーのRNase耐性検査
上記のように合成したリンカーSBP(rG)及びSBP(I)のRNase耐性検査を、大腸菌(E.coli)のペリプラズムに由来のRNase ONE(プロメガ社製)を用いて行った。RNase ONEは、A、C、G、Uの各RNAの3'末側のリン酸ジエステル結合を切断する活性を有するRNA分解酵素である。
上記のように合成したリンカーSBP(rG)及びSBP(I)のRNase耐性検査を、大腸菌(E.coli)のペリプラズムに由来のRNase ONE(プロメガ社製)を用いて行った。RNase ONEは、A、C、G、Uの各RNAの3'末側のリン酸ジエステル結合を切断する活性を有するRNA分解酵素である。
1pmolのSBP(rG)又はSBP(I)と、0.5μLのRNase ONE(10U/μL)と、1μLの10xRNase ONE reaction buffer(プロメガ社製))と、RNaseフリー水とを加えて混合し、10μLの混合液とした。
次にこの混合液を、37℃で30分間反応させ、反応産物をSDS-PAGE法にて分析した。電気泳動は、8Mの尿素を含有する12%ポリアクリルアミドゲルにて、200V、60℃の条件下で30分間行い、リンカー中のFITCを、Molecular Imager Pharos FX (Bio-Rad Laboratories, Inc.製)を用いて、波長488nmのレーザー光にて励起し、蛍光検出した。結果を図21Bに示す。
図21B中、第1レーンには、100bpのDNAラダー(プロメガ社製)をサイズマーカーとして1μLアプライした。第2レーンには未処理のSBP(rG)を0.5pmol、第3レーンにはRNase ONEで処理したSBP(rG)(図中、「puroFS」と示した。)を5μL、第4レーンには未処理のSBP(I)を0.5pmol、第5レーンにはRNase ONEで処理したSBP(I)を5μL、それぞれアプライした。
図22中、未処理のSBP(rG)をアプライした第2レーンとRNase ONEで処理したSBP(rG)をアプライした第3レーンとを比較すると、第3レーンでバンドの位置が低分子側にシフトしていた。このため、SBP(rG)はリボGサイトにてRNase ONEによって切断されたことが示された。一方未処理のSBP(I)をアプライした第4レーンとRNase ONEで処理したSBP(I)をアプライした第5レーンとを比較すると、バンドの位置の低分子側へのシフトは認められなかった。このため、SBP(I)はRNase ONEで切断されていないことが示された。
以上の検査により、SBP(I)は、旧来のSBP(rG)リンカーにはないリボヌクレアーゼ耐性を有することが示された。
以上の検査により、SBP(I)は、旧来のSBP(rG)リンカーにはないリボヌクレアーゼ耐性を有することが示された。
(比較例4)従来リンカー4の合成及び特性評価
(1)従来リンカー4の合成
従来リンカー4(図1D)は、(a)ピューロマイシンセグメント(図23A)、(b)ソラレン-アミノセグメント(図23B)、(c)アジドセグメント、及び(d)アルキン-ペプチド-ビオチンセグメントという4つのセグメントに分けて合成した。これらのうち、(a)と(b)とは主鎖の合成に使用し、(c)と(d)とは基質ユニットの合成に使用する。上記(a)~(c)のセグメントの合成に使用する特殊DNAの合成を、(株)ジーンワールドおよび(株)日本バイオサービスに委託した。(d)に使用するペプチド合成を(株)スクラムに委託した。上記の(a)ピューロマイシンセグメントは、上述した実施例2(2-1)で合成したものを使用した。
(1)従来リンカー4の合成
従来リンカー4(図1D)は、(a)ピューロマイシンセグメント(図23A)、(b)ソラレン-アミノセグメント(図23B)、(c)アジドセグメント、及び(d)アルキン-ペプチド-ビオチンセグメントという4つのセグメントに分けて合成した。これらのうち、(a)と(b)とは主鎖の合成に使用し、(c)と(d)とは基質ユニットの合成に使用する。上記(a)~(c)のセグメントの合成に使用する特殊DNAの合成を、(株)ジーンワールドおよび(株)日本バイオサービスに委託した。(d)に使用するペプチド合成を(株)スクラムに委託した。上記の(a)ピューロマイシンセグメントは、上述した実施例2(2-1)で合成したものを使用した。
(2)ソラレン-アミノセグメントの合成
以下の構造を有するソラレン-アミノセグメント(配列表の配列番号16)を合成した。ここで、(psoralen)には、Psoralen C6 Phosphoramidite((6-[4'-(Hydroxymethyl)-4,5',8-trimethylpsoralen]-hexyl-1-O-(2-cyanoethyl)-(N,N-diisopropyl)-phosphoramidite))を使用した。
