WO2012074029A1 - 無細胞翻訳系でFabを提示しうるポリヌクレオチド構築物ならびにそれを用いたFabの製造方法およびスクリーニング方法 - Google Patents
無細胞翻訳系でFabを提示しうるポリヌクレオチド構築物ならびにそれを用いたFabの製造方法およびスクリーニング方法 Download PDFInfo
- Publication number
- WO2012074029A1 WO2012074029A1 PCT/JP2011/077725 JP2011077725W WO2012074029A1 WO 2012074029 A1 WO2012074029 A1 WO 2012074029A1 JP 2011077725 W JP2011077725 W JP 2011077725W WO 2012074029 A1 WO2012074029 A1 WO 2012074029A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fab
- sequence
- chain
- amino acid
- dna
- Prior art date
Links
Images



























Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6854—Immunoglobulins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/46—Hybrid immunoglobulins
- C07K16/468—Immunoglobulins having two or more different antigen binding sites, e.g. multifunctional antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1041—Ribosome/Polysome display, e.g. SPERT, ARM
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1062—Isolating an individual clone by screening libraries mRNA-Display, e.g. polypeptide and encoding template are connected covalently
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1075—Isolating an individual clone by screening libraries by coupling phenotype to genotype, not provided for in other groups of this subclass
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6854—Immunoglobulins
- G01N33/6857—Antibody fragments
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
Definitions
- the present invention relates to a polynucleotide construct capable of presenting a Fab in a cell-free translation system, a kit containing the same, a Fab production method and a screening method using the same.
- Antibody is a glycoprotein produced by B cells and has the function of recognizing and binding molecules (antigens) such as proteins. Antibodies are produced in response to various internal and external stimuli (antigens), and in the defense mechanism of vertebrates by eliminating bacteria and viruses that have invaded the body in cooperation with other immunocompetent cells. It plays an important role.
- One type of B cell can only make one type of antibody, and one type of antibody can recognize only one type of antigen.
- millions to hundreds of millions of B cells each produce different antibodies and try to deal with any antigen. Collectively these are called immunoglobulins and are one of the most abundant protein components in the blood and constitute 20% by weight of total plasma protein. Antibodies are used as molecular target drugs and diagnostic agents because of their antigen specificity.
- a naturally occurring antibody molecule forms a Y-shaped basic structure by associating two polypeptide chains of L chain (light chain) and H chain (heavy chain).
- the lower half of the Y-shape consists of the Fc region
- the upper half of the Y-shape consists of two identical Fab regions.
- the front half of the Fab is called a variable region (V region), and the amino acid sequence is diverse so that it can bind to various antigens.
- the variable regions of the L and H chains are called VL and VH, respectively.
- the Fc side half of the Fab is called a constant region (C region), and there is little change in the amino acid sequence.
- the constant regions of L and H chains are called CL and CH1, respectively.
- CDR complementarity-determining region
- FR framework region
- Each VL and VH has three CDR (CDR1 to CDR3) 4 and four FR (FR1 to FR4) ⁇ ⁇ surrounding them.
- the sequence diversity of CDR1 and CDR2 in naive B cells not subjected to antigen stimulation is derived from the genomic DNA sequence (germline sequence).
- CDR3 sequences are newly formed by the recombination reaction of genomic DNA that occurs during the differentiation process of B cells.
- L chain CDR3 is formed by one recombination reaction (VJ recombination), whereas H chain CDR3 is formed through two recombination reactions (VD recombination, DJ recombination). Even in the same CDR3, H chain diversity is high.
- the six CDRs form one continuous surface involved in antigen binding at the front end of the Fab.
- the functional and structural unit involved in antigen binding in natural antibodies is Fab.
- VL, VH, CL, and CH1 that constitute Fab each form an independent domain as a three-dimensional structure, while obtaining higher stability by the interaction between the four domains.
- the Fc region is not directly involved in the binding of the antibody to the antigen, but is involved in various effector functions (eg, antibody dependent cellular cytotoxicity).
- the antibody library is expressed from DNA encoding the antibody library, brought into contact with the target antigen, and one that specifically binds to the target antigen is selected.
- the antibody is screened by repeating the cycle of amplifying the DNA encoding it. Since an antibody selected by a screening method using such display technology is accompanied by genetic information encoding its amino acid sequence, the selected antibody is immediately genetically engineered based on the genetic information encoding it. It can be prepared in large quantities. The amino acid sequence can also be easily clarified by analyzing genetic information.
- Non-patent Document 1 As an antibody screening method using display technology, there is phage display (Non-patent Document 1) reported by G. Smith et al. In 1985. Phage display is a technique in which a foreign protein is expressed as a fusion protein mainly in a coat protein of a filamentous phage, and a polynucleotide encoding the target foreign protein is selected. This method is widely used for selection of an antibody that specifically binds to an antigen molecule (Non-patent Documents 2 and 3). However, when constructing a phage library, a step of transforming E. coli is necessary, and this process limits the size of the library. In other words, due to the limitations of E.
- cell-free display systems such as ribosome display are technologies that associate proteins synthesized in cell-free translation systems with the polynucleotides that encode them. Screening of antibodies using this system has also been reported.
- Patent Document 1 what is actually produced here is the production of a single chain antibody (scFv) in which the heavy chain variable region (VH) and the light chain variable region (VL) are linked by a linker peptide. The method was not disclosed.
- ribosome display is generally a technology that associates one molecule of RNA with one molecule using the 3 'end of RNA, and the full-length peptide as the molecular weight of the protein to be synthesized increases.
- Non-patent Document 4 Fab is double-stranded, not only cis-association of H and L chains displayed on the same RNA, but also trans-association of H and L chains on different RNAs, screening efficiency There was also a risk of lowering.
- the present invention provides a method for efficiently expressing Fab in a cell-free translation system such as ribosome display, associating it with a polynucleotide encoding the same, and performing screening, and a polynucleotide construct therefor This is the issue.
- the present inventors have intensively studied to solve the above problems.
- a polynucleotide construct to be expressed in association with the nucleotide sequence information is prepared, and it is found that Fab screening can be efficiently performed in a cell-free translation system by performing ribosome display and / or CIS display using the polynucleotide construct.
- the present invention has been completed by finding a Ymacs method that dramatically improves the affinity of antibodies using this cell-free Fab display system.
- the present invention provides the following.
- (Polynucleotide construct used for Fab cell-free display method) [1] A Fab comprising a first strand coding sequence and a Fab second strand coding sequence. When introduced into a cell-free translation system containing a ribosome, the Fab encoded by itself is expressed without dissociation and is combined with the Fab. A polynucleotide construct capable of maintaining the body.
- polynucleotide construct (monocistronic)
- the Fab first chain expression cistron and the Fab second chain expression cistron including the ribosome binding sequence, the Fab first chain coding sequence or the Fab second chain coding sequence, and the scaffold coding sequence in this order are included.
- the first strand expression cistron of Fab has a ribosome stall sequence at its 3 ′ end, and the second strand expression cistron of Fab maintains a complex with its own encoded Fab at its 3 ′ end.
- polynucleotide construct used for ribosome / mRNA / CIS display method [4] The structure necessary to maintain the complex with the Fab encoded by itself is a ribosome stall sequence, puromycin or a derivative thereof, or a DNA binding protein coding sequence and a binding sequence of the DNA binding protein.
- the polynucleotide construct according to [2] or [3]. (Polynucleotide construct used for ribosome display method) [5] The polynucleotide construct according to [4], wherein the structure necessary for maintaining a complex with the Fab encoded by itself is a ribosome stall sequence.
- polynucleotide construct used for mRNA display method [9] The polynucleotide construct according to [4], wherein the structure necessary for maintaining the complex with the Fab encoded by itself is puromycin or a derivative thereof.
- polynucleotide construct used for CIS display method [10] The polynucleotide construct according to [4], wherein the structure necessary for maintaining a complex with the Fab encoded by itself is a DNA-binding protein coding sequence and a binding sequence of the DNA-binding protein.
- the DNA binding protein is RepA encoded by E. coli R1 Plasmid, and the binding sequence of the DNA binding protein is a CIS-ori sequence existing downstream of the RepA coding sequence on the same polynucleotide. The polynucleotide construct described.
- the library comprising a random sequence is a library comprising a single amino acid substitution in the complementarity determining region (CDR) of Fab first strand and / or Fab second strand, in [13] or [14] The polynucleotide construct described.
- Fab first chain coding sequence or Fab second chain coding sequence has one amino acid substitution for one position in CDR in the amino acid sequence of Fab first chain or Fab second chain of the parent antibody.
- a step of preparing a plurality of types of polynucleotide constructs according to [15] encoding an amino acid sequence containing the amino acid sequences so that one amino acid substitution is included at a plurality of positions in the CDRs of the Fab first strand and Fab second strand (II) a primary screening step of screening a plurality of high affinity Fabs by repeating the steps (i) to (iv) described in [16] using the plurality of types of polynucleotide constructs; (III) a step of analyzing the frequency of 1 amino acid substitution at each position in the CDRs of the Fab first chain and Fab second chain in a plurality of Fabs selected in the primary screening step; (IV) Combination of 1 amino acid substitution in which Fab first chain coding sequence and
- the in vitro translation system comprises independently purified factors.
- the in vitro translation system comprises at least one component selected from the group consisting of an initiation factor, an elongation factor, an aminoacyl tRNA synthetase, and a methionyl tRNA transformylase.
- the in vitro translation system does not contain a dissociation factor.
- the in vitro translation system is a cell extract containing ribosome.
- a method for producing a Fab comprising the step of introducing the polynucleotide construct according to any one of [1] to [15] into an in vitro translation system to generate a Fab.
- a kit for producing or screening a Fab comprising the polynucleotide construct according to any one of [1] to [15].
- step (III) a step of concentrating the primary library with target affinity using a protein display system; (IV) determining polynucleotide sequence information of the primary library-enriched sample obtained in step (III), (V) a step of extracting one frequently observed amino acid substitution from the base sequence information, (VI) constructing a secondary library containing a frequently observed combination of one amino acid substitution, and (VII) enriching the secondary library with target affinity using a protein display system, A method for maximizing target substance affinity of a target substance binding protein.
- step of determining polynucleotide sequence information of the primary library-enriched sample is performed using a next-generation sequencer.
- the target substance-binding protein is a full-length antibody or an antibody fragment such as scFv, Fab, scFab, etc., and the target substance-binding site is a so-called CDR region in which a variety of sequences are found in natural antibodies [24] Or the method of [25].
- the protein display system is ribosome display, CIS display, mRNA display, phage display, bacterial surface display, yeast cell surface display, higher eukaryotic cell surface display the method of.
- the target Fab can be screened by efficiently expressing the Fab without dissociating it from the polynucleotide in a cell-free display system. Since the method of the present invention can be performed in a cell-free translation system, the operation is simple and screening can be performed in a short period of time. In addition, it is easy to construct a large-scale library having a level of 10 12 or more, thereby enabling highly efficient screening. In addition, according to the Ymacs method of the present invention, a large number of Fabs whose affinity for an antigen is improved by several hundred to 1,000 times or more can be efficiently obtained as compared with error-prone PCR, CDR shuffling and the like.
- each symbol on mRNA or DNA means the following.
- “VL” coding sequence of Fab light chain variable region
- “CL” Fab L chain constant region coding sequence
- “VH” coding sequence of Fab heavy chain variable region
- “CH1” coding sequence of Fab heavy chain constant region
- “Linker” coding sequence of the linker peptide
- “RBS ( ⁇ )” ribosome binding site
- “Pu” puromycin or a derivative thereof
- Pro ⁇ ” Promoter
- “RNAP” RNA polymerase
- RSS (S) Ribosome stall sequence
- “LP (P)” Leader peptide sequence for secretory expression Schematic diagram in which Fab is displayed on a monocistronic polynucleotide construct (embodiment using ribosome display) (RBS ( ⁇ ) indicates a ribosome binding site; hereinafter the same).
- Schematic diagram in which Fab is displayed on monocistronic polynucleotide construct (embodiment using mRNA display).
- Schematic diagram in which Fab is displayed on a bicistronic polynucleotide construct an embodiment in which ribosome display is used for both 5′-side cistron and 3′-side cistron).
- Schematic diagram in which Fab is displayed on a bicistronic polynucleotide construct an embodiment in which a 5 ′ cistron uses a ribosome display and a 3 ′ cistron uses an mRNA display.
- Schematic diagram in which Fab is displayed on a bicistronic polynucleotide construct an embodiment in which a 5 ′ cistron uses a ribosome display and a 3 ′ cistron uses a CIS display.
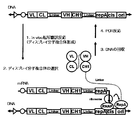
- Schematic diagram of Fab screening method ribosome display
- Schematic diagram of Fab screening method CIS display
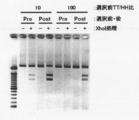
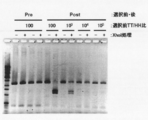
- the two left lanes are markers.
- BSA represents bovine serum albumin (control)
- stAv represents streptavidin.
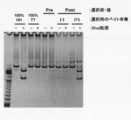
- Electrophoresis photograph showing the result of Fab-HHxho concentration (only in the center) with Bicistoronic Fab-PRD. WB indicates washing buffer. Electrophoresis photograph showing the result of Fab-HHxho enrichment (full length) with Bicistoronic Fab-PRD. Electrophoresis photograph showing the results of Fab-HHxho concentration (full length) with Monocistoronic Fab-PRD. Electrophoresis photograph (Western blot) showing the results of in vitro translation product detection of Monocistronic Fab-PRD. The figure which shows the relationship between the copy number and recovery rate of SecM in Monocistronic Fab-PRD.
- the figure which shows the construction method of the Ymacs-2 secondary library Schematic diagram of conversion from a Monocistronic secretory Fab expression vector to a Bicistronic Fab secretory expression vector.
- Electrophoresis photograph showing the results of Fab-HHxho concentration (full length) on CIS display. Electrophoresis photograph showing the results of examining the effect of transcription and translation reaction time on Fab-HHxho enrichment (full length) on CIS display.
- the polynucleotide construct of the present invention comprises a Fab first strand coding sequence and a Fab second strand coding sequence, and is expressed without dissociating the Fab encoded by itself when introduced into a cell-free translation system containing a ribosome, A polynucleotide construct capable of maintaining a complex with the Fab.
- Fab first chain and Fab second chain mean two chains constituting Fab, and usually one is Fab H chain and the other is Fab L chain. It may be a chimeric chain of chains.
- Fab H chain means a protein containing H chain variable region (VH) and H chain constant region 1 (CH1)
- Fab L chain means L chain variable region (VL) and L chain constant region (CL).
- VH H chain variable region
- CH1 H chain constant region 1
- CL L chain constant region
- Fab L chain means L chain variable region (VL) and L chain constant region (CL).
- maintaining a complex with Fab means that the Fab is expressed in a state linked to the polynucleotide construct, and the complex is maintained. It is also referred to as “Fab is presented on a polynucleotide”.
- the polynucleotide construct according to the first aspect of the present invention includes a ribosome binding sequence, a Fab first chain coding sequence, a linker peptide sequence, a Fab second chain coding sequence, and a scaffold coding sequence in this order monocistronic.
- the polynucleotide construct has a structure necessary for maintaining a complex with the Fab encoded by itself at the 3 ′ end of the coding region (cistron).
- Ribosome binding sequence means a sequence upstream of the initiation codon where ribosome binds to initiate translation, but when using E. coli-derived ribosome as a cell-free translation system, Shine-Dalgarno (SD It is preferable to use a sequence.
- SD sequence As an SD sequence, AGGAGGT is generally known. However, a modified sequence may be used as long as a ribosome can be bound.
- the ribosome binding sequence can be appropriately selected depending on the host, and is not limited to the SD sequence.
- Fab chain coding sequence As the Fab chain coding sequence, the sequence of the Fab common region has been disclosed (Sakano et al., Nature (1980) vol.286, p676, Ellison et al., Nucleic Acids Res. (1982) vol.10, p. 4071, Huck et al., Nucleic Acids Res. (1986) vol. 14, p. 1779, Hieter et al., J. Biol. Chem. (1982) vol. 257, p. 1516, and Max et al., Cell (1980) vol.22, p.197), primers can be designed based on these sequences and obtained by amplifying Fab H chain coding sequence and Fab L chain coding sequence.
- Fab chain coding sequence can be artificially synthesized in consideration of the codon bias and RNA processing of the host that is planned to be used for mass expression. When a specific Fab is expressed, the sequence may be amplified from the target template.
- a library containing a random Fab chain coding sequence may be used.
- a naive library can be used to obtain a new Fab that binds to the antigen of interest. If you want to optimize a specific Fab that has already been acquired for some purpose, you can use a focused library.
- the naive library aims to provide a very wide search space to obtain a new Fab that binds to the antigen of interest, and can be constructed in various formats as shown below. .
- a semi-synthetic naive library (Hoet et al., Nat. Biotechnol. (2005) by incorporating a natural sequence into the Fab L chain (VL-CL), an artificial sequence into the CDR 1-2 of the H chain, and a natural sequence into the CDR3. vol.23, p.344-348) can also be constructed.
- a fully artificial naive library (Knappik et al., J. Mol. Biol. (2000) vol.296, p.57-86) that artificially synthesizes the entire coding sequence while giving diversity to the CDR is constructed. You can also.
- the frequency of amino acids appearing at each amino acid position can be adjusted for each CDR length based on the analysis result of the natural CDR sequence.
- a focused library is based on the sequence of a specific parent Fab that has already been acquired, and various types including a non-mutant mutant as a main component can be constructed. Focused library maintains the antigen-binding properties of the parent Fab while improving affinity, optimizing specificity, humanizing animal-derived antibodies, removing inconvenient amino acids or sequences, improving stability, improving physical properties, etc. Can be used when optimizing for purposes. For example, Error-Prone PCR can be used to introduce one to several amino acid substitutions randomly and sporadically throughout the VH region and / or VL region (Boder et al. , Proc Natl Acad Sci USA (2000) vol.97, p.10701-10705).
- Error-prone PCR utilizes a phenomenon in which errors are likely to occur by adjusting the concentration of four types of deoxynucleotides as substrates and the type and concentration of divalent cations to be added.
- Using artificially designed mutagenesis primers one to several amino acid substitutions can be introduced while considering the amino acid composition, targeting only specific amino acids constituting the Fab (Rajpal et al Proc Natl Acad Sci USA (2005) vol.102, p.8466-8471).
- Using artificially designed mutagenesis primers out of a total of 6 CDRs, about 3 to 5 CDR sequences are maintained, but 1 to 3 CDR sequences are not amino acid units.
- the polynucleotide construct of the present invention is preferably a library containing one amino acid substitution in one or more amino acids in the complementarity determining region (CDR) of Fab first strand and / or Fab second strand.
- CDR complementarity determining region
- Such a library can be prepared by PCR or the like using a primer designed to introduce a single amino acid substitution.
- the sequence encoding the linker peptide connecting the Fab first chain coding sequence and the Fab second chain coding sequence is preferably a sequence encoding a water-soluble polypeptide consisting of about 15 to 120 amino acids. From the viewpoint of screening efficiency, a sequence encoding a water-soluble polypeptide consisting of 20 to 30 amino acids is more preferable. For example, an amino acid sequence in which Arg is introduced to enhance water solubility can be exemplified, but a sequence encoding a so-called GS linker mainly containing glycine and serine may be used.
- the sequence encoding the linker peptide may be sandwiched between protease recognition sequences, whereby the linker peptide is cleaved by the protease after the polynucleotide construct is translated into an amino acid sequence, and the natural Fab is presented.
- protease recognition sequence examples include DDDDK (SEQ ID NO: 31) which is an enterokinase recognition sequence, IEGR (SEQ ID NO: 32) which is a factor Xa recognition sequence, and the like.
- the scaffold coding sequence ligated to the 3 ′ side of the Fab chain coding sequence is a complex formed by translation of the first and second strands of Fab on ribosome, DNA and / or mRNA and folding them correctly. And a sequence encoding an amino acid sequence having a sufficient length as a scaffold for reacting with an antigen.
- the scaffold coding sequence is preferably a sequence encoding at least 15 amino acids, more preferably encoding 15 to 120 amino acids. It is preferable that the encoded scaffold sequence is highly water-soluble and does not take a special three-dimensional structure. Specifically, it includes a so-called GS linker mainly containing glycine and serine and a partial geneIII sequence of phage. Can be used.
- the structure necessary for maintaining the complex with the Fab chain encoded by itself includes, for example, a ribosome stall sequence (in this case, ribosome display) at the 3 ′ end of the cistron, or a DNA binding protein coding sequence.
- the Fab chain expressed by the binding sequence of the DNA-binding protein (in this case CIS display) or puromycin or a derivative thereof added at the 3 ′ end (in this case mRNA display) Forms a complex with the polynucleotide, and the amino acid sequences encoded by the nucleotide sequences of the first and second Fab chain coding sequences are physically associated with each other.
- Polynucleotide construct used for ribosome display method examples include a sequence encoding SecM of E. coli.
- the SecM sequence is also called a SecM stall sequence, and is a sequence that has been reported to cause translational arrest inside the ribosome (FXXXXWIXXXXGIRAGP: SEQ ID NO: 30).
- FXXXXWIXXXXGIRAGP SEQ ID NO: 30
- Two or more SecM sequences may be linked, preferably 2 to 4, and more preferably 2.
- a polyproline sequence such as diproline can also be used as a ribosome stall sequence, and may be used alone or in combination with a SecM sequence. It is preferable that a stop codon is arranged in alignment with the reading frame on the 3 ′ side of the ribosome stall sequence. Instead of adopting a ribosome stall sequence, it is also possible to perform ribosome display simply by deleting the stop codon.
- the Fab first strand coding sequence, the linker coding sequence, the Fab second strand coding sequence, the scaffold coding sequence, and the ribosome stall sequence are linked together in a reading frame.
- “linked together with a reading frame” means that these elements are linked so as to be translated as a fusion protein.
- the Fab first chain coding sequence, the linker coding sequence, the Fab second chain coding sequence, the scaffold coding sequence, and the ribosome stall sequence may be directly linked to each other, and a tag sequence or an arbitrary polypeptide sequence may be inserted between and before and after. It may be connected via.
- FIG. 1 shows an example of an embodiment using ribosome display in the polynucleotide construct according to the first embodiment of the present invention.
- SEQ ID NO: 19 promoter sequence (base numbers 9 to 31), ribosome binding sequence (SD sequence; base numbers 81 to 87), anti-Her2 Fab L chain coding sequence, FLAG tag, linker sequence (GS linker), FLAG Anti-Her2 Fab L + H chain expression cistron (base numbers 94 to 2220) including a tag, anti-Her2 Fab H chain coding sequence, His tag, scaffold sequence (GS linker) and ribosome stall sequence (secM + diproline) (base numbers 94 to 2220; amino acid sequence is SEQ ID NO: 20) shows the base sequence of the polynucleotide construct comprising SEQ ID NO: 21, promoter sequence (base numbers 9 to 31), ribosome binding sequence (SD sequence; base numbers 81 to 87), anti-TNF ⁇ R Fab L chain coding sequence, FLAG tag, linker
- Polynucleotide construct used for mRNA display method (monocistronic)
- puromycin or a derivative thereof may be used to form a complex of Fab and polynucleotide, and the amino acid sequence and the base sequence encoding it may be physically associated ( mRNA display). That is, puromycin or a derivative thereof is bound to the 3 ′ end of the polynucleotide construct via a spacer, and the C-terminal of the translation product is covalently bound to puromycin or a derivative thereof to form a complex of Fab and the polynucleotide.
- puromycin or a derivative thereof, a puromycin derivative such as puromycin, ribocytidylpuromycin, deoxycytidylpuromycin, deoxyuridylpuromycin and the like is particularly preferable.
- Examples of the spacer used for binding puromycin or a derivative thereof to the 3 ′ end of the polynucleotide construct include, for example, a high-molecular substance such as polyethylene or polyethylene glycol or a derivative thereof described in WO98 / 16636, an oligo Biopolymer substances such as nucleotides, peptides or derivatives thereof are used. Of these, polyethylene glycol is preferred.
- a streptavidin coding sequence is linked to the 3 ′ end of the polynucleotide construct, biotin is further bound to the 3 ′ end, and the streptavidin portion of the translated protein is combined with biotin. By binding, a complex of Fab and polynucleotide may be formed, and the amino acid sequence may be physically associated with the base sequence encoding it.
