WO2014007288A1 - プリフォーム用バインダー樹脂組成物、バインダー粒子、プリフォームおよび繊維強化複合材料 - Google Patents
プリフォーム用バインダー樹脂組成物、バインダー粒子、プリフォームおよび繊維強化複合材料 Download PDFInfo
- Publication number
- WO2014007288A1 WO2014007288A1 PCT/JP2013/068249 JP2013068249W WO2014007288A1 WO 2014007288 A1 WO2014007288 A1 WO 2014007288A1 JP 2013068249 W JP2013068249 W JP 2013068249W WO 2014007288 A1 WO2014007288 A1 WO 2014007288A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- preform
- resin composition
- binder resin
- molding
- resin
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/26—Layered products comprising a layer of synthetic resin characterised by the use of special additives using curing agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/38—Layered products comprising a layer of synthetic resin comprising epoxy resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B37/00—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding
- B32B37/06—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the heating method
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/02—Polycondensates containing more than one epoxy group per molecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/22—Di-epoxy compounds
- C08G59/24—Di-epoxy compounds carbocyclic
- C08G59/245—Di-epoxy compounds carbocyclic aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/68—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used
- C08G59/688—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used containing phosphorus
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/249—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs characterised by the additives used in the prepolymer mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0025—Crosslinking or vulcanising agents; including accelerators
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/34—Heterocyclic compounds having nitrogen in the ring
- C08K5/3442—Heterocyclic compounds having nitrogen in the ring having two nitrogen atoms in the ring
- C08K5/3445—Five-membered rings
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/96—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from other synthetic polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2305/00—Condition, form or state of the layers or laminate
- B32B2305/07—Parts immersed or impregnated in a matrix
- B32B2305/076—Prepregs
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2309/00—Parameters for the laminating or treatment process; Apparatus details
- B32B2309/02—Temperature
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2650/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G2650/28—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule characterised by the polymer type
- C08G2650/56—Polyhydroxyethers, e.g. phenoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/12—Applications used for fibers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
- C08L2205/025—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group containing two or more polymers of the same hierarchy C08L, and differing only in parameters such as density, comonomer content, molecular weight, structure
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2207/00—Properties characterising the ingredient of the composition
- C08L2207/04—Thermoplastic elastomer
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T156/00—Adhesive bonding and miscellaneous chemical manufacture
- Y10T156/10—Methods of surface bonding and/or assembly therefor
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31511—Of epoxy ether
- Y10T428/31515—As intermediate layer
Definitions
- the present invention relates to a binder resin composition used for preforms of reinforcing fibers, and preforms and fiber-reinforced composite materials using the same.
- Fiber reinforced composite materials composed of reinforced fibers and matrix resins can be designed using the advantages of reinforced fibers and matrix resins, so the applications are expanded to aerospace, sports, general industrial fields, etc. .
- the reinforcing fiber glass fiber, aramid fiber, carbon fiber, boron fiber, etc. are used.
- the matrix resin either a thermosetting resin or a thermoplastic resin is used, but a thermosetting resin that can be easily impregnated into the reinforcing fiber is often used.
- the thermosetting resin a resin composition obtained by adding a curing agent or a curing catalyst to an epoxy resin, an unsaturated polyester resin, a vinyl ester resin, a phenol resin, a bismaleimide resin, a cyanate resin, or the like is used.
- Fiber reinforced composite materials are manufactured by various methods.
- RTM Resin Transfer Molding
- the method is attracting attention as a method with excellent low-cost productivity.
- a preform in which a reinforcing fiber substrate is processed into a shape close to the desired product is prepared in advance, and this preform is placed in a mold and a liquid thermosetting resin is injected. Often done.
- a method for producing a preform As a method for producing a preform, several methods are known, such as a method for producing a three-dimensional braid from reinforcing fibers, and a method for stacking and stitching reinforcing fiber fabrics. There is known a method of laminating and shaping a sheet-like substrate such as a reinforcing fiber fabric using a hot-melt binder (tackifier).
- tackifier hot-melt binder
- Patent Document 1 discloses a resin composition having excellent adhesion between a thermoplastic resin and a reinforcing fiber made of an epoxy resin.
- a resin composition having excellent adhesion between a thermoplastic resin and a reinforcing fiber made of an epoxy resin.
- Patent Document 2 uses a resin composition having curing reactivity in which an epoxy resin such as liquid bisphenol A type epoxy resin is combined with a catalyst such as ethyltriphenylphosphonium acetic acid as a binder, and the binder is partially heated by heating.
- a resin composition for a binder that can increase the peel strength of the obtained preform by curing is disclosed.
- thermosetting is imparted to a mixture of a thermosetting resin and a thermoplastic resin.
- the fiber reinforced composite material obtained by molding this has greatly improved interlayer toughness, it is still necessary to raise and lower the temperature of the preform mold, and it does not lead to shortening of the preform production time. It was.
- JP 2005-194456 A Japanese National Patent Publication No. 8-509921 JP-T-2001-524171
- An object of the present invention is to improve the drawbacks of the prior art and to form a binder resin composition capable of fixing the base material in a short time without performing a temperature increase / decrease of the preform type, a reinforcing fiber base material using the binder resin composition, It is to provide a preform and a fiber reinforced composite material.
- a preform binder resin composition according to the first aspect of the present invention is a resin composition including a thermosetting resin [A], a thermoplastic resin [B], and a curing catalyst [C].
- the thermosetting resin [A] contains a bifunctional epoxy resin, and the content of the thermoplastic resin [B] is in the range of 10 to 100 parts by mass with respect to 100 parts by mass of the thermosetting resin [A].
- the curing catalyst [C] is at least one curing catalyst selected from an organic phosphorus compound, imidazole, and derivatives thereof.
- thermosetting resin [A] preferably contains a liquid bifunctional epoxy resin
- thermoplastic resin [B] has a hydroxyl group in the main chain. It is preferable.
- thermosetting resin [A] preferably contains a solid bifunctional bisphenol type epoxy resin.
- the curing catalyst [C] is preferably contained in an amount of 2 to 20 parts by mass with respect to 100 parts by mass of the thermosetting resin [A].
- the binder resin composition for preforms which concerns on 2nd this invention is a resin composition which contains thermosetting resin [A] and whose glass transition temperature is Tg1,
- Tg1 thermosetting resin [A]
- Tg2 glass transition temperature
- the resin composition includes a thermosetting resin [A], a thermoplastic resin [B], and a curing catalyst [C], and includes a thermosetting resin [ A] contains a bifunctional epoxy resin, the content of the thermoplastic resin [B] is in the range of 10 to 100 parts by mass with respect to 100 parts by mass of the thermosetting resin [A], and the curing catalyst [C] Is preferably at least one curing catalyst selected from organic phosphorus compounds, imidazoles and derivatives thereof.
- the combination of the molding time t and the molding temperature T is preferably within a range satisfying the formula (3), and a range satisfying the formula (4). More preferably, it is present within. 80 ⁇ T + 0.4t ⁇ 190 (3) 80 ⁇ T + 0.4t ⁇ 130 (4)
- the binder resin composition for preforms according to the present invention can include a preliminary reaction product of the thermosetting resin [A].
- the binder resin composition for preforms according to the present invention preferably has a particle form.
- the reinforcing fiber base material of the present invention has at least the above-mentioned binder resin composition for preform on the surface, and the preform of the present invention has a plurality of reinforcing fiber base materials laminated.
- the preform has a binder resin composition for preforms at least between the laminated layers, and the fiber-reinforced composite material of the present invention impregnates the liquid thermosetting resin composition with the preform. And cured.
- the preform production method of the present invention comprises the above-mentioned preform binder resin composition adhered to at least the surface of the reinforcing fiber base material to form a reinforcing fiber base.
- the reinforcing fiber base material is laminated and heated at an actual molding temperature TR in the range of 60 to 200 ° C. for 0.5 to 120 minutes, and the glass transition temperature of the preform binder resin composition satisfies the formula (5).
- the base material can be fixed in a short time without performing the temperature increase / decrease of the preform mold.
- a preform can be obtained.
- the demoldability when preforming the reinforcing fiber substrate is improved, and the preform can be removed from the preform mold without lowering the temperature of the preform mold after the preform is formed. Time can be shortened.
- the binder resin composition for preforms according to the first aspect of the present invention is a resin composition containing a thermosetting resin [A], a thermoplastic resin [B], and a curing catalyst [C], wherein the thermosetting resin [A] A] contains a bifunctional epoxy resin, the content of the thermoplastic resin [B] is in the range of 10 to 100 parts by mass with respect to 100 parts by mass of the thermosetting resin [A], and the curing catalyst [C] Is at least one curing catalyst selected from organic phosphorus compounds, imidazoles and derivatives thereof.
- thermosetting resin [A], the thermoplastic resin [B], and the curing catalyst [C] that contain the above-described constituents and have the above-mentioned contents are combined to form a thermosetting resin that is contained by heating during molding [
- a preforming binder resin composition having excellent adhesion to the reinforcing fiber substrate can be obtained while the curing reaction of A] proceeds in a short time.
- adheresiveness it means the adhesiveness of the preform binder resin composition to the reinforcing fiber substrate.
- thermosetting resin [A] contained in the binder resin composition proceeds in a short time, so that the binder resin composition is thermosetting when the reinforcing fiber substrate is set in a preform mold at a constant temperature. Since the resin [A] can be solidified by the curing reaction, the preform can be removed in a short time.
- thermosetting resin [A] contained in is referred to as “the curing reaction of the thermosetting resin [A] contained in the binder resin composition” as “the curing reaction of the binder resin composition”. "May be omitted.
- Solidification means that the binder resin composition is in a glass state and loses fluidity.
- thermosetting resin [A] in the present invention is a resin material that undergoes a curing reaction by heating to form a crosslinked structure, and is an epoxy resin, phenol resin, unsaturated polyester resin, vinyl ester resin, bismaleimide resin, cyanate resin.
- a resin composition in which a curing agent or a curing catalyst is added to a benzoxazine resin or the like can be used.
- the thermosetting resin [A] in the present invention preferably contains an epoxy resin from the viewpoint of adhesiveness and handleability, and more preferably contains an epoxy resin as a main component.
- the epoxy resin being the main component of the thermosetting resin means that the epoxy resin occupies 60% by mass or more in the thermosetting resin, and more preferably 80% by mass or more.
- An epoxy resin means a compound having two or more epoxy groups in one molecule. Such an epoxy resin may consist of only one kind of compound having an epoxy group, or may be a mixture of plural kinds.
- the epoxy resin examples include an aromatic glycidyl ether obtained from a phenol compound having a plurality of hydroxyl groups, an aliphatic glycidyl ether obtained from an alcohol compound having a plurality of hydroxyl groups, a glycidyl amine obtained from an amine compound, and a carboxyl having a plurality of carboxyl groups.
- examples include epoxy resins having epoxy groups such as glycidyl esters obtained from acid compounds as part of the glycidyl groups, and epoxy resins containing oxirane rings obtained by oxidizing unsaturated alicyclic compounds such as cyclohexene. It is done.
- the thermosetting resin [A] in the present invention includes a bifunctional epoxy resin because it is excellent in adhesiveness among epoxy resins.
- the bifunctional epoxy resin is an epoxy resin having two epoxy groups in one molecule of the epoxy resin. Since the bifunctional epoxy resin does not have an excessively high crosslinking density after curing the binder resin composition for preforms, excellent adhesiveness can be obtained.
- the thermosetting resin [A] in the present invention preferably contains a liquid bifunctional epoxy resin from the viewpoints of curing speed and extrusion kneadability among the bifunctional epoxy resins.
- the liquid bifunctional epoxy resin is an epoxy resin having a glass transition temperature lower than 20 ° C. and fluidity at room temperature, and having two epoxy groups in one molecule of the epoxy resin.
- normal temperature refers to 25 ° C. (hereinafter the same).
- the glass transition temperature is a value determined by differential scanning calorimetry (DSC) according to JIS K 7121: 1987.
- DSC differential scanning calorimetry
- An example of a measuring device that can be used for the above standard is Pyris1 DSC (manufactured by Perkin Elmer).
- the preform binder resin composition is collected in an aluminum sample pan and measured at a temperature increase rate of 40 ° C./min in a nitrogen atmosphere. The midpoint of displacement in the region where the baseline in the DSC curve thus obtained shifts to the endothermic side is adopted as the glass transition temperature.
- the thermosetting resin [A] in the present invention preferably contains a solid bifunctional epoxy resin from the viewpoint of excellent balance of curing reactivity, life, toughness, and heat resistance and flow adjustment.
- the solid bifunctional epoxy resin is an epoxy resin having a glass transition temperature of 20 ° C. or higher and no fluidity at room temperature, and having two epoxy groups in one molecule of the epoxy resin.
- the solid bifunctional epoxy resin is preferably used from the viewpoints of storage stability and flow adjustment, and the crosslink density after curing the preform binder resin composition does not become excessively high, so that excellent adhesiveness can be obtained.
- thermosetting resin [A] 30 to 80 parts by mass of liquid bifunctional epoxy resin and 20 to 70 parts by mass of solid bifunctional epoxy resin are contained in 100 parts by mass of thermosetting resin [A]. It is preferable.
