WO2015119166A1 - 植物の形質転換細胞の取得方法 - Google Patents
植物の形質転換細胞の取得方法 Download PDFInfo
- Publication number
- WO2015119166A1 WO2015119166A1 PCT/JP2015/053137 JP2015053137W WO2015119166A1 WO 2015119166 A1 WO2015119166 A1 WO 2015119166A1 JP 2015053137 W JP2015053137 W JP 2015053137W WO 2015119166 A1 WO2015119166 A1 WO 2015119166A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- dna
- marker gene
- plant
- transformation
- Prior art date
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8201—Methods for introducing genetic material into plant cells, e.g. DNA, RNA, stable or transient incorporation, tissue culture methods adapted for transformation
- C12N15/8209—Selection, visualisation of transformants, reporter constructs, e.g. antibiotic resistance markers
- C12N15/821—Non-antibiotic resistance markers, e.g. morphogenetic, metabolic markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8201—Methods for introducing genetic material into plant cells, e.g. DNA, RNA, stable or transient incorporation, tissue culture methods adapted for transformation
- C12N15/8209—Selection, visualisation of transformants, reporter constructs, e.g. antibiotic resistance markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/6895—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for plants, fungi or algae
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/001—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination
- C12N2830/002—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination inducible enhancer/promoter combination, e.g. hypoxia, iron, transcription factor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/001—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination
- C12N2830/002—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination inducible enhancer/promoter combination, e.g. hypoxia, iron, transcription factor
- C12N2830/003—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination inducible enhancer/promoter combination, e.g. hypoxia, iron, transcription factor tet inducible
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/001—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination
- C12N2830/005—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination repressible enhancer/promoter combination, e.g. KRAB
- C12N2830/006—Vector systems having a special element relevant for transcription controllable enhancer/promoter combination repressible enhancer/promoter combination, e.g. KRAB tet repressible
Definitions
- the present invention relates to a method for obtaining transformed cells of a plant and a method for producing a transformed plant.
- Plant Transformation When transforming plants, a technique for introducing DNA into plant cells that can maintain and express foreign genes in progeny seeds or vegetative propagation bodies is used. Using this technique, many monocotyledons and dicotyledons have actually been transformed.
- transformants such as Arabidopsis thaliana, rice and brachypodium are actively used for basic research.
- transformants have been produced not only in corn, rice, wheat, barley, sorghum, soybean, rapeseed, sunflower, cotton, potato, tomato, but also in fruits and other vegetables.
- sorghum sorghum
- soybean rapeseed
- sunflower cotton
- cotton potato
- tomato but also in fruits and other vegetables.
- fruits and other vegetables since it is possible to give a high-value-added trait by transformation, there is a great commercial interest.
- transformed corn, soybean, rapeseed and cotton are widely produced commercially.
- RNA silencing There are two types of homology-dependent gene silencing: post-transcription type (PTGS) and transcription type (TGS), both of which are mediated by double-stranded RNA. This is likely to occur when multiple copies are introduced in the direction or in the opposite direction, or when a partially deleted target DNA is introduced together.
- PTGS post-transcription type
- TGS transcription type
- TGS is triggered by epigenetics and generally involves DNA methylation via small RNA that is homologous to the promoter region upstream of the gene. This methylation is maintained even after somatic cell division and meiosis, and as a result, silencing is inherited to progeny (Eamens et al., 2008).
- the aspect of integration of the target DNA into the genome becomes more complex as the number of copies increases, and the analysis becomes difficult. Therefore, 1) evaluate the effect of the transgene in the transformant at the expression level, 2) evaluate the characteristics of the promoter and transcription factor in the transformant, 3) create a T-DNA tagging library, and 4) commercial.
- the target DNA is preferably introduced in 1-2 copies, and most preferably in a single copy.
- Plant transformation methods include physicochemical methods (direct DNA introduction method) such as particle gun method, electroporation method, electroinjection method, polyethylene glycol method and whisker method, and the functions of Agrobacterium bacteria.
- Biological methods to be used are known.
- direct DNA introduction method events such as introduction of the target DNA in a fragmented manner or introduction in a high-order copy (up to 100 copies when there are many) occur frequently (Butaye et al., 2005).
- multiple copies of the target DNA are introduced, they are often connected and integrated into the same gene locus (Kohli et al., 1998; Klein, 2010). Therefore, transformants exhibiting gene silencing phenomenon in which the target gene is not expressed frequently appear (Register et al., 1994).
- the target DNA is introduced by controlling the expression of genes in the pathogenic region (vir region) of the Ti or Ri plasmid.
- the target DNA is recognized by the interaction of the protein group encoded by the vir gene and the recognition and signal transduction of the interaction between the plant cell and the bacteria, the expression of the vir gene, the formation of the type IV secretion pathway, the T-DNA border repeat sequence It is introduced through many processes such as recognition, formation of T-DNA strands, transfer of T-DNA strands into plant cells and into the nucleus, and integration of T-DNA into the plant nucleus genome.
- the number of copies of the target DNA introduced is kept low (less than 10 copies) and is not fragmented and introduced (Shou et al., 2004; Butay et al., 2005). ).
- the Agrobacterium method is an excellent transformation method compared to the direct DNA introduction method, it still cannot sufficiently control the number of copies of DNA introduced into the genome. It is not uncommon for multiple copies of DNA to be introduced into the same locus (Wang and Waterhouse, 2000). Therefore, there are some differences between individuals in the expression level of the target gene, and there may be individuals with homology-dependent gene silencing (Butaye et al., 2005; Naga et al., 2005).
- the first is a method using a site-specific recombination system.
- a DNA fragment in which recognition sequences (lox) of site-specific recombinase are arranged in opposite directions on both sides of a desired DNA having a selection marker is introduced into a plant.
- a Cre expression cassette is introduced into another plant in advance.
- a T0 individual into which the target DNA has been introduced by multicopy ligation and a T0 individual expressing Cre are mated.
- oriRi of Agrobacterium rhizogenes is used as the replication origin of the vector having the T-DNA of the target DNA.
- Ye et al. (2011) used a binary vector that functions oriRi and a binary vector that functions oriV, which is the replication origin of IncP RK2 type, to obtain a vector corresponding to the outside of T-DNA in the resulting transformant.
- the frequency of individuals into which the target DNA was introduced in a single copy without including a backbone sequence was compared.
- the frequency of single-copy-introduced individuals with the oriRi vector was 38% -40% twice that of the oriV vector, 51% of the rapeseed transformant, and 51% of the rapeseed transformant.
- the production efficiency per test material taking into consideration the transformation efficiency in addition to the production frequency of backbone-free and single-copy transformants with the oriRi vector, was 0.96-1.36 times higher for soybean and 1 for rapeseed. .2 times and 0.82 times for corn. Even if the production efficiency per material is low, the oriRi binary vector is useful if the overall work efficiency and cost efficiency are high. Since transformants are costly and laborious for DNA extraction and copy number analysis, oriRi binary vectors may help improve overall efficiency.
- the third is a method of transforming a plant by retaining T-DNA on the Agrobacterium chromosome. (2010) incorporated a DNA region containing T-DNA into homologous recombination into the picA region on the chromosomes of Agrobacterium strains GV3101 and EHA101 and used it for maize transformation. And the frequency of the single copy introduction
- this method of retaining T-DNA on the Agrobacterium chromosome has a fundamental problem that the transformation efficiency is low, and the transformation efficiency of corn is about 1% for both EHA101 and GV3101 strains. (Oltmanns et al., 2010). Incidentally, the transformation efficiency by the Agrobacterium strain using the conventional binary vector was 9-12% for the EHA101 strain and 5-8% for the GV3101 strain. This method of retaining T-DNA on the Agrobacterium chromosome seems to be quite effective only in the Arabidopsis floral dip method using the GV3101 strain. The transformation efficiency is not significantly reduced to 0.9% compared to 1.6-2.1% in the conventional method, and the frequency of single copy introduced individuals is 16-35% in the conventional method.
- Co-transformation of foreign DNA in co-transformed plants is often used to obtain transformants into which only the target DNA has been introduced.
- Selectable marker genes and selectable drugs are very useful tools for obtaining transformed cells from plant tissues consisting mostly of non-transformed cells, but are basically unnecessary after transformants are obtained.
- co-transformation is mainly performed for the purpose of removing the selection marker gene in the next generation. That is, a target foreign DNA and a selectable marker gene are inserted into separate T-DNAs, and simultaneously introduced into plant cells via Agrobacterium. When the cell mass selected with the selection drug is regenerated, a transformant into which the target DNA is introduced together with the selection marker gene is obtained.
- the co-transformation method using Agrobacterium is a type in which two T-DNAs are separately arranged on two binary vectors in one strain, and two T-DNAs on one binary vector in one strain.
- a type in which DNA is arranged, a type in which two T-DNAs are separately arranged in each binary vector of two strains, and a type in which two right border sequences are arranged on one binary vector in one strain There are four types (Yau and Stewart, 2013).
- the co-transformation method is used not to eliminate the selection marker gene in the direct introduction method of DNA but to eliminate the prior work of linking the target DNA and the selection marker gene before gene introduction. This is because, as described above, the method for directly introducing DNA is because a plurality of foreign DNAs are easily incorporated into the same gene locus. In fact, in the direct introduction method of DNA, even if the co-transformation method is used for the purpose of eliminating the selection marker gene, the efficiency with which the selection marker gene can be removed in the next generation is very low (Yau and Stewart, 2013).
- a positive selection marker gene and a negative selection marker gene are arranged on the same T-DNA, and after obtaining a transformant by positive selection in the T0 generation, the selection marker is subjected to negative selection in the T1 generation.
- the T1 individual into which the gene has been introduced can be killed. The surviving T1 individual no longer contains the selection marker gene, and can simply determine the presence or absence of the target DNA by PCR or gene expression (Yau and Stewart, 2013).
- cells that contain the desired DNA and do not contain the selectable marker gene are removed, but some of the selected cells also have the desired DNA introduced therein. Thereafter, when the obtained tissue is transferred to a medium without a selective drug, a cell mass in which the desired DNA is integrated into the chromosome but the selective marker is not integrated into the chromosome is obtained. On the other hand, cells in which the selection marker gene is integrated into the chromosome are then killed by the expression of the negative selection marker. The desired plant body is obtained by redifferentiating the obtained cell cluster having only the desired DNA.
- Dutt et al. (2008) introduced only a desired DNA, eg, egfp (enhanced green fluorescent protein), using a large number of grape somatic embryos (specific figures not shown) as material. Obtained 5 individual plants.
- Dutt et al. (2008) stated that, as a result of analysis by real-time PCR, the number of copies of the egfp gene introduced into these 5 individuals was 1 single copy for 3 individuals, 2 copies for 1 individual, and 6 copies for 1 individual. . It seems that the efficiency is high because the proportion of individuals introduced by single copy is 60%. However, it was confirmed that 60% of the grape transformants obtained from the same research group by the usual transformation method were single copy. (Li et al., 2006).
- RNA silencing in plants yesterday, today, and tomorrow. Plant Physiol 147, 456-468. Hiei, Y. , And Komari, T. (2006). Improved protocols for transformation of indicia medium by by Agrobacterium tumefaciens. Plant Cell, Tissue and Organ Cul 85, 271-283. Klein, T. M.M. (2010). Particle Bombardment: An Established Weapon in the Arsenal of Plant Biotechnology. In Plant Transformation Technologies (Wiley-Blackwell), pp. 51-71. Kohli, A. , Leech, M.M. , Vain, P.M. Erasmus, D.C. A. , And Christou, P.M. (1998).
- a phenomenon called gene silencing in which the expression of the introduced gene is strongly suppressed often occurs.
- the aspect of integration of the target DNA into the genome becomes more complex as the number of copies increases, and the analysis becomes difficult.
- 1) the effect of the transgene is evaluated at the expression level in the transformant, 2) the characteristics of the promoter and transcription factor are evaluated in the transformant, and 3) a T-DNA tagging library is created. This is done for the purpose of producing a genetically modified plant for commercialization.
- it is desirable that the target DNA is introduced in 1-2 copies. Most desirable is to be introduced in a single copy.
- an object of the present invention is to provide a method for obtaining transformed plant cells, in which the number of copies of desired DNA introduced per plant cell is reduced.
- a method for obtaining transformed cells of a plant comprising the following steps: (A) a step of co-transforming a desired DNA and a first marker gene into a plant cell; and (b) from the transformed plant cell obtained in step (a), the desired DNA is present on the chromosome. Selecting a transformed cell that has been introduced and into which the first marker gene has not been introduced, However, the positive selection using the first marker gene does not include a step of removing transformed cells in which only desired DNA has been introduced into the chromosome, Said method.
