WO2014034274A1 - オリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼおよびアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法 - Google Patents
オリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼおよびアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法 Download PDFInfo
- Publication number
- WO2014034274A1 WO2014034274A1 PCT/JP2013/068549 JP2013068549W WO2014034274A1 WO 2014034274 A1 WO2014034274 A1 WO 2014034274A1 JP 2013068549 W JP2013068549 W JP 2013068549W WO 2014034274 A1 WO2014034274 A1 WO 2014034274A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mannosyl
- acetylglucosamine
- phosphorylase
- oligosaccharide
- asparagine
- Prior art date
Links
- 0 C[C@](C(*)=C1O)OC(CO)[C@]1(C)O Chemical compound C[C@](C(*)=C1O)OC(CO)[C@]1(C)O 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1048—Glycosyltransferases (2.4)
- C12N9/1051—Hexosyltransferases (2.4.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/18—Preparation of compounds containing saccharide radicals produced by the action of a glycosyl transferase, e.g. alpha-, beta- or gamma-cyclodextrins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/005—Glycopeptides, glycoproteins
Definitions
- the present invention relates to a newly discovered oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase and an oligosaccharide synthesis reaction catalyzed by the enzyme, and the core sugar chain structure mannosyl of an asparagine-linked glycoprotein.
- - ⁇ -1,4-N-acetylglucosamine and mannosyl- ⁇ -1,4-N-acetylglucosaminosyl- ⁇ -1,4-N-acetylglucosamine hereinafter referred to as mannosyl- ⁇ -1,4-chitobiose
- mannosyl- ⁇ -1,4-chitobiose mannosyl- ⁇ -1,4-chitobiose
- sugar chains are said to be the third chain after nucleic acids and proteins, and their functions have been elucidated rapidly in recent years.
- asparagine-linked sugar chains are known to bind to proteins and are deeply involved in life phenomena such as cell differentiation, aging, and immune responses, and diseases such as cancer, viral infection, and inflammation.
- elucidation of sugar chain functions at the molecular level and further application to drug discovery using sugar chains are expected.
- an object of the present invention is to efficiently produce a core sugar chain structure of an asparagine-linked glycoprotein.
- mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase As a result of diligent studies to achieve the above-mentioned problems, a novel mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase was discovered. Then, using an oligosaccharide synthesis reaction catalyzed by mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase, the core sugar chain structure of expensive asparagine-linked glycoprotein, mannosyl- ⁇ -1,4-N-acetylglucosamine, and We have found that mannosyl- ⁇ -1,4-chitobiose can be produced in one step and can be prepared in large quantities by scaling up the reaction system, and the present invention has been completed.
- the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase according to claim 1 of the present invention comprises ⁇ -mannose 1-phosphate and N-acetylglucosamine or N-acetylglucosamino.
- chitobiose syl- ⁇ -1,4-N-acetylglucosamine
- the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase according to claim 2 of the present invention is A) Actions It acts on ⁇ -mannose 1-phosphate and N-acetylglucosamine or chitobiose to produce the core sugar chain structure mannosyl- ⁇ of asparagine-linked glycoprotein.
- the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase described in claim 3 of the present invention has one amino acid sequence described in SEQ ID NO: 1 or one in the amino acid sequence described in SEQ ID NO: 1. Alternatively, it is characterized in that several amino acid residues have a deleted, substituted, added or inserted sequence.
- the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase according to claim 4 of the present invention is derived from Bacteroides thetaiotaomicron.
- a vector according to claim 5 of the present invention is a vector encoding the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase according to claim 3 or 4, or a DNA according to SEQ ID NO: 2. It is characterized by including.
- transformant according to claim 6 of the present invention is characterized by being transformed with the vector according to claim 5.
- a method for producing the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase according to claim 7 of the present invention includes: A method for producing the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase according to any one of claims 1 to 4, comprising the step of culturing the transformant according to claim 6. And a step of recovering mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase produced in the culturing step.
- the method for producing a core sugar chain structure of an asparagine-linked glycoprotein according to claim 8 of the present invention comprises ⁇ -mannose 1-phosphate, N-acetylglucosamine, and any one of claims 1 to 4.
- the method for producing a core sugar chain structure of an asparagine-linked glycoprotein according to claim 9 of the present invention includes ⁇ -mannose 1-phosphate, chitobiose, and any one of claims 1 to 4.
- a step of performing an oligosaccharide synthesis reaction in a solution containing the oligosaccharide synthase mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase and a step of recovering mannosyl- ⁇ -1,4-chitobiose
- the method for producing a core sugar chain structure of asparagine-linked glycoprotein according to claim 10 of the present invention is characterized in that the solution has a pH of 5.0 to 7.0.
- an asparagine bond is obtained using ⁇ -mannose 1-phosphate and N-acetylglucosamine or chitobiose as starting materials by an oligosaccharide synthesis reaction catalyzed by mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase.
- the core sugar chain structure mannosyl- ⁇ -1,4-N-acetylglucosamine or mannosyl- ⁇ -1,4-chitobiose of the type glycoprotein can be easily produced in one step.
- FIG. 2 is a chromatogram of mannosyl- ⁇ -1,4-N-acetylglucosamine of Example 2.
- 4 is a graph showing the yield of mannosyl- ⁇ -1,4-N-acetylglucosamine of Example 2.
- FIG. 2 is a chromatogram of mannosyl- ⁇ -1,4-chitobiose of Example 2.
- 4 is a graph showing the yield of mannosyl- ⁇ -1,4-chitobiose of Example 2.
- FIG. 2 is a graph showing the optimum pH of mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase prepared in Example 1.
- FIG. 2 is a graph showing the pH stability of mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase prepared in Example 1.
- FIG. 3 is a graph showing the temperature stability of mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase prepared in Example 1.