以下の構造を有するソラレン-アミノセグメント(配列表の配列番号16)を合成した。ここで、(psoralen)には、Psoralen C6 Phosphoramidite((6-[4'-(Hydroxymethyl)-4,5',8-trimethylpsoralen]-hexyl-1-O-(2-cyanoethyl)-(N,N-diisopropyl)-phosphoramidite))を使用した。
[配列16]
5'-(psoralen)-TACGACGATCTCGAACGAACCACCCCCGCCGCCCCCCG-(T-NH2)-CCT-3'
5'-(psoralen)-TACGACGATCTCGAACGAACCACCCCCGCCGCCCCCCG-(T-NH2)-CCT-3'
また、(T-NH2)には、Amino-Modifier C6 dT(5'-Dimethoxytrityl-5-[N-(trifluoro acetylaminohexyl)-3-acrylimido]-2'-deoxyUridine,3'-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite)を使用した。以上の試薬は、いずれもGlen Research社より購入した。これらの試薬を使用して、核酸自動合成機を用いてホスホロアミダイト法に従って反応させた(図23B)。
(3)アジドセグメントの合成
以下の構造を有するアジドセグメント(配列表の配列番号17)を合成した。ここで、(Spacer18)には、Spacer Phosphoramidite 18((18-0-Dimethoxytrityl hexaethyleneglycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite))を使用した。
以下の構造を有するアジドセグメント(配列表の配列番号17)を合成した。ここで、(Spacer18)には、Spacer Phosphoramidite 18((18-0-Dimethoxytrityl hexaethyleneglycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite))を使用した。
[配列17]
5'-CCCGTGGTTCGTTCGAGATCGTCGTAAA-3'
5'-CCCGTGGTTCGTTCGAGATCGTCGTAAA-3'
上記配列の3'端のAAAには-(Spacer18)-(C6)-(Azide)が結合しており、(C6)には、3'-Amino-Modifier C7 CPG 500(2-Dimethoxytrityloxymethyl-6-fluorenyl methoxycarbonylamino-hexane-1-succinoyl-long chain alkylamino-CPG)を使用した。
(Azide)には、アジドブチレートNHSエステル(4-Azido-butan-1-oic acid N-hydroxy succinimide ester)を使用した。以上の試薬は、いずれもGlen Research社より購入した。これらの試薬を使用して、核酸自動合成機を用いてホスホロアミダイト法に従って反応させ、アジドセグメントを合成した。
(Azide)には、アジドブチレートNHSエステル(4-Azido-butan-1-oic acid N-hydroxy succinimide ester)を使用した。以上の試薬は、いずれもGlen Research社より購入した。これらの試薬を使用して、核酸自動合成機を用いてホスホロアミダイト法に従って反応させ、アジドセグメントを合成した。
(4)アルキン-ペプチド-ビオチンセグメントの合成
以下の構造を有するアルキン-ペプチド-ビオチンセグメント(配列表の配列番号18)を合成した。以下の配列は、N末からC末に向かう構造として示した。ここで、Gly(Propargyl)には、Fmoc-Gly(Propargyl)-OHを使用した。(K-biotin)-NH2は、リジン残基のC末端をアミド化し、リジン残基の側鎖にビオチンを結合させて修飾したものである。
以下の構造を有するアルキン-ペプチド-ビオチンセグメント(配列表の配列番号18)を合成した。以下の配列は、N末からC末に向かう構造として示した。ここで、Gly(Propargyl)には、Fmoc-Gly(Propargyl)-OHを使用した。(K-biotin)-NH2は、リジン残基のC末端をアミド化し、リジン残基の側鎖にビオチンを結合させて修飾したものである。
[配列18]
Gly(Propargyl)-EDHVAHALAQ-(K-Biotin)-NH2
Gly(Propargyl)-EDHVAHALAQ-(K-Biotin)-NH2
下記式(I)で表わされるアジド
と、下記式(II)で表わされるアルキン
とを、1:100のモル比で混合し、CuSO4/Ascorbateを触媒として、室温にて64時間反応させて、下記式(III)で表わされる反応生成物を得た。
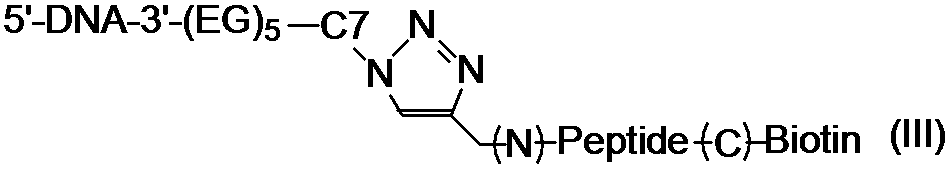
(5)アジドセグメントとアルキン-ペプチド-ビオチンセグメントとの架橋
