WO2012029577A1 - 単分子dnaから形成される環状dnaの作成方法 - Google Patents
単分子dnaから形成される環状dnaの作成方法 Download PDFInfo
- Publication number
- WO2012029577A1 WO2012029577A1 PCT/JP2011/068856 JP2011068856W WO2012029577A1 WO 2012029577 A1 WO2012029577 A1 WO 2012029577A1 JP 2011068856 W JP2011068856 W JP 2011068856W WO 2012029577 A1 WO2012029577 A1 WO 2012029577A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- adapter
- dna
- restriction enzyme
- dna molecule
- site
- Prior art date
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1096—Processes for the isolation, preparation or purification of DNA or RNA cDNA Synthesis; Subtracted cDNA library construction, e.g. RT, RT-PCR
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/64—General methods for preparing the vector, for introducing it into the cell or for selecting the vector-containing host
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2521/00—Reaction characterised by the enzymatic activity
- C12Q2521/30—Phosphoric diester hydrolysing, i.e. nuclease
- C12Q2521/301—Endonuclease
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/30—Oligonucleotides characterised by their secondary structure
- C12Q2525/307—Circular oligonucleotides
Definitions
- the present invention relates to a method for producing circular DNA formed only from single-molecule DNA, a novel adapter used in such a method, and a circular DNA production kit comprising the novel adapter.
- the present invention relates to a gene identification and / or detection method using the unimolecular circular DNA obtained by the above method.
- the present invention relates to a method for identifying and / or detecting a fusion gene that causes various pathological conditions.
- a conventional gene analysis method includes a vector method.
- the sequence of the full length of a gene obtained by incorporating the gene to be analyzed into a vector and growing it is determined by a sequencer.
- the vector method requires a culture operation, it is necessary to analyze the full length of the gene with a sequencer.
- Fig. 1 schematically shows an outline of gene analysis by the mate pair method.
- a base sequence for binding (restriction enzyme recognition site) is bound to both ends of an analysis target gene, and the target gene is circularized.
- the circularized gene is cut mainly at the restriction enzyme recognition site, usually using Type II restriction enzyme, and cut at 15 or more bases before and after the recognition site, preferably 25 bases or more and tens of bases or less.
- the base sequence of the amplified and excised partial gene is determined.
- a mate pair is a set of sequence data of base sequences obtained by decoding both ends of a single DNA fragment.
- a method of cutting out a gene with a certain number of bases a method that cuts a predetermined number of bases by cutting a site away from the recognition site using Type II restriction enzyme, and a linker by physically cutting circular DNA with Sonication etc.
- a method is used in which the fragments are collected with biotin attached to the DNA and the fragments are amplified by PCR and sequenced.
- a known gene in the mate pair method, can be identified by reading a certain base sequence before and after the binding portion in a circularized gene by binding both ends of DNA. Basically, if the base sequence of a part of the head portion and tail portion of a gene is read, the sequences can be reliably differentiated for each gene, so the mate pair method has been adopted as a reliable and simple gene analysis method ( Non-Patent Documents 1 and 2). In addition, the mate-pair method is applied to next-generation sequence analysis, and is becoming increasingly important with the advent of high-speed sequencers.
- DNA when DNA is circularized for gene analysis by the mate pair method, in addition to self-circularization with a single gene, DNA (single molecule), circularization with multiple DNA (multiple molecules) or multiple molecules (2 A linear bond of more than molecules).
- a linear molecule composed of a plurality of molecules is separated and removed from a cyclic molecule by a subsequent operation.
- a cyclic molecule composed of a plurality of molecules cannot be separated from a cyclic molecule composed of a single molecule and becomes a contaminant.
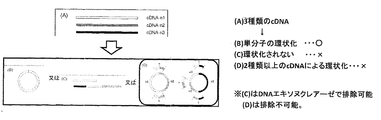
- Multi-molecule circulars inhibit individual gene analysis and greatly reduce analysis specificity for the reasons described below. Specifically, when three kinds of cDNAs are to be self-circulated as shown in FIG.
- the gene analysis by the mate pair method is to specify a gene by the base sequences of both ends of the target gene. Specifically, after binding adapters for circularization are bound to both ends of each gene, the genes are bound and circularized at both adapter sites, and then a fixed number of base sequences are formed around the adapter sites. Then, the gene is identified by cutting and analyzing the partial base sequence from each end of the initial gene as a result. Therefore, in the cyclized product of a plurality of molecules, there are a plurality of adapter sites, and both ends bound to the adapter are each one end of a different gene. Therefore, as described above, in gene analysis, the gene analysis is performed by cutting into a constant base sequence at both ends binding to the adapter by either of the two methods described above.
- the gene fragment for analysis obtained from the circularized product contains one end of each of the different genes, and one gene is not analyzed.
- the probability of circularization of multiple DNA molecules usually varies from several percent to several tens of percent, depending on the method, but in the analysis of known genes, it is recognized as an abnormal base sequence and can be almost excluded from the analysis sequence. As a result, complications arise, but the accuracy is only slightly reduced.
- the mate pair method is used to detect the presence of an abnormal gene such as a fusion gene from a group of normal genes, if multiple normal genes are circularized, it is determined that the abnormal gene is present. Therefore, the presence of an abnormal gene such as a fusion gene cannot be confirmed accurately.
- a fusion gene is a gene in which multiple (two) genes are joined together to construct a gene with a new function.
- cancer cells have abnormal chromosomal structures such as deletion, duplication, recombination, and translocation.
- Gene splitting and linking occur at the DNA level, and a fusion gene is formed when a structural gene is present at each breakpoint.
- fusion genes are lethal to cells, meaningless, and in many cases do not cause clinical problems.
- the fusion protein produced from the fusion gene inhibits the regulation of cell proliferation and thereby abnormally promotes cell proliferation, it becomes clinically prominent as a tumor or the like.
- the fusion gene was said to be expressed mainly in hematopoietic tumors, but in recent years, it has been estimated that the fusion gene is also involved in epithelial solid tumors (Non-patent Document 3). Among them, responsible fusion genes were discovered from prostate cancer and lung cancer (Non-patent Documents 4 and 5).
- fusion genes that is, confirmation of their presence
- cancers cancers
- rapid diagnosis of a disease state becomes possible by detecting a known fusion gene known to correspond to the disease state.
- discovery of new fusion genes will also lead to the discovery of drug discovery targets.
- Non-patent Document 6 chromosomal analysis has been limited for solid tumors, and analysis and confirmation of fusion genes has been extremely difficult, but recently, new methods such as cDNA functional expression analysis by Mano et al. Have been developed. However, this technique is still insufficient due to operational complexity and accuracy problems (Patent Document 1). Recently, various gene next-generation high-speed sequencers have been developed, and high-speed sequence analysis of genes has been remarkably advanced, and analysis in a short time is becoming possible. This has led to the search for fusion genes by high-speed, large-scale base sequence analysis of tumor genomes and genes (Non-patent Document 6).
- FIG. 4 A schematic diagram in the case of analyzing the fusion gene by the mate pair method is shown in FIG.
- FIG. 4 A schematic diagram in the case of analyzing the fusion gene by the mate pair method is shown in FIG.
- FIG. 4 there is a possibility that a plurality of cDNAs may form one circular DNA.
- sequence analysis using the mate pair method is performed, even if it is a normal gene, it appears as if it is a fusion gene. Results. If this is excluded because it does not exist in the conventional gene sequence, the fusion gene will be excluded as well, and it will be virtually impossible to confirm the presence of the fusion gene.
- an object of the present invention is to provide a method that enables one circular DNA to be prepared from one DNA.
- the present inventors can form a circular DNA consisting of only a single molecule by introducing an adapter having a specific structure containing a unique sequence recognition site into a single DNA molecule and performing two-step ligation. I found what I could do. The present inventors have found a method that ensures that only single-molecule DNA is circularized and does not cause circularization between multiple molecules by a plurality of DNAs.
- the present invention provides a method for producing circular DNA comprising circular DNA formed from single-molecule DNA and not including circular DNA formed from multiple-molecule DNA, comprising the following steps: 1) First-stage circularization adapter (A) is bound to one end of each target DNA molecule, and adapter (b) and adapter (A) for second-stage circularization are included at the other end ( B) binding step; where the adapter (B) binds to the DNA molecule via the adapter (b) side, and the adapter (A) in the adapter (B) is the DNA molecule and the adapter (b) Located outside the bond with, here, Adapter (A) contains a cleavage site that produces a cleavage end that binds non-specifically to the cleavage end of any adapter (A): Adapter (b) contains a cleavage site that produces a cleavage end that specifically binds only to the cleavage end from the same adapter (b); 2) a first cleavage step
- the adapter (b) specifically binds only to the cleavage ends derived from the same adapter (b), and thus linearization due to circularization of a plurality of DNA molecules substantially occurs.
- the DNA that is circularized without any change becomes a single molecule DNA.
- linear DNA molecules that are not circularized must be removed as described above.
- step 6 a very small amount of single-molecule DNA that was cleaved in step 5) and not recombined in part and multiple DNA molecules were circularized in most of step 3).
- step 5 it is single-molecule or multi-molecule DNA that could not be recombined after cleaving each adapter (b).
- the adapter (A) is a double-stranded DNA containing restriction enzyme sites whose cut ends recognize sequences complementary to each other, such as a restriction that recognizes a palindromic sequence. Double-stranded DNA containing enzyme sites.
- the present invention provides a method for producing a single circular DNA comprising the circular DNA formed from single-molecule DNA and not including circular DNA formed from a plurality of molecular DNAs, comprising the following steps: I will provide a: 1) First-stage circularization adapter (A) is bound to one end of each target DNA molecule, and adapter (b) and adapter (A) for second-stage circularization are included at the other end ( B) binding step; where the adapter (B) binds to the DNA molecule via the adapter (b) side, and the adapter (A) in the adapter (B) is the DNA molecule and the adapter (b) Located outside the bond with, here,
- the adapter (A) is a double-stranded DNA containing a restriction enzyme site that recognizes a palindromic sequence,
- the adapter (B) includes the adapter (b) and the adapter (A), and the adapter (b) includes the same nicking enzyme recognition site or restriction enzyme site oriented in the opposite directions and in the opposite
- a double-stranded DNA comprising a double-stranded DNA sequence having a sequence unique to each adapter (b) between the aligned nicking enzyme recognition sites or restriction enzyme sites, and oriented in the opposite direction
- the cleavage site recombines only with the cleavage site derived from the same adapter (b); 2) a first cleavage step of cleaving the DNA molecule obtained in step 1) with a restriction enzyme that recognizes a restriction enzyme site contained in the adapter (A); 3) a first-stage circularization step in which both ends of the DNA molecule obtained in step 2) are ligated and circularized; 4) A step of removing DNA in which the single molecule and the plurality of molecules bound in the step 3) are linearized without being circularized; 5) a second cleavage step of cleaving the circular DNA molecule obtained in step 3) and step 4) with the nicking enzyme or restriction enzyme that recognize
- the second aspect of the present invention is further formed by circularizing the adapter (b) by binding via partially different cleavage sequence sites (miss annealing) in the step 6).
