WO2013175936A1 - 脂環式ジエポキシ化合物の製造方法 - Google Patents
脂環式ジエポキシ化合物の製造方法 Download PDFInfo
- Publication number
- WO2013175936A1 WO2013175936A1 PCT/JP2013/062355 JP2013062355W WO2013175936A1 WO 2013175936 A1 WO2013175936 A1 WO 2013175936A1 JP 2013062355 W JP2013062355 W JP 2013062355W WO 2013175936 A1 WO2013175936 A1 WO 2013175936A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alicyclic
- group
- diepoxy compound
- compound
- formula
- Prior art date
Links
- MGAKFFHPSIQGDC-UHFFFAOYSA-N C1C(CC2OC2C2)C2C2OC12 Chemical compound C1C(CC2OC2C2)C2C2OC12 MGAKFFHPSIQGDC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/30—Tungsten
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D301/00—Preparation of oxiranes
- C07D301/02—Synthesis of the oxirane ring
- C07D301/03—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds
- C07D301/12—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds with hydrogen peroxide or inorganic peroxides or peracids
Definitions
- the present invention relates to a method for producing an epoxy compound from an olefin compound and hydrogen peroxide. More specifically, the present invention relates to a method for producing an alicyclic diepoxy compound from an alicyclic olefin compound in the presence of a solid support and a solid catalyst.
- diepoxy compounds having an alicyclic skeleton in the molecule are currently commercially available. These diepoxy compounds can be cured by reacting with various curing agents and curing catalysts. The cured product of this diepoxy compound can have the heat resistance, transparency, and good dielectric properties that are the characteristics of a resin using a compound having an alicyclic skeleton. It is useful as an intermediate to produce agents, adhesives, inks, sealant components or other compounds useful for a variety of end uses including pharmaceuticals and medical supplies.
- tetrahydroindene is by-produced when vinyl norbornene is synthesized by reaction of cyclopentadiene and 1,3-butadiene. In recent years, there has been a demand for effective use of tetrahydroindene.
- Patent Document 1 discloses a method for producing a diepoxide of tetrahydroindene, which is an epoxy compound having two alicyclic skeletons in a molecule, from tetrahydroindene. Therefore, an epoxy compound having an alicyclic skeleton having no ester group in the molecule is desired.
- an epoxy compound for example, a method of oxidizing an olefin with a peracid such as peracetic acid is known.
- peracid requires caution in handling, and a carboxylic acid in which an epoxy compound is present in the reaction system.
- ester forms are reduced by reacting with the acid, and the selectivity of the epoxy compound is reduced. It reacts easily with the epoxy group produced in step 1, the epoxy group is ring-opened to reduce the selectivity of the epoxy compound, and the post-treatment after the reaction is troublesome. Therefore, a production method that uses hydrogen peroxide, which is easy to handle and harmless water after the reaction, as an oxidizing agent has been attracting attention.
- a catalyst such as polyacids is used, and epoxidation is carried out by reacting olefins with hydrogen peroxide water using a halogenated hydrocarbon as a solvent.
- a method Patent Document 2 is known.
- halogen hydrocarbons are used, there are problems such as product halogen impurities and environmental impact.
- Patent Document 3 discloses a solid-phase oxidation reaction system comprising a solid support, a powder mixture of the solid catalyst for the oxidation reaction, an organic compound, and hydrogen peroxide.
- JP 2004-182648 A Japanese Unexamined Patent Publication No. 62-234550 International Publication No. 2008/093711
- An object of the present invention is to provide a method for producing an alicyclic diepoxy compound in a high yield by epoxidizing an alicyclic olefin compound at a high reaction rate.
- the present invention provides an alicyclic olefin compound represented by the following formula (2) in the presence of an alicyclic olefin compound represented by the following formula (2), a hydrogen peroxide solution, a solid support powder and a solid catalyst powder.
- the present invention relates to a method for producing an alicyclic diepoxy compound represented by the general formula (1), which comprises reacting hydrogen peroxide with hydrogen peroxide.
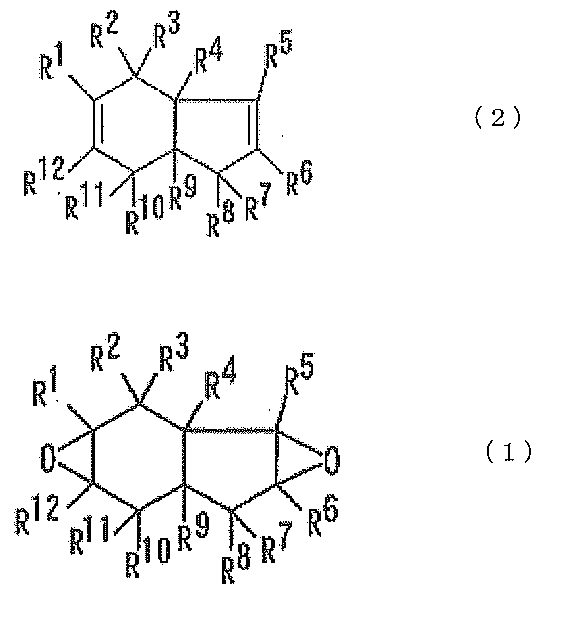
- R 1 to R 12 are each independently a hydrogen atom, a halogen atom, an alkyl group that may contain a halogen atom, or an alkoxy group that may have a substituent.
- the alicyclic olefin compound is a compound represented by the formula (4)
- the alicyclic diepoxy compound is a compound represented by the formula (3).
- the present invention relates to a method for producing a diepoxy compound.
- the present invention also provides an oxygen acid and a salt thereof, wherein the solid catalyst contains an oxide of a metal selected from the group consisting of tungsten, molybdenum and vanadium, a metal selected from the group consisting of tungsten, molybdenum and vanadium, and
- the present invention relates to a method for producing the above alicyclic diepoxy compound, wherein the alicyclic diepoxy compound is selected from the group consisting of oxides, halides and sulfates of elements selected from the group consisting of iron, manganese and ruthenium.
- the present invention is also characterized in that the solid catalyst is selected from the group consisting of oxides of tungsten or molybdenum, isopolyacids containing tungsten or molybdenum, and heteropolyacids containing tungsten or molybdenum. More particularly, the present invention relates to a method for producing an alicyclic diepoxy compound, wherein the solid catalyst is an isopolyacid containing tungsten.
- the present invention also provides the production of the alicyclic diepoxy compound, wherein the solid support is selected from the group consisting of phosphates, diatomaceous earth, silica, alumina, white porcelain clay, silica alumina, and calcium fluoride.
- the present invention relates to a method for producing the alicyclic diepoxy compound, wherein the solid support is apatite.
- an alicyclic diepoxy compound of the formula (1) having a high industrial value can be produced with a high reaction rate and yield, reduction of environmental burden due to waste water and organic solvent, and reuse of a catalyst.
- the solid catalyst and the solid support constituting the solid phase can be reused simply by drying after separation of the product, without requiring a step that complicates the reaction operation for reuse, and isolation of the product. And the recovery operation is easy. .