- Aspect 2 The method according to aspect 1 [aspect 3], wherein the first marker gene is a negative selection marker gene.
- a positive selection marker gene which is a second marker gene, is linked to the desired DNA used in step (a);
- Aspect 1-4 is carried out by a transformation method selected from the group consisting of the Agrobacterium method, particle gun method, electroporation method, electroinjection method, polyethylene glycol method and whisker method.
- a plant transformation method comprising the following steps: (A) a step of co-transforming a desired DNA and a first marker gene into a plant cell; and (b) from the transformed plant cell obtained in step (a), the desired DNA is present on the chromosome. Selecting a transformed cell that has been introduced and into which the first marker gene has not been introduced, However, the positive selection using the first marker gene does not include a step of removing transformed cells in which only desired DNA has been introduced into the chromosome, Said method.
- the method of the present invention is a technique that can eliminate cells in which multiple copies of the target DNA have been introduced at the early stage of the transformation system.
- a transformed plant into which only a single copy of the target DNA has been introduced can be preferably obtained at a frequency 1.3 times or more that of the prior art.
- the frequency of transformants introduced with 3 or more copies can be reduced to half or less of the prior art.
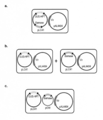
- FIG. 1 is a schematic diagram of the co-transformation method by the Agrobacterium method used in the multicopy cell exclusion technique.
- A. 1 strain 1 vector method LBA4404 (pLC41 GUS-HPTtra. Barnase), b. : Two strains mixing method, LBA4404 (pLC41 GUS-HPT) + LBA4404 (pLC41 Barnase), c.
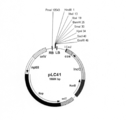
- FIG. 2 is a schematic diagram of the expression vector pLC41 GUS-HPT.
- FIG. 3 is a schematic diagram of the expression vector pLC41.
- FIG. 4 is a schematic diagram of the expression vector pLC41 Barnase.
- FIG. 5 shows the expression vector pLC41 GUS-HPT cotra. It is a schematic diagram of barnase.
- FIG. 6 is a schematic diagram of pGW.
- FIG. 7 is a schematic diagram of pGW Barnase.
- FIG. 8 is a schematic diagram of two types of T-DNA introduced.
- A. PLC41 GUS-HPT cotra. A T-DNA region common to Barnase and pLC41 GUS-HPT; b. : PLC41 GUS-HPT :: T-DNA region common to pGW Barnase, pLC41 Barnase and pGW Barnase
- FIG. 9 shows the number of introduced copies of the GUS-HPT fragment estimated as a result of Southern analysis.
- Control LBA4404 (pLC41 GUS-HPT) 1 strain 1 vector: LBA4404 (pLC41 GUS-HPT cotra.
- FIG. 10 shows the number of introduced copies of GUS-HPT fragments in tobacco transformants estimated by quantitative real-time PCR.
- FIG. 11 is a graph showing the introduced copy number of the GUS-bar fragment in maize transformants estimated by quantitative real-time PCR.
- the present invention relates to a method for obtaining transformed cells of a plant.
- the method of the present invention comprises the following steps: (A) a step of co-transforming a desired DNA and a first marker gene into a plant cell; and (b) from the transformed plant cell obtained in step (a), the desired DNA is present on the chromosome. Selecting a transformed cell that has been introduced and into which the first marker gene has not been introduced, However, the positive selection using the first marker gene does not include a step of removing transformed cells in which only desired DNA has been introduced into the chromosome, Said method.
- Plant The plant to be subjected to the method of the present invention is not particularly limited, and may be any monocotyledonous plant or dicotyledonous plant, including any plant such as algae, angiosperms and gymnosperms.
- the tissue to be used for transformation can be appropriately selected according to the type of plant and the transformation method used.
- the present invention includes (a) a step of cotransforming a desired DNA and a first marker gene into a plant cell.
- the “marker gene” in the present specification means a gene having a property that serves as an index for selecting a gene that has been introduced into a cell or a gene that has not been introduced into a cell.
- the desired DNA and the first marker gene are cotransformed, the desired DNA and the first marker gene are introduced into the genome on an occasional basis in a mixed state in the cell nucleus. It is thought. As a result of such transformation, “cells into which a low copy number of a gene has been introduced” and “cells into which a gene has been introduced with a large number of copies” are produced. “Is low as a probability, and many of them are” multicopy-introduced cells containing both a desired DNA and a marker gene ". Therefore, by selecting “a cell into which a desired DNA has been introduced and a marker gene has not been introduced” from the resulting transformed cell group, a “cell into which a desired DNA has been introduced in multiple copies” is greatly increased. Can be reduced.
- the “first marker gene” of the present invention is a gene different from the desired DNA, and it only has to have the property of removing the marker gene-expressing cell when expressed in the cell. That is, it may be a gene in which a gene that can be easily detected is introduced as an index of selection, and marker gene-expressing cells can be removed depending on the presence or absence of expression of the gene.
- negative selection means selective removal of cells in which a marker gene is expressed. Therefore, in other words, the first marker gene only needs to have a property that enables negative selection.
- the cell is artificially selected from the cell group, for example, using a tool such as tweezers or a scalpel under a microscope. Can be removed.
- marker genes include fluorescent protein genes such as green fluorescent protein (GFP), red fluorescent protein (DsRed), and luciferase gene, and genes of enzymes that catalyze a color reaction such as lacZ gene.
- drug resistance genes such as hygromycin resistance gene, gentamicin resistance gene, kanamycin resistance gene, ampicillin resistance gene, spectinomycin resistance gene, tetracycline resistance gene, bialaphos resistance gene, glyphosate resistance gene are also used as such marker genes Is possible.
- concentration of the drug added to the selection medium in the drug selection process is suppressed to such a level that the cells into which the drug resistance gene has not been introduced are not killed, the non-drug resistance gene-introduced cells are not killed but hardly proliferate. Due to the difference in appearance, it can be distinguished from drug-resistant transgenic cells.
- genes that allow visual identification of cells for example, genes for enzymes that catalyze a color reaction, drug resistance genes, have the property of enabling negative selection, and the first marker gene of the present invention It can be used as
- a negative selection marker gene can be preferably used as the “first marker gene” of the present invention.
- a marker gene having a property of selectively excluding cells including itself by its expression is referred to as a negative selection marker gene.
- a specific substance is added to a plant cell or a culture medium, this includes a substance that functions to selectively exclude cells including itself.
- the negative selection marker gene is not necessarily limited to a structural gene, and a non-structural gene other than a structural gene encoding a protein, such as an expression sequence of a non-coding RNA, may be used.
- the negative selectable marker gene is incorporated into the plant genome and expressed between the gene introduction treatment and the regeneration of the plant body, thereby inducing cell death, cell growth arrest, or abnormal tissue formation.
- a cell death-inducing negative selection marker gene is used as the marker gene, the marker gene-expressing cells are killed, so the work of removing the marker gene-expressing cells is unnecessary, and the working efficiency is greatly improved.
- the negative selection marker gene does not remain in the regenerated plant body.
- Those skilled in the art can appropriately select such a negative selection marker gene.
- the expression of the negative selection marker gene is preferably at a level that does not induce cell death in the transient expression stage.
- cells in which only the target DNA is integrated into the genome and the negative selection marker gene is transiently expressed can be produced. It becomes a cell containing only. Therefore, from the viewpoint of preventing a reduction in transformation efficiency, it is preferable not to kill such cells by transient expression of the negative selection marker gene.
- the toxicity of the negative selection marker is strong, it is preferable to suppress the toxicity by applying a promoter that reduces the expression of the marker gene.
- Such expression regulation can be appropriately performed by those skilled in the art based on known techniques. Although not limited, it is described in the Examples of the present application that the use of the nos promoter was suitable when the Barnase gene was used as the negative selection marker gene.
- the most widely used negative selection marker gene for plants is codA derived from E. coli (Yau and Stewart, 2013).
- codA encodes cytosine deaminase and converts non-toxic 5-fluorocytosine (5-FC) to toxic 5-fluorouracil (5-FU).
- the ornithine deacetylase gene argE of E. coli converts N-acetylphosphinotritin (N-acetyl-PPT), which is not toxic to plants, into herbicide-active phosphinotritin (PPT).
- Bacterial cytochrome P450sui converts a non-toxic herbicoid R4702 precursor to a cytotoxic herbicoid R4702.
- Diphtheria venom fragment A is toxic to plant cells, but not toxic to E. coli or Agrobacterium (Terada et al., 2002).
- the poly-A signal is not attached to the DT-A gene, the transiently expressed mRNA is immediately degraded, whereas when it is integrated into the genome, the nearby poly-A signal is It is expressed continuously using it.
- Barnase is a ribonuclease derived from Bacillus amyloliquefaciens. Both prokaryotes and eukaryotes are highly toxic. However, the toxicity to prokaryotes can be suppressed by interposing an intron in the gene (Burgess et al., 2002).
- these genes can be preferably used as a negative selection marker gene.
- the process of the present invention does not include a process of removing transformed cells in which only desired DNA has been introduced into the chromosome by positive selection using the first marker gene.
- the definition of positive selection is described in detail in the section "Second marker gene”. If positive selection is performed using the first marker gene, transformed cells in which only the desired DNA has been introduced into the chromosome are: The effect of the present invention cannot be obtained.
- the number of first marker genes used is not limited.
- one or two kinds of marker genes can be preferably used.
- Desired DNA is any DNA that is introduced into a cell, and is not particularly limited.
- the desired DNA is DNA introduced into a chromosome (genome) of a plant cell, and is not necessarily limited to a structural gene, and a nonstructural gene other than a structural gene encoding a protein may be used.
- a desired promoter and terminator may be linked to the “desired DNA”.
- the “desired DNA” can be of any length depending on the transformation method used. For example, when the Agrobacterium method is used, the length is preferably 0.1 kb to 50 kb, although not limited thereto.
- the mixing ratio of the desired DNA to be subjected to co-transformation and the first marker gene is not particularly limited, but is preferably between 3: 1 and 1: 5, more preferably between 2: 1 and 1: 3.
- the transformation method is not particularly limited as long as it is a method capable of transforming a plant, and can be appropriately selected according to the kind of plant.
- particle gun method electroporation method, electroinjection method, polyethylene glycol method, whisker method and other physicochemical methods (DNA direct introduction method) or biological methods such as Agrobacterium method (indirect DNA introduction) Method) can be preferably used.
- transformation genetic alteration of cells obtained as a result of introduction, transfer (and expression) of exogenous genetic material (exogenous DNA) is called transformation. It is also used as a term indicating a process for the change. The simultaneous transformation of two or more independent exogenous genetic materials is called co-transformation. Similarly, it is also used as a term indicating the process. Note that “independent” means that two or more exogenous genetic materials are not introduced as an integral DNA, but are introduced into cells as DNAs that can behave independently of each other.
- Agrobacterium-mediated co-transformation method is a type in which a plurality of T-DNAs are separately arranged on a plurality of binary vectors in one strain (one strain / multi-vector method), one binary vector in one strain A type in which two T-DNAs are arranged on top (one strain / one vector method), a type in which two T-DNAs are separately placed on each binary vector of two strains and mixed inoculated with both strains (two strain method), There are four types, one T-DNA in which two genes are constructed using two right border sequences on one binary vector of one strain (Yau and Stewart, 2013). Any type can be preferably implemented in the present invention.
- the two strain method is not particularly limited to two strains, and a plurality of T-DNAs can be separately placed and mixedly inoculated into each binary vector of a plurality of strains.
- the fourth type uses the double light border method.
- This is one of the selective marker removal methods when transformation is performed by the Agrobacterium method (Yau and Stewart, 2013). That is, T-DNA is constructed in the order of “RB (right border sequence) -positive selection marker-RB-desired DNA-LB (left border sequence)”, and “RB-positive selection marker-RB-desired”.
- This is a method in which “DNA-LB” and “RB-desired DNA-LB” are introduced into separate chromosomes of a plant and separated in the next generation. By applying this method, T-DNA can be converted into “RB-negative selection marker-RB-positive selection marker-desired DNA-LB” (or “RB-negative selection marker-RB-desired DNA-positive selection marker-LB”).
- the particle gun method is a method of introducing DNA into cells by ejecting at high speed a bullet obtained by coating DNA on metal fine particles such as gold or tungsten. It is also called Biolistic method, particle bombardment method, or microprojectile method.