- the core structure of an asparagine-linked sugar chain can be selectively produced by an oligosaccharide synthesis reaction that is the reverse reaction of the phosphorolysis reaction.
- mannosyl- ⁇ 1,4-N-acetylglucosamine can be produced.
- mannosyl- ⁇ -1,4-chitobiose is obtained. Can be manufactured.
- the mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention may have a chemical structure defined by the amino acid sequence shown in SEQ ID NO: 1.
- the mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention acts on ⁇ -mannose 1-phosphate and N-acetylglucosamine or chitobiose to produce the core sugar chain structure of asparagine-linked glycoprotein. If the enzyme produces mannosyl- ⁇ -1,4-N-acetylglucosamine or mannosyl- ⁇ -1,4-chitobiose, one or several amino acid residues in the amino acid sequence of SEQ ID NO: 1 are missing. Quality, substitution, addition or insertion. “Deletion, substitution, addition or insertion of amino acids” in this amino acid sequence can be carried out according to methods known to those skilled in the art (for example, mutagenesis and gene recombination methods).
- the mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention may be derived from any organism such as microorganisms, animals, plants, but preferably Bacteroides sp., Aristipes sp., Blautia sp., Cardisel Rosylptor, Capnocytophaga, Clostridium, Coprococcus, Dictyoglomus, Eubacterium, Faecaribacterium, Flavobacterium, Herpetosifon, Marinomonas, Mariprof Microorganisms belonging to the genera Particularly preferably Using an enzyme derived from Teroidesu-thetaiotaomicron.
- the vector of the present invention acts on ⁇ -mannose 1-phosphate and N-acetylglucosamine or chitobiose to act on the core sugar chain structure of asparagine-linked glycoprotein, mannosyl- ⁇ -1,4-N-acetylglucosamine or mannosyl
- Any vector containing a DNA encoding mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase that produces ⁇ -1,4-chitobiose may be used, and mannosyl- ⁇ -1,4-N described in SEQ ID NO: 1 -Acetylglucosamine phosphorylase, or mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase in which one or several amino acid residues are deleted, substituted, added or inserted in the amino acid sequence of SEQ ID NO: 1
- a vector containing the encoding DNA is preferred and is described in SEQ ID NO: 2.
- Vector containing the NA is especially preferred
- the expression vector of the present invention can be obtained by inserting the gene or DNA of SEQ ID NO: 2 or a modified form thereof into an appropriate vector.
- the expression vector of the present invention is not particularly limited as long as it can replicate in the host, and may be any of plasmid DNA, phage DNA, baculovirus such as AcMNPV, and the like.
- plasmid DNA include plasmids derived from E. coli (eg, pBR322, pUC118, etc.), plasmids derived from Bacillus subtilis (eg, pUB110, pTP5, etc.), yeast-derived plasmids (eg, YEp13, YEp24, YCp50, etc.), and the like.
- the expression vector of the present invention includes, for example, a replication origin, a selection marker, a promoter, an enhancer, so that the function of the gene encoding the protein of the present invention is exhibited.
- a terminator, a ribosome binding site, a polyadenylation signal and the like may be incorporated.
- replication origins for example, vectors derived from ColE1, R factor, and F factor are used for E.
- yeast vectors are derived from, for example, 2 ⁇ m DNA, ARS1, and vectors for animal cells are SV40, adeno. Virus-derived ones can be used.
- the promoter include trp promoter, lac promoter, PL promoter, PR promoter, etc. for vectors for E. coli, and yeast vectors include gal1 promoter, PHO5 promoter, PGK promoter, GAP promoter, ADH promoter, AOX1 promoter, etc.
- SR ⁇ promoter, SV40 promoter, LTR promoter, CMV promoter and the like can be used for the animal cell vector.
- vectors for Escherichia coli include kanamycin resistance gene, ampicillin resistance gene, tetracycline resistance gene, etc.
- yeast vectors include Leu2, Trp1, Ura3 gene, etc.
- animal cell vectors include neomycin resistance gene.
- Thymidine kinase gene, dihydrofolate reductase gene and the like can be used.
- a commercially available vector can be used as a vector into which the gene or DNA of SEQ ID NO: 2 or a modified product thereof is inserted. When such a vector is E.
- coli for example, when the host is yeast such as pET vector (Novagen), pTrxFUS vector (Invitrogen), pCYB vector (New England BioLabs), pCold vector (Takara Bio), etc., for example, pEP-1 expression
- yeast such as pET vector (Novagen), pTrxFUS vector (Invitrogen), pCYB vector (New England BioLabs), pCold vector (Takara Bio), etc.
- pEP-1 expression When a vector (manufactured by Stratagene), pAUR123 vector (manufactured by Takara Bio), pPIC vector (manufactured by Invitrogen) or the like is an animal cell, for example, a pMAM-neo expression vector (manufactured by CLONTECH), pCDNA3.
- pBK-CMV vector manufactured by Stratagene
- pBacPAK vector manufactured by CLONTECH
- pAcUW31 vector manufactured by CLONTECH
- pAcP (+) IE1 examples thereof include vectors (manufactured by Novagen).
- a tag may be incorporated into the expression vector of the present invention in order to facilitate detection and purification of the expressed protein. Examples of tags include glutathione-S-transferase, maltose binding protein, histidine (His6) and the like. Affinity purification becomes possible by using a carrier having affinity for these tags (a carrier chelated with nickel in the case of histidine).
- the insertion of a gene or the like into a vector includes, for example, a method in which the base sequence of the purified gene is cleaved with a suitable restriction enzyme, inserted into a restriction enzyme site or a multicloning site of a suitable vector DNA, and linked to the vector. Although it can be used, it is not limited to these. Moreover, you may insert what fused the gene which codes the protein of this invention, and the sequence which another protein codes.
- These expression vectors can be prepared from Escherichia coli or Agrobacterium by an alkali extraction method or a modified method thereof.
