WO2016101211A1 - 一种盐酸沙丙喋呤的制备方法 - Google Patents
一种盐酸沙丙喋呤的制备方法 Download PDFInfo
- Publication number
- WO2016101211A1 WO2016101211A1 PCT/CN2014/094961 CN2014094961W WO2016101211A1 WO 2016101211 A1 WO2016101211 A1 WO 2016101211A1 CN 2014094961 W CN2014094961 W CN 2014094961W WO 2016101211 A1 WO2016101211 A1 WO 2016101211A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrochloride
- reaction
- acyl
- sapropium
- deoxy
- Prior art date
Links
- 0 COC([C@@]([*+])[C@@](*)C(Cl)=O)=O Chemical compound COC([C@@]([*+])[C@@](*)C(Cl)=O)=O 0.000 description 5
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
- C07D475/02—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4
- C07D475/04—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4 with a nitrogen atom directly attached in position 2
Definitions
- the invention belongs to the technical field of drug synthesis, and particularly relates to the preparation of sapropium hydrochloride.
- the oxaprozin hydrochloride was developed by Merck and was launched in the United States and the European Union in 2007 and 2008 under the trade name Kuvan. This product can be used to treat hyperphenylalaninemia (HPA) caused by tetrahydrobiopterin (BH4) deficiency.
- HPA hyperphenylalaninemia
- BH4 tetrahydrobiopterin
- the oxaprozin hydrochloride can be obtained by hydrogenation of L-erythrobiopterin.
- the literature Liebigs Ann. Chem. 1989, 1267-1269 reports the preparation of L-erythrobiopterin starting from L-ribose. The preparation route is as follows:
- the reducing agent used in the route of the acid chloride to reduce the aldehyde is bis(triphenylphosphine) copper borohydride (I), which has a high price and is not favorable for the control of industrialization cost.
- the reaction temperature of the format reagent with carbonyl addition and lactone reduction is -78 ° C, and the energy consumption in industrial production is high.
- the post-treatment of the multi-step reaction uses silica gel column color The spectrum is purified and it is difficult to achieve industrialization. Therefore, this route has great disadvantages in terms of cost and operability in industrial production.
- the invention provides a preparation method of tetrahydrobiopterin hydrochloride.
- the method adopts cheap L-arabinose as a starting material, the reaction conditions are mild in each step, the operation is simple and easy, the materials used are cheap and easy to obtain, and the production cost is lowered.
- One embodiment of the present invention is to obtain an intermediate 5-deoxy-L-ribose (compound 5) by L-arabinose glycosidation, 5-position hydroxyiodo, reductive deiodination and glycosidic bond to obtain an intermediate 5-deoxy-L-ribose (compound 5).
- the target compound I oxaprozin hydrochloride
- the 5-position hydroxy iodide may be any means known to those skilled in the art, such as activation of the hydroxyl group at the 5-position and subsequent addition of iodine to the iodo reaction.
- any feasible synthetic route can be reported, such as Liebigs Ann. Chem. 1989, 1267-1269, WO2009/088979, etc., to obtain the final product, sapropene hydrochloride.
- One embodiment of the present invention is to obtain an intermediate 5-deoxy-L-ribose (compound 5) by L-arabinose glycosidation, 5-position hydroxyiodo, reductive deiodination and glycosidic bond to obtain an intermediate 5-deoxy-L-ribose (compound 5).
- Got To the target compound I oxaprozin hydrochloride. Among them, it is activated by a substituted sulfonyl chloride hydroxyl group, and then iodine is added to carry out an iodo reaction.
- the synthetic route for one embodiment of the scheme is as follows:
- R is hydrogen or a hydrocarbon of C1 to C8, and R' is a substituted sulfonyl chloride.
- the present invention includes, but is not limited to, the preparation methods shown in the above synthetic routes.
- a further technical solution of the present invention is an acyl-substituted L-erythrobiopter, which is then acylated with a primary amino group, and then deacylated to obtain an L-erythrobiopterin, which is reduced to obtain the target compound I, namely hydrochloric acid.
- the acyl substituted erythrobiopterin can be synthesized by the methods mentioned in the relevant literature.
- the synthetic route for one embodiment of the scheme is as follows:
- R' ⁇ is a substituted acyl group.
- the present invention includes, but is not limited to, the preparation methods shown in the above synthetic routes.
- the acyl-substituted erythrophysin can directly deacylate the product L-erythrobiopterin and then reduce to form sapphire hydrochloride.
- the acylation product has a certain purification effect by increasing the acylation step. And it is equivalent to the quality of the direct deacylated product L-erythrobio.
- the acyl substituted erythrobiopterin can be synthesized by the methods mentioned in the relevant literature.
- Another technical solution of the present invention is a method for preparing oxaprozin hydrochloride, which is obtained by hydrolyzing L-arabinose, 5-hydroxyl iodide and deiodinated glycosidic bonds to obtain an intermediate 5-deoxy-L- After the ribose (compound 5) is further reacted to form an acyl-substituted L-erythrobiopene, the acyl group is deacylated to obtain an L-erythrobiopterin, and the target compound I, i.e., sapropene hydrochloride, is obtained by reduction.
- the synthetic route for one embodiment of the scheme is as follows:
- R is hydrogen or a hydrocarbon of C1 to C8
- R' is a substituted sulfonyl chloride
- R' ⁇ is a substituted acyl group.
- the invention includes, but is not limited to, the preparation methods shown in the synthetic routes above.
- Another technical solution of the present invention is a method for preparing oxaprozin hydrochloride, which is obtained by hydrolyzing L-arabinose, 5-hydroxy iodine, and reducing deiodinated glycosidic bonds to obtain an intermediate 5-deoxy-L. -ribose (compound 5), after reacting to form an acyl-substituted L-erythrobiopeptide, acylating the primary amino group, and then removing the acyl group to obtain an L-erythrobiopterin, and reducing to obtain the target compound I, Saxoprofen hydrochloride.
- the synthetic route for one embodiment of the scheme is as follows:
- R is hydrogen or a hydrocarbon of C1 to C8;
- R' is a substituted sulfonyl chloride; and
- R' ⁇ is a substituted acyl group.
- This scheme includes, but is not limited to, the preparation methods shown in the above synthetic routes.
- the acyl-substituted L-erythrobiopterin (compound 9) is converted to the compound 9' by reaction with an acid anhydride or an acid chloride, followed by deacylation; the acylating agent is preferably an acid anhydride, more preferably acetic anhydride; deacylation
- the reaction conditions are the same as the direct deacylation conditions.
- Another technical scheme of the present invention is to obtain a key intermediate 5-deoxy-L-ribose by performing a glycosidation reaction, a 5-position hydroxy iodide, a reduction deiodination, and a glycosidic bond to remove an alkyl group by L-arabinose.