- metal fine particles such as gold or tungsten.
- Biolistic method, particle bombardment method, or microprojectile method gold fine particles are preferred because they have high specific gravity, high penetration into cells, and are chemically inactive and hardly cause harm to the living body.
- a high-pressure gas such as helium is mainly used for injection of metal fine particles.
- the injection strength of the metal fine particles can be adjusted by the gas pressure, the distance between the metal particles and the sample, etc., and can be introduced into various cells.
- two or more kinds of DNA are coated on metal fine particles, and cotransformation can be performed by introducing the DNA into cells as bullets.
- the electroporation method is a method for transformation by applying an electric pulse to the cell suspension to form a minute hole in the cell membrane and sending the DNA in the cell suspension into the cell.
- an electric pulse When plant cells are used as materials, it is common to use protoplasts obtained by decomposing and removing cell walls. However, it is also possible to transform using cells having cell walls, which is called electroinjection.
- co-transformation can be performed by dissolving two or more kinds of DNAs in a suspension and applying an electric pulse in the presence of plant cells.
- polyethylene glycol polyethylene glycol
- PEG polyethylene glycol
- the whisker method is a method of making a hole in a plant cell using a needle-like substance called a whisker and incorporating DNA into the cell.
- a whisker silicon carbide or aluminum borate is used.
- the same transformation method may be used for both, or different transformation methods may be used, but co-transformation is performed using the same transformation method. It is more efficient and more preferable.
- a positive selection marker gene may be linked to the desired DNA as the second marker gene.
- the desired DNA does not serve as an indicator of the success or failure of transformation, that is, when the desired DNA does not have a property as a marker gene, it is preferable to link a positive selection marker gene to the DNA.
- positive selection refers to selective selection of cells in which a marker gene is expressed, and a marker gene that can be used for positive selection is referred to as a positive selection marker gene.
- the marker gene is artificially selected from the cell group, for example, using a tool such as tweezers or a female under a microscope. Expression cells can be selected. Therefore, a gene having the property of imparting visually distinguishable properties such as fluorescence to a cell can be used as a positive selection marker gene.
- marker genes include fluorescent protein genes such as green fluorescent protein (GFP), red fluorescent protein (DsRed), and luciferase gene, genes of enzymes that catalyze a color reaction such as lacZ gene, and the like.
- the gene is incorporated into the plant chromosome genome and expressed during any period from gene transfer treatment to plant regeneration, thereby causing cell death, cell growth arrest, or abnormality. Genes that prevent events such as induction of tissue formation can also be suitably used. It is possible to efficiently perform positive selection by setting conditions in which cells expressing such a positive selection marker gene can grow and cells that do not express the positive selection marker gene cannot grow. it can.
- positive selection marker genes include, but are not limited to, hygromycin resistance gene, gentamicin resistance gene, kanamycin resistance gene, ampicillin resistance gene, spectinomycin resistance gene, tetracycline resistance gene and other antibiotic resistance genes, bialaphos.
- herbicide tolerance genes such as glyphosate tolerance gene, phosphorylated mannose isomerase (PMI) gene, 2-deoxyglucose-6-phosphatase gene and xylose isomerase gene, etc. Examples include genes to be given.
- Antibiotics and herbicides have a harmful effect on non-transformed cells of plants, but sugars such as mannose and xylose are not harmful to plants, although they are carbon sources that cannot be metabolized.
- a selection system that does not adversely affect non-transformed cells by combining carbon sources that cannot be originally metabolized by plants as a selection drug, and combining enzyme genes that convert these carbon sources into carbon sources that can be metabolized by plants.
- this system is sometimes referred to as a (selection narrow) positive selection system
- the selection marker is sometimes referred to as a (selection narrow) positive selection marker gene (Uppadya et al., 2010).
- positive selection in the present invention refers to selective selection of cells in which a marker gene is expressed, and a marker gene that can be used for positive selection is a positive selection marker gene. The content is not limited.
- a positive selection marker gene which is a second marker gene, is linked to the desired DNA used in step (a); Selecting a transformed cell having the desired DNA introduced into the chromosome in step (b) by positive selection using the second marker gene; Embodiments are also included.
- a positive selection marker gene that is a second marker gene is linked to the desired DNA means that the desired DNA and the second marker gene are linked, and both behave in transformation. Means that. Therefore, when the desired DNA is integrated into the chromosome of the plant cell by transformation, the second marker gene is also integrated. On the other hand, if the transformation fails and the desired DNA is not integrated into the chromosome of the plant cell, the second marker gene is also not integrated. In this respect, the second marker gene is co-transformed as a DNA independent of the desired DNA, and the position of the second marker gene in transformation is different from that of the first marker gene having a different behavior.
- Two marker genes are linked and sandwiched between a set of “RB” and “LB”.
- the first marker gene is sandwiched between “RB” and “LB” separate from the desired DNA.
- T-DNA is converted into “RB-negative selection marker-RB-positive selection marker-desired DNA-LB” (or “RB-negative selection marker-RB—desired DNA-positive”.
- the selection marker may be configured to be “LB”).
- a transformed cell refers to a cell in which a foreign gene has been incorporated into a chromosomal genome through a transformation step or a progeny thereof.
- a transformed cell in which a desired DNA is introduced into a chromosome and a first marker gene is not introduced is selected from the transformed plant cells obtained in the step (a). select.
- the negative selection marker gene is expressed in a cell in which the gene is introduced into the chromosome.
- transformed cells containing the marker gene can be removed by negative selection that causes cell death, cell growth arrest, or abnormal tissue formation.
- selection of transformed cells containing desired DNA when the desired gene is a marker gene, only cells having properties based on the expression of the marker gene are selected.
- the desired DNA is not an indicator of the success or failure of the transformation, that is, when the desired DNA is not a marker gene, it is preferable to link a positive selection marker gene to the DNA. Only cells having properties based on the expression of the positive selection marker gene are selected.
- Expression of each gene of the desired DNA, the first marker gene, and the second marker gene may be constitutive or inducible.
- inducible case for example, a specific compound given from outside described later It is possible to induce gene expression.
- Inducible expression can be performed by stress treatment such as high-temperature treatment and low-temperature treatment in addition to the specific compound given from the outside.
- the selection of transformed cells does not include selection with a selection marker in the progeny of transformed plants obtained by mating transformed plants.
- the expression time of the selection marker gene may be controlled using such a gene compound induction system with a specific compound.
- the expression time of the marker gene Is preferably delayed after it has been integrated into the chromosomal genome. In such a case, it is possible to control the expression time using a gene expression induction system using the following specific compound.
- the amino acid sequence of a region that binds to a bacterial cis sequence domain: domain
- the amino acid sequence of a region that binds to a specific compound and controls the activity of a transcription factor and transcriptional activation
- a chimeric trans transcription factor has been synthesized that fuses three regions with the amino acid sequence of the region.
- gene expression induction systems such as tetracycline and estradiol have been developed.
- Tetracycline induction system The expression of the tetracycline resistance operon (tet operon) present in the transposon Tn10 of E. coli is negatively expressed by the repressor TetR (amino acid sequence) and the operator tetO (5'-TCCCTATCAGTGGATAGGAGA-3 '). Is controlled. TetR binds to tetO and inhibits transcription in the absence of tetracycline, but dissociates from tetO in the presence of tetracycline. That is, tetracycline is an inducer of the tet operon.
- Tet operon The expression of the tetracycline resistance operon (tet operon) present in the transposon Tn10 of E. coli is negatively expressed by the repressor TetR (amino acid sequence) and the operator tetO (5'-TCCCTATCAGTGGATAGGAGA-3 '). Is controlled. TetR bind
- the TetR gene tetR is linked downstream of the promoter of a gene that is constitutively expressed in plants, and a plurality of tetOs are linked downstream of another promoter and the gene whose expression is to be induced is further linked downstream of it.
- This is a combination of The gene downstream of tetO is induced by administering tetracycline as an inducer.
- doxycycline is more inductive than tetracycline.
- Estradiol induction system transcriptional activity of VP16 (amino acid sequence) derived from herpes simplex virus (HSV: Herpes Simplex Virus), the 1-87 amino acid residue sequence of LexA, which is a repressor of the SOS regulon of E.
- HSV herpes Simplex virus
- coli Synthetic transcriptional activator XVE (amino acid sequence) produced by fusing the region (403-479 amino acid residue sequence) and the human estrogen receptor regulatory region (282-595 amino acid residue sequence) Is a transcription induction system in which multiple SOS boxes (5'-TACTGTATATATATACAGTA-3 '), an operator to which LexA binds, are arranged upstream of the TATA box (TATA box) of the CaMV 35S minimal promoter as a cis sequence to which XVE binds. It is.
- the CaMV 35S minimal promoter has almost no transcriptional activity in the absence of estradiol. However, when XVE and estradiol bind, XVE binds to SOS box and strongly induces the transcriptional activity of the downstream CaMV 35S minimal promoter. That is, it is a positive control system.
- Creating the present invention transformed plant further relates create a transformed plant.
- the method for producing a transformed plant of the present invention includes obtaining a transformed cell of a plant by the method for obtaining a transformed cell of the plant of the present invention; culturing the plant cell to obtain a plant body.
- the transformed cells are cultured.
- the method of culturing transformed cells to obtain a plant body can use any method according to the type of plant.
- the culture medium examples include a medium based on LS inorganic salts and N6 inorganic salts, specifically, for example, LSZ medium.
- the culture medium may contain a selection drug. “Cultivation” in this step refers to placing plant cells or plant tissues on a solidified culture medium or in a liquid culture medium and growing them at an appropriate temperature, light and dark conditions, and duration. .
- the form of the medium is not particularly limited as long as the medium components are sufficiently supplied to the plant tissue.
- the culture medium can be solidified using, for example, agarose.
- the culture temperature in this step can be appropriately selected, and is preferably 20 ° C-35 ° C, more preferably 25 ° C.
- the culture in this step is preferably performed under illumination for 16-24 hours / day, but is not limited thereto.
- the culture period in this step can also be appropriately selected, and is preferably 7 days to 21 days, more preferably 14 days.
- the present invention also relates to a plant transformation method.
- the method of the present invention comprises the following steps: (A) a step of co-transforming a desired DNA and a first marker gene into a plant cell; and (b) from the transformed plant cell obtained in step (a), the desired DNA is present on the chromosome. Selecting a transformed cell that has been introduced and into which the first marker gene has not been introduced, However, it does not include a step of removing transformed cells in which only desired DNA has been introduced into the chromosome by positive selection using the first marker gene.
- Example 1 Construction of co-transformation vector 1
- the HPT gene was used.
- the GUS gene was controlled by the 35S promoter, and nos was used as the terminator.
- the HPT gene was controlled by a maize ubiquitin (Ubi) gene promoter, and nos was used as a terminator.
- the first intron of the maize ubiquitin gene was placed upstream of the HPT translation region.
- Negative selection marker gene As the negative selection marker gene, Barnase gene of Bacillus amyloliquefaciens was used. Expression was controlled by the nos promoter, and 35S was used as the terminator. In addition, since the expression of Barnase also killed Escherichia coli and Agrobacterium, the fifth intron of the rice Rf-1 gene was mediated in the Barnase gene (Pnos-Barnase-T35S). Thereby, it can be set as the negative selection marker expressed only in a plant cell.
- the sequence in which the fifth intron of the Rf-1 gene of rice was interposed in the Barnase gene of Bacillus amyloliquefaciens used was SEQ ID NO: 1
- the sequence of the Barnase gene of Bacillus amyloliquefaciens was SEQ ID NO: 2
- the sequence of the 5th intron of the Rf-1 gene is shown in SEQ ID NO: 3.
- the base sequence of bases 180 to 288 of SEQ ID NO: 1 corresponds to the base sequence of the fifth intron of the rice Rf-1 gene of SEQ ID NO: 3.
- PLC41 GUS-HPT was constructed according to the following procedure. First, PCR for amplifying the GUS-HPT fragment was performed. Primer GUS-HPT in pSB34 F consisting of 3 ′ portion of Tnos located downstream of GUS using pSB34 (Hiei and Komari, 2006) as a template and a sequence encoding SpeI downstream thereof, and Tons 3 located downstream of HPT 'PCR was performed using a primer GUS-HPT in pSB34R consisting of a part and a sequence encoding KpnI downstream thereof.
- the obtained GUS-HPT fragment was double-digested with SpeI and KpnI and ligated to the pLC41 vector previously double-digested with XbaI and KpnI to obtain pLC41 GUS-HPT.