- the present invention provides an alicyclic olefin compound represented by the following formula (2) in the presence of an alicyclic olefin compound represented by the following formula (2), a hydrogen peroxide solution, a solid support powder and a solid catalyst powder. And a method for producing an alicyclic diepoxy compound represented by the general formula (1), wherein hydrogen peroxide is reacted with hydrogen peroxide.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 and R 12 are each independent.
- alkyl group an alkyl group having 1 to 10 carbon atoms is preferable, and an alkyl group having 1 to 4 carbon atoms is more preferable.
- examples of the substituent include a halogen atom and an alkoxy group.
- alkoxy group an alkoxy group having 1 to 10 carbon atoms is preferable, and an alkoxy group having 1 to 4 carbon atoms is more preferable.
- examples of the substituent include a halogen atom and an alkoxy group.
- R 1 to R 12 are each independently preferably a hydrogen atom, a fluorine atom, an alkyl group or an alkoxy group, more preferably a hydrogen atom or a fluorine atom, and even more preferably a hydrogen atom.
- a compound represented by the following formula (4) is preferably used to produce an alicyclic diepoxy compound represented by the following formula (3).
- the present invention uses a mixture of a powder of a solid support and a powder of a solid catalyst.
- a hydrogen peroxide solution and a compound of formula (2) is added to the powder of this mixture and bringing them into contact with each other, the formula ( The compound of 2) is oxidized to produce an alicyclic diepoxy compound of the formula (1).
- the production method of the present invention does not use peracid unlike the production method disclosed in Patent Document 1 and the like, post-treatment can be simplified and the environmental load can be greatly reduced. .
- no carboxylic acid is present in the system, the formation of ester bodies, alcohol bodies, etc. can be suppressed, and the selectivity for epoxidation is high.
- the diepoxy compound selectivity of the oxidation reaction is high and a complicated treatment step is not required, so that the diepoxy compound can be recovered without loss.
- a solid catalyst As the solid support, a solid catalyst, a hydrogen peroxide solution, a compound of the formula (2) is dispersed and has a property of not deteriorating and inhibiting an oxidation reaction, preferably a property of promoting an oxidation reaction.
- the powder of what it has is used. Specifically, phosphates such as apatite, diatomaceous earth (main component: silica), white ceramics (main component: silica alumina), clays such as hydrotalcite, fluorides such as calcium fluoride, silica, titania, alumina, etc. These oxides can be exemplified.
- a solid support selected from phosphates, diatomaceous earth, silica, alumina, white porcelain clay, silica alumina, and calcium fluoride is preferable, and a higher yield can be achieved.
- solid carriers selected from apatite, diatomaceous earth and calcium fluoride can achieve particularly high yields.
- apatite is a kind of calcium phosphate, and fluorine apatite, chlorapatite, carbonate apatite, hydroxyapatite, etc. are present as apatite-based minerals. Of these, hydroxyapatite and fluorapatite are preferably used.
- Diatomaceous earth is a soft rock or soil mainly made of diatom shell and mainly composed of silica.
- silica alumina, iron oxide, alkali metal oxides and the like are often included.
- a porous material having a high porosity and a cake bulk density of about 0.2 to 0.45 is often used.
- fired products are preferred, and freshwater diatomaceous earths are preferred, but other diatomaceous earths can also be used.
- Specific examples of such diatomaceous earth include those sold by Celite under the trade name Celite (registered trademark) and those sold by Eagle Pitcher Minerals under the trade name Ceratom. .
- what was baked with sodium carbonate etc. can also be used.
- the solid catalyst includes an oxide of a metal selected from the group consisting of tungsten, molybdenum and vanadium, an oxyacid and a salt thereof containing a metal selected from the group consisting of tungsten, molybdenum and vanadium, and iron, manganese and ruthenium.
- a metal selected from the group consisting of tungsten, molybdenum and vanadium
- iron, manganese and ruthenium examples thereof include oxides, halides and sulfates of elements selected from the group consisting of:
- Examples of the oxide of the metal selected from the group consisting of tungsten, molybdenum, and vanadium include WO 3 , MoO 3 , and V 2 O 5 .
- oxygen acids and salts thereof containing a metal selected from the group consisting of tungsten, molybdenum and vanadium include tungstates such as tungstic acid (H 2 WO 4 ) and Na 2 WO 4 , and molybdic acid (H 2 MoO 4).
- Molybdates such as Na 2 MoO 4
- vanadic acid vanadates such as NH 4 VO 3
- the isopolyacids and heteropolyacids containing tungsten, molybdenum, or vanadium are, for example, hybrids represented by Q 3 [PW 6 Mo 6 O 40 ], Q 7 [PV 4 Mo 8 O 40 ], and the like, 3 ⁇ PO 4 [W (O) (O 2 )] 4 ⁇ , Q 2 [W 2 O 3 (O 2 ) 4 ], etc. Represents a cation).
- heteroatoms of the heteropolyacids include phosphorus, boron, silicon, germanium, lanthanoid elements, manganese, nickel, iron, cobalt, and ruthenium.
- the counter cation of the isopolyacid salt or heteropolyacid salt includes tetrabutylammonium, butylammonium, benzyltrimethylammonium, cetyltrimethylammonium, cetylpyridinium organic cations, and ammonium, potassium, sodium, calcium, etc. Inorganic cations can be mentioned.
- isopolytungstic acids containing tungsten (NH 4 ) 6 W 7 O 24 , (NH 4 ) 10 [H 2 W 12 O 42 ], (CetylNMe 3 ) 7 (NH 4 ) 3 [H 2 W 12 O 42 ], (CetylNMe 3 ) 10 [H 2 W 12 O 42 ], (CetylPy) 9 (NH 4 ) [H 2 W 12 O 42 ], (CetylPy) 10 [H 2 W 12 O 42 ], (CetylPy) 4 [W 10 O 32 ], K 4 [W 10 O 32 ] and the like can be mentioned.
- heteropolytungstic acids containing tungsten examples include (CetylPy) 3 [PW 12 O 40 ]. , (CetylPy) 5 H 2 [ PW 11 O 39], there may be mentioned Na 9 [PW 9 O 34] or the like, further Phosphorus (P) in the hetero polytungstic acids, boron (B), silicon (Si), may be exemplified those replaced by germanium (Ge) or the like.
- CetylNMe 3 represents cetyltrimethylammonium
- CetylPy represents cetylpyridinium.
- examples of the oxygen acid containing molybdenum and salts thereof include compounds in which tungsten in the compounds exemplified as the oxygen acid containing tungsten and salts thereof is substituted with molybdenum.
- examples of the oxygen acid containing vanadium and salts thereof include compounds in which tungsten in the compounds exemplified as the oxygen acid containing tungsten and salts thereof is substituted with vanadium.
- a catalyst selected from the group consisting of oxides of tungsten or molybdenum, isopolyacids containing tungsten or molybdenum, and heteropolyacids containing tungsten or molybdenum is preferable.
- a catalyst selected from the group consisting of acids or heteropolyacids is preferred because high selectivity is obtained.
- oxides, halides, and sulfates of elements selected from the group consisting of iron, manganese, and ruthenium include FeCl 3 , MnSO 4 , RuCl 3 and the like.
- the solid catalyst does not need to be immobilized on the solid support, and it is only necessary to mix the solid catalyst powder and the solid support powder.