- FIG. 2 shows an example of an embodiment using an mRNA display in the polynucleotide construct according to the first embodiment of the present invention.
- CIS display Polynucleotide construct used for CIS display method (monocistronic) Also, instead of a ribosome stall sequence, a DNA-binding protein coding sequence and a binding sequence of the DNA-binding protein are used to form a complex of Fab and a polynucleotide, and the physical sequence of the amino acid sequence and the base sequence that encodes it. You may associate (CIS display: WO2004 / 22746).
- a DNA binding protein coding sequence and a binding sequence of the DNA binding protein are linked downstream of the scaffold sequence of the polynucleotide construct, and expressed as a fusion protein of Fab, scaffolding sequence and DNA binding protein, and the DNA binding protein Binds to a DNA binding protein binding sequence downstream of the 3 ′ cistron, thereby forming a complex of Fab and a polynucleotide, thereby making it possible to associate the amino acid sequence of the Fab chain with the base sequence encoding it.
- the DNA binding protein binds with the DNA binding protein that exists on the same DNA molecule without dissociating once during the transcription and translation reaction with the DNA molecule that becomes the template during transcription and translation.
- RepA protein having a cis-type binding mode is exemplified, and the RepA binding sequence includes a CIS sequence followed by an ori sequence (Proc. Natl. Acad. Sci. USA, vol. 101, p.2806-2810). , 2004 and special table 2005-537795).
- Other DNA binding proteins with a cis-type binding mode include RecC protein encoded by E. coli Ti plasmid (Pinto, et al., Mol. Microbiol.
- ⁇ X174 phage A protein (Francke, et al., Proc Natl Acad Sci USA (1972) vol.69, p.475-479), ⁇ phage Q protein (Echols, et al., Genetics (1976) vol.83, p .5-10) etc. may be used in combination with the binding sequence for each of the DNA binding proteins.
- the synthesized protein when using a cell-free translation system in which transcription and translation are well coupled, the synthesized protein is released in the vicinity of the transcription termination site, so the DNA binding domain of nuclear receptors such as estrogen receptor, Two- What is generally considered to be a DNA protein having a trans-type binding mode, such as the LexA and Gal4 DNA binding domains used in the Hybrid System, is used in combination with a binding sequence for each of the DNA binding proteins. Also good.
- FIG. 3 shows an example of an embodiment using a CIS display in the polynucleotide construct according to the first embodiment of the present invention.
- SEQ ID NO: 70 promoter sequence (base numbers 612 to 639), ribosome binding sequence (SD sequence; base numbers 672 to 675), anti-Her2 Fab L chain coding sequence, FLAG tag, linker sequence (GS linker), FLAG Anti-Her2 Fab L + H chain expression cistron (base number 689-3322; amino acid sequence is SEQ ID NO: 71), CIS including tag, anti-Her2 Fab heavy chain coding sequence, His tag, scaffold sequence (GS linker) and RepA coding sequence
- the base sequence of a polynucleotide construct containing -ori is shown.
- SEQ ID NO: 72 includes a promoter sequence (base numbers 612 to 639), a ribosome binding sequence (SD sequence; base numbers 672 to 675), an anti-TNF ⁇ R Fab L chain coding sequence, a FLAG tag, a linker sequence (GS linker), FLAG Anti-TNF ⁇ R Fab L + H chain expression cistron (base number 689-3328; amino acid sequence is SEQ ID NO: 73), CIS, including tag, anti-TNF ⁇ R Fab H chain coding sequence, His tag, scaffold sequence (GS linker) and RepA coding sequence, CIS
- the base sequence of the polynucleotide construct containing -ori is shown. However, it goes without saying that the polynucleotide construct of the present invention is not limited thereto.
- Polynucleotide construct (bicistronic)
- the polynucleotide construct according to the second aspect of the present invention comprises a Fab first strand expression cistron and a Fab second containing a ribosome binding sequence, a Fab first strand coding sequence or Fab second strand coding sequence, and a scaffold coding sequence in this order.
- a chain expression cistron a chain expression cistron.
- the Fab first chain expression cistron (5′-side Fab chain expression cistron) has a ribosome stall sequence at its 3 ′ end
- the Fab second-chain expression cistron (3′-side Fab chain expression cistron) has 3 'It has a structure necessary for maintaining a complex with the Fab encoded by itself on the terminal side.
- a ribosome stall sequence as described above, a DNA-binding protein coding sequence and a binding sequence of the DNA-binding protein, or puromycin or its Derivatives are exemplified.
- the Fab first strand expression cistron contains the Fab first strand coding sequence and the scaffold coding sequence in this order on the 3 ′ side of the ribosome binding sequence, and the Fab second strand expression cistron is on the 3 ′ side of the ribosome binding sequence.
- the Fab second strand coding sequence and the scaffold coding sequence are included in this order.
- the Fab first chain and the Fab second chain may be in the order of the H chain and the L chain, the order of the L chain and the H chain, or the chimeric chain of the H chain and the L chain, respectively.
- the length between the Fab first chain expression cistron and the Fab second chain expression cistron is such that the Fab first chain expression cistron is in a state where translation is once completed and the ribosome is stalled.
- the ribosome-binding sequence of cistron may have an interval that allows ribosomes to bind, but is preferably 50 to 200 bp.
- ribosome binding sequence and Fab chain coding sequence can be used.
- the H chain and L chain of Fab are translated on ribosome, DNA and / or mRNA and correctly folded to form a complex.
- Examples thereof include a sequence encoding an amino acid sequence having a sufficient length as a scaffold for reacting with an antigen.
- the scaffold coding sequence is preferably a sequence encoding at least 15 amino acids, more preferably encoding 15 to 120 amino acids.
- the encoded scaffold sequence is preferably a sequence that is highly water-soluble and does not take a special three-dimensional structure. Specifically, it includes a so-called GS linker mainly containing glycine and serine, a partial geneIII sequence of phage, and the like. Can be used.
- Polynucleotide construct used for ribosome display method (Bisictronic)
- Fab first strand or second strand 3 of Fab first strand expression cistron 'A ribosome stall sequence as described above is placed at the end.
- a stop codon is arranged in alignment with the reading frame on the 3 ′ side of the ribosome stall sequence.
- the Fab first strand coding sequence or Fab second strand coding sequence, the scaffold coding sequence, and the ribosome stall sequence are linked together in the reading frame.
- “linked together with a reading frame” means that these elements are linked so as to be translated as a fusion protein.
- the Fab first chain coding sequence or Fab second chain coding sequence, the scaffold coding sequence, and the ribosome stall sequence may be directly linked to each other, or may be linked via a tag sequence or an arbitrary polypeptide sequence between before and after. May be.
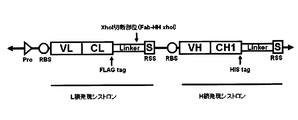
- FIG. 4 shows an example of an embodiment in which ribosome display is used for both the Fab first strand expression cistron and the Fab second strand expression cistron.
- SEQ ID NO: 16 includes promoter sequence (base numbers 9 to 31), ribosome binding sequence (SD sequence; base numbers 81 to 87), anti-TNF ⁇ receptor (TNF ⁇ R) Fab L chain coding sequence, FLAG tag, scaffold sequence ( An anti-TNF ⁇ R Fab L chain expression cistron (base numbers 94 to 1158; amino acid sequence is SEQ ID NO: 17) including a GS linker) and a ribosome stall sequence (secM + diproline); a ribosome binding sequence (SD sequence; base numbers 1191 to 1197); An anti-TNF ⁇ R Fab H chain expression cistron (base numbers 1264 to 2370; amino acid sequence SEQ ID NO: 18) including an anti-TNF ⁇ R Fab H chain coding sequence, a His tag, a scaffold sequence (GS linker) and a ribosome stall sequence (secM + diproline).
- the base sequence of the polynucleotide construct is shown. However, it goes without saying that the poly
- Polynucleotide construct for mRNA display method
- Fab second-chain expression cistron 3′-side Fab chain expression cistron
- a complex of Fab chain and polynucleotide is formed using puromycin or a derivative thereof, and an amino acid sequence and a base encoding the same. Sequences may be physically associated (mRNA display).
- the C terminus of the translation product Is covalently bound to puromycin or a derivative thereof to form a complex of the Fab chain and the polynucleotide, and the amino acid sequence of the Fab chain can be associated with the base sequence encoding it.
- FIG. 5 shows an example of an embodiment in which the Fab first strand expression cistron uses a ribosome display and the Fab second strand expression cistron uses an mRNA display in the polynucleotide construct according to the second embodiment of the present invention.
- Polynucleotide construct used for CIS display method (Bisictronic)
- Fab second strand expression cistron 3 ′ Fab chain expression cistron
- a complex of Fab chain and polynucleotide is formed using the DNA binding protein coding sequence and the binding sequence of the DNA binding protein, An amino acid sequence and a base sequence encoding the amino acid sequence may be physically associated (CIS display: WO2004 / 22746).
- CIS display WO2004 / 22746
- the binding of the DNA-binding protein coding sequence and the DNA-binding protein at the 3 ′ end of the Fab second-strand expression cistron that is, downstream of the Fab second-strand coding sequence and the scaffold sequence of the Fab second-strand expression cistron.
- Sequences are ligated, Fab second strand expression cistron is expressed as a fusion protein of Fab second strand, scaffold sequence and DNA binding protein, and DNA binding protein binds to DNA binding protein binding sequence downstream of Fab second strand expression cistron By doing so, a complex of the Fab second strand and the polynucleotide is formed, and the amino acid sequence of the Fab second strand can be associated with the base sequence encoding it.
- the DNA binding protein binds to a DNA molecule that becomes a template at the time of transcription and translation and binds to the binding sequence of the DNA binding protein that is present on the same DNA molecule without dissociating once during the transcription and translation reaction.
- RepA protein having a so-called cis-type binding mode is exemplified, and the RepA binding sequence includes a CIS sequence followed by an ori sequence (Proc. Natl. Acad. Sci. USA, vol. 101, p. 2806). -2810, 2004 and Special Table 2005-537795).
- Other DNA binding proteins with a cis-type binding mode include RecC protein encoded by E. coli Ti plasmid (Pinto, et al., Mol. Microbiol.
- ⁇ X174 phage A protein (Francke, et al., Proc Natl Acad Sci USA (1972) vol.69, p.475-479), ⁇ phage Q protein (Echols, et al., Genetics (1976) vol.83, p .5-10) etc. may be used in combination with the binding sequence for each of the DNA binding proteins.
- the synthesized protein when using a cell-free translation system in which transcription and translation are well coupled, the synthesized protein is released in the vicinity of the transcription termination site, so the DNA binding domain of nuclear receptors such as estrogen receptor, Two- What is generally considered to be a DNA protein having a trans-type binding mode, such as the LexA and Gal4 DNA binding domains used in the Hybrid System, is used in combination with a binding sequence for each of the DNA binding proteins. Also good.
- FIG. 6 shows an example of an embodiment in which the Fab first strand expression cistron uses a ribosome display and the Fab second strand expression cistron uses a CIS display in the polynucleotide construct according to the second embodiment of the present invention.
- the polynucleotide construct of the present invention may be mRNA or DNA that transcribes mRNA.
- “express Fab” means translation from mRNA to Fab protein
- “express Fab” means transcription from DNA to mRNA and It means translation of Fab protein from mRNA.
- the polynucleotide construct is DNA, it is preferable to further include a promoter sequence recognized by RNA polymerase for transcription of mRNA.
- the promoter can be appropriately selected according to the expression system to be used. For example, when an E. coli cell or a cell-free translation system derived from E. coli is used, promoters that function in E. coli such as T7 promoter, T3 promoter, SP6 promoter, or endogenous promoter in the E. coli genome are exemplified.
- the above polynucleotide construct may be incorporated into a plasmid vector, a phage vector, a virus vector, or the like.
- the type of vector can be appropriately selected according to the translation system or screening system used.
- the polynucleotide construct and a vector containing the polynucleotide construct can be prepared by a known genetic technique described in Molecular Cloning (Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001).
- either the Fab H chain coding sequence or the Fab L chain coding sequence may be arranged first. Place the Fab H chain coding sequence first (5 'side), translate the H chain first and wait nearby, and pairing immediately after completing the L chain translation, pairing H chains There is an advantage that the risk of pairing between L chains, which is said to occur more easily, can be reduced. On the other hand, if the Fab L chain coding sequence is placed first (5 'side), the L chain is first translated and waited nearby, and when the H chain translation is completed, pairing is performed immediately. It is possible to reduce the risk that the H chain, which is said to have poor physical properties and high risk of aggregation, proceeds to aggregation.
- the method for producing a Fab of the present invention includes a step of introducing the polynucleotide construct into a cell-free translation system containing a ribosome to generate Fab.
- the cell-free translation system include cell-free translation systems obtained from cells such as E. coli, yeast, and mammalian cells, but cell-free translation systems derived from E. coli are preferred.
- the cell-free translation system may be a cell extract obtained by extracting a ribosome-containing fraction from cells, or a reconstituted cell-free translation system composed of individually purified factors.
- the cell extract type cell-free translation system is generally prepared by crushing cells and adding an appropriate treatment to a cell extract called S30 from which unnecessary substances are removed by ultracentrifugation or the like of about 30,000 g.
- S30 a cell extract from which unnecessary substances are removed by ultracentrifugation or the like of about 30,000 g.
- E. coli Zubay, Annual Review of Genetics (1973) vol.7, p.267-287
- wheat germ Robotts, et al., Proc Natl Acad Sci U S A (1973) vol. 70, p. 2330-2334)
- rabbit reticulocytes Pelham, et al., Eur. J. Biochem. (1976) vol. 67, p. 247 -256 is generally used.
- S30 can be prepared using various mutants depending on the purpose.
- the SL119 strain Lesley, et
- a mutant mutant of RecD which is a subunit of the RecBCD complex (Exonuclease V). al., J. Biol. Chem. (1991) vol. 266, p. 2632-2638) can be used.
- Examples of the reconstituted cell-free translation system composed of individually purified factors include the PURE system described in JP2003-102495A and JP2008-271903A.
- This reconstituted cell-free translation system can more easily prevent nucleases and proteases from being mixed than a cell-free translation system using a cell extract, so that the efficiency of translation from mRNA to polypeptide can be increased.
- the CIS display since a relatively stable double-stranded DNA is used as a genetic medium in a cell extract, it is also possible to use a cell extract obtained by extracting a fraction containing ribosome from cells.
- RNA which is a genetic medium
- RNase RNase
- a reconstituted cell-free system composed of individually purified factors.
- a translation system is preferred.
- Ribosome display using such a PURE system is called “PURE ribosome display (PRD)”.
- Each factor can be obtained by purification from various cell extracts.
- Examples of the cell for purifying the factor include prokaryotic cells and eukaryotic cells.
- Prokaryotic cells can include E. coli cells, hyperthermophilic cells, or Bacillus subtilis cells.
- Eukaryotic cells can include yeast cells, plant cells, insect cells, or mammalian cells.
- the 50S subunit and the 30S subunit are composed of the following components, respectively.
- ribosomes can be isolated as complexes of each subunit.
- Purified ribosomes for example, in prokaryotic ribosomes, are complexed as 70S ribosomes composed of large and small subunits, or mixed with purified 50S subunits and 30S subunits, respectively. Refers to a complex.
- Ribosome (80S) Large subunit (60S) + Small subunit (40S) Therefore, when a cell-free translation system is composed of ribosomes derived from eukaryotic cells, it is possible to use ribosomes purified as 80S ribosomes. it can.
- factors other than ribosome added to the cell-free translation system include the following factors. These factors are not limited to those derived from prokaryotic cells such as Escherichia coli, but those derived from eukaryotic cells can also be used. These factors and methods for purifying the factors are known (Japanese Patent Laid-Open No. 2003-102495). Initiation factor (IF), Elongation Factor (EF), Aminoacyl-tRNA synthetase, Methionyl tRNA transformylase (MTF) Although a dissociation factor may also be included, it is preferable not to include a dissociation factor in order to stably maintain the protein-polynucleotide complex on the ribosome.
- IF Initiation factor
- EF Elongation Factor
- MTF Methionyl tRNA transformylase
- a dissociation factor may also be included, it is preferable not to include a dissociation factor in order to stably maintain the
- the initiation factor is a factor essential for or significantly promoting the formation of the translation initiation complex
- IF1, IF2 and IF3 are known as those derived from E. coli (Claudio O et al. (1990) Biochemistry, vol.29, p.5881-5889).
- Initiation factor IF3 promotes dissociation of 70S ribosomes into 30S and 50S subunits, a step necessary for initiation of translation, and other than formylmethionyl tRNA during the formation of the translation initiation complex. Inhibits the insertion of tRNA into the P site.
- Initiation factor IF2 binds to formylmethionyl tRNA and carries formylmethionyl tRNA to the P site of the 30S ribosomal subunit to form a translation initiation complex.
- Initiation factor IF1 promotes the functions of initiation factors IF2 and IF3.
- initiation factor derived from E. coli it can be used at, for example, 0.01 ⁇ M to 300 ⁇ M, preferably 0.04 ⁇ M to 60 ⁇ M.
- EF-Tu, EF-Ts and EF-G are known as elongation factors derived from E. coli.
- EF-Tu elongation factor EF-Tu
- GTP type binds aminoacyl tRNA and carries it to the A site of ribosome. When EF-Tu leaves the ribosome, GTP is hydrolyzed and converted to GDP.
- Elongation factor EF-Ts binds to EF-Tu (GDP type) and promotes conversion to GTP type (Hwang YW et al. (1997) Arch. Biochem.
- the elongation factor EF-G promotes the translocation reaction after the peptide bond formation reaction in the peptide chain elongation process (AgrawalRK et al, (1999) Nat. Struct. Biol., Vol.6, p.643). -647, Rodnina MW. Et al, (1999) FEMS Microbiology Reviews, vol.23, p.317-333).
- an elongation factor derived from E. coli it can be used at 0.005 ⁇ M to 200 ⁇ M, preferably 0.02 ⁇ M to 50 ⁇ M.
- Aminoacyl-tRNA synthetases is an enzyme that synthesizes aminoacyl tRNAs by covalently binding amino acids and tRNAs in the presence of ATP, and there is an aminoacyl tRNA synthetase corresponding to each amino acid.
- AAARS Aminoacyl-tRNA synthetases
- an aminoacyl-tRNA synthetase derived from E. coli it can be used at, for example, 0.01 ⁇ g / ml to 10,000 ⁇ g / ml, preferably 0.05 ⁇ g / ml to 5,000 ⁇ g / ml.
- Methionyl tRNA transformylase is an enzyme that synthesizes N-formylmethionyl (fMet) -initiated tRNA in which the amino group of methionyl-initiated tRNA has a formyl group in protein synthesis in prokaryotes. That is, the methionyl tRNA transformylase transfers the formyl group of FD to the amino group of the methionyl start tRNA corresponding to the start codon to form fMet-start tRNA (Ramesh V et al, (1999) Proc.Natl.Acad Sci. USA, vol.96, p.875-880).
- MTF does not exist in the protein synthesis system in eukaryotic cytoplasm, but exists in the protein synthesis system in eukaryotic mitochondria and chloroplasts.
- Preferred examples of MTF are those derived from E. coli, such as those obtained from E. coli K12. When MTF derived from E. coli is used, it can be used, for example, at 100 ⁇ U / ml to 1,000,000 ⁇ U / ml, preferably 500 ⁇ U / ml to 400,000 ⁇ U / ml.
- the activity to form 1 ⁇ pmol of fMet-initiated tRNA per minute is defined as 1 ⁇ U.
- formyl donor (FD) which is a substrate of MTF can be used at, for example, 0.1 ⁇ g / ml to 1000 ⁇ g / ml, preferably 1 ⁇ g / ml to 100 ⁇ g / ml.
- RNA polymerase for transcription to mRNA can be included. Specifically, the following RNA polymerase can be used. These RNA polymerases are commercially available. T7 RNA polymerase T3 RNA polymerase SP6 RNA polymerase When T7 RNA polymerase is used, for example, 0.01 ⁇ g / ml to 5000 ⁇ g / ml, preferably 0.1 ⁇ g / ml to 1000 ⁇ g / ml can be used. In the case of pursuing the efficiency of Fab-DNA complex formation by using a reconstructed cell-free translation system in CIS display, it is described in Nucleic Acid Research 2010, vol. 38, No.
- the cell-free translation system can further contain additional components in addition to factors for transcription and translation.
- additional components the following components can be shown, for example.
- Enzymes for regenerating energy in the reaction system Creatine kinase; Enzymes for degradation of inorganic pyrophosphate, such as myokinase; and nucleoside diphosphate kinase, produced by transcription and translation:
- the above-mentioned enzyme such as inorganic pyrophosphatase can be used at, for example, 0.01 ⁇ g / ml to 2000 ⁇ g / ml, preferably 0.05 ⁇ g / ml to 500 ⁇ g / ml.
- the cell-free translation system preferably contains amino acids, nucleoside triphosphates, tRNA, and salts. Furthermore, when the reaction system is derived from prokaryotic cells such as Escherichia coli, it preferably contains the methionyl tRNA transformylase and 10-formyl 5,6,7,8-tetrahydrofolic acid (FD).
- FD 10-formyl 5,6,7,8-tetrahydrofolic acid
- non-natural amino acids can also be used as amino acids. These amino acids are retained in tRNA by the action of aminoacyl tRNA synthetase constituting a cell-free translation system. Alternatively, amino acids can be charged to tRNA in advance and added to the cell-free translation system. Here, the charge of amino acid to tRNA means that tRNA is carried (carrying) to be used for translation reaction in ribosome. Introducing unnatural amino acids at specific codon sites in proteins by adding unnatural amino acids in the presence of artificial aminoacyl synthases that recognize unnatural amino acids or using tRNA charged with unnatural amino acids It becomes possible. When a natural amino acid is used, it can be used, for example, at 0.001 to 10 mM, preferably 0.01 to 2 mM.
- tRNA tRNA purified from cells such as Escherichia coli and yeast can be used. Artificial tRNA in which anticodons and other bases are arbitrarily changed can also be used (Hohsaka, Tet al. (1996) J. Am. Chem. Soc., Vol.121, p.34-40, Hirao I et al (2002) Nat. Biotechnol., Vol. 20, p.177-182). For example, by charging an unnatural amino acid to a tRNA having CUA as an anticodon, it is possible to translate a UAG codon that is originally a stop codon into an unnatural amino acid.
- an artificial aminoacyl-tRNA in which a tRNA having a 4-base codon as an anticodon is charged with an unnatural amino acid, it is possible to translate a non-naturally occurring 4-base codon into an unnatural amino acid (Hohsaka et al. (1999) J. Am. Chem. Soc., Vol. 121, p. 12194-12195).
- a method for producing such an artificial tRNA a method using RNA can also be used (Special Table 2003-514572).
- a protein into which an unnatural amino acid has been introduced can be synthesized in a site-specific manner.
- an E. coli tRNA mixed solution for example, it can be used at 0.1 A 260 / ml to 1000 A 260 / ml, preferably 1 A 260 / ml to 500 A 260 / ml.
- the reconstituted cell-free translation system can be prepared by adding each factor to a buffer that maintains pH 7-8 suitable for transcription and translation.
- a buffer that maintains pH 7-8 suitable for transcription and translation.
- the buffer include potassium phosphate buffer (pH 7.3) and Hepes-KOH (pH 7.6).
- Hepes-KOH pH 7.6
- it can be used, for example, at 0.1 m to 200 mM, preferably 1 mM to 100 mM.
- the cell-free translation system can also contain salts for the purpose of protecting factors and maintaining activity.
- Specific examples include potassium glutamate, potassium acetate, ammonium chloride, magnesium acetate, magnesium chloride, calcium chloride and the like. These salts are usually used at 0.01 to 1000 ⁇ m, preferably 0.1 to 200 ⁇ m.
- the cell-free translation system can contain other low-molecular compounds as an enzyme substrate and for the purpose of improving and maintaining the activity.
- substrates such as nucleoside triphosphates (ATP, GTP, CTP, UTP, etc.), polyamines such as putrescine, spermidine, reducing agents such as dithiothreitol (DTT), and creatine
- DTT dithiothreitol
- creatine A substrate for energy regeneration such as phosphoric acid can be added to the cell-free translation system.