- ⁇ hydroxy acylation protection
- docking with 2,4,5-triaminopyrimidinone and aromatization
- direct removal of acyl (or primary amino acylation followed by acyl removal) to obtain L-erythrobiopterin
- the L-erythrobiopterin is subjected to a reduction reaction to obtain sapropium hydrochloride.
- One embodiment of the present scheme synthetic route includes the following steps:
- R is hydrogen or a C1 to C8 hydrocarbon; R' is a substituted sulfonyl chloride; and R'' is a substituted acyl group.
- This scheme includes, but is not limited to, the preparation methods shown in the above synthetic routes.
- step 1 of the scheme the glycosidation reaction is carried out under acidic conditions with L-arabinose as a starting material.
- the alcohol suitable for the reaction in this step is an alcohol of C1 to C8, preferably methanol or ethanol.
- the ratio of L-arabinose to ROH is from 1:5 to 1:50, and is preferably from 1:15 to 1:30.
- the acid suitable for the reaction of this step is an alcohol solution of sulfuric acid, hydrochloric acid or hydrochloric acid, preferably sulfuric acid.
- the base suitable for the reaction in this step is sodium carbonate, sodium hydroxide, sodium methoxide, sodium ethoxide, sodium t-butoxide, potassium carbonate, potassium hydroxide or potassium t-butoxide, preferably sodium carbonate.
- the temperature suitable for the reaction in this step is from 0 ° C to 50 ° C, preferably from 20 ° C to 30 ° C.
- Step 2 The acetal 1 formed by the reaction of the previous step is esterified with a sulfonyl chloride under basic conditions to form a sulfonyl ester 2.
- the sulfonyl chloride suitable for the reaction of this step is p-toluenesulfonyl chloride, methylsulfonyl chloride or trifluoromethylsulfonyl chloride, preferably p-toluenesulfonyl chloride.
- the base suitable for the reaction in this step is triethylamine, diisopropylethylamine, 1,8-diazabicycloundec-7-ene, pyridine, 2,6-lutidine, 4-di Methylaminopyridine, preferably pyridine.
- Suitable solvents for the reaction of this step are acetonitrile, tetrahydrofuran, pyridine, preferably acetonitrile.
- Step 3 The sulfonyl ester 2 formed by the reaction of the previous step is substituted with an iodinating reagent in a polar aprotic solvent to form an iodine sugar 3.
- the iodinating reagent suitable for the reaction in this step is sodium iodide, potassium iodide or iodine, preferably sodium iodide.
- the polar aprotic solvent suitable for the reaction of this step is acetonitrile, tetrahydrofuran, acetone, methyl ethyl ketone, methyl isobutyl ketone, 2-pentanone, 3-pentanone, preferably butanone.
- step 4 the iodine 3 formed by the reaction of the previous step undergoes deiodination reaction under the catalysis of a metal catalyst to form deoxypentose 4.
- the metal catalyst suitable for the reaction of this step is Pd/C, Rh/C, Raney nickel, preferably Pd/C.
- the hydrogen pressure suitable for the reaction in this step is 0.1 to 1 MPa, preferably 0.1 MPa.
- Step 5 Deoxypentose 4 formed by the reaction of the previous step is hydrolyzed to remove 5-alkyl-de-arabinose under acidic conditions.
- the acid suitable for the reaction of this step is an alcohol solution of sulfuric acid, hydrochloric acid or hydrochloric acid, preferably sulfuric acid.
- the temperature suitable for the reaction of this step is from 20 ° C to 100 ° C, preferably 75 ° C.
- Step 6 5-deoxy-L-arabinose formed by the reaction of the previous step is condensed with phenylhydrazine under acidic conditions to form benzoquinone 6.
- the ratio of 5-deoxy-L-arabinose to phenylhydrazine suitable for the reaction of the present step is from 1:1 to 1:2, preferably 1:2.
- the purified solvent suitable for the reaction in this step is methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, isobutyl acetate, t-butyl acetate, petroleum ether, n-pentane, cyclopentane, n-hexane, A solvent of cyclohexane and n-heptane or a mixture of several solvents, preferably ethyl acetate and n-hexane.
- Step 7 is carried out by reacting product 6 with an acid anhydride or acid chloride to convert to the corresponding ester compound 7; preferably an acid anhydride, more preferably acetic anhydride; the reaction temperature of product 6 with acetic anhydride is 5 to 50 ° C, preferably 15 to 25 ° C .
- Step 8 is carried out by reacting product 7 with compound 8 (i.e., 2,4,5-triaminopyrimidinone or a salt thereof), and aromatization reaction is carried out without oxidizing to obtain product 9;
- suitable solvent for this step is methanol and A mixture of water;
- the aromatization agent is an oxidizing agent, and the oxidizing agent is preferably 30% hydrogen peroxide.
- Step 9 can be directly subjected to deacylation reaction to obtain product 10;
- suitable solvent for this step is a mixture of n-butanol and water, and the acid binding agent is an organic base, preferably triethylamine or diethylamine.
- Another embodiment of the present invention is to obtain a key intermediate 5-deoxy-L-ribose by aglycosylation reaction, hydroxyl activation, iodo, reduction, and glycosidic bond hydrolysis to remove the alkyl group, and then to form a key intermediate 5-deoxy-L-ribose.
- One embodiment of the present scheme synthetic route includes the following steps:
- R is hydrogen or a C1 to C8 hydrocarbon; R' is a substituted sulfonyl chloride; and R'' is a substituted acyl group.
- This scheme includes, but is not limited to, the preparation methods shown in the above synthetic routes.
- step 1 of the scheme the glycosidation reaction is carried out under acidic conditions with L-arabinose as a starting material.
- the alcohol suitable for the reaction in this step is an alcohol of C1 to C8, preferably methanol or ethanol.
- the ratio of L-arabinose to ROH is from 1:5 to 1:50, and is preferably from 1:15 to 1:30.
- the acid suitable for the reaction of this step is an alcohol solution of sulfuric acid, hydrochloric acid or hydrochloric acid, preferably sulfuric acid.
- the base suitable for the reaction in this step is sodium carbonate, sodium hydroxide, sodium methoxide, sodium ethoxide, sodium t-butoxide, potassium carbonate, potassium hydroxide or potassium t-butoxide, preferably sodium carbonate.
- the temperature suitable for the reaction in this step is from 0 ° C to 50 ° C, preferably from 20 ° C to 30 ° C.
- Step 2 The acetal 1 formed by the reaction of the previous step is esterified with a sulfonyl chloride under basic conditions to form a sulfonyl ester 2.