- the adapter (A) is a double-stranded DNA comprising a restriction enzyme site X that recognizes a palindromic sequence
- the adapter (B) is a double-stranded DNA composed of mutually complementary double-stranded DNAs having the following structure y 1 -Yy 2 -X: [In the structure, X is a double-stranded DNA that is a restriction enzyme site that recognizes a palindromic sequence; y 1 and y 2 are double stranded DNAs containing identical nicking enzyme recognition sites or restriction enzyme sites oriented in opposite directions to each other; Y is a double-stranded DNA sequence having a sequence unique to each DNA molecule to be circularized, n is an integer of 1 to 40, and N 1 to N n may be the same or different even if well, dAMP, dCMP, a deoxyribonucleotide selected from the group consisting of dGMP and dTMP, n
- y 1 and y 2 comprise a nicking enzyme recognition site, more preferably the nicking enzyme is Nb.BtsI with 6 base recognition or Nt.BsqQI with 7 base recognition.
- y 1 and y 2 may include restriction enzyme sites.
- the present invention provides an adapter for producing circular DNA comprising double-stranded DNAs complementary to each other having the following structure y 1 -Yy 2 -X:
- X is a double-stranded DNA that is a restriction enzyme site that recognizes a palindromic sequence
- y 1 and y 2 are double stranded DNAs containing identical nicking enzyme recognition sites or restriction enzyme sites oriented in opposite directions to each other
- Y is a double-stranded DNA sequence having a sequence unique to each DNA molecule to be circularized
- n is an integer of 1 to 40, and N 1 to N n may be the same or different even if well, dAMP, dCMP, a deoxyribonucleotide selected from the group consisting of dGMP and dTMP, n '1 ⁇ n' n , respectively to the n 1 ⁇ n n below:
- k is an integer from 1 to n].
- the adapter according to the third aspect of the present invention corresponds to the adapter (B) according to the second aspect of the present invention, and is composed of circular DNA formed from the unimolecular DNA of the present invention, and is formed from multiple molecular DNA. It is preferably used in a method for producing circular DNA that does not contain circular DNA.
- y 1 and y 2 comprise a nicking enzyme recognition site, more preferably the nicking enzyme is Nb.BtsI with 6 base recognition or Nt.BsqQI with 7 base recognition. is there.
- y 1 and y 2 may include restriction enzyme sites.
- the present invention further provides the adapter according to the third aspect described above in the fourth aspect (also simply referred to as adapter (B)), and the same restriction enzyme site as the restriction enzyme site in X contained in the adapter (B).
- a kit for producing a circular DNA comprising an adapter composed of a double-stranded DNA comprising (also simply referred to as adapter (A)) is provided.
- the present invention also provides, in the fifth aspect, a method for preparing a cDNA library using the method of the first or second aspect.
- the present invention further provides, in the sixth aspect, a method for identifying a gene by subjecting the circular DNA molecule obtained by using the method of the first or second aspect to the mate pair method, comprising the following steps: To: 1) Decoding a base sequence of 15 to 600 bases adjacent to both sides of the adapter (B) in the circular DNA molecule obtained by using the method of the first or second aspect; 2) A step of identifying a gene contained in a circular DNA molecule by comparing the decoded base sequence with sequences at both ends of a known gene.
- the base sequence to be decoded is 15 to 100 bases, more preferably 25 to 35 bases.
- the method of the present invention can be carried out even if 600 bases or more are decoded.
- the present invention further provides a method for detecting a fusion gene by subjecting the circular DNA molecule obtained by using the method of the first or second aspect to the mate pair method, which comprises the following steps: provide: 1) Decoding a base sequence of 15 to 600 bases adjacent to both sides of the adapter (B) in the circular DNA molecule obtained by using the method of the first or second aspect; 2) A step of comparing the decoded base sequence with the sequences at both ends of a known gene, where the genes contained in the circular DNA molecule are fusion genes when the genes at both ends correspond to known different genes Identified.
- the base sequence to be decoded is 15 to 100 bases, more preferably 25 to 35 bases.
- the method of the present invention can be carried out even if 600 bases or more are decoded.
- the known fusion gene when the sequences on both sides adjacent to both sides of the adapter (B) correspond to the ends on both sides of the known fusion gene, the known fusion gene is detected.
- a method for detecting a disease characterized by expression of the fusion gene using the fusion gene detected by the method of the seventh aspect as a marker is provided Is done.
- the sequences adjacent to both sides of the adapter (B) correspond to the end sequences of different genes and do not correspond to the ends of both sides of the known fusion gene, the gene contained in the circular DNA molecule is newly fused. Identified as a gene.
- the detected novel fusion gene is suitably used for drug discovery screening.
- the present invention in the circularization of DNA, by providing a circularization method of only single molecule DNA that substantially prevents circularization by a plurality of molecules, there is a problem of contamination in gene analysis such as mate pair analysis. This solves the problem and enables highly accurate analysis. Further, by applying the method of the present invention to detection / analysis of fusion genes, it becomes possible to analyze fusion genes with high accuracy and provide an effective diagnostic tool.
- FIG. 1 schematically shows an outline of gene analysis by the mate pair method.
- FIG. 2 schematically shows problems in gene analysis by the mate pair method.
- FIG. 3 shows a schematic diagram when the fusion gene is analyzed by the mate pair method.
- FIG. 4 schematically shows problems in analyzing a fusion gene by the mate pair method.
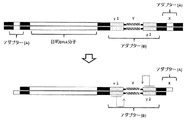
- FIG. 5 schematically shows the first cutting step of the two-stage cyclization method according to the present invention.
- FIG. 6 schematically shows the first cyclization, second cutting, and second cyclization steps of the two-stage cyclization method according to the present invention.
- FIG. 7 shows a schematic diagram when the method of the present invention is applied to cDNA synthesized from mRNA.
- FIG. 8 shows a schematic diagram when the method of the present invention is applied to a genomic library.
- FIG. 9 shows the experimental results of Example 4 that demonstrate that application of the method of the present invention reduces the amount of circularized DNA produced by multiple molecules.
- the present invention comprises a circular DNA formed from single-molecule DNA, comprising the following steps, and does not include circular DNA formed from multiple-molecule DNA.
- the first aspect of the present invention is a technique that substantially allows circularization from only one gene (DNA) and prevents circularization from a plurality of genes (DNA).
- the method of the first aspect of the present invention is characterized by two-stage cyclization.
- the method of the first aspect will be described in the order of steps.
- a first-step circularization adapter (A) is bound to one end of each target DNA molecule, and the adapter (b) and the adapter (A) are included at the other end.
- the adapter (B) is bound to the DNA molecule via the adapter (b) side, so that the adapter (A) in the adapter (B) is outside the bond between the DNA molecule and the adapter (b).
- the adapter (A) is bound to one end of each target DNA molecule, and the adapter (B) is bound to the other end, and the adapter (A) in the adapter (B) is located on the end side of the adapter (b).
- the adapter (A) is positioned at both ends of each target DNA molecule.
- the adapter (A) includes a cleavage site that generates a cleavage end that non-specifically binds to the cleavage end of any adapter (A), and the adapter (b) is a cleavage end derived from the same adapter (b). It contains a cleavage site that produces a cleavage end that specifically binds only. That is, the adapter (A) does not exhibit binding specificity, and can be non-selectively bound between the cleavage ends derived from any adapter (A). On the other hand, the adapter (b) has a different structure for each target DNA molecule, and can be recombined only with a cleavage end derived from the same adapter (b). It cannot be rejoined.
- Step 2) is a first cleavage step of cleaving the DNA molecule obtained in step 1) at the cleavage site of the adapter (A). Since the adapter (A) is bound to both ends of the DNA molecule obtained in step 1), the first cleavage step generates a cleavage end derived from the adapter (A) at both ends of each DNA molecule.
- Step 3) is a first-stage circularization step in which both ends of the DNA molecule obtained in Step 2) are combined to be circularized.
- the first-stage circularization is circularization by the adapter (A), and the cleavage ends derived from the adapter (A) generated at both ends of each DNA molecule are bonded to each other to cause circularization.
- the adapter (B) is incorporated into the cyclic molecule together with the adapter (A) during the first-stage cyclization.
- the function of the adapter (A) has no specificity and does not have selectivity due to rebinding of both ends of the target DNA molecule.
- Step 4) is a step of removing DNA in which a single molecule and a plurality of molecules that are linearized without being circularized in Step 3) are bound.
- Step 5) is a second cleavage step of cleaving the circular DNA molecule obtained in Step 3) and Step 4) at the cleavage site of the adapter (b). That is, cleavage is performed at the adapter (b) incorporated into each DNA molecule that has been circularized in step 3) to produce a linear molecule.
- cleavage ends derived from the adapter (b) are generated at both ends of each DNA molecule.
- the cleavage end generated by cleavage is substantially incapable of binding with a cleavage end derived from another cyclic molecule.
- Step 6) is a second-stage circularization step in which both ends of the DNA molecule obtained in Step 5) are combined and circularized. That is, there are cleavage ends derived from the adapter (b) at both ends of each DNA molecule obtained in step 5).
- the adapter (b) has a different configuration for each target DNA molecule. And can be recombined only with a cleavage end derived from the same adapter (b) and cannot be recombined with a cleavage end derived from a different adapter (b).
- Second-stage cyclization is performed by joining the cut ends. Due to the specificity of this adapter (b), only a single molecule of each target DNA molecule is circularized.
- the adapter (b) has binding specificity, and when the portion of each adapter (b) is cleaved, at the time of rebinding cyclization, only the portion that was bound in the original cyclized product is rebound. It cannot be recombined with other parts. Therefore, in the case of recombination cyclization in the case of cyclization of a plurality of molecules, there is nothing other than the recombination cyclization in the original plurality of molecules, and this probability is extremely low and substantially as described later. 0.
- the probability of cyclization with a single molecule is generally high from its concentration, resulting in substantially only unimolecular cyclization.
- the linear molecules that are not circularized can be separated and eliminated by the action of exonuclease.
- the structures of the adapters (A) and (b) are not particularly limited as long as they fulfill the above functions.
- examples of adapters (A) include those containing restriction enzyme sites that recognize palindromic structures
- examples of adapters (b) include at least 4 bases, preferably Is a double-stranded DNA composed of nucleic acids complementary to each other having 5 or more bases, and those that produce complementary single-stranded ends upon cleavage are exemplified.
- the adapters (A) and (b) are not limited to these.
- a substance that is cleaved and becomes a key and keyhole relation may be used.
- the method of the present invention can be carried out even in the case of a non-palindromic sequence cleavage site, in which case the upstream adapter (A1) and the downstream adapter (A2) are distinguished, and (A1) (A2) are It is set as the structure which mutually couple
- the circularized product of a plurality of genes (multiple molecules), which becomes a contaminant, is eliminated, and the circularized product of only a single molecule (one gene) is excluded. Can be created.
