WO2012161518A2 - RORα의 활성자로서의 신규한 thiourea 유도체 및 이를 함유하는 약학적 조성물 - Google Patents
RORα의 활성자로서의 신규한 thiourea 유도체 및 이를 함유하는 약학적 조성물 Download PDFInfo
- Publication number
- WO2012161518A2 WO2012161518A2 PCT/KR2012/004081 KR2012004081W WO2012161518A2 WO 2012161518 A2 WO2012161518 A2 WO 2012161518A2 KR 2012004081 W KR2012004081 W KR 2012004081W WO 2012161518 A2 WO2012161518 A2 WO 2012161518A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- carbon atoms
- thiourea
- rorα
- mmol
- Prior art date
Links
Images
















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C335/00—Thioureas, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C335/04—Derivatives of thiourea
- C07C335/06—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to acyclic carbon atoms
- C07C335/10—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to acyclic carbon atoms of an unsaturated carbon skeleton
- C07C335/12—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to acyclic carbon atoms of an unsaturated carbon skeleton the carbon skeleton containing six-membered aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/145—Amines having sulfur, e.g. thiurams (>N—C(S)—S—C(S)—N< and >N—C(S)—S—S—C(S)—N<), Sulfinylamines (—N=SO), Sulfonylamines (—N=SO2)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to a novel thiourea derivative or a pharmaceutically acceptable salt thereof for activating the ROR ⁇ gene and a pharmaceutical composition comprising the same.
- ROR ⁇ (or NR1F1, also known as RORA or RZR) is one of the steroid hormone receptor superfamily and is a transcription factor that regulates gene expression.
- ROR ⁇ consists of an N-terminal transactivation domain, a DNA-binding domain and a C-terminal ligand binding domain.
- ROR ⁇ binds to the ROR-binding response element (RORE) in the promoter of the target gene, where RORE is the hexanucleotide motif (5-AGGTCA-3 ') and the A / T rich sequence of the six base pairs preceding this hexanucleotide. (Jetten AM et al., (2001) Prog Nucleic Acid Res Mol Biol. 69: 205-47).
- ROR ⁇ is regulated by binding a ligand to the C-terminal ligand binding domain
- known ligands include CGP52608, one of cholesterol and cholesterol derivatives, melatonin and thiazolidinedione (Kallen J et al., (2002) Structure. 10: 1697-707 / Kallen J et al., (2004) J Biol Chem. 279: 14033-8 / Wiesenberg I et al., (1995) Nucleic Acids Res. 23: 327-33).
- X-ray crystallography revealed that cholesterol and cholesterol derivatives bind to the ligand binding domain of ROR ⁇ (Kallen JA et al., (2002) Structure.
- LXR ⁇ is a transcription factor whose activity is regulated by ligand and is a member of the nuclear hormone receptor superfamily (Baranowski M. (2008) J Physiol Pharmacol. Suppl 7: 31-55). LXR ⁇ is an important factor in lipid and carbohydrate metabolism and induces fatty acid biosynthesis in the liver by increasing the expression of the SREBP-1, FAS and SCD-1 genes (Joseph SB et al., (2002) J Biol Chem. 277: 11019-25). This increase in LXR ⁇ -mediated gene expression results in triglyceride accumulation in hepatocytes, which is a factor that causes fatty liver and hypertriglyceridemia.
- Fatty Acid synthase is an enzyme involved in the final stage of fatty acid biosynthesis and is also a target gene for LXR and SREBP-1 (Clarke SD. (1993) J Anim Sci. 71: 1957-65).
- ROR ⁇ is known to contribute to regulating genes important for lipid synthesis by crosstalk with LXR (Wada T et al., (2008) Exp Biol Med (Maywood).
- ACC is the main enzyme of the fatty acid production pathway that regulates the step of converting acetyl-CoA to malonyl-CoA (Tong L et al., (2006) J Cell Biochem. 99: 1476-88).
- Malonyl-CoA produced by ACC inhibits fatty acid oxidation by inhibiting CPT-1, an enzyme that plays a major role in fatty acid oxidation in mitochondria, while ACC phosphorylates serine residues by kinases such as AMPK. Inactivation and loss of fatty acid antioxidant function (Brownsey RW et al., (2006) Biochem Soc Trans. 34: 223-7).
- the present inventors synthesized thiourea-based new compounds selectively showing ROR ⁇ activity as a result of conducting structural activity studies using the conventional thiazolidinedione-based compound CGP52608 as a leading substance. . Accordingly, it is an object of the present invention to provide novel lipid accumulation inhibitors by synthesizing novel thiourea derivatives that exhibit ROR ⁇ activity.
- the present invention provides novel compounds or pharmaceutically acceptable salts thereof that activate the ROR ⁇ gene.
- the present invention also provides a lipid accumulation inhibitor comprising a novel compound or a pharmaceutically acceptable salt thereof that activates the ROR ⁇ gene as an active ingredient.
- the present invention also provides a pharmaceutical composition for preventing or treating metabolic disease or inflammatory disease, comprising a novel compound or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier that activates a pharmaceutically effective amount of the ROR ⁇ gene.
- the disease includes atherosclerosis, fatty liver, alcoholic fatty liver, hyperlipidemia and the like.
- the present invention also provides a method for preventing or treating a metabolic disease or inflammatory disease comprising administering a pharmaceutical composition.
- the compound according to the present invention is expected to be effective in the treatment and prevention of metabolic and inflammatory diseases through various in vivo functions of ROR ⁇ , and is particularly useful for the prevention and treatment of liver diseases through the regulation of cholesterol homeostasis and the inhibition of lipid synthesis. .
- Figure 3 shows the effect of JC1 compounds on the transcriptional activity of ROR ⁇ .
- Figure 4 shows the change in ROR ⁇ transcriptional activity according to the concentration of JC1-38, JC1-40 and JC1-42.
- Figure 6 shows the effect of JC1-38, JC1-40 and JC1-42 on the protein expression of LXR ⁇ and SREBP-1 and FAS.
- Figure 7 shows the effect of JC1-40 and JC1-42 on the phosphorylation of ACC.
- Figure 9 shows the effect of JC1-38, JC1-40 and JC1-42 on fatty acid uptake and ultra-low specific lipoprotein secretion of hepatocytes.
- FIG. 11 shows the effects of JC1-40 and JC1-42 on phosphorylation of AMPK and ACC and protein expression of LXR ⁇ , SREBP-1 and FAS in high fat diet-induced mouse fatty liver.
- FIG. 12 shows the effects of JC1-40 and JC1-42 on hepatic triglycerides and hepatic fat accumulation and body weight change in high-fat diet-induced mouse fatty liver.
- Figure 13 shows the results of blood biochemical analysis by JC1-40 and JC1-42 in a high fat diet-induced mouse fatty liver model.
- Figure 15 shows the effect of JC1-40 on the vascular proliferation after balloon dilation.
- Figure 16 shows the improvement effect of the mouse alcoholic fatty liver model of JC1-40.
- the present invention seeks to provide novel compounds or pharmaceutically acceptable salts thereof that activate the ROR ⁇ gene.
- the novel compounds or their pharmaceutically acceptable salts activate the expression of the ROR ⁇ gene, thereby reducing the gene expression of LXR ⁇ , SREBP-1 and FAS and increasing the inactivation of ACC, resulting in hepatocytes.
- a lipid accumulation inhibitor that inhibits lipid synthesis and induces fatty acid oxidation to inhibit lipid accumulation.
- the present invention also provides a pharmaceutical composition for the prophylaxis or treatment of metabolic or inflammatory diseases comprising a novel compound or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier activating a pharmaceutically effective amount of the ROR ⁇ gene. .
- the present invention also provides a method for preventing or treating a metabolic disease or inflammatory disease comprising administering the pharmaceutical composition to a subject.
- an activator activating the expressed ROR ⁇ gene means (a) activating expression of the ROR ⁇ gene; and / or (b) enhancing transcriptional activity of the expressed ROR ⁇ protein.
- LXR activators increase the expression of enzymes that induce lipid synthesis in the liver, which causes hyperlipidemia and fatty liver (Schultz JR et al., (2000) Genes Dev. 14: 2831-8 ).
- Fatty Acid synthase Fatty Acid synthase (FAS) is an enzyme involved in the final stage of fatty acid biosynthesis and is also a target gene for LXR and SREBP-1 (Clarke SD. (1993) J Anim Sci. 71: 1957-65).
- Fatty Acid synthase Fatty Acid synthase
- SREBP-1 SREBP-1
- oxidation of fatty acids is an equally important step in fatty acid biosynthesis in lipid metabolism.
- Acetyl-CoA carboxylase ACC is an enzyme that produces malonyl-CoA during the fatty acid biosynthesis step.
- the resulting malonyl-CoA acts to inhibit beta-oxidation of fatty acids in mitochondria in hepatocytes.
- ACC is inactivated upon phosphorylation by kinases such as AMPK (Brownsey et al., 2006). Inactivation of ACC weakens the fatty acid oxidation inhibiting function of these ACCs.
- the present inventors have found that an activator activating the ROR ⁇ gene can inhibit lipid accumulation in hepatocytes. It was observed that LXR protein expression was inhibited when ROR ⁇ 1 virus was infected to cells. Thus, the present inventors have activated the expression of the ROR ⁇ (or NR1F1, RORA or RZR) gene, which is a member of the steroid hormone receptor superfamily of transcription factors that regulate gene expression, the expressed ROR ⁇ is LXR ⁇ and SREBP-1 genes.
- the target gene is the ROR ⁇ gene.
- the present invention provides novel compounds or pharmaceutically acceptable salts thereof that activate the ROR ⁇ gene.
- the novel compound for activating the ROR ⁇ gene is a thiourea derivative having the thiazolidinedione-based compound CGP52608 as a leading substance (called the JC1 compound), and can be represented by the following formula (I).
- R 1 is hydrogen or an alkyl group having 1 to 3 carbon atoms
- R 2 is a hydrogen element, a halogen element, an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a nitro group, a hydroxyl group or a phenoxy group;
- R 3 is a hydrogen element, a halogen element, an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a nitro group, a hydroxyl group or a phenoxy group;
- R 4 is a hydrogen element, a halogen element, an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a nitro group, a hydroxyl group, a cyano group, a dimethylamino group, a methylsulfonylamide group, a trifluoromethyl group
- R 5 may be a hydrogen element, a halogen element, an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a nitro group, a hydroxyl group, or a phenoxy group.
- R 4 is an aryl group or a phenoxy group
- an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a trifluoromethyl group or t-butyl group may be substituted in an aromatic ring.
- R 3 and R 4 may be connected to a ring in the above substituent.
- R 2 to R 5 may be substituted at the same time with the same or different substituents.
- novel compounds of the present invention can be synthesized chemically by the method shown in the following schemes. However, this is merely for illustrative purposes, and the present invention is not limited thereto.
- R is a substituent of R 2 to R 5 in the above formula (I) is a hydrogen element, a halogen element, an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a nitro group, a hydroxyl group, a cyano group, Dimethylamino group, methylsulfonylamide group, trifluoromethyl group, vinyl benzene group, phenoxy group, benzo group, aryl group or phenylamine group; When an aryl group or a phenoxy group, the aromatic ring may be substituted with an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a trifluoromethyl group, and a t-butyl group.
- R is the same as above.
- the alkylation reaction is carried out on a hydroxyl group using K 2 CO 3 and alkyl halide on commercially available 5-bromo 2-cyano phenol.
- the coupling reaction is then performed by Suzuki coupling with aryl boronic acid under a palladium catalyst based on Cs 2 CO 3 .
- the cyan group at position 2 is reduced using LAH (Lithium Aluminum Hydride) to obtain benzylamine, and finally, N-methyl isothiocyanate is coupled to synthesize thiourea, a desired compound of formula (I).
- LAH Lithium Aluminum Hydride
- R is an alkyl group having 1 to 3 carbon atoms
- Ar is in the formula (I)
- R 4 is a vinyl benzene group or an aryl group
- the aryl group may be substituted with an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a trifluoromethyl group or t-butyl group in the aromatic ring.
- Thiourea is synthesized through nucleophilic addition of phenol using commercially available 2-fluorobenzaldehyde as a starting material and through a reductive amination reaction as in Scheme 2.
- the present invention activates the expression of the ROR ⁇ gene through a novel Formula (I) compound or a pharmaceutically acceptable salt thereof that activates the ROR ⁇ gene, thereby reducing gene expression of LXR ⁇ , SREBP-1 and FAS, It provides increased lipid inactivation, thereby inhibiting lipid synthesis in hepatocytes and inducing fatty acid oxidation to inhibit lipid accumulation.
- a novel Formula (I) compound or a pharmaceutically acceptable salt thereof that activates the ROR ⁇ gene, thereby reducing gene expression of LXR ⁇ , SREBP-1 and FAS, It provides increased lipid inactivation, thereby inhibiting lipid synthesis in hepatocytes and inducing fatty acid oxidation to inhibit lipid accumulation.
- the invention also provides for the prevention of metabolic or inflammatory diseases comprising a novel Formula (I) compound or a pharmaceutically acceptable salt thereof, a pharmaceutically acceptable carrier, and an adjuvant or diluent that activates a pharmaceutically effective amount of the ROR ⁇ gene or Provided is a therapeutic pharmaceutical composition.
- the pharmaceutical composition according to the present invention is expected to be effective in the treatment and prevention of metabolic diseases and inflammatory diseases, in particular can be used for the prevention and treatment of liver diseases through the regulation of cholesterol homeostasis and lipid synthesis inhibition.
- the liver disease may include fatty liver, alcoholic fatty liver and hyperlipidemia.
- novel compounds of the present invention such as JC1 compounds, which activate the ROR ⁇ gene, have been shown to prevent the formation of atherosclerotic plaques accompanied by the proliferation of vascular smooth muscle and the prevention of vascular restenosis by vascular smooth muscle proliferation following balloon therapy or stent surgery. It is expected to be applicable.
- the present invention also provides a method for preventing or treating a metabolic disease or inflammatory disease comprising administering the pharmaceutical composition to a subject.
- the subject refers to a subject in need of treatment of a disease, and more specifically, mammals such as primates, mice, rats, dogs, cats, horses, and cows, which are human or non-human. it means.
- the pharmaceutically effective amount may be variously adjusted according to the weight, age, sex, health condition, diet, time of administration, method of administration, excretion rate, and severity of disease. .
- Pharmaceutical dosage forms of the compounds of the present invention may be used in the form of their pharmaceutically acceptable salts, or may be used alone or in combination with other pharmaceutically active compounds as well as in a suitable collection.
- Salts of the compounds of the present invention are preferably pharmaceutically acceptable salts and may be prepared by currently known methods such as adding inorganic bases, organic bases, inorganic acids, organic acids, basic or acidic amino acids to the compounds according to the invention. Can be.
- the pharmaceutical composition of the present invention comprises a pharmaceutically acceptable carrier.
- the pharmaceutically acceptable carrier may include physiological saline, polyethylene glycol, ethanol, vegetable oil and isopropyl myristate, and the like, but is not limited thereto.
- the compounds of the present invention may be formulated in ointments or creams for topical application and may be formulated with conventional saline, water-soluble solvents such as 5% dextrose or non-aqueous solvents such as vegetable oils, synthetic fatty acid glycerides, higher fatty acid esters or propylene glycol.
- the compound may be formulated as an injection by dissolving, suspending or emulsifying the compound.
- Formulations of the present invention may include conventional additives such as solubilizers, isotonic agents, suspending agents, emulsifiers, stabilizers and preservatives.
- Preferred dosages of the compounds of the present invention vary depending on the condition and weight of the patient, the extent of the disease, the form of the drug, the route of administration and the duration, and may be appropriately selected by those skilled in the art.
- the compound of the present invention is administered at 0.001-100 mg / kg body weight per day, more preferably 0.01-30 mg / kg body weight. Administration may be administered once a day or may be divided several times.
- the compound of the present invention should be present in an amount of 0.0001 to 10% by weight, preferably 0.001 to 1% by weight based on the total weight of the total composition.
- the pharmaceutical composition of the present invention can be administered to mammals such as mice, mice, livestock, humans, and the like by various routes.
- the method of administration is not limited and may be administered, for example, by oral, rectal or intravenous, intramuscular, subcutaneous, intrauterine epidural or cerebrovascular (intracerbroventricular) injection.
- JC1 compounds preferably JC1-38, JC1-40 and JC1-42, have excellent activity in transcription of ROR ⁇ (see Experimental Examples 1 and 2).
- JC1 compounds increase the phosphorylation of ACC, thereby increasing the oxidation of fatty acids by promoting the inactivation of ACC (see Experimental Example 5), in another experimental example JC1 The compounds were found to inhibit lipid accumulation in hepatocytes (see Experimental Example 6).
- novel compounds that activate the ROR ⁇ gene such as JC1 compounds, may be applied to the treatment of metabolic or inflammatory diseases.
- Example 35 Method for preparing R-(+)-1- [1- (4-methoxy-phenyl) -ethyl] -3-methyl-thiourea (JC1-35)
- reaction liquid was poured and filtered.
- the filtered filtrate was acidified with 1N-HCl and extracted with a water layer, which was then basified with sodium bicarbonate and extracted with ethyl acetate. This process was repeated several times to obtain the desired 2-ethoxy-4-aryl-benzylamine.
- Example 45 Method for preparing (4'-tert-butyl-3-ethoxy-biphenyl-4-methyl) -thiourea (JC1-45)
- CV-1 cells (CCL-70) were purchased from the American Type Culture Collection (ATCC). CV-1 cells (4 ⁇ 10 4 cells / well) were seeded in 24-well culture plates and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% fetal bovine serum (FBS). CV-1 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, the cells were transformed with RORE-tk-Luc reporter plasmid (100 ng), ROR expression vector (5 ng) using calcium phosphate.