- a mixture of the solid catalyst and the solid support can be obtained by adding a solid catalyst powder to the solid support powder in advance and stirring and mixing the powders.
- the particle size of the solid catalyst powder and the solid support powder is not particularly limited, but an easily available powder having a particle size of about 5 to 100 ⁇ m can be used to obtain the effects of the present invention such as a high yield of the product. Can do.
- the amount of the solid catalyst is preferably 5 to 60% by mass of the solid support, more preferably 10 to 50% by mass. If the amount is less than 5% by mass, the reaction rate decreases, so that the compound of formula (1) cannot be obtained in good yield. Even if it exceeds 60 mass%, the yield is not improved, which is industrially disadvantageous.
- the compound of formula (2) to be oxidized and aqueous hydrogen peroxide are added to the mixture of the solid support powder and the solid catalyst powder obtained as described above.
- This addition is performed so that both are dispersed in the mixture and contact each other.
- mixing and stirring may be performed after the addition so as to improve the dispersion and contact between the two.
- the reaction may be carried out in a state where the mixture is allowed to stand, and mixing or stirring may be carried out.
- the amount of hydrogen peroxide water added can be used in the range of about 1 to 10 mmol as hydrogen peroxide per 1 mmol of the double bond site of the compound of formula (2), but is preferably 1.2 to 5 mmol. If it is less than 1 mmol, hydrogen peroxide is insufficient, and if it exceeds 10 mmol, the epoxy is ring-opened and the yield of the epoxy compound is lowered.
- the solid support and the solid catalyst can be used in the range of about 0.01 to 5 g with respect to 1 mmol of the compound of the formula (2), but 0.05 to 3.0 g is desirable.
- the concentration of the hydrogen peroxide solution used in the present invention is preferably 5 to 60% by mass, and more preferably 5 to 35% by mass.
- the generated epoxide is hydrolyzed to produce a by-product such as a diol and the selectivity of the target product is low.
- the selectivity is high and a high yield of the target product can be obtained.
- the hydrogen peroxide solution with a concentration of 35 to 60% by mass has a higher risk of handling the more restricted the transportation is, and the reaction system in a two-phase heterogeneous system is sufficient to avoid sudden heat generation and explosion.
- a reaction facility was required, in the present invention, the reaction can be performed more safely and a practical yield of the product can be obtained by the method of soaking hydrogen peroxide into the solid phase. .
- an organic solvent may be further added to the mixed powder of the solid support and the solid catalyst before or after the addition of the compound of the formula (2) and the hydrogen peroxide solution.
- an organic solvent By using an organic solvent, it is possible to suppress contact between the epoxy and water, thereby making it easier to prevent ring opening of the produced epoxy.
- the amount of the organic solvent to be added is in the range of 0 to 200% by mass with respect to the compound of the formula (2). If it exceeds 200% by mass, the reaction rate becomes slow, and the yield of the diepoxy compound of formula (1) decreases.
- organic solvents aliphatic hydrocarbons, aromatic hydrocarbons, alcohols, ethers, esters, ketones, nitriles, amides, and the like can be used.
- Preferred organic solvents are ethanol, ethyl acetate, hexane, toluene and the like, and particularly preferred is toluene.
- the reaction temperature of the oxidation reaction is preferably 0 to 50 ° C., more preferably 5 to 40 ° C. If it is less than 0 ° C., the reaction is slow, and if it exceeds 50 ° C., the yield decreases due to deactivation of the solid catalyst or ring opening of the epoxy.
- the reaction time is preferably 1 to 24 hours, more preferably 1 to 12 hours. If the reaction time is less than 1 hour, the progress of the reaction is not sufficient, so that the yield decreases. If the reaction time exceeds 24 hours, the productivity decreases.
- the conversion rate of the compound of formula (2) is preferably 80% or more, and the yield of the compound of formula (1) is preferably 50% or more.
- the method for isolating the generated diepoxy compound of formula (1) is not particularly limited, and examples thereof include a method of concentrating after solvent extraction.
- the chlorine content of the diepoxy compound of the formula (1) according to the present invention is preferably 100 mass ppm or less, and more preferably 10 mass ppm or less, because the moisture resistance reliability when a cured resin product is further improved. Is more preferable.
- the chlorine content is a value measured according to JIS standard K-7243-3. Specifically, a sample (epoxy compound) is dissolved in diethylene glycol monobutyl ether and heated to reflux with a potassium hydroxide alcohol solution. It is a value measured by subsaponification and performing potentiometric titration of a silver nitrate solution.
- the chlorine content of the epoxy compound can be reduced by distillation purification, and can also be reduced by methods such as alkaline aqueous solution washing and adsorbent treatment.
- the metal content of the diepoxy compound of the formula (1) of the present invention is preferably 100 ppm or less, more preferably 10 ppm or less, since the mechanical properties and electrical properties when a cured resin product is further improved. preferable.
- the metal content can be measured by inductively coupled plasma emission (ICP emission) analysis of a 10% toluene solution of the sample (epoxy compound).
- ICP emission inductively coupled plasma emission
- Optima4300DV manufactured by PerkinElmer, Inc. can be used as the measuring device.
- quantitative analysis can be performed using a calibration curve created using a commercially available metal standard solution.
- the metal content of the epoxy compound can also be reduced by distillation purification, and can also be reduced by methods such as alkaline aqueous solution washing and adsorbent treatment.
- Example 1 In a screw test tube, 1.0 g of apatite as a solid support and 0.085 g (0.015 mmol) of (CetylPy) 9 (NH 4 ) [H 2 W 12 O 42 ] as a solid catalyst were weighed and mixed well. . To these mixtures, 0.12 g (1.0 mmol) of tetrahydroindene and 0.23 g (2.4 mmol) of 35% aqueous hydrogen peroxide were added and stirred well, and then allowed to stand at 25 ° C. After standing at 25 ° C.
- Example 2 In a screw test tube, 0.50 g of apatite as a solid support and 0.085 g (0.015 mmol) of (CetylPy) 9 (NH 4 ) [H 2 W 12 O 42 ] as a solid catalyst were weighed and mixed well. . To these mixtures, 0.12 g (1.0 mmol) of tetrahydroindene and 0.23 g (2.4 mmol) of 35% aqueous hydrogen peroxide were added and stirred well, and then allowed to stand at 25 ° C. After standing at 25 ° C.
- Example 3 In a screw test tube, 2.0 g of apatite as a solid support and 0.056 g (0.010 mmol) of (CetylPy) 9 (NH 4 ) [H 2 W 12 O 42 ] as a solid catalyst were weighed and mixed well. . To these mixtures, 0.12 g (1.0 mmol) of tetrahydroindene and 0.23 g (2.4 mmol) of 35% aqueous hydrogen peroxide were added and stirred well, and then allowed to stand at 25 ° C. After standing at 25 ° C.
- reaction mixture was extracted with hexane (5 mL ⁇ 3 times), and the solvent was distilled off from the extract to obtain 0.12 g of tetrahydroindene diepoxide as a colorless transparent liquid.
- the yield was 81%.