- These low molecular weight compounds can be used usually in the range of 0.01 to 1000 ⁇ m, preferably 0.1 to 200 ⁇ m.
- the specific composition of the cell-free translation system is described by Shimizu et al. (Shimizu et al., Nat. Biotechnol. (2001) vol.19, p.751-755, Shimizu et al., Methods (2005) vol.36, p 299-304), or Ying et al. (Ying et al., Biochem. Biophys. Res. Commun. (2004) vol. 320, p. 1359-1364).
- the concentration of each factor can be appropriately increased or decreased according to the specific activity or purpose of the purified factor. For example, ATP can be increased if energy consumption increases. It is also possible to add a specific tRNA according to the frequency of use of the codon of the mRNA to be translated.
- the cell-free translation system can also include a group of proteins called molecular chaperones in the case of proteins that are difficult to form higher-order structures.
- molecular chaperones include cell-free translation systems to which Hsp100, Hsp90, Hsp70, Hsp60, Hsp40, Hsp10, low molecular weight Hsp, homologs thereof, and E. coli trigger factors are added.
- Molecular chaperone is a protein known to help the formation of higher-order protein structures in cells and prevent protein aggregation (Bukau and Horwich, Cell (1998) vol.92, p.351-366, Young). et. al., Nat. Rev. Mol. Cell Biol (2004) vol. 5, p. 781).
- the redox potential of the reaction solution is important because it forms a disulfide bond in the molecule. Therefore, DTT which is a reducing agent can be removed from the reaction solution, and further a cell-free translation system to which glutathione is added can be used. Furthermore, it is possible to use a cell-free translation system to which an enzyme that promotes disulfide bonds or recombines to correct bonds is added. Specifically, examples of such an enzyme include protein disulfide isomerase (PDI) present in ER of eukaryotic cells, DsbA and DsbC of E. coli.
- PDI protein disulfide isomerase
- Fab can be obtained by introducing a polynucleotide construct into the above cell-free translation system and performing a translation reaction.
- the obtained Fab can also be purified using an affinity column or the like.
- the screening method of the present invention comprises the steps of (i) introducing a polynucleotide construct encoding a library of Fabs into a cell-free translation system, synthesizing the Fab, and presenting the synthesized Fab on the polynucleotide encoding the Fab. , (Ii) contacting the Fab displayed on the polynucleotide with the antigen, (iii) selecting the target Fab that reacts with the antigen, and (iv) amplifying the polynucleotide encoding the target Fab. .
- Fab is displayed on ribosomes and / or mRNA based on given genetic information.
- ribosome display a polypeptide containing Fab is maintained with genetic information (mRNA) encoding ribosomes on mRNAs due to the presence of a ribosome stall sequence at the 3 'end. That is, a complex of three elements of mRNA-ribosome-polypeptide is formed.
- the C-terminal of the polypeptide containing Fab is shared by puromycin or a derivative thereof due to the presence of puromycin or a derivative thereof at the 3 ′ end of the polynucleotide construct (mRNA). It binds and maintains the link between the polypeptide containing the Fab and the mRNA that encodes it.
- a Fab-containing polypeptide is expressed as a fusion protein with a DNA-binding protein, and the DNA-binding protein binds to a target sequence on the DNA, thereby linking the Fab-containing polypeptide to the DNA that encodes it. Maintained.
- the genetic information is also recovered by selecting the Fab bound to the antigen and recovering the mRNA bound thereto.
- CDNA is synthesized from mRNA in a complex containing Fab and mRNA bound to the target antigen, amplified by PCR, and then subjected to transcription / translation reaction again. By repeating these steps, an antibody against the target antigen can be obtained.
- the scale of the Fab gene library is usually 1 ⁇ 10 8 or more, preferably 1 ⁇ 10 9 or more, more preferably 1 ⁇ 10 10 or more, and further preferably 1 ⁇ 10 12 or more.
- a Fab library expressed from the Fab gene library is brought into contact with a target substance (antigen), a Fab that binds to the target substance is selected from the Fab library, and a polynucleotide encoding it is amplified.
- the basic panning protocol is as follows. (1) Contact the Fab library with the target substance.
- a target substance may be bound to a carrier such as a bead, plate or column, and a sample containing a complex of Fab and polynucleotide may be contacted there (solid phase selection).
- the parent Fab and the antigen it is preferable to carry out the washing under conditions such that the bonds are maintained and weaker bonds are removed. (3) Collect the Fab that was not removed, that is, the Fab that was specifically bound to the target substance. (4) The operations (1) to (3) are repeated a plurality of times as necessary.
- RNA display In the case of ribosome display, mRNA display, and CIS display, when a series of steps is repeated, the polynucleotide in the complex containing the recovered polypeptide-polynucleotide is amplified before step (1).
- mRNA can be amplified by RT-PCR. DNA is synthesized by RT-PCR using mRNA as a template. The DNA can be transcribed again into mRNA and used for complex formation.
- Figures 7 and 8 show a series of Fab screening methods that transcribe mRNA from DNA, translate it to produce Fab, select the Fab bound to the antigen, collect the mRNA, and amplify it. The schematic diagram regarding an example of is shown.
- the polynucleotide encoding Fab that specifically binds to the target antigen is concentrated.
- the amino acid sequence information of the target Fab can be identified by analyzing the sequence of the obtained polynucleotide.
- an antibody having a further increased affinity can be obtained using the screening method of the present invention. That is, in the present invention, (I) An amino acid sequence in which the Fab first chain coding sequence or Fab second chain coding sequence contains one amino acid substitution for one position in the CDR in the amino acid sequence of the Fab first chain or Fab second chain of the parent antibody.
- a method for screening a Fab comprises a secondary screening step of screening the high affinity Fab by repeating the steps (i) to (iv) using the polynucleotide construct.
- the plural in step (I) may be two or more, preferably all amino acids in CDR.
- an amino acid sequence (parent sequence) of an antibody (parent antibody) against the target antigen is prepared.
- this amino acid sequence one amino acid in one of the CDR amino acids of the H chain (8 CDR-H1, 11 CDR-H2, 12 CDR-H3, 31 in total).
- a polynucleotide construct of the present invention comprising an H chain coding sequence having a substitution (preferably a single amino acid substitution in which all 20 natural amino acids including the parent amino acid appear) and the L chain coding sequence of the parent antibody is converted into the CDR of the H chain.
- one amino acid substitution preferably, one of the CDR amino acids of the light chain (7 CDR-L1, 6 CDR-L2, 6 CDR-L3, 19 in total)
- H chain library The H chain library and the L chain library are used as primary libraries, and using these, steps (i) to (iv) are repeated to screen for high affinity antibodies (primary screening).
- polynucleotide sequence encoding the high affinity antibody is enriched by the primary screening, a plurality of enriched sequences are analyzed, and amino acid substitutions frequently observed at each position of the CDRs of the H chain and L chain Is selected as a preferred amino acid substitution for improved affinity.
- next-generation sequencer used in contrast to the first-generation sequencer represented by the fluorescent capillary sequencer using the Sanger sequencing method actually includes various devices and technologies, and will continue to vary.
- first-generation sequencers are generally used for sequencing DNA cloned using a vector host system
- next-generation sequencers use vector host systems while analyzing DNA samples with diverse sequences.
- the DNA sequence present in the sample can be decoded at high speed without cloning the DNA.
- DNA sequencing using the first generation sequencer (i) integration of a DNA fragment into a vector such as a plasmid and transformation of a host such as E.
- DNA sequencing using next-generation sequencers DNA fragments containing various sequences are applied to amplification technologies such as single-molecule PCR methods such as Emulsion PCR and Bridge PCR, or high-sensitivity detection technologies such as single-molecule observation. By doing so, it is cloned on individual analysis spots existing on a large number of substrates on the analysis substrate, and sequence analysis is performed in a super parallel manner. Therefore, for example, the sequence of 10 6 to 10 9 clones formed on one analysis substrate can be determined by a single reaction and treatment regardless of the number of analysis clones.
- amplification technologies such as single-molecule PCR methods such as Emulsion PCR and Bridge PCR, or high-sensitivity detection technologies such as single-molecule observation.
- next-generation sequencer is based on the principle of determining a base sequence in a super-parallel manner by light detection such as fluorescence and luminescence using a sequential DNA synthesis method using DNA polymerase or DNA ligase.
- GS ⁇ FLX Roche Diagnostics
- Solexa Illumina
- SOLiD Applied Biosystems
- Polonator Polonator
- DNA synthesis is performed with DNA polymerase using one DNA molecule as a template, and the base sequence is determined by real-time observation of one molecule by detecting the reaction of each base with light such as fluorescence or luminescence.
- Helitos® (Helicos® Biosciences), SMRT ( Pacific Biosciences) and the like are known.
- the principle of determining base sequences in a super-parallel manner using a detection method other than light detection such as fluorescence and luminescence, called post-light sequencing, has been reported.
- Nanopore (Oxford Nanopore Technologies) is known.
- An amino acid sequence including a combination of 1 amino acid substitution of each amino acid of CDR of H chain and L chain obtained by primary screening is used as a secondary library.
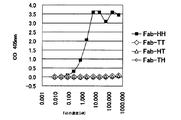
- the first amino acid of the first CDR (H1) of the H chain is preferably substituted with Ala or Thr, and the second amino acid is substituted with Ile or Leu.
- the library used for the secondary screening was Ala or Thr as the first amino acid, Ile or Leu as the second amino acid, and other amino acids were obtained in the primary screening.
- a secondary library is prepared by designing a base sequence so as to be a preferable combination of amino acids. In addition, it is good also considering the combination of the preferable variant amino acid obtained by the primary screening, and the amino acid of a parent sequence as a secondary library.
- steps (i) to (iv) are repeated, and secondary screening is performed, so that a large number of antibodies with significantly improved affinity for the antigen over the parent antibody are obtained. Can be acquired. It can be analyzed by SPR, ELISA, etc. that the affinity for the antigen is significantly improved compared to the parent antibody.
- the polynucleotide construct of the present invention can be used as a component of a kit for producing or screening a Fab.
- the principle of two-step affinity maturation consisting of a search for potential useful single amino acid substitutions using a primary library and an optimal combinatorial search for these useful single amino acid substitutions using a secondary library is based on the antibody Fab Not only fragments but also scFv, full-length antibodies, and other target binding proteins can be applied in combination with a wide range of protein display systems to improve target affinity.
- the 1-position library constructed in step (I) is divided into two or more groups and two or more types of primary libraries are used. May be used.
- the parent amino acid encoded by the parent antibody may be added to the secondary library constructed in (VI).
- examples of the target substance binding protein include antigen binding proteins and cytokines.
- Antigen-binding proteins include antibodies such as Fab, scFv, and full-length antibodies, but may be proteins that are not classified as antibodies as long as they specifically bind to antigens. It is also possible to introduce a mutation into the cytokine to enhance the binding to the receptor.
- the protein display system is not particularly limited as long as the protein and the polynucleotide encoding the protein are associated with each other.
- the protein display system is not limited to cell-free systems such as ribosome display, mRNA display, and CIS display as described above, but also phage display.
- Non-Patent Document 1 Non-Patent Document 1
- bacterial surface display Jose, Appl. Microbiol. Biotechnol. (2006) vol.69, p.607-614
- yeast surface display Yamadhaus et al., Nat. Biotechnol. (2003) vol. .21, p.163-70
- other eukaryotic cell surface displays Horlick, WO 2008/103475.
- Portions other than the target substance-binding protein coding region of the 1-position library polynucleotide can be configured according to known protein display vectors.
- the target substance binding protein coding region contained in the library polynucleotide may encode the full length of the target substance binding protein, or may encode a part of the target substance binding protein including the target substance binding site.
- the target substance binding protein wild type
- the target substance binding site is a region consisting of 10 amino acids
- a total of 10 1-position libraries with 20 amino acids appearing at one position from 1 to 10 are prepared.
- These 10 types of one-position libraries are combined to form a primary library, which is used to enrich with target substance affinity.
- the following steps (i ′) to (iv ′) are repeated to concentrate a high affinity sequence on the target substance.
- a target substance may be bound to a carrier such as a bead, a plate, or a column, and a sample containing the complex of the target substance-binding protein and the polynucleotide may be brought into contact therewith (solid phase selection). It is also possible to select in a liquid phase such that the target substance is biotinylated, bound to the target substance-binding protein, and the complex of the target substance and the target substance-binding protein is recovered with streptavidin magnetic bead beads. Further, solid phase selection and liquid phase selection may be combined. By conducting multiple rounds of screening, solid phase selection in the first half and liquid phase selection in the second half, high affinity proteins can be selected efficiently.
- washing term operation between (ii ′) and (iii ′). Washing is preferably performed under conditions such that the binding between the wild-type target substance binding protein and the target substance is maintained, and weaker binding is removed.
- polynucleotide sequence encoding the target substance high-affinity protein is enriched by the primary screening, the sequences of the multiple target substance binding sites are analyzed, and the frequency of each of the CDR positions of the H chain and L chain is high. Are selected as preferred amino acid substitutions for improving affinity.
- Polynucleotide sequence analysis may be performed by normal sequencing, but it can be comprehensively analyzed in large quantities without cloning the target substance-binding protein coding sequence amplified by screening, greatly reducing screening time. It is preferable to use a next-generation sequencer in that it can be used.
- next-generation sequencer used in contrast to the first-generation sequencer represented by the fluorescent capillary sequencer using the Sanger sequencing method actually includes various devices and technologies, and will continue to vary.
- first-generation sequencers are generally used for sequencing DNA cloned using a vector host system
- next-generation sequencers are designed to analyze vector samples with diverse sequences, The DNA sequence present in the sample can be decoded at high speed without using it to clone the DNA.
- DNA sequencing using the first generation sequencer (i) integration of a DNA fragment into a vector such as a plasmid and transformation of a host such as E. coli, (ii) cloning by isolation of host colonies; (iii) Cultivation of a number of clones and plasmid extraction according to the analysis scale, (iv) a sequencing reaction by PCR or the like using a plasmid or the like as a template, (v) Separation, detection and analysis of sequence reaction products using capillary electrophoresis, Through this process, the DNA sequence of each clone is determined.
- DNA sequencing using next-generation sequencers DNA fragments containing various sequences are applied to amplification technologies such as single-molecule PCR methods such as Emulsion PCR and Bridge PCR, or high-sensitivity detection technologies such as single-molecule observation. By doing so, it is cloned on individual analysis spots existing on a large number of substrates on the analysis substrate, and sequence analysis is performed in a super parallel manner. Therefore, for example, the sequence of 10 6 to 10 9 clones formed on one analysis substrate can be determined by a single reaction and treatment regardless of the number of analysis clones.
- next-generation sequencer is based on the principle of determining a base sequence in a super-parallel manner by light detection such as fluorescence and luminescence using a sequential DNA synthesis method using DNA polymerase or DNA ligase.
- GS ⁇ FLX Roche Diagnostics
- Solexa Illumina
- SOLiD Applied Biosystems
- Polonator Polonator
- DNA synthesis is performed with DNA polymerase using one DNA molecule as a template, and the base sequence is determined by real-time observation of one molecule by detecting the reaction of each base with light such as fluorescence or luminescence.
- Helitos® (Helicos® Biosciences), SMRT ( Pacific Biosciences) and the like are known.
- the principle of determining base sequences in a super-parallel manner using a detection method other than light detection such as fluorescence and luminescence, called post-light sequencing, has been reported.
- Nanopore (Oxford Nanopore Technologies) is known.
- each sequence can be used for secondary screening without cloning (cloning into a vector and expressing individually). Therefore, there is an advantage that the screening time can be greatly shortened. However, for the purpose of confirming the efficiency of the primary screening, it may be partially cloned and the binding property may be examined.
- one amino acid substitution at each amino acid of the target substance binding site obtained by sequence analysis in the primary screening is combined to obtain a secondary library.
- the result is that the first amino acid of the target substance binding site is preferably substituted with Ala or Thr, and the second amino acid is preferably substituted with Ile or Leu.
- the library used for the secondary screening is Ala or Thr as the first amino acid, Ile or Leu as the second amino acid, and the other amino acids are the preferred amino acids obtained in the primary screening.
- the sequence is designed to create a secondary library. Note that a combination of a preferable mutated amino acid obtained in the primary screening and the amino acid of the parent sequence may be used as the secondary library.
- steps (i ') to (iv') are repeated, and secondary screening is performed to obtain a large number of proteins with significantly improved affinity for the target substance over the parent protein.
- secondary screening is performed to obtain a large number of proteins with significantly improved affinity for the target substance over the parent protein.
- Example 1 Preparation of model Fab: As a Fab framework used in cell-free Fab display systems, it is one of the major antibody subclasses in humans and has excellent expression efficiency in E. coli (Kanappik et al., J Mol Biol. (2000) vol.296, p. 57-86), and results in a cell-free scFv display system were reported (Shibui et al., Appl Microbiol Biotechnol. (2009) vol.84, p.725-732), VL k subgroup I and VH subgroup III. A combination was selected.
- Fab-HH As a model Fab having the above-mentioned framework used for confirming the operation of the cell-free Fab display system, a human EGFR-2 (Her-2) reactive Fab (Carter et al., Proc Natl Acad Sci US A. (1992) vol. 89, p.4285-4289) was selected as Fab-HH. Another model Fab is the CDR1-3 region of Fab-HH L chain and H chain, and the sequence of TNF ⁇ R-reactive scFv found in Shibui et al.'S cell-free scFv display system (Shibui et al., Biotechnol. Lett (2009) vol.31, p.1103-1110) and replaced with Fab-TT.
- CDNAs encoding the VL domain and VH domain of these two types of model Fabs were artificially synthesized.
- cDNA encoding the CL domain and the CH1 domain was obtained by PCR using the genomic DNA of a human cancer cell line as a template. The obtained cDNA fragment was incorporated into an E. coli expression vector pTrc99A, and pTrc Fab-HH and pTrc Fab-TT were constructed as vectors for secreting and expressing model Fabs in Bicistronic.
- the combination of the original L chain and H chain was intentionally changed to produce Fab-HT (a combination of Her-2 reactive L chain and TNF ⁇ R reactive H chain) and Fab-TH (TNF ⁇ R reactive L chain).
- FIG. 9 shows a schematic structure of a unit that expresses Fab in Bicistronic in the pTrc Fab vector.
- the base sequence of pTrc Fab-HH is shown in SEQ ID NO: 1, and the encoded amino acid sequence is shown in SEQ ID NO: 2 and 3.
- the nucleotide sequence of pTrc Fab-TT is shown in SEQ ID NO: 4, and the encoded amino acid sequence is shown in SEQ ID NOs: 5 and 6.
- the base sequence of pTrc Fab-HT is shown in SEQ ID NO: 7, and the encoded amino acid sequences are shown in SEQ ID NOs: 8 and 9.
- the base sequence of pTrc Fab-TH is shown in SEQ ID NO: 10, and the encoded amino acid sequences are shown in SEQ ID NOs: 11 and 12.
- the Escherichia coli DH5 ⁇ strain carrying the pTrc Fab vector was cultured in LB medium containing 100 ⁇ g / ml ampicillin, and model Fab was expressed in the culture supernatant by adding IPTG at a final concentration of 0.1 mM in the logarithmic growth phase. . After culturing for 18 hours after adding IPTG, the culture supernatant was collected, and Ni-NTA agarose beads (QIAGEN) were used using the His tag in which the model Fab contained therein was added to the C-terminus of the H chain. Combined. The beads were washed and eluted with PBS containing imidazole at a final concentration of 250 mM.
- the eluted fraction was dialyzed against PBS to prepare a high purity model Fab.
- Samples were separated by 5-20% gradient gel SDS-PAGE and stained with Coomassie blue. The results are shown in FIG. It can be seen that the purity of the prepared model Fab is sufficient.
- FIG. 12 shows the results of contacting four types of model Fabs with two types of antigen proteins at high concentrations (7.1, 1.8, 2.9, and 5.8 ⁇ M in the order of HH, TT, HT, and TH). It can be seen that Fab-HH binds to Her-2 but not to TNF ⁇ R, and Fab-TT binds to TNF ⁇ R but not to Her-2.
- Example 3 Construction of a vector for Bicistronic Fab-PRD: It has an amino acid sequence very similar to the above Fab-HH, and Fellouse et al. (Fellouse et al., J. Mol. Biol. (2007) vol.373, p.924-940) will further improve the expression efficiency in E. coli.
- the reported sequence was designated as Fab-SS.
- a DNA fragment for Bicistronic Fab-PRD was artificially synthesized using an E. coli optimized codon. This was incorporated into a general-purpose phagemid vector pBluescript SK (+) to construct pGAv2-SS.
- pGAv2-HH was constructed by replacing the L chain CDR1-3 region and the H chain CDR1-3 region of pGAv2-SS with the corresponding region of pTrc Fab-HH.
- pGAv2-TT was constructed using pGAv2-SS and pTrc Fab-TT.
- pGAv2-HH xhoI was constructed by introducing an XhoI recognition site into the most downstream site of the GS linker downstream of the L chain of pGAv2-HH without changing the amino acid sequence.
- the schematic structure of the DNA fragment for Bicistronic Fab-PRD is shown in FIG.
- FIG. 14 shows the schematic structure of the DNA fragment for the Monocistronic Fab-PRD.
- Example 5 Preparation of template DNA: Template DNA fragments for in vitro transcription were amplified by PCR reaction using pGAv2-HH xhoI and pGAv2-TT for Bicistronic Fab-PRD and pGAv6-HH xhoI and pGAv6-TT for Monoocistronic Fab-PRD as templates. PrimeStarMax (Takara) was used as an enzyme for PCR, and a combination of PURE-rt-1F and PURE-3R, or a combination of SL-1F and SL-2R was used as a primer. The amplified template DNA fragment for in vitro transcription was purified by phenol / chloroform extraction and isopropanol precipitation.
- the sequences of template DNA fragments for in vitro transcription amplified from four types of vectors using the SL-1F and SL-2R primer sets are as follows.
- PGAv2-HH xhoI for Bicistronic Fab-PRD SEQ ID NO: 13 amino acid sequences are SEQ ID NOs: 14 (L) and 15 (H))
- pGAv2-TT SEQ ID NO: 16 amino acid sequence is SEQ ID NO: 17 (L), 18 (H)
- PGAv6-HH xhoI for Monocistronic Fab-PRD SEQ ID NO: 19 amino acid sequence is SEQ ID NO: 20
- pGAv6-TT SEQ ID NO: 21 amino acid sequence is SEQ ID NO: 22
- PURE-rt-1F (caatttcggtaatacgactcactatagggagaatttaggtgacactatagaagtg) (SEQ ID NO: 23)
- PURE-3R (caggtcagacgattggccttg) (SEQ ID NO: 24)
- SL-1F (caatttcggtaatacgactcactatagggagaccacaacggtttcccatttaggtgacactatagaagtg) (SEQ ID NO: 25)
- SL-2R (ccgcacaccagtaaggtgtgcggcaggtcagacgattggccttgatattcacaaacg) (SEQ ID NO: 26)
- Example 7 Translation of mRNA by a reconstructed cell-free translation system (PURE system): Translation factors and ribosomes that make up the PURE system are described in Shimizu (Shimizu et al., Nat Biotechnol. (2001) vol.19, p.751-755) and Ohashi (Ohashi et al., Biochem Biophys Res Commun. (2007). ) vol.352, p.270-276) and the like. The translation reaction was scaled to 20 ⁇ l using 2 p mole of mRNA and 20 p mole of ribosome.
- oxidized glutathione and protein disulfide isomerase 1 mM DTT was added to carry out a translation reaction under reducing conditions. After the translation reaction at 37 ° C. for 20 minutes, oxidized glutathione having a final concentration of 2.5 mM was added to neutralize DTT.
- Biotinylation of antigen and quality confirmation Her-2 and TNF ⁇ R (R & D systems) were each dissolved in PBS to 0.5 mg / ml. Prepare Sulfo-NHS-SS-Biotin (Thermo Scientific) as a biotinylation reagent at 0.6 mg / ml (for Her-2) and 4.0 mg / ml (for TNF ⁇ R) in PBS, and add 1.1 ⁇ l (Her- 2) and 3.6 ⁇ l (for TNF ⁇ R) were added to 100 ⁇ l of the antigen solution and reacted at room temperature for 1 hour. The reaction product was passed through a gel filtration spin column to remove unreacted biotinylation reagent.