- the second aspect of the present invention is a circular DNA comprising a circular DNA formed from a single molecule DNA and not including a circular DNA formed from a plurality of molecular DNAs, comprising the following steps: Provides a way to create: 1) First-stage circularization adapter (A) is bound to one end of each target DNA molecule, and adapter (b) and adapter (A) for second-stage circularization are included at the other end ( B) binding step; where the adapter (B) binds to the DNA molecule via the adapter (b) side, and the adapter (A) in the adapter (B) is the DNA molecule and the adapter (b) Located outside the bond with, here,
- the adapter (A) is a double-stranded DNA containing a restriction enzyme site that recognizes a palindromic sequence,
- the adapter (B) includes the adapter (b) and the adapter (A), and the adapter (b) includes the same nicking enzyme recognition site or restriction enzyme site
- a double-stranded DNA comprising a double-stranded DNA sequence having a sequence unique to each adapter (b) between the aligned nicking enzyme recognition sites or restriction enzyme sites, and oriented in the opposite direction
- the cleavage site recombines only with the cleavage site derived from the same adapter (b); 2) a first cleavage step of cleaving the DNA molecule obtained in step 1) with a restriction enzyme that recognizes a restriction enzyme site contained in the adapter (A); 3) a first-stage circularization step in which both ends of the DNA molecule obtained in step 2) are ligated and circularized; 4) A step of removing DNA in which the single molecule and the plurality of molecules bound in the step 3) are linearized without being circularized; 5) a second cleavage step of cleaving the circular DNA molecule obtained in step 3) and step 4) with the nicking enzyme or restriction enzyme that recognize
- step 6 circularized DNA formed by binding and circularizing the adapter (b) through partially different cleavage sites is obtained using an endonuclease. It is preferable to further include a step of decomposing.
- step 1) as schematically shown in FIG. 5, a first-stage circularization adapter (A) is bound to one end of each target DNA molecule, and the adapter (b) and the adapter are connected to the other end.
- the second-stage circularization adapter (B) containing (A) is bound.
- the adapter (B) binds to the DNA molecule via the adapter (b) side, and the adapter (A) in the adapter (B) is located outside the bond between the DNA molecule and the adapter (b). To do.
- the adapter (A) is a double-stranded DNA containing a restriction enzyme site that recognizes a palindromic sequence
- the adapter (B) includes the adapter (b) and the adapter (A)
- the adapter (b) Includes the same nicking enzyme recognition sites or restriction enzyme sites oriented in opposite directions, and between each of the same nicking enzyme recognition sites or restriction enzyme sites oriented in the opposite directions, each adapter (b)
- the double-stranded DNA comprising a double-stranded DNA sequence having a unique sequence in The cleavage site recombines only with the cleavage site derived from the same adapter (b).
- Adapter (A) This adapter is a sequence for cleaving with a restriction enzyme in the first step and then performing gene ligation in anticipation of circular DNA. Therefore, any restriction enzyme site that recognizes a palindromic sequence may be included. Preferably, it contains a restriction enzyme site that recognizes a rare gene sequence that does not cut the target DNA as much as possible. Examples of restriction enzyme sites contained in the adapter (A) include NotI and EcoRI.
- This nick-created enzyme recognition site or restriction enzyme site is for cleaving in the second stage to form a gene-cutting overhang specific for each molecule.
- the nicking enzyme cuts only one of the DNA strands, and the restriction enzyme cuts both double strands of the DNA strand at the same time, but can be introduced at any recognition site.
- the adapter (b) When the adapter (b) is cleaved by the same nicking enzyme recognition site or restriction enzyme site oriented in opposite directions, a cleavage site is generated, and this cleavage site is regenerated only with a cleavage site derived from the same adapter (b). It is to be combined.
- the length of the cleavage site is not limited in the case of a nicking enzyme, but in the case of a restriction enzyme, a restriction occurs as shown in the following example. Therefore, the nicking enzyme is preferable in order to freely adjust the length of the cleavage site.
- nicked enzyme recognition site In the case of the nick creation enzyme recognition site In case of restriction enzyme 1 In case of restriction enzyme 2 In case of restriction enzyme 3 In the case of this format, it is necessary to prepare two equivalent N sequences by using a primer extension method from a loop structure.
- the cleavage site is a portion corresponding to each molecule-specific gene cut overhang.
- the number of bases is limited when the restriction enzyme is used, but the length can be freely set with the nick preparation enzyme. To increase the specificity, the longer the number of bases at the cleavage site, the better. However, when the length is too long, the possibility of gene binding due to mismatch increases.
- the sequence of the cleavage site is a random combination and is designed to be different for each DNA molecule.
- an adapter (B) having the following sequence can be prepared by nucleic acid synthesis and used in the method of the present invention.
- Step 2) is a first cleavage step of cleaving the DNA molecule obtained in step 1) with a restriction enzyme that recognizes a restriction enzyme site contained in the adapter (A) (FIG. 5).
- Step 3) is a first-stage circularization step in which both ends of the DNA molecule obtained in step 2) are ligated and circularized (FIG. 6: first circularization).
- Step 4) is a step of removing DNA in which a single molecule and a plurality of molecules that are linearized without being circularized in Step 3) are bound.
- Step 5) is a second cleavage that cleaves the circular DNA molecule obtained in step 3) and step 4) with a nicking enzyme or restriction enzyme that recognizes the nicking enzyme recognition site or restriction enzyme site in adapter (b). It is a process.
- Step 6) is a second stage circularization step in which both ends of the DNA molecule obtained in Step 5) are ligated and circularized (FIG. 6: cleavage recirculation).
- FIG. 6 cleavage recirculation
- the adapter (A) is a double-stranded DNA containing a restriction enzyme site X that recognizes a palindromic sequence
- the adapter (B) has the following structure y 1 -Yy 2.
- a double-stranded DNA consisting of double-stranded DNAs complementary to each other having -X [In the structure, X is a double-stranded DNA that is a restriction enzyme site that recognizes a palindromic sequence; y 1 and y 2 are double stranded DNAs containing identical nicking enzyme recognition sites or restriction enzyme sites oriented in opposite directions to each other; Y is a double-stranded DNA sequence having a sequence unique to each DNA molecule to be circularized, n is an integer of 1 to 40, and N 1 to N n may be the same or different even if well, dAMP, dCMP, a deoxyribonucleotide selected from the group consisting of dGMP and dTMP, n '1 ⁇ n' n , respectively to the n 1 ⁇ n n below: Where k is an integer from 1 to n].
- n is 1 to 40, preferably n is 4 to 15, and more preferably n
- Y Y site is a pair of base sequences randomly combined. For example, if Y is composed of 8 base sequences, the combination is 4 to the 8th power. As a result, it is possible to specify (select) the formation of the circular pair, and to recombine only with the original pair. As a result, only a single DNA can be recircularized, and the Y part of other molecules has a different structure and cannot be recombined.
- N the number of nucleic acids in the Y portion
- the method for producing the Y portion of the adapter (B) can be basically produced by synthesizing the main chain by random sequential condensation that defines N (number) and does not define nucleic acid, and polymerase extension reaction using a primer of a common sequence. .
- y 1 and y 2 comprise a nicking enzyme recognition site, more preferably the nicking enzyme is Nb.BtsI.
- y 1 and y 2 may include a restriction enzyme site.
- the third aspect of the present invention is a suitable adapter (B) used in the method of the second aspect of the present invention, that is, the following structure y 1 -Yy 2 -X
- an adapter for producing circular DNA comprising double-stranded DNAs complementary to each other having: [In the structure, X is a double-stranded DNA that is a restriction enzyme site that recognizes a palindromic sequence; y 1 and y 2 are double stranded DNAs containing identical nicking enzyme recognition sites or restriction enzyme sites oriented in opposite directions to each other; Y is a double-stranded DNA sequence having a sequence unique to each DNA molecule to be circularized, n is an integer of 1 to 40, and N 1 to N n may be the same or different even if well, dAMP, dCMP, a deoxyribonucleotide selected from the group consisting of dGMP and dTMP, n '1 ⁇ n'
- y 1 and y 2 comprise a nicking enzyme recognition site, more preferably the nicking enzyme is Nb.BtsI or Nt.BsqQI. It is. y 1 and y 2 may include a restriction enzyme site.
- the fourth aspect of the present invention provides a kit for producing circular DNA comprising a suitable adapter (A) and adapter (B) used in the method of the second aspect of the present invention.
- a suitable adapter (A) and adapter (B) used in the method of the second aspect of the present invention comprising a suitable adapter (A) and adapter (B) used in the method of the second aspect of the present invention.
- the adapter (A) comprising a double-stranded DNA containing the same restriction enzyme site as the restriction enzyme site in X described in the third aspect
- the adapter (B) described in the third aspect A kit for producing circular DNA is provided.
- the kit for producing circular DNA of the present invention comprises an adapter (A) and an adapter (B), which are bound to both ends of the target DNA molecule, and the two-step circularization according to the first or second aspect of the present invention.
- the fifth aspect of the present invention provides a method for preparing a cDNA library using the two-step circularization method according to the first or second aspect of the present invention.
- a cDNA library containing circularized DNA consisting only of unimolecular DNA can be prepared.
- the sixth aspect of the present invention is to subject a circular DNA molecule obtained using the method according to the first or second aspect of the present invention to the mate pair method, comprising the following steps: Provides a method for identifying genes by: 1) Decoding a base sequence of 15 to 600 bases adjacent to both sides of the adapter (B) in the circular DNA molecule obtained by using the method according to the first or second aspect of the present invention; and , 2) A step of identifying a gene contained in a circular DNA molecule by comparing the decoded base sequence with sequences at both ends of a known gene.
- Step 1) of the method of the sixth aspect of the present invention comprises 15 each adjacent to both sides of the adapter (B) in the circular DNA molecule obtained using the method according to the first or second aspect of the present invention. It is a step of decoding a base sequence of not less than 600 bases.
- the base sequence to be decoded is 15 to 100 bases, more preferably 25 to 35 bases.
- the method of the present invention can be carried out even if 600 bases or more are decoded.
- the base sequence can be decoded using methods and sequencers well known to those skilled in the art.
- Step 2) of the method of the sixth aspect of the present invention is a step of identifying a gene contained in a circular DNA molecule by comparing the decoded base sequence with sequences at both ends of a known gene. If it is confirmed that the decoded base sequence corresponding to the both ends of the target DNA molecule is identical to the both ends of the known gene, the target DNA molecule is identified as the known gene. .
- the seventh aspect of the present invention is to subject the circular DNA molecule obtained by using the method according to the first or second aspect of the present invention to the mate pair method, comprising the following steps: Provides a method for detecting a fusion gene by: 1) Decoding a base sequence of 15 to 600 bases adjacent to both sides of the adapter (B) in the circular DNA molecule obtained by using the method according to the first or second aspect of the present invention; and , 2) A step of comparing the decoded base sequence with the sequences at both ends of a known gene, where the genes contained in the circular DNA molecule are fusion genes when the genes at both ends correspond to known different genes Identified.
- Step 1) of the method of the seventh aspect of the present invention comprises 15 each adjacent to both sides of the adapter (B) in the circular DNA molecule obtained using the method according to the first or second aspect of the present invention. It is a step of decoding a base sequence of not less than 600 bases.
- the base sequence to be decoded is 15 to 100 bases, more preferably 25 to 35 bases.
- the method of the present invention can be carried out even if 600 bases or more are decoded.
- the base sequence can be decoded using methods and sequencers well known to those skilled in the art.