- Example 4 In a screw test tube, 0.50 g of apatite as a solid support and 0.085 g (0.015 mmol) of (CetylPy) 9 (NH 4 ) [H 2 W 12 O 42 ] as a solid catalyst were weighed and mixed well. . To these mixtures, 0.12 g (1.0 mmol) of tetrahydroindene and 0.23 g (2.4 mmol) of 35% aqueous hydrogen peroxide were added and stirred well, and then allowed to stand at 15 ° C. After standing at 15 ° C.
- Example 5 In a screw test tube, 2.0 g of apatite as a solid support and 0.074 g (0.015 mmol) of (CetylPy) 10 [H 2 W 12 O 42 ] as a solid catalyst were weighed and mixed well. To these mixtures, 0.12 g (1.0 mmol) of tetrahydroindene and 0.23 g (2.4 mmol) of 35% aqueous hydrogen peroxide were added and stirred well, and then allowed to stand at 15 ° C. After standing at 15 ° C.
- Example 6 In a screw test tube, 0.50 g of apatite as a solid support and 0.074 g (0.015 mmol) of (CetylNMe 3 ) 7 (NH 4 ) 3 [H 2 W 12 O 42 ] as a solid catalyst were weighed well. Mixed. To these mixtures, 0.12 g (1.0 mmol) of tetrahydroindene and 0.23 g (2.4 mmol) of 35% aqueous hydrogen peroxide were added and stirred well, and then allowed to stand at 15 ° C. After standing at 15 ° C.
- the oil phase was separated and the aqueous phase was extracted with 2 ⁇ 2 ml of chloroform.
- the extract and the oil phase were mixed and washed with 10% aqueous sodium thiosulfate solution 2 ⁇ 8 ml, 5% aqueous sodium carbonate solution 2 ⁇ 8 ml, and water 2 ⁇ 8 ml.
- the solvent was distilled off from the washed mixture of the extract and the oil phase to obtain 0.24 g of tetrahydroindene diepoxide as a colorless transparent liquid. The yield was 7%.
- a highly industrial alicyclic diepoxy compound of formula (1) can be produced with a high reaction rate and yield.
Abstract
脂環式オレフィン化合物のエポキシ化を高い反応率で行い、高収率で脂環式ジエポキシ化合物を製造する方法として、下記式(2)で表わされる脂環式オレフィン化合物、過酸化水素水、固体担体の粉末及び固体触媒の粉末の共存下に、下記式(2)で表わされる脂環式オレフィン化合物と過酸化水素を反応させることを特徴とする一般式(1)で表わされる脂環式ジエポキシ化合物の製造方法を提供する。(式中でR1~R12は、それぞれ独立に、水素原子、ハロゲン原子、ハロゲン原子を含んでもよいアルキル基、又は置換基を有してもよいアルコキシ基である。)
Description
本発明は、オレフィン化合物と過酸化水素からエポキシ化合物を製造する方法に関する。より詳細には固体担体と固体触媒の存在下で、脂環式オレフィン化合物から脂環式ジエポキシ化合物を製造する方法に関するものである。
分子内に脂環骨格を持つジエポキシ化合物は、現在様々な種類のものが市販されている。これらジエポキシ化合物は種々の硬化剤および硬化触媒と反応させることにより硬化物が得られる。このジエポキシ化合物の硬化物は、脂環骨格を持つ化合物を用いた樹脂の特徴である耐熱性、透明性、良好な誘電特性を持たせることができ、これらジエポキシ化合物を用いた用途としては、コーティング剤、接着剤、インキ、シーラントの成分または医薬品および医療用品を含む種々の最終用途に有用な他の化合物を製造するための中間体として有用である。
ところで、従来、シクロペンタジエンと1,3-ブタジエンとの反応によってビニルノルボルネンを合成する際に、テトラヒドロインデンが副生することが知られている。そして近年、このテトラヒドロインデンの有効な利用法が求められている。
ところで、従来、シクロペンタジエンと1,3-ブタジエンとの反応によってビニルノルボルネンを合成する際に、テトラヒドロインデンが副生することが知られている。そして近年、このテトラヒドロインデンの有効な利用法が求められている。
例えば、特許文献1には、テトラヒドロインデンから、分子内に2個の脂環骨格を有するエポキシ化合物であるテトラヒドロインデンのジエポキシドを製造する方法が開示されている。そこで、分子内にエステル基を持たない脂環骨格を持つエポキシ化合物が望まれている。
エポキシ化合物の製造方法としては、例えばオレフィン類を過酢酸等の過酸で酸化する方法が知られているが、過酸は取扱いに注意を要し、エポキシ体が反応系内に存在するカルボン酸と反応することによりエステル体等が生成してエポキシ体の選択率が低下する、酸との反応性が高いとされる脂環式エポキシ化合物の製造においては、共存する有機酸が水の存在下で生成したエポキシ基と容易に反応し、エポキシ基が開環してエポキシ体の選択率が低下する、反応後の後処理が面倒である等の問題がある。そこで、取扱いが容易で、反応後には無害な水となる過酸化水素を酸化剤として用いる製造法が注目されてきている。
過酸化水素を用いてオレフィン類からエポキシ化合物を製造する方法としては、例えば、ポリ酸類等の触媒を用い、オレフィン類と過酸化水素水とをハロゲン化炭化水素を溶媒として反応させエポキシ化を行う方法(特許文献2)が知られている。しかしながら、ハロゲン炭化水素を用いているため、製品のハロゲン不純物、環境負荷等の問題がある。
特許文献3は固体担体及び前記酸化反応の固体触媒の粉末の混合物、有機化合物及び過酸化水素水からなる固相系酸化反応システムを開示する。
本発明は脂環式オレフィン化合物のエポキシ化を高い反応率で行い、高収率で脂環式ジエポキシ化合物を製造する方法を提供することを目的とする。
本発明は、下記式(2)で表わされる脂環式オレフィン化合物、過酸化水素水、固体担体の粉末及び固体触媒の粉末の共存下に、下記式(2)で表わされる脂環式オレフィン化合物と過酸化水素を反応させることを特徴とする一般式(1)で表わされる脂環式ジエポキシ化合物の製造方法に関する。
本発明はまた、脂環式オレフィン化合物が式(4)で表される化合物であり、脂環式ジエポキシ化合物が式(3)で表される化合物であることを特徴とする前記の脂環式ジエポキシ化合物の製造方法に関する。

本発明はまた、前記固体触媒が、タングステン、モリブデン及びバナジウムからなる群より選択された金属の酸化物、タングステン、モリブデン及びバナジウムからなる群より選択された金属を含有する酸素酸及びその塩類、並びに鉄、マンガン及びルテニウムからなる群より選択された元素の酸化物、ハロゲン化物及び硫酸塩からなる群より選択されることを特徴とする前記の脂環式ジエポキシ化合物の製造方法に関する。
本発明はまた、前記固体触媒が、タングステン又はモリブデンの酸化物、タングステン又はモリブデンを含有するイソポリ酸類、及びタングステン又はモリブデンを含有するヘテロポリ酸類からなる群より選択されることを特徴とする前記の脂環式ジエポキシ化合物の製造方法に関し、特に、前記固体触媒が、タングステンを含有するイソポリ酸類であることを特徴とする前記の脂環式ジエポキシ化合物の製造方法に関する。