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- HepG2 cells (1 ⁇ 10 5 cells / well) were seeded in 12-well plates and incubated overnight. Cells were transformed with LXRE-Luc (100 ng) or SRE-Luc (100 ng) reporter plasmids using Welfect-EXTM Plus (WelGENE Inc., Korea). 24 hours after transformation, the JC1 compound (100 ⁇ M), the solvent (control), and the like were treated. After 24 hours of treatment, luciferase activity was measured using an Analytical luminescence luminometer. To confirm transformation efficiency, luciferase activity was normalized using the activity of 200 ng of ⁇ -galactosidase ( ⁇ -gal) expression vector. The results are shown in Table 1 and shown in FIG.
- JC1 compound increased the transcriptional activity of ROR ⁇ .
- 13 JC1 compounds were shown to be more than two times more active than the solvent-treated control group, of which JC1-42 was observed to exhibit excellent activity of 460%.
- CV-1 cells (4 ⁇ 10 4 cells / well) were seeded in 24-well culture plates and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% fetal bovine serum (FBS).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- CV-1 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, RORE-Luc reporter (0.05 ⁇ g) and Flag-ROR ⁇ vector (0.005 ⁇ g) were transformed into CV-1 cells using calcium phosphate.
- CV-1 cells were treated with specific concentrations of JC1-38, JC1-40, and JC1-42 (1 ⁇ M, 10 ⁇ M) with excellent chemical stability, among the 14 JC1 compounds, especially the 14 JC1 compounds observed in Experimental Example 1 above. , 20 ⁇ M, 50 ⁇ M and 100 ⁇ M) and 100 ⁇ M of melatonin, the ligand of ROR ⁇ , for 24 hours. Cell lysates were then obtained, and luciferase activity was analyzed and determined using an analytical luminescence luminometer. Luciferase activity for transformation efficiency was normalized to ⁇ -gal activity, data is mean SD for two or three independent experimental results, and the results are shown in FIG. 4.
- HepG2 Human liver cancer cell line and HepG2 (ATCC HB 8065) were purchased from the American Type Culture Collection (ATCC). HepG2 cells (1.2 ⁇ 10 5 cells / well) were seeded in 24-well culture plates and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS (fetal bovine serum). HepG2 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, LXRE-Luc reporter (0.1 ⁇ g) was transformed into HepG2 cells using Welfect-EXTM Plus (WelGENE Inc., Korea).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- HepG2 cells were treated for 24 hours with specific concentrations of JC1-38, JC1-40 and JC1-42 (1 ⁇ M, 10 ⁇ M, 20 ⁇ M, 50 ⁇ M and 100 ⁇ M) and 1 ⁇ M of T17 (T0901317), the ligand of LXR ⁇ . .
- Cell lysates were then obtained, and analyzed and determined luciferase activity using an Analytical luminescence luminometer. Luciferase activity for transformation efficiency was normalized to ⁇ -gal activity, data is mean SD for three independent experimental results, and the results are shown in FIG. 5A.
- the transcriptional activity of LXR ⁇ increased to T17 decreased depending on the concentration of JC1-38, JC1-40 and JC1-42.
- all compounds were observed to reduce the T17-induced LXR ⁇ activity by more than 90% at a concentration of 100 ⁇ M.
- HepG2 cells were cultured under the same conditions, and after culturing, SRE-Luc reporter (0.1 ⁇ g) was transformed into HepG2 cells using Welfect-EXTM Plus (WelGENE Inc., Korea). After 24 hours of transformation, HepG2 cells were treated for 24 hours with 50 ⁇ M of each JC1-38, JC1-40 and JC1-42 compound and 1 ⁇ M of T17 (T0901317). Cell lysates were then obtained, and analyzed and determined luciferase activity using an Analytical luminescence luminometer. Luciferase activity for transformation efficiency was normalized to ⁇ -gal activity, data is mean SD for three independent experimental results, and the results are shown in FIG. 5B.
- HepG2 cells (1 ⁇ 10 6 cells / well) were seeded in 60-dish and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS (fetal bovine serum). HepG2 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, HepG2 cells were treated with 100 ⁇ M of each of JC1-38, JC1-40 and JC1-42 compounds and treated with AMPK activator metformin 2 mM for 24 hours as a positive control. After treatment the expression of proteins was analyzed by Western blotting assay.
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- HepG2 cells are destroyed for 30 minutes on ice in lysis buffer containing 50 mM NaCl, 50 mM Tris pH 7.4, 5 mM EDTA, 1% NP-40 and protease inhibitors, and centrifuged to dissociate total cell lysis. A solution was obtained. 20-30 ⁇ g protein obtained from whole cell lysate was transferred to 7-9% SDS-PAGE (sodium dodecylsulfate-polyacrylamide gel electrophoresis) and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA).
- SDS-PAGE sodium dodecylsulfate-polyacrylamide gel electrophoresis
- Blocking was performed with 5% or 10% (w / v) nonfat dry milk in PBS containing 0.1% Tween-20, ROR ⁇ (Affinity BioReagents), LXR ⁇ , SREBP-1, Santa Cruz Biotechnology (FAS), Reaction was performed with specific antibodies to phospho-ACC (Cell signaling) and ⁇ -tubulin (Calbiochem). Immunoreactive proteins were detected with Amersham ECL Western Blotting Detection Reagents using a horseradish peroxidase (HRP) -conjugated secondary antibody (Zymed Lab). Protein concentration was quantified by BCA (bicinchoninic acid) (Pierce) assay, and expression of ⁇ -tubulin was monitored as a control. The results are shown in Figure 6a.
- HRP horseradish peroxidase
- HepG2 cells were cultured under the same conditions, and after incubation, HepG2 cells were treated for 24 hours with JC1-40 (or JC1-42) and 1 ⁇ M T17 at specific concentrations (0 ⁇ M, 1 ⁇ M, 10 ⁇ M, 50 ⁇ M and 100 ⁇ M). . After treatment, expression of protein was analyzed by Western blotting assay and expression of ⁇ -tubulin was monitored as a control. The results are shown in FIGS. 6B and 6C.
- HepG2 cells (1 ⁇ 10 6 cells / well) were seeded in 60-dish and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS (fetal bovine serum). HepG2 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, HepG2 cells were treated with 100M of JC1-38 and JC1-40 (or 100 ⁇ M of JC1-38 and JC1-42), 2mM of metfornin and 2mM of AICIR for 24 hours. After treatment, expression of ROR ⁇ and phosphorylated ACC (pACC) protein was analyzed by Western blotting, and expression of ⁇ -tubulin was monitored as a control. The results are shown in Figure 7 (a, b).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- HepG2 cells (1 ⁇ 10 6 cells / well) were seeded in 60-dish and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS (fetal bovine serum). HepG2 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, HepG2 cells were treated with 100 ⁇ M of JC1-38, JC1-40 and JC1-42 for 24 hours. After treatment, the effect of real-time reverse transcriptase-polymerase chain reaction (RT-PCR) assay on transcriptional activity was analyzed.
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- MCAD forward: 5′-CTACCAAGTATGCCCTGGAAAG-3′SEQ ID NO 1, reverse: 5′-TGTGTTCACGGGCTACAATAAG-3′SEQ ID NO 2
- ACO1 forward: 5′-GGGCATGGCTATTCTCATTGC-3′SEQ ID NO 3, reverse: 5′-CGAACAAGGTCAACAGAAGTTAGGT -3'SEQ ID NO. 4
- ACO2 forward: 5'-GCGGACATGGCTACTCAAAGC-3'SEQ ID NO.
- JC1-38, JC1-40 and JC1-42 increase the transcriptional activity of MCAD, ACO1, ACO2, HMGCS2, CPT-1 and ACS involved in fatty acid oxidation in HepG2 cells. .
- HepG2 cells (1 ⁇ 10 6 cells / well) were seeded in 60-dish and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS (fetal bovine serum). HepG2 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, HepG2 cells were treated with 100 ⁇ M of JC1-38, JC1-40 and JC1-42 for 24 hours. After treatment, the effect of real-time reverse transcriptase-polymerase chain reaction (RT-PCR) assay on transcriptional activity was analyzed. PCR reactions were performed using specific primers for CD36 (FIG.
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- CD36 forward: 5′-GGAACTGTGGGCTCATTGC-3′SEQ ID NO 15, reverse: 5′-CATGAGAATGCCTCCAAACAC-3′SEQ ID NO 16
- MTTP forward: 5′-CCTTCATTCAGCACCTCA-3′SEQ ID NO 17, reverse: 5′-TGACAAGTGTCCCAGTGA -3'SEQ ID NO: 18
- ApoB100 forward: 5'-TAAATGGAGCACTTTTCAAG-3'SEQ ID NO: 19, reverse: 5'-GGAACAGCAGCAGTAGCG-3'SEQ ID NO: 20
- ⁇ -actin 5'-CGTGGGCCGCCCTAGGCACCA-3 ' 13, reverse: 5'-TTGGCTTAGGGTTCAGGGGGG-3'SEQ ID NO. 14).
- JC1-38, JC1-40 and JC1-42 reduce the transcriptional activity of CD36 involved in fatty liver uptake in HepG2 cells and MTTP involved in ultralow-weight lipoprotein secretion. It was observed that the transcriptional activity of and ApoB100 increased.
- HepG2 cells (3 ⁇ 10 5 cells / well) were seeded in 60-dish and incubated overnight in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS (fetal bovine serum). HepG2 cells were maintained at 37 ° C. in a humidified thermostat with 5% CO 2 and 95% air. After incubation, HepG2 cells were mixed with a fatty acid mixture of 50 ⁇ M JC1-40 (or JC1-42) in a 2: 1 ratio of 0.5 mM oleic acid and 0.5 mM palmitic acid as lipid-inducing substances (5 mM oleic acid).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- the leftmost line is the control group treated only with solvent
- the middle line is the experimental group treated with JC1-40 or JC1-42 with fatty acid
- the rightmost line is treated with fatty acid only
- FIG. 11 (a) 6-week-old test animal mice were fed a high fat diet containing safflower oil for 5 days. After 3 days of diet, JC1-40 and JC1-42 were diluted in 0.5% carboxymethyl cellulose to give high-fat diet containing safflower oil while orally administered at a concentration of 10 and 30 mg / kg / day for 2 days. After 5 days of diet, mice were sacrificed to obtain liver tissue and analyzed for protein expression by Western blotting.
- the cells were homogenized in lysis buffer containing 50 mM NaCl, 50 mM Tris pH 7.4, 5 mM EDTA, 1% NP-40, and protease inhibitors, and centrifuged to obtain total cell lysates. 20-30 ⁇ g protein obtained from whole cell lysate was transferred to 7-9% SDS-PAGE (sodium dodecylsulfate-polyacrylamide gel electrophoresis) and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA).
- SDS-PAGE sodium dodecylsulfate-polyacrylamide gel electrophoresis
- Blocking is done with 5% or 10% (w / v) nonfat dry milk in PBS containing 0.1% Tween-20, ROR ⁇ (Affinity BioReagents), LXR ⁇ , SREBP-1, Santa Cruz Biotechnology (FAS), Reaction was performed with specific antibodies against phospho-AMPK, phospho-ACC (Cell signaling) and ⁇ -tubulin (Calbiochem). Immunoreactive proteins were detected with Amersham ECL Western Blotting Detection Reagents using horseradish peroxidase (HRP) -conjugated secondary antibody (Zymed Lab). Protein concentration was quantified by BCA (bicinchoninic acid) (Pierce) assay and expression of ⁇ -tubulin was monitored as a control. The results are shown in Figure 11b.
- HRP horseradish peroxidase
- FIG. 11 (a) 6-week-old test animal mice were fed a high fat diet containing safflower oil for 5 days. After 3 days of diet, JC1-40 and JC1-42 were diluted in 0.5% carboxymethyl cellulose to give high-fat diet containing safflower oil while orally administered at a concentration of 10, 30 mg / kg / day. After 5 days of diet, mice were sacrificed to obtain liver tissue, and the amount of triglyceride in the liver was measured using EnzyChromTM Triglyceride Assay Kit (BioAssay Systems). As shown in FIG. 12 (a), it was observed that the amount of triglyceride in the liver increased by JC1-40 and JC1-42 due to the high fat diet.
- liver tissues of the mice were obtained and Oil-Red O staining was performed to confirm liver fat accumulation. As shown in Figure 12 (b), it was observed that increased liver fat accumulation due to high fat diet is reduced by JC1-40 and JC1-42.
- A7r5 a rat smooth muscle cell line, was purchased from the American Type Culture Collection (ATCC). A7r5 was incubated in 5% CO 2 using Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, penicillin (100 U / ml) and streptomycin (100 ⁇ g / ml). A7r5 cells (2 ⁇ 10 3 cells / well) were seeded in 96 well culture plates and incubated overnight. After incubation, A7r5 cells were treated with 100 ⁇ each of JC1-40 and JC1-42 compounds for 2, 4 and 6 days.
- ATCC American Type Culture Collection
- MTT (3-4,5-dimethylthiazol-2-yl) -2,5-diphenyl-tetrazolium bromide) 2 mg / ml 50 ⁇ l was treated for 4 hours, the medium was removed, and 200 ⁇ l of DMSO was added. After mixing the plate for 5 minutes, the absorbance was measured at 595 nm. Data are mean and SD values for three independent experimental results, the results of which are shown in FIG. 14.
- JC1-40 was administered to the white paper for one day, followed by balloon dilatation, followed by another 14 days.
- Balloon dilation was performed as follows. The cervix of the white paper was incised to expose the common carotid artery and to tie the internal branch at the branching to the external and internal branches.
- FIG. 15a is a cross-sectional view of the blood vessel of the control group
- Figure 15b shows a cross-sectional view of the test group administered with JC1-40.
- the control diet was pair-feeding by replacing maltose dextrin with the same calories based on the amount of feed consumed by the animals in the alcohol group instead of alcohol.
- JC1-40 was diluted orally at 10 mg / kg / day in 0.5% carboxymethyl cellulose for 2 weeks before the rats were sacrificed. Animals were fasted 12 hours after the end of the 5 week alcohol diet, and anesthesia was dissected to remove blood and liver.
- FIG. 16A GPT and GOT for measuring liver damage in plasma isolated from blood were significantly increased by about 50% in the alcohol group and the control group in the JC1-40 treated alcohol group. It was confirmed that the level was reduced.
- 16B and 16C blood triglyceride and cytokine MCP-1 levels were measured by ELISA. As a result, it was observed that the increased level in the alcohol group was significantly decreased in the JC1-40 treated alcohol group.
- the weight of the liver was divided by the weight of the rat, and the ratio of the liver to the weight was found to show that the proportion of the liver increased by alcohol intake was significantly reduced by the administration of JC1-40.
- FIG. 16E the expression of MCP-1 protein was analyzed by Western blotting.
- the present invention is expected to be effective in the treatment and prevention of metabolic and inflammatory diseases, and in particular, it is expected to be useful for the prevention and treatment of liver diseases through the regulation of cholesterol homeostasis and the inhibition of lipid synthesis.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Gastroenterology & Hepatology (AREA)
- Rheumatology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
Abstract
본 발명은 RORα 유전자를 활성화시키는 신규한 화합물 또는 그의 염에 관한 것이다. 구체적으로 본 발명은 thiazolidinedione의 하나인 CGP52608을 선도 물질로 하는 신규한 JC1 화합물 또는 이의 약제학적으로 허용가능한 염을 제공한다. 또한, 본 발명은 유효성분으로 상기 신규한 화합물을 포함하는 지질 축적 억제제를 제공한다. 본 발명은 대사성 질환 또는 염증성 질환 치료에 적용될 수 있을 것으로 기대된다.
Description
본 발명은 RORα 유전자를 활성화시키는 신규한 thiourea 유도체 또는 이의 약제학적으로 허용가능한 염 및 이를 포함하는 약학적 조성물 등에 관한 것이다.
RORα (또는 NR1F1, RORA or RZR로 알려짐)는 스테로이드 호르몬 수용체 슈퍼패밀리의 하나로, 유전자 발현을 조절하는 전사 인자이다. RORα는 N-말단 트랜스활성화 도메인, DNA-결합 도메인 및 C-말단 리간드 결합 도메인으로 이루어져 있다. RORα는 타겟 유전자의 프로모터에 있는 ROR-결합 반응 엘리먼트 (RORE)에 결합하며, RORE는 헥사뉴클레오타이드 모티프(5-AGGTCA-3')와 이 헥사뉴클레오타이드에 선행하는 6개 베이스페어의 A/T rich 서열을 포함한다(Jetten AM et al., (2001) Prog Nucleic Acid Res Mol Biol. 69:205-47). RORα는 리간드가 C-말단 리간드 결합 도메인에 결합함으로써 활성이 조절되며, 알려진 리간드로는 콜레스테롤과 콜레스테롤 유도체, 멜라토닌 및 thiazolidinedione의 하나인 CGP52608이 있다 (Kallen J et al.,(2002) Structure. 10:1697-707/Kallen J et al., (2004) J Biol Chem. 279:14033-8/Wiesenberg I et al., (1995) Nucleic Acids Res. 23:327-33). 엑스-레이 결정 분석법에 의해 콜레스테롤 및 콜레스테롤 유도체들이 RORα의 리간드 결합 도메인에 결합하는 것이 밝혀졌다(Kallen JA et al., (2002) Structure. 10:1697-707/Kallen J et al., (2004) J Biol Chem. 279:14033-8). 멜라토닌 역시 RORα에 특이적으로 결합해 RORα를 매개한 유전자 활성 조절을 일으키며, CGP52608 또한 이러한 멜라토닌과 경쟁적으로 RORα에 결합하는 합성 리간드임이 보고되었다 (Wiesenberg I et al., (1995) Nucleic Acids Res. 23:327-33). RORα는 아포리포단백질인 Apo A, Apo V 및 Apo C III를 발현하는 유전자들을 조절함으로서 말초 조직의 콜레스테롤을 간으로 운반하여 제거하는 기능을 한다(Vu-Dac N et al., (1997) J Biol Chem. 272:22401-4/Lind U et al., (2005) Biochem Biophys Res Commun. 330:233-41). 이것은 RORα가 콜레스테롤의 항상성 조절 및 지질 대사 관련 질병에 응용될 가능성을 시사한다.