- Step 2) of the method of the seventh aspect of the present invention is a step of comparing the decoded base sequence with the sequences at both ends of a known gene.
- the gene contained in the circular DNA molecule is identified as a fusion gene. That is, the portion corresponding to one end of the decoded base sequence corresponding to the both ends of the target DNA molecule is the same as one end of the known gene, and the portion corresponding to the other end is another known If it is the same as one end of the gene, the target DNA molecule is identified as a fusion gene composed of two known genes.
- the target DNA is detected as a known fusion gene.
- the relationship between the expression of a known fusion gene and a disease is known, it is possible to detect a disease characterized by the expression of the fusion gene by using the fusion gene detected by such a method as a marker. .
- the gene contained in the circular DNA molecule is a new fusion gene. Is identified.
- the novel fusion gene detected by such a method can be used in drug discovery screening.
- Example 1 Application of the method of the present invention to discovery of a fusion gene using cDNA synthesized from mRNA
- a complementary strand DNA is synthesized with reverse transcriptase using an oligonucleotide (1) having a polyT sequence complementary to the polyA site at the 3 ′ end of mRNA.
- a specific oligonucleotide sequence (2) is incorporated at the 5 ′ end of the mRNA as shown in FIG.
- DNA synthesis is performed from an oligonucleotide complementary to this specific sequence, or a cDNA library is prepared by PCR.
- the adapter (B) is introduced into the oligonucleotide (1) sequence having a polyT sequence complementary to the polyA site at the 3 ′ end of mRNA.
- a cDNA library is synthesized by adding an adapter (A) to the nucleotide (2) sequence, it automatically has the basic structure of the present invention, and a new sequence needs to be bound to the right end and the left end. You can proceed to the next step.
- a cDNA library is further amplified by PCR, a specific N base sequence is amplified if it is amplified as it is.
- a 5 'phosphate group-added primer is used as the upstream primer
- the downstream primer is amplified using a primer containing N bases, and the phosphate group primer is incorporated after PCR.
- the other strand is digested with ⁇ exonuclease, and then the primer-extension is performed again from the upstream primer to complete a cDNA library that maintains diversity by N bases.
- this process is not necessary for the adapter binding method, and the following adapter binding method is performed in order to introduce the adapter of the present invention after modification such as fragmentation of the library.
- restriction enzyme sequences can be introduced at the 3 ′ end and 5 ′ end, respectively, as shown in FIG. .
- the normal palindromic sequence can be used as a restriction enzyme, or a non-palindromic sequence can be used to distinguish the ends. It is also possible to bind the adapter to the shape of the protruding base. This method is desirable for introducing the adapter of the present invention after modifying the library.
- Example 2 Application of the method of the present invention to mate pair analysis of genomes
- a genome is targeted, the situation is different from that of a cDNA library.
- genomic fragments cannot distinguish between left and right DNA fragments. Therefore, the adapter at the left end and the adapter at the right end are both bound as shown in FIG. 8, and both adapters are bound appropriately, that is, only (a) in FIG.
- the sequence can be selected by inserting a non-palindromic sequence as a sequence, or by installing a plurality of restriction enzyme sites at the same site using a restriction enzyme (such as BstXI) containing the N region.
- a restriction enzyme such as BstXI
- the adapters (A) and (B) are added to both ends of the DNA molecule by using the following procedure, respectively.
- the adapter (A) is first bound to a plurality of DNA groups having a certain concentration determined according to the number of target DNA molecules.
- the amount of adapter (A) added is less than the target DNA molecular weight present.
- it may be the same as the number of target DNA molecules, or a single base overhang may be enzymatically created in a DNA fragment and a complementary adapter may be bound.
- the concentration of may be excessive.
- the adapter (A) is bound to the end of the DNA molecule.
- the adapter (B) is bound.
- the second circularization after nick cutting cannot be formed and is eliminated.
- the first cyclization is impossible as a single molecule and is removed.
- circularization with a plurality of DNAs can be formed (for example, two-molecule coupling by (b) type molecule and (c) type molecule). Since this DNA fragment originally has different N sequences at both ends, the second circularization after nick cutting is impossible and eliminated.
- BstXI is charged outside the restriction enzyme site of (A). That is, in the following restriction enzyme site BstXI site, CCANNNNNNTGG is incorporated, for example, EcoRI site so as to be CCAGAATTCTGG. That is, in this method, the restriction enzyme site of the adapter (A) contained in the adapter (A) and the adapter (B) is, for example, an EcoRI site.
- the sequence outside the EcoRI site is, for example, a BstXI recognition sequence. That is, the adapter (A) sequence is CCAGAATTCTGG.
- the adapter (A) included in the adapter (B) includes the EcoRI site, but does not incorporate the recognition sequence of BstXI so that it is not cleaved by BstXI.
- the adapters (A) are at both ends and are associated with each other, the circularization can be opened and eliminated by cutting with BstXI.
- the adapter (B) does not have a BstXI recognition sequence, so it is not cleaved by BstXI.
- Example 3 The possibility of gene recombination between the same genes in a multi-molecule circular DNA can be ignored sufficiently stochastically. The basis for this will be described below.
- Example 4 Preparation of circularized DNA by single molecule DNA In order to verify that the efficiency of circularization by single molecule DNA is increased by the two-step DNA ligation method and the noise of circularization by multiple DNA molecules is reduced, the following is performed. Such an experiment was conducted.