本発明はまた、前記固体担体が、リン酸塩類、珪藻土、シリカ、アルミナ、白陶土、シリカアルミナ及びフッ化カルシウムからなる群から選択されることを特徴とする前記の脂環式ジエポキシ化合物の製造方法に関し、特に、前記固体担体が、アパタイトであることを特徴とする前記の脂環式ジエポキシ化合物の製造方法に関する。
本発明によれば、工業的価値の高い式(1)の脂環式ジエポキシ化合物を高い反応率および収率で製造することができ、また排水や有機溶剤による環境負荷の軽減、触媒の再使用が可能である等の利点を有する。さらに、固相を構成する固体触媒と固体担体は、生成物の分離後、乾燥処理するだけで再使用でき、再使用に際して反応操作を複雑にする工程を必要とせず、かつ製造物の単離や回収操作が容易であるとの特徴も有する。
本発明の好適な実施形態について以下に説明する。
本発明は、下記式(2)で表わされる脂環式オレフィン化合物、過酸化水素水、固体担体の粉末及び固体触媒の粉末の共存下に、下記式(2)で表わされる脂環式オレフィン化合物と過酸化水素を反応させることを特徴とする一般式(1)で表わされる脂環式ジエポキシ化合物の製造方法である。

式(1)及び式(2)において、R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11及びR12は、それぞれ独立に、水素原子、ハロゲン原子、置換基を有していてもよいアルキル基、又は置換基を有していてもよいアルコキシ基を示す。
アルキル基としては、炭素数1~10のアルキル基が好ましく、炭素数1~4のアルキル基がより好ましい。アルキル基が置換基を有するとき、当該置換基としては、ハロゲン原子、アルコキシ基等が挙げられる。
アルコキシ基としては、炭素数1~10のアルコキシ基が好ましく、炭素数1~4のアルコキシ基がより好ましい。アルコキシ基が置換基を有するとき、当該置換基としては、ハロゲン原子、アルコキシ基等が挙げられる。
アルコキシ基としては、炭素数1~10のアルコキシ基が好ましく、炭素数1~4のアルコキシ基がより好ましい。アルコキシ基が置換基を有するとき、当該置換基としては、ハロゲン原子、アルコキシ基等が挙げられる。
R1~R12は、それぞれ独立に、水素原子、フッ素原子、アルキル基又はアルコキシ基であることが好ましく、水素原子又はフッ素原子であることがより好ましく、水素原子であることがさらに好ましい。
すなわち、脂環式オレフィン化合物としては、下記式(4)で表される化合物を用い、下記式(3)で表される脂環式ジエポキシ化合物を製造することが好ましい。

本発明は、固体担体の粉末及び固体触媒の粉末の混合物を用い、この混合物の粉末の中に、過酸化水素水と式(2)の化合物を添加して両者を接触させることにより、式(2)の化合物を酸化させ、式(1)の脂環式ジエポキシ化合物を製造することを特徴とする。本発明の製造方法は、前記特許文献1等に開示される製造法のように過酸を用いることがないので後処理を簡素することができ、環境負荷を大幅に低減することが可能である。また、系内にカルボン酸が存在しないのでエステル体、アルコール体等の生成を抑えることができ、エポキシ化の選択率が高い。さらに、本発明の製造方法によれば、酸化反応のジエポキシ化合物選択率が高く、かつ、複雑な処理工程を必要としないため、ジエポキシ化合物をロスなく回収することが可能である。
固体担体としては、固体触媒、過酸化水素水、式(2)の化合物を分散し、またこれらにより劣化せず、かつ酸化反応を阻害しない性質を有するもの、好ましくは酸化反応を促進する性質を有するものの粉末が用いられる。具体的には、アパタイトなどリン酸塩類、珪藻土〔主成分:シリカ〕、白陶土〔主成分:シリカアルミナ〕、ハイドロタルサイトなどクレイ類、フッ化カルシウムなどフッ化物類、シリカ、チタニア、アルミナなどの酸化物類を例示することができる。中でも、リン酸塩類、珪藻土、シリカ、アルミナ、白陶土、シリカアルミナ及びフッ化カルシウムから選択される固体担体が好ましく、より高い収率を達成できる。特に、アパタイト、珪藻土及びフッ化カルシウムから選択される固体担体は特に高い収率を達成できる。
ここで、アパタイトとは、リン酸カルシウムの一種であり、フッ素アパタイト、塩素アパタイト、炭酸アパタイト、水酸アパタイト等がリン灰石系鉱物として存在する。これらの中でも水酸アパタイト、フッ素アパタイトが好適に用いられる。
珪藻土とは、主に珪藻の殻からなる軟質の岩石又は土壌で、シリカを主成分とするが、シリカ以外にもアルミナ、酸化鉄、アルカリ金属の酸化物等が含まれていることが多い。又、ポーラスで高い空隙率を有し、ケーク嵩密度が0.2~0.45程度のものが用いられることが多い。珪藻土の中でも、焼成品が好ましく、又淡水産珪藻土が好ましいが、他の珪藻土を使用することも可能である。このような珪藻土の具体例としては、セライト社からセライト(登録商標)の商品名で販売されているものやイーグルピッチャーミネラルズ社よりセラトムの商品名で販売されているものを例示することができる。又、炭酸ナトリウム等とともに焼成したものも用いることができる。
固体触媒としては、タングステン、モリブデン及びバナジウムからなる群より選択された金属の酸化物、タングステン、モリブデン及びバナジウムからなる群より選択された金属を含有する酸素酸及びその塩類、並びに鉄、マンガン及びルテニウムからなる群より選択された元素の酸化物、ハロゲン化物及び硫酸塩を例示することができる。
タングステン、モリブデン及びバナジウムからなる群より選択された金属の酸化物としては、WO3、MoO3、V2O5を挙げることができる。タングステン、モリブデン及びバナジウムからなる群より選択された金属を含有する酸素酸及びその塩類としては、タングステン酸(H2WO4)、Na2WO4等のタングステン酸塩、モリブデン酸(H2MoO4)、Na2MoO4等のモリブデン酸塩、バナジン酸、NH4VO3等のバナジン酸塩、タングステン、モリブデン又はバナジウムを含有するイソポリ酸類及びその塩類、タングステン、モリブデン又はバナジウムを含有するヘテロポリ酸類およびその塩類を例示することができる。なお、前記タングステン、モリブデン又はバナジウムを含有するイソポリ酸類、ヘテロポリ酸類とは、Q3[PW6Mo6O40]、Q7[PV4Mo8O40]等として表される混成物や、Q3{PO4[W(O)(O2)]4}、Q2[W2O3(O2)4]等として表されるパーオキソ型化合物も含む意味である(式中、Qは対カチオンを表す。)。
前記ヘテロポリ酸類のヘテロ原子としては、リン、ホウ素、珪素、ゲルマニウム、ランタノイド系元素、マンガン、ニッケル、鉄、コバルト又はルテニウム等を挙げることができる。又、前記イソポリ酸の塩類又はヘテロポリ酸類の塩類の対カチオンとしては、テトラブチルアンモニウム、ブチルアンモニウム、ベンジルトリメチルアンモニウム、セチルトリメチルアンモニウム、セチルピリジニウムの有機カチオン類、及びアンモニウム、カリウム、ナトリウム、カルシウム等の無機カチオン類を挙げることができる。
より具体的には、タングステンを含有するイソポリタングステン酸類としては、(NH4)6W7O24、(NH4)10[H2W12O42]、(CetylNMe3)7(NH4)3[H2W12O42]、(CetylNMe3)10[H2W12O42]、(CetylPy)9(NH4)[H2W12O42]、(CetylPy)10[H2W12O42]、(CetylPy)4[W10O32]、K4[W10O32]等を挙げることができ、タングステンを含有するヘテロポリタングステン酸類としては、(CetylPy)3[PW12O40]、(CetylPy)5H2[PW11O39]、Na9[PW9O34]等を挙げることができ、さらに前記のヘテロポリタングステン酸類中のリン(P)を、ホウ素(B)、ケイ素(Si)、ゲルマニウム(Ge)等で置き換えたものを例示することができる。なお、式中のCetylNMe3はセチルトリメチルアンモニウムを、CetylPyはセチルピリジニウムを表す。
又、モリブデンを含有する酸素酸及びその塩類としては、前記のタングステンを含有する酸素酸及びその塩類として例示した化合物中のタングステンをモリブデンで置換した化合物を例示することができる。バナジウムを含有する酸素酸及びその塩類としては、前記のタングステンを含有する酸素酸及びその塩類として例示した化合物中のタングステンをバナジウムで置換した化合物を例示することができる。
前記の固体触媒の中でも、タングステン又はモリブデンの酸化物、タングステン又はモリブデンを含有するイソポリ酸類、及びタングステン又はモリブデンを含有するヘテロポリ酸類からなる群より選択される触媒が好ましく、特に、タングステンを含有するイソポリ酸類又はヘテロポリ酸類からなる群より選択される触媒が、高い選択率が得られるので好ましい。
鉄、マンガン及びルテニウムからなる群より選択された元素の酸化物、ハロゲン化物や硫酸塩としては、FeCl3、MnSO4、RuCl3等を挙げることができる。