10μLの25μM アジド-セグメントと、25μLの1mM アルキレンペプチド-ビオチンセグメントとを、60μLのt-ブタノール、5μLの0.5mM CuSO4、5μLの2.5mM アスコルビン酸エステルからなる溶液と混合し、室温で撹拌しながら64時間、架橋反応をさせた。得られた架橋反応物をリン酸バッファー(pH 7.2)で平衡化したマイクロバイオスピンカラム6 (Bio-rad社製)で脱塩し、脱塩された架橋反応物をポリアクリルアミドゲル電気泳動で分離し、DNAをSybrGold (invitrogen社製)で染色して解析した。
10μLの25μM アジド-セグメントと、25μLの1mM アルキレンペプチド-ビオチンセグメントとを、60μLのt-ブタノール、5μLの0.5mM CuSO4、5μLの2.5mM アスコルビン酸エステルからなる溶液と混合し、室温で撹拌しながら64時間、架橋反応をさせた。得られた架橋反応物をリン酸バッファー(pH 7.2)で平衡化したマイクロバイオスピンカラム6 (Bio-rad社製)で脱塩し、脱塩された架橋反応物をポリアクリルアミドゲル電気泳動で分離し、DNAをSybrGold (invitrogen社製)で染色して解析した。
結果を図23Cに示す。レーン1は、10bpのDNAステップラダー(プロメガ社製)、レーン2はアジド-セグメント、レーン3は架橋反応物を示す。この結果から、約70%の収率で、目的である架橋物(F)が得られていることが確認された(図23C)。
(6)ピューロマイシンセグメントとソラレン-アミノセグメントとの架橋
4mMのピューロマイシンセグメント5μLを、45μLの1Mリン酸バッファー(pH 9.0)と混合し、1M DTTを5μL加えて室温で1時間放置し、ピューロマイシンセグメントのジスルフィド基をチオール基に還元した。架橋反応を行う直前に、20mMのリン酸バッファー(pH 7.2)で平衡化したNAP-5 Columnを用いて、DTTの除去及び脱塩を行った。0.2Mのリン酸バッファー(pH 7.2)100μLに、10μLの1mMのソラレン-アミノセグメント、及び20μLの100mMの架橋剤(EMCS)を加えてよく攪拌し、37℃で30分間反応させた。
4mMのピューロマイシンセグメント5μLを、45μLの1Mリン酸バッファー(pH 9.0)と混合し、1M DTTを5μL加えて室温で1時間放置し、ピューロマイシンセグメントのジスルフィド基をチオール基に還元した。架橋反応を行う直前に、20mMのリン酸バッファー(pH 7.2)で平衡化したNAP-5 Columnを用いて、DTTの除去及び脱塩を行った。0.2Mのリン酸バッファー(pH 7.2)100μLに、10μLの1mMのソラレン-アミノセグメント、及び20μLの100mMの架橋剤(EMCS)を加えてよく攪拌し、37℃で30分間反応させた。
その後、常法に従ってエタノール沈殿を行って反応産物を沈殿させ、未反応のEMCSを除去した。沈殿を200μLの70%エタノールにて洗浄し、減圧下で乾燥させた。この反応産物を、上記のように還元したピューロマイシンセグメント溶液(約20nmol)に速やかに溶解させて、4℃で一晩放置した。この後に、終濃度が50mMになるようにDTTを加え、37℃で30分間放置し、チオール基の架橋反応を停止させ、上記2つのセグメントが架橋された架橋産物を含む反応溶液を得た。
得られた架橋産物を尿素変性ポリアクリルアミドゲル電気泳動(12%アクリルアミドゲル、8M尿素、60℃)で分離し、DNAをSybrGold(invitrogen社製)で染色して解析した。レーン1は10bp DNAステップラダー(プロメガ社製)、レーン2は架橋反応物の泳動結果である。ピューロマイシンセグメント及びソラレン-アミノセグメント以外に、これらの分子量の合計に匹敵するバンドが検出され、目的の架橋物(E)が得られていることが確認された。結果を図23Dに示す。
次いで、この架橋物(E)を、上記と同様にしてエタノールで沈殿させ、未反応のピューロマイシンセグメントを取り除き、さらに未反応のソラレン-アミノセグメント、及びそのEMCS架橋物を取り除くために、以下の条件でHPLCにより精製を行った。
(HPLC条件)
カラム:Symmetry300 C18, 5μm, 4.6 x 250mm (Waters 社製)
バッファーA:0.1M TEAA(酢酸トリメチルアンモニウム、和光純薬工業(株)製)
バッファーB:80%アセトニトリル(和光純薬工業(株)製、超純水で希釈したもの)
流速:0.5ml/分
濃度勾配:A及びBの混合バッファー濃度は、85:15→50:50(A:B)で変化させる(50分間)。
カラム:Symmetry300 C18, 5μm, 4.6 x 250mm (Waters 社製)
バッファーA:0.1M TEAA(酢酸トリメチルアンモニウム、和光純薬工業(株)製)
バッファーB:80%アセトニトリル(和光純薬工業(株)製、超純水で希釈したもの)
流速:0.5ml/分
濃度勾配:A及びBの混合バッファー濃度は、85:15→50:50(A:B)で変化させる(50分間)。