- Plasmid ⁇ A and Plasmid B Two types of plasmids, Plasmid ⁇ A and Plasmid B, were prepared. Each plasmid was cleaved with a restriction enzyme and then ligated as it was (i), and the one obtained by ligating the adapter of the present invention and ligated in two steps (ii) were compared.
- (i) and (ii) in the mixed solution contained (1) circular DNA by a single molecule of Plasmid A, (2) circular DNA by a single molecule of PlasmidPB, and (3) multiple molecules of PlasmaPA. It is predicted that (4) circular DNA by plural molecules of Plasmid B and (5) circular DNA by plural molecules including PlasmidPA and Plasmid B.
- the amount of (5) in the mixed solution produced from (i) and (ii) was evaluated as a representative, and quantitative PCR was performed with PCR primers specific to Plasmid A and Plasmid B.
- the amount of circularized DNA due to multiple molecules that are subject to noise as in (5) can be reduced to about 1/100 compared to the method in (i) by introducing the method of the present invention (ii). It was confirmed that.
- the present invention in the circularization of DNA, by providing a circularization method of only single molecule DNA that substantially prevents circularization by a plurality of molecules, there is a problem of contamination in gene analysis such as mate pair analysis. This solves the problem and enables highly accurate analysis. Further, by applying the method of the present invention to detection / analysis of fusion genes, it becomes possible to analyze fusion genes with high accuracy and provide an effective diagnostic tool.
Abstract
Description
1)各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程;ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する、
ここで、
アダプター(A)は、いずれのアダプター(A)の切断末端とも非特異的に結合する切断末端を生じさせる切断部位を含む:
アダプター(b)は、同一のアダプター(b)由来の切断末端にのみ特異的に結合する切断末端を生じさせる切断部位を含む;
2)アダプター(A)の切断部位において、工程1)で得られたDNA分子を切断する第一切断工程、
3)工程2)で得られたDNA分子の両末端を結合させて環状化させる第一段階環状化工程;
4)工程3)において、環状化せず直線状となった単分子および複数分子が結合したDNAを除去する工程;
5)アダプター(b)の切断部位において、工程3)および工程4)で得られた環状DNA分子を切断する第二切断工程、および、
6)工程5)で得られたDNA分子の両末端を結合させて環状化させる第二段階環状化工程。
第二段階環状化工程においては、アダプター(b)が同一のアダプター(b)由来の切断末端にのみ特異的に結合することから、複数のDNA分子の環状化による直線状化は実質的に生じなく環状化されるDNAは単分子DNAとなる。この時、環状化されない直線状のDNA分子は、上記同様に除去する必要がある。
工程6)において環状化されない直線状のDNAは、極めて微量の工程5)において切断された(b)部分の再結合がなされなかった単分子DNAと大半の工程3)で複数DNA分子が環状化し、工程5)において、各アダプター(b)の切断後、再結合できなかった、単分子または複数分子のDNAである。
1)各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程;ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する、
ここで、
アダプター(A)は、パリンドローム配列を認識する制限酵素サイトを含む二本鎖DNAであり、
アダプター(B)は、アダプター(b)と前記アダプター(A)を含み、アダプター(b)は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含み、その逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトの間に、個々のアダプター(b)に固有の配列を有する二本鎖DNA配列を含む二本鎖DNAであり、該逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトによりアダプター(b)が開裂した場合、該開裂部位は、同一のアダプター(b)由来の開裂部位とのみ再結合するものである;
2)アダプター(A)に含まれる制限酵素サイトを認識する制限酵素により、工程1)で得られたDNA分子を切断する第一切断工程、
3)工程2)で得られたDNA分子の両末端をライゲーションさせて環状化させる第一段階環状化工程;
4)工程3)において、環状化せず直線状となった単分子および複数分子が結合したDNAを除去する工程;
5)アダプター(b)におけるニック作成酵素認識サイトまたは制限酵素サイトを認識するニック作成酵素または制限酵素により、工程3)および工程4)で得られた環状DNA分子を切断する第二切断工程、および、
6)工程5)で得られたDNA分子の両末端をライゲーションさせて環状化させる第二段階環状化工程。
アダプター(B)は下記構造y1-Y-y2-Xを有する互いに相補的な二本鎖DNAからなる二本鎖DNAである:
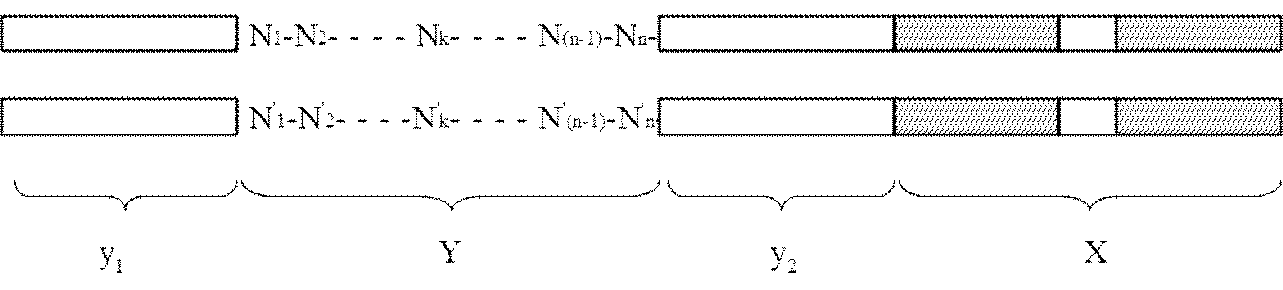
Xは、パリンドローム配列を認識する制限酵素サイトである二本鎖DNAであり;
y1およびy2は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含む二本鎖DNAであり;
Yは、環状化させる個々のDNA分子に固有の配列を有する二本鎖DNA配列であって、nは1以上40以下の整数であり、N1~Nnは、それぞれ同一であっても異なっていてもよく、dAMP、dCMP、dGMPおよびdTMPからなる群から選択されるデオキシリボヌクレオチドであり、N’1~N’nは、前記N1~Nnに対してそれぞれ下記:

上記構造において、nは1~40であり、好ましくは、nは4~15、さらに好ましくはnは5~10である。ただし、nが40を超えても本発明の方法を実施することができる。
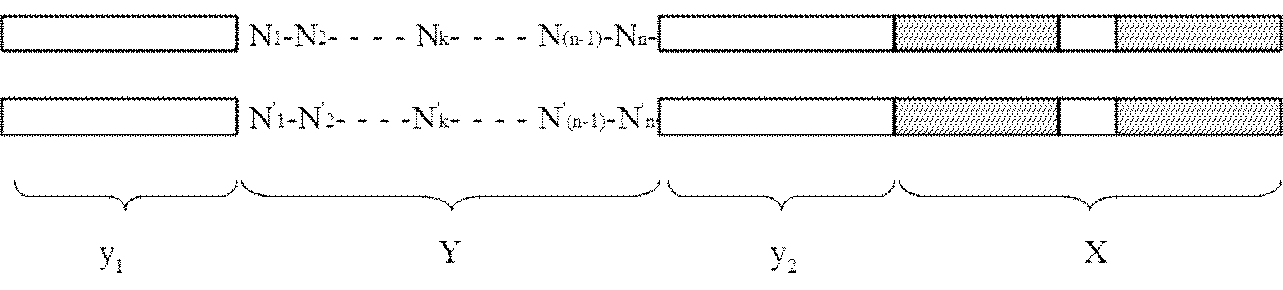
Xは、パリンドローム配列を認識する制限酵素サイトである二本鎖DNAであり;
y1およびy2は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含む二本鎖DNAであり;
Yは、環状化させる個々のDNA分子に固有の配列を有する二本鎖DNA配列であって、nは1以上40以下の整数であり、N1~Nnは、それぞれ同一であっても異なっていてもよく、dAMP、dCMP、dGMPおよびdTMPからなる群から選択されるデオキシリボヌクレオチドであり、N’1~N’nは、前記N1~Nnに対してそれぞれ下記:

上記構造において、nは1~40であり、好ましくは、nは4~15、さらに好ましくはnは5~10である。ただし、nが40を超えても本発明の方法を実施することができる。
1)上記第一または第二の態様の方法を用いて得られた環状DNA分子における、アダプター(B)の両側に隣接するそれぞれ15塩基以上600塩基以下の塩基配列を解読する工程;および、
2)解読した塩基配列を既知の遺伝子の両末端の配列と比較することにより、環状DNA分子に含まれる遺伝子を同定する工程。
好ましくは、解読する塩基配列は、15~100塩基、さらに好ましくは25~35塩基である。ただし、600塩基以上を解読しても本発明の方法を実施することができる。
1)上記第一または第二の態様の方法を用いて得られた環状DNA分子における、アダプター(B)の両側に隣接するそれぞれ15塩基以上600塩基以下の塩基配列を解読する工程;および、
2)解読した塩基配列を既知の遺伝子の両末端の配列と比較する工程、ここで、両末端の遺伝子が既知の相異なる遺伝子に対応する場合、環状DNA分子に含まれる遺伝子は融合遺伝子であると同定される。
好ましくは、解読する塩基配列は、15~100塩基、さらに好ましくは25~35塩基である。ただし、600塩基以上を解読しても本発明の方法を実施することができる。
本発明は、第一の態様において、以下の工程を含む、単分子DNAから形成される環状DNAからなり、複数分子DNAから形成される環状DNAを含まない、環状DNAの作成方法を提供する:
1)各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程;ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する、
ここで、
アダプター(A)は、いずれのアダプター(A)の切断末端とも非特異的に結合する切断末端を生じさせる切断部位を含む:
アダプター(b)は、同一のアダプター(b)由来の切断末端にのみ特異的に結合する切断末端を生じさせる切断部位を含む;
2)アダプター(A)の切断部位において、工程1)で得られたDNA分子を切断する第一切断工程、
3)工程2)で得られたDNA分子の両末端を結合させて環状化させる第一段階環状化工程;
4)工程3)において、環状化せず直線状となった単分子および複数分子が結合したDNAを除去する工程;
5)アダプター(b)の切断部位において、工程3)および工程4)で得られた環状DNA分子を切断する第二切断工程、および、
6)工程5)で得られたDNA分子の両末端を結合させて環状化させる第二段階環状化工程。
具体的には、本発明の第一の態様の方法は、2段階の環状化によることを特徴とする。
以下、第一の態様の方法を工程の順に説明する。
工程1)で得られたDNA分子はその両端にアダプター(A)が結合しているため、この第一切断工程により、各DNA分子の両端にアダプター(A)由来の切断末端が生じる。
本発明の第二の態様は、以下の工程を含む、単分子DNAから形成される環状DNAからなり、複数分子DNAから形成される環状DNAを含まない、環状DNAの作成方法を提供する:
1)各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程;ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する、
ここで、
アダプター(A)は、パリンドローム配列を認識する制限酵素サイトを含む二本鎖DNAであり、
アダプター(B)は、アダプター(b)と前記アダプター(A)を含み、アダプター(b)は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含み、その逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトの間に、個々のアダプター(b)に固有の配列を有する二本鎖DNA配列を含む二本鎖DNAであり、該逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトによりアダプター(b)が開裂した場合、該開裂部位は、同一のアダプター(b)由来の開裂部位とのみ再結合するものである;
2)アダプター(A)に含まれる制限酵素サイトを認識する制限酵素により、工程1)で得られたDNA分子を切断する第一切断工程、
3)工程2)で得られたDNA分子の両末端をライゲーションさせて環状化させる第一段階環状化工程;
4)工程3)において、環状化せず直線状となった単分子および複数分子が結合したDNAを除去する工程;
5)アダプター(b)におけるニック作成酵素認識サイトまたは制限酵素サイトを認識するニック作成酵素または制限酵素により、工程3)および工程4)で得られた環状DNA分子を切断する第二切断工程、および、
6)工程5)で得られたDNA分子の両末端をライゲーションさせて環状化させる第二段階環状化工程。
工程1)は、図5に模式的に示すように、各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程である。ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する。
アダプター(A):このアダプターは、第一段階目に制限酵素で切断し、その後に環状DNAを期待して遺伝子結合(ligation)を行うための配列である。従って、パリンドローム配列を認識する制限酵素サイトであれば如何なる制限酵素サイトを含むものでもよい。好ましくは、目的DNAをなるべく切断しないような稀な遺伝子配列を認識する制限酵素のサイトを含むものである。アダプター(A)に含まれる制限酵素サイトとしては、例えば、NotIやEcoRIが挙げられる。
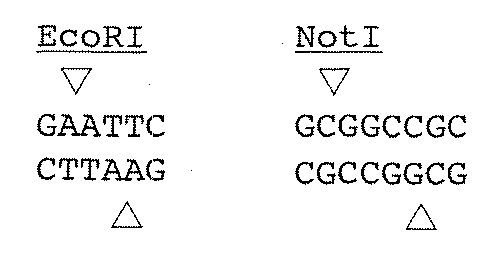
ニック作成酵素認識サイトの場合
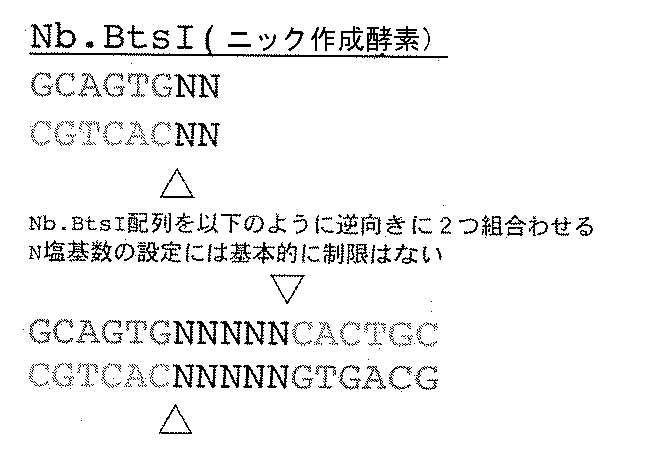