- the sulfonyl chloride suitable for the reaction of this step is p-toluenesulfonyl chloride, methylsulfonyl chloride or trifluoromethylsulfonyl chloride, preferably p-toluenesulfonyl chloride.
- the base suitable for the reaction in this step is triethylamine, diisopropylethylamine, 1,8-diazabicycloundec-7-ene, pyridine, 2,6-lutidine, 4-di Methylaminopyridine, preferably pyridine.
- Suitable solvents for the reaction of this step are acetonitrile, tetrahydrofuran, pyridine, preferably acetonitrile.
- Step 3 The sulfonyl ester 2 formed by the reaction of the previous step is substituted with an iodinating reagent in a polar aprotic solvent to form an iodine sugar 3.
- the iodinating reagent suitable for the reaction in this step is sodium iodide, potassium iodide or iodine, preferably sodium iodide.
- the polar aprotic solvent suitable for the reaction of this step is acetonitrile, tetrahydrofuran, acetone, methyl ethyl ketone, methyl isobutyl ketone, 2-pentanone, 3-pentanone, preferably butanone.
- step 4 the iodine 3 formed by the reaction of the previous step undergoes deiodination reaction under the catalysis of a metal catalyst to form deoxypentose 4.
- the metal catalyst suitable for the reaction of this step is Pd/C, Rh/C, Raney nickel, preferably Pd/C.
- the hydrogen pressure suitable for the reaction in this step is 0.1 to 1 MPa, preferably 0.1 MPa.
- Step 5 Deoxypentose 4 formed by the reaction of the previous step is hydrolyzed to remove 5-alkyl-de-arabinose under acidic conditions.
- the acid suitable for the reaction of this step is an alcohol solution of sulfuric acid, hydrochloric acid or hydrochloric acid, preferably sulfuric acid.
- the temperature suitable for the reaction of this step is from 20 ° C to 100 ° C, preferably 75 ° C.
- Step 6 5-deoxy-L-arabinose formed by the reaction of the previous step is condensed with phenylhydrazine under acidic conditions to form benzoquinone 6.
- the ratio of 5-deoxy-L-arabinose to phenylhydrazine suitable for the reaction of the present step is from 1:1 to 1:2, preferably 1:2.
- the purified solvent suitable for the reaction in this step is methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, isobutyl acetate, t-butyl acetate, petroleum ether, n-pentane, cyclopentane, n-hexane, A solvent of cyclohexane and n-heptane or a mixture of several solvents, preferably ethyl acetate and n-hexane.
- Step 7 is carried out by reacting product 6 with an acid anhydride or acid chloride to convert to the corresponding ester compound 7; preferably an acid anhydride, more preferably acetic anhydride; the reaction temperature of product 6 with acetic anhydride is 5 to 50 ° C, preferably 15 to 25 ° C .
- Step 8 is carried out by reacting product 7 with compound 8 (i.e., 2,4,5-triaminopyrimidinone or a salt thereof), and aromatization reaction is carried out without oxidizing to obtain product 9;
- suitable solvent for this step is methanol and A mixture of water;
- the aromatization agent is an oxidizing agent, and the oxidizing agent is preferably 30% hydrogen peroxide.
- Step 9 is carried out by reacting product 9 with an acid anhydride or acid chloride to convert to compound 9'; followed by deacylation;
- the acylating agent is preferably an acid anhydride, more preferably acetic anhydride;
- the deacylation reaction conditions are the same as the direct deacylation conditions.
- the acylation product has a certain purification effect by increasing the acylation step. However, it is equivalent to the mass of the direct deacylation product 10.
- the L-erythrophyte is reduced to obtain the target compound I, namely sapropine hydrochloride.
- the solvent suitable for the reaction in this step is water;
- the oxidizing agent is a metal oxidizing agent, preferably a palladium and platinum oxidizing agent, more preferably platinum dioxide;
- the acid binding agent is an organic base, preferably triethylamine, diisopropylethylamine, and Ethylamine, pyridine, substituted pyridine, etc., more preferably triethylamine, diisopropylethylamine, diethylamine, etc.; reaction pressure is 0.5 to 3 MPa, preferably 1 to 2.5 MPa, more preferably 1.5 to 2.0 MPa. .
- the product 1, 4 L of pyridine and 5 L of acetonitrile were added to the reaction flask and dissolved by mechanical stirring.
- the mixture was cooled by stirring, and a solution of 1.8 kg of p-toluenesulfonyl 5 L acetonitrile was added dropwise at a temperature of 0 to 5 ° C.
- the reaction was stirred at room temperature 20-25 ° C for 4 hours.
- the TLC monitors the reaction.
- the product 9' prepared in Example 11 was added to the reaction flask, 600 ml of purified water, 450 ml of n-butanol, and 120 ml of diethylamine were added, and the mixture was stirred and heated to 45-50 ° C for 16 hours. After the TLC reaction is completed, the layers are separated, and the aqueous layer is separated to obtain an aqueous solution of the product 10, which is directly reacted in the next step.