[규칙 제91조에 의한 정정 25.07.2012]

LXRα는 리간드에 의해 활성이 조절되는 전사인자로서, 핵 호르몬 수용체 슈퍼패밀리의 멤버이다(Baranowski M. (2008) J Physiol Pharmacol. Suppl 7:31-55). LXRα는 지질 및 탄수화물 대사에 중요한 인자로, SREBP-1, FAS 및 SCD-1 유전자의 발현을 증가시켜 간에서의 지방산 생합성을 유도한다(Joseph SB et al., (2002) J Biol Chem. 277:11019-25). LXRα을 매개한 이러한 유전자 발현의 증가로 인해 간세포 내 트리글리세라이드 축적이 일어나며, 이는 지방간 및 고트리글리세라이드혈증을 유발하는 요인이다. 다시말해, LXR 활성자에 의해 간에서 지질 합성을 유도하는 효소들의 발현이 증가되고, 이것이 고지질혈증 및 지방간을 일으킨다 (Schultz JR et al., (2000) Genes Dev. 14:2831-8). 이러한 효소들 가운데 Fatty Acid synthase (FAS)는 지방산 생합성의 마지막 단계에 관여하는 효소로서, LXR과 SREBP-1의 타겟 유전자이기도 하다 (Clarke SD. (1993) J Anim Sci. 71:1957-65). RORα가 LXR과의 크로스 토크로 지질 합성에 중요한 유전자들을 조절하는데 기여함이 알려져 있다 (Wada T et al., (2008) Exp Biol Med (Maywood).
ACC는 아세틸-CoA를 말로닐-CoA로 전환시키는 단계를 조절하는 지방산 생성 경로의 주 효소이다(Tong L et al., (2006) J Cell Biochem. 99:1476-88). ACC에 의해 생성된 말로닐-CoA는 미토콘드리아에서 지방산 산화가 일어나는 데 주요한 역할을 하는 효소인 CPT-1을 저해함으로서 지방산 산화를 억제하는 역할을 하고, ACC는 AMPK와 같은 키나아제에 의해 세린 잔기가 인산화되면 불활성화되어 지방산 산화 억제 기능을 잃게 된다 (Brownsey RW et al., (2006) Biochem Soc Trans. 34:223-7).
본 발명자 등은 지질 축적을 효과적으로 억제할 수 있는 물질을 개발하고자, 기존의 thiazolidinedione계 화합물 CGP52608을 선도 물질로 하여 구조활성연구를 수행한 결과, RORα에 선택적으로 활성을 나타내는 thiourea계 신규 화합물들을 합성하였다. 따라서, 본 발명의 목적은 RORα에 활성을 나타내는 신규thiourea 유도체를 합성하여 신규 지질 축적 억제제를 제공하는 것이다.
그러나, 본 발명이 이루고자 하는 기술적 과제는 이상에서 언급한 과제에 제한되지 않으며, 언급되지 않은 또 다른 과제들은 아래의 기재로부터 당업자에게 명확하게 이해될 수 있을 것이다.
본 발명은 RORα 유전자를 활성화시키는 신규한 화합물 또는 이의 약제학적으로 허용가능한 염을 제공한다.
또한 본 발명은 유효성분으로 RORα 유전자를 활성화시키는 신규한 화합물 또는 이의 약제학적으로 허용가능한 염을 포함하는 지질 축적 억제제를 제공한다.
또한 본 발명은 약제학적 유효량의 RORα 유전자를 활성화시키는 신규한 화합물 또는 이의 약제학적으로 허용가능한 염 및 약제학적으로 허용되는 담체를 포함하는 대사성 질환 또는 염증성 질환 예방 또는 치료용 약학적 조성물을 제공한다.
상기 질환은 동맥경화증, 지방간, 알코올성 지방간, 및 고지혈증 등을 포함한다.
또한 본 발명은 약학적 조성물을 투여하는 단계를 포함하는 대사성 질환 또는 염증성 질환의 예방 또는 치료 방법을 제공한다.
본 발명에 따른 화합물은 RORα의 다양한 생체내 기능을 통해 대사성 질환과 염증성 질환의 치료와 예방에 효과가 있을 것으로 기대되며 특히 콜레스테롤의 항상성 조절 및 지질 합성 억제를 통한 간 질환의 예방 및 치료에 유용하다.
도 1 및 도 2는 RORα 활성자 후보물질로서 합성된 JC1 화합물들의 화학 구조를 나타낸 것이다.
도 3은 JC1 화합물들이 RORα의 전사활성에 미치는 효과를 나타낸 것이다.
도 4는 JC1-38, JC1-40 및 JC1-42의 농도에 따른 RORα 전사 활성 변화를 나타낸 것이다.
도 5는 JC1-38, JC1-40 및 JC1-42가 LXRα와 SREBP-1의 전사 활성에 미치는 효과를 나타낸 것이다.
도 6은 JC1-38, JC1-40 및 JC1-42가 LXRα와 SREBP-1 및 FAS의 단백질 발현에 미치는 효과를 나타낸 것이다.
도 7은 JC1-40 및 JC1-42가 ACC의 인산화에 미치는 효과를 나타낸 것이다.
도 8은 JC1-38, JC1-40 및 JC1-42가 지방산 산화에 미치는 효과를 나타낸 것이다.
도 9는 JC1-38, JC1-40 및 JC1-42가 간세포의 지방산 흡수 및 초저비중지질단백 분비에 미치는 영향을 나타낸 것이다.
도 10은 JC1-40 및 JC1-42가 간세포의 지질 축적에 미치는 효과를 나타낸 것이다.
도 11은 JC1-40 및 JC1-42가 고지방식이-유도 마우스 지방간에서 AMPK와 ACC의 인산화 및 LXRα, SREBP-1 및 FAS의 단백질 발현에 미치는 효과를 나타낸 것이다.
도 12는 JC1-40 및 JC1-42가 고지방식이-유도 마우스 지방간에서 간 내 중성 지방과 간 내 지방 축적 및 체중 변화에 미치는 영향을 나타낸 것이다.
도 13은 고지방식이-유도 마우스 지방간 모델에서 JC1-40 및 JC1-42에 의한 혈액 생화학적 분석 결과를 나타낸 것이다.
도 14는 JC1-40 및 JC1-42가 혈관평활근세포 증식에 미치는 효과를 나타낸 것이다.
도 15는 JC1-40이 풍선 확장술 이후 혈관 내층 증식에 미치는 효과를 나타낸 것이다.
도 16는 JC1-40의 마우스 알코올성 지방간 모델에서의 개선 효과를 나타낸 것이다.
본 발명은 RORα 유전자를 활성화시키는 신규한 화합물 또는 이의 약제학적으로 허용가능한 염을 제공하고자 한다. 구체적으로, 상기 신규한 화합물 또는 이의 약제학적으로 허용가능한 염을 통해 RORα 유전자의 발현을 활성화시켜, LXRα, SREBP-1 및 FAS의 유전자 발현은 감소시키며, ACC의 불활성화를 증가시켜, 간세포에서의 지질 합성을 억제시키고, 지방산 산화를 유도하여 지질 축적을 억제시키는 지질축적 억제제를 제공하고자 한다. 또한 본 발명은 약제학적 유효량의 RORα 유전자를 활성화시키는 신규한 화합물 또는 이의 약제학적으로 허용가능한 염 및 약제학적으로 허용 가능한 담체를 포함하는 대사성 질환 또는 염증성 질환의 예방 또는 치료용 약학적 조성물을 제공한다. 또한 본 발명은 상기 약학적 조성물을 개체에 투여하는 단계를 포함하는 대사성 질환 또는 염증성 질환의 예방 또는 치료 방법을 제공한다. 본 발명에서, 표현 RORα 유전자를 활성화시키는 활성자 (activator)"는 (a) RORα 유전자의 발현 활성화; 및/또는 (b) 발현된 RORα 단백질의 전사 활성능 증진을 의미한다.
LXR 활성자에 의해 간에서 지질 합성을 유도하는 효소들의 발현이 증가되고, 이것이 고지질혈증 및 지방간을 일으킨다는 사실이 알려져 있다 (Schultz JR et al., (2000) Genes Dev. 14:2831-8). 이러한 효소들 가운데 Fatty Acid synthase (FAS)는 지방산 생합성의 마지막 단계에 관여하는 효소로서, LXR과 SREBP-1의 타겟 유전자이기도 하다 (Clarke SD. (1993) J Anim Sci. 71:1957-65). 또한, 지방산의 산화는 지질 대사에서 지방산 생합성 못지않게 중요한 단계이다. Acetyl-CoA carboxylase (ACC)는 지방산 생합성 단계 중 말로닐-CoA를 생성하는 효소로, 생성된 말로닐-CoA는 간세포 내 미토콘드리아에서 일어나는 지방산의 베타-산화를 억제하는 역할을 한다. ACC는 AMPK와 같은 키나아제에 의해 인산화되면 불활성화되는데, (Brownsey et al., 2006). ACC가 불활성화되면 이러한 ACC가 일으키는 지방산 산화 억제 기능이 약화된다.
본 발명자 등은 지질 축적을 효과적으로 억제할 수 있는 물질을 개발하고자 예의 연구 노력한 결과, RORα 유전자를 활성화시키는 활성자 (activator)가 간세포 내의 지질 축적을 억제할 수 있음을 발견하였다, 즉, 본 발명자 등은 세포에 RORα1 바이러스를 감염시켰을 때 LXR 단백질 발현이 억제되는 것을 관찰하였다. 따라서 본 발명자 등은 유전자 발현을 조절하는 전사 인자의 스테로이드 호르몬 수용체 슈퍼패밀리의 멤버인, RORα (또는 NR1F1, RORA or RZR로 알려짐) 유전자의 발현을 활성화시키면, 발현된 RORα는 LXRα 및 SREBP-1 유전자의 전사 활성을 감소시키고, LXRα, SREBP-1 및 FAS의 유전자 발현을 감소시키며, ACC의 불활성화를 증가시켜, 간세포에서의 지질 합성이 억제되고 지방산 산화가 유도되어 결국 지질 축적이 억제되는 분자적 기전을 발견하였다. 본 발명은 이러한 분자적 기전에 기초하여 완성되었다. 본 발명에 있어서, 타깃이 되는 유전자는 RORα 유전자이다.
이에, 본 발명은 RORα 유전자를 활성화시키는 신규한 화합물 또는 이의 약제학적으로 허용가능한 염을 제공한다.
RORα 유전자를 활성화시키는 신규한 화합물은 기존의 thiazolidinedione계 화합물 CGP52608을 선도 물질로 하는 thiourea 유도체로써(JC1 화합물 이라 함), 하기의 화학식(I)으로 표시할 수 있다.
[규칙 제91조에 의한 정정 25.07.2012]
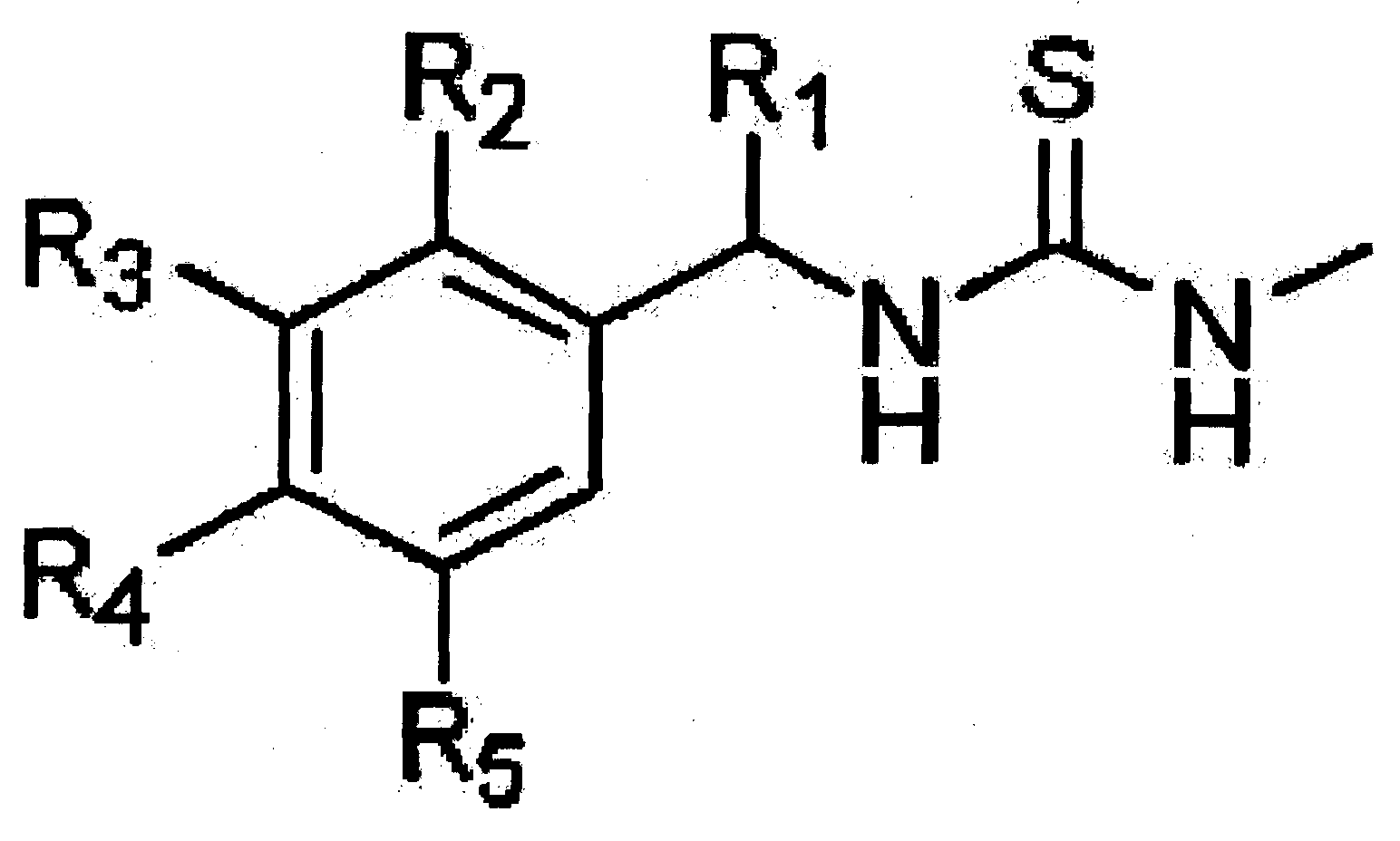
화학식(I)
상기 화학식(I)에서,
R1은 수소 또는 탄소수가 1 내지 3인 알킬기 이고;
R2는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기이고;
R3는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기이고;
R4는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기, 시아노기, 다이메틸아미노기, 메틸설포닐아마이드기, 트리플루오르메틸기, 바이닐 벤젠기, 페녹시기, 벤족시기, 아릴기 또는 페닐아민기 이고;
R5는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기 일 수 있다.
또한, 상기 R4가 아릴기, 페녹시기 일 때 방향족 환(aromatic ring)에 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 트리플루오르메틸기 또는 t-부틸기가 치환 될 수 있다.
또한, 상기 R3 와 R4 는 위와 같은 치환기에서 환으로 연결될 수 있다.
또한, 상기 R2 내지 R5 는 동일한 또는 다른 치환체로 동시에 치환 될 수 있다.
본 발명의 신규한 화합물은 하기의 반응식들에 도시된 방법에 의해 화학적으로 합성될 수 있다. 그러나, 이는 단지 예시를 들기 위한 것으로서, 본 발명이 이에 한정되는 것은 아니다.
[반응식1]
[규칙 제91조에 의한 정정 25.07.2012]

화학식(I)의 범위에 속하는 화합물은 치환된 벤질아민 형태의 기질을 [염의 경우 Triethylamine(TEA)적가] Diethyl ether 또는 Methylene Chloride 용매(시작물질이 녹지 않을 경우 메탄올이나 DMF(dimethylformamide)도 사용)하에서 N-methylisothiocyanate와 커플링하여 원하는 thiourea 모핵의 컴파운드를 합성한다.
R은 위 화학식(I)에서 R2 내지 R5의 치환체로 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기, 시아노기, 다이메틸아미노기, 메틸설포닐아마이드기, 트리플루오르메틸기, 바이닐 벤젠기, 페녹시기, 벤족시기, 아릴기 또는 페닐아민기 이며; 아릴기, 페녹시기 일 때 방향족 환에 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 트리플루오르메틸기, t-부틸기가 치환 될 수 있다.
[반응식2]
[규칙 제91조에 의한 정정 25.07.2012]

화학식(I)의 범위에 속하는 화합물은 벤잘데하이드 구조에 thiourea를 반응시켜 환원적 아민화 반응을 하여 다양한 치환기를 가진 thiourea 모핵의 컴파운드를 합성한다. 치환된 벤잘데하이드구조에 THF(tetrahydrofuran)용매 하에 ti(4)-isopropoxide와 N-methylthiourea를 사용하여 중간체를 만들고, 여기에 NaBH4를 사용하여 원하는 물질인 thiourea를 합성한다.
R은 위와 동일하다.