- the transformant of the present invention may be a transformant transformed by incorporating the expression vector of the present invention into a host.
- the host is not particularly limited as long as it can express the gene encoding the protein of the present invention.
- bacteria belonging to the genus Escherichia such as Escherichia coli, Bacillus genus such as Bacillus subtilis, Pseudomonas putida and Pseudomonas genus such as Saccharomyces cerevisiae S.
- the expression vector can be introduced into the host by a known method according to the type of the host, for example, calcium chloride method, calcium phosphate method, electroporation method, spheroplast method, lithium acetate method, lipofection method, etc. Is mentioned.
- gene transfer into each of the above host cells can be performed by a method not using a recombinant vector, such as a particle gun method.
- the method for producing mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention comprises culturing the transformant and the mannosyl- ⁇ -1,4-N-acetyl expressed in the culturing step. And a step of recovering glucosamine phosphorylase.
- the culturing method is performed according to a usual method used for culturing host cells.
- a medium for culturing a transformant using a microorganism such as Escherichia coli as a host, any medium that contains a carbon source, a nitrogen source, an inorganic salt, etc. that can be assimilated by the microorganism can be used.
- mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention is not particularly limited.
- the mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase is produced in cells or cells, the mannosyl- ⁇ -1,4-N-acetylglucosamine is disrupted by disrupting the cells or cells.
- the phosphorylase is recovered.
- the mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention is produced outside the cells or outside the cells, the culture solution is used as it is, or the cells or cells are removed by centrifugation or the like.
- mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase of the present invention can be isolated and purified from the culture.
- other proteins are inactivated by heat treatment or the like, so that it can be practically used as the mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase enzyme solution of the present invention.
- Mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase is not particularly limited, and an enzyme of any origin can be used.
- the usage form of this enzyme is not particularly limited, and various enzymes such as a bacterial cell extract, purified enzyme, and immobilized enzyme can be used.
- Saccharides used as starting materials may be of any origin.
- the concentration of carbohydrate added to the reaction system is not particularly limited, but is preferably about 0.1 to about 1000 g / L, more preferably about 0.1 to about 200 g / L.
- the concentration of mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase in the reaction solution is not particularly limited, but may be 0.76 to 76 ⁇ M, preferably 1.5 to 3.8 ⁇ M.
- the solution preferably has a pH of 5.0 to 7.0, particularly pH 5. 5 is preferred.
- the solution is not particularly limited, but an acetate buffer solution is preferable.
- the oligosaccharide synthesis reaction is preferably performed at 30 to 60 ° C., 30 ° C. is preferred.
- the reaction time is not particularly limited, but is preferably 30 to 60 minutes, and particularly preferably 60 minutes.
- the core sugar chain structure mannosyl- ⁇ -1,4-N-acetylglucosamine and mannosyl- ⁇ -1,4-chitobiose of the asparagine-linked glycoprotein produced by the above-mentioned oligosaccharide synthesis reaction include column chromatography, crystallization, etc. It is possible to isolate by known methods. Examples of column chromatography include, but are not limited to, size exclusion chromatography, silica gel column chromatography, ion exchange chromatography, ultrafiltration membrane separation, and reverse osmosis membrane separation. Crystallization methods include, but are not limited to, concentration, temperature reduction, and solvent addition (ethanol, methanol, acetone, etc.).
- a forward primer (SEQ ID NO: 3) and a reverse primer (SEQ ID NO: 4) for the BT1033 gene were designed and synthesized.
- the base sequence of the BT1033 gene is shown in SEQ ID NO: 2, and the amino acid sequence encoded by this base sequence is shown in SEQ ID NO: 1.
- the obtained amplified fragment was digested with restriction enzymes NdeI and XhoI, and then ligated to a commercially available gene expression plasmid pET-24a (manufactured by Novagen) similarly treated using a high efficiency ligation reagent Ligation high (manufactured by TOYOBO). . Further, an Escherichia coli competent cell DH5 ⁇ (manufactured by TOYOBO) was transformed with the ligation reaction solution, and an expression vector pET-24a containing DNA encoding BT1033 to which a His-tag consisting of 6-residue histidine was added at the C-terminus. was recovered.
- Escherichia coli BL21 (DE3) was transformed according to the method of Hanahan et al. (J. Mol. Biol., 1983, 166, 557-580).
- the transformant was inoculated into 200 mL of LB medium containing 50 ⁇ g / mL kanamycin, and induced culture was performed at 18 ° C. for 24 hours with an IPTG concentration of 0.1 mM.
- the cells recovered from the culture by centrifugation are suspended in 10 mL of 20 mM HEPES-NaOH buffer pH 7.5 containing 500 mM sodium chloride and 10% glycerol, disrupted by sonication, and then centrifuged to obtain the crude enzyme solution.
- the recombinant protein was purified by column chromatography using a His tag protein purification column HisTrapFF (manufactured by GE Healthcare).
- the obtained purified enzyme solution was dialyzed against 10 mM HEPES-NaOH buffer pH 7.0 and concentrated by ultrafiltration using a centrifugal filter unit Amicon Ultra-15 (Millipore). 6 mL of purified enzyme preparation was prepared.
- this protein was identified as a novel enzyme mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase by the following method, and the core sugar chain structure mannosyl- ⁇ of asparagine-linked glycoprotein. -1,4-N-acetylglucosamine and mannosyl- ⁇ -1,4-chitobiose were produced.
- the optimum pH of mannosyl- ⁇ -1,4-N-acetylglucosamine phosphorylase at 30 ° C. is around 5.5 (FIG. 3A), and the stable pH range is 4 ° C. for 24 hours under the condition of pH 4.5-10. .5 (FIG. 3B).
- the stability of the enzyme at pH 5.5 was up to 60 ° C. (FIG. 3C). It was found that the oligosaccharide synthesis reaction using this enzyme preferably has a pH of 5.0 to 7.0 and 30 to 60 ° C.