- This vector was introduced into Agrobacterium LBA4404 by electroporation to obtain LBA4404 (pLC41 GUS-HPT) (left in FIG. 1-b).
- FIG. 4 Construction of binary vector pLC41 Barnase (FIG. 4) Construction of pLC41 Barnase was carried out by the following procedure. First, a Pnos-Barnase-T35S fragment (hereinafter referred to as a Barnase fragment) was synthesized and cloned into the EcoRV site of the pUC57 vector (Barnase / pUC57). Next, PCR was performed to amplify the Barnase fragment.
- a Pnos-Barnase-T35S fragment hereinafter referred to as a Barnase fragment
- pLC41 was digested with XbaI, dephosphorylated, and ligated with a SpeI-digested pCR4TOPO / Barnase Barnase fragment to obtain pLC41 Barnase (FIG. 4).
- This vector was introduced into Agrobacterium LBA4404 by electroporation to obtain LBA4404 (pLC41 Barnase) (right of FIG. 1-b).
- Double T-DNA type binary vector pLC41 GUS-HPT cotra Creation of Barnase (FIG. 5)
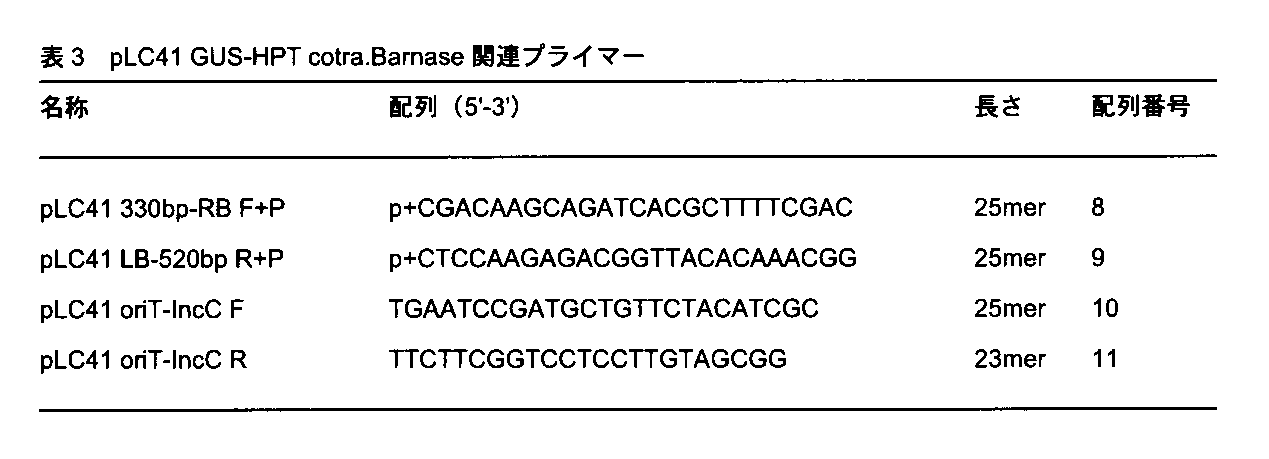
- pLC41 Barnase was used as a template, and RB-Barnase-LB from about 330 bp upstream of RB to about 520 bp downstream of RB, and pLC41 GUS-HPT from porc41 GUS-HPT to pLC41 GUS- from KorB to oriT PCR was performed to amplify HPT KorB to oriT.
- the PCR reaction for RB-Barnase-LB includes a 5 ′ terminal phosphorylated primer (pLC41 330 bp-RB F + P) consisting of a sequence encoding about 330 bp upstream portion of RB, and a sequence encoding about 520 bp downstream portion of LB.
- the 5 ′ terminal phosphorylated primer (pLC41 LB-520bp R + P) was used.
- primer pLC41 consisting of a sequence encoding downstream between oriT and IncC
- primer pLC41 consisting of a sequence encoding upstream between oriT-IncC and oriT-IncC oriT-IncCR was used.
- a PCR product of about 2280 bp was obtained for the RB-Barnase-LB fragment, and about 17000 bp for the pLC41 GUS-HPT KorB to oriT fragment.
- Patent document WO2007 / 148819 A1 contains pVGW2 which is a vector that can coexist with the binary vector pLC41 (Inc P type) in Agrobacterium and has pirBoN542D derived from pTiBo542. (Inc W type) is disclosed.
- the pVGW2 was modified to a simpler vector pGW (FIG. 6) from which virG N54D was removed.
- PCR was performed with pSaGW′EcT22I and M13 ( ⁇ 20) Fw primer set using pVGW2 as a template, and the resulting fragment was self-ligated to create a new cloning vector pGW for a ternary vector (FIG. 6).
- pGW can be maintained in the same Agrobacterium as a third vector (Turnary vector) together with a second vector (binary vector) pLC41, and T-DNA can be arranged separately in each vector. Therefore, it is possible to cotransform the target DNA linked with the positive selection marker gene and the negative selection marker gene using only one kind of Agrobacterium (FIG. 1-c).
- PCR was performed to add SpeI sites to both ends of the RB-Barnase-LB cassette using pLC41 Barnase as a template.
- a primer pLC41 330 bp-RB + SpeIF (10 pmol / ul) consisting of a sequence with a SpeI site added 330 bp upstream of RB and a primer pLC41 520 bp-LB + SpeI R consisting of a sequence with a SpeI site added 520 bp downstream of RB were used.
- a SpeI fragment of RB-Barnase-LB which is a 2300 bp PCR product, was obtained. This fragment was ligated to pGW previously digested with XbaI to obtain pGW Barnase in which the RB-Barnase-LB fragment was inserted in the positive direction.
- the completed pGW Barnase was introduced into Agrobacterium LBA4404 by electroporation simultaneously with pLC41 GUS-HPT to obtain LBA4404 (pLC41 GUS-HPT :: pGW Barnase) (FIG. 1-c).
- Example 2 Rice transformation material and method using co-transformation vector system
- Agrobacterium strain and vector to be tested A positive selection marker gene was ligated in the following three types of multi-copy cell exclusion co-transformation vector systems The target DNA and the negative selection marker gene were cotransformed.
- a. 1 strain 1 vector type: LBA4404 (pLC41 GUS-HPT cotra. Barnase) (FIG. 1-a)
- b. Two strains mixed type: LBA4404 (pLC41 GUS-HPT) + LBA4404 (pLC41 Barnase) (FIG. 1-b)
- LBA4404 pLC41 GUS-HPT :: pGW barnase
- Rice transformation method Yukihikari was used as the rice variety. Rice spikelets around 10 days after flowering cultivated in a greenhouse were collected. After sterilizing the immature seed from which the anther was removed with tweezers, an immature embryo having a length of 1.3 to 1.8 mm was collected under a stereomicroscope. These immature embryos were centrifuged for 10 minutes at a centrifugal acceleration of 20,000 ⁇ g.
- Agrobacterium was cultured on AB medium (Chilton et al., 1974) containing a selective drug according to the drug resistance of the strain for 3 days in the dark at 28 ° C., and then 1.0 ml of AA-inf medium (AA Major inorganic salts, B5 trace inorganic salts, B5 vitamins, AA amino acids, 0.1 mM acetosyringone, 20 g / l sucrose, 10 g / l glucose, 0.5 g / l vitamin assay casamino acids, pH 5.2) It became cloudy. The suspension concentration was adjusted to about 1.0 with an OD value of 660 nm.
- N6-As medium N6 inorganic salts and vitamins, 1 mg / l 2,4-D, 0.5 mg / l 6BA, 20 g / l sucrose, 10 g / l glucose, 0. 5 g / l proline, 0.5 g / l vitamin assay casamino acid, 8 g / l agarose Type I, 0.1 mM acetosyringone, pH 5.2
- immature embryos were placed with the scutellum side facing upward, A bacterial suspension was added dropwise. The co-culture was performed for 7 days in the dark at 25 ° C.
- nN6C medium N6 inorganic salts and vitamins, 1 mg / l 2,4-D, 0.5 mg / l 6BA, 20 g / l sucrose , 55 g / l sorbitol, 0.5 g / l proline, 0.5 g / l vitamin assay casamino acids, 5 g / l gellan gum, 250 mg / l cefotaxime, 100 mg / l carbenicillin, pH 5.8), 30 ° C., 5 Non-selective (resting) culture was performed for 10 days under light conditions of 1,000,000.
- nN6CH50 N6 inorganic salts and vitamins, 1 mg / l 2,4-D, 0.5 mg / l 6BA, 20 g / l sucrose, 55 g / l sorbitol, 0. 5 g / l proline, 0.5 g / l vitamin assay casamino acids, 5 g / l gellan gum, 250 mg / l cefotaxime, 100 mg / l carbenicillin, 50 mg / l hygromycin B, pH 5.8) placed on selective medium, 30 ° C. Selection culture with hygromycin was performed for 10-14 days under light conditions at 5,000 lx.
- the grown callus was regenerated into N6RH50 regeneration medium (N6 trace mineral salts and vitamins, 1/2 concentration N6 main inorganic salts, AA amino acids, 0.5 mg / l kinetin, 20 g / l sucrose, 30 g / l sorbitol, 0.5 g / L vitamin assay casamino acid, 4 g / l gellan gum, 50 mg / l hygromycin B, pH 5.8) and cultured for 14 days at 30 ° C. and 5,000 lx light conditions.
- the number of callus placed on the regeneration medium was one per subdivided immature embryo section. Thereby, each callus placed on the regeneration medium can be treated as an independent transformation event.
- N6FH50 rooting medium N6 trace mineral salts and vitamins, 1/2 concentration N6 main inorganic salts, AA amino acids, 20 g / l sucrose, 0.5 g / l vitamin assay casamino acid, 4 g / l gellan gum , 50 mg / l hygromycin B, pH 5.8, and cultured for 10-14 days under light conditions at 30 ° C. and 5,000 lx. The rooted plant was transplanted to a pot and cultivated in a greenhouse.
- FIG. 9 shows the result of Southern hybridization using an hpt probe. At least one band detected from each analyzed transformant showed a size of 6.7 kb or more. Detection of a band of 6.7 kb or more suggests that the entire GUS-HPT T-DNA has been introduced. In the control vector, 47% of the analyzed individuals were introduced with a single copy, whereas in the 1 strain 1 vector system, a single copy was introduced in 89% of the individuals more than twice. In addition, the remaining 11% of individuals showed introduction of 2 copies (FIG. 9).
- Example 3 Exclusion Material and Method of Multicopy Transformant by Barnase Utilization Cotransformation in Tobacco
- Transformation of Tobacco Seed of tobacco variety SR1 was sterilized with antiformin and rooting medium (1/2 concentration LS) Inoculated aseptically into inorganic salt, 1/2 concentration LS vitamin, 15 g / l sucrose, 3 g / l gellan gum, 250 mg / l cefotaxime, pH 5.8). After sowing, the cells were cultured under light conditions at 25 ° C. and grown until the cotyledons fully developed. A rectangular cotyledon section obtained by excising the tip and base of a fully expanded cotyledon with scissors was used for Agrobacterium infection.
- cotyledon slices were prepared from LSR liquid medium (LS inorganic salt, LS vitamin, 30 g / l sucrose, 0.5 g / l 2-morpholinoethanesulfonic acid monohydrate (MES), pH 5.8).
- the liquid medium was replaced with a suspension of Agrobacterium and immersed for 10 minutes.
- Agrobacterium suspension was prepared by culturing Agrobacterium on AB medium containing 50 mg / l kanamycin in the dark at 28 ° C. for 3 days and then suspending in LSR liquid medium. The suspension concentration was adjusted to 1.0 with an OD value of 660 nm.
- test was performed in two test groups, a control group inoculated with LBA4404 (pLC41 GUS-HPT) alone and a test group inoculated with two strains of LBA4404 (pLC41 Barnase) and LBA4404 (pLC41 GUS-HPT).
- the leaves were placed on an LSR solid medium containing 3 g / L gellan gum with the back of the leaf piece facing upward, and co-cultured for 2 days in the dark at 25 ° C.
- the leaf pieces were treated with LS-S medium (LS inorganic salt, LS vitamin, 10 mg / l 6- ( ⁇ , ⁇ -dimethylallylamino) purine (2 ip), 0.3 mg / l indole-3-acetic acid (IAA) , 30 g / l sucrose, 250 mg / l cefotaxime, pH 5.8) and cultured at 28 ° C. under light conditions for 2-4 days.