[반응식3]
[규칙 제91조에 의한 정정 25.07.2012]

상업적으로 구할 수 있는 5-브로모 2-시아노 페놀에 K2CO3와 alkyl halide를 사용하여 하이드록시기에 알킬화 반응을 수행한다. 그리고 Cs2CO3을 베이스로 팔라듐촉매 하에 아릴 보로닉 산을 사용하여 스즈키 커플링하여 커플링 반응을 수행한다. 2번 위치의 시안기를 LAH(Lithium Aluminium Hydride)를 사용하여 환원하여 벤질아민을 얻은 다음, 마지막으로 N-methyl isothiocyanate를 커플링하여 원하는 화학식(I) 화합물인 thiourea를 합성한다.
R은 탄소수가 1 내지 3인 알킬기 이며,
Ar은 위 화학식(I)에서 R4가 바이닐 벤젠기 또는 아릴기 이며; 아릴기는 방향족 환에 탄소수가 1 내지 3인 알킬기, 탄소수가 1내지 3인 알콕시기, 트리플루오르메틸기 또는 t-부틸기가 치환 될 수 있다.
[반응식4]
[규칙 제91조에 의한 정정 25.07.2012]

상업적으로 이용가능한 2-플루오르벤잘데하이드를 출발 물질로 하여 페놀을 nucleophilic addition하고, 반응식2와 같은 환원적 아민화 반응을 거쳐 thiourea를 합성한다.
또한, 본 발명은 RORα 유전자를 활성화시키는 신규한 화학식(I) 화합물 또는 이의 약제학적으로 허용가능한 염을 통해 RORα 유전자의 발현을 활성화시켜, LXRα, SREBP-1 및 FAS의 유전자 발현은 감소시키며, ACC의 불활성화를 증가시켜, 간세포에서의 지질 합성을 억제시키고, 지방산 산화를 유도하여 지질 축적을 억제시키는 지질축적 억제제를 제공한다.
또한 본 발명은 약제학적 유효량의 RORα 유전자를 활성화시키는 신규한 화학식(I) 화합물 또는 이의 약제학적으로 허용가능한 염, 약제학적으로 허용 가능한 담체, 및 보조제 또는 희석액을 포함하는 대사성 질환 또는 염증성 질환 예방 또는 치료용 약학적 조성물을 제공한다. 본 발명에 따른 약학적 조성물은 대사성 질환과 염증성 질환의 치료와 예방에 효과가 있을것으로 기대되며 특히 콜레스테롤의 항상성 조절 및 지질 합성 억제를 통한 간 질환의 예방, 치료를 위해 사용될 수 있다. 바람직하게, 상기 간 질환은 지방간, 알코올성 지방간 및 고지혈증 등을 포함할 수 있다. 이에 더하여, RORα 유전자를 활성화시키는 본 발명의 신규한 화합물, 예컨대 JC1 화합물들은 혈관평활근의 증식을 동반하는 죽상동맥경화반의 생성억제와 풍선요법 또는 스텐트 시술 후 혈관 평활근 증식에 의한 혈관 재협착의 방지에 적용될 수 있을 것으로 기대된다.
또한 본 발명은 상기 약학적 조성물을 개체에 투여하는 단계를 포함하는 대사성 질환 또는 염증성 질환 예방 또는 치료 방법을 제공한다.
본 발명에서 개체란 질병의 치료를 필요로 하는 대상을 의미하고, 보다 구체적으로는 인간 또는 비-인간인 영장류, 생쥐 (mouse), 쥐 (rat), 개, 고양이, 말 및 소 등의 포유류를 의미한다. 또한, 본 발명에서 약제학적 유효량은 환자의 체중, 연령, 성별, 건강상태, 식이, 투여시간, 투여방법, 배설율 및 질환의 중증도 등에 따라 그 범위가 다양하게 조절될 수 있음은 당업자에게 명백하다.
본 발명의 화합물의 약제학적 투여 형태는 이들의 약제학적 허용가능한 염의 형태로도 사용될 수 있고, 또한 단독으로 또는 다른 약제학적 활성 화합물과 결합 뿐만 아니라 적당한 집합으로 사용될 수 있다. 본 발명의 화합물의 염은 약제학적으로 허용 가능한 염이 바람직하며, 본 발명에 따른 화합물에 무기염기, 유기염기, 무기산, 유기산, 염기성 또는 산성 아미노산을 첨가하는 것과 같은 현재 공지된 방법에 의해서 제조될 수 있다.
본 발명의 약학적 조성물은 약제학적으로 허용 가능한 담체를 포함한다. 상기 약제학적으로 허용가능한 담체는 생리식염수, 폴리에틸렌글리콜, 에탄올, 식물성 오일 및 이소프로필미리스테이트 등을 포함할 수 있으며, 이에 한정되지는 않는다.
본 발명의 화합물은 국소 적용을 위해서 연고나 크림으로 제형화 할 수 있고, 일반적인 식염수, 5% 덱스트로스와 같은 수용성 용매 또는 식물성 오일, 합성 지방산 글리세라이드, 고급 지방산 에스테르 또는 프로필렌글리콜과 같은 비수용성 용매에 화합물을 용해시키거나, 현탁시키거나 또는 유화시켜 주사제로 제형화할 수 있다. 본 발명의 제형은 용해제, 등장화제(isotonic agents), 현탁화제, 유화제, 안정화제 및 방부제와 같은 종래의 첨가제를 포함할 수 있다.
본 발명의 화합물의 바람직한 투여량은 환자의 상태 및 체중, 질병의 정도, 약물 형태, 투여경로 및 기간에 따라 다르지만, 당업자에 의해 적절하게 선택될 수 있다. 그러나, 바람직하게는, 본 발명의 화합물을 1일 0.001~100 mg/체중kg으로, 보다 바람직하게는 0.01~30 mg/체중kg으로 투여한다. 투여는 하루에 한번 투여할 수도 있고, 여러번 나누어 투여할 수 있다. 약학 조성물에서 본 발명의 화합물은 전체 조성물 총 중량에 대하여 0.0001~10 중량%, 바람직하게는 0.001~1 중량%의 양으로 존재하여야 한다.
본 발명의 약학적 조성물은 쥐, 생쥐, 가축, 인간 등의 포유동물에 다양한 경로로 투여될 수 있다. 투여방법에는 제한이 없으며, 예를 들면, 경구, 직장 또는 정맥, 근육, 피하, 자궁내 경막 또는 뇌혈관(intracerbroventricular) 주사에 의해 투여될 수 있다.
본 발명의 일실험예에서는 JC1 화합물들, 바람직하게는 JC1-38, JC1-40 및 JC1-42가 RORα의 전사에 뛰어난 활성을 가지는 것으로 확인하였다(실험예 1 및 2 참조).
본 발명의 다른 일실험예에서는 상기 JC1 화합물들이 LXRα 및 SREBP-1의 전사활성을 감소시키는 것을 확인하였고(실험예 3 참조), 또 다른 일실험예에서는 LXRα, SREBP-1 및 FAS의 단백질 발현을 억제하는 것으로 확인하였다(실험예 4 참조). 다시말해, 본 발명의 JC1 화합물들이 지방산 합성을 일으키는 유전자들의 전사 활성 뿐 아니라 단백질 발현 또한 조절한다는 사실을 확인하였다.
본 발명의 또 다른 일실험예서는 상기 JC1 화합물들이 ACC의 인산화를 증가시켜, ACC의 불활성화를 촉진함으로써 지방산의 산화를 증가시키는 것으로 확인하였고(실험예 5 참조), 또 다른 일실험예에서는 JC1 화합물들이 간세포 내의 지질 축적을 억제시키는 것으로 확인하였다(실험예 6 참조).
상기 결과로부터, RORα 유전자를 활성화시키는 신규한 화합물, 예컨대 JC1 화합물들은 대사성 질환 또는 염증성 질환의 치료에 적용될 수 있을 것으로 기대된다.
이하, 본 발명의 이해를 돕기 위하여 바람직한 실시예를 제시한다. 그러나 하기의 실시예는 본 발명을 보다 쉽게 이해하기 위하여 제공되는 것일 뿐, 하기 실시예에 의해 본 발명의 내용이 한정되는 것은 아니다.
[실시예]
실시예 1. 1-(2-브로모-벤질)-3-메틸-thiourea (JC1-1) 제조방법
반응식 1과 같이 2-브로모벤질아민 (100mg, 0.54mmol)에 Methylen chloride 용매하에 TEA(TriEthylAmine, 0.09ml, 0.64mmol)을 넣은 후, N-methyl isothiocyanate(0.08ml, 1.18mmol)을 넣은 후 교반하였다. 그 후 시간이 지남에 따라 흰색 고체 결정이 석출되는 것을 볼 수 있었다. TLC(Thin layer chromatography) 상에 기질이 없어짐을 확인 후, 감압 여과기로 고체를 여과하여 백색 고체(101mg, 0.39mmol)를 얻었다. (수율: 72%)
1H-NMR (300MHZ, CDCl3) δ7.53 (m, 2H), 7.45(m, 1H) 7.15 (m, 1H), 4.78 (s, 2H), 2.99 (s, 3H)
실시예 2. 1-(4-브로모-벤질)-3-메틸-thiourea (JC1-2) 제조방법
상기 실시예 1과 같은 방법으로 4-브로모벤질아민을 사용하여 백색 고체(30mg, 0.12mmol)를 얻었다. (수율: 52%)
1H-NMR (300MHZ, CD3OD) δ7.46-7.42 (m, 2H), 7.24-7.21(d, J= 11.1 Hz, 2H), 4.67 (s, 2H), 2.93(s, 3H)
실시예 3. 1-(4-클로로-벤질)-3-메틸-thiourea (JC1-3) 제조방법
반응식 1과 같이 4-클로로벤질아민 (200mg, 1.41mmol)를 diethyl ether 용매(3ml)하에 N-methylisothiocyante (320mg, 3.10mmol)을 넣은 후 교반하였다. 그 후 시간이 지남에 따라 고체 결정이 석출되는 것을 볼 수 있었다. TLC 상에 기질이 없어짐을 확인 후, 감압 여과기로 고체를 여과하여 백색 고체(213.6mg, 1.00mmol)를 얻었다. (수율: 71%)
1H-NMR (300MHZ, CDCl3) δ7.32-7.24(m , 4H), 7.45(m, 1H) 4.67 (m, 2H), 2.97 (m, 3H)
실시예 4. 1-(4-메톡시-벤질)-3-메틸-thiourea (JC1-4) 제조방법
상기 실시예 3과 같은 방법으로 4-메톡시벤질아민을 사용하여 백색 고체(259.6mg, 1.23mmol)를 얻었다. (수율: 57%)
1H-NMR (300MHZ, CDCl3) δ7.20 (m , 2H), 6.86(m, 2H) 4.55 (m, 2H), 3.77 (m, 3H), 2.94 (m, 3H)
실시예 5. 1-(2, 4-다이메톡시-벤질)-3-메틸-thiourea(JC1-5) 제조방법
상기 실시예 3과 같은 방법으로 2,4-다이메톡시벤질아민을 사용하여 백색 고체(122.3mg, 0.51mmol)를 얻었다. (수율: 85%)
1H-NMR (300MHZ, CD3OD) δ7.24-7.15 (m, 1H), 6.43-6.40(m, 2H) 4.46 (s, 2H), 3.80-3.76 (d, J = 12.1 Hz, 6H), 2.93 (s, 3H)
실시예 6. 1-(3-메틸-벤질)-3-메틸-thiourea(JC1-6) 제조방법
상기 실시예 3과 같은 방법으로 3-메틸벤질아민을 사용하여 백색 고체(144mg, 0.74mmol)를 얻었다. (수율: 99%)
1H-NMR (300MHZ, CDCl3) δ7.36 (m, 2H), 7.21(m, 2H) 4.71 (s, 2H), 3.09 (d, 3H), 2.45 (s, 3H)
실시예 7. 1-(2-메틸-벤질)-3-메틸-thiourea(JC1-7) 제조방법
상기 실시예 3과 같은 방법으로 2-메틸벤질아민을 사용하여 백색 고체(167mg, 1.16mmol)를 얻었다. (수율: 99%)
1H-NMR (400MHZ, CD3OD) δ7.18(m, 4H), 4.59(s, 2H), 2.86(s, 3H), 2.27(s, 3H)
실시예 8. 1-(벤조[1,3]다이옥실-5-메틸)-3-메틸-thiourea(JC1-8) 제조방법
상기 실시예 3과 같은 방법으로 벤조[1,3]다이옥실-5-메틸아민을 사용하여 노란색 고체(132mg, 0.59mmol)를 얻었다. (수율: 89%)
1H-NMR (300MHZ, DMSO) δ7.13-6.75(m, 3H), 5.97(s, 2H), 4.53(s, 2H), 2.50(s, 3H)
실시예 9. 1-(4-하이드록시-3-메톡시-벤질)-3-메틸-thiourea (JC1-9) 제조방법
상기 실시예 1과 같은 방법으로 4-하이드록시-3-메톡시벤질아민을 사용하여 백색 고체(953mg, 4.21mmol)를 얻었다. (수율: 80%)
1H-NMR (300MHZ, DMSO) δ8.85 (s, 1H), 6.90(s, 1H) 6.70(s, 1H), 4.50 (s, 2H), 3.74(s, 3H) 2.83(s, 3H)
실시예 10. 1-(4-다이메틸아미노-벤질)-3-메틸-thiourea (JC1-10) 제조방법
상기 실시예 1과 같은 방법으로 4-다이메틸아미노벤질아민을 사용하여 노란색 고체(352mg, 1.58mmol)를 얻었다. (수율: 47%)
1H-NMR (400MHZ, DMSO) δ7.13(m, 2H), 6.68(m, 2H), 4.48(s, 2H), 2.85(m, 9H)
실시예 11. 1-(4-트리플루오르메틸-벤질)-3-메틸-thiourea (JC1-11) 제조방법
상기 실시예 3과 같은 방법으로 4-트리플루오르메틸벤질아민을 사용하여 노란색 고체(82mg, 0.36mmol)를 얻었다. (수율: 55%)
1H-NMR (400MHZ, CDCl3) δ7.61-7.28(d, J = 8.3 Hz, 2H), 6.68(d, J = 8.0 Hz 2H), 4.82-4.80(d, J = 5.0 Hz, 2H), 2.98-2.97(d, J = 5.0 Hz 3H)
실시예 12. 1-(4-나이트로-벤질)-3-메틸-thiourea (JC1-12) 제조방법
상기 실시예 1과 같은 방법으로 4-나이트로벤질아민을 사용하여 노란색 고체(132mg, 0.59mmol)를 얻었다. (수율: 89%)
1H-NMR (300MHZ, DMSO) δ8.20-8.17(d, J = 8.4 Hz, 2H), 7.53-7.50(d, J = 8.4 Hz 2H), 4.79(s, 2H), 2.83(s, 3H)
실시예 13. 1-(4-t-부틸-벤질)-3-메틸-thiourea (JC1-13) 제조방법
상기 실시예 3과 같은 방법으로 4-t-부틸벤질아민을 사용하여 백색 고체(144mg, 0.61mmol)를 얻었다. (수율: 99%)
1H-NMR (300MHZ, DMSO) δ7.38-7.35(d, J = 8.2 Hz, 2H), 7.53-7.50(d, J = 8.1 Hz, 2H), 4.61(s, 2H), 2.97-2.96(d, J = 4.6 Hz, 3H), 1.3(s, 9H)
실시예 14. 1-(4-메틸-벤질)-3-메틸-thiourea (JC1-14) 제조방법
상기 실시예 3과 같은 방법으로 4-메틸벤질아민을 사용하여 백색 고체(110mg, 0.57mmol)를 얻었다. (수율: 69%)
1H-NMR (300MHZ, CDCl3) δ7.29-7.21(m, 4H), 4.67(s, 2H), 3.03-3.01(d, J = 4.2 Hz, 3H), 2.42(s, 3H)
실시예 15. 1-(2, 3-다이메톡시-벤질)-3-메틸-thiourea (JC1-15) 제조방법
상기 실시예 3과 같은 방법으로 2,3-다이메톡시벤질아민을 사용하여 백색 고체(65mg, 0.27mmol)를 얻었다. (수율: 45%)
1H-NMR (300MHZ, CDCl3) δ7.27-7.05(m, 3H), 4.78(s, 2H), 4.09-4.07(d, J = 5.9 Hz, 6H), 3.16-3.15(d, J = 5.5 Hz, 3H)
실시예 16. 1-(2-에톡시-벤질)-3-메틸-thiourea (JC1-16) 제조방법
상기 실시예 3과 같은 방법으로 2-에톡시벤질아민을 사용하여 노란색 오일(128mg, 0.57mmol)을 얻었다. (수율: 86%)
1H-NMR (300MHZ, CD3OD) δ7.20-7.16(m, 2H), 6.90-6.82(m, 2H), 4.62(s, 2H), 4.08-4.01(q, J = 13.9 Hz, 2H), 2.92(s, 3H), 1.41-1.37(t, J = 7 Hz, 3H)
실시예 17. 1-(3-나이트로-벤질)-3-메틸-thiourea(JC1-17) 제조방법
상기 실시예 1과 같은 방법으로 3-나이트로벤질아민을 사용하여 노란색 고체(113mg, 0.50mmol)를 얻었다. (수율: 77%)
1H-NMR (300MHZ, CD3OD) δ8.18(s, 1H), 8.11-8.09(d, J = 7.7 Hz, 1H), 7.73-7.71(d, J = 7.5 Hz, 1H), 7.57-7.52(t, J = 7.9 Hz, 1H), 4.86(s, 2H), 2.95(s, 3H)
실시예 18. 1-(2-나이트로-벤질)-3-메틸-thiourea(JC1-18) 제조방법
상기 실시예 1과 같은 방법으로 2-나이트로벤질아민을 사용하여 노란색 고체(64mg, 0.28mmol)를 얻었다. (수율: 43%)
1H-NMR (300MHZ, CD3OD) δ8.01-7.99(m, 1H), 7.63-7.54(m, 2H), 7.46-7.40(m, 1H), 4.97(s, 2H), 2.90(s, 3H)
실시예 19. 1-(3, 4, 5-트리메톡시-벤질)-3-메틸-thiourea(JC1-19) 제조방법