- the present invention can be used in the medical drug discovery industry and the oligosaccharide manufacturing industry.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
【課題】効率的にアスパラギン結合型糖タンパク質のコア糖鎖構造を製造することを目的とする。 【解決手段】本発明は、マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼが触媒するオリゴ糖合成反応により、α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとを出発材料として、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースをワンステップで簡便に製造する方法を提供する。
Description
本発明は、新規に発見したオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼおよび前記酵素が触媒するオリゴ糖合成反応を用いた、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンおよびマンノシル-β-1,4-N-アセチルグルコサミノシル-β-1,4-N-アセチルグルコサミン(以下、マンノシル-β-1,4-キトビオースという)の製造方法に関する。
糖タンパク質が有する糖鎖の生体認識(細胞接着・抗原抗体反応・情報伝達・ウイルス感染など)への重要性については近年注目が集まるところである。糖鎖は、核酸、タンパク質に次ぐ第三の鎖といわれ、近年急速にその機能解明が進められている。その中で、アスパラギン結合型糖鎖は、タンパク質などに結合し、細胞分化、老化、免疫応答といった生命現象や、癌、ウイルス感染、炎症などの疾患に深く関与していることが知られている。さらに、分子レベルでの糖鎖機能の解明、さらには糖鎖を利用した創薬への応用が期待されている。
しかし、生体内での発現量が微量な糖鎖試料の調製は現在有機合成法に頼らざるを得ず、その困難さが糖鎖工学研究分野や糖鎖再生医療の進展を妨げている。例えば、アスパラギン結合型3糖は、従来は、有機合成法による煩雑な多段階反応で製造されており(特許文献1)、効率的な大量調製が困難であるため、非常に高額であるという問題点があった。そのため、糖鎖の簡便な製造法の確立は急務となっている。
そこで、本発明は上記問題点に鑑み、効率的にアスパラギン結合型糖タンパク質のコア糖鎖構造を製造することを目的とする。
上記課題を達成するため鋭意検討した結果、新規にマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを発見した。そしてマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼが触媒するオリゴ糖合成反応を用いて、高価なアスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンおよびマンノシル-β-1,4-キトビオースをワンステップで製造することができ、反応系のスケールアップによる大量調製も可能であることを見出し、本発明を完成させた。
すなわち、本発明の請求項1に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、α-マンノース1-リン酸と、N-アセチルグルコサミンまたはN-アセチルグルコサミノシル-β-1,4-N-アセチルグルコサミン(以下、キトビオースという)とに作用して、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成することを特徴とする。
また、本発明の請求項2に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、
以下の酵素学的性質を有することを特徴とする
a)作用
α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用して、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成する;
b)基質特異性
α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用する;
c)至適pH
30℃の条件下で、pH5.5;
d)温度安定性
pH5.5の条件下で、60℃まで安定;
e)pH安定性
4℃、24時間の条件下で、pH4.5-10.5で安定。
以下の酵素学的性質を有することを特徴とする
a)作用
α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用して、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成する;
b)基質特異性
α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用する;
c)至適pH
30℃の条件下で、pH5.5;
d)温度安定性
pH5.5の条件下で、60℃まで安定;
e)pH安定性
4℃、24時間の条件下で、pH4.5-10.5で安定。
また、本発明の請求項3に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、配列番号1記載のアミノ酸配列、または配列番号1記載のアミノ酸配列において、1つ若しくは数個のアミノ酸残基が、欠失、置換、付加、または挿入された配列を有することを特徴とする。
また、本発明の請求項4に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、バクテロイデス・シータイオタオミクロン由来であることを特徴とする。
また、本発明の請求項5に記載のベクターは、請求項3または4に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼをコードするベクターまたは配列番号2記載のDNAを含むことを特徴とする。
また、本発明の請求項6に記載の形質転換体は、請求項5記載のベクターにより形質転換されたことを特徴とする。
また、本発明の請求項7に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの製造方法は、
請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの製造方法であって、請求項6記載の形質転換体を培養する工程と、前記培養する工程において生産されたマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを回収する工程を含むことを特徴とする。
請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの製造方法であって、請求項6記載の形質転換体を培養する工程と、前記培養する工程において生産されたマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを回収する工程を含むことを特徴とする。
また、本発明の請求項8に記載のアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法は、α-マンノース1-リン酸と、N-アセチルグルコサミンと、請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを含む溶液中でオリゴ糖合成反応を行う工程と、マンノシル-β-1,4-N-アセチルグルコサミンを回収する工程を含むことを特徴とする。