開裂部位は、各分子特異的な遺伝子切断断端(overhang)に相当する部分である。上記のように制限酵素で作成する場合は塩基数に制限があるが、ニック作成酵素では自由に長さを設定することができる。特異性を高くするためには開裂部位の塩基数は長い程よいが、長すぎる時にはミスマッチで遺伝子結合する可能性が高くなる。ゲノムなど複雑性が増す場合には開裂部位の塩基数をより長く設定し、同時にミスマッチ結合(ミス・アニーリング)の可能性に対してエンドヌクレアーゼによる分解過程を追加することが望ましい。なお、開裂部位の配列はランダムな組合せとし、DNA分子1つ1つについて異なるものとなるよう設計する。
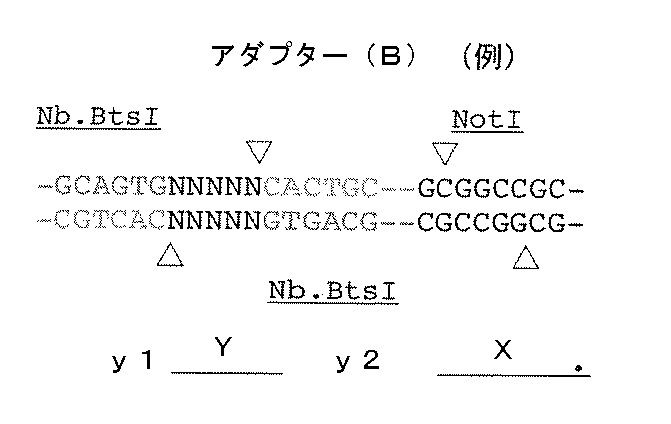
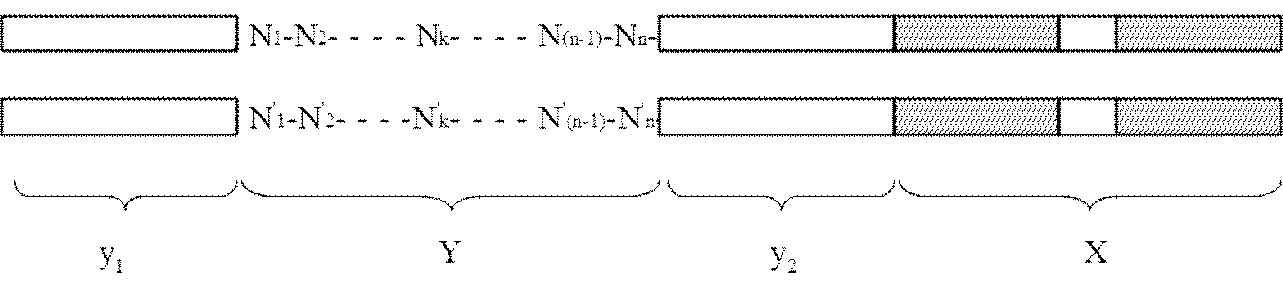
Xは、パリンドローム配列を認識する制限酵素サイトである二本鎖DNAであり;
y1およびy2は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含む二本鎖DNAであり;
Yは、環状化させる個々のDNA分子に固有の配列を有する二本鎖DNA配列であって、nは1以上40以下の整数であり、N1~Nnは、それぞれ同一であっても異なっていてもよく、dAMP、dCMP、dGMPおよびdTMPからなる群から選択されるデオキシリボヌクレオチドであり、N’1~N’nは、前記N1~Nnに対してそれぞれ下記:
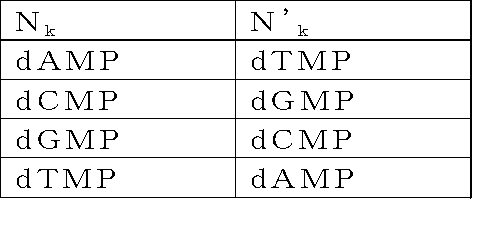
上記構造において、nは1~40であり、好ましくは、nは4~15、さらに好ましくはnは5~10である。ただし、nが40を超えても本発明の方法を実施することができる。
本発明の第三の態様は、上記本発明の第二の態様の方法において用いられる好適なアダプター(B)、即ち、下記構造y1-Y-y2-Xを有する互いに相補的な二本鎖DNAからなる環状DNA作成用アダプターを提供する:

Xは、パリンドローム配列を認識する制限酵素サイトである二本鎖DNAであり;
y1およびy2は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含む二本鎖DNAであり;
Yは、環状化させる個々のDNA分子に固有の配列を有する二本鎖DNA配列であって、nは1以上40以下の整数であり、N1~Nnは、それぞれ同一であっても異なっていてもよく、dAMP、dCMP、dGMPおよびdTMPからなる群から選択されるデオキシリボヌクレオチドであり、N’1~N’nは、前記N1~Nnに対してそれぞれ下記:
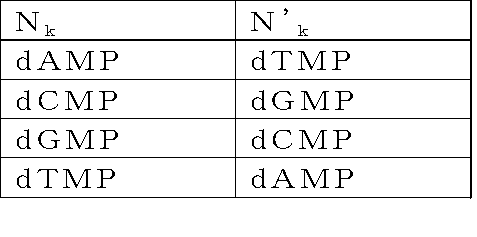
上記構造において、nは1~40であり、好ましくは、nは4~15、さらに好ましくはnは5~10である。ただし、nが40を超えても本発明を実施することができる。
本発明の第四の態様は、上記本発明の第二の態様の方法において用いられる好適なアダプター(A)およびアダプター(B)とを含む環状DNA作成用キット提供する。即ち、上記第三の態様に記載のXにおける制限酵素サイトと同一の制限酵素サイトを含む二本鎖DNAからなるアダプター(A)と、上記第三の態様に記載のアダプター(B)とを含む、環状DNA作成用キットを提供する。
本発明の第五の態様は、本発明の第一または第二の態様による二段階環状化方法を用いる、cDNAライブラリーの作成方法を提供する。本発明の第一または第二の態様による方法を直鎖状cDNAからなるライブラリーに適用することにより、単分子DNAのみからなる環状化DNAを含むcDNAライブラリーを作成することができる。
本発明の第六の態様は、以下の工程を含む、本発明の第一または第二の態様による方法を用いて得られた環状DNA分子をメイトペア法に供することにより遺伝子を同定する方法を提供する:
1)本発明の第一または第二の態様による方法を用いて得られた環状DNA分子における、アダプター(B)の両側に隣接するそれぞれ15塩基以上600塩基以下の塩基配列を解読する工程;および、
2)解読した塩基配列を既知の遺伝子の両末端の配列と比較することにより、環状DNA分子に含まれる遺伝子を同定する工程。
本発明の第七の態様は、以下の工程を含む、本発明の第一または第二の態様による方法を用いて得られた環状DNA分子をメイトペア法に供することにより融合遺伝子を検出する方法を提供する:
1)本発明の第一または第二の態様による方法を用いて得られた環状DNA分子における、アダプター(B)の両側に隣接するそれぞれ15塩基以上600塩基以下の塩基配列を解読する工程;および、
2)解読した塩基配列を既知の遺伝子の両末端の配列と比較する工程、ここで、両末端の遺伝子が既知の相異なる遺伝子に対応する場合、環状DNA分子に含まれる遺伝子は融合遺伝子であると同定される。
mRNAからcDNAライブラリーを合成する場合、現在使用されている最も一般的な方法(Clontech SMART cDNA method) では、図7のようにmRNAの3’末端のpolyAサイトに相補的なpolyT配列を有するオリゴヌクレオチド (1)を用いて、まず相補鎖DNAを逆転写酵素で合成する。合成終了時にmRNAの5’末端において図7のように特定オリゴヌクレオチド配列 (2)が取り込まれる。次にこの特定配列へ相補的なオリゴヌクレオチドからDNA合成を行う、あるいはPCRでcDNAライブラリーが作成される。
この場合、mRNAの3’末端のpolyAサイトに相補的なpolyT配列を有するオリゴヌクレオチド(1)配列にアダプター(B)を導入しておき、オリゴヌクレオチド(2)配列にアダプター(A)を付加しておくことでcDNAライブラリーが合成されたときには、自動的に本発明の基本形の構造になっており、右端左端に新たな配列を結合させる必要もなく、そのまま次のステップへ進むことができる。この方法で、さらにcDNAライブラリーをPCR増幅する際に、そのまま増幅したのではある特定のN塩基配列が増幅されてしまう。そのためN塩基の多様性を維持するため、例えば、上流primerとして5’リン酸基付加primerを使用し、下流primerにはN塩基を含むprimer を用いて増幅し、PCR後にリン酸基primerを取り込んだ側のストランドをλexonucleaseで分解し、再度上流primerからprimer-extensionを行い、N塩基による多様性を維持したcDNAライブラリーを完成させる、等の手続きを行う。但し、アダプター結合法であればこの過程は必要なく、ライブラリーの断片化などの修飾を行った上で本発明のアダプターを導入するためには、次のアダプター結合法を行う。
cDNAライブラリーを作成した場合、図7の(3)のように3’末端、5’末端にそれぞれ制限酵素配列を導入しておくことができる。この場合の制限酵素としては通常のパリンドローム配列を使用することも、末端を区別するためにノン・パリンドローム配列を使用することも可能であり、またアダプター結合を確実にするため平滑末端からA突出塩基の形状へアダプターを結合することも可能である。ライブラリーの修飾を行った上に本発明のアダプターを導入するためにはこちらの方法が望ましい。
ゲノムを対象とする場合はcDNAライブラリーとは状況が異なる。ゲノム断片にはcDNAと異なりDNA断片の左右を区別することができない。従って図8のように左端のアダプター、右端のアダプターを双方結合させて、双方のアダプターが適切に結合したもの、即ち図8の( a ) のみをPCR法やビオチン法、あるいは両端のX制限酵素配列としてノン・パリンドローム配列を挿入することや、N領域を含む制限酵素(BstXI等)により同一部位に制限酵素サイトを複数設置する方法等、で選択してゆくことができる。
基本的に、以下のような手法をとることによりアダプター(A)と(B)をDNA分子の両末端にそれぞれ付加する。第一に、目的DNA分子数に応じて、ある程度の濃度を決定した複数のDNA群に、先ず、アダプター(A)を結合させる。この場合、アダプター(A)の添加量は存在する対象DNA分子量よりも少なめとする。ただし、量論的には、対象DNA分子数と同数であっても、あるいは、DNA断片に一塩基突出を酵素的に作成し、相補的なアダプターを結合させてもよく、この場合は特にアダプターの濃度は過剰であっても構わない。これにより、アダプター(A)をDNA分子の末端に結合させる。次いで、アダプター(B)を結合させる。
図8の(a)タイプの分子の場合、第1環状化、ニックによる切断、第2環状化が問題なく行われ、問題なく目的の環状化DNAが作成される。
一方、図8の(b)タイプの分子の場合、第1環状化は、単分子としては不可であるため除去される。しかし複数DNAとの環状化は形成しうる(例えば、(b)タイプの分子と(c)タイプの分子による2分子結合)。この複数DNA分子は必ず結合先として(a)または(c)タイプの分子と結合することになる。従って、両側は必ずN配列のある特異配列との結合となる。このため、ニック切断後の第2環状化は形成不可となり排除される。
また、図8の(C)タイプの分子の場合、第1環状化は単分子としては不可で除去される。しかし複数DNAとの環状化は形成しうる(例えば、(b)タイプの分子と(c)タイプの分子による2分子結合)。このDNA断片はもともと両端に異なるN配列があるため、ニック切断後の第2環状化は不可であり排除される。
図8の(a) タイプの分子の場合、第1環状化、ニックによる切断、第2環状化が問題なく行われ、問題なく目的の環状化DNAが作成される。
一方、図8の(b)タイプの分子の場合、第1環状化は可能であるが、ニック配列を有さないためニック切断が行われずに環状化DNAのまま残ってしまい排除できない。これを防ぐ方法例として、下記ののBstXI法がある。
また図8の(c)タイプの分子の場合、第1環状化およびニックによる切断は可能であるが、もともと両端に異なるN配列があるため、ニック切断後の第2環状化は不可であり排除される。
両端にアダプター(A)が付加された場合、第一段階でライゲーションされるが、第二段階の開裂が起きないため、このままでは環状のままとどまるという問題を解決する方法として、アダプター(A)の制限酵素サイトを含む外側に例えばBstXIを仕込むことが挙げられる。つまり、下記制限酵素サイトBstXI部位において、CCANNNNNNTGGを、例えば、 CCAGAATTCTGGとなるようにEcoRIサイトを組み込む。

こうるすことにより、もしアダプター(A)同士が両端につき、それ同士が会合した場合、BstXIで切断することで、環状化を開き、排除することができる。もちろんアダプター(A)および(B)とが結合した場合にはアダプター(B)にはBstXIの認識配列がないため、BstXIでは開裂されない。
かかる可能性は確率論的に十分無視できるが、その根拠を以下に説明する。
確立論的に分離した遺伝子同士が再び会合する可能性を推定するため、(1)DNA分子の体積と反応溶液量(体積)で推定する場合、(2)反応溶液中の分子数から推定する場合(これらの時、Y部分のN塩基数が十分に長く特異性が高いと仮定しておく)、さらに(3)N塩基数が十分に長くはなく決まっている場合、の3つに分けて可能性を概算する。
まずDNA1分子を球状として体積を推定し、反応系で一度分離された分子同士が互いに球体として溶液中で会合する可能性を概算する。
一塩基の長さ:0.34 nm (0.34 x 10e-9 m = 3.4 x 10e-8 mm)
3kbp (3000 bp) のプラスミドの長さ:1 x 10e-4 mm
球体として占めると推定した体積は:4/3 x 3.14 x (1 x 10e-4) x (1 x 10e-4) x (1 x 10e-4) mm3 = 4 x 10e-12 mm3
一分子の体積を4 x 10e-12 mm3と仮定すると100 ul中には球体として:100 mm3 / 4 x 10e-12 mm3 = 2.5 x 10e13となる。
従って、均一であるとすると、ある球体に相補的とされる同等な球体が会合する可能性は、(1 / (2.5 x 10e13)) = 4 x 10e-14と極めて小さい。
一方、分子数を計算すると、
3kbpのプラスミドの分子量は:625 x 3000 = 1.8 x 10e6
1モルのプラスミド質量は:1 mol/L = 1.8 x 10e6 g / 1L = 1.8 x 10e12 ug / 1L
3 ugのプラスミドのモル数は:1 (mol/L) x 3 ug / (1.8 x 10e12) ug = 3 /1.8 x 10e-12 mol/L = 1.6 x 10e-12 mol/L = 1.6 x 10e-9 mol/ml
アボガドロ数を 6 x 10e23 とすると、3ugプラスミドを1ml中に溶かした時の分子数は:1.6 x 10e-9 mol/ml x 6 x 10e23 = 1.6 x 6 x 10e14 = 1 x 10e15 個
従って、100 ul の反応系に 3ugのプラスミドが存在すると:100 ul = 10 e-1 mm3であるので、1 x 10e14 分子数あることになる。
この分子数の溶液中である分子が、その分子に相補的とされる同等な分子に会合する可能性は、1 / 1 x 10e14と極めて小さい。
N塩基数が「5」である場合、同じ遺伝子断端同士を有して2つの同遺伝子が再結合する可能性は:((1/4) x (1/4) x (1/4) x (1/4) x (1/4)) x ((1/4) x (1/4) x (1/4) x (1/4) x (1/4)) = 1 x 10e-6
従ってこれらの概算から、複数のDNAが環状形成した後に分離した場合に、その際と同じDNA同士で再結合する可能性は極めて低く、十分に無視できると結論できる。
2段階DNA結合(ligation)法により単分子DNAによる環状化の効率を上げ、複数DNA分子による環状化のノイズを減少させることを検証するため以下のような実験を行った。
Claims (19)
- 以下の工程を含む、単分子DNAから形成される環状DNAからなり、複数分子DNAから形成される環状DNAを含まない、環状DNAの作成方法:
1)各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程;ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する、
ここで、
アダプター(A)は、いずれのアダプター(A)の切断末端とも非特異的に結合する切断末端を生じさせる切断部位を含む:
アダプター(b)は、同一のアダプター(b)由来の切断末端にのみ特異的に結合する切断末端を生じさせる切断部位を含む;
2)アダプター(A)の切断部位において、工程1)で得られたDNA分子を切断する第一切断工程、