- LS-S medium LS inorganic salt, LS vitamin, 10 mg / l 6- ( ⁇ , ⁇ -dimethylallylamino) purine (2 ip), 0.3 mg / l indole-3-acetic acid (IAA) , 30 g / l sucrose, 250 mg / l cefotaxime,
- the cells were transplanted to an LS-S medium containing 50 mg / l hygromycin, and hygromycin resistant cells were selected.
- Hygromycin-resistant shoots (shoots) obtained from the cut ends of the leaf pieces were cut from the leaf pieces and transplanted to a rooting medium containing 50 mg / l hygromycin and 250 mg / l cefotaxime.
- the rooted plant was transplanted to a large culture container (length: 77 mm ⁇ width: 77 mm ⁇ height: 97 mm) having the same medium composition and cultured until sampling. The thus obtained hygromycin-resistant plant was used as a transformant in the subsequent analysis.
- PCR primers and TaqMan MGB probe were designed by Primer Express (Life Technologies). The designed primers and probe names are as follows, and their sequences are shown in Table 6. NtBWC1-5F and NtBWC1-5R were used as primers for internal standard, and NtBWC1-5P was used as TaqMan MGB probe for internal standard.
- Hpt-2F and Hpt-2R were used as primers for target DNA, and Hpt-2P was used as a TaqMan MGB probe for target DNA. All real-time PCR experiments were performed according to the following program. That is, 95 ° C for 30 seconds once, 95 ° C for 5 seconds and 60 ° C for 34 seconds for 40 times. Fluorescence emission was monitored with a 60 ° C. extension step in each cycle.
- Example 2 co-transformation was carried out by inoculating two strains in rice, but the transformation efficiency decreased only as little as 10%.
- the reason why the transformation efficiency was significantly lower in tobacco is thought to be because the efficiency of two types of T-DNA integration into the genome of the same cell, that is, the co-transformation efficiency, is higher than that of rice.
- the proportion of tobacco transformants having a single copy of the target gene increased by more than 3 times.
- the transformation efficiency decreased to about one third.
- multicopy-introduced cells can be eliminated at an early stage of culturing, the labor for culturing and the cost and labor for preparing the medium can be greatly reduced.
- the method of co-transforming a negative selection marker is a useful technique that can efficiently obtain a transformant having a target gene in a single copy as a whole.
- Example 4 Exclusion Material and Method of Multicopy Transformant by Barnase Utilization Cotransformation in Corn 1) Transformation of Corn Aseptically from a plant grown in a greenhouse with a maize immature embryo (variety: A188) having a size of about 1.2 mm And immersed in liquid medium LS-inf (Ishida et al., 2007). After heat treatment at 46 ° C. for 3 minutes, immature embryos were washed once with the same liquid medium, and then centrifuged at 20,000 G for 10 minutes (4 ° C.). Agrobacterium inoculation was performed by immersing immature embryos in an Agrobacterium suspension.
- Immature embryos attached with Agrobacterium were placed on LS-AS co-culture medium (Ishida et al., 2007) and co-cultured for 3 days in the dark at 25 ° C.
- the Agrobacterium suspension was prepared by culturing Agrobacterium on a YP medium in the dark at 28 ° C. for 2 days and then suspending it in an LS-inf-As liquid medium containing 0.1 mM acetosyringone. .
- the suspension concentration was adjusted to 1.0 with an OD value of 660 nm.
- the test consisted of a control group inoculated with LBA4404 (pSB131) (Ishida et al., 1996) alone, as well as LBA4404 (pLC41) in which a superternary vector pVGW9 (patent document WO2014157541A1) was introduced into LBA4404 (pSB131) and LBA4404 (pLC41 Barnase). It was carried out in two test sections of the test section in which two strains of Barnase :: pVGW9) were mixed and inoculated.
- pSB131 is a super binary vector
- the T-DNA has an intron GUS gene controlled by a 35S promoter and a bar gene controlled by a 35S promoter.
- the bar gene is a phosphinothricin (PPT) resistance gene.
- PPT phosphinothricin
- LSD1.5B selection medium Ishida et al., 2007
- primary selection was performed for 10 days in the dark at 25 ° C.
- the grown callus was transplanted as it was to a 10 mg / l PPT-containing LSD1.5B selection medium and subjected to secondary selection for about 3 weeks.
- the callus grown in the second round selection was cut into small pieces, and the third round selection was performed for about 3 weeks on the same composition medium.
- the callus grown in the third selection was cut into small pieces, placed on LSZ regeneration medium (Ishida et al., 2007) containing 5 mg / l PPT, and cultured under light conditions at 25 ° C. Two weeks later, PPT-resistant redifferentiated plants were transplanted to an LSF rooting medium (Ishida et al., 2007) containing 5 mg / l PPT, and cultured under the same conditions until sampling. The PPT-resistant plant thus obtained was used as a transformant for the subsequent analysis.
- Quantitative real-time PCR was used to measure the copy number of T-DNA integrated in the genome of maize transformant.
- a multiplex PCR method was employed in which the target DNA region and the internal standard DNA region were amplified in the same well. The target DNA region was within the bar gene. E. coli from the leaves of each transformant.
- Z. N. A. Genomic DNA was extracted using (registered trademark) SP Plant DNA Kit and prepared to a concentration of 15.625 ng / ⁇ l.
- Real-time PCR was performed in triplicate in 96-well PCR plates using an Applied Biosystems® 7500 real-time PCR system and performed twice.
- PCR primers and TaqMan MGB probe were designed by PrimerExpress. The designed primers and probe names are as follows, and their sequences are shown in Table 8.
- Hmg-2F and Hmg-2R were used as internal standard primers, and Hmg-2P was used as an internal standard TaqMan MGB probe.
- Bar-1F and Bar-1R were used as primers for target DNA, and Bar-1P was used as a TaqMan MGB probe for target DNA.
- All real-time PCR experiments were performed according to the following program. That is, 95 ° C for 30 seconds once, 95 ° C for 5 seconds and 60 ° C for 34 seconds for 40 times. Fluorescence emission was monitored with a 60 ° C. extension step in each cycle.
- a standard curve was prepared using serial dilution series with 5 concentrations of genomic DNA (48, 24, 12, 6 and 3 ng / ⁇ l). The number of copies was calculated by the method of Yang et al. (2005).
- the proportion of individuals with two or more copies of GUS-HPT introduced was 37%, a decrease of 17% (FIG. 11). This is because, when multiple copies of the target gene GUS-bar are introduced into the same genome, the barnase gene, which is a negative selection marker, is easily introduced together, and such cells can be introduced at an early stage of culture due to the expression of Barnase. This is because it was excluded.
- the ratio of maize transformants having the target gene in a single copy increased to 1.3 times or more of the control group.
- the transformation efficiency decreased to approximately two thirds.
- multicopy-introduced cells can be eliminated at an early stage of culturing, as in the case of tobacco in Example 3, the labor for culturing and the cost and labor for preparing the medium could be greatly reduced in corn.
- the method of co-transforming a negative selection marker is a useful technique for efficiently obtaining a transformant having a target gene in a single copy even in maize.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Cell Biology (AREA)
- Plant Pathology (AREA)