- the content of the liquid bifunctional epoxy resin in 100 parts by mass of the thermosetting resin [A] is less than 30 parts by mass, the curing reactivity is lowered, so that the demoldability at the time of preforming is insufficient. (I.e., the mold cannot be removed unless the preform mold is cooled), and the viscosity of the binder resin composition increases, and it may be difficult to prepare the binder resin composition by extrusion kneading.
- the content of the liquid bifunctional epoxy resin in 100 parts by mass of the thermosetting resin [A] is more than 80 parts by mass, the glass transition temperature of the binder resin composition for preforms is lowered so that the storage stability is improved. May decrease.
- the toughness of the resin composition after molding is lowered, so that sufficient adhesive strength can be obtained. It is difficult, and the viscosity of the resin composition is lowered, and the storage stability may be lowered.
- the content of the solid bifunctional epoxy resin in 100 parts by mass of the thermosetting resin [A] is more than 70 parts by mass, the Tg after molding is difficult to increase and sufficient demolding property is obtained when forming into a preform. It is difficult to obtain, and fluidity is lowered. For example, when a preform binder resin composition is applied in the form of particles, it may not be sufficiently melted at the time of preform molding and adhesion may be lowered.
- a solid bifunctional bisphenol type epoxy resin as the solid bifunctional epoxy resin from the viewpoint of excellent balance of curing reactivity, life, toughness, and heat resistance, and flow adjustment.
- the solid bifunctional bisphenol type epoxy resin include bisphenol A type epoxy resin, bisphenol F type epoxy resin, bisphenol S type epoxy resin, bisphenol E type epoxy resin, bisphenol Z type epoxy resin, alkyl substitution products thereof, and halogen substitution. Solid bodies, hydrogenated substances and the like can be used, but are not limited thereto. Among these, a bisphenol A type epoxy resin excellent in balance of curing reactivity, life, toughness, and heat resistance can be suitably used.
- thermosetting resin [A] in the present invention may contain a polyfunctional epoxy resin in addition to the bifunctional epoxy resin from the viewpoint of increasing heat resistance and curing speed.
- Such polyfunctional epoxy resins can be broadly classified into glycidylamine type polyfunctional epoxy resins and non-glycidylamine type polyfunctional epoxy resins.
- Examples of the glycidylamine-type polyfunctional epoxy resin include tetraglycidyldiaminodiphenylmethane, triglycidylaminophenol, triglycidylaminocresol, tetraglycidylxylylenediamine, structural isomers thereof, halogens, alkyl-substituted products, and hydrogens thereof. An additive etc. are mentioned.
- triglycidylaminophenol or triglycidylaminocresol Commercially available products of triglycidylaminophenol or triglycidylaminocresol include “Sumiepoxy” (registered trademark) ELM100, “Sumiepoxy” (registered trademark) ELM120 (manufactured by Sumitomo Chemical Co., Ltd.), and “Araldide” (registered trademark). ) MY0500, “Araldide” (registered trademark) MY0510, “Araldide” (registered trademark) MY0600 (manufactured by Huntsman Advanced Materials), “jER” (registered trademark) 630 (manufactured by Mitsubishi Chemical Corporation), etc. Can be used.
- TTRAD (registered trademark) -X
- TTRAD registered trademark
- -C manufactured by Mitsubishi Gas Chemical Co., Ltd.
- non-glycidylamine type polyfunctional epoxy resins include phenol novolac type epoxy resins, cresol novolac type epoxy resins, triphenylmethane type epoxy resins, tetraphenylethane type epoxy resins, phenol aralkyl type epoxy resins, and naphthol aralkyl types.
- examples thereof include epoxy resins and dicyclopentadiene skeleton-containing epoxy resins.
- phenol novolac type epoxy resins include “jER” (registered trademark) 152, 154 (manufactured by Mitsubishi Chemical Corporation), “Epiclon” (registered trademark) N-740, N-770, N-775. (Above, manufactured by DIC Corporation).
- cresol novolac epoxy resins examples include “Epicron” (registered trademark) N-660, N-665, N-670, N-680, N-695 (above, manufactured by DIC Corporation), EOCN-1020. And EOCN-102S (Nippon Kayaku Co., Ltd.), YDCN-700, YDCN-701 (Nippon Steel Chemical Co., Ltd.) and the like.
- triphenylmethane type epoxy resins include “Tactix” (registered trademark) 742 (manufactured by Huntsman Advanced Materials), EPPN-501H, EPPN-502H (manufactured by Nippon Kayaku Co., Ltd.), and the like. It is done.
- Examples of commercially available tetraphenylethane type epoxy resins include “jER” (registered trademark) 1031 (manufactured by Mitsubishi Chemical Corporation), GTR1800 (manufactured by Nippon Kayaku Co., Ltd.), and the like.
- Examples of commercially available phenol aralkyl type epoxy resins include NC2000 series (manufactured by Nippon Kayaku Co., Ltd.), NC7000 series (manufactured by Nippon Kayaku Co., Ltd.), NC3000 series (manufactured by Nippon Kayaku Co., Ltd.), and the like. .
- Examples of commercially available dicyclopentadiene skeleton-containing epoxy resins include “Epiclon” (registered trademark) NC7200 series (manufactured by DIC Corporation), XD-1000 series (manufactured by Nippon Kayaku Co., Ltd.), and the like.
- thermoplastic resin [B] in the present invention is added to 100 parts by mass of the thermosetting resin [A] for the purpose of improving the adhesive strength in accordance with flow optimization and toughness improvement during preform molding of the preform binder resin composition. It is contained at a ratio of 10 to 100 parts by mass with respect to the mass.
- thermosetting resin [B] When the content of the thermoplastic resin [B] relative to 100 parts by mass of the thermosetting resin [A] is less than 10 parts by mass, the flow optimization at the time of preform molding and the resin toughness after curing are insufficient, and adhesion The strength may be insufficient.
- the content of the thermoplastic resin with respect to 100 parts by mass of the thermosetting resin [A] is more than 100 parts by mass, the Tg after curing is hardly increased and the demolding property becomes insufficient when forming into a preform. While becoming easy, fluidity
- thermoplastic resin [B] examples include polyamide, polycarbonate, polyacetal, polyphenylene oxide, polyphenylene sulfide, polyarylate, polyester, polyamideimide, polyimide, polyetherimide, and polyimide having a phenyltrimethylindane structure.
- Specific examples include polysulfone, polyethersulfone, polyetherketone, polyetheretherketone, polyaramide, polyethernitrile, polybenzimidazole, polyurethane, urea resin, polyvinyl acetal, polyvinyl formal, polyvinyl alcohol, and phenoxy resin.
- thermoplastic resin [B] polyvinyl alcohol having a hydroxyl group in the main chain, phenoxy resin, and the like can be suitably used as the thermoplastic resin [B].
- the thermoplastic resin [B] has a hydroxyl group in the main chain, the adhesive strength with the reinforcing fiber base is improved, and the effect of promoting the curing of the epoxy resin is obtained, and the preform time may be shortened.
- the preform binder resin composition according to the first aspect of the present invention is not only suitable for the purpose of optimizing the flow during molding and imparting adhesive strength and dimensional stability.
- a filler component such as an elastomer or particles can be blended.
- rubber particles and organic particles soluble in an epoxy resin, inorganic fillers, and the like can be suitably blended.
- the curing catalyst [C] in the present invention is included for the purpose of promptly smoothing the curing reaction by the single curing reaction of the thermosetting resin [A] and the bond formation with the curing agent.
- at least one curing catalyst selected from organic phosphorus compounds, imidazoles, and derivatives thereof is used.
- organic phosphorus compound examples include tributylphosphine, trioctylphosphine, tricyclohexylphosphine, triphenylphosphine, tribenzylphosphine, tri-o-tolylphosphine, tri-m-tolylphosphine, diphenylcyclohexylphosphine, 1,3- And bis (diphenylphosphino) propane.
- imidazole and derivatives thereof include imidazole, 2-ethylimidazole, 2-undecylimidazole, 2-heptadecylimidazole, 1,2-dimethylimidazole, 2-ethyl-4-methylimidazole, 1-benzyl- Examples include 2-phenylimidazole, 1-benzyl-2-methylimidazole, 1-cyanoethyl-2-methylimidazole, adducts of imidazole and epoxy compounds.
- the curing catalyst [C] in the present invention is preferably contained in an amount of 2 to 20 parts by mass with respect to 100 parts by mass of the thermosetting resin [A].
- the content of the curing catalyst [C] is less than 2 parts by mass with respect to 100 parts by mass of the thermosetting resin [A]
- the curing reactivity is low, and it may be impossible to remove the preform in a short time. is there.
- the content of the curing catalyst [C] relative to 100 parts by mass of the thermosetting resin [A] is more than 20 parts by mass, the curing reactivity may become too high, and the handleability may be lowered.
- a curing agent can be added to the preform binder resin composition of the present invention.
- the curing agent is a component that cures the resin by bonding with a thermosetting resin to form a three-dimensional network structure.
- an epoxy resin is a compound having an active group that can react with an epoxy group.
- Curing agents are roughly classified into amine-based, phenol-based, acid anhydride-based, and mercaptan-based curing agents. Specifically, dicyandiamide, aromatic polyamine, aliphatic amine, aminobenzoic acid esters, thiourea-added amine, hydrazide, etc. as amine series, bisphenol, phenol novolac resin, cresol novolak resin, polyphenol compound, etc.
- the acid anhydride type include phthalic anhydride, maleic anhydride, succinic anhydride, and carboxylic acid anhydride
- examples of the mercaptan type include polymercaptan and polysulfide resin.
- a temperature-latent curing catalyst and a curing agent as the curing catalyst and the curing agent. It is.
- a temperature-latent curing catalyst and curing agent a solid dispersion-heat curing type having low solubility in an epoxy resin near normal temperature, and a reactive group block in which a functional group having high reactivity is blocked with a functional group having low reactivity Broadly divided into types.
- Solid dispersion types include aliphatic amines, aromatic amines, dihydrazide compounds, amine adducts, 2-phenyl-4-methyl-5-hydroxymethylimidazole, and reactive block types include onium salts, boron halide / amine complexes, And vinyl ether block carboxylic acid.
- the binder resin composition for preform according to the second aspect of the present invention is a resin composition containing a thermosetting resin [A] and having a glass transition temperature of Tg1, in the range of 60 to 200 ° C. and (1 ) The molding time t at which the glass transition temperature rises to Tg2 shown in the formula (2) by heating at a molding temperature T ° C. satisfying the formula for a molding time t for 0.5 to 120 minutes.
- thermosetting resin [A] is preferably an epoxy resin from the viewpoints of adhesiveness and handleability.
- the molding temperature T in the second aspect of the present invention is set assuming the temperature at which the preform is molded, and is a temperature at which the preform binder resin composition can be cured and solidified.
- the molding temperature T is in the range of 60 to 200 ° C., preferably in the range of 80 to 150 ° C., preferably 90 to 130 ° C. from the viewpoint of curability and adhesive strength of the binder resin composition for preforms of the present invention. More preferably, it is the range.
- the fact that the molding temperature T can only be taken in a temperature range less than 60 ° C. means that the preform binder resin composition can be set only in a range where the molding temperature is less than 60 ° C.
- the resin composition at room temperature has high curing reactivity, and the handleability may be reduced.
- the fact that the molding temperature T can be taken only in a temperature range exceeding 200 ° C. indicates that the actual molding temperature at the time of molding can only be set in a range exceeding 200 ° C. Since the crosslinking density of the product obtained by curing the binder resin composition for use is increased, the adhesive strength is reduced.
- the molding temperature T needs to be able to take a range satisfying the glass transition temperature Tg1 of the preform binder resin composition and the formula (1) (Tg1 ⁇ T ⁇ Tg1 + 100).
- the molding temperature T can be set to a temperature higher than the glass transition temperature Tg1 of the binder resin composition for preforms, the molding is performed at a temperature higher than Tg1, thereby sufficiently melting the resin and forming the reinforcing fiber base material. It becomes possible to bond.
- the molding temperature T can only be set at a temperature exceeding the glass transition temperature Tg1 + 100 ° C. of the preform binder resin composition, if the molding is performed at a high temperature exceeding Tg1 + 100 ° C., the preform binder resin composition melts immediately.
- the relationship represented by the formula (1) is preferably Tg1 + 20 ⁇ T ⁇ Tg1 + 80, and more preferably Tg1 + 30 ⁇ T ⁇ Tg1 + 60.
- the molding time t in the second aspect of the present invention is set in consideration of the time for molding the preform, and is the time required for the preform binder resin composition to cure and solidify. .
- the molding time t needs to be in the range of 0.5 to 120 minutes, preferably in the range of 0.5 to 30 minutes, and more preferably in the range of 0.5 to 20 minutes.
- the fact that the molding time t can only be taken in an area shorter than 0.5 minutes means that the binder resin composition for preforms can be set only within a range where the actual molding time for heating during molding is less than 0.5 minutes. In such a case, since the binder resin composition is cured immediately after heating, it may be difficult to make the reinforcing fiber base completely conform to the shape of the preform type.
- the fact that the molding time can only be taken in an area longer than 120 minutes indicates that the actual molding time to be heated at the time of molding can be set only in a range exceeding 120 minutes. Since the time required for conversion becomes longer, productivity is lowered.