- Avidin (Calbiochem) was diluted to 2 ⁇ g / ml with PBS. This was added to the ELISA plate and left overnight at 4 ° C. to fix it to the plate. After washing the wells, biotinylated antigen was added and allowed to bind to avidin. After washing the wells, the high purity model Fab prepared in Example 1 was added and allowed to bind to the biotinylated antigen. After washing the wells, an HRP-labeled anti-FLAG antibody (Sigma) diluted 1000 times as an antibody for detection was added and bound to the FLAG tag added to the model Fab. After washing the wells, HRP chromogenic substrate was added.
- Fab selection by PRD In vitro selection of Fab by ribsome display method (PRD) using PURE system, referring to the method reported by Ohashi et al. Carried out. 30 ⁇ l of M-280 streptavidin magnetic beads (DYNAL) was washed, 5 pmol of the biotinylated antigen protein prepared in Example 8 was added and reacted at room temperature for 30 minutes, and the bait protein was fixed to the magnetic beads. The magnetic beads were washed and used as selection beads. Preclear beads without immobilizing bait protein were prepared in the same manner.
- PRD ribsome display method
- Fab-HHxho As a template DNA or mRNA, a small amount of Fab-HHxho was added to Fab-TT at an arbitrary content to prepare a sample before selection. Using this, an in vitro translation product was prepared by the method described in Example 7. This was mixed with preclear beads and reacted at 4 ° C. for 30 minutes to recover the supernatant. The supernatant was mixed with selection beads and reacted at 4 ° C. for 30 minutes to recover magnetic beads. After washing the magnetic beads, 100 ⁇ l of the eluate was added and reacted at room temperature for 10 minutes.
- the eluate is based on (50 mM Tris-Cl pH 7.6, 150 mM NaCl, 10 ⁇ g / ml budding yeast RNA), with a final concentration of 100 mM DTT for reducing elution and a final concentration of 50 mM EDTA for chelate elution did.
- Reduced elution is by reducing and cleaving the SS bond in the biotin linker
- chelate elution is by dissociating the magnesium ion-dependent display molecule complex (mRNA-ribosome-polypeptide tripartite complex), via Fab.
- the mRNA bound to the magnetic beads is eluted.
- MRNA was purified from the eluted sample with phenol / chloroform extraction and isopropanol precipitation, or with the RNaeasy RNA purification kit (QIAGEN).
- the eluted mRNA was converted to cDNA by a reaction using SuperScript III (Invitrogen) as a reverse transcriptase.
- PURE-2R or PURE-3R was used as a primer for reverse transcription reaction.
- the cDNA was amplified by PCR reaction using PrimeStarMax (Takara), then purified by phenol / chloroform extraction and isopropanol precipitation, and selected as a DNA sample.
- PrimeStarMax Takara
- a combination of PURE-rt-1F and PURE-3R or a combination of SL-1F and SL-2R was used as a PCR primer for amplifying the full-length mRNA sequence.
- L-CDRex-1F (gacgattggccttgatattcacaacg) (SEQ ID NO: 27)
- L-CDRex-1F (attaaacgtaccgttgcagcaccgagc) (SEQ ID NO: 28)
- H-CDRex-2R (tgagcctccaggctgaaccagaccac) (SEQ ID NO: 29)
- TT / HH ratio 10
- FIG. 18 shows the results of selecting Fab-HH from a DNA sample having a TT / HH ratio of 100 to 100,000 using a Monostronic Fab-PRD.
- the TT / HH ratio was 100 to 1000, an increase in XhoI sensitivity was observed, indicating that Fab-HH was specifically concentrated.
- Example 11 Confirmation of in vitro translation product of Monostronic Fab: A cell-free translation product was prepared using the mRNA for Monocistronic Fab-PRD by the method described in Example 7. The translation product was separated by 5-20% gradient gel SDS-PAGE, transferred to a PVDF membrane, and Western blotting was performed using an HRP-labeled anti-FLAG antibody (Sigma) diluted 1000 times as a detection antibody. The results are shown in FIG. Although the full-length translation product of scFab-TT can be confirmed, it can be seen that the ratio of the full-length translation product is extremely small compared to the translation amount of scFv-TT used as a positive control.
- Monocistronic Fab-PRD was performed by solid phase selection described in Example 9, and the recovered reverse transcription product was quantified by quantitative PCR using a template DNA fragment having a known concentration as a sample for a calibration curve.
- the recovery rate in the upstream region near CDR-L1 and the downstream region near CDR-H3 (number of 1st strand cDNA molecules / injected)
- the number of RNA molecules was determined. The results are shown in FIG. Increasing the SecM sequence to 2 copies improved the recovery rate of Fab-HH by Fab-PRD by about 10 times.
- Example 13 Relationship between linker length and concentration factor:
- the template DNA used in the Monocistronic Fab-PRD has a total of two linker sequences between the L chain and H chain and between the H chain and SecM.
- the L chain to H chain linker The length is 128 amino acids, and the linker length between H chain and SecM is 123 amino acids.
- a v6.5 fragment with two copies of SecM sequence for Fab-HH xhoI and Fab-TT was used as the basic structure, and an H chain-SecM linker A v7 fragment with a minimized length and a v8.1 fragment with a minimized L-chain-H chain linker length were prepared.
- the linker length between the H chain and SecM in the v7 fragment was 20 amino acids between the cysteine of the last amino acid of the H chain and the alanine of the first amino acid of SecM.
- the linker length between the L chain and the H chain in the v8.1 fragment was 30 amino acids between the cysteine at the C terminal of the L chain and the glutamic acid at the N terminal of the H chain.
- the recovery rates of Fab-HH and Fab-TT were measured by quantitative PCR, respectively, and the concentration factor (Fab-HH recovery rate / Fab-TT recovery rate) was determined.
- a detection primer used for quantitative PCR a downstream primer near CDR-H3 was used. The results are shown in FIG.
- Downstream primer for FabHH HH-4F ccagggaactttggttactgtttc (SEQ ID NO: 35)
- Model-4R ctttaaccagacaacccagtgc (SEQ ID NO: 36)
- Downstream primer for Fab-TT TT-5F gcaccctggttaccgtgag (SEQ ID NO: 49)
- Model-4R ctttaaccagacaacccagtgc (SEQ ID NO: 36)
- the sequences of the linker portions of v7, v8.1 and DNA fragment used for Fab-PRD are shown in SEQ ID NOs: 50 and 52, respectively.
- Example 14 Examination of minimum copy number required for Monostronic Fab-PRD: Fab-HH xhoI and Fab-TT v7 fragment (Example 13) was prepared as template DNA, and Fab-HH template DNA was prepared with a carrier solution containing 0.125 mg / ml yeast RNA and 7 ⁇ 10 9 molecules / ⁇ l Fab-HH template DNA. A 10-fold dilution series of TT template DNA was prepared. 1 x 10 6 Fab-TT molecule template DNA / ⁇ l to 1 x 10 -5 Fab-TT molecule template DNA / ⁇ l of DNA sample amplified by single molecule PCR using Fab-TT specific primers Were separated by 5-20% gradient PAGE.
- a Fab-TT specific band is detected from PCR that contains one or more Fab-TT molecules as template DNA, and no Fab-TT specific band is detected from PCR that contains 0.1 or less Fab-TT molecules. It was confirmed. 400 molecule equivalents were collected from a diluted sample of Fab-TT template DNA whose quality was confirmed by single molecule PCR, and added to 1 ⁇ 10 12 molecules of Fab-HH template DNA. RNA was synthesized from this and used as RNA before selection. Pre-selection RNA was injected into Fab-PRD and an attempt was made to recover Fab-TT equivalent to 400 molecules. The translation reaction was carried out at 30 ° C for 40 minutes on a 20 ⁇ l scale.
- 25 ⁇ l of M-280 streptavidin magnetic bead pellet was added to recover Fab-TT.
- RNA eluted from M-280 beads with DTT was selected as RNA after selection.
- Fab-TT fragments (lanes 1 and 2 in Fig. 22) are used with primers specific to Fab-TT CDR-H3, and Fab-HH and Fab-
- the core fragments (lanes 4 and 5 in FIG. 22) containing important genetic information from CDR-L1 to CDR-H3 with primers recognizing the common DNA sequence of both TT were each amplified by RT-PCR.
- the band of the core DNA fragment was recovered from a 5-20% gradient gel and reamplified by 2nd PCR (lanes 7 and 8 in FIG. 22).
- the Fab-TT fragment was detected not only from the pre-selection sample but also from the post-selection sample. Therefore, it was considered that the core fragment was successfully recovered from 400 molecules of Fab-TT.
- the concentration ratio of Fab-TT was measured by quantitative PCR. As a result of quantifying the Fab-TT fragment and the core fragment independently and calculating the concentration ratio (relative amount of Fab-TT in the sample after selection / relative amount of Fab-TT in the sample before selection), the core DNA fragment after selection (FIG. 22).
- T1P-F gttttactattgaacgttatgcgatgggt
- T1P-R cgtagtacataccgttcgggtagttag (SEQ ID NO: 55)
- Primer for core region amplification common to Fab-HH and Fab-TT Core-1F gcgcaagcgttggtgatc (SEQ ID NO: 56)
- Core-1R gctcggacctttggtgcttg (SEQ ID NO: 57)
- Example 15 Construction of Ymacs-1 primary library (first half of affinity maturation): As an application example of Fab-PRD, we attempted affinity maturation of Fab-TT, one of the model Fabs. The antigen-antibody binding interface has more than 100 van der Waals forces, several hydrogen bonds and salt bridges. To achieve global optimization of these interaction networks, search for mutations in the CDR that may be useful for improving affinity in the first stage, and search for the optimal combination of these mutations in the second stage. The original two-step strategy called Ymacs was adopted.
- FabTT Ymacs-1 L1-1 tgtcgtgcaagccagNNKattaaaattat (SEQ ID NO: 58) (primer to insert 1 amino acid substitution into the first amino acid of L chain CDR1)
- FabTT Ymacs-1 H3-12 atgtactacgttatgNNKtattggggtcag (SEQ ID NO: 59) (primer to insert 1 amino acid substitution into the 12th amino acid of H chain CDR3) 50 mutagenesis downstream DNA fragments were independently synthesized by PCR using pGAv6.5-Fab-TT as a template and the above 50 types of single amino acid substitution-introduced forward primers combined with PURE-3R primers.
- the PCR product was separated on a 1% agarose gel, and the band was confirmed by ethidium bromide staining.
- a common constant sequence upstream DNA fragment was synthesized by PCR as a template in a reaction in which the reverse strand of the mutagenized downstream DNA fragment was extended in the upstream direction to lengthen it.
- the upstream fragment of the constant sequence was synthesized by PCR using PURE-rt-1F and H3checkR Not TA6 as primers so as to have a misannealed region on the 3 terminal side in order to avoid the full length of its own forward strand,
- the PCR product was separated on a 1% agarose gel, and the band was confirmed by ethidium bromide staining.
- PURE-rt-1F caatttcggtaatacgactcactatagggagaatttaggtgacactatagaagtg (SEQ ID NO: 60)
- H3checkR Not TA6 tatatatatatagcggccgcagaactgccggaaaggtatg (SEQ ID NO: 61)
- FIG. 2 An outline of the synthesis of a 1-position 1-amino acid substitution library is shown in FIG. 2 amino acid substitution introduction sites are illustrated, the most upstream and the most downstream.
- a constant sequence upstream DNA fragment (A and B in the figure) and a mutagenized downstream DNA fragment (C and D in the figure) were mixed, and asymmetric PCR was performed in the presence of a PURE-rt-1F primer (E in the figure).
- E PURE-rt-1F primer
- each of the Ymacs-1 primary libraries is composed of 19 types of L-chains and 31 types of 1-position 1-amino acid substitution libraries.
- Example 16 Concentration of Ymacs-1 primary library and next-generation sequence analysis (second half of the first stage of affinity maturation): As the work in the latter half of the first stage, the L-chain and H-chain Ymacs-1 primary libraries were independently enriched with Fab-PRD, and the results of the next-generation sequence analysis of the obtained DNA samples suggested that it would be useful in CDR. Identified mutations.
- the enrichment of the Ymacs-1 primary library by Fab-PRD used both the solid phase selection shown in Example 9 and the liquid phase selection shown in Example 14. In round 1, solid phase selection was performed, and in round 2, washing was performed for a long time (13 h) after solid phase selection.
- the base sequence data of the H chain CDR1-3 sequence 2288 reads was recovered. From these, the L chain 468 lead and the H chain 1293 lead having a continuous ORF in the CDR1-3 region were selected as effective leads, and the L chain 19 amino acids and H chain 31 amino acids constituting the CDR, a total of 50 amino acids. Mutations at CDR positions were counted. The counts of each mutation are summarized in a matrix table in which 20 amino acids are arranged in the vertical direction and a total of 50 CDR positions are arranged in the horizontal direction, and are shown in FIG. The parent amino acid at each CDR position was excluded, and a mutation with a large count was regarded as a potential useful mutation.
- Example 17 Construction of Ymacs-secondary library (first half of affinity maturation): As an operation in the first half of the second stage, a Ymacs-2 secondary library was constructed by combining useful mutations identified in Example 16. A total of 21 mutations in 12 CDR positions were employed as useful mutations, and a mixed base codon capable of encoding the parent amino acid and the useful mutant amino acid was determined at each CDR position. Depending on the mixed base codon, an unintended amino acid other than the parent amino acid and the useful variant amino acid is included. The useful mutations and mixed base codons employed are summarized in FIG. The theoretical diversity of Ymacs-2 secondary library is 1.9 x 10 7 at the nucleic acid level and 4.9 x 10 6 at the protein level.
- FabTT Ymacs-2 L1-Fwd tgtcgtgcaagccaggatattaaaaattatttgWCTtggtatcaacaacaa (SEQ ID NO: 62) (L chain CDR1) FabTT Ymacs-2 L2-Fwd: gccccgaagccactgatttatGSTggttctaaccgccaatctggagttcct (SEQ ID NO: 63) (L chain CDR2) FabTT Ymacs-2 L3-Fwd: acctattattgccaacaaactKMTRNMtaccctatcacctttggccag (SEQ ID NO: 64) (L chain CDR3) FabTT Ymacs-2 H1-Fwd: agctgtgcagcaagcggttttASAattGRGcgttatgcgatgRSTtgggtgcgtcaggct (SEQ
- Example 18 Enrichment of Ymacs-2 secondary library and affinity analysis of clones (second half of affinity maturation): In the latter half of the second stage, the Ymacs-2 secondary library was enriched with Fab-PRD, and the resulting DNA was cloned into a secretory expression vector of E. coli. Using ELISA and SPR (ProteOn XPR36, BioRad) to narrow down high-affinity clones, the KD of two representative Fab-TT mutant clones (Ymacs # 10 and # 19 in Figures 29-32) was set to KinExA (KinExA3200, Sapidyne) ).
- Concentration of the Ymacs-2 secondary library by Fab-PRD was performed by liquid phase selection for all 5 rounds. The selection pressure was increased by gradually lowering the bait concentration to 100 nM, 60 nM, 20 nM, 1 nM, 100 pM or 10 pM from round 1 to round 5.
- the core DNA fragment recovered in round 5 was cloned in the form of replacing the Fab-HH core region in the monocistronic scFab-HH expression vector (FIG. 28).
- An ELISA using TNF ⁇ R as an antigen was performed using 48 clones of randomly selected culture supernatants.
- ELISA using TNF ⁇ R as an antigen was performed using 96 clones of randomly picked culture supernatants, and the CDR sequences of the ELISA hit clones were determined (FIG. 29).
- ELISA hit clones were screened by SPR with a binding time of 1 minute and a dissociation time of 30 minutes, and high affinity clones were narrowed down using Koff as an index (FIG. 30).
- the KD measurement experiment of Ymacs # 19 with KinExA was performed at two Fab concentrations of 35 pM and 350 pM.
- the concentration of antigen TNF ⁇ R was a 2-fold dilution series from 2 nM to 976 fM for 35 pM Fab, and a 2-fold dilution series from 5 nM to 2.44 pM for 350 pM Fab.
- the sample was incubated at room temperature for 48 hours until the antigen-antibody reaction reached equilibrium, and the presence of free Fab after reaching equilibrium was quantified with KinExA using TNF ⁇ R-immobilized azlactone beads.
- Fab selection by CIS display method We selected Fabs by the CIS display method with reference to the report by Odegrip et al. (Odegrip et al., Proc Natl Acad Sci USA (2004) vol. 101, p. 2806-2810).
- a promoter sequence for Escherichia coli RNA polymerase and a RepA-CIS-ori sequence were added by overlapping extension-PCR upstream and downstream of the scFab coding region in the v7 fragment to obtain a v10.1 fragment. Since the RepA coding sequence was different from the sequence reported by Odegrip et al. And the GenBank V00351 sequence, the V00351 sequence was adopted.
- a mixture of Fab-HH xhoI and Fab-TT v10.1 fragment at TT / HH ratio 10 was used as template DNA for in vitro transcription and translation reaction.
- Purified template DNA (1.2 ⁇ g) is put into an E. coli-derived S30-based transcription and translation system (25 ⁇ L, derived from E. coli SL119, for linear DNA, L1030, Promega), and a display molecule complex is synthesized at 30 ° C. for 40 minutes. did.
- Fab-HH was recovered by liquid phase selection similar to Example 14, and the full length was obtained by PCR using the eluted DNA as a template and Cis-1F and Cis-6R as primers. The fragment was amplified.
- the operation of the Fab-HH selection system was confirmed by the CIS display method. The results are shown in FIG. Compared to the DNA sample before selection, no increase in XhoI sensitivity was observed when selection was performed without adding biotinylated Her-2. On the other hand, when selection was performed by adding biotinylated Her-2, an increase in XhoI sensitivity was observed.
- Cis-1F cagttgatcggcgcgagatttaatcgccgc (SEQ ID NO: 68)
- Cis-6R cgtaagccggtactgattgatagatttcaccttacccatc (SEQ ID NO: 69)
- the present invention is useful in fields such as genetic engineering and protein engineering.