HPLCにより分画された生成物を、再度、12%アクリルアミドゲル(8M尿素、60℃)の電気泳動にて検出し、目的の画分を減圧下で濃縮した後、上記と同様にしてエタノールで沈殿させた。この後、沈殿物を水で溶解して50μMとした。
(7)架橋物(E)と架橋物(F)との光架橋
1pmolの架橋物(E)と1.2pmolの架橋物(F)を、20mM Tris-HCl (pH 8.0)と100mM NaCl を含む溶液中で混合し、その後60℃に加熱し、10分かけて25℃まで冷却した。次いで、試料にキセノンランプを光源とする365nm(2W/cm2)の光を20分間照射し、試料を露光させて露光試料とした。この露光試料を、上記と同じ条件の下で、尿素変性ポリアクリルアミドゲル電気泳動で分離し、SybrGold (invitrogen社製)でDNAを染色し解析した。結果を図23Eに示す。
1pmolの架橋物(E)と1.2pmolの架橋物(F)を、20mM Tris-HCl (pH 8.0)と100mM NaCl を含む溶液中で混合し、その後60℃に加熱し、10分かけて25℃まで冷却した。次いで、試料にキセノンランプを光源とする365nm(2W/cm2)の光を20分間照射し、試料を露光させて露光試料とした。この露光試料を、上記と同じ条件の下で、尿素変性ポリアクリルアミドゲル電気泳動で分離し、SybrGold (invitrogen社製)でDNAを染色し解析した。結果を図23Eに示す。
図23Eに示すように、露光試料では長鎖長側に移動した電気泳動バンドが検出され、架橋物(G)が生成されたことを確認した。HPLC分析の結果でも、保持時間31.819分の位置に架橋物が溶出されたことが確認された。
本発明は、分子標的型ペプチド医薬、低分子化抗体及び抗体様タンパク質を用いた医薬等の技術分野で有用である。
配列番号1:本発明のcnvKリンカーの主鎖の塩基配列
配列番号2:プライマー(T7Ωnew)の塩基配列
配列番号3:プライマー(NewYtag)の塩基配列
配列番号4:プライマー(Newleft)の塩基配列
配列番号5:Aタンパク質のBドメインの塩基配列
配列番号6:VHHライブラリコンストラクト
配列番号2:プライマー(T7Ωnew)の塩基配列
配列番号3:プライマー(NewYtag)の塩基配列
配列番号4:プライマー(Newleft)の塩基配列
配列番号5:Aタンパク質のBドメインの塩基配列
配列番号6:VHHライブラリコンストラクト
配列番号7:試験管内淘汰法で使用するランダムライブラリDNA配列
配列番号8:ΩRT-Lnew
配列番号9:NewYtag(22mer)
配列番号10:PDOのDNA配列
配列番号11:FLAGの塩基配列
配列番号12:従来リンカーのHybriセグメントの塩基配列
配列番号8:ΩRT-Lnew
配列番号9:NewYtag(22mer)
配列番号10:PDOのDNA配列
配列番号11:FLAGの塩基配列
配列番号12:従来リンカーのHybriセグメントの塩基配列
配列番号13:従来リンカーの主鎖の塩基配列
配列番号14:従来リンカーのI-hybriセグメント
配列番号15:従来リンカーのrG-Hybriセグメントの塩基配列
配列番号16:従来リンカーのソラレン-アミノセグメントの塩基配列
配列番号17:従来リンカーのアジドセグメントの塩基配列
配列番号18:従来リンカーの側鎖のビオチンセグメントの塩基配列
配列番号14:従来リンカーのI-hybriセグメント
配列番号15:従来リンカーのrG-Hybriセグメントの塩基配列
配列番号16:従来リンカーのソラレン-アミノセグメントの塩基配列
配列番号17:従来リンカーのアジドセグメントの塩基配列
配列番号18:従来リンカーの側鎖のビオチンセグメントの塩基配列
Claims (15)
- 主鎖と側鎖とを有するリンカーであって、
前記主鎖は、
所定の塩基配列を有し、前記主鎖の5’末端に位置し固相との結合を形成するための固相結合部位と;
前記固相結合部位ごと前記固相を切り離すための固相切断部位と;
前記側鎖を連結するための側鎖連結部位と;
前記側鎖連結部位と前記固相切断部位との間に位置し、前記主鎖と相補的な配列を有するmRNAを、前記主鎖と光架橋によって連結する高速光架橋部位と;
前記側鎖連結部位に隣接し、前記主鎖の3’末端に位置する逆転写開始領域と;
を備え、
前記側鎖は、蛍光標識と、遊離末端に位置するタンパク質結合部位と、前記主鎖の側鎖連結部位に連結される連結形成部位とを備え、
前記側鎖は前記連結形成部位で前記側鎖連結部位に連結されている、試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。 - 前記固相切断部位は、デオキシイノシン、リボG又はリボピリミジンからなる群から選ばれる塩基で構成される、ことを特徴とする請求項1に記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。