固体触媒は固体担体に固定化する必要はなく、単に固体触媒の粉末と固体担体の粉末を混合するだけでよい。例えば、予め固体担体の粉末に固体触媒の粉末を添加し、粉末同士を攪拌混合する方法により、固体触媒と固体担体との混合物を得ることができる。固体触媒の粉末及び固体担体の粉末の粒度は特に限定されないが、入手の容易な粒径5~100μm程度の粉末を用いることができ、生成物の高い収率等、本発明の効果を得ることができる。
固体触媒の量は、固体担体の5~60質量%であることが好ましく、より好ましくは10~50質量%である。5質量%未満では反応速度が低下するため式(1)の化合物を収率よく得られない。60質量%を超えて用いても収率は向上せず、工業的に不利となる。
固体触媒の量は、固体担体の5~60質量%であることが好ましく、より好ましくは10~50質量%である。5質量%未満では反応速度が低下するため式(1)の化合物を収率よく得られない。60質量%を超えて用いても収率は向上せず、工業的に不利となる。
次に、前記のようにして得られた固体担体の粉末と固体触媒の粉末の混合物に、酸化対象の式(2)の化合物及び過酸化水素水を添加する。この添加は、両者が前記混合物中に分散し互いに接触するように行われるが、例えば、両者の分散及び互いの接触を良好にするように、添加後混合撹拌を行ってもよい。その後は、この混合物を静置した状態で反応を行ってもよく、混合や撹拌を行ってもよい。
過酸化水素水の添加量は、式(2)の化合物の二重結合部位1mmolに対して、過酸化水素として1~10mmol程度の範囲で用いることができるが、1.2~5mmolが望ましい。1mmol未満では過酸化水素が不足し、10mmolを超えるとエポキシが開環してエポキシ化合物の収率が下がる。
固体担体及び固体触媒は、式(2)の化合物1mmolに対して、0.01~5g程度の範囲で用いることができるが、0.05~3.0gが望ましい。
固体担体及び固体触媒は、式(2)の化合物1mmolに対して、0.01~5g程度の範囲で用いることができるが、0.05~3.0gが望ましい。
本発明に用いる過酸化水素水の濃度は、好ましくは5~60質量%であり、さらに好ましくは5~35質量%である。過酸化水素を用いるエポキシ化合物の製造法では、一般に低濃度の過酸化水素水を用いた場合は、生成したエポキシドが加水分解されてジオールなど副生成物が生成し目的生成物の選択率が低くなるが、本発明の製造方法では、低濃度の過酸化水素水を用いた場合でも選択率が高く目的生成物の高い収率が得られる。
また、同じく35~60質量%濃度の過酸化水素水は、輸送規制があるほど取り扱いの危険性が高く、二相不均一系での反応システムでは急激な発熱や爆発を回避するために十分な反応設備を必要としたが、本発明では、過酸化水素水を固相に染み込ませる手法により、反応をより安全に行うこともでき、かつ生成物の実用的な収率も得られることができる。
また、同じく35~60質量%濃度の過酸化水素水は、輸送規制があるほど取り扱いの危険性が高く、二相不均一系での反応システムでは急激な発熱や爆発を回避するために十分な反応設備を必要としたが、本発明では、過酸化水素水を固相に染み込ませる手法により、反応をより安全に行うこともでき、かつ生成物の実用的な収率も得られることができる。
本発明においては、前記の固体担体と固体触媒の混合粉末に、式(2)の化合物及び過酸化水素水を添加する前後、あるいは同時に、さらに有機溶剤を添加してもよい。有機溶剤を使用することでエポキシと水の接触を抑えることができ、これにより生成したエポキシの開環を防止しやすくなる。添加する有機溶剤の量は、式(2)の化合物に対して0~200質量%の範囲である。200質量%を超えると、反応速度が遅くなり、式(1)のジエポキシ化合物の収率が低下する。
有機溶剤の種類としては、脂肪族炭化水素、芳香族炭化水素、アルコール類、エーテル類、エステル類、ケトン類、ニトリル類、アミド類等を用いることができる。好ましい有機溶剤はエタノール、酢酸エチル、ヘキサン、トルエン等であり、特にトルエンが好ましい。
本発明において、酸化反応の反応温度は0~50℃であることが好ましく、5~40℃がより好ましい。0℃未満では反応が遅く、50℃を超えると固体触媒の失活やエポキシの開環により収率が低下する。
また、反応時間は1~24時間であることが好ましく、1~12時間がより好ましい。1時間未満では反応の進行が十分でないため収率が低下し、24時間を超えると生産性が低下する。
また、反応時間は1~24時間であることが好ましく、1~12時間がより好ましい。1時間未満では反応の進行が十分でないため収率が低下し、24時間を超えると生産性が低下する。
本発明において、式(2)の化合物の転化率は好ましくは80%以上であり、式(1)の化合物の収率は好ましくは50%以上である。
生成した式(1)のジエポキシ化合物の単離の方法については特に限定されず、例えば、溶剤抽出の後に濃縮する方法が挙げられる。
本発明の式(1)のジエポキシ化合物の塩素含量は、樹脂硬化物を作製した場合の耐湿信頼性が一層向上することから、100質量ppm以下であることが好ましく、10質量ppm以下であることがより好ましい。なお、塩素含量は、JIS規格K-7243-3に準拠して測定される値であり、具体的には、ジエチレングリコールモノブチルエーテルに試料(エポキシ化合物)を溶解させ、水酸化カリウムアルコール溶液で加熱還流下けん化し、硝酸銀溶液の電位差滴定を行うことで測定される値である。
エポキシ化合物の塩素含量は、蒸留精製で低減することもでき、アルカリ水溶液洗浄、吸着剤処理等の方法で低減することもできる。
エポキシ化合物の塩素含量は、蒸留精製で低減することもでき、アルカリ水溶液洗浄、吸着剤処理等の方法で低減することもできる。
本発明の式(1)のジエポキシ化合物の金属含量は、樹脂硬化物を作製した場合の機械特性や電気特性が一層向上することから、100ppm以下であることが好ましく、10ppm以下であることがより好ましい。なお、金属含量は、試料(エポキシ化合物)の10%トルエン溶液を、誘導結合プラズマ発光(ICP発光)分析することにより測定することができる。測定装置は、例えばパーキンエルマー社のOptima4300DVなどが使用できる。この測定では、定性分析で検出された金属種において、それぞれ市販の金属標準液を使用して作成した検量線を用い、定量分析を行うことができる。
エポキシ化合物の金属含量は、蒸留精製で低減することもでき、アルカリ水溶液洗浄、吸着剤処理等の方法で低減することもできる。
エポキシ化合物の金属含量は、蒸留精製で低減することもでき、アルカリ水溶液洗浄、吸着剤処理等の方法で低減することもできる。
以下、実施例により本発明をより具体的に説明するが、本発明は実施例に限定されるものではない。
(実施例1)
ねじ口試験管に、固体担体であるアパタイト1.0g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.085g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、25℃で静置した。25℃で2時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.14gを得た。収率は93%であった。
ねじ口試験管に、固体担体であるアパタイト1.0g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.085g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、25℃で静置した。25℃で2時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.14gを得た。収率は93%であった。
(実施例2)
ねじ口試験管に、固体担体であるアパタイト0.50g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.085g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、25℃で静置した。25℃で2時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.14gを得た。収率は89%であった。
ねじ口試験管に、固体担体であるアパタイト0.50g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.085g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、25℃で静置した。25℃で2時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.14gを得た。収率は89%であった。
(実施例3)
ねじ口試験管に、固体担体であるアパタイト2.0g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.056g(0.010mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、25℃で静置した。25℃で6時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.12gを得た。収率は81%であった。