상기 실시예 3과 같은 방법으로 3,4,5-트리메톡시벤질아민을 사용하여 백색 고체(126mg, 0.47mmol)를 얻었다. (수율: 92%)
1H-NMR (300MHZ, DMSO) δ6.64(d, J = 2.2 Hz, 2H), 6.36-6.34(t, J = 2.2 Hz, 2H), 4.62(s, 2H), 3.74(s, 6H), 2.94(s, 3H)
실시예 20. 1-(3, 5-다이메톡시-벤질)-3-메틸-thiourea(JC1-20) 제조방법
상기 실시예 3과 같은 방법으로 3,5-다이메톡시벤질아민을 사용하여 백색 고체(101mg, 0.42mmol)를 얻었다. (수율: 70%)
1H-NMR (300MHZ, CD3OD) δ6.48-6.47(d, 2H), 4.55(s, 2H), 3.74(s, 6H), 3.62(s, 3H), 2.84(s, 3H)
실시예 21. 1-(2, 4-다이클로로-벤질)-3-메틸-thiourea (JC1-21) 제조방법
상기 실시예 3과 같은 방법으로 2,4-다이클로로벤질아민을 사용하여 백색 고체(101mg, 0.54mmol)를 얻었다. (수율: 94%)
1H-NMR (300MHz, CDCl3): δ10.04(s, 1H), 8.13(d, 2H, J=8.40 Hz), 7.95(m, 3H), 7.43(d, 1H, J=3.30)
실시예 22. 1-(3, 4-다이하이드록시-벤질)-3-메틸-thiourea (JC1-22) 제조방법
반응식 1과 같이 3,4-다이하이드록시벤질아민(50mg, 0.23mmol)를 Dimethylforamide 용매하에서 TEA(TriEthylAmine, 0.04ml, 0.27mmol)을 넣은 후, N-methylisothiocyante(0.03ml, 0.50mmol)을 넣은 후, 교반하였다. TLC 상에 기질이 없어짐을 확인 후, 용매를 날리고 Methylen chloride를 사용하여 감압 여과기로 고체를 여과하여 노란색 고체(29mg, 0.14mmol)를 얻었다. (수율: 58%)
1H-NMR (300MHZ, CD3OD) δ6.76-6.60(m, 3H), 4.50(s, 2H), 2.98(s, 3H)
실시예 23. 1-(2, 4-다이메톡시-벤질)-3-메틸-thiourea (JC1-23) 제조방법
상기 실시예 3과 같은 방법으로 2,4-다이메톡시벤질아민을 사용하여 백색 고체(122.3mg, 0.51mmol)를 얻었다. (수율: 85%)
1H-NMR (300MHZ, CD3OD) δ7.24-7.15 (m, 1H), 6.43-6.40(m, 2H) 4.46 (s, 2H), 3.80-3.76 (d, J = 12.1 Hz, 6H), 2.93 (s, 3H)
실시예 24. 1-(4-페닐-벤질)-3-메틸-thiourea(JC1-24) 제조방법
상기 실시예 3과 같은 방법으로 4-페닐벤질아민을 사용하여 백색 고체(364mg, 1.42mmol)를 얻었다. (수율: 87%)
1H-NMR (300MHz, CDCl3): δ8.61(dd, 1H, J=1.37, 4.67 Hz), 7.96(d, 2H, J=8.43 Hz), 7.83(m, 4H), 7.57(d, 2H, J=8.79 Hz), 7.38(d, 2H, J=8.61 Hz), 7.26(m, 1H), 1.31(s, 9H)
실시예 25. 1-(3-하이드록시-4-메톡시-벤질)-3-메틸-thiourea(JC1-25) 제조방법
상기 실시예 1과 같은 방법으로 3-하이드록시-4-메톡시벤질아민을 사용하여 백색 고체(472mg, 2.09mmol)를 얻었다. (수율: 64%)
1H-NMR (300MHZ, CD3OD) δ6.82-6.68(m, 3H), 4.50(s, 2H), 3.77(s, 3H), 2.89(s, 3H)
실시예 26. 1-(4-시아노-벤질)-3-메틸-thiourea(JC1-26) 제조방법
상기 실시예 1과 같은 방법으로 4-시나오벤질아민을 사용하여 노란색 고체(100mg, 0.49mmol)를 얻었다. (수율: 82%)
1H-NMR (300MHZ, CDCl3) δ7.79-7.76 (d, J = 8.2 Hz, 2H), 7.45-7.43(d, J = 8.1 Hz 5H) 4.74(s, 2H) 2.83(s, 4H)
실시예 27. 1-(3, 4-다이클로로-벤질)-3-메틸-thiourea(JC1-27) 제조방법
상기 실시예 3과 같은 방법으로 3,4-다이클로로벤질아민을 사용하여 백색 고체(56mg, 0.23mmol)를 얻었다. (수율: 39%)
1H-NMR (300MHZ, CD3OD) δ7.47-7.42(m, 2H), 7.79-7.76 (dd, J = 8.3 Hz, 1H), 4.66(s, 2H), 2.84(s, 3H)
실시예 28. 1-(3-클로로-벤질)-3-메틸-thiourea(JC1-28) 제조방법
상기 실시예 3과 같은 방법으로 3-클로로벤질아민을 사용하여 백색 고체(156mg, 0.70mmol)를 얻었다. (수율: 90%)
1H-NMR (400MHZ, DMSO) δ7.36-7.23(m, 4H), 7 4.70(s, 2H), 2.93(s, 3H)
실시예 29. 1-(3-플루오르-벤질)-3-메틸-thiourea(JC1-29) 제조방법
상기 실시예 3과 같은 방법으로 3-플루오르벤질아민을 사용하여 노란색 고체(269mg, 1.36mmol)를 얻었다. (수율: 75%)
1H-NMR (300MHZ, CD3OD) δ7.38-7.30(m, 1H), 7.13-7.00(m, 3H), 4.67(s, 2H), 2.83(s, 3H)
실시예 30. 1-(4-플루오르-벤질)-3-메틸-thiourea(JC1-30) 제조방법
상기 실시예 3과 같은 방법으로 4-플루오르벤질아민을 사용하여 백색 고체(64mg, 0.32mmol)를 얻었다. (수율: 81%)
1H-NMR (300MHZ, CD3OD) δ7.35-7.30(m, 2H), 7.05-6.99(m, 2H)4.68(s, 2H), 2.93(s, 3H)
실시예 31. 1-(3, 4-다이플루오르-벤질)-3-메틸-thiourea(JC1-31) 제조방법
상기 실시예 3과 같은 방법으로 3,4-다이플루오르벤질아민을 사용하여 백색 고체(84mg, 0.39mmol)를 얻었다. (수율: 55%)
1H-NMR (400MHZ, DMSO) δ7.40-7.13(m, 3H), 4.63(s, 2H), 2.83(s, 3H)
실시예 32. 1-(2, 4-다이플루오르-벤질)-3-메틸-thiourea(JC1-32) 제조방법
상기 실시예 3과 같은 방법으로 2,4-다이플루오르벤질아민을 사용하여 노란색 고체(199mg, 0.92mmol)를 얻었다. (수율: 99%)
1H-NMR (400MHZ, CD3OD) δ7.42-7.37(m, 1H), 6.93-6.88(m, 2H), 4.72(s, 2H), 2.94(s, 3H)
실시예 33. 1-(2, 3-다이클로로-벤질)-3-메틸-thiourea (JC1-33) 제조방법
상기 실시예 3과 같은 방법으로 2,3-다이클로로벤질아민을 사용하여 백색 고체(107mg, 0.43mmol)를 얻었다. (수율: 75%)
1H-NMR (300MHZ, DMSO) δ7.54-7.51(m, 1H), 7.36-7.31(t, J = 7.8 Hz, 1H), 7.24-7.21 (d, J = 7.7 Hz, 1H), 4.72(s, 2H), 2.84(s, 3H)
실시예 34. 1-(2-클로로-벤질)-3-메틸-thiourea (JC1-34) 제조방법
상기 실시예 3과 같은 방법으로 2-클로로벤질아민을 사용하여 노란색 고체(134mg, 1.60mmol)를 얻었다. (수율: 88%)
1H-NMR (300MHZ, DMSO) δ7.38-7.20(m, 4H, 4.79(s, 2H), 2.96(s, 3H)
실시예 35. R-(+)-1-[1-(4-메톡시-페닐)-에틸]-3-메틸-thiourea(JC1-35) 제조방법
상기 실시예 3과 같은 방법으로 (R)-(+)-4-메톡시-a-메틸벤질아민을 사용하여 노란색 고체(181mg, 0.80mmol)를 얻었다. (수율: 99%)
1H-NMR (300MHZ, DMSO) δ7.17-7.14(d, J = 8.6 Hz, 2H), 6.88-6.85(d, J = 8.6 Hz, 1H), 5.33(s, 1H), 3.71(s, 3H), 2.83-2.81(d, J = 4.2Hz, 3H), 1.38-1.36(d, J = 6.8 Hz, 3H)
실시예 36. S-(-)-1-[1-(4-메톡시-페닐)-에틸]-3-메틸-thiourea(JC1-36) 제조방법
상기 실시예 3과 같은 방법으로 (S)-(-)-4-메톡시-a-메틸벤질아민을 사용하여 노란색 오일(170mg, 0.76mmol)을 얻었다. (수율: 99%)
1H-NMR (300MHZ, DMSO) δ7.17-7.14(m, 2H), 6.79-6.74(m, 2H), 5.21(s, 1H), 3.66(s, 3H), 2.82(s, 3H), 1.37-1.34(d, J = 6.8 Hz, 3H)
실시예 37. N-4-[(3-메틸-thioureido)-메틸]-페닐-메탄술폰아마이드(JC1-37) 제조방법
메틸아민염화수소 염(1eq)에 DMF 용매 하에 TEA(TriEthylAmine, 1.2eq) 4-metylaminobenzylisothiocyanate(1eq)를 넣은 후, 교반하였다. TLC 상에 기질이 없어짐을 확인 후, DMF를 날렸다. 그 후 에틸아세테이트로 희석한 후 브라인을 사용하여 씻어낸 후 감압증발하여 얻은 잔사로 컬럼 크로마토그래피(헥산:에틸 아세테이트 = 3:1)를 실시하였다. (수율: 80%)
1H-NMR (300MHZ, CD3OD) δ77.31-7.28(m, 2H), 7.22-7.18(m, 2H), 4.67(s, 2H), 2.98(s, 3H), 2.85(s, 3H)
실시예 38. 1-메틸-3-(4-피리디닐-2-벤질)-thiourea(JC1-38) 제조방법
반응식 2와 같이 두 가지 둥근 바닥 플라스크에 4-(2-피리딜)벤잘데하이드(97%, 1000mg, 5.46mmol)와 N-methylthiourea(4921mg, 54.6mmol)을 넣은 후, 감압하여 아르고 기체를 치환 시켰다. 그 후 무수 THF(Tetrahydrofuran, 20ml)을 용매로 넣고, 냉동 보관되어 있던 Ti(OiPr)4(2.72ml, 9.28mmol)을 넣어서 환류시켰다. TLC 상에서 기질이 다 없어지면 반응 용기를 천천히 식힌 후, 소디윰보로하이드라이드(103mg, 2.73mmol)을 넣어주었다. 그리하여 노란색의 원하는 프로덕트(512.9mg, 2mmol)를 얻었다. (수율: 37%)
1H-NMR (300MHZ, CDCl3) δ8.67-8.65(d, J = 5.0 Hz, 1H), 7.95-7.92(d, J = 8.2 Hz, 2H), 7.78-7.68(m, 2H), 7.42-7.39(d, J = 8.0 Hz, 2H), 7.21(s, 1H), 4.71(s,2H), 2.99(s, 3H)
실시예 39. 1-(2-알릴옥시-벤질)-3-메틸-thiourea (JC1-39) 제조방법
상기 실시예 37과 동일한 방법으로 2-알릴옥시벤잘데하이드를 사용하여 백색의 고체(330.2mg, 1.40mmol)을 얻었다. (수율: 45%)
1H-NMR (300MHZ, CDCl3) δ7.29-7.20(m, 2H), 6.98-6.74(m, 2H), 6.11-5.99(m, 1H), 5.43-5.27(m, 2H), 4.57(s, 2H), 4.55(s,2H), 2.91(s, 3H)
실시예 40. 1-(4-벤질옥시-벤질)-3-메틸-thiourea(JC1-40) 제조방법
상기 실시예 37과 동일한 방법으로 4-벤질옥시벤잘데하이드를 사용하여 노란색의 고체(202.7mg, 0.71mmol)을 얻었다. (수율: 30%)
1H-NMR (300MHZ, CD3OD) δ7.44-7.28 (m, 5H), 7.23-7.20(d, J = 8.4 Hz, 2H), 6.98-6.94(m, 2H), 5.07(s, 2H), 4.55(s, 2H), 2.82(s, 3H)
실시예 41. 1-(3-벤질옥시-4-메톡시-벤질)-3-메틸-thiourea(JC1-41) 제조방법
상기 실시예 37과 동일한 방법으로 3-벤질옥시-4-메톡시벤잘데하이드를 사용하여 백색의 고체(263.4mg, 0.83mmol)을 얻었다.(수율: 39%)
1H-NMR (300MHZ, DMSO) δ7.46-7.32 (m, 5H), 7.05(s, 1H), 6.92-6.90(d, J = 8.3 Hz, 1H), 6.84-6.81(d, J = 8.2 Hz, 1H), 5.02(s, 2H), 4.52(s, 2H), 3.73(s, 3H), 2.81(s, 3H)
실시예 42. 1-(4-페녹시-벤질)-3-메틸-thiourea(JC1-42) 제조방법
상기 실시예 37과 동일한 방법으로 4-페녹시벤잘데하이드를 사용하여 노란색의 고체(178.9mg, 0.66mmol)을 얻었다.(수율: 65%)
1H-NMR (300MHZ, CDCl3) δ7.33-7.23 (m, 4H), 7.11-7.06(m, 1H), 6.98-6.92(m, 4H), 4.62(s, 2H), 2.96-2.94(d, J= 4.6 Hz, 3H)
실시예 43. 1-메틸-3-나프탈렌-2-메틸-thiourea(JC1-43) 제조방법
2-브로모메틸 나프탈렌(200mg, 0.90mmol)에 메탄올 용매 하에 N-methyl thiourea( 163.1mg, 1.8mmol)를 넣은 후, 교반하였다. TLC 상에 기질이 없어짐을 확인 후, 메탄올를 감압증류하였다. 최소량의 메탄올로 다시 녹인 후 에틸아세테이트로 희석한 후 브라인으로 씻어주었다. 이 때 흰색의 고체는 여과하였다. 감압증류 후 남은 잔사를 칼럼크로마토그래피(에틸아세테이트:메탄올=7:1)를 실시하여 백색고체( 118.0mg, 0.51mmol)를 얻었다.(수율: 57%)
1H-NMR (400MHZ, DMSO) δ7.91-7.86 (m, 4H), 7.55-7.51(m, 3H), 4.71(s, 2H), 2.83(s, 3H)
<반응식3에 대한 일반적인 실험방법>
(실험 방법 1) 2-에톡시-4-브로모-벤조나이트릴
둥근바닥 플라스크에 4-플루오르-2-하이드록시벤조나이트릴( 50mg, 0.32mmol)과 DMF(2ml)을 넣어서 교반 후, 곱게 간 포타슘카보네이트(133mg, 0.96mmol)을 넣고나서 아이오도에탄(0.03ml, 0.39mmol)을 넣어준 후 교반 시켰다. TLC로 기질이 없어짐을 확인 후, DMF를 감압증류하고 에틸아세테이트로 희석한 후 브라인으로 씻어주었다. 감압증류 후 얻은 잔사로 칼럼크로마토그래피(헥산:에틸아세테이트5:1)를 실시하여 2-에톡시-4-브로모-벤조나이트릴(53.8mg)을 얻었다. (수율: 94%)
(실험 방법 2) 2-에톡시-4-아릴-벤조나이트릴
상기의 실험방법 1로 얻은 1-메틸-3-나프탈렌-2-메틸-thiourea (50 mg, 0.22mmol)와아릴 보로닉 산(32.4mg, 0.27mmol), 팔라듐(Ⅱ) 아세테이트(0.5mg. 0.0002mmol), p-아세토아세트아니시다이드(1.3mg, 0.007mmol), 세슘카보네이트(61.1mg, 0.44mmol)에 DMF(1ml)을 넣어 80℃로 워밍하여 교반하였다. 반응 액의 색이 갈색으로 변하는 것을 볼 수 있었다. TLC로 기질이 사라짐을 확인 후, DMF를 감압증류하였다. 고체로 석출되는 것은 여과하고 여액을 감압 증류하여 얻은 잔사로 칼럼크로마토그래피를 실시하여 2-에톡시-4-아릴-벤조나이트릴를 분리하여 얻었다.
(실험 방법 3) 2-에톡시-4-아릴-벤질아민
환류장치와 두 가지 둥근바닥 플라스크를 오븐에 건조한 후에 열을 식히고 장치한 후 감압하였다. 그 후 아르곤 기체 치환 상태에서 리튬알루미늄하이드라이드(212.9mg, 5.61mmol)와 무수 다이 이소프로필 이써(20ml)를 넣은 후 환류시켰다. 그 후 상기의 실험 방법 2에서 얻어진 2-에톡시-4-아릴-벤조나이트릴 (1.87mmol)을 용매에 녹여 canula를 통해서 용기에 넣어 교반하였다. TLC로 기질이 사라짐을 확인 후, 1N-HCl을 중성이 될 때까지 넣어주고 교반하였다. 그리고 로쉘 염를 적당량 넣어 30분 가량 교반하였다. 그리고 감압 여과기에 셀라이트를 깐 후에, 반응액을 부어서 여과하였다. 걸러진 여액을 1N-HCl로 산성화하여 물층으로 추출해내고, 이것을 다시 소듐바이카보네이트를 사용하여 염기성화하여 에틸아세테이트로 추출하였다. 이 과정을 여러 번 반복하여 원하는 2-에톡시-4-아릴-벤질아민를 얻어냈다.