- An aqueous solution of the product 10 prepared in Example 12 was added to the hydrogenation vessel, and 80 ml of triethylamine, 3 g of platinum dioxide was added thereto with stirring, and vacuum was applied thereto, and the pressure was controlled to 1.5 MPa, and the reaction was stirred at 35 ° C for 20 hours. After filtration, the filtrate was added to 45 ml of n-butanol for 5 min, and the mixture was allowed to stand to give an aqueous solution of product I. After cooling at 10 ° C, 300 ml of concentrated hydrochloric acid was added dropwise, and the aqueous solution was concentrated under reduced pressure to dryness.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Saccharide Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明属于药物合成技术领域,具体涉及盐酸沙丙喋呤的制备方法。该方法将L-阿拉伯糖糖苷化、5位羟基碘代、还原脱碘、糖苷键水解脱除烷基得到5-脱氧-L-核糖,再经反应得到L-赤式生物喋呤,还原L-赤式生物喋呤得到盐酸沙丙喋呤。该制备方法各步反应条件温和,操作简单易行;采用的物料价廉易得,降低了成本。
Description
本发明属于药物合成技术领域,具体涉及盐酸沙丙喋呤的制备。
盐酸沙丙喋呤由Merck公司研发,于2007年和2008年,分别在美国及欧盟上市,商品名为Kuvan。该产品可以用于治疗四氢生物喋呤(BH4)缺乏症所导致的高苯丙氨酸血症(HPA)。结构式如下:

化学名称为:(6R)-2-氨基-6-[(1R,2S)-1,2-二羟基丙基]-5,6,7,8-四氢-4(1H)-喋呤酮二盐酸盐。
盐酸沙丙喋呤可由L-赤式生物喋呤进行氢化得到的。文献Liebigs Ann.Chem.1989,1267-1269报道的了以L-核糖为起始物料制备L-赤式生物喋呤的方法,制备路线如下图:

该方法虽然简单易行,是一条较好的制备路线,但缺点在于起始物料L-核糖价格较高,因而导致盐酸沙丙喋呤成本很高。
文献Helv.Chim.Acta,1985,1639-1643、US2011218339A等报道制备L-赤式生物喋呤的方法是由腙类化合物6进行乙酰化反应的产物与2,4,5-三氨基嘧啶酮在甲醇/水/吡啶体系中进行环合,然后采用碘试剂进行芳构化反应得到乙酰化的L-
赤式生物喋呤,再经过水解脱乙酰基得到L-赤式生物喋呤。反应方程式如下所示:

其中化合物6做为关键中间体,其制备方法的报道有许多。文献J.Am.Chem.Soc.1974,6781-6782、J.Am.Chem.Soc.1976,2301-2307等报道的方法以L-鼠李糖为原料,与乙硫醇反应生成相应的缩硫醛,用氧化剂将缩硫醛氧化为砜,砜在碱性条件下脱除一个碳得到5-脱氧-L-阿拉伯糖,5-脱氧-L-阿拉伯糖与苯肼反应得到关键中间体式6。合成路线如下所示:

虽然这一方法被多次改进和完善,但使用的乙硫醇具有特殊的恶臭,需要使用除臭装置,其较低的沸点也给生产带来诸多不便。
文献J.Org.Chem.1996,8699-8700报道了以L-酒石酸为起始原料,经羟基保护、羧基转化、还原、加成、脱保护得到5-脱氧-L-核糖、5-脱氧-L-阿拉伯糖与苯肼缩合得到关键中间体,合成路线如下所示:
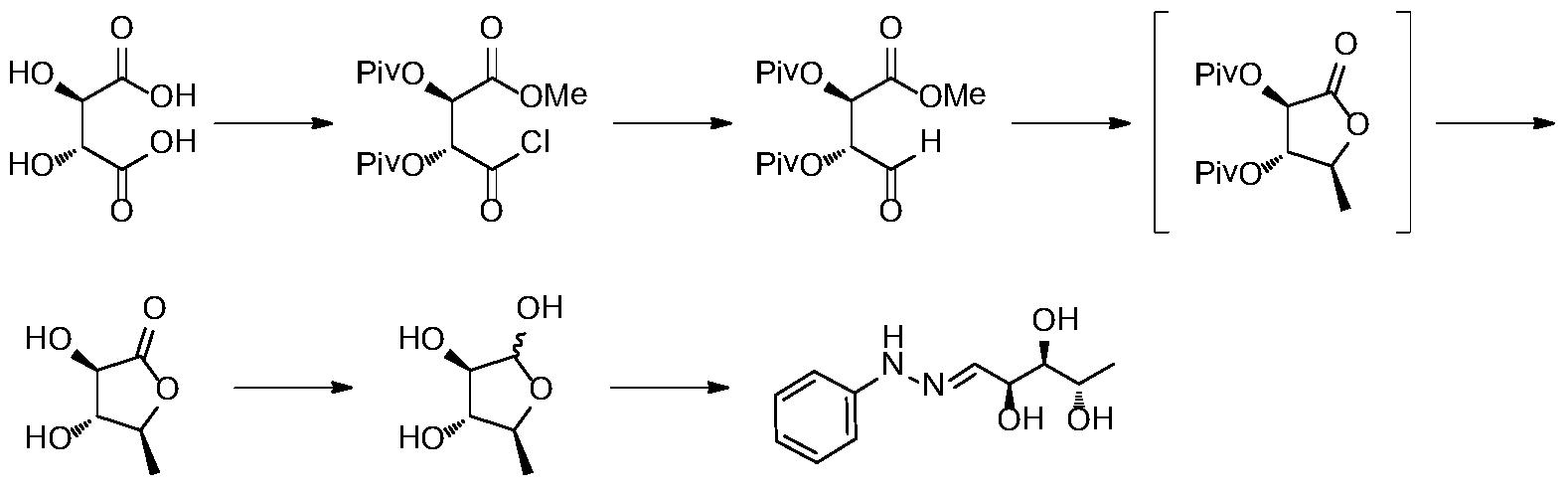
该路线在酰氯还原醛时所用还原剂为双(三苯基膦)硼氢化铜(I),该还原试剂价格较高,不利于工业化成本的控制。格式试剂与羰基加成及内酯还原时反应温度为-78℃,工业生产时能耗较高。此外,多步反应的后处理使用硅胶柱色
谱纯化,难以实现工业化。因此这条路线无论从成本上还是操作性上在工业化生产时都存在较大的不利因素。
文献CN201010151443.2报道了以L-阿拉伯糖为起始物料,经过多步反应得到L-赤式生物喋呤。该制备路线如下:

在重现该制备方法中,我们发现在对中间体1直接进行还原脱磺酰反应制备中间体2存在收率低,产品纯度低,产品不易纯化的缺点。因此有必要找到一种简单可行低成本的制备路线。
发明内容:
本发明提供了盐酸四氢生物喋呤的制备方法。该方法以价格便宜的L-阿拉伯糖为起始原料,各步反应条件温和,操作简单易行,采用的物料价廉易得,降低了生产成本。
本发明的一个技术方案为将L-阿拉伯糖糖苷化、5位羟基碘代、还原脱碘并糖苷键水解脱除烷基得到中间体5-脱氧-L-核糖(化合物5),再经过反应得到目标化合物I,即盐酸沙丙喋呤。本发明中5位羟基碘代可以使用对5位羟基进行活化然后加入碘进行碘代反应等本领域技术人员所知的任何手段。从中间体5-脱氧-L-核糖(化合物5)可以采用如Liebigs Ann.Chem.1989,1267-1269,WO2009/088979等报道任何可行的合成路径得到终产品盐酸沙丙喋呤。
本发明的一个技术方案为将L-阿拉伯糖糖苷化、5位羟基碘代、还原脱碘并糖苷键水解脱除烷基得到中间体5-脱氧-L-核糖(化合物5),再经过反应得
到目标化合物I,即盐酸沙丙喋呤。其中,用取代的磺酰氯羟基活化,然后加入碘进行碘代反应。本方案的一个实施方案合成路线如下所示:

这里,R为氢或C1~C8的烃类,R`为取代的磺酰氯。本发明包括但不限于上述合成路线所示制备方法。
本发明的又一个技术方案为先生成酰基取代的L-赤式生物喋呤,然后将伯胺基酰化,再脱去酰基得到L-赤式生物喋呤,还原得到目标化合物Ⅰ,即盐酸沙丙喋呤。酰基取代的赤式生物喋呤可以用相关文献提到的方法合成。本方案的一个实施方案合成路线如下所示:

这里,R``为取代的酰基。本发明包括但不限于上述合成路线所示制备方法。
酰基取代的赤式生物喋呤可以直接脱酰化产物L-赤式生物喋呤,再还原生成盐酸沙丙喋呤。但通过增加酰化步骤对酰化产品有一定的提纯作用。且与直接脱酰化产物L-赤式生物喋呤质量相当。酰基取代的赤式生物喋呤可以用相关文献提到的方法合成。
本发明的又一个技术方案是制备盐酸沙丙喋呤的方法,将L-阿拉伯糖糖苷化、5位羟基碘代、脱碘并糖苷键水解脱除烷基得到中间体5-脱氧-L-核糖(化合物5),再经过反应生成酰基取代的L-赤式生物喋呤后,脱去酰基得到L-赤式生物喋呤,还原得到目标化合物I,即盐酸沙丙喋呤。本方案的一个实施方案合成路线如下所示:

其中,R为氢或C1~C8的烃类,R`为取代的磺酰氯,R``为取代的酰基。本发明包括但不限于上述合成路线所示的制备方法。
本发明的又一个技术方案是制备盐酸沙丙喋呤的方法,将L-阿拉伯糖糖苷化、5位羟基碘代、还原脱碘并糖苷键水解脱除烷基得到中间体5-脱氧-L-核糖(化合物5),再经过反应生成酰基取代的L-赤式生物喋呤后,将伯胺基酰化,再脱去酰基得到L-赤式生物喋呤,还原得到目标化合物I,即盐酸沙丙喋呤。本方案的一个实施方案合成路线如下所示:

这里,R为氢或C1~C8的烃类;R`为取代的磺酰氯;R``为取代的酰基。本方案包括但不限于上述合成路线所示制备方法。酰基取代的L-赤式生物喋呤(化合物9)经与酸酐或酰氯反应,转化为化合物9`,再进行脱酰化反应;酰化试剂优选为酸酐,更优选为醋酸酐;脱酰化反应条件同直接脱酰条件。
本发明的另一个技术方案是将L-阿拉伯糖经糖苷化反应、5位羟基碘代、还原脱碘、糖苷键水解脱除烷基反应得到关键中间体5-脱氧-L-核糖,再经过成
腙、羟基酰化保护、与2,4,5-三氨基嘧啶酮对接以及芳构化、直接脱除酰基(或先伯胺基酰化再脱除酰基)得到L-赤式生物喋呤,L-赤式生物喋呤经过还原反应得到盐酸沙丙喋呤。本方案的一个实施方案合成路线包括以下步骤:

其中R为氢或C1~C8的烃类;R`为取代的磺酰氯;R``为取代的酰基。本方案包括但不限于上述合成路线所示的制备方法。
方案中步骤1以L-阿拉伯糖为起始原料与醇在酸性条件下发生糖苷化反应。适合本步反应的醇为C1~C8的醇类,优选甲醇、乙醇。L-阿拉伯糖与ROH的比例为1:5~1:50均可以反应,优选为1:15~1:30。
适合本步反应的酸为硫酸、盐酸或盐酸的醇溶液,优选为硫酸。
适合本步反应的碱为碳酸钠、氢氧化钠、甲醇钠、乙醇钠、叔丁醇钠、碳酸钾、氢氧化钾、叔丁醇钾,优选为碳酸钠。
适合本步反应的温度为0℃~50℃,优选为20℃~30℃。
步骤2用前一步反应生成的缩醛1与磺酰氯在碱性条件下,发生酯化反应生成磺酰酯2。
适合本步反应的磺酰氯为对甲基苯磺酰氯、甲基磺酰氯、三氟甲基磺酰氯,优选为对甲基苯磺酰氯。
适合本步反应的碱为三乙胺、二异丙基乙基胺、1,8-二氮杂二环十一碳-7-烯、吡啶、2,6-二甲基吡啶、4-二甲氨基吡啶,优选为吡啶。
适合本步反应的溶剂为乙腈、四氢呋喃、吡啶,优选为乙腈。
步骤3用前一步反应生成的磺酰酯2在极性非质子性溶剂中与碘化试剂发生取代反应生成碘代糖3。
适合本步反应的碘化试剂为碘化钠、碘化钾、碘,优选为碘化钠。
适合本步反应的极性非质子性溶剂为乙腈、四氢呋喃、丙酮、丁酮、甲基异丁酮、2-戊酮、3-戊酮,优选为丁酮。
步骤4用前一步反应生成的碘代糖3在金属催化剂催化下发生脱碘反应生成脱氧戊糖4。
适合本步反应的金属催化剂为Pd/C,Rh/C,雷尼镍,优选为Pd/C。