ねじ口試験管に、固体担体であるアパタイト2.0g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.056g(0.010mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、25℃で静置した。25℃で6時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.12gを得た。収率は81%であった。
(実施例4)
ねじ口試験管に、固体担体であるアパタイト0.50g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.085g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、15℃で静置した。15℃で3時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.15gを得た。収率は98%であった。
ねじ口試験管に、固体担体であるアパタイト0.50g及び固体触媒である(Cety1Py)9(NH4)[H2W12O42]0.085g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、15℃で静置した。15℃で3時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.15gを得た。収率は98%であった。
(実施例5)
ねじ口試験管に、固体担体であるアパタイト2.0g及び固体触媒である(Cety1Py)10[H2W12O42]0.074g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、15℃で静置した。15℃で3時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.15gを得た。収率は94%であった。
ねじ口試験管に、固体担体であるアパタイト2.0g及び固体触媒である(Cety1Py)10[H2W12O42]0.074g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、15℃で静置した。15℃で3時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.15gを得た。収率は94%であった。
(実施例6)
ねじ口試験管に、固体担体であるアパタイト0.50g及び固体触媒である(CetylNMe3)7(NH4)3[H2W12O42]0.074g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、15℃で静置した。15℃で3時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.14gを得た。生成物の収率(ジエポキシド収率)は95%であった。
ねじ口試験管に、固体担体であるアパタイト0.50g及び固体触媒である(CetylNMe3)7(NH4)3[H2W12O42]0.074g(0.015mmol)を秤取り、よく混合した。これらの混合物に、テトラヒドロインデン0.12g(1.0mmol)、35%過酸化水素水0.23g(2.4mmol)を加えてよく攪拌した後、15℃で静置した。15℃で3時間静置後、反応混合物をヘキサン(5mL×3回)で抽出し、抽出液から溶媒を留去したところ、無色透明の液体としてテトラヒドロインデンジエポキシド0.14gを得た。生成物の収率(ジエポキシド収率)は95%であった。
(比較例1)
固体触媒であるH3PW12O40・nH2O 0.17g(0.05mmol)に30%過酸化水素水5.74g(50mmol)を加え、60℃で30分間攪拌して溶解させた。
冷却管を備えたなす型フラスコにテトラヒドロインデン2.8g(23mmol)、セチルピリジニウムクロライド一水和物0.051g(0.15mmol)、クロロホルム20mlを加え、溶解させた。これらの混合物に前記固体触媒の過酸化水素水溶液を全量加え、25℃で40分間攪拌した。油相を分離し、水相をクロロホルム2×2mlを用いて抽出した。抽出液と油相とを混合し、10%チオ硫酸ナトリウム水溶液2×8ml、5%炭酸ナトリウム水溶液2×8ml、水2×8mlで洗浄した。洗浄された抽出液と油相の混合物から溶媒を留去し、無色透明の液体としてテトラヒドロインデンジエポキシド0.24gを得た。収率は7%であった。
固体触媒であるH3PW12O40・nH2O 0.17g(0.05mmol)に30%過酸化水素水5.74g(50mmol)を加え、60℃で30分間攪拌して溶解させた。
冷却管を備えたなす型フラスコにテトラヒドロインデン2.8g(23mmol)、セチルピリジニウムクロライド一水和物0.051g(0.15mmol)、クロロホルム20mlを加え、溶解させた。これらの混合物に前記固体触媒の過酸化水素水溶液を全量加え、25℃で40分間攪拌した。油相を分離し、水相をクロロホルム2×2mlを用いて抽出した。抽出液と油相とを混合し、10%チオ硫酸ナトリウム水溶液2×8ml、5%炭酸ナトリウム水溶液2×8ml、水2×8mlで洗浄した。洗浄された抽出液と油相の混合物から溶媒を留去し、無色透明の液体としてテトラヒドロインデンジエポキシド0.24gを得た。収率は7%であった。
本発明により、工業的価値の高い式(1)の脂環式ジエポキシ化合物を高い反応率および収率で製造することができる。
Claims (7)
- 下記式(2)で表わされる脂環式オレフィン化合物、過酸化水素水、固体担体の粉末及び固体触媒の粉末の共存下に、下記式(2)で表わされる脂環式オレフィン化合物と過酸化水素を反応させることを特徴とする一般式(1)で表わされる脂環式ジエポキシ化合物の製造方法。

- 前記脂環式オレフィン化合物が式(4)で表される化合物であり、前記脂環式ジエポキシ化合物が式(3)で表される化合物であることを特徴とする請求項1に記載の脂環式ジエポキシ化合物の製造方法。

- 前記固体触媒が、タングステン、モリブデン及びバナジウムからなる群より選択された金属の酸化物、タングステン、モリブデン及びバナジウムからなる群より選択された金属を含有する酸素酸及びその塩類、並びに鉄、マンガン及びルテニウムからなる群より選択された元素の酸化物、ハロゲン化物及び硫酸塩からなる群より選択されることを特徴とする請求項1または2に記載の脂環式ジエポキシ化合物の製造方法。
- 前記固体触媒が、タングステン又はモリブデンの酸化物、タングステン又はモリブデンを含有するイソポリ酸類、及びタングステン又はモリブデンを含有するヘテロポリ酸類からなる群より選択されることを特徴とする請求項1~3のいずれかに記載の脂環式ジエポキシ化合物の製造方法。
- 前記固体触媒が、タングステンを含有するイソポリ酸類であることを特徴とする請求項1~4のいずれかに記載の脂環式ジエポキシ化合物の製造方法。
- 前記固体担体が、リン酸塩類、珪藻土、シリカ、アルミナ、白陶土、シリカアルミナ及びフッ化カルシウムからなる群から選択されることを特徴とする請求項1~5のいずれかに記載の脂環式ジエポキシ化合物の製造方法。
- 前記固体担体が、アパタイトであることを特徴とする請求項1~6のいずれかに記載の脂環式ジエポキシ化合物の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/402,376 US9212188B2 (en) | 2012-05-22 | 2013-04-26 | Method for producing alicyclic diepoxy compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-116389 | 2012-05-22 | ||
| JP2012116389A JP2013241374A (ja) | 2012-05-22 | 2012-05-22 | 脂環式ジエポキシ化合物の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013175936A1 true WO2013175936A1 (ja) | 2013-11-28 |
Family
ID=49623635
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/062355 WO2013175936A1 (ja) | 2012-05-22 | 2013-04-26 | 脂環式ジエポキシ化合物の製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9212188B2 (ja) |
| JP (1) | JP2013241374A (ja) |