- 前記高速光架橋部位はシアノビニルカルバゾール化合物で構成される、ことを特徴とする請求項1又は2に記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。
- 前記シアノビニルカルバゾール化合物は3-シアノビニルカルバゾールである、ことを特徴とする請求項3に記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。
- 前記固相結合部位は、ビオチン、ストレプトアビジン、アルキン、クリックケミストリーによるアジ化物、アミノ基、N-ヒドロキシスクシンイミドエステル(NHS)、SH基、及びAuからなる群から選ばれるいずれかの化合物と、前記化合物に結合したポリAとで構成される、ことを特徴とする請求項1~4のいずれかに記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。
- 前記タンパク質結合部位はピューロマイシン又はその類縁化合物で構成されている、ことを特徴とする請求項1~5のいずれかに記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。
- 前記ピューロマイシンの類縁化合物は、3’-N-アミノアシルピューロマイシン及び3’-N-アミノアシルアデノシンアミノ酸のヌクレオシドからなる群から選ばれるいずれかの化合物である、ことを特徴とする請求項6に記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカー。
- 請求項1に記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーの主鎖と所望のmRNAとを相補結合させる相補結合形成工程と;
前記相補結合が形成された主鎖とmRNAとに、300~400nmの波長の光を0.5~5分間照射して光架橋を形成させる光架橋工程と;
前記リンカーに結合されたmRNAを無細胞翻訳系で翻訳し、前記mRNAに対応するタンパク質を前記リンカーのタンパク質結合部位に結合させ、mRNA-タンパク質の融合体を形成する融合体形成工程と;
前記融合体を固相に結合させる固相結合工程と;
前記融合体に含まれるmRNAを逆転写し、前記融合体と逆転写されたcDNAとの結合体を形成する逆転写工程と;
前記結合体を固相切断部位から切断し、所望のcDNAディスプレイ法を選択する選択工程と;
を備える、試験管内淘汰方法。 - 前記固相は、ストレプトアビジン又はアビジンでコーティングされた磁性粒子である、ことを特徴とする請求項8に記載の試験管内淘汰方法。
- 前記選択工程における前記結合体の切断は、エンドヌクレアーゼV、RNase T1、及びRNase Aからなる群から選ばれるいずれかの酵素で行う、ことを特徴とする請求項8又は9に記載の試験管内淘汰方法。
- 前記高速光架橋型共用リンカーの主鎖は、糖鎖抗原を認識する配列を有する、ことを特徴とする、請求項8~10のいずれかに記載の試験管内淘汰方法。
- 請求項1に記載の試験管内淘汰及び分子間相互作用解析のための高速光架橋型共用リンカーの主鎖と所望のmRNAとを相補結合させる相補結合形成工程と;
前記相補結合が形成された主鎖とmRNAとに、300~400nmの波長の光を0.5~5分間照射して光架橋を形成させる光架橋工程と;
前記リンカーに結合されたmRNAを無細胞翻訳系で翻訳し、前記mRNAに対応するタンパク質を前記リンカーのタンパク質結合部位に結合させ、mRNA-タンパク質の融合体を形成する融合体形成工程と;
前記融合体をRNAで消化してリンカー-タンパク質融合体とする、リンカー-タンパク質融合体形成工程と;
前記リンカー-タンパク質融合体を固相に結合させる固相結合工程と;
前記リンカー-タンパク質融合体を所定の条件下で前記固相から溶出させ、精製する精製工程と;
を備える親和性測定用リンカー-タンパク質の作製方法。 - 前記固相は、ストレプトアビジン又はアビジンでコーティングされた磁性粒子である、ことを特徴とする請求項12に記載の親和性測定用リンカー-タンパク質の作製方法。
- 前記精製工程は、0~100mM NaClを含む水溶液中、室温にて行う、ことを特徴とする請求項12又は13に記載の親和性測定用リンカー-タンパク質の作製方法。
- 請求項12~14のいずれかに記載の方法で作製された親和性測定用リンカー-タンパク質。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017510169A JP6506838B2 (ja) | 2015-03-31 | 2016-03-31 | 試験管内淘汰及び分子間相互作用解析用高速光架橋型共用リンカー、そのリンカーを用いた試験管内淘汰方法 |
| US15/719,979 US20180100146A1 (en) | 2015-03-31 | 2017-09-29 | High-speed photo-cross-linking linker for molecular interaction analysis and in vitro selection, and in vitro selection method using linker |