また、本発明の請求項9に記載のアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法は、α-マンノース1-リン酸と、キトビオースと、請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを含む溶液中でオリゴ糖合成反応を行うステップと、マンノシル-β-1,4-キトビオースを回収するステップを含むことを特徴とする。
また、本発明の請求項10に記載のアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法は、前記溶液が、pH5.0~7.0であることを特徴とする。
本発明によれば、マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼが触媒するオリゴ糖合成反応により、α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースを出発材料として、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースをワンステップで簡便に製造することができる。
本発明の方法によれば、加リン酸分解反応の逆反応であるオリゴ糖合成反応により、アスパラギン結合型糖鎖のコア構造を選択的に製造できる。
具体的には、α-マンノース1-リン酸と、N-アセチルグルコサミンと、マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを含む溶液中でオリゴ糖合成反応を行うことにより、マンノシル-β-1,4-N-アセチルグルコサミンを製造することができる。
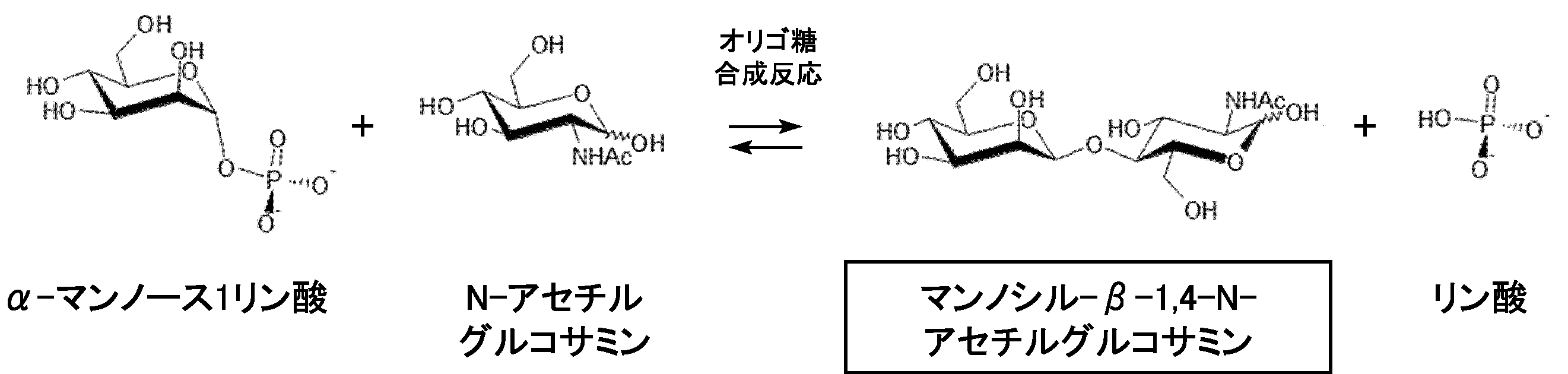
また、α-マンノース1-リン酸と、キトビオースと、マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを含む溶液中でオリゴ糖合成反応を行うことにより、マンノシル-β-1,4-キトビオースを製造することができる。

本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、配列番号1に示すアミノ酸配列で規定される化学構造を持つものでもよい。なお、本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用してアスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成する酵素であれば、配列番号1のアミノ酸配列において、一つ若しくは数個のアミノ酸残基が、欠質、置換、付加若しくは挿入されていてもよい。このアミノ酸配列における「アミノ酸の欠失、置換、付加若しくは挿入」は、当業者に公知の方法(例えば、突然変異誘発法や遺伝子組み換え法)に従って実施できる。
本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは、微生物、動物、植物等どのような生物に由来するものでも良いが、好ましくはバクテロイデス属、アリスティペス属、ブラウティア属、カルジセルロシルプトル属、カプノサイトファーガ属、クロストリジウム属、コプロコッカス属、ディクチオグロムス属、ユーバクテリウム属、ファエカリバクテリウム属、フラボバクテリウム属、ヘルペトシフォン属、マリノモナス属、マリプロフンドゥス属、マルビンブリャンティア属、オリバクテリウム属、パエニバチスル属、パラバクテロイデス属、プレボテラ属、ロゼブリア属、ルミノコッカス属、テルモトガ属、テルモシフォ属、ウェルコミクロビアエ属、ビブリオ属に属する微生物、とりわけ好ましくはバクテロイデス・シータイオタオミクロン由来の酵素を用いる。
本発明のベクターは、α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用してアスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成するマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼをコードするDNAを含むベクターであればよく、配列番号1記載のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼ、または配列番号1のアミノ酸配列において、一つ若しくは数個のアミノ酸残基が、欠質、置換、付加若しくは挿入されているマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼをコードするDNAを含むベクターが好ましく、配列番号2記載のDNAを含むベクターがとりわけ好ましい。
本発明の発現ベクターは、前記遺伝子もしくは配列番号2のDNAまたはその修飾体を適当なベクターに挿入することによって得ることができる。本発明の発現ベクターは、宿主中で複製可能なものであれば、特に制限されるものではなく、例えば、プラスミドDNA、ファージDNA、AcMNPVなどのバキュロウイルス等のいずれであってもよい。プラスミドDNAとしては、大腸菌由来のプラスミド(例えばpBR322、pUC118等)、枯草菌由来のプラスミド(例えばpUB110、pTP5等)、酵母由来のプラスミド(例えばYEp13、YEp24、YCp50等)などが挙げられ、ファージDNAとしてはλファージ等が挙げられる。さらに、レトロウイルスまたはワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクター等を用いることもできる。また、本発明のタンパク質をコードする遺伝子の機能が発揮されるように、本発明の発現ベクターには本発明のタンパク質をコードする遺伝子のほか、例えば、複製開始点、選択マーカー、プロモーター、エンハンサー、ターミネーター、リボソーム結合部位、ポリアデニル化シグナル等を組み込んでいてもよい。複製開始点としては、大腸菌用ベクターには、例えばColE1,R因子,F因子由来のものが、酵母用ベクターには、例えば2μmDNA,ARS1由来のものが、動物細胞用ベクターには、SV40,アデノウイルス由来のものが用いることができる。プロモーターとしては、大腸菌用ベクターには、trpプロモーター、lacプロモーター、PLプロモーター、PRプロモーター等が、酵母用ベクターには、gal1プロモーター、PHO5プロモーター、PGKプロモーター、GAPプロモーター、ADHプロモーター、AOX1プロモーター等が、動物細胞用ベクターには、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMVプロモーター等が用いることができる。選択マーカーとしては、大腸菌用ベクターには、カナマイシン耐性遺伝子、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子等が、酵母用ベクターには、Leu2,Trp1,Ura3遺伝子等が、動物細胞用ベクターには、ネオマイシン耐性遺伝子、チミジンキナーゼ遺伝子、ジヒドロ葉酸還元酵素遺伝子等が用いることができる。また、前記遺伝子もしくは配列番号2のDNAまたはその修飾体を挿入するベクターとして、商業的に入手可能なものを使用することができるが、そのようなベクターには、宿主が大腸菌である場合は、例えばpETベクター(Novagen社製)、pTrxFUSベクター(Invitrogen社製)、pCYBベクター(New England BioLabs社製)pColdベクター(タカラバイオ社製)等が、宿主が酵母である場合は、例えばpEP-1発現ベクター(Stratagene社製)、pAUR123ベクター(タカラバイオ社製)、pPICベクター(Invitrogen社製)等が、また宿主が動物細胞である場合は、例えばpMAM-neo発現ベクター(CLONTECH社製)、pCDNA3.1ベクター(Invitrogen社製)、pBK-CMVベクター(Stratagene社製)等が、宿主が昆虫細胞である場合は、例えばpBacPAKベクター(CLONTECH社製)、pAcUW31ベクター(CLONTECH社製)、pAcP(+)IE1ベクター(Novagen社製)等がそれぞれ挙げられる。また、本発明の発現ベクターには、発現されるタンパク質の検出および精製を容易にするためにタグを組み込んでもよい。タグの例としては、例えば、グルタチオン-S-トランスフェラーゼ、マルトース結合プロテイン、ヒスチジン(His6)等が挙げられる。これらのタグに親和性のある担体(ヒスチジンであればニッケルをキレートした担体)を用いることにより、アフィニティー精製が可能となる。