(실험 방법 4) 1-(2-에톡시-4-아릴-벤질)-3-메틸-thiourea
상기의 실시예 1과 동일한 방법으로 실험방법 3에서 얻은 2-에톡시-4-아릴-벤질아민을 사용하여 1-(2-에톡시-4-아릴-벤질)-3-메틸-thiourea을 얻었다.
실시예 44. 1-(3-에톡시-바이페닐-4-메틸)-3-메틸-thiourea(JC1-44) 제조방법
상기 실험 방법 1,2,3,4를 거쳐 실험을 수행하였으며 실험방법 2에서 페닐 보로닉 산을 사용하여 원하는 노란색의 고체(348.2mg, 1.16mmol)를 얻었다. (전제 수율: 79%)
1H-NMR (400MHZ, CD3OD) δ7.40-7.37(d, 2H), 7.43-7.38(m, 2H), 7.33-7.26(m, 2H), 7.14-7.12(m, 2H), 4.33(s, 2H), 4.19-4.12(q, J = 13.9 Hz, 2H), 2.71(s, 3H), 1.48-1.43(t, J = 7.0 Hz, 3H)
실시예 45. (4'-tert-부틸-3-에톡시-바이페닐-4-메틸)-thiourea(JC1-45) 제조방법
상기 실험 방법 1,2,3,4를 거쳐 실험을 수행하였으며 실험방법 2에서 4-tert-부틸페닐 보로닉 산을 사용하여 원하는 노란색의 액체 (55.6mg, 0.16mmol)를 얻었다. (전제 수율: 32%)
1H-NMR (300MHZ, CDCl3) δ7.49-7.42(m, 4H), 7.33-7.30(d, J = 7.7 Hz, 1H), 7.13-7.10(m, 1H), 7.04(s, 1H), 4.61(s, 2H), 4.17-4.10(q, J = 13.9 Hz, 2H), 2.97-2.96(d, J = 3.8 Hz, 3H), 1.48-1.43(t, J= 7.0 Hz, 3H), 1.34(s, 9H)
실시예 46. 1-(3,3'-다이에톡시-바이페닐-4-메틸)-3-메틸-thiourea(JC1-46) 제조방법
상기 실험 방법 1,2,3,4를 거쳐 실험을 수행하였으며 실험방법 2에서 4-에톡시페닐 보로닉 산을 사용하여 원하는 합성물 (444mg, 1.29mmol)를 얻었다. (전제 수율: 45%)
1H-NMR (300MHZ, CDCl3) δ7.50-7.48(m, 2H), 7.37-7.32(m, 2H) 7.27-7.23(m, 3H), 7.09-7.05(m, 3H), 4.57(s, 2H), 4.19-4.12(t, J= 13.9 Hz, 3H), 2.99-2.80(d, J = 4.41 Hz, 3H), 1.59(s, 3H), 1.50-1.45(t, J = 7 Hz, 3H)
실시예 47. 1-(2-에톡시-4-스티릴-벤질)-3-메틸-thiourea(JC1-47) 제조방법
상기 실험 방법 1,2,3,4를 거쳐 실험을 수행하였으며 실험방법 2에서 4-스티릴 보로닉 산을 사용하여 원하는 노란색 고체(30mg, 0.09mmol)를 얻었다. (전제 수율: 8%)
1H-NMR (300MHZ, CDCl3) δ7.50-7.48(m, 2H), 7.37-7.32(m, 2H) 7.27-7.23(m, 3H), 7.09-7.05(m, 3H), 4.57(s, 2H), 4.19-4.12(t, J = 13.9 Hz, 3H), 2.99-2.80(d, J = 4.4 Hz, 3H), 1.59(s, 3H), 1.50-1.45(t, J = 7 Hz, 3H)
실시예 48. 1-(3-에톡시-4'-트리플루오르메틸-바이페닐-4-메틸)-3-메틸-thiourea(JC1-48) 제조방법
상기 실험 방법 1,2,3,4를 거쳐 실험을 수행하였으며 실험방법 2에서 4-트리플루오르페닐 보로닉 산을 사용하여 원하는 합성물 (93mg, 0.25mmol)를 얻었다. (전제 수율: 24%)
1H-NMR (300MHZ, CDCl3) δ7.62-7.54(dd, J = 13.4 Hz, 4H), 7.34-7.31(d, J = 13.4 Hz, 1H), 7.08-7.05(m, 1H), 6.98(s, 1H), 4.59(s, 2H), 4.14-4.05(q, J = 13.9 Hz, 2H), 2.93-2.91(d, J = 4.2 Hz, 3H), 1.44-1.40(t, J = 7 Hz, 3H)
실시예 49. 1-메틸-3-(3-페녹시-벤질)-thiourea(JC1-50) 제조방법
상기 실시예 37과 동일한 방법으로 3-페녹시 벤잘데하이드(97%, 500mg, 2.52mmol)를 사용하여 원하는 화합물 (572.7mg, 2.1 mmol)을 얻었다. (수율: 84%)
1H-NMR (400MHZ, CD3OD) δ7.33-7.25(m, 2H), 7.09-7.05(m, 2H), 6.96-6.95(m, 3H), 6.84-6.82(dd, J = 4.8 Hz, 1H), 4.68(s, 2H), 2.92(s, 3H)
실시예 50. 1-메틸-3-(2-페녹시-벤질)-thiourea(JC1-51) 제조방법
상기 실시예 37과 동일한 방법으로 2-페녹시 벤잘데하이드(900mg, 3.94 mmol)를 사용하여 원하는 화합물 (255.7mg, 0.94 mmol)을 얻었다. (수율: 24%)
1H-NMR (300MHZ, CD3OD) δ7.40-7.31(m, 3H), 7.26-7.21(m, 1H), 7.13-7.06(m, 2H), 6.98-6.95(m, 2H), 6.85-6.82(m, 1H), 4.72(s, 2H), 2.90(s, 3H)
(부가 실험방법) 2-페녹시 벤잘데하이드
반응식 4에서와 같이 상업적으로 구할 수 있는 2-플루오르벤잘데하이드 (200mg, 1.61 mmol)를 출발 물질로 하였다. DMF (3ml) 용매하에서 포타슘카보네이트(245mg, 1.77 mmol)를 베이스로 하여 페놀(152mg, 1.61 mmol)로 친핵성 첨가반응을 수행하여 원하는 2-페녹시 벤잘데하이드 (183mg, 0.92 mmol)를 합성하였다. (수율: 57%)
실시예 51. 1-[4-(4-플루오르-페녹시)-벤질]-3-메틸-thiourea(JC1-52) 제조방법
상기 실시예 37과 동일한 방법으로 4-(4-플루오르페녹시) 벤잘데하이드(500mg, 2.31mmol)를 사용하여 원하는 화합물 (447mg, 1.56 mmol)을 얻었다. (수율: 67%)
1H-NMR (300MHZ, CDCl3) δ7.28-7.26(m, 2H), 7.05-6.91(m, 6H), 4.64-4.62(d, J = 4.7 Hz, 2H), 2.98-2.97(d, J = 5.0 Hz, 3H)
실시예 52. 1-[4-(4-메톡시-페녹시)-벤질]-3-메틸-thiourea(JC1-53) 제조방법
상기 실시예 37과 같은 방법으로 4-(4-메톡시페녹시) 벤잘데하이드(500mg, 2.19mmol)를 사용하여 원하는 화합물 (553.2mg, 1.56 mmol)을 얻었다. (수율: 84%)
1H-NMR (400MHZ, CDCl3) δ7.24-7.20(m, 2H), 6.93-6.84(m, 6H), 4.58(s, 2H), 2.93(s, 3H)
[실험예]. 생물학적 효능 검사
실험예 1. Thiourea계 화합물인 JC1 화합물이 RORα의 전사활성에 미치는 효과
CV-1 세포(CCL-70)는 ATCC (American Type Culture Collection)으로부터 구입하였다. CV-1 세포 (4 x 104 세포/웰)를 24-웰 배양 플레이트에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. CV-1 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, 칼슘 포스페이트를 이용하여 세포를 RORE-tk-Luc 리포터 플라스미드 (100 ng), ROR 발현 벡터 (5 ng)로 형질전환시켰다.
HepG2 세포 (1 x 105 세포/웰)를 12-웰 플레이트에 씨딩하고 하룻밤동안 배양하였다. 세포를 Welfect-EXTM Plus (WelGENE Inc., Korea)를 이용하여 LXRE-Luc (100 ng) 또는 SRE-Luc (100 ng) 리포터 플라스미드로 형질전환시켰다. 형질 전환 24시간 후, JC1 화합물(100μM ), 용매(대조군) 등을 처리하였다. 처리한 지 24시간 후, 루시퍼라아제 활성을 Analytical luminescence luminometer를 이용하여 측정하였다. 형질전환 효율을 확인하기 위하여, 200 ng의 β-galactosidase (β-gal) 발현 벡터의 활성을 이용하여 루시퍼라아제 활성을 표준화하였다. 이에 대한 결과는 표 1과 같으며, 표 1을 도식화하여 도 3에 나타내었다.
표 1
| 화합물 | Fold Activation | 화합물 | Fold Activation |
| 1 | 3.4 | 28 | 0.7 |
| 2 | 1.4 | 29 | 1.1 |
| 3 | 2.6 | 30 | 2.5 |
| 4 | 3.4 | 31 | 0.8 |
| 5 | 1.1 | 32 | 0.9 |
| 6 | 1.1 | 33 | 0.7 |
| 7 | 0.7 | 34 | 0.8 |
| 8 | 1.7 | 35 | 0.7 |
| 9 | 1.4 | 36 | 0.6 |
| 10 | 1.3 | 37 | ND |
| 11 | 2.3 | 38 | 1.9 |
| 12 | 1.6 | 39 | 1.8 |
| 13 | 0.9 | 40 | 3.6 |
| 14 | 3.9 | 41 | 2.5 |
| 15 | 1.5 | 42 | 4.6 |
| 16 | 1.4 | 43 | 0.7 |
| 17 | 0.9 | 44 | 1.2 |
| 18 | 2.1 | 45 | 1.0 |
| 19 | 2.2 | 46 | ND |
| 20 | 1.0 | 47 | 1.2 |
| 21 | 0.6 | 48 | 1.0 |
| 22 | 0.6 | 49 | ND |
| 23 | 2.9 | 50 | ND |
| 24 | 0.2 | 51 | 1.3 |
| 25 | 1.9 | 52 | 1.5 |
| 26 | ND | 53 | 2.1 |
| 27 | 1.0 |
ND; not determined
표 1 및 도 3에 나타난 바와 같이, JC1 화합물은 RORα의 전사활성을 증가시켰다. 특히 용매만 처리한 대조군에 비해 2배 이상으로 활성을 나타낸 13개의 JC1 화합물이 관찰되었고, 이 중 JC1-42는 460%의 뛰어난 활성을 나타내는 것으로 관찰되었다.
실험예 2. JC1-38, JC1-40 및 JC1-42가 농도에 따라 RORα의 전사활성에 미치는 효과
CV-1 세포 (4 x 104 세포/웰)를 24-웰 배양 플레이트에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. CV-1 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, 칼슘 포스페이트를 이용하여 RORE-Luc 리포터 (0.05 μg)와 Flag-RORα 벡터 (0.005 μg)를 CV-1 세포에 형질전환시켰다. 형질전환 24시간 후, CV-1 세포를 JC1 화합물, 특히 상기 실험예 1에서 관찰된 14개의 JC1 화합물 중, 화학적 안정성이 뛰어난 JC1-38, JC1-40 및 JC1-42의 특정 농도(1μM, 10μM, 20μM, 50μM 및 100μM) 및 RORα의 리간드인 멜라토닌 100 μM로 24시간 동안 처리하였다. 이후 세포 용해물을 수득하고,Analytical luminescence luminometer를 이용하여 루시퍼라아제 활성을 분석하고 결정하였다. β-gal 활성으로 형질전환 효율에 대한 루시퍼라아제 활성을 표준화하였고, 데이터는 2개나 3개의 독립된 실험 결과에 대한 평균 SD이며, 그 결과를 도 4에 나타내었다.
도 4에 나타난 바와 같이, RORα의 전사활성은 JC1-38, JC1-40 및 JC1-42의 농도에 의존적으로 증가하였다. 저농도(1μM, 10μM 및 20μM)의 JC1 화합물(JC1-38, JC1-40 및 JC1-42)에 의해서는 RORα 활성이 미미한 증가를 보였으나, 50-100 μM 범위의 농도로 처리하였을 때는 멜라토닌이 나타내는 활성 이상의 효과를 나타내는 것으로 관찰되었다.
실험예 3. JC1-38, JC1-40 및 JC1-42가 LXRα 및 SREBP-1의 전사활성에 미치는 효과
인간 간암세포주 및 HepG2(ATCC HB 8065)는 ATCC (American Type Culture Collection)으로부터 구입하였다. HepG2 세포(1.2 x 105 세포/웰)를 24-웰 배양 플레이트에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. HepG2 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, Welfect-EXTM Plus (WelGENE Inc., Korea)를 이용하여 LXRE-Luc 리포터 (0.1 μg)를 HepG2 세포에 형질전환시켰다. 형질전환 24시간 후, HepG2 세포를 JC1-38, JC1-40 및 JC1-42의 특정 농도(1μM, 10μM, 20μM, 50μM 및 100μM) 및 LXRα의 리간드인 T17(T0901317) 1μM로 24시간 동안 처리하였다. 이후 세포 용해물을 수득하고, Analytical luminescence luminometer를 이용하여 루시퍼라아제 활성을 분석하고 결정하였다. β-gal 활성으로 형질전환 효율에 대한 루시퍼라아제 활성을 표준화하였고, 데이터는 3개의 독립된 실험 결과에 대한 평균 SD이며, 그 결과를 도 5a에 나타내었다.
도 5a에 나타난 바와 같이, T17으로 증가된 LXRα의 전사 활성이 JC1-38, JC1-40 및 JC1-42의 농도에 의존적으로 감소하였다. 특히, 100 μM의 농도에서 세 화합물 모두 T17으로 유도된 LXRα의 활성을 90%이상 감소시키는 것을 관찰하였다.
이에 더하여, 동일한 조건으로 HepG2 세포를 배양하고, 배양 후 Welfect-EXTM Plus (WelGENE Inc., Korea)를 이용하여 SRE-Luc 리포터 (0.1 μg)를 HepG2 세포에 형질전환시켰다. 형질전환 24시간 후, HepG2 세포를 50μM의 각각의 JC1-38, JC1-40 및 JC1-42 화합물 및 1μM의 T17(T0901317) 로 24시간 동안 처리하였다. 이후 세포 용해물을 수득하고, Analytical luminescence luminometer를 이용하여 루시퍼라아제 활성을 분석하고 결정하였다. β-gal 활성으로 형질전환 효율에 대한 루시퍼라아제 활성을 표준화하였고, 데이터는 3개의 독립된 실험 결과에 대한 평균 SD이며, 그 결과를 도 5b에 나타내었다.
도 5b에 나타난 바와 같이, JC1-38, JC1-40 및 JC1-42는 T17으로 증가된 SREBP-1의 전사 활성을 감소시키는 것을 명백히 알 수 있었다.
실험예 4. JC1-38, JC1-40 및 JC1-42가 LXRα, SREBP-1 및 FAS의 단백질 발현에 미치는 효과
HepG2 세포(1 x 106 세포/웰)를 60- 디쉬에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. HepG2 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, HepG2 세포를 100μM의 각각의 JC1-38, JC1-40 및 JC1-42 화합물로 처리하였고, 양성 대조군으로서 AMPK 활성자인 metformin 2mM을 24시간 처리하였다. 처리 후 웨스턴 블롯팅 분석법으로 단백질의 발현을 분석하였다. 즉, 처리 후, 50 mM NaCl, 50 mM Tris pH 7.4, 5 mM EDTA, 1% NP-40 및 프로테아제 억제제를 포함하는 용해 완충액에서 얼음 위 30분 동안 HepG2 세포를 파괴하고, 원심분리하여 전체 세포 용해액을 얻었다. 전체 세포 용해액으로부터 얻은 20-30 μg 단백질을 7-9% SDS-PAGE (sodium dodecylsulfate-polyacrylamide gel electrophoresis) 하고, 폴리비닐리덴 디플루오리드 막 (Millipore, Bedford, MA, USA)에 전이시켰다. 0.1% Tween-20을 포함하는 PBS내의 5% 또는 10% (w/v) 비지방 건조 밀크로 블롯킹을 실시하고, RORα (Affinity BioReagents), LXRα, SREBP-1, FAS (Santa Cruz Biotechnology), phospho-ACC (Cell signaling), α-tubulin (Calbiochem)에 대한 특정 항체와 반응시켰다. HRP (horseradish peroxidase)-접합 2차 항체 (Zymed Lab)를 이용, 면역반응성 단백질을 Amersham ECL Western Blotting Detection Reagents로 검출하였다. 단백질 농도는 BCA (bicinchoninic acid) (Pierce) 분석으로 정량화하였고, α-튜블린의 발현을 대조군으로 모니터링하였다. 그 결과를 도 6a에 나타내었다.