| US18/368,817 US20240002838A1 (en) | 2015-03-31 | 2023-09-15 | High-speed photo-cross-linking linker for molecular interaction analysis and in vitro selection, and in vitro selection method using linker |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-072810 | 2015-03-31 | ||
| JP2015072810 | 2015-03-31 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/719,979 Continuation US20180100146A1 (en) | 2015-03-31 | 2017-09-29 | High-speed photo-cross-linking linker for molecular interaction analysis and in vitro selection, and in vitro selection method using linker |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016159211A1 true WO2016159211A1 (ja) | 2016-10-06 |
Family
ID=57004394
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/060611 WO2016159211A1 (ja) | 2015-03-31 | 2016-03-31 | 試験管内淘汰及び分子間相互作用解析用高速光架橋型共用リンカー、そのリンカーを用いた試験管内淘汰方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (2) | US20180100146A1 (ja) |
| JP (1) | JP6506838B2 (ja) |
| WO (1) | WO2016159211A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021182587A1 (ja) * | 2020-03-12 | 2021-09-16 | 株式会社Epsilon Molecular Engineering | 光架橋塩基を活用した、新規ペプチドアプタマー探索用標的モジュール、新規ペプチドアプタマー探索用リンカー及びそれらを用いる新規ペプチドアプタマーの探索方法 |
| WO2022102791A1 (ja) * | 2020-11-16 | 2022-05-19 | 国立大学法人埼玉大学 | 新規抗原検出用リンカー及びそれを用いた抗原検出方法 |
| WO2023188623A1 (ja) * | 2022-03-29 | 2023-10-05 | 株式会社トクヤマ | リンカー、およびキット |
| JP7475056B2 (ja) | 2019-01-30 | 2024-04-26 | 国立大学法人北陸先端科学技術大学院大学 | 光応答性ヌクレオチドアナログの製造方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110824168B (zh) * | 2018-08-10 | 2023-06-09 | 天津医科大学 | 一种多肽探针及其在翻译后修饰的结合蛋白鉴定中的应用 |
| CN114585736B (zh) * | 2020-10-27 | 2023-05-02 | 北京寻因生物科技有限公司 | 一种嘌呤霉素连接子及其在体外核酸展示肽合成中的应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011087573A (ja) * | 2009-09-24 | 2011-05-06 | Saitama Univ | cDNA/mRNA−タンパク質連結体の効率的合成法 |
| WO2014142020A1 (ja) * | 2013-03-13 | 2014-09-18 | 国立大学法人東京大学 | 核酸リンカー |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1816192B1 (en) * | 2004-10-15 | 2012-12-12 | National Institute of Advanced Industrial Science and Technology | LINKER FOR CONSTRUCTING mRNA-PUROMYCIN-PROTEIN CONJUGATE |