- the glass transition temperature Tg1 of the binder resin composition for preforms according to the second invention preferably satisfies the above formula (1) and is in the range of 40 to 100 ° C.
- the glass transition temperature Tg1 is lower than 40 ° C., the viscosity becomes too low at the time of molding, and the flow becomes large, so that the fixation is insufficient and storage stability may be problematic.
- the glass transition temperature Tg1 exceeds 100 ° C., the resin composition is difficult to melt, so that the adhesive strength becomes insufficient and a problem may occur in storage stability.
- the binder resin composition for preforms according to the second aspect of the present invention is heated at a molding temperature T satisfying the above conditions over a molding time t in the above-described range, so that the glass transition temperature Tg2 is the above (2). It is necessary that a combination of the molding time t and the molding temperature T satisfying the formula (T ⁇ Tg2 ⁇ T + 30) is present, and it is preferable that a combination of the molding time t and the molding temperature T satisfying T + 10 ⁇ Tg2 ⁇ T + 30 is present. .
- the glass transition temperature Tg2 is lower than the actual molding temperature, no matter how the conditions are adjusted during molding.
- the glass transition temperature Tg2 exceeds the actual molding temperature by more than 30 ° C.
- the preform binder resin composition is not sufficiently solidified, and therefore deforms when the preform is removed from the mold, and the dimensional accuracy of the resulting preform. Is insufficient.
- the glass transition temperature Tg2 exceeds the actual molding temperature by more than 30 ° C., the crosslinking density of the resin composition becomes too high, and sufficient adhesive strength cannot be obtained.
- the glass transition temperature Tg2 satisfies the above formula (2) and is preferably in the range of 100 to 150 ° C., more preferably 100 to 130 ° C.
- the glass transition temperature Tg2 is lower than 100 ° C., the resin composition is not sufficiently solidified, so that it deforms when the preform is removed from the mold, and the dimensional accuracy of the obtained preform tends to be insufficient.
- the glass transition temperature Tg2 exceeds 150 ° C., it is difficult to obtain sufficient adhesive strength because the crosslink density of the resin composition increases.
- the binder resin composition for preforms according to the second aspect of the present invention is a resin composition containing a thermosetting resin [A], a thermoplastic resin [B], and a curing catalyst [C], wherein the thermosetting resin [A] A] contains a bifunctional epoxy resin, the content of the thermoplastic resin [B] is in the range of 10 to 100 parts by mass with respect to 100 parts by mass of the thermosetting resin [A], and the curing catalyst [C] Is a resin composition that is at least one curing catalyst selected from an organic phosphorus compound, imidazole, and derivatives thereof, that is, a preform binder resin composition according to the first aspect of the present invention. It is preferable because of its applicability.
- the combination of the molding time t and the molding temperature T is preferably within a range satisfying the formula (3), and a range satisfying the formula (4). More preferably, it is present within. In order to adjust the combination of the molding time t and the molding temperature T so as to be within such a range, first, the selection of the curing catalyst is effective, and if necessary, fine adjustment with other components may be performed.
- a molding time t and the molding temperature T satisfy the formula (3) with a composition in which a polyfunctional epoxy resin is combined with dicyandiamide as a curing agent and 2,4-tolylenebis (1,1-dimethylurea) as a curing catalyst. It can be adjusted so that the combination exists, and there exists a combination in which the molding time t and the molding temperature T satisfy the formula (4) in the composition in which the solid bifunctional epoxy resin is combined with the imidazole adduct as the curing catalyst. Can be adjusted.
- the curing reaction hardly proceeds in the process from the preparation of the resin composition to the molding of the preform, that is, the thermal stability is high.
- the curing reaction proceeds in the process before preform molding, the preform binder resin composition is not sufficiently melted during preform molding, and the adhesive strength between the layers of the obtained preform decreases. There is a case.
- the preform binder resin composition of the present invention can be preliminarily reacted to partially contain a pre-reaction product of a thermosetting resin, and can increase the glass transition temperature.
- the term “preliminary reaction” as used herein means that a part of the curing reaction of the thermosetting resin in the preform binder resin composition is allowed to proceed. Thereby, the storage stability of the binder resin composition for preforms may be improved. Moreover, since the flow by the heating at the time of preform shaping
- the preliminary reaction of the preform binder resin composition may be performed at the time of resin preparation, or may be performed after spraying on the reinforcing fiber base.
- the form of the binder resin composition of the present invention is not particularly limited, and films, tapes, long fibers, short fibers, spun yarns, woven fabrics, knits, non-woven fabrics, nets, particles, etc. may be employed. it can. Among these, the particle form can be particularly preferably used.
- the binder resin composition in the form of particles may be referred to as binder particles.
- the average particle size is preferably 10 to 1000 ⁇ m.
- the average particle diameter refers to the volume average particle diameter.
- the adhesive strength and workability may be lowered.
- the average particle size is larger than 1000 ⁇ m, the reinforced fiber swells when formed into a preform, resulting in a decrease in mechanical properties of the fiber reinforced composite material, and it becomes difficult to dissolve the particles in the liquid thermosetting resin. Problems such as reduced chemical resistance may occur.
- the average particle size of the binder particles can be measured using, for example, a laser diffraction type particle size distribution meter.
- the preform binder resin composition of the present invention is used by adhering to a reinforcing fiber or a reinforcing fiber substrate.
- a reinforcing fiber carbon fiber, glass fiber, aramid fiber, metal fiber, or a combination thereof can be used.
- carbon fibers can be suitably used because they are excellent in lightness and strength.
- the reinforcing fiber may be either a short fiber or a continuous fiber, or both may be used in combination.
- a fiber-reinforced composite material having a high volume content hereinafter referred to as high Vf
- the reinforcing fiber may be used in the form of a strand, but a reinforcing fiber substrate obtained by processing the reinforcing fiber into a mat, woven fabric, knit, braid, unidirectional sheet or the like is preferably used. Among them, a woven fabric which is easy to obtain a high Vf fiber-reinforced composite material and excellent in handleability is preferably used.
- Plain weaving, satin weaving, twill weaving, non-crimp cloth, etc. can be selected as appropriate as the weaving structure used for the preform, but plain weaving or twill weaving is used when the texture is shown on the design surface by clear painting. And the design is high. Also, satin weave and twill weave are good for draping and are therefore preferably used when shaping a three-dimensional shape with a deep depth.
- the ratio of the net volume of the reinforcing fibers to the apparent volume of the fabric is defined as the fabric filling rate.
- the filling rate of the woven fabric is obtained from the weight per unit area W (unit: g / m 2 ), the thickness t (unit: mm), and the density ⁇ f (unit: g / cm 3 ) of the reinforcing fiber by the formula of W / (1000 t ⁇ ⁇ f). It is done.
- the basis weight and thickness of the woven fabric are determined in accordance with JIS R 7602: 1995. The higher the woven fabric filling rate, the easier it is to obtain a fiber reinforced composite material having a high Vf. Therefore, the woven fabric filling rate is 0.10 to 0.85, preferably 0.40 to 0.85, more preferably 0.50. It is preferably in the range of ⁇ 0.85.
- the fiber volume content Vf is preferably in the range of 40 to 85%, preferably 45 to 85%.
- the fiber volume content Vf of the fiber reinforced composite material referred to here is a value defined and measured by the following in accordance with ASTM D3171 (1999). It refers to the state after injecting and curing the curable resin. That is, the measurement of the fiber volume content Vf of the fiber reinforced composite material can be expressed by the following (formula 1) from the thickness h of the fiber reinforced composite material.
- Fiber volume content Vf (%) (Af ⁇ N) / ( ⁇ f ⁇ h) / 10 ...
- Af fiber base material one ⁇ 1 m 2 per weight (g / m 2)
- N Number of laminated fiber substrates (sheets)
- ⁇ f density of reinforcing fiber (g / cm 3 )
- h Thickness (mm) of the fiber reinforced composite material (test piece).
- the weight Af per fiber substrate / m 2 , the number N of laminated fiber substrates, and the density ⁇ f of the reinforcing fiber are not clear, a combustion method or a nitric acid decomposition method based on JIS K 7075: 1991, sulfuric acid
- the fiber volume content of the fiber reinforced composite material is measured by any of the decomposition methods.
- the density of the reinforcing fiber used in this case a value measured based on JIS R 7603: 1999 is used.
- the preform binder resin composition of the present invention is preferably used by adhering to at least the surface of the reinforcing fiber substrate.
- the deposition amount in the case of attaching to the surface, on one or both sides, one side per 0.5 ⁇ 30g / m 2, it is preferable that preferably attached in the 1 ⁇ 10g / m 2 basis weight. If the adhesion amount is less than 0.5 g / m 2 , the effect of fixing the shape and increasing the toughness is small, and if it is more than 30 g / m 2 , the apparent volume of the reinforcing fiber strand or the reinforcing fiber base becomes large. There may be disadvantages such as difficulty in producing a fiber-reinforced composite material having a large volume content of reinforcing fibers, or poor impregnation of the thermosetting resin.
- the preform of the present invention is formed by laminating a reinforcing fiber base having at least the above-described binder resin composition for preform on the surface and fixing the form.
- the reinforced fiber base material has a preform binder resin composition on at least one surface of the reinforcing fiber base material, and a laminate having a preform binder resin composition at least between the lamination layers by laminating a plurality thereof. Is obtained.
- the binder resin composition is cured and the form is fixed by fixing between the substrates, whereby a preform having at least the binder resin composition for preforms between the laminated layers is obtained.
- a preform can be produced by cutting a sheet-like reinforcing fiber base to which a binder resin composition is adhered into a predetermined shape, laminating it on a mold, and applying appropriate heat and pressure.
- the pressurizing means may be a press, or a method of enclosing with a vacuum bag film and sucking the inside with a vacuum pump and pressurizing with atmospheric pressure.
- the binder resin composition for preform of the present invention is attached to at least the surface of the reinforcing fiber base material to form a reinforcing fiber base, and then the reinforcing fiber base is laminated.
- the glass transition temperature Tg2 after heating that satisfies the formula is raised to obtain a preform.
- the actual molding temperature TR when molding a preform using the binder resin composition for preform of the present invention is a temperature when molding the preform, and the binder resin composition for preform is cured and solidified. It is the temperature to make.
- the actual molding temperature TR is in the range of 60 to 200 ° C., and preferably in the range of 80 to 150 ° C., from the viewpoint of curability and adhesive strength of the preform binder resin composition of the present invention. More preferably, it is in the range of ° C.
- the curing reaction at the time of preforming does not proceed sufficiently, so that the demolding property is lowered and the adhesive strength is insufficient.
- the actual molding temperature TR exceeds 200 ° C.
- the glass transition temperature of the product obtained by curing reaction of the preform binder resin composition is less likely to be higher than the actual molding temperature TR, and the demolding property is lowered.
- the cross-linking density of the product obtained by curing reaction of the resin composition is increased, so that the adhesive strength is lowered.
- the actual molding temperature TR is set to a temperature higher than the glass transition temperature Tg1 of the preform binder resin composition so as to satisfy the formula (5), thereby sufficiently melting the resin and reinforcing the fiber base material. It becomes possible to adhere to.
- the composition immediately melts and penetrates into the reinforcing fibers, so that the adhesion between the reinforcing fibers decreases. There is a case.
- the preform binder resin composition after becoming a preform is lower than the actual molding temperature TR, the preform binder resin composition is not sufficiently solidified. Deformed when the mold is removed from the mold, resulting in insufficient dimensional accuracy of the preform. Moreover, when glass transition temperature Tg2 exceeds actual molding temperature TR + 30 degreeC, the crosslinking density of a resin composition becomes high too much and sufficient adhesive strength is not obtained.
- the actual molding time tR to be heated when molding the preform using the binder resin composition for preform of the present invention needs to be in the range of 0.5 to 120 minutes, and is in the range of 0.5 to 30 minutes. Preferably, the range is 0.5 to 20 minutes.
- the actual molding time tR is shorter than 0.5 minutes, it becomes difficult to sufficiently cure the preform binder resin composition, so that the preform demoldability is lowered.
- the actual molding time tR is longer than 120 minutes, the time for the preforming cycle becomes longer, and the productivity is lowered.
- the preform of the present invention has excellent demolding property, high dimensional accuracy, and can exhibit sufficient adhesive strength even when the molding temperature is substantially constant.
- the substantially constant temperature usually means a temperature fluctuation within ⁇ 5 ° C.
- a fiber reinforced composite material can be produced by impregnating a preform produced using the preform binder resin composition of the present invention with a liquid thermosetting resin and curing the liquid thermosetting resin. When the liquid thermosetting resin is cured, the binder resin composition is usually further cured.
- the production method of the fiber reinforced composite material in the present invention is not particularly limited, but a molding method using a two-component resin such as a hand layup method or an RTM method is preferably used.
- the RTM molding method is particularly preferably used from the viewpoints of productivity and the shape freedom of the molded body.
- the RTM molding method is a method in which a liquid thermosetting resin is injected and impregnated into a reinforcing fiber base disposed in a mold and cured to obtain a reinforcing fiber composite material.
- the liquid thermosetting resin is composed of a liquid resin mainly composed of a monomer component and a curing agent or a curing catalyst for polymerizing the monomer component by three-dimensionally crosslinking the monomer component.