适合本步反应的氢气压力为0.1~1MPa,优选0.1MPa。
步骤5用前一步反应生成的脱氧戊糖4在酸性条件下糖苷键水解脱除烷基生成5-脱氧-L-阿拉伯糖。
适合本步反应的酸为硫酸、盐酸或盐酸的醇溶液,优选为硫酸。
适合本步反应的温度为20℃~100℃,优选为75℃。
步骤6用前一步反应生成的5-脱氧-L-阿拉伯糖在酸性条件下与苯肼发生缩合反应生成苯腙6。
适合本步反应的5-脱氧-L-阿拉伯糖与苯肼的比例为1:1~1:2,优选为1:2。
适合本步反应的精制溶剂为乙酸甲酯、乙酸乙酯、乙酸异丙酯、乙酸正丁酯、乙酸异丁酯、乙酸叔丁酯、石油醚、正戊烷、环戊烷、正己烷、环己烷和正庚烷的一种溶剂或几种溶剂的混合物,优选为乙酸乙酯和正己烷。
步骤7用产物6与酸酐或酰氯反应,转化为相应的酯类化合物7;优选为酸酐,更优选为醋酸酐;产物6与醋酸酐的反应温度为5~50℃,优选为15~25℃。
步骤8用产物7与化合物8(即2,4,5-三氨基嘧啶酮或其盐类)反应,不分离再在氧化剂作用下芳构化反应得到产物9;适合本步反应溶剂为甲醇和水的混合物;芳构化试剂为氧化剂,氧化剂优选为30%的双氧水。
步骤9用产物9可以直接进行脱酰化反应得到产物10;适合本步反应溶剂为正丁醇和水的混合物,缚酸剂为有机碱,优选为三乙胺、二乙胺。
本发明的另一个实施方案是将L-阿拉伯糖经糖苷化反应、羟基活化、碘代、还原、糖苷键水解脱除烷基反应得到关键中间体5-脱氧-L-核糖,再经过成腙、羟基酰化保护、与2,4,5-三氨基嘧啶酮对接以及芳构化、全酰化再脱除酰基得到L-赤式生物喋呤,再还原反应得到盐酸沙丙喋呤。本方案的一个实施方案合成路线包括以下步骤:

其中R为氢或C1~C8的烃类;R`为取代的磺酰氯;R``为取代的酰基。本方案包括但不限于上述合成路线所示的制备方法。
方案中步骤1以L-阿拉伯糖为起始原料与醇在酸性条件下发生糖苷化反应。适合本步反应的醇为C1~C8的醇类,优选甲醇、乙醇。L-阿拉伯糖与ROH的比例为1:5~1:50均可以反应,优选为1:15~1:30。
适合本步反应的酸为硫酸、盐酸或盐酸的醇溶液,优选为硫酸。
适合本步反应的碱为碳酸钠、氢氧化钠、甲醇钠、乙醇钠、叔丁醇钠、碳酸钾、氢氧化钾、叔丁醇钾,优选为碳酸钠。
适合本步反应的温度为0℃~50℃,优选为20℃~30℃。
步骤2用前一步反应生成的缩醛1与磺酰氯在碱性条件下,发生酯化反应生成磺酰酯2。
适合本步反应的磺酰氯为对甲基苯磺酰氯、甲基磺酰氯、三氟甲基磺酰氯,优选为对甲基苯磺酰氯。
适合本步反应的碱为三乙胺、二异丙基乙基胺、1,8-二氮杂二环十一碳-7-烯、吡啶、2,6-二甲基吡啶、4-二甲氨基吡啶,优选为吡啶。
适合本步反应的溶剂为乙腈、四氢呋喃、吡啶,优选为乙腈。
步骤3用前一步反应生成的磺酰酯2在极性非质子性溶剂中与碘化试剂发生取代反应生成碘代糖3。
适合本步反应的碘化试剂为碘化钠、碘化钾、碘,优选为碘化钠。
适合本步反应的极性非质子性溶剂为乙腈、四氢呋喃、丙酮、丁酮、甲基异丁酮、2-戊酮、3-戊酮,优选为丁酮。
步骤4用前一步反应生成的碘代糖3在金属催化剂催化下发生脱碘反应生成脱氧戊糖4。
适合本步反应的金属催化剂为Pd/C,Rh/C,雷尼镍,优选为Pd/C。
适合本步反应的氢气压力为0.1~1MPa,优选0.1MPa。
步骤5用前一步反应生成的脱氧戊糖4在酸性条件下糖苷键水解脱除烷基生成5-脱氧-L-阿拉伯糖。
适合本步反应的酸为硫酸、盐酸或盐酸的醇溶液,优选为硫酸。
适合本步反应的温度为20℃~100℃,优选为75℃。
步骤6用前一步反应生成的5-脱氧-L-阿拉伯糖在酸性条件下与苯肼发生缩合反应生成苯腙6。
适合本步反应的5-脱氧-L-阿拉伯糖与苯肼的比例为1:1~1:2,优选为1:2。
适合本步反应的精制溶剂为乙酸甲酯、乙酸乙酯、乙酸异丙酯、乙酸正丁酯、乙酸异丁酯、乙酸叔丁酯、石油醚、正戊烷、环戊烷、正己烷、环己烷和正庚烷的一种溶剂或几种溶剂的混合物,优选为乙酸乙酯和正己烷。
步骤7用产物6与酸酐或酰氯反应,转化为相应的酯类化合物7;优选为酸酐,更优选为醋酸酐;产物6与醋酸酐的反应温度为5~50℃,优选为15~25℃。
步骤8用产物7与化合物8(即2,4,5-三氨基嘧啶酮或其盐类)反应,不分离再在氧化剂作用下芳构化反应得到产物9;适合本步反应溶剂为甲醇和水的混合物;芳构化试剂为氧化剂,氧化剂优选为30%的双氧水。
步骤9用产物9经与酸酐或酰氯反应,转化为化合物9`;再进行脱酰化反应;酰化试剂优选为酸酐,更优选为醋酸酐;脱酰化反应条件同直接脱酰条件。
通过增加酰化步骤对酰化产品有一定的提纯作用。但与直接脱酰化产物10质量相当。
L-赤式生物喋呤经还原得到目标化合物I,即盐酸沙丙喋呤。适合本步反应溶剂为水;氧化剂为金属类氧化剂,优选为钯和铂类氧化剂,更优选为二氧化铂;缚酸剂为有机碱,优选为三乙胺、二异丙基乙胺、二乙胺、吡啶、取代的吡啶等,更优选为三乙胺、二异丙基乙胺、二乙胺等;反应压力为0.5~3MPa,优选为1~2.5MPa,更优选为1.5~2.0MPa.