- Fabs obtained by the method of the present invention are useful in fields such as diagnosis, medicine and research.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Plant Pathology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicinal Chemistry (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Peptides Or Proteins (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
(Fab無細胞系ディスプレイ法に供するポリヌクレオチド構築物)
[1]Fab 第1鎖コード配列とFab 第2鎖コード配列を含み、リボゾームを含む無細胞翻訳系に導入されたときに自身がコードするFabを解離させることなく発現し、該Fabとの複合体を維持しうるポリヌクレオチド構築物。
(ポリヌクレオチド構築物(モノシストロニック))
[2]リボゾーム結合配列、Fab 第1鎖コード配列、リンカーペプチドをコードする配列、Fab 第2鎖コード配列、および足場コード配列をこの順にモノシストロニックに含む[1]に記載のポリヌクレオチド構築物であって、その3’末端に自身がコードするFabとの複合体を維持するために必要な構造を有するポリヌクレオチド構築物。
(ポリヌクレオチド構築物(バイシストロニック))
[3]リボゾーム結合配列、Fab 第1鎖コード配列またはFab 第2鎖コード配列、および足場コード配列をこの順に含む、Fab 第1鎖発現シストロンおよびFab 第2鎖発現シストロンを含む[1]に記載のポリヌクレオチド構築物であって、Fab 第1鎖発現シストロンはその3’末端にリボゾームストール配列を有し、Fab 第2鎖発現シストロンはその3’末端に自身がコードするFabとの複合体を維持するために必要な構造を有するポリヌクレオチド構築物。
(リボゾーム/mRNA/CISディスプレイ法に供するポリヌクレオチド構築物)
[4]自身がコードするFabとの複合体を維持するために必要な構造が、リボゾームストール配列、ピューロマイシンもしくはその誘導体、またはDNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列である、[2]または[3]に記載のポリヌクレオチド構築物。
(リボゾームディスプレイ法に供するポリヌクレオチド構築物)
[5]自身がコードするFabとの複合体を維持するために必要な構造が、リボゾームストール配列である、[4]に記載のポリヌクレオチド構築物。
[6]リボゾームストール配列がSecM配列、ジプロリン配列、またはこれらの両方である、[5]に記載のポリヌクレオチド構築物。
[7] リボゾームストール配列がSecM配列の2~4個の繰り返しである、[5]~[6]のいずれかに記載のポリヌクレオチド構築物。
[8]リボゾームストール配列の3’側に終止コドンが存在する、[4]~[7]のいずれかに記載のポリヌクレオチド構築物。
(mRNAディスプレイ法に供するポリヌクレオチド構築物)
[9]自身がコードするFabとの複合体を維持するために必要な構造が、ピューロマイシンもしくはその誘導体である[4]に記載のポリヌクレオチド構築物。
(CISディスプレイ法に供するポリヌクレオチド構築物)
[10]自身がコードするFabとの複合体を維持するために必要な構造が、DNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列である、[4]に記載のポリヌクレオチド構築物。
[11]DNA結合蛋白質が、転写翻訳時に鋳型となったDNA分子と、転写翻訳反応中に一度も解離することなく、同一DNA分子上に存在する該DNA結合蛋白質の結合配列と結合するシス型結合蛋白質である、[10]に記載のポリヌクレオチド構築物。
[12]DNA結合蛋白質が大腸菌R1 Plasmid にコードされるRepAであり、該DNA結合蛋白質の結合配列が同ポリヌクレオチド上でRepAコード配列の下流に存在するCIS-ori配列である、[10]に記載のポリヌクレオチド構築物。
[13]Fab 第1鎖コード配列およびFab 第2鎖コード配列がランダム配列を含むライブラリーである、[1]~[12]のいずれかに記載のポリヌクレオチド構築物。
[14]ランダム配列を含むライブラリーが、(i)ナイーブライブラリーまたは(ii) フォーカスドライブラリーである[13]に記載のポリヌクレオチド構築物。
[15]ランダム配列を含むライブラリーが、Fab 第1鎖および/またはFab 第2鎖の相補性決定領域(CDR)内に1アミノ酸置換を含むライブラリーである、[13]または[14]に記載のポリヌクレオチド構築物。
[16](i)[1]~[15]のいずれかに記載のポリヌクレオチド構築物を無細胞翻訳系に導入してFabを合成し、合成されたFabをそれをコードするポリヌクレオチド上に提示する工程、
(ii)ポリヌクレオチド上に提示されたFabと抗原を接触させる工程、
(iii)抗原と反応する目的Fabを選択する工程、および
(iv)目的Fabをコードするポリヌクレオチドを増幅する工程を含む、Fabのスクリーニング方法。
[17](I)Fab 第1鎖コード配列またはFab 第2鎖コード配列が、親抗体のFab 第1鎖またはFab 第2鎖のアミノ酸配列においてCDR内の1つの位置に対して1アミノ酸置換を含むアミノ酸配列をコードする、[15]に記載のポリヌクレオチド構築物を、Fab 第1鎖およびFab 第2鎖のCDR内の複数の位置に対して1アミノ酸置換が含まれるように複数種類用意する工程、
(II)該複数種類のポリヌクレオチド構築物を用いて前記[16]に記載の工程(i)~(iv)を繰り返して高親和性Fabを複数スクリーニングする一次スクリーニング工程、
(III)一次スクリーニング工程で選択された複数のFabにおけるFab 第1鎖およびFab 第2鎖のCDR内の各位置での1アミノ酸置換の頻度を解析する工程、
(IV)Fab 第1鎖コード配列およびFab 第2鎖コード配列が、親抗体のFab 第1鎖およびFab 第2鎖の配列においてCDR内の各位置に一次スクリーニングで同定された1アミノ酸置換の組み合わせを含むアミノ酸配列をコードする、[15]に記載のポリヌクレオチド構築物を用意する工程、
(V)該ポリヌクレオチド構築物を用いて前記[16]に記載の工程(i)~(iv)を繰り返して高親和性Fabをスクリーニングする二次スクリーニング工程
を含む、[16]に記載のFabのスクリーニングを行う方法。
[18]インビトロ翻訳系が、独立に精製された因子からなる、[16]または[17]に記載の方法。
[19]インビトロ翻訳系が、開始因子、伸長因子、アミノアシルtRNA合成酵素、およびメチオニルtRNAトランスフォルミラーゼからなる群から選択される少なくとも1つの成分を含む、[18]に記載の方法。
[20]インビトロ翻訳系が解離因子を含まない、[18]または[19]に記載の方法。
[21]インビトロ翻訳系が、リボゾームを含む細胞抽出液である、[16]または[17]に記載の方法。
[22][1]~[15]のいずれかに記載のポリヌクレオチド構築物をインビトロ翻訳系に導入してFabを生成させる工程を含む、Fabの製造方法。
[23][1]~[15]のいずれかに記載のポリヌクレオチド構築物を含む、Fabを製造またはスクリーニングするためのキット。
[24](I)標的物質結合蛋白質における標的物質結合部位を構成する全アミノ酸ポジションについて、1アミノ酸ポジションごとに天然アミノ酸全20種類にランダム化する1ポジションライブラリーを、アミノ酸ポジションの数だけ構築する工程、
(II)これらの1ポジションライブラリーを全てあるいは適当な単位で統合して1次ライブラリーを構築する工程、
(III)蛋白質ディスプレイ系を用いて1次ライブラリーを標的親和性で濃縮する工程、
(IV)工程(III)で得られた1次ライブラリー濃縮サンプルのポリヌクレオチド配列情報を決定する工程、
(V)塩基配列情報から高頻度に観察される1アミノ酸置換を抽出する工程、
(VI)高頻度に観察される1アミノ酸置換の組み合わせを含む2次ライブラリーを構築する工程、および
(VII)蛋白質ディスプレイ系を用いて2次ライブラリーを標的親和性で濃縮する工程、
を含む、標的物質結合蛋白質の標的物質親和性を最大化する方法。
[25]1次ライブラリー濃縮サンプルのポリヌクレオチド配列情報を決定する工程が次世代シークエンサーを用いて行われる、[24]の方法。
[26]標的物質結合蛋白質が完全長抗体あるいはscFv、Fab、scFab等の抗体断片であり、標的物質結合部位が天然の抗体で多様性に富む配列が認められる、いわゆるCDR領域である[24]または[25]の方法。
[27]蛋白質ディスプレイ系が、リボゾームディスプレイ、CISディスプレイ、mRNAディスプレイ、ファージディスプレイ、バクテリア表面ディスプレイ、酵母細胞表面ディスプレイ、高等真核生物の細胞表面ディスプレイ、である[24]~[26]のいずれかの方法。
「VL」:FabのL鎖可変領域のコード配列;
「CL」:FabのL鎖定常領域のコード配列;
「VH」:FabのH鎖可変領域のコード配列;
「CH1」:FabのH鎖定常領域のコード配列;
「Linker」:リンカーペプチドのコード配列;
「RBS(○)」:リボゾーム結合部位;
「Pu」:ピューロマイシン、若しくはその誘導体;
「Pro △」:プロモーター;
「RNAP」:RNAポリメラ-ゼ
「RSS(S)」:リボゾームストール配列
「LP(P)」:分泌発現用リーダーペプチド配列
本発明のポリヌクレオチド構築物は、Fab 第1鎖コード配列とFab 第2鎖コード配列を含み、リボゾームを含む無細胞翻訳系に導入されたときに自身がコードするFabを解離させることなく発現し、該Fabとの複合体を維持しうるポリヌクレオチド構築物である。ここで、Fab 第1鎖およびFab 第2鎖とは、Fabを構成する2つの鎖を意味し、通常は一方がFab H鎖であり、他方がFab L鎖であるが、それぞれH鎖とL鎖のキメラ鎖であってもよい。なお、Fab H鎖とはH鎖可変領域(VH)およびH鎖定常領域1(CH1)を含む蛋白質を意味し、Fab L鎖とはL鎖可変領域(VL)およびL鎖定常領域(CL)を含む蛋白質を意味する。
また、「Fabとの複合体を維持する」とは、Fabがポリヌクレオチド構築物とリンクした状態で発現され、その複合体が維持されることを意味する。なお、「ポリヌクレオチド上にFabが提示される」ともいう。
本発明の第1の態様にかかるポリヌクレオチド構築物は、リボゾーム結合配列、Fab 第1鎖コード配列、リンカーペプチド配列、Fab 第2鎖コード配列、および足場コード配列をこの順にモノシストロニックに含む。そして、該ポリヌクレオチド構築物はコード領域(シストロン)の3’末端に、自身がコードするFabとの複合体を維持するために必要な構造を有する。
なお、特定のFabを発現させる場合は目的の鋳型から配列を増幅して用いればよい。また、目的抗原に対するFabをスクリーニングする場合はランダムなFab鎖コード配列を含むライブラリーを使用すればよい。目的抗原に結合するFabを新規に取得したい場合は、ナイーブライブラリーを使用することができる。また既に取得済みの特定のFabを何らかの目的で最適化したい場合は、フォーカスドライブラリーを使用することができる。
ナイーブライブラリーは目的抗原に結合するFabを新規に取得するために、非常に広範な探索空間を提供することを目的とし、以下に例を示すように様々な形式のものを構築することができる。例えばB細胞から採取したmRNAを出発材料とした逆転写PCRでFabを構成するVH領域および/またはVL領域のcDNA断片を増幅して使用することで、天然B細胞が作り出す抗体多様性を利用した天然ナイーブライブラリー(Clackson et al., Nature (1991) vol.352, p.624-628)を構築できる。また、VH領域および/またはVL領域のうちフレームワーク部分を人工的に合成し、CDR領域に多様な天然のCDR配列を組み込むことで半合成ナイーブライブラリー(Soderlind et al., Nat. Biotechnol. (2000) vol.18, p.852-856)を構築できる。あるいはFabのL鎖(VL-CL)に天然配列、H鎖のCDR1-2に人工配列、CDR3に天然配列を組み込むことで半合成ナイーブライブラリー(Hoet et al., Nat. Biotechnol. (2005) vol.23, p.344-348)を構築することもできる。さらにCDRに多様性を与えつつコード配列の全領域を人工合成する完全人工ナイーブライブラリー(Knappik et al., J. Mol. Biol. (2000) vol.296, p.57-86)を構築することもできる。CDR配列を人工合成する場合は、天然のCDR配列の解析結果に基づき、CDRの長さごとに、各アミノ酸ポジションに出現するアミノ酸の頻度を調整することもできる。
例えばVH領域および/またはVL領域全体に、ランダムかつ散発的に1個~数個程度の1アミノ酸置換を導入するためにエラープローンPCR(Error-Prone PCR)を用いることができる(Boder et al., Proc Natl Acad Sci U S A (2000) vol.97, p.10701-10705)。エラープローンPCRは、基質である4種類のデオキシヌクレオチドの濃度や、添加する二価カチオンの種類や濃度を調節することによってエラーが起こり易くなる現象を利用している。人工的に設計した変異導入プライマーを用いて、Fabを構成する特定のアミノ酸だけを狙って、アミノ酸組成に考慮しながら1個~数個程度の1アミノ酸置換を導入することもできる(Rajpal et al., Proc Natl Acad Sci U S A (2005) vol.102, p.8466-8471)。また人工的に設計した変異導入プライマーを用いて、合計6個のCDRのうち、3~5個程度のCDR配列を維持したまま、1~3個程度のCDRの配列を、1アミノ酸単位ではなくCDR配列単位でランダム化することもできる(Lee et al., Blood (2006) vol.108, p.3103-3111)。またL鎖とH鎖のうち片方を固定し、多様な天然配列を利用して他方をランダム化することもできる(Kang et al., Proc Natl Acad Sci U S A (1991) vol.88, p.11120-11123)。
このようなポリヌクレオチド構築物のライブラリーを用いてスクリーニングを行うことにより、目的の抗原に対する抗体を得ることができる。
前記リンカーペプチドをコードする配列はプロテアーゼ認識配列に挟まれていてもよく、それにより、前記ポリヌクレオチド構築物がアミノ酸配列に翻訳された後にプロテアーゼによってリンカーペプチドが切断され、天然型Fabが提示される。プロテアーゼ認識配列としては、エンテロキナーゼ認識配列であるDDDDK(配列番号31)、ファクターXa認識配列であるIEGR(配列番号32)などが例示される。
リボゾームストール配列としては、大腸菌のSecMをコードする配列が例示される。SecM配列は、SecM stall配列とも呼ばれ、リボゾーム内部で翻訳アレストを起こすと報告されている配列である(FXXXXWIXXXXGIRAGP:配列番号30)。この配列を導入することで、mRNA、リボゾーム、融合蛋白質の複合体を効率よく維持できる(Nakatogawa et al., Mol.Cell (2006) vol.22, p.545-552)ので、Fab鎖とそれをコードする塩基配列の対応付けができる。SecM配列は2個以上連結してもよく、2~4個が好ましく、2個がより好ましい。
また、ジプロリンなどのポリプロリン配列もリボゾームストール配列として使用することができ、単独で用いてもよいし、SecM配列と組み合わせてもよい。
リボゾームストール配列の3’側には終止コドンが読み枠を合わせて配置されることが好ましい。
なお、リボゾームストール配列を採用する代わりに単に終止コドンを欠落させてリボゾームディスプレイを行うことも可能である。
配列番号19に、プロモーター配列(塩基番号9~31)と、リボゾーム結合配列(SD配列;塩基番号81~87)と、抗Her2 Fab L鎖コード配列、FLAGタグ、リンカー配列(GSリンカー)、FLAGタグ、抗Her2 Fab H鎖コード配列、Hisタグ、足場配列(GSリンカー)およびリボゾームストール配列(secM+ジプロリン)を含む抗Her2 Fab L+H鎖発現シストロン(塩基番号94~2220;アミノ酸配列は配列番号20)を含むポリヌクレオチド構築物の塩基配列を示す。
配列番号21に、プロモーター配列(塩基番号9~31)と、リボゾーム結合配列(SD配列;塩基番号81~87)と、抗TNFαR Fab L鎖コード配列、FLAGタグ、リンカー配列(GSリンカー)、FLAGタグ、抗TNFαR Fab H鎖コード配列、Hisタグ、足場配列(GSリンカー)およびリボゾームストール配列(secM+ジプロリン)を含む抗TNFαR Fab L+H鎖発現シストロン(塩基番号94~2226;アミノ酸配列は配列番号22)を含むポリヌクレオチド構築物の塩基配列を示す。
ただし、本発明のポリヌクレオチド構築物はこれらに限定されないことは言うまでもない。
また、リボゾームストール配列の代わりに、ピューロマイシンまたはその誘導体を利用してFabとポリヌクレオチドとの複合体を形成させ、アミノ酸配列とそれをコードする塩基配列の物理的対応付けをしてもよい(mRNAディスプレイ)。すなわち、ポリヌクレオチド構築物の3’末端にスペーサーを介してピューロマイシンまたはその誘導体を結合させ、翻訳産物のC末端がピューロマイシンまたはその誘導体に共有結合することによりFabとポリヌクレオチドとの複合体が形成され、Fab鎖のアミノ酸配列とそれをコードする塩基配列の対応付けが可能となる。
ここで、ピューロマイシンまたはその誘導体としては、ピューロマイシン、リボシチジルピューロマイシン、デオキシシチジルピューロマイシン、デオキシウリジルピューロマイシンなどのピューロマイシン誘導体が特に好ましい。
なお、mRNAディスプレイの別の態様として、ポリヌクレオチド構築物の3’末端側にストレプトアビジンコード配列を連結させ、さらに、3’末端にビオチンを結合させて、翻訳された蛋白質のストレプトアビジン部分がビオチンと結合することにより、Fabとポリヌクレオチドとの複合体を形成させ、アミノ酸配列とそれをコードする塩基配列の物理的対応付けをしてもよい。
また、リボゾームストール配列の代わりに、DNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列を利用してFabとポリヌクレオチドとの複合体を形成させ、アミノ酸配列とそれをコードする塩基配列の物理的対応付けをしてもよい(CISディスプレイ: WO2004/22746)。具体的には、ポリヌクレオチド構築物の足場配列の下流にDNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列を連結させ、Fabと足場配列とDNA結合蛋白質との融合蛋白質として発現させ、DNA結合蛋白質が3’側シストロン下流のDNA結合蛋白質結合配列に結合することによりFabとポリヌクレオチドとの複合体が形成されFab鎖のアミノ酸配列とそれをコードする塩基配列の対応付けが可能となる。ここで、DNA結合蛋白質としては、DNA結合蛋白質が転写翻訳時に鋳型となったDNA分子と転写翻訳反応中に一度も解離することなく同一DNA分子上に存在する該DNA結合蛋白質の結合配列と結合する、シス型の結合様式をもつRepA蛋白質が挙げられ、RepA結合配列としてはCIS配列とそれに続くori配列が挙げられる(Proc. Natl. Acad. Sci. U. S. A., vol.101, p.2806-2810, 2004および特表2005-537795)。シス型の結合様式をもつ他のDNA結合蛋白質として、大腸菌Ti plasmid にコードされるRecC蛋白質(Pinto, et al., Mol. Microbiol. (2011) vol.81, p.1593-1606)、φX174ファージのA蛋白質(Francke, et al., Proc Natl Acad Sci U S A (1972) vol.69, p.475-479)、λファージのQ蛋白質(Echols, et al., Genetics (1976) vol.83, p.5-10)等を、該DNA結合蛋白質それぞれに対する結合配列と組み合わせて用いてもよい。また、転写と翻訳がよく共役する無細胞翻訳系を用いる場合は、合成された蛋白質が転写終結部位の近傍でリリースされるため、エストロゲン受容体などの核内受容体のDNA結合ドメイン、Two-Hybrid Systemに使用されるLexAやGal4のDNA結合ドメインのように、一般的にトランス型の結合様式を持つDNA蛋白質と考えられているものを、該DNA結合蛋白質それぞれに対する結合配列と組み合わせて用いてもよい。
配列番号70に、プロモーター配列(塩基番号612~639)と、リボゾーム結合配列(SD配列;塩基番号672~675)と、抗Her2 Fab L鎖コード配列、FLAGタグ、リンカー配列(GSリンカー)、FLAGタグ、抗Her2 Fab H鎖コード配列、Hisタグ、足場配列(GSリンカー)およびRepAコード配列を含む抗Her2 Fab L+H鎖発現シストロン(塩基番号689~3322;アミノ酸配列は配列番号71)、CIS-ori(塩基番号3326~4100)を含むポリヌクレオチド構築物の塩基配列を示す。
配列番号72に、プロモーター配列(塩基番号612~639)と、リボゾーム結合配列(SD配列;塩基番号672~675)と、抗TNFαR Fab L鎖コード配列、FLAGタグ、リンカー配列(GSリンカー)、FLAGタグ、抗TNFαR Fab H鎖コード配列、Hisタグ、足場配列(GSリンカー)およびRepAコード配列を含む抗TNFαR Fab L+H鎖発現シストロン(塩基番号689~3328;アミノ酸配列は配列番号73)、CIS-ori(塩基番号3332~4106)を含むポリヌクレオチド構築物の塩基配列を示す。
ただし、本発明のポリヌクレオチド構築物はこれらに限定されないことは言うまでもない。
本発明の第2の態様にかかるポリヌクレオチド構築物は、リボゾーム結合配列、Fab 第1鎖コード配列またはFab 第2鎖コード配列、および足場コード配列をこの順に含むFab 第1鎖発現シストロンおよびFab 第2鎖発現シストロンを含む。そして、Fab 第1鎖発現シストロン(5'側のFab鎖発現シストロン)はその3’末端にリボゾームストール配列を有し、Fab 第2鎖発現シストロン(3'側のFab鎖発現シストロン)はその3’末端側に自身がコードするFabとの複合体を維持するために必要な構造を有する。ここで、自身がコードするFabとの複合体を維持するために必要な構造としては、上記のようなリボゾームストール配列、DNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列、またはピューロマイシンもしくはその誘導体が例示される。
なお、Fab 第1鎖発現シストロンとFab 第2鎖発現シストロンとの間の長さは、Fab 第1鎖発現シストロンで翻訳が一旦終了し、リボゾームがストールされている状態で、Fab 第2鎖発現シストロンのリボゾーム結合配列にリボゾームが結合できる程度の間隔があればよいが、好ましくは50~200bpである。
本発明の第2の態様にかかるポリヌクレオチド構築物においては、リボゾーム上で、mRNAとポリペプチド(Fabの第1鎖または第2鎖)の複合体を形成させるため、Fab 第1鎖発現シストロンの3’末端に上述したようなリボゾームストール配列を配置する。なお、リボゾームストール配列の3’側には終止コドンが読み枠を合わせて配置されることが好ましい。リボゾームストール配列を採用する代わりに単に終止コドンを欠落させてリボゾームディスプレイを行うことも可能である。
配列番号13に、プロモーター配列(塩基番号9~31)と、リボゾーム結合配列(SD配列;塩基番号81~87)と、抗Her2 Fab L鎖コード配列、FLAGタグ、足場配列(GSリンカー)およびリボゾームストール配列(secM+ジプロリン)を含む抗Her2 Fab L鎖発現シストロン(塩基番号94~1158;アミノ酸配列は配列番号14)と、リボゾーム結合配列(SD配列;塩基番号1191~1197)と、抗Her2 Fab H鎖コード配列、Hisタグ、足場配列(GSリンカー)およびリボゾームストール配列(secM+ジプロリン)を含む抗Her2 Fab H鎖発現シストロン(塩基番号1264~2364;アミノ酸配列は配列番号15)を含むポリヌクレオチド構築物の塩基配列を示す。
配列番号16に、プロモーター配列(塩基番号9~31)と、リボゾーム結合配列(SD配列;塩基番号81~87)と、抗TNFα受容体(TNFαR) Fab L鎖コード配列、FLAGタグ、足場配列(GSリンカー)およびリボゾームストール配列(secM+ジプロリン)を含む抗TNFαR Fab L鎖発現シストロン(塩基番号94~1158;アミノ酸配列は配列番号17)と、リボゾーム結合配列(SD配列;塩基番号1191~1197)と、抗TNFαR Fab H鎖コード配列、Hisタグ、足場配列(GSリンカー)およびリボゾームストール配列(secM+ジプロリン)を含む抗TNFαR Fab H鎖発現シストロン(塩基番号1264~2370;アミノ酸配列は配列番号18)を含むポリヌクレオチド構築物の塩基配列を示す。
ただし、本発明のポリヌクレオチド構築物はこれらに限定されないことはいうまでもない。
なお、Fab 第2鎖発現シストロン(3'側のFab鎖発現シストロン)については、ピューロマイシンまたはその誘導体を利用してFab鎖とポリヌクレオチドの複合体を形成させ、アミノ酸配列とそれをコードする塩基配列の物理的対応付けをしてもよい(mRNAディスプレイ)。すなわち、3'側のシストロンの末端、すなわち、ポリヌクレオチド構築物の3’末端にスペーサーを介してピューロマイシンまたはその誘導体を結合させ、3'側のシストロンが翻訳されたときに、翻訳産物のC末端がピューロマイシンまたはその誘導体に共有結合することによりFab鎖とポリヌクレオチドの複合体を形成させ、Fab鎖のアミノ酸配列とそれをコードする塩基配列の対応付けが可能となる。
また、Fab 第2鎖発現シストロン(3'側のFab鎖発現シストロン)については、DNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列を利用してFab鎖とポリヌクレオチドの複合体を形成させ、アミノ酸配列とそれをコードする塩基配列の物理的対応付けをしてもよい(CISディスプレイ: WO2004/22746)。具体的には、Fab 第2鎖発現シストロンの3'末端側、すなわち、Fab 第2鎖発現シストロンのFab第2鎖コード配列と足場配列の下流にDNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列を連結させ、Fab 第2鎖発現シストロンをFab第2鎖と足場配列とDNA結合蛋白質との融合蛋白質として発現させ、DNA結合蛋白質がFab 第2鎖発現シストロン下流のDNA結合蛋白質結合配列に結合することによりFab第2鎖とポリヌクレオチドの複合体を形成させ、Fab第2鎖のアミノ酸配列とそれをコードする塩基配列の対応付けが可能となる。ここで、DNA結合蛋白質としては、DNA結合蛋白質が転写翻訳時に鋳型となったDNA分子と転写翻訳反応中に一度も解離することなく同一DNA分子上に存在する該DNA結合蛋白質の結合配列と結合する、いわゆるシス型の結合様式をもつRepA蛋白質などが挙げられ、RepA結合配列としてはCIS配列とそれに続くori配列が挙げられる(Proc. Natl. Acad. Sci. U. S. A., vol.101, p.2806-2810, 2004および特表2005-537795)。シス型の結合様式をもつ他のDNA結合蛋白質として、大腸菌Ti plasmid にコードされるRecC蛋白質(Pinto, et al., Mol. Microbiol. (2011) vol.81, p.1593-1606)、φX174ファージのA蛋白質(Francke, et al., Proc Natl Acad Sci U S A (1972) vol.69, p.475-479)、λファージのQ蛋白質(Echols, et al., Genetics (1976) vol.83, p.5-10)等を、該DNA結合蛋白質それぞれに対する結合配列と組み合わせて用いてもよい。また、転写と翻訳がよく共役する無細胞翻訳系を用いる場合は、合成された蛋白質が転写終結部位の近傍でリリースされるため、エストロゲン受容体などの核内受容体のDNA結合ドメイン、Two-Hybrid Systemに使用されるLexAやGal4のDNA結合ドメインのように、一般的にトランス型の結合様式を持つDNA蛋白質と考えられているものを、該DNA結合蛋白質それぞれに対する結合配列と組み合わせて用いてもよい。
本発明のFabの製造方法は上記ポリヌクレオチド構築物をリボゾームを含む無細胞翻訳系に導入してFabを生成させる工程を含む。無細胞翻訳系としては大腸菌、酵母、哺乳動物細胞等の細胞から得られた無細胞翻訳系が例示されるが、大腸菌由来の無細胞翻訳系が好ましい。
上記CISディスプレイの場合は遺伝媒体として細胞抽出液中でも比較的安定な二本鎖DNAを用いるのでリボゾームを含む画分を細胞から抽出して得られる細胞抽出液を用いることも可能である。
リボゾームディスプレイの場合は、細胞抽出液中に大量に存在するRNA分解酵素により遺伝媒体であるRNAが分解される危険性があるため、個別に精製された因子から構成された再構成型の無細胞翻訳系が好ましい。このようなPUREシステムを用いたリボゾームディスプレイは「PURE ribosome display(PRD)」と呼ばれている。
リボゾームは、リボゾームRNAと種々のリボゾーム蛋白質とで構成される巨大な複合体であり、大小2つのサブユニットからなる。リボゾームやそれを構成するサブユニットは、ショ糖密度勾配などによって相互に分離することができ、その大きさは、沈降係数によって表される。具体的には、原核生物においては、リボゾームとそれを構成するサブユニットは、それぞれ次のような大きさを有する。大腸菌などの原核生物は容易に大量培養することができるので、大腸菌などの原核生物は、リボゾームを大量に調製するうえで、好ましい生物である。