- the liquid resin is preferably an epoxy resin from the viewpoint of curing reactivity and compatibility of the binder resin composition of the present invention.
- epoxy resins include aromatic glycidyl ethers obtained from phenols having a plurality of hydroxyl groups, aliphatic glycidyl ethers obtained from alcohols having a plurality of hydroxyl groups, glycidyl amines obtained from amines, and carboxylic acids having a plurality of carboxyl groups.
- examples thereof include glycidyl esters and epoxy resins having an oxirane ring.
- aliphatic polyamines aromatic polyamines, acid anhydrides, imidazoles, Lewis acid complexes and the like are suitable, and they are appropriately selected and used depending on the intended use.
- the mold temperature at the time of heat curing may be the same as the mold temperature at the time of injection of the liquid thermosetting resin, but in the case of curing at a low temperature, the rigidity of the fiber reinforced composite material is not deformed at the time of demolding. Since it may take time to advance the curing until it is obtained, it is preferable to select a temperature higher than the mold temperature at the time of injection, for example, a range of 60 to 180 ° C. is preferable.
- Epoxy resin YD-128 (manufactured by Nippon Steel & Sumikin Chemical Co., Ltd.): Liquid bifunctional bisphenol A type epoxy resin, epoxy equivalent 189 “JER” (registered trademark) 1001 (manufactured by Mitsubishi Chemical Corporation): solid bifunctional bisphenol A type epoxy resin, epoxy equivalent 475, glass transition temperature 35 ° C.
- JER registered trademark
- 1004 solid bifunctional bisphenol A type epoxy resin, epoxy equivalent 975, glass transition temperature 60 ° C
- ELM434 registered trademark
- ELM434 tetraglycidyldiaminodiphenylmethane
- EPICLON registered trademark
- N-695 manufactured by DIC Corporation
- solid cresol novolac type epoxy resin epoxy equivalent 214
- Benzoxazine resin / Pd type benzoxazine resin manufactured by Shikoku Kasei Kogyo Co., Ltd.
- Thermoplastic resin YP-50 (manufactured by Nippon Steel & Sumikin Chemical Co., Ltd.): Phenoxy resin, weight average molecular weight 70000, glass transition temperature 88 ° C "Sumika Excel” (trademark registration) PES5003P (manufactured by Sumitomo Chemical Co., Ltd.): polyethersulfone, weight average molecular weight 47300 Hardener HF-3M (Maywa Kasei Co., Ltd.): Phenol novolac resin (softening point 96 ° C) “JER Cure” (registered trademark) DICY7 (manufactured by Mitsubishi Chemical Corporation): finely pulverized dicyandiamide (melting point: 210 ° C.) 3,3′-DAS (Mitsui Chemical Fine Co., Ltd.): 3,3′-diaminodiphenyl sulfone (melting point: 170 ° C.) Curing catalyst “C
- a binder resin composition for preform Were mixed uniformly to prepare a binder resin composition for preform. Further, the preform binder resin compositions B, C, and D in Table 1 are heat-treated in the manner described later to advance a partial curing reaction of the epoxy resin in each resin composition, thereby preserving the epoxy resin. Preform binder resin compositions B ′, B ′′, B ′ ′′ and C ′ and D ′, D ′′ containing the reaction product were used. Such preform resin compositions B ′, B ′′, B ′ ′′ and C ′ and D ′, D ′′ for preforms are compared with the binder resin compositions B, C and D for preforms before heat treatment. The glass transition temperature was high.
- Extrusion kneading of binder resin composition Small extruder (S1KRC kneader, Kurimoto Co., Ltd.) under the temperature conditions where the curing reaction of epoxy resin, thermoplastic resin and curing catalyst does not proceed substantially with the raw materials and blending ratios listed in Table 4. Extrusion kneading was carried out using The epoxy resin and the thermoplastic resin were first extruded and kneaded at a compatible temperature, and a curing catalyst was added later, and kneaded under a temperature condition in which the curing reaction did not substantially proceed.
- a binder resin composition that is stable at a kneader current value of 6 A or less during extrusion kneading is A, B is 6 to 7 A, B is 7 A or more, and the extruder is stopped when torque is over, and C. did.
- binder particles The prepared binder resin composition for preforms was freeze-pulverized using liquid nitrogen using a hammer mill (PULVERIZER, manufactured by Hosokawa Micron Co., Ltd.) with a screen having a pore size of 1 mm to obtain particles. . By passing the particles through a sieve having an opening size of 212 ⁇ m, coarse particles were eliminated, and preform binder particles were obtained.
- a hammer mill PULVERIZER, manufactured by Hosokawa Micron Co., Ltd.
- Preform Demoldability Evaluation The preform produced as described above was evaluated according to the degree of deformation of the preform immediately after demolding from the preform mold. The sag of the preform in the longitudinal direction of a preform having a length of 150 mm and a width of 50 mm was measured by its own weight.
- Example 1 According to the blending ratio in Table 1, the glass transition temperature of the preform binder resin composition prepared as described above was measured. Further, using the preform binder resin composition, the molding conditions shown in Table 2 were adopted to produce a cured resin plate as described above, and the glass transition temperature thereof was measured. Also, using the preform binder resin composition, the binder particles prepared so as to have the average particle size shown in Table 2, and using the molding conditions shown in Table 2, the preform was prepared as described above. In Example 1, the mold release property evaluation and the adhesive strength evaluation were performed. As shown in Table 2, the preform binder resin composition was subjected to an actual molding temperature of 65 ° C., which is 5 ° C. higher than the glass transition temperature.
- the glass transition temperature of the resin composition increased from 60 ° C to 70 ° C. As the glass transition temperature rose above the actual molding temperature, the preform could be removed. When the actual molding temperature was 65 ° C., the degree of curing of the resin composition was lower than other levels, but the adhesive strength was acceptable.
- Examples 2 to 4 as shown in Table 2, by setting the actual molding temperatures to 80 ° C., 90 ° C., and 110 ° C., the glass transition temperatures of the resin compositions are changed from 60 ° C. to 90 ° C. and 110 ° C., respectively. The temperature rose to 129 ° C. Since the glass transition temperature was higher than the actual molding temperature, the preform demoldability was good.
- the adhesive strength by increasing the actual molding temperature, the curing of the binder resin composition for preform progressed, and a more excellent value was obtained. Further, by increasing the actual molding temperature, molding was possible in a shorter time. In particular, even when the actual molding time was 5 minutes when the actual molding temperature was 110 ° C., the results of excellent adhesive strength were obtained.
- Example 5 After preparing the preform binder resin composition D as described above in accordance with the blending ratio in Table 1, a part of the epoxy resin in the resin composition is allowed to proceed by heating at 80 ° C. for 120 minutes. Thus, a preform binder resin composition D ′ containing a pre-reaction product of an epoxy resin was obtained. The glass transition temperature of the resin composition D ′ was increased to 90 ° C. Preparation and evaluation of preform binder resin composition, and preparation and evaluation of preform in the same manner as in Example 1 except that the resin composition D ′ was used and the average particle diameter and molding conditions shown in Table 2 were adopted. Went.
- the glass transition temperature of the resin composition increased from 90 ° C to 160 ° C. Since the glass transition temperature was higher than the actual molding temperature, the preform demoldability was good. On the other hand, when the glass transition temperature after curing of the preform binder resin composition is increased to 160 ° C., the crosslink density of the product obtained by curing reaction of the resin composition is increased, so that the adhesive strength is slightly reduced. It was an acceptable level.
- Example 1 Except that the molding conditions shown in Table 2 were adopted, the preparation and evaluation of a preform binder resin composition and the production and evaluation of a preform were performed in the same manner as in Example 1.
- the resin was not melted and the reinforcing fiber base materials could not be fixed to each other at 50 ° C., which is an actual molding temperature lower than the glass transition temperature, for 3 hours.
- Example 6 Preparation and evaluation of preform binder resin composition and evaluation, and preparation and evaluation of preform in the same manner as in Example 1 except that the blending ratio of Table 1 was used and the average particle diameter and molding conditions shown in Table 2 were adopted. Went.
- Example 7 After preparing the preform binder resin composition B as described above in accordance with the blending ratio in Table 1, the curing reaction of a part of the epoxy resin in the resin composition is advanced by heating at 90 ° C. for 30 minutes. Thus, a preform binder resin composition B ′ containing an epoxy resin pre-reaction product was obtained. The glass transition temperature of the resin composition B ′ was increased to 90 ° C. Using the resin composition B ′, except for employing the average particle diameter and molding conditions shown in Table 2, the preparation and evaluation of a preform binder resin composition in the same manner as in Example 1, and the preparation of the preform, Evaluation was performed.
- Example 8> After preparing the preform binder resin composition B as described above in accordance with the blending ratio in Table 1, the curing reaction of a part of the epoxy resin in the resin composition is advanced by heating at 90 ° C. for 60 minutes. Thus, a preform binder resin composition B ′′ containing an epoxy resin pre-reaction product was obtained. The glass transition temperature of the resin composition B ′′ increased to 105 ° C. Preparation of a binder resin composition for preform, evaluation, and preparation of a preform in the same manner as in Example 1 except that the resin composition B '' was used and the average particle diameter and molding conditions shown in Table 2 were adopted. And evaluated.
- a preform binder resin composition having a glass transition temperature of 105 ° C. was molded under conditions of an actual molding temperature of 110 ° C. and an actual molding time of 120 minutes. The transition temperature rose to 125 ° C. As a result of the glass transition temperature exceeding the actual molding temperature by 15 ° C., the preform has good mold release properties. On the other hand, the resin hardly melted and did not adhere sufficiently to the reinforcing fiber substrate, so the adhesive strength was slightly low but acceptable.
- Example 3 Preparation and evaluation of a preform binder resin composition, and preparation and evaluation of a preform in the same manner as in Example 1 except that the blending ratios shown in Table 1 were used and the average particle size and molding conditions shown in Table 3 were adopted. Went.
- Example 9 as shown in Table 3, even when the actual molding temperature of the preform binder resin composition was 120 ° C. and the actual molding time was 1 minute, the glass transition temperature of the resin composition was 60 ° C. Therefore, the mold release property of the preform was at an acceptable level. Since the resin composition was sufficiently cured, the adhesive strength was at a sufficient level.
- Example 10 as shown in Table 3, when the actual molding temperature of the preform binder resin composition was 110 ° C. and the actual molding time was 120 minutes, the glass transition temperature of the resin composition was up to 130 ° C. Since it rose, the demolding property of the preform was good. As for the adhesive strength, an excellent value was obtained because the curing reaction proceeded sufficiently by increasing the actual molding time.
- Example 11 as shown in Table 3, even if the glass transition temperature after molding of the preform binder resin composition is 90 ° C., it is higher than the actual molding temperature of 80 ° C. Was enough. Also, the adhesive strength was within an acceptable range.
- Example 12 when the glass transition temperature after molding was increased to 100 ° C., the curing reaction proceeded, and excellent values of adhesive strength and demoldability were obtained.
- Example 13 by combining the polyfunctional epoxy, the amine curing agent, and the urea curing catalyst, as shown in Table 3, the glass transition temperature after molding of the preform binder resin composition is increased to 140 ° C. However, although the adhesive strength was low because curing did not proceed sufficiently, it was at an acceptable level. Further, the demoldability was good.
- Examples 14 and 15> After preparing the preform binder resin composition D as described above according to the blending ratio in Table 1, the curing reaction of a part of the epoxy resin in the resin composition is advanced by heating at 120 ° C. for 60 minutes. Thus, a preform binder resin composition D ′′ containing an epoxy resin pre-reaction product was obtained. The glass transition temperature of the resin composition D ′′ was increased to 140 ° C. Preparation of a binder resin composition for preform, evaluation, and preparation of a preform in the same manner as in Example 1 except that the resin composition D ′′ was used and the average particle diameter and molding conditions shown in Table 3 were adopted. And evaluated.
- Example 14 as shown in Table 3, by setting the actual molding temperature to 180 ° C., the glass transition temperature after molding of the binder resin composition for preforms increased to 200 ° C., and the molecular crosslinking density was increased. Since it was high, the adhesive strength was low but acceptable. Further, the demoldability was good.
- Example 15 as shown in Table 3, by setting the actual molding temperature to 200 ° C., the glass transition temperature after molding of the binder resin composition for preforms was increased to 215 ° C., and the molecular crosslinking density was increased. Since it became high, the adhesive strength was low but acceptable. Moreover, it was a level which has no problem about demoldability.
- ⁇ Reference Example 4> Preparation and evaluation of a preform binder resin composition, and preparation and evaluation of a preform in the same manner as in Example 1 except that the blending ratios shown in Table 1 were used and the average particle size and molding conditions shown in Table 3 were adopted. Went.
- a preform binder resin composition C was prepared as described above.
- the glass transition temperature of the resin composition C was 10 ° C.
- a part of the epoxy resin in the resin composition is allowed to proceed by heating at 80 ° C. for 30 minutes to generate a preliminary reaction of the epoxy resin.
- Preform binder resin composition C ′ containing the product.
- the glass transition temperature of the resin composition C ′ was increased to 30 ° C.