- Analytical Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Botany (AREA)
- Mycology (AREA)
- Immunology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Breeding Of Plants And Reproduction By Means Of Culturing (AREA)
Abstract
Description
植物を形質転換する際には、稔実種子の後代または栄養繁殖体において外来遺伝子を維持・発現させることのできる、植物細胞へのDNA導入技術が利用される。この技術を用いて、実際に多くの単子葉植物や双子葉植物が形質転換されてきた。
このような状況が背景にあるため、目的とするDNAを低コピーで導入する方法も数件報告されている。1つ目が、部位特異的組換えシステムを用いた方法である。まず、選抜マーカーを有する所望のDNAの両側に、部位特異的組換え酵素の認識配列(lox)を逆向きで配置したDNA断片を植物へ導入する。また予めCre発現カセットを別の植物へ導入しておく。次に、目的DNAが多コピー連結導入されたT0個体とCreを発現しているT0個体とを交配する。得られたF1個体では、Creが発現すると多コピー連結導入されたDNA領域から、その内側の順方向lox配列で挟まれたDNA領域だけがすべて切り出される。その結果、残存する両端のDNAが結合しシングルコピー分の目的DNAだけを有する細胞が得られる。その細胞が生殖細胞へ分化することにより、目的DNAがシングルコピーまたはシングルコピーホモとなったF2個体を得ることができる(Srivastava et al.,1999;De Buck et al.,2007)。しかしながら、交配による方法は、目的個体が得られるまで時間が掛かるという大きな欠点がある。また、エピジェネティックなTGSが発動した場合には、Cre-lox反応によりコピー数が減っても、所望のDNAの発現は抑制されたままとなる。
植物における外来DNAの共形質転換(co-transformation)は、目的DNAのみが導入された形質転換体を得るために用いられることが多い。選抜マーカー遺伝子と選抜薬剤は、ほとんどが非形質転換細胞からなる植物組織から形質転換細胞を得るための大変有用なツールであるが、形質転換体が得られた後は基本的には不要となる。アグロバクテリウム法では、共形質転換は主として選抜マーカー遺伝子を次世代で除去する目的で行われる。すなわち、目的とする外来DNAと選抜マーカー遺伝子を、それぞれ別々のT-DNA上に挿入し、アグロバクテリウムを介して同時に植物細胞に導入する。選抜薬剤で選ばれた細胞塊を再生させると選抜マーカー遺伝子とともに目的DNAが導入された形質転換体が得られる。もし、目的DNAと選抜マーカー遺伝子が別々の染色体に導入されていれば、遺伝的分離により、次世代において目的DNAは有するが選抜マーカー遺伝子が排除された個体を得ることができる(Yau and Stewart,2013)。また、アグロバクテリウムを介する共形質転換法には、1菌株中の2つのバイナリーベクター上へ2つのT-DNAを別々に配置したタイプ、1菌株中の1つのバイナリーベクター上へ2つのT-DNAを配置したタイプ、2菌株の各バイナリーベクターへ2つのT-DNAを別々に配置し両菌株で混合接種するタイプ、1菌株の1つのバイナリーベクター上へ2つの右ボーダー配列を配置したタイプの4種類がある(Yau and Stewart,2013)。
[態様1]
植物の形質転換細胞の取得方法であって、以下の工程:
(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程;及び
(b)工程(a)で得られた形質転換植物細胞の中から、染色体に、所望のDNAが導入され、かつ、第1マーカー遺伝子が導入されていない形質転換細胞を選択する工程
を含む、
但し、前記第1マーカー遺伝子を用いたポジティブ選抜によって、所望のDNAのみが染色体に導入された形質転換細胞が除去される工程は含まない、
前記方法。
[態様2]
第1マーカー遺伝子が、ネガティブ選抜マーカー遺伝子である、態様1に記載の方法
[態様3]
工程(a)において共形質転換に用いる所望のDNAと第1マーカー遺伝子の混合比率が3:1-1:5である、態様1又は2のいずれか1項に記載の方法
[態様4]
工程(a)に用いる所望のDNAに、第2のマーカー遺伝子であるポジティブ選抜マーカー遺伝子が連結されており、
工程(b)の染色体に所望のDNAが導入された形質転換細胞の選択を、第2マーカー遺伝子を用いたポジティブ選抜によって行う、態様1-3のいずれか1項に記載の方法
[態様5]
工程(a)が、アグロバクテリウム法、パーティクルガン法、エレクトロポレーション法、エレクトロインジェクション法、ポリエチレングリコール法及びウィスカー法からなる群から選択される、形質転換方法によって行われる、態様1-4のいずれか1項に記載の方法
[態様6]
態様1-5のいずれか1項に記載の方法で植物の形質転換細胞を取得し;
前記植物細胞を培養して植物体を得る、
ことを含む、形質転換植物の作成方法。
[態様7]
植物の形質転換方法であって、以下の工程:
(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程;及び
(b)工程(a)で得られた形質転換植物細胞の中から、染色体に、所望のDNAが導入され、かつ、第1マーカー遺伝子が導入されていない形質転換細胞を選択する工程
を含む、
但し、前記第1マーカー遺伝子を用いたポジティブ選抜によって、所望のDNAのみが染色体に導入された形質転換細胞が除去される工程は含まない、
前記方法。
(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程;及び
(b)工程(a)で得られた形質転換植物細胞の中から、染色体に、所望のDNAが導入され、かつ、第1マーカー遺伝子が導入されていない形質転換細胞を選択する工程
を含む、
但し、前記第1マーカー遺伝子を用いたポジティブ選抜によって、所望のDNAのみが染色体に導入された形質転換細胞が除去される工程は含まない、
前記方法。
本発明の方法の対象となる植物は特に限定されず、藻類、被子植物、裸子植物など任意の植物を含み、単子葉植物または双子葉植物であってよい。形質転換に供試する組織は、植物の種類や用いる形質転換方法に応じて適宜選択することができる。
本発明は、(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程を含む。本明細書における「マーカー遺伝子」とは、当該遺伝子が細胞に導入されたものあるいは導入されなかったものを選抜するための指標となる性質を有する遺伝子を意味する。
「所望のDNA」は、細胞への導入を行う任意のDNAであり、特段に限定されない。所望のDNAは植物細胞の染色体(ゲノム)に導入されるDNAであり、必ずしも構造遺伝子に限定されるものではなく、タンパク質をコードする構造遺伝子以外の非構造遺伝子を用いてもよい。「所望のDNA」には、所望のプロモーター及びターミネーターを連結してもよい。「所望のDNA」は、使用される形質転換方法に応じた任意の長さのものを使用可能である。例えば、アグロバクテリウム法を用いる場合、限定されるものではないが、0.1kb-50kbの長さのものが好ましい。
本発明において形質転換方法は、植物を形質転換できる方法であれば特に限定されず、植物の種類に応じて適宜選択することができる。例えば、パーティクルガン法、エレクトロポレーション法、エレクトロインジェクション法、ポリエチレングリコール法、ウィスカー法などの物理化学的方法(DNAの直接導入法)あるいはアグロバクテリウム法などの生物学的方法(DNAの間接導入法)を好ましく用いることができる。
本明細書において、外因性遺伝物質(外因性DNA)の導入、編入(および発現)の結果、得られる細胞の遺伝的な変更のことを形質転換と呼ぶ。また、その変更のための工程(プロセス)を指す言葉としても用いられる。そして、独立した二つ以上の外因性遺伝物質を同時に形質転換することを共形質転換(Co-transformation)と呼ぶ。同様に、その工程を指す言葉としても用いられる。なお、「独立した」とは、二つ以上の外因性遺伝物質が、一体のDNAとして導入されるのではなく、それぞれ独立して挙動可能なDNAとして、細胞に導入されることを意味する。
なお、所望のDNAに、第2のマーカー遺伝子としてポジティブ選抜マーカー遺伝子を連結してもよい。特に所望のDNAが形質転換の成否の指標とならない場合、即ち、所望のDNAがマーカー遺伝子としての性質を有していない場合、当該DNAにポジティブ選抜マーカー遺伝子を連結することが好ましい。
工程(a)に用いる所望のDNAに、第2のマーカー遺伝子であるポジティブ選抜マーカー遺伝子が連結されており、
工程(b)の染色体に所望のDNAが導入された形質転換細胞の選択を、第2マーカー遺伝子を用いたポジティブ選抜によって行う、
態様も含む。
本明細書において、形質転換細胞とは、外来遺伝子が形質転換工程を経て染色体ゲノムに組み込まれた細胞あるいはその後代をいう。
1)第1マーカー遺伝子が入っている形質転換細胞を除去し、そこから所望のDNAが入っている形質転換細胞を選択する;
2)所望のDNAが入っている形質転換細胞を選択し、そこから第1マーカー遺伝子が入っている形質転換細胞を除去する;あるいは
3)第1マーカー遺伝子が入っている形質転換細胞の除去と、所望のDNAが入っている形質転換細胞を選択、とを同時に行う
のいずれの態様でもよい。
外部から与えた化合物によって遺伝子発現を誘導する方法がある。本発明の方法において、このような特異的化合物による遺伝子発現誘導系を用いて、選抜マーカー遺伝子の発現時期を制御してもよい。特に第1マーカー遺伝子として、一過性レベルの発現で細胞死または細胞増殖停止を誘起するような強力なネガティブ選抜マーカー遺伝子を使用する場合には、形質転換効率の観点から、マーカー遺伝子の発現時期を染色体ゲノムに組み込まれた後へと遅らせることが好ましい。このような場合、下記の特異的化合物による遺伝子発現誘導系を用いて、発現時期を制御することが可能である。
本発明は、さらに形質転換植物の作成方法に関する。本発明の形質転換植物の作成方法は、本発明の植物の形質転換細胞を取得する方法によって、植物の形質転換細胞を取得し;前記植物細胞を培養して植物体を得る、ことを含む。
本発明はまた、植物の形質転換方法に関する。本発明の方法は、以下の工程:
(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程;及び
(b)工程(a)で得られた形質転換植物細胞の中から、染色体に、所望のDNAが導入され、かつ、第1マーカー遺伝子が導入されていない形質転換細胞を選択する工程を含み、
但し、前記第1マーカー遺伝子を用いたポジティブ選抜によって、所望のDNAのみが染色体に導入された形質転換細胞が除去される工程は含まない。
1)目的DNAおよびポジティブ選抜マーカー遺伝子
目的DNAとしてヒマのカタラーゼ遺伝子の第1イントロンが介在するβ―グルクロニダーゼ(GUS)遺伝子を、ポジティブ選抜マーカー遺伝子としてハイグロマイシン耐性(HPT)遺伝子を用いた。GUS遺伝子は35Sプロモーターで制御し、ターミネーターにはnosを用いた。HPT遺伝子はトウモロコシユビキチン(Ubi)遺伝子のプロモーターで制御し、ターミネーターにはnosを用いた。なお、HPT翻訳領域の上流には、トウモロコシのユビキチン遺伝子の第1イントロンを配置した。
ネガティブ選抜マーカー遺伝子には、バチルス・アミロリケファシエンスのBarnase遺伝子を用いた。発現はnosプロモーターで制御し、ターミネーターには35Sを用いた。また、Barnaseの発現は、大腸菌やアグロバクテリウムも致死させるため、Barnase遺伝子中にイネのRf-1遺伝子の第5イントロンを介在させた(Pnos-Barnase-T35S)。これにより、植物細胞内のみで発現するネガティブ選抜マーカーとすることができる。用いたバチルス・アミロリケファシエンスのBarnase遺伝子中にイネのRf-1遺伝子の第5イントロンを介在させた配列を配列番号1、バチルス・アミロリケファシエンスのBarnase遺伝子の配列を配列番号2,イネのRf-1遺伝子の第5イントロンの配列を配列番号3に示した。配列番号1の塩基180-288の塩基配列が配列番号3のイネのRf-1遺伝子の第5イントロンの塩基配列に相当する。
国際出願公開WO2007/148819 A1に記載されているコスミドベクターpLCleo(別名pLC41GWH)は、複製開始点oriVを有するIncPプラスミドである。oriVは大腸菌およびアグロバクテリウムの双方で機能する。pLC41GWHのEcoRI-PmeI断片およびマルチクローニングサイトを有するpSB11(Komari et al.,1996)のEcoRI-PmeI断片をライゲーションすることにより、T-DNA上にマルチクローニングサイトのみを有するバイナリーベクターpLC41(図3)を得た。

pLC41 Barnaseの構築は、以下の手順で行った。まず、Pnos-Barnase-T35S断片(以下、Barnase断片)について合成を行い、pUC57ベクターのEcoRVサイトにクローニングした(Barnase/pUC57)。次に、Barnase断片を増幅するPCRを行った。鋳型としてBarnase/pUC57、プライマーとしてM13シークエンシングプライマー領域をコードするM13/pUC 24merとpUC57のマルチクローニングサイトの3’部分にAvrIIをコードする配列を加えたpUC57 476(ArII) long Rを用いた。その結果、1120bpのPCR産物を得た。このBarnase断片をベクターpCR4 TOPO(Invitrogen社製)にTAクローニングし、pCR4TOPO/Barnaseを得た。

次に、pLC41 Barnaseを鋳型としてRBの約330bp上流からLBの約520bp下流までのRB-Barnase-LB、またpLC41 GUS-HPTを鋳型としてKorBからoriTまでのpLC41 GUS-HPT KorB to oriTを増幅するPCRを行った。RB-Barnase-LBのためのPCR反応には、RBの約330bp上流部分をコードする配列からなる5’末端リン酸化プライマー(pLC41 330bp-RB F+P)、LBの約520bp下流部分をコードする配列からなる5’末端リン酸化プライマー(pLC41 LB-520bp R+P)を用いた。pLC41 GUS-HPT KorB to oriTのためのPCR反応には、oriT-IncC間を下流方向にコードする配列からなるプライマーpLC41 oriT-IncC FおよびoriT-IncC間を上流方向にコードする配列からなるプライマーpLC41 oriT-IncC Rを用いた。その結果、RB-Barnase-LB断片では約2280bp、pLC41 GUS-HPT KorB to oriT断片では約17000bpのPCR産物を得た。
特許文献WO2007/148819 A1には、アグロバクテリウムの中でバイナリーベクターpLC41(Inc P型)と共存可能なベクターでpTiBo542由来のvirG N54Dを有する pVGW2(Inc W型)が開示されている。本実施例では、このpVGW2からvirG N54Dを除去したよりシンプルなベクターpGW(図6)へと改変した。具体的には、pVGW2を鋳型にpSa5‘EcT22IとM13(-20)FwのプライマーセットでPCRを行い、得られたフラグメントをセルフライゲーションさせてターナリーベクター用の新規クローニングベクターpGWを作成した(図6)。pGWは、第2のベクター(バイナリーベクター)pLC41とともに第3のベクター(ターナリーベクター)として同一アグロバクテリウム中で保持することができ、それぞれのベクターに別々にT-DNAを配置ことができる。したがって、1種類のアグロバクテリウムのみを用いてポジティブ選抜マーカー遺伝子を連結した目的DNAとネガティブ選抜マーカー遺伝子を共形質転換することができる(図1-c)。pLC41 Barnaseを鋳型にしてRB-Barnase-LBカセットの両端にSpeIサイトを付加するためのPCRを行った。PCR反応には、RBの330bp上流にSpeIサイトを付加した配列からなるプライマー pLC41 330bp-RB+SpeI F(10pmol/ul)およびLBの520bp下流にSpeIサイトを付加した配列からなるプライマーpLC41 520bp-LB+SpeI Rを用いた。その結果、2300bpのPCR産物であるRB-Barnase-LBのSpeI断片を得た。この断片を、あらかじめXbaIで消化しておいたpGWにライゲーションし、RB-Barnase-LB断片が正の向きで挿入されたpGW Barnaseを得た。
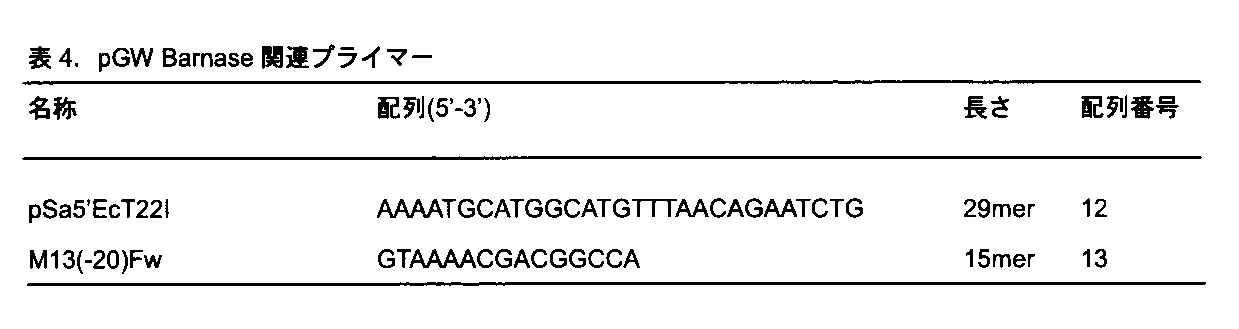
材料および方法
1)供試アグロバクテリウム菌株およびベクター
下記の3種類の多コピー細胞排除共形質転換ベクターシステムでポジティブ選抜マーカー遺伝子を連結した目的DNAとネガティブ選抜マーカー遺伝子の共形質転換を行った。
a.1菌株1ベクター型:LBA4404(pLC41 GUS-HPT cotra.Barnase) (図1-a)