リボゾーム(70S)=大サブユニット(50S)+小サブユニット(30S)
分子量: 約2.5x106約1.6x106 約0.9x106
50Sサブユニット;L1~L34の34種類の蛋白質(リボゾーム蛋白質)
23S RNA(約3200ヌクレオチド)
5S RNA(約120ヌクレオチド)
30Sサブユニット;
S1~S21の21種類の蛋白質(リボゾーム蛋白質)
16S RNA(約1540ヌクレオチド)
つまり各サブユニットは、これらの成分からなる複合体として単離されうる。更にリボゾームは、各サブユニットの複合体として単離されうる。精製されたリボゾームとは、たとえば原核生物由来のリボゾームにおいては、大小のサブユニットからなる70Sリボゾームとして精製された複合体、または、それぞれ精製された50Sサブユニットと30Sサブユニットを混合してできた複合体を指す。
開始因子 (Initiation Factor; IF)、
伸長因子 (Elongation Factor; EF)、
アミノアシルtRNA合成酵素、
メチオニルtRNAトランスフォルミラーゼ(MTF)
なお、解離因子も含んでよいが、リボゾーム上で蛋白質とポリヌクレオチドの複合体を安定に維持するためには解離因子を含まないことが好ましい。
T7 RNAポリメラーゼ
T3 RNAポリメラーゼ
SP6 RNAポリメラーゼ
T7RNAポリメラーゼを使用した場合、例えば、0.01μg/ml~5000μg/ml、好ましくは、0.1μg/ml~1000μg/mlで使用できる。またCISディスプレイにおいて再構築型の無細胞翻訳系を用いることで、FabとDNAの複合体形成の効率を追及する場合には、Nucleic Acid Research 2010, vol. 38, No. 13, e141に記載されているように、精製された大腸菌の内因性RNAポリメラーゼを添加することもできる。また、Nucleic Acid Research 1988, vol. 16, No. 14, 6493に記載されているように、精製された大腸菌の転写終結因子であるrho蛋白質を添加することもできる。
反応系においてエネルギーを再生するための酵素:
クレアチンキナーゼ;
ミヨキナーゼ;および
ヌクレオシドジフォスフェートキナーゼなど
転写・翻訳で生じる無機ピロリン酸の分解のための酵素:
無機ピロフォスファターゼなど
上記酵素は、例えば、0.01μg/ml~2000μg/ml、好ましくは、0.05μg/ml~500μg/mlで使用できる。
本発明のスクリーニング方法は、(i)Fabのライブラリーをコードするポリヌクレオチド構築物を無細胞翻訳系に導入してFabを合成し、合成されたFabをそれをコードするポリヌクレオチド上に提示する工程、(ii)ポリヌクレオチド上に提示されたFabを抗原と接触させる工程、(iii)抗原と反応する目的Fabを選択する工程、および(iv)目的Fabをコードするポリヌクレオチドを増幅する工程を含む。
Fab遺伝子ライブラリーの規模は、通常1×108以上、好ましくは1×109以上、より好ましくは1×1010以上、さらに好ましくは1×1012以上である。
標的物質(抗原)に前記Fab遺伝子ライブラリーから発現したFabライブラリーを接触させ、Fabライブラリーから標的物質に結合するFabを選択し、それをコードするポリヌクレオチドを増幅する。
(1)標的物質にFabライブラリーを接触させる。ビーズ、プレートやカラム等の担体に標的物質を結合させ、ここにFabとポリヌクレオチドの複合体を含む試料を接触させてもよい(固相選択)。また、標的物質をビオチン化し、これをFabと結合させ、標的物質とFabの複合体をストレプトアビジン磁気ビーズビーズで回収するような、液相での選択も可能である。また、固相選択と液相選択を組み合わせてもよい。スクリーニングを複数ラウンド行い、前半は固相選択、後半は液相選択を行うことで、高親和性抗体を効率よく選択することができる。
(2)標的物質に結合しなかった、ライブラリーに含まれていたその他のFabを除去する。例えば、洗浄により除去することができる。洗浄は通常の抗原抗体反応の洗浄に使用される洗浄液を使用することができるが、後述するような親Fabより親和性の高いFabを得るためのスクリーニングを行う場合には、親Fabと抗原との結合が維持され、それより弱い結合が除かれるような条件で洗浄を行うことが好ましい。
(3)除去されなかったFab、すなわち標的物質に特異的に結合していたFabを回収する。
(4)必要に応じ(1)から(3)の操作を複数回繰り返す。
すなわち、本発明においては、
(I)Fab 第1鎖コード配列またはFab 第2鎖コード配列が、親抗体のFab 第1鎖またはFab 第2鎖のアミノ酸配列においてCDR内の1つの位置に対して1アミノ酸置換を含むアミノ酸配列をコードする、本発明のポリヌクレオチド構築物を、Fab 第1鎖およびFab 第2鎖のCDR内の複数の位置に対して1アミノ酸置換が含まれるように複数種類用意する工程、
(II)該複数種類のポリヌクレオチド構築物を用いて前記工程(i)~(iv)を繰り返して高親和性Fabを複数スクリーニングする一次スクリーニング工程、
(III)一次スクリーニング工程で選択された複数のFabにおけるFab 第1鎖およびFab 第2鎖のCDR内の各位置での1アミノ酸置換を解析する工程、
(IV)Fab 第1鎖コード配列およびFab 第2鎖コード配列が、親抗体のFab 第1鎖およびFab 第2鎖の配列においてCDR内の各位置に一次スクリーニングで同定された1アミノ酸置換の組み合わせを含むアミノ酸配列をコードする、本発明のポリヌクレオチド構築物を用意する工程、
(V)該ポリヌクレオチド構築物を用いて前記工程(i)~(iv)を繰り返して高親和性Fabをスクリーニングする二次スクリーニング工程
を含む、Fabのスクリーニングを行う方法が提供される。
なお、工程(I)における複数とは2以上であればよいが、好ましくはCDR内の全てのアミノ酸である。
ただし、本発明のスクリーニング方法はこの態様に限定されない。
まず、目的の抗原に対する抗体(親抗体)のアミノ酸配列(親配列)を用意する。そして、図24のように、このアミノ酸配列において、H鎖のCDRのアミノ酸(CDR-H1 8個、CDR-H2 11個、CDR-H3 12個、合計31個)のうちの1つにおいて1アミノ酸置換(好ましくは親アミノ酸を含めた天然20アミノ酸全てが出現する1アミノ酸置換)を有するようなH鎖コード配列と親抗体のL鎖コード配列を含む本発明のポリヌクレオチド構築物をH鎖のCDRのアミノ酸(合計31個)の数だけ用意する(H鎖ライブラリー)。
また、親抗体のアミノ酸配列において、L鎖のCDRのアミノ酸(CDR-L1 7個、CDR-L2 6個、CDR-L3 6個、合計19個)のうちの1つにおいて、1アミノ酸置換(好ましくは親アミノ酸を含めた天然20アミノ酸全てが出現する1アミノ酸置換)を有するようなL鎖コード配列と親抗体のH鎖コード配列を含むポリヌクレオチド構築物をL鎖のCDRのアミノ酸(合計19個)の数だけ用意する(L鎖ライブラリー)。
上記H鎖ライブラリーとL鎖ライブラリーを一次ライブラリーとし、これを用いて工程(i)~(iv)を繰り返し、高親和性抗体のスクリーニングを行う(1次スクリーニング)。
1次スクリーニングによって高親和性抗体をコードするポリヌクレオチド配列が濃縮されているので、濃縮された複数の配列を解析し、H鎖およびL鎖のCDRの各位置について高頻度に観察されるアミノ酸置換を、親和性向上のために好ましいアミノ酸置換として選択する。
ここで、Sanger シークエンシング法を利用した蛍光キャピラリーシークエンサーに代表される第1世代シークエンサーと対比させて使われている次世代シークエンサーという用語は、実際には多様な機器や技術を含み、今後も様々な形態のものが考案されると考えられる(Mardis, Annu. Rev. Genom. Human Genet. (2008) vol.9, p.387-402およびPersson et al., Chem. Soc. Rev. (2010) vol.39, p.985-999)。
第1世代シークエンサーが一般にベクター宿主システムを用いてクローン化したDNAの配列決定に利用されるのに対し、次世代シークエンサーは多様な配列をもつDNAサンプルを解析対象としながらも、ベクター宿主システムを用いてDNAのクローン化をすることなく、サンプル中に存在するDNA配列を高速に解読できる。
第1世代シークエンサーを用いたDNA配列決定では、
(i)プラスミド等のベクターへのDNA断片の組み込みと大腸菌等の宿主の形質転換、
(ii)宿主コロニーの単離によるクローン化、
(iii)解析規模に応じた数のクローンの培養およびプラスミドの抽出、
(iv)プラスミド等を鋳型としたPCR等によるシークエンス反応、
(v)キャピラリー電気泳動等を用いたシークエンス反応産物の分離・検出・解析、
という過程を経て個々のクローンのDNA配列を決定する。多数のクローンのDNA配列を一度に決定する場合は、クローン化以降の(ii)~(v)の工程で、クローンごとに独立した反応・処理が必要となり、解析クローン数の規模に比例した作業量が必要となる。
一方、次世代シークエンサーを用いたDNA配列決定では、多様な配列を含むDNA断片を、Emulsion PCRやBridge PCR といった単分子PCR法等の増幅技術、あるいは1分子観察等の高感度検出技術、を応用することで解析用基板上に大規模数存在する個々の解析スポット上にクローン化して超並列的に配列解析する。このため、解析クローン数の規模に関わらず単一の反応・処理で、例えば1つの解析用基板上に形成された106~109のクローンの配列を決定できる。
(I)標的物質結合蛋白質における標的物質結合部位を構成する全アミノ酸ポジションについて、1アミノ酸ポジションごとに天然アミノ酸全20種類にランダム化する1ポジションライブラリーを、アミノ酸ポジションの数だけ構築する工程、
(II)これらの1ポジションライブラリーを全てあるいは適当な単位で統合して1次ライブラリーを構築する工程(1次ライブラリーポリヌクレオチド構築物を用意する工程)、
(III)蛋白質ディスプレイ系を用いて1次ライブラリーを標的親和性で濃縮する工程、
(IV)(III)で得られた1次ライブラリー濃縮サンプルのポリヌクレオチド配列情報を決定する工程、
(V)塩基配列情報から高頻度(好ましくは2以上)に観察される1アミノ酸置換を抽出する工程、
(VI)高頻度に観察される1アミノ酸置換の組み合わせを含む2次ライブラリーを構築する工程(2次ライブラリーポリヌクレオチド構築物を用意する工程)、および
(VII)蛋白質ディスプレイ系を用いて2次ライブラリーを標的親和性で濃縮する工程、
を含む、標的物質結合蛋白質の標的物質親和性を最大化する方法(標的物質親和性が向上した変異型標的物質結合蛋白質を得る方法)が提供される。
なお、工程(II)で適当な単位で統合して1次ライブラリーを構築する場合、工程(I)で構築した1ポジションライブラリーを2群以上に分けて、1次ライブラリーを2種類以上使用してもよい。また(VI)で構築する2次ライブラリーには、高頻度に観察される1アミノ酸置換に加え、親抗体がコードしていた親アミノ酸を追加してもよい。
抗原結合蛋白質としては、Fab、scFv、完全長抗体などの抗体が挙げられるが、抗原に特異的に結合するものである限り、抗体に分類されない蛋白質であってもよい。
また、サイトカインに変異を導入し、受容体に対する結合性を高めることもできる。
例えば、標的物質結合蛋白質(野生型)において、標的物質結合部位が10アミノ酸からなる領域である場合、1~10のうちの1つのポジションに20アミノ酸が出現する1ポジションライブラリーを合計10種類用意する。
この10種類の1ポジションライブラリーを合わせて1次ライブラリーとし、これを用いて、標的物質親和性で濃縮する。具体的には、下記の工程(i')~(iv')を繰り返して標的物質に高親和性の配列を濃縮する。
(i')ライブラリーを蛋白質ディスプレイ系に導入して標的物質結合蛋白質を発現させ、該標的物質結合蛋白質をコードするポリヌクレオチド上で提示させる工程、
(ii')標的物質と標的物質結合蛋白質を接触させる工程、
(iii')標的物質と反応する目的標的物質結合蛋白質を選択する工程、および
(iv')目的標的物質結合蛋白質をコードするポリヌクレオチドを増幅する工程、を繰り返す(1次スクリーニング)。
これらの工程は、通常の蛋白質ディスプレイにおける操作と同様にして行うことができる。
工程(ii')は、ビーズ、プレートやカラム等の担体に標的物質を結合させ、ここに標的物質結合蛋白質とポリヌクレオチドの複合体を含む試料を接触させてもよい(固相選択)。また、標的物質をビオチン化し、これを標的物質結合蛋白質と結合させ、標的物質と標的物質結合蛋白質の複合体をストレプトアビジン磁気ビーズビーズで回収するような、液相での選択も可能である。また、固相選択と液相選択を組み合わせてもよい。スクリーニングを複数ラウンド行い、前半は固相選択、後半は液相選択を行うことで、高親和性蛋白質を効率よく選択することができる。
非特異的結合を除くため、(ii')と(iii')の間に洗浄項操作を行うことが好ましい。洗浄は、野生型標的物質結合蛋白質と標的物質との結合が維持され、それより弱い結合が除かれるような条件で行うことが好ましい。
ポリヌクレオチドの配列解析は通常のシークエンシングによって行ってもよいが、スクリーニングによって増幅された標的物質結合蛋白質コード配列をクローン化することなく、大量に網羅的に配列解析でき、スクリーニング時間が大幅に短縮できるという点で、次世代シークエンサーを用いることが好ましい。
ここで、Sanger シークエンシング法を利用した蛍光キャピラリーシークエンサーに代表される第1世代シークエンサーと対比させて使われている次世代シークエンサーという用語は、実際には多様な機器や技術を含み、今後も様々な形態のものが考案されると考えられる(Mardis, Annu. Rev. Genom. Human Genet. (2008) vol.9, p.387-402およびPersson et al., Chem. Soc. Rev. (2010) vol.39, p.985-999)。
第1世代シークエンサーが一般にベクター宿主システムを用いてクローン化したDNAの配列決定に利用されるのに対し、 次世代シークエンサーは、多様な配列をもつDNAサンプルを解析対象としながらも、ベクター宿主システムを用いてDNAのクローン化をすることなく、サンプル中に存在するDNA配列を高速に解読できる。
第1世代シークエンサーを用いたDNA配列決定では、
(i)プラスミド等のベクターへのDNA断片の組み込みと大腸菌等の宿主の形質転換、
(ii)宿主コロニーの単離によるクローン化、
(iii)解析規模に応じた数のクローンの培養およびプラスミドの抽出、
(iv)プラスミド等を鋳型としたPCR等によるシークエンス反応、
(v)キャピラリー電気泳動等を用いたシークエンス反応産物の分離・検出・解析、
という過程を経て個々のクローンのDNA配列を決定する。多数のクローンのDNA配列を一度に決定する場合は、クローン化以降の(ii)~(v)の工程で、クローンごとに独立した反応・処理が必要となり、解析クローン数の規模に比例した作業量が必要となる。
一方、次世代シークエンサーを用いたDNA配列決定では、多様な配列を含むDNA断片を、Emulsion PCRやBridge PCR といった単分子PCR法等の増幅技術、あるいは1分子観察等の高感度検出技術、を応用することで解析用基板上に大規模数存在する個々の解析スポット上にクローン化して超並列的に配列解析する。このため、解析クローン数の規模に関わらず単一の反応・処理で、例えば1つの解析用基板上に形成された106~109のクローンの配列を決定できる。
この二次ライブラリーを用いて、工程(i')~(iv')を繰り返し、2次スクリーニングを行うことにより、親蛋白質よりも標的物質に対する親和性が顕著に向上した蛋白質を多数取得することができる。親蛋白質よりも標的物質に対する親和性が顕著に向上したことは、SPRやELISAなどによって解析することができる。
無細胞Fabディスプレイ系で用いるFabのフレームワークとして、ヒトにおける主要な抗体サブクラスのひとつであり、大腸菌での発現効率に優れ(Kanappik et al., J Mol Biol. (2000) vol.296, p.57-86)、無細胞scFvディスプレイ系での実績が報告された(Shibui et al., Appl Microbiol Biotechnol. (2009) vol.84, p.725-732)、VL k subgroup IおよびVH subgroup IIIの組合せを選択した。無細胞Fabディスプレイ系の動作確認に用いる上記フレームワークをもつモデルFabとして、Carterらが報告したヒトEGFR-2(Her-2)反応性のFab(Carter et al., Proc Natl Acad Sci U S A. (1992) vol. 89, p.4285-4289)を選択し、Fab-HHとした。
もうひとつのモデルFabとしてFab-HHのL鎖およびH鎖のCDR1-3領域を、渋井らの無細胞scFvディスプレイ系で見出されたTNFαR反応性scFvの配列(Shibui et al., Biotechnol. Lett. (2009) vol.31, p.1103-1110)で置換し、Fab-TTとした。
これら2種類のモデルFabのVLドメインおよびVHドメインをコードするcDNAを人工合成した。またCLドメイン、CH1ドメインをコードするcDNAをヒト癌細胞株のゲノムDNAを鋳型としたPCRで取得した。得られたcDNA断片を、大腸菌発現ベクターpTrc99Aに組み込み、モデルFabをBicistronicに分泌発現するベクターとして、pTrc Fab-HH、pTrc Fab-TTを構築した。また本来のL鎖とH鎖の組合せを意図的に変更してFab-HT(Her-2反応性のL鎖とTNFαR反応性のH鎖の組合せ)およびFab-TH(TNFαR反応性のL鎖とHer-2反応性のH鎖の組合せ)とし、これらを発現するpTrc Fab-HTおよびpTrc Fab-THを合わせて構築した。このとき、L鎖のC末端にmycタグを、H鎖のC末端に2連続FLAGタグおよびHisタグを付加した。pTrc Fabベクター中のBicistronicに Fabを発現するユニットの概略構造を図9に示す。
モデルFabの抗原蛋白質である、Her-2およびTNFαR(R&D systems社)をPBSで1μg/mlに希釈した。これをELISAプレートに添加し4℃で一晩放置することでプレートに固定した。ウェルを洗浄後、モデルFabを添加し抗原に結合させた。ウェルを洗浄後、検出用抗体として1000倍希釈したHRP標識抗FLAG抗体(シグマ社)を添加し、モデルFabに付加したFLAGタグに結合させた。ウェルを洗浄後、HRP用発色基質を添加した。適度な発色が得られたところで停止液を添加し、OD405nmにて吸光度を測定した。抗原蛋白質にHer-2を用いて4種類のモデルFabを広い濃度域でタイトレーションした結果を図11に示す。Fab-HHがHer-2に強く結合(ED50=2nM)することがわかる。次に2種類の抗原蛋白質に4種類のモデルFabを高濃度(HH、TT、HT、THの順に7.1、1.8、2.9、5.8μM)で接触させた結果を図12に示す。Fab-HHがHer-2には結合するがTNFαRには結合しないこと、またFab-TTがTNFαRには結合するがHer-2には結合しないことがわかる。さらにハイブリッドFab であるFab-HTおよびFab-THがどちらの抗原にも結合しないことから、Fab-HHのHer-2に対する結合およびFab-TTのTNFαRに対する結合は、L鎖H鎖相互依存的であり、正しいL鎖とH鎖の組み合わせが抗原蛋白質との結合に必要であることがわかる。
上記のFab-HHと酷似したアミノ酸配列をもち、Fellouseら(Fellouse et al., J. Mol.Biol. (2007) vol.373, p.924-940)により大腸菌での発現効率がさらに向上すると報告された配列をFab-SSとした。この配列を無細胞ディスプレイ系で提示するために、大腸菌最適化コドンを用いてBicistronic型Fab-PRD用のDNA断片を人工合成した。これを汎用ファージミドベクターpBluescript SK(+)に組み込むことでpGAv2-SSを構築した。次にpGAv2-SSのL鎖CDR1-3領域およびH鎖CDR1-3領域を、pTrc Fab-HHの対応する領域と置換することでpGAv2-HHを構築した。同様にpGAv2-SSとpTrc Fab-TTを用いてpGAv2-TTを構築した。次にpGAv2-HHのL鎖下流のGSリンカー最下流部位にアミノ酸配列を変えずにXhoI認識部位を導入することでpGAv2-HH xhoIを構築した。Bicistronic型Fab-PRD用のDNA断片の概略構造を図13に示す。
Bicistronic型Fab-PRD用のpGAv2-HH xhoIとpGAv2-TTの中の、L鎖下流のリボゾームストール配列の先頭アミノ酸からH鎖の最初のメチオニンまでの領域を、FLAGタグをコードする配列に置換し、FLAGタグを介してL鎖下流GSリンカーのC末端をインフレームでH鎖と接続した構造をもつ、Monocistronic型Fab-PRD用のベクター、pGAv6-HH xhoIとpGAv6-TTを構築した。Monocistronic型Fab-PRD用のDNA断片の概略構造を図14に示す。
Bicistronic型Fab-PRD用にpGAv2-HH xhoIおよびpGAv2-TT 、Monocistronic型Fab-PRD用にpGAv6-HH xhoIおよびpGAv6-TTを鋳型として、PCR反応によりin vitro転写用鋳型DNA断片を増幅した。PCR用酵素としてPrimeStarMax(タカラ社)を使用し、プライマーとしてPURE-rt-1FおよびPURE-3Rの組合せ、またはSL-1FおよびSL-2Rの組合せを使用した。増幅したin vitro転写用鋳型DNA断片はフェノール・クロロホルム抽出、イソプロパノール沈殿で精製した。in vitro転写用鋳型DNAの濃度は260nmの吸光度から1 OD=5 0μg/mlとして算出した。4種類のベクターからSL-1FおよびSL-2R のプライマーセットで増幅したin vitro転写用鋳型DNA断片の配列は以下のとおりである。
Bicistronic型Fab-PRD用
pGAv2-HH xhoI 配列番号13(アミノ酸配列は配列番号14(L),15(H))
pGAv2-TT 配列番号16(アミノ酸配列は配列番号17(L),18(H))
Monocistronic型Fab-PRD用
pGAv6-HH xhoI 配列番号19(アミノ酸配列は配列番号20)
pGAv6-TT 配列番号21(アミノ酸配列は配列番号22)
PURE-rt-1F:(caatttcggtaatacgactcactatagggagaatttaggtgacactatagaagtg)(配列番号23)
PURE-3R:(caggtcagacgattggccttg)(配列番号24)
SL-1F:(caatttcggtaatacgactcactatagggagaccacaacggtttcccatttaggtgacactatagaagtg)(配列番号25)
SL-2R:(ccgcacaccagtaaggtgtgcggcaggtcagacgattggccttgatattcacaaacg)(配列番号26)
mRNA合成は500ngの鋳型DNA断片と2.7μgの精製T7 RNA polymeraseを用いて50μlスケールで実施した。37度60分の合成反応の後、RNase free DNase(Promega社)を1μl添加し、37度10分の反応で鋳型DNAを分解した。mRNAはフェノール・クロロホルム抽出、イソプロパノール沈殿で精製した後、30μlのNuclease free water(Promega社)で溶解した。mRNAの濃度は260nmの吸光度から1 OD=40 μg/mlとして算出した。
PUREシステムを構成する翻訳因子群およびリボゾームは、清水(Shimizu et al., Nat Biotechnol. (2001) vol.19, p.751-755)や大橋(Ohashi et al., Biochem Biophys Res Commun. (2007) vol.352, p.270-276)らの報告に記載の方法で調製した。翻訳反応は2 p moleのmRNAと20 p moleのリボゾームを用いて20μlスケールとした。酸化還元条件に関しては、酸化型および還元型グルタチオン、蛋白質ジスルフィドイソメラーセを除き、1mMのDTTを添加し還元条件下での翻訳反応とした。37度20分の翻訳反応の後に終濃度2.5mMの酸化型グルタチオンを添加してDTTを中和した。
Her-2およびTNFαR(R&D systems社)をPBSで各々0.5mg/mlに溶解した。ビオチン化試薬としてSulfo-NHS-SS-Biotin(サーモサイエンティフィック社)をPBSで0.6 mg/ml (Her-2用)および4.0 mg/ml(TNFαR用)にて調製し、1.1μl(Her-2用)および3.6μl(TNFαR用)を抗原溶液100μlに添加して室温で1時間反応させた。反応産物をゲル濾過スピンカラムに通して未反応のビオチン化試薬を除去した。アビジン(カルビオケム社)をPBSで2μg/mlに希釈した。これをELISAプレートに添加し4℃で一晩放置することでプレートに固定した。ウェルを洗浄後、ビオチン化抗原を添加しアビジンに結合させた。ウェルを洗浄後、実施例1で調製した高純度モデルFabを添加しビオチン化抗原に結合させた。ウェルを洗浄後、検出用抗体として1000倍希釈したHRP標識抗FLAG抗体(シグマ社)を添加し、モデルFabに付加したFLAGタグに結合させた。ウェルを洗浄後、HRP用発色基質を添加した。適度な発色が得られたところで停止液を添加し、OD405nmにて吸光度を測定した。得られた結果を図15に示す。2種類のビオチン化抗原がアビジンと対応するモデルFabに同時反応性をもつことがわかる。
大橋らが報告した方法(Ohashi et al., Biochem Biophys Res Commun. (2007) vol.352, p.270-276)を参考に、PUREシステムによるribsome display法(PRD)によるFabのin vitro選択を実施した。M-280ストレプトアビジン磁気ビーズ(DYNAL社)30μlを洗浄し、実施例8で調製したビオチン化抗原蛋白質を5 p mole加えて室温で30分反応させ、磁気ビーズにベイト蛋白質を固定した。磁気ビーズを洗浄し選択用ビーズとした。同様の操作でベイト蛋白質を固定しないプレクリア用ビーズを調製した。
PURE-2R:(gacgattggccttgatattcacaaacg)(配列番号27)
L-CDRex-1F:(attaaacgtaccgttgcagcaccgagc)(配列番号28)
H-CDRex-2R:(tgagcctccaggctgaaccagaccac)(配列番号29)
選択前と選択後のDNAサンプルをXhoIで消化し、1%アガロースゲルで分離後、エチジウムブロマイドで染色した。選択前のDNAサンプルはFab-TT が主成分となるためXhoIでほとんど切断されないが、選択後のDNAサンプルはFab-HH xhoの含有率が上がるため、XhoI感受性が増すと考えられる。このようにDNAサンプルのXhoI感受性の増加によりFab-HHの濃縮を確認した。以下、Her-2をベイト蛋白質としたシングルラウンドのBicistronic型あるいはMonocistronic型Fab-PRDでFab-HHを選択した結果を示す。
実施例7に記載した方法でMonocistronic型Fab-PRD用のmRNAを用いて無細胞翻訳産物を調製した。翻訳産物を5-20%グラジエントゲルSDS-PAGEで分離後、PVDF膜に転写し、検出用抗体として1000倍希釈したHRP標識抗FLAG抗体(シグマ社)を用いてウエスタンブロッティングを実施した。結果を図19に示す。scFab-TTの全長翻訳産物が確認できるが、全長翻訳産物の比率が陽性対照に使用したscFv-TTの翻訳量に比べて極端に少ないことがわかる。
pGAv6-HH xhoIを鋳型としたPCRでリボゾームストール配列(SecM配列)を2コピーに増やしたDNA断片を合成し、v6.5断片とした。同様のPCRで、SecM配列を3コピーもつv6.5-S3断片、SecM配列を4コピーもつv6.5-S4断片を調製した。調製した鋳型DNA断片(100% Fab-HH)を用いて、Monocistronic型Fab-PRDによるFab-HHの回収率を測定した。Monocistronic型Fab-PRDは実施例9に記載の固相選択にて実施し、回収した逆転写産物の定量は、濃度既知の鋳型DNA断片を検量線用サンプルとした定量PCRにて実施した。定量PCRに用いる検出用プライマーとして上流域プライマーと下流域プライマーの2種類を準備し、CDR-L1付近の上流域とCDR-H3付近の下流域における回収率(1st strand cDNAの分子数/投入したRNAの分子数)をそれぞれ求めた。結果を図20に示す。SecM配列を2コピーに増やすことでFab-PRDによるFab-HHの回収率が約10倍向上した。本実験ではSecM配列を3コピー以上に増やしても更なる回収率の向上は認められなかったが、翻訳反応時間を長くするなど実験条件を変更した場合は、SecMを 3コピー以上にすることで回収率が更に上昇する可能性もあると考えられる。
定量PCRで使用したプライマーを以下に示す。
FabHH用上流域プライマー
RT-1F:gatattcagatgacccagagcccgagc(配列番号33)
RT-1R:cagcttcggggcttttcctgg(配列番号34)
FabHH用下流域プライマー
HH-4F:ccagggaactttggttactgtttc(配列番号35)
Model-4R:ctttaaccagacaacccagtgc(配列番号36)
Fab-PRDに使用した、v6.5、v6.5-S3、v6.5-S4、DNA断片のSecM領域以降の配列を、それぞれ配列番号37、40、44に示す。