- preform molding was performed at an actual molding temperature of 100 ° C. with the blending amount of YP50 being a phenoxy resin being 10 parts by mass, 60 parts by mass, and 100 parts by mass. Good preform demoldability was obtained in each actual molding time within 20 minutes. As the blending amount of the phenoxy resin was increased, the adhesive strength was improved and an excellent adhesive strength was obtained.
- Comparative Example 1 As shown in Table 4, when YP-50, which is a phenoxy resin, is not included and the initial Tg is as low as 0 ° C., the resin composition flows during molding, so that sufficient adhesive strength is obtained. I could't.
- Example 21 In Comparative Example 2, as shown in Table 4, curing was performed at an actual molding temperature of 100 ° C. and an actual molding time of 25 minutes with a blending amount of phenoxy resin YP-50 of 120 parts by mass. Only the temperature rose to ° C, and the demoldability was insufficient. Also, extrusion kneading was difficult.
- Example 21 As shown in Table 4, preform molding was performed at an actual molding temperature of 120 ° C. with a blending amount of 2E4MZ as a curing catalyst being 2 parts by mass. Good preform demoldability was obtained in an actual molding time of 13 minutes.
- Example 22 preform molding was performed at an actual molding temperature of 85 ° C. with a blending amount of 1,2-DMZ as a curing catalyst being 20 parts by mass. Good preform demoldability was obtained in an actual molding time of 13 minutes.
- Comparative Example 3 As shown in Table 4, preform molding was performed at an actual molding temperature of 130 ° C. with 3,3′-DAS as the curing agent and no curing catalyst. An actual molding time of 300 minutes was required until sufficient preform demoldability was obtained, and the curing reactivity was insufficient.
- Example 23 In Example 23, as shown in Table 4, PES5003P, which is polyethersulfone, was used as the thermoplastic resin, and preform molding was performed at an actual molding temperature of 100 ° C. The actual molding time required 30 minutes, and the curability was lowered as compared with the case where a phenoxy resin was used as the thermoplastic resin. Moreover, although sufficient adhesive strength was obtained, the value of adhesive strength was low compared with the phenoxy resin which has a hydroxyl group in the principal chain of a thermoplastic resin.
- Example 24 As shown in Table 5, preform molding was performed under the conditions of an actual molding temperature of 100 ° C. and an actual molding time of 12 minutes with the blending amount of jER1001 which is a solid bisphenol type epoxy resin being 20 parts by mass. Demoldability and adhesive strength were good values.
- preform molding was performed at an actual molding temperature of 100 ° C. with the blending amount of the solid bisphenol type epoxy resin being 30 parts by mass and 70 parts by mass. In both cases, good values were obtained for both demolding and adhesive strength.
- Example 27 as shown in Table 5, preform molding was performed at an actual molding temperature of 100 ° C. with the blending amount of the solid bisphenol type epoxy resin being 10 parts by mass. Since the blending amount of the solid bisphenol-type epoxy resin was small and the crosslinking density was high, the adhesive strength was low, but acceptable results were obtained.
- Example 28 as shown in Table 5, preform molding was performed at an actual molding temperature of 100 ° C. with a blending amount of the solid bisphenol type epoxy resin being 90 parts by mass. Although the blending amount of the solid bisphenol-type epoxy resin is large and the glass transition temperature rises only up to 100 ° C., an acceptable demolding property was obtained. The extrusion kneadability was in an acceptable range.
- Example 29 as shown in Table 5, preform molding was performed at an actual molding temperature of 100 ° C. with the blending amount of the solid bisphenol type epoxy resin being 0 parts by mass and the liquid bisphenol type epoxy resin being 100 parts by mass. Since the crosslink density increased, the adhesive strength decreased but acceptable results were obtained.
- Example 30 as shown in Table 5, preform molding was performed at an actual molding temperature of 100 ° C. with the blending amount of the solid bisphenol type epoxy resin being 100 parts by mass. Tg did not rise to the actual molding temperature, and the demoldability decreased, but was in an acceptable range.
- Example 31 preform molding was performed at an actual molding temperature of 65 ° C. with a blending amount of 1,2-DMZ as a curing catalyst being 20 parts by mass. Good preform demoldability was obtained in an actual molding time of 40 minutes. Since the Tg after curing was 70 ° C., the adhesive strength was a low value, but was in an acceptable range.
- preform molding was performed using a benzoxazine resin as a main component as a thermosetting resin at an actual molding temperature of 150 ° C. and an actual molding time of 10 minutes. Good demoldability and adhesive strength were obtained.
- Example 33 Solid cresol novolac type epoxy resin N-695 was added to the epoxy component, and preform molding was performed at an actual molding temperature of 100 ° C. Good mold release properties and adhesive strength were obtained in an actual molding time of 10 minutes.
Abstract
Description
Tg1<T≦Tg1+100 ・・・(1)
T≦Tg2≦T+30 ・・・(2)
第2の本発明に係るプリフォーム用バインダー樹脂組成物において、前記熱硬化性樹脂[A]がエポキシ樹脂であることが好ましい。
80≦T+0.4t≦190 (3)
80≦T+0.4t≦130 (4)
本発明に係るプリフォーム用バインダー樹脂組成物は、熱硬化性樹脂[A]の予備反応生成物を含むことができる。
TR-100≦Tg1<TR ・・・(5)
TR≦Tg2≦TR+30 ・・・(6)
T≦Tg2≦T+30 ・・・(2)
第2の本発明に係るプリフォーム用バインダー樹脂組成物は、熱硬化性樹脂[A]が接着性、取り扱い性の観点からエポキシ樹脂であることが好ましい。
80≦T+0.4t≦190 ・・・(3)
80≦T+0.4t≦130 ・・・(4)