b.2菌株混合型:LBA4404(pLC41 GUS-HPT) + LBA4404(pLC41 Barnase) (図1-b)
c.1菌株2ベクター型:LBA4404(pLC41 GUS-HPT:: pGW Barnase) (ターナリーベクターシステム)(図1-c)
イネ品種には、ゆきひかりを用いた。温室で栽培した開花後10日前後のイネの穂を採集した。頴をピンセットで除去した未熟種子を殺菌した後、実体顕微鏡下で1.3‐1.8mm長の未熟胚を採集した。これらの未熟胚に、20,000xgの遠心加速度で10分間の遠心処理を行った。アグロバクテリウムは、菌株の薬剤耐性に応じた選抜薬剤を含むAB培地(Chilton et al.,1974)上で、28℃暗黒下で3日間培養した後、1.0mlのAA-inf培地(AA主要無機塩類、B5微量無機塩類、B5ビタミン、AAアミノ酸、0.1mM アセトシリンゴン、20g/l ショ糖、10g/l グルコース、0.5g/l ビタミンアッセイカザミノ酸、pH5.2)中へ懸濁した。懸濁濃度は、660nmのOD値で約1.0に調整した。なお、2菌株を混合接種する場合は、2菌株ともOD値で約1.0に調整したのち、混合し接種した。次に未熟胚を、共存培養用のN6-As培地(N6無機塩類およびビタミン、1mg/l 2,4-D、0.5mg/l 6BA、20g/l ショ糖、10g/l グルコース、0.5g/l プロリン、0.5g/l ビタミンアッセイカザミノ酸、8g/l アガロース Type I、0.1mM アセトシリンゴン、pH5.2)上に、胚盤側を上向きにして未熟胚を置床し、アグロバクテリウムの懸濁液を滴下した。共存培養は、25℃暗黒下で7日間行った。
温室で栽培したハイグロマイシン耐性形質転換体の葉からフェノール‐クロロホルム抽出法によりゲノムDNAを抽出した。ゲノムDNAをKpnIで消化後、TypeIIアガロース(SIGMA社)を用いてTAE緩衝液中でアガロース電気泳動を行った。ナイロンメンブレンへアルカリブロッティングによる転写を行った後、hptをプローブとしてサザンハイブリダイゼーションを行った(図8-a)。また、1菌株1ベクター型のLBA4404(pLC41 GUS-HPT cotra.Barnase)(図1-a)を用いて得たイネ形質転換体51個体については、SalIで消化後、Barnaseをプローブとしたサザン解析を行った(図8-b)。
1)イネの形質転換結果
何れの試験区においても形質転換体は得られたが、対照区のGUS-HPTのみを導入した場合に比べて、Barnaseを共形質転換した試験区では、形質転換効率が低くなった(表5)。これは、ネガティブ選抜マーカーであるBarnaseがGUS-HPTと共にイネの同一細胞の染色体へ組み込まれ、Barnaseの発現によりその細胞が致死したことによるものである。Barnaseによるイネの細胞死は、胚盤表面の細胞が部分的に明らかな褐変を呈する形で現れる。これは、アグロバクテリウムを接種した日から10日-20日で観察されるようになる。まだ、選抜培養を開始する以前の段階であり、褐変はハイグロマイシンによる影響ではない。また、7日間の共存培養直後は、未熟胚の胚盤細胞での一過性GUS活性は妨げられることはなく、基質である5-ブロモ-4-クロロ-3-インドリル-β-D-グルクロン酸(X-Gluc)溶液を処理すると、対照ならびに3種試験区とも同等のGUS活性を示した。これは、Barnaseは、一過性発現のレベルでは、細胞を致死させないことを示している。

1菌株1ベクター:LBA4404(pLC41 GUS-HPT cotra.Barnase)
2菌株混合:LBA4404(pLC41 GUS-HPT) + LBA4404(pLC41 Barnase)
1菌株2ベクター:LBA4404(pLC41 GUS-HPT:: pGW Barnase)
hptプローブを用いてサザンハイブリダイゼーションを行った結果を図9に示した。解析した各々の形質転換体から検出されたバンドは、少なくとも1本が6.7kb以上のサイズを示した。6.7kb以上のバンドの検出は、GUS-HPTのT-DNA全体が導入されていることを示唆している。対照のベクターでは、解析個体のうち47%がシングルコピー導入であったのに対し、1菌株1ベクターシステムでは、2倍以上の89%の個体においてシングルコピー導入であった。また、残りの11%の個体はすべて2コピー導入を示した(図9)。2菌株混合システムと1菌株2ベクターシステムでは、シングルコピー個体の割合にほとんど差がなく、それぞれ67%および65%であり、対照の1.43倍および1.38倍へと増加した(図9)。このように、試験区では、顕著に低コピー導入個体の割合が増加したが、これは、共形質転換において、同一細胞の染色体へGUS-HPTが多コピー導入される場合には、ネガティブ選抜マーカーのBarnase遺伝子も一緒に導入される傾向があることを示している。Wang and Waterhouse(2000)は、アグロバクテリウム法で得たイネ形質転換体において、T-DNA同士が順方向にあるいは互いに逆方向に繰り返し連結された形で、同一遺伝子座に組み込まれている個体が多いことを報告している。ネガティブ選抜マーカー遺伝子との共形質転換は、そのような個体を生む細胞を致死させているものと推測される。
ネガティブ選抜マーカー遺伝子と、ポジティブ選抜マーカー遺伝子を含む目的DNAの共形質転換により、多コピー導入細胞を培養の初期に排除できることが分った。実施例では、3種類のアグロバクテリウムのベクターシステムを用いたが、このシステムに限らず、幅広い形質転換系に用いることができると容易に推測される。パーティクルガンなどのDNAの直接導入法についても共形質転換を通じて適用できることは、そのメカニズムから明らかである。直接導入法は、もともと多コピー導入の頻度が高いので、低コピー化には非常に有効である。
材料および方法
1)タバコの形質転換
タバコ品種SR1の種子をアンチホルミンにより殺菌処理し、発根培地(1/2濃度LS無機塩、1/2濃度LSビタミン、15g/l ショ糖、3g/l ジェランガム、250mg/l セフォタキシム、pH5.8)へ無菌的に播種した。播種後、25℃明条件下で培養し、子葉が完全に展開するまで育成した。完全展開した子葉の先端と基部をハサミで切除することによって得られる長方形の子葉切片を、アグロバクテリウムの感染に用いた。アグロバクテリウムの接種は、子葉切片をLSR液体培地(LS無機塩、LSビタミン、30g/l ショ糖、0.5g/l 2‐モルフォリノエタンスルホン酸一水和物(MES)、pH5.8)が入ったシャーレに集めたのち、当該液体培地をアグロバクテリウムの懸濁液に置き換えて10分間浸漬することにより行った。アグロバクテリウム懸濁液は、50mg/l カナマイシンが入ったAB培地上で28℃暗黒下においてアグロバクテリウムを3日間培養した後、LSR液体培地に懸濁することにより作成した。懸濁濃度は、660nmのOD値で1.0に調整した。試験は、LBA4404(pLC41 GUS-HPT)を単独接種する対照区、ならびにLBA4404(pLC41 Barnase)とLBA4404(pLC41 GUS-HPT)の2菌株を混合接種する試験区の2試験区で実施した。
タバコ形質転換体のゲノムに組み込まれたT-DNAのコピー数の測定には、定量的リアルタイムPCRを用いた。ターゲットDNA領域と内部標準DNA領域を同一ウェル内で増幅するマルチプレックス PCR法を採用した。ターゲットDNA領域はHPT遺伝子内とした。各形質転換体の葉からE.Z.N.A.(登録商標) SP Plant DNA Kit(Omega Bio-tek社)を用いてゲノムDNAを抽出し、12.5ng/μlの濃度に調製した。Applied Biosystems(登録商標)7500 リアルタイムPCRシステム(Life Technologies社)を用いて、96穴PCRプレート中で、リアルタイムPCRを3反復行い、これを2回実施した。PCR反応液には、25μl Premix Ex. Taq(TaKaRa社)と5μl テンプレートDNAと0.3-0.4μMのプライマー、および0.2-0.24μM のTaqMan MGBプローブ(Life Technologies社)を含み、全量は50μlとした。PCRプライマーとTaqMan MGBプローブは、PrimerExpress(Life Technologies社)によって設計した。設計したプライマーとプローブ名は以下のとおりであり、その配列は表6に示した。内部標準用プライマーとしてNtBWC1-5FおよびNtBWC1-5Rを、内部標準用TaqMan MGBプローブとしてNtBWC1-5Pを用いた。ターゲットDNA用プライマーとしてHpt-2FおよびHpt-2Rを、ターゲットDNA用TaqMan MGBプローブとしてHpt-2Pを用いた。すべてのリアルタイムPCR実験は、以下のプログラムに従い実施した。すなわち、95℃で30秒間を1回、95℃5秒間と60℃34秒間を40回である。蛍光発光は、各サイクルにおける60℃の伸長ステップでモニターされた。

1)タバコの形質転換結果
対照区のGUS-HPTのみを導入した場合に比べて、Barnase遺伝子を2菌株混合法で共形質転換した試験区では、形質転換効率が3分の1程度まで低くなった(表7)。形質転換効率の低下は、ネガティブ選抜マーカーであるBarnase遺伝子がGUS-HPT遺伝子と共にタバコの同一細胞のゲノムへ組み込まれ、Barnaseの安定発現によりその細胞が致死したことによるものと考えられる。試験区では、ハイグロマイシンを含むLS-S培地で選抜する段階で、葉片からのハイグロマイシン耐性の苗条の形成効率が3分の1程度に低下した。そのため、苗条を発根培地へ移植したり発根培地を作成したりする労力が大きく削減された。

定量的リアルタイムPCR法によるタバコへの導入DNAのコピー数を解析した。その結果を図10に示した。対照区であるLBA4404(pLC41 GUS-HPT)の単独接種では、解析個体のうち10%でGUS-HPTがシングルコピーで導入されており、残りの90%は複数コピーで導入されていた。これに対し、Barnase遺伝子を2菌株混合接種法で共形質転換した試験区では、対照の3倍以上の34%の個体においてGUS-HPTがシングルコピーで導入されていた(図10)。その一方で、GUS-HPTが3コピー以上導入された個体の割合は大きく減少していた(図10)。これは、同一ゲノムへ目的遺伝子であるGUS-HPTが多コピー導入される場合には、ネガティブ選抜マーカーであるBarnase遺伝子も一緒に導入されやすく、そのような細胞がBarnaseの発現により培養の早期段階で排除されたためである。
材料および方法
1)トウモロコシの形質転換
大きさ約1.2mmのトウモロコシ未熟胚(品種:A188)を温室栽培した植物から無菌的に取り出し、液体培地LS-inf(Ishida et al.,2007)に浸漬した。46℃、3分間の熱処理後、同液体培地で未熟胚を1回洗浄した後、20,000Gで10分間(4℃)の遠心処理を行った。アグロバクテリウムの接種は、未熟胚をアグロバクテリウム懸濁液に浸漬することにより行った。アグロバクテリウムが付着した未熟胚をLS-AS共存培地(Ishida et al.,2007)上に置床し、25℃暗黒下で、3日間の共存培養を行った。アグロバクテリウム懸濁液は、アグロバクテリウムをYP培地上で28℃暗黒下において2日間培養した後、0.1mMアセトシリンゴンを含むLS-inf‐As液体培地に懸濁することにより作成した。懸濁濃度は、660nmのOD値で1.0に調整した。試験は、LBA4404(pSB131)(Ishida et al.,1996)を単独接種する対照区、ならびにLBA4404(pSB131)とLBA4404(pLC41 Barnase)にスーパーターナリーベクターpVGW9(特許文献WO2014157541A1)を導入したLBA4404(pLC41 Barnase::pVGW9)の2菌株を混合接種する試験区の2試験区で実施した。pSB131はスーパーバイナリーベクターで、T-DNAには、35Sプロモーター制御のイントロンGUS遺伝子と35Sプロモーター制御のbar遺伝子を有する。bar遺伝子は、ホスフィノスリシン(PPT)耐性遺伝子である。
トウモロコシ形質転換体のゲノムに組み込まれたT-DNAのコピー数の測定には、定量的リアルタイムPCRを用いた。ターゲットDNA領域と内部標準DNA領域を同一ウェル内で増幅するマルチプレックス PCR法を採用した。ターゲットDNA領域はbar遺伝子内とした。各形質転換体の葉からE.Z.N.A.(登録商標) SP Plant DNA Kitを用いてゲノムDNAを抽出し、15.625ng/μlの濃度に調製した。Applied Biosystems(登録商標)7500 リアルタイムPCRシステムを用いて、96穴PCRプレート中で、リアルタイムPCRを3反復行い、これを2回実施した。PCR反応液には、25μl Premix Ex. Taqと5μlテンプレートDNAと0.3μMのプライマー、および0.2μM のTaqMan MGBプローブを含み、全量は50μlとした。PCRプライマーとTaqMan MGBプローブは、PrimerExpressによって設計した。設計したプライマーとプローブ名は以下のとおりであり、その配列は表8に示した。

1)トウモロコシの形質転換結果
対照区のGUS-barのみを導入した場合に比べて、Barnase遺伝子を2菌株混合法で共形質転換した試験区では、形質転換効率が3分の2程度まで低くなった(表9)。形質転換効率の低下は、ネガティブ選抜マーカーであるBarnase遺伝子がGUS-bar遺伝子と共にトウモロコシの同一細胞のゲノムへ組み込まれ、Barnaseの安定発現によりその細胞が致死したことによるものと考えられる。試験区では、2次選抜の終了時点で、PPT耐性カルスを生出した未熟胚の割合が3分の2程度に低下した。そのため、カルスの小片を切り取り3次選抜培地へ移植したり、3次選抜後のカルスの小片を再分化培地へ移植したり、再分化個体を発根培地へ移植したりする労力が3分の1削減された。トウモロコシの形質転換では、この間の労力が全体の労力の85%を占めるため、28%の労力が削減されたことになる。

定量的リアルタイムPCR法によるトウモロコシへの導入DNAのコピー数を解析した。その結果を図11に示した。対照区であるLBA4404(pSB131)の単独接種では、解析個体の46%においてGUS-barがシングルコピーで導入されおり、残りの54%は複数コピーで導入されていた。これに対し、Barnase遺伝子を2菌株混合接種法で共形質転換した試験区では、対照より17%多い63%の個体でGUS-barがシングルコピーで導入されていた(図11)。その一方で、GUS-HPTが2コピー以上導入された個体の割合は37%となり17%減少した(図11)。これは、同一ゲノムへ目的遺伝子であるGUS-barが多コピー導入される場合には、ネガティブ選抜マーカーのBarnase遺伝子も一緒に導入されやすく、そのような細胞がBarnaseの発現により培養の早期段階で排除されたためである。
Claims (7)
- 植物の形質転換細胞の取得方法であって、以下の工程:
(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程;及び
(b)工程(a)で得られた形質転換植物細胞の中から、染色体に、所望のDNAが導入され、かつ、第1マーカー遺伝子が導入されていない形質転換細胞を選択する工程
を含む、
但し、前記第1マーカー遺伝子を用いたポジティブ選抜によって、所望のDNAのみが染色体に導入された形質転換細胞が除去される工程は含まない、
前記方法。 - 第1マーカー遺伝子が、ネガティブ選抜マーカー遺伝子である、請求項1に記載の方法
- 工程(a)において共形質転換に用いる所望のDNAと第1マーカー遺伝子の混合比率が3:1-1:5である、請求項1又は2のいずれか1項に記載の方法
- 工程(a)に用いる所望のDNAに、第2のマーカー遺伝子であるポジティブ選抜マーカー遺伝子が連結されており、
工程(b)の染色体に所望のDNAが導入された形質転換細胞の選択を、第2マーカー遺伝子を用いたポジティブ選抜によって行う、請求項1-3のいずれか1項に記載の方法 - 工程(a)が、アグロバクテリウム法、パーティクルガン法、エレクトロポレーション法、エレクトロインジェクション法、ポリエチレングリコール法及びウィスカー法からなる群から選択される、形質転換方法によって行われる、請求項1-4のいずれか1項に記載の方法
- 請求項1-5のいずれか1項に記載の方法で植物の形質転換細胞を取得し;
前記植物細胞を培養して植物体を得る、
ことを含む、形質転換植物の作成方法。 - 植物の形質転換方法であって、以下の工程:
(a)所望のDNAと第1マーカー遺伝子とを、植物細胞に共形質転換する工程;及び
(b)工程(a)で得られた形質転換植物細胞の中から、染色体に、所望のDNAが導入され、かつ、第1マーカー遺伝子が導入されていない形質転換細胞を選択する工程
を含む、
但し、前記第1マーカー遺伝子を用いたポジティブ選抜によって、所望のDNAのみが染色体に導入された形質転換細胞が除去される工程は含まない、
前記方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2015215536A AU2015215536B2 (en) | 2014-02-06 | 2015-02-04 | Method for obtaining transformed cells of plant |