现通过以下实施例来进一步描述本发明的有益效果,应理解为这些实施例
仅用于例证的目的,不限制本发明的范围,同时本领域普通技术人员根据本发明所做的显而易见的改变和修饰也包含在本发明范围之内。
实施例1:产物1的制备
向反应瓶中加入10L无水甲醇,机械搅拌下加入起始原料L-阿拉伯糖1.5kg。水浴下滴加250g浓硫酸,搅拌反应20-24小时。TLC监控反应完毕,向反应体系中加入350g碳酸钠。搅拌至pH=7-8,过滤。滤液35℃-40℃减压浓缩尽干得油状物1.64kg,收率~100%。
实施例2:产物2的制备
向反应瓶中加入产物1、4L吡啶和5L乙腈,机械搅拌溶解。搅拌降温,控温0-5℃滴加1.8kg对甲苯磺酰的5L乙腈溶液。滴加完毕,室温20-25℃搅拌反应4小时。TLC监控反应完毕。
浓缩,向浓缩剩余物中加入12L乙酸乙酯和5L水,搅拌分层。有机层用1mol/L的盐酸、饱和的碳酸氢钠、饱和盐水洗涤,干燥。过滤,滤液浓缩得淡黄色油状物1.7kg,收率56.3%。
实施例3:产物3的制备
向10L反应瓶中加入1.2kg产物2,用6L丁酮溶解,搅拌加入840g碘化钠。加入完毕升温回流12小时,TLC检测反应完毕。降温至室温,过滤,滤液减压浓缩得油状物。用乙酸乙酯溶解,用水洗涤,水层加入乙酸乙酯提取。合并有机层,用饱和盐水洗涤,干燥,过滤,浓缩得900g淡黄色油状物产物3,收率87.4%。
实施例4:产物4的制备
向20L反应瓶中加入用9L甲醇溶解的900g产物3,332g三乙胺,45g10%Pd/C,抽真空,氢气置换2次,控温25-30℃常压氢化16小时。TLC检测反应完毕,过滤,滤液减压浓缩尽干得剩余物。向残余物中加4L乙酸乙酯析出白色固体。降温0℃搅拌30min,过滤。滤液加入2L的0.4mol/L的硫酸溶液,分层。水层用50mL乙酸乙酯洗涤一次,得到产物4的水溶液(约有250g)直接下一步反应。
实施例5:产物5的制备
将产物4的水溶液加入反应瓶中,升温75℃反应反应3小时,TLC检测反应(DCM:MeOH=10:1)反应完毕。降温至室温,加入100mL乙酸乙酯洗涤,分出水层得产物5,即5-脱氧-L-阿拉伯糖的水溶液约213g,直接下一步反应。
实施例6:产物6的制备
在氮气下向反应瓶中加入2.5L乙酸乙酯,170g苯肼,搅拌降温5-10℃(避光)滴加产物5的水溶液。保温反应1小时,然后升温至20-25℃反应30min,TLC检测反应完毕,分层。水层用乙酸乙酯提取,合并有机层。饱和盐酸洗涤,有机相用无水硫酸钠干燥,过滤。
在氮气下向反应瓶中加入产物6的乙酸乙酯溶液,搅拌下缓慢加入8L石油醚。加入完毕,降温至-5--10℃搅拌析晶1小时,过滤,得到米黄色固体。30-35℃减压干燥得到干品约250g,收率71.4%。
实施例7:产物7的制备
向反应瓶中加入2.5L乙酸乙酯,250g产物6。搅拌下加入30g DMAP。降温15℃下滴加400ml醋酐,加入完毕控温20-25℃搅拌反应3小时。TLC监控反应完毕,降温15℃下加入盐酸溶液,分层。有机层用饱和盐酸、饱和碳酸氢钠洗涤。分出有机相,干燥,过滤浓缩得到371g红棕色油状物,收率95%。
实施例8:产物9的制备
向反应瓶中加入220g产物8,2.2L纯净水。搅拌下,加入500g产物7的5L甲醇溶液,1.5L水溶解的无水高氯酸锂150g。加入完毕,控温30-32℃搅拌反应20小时。反应完毕,过滤。滤液控温15℃-20℃滴加1L 30%的双氧水,加入完毕20℃保温反应6小时,析出固体,过滤,35-40℃鼓风干燥得到215g棕黄色产物9,收率47%。
实施例9:产物10的制备
向反应瓶中加入80g产物9,纯净水400ml,正丁醇300ml,二乙胺80ml加入完毕搅拌升温至45-50℃反应16小时。TLC反应完毕,分层,分出水层得到产物10水溶液,直接下一步反应。
实施例10:产物Ⅰ的制备
向氢化釜中加入产物10的水溶液,搅拌下加入50ml三乙胺,二氧化铂2g,
抽真空,氢气置换三次,压力控制为1.5MPa,35℃搅拌反应20小时。过滤,滤液加入30ml正丁醇搅拌5min,静置分层,得到产物Ⅰ的水溶液。降温10℃下滴加200ml浓盐酸,水溶液减压浓缩近干。向粗品中加入95%乙醇500ml升温至55-60℃搅拌1小时,然后降温至35℃搅拌2小时,过滤,滤饼干燥得产物Ⅰ35g。
实施例11:产物9`的制备
向反应瓶中加入1.25L乙酸乙酯,125g产物9。搅拌下加入15g DMAP。降温15℃下滴加200ml醋酐,加入完毕控温20-25℃搅拌反应3小时。TLC监控反应完毕,降温15℃下加入盐酸溶液,分层。有机层用饱和盐酸、饱和碳酸氢钠洗涤。分出有机相,干燥,过滤浓缩得到125.8g油状物。
实施例12:产物10的制备
向反应瓶中加入实施例11制备的产物9`,纯净水600ml,正丁醇450ml,二乙胺120ml加入完毕搅拌升温至45-50℃反应16小时。TLC反应完毕,分层,分出水层得到产物10水溶液,直接下一步反应。
实施例13:产物Ⅰ的制备
向氢化釜中加入实施例12制备的产物10的水溶液,搅拌下加入80ml三乙胺,二氧化铂3g,抽真空,氢气置换三次,压力控制为1.5MPa,35℃搅拌反应20小时。过滤,滤液加入45ml正丁醇搅拌5min,静置分层,得到产物Ⅰ的水溶液。降温10℃下滴加300ml浓盐酸,水溶液减压浓缩近干。向粗品中加入95%乙醇750ml升温至55-60℃搅拌1小时,然后降温至35℃搅拌2小时,过滤,滤饼干燥得产物Ⅰ48.9g。
Claims (10)
- 一种盐酸沙丙喋呤的制备方法,其中盐酸沙丙喋呤结构式如下:
 其特征在于:将L-阿拉伯糖糖苷化、5位羟基碘代、还原脱碘、糖苷键水解脱除烷基得到中间体5-脱氧-L-核糖,再经过反应生成盐酸沙丙喋呤。
其特征在于:将L-阿拉伯糖糖苷化、5位羟基碘代、还原脱碘、糖苷键水解脱除烷基得到中间体5-脱氧-L-核糖,再经过反应生成盐酸沙丙喋呤。 - 一种盐酸沙丙喋呤的制备方法,其特征在于:制备得到酰基取代的L-赤式生物喋呤后,将伯胺基进行酰化再脱去所有酰基得到L-赤式生物喋呤,然后还原得到目标化合物I,即盐酸沙丙喋呤。
- 根据权利要求1所述的盐酸沙丙喋呤的制备方法,其特征在于:得到中间体5-脱氧-L-核糖后经过反应生成酰基取代的L-赤式生物喋呤后脱去酰基得到L-赤式生物喋呤,然后还原得到目标化合物I,即盐酸沙丙喋呤。
- 根据权利要求1所述的盐酸沙丙喋呤的制备方法,其特征在于:得到中间体5-脱氧-L-核糖后经过反应生成酰基取代的赤式生物喋呤后,将伯胺基进行酰化,再脱去所有酰基得到L-赤式生物喋呤,然后还原得到目标化合物I,即盐酸沙丙喋呤。
- 根据权利要求1所述的盐酸沙丙喋呤的制备方法,其特征在于:得到中间体5-脱氧-L-核糖后成腙、羟基乙酰化、与2,4,5-三氨基嘧啶酮对接以及芳构化、脱除酰基得到L-赤式生物喋呤,再经过还原反应得到盐酸沙丙喋呤。
- 根据权利要求1所述的盐酸沙丙喋呤的制备方法,其特征在于:得到中间体5-脱氧-L-核糖后成腙、羟基乙酰化、与2,4,5-三氨基嘧啶酮对接以及芳构化、伯胺基酰化,再脱去所有酰基得到L-赤式生物喋呤,再经过还原得到盐酸沙丙喋呤。
- 根据权利要求1或3或4或5或6所述的制备方法,其特征在于羟基活化所用试剂为对甲基苯磺酰氯或甲基磺酰氯或三氟甲基磺酰氯。
- 根据权利要求7所述的制备方法,其特征在于羟基活化所用试剂为对甲基苯磺酰氯。
- 根据权利要求2或3或4或5或6所述的制备方法,其特征在于酰基取代的赤式生物喋呤的酰基取代基为乙酰基。
- 根据权利要求9所述的制备方法,其特征在于酰基取代的赤式生物喋呤的酰基取代基为乙酰基。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2014/094961 WO2016101211A1 (zh) | 2014-12-25 | 2014-12-25 | 一种盐酸沙丙喋呤的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2014/094961 WO2016101211A1 (zh) | 2014-12-25 | 2014-12-25 | 一种盐酸沙丙喋呤的制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016101211A1 true WO2016101211A1 (zh) | 2016-06-30 |
Family
ID=56148941