| EP3450554B1 (en) * | 2016-03-30 | 2022-09-21 | Epsilon Molecular Engineering Inc. | High-speed in vitro screening method |
-
2016
- 2016-03-31 JP JP2017510169A patent/JP6506838B2/ja active Active
- 2016-03-31 WO PCT/JP2016/060611 patent/WO2016159211A1/ja active Application Filing
-
2017
- 2017-09-29 US US15/719,979 patent/US20180100146A1/en not_active Abandoned
-
2023
- 2023-09-15 US US18/368,817 patent/US20240002838A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011087573A (ja) * | 2009-09-24 | 2011-05-06 | Saitama Univ | cDNA/mRNA−タンパク質連結体の効率的合成法 |
| WO2014142020A1 (ja) * | 2013-03-13 | 2014-09-18 | 国立大学法人東京大学 | 核酸リンカー |
Non-Patent Citations (5)
| Title |
|---|
| MOCHIZUKI Y. ET AL.: "A versatile puromycin- linker using cnvK for high-throughput in vitro selection by cDNA display", JOURNAL OF BIOTECHNOLOGY, vol. 212, 28 August 2015 (2015-08-28), pages 174 - 180, XP029284068 * |
| MOCHIZUKI Y. ET AL.: "Increasing the library size in cDNA display by optimizing purification procedures", BIOLOGICAL PROCEDURES ONLINE, vol. 15, no. 7, 2013, pages 1 - 5, XP021152377 * |
| MOCHIZUKI Y. ET AL.: "One-Pot Preparation of mRNA/cDNA Display by a Novel and Versatile Puromycin-Linker DNA", ACS COMBINATORIAL SCIENCE, vol. 13, no. 5, 2011, pages 478 - 485, XP055277429 * |
| NEMOTO N. ET AL.: "Versatile C-Terminal Specific Biotinylation of Proteins Using Both a Puromycin-Linker and a Cell -Free Translation System for Studying High-Throughput Protein- Molecule Interactions", ANALYTICAL CHEMISTRY, vol. 86, no. 17, 2 September 2014 (2014-09-02), pages 8535 - 8540, XP055316565 * |
| USNO S. ET AL.: "Improvement of a puromycin- linker to extend the selection target varieties in cDNA display method", JOURNAL OF BIOTECHNOLOGY, vol. 162, no. 2-3, 2012, pages 299 - 302, XP055277425 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7475056B2 (ja) | 2019-01-30 | 2024-04-26 | 国立大学法人北陸先端科学技術大学院大学 | 光応答性ヌクレオチドアナログの製造方法 |
| WO2021182587A1 (ja) * | 2020-03-12 | 2021-09-16 | 株式会社Epsilon Molecular Engineering | 光架橋塩基を活用した、新規ペプチドアプタマー探索用標的モジュール、新規ペプチドアプタマー探索用リンカー及びそれらを用いる新規ペプチドアプタマーの探索方法 |
| WO2022102791A1 (ja) * | 2020-11-16 | 2022-05-19 | 国立大学法人埼玉大学 | 新規抗原検出用リンカー及びそれを用いた抗原検出方法 |
| WO2023188623A1 (ja) * | 2022-03-29 | 2023-10-05 | 株式会社トクヤマ | リンカー、およびキット |