Monocistronic型Fab-PRDで使用する鋳型DNAにはL鎖-H鎖間とH鎖-SecM間に計2つのリンカー配列が存在し、実施例12のv6.5断片ではL鎖-H鎖間リンカー長が128アミノ酸、 H鎖-SecM間リンカー長が123アミノ酸となっている。Monocistronic型Fab-PRDの濃縮倍率に与えるリンカー長の影響を検討するために、Fab-HH xhoIとFab-TTについてSecM配列を2コピーもつv6.5断片を基本構造とし、H鎖-SecM間リンカー長を最小化したv7断片と、L鎖-H鎖間リンカー長を最小化したv8.1断片を調製した。
v7断片におけるH鎖-SecM間リンカー長は、H鎖の最終アミノ酸のシステインからSecMの先頭アミノ酸のアラニンの間のアミノ酸数を20とした。v8.1断片におけるL鎖-H鎖間リンカー長はL鎖のC末端のシステインからH鎖のN末端のグルタミン酸の間のアミノ酸数を30とした。それぞれの構造で鋳型DNAをTT/HH比=100で混合し、実施例9に記載の固相選択によるFab-PRDでFab-HH xhoIを選択した。得られた逆転写産物をサンプルとしてFab-HHおよびFab-TTの回収率をそれぞれ定量PCRで測定し、濃縮倍率(Fab-HHの回収率/Fab-TTの回収率)を求めた。定量PCRに用いる検出用プライマーはCDR-H3付近の下流域プライマーを使用した。結果を図21に示す。H鎖-SecM間リンカーおよびL鎖-H鎖間リンカーのいずれを最小化した場合でも、実用的な濃縮倍率が確認されたことから、リンカー長に関しては状況に応じて幅広い選択が可能と考えられる。
定量PCRで使用したプライマーを以下に示す。
FabHH用下流域プライマー
HH-4F:ccagggaactttggttactgtttc(配列番号35)
Model-4R:ctttaaccagacaacccagtgc(配列番号36)
Fab-TT用下流域プライマー
TT-5F:gcaccctggttaccgtgag(配列番号49)
Model-4R:ctttaaccagacaacccagtgc(配列番号36)
Fab-PRDに使用した、v7、v8.1、DNA断片のリンカー部分の配列をそれぞれ配列番号50、52に示す。
Fab-HH xhoIとFab-TTのv7断片(実施例13)を鋳型DNAとして調製し、0.125mg/ml yeast RNAと7 x 109分子/μlのFab-HH鋳型DNAを含むキャリア溶液でFab-TT鋳型DNAの10倍希釈系列を調製した。1 x 106 Fab-TT分子の鋳型DNA /μlから1 x 10-5Fab-TT分子の鋳型DNA /μlのDNAサンプルをFab-TT特異的プライマーを用いた1分子PCRで増幅し、PCR産物を5-20%グラジエントPAGEで分離した。鋳型DNAとしてFab-TT分子が1分子以上含まれるPCRからはFab-TT特異的なバンドが検出され、Fab-TT分子が0.1分子以下のPCRからはFab-TT特異的なバンドが検出されないことを確認した。
1分子PCRによる品質確認済みFab-TT鋳型DNAの希釈サンプルから400分子相当を分取し、1x1012分子のFab-HH鋳型DNAに加えた。ここからRNAを合成し、選択前RNAとした。選択前RNAをFab-PRDに投入し、400分子相当のFab-TTの回収を試みた。翻訳反応は20μlスケールで30℃40分とし、反応後1μMのビオチン化TNFαRを含むWBTBR buffer(50mM Tris-HCl pH=7.6, 90mM NaCl, 50mM Mg(OAc)2, 5mg/ml BSA, 1.25mg/ml yeast RNA, 0.5% Tween20, 0.04U/μl RNase Inhibitor)を等量混合し、4℃で一晩静置した。これに25μl分のM-280ストレプトアビジン磁気ビーズビーズペレットを添加しFab-TTを回収した。M-280ビーズからDTTで溶出したRNAを選択後RNAとした。Fab-PRDによる選択前と選択後のRNAを逆転写し、Fab-TT特異的なCDR-H3付近のプライマーでFab-TT断片(図22のレーン1と2)を、さらにFab-HHとFab-TT双方の共通DNA配列を認識するプライマーで CDR-L1からCDR-H3までの重要な遺伝情報を含むコア断片(図22のレーン4と5)を、それぞれRT-PCRで増幅した。コアDNA断片のバンドを5-20%グラジエントゲルから回収し2nd PCRで再増幅した(図22のレーン7と8)。
2nd PCRで得たコアDNA断片を鋳型とし、Fab-TT特異的プライマーを用いたPCRでFab-TT断片を増幅したところ、選択前サンプルのみならず、選択後サンプルからもFab-TT断片が検出されたことから、400分子のFab-TTからコア断片の回収に成功したと考えた。次にこの回収がFab-PRDによる特異的なものであることを確認するために、Fab-TTの濃縮倍率を定量PCRで測定した。Fab-TT断片とコア断片を独立して定量し、濃縮倍率(選択後サンプル中Fab-TT相対量/選択前サンプル中Fab-TT相対量)を算出した結果、選択後コアDNA断片(図22のレーン7)では選択前コアDNA断片(図22のレーン8)に対しFab-TTの含量が582倍上昇していた(濃縮倍率=582倍)。これらの結果から、ライブラリー中に存在する標的結合クローンは少なくとも400分子以上あればFab-PRDで特異的に回収できることが示唆された。この回収率はファージディスプレイ系に匹敵すると考えられる。
使用したプライマーを以下に示す。
Fab-TT特異的プライマー
T1P-F:gttttactattgaacgttatgcgatgggt(配列番号54)
T1P-R:cgtagtacataccgttcgggtagttag(配列番号55)
Fab-HHとFab-TTに共通のコア領域増幅用プライマー
Core-1F:gcgcaagcgttggtgatc(配列番号56)
Core-1R:gctcggacctttggtgcttg(配列番号57)
Fab-PRDの応用例として、モデルFabの一つであるFab-TTの親和性成熟を試みた。抗原抗体の結合界面は100個以上のファンデルワールス力、数個の水素結合や塩橋が存在する。これらの相互作用ネットワークの全体最適化を実現するために、第一段階で親和性向上に有用と思われるCDR中の変異を検索し、第二段階でこれらの変異の最適な組み合わせを検索する、Ymacsと呼ぶ本来的二段階戦略を採用した。
第一段階前半の作業として、Fab-TTのCDRを構成する全50のアミノ酸の1つ1つを20種類のアミノ酸にランダム化しながら、50アミノ酸分のCDRポジションをマトリクススキャンするためのライブラリー(Ymacs-1次ライブラリー)を構築した。
まずFab-TTのCDRを構成する全50のアミノ酸を1つずつNNKコドンで置換するための1アミノ酸置換導入forwardプライマーを全50種類合成した。これら1アミノ酸置換導入forwardプライマーはNNKコドンの上流に15bp、下流に12bpのアニーリング領域をもつよう設計した。例として50箇所の1アミノ酸置換導入部位の最上流部位と最下流部位に対応するプライマーを以下に示す。
FabTT Ymacs-1 L1-1:tgtcgtgcaagccagNNKattaaaaattat(配列番号58)(L鎖CDR1の1番目のアミノ酸に1アミノ酸置換を入れるためのプライマー)
FabTT Ymacs-1 H3-12:atgtactacgttatgNNKtattggggtcag(配列番号59)(H鎖CDR3の12番目のアミノ酸に1アミノ酸置換を入れるためのプライマー)
pGAv6.5-Fab-TTを鋳型とし上記50種類の1アミノ酸置換導入forwardプライマーをPURE-3Rプライマーと組み合せたPCRで50種類の変異導入下流DNA断片を独立して合成した。PCR産物を1%アガロースゲルで分離後、エチジウムブロマイド染色でバンドを確認した。次に変異導入下流DNA断片のreverse鎖を上流方向に伸長させて全長化する反応で鋳型となる、共通の定常配列上流DNA断片をPCRで合成した。定常配列上流DNA断片は、自身のforward鎖が全長化するのを回避する目的で3末端側にミスアニール領域をもつよう、PURE-rt-1F とH3checkR Not TA6をプライマーとしたPCRで合成し、PCR産物を1%アガロースゲルで分離後、エチジウムブロマイド染色でバンドを確認した。
PURE-rt-1F:caatttcggtaatacgactcactatagggagaatttaggtgacactatagaagtg(配列番号60)
H3checkR Not TA6:tatatatatatagcggccgcagaactgccggaaaggtatg(配列番号61)
1ポジション1アミノ酸置換ライブラリーをL鎖とH鎖にグループ化して等量混合し、L鎖Ymacs-1次ライブラリー、H鎖Ymacs-1次ライブラリーとした。図24に示すように、Ymacs-1次ライブラリーはそれぞれL鎖は19種類、H鎖は31種類の1ポジション1アミノ酸置換ライブラリーから構成される。
第一段階後半の作業として、L鎖とH鎖のYmacs-1次ライブラリーを独立してFab-PRDで濃縮し、得られたDNAサンプルの次世代シークエンス解析結果から、CDR中の有用と思われる変異を同定した。
Fab-PRDによるYmacs-1次ライブラリーの濃縮は実施例9で示した固相選択と実施例14で示した液相選択を併用した。ラウンド1では固相選択を実施し、ラウンド2では固相選択後、長時間(13h)の洗浄を実施した。最終ラウンドのラウンド3ではベイト濃度1nMで液相選択を実施した。全ラウンド1x1012分子のRNAを20μlスケールで翻訳した。逆転写PCRでは実施例14で示したコアDNA断片を回収した。コアDNA断片に適当な上流断片、下流断片を加え、PURE-rt-1FとPURE-3Rをプライマーとしたoverlapping extension-PCRで全長DNA断片を合成し、これを精製後に次のラウンドの鋳型DNAとして使用した。
ラウンド3で得られたコアDNA断片のL鎖、H鎖それぞれのCDR1-3領域の塩基配列をRoche GS FLX次世代シークエンサーで解析し、L鎖ライブラリー由来サンプルからL鎖CDR1-3配列1812リード、H鎖ライブラリー由来サンプルからH鎖CDR1-3配列2288リードの塩基配列データを回収した。この中から、CDR1-3領域において連続したORFをもつ、L鎖468リード、H鎖1293リードを有効リードとして選抜し、CDRを構成するL鎖19アミノ酸、H鎖31アミノ酸、合計50アミノ酸分のCDRポジションにおける変異を集計した。各変異のカウント数を、20種類のアミノ酸を縦方向、合計50のCDRポジションを横方向に配したマトリクス表にまとめ図25に示した。各CDRポジションにおける親アミノ酸は除外し、カウント数が大きい変異をポテンシャルな有用変異とした。
第二段階前半の作業として、実施例16で同定した有用変異を組み合わせることで、Ymacs-2次ライブラリーを構築した。有用変異として計12のCDRポジションにおける計21の変異を採用し、各CDRポジションで親アミノ酸と有用変異アミノ酸をコードできる混合塩基コドンを決定した。混合塩基コドンによっては、親アミノ酸と有用変異アミノ酸以外の意図しないアミノ酸が入る。採用した有用変異と混合塩基コドンを図26にまとめた。Ymacs-2次ライブラリーの理論多様性は核酸レベルで1.9 x 107、蛋白質レベルで4.9 x 106となる。12のCDRポジションは6つのCDR全てに分散したため、1つのCDRごとに計6つの変異導入forwardプライマーを合成した。これらを用いて図27に示す断片2から断片7の計6本の変異導入DNA断片を合成し、これに断片1を加えた計7本のDNA断片をoverlapping extension反応による連結で全長化し、PCR反応で増幅した。得られたPCR産物を精製後、設計通りの混合塩基で変異導入部位がランダム化されていることを確認し、Ymacs-2次ライブラリーとした。
以下に変異導入forwardプライマーを示した。
FabTT Ymacs-2 L1-Fwd : tgtcgtgcaagccaggatattaaaaattatttgWCTtggtatcaacaacaa(配列番号62)(L鎖CDR1)
FabTT Ymacs-2 L2-Fwd : gccccgaagccactgatttatGSTggttctaaccgccaatctggagttcct(配列番号63)(L鎖CDR2)
FabTT Ymacs-2 L3-Fwd : acctattattgccaacaaactKMTRNMtaccctatcacctttggccag(配列番号64)(L鎖CDR3)
FabTT Ymacs-2 H1-Fwd : agctgtgcagcaagcggttttASAattGRGcgttatgcgatgRSTtgggtgcgtcaggct(配列番号65)(H鎖CDR1)
FabTT Ymacs-2 H2-Fwd : ggcctggaatgggttggtacgatttatcctKDSRSCgattatRBYgattatgccgatagc(配列番号66)(H鎖CDR2)
FabTT Ymacs-2 H3-Fwd : tactactgcgctcgctctaactacccgaacggtMTGKRCtacgttatggaatat(配列番号67)(H鎖CDR3)
第二段階後半の作業として、Ymacs-2次ライブラリーをFab-PRDで濃縮し、得られたDNAを大腸菌の分泌発現ベクターにクローン化した。ELISAおよびSPR(ProteOn XPR36、BioRad社)で高親和性クローンを絞込み、Fab-TT変異体の代表2クローン(図29~32中のYmacs#10、 #19)のKDをKinExA(KinExA3200、Sapidyne社)で測定した。
Fab-PRDによるYmacs-2次ライブラリーの濃縮は全5ラウンドを液相選択にて実施した。ラウンド1から5にかけてベイト濃度を、100nM、60nM、20nM、1nM、100pMまたは10pMと徐々に下げることで、選択圧を上げていった。ラウンド5で回収したコアDNA断片を、monocistronicなscFab-HH発現ベクター中のFab-HHのコア領域と置換する形でクローン化した(図28)。ランダムにピックアップした48クローンの培養上清を用いてTNFαR を抗原としたELISAを実施した。ELISAのヒット率が50%程度であることを確認した後、残りの数百コロニーをポリクローナルな状態で一括して回収しプラスミドを精製した。このプラスミドがコードするscFab-TT変異体のL鎖-H鎖間のGSリンカーの領域を、Bicistronic発現用の非翻訳配列-分泌シグナル断片で置換することで、発現様式をmonocistronic 型からBicistronic型に変換した(図28)。この操作によりscFabではなく天然型FabとしてFab-TT変異体を発現するベクターを構築し、再度クローン化した。ランダムにピックアップした96クローンの培養上清を用いてTNFαR を抗原としたELISAを実施し、ELISAのヒットクローンについてCDRの配列を決定した(図29)。ELISAヒットクローンについては結合時間1分、解離時間30分のSPRによるスクリーニングを実施し、Koffを指標に高親和性クローンを絞り込んだ(図30)。親FabのFab-TTと、親和性向上変異体であるYmacs #10について、SPRによる親和性測定を試みた結果、Fab-TTがKD = 7.28 x 10-9、Ymacs #10がKD < 1.57 x 10-11とそれぞれ算出され、約460倍の親和性向上が観察された(図31)。次に高度に親和性が向上しているYmacs#10およびYmacs #19の2クローンについてKinExAでの親和性測定を試みた結果、Ymacs #10がKD = 1.87 x 10-11、Ymacs #19がKD = 3.45 x 10-12とそれぞれ算出され、Ymacs #19で約2100倍の親和性向上が観察された(図32)。
KinExAによるYmacs #19のKD測定実験は、35pMと350pMの2つのFab濃度で実施した。抗原であるTNFαRの濃度は35pMのFabに対し2nMから976fMまでの2倍希釈系列、また350pMのFabに対し5nMから2.44pMまでの2倍希釈系列とした。抗原抗体反応が平衡に達するまでサンプルを室温で48時間インキュベートし、TNFαR 固相化アズラクトンビーズを用いたKinExAで平衡到達後のフリーのFabの存在率を定量した。フリーのFabの存在率(縦軸)を抗原濃度(横軸)に対してプロットし、Fabの濃度ごとに2本の用量-反応曲線を描いた。これをKinExA Pro Softwareでグローバルフィッティング処理しKDを算出した。
Odegripらの報告(Odegrip et al., Proc Natl Acad Sci U S A. (2004) vol. 101, p. 2806-2810)を参考にCISディスプレイ法によるFabの選択を試みた。図8のように、v7断片中のscFabコード領域の上流および下流にそれぞれ、大腸菌RNAポリメラーゼ用プロモータ配列およびRepA-CIS-ori配列をoverlapping extension-PCRで付加しv10.1断片とした。RepAコード配列はOdegripらが報告した配列とGenBank V00351の配列が異なっていたため、V00351の配列を採用した。Fab-HH xhoI とFab-TTのv10.1断片をTT/HH比=10で混合したものをin vitro転写翻訳反応の鋳型DNAとした。精製済み鋳型DNA(1.2μg)を大腸菌由来S30ベースの転写翻訳系(25μL、大腸菌SL119株由来、直鎖DNA用、L1030、Promega社)に投入し、30℃40分でディスプレイ分子複合体を合成した。翻訳反応後はOdegripらの報告を参考に、実施例14と同様の液相選択でFab-HHを回収し、溶出したDNAを鋳型とし、Cis-1FとCis-6RをプライマーとしたPCRで全長断片を増幅した。XhoI感受性を指標とした実施例10と同様の評価系で、CISディスプレイ法によるFab-HH選択系の動作確認を実施した。結果を図33に示す。選択前のDNAサンプルと比較し、ビオチン化Her-2を添加せずに選択を実施した場合はXhoI感受性の増加が認められなかった。一方で、ビオチン化Her-2を添加して選択を実施した場合、XhoI感受性の増加が認められた。これらの結果から、Fab-HHが特異的に濃縮されたことがわかる。
さらに、Fab-HH xhoIとFab-TTのv10.1断片をTT/HH比=100で混合したものを用いて同様の実験を実施し最適な転写翻訳時間を検討した。結果を図34に示す。検討した全ての転写翻訳時間においてFab-HHの特異的な濃縮は認められたが、転写翻訳時間10分では不十分であった濃縮倍率が転写翻訳時間20分~40分にかけて十分高まり、転写翻訳時間80分~120分にかけて次第に低下する傾向を示した。これらの結果から、転写翻訳時間として20分~40分が最適と考えられた。
v10.1断片の全長増幅用プライマーの配列を以下に示す。
Cis-1F
cagttgatcggcgcgagatttaatcgccgc(配列番号68)
Cis-6R
cgtaagccggtactgattgatagatttcaccttacccatc(配列番号69)
Claims (27)
- Fab第1鎖コード配列とFab第2鎖コード配列を含み、リボゾームを含む無細胞翻訳系に導入されたときに自身がコードするFabを解離させることなく発現し、該Fabとの複合体を維持しうるポリヌクレオチド構築物。
- リボゾーム結合配列、Fab 第1鎖コード配列、リンカーペプチドコード配列、Fab 第2鎖コード配列、および足場コード配列をこの順にモノシストロニックに含む請求項1に記載のポリヌクレオチド構築物であって、その3’末端に自身がコードするFabとの複合体を維持するために必要な構造を有するポリヌクレオチド構築物。
- リボゾーム結合配列、Fab 第1鎖コード配列またはFab 第2鎖コード配列、および足場コード配列をこの順に含む、Fab 第1鎖発現シストロンおよびFab 第2鎖発現シストロンを含む請求項1に記載のポリヌクレオチド構築物であって、Fab 第1発現シストロンはその3’末端にリボゾームストール配列を有し、Fab 第2鎖発現シストロンはその3’末端に自身がコードするFabとの複合体を維持するために必要な構造を有するポリヌクレオチド構築物。
- 自身がコードするFabとの複合体を維持するために必要な構造は、リボゾームストール配列、ピューロマイシンもしくはその誘導体、またはDNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列である、請求項2または3に記載のポリヌクレオチド構築物。
- 自身がコードするFabとの複合体を維持するために必要な構造が、リボゾームストール配列である、請求項4に記載のポリヌクレオチド構築物。
- リボゾームストール配列がSecM配列、ジプロリン配列、またはこれらの両方である、請求項5のいずれか一項に記載のポリヌクレオチド構築物。
- リボゾームストール配列がSecM配列の2~4個の繰り返しである、請求項5または6に記載のポリヌクレオチド構築物。
- リボゾームストール配列の3’側に終止コドンが存在する、請求項4~7のいずれか一項に記載のポリヌクレオチド構築物。
- 自身がコードするFabとの複合体を維持するために必要な構造が、ピューロマイシンもしくはその誘導体である、請求項4に記載のポリヌクレオチド構築物。
- 自身がコードするFabとの複合体を維持するために必要な構造が、DNA結合蛋白質コード配列と該DNA結合蛋白質の結合配列である、請求項4に記載のポリヌクレオチド構築物。
- DNA結合蛋白質が、転写翻訳時に鋳型となったDNA分子と、転写翻訳反応中に一度も解離することなく、同一DNA分子上に存在する該DNA結合蛋白質の結合配列と結合するシス型結合蛋白質である、請求項10に記載のポリヌクレオチド構築物。
- DNA結合蛋白質が大腸菌R1 Plasmid にコードされるRepAであり、該DNA結合蛋白質の結合配列が同ポリヌクレオチド上でRepAコード配列の下流に存在するCIS-ori配列である、請求項10に記載のポリヌクレオチド構築物。
- Fab 第1鎖コード配列およびFab 第2鎖コード配列がランダム配列を含むライブラリーである、請求項1~12のいずれか一項に記載のポリヌクレオチド構築物。
- ランダム配列を含むライブラリーが、(i)ナイーブライブラリーまたは(ii) フォーカスドライブラリーである、請求項13に記載のポリヌクレオチド構築物。
- ランダム配列を含むライブラリーが、Fab 第1鎖および/またはFab 第2鎖の相補性決定領域(CDR)内に1アミノ酸置換を含むライブラリーである、請求項13または14に記載のポリヌクレオチド構築物。
- (i)請求項1~15のいずれか一項に記載のポリヌクレオチド構築物を無細胞翻訳系に導入してFabを合成し、合成されたFabをそれをコードするポリヌクレオチド上に提示する工程、
(ii)ポリヌクレオチド上に提示されたFabを抗原と接触させる工程、
(iii)抗原と反応する目的Fabを選択する工程、および
(iv)目的Fabをコードするポリヌクレオチドを増幅する工程を含む、Fabのスクリーニング方法。 - (I)Fab 第1鎖コード配列またはFab 第2鎖コード配列が、親抗体のFab 第1鎖またはFab 第2鎖のアミノ酸配列においてCDR内の1つの位置に対して1アミノ酸置換を含むアミノ酸配列をコードする、請求項15に記載のポリヌクレオチド構築物を、Fab 第1鎖およびFab 第2鎖のCDR内の複数の位置に対して1アミノ酸置換が含まれるように複数種類用意する工程、
(II)該複数種類のポリヌクレオチド構築物を用いて前記工程(i)~(iv)を繰り返して高親和性Fabを複数スクリーニングする一次スクリーニング工程、
(III)一次スクリーニング工程で選択された複数のFabにおけるFab 第1鎖およびFab 第2鎖のCDR内の各位置での1アミノ酸置換を解析する工程、
(IV)Fab 第1鎖コード配列およびFab 第2鎖コード配列が、親抗体のFab 第1鎖およびFab 第2鎖の配列においてCDR内の各位置に一次スクリーニングで同定された1アミノ酸置換の組み合わせを含むアミノ酸配列をコードする、請求項15に記載のポリヌクレオチド構築物を用意する工程、
(V)該ポリヌクレオチド構築物を用いて前記工程(i)~(iv)を繰り返して高親和性Fabをスクリーニングする二次スクリーニング工程
を含む、請求項16に記載のFabのスクリーニングを行う方法。 - インビトロ翻訳系が、独立に精製された因子からなる、請求項16または17に記載の方法。
- インビトロ翻訳系が、開始因子、伸長因子、アミノアシルtRNA合成酵素、およびメチオニルtRNAトランスフォルミラーゼからなる群から選択される少なくとも1つの成分を含む、請求項18に記載の方法。
- インビトロ翻訳系が解離因子を含まない、請求項18または19に記載の方法。
- インビトロ翻訳系が、リボゾームを含む細胞抽出液である、請求項16または17に記載の方法。
- 請求項1~15のいずれか一項に記載のポリヌクレオチド構築物をインビトロ翻訳系に導入してFabを生成させる工程を含む、Fabの製造方法。
- 請求項1~15のいずれか一項に記載のポリヌクレオチド構築物を含む、Fabを製造またはスクリーニングするためのキット。
- (I)標的物質結合蛋白質における標的物質結合部位を構成する全アミノ酸ポジションについて、1アミノ酸ポジションごとに天然アミノ酸全20種類にランダム化する1ポジションライブラリーを、アミノ酸ポジションの数だけ構築する工程、
(II)これらの1ポジションライブラリーを全てあるいは適当な単位で統合して1次ライブラリーを構築する工程、
(III)蛋白質ディスプレイ系を用いて1次ライブラリーを標的親和性で濃縮する工程、
(IV)1次ライブラリー濃縮サンプルのポリヌクレオチド配列情報を決定する工程、
(V)塩基配列情報から高頻度に観察される1アミノ酸置換を抽出する工程、
(VI)高頻度に観察される1アミノ酸置換の組み合わせを含む2次ライブラリーを構築する工程、および
(VII)蛋白質ディスプレイ系を用いて2次ライブラリーを標的親和性で濃縮する工程、
を含む、標的物質結合蛋白質の標的物質親和性を最大化する方法。 - 1次ライブラリー濃縮サンプルのポリヌクレオチド配列情報を決定する工程が次世代シークエンサーを用いて行われる、請求項24に記載の方法。
- 標的物質結合蛋白質が完全長抗体あるいは抗体断片であり、標的物質結合部位がCDR領域である、請求項24または25に記載の方法。
- 蛋白質ディスプレイ系が、リボゾームディスプレイ、CISディスプレイ、mRNAディスプレイ、ファージディスプレイ、バクテリア表面ディスプレイ、酵母細胞表面ディスプレイ、または高等真核生物の細胞表面ディスプレイである、請求項24~26のいずれか一項に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/990,654 US9753040B2 (en) | 2010-12-01 | 2011-11-30 | Polynucleotide construct capable of displaying fab in a cell-free translation system, and method for manufacturing and screening fab using same |
| EP11845270.5A EP2647704B1 (en) | 2010-12-01 | 2011-11-30 | Polynucleotide construct capable of presenting fab in acellular translation system, and method for manufacturing and screening fab using same |
| JP2012546923A JP5920835B2 (ja) | 2010-12-01 | 2011-11-30 | 無細胞翻訳系でFabを提示しうるポリヌクレオチド構築物ならびにそれを用いたFabの製造方法およびスクリーニング方法 |
| US15/693,738 US20180017573A1 (en) | 2010-12-01 | 2017-09-01 | Polynucleotide construct capable of displaying fab in a cell-free translation system, and method for manufacturing and screening fab using same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-268763 | 2010-12-01 | ||
| JP2010268763 | 2010-12-01 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/990,654 A-371-Of-International US9753040B2 (en) | 2010-12-01 | 2011-11-30 | Polynucleotide construct capable of displaying fab in a cell-free translation system, and method for manufacturing and screening fab using same |
| US15/693,738 Division US20180017573A1 (en) | 2010-12-01 | 2017-09-01 | Polynucleotide construct capable of displaying fab in a cell-free translation system, and method for manufacturing and screening fab using same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012074029A1 true WO2012074029A1 (ja) | 2012-06-07 |
Family
ID=46171954
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/077725 WO2012074029A1 (ja) | 2010-12-01 | 2011-11-30 | 無細胞翻訳系でFabを提示しうるポリヌクレオチド構築物ならびにそれを用いたFabの製造方法およびスクリーニング方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US9753040B2 (ja) |