例えば、多官能エポキシ樹脂に硬化剤であるジシアンジアミドと硬化触媒である2,4-トリレンビス(1,1-ジメチルウレア)を組み合わせた組成で成形時間tと前記成形温度Tが(3)式を満たす組み合わせが存在する様に調整することができ、固形2官能エポキシ樹脂に硬化触媒であるイミダゾールアダクトを組み合わせた組成で成形時間tと前記成形温度Tが(4)式を満たす組み合わせが存在する様に調整することができる。
・・・(式1)
Af:繊維基材1枚・1m2当たりの重量(g/m2)
N:繊維基材の積層枚数(枚)
ρf:強化繊維の密度(g/cm3)
h:繊維強化複合材料(試験片)の厚み(mm)。
TR-100≦Tg1<TR (5)
TR≦Tg2≦TR+30 (6)
本発明のプリフォーム用バインダー樹脂組成物を用いてプリフォームを成形する際の実成形温度TRは、プリフォームを成形する際の温度であり、プリフォーム用バインダー樹脂組成物を硬化反応して固化させる温度である。かかる実成形温度TRは、本発明のプリフォーム用バインダー樹脂組成物の硬化性および接着強度の観点から60~200℃の範囲であり、80~150℃の範囲であることが好ましく、90~130℃の範囲であることがより好ましい。かかる実成形温度TRが60℃に満たない場合、プリフォーム化の際の硬化反応が十分に進行しないため、脱型性が低下するとともに接着強度が不足する。一方、かかる実成形温度TRが200℃を越える場合、プリフォーム用バインダー樹脂組成物を硬化反応させた物のガラス転移温度が実成形温度TRよりも高くなりにくく脱型性が低下し、プリフォーム用バインダー樹脂組成物のガラス転移温度が実成形温度TR以上に高くなった場合でも、当該樹脂組成物を硬化反応させた物の架橋密度が高くなるため接着強度が低くなる。
各実施例の樹脂組成物を得るために、以下の樹脂原料を用いた。なお、表1、表4および表5の樹脂組成物の含有割合の単位は、特に断らない限り「質量部」を意味する。
・YD-128(新日鉄住金化学(株)製):液状2官能ビスフェノールA型エポキシ樹脂、エポキシ当量189
・“jER”(登録商標)1001(三菱化学(株)製):固形2官能ビスフェノールA型エポキシ樹脂、エポキシ当量475、ガラス転移温度35℃
・“jER”(登録商標)1004(三菱化学(株)製):固形2官能ビスフェノールA型エポキシ樹脂、エポキシ当量975、ガラス転移温度60℃
・“スミエポキシ”(登録商標)ELM434(住友化学(株)製):テトラグリシジルジアミノジフェニルメタン、エポキシ当量120
・“EPICLON”(登録商標)N-695(DIC(株)製):固形クレゾールノボラック型エポキシ樹脂、エポキシ当量214
ベンゾオキサジン樹脂
・P-d型ベンゾオキサジン樹脂(四国化成工業(株)製):固形P-d型ベンゾオキサジン樹脂、分子量434、軟化点75℃
熱可塑性樹脂
・YP-50(新日鉄住金化学(株)製):フェノキシ樹脂、重量平均分子量70000、ガラス転移温度88℃
・“スミカエクセル”(商標登録)PES5003P(住友化学(株)製):ポリエーテルスルホン、重量平均分子量47300
硬化剤
・HF-3M(明和化成(株)製):フェノールノボラック樹脂(軟化点96℃)
・“jERキュア”(登録商標)DICY7(三菱化学(株)製):ジシアンジアミド微粉砕物(融点:210℃)
・3,3’-DAS(三井化学ファイン(株)製):3,3’-ジアミノジフェニルスルホン(融点:170℃)
硬化触媒
・“キュアダクト”(登録商標)P-0505(四国化成工業(株)製):イミダゾールアダクト
・“キュアダクト”(登録商標)L-07N(四国化成工業(株)製):上記P-0505の保存安定化成分
・“キュアゾール”(登録商標)2E4MZ(四国化成工業(株)製):2-エチル-4メチルイミダゾール
・“キュアゾール”(登録商標)1,2-DMZ(四国化成工業(株)製):1,2-ジメチルイミダゾール
・TPP(ケイ・アイ化成(株)製):トリフェニルホスフィン(融点80℃)
・“オミキュア”(登録商標)24(PTIジャパン(株)製):2,4-トリレンビス(1,1-ジメチルウレア)
・DY9577(ハンツマン・アドバンスド・マテリアルズ(株)製):三塩化ホウ素オクチルアミン錯体
2.バインダー樹脂組成物の調製
表1および表4に記載した原料と配合比で熱硬化性樹脂、熱可塑性樹脂、硬化剤、硬化触媒を硬化反応が実質的に進まない温度/時間条件にて加熱攪拌により均一に混合し、プリフォーム用バインダー樹脂組成物を調製した。さらに、表1のプリフォーム用バインダー樹脂組成物B、CおよびDについては、後述の要領で熱処理を行い、各樹脂組成物中のエポキシ樹脂の一部の硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物B'、B''、B'''およびC'およびD'、D''とした。かかるプリフォーム用バインダー樹脂組成物B'、B''、B'''およびC'およびD'、D''は、熱処理前のプリフォーム用バインダー樹脂組成物B、CおよびDに比較して、ガラス転移温度が高くなっていた。
表4に記載した原料と配合比でエポキシ樹脂、熱可塑性樹脂、硬化触媒を硬化反応が実質的に進まない温度条件にて小型押出機(S1KRCニーダー、(株)栗本鐵工所)を使用して押出混練を行った。エポキシ樹脂と熱可塑性樹脂は相溶する温度で先に押出混練を行い、硬化触媒を後から加えて硬化反応が実質的に進まない温度条件で混練した。押出混練性の評価として、押出混練時にニーダー電流値が6A以下で安定するバインダー樹脂組成物をA、6~7AであるものをB、7A以上となりトルクオーバーで押出機が停止したものをCとした。
プレス装置下面に、一辺50mmの正方形をくり抜いた、厚さ2mmの銅製スペーサーを設置し、プレスの温度を所定の実成形温度に設定し、エポキシ樹脂組成物をスペーサーの内側に注ぎ、プレスを閉じた。所定の実成形時間を経過後にプレスを開け、樹脂硬化板を得た。
樹脂組成物および所定の実成形温度で硬化させた樹脂硬化板を試料として、JIS K 7121:1987に従って、示差走査熱量計(DSC)を用いて中間点ガラス転移温度を測定した。測定装置にはPyris1 DSC(Perkin Elmer製)を使用した。アルミサンプルパンに5~10mgの試料を採取し、窒素雰囲気下で-30~300℃の温度範囲、40℃/minの昇温速度で測定を行い、DSC曲線が吸熱側に階段状変化を示す部分の中間点ガラス転移温度を求めた。
調製したプリフォーム用バインダー樹脂組成物をハンマーミル(PULVERIZER、ホソカワミクロン(株)製)にて、孔サイズ1mmのスクリーンを使用し、液体窒素を用いて凍結粉砕して粒子を得た。かかる粒子を目開きサイズ212μmの篩いに通すことで粗大な粒子を排除し、プリフォーム用バインダー粒子を得た。
レーザー解析・散乱式粒子径・粒度分布測定装置MT3300II(日機装(株)製)を用い、取り込み回数500回で測定した。
前述のように作製したバインダー粒子を炭素繊維織物に6g/m2の散布量で片面に散布した後、表面に遠赤ヒーターを用いて加熱してバインダー樹脂組成物を付与した強化繊維基材を得た。バインダー樹脂組成物を付与した強化繊維基材を一方は散布面、他方は非散布面が接着する方向となるように2枚重ね、50kPaの圧力を加えながら各々の実成形温度、実成形時間で加熱することによりプリフォームを作製した。
上記のように作製したプリフォームをプリフォーム成形型から脱型した直後のプリフォームの変形度合に応じて評価した。長さ150mm、幅50mmのプリフォームの長手方向端部の自重での垂れ下がりを測定した。
各々の実成形温度、実成形時間で作製したプリフォームについて、強化繊維基材間の引き剥がし試験を行った。試験はJIS K 6854:1977に従い、インストロン万能試験機(インストロン製)を用いて実施した。試験片は作製したプリフォームを長さ150mm(接着部分は100mm)、幅25mmにカットして仕上げた。同一試験に用いる試験片の数は5とし、試験結果にはその平均値を使用した。引っ張り速さは50mm/分とした。
表1の配合比に従って、前記したようにして調製したプリフォーム用バインダー樹脂組成物のガラス転移温度を測定した。また、そのプリフォーム用バインダー樹脂組成物を用いて、表2に示す成形条件を採用して前記のようにして樹脂硬化板を作製し、そのガラス転移温度を測定した。また、そのプリフォーム用バインダー樹脂組成物を用いて表2に示す平均粒径を有するように作製したバインダー粒子を用い、表2に示す成形条件を採用して前記したようにしてプリフォームを作製し、それについて脱型性評価および接着強度評価を行った
実施例1では、表2に示したように、プリフォーム用バインダー樹脂組成物をガラス転移温度よりも5℃高い実成形温度65℃で実成形時間120分間の条件で成形することにより、当該樹脂組成物のガラス転移温度が60℃から70℃まで上昇した。ガラス転移温度が実成形温度以上に上昇したことにより、プリフォームは脱型可能であった。実成形温度が65℃では、当該樹脂組成物の硬化の程度は他の水準に比べて低いものの、接着強度は許容できるレベルであった。
表1の配合比に従い、前記したようにしてプリフォーム用バインダー樹脂組成物Dを調製した後、80℃、120分の加熱により当該樹脂組成物中のエポキシ樹脂の一部の硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物D'とした。当該樹脂組成物D'のガラス転移温度は90℃まで上昇していた。当該樹脂組成物D'を用い、表2の平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表2の成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比を用い、表2に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比に従って、前記したようにしてプリフォーム用バインダー樹脂組成物Bを調製した後、90℃、30分の加熱により当該樹脂組成物中のエポキシ樹脂の一部の硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物B'とした。当該樹脂組成物B'のガラス転移温度は90℃まで上昇していた。当該樹脂組成物B'を用い、表2に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比に従って、前記したようにしてプリフォーム用バインダー樹脂組成物Bを調製した後、90℃、60分の加熱により当該樹脂組成物中のエポキシ樹脂の一部の硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物B''とした。当該樹脂組成物B''のガラス転移温度は105℃まで上昇していた。当該樹脂組成物B''を用い、表2に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比に従い、前記したようにしてプリフォーム用バインダー樹脂組成物Bを調製した後、90℃、90分の加熱により当該樹脂組成物中のエポキシ樹脂の一部硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物B'''とした。当該樹脂組成物B'''のガラス転移温度は120℃まで上昇していた。当該樹脂組成物B'''を用い、表2に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比を用い、表3に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比に従って、前記したようにしてプリフォーム用バインダー樹脂組成物Dを調製した後、120℃、60分の加熱により当該樹脂組成物中のエポキシ樹脂の一部の硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物D''とした。当該樹脂組成物D''のガラス転移温度は140℃まで上昇していた。当該樹脂組成物D''を用い、表3に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比を用い、表3に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表1の配合比に従って、前記したようにしてプリフォーム用バインダー樹脂組成物Cを調製した。当該樹脂組成物Cのガラス転移温度は10℃であった。また、同様にしてプリフォーム用バインダー樹脂組成物Cを調製した後、80℃、30分の加熱により当該樹脂組成物中のエポキシ樹脂の一部の硬化反応を進行させてエポキシ樹脂の予備反応生成物を含むプリフォーム用バインダー樹脂組成物C'とした。当該樹脂組成物C'のガラス転移温度は30℃まで上昇していた。それぞれのプリフォーム用バインダー樹脂組成物を用い、表3に示す平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
表4に示す配合比、平均粒径および成形条件を採用した以外は、実施例1と同様にしてプリフォーム用バインダー樹脂組成物の調製、評価、およびプリフォームの作製、評価を行った。
〈実施例21、22〉
実施例21では、表4に示したように、硬化触媒である2E4MZの配合量を2質量部として実成形温度120℃でプリフォーム成形を行った。13分の実成形時間で良好なプリフォームの脱型性が得られた。
〈比較例3〉