Also Published As
| Publication number | Publication date |
|---|---|
| US20180100146A1 (en) | 2018-04-12 |
| JPWO2016159211A1 (ja) | 2018-02-01 |
| US20240002838A1 (en) | 2024-01-04 |
| JP6506838B2 (ja) | 2019-05-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20240002838A1 (en) | High-speed photo-cross-linking linker for molecular interaction analysis and in vitro selection, and in vitro selection method using linker | |
| AU2008275915B2 (en) | Method for generating aptamers with improved off-rates | |
| US7947447B2 (en) | Method for generating aptamers with improved off-rates | |
| CA2611198C (en) | Methods for the selection of aptamers | |
| EP1477561A2 (en) | Molecule assigning genotype to phenotype and use thereof | |
| JP7026248B2 (ja) | 二本鎖dnaを増幅するための方法およびキット | |
| EP2589657A9 (en) | Method for detection of target molecule | |
| US20140235508A1 (en) | Nucleic acid linker | |
| JP2008253176A (ja) | 高親和性分子取得のためのリンカー | |
| JP2023551072A (ja) | Rnaおよびdna修飾の多重プロファイリング | |
| JP5733784B2 (ja) | cDNA/mRNA−タンパク質連結体の効率的合成法 | |
| US20190093156A1 (en) | Methods of identifying multiple epitopes in cells | |
| JP2011217708A (ja) | 核酸リンカー | |
| WO2013065782A1 (ja) | タンパク質固定化固相、及びポリヌクレオチド固定化固相、並びに、核酸回収方法 | |
| EP3262185B1 (en) | Dna display and methods thereof | |
| JP6478392B2 (ja) | 核酸リンカー | |
| WO2022102791A1 (ja) | 新規抗原検出用リンカー及びそれを用いた抗原検出方法 | |
| WO2020102287A1 (en) | Compositions and methods for the synthesis and identification of covalent aptamers | |
| WO2021182587A1 (ja) | 光架橋塩基を活用した、新規ペプチドアプタマー探索用標的モジュール、新規ペプチドアプタマー探索用リンカー及びそれらを用いる新規ペプチドアプタマーの探索方法 | |
| JP2015227806A (ja) | 磁性体ゲルビーズを用いた標的分子の高感度検出方法及び検出用キット | |
| KR101069590B1 (ko) | Cea에 특이적으로 결합하는 단일 가닥 dna 압타머 | |
| JP5858415B2 (ja) | mRNA/cDNA−タンパク質連結体作製用リンカーとそれを用いたヌクレオチド−タンパク質連結体の精製方法 | |
| JP6194894B2 (ja) | 核酸リンカー | |
| WO2013094080A1 (ja) | ヒト心筋由来トロポニンiに特異的にかつ速やかに結合できる組み換えタンパク質 | |
| KR101069591B1 (ko) | Cea에 특이적으로 결합하는 단일 가닥 dna 압타머 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16773091 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2017510169 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16773091 Country of ref document: EP Kind code of ref document: A1 |