| EP (1) | EP2647704B1 (ja) |
| JP (1) | JP5920835B2 (ja) |
| WO (1) | WO2012074029A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014106004A3 (en) * | 2012-12-28 | 2014-08-28 | Abbvie, Inc. | High-throughput system and method for identifying antibodies having specific antigen binding activities |
| WO2014189768A1 (en) * | 2013-05-19 | 2014-11-27 | The Board Of Trustees Of The Leland | Devices and methods for display of encoded peptides, polypeptides, and proteins on dna |
| US9133452B2 (en) | 2010-06-23 | 2015-09-15 | The University Of Tokyo | High-speed maturation method for an oligonucleotide library for the purpose of preparing a protein library |
| US9458244B2 (en) | 2012-12-28 | 2016-10-04 | Abbvie Inc. | Single chain multivalent binding protein compositions and methods |
| JP2018517899A (ja) * | 2015-05-21 | 2018-07-05 | フル スペクトラム ジェネティクス, インコーポレイテッド | タンパク質の特徴を改善する方法 |
| WO2018168999A1 (ja) * | 2017-03-17 | 2018-09-20 | 国立研究開発法人理化学研究所 | Rna分子とペプチドとの複合体の製造方法、及びその利用 |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014176327A2 (en) * | 2013-04-25 | 2014-10-30 | Sutro Biopharma, Inc. | Selection of fab fragments using ribosomal display technology |
| WO2014176439A1 (en) | 2013-04-25 | 2014-10-30 | Sutro Biopharma, Inc. | The use of lambda-gam protein in ribosomal display technology |
| US20160312212A1 (en) * | 2015-04-23 | 2016-10-27 | Roche Sequencing Solutions, Inc. | Cow antibody scaffold polypeptide method and composition |
| EP3475700A4 (en) | 2016-06-26 | 2020-03-04 | Gennova Biopharmaceuticals Limited | PHAGE ANTIBODY PRESENTATION BANK |
| US11267859B2 (en) | 2017-06-09 | 2022-03-08 | The Regents Of The University Of Colorado, A Body Corporate | GM-CSF mimetics and methods of making and using same |
| WO2019075098A1 (en) * | 2017-10-13 | 2019-04-18 | The Regents Of The University Of California | TRANSLATION BLOCKING COMPOSITIONS AND METHODS OF USE |
| US10320283B1 (en) * | 2018-01-26 | 2019-06-11 | Universal Lighting Technologies, Inc. | Resonant converter with pre-charging circuit for startup protection |
| US11174479B2 (en) | 2018-08-13 | 2021-11-16 | The Board Of Trustees Of The Leland Stanford Junior University | Devices and methods for display of encoded peptides, PolyPeptides, and proteins on DNA |
| WO2020215044A1 (en) * | 2019-04-17 | 2020-10-22 | The Regents Of The University Of California | Small molecule-based control of immune cell receptor expression |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016636A1 (fr) | 1996-10-17 | 1998-04-23 | Mitsubishi Chemical Corporation | Molecule permettant d'homologuer un genotype et un phenotype, et utilisation de celle-ci |
| JP2003102495A (ja) | 2000-12-28 | 2003-04-08 | Post Genome Institute Co Ltd | invitro転写/翻訳系によるペプチド等の製造方法 |
| JP2003514572A (ja) | 1999-11-24 | 2003-04-22 | ザ リサーチ ファウンデイション オブ ステイト ユニバーシティー オブ ニューヨーク | アミノアシル化活性を有した触媒RNAs |
| WO2004022746A1 (en) | 2002-09-06 | 2004-03-18 | Isogenica Limited | In vitro peptide expression libraray |
| JP2005535301A (ja) * | 2002-06-03 | 2005-11-24 | ジェネンテック・インコーポレーテッド | 合成抗体ファージライブラリー |
| WO2008103475A1 (en) | 2007-02-20 | 2008-08-28 | Anaptysbio, Inc. | Somatic hypermutation systems |
| JP2008271903A (ja) | 2007-05-02 | 2008-11-13 | Univ Of Tokyo | リボソームディスプレイ |
| JP2011182794A (ja) | 2003-06-27 | 2011-09-22 | Bioren Inc | ルックスルー突然変異誘発 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE521704T1 (de) | 2007-03-01 | 2011-09-15 | Symphogen As | Verfahren zur klonierung verwandter antikörper |
-
2011
- 2011-11-30 JP JP2012546923A patent/JP5920835B2/ja not_active Expired - Fee Related
- 2011-11-30 US US13/990,654 patent/US9753040B2/en not_active Expired - Fee Related
- 2011-11-30 WO PCT/JP2011/077725 patent/WO2012074029A1/ja active Application Filing
- 2011-11-30 EP EP11845270.5A patent/EP2647704B1/en not_active Not-in-force
-
2017
- 2017-09-01 US US15/693,738 patent/US20180017573A1/en not_active Abandoned
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016636A1 (fr) | 1996-10-17 | 1998-04-23 | Mitsubishi Chemical Corporation | Molecule permettant d'homologuer un genotype et un phenotype, et utilisation de celle-ci |
| JP2003514572A (ja) | 1999-11-24 | 2003-04-22 | ザ リサーチ ファウンデイション オブ ステイト ユニバーシティー オブ ニューヨーク | アミノアシル化活性を有した触媒RNAs |
| JP2003102495A (ja) | 2000-12-28 | 2003-04-08 | Post Genome Institute Co Ltd | invitro転写/翻訳系によるペプチド等の製造方法 |
| JP2005535301A (ja) * | 2002-06-03 | 2005-11-24 | ジェネンテック・インコーポレーテッド | 合成抗体ファージライブラリー |
| WO2004022746A1 (en) | 2002-09-06 | 2004-03-18 | Isogenica Limited | In vitro peptide expression libraray |
| JP2005537795A (ja) | 2002-09-06 | 2005-12-15 | イソゲニカ リミテッド | イン・ビトロでのペプチド発現ライブラリー |
| JP2011182794A (ja) | 2003-06-27 | 2011-09-22 | Bioren Inc | ルックスルー突然変異誘発 |
| WO2008103475A1 (en) | 2007-02-20 | 2008-08-28 | Anaptysbio, Inc. | Somatic hypermutation systems |
| JP2008271903A (ja) | 2007-05-02 | 2008-11-13 | Univ Of Tokyo | リボソームディスプレイ |
Non-Patent Citations (66)
| Title |
|---|
| "Molecular Cloning", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| AGRAWAL RK ET AL., NAT. STRUCT. BIOL, vol. 6, 1999, pages 643 - 647 |
| BODER ET AL., PROC NATL ACAD SCI USA, vol. 97, 2000, pages 10701 - 10705 |
| BUKAU; HORWICH, CELL, vol. 92, 1998, pages 351 - 366 |
| CAMPBELL ET AL., MOL. IMMUNOL., vol. 29, 1992, pages 193 |
| CARTER ET AL., PROC NATL ACAD SCI U S A., vol. 89, 1992, pages 4285 - 4289 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLAUDIO 0 E, BIOCHEMISTRY, vol. 29, 1990, pages 5881 - 5889 |
| COOMBER, METHOD MOL. BIOL., vol. 178, 2002, pages 133 - 145 |
| ECHOLS ET AL., GENETICS, vol. 83, 1976, pages 5 - 10 |
| ELLISON ET AL., NUCLEIC ACIDS RES., vol. 10, 1982, pages 4071 |
| FELDHAUS ET AL., NAT. BIOTECHNOL., vol. 21, 2003, pages 163 - 70 |
| FELLOUSE ET AL., J. MOL. BIOL., vol. 373, 2007, pages 924 - 940 |
| FRANCKE ET AL., PROC NATL ACAD SCI USA, vol. 69, 1972, pages 475 - 479 |
| FRANCKE ET AL., PROC NATLACAD SCI USA, vol. 69, 1972, pages 475 - 479 |
| FRANCKLYN C ET AL., RNA, vol. 3, 1997, pages 954 - 960 |
| G. P. SMITH ET AL., SCIENCE, vol. 228, 1985, pages 1315 - 1317 |
| HIETER E, J. BIOL. CHEM., vol. 257, 1982, pages 1516 |
| HIRAO I ET AL., NAT. BIOTECHNOL., vol. 20, 2002, pages 177 - 182 |
| HOET ET AL., NAT. BIOTECHNOL., vol. 23, 2005, pages 344 - 348 |
| HOHSAKA ET AL., J. AM. CHEM. SOC., vol. 121, 1999, pages 12194 - 12195 |
| HOHSAKA, T ET AL., J. AM. CHEM. SOC., vol. 121, 1996, pages 34 - 40 |
| HUCK ET AL., NUCLEIC ACIDS RES., vol. 14, 1986, pages 1779 |
| HWANG YW ET AL., ARCH. BIOCHEM. BIOPHYS., vol. 348, 1997, pages 157 - 162 |
| J. MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 554 |
| JOSE, APPL. MICROBIOL. BIOTECHNOL., vol. 69, 2006, pages 607 - 614 |
| KANAPPIK ET AL., J MOL BIOL., vol. 296, 2000, pages 57 - 86 |
| KANG ET AL., PROC NATL ACAD SCI USA, vol. 88, 1991, pages 11120 - 11123 |
| KNAPPIK ET AL., J. MOL. BIOL., vol. 296, 2000, pages 57 - 86 |
| LEE ET AL., BLOOD, vol. 108, 2006, pages 3103 - 3111 |
| LESLEY ET AL., J. BIOL. CHEM., vol. 266, 1991, pages 2632 - 2638 |
| M. HUST ET AL., BMC BIOTECHNOLOGY, vol. 7, 2007, pages 14 |
| MARDIS, ANNU. REV. GENOM. HUMAN GENET., vol. 9, 2008, pages 387 - 402 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1991, pages 581 |
| MASAKI UEMURA ET AL.: "Gosei CDR Library ni yoru Kotai no Shinwasei Zokyo-Tokuisei Henkan eno Approach", JOURNAL OF JAPANESE BIOCHEMICAL SOCIETY, 2009, pages 4T15P-6, XP008168727 * |
| MAX ET AL., CELL, vol. 22, 1980, pages 197 |
| NAKATOGAWA ET AL., MOL. CELL, vol. 22, 2006, pages 545 - 552 |
| NUCLEIC ACID RESEARCH, vol. 16, no. 14, 1988, pages 6493 |
| NUCLEIC ACID RESEARCH, vol. 38, no. 13, 2010, pages E141 |
| ODEGRIP ET AL., PROC NATL ACAD SCI U S A., vol. 101, 2004, pages 2806 - 2810 |
| OHASHI E, BIOCHEM BIOPHYS RES COMMUN., vol. 352, 2007, pages 270 - 276 |
| OHASHI ET AL., BIOCHEM BIOPHYS RES COMMUN., vol. 352, 2007, pages 270 - 276 |
| PAPE T ET AL., EMBO J, vol. 17, 1998, pages 7490 - 7497 |
| PELHAM ET AL., EUR. J. BIOCHEM., vol. 67, 1976, pages 247 - 256 |
| PERSSON ET AL., CHEM. SOC. REV., vol. 39, 2010, pages 985 - 999 |
| PINTO ET AL., MOL. MICROBIOL., vol. 81, 2011, pages 1593 - 1606 |
| PROC. NATL. ACAD. SCI. U. S. A., vol. 101, 2004, pages 2806 - 2810 |
| PROTEINS, NUCLEIC ACIDS AND ENZYMES, vol. 39, 1994, pages 1215 - 1225 |
| RAJPAL ET AL., PROC NATL ACAD SCI U S A, vol. 102, 2005, pages 8466 - 8471 |
| RAMESH V ET AL., PROC. NATL. ACAD. SCI. USA, vol. 96, 1999, pages 875 - 880 |
| ROBERTS ET AL., PROC NATL ACAD SCI USA, vol. 70, 1973, pages 2330 - 2334 |
| RODNINA MW. ET AL., FEMS MICROBIOLOGY REVIEWS, vol. 23, 1999, pages 317 - 333 |
| SAKANO ET AL., NATURE, vol. 286, 1980, pages 676 |
| See also references of EP2647704A4 |
| SHIBUI ET AL., APPL MICROBIOL BIOTECHNOL., vol. 84, 2009, pages 725 - 732 |
| SHIBUI ET AL., BIOTECHNOL. LETT., vol. 31, 2009, pages 1103 - 1110 |
| SHIMIZU ET AL., METHODS, vol. 36, 2005, pages 299 - 304 |
| SHIMIZU ET AL., NAT BIOTECHNOL., vol. 19, 2001, pages 751 - 755 |
| SHIMIZU ET AL., NAT. BIOTECHNOL., vol. 19, 2001, pages 751 - 755 |
| SODERLIND ET AL., NAT. BIOTECHNOL., vol. 18, 2000, pages 852 - 856 |
| SUMIDA T. ET AL.: "Bicistronic DNA display for in vitro selection of Fab fragments.", NUCLEIC ACIDS RES., vol. 37, no. 22, 2009, pages E147, XP002695888 * |
| T. SUMIDA ET AL., NUCLEIC ACID RESEARCH, vol. 37, no. 22, 2009, pages 147 |
| WELSCHOF ET AL., J. IMMUNOL. METHODS, vol. 179, 1995, pages 203 |
| YING ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 320, 2004, pages 1359 - 1364 |
| YOUNG ET AL., NAT. REV. MOL. CELL BIOL, vol. 5, 2004, pages 781 |
| ZUBAY, ANNUAL REVIEW OF GENETICS, vol. 7, 1973, pages 267 - 287 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9133452B2 (en) | 2010-06-23 | 2015-09-15 | The University Of Tokyo | High-speed maturation method for an oligonucleotide library for the purpose of preparing a protein library |
| WO2014106004A3 (en) * | 2012-12-28 | 2014-08-28 | Abbvie, Inc. | High-throughput system and method for identifying antibodies having specific antigen binding activities |
| US9458244B2 (en) | 2012-12-28 | 2016-10-04 | Abbvie Inc. | Single chain multivalent binding protein compositions and methods |
| US9915665B2 (en) | 2012-12-28 | 2018-03-13 | Abbvie Inc. | High-throughput system and method for identifying antibodies having specific antigen binding activities |
| WO2014189768A1 (en) * | 2013-05-19 | 2014-11-27 | The Board Of Trustees Of The Leland | Devices and methods for display of encoded peptides, polypeptides, and proteins on dna |
| JP2018517899A (ja) * | 2015-05-21 | 2018-07-05 | フル スペクトラム ジェネティクス, インコーポレイテッド | タンパク質の特徴を改善する方法 |
| JP2021061852A (ja) * | 2015-05-21 | 2021-04-22 | フル スペクトラム ジェネティクス, インコーポレイテッド | タンパク質の特徴を改善する方法 |
| JP7109605B2 (ja) | 2015-05-21 | 2022-07-29 | フル スペクトラム ジェネティクス, インコーポレイテッド | タンパク質の特徴を改善する方法 |
| WO2018168999A1 (ja) * | 2017-03-17 | 2018-09-20 | 国立研究開発法人理化学研究所 | Rna分子とペプチドとの複合体の製造方法、及びその利用 |
| JPWO2018168999A1 (ja) * | 2017-03-17 | 2020-02-27 | 株式会社CUBICStars | Rna分子とペプチドとの複合体の製造方法、及びその利用 |
| US11286481B2 (en) | 2017-03-17 | 2022-03-29 | Cubicstars, Inc. | Method for producing complex of RNA molecule and peptide, and utilization thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5920835B2 (ja) | 2016-05-18 |
| JPWO2012074029A1 (ja) | 2014-05-19 |
| US20130288908A1 (en) | 2013-10-31 |
| US20180017573A1 (en) | 2018-01-18 |
| EP2647704A4 (en) | 2014-12-03 |
| EP2647704B1 (en) | 2017-11-01 |
| EP2647704A1 (en) | 2013-10-09 |
| US9753040B2 (en) | 2017-09-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5920835B2 (ja) | 無細胞翻訳系でFabを提示しうるポリヌクレオチド構築物ならびにそれを用いたFabの製造方法およびスクリーニング方法 | |
| EP2670847B1 (en) | Methods and materials for enhancing functional protein expression in bacteria | |
| JP6073038B2 (ja) | 安定化された免疫グロブリン可変ドメイン選別方法及び選別されたドメインの応用 | |
| He et al. | Ribosome display: cell-free protein display technology | |
| JP2013517761A (ja) | 酵母細胞の表面上にポリペプチドをディスプレイするための方法および組成物 | |
| JP2015531597A5 (ja) | ||
| GB2422606A (en) | Method of in vitro protein evolution to improve stability | |
| JP2023518900A (ja) | 抗原特異的結合ポリペプチド遺伝子ディスプレイベクターの構築方法および用途 | |
| Villemagne et al. | Highly efficient ribosome display selection by use of purified components for in vitro translation | |
| DK2989239T3 (en) | Selection of Fab fragments using ribosome display technology | |
| JP2008271903A (ja) | リボソームディスプレイ | |
| KR102194203B1 (ko) | 항체 나이브 라이브러리의 생성 방법, 상기 라이브러리 및 그 적용(들) | |
| JP2011178691A (ja) | カテニン結合プローブとその用途 | |
| JP2009112286A (ja) | 標的ポリペプチドに結合するポリペプチドを選択する方法 | |
| KR102216032B1 (ko) | 합성 항체 라이브러리의 생성 방법, 상기 라이브러리 및 그 적용(들) | |
| US20160002317A1 (en) | Cytoplasmic Expression of Fab Proteins | |
| Liu et al. | High-throughput reformatting of phage-displayed antibody fragments to IgGs by one-step emulsion PCR | |
| Chance et al. | Eukaryotic ribosome display for antibody discovery: A review | |
| JP6293829B2 (ja) | 安定化された免疫グロブリン可変ドメイン選別方法及び選別されたドメインの応用 | |
| Kulmala | UNVEILING THE SECRETS OF DNA | |
| Douthwaite et al. | Accelerated protein evolution using ribosome display |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11845270 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012546923 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011845270 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011845270 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13990654 Country of ref document: US |