比較例3では、表4に示したように、硬化剤を3,3’-DASとして硬化触媒を配合せずに、実成形温度130℃でプリフォーム成形を行った。十分なプリフォームの脱型性が得られるまで実成形時間300分かかり、硬化反応性が不足した。
実施例23では、表4に示したように、熱可塑性樹脂としてポリエーテルスルホンであるPES5003Pを使用し、実成形温度100℃でプリフォーム成形を行った。実成形時間に30分を要し、熱可塑性樹脂としてフェノキシ樹脂を用いた場合と比較して硬化性は低下した。また、十分な接着強度は得られるが、熱可塑性樹脂の主鎖に水酸基を有するフェノキシ樹脂と比較すると、接着強度の値は低くなった。
実施例24では、表5に示したように、固形ビスフェノール型エポキシ樹脂であるjER1001の配合量を20質量部として実成形温度100℃、実成形時間12分の条件でプリフォーム成形を行った。脱型性、接着強度は良好な値であった。
〈実施例32〉
実施例32では、熱硬化性樹脂として、ベンゾオキサジン樹脂を主成分として、実成形温度150℃、実成形時間10分でのプリフォーム成形を行った。良好な脱型性、接着強度が得られた。
〈実施例33〉
実施例33では、固形クレゾールノボラック型エポキシ樹脂N-695をエポキシ成分に加え、実成形温度100℃でプリフォーム成形を行った。実成形時間が10分で良好な脱型性、接着強度が得られた。

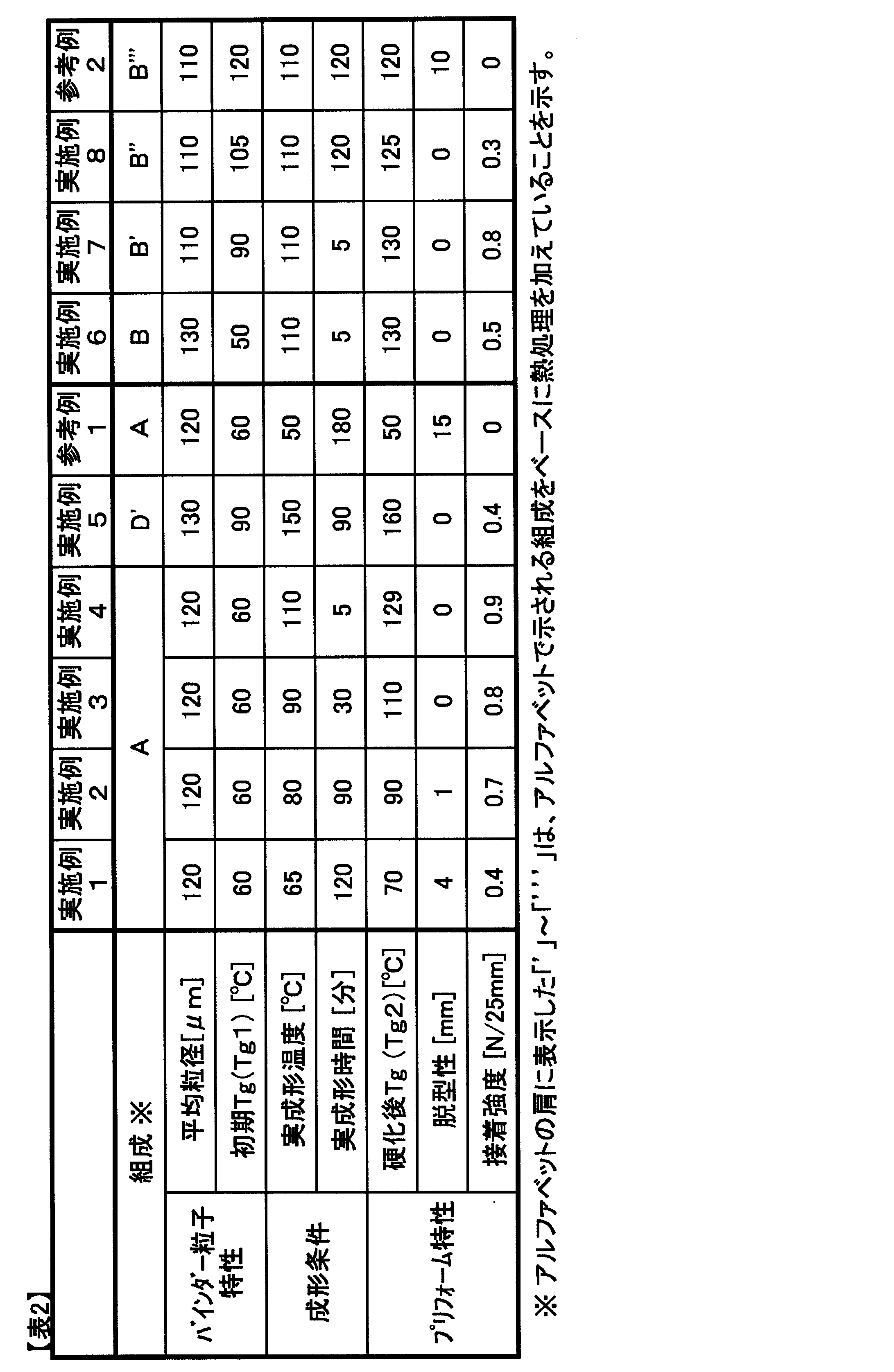
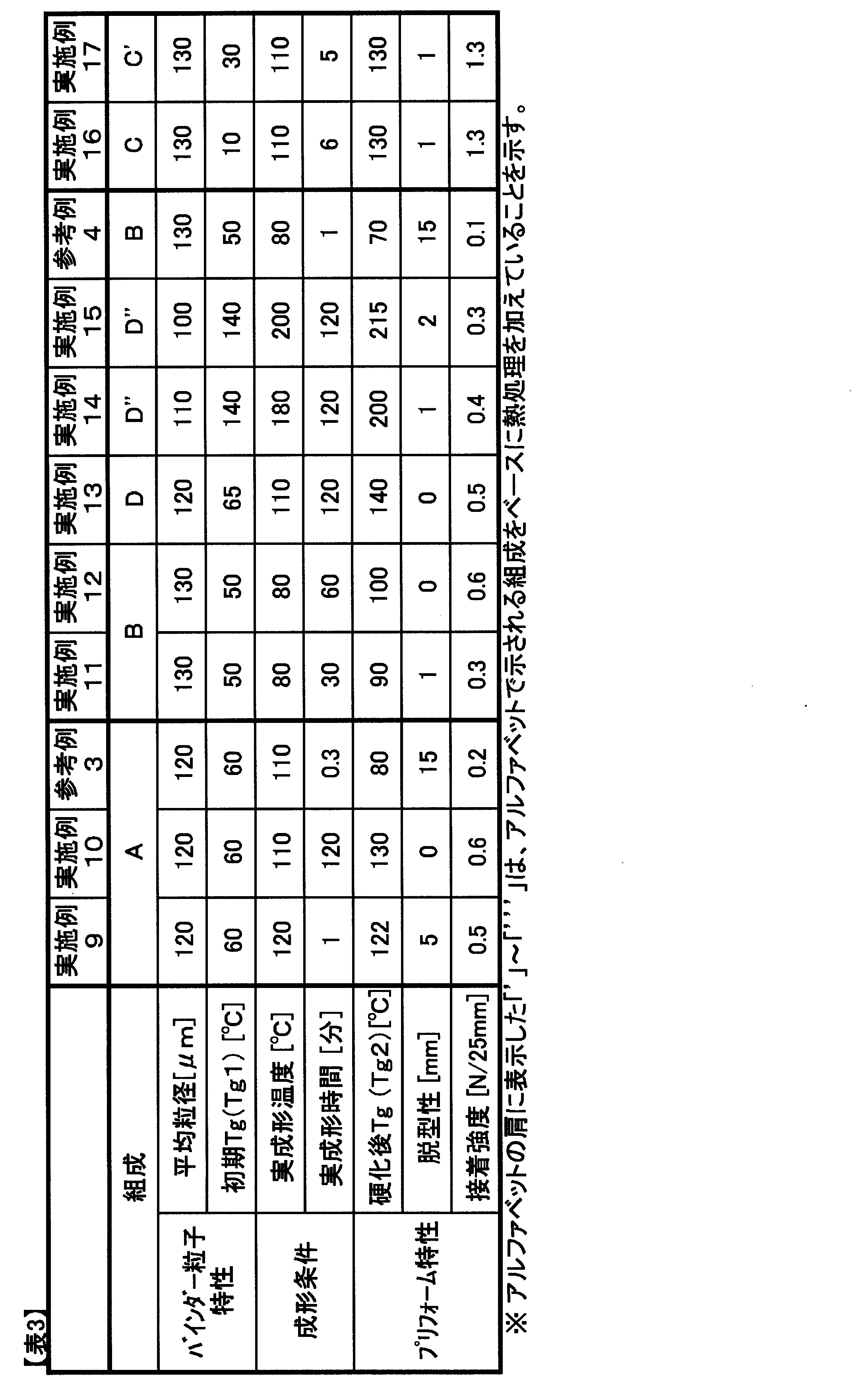
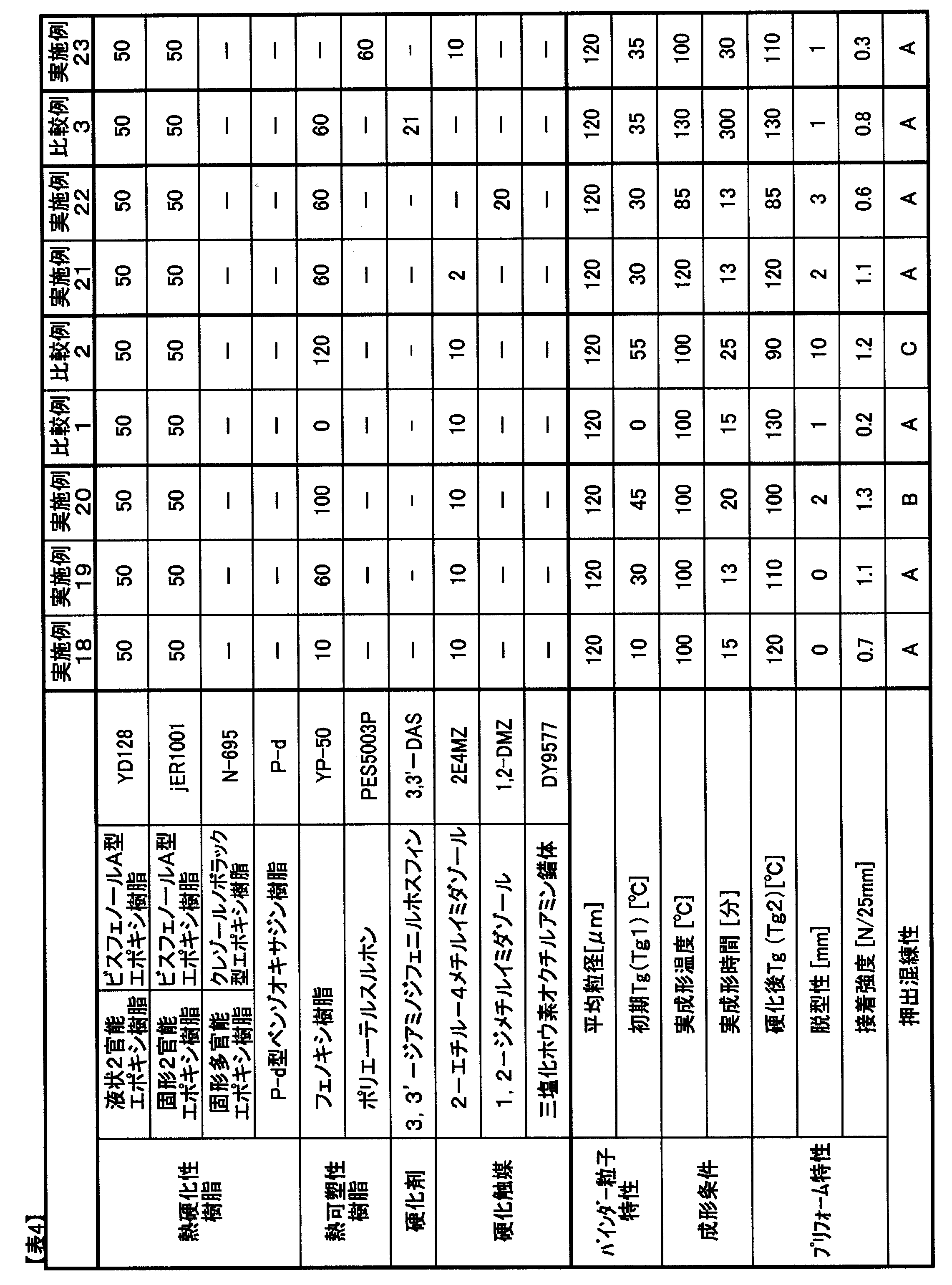
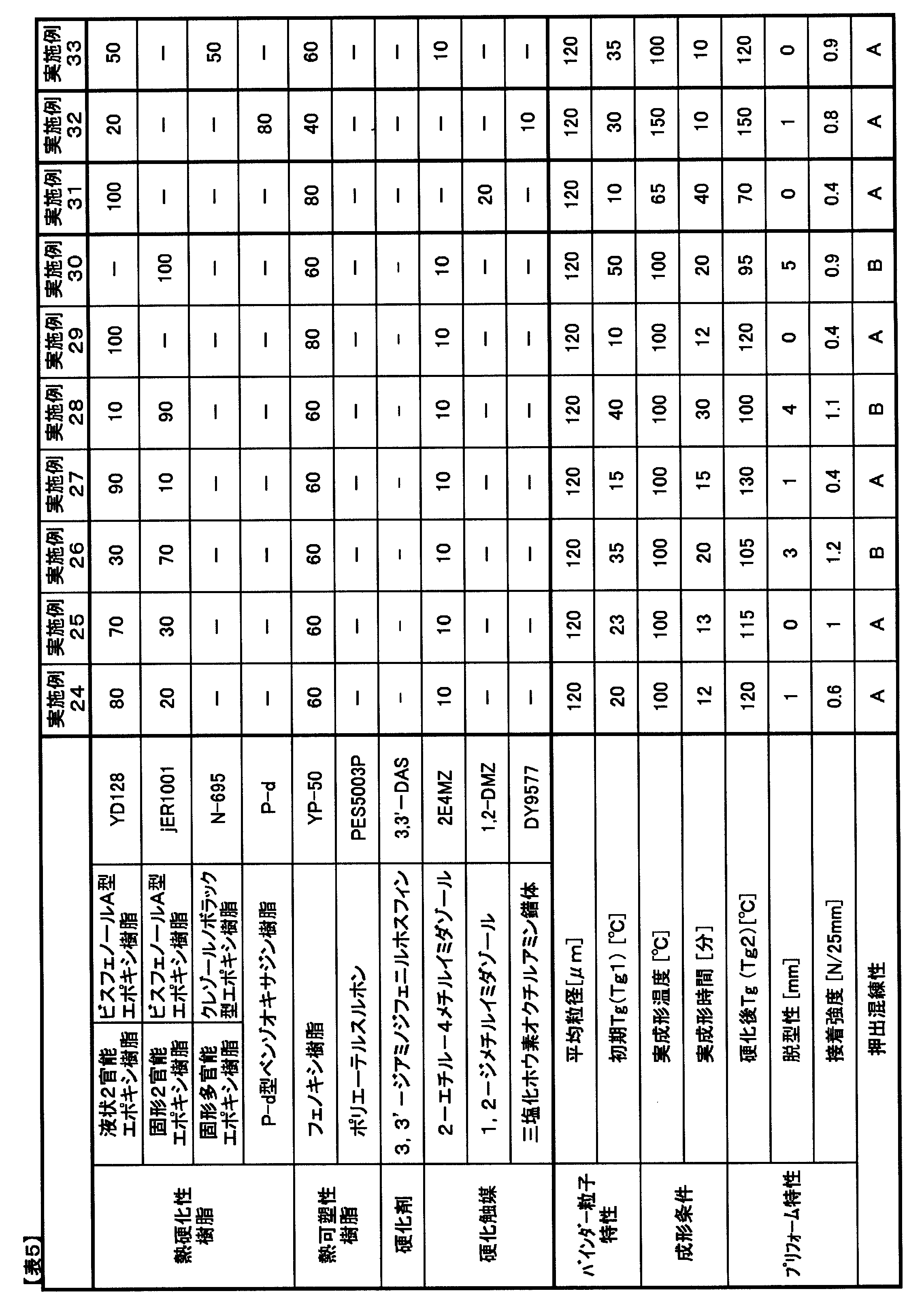
Claims (17)
- 熱硬化性樹脂[A]、熱可塑性樹脂[B]および硬化触媒[C]を含む樹脂組成物であって、熱硬化性樹脂[A]が2官能エポキシ樹脂を含み、熱可塑性樹脂[B]の含有量が熱硬化性樹脂[A]100質量部に対して10~100質量部の範囲であり、かつ、硬化触媒[C]が、有機リン化合物、イミダゾールおよびこれらの誘導体から選ばれる少なくとも1つの硬化触媒であるプリフォーム用バインダー樹脂組成物。
- 前記熱硬化性樹脂[A]が液状2官能エポキシ樹脂を含む請求項1に記載のプリフォーム用バインダー樹脂組成物。
- 前記熱可塑性樹脂[B]が主鎖に水酸基を有する請求項1または2に記載のプリフォーム用バインダー樹脂組成物。
- 前記熱硬化性樹脂[A]100質量部中に、液状2官能エポキシ樹脂が30~80質量部、固形2官能エポキシ樹脂が20~70質量部含まれる、請求項1~3のいずれかに記載のプリフォーム用バインダー樹脂組成物。
- 前記熱硬化性樹脂[A]が固形2官能ビスフェノール型エポキシ樹脂を含む請求項1~4のいずれかに記載のプリフォーム用バインダー樹脂組成物。
- 前記硬化触媒[C]が前記熱硬化性樹脂[A]100質量部に対して2~20質量部含まれる請求項1~5のいずれかに記載のプリフォーム用バインダー樹脂組成物。
- 熱硬化性樹脂[A]を含み、ガラス転移温度がTg1である樹脂組成物であって、60~200℃の範囲でかつ(1)式を満たす、ある成形温度T℃で、0.5~120分の範囲の、ある成形時間t分間、加熱することで、ガラス転移温度が(2)式に示すTg2に上昇する成形時間tと成形温度Tの組み合わせが存在するプリフォーム用バインダー樹脂組成物。
Tg1<T≦Tg1+100 ・・・(1)
T≦Tg2≦T+30 ・・・(2) - 前記熱硬化性樹脂[A]がエポキシ樹脂である請求項7に記載のプリフォーム用バインダー樹脂組成物。
- 前記樹脂組成物が、熱硬化性樹脂[A]、熱可塑性樹脂[B]および硬化触媒[C]を含み、熱硬化性樹脂[A]が2官能エポキシ樹脂を含み、熱可塑性樹脂[B]の含有量が熱硬化性樹脂[A]100質量部に対して10~100質量部の範囲であり、かつ、硬化触媒[C]が、有機リン化合物、イミダゾールおよびこれらの誘導体から選ばれる少なくとも1つの硬化触媒である請求項7または8に記載のプリフォーム用バインダー樹脂組成物。
- 前記成形時間tと前記成形温度Tの組み合わせが(3)式を満たす範囲内に存在する請求項7~9のいずれかに記載のプリフォーム用バインダー樹脂組成物。
80≦T+0.4t≦190 ・・・(3) - 前記成形時間tと前記成形温度Tの組み合わせが(4)式を満たす範囲内に存在する請求項7~10のいずれかに記載のプリフォーム用バインダー樹脂組成物。
80≦T+0.4t≦130 ・・・(4) - 熱硬化性樹脂[A]の予備反応生成物を含む、請求項1~11のいずれかに記載のプリフォーム用バインダー樹脂組成物。
- 粒子形態を有する請求項1~12のいずれかに記載のプリフォーム用バインダー樹脂組成物。
- 請求項1~13のいずれかに記載のプリフォーム用バインダー樹脂組成物を少なくとも表面に有する強化繊維基材。
- 複数の強化繊維基材が積層されたプリフォームであって、請求項1~13のいずれかに記載のプリフォーム用バインダー樹脂組成物を少なくとも積層層間に有するプリフォーム。
- 請求項15に記載のプリフォームに液状熱硬化性樹脂を含浸させ、硬化させてなる繊維強化複合材料。
- 請求項1~13のいずれかに記載のプリフォーム用バインダー樹脂組成物を強化繊維基材原反の少なくとも表面に付着させて強化繊維基材とした後、該強化繊維基材を積層し、60~200℃の範囲の実成形温度TRで0.5~120分加熱し、前記プリフォーム用バインダー樹脂組成物のガラス転移温度を(5)式を満たす加熱前のガラス転移温度Tg1から(6)式を満たす加熱後のガラス転移温度Tg2に上昇せしめてプリフォームを得るプリフォームの製造方法。
TR-100≦Tg1<TR ・・・(5)
TR≦Tg2≦TR+30 ・・・(6)
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/410,439 US10053575B2 (en) | 2012-07-05 | 2013-07-03 | Binder resin composition for preform, binder particle, preform, and fiber-reinforced composite material |
| JP2013531802A JP6330327B2 (ja) | 2012-07-05 | 2013-07-03 | Rtm成形法用プリフォーム用のバインダー樹脂組成物を用いたrtm成形法用強化繊維基材、rtm成形法用プリフォームおよび繊維強化複合材料 |
| EP13813642.9A EP2871214B1 (en) | 2012-07-05 | 2013-07-03 | Binder resin composition for preform, binder particle, preform, and fiber-reinforced composite material |
| CN201380033681.5A CN104395399B (zh) | 2012-07-05 | 2013-07-03 | 预成型体用粘合剂树脂组合物、粘合剂粒子、预成型体及纤维增强复合材料 |
| KR1020157001073A KR102050341B1 (ko) | 2012-07-05 | 2013-07-03 | 프리폼용 바인더 수지 조성물, 바인더 입자, 프리폼 및 섬유 강화 복합 재료 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-151102 | 2012-07-05 | ||
| JP2012151102 | 2012-07-05 | ||
| JP2013036672 | 2013-02-27 | ||
| JP2013-036672 | 2013-02-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014007288A1 true WO2014007288A1 (ja) | 2014-01-09 |
Family
ID=49882040
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/068249 WO2014007288A1 (ja) | 2012-07-05 | 2013-07-03 | プリフォーム用バインダー樹脂組成物、バインダー粒子、プリフォームおよび繊維強化複合材料 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10053575B2 (ja) |
| EP (1) | EP2871214B1 (ja) |
| JP (1) | JP6330327B2 (ja) |
| KR (1) | KR102050341B1 (ja) |
| CN (1) | CN104395399B (ja) |
| WO (1) | WO2014007288A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015079917A1 (ja) * | 2013-11-29 | 2015-06-04 | 東レ株式会社 | 強化繊維織物基材、プリフォームおよび繊維強化複合材料 |
| WO2017104445A1 (ja) | 2015-12-17 | 2017-06-22 | 東レ株式会社 | プリフォーム用バインダー樹脂組成物、バインダー粒子、強化繊維基材、プリフォームおよび繊維強化複合材料 |
| WO2018117214A1 (ja) * | 2016-12-21 | 2018-06-28 | 三菱ケミカル株式会社 | 硬化性樹脂組成物、並びにこれを用いたフィルム、成形品、プリプレグ及び繊維強化プラスチック |
| US10792869B2 (en) | 2015-02-05 | 2020-10-06 | Toray Industries, Inc. | Preform, fiber-reinforced composite material, and method of manufacturing fiber-reinforced composite material |
| JP2020164672A (ja) * | 2019-03-29 | 2020-10-08 | 帝人株式会社 | バインダー樹脂組成物、プリフォーム、並びに繊維強化複合材料、及び繊維強化複合材料の製造方法 |
| WO2023058546A1 (ja) * | 2021-10-06 | 2023-04-13 | 東レ株式会社 | プリプレグ、繊維強化樹脂成形体、および一体化成形品 |
| WO2024080357A1 (ja) * | 2022-10-14 | 2024-04-18 | Art&Tech株式会社 | Pp-frp部材およびその製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2814875A1 (en) * | 2012-02-15 | 2014-12-24 | Dow Global Technologies LLC | Pre-molding article from thermoset polymer dispersions |
| JP5768893B2 (ja) * | 2012-08-20 | 2015-08-26 | 三菱レイヨン株式会社 | エポキシ樹脂組成物及びこれを用いたフィルム、プリプレグ、繊維強化プラスチック |
| US9941450B2 (en) * | 2015-06-18 | 2018-04-10 | Articulated Technologies, Llc | Roll-to-roll fabricated light sheet and encapsulated semiconductor device |
| GB201514516D0 (en) * | 2015-08-14 | 2015-09-30 | Cytec Ind Inc | Fast-cure pre-preg |
| EP4335557A2 (en) | 2017-05-23 | 2024-03-13 | Alpha Assembly Solutions Inc. | Graphene enhanced and engineered materials for membrane touch switch and other flexible electronic structures |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08509921A (ja) | 1993-05-07 | 1996-10-22 | ザ ダウ ケミカル カンパニー | 改良された樹脂トランスファー成形法 |
| JP2001524171A (ja) | 1997-05-06 | 2001-11-27 | サイテク・テクノロジー・コーポレーシヨン | 成形法のための予備成形物およびそのための樹脂 |
| JP2005194456A (ja) | 2004-01-09 | 2005-07-21 | Toray Ind Inc | プリフォーム作製用バインダー組成物、強化繊維基材、プリフォームおよび繊維強化複合材料の製造方法 |
| JP2011144361A (ja) * | 2009-12-14 | 2011-07-28 | Ajinomoto Co Inc | 樹脂組成物 |