ベクターへの遺伝子等の挿入は、例えば、精製された遺伝子の塩基配列を適当な制限酵素で切断し、適当なベクターDNAの制限酵素部位またはマルチクローニングサイトに挿入してベクターに連結する方法などを用いることができるが、これらに限定されない。また、本発明のタンパク質をコードする遺伝子と他のタンパク質のコードする配列を融合したものを挿入してもよい。これら発現ベクターは、大腸菌やアグロバクテリウムからアルカリ抽出法またはその変法等により調製できる。
本発明の形質転換体は、前記本発明の発現ベクターを宿主に組み込むことにより形質転換された形質転換体であれば良い。なお、宿主としては、本発明のタンパク質をコードする遺伝子を発現できるものであれば、特に制限されるものではない。例えば、大腸菌(Escherichia coli)等のエシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)等のバチルス属、シュードモナス・プチダ(Pseudomonas putida)等のシュードモナス属に属する細菌、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)およびピキア・パストリス(Pichia pastoris)等の酵母、サル細胞COS-7、Vero、チャイニーズハムスター卵巣細胞(CHO細胞)、マウスL細胞、ヒトGH3、ヒトFL細胞等の動物細胞、あるいはSf9、Sf21等の昆虫細胞が挙げられる。
宿主への発現ベクターの導入は、宿主の種類に応じて公知の方法で行うことができ、例えば、塩化カルシウム法、リン酸カルシウム法、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法、リポフェクション法等が挙げられる。また、上記の各宿主細胞への遺伝子導入は、組換えベクターによらない方法、例えばパーティクルガン法等も用いることができる。
本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの製造方法は、前記形質転換体を培養する工程と、前記培養する工程において発現した前記マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを回収する工程とを含む製造方法である。前記培養する方法は、宿主細胞の培養に用いられる通常の方法に従って行われる。大腸菌等の微生物を宿主とした形質転換体を培養する培地としては、微生物が資化し得る炭素源、窒素源、無機塩類などを含有し、形質転換体の培養を効率的に行えるものであれば、天然培地、合成培地などのいずれを用いてもよい。本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの回収は、特に制限されない。前記マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼが菌体内または細胞内に生産される場合には、菌体または細胞を破砕することによって前記マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを回収する。また、本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼが菌体外または細胞外に生産される場合には、培養液をそのまま使用するか、遠心分離などにより菌体または細胞を除去した後、タンパク質の単離精製に用いられる一般的な生化学的方法、例えば、硫酸アンモニウム沈殿、ゲルクロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、アフィニティークロマトグラフィーなどを単独でまたは適宜組み合わせて用いることにより、培養物中から本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを単離精製できる。なお、培養液をそのまま使用する場合、熱処理などにより他のタンパク質が失活するので、実質上、本発明のマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼ酵素液として使用できる。
マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼは特に限定されるものではなく、いかなる起源の酵素を用いることも可能である。この酵素の使用形態は特に限定されるものではなく、菌体抽出液、精製酵素、固定化酵素など種々のものを利用することができる。本発明の好適な実施形態によれば、上記反応に関わる酵素を固定化したバイオリアクターカラムを用いて、固定化酵素リアクターとして反応を行うことも可能である。
出発原料として用いる糖質はいかなる起源のものであっても良い。反応系に加える糖質濃度は特に限定されるものではないが、好ましくは約0.1~約1000g/Lであり、より好ましくは約0.1~約200g/Lである。
反応液中でのマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの濃度は特に限定されないが、0.76~76μM、好ましくは、1.5~3.8μMで使用し得る。
マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの30℃における至適pHは5.5付近であることから、前記溶液は、pH5.0~7.0であることが好ましく、特にpH5.5が好ましい。前記溶液としては、特に限定されるものではないが、酢酸緩衝溶液が好適である。
また、マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼのpH5.5における温度安定性は60℃までであることから、オリゴ糖合成反応は、30~60℃で行うことが好ましく、特に、30℃が好ましい。
また、反応時間は、特に限定されるものではないが、30~60分が好ましく、特に、60分が好ましい。
上記オリゴ糖合成反応により製造されたアスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンおよびマンノシル-β-1,4-キトビオースは、カラムクロマトグラフィーや結晶化等の公知の方法により単離することが可能である。カラムクロマトグラフィーとして、これに限定されるものではないが、サイズ排除クロマトグラフィー、シリカゲルカラムクロマトグラフィー、イオン交換クロマトグラフィー、限外濾過膜分離、逆浸透膜分離が含まれる。結晶化方法としては、これに限定されるものではないが、濃縮、温度低下、溶媒添加(エタノール、メタノール、アセトンなど)が含まれる。
なお、本発明は上記実施形態に限定されるものではなく、本発明の思想を逸脱しない範囲で種々の変形実施が可能である。
次に、本発明を実施例により詳しく説明するが、本発明はこれらにより限定されるものではない。
バクテロイデス・シータイオタオミクロン(Bacteroides thetaiotaomicron)のゲノム情報を基に、BT1033遺伝子に対するフォーワードプライマー(配列番号3)およびリバースプライマー(配列番号4)を設計し、合成した。BT1033遺伝子の塩基配列を配列番号2に、またこの塩基配列にコードされているアミノ酸配列を配列番号1に示す。