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/094961 WO2016101211A1 (zh) | 2014-12-25 | 2014-12-25 | 一种盐酸沙丙喋呤的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2016101211A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109776540A (zh) * | 2017-11-15 | 2019-05-21 | 北京启慧医疗器械有限公司 | 一种盐酸沙丙蝶呤的制备方法 |
| CN113845441A (zh) * | 2021-10-21 | 2021-12-28 | 北京阳光诺和药物研究股份有限公司 | 一种四氢生物蝶呤关键中间体的制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008099560A1 (ja) * | 2007-02-16 | 2008-08-21 | Masahiro Kajiwara | アルドース誘導体の製造方法 |
| CN101792445A (zh) * | 2010-04-16 | 2010-08-04 | 北京理工大学 | L-赤型生物喋呤的制备方法 |
| CN101792446A (zh) * | 2010-04-21 | 2010-08-04 | 北京理工大学 | L-赤型生物喋呤的制备方法 |
| CN102939298A (zh) * | 2010-06-15 | 2013-02-20 | 迪法玛弗朗西斯有限公司 | 制备蝶啶衍生物的方法 |
-
2014
- 2014-12-25 WO PCT/CN2014/094961 patent/WO2016101211A1/zh active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008099560A1 (ja) * | 2007-02-16 | 2008-08-21 | Masahiro Kajiwara | アルドース誘導体の製造方法 |
| CN101792445A (zh) * | 2010-04-16 | 2010-08-04 | 北京理工大学 | L-赤型生物喋呤的制备方法 |
| CN101792446A (zh) * | 2010-04-21 | 2010-08-04 | 北京理工大学 | L-赤型生物喋呤的制备方法 |
| CN102939298A (zh) * | 2010-06-15 | 2013-02-20 | 迪法玛弗朗西斯有限公司 | 制备蝶啶衍生物的方法 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109776540A (zh) * | 2017-11-15 | 2019-05-21 | 北京启慧医疗器械有限公司 | 一种盐酸沙丙蝶呤的制备方法 |
| CN109776540B (zh) * | 2017-11-15 | 2021-07-06 | 北京启慧生物医药有限公司 | 一种盐酸沙丙蝶呤的制备方法 |
| CN113845441A (zh) * | 2021-10-21 | 2021-12-28 | 北京阳光诺和药物研究股份有限公司 | 一种四氢生物蝶呤关键中间体的制备方法 |
| CN113845441B (zh) * | 2021-10-21 | 2024-01-02 | 北京阳光诺和药物研究股份有限公司 | 一种四氢生物蝶呤关键中间体的制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102068381B1 (ko) | 담즙산 유도체의 제조 방법 | |
| WO2020177240A1 (zh) | 鹅去氧胆酸及其制备方法 | |
| AU2018383864B2 (en) | Method for synthesis of Roxadustat and intermediate compounds thereof | |
| JP5153334B2 (ja) | L−ビオプテリンの製造方法 | |
| Minagawa et al. | Stachyflin and acetylstachyflin, novel anti-influenza a virus substances, produced by Stachybotrys sp. RF-7260 II. Synthesis and preliminary structure-activity relationships of Stachyflin derivatives | |
| WO2018233477A1 (zh) | 一种β-内酰胺类化合物羧基及羟基保护基脱除方法 | |
| US20230339840A1 (en) | Preparation method for water-soluble magnolol derivatives and honokiol derivatives and intermediates thereof, and related monohydroxy protection intermediates | |
| WO2016101211A1 (zh) | 一种盐酸沙丙喋呤的制备方法 | |
| AU2010328480A1 (en) | Process for synthesis of intermediates useful for making substituted indazole and azaindazole compounds | |
| Zhu et al. | Concise synthesis of zanamivir and its C4-thiocarbamido derivatives utilizing a [3+ 2]-cycloadduct derived from d-glucono-δ-lactone | |
| ES2548992T3 (es) | Un enfoque quimio-enzimático para la síntesis de pimecrolimus | |
| RU2477285C1 (ru) | Способ получения 3-ацетата-28-сульфата бетулина | |
| KR101230678B1 (ko) | E1 활성화 억제제인 mln4924의 제조방법 | |
| CN108299466B (zh) | 一种改进的度鲁特韦合成方法 | |
| WO2018233461A1 (zh) | 一种拉氧头孢羧基及羟基保护基的脱除方法 | |
| WO2016189542A1 (en) | Novel process for the preparation of sapropterin dihydrochloride and its key intermediate, l-biopterin | |
| CN108623602A (zh) | 一种制备和纯化依鲁替尼的方法 | |
| CN107325133A (zh) | 一种1,2,3‑三‑o‑乙酰基‑5‑脱氧‑d核糖的合成方法 | |
| CN103923135B (zh) | 一种氘代5-羟基色氨糖苷衍生物及其制备方法 | |
| CN112939814A (zh) | 一种氘代达卡他韦中间体的制备方法 | |
| RU2482125C1 (ru) | Способ получения натриевой соли 3-сульфата аллобетулина | |
| CN111170932A (zh) | 一种2-氨甲基-5-三氟甲基吡啶盐的制备方法 | |
| CN106831774B (zh) | 一种(6s,7s)-9-叔丁氧羰基-7-(三氟甲基)-2,9-二氮杂螺[5.5]十一烷的合成方法 | |
| BR112021014815A2 (pt) | Método para preparação de 4-amino-5-metilpiridona | |
| WO2024022426A1 (zh) | 一种4-(三甲基铵)戊酸的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14908779 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14908779 Country of ref document: EP Kind code of ref document: A1 |