| JP2012019240A (ja) * | 2011-10-12 | 2012-01-26 | Sumitomo Bakelite Co Ltd | 樹脂組成物、基材付き絶縁シート、及び、多層プリント配線板 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5288150B2 (ja) | 2005-10-24 | 2013-09-11 | 株式会社スリーボンド | 有機el素子封止用熱硬化型組成物 |
| CN101910493B (zh) * | 2008-01-11 | 2013-07-24 | 东丽株式会社 | 弯曲形状强化纤维基材、及使用其的层合体、预成型体、纤维强化树脂复合材料和它们的制造方法 |
| EP2256163B1 (en) * | 2008-03-25 | 2018-11-14 | Toray Industries, Inc. | Epoxy resin composition, fiber-reinforced composite material and method for producing the same |
| KR101383434B1 (ko) * | 2008-07-31 | 2014-04-08 | 세키스이가가쿠 고교가부시키가이샤 | 에폭시 수지 조성물, 프리프레그, 경화체, 시트상 성형체, 적층판 및 다층 적층판 |
| CN102165010A (zh) * | 2008-09-29 | 2011-08-24 | 东丽株式会社 | 环氧树脂组合物、预浸料坯及纤维增强复合材料 |
| JP5195454B2 (ja) | 2009-01-22 | 2013-05-08 | 味の素株式会社 | 樹脂組成物 |
-
2013
- 2013-07-03 US US14/410,439 patent/US10053575B2/en active Active
- 2013-07-03 KR KR1020157001073A patent/KR102050341B1/ko active IP Right Grant
- 2013-07-03 CN CN201380033681.5A patent/CN104395399B/zh not_active Expired - Fee Related
- 2013-07-03 EP EP13813642.9A patent/EP2871214B1/en active Active
- 2013-07-03 JP JP2013531802A patent/JP6330327B2/ja not_active Expired - Fee Related
- 2013-07-03 WO PCT/JP2013/068249 patent/WO2014007288A1/ja active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08509921A (ja) | 1993-05-07 | 1996-10-22 | ザ ダウ ケミカル カンパニー | 改良された樹脂トランスファー成形法 |
| JP2001524171A (ja) | 1997-05-06 | 2001-11-27 | サイテク・テクノロジー・コーポレーシヨン | 成形法のための予備成形物およびそのための樹脂 |
| JP2005194456A (ja) | 2004-01-09 | 2005-07-21 | Toray Ind Inc | プリフォーム作製用バインダー組成物、強化繊維基材、プリフォームおよび繊維強化複合材料の製造方法 |
| JP2011144361A (ja) * | 2009-12-14 | 2011-07-28 | Ajinomoto Co Inc | 樹脂組成物 |
| JP2012019240A (ja) * | 2011-10-12 | 2012-01-26 | Sumitomo Bakelite Co Ltd | 樹脂組成物、基材付き絶縁シート、及び、多層プリント配線板 |
Non-Patent Citations (2)
| Title |
|---|
| ASTM D3171, 1999 |
| JIS R 7602, 1995 |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015079917A1 (ja) * | 2013-11-29 | 2015-06-04 | 東レ株式会社 | 強化繊維織物基材、プリフォームおよび繊維強化複合材料 |
| US10400077B2 (en) | 2013-11-29 | 2019-09-03 | Toray Industries, Inc. | Reinforcing fiber fabric substrate, preform, and fiber-reinforced composite material |
| US10792869B2 (en) | 2015-02-05 | 2020-10-06 | Toray Industries, Inc. | Preform, fiber-reinforced composite material, and method of manufacturing fiber-reinforced composite material |
| US20180340047A1 (en) * | 2015-12-17 | 2018-11-29 | Toray Industries, Inc. | Binder resin composition for preform, binder particles, reinforcing fiber base material, preform, and fiber reinforced composite material |
| WO2017104445A1 (ja) | 2015-12-17 | 2017-06-22 | 東レ株式会社 | プリフォーム用バインダー樹脂組成物、バインダー粒子、強化繊維基材、プリフォームおよび繊維強化複合材料 |
| JPWO2018117214A1 (ja) * | 2016-12-21 | 2018-12-20 | 三菱ケミカル株式会社 | 硬化性樹脂組成物、並びにこれを用いたフィルム、成形品、プリプレグ及び繊維強化プラスチック |
| WO2018117214A1 (ja) * | 2016-12-21 | 2018-06-28 | 三菱ケミカル株式会社 | 硬化性樹脂組成物、並びにこれを用いたフィルム、成形品、プリプレグ及び繊維強化プラスチック |
| US11161975B2 (en) | 2016-12-21 | 2021-11-02 | Mitsubishi Chemical Corporation | Curable resin composition, and film, molded article, prepreg, and fiber-reinforced plastic using said curable resin composition |
| JP2020164672A (ja) * | 2019-03-29 | 2020-10-08 | 帝人株式会社 | バインダー樹脂組成物、プリフォーム、並びに繊維強化複合材料、及び繊維強化複合材料の製造方法 |
| JP7431508B2 (ja) | 2019-03-29 | 2024-02-15 | 帝人株式会社 | バインダー樹脂組成物、プリフォーム、並びに繊維強化複合材料、及び繊維強化複合材料の製造方法 |
| WO2023058546A1 (ja) * | 2021-10-06 | 2023-04-13 | 東レ株式会社 | プリプレグ、繊維強化樹脂成形体、および一体化成形品 |
| JP7334867B1 (ja) * | 2021-10-06 | 2023-08-29 | 東レ株式会社 | プリプレグ、繊維強化樹脂成形体、および一体化成形品 |
| WO2024080357A1 (ja) * | 2022-10-14 | 2024-04-18 | Art&Tech株式会社 | Pp-frp部材およびその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104395399B (zh) | 2018-12-07 |
| JP6330327B2 (ja) | 2018-05-30 |
| EP2871214A1 (en) | 2015-05-13 |
| US20150322257A1 (en) | 2015-11-12 |
| EP2871214A4 (en) | 2015-12-02 |
| KR102050341B1 (ko) | 2019-11-29 |
| JPWO2014007288A1 (ja) | 2016-06-02 |
| CN104395399A (zh) | 2015-03-04 |
| KR20150036085A (ko) | 2015-04-07 |
| US10053575B2 (en) | 2018-08-21 |
| EP2871214B1 (en) | 2017-04-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6330327B2 (ja) | Rtm成形法用プリフォーム用のバインダー樹脂組成物を用いたrtm成形法用強化繊維基材、rtm成形法用プリフォームおよび繊維強化複合材料 | |
| JP5954441B2 (ja) | 強化繊維織物基材、プリフォームおよび繊維強化複合材料 | |
| JP6856157B2 (ja) | シートモールディングコンパウンド、および繊維強化複合材料 | |
| KR102081662B1 (ko) | 에폭시 수지 조성물, 프리프레그 및 탄소 섬유 강화 복합 재료 | |
| JP5768893B2 (ja) | エポキシ樹脂組成物及びこれを用いたフィルム、プリプレグ、繊維強化プラスチック | |
| EP2794735B1 (en) | Improvements in or relating to fibre reinforced composites | |
| EP3102621B1 (en) | Amino benzoates or benzamides as curing agents for epoxy resins | |
| WO2013081060A1 (ja) | エポキシ樹脂組成物、プリプレグ、繊維強化複合材料とその製造方法 | |
| JP2019167429A (ja) | エポキシ樹脂組成物、プリプレグ、炭素繊維強化複合材料及びこれらの製造方法 | |
| KR102512809B1 (ko) | 섬유강화 복합재료용 에폭시 수지 조성물 및 이를 이용한 프리프레그 | |
| WO2017222339A1 (ko) | 섬유강화 복합재료용 에폭시 수지 조성물 및 이를 이용한 프리프레그 | |
| JP7431508B2 (ja) | バインダー樹脂組成物、プリフォーム、並びに繊維強化複合材料、及び繊維強化複合材料の製造方法 | |
| EP2812393B1 (en) | Epoxy resin formulations for textiles, mats and other fibrous reinforcements for composite applications | |
| WO2021020045A1 (ja) | 繊維強化複合材料用エポキシ樹脂組成物およびプリフォームならびに繊維強化複合材料 | |
| JP2020097694A (ja) | 繊維強化複合材料用エポキシ樹脂組成物および、繊維強化複合材料用プリフォームならびに繊維強化複合材料 | |
| WO2017104445A1 (ja) | プリフォーム用バインダー樹脂組成物、バインダー粒子、強化繊維基材、プリフォームおよび繊維強化複合材料 | |
| TWI720404B (zh) | 預浸材及纖維強化複合材料 | |
| JP2023149496A (ja) | エポキシ樹脂組成物、プリプレグ、繊維強化複合材料及びその製造方法 | |
| JP2015108052A (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| CN111936564A (zh) | 预浸料坯及其制造方法 | |
| JP2017095537A (ja) | プリプレグ、プリプレグの製造方法及び炭素繊維強化複合材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2013531802 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13813642 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14410439 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20157001073 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013813642 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013813642 Country of ref document: EP |