バクテロイデス・シータイオタオミクロンのゲノムDNAを鋳型とし、上記のプライマー及びKOD plus polymerase(TOYOBO社製)を用い、95℃に2分間保持したのち、95℃で30秒間、58℃で30秒間、68℃で1分30秒間のサイクルを45回繰り返してPCR反応を行い、最後に68℃に5分間保持した。その結果、985bpの増幅断片が得られた。このPCRで増幅されるDNA断片は、5’末端にNdeIサイトを、3’末端にXhoIサイトをそれぞれ有するBT1033をコードするDNAである。
得られた増幅断片を制限酵素NdeI及びXhoIで消化後、同様に処理した市販の遺伝子発現用プラスミドpET-24a(ノバジェン社製)に高効率ライゲーション試薬Ligation high(TOYOBO社製)を用いて連結した。さらに、ライゲーション反応液を用いて大腸菌コンピテントセルDH5α(TOYOBO製)を形質転換し、C末端に6残基のヒスチジンからなるHisタグが付加されたBT1033をコードするDNAを含む発現ベクターpET-24aを回収した。
この発現ベクターpET-24aを用いて、大腸菌BL21(DE3)をHanahanらの方法(J.Mol.Biol.、1983年、第166巻、第557-580頁)に従って形質転換した。形質転換体を50μg/mLのカナマイシンを含むLB培地200mLに植菌し、IPTG濃度を0.1mMとして誘導培養を18℃で24時間行った。培養液から遠心分離で回収した菌体を10mLの500mM塩化ナトリウムおよび10%グリセロールを含む20mMHEPES-NaOH緩衝液pH7.5に懸濁し、超音波処理により破砕した後、遠心分離後によって粗酵素液を得た。組換えタンパク質の精製は、Hisタグタンパク質精製用カラムHisTrapFF(GEヘルスケア社製)を用いたカラムクロマトグラフィーにより行った。得られた精製酵素溶液を、10mM HEPES-NaOH緩衝液pH7.0に対して透析を行い、遠心式フィルターユニットアミコンウルトラ-15(ミリポア社製)を用いた限外濾過によって濃縮することで、3.6mLの精製酵素標品を調製した。
得られた精製酵素標品を用い、以下に示す方法によって本タンパク質を新規酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼと同定し、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンおよびマンノシル-β-1,4-キトビオースを生成した。
50mM糖供与体(α-マンノース1-リン酸)、50mM糖受容体(N-アセチルグルコサミンまたはキトビオース)、精製酵素(それぞれ1.5、3.8μM)を含む40mM酢酸緩衝液(pH5.5)中で酵素反応を30℃、1時間行った。各反応液をアンバーライトMB3(オルガノ社製)で脱塩後、ショウデックスアサヒパック NH2P-50 4Eカラムによる75%アセトニトリルを溶媒とした高速液体クロマトグラフィーにより、それぞれ二糖画分(図1A)および三糖画分(図2A)を単離した。精製後の収量は共に2mgであった。それぞれの生成物をNMRにより分析したところ、マンノシル-β-1,4-N-アセチルグルコサミンおよびマンノシル-β-1,4-キトビオースであることを確認した。
40mM酢酸緩衝液(pH5.5)中、2mMのα-マンノース1-リン酸および糖受容体を用いて、合成反応時に生成するリン酸をモリブデンブルー法(J.Biol.Chem.1946、162、421-428)により定量した。上記条件下に毎分1μモルのリン酸を生成する活性を1ユニットと定義した。その結果、N-アセチルグルコサミンおよびキトビオースを糖受容体としたときの活性はそれぞれ20および1.9ユニット/mgであった。
マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの30℃における至適pHは5.5付近であり(図3A)、安定pH範囲は4℃,24時間の条件下でpH4.5-10.5であった(図3B)。本酵素のpH5.5における安定性は60℃までであった(図3C)。本酵素を用いたオリゴ糖合成反応は、pH5.0~7.0、30~60℃が好ましいことがわかった。
以上のように本発明は、医療創薬産業およびオリゴ糖製造業で利用できる。
Claims (10)
- α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用して、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成することを特徴とするオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼ。
- 以下の酵素学的性質を有する請求項1記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼ:
a)作用
α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用して、アスパラギン結合型糖タンパク質のコア糖鎖構造マンノシル-β-1,4-N-アセチルグルコサミンまたはマンノシル-β-1,4-キトビオースを生成する;
b)基質特異性
α-マンノース1-リン酸と、N-アセチルグルコサミンまたはキトビオースとに作用する;
c)至適pH
30℃の条件下で、pH5.5;
d)温度安定性
pH5.5の条件下で、60℃まで安定;
e)pH安定性
4℃、24時間の条件下で、pH4.5-10.5で安定。 - 配列番号1記載のアミノ酸配列、または配列番号1記載のアミノ酸配列において、1つ若しくは数個のアミノ酸残基が、欠失、置換、付加、または挿入された配列を有する請求項1または2に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼ。
- バクテロイデス・シータイオタオミクロン由来の請求項1~3のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼ。
- 請求項3または4に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼをコードするベクターまたは配列番号2記載のDNAを含むベクター。
- 請求項5記載のベクターにより形質転換された形質転換体。
- 請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼの製造方法であって、請求項6記載の形質転換体を培養する工程と、前記培養する工程において生産されたマンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを回収する工程を含む製造方法。
- α-マンノース1-リン酸と、N-アセチルグルコサミンと、請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを含む溶液中でオリゴ糖合成反応を行う工程と、マンノシル-β-1,4-N-アセチルグルコサミンを回収する工程を含むアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法。
- α-マンノース1-リン酸と、キトビオースと、請求項1~4のいずれか1項に記載のオリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼを含む溶液中でオリゴ糖合成反応を行う工程と、マンノシル-β-1,4-キトビオースを回収する工程を含むアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法。
- 前記溶液がpH5.0~7.0である請求項8または9に記載のアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012190474A JP6000758B2 (ja) | 2012-08-30 | 2012-08-30 | オリゴ糖合成酵素およびアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法 |