도 6a에 나타난 바와 같이, JC1-40과 JC1-42에 의해 LXRα와 SREBP-1 및 FAS의 단백질 발현이 감소되었고, JC1-38에 의해 LXRα와 SREBP-1 단백질의 발현이 감소되는 것을 관찰하였다.
이에 더하여, 동일한 조건으로 HepG2 세포를 배양하고, 배양 후, HepG2 세포를 특정 농도(0μM, 1μM, 10μM, 50μM 및 100μM)의 JC1-40(또는 JC1-42) 및 1μM의 T17로 24시간 처리하였다. 처리 후, 웨스턴 블롯팅 분석법으로 단백질의 발현을 분석하였고, α-튜블린의 발현을 대조군으로 모니터링하였다. 그 결과를 도 6b와 도 6c에 나타내었다.
도 6b에 나타난 바와 같이, T17에 의해 증가된 SREBP-1 및 FAS 발현이 JC1-40의 농도에 의존적으로 감소하였다. 또한 도 6c에 나타난 바와 같이, T17에 의해 증가된 LXRα, SREBP-1 및 FAS 발현이 JC1-42의 농도에 의존적으로 감소하였다. 특히, JC1-40 및 JC1-42는 50-100 μM 범위의 농도에서 단백질의 발현을 강하게 억제한다는 것을 관찰하였다(도 6b 및 도 6c 참조).
실험예 5. JC1-38, JC1-40 및 JC1-42가 ACC의 인산화에 미치는 효과
HepG2 세포(1 x 106 세포/웰)를 60- 디쉬에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. HepG2 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, HepG2 세포를 100M의 JC1-38와 JC1-40(또는 100μM의 JC1-38와 JC1-42), 2mM의 metfornin 및 2mM의 AICIR을 24시간 처리하였다. 처리 후 RORα와 인산화된 ACC(pACC)단백질의 발현을 웨스턴 블롯팅으로 분석하였고, α-튜블린의 발현을 대조군으로 모니터링하였다. 그 결과를 도 7(a, b)에 나타내었다.
도 7(a, b)에 나타난 바와 같이, ACC의 인산화는 JC1-40과 JC1-42에 의해 증가된다.
실험예 6. JC1-38, JC1-40 및 JC1-42가 간세포 내의 지방산 산화에 미치는 영향
HepG2 세포(1 x 106 세포/웰)를 60- 디쉬에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. HepG2 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, HepG2 세포를 100μM의 JC1-38, JC1-40과 JC1-42를 24시간 처리하였다. 처리 후 real-time RT-PCR (real-time reverse transcriptase-polymerase chain reaction; 정량 중합효소 연쇄반응) 분석법으로 전사 활성에 미치는 효과를 분석하였다. PCR반응은 지방산 산화에 관여하는 MCAD, ACO1, ACO2, HMGCS2, CPT-1과 ACS에 다음과 같이 각각 특이적인 primer를 사용해서 수행하였다; MCAD (forward: 5′-CTACCAAGTATGCCCTGGAAAG-3′서열번호1, reverse: 5′-TGTGTTCACGGGCTACAATAAG-3′서열번호2), ACO1 (forward: 5′-GGGCATGGCTATTCTCATTGC-3′서열번호3, reverse: 5′-CGAACAAGGTCAACAGAAGTTAGGTTC-3′서열번호4), ACO2 (forward: 5′-GCGGACATGGCTACTCAAAGC-3′서열번호5, reverse: 5′-GCAGTGCACCTTAGCAGCCTG-3′서열번호6), HMGCS2 (forward: 5′-GGAACCCATATGGAGAATGTGT-3′서열번호7, reverse: 5′-TCCTGAGAGGCCTTTAGAAGTG-3′서열번호8), CPT1 (forward: 5′-AGACGGTGGAACAGAGGCTGAAG-3′서열번호9, reverse: 5′-TGAGACCAAACAAAGTGATGATGTCAG-3′서열번호10), ACS (forward: 5′-AGCAGAGCTTCGCAGCGGC-3′서열번호11, reverse: 5′-TCTGCTGTTTTCGCTGGGTCC-3′서열번호12) 과 β-actin (5′-CGTGGGCCGCCCTAGGCACCA-3′서열번호13, reverse: 5′-TTGGCTTAGGGTTCAGGGGGG-3′서열번호14).
도 8에 나타난 바와 같이, JC1-38, JC1-40및 JC1-42에 의해 HepG2세포에서 지방산 산화에 관여하는 MCAD, ACO1, ACO2, HMGCS2, CPT-1및 ACS의 전사 활성이 증가한다는 것을 관찰하였다.
실험예 7. JC1-38, JC1-40 및 JC1-42가 간세포의 지방산 흡수 및 초저비중지질단백 분비에 미치는 영향
HepG2 세포(1 x 106 세포/웰)를 60- 디쉬에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. HepG2 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, HepG2 세포를 100μM의 JC1-38, JC1-40과 JC1-42를 24시간 처리하였다. 처리 후 real-time RT-PCR (real-time reverse transcriptase-polymerase chain reaction; 정량 중합효소 연쇄반응) 분석법으로 전사 활성에 미치는 효과를 분석하였다. PCR반응은 지방간 흡수 (fatty acid uptake) 에 관여하는 CD36 (도 9a)과 초저비중지질단백 분비에 관여하는 MTTP와 ApoB100 (도 9b) 에 다음과 같이 각각 특이적인 primer를 사용해서 수행하였다; CD36 (forward: 5′-GGAACTGTGGGCTCATTGC-3′서열번호15, reverse: 5′-CATGAGAATGCCTCCAAACAC-3′서열번호16), MTTP (forward: 5′-CCTTCATTCAGCACCTCA-3′서열번호17, reverse: 5′-TGACAAGTGTCCCAGTGA-3′서열번호18), ApoB100 (forward: 5′-TAAATGGAGCACTTTTCAAG-3′서열번호19, reverse: 5′-GGAACAGCAGCAGTAGCG-3′서열번호20), 과 β-actin (5′-CGTGGGCCGCCCTAGGCACCA-3′서열번호13, reverse: 5′-TTGGCTTAGGGTTCAGGGGGG-3′서열번호14).
도9 (a,b)에 나타난 바와 같이, JC1-38, JC1-40및 JC1-42에 의해 HepG2세포에서 지방간 흡수에 관여하는 CD36의 전사 활성이 감소한다는 것과 초저비중지질단백 분비에 관여하는 MTTP와 ApoB100의 전사 활성이 증가한다는 것을 관찰하였다.
실험예 8. JC1-40 및 JC1-42가 간세포 내의 지질 축적에 미치는 효과
HepG2 세포(3 x 105 세포/웰)를 60- 디쉬에 씨딩하고, 10% FBS (fetal bovine serum)를 함유하는 DMEM (Dulbecco's modified Eagle's medium) 배지에서 하룻밤동안 배양하였다. HepG2 세포는 5% CO2 및 95% 공기를 갖는 함습 항온기에서 37℃로 유지하였다. 배양 후, HepG2 세포에 50μM의 JC1-40(또는 JC1-42)을 지질 생성 유도 물질로서 0.5 mM의 올레익산과 0.5 mM의 팔미틱산을 2:1 비율로 섞은 지방산 믹스처(5 mM의 oleic acid와 5 mM의 palmitic acid는 1% fatty acid-free bovine serum albumin (BSA)이 포함된 배지에서 섞어 최종 농도로 희석하여 사용함)와 함께 3일간 처리하였다. 3일 후, 세포 용해액을 수득하여 1 μg/ml의 나일 레드로 15분간 상온에서 염색하고, 트리글리세라이드 농도를 FACSCaliburTM machine (BD Biosciences)을 이용하여 플로우 사이토메트리법으로 측정하였다. 형광 방출 파장은 500 nm및 580 nm 사이에서 측정하였다. 그 결과를 도 10(a, b)에 나타내었다.
도 10(a, b)에 나타난 바와 같이, 가장 왼쪽 선은 용매만 처리한 대조군, 중간 선은 JC1-40또는 JC1-42를 지방산과 함께 처리한 실험군, 그리고 가장 오른쪽 선은 지방산만 처리한 대조군을 나타내고, JC1-40 또는 JC1-42 처리에 의해 간세포내의 지질의 축적이 현저히 감소함을 관찰하였다.
실험예 9. JC1-40 및 JC1-42가 고지방식이-유도 마우스 지방간에서 AMPK와 ACC의 인산화 및 LXRα, SREBP-1 및 FAS의 단백질 발현에 미치는 효과
도 11(a)에 나타난 바와 같이, 6주령의 검정 실험 동물 마우스에 5일 동안 홍화유를 포함하는 고지방식이를 하였다. 식이 3일 후에 JC1-40과 JC1-42을 0.5% carboxymethyl cellulose에 희석하여 10, 30 mg/kg/day의 농도로 2일간 구강투여하면서 홍화유를 포함하는 고지방식이를 하였다. 식이 5일이 지난 후, 마우스를 희생시켜 간 조직을 얻어 웨스턴 블롯팅 분석법으로 단백질의 발현을 분석하였다. 즉, 처리 후, 50 mM NaCl, 50 mM Tris pH 7.4, 5 mM EDTA, 1% NP-40 및 프로테아제 억제제를 포함하는 용해 완충액에서 균질화하고, 원심분리하여 전체 세포 용해액을 얻었다. 전체 세포 용해액으로부터 얻은 20-30 μg 단백질을 7-9% SDS-PAGE (sodium dodecylsulfate-polyacrylamide gel electrophoresis) 하고, 폴리비닐리덴 디플루오리드 막 (Millipore, Bedford, MA, USA)에 전이시켰다. 0.1% Tween-20을 포함하는 PBS내의 5% 또는 10% (w/v) 비지방 건조 밀크로 블롯킹을 실시하고, RORα (Affinity BioReagents), LXRα, SREBP-1, FAS (Santa Cruz Biotechnology), phospho-AMPK, phospho-ACC (Cell signaling), α-tubulin (Calbiochem)에 대한 특정 항체와 반응시켰다. HRP (horseradish peroxidase)-접합 2차 항체 (Zymed Lab)를 이용, 면역반응성 단백질을 Amersham ECL Western Blotting Detection Reagents로 검출하였다. 단백질 농도는 BCA (bicinchoninic acid) (Pierce) 분석으로 정량화하였고, α-튜블린의 발현을 대조군으로 모니터링하였다. 그 결과를 도 11b에 나타내었다.
도 11b에 나타난 바와 같이, AMPK와 ACC의 인산화가 JC1-40과 JC1-42에 의해 증가하는 것과 고지방식이로 인해 증가한 LXRα와 SREBP-1 및 FAS의 단백질 발현이 JC1-40 및 JC1-42에 의해 감소되는 것을 관찰하였다.
실험예 10. JC1-40 및 JC1-42가 고지방식이-유도 마우스 지방간에서 간 내 중성 지방과 간 내 지방 축적 및 체중 변화에 미치는 영향
도 11(a)에 나타난 바와 같이, 6주령의 검정 실험 동물 마우스에 5일 동안 홍화유를 포함하는 고지방식이를 하였다. 식이 3일 후에 JC1-40및 JC1-42을 0.5% carboxymethyl cellulose에 희석하여 10, 30 mg/kg/day의 농도로 2일간 구강투여하면서 홍화유를 포함하는 고지방식이를 하였다. 식이 5일이 지난 후, 마우스를 희생시켜 간 조직을 얻어 EnzyChromTM Triglyceride Assay Kit (BioAssay Systems)를 이용하여 간 내 중성 지방량을 측정하였다. 도 12(a)에 나타난 바와 같이, 고지방식이로 인해 증가한 간 내 중성 지방량이 JC1-40 및 JC1-42에 의해 감소되는 것을 관찰하였다.
이에 더하여 마우스의 간 조직을 얻어 간 내 지방 축적을 확인할 수 있는 Oil-Red O 염색을 수행하였다. 도 12(b)에 나타난 바와 같이, 고지방식이로 인해 증가한 간 내 지방 축적이 JC1-40 및 JC1-42에 의해 감소되는 것을 관찰하였다.
고지방식이 5일이 지난 후, 마우스의 체중을 측정해본 결과, 도 12(c)에 나타난 바와 같이 고지방식이로 인해 증가한 마우스의 체중이 JC1-40및 JC1-42에 의해 감소되는 것을 관찰하였다.
실험예 11. 고지방식이-유도 마우스 지방간 모델에서 JC1-40 및 JC1-42에 의한 혈액 생화학적 분석
도 11(a)에 나타난 바와 같이, 6주령의 검정 실험 동물 마우스에 5일 동안 홍화유를 포함하는 고지방식이를 하였다. 식이 3일 후에 JC1-40 및 JC1-42을 0.5% carboxymethyl cellulose에 희석하여 10, 30 mg/kg/day의 농도로 2일간 구강투여하면서 홍화유를 포함하는 고지방식이를 하였다. 식이 5일이 지난 후, 마우스를 희생시켜 혈액을 체취하여 serum을 분리하였다.
도 13에 나타난 바와 같이, 혈중 포도당과 혈중 전체 콜레스테롤의 양은 고지방식이와 비교하여 JC1-40 및 JC1-42에서 차이가 없는 것을 관찰하였으며, 혈중 중성지방의 양은 고지방식이에 의해 증가하였던 것이 JC1-40과 JC1-42에 의해 감소하는 것을 관찰하였다. 또한 고비중지질단백의 양은 고지방식이에 비해서 30 mg/kg/day를 처리한 JC1-40 및 JC1-42에 의해 증가하는 것을 관찰하였다. 간기능을 나타내는 GPT와 GOT는 고지방식이에 비해서 30 mg/kg/day를 처리한 JC1-40군에서 감소하는 것을 관찰하였다.
실험예 12. JC1-40 및 JC1-42가 혈관평활근세포 증식에 미치는 효과
rat smooth muscle cell line인 A7r5는 ATCC (American Type Culture Collection)으로부터 구입하였다. A7r5는 10% fetal bovine serum, penicillin (100 U/ml)과 streptomycin (100 μg/ml)이 첨가된 Dulbecco's modified Eagle's medium을 사용하여 5% CO2에서 배양하여 실험에 사용하였다. A7r5 세포(2 x 103 세포/웰)을 96웰 배양 플레이트에 씨딩하고, 하룻밤 동안 배양하였다. 배양 후, A7r5 세포에 JC1-40 및 JC1-42 화합물 각 100 μΜ을 2, 4 및 6일 동안 처리하였다. 이후 MTT (3-4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) 2 mg/ml 50 μl를 4 시간 처리한 후, medium을 제거하고 DMSO 200 μl를 넣었다. 플레이트를 5 분 정도 섞어준 후, 595 nm에서 흡광도를 측정하였다. 데이터는 3개의 독립된 실험 결과에 대한 평균과 SD 값이며, 그 결과를 도 14에 나타내었다.
도 14에 나타난 바와 같이, JC1-40 및 JC1-42 처리에 의해 혈관 평활근세포의 세포 증식이 감소하는 것을 관찰하였다 (도 14 참조).
실험예 13. JC1-40 이 풍선 확장술 이후 혈관 내층 증식에 미치는 효과
JC1-40에 의한 혈관 협착의 저해를 알아보기 위해 백서에 일 동안 JC1-40을 투여한 후 풍선확장술을 실시하고 그 이후 다시 14일 동안 투여하였다. 시험군은 JC1-40을 5 mg/kg (in 0.5% carboxymethyl cellulose, 2 ml/kg) (n=5)의 농도로 경구투여 하였으며, 대조군은 0.5% carboxymethlycellulose, 2 mg/kg (n=5)의 농도로 경구투여 하였다. 풍선 확장술은 다음과 같이 실시하였다. 백서의 경부를 절개하여 common carotid artery를 노출시키고 external branch와 internal branch로 갈라지는 부위에서 internal branch를 묶었다. External branch를 일부 절개하여 2F Forgathy balloon catheter로 3회 ballooning하여 혈관에 손상을 주었다. 이후 balloon catheter를 빼내고 절개부위를 묶었다. 풍선 확장술을 실시한 후, 14일 동안 약물을 투여하고 동물을 희생하여 손상부위를 포함하는 동맥 부위을 절단하였다. 얻어진 JC1-40시험군과 대조군의 혈관을 section하여 혈관 내층을 H&E (Hematoxylin and Eosin) 염색한 뒤, JC1-40에 의한 혈관내층 증식 억제 효과를 확인하여 도 15에 나타내었다. 도 15a 는 대조군의 혈관 단면이며, 도 15b는 JC1-40을 투여한 시험군의 혈관 단면을 나타낸 것이다.
도 15에 나타난 바와 같이, 풍선 확장술에 의한 혈관 내층의 증식이 JC1-40투여에 의해 억제되는 것을 관찰하였다 (도 15 참조).
상기 실험예 12 및 실험예 13의 결과로부터, RORα 유전자를 활성화시키는 신규한 화합물, 예컨대 JC1 화합물들은 혈관평활근의 증식을 동반하는 죽상동맥경화반의 생성억제와 풍선요법 또는 스텐트 시술 후 혈관 평활근 증식에 의한 혈관 재협착의 방지에 적용될 수 있을 것으로 기대된다.