| WO (1) | WO2013175936A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106661046A (zh) * | 2014-04-25 | 2017-05-10 | 捷客斯能源株式会社 | 具有双螺降冰片烷结构的脂环式二环氧化合物、其制造方法及其用途 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111097525A (zh) * | 2018-10-25 | 2020-05-05 | 中国石油化工股份有限公司 | 用于烯烃环氧化反应固体酸催化剂和制备方法及应用 |
| DE102022106647A1 (de) | 2022-03-22 | 2023-09-28 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Niedertemperaturhärtende Massen auf Basis von Glycidylethern |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003238545A (ja) * | 2002-02-21 | 2003-08-27 | Nippon Shokubai Co Ltd | エポキシ化合物の製造方法 |
| WO2008093711A1 (ja) * | 2007-01-31 | 2008-08-07 | Osaka University | 固相系酸化反応システム |
| JP2010235649A (ja) * | 2009-03-30 | 2010-10-21 | Sanyo Chem Ind Ltd | 精製エポキシ樹脂の製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62234550A (ja) | 1985-12-24 | 1987-10-14 | San Petoro Chem:Kk | 触媒およびその使用法 |
| GB9114127D0 (en) * | 1991-06-29 | 1991-08-14 | Laporte Industries Ltd | Epoxidation |
| JP2004182648A (ja) | 2002-12-03 | 2004-07-02 | Daicel Chem Ind Ltd | 脂環式ジエポキシ化合物の製造方法 |
-
2012
- 2012-05-22 JP JP2012116389A patent/JP2013241374A/ja active Pending
-
2013
- 2013-04-26 US US14/402,376 patent/US9212188B2/en not_active Expired - Fee Related
- 2013-04-26 WO PCT/JP2013/062355 patent/WO2013175936A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003238545A (ja) * | 2002-02-21 | 2003-08-27 | Nippon Shokubai Co Ltd | エポキシ化合物の製造方法 |
| WO2008093711A1 (ja) * | 2007-01-31 | 2008-08-07 | Osaka University | 固相系酸化反応システム |
| JP2010235649A (ja) * | 2009-03-30 | 2010-10-21 | Sanyo Chem Ind Ltd | 精製エポキシ樹脂の製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| MATOBA, Y. ET AL.: "Epoxidation of cyclic diolefins with hydrogen peroxide catalyzed by areneseleninic acid", JOURNAL OF JAPAN PETROLEUM INSTITUTE, vol. 26, no. 5, 1983, pages 349 - 354 * |
| OKOVYTYY, S.I. ET AL.: "Identification of the stereoisomers of tetrahydroindene diepoxide by the 1H and 13C NMR characteristics: A combined experimental and theoretical study", JOURNAL OF MOLECULAR STRUCTURE: THEOCHEM, vol. 730, no. 1-3, 2005, pages 125 - 132 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106661046A (zh) * | 2014-04-25 | 2017-05-10 | 捷客斯能源株式会社 | 具有双螺降冰片烷结构的脂环式二环氧化合物、其制造方法及其用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013241374A (ja) | 2013-12-05 |
| US20150141675A1 (en) | 2015-05-21 |
| US9212188B2 (en) | 2015-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5376505B2 (ja) | 固相系酸化反応用混合物 | |
| KR100937891B1 (ko) | 디올레핀 화합물의 선택적 산화에 의한 2관능성 에폭시모노머의 제조방법 | |
| EP2383264B1 (en) | Epoxy compound production method | |
| KR101236376B1 (ko) | 에폭시 화합물의 제조 방법 | |
| WO2013175936A1 (ja) | 脂環式ジエポキシ化合物の製造方法 | |
| WO2010110151A1 (ja) | エポキシ化合物の製造方法 | |
| JPWO2011078060A1 (ja) | グリシジルエーテル化合物の製造方法及びモノアリルモノグリシジルエーテル化合物 | |
| JP4013334B2 (ja) | エポキシシクロドデカジエンの製造方法 | |
| JP4118642B2 (ja) | 環状オレフィンのエポキシ化方法 | |
| JP5979632B2 (ja) | エポキシ化合物の製造方法 | |
| JP5979633B2 (ja) | エポキシ化合物の製造方法 | |
| US9783548B2 (en) | Production method for epoxy compound using solid catalyst | |
| JP4998977B2 (ja) | ジオレフィン化合物の選択酸化による二官能性エポキシモノマーの製造方法 | |
| US9266899B2 (en) | Method for producing epoxy compound | |
| JP4178351B2 (ja) | 1,2−エポキシ−5,9−シクロドデカジエンを製造する方法 | |
| JP2003096079A (ja) | オキセタン環を有する脂環式エポキシ化合物の製造方法 | |
| JP2003238545A (ja) | エポキシ化合物の製造方法 | |
| JP2005104902A (ja) | エポキシ化合物の製造方法 | |
| Kaczmarczyk et al. | Important Parameters of Epoxidation of 1, 4‐bis (allyloxy) butane in Aqueous‐Organic Phase Transfer Catalytic System | |
| JP2010180362A (ja) | 多官能エポキシポリマーの製造方法 | |
| JP2010001256A (ja) | エポキシ化合物の製造方法 | |
| JP2012056883A (ja) | ジクロロエポキシブタンの製造方法 | |
| PL213050B1 (pl) | Nowy związek 1-glicydoloksy-4-hydroksybutan i sposób wytwarzania 1-glicydoloksy-4-hydroksybutanu |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13793315 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14402376 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13793315 Country of ref document: EP Kind code of ref document: A1 |