3)工程2)で得られたDNA分子の両末端を結合させて環状化させる第一段階環状化工程;
4)工程3)において、環状化せず直線状となった単分子および複数分子が結合したDNAを除去する工程;
5)アダプター(b)の切断部位において、工程3)および工程4)で得られた環状DNA分子を切断する第二切断工程、および、
6)工程5)で得られたDNA分子の両末端を結合させて環状化させる第二段階環状化工程。 - 以下の工程を含む、単分子DNAから形成される環状DNAからなり、複数分子DNAから形成される環状DNAを含まない、環状DNAの作成方法:
1)各目的DNA分子の一方の末端に第一段階環状化用アダプター(A)を結合させ、他方の末端に、アダプター(b)と前記アダプター(A)を含む第二段階環状化用アダプター(B)を結合させる工程;ここで、アダプター(B)は、DNA分子にアダプター(b)側を介して結合して、アダプター(B)中のアダプター(A)は、DNA分子とアダプター(b)との結合の外側に位置する、
ここで、
アダプター(A)は、パリンドローム配列を認識する制限酵素サイトを含む二本鎖DNAであり、
アダプター(B)は、アダプター(b)と前記アダプター(A)を含み、アダプター(b)は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含み、その逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトの間に、個々のアダプター(b)に固有の配列を有する二本鎖DNA配列を含む二本鎖DNAであり、該逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトによりアダプター(b)が開裂した場合、該開裂部位は、同一のアダプター(b)由来の開裂部位とのみ再結合するものである;
2)アダプター(A)に含まれる制限酵素サイトを認識する制限酵素により、工程1)で得られたDNA分子を切断する第一切断工程、
3)工程2)で得られたDNA分子の両末端をライゲーションさせて環状化させる第一段階環状化工程;
4)工程3)において、環状化せず直線状となった単分子および複数分子が結合したDNAを除去する工程;
5)アダプター(b)におけるニック作成酵素認識サイトまたは制限酵素サイトを認識するニック作成酵素または制限酵素により、工程3)および工程4)で得られた環状DNA分子を切断する第二切断工程、および、
6)工程5)で得られたDNA分子の両末端をライゲーションさせて環状化させる第二段階環状化工程。 - 請求項2に記載の工程6)において、アダプター(b)が部分的に異なる切断配列部位を介して結合して環状化することにより形成される環状化DNAを、エンドヌクレアーゼを用いて分解する工程をさらに含む請求項2に記載の方法。
- アダプター(A)がパリンドローム配列を認識する制限酵素サイトXを含む二本鎖DNAであり、
アダプター(B)が下記構造y1-Y-y2-Xを有する互いに相補的な二本鎖DNAからなる二本鎖DNAである請求項2記載の方法:

Xは、パリンドローム配列を認識する制限酵素サイトである二本鎖DNAであり;
y1およびy2は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含む二本鎖DNAであり;
Yは、環状化させる個々のDNA分子に固有の配列を有する二本鎖DNA配列であって、nは1以上40以下の整数であり、N1~Nnは、それぞれ同一であっても異なっていてもよく、dAMP、dCMP、dGMPおよびdTMPからなる群から選択されるデオキシリボヌクレオチドであり、N’1~N’nは、前記N1~Nnに対してそれぞれ下記:

- y1およびy2がニック作成酵素認識サイトを含む請求項4記載の方法。
- ニック作成酵素がNb.BtsIまたはNt.BsqQIである請求項5記載の方法。
- y1およびy2が制限酵素サイトを含む請求項5記載の方法。
- 下記構造y1-Y-y2-Xを有する互いに相補的な二本鎖DNAからなる環状DNA作成用アダプター:
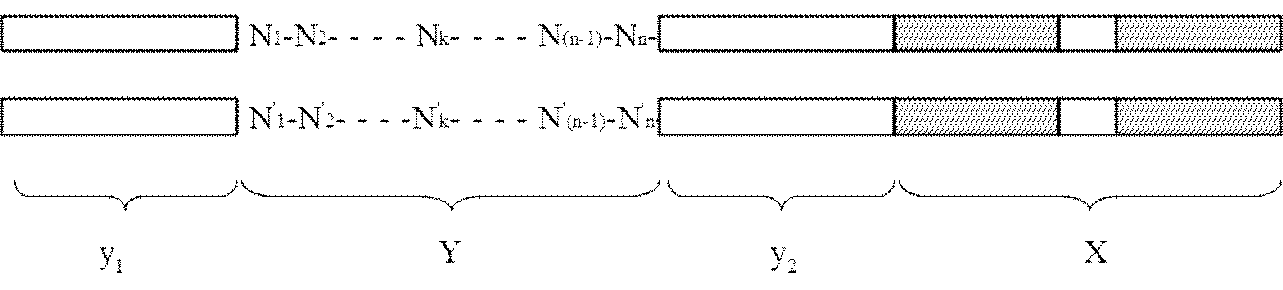
Xは、パリンドローム配列を認識する制限酵素サイトである二本鎖DNAであり;
y1およびy2は、互いに逆向きに配向した互いに同一のニック作成酵素認識サイトまたは制限酵素サイトを含む二本鎖DNAであり;
Yは、環状化させる個々のDNA分子に固有の配列を有する二本鎖DNA配列であって、nは1以上40以下の整数であり、N1~Nnは、それぞれ同一であっても異なっていてもよく、dAMP、dCMP、dGMPおよびdTMPからなる群から選択されるデオキシリボヌクレオチドであり、N’1~N’nは、前記N1~Nnに対してそれぞれ下記:

- y1およびy2がニック作成酵素認識サイトを含む請求項8記載のアダプター。
- ニック作成酵素がNb.BtsIまたはNt.BsqQIである請求項9記載のアダプター。
- y1およびy2が制限酵素サイトを含む請求項8記載のアダプター。
- 請求項8に記載のXにおける制限酵素サイトと同一の制限酵素サイトを含む二本鎖DNAからなるアダプター(A)と、請求項8~10のいずれかに記載のアダプター(B)とを含む、環状DNA作成用キット。
- 請求項1から7いずれか記載の方法を用いる、cDNAライブラリーの作成方法。
- 以下の工程を含む、請求項1から7いずれか記載の方法を用いて得られた環状DNA分子をメイトペア法に供することにより遺伝子を同定する方法:
1)請求項1から7いずれか記載の方法を用いて得られた環状DNA分子における、アダプター(B)の両側に隣接するそれぞれ15塩基以上600塩基以下の塩基配列を解読する工程;および、
2)解読した塩基配列を既知の遺伝子の両末端の配列と比較することにより、環状DNA分子に含まれる遺伝子を同定する工程。 - 以下の工程を含む、請求項1から7いずれか記載の方法を用いて得られた環状DNA分子をメイトペア法に供することにより融合遺伝子を検出する方法:
1)請求項1から7いずれか記載の方法を用いて得られた環状DNA分子における、アダプター(B)の両側に隣接するそれぞれ15塩基以上600塩基以下の塩基配列を解読する工程;および、
2)解読した塩基配列を既知の遺伝子の両末端の配列と比較する工程、ここで、両末端の遺伝子が既知の相異なる遺伝子に対応する場合、環状DNA分子に含まれる遺伝子は融合遺伝子であると同定される。 - アダプター(B)の両側に隣接する両側の配列が、既知融合遺伝子の両側の末端に対応する、請求項15に記載の融合遺伝子の検出方法。
- 請求項15に記載の方法により検出された融合遺伝子をマーカーとして用いる、該融合遺伝子の発現を特徴とする疾患を検出する方法。
- アダプター(B)の両側に隣接する配列が、相異なる遺伝子の末端配列に対応し、かつ既知融合遺伝子の両側の末端には対応しないことにより、環状DNA分子に含まれる遺伝子が新規融合遺伝子であると同定される請求項15に記載の方法。
- 請求項18に記載の方法により検出された新規融合遺伝子の創薬スクリーニングにおける使用。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012531803A JP5780527B2 (ja) | 2010-09-02 | 2011-08-22 | 単分子dnaから形成される環状dnaの作成方法 |
| EP11821594.6A EP2620497B1 (en) | 2010-09-02 | 2011-08-22 | Method for producing circular dna formed from single-molecule dna |
| KR1020137006174A KR101583589B1 (ko) | 2010-09-02 | 2011-08-22 | 단분자 dna로 형성되는 환상 dna의 작성 방법 |
| US13/819,396 US8962245B2 (en) | 2010-09-02 | 2011-08-22 | Method for producing circular DNA formed from single-molecule DNA |
| CN201180042478.5A CN103119162B (zh) | 2010-09-02 | 2011-08-22 | 用于产生由单分子dna形成的环状dna的方法 |
| US14/594,458 US9434941B2 (en) | 2010-09-02 | 2015-01-12 | Method for producing circular DNA formed from single-molecule DNA |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010196719 | 2010-09-02 | ||
| JP2010-196719 | 2010-09-02 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/819,396 A-371-Of-International US8962245B2 (en) | 2010-09-02 | 2011-08-22 | Method for producing circular DNA formed from single-molecule DNA |
| US14/594,458 Continuation US9434941B2 (en) | 2010-09-02 | 2015-01-12 | Method for producing circular DNA formed from single-molecule DNA |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012029577A1 true WO2012029577A1 (ja) | 2012-03-08 |
Family
ID=45772675
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/068856 WO2012029577A1 (ja) | 2010-09-02 | 2011-08-22 | 単分子dnaから形成される環状dnaの作成方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8962245B2 (ja) |
| EP (1) | EP2620497B1 (ja) |
| JP (1) | JP5780527B2 (ja) |
| KR (1) | KR101583589B1 (ja) |
| CN (1) | CN103119162B (ja) |
| WO (1) | WO2012029577A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013031700A1 (ja) * | 2011-08-31 | 2013-03-07 | 学校法人 久留米大学 | Dna分子の環状化において単分子による環状化dnaのみを選別する方法 |
| JP2015521468A (ja) * | 2012-06-18 | 2015-07-30 | ニューゲン テクノロジーズ, インコーポレイテッド | 望まれない核酸配列のネガティブ選択のための組成物および方法 |
| US9249460B2 (en) | 2011-09-09 | 2016-02-02 | The Board Of Trustees Of The Leland Stanford Junior University | Methods for obtaining a sequence |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2620497B1 (en) * | 2010-09-02 | 2017-10-11 | Kurume University | Method for producing circular dna formed from single-molecule dna |
| EP3015554A1 (en) * | 2014-10-31 | 2016-05-04 | Rheinische Friedrich-Wilhelms-Universität Bonn | Gene expression analysis |
| US20170349893A1 (en) * | 2014-11-26 | 2017-12-07 | Bgi Shenzhen | Method and reagent for constructing nucleic acid double-linker single-strand cyclical library |
| US10479991B2 (en) | 2014-11-26 | 2019-11-19 | Mgi Tech Co., Ltd | Method and reagent for constructing nucleic acid double-linker single-strand cyclical library |
| EP3237616A1 (en) * | 2014-12-24 | 2017-11-01 | Keygene N.V. | Backbone mediated mate pair sequencing |
| CN106148323B (zh) * | 2015-04-22 | 2021-03-05 | 北京贝瑞和康生物技术有限公司 | 一种用于构建alk基因融合突变检测文库的方法和试剂盒 |
| EP3382034A1 (en) * | 2017-03-31 | 2018-10-03 | Rheinische Friedrich-Wilhelms-Universität Bonn | Gene expression analysis by means of generating a circularized single stranded cdna library |
| WO2021214258A1 (en) | 2020-04-22 | 2021-10-28 | Fabmid | Methods for circularizing linear double stranded nucleic acids |
| WO2023072958A1 (en) | 2021-10-25 | 2023-05-04 | Fabmid | Methods for circularizing linear double stranded nucleic acids and the products thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008295444A (ja) * | 2006-10-11 | 2008-12-11 | Astellas Pharma Inc | Eml4−alk融合遺伝子 |