| CN201580007421.XA CN105960458B (zh) | 2014-02-06 | 2015-02-04 | 转化的植物细胞的取得方法 |
| US15/112,343 US11136587B2 (en) | 2014-02-06 | 2015-02-04 | Method of obtaining transformed plant cell |
| BR112016016772A BR112016016772A2 (pt) | 2014-02-06 | 2015-02-04 | Método para obter células vegetais transformadas |
| EP15745845.6A EP3103870B1 (en) | 2014-02-06 | 2015-02-04 | Method for obtaining transformed cells of plant |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JPPCT/JP2014/052765 | 2014-02-06 | ||
| PCT/JP2014/052765 WO2015118640A1 (ja) | 2014-02-06 | 2014-02-06 | 植物の形質転換細胞の取得方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015119166A1 true WO2015119166A1 (ja) | 2015-08-13 |
Family
ID=53777473
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/052765 WO2015118640A1 (ja) | 2014-02-06 | 2014-02-06 | 植物の形質転換細胞の取得方法 |
| PCT/JP2015/053137 WO2015119166A1 (ja) | 2014-02-06 | 2015-02-04 | 植物の形質転換細胞の取得方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/052765 WO2015118640A1 (ja) | 2014-02-06 | 2014-02-06 | 植物の形質転換細胞の取得方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11136587B2 (ja) |
| EP (1) | EP3103870B1 (ja) |
| JP (1) | JPWO2015118640A1 (ja) |
| CN (1) | CN105960458B (ja) |
| AU (1) | AU2015215536B2 (ja) |
| BR (1) | BR112016016772A2 (ja) |
| WO (2) | WO2015118640A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11180770B2 (en) | 2017-03-07 | 2021-11-23 | BASF Agricultural Solutions Seed US LLC | HPPD variants and methods of use |
| US11371056B2 (en) | 2017-03-07 | 2022-06-28 | BASF Agricultural Solutions Seed US LLC | HPPD variants and methods of use |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210032638A1 (en) * | 2019-07-29 | 2021-02-04 | The University Of Birmingham | Plasmid curing |
| WO2022026395A2 (en) * | 2020-07-31 | 2022-02-03 | Inari Agriculture Technology, Inc. | Excisable plant transgenic loci with signature protospacer adjacent motifs or signature guide rna recognition sites |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999001563A1 (en) * | 1997-06-30 | 1999-01-14 | Mogen International N.V. | Plasmids for plant transformation and method for using the same |
| WO2004092390A2 (en) * | 2003-04-09 | 2004-10-28 | Monsanto Technology Llc | Dna constructs and methods to enhance the production of commercially viable transgenic plants |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2239332A3 (en) * | 2001-07-06 | 2011-01-19 | Monsanto Technology LLC | Methods for enhancing segregation of transgenes in plants and compositions thereof |
| WO2008001414A1 (fr) | 2006-06-23 | 2008-01-03 | Japan Tobacco Inc. | Vecteur cosmide destiné à transformer une plante et son procédé d'utilisation |
| EP3023499A1 (en) | 2008-07-16 | 2016-05-25 | Monsanto Technology LLC | Methods and vectors for producing transgenic plants |
-
2014
- 2014-02-06 JP JP2014514945A patent/JPWO2015118640A1/ja active Pending
- 2014-02-06 WO PCT/JP2014/052765 patent/WO2015118640A1/ja active Application Filing
-
2015
- 2015-02-04 AU AU2015215536A patent/AU2015215536B2/en not_active Expired - Fee Related
- 2015-02-04 BR BR112016016772A patent/BR112016016772A2/pt not_active IP Right Cessation
- 2015-02-04 CN CN201580007421.XA patent/CN105960458B/zh not_active Expired - Fee Related
- 2015-02-04 EP EP15745845.6A patent/EP3103870B1/en active Active
- 2015-02-04 WO PCT/JP2015/053137 patent/WO2015119166A1/ja active Application Filing
- 2015-02-04 US US15/112,343 patent/US11136587B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999001563A1 (en) * | 1997-06-30 | 1999-01-14 | Mogen International N.V. | Plasmids for plant transformation and method for using the same |
| WO2004092390A2 (en) * | 2003-04-09 | 2004-10-28 | Monsanto Technology Llc | Dna constructs and methods to enhance the production of commercially viable transgenic plants |
Non-Patent Citations (3)
| Title |
|---|
| GALLEGO M.E. ET AL.: "Positive-negative selection and T-DNA stability in Arabidopsis transformation", PLANT MOL. BIOL., vol. 39, no. 1, January 1999 (1999-01-01), pages 83 - 93, XP002193570 * |
| KONDRAK M. ET AL.: "Generation of marker- and backbone-free transgenic potatoes by site- specific recombination and a bi-functional marker gene in a non-regular one-border agrobacterium transformation vector", TRANSGENIC. RES., vol. 15, no. 6, December 2006 (2006-12-01), pages 729 - 737, XP019454972 * |
| See also references of EP3103870A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11180770B2 (en) | 2017-03-07 | 2021-11-23 | BASF Agricultural Solutions Seed US LLC | HPPD variants and methods of use |
| US11371056B2 (en) | 2017-03-07 | 2022-06-28 | BASF Agricultural Solutions Seed US LLC | HPPD variants and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3103870A4 (en) | 2017-09-20 |
| US11136587B2 (en) | 2021-10-05 |
| BR112016016772A2 (pt) | 2017-10-03 |
| JPWO2015118640A1 (ja) | 2017-03-23 |
| AU2015215536A1 (en) | 2016-08-25 |
| WO2015118640A1 (ja) | 2015-08-13 |
| EP3103870A1 (en) | 2016-12-14 |
| CN105960458A (zh) | 2016-09-21 |
| AU2015215536B2 (en) | 2021-02-18 |
| US20160340682A1 (en) | 2016-11-24 |
| EP3103870B1 (en) | 2020-10-21 |
| CN105960458B (zh) | 2019-09-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Barampuram et al. | Recent advances in plant transformation | |
| JP2000083680A (ja) | 光誘導型プロモ―タ―の制御下に置かれた不定芽再分化遺伝子を選抜マ―カ―遺伝子とする植物への遺伝子導入方法及びこれに用いる植物への遺伝子導入用ベクタ― | |
| KR20140107419A (ko) | 애그로박테리움을 사용하는 개선된 형질전환 방법 | |
| Yamchi et al. | Proline accumulation in transgenic tobacco as a result of expression of Arabidopsis Δ 1-pyrroline-5-carboxylate synthetase (P5CS) during osmotic stress | |
| WO2015119166A1 (ja) | 植物の形質転換細胞の取得方法 | |
| US20240002870A1 (en) | Rapid transformation of monocot leaf explants | |
| CA3128376A1 (en) | Plant explant transformation | |
| AU2017329739A1 (en) | Targeted genome optimization in plants | |
| WO2023056236A1 (en) | Seedling germination and growth conditions | |
| US7238854B2 (en) | Method of controlling site-specific recombination | |
| US20240191248A1 (en) | Method for rapid genome modification in recalcitrant plants | |
| AU2010211450B2 (en) | Plant transformation using DNA minicircles | |
| US20060143737A1 (en) | Method of controlling gene silencing using site specific recombination | |
| US20170240911A1 (en) | Agrobacterium-mediated site specific integration | |
| JP5186683B2 (ja) | 形質転換植物体の作出方法及びその利用 | |
| JP4179217B2 (ja) | 新規ベクター及びこのベクターを用いて行う植物形質転換体の作出方法 | |
| JP4595631B2 (ja) | 選抜マーカー遺伝子の影響が排除された遺伝子導入細胞、組織又は植物の作成方法 | |
| KR101040579B1 (ko) | 스트레스 유도성 자가-절단형 식물형질전환 벡터 및 이를이용한 선발 마커 프리 식물체의 제조방법 | |
| Thomson | Genetic engineering of plants | |
| Lu et al. | Ethanol inducible isopentenyl transferase as a high efficiency marker for tobacco transformation | |
| Kumar et al. | Antibiotic Resistance Marker-Free Transgenic Plants | |
| Hegedus | 10 pORE Modular Vectors for Plant Transformation | |
| Akbudak | Applications of site-specific recombination systems in transgene expression and marker gene removal |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15745845 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015745845 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015745845 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15112343 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016016772 Country of ref document: BR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2015215536 Country of ref document: AU Date of ref document: 20150204 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| ENP | Entry into the national phase |
Ref document number: 112016016772 Country of ref document: BR Kind code of ref document: A2 Effective date: 20160720 |