실험예 14. JC1-40 의 마우스 알코올성 지방간 모델에서의 개선 효과
8 주령 검정 실험 동물 수컷 쥐 (C57BL/6)를 1) 대조군 (n=8), 2) JC1-40을 투여한 대조군 (n=8), 3) 알코올군 (n=12), 그리고 4) JC1-40을 투여한 알코올군 (n=12)으로 나누어 Lieber-DeCarli liquid diet (Dyets Inc, Bethlehem, PA, USA) 를 공급하였다. Lieber-DeCarli liquid diet는 가루 상태로 구입한 후 매일 아침 물을 첨가하여 액상 상태로 만들어 사료 1 ml 당 1 kcal 가 되도록 제조하였다. 알코올군은 2주의 적응기간 동안 알코올을 0.5%씩 점진적으로 증가시켜 공급하였고, 이 후 5주 동안은 5% 알코올이 함유된 사료를 공급하였다. 대조군의 사료는 알코올 대신 알코올군의 동물이 섭취한 사료량을 기준으로 같은 열량의 maltose dextrin으로 대체하여 pair-feeding 하였다. 5주간의 5% 알코올 섭취기간 중, 쥐를 희생시키기 전 2주 동안 체중에 따라 JC1-40을 10 mg/kg/day 농도로 0.5% carboxymethyl cellulose에 희석하여 매일 1회 구강투여하였다. 5 주간의 알코올 식이기간이 끝난 후 동물을 12 시간 fasting시키고, 마취 후 복부를 절개하여 혈액과 간을 적출하였다.
도 16a 에 나타난 바와 같이 혈액에서 분리해 낸 혈장에서 간 손상도를 측정하기 위한 GPT와 GOT를 측정한 결과, 알코올군에서 대조군보다 약 50% 정도 유의적으로 증가되었으며 JC1-40 처리 알코올군에서는 대조군 수준으로 감소되어 있는 것을 확인하였다. 도 16b와 도 16c에서는 혈중 중성지방과 사이토카인 MCP-1레벨을 ELISA 방법으로 측정한 결과, 알코올군에서 증가된 레벨이 JC1-40처리알코올 군에서는 유의적으로 감소되어 있는 것을 관찰하였다. 더 16d에서 나타난 바와 같이 간의 무게를 쥐의 체중으로 나누어 체중에서 간이 차지하는 비율을 알아본 결과, 알코올 섭취에 의해 증가된 간의 비율이 JC1-40투여에 의해 매우 유의적으로 감소되고 있는 것이 확인되었다. 도 16e에서는 웨스턴 블롯팅 분석법을 통해 MCP-1 단백질의 발현을 분석한 결과, 알코올군에서 증가되어 있던 MCP-1 발현이 JC1-40투여에 의해 대조군 수준으로 감소되어 있었다. 조직 염색법 (Hematoxyllin & Eosin) 을 통해 간 조직의 형태학적 변화를 알아본 결과에서는 알코올로 유도된 지방 소포의 축적이 JC1-40 투여에 의해 현저히 감소되는 것을 관찰하였다 (도 16f 참조).
전술한 본 발명의 설명은 예시를 위한 것이며, 본 발명이 속하는 기술분야의 통상의 지식을 가진 자는 본 발명의 기술적 사상이나 필수적인 특징을 변경하지 않고서 다른 구체적인 형태로 쉽게 변형이 가능하다는 것을 이해할 수 있을 것이다. 그러므로 이상에서 기술한 실시예 및 실험예들은 모든 면에서 예시적인 것이며 한정적이 아닌 것으로 이해해야만 한다.
본 발명은 대사성 질환과 염증성 질환의 치료와 예방에 효과가 있을 것으로 기대되며 특히 콜레스테롤의 항상성 조절 및 지질 합성 억제를 통한 간 질환의 예방 및 치료에 유용할 것으로 기대된다.
<110> SNU R&DB FOUNDATION
<120> New Thiourea derivatives as RORa Activators, and pharamaceutical
compositions containing the same
<130> PCT01471
<150> 1020110048455
<151> 2011-05-23
<150> 1020110128903
<151> 2011-12-05
<150> 1020120054540
<151> 2012-05-23
<160> 20
<170> KopatentIn 2.0
<210> 1
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> MCAD primer seqeunce(forward)
<400> 1
ctaccaagta tgccctggaa ag 22
<210> 2
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> MCAD primer seqeunce(reverse)
<400> 2
tgtgttcacg ggctacaata ag 22
<210> 3
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> ACO1 primer seqeunce(forward)
<400> 3
gggcatggct attctcattg c 21
<210> 4
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> ACO1 primer seqeunce(reverse)
<400> 4
cgaacaaggt caacagaagt taggtt 26
<210> 5
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> ACO2 primer seqeunce(forward)
<400> 5
gcggacatgg ctactcaaag c 21
<210> 6
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> ACO2 primer seqeunce(reverse)
<400> 6
gcagtgcacc ttagcagcct g 21
<210> 7
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> HMGCS2 primer seqeunce(forward)
<400> 7
ggaacccata tggagaatgt gt 22
<210> 8
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> HMGCS2 primer seqeunce(reverse)
<400> 8
tcctgagagg cctttagaag tg 22
<210> 9
<211> 23
<212> DNA
<213> Artificial Sequence
<220>
<223> CPT1 primer seqeunce(forward)
<400> 9
agacggtgga acagaggctg aag 23
<210> 10
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> CPT1 primer seqeunce(reverse)
<400> 10
tgagaccaaa caaagtgatg atgtcag 27
<210> 11
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> ACS primer seqeunce(forward)
<400> 11
agcagagctt cgcagcggc 19
<210> 12
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> ACS primer seqeunce(reverse)
<400> 12
tctgctgttt tcgctgggtc c 21
<210> 13
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> B-actin primer seqeunce(forward)
<400> 13
cgtgggccgc cctaggcacc a 21
<210> 14
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> B-actin primer seqeunce(reverse)
<400> 14
ttggcttagg gttcaggggg g 21
<210> 15
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> CD36 primer seqeunce(forward)
<400> 15
ggaactgtgg gctcattgc 19
<210> 16
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> CD36 primer seqeunce(reverse)
<400> 16
catgagaatg cctccaaaca c 21
<210> 17
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> MTTP primer seqeunce(forward)
<400> 17
ccttcattca gcacctca 18
<210> 18
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> MTTP primer seqeunce(reverse)
<400> 18
tgacaagtgt cccagtga 18
<210> 19
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> ApoB100 primer seqeunce(forward)
<400> 19
taaatggagc acttttcaag 20
<210> 20
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> ApoB100 primer seqeunce(reverse)
<400> 20
ggaacagcag cagtagcg 18
Claims (11)
- [규칙 제91조에 의한 정정 25.07.2012]
하기 화학식(I)으로 표시되는 화합물 또는 이의 약제학적으로 허용가능한 염: [I]상기 화학식(I)에서,R1은 수소 또는 탄소수가 1 내지 3인 알킬기 이고;R2는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기이고;R3는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기이고;R4는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기, 시아노기, 다이메틸아미노기, 메틸설포닐아마이드기, 트리플루오르메틸기, 바이닐 벤젠기, 페녹시기, 벤족시기, 아릴기 또는 페닐아민기 이고;R5는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기 이다.
[I]상기 화학식(I)에서,R1은 수소 또는 탄소수가 1 내지 3인 알킬기 이고;R2는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기이고;R3는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기이고;R4는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기, 시아노기, 다이메틸아미노기, 메틸설포닐아마이드기, 트리플루오르메틸기, 바이닐 벤젠기, 페녹시기, 벤족시기, 아릴기 또는 페닐아민기 이고;R5는 수소원소, 할로겐원소, 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 나이트로기, 하이드록시기 또는 페녹시기 이다. - 제1항에 있어서,상기 R4가 아릴기 또는 페녹시기 일 때 방향족 환(aromatic ring)에 탄소수가 1 내지 3인 알킬기, 탄소수가 1 내지 3인 알콕시기, 트리플루오르메틸기 또는 t-부틸기로 치환 되는, 화합물.
- 제1항에 있어서,상기 R3 및 R4 는 환으로 연결되는, 화합물.
- 제1항에 있어서,상기 R2 내지 R5 는 동일한 또는 다른 치환체로 동시에 치환 되는, 화합물.
- 제1항 내지 제4항 중 어느 한 항에 따른 화합물 또는 이의 약제학적으로 허용가능한 염을 유효성분으로 포함하는 지질 축적 억제제.
- 제1항 내지 제4항 중 어느 한 항에 따른 약제학적 유효량의 화합물 또는 이의 약제학적으로 허용가능한 염 및 약제학적으로 허용 가능한 담체를 포함하는 대사성 질환 또는 염증성 질환 예방 또는 치료용 약학적 조성물.
- 제6항에 있어서,상기 대사성 질환은 동맥경화증인, 조성물.
- 제6항에 있어서,상기 대사성 질환은 동맥경화증인, 조성물.
- 제8항에 있어서,상기 질병은 고지혈증 또는 지방간인, 조성물.
- 제8항에 있어서,상기 질병은 알코올성 지방간인, 조성물.
- 제6항에 따른 약학적 조성물을 개체에 투여하는 단계를 포함하는 대사성 질환 또는 염증성 질환 예방 또는 치료 방법.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/759,588 US10407388B2 (en) | 2011-05-23 | 2012-05-23 | Thiourea derivatives as activators of RORα and pharmaceutical composition containing same |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20110048455 | 2011-05-23 | ||
| KR10-2011-0048455 | 2011-05-23 | ||
| KR10-2011-0128903 | 2011-12-05 | ||
| KR1020110128903A KR20120130678A (ko) | 2011-05-23 | 2011-12-05 | RORα의 활성자로서의 신규한 thiourea 유도체 및 이를 함유하는 약학적 조성물 |
| KR10-2012-0054540 | 2012-05-23 | ||
| KR1020120054540A KR101450960B1 (ko) | 2011-05-23 | 2012-05-23 | RORα의 활성자로서의 신규한 thiourea 유도체 및 이를 함유하는 약학적 조성물 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012161518A2 true WO2012161518A2 (ko) | 2012-11-29 |
| WO2012161518A3 WO2012161518A3 (ko) | 2013-01-17 |
Family
ID=47217904
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2012/004081 WO2012161518A2 (ko) | 2011-05-23 | 2012-05-23 | RORα의 활성자로서의 신규한 thiourea 유도체 및 이를 함유하는 약학적 조성물 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US10407388B2 (ko) |
| KR (1) | KR101450960B1 (ko) |
| WO (1) | WO2012161518A2 (ko) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101581665B1 (ko) | 2014-08-08 | 2015-12-31 | 주식회사 에이치엘앤피 | 피부질환 치료를 위한 광선치료기 |
| KR20160132534A (ko) | 2015-05-11 | 2016-11-21 | 경북대학교병원 | RORα 활성조절제를 유효성분으로 함유하는 암의 예방 또는 치료용 약학적 조성물 |
| WO2017026830A1 (ko) * | 2015-08-12 | 2017-02-16 | 서울대학교 산학협력단 | 티오우레아 유도체를 포함하는 자가면역질환 예방 또는 치료용 약학적 조성물 |
| US11390582B2 (en) * | 2019-11-26 | 2022-07-19 | Therasid Bioscience Inc. | Polymorphs of 1-(4-benzyloxy-benzyl)-3-methyl-thiourea |
| US10995066B1 (en) | 2019-11-26 | 2021-05-04 | Therasid Bioscience Inc. | Method for preparing novel crystalline forms of 1-(4-benzyloxy-benzyl)-3-methyl-thiourea |
| CN113181150A (zh) * | 2021-04-29 | 2021-07-30 | 新乡医学院第二附属医院 | Sr1078在制备治疗酒精性肝损伤药物方面的应用、药物组合物 |
| WO2022245067A1 (ko) * | 2021-05-17 | 2022-11-24 | 서울대학교 산학협력단 | RORα의 활성자 JC1-40을 포함하는 근감소증 예방, 개선, 또는 치료를 위한 약학적 조성물 |
| KR20230071841A (ko) | 2021-11-15 | 2023-05-24 | 서울대학교산학협력단 | RORα의 활성자로서의 신규한 티오우레아 유도체 및 이를 포함하는 약학적 조성물 |
| KR102417640B1 (ko) | 2022-05-09 | 2022-07-06 | (주)테라시드바이오사이언스 | 티오우레아 유도체를 포함하는 섬유화 질환의 예방 또는 치료용 조성물 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3090810A (en) * | 1961-01-11 | 1963-05-21 | Baxter Laboratories Inc | Benzyl thioureas |
| US3949089A (en) * | 1969-06-23 | 1976-04-06 | Burroughs Wellcome Co. | Substituted guanidine compounds as antifibrillatory agents |
| US3968243A (en) * | 1970-06-05 | 1976-07-06 | Burroughs Wellcome Co. | Substituted guanidine compounds in the treating of arrythmias |
| US20020147365A1 (en) * | 2000-05-10 | 2002-10-10 | The Fanning Corporation | 3-methoxybenzyl thiourea derivatives and improved lipid compositions containing same |
| US20080286231A1 (en) * | 2007-04-18 | 2008-11-20 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
-
2012
- 2012-05-23 US US15/759,588 patent/US10407388B2/en active Active
- 2012-05-23 KR KR1020120054540A patent/KR101450960B1/ko active IP Right Grant
- 2012-05-23 WO PCT/KR2012/004081 patent/WO2012161518A2/ko active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3090810A (en) * | 1961-01-11 | 1963-05-21 | Baxter Laboratories Inc | Benzyl thioureas |
| US3949089A (en) * | 1969-06-23 | 1976-04-06 | Burroughs Wellcome Co. | Substituted guanidine compounds as antifibrillatory agents |
| US3968243A (en) * | 1970-06-05 | 1976-07-06 | Burroughs Wellcome Co. | Substituted guanidine compounds in the treating of arrythmias |
| US20020147365A1 (en) * | 2000-05-10 | 2002-10-10 | The Fanning Corporation | 3-methoxybenzyl thiourea derivatives and improved lipid compositions containing same |
| US20080286231A1 (en) * | 2007-04-18 | 2008-11-20 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
Non-Patent Citations (2)
| Title |
|---|
| HAN, HO GYU ET AL.: 'Synthesis of 2-Benzylimino-1,3-thiazolines and Their Structure Determination' JOURNAL OF THE KOREAN CHEMICAL SOCIETY vol. 45, 2001, pages 612 - 615 * |
| KIM, JEONGHAN ET AL.: 'Liquid Crystalline Properties of Polyguanidines' MARCROMOLECULES vol. 37, 2004, pages 8286 - 8292 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2012161518A3 (ko) | 2013-01-17 |
| KR101450960B1 (ko) | 2014-10-22 |
| US10407388B2 (en) | 2019-09-10 |
| KR20120130733A (ko) | 2012-12-03 |
| US20180265462A1 (en) | 2018-09-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012161518A2 (ko) | RORα의 활성자로서의 신규한 thiourea 유도체 및 이를 함유하는 약학적 조성물 | |
| WO2011043568A2 (en) | Novel compounds effective as xanthine oxidase inhibitors, method for preparing the same, and pharmaceutical composition containing the same | |
| WO2016032120A1 (ko) | 신규한 아미노-페닐-설포닐-아세테이트 유도체 및 이의 용도 | |
| WO2021256861A1 (ko) | 신규한 산 분비 억제제 및 이의 용도 | |
| AU2019310508B2 (en) | 1,3,4-oxadiazole derivative compounds as histone deacetylase 6 inhibitor, and pharmaceutical composition comprising the same | |
| WO2021118318A2 (ko) | 신규한 인돌 유도체 및 이의 용도 | |
| WO2017018751A1 (ko) | Blt 저해 활성을 갖는 신규 화합물 및 이를 유효성분으로 포함하는 염증성 질환 예방 또는 치료용 조성물 | |
| WO2012118308A9 (ko) | 피라졸 유도체를 포함하는 심혈관 질환 예방 및 치료용 조성물. | |
| WO2013043002A1 (en) | Imide-containing benzothiazole derivative or its salt and pharmaceutical composition comprising the same | |
| WO2013105753A1 (en) | Substituted piperidine derivatives and methods for preparing the same | |
| WO2011122815A2 (en) | Novel quinoxaline derivatives | |
| WO2015060613A1 (en) | Novel antifungal oxodihydropyridinecarbohydrazide derivative | |
| WO2012148140A2 (ko) | 혈관 신생 억제 및 항산화 효과를 가지는 이미다졸계 알칼로이드 유도체 및 이의 제조방법 | |
| WO2018008989A1 (ko) | 벤조[d]싸이아졸 유도체 또는 그의 염 및 이를 포함하는 약학 조성물 | |
| WO2014065575A1 (ko) | 4,5-다이하이드로-1h-피라졸 유도체 또는 그의 염 및 이를 포함하는 약학 조성물 | |
| WO2019235879A1 (ko) | 신규 mTOR 저해제를 포함하는 암 예방 또는 치료용 조성물 | |
| WO2021096314A1 (ko) | 신규한 벤즈이미다졸 유도체 및 이의 용도 | |
| WO2017217741A1 (ko) | 신규한 브롬화 퓨라논 유도체, 이의 제조방법 및 이를 유효성분으로 함유하는 약학적 조성물 | |
| WO2023085894A1 (ko) | RORα의 활성자로서의 신규한 티오우레아 유도체 및 이를 포함하는 약학적 조성물 | |
| WO2020242245A1 (ko) | 프탈라진온 화합물 및 이들의 용도 | |
| WO2018194372A1 (ko) | 신규 sirt 1 활성화제 및 이의 의학적 용도 | |
| WO2022235097A1 (ko) | 신규한 피라졸로[3,4-b]피리딘 유도체를 포함하는 비만 및 당뇨병을 비롯한 대사성 질환 또는 비알콜성 지방간염의 예방 또는 치료용 약학 조성물 | |
| WO2022119090A1 (ko) | 5-ht7 세로토닌 수용체 활성 저해용 바이페닐 피롤리딘 및 바이페닐 다이하이드로이미다졸 유도체 및 이를 유효성분으로 포함하는 약학 조성물 | |
| WO2017119570A1 (ko) | 티아졸리딘디온 유도체 및 그의 용도 | |
| WO2023090908A1 (ko) | 신규 플라보노이드 유도체 및 이를 유효성분으로 포함하는 대사성 질환의 예방 또는 치료용 약학 조성물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12790167 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12790167 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15759588 Country of ref document: US |