| JP2008545448A (ja) * | 2005-06-06 | 2008-12-18 | 454 ライフ サイエンシーズ コーポレイション | 両末端配列決定(pairedendsequencing) |
| WO2009098037A1 (en) * | 2008-02-05 | 2009-08-13 | Roche Diagnostics Gmbh | Paired end sequencing |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04303303A (ja) | 1991-03-29 | 1992-10-27 | Fuji Heavy Ind Ltd | 高層ビルのごみ容器輸送装置 |
| US20080096255A1 (en) | 2004-07-02 | 2008-04-24 | Matthias Harbers | Method for Preparing Sequence Tags |
| WO2007145612A1 (en) | 2005-06-06 | 2007-12-21 | 454 Life Sciences Corporation | Paired end sequencing |
| WO2009032167A1 (en) | 2007-08-29 | 2009-03-12 | Illumina Cambridge | Method for sequencing a polynucleotide template |
| US7897344B2 (en) | 2007-11-06 | 2011-03-01 | Complete Genomics, Inc. | Methods and oligonucleotide designs for insertion of multiple adaptors into library constructs |
| US8352194B1 (en) | 2008-06-17 | 2013-01-08 | University Of South Florida | Method to identify cancer fusion genes |
| EP2487250A4 (en) | 2009-10-06 | 2013-03-20 | Fujirebio Kk | METHOD FOR DETECTION OF A FUSION GENE |
| JP5861244B2 (ja) | 2010-06-22 | 2016-02-16 | 公益財団法人がん研究会 | 新規ros1融合体の検出法 |
| JPWO2012014795A1 (ja) | 2010-07-26 | 2013-09-12 | アステラス製薬株式会社 | 新規ret融合体の検出法 |
| EP2620497B1 (en) * | 2010-09-02 | 2017-10-11 | Kurume University | Method for producing circular dna formed from single-molecule dna |
| WO2013006195A1 (en) | 2011-07-01 | 2013-01-10 | Htg Molecular Diagnostics, Inc. | Methods of detecting gene fusions |
| CA2848369A1 (en) | 2011-08-04 | 2013-02-07 | National Cancer Center | Fusion gene of kif5b gene and ret gene, and method for determining effectiveness of cancer treatment targeting fusion gene |
-
2011
- 2011-08-22 EP EP11821594.6A patent/EP2620497B1/en not_active Not-in-force
- 2011-08-22 US US13/819,396 patent/US8962245B2/en not_active Expired - Fee Related
- 2011-08-22 WO PCT/JP2011/068856 patent/WO2012029577A1/ja active Application Filing
- 2011-08-22 JP JP2012531803A patent/JP5780527B2/ja active Active
- 2011-08-22 KR KR1020137006174A patent/KR101583589B1/ko not_active IP Right Cessation
- 2011-08-22 CN CN201180042478.5A patent/CN103119162B/zh not_active Expired - Fee Related
-
2015
- 2015-01-12 US US14/594,458 patent/US9434941B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008545448A (ja) * | 2005-06-06 | 2008-12-18 | 454 ライフ サイエンシーズ コーポレイション | 両末端配列決定(pairedendsequencing) |
| JP2008295444A (ja) * | 2006-10-11 | 2008-12-11 | Astellas Pharma Inc | Eml4−alk融合遺伝子 |
| JP4303303B2 (ja) | 2006-10-11 | 2009-07-29 | アステラス製薬株式会社 | Eml4−alk融合遺伝子 |
| WO2009098037A1 (en) * | 2008-02-05 | 2009-08-13 | Roche Diagnostics Gmbh | Paired end sequencing |
Non-Patent Citations (7)
| Title |
|---|
| "Shikkan Idenshi no Tansaku to Chokosoku Sequence", JIKKEN IGAKU EXTRA EDITION, vol. 27, no. 12, 2009, pages 113.1929 - 143.1959 |
| BASHIR ET AL., PLOS COMPUTATIONAL BIOLOGY, vol. 4, no. 4, April 2008 (2008-04-01), pages EL000051 |
| CHINNAIYAN ET AL., SCIENCE, vol. 310, 2005, pages 644 - 648 |
| MITELMAN ET AL., NATURE GENETICS, vol. 36, no. 4, 2004, pages 331 - 334 |
| RUAN Y. ET AL.: "Multiplex parallel pair-end-ditag sequencing approaches in system biology", WILEY INTERDISCIP. REV. SYST. BIOL. MED., vol. 2, no. 2, 2010, pages 224 - 234, XP002680213 * |
| SODA ET AL., NATURE, vol. 448, 2007, pages 561 - 566 |
| TANPAKUSHITSU KAKUSAN KOSO, August 2009 (2009-08-01), pages 1233 - 1247,1271-1275 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013031700A1 (ja) * | 2011-08-31 | 2013-03-07 | 学校法人 久留米大学 | Dna分子の環状化において単分子による環状化dnaのみを選別する方法 |
| US9416358B2 (en) | 2011-08-31 | 2016-08-16 | Kurume University | Method for exclusive selection of circularized DNA from monomolecular DNA in circularizing DNA molecules |
| US9249460B2 (en) | 2011-09-09 | 2016-02-02 | The Board Of Trustees Of The Leland Stanford Junior University | Methods for obtaining a sequence |
| GB2496016B (en) * | 2011-09-09 | 2016-03-16 | Univ Leland Stanford Junior | Methods for obtaining a sequence |
| US9725765B2 (en) | 2011-09-09 | 2017-08-08 | The Board Of Trustees Of The Leland Stanford Junior University | Methods for obtaining a sequence |
| JP2015521468A (ja) * | 2012-06-18 | 2015-07-30 | ニューゲン テクノロジーズ, インコーポレイテッド | 望まれない核酸配列のネガティブ選択のための組成物および方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20130101508A (ko) | 2013-09-13 |
| EP2620497A1 (en) | 2013-07-31 |
| US20150197745A1 (en) | 2015-07-16 |
| CN103119162A (zh) | 2013-05-22 |
| JPWO2012029577A1 (ja) | 2013-10-28 |
| US20130164757A1 (en) | 2013-06-27 |
| US8962245B2 (en) | 2015-02-24 |
| KR101583589B1 (ko) | 2016-01-08 |
| CN103119162B (zh) | 2016-09-07 |
| EP2620497A4 (en) | 2014-02-19 |
| US9434941B2 (en) | 2016-09-06 |
| EP2620497B1 (en) | 2017-10-11 |
| JP5780527B2 (ja) | 2015-09-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5780527B2 (ja) | 単分子dnaから形成される環状dnaの作成方法 | |
| AU2018212756B2 (en) | Construction of next generation sequencing (NGS) libraries using competitive strand displacement | |
| CN108431233B (zh) | Dna文库的高效率构建 | |
| JP7008407B2 (ja) | ヌクレアーゼ、リガーゼ、ポリメラーゼ、及び配列決定反応の組み合わせを用いた、核酸配列、発現、コピー、またはdnaのメチル化変化の識別及び計数方法 | |
| EP4043584A1 (en) | Improved adapters, methods, and compositions for duplex sequencing | |
| US20190078150A1 (en) | Methods and Kits for Tracking Nucleic Acid Target Origin for Nucleic Acid Sequencing | |
| US20210363570A1 (en) | Method for increasing throughput of single molecule sequencing by concatenating short dna fragments | |
| US20230017673A1 (en) | Methods and Reagents for Molecular Barcoding | |
| JP2023513606A (ja) | 核酸を評価するための方法および材料 | |
| JP6066209B2 (ja) | Dna分子の環状化において単分子による環状化dnaのみを選別する方法 | |
| JP2023540782A (ja) | 遺伝子ライブラリーの高効率構築および遺伝子解析のためのアダプターおよび方法 | |
| US20210147926A1 (en) | Construction of next generation sequencing (ngs) libraries using competitive strand displacement | |
| KR20240004397A (ko) | 다중 라이브러리의 동시 유전자 분석을 위한 조성물 및 방법 | |
| WO2022251510A2 (en) | Oligo-modified nucleotide analogues for nucleic acid preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180042478.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11821594 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012531803 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13819396 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137006174 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011821594 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011821594 Country of ref document: EP |