| JP2012-190474 | 2012-08-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014034274A1 true WO2014034274A1 (ja) | 2014-03-06 |
Family
ID=50183096
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/068549 WO2014034274A1 (ja) | 2012-08-30 | 2013-07-05 | オリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼおよびアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP6000758B2 (ja) |
| WO (1) | WO2014034274A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014199948A1 (ja) * | 2013-06-11 | 2014-12-18 | 国立大学法人新潟大学 | β-マンノシドの製造方法 |
| WO2015014973A3 (en) * | 2013-07-31 | 2015-04-02 | Institut National De La Recherche Agronomique | Use of specific glycoside phosphorylases for the implementation of phosphorolysis or reverse phosphorolysis reactions |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005063782A1 (ja) * | 2003-12-26 | 2005-07-14 | Shionogi & Co., Ltd. | アスパラギン結合型糖タンパク質コア糖鎖構造の合成 |
-
2012
- 2012-08-30 JP JP2012190474A patent/JP6000758B2/ja active Active
-
2013
- 2013-07-05 WO PCT/JP2013/068549 patent/WO2014034274A1/ja active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005063782A1 (ja) * | 2003-12-26 | 2005-07-14 | Shionogi & Co., Ltd. | アスパラギン結合型糖タンパク質コア糖鎖構造の合成 |
Non-Patent Citations (1)
| Title |
|---|
| DATABASE GENBANK 8 February 2011 (2011-02-08), XU, J. ET AL.: "Definition: Glycosidase, PH117-related [Bacteroides thetaiotaomicron VPI-5482]", accession no. A076140 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014199948A1 (ja) * | 2013-06-11 | 2014-12-18 | 国立大学法人新潟大学 | β-マンノシドの製造方法 |
| WO2015014973A3 (en) * | 2013-07-31 | 2015-04-02 | Institut National De La Recherche Agronomique | Use of specific glycoside phosphorylases for the implementation of phosphorolysis or reverse phosphorolysis reactions |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6000758B2 (ja) | 2016-10-05 |
| JP2014045704A (ja) | 2014-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2100967B1 (en) | Method for producing lacto-n-biose i or galacto-n-biose | |
| CN107604023A (zh) | 岩藻糖基转移酶及其应用 | |
| KR101812018B1 (ko) | 단당류 생산 방법 | |
| Lin et al. | Characterization of a thermophilic L-rhamnose isomerase from Thermoanaerobacterium saccharolyticum NTOU1 | |
| CN107278232B (zh) | 由核苷酸活化的糖产生游离形式的单糖的发酵方法 | |
| CN114369584B (zh) | 重组人源岩藻糖基转移酶变异体及应用 | |
| JP2005198655A (ja) | セファロスポリンcアシラーゼ | |
| CN107779461B (zh) | 一种引入多胺标签的基因改造方法,脂肪酶的可溶表达及生物仿生固定化方法 | |
| EP2948546B1 (en) | A method of production of rare disaccharides | |
| WO2019095161A1 (zh) | 转氨酶突变体及其应用 | |
| WO2014034274A1 (ja) | オリゴ糖合成酵素マンノシル-β-1,4-N-アセチルグルコサミンホスホリラーゼおよびアスパラギン結合型糖タンパク質のコア糖鎖構造の製造方法 | |
| EP2948545B1 (en) | Method of production of monosaccharides | |
| WO2023006109A1 (zh) | 鼠李糖高度专一的糖基转移酶及其应用 | |
| JP2010512741A (ja) | 新規n−アセチルグルコサミン−2−エピメラーゼ及びcmp−n−アセチルノイラミン酸の製造方法 | |
| CN113699131B (zh) | 一种α-环糊精葡萄糖基转移酶突变体及其应用 | |
| WO2014199948A1 (ja) | β-マンノシドの製造方法 | |
| JP6678483B2 (ja) | オリゴ糖の製造方法 | |
| JP6033632B2 (ja) | セロビオン酸ホスホリラーゼを用いた酸性βグルコシル二糖の製造方法 | |
| WO2014103659A1 (ja) | グルコシル-α-1,2-グリセロールホスホリラーゼ及びそれを用いたグルコシル-α-1,2-グリセロールの製造方法 | |
| KR102613552B1 (ko) | 목적 단백질의 수용성 및 열안정성을 증가시키기 위한 태그 단백질 및 이를 포함하는 융합 단백질 | |
| JP2019017259A (ja) | フコース含有糖鎖を特異的に切断するエンドグリコシダーゼ | |
| JP6033621B2 (ja) | オリゴ糖合成酵素並びにβ−1,2−マンノビオース及びその誘導体の製造方法 | |
| WO2019035482A1 (ja) | エピメリ化活性を有するタンパク質 | |
| CN108179160B (zh) | 一种植烷醇连接的高甘露糖型寡糖的制备方法 | |
| CN110951717B (zh) | 一种l-阿拉伯糖异构酶异构体及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13833723 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13833723 Country of ref document: EP Kind code of ref document: A1 |